Campo da Invenção
[0001] A presente invenção se refere ao campo de composições farmacêuticas com liberação de ingrediente ativo controlada para ingredientes ativos com boa solubilidade em água.
Técnica Anterior
[0002] O EP-A 0 463 877 descreve composições farmacêuticas com liberação de ingrediente ativo retardada consistindo em um núcleo com um ingrediente farmacêutico ativo e uma película de revestimento em monocamada que compreende um sal repelente de água e um copolímero insolúvel em água de acrilato de etila, metacrilato de metila e cloreto de metacrilato de trimetilamonioetila. O sal repelente de água pode ser, por exemplo, estearato de Ca ou estearato de Mg. Gráficos de liberação sigmoidal são obtidos.
[0003] Os EP-A 0 225 085, EP-A 0 122 077 e EP-A 0 123 470 descrevem o uso de ácido orgânico em núcleos de medicamento que são providos com vários revestimentos de soluções orgânicas. Características de liberação essencial mente sigmoidal resultam. O EP-A 0 463 370 descreve composições farmacêuticas com liberação de ingrediente ativo retardada consistindo em um núcleo com um ingrediente farmacêutico ativo e um ácido orgânico e uma película de revestimento externa que foi aplicada através de pulverização aquosa e é um copolímero de acrilato de etila, metacrilato de metila e cloreto de metacrilato trimetilamonioetila. Neste caso, gráficos de liberação sigmoidal são da mesma maneira obtidos.
[0004] A EP 1 117 387 B1 descreve um sistema similar ao EP-A 0436 370. A diferença essencial é o uso dos ácidos orgânicos na forma de sal, sendo então possível ter uma influência benéfica em particular sobre a duração da fase lag e sobre o declive dos gráficos de liberação sigmoidal. Auxiliares de processamento possíveis que são mencionados são inter aliasílica moída e formadores de poro.
Problema e Solução
[0005] Com um grande número de formas farmacêuticas e ingredientes ativos é terapeuticamente válido ter características de liberação sigmoidal com uma fase inicial com liberação de ingrediente ativo retardada (fase lag), uma fase de liberação principal subsequente (fase de pulso) e uma fase acelerada ("run-out").A intenção geral é que a liberação de ingrediente ativo na fase lag seja a mais baixa possível e a liberação do ingrediente ativo na fase de pulso subsequente seja a mais rápida possível. Este objetivo é atingido em parte pelos ensinamentos técnicos do EP-A 0 436 370 e da EP 1 117 387 B1. Na EP 1 117 387 B1, bons resultados são obtidos em particular com acetato de sódio. Há, no entanto, uma necessidade contínua de aperfeiçoamento. Um problema foi considerado como sendo desenvolver mais as preparações farmacêuticas com liberação de ingrediente ativo sigmoidal que são descritas no EP-A 0 436 370 e na EP 1 117 387 B1 a fim de se aproximar dos objetivos mencionados acima.
[0006] O problema foi resolvido através de uma preparação farmacêutica compreendendo um núcleo com um ingrediente ativo e com um ácido orgânico e/ou o sal de um ácido orgânico um revestimento que envolver o núcleo e que compreende um teor de polímero de copolímeros de (met)acrilato que não têm mais do que 15% em peso de grupos catiônicos ou aniônicos, e que compreende pelo menos 60% em peso de um copolímero de (met)acrilato que é composto de unidades polimerizadas radicalmente livres de 93 a 98% em peso de ésteres Ci- a C4-alquílicos de ácido acrílico ou metacrílico e 7 a 2% em peso de monômeros de (met)acrilato tendo um grupo amónio quaternário no radical alquila, sendo que o ingrediente ativo tem uma solubilidade em água de pelo menos 10 g/L a 20° C e o revestimento compreende partículas de dióxido de silício tendo um tamanho de partícula médio na faixa de 1 a 50 pm.
Implementação da Invenção Núcleos (a)
[0007] No caso mais simples, o núcleo pode ser composto apenas do ingrediente ativo e do ácido orgânico e/ou do sal do ácido orgânico, mas geralmente ele compreende adicionalmente um veículo, por exemplo, "nonpareil", e excipientes farmacêuticos tal como, por exemplo, sílica coloidal ou polivinilpirrolidona (PVP).
[0008] O núcleo (a) pode consistir em, por exemplo: ingrediente ativo em uma quantidade de 97,5% a 2,5, de preferência 80 a 5% em peso com base no peso do núcleo um ácido orgânico e/ou um ou mais sais de ácidos orgânicos em uma quantidade de 2,5 a 97,5, de preferência 5 a 80, em particular 10 a 50 % em peso com base no peso do núcleo excipientes opcionalmente farmacêuticos em uma quantidade de 0 a 95, de preferência 10 a 50% em peso com base no peso do núcleo opcionalmente um veículo com uma proporção do peso do núcleo de 0 a 95, de preferência 10 a 50, % em peso.
[0009] Os núcleos podem ser produzidos, por exemplo, através de compressão direta, extrusão e subsequente arredondamento, granulação a úmido ou a seco ou peletização direta (por exemplo, em discos) ou através de ligação de pós (colocação em camada de pó) sobre contas livres de ingrediente ativo (nonpareils) ou partículas contendo ingrediente ativo.
[00010] Os excipientes farmacêuticos que estão contidos em adição ao ingrediente ativo podem ser, por exemplo, ligantes tal como celulose e seus derivados, polivinilpirrolidona (PVP), gelatina, (met)acrilatos, amido e derivados dos mesmo ou açúcares.
[00011] Os núcleos podem ser homogêneos ou ter uma estrutura em camada, caso em que o ingrediente ativo está de preferência localizado na camada externa. É particularmente preferido que o ácido orgânico e/ou o sal do ácido orgânico forme a camada externa do núcleo.
Ácidos Orgânicos
[00012] Os ácidos orgânicos empregados devem ser toxicologicamente aceitáveis e úteis em medicamentos. O tipo preferido depende da formulação específica. Ácidos orgânicos tal como ácido cítrico, ácido fumárico, ácido fórmico, ácido succínico, ácido acético, ácido maléico, ácido tartárico, ácido glutárico ou ácido láctico são preferidos.
[00013] Ácido succínico é particularmente adequado para os propósitos da invenção. Ácido cítrico é, a princípio, da mesma maneira adequado, embora os perfis de liberação obtidos em meios tamponados que correspondem aproximadamente a condições fisiológicas não sejam tão baixos quanto com succinato. Ácido acético pode ocasionalmente levar a problemas de estabilidade que podem aparecer durante armazenamento das formas farmacêuticas. Nenhum de tais problemas é conhecido com o uso de ácido succínico.
[00014] O tipo de ácido controla o declínio do gráfico de liberação de ingrediente ativo especialmente em gráficos de liberação sigmoidal.
[00015] Os ácidos orgânicos podem estar presentes nas formulações de acordo com a invenção de preferência como camada externa do núcleo, ligados por ligantes. Eles podem ser aplicados através de pulverização a partir da solução ou através de liberação de pó com adição simultânea de solução ligante.
[00016] No entanto, em casos individuais, variantes em que o ingrediente ativo é aplicado a uma mistura com ácidos orgânicos, ou uma camada de vedação é aplicada entre a camada de ingrediente ativo e a camada de sal, são também válidos. O ácido orgânico pode ser também aplicado por último ao núcleo, de modo que ele forme a camada externa.
[00017] A quantidade do(s) ácido(s) orgânico(s) como uma proporção do peso do núcleo pode ser 2,5% em peso a 97,5% em peso, de preferência 5 a 80% em peso, em particular 10 a 50% em peso.
Sais de Ácidos Orgânicos
[00018] Sais de ácidos orgânicos são preferidos aos ácidos orgânicos. Na maioria dos casos, uma liberação de ingrediente ativo menor durante a fase lag e subsequentemente uma liberação de ingrediente ativo mais rápida são observadas quando do uso de ácidos orgânicos em forma de sal comparado aos ácidos orgânicos.
[00019] Os sais empregados de ácidos orgânicos devem ser toxicologicamente aceitáveis e úteis em medicamentos. Sais de metal alcalino (amónio, lítio, sódio, potássio) são preferidos. O tipo preferido depende da formulação específica; além da funcionalidade de acordo com a invenção, no entanto, os efeitos farmacológicos dos íons devem ser também levados em consideração. Sais de ácidos orgânicos fracos tal como ácido succínico, ácido cítrico, ácido fumárico, ácido fórmico, ácido acético, ácido maléico, ácido tartárico, ácido glutárico ou ácido láctico são preferidos.
[00020] Succinato de sódio é particularmente adequado para os propósitos da invenção. Citrato de sódio é, a princípio, da mesma maneira adequado, embora os perfis de liberação obtidos em meios tamponados que correspondem aproximadamente a condições fisiológicas não sejam tão baixos quanto com succinato de sódio. Acetato de sódio pode ocasionalmente levar a problemas de estabilidade que pode aparecer durante armazenamento das formas farmacêuticas. Nenhum de tais problemas é conhecido ainda com o uso de succinato de sódio.
[00021] O tipo de ácido controla o declínio do gráfico de dose de ingrediente ativo especialmente em gráficos de liberação sigmoidal.
[00022] Os sais podem estar presentes nas formulações de acordo com a invenção como camada externa do núcleo, ligados por ligantes. Eles podem ser aplicados através de pulverização a partir da solução ou através de liberação em pó com adição simultânea de solução ligante.
[00023] No entanto, em casos individuais, variantes em que o ingrediente ativo é aplicado a uma mistura com os sais, ou uma camada de vedação é aplicada entre a camada de ingrediente ativo e a camada de sal, são também válidos. O sal do ácido orgânico pode ser também aplicado ao núcleo, de modo que ele forma a camada externa.
[00024] A quantidade dos sais do(s) ácido(s) orgânico(s) como uma proporção do peso do núcleo pode ser 2,5% em peso a 97,5% em peso, de preferência 5 a 80% em peso, em particular 10 a 50% em peso.
Revestimento b)
[00025] O revestimento b) consiste em um ou mais copolímeros de (met)acrilato, partículas de SÍO2 e em que apropriado excipientes farmacêuticos convencionais tal como, por exemplo, plasticizantes, pigmentos, agentes umectantes, agentes de liberação do molde, etc. O revestimento externo de preferência envolve o núcleo diretamente sem camadas adicionais estando presentes entre o núcleo e a película de revestimento.
[00026] O teor de polímero do revestimento é convertido em uma película junto com as partículas de SÍO2 contidas e excipientes adicionais tal como, por exemplo, plasticizantes que estão presentes em que apropriado, e forma um revestimento ou película de revestimento contínuo. O revestimento ou película de revestimento em sua totalidade controla, junto com o ácido orgânico e/ou os sais do mesmo presentes no núcleo, a liberação de ingrediente ativo.
Teor de Polímero do Revestimento b)
[00027] O teor de polímero do revestimento b) compreende pelo menos 60% em peso, de preferência 85 a 95% em peso, de um ou opcionalmente também mais copolímeros de (met)acrilato de unidades de monômero polimerizadas radicalmente livres consistindo em 93 a 98% em peso de Ci- a C4-alquil ésteres de ácido acrílico ou de ácido metacrílico e 7 a 2% em peso de monômeros de (met)acrilato tendo um grupo amónio quaternário no radical alquila (tipo EUDRAGIT® RS). O teor de polímero do revestimento pode também em que apropriado consistir em 100% do tipo de polímero acima.
[00028] O teor de polímero do revestimento pode perfazer de preferência de 10 a 200, de preferência 20 a 100% em peso com base no peso do núcleo.
[00029] O teor de polímero do revestimento baseado no revestimento deve perfazer pelo menos 50% em peso. O revestimento pode, em que apropriado, consistir apenas em ditos copolímeros de (met)acrilato e no teor de SiO2. No entanto, geralmente, o revestimento vai compreender em adição ao teor de SiO2 aditivos farmaceuticamente comuns adicionais tal como, por exemplo, plasticizantes ou pigmentos.
[00030] O princípio da invenção é baseado em uma interação presumida entre os ingredientes essenciais do núcleo e os ingredientes essenciais do revestimento.
[00031] O efeito de acordo com a invenção surpreendentemente acontece apenas com os ingredientes ativos que têm uma solubilidade em água de pelo menos 10 g/L, de preferência pelo menos 30 g/L, com mais preferência pelo menos 100 g/L. A fim de assegurar esta interação, o dito copolímero de (met)acrilato deve ser pelo menos 50% em peso envolvido na estrutura do revestimento a fim de obter a interação desejada. Tais copolímeros de (met)acrilato estão comercial mente disponíveis e têm sido usados por um longo tempo para revestimentos de diminuição de liberação. Eles são praticamente insolúveis em água. Eles podem ser usados sozinhos ou em uma mistura com outros copolímeros de (met)acrilato.
[00032] A fim de obter de acordo com a invenção características de liberação de ingrediente ativo sigmoidal, o teor de polímero do revestimento b) deve consistir em pelo menos 60, de preferência pelo menos 85, % em peso ou 100% em peso do dito tipo de copolímero (tipo Eudragit® RS).
[00033] Ésteres C1- a C4-alquílicos preferidos de ácido acrílico ou de ácido metacrílico são acrilato de metila, acrilato de etila, acrilato de butila, metacrilato de butila e metacrilato de metila.
[00034] O monômero de (met)acrilato particularmente preferido tendo um grupo amónio quaternário é cloreto de metacrilato de 2- trimetilamonioetila.
[00035] Um copolímero correspondente pode ser composto, por exemplo, de 50 a 70% em peso de metacrilato de metil, 20 a 40% em peso de acrilato de etila e 7 a 2% em peso de cloreto de metacrilato de trimetilamonioetila.
[00036] Um copolímero preferido compreende 65% em peso de metacrilato de metila, 30% em peso de acrilato de etila e 5% em peso de cloreto de metacrilato de 2-trimetilamonioetila (EUDRAGIT® RS).
Misturas de Copolímeros de (Met)Acrilato
[00037] O teor de polímero do revestimento pode também estar na forma de uma mistura de copolímeros de (met)acrilato. Os copolímeros de (met)acrilato usados em adição para a mistura não devem ter mais do que 15% em peso de grupos catiônicos ou aniônicos. Com um teor de mais de 15% em peso de grupos catiônicos ou aniônicos, isto é, grupos básicos ou grupos ácidos, no radical alquila, as interações dos componentes uns com os outros são influenciadas de uma maneira indesejável ou dificilmente previsível.
[00038] No caso de uma mistura, a proporção do copolímero de (met)acrilato das unidades de monômero polimerizadas radicalmente livre consistindo em 93 a 98% em peso de ésteres C1- a C4-alquílicos de ácido acrílico ou de ácido metacrílico e 7 a 2% em peso de monômeros de (met)acrilato tendo um grupo amónio quaternário no radical alquila (tipo EUDRAGIT® RS) é pelo menos 60, de preferência 85 a 95, % em peso, em cada caso com base no peso do núcleo. A proporção do(s) polímero(s) misturado(s) é até 40% em peso, de preferência 5-15% em peso, com as proporções dos polímeros misturados totalizando 100% em peso.
[00039] Um copolímero de (met)acrilato adequado para uma mistura pode ser composto de, por exemplo, unidades de monômero polimerizadas radicalmente livres de 85 a menos do que 93% em peso de ésteres C1- a C4-alquílicos de ácido acrílico ou de metacrílico e mais do que 7 a 15% em peso de monômeros de (met)acrilato tendo um grupo amónio quaternário no radical alquila. Tais copolímeros de (met)acrilato estão comercial mente disponíveis e têm sido usados por um longo tempo para revestimentos de diminuição de liberação (tipo EU DRAG IT® RL). A proporção na mistura pode ser de até 40% em peso, de preferência 5 a 15% em peso.
[00040] Um copolímero especificamente adequado para uma mistura compreende, por exemplo, 60% em peso de metacrilato de metila, 30% em peso de acrilato de etila e 10% em peso de cloreto de metacrilato de 2-trimetilamonioetila (EUDRAGIT® RL).
[00041] Um copolímero de (met)acrilato adequado adicional para uma mistura consiste em 95 a 100, em particular mais do que 95 a 100, % em peso de ésteres C1- a C4-alquílicos de ácido acrílico ou metacrílico e até 5% em peso, ou 0 a 5, em particular 0 a menos do que 5, % em peso de ácido acrílico ou metacrílico. Tais copolímeros de (met)acrilato estão comercialmente disponíveis (tipo EUDRAGIT® NE).
[00042] O teor de copolímero de (met)acrilato da película de revestimento externa b) pode ser, por exemplo, uma mistura de:
[00043] 60 a 99, de preferência 85 a 95, % em peso de um copolímero de (met)-acrilato que consiste em 93 a 98% em peso de C1- a C4-alquil ésteres de ácido acrílico ou de ácido metacrílico e 2 a 7% em peso de monômeros de (met)acrilato tendo um grupo de amónio quaternário no radical alquila, e 1-40, de preferência 5 a 15, % em peso de um copolímero de (met)-acrilato que é composto de 85 a menos do que 93% em peso de ésteres C1- a C4-alquilícos de ácido acrílico ou de metacrílico e mais do que 7 a 15% em peso de monômeros de (met)acrilato tendo um grupo amónio quaternário no radical alquila.
[00044] Ésteres C1- a C4-alquilícos preferidos de ácido acrílico ou de metacrílico são acrilato de metila, acrilato de etila, acrilato de butila, metacrilato de butila e metacrilato de metila.
[00045] O monômero de (met)acrilato particularmente preferido tendo um grupo de amónio quaternário é cloreto de metacrilato de 2-trimetilamonioetila.
Preparação dos Copolímeros de (Met)Acrilato em Geral
[00046] Os copolímeros de (met)acrilato podem ser obtidos de uma maneira conhecida per se através de polimerização de massa, solução, conta ou emulsão radicalmente livres. Eles podem estar na forma de, por exemplo, grânulos extrudados, pó moído, solução ou dispersão.
Revestimentos
[00047] A liberação de polímero depende do tamanho e da superfície dos núcleos, da solubilidade dos ingredientes ativos e do perfil de liberação desejado. O teor de polímero do revestimento com base no peso do núcleo pode ser 10 a 200, de preferência 15 a 100% em peso.
[00048] Os revestimentos podem ser aplicados a uma pluralidade de camadas ou como mistura. Misturas dos polímeros permitem que gradientes particulares sejam postos na segunda fase do perfil de liberação. O teor de grupos amónio quaternário no revestimento controla a permeabilidade e então a taxa de difusão de substâncias dissolvidas (McGinity, Ed., Aqueous Polymeric Coatings for Pharmaceutical Dosage Forms, Marcel Dekker, Inc., Capítulo 4, pp. 208-216). Uma proporção maior de grupos amónio quaternário hidrofílicos significa uma taxa de liberação mais rápida. Uma possibilidade adicional para controle de liberação de ingrediente ativo na segunda fase do perfil de liberação é obtida desta maneira.
Camadas Externas Adicionais
[00049] A preparação de acordo com a invenção pode adicionalmente ser envolvida por um copolímero de (met)acrilato que compreende 5 a 60% em peso de resíduos de ácido metacrílico. É possível desta maneira prover a preparação com uma cobertura que é resistente a suco gástrico mas solúvel em suco intestinal.
[00050] Também adequados são copolímeros de (met)acrilato aniônicos compostos de 40 a 60% em peso de ácido metacrílico e 60 a 40% em peso de metacrilato de metila ou 60 a 40% em peso de acrilato de etila (tipos EUDRAGIT® L ou EUDRAGIT® L100-55).
[00051] EUDRAGIT® L é um copolímero de 50% em peso de metacrilato de metila e 50% em peso de ácido metacrílico.
[00052] EUDRAGIT® L 100-55 é um copolímero de 50% em peso de acrilato de etil e 50% em peso de ácido metacrílico. EUDRAGIT® L 30D-55 é uma dispersão compreendendo 30% em peso de EUDRAGIT® L 100-55.
[00053] Da mesma maneira adequados são copolímeros de (met)acrilato aniônicos compostos de 20 a 40% em peso de ácido metacrílico e 80 a 60% em peso de metacrilato de metila (tipo EUDRAGIT® S).
[00054] Particularmente adequados são copolímeros de (met)acrilato consistindo em 10 a 30% em peso de metacrilato de metila, 50 a 70% em peso de metacrilato de metila e 5 a 15%, de preferência 8 a 12% em peso de ácido metacrílico (tipo EUDRAGIT® FS).
[00055] EUDRAGIT® FS é um copolímero de 25% em peso de metacrilato de metila, 65% em peso de acrilato de metila e 10% em peso de ácido metacrílico. EUDRAGIT® FS 30 D é uma dispersão compreendendo 30% em peso de EUDRAGIT® FS.
[00056] Adicionalmente adequado para os propósitos da invenção é um copolímero (vide WO 2003/072087) que é composto de 20 a 34% em peso de ácido metacrílico e/ou ácido acrílico, 20 a 69% em peso de acrilato de metila e 0 a 40% em peso de acrilato de etila e/ou em que apropriado 0 a 10% em peso de monômeros adicionais capazes de copolimerização vinílica, contanto que a temperatura de transição vítrea do copolímero de acordo com ISO 11357-2, subclausa 3.3.3, não seja mais do que 60° C. Este copolímero de (met)acrilato é particularmente adequado, por causa de seu bom alongamento em propriedades de quebra, para compressão de péletes em comprimidos.
[00057] O copolímero é em particular composto de unidades polimerizadas radicalmente livres de: 20 a 34, de preferência 25 a 33, particularmente de preferência 28 a 32% em peso de ácido metacrílico ou ácido acrílico, com preferência para ácido metacrílico, 20 a 69, de preferência 35 a 65, particularmente de preferência 35 a 55% em peso de acrilato de metila e onde apropriado 0 a 40, de preferência 5 a 35, particularmente de preferência 15 a 35, % em peso de etil acrilato junto, contanto que a temperatura de transição vítrea do copolímero (medida sem plasticizante adicionado com um teor de monômero residual (REMO) de menos do que 100 ppm, taxa de aquecimento 10° C/min, atmosfera de nitrogênio) de acordo com ISO 11357-2, subclasse 3.3.3 (Tmg), não seja mais do que 60, de preferência 40 a 60, particularmente de preferência 45 a 55° C.
[00058] O copolímero consiste em preferência substancialmente a exclusivamente nos monômeros de ácido metacrílico, acrilato de metila e acrilato de etila na proporção quantitativa indicada acima.
[00059] No entanto, é possível ainda, sem levar a um dano das propriedades essenciais, que pequenas quantidades na faixa de a partir de 0 a 10, por exemplo, 1 a 5% em peso dos monômeros adicionais capazes de copolimerização vinílica, tal como, por exemplo, metacrilato de metila, metacrilato de butila, acrilato de butila ou metacrilato de hidroxietila, estejam presentes.
[00060] É também possível empregar misturas dos ditos copolímeros para ajustar perfis de liberação ou sítios de liberação específicos.
[00061] Temperatura de transição vítrea significa aqui em particular a temperatura de ponto médio Tmg especificada na ISO 11357-2, subclasse 3.3.3. A medição acontece sem plasticizante adicionado, com teores de monômero residual (REMO) de menos do que 100 ppm, com uma taxa de aquecimento de 10° C/min e sob uma atmosfera de nitrogênio.
[00062] Os copolímeros são obtidos de uma maneira conhecida per se através de polimerização de massa, solução, conta ou emulsão por radical livre. Eles devem ser trazidos antes do processamento para a faixa de tamanho de partícula de acordo com a invenção através de processos de moagem, secagem ou pulverização adequados. Isto pode acontecer através de moagem simples de péletes extrudados e esfriados ou corte a quente.
[00063] O uso de pós pode ser vantajoso, especialmente no caso de mistura com pós ou líquidos adicionais. Itens adequados de aparelho para produção dos pós são familiares à pessoa versada na técnica, por exemplo, moedores a jato de ar, moedores de disco com pino, moedores de compartimento. É também possível onde apropriado incluir etapas de peneiramento apropriadas. Um moedor adequado para quantidades industriais grandes é, por exemplo, um moedor de jato oposto (Multi No. 4200) que é, por exemplo, operado com uma pressão de cerca de 0,6 mPa (6 bar).
[00064] Adicionalmente adequado para os propósitos da invenção são copolímeros (vide WO 2004/096185) compostos de 20 a 33% em peso de ácido metacrílico e/ou ácido acrílico, 5 a 30% em peso de acrilato de metila e 20 a 40% em peso de acrilato de etila e mais de 10 a 30% em peso de metacrilato de butila e em que apropriado 0 a 10% em peso de monômeros adicionais capazes de copolimerização vinílica, em que as proporções dos monômeros somam 100% em peso, contanto que a temperatura de transição vítrea do copolímero de acordo com ISO 11357-2, subclasse 3.3.3 (temperatura de ponto médio Tmg), seja 55 a 70° C.
[00065] Copolímeros deste tipo são particularmente adequados, por causa de suas boas propriedades mecânicas, para compressão de péletes em comprimidos.
[00066] O copolímero acima mencionado é composto em particular de unidades polimerizadas radicalmente livres de 20 a 33, de preferência 25 a 32, particularmente de preferência 28 a 31% em peso de ácido metacrílico ou ácido acrílico, com preferência para ácido metacrílico, 5 a 30, de preferência 10 a 28, particularmente de preferência 15 a 25% em peso de acrilato de metila, 20 a 40, de preferência 25 a 35, particularmente de preferência 28 a 32% em peso de acrilato de etila, e mais de 10 a 30, de preferência 15 a 25, particularmente de preferência 18 a 22% em peso de metacrilato de butila, juntas, em que a composição do monômero é escolhida de modo que a temperatura de transição vítrea do copolímero seja 55 a 70° C, de preferência 59 a 66, particularmente de preferência 60 a 65° C.
[00067] É também possível empregar misturas dos ditos copolímeros para ajustar os perfis de liberação ou sítios de liberação específicos.
[00068] Temperatura de transição vítrea significa aqui em particular a temperatura de ponto médio Tmg especificada na ISO 11357-2, subclausa 3.3.3. A medição acontece sem plasticizante, com teores de monômero residual (REMO) de menos do que 100 ppm, com uma taxa de aquecimento de 10° C/min e sob uma atmosfera de nitrogênio.
[00069] O copolímero consiste de preferência substancialmente a exclusivamente, até o ponto de 90, 95 ou 99 a 100% em peso, dos monômeros de ácido metacrílico, acrilato de metila, acrilato de etila e metacrilato de butila nas faixas quantitativas indicadas acima.
[00070] É, no entanto, possível ainda, sem necessariamente levar a um prejuízo das propriedades essenciais, que pequenas quantidades na faixa a partir de 0 a 10, por exemplo, 1 a 5% em peso de monômeros adicionais capazes de copolimerização vinílica, tal como, por exemplo, metacrilato de metila, acrilato de butila, metacrilato de hidroxietila, vinilpirrolidona, ácido vinil malônico, estireno, álcool vinílico, acetato de vinila e/ou derivados dos mesmos estejam presentes.
Partículas de Dióxido de Silício
[00071] O revestimento que envolve o núcleo compreende partículas de dióxido de silício tendo um tamanho de partícula médio na faixa a partir de 1 a 50 pm. As partículas de dióxido de silício presentes no revestimento também assumem a função de agente de não-pegajosidade. O efeito de acordo com a invenção, em particular uma diminuição da fase de pulso para menos do que 4 horas, acontece no uso de acordo com este tipo de partículas no revestimento. No uso de partículas de SÍO2 de tamanho de partícula médio diferente ou no uso de outros agentes de liberação do molde tal como, por exemplo, talco ou glicerol monoestearato (GMS) sozinho, os efeitos vantajosos da invenção surpreendentemente não parecem acontecer (vide exemplos).
[00072] O revestimento compreende partículas de dióxido de silício (partículas de SiO2) tendo um tamanho de partícula médio d50, que pode ser medido, por exemplo, por meio de difração a laser de acordo com ISO 13320-1, na faixa a partir de 1 a 50, de preferência a partir de 1 a 30, particularmente de preferência 1 a 10 pm. Os melhores resultados são alcançados com SiO2 precipitado e moído, por exemplo, produzido através do processo sol-gel. Este tipo de dióxido de silício é também designado de acordo com a Farmacopéia Alemã, DAB 1999, como Silicii dioxidium praecipitatum. Preferência é dada com certeza a produtos ou partículas de dióxido de silício de qualidade farmacêutica comprovada ou em qualidade farmacêutica que se obedeça às exigências de DAB 1999 em relação à pureza.
[00073] Inadequado para os propósitos da invenção é SiO2 coloidal do tipo Aerosil®, que é produzido por um processo de chama e geralmente tem tamanhos de partícula médios na faixa abaixo de 100 nm. O último pode, no entanto, ser empregado de forma não-crítica, por exemplo, como excipiente para formulação dos núcleos.
[00074] Quantidades preferidas empregadas das partículas de dióxido de silício são 5 a 50, particularmente de preferência 10 a 40, e especial mente 10 a 30% em peso de SiO2 com base no peso seco do(s) copolímero(s) de (met)acrilato no revestimento.
Excipientes Farmaceuticamente Comuns Adicionais
[00075] O núcleo e/ou o revestimento pode compreender excipientes farmaceuticamente comuns adicionais.
[00076] Aditivos adicionais servem em particular como auxiliares de processamento e pretendem assegurar um processo de produção confiável e reproduzível e estabilidade em armazenamento a longo prazo bom. Eles podem influenciar a permeabilidade dos revestimentos, que podem ser utilizados onde apropriado como parâmetro de controle adicional.
[00077] Os núcleos podem ser produzidos, por exemplo, através de compressão direta, extrusão e subsequente arredondamento, granulação a úmido ou a seco ou peletização direta (por exemplo, em discos) ou através de ligação de pós (colocação em camada de pó) sobre contas livres de ingrediente ativo (nonpareils) ou partículas contendo ingrediente ativo. Os excipientes farmacêuticos que estão presentes em adição ao ingrediente ativo podem ser, por exemplo, ligantes tal como celulose e seus derivados, polivinilpirrolidona (PVP), gelatina, (met)acrilatos, amido e derivados dos mesmos ou açúcares.
Plasticizantes:
[00078] Plasticizantes podem estar presentes em particular no revestimento ou nos copolímeros de (met)acrilato do revestimento. Substâncias adequadas como plasticizantes geralmente têm um peso molecular entre 100 e 20 000 e compreendem um ou mais grupos hidrofílicos na molécula, por exemplo, grupos hidroxila, éster ou amónio. Eles são frequentemente ésteres que são líquidos em temperatura ambiente: citratos, ftalatos, sebacatos ou óleo de rícino. Exemplos de plasticizantes adequados são citratos de alquila, por exemplo, citrato de trietila, glicerol ésteres, ftalatos de alquila, sebacatos de alquila, ésteres de sacarose, ésteres de sorbitano, sebacato de dietila, sebacato de dibutila e polietileno glicóis 4000 a 20 000. Plasticizantes preferidos são citrato de trietila e citrato de acetil trietila. Plasticizantes podem estar presentes, por exemplo, em quantidades de a partir de 5 a 25% em peso com base no polímero ou, onde apropriado, os polímeros do revestimento.
Agentes de não-peqaiosidade:
[00079] As partículas de dióxido de silício presentes no revestimento também assumem a função como agente de não- pegajosidade. Então, normalmente e de preferência, quaisquer agentes de não-pegajosidade adicionais são necessários ou presentes. No entanto, o possível uso adicional de agentes de não- pegajosidade adicionais não é precludido.
[00080] Essas substâncias, que geralmente têm propriedades lipofílicas, podem ser adicionadas às suspensões de spray e previnem, em adição ao SiO2 que está presente de acordo com a invenção, aglomeração dos núcleos durante o revestimento da película. É possível empregar, por exemplo, talco ou emulsificantes não-iônicos tal como, por exemplo, monoestearato glicerol, tendo um HLB entre 3 e 8. A quantidade pode estar entre 1 e 100% em peso com base no polímero. No entanto, deve sempre tomar cuidado para que nenhum prejuízo do perfil de liberação que é desejado de acordo com a invenção aconteça.
Excipientes adicionais:
[00081] Excipientes farmaceuticamente comuns adicionais que podem ser adicionados de uma maneira conhecida per se são, por exemplo, estabilizadores, corantes, antioxidantes, agentes umectantes, formadores de poro, pigmentos, agentes de brilho, etc.
Aplicação do Revestimento de Película
[00082] O processo de aplicação acontece por meio de aplicação por spray a partir da solução orgânica, ou dispersões aquosas através de fusão ou através de aplicação de pó direta. É crucial para implementação neste caso que revestimentos livres de poro, uniformes, sejam produzidos.
[00083] Quanto a processos de aplicação da técnica anterior, vide, por exemplo, Bauer, Lehmann, Osterwald, Rothgang, "Überzogene Arzneiformen"Wissenschaftliche Verlagsgesellschaft mbH Stuttgart, Capítulo 7, pp. 165-196.
[00084] Propriedades relevantes, testes necessários e especificações para a aplicação são listados em farmacopéias.
[00085] Detalhes devem ser encontrados em livros comuns, por exemplo: Voigt, R. (1984); Lehrbuch der pharmazeutischen Technologie; Verlag Chemie Weinheim - Beerfield Beach/Florida - Basle. Sucker, H., Fuchs, P., Speiser, P.: Pharmazeutische Technologie, Georg Thieme Verlag Stuttgart (1991), especialmente Capítulos 15 e 16, pp. 626-642. Gennaro, A., R. (Editor), Remington’s Pharmaceutical Sciences, Mack Publishing Co., Easton Pennsylvania (1985), Capítulo 88, p. 1567-1573. List, P.H. (1982): Arzneiformenilehre, Wissenschaftliche Verlagsgesellschaft mbH, Stuttgart.
Ingredientes Ativos (Substâncias Bioativas)
[00086] A invenção é adequada para ingredientes ativos que têm uma solubilidade em água de pelo menos 10 g/L, de preferência pelo menos 30 g/L, particularmente de preferência de pelo menos 50 g/L, especialmente de preferência de pelo menos 100 g/L, 200 g/L, 300 g/L ou 400 g/L a 20° C (solubilidade em água com base em métodos padrão tal como, por exemplo, Pharmeuropa - Technical Guide for the Elaboration of Monographs, 3a Edição (1999), Capítulo IV, Apêndice IV, com agitação vigorosa por 1 minuto, deixando descansar por 15 minutos a 20° C em água purificada). Os efeitos vantajosos da invenção surpreendentemente parecem não acontecer com ingredientes ativos tendo solubilidade menor em água, tal como, por exemplo, teofilina, com uma formulação que é de outro modo como reivindicado.
[00087] As substâncias farmacêuticas empregadas no contexto da invenção pretendem ser usadas sobre ou no corpo humano ou animal a fim de curar, aliviar, prevenir ou diagnosticar doenças, condições, dano físico ou sintomas patológicos; revelar a condição, o estado ou as funções do corpo ou estados mentais; substituir substâncias ativas ou fluidos do corpo produzidos pelo corpo humano ou animal; evitar, eliminar ou tornar menos prejudiciais patógenos, parasitas ou substâncias exógenas, ou influenciar a condição, o estado ou as funções do corpo, ou estados mentais.
[00088] Substâncias farmacêuticas convencionais podem ser encontradas em trabalhos de referência tal como, por exemplo, o Rote Liste ou o Merck Index. É possível de acordo com a invenção empregar todos os ingredientes ativos que cumprem com o efeito terapêutico desejado no sentido definido acima e têm estabilidade térmica adequada.
[00089] A preparação farmacêutica pode compreender, por exemplo, um ou mais dos ingredientes ativos que seguem com uma solubilidade em água de pelo menos 10 g/L a 20° C, onde apropriado na forma dos sais farmaceuticamente empregados, solúveis em água: acebutolol, amitriptilina, aripiprazol, atenolol, atropina, betaxolol, bisoprolol, bupavacaína, buproprion, butabarbital, carteolol, carvedilol, cefazolina, cefotaxima, clorfenaramina, clorpromazina, clindamicina, codeína, diltiazem, dimercaprol, difenidramina, dopamina, doxilamina, duloxetina, flexainida, fluoxetina, flufenazina, flurazepam, gentamicina, hidralazina, hidrocortisona, hidroquinona, hiosciamina, isoniazid, isoproterenol, canamicina, labetolol, linisoprila, metipranolol, mexiletina, morfina, nadolol, neomicina, norepinefrina, north pti li na, ondansetron, oxprenolol, oximetazolina, oximorfona, paroxetina, pembutolol, fenilefrina, pindolol, prednisolona, primaquina, propanolol, pirrocaína, sotalol, sulfadiazina, tamoxifeno, terbutalina, timolol, tramadol, trazodona, triflupromazina, tetraciclina, tubocurarina, venlafaxina e/ou verapamila. Preferência particular é dada a esses ingredientes ativos na forma dos sais farmaceuticamente empregados solúveis em água.
[00090] Ingredientes ativos particularmente preferidos para os propósitos da invenção são: cloridrato de fenilefrina e sulfato de terbutalina.
Formas de Administração e Modalidades Adicionais
[00091] É possível a princípio que as formas farmacêuticas descritas sejam usadas diretamente através de administração oral. No entanto, etapas de processamento adicionais de preferência seguem para formas multipartículas (forma de dosagem multiunidade):
[00092] Formas farmacêuticas revestidas produzidas de acordo com a invenção podem ser aplicadas como doses únicas em cápsulas de gelatina e bolsas (sachês) ou em recipientes multidose adequados com dispositivo de medição. Ingestão acontece em forma sólida ou suspensa em líquidos.
[00093] Compressão de grânulos, onde apropriado após misturas de excipientes adicionais, resulta em comprimidos que desintegram após ingestão e liberam as subunidades de liberação lenta. É da mesma maneira possível embutir aglomerados em polietileno glicol ou lipídeos para produzir supositórios ou formas farmacêuticas vaginais.
[00094] Os revestimentos externos podem adicionalmente ser combinados ou revestidos também com revestimentos da técnica anterior adicionais. Neste caso especial, o revestimento externo b) não é o revestimento mais externo. Adequados para este propósito são em particular copolímeros de (met)acrilato que compreendem 10 a 60% em peso de resíduos de ácido metacrílico e são de outro modo compostos, por exemplo, metacrilato de metila e/ou acrilato de etila (tipo EUDRAGIT® L ou S). É possível desta maneira em combinação com as formulações de acordo com a invenção adicionalmente obter propriedades ou formulações de mascaramento de gosto para liberações direcionadas ao colo.
Uso
[00095] A preparação ou composição farmacêutica de acordo com a invenção pode ser usada para produzir uma preparação ou composição farmacêutica ou uma forma farmacêutica para ingredientes ativos que têm uma solubilidade em água de pelo menos 10 g/L a 20° C, com a forma farmacêutica mostrando características de liberação de ingrediente ativo sigmoidal com uma fase lag, uma fase de pulso e uma fase acelerada, caracterizado por uma liberação de ingrediente ativo no aparelho de pá a 100 rpm em tampão de pH 6,8 de acordo com a Farmacopéia Européia de aproximadamente 10% durante a fase lag e uma liberação de ingrediente ativo subsequente de aproximadamente mais 80% dentro de menos de 4 horas na fase de pulso.
[00096] Para liberação de ingrediente ativo de acordo com USP, vide, em particular, USP 28-NF23, Capítulo Geral <711>, Dissolution, Aparelho 2 (Pá), Método <724>"Delayed Release (Enteric Coated) Articles-General General Drug Release Standard", Método B (100 rpm, 37° C), mas com tampão de pH 6,8 de acordo com a Farmacopéia Européia.
[00097] As características de liberação de ingrediente ativo sigmoidal são suficientemente bem conhecidas da pessoa versada na técnica, por exemplo, das EP-A 0 463 877, EP 1 117 387 B1 e EP-A 0 436 370.
Formas Farmacêuticas
[00098] A preparação de acordo com a invenção é adequada de uma maneira conhecida para produção de formas farmacêuticas. A preparação pode estar presente, por exemplo, em forma de pélete que pode ser processado por meio de excipientes farmaceuticamente comuns e de uma maneira conhecida per se para formas farmacêuticas multiparticuladas, em particular para comprimidos contendo pélete, minicomprimidos, cápsulas, sachês ou pós reconstituíveis.
[00099] A preparação pode de preferência ser comprimida na forma de életes, por exemplo, para dar um comprimido.
[000100] A preparação pode, por exemplo, em particular estar também na forma de péletes ou minicomprimidos que são introduzidos em uma cápsula de gelatina e envolvidos por ela.
Exemplos Copolímeros Usados Copolímero 1:
[000101] 65% em peso metacrilato de metila, 30% em peso acrilato de etila e 5% em peso de cloreto de metacrilato de 2- trimetilamonioetila (EUDRAGIT® RS).
Copolímero 2:
[000102] 60% em peso de metacrilato de metila, 30% em peso de acrilato de etila e 10% em peso de cloreto de metacrilato de 2- trimetilamonioetil (EUDRAGIT® RL).
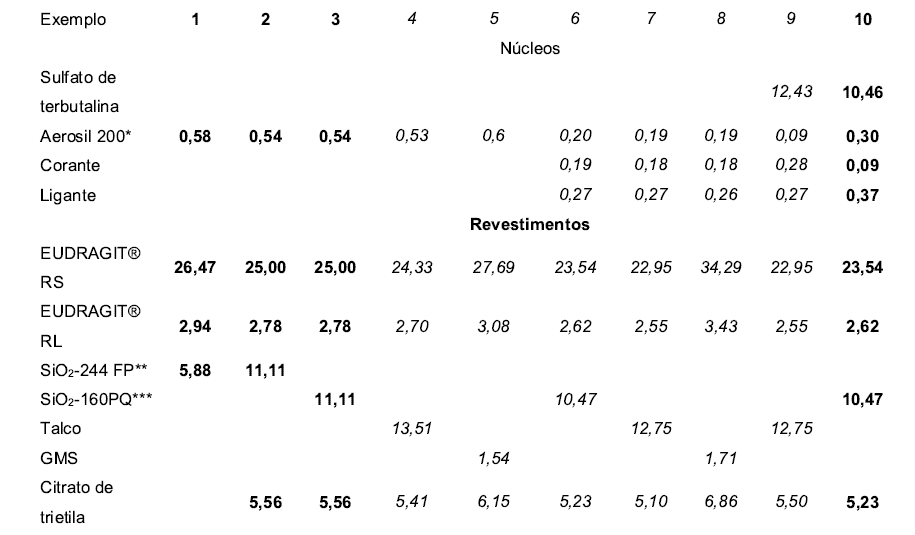
[000103] Os dados na Tabela 1 são baseados em matéria seca.
Solubilidade dos ingredientes ativos em água:
[000104] Solubilidade em água com base na Pharmeuropa - Technical Guide for the Elaboration of Monographs, 3a Edição (1999), Capítulo IV, Apêndice IV, com agitação por 15 minutos, mas a 20° C.
[000105] Teofilina: solubilidade em água = 8, g/L a 20° C.
[000106] Cloridrato de fenilefrina: solubilidade em água = 500 g/L a 20° C.
[000107] Sulfato de terbutalina: solubilidade em água = 500 g/L a 20° C. Tabela 1
[000108] Exemplos 1-3,10 = De acordo com a invenção;
[000109] Exemplos 4 a 9 = Exemplos comparativos Todos os dados em % em peso
[000110] *= Sílica coloidal, qualidade farmacêutica, tamanho de partícula médio de cerca de 12 nm
[000111] **= SÍO2-244FP = Syloid® 244 FP = Sílica precipitada, qualidade farmacêutica, tamanho de partícula médio cerca de 3 pm
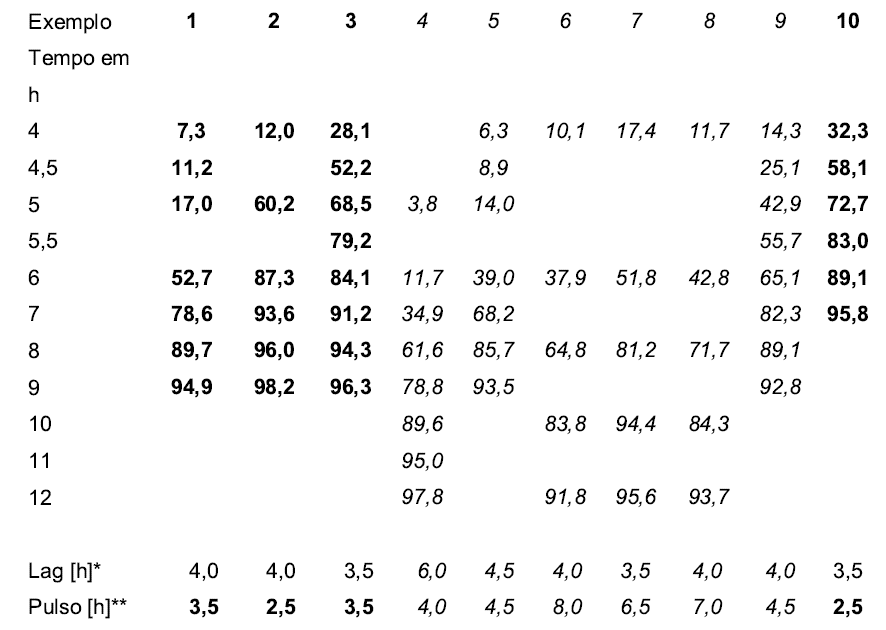
[000112] ***= SÍO2-160PQ = Sipernat® 160PQ (Degussa AG) = Sílica precipitada, qualidade farmacêutica, tamanho de partícula médio cerca de 11 pm. Tabela 2: Liberação de ingrediente ativo em meio tamponado de acordo com USP em [%]
[000113] Os exemplos 1-3, 10 = De acordo com a invenção;
[000114] Exemplos 4-9 = Exemplos comparativos
[000115] *=Lag [h]: indica o tempo da fase lag em horas, em que até 10% do ingrediente ativo são liberados.
[000116] **=Pulso [h]: indica o tempo da fase de pulso em horas, em que cerca de mais 80% do ingrediente ativo são liberados. A fase de pulso nos exemplos 1, 2, 3 e 10 de acordo com a invenção é diminuída para menos do que 4 horas.