A presente invenção refere-se a comprimidos farmacêuticos compreendendo baixas doses de um analgésico 5 de opióide bem como a um método para preparar o mesmo. Os comprimidos da invenção são particularmente úteis para administração sublingual dos ingredientes ativos.
A presente invenção refere-se ainda a um método de tratar dor que emprega os comprimidos da invenção.
ANTECEDENTES DA INVENÇÃO
A administração sublingual tem uma vantagem para ingredientes ativos que, quando dados por via oral, estão sujeitos a metabolismo na primeira passagem substancial e degradação enzimática através do figado, resultando em 15 rápida metabolização e perda de atividade terapêutica relacionada à atividade das enzimas do figado que convertem a molécula em metabolites inativos, ou cuja atividade é diminuida devido a essa bioconversão.
A via sublingual é capaz de produzir um inicio 20 rápido de ação devido à permeabilidade e vascularização consideráveis da mucosa da boca. No caso de administração oral, o comprimido é engolido e distribuição sistêmica da droga ocorre somente no nivel da mucosa gastrointestinal, isto é, posteriormente.
Além disso, a administração sublingual podepermitir também a administração de ingredientes ativos que não são normalmente absorvidos no nivel da mucosa do estômago ou mucosa digestiva após administração oral, ou alternativamente que são parcial ou totalmente degradados 30 em meio ácido após ingestão do comprimido.
Uma alternativa para administração sublingual é a administração por transmucosa através da mucosa da bochecha. Citrato de fentanil, um analgésico de opióide, é atualmente disponível na forma de um pirulito para administração por transmucosa que é comercializado sob o nome comercial Actiq®. 0 problema ligado a essa forma específica é que o paciente deve manter o pirulito na boca 5 por pelo menos 15 minutos para obter a quantidade desejada de fentanil. Além disso, a quantidade de fentanil absorvida é dependente da frequência de engolir saliva e desse modo muito dependente do paciente. É desse modo difícil checar precisamente a quantidade absorvida de fentanil. E 10 finalmente, a absorção através da mucosa da bochecha é geralmente menos eficiente do que a ...absorção sublingual. Por esse motivo, pensa-se que seria vantajosa a formulação de analgésicos de opióide, como fentanil, na forma de comprimidos sublinguais. Os comprimidos sublinguais conhecidos a partir da técnica anterior, são normalmente preparados por compressão direta de uma mistura de pós que compreendem o ingrediente ativo e excipientes para compressão, como diluentes., aglutinantes, agentes desintegrantes e adjuvantes.
Em um método de preparação alternativo, o ingrediente ativo e os excipientes de compressão podem ser granulados a seco ou úmidos antecipadamente. Nesse caso, o ingrediente ativo é distribuído por toda a massa do comprimido. WO 00/16750 descreve um comprimido para uso sublingual que desintegra rapidamente e compreende uma mistura ordenada na qual o ingrediente ativo tem a forma de micropartículas que aderem à superfície de partículas solúveis em água que são substancialmente maiores em 30 tamanho, constituindo um suporte para as micropartículas ativas, a composição também compreendendo um agente mucoadesivo. WO 00/57858 descreve um comprimido para uso sublingual, compreendendo um ingrediente ativo combinado com um sistema efervescente destinado a promover absorção, e também um modificador de pH.
A administração sublingual é uma via de administração que tem certos limites devido ao tamanho da cavidade sublingual na qual o comprimido é colocado, ao volume limitado de saliva para solubilizar o ingrediente ativo ou então à quantidade limitada de ingrediente ativo 10 que pode cruzar a mucosa da boca.
Devido a esses limites, comprimidos nos quais o ingrediente ativo é distribuído uniformemente na massa do comprimido têm certas desvantagens que a presente invenção tem como objetivo resolver. Uma primeira desvantagem desses comprimidos nos quais o ingrediente ativo é disperso na massa é a dependência que existe entre o tamanho do comprimido e a dosagem do ingrediente ativo. Desse modo, se for destinado a fornecer comprimidos de várias dosagens, será necessário 20 ter comprimidos de vários tamanhos.
Pode ser, portanto, que o tamanho do comprimido contendo a dose mais elevada, em particular seu diâmetro, não mais é apropriado para administração sublingual. Isso pode forçar aqueles versados na técnica a 25 modificar a fórmula do comprimido contendo a dose mais elevada, em particular de modo a adaptar seu tamanho a uso sublingual, o que significa, no fim, ter comprimidos com fórmulas qualitativa e/ou quantitativa diferentes para um mesmo ingrediente ativo, que não é economicamente 30 desejável, nem desejável em termos de segurança.
Além disso, a administração sublingual requer o uso de um ingrediente ativo de tamanho especifico de partícula, normalmente consistindo em uma população para a qual o diâmetro é menor do que 10 μm, preferivelmente menor do que 5 μm, como medido pelas técnicas normais, por exemplo por difração a laser.
Essa escolha tem como objetivo assegurar 5 solubilização rápida e completa do ingrediente ativo na saliva e permitir passagem sistêmica imediata e suficiente de modo a obter um efeito instantâneo, que é altamente desejável para o controle de dor.
Agora, o uso de partículas desse tamanho em 10 comprimidos significa que também é necessário adaptar o tamanho de partícula dos excipientes que constituem a massa do comprimido e definir muito precisamente os parâmetros de mistura para a massa pulverulenta, para obter uma mistura ordenada na qual o ingrediente ativo é uniformemente 15 distribuído, sem testemunhar o surgimento de um fenômeno de segregação na tremonha de alimentação da prensa de comprimido, que seria propensa a comprometer a uniformidade do conteúdo dos comprimidos durante a compressão. O risco de surgimento de um fenômeno de 20 segregação é adicionalmente aumentado quando a dose unitária de ingrediente ativo em cada comprimido é baixa. Esse é, por exemplo, o caso com fentanil, para a qual a dose unitária é geralmente menor do que um miligrama até alguns miligramas., É então difícil obter uma uniformidade aceitável de teor para a mesma batelada por toda a etapa de compressão, o ingrediente ativo sendo então altamente diluído na mistura pulverulenta de excipientes.
Para comprimidos para administração sublingual 30 nos quais o ingrediente ativo é uniformemente disperso na massa, a liberação do ingrediente ativo também depende da taxa de desintegração do comprimido.
A técnica anterior descreve comprimidos que desintegram rapidamente, apropriados para administração sublingual, nos quais o ingrediente ativo é distribuído na massa do comprimido.
É conhecido que esses comprimidos têm normalmente uma dureza baixa, frequentemente menor do que 40 N, e apresentam uma friabilidade que é demasiadamente grande, de tal modo que devem ser manipulados com cuidado.
No caso de um comprimido tendo uma dureza maior, 10 o tempo de desintegração é aumentado, de tal modo que o comprimido se desgasta gradualmente enquanto libera o ingrediente ativo a partir da superfície externa do comprimido para seu núcleo.
Portanto é particularmente vantajoso ter uma 15 formulação para administração sublingual que pode t j rapidamente liberar o ingrediente ativo e permitir absorção imediata do mesmo, sem essa liberação ser dependente da taxa de desintegração ou da dureza do comprimido.
Quando o tablete é destinado a desintegrar I 20 rapidamente e sem mastigar, a desintegração leva à formação I de uma polpa ou de uma suspensão.que pode ser engolida sem a intenção.
A viscosidade da polpa ou suspensão, que é relacionada ao uso de agentes de desintegração ou de 25 agentes de intumescimento destinados a acelerar a desintegração, pode causar um refluxo ao engolir. Consequentemente, parte do ingrediente ativo é engolida antes de ser absorvida através da mucosa da boca. ’ A absorção no nivel da mucosa da boca, na qual a* 30 biodisponibilidade do ingrediente ativo diretamente depende, é portanto dependente da natureza dos excipientes utilizados nesse tipo de formulação de desintegração rápida.
SUMÁRIO DA INVENÇÃO
O requerente demonstrou que é possível remediar todas essas desvantagens por intermédio de um comprimido compreendendo uma dose baixa de ingrediente ativo formado 5 de microgrânulos. Os comprimidos da invenção são particularmente úteis para administração sublingual dos ingredientes ativos.
DESCRIÇÃO DETALHADA DA INVENÇÃO
O comprimido sublingual, de acordo com a 10 invenção, compreende um diluente diretamente compressível na forma de núcleos neutros, onde os núcleos neutros são revestidos com pelo menos uma camada que compreende uma dose baixa de um ingrediente ativo (também mencionado como "camada ativa" a seguir) e onde o ingrediente ativo é um 15 analgésico de opióide apropriado para administração sublingual.
No contexto da presente invenção, o termo "núcleos neutros" é entendido como significando essencialmente grânulos esféricos compreendendo sacarose e 20 amido.
Núcleos neutros particularmente valiosos no contexto da invenção compreende menos de 91,5% de sacarose.
A presente invenção emprega, vantajosamente, partículas esféricas, assegurando boa fluidez e boa 25 homogeneidade de mistura a ser formada em comprimidos.
As excelentes propriedades reológicas dos núcleos neutros torna os mesmos bons candidatos como excipientes de compressão direta. O tempo de fluxo dos núcleos neutros sob as condições do teste descrito na European Pharmacopoeia é 30 bem menor do que 10 segundos. Essa propriedade torna possível alimentação muito eficiente das prensas de comprimidos. Além disso, os núcleos neutros têm um volume de compactação muito baixo. * Os núcleos neutros têm a vantagem de constituir um excipiente de compressão direta que não gera poeira. Finalmente, os núcleos neutros têm um tempo de dissolução muito menor do que 15 minutos. Além disso, a presente invenção torna possivel evitar os problemas de separação geralmente observados em compressão direta visto que todas as partículas a serem formadas em comprimidos têm o mesmo tamanho. Os núcleos neutros têm um tamanho entre 100 e 10 2000 μm, preferivelmente entre 150 e 600 μm, ou preferivelmente entre 150 e 500 μm, por exemplo 180 a 250 μm ou 400 a 500 μm. Ingredientes ativos que são vantajosos no comprimido, de acordo com a invenção, são os ingredientes ativos que são apropriados para administração bucal ou sublingual devido a suas características farmacocinéticas, em particular quando o ingrediente ativo tem uma janela de absorção na cavidade bucal, ou um efeito do primeiro passo considerável através do figado que requer uma via de administração alternativa à administração oral convencional, ou alternativamente quando se busca obter um efeito sistêmico muito rápido para superar os efeitos de um ataque como um ataque de angina do peito, um ataque de ansiedade, dor aguda, uma crise alérgica, uma crise de asma, ou um ataque de abstinência, causado por exemplo, por abstinência de álcool ou opiáceo . Os exemplos de tais ingredientes ativos são fornecidos na publicação "Oral Mucosal Drug delivery", Hao Zhang e outros, Clin. Pharmacokinet. 2002, 41, (9) 661-680. Na presente invenção o ingrediente ativo é escolhido entre analgésicos de opióide apropriados para administração sublingual. Os exemplos de analgésicos deopióide apropriados compreendem buprenorfina, norbuprenorfina, fentanil, metadona, levorfanol, morfina, hidromorfona, codeina oximorfona, oxicodona, hidrocodona e seus sais farmaceuticamente aceitáveis.
Na presente invenção, "fentanil" como ingrediente ativo, significa fentanil e seus derivados.
Derivados de fentanil compreendem alfentanil, sufentanil e remifentanil. O termo "sais farmaceuticamente aceitáveis" significam os derivados dos compostos descritos nos guais o composto de base farmaceuticamente ativo é convertido em seu sal com uma base ou ácido, exemplos de sais farmaceuticamente ativos compreendendo em particular sais de ácido orgânico e inorgânico de residuos básicos, como aminas, derivados de metal alcalino ou sais orgânicos de residuos ácidos, como ácidos carboxilicos e similares.
Os exemplos de sais farmaceuticamente aceitáveis de fentanil compreendem citrato de fentanil e cloridrato de fentanil. Os exemplos de sais farmaceuticamente aceitáveis de derivados de fentanil compreendem cloridrato de alfentanil, citrato de sufentanil e cloridrato de remifentanil. Ingredientes ativos preferidos são base de fentanil, citrato de fentanil, alfentanil, cloridrato de alfentanil, sufentanil, citrato de sufentanil, remifentanil, cloridrato de remifentanil. Ingredientes ativos podem ser utilizados em qualguer forma polimórfica, em forma racêmica ou na forma de seus enantiômeros ou na forma de mistura de 30 enantiômeros. O ingrediente ativo pode estar na forma de um pó, por exemplo, um pó micronizado ou de microcristais. I • A quantidade de ingrediente ativo é preferivelmente menor do que 20 mg/comprimido, mais preferivelmente menor do que 10 mg/comprimido, por exemplo 0,1 a 10 mg/comprimido, preferivelmente 0,1 a 2 mg/comprimido. A quantidade de ingrediente ativo é ajustada nos limites acima, de acordo com o tipo de ingrediente ativo.
Dados que os comprimidos são de dose baixa, não é normalmente necessário adicionar excipientes adicionais à 10 composição de revestimento compreendendo o ingrediente ativo aplicado sobre os núcleos neutros (também mencionado a seguir como a "camada ativa"). Os microgrânulos são preferivelmente compostos de um núcleo neutro, em cuja superfície o ingrediente ativo é adsorvido. Em uma modalidade preferida a camada ativa é isenta de qualquer excipiente. Se, apesar de tudo, os excipientes provarem ser preferíveis na realização da aplicação da camada ativa sobre os núcleos neutros, a escolha de sua respectiva 20 composição e quantidade será tal que não modificam substancialmente as propriedades de formação de comprimido dos núcleos neutros ou modificam a liberação do ingrediente ativo. Os excipientes farmaceuticamente aceitáveis 25 presentes opcionalmente na camada ativa são escolhidos entre aglutinantes, agentes solúveis, tensoativos, promotores de absorção, agentes bioadesivos, agentes antiestáticos, pares de base/ácido que produzem uma ’ efervescência, edulcorantes, aromatizantes, substâncias • 30 corantes e misturas dos mesmos. O aglutinante, que está opcionalmente presente na camada ativa, é utilizado em proporções que podem variar até 95% em peso em relação ao peso seco do revestimento, preferivelmente até 30% em peso em relação ao peso seco da camada ativa. Seu papel é ligar o ingrediente ativo ao núcleo neutro sem perda de material, ou formar uma camada 5 homogênea de ingrediente ativo e outros excipientes, distribuídos uniformemente em torno do núcleo neutro. O aglutinante é preferivelmente escolhido de polimeros que são hidrofilicos e/ou solúveis no pH da saliva, de modo a permitir uma liberação instantânea do 10 ingrediente ativo, como polivinil pirrolidonas e polimeros baseados em celulose, polimeros acrílicos e polietileno glicóis. A polivinil pirrolidona pode ser escolhida dos polimeros tendo uma massa molecular entre 10 000 e 50 000. O polimero baseado em celulose é escolhido entre derivados hidroxilados, por exemplo hidroxipropil- metilcelulose, hidroxipropilcelulose, hidroximetilcelulose, ftalato de hidroxipropilmetilcelulose e acetossuccinato de hidroxipropilmetilcelulose. O hidroxipropilmetilcelulose preferido é escolhido entre aqueles para os quais a viscosidade aparente (solução aquosa a 2% m/m a 20°C, método USP) está entre 2,4 e 18 cP, e ainda mais preferivelmente entre 2,4 e 5 cP. O polietileno glicol preferido é escolhido a partir daqueles para os quais a massa molecular nominal é 4000 ou 6000 g/mol. O agente solúvel, que pode estar opcionalmente presente na camada ativa, é utilizado em uma proporção que 30 pode variar até 90% em peso, preferivelmente entre 1% e 60%, e ainda mais preferivelmente entre 30 e 60% em peso, calculado em relação ao peso seco da camada ativa.
Esse agente solúvel é utilizado em particular para melhorar a solubilização do ingrediente ativo por acelerar a solubilização do revestimento compreendendo o ingrediente ativo. O agente solúvel pode ser escolhido do grupo de açúcares como sacarose, lactose ou dextrose, de polióis como manitol, sorbitol ou lactitol, ou então de sais inorgânicos como cloreto de sódio. O tensoativo, que está opcionalmente presente na 10 camada ativa, pode ser escolhido entre agentes catiônico, aniônico, não iônico ou anfotéricos, individualmente ou como mistura. O tensoativo pode ser escolhido, por exemplo, a partir de compostos como lauril de sulfato de sódio, o 15 monooleato, o monolaurato, o monopalmitato, o monoestearato, o trioleato, o triestearato ou qualquer outro éster de sorbitano polioxietilenado, preferivelmente Tween® 20, 40, 60 ou 80, glicerideos de ácidos graxos polioxietilenados, esses ácidos graxos sendo saturados ou 20 insaturados e compostos de pelo menos 8 átomos de carbono, poloxâmeros, como poloxâmero 188, copolimeros de bloco de óxido de etileno/óxido de propileno, como Pluronic® F68 ou F87, lecitina, álcool de estearila, álcool de cetearila, colesterol, óleo de ricino polioxietilenado, éteres 25 polioxietilenados de álcool graxo, como os produtos Brij® e estearatos polioxietilenados. O tensoativo está vantajosamente presente em uma proporção que pode variar até 20%, preferivelmente entre 0,1 e 20% em peso em relação ao peso seco total da camada 30 ativa. Os promotores de absorção, que estão opcionalmente presentes na camada ativa, são compostos que tornam possível melhorar a absorção do ingrediente ativo através das paredes da cavidade bucal até o fluxo sanguíneo.
Esses compostos podem ser escolhidos do grupo que 5 compreende, por exemplo, sulfato de lauril de sódio, caprato de sódio ou quitosanas, e também inibidores de P- glicoproteína (P-gp), como polisorbato 80, Cremophor® EL (óleo de rícino hidrogenado) ou Solutol® HS-15 (PEG-HS ou polietileno glicol-660 12-hidroxiestearato). Os agentes bioadesivos, que estão opcionalmente presentes na camada ativa, podem ser escolhidos do grupo que compreende, por exemplo, carbômeros, carboxi- metilcelulose de sódio, alginato de sódio, hidroxi- propilmetilcelulose, hidroxipropilcelulose, hidroxietil- 15 celulose, etilcelulose, gelatina, goma guar, óxido(s) de polietileno (nome comercial Polyox®) e dextrano. O agente antiestático, que está opcionalmente presente na camada ativa, pode ser escolhido do grupo que consiste em sílica coloidal e preferivelmente sílica 20 precipitada, talco micronizado e misturas dos mesmos. O agente antiestático é utilizado em uma proporção que pode variar até 60% em peso, calculado em relação ao peso seco do revestimento aplicado em torno do núcleo comprimido. O par de ácido/base que produz uma efervescência, que está opcionalmente presente na camada ativa, é formado de um agente alcalino e um agente ácido que são escolhidos daqueles que são farmaceuticamente aceitáveis, de tal modo que, na presença de água, permitem a liberação de um gás.
A vantagem de utilizar uma mistura efervescente no revestimento compreendendo o ingrediente ativo é aquela de facilitar a dissolução rápida da camada ativa formada em torno dos microgrânulos após contato com saliva, e desse modo obter, através da liberação de um gás farmaceuticamente aceitável e indução de um micro pH bucal, solubilização rápida do ingrediente ativo nas membranas 5 mucosas bucal ou sublingual e uma distribuição de droga sistêmica aperfeiçoada enquanto ao mesmo tempo aperfeiçoa as propriedades organolépticas de modo a diminuir a sensação do ingrediente ativo na cavidade bucal, ou induzir um sabor levemente ácido agradável. O agente ácido é um composto doador de prótons que pode reagir com um agente alcalino de modo a formar um gás que causa a efervescência do liquido no qual esse gás é liberado. O agente ácido pode consistir em qualquer ácido 15 inorgânico ou orgânico, na forma de um ácido livre, um anidrido ácido ou um sal de ácido.
Esse ácido é escolhido a partir do grupo que compreende em particular ácido tartárico, ácido citrico, ácido maléico, ácido fumárico, ácido málico, ácido adipico, 20 ácido succinico, ácido láctico, ácido glicólico, ácidos alfa-hidróxi, ácido ascórbico e aminoácidos, e também os sais e derivados desses ácidos. O agente alcalino consiste em um composto capaz de gerar um gás por reação com um composto doador de 25 prótons. 0 gás formado é dióxido de carbono, oxigênio ou qualquer outro tipo de gás biocompativel. O agente alcalino é escolhido do grupo que compreende carbonato de potássio, carbonato de litio, carbonato de sódio, carbonato de cálcio, carbonato de 30 amónio, carbonato L-lisina, carbonato arginina, carbonato de glicina de sódio, carbonatos de sódio de aminoácidos, perborato de sódio anidro, perborato efervescente, monoidrato de perborato de sódio, percarbonato de sódio, dicloroisocianurato de sódio, hipoclorito de sódio, hipoclorito de cálcio e misturas dos mesmos.
No contexto da presente invenção, o termo "carbonato" pretende significar, sem distinção, carbonatos, 5 sesquicarbonatos e carbonatos de hidrogênio.
As quantidades respectivas de agente ácido e de agente alcalino são ajustadas de tal modo que a reação entre o agente alcalino e os prótons liberados pelo ácido permitem a geração de uma quantidade suficiente de gás para 10 obter uma efervescência satisfatória. O agente ácido individualmente ou agente alcalino individualmente pode ser utilizado em quantidade adaptada no revestimento para assegurar que o ingrediente ativo estará principalmente sob forma de base, isto é, fração 15 absorvivel.
Edulcorantes apropriados, que estão opcionalmente presentes na camada ativa, podem ser selecionados do grupo que compreende em particular aspartame, acesulfame de potássio, sacarinato de sódio, neoesperidina 20 dihidrocalcona, sucralose, glicirrizinato de monoamônio e misturas dos mesmos.
Aromatizantes e substâncias corantes apropriadas, que estão opcionalmente presentes na camada ativa, são aquelas comumente utilizadas em farmácia para a preparação 25 de comprimidos.
A incorporação de edulcorante(s) e/ou aromatizantes no revestimento dos núcleos neutros da invenção é particularmente vantajosa para mascarar o amargor de certos ingredientes ativos.
As substâncias corantes são aquelas normalmente utilizadas em farmácia. A substância corante é utilizada em uma proporção que pode variar até 1% em peso, calculado em relação ao peso seco da camada aplicada em torno do núcleo neutro.
Em uma modalidade especifica, a camada ativa não compreende edulcorantes nem aromatizantes.
A composição de excipientes na camada ativa é 5 ajustada de tal modo que o ingrediente ativo solubiliza totalmente quando o comprimido desintegra.
Os núcleos neutros compreendendo uma camada ativa (definida abaixo como "núcleo(s) ativo(s)") podem ser opcionalmente revestidos com uma camada que compreende um 10 composto modificador de pH, a camada sendo denominada camada modificadora de pH.
De acordo com uma modalidade, a camada modificadora de pH pode estar presente acima ou sob a camada ativa.
De acordo com outra modalidade, o composto modificador de pH pode estar presente na camada ativa.
A camada modificadora de pH permite a provisão de um pH ácido ou alcalino local quando o comprimido é colocado na cavidade bucal que aumenta a absorção de 20 ingrediente ativo pela mucosa.
Compostos modificadores de pH apropriados compreendem ácido citrico e citrato de sódio ou citrato de potássio, hidróxido de sódio, monoetanolamina, dietanolamina, bicarbonato de sódio ou bicarbonato de 25 potássio, fosfato de sódio, ácido tartárico, ácido propiônico, ácido láctico, ácido málico e glutamato monossódico.
A escolha do composto modificador de pH depende da natureza do ingrediente ativo utilizado. No caso onde 30 absorção do ingrediente ativo pela mucosa bucal é intensificada sob condições alcalinas, um composto alcalino será utilizado como composto modificador de pH. No caso onde absorção do ingrediente ativo pela mucosa bucal é
aumentada sob condições ácidas, um composto ácido será utilizado como composto modificador de pH.
A composição da camada ' modificadora de pH é ajustada de tal modo que quando o ingrediente ativo solubiliza totalmente após contato com a saliva, o pH em torno da área bucal é ajustado em um valor favorável para a absorção sublingual do ingrediente ativo.
A camada modificadora de pH pode compreender, opcionalmente, excipientes idênticos àqueles presentes na 10 camada ativa.
No caso de fentanil como ingrediente ativo, a camada modificadora de pH opcional é uma camada alcalina que compreende um composto alcalino.
De acordo com uma modalidade, a camada de revestimento alcalina pode estar presente acima ou sob a I camada de fentanil. [ De acordo com outra modalidade, o composto alcalino pode estar presente na camada de fentanil.
A camada alcalina permite a provisão de um pH alcalino local quando o comprimido é colocado na cavidade bucal que aumenta a absorção de fentanil através da mucosa.
A camada alcalina pode compreender, opcionalmente, excipientes idênticos aqueles presentes na camada que compreende fentanil. O composto alcalino é vantajosamente selecionado do grupo que compreende tris, tartrato, acetato, fosfato, e preferivelmente fosfato dissódico anidro e misturas dos mesmos.
A invenção também se refere a um método de preparar os núcleos neutros do comprimido da invenção. O método compreende aplicar uma camada ativa nos núcleos neutros por pulverizar uma solução, suspensão ou dispersão coloidal compreendendo um ingrediente ativo • apropriado para administração sublingual e, opcionalmente, pelo menos um excipiente farmaceuticamente aceitável, sobre os núcleos neutros. Os ingredientes ativos e excipientes são aqueles 5 descritos acima em relação ao comprimido da invenção.
Em uma modalidade preferida, o ingrediente ativo é fentanil ou um sal farmaceuticamente aceitável do mesmo, em qualquer forma polimórfica, em forma racêmica ou enantiomérica.
A camada ativa é distribuída uniformemente através da superfície dos microgrânulos neutros. Em particular, para substâncias insolúveis em água, é possivel formar uma camada na qual o ingrediente ativo esteja na forma de uma dispersão sólida, obtida por 15 co-precipitação do ingrediente ativo com um polímero hidrofilico.
A pulverização pode ser realizada em um tambor perfurado ou em um revestidor de leito fluidizado. Em uma modalidade preferida, a pulverização da camada ativa sobre 20 os microgrânulos neutros é realizada em um tambor perfurado, em particular em um tambor perfurado tendo seções com perfis triangulares, paralelos entre si e definindo as aberturas entre os mesmos, como aquele descrito no pedido de patente EP 1044064.
A composição de revestimento é pulverizada na forma de uma solução, uma suspensão ou uma dispersão coloidal em um solvente orgânico ou aquoso, ou misturas dos mesmos, e é então seca. O solvente orgânico pode ser escolhido entre * 30 etanol, isopropanol, tetraidrofurano, éter isopropilico, acetona, metiletilcetona, cloreto de metileno ou uma mistura desses solventes.
Água purificada é o solvente preferivelmente utilizado se o revestimento for isento de agentes efervescentes; por outro lado, um solvente orgânico tem de ser utilizado quando a composição pulverizada contém um par 5 ácido/base efervescente.
Em uma modalidade, o método de preparar os núcleos neutros compreende uma etapa de aplicar uma camada modificadora de pH nos núcleos, preferivelmente por pulverizar uma solução, suspensão ou dispersão coloidal 10 compreendendo um composto modificador de pH e opcionalmente pelo menos um excipiente farmaceuticamente aceitável, sobre os núcleos neutros.
No caso onde o ingrediente ativo é fentanil ou um sal farmaceuticamente aceitável do mesmo, o composto 15 modificador de pH é um composto alcalino e a camada modificadora de pH uma camada alcalina.
A camada modificadora de pH pode ser aplicada simultaneamente com a camada ativa ou acima ou sob a camada ativa. A pulverização da camada modificadora de pH pode ser realizada, por exemplo, em um tambor perfurado ou em um revestidor de leito fluidizado. Em uma modalidade preferida, a pulverização da camada modificadora de pH sobre os núcleos neutros é 25 realizada em um tambor perfurado, em particular em um tambor perfurado tendo seções com perfis triangulares, paralelos entre si e definindo as aberturas entre os mesmos, como aquele descrito no pedido de patente EP 1044064.
A escolha do equipamento torna possivel controlar a aplicação da camada modificadora de pH e evitar qualquer fenômeno de aderência, ligado à natureza do ingrediente ativo e dos excipientes da composição de revestimento modificadora de pH, e aos vários parâmetros do método (temperatura, pressão de ar, taxa de fluxo de solução).
A composição de revestimento contendo o composto modificador de pH é pulverizada na forma de uma solução, 5 uma suspensão ou uma dispersão coloidal em um solvente orgânico ou aquoso, ou misturas dos mesmos, e é então seco.
A quantidade de aglutinante é ajustada, de acordo com a natureza e a quantidade de ingrediente ativo pulverizado sobre núcleos neutros.
Em particular, para substâncias insolúveis em água, é possivel formar uma camada na qual o ingrediente ativo tem a forma de uma dispersão sólida, obtida por co- precipitação do ingrediente ativo com um polimero hidrofilico. O solvente utilizado para a aplicação do revestimento ’ será geralmente água ou qualquer outro solvente autorizado com um estágio de secagem apropriado.
Água purificada é o solvente preferivelmente utilizado se o revestimento estiver isento de agentes 20 efervescentes; por outro lado, um solvente orgânico tem de ser utilizado quando a composição pulverizada contém um par ácido/base efervescente. O solvente orgânico pode ser escolhido entre etanol, isopropanol, tetraidrofurano, éter isopropilico, 25 acetona, metiletilcetona, cloreto de metileno ou uma mistura desses solventes. O comprimido sublingual da invenção pode ser preparado por intermédio de um método que compreende pelo menos as seguintes etapas: 1. preparação de microgrânulos compreendendo uma camada que compreende um ingrediente ativo por pulverizar uma solução ou uma suspensão compreendendo um ingrediente ativo apropriado para administração sublingual e, opcionalmente, pelo menos um excipiente farmaceuticamente aceitável, sobre os núcleos neutros; e 2. compressão dos microgrânulos obtidos na etapa 1 de modo a obter um comprimido. Em uma modalidade, a etapa 1 do método de preparação dos comprimidos da invenção compreende uma etapa de aplicar uma camada modificadora de pH aos núcleos.
A camada modificadora de pH pode ser aplicada simultaneamente com a camada ativa ou acima ou sob a camada 10 ativa. O método da invenção é vantajoso em termos de segurança uma vez que evita a manipulação de ingredientes ativos na forma de misturas pulverulentas, como é o caso em etapas de granulação e/ou compressão convencionais, e 15 permite que o produto esteja contido utilizando o ingrediente ativo na forma de uma solução ou suspensão pulverizada.
Especialmente no caso de ingredientes ativos altamente tóxicos, será reconhecido que o método da 20 invenção evita a manipulação dessas substâncias na forma de misturas pulverulentas, como é o caso nas etapas de granulação e/ou compressão tradicionais, e permite que o ingrediente ativo altamente tóxico esteja contido utilizando o ingrediente ativo na forma de uma solução ou 25 suspensão pulverizada.
As condições e detalhes da etapa 1 do método de preparação dos comprimidos da invenção são como descrito acima em relação ao método de preparação dos núcleos neutros da invenção. Opcionalmente um ou mais lubrificantes podem ser adicionados na etapa de compressão aos microgrânulos obtidos na etapa 1. O(s) lubrificante 4s) pode(m) estar presentes em uma quantidade menor do que 1% em peso com relação ao peso do comprimido, preferivelmente entre 0,10 e 0,75% em peso, mais preferivelmente da ordem de 0,10% a 0,50% em peso, do 5 comprimido. O lubrificante torna possivel reduzir fricção entre partículas e entre partículas e o molde de prensa. Também torna possivel reduzir adesão das partículas às pinças e obter um grau de brilho. O lubrificante é 10 escolhido, por exemplo, a partir de magnésio, estearato de zinco ou cálcio, talco, Aerosil®, ácido esteárico,estearil fumarato de sódio e PEGs. Os comprimidos sublinguais da presente invenção apresentam uma uniformidade em massa muito menor do que 5% 15 e da ordem de 1% para tabletes com uma massa da ordem de 300 a 500 mg, uma friabilidade menor do que 1%, um tempo de dissolução a 37°C menor do que 15 minutos, e uma dureza da ordem de 0 a 200 N. Esses parâmetros podem ser ajustados pela ação dos parâmetros de formação de comprimidos. A força de compressão aplicada na etapa 2 está vantajosamente entre 5 e 50 kN quando a área superficial de compressão é 1 cm2 (isto é, 50 a 500 MPa) , preferivelmente entre 10 e 30 kN. A força de compressão é ajustada de modo a obter um comprimido cuja dureza está preferivelmente 25 entre 10 e 180 N, mais preferivelmente entre 15 e 100 N, medido de acordo com o método da European Pharmacopoeia (2.9.8). Preferivelmente, a dureza do comprimido da invenção é ajustada de modo a obter uma friabilidade, 30 medida de acordo com o método da European Pharmacopoeia, menor do que 1% em peso.
Uma vantagem dos comprimidos de acordo com a invenção é que têm um tempo de desintegração menor do que 15 min., preferivelmente 5 a 15 min. O tempo de desintegração é medido ao vivo colocando o comprimido revestido na cavidade sublingual, e medindo, utilizando um cronômetro o tempo que passa entre o 5 inicio da medição e o momento quando o comprimido revestido se desintegrou totalmente sob a ação de saliva e sem mastigar, de modo a formar somente uma polpa viscosa, o paciente não tendo de usar, durante todo esse tempo, qualquer ação das mandíbulas. Os comprimidos da invenção podem ter um diâmetro entre 2 e 14 mm e um formato redondo, oval, retangular ou outro pode ter uma superfície plana, convexa, ou outra, e pode ter opcionalmente gravação. Preferivelmente, os comprimidos da invenção têm 15 um formato biconvexo redondo, que é um formato vantajoso para o processo de formação de comprimido e o contato do comprimido revestido com saliva quando o comprimido é colocado na cavidade bucal.
De acordo com uma modalidade especifica, os 20 comprimidos sublinguais de acordo com a invenção podem ser revestidos com filme, para aperfeiçoar sua aparência ou mascarar a cor ou proteger o ingrediente ativo contra luz, umidade ou oxigênio no ar.
De acordo com uma outra modalidade da invenção, é 25 possivel utilizar agentes de coloração no revestimento do comprimido como um código para indicar o tipo e dosagem de ingrediente ativo. Na realidade, qualquer que seja a dosagem, o tamanho dos comprimidos pode ser igual. Para fazer a diferença entre dosagens diferentes, uma cor 30 especifica pode ser associada a uma dosagem especifica.
Em uma modalidade particularmente vantajosa, o método para preparar um comprimido sublingual, de acordo com a invenção, compreende pelo menos as seguintes etapas: 1. preparação de microgrânulos compreendendo um analgésico de opióide apropriado para administração sublingual, preferivelmente fentanil como ingrediente ativo e, opcionalmente excipientes farmaceuticamente aceitáveis, 5 sobre os núcleos neutros; e 2.compressão dos microgrânulos obtidos na etapa 1, opcionalmente com um lubrificante, de modo a obter um comprimido; em que o termo "fentanil" deve ser entendido como 10 definido acima.
As condições e detalhes das etapas 1 e 2 dessa modalidade são como descrito acima em relação ao método geral para preparar comprimidos, de acordo com a invenção.
Opcionalmente, o método de preparar os 15 comprimidos da invenção compreende pulverizar uma solução ou suspensão também compreendendo um composto alcalino sobre os núcleos neutros. A etapa pode ser realizada diretamente sobre os núcleos neutros (antes da etapa 1), ou simultaneamente com a etapa de pulverização do revestimento 20 contendo o ingrediente ativo (simultaneamente com a etapa 1), ou sobre o núcleo ativo obtido a partir da etapa 1.
De acordo com uma opção especifica, os comprimidos sublinguais contendo fentanil de acordo com a invenção podem ser revestidos por filme, para melhorar sua 25 aparência ou mascarar a cor ou proteger o ingrediente ativo contra luz, umidade ou oxigênio no ar.
A presente invenção também é dirigida a uma pré- mistura de formação de comprimidos que consiste em uma composição contendo 99 a 100% em peso de núcleos neutros 30 revestidos com pelo menos uma camada ativa que compreende uma dose baixa de um analgésico de opióide apropriado para administração sublingual e 0 a 1% em peso de um lubrificante. A composição é destinada a ser submetida à compressão direta. Os núcleos revestidos da pré-mistura de formação de comprimido, de acordo com a invenção, podem compreender uma camada modificadora de pH como descrito aqui acima em 5 relação aos núcleos neutros que compreende uma camada ativa e sua preparação. O ingrediente ativo representa, preferivelmente menos de 5% em peso dos núcleos neutros.
Em uma modalidade preferida, o analgésico de 10 opióide é fentanil, onde o termo "fentanil" deve ser entendido como definido acima.
A presente invenção também é dirigida a um processo para a preparação dos comprimidos sublinguais da invenção por compressão direta da pré-mistura de formação 15 de comprimidos acima. De acordo com esse processo, a força de compressão está vantajosamente entre 5 e 50 kN quando a área superficial de compressão é 1 cm2 (isto é, 50 a 500 MPa), preferivelmente entre 10 e 30 kN.
Além disso, a presente invenção refere-se a um 20 método de tratar dor que compreende introduzir na cavidade bucal de um paciente uma quantidade terapeuticamente eficaz de um ingrediente ativo na forma de um comprimido sublingual onde o ingrediente ativo é selecionado do grupo que compreende analgésicos de opióide apropriados para 25 administração sublingual, como buprenorfina, nor- buprenorfina, fentanil, alfentanil, sufentanil, remifentanil, metadona, levorfanol, morfina, hidromorfona, codeina oximorfona, oxicodona, hidrocodona, e seus sais farmaceuticamente aceitáveis. Ingredientes ativos particularmente preferidos são fentanil, citrato de fentanil, alfentanil, cloridrato de alfentanil, sufentanil, citrato de sufentanil, remifentanil, cloridrato de remifentanil.
Em uma modalidade particularmente vantajosa, o método de tratar dor de acordo com a presente invenção compreende introduzir na cavidade bucal de um paciente, uma quantidade terapeuticamente eficaz de um comprimido 5 sublingual compreendendo um diluente diretamente compressivel na forma de núcleos neutros, onde os núcleos neutros são revestidos com uma dose baixa de fentanil como ingrediente ativo, onde o termo "fentanil" deve ser entendido como definido acima. O método de tratar dor, de acordo com a presente invenção, é particularmente útil para tratar dor episódica, em particular dor episódica de câncer. É particularmente apropriado para tratar pacientes que já estão recebendo e que são tolerantes à terapia de opióide para sua dor 15 persistente associada. Pacientes considerados tolerantes a opióide são aqueles que estão tomando pelo menos 60 mg de morfina/dia, pelo menos 25 μg de fentanil transdérmico/hora, pelo menos 30 mg de oxicodona diariamente, pelo menos 8 mg de 20 hidromorfona oral diariamente ou uma dose equianalgésica de outro opióide por uma semana ou mais tempo.
A invenção também se refere ao uso de um analgésico de opióide apropriado para administração sublingual, como buprenorfina, nor-buprenorfina, fentanil, 25 alfentanil, sufentanil, remifentanil, metadona, levorfanol, morfina, hidromorfona, codeina oximorfona, oxicodona, hidrocodona e seus sais farmaceuticamente aceitáveis, para a fabricação de um comprimido sublingual de acordo com a invenção. Fentanil e seus derivados em qualquer forma polimórfica, em forma racêmica ou na forma de seus enantiômeros ou na forma de misturas de enantiômeros, na forma de base ou na forma de um sal farmaceuticamente aceitável são princípios ativos preferidos. Princípios ativos particularmente preferidos são fentanil, citrato de fentanil, alfentanil, cloridrato de alfentanil, sufentanil, citrato sufentanil, remifentanil, cloridrato de remifentanil. Os comprimidos sublinguais desse modo obtidos, de acordo com a invenção, são particularmente úteis para tratar dor episódica, em particular dor episódica de câncer. É particularmente apropriado para tratar pacientes que já estão recebendo e que são tolerantes à terapia de opióide para sua 10 dor persistente associada. A invenção será entendida mais claramente a partir do seguinte exemplo, sem que o último limite de modo algum o escopo da invenção.
EXEMPLOS
Nos exemplos a seguir, os produtos mencionados abaixo são utilizados: Núcleos neutros "Neutres SP" (400 - 500 μm) disponíveis a partir de NPPHARM. Núcleos neutros "NPTAB 200" (180 - 250 μm) disponível a partir de NPPHARM. HPMC 603 (hidroxipropilmetilcelulose) disponível sob a marca registrada Pharmacoat 603 da Shin-Etsu. PEG 6000 disponível sob a marca registrada Renex PEG 6000 da Quimasso. Estearato de magnésio disponível da Quimdia. As percentagens dadas são expressas em peso. Exemplo 1
1 - Revestimento dos núcleos neutros
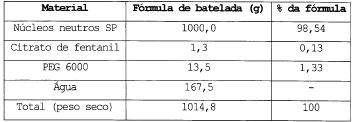
A solução de revestimento de citrato de fentanil e 30 PEG 6000 em água é pulverizada, em um revestidor de leito fluidizado, sobre 1000 g de núcleos neutros SP. A fórmula dos microgrânulos revestidos é dada na tabela 1. Tabela 1
Os microgrânulos revestidos são lubrificados com 0,12% de estearato de magnésio. Os microgrânulos são então comprimidos em uma prensa alternativa (Frogerais OA) equipada com pinças chanfradas planas redondas, com 11 mm de diâmetro. Os comprimidos obtidos, de acordo com a invenção, têm uma dosagem unitária de 0,63 mg de citrato de fentanil, isto é, de 0,4 10 mg de base de fentanil.
Exemplo 2
1 - Revestimento dos núcleos neutros com citrato de fentanil
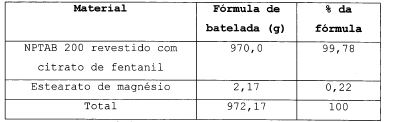
A solução de revestimento de citrato de fentanil e HPMC 603 em água é pulverizada, em um revestidor de ar fluidificado, sobre 700 g de núcleos neutros NPTAB 200. A fórmula dos microgrânulos revestidos é dada na tabela 1. Tabela 2
Os microgrânulos revestidos são lubrificados com 0,22% de estearato de magnésio. Os microgrânulos são então comprimidos em uma prensa alternativa (SVIAC PR12) equipada com pinças côncavas redondas, com 5,5 mm de diâmetro. A fórmula dos comprimidos obtidos, de acordo com a invenção, é dada na tabela 3. Tabela 3
Os comprimidos obtidos, de acordo com a invenção,têm uma dosagem unitária de 0,63 mg de citrato de fentanil, isto é, de 0,4 mg de base de fentanil.
Um Estudo de Biodisponibilidade Comparativa de 15 dose única de comprimidos preparados de acordo com o exemplo 2 versus Actiq® 0,4 mg foi executado em
Voluntários saudáveis do sexo masculino sob condições de jejum. O objetivo desse estudo piloto foi avaliar a 20 biodisponibilidade relativa de dose única das duas formulações em voluntários saudáveis do sexo masculino sob condições de jejum. Os comprimidos preparados, de acordo com o exemplo 2 e o produto de referência que foram administrados 25 a 10 pacientes e Cmax, Tmax e AUC foram medidos. O produto de referência é uma formulação de citrato de fentanil (matriz de droga sólida em um cabo) projetado para facilitar a absorção por transmucosa e comercializado no mundo inteiro sob a marca registrada Actiq®.
Tanto a invenção como o produto de referência 5 contem citrato de fentanil em uma quantidade equivalente a 0,4 mg de base de fentanil.
Pontos de amostragem de sangue: antes de dosagem e nos seguintes tempos posteriormente em cada periodo: 5, 10, 15, 20, 25, 30, 35, 40, 45, 50, 60, 75, 90 minutos e em 10 2, 3, 4, 6, 8, 12 e 24 horas pós-dose.
Os parâmetros farmacocinéticos AUC 0-t, AUCoc, Cmax, tmax foram calculados para fentanil em plasma.
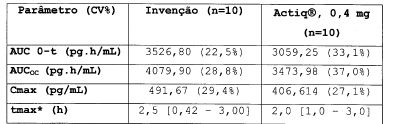
Médias geométricas são dadas na tabela 4:Tabela 4
15 *mediano [faixa]
Relações médias da invenção versus Actiq® são calculadas na tabela 5 abaixo.Tabela 5
A invenção mostra biodisponibilidade similar e Cmax do que Actiq®.