BRPI0811601A2 - composto, método para tratar um distúrbio mediado por jnk em um paciente que tem um distúrbio mediado por jnk, composição farmacêutica, uso do composto e medicamento que compreende o composto. - Google Patents
composto, método para tratar um distúrbio mediado por jnk em um paciente que tem um distúrbio mediado por jnk, composição farmacêutica, uso do composto e medicamento que compreende o composto. Download PDFInfo
- Publication number
- BRPI0811601A2 BRPI0811601A2 BRPI0811601A BRPI0811601A BRPI0811601A2 BR PI0811601 A2 BRPI0811601 A2 BR PI0811601A2 BR PI0811601 A BRPI0811601 A BR PI0811601A BR PI0811601 A BRPI0811601 A BR PI0811601A BR PI0811601 A2 BRPI0811601 A2 BR PI0811601A2
- Authority
- BR
- Brazil
- Prior art keywords
- compound
- phenyl
- oxo
- methyl
- mixture
- Prior art date
Links
- 150000001875 compounds Chemical class 0.000 title claims abstract description 157
- 238000000034 method Methods 0.000 title claims abstract description 43
- 230000001404 mediated effect Effects 0.000 title claims abstract description 20
- 239000008194 pharmaceutical composition Substances 0.000 title claims abstract description 11
- 239000003814 drug Substances 0.000 title claims abstract description 8
- -1 compound compounds Chemical class 0.000 claims abstract description 151
- 150000003839 salts Chemical class 0.000 claims abstract description 13
- 108010055717 JNK Mitogen-Activated Protein Kinases Proteins 0.000 claims description 45
- 102000019145 JUN kinase activity proteins Human genes 0.000 claims description 45
- 125000000217 alkyl group Chemical group 0.000 claims description 39
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 30
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims description 28
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 19
- 208000035475 disorder Diseases 0.000 claims description 16
- 125000003545 alkoxy group Chemical group 0.000 claims description 15
- 229910052801 chlorine Inorganic materials 0.000 claims description 15
- 125000000753 cycloalkyl group Chemical group 0.000 claims description 15
- 229910052739 hydrogen Inorganic materials 0.000 claims description 12
- 238000011282 treatment Methods 0.000 claims description 11
- 238000004519 manufacturing process Methods 0.000 claims description 10
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 9
- 229910052731 fluorine Inorganic materials 0.000 claims description 8
- 125000001072 heteroaryl group Chemical group 0.000 claims description 8
- 125000000623 heterocyclic group Chemical group 0.000 claims description 8
- 206010039073 rheumatoid arthritis Diseases 0.000 claims description 8
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 claims description 7
- 125000002947 alkylene group Chemical group 0.000 claims description 6
- 125000004093 cyano group Chemical group *C#N 0.000 claims description 4
- 125000002993 cycloalkylene group Chemical group 0.000 claims description 4
- 125000000592 heterocycloalkyl group Chemical group 0.000 claims description 3
- 125000006588 heterocycloalkylene group Chemical group 0.000 claims description 3
- 239000000546 pharmaceutical excipient Substances 0.000 claims description 3
- 229910052740 iodine Inorganic materials 0.000 claims description 2
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 claims description 2
- CYRMSUTZVYGINF-UHFFFAOYSA-N trichlorofluoromethane Chemical compound FC(Cl)(Cl)Cl CYRMSUTZVYGINF-UHFFFAOYSA-N 0.000 claims description 2
- 125000001475 halogen functional group Chemical group 0.000 claims 4
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 202
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 177
- 239000000203 mixture Substances 0.000 description 162
- 235000019439 ethyl acetate Nutrition 0.000 description 99
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 84
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 81
- 239000002253 acid Substances 0.000 description 74
- 239000007787 solid Substances 0.000 description 72
- 230000015572 biosynthetic process Effects 0.000 description 70
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 66
- 150000004702 methyl esters Chemical class 0.000 description 66
- 238000003786 synthesis reaction Methods 0.000 description 66
- 230000002829 reductive effect Effects 0.000 description 59
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 48
- 239000000243 solution Substances 0.000 description 48
- 239000000047 product Substances 0.000 description 45
- 239000000843 powder Substances 0.000 description 43
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 43
- 239000011541 reaction mixture Substances 0.000 description 42
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 41
- 238000003756 stirring Methods 0.000 description 37
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 27
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 26
- 238000002360 preparation method Methods 0.000 description 26
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 25
- 239000012044 organic layer Substances 0.000 description 24
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 23
- 238000001035 drying Methods 0.000 description 20
- 230000008569 process Effects 0.000 description 19
- 238000009472 formulation Methods 0.000 description 18
- 239000012267 brine Substances 0.000 description 17
- 238000006243 chemical reaction Methods 0.000 description 17
- 239000000460 chlorine Substances 0.000 description 17
- 239000000377 silicon dioxide Substances 0.000 description 17
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 17
- 239000002904 solvent Substances 0.000 description 17
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 16
- 239000004480 active ingredient Substances 0.000 description 16
- 239000010410 layer Substances 0.000 description 16
- 239000003921 oil Substances 0.000 description 16
- 235000019198 oils Nutrition 0.000 description 16
- 238000010992 reflux Methods 0.000 description 16
- 210000004027 cell Anatomy 0.000 description 15
- 239000000284 extract Substances 0.000 description 15
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 14
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 14
- 239000006260 foam Substances 0.000 description 14
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 13
- 241000700159 Rattus Species 0.000 description 13
- 239000004615 ingredient Substances 0.000 description 13
- CTSLXHKWHWQRSH-UHFFFAOYSA-N oxalyl chloride Chemical compound ClC(=O)C(Cl)=O CTSLXHKWHWQRSH-UHFFFAOYSA-N 0.000 description 13
- 201000010099 disease Diseases 0.000 description 12
- 238000003818 flash chromatography Methods 0.000 description 12
- 230000005764 inhibitory process Effects 0.000 description 12
- KWGKDLIKAYFUFQ-UHFFFAOYSA-M lithium chloride Chemical compound [Li+].[Cl-] KWGKDLIKAYFUFQ-UHFFFAOYSA-M 0.000 description 12
- 238000000746 purification Methods 0.000 description 12
- 238000004809 thin layer chromatography Methods 0.000 description 12
- PAYRUJLWNCNPSJ-UHFFFAOYSA-N Aniline Chemical compound NC1=CC=CC=C1 PAYRUJLWNCNPSJ-UHFFFAOYSA-N 0.000 description 10
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 10
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 10
- 239000002775 capsule Substances 0.000 description 10
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Substances [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 10
- 239000011734 sodium Substances 0.000 description 10
- 239000007858 starting material Substances 0.000 description 10
- 239000003826 tablet Substances 0.000 description 10
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 9
- 239000002552 dosage form Substances 0.000 description 9
- 230000000694 effects Effects 0.000 description 9
- 150000002148 esters Chemical class 0.000 description 9
- 239000000706 filtrate Substances 0.000 description 9
- IKHGUXGNUITLKF-UHFFFAOYSA-N Acetaldehyde Chemical compound CC=O IKHGUXGNUITLKF-UHFFFAOYSA-N 0.000 description 8
- YNPNZTXNASCQKK-UHFFFAOYSA-N Phenanthrene Natural products C1=CC=C2C3=CC=CC=C3C=CC2=C1 YNPNZTXNASCQKK-UHFFFAOYSA-N 0.000 description 8
- 230000004913 activation Effects 0.000 description 8
- 125000004432 carbon atom Chemical group C* 0.000 description 8
- 125000005843 halogen group Chemical group 0.000 description 8
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 7
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 description 7
- YLQBMQCUIZJEEH-UHFFFAOYSA-N Furan Chemical compound C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 7
- 241001465754 Metazoa Species 0.000 description 7
- ZMANZCXQSJIPKH-UHFFFAOYSA-N N,N-Diethylethanamine Substances CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 7
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 7
- 229910052799 carbon Inorganic materials 0.000 description 7
- 239000003153 chemical reaction reagent Substances 0.000 description 7
- ZXUQEPZWVQIOJE-UHFFFAOYSA-N methyl 2-chloro-2-oxoacetate Chemical compound COC(=O)C(Cl)=O ZXUQEPZWVQIOJE-UHFFFAOYSA-N 0.000 description 7
- 229910052757 nitrogen Inorganic materials 0.000 description 7
- 239000002798 polar solvent Substances 0.000 description 7
- 239000000725 suspension Substances 0.000 description 7
- SNLMOXFUCILIPL-UHFFFAOYSA-N 1,8-naphthyridine-2-carboxylic acid Chemical compound C1=CC=NC2=NC(C(=O)O)=CC=C21 SNLMOXFUCILIPL-UHFFFAOYSA-N 0.000 description 6
- HZAXFHJVJLSVMW-UHFFFAOYSA-N 2-Aminoethan-1-ol Chemical compound NCCO HZAXFHJVJLSVMW-UHFFFAOYSA-N 0.000 description 6
- FPQQSJJWHUJYPU-UHFFFAOYSA-N 3-(dimethylamino)propyliminomethylidene-ethylazanium;chloride Chemical compound Cl.CCN=C=NCCCN(C)C FPQQSJJWHUJYPU-UHFFFAOYSA-N 0.000 description 6
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 6
- 101000950695 Homo sapiens Mitogen-activated protein kinase 8 Proteins 0.000 description 6
- 102100037808 Mitogen-activated protein kinase 8 Human genes 0.000 description 6
- SECXISVLQFMRJM-UHFFFAOYSA-N N-Methylpyrrolidone Chemical compound CN1CCCC1=O SECXISVLQFMRJM-UHFFFAOYSA-N 0.000 description 6
- GLUUGHFHXGJENI-UHFFFAOYSA-N Piperazine Chemical compound C1CNCCN1 GLUUGHFHXGJENI-UHFFFAOYSA-N 0.000 description 6
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 6
- 150000001408 amides Chemical class 0.000 description 6
- 206010003246 arthritis Diseases 0.000 description 6
- 125000003118 aryl group Chemical group 0.000 description 6
- 238000003556 assay Methods 0.000 description 6
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 6
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 6
- 239000000839 emulsion Substances 0.000 description 6
- 125000001188 haloalkyl group Chemical group 0.000 description 6
- 238000010438 heat treatment Methods 0.000 description 6
- 230000004054 inflammatory process Effects 0.000 description 6
- 108090000623 proteins and genes Proteins 0.000 description 6
- 125000000472 sulfonyl group Chemical group *S(*)(=O)=O 0.000 description 6
- 230000008961 swelling Effects 0.000 description 6
- 208000024891 symptom Diseases 0.000 description 6
- LMDZBCPBFSXMTL-UHFFFAOYSA-N 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide Substances CCN=C=NCCCN(C)C LMDZBCPBFSXMTL-UHFFFAOYSA-N 0.000 description 5
- 102100023033 Cyclic AMP-dependent transcription factor ATF-2 Human genes 0.000 description 5
- 101000974934 Homo sapiens Cyclic AMP-dependent transcription factor ATF-2 Proteins 0.000 description 5
- 101000997829 Homo sapiens Glial cell line-derived neurotrophic factor Proteins 0.000 description 5
- 206010061218 Inflammation Diseases 0.000 description 5
- WMFOQBRAJBCJND-UHFFFAOYSA-M Lithium hydroxide Chemical compound [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 description 5
- GSEJCLTVZPLZKY-UHFFFAOYSA-N Triethanolamine Chemical compound OCCN(CCO)CCO GSEJCLTVZPLZKY-UHFFFAOYSA-N 0.000 description 5
- 150000001412 amines Chemical class 0.000 description 5
- 238000010171 animal model Methods 0.000 description 5
- 208000006673 asthma Diseases 0.000 description 5
- 239000010949 copper Substances 0.000 description 5
- 125000001316 cycloalkyl alkyl group Chemical group 0.000 description 5
- 239000001257 hydrogen Substances 0.000 description 5
- 125000004435 hydrogen atom Chemical class [H]* 0.000 description 5
- 150000007529 inorganic bases Chemical class 0.000 description 5
- 239000007788 liquid Substances 0.000 description 5
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 5
- 239000000463 material Substances 0.000 description 5
- 150000007530 organic bases Chemical class 0.000 description 5
- 229910000027 potassium carbonate Inorganic materials 0.000 description 5
- 239000002244 precipitate Substances 0.000 description 5
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 5
- 229920006395 saturated elastomer Polymers 0.000 description 5
- 239000007921 spray Substances 0.000 description 5
- 208000001072 type 2 diabetes mellitus Diseases 0.000 description 5
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 4
- FLBAYUMRQUHISI-UHFFFAOYSA-N 1,8-naphthyridine Chemical compound N1=CC=CC2=CC=CN=C21 FLBAYUMRQUHISI-UHFFFAOYSA-N 0.000 description 4
- 125000000954 2-hydroxyethyl group Chemical group [H]C([*])([H])C([H])([H])O[H] 0.000 description 4
- XASOHFCUIQARJT-UHFFFAOYSA-N 8-methoxy-6-[7-(2-morpholin-4-ylethoxy)imidazo[1,2-a]pyridin-3-yl]-2-(2,2,2-trifluoroethyl)-3,4-dihydroisoquinolin-1-one Chemical compound C(N1C(=O)C2=C(OC)C=C(C=3N4C(=NC=3)C=C(C=C4)OCCN3CCOCC3)C=C2CC1)C(F)(F)F XASOHFCUIQARJT-UHFFFAOYSA-N 0.000 description 4
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 4
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 4
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 4
- 239000012825 JNK inhibitor Substances 0.000 description 4
- 241000124008 Mammalia Species 0.000 description 4
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 4
- 241000699670 Mus sp. Species 0.000 description 4
- 206010028980 Neoplasm Diseases 0.000 description 4
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 4
- 239000004698 Polyethylene Substances 0.000 description 4
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 4
- KAESVJOAVNADME-UHFFFAOYSA-N Pyrrole Chemical compound C=1C=CNC=1 KAESVJOAVNADME-UHFFFAOYSA-N 0.000 description 4
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 4
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 4
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 4
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 4
- YKIOKAURTKXMSB-UHFFFAOYSA-N adams's catalyst Chemical compound O=[Pt]=O YKIOKAURTKXMSB-UHFFFAOYSA-N 0.000 description 4
- HUMNYLRZRPPJDN-UHFFFAOYSA-N benzaldehyde Chemical compound O=CC1=CC=CC=C1 HUMNYLRZRPPJDN-UHFFFAOYSA-N 0.000 description 4
- 210000004369 blood Anatomy 0.000 description 4
- 239000008280 blood Substances 0.000 description 4
- 201000011510 cancer Diseases 0.000 description 4
- 239000000969 carrier Substances 0.000 description 4
- 239000002361 compost Substances 0.000 description 4
- 230000034994 death Effects 0.000 description 4
- 239000002270 dispersing agent Substances 0.000 description 4
- 238000001914 filtration Methods 0.000 description 4
- 239000003112 inhibitor Substances 0.000 description 4
- 230000002401 inhibitory effect Effects 0.000 description 4
- 238000002347 injection Methods 0.000 description 4
- 239000007924 injection Substances 0.000 description 4
- NOESYZHRGYRDHS-UHFFFAOYSA-N insulin Chemical compound N1C(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(NC(=O)CN)C(C)CC)CSSCC(C(NC(CO)C(=O)NC(CC(C)C)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CCC(N)=O)C(=O)NC(CC(C)C)C(=O)NC(CCC(O)=O)C(=O)NC(CC(N)=O)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CSSCC(NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2C=CC(O)=CC=2)NC(=O)C(CC(C)C)NC(=O)C(C)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2NC=NC=2)NC(=O)C(CO)NC(=O)CNC2=O)C(=O)NCC(=O)NC(CCC(O)=O)C(=O)NC(CCCNC(N)=N)C(=O)NCC(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC(O)=CC=3)C(=O)NC(C(C)O)C(=O)N3C(CCC3)C(=O)NC(CCCCN)C(=O)NC(C)C(O)=O)C(=O)NC(CC(N)=O)C(O)=O)=O)NC(=O)C(C(C)CC)NC(=O)C(CO)NC(=O)C(C(C)O)NC(=O)C1CSSCC2NC(=O)C(CC(C)C)NC(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CC(N)=O)NC(=O)C(NC(=O)C(N)CC=1C=CC=CC=1)C(C)C)CC1=CN=CN1 NOESYZHRGYRDHS-UHFFFAOYSA-N 0.000 description 4
- 239000007937 lozenge Substances 0.000 description 4
- HWYHDWGGACRVEH-UHFFFAOYSA-N n-methyl-n-(4-pyrrolidin-1-ylbut-2-ynyl)acetamide Chemical compound CC(=O)N(C)CC#CCN1CCCC1 HWYHDWGGACRVEH-UHFFFAOYSA-N 0.000 description 4
- 210000002569 neuron Anatomy 0.000 description 4
- 230000037361 pathway Effects 0.000 description 4
- 229960005141 piperazine Drugs 0.000 description 4
- 239000003586 protic polar solvent Substances 0.000 description 4
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 4
- 238000000926 separation method Methods 0.000 description 4
- 239000003381 stabilizer Substances 0.000 description 4
- 125000001424 substituent group Chemical group 0.000 description 4
- 239000000829 suppository Substances 0.000 description 4
- 239000000375 suspending agent Substances 0.000 description 4
- 230000001225 therapeutic effect Effects 0.000 description 4
- 239000002562 thickening agent Substances 0.000 description 4
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 4
- CQHYICHMGNSGQH-UHFFFAOYSA-N 1,3-oxazole-2-carboxylic acid Chemical compound OC(=O)C1=NC=CO1 CQHYICHMGNSGQH-UHFFFAOYSA-N 0.000 description 3
- DURPTKYDGMDSBL-UHFFFAOYSA-N 1-butoxybutane Chemical compound CCCCOCCCC DURPTKYDGMDSBL-UHFFFAOYSA-N 0.000 description 3
- 125000004200 2-methoxyethyl group Chemical group [H]C([H])([H])OC([H])([H])C([H])([H])* 0.000 description 3
- PCPUKVSTMLHXQF-UHFFFAOYSA-N 4-formylbenzenesulfonamide Chemical compound NS(=O)(=O)C1=CC=C(C=O)C=C1 PCPUKVSTMLHXQF-UHFFFAOYSA-N 0.000 description 3
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 3
- 102000004127 Cytokines Human genes 0.000 description 3
- 108090000695 Cytokines Proteins 0.000 description 3
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 3
- 238000008157 ELISA kit Methods 0.000 description 3
- 102000004190 Enzymes Human genes 0.000 description 3
- 108090000790 Enzymes Proteins 0.000 description 3
- 108010010803 Gelatin Proteins 0.000 description 3
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 3
- 101000950669 Homo sapiens Mitogen-activated protein kinase 9 Proteins 0.000 description 3
- QIGBRXMKCJKVMJ-UHFFFAOYSA-N Hydroquinone Chemical compound OC1=CC=C(O)C=C1 QIGBRXMKCJKVMJ-UHFFFAOYSA-N 0.000 description 3
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 3
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 3
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 3
- 235000010643 Leucaena leucocephala Nutrition 0.000 description 3
- 240000007472 Leucaena leucocephala Species 0.000 description 3
- 102100037809 Mitogen-activated protein kinase 9 Human genes 0.000 description 3
- 241000699666 Mus <mouse, genus> Species 0.000 description 3
- 229910004809 Na2 SO4 Inorganic materials 0.000 description 3
- 239000002202 Polyethylene glycol Substances 0.000 description 3
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 3
- NBBJYMSMWIIQGU-UHFFFAOYSA-N Propionic aldehyde Chemical compound CCC=O NBBJYMSMWIIQGU-UHFFFAOYSA-N 0.000 description 3
- 241000283984 Rodentia Species 0.000 description 3
- 229920002472 Starch Polymers 0.000 description 3
- 108060008682 Tumor Necrosis Factor Proteins 0.000 description 3
- DPXJVFZANSGRMM-UHFFFAOYSA-N acetic acid;2,3,4,5,6-pentahydroxyhexanal;sodium Chemical compound [Na].CC(O)=O.OCC(O)C(O)C(O)C(O)C=O DPXJVFZANSGRMM-UHFFFAOYSA-N 0.000 description 3
- 125000002252 acyl group Chemical group 0.000 description 3
- 239000002671 adjuvant Substances 0.000 description 3
- 230000006907 apoptotic process Effects 0.000 description 3
- 230000002917 arthritic effect Effects 0.000 description 3
- YCOXTKKNXUZSKD-UHFFFAOYSA-N as-o-xylenol Natural products CC1=CC=C(O)C=C1C YCOXTKKNXUZSKD-UHFFFAOYSA-N 0.000 description 3
- 125000004429 atom Chemical group 0.000 description 3
- 230000037396 body weight Effects 0.000 description 3
- 125000003917 carbamoyl group Chemical group [H]N([H])C(*)=O 0.000 description 3
- 239000003054 catalyst Substances 0.000 description 3
- 239000003795 chemical substances by application Substances 0.000 description 3
- 208000037976 chronic inflammation Diseases 0.000 description 3
- 229910052802 copper Inorganic materials 0.000 description 3
- 230000006866 deterioration Effects 0.000 description 3
- 206010012601 diabetes mellitus Diseases 0.000 description 3
- ZUOUZKKEUPVFJK-UHFFFAOYSA-N diphenyl Chemical compound C1=CC=CC=C1C1=CC=CC=C1 ZUOUZKKEUPVFJK-UHFFFAOYSA-N 0.000 description 3
- 229940079593 drug Drugs 0.000 description 3
- 239000003937 drug carrier Substances 0.000 description 3
- 229940088598 enzyme Drugs 0.000 description 3
- 235000019441 ethanol Nutrition 0.000 description 3
- 230000006870 function Effects 0.000 description 3
- 239000007789 gas Substances 0.000 description 3
- 239000000499 gel Substances 0.000 description 3
- 239000008273 gelatin Substances 0.000 description 3
- 229920000159 gelatin Polymers 0.000 description 3
- 235000019322 gelatine Nutrition 0.000 description 3
- 235000011852 gelatine desserts Nutrition 0.000 description 3
- 230000002068 genetic effect Effects 0.000 description 3
- 239000011521 glass Substances 0.000 description 3
- 229910052736 halogen Inorganic materials 0.000 description 3
- 150000002367 halogens Chemical class 0.000 description 3
- 125000005842 heteroatom Chemical group 0.000 description 3
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 3
- 230000002757 inflammatory effect Effects 0.000 description 3
- SNHMUERNLJLMHN-UHFFFAOYSA-N iodobenzene Chemical compound IC1=CC=CC=C1 SNHMUERNLJLMHN-UHFFFAOYSA-N 0.000 description 3
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 3
- 239000008101 lactose Substances 0.000 description 3
- GRVDJDISBSALJP-UHFFFAOYSA-N methyloxidanyl Chemical group [O]C GRVDJDISBSALJP-UHFFFAOYSA-N 0.000 description 3
- 239000007922 nasal spray Substances 0.000 description 3
- 229940097496 nasal spray Drugs 0.000 description 3
- 238000013116 obese mouse model Methods 0.000 description 3
- 239000012074 organic phase Substances 0.000 description 3
- 229910052760 oxygen Inorganic materials 0.000 description 3
- QNGNSVIICDLXHT-UHFFFAOYSA-N para-ethylbenzaldehyde Natural products CCC1=CC=C(C=O)C=C1 QNGNSVIICDLXHT-UHFFFAOYSA-N 0.000 description 3
- 229920001223 polyethylene glycol Polymers 0.000 description 3
- 229920006316 polyvinylpyrrolidine Polymers 0.000 description 3
- 235000011181 potassium carbonates Nutrition 0.000 description 3
- NLKNQRATVPKPDG-UHFFFAOYSA-M potassium iodide Chemical compound [K+].[I-] NLKNQRATVPKPDG-UHFFFAOYSA-M 0.000 description 3
- 108090000765 processed proteins & peptides Proteins 0.000 description 3
- 235000018102 proteins Nutrition 0.000 description 3
- 102000004169 proteins and genes Human genes 0.000 description 3
- 150000003254 radicals Chemical class 0.000 description 3
- 229930195734 saturated hydrocarbon Natural products 0.000 description 3
- 230000011664 signaling Effects 0.000 description 3
- 239000000741 silica gel Substances 0.000 description 3
- 229910002027 silica gel Inorganic materials 0.000 description 3
- 239000002002 slurry Substances 0.000 description 3
- 229910052708 sodium Inorganic materials 0.000 description 3
- 239000008107 starch Substances 0.000 description 3
- 235000019698 starch Nutrition 0.000 description 3
- 239000000758 substrate Substances 0.000 description 3
- 229940124530 sulfonamide Drugs 0.000 description 3
- 150000003456 sulfonamides Chemical class 0.000 description 3
- 229910052717 sulfur Inorganic materials 0.000 description 3
- 230000004083 survival effect Effects 0.000 description 3
- 238000012360 testing method Methods 0.000 description 3
- 210000001519 tissue Anatomy 0.000 description 3
- 239000003039 volatile agent Substances 0.000 description 3
- GLGNXYJARSMNGJ-VKTIVEEGSA-N (1s,2s,3r,4r)-3-[[5-chloro-2-[(1-ethyl-6-methoxy-2-oxo-4,5-dihydro-3h-1-benzazepin-7-yl)amino]pyrimidin-4-yl]amino]bicyclo[2.2.1]hept-5-ene-2-carboxamide Chemical compound CCN1C(=O)CCCC2=C(OC)C(NC=3N=C(C(=CN=3)Cl)N[C@H]3[C@H]([C@@]4([H])C[C@@]3(C=C4)[H])C(N)=O)=CC=C21 GLGNXYJARSMNGJ-VKTIVEEGSA-N 0.000 description 2
- IUSARDYWEPUTPN-OZBXUNDUSA-N (2r)-n-[(2s,3r)-4-[[(4s)-6-(2,2-dimethylpropyl)spiro[3,4-dihydropyrano[2,3-b]pyridine-2,1'-cyclobutane]-4-yl]amino]-3-hydroxy-1-[3-(1,3-thiazol-2-yl)phenyl]butan-2-yl]-2-methoxypropanamide Chemical compound C([C@H](NC(=O)[C@@H](C)OC)[C@H](O)CN[C@@H]1C2=CC(CC(C)(C)C)=CN=C2OC2(CCC2)C1)C(C=1)=CC=CC=1C1=NC=CS1 IUSARDYWEPUTPN-OZBXUNDUSA-N 0.000 description 2
- MZOFCQQQCNRIBI-VMXHOPILSA-N (3s)-4-[[(2s)-1-[[(2s)-1-[[(1s)-1-carboxy-2-hydroxyethyl]amino]-4-methyl-1-oxopentan-2-yl]amino]-5-(diaminomethylideneamino)-1-oxopentan-2-yl]amino]-3-[[2-[[(2s)-2,6-diaminohexanoyl]amino]acetyl]amino]-4-oxobutanoic acid Chemical compound OC[C@@H](C(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCN=C(N)N)NC(=O)[C@H](CC(O)=O)NC(=O)CNC(=O)[C@@H](N)CCCCN MZOFCQQQCNRIBI-VMXHOPILSA-N 0.000 description 2
- MIOPJNTWMNEORI-GMSGAONNSA-N (S)-camphorsulfonic acid Chemical compound C1C[C@@]2(CS(O)(=O)=O)C(=O)C[C@@H]1C2(C)C MIOPJNTWMNEORI-GMSGAONNSA-N 0.000 description 2
- DEVSOMFAQLZNKR-RJRFIUFISA-N (z)-3-[3-[3,5-bis(trifluoromethyl)phenyl]-1,2,4-triazol-1-yl]-n'-pyrazin-2-ylprop-2-enehydrazide Chemical compound FC(F)(F)C1=CC(C(F)(F)F)=CC(C2=NN(\C=C/C(=O)NNC=3N=CC=NC=3)C=N2)=C1 DEVSOMFAQLZNKR-RJRFIUFISA-N 0.000 description 2
- QRAIXSHHSMHLNM-UHFFFAOYSA-N 1,4-dihydroquinoline-2-carboxylic acid Chemical compound C1=CC=C2NC(C(=O)O)=CCC2=C1 QRAIXSHHSMHLNM-UHFFFAOYSA-N 0.000 description 2
- IIZPXYDJLKNOIY-JXPKJXOSSA-N 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCC\C=C/C\C=C/C\C=C/C\C=C/CCCCC IIZPXYDJLKNOIY-JXPKJXOSSA-N 0.000 description 2
- QEBYEVQKHRUYPE-UHFFFAOYSA-N 2-(2-chlorophenyl)-5-[(1-methylpyrazol-3-yl)methyl]-4-[[methyl(pyridin-3-ylmethyl)amino]methyl]-1h-pyrazolo[4,3-c]pyridine-3,6-dione Chemical compound C1=CN(C)N=C1CN1C(=O)C=C2NN(C=3C(=CC=CC=3)Cl)C(=O)C2=C1CN(C)CC1=CC=CN=C1 QEBYEVQKHRUYPE-UHFFFAOYSA-N 0.000 description 2
- ZYHQGITXIJDDKC-UHFFFAOYSA-N 2-[2-(2-aminophenyl)ethyl]aniline Chemical group NC1=CC=CC=C1CCC1=CC=CC=C1N ZYHQGITXIJDDKC-UHFFFAOYSA-N 0.000 description 2
- ZNQVEEAIQZEUHB-UHFFFAOYSA-N 2-ethoxyethanol Chemical compound CCOCCO ZNQVEEAIQZEUHB-UHFFFAOYSA-N 0.000 description 2
- DFRAKBCRUYUFNT-UHFFFAOYSA-N 3,8-dicyclohexyl-2,4,7,9-tetrahydro-[1,3]oxazino[5,6-h][1,3]benzoxazine Chemical compound C1CCCCC1N1CC(C=CC2=C3OCN(C2)C2CCCCC2)=C3OC1 DFRAKBCRUYUFNT-UHFFFAOYSA-N 0.000 description 2
- YOXPNCGDCLXSFG-MDWZMJQESA-N 4-[(e)-3-(2-anilino-4-chlorophenyl)-3-oxoprop-1-enyl]benzoic acid Chemical compound C1=CC(C(=O)O)=CC=C1\C=C\C(=O)C1=CC=C(Cl)C=C1NC1=CC=CC=C1 YOXPNCGDCLXSFG-MDWZMJQESA-N 0.000 description 2
- CVICEEPAFUYBJG-UHFFFAOYSA-N 5-chloro-2,2-difluoro-1,3-benzodioxole Chemical group C1=C(Cl)C=C2OC(F)(F)OC2=C1 CVICEEPAFUYBJG-UHFFFAOYSA-N 0.000 description 2
- AHSPOVNPXMDIEL-UHFFFAOYSA-N 6-methyl-n-phenylpyridin-2-amine Chemical compound CC1=CC=CC(NC=2C=CC=CC=2)=N1 AHSPOVNPXMDIEL-UHFFFAOYSA-N 0.000 description 2
- JRLTTZUODKEYDH-UHFFFAOYSA-N 8-methylquinoline Chemical group C1=CN=C2C(C)=CC=CC2=C1 JRLTTZUODKEYDH-UHFFFAOYSA-N 0.000 description 2
- 208000024827 Alzheimer disease Diseases 0.000 description 2
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 2
- 241000416162 Astragalus gummifer Species 0.000 description 2
- KXDAEFPNCMNJSK-UHFFFAOYSA-N Benzamide Chemical compound NC(=O)C1=CC=CC=C1 KXDAEFPNCMNJSK-UHFFFAOYSA-N 0.000 description 2
- 239000005711 Benzoic acid Substances 0.000 description 2
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 2
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 2
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 2
- 102000008186 Collagen Human genes 0.000 description 2
- 108010035532 Collagen Proteins 0.000 description 2
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 2
- QUSNBJAOOMFDIB-UHFFFAOYSA-N Ethylamine Chemical compound CCN QUSNBJAOOMFDIB-UHFFFAOYSA-N 0.000 description 2
- 208000009386 Experimental Arthritis Diseases 0.000 description 2
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 2
- AEMRFAOFKBGASW-UHFFFAOYSA-N Glycolic acid Chemical compound OCC(O)=O AEMRFAOFKBGASW-UHFFFAOYSA-N 0.000 description 2
- HTTJABKRGRZYRN-UHFFFAOYSA-N Heparin Chemical compound OC1C(NC(=O)C)C(O)OC(COS(O)(=O)=O)C1OC1C(OS(O)(=O)=O)C(O)C(OC2C(C(OS(O)(=O)=O)C(OC3C(C(O)C(O)C(O3)C(O)=O)OS(O)(=O)=O)C(CO)O2)NS(O)(=O)=O)C(C(O)=O)O1 HTTJABKRGRZYRN-UHFFFAOYSA-N 0.000 description 2
- 101001050288 Homo sapiens Transcription factor Jun Proteins 0.000 description 2
- WTDHULULXKLSOZ-UHFFFAOYSA-N Hydroxylamine hydrochloride Chemical compound Cl.ON WTDHULULXKLSOZ-UHFFFAOYSA-N 0.000 description 2
- 206010020751 Hypersensitivity Diseases 0.000 description 2
- 102000004877 Insulin Human genes 0.000 description 2
- 108090001061 Insulin Proteins 0.000 description 2
- 108010034219 Insulin Receptor Substrate Proteins Proteins 0.000 description 2
- 206010022489 Insulin Resistance Diseases 0.000 description 2
- 102100025087 Insulin receptor substrate 1 Human genes 0.000 description 2
- 108090001005 Interleukin-6 Proteins 0.000 description 2
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 2
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 2
- FEIOASZZURHTHB-UHFFFAOYSA-N Methyl-p-formylbenzoate Natural products COC(=O)C1=CC=C(C=O)C=C1 FEIOASZZURHTHB-UHFFFAOYSA-N 0.000 description 2
- 208000012902 Nervous system disease Diseases 0.000 description 2
- 208000025966 Neurological disease Diseases 0.000 description 2
- DFPAKSUCGFBDDF-UHFFFAOYSA-N Nicotinamide Chemical compound NC(=O)C1=CC=CN=C1 DFPAKSUCGFBDDF-UHFFFAOYSA-N 0.000 description 2
- 208000018737 Parkinson disease Diseases 0.000 description 2
- 108091000080 Phosphotransferase Proteins 0.000 description 2
- ATUOYWHBWRKTHZ-UHFFFAOYSA-N Propane Chemical compound CCC ATUOYWHBWRKTHZ-UHFFFAOYSA-N 0.000 description 2
- LCTONWCANYUPML-UHFFFAOYSA-N Pyruvic acid Chemical compound CC(=O)C(O)=O LCTONWCANYUPML-UHFFFAOYSA-N 0.000 description 2
- 241000219061 Rheum Species 0.000 description 2
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 2
- 208000006011 Stroke Diseases 0.000 description 2
- RAHZWNYVWXNFOC-UHFFFAOYSA-N Sulphur dioxide Chemical compound O=S=O RAHZWNYVWXNFOC-UHFFFAOYSA-N 0.000 description 2
- 210000001744 T-lymphocyte Anatomy 0.000 description 2
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 2
- 229920001615 Tragacanth Polymers 0.000 description 2
- 108091023040 Transcription factor Proteins 0.000 description 2
- 102000040945 Transcription factor Human genes 0.000 description 2
- 102100023132 Transcription factor Jun Human genes 0.000 description 2
- 102000000852 Tumor Necrosis Factor-alpha Human genes 0.000 description 2
- LNUFLCYMSVYYNW-ZPJMAFJPSA-N [(2r,3r,4s,5r,6r)-2-[(2r,3r,4s,5r,6r)-6-[(2r,3r,4s,5r,6r)-6-[(2r,3r,4s,5r,6r)-6-[[(3s,5s,8r,9s,10s,13r,14s,17r)-10,13-dimethyl-17-[(2r)-6-methylheptan-2-yl]-2,3,4,5,6,7,8,9,11,12,14,15,16,17-tetradecahydro-1h-cyclopenta[a]phenanthren-3-yl]oxy]-4,5-disulfo Chemical compound O([C@@H]1[C@@H](COS(O)(=O)=O)O[C@@H]([C@@H]([C@H]1OS(O)(=O)=O)OS(O)(=O)=O)O[C@@H]1[C@@H](COS(O)(=O)=O)O[C@@H]([C@@H]([C@H]1OS(O)(=O)=O)OS(O)(=O)=O)O[C@@H]1[C@@H](COS(O)(=O)=O)O[C@H]([C@@H]([C@H]1OS(O)(=O)=O)OS(O)(=O)=O)O[C@@H]1C[C@@H]2CC[C@H]3[C@@H]4CC[C@@H]([C@]4(CC[C@@H]3[C@@]2(C)CC1)C)[C@H](C)CCCC(C)C)[C@H]1O[C@H](COS(O)(=O)=O)[C@@H](OS(O)(=O)=O)[C@H](OS(O)(=O)=O)[C@H]1OS(O)(=O)=O LNUFLCYMSVYYNW-ZPJMAFJPSA-N 0.000 description 2
- DGEZNRSVGBDHLK-UHFFFAOYSA-N [1,10]phenanthroline Chemical compound C1=CN=C2C3=NC=CC=C3C=CC2=C1 DGEZNRSVGBDHLK-UHFFFAOYSA-N 0.000 description 2
- 239000000443 aerosol Substances 0.000 description 2
- 238000013019 agitation Methods 0.000 description 2
- 208000026935 allergic disease Diseases 0.000 description 2
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 2
- 125000004397 aminosulfonyl group Chemical group NS(=O)(=O)* 0.000 description 2
- 239000007864 aqueous solution Substances 0.000 description 2
- 239000007900 aqueous suspension Substances 0.000 description 2
- 230000008901 benefit Effects 0.000 description 2
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 2
- 239000004305 biphenyl Substances 0.000 description 2
- 235000010290 biphenyl Nutrition 0.000 description 2
- 230000000903 blocking effect Effects 0.000 description 2
- 210000004556 brain Anatomy 0.000 description 2
- 239000000872 buffer Substances 0.000 description 2
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 2
- 239000001768 carboxy methyl cellulose Substances 0.000 description 2
- 208000015114 central nervous system disease Diseases 0.000 description 2
- NEHMKBQYUWJMIP-UHFFFAOYSA-N chloromethane Chemical compound ClC NEHMKBQYUWJMIP-UHFFFAOYSA-N 0.000 description 2
- 238000004587 chromatography analysis Methods 0.000 description 2
- 230000006020 chronic inflammation Effects 0.000 description 2
- 229920001436 collagen Polymers 0.000 description 2
- 239000003086 colorant Substances 0.000 description 2
- 239000002131 composite material Substances 0.000 description 2
- 229940125758 compound 15 Drugs 0.000 description 2
- 229940125782 compound 2 Drugs 0.000 description 2
- 229940125807 compound 37 Drugs 0.000 description 2
- 229940127573 compound 38 Drugs 0.000 description 2
- 239000007822 coupling agent Substances 0.000 description 2
- 239000006071 cream Substances 0.000 description 2
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 2
- 230000006378 damage Effects 0.000 description 2
- 238000011161 development Methods 0.000 description 2
- 239000003085 diluting agent Substances 0.000 description 2
- XBDQKXXYIPTUBI-UHFFFAOYSA-N dimethylselenoniopropionate Natural products CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 2
- 239000000975 dye Substances 0.000 description 2
- 238000006911 enzymatic reaction Methods 0.000 description 2
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 2
- 238000001704 evaporation Methods 0.000 description 2
- 230000008020 evaporation Effects 0.000 description 2
- 230000003492 excitotoxic effect Effects 0.000 description 2
- 231100000063 excitotoxicity Toxicity 0.000 description 2
- 239000000796 flavoring agent Substances 0.000 description 2
- 239000001530 fumaric acid Substances 0.000 description 2
- 239000003365 glass fiber Substances 0.000 description 2
- 239000008103 glucose Substances 0.000 description 2
- 229930195712 glutamate Natural products 0.000 description 2
- 230000036541 health Effects 0.000 description 2
- 229960002897 heparin Drugs 0.000 description 2
- 229920000669 heparin Polymers 0.000 description 2
- 150000002430 hydrocarbons Chemical group 0.000 description 2
- 238000005984 hydrogenation reaction Methods 0.000 description 2
- 238000001727 in vivo Methods 0.000 description 2
- 230000006698 induction Effects 0.000 description 2
- 239000012442 inert solvent Substances 0.000 description 2
- 208000027866 inflammatory disease Diseases 0.000 description 2
- 238000001802 infusion Methods 0.000 description 2
- 229940125396 insulin Drugs 0.000 description 2
- 239000000543 intermediate Substances 0.000 description 2
- 230000000302 ischemic effect Effects 0.000 description 2
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 2
- 150000002576 ketones Chemical class 0.000 description 2
- JVTAAEKCZFNVCJ-UHFFFAOYSA-N lactic acid Chemical compound CC(O)C(O)=O JVTAAEKCZFNVCJ-UHFFFAOYSA-N 0.000 description 2
- 239000000787 lecithin Substances 0.000 description 2
- 235000010445 lecithin Nutrition 0.000 description 2
- 229940067606 lecithin Drugs 0.000 description 2
- 230000000670 limiting effect Effects 0.000 description 2
- 229910052744 lithium Inorganic materials 0.000 description 2
- 239000006210 lotion Substances 0.000 description 2
- 235000019359 magnesium stearate Nutrition 0.000 description 2
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 2
- 239000011976 maleic acid Substances 0.000 description 2
- 238000005259 measurement Methods 0.000 description 2
- 238000002844 melting Methods 0.000 description 2
- 230000008018 melting Effects 0.000 description 2
- 239000012528 membrane Substances 0.000 description 2
- 208000030159 metabolic disease Diseases 0.000 description 2
- 229910021645 metal ion Inorganic materials 0.000 description 2
- 229940098779 methanesulfonic acid Drugs 0.000 description 2
- LXCFILQKKLGQFO-UHFFFAOYSA-N methylparaben Chemical compound COC(=O)C1=CC=C(O)C=C1 LXCFILQKKLGQFO-UHFFFAOYSA-N 0.000 description 2
- 239000002480 mineral oil Substances 0.000 description 2
- 235000010446 mineral oil Nutrition 0.000 description 2
- 210000003928 nasal cavity Anatomy 0.000 description 2
- 230000004770 neurodegeneration Effects 0.000 description 2
- 208000015122 neurodegenerative disease Diseases 0.000 description 2
- 239000011570 nicotinamide Substances 0.000 description 2
- 229960003966 nicotinamide Drugs 0.000 description 2
- 239000002674 ointment Substances 0.000 description 2
- PIDFDZJZLOTZTM-KHVQSSSXSA-N ombitasvir Chemical compound COC(=O)N[C@@H](C(C)C)C(=O)N1CCC[C@H]1C(=O)NC1=CC=C([C@H]2N([C@@H](CC2)C=2C=CC(NC(=O)[C@H]3N(CCC3)C(=O)[C@@H](NC(=O)OC)C(C)C)=CC=2)C=2C=CC(=CC=2)C(C)(C)C)C=C1 PIDFDZJZLOTZTM-KHVQSSSXSA-N 0.000 description 2
- 230000036542 oxidative stress Effects 0.000 description 2
- 229910052763 palladium Inorganic materials 0.000 description 2
- FJKROLUGYXJWQN-UHFFFAOYSA-N papa-hydroxy-benzoic acid Natural products OC(=O)C1=CC=C(O)C=C1 FJKROLUGYXJWQN-UHFFFAOYSA-N 0.000 description 2
- 238000007911 parenteral administration Methods 0.000 description 2
- FDPIMTJIUBPUKL-UHFFFAOYSA-N pentan-3-one Chemical compound CCC(=O)CC FDPIMTJIUBPUKL-UHFFFAOYSA-N 0.000 description 2
- 239000000825 pharmaceutical preparation Substances 0.000 description 2
- 230000000144 pharmacologic effect Effects 0.000 description 2
- 230000026731 phosphorylation Effects 0.000 description 2
- 238000006366 phosphorylation reaction Methods 0.000 description 2
- 102000020233 phosphotransferase Human genes 0.000 description 2
- 239000006187 pill Substances 0.000 description 2
- HDOWRFHMPULYOA-UHFFFAOYSA-N piperidin-4-ol Chemical compound OC1CCNCC1 HDOWRFHMPULYOA-UHFFFAOYSA-N 0.000 description 2
- 229920000136 polysorbate Polymers 0.000 description 2
- 235000015320 potassium carbonate Nutrition 0.000 description 2
- 230000000770 proinflammatory effect Effects 0.000 description 2
- 230000000069 prophylactic effect Effects 0.000 description 2
- 125000001501 propionyl group Chemical group O=C([*])C([H])([H])C([H])([H])[H] 0.000 description 2
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 2
- 125000006239 protecting group Chemical group 0.000 description 2
- 125000003226 pyrazolyl group Chemical group 0.000 description 2
- 239000011347 resin Substances 0.000 description 2
- 229920005989 resin Polymers 0.000 description 2
- CVHZOJJKTDOEJC-UHFFFAOYSA-N saccharin Chemical compound C1=CC=C2C(=O)NS(=O)(=O)C2=C1 CVHZOJJKTDOEJC-UHFFFAOYSA-N 0.000 description 2
- 235000019204 saccharin Nutrition 0.000 description 2
- 229940081974 saccharin Drugs 0.000 description 2
- 239000000901 saccharin and its Na,K and Ca salt Substances 0.000 description 2
- YGSDEFSMJLZEOE-UHFFFAOYSA-N salicylic acid Chemical compound OC(=O)C1=CC=CC=C1O YGSDEFSMJLZEOE-UHFFFAOYSA-N 0.000 description 2
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 2
- 235000017557 sodium bicarbonate Nutrition 0.000 description 2
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 2
- 235000019812 sodium carboxymethyl cellulose Nutrition 0.000 description 2
- 229920001027 sodium carboxymethylcellulose Polymers 0.000 description 2
- 239000011780 sodium chloride Substances 0.000 description 2
- 239000000126 substance Substances 0.000 description 2
- 238000006467 substitution reaction Methods 0.000 description 2
- 239000011593 sulfur Substances 0.000 description 2
- 230000002459 sustained effect Effects 0.000 description 2
- 230000009897 systematic effect Effects 0.000 description 2
- 239000011975 tartaric acid Substances 0.000 description 2
- 235000002906 tartaric acid Nutrition 0.000 description 2
- 125000001544 thienyl group Chemical group 0.000 description 2
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 2
- 238000011200 topical administration Methods 0.000 description 2
- 239000000196 tragacanth Substances 0.000 description 2
- 235000010487 tragacanth Nutrition 0.000 description 2
- 229940116362 tragacanth Drugs 0.000 description 2
- 239000003981 vehicle Substances 0.000 description 2
- 239000008215 water for injection Substances 0.000 description 2
- UAOUIVVJBYDFKD-XKCDOFEDSA-N (1R,9R,10S,11R,12R,15S,18S,21R)-10,11,21-trihydroxy-8,8-dimethyl-14-methylidene-4-(prop-2-enylamino)-20-oxa-5-thia-3-azahexacyclo[9.7.2.112,15.01,9.02,6.012,18]henicosa-2(6),3-dien-13-one Chemical compound C([C@@H]1[C@@H](O)[C@@]23C(C1=C)=O)C[C@H]2[C@]12C(N=C(NCC=C)S4)=C4CC(C)(C)[C@H]1[C@H](O)[C@]3(O)OC2 UAOUIVVJBYDFKD-XKCDOFEDSA-N 0.000 description 1
- AOSZTAHDEDLTLQ-AZKQZHLXSA-N (1S,2S,4R,8S,9S,11S,12R,13S,19S)-6-[(3-chlorophenyl)methyl]-12,19-difluoro-11-hydroxy-8-(2-hydroxyacetyl)-9,13-dimethyl-6-azapentacyclo[10.8.0.02,9.04,8.013,18]icosa-14,17-dien-16-one Chemical compound C([C@@H]1C[C@H]2[C@H]3[C@]([C@]4(C=CC(=O)C=C4[C@@H](F)C3)C)(F)[C@@H](O)C[C@@]2([C@@]1(C1)C(=O)CO)C)N1CC1=CC=CC(Cl)=C1 AOSZTAHDEDLTLQ-AZKQZHLXSA-N 0.000 description 1
- ZFSXKSSWYSZPGQ-TYSVMGFPSA-N (1r,2r)-2-aminocyclopentan-1-ol;hydrochloride Chemical class Cl.N[C@@H]1CCC[C@H]1O ZFSXKSSWYSZPGQ-TYSVMGFPSA-N 0.000 description 1
- ABJSOROVZZKJGI-OCYUSGCXSA-N (1r,2r,4r)-2-(4-bromophenyl)-n-[(4-chlorophenyl)-(2-fluoropyridin-4-yl)methyl]-4-morpholin-4-ylcyclohexane-1-carboxamide Chemical compound C1=NC(F)=CC(C(NC(=O)[C@H]2[C@@H](C[C@@H](CC2)N2CCOCC2)C=2C=CC(Br)=CC=2)C=2C=CC(Cl)=CC=2)=C1 ABJSOROVZZKJGI-OCYUSGCXSA-N 0.000 description 1
- YHFYRVZIONNYSM-RFZPGFLSSA-N (1r,3r)-3-aminocyclopentan-1-ol Chemical compound N[C@@H]1CC[C@@H](O)C1 YHFYRVZIONNYSM-RFZPGFLSSA-N 0.000 description 1
- SZUVGFMDDVSKSI-WIFOCOSTSA-N (1s,2s,3s,5r)-1-(carboxymethyl)-3,5-bis[(4-phenoxyphenyl)methyl-propylcarbamoyl]cyclopentane-1,2-dicarboxylic acid Chemical compound O=C([C@@H]1[C@@H]([C@](CC(O)=O)([C@H](C(=O)N(CCC)CC=2C=CC(OC=3C=CC=CC=3)=CC=2)C1)C(O)=O)C(O)=O)N(CCC)CC(C=C1)=CC=C1OC1=CC=CC=C1 SZUVGFMDDVSKSI-WIFOCOSTSA-N 0.000 description 1
- QBYIENPQHBMVBV-HFEGYEGKSA-N (2R)-2-hydroxy-2-phenylacetic acid Chemical compound O[C@@H](C(O)=O)c1ccccc1.O[C@@H](C(O)=O)c1ccccc1 QBYIENPQHBMVBV-HFEGYEGKSA-N 0.000 description 1
- GHYOCDFICYLMRF-UTIIJYGPSA-N (2S,3R)-N-[(2S)-3-(cyclopenten-1-yl)-1-[(2R)-2-methyloxiran-2-yl]-1-oxopropan-2-yl]-3-hydroxy-3-(4-methoxyphenyl)-2-[[(2S)-2-[(2-morpholin-4-ylacetyl)amino]propanoyl]amino]propanamide Chemical compound C1(=CCCC1)C[C@@H](C(=O)[C@@]1(OC1)C)NC([C@H]([C@@H](C1=CC=C(C=C1)OC)O)NC([C@H](C)NC(CN1CCOCC1)=O)=O)=O GHYOCDFICYLMRF-UTIIJYGPSA-N 0.000 description 1
- JNYAEWCLZODPBN-JGWLITMVSA-N (2r,3r,4s)-2-[(1r)-1,2-dihydroxyethyl]oxolane-3,4-diol Chemical compound OC[C@@H](O)[C@H]1OC[C@H](O)[C@H]1O JNYAEWCLZODPBN-JGWLITMVSA-N 0.000 description 1
- VIJSPAIQWVPKQZ-BLECARSGSA-N (2s)-2-[[(2s)-2-[[(2s)-2-[[(2s)-2-[[(2s)-2-[[(2s)-2-acetamido-5-(diaminomethylideneamino)pentanoyl]amino]-4-methylpentanoyl]amino]-4,4-dimethylpentanoyl]amino]-4-methylpentanoyl]amino]propanoyl]amino]-5-(diaminomethylideneamino)pentanoic acid Chemical compound NC(=N)NCCC[C@@H](C(O)=O)NC(=O)[C@H](C)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(C)(C)C)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCNC(N)=N)NC(C)=O VIJSPAIQWVPKQZ-BLECARSGSA-N 0.000 description 1
- WWTBZEKOSBFBEM-SPWPXUSOSA-N (2s)-2-[[2-benzyl-3-[hydroxy-[(1r)-2-phenyl-1-(phenylmethoxycarbonylamino)ethyl]phosphoryl]propanoyl]amino]-3-(1h-indol-3-yl)propanoic acid Chemical compound N([C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)O)C(=O)C(CP(O)(=O)[C@H](CC=1C=CC=CC=1)NC(=O)OCC=1C=CC=CC=1)CC1=CC=CC=C1 WWTBZEKOSBFBEM-SPWPXUSOSA-N 0.000 description 1
- QFLWZFQWSBQYPS-AWRAUJHKSA-N (3S)-3-[[(2S)-2-[[(2S)-2-[5-[(3aS,6aR)-2-oxo-1,3,3a,4,6,6a-hexahydrothieno[3,4-d]imidazol-4-yl]pentanoylamino]-3-methylbutanoyl]amino]-3-(4-hydroxyphenyl)propanoyl]amino]-4-[1-bis(4-chlorophenoxy)phosphorylbutylamino]-4-oxobutanoic acid Chemical compound CCCC(NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](Cc1ccc(O)cc1)NC(=O)[C@@H](NC(=O)CCCCC1SC[C@@H]2NC(=O)N[C@H]12)C(C)C)P(=O)(Oc1ccc(Cl)cc1)Oc1ccc(Cl)cc1 QFLWZFQWSBQYPS-AWRAUJHKSA-N 0.000 description 1
- FNHHVPPSBFQMEL-KQHDFZBMSA-N (3S)-5-N-[(1S,5R)-3-hydroxy-6-bicyclo[3.1.0]hexanyl]-7-N,3-dimethyl-3-phenyl-2H-1-benzofuran-5,7-dicarboxamide Chemical compound CNC(=O)c1cc(cc2c1OC[C@@]2(C)c1ccccc1)C(=O)NC1[C@H]2CC(O)C[C@@H]12 FNHHVPPSBFQMEL-KQHDFZBMSA-N 0.000 description 1
- UDQTXCHQKHIQMH-KYGLGHNPSA-N (3ar,5s,6s,7r,7ar)-5-(difluoromethyl)-2-(ethylamino)-5,6,7,7a-tetrahydro-3ah-pyrano[3,2-d][1,3]thiazole-6,7-diol Chemical compound S1C(NCC)=N[C@H]2[C@@H]1O[C@H](C(F)F)[C@@H](O)[C@@H]2O UDQTXCHQKHIQMH-KYGLGHNPSA-N 0.000 description 1
- OOKAZRDERJMRCJ-KOUAFAAESA-N (3r)-7-[(1s,2s,4ar,6s,8s)-2,6-dimethyl-8-[(2s)-2-methylbutanoyl]oxy-1,2,4a,5,6,7,8,8a-octahydronaphthalen-1-yl]-3-hydroxy-5-oxoheptanoic acid Chemical compound C1=C[C@H](C)[C@H](CCC(=O)C[C@@H](O)CC(O)=O)C2[C@@H](OC(=O)[C@@H](C)CC)C[C@@H](C)C[C@@H]21 OOKAZRDERJMRCJ-KOUAFAAESA-N 0.000 description 1
- YQOLEILXOBUDMU-KRWDZBQOSA-N (4R)-5-[(6-bromo-3-methyl-2-pyrrolidin-1-ylquinoline-4-carbonyl)amino]-4-(2-chlorophenyl)pentanoic acid Chemical compound CC1=C(C2=C(C=CC(=C2)Br)N=C1N3CCCC3)C(=O)NC[C@H](CCC(=O)O)C4=CC=CC=C4Cl YQOLEILXOBUDMU-KRWDZBQOSA-N 0.000 description 1
- STPKWKPURVSAJF-LJEWAXOPSA-N (4r,5r)-5-[4-[[4-(1-aza-4-azoniabicyclo[2.2.2]octan-4-ylmethyl)phenyl]methoxy]phenyl]-3,3-dibutyl-7-(dimethylamino)-1,1-dioxo-4,5-dihydro-2h-1$l^{6}-benzothiepin-4-ol Chemical compound O[C@H]1C(CCCC)(CCCC)CS(=O)(=O)C2=CC=C(N(C)C)C=C2[C@H]1C(C=C1)=CC=C1OCC(C=C1)=CC=C1C[N+]1(CC2)CCN2CC1 STPKWKPURVSAJF-LJEWAXOPSA-N 0.000 description 1
- YSGQGNQWBLYHPE-CFUSNLFHSA-N (7r,8r,9s,10r,13s,14s,17s)-17-hydroxy-7,13-dimethyl-2,6,7,8,9,10,11,12,14,15,16,17-dodecahydro-1h-cyclopenta[a]phenanthren-3-one Chemical compound C1C[C@]2(C)[C@@H](O)CC[C@H]2[C@@H]2[C@H](C)CC3=CC(=O)CC[C@@H]3[C@H]21 YSGQGNQWBLYHPE-CFUSNLFHSA-N 0.000 description 1
- BJEPYKJPYRNKOW-REOHCLBHSA-N (S)-malic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O BJEPYKJPYRNKOW-REOHCLBHSA-N 0.000 description 1
- SCYULBFZEHDVBN-UHFFFAOYSA-N 1,1-Dichloroethane Chemical compound CC(Cl)Cl SCYULBFZEHDVBN-UHFFFAOYSA-N 0.000 description 1
- HORKYAIEVBUXGM-UHFFFAOYSA-N 1,2,3,4-tetrahydroquinoxaline Chemical compound C1=CC=C2NCCNC2=C1 HORKYAIEVBUXGM-UHFFFAOYSA-N 0.000 description 1
- LTWIBTYLSRDGHP-HCURTGQUSA-N 1,2-bis[(e)-(4-chlorophenyl)methylideneamino]guanidine;hydrochloride Chemical compound [Cl-].C=1C=C(Cl)C=CC=1\C=N\N=C(/N)N\[NH+]=C\C1=CC=C(Cl)C=C1 LTWIBTYLSRDGHP-HCURTGQUSA-N 0.000 description 1
- IRFSXVIRXMYULF-UHFFFAOYSA-N 1,2-dihydroquinoline Chemical compound C1=CC=C2C=CCNC2=C1 IRFSXVIRXMYULF-UHFFFAOYSA-N 0.000 description 1
- KKHFRAFPESRGGD-UHFFFAOYSA-N 1,3-dimethyl-7-[3-(n-methylanilino)propyl]purine-2,6-dione Chemical compound C1=NC=2N(C)C(=O)N(C)C(=O)C=2N1CCCN(C)C1=CC=CC=C1 KKHFRAFPESRGGD-UHFFFAOYSA-N 0.000 description 1
- HSOQBSYABQUNQV-UHFFFAOYSA-N 1,4-dihydro-1,8-naphthyridine Chemical compound C1=CC=C2CC=CNC2=N1 HSOQBSYABQUNQV-UHFFFAOYSA-N 0.000 description 1
- ROBKVGDBQLURAF-UHFFFAOYSA-N 1,4-dihydroquinoline Chemical compound C1=CC=C2CC=CNC2=C1 ROBKVGDBQLURAF-UHFFFAOYSA-N 0.000 description 1
- YJWZHHLRVPCSGP-UHFFFAOYSA-N 1-(2-amino-4-chlorophenyl)ethanone Chemical compound CC(=O)C1=CC=C(Cl)C=C1N YJWZHHLRVPCSGP-UHFFFAOYSA-N 0.000 description 1
- GETKKXOYEMMPBP-UHFFFAOYSA-N 1-(2-anilino-4-chlorophenyl)-3-[4-(4-hydroxypiperidin-1-yl)sulfonylphenyl]propan-1-one Chemical compound C1CC(O)CCN1S(=O)(=O)C(C=C1)=CC=C1CCC(=O)C1=CC=C(Cl)C=C1NC1=CC=CC=C1 GETKKXOYEMMPBP-UHFFFAOYSA-N 0.000 description 1
- SHMWLYUKXRLPJV-UHFFFAOYSA-N 1-(2-chloro-6-methylpyridin-3-yl)ethanone Chemical compound CC(=O)C1=CC=C(C)N=C1Cl SHMWLYUKXRLPJV-UHFFFAOYSA-N 0.000 description 1
- PUUYGMZERWRIDS-UHFFFAOYSA-N 1-(4-chloro-2-nitrophenyl)ethanone Chemical compound CC(=O)C1=CC=C(Cl)C=C1[N+]([O-])=O PUUYGMZERWRIDS-UHFFFAOYSA-N 0.000 description 1
- PVOAHINGSUIXLS-UHFFFAOYSA-N 1-Methylpiperazine Chemical compound CN1CCNCC1 PVOAHINGSUIXLS-UHFFFAOYSA-N 0.000 description 1
- PWMWNFMRSKOCEY-UHFFFAOYSA-N 1-Phenyl-1,2-ethanediol Chemical compound OCC(O)C1=CC=CC=C1 PWMWNFMRSKOCEY-UHFFFAOYSA-N 0.000 description 1
- KQZLRWGGWXJPOS-NLFPWZOASA-N 1-[(1R)-1-(2,4-dichlorophenyl)ethyl]-6-[(4S,5R)-4-[(2S)-2-(hydroxymethyl)pyrrolidin-1-yl]-5-methylcyclohexen-1-yl]pyrazolo[3,4-b]pyrazine-3-carbonitrile Chemical compound ClC1=C(C=CC(=C1)Cl)[C@@H](C)N1N=C(C=2C1=NC(=CN=2)C1=CC[C@@H]([C@@H](C1)C)N1[C@@H](CCC1)CO)C#N KQZLRWGGWXJPOS-NLFPWZOASA-N 0.000 description 1
- WZZBNLYBHUDSHF-DHLKQENFSA-N 1-[(3s,4s)-4-[8-(2-chloro-4-pyrimidin-2-yloxyphenyl)-7-fluoro-2-methylimidazo[4,5-c]quinolin-1-yl]-3-fluoropiperidin-1-yl]-2-hydroxyethanone Chemical compound CC1=NC2=CN=C3C=C(F)C(C=4C(=CC(OC=5N=CC=CN=5)=CC=4)Cl)=CC3=C2N1[C@H]1CCN(C(=O)CO)C[C@@H]1F WZZBNLYBHUDSHF-DHLKQENFSA-N 0.000 description 1
- ONBQEOIKXPHGMB-VBSBHUPXSA-N 1-[2-[(2s,3r,4s,5r)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]oxy-4,6-dihydroxyphenyl]-3-(4-hydroxyphenyl)propan-1-one Chemical compound O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1OC1=CC(O)=CC(O)=C1C(=O)CCC1=CC=C(O)C=C1 ONBQEOIKXPHGMB-VBSBHUPXSA-N 0.000 description 1
- UNILWMWFPHPYOR-KXEYIPSPSA-M 1-[6-[2-[3-[3-[3-[2-[2-[3-[[2-[2-[[(2r)-1-[[2-[[(2r)-1-[3-[2-[2-[3-[[2-(2-amino-2-oxoethoxy)acetyl]amino]propoxy]ethoxy]ethoxy]propylamino]-3-hydroxy-1-oxopropan-2-yl]amino]-2-oxoethyl]amino]-3-[(2r)-2,3-di(hexadecanoyloxy)propyl]sulfanyl-1-oxopropan-2-yl Chemical compound O=C1C(SCCC(=O)NCCCOCCOCCOCCCNC(=O)COCC(=O)N[C@@H](CSC[C@@H](COC(=O)CCCCCCCCCCCCCCC)OC(=O)CCCCCCCCCCCCCCC)C(=O)NCC(=O)N[C@H](CO)C(=O)NCCCOCCOCCOCCCNC(=O)COCC(N)=O)CC(=O)N1CCNC(=O)CCCCCN\1C2=CC=C(S([O-])(=O)=O)C=C2CC/1=C/C=C/C=C/C1=[N+](CC)C2=CC=C(S([O-])(=O)=O)C=C2C1 UNILWMWFPHPYOR-KXEYIPSPSA-M 0.000 description 1
- QXOGPTXQGKQSJT-UHFFFAOYSA-N 1-amino-4-[4-(3,4-dimethylphenyl)sulfanylanilino]-9,10-dioxoanthracene-2-sulfonic acid Chemical compound Cc1ccc(Sc2ccc(Nc3cc(c(N)c4C(=O)c5ccccc5C(=O)c34)S(O)(=O)=O)cc2)cc1C QXOGPTXQGKQSJT-UHFFFAOYSA-N 0.000 description 1
- XHTUOWXUBNMVEU-UHFFFAOYSA-N 1-benzoyl-5-ethyl-5-(3-methylbutyl)-1,3-diazinane-2,4,6-trione Chemical group O=C1C(CC)(CCC(C)C)C(=O)NC(=O)N1C(=O)C1=CC=CC=C1 XHTUOWXUBNMVEU-UHFFFAOYSA-N 0.000 description 1
- CWLUFVAFWWNXJZ-UHFFFAOYSA-N 1-hydroxypyrrolidine Chemical compound ON1CCCC1 CWLUFVAFWWNXJZ-UHFFFAOYSA-N 0.000 description 1
- DZNNIRHFSWTOJH-UHFFFAOYSA-N 1-methyl-4-oxoquinoline-2-carboxylic acid Chemical compound C1=CC=C2N(C)C(C(O)=O)=CC(=O)C2=C1 DZNNIRHFSWTOJH-UHFFFAOYSA-N 0.000 description 1
- PLRACCBDVIHHLZ-UHFFFAOYSA-N 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine Chemical compound C1N(C)CCC(C=2C=CC=CC=2)=C1 PLRACCBDVIHHLZ-UHFFFAOYSA-N 0.000 description 1
- XVCNQQAWVICUCY-UHFFFAOYSA-N 1-phenyl-4H-quinoline-2-carboxylic acid Chemical compound C1(=CC=CC=C1)N1C(=CCC2=CC=CC=C12)C(=O)O XVCNQQAWVICUCY-UHFFFAOYSA-N 0.000 description 1
- UBNPTJSGEYBDCQ-UHFFFAOYSA-N 1-phenylethanone Chemical class CC(=O)C1=CC=CC=C1.CC(=O)C1=CC=CC=C1 UBNPTJSGEYBDCQ-UHFFFAOYSA-N 0.000 description 1
- QWENRTYMTSOGBR-UHFFFAOYSA-N 1H-1,2,3-Triazole Chemical compound C=1C=NNN=1 QWENRTYMTSOGBR-UHFFFAOYSA-N 0.000 description 1
- YBYIRNPNPLQARY-UHFFFAOYSA-N 1H-indene Natural products C1=CC=C2CC=CC2=C1 YBYIRNPNPLQARY-UHFFFAOYSA-N 0.000 description 1
- PMZDQRJGMBOQBF-UHFFFAOYSA-N 1H-quinolin-4-one Natural products C1=CC=C2C(O)=CC=NC2=C1 PMZDQRJGMBOQBF-UHFFFAOYSA-N 0.000 description 1
- NQUIRWXTTAMWIU-UHFFFAOYSA-N 1h-1,8-naphthyridin-4-one Chemical compound C1=CC=C2C(O)=CC=NC2=N1 NQUIRWXTTAMWIU-UHFFFAOYSA-N 0.000 description 1
- ZPOROQKDAPEMOL-UHFFFAOYSA-N 1h-pyrrol-3-ol Chemical compound OC=1C=CNC=1 ZPOROQKDAPEMOL-UHFFFAOYSA-N 0.000 description 1
- KYNFOMQIXZUKRK-UHFFFAOYSA-N 2,2'-dithiodiethanol Chemical compound OCCSSCCO KYNFOMQIXZUKRK-UHFFFAOYSA-N 0.000 description 1
- LTMRRSWNXVJMBA-UHFFFAOYSA-L 2,2-diethylpropanedioate Chemical compound CCC(CC)(C([O-])=O)C([O-])=O LTMRRSWNXVJMBA-UHFFFAOYSA-L 0.000 description 1
- LFEXGBPVZBXATF-UHFFFAOYSA-N 2,3-dimethylbenzenesulfonamide Chemical compound CC1=CC=CC(S(N)(=O)=O)=C1C LFEXGBPVZBXATF-UHFFFAOYSA-N 0.000 description 1
- FFRBMBIXVSCUFS-UHFFFAOYSA-N 2,4-dinitro-1-naphthol Chemical compound C1=CC=C2C(O)=C([N+]([O-])=O)C=C([N+]([O-])=O)C2=C1 FFRBMBIXVSCUFS-UHFFFAOYSA-N 0.000 description 1
- IKWJKXMMTRXCDI-UHFFFAOYSA-N 2,5-dimethylpyrrole Chemical compound CC1=CC=C(C)[N]1 IKWJKXMMTRXCDI-UHFFFAOYSA-N 0.000 description 1
- WGFNXGPBPIJYLI-UHFFFAOYSA-N 2,6-difluoro-3-[(3-fluorophenyl)sulfonylamino]-n-(3-methoxy-1h-pyrazolo[3,4-b]pyridin-5-yl)benzamide Chemical compound C1=C2C(OC)=NNC2=NC=C1NC(=O)C(C=1F)=C(F)C=CC=1NS(=O)(=O)C1=CC=CC(F)=C1 WGFNXGPBPIJYLI-UHFFFAOYSA-N 0.000 description 1
- FQMZXMVHHKXGTM-UHFFFAOYSA-N 2-(1-adamantyl)-n-[2-[2-(2-hydroxyethylamino)ethylamino]quinolin-5-yl]acetamide Chemical compound C1C(C2)CC(C3)CC2CC13CC(=O)NC1=CC=CC2=NC(NCCNCCO)=CC=C21 FQMZXMVHHKXGTM-UHFFFAOYSA-N 0.000 description 1
- VCUXVXLUOHDHKK-UHFFFAOYSA-N 2-(2-aminopyrimidin-4-yl)-4-(2-chloro-4-methoxyphenyl)-1,3-thiazole-5-carboxamide Chemical compound ClC1=CC(OC)=CC=C1C1=C(C(N)=O)SC(C=2N=C(N)N=CC=2)=N1 VCUXVXLUOHDHKK-UHFFFAOYSA-N 0.000 description 1
- MOMFXATYAINJML-UHFFFAOYSA-N 2-Acetylthiazole Chemical group CC(=O)C1=NC=CS1 MOMFXATYAINJML-UHFFFAOYSA-N 0.000 description 1
- OXQGTIUCKGYOAA-UHFFFAOYSA-N 2-Ethylbutanoic acid Chemical compound CCC(CC)C(O)=O OXQGTIUCKGYOAA-UHFFFAOYSA-N 0.000 description 1
- PYRKKGOKRMZEIT-UHFFFAOYSA-N 2-[6-(2-cyclopropylethoxy)-9-(2-hydroxy-2-methylpropyl)-1h-phenanthro[9,10-d]imidazol-2-yl]-5-fluorobenzene-1,3-dicarbonitrile Chemical compound C1=C2C3=CC(CC(C)(O)C)=CC=C3C=3NC(C=4C(=CC(F)=CC=4C#N)C#N)=NC=3C2=CC=C1OCCC1CC1 PYRKKGOKRMZEIT-UHFFFAOYSA-N 0.000 description 1
- FMKGJQHNYMWDFJ-CVEARBPZSA-N 2-[[4-(2,2-difluoropropoxy)pyrimidin-5-yl]methylamino]-4-[[(1R,4S)-4-hydroxy-3,3-dimethylcyclohexyl]amino]pyrimidine-5-carbonitrile Chemical compound FC(COC1=NC=NC=C1CNC1=NC=C(C(=N1)N[C@H]1CC([C@H](CC1)O)(C)C)C#N)(C)F FMKGJQHNYMWDFJ-CVEARBPZSA-N 0.000 description 1
- VVCMGAUPZIKYTH-VGHSCWAPSA-N 2-acetyloxybenzoic acid;[(2s,3r)-4-(dimethylamino)-3-methyl-1,2-diphenylbutan-2-yl] propanoate;1,3,7-trimethylpurine-2,6-dione Chemical compound CC(=O)OC1=CC=CC=C1C(O)=O.CN1C(=O)N(C)C(=O)C2=C1N=CN2C.C([C@](OC(=O)CC)([C@H](C)CN(C)C)C=1C=CC=CC=1)C1=CC=CC=C1 VVCMGAUPZIKYTH-VGHSCWAPSA-N 0.000 description 1
- GOJUJUVQIVIZAV-UHFFFAOYSA-N 2-amino-4,6-dichloropyrimidine-5-carbaldehyde Chemical group NC1=NC(Cl)=C(C=O)C(Cl)=N1 GOJUJUVQIVIZAV-UHFFFAOYSA-N 0.000 description 1
- YSUIQYOGTINQIN-UZFYAQMZSA-N 2-amino-9-[(1S,6R,8R,9S,10R,15R,17R,18R)-8-(6-aminopurin-9-yl)-9,18-difluoro-3,12-dihydroxy-3,12-bis(sulfanylidene)-2,4,7,11,13,16-hexaoxa-3lambda5,12lambda5-diphosphatricyclo[13.2.1.06,10]octadecan-17-yl]-1H-purin-6-one Chemical compound NC1=NC2=C(N=CN2[C@@H]2O[C@@H]3COP(S)(=O)O[C@@H]4[C@@H](COP(S)(=O)O[C@@H]2[C@@H]3F)O[C@H]([C@H]4F)N2C=NC3=C2N=CN=C3N)C(=O)N1 YSUIQYOGTINQIN-UZFYAQMZSA-N 0.000 description 1
- TVTJUIAKQFIXCE-HUKYDQBMSA-N 2-amino-9-[(2R,3S,4S,5R)-4-fluoro-3-hydroxy-5-(hydroxymethyl)oxolan-2-yl]-7-prop-2-ynyl-1H-purine-6,8-dione Chemical compound NC=1NC(C=2N(C(N(C=2N=1)[C@@H]1O[C@@H]([C@H]([C@H]1O)F)CO)=O)CC#C)=O TVTJUIAKQFIXCE-HUKYDQBMSA-N 0.000 description 1
- DQDYIKGGALFGSJ-UHFFFAOYSA-N 2-anilino-4-fluorobenzoic acid Chemical compound OC(=O)C1=CC=C(F)C=C1NC1=CC=CC=C1 DQDYIKGGALFGSJ-UHFFFAOYSA-N 0.000 description 1
- RRKPMLZRLKTDQV-UHFFFAOYSA-N 2-bromo-4-fluorobenzoic acid Chemical compound OC(=O)C1=CC=C(F)C=C1Br RRKPMLZRLKTDQV-UHFFFAOYSA-N 0.000 description 1
- LCBXSXVFOUKYDM-UHFFFAOYSA-O 2-hydroperoxyethyl(trimethyl)azanium Chemical compound C[N+](C)(C)CCOO LCBXSXVFOUKYDM-UHFFFAOYSA-O 0.000 description 1
- LFOIDLOIBZFWDO-UHFFFAOYSA-N 2-methoxy-6-[6-methoxy-4-[(3-phenylmethoxyphenyl)methoxy]-1-benzofuran-2-yl]imidazo[2,1-b][1,3,4]thiadiazole Chemical compound N1=C2SC(OC)=NN2C=C1C(OC1=CC(OC)=C2)=CC1=C2OCC(C=1)=CC=CC=1OCC1=CC=CC=C1 LFOIDLOIBZFWDO-UHFFFAOYSA-N 0.000 description 1
- HNTZKNJGAFJMHQ-UHFFFAOYSA-N 2-methylpyridine-3-carboxylic acid Chemical compound CC1=NC=CC=C1C(O)=O HNTZKNJGAFJMHQ-UHFFFAOYSA-N 0.000 description 1
- JTWHVBNYYWFXSI-UHFFFAOYSA-N 2-nitro-1-phenylethanone Chemical group [O-][N+](=O)CC(=O)C1=CC=CC=C1 JTWHVBNYYWFXSI-UHFFFAOYSA-N 0.000 description 1
- VOKLUGVCKPXDJE-UHFFFAOYSA-N 2-oxo-n-phenylpropanamide Chemical compound CC(=O)C(=O)NC1=CC=CC=C1 VOKLUGVCKPXDJE-UHFFFAOYSA-N 0.000 description 1
- AUUTXOKCFQTKPL-UHFFFAOYSA-N 2-oxopropanoyl chloride Chemical compound CC(=O)C(Cl)=O AUUTXOKCFQTKPL-UHFFFAOYSA-N 0.000 description 1
- DOZZSWAOPDYVLH-UHFFFAOYSA-N 2-phenylpropanamide Chemical compound NC(=O)C(C)C1=CC=CC=C1 DOZZSWAOPDYVLH-UHFFFAOYSA-N 0.000 description 1
- 125000004105 2-pyridyl group Chemical group N1=C([*])C([H])=C([H])C([H])=C1[H] 0.000 description 1
- QBWKPGNFQQJGFY-QLFBSQMISA-N 3-[(1r)-1-[(2r,6s)-2,6-dimethylmorpholin-4-yl]ethyl]-n-[6-methyl-3-(1h-pyrazol-4-yl)imidazo[1,2-a]pyrazin-8-yl]-1,2-thiazol-5-amine Chemical compound N1([C@H](C)C2=NSC(NC=3C4=NC=C(N4C=C(C)N=3)C3=CNN=C3)=C2)C[C@H](C)O[C@H](C)C1 QBWKPGNFQQJGFY-QLFBSQMISA-N 0.000 description 1
- WFOVEDJTASPCIR-UHFFFAOYSA-N 3-[(4-methyl-5-pyridin-4-yl-1,2,4-triazol-3-yl)methylamino]-n-[[2-(trifluoromethyl)phenyl]methyl]benzamide Chemical compound N=1N=C(C=2C=CN=CC=2)N(C)C=1CNC(C=1)=CC=CC=1C(=O)NCC1=CC=CC=C1C(F)(F)F WFOVEDJTASPCIR-UHFFFAOYSA-N 0.000 description 1
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 1
- 125000004180 3-fluorophenyl group Chemical group [H]C1=C([H])C(*)=C([H])C(F)=C1[H] 0.000 description 1
- 125000006497 3-methoxybenzyl group Chemical group [H]C1=C([H])C(=C([H])C(OC([H])([H])[H])=C1[H])C([H])([H])* 0.000 description 1
- XJCVRTZCHMZPBD-UHFFFAOYSA-N 3-nitroaniline Chemical compound NC1=CC=CC([N+]([O-])=O)=C1 XJCVRTZCHMZPBD-UHFFFAOYSA-N 0.000 description 1
- 125000003349 3-pyridyl group Chemical group N1=C([H])C([*])=C([H])C([H])=C1[H] 0.000 description 1
- SDPWBDGMNVKHMB-UHFFFAOYSA-N 4-(2-hydroxyethylsulfanyl)benzaldehyde Chemical compound OCCSC1=CC=C(C=O)C=C1 SDPWBDGMNVKHMB-UHFFFAOYSA-N 0.000 description 1
- WYFCZWSWFGJODV-MIANJLSGSA-N 4-[[(1s)-2-[(e)-3-[3-chloro-2-fluoro-6-(tetrazol-1-yl)phenyl]prop-2-enoyl]-5-(4-methyl-2-oxopiperazin-1-yl)-3,4-dihydro-1h-isoquinoline-1-carbonyl]amino]benzoic acid Chemical compound O=C1CN(C)CCN1C1=CC=CC2=C1CCN(C(=O)\C=C\C=1C(=CC=C(Cl)C=1F)N1N=NN=C1)[C@@H]2C(=O)NC1=CC=C(C(O)=O)C=C1 WYFCZWSWFGJODV-MIANJLSGSA-N 0.000 description 1
- IMLXLGZJLAOKJN-UHFFFAOYSA-N 4-aminocyclohexan-1-ol Chemical compound NC1CCC(O)CC1 IMLXLGZJLAOKJN-UHFFFAOYSA-N 0.000 description 1
- 125000004217 4-methoxybenzyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1OC([H])([H])[H])C([H])([H])* 0.000 description 1
- PSVPUHBSBYJSMQ-UHFFFAOYSA-N 4-methylsulfonylbenzaldehyde Chemical compound CS(=O)(=O)C1=CC=C(C=O)C=C1 PSVPUHBSBYJSMQ-UHFFFAOYSA-N 0.000 description 1
- YYROPELSRYBVMQ-UHFFFAOYSA-N 4-toluenesulfonyl chloride Chemical compound CC1=CC=C(S(Cl)(=O)=O)C=C1 YYROPELSRYBVMQ-UHFFFAOYSA-N 0.000 description 1
- VKLKXFOZNHEBSW-UHFFFAOYSA-N 5-[[3-[(4-morpholin-4-ylbenzoyl)amino]phenyl]methoxy]pyridine-3-carboxamide Chemical compound O1CCN(CC1)C1=CC=C(C(=O)NC=2C=C(COC=3C=NC=C(C(=O)N)C=3)C=CC=2)C=C1 VKLKXFOZNHEBSW-UHFFFAOYSA-N 0.000 description 1
- RSIWALKZYXPAGW-NSHDSACASA-N 6-(3-fluorophenyl)-3-methyl-7-[(1s)-1-(7h-purin-6-ylamino)ethyl]-[1,3]thiazolo[3,2-a]pyrimidin-5-one Chemical compound C=1([C@@H](NC=2C=3N=CNC=3N=CN=2)C)N=C2SC=C(C)N2C(=O)C=1C1=CC=CC(F)=C1 RSIWALKZYXPAGW-NSHDSACASA-N 0.000 description 1
- GDUANFXPOZTYKS-UHFFFAOYSA-N 6-bromo-8-[(2,6-difluoro-4-methoxybenzoyl)amino]-4-oxochromene-2-carboxylic acid Chemical compound FC1=CC(OC)=CC(F)=C1C(=O)NC1=CC(Br)=CC2=C1OC(C(O)=O)=CC2=O GDUANFXPOZTYKS-UHFFFAOYSA-N 0.000 description 1
- CTAIEPPAOULMFY-UHFFFAOYSA-N 6-methoxypyridine-3-carbaldehyde Chemical compound COC1=CC=C(C=O)C=N1 CTAIEPPAOULMFY-UHFFFAOYSA-N 0.000 description 1
- WDYVUKGVKRZQNM-UHFFFAOYSA-N 6-phosphonohexylphosphonic acid Chemical compound OP(O)(=O)CCCCCCP(O)(O)=O WDYVUKGVKRZQNM-UHFFFAOYSA-N 0.000 description 1
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 1
- BSFODEXXVBBYOC-UHFFFAOYSA-N 8-[4-(dimethylamino)butan-2-ylamino]quinolin-6-ol Chemical compound C1=CN=C2C(NC(CCN(C)C)C)=CC(O)=CC2=C1 BSFODEXXVBBYOC-UHFFFAOYSA-N 0.000 description 1
- 101150111329 ACE-1 gene Proteins 0.000 description 1
- RZVAJINKPMORJF-UHFFFAOYSA-N Acetaminophen Chemical compound CC(=O)NC1=CC=C(O)C=C1 RZVAJINKPMORJF-UHFFFAOYSA-N 0.000 description 1
- 208000036065 Airway Remodeling Diseases 0.000 description 1
- 241000156724 Antirhea Species 0.000 description 1
- 102100021569 Apoptosis regulator Bcl-2 Human genes 0.000 description 1
- 206010003694 Atrophy Diseases 0.000 description 1
- 241000972773 Aulopiformes Species 0.000 description 1
- KHBQMWCZKVMBLN-UHFFFAOYSA-N Benzenesulfonamide Chemical compound NS(=O)(=O)C1=CC=CC=C1 KHBQMWCZKVMBLN-UHFFFAOYSA-N 0.000 description 1
- 241000283690 Bos taurus Species 0.000 description 1
- 208000003174 Brain Neoplasms Diseases 0.000 description 1
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 1
- 206010066091 Bronchial Hyperreactivity Diseases 0.000 description 1
- JQUCWIWWWKZNCS-LESHARBVSA-N C(C1=CC=CC=C1)(=O)NC=1SC[C@H]2[C@@](N1)(CO[C@H](C2)C)C=2SC=C(N2)NC(=O)C2=NC=C(C=C2)OC(F)F Chemical compound C(C1=CC=CC=C1)(=O)NC=1SC[C@H]2[C@@](N1)(CO[C@H](C2)C)C=2SC=C(N2)NC(=O)C2=NC=C(C=C2)OC(F)F JQUCWIWWWKZNCS-LESHARBVSA-N 0.000 description 1
- KCBAMQOKOLXLOX-BSZYMOERSA-N CC1=C(SC=N1)C2=CC=C(C=C2)[C@H](C)NC(=O)[C@@H]3C[C@H](CN3C(=O)[C@H](C(C)(C)C)NC(=O)CCCCCCCCCCNCCCONC(=O)C4=C(C(=C(C=C4)F)F)NC5=C(C=C(C=C5)I)F)O Chemical compound CC1=C(SC=N1)C2=CC=C(C=C2)[C@H](C)NC(=O)[C@@H]3C[C@H](CN3C(=O)[C@H](C(C)(C)C)NC(=O)CCCCCCCCCCNCCCONC(=O)C4=C(C(=C(C=C4)F)F)NC5=C(C=C(C=C5)I)F)O KCBAMQOKOLXLOX-BSZYMOERSA-N 0.000 description 1
- BQXUPNKLZNSUMC-YUQWMIPFSA-N CCN(CCCCCOCC(=O)N[C@H](C(=O)N1C[C@H](O)C[C@H]1C(=O)N[C@@H](C)c1ccc(cc1)-c1scnc1C)C(C)(C)C)CCOc1ccc(cc1)C(=O)c1c(sc2cc(O)ccc12)-c1ccc(O)cc1 Chemical compound CCN(CCCCCOCC(=O)N[C@H](C(=O)N1C[C@H](O)C[C@H]1C(=O)N[C@@H](C)c1ccc(cc1)-c1scnc1C)C(C)(C)C)CCOc1ccc(cc1)C(=O)c1c(sc2cc(O)ccc12)-c1ccc(O)cc1 BQXUPNKLZNSUMC-YUQWMIPFSA-N 0.000 description 1
- 101100452593 Caenorhabditis elegans ina-1 gene Proteins 0.000 description 1
- 101100454194 Caenorhabditis elegans mei-1 gene Proteins 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- 241000282472 Canis lupus familiaris Species 0.000 description 1
- 241000283707 Capra Species 0.000 description 1
- 239000004215 Carbon black (E152) Substances 0.000 description 1
- UGFAIRIUMAVXCW-UHFFFAOYSA-N Carbon monoxide Chemical compound [O+]#[C-] UGFAIRIUMAVXCW-UHFFFAOYSA-N 0.000 description 1
- 102000011727 Caspases Human genes 0.000 description 1
- 108010076667 Caspases Proteins 0.000 description 1
- 241000700198 Cavia Species 0.000 description 1
- 241000282693 Cercopithecidae Species 0.000 description 1
- 102000001327 Chemokine CCL5 Human genes 0.000 description 1
- 108010055166 Chemokine CCL5 Proteins 0.000 description 1
- 102000029816 Collagenase Human genes 0.000 description 1
- 108060005980 Collagenase Proteins 0.000 description 1
- 229940126657 Compound 17 Drugs 0.000 description 1
- 229940126639 Compound 33 Drugs 0.000 description 1
- 206010011224 Cough Diseases 0.000 description 1
- 208000011231 Crohn disease Diseases 0.000 description 1
- 229920002785 Croscarmellose sodium Polymers 0.000 description 1
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 1
- RGHNJXZEOKUKBD-UHFFFAOYSA-N D-gluconic acid Natural products OCC(O)C(O)C(O)C(O)C(O)=O RGHNJXZEOKUKBD-UHFFFAOYSA-N 0.000 description 1
- PAPNRQCYSFBWDI-UHFFFAOYSA-N DMP Natural products CC1=CC=C(C)N1 PAPNRQCYSFBWDI-UHFFFAOYSA-N 0.000 description 1
- 239000004375 Dextrin Substances 0.000 description 1
- 229920001353 Dextrin Polymers 0.000 description 1
- 239000004338 Dichlorodifluoromethane Substances 0.000 description 1
- 241000790917 Dioxys <bee> Species 0.000 description 1
- SNRUBQQJIBEYMU-UHFFFAOYSA-N Dodecane Natural products CCCCCCCCCCCC SNRUBQQJIBEYMU-UHFFFAOYSA-N 0.000 description 1
- 101100456896 Drosophila melanogaster metl gene Proteins 0.000 description 1
- 102100023274 Dual specificity mitogen-activated protein kinase kinase 4 Human genes 0.000 description 1
- 102100023332 Dual specificity mitogen-activated protein kinase kinase 7 Human genes 0.000 description 1
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 1
- LVGKNOAMLMIIKO-UHFFFAOYSA-N Elaidinsaeure-aethylester Natural products CCCCCCCCC=CCCCCCCCC(=O)OCC LVGKNOAMLMIIKO-UHFFFAOYSA-N 0.000 description 1
- 241000283086 Equidae Species 0.000 description 1
- 102000007665 Extracellular Signal-Regulated MAP Kinases Human genes 0.000 description 1
- 108010007457 Extracellular Signal-Regulated MAP Kinases Proteins 0.000 description 1
- 241000282326 Felis catus Species 0.000 description 1
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 1
- ALIVXCSEERJYHU-UHFFFAOYSA-N Flurbiprofen axetil Chemical compound FC1=CC(C(C)C(=O)OC(OC(C)=O)C)=CC=C1C1=CC=CC=C1 ALIVXCSEERJYHU-UHFFFAOYSA-N 0.000 description 1
- 206010018338 Glioma Diseases 0.000 description 1
- RGHNJXZEOKUKBD-SQOUGZDYSA-N Gluconic acid Natural products OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C(O)=O RGHNJXZEOKUKBD-SQOUGZDYSA-N 0.000 description 1
- 102000018899 Glutamate Receptors Human genes 0.000 description 1
- 108010027915 Glutamate Receptors Proteins 0.000 description 1
- WHUUTDBJXJRKMK-UHFFFAOYSA-N Glutamic acid Natural products OC(=O)C(N)CCC(O)=O WHUUTDBJXJRKMK-UHFFFAOYSA-N 0.000 description 1
- 229910004373 HOAc Inorganic materials 0.000 description 1
- 241000152447 Hades Species 0.000 description 1
- 241000282412 Homo Species 0.000 description 1
- 101000971171 Homo sapiens Apoptosis regulator Bcl-2 Proteins 0.000 description 1
- 101000883515 Homo sapiens Chitinase-3-like protein 1 Proteins 0.000 description 1
- 101001115395 Homo sapiens Dual specificity mitogen-activated protein kinase kinase 4 Proteins 0.000 description 1
- 101000624594 Homo sapiens Dual specificity mitogen-activated protein kinase kinase 7 Proteins 0.000 description 1
- 101100457330 Homo sapiens MAPK10 gene Proteins 0.000 description 1
- 101000628949 Homo sapiens Mitogen-activated protein kinase 10 Proteins 0.000 description 1
- 101000668058 Infectious salmon anemia virus (isolate Atlantic salmon/Norway/810/9/99) RNA-directed RNA polymerase catalytic subunit Proteins 0.000 description 1
- 108010002352 Interleukin-1 Proteins 0.000 description 1
- 101800003050 Interleukin-16 Proteins 0.000 description 1
- 102000049772 Interleukin-16 Human genes 0.000 description 1
- 108090001007 Interleukin-8 Proteins 0.000 description 1
- LNQCUTNLHUQZLR-VNPYQEQNSA-N Iridin Natural products O(C)c1c(O)c2C(=O)C(c3cc(OC)c(OC)c(O)c3)=COc2cc1O[C@H]1[C@@H](O)[C@@H](O)[C@H](O)[C@H](CO)O1 LNQCUTNLHUQZLR-VNPYQEQNSA-N 0.000 description 1
- 229940118135 JNK inhibitor Drugs 0.000 description 1
- 108010014906 JNK-interacting protein 1 (153-163) Proteins 0.000 description 1
- 108010075654 MAP Kinase Kinase Kinase 1 Proteins 0.000 description 1
- 108010075647 MAP Kinase Kinase Kinase 4 Proteins 0.000 description 1
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 1
- 241000407429 Maja Species 0.000 description 1
- 102000002274 Matrix Metalloproteinases Human genes 0.000 description 1
- 108010000684 Matrix Metalloproteinases Proteins 0.000 description 1
- 229920000168 Microcrystalline cellulose Polymers 0.000 description 1
- 102100026931 Mitogen-activated protein kinase 10 Human genes 0.000 description 1
- 102100033115 Mitogen-activated protein kinase kinase kinase 1 Human genes 0.000 description 1
- 102100033060 Mitogen-activated protein kinase kinase kinase 4 Human genes 0.000 description 1
- 244000131360 Morinda citrifolia Species 0.000 description 1
- YNAVUWVOSKDBBP-UHFFFAOYSA-N Morpholine Chemical compound C1COCCN1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 description 1
- 102100021339 Multidrug resistance-associated protein 1 Human genes 0.000 description 1
- 101001034845 Mus musculus Interferon-induced transmembrane protein 3 Proteins 0.000 description 1
- 101001135571 Mus musculus Tyrosine-protein phosphatase non-receptor type 2 Proteins 0.000 description 1
- LVDRREOUMKACNJ-BKMJKUGQSA-N N-[(2R,3S)-2-(4-chlorophenyl)-1-(1,4-dimethyl-2-oxoquinolin-7-yl)-6-oxopiperidin-3-yl]-2-methylpropane-1-sulfonamide Chemical compound CC(C)CS(=O)(=O)N[C@H]1CCC(=O)N([C@@H]1c1ccc(Cl)cc1)c1ccc2c(C)cc(=O)n(C)c2c1 LVDRREOUMKACNJ-BKMJKUGQSA-N 0.000 description 1
- AVYVHIKSFXVDBG-UHFFFAOYSA-N N-benzyl-N-hydroxy-2,2-dimethylbutanamide Chemical compound C(C1=CC=CC=C1)N(C(C(CC)(C)C)=O)O AVYVHIKSFXVDBG-UHFFFAOYSA-N 0.000 description 1
- MBBZMMPHUWSWHV-BDVNFPICSA-N N-methylglucamine Chemical compound CNC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO MBBZMMPHUWSWHV-BDVNFPICSA-N 0.000 description 1
- QOVYHDHLFPKQQG-NDEPHWFRSA-N N[C@@H](CCC(=O)N1CCC(CC1)NC1=C2C=CC=CC2=NC(NCC2=CN(CCCNCCCNC3CCCCC3)N=N2)=N1)C(O)=O Chemical compound N[C@@H](CCC(=O)N1CCC(CC1)NC1=C2C=CC=CC2=NC(NCC2=CN(CCCNCCCNC3CCCCC3)N=N2)=N1)C(O)=O QOVYHDHLFPKQQG-NDEPHWFRSA-N 0.000 description 1
- 239000007832 Na2SO4 Substances 0.000 description 1
- 241001644525 Nastus productus Species 0.000 description 1
- 101100062121 Neurospora crassa (strain ATCC 24698 / 74-OR23-1A / CBS 708.71 / DSM 1257 / FGSC 987) cyc-1 gene Proteins 0.000 description 1
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 1
- 241000746935 Nolina Species 0.000 description 1
- DEQZXQPMEVYFQE-UHFFFAOYSA-N O1COC=C1.C(C1=CC=CC=C1)(=O)O Chemical compound O1COC=C1.C(C1=CC=CC=C1)(=O)O DEQZXQPMEVYFQE-UHFFFAOYSA-N 0.000 description 1
- PSAPEVDCUIUGGM-UHFFFAOYSA-N OC(=O)C1=CC(=O)C2=CC=CN=C2N1C1=CC=CC=C1 Chemical compound OC(=O)C1=CC(=O)C2=CC=CN=C2N1C1=CC=CC=C1 PSAPEVDCUIUGGM-UHFFFAOYSA-N 0.000 description 1
- 208000008589 Obesity Diseases 0.000 description 1
- 108700020796 Oncogene Proteins 0.000 description 1
- 241000283973 Oryctolagus cuniculus Species 0.000 description 1
- ZCQWOFVYLHDMMC-UHFFFAOYSA-N Oxazole Chemical compound C1=COC=N1 ZCQWOFVYLHDMMC-UHFFFAOYSA-N 0.000 description 1
- 241000282579 Pan Species 0.000 description 1
- 101150003085 Pdcl gene Proteins 0.000 description 1
- 241001494479 Pecora Species 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- IWYDHOAUDWTVEP-UHFFFAOYSA-N R-2-phenyl-2-hydroxyacetic acid Natural products OC(=O)C(O)C1=CC=CC=C1 IWYDHOAUDWTVEP-UHFFFAOYSA-N 0.000 description 1
- 101000648290 Rattus norvegicus Tumor necrosis factor Proteins 0.000 description 1
- 125000000066 S-methyl group Chemical group [H]C([H])([H])S* 0.000 description 1
- 101100402895 Schizosaccharomyces pombe (strain 972 / ATCC 24843) met11 gene Proteins 0.000 description 1
- 229920002684 Sepharose Polymers 0.000 description 1
- PNUZDKCDAWUEGK-CYZMBNFOSA-N Sitafloxacin Chemical compound C([C@H]1N)N(C=2C(=C3C(C(C(C(O)=O)=CN3[C@H]3[C@H](C3)F)=O)=CC=2F)Cl)CC11CC1 PNUZDKCDAWUEGK-CYZMBNFOSA-N 0.000 description 1
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-N Succinic acid Natural products OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 1
- 241000282887 Suidae Species 0.000 description 1
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 1
- 208000018359 Systemic autoimmune disease Diseases 0.000 description 1
- 108700012920 TNF Proteins 0.000 description 1
- DHXVGJBLRPWPCS-UHFFFAOYSA-N Tetrahydropyran Chemical compound C1CCOCC1 DHXVGJBLRPWPCS-UHFFFAOYSA-N 0.000 description 1
- 244000299461 Theobroma cacao Species 0.000 description 1
- 235000009470 Theobroma cacao Nutrition 0.000 description 1
- 239000007983 Tris buffer Substances 0.000 description 1
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 1
- LJOOWESTVASNOG-UFJKPHDISA-N [(1s,3r,4ar,7s,8s,8as)-3-hydroxy-8-[2-[(4r)-4-hydroxy-6-oxooxan-2-yl]ethyl]-7-methyl-1,2,3,4,4a,7,8,8a-octahydronaphthalen-1-yl] (2s)-2-methylbutanoate Chemical compound C([C@H]1[C@@H](C)C=C[C@H]2C[C@@H](O)C[C@@H]([C@H]12)OC(=O)[C@@H](C)CC)CC1C[C@@H](O)CC(=O)O1 LJOOWESTVASNOG-UFJKPHDISA-N 0.000 description 1
- PSLUFJFHTBIXMW-WYEYVKMPSA-N [(3r,4ar,5s,6s,6as,10s,10ar,10bs)-3-ethenyl-10,10b-dihydroxy-3,4a,7,7,10a-pentamethyl-1-oxo-6-(2-pyridin-2-ylethylcarbamoyloxy)-5,6,6a,8,9,10-hexahydro-2h-benzo[f]chromen-5-yl] acetate Chemical compound O([C@@H]1[C@@H]([C@]2(O[C@](C)(CC(=O)[C@]2(O)[C@@]2(C)[C@@H](O)CCC(C)(C)[C@@H]21)C=C)C)OC(=O)C)C(=O)NCCC1=CC=CC=N1 PSLUFJFHTBIXMW-WYEYVKMPSA-N 0.000 description 1
- SMNRFWMNPDABKZ-WVALLCKVSA-N [[(2R,3S,4R,5S)-5-(2,6-dioxo-3H-pyridin-3-yl)-3,4-dihydroxyoxolan-2-yl]methoxy-hydroxyphosphoryl] [[[(2R,3S,4S,5R,6R)-4-fluoro-3,5-dihydroxy-6-(hydroxymethyl)oxan-2-yl]oxy-hydroxyphosphoryl]oxy-hydroxyphosphoryl] hydrogen phosphate Chemical compound OC[C@H]1O[C@H](OP(O)(=O)OP(O)(=O)OP(O)(=O)OP(O)(=O)OC[C@H]2O[C@H]([C@H](O)[C@@H]2O)C2C=CC(=O)NC2=O)[C@H](O)[C@@H](F)[C@@H]1O SMNRFWMNPDABKZ-WVALLCKVSA-N 0.000 description 1
- 230000002159 abnormal effect Effects 0.000 description 1
- 229960000583 acetic acid Drugs 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 230000003213 activating effect Effects 0.000 description 1
- 239000013543 active substance Substances 0.000 description 1
- 150000001263 acyl chlorides Chemical class 0.000 description 1
- 125000004442 acylamino group Chemical group 0.000 description 1
- 125000004423 acyloxy group Chemical group 0.000 description 1
- 239000000853 adhesive Substances 0.000 description 1
- 230000001070 adhesive effect Effects 0.000 description 1
- 230000004520 agglutination Effects 0.000 description 1
- 239000000556 agonist Substances 0.000 description 1
- 150000001335 aliphatic alkanes Chemical group 0.000 description 1
- 230000000172 allergic effect Effects 0.000 description 1
- 230000007815 allergy Effects 0.000 description 1
- 229940060585 alora Drugs 0.000 description 1
- VLSMHEGGTFMBBZ-UHFFFAOYSA-N alpha-Kainic acid Natural products CC(=C)C1CNC(C(O)=O)C1CC(O)=O VLSMHEGGTFMBBZ-UHFFFAOYSA-N 0.000 description 1
- BJEPYKJPYRNKOW-UHFFFAOYSA-N alpha-hydroxysuccinic acid Natural products OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 description 1
- VREFGVBLTWBCJP-UHFFFAOYSA-N alprazolam Chemical compound C12=CC(Cl)=CC=C2N2C(C)=NN=C2CN=C1C1=CC=CC=C1 VREFGVBLTWBCJP-UHFFFAOYSA-N 0.000 description 1
- 229910052782 aluminium Inorganic materials 0.000 description 1
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 1
- WNROFYMDJYEPJX-UHFFFAOYSA-K aluminium hydroxide Chemical compound [OH-].[OH-].[OH-].[Al+3] WNROFYMDJYEPJX-UHFFFAOYSA-K 0.000 description 1
- 229910021529 ammonia Inorganic materials 0.000 description 1
- 230000033115 angiogenesis Effects 0.000 description 1
- IMHBYKMAHXWHRP-UHFFFAOYSA-N anilazine Chemical compound ClC1=CC=CC=C1NC1=NC(Cl)=NC(Cl)=N1 IMHBYKMAHXWHRP-UHFFFAOYSA-N 0.000 description 1
- 150000001448 anilines Chemical class 0.000 description 1
- 125000002490 anilino group Chemical group [H]N(*)C1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 description 1
- 230000002424 anti-apoptotic effect Effects 0.000 description 1
- 239000008135 aqueous vehicle Substances 0.000 description 1
- 229910052786 argon Inorganic materials 0.000 description 1
- 150000004945 aromatic hydrocarbons Chemical group 0.000 description 1
- 235000021311 artificial sweeteners Nutrition 0.000 description 1
- 239000012298 atmosphere Substances 0.000 description 1
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 1
- 208000010668 atopic eczema Diseases 0.000 description 1
- 230000037444 atrophy Effects 0.000 description 1
- 230000002238 attenuated effect Effects 0.000 description 1
- 230000001363 autoimmune Effects 0.000 description 1
- 210000001099 axilla Anatomy 0.000 description 1
- XTKDAFGWCDAMPY-UHFFFAOYSA-N azaperone Chemical compound C1=CC(F)=CC=C1C(=O)CCCN1CCN(C=2N=CC=CC=2)CC1 XTKDAFGWCDAMPY-UHFFFAOYSA-N 0.000 description 1
- 125000002785 azepinyl group Chemical group 0.000 description 1
- 125000003828 azulenyl group Chemical group 0.000 description 1
- 108700041737 bcl-2 Genes Proteins 0.000 description 1
- 230000003542 behavioural effect Effects 0.000 description 1
- 230000009286 beneficial effect Effects 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-N benzenesulfonic acid Chemical compound OS(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-N 0.000 description 1
- 229940092714 benzenesulfonic acid Drugs 0.000 description 1
- 125000003785 benzimidazolyl group Chemical group N1=C(NC2=C1C=CC=C2)* 0.000 description 1
- 235000010233 benzoic acid Nutrition 0.000 description 1
- 229960004365 benzoic acid Drugs 0.000 description 1
- LDECUSDQMXVUMP-UHFFFAOYSA-N benzyl 3-[6-[[2-(butylamino)-1-[3-methoxycarbonyl-4-(2-methoxy-2-oxoethoxy)phenyl]-2-oxoethyl]-hexylamino]-6-oxohexyl]-4-methyl-2-oxo-6-(4-phenylphenyl)-1,6-dihydropyrimidine-5-carboxylate Chemical compound O=C1NC(C=2C=CC(=CC=2)C=2C=CC=CC=2)C(C(=O)OCC=2C=CC=CC=2)=C(C)N1CCCCCC(=O)N(CCCCCC)C(C(=O)NCCCC)C1=CC=C(OCC(=O)OC)C(C(=O)OC)=C1 LDECUSDQMXVUMP-UHFFFAOYSA-N 0.000 description 1
- HSDAJNMJOMSNEV-UHFFFAOYSA-N benzyl chloroformate Chemical compound ClC(=O)OCC1=CC=CC=C1 HSDAJNMJOMSNEV-UHFFFAOYSA-N 0.000 description 1
- 125000001584 benzyloxycarbonyl group Chemical group C(=O)(OCC1=CC=CC=C1)* 0.000 description 1
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 1
- 125000002619 bicyclic group Chemical group 0.000 description 1
- MUALRAIOVNYAIW-UHFFFAOYSA-N binap Chemical compound C1=CC=CC=C1P(C=1C(=C2C=CC=CC2=CC=1)C=1C2=CC=CC=C2C=CC=1P(C=1C=CC=CC=1)C=1C=CC=CC=1)C1=CC=CC=C1 MUALRAIOVNYAIW-UHFFFAOYSA-N 0.000 description 1
- 125000006267 biphenyl group Chemical group 0.000 description 1
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 1
- 229910052794 bromium Inorganic materials 0.000 description 1
- GCSVCUMDOQKEMT-UHFFFAOYSA-N butan-1-amine;hydrofluoride Chemical compound [H+].[F-].CCCCN GCSVCUMDOQKEMT-UHFFFAOYSA-N 0.000 description 1
- DNSISZSEWVHGLH-UHFFFAOYSA-N butanamide Chemical compound CCCC(N)=O DNSISZSEWVHGLH-UHFFFAOYSA-N 0.000 description 1
- KDYFGRWQOYBRFD-NUQCWPJISA-N butanedioic acid Chemical compound O[14C](=O)CC[14C](O)=O KDYFGRWQOYBRFD-NUQCWPJISA-N 0.000 description 1
- 235000014121 butter Nutrition 0.000 description 1
- 239000001582 butter acid Substances 0.000 description 1
- 239000011575 calcium Substances 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- AXCZMVOFGPJBDE-UHFFFAOYSA-L calcium dihydroxide Chemical compound [OH-].[OH-].[Ca+2] AXCZMVOFGPJBDE-UHFFFAOYSA-L 0.000 description 1
- 239000000920 calcium hydroxide Substances 0.000 description 1
- 229910001861 calcium hydroxide Inorganic materials 0.000 description 1
- 150000001721 carbon Chemical group 0.000 description 1
- 239000001569 carbon dioxide Substances 0.000 description 1
- 229910002092 carbon dioxide Inorganic materials 0.000 description 1
- 229910002091 carbon monoxide Inorganic materials 0.000 description 1
- 150000001733 carboxylic acid esters Chemical group 0.000 description 1
- 150000001768 cations Chemical class 0.000 description 1
- 230000030833 cell death Effects 0.000 description 1
- 230000003915 cell function Effects 0.000 description 1
- 230000010261 cell growth Effects 0.000 description 1
- 230000012292 cell migration Effects 0.000 description 1
- 230000004663 cell proliferation Effects 0.000 description 1
- 230000001413 cellular effect Effects 0.000 description 1
- 239000001913 cellulose Substances 0.000 description 1
- 229920002678 cellulose Polymers 0.000 description 1
- 235000010980 cellulose Nutrition 0.000 description 1
- 230000008859 change Effects 0.000 description 1
- 239000007795 chemical reaction product Substances 0.000 description 1
- 125000001309 chloro group Chemical group Cl* 0.000 description 1
- 230000001684 chronic effect Effects 0.000 description 1
- 208000037893 chronic inflammatory disorder Diseases 0.000 description 1
- 229940110456 cocoa butter Drugs 0.000 description 1
- 235000019868 cocoa butter Nutrition 0.000 description 1
- 229960002424 collagenase Drugs 0.000 description 1
- 238000004440 column chromatography Methods 0.000 description 1
- 239000000306 component Substances 0.000 description 1
- 229940125904 compound 1 Drugs 0.000 description 1
- 229940125773 compound 10 Drugs 0.000 description 1
- 229940125797 compound 12 Drugs 0.000 description 1
- 229940126543 compound 14 Drugs 0.000 description 1
- 229940126142 compound 16 Drugs 0.000 description 1
- 229940126208 compound 22 Drugs 0.000 description 1
- 229940125833 compound 23 Drugs 0.000 description 1
- 229940125846 compound 25 Drugs 0.000 description 1
- 229940125851 compound 27 Drugs 0.000 description 1
- 229940127204 compound 29 Drugs 0.000 description 1
- 229940126214 compound 3 Drugs 0.000 description 1
- 229940125877 compound 31 Drugs 0.000 description 1
- 229940126540 compound 41 Drugs 0.000 description 1
- 229940125936 compound 42 Drugs 0.000 description 1
- 229940125844 compound 46 Drugs 0.000 description 1
- 229940127113 compound 57 Drugs 0.000 description 1
- 238000013270 controlled release Methods 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- 238000001816 cooling Methods 0.000 description 1
- 239000013256 coordination polymer Substances 0.000 description 1
- 230000008878 coupling Effects 0.000 description 1
- 238000010168 coupling process Methods 0.000 description 1
- 238000005859 coupling reaction Methods 0.000 description 1
- 229960001681 croscarmellose sodium Drugs 0.000 description 1
- 235000010947 crosslinked sodium carboxy methyl cellulose Nutrition 0.000 description 1
- 238000002425 crystallisation Methods 0.000 description 1
- 230000008025 crystallization Effects 0.000 description 1
- 125000004122 cyclic group Chemical group 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- JMYVMOUINOAAPA-UHFFFAOYSA-N cyclopropanecarbaldehyde Chemical compound O=CC1CC1 JMYVMOUINOAAPA-UHFFFAOYSA-N 0.000 description 1
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 1
- XUJNEKJLAYXESH-UHFFFAOYSA-N cysteine Natural products SCC(N)C(O)=O XUJNEKJLAYXESH-UHFFFAOYSA-N 0.000 description 1
- 235000018417 cysteine Nutrition 0.000 description 1
- 238000000354 decomposition reaction Methods 0.000 description 1
- 230000003247 decreasing effect Effects 0.000 description 1
- 230000002950 deficient Effects 0.000 description 1
- 235000019425 dextrin Nutrition 0.000 description 1
- 239000008121 dextrose Substances 0.000 description 1
- 125000004663 dialkyl amino group Chemical group 0.000 description 1
- MHDVGSVTJDSBDK-UHFFFAOYSA-N dibenzyl ether Chemical compound C=1C=CC=CC=1COCC1=CC=CC=C1 MHDVGSVTJDSBDK-UHFFFAOYSA-N 0.000 description 1
- PXBRQCKWGAHEHS-UHFFFAOYSA-N dichlorodifluoromethane Chemical compound FC(F)(Cl)Cl PXBRQCKWGAHEHS-UHFFFAOYSA-N 0.000 description 1
- 235000019404 dichlorodifluoromethane Nutrition 0.000 description 1
- 235000005911 diet Nutrition 0.000 description 1
- 230000000378 dietary effect Effects 0.000 description 1
- 235000014113 dietary fatty acids Nutrition 0.000 description 1
- 125000005043 dihydropyranyl group Chemical group O1C(CCC=C1)* 0.000 description 1
- 125000002147 dimethylamino group Chemical group [H]C([H])([H])N(*)C([H])([H])[H] 0.000 description 1
- 208000016097 disease of metabolism Diseases 0.000 description 1
- 239000006185 dispersion Substances 0.000 description 1
- 238000004821 distillation Methods 0.000 description 1
- 238000009826 distribution Methods 0.000 description 1
- VHJLVAABSRFDPM-QWWZWVQMSA-N dithiothreitol Chemical compound SC[C@@H](O)[C@H](O)CS VHJLVAABSRFDPM-QWWZWVQMSA-N 0.000 description 1
- 125000003438 dodecyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 238000012377 drug delivery Methods 0.000 description 1
- 239000003596 drug target Substances 0.000 description 1
- 238000010828 elution Methods 0.000 description 1
- 239000003995 emulsifying agent Substances 0.000 description 1
- 230000001804 emulsifying effect Effects 0.000 description 1
- 239000002702 enteric coating Substances 0.000 description 1
- 238000009505 enteric coating Methods 0.000 description 1
- 230000007613 environmental effect Effects 0.000 description 1
- 238000001952 enzyme assay Methods 0.000 description 1
- 210000003979 eosinophil Anatomy 0.000 description 1
- 210000002615 epidermis Anatomy 0.000 description 1
- 210000002919 epithelial cell Anatomy 0.000 description 1
- CCIVGXIOQKPBKL-UHFFFAOYSA-M ethanesulfonate Chemical compound CCS([O-])(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-M 0.000 description 1
- LVGKNOAMLMIIKO-QXMHVHEDSA-N ethyl oleate Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OCC LVGKNOAMLMIIKO-QXMHVHEDSA-N 0.000 description 1
- 229940093471 ethyl oleate Drugs 0.000 description 1
- 238000011156 evaluation Methods 0.000 description 1
- 238000002474 experimental method Methods 0.000 description 1
- 239000000194 fatty acid Substances 0.000 description 1
- 229930195729 fatty acid Natural products 0.000 description 1
- ZWJINEZUASEZBH-UHFFFAOYSA-N fenamic acid Chemical compound OC(=O)C1=CC=CC=C1NC1=CC=CC=C1 ZWJINEZUASEZBH-UHFFFAOYSA-N 0.000 description 1
- 239000011152 fibreglass Substances 0.000 description 1
- 235000019634 flavors Nutrition 0.000 description 1
- 239000012530 fluid Substances 0.000 description 1
- 125000003983 fluorenyl group Chemical group C1(=CC=CC=2C3=CC=CC=C3CC12)* 0.000 description 1
- DWYMPOCYEZONEA-UHFFFAOYSA-L fluoridophosphate Chemical compound [O-]P([O-])(F)=O DWYMPOCYEZONEA-UHFFFAOYSA-L 0.000 description 1
- 239000011737 fluorine Substances 0.000 description 1
- 235000013355 food flavoring agent Nutrition 0.000 description 1
- 238000004108 freeze drying Methods 0.000 description 1
- 230000002518 glial effect Effects 0.000 description 1
- 239000000174 gluconic acid Substances 0.000 description 1
- 235000012208 gluconic acid Nutrition 0.000 description 1
- 235000013922 glutamic acid Nutrition 0.000 description 1
- 239000004220 glutamic acid Substances 0.000 description 1
- 235000011187 glycerol Nutrition 0.000 description 1
- 239000008187 granular material Substances 0.000 description 1
- 235000021552 granulated sugar Nutrition 0.000 description 1
- 150000004820 halides Chemical class 0.000 description 1
- 125000004404 heteroalkyl group Chemical group 0.000 description 1
- 125000004415 heterocyclylalkyl group Chemical group 0.000 description 1
- 239000008240 homogeneous mixture Substances 0.000 description 1
- 235000012907 honey Nutrition 0.000 description 1
- 102000054350 human CHI3L1 Human genes 0.000 description 1
- 229930195733 hydrocarbon Natural products 0.000 description 1
- 230000007062 hydrolysis Effects 0.000 description 1
- 238000006460 hydrolysis reaction Methods 0.000 description 1
- 230000009610 hypersensitivity Effects 0.000 description 1
- 125000002636 imidazolinyl group Chemical group 0.000 description 1
- 208000026278 immune system disease Diseases 0.000 description 1
- 239000007943 implant Substances 0.000 description 1
- 238000000338 in vitro Methods 0.000 description 1
- 238000010348 incorporation Methods 0.000 description 1
- 238000011534 incubation Methods 0.000 description 1
- 125000003453 indazolyl group Chemical group N1N=C(C2=C1C=CC=C2)* 0.000 description 1
- 125000003454 indenyl group Chemical group C1(C=CC2=CC=CC=C12)* 0.000 description 1
- 125000001041 indolyl group Chemical group 0.000 description 1
- 230000001939 inductive effect Effects 0.000 description 1
- 230000008595 infiltration Effects 0.000 description 1
- 238000001764 infiltration Methods 0.000 description 1
- 230000000977 initiatory effect Effects 0.000 description 1
- 230000003914 insulin secretion Effects 0.000 description 1
- 230000003993 interaction Effects 0.000 description 1
- 238000001361 intraarterial administration Methods 0.000 description 1
- 238000007918 intramuscular administration Methods 0.000 description 1
- 238000007912 intraperitoneal administration Methods 0.000 description 1
- 238000001990 intravenous administration Methods 0.000 description 1
- 239000011630 iodine Substances 0.000 description 1
- INQOMBQAUSQDDS-UHFFFAOYSA-N iodomethane Chemical compound IC INQOMBQAUSQDDS-UHFFFAOYSA-N 0.000 description 1
- 150000002500 ions Chemical class 0.000 description 1
- LNQCUTNLHUQZLR-OZJWLQQPSA-N iridin Chemical compound OC1=C(OC)C(OC)=CC(C=2C(C3=C(O)C(OC)=C(O[C@H]4[C@@H]([C@@H](O)[C@H](O)[C@@H](CO)O4)O)C=C3OC=2)=O)=C1 LNQCUTNLHUQZLR-OZJWLQQPSA-N 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- 125000002183 isoquinolinyl group Chemical group C1(=NC=CC2=CC=CC=C12)* 0.000 description 1
- 125000004628 isothiazolidinyl group Chemical group S1N(CCC1)* 0.000 description 1
- 125000001786 isothiazolyl group Chemical group 0.000 description 1
- 239000000644 isotonic solution Substances 0.000 description 1
- 125000003965 isoxazolidinyl group Chemical group 0.000 description 1
- 125000000842 isoxazolyl group Chemical group 0.000 description 1
- ZLVXBBHTMQJRSX-VMGNSXQWSA-N jdtic Chemical compound C1([C@]2(C)CCN(C[C@@H]2C)C[C@H](C(C)C)NC(=O)[C@@H]2NCC3=CC(O)=CC=C3C2)=CC=CC(O)=C1 ZLVXBBHTMQJRSX-VMGNSXQWSA-N 0.000 description 1
- 210000005067 joint tissue Anatomy 0.000 description 1
- VLSMHEGGTFMBBZ-OOZYFLPDSA-N kainic acid Chemical compound CC(=C)[C@H]1CN[C@H](C(O)=O)[C@H]1CC(O)=O VLSMHEGGTFMBBZ-OOZYFLPDSA-N 0.000 description 1
- 229950006874 kainic acid Drugs 0.000 description 1
- 239000004310 lactic acid Substances 0.000 description 1
- 235000014655 lactic acid Nutrition 0.000 description 1
- 208000032839 leukemia Diseases 0.000 description 1
- 230000037356 lipid metabolism Effects 0.000 description 1
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 1
- RENRQMCACQEWFC-UGKGYDQZSA-N lnp023 Chemical compound C1([C@H]2N(CC=3C=4C=CNC=4C(C)=CC=3OC)CC[C@@H](C2)OCC)=CC=C(C(O)=O)C=C1 RENRQMCACQEWFC-UGKGYDQZSA-N 0.000 description 1
- 239000000314 lubricant Substances 0.000 description 1
- 210000004698 lymphocyte Anatomy 0.000 description 1
- 239000011777 magnesium Substances 0.000 description 1
- 229910052749 magnesium Inorganic materials 0.000 description 1
- ZLNQQNXFFQJAID-UHFFFAOYSA-L magnesium carbonate Chemical compound [Mg+2].[O-]C([O-])=O ZLNQQNXFFQJAID-UHFFFAOYSA-L 0.000 description 1
- 239000001095 magnesium carbonate Substances 0.000 description 1
- 229910000021 magnesium carbonate Inorganic materials 0.000 description 1
- CCERQOYLJJULMD-UHFFFAOYSA-M magnesium;carbanide;chloride Chemical compound [CH3-].[Mg+2].[Cl-] CCERQOYLJJULMD-UHFFFAOYSA-M 0.000 description 1
- 239000001630 malic acid Substances 0.000 description 1
- 235000011090 malic acid Nutrition 0.000 description 1
- 229960002510 mandelic acid Drugs 0.000 description 1
- 230000007246 mechanism Effects 0.000 description 1
- 125000005358 mercaptoalkyl group Chemical group 0.000 description 1
- 229910052751 metal Inorganic materials 0.000 description 1
- 239000002184 metal Substances 0.000 description 1
- 230000001394 metastastic effect Effects 0.000 description 1
- 206010061289 metastatic neoplasm Diseases 0.000 description 1
- 125000005948 methanesulfonyloxy group Chemical group 0.000 description 1
- PAUWJGNLAVWRMD-UHFFFAOYSA-N methyl 1,8-naphthyridine-2-carboxylate Chemical compound C1=CC=NC2=NC(C(=O)OC)=CC=C21 PAUWJGNLAVWRMD-UHFFFAOYSA-N 0.000 description 1
- VMVNZNXAVJHNDJ-UHFFFAOYSA-N methyl 2,2,2-trifluoroacetate Chemical compound COC(=O)C(F)(F)F VMVNZNXAVJHNDJ-UHFFFAOYSA-N 0.000 description 1
- UQSKWEFWQGNUKQ-UHFFFAOYSA-N methyl 3-[(4-acetamidophenyl)methyl]-7-chloro-4-oxo-1-phenylquinoline-2-carboxylate Chemical compound O=C1C2=CC=C(Cl)C=C2N(C=2C=CC=CC=2)C(C(=O)OC)=C1CC1=CC=C(NC(C)=O)C=C1 UQSKWEFWQGNUKQ-UHFFFAOYSA-N 0.000 description 1
- QBIVXUQYJUTFQP-UHFFFAOYSA-N methyl 3-[[4-(methylcarbamoyl)phenyl]methyl]-4-oxo-1-phenylquinoline-2-carboxylate Chemical compound C1=CC(C(=O)NC)=CC=C1CC(C(C1=CC=CC=C11)=O)=C(C(=O)OC)N1C1=CC=CC=C1 QBIVXUQYJUTFQP-UHFFFAOYSA-N 0.000 description 1
- XVRPKRUSQCWQCU-UHFFFAOYSA-N methyl 4-oxo-1-phenylquinoline-2-carboxylate Chemical compound COC(=O)C1=CC(=O)C2=CC=CC=C2N1C1=CC=CC=C1 XVRPKRUSQCWQCU-UHFFFAOYSA-N 0.000 description 1
- ZJKMTTJERBJPFI-UHFFFAOYSA-N methyl 7-chloro-3-[[4-(dimethylsulfamoyl)phenyl]methyl]-4-oxo-1-phenylquinoline-2-carboxylate Chemical compound O=C1C2=CC=C(Cl)C=C2N(C=2C=CC=CC=2)C(C(=O)OC)=C1CC1=CC=C(S(=O)(=O)N(C)C)C=C1 ZJKMTTJERBJPFI-UHFFFAOYSA-N 0.000 description 1
- ZBELDPMWYXDLNY-UHFFFAOYSA-N methyl 9-(4-bromo-2-fluoroanilino)-[1,3]thiazolo[5,4-f]quinazoline-2-carboximidate Chemical compound C12=C3SC(C(=N)OC)=NC3=CC=C2N=CN=C1NC1=CC=C(Br)C=C1F ZBELDPMWYXDLNY-UHFFFAOYSA-N 0.000 description 1
- 229920000609 methyl cellulose Polymers 0.000 description 1
- 229940050176 methyl chloride Drugs 0.000 description 1
- 239000004292 methyl p-hydroxybenzoate Substances 0.000 description 1
- 235000010270 methyl p-hydroxybenzoate Nutrition 0.000 description 1
- 239000001923 methylcellulose Substances 0.000 description 1
- 235000010981 methylcellulose Nutrition 0.000 description 1
- 229960002216 methylparaben Drugs 0.000 description 1
- 125000004170 methylsulfonyl group Chemical group [H]C([H])([H])S(*)(=O)=O 0.000 description 1
- 239000008108 microcrystalline cellulose Substances 0.000 description 1
- 235000019813 microcrystalline cellulose Nutrition 0.000 description 1
- 229940016286 microcrystalline cellulose Drugs 0.000 description 1
- 238000013508 migration Methods 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 238000007479 molecular analysis Methods 0.000 description 1
- 125000002950 monocyclic group Chemical group 0.000 description 1
- 125000002757 morpholinyl group Chemical group 0.000 description 1
- 238000010172 mouse model Methods 0.000 description 1
- 239000002324 mouth wash Substances 0.000 description 1
- 230000036457 multidrug resistance Effects 0.000 description 1
- 108010066052 multidrug resistance-associated protein 1 Proteins 0.000 description 1
- 210000003098 myoblast Anatomy 0.000 description 1
- APVPOHHVBBYQAV-UHFFFAOYSA-N n-(4-aminophenyl)sulfonyloctadecanamide Chemical compound CCCCCCCCCCCCCCCCCC(=O)NS(=O)(=O)C1=CC=C(N)C=C1 APVPOHHVBBYQAV-UHFFFAOYSA-N 0.000 description 1
- SYSQUGFVNFXIIT-UHFFFAOYSA-N n-[4-(1,3-benzoxazol-2-yl)phenyl]-4-nitrobenzenesulfonamide Chemical class C1=CC([N+](=O)[O-])=CC=C1S(=O)(=O)NC1=CC=C(C=2OC3=CC=CC=C3N=2)C=C1 SYSQUGFVNFXIIT-UHFFFAOYSA-N 0.000 description 1
- YGBMCLDVRUGXOV-UHFFFAOYSA-N n-[6-[6-chloro-5-[(4-fluorophenyl)sulfonylamino]pyridin-3-yl]-1,3-benzothiazol-2-yl]acetamide Chemical compound C1=C2SC(NC(=O)C)=NC2=CC=C1C(C=1)=CN=C(Cl)C=1NS(=O)(=O)C1=CC=C(F)C=C1 YGBMCLDVRUGXOV-UHFFFAOYSA-N 0.000 description 1
- FVLVBVSILSHUAF-UHFFFAOYSA-N n-benzyl-3,5-dimethyl-n-propan-2-ylbenzamide Chemical compound C=1C(C)=CC(C)=CC=1C(=O)N(C(C)C)CC1=CC=CC=C1 FVLVBVSILSHUAF-UHFFFAOYSA-N 0.000 description 1
- 125000001280 n-hexyl group Chemical group C(CCCCC)* 0.000 description 1
- 125000001624 naphthyl group Chemical group 0.000 description 1
- 235000021096 natural sweeteners Nutrition 0.000 description 1
- IOMMMLWIABWRKL-WUTDNEBXSA-N nazartinib Chemical compound C1N(C(=O)/C=C/CN(C)C)CCCC[C@H]1N1C2=C(Cl)C=CC=C2N=C1NC(=O)C1=CC=NC(C)=C1 IOMMMLWIABWRKL-WUTDNEBXSA-N 0.000 description 1
- 210000001577 neostriatum Anatomy 0.000 description 1
- 230000016273 neuron death Effects 0.000 description 1
- 210000000440 neutrophil Anatomy 0.000 description 1
- 235000005152 nicotinamide Nutrition 0.000 description 1
- 229910017604 nitric acid Inorganic materials 0.000 description 1
- 125000004433 nitrogen atom Chemical group N* 0.000 description 1
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 1
- 229910000069 nitrogen hydride Inorganic materials 0.000 description 1
- 239000002687 nonaqueous vehicle Substances 0.000 description 1
- 235000017524 noni Nutrition 0.000 description 1
- 231100000252 nontoxic Toxicity 0.000 description 1
- 230000003000 nontoxic effect Effects 0.000 description 1
- 235000020824 obesity Nutrition 0.000 description 1
- 125000002347 octyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 235000008390 olive oil Nutrition 0.000 description 1
- 239000004006 olive oil Substances 0.000 description 1
- 238000003305 oral gavage Methods 0.000 description 1
- 239000010502 orange oil Substances 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 150000002895 organic esters Chemical class 0.000 description 1
- 125000000962 organic group Chemical group 0.000 description 1
- 239000003960 organic solvent Substances 0.000 description 1
- AICOOMRHRUFYCM-ZRRPKQBOSA-N oxazine, 1 Chemical compound C([C@@H]1[C@H](C(C[C@]2(C)[C@@H]([C@H](C)N(C)C)[C@H](O)C[C@]21C)=O)CC1=CC2)C[C@H]1[C@@]1(C)[C@H]2N=C(C(C)C)OC1 AICOOMRHRUFYCM-ZRRPKQBOSA-N 0.000 description 1
- 125000000160 oxazolidinyl group Chemical group 0.000 description 1
- 125000002971 oxazolyl group Chemical group 0.000 description 1
- KJIFKLIQANRMOU-UHFFFAOYSA-N oxidanium;4-methylbenzenesulfonate Chemical compound O.CC1=CC=C(S(O)(=O)=O)C=C1 KJIFKLIQANRMOU-UHFFFAOYSA-N 0.000 description 1
- CFHIDWOYWUOIHU-UHFFFAOYSA-N oxomethyl Chemical group O=[CH] CFHIDWOYWUOIHU-UHFFFAOYSA-N 0.000 description 1
- MUMZUERVLWJKNR-UHFFFAOYSA-N oxoplatinum Chemical compound [Pt]=O MUMZUERVLWJKNR-UHFFFAOYSA-N 0.000 description 1
- 239000001301 oxygen Substances 0.000 description 1
- 125000004430 oxygen atom Chemical group O* 0.000 description 1
- 239000002245 particle Substances 0.000 description 1
- 239000006072 paste Substances 0.000 description 1
- 230000008506 pathogenesis Effects 0.000 description 1
- 239000001814 pectin Substances 0.000 description 1
- 235000010987 pectin Nutrition 0.000 description 1
- 229920001277 pectin Polymers 0.000 description 1
- 239000003961 penetration enhancing agent Substances 0.000 description 1
- 125000001147 pentyl group Chemical group C(CCCC)* 0.000 description 1
- 125000005010 perfluoroalkyl group Chemical group 0.000 description 1
- 125000005342 perphosphate group Chemical group 0.000 description 1
- RLZZZVKAURTHCP-UHFFFAOYSA-N phenanthrene-3,4-diol Chemical compound C1=CC=C2C3=C(O)C(O)=CC=C3C=CC2=C1 RLZZZVKAURTHCP-UHFFFAOYSA-N 0.000 description 1
- 125000005561 phenanthryl group Chemical group 0.000 description 1
- XAMUDJHXFNRLCY-UHFFFAOYSA-N phenthoate Chemical compound CCOC(=O)C(SP(=S)(OC)OC)C1=CC=CC=C1 XAMUDJHXFNRLCY-UHFFFAOYSA-N 0.000 description 1
- UYWQUFXKFGHYNT-UHFFFAOYSA-N phenylmethyl ester of formic acid Natural products O=COCC1=CC=CC=C1 UYWQUFXKFGHYNT-UHFFFAOYSA-N 0.000 description 1
- 125000003170 phenylsulfonyl group Chemical group C1(=CC=CC=C1)S(=O)(=O)* 0.000 description 1
- 239000002953 phosphate buffered saline Substances 0.000 description 1
- 125000005499 phosphonyl group Chemical group 0.000 description 1
- SIOXPEMLGUPBBT-UHFFFAOYSA-N picolinic acid Chemical compound OC(=O)C1=CC=CC=N1 SIOXPEMLGUPBBT-UHFFFAOYSA-N 0.000 description 1
- 125000003386 piperidinyl group Chemical group 0.000 description 1
- 125000005936 piperidyl group Chemical group 0.000 description 1
- BASFCYQUMIYNBI-UHFFFAOYSA-N platinum Substances [Pt] BASFCYQUMIYNBI-UHFFFAOYSA-N 0.000 description 1
- 229910003446 platinum oxide Inorganic materials 0.000 description 1
- 229920000747 poly(lactic acid) Polymers 0.000 description 1
- 229920000573 polyethylene Polymers 0.000 description 1
- 239000004626 polylactic acid Substances 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- SCVFZCLFOSHCOH-UHFFFAOYSA-M potassium acetate Chemical compound [K+].CC([O-])=O SCVFZCLFOSHCOH-UHFFFAOYSA-M 0.000 description 1
- BDAWXSQJJCIFIK-UHFFFAOYSA-N potassium methoxide Chemical compound [K+].[O-]C BDAWXSQJJCIFIK-UHFFFAOYSA-N 0.000 description 1
- 238000001556 precipitation Methods 0.000 description 1
- 239000003755 preservative agent Substances 0.000 description 1
- 230000002335 preservative effect Effects 0.000 description 1
- 208000030266 primary brain neoplasm Diseases 0.000 description 1
- 230000035755 proliferation Effects 0.000 description 1
- BDERNNFJNOPAEC-UHFFFAOYSA-N propan-1-ol Chemical compound CCCO BDERNNFJNOPAEC-UHFFFAOYSA-N 0.000 description 1
- LFULEKSKNZEWOE-UHFFFAOYSA-N propanil Chemical compound CCC(=O)NC1=CC=C(Cl)C(Cl)=C1 LFULEKSKNZEWOE-UHFFFAOYSA-N 0.000 description 1
- 239000003380 propellant Substances 0.000 description 1
- 229940080818 propionamide Drugs 0.000 description 1
- 235000019260 propionic acid Nutrition 0.000 description 1
- WGYKZJWCGVVSQN-UHFFFAOYSA-N propylamine Chemical group CCCN WGYKZJWCGVVSQN-UHFFFAOYSA-N 0.000 description 1
- 125000001042 pteridinyl group Chemical group N1=C(N=CC2=NC=CN=C12)* 0.000 description 1
- 230000002685 pulmonary effect Effects 0.000 description 1
- 239000012264 purified product Substances 0.000 description 1
- 125000004309 pyranyl group Chemical group O1C(C=CC=C1)* 0.000 description 1
- 125000003373 pyrazinyl group Chemical group 0.000 description 1
- PBMFSQRYOILNGV-UHFFFAOYSA-N pyridazine Chemical compound C1=CC=NN=C1 PBMFSQRYOILNGV-UHFFFAOYSA-N 0.000 description 1
- 125000002098 pyridazinyl group Chemical group 0.000 description 1
- VHNQIURBCCNWDN-UHFFFAOYSA-N pyridine-2,6-diamine Chemical compound NC1=CC=CC(N)=N1 VHNQIURBCCNWDN-UHFFFAOYSA-N 0.000 description 1
- 125000004076 pyridyl group Chemical group 0.000 description 1
- 125000000714 pyrimidinyl group Chemical group 0.000 description 1
- 239000002510 pyrogen Substances 0.000 description 1
- 125000000719 pyrrolidinyl group Chemical group 0.000 description 1
- 229940107700 pyruvic acid Drugs 0.000 description 1
- 125000002294 quinazolinyl group Chemical group N1=C(N=CC2=CC=CC=C12)* 0.000 description 1
- IUVKMZGDUIUOCP-BTNSXGMBSA-N quinbolone Chemical compound O([C@H]1CC[C@H]2[C@H]3[C@@H]([C@]4(C=CC(=O)C=C4CC3)C)CC[C@@]21C)C1=CCCC1 IUVKMZGDUIUOCP-BTNSXGMBSA-N 0.000 description 1
- 125000002943 quinolinyl group Chemical group N1=C(C=CC2=CC=CC=C12)* 0.000 description 1
- 125000001567 quinoxalinyl group Chemical group N1=C(C=NC2=CC=CC=C12)* 0.000 description 1
- 125000004621 quinuclidinyl group Chemical group N12C(CC(CC1)CC2)* 0.000 description 1
- 230000001105 regulatory effect Effects 0.000 description 1
- 210000002345 respiratory system Anatomy 0.000 description 1
- 125000006413 ring segment Chemical group 0.000 description 1
- 229960004889 salicylic acid Drugs 0.000 description 1
- 235000019515 salmon Nutrition 0.000 description 1
- 230000019491 signal transduction Effects 0.000 description 1
- 229920002379 silicone rubber Polymers 0.000 description 1
- 239000004945 silicone rubber Substances 0.000 description 1
- 210000003491 skin Anatomy 0.000 description 1
- 150000003384 small molecules Chemical class 0.000 description 1
- 210000002460 smooth muscle Anatomy 0.000 description 1
- WRIKHQLVHPKCJU-UHFFFAOYSA-N sodium bis(trimethylsilyl)amide Chemical compound C[Si](C)(C)N([Na])[Si](C)(C)C WRIKHQLVHPKCJU-UHFFFAOYSA-N 0.000 description 1
- 229910000029 sodium carbonate Inorganic materials 0.000 description 1
- 239000012312 sodium hydride Substances 0.000 description 1
- 229910000104 sodium hydride Inorganic materials 0.000 description 1
- 229910052938 sodium sulfate Inorganic materials 0.000 description 1
- 235000011152 sodium sulphate Nutrition 0.000 description 1
- 239000008279 sol Substances 0.000 description 1
- 239000011343 solid material Substances 0.000 description 1
- 239000012265 solid product Substances 0.000 description 1
- 239000012453 solvate Substances 0.000 description 1
- 239000000600 sorbitol Substances 0.000 description 1
- 241000894007 species Species 0.000 description 1
- 230000003595 spectral effect Effects 0.000 description 1
- 230000000087 stabilizing effect Effects 0.000 description 1
- 210000000130 stem cell Anatomy 0.000 description 1
- 239000008223 sterile water Substances 0.000 description 1
- 230000035882 stress Effects 0.000 description 1
- 238000007920 subcutaneous administration Methods 0.000 description 1
- 235000000346 sugar Nutrition 0.000 description 1
- 125000001174 sulfone group Chemical group 0.000 description 1
- 150000003457 sulfones Chemical class 0.000 description 1
- RWSOTUBLDIXVET-UHFFFAOYSA-O sulfonium Chemical compound [SH3+] RWSOTUBLDIXVET-UHFFFAOYSA-O 0.000 description 1
- YBBRCQOCSYXUOC-UHFFFAOYSA-N sulfuryl dichloride Chemical compound ClS(Cl)(=O)=O YBBRCQOCSYXUOC-UHFFFAOYSA-N 0.000 description 1
- 230000008093 supporting effect Effects 0.000 description 1
- 239000004094 surface-active agent Substances 0.000 description 1
- 238000001356 surgical procedure Methods 0.000 description 1
- 238000013268 sustained release Methods 0.000 description 1
- 239000012730 sustained-release form Substances 0.000 description 1
- 230000007470 synaptic degeneration Effects 0.000 description 1
- 239000006188 syrup Substances 0.000 description 1
- 235000020357 syrup Nutrition 0.000 description 1
- 230000009885 systemic effect Effects 0.000 description 1
- 239000000454 talc Substances 0.000 description 1
- 229910052623 talc Inorganic materials 0.000 description 1
- 235000018553 tannin Nutrition 0.000 description 1
- 239000001648 tannin Substances 0.000 description 1
- 229920001864 tannin Polymers 0.000 description 1
- 239000011269 tar Substances 0.000 description 1
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- IRFHMTUHTBSEBK-QGZVFWFLSA-N tert-butyl n-[(2s)-2-(2,5-difluorophenyl)-3-quinolin-3-ylpropyl]carbamate Chemical compound C1([C@H](CC=2C=C3C=CC=CC3=NC=2)CNC(=O)OC(C)(C)C)=CC(F)=CC=C1F IRFHMTUHTBSEBK-QGZVFWFLSA-N 0.000 description 1
- SFLXUZPXEWWQNH-UHFFFAOYSA-K tetrabutylazanium;tribromide Chemical compound [Br-].[Br-].[Br-].CCCC[N+](CCCC)(CCCC)CCCC.CCCC[N+](CCCC)(CCCC)CCCC.CCCC[N+](CCCC)(CCCC)CCCC SFLXUZPXEWWQNH-UHFFFAOYSA-K 0.000 description 1
- 125000001412 tetrahydropyranyl group Chemical group 0.000 description 1
- RWRDLPDLKQPQOW-UHFFFAOYSA-N tetrahydropyrrole Natural products C1CCNC1 RWRDLPDLKQPQOW-UHFFFAOYSA-N 0.000 description 1
- 125000000147 tetrahydroquinolinyl group Chemical group N1(CCCC2=CC=CC=C12)* 0.000 description 1
- 125000001113 thiadiazolyl group Chemical group 0.000 description 1
- 125000006089 thiamorpholinyl sulfoxide group Chemical group 0.000 description 1
- 125000001984 thiazolidinyl group Chemical group 0.000 description 1
- 125000000335 thiazolyl group Chemical group 0.000 description 1
- 125000003396 thiol group Chemical group [H]S* 0.000 description 1
- 125000004568 thiomorpholinyl group Chemical group 0.000 description 1
- 125000003944 tolyl group Chemical group 0.000 description 1
- 230000000699 topical effect Effects 0.000 description 1
- 239000012049 topical pharmaceutical composition Substances 0.000 description 1
- 230000037317 transdermal delivery Effects 0.000 description 1
- 238000012546 transfer Methods 0.000 description 1
- 238000011269 treatment regimen Methods 0.000 description 1
- 229940029284 trichlorofluoromethane Drugs 0.000 description 1
- 229940086542 triethylamine Drugs 0.000 description 1
- 102000003390 tumor necrosis factor Human genes 0.000 description 1
- 238000001291 vacuum drying Methods 0.000 description 1
- 235000015112 vegetable and seed oil Nutrition 0.000 description 1
- 239000008158 vegetable oil Substances 0.000 description 1
- 239000011345 viscous material Substances 0.000 description 1
- 229910052725 zinc Inorganic materials 0.000 description 1
- 239000011701 zinc Substances 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D215/00—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems
- C07D215/02—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom
- C07D215/16—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D215/48—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/47—Quinolines; Isoquinolines
- A61K31/4709—Non-condensed quinolines and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/06—Antiasthmatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
- A61P25/16—Anti-Parkinson drugs
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/06—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/06—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/12—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Pharmacology & Pharmacy (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Biomedical Technology (AREA)
- Neurosurgery (AREA)
- Diabetes (AREA)
- Neurology (AREA)
- Obesity (AREA)
- Pulmonology (AREA)
- Hematology (AREA)
- Rheumatology (AREA)
- Immunology (AREA)
- Emergency Medicine (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Pain & Pain Management (AREA)
- Psychiatry (AREA)
- Child & Adolescent Psychology (AREA)
- Endocrinology (AREA)
- Hospice & Palliative Care (AREA)
- Psychology (AREA)
- Physical Education & Sports Medicine (AREA)
- Epidemiology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Plural Heterocyclic Compounds (AREA)
- Quinoline Compounds (AREA)
Abstract
composto, método para tratar um distúrbio mediado por jnk em um paciente que tem um distúrbio mediado por jnk, composição farmacêutica, uso do composto e medicamento que compreende o composto compostos da fórmula i ou um sal farmaceuticamente aceitável dos mesmos são moduladores efetivos de jnk: em que x , y, z, r4, r5 , r6 e r7 são tais como definidos neste contexto.
Description
COMPOSTO, MBTODO PÃM mm W DISTNRBTO MEDXADO.^Og_„ JNK EM UM PACIENTE QUE TEM W DISTÚRBIO MEDIAD0„^Og„
JKR, COMPOSIÇÃO FOMACtOTICA, OSO BO COMPOSTO EMEPICA^
MENTO QUECOMFREENDE 0 COMPOSTO
Refere-se a presente invenção de urn modo geral aos campos da química medicinal e tratamento de oistúrbios inflamatdrios. Maia particularmente, a invenção refere-se a inibidores de pequenas moléculas de INK., métodos e formulações para inibirem JNK e tratamento de distúrbios mediados por JNKf e assemelhados.
As quinases c-Jun N-terminais (<JNKs) são. meivbros da familia das guinases de proteínas mitogenativadas juntamente com p38 e guinases reguladas por sinal ext.racelular (ERKs; . Identificaram-se trás genes 15 distintos (ink.l, jnk2 e jnk3) que codificam variantes de. 10 junções (Ip and Davis, Curr. Opin. Cell Bio.L· (1998} 10:205-19}. O JNK1 e JNK2 são expressos em uma ampla variedade de tecidos, enquanto que <TNK3 é expresso principalmente em neurônios, e em uma extensão menor no coração e em testes (Yang et al < , .Mature (129 /) .309:855 -70} , Os membros da família das <JNK são ativados por oltoquinas pré-ínflamatõrias tais como o fator de necrose de tumor <x (TNF-u) e interleucina 16 (Us-
| 16 l < | bem | como | e s t resees amb ien tais. | A a t r va ç ã o d a s | |
| 5 | uNKs | ê me; | liada | por suas qu leases de | montante, MKK4 e |
| MKK7 | * V Λ- | f osfí | >rilação dupla de Thr-1 | 33 e Tyr-18S (De- | |
| ri ή a | rd et | al., | Cell. (.1: S.M.} 7 6 Í10 2 S - 3 7 | } , Demon&trou-se |
que MKK.4 e MMK7 podem ser ativadas pelas diversas qui2/149 cases de montanta, que inclui MEKK1 e MEKK4, na dependência das estímulos externos e contexto celurar (Boyle et al. f Arthritis Rheum (2003} 4:8:2450-24) . A especificidade da sinalização de JNK é conseguida pela formação 5 de um complexo de sinaliração específico de d$K que contêm múltiplos componentes da cascata da quinases pelo uso de proteínas de tablado chamadas proteínas de interação de JNK (Yasuda et al., Mol. Cell. Biol(1999) 19:7245-54). As JBKs mostraram desempenhar fun10 çdes importantes em .inflamação, funções de células T, apoptose e sobrevivência celular por substratos específicos,. que inclui fatores de transcrição tais como cJun, o componente da família da proteína-1 ati.vadora (API) ,< e ATF2, bem como fatores de não transcrição,. 15 tais como XRS-1 e Bcl-2 (Wanning and Davis, hat. Rev.
Drug Díscov. (2003) 2:554-55). Acredita-se que a sobreativação de JNK constitua um mecanismo importante nas enfermidades auto-imunes, inflamatóri.as, neurológicas, bem com© câncer.
A artrite reumatôide (RA) é uma enfermidade auto- imune sistêmica caracterizada por inflamação crônica das juntas. Adicionalmente à inchação e dor das juntas causada pelo processo inflamatório, a .maior parte dos pacientes de RA desenvolvem por ultimo dano e ueforma25 ção debi.litante das juntas.. Diversas linhas de evidencia farmacolõgica e genética prementes em modelos celulares e de animais sugerem fortemente a relevância e importância da JNK ativada na patogênese da BA. Prí
3/149 melra, a ativação normal de JNK foi detectada nas juntas artríticas humanas a partir de pacientes de PA (Schett et ai., Arthritis Rheum {3000) 43:2501-12) e fias juntas- artríticas de roedores provenientes de modelos 5 de artrite de animais (Han et al > f dl Clin. Invest .
(2001) 108:73-81). Além disso, a inibição de. ativação de JNK per inibidores de JNK seletivos bloqueou citoquinas prõ-inflamatórias e a produção de MMP em smoviocitos, maerôfagos e linfocítos humanos (Han et al. , 10 (200'1) supra). De acordo com um aspecto importante, a administração dos inibidores de UNK seletivos em ratos com artrite adjuvante (Han et al., (2001) supra) ou em camundangos com artrite induzida por colégeno «Sailiard et al., J Med Ch&jfí. (2005) 14:4596-607) protegeu efeti15 vamente as juntas contra destruição e reduziu de forma significativa inchação das patas, pela inibição da expressão de oitoguina e colagenaseA asma é uma enfermidade inflamatôria crônica das vias aéreas, caracterizada pela presença de um pro20 cesso inflamatôrio celular e por hipersensibilida.de brônquica associada son· mudanças estruturais das vias aéreas (Bradley et al < , d< Allergy Clin. Aim?uno 1..
(1991) 88:66'1-74). Este distúrbio mostrou ser provocado por muitos tipos de células nas vias aéreas .incluxn25 do linfocítos T, eosinófilos, células haste, neutrófrlos e células epiteliais (Bousquet et al., Am. J. Respir. Crit. Care Med< (2000) 161:1720-45). As JNKs surgiram como objetos terapêuticos promissores para a asma
4/149 baseados nos recentes estudos de prova-de-conceito: ficou demonstrado gue os inibidores de JNK bloquearam de forma significativa a produção de RANTES em células lisas das vias aéreas humanas ativadas (Kujime et al., J'.
Immunol. (2000) 164:3222-28). Mais importante, os inibidores de JNK mostraram boa eficácia em modelos crônicos de rasto e camundongo quanto às suas capacidades de reduzirem a infiltração celular, inflamação, hiperse.na.ibi 1 idade, proliferação no musculo liso, e produção IQ de IgE (Nath et al., E’u.r. J. Pharmacol. (2005) 506:27333; Eynott et &1., Bx‘> J, Pharmacol. (2GO 3) 140:137380)> Estas observações sugerem importantes funções das JNKs na inflamação alérgica, processo de remodelagem das vias aéreas associado oom a hiper-sensibilidade.
Portanto, espera-se que o bloqueio da atividade de JNK seja benéfico para o tratamento da asma.
A diabetes do Tipo 2 e a enfermidade metabolica mais séria e preponderante caracterizada por resistência a insulina e deterioração de secreção de insulina 20 como um resultado de metabolismo de lipídeos anormal e inflamação crônica de baixo nível associados com estresse oxidaste. Reportou-se que a atividade de JNK é anormalmente elevada em vãrios tecidos objeto diabéticos sob condições obesas e diabéticas (Hirosumi et al-, 25 Nature (2022; 420:333-36; KanetO, Kxpert. Qpin. Ther.
Targets (2005) 9:581-92). A ativação do caminhe das JNK por citoquinas pró-ínflamatorlas e estresses oxidant es regula negativamente a sinalização de insulina
5/149 por intermédio da fosforilação do substrato-1 receptor de insulina (IRS-1) no Ser!ii\ portanto contribui para a resistência a insulina e tolerância à glicose (Hirosumi et al., Nature (2002? supra? Lee et ai., 7. Bial* Chem, (2003) 278;2896-902; Nafeata.nl st al., J. Biol. Chem. (2004) 279:45803--09)- Evidência genética imperiosa provêm de estudes de modelo de animal elegante utilizandose camundongos ink'·' cruzados com seja camundongos obesos genéticos (ob/ob) ou camundongos obesos dietéticos. A perda de JNK1 (JNK1’Z 3 , mas não funções JNK.2 (jnk2 ? } , protegeu camundongos obesos de ganhos corpéreos, níveis de glicose do sangue de condição estável aumentados, e níveis de insulina de plasma diminuídos (Hírosumi et al., Nature (2002} supra). Estes estudos demonstraram a utilidade potencial do inibidor de JHK no tratamento de obesidade/diabetes do tips© 2.
As enfermidades neurodegenerativas, tais como de Alzheimer (AD) , Parkinson (PD? e Derrame Cerebral são enfermidades de CNS caracterizadas por perda sinâptica, atrofia ueurônica e óbito. O caminho da JNK que conduz à ativação de o-Jun demonstrou desempenhar uma função causativa na apoptose de neurônios embrionários primários isolados e lixxhas de células neurônicas múltiplas quando da indução de uma variedade de estímulos (fôozyczko-Coyxxe et al. # Cuzr. Drug Targets Ch'S Neurol. Disord. (2002) 1:31-49-. Observou-se superativação de JNK em cérebros humanos de pacientes de Ah (Pei et al.# Alshe.ime.rs Dis. (2001) 3:41-48) ou seções de cêre
6/149 bros; de roedores derivados de modelos de animais de enfermidades neurodegenerativas (Saparito et al., J. Neurochenn <2000; 75:1200-08). Por exemplo, detectaram-se fosfo-UNKs aumentadas nos cérebros após a morte prove-
| nientes de pacientes | A adm.ini st | ração de peptí- | ||
| deo inibidor de JNK | (peptídeo | JIP-l) no | modelo de roe- | |
| dor de AD induzido | por adm.i ni s t ração | de peptídeo β- |
amilôide preveniu a deterioração da plasticidade einápti ca. Nos modelos de animais de PI> (modelo de NPTF)x 10 observaram-se fosfo-MKK4 e fosfo-JNKs elevados concomitant emente com st morte de células neurônicas. A transferência de genes adeno-v.ira.is de pent ideo inibidor de
JSX (peptídeo de JIP-1) no estriato de eamundongos atenuou a deterioração oomportamental pela inibição de JNK 15 mediada por MPTP, ativação de c-Jun e caspase, bloqueando desta forma a morte de células na substância nigra (Xia et ai., Proc. Natl. Acad. S’ci. ££?A. (2001)
58:104 3.3-38), Adicionalmente, no modelo animal de ataque isquêmico induzido por excitotoxicidade de glutama20 to, os camundongos deficientes em JNK3, mas não em JNK1 ou 0NK2, foram resistentes a insulto mediado por ácido cainíco (agonists, receptor de glutamato) ou morte neurônios (Yang et al. . Nature (1997} 385:865-70). Estes dados sugerem que a JNK.,3 foi responsável principa.lmen.te 25 pela excitotoxicidade de glutamato, um componente importante nas condições isquêmicas. Tomados em conjunto, as dados apresentaram-se sugerindo JNKs como objetivos atraentes para múltiplas enfermidades de CNS associadas oom a morte de células netwônícas.
O crescimento, proliferação e migração celular descontrolados juntamente com angiogênese desregulada conduzem à. formação de tumores malignos. 0 caminho de 5 t.ransdução de sinal de JJíK pode não agir exclusivamente na apoptose, com a ativação de JNK sustentada, conduzindo à ativação de API implicou recentemente na contribuição para a sobrevivência celular de tipos de câncer específicos,, tais como tumores de células gliais e lin~ 10 foblastos B transformados por BCL--ABL {Antonyak et al.,
Oncogene (2002) 21x5038-46; Hess et al., &rat. Genet. (2002) 32x201-05). rto caso de tumores de células gliais, constatou-se atividade de JNK/A..B1. aumentada na maior parte das amostras de tumores de cérebro primá·· 15 rios. Para os linfoblastos B transformados, o BCL-ABL mostrou ativar o caminho da JIJK que por sua vez regulem a expressão do gene bcl-2 anti-apoptõtico. Sob um aspecto interessante, a resistência a muiti-drogas e hipex-proliferação observadas em pacientes de AML refra20 tãrios ao tratamento foi casualmente associada à atividade de JNK sustentada presente nestas amostras de AML(Cripe et al. , Leukemia (2002) 16:799-812). A ativação de JNK em células leucêmicas resultou na expressão induzida de bombas de ef.lu.xo, tais como mdrl e MRP1, res20 ponsãveis pela resistência a multi-drogas, Também, os genes com um benefício de sobrevivência em .resposta a estresse ox.ida.nte, incluindo glutationa-S-transferase π e sintase de γ-g.lutami.l cisteína, foram também aumenta /14 8 dos pelo caminho de JMK ativada. Consequentemente, os moduladores de JNK são de utilidade no tratamento de uma variedade de enfermidade» e/ou condições.
Um aspecto da invenção consiste em um composto da formula It

em que
X é CR1: ou M;
Y é ~C(O|RJ, heteroarila de 5~ele.mentos, ou heterocicilia de 5- elementos j
X ê fenila, cicloalquila, heterociclila ou heteroarila, e é substituído com R5, e R2;
R: e R2 são cada um .independentemente H, halo, CH, al~ quila inferior, ou -Y^-Y^-Y-?^, ou Rx e R<: em conjunto formam -0(CH3} a0~ f onde n é 1 ou 2;
Y1 ê -O-, -C(O)-« -C(O)O~, -CfOjRR*-, -RRSC(O)-, --8-, 80ΰ-, ou uma. 1 igação:
Y2 é cicloalquileno, heterocicloalquileno, alquileno .inferior ou uma ligação;
Y5 é -O-, -e(0}-» ~C(OÍO-, -C{Ç))dRs-f -NP?C(O) ·, -SO3-, ou uma ligação;
P? é H, al.qu.ila inferior, alcoxila. inferior, cicloalquila, heterocicloaiquiia, ou em que Rs outro que não H é opcional men te substituído com aiquila inferior, halo, -CF;}, ou -OH;
9/14 9
R* e R!:! sã» cada, um Índependentemente H ou alquila infer ior;
R? é OH, alquila .inferior, alcoxila inferior, (alcoxila inferior) -alcoxila inferior, ou -NR?RJ%
R.'$ é alquila inferior, fenila, heterociclila, ciclcalquila, heterocicloalgui.la, ou heteroarila, e é- opcionalmente substituído com alquila inferior, hidroxila, alco.xi.la inferior, halo, nitro, amino, ciano, ou halo-alquila .inferior;
R': e R* são cada um Índependentemente H, halo, ciano, alquila inferior, -CF^, alcoxila inferior, -OC.KF-, NO?, OU -NR.!iR: \;
Ri é H, F, Cl, metila, ou OH;
EiX ê H, alquila inferior, cicloalquila inferior, ou fen.ila;
ou urn sal farmaceuticamente aceitável do mesmo.
Outro aspecto da invenção- consiste em uma composição farmacêutica que compreende um composto da fórmula I e um excipiente farmaceutícamente aceitável.
Qutro aspecto da invenção consiste em um método para modular atividade de JNK, que compreende contactar uma célula que expressa JNK coa uma composto da fórmula I .·
Outro aspecto da invenção consiste em um método para tratar inflamação, que compreende administrar a um paciente com necessidade do mesmo uma quantidade efetiva de um composto da fórmula I.
Todas as publicaçdes citadas nesta exposição /1 <1 9 ficam incorporadas neste contexto por referência na sua. cotaiidade.
Anão ser que de outro modo estabelecido, os termos seguintes usados neste pedido, incluindo o relatório e as reivindicações, têm as definições que estão expostas em seguida. Deve ser observado que, quando usadas no relatório e nas reivindicações anexas, as formas do singular “um, “uma, e *o, aw incluem referências do plural, a não ser que o contexto determine ci.arame.nte de outro modo.
“Alquila significa a metade de hidrocarboneto saturado linear ou ramificado monovalente, que consiste de somente de átomos de carbono e hidrogênio, tendo de u.m a doze átomos de carbono. “Alquila inferior refere-se a um grupo alquila de um a seis átomos de carbono, isto ê alquila C^-Cg. Exemplos de grupos alquila incluem, sendo que não se fica .limitado aos mesmos, metila, etíla, propila, ísopropila, isobutíla, secbutila, terc-butila, pentila, n-hexila, octila, dodecíla, e assemelhados. “Alquila ramificada refere-se a uma metade de alquila que. cem pelo menos uma ramificação, per exemplo, isopropíla, isobutíla, terc-buclia, e assemelhados. De forma assemelhada, “alcoxila inferior refere-se a uma metade da forma -OR, e acila referese a uma metade da forma -C(O)R, em que R é alquila inferior .
“Alquileno significa uma metade de hidrocarboneto divalente saturado linear de um a seis ãtomos de carbono cm um radical de hidrocarboueto divalente, saturado ramificado de três a seis átomos de carbono, por exemplo, met!leno, etileno, 2,2-dimetiletilejio, propileno, 2--metilpropieno, butileno, pentile.no, e assemelhados.. «Cicloalquileno significa uma metade de hidrocarbonato divalent a de três a. oito átomos de carbono que incorpora um anel carboxílíco. Metades de Excicloalquileno exemplifioativas incluem, sem limitação.

:1C assemelhados. «Keterocicloalquileno significa uma metade de cicloalquileno em que um, dois, ou três átomos de carbono são substituídos com heteroâtomos (0# h, ou S; , onde a metade de heterocícloalquileno ainda contém pelo menos dois átomos de carbono. Metades de heterooiloalquileno exemplificativas incluem, sem limitação, assemelhados.
Dioxi alquileno significa uma metade divalence da fórmula em que R e alquileno tal como definido neste contexto.
«Arila* significa uma metade de hidrocarboneto aromático oíclico monovalente que consiste de um anel aromático mono-, bi- ou tricíclico, O grupo arila pode ser opelonalmente substituído tal como definido neste 25 contexto. Exemplos de metade arila incluem, sendo que
12/14« não se fica limitada aos mesmos, fenila opcionalmente substituído, naftila, fenantr.il, fluorenil, indenil, pentalenil, azuleníl, oxídifenila, bifenila, metíIened 1 f eni 1 a, aminod i fenil, di f eni .1 sul f í dl 1, d if eni 1 su 1 f o nil, dífenil-isopropilidenil, benzodioxanil, benzeiuranil, benzodíoxilil, benzopíraníl, benzoxazinil, benzoxazi.noni.1, benzopiperadinil, benzopiperazin.il, benzopirrolidinil, benzomor fali nil, metilenodiaxí fenil, etí lenodíoxifenil, e assemelhadas, incluindo os seus derivados parcialmente hídrogenados.
Cicloalquila significa uma metade c&rbacíclica. saturada monovalente que consiste de anéis mano- ou bicíclicos. A aiclaa.Iqu.ila pode ser opcionalmente substituída com um ou mais substituintes, em que cada um substituinte é independentemente hidroxila... alquila, aluoxíla, halo, haloalquila, ami.no, ffjQnaalquilami.no, ou dialquilamino, a não ser que de outro modo específicamente indicada. Exemplos de metades de cialoalquila incluem,, sendc que não se fica li.mit.uado aos mesmos, cíclopropil, oiulobut.il, ciolopentil, ciclohexil, aíclaeptil, e assemelhadas, incluindo os seus derivados par c.i a Iment e, não- satur ados..
Cicloalguilalquila*' significa uma metade da formula Εϊ!, ande R* é alquil.eno e é cieloalquila tal como definido neste contexto.
Heteraalquila” significa uma metade alquila tal coma definida neste contexto, incluindo um alquila Q--C-? ramificado, em que um, dois ou três átomos de hi
13/149 drogêxxio foram substituídos com um substituinte selecionado independentemento a partir do grupo que consrste de ~0P?, -~NP?R\ e -S (Cú ,.R'J (onde n é um inteiro de 0 a .2) , com a compreensão de que o ponto de vinculação do radical heteroalquila ê através de um átomo de carbono, em que R! ê hidrogênio, acila, alquila, cicloalquila, ou cicloalquilalquíla; R* e R!' são independeu temente um do outro hidrogênio, acila, alquila, cicloalquila, ou cicloalquiialquila; e quando n é 0, Rô ê hidrogênio, alquila, cicloalquila, ou ciclcalquilalquila; quando n è 1, R^ é alquxla, cicloalquila, ou cxcioau.quila^quiiaí a quando n é 2, R“ é alquila, cicloalquila, cicloalquilalquila, amino, acilamino, monoalquilami.no, ou dialquil.ami.no. Exemplos representativos incluem, sendo que não se fica limitado aos mesmos, 2-hidroxiet.il, 3hldroxipropil, 2-hidrcxi-l-hidroxi.metiletil, 2,3 diídroxipropil, 1 - hidroximet ii-et ii, 3 hidroxibut il, 2,3 ~di idroxibutil, 2 -hidroxi 1-metilpropil, 2aminoet.il, 3 aminopropil, 2 -met i Isul f onil et .11, aminos sulf on.iimetii, aminossulf oniletil, aminossulfonilpropil, metilaminossulfo-nilmeti1, metilaminossulfoniletil, metilaminassulfonilpropil, e assemelhados.
,KHeteroarila* .significa uma metade monocíclica ou bioíclioa de 5 a 12 átomos de anel que têm pelo menos um anel aromático que contém um, dois, ou três heteroátomos de anel selecionados a partir de te O, ou S, sendo os átomos de anel remanescentes C, oom a compreensão de que o ponto de vinculação do radical heteroa14/149 ri la serã an um anel aromática. O anel de heteroarrla pode ser opcicna.lmente substituída tal coao definioo neste contexto. Exemplos de metade de heternarila incluem, sendo que não se fica limitado aos mesmos, imi5 dazol.il opcionalmente substituído, oxazolil, isoxazolil, tiazol.il , isotíazolil, oxadiasolil, tiadiazolil, pírazinil, tienil, tiofenil, furan.il, piranil, piridinil, pirrol.il, pirazolil, pirimidil, piridazin.il, quinoli ni1, isoquinolini1, benzofuri1, her zofurani1, ben 10 zot.iof.enil, benzotiopiran.il, benzimidazolil, benzoxazolil, benzooxadiazolii, benzctiazolil, benzotiadiazolil, benzap.irani.1, indolil, isuindolil, indazolil, triazol.il, triazin.il, quinoxalínil, purinil, quinazolinil, quinolisinil, naftiridin.il, pteridinil, oarbazolíl, a15 zepin.il, diazepinil, acridlnil e assemelhados, incluindo os seus derivados parcialmente hidrogenados.
Qs termos -'halo, ’’halogênio, e halogeneto são usados permutavelmente neste contexto e referem-se a flúor, cloro, bromo, e iodo.
Haloalquila signifi.ua alquíla tal como definido anteriormente neste contexto em que um ou maia hi drogênio foi substituído com α mesmo ou halogênio diferente. Haloalquilas exemplif.inativos incluem -CIECi, CHjCFj, - CBsCCl>, perfluoroalquila (par exemplo, - CF;}) , e assem.nl:hados.
Heterociclila** significa uma metade saturada monovalente, que consiste de um a dois anéis, incorporando um, dois, ou três au quatro heteroátomos (sele
15/149 oionados a partir de nitrogênio, oxigênio ou enxofre). 0 anel de heterociclila pode ser opcionalmente substituído tal como definido anteriormente. Exemplos de metades de heterociclila incluem, sendo que não se fica 5 limitado aos meemos, piperídiníl opcionalmente substituído, plperazinil, homapiperazin.il, azepinil, pirrolidin.il, pirazolidinll, ímidazolinil, imidazolidlnil, pi~ ridinil, piridazinilf pirimldinll, oxazolidinil, isoxazolidinil, morfolinil, tiazolidiniI, isotíazolidínil, 10 quinuclidinilf quinolin11, ísoquínoliníl, benzimidazo111, tíad.iazolilídiníl, benzotíazolidínil. benzoazdi lidin.il, dihidrofur.il, tetrahidrofuril, diidropiranil, tetraidropiranil, tiamorfolinil> tiamorfolinilsulfoxido, t iamorf olinil. sul fona, diidroquínalinildiidroiso 15 quinolinil, tetraidro-quinolinil, tetraidrisaquinolinil,, e assemelhados.
’Opcionalmente substituído significa um substituinte que é substituído independentemente com zero a três subat.inui.ntes selecionados a partir de alquila in20 ferlor, halo, OH, ciano, amino, nitro, alcoxila inferior, ou haloalquila inferior.
'Grupo retirante significa um grupo com o significado convencionaimente associado com ele em química orgânica sintética, isto ê, um átomo ou grupo desloca 2S vel sob condições de reação de substituição. Exemplos de grupos retirantes incluem, sendo que não se fica limitado aos mesmos, halogênio, alca.no- ou arilenossulfoniloxila, tais como metanossulfoniloxila, etanos16/149 sulfoniloxila. tiometila, benzenossulfoniloxila, tosiloxila, e tien11 axila, dialofosfinoiloxila, benziloxila opcionalmente substituído, isopropiloxila, aciloxila, e assemelhadosà
S Opcional ou «opcional mea te* significa que o evento ou circunstância subsequentemente descrito pode,
| mas não precisa ocorn | ?.r, e que a descrição inclui casos |
| onde o evento ou cir | cunstância ocorre e casos em que |
| ele não ocorre. | |
| Ente rm i d ade | e «Condição enferma significa |
| qualquer enfermidade, | condição, sintoma, distúrbio ou |
indicação.
Solvente orgânico inerte'1’ ou solvente inerte significa que o solvente é inerte sob as condições da 15 reação que está sendo descrita em conjunto com as mesmas, incluindo por exemplo, benzene, toluene, acetonit rila, tetraidrofurano, N,N-dimet ilformamida, clorofórmio, cloreto de metileno ou diclorometano, dicloroetano, éter dietílico, acetato de etíla, acetona, metil 20 etil cetona, metanol, etanol, propanol, isopropanol, te.rc-butanol, dioxana, piridina, e assemelhados. A não ser que especificado em contrário, os solventes usados nas reações da presente invenção são constituídos por solventes inertes.
«Farmaceuticamente aceitável significa que é
d.e utilidade na preparação de uma composição farmacêutica que é de uma maneira geral segura, não tóxica, e nem biologicamente ou de outro modo indesejável e in clui que é aceitável para o uso humano, bem como vete£ X HciX' XG
Saía farmaceuticamente aceitáveis de um composto significam sais que são farmaceuticamente aceitáveis, tais como definidos neste contexto, e que possuem a atividade farmacológica desejada do composto de origem. Esses sais incluem:
Eais de adição ácida, formados com ácidos inorgânicos, tais como ácido clorídrico ácido bromidrico, ácido sulfúrico, ácido nítrico, ácido fosfôrico, e assemelhados; ou formados com ácidos orgânicos, tais como ácido acétíco, ácido benzenossulfônica, ácida benzõico, ácido canforsulfônico, ácido cítrico, ácido etanossulfônico, ácida fumáríca, ácido glicoeptônicu, ácido gluconic©, ácido glutâmico, ácido glicõl.ioo, ácido hidroxinaftoico, ácido 2-hidroxietanossulfônioo, ácido láctico, ácido maléica, ácido málico, ácido malônico, ácido mandé.l.ico, ad metano-sulfônico, ad mucônico, ácido 2-naftaienossulfônico, ácido propíônico, ácido salicíliuo, ácido succínico, ácido tartárico, ácido ptoluenossulfônico, ácido, tr.imeh.i.lacêt.ico, e outros ass eme 1 hades; ou
Sais formados quando um próton ácido presente na composto de origem ou é substituído por um íon de metal, por exemplo, urn íon de metal alcalina, um ion alcalino·-terroso, ou um fan de alumínio; ou coordenados com uma base orgânica ou inorgânica, As bases orgânicas aceitáveis incluem d.ietanolam.ina, etanolamina, N
18/149 metílglucamina, trietanolamina, trometamiua, e assemelhados, As bases inorgânicas aceitáveis incluem hidróxido de alumínio, hidróxido de cálcio, hidróxido de potássio, carbonato de sódio, hidróxido de sódio, e assemelhados.
Os sais farttôceuticamente aceitáveis preferidas são os sais formados a partir de ácido acético, ácido clorídrico, ácido sulfurlco, ácido metanossulfônico, ácido maléico, ácido fosforico, ácido tartárico, ácido cítrico, sódio, potássio, cálcio» zinco, e magnésio.
'Grupo protetor ou grupo de proteção significa o grupo que bloqueia se.letivamente um local reativo em um composto multifuncional, de. forma tal que unia reação química pode ser realizada seletivamente em um outro sítio reativo não protegido no significado convenciona.Ime.nte associado com ele na química sintética. Determinados processos desta invenção baseiam-se nos grupos de proteção para bloquear os átomos de nitrogênio e/ou oxigênio reativos presentes nos reagentes, Por exemplo, os termos '’grupo de proteção amíno” e grupo de proteção de nitrogênio” são usados permuta·· velmente neste contexto e referem-se àqueles grupos orgânicos destinados a proteger o átomo de .nitrogênio contra reações indesejáveis durante os procedimentos sintéticos. Grupos de proteção de nitrogênio exemplificativos incluem, sendo que não se fica limitado aos mesmos, triflueroacetil, acetamido, benzil (Bn), benziIcxicarbonil (carhubenzi.iox.i, CBZ) f ρ me toxibenzi loxicarbon i 1, p - nit robenz i loxicarbon! 1, fere -butoxicarbonil (BQC), e assemelhados. Aqueles versados na técnica saberão como selecionar ura grupo para facilidade de remoção e para a capacidade de compreender as reaçòes expostas era seguida.
Paciente' signif ica mamíferos e não-mamíferos. Mamíferos significa qualquer membro da classe mamífera incluindo, sendo que não se fica limitado sos mesmos, seres humanos,· primatas não-humanos, tais como chimpanzés e outros macacos e espécies siralescas; animais de fazenda tais como gado vacum, equinos,, carneiros, caprídeoa, e suínos; animas domésticos tais como coelhos, cães, e gatos; animais de laboratório, incluindo roedores, tais como ratos, camundongos, e porcos da guiné; e assemelhados. Exemplos de não mamíferos incluem, sendo que não se fica limitado aos mesmos, aves, e assemelhados. 0 termo paciente não indica qualquer idade ou sexo em particular.
Quantidade terapeuticamente efetiva. significa uma quantidade de um composto que, quando administrada a um paciente para tratamento de uma condição enferma, é suficiente para realizar esse tratamento para a condição enferma. A quantidade terapeuticamento efetiva será variável na dependência do composto, da condição da enfermidade que está sendo tratada, da seriedade da enfermidade que está sendo tratada, da. idade e da saude relativa do paciente, da via e forma de administração, do critério do medico ou pratico veterinário atendente,
Ο /14.9 e outros fatores-, «Tratar ou tratamento de uma condição enferma indui;
{f; prevenir a condição enferma, isto é, fazer com que os sintomas clínicos da condição enferma não se desenvolva em um paciente que possa estar exposto ou pre disposto à condição enferma, mas que ainda não experimentou ou mostra sintomas da condição enferma.
(11} inibir a condição enferma, ou seja, interromper o desenvolvimento da condição enferma ou seus sintomas clínicos, ou (ill) aliviar a condição enferma f ou seja, provocar a regressão temporária ou permanente ou seus sintomas clínicoa.
Ps termos “tratar, contactar e fazer reagir quando se faz referência a uma reação química significa adicionar ou misturar dois cu mais reagentes sob condições apropriadas para produzir o produto indicado e/ou desejado. Deverá ser apreciado que a reação que produz o produto indicado e/ou desejado pode não resultar necessariamente diretamente da combinação dos de dois reagentes que foram inicialmente adicionados, ou seja, poderá haver um ou mais intermediários que são produzidos na mistura que por ultime conduz à formação do produto indicado e/ou desejado.
Um distúrbio mediado por JMK. refere-se a uma condição enferma que ê provocada por atividade ou expressão elevada de JNK, e que pode ser aliviada pela inibição da atividade da. <.TNK. Exemplas de distúrbios mediados por JNK incluem artrite reumatoide, /asma, diabetes do tipo 2, doença de Alzheimer, doença de Parkinson, hiper-proliferação celular, e assemelhados♦ i A invenção proporeiona compostos e composições para o tratamento de distúrbios inflamatdrios, e métodos para o tratamento de distúrbios mediados por JNK.
Nos compostos da invenção presentemente preferidos, R15 e R são, cada um deles, H. Em outros compos10 tos preferidos da invenção, Rn também é K.
Em alguns compostos presentemente preferidos da invenção, R'1 é halo, alquila inferior, ou trifluoromeCl leu
Em alguns compostos presentemente, preferidos da.
.invenção, R* é fenila ou heteroarila. Em alguns compostos presentemente preferidos, ¥ é -C{Q)R3, onde R;i ê a 1 c ox 11 a i n f e r i o r .
Em um grupo de compostos presentemente preferidos da invenção, Z é fenila e P/‘ é H. Em alguns compos20 tos pre sent.e.mente preferidos, R! é -Y'-Y^-Y'-R*, onde V* è sulfonila. Em alguns compostos presentemente preferidos deste grupo, Y2 é heterocicloalquileno e ¥*' ê -O-< Em outros compostos present emente preferidos, Yz é a.l quileno inferior e ¥ é uma ligação. Em outros compos25 tos preferidos, Ύ é
Em outro grupo de compostos presentemente preferidos da invenção, Z ê heteroeiolila,
Em outra concretização a presente invenção pro22/149 porciona um composto de fórmula I em que ¥ ê -C (O) R:;, e R·5 é metoxila. Ainda em outra concretização a presente invenção proporciona um composto de fórmula I em que V ê -C(Q)R5, F? é metoxila e R7 é K. Ainda de acordo com S outra concretização a presente invenção proporciona um composto de formula I em que ¥ é ~C(O)R', P? é metóxila, R7 e H, Z é fenila e R* ê H. Ainda de acordo com outra concretização a presente invenção proporciona um compos to de fórmula X em que Y é ~Ό(Ο)Α5, R-> e metóxi10 la, R; é H, Z é fenila, R'? è H e R6 ê H, Ainda de acordo com outra concretização a presente invenção proporciona um composto de fórmula I em que V e -C(OjR\ R'? é metoxila, R7 é H, £ é fenila, R',: é H, R!> é H e R* é selecionado a partir do grupo que consiste de H, F, Cl, tie, e 15 CP; < Ainda de acordo com outra concretização a presente invenção proporciona um composto da fórmula I em que ¥ é -C<O)P?, R'· é metoxila, R? é H, ã é fenila, R? é H, R?
é H, R;1 é selecionado a partir dc- grupo que consiste de H, F, Cl, Me, e CPj e X ê CK ou N, Ainda de acordo com 20 outra concretização a presente invenção proporciona um composto da fórmula I em que ¥ é -CíO/R*, R* é metoxila, R.7 é Η, Z é fenila, R;: é H, R* é H, R;i é selecionado a partir do grupo que consiste de H, F, Cl, Me, e CF-S, X ê C.H OU N e R1 ê -Y^-Y^-Y^-R8, em que Y1 é S0&, Ainda 25 de acordo com outra concretização a presente invenção proporciona um composto de fórmula X em que Y é C-Q)RS, R': é metóxila, R? ê Η, X é fenila, R: é H, R” é H, R* é .selecionado a partir do grupo que consiste de
H, F( Cl, Me. e CFj, X é CH ou N e R! é -Y; -Y-''/-Rf, em que Y1 é SO2, ¥2 é a ligação, Y* é a ligação, e R* é alquila inferior, ou alquila inferior substituído por hidroxila, alcoxila inferior substituído por hidroxila, cicloalquila substituído por hidroxila, heterocícloalquila substituído por hidroxila, ou •••NR'*Riv substituído por hidroxila.
Ainda em outra concretização a presente invenção proporciona um composto de fórmula X em. que Y £ CÍOlR, R'> é metoxila, R' e H e X é selecionado a partir do grupo que consiste de piperidil, pirrolidil, ciclo·· propil, pirazolil, piperaxinil, morf.ol.ino, e pirimidini 1
Outro aspecto da invenção compreende uma composição farmacêutica, que compreende um composto da Fórmula X em combinação com um excipiente farmaceuticamente aceitável.
Outro aspecto da invenção compreende um método para tratar uma enfermidade mediada por JNK, que compreende administrar unia quantidade efetiva de um composto da Fórmula I a um paciente com necessidade do mesmo. Um método da invenção presentemente preferida é o método de tratar uma enfermidade mediada por JNK selecionada a partir do grupo que consiste de artrite reumatóide e doença de Crohn.
Outro aspecto da invenção compreenda um método para inibir a atividade de JKK, que compreende contactar a dita JNK cem uma quant idade efetiva de um compos··
24/14 9 to da -Fórmula- IQs materiais de partida e os intermediários dos esquemas de reações sintéticas podem ser isolados e purificados se desejado utilizando-se técnicas convencio5 nais, incluindo, sendo que não se fica limitado às mesmas, filtragem, destilação, cristalização, cromatografia, e assemelhados. Tais materiais podam ser caracter! z.ados uti 11.zando se meios convene 1 ona is, incluindo constantes físicas e dados espectrais*
A não ser que. especificado em contrário, as reações descritas neste contexto preferentemente são conduzidas sob atmosfera inerte, sob pressão atmosférica, a uma faixa, de temperatura de reação entre cerca de 78°C até cerca de 180C, e com maior preferência e .5 eonvenientemsnte sob temperatura do recinto (ou ambiente} , por exemplo, cerca de 20“C,
Os compostos da presente invenção são preparados de acordo com os esquemas ilustrados adiante, e mediante a utilização des procedimentos que se encontram !0 expostos nos exemplos - Nos esquemas que se apresentam em seguida estão expostas algumas rotas sintéticas possíveis que conduzem aos compostos objeto da presente invenção. Os radicais R1, R'', R\ R'h £?, e R? são tais como definidos anteriormente, a não ser que estejam es5 pecif içados de out ra forma.
ESQUEMA. I:

nitroacetofenona substituída na presença de uma base .inorgânica., tal. ccao NaOH., em um solvente prótico polar tal como uma mistura de metanol/água, para proporcionar a correspondente cetoxia u, p-· não -saturada da fórmula (I) . Esta produto pode ser então reduzido para a correspondente anilina saturada (XX) por hidrogenação na presença de um catalisador, tal como oxido de platina (IV; em um solvente polar tal como metanol ou tetraidrofurano ou uma mistura dos expostos retro como descrito na Etapa B, A anilina (XX} pode ser acoplada com iodobenxeno por aquecimento a 160*0 na presença de cobre (0) , iodeto de potássio e carbonato de potássio em um solvente polar tal como éter butílico, conforme des··
26/149 crito na Etapa C. Na Etapa a anilína da fórmula (XV) pode ser adiada por aquecimento sob reflexo na presença de um cloreto de adia, tal como cloreto de oxalii media, em um solvente apoiar, tal como tolueuo. A amida (XV) pode cíclisar-se por aquecimento sob refluxo em um solvente prático polar, tal como metanol, na presença de uma base .inorgânica, tal como carbonato de potássio., como descrito na Etapa E para proporcionar a cor--

respondente X,4-diidroquinolina♦
ESQUEMA IIí

Na Etapa A, um benaaldeída substituído é con densado com uma acetofenona substituída acetofenona. ou
I-piridin--3-.il--etanona (V), na presença de uma base i norgânica, tal con© NaQH, em um solvente prótico polar tal corno MeOH, para proporcionar a correspondente cetona. α,β-não-saturada da fórmula (VX) . Este produto pode ser reduzido para uma cetona saturada correspondente 5 por hidrogenação na presença de ura catalisador, tal como oxido de platina(IV), em uma mistura de solventes polares, tais como etanal e EtOAc, conforme descrito na Stapa. 8. Quando R: ou são cloro, esta metade pode ser simultaneamente reduzida utilizando-se paládio em 0 carbono como um catalisador em vez de õxido de pia tina (IV). Na. etapa C, a anilina (VII) pode ser acilada por aquecimento sob reflux» na presença de um cloreto de a oil a, cloreto, tal corao cloreto de oxa.líl met 11 a em um solvente apoiar, tal como toluene, A amlda (VIII) 5 pode ser ciclizada por aquecimento sob refluxo era um solvente prótico polar, tal como metanol, na presença de uma base inorgânica, tal como carbonato d.e potássio, tal como descrito na Etapa D para proporcionar uma 1,4diid.roquinoli.aa correspondente. Quando R' é uma metade 0 de éster car.boxi.lícc· na Etapa A de acoplamento, tanto o éster quanto o ácido carbox11ico correspondente podem ser formados por hidrólise; o ácido pode ser então acoplado coa: uma arai na ΝΗΒΐ'Ά3' na presença de agentes de acoplamento, tais como EDCI e BOP e uma base orgânica, is. tal como diisoprcpiletilamina em um solvente polar, tal como tetraidrofurano. A amida. da formula genérica (XX) pode ser submetida às mesmas etapas descritas anteriormente de B até P para proporcionar uma. 1,4 -diidroquinolina correspondente.
ESQUEMA 111;

diidroquinolina da fórmula genérica (X) pods ser hidrolisada para um ácido earboxilico (XI) correspondente na presença. de uma base Inorgânica, tal como hidróxido de lit io, em um solvente prõtico polar, tai como metanol., conforme descrito na Etapa A, O ácido earboxilico (XX) 10 pode ser acoplado com uma amina NHR?R4V na presença de um agente de acoplamento, tal como BOP, e uma base inorgânica, tal como diisopropiletilamina, em um solvente polar, tal como tetraidrof.urano, para proporcionar uma amida correspondente, tal como descrita na Etapa E.
Quando R5 é uma metade mercaptoalquila, a 1,4diidroquinolina da. fórmula (XX) pode ser oxidada para
9 / 14 9 uma sulfona correspondente utilizando-se como em uma mistura de solventes polares, tais come tetraídroíuranQ, metanol e água conforme descrito na Etapa C.
A Tabela seguinte relaciona os compostos repre5 sentativos da invenção:
TABELA 1; COMPOSTOS EXEMPLIPICATIVOS
Estrutura | Dados





34/149


36/149
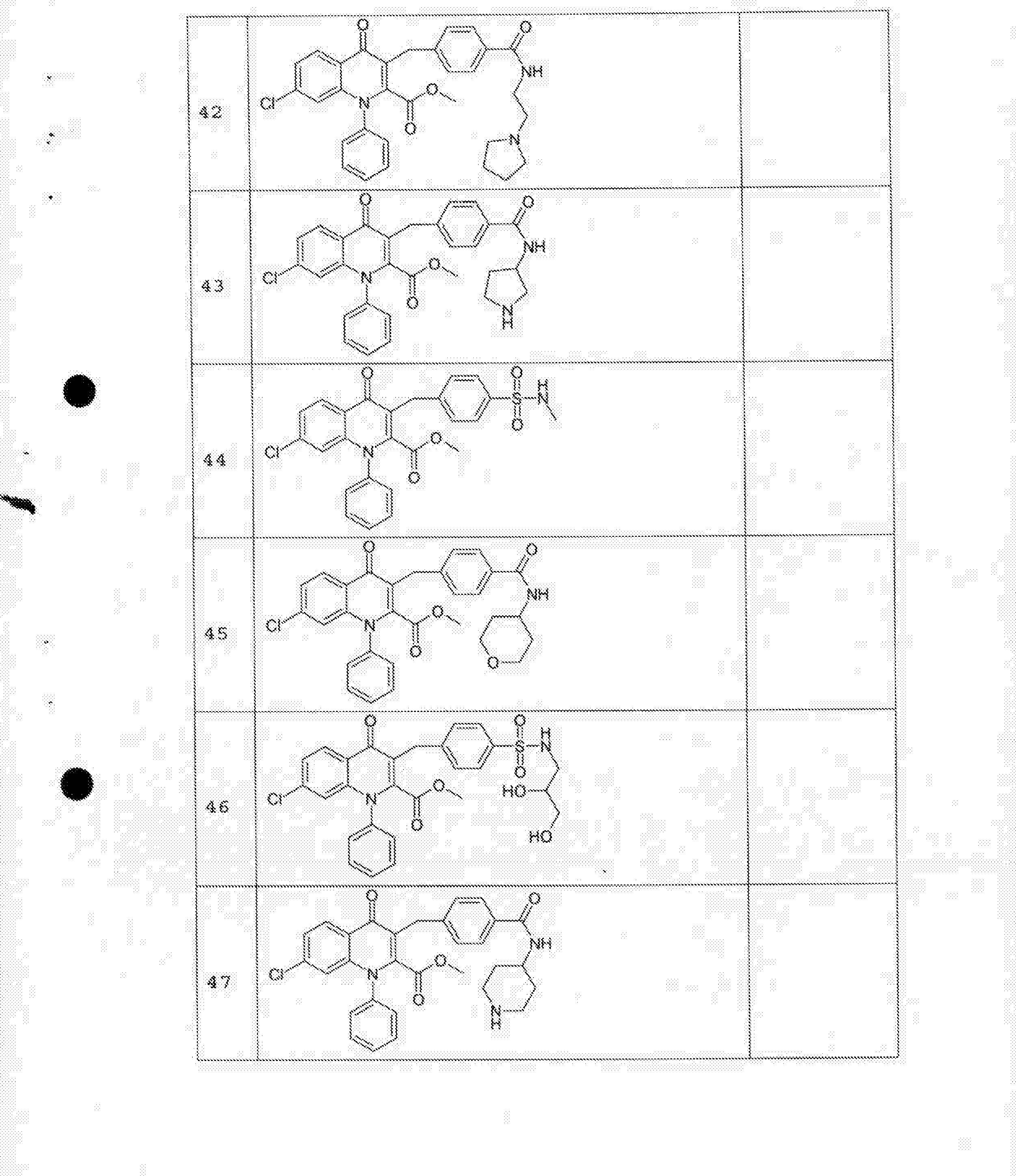
37,/149

Ο,.
3S/149

38/14 9


41/149


43/14'3


45/149
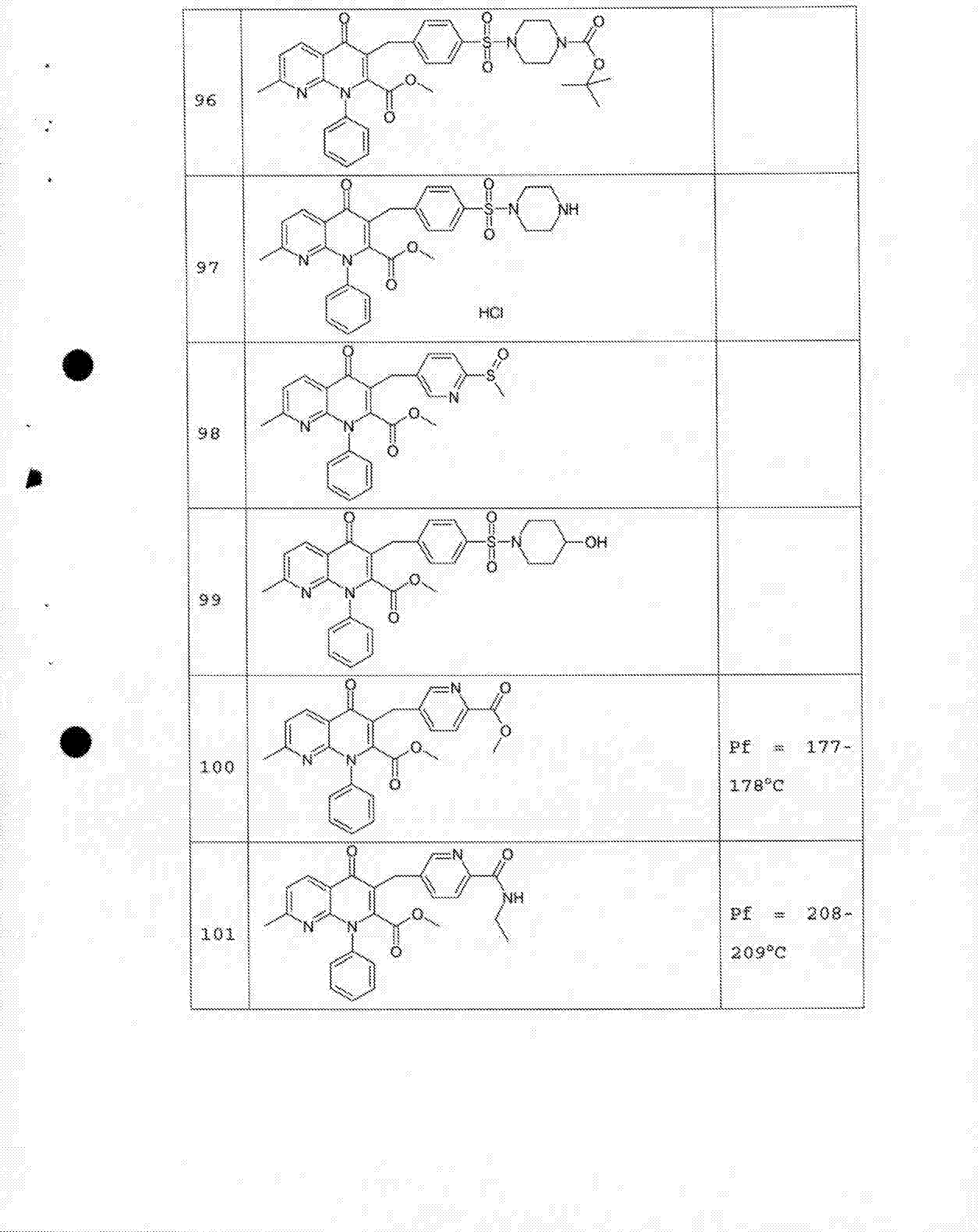
6 / 14 9
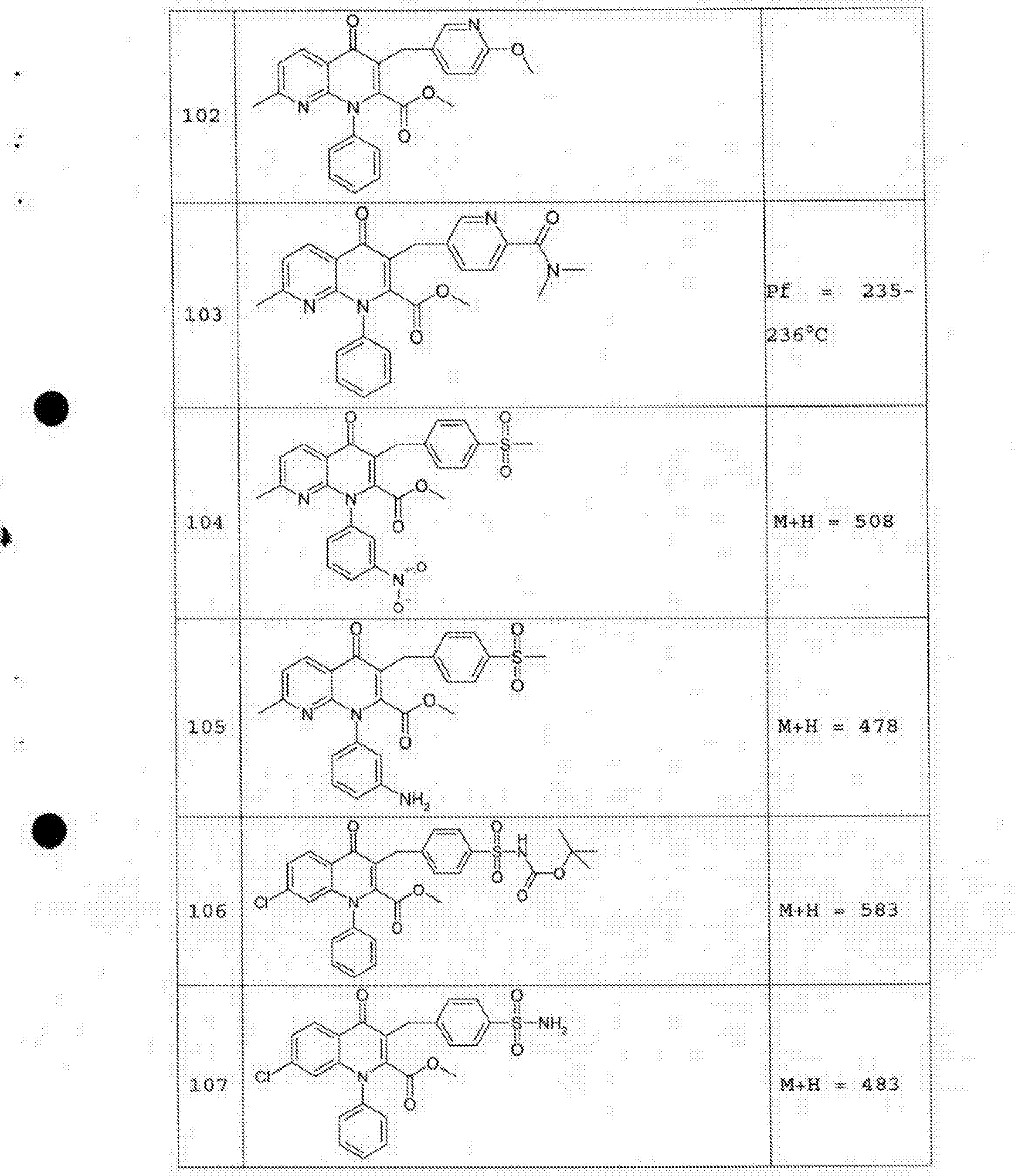
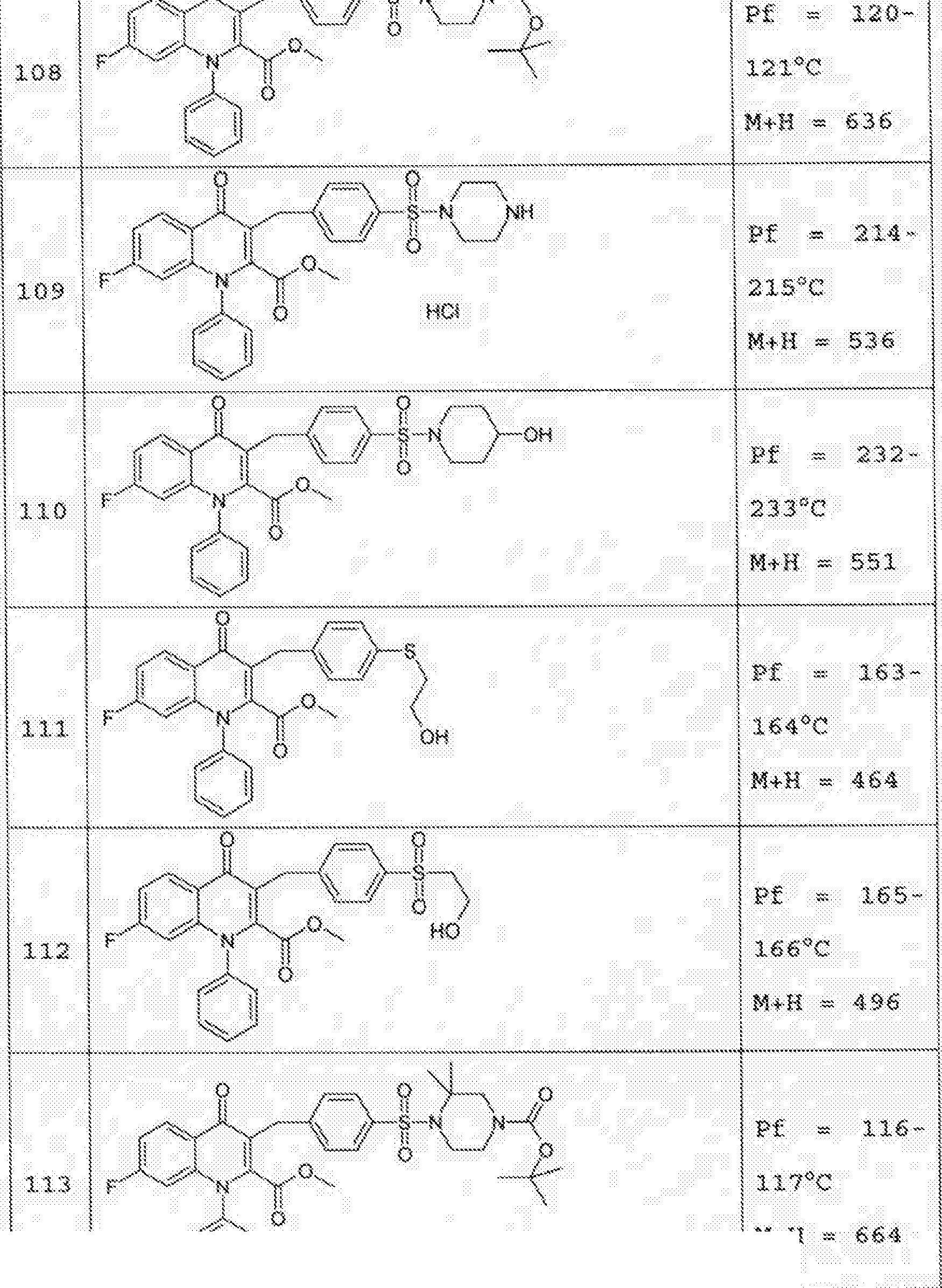
Μί-Η
48/149

49/

50/14$

51/149


Qs composton desta invenção são mcdu'iadores de jfiK e cesso tal espera-se que sejam efetivos no tratamento de uma ampla gama de distúrbios mediados por JNK, 5 Distúrbios mediados por JNK exemplifinativos incluem, sendo que não se fica limitado aos mesmos, distúrbios auto-imunes, distúrbios i.nf l&matôrios, distúrbios metabôlicos, doenças neurológicas, e câncer. Consequentemente, os compostos da invenção podem ser usados para tratar um ou mais de tais distúrbios. Em. algumas concretizações, os compostos da invenção podem ser usados para tratar um distúrbio mediado por JNK, tal como artrite raumatõide, asma, diabetes do tipo II, doença de Alzheimer, doença de Parkinson ou acidente vascular cerebral .,
A .invenção incluí composições farmacêuticas que compreendem pelo menos um composto da presente invenção. ou um isômero individual, mistura racêmica ou nãoracêmica de isômeros ou um sal ou solvato farmacêutica mente aceitável doa: mesmos, em conjunto com pelo menos um carreador farmaceuticamente aceitável, e opcionalmente outros ingredientes terapêuticos e/ou profilãtiDe uma maneira geral, os compostos da invenção serão administrados em uma quantidade terapeuticamente efetiva por meio de qualquer uma das modalidades de administração aceitas para agentes que servem a utilidades assemelhadas. As faixas de dosagem que são adequadas situam-se tipicamente em 1-500 mg diariamente, preferentementa 1-100 mg diariamente, e cora maior preferência 1-30 mg diariamente, na dependência de numerosos fatores, tais como a severidade da enfermidade a ser tratada, a idade e saúde relativa do paciente, a potência do composto usado, a. via. e forma de adminis
54/149 tração, a indicação no sentida da qual a administração é encaminhada, e as preferências e experiência do clínico envoivido. Aquele normalmente versada na técnica do tratamento de tais enfermidades será, capaz, sem ex5 perimencação .indevida e com base no conhecimento pesso al e na exposição desta invenção, apurar uma quantidade terapeuticamente efetiva des compostos da presente invenção para uma determinada enfermidade.
Os compostos da invenção podem ser administra0 dos como formulações farmacêuticas incluindo aquelas adequadas para administração oral {incluindo bucal e sublingual}, retal, nasal, tópica, pulmonar, vaginal, ou parenteral {incluindo intramuscular, intra-arterial, intra-espinal, subcutânea e intravenosa) on em uma far5 ma adequada para administração por inalação ou insuflação. De uma maneira geral, a forma de administração preferida ê oral utilizando-se um regime de dosagem conveniente diariamente que pode ser ajustado de acorda com o grau de doença.
| 0 | Um composto ou compos | tos da invenção. | em con- | |
| junto | com um ou mais adjuvantes, carreadores. | ou dilu- | ||
| entes | convencionais, podem ser | levado à forma < | ie campo | |
| siçõe; | s farmacêuticas e de das; | sgens unitárias. | As com- |
posições farmacêuticas e formas de dosagem unitária podem ser compreendidas de ingredientes convencionais em proporções convencionais, com ou sem compostos ou princípios ativas adicionais, e as formas de dosagem unitária podem conter qualquer quantidade efetiva adequada do ingrediente ativo comen.suredo com a faixa de dosagem pretendida diariamente a ser empregada. As composições farmacêuticas podem ser empregadas como sólidos, tars como comprimidos ou cápsulas preenchidas, semi-sólidos, pôs, formulações de liberação sustentada, ou líquidos, tais como soluções, suspensões, emulsões, elixires, ou cápsulas preenchidas para o uso oral; ou na forma de suposítórios para administração retal ou vaginal; ou na forma de soluções injetáveis estéreis para uso parente30 ral.
As formulações que contêm cerna de jam (1) mg de ingrediente ativo ou, mais amplamente, cerca de 0,01 até cerca de cem (100) mg, por comprimido, são consequentemente, formas de dosagem 'unitária representativas 15 adequadas.
Os compostos da invenção podem ser formulados em uma ampla variedade de formas de dosagem para administração oral. As composições farmacêuticas e formas de dosagem podem compreender um composto ou compostos 20 da presente invenção ou seus saís farmaceuticamente aceilaveis como o componente ativo. Os carreadores farmaceuticamente aceitáveis podem estar na forma sólida ou líquida. Os preparados na forma sólida incluem pos, comprimidos, pílulas, cápsulas, supositóríos, e granu25 lados dispersâveis. Um carreador sólido pode ser uma ou mais substâncias que podem também funcionar como diluentes, agentes aromatizantes, solubilizantes, lubrificantes, agentes de suspensão, aglutinant.es, preserva
t.Ivos, agentes de desintegração de comprimidos, ou urn material de encapsulamento. Mos pôs, o carteador de
| uma maneira | é um sólido | f inamente d ivididc | t que é | ||
| uma mistura | COi^ O | componente | ativo finamente dix | ridido. | |
| Nos comprimi | dos, o | componente | ativo geralmente é | mistu- |
rado com o carreador tendo a capacidade de aglutinação necessária em proporções adequadas e compactado na forma e dimensão desejada. Os pôs e comprimidos preferentemente que contêm entre cerca de um (1? até cerca de 10 setenta (70) por cento do composto ativo. Os carreadores que são adequados incluem, sendo que não se fica limitado aos mesmos, carbonato de magnésio, estearato d.e magnésio, talco, açúcar, lactose, pectina, dextrins, amido, gelatina, tragacanto, matilcelulose, carbo15 ximetilcelulose de sódio, uma cera de baixe- ponto de fusão, manteiga de cacau, e assemelhados. O termo preparação destina-se a incluir a formulação do composto ativo com material de encapaular como carreador, proporcionando tmia cápsula em que o componente ativo, 20 com ou sem car re adores, é circundado por um carreador, que se encontra em associação com ela. L>e forma assemelnada, incluem-se encapsulados e pastilhas. Os comprimidos, pôs, cápsulas, pílulas, encapsulates e pastilhas podem estar como formas sólidas adequadas para ad25 ministração oral.
Outras formas adequadas para administração oral incluem preparados de forma líquida incluindo emulsões, xaropes, elixires, soluções aquosas, suspensões aquosas gu preparadas de forma sólida que se destinam a ser convertidos rapidamente antes do uso para preparados da forma liquida. Emulsões podem ser preparadas em soluções, por exemplo, em soluções de prap.ileno glicol aquosas ou podem conter agentes emulsionadores, por exemplo, tais coma lecitina, monocleata de sorbitan, ou acácia. As soluções aquosas podem ser preparadas por dissolução da componente ativo em água e adição de corantes, aromatizant.es, estabilizadores, e agentes de espessamento adequados. As suspensões aquasas podem ser preparadas por dispersão do componente ativo finamente dividido em água com material viscoso, tais camo gomas saturais ou sintéticas, resinas, met11 celulose, carboximetilcelulose de sódio, e outros agentes de suspensão amplamente conhecidos. Os preparados de forma sólida incluem soluções, suspensões, e emulsões, e podem conter, adicíonalmente ao componente ativo, corantes, aromatizantes, estabilizante», tampões, adoçantes artificiais e naturais, dispersantes, espessantes, agentes de solubilizaçao, e assemelhados.
Os compostos da invenção podem ser formulados para, administração parenteral (por exemplo, por injeção, por exemplo, injeção de bolo ou infusão contínua} e podem aa.r apresentados ns forma de dose unitária em ampolas, seringas previamente preenchidas, infusão de pequenos volume ou em recipientes de várias doses com um preservativo adicionado. As composições podem apresentar-se em formas tais como suspensões, soluções, ou
58/149 emulsões em veículos oleosos ou aquosos, por exemplo, soluções em polietíleno glicol aquoso. Exemplos de carxeadores., diluentes, solventes ou veículo# oleosos ou não aquosos incluem propileno glícol, políetileno qlicol, õleos vegetais (por exemplo, óleo de oliva}, e. êsteres orgânicos injetáveis (pox' exemplo, et.il oleato) , e podem conter agentes de formulação tais como agentes de preservação, umedecimento, emulsionamexxto ou suspensão, estabilização e/ou dispersão. Alternativamente, o ingrediente ativo pode estar na forma de pó, obtido por isolamento «cético de sólido estéril ou por liofilização a partir de solução para constituição antes do uso com um veículo adequado, por exemplo, água estéril, isenta de substância píretogênica.
Os compostos da invenção podem ser formulados para administração tópica a epiderme como ungüentos, cremes e loções, ou como um emplastro transdérmico> Os ungüentos e cremes, por exemplo, podem ser formulados com uma base aquosa ou oleosa com a adição de agentes de espessamento e/ou gelifícação adequados. As loções podem ser formuladas com uma base aquosa ou oleosa e de uma maneira, geral também? contendo um ou mais agentes de dispersão, agentes de estabilização, agentes de dispersão, agentes de suspensão, «gentes de espessarnento, ou agentes de coloração, Formulações que são adequadas para administração tópica na boca incluem pastilhas que compreendem agentes ativos em uma base aromatizada usualmente sacarina e acácia ou tragacanto; pastilhas que
59/149 compreendem o ingrediente ativa em uma base xnerte tai. come;· gelatina anda glicerina ou sacarina a acácia; e colutôrios que compreendam o ingrediente ative em um carreador líquido adequado.
S 0a compostos da invenção também podem ser formulados para administração como supositôrios. Uma cera de baixo ponto de fusão, tal como uma mistura de glicerídeos de ácidos graxos cu manteiga, de cacau é primeiro fundida e o componente ativo é dispersado homogeneamen~ 10 te, por exemplo, mediante agitação, A mistura, homogênea fundida é então vazada em moldes dimensionados de forma conveniente, deixada esfriar, e solidificar.
Os compostos da invenção podem ser formulados para administração vaginal. Fessários, tampões, creases, IS gels, mente ci dos ra administração nasal, As soluções ou suspensões são .aplicadas diretamente à cavidade nasal por meios convencionais, por exemplo, com. um conta-gotas, pipet a ou spray. As formulações poderão ser proporcionadas em uma forma de dose única ou. de várias doses. Neste último caso de um conta-gotas ou pipeta, isto poderá ser 25 obtido mediante a administração pelo paciente de um volume predeterminado, apropriado, da solução ou suspensão. No caso de uma aspersão, esta poderá ser obtida, por exemplo, por meio de uma bomba de spray com medidor pastas, espumas ou sprays que coutem aoieiunaiao ingrediente ativo esses carreadores são condena técnica como sendo apropriados.
Os compostos em apreço podem ser formulados pa60/149 de atorn!cação,
Qs compostos da invenção podem ser formulados para administração por aerossol, particularmente para o trato respiratório e incluindo admmistraçao mtrana5 sal. O composto terâ de uma maneira geral, uma. pequena dimensão de partícula, por exemplo, da ordem de cinco (5) micrometres ou menos. Essa dimensão de parti.cu.xa pode, ser obtida por meios conhecidos na técnica, por exemplo, por micronização. 0 ingrediente ativo é prole porcionado em uma embalagem pressurizada com um propelente adequado, tal como um. clorof xuorocarbo.net o (CFC? , por exemplof diclorodifluorornetano, triclcrofluorometano, ou dlclorotetrafluoxoeta.no, ou dióxido de carbono ou outro gás adequado. 0 aerossol também pode conter 15 convenientemente um tensoativo, tal como lecitina. A dose da droga pode ser controlada por uma válvula de medição. Alternativamente, os ingredientes ativos podem ser proporcionados na forma de um pó seco, por e~ xemplo, uma mistura de um pó do composto em uma base de 20 pó adequada, tal oomo lactose, amido, derivados de amido tais como hidroxíprnpilmetil celulose e polyvínilpirrolidina (PV»). O carteador de pó formará um gel na cavidade nasal* A composição de pó poderá ser apresentada na forma de do.se unitária, por exemplo, em câpsu25 las ou cartuchos de, por exemplo, gelatina ou pacotes de vesicatõrio a partir dos quais o pó pode ser administrado por meio de um inalador.
Quando desejado, formulações podem ser prepara
61/149 das cam. revestimentos entéricos adaptados para administração de liberação sistemática ou controlada do ingrediente ativo. Por exemplo, os compostos da presente invenção podem ser formulados em dispositivas de disb trifcuição de droga transdêrmica ou subeutânea. Estes sistemas de distribuição são vantajosos quando é necessária liberação sistemática do composta e guanao a anuência do paciente com um regime de tratamento é crucial. Os compostas nos sistemas de distribuição transiu dérmica são frequentemente vinculados a u® suporte sólida de adesivo para, a pele. 0 composto da interesse também pode ser combinado com. um intensificador de penetração, por exemplo, Aaone (l-dodeoilazaciclo-heptan2-ona). Sistemas de distribuição de liberação sistemâIS tica são inseridos de forma aubcutãnea na camada subdérmica por meio de cirurgia ou injeção. Os implantes subdérmicos encapsulam. o composto em uma membrana solúvel em lipídeos, por exemplo, borracha de silicone, ou um po.li.me.ro .biodegradável, por exemplo, ácido polilâc20 tico.
Os preparados farmacêuticos estão preferencemente em formas de dosagem unitária, Nessa forma o preparado é subdividido em doses unitárias que contêm quantidades apropriadas do componente ativo. A forma 25 de dosagem unitária pode ser um preparado disposto em pacotes, a embalagem contenda quantidades distintas do preparado, tais como comprimidos, cápsulas e pós acondicionados em frascos ou ampolas. Também, a forma, de dosagem unitária pode ser a própria cápsula, comprimido, encapsulado, ou pastilha, ou ela pode ser constituída por um. número apropriado de qualquer um destes na forma acondicionada <
Outros uarreadores farmacêuticos adequados e suas formulações encontram-se descritos em Remington.The Science and Practice de Pharmacy 1.995, editado per Martin, Mack. Publishing Company, 19th edition, Easton, Pennsylvania. Formulações farmacêuticas representati vas que contêm um composto da presente invenção encont rarase deseritos ad iante.
Objetivos, vantagens, e aspectos novos adicionais desta invenção serão evidentes para aqueles versados na técnica quando do exame dos seus exemplos .seguintes, os quais não devem ser considerados como limitativos.
EXEMPLOS
Os preparados e exemplos seguintes são apresentados com a finalidade de permitir àqueles versados na técnica compreenderem mais amplamente e porem em prática a presente invenção. Os mesmos não deverão ser considerados como limitatívos do escopo da invenção, mas me.ramente como sendo ilustrativos e representativos da
AcOH: Ácido acético; BINAP; 2,2'-bis(difenilfosfino) 1,1‘ -binai111: 80P: Hexa.f luorofosfato de bensot.ri.azol:1 - iloxi tris {dime ti lamino) fosfonlo; BusOt éter dibutílico; DCM: Diclorometano/Cloreto de metíleno; ΕΊΧ-ΈΑ; Dli
03/149 soprapiletilamina; DMF: Ν,Ν-dimetilformamida; EDCI cloridrato de 1- (3-Dimetilaminopropil) -3-etilcarbodiimida; Et:íO: Dietil éter; Et OH: Etanol/Álcool etilico; EtOAc: Acetato de etila; HOBt: 1-Hidroxibenzatriazol; MeQHt Metanol/álcool metílico; MW: Microondas; HMP; 1Metil-2-pirroliâinona; RTí Temperatura ambiente; NaHCOS: bicarbonate de sódio; TEA; Triet.il amina; THE: Tetraídrafurano; TLC: Cromat.ograf.ia de camada fina. Preparação ls Síntese de 1-{2~amino~4-clorofe&il} etanema
A síntese de 1 - (2-amino-4-clorofenil) -etanona f o X jC ’f? Λ- X zada de acorde com o processo ilustrado no Esquema 1 exposta em seguida.

ESQUEMA 1
Etapa A; Síntese de 1-(4-clcro-2-nitro-feníl) etanana: A uma solução de ácido á-cloro-S-nítrcbenzóico (30,0 g, 0,15 mol) em THE (400 ml) adicionou-se cloreto de oxalíla (26 ml, 0,3 mel) a 0”C, seguida par DMF (2 gotas) . Depois de agitação durante 10 min a 0!>C, removeu-se o banho de gele e aqueceu.-se a mistura de reação sob refluxo durante 3 h. A mistura resultante foi então refrigerada e evaporada sob pressão reduzida. Carregouse um segundo balão de vidro de funda redondo de 1 1
2K< cam malonato de díetil (22,8 ml, 0,15 mol) s THE (130 ml) : adicionou-se então hidreto de sódio (dispersão a
60% em õleo mineral, 7,2 g, 0,18 mol) a 0C, par partes durante urn periodo de 30 minutos. Removeu~se então o banho de gelo, e a mistura ioi agues?ida para ret luxo durante 3 h. Adicionou-se o cloreto de acila, dissolví.S do em 2 partes de THE, e a mistura resultante foi aquecida so.b refluxo durante 3 h e então submetida a agitação sob RT durante a noite, A mistura de reação tor d.tvi.did.a entre água e EtOAc; a camada, orgânica foi separada e a camada aquosa foi extraída duas vezes com Et.010 Ac, Os extratos orgânicos combinados foram lavados com salmoura, submetidos a secagem sobre sulfato de sódio anídrico, filtrados e evaporados sob pressão reduzida.
Adicionou-se ao resíduo uma mistura de AcOK glacial (5Q ml} e ácido sulfúnico (20% em âgua, 50 ml}, e a mistura 15 resultante foi aquecida sob refluxo durante 6 h e então submetida a agitação a 80°C durante a noite, A mistura de reação foi hasificada para pH 10 pela adição de NaOH {aq) e então foi extraída 3 vezes cora EtOAc, Os extratos orgânicos combinados foram lavados com salmoura, 20 submetidos a secagem sobre sulfato de sódio anídrico, filtrados e evaporados sob pressão reduzida, O resíduo bruto foi purificado por cromatografi.a instantânea ÇEt.GAo/hexano) para proporcionar 7,1 g de 1-(4-cloro-Επί t ro-fenil) -etanona.
Preparação 2; Síntese d® 4~H-metll-piperazxna-1sulfcniI} -benzaldsído
A síntese de 4- (4-rnetil-piperazina-l-sulfonil)
-benxaldeido foi realizada de acordo com o processo i149
1us trado no Esquema 2.
CHO0 hh
....¼. «« .«'« if '>; d ': ff YY \\ N 4 l|. * I: I I o i j 'NY' v<·' OHC^’ X<::>s ;t* ................ ' .............
SOX:· CH.
BSQWMA 2
A uma solução de cloreto de 4form!lbenzenossulfon.il (1,0 g) em DCM (10 ml) adicionou-se MaHCOi (saturado aquoso, 10 ml) seguido por 1met.il-piper az i.na (0,6 ml). A mistura de reação foi vigorosamente submetida a agitação durante 1 h, a camada orgânica separada, e a camada aquosa extraída com DOM (2x 20 ml). Os extratos orgânicos combinados foram lavados com salmoura, submetidos a secagem sobre anídrico SJaaSOo filtrados e evaporados sob pressão reduzida para proporcionarem 4- (4-metil -piperazina-1-su.lfoni.l) benzaldeido {1,28 g) sem qualquer outra purificação.
Da mesma maneira, utilizando-se os materiais de partida apropriados, prepararam-se os seguintes compostOS:
M- {2,2 -dimet.il- [1,3] dioxolan-4 -ilmetil) -4-formil benzenossul f onamida >· (Morfol ..ina-4.-sulfoni 1} -benza Ide ido;
- Formil-xV-metí 1 -benzenossul f onamida;
-Formil-At 2V~dímetilbenzenossulfonamida;
-Formil(2-hidroxi-etil) -IV-metil - benzenossulfonamída; e
K- [2- (terc-But.il-dimetil• silaniloxi) -etil] -4-formil benzenes sul f onamida (para a síntese de amina vide Bío66/149 erg, A Med. C’hem. (20Q51 13;. 11) : :3 601--39, ; e
Ester tern-butHico de ácido 4-- (4 -zormii benzenossulfonil) -piperazina-1 -carboxHico.
Preparação 3t Siutes® de 1-(4-daro-2-fesiilamxnof eni 1) - e tanm
A síntese de 1 -(4-cloro~2-fenilamino-fenil} stanona foi realizada de acordo com o processo ilustrado no Esquema. 3...
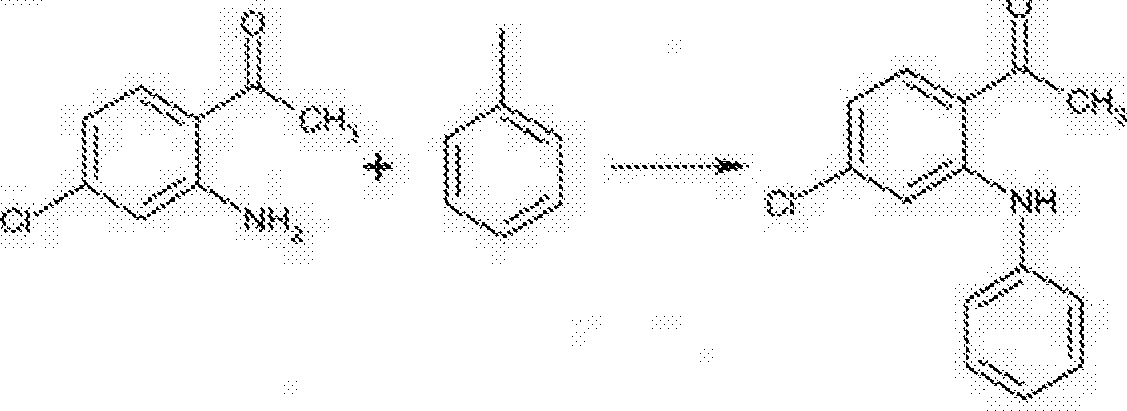
í TOWÍMÀ 3 .v IjÍ Xm JfluOrna mistura de I -{2-amino--4~clcrufen.il) etanona. (1,85 g), põ de cobre (140 mg), Kl (360 mg}, K2CQ3 (3,06 g} e iodobenzeno (4,B ml) em éter dibut.ilίσο í40 ml} foi aquecida a 16Ο'·'ϋ (temperatura de banho de oleo) durante « noite. .Adicionou-se mais põ de cobre (14 0 mg), KJ. (360 mg) , (3,08g) e iudobenzenc (4,9 ml) e a mistura de reação foi aquecida sob refluxo durante a noite. A mistura resultante foi então refrigerada e concentrada sob pressão reduzida. O resíduo foi dividido entre ãgua e acetato de etila, a camada orgânica foi separada e a camada aquosa foi extraída com acetato de etila (2x 50 ml). Os extratos orgânicos combinados foram lavados com salmoura, submetidos a secagem sobre sulfato de sõdio anídrico, filtrados e eva5 parados sob pressão reduzida. O resíduo bruto foi puri
67/149 ficado por cromatograf1a instantânea (EtOAc/hexano} para proporcionar 2,2 g de 1- (4 -cloro-2-fenilaminofeni1) -e tanona..
Eequindo-se o procedimentos descrito anteriormente e com materiais de partida apropriados, prepararam-se os seguintes compostos;
-(2 -Fenilamino-feni1) -etanona;
- í 4 -Met oxi - 2 - f en 1 1 ami.no f eni 1) - et anona;
- (5 - Cloro- 2 - fenil ami.no - f en 11) -etanona;
·· (5 - Met ox 1. - 2 - f eni lamino - f eni 1) - et anona;
- (2 - Fen ilamino-p.l r id in · 3 - i 1) et anona; MS 213 [M+HF ; e
- (6 -Met í 1. - 2 - f eni lamino · pirid in - 3 -11) - e tanona.
Preparação 4$ Síntese de 4-(2-.hidroxi -etilsulfanil)
- ben.® a ide ido
A síntese de 4 - (2-hidróxi-et 11 sulfanil’} benzaldeído foi realizada de acordo com o processo ilustrade no Esquema 4.

ESQUEMA 4
A uma mistura de 4 -iodobenzaldeído (1,0 g) e pd de cobre (0) (2,34 g) adicionou-se DMF (10 ml; f and a mistura resultante foi. aquecida a 14CTC sob argônio durante 3 h. Adicionou-se dissulfureto de 2-hidróxietil (0,32 ml}, e a mistura resultante foi submetida a agitação a 140cC durante a noite sob argênio. A mistura de reação foi refrigerada e filtrada através de papel de filtro de fibra de vidro. O filtrado foi evaporado sob alto vácuo e o resíduo purificado per cromatografia instantânea (EtOAc/ hexano) para proporcionar 4-(2hidroxi-etil-sulfanil) -benzaldeído (0,784 g, rendimento quantitativo) ..
Preparação 5: Sínteae de 3-Amino-3,N,N-trimatilhutiramida
A síntese de 3 -Ami.no- 3, Ν', N-trimetil -butiramida foi realizada de acordo com o processo ilustrado no Esquema 5.

Etapa A: Síntese de ácido 3-benziloxicarbonilamino- 3 -met 11 -butí rico ,. Dissolveu-se ácido 3amino-3-metll-butírioo (2,40 g, 20,49 mmol) em NaOH (2 M aq, 35 ml), a mistura resultante refrigerada para 0f’C, e adicionou-se cloroformato de benzíla (5,77 ml, 40,97 mmol), A. mistura de reação foi submetida a agitação vigorosamente a Q*C durante 1 h e sob RT durante 3 h. Adicionou-se então Et-0 (50 ml), e as camadas foram separadas. A oamada orgânica foi descartada. A camada aguosa foi. acidulada para pH 2, adicionou-se salmoura, e a mistura resultante foi extraída com EtOAcu Qs ex tratas orgânicos combinadas foram submetidos a secagem sobre Na^SQ4 anídrioo, filtrados e evaporados sob pres €9/149 são reduzida para proporcionarem ácido 3 benziloxicarboni1-amino-3-metil-butírico (2,39 g, 46% de rendimento) sem outra purificação.
Etapa S: Síntese de éster benzilíco de ácido (2~dimetilcarbamoíl-l,1-dimetileti.l) -carbâmíco. Adicionaram-se HO8t (1,242 g, 9,193 mmol) e EDCI (2,35 g, 1.2,26 jnmol) a uma solução de ácido 3benziloxicarbonilamino-3-m.etil-butírico (1,54 g, δ, 129 mmol) em DCM (45 ml) a 0eC. A mistura resultante foi .0 submetida a agitação a 0’C durante 30 min, e adicionouse uma solução de MesNH {2 M em THE, 1.2,26 ml, 24,51. mmol) - A mistura de reação foi aquecida para a RT e submetida a agitação durante a noite. 0 solvente foi evaporado sob pressão reduzida, e o resíduo dividido „5 entre EtOAc e uma mistura de água, salmoura e NaHCCq (sat ag) . A. camada orgânica foi separada, lavada com HC1 (1 ΓΊ) , submetida a secagem sobre NgSO<. anídrico, filtrada e evaporada sob pressão reduzida. O residue bruto foi purificado por cromatografia instantânea (E~ í0 tOAc/hexano) para proporcionar éster benzíiico de ácido (2-dimeti lcarbamoi.1 -1,1 - dimetil-etil) -carbâmlco (1,14 g, 67% de rendimento) .
Etapa C: Síntese de 3-amino- 3, N, N-1rimet11 butiramida. Uma mistura de êster benzílico de ácido (2dimetil-carbamoíl-1,1-dimetiletil) -carbâmíco (1,14 gj e Pd/C (10%, 114 mg) foi. submetida a agitação sob H2 (pressão de balão? durante 1.0 h sob KT, A mistura de reação foi filtrada através de um chumaço de CELITE^8 e o filtrado evaporado sob pressão reduzida para proporcionar 3-amino~3,N,N-trimet11-butiramida (366 mg, 62% de rendimento).Preparação 6: Síntese de 1-(2-((3.R,2R) 2-Hxdroxi-ciclopentilamino) -6-metil -piridin-3-ill etanona
A síntese de 1 - [2-((1.R, 2R) ••2-hidroxi-ciclopenti.land.no) -6-metil -piridin-3-il] -etancna foi realizada de acordo com. o processo ilustrado no Esquema 6 .
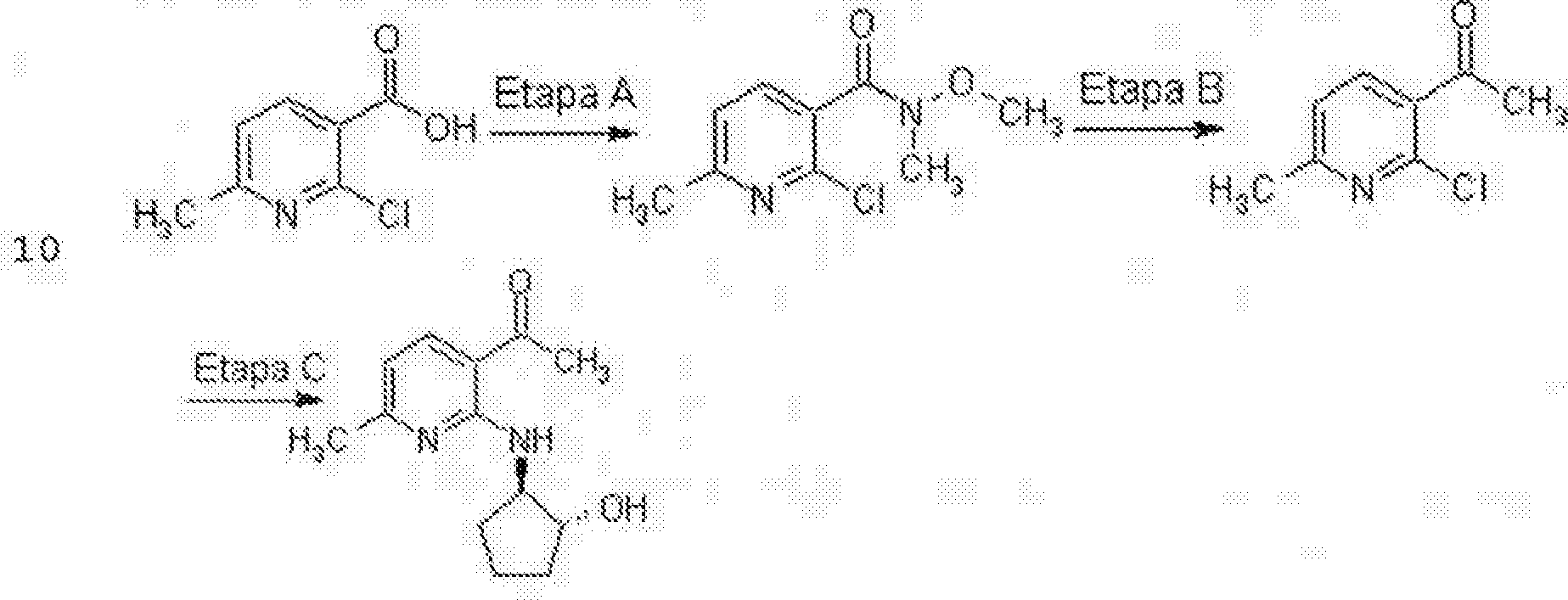
ESQUEMA 6
Etapa Aí Síntese de 2-cloro~N---metoxi--6, Ndimetil-nicotinamida. Uma mistura de ácido 2-cloro-615 metil-nicotíníco (2 g, 12 mmol), cloridrato de N,0dimetilidroxilamina (1,14 g, 12 mmol), EDCI (2,68 g, 14 mmol}, HOBt (81 mg, 16 mmol) e DIPEA (1 ml} em DCM (100 ml) foi submetida a agitação sob RT durante a noite. A mistura de reação foi então dividida entre água e DCM, a camada orgânica foi separada, submetida a secagem sobre NajSOí anídrico, filtrada, e evaporada sob pressão reduzida, O resíduo bruto foi purificado por crosiatogrsfia instantânea (EtOAc/hexano, gradiente a partir ?1/149 de 0 até 25% em 25 minutos} para proporcionar 2,4 g de 2-clora-ZV-metox.i 6, áf-dimet i l-nicotinamida na torma de um. sólido branco.
Etapa 8- Síntese de 1-(2-cloro-6-metil pirídin-3-il) -etanona. Adicionou-se lentamente uma solução de cloreto de met.il magnésio {3 M em TKE, 10 ml) a (PC a uma solução de 2-cloro-??-metoxi“6,jVdimetil··.nicotinamide (2,4 g) em THF (100 ml) . A mistura de reação foi submetida a agitação a (PC durante 1 h, então resfriada rapidamente com água. A mistura resultante foi extraída com EtOAc, os orgânicos combinados submetidas a secagem sobre Na3SO..; anídrico, filtrados e evapcrados sob pressão reduzida. O resíduo bruto foi. purificado por cromatografia instantânea (EtO15 Ac/hexano, gradiente de 0 a 25% em 25 minutas) para proporeionar 1 - (2 - c 1 oro - 5 -met 1. Ipi r id in -3-11} - etanona (0,25 g) na forma de um ôleo castanho.
Etapa C: Síntese de 1 [2~ ((IR,2R) -2-hidróxicielopentilamino) -6-metil -piridin-3-il] -etanona.
.20 Tratou-se sal de (1R,2R) -2-amino-ciclopentanol HC1 (1,0 g} com K5CO5 (sat aq) e a mistura foi extraída 3x com EtOAc. Cs extratos orgânicos combinados foram submetidos a secagem sabre da;<SO;i anídrico, filtrados, e evaporados sob pressão reduzida. 0 produto foi combina25 do com 1-(2-claro-6-metil -piridin-3-il} -etanona (0,4 g) e 1-metil-2-pirrolidínona (1 ml}, e. a mistura foi aquecida para lBOyC em um reator de microondas durante 2 η, A mistura de reação foi então dividida entre, água
72/149 © EtOAc, a camada orgânica separada e lavadas 3* com âgua, submetida a secagem sobre Na-iSO^ anídrico, fi.ltrada, e evaporada sob pressão reduzida, O resíduo bruto foi purificado por cxomatografi.a instantânea (EtQ5 Ac/hexano, gradiente de 0 até 25% em 20 mm) pana proporcionar 1- (2- ((1R.< 2R) '-2-hidroxi-ciolopentílamino) 5-metilpirídin-3-il] -etanona (507 mg, 92% de rendimento) . MS - 235 [M+Hf.
Utilizando-se o procedimento descrito anterior1G mente e os materiais de partida apropriados, prepararam-se os seguintes compostos:
1- [2 - (3-H.idroxi - clcloexilamino) - 5 -metil -pirid.in- 3 11] -etanonaj
- (2 Clclopentilamino-6~met.il -p.iridín-3-il) -etanona;
3.5 1- [t-Metil -2-- (tetraidro-piran-4-.ilamlno) -piridin-3.11] -etanona;
1- [2- ((1R,3R) - 3-Hidrox l-ciclcpent.il amino) -6-metil pir.id.in--3-.il] etanona (utilizando-se (IR,3R) -3-aminociclopentanol disponível comercialmente a partir da A~ 20 FID Therapeutics);
- (2 - lsoprop.ilamino - 6-metil. -plridin- 3- .11) -etanona,:
1- [2- (1,1 -líioxo-t.etraidro-11”-1iofen-3-ilamino) -6~ met.il -p.irid.in-3- ill -etanona;
1- (2-Ciclopropilamino-6-metil -piridin-3 -1.1) -etanona;
1-[2-{2-Metóxi-1-metiletilamino) -6-metilpiridin-3 -il]
-etanona;
.1. - (2 -Metilaminof enil) -etanona;
1- (2-Ciclobutilaxnino-6 -metil -piridi.n~3--.il) -etanona;
73/14 9
-3 -Hidróxi ciclopentilamino) - 6-metil piridin-3-ilJ
-etanona;
- 3 - Hidróxi - c 1 c lopent í lami no) 6 - met 11 piridin-3 11]
-etanona; e
1- (.2- ( (1R.3R)
- 3 - Hidroximeti 1 -c.lolopenti lamino) - 6 metil -piridin-3-il] -etanona.
Preparação 7: Eíntese de 4-Formil-benzenossulfonamida
A síntese de 4 -formil-benzenossulfonamida foi
1HO one
Uma solução de amônia (0,5 M em 1,4 - di oxana., ml * f o 1 adi cionada a cloreto de 4-form.ilbenzenossulfonil (0,5 g) , seguido por DCM (10 ml).
mistura, de reação foi submetida a agitação vigorosamen te durante 3 h sob RT, então concentrada sob pressão reduzida, o resíduo bruto foi puri f1cado por crematografia instantânea para proporcionar
4-formilbenzenossulfonamida. MS ~ 183,9 [M-H]
Exemplo li
Eíntese de âster mstílloo de ácido 7 Cloro-3-[4-(4-met. 11-piperazine-· 1-sul fonil) benzill -4-axo-l-Êsnil-l,4“diidroquinolina-2 c&rboxílico
4- (4 -meti.1 -piperazina -1 sulfonil) -benzí
.......:K κ: ........
* 74/149 fenil-1,4-diidroquinolina-2 -carboxílico í composto 30) foi realizada de acordo com o processo ilustrado no Esquema 8;

BSQOTKA 8
Etapa At A uma solução de 4 (4-metilp ípuras ina · 1 sul ford 1) bensaldeído (1,23 g) e 1 (4 cloro-2-nitrofenil)etanona (0,938 g) in MeOH (50 ml) adicionou-·se NaOH (2 M aq, 4,3 ml), e a mistura resultante foi submetida a agitação durante 15 min. O sólido castanho claro que se desmanchou para fora da solução foi coletado por filtragem e lavado com água. 0 materi al sólido foi recolhido em MeOH, e a mistura evaporada sob pressão reduzida para se remover azeotropicamente a água residual. Q resíduo sólido foi submetido a secagem sob pressão reduzida para proporcionar (E) -1-(4-cloro-
2-nitro-fenil) -3- [4- (4-metil-piperazi.ua 1 -sulfonil) fenil] -propenona (1.20 g) sem qualquer outra purifica- ção.
Etapa B; Uma mistura de. (E) -1 - (4-cloro-2 nitrofenil) - 3 - (4 - (4 -metil-piperazine- l-sulfan.il) fenii] -propenona (1,167 g) e õxido de platina(IV) (50 mg) em uma. mistura da EtOH e THF (10/1, 110 ml) fox 5 submetida a agitação sob (pressão de balão) durante a noite. A mistura de reação foi então filtrada, e o filtrado foi evaporado sob pressão reduzida para proporcionar 1- (2 amine-4-cloro-f.en.il) -3- [4- (4-met.ilpiperaz.ina-1-sulfonil) -fenil] -propan-1-ona (1,28 g) 10 na forma de um sólido amarelo.
Etapa C; Uma mistura. de 1-(2-amino-4 clorafenil) -3- [4- {4-met 1.1 -piperazina-l~sulfon.il) fenil] -propan-l-ona (1,268 g) , pó de cobre (38 mg), Kl (99 mg), K;;CQ> (829 mg) e lodobenseno (1,35 ml) em di15 butil éter (25 ml) foi aquecida a 160’0 (temperatura do banho de óleo) durante a noite. Acrescentaram-se nó de cobre (38 mg) , Kl (99 mg) , K2CO3 (829 mg) e iodobenseno (1,.35 ml) adicionais e a mistura resultante foi novamente submetida a agitação a 168*0 durante a noite. A 20 mistura de reação foi então refrigerada e concentrada sob pressão reduzida. 0 residue foi. dividido entre ãgua e EtOAc, a camada orgânica foi separada, e a. camada aquosa foi extraída 2* com EtOAc (50 ml) . Os extratos orgânicos combinados foram lavadas com salmoura, subme25 tidos a secagem sobre Na^SO^ anidrico, filtrados, e evaporados sab pressão reduzida. O resíduo bruto fai purificado por cromatografia instantânea (DCN/MeOH,
100/O durante 5 minutos,. 98/2 a partir do minuto 8 até
20) para proporcionar 1-(4-cloro-2-fenil-amino-fenil) -
3- [4- (4-met.il-piperazina-l~sulfon.il) -fenil] -propan-1ona (1,02 g; 68% de rendimento), MS - 498,0 Ifb-Hj ’.
Etapa D: Adicionou-se cloreto de metil oxairi (1,6 ml) a uma solução de 1 - (4 cloro - 2 - f en.il - amino f eni 1) - 3 - [4 - (4 -met i 1 -piperaz1na 1 - sulf oni 1) - f eni 1 ] propan-l~ona (0,86 3 g, 1,1 mmoi) em tolue.no (40 ml). A mistura de reação foi aquecida para refluxo durante a noite, então evaporada, sob pressão reduzida para pro10 porciosar éster metílico de ácido JV-{á-oloro-2-(3- [4(4-metil-piperasina-1-sulfonil) -fenil] -propionil} fenil) ~?í-fenil-oxalâmico sem qualquer outra purificação ..
Etapa E; Aqueceu-se uma mistura de éster metí15 lico de doido &(5-cioro-2-{3-[4-(4-metil-piperazina-1 sulfonil) -fenil] -propionil} -fenil) -fbfeuiloxalâmico (1,1 mmoi) e K^CO:· (282 mg ) em MeOH (60 ml) sob refluxo durante 1 h. A mistura de reação foi então refrigerada e evaporada sob pressão reduzida. 0 resíduo 20 foi dividido entre água e EtOAc, a camada orgânica separada, e a camada aquosa extrai da com EtC-Ac (2x 25 ml) . Os extratos orgânicos combinados foram lavados com salmoura, submetidos a secagem sobre Naj.SCd anidrico, filtrados, e evaporados sob pressão reduzida para 25 proporcionarem um óleo castanho alaranjado. 0 residues bruto foi purificado duas vezes por cromatografia instantânea (bCM/MeQH) para proporcionar éster metílico de ácido ?-cloro~3- (4- (4-metil-pipexazina-l-sulfon.il; ben z i.l 1 -- 4 - oxo -1 - f eni 1 -1,4 -di - hidroqui nol i na - 2 carboxilico na forma de um sólido castanho claro. MS « Sd» [Μ+ΗΓ; PF «: .223,3-226,6°C.
Utilizando-se o procedimento descrito anteriormente, substituindo--se os materiais de partida apropriados, prepararam-se os seguintes compostos♦
Éster metílioo de ácido 7-cloro-3- [4 - (morfolin.a-4~ sulfon.il) -benzil] -4-oxo- 1-fenil -1,4-diídroquinolina2-carboxilico (pó incolor), MS =- 553,0 (Η+ΗΓ; PF 237,9-238,8“C (composto 31); e
Éster metí.lico de ácido 7-cloro-3-(4-dimetilsulfamo.il benzil) -4-oxo-1-fenil-l,4-díidroquinolina-2carboxílico (pó castanho claro) (composto 32} , MS '511,0 [MW] *; PF a 218,1 -219,5*0*
Exemplo 2s Síntese d® éster metílico de ácido 7cloro-3-[4-(2,3“diidroxi-propxlsulfamoil) benzil) ~4 -oxo~l-Senil-1,4 -diidroqui.nolina-2~ carboxílíco
A síntese de éster metílíco de ácido 7-cloro-3(4 - (2,3 - d i i droxi ·· pr op i 1 sul f arnoí. 1) - be n z 11 ] --4- oxo -1 f ení1-1,4-dlidroquinolina-2 -carboxí1ico (composto 46) foi realizada de acordo com o processo ilustrado no Esquema 9.
’8/143


ESQUEMA 9

Etapa A: Adicionou··se
1,8 mmol} a uma solução de (2,2-dimetil- [1,3] dioxo-4-form!1-benzenossulfonami.da (305 mg mmol) e 1 - (4 ••clGro-2-fenilamino-f.en.il) -etanona (250 mg, 1 mmol) em MeOH (10 ml), e a mistura resultante foi submetida a .agitação sob RT durante a noite. Adicionouse uma segunda alíquota de JíaCH (2 M aq, 0,1 ml), e a mistura de reação foi novamente submetida a agitação sob 8’1 durante a noite. A mistura resultante foi evaporada, sob pressão reduzida, e o resíduo bruto dividido entre ãgua e EtOAc. A camada orgânica foi separada, e a camada aquosa foi extraída duas vezes com EtOAc. Os extratos orgânicos combinados foram lavados com salmon5 ra, submetidos a. secagem sobre Bag-liA anídrico, filtrados e evaporadas sob pressão reduzida paru proporcionarem um óleo. Q resíduo bruto foi purificado duas vezes por cromatograf ia instantânea (EtOAc/hexano) para proporcionar 4 · [(E) -3-(4-cloro-2-fenilamino-fenil) -Βίο oxo-propenil ] - M- (2,2 -dimeti 1 [ 1,3] dí-oxolan-4 - ilmeti 1}
-benzenossulfonamida (70 mg) <
Etapa B: Adiolanou-se óxido de platína(IV) (50 mg} a 4 - [ (E) - 3 - (4 - c.I oro - 2 ·· f eni 1 - ami no - f en i 1) ·· 3 oxopropeni 11 Λ’ (2,2 -dimet .11- [ 1,3} d ioxolan· 4 - ilmet i 1)
-benzenossulfonamida (66 mg) em uma mistura de EtOH e
EtOAc ilrl, 50 ml) e a mistura resultante foi submetida a agitação safc H;i (pressão de balão) durante 1,5 h. A mistura de reação foi então filtrada através de papel de fibra de vidro em um funil Buchner. 0 filtrado foi 20 evaporado sob pressão reduzida para proporcionar 52 mg de uma mistura 2:1 (pur WR) do di-ol desprotegido: 4[3 - (4··cloro-2 -fenilaminofeníl) -3~axuprap.il] -H- (2,3diidroxi-propil) -benzenossulfonamida, e o composto protegido - 4 - [3 - (4 ··claro-2~ f enil-amirio-fenil) -325 oxapro-pil] -ff-(2,2-dimetil-[1,3] díoxclan-4-ilmetil) benzenes-sulfanamide.
Etapa C: Adicionou-se cloreto de met11 oxalíl (80 μΐ) a uma mistura 2:1 de 4-[3-(4~cloro--2-fenilaminofeni I) - 3 - oxopropil I N- (2,3 - di.i dr oxiprop i 1) benxe-nassulf onamida e 4- [3 · (4 - cloro- -2 - fenilaminofen.il) -3 -oxo-propil 1 ~Ay- (2,2-dimetil · (1,3] dioxo.ian-4 ilmet.il) -benzenossulfonamida (25 mg) em toluene (5 5 ml) , e a mistura resultante aquacade até 120<;C durante
1,5 h. A. mistura de reação foi evaporada sob pressão reduzida para proporcionar éster metílica de ácido N( 5 - cloro 2 - {3 · (4 - (2,3 - dl -hidróxi -prop! Isulfamoil) fenil] -propionil? ··fenil) -ür-feníl-oxalâmico e o com10 posto protegido, éster metílicu de ácido N- (5-cloro-2{3-(4-(2,2-dimetil-[1,3] dioxolan-4 -ilmetilsulfamoí1) fenil] -propienil} -fenil) -N-fenil-oxalâmico, que foi usado sem qualquer outra purificação.
Etapa D: A KSCQ?, (12 mg} adicionou-se a mistura 15 de éster metílico de ácido N-(S-cloro-2-{3-(4-(2,3dihidroxí-propilsulfamoil) -fenil] -propionil) -fenil)
- Kr-fenil -oxalâmico e éster metílico de ácido N- (5cloro-2- {3- [4- (2,2-dimet.il - (1,3] dioxolan-4ilmetilsulfamoil) -fenil] -propionil) -fenil) -N-fenil20 oxalâmico (0,05 mmol; em MeOH (5 ml), e a mistura resultante foi submetida a agitação a 80’C durante 1,5 h.
A mistura de reação foi então refrigerada e evaporada sob pressão reduzida.. O resíduo foi dividido entre agua e EtOAc, a camada orgânica separada, e a camada aquosa 25 extraída com EtOAc (2x 25 ml}. Os extratos orgânicos combinados foram .lavados com salmoura, submetidos a secagem sobre NasS0$, filtrados e evaporados sob pressão reduzida. 0 resíduo bruto foi purificada duas vezes por
81/149
TLC de preparação (DCM/MeOH, 5:1} e então (DCM/MeOH, 9S:5) para proporcionar éster metílico de ácido ?cloro-3 - [ 4 - (2,3 -di idroxi - propil sulf email) -benzil} - 4 oxo-1-fenil-1,4~diidroquinolina-2-carboxilico na forma de um pó castanha claro. MS - 556,8 [MtH0 e 554,9 [ΜΗ'} -,
Utílizando-se o procedimento exposto anteriormente, substituindo-se oe materiais de partida apropriados, prepararam-se os seguintes compostos;
É s t e r rae t í 1 i c o de â c i do 7 · c .1 or o - 3 - [ 4 - (2 hídroxíetilsulfanil) -bensilj -4 -oxo-1-fenil-l, 4 diidroquinolina-2-carboxilico (pó amarelo claro); MS « 479,8 [M-+H]’; PF - 204 , 0-205,0 *C (composto 55);
Ester metílico de ácido 7-d oro-3-(4-metilsdfamoilbenzil) -4- oxo-1 - f eni.1 -1,4 díidroquincline-2carbcxílico {pó amarelo claro·; MS - 497 [Μ+ΗΓ (composto 44);
Éster metílico de ácido 7cloro-3-{4-metoxicarbonrlbenzil) - 4 -oxo- 1-fenil -1,4-diidroquinoli.na·· 2 ca.rboxfl.ico (sólido incolor); MS = 462 PF =
194,7 - 1.95,2 * C (COTUpO S t O 10) ;
Éster metilioo de ácido 7-cloro-3 -(3-metoxicarbon!1benz11) -4-oxo-1-fenil-1,4 -diidroquinclina -2carboxílíco (pó branco) ; MS === 462 (M+H]*; PF - 183185C (composto 23);
Éster metílico de ácido 7-cloro-,3-(4-metoxi-benzil) -4oxo -1 - fen i 1 -1,4 - d i i.dr oqu i nol i n a - 2 ca r box í 11. c o í pó incolor); MS ~ 434 [M+H]'; PF - 198-2GÜ’C (composto
19) ;
Ester metílico de ácido 3-benzo[I,3]dioxol-5-ilmetil-4oxo-1-fenil-1,4-diidroquinolina-2-carboxílico (pô amarelo claro); MS * 414 [M+Hf (composto 2);
Éster metí liou de soldo 3-(4-raetanossulfon.il-benzil) 4 - oxo-1 - fenil-1,4 -diidroqu.inol.ina - 2-carboxíl ico (pó incolor); MS - 44 8 [M + H] f (composto 4);
Éster me tí lico de ácido 7 - cloro 3-(3,4 -diraetoxi benzil)
- 4 -oxo-1-fenil-1,4-diidroguinolina-2 carboxílico (solido castanho claro); MS - 464 (M*H] ’ PF 153,0-154,5C (compost o 21) ;
Éster metílico de ácido 7-cloro-3-(3-metóxi-benzil) -4oxo-1 -fenil-1,4-dildroquinolina-2-carboxílico (pó ::: 434 [MeH] ; PF » 178,5-180,9C ícomde ácido 7-cloro-S- (4-me.taaossulfonil-4 -oxo-1 -f eni1-1,4 -dildroquinolina- 2 (sólido branco) ; MS ® 4.82 PF 215,5-219,0*C {composto 9) ;
Éster metílico de ácido 3-benzo[1,3]dioxol-5-ilmetil-7me toxi - 4 - oxo-1 - f.en.i 1 -1,4 - d iidroquinol ina - 2 carboxílico (pó incolor); MS » 444 [M*H]*; PF « 171,0-17 2, 1 ° C {compos to 6);
Ester metilico de ácido 3benzo[l, 3] dioxc<l-5-ilmetil-6cloro-4 - oxo-1 - fenil -1,4-diidroquinol.ina-2 carboxílico (pó incolor); MS - 448 + ; PS 172,2 -177,7* C (composto 7) ;
Ester metí lico de ácido 3-benzo [1,3] dioxol - 5 - ilm.etil -6 brancoí; MS posto 20);
É s t e r me. t í 1 i u o benzil) carboxí1ico metoxi~4-oxo- 1-fenil 1 , 4~diidroquínolina“2carboxílico (solido castanho); MS « <44 [Mí-H]”; PF ~: 171,9-173,5 ’ C (CompostO 5);
Ester' metílico de ácido 1-((1R,2R) -2-hidróxiη ciclopenti 1) · 3 (4-metanossultonil-benzi.l} - Z~ met 11 - 4 - oxo-1 ,· 4 -diidro- (1,8 ] naf tí r idina -2carboxílico (sólido amarelo},- MS « 471 [M-í-S]; PF « 200,2 -204,6*0 (compostO 48) ;
Ester metílico de ácido 3-(4-metanossulfonii-benzí1;
4 -oxo-1 - feni 1 -1,4 -diidro··· [1,8] naf tiridina-2 carboxílico (sólido incolor}; MS ~ 449 [M+H]:; PF 235-237«C {composto 11);
Ester metílico de ácido 3-(4-metanossulfeníl-benzil)
- met. i 1 - 4 -oxo-1 - fen il -1,4 - di idro - [1,8)- naft ir idina 15 2 carboxílico (sólido amarelo? ; MS - 463 [M-M-lp; PF ® 202-204C {composto 35} ;
êster metí.Iicu de ácido 1-(3-hidróxi-cicloexil) -3-(4metanoesulfonil-benzil) -7-me ti 1 -ã-oxo-l, 4 - diidro·· [1,8] naftiridina -2-carboxílico (sólido castanho);
MS « 435 PP - 196,6-198*0 (composto 51);
Êster metílico de ácido l-ciclopentil-3(4-metanossul“ f oni 1 - benz i 1) - ? - me t i 1 4 · oxo -1,4 ~ di idr o [ 1,8.1 naftiridina-2-carboxílico (espuma incolor); MS ~ 455 [M-íH] ’; PF ~ 75,0-77,2*0 (composto 28) ;
êster metílico de ácido 3-(4-metanoesulfonil~henx.il) · 7~met. il- 4 oxo -1 - (tetxaidropira.u-4 -11) 1,4 -diidro- [1,8] -naftiridina-2“Carboxílíco (espuma amarela}; MS - 471 [M+HJ*; PF ~ 207,2-208,2”C (composto SO);
84/149
Éster metí lice de ácido 1-{(1R,3R) - 3-hidróxi ·· cidopentil) -3- <4 -met anos sul fenil -benzil.) -7metil-4-oxo «1,4-diidro[l, 8] naftiridina-2carboxílico (sólido amarelo) ; MS - 471 [M-t-H]*; PF :::
195,1-197,8”C (composto 54);
Ester metílico de ácido 1- isoprcpil- 3 (4··· metanossulfenil -benzil) -7-metil-4-oxo-1, 4-dridro- [1.8] -naftiridina -2-carboxílico (sólido amarelo claro); MS - 429 RF - 73,0-75,5C {composto
34} ;
Éster metílico de ácido 1 ·· (1, l-dioxo~t.etraídrotiofen~3 il) -3- (4•metanossulfonil-benzil) -7~metil~4-oxo1,4-diidro-[1,8] naftirídina-2-carboxílico (espuma amarelo claro); .MS « 505 [Μ+ΗΓ; PF - 121,1 123,3 «Ç.
L5 (composto 38);
Ester metílico de ácido l-ciclopropil-ü-(4-metanoss u 1 f on i 1 ·· benz i 1) - Ί ~me 11.1 - 4 - oxo -1,4-diidro - (1,8] naftiridina-2-carboxílico (espuma castanho claro); MS - 427 [M*H]+; PF -= 73,9-78,8®C (composto 33) ;
2G Éster metílico de ácido 3-(4-metanossulfonil~benzll) 1-(1-metoxi-prop-2 il) -7~metíl-4~oxo -1,4-diidro- (1.8] -naftirídina-2 carboxílico (sólido amarelo); MS - 459 [M+K] '; PF - 125,2-126,6^ {composto S3);
Êster metί1i co de ác ido 3-benzo[1,3 Jdioxol- 5 -íImet i1-12 b metil-4-oxo-1,4~di idroquinolina-2-carboxílico (pó branco)·; MS ~ 352 [M+H] h (composto 3);
Et 11 éster de ácido 3-(4-metanossnlfonil-benzíl) -1metil-4-oxo-1,4-dildroquinolina-2-carboxílico (pó
85/149 amarela) .: MS - 400 [M+H] * (composto 1) ;
Éster metxlico de áai.do 3- (4 -acetilaminobenzil) -7cloro-4 -oxo-1-fenil-1,4-diídroquinol.ina-2carboxxlico (p6 branco); MS » 4 81 IM-rH]’; pf - 15?5 159C- (composto 22)
Éster metálico de ácido benao[1,3]dioxol -S-ilmeti.1-7cloro-4 oxo-l- fenil -1 < 4-d.i idroquinolina~ 2 ·· carboxílico (pó incolor); MS ~ 448 [M*K]*; PF 228,8-230, 1;>C (composto 8) ;
Éster metxlico de ácido 7-cloro-3-(4 - i (2-hidroxietí.i.) met í 1- sulfamoil] -benzil) -4-oxo-1-fenil-1,4 diidroquinolina·· 2carboxxlico (p6 incolor); MS ~ 541 ÍM-íH]*; PF » 95,2-100,2*0; M-í-H 541 {COmpUStO
59) ;
Éster metílxco de ácido 3-{4-[2-{terc-butxl-dimet.ilsilaniloxi) -etilsulfamoil] -benzil} -7-cloro-4oxD-1-fenil-1,4 - d.iidroguinolina-2-carboxílice; MS 641 [M+H]* (composto 60);
Éster metl.li.co de ácido 7-cloro-3 ·· [4- (metoxioxalil 20 sulfamoil) -benzi 1 ] - 4 oxo-1 -fenil-1,4 -di.idroquínolina-2-carboxílico (po amarela); MS - 569 (M4-HP; PF ::: 151,0-154,0’C (composto 64);
Éster metílico de ácido 3· [4-(4-terc-bntoxícarbonilpiperazxna-l-sulfonil) -bens.il] -7-cloro~4-oxo-1 25 fenil-1,4 •di.idxoquinolina-2-carbox.ílico (pô incolor) ; MS - 6 52 (M+H]'' {composta 63);
Éster metílico de ácido 1-((18,35} -3 -Hidróxicíclopentil) -3-(4-met anossulfonil-benzil)
86/148 mct.il. -4-oxo ~1 f 4-diidro - [1,. 8] naftxridina -2carboxllico (espuma incolor) ; MS - 4’’3. [Μ+Η.Γ; PF 183,3-18 7,5* C (oomposto 62);
Éster metílico de ácido 1-ciclobutil - 3-(4 me t anossul fon.il - benz i 1) - 7 -met i 1 - 4 - oxo-1,4 - d 1 idro -- [1,8] -naftiridina-2-carboxflino (espuma amarelo claro}; MS 441 [M+H] *; PF - 90,9-93,3eC (composto 88} ;
Ester metílico de ácido 1-((18,33) -3-hidroxi~ 10 ciclopentil) - 3 (4 -metanos sulf on 11 -benzil) -7met il - 4 - oxo -1,4 -di idro - [ 1,81 naf t ir idi na 2 carbox.ilico (espuma amarelo claro); MS - 471 [M+H]*; PF ::: 118,3-119,7*0 (composto 71);
Éster metí 1 ico de ácido X- ({IR, 3R} - 3 - hidroximeti.1 13 ciclopentil) --3 - i4-metanossulfoni 1-benz 11)-7met.il-4-0X0 -1,4-01101-0-11,8] naf tiridina-2carboxílico (sólido castanho claro) ; MS ~486 [M+H]*; PF - 161,8-164,7”C (composto 57);
Éster metílico de ácido 1 - (3-hidroxiciclopenti.1} -3--{r 20 metanossulfonilbenzil) -7-metil-4-oxo-1,4-diid.ro[:1,8.] naftiridina-2-carboxilico (composto 61);
Éster metílico de ácido 3- [4- (2-hidroxi-etil.carbama.il) -.benzil] -7-metil -4-oxo-1- fenil-1,4-diidro- (1,8] naftiridina-2-carboxilico (espuma incolor); MS
472 [Μ*ΗΓ; PF - 191,7-182,8'0 (composto 68);
Éster metílico de ácido 3 - (4 -metoxicarbonil-henz.il; -7metíl-4 -oxo-1 -fenil -X,4-diid.ro [1,8] naf.tiridina-2carboxflico (composto 731;
7/149
Ester raetilico de ácido 1- (4~f Ixxorofenil) -3-(4metanossulfonilbenzil) -7-metil-4-oxo-1,4 -dridro- [1,8] naftiridina-2-carboxílico (composto 74);
Acid© 3-(4--metoxicarbonil-benzil) - 7-metil-4-exo-1 fenil -1,4-diidro[l, 8] naftiridina -2-ca.rbox.ilico (composCO 75};
Êster met5.11.00 de ácido 3-[4 -(4-hidroxicxclo~ exi Icarbamoil) benzil j - 7 -met 11 - 4 -oxo-1 - fenil -1... 4 diidro- fl,S] -naftiridina-2-carboxílico (compost©
76} ;
Ester metilico de ácido 1-(3,4-difluorofenil) -3-(4metanossulfonilbenzil) -7-metil -4-oxo -lf 4 -diidro·[l,8j naf tiridina-2-ca.rboxílico (composto 77);
Éster metilico de ácido 3-(4-aceti.lbenzil) -7-metil~4~ oxo X-.fen.il -1,4-diidro[1 f 8] naftiridina ~2carboxilico (composto 79);
7-cloro-3-[4 -(4-metilpiperazin-1-sulfon.il) benzil] -1fenil-2-propionil-1H-quinolin-4-ona {composto 80)
p.f :;i >3:0.0·*C; 564:,Éster metilico de ácido 1- (3-fluorofenil) -3-(4metanossulfoni.lbenzil) - 7 - met 11 -4 -oxo-1,4 -dlldro ll., 8] naftiridina-2-carboxilico (composto 81);
Ester metilico de ácido 7-metil-4~oxo~l~fer!.il-3pirimidin-5- ilmetil-X, 4.-diidro U, 8.] naf.tlridi.ua-2carboxilico (composto 82.) ;
Êster metilico de ácido 3-bsnzil-7-metil-4-cxo-l-fenil-1,4-diidro(1,8] naftiridina-2-carboxxlico (composto
91) ;
88/149
Ester metílico de ácido 3-(4-(2-hidroxietanossulfonil) benzil] -7-met11 - 4 -oxo-1-fenil-1< 4-di idro- (1,8] -r.a.ftiridina~2-carhoxilico (composto 93)· e
Éster metxlico de ácido 3-(4-(4- t-butoxicarbon115 piperazina -1 -- sulf anil) benzil] -7--metiI-4-oxo-1 fení 1 -1,4- diidro[1>8] naftiridina -2--carboxilloo (composto 9β): ,.
Exemplo 3; Síntese de ester metálico de ácido 7«10X0-3-[4-(2-hidroxietanossulfonll) -benzil] 10 - 4 -oxo --1- f ení 1-1,4 - di idroquinolina ~ 2 carboxílico
Realizou-se a síntese de ester metálico d® ácido 7-cloro-3 (4 - (2-hidroxietanossulfonil) -benzí 1] -4oxo-1 -fení 1 ~ 1,4 -düdroquinolina-2-carboxilico (composto 15 56} de acordo com o processo ilustrado no Esquema 10.

ESQUEMA 10
A éster metíliso de ácido 7-clorO“3-[4 -(2hádroxi-etilsulfanil) -benzil] -4-oxo-l-feniI-1,4 diidro-quinolina-2-carboxílíco (108 mg) em uma mistura de THE e MeOH (1:5, 12 ml) adicionou-se uma solução de OXQNE™ (294 mg) em água (3 ml) a CTC. A mistura de re
30/149 ação foi submetida a agitação durante 1,5 h, e adicionou-se NaHCOj <sat aq, 20 ml) - A mistura resultante foi extraída com EtOAc (3* 25 ml). Os extratos orgânicos combinados foram .lavados com salmoura, submetidos a secagem sobre Nas80o filtrados e evaporados sob pressão reduzida para proporcionar um óleo castanho amarelado. O resíduo bruto foi purificado por cromatografia instantânea (EtOAc/-hexano) para proporcionar éster metílico de ãcido 7 -cloro-3-[4 -(210 hidroxietanossu.1 fonil) -benzil) -4 -oxo-1 feni 1-1,4diidro-quinolina-2~carboxíliso (23,6 mg) na forma de um pó incolor, MS - 512 [M-í-H] '; PF - 172,2-173,5,:C. Exemplo 4í Síntese de éster metílico de aside 3(4-(4-metil-pipsrazlna-l~sulfoniI) -benzil] 15 4-oxo-l-fenil-l,4-diidrcguinclina-2carboxílloo
A síntese de éster metílico de ácido 3-(4-(4met i1-piperazina-1 -sulfon il) -benzil] -4-oxo-1-feni1-
I, 4-diidroquínolina -2-carboxílico (composto 29) foi realizada de acordo com o processo ilustrado no Esquema
II.

90/149

ESQUEMA 11
Etapa A: Uma mistura de (E) 1~ (4-cloro··2 n i tro ·· f anil) ·· 3 - [ 4 - (4 -me ti 1 ~ p i perazi na - 1 - sulf oni 1) fenil] -propenona (1,13 g) e paládio(8} em carbono (10%, 100 mg) em uma mistura de EtOH e THE (10:1, 250 ml) foi submetida a agitação sob H2 (pressão de balão) durante a noite. A mistura de reação foi então filtrada através de papel de filtro de fibra de vidro, e o filtrado evaporado sob pressão reduzida para, proporcionar 1-(2-amino-fenil) -3-[4- (4~metil~piperazina-l-sulfonil) -fen.il) -propan-l-ona (1/18 g) na forma de um sólido branco.
Etapa B: Uma mistura de 1-{2-aminofenil) -3-[4(4-met il.piperazi.na -1 - sulfonil} ~ fen.il] -propan- :i-ona (1,16 g) , pô de cobre (90 mg). Kl (19,7 mg), K;iCO:'; (496 mg) e iodobenze.no (1,3 ml) em dibut.ll éter (25 ml) fed aquecida a 160 :>C (temperatura de banho de óleo) durante a noite. Adicionou-se uma segunda alíquota de Kl (.19,7
91/149 mg) , K^COj (498 mg) e de iodobenzeno (1,30 ml), e a mistura resultante novamente aquecida a 160*0 durante a noite. A mistura, de reação foi então refrigerada e concentrada sob pressão reduzida. G resíduo foi dividido 5 entre água (50 ml) e EtOAc (50 ml) , a camada orgârmoa.
separada, e a camada açposa foi extraída com EtOAc (2x ml). Os extratos orgânicos combinados foram lavados com salmoura, submetidos a secagem sobre filtrados e evaporados sob pressão reduzida. O resídua i 0 bruta foi purif icado por cromatograf ia instantânea (DOM/MeOH, 98;2 durante 5 minutos, 95:5 durante de. minutos 10 atê 15) para proporcionar 3- (4- (4-metilp.iperazina-1-sulfonil) -fenil.) -1- (2-fen.ilamin.o£enil) propan-l-ona (258 mg). MS = 484,0 [M-*H]\
Etapa C; Adicionou-ae cloreto de metil oxalil (0,8 ml) a uma solução de 3- (4 -(4-metilpíperazina-1sulfoniij -fenil] -1- (2- fenilaminofenil) --propan-l-ona (0,237 g, 0,5 mmoi) era toluene (20 ml). .9 rn.istu.ra de reação foi aquecida sob refluxo durante a noite, então evaporada sob pressão reduzida para proporcionar éster metálica de ácido .itf- (2- (3 - {4 - (4-metílpiperazina-1 sulíonil) -fenil] -propionil} -fenil) -N-feníloxalâmico, que foi usado sem qualquer outra purificação. MS « 550,0 ΙΜ+ΗΓ.
Etapa Pr Aqueceu-se uma mistura de éster metí1.ico de âcido 17- (2- {3 - [4 - (4 -met íIpiperazína-1 - suif oni 1) •••fenil] -propionil} -fenil) -jtf-fenil-oxalâmico (ca. 0,5 mmoi) e (1.28 mg ) em MeOH (39 ml) sob refluxo du rants 1 h. A mistura de reação foi então refrigerada, e evaporada sob pressão reduzida. O resíduo foi dividido entre água (25 ml) e EtOAc (25 ml) , a camada orgânzca foi separada, e a camada aguosa extraída com EtOAc (2*
25 ml) . Os extratos orgânicos combinados foram lavados com salmoura, submetidos a. secagem sobre Ea3SO4, filtrados e concentrados sob pressão reduzida, 0 resíduo bruto foi purificado duas vezes por oromatografia instantânea (DCM/MeOH) e uma vez por TbC de preparação IO (DCM/MeQH, 95:5) para proporcionar éster metílíco de ácido 3- [4-(4-metilpiperazina-l-sulfoníl) -benzil.) -4oxo-1-f eni1-1,4-di idroquinolina- 2 - carboxi1í co na forma de um solido castanho claro. ES ~ 532,0 >M+H] \ PF -: 21Θ , 0-212,0P2.
Exemplo 5: Síntese de êster metílíco de ácido 3(4-carboxi-benzil) -7-elorc-4-oxo-l-fenil1,4-diidro-quineiina- 2 -oerboxílico
A síntese de éster metílíco de ácido 3-(4carboxi-benzil) - 7 - cloro-4 -oxo-1-fenil-1,4 -díidroquino20 1 ina-2-oa.rbo.xí.li co (composto 12) foi realizada de acordo com o processo ilustrado no Esquema 12.

ESQUEMA 12
Uma mistura de éster metílíco de ácido 7-cloro
93/149
3-- (4 -metoxicarbonil-benzil} --4-oxo-l-fen.il.--1,4-diidro·· quinol.ina-2·· carboxilico (composto 10, 31 mg) e LÍOH (1 M aq, 1 ml) em MeOH (10 ml) foi submetida a agitação sob R.T durante a noite. A mistura de reação foi então ; submetida a agitação a 50C durante 3 h, então extraída cem EtOAc. A camada orgânica foi separada, a camada aquosa acidulada por adição de AcOH (aq) , e a mistura resultante extraída com EtOAc* Os extratos orgânicos combinados foram submetidos a secagem sobre Na^SO^, ) filtrados e evaporados sob pressão reduzida. 0 resíduo bruto foi purificado por TLC de preparação (hexano/acetona, 70:30 *1% AcOH) para proporcionar 11 mg de êster metílico de ácido éster metílico de ácido 3-{4carboxibenzil) -7-cloro-4-oxo-l-fenil-1,4 · diídroquinolina-2-carboxilico na forma de um sólido incolor. MS = 447; PF « 258,0-2él,0ôC.
Exemplo ê; Síntese de ester metílico d® ácido 7cloro-3-[4-(2-dimetilcarbamoil-l*1disaetiletil-carb&moil) -benzill -4-oxo-l0 feníl-1,4~diidroguiuoliua~2“carboxílico
A síntese de éster metílico de ácido 7-cloro-3[4-(2 -dimetilcarbamoil-1,1-dimetiletiluarbamoil> ben z,i 1 ] - 4 - oxo -1 - f en i 1 -1,4 - di idroqu inol í na - 2 -carboxilico (composto 38) foi realizada de acordo com o S proceaeo ilustrado no Esquema 1394/149

ESQOEMA 13
Uma mistura de éster metílieu de ácida 3~{4~ carboxi-benzil} -7-cloro~4-oxo-1 -fenil-1,4-diidroquino5 lina-2-carboxílico (75 mg, 0,17 mmol), 3-amino-Β,Α,Μtrímetil -butiramida (24 mg, 0,17 mmol), BOP (ISO mg,
0,34 mmol..) e DIPEA (.1 ml) an THF (15 ml) foi submetida a agitação sob RT durante a noite. A mistura de reação foi dividida entre água e EtOAc, a camada orgânica se10 parada, ’.lavada com agua, submetida a secagem sobre
Na^SCb, filtrada, © evaporada sub pressão reduzida. Q resíduo bruto foi purificada por TLC de preparação (EtQAc/hexano, 75;25) para proporcionar éster metílicu d.e ácido 7 - cloro-3-[4-(2-dimet iXcarbamoil-1,1-dimet il ls etilcarbamail} -benzí1] -4-ox.o-1 -fenil 1,4 -diidraquínol.ina-2-carboxí.lico (71 mg) na forma de uma espuma de car castanho clara. MS ~ 574 [M*H]’’; PF - 84,1ytilizando-se u procedimento descrito anterior20 mente e os materiais de partida apropriados, prepararam-se os seguintes compostos:
Éster metilico de ácido 7-cloro-4-oxo-1 -fenil-3 -{4(((S) -1-pirrulidin-2 -ilmetil) -carbamaí1] -benzil} -1,4-dlidroquinolina-2 carboxílico (espuma castanho 25 claro), MS » 530 ; PF » 124-138 “C (composto
91/149
39) ;
Éster metílico de ácido 3- [4-(2-amino-2-metiipropil carbamoil) -benzil] - ? -d oro-4-oxo-1 fenil 1,4-diidxoquinolina-2-carboxílico (sólido castanho 5 claro) , MS - 518 [M+Rj ' ; PP ::: 210,6-212, ?eC .composto 52) ;
Éster metílico de ácido 7-cloro-4-oxo-l-fenil-3-[4 (p1rroli d in- 3 -i1carbamoi1) -benzí1] -1,4 -d iidroquinolina-2-carboxílico (espuma amarelo claro), MS 10 - 518 [Μ*ΗΓ; PF - 98.0-105.0wC (composto 43);
Ester metílico de ácido 7-cloro-4-oxo-1-fenil-3-[4(pipe rid in-4 -iIcarbamoi1) -bena ilJ -1,4-di1dr oquinoline-2-carboxílico (sólido castanho), MS « 530 [Μ+ΗΓ; PF » 221,5-234.3°C (composto 47);
Éster metílico de ácido 7-oloro-3-(4 ·· (2dimet Í laminoeti .Icarbamoil) -benzi 1 ] - 4-oxo-1 - f eni 1 1,4 - d 1 idro ··quinol ina - 2 - carboxi lico (espuma amarei ο claro?, MS = 518 [M-f-Hj*; PF = 218.0-223.0’C (composto 41); and
7-Cloro-4-oxo-1 -fenil- 3 -[4 -(2-pirrolidin-1-iletilcarbamoil) -.benzil] -1,4-diidro-quinolina-2carboxilico metil ester (espuma amarelo claro), MS --= 544 [Μ+ΗΓ; PF == 1E5,0-196,0cC (composto 42).
Exemplo 7t Síntese de ester metílico da ácido 725 cloro-3~[4-(2-hidroxietilcarbamoil) -benzil]
-4-oxo-1-fenil-1,4-di idroquinolina-2 carbexilico (composto 18)
A síntese de éster metílion de ácido 7-cloro-3
96/149 [4 - (2~hidroxietiIcarbamoil) -benzil] -4 oxa-l-ienill,4--diidroquinolina-2-carboxílico (composto 18) foi realizada de acordo com o processo ilustrado no Esquema
14.

ESQOTMA 14
Wã mistura de éster met11íco de ácido 3 - (4 carboxibenzil) -7-cloro-4-oxo-l-feuil ·· 1,4 -diidroquino10 lina-2-carboxílico (167 mg, 0,37 mmol}, 2-aminoetanol (34 mg, 0,56 mmol), HGBt (50 mg} , EDCI Í142 mg, 0,74 mmol) e DIPEA (0,5 ml) em DCM (10 ml} e DMF (0,4 ml) foi submetida a agitação sob RT durante 2 h< A mistura de reação foi então dividida entre água e EtOAc. A ca15 made orgânica foi separada, lavada duas vezes com água, submetida a secagem sobre Na;;S€q# filtrada e evaporada sob pressão reduzida. 0 resíduo bruto foi purificado duas vezes por flash cromatografia para proporcionar és ter met í I i co de áci do 7 - c loro - 3 - [4 - (2 - hi dr oxi - et ?.. ί. 20 carbamoil} -benzí11 -4 - oxo·· 1 - fenil -1,4 -dildroquinolina-2-carboxilico (99 mg) na forma de um po incolor. MS «= 491 (Μ-Ι-ΗΓ; PF » 162-163,5°C<
Da mesma maneira, utilizando-se os materiais de partida apropriados, prepararam se os segtd.nt.es compos9'7/149 tOS c
Éster met11ice de ácido 7-cloro-3-[4 -{1,j-dihidruxx· prop-2-ilcarbamoil) -benzil] -4 -oxo- 1-fenil-l,4 diidra-quinolina-2-carboxílico ou {sólido castanho t claro), MS = 521 ; PF 110-112*£ (composto
24) ;
Éster raetílica de ácido 7-cloro-3- [4- (2-martolin-4-iretilaarbamo.il) -benzil] -4 ·· oxo-l-fen.il -1,4-diidroquinolína-2-carboxílico (sólido castanho ciaio),
ÍO MS ~ 590 [MtK]*; PF ~ 1O2~1Q6*C (composto 26);
Éster raetílica de ácido 7-cloro-3-[4-<2,3-dridroxipropilcarbamoíl) -benz.il] -4-oxa-l-fenil-1< 4díídra-quinolina-2-carboxílico (pó amarela claro}, MS ::: 521 [M+H]*; PF === 98,0-103,9*C (composto 25);
Éster metílico de ácido 7-cloro-3-(4 -M,N-dimet.il earbamoll -benzil] -4 -oxo-1-fenil 1,4-díidroqui nolina-2-carboxílico (pó amarelo claro) , MS == 4?S [MtH]·*; PF = 227,3-228,1*9.’ (composto 16);
Éster metílico de ácido 7-claro-4-oxo-1-fenil-3-[428 (piperazina -1-carboni1} -benzil] -1,4 dildroquinolina -2-carboxílico, cloridrato de sai (composto 17) (pó amarelo) (o sal foi gerado pela adição de uma solução de HC1 em Et^O a uma solução de éster metílico de ácido 7-eloro-4-oxo-1 -fenzl-325 [4- (p.ipexazina-1-carbanil) -benzil] -1,4dlidroquinolina-2-carboxílico em EtOAc) , MS =- 515 [MfH]+; PF - 161 -16 4 * C;
Ác i do 7 - e 1 oro - 3 - [ 4 (2 - h idrox 1 - et 1 lua.rbaraaí 1) -ben z i 1 ] ·
- oxo-1 fen 11 -1 ,< 4 diidro - qni nol Ina - 2 -· carboxi 1 loo (compost o 49);
Éster metílico de ácido 3-(4 - (2 -hidroxi-2··metilprop.il·· carbamoi 1) - benz i 1] 7 -me ti1 - 4 - oxo -1 - f eni 1-1,45 diidro (1,8] uaftixidina -2-carboxilico (composto 7BH e
Éster metílic© de ácido 3-[4 - (2-acetoxietilcarbamoi.1} benzil] ··7-cloro-4 -oxo-1 fenil -1,4diidroqul-nolina-2-carboxílico (composto 95).
IQ Exemplo 8s Síntese de éster metílino de âcído 7~ Cloro-3~[4-(4-hidzcxiciclcexiloarbamoil) benzil] - 4-oxo-1-fenil -l^-diidroquinolina2-carboxili©©
A síntese de éster metílice de ácido 7-cloro-315 [4-(4-hidróxicicloexilcarbamoil) -benzil] -4-oxo-lfenil-l, 4 -diidxoquinolina-2-carboxilicQ (composto 37} foi realizada de acordo com o processo ilustrado no Esquema 15.

Etapa A; Uma mistura de 1-(4-cloro-2
99/149 fenilaminofenil) -etanona (1,7 g, 6,9 mmol). éster metílico de ácido 4-formíl-benzóico (1,14 g, 6,9 mmol) e NaOH (2 M aq, 2 ml) In MeOH (30 ml) foi submetida a agitação sob RT durante a noite. O precipitado formado foi coletado por filtragem e submetido a secagem era ura forno a vácuo para proporcionar 0,7 g de éster metí la. co de ácido 4-[(E) -3-(4-cloro-2-fenilamino-fenil) -3-oxopropenil] -benzôico na forma de um solido cor de laranja. Os licores mãe foram acidulados por adição de HOAc glacial (99%) para pH 5, e a mistura resultante foi extraída com EtOAc. Os extratos orgânicos combinados foram submetidos a secagem sobre Na^SO*, filtrados e evaporados sob pressão reduzida para proporcionarem ácido 4- [ (E) -3- (4-cloro-2-fenilamino~fenil} - 3-oxo-prapen.il 1 -benzõica (350 mg).
Etapa B: Uma mistura de ácido 4-[(E) -3-(4cloro-2-fenilamino-fenil} - 3 -oxo-propenil] -benzóica (0,2 g, 0,53 mmol) f 4 -amino-cicloexanol (61 mg, 0,53 mmol) f ED€I (152 m$j, 0,8 ramol), HOBt (25 mg), ΠΤΡΕΑ (1 ml) em THF (50 ml) foi submetida a .agitação sab RT durante a noite. A mistura resultante foi evaporada sob pressão reduzida, e o resíduo dividido entre água e EtüAc. Q precipitado resultante foi coletado por filtragem e submetido a secagem em um forno a vácuo, o filtrado separado, a camada orgânica submetida a secagem sobre Na2SO,., filtrado e evaporada sob pressão reduzida. Q resídua foi purificada por TLC de preparação (EtOAc/hex.ano, 75:25) e combinado com o sólido coletado
100/149 para proporcionar 4 -- [ (E> -3- (4-cloro-2~fenilaminofenil) -3-oxo-propenil] - N- (4-hidroxi-cicIoexil) benzami-da (197 mg) . MS - 475 [M-s-H] *,
Etapa C; Uma mistura de 4-[(E) -3-(4 -cloro-25 f enilaminc-fen 1.1) -3 oxo-propenil] -N- (4 -hidroxicicloexil) -benzamida (:197 mg) e oxido de plat ins(V) (50 mg) em uma mistura de EtOH, THE e acetona (100/50/100 ml) foi submetida a agitação sob Hs (pressão de balão) sob RT durante I h. A mistura resultante foi filtrada, β o filtrado concentrado sob pressão reduzida para proporcionar 4- [3- (4-cloro-2-fenílamino feníl) ~3-ox.o--prop.il] -N- (4~hidrox.i-cicloex.il) benzamida (190 mg).
Etapa D: Uma mistura de 4-[3-(4-cloro-215 fenilamino-feníl) -3-oxo-propil] -·#- (4-hidróxicicloexil) -benzamida {190 mg) e cloreto de met11 oxalíl (0,4 ml) em tc-.lu.eno (40 ml) foi aquecida sob refluxo durante a noite, então concentrada sob pressão reduzida. 0 resíduo foi dissolvido em MeOH (30 ml), adicio20 nou-se K2CO:i (100 mg) , e a mistura resultante aquecida sob refluxo durante 10 min. A mistura de reação foi então refrigerada, filtrada, e o filtrado evaporado sob pressão reduzida. 0 resíduo bruto foi purificado por TLC de preparação (EtOAc) para proporcionar êster metí25 lieo de ácido 7-cloro-3 -[4-(4-hidróxi ci cl oex i 1 ca rbamoil} - ben zil ] - 4 ~ox.o - 1 - f eni 1-1,4diídrc'qu.ínolina-2-carboxílico (composto 37) (63 mg) na forma se um sólido incolor. MS ~ 545 (M+K)*; PF 150,6-152,5’C, üt 1.11 zanda se ο ρ rosed i menta descx 1 to ant er i or mente e os materiais de partida apropriados, prepararam-se os seguintes compostos:
Éster metílico de ácido 7-cloro-4-oxa-l-feníl-3-[4(tetraidropiran -4-.i.lcarbamnil) •nenzíx] -1,4diidroquinclina -2-carboxílico {sólido amarelo) ; MS = 531 FE ~ 209-213’C (composto 45);
Éster metílioo de ácido 7-cloro-3- [4 - {2-hxdroxi --2 10 met íIprapíicarbamoi 1) -benzil) -4-oxo-l- fenx 1 -1,4dí idroqui nol i na - 2 - carbox.í 1. ico {só1i do cast anho claro); MS benzil)
51.9 [MeH] PE « 2 82 -286°C (composto
Ès c er me t, .í 11 co de ácido 7-cloro-3 -(4-etiIcarbamoil
- 4 -oxo-1 f eni 1 -1,4 -di idxoguinol ina - 2 carboxílico {sólido castanho claro); MS ~ 475
ΙΜ+ΗΓ; PF Ê s te r met í1i co benzil) -4 ,ca x box 1 x. .x co [Μ*ΗΓ; PE =
És ter me t ϊ .1. ico car bamai1)
209,0-212.t0C (composto 15};
de ác ido '? - c 1 oro - 3 - (4 - met i icarbamai 1 oxo-1 - funil -1,4 - dixdroquinollna - 2 {sólido castanha alaro) ; MS =- 461
203,3-205,5<!C (composta 14);
de ácido 7-cloro-3-[3-(2-hidroxietil-benzil ] -4 -oxo~ 1 - f eni .1 -1,4 -diídroquinolina-2-carboxílico (sólido castanho claro); MS
- 4 91 [Μ*ΗΓ; PF - 216-218<!C (composto 27); e
Ester metílico de ácido 3-(4-metilcarbamo.il-benzil) -4oxo-1-feni1 -1,4-díídroquinolina-2 -carboxílico castanho) (composta 13); MS - 427 PF '102 / 14 9
199,3 -200, 3*0’.
Exemplo 9: Síntese de éster metxlico de ácido 7~
Cloro-3(4-(2-hidróxi-atiísulfamoil) -benzil] -4-oxo-1-fenxlc-1,4-diidro-quinclina-2 S carboxílioo
A Síntese de éster metílico de ácido 7-cloro-3[4-(2-hidroxi-etxlsulfamoil} -benzilj “4-oxq-1-fenil1,4-diidro-quinolina -2-earboxílico (composto 65} foi realizada de acordo com o processe ilustrado no Esquema .0 16.

ESQUEMA 16
Adicionou-se uma solução de fluoretu de retrais butilamônio (1 M em THE, 0,26 ml) a uma solução de éster metílico de ácido 3-{4~ [2-(terc-butil-dímetil ·· sílaniloxi) -etílsulfamoil] -benzil) -7-cloro-4oxo-lfeni.l-l,4-diidroquinolina-2carboxílico (113 mg) em THE (5 ml) e a mistura resultante foi submetida a agitação 20 durante 1 h. A mistura de reação foi dividida entre EtOAc e água, a camada orgânica separada e a camaaa a~ quosa foi extraída com EtOAc (2x 25 ml) . Gs extratos orgânicos combinados foram lavados com salmoura, submetidos a secagem sobre Ma;:5Cg anídrico, filtrados e eva25 porados sob pressão reduzida. O resídua solido amarelo
1G3/149 foi purificado por flash cromatografia (DCM/MeOK, 90/10) para proporcionar éster metílico de ácido 7cloro-3-(4 -(2-hidróxi-etilsulfamail) -benzil] -4-oxo-lfenil 1,4-d.í idro-quinei ina-2-carbaxílico na forma de um põ incolor. MS - 527 [Μ+Η]’’; FF - 191,5-193,1*1·’Exemplo 10; Síntese de triflnoroacetato de éster metílico de ácido 7-Cloro-4~oxo-l-fenil-3-[4(piperazina -l-sulfanil) -benzil] »1,4diidroquínoliaa-2-carboxílico
A síntese de trifluoroacetato de éster metílico de ácido ?-cloro-4-oxo-1-fenil-3-[4-(piperazina-isu 1 f on.i 1) -beu zi 1) -1,4 - d ii droquinol ina- 2 - carboxí 1 i co (composto 66) foi realizada de acordo com o processo ilustrado no Esquema 17.

ESQUEMA 17
Adicionou-se uma mistura de TEA e DCM (1:4, ml? a éster metílico de. ácido 3-- (4- (4 - t-butoxicarboni 1. - p i perazi na -1 - sul f on i 1) ·· benzil ] - 7 - cloro - 4 oxo-l-fenil-l, 4-diidroquinallna-2-carboxí .liso (48 mg, 0,07 mmol) e a mistura resultante submetida a agitação sob ET durante 2 h. A mistura de reação foi evaporada sob pressão reduzida, e o resíduo sólido oleosa foi pu
104/149 r i f icado por flash cromatografia (DCM/MeOH, 100:0 até 90:10') para proporcionar êster metílico de ácido 7cloro-4-oxo -1-fenil3- (4- (piperazina · 1-sulfon.il ) benzil] 1,4-diidro -quinolina -2-carboxílico trifluo5 roacetato na forma de um pó incolor. MS ~ 552 J ; PE - 203,0-205,0“C.
Biscloridrata de éster metílico de ácida 7cloro-4-oxo-1-fenil- 3 -(4 -(piperazina-1 -sulfonil) ben z 11 ] -1.4 -di i d roqu 1 nol ina -2 - carbox í 1.1 co (pó incolor ) 10 (composto 67) pode ser preparado utilizando-se HC1 em 1,4-dioxana em vez de TEA em DCM. MS ~ 552 (M+Hj*; PF ~ 214,0 - 21S 5 * C ,
De forma assemelhada, procedendo-se como descrito ant.eriarme.nte e substituindo-se ©s reagentes a15 propriados, preparou-se o compost© éster metílie© de ácido 7-met 11 -4-oxo -L-fenil-3- [4- (pipera.ziua -1sulfonil) benzil] -1,4-diidro-[1,8] -naftiridina-2 carboxílico (composto 97}.
Exemplo 11: Síntese de éster metí lí oes de ácido 320 (4-(2-Bramoacetil) -benzil] -7-metil-4-oxo-l£enil~1,4-diidro-[1,8] naftizidiaa-2carboxílino
A síntese de éster metálico de ãcido 3-(4-(2bromoacetil} -benzil] -7-m.etil -4-oxo-1 -fenil -1,425 diidro-(1,8] naftiridina -2-carboxílico (composto 88) foi realizada de acordo com o processo ilustrado no Esquema 18:
105/149

EEQWMA 18
Adieionou-se tribrometo de tetrábutilamônio (72 mg, 0,15 mmol) a uma solução de êster metílico de ácido 3- (4-acetilbenzil) -7-metil-4-oxo-I-fenil-1,4-di.idro[1,8] naftiridina-2--carboxílico (53 mg, 0,12 mmol) em MeOH (8 ml) , e a mistura resultante submetida a agitação sob ET durante a noite. A mistura de reação foi concentrada sob pressão reduzida, e o resíduo bruto purificado por TLC de preparação (EtOAc/hexano, 10/90} para proporcionar 23 de ester metílico de ácido 3- [4 (2-bromoaceti1) -benzil] - 7-meti1-4 -oxo-1 - feni1-1,4 diidro-11,8] naftiridina-2-carboxilico na forma de um p 6 1 nco 10 r. MS - 5 0 7 [ Μ * H } * ; P F - 189,0-191,1C.
Exemplo 12í Síntese de éster metílico de ácido 3H-{2-Hidroxiacetil} -benzil] -7~metil~4-exol-ferdl-l,4-diidra-[1,8] naftíridina-2carboxílico
A síntese de éster metílico de ácido 3-(4-(2hidroxiaceti1} -benzil] - 7-metί1-4 -oxo-1-f eni1-1,4 d1idro-[1,81 nafti rIdina -2-carboxi1ico (composto 90) fox realizada conforme ilustrada no Esquema 19.
106/149

ERQUWA 19
Uma mistura de éster metílico de ácido 3-14-(2bromo - ace t11} - ben z i 1 ] - 7 - met i 1 - 4 oxo -1 - f en i 1 -1,4 5 díídro-[l,8] naftiridina -2-carboxílico (38 mg, 0.075 mmol) e .formata de sódio (28,5 mg, 5 eq>) em EtOH (15 ml) foi aquecida a 80<!C durante a noite. A mistura de reação foi concentrada sob pressão reduzida e filtrada. O resíduo bruto foi purificado por TLC de preparação (EtUAc/hexano, 60/40} para proporcionar 14 mg de éster metílico de ácido 3-[4 (2-hidrc-xi-acetil) -benzil; -7metil-4-oxo~1 -f enil-1,4-diidro-· [1,8] nattiridina-2oarboxílico na forma de um pô incolor. MS ~ 443 [M+Hj ’; PP ® 189,9-192,9C.
Exemplo 13; Síntese de éster metílico de ácido 3[4-(2-Hid.roxi -2-matilpxepilsulfona®oiX) benzil] -4«oxo-?-cloro -1-fenil -1,4díidroquinolina -2-carboxílico
Preparou-se éster metílico de ácido 3-(4-(229 hidroxi- 2-metilpropilsulfonamoil)benzil) -4 -oxo-7cloro-1-fenil-1,4-di ídroquinolina-2-carboxílico (composto 85) conforme ilustrado no Esquema 20.




ESQUEMA 20
Etapa Αϊ A uma solução de cloreto de 4~form.ilbenzeno sulfonil {1,0 g) em MeQH adicionou··se NaHCOB (0,435 g) # seguido por 1 - amino 2 -met. i Ipropan - 2 -ol (0,565 g) . Ά mistura de reação foi. submetida a agitação durante 1 h sob RT, então filtrada através de Cellte e o solvente removido sob pressão reduzida para, propor
108/149 cíonar 4 - form! 1 -N- (2 - hidroxi ·· 2 -met i 1 prop 11) ··benzene sul fonamida.
Etapa Bi A uma mistura de l-\4Clorc-2fenilaminofen.il )etancna (0,401 g) e 4-formil-N-(2hidroxi-2-met ilpropil) -benzene sulfouamida (>J,42u g; em MeOH (10 ml) adicionou-se Na.OH (1,39 mi, 2 N, aq) , e a mistura submetida a agitação durante a noite sob RT0 produto foi concentrado sob pressão reduzida, lavado com EtOAc, Hs0 e salmoura, então submetido a secagem sobre JíasSO*. O produto parcialmente purificado foi cromatog rafado em silica anídriua (DCM durante 0~.s. v min, então MeOH/DCM durante 11-30 min) para proporcionar sulfonamida de 4-[3-(4-cloro-2-fenilam.inofenxl) -3oxc-propeni11 ~N-(2-hid r oxi- 2-metiIpropi1) -benzeno (0,540 g} .
Etapa C: A uma solução de sulfonamida de 4-(3(4 - cloro-2 - f eni laminof eni 1) -3-oxo-propeni.l] -14- (2hidroxi-2-metilpropiI) - benzene (0,.535 g) em EtOAc (100 ml) e StOH (50 ml) adicionou-se Pt(XV)O3, e a mistura submetida a agitação sob H2 durante 1.5 h, O pioduto foi filtrado e concentrado sob pressão reduzida para proporcionar sulfonamida de 4-[3-(4-cloro-2-tenrlaminofen.il) - 3 -oxo-propil] -E- (2-hidrox.i-.2- met ilprop.il) -benzeno na forma de um sólido oleoso amarelo.
Etapa Bí A sulfonamida de 4-í3~(4-cloro-2feni1aminofeni1) - 3-oxo-propil 1 -H-(2-hidrox i-2-met11propil) -benzeno (0,150 g) adicionou-se toxueno (1b ml), seguido por cloreto de metil oxalil (0,6 ml), e a
109/149 mistura foi aquecida para 80“C durante 3 h para proporcionar éster metilíco de ácido N- (;>-cloro-2 - (3~ .[4- (,2.·· hídroxi-2-metilpropilsulfamoil) -feníl] -propxonil} fenil) -N-f.en.il· -oxalâmico. 0 produto foi usado sem qualquer outra purificação.
Etapa E: A uma solução de ester metílico de á~ cído N- {S-cloro-2-{3- [4- (2-h.idrox.i-2-metil.prapilsulfamoil) -fen’ll·] -propionil) -fenil) -N-fenil-oxalâmico em toluene adicionou-se K2CO3 ; 0,073 g) em MebH 110 ml), e a mistura foi submetida a agitação a 80eC durante 2 h. 0 produto foi lavado com EtOAc, Hs0, e salmoura, submetido a secagem sobra Na/SO.j, e cromatogratacu em silica anídríea com 3% MeOH/DCM. O produto foi finalmente purificado em silica anídrica com 5% MeOH/DCM para proporcionar éster metílico de ácido 3-(4-(2hi d r oxi - 2 - me 11 Ipropi 1 sul f onamoil) - benz ill - 4 - 0x0 - 7 ~ cloro-1-fen.il -1,4 ••diidroquinolína^S^carboxíl.ico (composto 85) (Ms-H 855,1; PF - 240, 0-242,6 C? .
Exemplo 14; Síntese de 3-[4~(2-Eidr<?x.i-2~m.etilpro~ pilsulfonamaillbenzil] -4-oxo-7-cloro-lfsnil-2-acetil-1,4 -diidrequinoli»a
A síntese de 3-|4~(2-nidroxr-2met i ipropiIsulf onamoi 1) benzil] -4 - oxo- 7 - cloro-1 - f e.ni 1 2-acetil-1,4-diidroquinolina (composto 88} foi realizada tal como ilustrada no Esquema
110/149

ESQUEMA 21
Etapa A: A ácido pirúvico (12,2 g) adicionou-se metil-a,a-dicloromet11 éter (12,3 ml} lentamente durante 20 min. A mistura de reação foi então aquecida para 50*0 durante 30 min., e refrigerada para a RT durante a noite. Adicionou-se lentamente uma alíquota adicional de metilα-diclorametil éter >4 ml) , a a mistura foi aquecida para 50*C durante 2 h, então refrigerada para 2 0<:C< Os materiais de partida que não reagiram foram removidos por evaporação sob pressão reduzida para proporcionar cloreto de 2-oxoprupion.il.
A 4 - [3 - (4 - claro- 2 - f enilam.í nof enil} 3 -oxo-pro38 pH] -M- (2-.hldrnxi-2-metil-prap.il} -benzenossulfonamida (0,138 g) em toluene (10 ml) adicionou-se cloreto de 2oxao.ropion.il (0,3 ml), e a mistura aquecida para 120*C durante 3 h. para formar N-(8-claru-2-{3-[ã~{2h.idruxi·· 2-metilpropilsulfama.il) -fenil] -prapionil} -fenil) -220 oxo-N-fenil -propionamida, que foi usado sem, qualquer outra purifícação.
Etapa B í A N~ (3 - cloro - 2 - { 3 - [ 4 - (2 - h Idroxi 2 111/149 me til - prop! 1 - sul f amoi 1; - f eni 1 ] propiom 1} - f eni.* - 2 oxo-N-· fenil-propionamida era MeOB adicxonou-se -10,.067 g) , e a mistura de reação foi aquecida para 8t>C durante 2 h. Acrescentou-se uma. alíquota adicional de K2CO3 (1 g), e a mistura aquecida durante um adicional de 2 h. 0 produto foi deixado esfriar para a RT, então coletado em Et0Ac/H?0, lavado 3x com EtOAc, salmoura, e submetido a secagem sobre NasSO^. O produto bruto fui então croraatografado em silica prep (5% MeQH/DCM) para p r opor c i on a r 3 - í 4 - 12 - n i dr ox i - 2 - me t .11 - prop 11 sulfonaraoi.1.) benzil] -4 -oxo-7-cloro -1-i em. 1-2 -anetii
1f4-díldroqulnolina (composto 88). MS - 5 38 IM+Hj bn forma assemelhada, procedendo como exposto anteriormente e substituindo-se os reagentes apropriados, preparou-se o composto 4 - (7-cloro-4-oxo-1 tenil- 2prop ionil -1,4 -di idroqu inol in - 3 i Imet i '1) -N - (2 - hidróxi 2-metilpropil) -banzenossulfonamida (composto 86, M+H 553) .
Exemplo 15 s Síntese de éster metili.cn de ácido 7Cloro-3-[4-H-hidroxipiperidina-l-sulfoníl) benzil] -4-oxe-1-fenil-1,4-di idroquinolina-2carboxílic© (composto 69)
Etapa As A cloreto de 4-forrai1 -benzenessulfenil (1,.0 g) i.n DCM (10 mi) adicionou-se 4-hidroxipiperidina (0,884 g) , seguido por NaKCOj (10 ml, a.q sat'd) e a mistura foi submetida a agitação durante 2 h sob ET. A camada aquosa foi lavada com DCM (2x 25 ml) , e as partes orgânicas combinadas lavadas com salmoura, submetí das a secagem sobre Na2SO,s, filtradas, e o soivente removido sob pressão reduzida para proporcionar 4-(4hidroxiniperidina- l~suifonil}benzaldeídc> 0 predate ».o.i purificado em silica anídrica (10% MeOH/DCM). bUH 269.9
Etapa 8t A i-(4-cloro-2-.fenilaminofenil)etanona (0,500 g) e 4-(4-hidroxip.iperidina-l·· sulfonil)benzaIdexdo (0.54 8 g) em MeOH (10 ml) adicionou-se MaOH (1,65 ml, 2 U aq) , e a mistura deixada sob agitação durante a noite sob RT, O produto soltou-se da 10 solução, e foi coletado em um funil de vidro sínterizado, enxaguado com água, e recolhido em MeOH. A água e
MeOH foram removidos sob pressão reduzida para proporc tonarem 1 - (4 - c 1 oro ··· 2 - f en i I amino f en i 1) - 3 ~ 14 - (4 hidroxipiperídina-l-sulfonil) -fenilIpropenona. O pro15 duto foi reduzido tal como descrito no Exemplo 13, Etapa C, para produzir 1 - (4-eloro-2-fen.ilamino-fenil) ~3~ [4 -(4 -h idroxipiperidina-1-su1f oni1) -feni11 propan-1 Etapa C» A 1-(4-cloro~2-fenilamino~fen.il) -32 0 [ 4 ·· (4 - h idroxipi per idi na -1 - sul f oni 1) - f ení 1 ] propan··· 1 -ona (0,200 g) em toluene (20 ml) adicionou-se cloreto de met 11 oxalil (0,64 ml) * A mistura toi aquecida para
120’C durante 3 h, então submetida a agitação durante a noite sob RT. O produto, éster metílico de ácido M-(525 cloro-2-{3- [4- (4-hídroxipiperidina-l~su.lfoníl) -fenil]
-oropionil) -fenil) -N~fenil-oxalámxco, foi usado sem qualquer outra purificação.
Etapa »í A éster metílico de ácido N- (5-cloro113/149
2- {3» (4- (4--hídroxípiperídina·· 1 -sulfcn.il} -fen.il) prop.io.nil.} -fenil) -M-feníl-axalâmico {0,40 g) em MeOH (15 ml) adi.cionou-se K2CO3 (0,096 g) , e a mistura fol aquecida para 80*C durante 1.,5 h, então concentrada. 0 residue foi. recolhido em EtQAc/H20, e a camada aquosa lavada oom EtOAc (2* 25 ml). As camadas orgânicas cmbinadas foram submetidas a secagem sobre Na^SO*, solvente removido sob pressão reduzíca. O resíduo foi cxomatografado em. silica anídries (20% EtOAc/hexanoa 01C 5 min; 80% EtOAc/hexanos 6-10 min; 100% EtQAc 11-40 min) para proporcionar ester metílico de ácido 7-cloro3- [4- (4-h.idroxipiperid.iua-1 -sulfonil) -benzil] -4-oxo1-fenil-1,4 -díidroquinolina-2-carboxílicc (composto 6.9). M+H 567; PF - 170,0-171.0*0.
De uma forma assemelhada, procedendo-se como descrito anteriormente e com substituição de reagents apropriada, preparou-se o composto 3- (4- (4-hídroxxp.':.per id ina -1 - sul f on 11} benzi 1 ] - 7 - met i.l - 4 oxo -1 - f en il 1,4 diidro(l,«] naftiriãina-2··carboxílicc met.il éster (com20 pOStQ 9:9} .
Exemplo 16 í Síntese de éster metílico de ácido 7Cloro-3-[4-(3-hidraxípirrolidine-l-sulfonil) -benz ill -4 -oxo-1-feni1-1,4-diidroqu inol1na2-carboxilico (composto 87)
2b Etapa As A cloreto de 4-fcrmílbenzenossulfonil.
(1,0 g) em MeQH (10 ml) adicionou-se MaHCO-, (0,425 g, s) , seguido por S-{-) -3-h.idroxipirrolid.ina (0,533 g) , e a mistura foi. submetida a agitação sob RT durante 1
114/149
h. O produto foi filtrado através de celite, concentrado para proporcionar um õleo, e cromatografado em silica (3% MeOH/DCM) parti proporcionar 4-(3-hidroxip.írrolidina-1-sulf.onil} -benzaldeido (1,14 g) .
Etapa Bi A i-(4-cloro-2-fenilaminofenil)etanona (0,500 g) e 4 (3-hidroxipírrol.idine-l·sulfonil) -benzaldefdo (0,519 g) em MeOH (10 mi) aoicionou-se NaOH (1,68 nil, 2 N aq) , e a mistura foi submetida a agitação durante a noite sob RT. 0 produto vermelho alaranjado soltou-se da solução, e fo.i coletado em um funil de vidro sinterizado, enxaguado com agua, e coletado em MeOH. O produto foi concentrado, então cromatografado em silica anidríca (100% DCM 0-10 min; 2% MeOH/DCM 11-30 min) para proporcionar l-(415 cloro-2- fenilamino-fenil.) - 3 (4- (3-hidroxi-pirrolxdina-
1- sulfoníl) -fenil] -propenona (0,588 g) .
Etapa Cs Procedendo-se conforme descrito no fíxemplo 13, Etapa C, hidrogenou-se 1-(4-cloro-2feni.lamíiio -fenil) -3-- -4-(3-hidroxi -pxrrolrdina-120 sulfon.il) -fenil] -propencna para produzir 1-(4-cloro··
2- funílamino -fenil) -3- (4- (3-h.idroxipirrolidína-1aulfonil) fenil] -propan-l-ona na forma de um sólido oleoso, amarelo.
Etapa Di A 1-(4-cloro-2-fenilamino-fenil) -325 (4 (3-hldroXÍpirrali.di.na-l - sulfon.il) -fenil] -propau- -1ona (0,150 g) em tolueno adicionou-se cloreto de metil oxalil .(0,6..ml}.. A mistura de reação foi aquecida para <W“C durante 3 h, então submetida a agitação durante a
11.5/149 noite sob RT para produzir éster metílico de ácxdo N(5-cloro-2~{3- Í.4 (2-hidroxipiroolidina-l-sulfonil) fenil]propion!!} -fenil) -N-fenil-oxalâmico, que foi usado sem qualquer outra purificação.
Etapa Ex Seguindo-se o procedimento exposto no Exemplo 14, Etapa B, ciclizou-se éster metílico de ácido H-(5-cloro-2-(3-(4-(2-hidroxipyroolidine-l-sulfonil) fenil] propionil} -fenil) -N-fenil·-oxalâmico para formar éster metílico de ácido 7-cloro-3-(4-(3hidroxipirrolidina-1-sulfonil) -benzil! -4-oxo-1-fenil:1 (4-diidroquinollna-2-carboxílico (composto 87), o produto foi cromatografado em silica anídrxca (3% MeCH/DUM 0-10 min; 10% MepH/DCM 11-20 min) t então recromatografado em silica pura com 5% MeOH/DCM. 5t3;
PF « 135,0-136,0*C.
Exemplo 17: Síntese de 2-Acetil-7-cloro-3-[4-(4hidroxipiperidina-l-sulfonil) -benzil] -1fenil-IE-quinolin-4-ona (composto 70)
Etapa Ax A 1 - (4 - cloro- 2 - f enilamino- fenil) - 3 [4-(4-hidróxi-piperidina-1-sulfonil) -fenil] -propan-1 ona (0,200 g) em toluene (20 ml) adicionou-se cloreto de 2-oxopropanoil >0,427 g) . A mistura de reação for aquecida para 12QeC durante 3 h, então submetida a agitação sob RT durante a noite para produzir M-(5-cloro·· 2-(3-(4-(4 -hidroxlpiperidina-1-sulf onil) -f eniX] propio-nil) -fenil) -2-oxo-N-fenil-propionamida, que foi usado sem qualquer outra, purificação.
Etapa 8: A N-(5-cloro-2-{3-[4-(4116/149 hidroxipiperídina-1 · sul fonil) -fenxi] -propioni* ?
fenil} -2-oxo-N-£snil-propionamide em hteOH adicionou-se K3COi (0,096 g) , e a mistura foi aquecida para 80*C durante 1,5 h, então concentrada. O resíduo foi recolhido em EtOAc/-H3O, e a camada aquosa lavada com EtOAc ml) .. As camadas orgânicas combinadas foram submetidas a secagem sobre Na;;SO..<, e o solvente removido sub pressão reduzida. O resíduo foi cromatografado em silica anídrica (20% EtQAc/hexanos 0-5 min; 60% EtO10 Ac/hexanos 15-20 min; 100% .EtOAc 22-35 min) para proporcionar 2 -acer.il -7-cloro-3- [4 - (4-hidroxipiperi-dina 1- sultoni1) -benzí1] -1-feni1-1H -quinolin-4 -ona {composto 70} < PF « 145,5-147,2*0; M*H 551.
De forma as some .1 hada, procedendo-se conforme exposta anteriormente e substituindo-se os reagentes apropr i ados, preparou - se o composta 2 - acet il - 7 - cl aro - 3 [4~. (4-metilpiperazína-l-sulfonil) -benzil] - 1-fenil- 1H-quinoli n-4 - ona {compos to 72, PF ~ 206,3-2 08,0° C; Μ 550}
Exemplo 18: Síntese de éster metílico da ácido 3(g-cloro-pir.idin-3-imstíX) -7-metil-4-oxo-lfenil-l,4-diidro[l,8] naftiridína-2-carboxílico (composto 83)
Etapa A: Uma solução de 2-cloro-3-acetii-6·· me ti ipiridi na (6,0 g) in 1,4-dioxana í6« ml) fox c®cx* nada cam ácido cânfora sulfônico (20,7 g) e deixou-se sob agitação em um tubo vedado até ficar homogêneo. A mistura foi aquecida para 70eC, adicioxiou-se anflina
117/149 (5,0 g) , e continuou-se cam o aquecimento até todos cs sólidos de precipitação serem xedrssolvicos. Monitorouse o croqresso da reação por resfriamento rápido oe a mostras em Et.OAc/NaHCO3 (sat'd aq) e examinando-se por 5 TL-C (1:2 EtOAc/hexana) . A reação foi concluída depois de 1,5 h, e foi esfriada rapidamente com EtOAc/NaHCQj (ear'd aq), submetendo-se a agitação até cessar o desprendimento de gás < A camada organica foi lavada com MaHCOj (sat'd aq;, filtrada, concentrada, e croraatogra18 fada em silica (1:4 EtOAc/hexano) para proporcionar 2f eni lam ina - 3 - ace t i 1 - 6 - pi r idi na (6,2 g) na t erma de um solido amarelo. M+H - 227,
Etapa B: Dissolveu-se z-fenilamino-3-acetil-6piridina (1,0 g) em MeOH (20 ml) utilizando-se um soni15 cador, seguida por 2-cloro-5-form!iprridina (1.1 g) e
NaOH (2 M, 3,5 ml), A mistura foi deixada, sob agitação sob RT, formando-se um precipitado vermelho.. Q produto foi filtrado, lavado cam MeOH/HjO (2:1, 10 ml), e submetido a secagem sob vácuo para proporcionar l-(620 metil-2-fenilamino-piridiu- 3-il) -3·· (6-cloropiridin-311) -propenona (1,45 g) na forma de um sólido laranj a/verme1ho < M<K - 3 50.
Etapa Cx A uma solução de 1-(e-raetíl-2fenilamino-piri.din 3 -11) - 3 - (6 -cloropiridin-3 11) propenona (1,15 g) em EtOAc quente (40 ml) adicionou-se Ptü3. (0,04 g) , depois do que o vasa foi evacuado e preenchido cam (2x) sub pressão de balão. A mistura ae reação foi aquecida para 60*C durante 2,5 h, então re~
118/149 frigerada para a RT, filtrada através de celíte, lavada cosa THF/-EtOH, concentrada, e cromatogratada em aplica (EtQAc/hexano 1:1) para proporcionar '1 -· (6-metii -2fenílamino-piridin-3-il) -3-· í6-cloropiridin-3-il} propan-l-ona (1,0 g) na forma de um sólido amarelo. M*H ::: 352.
Etapa ΣΗ A uma solução de 1- (6-metil-2fenilamiuo-piridin-3 -il) -3 -(6--cloropirídin-3-il) propan-l-ona (0,9 g) in toluene (5 ml) e THE (10 ml) 10 adicionou-se cloreto de metil oxalil (0,56 g), e a mistura foi aquecida para 80*0 durante 2,5 h. O produto foi concentrado para uma solução vermelho escuro de ester metílico de ácido 8-{-3-[3-(6-cloropiridin-3-il) prop!anil] -6-metil -piridin-2-il} -E-fenil-oxalâmico em tolueno, que foi usada sem qualquer outra purificação.
Etapa Es A uma solução de éster metílico de âcido N-(-3-[3-(6~cloropiridin-3-il) -propicnil] -6~ metil --piridin-2-il} -K-fenil-oxalâmico em tolueno adi20 cionou-se MeOH, e a mistura colocada, em usr banho quente preaquecido a 90*0. Adicionou-se KpXh (0,9 g) ? que produziu, uma mudança de cor imediata. A mistura foi refrigerada para a RT, diluída com EtOAc, e lavada com bid (0,3%, em peso, aq, 2x) . camada orgânica foi .25 submetida a secagem sobre MgSO4, filtrada, e concentrada» O produto foi dissolvido em EtOAc quente, e deixado esfriar para a RT. O precipitado resultante foi filtrado e lavado com EtOAc para proporcionar éster metílico de ácido 3-(6-cloro-piridin-3-imetil) -?-metil-4-oxo-lfeni1 -1,4 - diidro [1> Si naftiridina-2 -carboxílico (composto S3 5 (0,14 g) na forma de um solido bronzeado. M+H :=: 420, PF - 220,0-220,5.
Exemplo 19s Síntese de áster metílico de ácido 3(Piridin -3-imefcil) -7-metil-4-oxo~l-fenil1,4~diidro[1,81 -naffciridina -2-carboxílico (Composto 84)
Etapa Aí Adicionou-se 1-(6-metil-2--feBÍiaminopiri.din-.3-il) -3 - (6-cloroplridin-3-il) -propenona (0,30 g) a EtOH (3 ml} , seguido por THE (6 ml) , toluene (6 ml), EtQH adicional (cerca de .2 ml), e Pd/C (10%, 0,02 g) . O vaso foi evacuado, então reenchido com. H2 (2x) , envelhecido durante 1 h sob RT, então a 60’C durante 3 h. A mistura foi deixada esfriar para a RT durante a noite, então adicionou-se uma alíquota adicional de Pd/C (10%, G.02 g) , a mistura foi aquecida para 75*C durante 8 h, então deixada esfriar para a RT durante a noite. G produto foi filtrado através de celite, lavado com EtOH, concentrado, e cromatografa.de em silica (1x1 EtOAc/hex para 5% MeOH/EtOAc/hex) para proporcionar 1(6 -met il - 2 - fen 11 ami.no - p ir i dí n - 3 - il) - 3 -pir idin - 3 ilpropan-1-ona (0,14 g) na forma de um óleo amarelo, EUH =:: 318 .
Etapa Bt Procedendo-se conforme exposto no Exemplo 13, Etapas Ώ e E, produziu-se o composto enter metílico de ácido 3-{piridin-3-imetil) -7-metil-4~oxo1- fed1-1,4-diidro-[1,8] -nafti r idina- 2 -carbox ί1ico
3.20 / 14 3 (composto 84 j (28 mg). M-í-M - 386.
Exemplo 2£k Síntese de éster metílico de ácida 3~ (6 - sãs t axi ca rboni 1 -piridin - 3 > i Ime 111) ~ 7 ~ ^etil-4-oxo-l-fenil-l,4-diidraIl, 8] naftiri5 dina-2-carbaxílico (Composto 100)
Etapa At Colocou-se em uma bomba uma mistura, de 1 - (8-metil -2 -fenilamino-pi.ri.din-3-.il) -3- (6 cloropiridin-3 il) -propan-l-ona (1, â g) ,. Et;;N (lfu ml) f MeOl-l (30 ml.) , e (R-BINAR) PdCl;< (43 mg). Purgou-se 10 então o ar da bomba., pressurizou-se para 344,7 kPa (SO pai) com monóxido de carbono, e aqueceu-se para 100 durante 3,25 h. A bomba foi então refrigerada para 50*C, aberta, e deixou-se esfriar para a ET. O produto sólido foi recolhido em DCM, a cristalizado a partir de 15 DCM/MeüH para proporcionar éster metílico de ácido 5[3- (6-metil-2-fenílamino-piridin 3-i 1) -3-oxo-propil] piridina-2-carboxílico (1,7 g) na forma de um solido amarelo. M-s-H - 376.
Etapa Bs Dissolveu-se éster metílico de áciuo 20 5-[3-(6-metll-2-fenílamino-piridin-3“il) -3oxo-prop.il] •pirldina-2-carboxilico (1,7 g) em tolueno (15 ml) e THF (30 ml) sob Ns a 80ftC, Adicionou-se cloreto de monometil oxalíl (2,2 g) , e a mistura foi aquecida para 80’C durante 3,5 h, tornando-se de uma cor vermelho ôs.25 curo/preto. A mistura foi refrigerada para a RT e concentrada para um sólido, então dissolvida em MeOH e aquecida novamente para. 80 &C. Adicionou-se RzCOj (0>-8 g) , a mistura foi aquecida para 80&C durante 30 min,
121/149 acrescentou-se uma parte adicional, de K^CCh >0,8 g; , e a mistura fui aquecida outra 1 h. 0 produto tor coletado em EtOAc e Η?0, a fase organíca .lavada -<.om .HsQ (lx) t submetida a secagem sobre MgSCg, filtrada, con5 centrada, e cromatugrafada em silica (2:3 EtOAc/ hexa.ru? para 100% EtOAc} para proporcionar éster metílico de ã c ido 3 - (6 - metoxí carbon! 1 -pi r ídin - 3 - timet 11} - 7 ·· met. x 1. ·
- oxo -1 - f en 1 1-1,4- d i i d r o [ 1,8] - n a f t .1 r i d i na - 2 carboxílico (composto 190} (0,38 g) na forma ae um so10 lido amarelo. M-*H ~ 444, PF - 177,9 - I?8,0”C.
Exemplo 21í Síntese de ester matilxeo de acxdo 3(6»M«tanossulfonil-piridin-3-imeti1) -7metí1-4-oxo-1-fanil-1,4-diidroí1,8] naftiri~ dí na~2 -earboxí1ico (Composto 92)
Etapa Aj Dissolveu-se 1-(Smetil-2-tenxlaminopiridin-3-11 5 -3- {6-cloropiridin-3-.il) -prupa.n- 1-ona (0,42 g) em NMP (3 ml), adicionou-se NaSMe (0,34 g) , e a mistura foi aquecida para 150 C em um mxcroondas durante 1 h. O produto foi coletado em EtOAc/H^O, a cama20 da orgânica, lavada com. LiCl (2x, 0,3%, em peso, aq; , submetido a secagem sobre MgSO*, filtrado, e concentrado na forma de um óleo vermelha para proporcionar 3-(6met capt op iridin - 3 11) -1 - í 6 -me t í 1 - 2 - f eni 1 - ami nopir id an 3-1.1} -prupan-l-ona (0,4 g) .
Etapa Dissolveu-se 3 (6-mercapcopiridin-3il) -1- (6-met.il-2- fenilaminopirídin-3-il) -propan.-l-o.ua (0,.4 g} em D.MF {5 ml} , adicionou-se iode to de metila (0,05 ml) a, seguido por K3COà (0,138 g) . A mistura de
122/149 reação fed deixada assentar sob RT durante 2,5 h, então trabalhada era EtOAc/H/J. A camada orgânica foi lavada cora Li.Cl (2x, o, 3%, peso, aq) , submetida a secagem sobre MgSOs, filtrada, concentrada, e cromatogratada sobre silica (12 g, 1x2 EtOAc/hexano) para proporcionar
- (6 - me t i 1 - 2 - f en 11 ami no - p i r idin - 3 - i 1) - 3 ~ (6 - me t i 1 - sulfanil-piridin-3-íl) -propan-l-ona (0,23 g) > «=
264>
Etapa Cs Dissolveu-se 1 - (6-raetil-2-fenilami.nopiridin-3-iI) - 3 -(6-met11sulfani1-piridin-3 -11) propan-l-ona (0,23 g) em DMF (5 ml), e adicionou-se Oxone (0,32 g) . A mistura foi submetida a agitação sob RT durante a noite< Acrescentou-se uma pequena quantidade adicional de Oxone, e a mistura foi submetida a agitação um adicional de 3 h, então trabalhada com EtOAu/HsO, a camada orgânica lavada cora LiCl (2x, 0,3%, era peso, aq) , submetida a secagem sobre MgiSO*, e concentrada para proporcionar uma mistura 2:1 de l-(6-metil-
- f enilamino-piridin- 3 -11} -3 - (6 - met ilsulf onir -pi.ri.cin3-il) -propan-l-ona e 1- (6-metil-2-fenilamino-piridin-
-il) -3 -{6 -metilsulftail-piridin-3-11) -propan-1-ona (0,22 g) na forma de um óleo vermelho-laranja, que foi usado sem separação, Mdí » 396 (sulfcn.il) e 380 fsulfinil) Etapa Dí Dissolveu -se uma mistura 2x1 de l-(6me t i I - 2 - f en i 1 am ino -piridin-3 - i 1) 3 - (6 - me t i 1 su 1 f on 11piridin-3-.il) -propan-l-ona e 1- (6-metil-2-fenilarainopiridin-3-il) -3 - (6-metil-sulf.inil -piridin-3-.il)
121/149 prepan--1-ona (0,22 g) em TKF (3 ml) e toluene (1,5 ml)» e adicionou-se cloreto de monomet11 oxalil (0,27 g) . A mistura foi aquecida para 80’C durante 2,5 h, concentrada para urn semi-sólido, recolhida em MeGH, e adic.ro·· nou-se K3CO3 (0,22 g) > Esta mistura foi aquecida para 80!íC durante 15 min, então refrigerada para a RT> p produto foi trabalhado em EtOAc/H.aO, a camada orgânica lavada com LiCl (2χ, 0,3%, peso, aq) , submetida a secagem sobre MgSO.«, e purificada por TLC de prep (1:1 EW~
Ac/he.xano) para proporcionar éster metílico de ãcido 3(8 - met anos sul. f oni 1 · p i r idi n- 3 - imet.il) - 7 - met i 1 - 4 oxo -1 fenil-1,4-diidro-[1,8] -naftiridina-2-carboxilico (composto 92) (8 mg) na forma de um sólido amarelo. tUH 484, PF - 217-218*0,
Exemplo 22t Bíntese de éster metílisa de ácido 3(5-metanassulfani1-piridle-3-imeti1) ~7~ metil“4-oxo-1-femil-l,4-diidxo-[1,8) naftiridina~2-oarbox£liao (Composto 34)
Etapa A; Procedendo conforme exposto no Exemplo
21, Etapa. D, fez-se reagir 1-(8-metil-2-fenilam.inopi.rid.in - 3 il) - 3- (6-metilsulfanil-piridin-3 -11) prapan-l-ona (0,2 g) com cloreto de metil oxalil (8,27 g) e K.3CO5 (0f2 g) em toluene (2 ml) e THF (4 ml) para formar éster metílico de ácido 3- (8-metanossulfanil · piridin-3-imetil) - 7-metil-4-oxo-1-fenil-1,4-diidro[1,8] -na£tiridina-2“carboxílíco (composto 94] (0,12
g) .. M+H 4.32, PP « 185,0-186,5*0.
Exempla 23: Síntese de ester mstílicc de ácido 3124/149 (6~metanossulfinilpiridín-3-ilmetil) -7metil-^oxo-l-fsnil-l, 4-diidroU,8] naftiridina-2-carhoxílico (Composto 98)
Etapa A; Dissolveu-se o composto 94 (9,12 g) em
DMF (5 ml), adicionou-se Oxone (0,14 g) , e a mistura foi submetida a agitação durante 30 mm. 0 produto roí separado entre EtOAc e HSO, a camada orgânica lavada com LiCl (2x, 0,3%, peso, aq} , submetida a. secagem sobre MgSO..., e purificada por TI»C prep (1;1 EtOAc/hexano?
para proporcionar éster metílico de ácido 3--(6metanossul f inil pirldin-3-imet 11) -7-metrl -4 -oxo-1 fenil-1,4-diidro-(1,81 -naftiridina-2-carboxílico (composto 96} (5 mg) na forma de um sólido amarelo. M+H 44 8. ............ ........
Exemplo 24 s Síntese de estar metílíoo de ácido 3CS-dimetiXcarbamoil-piridin-S-ilmetil} -7~ met il-4-oxo-1-fenil-1,4-diidro(1,81naftiridina-2-carboxílico (Composto 103)
Etapa A; A éster metílico de ácido 3-(6-20 metoxicarbonil -piridin -3-ilmetil) -7-metil-4-oxo-lfenil -1,4-di idro-[1,8] na f ti.ridina- 2-carboxí1ico {cora posto 100} (0,2 g) em THE (10 ml) adicionou--se LiOH'HsO (0,06 g) and H?O (2 ml), e a mistura foi submetida, a. agitação sob R.T durante 1 h. 0 produto foi recolhido em
2S EtOAc, lavado com H;;0 e HC1 (2M, 2 ml), filtrado, e trabalhado com EtQAc e H2O. A camada orgânica foi submetida ά secagem sobre MgSO$, e concentrada para um õleo amarelo. O óleo foi recolhido em EtOAc, .lavada com
H?O, submetido a secagem sobre MgSO«, e concentrado para um sólido amarelo para proporcionar éster metílico de ácido 3 (6-carboxi-piridin - 3-i Imet 11) - 7-met í 1-4oxo-1-fenil-114 -diidro (1,8] naftiridina-2-carboxílico (C,11 g) . - 430 >
Etapa Bi A éster metílico de ácido 3-(ucarboxi- pirídin- 3 -11 met 11) - 7 -metrl-4 -oxo-1 feni 1-; 3 diidro-[1,B] -naftiridina-2-carboxílico (0,095 g) em THF (6 ml) adicionou-se BOP (0,11 g) , seguido por DMA (0,15 ml, 2M em THF) e Base de Hunig (etil-diisopropilamina) (0,042 ml). Sólidos começaram a precipitar- set adicionou··se DCM até todos os sólidos se dissolverem, e obteve-se uma a mistura de reação homogênea. A mistura foi submetida a agitação sob RT durante 1 h, então acrescentou-se uma alíquota adicional oe BOP, base de Hunig, e DMA, e a mistura foi submetida a agitação sob RT durante a noite.. O produto foi trabalhado com EtOAc e Hs0, lavado com NaHCO, (aq) e H:iQ, e submetido a secagem sobre MgSG*< 0 produto foi concentrado, aplicado a uma coluna de gel d.e silica com EtOAc, e submetido a eluição com EtOAc/MeOH, e concentrado para proporcionar éster metílico de ácido 3- *.6-dime t i 1 · carbamoi 1 -pirídi n. · 3 i Ime ti 1) 7 -met i 1 - 4 - oxc 1 fenil 1 ,· 4 -diidro- (1,8] naf tiridina -2-carboxílico (composto 103) (80 mg) na forma de um sólido amarelo pálido. M*H - 457? PF « 23S,O-236,OeC.
Exemplo 25s Síntese de éster metílico de ãcidc 3(5-etilcarbamoil-piridin-3-ilmetil) -7-metil126/3.49
4-oxo-l-fenxl-l,4-diidro [1,8] naftiridina-Scarboxxlíca (Composto 101)
Ά deter metílico de ácido 3 ·· (6-carboxí-piridin3-ilmetxl) -7-metíl-4-oxo-l-feníl~l,4-diídro- [I, ?>] naftirxdina-2-carboxílico (8,11 g) esi DCM adicionou-se BOP (0,14 g) , seguido por etilamina (0,03.6 g) e Base de Hunig (0,06 ml), e a mistura fed submetida a agitação sob RT durante a noite. O produto foi recolhido es: EtOAc e HSO, lavado com NaHCO., (aq) , e submetido a secagem sobre M.gEQj, então filtrado, concentrado, e purificado por TLC prep utilizando-se 100% EtOAc, para proporcionar éster metílico de ácido 3-(6-etii.carbamoxl-piridm3 - i Imet il} · 7 -met 11 - 4 - oxo -- 3. · fenil - X, 4 - d 1 idro 11,8 ] naftiridins-2-carboxílico (composto' 101) na forma de um solido incolor. - 457.
Exemplo 25 s Síntese de éster metílico de ácido 3(6-Metoxipiridin-3-ílmetil) -7-metí1 -4-cxo1-fenil -1,4-dxídrc[X,8] naftiridxna -2» carboxílica (Composto 102)
Etapa A; A X (6-meti.l-2 -fenilaminopiridín~3~il)
-etanona (0,5 g) in MeOH (12 ml.) adicionou-se carboxaldei do de 6-metoxí-3~piridina (0,5 g) , seguido por MaílH (2 M, 1,75 ml), e a mistura, foi submetida a agitação sob RT durante a noite. O sólido que se precipitou foi filtrado, lavado com MeOH, e submetido a secagem sob vácuo para proporcionar 3-(S-metoxi-piridin-S-íl) -1(6 - me t x 1 -2 ~ feni1- ami no - p .1 r i dí n - 3 · í 1} -pr open ona (0,71
g) na forma de um sólido amarelo açafrão brilhante, M-s-H := 34 6.
Etapa E: A 3- (6-metOXÍ-pxriain-3 ~ 11) -r~’bmetil-2-fenil-amino-pirxdxn-3-xl) -propenona (0,71 g) em THE (20 ml; e EtOH (10 ml) adicionou-se uma pequena quantidade de Pd/C. O balão de vidro foi evacuado, reenchido com Hj duas vezes, e deixado sob agitação durante a noite sob RT. Q produto foi filtrado atxavéb de cell, te, lavado com EtOH, concentrado, submetido a eluiçao a partir de coluna de gel de silica com .30:/0 EtOAo/hexano. As frações que continham produto foram coletadas e concentradas para proporcionar 3-(6-metox.ipi r id in - 3 il) 1 - (6-met 11-2- fenil - amino - pi r id in···.» il j prooan-l-ona (0,70 g) na forma de um sólido amareis pálido. Μ·*Η 34 8.
Etapa C: A 3-(6-metoxi-piridin-3-il) -1-(6met il - 2 - fenil - amino- pir idin-3~.il) ·· propan - l~ona ( 0, /U
q) em THE e toluene adicionou-se cloreto de metil oxa111 (0,99 g) ,· seguido por TEA (0,5 ml) para solubrlizar os sólidos que se precipitaram da solução, A pasta fluida resultante, foi aquecida para 80C durante 3 h, então concentrada e re-suspensa em MeOH. K2COS (0,70 g) adicionou-se, a a mistura foi submetida a agitação a 80“C durante 30 min. 0 produto foi trabalhado com EtOAc e H:;O, e concentrado para um solido amarelo. O sólido foi recolhido em MeOH, aquecida para 70*0, acrescentouse uma alíquota adicional de K3CO3 (0,70 g) , e a mistura foi. submetida a agitação durante. 90 min. O produto foi trabalhado com EtOAc e H2O, concentrado, submetido
128/:149 a secagem sobre MgSCq, eluído a partir de uma -coluna de gel de silica (30; 70 EtOAc/hexano), e concentrado para pronorcionar éster metílico ce ácido 3-',8metoxipiridin- 3~ilm.et.il) - 7-metil~4 -ox.o-l - fers.il - x f <*5 diidro[1,81 naftiridina-2-uarboxílico (composto 102) (1.4 0 mg) na forma de um sólido amarelo. M*H - 416; PF 176,0-177,0’C.
Exemplo 27í Síntese de ester metílico de ácido 7~ £luorc-3-(4-metanossulfonil-piperidin-11Q ilmetil) -4-oxo-l-fsuil-l,4-diídre-quinolína2-carboxílico (Composto 126}
Etapa At Combinaram-se ácido 2-bromo-4fluorobenzóico (2Q g; , anilina (10,2 g,> , R^Cíl; (13,.9 g) , oxido de Cu(I) (Aldrich 20822, 648 mg) e pó de Cu 15 [3 μ, 560 mg) em etoxi-etan.ol (3 0 ml) e aqueceram-se sob refluxo (130-135*0 durante 4 h sob . A reação foi considerada completa por LCMS e ThC. A mistura, fox refrigerada para a RT, diluída com Hs0 (30 ml), e neutralizada para pH 7 com HC.i conc., formando um precipi20 tado. O produto foi. filtrado, lavado com K20, e submetido a secagem a 55 C durante 3 d em um forno a vácuo para proporcionar ácido 4 -fluoro-2-fenilamino-benzóico (17.7 g;.
Etapa Bs Dissolveu-se ácido 4-fluoro-225 fenilamino-benzóico (17,7 g) em. DMF (180 ml), e foi submetido a agitação cora 1,1'-carbonildilmicazol <14,91 g) sob bb a 60cC durante .30 min. Adicionou-se cloridrato de N.G-dimetil-hidroxilamina (8,97 g), e prosseguiu
129/149 p r αρ o r c 1 ona r.
benzam ida. í 11 se com a agitação a 60*C dux ante 4 h. O DMF foi então removido sob alto vácuo, e o resíduo preto dividido em EtOAc e salmoura, e cromatografado (EtOAc/hexano} para
-fluoro-lí-me taxi-h-met 11-2- tem 1 - aminog) <
C? Dissolveu-se á-fluoro-K-metcxi-hmet.il-2-f eni 1-ami no-ben zami da (12,5 g; em THF (lou m.·. s a 0<:C, a adicionou-se lentamente vinil-MgEr (84 ml, 1 M em THE) . A mistura foi submetida a agitação durante 2 h a O’C( então resfriada rapidamente com HC1 (0,5 M, 200 ml). O produto foi extraído com EtOAc, e cromatografado com EtOAc/hexano (10-80%) para proporcionar 1-(4fluoro-2-fenilamino-fenil) -propenona (4,0 g) na forma de um óleo amarela.
Etapa D; A uma solução de 1- (4 - fiuoro-2-fenilamlno-fenil) -propeno.ua (480 mg) em Et OH (8 ml) adicionou-se K2CO3 (~150 mg) e 4-met 11 sulfonilpiperidina (326 mg) . A mistura foi submetida a agitação durante 24 h,< diluída com EtOAc, e filtrada. O produto foi recuperado por cromatografia (0-30% MeOH/DCM) para proporcionar 1{4-fluoro-2-fenilamino-fenil) -3-(4-metanossulfoni1 piperidin-1-11) -propan-l-ona (598 mg) na forma de um solido amarelo,
Etapa E: A uma solução a 8*0 de l-(4-fluoro-2fenílamino-fenil) -3- (4-metanossulfan.il-piper.idin-l-il) -propan-l-ona (500 mg) em THF (8 ml) sob tb adicionouse UaHMDS Í3(l ml, 1 M em THE). Depois de 5 min, adicionou- se ester metílico da ácido cloro-oxoacético
Ο / '14 9 (0,28 ml), e a mistura fol submet ida a agitaçao durante h a 0®C, então durante 1 h sub RT < A mistura de reação foi resfriada rapidamente cora NH^Cl (sat 'd aq) , extraída com EtOAc, e cromatografada (0-20% MeOH/DCM) pa~ ra proporcionar éster metilico de ácido 7-f lucro-3- (4~ met anossul funil -p iperidin-1 - ilraet 11} - 4 ~ oxo -1 - teu.il
- di idro ·· qu inol i na - 2 - earbox 11 i co (301 mg, Compos to 1.20). M+H - 4 73: PR - 190,0 - 191,0 ’C.
Exemplo 28í Síntese de 2-metnxi-etil éster de ácido ; 0 3-benzil-7-£luoto-4-oxo-1-fes il-1,4- di idro quinolina~2-carbox£licG (Composto 130}
Etapa A: A uma solução a 0°C de 4-fluora-Nmetóxi-N-raetíl-E-fenil-amino-benzamida (10 g) em THE (80 ml) adicionou-se lentamente fenetil-MgBr (91,2 ml,
1 M em THF), e a mistura foi submetida a agitação a 0eC durante 30 min, seguida por 1 h sob RT. A mistura de reação fci então resfriada rapidamente com NH4C1 (sat'd aq) .. extraída com EtOAc, e cromatografada (0-40% EtOAc/DCM) para proporcionar 1- (4-f.luoro-2 - fenilamino20 fenil) -3-feniI-prspan-I-ona (11,34 g) na forma de um sóiido amare1o.
Etapa Bs A. uma solução de cloreto de { [5fluoro-2 (3-fenil -propionil) -fenil.) -fenil-amino) oxo-acetil (540 mg) em THF (3 ml) adicionou-se 225 metóxi eta.uol (0,12 ml), seguido por Et;?N (0,22 ml). A. mistura de reação tornou-se uma pasta fluida de cor araarelo pálido: acrescentou-se THE adicional (2 ml; , e deixou-se a mistura assentar durante 1 h. A. mistura foi
121/149 então resfriada rapidamente com NH^Cl (sat'd aq) .< extraída com EtOAc, lavada com salmoura, e concentrada para -proporcionar 2-metoxi-etil éster de ácido N-Lbf lucro-2 ( 3 - fenil -propionil) - fenil] -N- feni1-oxal ãmxco (504 mg) na forma de um óleo.
Etapa Ct A uma solução de 2-metoxi-etil éster de ácido N- [5- flucra-2- (3-fenil-propionil) -fenil] -Nfenil-oxalâmico (500 mg) em MeOH <10 ml) adicionou-se K-CCg (-150 mg), e a mistura de reação foi aquecida sob 10 refluxo sob durante 1,5 h. A mistura foi então refrigerada, os sais filtrados, e o solvente evaporado in vacuo. O resíduo foi cromatografado (0-20% MeOH/DCM) para, proporcionar 2-metoxi-etxl éster de ácido 3benzil-7-fluo.ro- 4 -oxo- 1-fenil-l , 4- -diídro-quinolina-215 carboxílico (Composto 130). - 4 32; PP ~ 3.68, o1:69, 0°C.
Exemplo 29: Síntese de 3-benzil~7~cl©ro-2“Oxazol~2il-l-fanil-lH-quinolin-á-ona (Composto 140)
Etapa Aí Colocou-se em suspensão ácido oxazol20 2-carboxílico (0,5 g) em DCM (3.5, ml), e adicionaram-se gotas de DMF sob N2. A.dicionou-se lentamente cloreto de oxalil (674 mg), e a mistura de reação foi submetida a agitação até cessar o horbulhamentc (cerca de 2 h). o solvente foi removido in vácuo, e o resíduo recolhido em toluene (15 ml)* Adicionou-se i-(4-cloro-2fenilamino-fenil) -3-fanil-propan-1-ona (1.,48 g) , e a mistura foi aquecida sob refluxo durante 20 h. 0 produto foi refrigerada, a solvente removido in vacuo, e o
132/149 resíduo cromatografado (0-20% MeOHtDCM) para proporcionar [5-cloro··2 - (3-fenil-propionil) -fenil] -fenil-amida de ácido oxazol-2-carboxílico (697 mg) na forma de um põ amarelo pálido.
Etapa Et Dissolveu-se [5~clora-2-(3-fenilnropíonil) -fenil] -fenil-amída de ácido oxazol-2carboxílico (387 mg) em MeOH seco (2U mi.) and K^CO,? (100 mg}, e aqueceu-se sob refluxo durante 1,5 h, (3 produto foi então refrigerada, o solvente evaporado, e 10 o resíduo cromatografado (0-30% MeOH/DCM) para proporcionar 3 - benz i 1 - 7 - c 1 oro - 2 - oxazol - .2 - i .1 · 1 - tem 1 - 1H quinolín-4--ona (Composto 140, 233 mg), PF ~ 219,0220, (FC.
Exemplo 30í Síntese de 3-(l-metil-lH-pirazol-415 ilxaetil} -1-feníl-7-trifluoro-metil-lH- [1, 8] naftiridin-4-ona (Composto 129)
Etapa At Misturou-se 1 -(2-fenilamino-6trifluoremetil -piridin -3--Í1) -eta.nona (0,1 g) com 1metil-IH-pirazol-4-carbaldeído (39 mg} e NaNH>: (2 N, 2 20 ml) em MeOH (10 ml) e submeteu-se a agitação durante a noite sob RT. O sólido laranja claro que se formou foi filtrado, e submetido a secagem em um forno a vácuo para proporeionar 3 - (1 -metil 1H -pirazo 1 -4 - í 1) -1 - (2 fenil-amíno-6-trifluorometil -piridin-3-il) -propenona 25 (45 mg),
Etapa Bt Dissolveu-se 3-{l-metil-lH-pirazol-411} -1- (2-fenílamino-6-trifluoromet.il -piridin-3-il) ·· propenona (45 mg) em EtOAc (25 ml), adicionou-se Fd/C
133/149 (10%, 30 mq) , e a material de partida hidrogeuacij com H·.: (balão) durante 1 h. Q produto, 3~ !.1 -meti*.- χΗp i r a zol. - 4 -i1) -1-(2 - f en i1 am ino 8 -1 r i f. 1 uorome t 11 piridin-3-il) -propan-1-ana, foi filtrado, concentrado, e usado sem qualquer outra purificação.
Etapa C í Dissolveu- se 3--(1 -met i 1 - 1H pi ra sol - 4 11} --1-- (2“fenilamino-6-trifluorometil -pxridin-.s·· xl; propan-l-ona (-45 rag) era toluene (10 ml) com éster metílico de ácido cloro-ozo-acétxco (0,2 ml; e aguecxda para 110*0 durante 72 h, então re.frigerou-se< O produto foi concentrado in vácuo, então dissolvido em MeOH (j.0 ra.i) cura KaCOx (0,2 g) , e aquecida para 50 *C durante 15 min. Ό produto foi refrigerado, filtrado, e concentrado para proporcionar éster metxlico de ácido 3-{l-raetxl1H --pxrarol-4-iIraetil) -1-4 -oxo-f enx1 - 7 -trifluorometi1l.H-[l,8j naftiridína-2-carboxílico (Composto 129} . M+H :== 443.
Exemple 31s Síntese de éster metálico de ácido ΙΟ -assino fenil) -3-benzil -7-metil -4-oxo-l,4diidro -[1,81 ~naftiridiua-2-carboxilioo (Cosspnsto 105)
Etapa As Misturou-se 1- (2-cloro-8 -meti.1 píri.din-3-il) -etanona (0,4 g) com 3-nitro-fenilamina (0,33 g) , Pd (OAC)S (16 mg), ΒΙΝΆΡ (45 mg), CaCO:} (0,25 g) , e Et.3N (0,2 ml) em toluene (10 ml), e aqueceu-se para 100GC durante 4 d. O produto foi então filtrado e concentrado para proporcionar 1 - [8 -metí 1-2-(3 -nit.rofenilamino) -piridin-3-il] etanona (0,2 g).
134/149
Etapa Bs Misturou-se 1-[6-metil- 2 -{3 η í t rofeni lamino) -pi ri d in - 3 -11 ] ·· etanona (0,2 g ,> com 4 metanossulfonil-benzaldeído (0,135 g) e □ S. 2 ml) em MeOH (10 ml), e submeteu-se a agitação durante a noite sob RT. 0 sólido amarelo que se formou foi filtrado para proporcionar 3- (4-metanossulfonil~fen.il) -1[6 -metil- 2 - (3-ni trofenilamino) -pirldin- 3-il] propenona (0,33 g).
Etapa Cs Hidrogenou-se 3 (4-metanossul fonír10 fenil) -1- [6-metil-2- (3-nitrofenilamino) -piridin-3-il]
-propenona {125 mg) com RhCl(PPhs)< (30 mg) em tolneno (20 ml) utilizando-se H?_ a 455,0 kPa (66 psi) , a 60'C durante 7 h, para proporcionar 3-{4-metanossulfonil~ fenil) -1- [6-metil -2- (3 -nitro-fenilamino) -piridin-3·· 15 11] -propan-l-ona (-100 mg).
Etapa Di Dissolveu-se 3- (4-metanossulfon.ilfenil) -1- [6-metil-2·· (3 nitro-fenilamino) -plridin-3il] -propan i-ona (0,1 g) em THF (5 mi). Adiozonou-se estar metílico de ácido cloro-oxo-acético (0,1 ml), e a 20 mistura foi aquecida por microondas a 80‘C durante 1 h.
produto foi concentrado, o resíduo recolhido em MeOH (5 ml), adicionou-se Κ»α>Λ (0,1 g) , fô a mistura aquecida para 50;íC durante 15 min. 0 produto foi refrigerado e concentrado, e purificado por TLC prep (50% EtOAc/hex.a.no) para proporcionar éster metíi-ico de àcioo 3benzil- 7-metil-1-(3-nitro-fenil) -4-oxo-X,4-ei idro[1.,8] -naft iridina-2-carboxílico (Composto 104, -20 mg). Mí-H - 508.
135/149
Etapa E: Dissolveu-se o composto 104 (15 mg) em EtOAc (IO ml) com Pc/C (10%, 5 mg) , e hídrogenou-se u~ tilizando--se H2 (balão) durante 2 h. 0 produto foi filtrado, concentrado, e purificado por TLC prop (50/50 5 EtOAc/hexano), então 10% MeOH/DCM para proporcionar éster metílico de ácido 1· (3-aminofen.il) -3-benzíl·-'?metil-4-οχα-Ι,4-diidro[1,8] naftiridina-2-carbaxilico (Composto 105, 2,4 mg). - 478.
Exempla 32s Síntese de 1-(6-azdno-piridin-2-il) -3lO benzí 1-7 -alora~2-axazal- 2 ~ il ~ 1H ~ quinai in~ 4 aaa (Composto 141)
Etapa A: Aqueceu··se uma mistura de ácido 2bromo-4-clarobenzÓico (5 g) , 2,6-diaminopiridina (8,95 g) f KàCOa (3,18 g> . pó de Cu (0,13 g) e óxido de Cu(I) 15 (0,15 g) em etoxietanol (10 ml) at 130eC durante 2 h, até a reação ser concluída (tal como donfirmado por tic) . A mistura foi refrigerada, para a RT, e adicionouse água (78 ml) e carbono ativado, e a. mistura fai submetida a agitação durante 3 h. O produto fox então £1120 Irado, e adicionou-se HC1 (4 M) atê ser alcançado o pH
7. O precipitada castanho resultante foi filtrado e submetida a secagem em um forno (50<5C, pressão ~ 30. mbar) durante a noite para proporcionar ácido 2-(5amino-piridin-2-ilamino) 4-olarobenzoico (2,4 g) ♦
Etapa Bj Aqueceu-se uma parta fluida de ácido
2- (6-ajKjino-pirid.in~2-ilamino) -4-clorobenzóicc (2,4 g) e 2,5 -haxan.odio.na (1,04 g) e ácido p-toluenossulfôníco (0,15 g) sab refluxo em um purgador Dean-Stark durante
S4 h. A mistura resultante fax refrigerada para a RT e filtrada, então foi submetida a agitação cos Η,Ο <,50 ml) durante a noite,, filtrada, e submetida a secagem para proporcionar ácido 4-cloro-2·· |6- (2,S-aimetxxpxrrol-l-il) -piridiu-2-ilamino’J -benzóxco (2,4 g) .
E tapa C* -Submeteu-se- a agi tação uma mistura ne ácido 4-eloro--2- [8- (2, S -dímetíl -pirrol-1-ir) --piridin2-ilaminoj -benzôico (4,2 g) em DMF (25 ml) e carbonxldiimida (2,4 g) a 6Q°C durante 30 min, e adicionou-se M-metóxi -N-metilamina (1,41 g) < A mistura foi submetida a agitação a SO^C durante 16 h, então os voláteis eva norados, e a reel.duo dividida entre EtOAc e H^O, então entre EtOAc e HQ1 (0,1 N aq) . A fase orgânica foi adsorvida em silica gel e cromatografada. Gromatografia em silica (80 g) contra he.xano/EtOAc (100:0 até 50; 50) proporcicsnou 4·Ήογο-2- [t- (2,5-dimetíl-pirrol 1-xlj piridin-2-1lamino] -N-metoxi-^-meti 1 -benzamida (~1,6
g) ·
Etapa X>; Refrigerou-se uma solução de 4-cioro2 - (6- (2,5-dimetil-pirrol 1-il) -piridin-2 ilamino] -Nmetóxi 14 -metil -benz&mída (1,55 g) em THF (20 ml) em um banha de gela, e adicionou-se. solução de fenetxlGrignard (10 ml, 1 M em THF) par meio de seringa. A mistura foi submetida a agitação durante a noite sob RT, resfriada rapidamente com NH4CI (aq), extraída com EtOAc, e adsorvida em silica. 0 produto fui cromatografa.de em silica (80 g) (hexano/EtOAc 100:0 para 50:50) (ou. utilizando-se DCM/MeC-R) para proporcionar 1 -{4137/149 cloxo-2~ [6- Í2( 5 -dimer il-pirrol 1-11) -p.irrdin-2i 1 amino] - fen 11} ·· 3 - fen 11 - proper. -1 >ona.
Etapa Es Tratou-se. uma mistura de ácido oxazol2-carboxílico (147 mg) e DMF (2 gotas) em DCM (1 ml) com cloreto de oxalil (127 μΐ). O desprendimento de gás cessou depois de 2 h. Adicionou-se CHClj (2 ml), removeram~se os voláteis em um forno a. vácuo «40C, 290 mbar) , e o óleo residual foi dissolvido em THF (2 ml) para formar a solução A*. Tratou-se uma mistura de 110 { 4-elo.ro--2 - [6- ( 2 < 5-dimetli-pixrol-1 - il) --píridi.n- 2 ilamino] -fenil} -3--fen.il-propan-1 -ona {430 mg) em THF (2 ml) com UME (2 gotas) e adicionou-se gota a gota uma solução de NaHMDS (2,5 ml, 1 M em THE). Adicionou-se a solução A (2 ml) gota a gota, e a mistura foi submetida
a. agitação sob RT durante 12 h. 0 produto foi traba.1 nado em EtOAc/HaO e cromatografado (DOí/MeOH 100:0 até 70:30) para proporcionar 3-benzil~7~cloro-1-[6-(2,5d 1 me t i 1 - p i r r ol --1-11) - p i r i d i n - 2 -- i 1 j - 2 - oxa zo 1 - 2 - i 1 -1H quinol in-4 -ona .
Etapa Fí Uma solução de 3-benzil-7-cloxo-1-[6(2,5-dimetil-pirrol-l-il) -piridín-2-ilj -2-oxazoi-2il -lH-quinolin-4-ona (640 mg) em EtQH (3,5 ml) e ãgua {1,3 ml) e cloridrato de hidroxilamina (450 mg) foi submetida, a agitação durante 48 h a 80'?C. Depois ae e25 vaporação dos voláteis, o resíduo castanho escuro foi dividido em EtOAc e H30, e a fase orgânica adsorvida em sí1ica para proporcionar 1 -(6-amino-pi ridin -2 -i1) - 3 benzil-7~cloro-2-oxazol -2-il-lH-quinolin-4-ona (Compos
138/149 to 141). Μ*Η ~ 428; PF - 279,3 - 882(8ύό.
Exemplo 33: Síntese de ester metxlico de ácido 3eiclopxopilmatil~7-metil-4-ox©-l-feuil-l,4'diidro [1,81 naftxridina -S-carboxxlico (composto 142)
Etape As A 1-(6-metil-2-fen.ilamiuc-pir-idin-311) -etanona (0,7 g) dissolvido em MeOH quente (15 ml) adicionou-se ciclopropanocarbaldeído (0,4 g) e NaOH (2 M, 2,5 ml), e a mistura foi aquecida para »0°C durante a noite. A mistura foi então refrigerada para a RT, dx~ .0 vidida entre EtOAc e H,>0, e a camada organise lavada com LiCl (8,3%, peso, aq) , submetida a secagem sobre MgSOí, filtrada, e concentrada, O produto foi purificado por cromatografia de coluna (1:4 EtQAc: hexa.no) , e concentrado para um óleo amarelo para proporcionar 15 ciclopropil-1- (t-metil-S-fenilamino-piridin-3-il) propenona (0,32 g) .
Etapa Bs Di ssol veu - se 3 c i c 1 op r op i 1 -1 - (t - me 1112-feuilamóno-piridin-3-il} -propenona (8,32 g) em E~ tOH/THF/EtOAc (12 ml), e adicionou-se Pd/C (0,02 g, 20 10%, peso) . O balão de vidro foi evacuado e repreenchído duas vezes com H2. Depois de 2 h, a mistura foi filtrada através de cellte, concentrada, e purificada por coluna utilizando-se 1:8 EtOAc/hexano para proporcionar 3-cicloprop.il -1 - (t-metil -2 -fenílamxno'28 pirid.in-3-il) -propan-1-ona (0,34 g) na forma de um sólido amarelo.
Etapa Cs Dissolveu-se 3-ciclopropíl-l-(tmetil~ 2~fenílamino~piridin-3-11} -propan-1-ona (0.3 g} em THF
139/149 e toluene (10 ml), adicionou-se cloreto de oxalxl (0,3 g) . e aqueceu-se a mistura a S0'’C durante 2,5 h. A tura fax então deixada esfriar para a RT durante a noite, então παν amen te aquecida para 80'C durante 2,. τ> h, e 5 refrigerada para a RT para proporcionar éster metílico de ácido N~[3-(3-ciclopropil-propionii) -6-raetil piridin-2-il] -R-fenil oxalãmico,
Etapa »: A éster metílico de ácido N-[3-(3cxclopropil -propionil) -6-metil -piridin-2-il] -Nfenil -oxalâmioo (-0,3 g; dissolvido em tolueno adicionou-se MeOH (5 ml? e K?,CO; (0,3 g) . A mistura foi aquecida para 80°C durante 30 min, refrigerada para a RT, então recolhida, em EtOAc e lavada 2X coat LiCl (0.3%, peso, aqj . O produto foi submetido a secagem sobre MgIS SO4t filtrado e concentrado, então purificado por TIA' prop {1:4 EtOAc/hexano) para proporcionar éster metílice de ácido 3-ciclopropilmetil -7-metil-4-oxo~l~fen.il 1,4-diidro [1,8] naftiridina -2-carbaxÍlico (composto 142) .
Exemplo 34t Formulações
Preparados farmacêuticos para distribuição por várias vias são formuladas conforme ilustradas na Tabelas seguintes. Ingrediente ativo ou ''Composto atxvo tal αοβ» usado nas tabelas significa um ou. mais dos 25 compostos da Fórmula I.
Composição para Administração Oral j
{ Ingrediente ................................................
( Ingrediente ativo J 20,0%................................................................................
140/14 9
| Laotose | 79,5% ................................................ |
| Estearato de magnésio | U , 5%................................................................... :.......... |
Os ingredientes sãe mist'axados e distribuídos era cápsulas que contêm cerca de 100 mg cada uma; u.ma cápsula será aproximadamente uma dosagem diária total.
Composição para Administração Oral ..............................................
| Ingrediente | % psso/peso |
| Ingrediente at ivo | 20,0% ....................................... |
| Estearato de Magnésio | 0,5% .............................. ... |
| Croscarmelose sód ica | 2,1)1 ..................................................; |
| Lactose | 76,5% ............................................ |
| PVP (polivinllpirrolidina) | 1,0% |
Os ingredientes são combinados e granulados utilitando··se ura solvente, tal como metanol. A formula·· ção é então submetida a secagem e levaaa à forma de comprimidos {que contêm cerca de 20 mg decomposto ativo) com urna máquina de formar comprimidos apropriada.
| Ingrediente | Quantidade |
| Composto ativo | 1,0 g ................................. |
| Ãcido Fumárico | ü„ 5 g |
| Cloreto de sódio | „JL..L.SL.................................. |
| Met 1.1 paraben | ........................................ |
| Prep i1 paraben | 0,05 g· |
| Açúcar granulado | 25,5 g ............ ............................... |
| Sorbitol (solução a 70%} | 12,85 g |
| Veegura K (Vanderbilt Co. <· | __LA.J................................................. |
141/149
| j Aromat.izante | J 0,035 ml ................ j |
| --------------- | |
| iCorantes ________ | __1 0,5 m$3 _______________________ |
| | Água dest liada ____ | [q.s. para 100 ml__ |
| Os ingredlentes | são misturadas para formarem |
| uma suspensão para admin: | .stração oral. |
Formulação Parenteral ΐIngrediente_____ j Ingrediente ativo iCloreto de sódio
I............................
Agua para injeção__

ί100
Q ingrediente ativo é dissolvido em uma parte da água para injeção. Uma quantidade suficiente de cloreto de sódio é então adicionada com agitação para preparar a solução isotônica. A solução é misturada até ao peso com o restante da água para injeção, filtrada através de um filtro de membrana de 0,2 micron, e embalada sob condições estéreis.
Formulação de supositórios... ......................................
J í ί Ingrediente............................................ i % peso/pseo........................
pügredients ativo .................................|1.% .......................................
I Polyethylene glyeo.1 1000.................... 74,5¾.....................................
i Polyethylene glycol 4 000 _______ [24,5%.............................
Os ingredientes são fundidos em conjunto e mis··· ttrados em ura banho de vapor, e vazados em moldes que contêm 2,5 g de peso total.
Formulação Tópica
142/149
| Ingredientes | gramas |
| Comoosto st iva | 0,2-2 |
| Span 50 | 2 |
| Tween βί) | 2 |
| Óleo mineral | 5 |
| Pfòtrclato | 10 |
| Metil paraben | 0.15 |
| Propll paraben | 0,05 |
| BHA (hidróxi anisol butilado) | 0,01 |
| Apua | q,s. 100 |
Todos os ingredientes, exceto a água, são combinados e aquecidos a cerca de 60<!C cara agitação. Adiciona-se uma quantidade de água suficiente a cerca de 6ô°C com agitação vigorosa para emulsionar os ingredi5 entes, e adiciona-se então água q.s. cerca de 100 g.
Formulação de Spray Masal
Preparam-se várias suspensões aquosas que contêm entre cerca de 0,025-0,5 por cento de composto ativo como formulações de spray nasal. As formulações cp10 cicnalmente contêm ingredientes ativos tais como, por exemplo, celulose microcristalina, carboximetilcelulose de sõ-dio, dextrose, e assemelhados, Ãciao clorídrico também pode ser adicionado para ajustar o pH. As formulações de spray nasal pode ser distribuí15 das por meio de uma bomba de medição de spray nasal que distribuí tipicamente cerca de 50-100 μΐ de formulação por cada acionamento. Um programa de dosagem típico e de 2-4 sprays a cada 4-12 h.
Exemplo 35; Ensaio de JEK in vitro
Mediu-se a atividade de JNK por fosforilação de GST-ATF2 (19-96) com [y~!5Pl ATP. A reação de enzima foi conduzida sob concentrações Km de ATP e o substrato no 5 volume final de 4 0 pl era tampão conteúdo 25 mM HEPE8, pH 7,5, 2 mM ditiotreitol, 150 mM NaCi, 20 raM MgCxj, 0,001% Tween® 20, 0,1% BSA e 10% DMSO. Ensaio de JNK2C12 humana contém enzima ΙπΜ, 1 μΜ ATF2, 8 μΜ ATP com luCi ly-J3P] ATP. O ensaio de üNKlul humana contém IO enzima 2 nM, IpM ATF2, 6 pM ATP com 1 pCi ft-^Pl ATP. O ensaio de JNK3 humana (Upstate biotech 414-501M) contém enzima 2 nM, 1 pM ATF2, 4 pM ATP com 1 p€i íy-'í5P] ATP. 0 ensaio de enzimas foi realizado na presença ou ausência de várias concentrações de composta, Pré-incubaram~ i.5 se JNK e composto durante 10 min., seguido por iniciação de reação enzimâtica mediante adição de ATP e do substrato, A misturai de reação foi incubada a 3 0<:C durante 30 raio. Ao final da incubação, a reação foi concluída por transferência de 25 pl da mistura de reação 20 para 150 pl de 10% de pasta fluida de glutatlona Sepharose® (Amershams # 27-4574-01) contendo 135 mM de EDTA. O produto de reação foi captado na resina de afmxdaae, e lavado em uma placa de filtragem (Mil1.ipore, MABVNGB50) cuia salina tamponada per fosfato durante seis 25 vezes para se remover o radionucleotideo livre. A incorporação de JJP na ATF2 foi quantificada era ura contador de cintilação de míoroplaquetas (Packard Topeount). A potência de inibição do composto na CTN.K foi. medida
144/149
| doi valor de IC-x, geruuo a partn de dez | curvas de ini - |
| feição de concentração ajustadas no | modelo de 3- |
| parâmetros: % de inibição | Máximo/(1* |
| {ICm./ [Inibidor] }tovia) . Os dados foram | analisados no |
Microsoft Excel para avaliação dos parâmetros. Os resultados encontram-se expostos na Tabela 2 seguinte:
Tabela 2í Composto XC^s's vs. JNK1 © JNK2
Composto JNK1 (pb JNK2 (μΜ)
| 66 0,018'1 | 0,0445 |
| 69 0,0184 | 0,047 |
| 59 0,7226 | 0,0614 |
| 67 0,0304 | 0,0485 |
| 65 0,0328 | 0,0957 |
| 8? 0,0334 | 0,0832 |
| 30 0,0355 | 0,0863 |
| 32 0,0445 | 0,1289 |
| 46 0,0454 | 0,1367 |
| 85 0,0481 | 0,1178 |
| 44 0,0491 | 0,1682 |
| 31 0,0529 | 8,1812 |
| 63 0,0987 | 0, 212 |
| 6’0 0,1342 | 0,3988 |
| 72 0,1544 | 0,3362 |
| 80 0,1.931 | 0,4773 |
| 88 0,2454 | 0,84 02 |
| 64 0,6032 | 2,0554 |
| 60 1,5324 | 4,9829 |
| 100 01 48 |
5/14.9
| Composta | OTK1 ίμΜ) | JNK.2 <pW |
| 1 1 0 | 0,0502 | 0,2014 |
| 117 | 0,032 | 0,0867 |
| 129 | 0,2523 | 0,5631. |
| 135 | 0,2496 | 2,103 |
| 1.41 | 0,612 | 2,086 |
Exempla 36: Ensaio de Produção de XL-6 induzido por TNFa em Ratos ín vivo
Deixou-se que ratas fêmeas Wistar-Ran adquiridas a partir de Charles River Laboratories tossem aclimatadas durante uma semana antes do uso e alcançassem um neso corpõreo aproximado de 101-130 g. AdminiStrouse aos ratos composto de teste (N « 8 por composto) via gavagem oral 30 min antes de uma provocação ^.n^xapex itoneal de 0,5 pg de TNF-α de rato recombinante (Biosource). Coletou-se sangue via cardiacentese 98 min depois da provocação com TJJF-α. Preparou-se plasma utilizando-se tubos de separação de lit.ro heparrna vBD mxczotainer) e congelados a -80*C até serem analisados. Os uiveis de XL-6 foram determinados utilizando-se um kit ELISA IL-6 específico para rato (Bíosource). Determinou-se o percentual de inibição e os valores de EDs& (calculadas coma a dose da composto sob a qual a produção de THF-« ê 50% do valor de controle) . Os resultados estão ilustrados na Tabela .3 seguinte·.
Tabela 3: Inibição de Produção de XL-6
Composto Dose (mg/Kg) Inibição de XL-6 (%)
3 58
0 30 48
Exemplo 37ϊ Ensaio de Produção de XL»6 induzido por Wa em Ratos in vivo
Deixou-se que ratas fêmeas Wistar-Han aquiridas a partir de Charle® River Laboratories fossem aclimatadas durante uma semana antes do uso e alcançassem um peso corpôreo aproximado de 13.4-132 g. Administrou-se aos ratos composto de teste IS (N - 8 por dose) de forma subcutânea 30 min antes de uma provocação intraperi10 toneal de 0,5 pg da TNF-u de rato recombrnante (Binsource) . Coletou-se sangue via cardiocente.se 90 min depois da provocação com TNF-α. Preparou-se plasma uti.lizando-se tubos de separação de l.ítio heparina (BD microt.ain.er) e congelados a -80’C até serem analisados.
Qs níveis de IL-6 foram determinados utilizando-se um kit ELISA IL-8 específico para rato (Biosource). Determinou-se o percentual de inibição e os valores de EDSÍI (calculados como a dose de composto sob a qual a produção de TN.F-a ê 50% do valor de controle) < Cs resultados 28 estão ilustrados na Tabela 4 seguinte:
Tabela 4$ Inibição de Produção de XL-6
Dose (mg/Kg) δ Inibição (%) p vs. veículo
0.03 NS NE
0,1 NS NS
0,3 30,01. 0,01
Dose üag/Kg)
1,. 0
3,0
Inibição (¾)
47.83
54,85
71.15 ρ w. veicule
0.002
0,0003
0.,0002
Exemplo 38: Artrite induzida por Colãgeno ® Koedor
Deixou-se que ratas fêmeas Wistar-Han aquxridaa <% X' L, X.X de Charles River Laboratories com 7-8 semanas fossem aclimatadas durante uma semana antes do cançassem um peso corpôreo aproxxmauo de 120 140 g. Do dia 0 do estudo, os ratos são preparados de forma intradermica (i.d.) em vários locais nas costas com uma emulsão de 100 ng de Sovine Type xí Colragen (Chondrex) em Incomplete Freund's adjuvants {IFA; total de 0,1 ml em 2-3 locais) . A indução de artrite é geralmente observada 12-3.4 dias em relação ao prepare;
entretanto uma injeção de intensífioador de 100 qg de colágeno/IFA é aplicada em torno de 7-10 dias (i.d. até 15 um total de 0,1 ml) na .base da cauda ou um local alternado nas costas para sincronizar a. indução da enfermidade. A dosagem do composto pode ser profilática (partindo-se da hora da intensificação ou 1-2 dias antes) ou terapêutica (Iniciando depois da. intensífreação e coincidindo com as contagens iniciais de enfermidade de
1-2 - vide contagem clínica adiante) * Os animais são avaliados quanto ao desenvolvimento e progressão da enfermidade durante os 23. dias seguintes.
Os ratos são avaliados utilizando-se um ai sterna
148/149 de contagem (descrito adiante), medições de volume de patas utilizando-se um pletismômetro para cada pata, ou medindo-se a espessura da pata ou da junta com *.»m cal.·, bre. As medições de linha de base são realizadas no dia 0, e começando - se novamente nus primeiros sinais de inchação durante ate três vezes por semana até ao final da experiência. A contagem é avaliada como se segue para cada pata:
1~ inchação e/ou vermelhidão da pata ou de um dedo.
2=:: inchação Ms duas ou mais juntas.
3® inchação flagrante da pata com mais de duas j untas envoividas.
4::: artrite séria de toda a pata e dedos.
Q índice artrítico para cada rato é avaliado pela adição das quatro contagens das patas individuais, dando-se uma contagem máxima de 18. Com a finalicade de medir em série o início e a progressão na doença, o volume das patas posteriores também é determinado atra20 vés do uso de um pletismômetro.
Ao final do estudo, as patas traseiras (e outros tecidos) são colhidos para determinação de peso, histelogia, análise celular e/ou molecular. Adicionaimente, sangue é coletado via cardiocentese, o piasma e 26 preparado utilizando-se tubos de separação de heparma de li tio (BD microtainer) e congelado a -7O’C até ser analisado. Uiveis inflamatórios de citoquina (por exemplo, TNF-α, IL-1 e XL-6) a partir do plasma ou pro
149/148 veníentes do tecido de junta homogeneizado são determinados utilizando-se kits ELISA específicos de ratos (RW) . O nível de proteção ou de inibição da enfermidade ê determinado como um composto de mudanças em con5 tagens clínicas, volumes de patas e histopatologia comparados a animais de controle.
Muito embora a presente invenção fosse descrita com referência a concretizações específicas da mesma, será compreendido por aqueles versados na técnica que 0 várias alterações podem ser realizadas e equivalences podem ser substituídos sem escapar do espirito verdadeiro e escopo da invenção. Alêm disso, muitas modificações nodem ser feitas para adaptar uma situação, material, composição de material, processo, etapa ou eta,§ pas de processo, ao espirito objetivo e escopo da presente invenção. Pretende-se que todas essas modificações fiquem situadas dentro do escopo das reivindicações apresentadas em anexo.
Claims (12)
- REIVINDICAÇÕES1 - Composto, caracterizado por compreender a fórmula IR7 O
 em queX é CR11 ou N;Y é —C(O)R3, heteroarila de 5-elementos, ou heterociclila de 5-elementos;Z é fenila, cicloalquila, heterociclila ou heteroarila, e é substituído com R1 e R2;R e R são, cada um, independentemente, H, halo, CN, alquila inferior, ou -Y1-Y2-Y3-R8, ou R1 e R2 em conjunto formam -O(CH2)nO-, onde n é 1 ou 2;Y1 é -O-, -C(O)~, -C(O)O-, -C(O)NR9-, -NR9C(O)-, -S-, SO2-, ou uma ligação;Y e cicloalquileno, heterocicloalquileno, alquileno inferior ou uma ligação;Y3 é -O-, —C (O) —, -C(O)O-, -C(O)NR9-, -NR9C(O)-, -SO2-, ou uma ligação;R e H, alquila inferior, alcoxila inferior, cicloalquila, heterocicloalquila, ou -NR9R10, em que R8 outro que não seja H é, opcionalmente, substituído com alquila inferior, halo, -CF3, ou -OH;R9 e R10 são, cada um, independentemente, H ou alquila inferior;
em queX é CR11 ou N;Y é —C(O)R3, heteroarila de 5-elementos, ou heterociclila de 5-elementos;Z é fenila, cicloalquila, heterociclila ou heteroarila, e é substituído com R1 e R2;R e R são, cada um, independentemente, H, halo, CN, alquila inferior, ou -Y1-Y2-Y3-R8, ou R1 e R2 em conjunto formam -O(CH2)nO-, onde n é 1 ou 2;Y1 é -O-, -C(O)~, -C(O)O-, -C(O)NR9-, -NR9C(O)-, -S-, SO2-, ou uma ligação;Y e cicloalquileno, heterocicloalquileno, alquileno inferior ou uma ligação;Y3 é -O-, —C (O) —, -C(O)O-, -C(O)NR9-, -NR9C(O)-, -SO2-, ou uma ligação;R e H, alquila inferior, alcoxila inferior, cicloalquila, heterocicloalquila, ou -NR9R10, em que R8 outro que não seja H é, opcionalmente, substituído com alquila inferior, halo, -CF3, ou -OH;R9 e R10 são, cada um, independentemente, H ou alquila inferior; - 2/3R3 é OH, alquila inferior, alcoxila inferior, (alcoxila inferior)-alcoxila inferior, ou -NR9R10;R4 é alquila inferior, fenila, heterociclila, cicloalquila, heterocicloalquila, ou heteroarila, e é opcionalmente substituído coin alquila inferior, hidroxila, alcoxila inferior, halo, nitro, amino, ciano, ou halo-alquila inferior;R5 e R6 são, cada um, independentemente, H, halo, ciano, alquila inferior, “CF3, alcoxila inferior, OCHF2, -NO2, ou -NR9R10;R7 é H, F, Cl, metila, ou OH;R11 é H, alquila inferior, cicloalquila inferior, ou fenila;ou um sal farmaceuticamente aceitável do mesmo.2 - Composto, de acordo com a reivindicação 1, caracterizado por Y ser -C(O)R3, e R3 ser metoxila.
- 3 - Composto, de acordo com a reivindicação 2, caracterizado por R7 ser H.
- 4 - Composto, de acordo com a reivindicação 3, caracterizado por Z ser fenila, e R2 ser H.
- 5 - Composto, de acordo com a reivindicação 4, caracterizado por R6 ser H.
- 6 - Composto, de acordo com a reivindicação 5, caracterizado por R5 ser selecionado do grupo que consiste em H, F, Cl, Me, e CF3.
- 7 - Composto, de acordo com a reivindicação 6, caracterizado por X ser CH ou N.
- 8 - Composto, de acordo com a reivindicação 7,3/3 caracterizado por R1 ser -Y1-Y2-Y3-R8, e Y1 ser S02.
- 9 - Método para tratar um distúrbio mediado por JNK em um paciente que tem um distúrbio mediado por JNK, caracterizado por o dito método compreender admi-5 nistrar a um paciente com necessidade do mesmo uma quantidade efetiva de um composto de fórmula I conforme definido na reivindicação 1.
- 10 - Composição farmacêutica, caracterizada por compreender um composto de fórmula I, conforme definido10 na reivindicação 1, e um excipiente farmaceuticamente aceitável.
- 11 - Uso de um composto conforme definido na reivindicação 1, caracterizado por ser para a manufatura de um medicamento para o tratamento de um distúrbio15 mediado por JNK, como artrite reumatóide.
- 12 - Medicamento, caracterizado por compreender um composto da fórmula I para o tratamento de um distúrbio mediado por JNK, como artrite reumatóide.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US93005407P | 2007-05-14 | 2007-05-14 | |
| PCT/EP2008/055807 WO2008138920A1 (en) | 2007-05-14 | 2008-05-13 | Dihydroquinone and dihydronaphthridine inhibitors of jnk |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| BRPI0811601A2 true BRPI0811601A2 (pt) | 2019-08-27 |
Family
ID=39677349
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| BRPI0811601A BRPI0811601A2 (pt) | 2007-05-14 | 2008-05-13 | composto, método para tratar um distúrbio mediado por jnk em um paciente que tem um distúrbio mediado por jnk, composição farmacêutica, uso do composto e medicamento que compreende o composto. |
Country Status (17)
| Country | Link |
|---|---|
| US (1) | US8163906B2 (pt) |
| EP (1) | EP2148862B1 (pt) |
| JP (1) | JP5160638B2 (pt) |
| KR (1) | KR101176711B1 (pt) |
| CN (1) | CN101679270B (pt) |
| AR (1) | AR066531A1 (pt) |
| AT (1) | ATE551326T1 (pt) |
| AU (1) | AU2008250049A1 (pt) |
| BR (1) | BRPI0811601A2 (pt) |
| CA (1) | CA2686385C (pt) |
| CL (1) | CL2008001393A1 (pt) |
| ES (1) | ES2382137T3 (pt) |
| IL (1) | IL201686A0 (pt) |
| MX (1) | MX2009012222A (pt) |
| RU (1) | RU2466993C2 (pt) |
| TW (1) | TW200908974A (pt) |
| WO (2) | WO2009015917A2 (pt) |
Families Citing this family (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8828924B2 (en) * | 2009-05-14 | 2014-09-09 | University Of Maryland, Baltimore | Methods of treating a diabetic embryopathy |
| UA108363C2 (uk) | 2009-10-08 | 2015-04-27 | Похідні імінотіадіазиндіоксиду як інгібітори bace, композиція на їх основі і їх застосування | |
| CN101985424B (zh) * | 2010-09-21 | 2013-05-29 | 浙江工业大学 | 一种合成邻硝基苯乙酮类化合物的方法 |
| US8471027B2 (en) * | 2011-04-06 | 2013-06-25 | Hoffmann-La Roche Inc. | Adamantyl compounds |
| BR112014000499A2 (pt) * | 2011-07-12 | 2017-01-10 | Hoffmann La Roche | compostos de quinolona de aminometila |
| US10059663B2 (en) | 2013-08-29 | 2018-08-28 | Kyoto Pharmaceutical Industries, Ltd. | Aromatic compound and use thereof |
| CN106008479B (zh) * | 2015-03-06 | 2020-01-10 | 南京圣和药业股份有限公司 | 作为磷脂酰肌醇3-激酶δ抑制剂的取代嘧啶类化合物及其应用 |
| CN105936635B (zh) * | 2015-03-06 | 2019-06-21 | 南京圣和药业股份有限公司 | 作为磷脂酰肌醇3-激酶δ抑制剂的化合物及其应用 |
| NZ739090A (en) | 2015-10-02 | 2025-06-27 | Hoffmann La Roche | Bispecific antibodies specific for pd1 and tim3 |
| CN107602465A (zh) * | 2016-07-12 | 2018-01-19 | 重庆大学 | 一种喹诺酮类衍生物及其应用 |
| RU2761377C2 (ru) | 2017-04-03 | 2021-12-07 | Ф. Хоффманн-Ля Рош Аг | Иммуноконъюгаты антитела к pd-1 с мутантом il-2 или с il-15 |
| TWI690538B (zh) | 2017-04-05 | 2020-04-11 | 瑞士商赫孚孟拉羅股份公司 | 特異性結合至pd1至lag3的雙特異性抗體 |
| EP4125861A4 (en) * | 2020-04-03 | 2024-07-10 | The Regents of the University of California | PROTEIN-PROTEIN INTERACTION STABILIZERS |
Family Cites Families (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| FR2733233B1 (fr) * | 1995-04-20 | 1997-05-30 | Roussel Uclaf | Nouveaux derives de quinoleine, leur procede de preparation, les nouveaux intermediaires obtenus, leur application a titre de medicaments et les compositions pharmaceutiques les renfermant |
| US6080757A (en) | 1996-06-06 | 2000-06-27 | Pfizer Inc | Antibiotic quinolones and derivatives |
| EP0938477A4 (en) | 1996-11-13 | 1999-12-29 | Cephalon Inc | BENZOTHIAZOLO AND RELATED HETEROCYCLIC GROUPS CONTAINING CYSTEIN AND SERINE PROTEASE INHIBITORS |
| AR035016A1 (es) * | 1999-08-25 | 2004-04-14 | Takeda Chemical Industries Ltd | Composicion de azol promotor de produccion/secrecion de neurotrofina, compuesto prodroga del mismo, composicion farmaceutica que lo comprende y uso del mismo para preparar esta ultima. |
| AU2002305926A1 (en) * | 2001-02-05 | 2002-10-08 | Exegenics Inc. | Cysteine protease inhibitors |
| US7973064B2 (en) * | 2001-11-27 | 2011-07-05 | The Board Of Trustees Of The University Of Illinois | Method and composition for potentiating an opiate analgesic |
| SE0104331D0 (sv) * | 2001-12-19 | 2001-12-19 | Astrazeneca Ab | Novel compounds |
| AU2003211931A1 (en) * | 2002-02-13 | 2003-09-04 | Takeda Chemical Industries, Ltd. | Jnk inhibitor |
| WO2004014388A1 (en) | 2002-08-13 | 2004-02-19 | Warner-Lambert Company Llc | 6,6-fused heteroaryl derivatives as matrix metalloproteinase inhibitors |
| RU2275361C3 (ru) * | 2002-11-20 | 2021-10-01 | Джапан Тобакко Инк. | Соединение 4-оксохинолина и его применение в качестве ингибитора вич интегразы |
| WO2005014576A1 (ja) * | 2003-08-12 | 2005-02-17 | Takeda Pharmaceutical Company Limited | イソキノリノン誘導体、その製造法および用途 |
| WO2005091857A2 (en) * | 2004-03-12 | 2005-10-06 | Bayer Pharmaceuticals Corporation | 1,6-naphthyridine and 1,8-naphthyridine derivatives and their use to treat diabetes and related disorders |
| JO2959B1 (en) | 2007-05-14 | 2016-03-15 | جانسين فارماسوتيكا ان. في | Mono-hydrochloric salts for histone dacetylase inhibitor |
-
2008
- 2008-05-05 WO PCT/EP2008/055470 patent/WO2009015917A2/en not_active Ceased
- 2008-05-12 AR ARP080101996A patent/AR066531A1/es unknown
- 2008-05-13 RU RU2009146124/04A patent/RU2466993C2/ru not_active IP Right Cessation
- 2008-05-13 AT AT08759523T patent/ATE551326T1/de active
- 2008-05-13 BR BRPI0811601A patent/BRPI0811601A2/pt not_active IP Right Cessation
- 2008-05-13 AU AU2008250049A patent/AU2008250049A1/en not_active Abandoned
- 2008-05-13 JP JP2010507896A patent/JP5160638B2/ja not_active Expired - Fee Related
- 2008-05-13 CN CN2008800158094A patent/CN101679270B/zh not_active Expired - Fee Related
- 2008-05-13 CA CA2686385A patent/CA2686385C/en not_active Expired - Fee Related
- 2008-05-13 WO PCT/EP2008/055807 patent/WO2008138920A1/en not_active Ceased
- 2008-05-13 CL CL2008001393A patent/CL2008001393A1/es unknown
- 2008-05-13 ES ES08759523T patent/ES2382137T3/es active Active
- 2008-05-13 TW TW097117583A patent/TW200908974A/zh unknown
- 2008-05-13 EP EP08759523A patent/EP2148862B1/en not_active Not-in-force
- 2008-05-13 KR KR1020097025880A patent/KR101176711B1/ko not_active Expired - Fee Related
- 2008-05-13 MX MX2009012222A patent/MX2009012222A/es active IP Right Grant
- 2008-05-14 US US12/152,432 patent/US8163906B2/en not_active Expired - Fee Related
-
2009
- 2009-10-22 IL IL201686A patent/IL201686A0/en unknown
Also Published As
| Publication number | Publication date |
|---|---|
| CN101679270A (zh) | 2010-03-24 |
| KR101176711B1 (ko) | 2012-08-23 |
| CN101679270B (zh) | 2013-05-01 |
| WO2009015917A2 (en) | 2009-02-05 |
| MX2009012222A (es) | 2009-12-01 |
| ATE551326T1 (de) | 2012-04-15 |
| CA2686385C (en) | 2016-01-12 |
| AR066531A1 (es) | 2009-08-26 |
| TW200908974A (en) | 2009-03-01 |
| CA2686385A1 (en) | 2008-11-20 |
| EP2148862B1 (en) | 2012-03-28 |
| WO2008138920A1 (en) | 2008-11-20 |
| KR20100007987A (ko) | 2010-01-22 |
| JP2010527341A (ja) | 2010-08-12 |
| ES2382137T3 (es) | 2012-06-05 |
| EP2148862A1 (en) | 2010-02-03 |
| CL2008001393A1 (es) | 2009-05-22 |
| IL201686A0 (en) | 2010-05-31 |
| US8163906B2 (en) | 2012-04-24 |
| RU2009146124A (ru) | 2011-06-20 |
| AU2008250049A1 (en) | 2008-11-20 |
| US20080287458A1 (en) | 2008-11-20 |
| RU2466993C2 (ru) | 2012-11-20 |
| JP5160638B2 (ja) | 2013-03-13 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| BRPI0811601A2 (pt) | composto, método para tratar um distúrbio mediado por jnk em um paciente que tem um distúrbio mediado por jnk, composição farmacêutica, uso do composto e medicamento que compreende o composto. | |
| CN102131801B (zh) | 1,2-二取代的杂环化合物 | |
| KR101152162B1 (ko) | 치환된 피리미딘 및 jnk 조절제로서 그의 용도 | |
| BR112018007021B1 (pt) | Composto, composição compreendendo os ditos compostos e uso dos mesmos para tratar fibrose cística | |
| PT2841437T (pt) | Derivados de imidazotiadiazol e imidazopirazina como inibidores de recetor 4 ativado por protease (par4) para o tratamento de agregação plaquetária | |
| US20190000807A1 (en) | Organic compounds | |
| CN108349981A (zh) | 新型的吡唑并[3,4-d]嘧啶化合物或其盐 | |
| JP2013519724A (ja) | Nadphオキシダーゼ阻害剤としてのピラゾロピペリジン誘導体 | |
| KR20140143796A (ko) | Wnt 신호 경로의 인다졸 저해제들 및 이의 치료요법적 용도 | |
| HUE030067T2 (en) | Bicyclic pyrazinone derivatives | |
| EP2799437A1 (en) | Quinoline and cinnoline compounds and use thereof | |
| BR112015030399B1 (pt) | Derivados heterocíclicos, uso dos referidos derivados e composição farmacêutica para a prevenção ou tratamento de doenças associadas com a ativação da proteína stat3 | |
| CN111655693B (zh) | 抑制瞬时型感受器电位a1离子通道 | |
| BR112020006239A2 (pt) | composto de griseofulvina e uso farmacêutico do mesmo | |
| CN111548345A (zh) | 一类苯并咪唑类衍生物及其制备方法和应用 | |
| TR201900735T4 (tr) | Yeni kaynaşık imidazobenzotiyazol bileşikleri. | |
| CN103965107B (zh) | 2-芳基取代喹啉类化合物及其用途 | |
| JP7836137B2 (ja) | 芳香族複素環系化合物の結晶形、その組成物、製造方法及びその応用 | |
| CN114685519B (zh) | 一类吡喃并咔唑生物碱衍生物及其治疗阿尔茨海默症的用途 | |
| CN112047941A (zh) | 一种化合物及其制备由Flt3/c-Met激酶高表达所引起疾病的药物的应用 | |
| CN119930563B (zh) | 一种瞬时受体电位阳离子通道trpv3抑制剂及其用途 | |
| TW202448441A (zh) | 脯氨醯羥化酶抑制劑及其用途 | |
| CN111393363A (zh) | 4-苯氧基喹啉并n-磺酰脒类化合物及其制备方法和用途 | |
| CN104003990A (zh) | 杂环胺类Hedgehog信号通路抑制剂 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| B06F | Objections, documents and/or translations needed after an examination request according [chapter 6.6 patent gazette] | ||
| B07D | Technical examination (opinion) related to article 229 of industrial property law [chapter 7.4 patent gazette] |
Free format text: DE ACORDO COM O ARTIGO 229-C DA LEI NO 10196/2001, QUE MODIFICOU A LEI NO 9279/96, A CONCESSAO DA PATENTE ESTA CONDICIONADA A ANUENCIA PREVIA DA ANVISA. CONSIDERANDO A APROVACAO DOS TERMOS DO PARECER NO 337/PGF/EA/2010, BEM COMO A PORTARIA INTERMINISTERIAL NO 1065 DE 24/05/2012, ENCAMINHA-SE O PRESENTE PEDIDO PARA AS PROVIDENCIAS CABIVEIS. |
|
| B08F | Application dismissed because of non-payment of annual fees [chapter 8.6 patent gazette] |
Free format text: REFERENTE A 12A ANUIDADE. |
|
| B08K | Patent lapsed as no evidence of payment of the annual fee has been furnished to inpi [chapter 8.11 patent gazette] |
Free format text: REFERENTE AO DESPACHO 8.6 PUBLICADO NA RPI 2566 DE 2020-03-10 |
|
| B350 | Update of information on the portal [chapter 15.35 patent gazette] |