BRPI0918802A2 - uso de (s)-n-[[3-[3-fluoro-4-(4-tiomorfolinil)fenil]-2-oxo-5-oxazolidinil] metil] acetamida e composição farmacêutica - Google Patents
uso de (s)-n-[[3-[3-fluoro-4-(4-tiomorfolinil)fenil]-2-oxo-5-oxazolidinil] metil] acetamida e composição farmacêutica Download PDFInfo
- Publication number
- BRPI0918802A2 BRPI0918802A2 BRPI0918802A BRPI0918802A BRPI0918802A2 BR PI0918802 A2 BRPI0918802 A2 BR PI0918802A2 BR PI0918802 A BRPI0918802 A BR PI0918802A BR PI0918802 A BRPI0918802 A BR PI0918802A BR PI0918802 A2 BRPI0918802 A2 BR PI0918802A2
- Authority
- BR
- Brazil
- Prior art keywords
- formula
- compound
- tuberculosis
- treatment
- pharmaceutically acceptable
- Prior art date
Links
- GJYBCANWJZUNKE-HNNXBMFYSA-N CC(NC[C@@H](CN1c(cc2)cc(F)c2N2CCSCC2)OC1=C)=O Chemical compound CC(NC[C@@H](CN1c(cc2)cc(F)c2N2CCSCC2)OC1=C)=O GJYBCANWJZUNKE-HNNXBMFYSA-N 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/54—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one sulfur as the ring hetero atoms, e.g. sulthiame
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/54—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one sulfur as the ring hetero atoms, e.g. sulthiame
- A61K31/541—Non-condensed thiazines containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/42—Oxazoles
- A61K31/421—1,3-Oxazoles, e.g. pemoline, trimethadione
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4409—Non condensed pyridines; Hydrogenated derivatives thereof only substituted in position 4, e.g. isoniazid, iproniazid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/47—Quinolines; Isoquinolines
- A61K31/4709—Non-condensed quinolines and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/496—Non-condensed piperazines containing further heterocyclic rings, e.g. rifampin, thiothixene or sparfloxacin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/4965—Non-condensed pyrazines
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/5375—1,4-Oxazines, e.g. morpholine
- A61K31/5377—1,4-Oxazines, e.g. morpholine not condensed and containing further heterocyclic rings, e.g. timolol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
- A61P31/06—Antibacterial agents for tuberculosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Medicinal Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- Epidemiology (AREA)
- General Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Engineering & Computer Science (AREA)
- Pulmonology (AREA)
- Communicable Diseases (AREA)
- Oncology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Abstract
terapia de combinação para tuberculose a presente invenção refere-se a métodos de tratamento de tuberculose, incluindo variedades resistentes a multifármaco e tuberculose latente_ mais particularmente, a presente invenção refere-se a um método de tratamento de tuberculose em um mamífero compreendendo administrar a referidos mamíferos em necessidade do mesmo uma quantidade eficaz de um composto de fórmula (i), (s)-n-[[3-[3-fluoro-4-(4-tiomorfolinil)fenil]-2-oxo- 5-oxazolidiníl]metil]acetamida, ou um sal farmaceuticamente aceitável do mesmo em combinação com pelo menos dois agentes úteis no tratamento de tuberculose. a presente invenção também se refere a uma composição farmacêutica compreendendo uma quantidade terapeuticamente eficaz de um composto de fórmula (i) ou um sal ou solvato farmaceuticamente aceitá- vel do mesmo, (ii) uma quantidade terapeuticamente eficaz de pelo menos um agente útil no tratamento de tuberculose e (iii) um ou mais transportado-res ou veículos farmaceuticamente aceitáveis.
Description
COMBINAÇÃO PARA TUBERCULOSE.
Campo da Invenção
A presente invenção refere-se a métodos de tratamento de tu5 beroulose, incluindo variedades resistentes a multifármaca e tuberculose latente. Mais páttteularmente, a presente invenção refere-se a um método de tratamento de tuberculose em um mamífero compreendendo administrar ao retendo mamífero em necessidade do mesmo uma quantidade eficaz de urn composto de fórmula (I), (S)-N-[[3-[3-fluoro-4-(4-tiomorfolinil)fenil]~2-oxo-510 oxãzolidiníl] metii] acetamido, ou um sai farmaceuticamente aceitável do mesmo em combinação com pelo menos dois agentes úteis no tratamento de tuberculose. A. presente invenção também se refere a uma composição t farmacêutica compreendendo: i) uma quantidade terapeuticamente efetiva de um composto de fórmula (I) ou um sal farmaceuticamente aceitável do mesmo, (li) uma quantidade terapeuticamente eficaz de pelo menos um agente útil no tratamento de tuberculose e (ili) um ou mais transportadores ou veículos farmaceuticamente aceitáveis.
Antecedentes da Invenção
A tuberculose (TB) mata aproximadamente 1,6 milnào de pesso20 as ac redor do mundo a cada ano, tornando-a a segunda condução de óbito de adultos aíòm do H!V.. Quase 500.000 novos casos de TB resistente a muitifármaco (MDR) ocorrem a cada ano. e a emergência recente de TB extensivamente resistente a fánmaco (XDR-TB) pressagia novas epidemias de TB não tratãvel. (Ver Dorman, S. E. et al, Nat.Med 13:295-298 2007, Zignoí, M.
et ai., 3 Infect. Dis 194:478-480. 2008). Novos fármacosfármacos com atividade antituberculose potente, especialmente contra persistidores de não multiplicação, são necessárias para encurtar a duração de tratamento para TB e. desse modo, facilitar a implementação global de terapia díretamente observada. (Ver O'Brien, R. J et al., Am.J.Respir. Crit Care Med. 163.1085··
1053,2001).
Atualmente, o tratamento de tuberculose sensível a fármaco consiste em administrar uma combinação de pelo menus os seguintes fár
2/63 macos, isoniazid, rifampin a pirazinamida, Para tratamento eficaz, ca fàrmanas acima mencionados são dados a um paciente em uma fase inicial de tratamento por 8 semanas, durante as quais os fármacos são usados em combinação para matar a população que se multiplica rapidamente de My5 cobacíencrm febemt/iose, bem como para prevenir a emergência de resistência a fármaco. A fase inicial de tratamento é seguida por uma fase de continuação ou de esterilização pm 18 semanas durante as quais dois ou mais fármacos de esterilização (por exemplo, isoniazid e rifampin) sãs dados para matar a população de que divide intermitentemante (persistidores de 10 nâo multiplicação) de Mycobac/enum fudercufose.
Enquanto a combinação acima mencionada de fármacos juntos proporciona tratamento contra infecção de Mycobacterium tub&rciriose sensível em 4 a 6 meses de tempo, tal terapia de combinação não ê sempre bem sucedida, espacialmente em pacientes que abrigam cepas resistentes a 15 fármaco. Também, a longa duração de tratamento consistindo amseis meses pode conduzir a efeitos colaterais desagradáveis, Adicionalrnente. a concordância com o curso relativamente longo de tratamento é geraimente pobre. Tal discordância pode conduzir a falha de tratamento resultando no desenvolvimento de resistência a fámaco.
As oxazolidinonas compreendem urna ciasse de inibidores de síntese de proteína que bloqueiam translação por prevenção da formação do complexo de iniciação (para mecanismo, ver K, Leach et al., Molecular Ceil, 26:4, 480-462, 2007). Linezolid (L.ZD, Zyvox), a única oxazoiidinona comercializada, tem atividade contra bactéria Gram-positiva e è atualmente apro25 vada para uso em pele complicada e infecções de estrutura de pele e pneumonia adquirida em hospital (inserto da acondrcionamento Zyvox^). Contudo, ela também é ativa contra muitas espécies micobacteriais, incluindo Mycoíbacteríum fubemu/use, para qual seu MIC varia de 0,126-1 gg/rnl, cem um MIC50 de 0,5 pg/ml e um MIC&> de 1 pg/mL. (Ver Aloala., L., et al., Anti30 microb Agents Chemother 47:416-417. 2003: Cynamon, Μ, H., et al., Antímícrob Agents Chemother 43.1189-1191, 1999; Fattorini, L.. et ai., Anfimierob Agents Chemother 47.360-362, 2003). Como um resultado, LZD foi usado
3/63 fora das indicações etiquetadas para tratar eases recalcitrantes de MDR- e XDR-TB. Embora várias series de casas sugiram que LZD pode contribuir para conversão de cultura da saliva bem sucedida em tal caso, sua atividade individual em pacientes com TB e sua contribuição precisa a regimes de 5 combinação permanecem não claras. Estes estudos também demonstram que a duração de administração de LZD pode ser limitada por toxicidade hematológíca e neurológica que pode ocorrer com administração de longo prazo, (Ver Fortun, J.. et al, 56:160-185. 2005: Park. I. R.. et al.. Antimícrob Chemolher 58:701-704, 2006: Zignol. et ai... J Infect.Dis 194:479-485, 10 2006). Portanto, novas oxazolidinonas com atividade mate patente te wo contra A4ycoãacfete.rm iubereteoae e risco mate baixo de toxicidade corn administração prolongada são desejáveis.
As oxazolidinonas com atividade mais potente contra Mycubacfertem íuderuteose foram descritas anteriormente. (Ver Barbachyn. M. R.. et 15 al, J Med Chem. 39:680-685, 1996: Sood, R., et al, Antimicrob Agents Chemcther 49:4351-4353, 2005). A atividade de antituberculose do composto de fórmula (l). (S}-N-fi3-[3-fIuoroA-(4-tiomorfoiinil)feníl}-2-oxo-5uxazolídind] metíl] acetemida. tei primeiro descrita em 1996. (Ver Barbachyn, M. R.. et ai.. J Med Chem. 39:680-685, 1996). Experimentos subsequentes 20 em um modela de murina encontram o composto de fórmula (I) a ser mais ativo do que LZD quando ambas as fármacos foram administradas a 100 mg/kg, mas a relevância clinica desta dose de LZD não foi estabelecida e as atividades do composto de fórmula (I), e LZD não foram dararnente diferentes quando comparado a doses mais baixas. (Ver Cynarnon, Μ. H.. et al., 25 Anf.imiorob Agents ühemother 43:1189-1191, 1999). Além disso, embora o composto de fórmula (I) pareça ter atividade modesta quando combinado com rifampin (RIF) em um modelo de infecção agudo (precoce), o composto de? fórmula <l> não tinha atividade adicional quando oombmados corn isoniazid ísNH}. (Cynamon, Μ. H., et al., Anhmlcrob Agents Chemother 43.118930 1191, 1999 ) Mais impodanfemente, a atividade do composto de fórmula (l).
se sozinho ou em combinação com RIF ou INH, foi somente avaliada sobre as 4 semanas iniciais de tratamento que ê insuficiente para avaliar a atívida
4/63 de de um composto ou combinação de agentes contra persislidores de não multiplicação que, por sua vez, ultimamente determina a duração de tratamento necessário para cura (isto é. prevenção de relapso após completação de trata mento)
Consequentemente, existe uma necessidade urgente de desenvolver regimes mais recentes que possam ser usadas para prevenir, tratar e/ou reduzir tuberculose e/ou eliminar a ameaça de tuberculose resistente a rnultifãrmaco ou encurtar a duração de tratamento,
Bumánp,da In venção
A presente invenção refere-se a um método de tratamento de tuberculose em um mamífero compreendendo administrar a um referido mamífero em necessidade do mesmo uma quantidade eficaz de um composto de fórmula (I) ou um sal farmaceuticamente aceitável do mesmo:

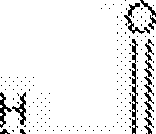

(0 em combinação com pelo menos dois agentes úteis no tratamento der tuberculose.
Em urna concretização, os pelo menos dois agentes são selecionados a partir do grupo consistindo em isoniazid, rifampin, nfapentina, rifabutin, pirazinamida, etambutoi, estreptomicina, canamicina, amicacin, 20 moxifloxacin, gatifioxacin. Isvctloxacm, ofloxacin, ciprofloxacin, capreomicin, etionamida, ciclossenna, ácido para-aminussaiicllicc. tiacetazena, claritromi·· cin, amoxilina-ácido clavulânico, ímípenem, meropenem, viomícin, terizidona, clofazlmina, TMC207, PA-824, OPC-7683, LL-3858 e SQ-109.
Em outra concretização, um dos referidos pelo menus doss agen25 ies é selecionada a partir do grupo consistindo em, isomazid, rifampin, rifapeniina, rifabuhn, pirazinamida. moxifloxacin, gatifloxscin, levofloxacin, ofloxacin, ciprofloxacin, clofazimine e etambutol
Em ainda outra concretização, um dos referidos pelo menos dois
5/63 agentes è pirazlnamida.
Era alada outra concretização, ura dos referidos pelo menos dois agentes è rifampin.
Em ainda outra concretização, um dos referidos pelo menos dois 5 agentes ê rifapentina.
Em ainda outra concretização, um des referidos pelo menos dois agentes è PA824..
Em ainda outra concretização, um dos referidos pelo menos dois agentes é OPC -67683
Em ainda outra concretização, um dos referidos pelo menos dois agentes é TMC-207.
Em ainda outra concretização, um dos referidos pelo menos dois agentes é selecionado a partir do grupo consistindo em moxifloxacin, gatiflO xacin, tevoflexscin, e ofloxacin.
Em ainda outra concretização, um dos referidos pelo menos dois agentes é moxifloxacin.
Em urna concretização especifica, referidos pela menos dors agentes são pirazinamida e rifampin.
Em ainda outra concretização específica, referidos pelo menos 20 dois agentes são pirazinamida, rifampin e isoniazid.
Em ainda outra concretização especifica, referidas pelo menos dois agentes são pirazinamida e rifapentina.
Em ainda outra concretização especifica, referidas pelo menos dois agentes são pirazinamida, rifapentlna, e isoniazid.
Em uma concretização especifica, referidos pela menos dois agentes são pirazinamida e moxifioxacin..
Em ainda outra concretização específica, referidos pelo menus dois agentes são pirazinamida, moxifluxacin. e rifampin.
Em ainda outra concretização especifica, referidas pelo menus 30 dois agentes sâa pirazinamida. moxifioxacin, e rifepentína.
Em ainda outra concretização específica, referidas pelo menus dois agentes são PA-824 e pirazinamida.
6/63
Em ainda outra coftetefeação específica, referidos· peto menos dais agentes são PA-824, pirazínamida © um agente selecionado a partir do grupo consistindo emestreptomicina, canamicina, amícacin e capreornicin.
Em ainda outra concretização específica, referidos peto menos dois agentes são PA-824 e moxifloxacin.
Em ainda outra concretização específica, referidos peto menos dois agentes são PA-824, moxiftoxacin e urn agente selecionado a partir de grupo consistindo em estreptomicína, canarn-icína, amicacin e capreomicin.
Em ainda outra concretização específica, referidos pelo menos D dois agentes são OPC-67683 e pírazinamrda.
Em ainda outra concretização específica, referidos pelo menos dois agentes são OPC-67683, pírazinamida e um agente selecionado a partir do grupe consistindo ern estreptomicina, canamicma, amícacin e capreomicin.
Em ainda outra concretização específica, referidos pelo menos dois agentes são OPC-67683 e moxifioxaoin.
Em ainda outra concretização especifica, referidos pelo menos dois agentes são OPC-67683, moxifioxadn e um agente selecionado a partir do grupo consistindo ern estreptomicina, canamícina, amicacin e capreomi0 cín.
Em ainda outra concretização específica, referidos pelo menos dois agentes são TMC-207 e pirazinamida.
Em ainda outra concretização específica, referidos pelo menos dois agentes são TMC-207, pirazinamida e moxifloxacin ou isoniazid,
Em ainda outra concretização, referida tuberculose compreende tuberculose ativa ou tuberculose latente.
Em ainda outra concretização, referida tubercuíuse ativa compreende tuberculose sensível a fármaco, resistente a monofármaco, resistente multífàrmacc- (MDR) ou tuberculose extensrvamente resistente a tár0 maco (XDR).
Em ainda outra concretização, o método da presente invenção erradica completamente tuberculose sensível a fãrmaco. tuberculose rests
7/63 tents a monofàrmaoo. tuberculose resistente a muhífârmaco, e tuberculose extensivamente resistente a fàrmace (XDR) em completação do tratamento.
Em ainda outra concretização, referida tuberculose é causada por uma infecção de Mycobacterium selecionada a partir do grupo consistin·· 5 do em Mycobacterium tebercutese, Mycobacterium botes ou outras espécies de micobacteriais relacionadas.
Em ainda outra concretização, o método da presente invenção prevení relapso da infecção de Mycobacterium apôs oompieteção do tratamento.
Em ainda outra concretização. o composto de fórmula (I) e sais farmaceuticamente aceitáveis destes é administrado oralmente.
Em ainda outra concretização, referidos pelo menos dois agentes são cada um administrado oralmente.
Em ainda outra concretização, referidos pelo menos dois agem 15 íes são administrados juntos em uma composição.
Em ainda outra concretização, referidos pelo menos dois agentes são administrados separadamente
Em ainda outra concretização, referidos pelo menos dois agentes são administrados juntos e outro agente útil para o tratamento de tuber20 ouiose é administrado separadamente.
Em ainda outra concretização, o composto de fórmula (!) e sais farmaceuticamente aceitáveis destes é administrado uma vez por dia (QD) ou duas vezes por dia (BID).
Em ainda outra concretização, o composto de fórmula (I) e sais farmaceuticamente aceitáveis destes é administrado uma vez. por semana, duas vezes por semana, très vezes por semana ou todo dia
Em uma concretização, o composto de formula (!) ou um sal farmaceuticamente aceitável do mesmo é administrado entre cerca de 10 mg a cerca de 2000 mg.
Em ainda outra concretização, o composto de fórmula (I), ou um sai farmaceuticamente aceitável do mesmo, é administrado entre cerca de 250 mg a cerca de 1000 mg
8/63
Em ainda outra concretização, o composto de fórmula (l>, ou um sai farmaceuticamente aceitável do mesmo, é administrado entra cerca de 600 mg a cerca de 1000 mg.
Em ainda outra concretização, o composto de fórmula (I) ou um sal farmaceuticamente aceitável do mesmo é administrado entre cerca de 25 mg a cerca de 1000 mg.
Em ainda outra concretização, o composto de fórmula (1) ou um sal farmaceuticamente aceitável do mesmo é administrado entre cerca de 50 mg a cerca de 500 mg.
Em ainda outra concretização, o composto de fórmula (I) nu um sal farmaceuticamente aceitável do mesmo é administrado entre cerca de 100 mg a cerca de 500 mg.
A presente invenção também se refere a um método de tratamento de tuberculose em um mamífero apôs referido mamífero ter suportado uma fase inioíai de tratamento compreendendo administrar a referido mamífero em necessidade do mesmo uma quantidade efetiva de um composto de fórmula (I) ou urn sal farmaceuticamente aceitável do mesma

cm combinação corn pelo menos um agente útil no tratamento de tuberculose.
Em uma concretização, referido peio menos um agente é selecionado a partir do grupo consistindo em rifampin, rif.apentina. rifabutin, pirazinamida, eíambutol, estraptomicina, canamicina, amicaoín, mcxifioxacin, gatífloxacin. levofloxacin, ofloxacin, ciprofloxacin, capreomicin, ebonamida, cicíosserina, ácido para-aminossalieílíco, tlacetazona. claritromlcin, amsxíli· na-ácido oiavulânico, imipenem. meropenem. clofazimine. viomicin, terizidona, TMC207, PA-824, CPC-7683, LL-3858 e SQ-109.
Em ainda outra concretização, referido pelo menos um agente ê
9/63 selecionado a partir do grupo consistindo em rifampin, ritapentina. rifabutin, pirazínamída. moxifloxacin, gatifloxacin, levofloxacin, ofloxacin, o etambutol.
Em uma concretização especifica, referido pelo menos um agen te ê pírazinamida.
Em uma concretização especifica, referido pelo menos um agente é rifampin.
Em uma concretização específica, referido pelo menos um agente é rifapentína.
Em uma concretização específica., referido pelo menos um agsn10 teéPA-824..
Em uma concretização específica, referido pelo menos um agente é QPC-7883.
Em uma concretização específica, referido pelo menos um agenteéTMC-207;
Em uma concretização específica, referido pelo menos um agente é selecionado a partir do grupo consistindo em moxiHoxaoia, gatifloxacin. levofloxacín, e ofloxacin.
Em urna concretização específica, referido pelo menus um agen te é rnoxitluxaciã;
Em ainda outra concretização, referida tuberculose compreende tuberculose ativa ou tuberculose latente.
Ern ainda outra concretização., referida tuberculose ativa compreende tuberculose sensível a fármaco, tuberculose resistente a monofàrmaco, tuberculose? resistente? a multifármaco (MDR) ou tuberculose extensi25 vamente resistente a fármaco (XDR).
Em ainda outra concretização, o método da presente invenção erradica oompletamente tuberculose sensível a fármaco, tuberculose resistente a monofármaco, tuberculose resistente a multifármaco e tuberculose extensivamente resistente a fármaco (XDR) na completação do tratamento.
Em ainda outra concretização, referida tuberculose è causada por uma infecção de Mycobactebum selecionada a partir do grupo consistindo em Mycobacterium fubercu/ose, Mycobacterium botes ou outras espécies
10/63 m icobacte riais reladonad as.
Em ainda outra concretização, o método da presente invenção previne relapso de infecção de Mycodacferw? após compietaçâo do tratamento.
Em ainda outra concretização, o composto de fórmula (I) e sais farmaceuticamente aceitáveis destes é administrado oralmente.
Em ainda outra concretização, referido pelo menos um agente é administrado oralmente.
Em ainda outra concretização, o composto de formula (!) e sais farmaceuticamente aceitáveis destes e referido peto menos um agente são administrados juntos em uma composição.
Em ainda outra concretização, o composto de fórmula (!) e sais farmaceuticamente aceitáveis destes e refendo paio menos um agente são ad ministrados separada men te.
Em ainda outra concretização, o composto de fórmula (!) e sais farmaceuticamente aceitáveis destes é administrado uma vez por dia (QD) ou duas vozes por dia (BID).
Em ainda outra concretização, o composto de fórmula (!) e sais farmaceuticamente aceitáveis destes é administrado uma vez por semana,, duas vezes por semana, três vezes por semana, ou todo dia.
Em uma concretização, o composto de fórmula (!) ou um sal farmaceuticamente aceitável do mesmo ê administrado entre cerca de 10 mg a cerca de 2000 mg.
Em ainda outra concretização, o composto de fórmula (!) ou um sal famraceuticamente aceitável do mesmo ê administrado entre cerca de 25 mg a cerca de 1000 mg.
Em atnda outra concretização, o composto de formula (!) ou um sal farmaceuticamente aceitável do mesmo é administrado entre cerca de 50 mg a cerca de 500 mg.
Em ainda outra concretização, o composto de fórmula (I) ou um sal farmaceuticamente aceitável do mesmo è administrado entre cerca de WG mg a cerca de 500 mg
A presente invenção também se refere a uma composição farmacêutica compreendendo:
uma quantidade terapeutícamente eficaz de um composto de fórmula (t) ou um sai famiaceuticamente aceitável' do mesmo:
Q

(ii) uma quantidade terapeutícamente eficaz de pelo menos um agente útil no tratamento de tuberculose, e (üí) um ou mais transportadores ou veículos farmaceuticamente aceitáveis.
Em ainda outra concretização, referido pelo menos um agente é selecionado a partir do grupo consistindo em isoniazid, rifampin, rifapentina, rifabutin, pirazinamida, etambutol, estreptomicina, canamicina, amicacin, moxifloxacin. gatifloxacin, ievofioxacin, ofloxacin, ciprofloxacin, oapreomicin, etionamida, cícíosserína, ácida para- ami nossa I icih'co, tiacetazona, claritromi15 cin, clofazimine, arnoxílina-àcíde clavulânico, imipenem, meropenem. viomicin, tenzidona, clofazimine, TMC2Ü7. PA-824, ÜPC-7683, LL-3858 e SQIDG.
Em ainda outra concretização, referido pelo menos um agente è selecionado a partir do grupo consistindo emisoníazid, rifampin, rifapentina, 20 rifabutin, pirazinamida, moxifloxacin, gatifloxacin, ievofloxacíra ofloxacin, e etambutol.
Em uma concretização especifica, referido pelo menos um agente è pirazinamida
E.m uma concretização específica, referido pelo menos um agen25 te é rifampin.
Em uma concretização específica, referido pelo menos um agen te é rifapentina.
Em uma concretização específica, referido pelo menos um agente è isoniazid.
Em unia concretização específica.. referida peto menos um agen' te é PA-824,
Em urna concretização específica, referida pelo menos um agen5 te é OFQ-7683,
Em urna concretização específica, referido pelo menos um agente é TMC-207.
Ern uma concretização específica, referido pelo menus urn agente é selecionado a partir do grupo consistindo em moxrfióxacin, gatifloxacin, 10 levafluxacin, e ofloxacin.
Ern uma concretização especifica, referido pelo menus um agen te ê rnoxifloxacin.
Em asida outra concretização, a composição farmacêutica compreende cerca de W mg a cerca de 2000 mg de composto de fórmula (I) ou 15 um sal farmaceuticamente aceitável do mesmo.
Em ainda outra concretização, a composição farmacêutica compreende cerca de 250 mg a cerca de 1000 mg de composto de fórmula (!) ou um sal farmaceuticamente aceitável do mesmo.
Em ainda outra concretização, a composição farmacêutica com 20 preende cerca de 600 mg a cerca de 1000 mg de composta de fórmula (!) ou um sai farmaceuticamente aceitável do mesmo.
Em amda outra concretuação, a composição farmacêutica com preende cerca de 2.5 mg a cerca de 1000 mg de composto de fórmula fl) ou um sal farmaceuticamente aceitável do mesmo.
Em ainda outra concretização, a composição farmacêutica compreende cerca de 50 mg a cerca de 500 mg de composto de fórmula (!) eu um sal farmaceuticamente aceitável do mesmo.
Adicionalmente, qualquer formulação, incluindo as combinações abaixo, pode conter de 250 mg a 1000 mg do composto de formula (I) ou um 30 sal farmaceuticamente aceitável do mesmo ou de 600 mg a 1000 mg de composto de fórmula (!) ou um sal farmaceuticamente aceitável do mesmo.
Em ainda outra concretização, a composição farmacêutica com
13/63 preende cerca de 100 mg a cerca de 500 mg de composto de fórmula (I) ou um sai farmaceuticamente aceitável do mesmo.
Em ainda outra concretização, a composição farmacêutica compreende cerca de 100 mg a cerca de 500 mg de composto de fórmula (!) ou 5 um sal farmaceuticamente aceitável do mesmo e cerca de 600 mg de rifampin.
Em ainda outra concretização, a composição farmacêutica compreende cerca de 100 mg a cerca de 500 mg da composto da fórmula (I) ou um sai farmaceuticamente aceitável do mesmo a cerca de 300 mg de isonia10 zid
Em ainda outra concretização, a composição farmacêutica compreende cerca de 100 mg a cerca de 500 mg de composto de fórmula (I) ou um sal farmaceuticamente aceitável do mesmo e cerca de 300 mg de isoniazid e cerca de 600 mg de rifampin.
Em ainda outra concretização, a composição farmacêutica compreende cerca de 100 mg a cerca de 500 mg de composto de fórmula (I) ou um sal farmaceuticamente aceitável do mesmo, cerca de 300 mg de isoniazid, cerca de 600 mg de rifampin e cerca de 20-25 mg/kg a cerca de 50-70 mg/kg de pozinamida.
Em ainda outra concretização, a composição farmacêutica compreende cerca de 100 mg a cerca de 500 mg de um composto de fórmula (I) ou um sai farmaceuticamente aceitável do mesmo e cerca de 10-15 mg/kg a cerca de 20-30 mg/kg de isoniazid..
Em ainda outra concretização, a composição farmacêutica com25 preende cerca de 100 mg a cerca de 500 mg de um composto de fórmula (I) ou um sai farmaceuticamente aceitável do mesmo e cerca de 10-15 mg/kg a cerca de 20-30 mg/kg de isoniazid e cerca de 10 mg/kg a cerca de 20 mg/kg de rifampin
Em ainda outra concretização, a composição farmacêutica com30 preende cerca de 100 mg a cerca de 500 mg de um composto de fórmula (I) ou um sal farmaceuticamente aceitável do mesmo, cerca de 10-15 mg/kg a cerca de 20-30 mg/kg de isoniazid, cerca da 10 mg/kg a cerca de 20 mg/kg de rifampin e cerca de 15-30 mg&g a cerca de 50 mgZkg de pirazinamida.
Em ainda outra concretização, a composição farmacêutica compreende cerca de 50 mg a cerca de 250 mg de um composto de fórmula (I) ou um sal farmaceuticamente aceitável do mesmo, cerca de 75 mg de isoniazid, cerca de 150 mg de rifampin e cerca de 400 mg de pirazinamida.
Em ainda outra concretização, a composição farmacêutica compreende cerca de 25 mg a cerca de 250 mg de um composto de fórmula (l) ou um sal farmaceuticamente aceitável do mesmo, cerca de 300 mg de rifampin.
A presente invenção também se refere a um artigo de fabricação compreendendo:
uma composição acondicionada compreendendo:
(a)
i) uma quantidade terapeuticamente eficaz de um composto de fórmula (li ou um sal farmaceuticamente aceitável do mesmo;
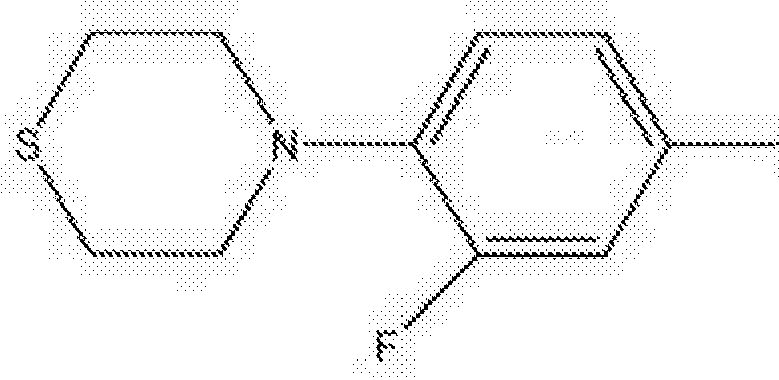

(iò uma quantidade terapeuticamente eficaz de paio menos um agente útil no tratamento de tuberculose, e (iii) um ou mais Iran sported oras ou veículos farmaceuticamente aceitáveis.
(b) um inserto proporcionando Instruções para administração da composição acondicionada de (a) para tratar tuberculose, e (o) um recipiente para (a) e (b).
A presente invenção tambérn se refere a um acondicionamento farmacêutico para tratamento de tuberculose em um mamífero que compreende um composto de fórmula (!) ou um sai farmaceuticamente aceitável do mesmo.
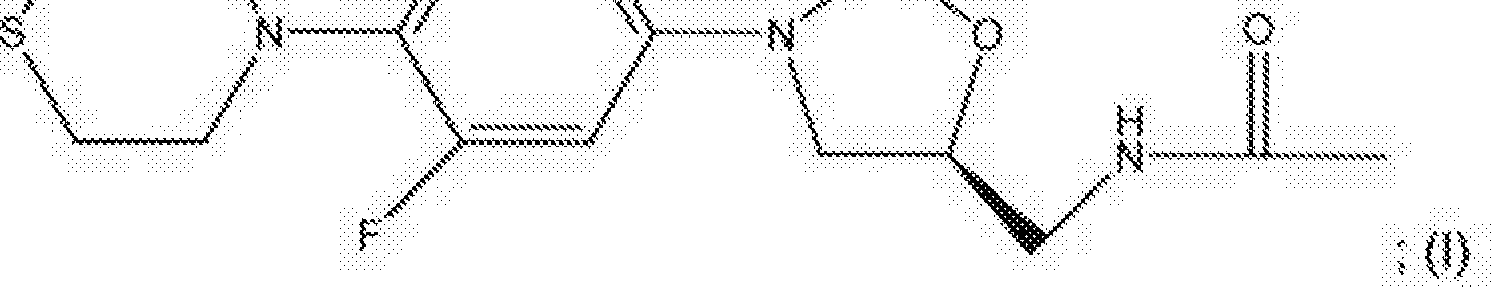
e um inserto proporcionando instruções para administração da referida com. posição em combinação com paio menos um agente útil no tratamento de ;. tubarcutese;
Estes e outros aspectos, vantagens e características da invenção tomar-se-ão aparentes pela seguinte descrição detalhada da invenção.
Descrição Detalhada da Invenção
A presente invenção refere-se a um método de tratamento de tuberculose em um mamífero compreendendo administrar a referido mamífe10 ro uma quantidade eficaz de um composto de fórmula (I) ou um sal farmaceutioamente aceitável da mesmo.
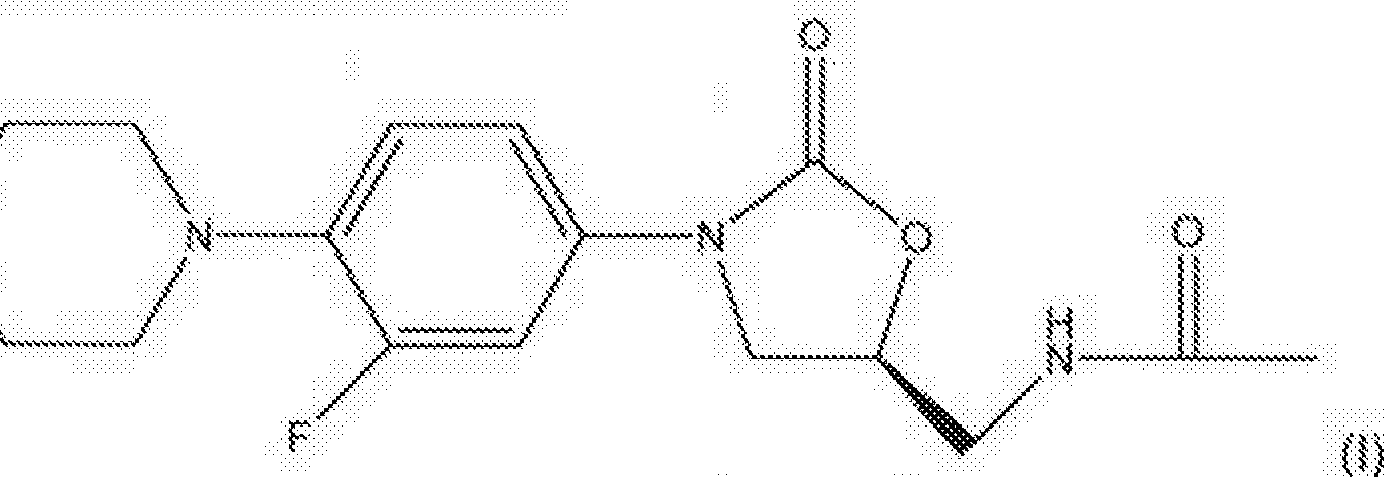
em combinação corn pelo menus dois agentes úteis no tratamento de tuberculose. ............ ............ ..........
Q composto de fórmula (I) da invenção è descrito na Patente
U.S. No. 5.880.118, (incorporada em sua totalidade aqui por referência) no Exemplo 1, (Sj“N“[[3“[3-f!uoro-4-(4 tiornorfolíníl)fenil]-2-oxo-5-oxazolidinil] metil] acetamide. Conforme descrito em maiores detalhes abaixo, o composto de fórmula (I) da invenção pode ser administrado como a base livre ou na 20 forma de urn sal do mesmo..
A frase sal tis) farmaceuticamente aceitável (is), conforme aqui usada, a menos que de outro modo indicado, inclui sais de grupos acidicos ou básicos que podem estar presentes nos compostos de fórmula (I).
Por exemplo, os compostos de fórmula (I) que são básicos na
16/63 natureza são capazes de formar urna ampla variedade de sais num vários ácidos inorgânicos e orgânicos. Os ácidos que podem ser usados para preparar sais de adição de ácido farmaceuticamenie aceitáveis de tais compostas básicos daqueles que formam sais de adição de ácido não tóxicos, isto è, 5 saís contendo âníons farmacofogíoamente aceitáveis, tais coto cloridrafo, bromidrato, iodidrato, nitrato, sulfato., bissulfato, fosfato, fosfato ácido, isonicotinato. Isolate, salieiiato, nitrato, nitrato ácido, tartarato. panfotenato, bitartarato, asoorbato. succinate, maleate, fumarate, gluconate, glucuronato, sacarato, formiato, benzoate, glutamate, metanossulfonato, eianossulfonafo, 10 benzenossulfonato, p-toíuenossuifonato e pamoato [isto é, sais de 1.1'metiieno-bis-(2-hidróxi-3-naftoato)}
Exemplos de sais incluem, mas não são limitados a, sais de acetato, acrilato, benzenossulfonato, benzoato (tais como clorobenzoato, metílbenzeato, dinitrobenzoato, hidroxibenzeato, e metoxibenzoato), bicarbonato, 15 bissulfato, bissulfito, bitanarato, borate, brometo, buKno-1,4-dieato, edeiato de cálcio, cansilato, cloreto, caproato, caprilato, nitrato, deeanoato, dl· hidrogenfosfonato, edetato, edisíiato, estolato, esilato, etilsuocinato, formiato, fumarato, gluceptate, gluconato, glutamate, glicolato, glicohíars-anilato, heptanoato, hexino-1<6-dioato. hexiiresorcinato, hidrabamina, bromidrato, oiori20 drato, γ-hidroxibutirato, iodeto, isebutirato, isotionato, lactate, lacfobíenato, laurato, maiato, maleate, malenato, mandelate, mesilate, met afestalo. metano-suifonato, metiteulfato, mono-hidrogenofosfato, mucato, napsilato, naftaieno-l-sulfonate, naftaieno-2-sulfoaafo, nitrato, oleato, oxalate, pamoate (embonato), palmitate, panfotenato, fenilacetatos, fenilbubrato, fenilpropiona25 to, ftalato, fosfato/di'tosfato, poligalaeturonato, prepanossuKonato, propionate, pmpiolato, pirofosfato, plrossuifoto, salícilato, estearato, aubacetate, suberato, succinate, sulfato, sulfonato. sulfite, tanato, tertarato. fecclato, tesiiato, trietiodode. e valerate.
A invenção também se refere a sais de adição de base dos 30 compostos de fórmula (lí. As bases químicas que podem ser usadas como reagentes para preparar sais de base farmaceuticamonte aceitáveis dos compostos de fórmula (!) que são acídicos na natureza são aqueles que formam sais de bases não tóxicas com tais compostos. Tais sass de base não tóxicos incluem, mas não são limitados a, aqueles derivados de tais cátions farmacologicamente aceitáveis tars como nations de metal alcalino (por exemplo, potássio e sódio) e cations de metal aícalinoterroso (por exemplo, cálcio e magnésio), sais de adição de amônia ou amina solúvel em água tais como N-metilglucamina-Cnmglumina), e o alcanolamônia inferior e outros saís de base de aminas orgânicas farmaceuticamente aceitáveis
Hemissais de ácidos e bases podem também ser formados, por exemplo, sais de hemissulfato e hemicáfcio
Para uma revisão dos sais adequados, ver Handbook of Pharmaceutical Salts: Properties. Selection, end Use by Stahl e Wermuth (WileyVCH. 2002). Métodos para produção de sais farmaceuticamente aceitáveis de compostos da fórmula (I) da invenção são conhecidos a um versado na técnica.
Conforme aqui usado os termos !!!Ifórmuia (IΓ e ’’fórmula (I) ou sais farmaceuticamente aceitáveis dos mesmos” são definidos para incluir todas as formas do composto de fórmula (I), incluindo isômeros, formas cristalinas e nâo cristalinas, isomorfos. polimorfos, metabolites, solvates, hidrates e profàrmacos dos mesmos.
O termo “solvato é usado aqui para descrever uma combinação não covalente ou facilmente reversível entre solvente e solute, ou meio de dispersão e fase dispersa. Serà compreendido que o solvato pode estar na forma de um sólido, pasta fluida (por exemplo, uma suspensas ou dispersão), ou solução. Exemplos não íimitativos de solventes incluem etanol, metanol, propanol, acetonitrila. dirnetií éter, dietil éter, tetraidrofurano, metileno cloreto e água. O termo hidrato é empregado quando referido solvente é àgua.
Um sistema de classificação atualmente aceitável para hidrates orgânicos è um que define local isolado, ou hidmtos de canal - ver Polymorphism in Pharmaceutical Solids by K. R. Morris (Ed. H. G. Brittain. Marsel Dekker, 1995). Hidretos de local isolados são uns em que as moléculas de água são isoladas de contato direto entre si por intervenção de moléculas
18/83 orgânicas. Em hidratos de canal, as moléculas de água assentam em canais de trelíça under ales estão próximos a outras moléculas de água.
Quando o solvente ou a agua são íntimamente ligados, o complexo terá uma estequio.mehia bem definida independente da umidade.
Quando, contudo, o solvente ou água são fracamente ligados, como nos solvates de canal e compostos hígroscópícos, o teor de água/soívente será dependente das condições de umidade e secagem. Em tais casos, a não esteqraometrla será a norma.
A invenção também se refere a profàrmaoos dos compostos de 10 fórmula (I). Desse modo, certos derivados de compostos de fórmula (I) que podem ter pouca ou nenhuma atividade farmaonlógica podem, quando administrados em ou no corpo, ser convertidos em compostos de fórmula (I) tendo a atividade desejada, por exemplo, por divagem hldrofitica. Tais derivados são referidos coma profàrmaoos. Informação adicional no uso de 15 profármacos pode ser encontrada em Pro-drugs as Novel Delivery Systems.
Vol 1-4. ACS Symposium Series (T. Higuchi e W. Stella) e Bio-reversible Carriers in Drug Design, Pergamon Press, 1087 (Ed. E. B. Roche, American Pharmaceutical Association).
Profárrnacas, de acordo cu.m a invenção, podem, por exemplo.
ser produzidos por substituição de funcionalidades apropriadas presentes nos compostos de fórmula (l) com certas porções conhecidas ao versado na técnica como 'pró-porções' conforme descrito, por exempla, em Design of Prodrugs by H. Bundgaard (Elsevier, 1885).
Alguns exemplos não hmitativos de profárrnacos de acordo com 25 a invenção incluem:
(i) onde o composto de fórmula (I) contém uma funcionalidade de ácido carboxüico que è functonalíxada ern um grupo instável metabclicamente adequado (ésteres, carbamates, etc.) de composto de fórmula (i):
(ii) onde o composto de fórmula (I) contém uma funcionalidade de álcool que é funcionahzada em grupa instável metabolicamente adequado (éteres, ésteres, carbamates, acetais, catais, etc.) de composto de fórmula (iii) onde o composto de formula (I) contém uma funcionalidade de amino primária ou secundária, ou uma amida que é funcionalízada em um grupo instável metabolicamente adequado, por exemplo, um grupo hídrolízávei (amides, carbamates, urèias. fosfbnatos, sulfonates, etc.) de composto de fórmula (I).
Exemplos adicionais de grupos de substituição de acorde com os exemplos precedentes e exemplos de outros tipos de profármaco podem ser encontrados nas referências antes mencionadas.
Os compostos da fórmula (í) da invenção podem exibir o fenômeno de tautomerismo e isomerísmo estruturai. Por exemplo, os compostos de fórmula (I) da invenção podem existir em várias formas tautomèncas. incluindo a forma enol e ímina. e a forma ceio e enarnina e isômeros geométricos e misturas destes, Todas tais formas taufoméricas são incluídas dentro do escopo dos compostos da fórmula (l) da invenção. Os tautômeros existem como misturas de um conjunto tautomèrico na solução. Na forma sólida, usualmente um íautôrnero predomina. Mesmo embora um tautômero possa ser descrito, a presente Invenção inclui todos os tautômeros dos compostos de fórmula (0 da invenção.
A presente Invenção também inclui compostos isotopioamente etiquetados, que são idênticos àqueles recitados na fórmula (I) acima, mas para o fate que um ou mais átomos são substituídos por um átomo tendo uma massa atômica ou número da massa diferente da massa atômica ou número de massa usualrnenle encontrado na natureza Exemplos de isotopes que podem ser incorporados nos compostos de fórmula (I) incluem isótopos de hidrogênio, carbono, nitrogênio, oxigênio, fósforo, flúor e cloro, tais como, mas não limitados a, ?H, ΛΗ, ^C, UC, ^N, '^O, ‘U'S e 5ÔP. Certos compostos isotopicamente etiquetados de fórmula (I) da invenção, por exemplo. aqueles em que isótopos radioativos tais como 'SH e :4C são incorporados, são útess em ensaios de distribuição de tecido da substrato e/ou fármaco. Isótopos tritiatados, isto é< Ή e carbono-14, isto é. ''C, isótopos são particularmente preferidos por sua facilidade de preparação e detectabilidade. Adlcionaimente, substituição com isótopos mais pesados tais como
20763 deutèrio. isto è. “H podam proporcionar certas vantagens terapêuticas resultantes de estabilidade metebólica maior, por exemplo, meia-vida aumentada ' in vivo ou requerimentos de dosagem reduzidos e, consequentemente, podem ser preferidos em algumas circunstâncias. Os compostos isutopícamen5 te etiquetados de fórmula (I) da invenção podem geralmente ser preparados por efetuação dos procedimentos descritos nos Esquemas e/ou nos Exemplos e Preparações abaixo, por substituição de um reagente isotopicamente etiquetado para um reagente nau isotopicamente etiquetado.
Os compostos de fórmula (1) da invenção podem exibir polimor10 fismo. Os pulimorflsmus dos compostos de fórmula (I) da invenção podem ser preparados por cristalização de um composto de fórmula (I) da invenção sub várias condições, Por exemplo, foram empregados vários solventes (incluindo agua) ou misturas de solvente diferentes para recristalização; custelização em temperaturas diferentes; vários modos de resfriamento variando 15 de resfriamento muito rápido a muito lento durante cristalização. Os polimorfos podem também ser obtidos por aquecimento ou fusão da um composto de fórmula (I) da invenção, seguido por resfriamento gradual ou rápido. A presença de pclimorfismos pode ser determinada por espeotroscopia de RMN de sonda sólida, espeoiroscnpia de IV. calonmetna de varredura dite20 rencial. difração de raias X da energia c-u outras tais técnicas.
A quantidade mínima do composto de formate (i) da invenção a ser administrado é uma quantidade eficaz. O termo quantidade eficaz significa a quantidade de um composto de formula (I) da invenção que previne o começo de. alivia os sintomas de, cessa a progressão de, /ou elimina uma 25 infecção de TB em um mamífero, por exemplo, um ser humano.
A quantidade terapeutícamente eficaz do composto de fórmula (I) da invenção Foi verificada possuir as propriedades antítuberculares desejadas descritas abaixo. Contudo, um efeito sinergistice é observado quando O composto de fórmula (I) da invenção é administrado em combinação com 30 pelo menos dois agentes úteis no tratamento de tuberculose.
Par efeito sinergístico ê significativo que o efeito terapêutico de administrar um composto de fórmula (!) da invenção e os peto menos dois
21/63 agentes úteis no tratamento de tuberculose é maior do que o efeito terapêutico obtido na administração da quantidade eficaz de ou composto da fórmula (I) da invenção sozinho, ou a quantidade terapêutica eficaz dos peto menos dois agentes úteis no tratamento de tuberculose administrada individu5 almaate ou em noaibinação.
Tal sinergia é vantajosa em que ela pode permitir administração de cada um dos componentes na combinação em uma quantidade menor do que sena requerida administrada individualmente que pode reduzir a probabilidade de eventos adversos ou efeitos colaterais desagradáveis. Alternati10 vamente. tal sinergia pode encurtar a duração de tratamento de TB
Desse modo, a administração de ambos o composta de fórmula (I) da invenção e os pelo menos dois agentes úteis no tratamento de tuberculose, por exemplo, pelo menus dois dos compostos selecionados a partir do grupo consistindo em Isoniazid, rifampin, nfapentina, rifabutin, pirazinami15 da, moxifloxacin, gatitfoxacin. levotloxaoin. ofloxacin, ciprofloxacin, e etambutol foi verificada produzir um efeito que resulta em tratamento aperfeiçoado de tuberculose conforme comparado ao efeito quando o composto de fórmula (!) da invenção sozinho, quando os pelo menos dois agentes úteis no tratamento de tuberculose administrado individualmente ou quando os pelo 2G menos dois agentes úteis no tratamento de tuberculose são administrados em combinação um com o outro.
Em uma concretização da invenção, administração de ambos o composto de fórmula (I) da invenção ou um sal farmaceuticamente aceitável do mesmo e pelo menos dois dos compostos selecionados a partir do grupo 25 consistindo emisonlazid, rifampin, rifapentina. rifabutin, pírazinamida. moxifloxacin, gatifloxacin, íevofloxacin. ofloxacin, ciprofloxacin, e etambutoi foi verificada produzir um efeito que resulta na erradicação complete de tuberculose comparada com erradicação incompleta quando o composto de fórmula (I) da invenção ou os pela menos dois agentes úteis ne tratamento de 30 tuberculose são administrados individualmente ou em combinação um com o outro.
O termo ’’erradicação complete significa que miccbacféria não
22/63 cultivãvel pode ser observada no õrgão-alvo, isto è, pulmões des mamíferas infectados apôs o regime de tratamento corn a combinação da presente in venção. É notado que no final do tratamento de mamíferos infectados com o regime de fármaco existente, isto é, a combinação dos pelo menos dois a5 gentes úteis no tratamento de tuberculose, isomazíd, pirazinamída e rifampin, uma quantidade signíficantemento cultivável de bacilo da tuberculose é recuperada do órgão-alvo, isto é, pulmões isto é evidente dos dados na Ta bela 3 abaixo. É adicionalrnente notado que mesmo apôs tratamento por 4 meses com o padrão de cuidado, 2 meses de isoniazid, pirazinamida e ri10 famprn. seguido por 2. meses de isoniazid e rifampin. 90% de relapsa de camundongos após compietação de tratamento, significando que bacilo viável permanece na compleiaçao de tratamento, mesmo se eles não podem ser cultivados no momento da completação de tratamento, que é evidente a partir dos dados na Tabela 4,
Em uma concretização da presente invenção, os pelo menos dois agentes úteis no tratamento de tuberculose usados em conjunto com um composto de fórmula (I) e composições farmacêuticas da invenção aqui descritos são como segue: isoniazid, rifampin, rifaperrtina, rifabutin, pirazinamida, etambutol, estreptomicina, canamicina, amicacin, moxifloxacin, gati20 fioxaoin, levofloxacin, ofloxacin, ciprofloxacin, capreumicin, etionamsda, ciclossenna, ácido para-aminossalicihco, tiacetazoua, claritramicin, amoxilinaáddo clavuiânico, irnipenem, meropenern, clofazimine, viomicin, terizidona. TMC207, PA-824, OPC-7683, LL-3858 e SQ-109.
Em outra concretização, urn dos referidos pelo menos dois agen25 tes é selecionado a partir do grupo consistindo em isoniazid, rifampsn, rifapentína. rifabutin, pirazinamida. moxifloxacin. gatifloxacin, levofloxaoin. ofloxacin, ciprofloxacin, e etambutul.
Em ainda outra concretização, um dos referidos peto menos dois agentes ê pirazinamida.
Em ainda nutra concretização, um dos referidos pela menus dais agentes é rifampin.
Em ainda outra concretização, um dos referidas pelo menus dais
23/83 agentes ê rifapentina.
Em ainda outra concretização, um dos referidos pelo menos dois agentes é PA-824.
Em ainda outra concretização, um dos referidos pelo menos dois 5 agentes é OPC-67683
Em ainda outra concretização, um dos referidos pelo menos dois agentes è TMC-207.
Em ainda outra concretização, um dos referidos pelo menos dois agentes é selecionado a partir do grupo consistindo em moxifloxacin. gatiflo10 xaan, levofloxacín. e ofloxacin.
Em ainda outra concretização um dos referidos pelo menos dois agentes è moxifloxacin..
Em uma concretização específica, referidos pelo menos dois agentes são pirazmamtda e rifampin.
Em ainda outra concretização especifica, referidos pelo menus dois agentes são pirazinamida, rifampin e isoniazid.
Em ainda outra concretização específica, referidos pelo menos dois agentes são pirazioamida e rifapentina.
Em ainda outra concretização específica, referidos pelo menus 20 dois agentes são pirazinamlda, rifapentina. e isoniazid.
Em uma concretização específica, referidos pelo menos dois agentes são pirazinamlda e moxifloxacin.
Em ainda outra concretização específica, referidos pelo menus dois agentes é pirazinamída. moxifloxacin, e rifampin.
Em ainda outra concretização especifica, referidos pelo menos dois agentes são pirazinamlda, moxifloxacin, e rifapentina
Em ainda outra concretização especifica, referidos pelo menus dois agentes são PA-824 e pirazinamida.
Em ainda outra concretização especifica, referidos pelo menus 30 dois agentes são PA-824, pirazmamida e um agente selecionado a partir do grupo consistindo emestreptomicina, canamicina, amicacin e capreurnidn.
Em ainda outra concretização específica, referidos pelo menus
24/63 dois agentes são PA-824 e moxífloxacin.
Em ainda outra concretização específica, referidos pelo menos dois agentes são PA-824, rnoxifloxâcin e um agente selecionado a partir do grupo consistindo emestreptornioina. canamicina, amicacm e oapreomicln.
Em ainda outra concretização específica, referidos pelo menos dois agentes são OPC-67683 e pírazinamida.
Ern ainda outra concretização específica, referidos pelo menos dais agentes são 0PC-67683, pírazinamida a um agente selecionado a partir do grupo consistindo em estreptomicina, canamicina, amicacin e capreomi10 cin
Em ainda outra concretização específica, referidos pelo menus dois agentes são OPC-67683 e moxifloxacin.
Em ainda nutra concretização especifica, referidos pelo menos dois agentes são OPC-67683, moxifloxacin e um agente selecionado a partir 15 do grupo consistindo em estreptomicina, canamicina, amicacin e oapreomicin
Em ainda outra concretização especifica, referidos pelo menos dois agentes são TMC-207 e pírazinamida.
Em ainda outra concretização específica, referidos pele menos 20 dois agentes são TMC-207, pírazinamida e moxiHoxaoin ou isoniazid.
> Foi verificado que um efeito sinergístico è observado quando o composta de fórmula (i) da invenção è administrado em combinação com pelo menos um agente útil no tratamento de tuberculose, por exemplo, rifampin, durante a fase de continuação (esterilização} de tratamento (após a 25 fase iniciai de tratamento) quando bactérias de não multiplicação persistentes são prevalecentes. Tal sinergia è vantajosa em que ela pode encurtar a duração de tratamento para TB. Mais slgníficantemente, os dados abaixo mostram que na administração de um composto de fórmula (I) em combinação oom pelo menos dois agentes úteis para o tratamento de TB pode en30 curtar a duração de terapia para TB suscetível a fãrmaco, TB resistente a rnonofârmaco e TB resistente a multífârmaco.
Em uma concretização da presente invenção, os métodos da
25/53 presente invenção proporcionam tratamento de um mamífero após o mamífero ter suportado uma fase inicial de tratamento compreendendo pelo menos um agente útil ao tratamento de tuberculose usado em conjunto som um composto de formula (!).
< : : : : : : : : : : :: ::
Em outra concretização, o peto menos um agente útil no tratamento de tuberculose selecionado a partir do grupo consistindo emrifampin, rifapentina. rifabutin, pirazinamida, etambuíol, estreptomícina, canamicina, amicacin, mnxrfloxacin, gatífloxacín, levofloxacin. ofloxacin, ciprofloxacin, capreomicln, etionamlda, ciclosserina, ácido para aminossaiicílicu, tianeta10 zona, clantromicín, amoxílína-ácido clavulâníco. ímípenem, meropenem, clofazimine, viomicín, terizidona, TMC207, PA-824, OPC-7583, LL-3858 e SQfim ainda outra concretização, referido pelo menos um agente é selecionado a partir do grupo consistindo em rifampin, rifapentina, nfabutin.
pirazinamida, moxífloxacin, gatifioxaoin, levofloxacin, ofloxacin, e etambutol.
Em uma concretização específica, referido pelo menos um agente é pirazinamida.
Em uma concretização especifica, referido pelo menos um agente é rifampin.
Em uma concretização especifica, referido pelo menos um agente à nfapentma.
Em uma concretização especifica, referida pela menos um agente è PA-824
Em uma concretização específica, referido pelo menos um agen25 te è OPC-7683.
Em uma concretização específica, retendo pelo menos um agente è TMC-2Q7.
Em uma concretização específica, retendo pelo menos um agente é selecionado a partir do grupo consistindo em moxifloxacin, gatifloxacin, 30 íevofloxacin, e ofloxacin.
Em uma concretização específica, referido pelo menos um agente é moxifloxacin.
26/63
Os métodos e as composições da invenção são particularmente afetivos contra tuberculose incluindo tuberculose alive e tuberculose latente. Em um exemplo., a tuberculose ativa compreende tuberculose sensível a fármaco, tuberculose resistente a monofármaco. tuberculose resistente a 5 multifármaco e tuberculose extensivamente resistente a fármaco. Ern outro exemplo, a presente invenção proporciona um método para erradicar com pletamente tuberculose sensível a fármaco, tuberculose resistente a monofármaco. tuberculose resistente a multifármaco, e tuberculose extensivamente resistente a fármaco (XDR) na completação do tratamento.
Em adição, os métodos e a composição da invenção podem ser usados em conjunto com testes diagrtotíccxs para identificar a tuberculose no mamífero que é conhecida àqueles versados, na técnica. Por exemplo, os métodos e as composições da invenção podem ser usadas em conjunta com o assim denominado ensaio de linha de sonda (Hain Life-science GmbH) que pode ser usado para identificar genes ligados corn resistência a rifampin e isoniazid para indicar tuberculose resistente a multifármaco e/ou tuberculose extensivamente resistente a fármaco. Outros ensaias podem também ser usadas em conjunto com os métodos e as composições da invenção, que são conhecidos àqueles versados na técnica.
Em outra concretização, os métodos e as composições da invenção sâa particularmente eficazes contra tuberculose causada por uma infecção de Mycobacterium selecionada a partir do grupo consistindo em Mycobacterium fubemufose, Mycobacterium bows ou outras espécies relacionadas à micobactenana, que seriam conhecidas por um versado na técni25 ca. Em um exempla, a presente invenção proporciona métodos e composições para prevenir relapso da infecção de Mycobacterium após compíetação do tratamento:.
Tal combinação poda ser para uso simultâneo, separada ou sequencial Em uma concretização, cs pelo menos dois agentes (uu o pelo 30 menos urn agente; úteis para o tratamento de tuberculose sâo administrados antes da administração do composto de fórmula (I) da invenção. Em outra concretização, os pelo menos deis agentes (ou o pelo menus um agente) úteis para o tratamento de tuberculose são administrados apôs administração do composto de fórmula (I) da invenção. Em outra concretização, os pelo menos dois agentes (ou o pelo menos um agente) úteis para o tratamento de tuberculose são administrados a cerca do mesmo tempo da administração do composto de fórmula (I) da invenção.
A administração separada de cada composto, em tempos diferentes e por rotas diferentes, em alguns casos seria vantajoso. Desse modo, os componentes na combinação, isto é; o composto de fórmula (I) da invenção e os pelo menos dois agentes (ou o pelo menos urn agente) no tratamento de tuberculose não necessita necessariamente administrados em essencíalmente o mesmo tempo ou em qualquer' ordem. A administração pode ser regulada que o efeito de farmacocinètica de pico de um composto coincide com o efeito de farmacocinética de pico do outro.
Todos os ingredientes ativos podem ser formulados nas formas da dosagem separadas ou individuais que podem ser coadministradas uma após a outra, Gutra opção é que se a cota de administração é a mesma (por exemplo, oral), dois ou mais dos compostos atives podem ser formuladas em uma forma simples para coadmínístração, ambos os métodos de caadministração, contudo, sendo parte do mesmo tratamento ou regime terapêutim.
Agentes preferidas para o tratamento de tuberculose podem ser conforme segue: isoniazid, rifampin, dfapentina, rifabutin, pirazinamida, etambutol. estreptomiclna, cariarnicina, amicacm, maxifloxacin, gatifloxacin. levofloxacin, ofloxacin, ciprofloxacin, capreomioin, etionamida, ciclosserina, ácido para-amiriossalidiico, tiacetazona, ctaritromicin, amcxillna-ácido clavulânlco, imipenem, meropenem, ciafazimma, viomicm, terizidona, TMC207, PA-824. OPC-7683, Lt.-3858 e SQ-109. Os agentes úteis para o tratamento de tuberculose podem ser usados na presente invenção em uma variedade de forma, incluindo forma ácida, forma de sai, racemates, enanbômeros, solvatos. e tautômeros. Os agentes úteis para o tratamento de tuberculose podem ser administrados por qualquer rota útil para administrar referidos agentes, que são conhecidas ao versado na técnica.
A invenção também se refere a composições da invenção que compreendem (i) uma quantidade terapeutioameate eficaz de um composto de fórmula (I) ou um sal farmaceuticamente aceitável do mesmo, (ií) pelo menos urn agente útil no tratamento de tuberculose e (iii) transportadores ou 5 veículos fermaceutícamente aceitáveis (daqui por diante as composições da invenção**).
As composições da invenção que são adequadas para administração a um paciente em necessidade do mesmo (por exemplo, urn ser humane) são também referidas aqui como composições farmacêuticas da in10: veação.
As composições farmacêuticas da invenção podem estar em qualquer forma adequada para administração a um paciente. Por exemplo, as composições farmacêuticas da invenção podem estar na forma adequada para administração oral tais como oompnmido, cápsula, pílula, pó. formula15 ções de liberação sustentada, solução, e suspensão; para injeção parenteral como uma solução estéril, suspensão ou emulsão; para administração tópica como um unguento ou creme; ou para administração ratai como um suposítório. As composições farmacêuticas da invenção podam estar em formas de dosagem unitárias para administração única de dosagens precisas.
Formas de administração parenteral exemplaras incluem soluções ou suspensões de compostos ativos em soluções aguosas estéreis, por exempla, propileno glicol aquoso ou soluções de dextrose. Tais formas de dosagem podem ser adeguadamente tamponadas, se desejado.
Em uma concretização, as composições farmacêuticas da ín25 venção podem estar na forma de uma forma de dosagem oral. Exemplos não hmitativos de formas de dosagem orais Incluem tais como, por exemplo, comprimidos mastigáveis, cápsulas, pílulas, pastilha em forma de losango, trocísuos. sacnês, pôs, xaropes, ehxires. soluções a suspensões, e similares, de acordo com a pratica farmacêutica padrão. Em outra concretização, as 30 composições farmacêuticas da invenção podem também ser distribuídas diretamente a um trato gastrointestinal do paciente através de um tubo nasogástric-u.
29/63
O composto de fórmula (I) da invenção estará presente na composição farmacêutica da invenção em uma quantidade suficiente para pro' porcionar a quantidade de dosagem desejada na faixa aqui descrita. A razão proporcional de composto de fórmula (I) da invenção para excipientes dependerá naturalmente da natureza química, solubifidade e estabilidade dos ingredientes ativos, bem como da forma de dosagem contemplada. Tipicamente, composições farmacêuticas da presente invenção podem conter cerca de 2ü% a coroa de 99% do composto de formula (I) da invenção em peso.
Em uma concretização, o composto de fórmula (I) e sais farmaceuticamente aceitáveis destes è administrado entre cerca de 10 mg a cerca de 2000 mg. Em ainda outra concretização, o composto de fórmula 0) e sais farmaceuticamente aceitáveis dos mesmos è administrado entre cerca de 25 mg a cerca de 1000 mg.
Em ainda outra concretização, o composto de fórmula (I) e sais farmaceuticamente aceitáveis dos mesmos è administrado entre cerca de 50 mg a cerca de 500 mg.
Em ainda outra concretização, o composto de fórmula (í) e sais farmaceuticamente aceitáveis dos mesmos é administrado entre cerca de 100 mg a cerca de 500 mg.
Técnicas para formulação e administração do composto de fórmula (li da presente invenção e as composições da invenção podem ser encontradas em Remington: the Science and Practice of Pharmacy, 19th ed.. Mack Pub. Co., Easton, Pa. (1995).
O termo ‘’excipiente” significa am material inerte que é combinado com o composto de fórmula (I) para produzir uma composição farmacêutica ou forma de dosagem de fármaco oral.
O termo “excipiente farmaceuticamente aceitável significa que o exaipíeníe deve ser compatível com outros ingredientes da composição, e não nocivos ao recipiente dos mesmos. Os exciptentes farmaceuticamente aceitáveis são escolhidos com base na forma de dosagem pretendida.
Os comprimidos, pílulas, cápsulas, e similares podem conter excipientes selecionados da líganies tais como polívinilpírroíldcna, hldroxípro· pllmetil celulose (HPMC), hidroxlpropilcelulose (HPC), sucrose, gelatina, acácia, goma tragacanto, ou amido do milho; cargas tais como celulose mi' crocristalina, lactose, citrato de sódio, carbonato de cálcio, fosfato de cálcio dibãsioc. glicina e amido; desiníegrantes tais como amido de milho, amido de batata, ácido alginico, glicolato de amido da sódio, croscatmelose de sódio e certos silicates complexos; lubrificantes tais como estearato de magnésia. laurü sulfato de sódio e talco; e adoçantes tais como sucrose, lactose ou sacarina. Quando uma forma unitária de dosagem é uma cápsula, ela pode conter, em adição a materiais do tipo acima., um transportador liquido tal como òleo graxo. Vários outros materiais podam estar presentes como revestimentos ou para modificar a forma física da unidade de dosagem. Par exempío. comprimidos podem ser revestidos com verniz, açúcar, ou ambos.
No caso de suspensões orais pediátricas e sachêa, estes excipientes podem compreender auxiliadoras de suspensão tais como gama xantham ou hidroxipropilmetilcelulose, deslizantes tais corno silica coloidai, diluentes e agentes de massa tais como dióxido de siiício.. aromatizastes tais como goma de mascar, laranja, banana, framboesa e xarope de ouro ou misturas dos mesmos, adoçantes tais como aspartame ou açúcar, e estabilizadores tal como ácido suocinico. Pò ou formulações granulates tais como formulações de suspensão pediátricas e sachês, podem ser fabricados usando técnicas que são geralmente convencionais no campo de fabricação de formulações farmacêuticas de formulações secas para reconstituição em tais suspensões. Por exemplo, uma técnica adequada é aquela de misturar ingredientes pulverizados secos ou granulados.
Tipicamente, urna dose diária eficaz (isto é, dosagem total anima de cerca de 24 horas) do composto de fórmula (l) da invenção para adultos é cerca de 10 mg a cerca de 2000 mg; cerca de 25 mg a cerca de 1000 mg; cerca de 50 mg a cerca de 500 mg; e 100 mg a cerca de 500 mg com ou sem alimentação. Ern alguns casos, pode ser necessário usar dosagens fora dos mesmos hmites.
Uma dosagem diána do composto de fórmula (I) da invenção é vsualrnente administrada de 1 a 4 vezes diariamente em doses Iguais.
Em uma concretização, uma dose única de composto de fórmula (I) dai invenção è administrada por dia (isto é, em intervalos de cerca de 24 horas) (isto é, QD): em outra concretização, duas doses de composto de fórmula (I) da invenção são administradas por dia (isto é, BID}; em outra concretização, três doses de composto de fórmula (I) da invenção são administrado por dia (isto è: TID); e: em outra concretização, quatro doses de composto de fórmula (l) da invenção são administradas por dia (isto é, (QID); em outra concretização, uma dose única de composto de fórmula (l) da invenção é admmístrada todo dua (isto é, em intervalos de 48 floras), em outra concretização, uma dose única de composto de fórmula (I) da invenção é administrada duas vezes por semana: em outra concretização, uma dose única da composto de fórmula (I) da invenção ê administrada três vezes por semana.
Em uma concretização, a dose eficaz do composto de fórmula (!) da invenção è administrada BID em intervalos de cerca de 12 horas.
Em outra concretização, a dose eficaz do composto de formula (I) da invençoo é administrada TID em intervalos de cerca de 8 horas.
Em outra concretização, a dose eficaz do composto de fórmula (0 da invenção é administrada DID em intervalos de cerca de 6 horas.
Em uma concretização, uma dose eficaz do composto de fórmula (h da invenção é cerca de 25 mg a cerca de 1000 mg que é administrada BID em intervalos de cerca de 12 horas.
Administração oral é preferida.
A composição farmacêutica da invenção em uma combinação de dose fixa compreendendo um composto de fórmula (i) da invenção e pelo menos um agente útil para o tratamento de tuberculose e transportadores farmaoeuticamente aceitáveis pode ser preparada par métodos convencionais na técnica. Por exemplo, uma forma de comprimido da combinação pode ser preparada par qualquer versado na técnica.
Alguns exemplos da presente invenção são combinações e as composições farmacêuticas que envolvem as seguintes misturas não limitativas:
a) urn composto de fórmula (I) ou um sal farmaceuticamente aceüável du mesmo e pirazinamida:
b) urn composta de fórmula (!) gu um sal farmaceuticamente aceítável do mesmo e rifampin;
c) um composto de fórmula (1) ou um sal farmaceuticamente a ceitável do mesma e rifapentina;
d) um composto de fórmula (!) ou um sal farmaceuticamente aofôitável do mesma, e PA-824;
e) um composto de fórmula (I) ou um sal farmaceuticamente οι 0 ceitável do mesma, e TMG-207;
f) um composto de fórmula (!) ou urn sal farmaceuticamente aceitável do mesmo, e um agente selecionado a partir do grupo consistindo emmoxiftcxacirr gatifloxacin, levofloxacin, ofloxacin moxifloxacir?;
g) um composto de fórmula (l) ou um sal farmaceuticamente a15 ceitável do mesmo, isoniazid e rifampin:
h) urn composto de fórmula (l) ou um sal farmaceuticamente aceitáve! do mesmo, isoniazid, rifampm e plrazinamida; e
í) urn composto de fórmula (I) ou urn sal farmaceuticamente aceitável do mesmo, rifampin e plrazinamida,
Outros exemplos da presente invenção são combinações e as composições farmacêuticas que envolvem as seguintes misturas não limitativas,
i) cerca de 100 mg a cerca de 500 rng de um composto de fórmula (I) ou um sal farmaceuticamente aceitável do mesmo e cerna de 000 mg de rifampin;
k) cerca de 100 mg a cerca de 500 mg da um composto de fórmula (!) gu um sal farmaceuticamente aceitável do mesmo e cerca de 300 mg de isoniazid:
l) cerca de 100 mg a cerca de 500 rng de um composto de fór- mula (I) ou um sal farmaceuticamente aceitável do mesmo e cerca de 300 mg de isoniazid e cerca de 600 rng de rifampin:
m) cerca de 100 mg a cerca de 500 mg de um composto de fór
33/63 mula 0) au um saí farmaceuticamente aceitável do mesmo, cerna de 300 mg de isoniazid, cerca de 600 mg de rifampin e cerca de 20-25 mg/kg a cerca de 50-70 mg/kg de pirazinamida;
n) cerca de 100 mg a cerca de 500 mg de um campaste de fór5 mute (I) eu um sal farmaceuticamente aceitável do mesmo e cerca de W mg/kg a cerca de 20 mg/kg de rifampin;
c) cerca de 100 mg a cerca de 500 mg de um composto de fórmula (I) ou um sal farmaceuticamente aceitável da mesma e cerca de 10-15 mg/kg a cerca de 20-30 mg/kg de isoniazid,
p) cerca de 100 mg a cerca da 500 mg de um composto de fórmula (I) ou um sal farmaceuticamente aceitável de mesmo e cerca de 10-15 mg/kg a cerca de 20-30 mg/kg de isoniazid e cerca de 10 mg/kg a cerca de 20 mg/kg de rifampin,
q) cerca de 100 mg a cerca de 500 mg de um composto de fór15 mula (I) ou um sai farmaceuticamente aceitável do mesmo, cerca de 10-15 mg/kg a cerca de 20-30 mg/kg de isomazid, cerca de 10 mg/kg a cerca de 20 mg/kg de rifampin, e cerca de 15-30 mg/kg a cerca de 50 mg/kg de pirazinamida:
Ó cerna de 50 mg a cerca de 250 mg de um composto de fórmu 20 la (!) ou um sai farmaceuticamente aceitável do mesmo, cerca de 75 mg de isoniazid, cerca de 150 mg de rifampin e cerca de 400 mg de pirazinamida; e
s) cerca de 25 mg a cerca de 250 mg de um composto de fórmula (I) ou um sai farmaceuticamente aceitável do mesmo, e cerca de 300 mg de rifampin,
Os compostos de fórmula (I) da presente invenção são prontamente preparados de acorde com métodos sintéticos familiares àqueles técnicos no assunto. O composto de fórmula (I) da invenção pede ser preparado em uma maneira similar àquela descrita para a preparação de Exemplo 1 descrita na seção de Exemplas na Patente U.S. Να. 5.880.118. Em adição, ο composto de formula (i) pode ser preparado pelos processos colocados nas Publicações Internacionais WO97/37980 e W099/2.4393, ambas das quais são aqui incorporadas per referência.
34/63
Outro exemplo da preparação de (S)- AE[[3{3-flutoro-4“(4·· ticmcrfoliailjfemlJ-ã-oxu-S-oxazulidimi] metil] aceiamida é conforme segue:
O esquema 1 ilustra uma sequência sintética gerai para preparação das campastes da presente invenção.
ESQUEMA I
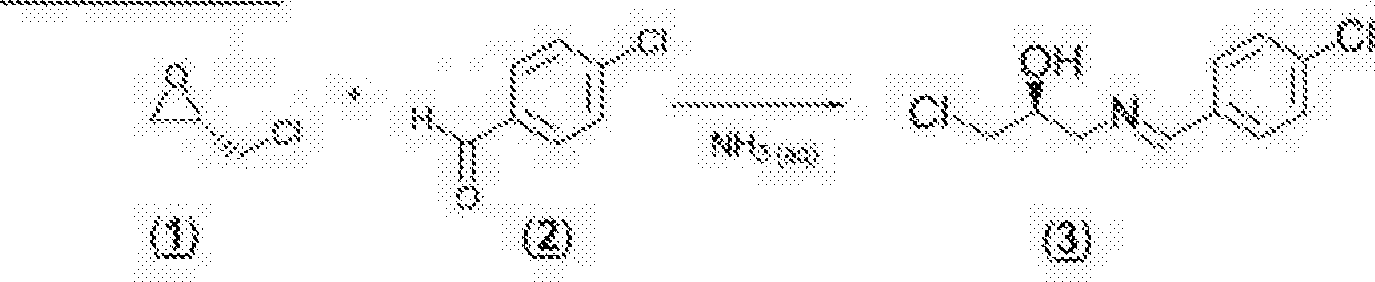


V Η I ' NH;.: I / 1 Λ. HCd lu W
O esquema 1 ilustra um método de sintetização de compostos de fórmula (I) da invenção em uma síntese de muitietapa via um composto de fórmula 6. Referindo-se ao Esquema l.: a síntese começa cam a formação de intermediário (3) por reação de (St-epicloroidrina (1) com uma mistura do derivada de benzaideído apropriadamente substituído (2) (preferivelmente 0,5 a 2 eq, mais preferivelmente 1 eq) e amônia aquosa (preferivelmente 0,5 a 3 eq, mais preferivelmente 1,5 eq). A reação é melhor realizada em ambos solventes próticos e aprótícos não nucleofilicos e solventes inertes tais como afcaòís (incluindo alcoóis C<.C$ ramificados e lineares e pohóís), éteres (in oluindo MTBE. THF, a outros éteres C%C§ lineares, ramificados e cíclicos), bem como solventes olorinatados tais como cloreto de metiteno. MTBE é urn solvente preterido. As temperaturas em urna faixa de cerca de 15 a cerca de 6(h:C são preferidas, mais preferivelmente entre 30 a 50X. Após isolamento 5 extratíve e concentração, a porção de smina (3) ê obtida. Ela é então cristalizada de uma segunda fase liquida, na presença de solventes de hidrocarboneto não polares tais corno. MS não limitados a, alcanas, misturas de alcanas (hexane, heptane, octano, feo-octano e misturas de alcano comercial mente disponíveis), opcionalmente na presença de solventes polares apróib 10 cos. preferivelmente solventes etereais tais como MTBE ou solventes aromáticos tais como toluene nu solventes olorinatados tais como cloreto de metilerio ou misturas dos mesmos. Solventes preteridos são uma mistura de MTBE e heptane nu urna mistura de toluene e heptane. Q processo de cristalização pode ser conduzido a uma temperatura em uma faixa de tempera15 iura ambiente (cerca de 18-25!>C) a cerca de 55'C, preferivelmente em uma faixa de 30 a 50*0, mais preferivelmente em uma faixa de 33 a 45:'C, Esta cristalização proporciona surpreendentemente aito rendimento e proporciona pureza enantiornénca significantemenle aperfeiçoada após isolamento por fíltração. (St-epiCluroidnna (1) e derivado de benzaideído (2) são comercial20 mente disponíveis ou podem ser produzidas por métodos bem conhecidos àqueles técnicos no assunta.
A porção de imina substituída (3) é acoplada com carbamafo (4) (que é conhecido àquela versado na técnica, por exemplo, ver J. Mad Chem,, 1595, 35, (3), 680-585 e também Exemplo 2 abaixo, (preferivelmente 25 1 a 3 eq. mais preferivelmente 1.5 a 2 eq)) para proporcionar a (3>
oxazolidinona imina (5) correspondente. A reação è efetuada preferivelmente a uma temperatura em uma faixa da temperatura ambiente a cerca de 65 C na presença de uma base com pKa maior do que 12, preferivelmente uma base alcóxida terciária, mais preferivelmente litso fem-butòxido e um solvente 36 aprótico não nucleofllico (preferivelmente DMF”, DMAc. acetonitrila. éteres
CvCç lineares, ramificados e cíclicos e/ou solventes clorinatados e/ ou mistu ras dos mesmos solventes, mais preferivelmente MTBE ou cloreto de meti
36/63 lone), Mato preferivelmente, a temperatura é de cerca de 30-60*0 e o tempo de reação é 2 a 24 haras. Preferivelmente, a (S^-oxazolidinona Imina (5), apôs uma operação de extração aquosa, é isolada por filtração de uma mistura 1:1 de um éter (incluindo MTBEi. THE, e outros éteres CrC§líneares5 5 ramificados e cíclicos) e água, mass preferivelmente MI BE. Aítematlvamen te, (5) é isolado após uma operação extrafiva equosa, por filtração ou crista lização de um álcool (inciumdo alcoóis (T-Celineares, ramificados e poiióis); mato preferivelmente iscpropanoL llidròlise do composto (5) com uma solução acídica aquosa proporciona um composto (6) e acilação subsequente 10 proporciona o composto bruto (7). O composto (5) é melhor hidroltoado com uma mistura de água e um ácido iode tal como ácido hidroclórico e o sub produto banzaldeído substituído è removido por extração corn um solvente orgânico imisoível em água (preferivelmente tolueno. MTBE, cloreto de metileno ou etil acetate), mais preferivelmente etil acetato. A solução aquosa re15 sultante de amlna olcridrato (6) é preferivelmente acilatada earn anidrido aoético, preferivelmente na presença de agua e em solvente orgânico imisaivel em água (mais preferivelmente cloreto de metiíeno). A conversão de amina clondrato (6) em composto (7) é bem conhecida na literatura. (Ver Bríckner, S.J. e?. a/. J Med. Cáem. 1996 39 (3) 673-679, Patente U.S.
5.837.870. US 5.688.792)
Os exemplos providos abaixo ilustram adicionalmente os compostos de fórmula (í) da invenção, composições da invenção e métodos de use do composto da invenção. E para ser compreendido que c escopo da presente Invenção não é limitado de qualquer modo peio escopo dos seguln25 tes exemplos e preparações.
Exemplo 1
.....de........(6) -1 -ctoro--3-[(4-çigrg:A~benzüiddno)'^rninglprgpan--2-gl
MétodoA
Um frasco de fundo redondo de trés gargalos de 51 equipado com um agitador mecânica, termopar, condensado?' de refluxo e manta de aquecimento é carregado com 4-clorobenzaldeidc (351,0 g, 2,5 mols, 1,0 eq.). MTBE (1,5 L) ú, em seguida, carregado ac funde redondo para dar urna solução homogênea. Amônia aquosa (28 % em peso, 252,98 mL. 3.75 mofe,
1,5 eq.) è adicionada em uma porção única resultando em um precipitado branco que se transforma em uma pasta fluida delgada dentro de 15 minutos 5 de agitação. (SX*>epiclorohidnna (> 99 % ee, 198,0 mL, 2,5 mofe, 1,0 eq,) é, em seguida, vagarosamente carregada no vaso. Após 40 minutos, os conteúdos são, em seguida, vagarosamente aquecidos a 43':C. A reação è agi tada a 40*Q por 18 horas em cujo tempo 8,4% de área de apicorohídrina permaneceram por GC. Após resfriamento, a mistura de reação è transferida 10 para um funü de separação e as camadas são separadas. A camada aquosa inferior è descartada. A camada orgânica è transferida para um frasco de fundo redondo de 3L. concentrada ?r? vácuo a cerca da metade do volume (800-900 mL) ern cuso tempo /so-octans é vagarosamente adicionado de um tubo de alimentação (-75Ü ml.) até turvação ser observada. A mistura bífási15 ca é semeada com -4 mg do composto do título, A reação é resfriada uom um banho de gelo por 45 minutos enquanto agitação. O precipitado é coleta do e enxaguado com .feu-ootano trie (500 ml..). O sólido è secado por 18 horas a 50*C sob vácuo para dar 345,19 g (59% de rendimento) do composto do titulo como um sólido branco. Ensaie- GC: 190%, 99,7% se por Chiral 20 SFC). GC (condições: coluna - 30 metros HP-1, 0,25 mm de ID e 0.25 micron der película e 103,35 KPa (15 psi) de pressão principal. 1,0 pi de tamanha de injeção; T-n! ~ 70X, rampa de 20%Umi.n) Tr (epicloroidrina) ~ 2,4 min, Tr (4-clorpbenzaldeído) ~ 4,8 min e (composto do titulo) ~ 9,7 min; condições de HPLC: Chiraípak AD-H 250 mm X 4,6 mm coluna, eluindo com 70% 25 de COjv 30% MeOH a 3,0 mL/min, detectando a 255 nm. (composto do título] 3,9 min; TR (enantiomers de composto do título) ~ 2,8 min: Ή RMN (400 MHz. CDCI.Ú δ 3,69 (bs, 2 H), 3,80 (m, 2 H). 4,15 (s, 1 H), 7,41 (d, J « 8 Hz, 2 H), 7,69 (d, J 8 Hz, 2 H), 8,33 (s„ 1 H); 5SC RMN (CDCh) õ 47,05, 63,09, 70,82, 128,93, 129,39, 134,08, 137,07, 162.30: IV (Pèlete KBr) 1630 30 cm'\
M.étgdp.B
Um frasco de fundo redondo de três gargalos de 51 equipado
38/63 com um agitador mecânico, termupar, condensador de refluxo e manta de aquecimento é carregado com 4-olorobenzaldeido (375 g, 2,67 mols, 1,0 ' eq.).. Metanol (0.75 L) é adicionado para dar uma solução homogênea após aquecimento de 10 a 23*C. Amônia aquosa {28,4 % em peso, 264 ml.., 3,95 mols, 1,5 eq.) é adicionada em uma porção única resultando em uma solução bifásica se formando após agitação por 15 minutos a 23 a 26*C, ($)-(·*)epicloroídrina (99,3 % ee. 207 mL, 2,64 mots. 1,0 eq.) é, em seguida, adicionada em uma porção, A mistura de reação é agitada a 23-24*0 por 18 horas, em seguida aquecida aí 40 a 45*0 e agitada por 2,5 horas ern cujo tempo 0,26% de area de (S) epichoroidrina permanece por GC (condições de GC, 0.050 ml., de mistura de reação em 1 mi., de acetonitrila, injeta 1 microlitro; 15 M DB-1 coluna, 0,25 mm de Dl e 0,25 micron de película e 103.35 KPa (15 psi) de pressão principal, 1,0 pl de tamanho de injeção: ~ 38*0, rampa de 10’*C/min) T?> (epiolarohidrina) ~ 1,1 min, TK (4-clorobenzaldeido) - 6,9 min e ϊ'η (composto do titulo) ~ 16,0 min). A mistura è concentrada to vacuo a um volume total da 1250 ml... Tolueno (250 ml.) é adicionada e a mistura concentrada to vacuo a run volume total de 1250 ml. Tolueno (250 mL) é adicionado e a mistura concentrada to vacuo a um volume total de 1145 mL. Tolueno (355 mL) é adicionado e a mistura concentrada to vacuo a um volume total de 900 ml... Toluene (600 ml.) è adicionada e a mistura concentrada to vacuo a um volume total de 1120 ml.. Enquanto mantendo-se 45 a 50*0, heptano (1500 ml.) è adicionado. A solução bifásica resultante é resfriada a 45*C e semeada. A mistura é, em seguida, adicionalmente resfriada a 38*C por 1/2 hora enquanto serneadura após 1 grau de resfriamento, A mistura é, em seguida, adicionalmente permitida resfriar vagarosamente a 23Tz por 16 horas. Os cristais brancos são. ern seguida, coletados por filtração a vácuo e lavados com heptane à temperatura ambiente (180 ml), O produto é secado em uma corrente de nitrogênio para dar o composto do titulo (431,57 g, 70,4%). Area de HPLC 95 % de área (Kromasil 150 mm .X
4,6 mm coluna, 254 nm, taxa de fluxo 1,5 mL/ min: A ~ I960 ml., água * 0,52 ml de ácido trifluoroacético ·*· 1,20 ml, de trletilamma; B ~ acetonitrila; Isocrático 47; 53 A: B por 5 min, em seguida, gradiente a 100% B por 5 min de
39/63 [composto do título] ~ 2,1 mm; 1% (4-ctorobenzaldeido) ~ .2,3 mini; 99,72% ee per Chiral SFC, Condições de Chiral HPLC: Chiralpak AD-H 250 mm X
4,6 mm coluna, eluindo cum 70% de C(>>/ 30% de MeOH a 3,0 mtfmin, detectando a 255 non (composto do titulo] ~ 3,9 min; Tp (enantiôrnero de composto du titulo) ~ 2,8 min. Ή RMN (400 MHz, CDCl*) δ 3,69 (bs, 2 H), 3,80 (m, 2 H), 4,15 (s, 1 H), 7,41 (d. ,J ~ 8 Hz, 2 H), 7,69 (d, 4 8 Hz, 2 H), 8,33 (s, 1 H); nC RMN (CDCh) Õ 47,05, 63,09, 70,82, 128,93, 129,39, 134,08, 137,07, 162,30.
M^gdoG'
Um frasco de fundo redondo de três gargalos de 51 equipado oom um agitador mecânico, termopar, condensador de refluxu e mania de aquecimento è carregado com 4-clurobenz.aldeido (375 g.< 2,67 mais, 1,0 eq,). MTBE (1,50 L) è, em seguida, adicionado para dar uma solução homogênea após aquecimento de 9 a 24 SC. Amôniã aquosa (28,4 % em peso, 265 ml.., 3,97 mois, 1,5 eq.) è adicionada em uma porção única resultando em uma solução bifáslca que se forma após agitação por 15 minutos a 23 a 26*C. (S)-(r )-epiOlorchidrína (99,3 % ae, 209 ml: 2,67 rnols, 1,0 eq,) é. em seguida, adicionada em uma porção, A mistura de reação é agitada a 2324°C por 3 dias. As fases são separadas e a fase superior concentrada sub pressão atmosférica de 2000 a 1000 ml de volume total (ponto de ebulição de 58 a 67*C). Enquanto mantendo 45 a 50“C, heptano (1700 ml) é adicionado A solução bifásica resultante é resfriada a 45*C e semeada. A mistura é, em seguida, adicionalrnente resfriada a 38X por 1/2 hora enquanto semeadura após todo 1 grau de resfriamento, A mistura é, em seguida, adioionalmente permitida r esfriar vagarusamente a 23CC por 1 hora. Cs cristais pesados brancos de neve são, em seguida, coletados por filtração a vácuo e lavadas corn heptano à temperatura ambiente {180 ml). O produto è secada em uma corrente de nitrogênio para dar o composto do título (462,43 g, 74,7%). HPLC 94 % de área [Kromasii 150 mm X 4,6 mm coluna, 254 nm, taxa de fluxo 1,5 ml/ mm; A ~ 1000 ml água * 0,52 mL de ácido trifluoroacático + 1,20 ml. de trietílamina; B ~ acetonitrila; Isocrátíco 47; 53 A; 8 por 5 min. em seguida, gradiente a 196% de B por 5 min. (composto do título] ~
40/63
2,1 min; TR (4-darnbenzaldeido) ~ 2,3 min]; 99.92% ee per Chiral BFG, Chiral HPLC condições· Chkaipak AD H 250 mm X 4,6 mm coluna, eluindo com 70% de CO?/ 30% de MeOH a 3,0 mLZmin. detectando a 255 nm. 1« (compesto do título] ~ 3,9 min; Tr (enanllumero de composto do titulo) -- 2,8 min;
Ή RMN (400 MHz. CDCM d 3,69 (bs, 2 H), 3,80 (m, 2 H), 4,15 (s, 1 H), 7,41 (d, J ~ 8 Hz, 2 H), 7,69 (d, J 8 Hz, 2 H), 8,33 (s, 1 H); nG RMN (CDCI3) δ 47,05, 63,09, 70,82, 128,93, 129,39, 134.08, 137.07, 162,30
Exemplo 2
ElOPgmo . de benzil éster de ácido í34luoro-4-morfolin-4-íl10 fenilj carbâmiço
O composto do titulo pode ser preparado de acordo com o método descrito em J. Med. Chem., 1998, 39, (3), 680-685 e representado no ESQUEMA It
ESQUEMA lí

A B ®
Métodos adicionais para a conversão de intermediário A 3fluoro-4-tiomorfolin-4~ilanisina (B) são providos.
Método A
4-(2-5íuoro-4-nitroíenil)tk?rnorf<}lina (A, 250 g.. 1,03 mol) foi car20 regada em uma mistura de dioxano (1400 mL), EtOH (1000 mL) e água (600 ml) em um frasco de fundo redondo de três gargalos de 5000 mL equipado com um agitador mecânico. Na mistura agitada foi carregado cloreto de amônia (166 g, 3,1 mols), seguido por pó de ferro (247 g. 4,25 muls), cada em porção única. A reação foi aquecida a reflexo com agitação vigorosa. A rea25 çâo fui aquecida a refluxo por um total de 16 horas e foi, em seguida, permitida resfriar à temperatura ambiente. A mistura escura foi diiuida com EtOAc (800 mL), filtrada através de uma almofada de celite, e concentrada fe vácuo a um resíduo pastoso. O resíduo foi dividido entre salmoura (1000 ml.) e diclorometano (750 ml..) Uma filtração através de celite removeu particulados 30 que estavam interferindo com a separação de fase. A camada aquosa foi,
4=/63 em seguida, extraída cem díclorometano adicional (750 ml). As camadas orgânicas combinadas foram secadas sobre carbonato de potássio anidro e concentradas fo vácuo para dar 225 g de urn sólido escuro. O material bruto foi dissolvido em diclorometano (W00 mL), tratado com 200 g de sílica-gel 5 (malha 230-40()} e a mistura foi concentrada para secagem. O tampão foi filtrado sobre 500 g de sílica-gel (malha 230-400, acendicionado como uma pasta fluida com 2.0% de EtOAc/hexano) eluindo com 20-30% de EtQAc/hexano, enquanto coletando frações de 1000 ml... Trações 3-11 foram combinadas e concentradas para dar 3-fluoro-4tiomarfolin-4-itanilina (B, 232 10 g. 106% de rendimento} cerno um sólido esbranquiçado. 'H RMN indicou o material desejado junto com traço de solventes residuais para cantar mais do que recuperação teórica. ;H RMN (400 MHz, CDOI3): δ 2,8 (m, 4H), 3,2 (m, 4H}, 3,6 (s, 2H), 6;4 (m, 2H), 6.8 (m, 1H).
MétodpB
Um frasco agitador' de 2000 ml.. Part foi carregado com 5'% de paládio suifidido em carbons (Johnson Matthey tipo A103038-5. 18 g) e 4-(2fluoro-A-fotrofenilttiemorfulina (A. 60 g, 0,25 mol). A mistura foi suspensa em MeOH (1056 mL) e a reação foi hídrogenada a 344,50 KPa (50 PSI) por 7 horas. O catalisador foi removido por' fíltraçâo através de celite e a massa de 20 filtre foi lavada bem com MeOH fresco. O filtrada cinza claro foi concentrado m vácuo para dar 3-fluoro-4-tícmorfolir>-4-ilanilina (B, 51,3 g, 98% de rendimento) como um solido cinza. ’H RMN (400 MHz, ODCT<T ó 2,8 (m, 4H), 3,2 (m. 4H)„ 3,6 (s. 2H), 6,4 (m, 2H), 6,8 (m, 1H).
2.5 Plftparaç^. de.J5g}-fon(4-:alorgbenzilldeno],.. aminol metii)·· 3-(3 fíuorg-.4..tiornprfolin-4-ílfenil)-1i3“Oxazoíidin-2-ona
Q composto do título no Exemplo 2 (194 g., 0,56 mui), e o composto do título do Exemplo 1 (195 g, 0,84 mol), e ferc-butôxido de iitio(116 g, 1,4 mal) foram carregadas em um frasco de fondo redondo de três gargalos 30 de 3000 ml sob nitrogênio. Os reagentes foram fluidizados com fem-butil éter de metila (1200 ml..) e a mistura foi aquecida a 56*C e agitada por 2 horas conic um sólido amarelo gradualmente formado. A reação foi resfriada à
42/03 temperatura ambiente, a diluída com '1200 mL de água, A mistura foi, em seguida, agitada vigorosamante por 60 minutos como o sólido mudada de amarelo escuro para um sólido amarelo mais pálida, A mistura foi resfriada a KFC, filtrada, e a massa de filtro foi lavada com I'ercbutil éter da metila frio 5 (450 mL). O sólido amarela claro resultante foi secado em ar per 30 minutos, em seguida colocado ern urn forno a vácuo e secado a 4O'-‘C durante a noite para proporcionar o composto do título (243 g, 99% de rendimento). ’H RMN (400 MHz. CDCla): δ 2.8 (m, 4H), 3,2 (m, 4H)„ 3,9 (ms 2.H), 4,1 (m, 2H); 5,0 (m, 1H), 6,9 (rn, 1H), 7,2 (rn, 1H), 7,4 (m, 3H), 7,6 (m, 2H), 8,4 (s, 1H).
fô- Exernglg.4
Preparação de /V-8(58)-3-(3-fiuoru-4-tiomorfolin-4-ilfe-ml)~2-oxoO composto do titulo no Exemplo 3 (243 g. 0,56 mol) for combinado corn EtOAc (1300 ml.) e água (1300 ml.) em um frasco de fundo ιοί 5 dondo de três gargalos de 6000 ml., equipado com um agitador mecânico.. A mistura foi tratada gota a gota com HCI a 12N (140 mL, 1,68 mol) e a mistura foi agitada vígurosamenie por 1 hora à temperatura ambiente. As camadas foram separadas e a camada aquosa foi lavada com EtOAc (1 x 500 mt), A solução aquosa resultante contendo cloridraío de (S)-5-(aminometil)-3-(3~ 20 fíuoru--4-tíomorfoiinofenil) oxazolidin-2-ona fos combinada com uma mistura de diclorometano (1808 mL) e MeOH (120 mL), e a mistura vigorosamente agitada foi carregada com anidrido acético (132 ml. 1,4 mol) em uma porção e subsequentemente tratada gota a gota com NaQH a 10 N (200 mL. 2,0 mois) por 15 minutos.. Uma mistura de reação extremamente espessa reste25 tou da adição da base, que se adeigaçcu gradualmente á medida que o pH se elevou e a acilação progrediu rapidamente. A reação foi agitada vigorosamente por 1 hora apôs a mistura difundida em duas toses. Neste momento, NaOH a 10 M (160 mL, 1.6 mol) foi adicionada gota a gota sã mistura até que o pH foi estável a 7. As camadas foram separadas, a camada aquosa foi 30 extraída com diclorometano (250 mL), e as camadas orgânicas combinadas forarn secadas sobre carbonato de potássio anidro. Gs voláteis foram removidos to vácuo para dar um sólido esbranquiçado que foi titulado com metil
43/63 terc-butil éter (250 ml), coletado, e senado m vacw para dar composto do tífolo (5) (L86;1 g, 94% de rendimento) corno um sólido branco fino com mais do que 98% de pureza de HPLC (tempo de retenção -- 3,93 minutos, condições de HPLC reportadas abaixo),
O sólido bruto foi dissolvido em 6% de metanol quente em diciorometano (1250 mL) em um frasco de fundo redondo de três gargalos de 5QÔ0 mL. equipado com um agitador mecânico. A solução foi aquecida a refluxo, diluída por adição de porção-a-porção (500 ml) de 2500 mL de isopropanol (IPA), e, de modo a manter reflexo, a temperatura foi elevada para 50-70*0. Na completaçào desta adição de IPA, o condensador de reflexo foi colocado corn uma cabeça de destilação de trajetória curta e destilação foi continuada em um frasco resfriado. Durante destilação, uma porção de 500 ml. de IPA frasco foi adicionada após 500 mL de destilado ser coletado para manter entre 2000 e 2500 mL de IPA presente em iodos os tempos. Após esta adição (temperatura interna do frasco caiu para 60°C), a mistura tomouse levemente turva e permaneceu assim para o equilíbrio da destilação, tornando-se aumentadamente turva conforme a temperatura do destilado excedeu 7C*C; a matéria particulada apareceu conforme AA temperatura do destilado excedeu 75'C. O controlador de temperatura foi aumentado para 85':'G o mantido all até a conclusão da destilação. Quando o destilado era claramente isopropanol sozinho (82-83^0), o volume foi reduzido para 2500 ml., de IPA quente, a manta de aquecimento foi removida, agitação foi descontinuada, e a pâ foi removida do frasco. A mistura foi permitida continuar a cristalizar conforme o frasco resfria. O sólido cristalino branco foi, em seguida, coletado por fíltração, lavado com tera-butil éter de metila (250 mL), e secado to vacuo a 4CPC para proporcionar 180 g (91% de rendimento) do composta do titulo em mass do que 99% de pureza de HPLC (tempo de re tenção ~ 3.93 minutos, condições de HPLC reportadas abaixo). RMN (400 MHz, DMSO-ds,·)·. δ 1,8 (s, 3H), 2,7 (m, 4H). 3,2 (rn, 4H), 3,4 (m, 2H), 3,7 (m, 1H), 4,7 (m, 1H), 7,1 (m, 1H), 7,15 (m, 1H), 7,2 (m, 1H), 0,2 (m, 1H>. Espec. de Massa. 0^Η^ΡΝ3Ο3δ: m/z 354,1 (M+1).
As condições de HPLC para analises mencionadas no texto: HP
44/63
Series 1100; Coluna; Symmetry CB 5 uM 4.6 x 50 mm; Taxa de fluxo 1,2 mt./mrn; Solvente A; água com 0,1% de ácido fórmico, Solvente B: acetonitrila com 0,1% de ácido fórmico: Volume de injeção - 10 uL de 1 mg/ml de (acetonitrila); Gradiente: Solvente B 0-100% por 7 minutos, em seguida
100% de B por 1 minute; comprimento de onda ~ 254 nm.
EXEMPLOS BIOLÓGICOS
As seguintes abreviações são usadas nos seguintes Exemplos e têm as definições indicadas, a menos que de outro mudo notado: CFU é trnidade de formação de colônia; INH é isoniazid; RIF è rifampin; PZA é pha10 zinamida, M.XF é rnuxlfloxadn, e LZD ê linezolíd.
A cepa bacleriana Mycoóactenum foõercrrfose H37Rv foi passada em camundongos congelados em alíquotas de 1 ml e armazenada a 80X antes de uso. Para cada infecção, ama alíquota foi descongelada e eub-cultivada em caldo Middlebrook 7H9 suplementada com 10% de ácido 15 oteícu-albumina-dextrose-catalase (OADCi (Difoo, Detroit, Ml) e 0,0536 de
Tween 80 (Sigma, St. Louis MO). Camundongos fêmeas BALB/c (Charles River, Wilmington, MA), de quatro a seis semanas de idade foram infectados via aerossol usando o Inhalation Exposure System (Gias-col inc, Terre Haute, IN) e uma cultura de caldo de fase de log com uma densidade ótica de 20 aproximadamente 1,0 a 600 nm. Os camundongos foram randomizados em grupos de tratamento (5 camundongos por grupo por ponto de tempo) após infecção de aerossol. Os camundongos não tratados foram rotineiramente sacrificados (i) na dia após infecção para determinar a numero de CFU im plantada nus pulmões cs (ii) no dia de iniciação de tratamento para determi25 ear a contagem de CFU de prè-fratamento.
O composto de fórmula (I) da invenção e LZD (obtidos de Pfizer Inn. Ann Arbor, Mi e Groton, CT.} usados nos testes descritos abaixo foi suspenso em uma solução composta de 536 de poiletileno glicni-200 (PEG200) e 95% de metücelulose (0,536) em água destilada. MXF foi obtido de 30 Bayer (Rolling Meadows. IL), PZA foi obtido de Fisher, e INF1 e RIF foram obtidos de Sigma. As soluções de estoque foram preparadas semanalmente usando-se água destilada. Todas as soluções de antibiótico foram armazenadas a 4<:C.
Exceto nade de outro modo indicado, os antibióticos foram administrados uma vez diariamente. cinco dias por sernana em 0,2 ml por ingestão. Ambas as suspensões de oxazolidinona foram sonicadas brevemente antes do uso e sacudidas entre doses. RIF foi dado 1 hora antes da administração de outros fàrmacos para evitar uma interação farmaaocinétma adversa.
Exemplo 5
O MIC do composto de fórmula (?) e I..ZD foi determinado pelo método de diluição ágar ern Middlebrook 7H11 agar suplementado com 10% de QADC (Bedon-Dickinsun, Sparks. MD}. Placas contenda concentrações duas vezes em série do composta de formula (l) e I.zlD variando de 0,125 a 4 pg/mL foram inoculadas com aproximadamente 5x106 CFU de Mycobacterium fubercu/ose H37Rv. CFU foram contadas apôs 28 dias de incubação a 37*C com 5% de CO^ ambiente. O MIO for definido como a concentração mais baixa para inibir pele menos 99% de crescimento baderiano.
O MIC do composto de fórmula (l) e 1.21D contra Mycobacterium tobemufose H37Rv foi 0.25 pg/mL
Atividade de variação de dose do aomppstp de formula (1) e LZD em. um modelo de infecção estabelecido
Camunda.ngos foram infectadas pela rota de aerossol com 4,44 ± 0,04 iagni de CFU, Começando 13 dias após infecção de aerossol quando a contagem de CFU de pulmão média foi 7,49 ± 0,11 fog^ e o peso de baço médio foi 105 ± 11 mg, os camundongos de controle receberam um dos seguirdes tratamentos: INH a 25 mg/kg, LZD a 2.5 mg/kg a 260 mg/kg (dose única e 130 mg/kg duas vezes diariamente), e composto de fórmula (I) 25 mg/kg a 100 mg/kg doses, No final do período do tratamento, 28 dias mais tarde, somente 2 de 5 carnundonges nao tratados permaneceram vivos. O pesa de baço médio entre estes camundongos vivos tinha aumentado para 237 ± 16 mg.
Em contraste, a monoterapia corn INH a 25 mg/kg preveniu es46/53 píenomegalia (peso de baço médio - 100 ± 1 mg) e morte. Uma diminuição dependente da dosa no peso de baço relativo a controles não tratados foi ' observada com doses aumentadas de LZD entre 25 mg/kg (200 ± 47 mg) e
130 mg/kg (102 f 5 mg). Contudo, baços de camundongos tratados com
I..ZD 260 mg/kg (se como uma dosa única ou 130 mg/kg duas vezes diariamente) foram maiores do que esperado (190 ± 32 mg) e similares em tamanho de baços de camundongos tratados com LZD 25 mg/kg/dia, Devido ao número e tamanho de lesões de pulmão diminuir corn doses diárias aumentadas de LZD incluindo 260 mg/kg, a esplenomegalia observada em camun10 dongas recebendo 260 mg/kg/dia não foi sentida ser devido à atividade antituberculose reduzida. O tratamento com o composto da fórmula (I) preveniu espleuomegalia em todas as doses administradas, incluindo a dose de 25 mg/kg (peso de baço médio ”113 ± 17 mg).
Os camundongos não tratados sobreviventes expenmentaram um aumento nas contagens de CFU a quase 8 lagw- O tratamento com INH reduziu a contagem da CFU de pulmão media para 5.62 log^. para uma morte iog de 1,87 comparada ao valor de linha da base. 'Todos os regimes incluindo o composto de fórmula (I) resultaram em uma redução significaste na contagem de CFU média de linha de base (p<0,01), começando com 20 uma redução de 0.78 log^ com 2.5 mg/kg. Em 50 mg/kg, o composto de fórmula (!) exibiu atividade bactericida (definida pelo oriténo de redução de 2leg) por redução da contagem de CFU média para 5,28 log^, por uma morte log de 2,21, maior do que observada com 1NH. Embora LZD também revele atividade dependente de dose, sua atividade foi mais limitada do que aquela 25 do composto de fórmula (I). Semente em dosas de LZD 4100 mg/kg foi uma redução signrfrcante a partir da contagem de CFU de linha de base demonstrada (p<0,01). Mesmo na dose mais alta testada, 260 rng/kg, LZD nãa em centrou a critério de redução de 2-log definindo atividade bactericide, produzindo uma morte log de semente 1,46, O composto de fórmula (!) foi signifi30 cantemente mais ativo do que LZD em cada dose testada,
O tratamento cem o composto de fórmula (I) a 2.5 mg/kg resultou em uma contagem de CFU mais baixa do que aquela observada após trata
47/63 mento com LZD a 25 nu 50 mg/kg (ρ<Ό,01) e náo sigmficantemente diferente daquela observada após tratamento com LZD a 100 ou 130 mg/kg. A 50 e
100 mg/kg, o composto de fórmula (I) for mais ativo do que qualquer dose de
LZD (p<0.00l). A divisão da dose diána total do composto de fórmula (!) e 5 LZD em duas doses diárias não tinha efeito claro (isto é, positivo ou negativo) na atividade (Tabela 1).
Tabela 1
| Fàrmaco | Regime de dosagem diária | Contagem de CFU logMédia (± SD) no: Dia 0 | Dia 28 |
| Nenhuma | Nenhum | 7,49 ± 0,11 | 7,92 ± 0,03 |
| INH | 25 mg/kg uma vez | 5,62 ± 0,20 | |
| Composto | 25 mg/kg uma vez | 6,71 ± 0,28 | |
| de Fórmula | 25 mg/kg duas vezes | 5,51 ±0,14 | |
| | úlí | | 50 mg/kg uma vez | 5.28 ± 0,53 | |
| 5G mg/kg duas vezes | 4,89 ± 0,27 | ||
| 100 mg/kg uma vez | 5.07 ± 0.14 ' | ||
| LZD | 25 mg/kg uma vez. | 7,55 ± 0,24 | |
| 50 mg/kg uma vez | 7M ± (1T1 ; | ||
| 100 mg/kg uma vez | : 67S ± ai4 | ||
| 130 mg/kg uma vez | 07 ±0,13 Ί | ||
| 130 mg/kg duas vezes | 6.02 ±0,17 | ||
| 260 mg/kg uma vez | | 6.03 ± 0,12 |
Consequentemente, o composto de fórmula (1) demonstrou que ele è significantemente mais ativo do que a do,se humana equivalente (isto ê, 10 100 mg/kg) de LZD, a ómoa oxazoikiinona clínicamente disponível que está sendo usada sem etiqueta para o tratamento de MDR- e XDR-TB.
Para determinar os perfis farmacocinéticos de dose única e estado constante do composto de formula (l) e LZD no modelo de murine, um 15 sub-estudo foi efetuado no estudo de variação de dose anima usando camundongos tratados com o composto de fórmula 0) a 180 mg/kg uma vez diariamente nu LZD a 130 mg/kg urna vez eu duas vezes diariamente. Três camundcngos por grupo foram sacrificados a 0,5, 1. 2, 4, 8 e 24 homo apõe a primeira dose de tratamento (01) e nos pontos de tempo antes menciona- des com a adição de urn ponto da tempo de 9 horas (isto é. 1 hora após a segunda dose diária) no Dia 24 (024) de tratamento. Os camundongos fo§ ram anestesiados corn clorofórmio e exsanguinados por punção cardíaca. O sangue total foi coletado em gelo e. em seguida, centrifugado para obter soro. Acetonitrila foi adicionada ás amostras antes de armazenagem em tubos de capa de rosca gaxetada a -2CFC. As amostras em D24 foram coletadas e processadas na mesma maneira e armazenadas a -2íTC antes de serem 10 enviadas juntas corn as amostras Dl para Pfizer (Groton, CT) para determinação de concentrações do fármaco. O sobrenadante foi injetado (10 p.L de volume de injeção) no l.C (Shimadzu SCL-10A, Kyoto. Japan; - MSZMS (Sciex API 3000, Applied Biosystems Group, Foster City. CA) usando uma coluna Hypersil 018 (5p.m. 50 x 2,1 mm. Thermo Electron Corp, Waltham, MA) e 15 um gradiente de etapa consistindo em uma fase móvel de A: água com 0,05% 5m M de fomfiato de amônia e B. aceionitríla/água/ÓmM de formiate de amônia (80:.20:0.05%). A ionização foi de modo de ion de eletropulverização posãiva. Os dados bioanaliticos foram capturados usando-se Analyst (Version 1.4 1; Applied Biosystems Group,. Foster City, CA). Os cálculos far20 macocínéticos foram baseados nas concentrações de soro médias e realizados usando a aproximação não comporíamental (regra trapezoidal linear para cálculo de AUC corn o auxilio de Watson 7.2 Bioanalyfioal DIMS [Thermo Electron Corp, Waltham,. MA)). Nos calculus da AUC para 024. o nível de plasma de anaiitos no tempo zero foi ajustado à concentração de 24 ho25 ras Concentrações de 0 foram usadas para cálculos cinéticas para todos os resultados abaixo do limite inferior de quantificação (5 ng/ml).
Os valores de parâmetro famiacocinétice para o composto de fórmula (I) e LZD são apresentados na Tabela '2, A dose diária de 130 mg/kg de LZD produziu uma AUC de estado constante de 379 pg-h/ml, aproxima30 damente 5035 mais alta do que a AUC de estada constante antecipada observada em seres humanos administrados 600-625 mg aralmeote duas vezes diariamente, 215-294 pg-h/ml (Ver Gee. T. et a: . Anfimicrub Agents
49/83
Chemother 45.1843-1848, 2001; Stalker DJ et al, J Antimicrob Cb.emottter 51:1239-46, 2003.) Baseado nas cineticas lineares nesta Faixa de dose, a dose de 100 mg/kg de LZD no camundongo representa a extremidade superior da faixa de ABC de estada constante em humanos, mas com a CWÍ!.< que è aproximadamente 3 vezes mais alta do que aquela obtida em humanos. Conforme esperado existe metabolismo de primeira passagem signifieante do composto de fórmula (I) para o metabolite de sulfòxido maior e o metabolite de sulfona menor (Barbachyn et al, J Med Ghent 39:680-685, 1996). Devido a cada um des mesmos metabolites terem um MIC-^ de 0,5 pg/mL contra M tuberculose, perto do MÍC^ de 0,25 pg/mL para o composto de fórmula (l), a concentração da cada metabolite foi adicionada àquela da origem para as análises farmacocinéticas. .A AUG de estado constante do composto da fórmula (l) e seus metabolites observados com a dose diária de 100 mg/kg foi aproximadamente 3 vezes mais baixa do que aquela observada com LZD a 130 mg/kg. O fato que a dose de 25 mg/kg do composto de fórmula (l) foi tão ativa quanto esta dose de LZD sugere que o composto de fórmula (I) é 12 vezes mais potente è? wo do que I.Z.D apesar dos MICs si milares para ambos os compostos.
Tabela 2
| i Regime | GfnàK (gy/mL) | AUCq.?4 (pg-h/ml.) |
| Í Composto de fórmula (1) 100 mg/kg | ||
| | Dia 1 | i 6,07 | |
| | Dia 24 | 1 4,32 | 8,74 |
| I Metabólito de sulfòxido | ||
| | Dia 1 | | 20,0 | 37,8 |
| | Dia 24 | j 16,9 | 98,8 |
| | Metabolite de sulfona | ||
| r Dia 1 | | 0,79 | |
| | Dia 24 | 1 0,66 | 9.73 |
| i Composto de fórmula (1) * rnelabóli - | ITf Z ............ ) | |
| 1 tos | ||
| 1 Dia 1 | 1 24,1 | 47,8 |
| 1 Dia 24 | 1 21,7 | 117 |
50/63
| Regime | C-rm (pg/rnL) | AUCq,?4 (pg-h/ml) |
| J130 mg/kg | ||
| Dia 1 | 84.9 | 184 |
| Dia 24 | 58,4 | 379 |
Camundongos foram infectados com aerossol com 3,89 ± 0.19 * CFU log 10 . Começando 14 dias após infecção cem aerossol quando a coo5 togem de CFU de pulmão média foi 7,37 ±0,05 íogw> camundongos de controle receberam um dos seguintes tratamentos: INH (25 mg/kg) sozinho. RIF (10 mg/kg) sozinho, RIF-i-PZA (150 mg/kg), RIF'HNH*PZA, MXF (100 mg/kg) ±PZA ou RÍF*MXF±PZA. Os camundongos de teste receberam o composto de fórmula (I) (100 mg/kg) sozinho ou adicionado a cada um dos regime de 10 controle. Camundongos adicionais foram não tratados par servirem como controles negativos. Qs camundongos foram sacrificados após 4 e 8 semanas de tratamento para avaliação de pesos de baço e contagens de CFU de pulmão. Os camundongos de controle não tratados morreram durante o segundo mês do experimento.
O efeito sinergistícc nas contagens de CFU de pulmão de adição do composto de formula (I) para uma variedade de 1- 2- e 3 regimes de fármaco é evidente na Tabela 3.
Tabela 3
| Contagem de CFU log 10 de pulmão (±SD) | |||||
| Regime | Antes de tratamento | Após tratamento com o regime indicado | Após tratamento com o regime indicado mais o composto de fórmula (1) | ||
| iuu mc | img/rna | ||||
| Dia 0 | Dia 23 | Dia 56 | Dia 28 | Dia 58 | |
| Sem tratamento | 7,37 ± 0,12 | 7,76 ± 0,09 | spidl | 4,72 ±0.17 | 2.70 ± 0,29 |
| INH | 5,79 ± 0,14 | 4.53 ± 0,24 | 4.33 ± 0,29 | 2,37 ±0.23 | |
| RIF | 5,74 ±0,17 | 4.85 ± 0,24 | 3.73 ±0,19 | 1.29 ±0.22 | |
| RIF-PZA | 3,97 ±0.11 | 1,05 ±0,44 | 3.04 ± 0,22 | 0,50 ± 0.33 | |
| RIF-INH- PZA | 4,38 ± 0,89 | 2.47 ±0,18 | 3.54 ± 0,24 | 0,47 ± 8,20 |
/63
| Γ ------------------------ Regime | Contagem de CFU logl 0 de pulmão (±SD) | ||||
| Antes de tratamento | Após tratamento com o regime indicado | Após tratamento com o regime indicado mais o composto de fórmula (h 1(H) mg/kg/día | |||
| Dia 0 | Dia 28 | Dia 56 | Dia 28 | Dia 56 | |
| RIF-MXF- PZA MXF-PZA | 3.72 Á 0,20 | 0,61 ± 0,38 | 3,41 ±0,18 | 0.12 ± 0,27 | |
| 5,10 ±0,13 | 3,17 ± 0,28 | 3.33 ±0,33 | 0.93 ± 0,34 |
A monoterapie corn o composto de fórmula (I) da invenção resultou em uma redução de 2,65 tog-^ em contagens de CFU da linha de base para 4,72 log^ sebre as primeiros 28 dias, onde monoterapia de INH e RIF reduziu as contagens de CFU de pulmão par 1,58 e 1,63 log50 a 5,79 e 5.74 lug·,;.·;, respectivamente. Maroadamente, e composto de fórmula (i) da invenção continuou a exercer atividade bacteriada durante o segunda mês de tratamento, reduzindo a contagem de CFU paca 2,70 CPU lugW maior deque a atividade de INH ou RIF sozinho e similar á contagem de CFU em carnundongos tratados corn o regime de combinação de primeira linha padrâa de RIF-INH-PZA que mduzir a contagem de CFU para 2,47 log$i-;, A combinação de INH a o composto de fórmula (I) da invenção não foi mais ativa do que o composto de fórmula (I) sozinha, enquanto combinando RIF com o composto de fórmula (i) tem-se um efeito multiplicative, resultando em uma contagem de CFU de pulmão média perto de 30 vezes mais baixa do que aquela observada com o composto de fórmula (I) sozinha. A combinação de RIF-PZA tinha atividade que reduziu a contagem de CFU de putrnào média para 1.05.
Neste exemplo, a adição de INH a RIF-PZA tinha um efeito an~ tagonístico signíficante, resultando em contagens de CFU de pulmão medias de 1,05 ± 0,44 e 2,47 ± 0,18 após 2 meses de tratamento com RIF-PZA e RÍF-INH-PZA, respectivamente. Este efeito podia falsamente sugerir que substituindo-se um novo fármaco por INH no regime de RIF-INH-PZA tem um efeito benéfico signíficante, mesmo se o novo fármaco far camptetamente inativa, simplesmente porque o efeito antagonístico de INH foi removido.
52/63
Neste exemplo., contudo, o composto da fórmula (I) da Invenção tinha sirnilarmente efeitos fones se ele fosse adicionado a RIF-INH-PZA ou substituído por INH (isto é, RIF-PZA- composto de fórmula (I). Após 2 meses de tratamento, as contagens de CFU de pulmão médias foram 0,47 ± 0,20 e 0.50 ± 0,33 em camundongos tratadas com RIF-INH-PZA-composto de fórmula (I) a RIF-PZA-oampoetu de fórmula (I). respectivamente, resultando em uma contagem de CPU de pulmão média aproximadamente 3,6 vezes mais baixa do que a contagem de CFU de pulmão média em camundongos que receberam RIPPZA sozinho (p<0,05); proporcionando evidência adicional que o beneficio de substituição do composto de fórmula (l) da invenção para INH não vam da remoção da influência antagonlstíca de INH. O composto de fórmula (I) da invenção também aperfeiçoa a atividade do regime de RIF-MXF-PZA O tratamento com RIF-MXF-PZA por 2 meses resultou em urna contagem da CFU de pulmão média de 0,61 ± 0,38, eom 1 de 5 camundongos de cultura negativa, enquanto o tratamento com RiF-MXF-PZA e o composta de fórmula (I) resultou em uma contagem de CFU média de 0,12 ± 0,27, cam 4 fara de 5 camundongos de cultura negativa erradicação completa) (p- 0,05 por diferença ®m contagens de CFU médias). A diferença fes de significância da estatística marginal, provavelmente devido a baixas contagens de CFU, mas implica que o composta de fórmula (I) da invenção pude adicionaimente apedéiçoar a atividade de esterilização de regime para prevenir relapsa. Finalmente. o tratamento com MXF-PZA-composto de fórmula (I) por 2 meses resultou em uma contagem de CFU de pulmão média de 0.93 ± 0,34, resultando em uma contagem de CFU de pulmão média quase 175 vezes mais baixa do que aquela observada sem o composto de fórmula (I) demonstrando que o composto de fórmula (I) da invenção è capaz de substituir RIF rio regime de RIF-MXF-PZA de encurtamento de tratamento sem diminuir a atividade do regime.
Exemplo 7
Atividade to vfeo da combinação após administração prolongada e seguimento
Camundongos foram infectadas com aerossol com 4,45 ± 0,05
53/63
CFU tog10 . Começando 14 dias após infecção com aerossol quando a contagem de CFU de pulmão média foi 7.92 ± 0,15 fogio. os camundongos de controle receberam RIF-INH-PZA por 8 semanas, seguida par RIF-INH por 8 semanas. Um grupo de camundongos de reste recebeu o mesmo regime para qual o composto de fórmula (I) (160 mg/Xg) foi adicionado para a duração total de 16 semanas, adicionado para as primeiras 8 semanas somente, eu adicionada para a duração total de 16 semanas com a remoção de INH apôs as primeeas 8 semanas. Outra grupo de camundongos de teste recebeu o regime de controle ao qual LZD foi adicionado para a duração total de 16 semanas ou adicionado para as primeiras 8 semanas somente. Camundongas adicionais foram não tratados para servirem como controles negativos. Os carnundongos foram sacrificados após 8, 12 e 16 semanas de tratamento para avaliação de contagens de CFU de pulmão. Camundongos adicionais foram mantidos sem tratamento por 12 semanas após completaçac de 12 ou 16 semanas da tratamento. Os eamundongos de controle não tratados morreram durante o segundo mês do experimento,
O efeito sinergístico nas contagens de CFU de pulmão per adição do composto de fórmula (I) (Abreviado cerno >!Uf>) ao regime de controle durante as primeiras 8 semanas é evidente na Tabela 4. O efeito baatericida do regime de controle foi aumentado por 2 ordens de grandeza, Este efeito é guantitatlvamente e quahtativamente diferente do efeito antaganístico de adiçãu de LZD
Tabela 4
Regime i 0 dia
Não tratado
7.92 ± 0,15
I 2 mu, RIF-INH-PZA r I mo. RIF-INH | mo. RIF-INH-PZA | i U * 2 mo. RIF-INH-U i semanas
3,17 ± 0,27
Contagens de CFU log<r> de pulmão (± SDi após tratamento por:
| Proporção (%) de camundongos com relapsa positivo de cultura após tratamento por; | |
| 12 sema- nas | 16 sema- nas |
| 9 de 2.0 (45%) | 18 de 20 (90%) 1 de 20 (5%) |
54/63
Regime l Contagens da CPU log^ | Proporção (%) de ca- i
Ϊ do pulmão (± SIT) após I mundongos com relapso i
Ϊ tratamento por; i positivo de cultura após i
I Í tratamento por
| | 2 rno. RIF INH-PZA· | U t-2mo RIF-U | | 0 dia | 8 semanas i 12. soma- i | OãS | f 7 de 20 [ | (35%) ΐ | 16 semanas 1 de 20 (5%) | |
| 2 mo. RIF4NH-PZA- | i 17 de 20 | | 7 de 20 | |
| U * 2 mo, RIF-INH | | j (85%) | | (35%) | | |
| 2 mo. RF-INH-PZA- | | 4.28 ± 0,24 i | 20 de 20 | |
| LZD-i-2 mo. RIF- i | (100%) | ||
| INH-LZD | | |||
| 2 mo, RIFINH-PZA- | | 20 de 20 | | ||
| LZD * 2 mo. RIF-INH i | (100%) | |
Tratamento com regime incluindo o composto de fórmula (I) (abreviado como :’U na Tabeia 4) fol associado a uma probabilidade mferior de relapso (a medida de padrão de ouro de erradicação completa) após complefação de 12 e 16 semanas de tratamento comparado a oamundongos 5 que recebem outros regime. Após 1.2 semanas de tratamento com o regime de controle todos os camundongos permaneceram positivos a cultura e seda esperado exibirem uma taxa de relapso de 106%. isto ê adicíonalmente suportada por uma taxa de relapso de 90% após 16 semanas de tratamento.
Em contraste, grupos que receberam um regime incluindo o 10 composto de fórmula (I) para o total de 16 semanas tem uma taxa de relapso média de 40% após 12 semanas e 5% após 16 semanas. Quando o composto de fórmula (I) foi adicionado ao regime de controle por somente as primeiras 8 semanas, 85% e 35% dos oamtmdongos com relapso após durações de tratamento total de 12 e 16 semanas, respectivamente. Por outro 15 lado, a adição de I..ZD foi antagonistic^ e preveniu erradicação completa em qualquer camundongo. Estes dados demonstram que o composto de fórmula (I) tem efeito sínergistíco quando combinado com o regime de primeira linha padrão e è capaz de encurtar a duração de tratamento por 1 a 2 meses sem eficácia de sacrifício.
Consequentemente, o composto de fórmula (1) tem efeito síner
55/63 glstico quando combinado com pelo monas dois agentes úteis para o tratamento de tuberculose. Mais importantemente, a combinação do composto de fórmula (I) com pela menos dois agentes para o tratamento de tuberculose aumenta dramaticamente a atividade bactencida, sugerindo que ela pode encurtar a duração de quimioterapia para TB suscetível a fánnaco, bem como MDR-TB
Exernpto.8 agentes de primeira tinha padrão, rifampin, isoniazid, e pirazln·
O Exemplo 8 descreve os resultados de um estudo sao que camundongcs foram infectados oom Mycobacterium te.bercteose similar á maneira descrita no Exemplo 6 acima. Diferente dos controles, iodos os camundongos foram tratados com os seguintes três fármacos:
1) rifampin, 10mg/kg(R),
2) isoniazid, 25mg/kg(N), e
3) pirazinamida, 1S0mg/kg(Z).
Os camundongos foram divididos em oito grupos (8) a seis dos grupos receberam dosas variantes do composto de Fórmula iftH. As doses variaram de 12,5 mg/kg a 160 mg/kg e são listadas na Tabela 5 abaixo..
O estudo foi efetuado da seguinte maneira. Camundongos de seis semanas da idade fêmeas BALB/c foram infectados por rota de aerossol com Myoubaotenuvn teheroteose H37Rv, Naquele dia (D· 17), 5 camundongcs foram sacrificados para determinar o e número de CFIJ implantada nos pulmões. Dezessete dias mais tarde (DO), 5 camundongos adicionais foram sacrificados para determinar a contagem de CFU de tinha de base nos pulmões. Os camundongos remanescentes foram randomizados para grupos de tratamento, conforme indicado na Tabela 5, e iniciados no regime de fárrnacc especificado. Q tratamento foi administrado uma vez diariamente, 5 dias por semana, exceto para a grupo 8. que recebeu três tratamentos sernanalmente (3/7). Na conclusão do periodo de teste de 4 semanas, todos os animais foram sacrificados para contagens de CFU de pulmão, exceto os 80
56/63 animais da Grupo 4 abaixo Estes animais foram utilizados em um protocolo do 8 semanas que è descrito abaixo no Exempla BB,
Tabela 5
| Regime | No. de Carnundongos | Contagem de CFU Dia (-)17 | Contagem de CFU Dia ' lit | Contagem de CFU 4 semanas |
| Controles | ||||
| 1) Não tratadas | 15 | 3,50 ± 0.23 | 7.71 ± 0,06 | Morte |
| 2) RHZ | j|! | 4,71 ±0,17 | ||
| Regime de Teste | ||||
| 3)RHZU !2 a | 5 | G / | 4,51 ± 0,16 | |
| 4) RHZU^ | 85 | 4,19 ±0,26 | ||
| 5) RHZUS0 | 5 | 4.32 ± 0,26 | ||
| 6) RHZUvxi | ±™...................... | 4.20 ± 0,25 | ||
| 7) RHZUvrs | 5 | 4.01 ± 0,21 | ||
| ........i RHZUK(3/7) | 5 __ | _______________________ | 4,88 ± 0,33 |
Conforme representado acima, α composto de formula I de5 monstrou um efeito dependente de dose nus números de CFU A adição deste composto ao regime padrão de RHZ produziu redução adicional no número de colônias de TB contidas dentro do pulmão.
ExerngíoSB sMM
Estudo
Oitenta dos camundongos do Grupo 4 (no Exemplo BA) que tinham recebido 25 mg/kg dc- composto de fórmula í (U) junto com rifampin(R), isoniazid(H), e pirazmamsda(Z) foram avaliados na segunda fase do estudo. Eles foram transferidos para um dos grupos descritos abaixo na Tabela 6 e o estudo foi continuado por um adicional de 4 semanas (isto é, 8 semanas de resultados).
57/63
| Tabela 6 | |
| Regime de Fàrrnaco | No. de Camundongos |
| 1) Nenhum tratamento | 5 |
| 2): RH ........ | 7:7 |
| 3)R ' | Cs ST...... |
| :: 1Π:||Ι|ΙΖιί''Σ | |·····^ |
| U dado com ou sem R. | |
| 5) UW*LR | ......... .....9' |
| 7 ;;3^7::K^ |
7).i§sw. I .................' '
7) UfjO'M-R ?: 5
........ 3) tJ^te/R......................................|·............ 5
.................. ...........f —
1Q)Ü5O(3/7)4CR ].....................................................5
Conforme descrito acima. 5 animais serviram como o controle e nenhum fármaco adicionas foi dado. A cinco foi dada a combinação de rifampin(R) e isoniazid (H). cinco dada isoniazid (H) ou rifampin(R) apenas. Dos 5 doze (12) grupos remanescentes, todos foram dados o composto de fórmula (I) em dosas variando de 12,5 mg/kg a 180 mg/kg. Seis dos mesmos grupos foram também dosados com rifampin (R). A rifampin e isoniazid foram dosadas na mesma maneira e na mesma quantidade como no Exemplo 8A.
Ma conclusão do experimento de oito (8) semanas, todos os a10 nimais foram sacrificados e as contagens de CFU no pulmão foram obtidas.
Os seguintes resultados foram obtidas:
Tabela?
| i Tratamento | CFU no Pulmão Ϊ |
| í 1) Não tratado | 5,49 :t 0.23 | |
| i 2) tsoniazid(H) | 4,46*0.37 i |
| i 3) Rifampin(R) | 4.18±0.55 j ____________________$ |
| I <ReR | 3,84 ± 0.48 | |
Na iniciação desta segunda fase, os oitenta animais tinham uma
CFU media de 4.19 * 0,26 (baseada nos outros oamundongos do Grupo 4 15 que foram sacrificados). A 'Tabela 7 mostra os resultados obtidos com grupos de controle, a rifampin(R) somente, ísoniazíd(K) somente e a combinação de ambas isoniazid(H) e rifampin(R).
58/63
A Tabela 8 mostra os resultados obtidos som os camundongos que receberam o composto de fórmula (I) (U) sozinho ou em combinação com rifampin(R).
Tabela 8
| Dose de U | CFU-U somente | CFU-Ue R |
| 4,99 ±0,19 | 3,42 ± 0,40 | |
| U;>& | 4,26 ± 0,24 | 2,83 ±0.24 |
| 3,56^:0,15/ | 2,49 ±0.31 | |
| Üibç | ............2,8810,34 | ...................t,54-±ÍÍ5':................ |
| Í,42±0,31 | 1^6013 | |
| ............03............ | 4.21 ± 0,25 .......................?....................;_____________________ | ......... |
O composto de Fórmula l (U) demonstrou atividade bauterioida dependente de dose contra bacilo da tuberculose persistente que permaneceu viável após as 4 semanas iniciais de tratamento com RHZU^. Os seguintes efeitos foram observados com monoterapia de LJ: um efeito inibitorio de crescimento a 12,5 mg/kg, um efeito bactenostãtioo a 2.5 mg/kg, uma redução de >0,5 log a 50 mg/kg, uma redução de >1 log a 100 mg/kg, e uma redução de quase 2. log a 160 mg/kg. A combinação de rifampin(R) e o composto de fórmula (i) U foi slriergistica e se comprovou ter forte atividade bactericide contra persistídores. Quando o composto de fórmula (I) U foi administrado em doses P25 mg/kg, a combinação dele e rifampin R foi mais eficaz do que o regime padrão de rífampin(R) e isoniazid (H), 0s resultados com U somente suporta a conclusão que regimes contendo U podem ser capazes de encurtar signlficantemente a duração de tratamento para ambas tuberculose resistente a muliifârmaco e tuberculose extensivamente resistente a fárrnaco.
Exemplo 8C .Ç..?tydoVarian^çgmjD^
O protocolo descrito acima no Exemplo SA e 8B foi efetuado em uma ocasião alternativa. As únicas mudanças substantivas foram o número de animais por grupo e a periodo de incubação inicial. A Tabela 9 proporciona os resultados obtidos
59/63 l abels 9
| Regime | No. de Camundongos | Contagem de CFU Dia j/llll·................ | Contagem de CFU Die 0 | Contagem de CFU 4 semanas |
| Controles | ||||
| 1) Não tratados | 12 | 3,26 ±0.14 | 6,28 ± 0,08 | 7,2310,15 |
| 2) RHZ | 4 | 4,5810.15 | ||
| Regime de teste | ||||
| DRHZU^o | 4 | 4,1610,28 | ||
| 2) RHZUW | 64 | 3,93 1 0.21 | ||
| 3) RHZUso | 4 | 4,30 1 0.27 | ||
| 4) RHZU^ | 4 | 4,3910,23 | ||
| 5) RHZU^.s | 4 | 4,3010.25 | ||
| 6} RHZUs | 4 | 4.39 |
Os testes continuaram por um adicional de 4 semanas com 50 dos camundongcs de Grupo de Teste 2 irnedialamente acima. Os camtindongas ou receberam u sozinho, em doses variantes, eu a combinação de U 5 e R, Os seguintes resultados foram obtidos.
| Regime ;T::: ::: ,4________________________________________________________________I | No. de Camundongos | Contagem de CFU 8 semanas |
| Centrales | ||
| 1) Não tratados | 4 | :4^111,17 |
| 2) R | 4 | 4,1610.65 |
| 3) RH | 4 | 3,00 i: 0.16 |
Regime de Teste
| w | 4 | 2,90 1 0.32 |
| 2} RU^ | 2,1610.20 | |
| 3) U ?χ | 4 | 3,8610.53 |
| 4) RLW | 4 | 2,3510,21 |
| 5) | 4 | 4,5910.28 |
| 6} FW | 4 | 3,1710.76 |
| 7) | 4 | 5,27 10.20 |
| 8) RUSS | 4 | 4,27 10<33 |
| 9)U52.S | 4 | 5,30 10.08 |
60/63
| Regime | í 5 | èo. de Camundongos ΐ C | onfagem de CFU 8 |
| ..................................... ................. | semanas | ||
| 10) RUi?í | 2 22% | 4 1 | 3,62 ± 0,40 |
| 11) us.^. 12) RD6 | .........4— ......,..,1..,..,. | 4 j 4 j | 4,93 ± 0,32 4.33^0,16 |
Conforme representado acima, u composto de formula I demonstrou um efeito dependente de dose nos números da CPU. A adição deste composto ao regime padrão de nfampin(R) a isoniazid (H) e rifampin (R) sozinhas produziu redução adicional no número de adornas de TB conte 5 das dentro do pulmão em ambas 4 e 8 semanas
Exemplo 9
No Exemplo 9, o impacto do composto de fórmula I nos fármaoos antituberculose de primeira e de segunda linhas existentes fui avaliado. Duas doses do composto foram avaliadas, 25 mg/kg e 100 mg/kg, Os se10 guintes agentes antituberculose padrão foram avaliados nas doses especificadas'.
1) isoniazid, lOmg/kg (H)
2) rifampin, IQrng/kg (R)
3) pírazinaniíde, 150mg/kg: (Z)
4) etambutol 100mg/kg (Eb)
5) moxifloxacin lOOmg (M)
6) etionamida 50mg/kg (Et)
7) arnicacin 1S0mg/kg subcataaeameaie. (A)
8) eiclossarina 25Ômg/kg duas vezes diariamente (Cs)
9) ácido para-aminessalioílico 750 mg/kg (Ps)
10) capreomicin 125mg/kg subcutaneamente (Cap)
11) clofaz.imina 20mg/kg (C)
O estudo foi efetuado da seguinte maneira:
Métodos (3amun.dpn.dOS· fêmeas BALB/c, 5 semanas de idade infeooãq· infecção com aerossol com -104 CFU M, tubermufose
H37Rv; seguido por randomizaçâo para grupo de tratamento
Tratamento: iniciado em dias 14 apôs infecção (D0) quando as /63 contagens de CFU nos pulmões do camundongu foi -IO*5 CFU.
Todos os fármacos foram dados uma vez diariamente (exceto ciulossenna -- duas vezes diariamente) pela rota oral (exceto amrcacin e capreomicm - injeção subcutaneameute). 5 dias por semana.
Sacnflcios do Çamundongo para contagens de CFU nos pulmões: De acordo com o esquema experimental 4 camundongos foram sacrificados no dia após infecção (D-13) e 2 semanas mais tarde (DO) para avaliar o número de CFU implantada e as contagens de QFU de linha de base, respectivamente, A atividade da cada regime foi ser avaliada por contagens de CFU de pulmão após 4 semanas de tratamento.
Besulíadõs
Camundongos foram infectados com ~ 4,5 CFU log 10 .. Quatorze dias mais tarde a contagem de CFU de pulmão na iniciação de tratamento no dia zero fui aproximadamente 8,3 log^· Os camundongos não tratadas morreram dentro das primeiras 3 semanas de infecção (Tabela 11), A sobre vivência dos camundongos tratados com pirazinamlda (Z), cíclosserina (Cs), e caprsornícin (Cap) não foi significantemente diferente dos controles não tratados, O tratamento com ácido para-aminossaliciHco (Ps) retardou, mas não impediu mortalidade. O tratamento com etambutol (Eh) e clofazímina (C) impediu alguma, mas não toda mortalidade. O tratamento com isoniazid (H), rifampin (R), muxifloxaoin (M), amicacin (A) e etinnamida (Et) impediu morte com o tratamento com o composto de fórmula I (D) em qualquer dose, se sozinha ou em combinação com outros fármacos.
abela 11
JWWWWTOWWWWWWft........
| i Grupo de tratamento | Proporção (%) que sobreviveu a Dia 42 pós-infecção | Tempo para 50% de mortalidade (dias) |
| | Qualquer regime uon: tendo U | 4/4 (100%) | N/A |
| | H, R, M, A ou Et | 4/4 (100%) | N/A |
| 3/4 (75%) | WA | |
| 1 Eb | 2/4 (50%) | 30 |
| i Ps | 0/4 (0%) | ............................ |
| 1 cap | 0/4 (0%) | 22 |
62/83
| Grupo de tratamento | Proporção (%) que sobrevi- | Tempo para 50% der |
| veu a Dia 42 põs-infecção | mortalidade (dias) | |
| Cs | 0/4 (0B | 22 ....... |
| ........................................ | .....'T:1Í;2..... '''I; | |
| Nenhum tratamento | 0/4(0%) | 21 |
Em adição à medição de sobrevivência, contagens de CFU de pulmão foram também determinadas conforme descrito acima e são reporta·· - das abaixo na Tabela 12
Tabela 12
| Contagem | de CFU de pulmão Média log·. | ' (±) S.Ü. | |
| Regime ; na iniciação | apôs 4 semanas de tratamento com: | ||
| í do tratamento | Regime so- | Regime + U | Regime + U |
| (D0) | zinho | 25 mg/kg | 100 mg/kg |
| 1) Não ira- ; 8.36 ±0.10 fados | N/A | 7,17 ± 0,23 | 5.40 ±0,10 |
| ............ | 7,01 ± 0,14 | 6.22 ± 0,06 | .....5,791 0,33...... |
| 6,23 + 0,11 | 5.91 ± 0.21 | 5,02 ± 0,10 | |
| ..... | N/A | 4,73 ±0,38 | < 3.59** |
| 5) Eb | 7,41 1:0,57* | 6,86 + 0,16 | 5,50 + 0.13 |
| 6)M | 6,49 + 0,1.2 | 7,08 ± 0,10 | 5.33 + 0,30 . < ' ' . '· |
| 7) Et | 7,19 i: 0,04 | 7.18 ±0.18 | 5,46 + 0.15 |
| 8)A 1 | 7,B.±©4 | 6.78 ± 0.16 | 5,18 ±0,17 |
| .......N/A............. | 7.19 ± 0,21 | 5,68 ±0.11 | |
| 10) Ps | N/A | 6,87 ti 0,2 Γ* | 5.39 ±0,32 |
| 11) Cap I ^...........,.D..^.^___.^%X<sssssssss%ss%sssssssssssssssssssssss<sssssssh | N/A | 6.81 + 0.13 | 5,35 + 0,17 |
| 12)0 í | 5,92 ± 0,24** | 6.25 + 0.20 | 4,91+0,14 |
N/A, não disponível devido á mede em 4 de 4 camundongos antes W4;
* 2 de 4 camundongos sobreviveram:
** 3 de 4 carnundongos sobreviveram;
as contagens de CPU foram abaixo do limite inferior de detecção de 3,40 para 2 de 4 camundongos
Uma revisão dos dados mostra que a adição da composto de
Fórmula 1 (U) reduz sigmficantemente a carga bacteriana em uma maneira dependente da dose quando adicionada a fàrmacos antituberculares padrão. Estes resultados sugerem que o composto de formula I pode ser usado para
63/63 aperfeiçoar a eficiência de regimes de tratamento para tuberculose resistente a tarmacs (ou resistente a multifármaco ou extensivamente resistente a fdrmaco).
Todas as patentes, pedidos, publicações, métodos de teste, literatura, e outras materiais aqui citados são, desse modo, incorporadas aqui por referência ern suas fatalidades.
Claims (10)
- REIVINDICAÇÕES1. Uso de um composto da fórmula (I) ou um sal farmaceutica- mente aceitável do mesmo:o
 em combinação com pelo menos dois agentes antituberculina, caracterizado pelo fato de ser para a fabricação de um medicamento para o tratamento de tuberculose.
em combinação com pelo menos dois agentes antituberculina, caracterizado pelo fato de ser para a fabricação de um medicamento para o tratamento de tuberculose. - 2. Uso de acordo com a reivindicação 1, caracterizado pelo fato de que referidos pelo menos dois agentes são selecionados a partir do grupo consistindo em isoniazid, rifampin, rifapentina, rifabutin, pirazinamida, etambutol, estreptomicina, canamicina, amicacin, moxifloxacin, gatifloxacin, levofloxacin, ofloxacin, ciprofloxacin capreomicin, etionamida, ciclosserina, ácido para-aminossalicilico, tiacetazona, claritromicin, amoxilina-ácido clavulânico, imipenem, meropenem, viomicin, terizidona, TMC207, PA-824, OPC-7683, LL-3858 e SQ-109.
- 3. Uso de acordo com a reivindicação 1, caracterizado pelo fato de que um dos referidos pelo menos dois agentes é selecionado a partir do grupo consistindo em isoniazid, rifampin, rifapentina, rifabutin, pirazinamida, moxifloxacin, gatifloxacin, levofloxacin, ofloxacin, ciprofloxacin, e etambutol.
- 4. Uso de acordo com a reivindicação 1, caracterizado pelo fato de que um dos referidos pelo menos dois agentes é selecionado a partir do grupo consistindo em pirazinamida, rifampin, rifapentina e isoniazid.
- 5. Uso de um composto de fórmula (I) ou um sal farmaceuticamente aceitável do mesmo:o
 em combinação com pelo menos um agente antituberculina, caracterizado pelo fato de ser para a fabricação de um medicamento para tratamento de tuberculose após um indivíduo ter suportado uma fase inicial de tratamento para tuberculose.
em combinação com pelo menos um agente antituberculina, caracterizado pelo fato de ser para a fabricação de um medicamento para tratamento de tuberculose após um indivíduo ter suportado uma fase inicial de tratamento para tuberculose. - 6. Uso de acordo com a reivindicação 5, caracterizado pelo fato de que o referido pelo menos um agente é selecionado a partir do grupo consistindo em rifampin, rifapentina, rifabutin, pirazinamida, etambutol, estreptomicina, canamicina, amicacin, moxifloxacin, gatifloxacin, levofloxacin, ofloxacin, ciprofloxacin, capreomicin, etionamida, ciclosserina, ácido paraaminossalicílico, tiacetazona, claritromicin, amoxilina-ácido clavulânico, imipenem, meropenem, viomicin, terizidona, TMC207, PA-824, OPC-7683, LL3858eSQ-109.
- 7. Uso de acordo com a reivindicação 5, caracterizado pelo fato de que o referido indivíduo é afligido por uma condição selecionada a partir do grupo consistindo em tuberculose sensível a fármaco, tuberculose resistente a monofármaco, tuberculose resistente a multifármaco (MDR) e tuberculose resistente extensivamente a fármaco (XDR).
- 8. Composição farmacêutica, caracterizada pelo fato de que compreende:i) uma quantidade terapeuticamente eficaz de um composto de fórmula (I) ou um sal farmaceuticamente aceitável do mesmo:o
 (ii) uma quantidade terapeuticamente eficaz de pelo menos um agente útil no tratamento de tuberculose e, (iii) um ou mais transportadores ou veículos farmaceuticamente aceitáveis.
(ii) uma quantidade terapeuticamente eficaz de pelo menos um agente útil no tratamento de tuberculose e, (iii) um ou mais transportadores ou veículos farmaceuticamente aceitáveis. - 9. Composição farmacêutica de acordo com a reivindicação 8, caracterizada pelo fato de que o referido pelo menos um agente é selecionado a partir do grupo consistindo em isoniazid, rifampin, rifapentina, rifabu- tin, pirazinamida, etambutol, estreptomicina, canamicina, amicacin, moxifloxacin, gatifloxacin, levofloxacin, ofloxacin, ciprofloxacin, capreomicin, etionamida, ciclosserina, ácido para-aminossalicilico, tiacetazona, claritromicin, amoxilina-ácido clavulânico, imipenem, meropenem, viomicin, terizidona, 5 TMC207, PA-824, OPC-7683, LL-3858 e SQ-109.
- 10. Composição farmacêutica de acordo com a reivindicação 8 ou 9, caracterizada pelo fato de que o referido pelo menos um agente é selecionado a partir do grupo consistindo em isoniazid, rifampin, rifapentina, rifabutin, pirazinamida, moxifloxacin, gatifloxacin, levofloxacin, ofloxacin, e 10 etambutol.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US9387908P | 2008-09-03 | 2008-09-03 | |
| PCT/IB2009/053796 WO2010026526A1 (en) | 2008-09-03 | 2009-08-31 | Combination therapy for tuberculosis |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| BRPI0918802A2 true BRPI0918802A2 (pt) | 2019-09-17 |
Family
ID=41279484
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| BRPI0918802A BRPI0918802A2 (pt) | 2008-09-03 | 2009-08-31 | uso de (s)-n-[[3-[3-fluoro-4-(4-tiomorfolinil)fenil]-2-oxo-5-oxazolidinil] metil] acetamida e composição farmacêutica |
Country Status (14)
| Country | Link |
|---|---|
| US (3) | US20110190199A1 (pt) |
| EP (1) | EP2340022B1 (pt) |
| JP (1) | JP5661037B2 (pt) |
| KR (1) | KR101318189B1 (pt) |
| CN (1) | CN102143748A (pt) |
| AU (1) | AU2009288820A1 (pt) |
| BR (1) | BRPI0918802A2 (pt) |
| CA (1) | CA2735229C (pt) |
| IL (1) | IL211293A (pt) |
| MX (1) | MX2011002348A (pt) |
| NZ (1) | NZ591169A (pt) |
| RU (1) | RU2484819C2 (pt) |
| WO (1) | WO2010026526A1 (pt) |
| ZA (1) | ZA201101742B (pt) |
Families Citing this family (35)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| RU2464016C2 (ru) * | 2010-06-11 | 2012-10-20 | Ольга Германовна Челнокова | Способ лечения туберкулеза |
| ES2395304B1 (es) * | 2011-05-20 | 2014-01-16 | Interquim, S.A. | Procedimiento de obtención de una tiofen-2-carboxamida. |
| CN102786516B (zh) * | 2012-08-21 | 2014-10-01 | 湖南师范大学 | 一种利伐沙班的合成方法 |
| CN103869040B (zh) * | 2012-12-14 | 2016-12-28 | 上海科胜药物研发有限公司 | 一种特立齐酮中d‑环丝氨酸含量的测定分析方法 |
| US20170010272A1 (en) * | 2014-02-18 | 2017-01-12 | Stc, Unm | Rapid phenotype tests for antitubercular drug sensitivity and resistance |
| WO2016099233A2 (ru) * | 2014-10-20 | 2016-06-23 | Товарищество С Ограниченной Ответственностью "Фармацевтическая Компания "Ромат" | Фармацевтическая композиция для лечения туберкулеза |
| CN107249691A (zh) * | 2014-11-03 | 2017-10-13 | 加利福利亚大学董事会 | 用于肺结核治疗的多药疗法 |
| AU2016212116B2 (en) * | 2015-01-27 | 2021-07-15 | Janssen Pharmaceutica Nv | Dispersible compositions |
| WO2017066053A1 (en) * | 2015-10-14 | 2017-04-20 | The Global Alliance For Tb Drug Development, Inc. | Combination antibacterial composition and short-course antibacterial regimen |
| WO2017066964A1 (en) * | 2015-10-22 | 2017-04-27 | Merck Sharp & Dohme Corp. | Oxazolidinone compounds and methods of use thereof as antibacterial agents |
| BR102015027449B1 (pt) * | 2015-10-29 | 2021-02-17 | Fundação Oswaldo Cruz | composição farmacêutica |
| RU2627611C2 (ru) * | 2015-10-30 | 2017-08-09 | Акционерное общество "Фармасинтез" (АО "Фармасинтез") | Противотуберкулезная фармацевтическая композиция, содержащая тиоацетазон |
| US10472687B2 (en) | 2016-01-08 | 2019-11-12 | Institute For Systems Biology | Methods to identify antituberculosis compounds |
| RU2650633C2 (ru) * | 2016-06-02 | 2018-04-16 | Государственное бюджетное учреждение Республики Саха (Якутия) "Научно-практический центр "Фтизиатрия" | Способ лечения инфильтративного туберкулеза легких с множественной лекарственной устойчивостью |
| CN105853461A (zh) * | 2016-06-14 | 2016-08-17 | 黄峰 | 一种治疗肺结核的药物组合 |
| CN106580915B (zh) * | 2016-10-13 | 2019-11-08 | 中国人民解放军第三〇九医院 | 一种载利福喷丁和利奈唑胺的缓释微球及其制备方法和用途 |
| CN106581048A (zh) * | 2016-12-07 | 2017-04-26 | 郑州郑先医药科技有限公司 | 一种治疗肺结核的西药组合及其制备方法 |
| CN107243008B (zh) * | 2017-04-13 | 2021-03-30 | 中国科学院广州生物医药与健康研究院 | 吡唑并[1,5-a]吡啶类化合物的新应用及一种治疗脓肿分枝杆菌感染的组合物 |
| WO2018190680A1 (ko) * | 2017-04-13 | 2018-10-18 | 충남대학교산학협력단 | 세포내 결핵균 제어를 위한 뉴트린-3α 및 피53 발현 조절 조성물 또는 방법 |
| KR102082285B1 (ko) * | 2017-04-13 | 2020-02-28 | 충남대학교산학협력단 | 세포내 결핵균 제어를 위한 Nutlin-3α (뉴트린-3α) 및 p53 발현 조절 조성물 또는 방법 |
| WO2018191628A1 (en) * | 2017-04-14 | 2018-10-18 | The Regents Of The University Of California | Multi-drug therapies for tuberculosis treatment |
| RU2702607C1 (ru) * | 2018-07-25 | 2019-10-08 | Федеральное государственное бюджетное учреждение "Новосибирский научно-исследовательский институт туберкулеза" Министерства здравоохранения Российской Федерации (ФГБУ "ННИИТ" Минздрава России) | Способ лечения туберкулеза мочевого пузыря |
| CN109223778B (zh) * | 2018-11-16 | 2021-07-27 | 上海市肺科医院 | C24h24n6o2s3在制备抗结核菌药物中的用途 |
| CN110441434A (zh) * | 2019-08-29 | 2019-11-12 | 重庆华邦胜凯制药有限公司 | Gc-ms分离测定对氨基水杨酸中相关杂质的方法 |
| US20230132088A1 (en) * | 2020-03-18 | 2023-04-27 | The Board Of Trustees Of The Leland Stanford Junior University | Detection of gene polymorphisms |
| WO2021184339A1 (en) * | 2020-03-20 | 2021-09-23 | Merck Sharp & Dohme Corp. | Oxazolidinone compound and methods of use thereof as an antibacterial agent |
| WO2021257461A1 (en) * | 2020-06-15 | 2021-12-23 | Mylan Laboratories Limited | Combination antibacterial composition and method for antibacterial therapy |
| WO2021257466A1 (en) * | 2020-06-15 | 2021-12-23 | The Global Alliance For Tb Drug Development, Inc. | Combination antibacterial composition and method for antibacterial therapy |
| AU2022214530B2 (en) * | 2021-02-01 | 2025-12-11 | The Global Alliance For Tb Drug Development, Inc. | Pretomanid amorphous form |
| CN113730550B (zh) * | 2021-04-06 | 2023-08-18 | 中国医学科学院医药生物技术研究所 | 博宁霉素在治疗耐药性结核病中的应用 |
| CN114354804B (zh) * | 2021-12-31 | 2025-03-18 | 深圳市第三人民医院(深圳市肝病研究所) | 试剂盒和样本中抗结核药物及其代谢物的检测方法 |
| CN117045657A (zh) * | 2022-05-12 | 2023-11-14 | 广州嘉越医药科技有限公司 | 一种药物组合及其应用 |
| US20260062394A1 (en) * | 2022-08-10 | 2026-03-05 | Merck Sharp & Dohme Llc | Processes for preparing oxazolidinone compounds |
| US11977085B1 (en) | 2023-09-05 | 2024-05-07 | Elan Ehrlich | Date rape drug detection device and method of using same |
| CN120004814B (zh) * | 2025-02-13 | 2025-08-12 | 山西立业制药有限公司 | 一种氯法齐明乙醇晶型化合物及其制备方法和应用、药物组合物 |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5688792A (en) * | 1994-08-16 | 1997-11-18 | Pharmacia & Upjohn Company | Substituted oxazine and thiazine oxazolidinone antimicrobials |
| US8394839B2 (en) * | 2007-08-21 | 2013-03-12 | Stc.Unm | Rationally improved isoniazid and ethionamide derivatives and activity through selective isotopic substitution |
-
2009
- 2009-08-31 BR BRPI0918802A patent/BRPI0918802A2/pt not_active IP Right Cessation
- 2009-08-31 EP EP09787058.8A patent/EP2340022B1/en not_active Revoked
- 2009-08-31 CA CA2735229A patent/CA2735229C/en not_active Expired - Fee Related
- 2009-08-31 AU AU2009288820A patent/AU2009288820A1/en not_active Abandoned
- 2009-08-31 US US13/061,233 patent/US20110190199A1/en not_active Abandoned
- 2009-08-31 MX MX2011002348A patent/MX2011002348A/es active IP Right Grant
- 2009-08-31 WO PCT/IB2009/053796 patent/WO2010026526A1/en not_active Ceased
- 2009-08-31 CN CN200980134310XA patent/CN102143748A/zh active Pending
- 2009-08-31 KR KR1020117007672A patent/KR101318189B1/ko not_active Expired - Fee Related
- 2009-08-31 JP JP2011525657A patent/JP5661037B2/ja not_active Expired - Fee Related
- 2009-08-31 RU RU2011108026/15A patent/RU2484819C2/ru not_active IP Right Cessation
- 2009-08-31 NZ NZ591169A patent/NZ591169A/xx not_active IP Right Cessation
-
2011
- 2011-02-17 IL IL211293A patent/IL211293A/en not_active IP Right Cessation
- 2011-03-07 ZA ZA2011/01742A patent/ZA201101742B/en unknown
-
2015
- 2015-10-06 US US14/876,311 patent/US20160220578A1/en not_active Abandoned
-
2017
- 2017-07-10 US US15/645,254 patent/US20170368070A1/en not_active Abandoned
Also Published As
| Publication number | Publication date |
|---|---|
| MX2011002348A (es) | 2011-06-27 |
| JP5661037B2 (ja) | 2015-01-28 |
| US20160220578A1 (en) | 2016-08-04 |
| ZA201101742B (en) | 2011-11-30 |
| AU2009288820A1 (en) | 2010-03-11 |
| KR20110063518A (ko) | 2011-06-10 |
| RU2484819C2 (ru) | 2013-06-20 |
| JP2012502017A (ja) | 2012-01-26 |
| CN102143748A (zh) | 2011-08-03 |
| NZ591169A (en) | 2012-12-21 |
| US20110190199A1 (en) | 2011-08-04 |
| EP2340022B1 (en) | 2013-11-20 |
| KR101318189B1 (ko) | 2013-10-16 |
| RU2011108026A (ru) | 2012-10-10 |
| IL211293A0 (en) | 2011-04-28 |
| IL211293A (en) | 2016-08-31 |
| US20170368070A1 (en) | 2017-12-28 |
| EP2340022A1 (en) | 2011-07-06 |
| WO2010026526A1 (en) | 2010-03-11 |
| CA2735229A1 (en) | 2010-03-11 |
| CA2735229C (en) | 2014-01-28 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| BRPI0918802A2 (pt) | uso de (s)-n-[[3-[3-fluoro-4-(4-tiomorfolinil)fenil]-2-oxo-5-oxazolidinil] metil] acetamida e composição farmacêutica | |
| US10717735B2 (en) | Solid forms of a compound for modulating kinases | |
| CA2910756C (en) | Compounds and methods of treating infections | |
| JP7045985B2 (ja) | Ezh2阻害剤を用いた髄芽腫の処置方法 | |
| JP5925065B2 (ja) | 医薬組成物 | |
| HK1252318A1 (zh) | 抗菌治疗剂和预防剂 | |
| CN108135887A (zh) | 噁唑烷酮化合物及其作为抗菌剂的使用方法 | |
| JP2022082753A (ja) | 組み合せ抗細菌組成物および短期抗細菌レジメン | |
| CN110831630A (zh) | 组合疗法 | |
| US11370787B2 (en) | Pyrazole compounds, formulations thereof, and a method for using the compounds and/or formulations | |
| TWI799814B (zh) | 噁唑啶酮化合物及其做為抗菌劑之使用方法 | |
| US20140249155A1 (en) | Spectinamides as anti-tuberculosis agents | |
| US20230105108A1 (en) | Compounds for the treatment of bacterial infections and potentiation of antibiotics | |
| BR112016029619B1 (pt) | Compostos da classe de fluoroquinolonas, composição farmacêutica e medicamento compreendendo tais compostos e usos de tais compostos | |
| US12357623B2 (en) | Method for treating a disease or condition using a pyrazole compound or formulation thereof | |
| US20120231994A1 (en) | Antagonism of human formyl peptide receptor for treatment of disease | |
| CN117858708A (zh) | F-atp合酶抑制剂用于治疗脓肿分枝杆菌疾病的发现 | |
| TW201002321A (en) | Pneumonia treatment | |
| CN102770416A (zh) | 美他沙酮共晶体 | |
| JP2007126452A (ja) | 医薬組成物 | |
| HK1155971A (en) | Combination therapy for tuberculosis | |
| JPWO2016093203A1 (ja) | 二環式含窒素芳香族ヘテロ環アミド化合物を有効成分とする医薬組成物 | |
| HK40019086A (en) | Combination therapy | |
| WO2010122456A1 (en) | Compositions of sulopenem and use for treating tuberculosis | |
| JP2019064921A (ja) | C−4”位置換マクロライド化合物の結晶形及びそれらの製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| B08K | Patent lapsed as no evidence of payment of the annual fee has been furnished to inpi [chapter 8.11 patent gazette] |
Free format text: EM VIRTUDE DO ARQUIVAMENTO PUBLICADO NA RPI 2542 DE 24-09-2019 E CONSIDERANDO AUSENCIA DE MANIFESTACAO DENTRO DOS PRAZOS LEGAIS, INFORMO QUE CABE SER MANTIDO O ARQUIVAMENTO DO PEDIDO DE PATENTE, CONFORME O DISPOSTO NO ARTIGO 12, DA RESOLUCAO 113/2013. |
|
| B350 | Update of information on the portal [chapter 15.35 patent gazette] |