CAMPO DA INVENÇÃO
A presente invenção refere-se a um processo para a obtenção de uma estatina, bem como a determinados compostos utilizados como intermediários no referido processo. Preferivelmente, a presente invenção refere-se a um processo para a obtenção de atorvastatina cálcica, bem como aos compostos 2-[2-(4-fluorofenil)-2-oxo-l-feniletil]-4- metil-3-oxo-N-fenilpentanamida, 1-[(R)-6-((S)-2,2-dimetil-1,3-dioxolan-4-il)-3-hidroxi-5-oxo-hexil]-5-(4-fluor- fenil)-2-isopropil-N,4-difenil-lH-pirrol-3-carboxamida, 1- *( (3R7 5R) -6- ( (~S)~-2,2-dimet'il‘-l”, 3-dioxolan-4-il) -3, 5- diidroxiexil)-5-(4-fluorofenil) -2-isopropil-N,4-difenil-lH- pirrol-3-carboxamida e 5-(4-fluorofenil)-2-isopropil-N,4- difenil-1-((3R,7S)-3,5,7,8-tetraidroxioctil)-lH-pirrol-3- carboxamida, os quais são utilizados como intermediários no referido processo.
Além disso, a presente invenção refere-se ao método para a obtenção de uma estatina, preferivelmente atorvastatina cálcica.
A presente invenção refere-se ainda às estatinas obtidas a partir do método para a obtenção de uma estatina, preferivelmente atorvastatina cálcica.
FUNDAMENTOS DA INVENÇÃO
A atorvastatina cálcica é uma estatina indicada como adjunto à dieta para o tratamento de pacientes com níveis elevados de colesterol total, LDL-colesterol, apolipoproteina B e triglicerídeos, para aumentar os níveis de HDL-colesterol em pacientes com hipercolesterolemia primária e hiperlipidemia combinada, níveis elevados de triglicerídeos séricos e para pacientes com disbetalipoproteinemia que não respondem de forma adequada à dieta. A atorvastatina também é indicada para a redução do colesterol total e do LDL-colesterol em pacientes com hipercolesterolemia familiar homozigótica.” ” A’ atórvásfatínã é 'um* agente hipolipemiante inibidor seletivo e competitivo da 3-hidroxi-3-metil-glutaril- coenzima A (HMG-CoA) redutase, a enzima limitante responsável pela conversão da HMG-CoA a mevalonato, um precursor dos esteróis, inclusive do colesterol. A droga diminui os níveis plasmáticos de colesterol e lipoproteínas através da inibição da HMG-CoA redutase e da síntese do colesterol no fígado, aumentando o número de receptores de LDL hepáticos na superfície da célula, aumentando a absorção e o catabolismo do LDL.
De acordo com o estado da técnica, observa-se que a molécula de atorvastatina possui, de maneira geral, duas rotas sintéticas principais.
A primeira rota desenvolvida é completamente linear e constitui-se de 12 etapas, partindo do 4-metil-3-oxo- pentanoato de metila, com um rendimento global de 4,2%.
Em face do baixo rendimento global da referida rota e levando-se em consideração os aspectos econômicos da sintese, foi desenvolvida posteriormente uma rota sintética convergente, onde são preparados separadamente dois fragmentos chave: (a) a 2-[2-(4-fluorofenil)-2-oxo-l- feniletil]-4-metil-3-oxo-N-fenilpentanamida, uma 1,4- dicetona, a qual é preparada em 3 etapas a partir do 4- metil-3-oxo-pentanoato de metila em um rendimento global de ‘33,4%,r 'e " (b) õ 'acetato ' de terc-butil 2-(?4R,6R)-6-(2- aminoetil)-2,2-dimeti1-1,3-dioxan-4-il), um amino éster, o qual pode ser preparado a partir do 4-cloro-3-oxo-butanoato de etila em 6 etapas com um rendimento global de 27%. Alternativamente, o referido amino éster pode ser preparado em 8 etapas, partindo do ácido L-(-)málico, com um rendimento global de 22%. Por fim, os dois fragmentos são conectados através de uma reação de condensação de Pall- Knorr.
De modo a facilitar a compreensão das principais rotas de sintese da atorvastatina existentes no estado da técnica, a seguir serão descritos diversos documentos envolvendo processos para a obtenção de atorvastatina cálcica, bem como seus intermediários.
Os documentos de patente US 4,647,576 e US 4,681,8935 descrevem um processo para a obtenção de lactona de atorvastatina (5') usando uma rota sintética linear. Nesse processo, os inventores descrevem a adição de dienolato de acetoacetato de etila (2) ao aldeido pirrólico (1),produzindo a respectiva hidroxicetona na forma de sua10 mistura racêmica (3 e 3') (Esquema 1) . A redução da hidroxicetona (3, 3' ) com borohidreto de sódio (NaBH4) na presença de uma trialquilborana (n-Bu^B) fornece o respectivo diol (4 e 4') (Esquema 2).Esquema- 1
Esquema 2
Embora o processo descrito nos documentos de patente - 5 —em ’questão forneça "os compostos' desejados, ~elé“ apresenta alguns problemas técnicos, a saber:- As reações envolvidas neste processo são muito dificeis de ser conduzidas em larga escala, pois os reagentes são altamente pirofóricos (NaH e n-BuLi), perigosos e de 10 difícil manipulação. Além disso, os reagentes empregados são de alto custo; e- Os compostos obtidos (4 e 4') devem ser separados por resolução seguida de cristalização fracionada para a obtenção do produto enantiomericamente puro (5') . Este15 processo é dispendioso, necessita de um longo tempo de operação e, ao final, o rendimento é sempre menor que 50%. Estes fatores acabam por elevar consideravelmente o custo operacional do processo tecnológico.
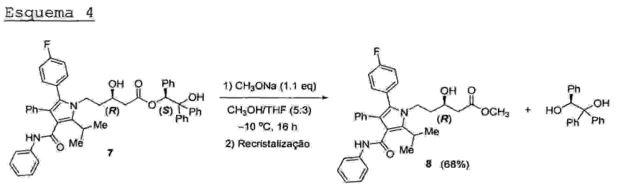
O processo para a produção de atorvastatina cálcica descrito nos Esquemas 3-8 do documento de patente US 5,273,995 consiste em uma rota completamente linear. O aldeido pirrólico (6) é submetido à reação aldólica estereosseletiva com acetato de (S)-2-hidróxi-l,2,2- trifeniletila (A*) em THE' a -78°C via enolato de magnésio com controle de quelação, fornecendo o respectivo β- hidroxi-éster (7) em 73% de rendimento após recristalização, com uma diastereoseletividade de 86:14 em favor do isômero (R,S) (Esquema 3).Esquema 3
A reação de transesterificação do β-hidroxi éster (7) com metóxido de sódio em THE’/metanol a baixa temperaturafornece o respectivo éster metilico (8) em 68% derendimento após recristalização, liberando o auxiliarquiral, (S)-1,1,2-trifeniletano-1,2-diol, o qual pode ser reutilizado (Esquema 4),Esquema 4
Na etapa seguinte, a reação de condensação de Claisen5 cruzada entre o enolato de litio derivado do acetato de terc-butila (THF a baixa temperatura) e o β-hidroxi éster (8) fornece o respectivo β-ceto-δ-hidroxi-0ster (9) em 77%de rendimento isolado -(Esquema 5)*. *
A reação de redução estereosseletiva do β-ceto-δ- hidroxi-éster (9) na presença de trietilborana e hidreto de boro e sódio sob as condições de Narasaka, a baixa temperatura, fornece o respectivo (R,R)-β-δ-dihidroxi éster 15 syn (10) em 83% de rendimento (Esquema 6).Esquema 6
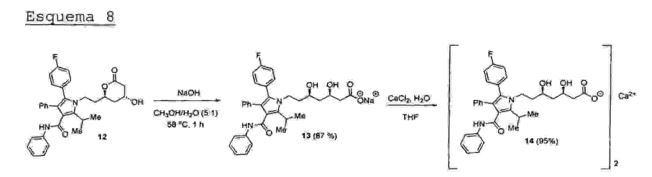
O (R,R)-β-δ-dihidroxi éster syn (10) é submetido a uma reação de hidrólise básica seguida de acidificação para 5 fornecer o respectivo β-δ-dihidroxi-acido syn (11), o qual é imediatamente aquecido em refluxo de tolueno com remoção azeotrópica de água para produzir a respectiva β-hidroxi-δ- "lactona- (lactfõna" dã atorvastatina) (12) em 82% de rendimento isolado após recristalização (Esquema 7).
O tratamento da β-hidroxi-δ-lactona (12) com hidróxido de sódio em metanol/água promove a abertura da lactona, fornecendo o respectivo carboxilato de sódio (13) (sal de 15 sódio da atorvastatina) em 87% de rendimento. Por fim, a reação do sal de sódio (13) com cloreto de cálcio em meioaquoso fornece a atorvastatina cálcica (14) em 95% de rendimento isolado após recristalização (Esquema 8).
O processo acima descrito possui alguns problemas 5 técnicos quando realizado em larga escala, os quais são:- A reação aldôlica (Esquema 3) tem baixa estereoseletividade (relação R,S: S,S = 84:16). Neste caso, um processo adicional de recristalização é necessário para a obtenção do produto final puro. Além disso, o rendimento 10 desta etapa de recristalizaçâo é baixo (menos de 40%), tornando o“ processo global menos eficiente;- É necessária a purificação do éster metilico (8) em coluna cromatográfica de silica gel. Esta operação unitária torna o processo muito mais caro devido ao custo da sílica 15 e dos solventes;- 0 auxiliar quiral (A*) (Esquema 3) (acetato de (S)-2- hidróxi-1,2,2-trifeniletila) é um reagente de alto custo, aumentando muito o custo do processo;- etapa de transesterificação (Esquema 4) emprega20 metóxido de sódio (CHaONa) como base, o qual é um reagente caro, higroscópico e corrosivo. Além disso, o metóxido de sódio, sendo uma base forte, pode gerar subprodutos indesej ados;- A etapa da reação de condensação de Claisen cruzada (Esquema 5) emprega n-butil-litio(n-BuLi) para a geração do LDA (diisopropil amideto de lítio). Este reagente é caro e altamente inflamável, o que representa um grande risco operacional em uma planta industrial; e- A realização da reação aldólica (Esquema 3) necessita da obtenção prévia do brometo de magnésio (MgBrz) a partir de magnésio metálico e bromo elementar, que é um reagente altamente tóxico e corrosivo, tornando seu uso problemático em escala industrial.
Os documentos de patente US 5,003,080; US 5,097,045; US 5*, 124/482; US *5,14 9', 8 37 ;* US’ 5; 21'6,174'; US’S, 245* 047 e US 5,280,126 descrevem a obtenção de lactona de atorvastatina (12) através de uma rota convergente (Esquemas 9 e 10). Inicialmente, o epóxido (15) é tratado com cianeto de potássio, seguido da reação com 2,2-dimetoxipropano (2,2- DMP) para fornecer o acetonideo (16) . A olefina (16) é tratada com ozônio, seguido da oxidação com trióxido de cromo para fornecer o ácido carboxilico (17), o qual é tratado com terc-butanol na presença de 4-DMAP (4- dimetilaminopiridina) e DCC (diciclo-hexilcarbodiimida) esubmetido à hidrogenação catalítica (H2 a 50 psi e óxido de platina (PtOz) como catalisador)éster (18) (Esquema 9).Esquema 9

Esta rota, embora forneça os compostos com a pureza10 desejada, possui alguns inconvenientes e problemas técnicosgraves, conforme descrito a seguir:- A etapa de redução da nitrila (17) (Esquema 9) emprega uma reação de hidrogenação catalítica com hidrogênio gasoso sob alta pressão (50 psi) e óxido de platina (PtOz) como 15 catalisador. Neste caso, o uso de hidrogênio gasoso é problemático devido ao fato de ser um gás altamente inflamável e, além disso, o catalisador (PtO2) tem um alto custo;- A rota sintética envolve o uso de intermediários contendo grupos de proteção e materiais de partida de alto custo. Na obtenção do material de partida (15) emprega-se n-butil- litio (n-BuLi) , o qual é caro e altamente inflamável, representando um grande risco operacional em uma planta industrial;- O processo emprega gás ozônio (O3) para realizar a reação de clivagem de olefina (16). O gás ozônio é altamente tóxico e necessita de equipamentos especiais para a sua geração. Estes fatores contribuem negativamente em um ‘prõcèsso industrial; e- O processo emprega cianeto de potássio (KCN) e trióxido ■ de -cromo (CrOi)*, “ quê s'ãó reagentes altamente tóxicos e, portanto, dificeis de serem empregados em escala industrial, levando-se em conta os aspectos ambientais.
Os documentos de patente US 5,103,024 e US 5,248,793 descrevem a obtenção do amino éster (18), o qual é um intermediário chave na sintese de atorvastatina cálcica (14) em um processo que utiliza sulfonatos arilicos (21) e cianeto de sódio (NaCN) em dimetilsulfóxido (DMSO) como solvente (Esquema 11). Esquema 11
Embora a rota descrita nos documentos de patente US5,103,024 e US 5,248,793 forneça os compostos com pureza desejada, a mesma possui alguns problemas técnicos, 5 conforme detalhado abaixo:
O processo- emprega cianeto de sódio (NaCN)-, o- qual é altamente tóxico e, portanto, difícil de ser utilizado eme_scala industrial, dey_ido_à geração de resíduos;.
Os sulfonatos arílicos (ArSOzCl) empregados neste10 processo possuem custo elevado;
O uso de dimetilsulfóxido (DMSO) como solvente é particularmente problemático por ser um solvente de alta toxicidade e alto ponto de ebulição;- A última etapa (Esquema 11) consiste em uma reação de 15 hidrogenação catalítica, empregando hidrogênio molecularsob alta pressão (50 psi) e níquel de Raney como catalisador, o qual é muito pirofórico e perigoso; e- 0 álcool (20) é preparado por um processo biotecnológico que emprega 7 etapas, resultando em um custo de produção relativamente alto.
O documento de patente US 5,155,251 trata da obtenção do amino éster quiral (18) através de uma sequência linear, conforme mostrado nos Esquemas 12-14.
Inicialmente, o (S)-4-cloro-3-hidróxi-butanoato de etila (24) é submetido à reação com NaCN em etanol aquoso, fornecendo o respectivo (R)-4-ciano-3-hidróxi~butanoato de etila (25) em 57% de rendimento após destilação sob pressão reduzida (Esquema 12). Esquema 12
Na etapa seguinte, o ciano éster (25) é submetido a uma sequência de duas reações sem purificação. Inicialmente, realiza-se uma reação de condensação de Claisen cruzada, onde o ciano éster (25) reage com o enolato de litio derivado do acetato de terc-butila em THE, fornecendo o respectivo (R)-6-ciano-5-hidróxi-3-oxo- h.exanoato (26) , que não é isolado (Esquema 13) . Na etapa subsequente, o β-ceto éster (26) é reduzido seletivamente com hidreto de boro e sódio (NaBH«) na presença de dietil- metóxi-borana (B(OMe)Et2), fornecendo o respectivo diol
Na etapa seguinte (Esquema 14), o (3R,5R)-6-ciano-3, 5-dihidroxihexanoato de terc-butila (27) é tratado com 2,2- dimetoxipropano em acetona na presença de uma quantidade catalitica de ácido metanossulfônico para fornecer o respectivo ciano éster acetonideo (23) em 53% de rendimento (Esquema 14). O produto (23) é um sólido branco altamentecristalino, que é purificado por recristalização. Na últimaetapa reacional, o ciano éster (20) é hidrogenado napresença de níquel de Raney, fornecendo o respectivo aminoéster (18) como um óleo viscoso incolor em 8 5% derendimento.
O processo descrito no documento US 5,155,251, emboraforneça os compostos com pureza desejada, possui alguns problemas técnicos importantes, ,os quais são enumerados abaixo: - 0 (S)-4—cloro-3-hidróxi-butanoato de etila (24) é um reagente de alto custo e que faz com que o processo torne- se mais caro;- O cianeto de sódio (NaCN) empregado na segunda etapa do processo é um reagente altamente tóxico e venenoso, o qual requer procedimentos especiais para sua manipulação e manejo de residues;
Na obtenção do LDA (diisopropil amideto de lítio) (Esquema 13) emprega-se n-butil-lítio (n-BuLi), o qual é caro e altamente inflamável, representando um grande risco operacional em uma planta industrial. Além disso, um grande excesso de LDA e acetato de t-butila (t-BuOAc) (4 equivalentes pára* cada reagente) deve ser empregado para que o processo tenha rendimento satisfatório; e - A última etapa do processo (Esquema 14) consiste em uma reação de hidrogenação catalítica, empregando hidrogênio gasoso sob alta pressão (50 psi) e níquel de Raney como catalisador. 0 gás hidrogênio é potencialmente inflamável e o níquel de Raney é muito pirofórico e este processo requer aparelhagem especial para ser realizado em escala industrial.
Os documentos de patente US 5,298,627; US 5,397,792;
US 5,342,952; US 5,446,054; US 5,470,981; US 5,489,690; US 5,489,691 e US 5,510,488 descrevem um processo para a obtenção de lactona da atorvastatina (12) empregando uma rota convergente, conforme mostrado nos Esquemas 15-19. Na primeira etapa reacional, o ciano éster (25) é submetido à i acetamide (28), (Esquema 15).R1 = R2 = Me. Et, Ph, Bn R1, R2 = -(CH2)5-
Na etapa subsequente, a β-ceto amida (29) é reduzida seletivamente com hidreto de boro e sódio na presença de dietil-metóxi-borana (B(OMe)Eta), fornecendo o respectivo diol 1,3-syn (30) com alta diastereoseletividade (Esquema 16) .Esquema 16OH O O NaBH4l MeOH OH OH O
Na etapa seguinte, o diol 1,3-syn (30) é tratado com2,2-dimetoxipropano em acetona na presença de quantidade catalitica de ácido metanossulfônico para fornecer o respectivo ciano acetonídeo (23) (Esquema 17) . Na etapa posterior, o ciano acetonídeo (23) é hidrogenado na presença de níquel de Raney, fornecendo o respectivo amino acetonídeo (24) (Esquema 17).Esquema 17
A reação entre 1,4-dicetona (4) e o amino acetonídeo (24) na presença de ácido piválico como catalisador, em umsistema ternário de solventes sob refluxo de 32 horas e (25) (Esquema 18) .Esquema 18
(25) é tratado com HC1 aquoso em metanol para fornecer o respectivo diol (26), o qual é tratado subsequentemente com hidróxido de sódio, seguido de neutralização e aquecimento em tolueno para finalmente produzir a lactona de Esquema 19
O processo descrito nos documentos de patente em questão apresenta alguns problemas técnicos, a saber:- Na primeira etapa (Esquema 15) , emprega-se n-BuLi na obtenção do diisopropilamideto de litio (LDA), o qual é caro e altamente pirofórico, representando um grande risco operacional em uma planta industrial;- Na quarta etapa do processo (Esquema 17), emprega-se uma reação de hidrogenação catalitica, com hidrogênio gasoso sob alta pressão (50 psi) e niquel de Raney como catalisador. O gás hidrogênio é potencialmente inflamável e o niquel de Raney é muito pirofórico e este processo requer aparelhagem especial para ser realizado em escala industrial; e- 0 processo emprega tolueno como solvente, o qual possui elevada toxicidade. Este fato pode gerar problemas no que se refere à sua manipulação em grande escala.
O documento de patente US 6,476,235 descreve umprocesso para a obtenção de lactona da atorvastatina (12) através de uma rota convergente estereosseletiva, conformemostrado nos Esquemas 20-26.
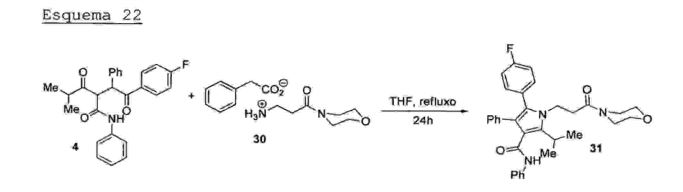
Na primeira etapa, o ciano éster (27) é tratado com morfolina em MTBE (metil-terc-butil éter) para fornecer a5 respectiva ciano amida (28) , a qual é imediatamente submetida à reação de hidrogenação catalitica (50 psi de hidrogênio) na presença de catalisador de platina (Pt/C) em meio ácido, fornecendo o respectivo hidrocloreto de amina(29) (Esquema 20).10 Esquema 20
Na etapa seguinte, o hidrocloreto (29)_é_desprotonado com metóxido de sódio (MeONa) e submetido à reação com ácido fenilacético para fornecer o respectivo carboxilato de amónio (30) (Esquema 21) . Esquema 21_ 9 1) MeONa, MeOH 9
A reação de Pall-Knor entre a 1,4-dicetona (4) e o carboxilato de amónio (30) sob refluxo de THF com remoção20 azeotrópica de água fornece o respectivo pirrol (31) (Esquema 22).Esquema 22
Na sequência reacional, o amido pirrol (31) é 5 submetido à reação com o dienolato de sódio-lítio do acetoacetato de etila (32), à baixa temperatura, para (Esquema 23).
10 A 1,3-dicetona (33) é submetida à redução seletiva sobpressão de gás hidrogênio na presença de um catalisador de rutênio quiral (RuCl2(DMF) (R) (+ )-Cl-MeO-BIPHEP) em meio ácido. O 1,3-diol (34) é obtido, neste caso, na forma da mistura diastereoisomérica 1:1 dos isômeros syn/anti15 (Esquema 24).
Na etapa reacional seguinte, o éster-1,3-diol (34) é submetido à hidrólise básica (KOH), seguido de acidificação com HC1, lactonização e eliminação empregando condições5 reacionais drásticas (refluxo de tolueno, 110°C, anidridoacético, 60°C) para fornecer a respectiva lactona ct, β-insaturada (35) (Esquema 25).
Na última etapa reacional, a lactona α, β-insaturada(35) é tratada com álcool benzílico na presença de base (NaOH), seguido de neutralização e hidrogenação catalítica (H2 a 50 psi, empregando hidróxido de paládio suportado em carbono) para fornecer a lactona de atorvastatina (12) após15 8 etapas reacionais (Esquema 26).
Embora o processo descrito no documento de patente US6,476,235 forneça os compostos desejados, o mesmo apresenta grandes problemas técnicos operacionais, conforme é descrito a seguir:- A sequência reacional linear de 8 etapas emprega tempos reacionais longos. Além disso, os compostos são produzidos sem nenhuma diastereoseletividade, ou seja, o composto (34) é obtido na forma da mistura 1:1 dos isômeros syn/anti. Do ponto de" vista sTntético é~~ econdmicdy esfes “fatores representam uma grande desvantagem;
O processo emprega reações de hidrogenação a altas pressões (Esquema 24), com tempos reacionais longos, fazendo com que estes procedimentos sejam impraticáveis na produção em larga escala, onde os aspectos de segurança, eficiência e custo são críticos;
O catalisador de rutênio quiral empregado (RUC12(DMF) (R) (+) -Cl-MeO-BIPHEP) é extremamente caro para utilização em larga escala;- 0 processo emprega base forte corrosiva (MeONa), n-BuLi e hidreto de sódio (NaH), os quais sâo caros e altamente inflamáveis, o que representa um grande risco operacional em uma planta industrial; e- 0 processo utiliza morfolina (Esquema 20), que é uma amina altamente tóxica, podendo ocasionar problemas no que se refere à sua manipulação em larga escala.
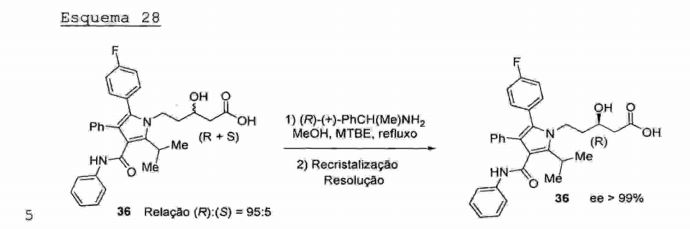
O processo para a produção de atorvastatina cálcica (14) descrito no documento de patente US 2009/0131683 é semelhante ao processo descrito no documento US 5,273,995 (Esquemas 27-29). O processo revelado no documento de patente US 2009/0131683 consiste em uma rota completamente linear/ O aldeido pirrólico (6) é submetido à reação aldólica estereosseletiva com acetato de (S)-2-hidróxi- 1,-2,2-trifenrletil’a (A*) em* THF-a -78°C'~via enolãto de litio na presença de um co-solvente quelante (2,2- dimetoxietano - 2,2-DME), fornecendo o respectivo β-hidroxi-ácido (36) após reação de hidrólise ácida. Esquema 27
Na etapa seguinte do processo, o β-hidroxi-0cido (36) é resolvido empregando (R)-(+)-metilfornecer o respectivo p-hidroxi-ácido (36) com excesso Esquema 28
Após a obtenção do p-hidroxi-ácido (36), uma sequênciacompletamente Linear de 4 etapas _é necessária para, aobtenção da lactona de atorvastatina (12) e mais 2 etapasaté a atorvastatina cálcica (14) (Esquema 29).10 Esquema 29
O processo acima descrito referente ao documento de patente US 2009/0131683 possui alguns problemas técnicos quando realizado em larga escala, quais sejam:15 - 0 auxiliar quiral (A*) (Esquema 27) (acetato de (S)-2-hidróxi-1,2,2-trifeniletila) é um reagente de alto custo, aumentando muito o custo do processo quando aplicado em escala industrial;
Na obtenção do LDA (diisopropil amideto de litio) (Esquema 27) emprega-se n-BuLi, o qual é caro e altamente inflamável, o que representa um grande risco operacional em uma planta industrial;- Neste processo, uma etapa de resolução (Esquema 28) empregando (R)-(+)metil benzil amina é necessária para a obtenção do produto final com grau de pureza satisfatória. Este fator torna o processo mais caro, devido ao custo do reagente de resolução; e- A sequência reacional completamente linear resulta em rendimento global baixo (> 10%).
O documento de~"patenCe US*"2009/0209612’ descreve a obtenção de atorvastatina cálcica (14) empregando uma rota convergente, conforme mostrado nos Esquemas 30-33. Na primeira etapa, o β-cetoester (37) é submetido à reação de Knoevenagel com benzaldeido, fornecendo o respectivo β- cetoéster-a,β-insaturado (38). Na etapa seguinte, o β- cetoéster (38) é submetido à reação de Stetter, fornecendo a respectiva 1,4-dicetona (39), a qual é purificada em uma coluna cromatográfica (Esquema 30). Esquema 30
5 mistura ternária de solventes. O produto (40) obtido neste
15 anilina em benzeno como solvente, produzindo a amida (42) após purificação em coluna cromatográfica de sílica (Esquema 32) .
O éster acetonideo (42) é submetido á hidrólise ácida
(HC1) seguida de hidrólise básica, neutralização e extração com acetato de etila, para fornecer o respectivo ácido 5 carboxilico (43) , o qual é purificado em uma coluna cromatográfica de silica. O ácido carboxilico (4 3) é, por fim, tratado com solução de hidróxido de sódio seguido de tratamento com acetato de cálcio para produzir a 'atorvastatina cálcica ~(14) (Esquema 33).10 Esquema 33
O processo acima descrito, revelado no documento US2009/0209612, possui alguns problemas técnicos que j15 dificultam a sua aplicação em escala industrial, conforme descrito a seguir:- Neste processo, há 3 intermediários - (40), (42) e (43) -que foram purificados em coluna cromatográfica de silica.
Esta operação unitária é totalmente inviável em larga escala, devido ao consumo excessivo de solventes e silica, que são insumos de alto custo;- Na reação de obtenção do composto (42) (Esquema 32), os inventores usaram benzeno como solvente, o qual é potencialmente tóxico e carcinogênico;- 0 processo emprega uma reação de hidrogenação a alta pressão (H2 a 40 psi) (Esquema 32), o que requer equipamentos especiais, bem como adoção de critérios de segurança operacional na planta piloto; e- 0 amino éster quiral (18) empregado no processo possui um alto custo e sua obtenção requer uma sequência” r’eacional linear de 6 etapas.‘ O~ documento ^de~ patente W0‘—2004/014896“‘descreve a obtenção de lactona de atorvastatina (12) empregando complexos quirais de rutênio em reações de redução, conforme mostrado nos Esquemas 34-35.
Inicialmente, a 1,3-dicetona (44) é submetida à reação de redução estereoseletiva na presença do catalisador de rutênio (45) e ácido fórmico como redutor, fornecendo o respectivo diol éster (46) em uma relação syn/anti = 6:1 (Esquema 34) . Esquema 34
Na última etapa do processo, o éster (46) é submetidoà hidrólise básica, seguida de neutralização e lactonização5 em refluxo de tolueno com remoção azeotrópica de água. Alactona de atorvastatina (12) é obtida em excessoenantiomérico de apenas 85% (Esquema 35).Esquema 35
O processo descrito no documento WO 2004/014896 possuialgumas desvantagens, a saber:- 0 catalisador de rutênio (45) possui custo excessivamente alto, tornando o processo economicamente desfavorável;- O diol éster (46) é obtido com baixa seletividade15 (relação syn/anti= 6:1), o que resulta em operaçõesunitárias adicionais para a obtenção do isômero syn (46) na sua forma enantiomericamente pura. Como consequênciadireta, o processo torna-se mais caro; e- Na última etapa do processo, a reação de lactonização emprega tolueno como solvente, o qual é um solvente de elevada toxicidade.
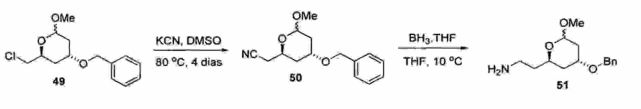
O documento de patente WO 2005/012246 descreve a obtenção de lactona de atorvastatina (12) empregando uma rota convergente, conforme mostrado nos Esquemas 36-40. Naprimeira etapa, o cloro lactol (47) é protegido na forma de acetal metilico (48), o qual é submetido à reação com brometo de benzila para proteção do álcool secundário (49) "(Esquema 36) .Esquema 36
O cloro acetal (49) é tratado com cianeto de potássio (KCN) em dimet ilsulfóxido (DMSO) durante 4 dias para fornecer a respectiva nitrila (50), a qual é imediatamente reduzida com BH3.THF para a obtenção da respectiva amina primária (51) (Esquema 37).
Esquema 37
Na sequência reacional, a 1,4-dicetona (52) ésubmetida à reação de Pall-Knorr com a amina (51) sob 5 catálise ácida, fornecendo o respectivo pirrol (53) (Esquema 38) . Esquema 38
(54), a qual é purificada em coluna cromatográf ica de silica. Na etapa seguinte, o acetal metilico (54) é hidrogenado para desproteção da hidroxila primária, produzindo o álcool (55) (Esquema 39).15 Esquema 39
hidrólise ácida para a obtenção do lactol de atorvastatina (56) , o qual é posteriormente oxidado com o reagente de Dess-Martin periodinana (DMP) em diclorometano como solvente, para então produzir a lactona de atorvastatina 5 (12) (Esquema 40).Esquema 40
Na última etapa do processo, o tratamento da lactonade atorvastatina (12) com hidróxido de cálcio aquoso10 fornece a respectiva atorvastatina cálcica (Esquema 41).
O processo descrito no documento de patente WO2005/012246 possui alguns problemas técnicos que15 comprometem a sua aplicação em escala industrial, conformeé detalhado abaixo: inventores empregam hidreto de sódio (NaH) e brometo de benzila, os quais são reagentes problemáticos: o hidreto de sódio é altamente pirofórico e o brometo de benzila é lacrimejante e altamente tóxico para a manipulação em larga escala;
A rota sintética é longa e envolve o uso de intermediários contendo grupos de proteção, empregando materiais de partida de alto custo. Além disso, a sequência reacional é longa e algumas reações necessitam de longo tempo (por exemplo, 4 dias) para se completar (Esquema 37); - A sintese da nitrila (50) (Esquema 37) emprega cianeto de potássio (KCN) e dimetilsulfóxido (DMSO), os quais são reagentes de alta toxicidade;A' obtenção da facfoná- de af orvasf at'iná~ (1’2) ~a "partir' dõ lactol (Esquema 40) emprega uma reação de oxidação com o reagente de Dess-Martin periodinana (DMP), o qual é extremamente caro para uso em larga escala. Além disso, os inventores usam diclorometano (CH2CI2) como solvente, o qual é tóxico e altamente nocivo ao meio ambiente; e - Neste processo, o composto pirrólico (54) (Esquema 39) foi purificado em coluna cromatográfica de silica. Esta operação unitária é totalmente inviável na sintese em larga escala, devido ao consumo excessivo de solventes e silica,os quais são insumos de alto custo. 0 documento de patente WO 2007/029216 descreve aobtenção de lactona de atorvastatina (12) empregando umarota parcialmente linear, conforme mostrado nos Esquemas42-44. Inicialmente, o ciano éster (23) é hidrogenado na5 presença de níquel de Raney, fornecendo o respectivo amino éster (18) como um óleo viscoso incolor em 85% de rendimento após purificação por destilação sob pressão reduzida (Ponto de ebulição = 125-135°C/0,5 mmHg) (Esquema 42) .10 Esquema 42
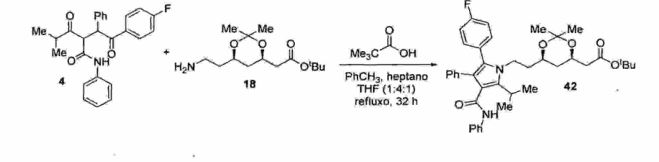
O' trataméntõ da 1 ,*4-dicetona (4) com "o amino éster(18) na presença de ácido piválico em um sistema ternário de solventes sob refluxo de 32 horas fornece o respectivo 15 acetonídeo da atorvastatina (42) em 61% de rendimento isolado após recristalização (Esquema 43).Esquema 43
Na etapa seguinte, o acetonídeo de atorvastatina (42) é tratado com HC1 (IN) em metanol como solvente a temperatura ambiente, fornecendo o respectivo diol éster, o qual é mantido em solução. O tratamento subsequente do diol 5 éster com solução de NaOH aquoso a 10% fornece o respectivo carboxilato de sódio, o qual é tratado com HC1 para a obtenção do diol ácido (43). A lactona de atorvastatina (12) é obtida através do tratamento do diol ácido (43) com tolueno sob refluxo e remoção azeotrópica de água (Esquema 10 44 ) .
O processo descrito no documento de patente WO 2007/029216 possui alguns problemas técnicos, quais sejam:15 - 0 processo emprega uma reação de hidrogenação a altapressão (H2 a 50 psi) (Esquema 42) , o que requer equipamentos especiais, bem como adoção de critérios rigorosos de segurança operacional na planta piloto;- 0 catalisador empregado na reação de hidrogenação (Ni 20 Raney) é altamente pirofórico (Esquema 42); - A obtenção do amino éster (18) demanda uma sequência reacional de 5 etapas e emprega reagentes de alto custo. Além disso, o amino éster (18) necessita de purificação prévia antes de ser empregado na etapa posterior; e- A etapa de lactonização (Esquema 44) emprega condições reacionais drásticas e altas temperaturas (refluxo de tolueno: 110°C) .
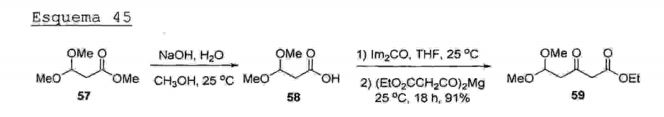
O documento de patente europeu EP 1.834.944 descreve a obtenção de alguns intermediários para a síntese de atorvastatina cálcica, bem como a obtenção da lactona de atorvastatina (12) e do lactol de atorvastatina. Na primeira etapa, o éster (57) é submetido a uma reação de hidrólise básica, fornecendo o respectivo ácido carboxilico ('58)."Nà sequência"-rèacio"nal, o ácido carboxilico (58) é homologado em dois carbonos para produzir o respectivo β- cetoéster (59), conforme mostrado no Esquema 45. Esquema 45
0 β-cetoester (59) é reduzido estereosseletivamente, empregando hidrogenação catalítica na presença de catalisador quiral de rutênio (RuCl?(R)-BINAP), fornecendoassim o respectivo β-hidroxi-6ster (60). A reação de condensação de Claisen cruzada do β-hidroxi-ester (60) como enolato de lítio derivado do acetato de terc-butila 46)Esquema 46
Na etapa reacional seguinte, o 5-hidroxi-p-ceto-éster (61) é reduzido seletivamente com borohidreto de sódio e dietilmetoxiborana a baixa temperatura, seguido de proteção 10 do-dioí intermediário' com 2,'2-di'me’toxipropano (2,2-DMP) sob catálise ácida, produzindo o respectivo acetonídeo syn (62) JEsquema .4 7.) .. - - - - - - - - - - - -- -- - — - -Esquema 471) NaBH4, Et2BOMe THF, MeOH-78 °C2) 2,2-DMP p-TsOH 25°C0 éster acetonídeo (62) é reduzido com hidreto de lítio e alumínio (LÍA1H4) para a obtenção do respectivo álcool primário, o qual é posteriormente tratado com cloreto de tosila (Ts-Cl), na presença de trietilamina,para fornecer o respectivo composto tosilato (63). Na etapa seguinte, o tosilato (63) é submetido à reação com azida desódio (NaN3) em dimetilsufóxido, fornecendo a azidaalquilica (64) (Esquema 4B).
Na sequência reacionai, a azida alquilica (64) é reduzida sob hidrogenação catalítica para produzir a respectiva amina primária (65) (Esquema 49).Esquema 49 - - *
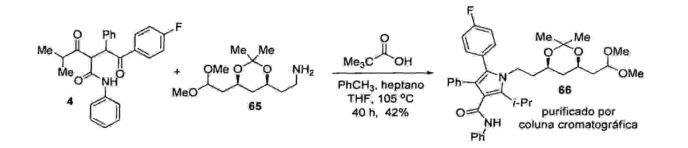
Na etapa seguinte, a 1,4-dicetona (4) é tratada com a amina (65) na presença de ácido piválico como catalisador em um sistema ternário de solventes sob refluxo durante 40 h. 0 respectivo composto pirrólico (66) foi obtido em apenas 42% de rendimento isolado, após purificação em coluna cromatográfica de sílica (Esquema 50).
e brometo de benzila para a proteção da hidroxila secundária na forma de éter benzilico (69) (Esquema 52). Na etapa seguinte, o acetal metilico (69) é tratado com ácido acético para produzir o respectivo lactol (50) (Esquema 52) .
Na etapa reacional posterior, o lactol protegido (50) é oxidado com dióxido de manganês (MnOa) para produzir a respectiva lactona (51) (Esquema 53) . Por fim, a reação de 5 hidrogenação catalítica (H2 a 1 bar - Pd/C) da lactona (51)produz a lactona de atorvastatina (12) (Esquema 53).Esquema 53
O processo descrito no documento de patente europeu EP1.834.944 possui vários problemas técnicos que comprometemo seu emprego em escala industrial, conforme é descritoabaixo: - O processo emprega uma reação de hidrogenação a alta pressão (Hz a 50 bar) (Esquema 46) , o que requer equipamentos especiais, bem como a adoção de critérios rigorosos de segurança operacional na planta piloto;
O catalisador de rutênio quiral (RUC12 (R) -BINAP) empregado na reação de hidrogenação (Esquema 46), embora forneça resultados satisfatórios, é extremamente caro para emprego em larga escala;- O processo emprega n-butil lítio (n-BuLi) para a geração do diisopropil araideto de lítio (LDA) (Esquema 46) . O n- BuLi possui um custo elevado e é altamente pirofórico, o que representa um grande risco operacional em uma planta piloto onde é empregado em grandes volumes;-'Na reação dê redução dò éster (62) (Esquema~ 4 8)“ pa"ra õ seu respectivo álcool, empregou-se hidreto de lítio e alumínio (LÍAIH4). Este reagente é particularmente perigoso quando no estado puro, pois é altamente pirofórico, podendo inflamar-se espontaneamente no ar. Isto representa um grande risco de acidente durante a sua manipulação. Além disso, o LÍAIH4 tende a gerar grandes quantidades de sal e hidróxido de alumínio, os quais dificultam a filtração e a remoção do produto do meio reacional;- Na reação de obtenção do éter benzílico (69) (Esquema52), emprega-se hidreto de sódio (NaH) e brometo de benzila (Bn-Br) , os quais são reagentes muito problemáticos: o hidreto de sódio é altamente pirofórico e o brometo de benzila é lacrimejante e altamente tóxico para a manipulação em escala industrial;- A reação de Pall-Knorr para a obtenção do acetonideo pirrólico (66) (Esquema 50) apresenta um tempo reacional longo e tem como resultado um rendimento muito baixo (apenas 42%). Isso significa que maiores quantidades de materiais de partida devem ser utilizados para a obtenção da massa desejada de produto. Além disso, o acetonideo pirrólico (66) é purificado em coluna cromatográfica de silica, uma operação unitária praticamente impossível de ser aplicada em escala industrial; eA~ rota ~ de“ síntese empregada ~ neste- processo" é~ relativamente longa (18 etapas), o que demanda maior tempo operacional e um maior número de operações unitárias, empregando etapas de proteção e desproteção. Estes fatores tendem a pesar negativamente na escolha da rota sintética ideal.
Em vista do acima exposto, seria útil se a técnica dispusesse de um processo sintético mais eficiente para a obtenção de estatinas, preferivelmente atorvastatina cálcica, sem a utilização de reagentes pirofóricos, taiscomo n-butil lítio, empregando matéria prima de menor custo e obtida a partir de fontes renováveis. SUMÁRIO DA INVENÇÃO
De acordo com um primeiro aspecto da presente invenção, é fornecido um processo para a obtenção de atorvastatina cálcica.
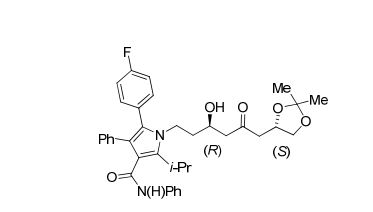
De acordo com um segundo aspecto da presente invenção, o composto 1-[(R)-6-((S)-2,2-dimetil-1,3- dioxolan-4-il)-3-hidroxi-5-oxo-hexil]-5-(4-fluor-fenil)-2- isopropil-N,4-difenil-lH-pirrol-3-carboxamida é fornecido como um dos intermediários no referido processo para a obtenção de atorvastatina cálcica.
De acordo com um terceiro aspecto da presente invenção, o composto 1- ( (3R,5R)-6-( (S)-2,2-dimetil-l,3- droxol*an-4-ir) -375-diidroxi’exir) -5~ (4-fluõrofenil') -2‘=: isopropil-N, 4-difenil-lH-pirrol-3-carboxamida é fornecido como um dos intermediários no referido processo para a obtenção de atorvastatina cálcica.
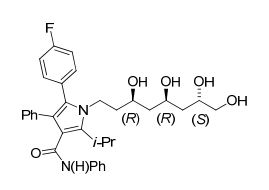
De acordo com um quarto aspecto da presente invenção, o composto 5-(4-fluorofenil)-2-isopropil-N,4-difenil-l- ( (3R,7S)-3,5,7,8-tetraidroxioctil) -lH-pirrol-3-carboxamida é fornecido como um dos intermediários no referido processo para a obtenção de atorvastatina cálcica.
De acordo com um quinto aspecto da presente invenção, é fornecido o uso do composto 1-[(R)-6-((S)-2,2-dimetil- 1,3-dioxolan-4-il)-3-hidroxi-5-oxo-hexil]-5-(4-fluor- fenil)-2-isopropil-N, 4-difenil-lH-pirrol-3-carboxamida.
De acordo com um sexto aspecto da presente invenção, é fornecido o uso do composto 1-((3R,5R)-6-((S)-2,2- dimetil-1,3-dioxolan-4-il)-3, 5-diidroxiexil)-5 - ( 4 - fluorofenil)-2-isopropil-N, 4-difenil-lH-pirrol-3- carboxamida.
De acordo com um sétimo aspecto da presente invenção, é fornecido o uso do composto 5-(4-fluorofenil)-2- isopropil-N,4-difenil-l-((3R,7S)-3,5,7,8- ■ tetraidroxioctil)-lH-pirrol-3-carboxamida.
De acordo com um oitavo aspecto da presente invenção, , jè, fornecida a atorvastatina cálcica obtida através do processo do primeiro aspecto da dita invenção.
De acordo com um nono aspecto da presente invenção, é fornecido um método para a obtenção de uma estatina.
De acordo com um décimo aspecto da presente invenção, são fornecidos compostos intermediários no referido método para a obtenção de uma estatina.
De acordo com um undécimo aspecto da presente invenção, é fornecido o uso dos compostos intermediários mencionados acima.
De acordo com um duodécimo aspecto da presenteinvenção, é fornecida uma estatina obtida através do método do nono aspecto da dita invenção.
BREVE DESCRIÇÃO DAS FIGURAS
A Figura 1 em anexo apresenta o espectro de RMN de H1 da 4-metil-3-oxo-N-fenilpentanamida (2) em DMSO D6 a 250 MHz.
A Figura 2 em anexo apresenta o espectro de RMN de 13C da 4-metil-3-oxo-Aí-fenilpentanamida (2) em DMSO D6 a 63 MHz.
A Figura 3 em anexo apresenta o espectro de RMN de H1 da (Z) -2-benzilideno-4-metil-3-oxo-N-fenilpentanamida (3) em acetona D6 a 250 MHz.
A Figura 4 em anexo apresenta o espectro de RMN de I3C da (Z)-2-benzilideno-4-metil-3-oxo-W-fenilpentanamida (3) em acetona D6 a 63 MHz. ' ’
A Figura 5 em anexo apresenta o espectro de RMN de H1 da 2 - [2-(4-fluorofenil)-2-oxo-l-feniletil]-4-meti1-3-oxo-W- fenilpentanamida (4) em DMSO D6 a 250 MHz.
A Figura 6 em anexo apresenta o espectro de RMN de 13C da 2-[2-(4-fluorofenil)-2-oxo-l-feniletil]-4-metil-3-oxo-N- fenilpentanamida (4) em CDCI3 a 63 MHz.
A Figura 7 em anexo apresenta o espectro de RMN de H1 da [5-(4-fluorofenila)-1-(3-hidroxipropil]-2-isopropil-N, 4- difenil-lH-pirrol-3-carboxamida (5) em CDCI3 a 250 MHz.
A Figura 8 em anexo apresenta o espectro de RMN de 13C da [5-(4-fluorofenila)-l-(3-hidroxipropilJ -2-isopropil-AZ, 4- difenil-lH-pirrol-3-carboxamida (5) em CDClj a 63 MHz.
A Figura 9 em anexo apresenta o espectro de RMN de XH da (S)-1-(2,2-dimetil-l,3-dioxolan-4-ila)propan-2-ona (11) 5 em CDCI3 a 250 MHz.
A Figura 10 em anexo apresenta o espectro de RMN de 13C da (S)-1-(2,2-dimetil-l, 3-dioxolan-4-ila)propan-2-ona (11) em CDC13 a 63 MHz.
A Figura 11 em anexo apresenta o espectro de RMN de 10 da 1-[(R)-6-((S)-2,2-dimetil-l, 3-dioxolan-4-il)-3-hidroxi- 5-oxo-hexil]-5-(4-fluor-fenil) -2-isopropil-W, 4-difenil-lH- pirrol-3-carboxamida (13) em CèDg a 250 MHz.
A Figura 12 em anexo apresenta o espectro de RMN de 13C da 1-[ (R)-6- ( (5) -2,2-dimetil-l, 3-dioxol*an-'4-i‘l) -3^~ • 15 hidroxi-5-oxo-hexil]-5-(4-fluor-fenil)-2-isopropil-N, 4- difenil-lH-pirrol-3-carboxamida (13) em CDCI3 a 63 MHz.
A Figura 13 em anexo apresenta o espectro de RMN de H1 da l-((3R,5R)-6-((S)-2, 2-dimetil-l,3-dioxolan-4-il)-3,5- diidroxihexil)-5-(4-fluorofenil)-2-isopropil-N,4-difenil- 20 lH-pirrol-3-carboxamida (14) em C6D6 a 250 MHz.
A Figura 14 em anexo apresenta o espectro de RMN de l3C da 1-((3R,5R)-6-((S)-2,2-dimetil-l,3-dioxolan-4-il)- 3,5-diidroxihexil)-5-(4-fluorofenil)-2-isopropil-N,4-difenil-lH-pirrol-3-carboxamida (14) em CgDe a 63 MHz.
A Figura 15 em anexo apresenta o espectro de RMN de H1 da 5- (4-fluorofeni1)-2-isopropil-N, 4-difenil-l-((3R,5R,7S)- 3,5, 7,8-tetraidroxioctil)-lH-pirrol-3-carboxamida (15) em MeOD a 250 MHz.
A Figura 16 em anexo apresenta o espectro de 13C da 5- (4-fluorfenil)-2-isopropil-N, 4-difenil-l-((3R,5R,7S)- 3, 5,7,8-tetraidroxioctil)-lH-pirrol-3-carboxamida (15) em MeOD a 63 MHz.
A Figura 17 em anexo apresenta o espectro de RMN de H1 da l-(2-((2R,4R)-4,6-diidroxitetraidro-2H-piran-2-i1)etil)- 5- (4-fluorfenil)-2-isopropil-N, 4-difenil-lH-pirrol-3- carboxamida (26) em CDC13 a 250 MHz.
A Figura 18 em anexo apresenta o espectro de RMN de 13C da 1-(2-( (2R, 4R)-4,6-diidroxitetraiciro-2H-piran-2= il)etil)-5-(4-fluorofenil)-2-isopropil-N,4-difenil-1H- pirrol-3-carboxamida (26) em CDC13 a 63 MHz.
A Figura 19 em anexo apresenta o espectro de RMN de H1 da 5- (4-fluorfenil)-l-(2-((2R,4R)-4-hidroxi-6-oxotetraidro- 2H-piran-2-il)etil)-2-isopropil-N, 4-difenil-IH-pirrol-3- carboxamida (31) em CDCI3 a 250 MHz.
A Figura 20 em anexo apresenta o espectro de RMN de 13C da 5-(4-fluorfenil)-1-(2-((2R,4R)-4-hidroxi-6-oxotetrahidro-2H-piran-2-il) etil)-2-isopropil-N,4-difenil-lH-pirrol-3-carboxamida (31) em CDC13 a 63 MHz.
A Figura 21 em anexo apresenta o espectro de RMN de H1 da atorvastatina cálcica (41) em DMSO-D6 a 250 MHz.
A Figura 22 em anexo apresenta o espectro de RMN de 13C da atorvastatina cálcica (41) em DMSO-D6 a 63 MHz.
A Figura 23 em anexo apresenta o difratograma da atorvastatina cálcica amorfa (41).
DESCRIÇÃO DETALHADA DA INVENÇÃO
A presente invenção trata de um processo para a obtenção de atorvastatina cálcica e seus intermediários.De modo a facilitar a compreensão de todas as etapas do referido processo, o mesmo foi dividido em quatro partes. Na primeira parte, estão descritas as etapas para a obtenção da 2-[2-(4-fluorofeni1)-2-oxo-l-feniletil ]-4- metil=3-oxo-N-fenilpentanamÍda (4), primeiro intermediário chave para a síntese de atorvastatina cálcica (41). Na segunda parte, estão descritas as etapas para a obtenção da 5- (4-fluorofenil)-2-isopropil-l-(3-oxopropil)-N,4-difenil-lH-pirrol-3-carboxamida (6), a partir da 2—[2—(4— fluorofenil)-2-oxo-l-feniletil]-4-metil-3-oxo-N- fenilpentanamida (4) obtida na primeira parte do referido processo. Na terceira parte, estão descritas as etapas para a obtenção da (S)-1-(2,2-dimetil-l,3-dioxolan-4- ila)propan-2-ona (11) a partir do ácido (L)-málico (7). Por fim, na quarta parte estão descritas as etapas referentes à junção dos fragmentos (6) e (11) e a síntese final da atorvastatina cálcica (41). Cabe aqui ressaltar que um objetivo da presente invenção foi a obtenção de atorvastatina cálcica na forma amorfa, a qual é a forma farmacologicamente ativa.Primeira Parte - Obtenção da 2-[2-(4-fluorofenil)-2-oxo-l- feniletil]-4-metil-3-oxo-N-fenilpentanamida (4)
Conforme mencionado anteriormente, a primeira parte do processo para obtenção de atorvastatina da presente invenção compreende a obtenção da 2-[2-(4-fluorofenil)-2- oxo-l-feniletil]-4-metil-3-oxo-N-fenilpentanamida (4). A referida primeira parte compreende três etapas, de acordo
fenilpentanamida (2)
A etapa 1 consiste na reação do 4-metil-3-oxo pentanoato de metila (1) com excesso de anilina (0,8 a 2,0 equivalentes, preferivelmente 1,2 equivalente) em presença de NaOH como catalisador em uma concentração que varia de 0,5 a 20 mol%, preferivelmente 2 mol%, na ausência de solvente e com remoção concomitante de metanol, tendo como produto a 4-metil-3-oxo-N-fenilpentanamida (2) (Esquema tI) . 0 referido produto é purificado apenas por extração com HC1 aquoso (1%), podendo ser empregado diretamente na etapa 2 sem purificação prévia. A reação da referida etapa 1 é conduzida durante um período de 12 a 24 horas, preferivelmente durante 16 horas, a uma temperatura que varia entre 90°C e 160°C, preferivelmente 135°C.Esquema I:
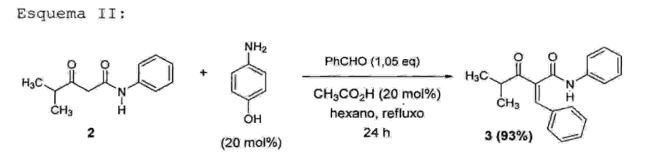
Cabe ressaltar que a reação da referida etapa 1 fornece um rendimento na faixa de 85% a 95%, preferivelmente de cerca de 92%, o qual é superior ao rendimento descrito no estado da técnica (63%). Além disso, a reação descrita na etapa 1 da presente invenção prevê a utilização de uma menor quantidade de anilina, além de utilizar NaOH como catalisador ao invés de etilenodiamina, que é mais cara e muito mais tóxica. Adicionalmente, a reação da presente invenção ocorre na ausência de solvente. • Etapa 2 - obtenção da 2-benzilideno-4-metil-3-oxo-N- fenilpentanamida (3)
A etapa 2 consiste na reação de condensação da 4- metil-3-oxo-N-fenilpentanamida (2) com aldeído benzóico (0,9 a 2,0 equivalentes, preferivelmente 1,05 equivalente)sob refluxo em hexano e remoção azeotrópica de água na presença de p-aminofenol e ácido acético comocatalisadores, em uma concentração que varia de 5 mol% a 40 mol%, preferivelmente 20 mol%, tendo como produto a 2- benzilideno-4-metil-3-oxo-N-fenilpentanamida (3) (Esquema II). 0 referido produto (3) é purificado por simples lavagem com hexano a 60°C, seguido de resfriamento, lavagem com água, filtração e secagem sob alto vácuo. 0 produto ~f3) é um sólido branco' com ponto dê" fusão entre preferivelmente de cerca de 93%, o qual é superior ao rendimento descrito no estado da técnica (77%). Além disso, a reação descrita na etapa 2 da presente invenção prevê a utilização de p-aminof enol, o qual é um catalisador mais barato em comparação com aquele comumente empregado nas reações descritas no estado da técnica (β- alanina).
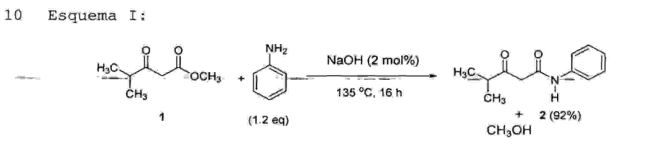
• Etapa 3 - obtenção da 2-[2-(4-fluorofenil)-2-oxo-l- feniletil)-4-metil-3-oxo-N-fenilpentanamida (4)
A etapa 3 consiste em uma reação de Stetter entre a 2-benzilideno-4-metil-3-oxo-N-fenilpentanamida (3) e 4- fluor-benzaldeído (0,9 a 2,0 equivalentes, preferivelmente 1,05 equivalente), na ausência de solvente e na presença dê qãanÇidacie 'catalítica ~de ~~ brometo" de 3-etil-5-(2- hidroxietil)-4-metil-3-tiazólio em uma concentração que varia entre 10 mol% e 30 mol%, preferivelmente 20 mol%, e trietilamina (0,3 a 3,0 equivalentes, preferivelmente 1,0 equivalente) como base, tendo como produto a 2—[2—(4 — fluorofenil)-2-oxo-l-feniletil]-4-metil-3-oxo-N- fenilpentanamida (4) (Esquema III) em um rendimento que varia de 60% a 90%, preferivelmente de 7 3%, após recristalização em isopropanol, etanol ou n-butanol, preferivelmente isopropanol. A reação da referida etapa 3 é conduzida durante um período de 12 a 24 horas, preferivelmente durante 16 horas.Esquema III:
Cabe ressaltar que a reação da referida etapa 35 fornece um rendimento superior ao rendimento descrito noestado da técnica (69%). Além disso, a reação descrita naetapa 3 da presente invenção é realizada sem solvente,pois não utiliza etanol, o qual é comumente empregado nas reações'descritas nb estado" da "técnica.10 É importante mencionar que as três etapas da primeiraparte do processo de-obtenção de atorvastatina" cálcica(41) da presente invenção fornecem um rendimento global na faixa de 50% a 65%, preferivelmente de cerca de 62,4%, o qual é superior àquele descrito no estado da técnica 15 (33,4%). Adicionalmente, é importante mencionar que asreações envolvidas nessa primeira parte reduzem o uso de solvente e de reagentes importantes, além de utilizar reagentes mais baratos.Segunda Parte - Obtenção da 5-(4-fluorofenil)-2-isopropil- 20 1- (3-oxopropil)-N,4-difenil-lH-pirrol-3-carboxamida (6)Conforme mencionado anteriormente, a segunda parte do processo para obtenção de atorvastatina da presente invenção compreende a obtenção da 5-(4-fluorofenil)-2- isopropil-1-(3-oxopropil)-N, 4-difenil-lH-pirrol-3- carboxamida (6). A referida segunda parte compreende duas 5 etapas, de acordo com a descrição a seguir.• Etapa 4 - obtenção da [5-(4-fluorofenila)-1- (3-hidroxipropil]-2-isopropil-N, 4-difenil-lH-pirrol-3- carboxamida (5)
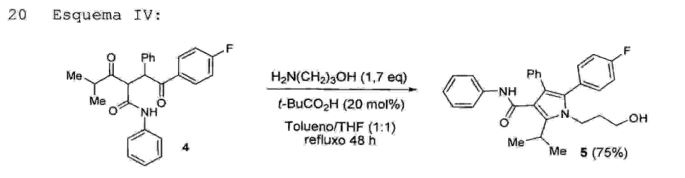
A etapa 4 consiste no tratamento da 2-[2-(4- 10 f luorofenil)-2-oxo-l-feniletil]-4-metil-3-oxo-N-fenilpentanamida (4) com 1,0 a 3,0 equivalentes, preferivelmente 1,7 equivalente de 3-amino-l-propanol, sobcatálise de ácido piválico (5 mol% a 40 mol%,preferivelmente 20~ mõl%)T ém uma “ misTurá" lil 'cie15 tolueno/THF sob refluxo, com remoção azeotrópica de águadurante um período de 24 a 56 horas, preferivelmente de t48 horas, tendo como produto a [5-(4-fluorofenila)-1-(3-hidroxipropil]-2-isopropil-N, 4-difenil-lH-pirrol-3-carboxamida (5) (Esquema IV).Esquema IV:
Cabe ressaltar que a reação da referida etapa 4 fornece um rendimento na faixa de 50% a 80%, preferivelmente de cerca de 75%, o qual é superior ao rendimento descrito no estado da técnica (35%). Além 5 disso, a reação descrita na etapa 4 da presente invenção não utiliza heptano como solvente, o que representa uma vantagem do ponto de vista operacional.• Etapa 5 - obtenção da 5-(4-fluorofenil)-2-isopropil-1- ( 3-oxopropil)-N,4-difenil-lH-pirrol-3-carboxamida 10 (6)
A etapa 5 consiste em uma reação de oxidação seletiva da [5- (4-fluorofenila)-1-(3-hidróxipropil]-2-isopropi1- N, 4-difenil-lH-pirrol-3-carboxamida (5) sob as condições ■" ‘ de ~ Swe~rn~ "fclo'retd’ ~de oxalila, dimetil sulfóxido e 15 trietilamina) para fornecer a 5-(4-fluorofenil)-2- isopropil-1-(3-oxopropil)-N, 4-difenil-lH-pirrol-3- carboxamida (6) (Esquema V). Esquema V:
Primeiramente, o composto (5) dissolvido em diclorometano é reagido com cloreto de oxalila e dimetil sulfóxido a uma temperatura que varia entre -78°C e -40°C, preferivelmente -50°C, durante um periodo de 0,5 a 3 horas, preferivelmente 1 hora. Em seguida, trietilamina é adicionada lentamente à mistura reacional obtida na primeira sub-etapa para fornecer o composto (6).
Cabe ressaltar que a reação da etapa 5 fornece um rendimento na faixa de 60% a 90% e é realizada conforme descrita no estado da técnica (documento de patente indiano 2005-K0485) , onde é previsto um rendimento de cerca de 84%.Terceira Parte - Obtenção da (S)-1-(2,2-dimetil-l, 3- dioxolan-4-ila)propan-2-ona (11)
Conforme mencionado anteriormente, a terceira parte do processo para obtenção de atorvastatina da presente invenção compreende a obtenção da (S)-1-(2,2-dimetil-l,3- dioxolan-4-ila)propan-2-ona (11) a partir do ácido L-(S)- málico (7). O dito ácido é um substrato orgânico quiral de origem natural e disponível comercialmente a um custo relativamente baixo. A referida terceira parte compreende três etapas, de acordo com a descrição a seguir.• Etapa 6 - obtenção do (S)-malato de dimetila (8)
A etapa 6 consiste na transformação do ácido (L) - málico (7) em seu éster metilico correspondente ((S)~ malato de dimetila (8)), através de uma reação deesterificação com metanol na presença de quantidade a catalítica de ácido sulfúrico sob refluxo durante umJ 5 período de 12 a 36 horas, preferivelmente 24 horas (Esquema VI). É importante salientar que o produto (S)- malato de dimetila (8) não necessita de purificação, podendo ser empregado diretamente na etapa 7 do processo da presente invenção em sua forma bruta, o que reduz uma 10 operação unitária no referido processo.Esquema VI:
Cabe ainda ressaltar que a reação da etapa 6 fornece um rendimento na faixa de 80% a 95%, preferivelmente 91%, 15 o qual é superior ao rendimento descrito no estado da técnica (90%) .• Etapa 7 - obtenção do (S)-metil 2-(2,2-dimetil-l,3- dioxolan-4-ila) (10)A etapa 7 consiste na transformação do (S)-malato de 20 dimetila (8) em (S)-metil 2-(2,2-dimetil-l,3-dioxolan-4-através de uma reação de redução regiosseletiva empregando BH3-SMe2 (0,8 equivalente a 2,0 equivalentes, preferivelmente 1,05 equivalente) e NaBH4 (1 molí a 20mol%, preferivelmente 5 mol%) na presença de THE’, tendocomo produto inicial o diol éster (9), o qual não épurificado. 0 referido diol éster (9) é empregado diretamente na reação de cetalização com 2,2-dimetoxipropano em acetona, na presença de quantidade catalítica de p-TsOH (Esquema VII). A referida reação de cetalização é conduzida a uma temperatura de 0°C a 50°C, preferivelmente 25°C, durante um período de 2 a 10 horas, preferivelmente 4 horas. A (S)-metil 2-(2,2-dimetil-l,3- dioxolan-4-ila) (10) pode ser facilmente" purificada*’ pordestilação sob pressão reduzida, embora também possa ser empregada na etapa—seguinte sem-purificação■ prévia-. 15Esquema VII:


Cabe ainda ressaltar que a reação da etapa 7 fornece um rendimento na faixa de 60% a 80%, preferivelmente 75%, o qual é superior ao rendimento descrito no estado da dioxolan-4-ila)propan-2-ona (11)
A etapa 8 consiste na reação da (S)-metil 2-(2,2- dimetil-1, 3-dioxolan-4-ila) (10) com MeMgCl (3,0 a 6,0 equivalentes, preferivelmente 4,0 equivalentes) na presença de hidrocloreto de metil-metoxi amina (MeN(OMe)H.HC1) em THF, tendo como produto a (S)-l-(2,2- dimetil-1,3-dioxolan-4-ila)propan-2-ona (11) via amida de Weinreb (Esquema VIII). A reação da referida etapa 8 é conduzida em uma temperatura na faixa de -50°C a 35°C, preferivelmente na faixa de -10°C a 25°C, durante um periodo de 4 a 16 horas, preferivelmente 8 horas. A referida (S)-i-(2,2-dimetil-lt 3-diõxolan-4-ila)propan-2- ona (11) pode ser facilmente purificada por destilação, embora também possa'‘ser"empregada—na—etapa- seguinte sem purificação prévia.Esquema VIII:
Rendimento da Literatura 90%
Cabe ainda ressaltar que a reação da etapa 8 fornece um rendimento na faixa de 80% a 95%, preferivelmente 92%, o qual é superior ao rendimento descrito no estado da técnica (90%) .
É importante mencionar que as três etapas da terceira parte do processo de obtenção de atorvastatina cálcica (41) da presente invenção fornecem um rendimento global de 55% a 65% e são realizadas conforme descrito no estado da técnica (Doroh, B.; Sulikowski, G. A. Org. Lett. 2006, 8, 903), onde é previsto um rendimento global de cerca de 62,8%.Quarta Parte - Junção dos fragmentos (6) e (11) e síntese final da atorvastatina cálcica (41)
Conforme mencionado anteriormente, a quarta parte do processo para obtenção de atorvastatina da presente invenção compreende as etapas referentes à junção dos fragmentos (6) e (11) e a síntese final da atorvastatina cálcica (41) .
As reações descritas a seguir correspondem à obtenção do lactol da atorvastatina, l-(2-((2R,4R)-4,6- dihidroxitetraidro-2H-piran-2-i1)etil)-5-(4-fluorofenil)- 2-isopropil-N, 4-difenil-lH-pirrol-3-carboxamida (26), e da lactona da atorvastatina, 5-(4-fluorofenil)-1-(2-((2R,4R)- 4-hidroxi-6-oxotetraidro-2H-piran-2-il) etil)-2-isopropil- N, 4-difenil-lH-pirrol-3-carboxamida (31), empregando uma reação aldólica. A 1-(2-((2R, 4R)-4,6-dihidroxitetraidro- 2H-piran-2-il)etil)-5-(4-fluorofenil)-2-isopropil-N,4- difenil-lH-pirrol-3-carboxamida (26) e a 5-(4- fluorofenil)-1-(2-((2R,4R)-4-hidroxi-6-oxotetraidro-2H- piran-2-il)etil)-2-isopropil-N, 4-difenil-lH-pirrol-3- carboxamida (31) foram produzidas através de uma rota sintética mais eficiente, onde os intermediários foram obtidos com maior estereosseletividade empregando-se menor número de etapas em relação às metodologias até então descritas no estado da técnica. Os compostos (26) e (31) são intermediários chave de grande importância na síntese da atorvastatina cálcica (41).• Etapa 9 - obtenção da 1-[(R)-6-((S)-2,2-dimetil-l,3- dioxolan-4-il)-3-hidroxi-5-oxo-hexil]-5-(4-fluorfenil) -2-isopropil-N,4-difenil-lH-pirrol-3- carboxamida (13)
A etapa 9 cδnslste," Tnicraimente, no tratamento da (S)-1-(2,2-dimetil-l,3-dioxolan-4-ila)propan-2-ona (11)com cloro dicicloexil borana (c-Hex∑BCl) em Et2O seguido de trietilamina em uma temperatura de -50°C a 25°C, preferivelmente 0°C, para geração do enolato de boro cinético durante um período de 10 minutos a 3 horas, preferivelmente 1 hora. Na sequência, a reação é resfriada em uma faixa de temperatura de -90°C a -5°C, preferivelmente -78°C, e a 5-(4-fluorofenil)-2-isopropil- 1-(3-oxopropil)-N, 4-difenil-lH-pirrol-3-carboxamida (6) é adicionada lentamente durante um período de 30 minutos a 8 horas, preferivelmente 2 horas, tendo como produto final a 1-[(R)-6-((S)-2,2-dimetil-l, 3-dioxolan-4-il)-3-hidroxi-5-oxo-hexil]-5-(4-fluor-fenil)-2-isopropil-N,4-difenil-lH- pirrol-3-carboxamida (13) (Esquema IX).Esquema IX:
É importante ressaltar que a reação da referida etapa 9 fornece^ um rendimento de _50% a 85%, preferivelmente de cerca de 79%, com uma estereosseletividade (relação R,S/S,R) de cerca de 92:08 na geração do primeiro centro estereogênico da molécula. Tanto o rendimento quanto a estereosseletividade obtidos são superiores aos descritos no estado da técnica (63% de rendimento e 86:14 de estereosseletividade). Além disso, a reação descrita na etapa 9 da presente invenção não emprega o auxiliar quiral acetato de (S)-2-hidroxi-l, 2,2-trifeniletila, o qual possui um custo elevado, bem como não utiliza n-BuLi (inflamável) nem diisopropilamina (alto custo). Estes três compostos são amplamente utilizados nas reações aldólicas descritas no estado da técnica. Cabe ainda mencionar que a reação descrita na etapa 9 da presente invenção já prevê a adição de todos os carbonos da estrutura da atorvastatina e, após a obtenção do composto (13), apenas mais 5 etapas reacionais fornecem a atorvastatina cálcica (41), ao passo que no estado da técnica são necessárias mais 7 etapas para a obtenção da atorvastatina cálcica.
Em outras palavras, a reação da etapa 9 acima descrita emprega reagentes de menor custo, de fácil manipulação e baixa toxicidade e, portanto, mais adequados à sintese em larga escala empregada em processos industriais.• Etapa 10 - obtenção da 1-((3R,5R)-6-((S)-2,2-dimetil- 1,3-dioxolan-4-il)-3,5-diidroxiexil)-5-(4-f luõrof enil)~2-isopfôpi 1-N, 4 “dif enil-*lH-pirrõl“ 3- carboxamida (14)
A etapa 10 consiste na reação de redução diastereosseletiva da 1-f(R)-6-((S)-2,2-dimetil-l,3- dioxolan-4-il)-3-hidroxi-5-oxo-hexil]-5-(4-fluor-fenil)-2- isopropil-N, 4-difenil-lH-pirrol-3-carboxamida (13)conforme as condições descritas em Narasaka, K. ; Pai, F. C. Tetrahedron 1984, 40, 2233 e Chen, K. M. ; Hardtmann, G. E.; Prasad, K.; Repic, Shapiro, M. J. Tetrahedron Lett. 1987, 28, 155 (0,8 equivalente a 2,0 equivalentes, preferivelmente 1, 1 equivalente de NaBHa e Et∑BOMe em uma faixa de temperatura de -95°C a -5°C, preferivelmente - 78°C, durante um período de 2 horas a 10 horas, preferivelmente 4 horas, na presença de uma mistura 4:1 de THF e metanol), fornecendo como produto a l-((3R,5R)-6- ((S)-2,2-dimetil-l,3-dioxolan-4-il)-3,5-diidroxiexil)-5- (4-fluorofenil)-2-isopropil-N, 4-difenil-lH-pirrol-3- carboxamida (14) (Esquema X).

É importante ressaltar que a reação da referida etapa10 fornece um rendimento ha faixa~~de 60% ’ ka— 90%, preferivelmente 87%, com uma relação díastereoisomérica > 95:5 em favor do diasteoisômero desejado 1,3-syn.• Etapa 11 - obtenção da 5-(4-fluorofenil)-2-isopropil- N, 4-difenil-l-((3R,7S)-3, 5, 7,8-tetraidroxioctil)-1H- pirrol-3-carboxamida (15)
A etapa 11 consiste na reação de hidrólise ácida (HC1 IN e H2O/MeOH ou H2O/THF) da 1-( (3R, 5R)-6-( (S)-2,2-dimetil- 1,3-dioxolan-4-il)-3,5-diidroxiexil)-5-(4-fluorofenil)-2- isopropil-N, 4-difenil-lH-pirrol-3-carboxamida (14), tendo como produto a 5-(4-fluorofenil)-2-isopropil-N,4-difenil- 1-((3R,7S)-3,5,7,8-tetraidroxioctil)-lH-pirrol-3- carboxamida (15) (Esquema XI). A reação da referida etapa 11 é conduzida em uma faixa de temperatura de -10°C a 50°C, preferivelmente 25°C, durante um periodo de 2 a 8 horas, preferivelmente 4 horas. A referida 5-(4- fluorofenil)-2-isopropil-N, 4-difenil-l-( (3R,7S)-3,5,7,8- tetraidroxioctil)-lH-pirrol-3-carboxamida (15) pode ser empregada na etapa reacional seguinte sem purificação prévia, uma vez que é obtida em alta pureza, mesmo em seu estado bruto.Esquema XI:
Cabe aqui ressaltar que a reação da referida etapa 11 fornece um rendimento na faixa de 8 0% a 97%, preferivelmente 94%.• Etapa 12 - obtenção da 1-(2-((2R,4R)-4,6-dihidroxitetraidro-2H-piran-2-il)etil)-5-(4- fluorofenil)-2-isopropil-N, 4-difenil-lH-pirrol-3- carboxamida (26)
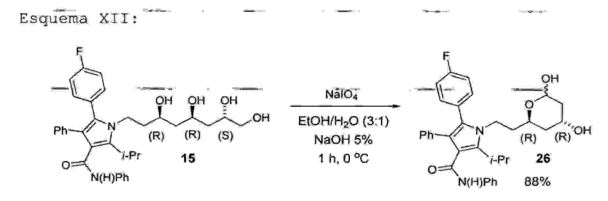
A etapa 12 consiste na reação de clivagem da 5-(4- fluorofenil)-2-isopropil-N, 4-difenil-l-((3R,7S)-3,5,7,8- tetraidroxioctil)-lH-pirrol-3-carboxamida (15) com periodato de sódio (NaIC>4) em condições muito suaves (etanol/água na proporção de 3:1 e NaOH na faixa de 1% a 20%, preferivelmente 5%), fornecendo como produto a l-(2- ( (2R, 4R) - 4 ,6-dihidroxitetraidro-2H-piran-2-il)etil)-5-(4- fluorofenil)-2-isopropil-N, 4-difenil-lH-pirrol-3- carboxamida (26) (Esquema XII) . A reação da referida etapa 12 é conduzida em uma faixa de temperatura de -20°C a 15°C, preferivelmente 0°C, durante um periodo de 30 minutos a 3 horas, preferivelmente 1 hora.Esquema XII: '
Cabe aqui ressaltar que a reação da referida etapa 12 fornece um rendimento na faixa de 75% a 95%, preferivelmente 88%. Deste modo, a 1-(2-((2R,4R)-4,6- dihidroxitetraidro-2H-piran-2-il) etil)-5-(4-fluorofenil)- 2-isopropil-N,4-difenil-lH-pirrol-3-carboxamida (26) foi obtida após 9 etapas com um rendimento global na faixa de 15% a 25%, preferivelmente 22,3%. • Etapa 13 — obtenção da 5-(4-fluorofenil)-1-(2-( (2R, 4 R) -4-hidroxi-6-oxotetraidro-2H-piran-2- il)etil)-2-isopropil-N, 4-difenil-lH-pirrol-3- carboxamida (31)
A etapa 13 consiste na reação de oxidação seletiva da 1- (2- ( (2R,4R)-4,6-dihidroxitetraidro-2H-piran-2-il)etil)- 5 -(4-fluorofenil)-2-isopropil-N,4-difenil-lH-pirrol-3- carboxamida (26) na presença de excesso de dióxido de manganês (MnOa) ativado (10 a 20 equivalentes, preferivelmente 15 equivalentes) e diclorometano, fornecendo como produto a 5-(4-fluorofenil)-1-(2-((2R,4R)- 4-hidroxi-6-oxotetraidro-2H-piran-2-il)etil)-2-isopropi1- N, 4-difenil-lH-pirrol-3-carboxamida (31) (Esquema XIII) . A reação “da referida'- etapa’ 13 “é conduzida" em uma fãixà cie temperatura de 0°C a 40°C, preferivelmente a 25°C, durante um periodo de 24 a 56 horas, preferivelmente 48 horas.

Cabe aqui ressaltar que a reação da referida etapa 13fornece um rendimento na faixa de 78% a 98%, preferivelmente 95%.• Etapa 14 - obtenção da atorvastatina cálcica (41)A etapa 14 consiste no tratamento da 5-(4-f luorofenil)—1—(2—( (2R,4R)-4-hidroxi-6-oxotetraidro-2H-5 piran-2-il)etil)-2-isopropil-N, 4-difenil-lH-pirrol-3-carboxamida (31) com hidróxido de sódio 10% na presença de MeOH em uma faixa de temperatura de 4 5°C a 65°C, preferivelmente 55°C, durante um periodo de 40 minutos a 3 horas, preferivelmente 1 hora, seguido do tratamento do10 sal resultante com acetato de cálcio em uma faixa de temperatura de 40°C a 60°C, preferivelmente 50°C, durante um periodo de 40 minutos a 3 horas, preferivelmente '1como produto final a atorvastatinacálcica (41) .15 Esquema XIV:
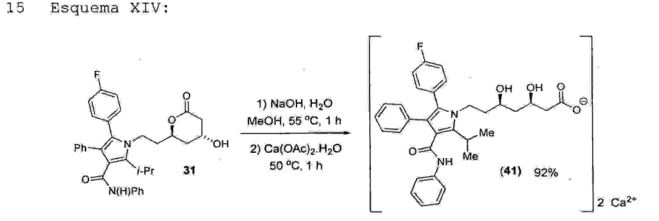
Cabe aqui ressaltar que a reação da referida etapa 14fornece um rendimento na faixa de 85%preferivelmente 92%. Deste modo, atorvastatina (41) foi obtida após 12 etapas com um rendimento global na faixa de 15% a 22%, preferivelmente 19,5%.
Adicionalmente, a presente invenção trata dos compostos l-[(R)-6-((S)-2,2-dimetil-l, 3-dioxolan-4-il)-3- hidroxi-5-oxo-hexil]-5-(4-fluor-fenil)-2-isopropil-N,4- difenil-lH-pirrol-3-carboxamida (13), 1-((3R,5R)-6-( (S)-2,2-dimetil-l,3-dioxolan-4-il) -3, 5-diidroxiexil)-5-(4- fluorofenil)-2-isopropil-N,4-difenil-lH-pirrol-3- carboxamida (14) e 5-(4-fluorofenil)-2-isopropil-N,4-difenil-l-((3R,7S)-3,5,7,8-tetraidroxiocti1)-lH-pirrol-3- carboxamida (15), os quais são obtidos através das etapas 9, 10 e 11, respectivamente, e são utilizados comointermediários no processo para a obtenção de atorvastatina cálcica da presente" invenção."
Além disso, a presente invenção trata do uso dos ditos intermediários nas etapas 10, 11 e 12 do processo para a obtenção de atorvastatina cálcica da presente invenção.
Ademais, a presente invenção trata da atorvastatina cálcica obtida através do processo para a obtenção de atorvastatina cálcica da presente invenção.
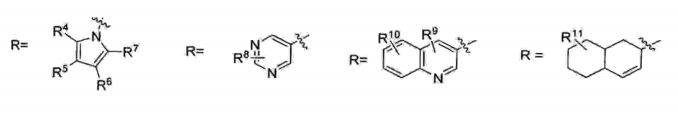
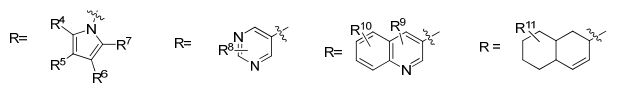
A presente invenção trata ainda de um método para a obtenção de uma estatina (por exemplo, sinvastatina, lovastatina, pravastatina, rosuvastatina ou pitavastatina) e seus intermediários.
Uma vez que outras estatinas compartilham a mesma cadeia lateral da atorvastatina, o processo para obtenção de atorvastatina cálcica da presente invenção pode ser5 estendido para outras estatinas, conforme será descrito a seguir.• Etapa 1 - reação aldólica
Na reação aldólica da etapa 1 o composto II é tratado com uma dialquil haloborana, seguida do resfriamento da10 reação e adição lentamente do composto I:
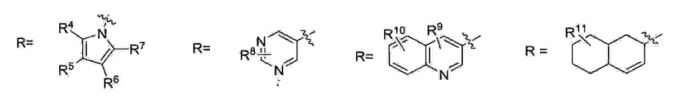
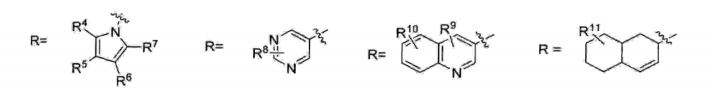
onde: X=YX= CH2, CH 15 Y= CH2, CHR pode ter um número muito grande de possibilidades, tal como, por exemplo:
20 R1 pode ser igual a R2 (R1=R2) ou R1 pode ser diferente de R2 (R1#R2)R1 e R2 = H ou CnH2n*i (com n variando de 1 a 10)R1 e R2 = H, anel aromático (CgHs- representado de maneira genérica por Ar) ou heteroaromático (hetAr) com diferentes 5 substituintes no anel aromático ou heteroaromático.
• Etapa 2 - reação de redução de Narazaka
Na reação da etapa 2 o composto III é reduzido de maneira diastereosseletiva empregando uma dialquil 10 alcoxiborana e tetraidreto de boro para fornecer o composto IV:
onde: X=Y 15 X= CH2, CHY= CH2, CHR pode ter um número muito grande de possibilidades, talcomo, por exemplo:
R1 pode ser igual a R2 (R1=R2) ou R1 pode ser diferente deR2 (RX#R2)5 R1 e R2 = H ou CnH2n+i (com n variando de 1 a 10)R1 e R2 = H, anel aromático (C6H5- representado de maneiragenérica por Ar) ou heteroaromático (hetAr) com diferentessubstituintes no anel aromático ou heteroaromático.
Etapa 3 - reação de hidrólise ácidaA reação da etapa 3 corresponde à hidrólise, em meiopara a obtenção do composto V:
R pode ter um número muito grande de possibilidades, tal como, por exemplo:
R1 pode ser igual a R2 (Rr=R2) ou R1 pode ser diferente deR1 e R2 = H, anel aromático (CgHs- representado de maneira genérica por Ar) ou heteroaromático (hetAr) com diferentes substituintes no anel aromático ou heteroaromático.
Etapa 4 - reação de divagem para obtenção do lactolde estatina
A reação da etapa 4 corresponde a divagem do composto V com sal de periodato de metal alcalino para fornecer o composto VI:
onde: X=Y Y= CH2, CHR pode ter um número muito grande de possibilidades, tai como, por exemplo:
R1 pode ser igual a R2 (Rl=R2) ou R1 pode ser diferente deR2 (RX#R2)R1 e R2 = H ou CnHzπ*! (com n variando de 1 a 10)R1 e R2 = H, anel aromático (C6H5- representado de maneira genérica por Ar) ou heteroaromático (hetAr) com diferentes 10 substituintes no anel aromático ou heteroaromático.
• Etapa 5 - reação de oxidação seletiva do lactol de estatinaA reação da etapa 5 corresponde a uma oxidação15 seletiva do composto VI na presença de excesso de oxidante metálico ativado para fornecer o composto VII:
onde: X=YX= CH2, CHY= CH2, CHR pode ter um número muito grande de possibilidades, tal5 como, por exemplo:R= Ar, HetAr, alquila
• Etapa 6 - reação de obtenção da estatina
Na reação da etapa 6 o composto VII obtido é tratado10 com uma base, e em seguida o sal resultante é tratado com um sal de cálcio para fornecer uma estatina (VIII):
onde: X=YX= CH2, CH15 Y= CH2, CHR pode ter um número muito grande de possibilidades, talcomo, por exemplo: R= Ar, HetAr, alquila
Adicionalmente, a presente invenção trata dos compostos obtidos através das etapas 1, 2 e 3, os quais são utilizados como intermediários no método para a obtenção de uma estatina da presente invenção.
Além disso, a presente invenção trata do uso dos ditos intermediários nas etapas 2, 3 e 4 do método para a obtenção de uma estatina da presente invenção.
Ademais, a presente invenção trata das estatinas obtidas através do processo para a obtenção de uma estatina da presente invenção.
Embora tenham sido mostradas apenas modalidades preferidas da presente invenção, será entendido que várias omissões, substituições e alterações nos processos conforme aqui apresentados podem ser feitas por um técnico versado no assunto, sem se afastar do espírito e escopo da presente invenção.
É expressamente previsto que todas as combinações de elementos que desempenham a mesma função substancialmente da mesma forma para alcançar os mesmos resultados estão dentro do escopo da invenção. Substituições de elementos de uma modalidade descrita por outros são também totalmente pretendidas e contempladas.
A invenção será ilustrada a seguir por diversos exemplos, que não devem ser considerados limitativos da mesma. EXEMPLOS
Exemplo 1: Procedimento para a obtenção da 4-metil-3- oxo-AJ-fenilpentanamida (2). A uma mistura do 4-metil-3-oxo pentanoato de metila (1) (1,0 equivalente, 100,0 g, 700 mmol) e NaOH (0,56 g, 2 mol%) foi adicionado anilina (1,2 equivalente, 78,1 g, 840 mmol) . A mistura reacional foi aquecida até 135°C e mantida por 16 horas, com remoção concomitante de metanol, sendo monitorada por cromatográfia em camada delgada. A reação foi resfriada a temperatura ambiente e adicionou-se lentamente a 0°C HC1 1% até pH = 6 e, após, água (500 mL) . A mistura reacional foi extraída com acetato de etila (2x 500 mL) e lavou-se a fase orgânica com água (500 mL). A fase orgânica foi seca com sulfato de magnésio anidro e então concentrada sob vácuo fornecendo a 4-metil-3-oxo-N-fenilpentanamida (2) como um óleo amarelo viscoso em 92% de rendimento (130,0 g) na forma pura. (Lit. p.e. - 261-264 °C), RMN XH (250 MHz, DMSO-G?6) δ 1,03 (d, J = 7,0 Hz, 6H); 2,77-2,83 (m, 1H) , 3,61 (s, 2H) ; 7,04 (t, J = 8,1 Hz, 1H); 7,30 (t, 8,1 Hz, 2H) ; 7,58 (d, J = 8,1 Hz, 2H); 10,07 (s, 1H) (FIGURA 1). RMN 13C (62 MHz, DMSO-de) δ 17,5; 40,3; 49,4; 118,9; 123,3; 128,6; 138,8; 165,1; 208,2 (FIGURA 2) .
Exemplo 2: Procedimento para a obtenção da 2-5 benzilideno-4-metil-3-oxo-N-fenilpentanamida (3) (Reação de Knoevenagel) . Uma mistura da 4 -metil-3-oxo-AT-fenilpentanamida (2) (1,0 equivalente, 100 g, 487,2 mmol), p-aminofenol (10,0 g, 96 mmol, 20 mol%), aldeído benzóico (1,05 equivalente; 53,0 g; 504 mmol) e ácido acético (5,710 g; 96 mmol; 20 mol%) em hexano (1 L) foi mantida sob refluxo durante 24 horas. O sólido obtido foi filtrado elavado com n-hexano (1 L) , seguido por lavagem com águadestilada (1,5 L) e secagem sob alto vácuo durante 48 horas fornecendo’ a 2-benziíideno-4-metil-3-oxo-N-fenilpentanamida15 (3) em 93% de rendimento (132,8 g) . P.f. = 190-193 °C. RMNXH (250 MHz, DMSO-dβ) δ 1.12 (d, J= 7,1 Hz, 6H); 3,30-3,47 (m, 1H); 7,08 (t, J=8,0Hz, 1H); 7,30-7,41’(m, 5H); 7,66- 7,70 (m, 5H); 9,61 (s, 1H) (FIGURA 3). RMN 13C (62,5 MHz, DMSO-dg) δ 18,3; 36,0; 121,1; 124,2; 128,3; 129,5; 130,3;20 132,7; 135,1; 136,0; 137,3; 140,2; 165,3; 202,8 (FIGURA 4).Exemplo 3: Procedimento para a obtenção da 2-[2-(4- fluorofenil)-2-oxo-l-feniletil]-4-methil-3-oxo-N- fenilpentanamida (4) (Reação de Stetter) . Uma mistura da 2- benzilideno-4-metil-3-oxo-N-fenilpentanamida (3) (1/0 equivalente; 100,0 g; 341 mmol), brometo de 3-etil-5-(2- hidroxietil)-4-metil-3-tiazolio (0,2 equivalente; 17,1 g; 68,2 mmol), trietilamina (1,0 equivalente; 48 mL; 341 mmol) e 4-flúor-benzaldeido (1,05 equivalente; 44,2 g; 358 mmol) foi aquecida a 7 5 °C sob atmosfera de argônio e sob agitação vigorosa. A reação foi monitorada por cromatográfia em camada delgada (CCD) até consumo da 2- benzilideno-4-metil-3-oxo-W-fenilpentanamida (3) (16horas). Adicionou-se 500 mL de isopropanol e a mistura reacional foi mantida a temperatura ambiente por 4 horas. O sólido obtido foi filtrado a vácuo e lavado com 1 L de água. A 2-[2-(4-fluorofenil)-2-oxo-l-feniletil]-4-metil-3- oxo-W-fenilpentanamida (4) foi recristalizada com isopropanol quente (60-65 °C) seguido de filtração esecagem sob alto vácuo durante 24 horas, fornecendo um sólido branco cristalino (P.f. = 205-208 °C, Lit. 206-209 °C) em 75% de rendimento (106,7 g) . RMN *H (250 MHz, DMSO- d6) δ 0,92 (d, J = 6,5 Hz, 3H) ; 1,15 (d, J = 7,5 Hz, 3H) ; 2,83-2,94 (m, 1H); 4,87 (d, J = 11 Hz, 1H) ; 5,42 (d, J = 11 Hz, 1H); 6, 97-7,40 (m, 12H) ; 8,13 (d, J = 8,2 Hz, 2H); 10,18 (s, 1H) (FIGURA 5). RMN 13C (62,5 MHz, DMSO-d6) δ 18,1; 18,6; 40,9; 54,1; 64,1; 115,4; 120, 6; 124,7; 128,0; 128,8; 129,4; 131,5; 132,1; 132,2; 135,2; 136,7; 165,5; Exemplo 4: Procedimento para a obtenção da [5-(4- fluorofenila)-1-(3-hidroxipropil]-2-isopropil-N, 4-difenil- lH-pirrol-3-carboxamida (5) (Reação de Pall-Knorr) . A uma solução da 2-[2-(4-fluorofenil)-2-oxo-l-feniletil]-4-metIl-5 3-oxo-N-fenilpentanamida (4) (20,0 g; 47,8 mmol; 1,0equivalente) e 3-amino-l-propanol (6,1 g; 81,2 mmol; 1,7 equivalente) em tolueno/THF (1:1) (100 mL) adicionou-sequantidade catalítica de ácido piválico (0,9 g; 9,5 mmol;20 mol%) e a mistura foi mantida sob refluxo durante 48 h10 com remoção azeotrópica de água. Resfriou-se à temperatura ambiente e a reação foi extraída com acetato de etila (3 x 100 mL) . O produto (5) foi obtido na forma de um sólido branco após recristalização em isopropanol/hexano em 75% de rendimento (16,3 gj . Uma amostra analítica do produto (5)15 foi purificada por coluna cromatográfica (hexano/acetato 8:2). RMN (250 MHz, CDC13) δ 1,53 (d, J = 7,0 Hz; 6H) ; 1,71-1,82 (m, 2H) ; 3,46-3,61 (m, 3H) ; 3,99 (t, j = 7,7 Hz; 2H); 6,87 (s, 1H); 6,95-7,21 (m, 14H) (FIGURA 7). RMN 13C (62,5 MHz, CDCI3) δ 21,7; 26,1; 34,3; 41,7; 59,8; 115,2;20 119,6; 121,8; 123,5; 126,5; 128,0; 128,2; 128,3; 128,8;130,4; 133,0; 133,2; 134,6; 138,3; 141,4; 160,2; 164,8 (FIGURA 8).\ Exemplo 5: Procedimento para a obtenção da 5-(4-fluorofenil)-2-isopropil-l-(3-oxopropil)-N,4-difenil-lH- pirrol-3-carboxamida (6) (Reação de oxidação de Swern). A uma solução de cloreto de oxalila (5,0 g; 39,3 mmol; 1,2 equivalente) em CH2CI2 (150 mL) anidro sob atmosfera de argônio a -50 °C adicionou-se gota a gota DMSO (4,8 mL; 65,5 mmol; 2,0 equivalentes) . Após 20 minutos a -50 °C adicionou-se a [5-(4-fluorofenila)-1-(3-hidroxipropil]-2- isopropil-N, 4-difenil-l/i-pirrol-3-carboxamida (5) (15,0 g; 32,8 mmol; 1,0 equivalente) dissolvido em CH2CI2 (30 mL) . A mistura reacional foi mantida a -50 °C por 1 hora e, após, adicionou-se Et3N (18,4 mL; 131,2 mmol; 4,0 equivalentes) lentamente- A.reação foi mantida durante mais 1 hora a temperatura ambiente e extraiu-se com acetato de etila (3 x 100 mL) . A 5-(4-fluorofenil)-2-isopropil-l-(3-oxopropil)- N, 4-difenil-lH-pirrol-3-carboxamida (6) foi purificada por recristalização em isopropanol/hexano, sendo obtida na forma de um sólido branco em 84% de rendimento (12,0 g) . Uma amostra analítica foi purificada " por * coluna cromatográfica de silica, eluindo-se com hexano/acetato 9:1. RMN XH (250 MHz, CDCI3) δ 1,51 (d, J = 7,0 Hz, 6H) ;2,67 (t, J = 7,5 Hz; 2H) ; 3,61 (quint, J = 7,2 Hz, 1H);4,25 (t, J = 7,5 Hz; 2H); 6,85 (s, 1H); 6,95-7,21 (m, 14H);9,58 (s, 1H) . RMN 13C (62,5 MHz, CDCI3) δ 20,3; 25,1; 42,8;44,5; 115,3; 119,8; 121,9; 123,6; 126,8; 128,1; 128,2;128,3; 128,9; 130,5; 133,2; 133,4; 134,7; 138,5; 141,5; 160,7; 202,2,
Exemplo 6: Procedimento para a obtenção do (S)-malato de dimetila (8). A uma mistura de ácido (L)(-)-málico (30,0 g; 223 mmol) em metanol anidro (300 mL) foi adicionado quantidade catalítica de ácido sulfúrico (5 mL) . A solução resultante foi mantida sob refluxo durante 24 horas. Destilou-se o metanol até obter um volume final de aproximadamente 50 mL, Adicionou-se à mistura solução saturada de NaHCOs até pH = 8 (100 mL) . Extraiu-se a reação com acetato de etila (3 x 200 mL) . A fase orgânica foi coletada e seca com NajSC^ anidro. Evaporou-se o solvente em evaporador rotatório (40°C/80 mbar) e o solvente remanescente foi removido em bomba de alto vácuo (1-3 mmHg) durante 18 horas, O (S)-malato de dimetila (8) foi obtido em 91% de rendimento (32,8 g) como um óleo amarelo pálido o qual foi empregado na próxima etapa sem purificação prévia. RMN (250 MHz, ^CDClj) δ 1,23 (t, J “= ’7,2 Hz, -3H> ; -1 ,-29 (t, J = 7,2 Hz, 3H), 2,73-2,90 (m, 2H); 3,31 (s, 1H); 4,13 (q, J = 7,2 Hz, 2H); 4,30 (q, J = 7,2 Hz, 2H); 4,49 (t, J = 4,75 Hz, 1H) . RMN 13C (62,5 MHz, CDC13) δ 13,9 (2C) ; 38,6; 60,7; 61,7; 67,1; 170,3; 173,2,
Exemplo 7; Procedimento para a obtenção do acetato de (S)-metil 2-(2,2-dimetil-l,3-dioxolan-4-ila) (10). A uma solução de (S)-malato de dimetila (8) (1,0 equivalente; 30,0 g; 185,1 mmol) em THF anidro (200 mL) a 20 °C sob atmosfera de argônio foi adicionado BH3.SMe2 (lOM) (1,05 equivalente; 16,5 mL; 165 mmol) lentamente durante 25 minutos. A solução foi mantida sob agitação durante 305 minutos a 20°C até cessar a liberação de hidrogênio. A temperatura reacional foi reduzida para 10°C e a solução foi mantida por 10 minutos a esta temperatura. Em seguida, adicionou-se, de uma só vez, NaBH4 (7,8 mmol; 0,305 g; 5 mol%) e a mistura reacional foi mantida sob agitação10 intensa durante 1 hora a 20°C. A reação foi monitorada por cromatográfia em camada delgada até o consumo total do (S)- malato de dimetila (8) . Adicionou-se à mistura reacional metanol anidro (150 mL) lentamente e a mistura foi mantida sob agitação durante 30 minutos a 20°C. Remòveu-se o 15 solvente completamente em evaporador rotatório e, após, nabomba de alto vácuo durante 4 horas. O residuo foi dissolvido em uma mistura de“ acetona - (70 mL) - e -2,2- dimetoxipropano (70 mL) . A esta mistura adicionou-se quantidade catalítica de p-TsOH (1,3 g) e deixou-se sob 20 agitação durante 4 horas a temperatura ambiente. A reaçãofoi extraída com acetato de etila (3 X 70 mL) e o resíduo foi submetido a destilação sob pressão reduzida (p. e. = 74 °C/6 mbar). 0 (S)-metil 2-(2,2-dimetil-l,3-dioxolan-4-ila) (10) foi obtido na forma de um líquido incolor em 75% de rendimento (24,1 g; 138,8 mmol). RMN XH (250 MHz, CDC13) δ 1,35 (s, 3H); 1,41 (s, 3H); 2,52 (dd, J = 15,0 Hz; 7,5 Hz; 1H); 2,72 (dd, J = 15,0 Hz; 7,5 Hz, 1H); 3,65 (dd, J= 10,0 Hz; 7,5 Hz; 1H); 3,70 (s, 3H); 4,15 (dd, J = 8,7 Hz; 7,5 Hz; 1H) ; 4,47 (quint, J = 7,5 Hz; 1H) . RMN 13C (62,5 MHz, CDCI3) δ 25,4; 26,8; 38,7; 51,7; 69,1; 72,0; 109,2; 171,0.
Exemplo 8: Procedimento para a obtenção da (S)-l-(2,2- dimetil-1,3-dioxolan-4-ila)propan-2-ona (11). A uma solução do (S)-metil 2-(2,2-dimetil-1,3-dioxolan-4-ila) (10) (22,0g; 125,6 mmol; 1,0 equivalente) e Me(OMe)NH.HC1 (15,8 g; 162,6 mmol; 1,3 equivalente) em THF (200 mL) a -10 °C foi adicionada lentamente uma solução de MeMgCl (3M) em THF (168 mL; 504 nunol; 4,0 equivalentes) durante 50 minutos. Após 1 h de reação a -5 °C, a mistura foi mantida sob agitação por 8 h a temperatura ambiente. A reação foi extraída com acetato de etila (3 x 18 0 mL) e o resíduo—foi submetido a destilação sob pressão reduzida (p.e.= 68 °C/7 mmHg). A (S)-1-(2,2-dimetil-l,3-dioxolan-4-ila)propan-2-ona (11) foi obtida na forma de um líquido amarelo pálido em 92% de rendimento (18 g; 115 mol). RMN XH (250 MHz, CDCI3) δ 1,34 (s, 3H) ; 1,40 (s, 3H) ; 2,19 (s, 3H); 2,60 (dd, J = 17,5 Hz, 7,0 Hz, 1H); 2,89 (dd, 17,5 Hz; 6,0 Hz; 1H);3,53 (dd, J = 7,5 Hz; 6,7 Hz; 1H); 4,17 (dd, J = 7,5 Hz; 6,7 Hz; 1H) ; 4,45 (quint; J = 6,7 Hz; 1H) (FIGURA 9). RMN 13C (62,5 MHz, CDClj) δ 25,3; 26,8; 30,5; 47,7; 69,3; 71,6; 108,7; 206,2 (FIGURA 10).
Exemplo 9: Procedimento para a obtenção da l-[(R)-6-5 ((S)-2,2-dimetil-l, 3-dioxolan-4-il) -3-hidroxi-5-oxo-hexil]-5- (4-fluor-fenil)-2-isopropil-N, 4-difenil-lH-pirrol-3- carboxamida (13) (Reação aldólica). A uma solução da (S)-l- (2,2-dimetil-l, 3-dioxolan-4-ila)propan-2-ona (11) (3,5 g;22 mmol; 2,0 equivalentes) em Et2O anidro (120 mL) sob10 atmosfera de argônio a 0°C foi adicionado, gota a gota, c- Hex2BCl (9,5 mL; 44 mmol; 4,0 equivalentes) e logo após adicionou-se trietilamina (10,8 mL; 77 mmol; 4,5 equivalentes). A mistura reacional foi mantida sob agitação durante 1 hora a 0°C, para geração dõ enolato de boro. Após15 este tempo, a reação foi resfriada a -78°C e 5—(4 — fluorofenil)-2-isopropil-l-(3-oxopropil)-N,4-difenil-lH- pirrol-3-carboxamida (6) (5,0 g; 11 mmol; 1,0—equivalente)dissolvida em Et2O (20 mL) foi adicionada gota a gota à solução do enolato de boro previamente formado. A reação20 foi mantida por 2 h a -78°C e, após, foi finalizada pela adição de metanol (40 mL) e aquecida a temperatura ambiente. O solvente foi removido sob pressão reduzida e o residuo foi purificado por recristalização em isopropanol/hexano. A 1-[(R)-6-((S)-2,2-dimetil-l,3- dioxo Lan-4-il)-3-hidroxi-5-oxo-hexil]-5-(4-fluor-fenil)-2- isopropil-N, 4-difenil-lH-pirrol-3-carboxamida (13) foiobtida como um sólido branco cristalino em rendimento de 79% (5,3 g) . Uma amostra analítica foi purificada por5 coluna cromatográfica de silica (hexano/acetato 7:3). RMN lH (250 MHz, C6D6) δ 1,37 (s, 3H) ; 1,46 (s, 3H) ; 1,50-1,63(m, 2H); 1,73-1,80 (m, 1H) ; 1,86 (d, 7,0 Hz; 6H) ; 1,91-2,03 (m, 2H); 2,32 (dd, J = 15,0 Hz; 6,7 Hz; 1H); 2,85 (s,1H) ; 3,33 (t, <J = 7,5 Hz; LH) ; 3,70-3, 79 (m, 2H) ; 3,88-3,9610 (m, 2H); 4,13-4,33 (m, 2H); 6,77-7,32 (m, 13H, 7,48 (d, J =8,0 Hz, 2H) (FIGURA 11). RMN 13C (62,5 MHz, C6D6) δ 21,8; 22,0"; 25,-4; 26,6; 26,9; 38,1; 4ÍT3~47>1; 49,3; 64,7; 69,'2~T 71,6; 108,9; 115,4; 116,7; 119,2; 122,3; 123,4; 126,6;127,7; ’127,9; 128,8; 130,6; 133,4; 135,1; 139,4; 141,7;“'15 161,5; 163,4; 164,6; 208,1 (FIGURA 12). Massa de alta resolução: 613, 3078 (M+H) (C37H41FN2O5 + H) .
Exemplo 10: —Rrocedimento para a obtenção da 1-( (3R,5R)-6-((S)-2, 2-dimetil-l, 3-dioxolan-4-il)-3,5-diidroxiexil)-5-(4-fluorofenil) -2-isopropil-N, 4-difenil-lH-20 pirrol-3-carboxamida (14) (Redução de Narazaka). A uma solução da 1-[(P)-6-((S)-2,2-dimetil-l,3-dioxolan-4-il)-3- hidroxi-5-oxo-hexil]-5-(4-fluor-fenil) -2-isopropil-N, 4- difenil-lH-pirrol-3-carboxamida (13) (5,0 g, 8 mmol) emTHF/MeOH (4:1) (50 mL) sob atmosfera de argônio a -78°C adicionou-se o Et2B(OMe) (1,5 mL; 8,8 mmol; 1,1 equivalente) gota a gota. Após 40 minutos, adicionou-se NaBHq (0,42 g; 1,1 equivalente) e a mistura reacional foi mantida a -78°C por 4 h. A reação foi finalizada pela 5 adição de ácido acético glacial (20 mL), metanol (40 mL) e H2O2 30% (15 mL) . Após agitação durante 30 minutos, amistura foi extraída com acetato de etila (3 x 60 mL) e o resíduo foi purificado por coluna cromatográfica de sílica (hexano/acetato 8:2). A 1-((3R,5R)-6-((S)-2,2-dimetil-l,3- 10 dioxolan-4-il)-3,5-diidroxiexil)-5-(4-fluorofenil)-2-isopropil-N,4-difenil-lH-pirrol-3-carboxamida (14) foi obtida na forma de um óleo viscoso incolor em 87% de rendimento (4,2 g; 6,9 mmol). 0 composto (14) foi empregadona próxima etapa reacional sèm purificação prévia. RMN XH 15 (250 MHz, C6D6) Õ 0,98 (m, 2H) , 1,29-1,41 (m, 8H) , 1,73 (m, 2), 1,86 (d, J = 7,2 Hz, 6H) , 3,39 (m, 1H), 3,66 (m, 1H) , 3,.85. (m, 3H) , 3,93-4,11 _(m, 4H).,_6,80 _(s, 1H) 6, 90-1, 46(m, 14H) (FIGURA 13). RMN 13C (62,5 MHz, C6D6) δ 21,9; 22,1; 25,5; 26,6; 26,8; 39,9; 40,5; 41,4; 42,9; 69,2; 69,8; 70,1;20 73,2; 108,*9; 115,4; 115,6; 116,6; 119,4; 122,3; 123,5;126,6; 127,7; 127,9; 128,8; 128,9; 130,6; 133,4; 133,5;135,1; 139,2; 139,3; 141,6; 161,4; 163,4; 164,9 (FIGURA14). Massa de alta resolução: 615,3234 (M+H) (C37H43FN2O5 +H) .
Exemplo 11: Procedimento para a obtenção da 5-(4- fluorofenil)-2-isopropil-N, 4-difenil-l-((3R,7S)-3,5,7,8- tetraidroxioctil)-lH-pirrol-3-carboxamida (15). A uma solução da 1-[(3R,5R)-6-((S)-2,2-dimetil-l,3-dioxolan-4- il) - 3,5-diidroxiexil]-5-(4-fluorofenil)-2-isopropil-M, 4- difenil-lH-pirrol-3-carboxamida (14) (4,0 g, 6,5 mmol) emTHF (40 mL) a 25°C adicionou-se solução aquosa de HC1 (IN) (6,4 mL ) . A mistura reacional foi mantida sob agitação durante 4 horas a temperatura ambiente. Adicionou-se solução saturada de NaHCOa (50 mL) . A mistura reacional foi extraída com acetato de etila (3 x 70 mL) , A 5-(4- fluorofenil)-2-isopropil-N, 4-difeni1-1-((3R,7S)-3,5,7,8- tetraidroxioctil)-lH-pirrol-3-carboxamida (15) foi obtida na forma de um óleo viscoso incolor em 94% de rendimento (3,4 g; 6,1 mmol). O composto (15) foi empregado na próxima etapa reacional sem purificação prévia. RMN :H (250 MHz, MeOD) 5 1,17—l-; 28 (m, 4H) , 1,49 (d, 7-,-Q Hz,- 6H) , 1,60-1,78 (m, 2H) , 3, 37-3, 45 (m, 3H), 3,65 (m, 1H) , 3,78-4,00 (m, 4H), 7,02-7,30 (m, 14H) (FIGURA 15). RMN 13C (62,5 MHz, MeOD) δ 22,7; 27,6; 30,7; 40,3; 41,9; 42,2; 45,4; 47,9; 67,8; 67,9; 68,9; 70,0; 116,1; 118,0; 121,4; 123,3; 125,1; 126,8; 128,8; 129,6; 130,2; 134,6; 134,8; 136,3; 139,0; 139,8; 161,8; 165,7; 169,5 (FIGURA 16). Massa de altaresolução: 575, 2921 (M+H) (Ca^HjgFNzOs +■ H).
Exemplo 12: Procedimento para a obtenção da l-(2- ( (2R, 4R) -4,6-dihidroxitetraidro-2H-piran-2-il)etil)-5- (4- fluorofenil)-2-isopropil-N, 4-difenil-lH-pirrol-3- carboxamida (26). A uma solução da 5-(4-fluorofenil)-2- isopropil-N,4-difenil-1-((3R,7S)-3,5,7,8-tetraidroxioctil)- lH-pirrol-3-carboxamida (15) (3,1 g, 5,4 mmol, 1,0equivalente) em etanol (50 mL) a 0°C adicionou-selentamente uma solução de NaIO4 (3,4 g; 16,2 mmol; 3,0 equivalentes) e NaOH (11 mg; 0,27 mmol; 5 mol%) em 16 mL de H2O. A mistura reacional foi mantida sob agitação por 1 hora a 0°C. Adicionou-se solução saturada de NH4CI (50^mL). A mistura foi extraída com acetato de etila (3 x 50 mL) . A l-(2-((2R,4R)-4,6-dihidroxitetraidro-2H-piran-2-il)etil)-5- (4-fluorofenil)-2-isopropil-N, 4-difenil-lH-pirrol-3- carboxamida (26) foi obtida na forma de um óleo viscoso incolor em 88% de rendimento (2,5 g; 4,7 mmol). O composto (26) foi empregado ~ na próxima etapa reacional sem purificação prévia. Uma amostra analítica foi purificada por coluna cromatográfica de sílica (hexano/acetato de etila 3:7). RMN rH (250 MHz, CDC13) δ 1,52 (d, J = 7,0 Hz, 6H), 1,63-1,93 (m, 6H), 3,15 (d, J = 5,7 Hz, 1H), 3,46-3,60 (m, 1H), 3,82-3,94 (m, 1H); 4,00-4,26 (m, 3H), 4,27 (d, J = 7,5 HZ, 1H) , 5,19 (s, 1H), 6,88 (s, 1H) , 6,95-7,20 (m, 14H) (FIGURA 17). RMN 13C [62,5 MHz, CDC13) δ 21,6; 21,8; 26,1; 35,Of 37,4; 37,7; 41,4; 92,6; 115,2; 115,3; 115,5; 119,6; 121,8; 123,6; 126,5; 128,3; 128,6; 128,8; 130,4; 133,0;133,1; 134,6; 138,2; 141,3; 165,0 (FIGURA 18). Massa de alta resolução: 543,2659 (M+H) (C33H35FN2O4 + H) ., 5
Exemplo 13: Procedimento para a obtenção da 5-(4- fluorofenil)-1-(2-((2R,4R)-4-hidroxi-6-oxotetraidro-2H- piran-2-il)etil)-2-isopropil-N, 4-difenil-lH-pirrol-3- carboxamida (31) (lactona da atorvastatina). A uma solução10 da 1-(2-((2R,4R)-4,6-dihidroxitetraidro-2H-piran-2-il)etil)-5-(4-fluorofenil)-2-isopropil-AZ,4-difenil-lH- pirrol-3-carboxamida (26) (lactol da atorvastatina) (2,2 g, 3,9 mmol) em CH2C12 (15 mL) adicionou-se MnO2 ativado (5,1'g; 58,5 mmol; 15,0 equivalentes). A suspensão foi mantida 15 sob agitação durante 48 horas a temperatura ambiente. Amistura foi filtrada e o solvente evaporado. A 5-(4------ fluorofenil)-1-(2-((2R)4R)-4-hidroxi-6-oxotetraidro-2H- piran-2-il)etil)-2-isopropil-N, 4-difenil-lH-pirrol-3- carboxamida (31) (lactona da atorvastatina) foi obtida em 20 95% de rendimento (2,1 g) como um sólido branco cristalinoapós recristalizaçào em hexano/isopropanol. P.f.= 160-162°C, [a]D20 = + 27,00 (c = 1, CHCI3) . RMN XH (250 MHz, CDC13)õ 1,49-1,53 (m, 6H); 1,58-1,90 (m, 4H); 2,48-2,65 (m, 2H);2,89 (s, 1H, OH); 3, 47-3, 58 (m, 1H) ; 3,95-4,24 (m, 3H) ; 4,48-4,56 (m, 1H); 6,91 (s, 1H, NH) ; 6/99-7,21 (m, 14)(FIGURA 19). RMN 13C (62,5 MHz, CDC13) δ 21,7; 22,0; 26,1; 35,5; 37,1; 38,4; 40,7; 62,2; 73,1; 115,4; 115,6; 115,7; 119,8; 122,0; 123,8; 126,6; 127,8; 127,9; 128,3; 128,7;128,8; 130,3; 133,1; 134,3; 138,0; 141,2; 160,3; 164,3;165,0; 169,8 (FIGURA 20).Exemplo 14: Procedimento para a obtenção daatorvastatina cálcica (41). A uma solução da 5-(4-fluorofenil)-1-(2-((2R, 4R)-4-hidroxi-6-oxotetraidro-2H-piran-2-il)etil)-2-isopropil-N, 4-difenil-lH-pirrol-3- carboxamida (31) (lactona da atorvastatina) (1,0 equivalente;. 2,0 g, 3,6 mmol) em metanol adicionou-se solução aquosa de NaOH 10% até o pH = 12 e a mistura resultante foi mantida sob agitação por 1 hora a 55°C. Após o término da reação (conforme análise por CCD) , a reação foi concentrada a 1/3 do volume inicial e foi adicionada água destilada/deionizada (100 mL), metanol (20 mL) e acetato de etila (100 mL) . Separaram-se as fases e a fase aquosa foi lavada com acetato de etila (2 x 50 mL). A fase aquosa foi separada, o pH foi ajustado para 8,0 através da adição de solução aquosa de HC1 (IN) e foi então aquecida a 50°C. A mistura resultante foi tratada com uma solução de acetato de cálcio monoidratado (0,35 g; 1,98 mmol; 0,55 equivalente) em água destilada/deionizada e a mistura foi aquecida a 50°C. A reação foi mantida sob agitação durante 1 hora e, após, foi lentamente resfriada a 30°C durante 4 horas. 0 produto precipitado (31) foi filtrado e lavado com metanol/HjO (1:1). A atorvastatina cálcica (31) foi obtida 5 na forma de um sólido branco amorfo em 92% de rendimento(1,9 g) após recristalização em THF/MeOH/HzO e secagem sob alto vácuo a 50°C durante 24 horas. [otJD20 = -2,8° (c = 1,0;DMSO). RMN (250 MHz, DMSO-d6) δ 1,21-1,68 (m, 4H) ; 1,34(d, J = 7,2 Hz; 6H); 1,76-2,11 (m, 2H) ; 3,19-4,10 (m, 7H) ;10 6,85-7,62 (m, 14H); 9,77 (s, 1H) (FIGURA 21). RMN 13C (62,5MHz, DMSO-dé) δ 21,7; 26,4; 29,7; 34,2; 39,4; 41,0; 42,7; - I- - -’ 67,δ’; " 115,2; 116,9; 120,3; 122,1; 124,0; 124,9; 125,7;127,7; 128,4; 129,7; 133,5; 135,1; 137,8; 138,0; 138,6;161,6; 163,5; 168,3. IV (cm-1) 3418; 3062; 2961; 2873;15 2252; 1793; 1660; 1528; 1480; 911 (FIGURA 22).