CAMPO DA INVENÇÃO
[001] A presente invenção trata de formulações farmacêuticas contendo lactonas sesquiterpênicas da classe dos furanoeliangolidos constituídas principalmente de nanocápsulas poliméricas que aumentam a eficácia dessa classe de bioativos em doenças parasitárias e em tumores. Mais especificamente, esta invenção trata de uma nova forma de tratamento de infecções causadas pelos parasitas protozoários da família Trypanosomatidae, particularmente os causadores da doença de Chagas, das Leishmanioses e da Tripanossomíase Africana e também para o tratamento do câncer.
[002] A presente invenção descreve a técnica de preparo de formulações de nanocápsulas constituídas de polímeros contendo a associação de ativos da classe dos furanoeliangolidos com ação anti-protozoário e antitumoral. Mais especificamente, a título de exemplo, foi utilizada a substância química licnofolida, uma lactona sesquiterpênica lipofílica pertencente à classe dos furanoeliangolidos que demonstrou alta atividade e eficácia no tratamento de infecções experimentais em camundongos e atividade antitumoral in vitro. Esta invenção descreve assim, as metodologias de obtenção destas formulações contendo as substâncias em questão.
[003] Objetiva-se a utilização destes produtos no tratamento da doença de Chagas, das Leishmanioses, da Tripanossomíase Africana e também no tratamento de tumores. Estas formulações de uso farmacêutico podem ser utilizadas em humanos e em animais, com potencial efeito tóxico reduzido e aumento da eficácia em relação às formas farmacêuticas convencionais; administradas na forma de formulações por via parenteral (via intravenosa, intramuscular, intradérmica e subcutânea), oral, tópica, retal, inalatória, intranasal, transdérmica, bucal dentre outras. Utilizando-se estas formulações a eficácia desses novos bioativos pode ser aumentada, devido à alteração da biodistribuição atribuída àsnanocápsulas aqui mencionadas e descritas.
[004] Além disso, os bioativos na sua forma livre, tais como são administrados nas formulações comercializadas clássicas, estão disponíveis para atingir o sangue a partir do local de administração, podendo, portanto, atingir outros tecidos que não os afetados pelos microorganismos causadores das doenças supracitadas e pelas células tumorais. O fármaco na forma nanoencapsulada fica mais restrito aos sítios de ação no tecido e nas células alvo da infecção pelos agentes causadores dessas doenças infecciosas e nas massas celulares tumorais. Graças aos componentes das formulações, sob tema desta invenção e do método de produção das formulações nanoestruturadas aqui apresentadas, os bioativos associados a essas nanocápsulas poliméricas, podem assim atingir especificamente o alvo, e serem liberados lentamente no sítio de ação, evitando perdas por inativações dos bioativos em outros tecidos não-alvo do tratamento. Desta maneira, as doses podem ser reduzidas o que reduz os efeitos adversos e aumenta a eficácia devido ao direcionamento preferencial para os sítios acometidos por essas doenças no corpo.
BACKGROUND DA INVENÇÃO
[005] Um grande número de lactonas sesquiterpênicas e suas origens são descritas em “The Biogenesis and Chemistry of Sequiterpene Lactones,” in W.Herz et al., (eds.), Prog. Chem. Org. Nat. Prod. Springer-Verlag, vol.38, pp. 47-390 (1979). As lactonas sesquiterpênicas contêm grupos funcionais alquilantes responsáveis por suas principais ações biológicas, bem como por seus efeitos tóxicos tais como α, β-ciclopentenona insaturada e a a-metileno—Y— lactona (Rodriguez et al., Phytochemistry, 15, 1573-158, 1976 e Picman Biochemical Systematics and Ecology, 14, 225-281, 1986) que atuam alquilando nucleófilos biológicos, tais como L—cisteína ou enzimas contendo sulfidrilas por adição do tipo Michael (Schmidt, Bioorg. Med. Chem., 5, 645,1997). O mecanismo de ação está provavelmente associado à inibição de enzimas lisossomais, que conduzem por sua vez, à inibição da síntese do DNA e, portanto, à diminuição do crescimento anárquico de células neoplásicas, bem como a multiplicação de microorganismos unicelulares, tais como os protozoários da família Tripanosomatidae. A presença de outros grupos funcionais tais como epóxido, hidroxila, cloridrina, e ésteres também podem ser responsáveis pela ação biológica das lactonas sesquiterpênicas. No entanto, outros fatores como a lipofilicidade e geometria molecular, também podem influenciar a atividade das lactonas sesquiterpênicas (Schmidt, Bioorg. Med. Chem., 5, 645,1997).
[006] A literatura científica é abundante em estudos que descrevem a atividade antitumoral desta classe de compostos. Entre eles estão; Lee et al.,(Science, 196, 533—536, 1977) que sugeriram que a ciclopentenona, presente em guaianolidas, também uma lactona sesquiterpênica, é fundamental para atividade citotóxica in vitro contra células tumorais. Kupchan et al., (Journal of Medicinal Chemistry, 14, 1147-1152,1971) relatou a relação estrutura-atividade de várias lactonas sesquiterpênicas citotóxicas, mostrando que o grupamento α-metileno-Y-lactona é essencial para a atividade citotóxica dentre outros grupos tais como, ésteres insaturados e ciclopentenona.
[007] Dentre outros estudos que relatam a atividade antitumoral estão: Cassady, (Journal of Medicinal Chemistry, 21, 815-819,1978), Howie et al., (Journal of Medicinal Chemistry, 19, 309-313, 1976), Saúde, (Estudo químico e atividade tripanossomicida de Lychnophora trichocarpha Spreng. Dissertação de Mestrado-Programa de Pós-Graduação em Química, UFMG, 136 p., 1994), Waddel et al., (Journal of Pharmaceutical Sciences, 68, 715-718, 1979). A patente européia WO 9506476 intitulada: “Métodos e composicões para tratamento de disordens neoplásicas” trata de lactonas sesquitepênicas da classe dos guaianolidas. Outra patente européia, WO 0267964 A1, trata da composição e métodos para prevenção de câncer e tratamento derivado de Inula britanica (família Asteraceae), com efeitos apoptóticos relacionados a duas lactonas sesquiterpênicas isoladas: 1-0-acetil-britanilactona ou 1,6-0,0- diacetilbritanillactona. A patente, WO 2006134606 A2, relata a atividade antitumoral de lactonas sesquitêpenicas, isoladas da planta Sphaeranthus indicus (família Asteraceae), administradas em formulação farmacêutica oral e injetável.
[008] Dentre outras atividades das lactonas sesquiterpênicas destacam-se ainda a atividade antimicrobiana, especialmente a atividade antibacteriana, descrita por Giesbrescht et al., (Química Nova, 13, 312-314, 1990) e Saúde et al,.(Revista Brasileira de Farmacognosia, 12(1), 7-10, 2002); e a atividade frente a diferentes protozoários, tais como a dos compostos sesquiterpenóides, germacranolida ou guaianolida em formulações farmacêuticas para tratamento das leismanioses (WO 0158888 A1) e com atividade anti-T. cruzi in vitro relatados por Chiari et al., (Transactions of the Royal Society of Tropical Medicine and Hygiene, 85(3), 372-374, 1991) e Oliveira et al., (Phytotherapy Research, 10(4), 292-295, 1996).
[009] Existe na terapêutica atual um exemplo importante de fármaco ativo contra os protozoários causadores da malária (família Plasmodiidae) e também ativos contra certas formas de tumores, como é o caso da lactona sesquiterpênica, artemisinina e seus derivados. Esse fato reforça a existência de mecanismos de ação para esta classe de substâncias que são comuns para a inibição da multiplicação celular, tanto em parasitos quanto em células neoplásicas (He et al., PLoS ONE, 6 (8), 2011; Mullard, Nature Reviews Drug Discovery, 10 (2), 82-85, 2011; Ghantous, et al. Drug Discovery Today, 15, 668-678, 2010).
[0010] Destaca-se também a atividade anti-inflamatória (Abad, et al. Planta Med., 60(3), 228-231, 1994) relacionada às lactonas sesquiterpênicas, como relacionada nas patentes americana (US 5,905,089) e na patente européia (WO 2007053271 A2) que descrevem formulações de extrato de Cichorium intybus L. que demonstra elevada eficácia antiinflamatória in vivo e in vitro.
[0011] As lactonas sesquiterpênicas apresentam elevado interesse também como potenciais substâncias químicas para tratamento do câncer. Algumas revisões sobre o tema situam a importância desta classe de compostos em estudos pré-clínicos e clínicos (Ghantous et al., What made sesquiterpene lactones reach cancer clinical trials? Drug Discovery Today 15,668-678, 2010). As lactonas sesquiterpênicas são promissoras candidatas na descoberta de novos fármacos para o câncer devido à sua maior seletividade para células tumorais e para células tronco, em fase acelerada de mitose, o que as coloca em posição privilegiada para uso clínico. Entretanto, certas desvantagens devem ser contornadas antes do uso desta classe de compostos; tais como a obtenção de fontes naturais, a baixa biodisponibilidade devido à alta lipolificidade, as fortes interações com proteínas plasmáticas, perfis toxicológicos desfavoráveis e interação inespecífica com grupamentos tióis de enzimas e proteínas vitais (Ghantous, et al. Drug Discovery Today, 15, 668678, 2010).
[0012] Deste modo é importante a associação das lactonas sesquiterpênicas furanoeliangolidos com sistemas carreadores de fármacos tecnologicamente delineados para melhorar a biodisponibilidade dessas sustâncias, redução das inativações e das reações inespecíficas durante o trajeto até o alvo biológico e da toxicidade. A associação dos furanoeliangolidos com carreadores de fármacos, como descrito nesta invenção, pode aumentar a interação dessas substâncias com os tecidos alvo da ação, sejam eles tecidos infectados com parasitas da família Trypanosomatidae, particularmente os causadores da doença de Chagas, das Leishmanioses e da Tripanossomíase Africana ou podem também ser utilizados no tratamento de tumores. O desenvolvimento tecnológico de formulações adequadas para encapsulamento de lactonas sesquiterpênicas furanoeliangolidos é primordial para melhoria da atividade biológica geral desses compostos.
[0013] A família Trypanosomatidae inclui parasitas de elevada relevância médica e veterinária. Entre os principais agentes causadores de doenças com importância epidemiológica mundial nos seres humanos e nos animais, podemos citar: 1) Trypanosoma cruzi, causador da doença de Chagas ou Tripanossomíase Americana; 2) Trypanosoma brucei, causador da doença do sono ou Tripanossomíase Africana, cujos agentes etiológicos são atualmente considerados como subespécie do T. brucei: T.brucei gambiense, T.brucei rhodesiense, T.brucei evansi, T.brucei brucei, T.brucei equiperdum. 3) As espécies do gênero Leishmania que são responsáveis pelas leishmanioses em todo mundo. Devido a caracteres epidemiológicos, clínicos, imunológicos, e genéticos as leishmânias são agrupadas em “complexos” assim: Complexo “Leishmania brasiliensis”: compreendendo as espécies: L. braziliensis, L. guyanensis, L. panamensis; complexo “Leishmania mexicana”: abrangendo as espécies: L. mexicana, L. pifanoi, L. amazonensis, L. venezuelensis e L. garnhame e complexo “Leishmania donovani”: com L. donovani e L.infantum (mesma espécie de L. chagasi).
[0014] Existe um grande número de compostos empregados como quimioterápicos para doenças infecto-contagiosas causadas por protozoários, dentre elas a doença de Chagas, doença do sono e as leishmanioses. Entretanto, estas espécies de parasitos apresentam resistência natural à grande parte das substâncias atualmente utilizadas na terapêutica ou desenvolvem resistência ao tratamento sob a pressão medicamentosa, o que leva à necessidade constante de descoberta de novos agentes quimioterápicos.
[0015] A doença de Chagas é uma doença parasitária causada pelo protozoário Trypanosoma cruzi, doença tropical exclusiva do continente americano, presente desde o sul dos Estados Unidos até Argentina, especialmente na América Latina e representa uma das seis maiores doenças tropicais. Segundo a Organização Mundial de Saúde, são estimados entre 12-14 milhões de pessoas infectadas pelo T. cruzi na América, principalmente em países americanos de colonização ibérica (WHO, 2009). A incidência anual é de 41.200 novos casos e 12.500 mortes ocorrem como conseqüência dos danos irreversíveis ao coração e ao trato digestivo. Uma população em torno de 28 milhões vive atualmente em áreas de risco (WHO, 2009). Estudos mais recentes revelam que a prevalência da doença de Chagas se estende além dos países endêmicos, o que é resultante principalmente da imigração de latino americanos e da transfusão sanguínea (Schimunis, Mem. Inst. Oswaldo Cruz 102 (Suppl.I), 75-85 2007). O T. cruzi apresenta uma grande diversidade genética, sendo divididos em seis grupos ou DTUs (discrete typing units) (T. cruzi I a T. cruzi VI) aparentemente associados as suas características epidemiológicas, biológicas bem como a susceptibilidade ou resistência a fármacos (Zingales, et al. Mem Inst Oswaldo Cruz., 104(7):1051-4; 2009). Um ponto agravante desta endemia do ponto de vista social está relacionado à sua elevada prevalência, principalmente na população economicamente menos favorecida, e extensa distribuição geográfica dos parasitos e vetores, somadas à alta morbidade e letalidade observadas na forma clínica cardíaca (Uchoa et al. Caderno de Saúde Pública, 18(1), 71-79, 2002). O principal mecanismo de transmissão desta doença se dá por insetos vetores ou triatomíneos da família Reduvidae e ocorre por contaminação com tripomastigotas metacíclicos presentes nas suas fezes ou urina eliminados durante ou após o repasto sanguíneo. Sua transmissão se dá ainda por transfusão sanguínea, congênita e oral, dentre outros mecanismos de menor importância epidemiológica, tais como transplantes e acidentes laboratoriais. Após a infecção e um subsequente período de incubação, tem-se início a fase aguda da doença de Chagas. Na ausência de tratamento específico, seus sintomas podem persistir por até quatro meses, com taxa de mortalidade variando de 2-8%, principalmente em crianças. Em casos mais graves surgem à miocardite intensa e a meningoencefalite, muito associadas à mortalidade desta doença. Essa fase da doença é muitas vezes imperceptível ou oligossintomática na grande maioria dos casos, com manifestações clínicas diversas tais como: febre, edema, mialgia, hipertrofia dos linfonodos, hepatomegalia, esplenomegalia, insuficiência cardíaca e sintomas neurológicos. Nesta fase ocorrem os fenômenos de porta de entrada inicial do parasito, tais como o edema palpebral (sinal de Romana) e subcutâneo (chagoma de inoculação), que auxiliam muito na suspeita clínica da doença de Chagas. Com a instalação da resposta imune do hospedeiro, as manifestações da fase aguda regridem e instala-se gradativamente a fase crônica, na qual a parasitemia e o parasitismo são escassos, o que persiste por toda a vida do indivíduo.
[0016] Já a fase crônica pode ser subdividida em forma indeterminada, cardíaca, digestiva ou mista. A forma indeterminada é definida como um estado no qual o indivíduo apresenta-se assintomático durante cerca de 10 a 20 anos, ou mesmo pelo resto de sua vida. Cerca 30 a 40% dos indivíduos infectados evoluem para as formas sintomáticas da doença de Chagas crônica. A forma cardíaca que ocorre em cerca de 30% casos é caracterizada por sinais e sintomas de insuficiência cardíaca e alterações eletrocardiográficas de gravidade variável. Ela é responsável pela maioria dos óbitos decorrentes da infecção chagásica, sendo considerada a forma clínica mais grave da doença pela ocorrência freqüente do fenômeno de morte súbita. A forma digestiva (10% dos casos) é caracterizada por alterações morfológicas e funcionais do esôfago e do cólon, que levam a dificuldades na deglutição, peristaltismo e defecação. Pacientes com a forma mista apresentam sintomatologia cardíaca e digestiva associadas (Prata, Lancet, 1(2), 92-100, 2001). Os fatores envolvidos na patogênese da doença de Chagas ainda não são completamente conhecidos e durante muitos anos os estudos referentes à eficácia de substâncias químicas frente ao parasito foram relegados a segundo plano, sendo a autoimunidade considerada o principal mecanismo responsável pelo desencadeamento das lesões que acometem os indivíduos infectados (Cunha- Neto, et al. Proc. Natl. Acad. Sci. U.S.A 92, 3541-3545, 1995). Até meados da década de 90, o tratamento específico era indicado principalmente nos casos agudos (The National Health Foudantion of Brazil, 1996). Entretanto, com as crescentes evidências de que a persistência do parasito representa um fator importante no desenvolvimento da doença (Tarleton, Trends Parasitol. 19, 447451, 2003) e que o tratamento atua beneficamente no prognóstico e evolução clínica, mesmo nos casos não curados (Andrade et al. Mem. Inst. Oswaldo Cruz 86, 187-200, 1991 e Viotti et al., Ann. Intern. Med. 144, 724-734, 2006), vários grupos de especialistas em quimioterapia da doença de passaram a recomendá-lo a todo paciente soropositivo, em ambas as fases de infecção, com o objetivo de erradicá-la, evitar o agravamento das lesões e consequentemente auxiliar na interrupção da transmissão do parasito (Consenso Brasileiro em Doença de, 2005).
[0017] No tratamento etiológico da doença de Chagas apenas dois compostos têm sido utilizados: o (Rochagan® da Roche) e o nifurtimox (Lampit® da Bayer). O primeiro teve sua produção interrompida pela Roche ficando o Laboratório LAFEPE em PE, Brasil, responsável pela sua produção. Atualmente o único medicamento utilizado no Brasil é o produzido pelo Laboratório Público, LAFEPE. O nifurtimox também teve sua produção descontinuada. Ambos apresentam eficácia terapêutica em 50 a 100% dos casos apenas na fase aguda da doença e em infecções recentes (menos de cinco anos). Entretanto, não existe no momento medicamentos com boa eficácia para utilização na fase crônica tardia da doença, presente na grande maioria da população infectada. No Brasil, por exemplo, os resultados de tratamento humano nesta fase variaram de 0 a 19% (Coura, J.R. e Castro, S.L. Mem. Inst. Oswaldo Cruz, 97(1), 3-24, 2002.). O efeito do nifurtimox nesses casos é parcial (0 a 19%) em função da resistência natural das diferentes populações do parasito ao tratamento.
[0018] O benzonidazol é um composto heterocíclico bastante tóxico, especialmente, quando administrado em esquema posológico prolongado, como o recomendado no caso da doença de Chagas. Os índices terapêuticos do benzonidazol são baixos e este fármaco é recomendado em doses diárias inferiores a 300 mg/dia devido ao pequeno índice terapêutico, ou seja, a dose preconizada para um adulto é de 5 mg/Kg/dia até 60 dias. Se por ventura, um adulto tiver que fazer uso de mais que 300 mg/dia em função de seu peso elevado, a duração do tratamento deverá aumentar, ou seja ir além dos 60 dias, mas jamais superar os 300 mg/dia. Portanto, o tratamento exige cuidadosa atenção para o manejo de reações adversas que ocorrem em cerca de 30 a 40% dos pacientes, em gravidade variável. Os principais efeitos adversos são as freqüentes manifestações cutâneas, tais como hipersensibilidade, dermatite com erupções cutâneas, edema generalizado, além de manifestações generalizadas, tais como febre, a linfoadenopatia, dor articular e muscular, depressão da medula óssea (neutropenia, agranulocitose e púrpura trombocitopênica) e polineuropatia periférica. Em alguns pacientes, tais reações adversas são intensas exigindo a interrupção temporária ou definitiva do tratamento, em função do quadro clínico e laboratorial.
[0019] Além do quadro de reações adversas descrito acima com a terapia preconizada para a doença de Chagas acima supracitada, outro grande problema encontrado na quimioterapia atual é a dificuldade de eliminação do T. cruzi, em grande parte dos indivíduos infectados. A localização intracelular do parasita na forma amastigota se constitui na mais difícil etapa para ação dos fármacos, pois as concentrações intracelulares são geralmente insuficientes para eliminação total do parasito, mas suficientes para indução dos efeitos tóxicos.
[0020] Neste cenário complexo, novas possibilidades terapêuticas com substâncias ativas formuladas para tratamento dos pacientes na fase crônica da doença de Chagas, com vistas à cura parasitológica, até então dificilmente alcançada são urgentemente necessárias, visando à redução da sintomatologia e melhoria do prognóstico da doença (Coura e Castro, Mem. Inst. Oswaldo Cruz, 97(1), 3-24, 2002). Esta urgência é reforçada por descobertas relativamente recentes de grande ocorrência de casos novos da doença de Chagas decorrentes das atividades extrativistas existentes na região amazônica (Coura, Rev Soc Bras Med Trop, 39 (Suppl 3), 113-117, 2006) e do grande número de indivíduos infectados em outros países como conseqüência do fenômeno migratório (Schmunis, Mem. Inst. Oswaldo Cruz 102 (Suppl.I), 7585, 2007). A conhecida existência de um grande contingente de indivíduos infectados são também motivações para o interesse e descoberta de novos fármacos para esta enfermidade, além também de outras doenças causadas por parasitos da família Trypanosomatidae, tais como a Leishmaniose e a Tripanossomíase Africana.
[0021] Por outro lado, outra aplicação da invenção relatada neste documento é o tratamento do câncer. O câncer é o resultado de uma série de alterações nos mecanismos de reprodução celular que controlam seu crescimento. Essas alterações provocam crescimento celular desordenado e contínuo, inferindo nessas células anormais a capacidade de invadir tecidos e órgãos adjacentes, bem como a capacidade de migrar e se alojar em outras regiões do organismo (metástase). O câncer pode surgir de qualquer tipo de célula e de qualquer tipo de tecido, não sendo uma doença única, mas um grande número de doenças caracterizadas pelo seu local de origem. Dividindo-se rapidamente, estas células anormais tendem a ser muito agressivas e incontroláveis, determinando a formação de tumores ou neoplasias (acúmulo de células cancerosas). Existem centenas de formas distintas, sendo três os principais subtipos: os sarcomas, os carcinomas e os linfomas (leucemias). O câncer é a segunda maior causa de morte de adultos no mundo ocidental e é uma das principais causas de morte por doença em crianças de 1 a 14 anos. Segundo dados da Organização Mundial da Saúde, 2009, aproximadamente 7,6 milhões de pessoas morreram no mundo vítimas de alguma neoplasia. É um crescente problema de saúde pública mundial com incidência de 6 milhões de novos casos a cada ano. A terapia do câncer pode envolver uso de tratamento múltiplo incluindo: cirurgia, radioterapia, quimioterapia, imunoterapia e ainda terapias alternativas. O determinante para a escolha do tratamento dependerá do local, tamanho e fase do tumor, bem como o estado de saúde global do paciente.
[0022] O tratamento do câncer tem sido o principal objetivo da pesquisa e desenvolvimento de compostos e formulações pelas indústrias farmacêuticas mundiais nas últimas três décadas. Como resultado, muitas abordagens na terapia do câncer têm sido descobertas e investigadas. Neste contexto, fármacos com reconhecida atividade anti-T. cruzi, como o nifurtimox, um nitroderivado heterocíclico, utilizado no tratamento da doença de Chagas desde a década de 70, foi proposto na patente (WO 2007108947 A2) como medicamento para tratamento do câncer, especialmente de neuroblastoma, e como inibidores da angiogênese. Na presente invenção também as substâncias testadas in vivo com eficácia contra o T. cruzi, foram testadas in vitro em diferentes linhagens de células tumorais e também tiveram atividade comprovada em várias delas. (vide Tabela 13.).
[0023] Nas últimas três décadas, tem havido notável aumento do uso de plantas medicinais para o tratamento de várias doenças (Newman e Cragg, J. Nat. Prod., 70, 461-467, 2007).
[0024] A descoberta do taxol, da vincristina, da vimblastina, da camptotecina, substâncias de origem natural, com ação antitumoral, representou um grande avanço para o tratamento do câncer utilizando produtos naturais. Entretanto, esses compostos também apresentam efeitos adversos freqüentes, tais como: alopecia, depressão da medula óssea, bradicardia, hipersensibilidade, disfunção hepática dentre outros.
[0025] Para superar esses desafios, é necessário o desenvolvimento tecnológico de formulações farmacêuticas contendo compostos de origem natural, com amplo espectro de ação antineoplásica, mais que sejam mais seletivos, mais eficazes para vários tipos de tumor como, por exemplo: o câncer de mama, o câncer de próstata, tumores de cólon, neuroblastoma, linfomas, tumores de fígado, pele e as leucemias.
[0026] Existem atualmente limitadas opções para administração de substâncias altamente lipofílicas, como é o caso das lactonas sesquiterpênicas, tanto por via oral, quanto por via parenteral, devido à baixa solubilidade aquosa desses compostos. Esta classe de compostos apresenta, portanto, problemas de solubilidade, os quais reduzem a biodisponibilidade e muitas vezes mascarando a atividade destas substâncias in vivo, devido a um perfil farmacocinético desfavorável. Sendo assim, as formulações farmacêuticas tecnologicamente desenvolvidas nesta invenção para associação efetiva de lactonas sesquiterpênicas da classe dos furanoeliangolidos, podem ser classificadas como nanocápsulas de tamanho submicrométrico para serem administrados por diferentes vias, a saber: via parenteral (via intravenosa, intramuscular, intradérmica e subcutânea), oral, tópica, retal, inalatória, intranasal, transdérmica, bucal dentre outras. A descrição técnica desta invenção compreende principalmente formulações constituídas de polímeros biocompatíveis e alguns deles biodegradáveis, onde as substâncias ativas estão associadas e dispersas tanto em membranas poliméricas quanto em fases oleosas ou pastosas graxas localizadas no interior de cápsulas de tamanho superior a 50 nm e inferior a 1000nm, as quais podem ser chamadas nanocápsulas poliméricas. Fazem parte também das reinvidicações desta invenção as misturas de solventes biocompatíveis em proporções apropriadas para a solubilização das lactonas sesquiterpênicas furanoeliangolidos listados nesta invenção, os quais foram utilizados nos estudos pré-clínicos para solubilização das substâncias bioativas para efeito de comparação com os efeitos biológicos das formulações nanoestrutradas.
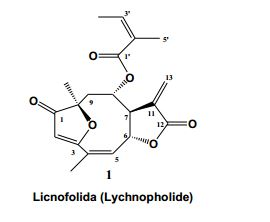
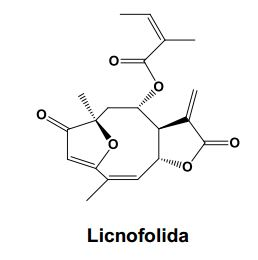
[0027] Conceitualmente as nanocápsulas são constituídas por um núcleo oleoso, envolto por uma parede polimérica com surfactantes lipofílicos e/ou hidrofílicos localizados na interface entre a fase aquosa da superfície das nanocápsulas (Legrand et al. S.T.P. Pharma Sci., 9, 411-418,1999). Estas formulações são a escolha para administração intravenosa de substâncias lipofílicas, e podem ser formadas por polímeros estáveis com baixa toxicidade que consistem de materiais biocompatíveis e biodegradáveis, os quais são degradados in vivo em fragmentos menores e facilmente excretados. As nanocápsulas foram descritas pela primeira vez na patente francesa FR2608942A1. As nanocápsulas presentes nesta invenção apresentam elevada capacidade de encapsulamento de lactonas sesquiterpênicas furanoeliangolidos, como exemplificado para a substância licnofolida representada abaixo (1).
[0028] De acordo com Kupchan e colaboradores, 1971, a presença da porção a-metileno-y-lactona é essencial para a atividade citotóxica dessa classe de substâncias, e que a presença de grupos tais como ésteres insaturados, ciclopentenona ou α-metileno-δ-lactona aumentam a citotoxicidade. O mecanismo das atividades citotóxica e antitumoral ocorre através de reações de a-metileno-y-lactonas e outros sistemas conjugados com grupos sulfidrila das enzimas que controlam a divisão celular, inibindo assim a síntese de proteínas e do DNA (Kupchan et al. Journal of Medicinal Chemistry, 14, 11471152,1971) Os agentes ativos da presente invenção tem uma estrutura da a- metileno-y-lactona (I) e da furanona (II) como grupos responsáveis pela atividade biológica, como representado abaixo.
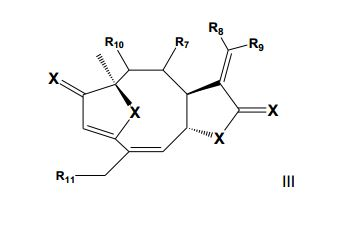
[0029] As substâncias químicas ativas associadas as nanocápsulas com as atividades biológicas descritas nesta invenção podem ser quaisquer entidades químicas com estrutura química I ou II, quando cada ou qualquer dos R1,R2, R3, R4, R5 e R6 é (1) um substituinte selecionado a partir de grupos consistindo de hidrogênio (-H), halogênio (por exemplo, flúor, cloro ou bromo), hidroxila (-OH), alcoxila (-OR’), acila (-COR’) , carboxila (-CO2H), ésteres carboxílicos (-CO2R’), amida ((-CONR2’), amina (-NR2’), nitro (-NO2), nitroso (-NO), azo (N=N-), diazônio (-N2+), azida (-N3), hidrazina (-NR’-NR’2), ciano (-CN), isociano (CN-), cianato (NCO-), isocianato (OCN-), tioéter (-SR’), tiol (-SH), sulfóxido (-SOR’), sulfona (S(O)2R’), ácido sulfônico (HO3S-), ésteres sulfonila (R’O3S-), ácido sulfínico (HO2S-), ésteres sulfinila (R’O2S-), ácido sulfênico (HOS-), ésteres sulfenila (R’OS-), onde R’ é um grupo alquil, alquenil ou alquinil de 1 a 5 carbonos; (2) um radical hidrocarboneto insaturado, alifático saturado, alicíclico ou aromático possuindo de 1 a 50 átomos de carbono, preferencialmente 1 a 25 átomos de carbono, e mais preferencialmente 1 a 20 átomos de carbono, que podem ser substituído com um ou mais dos substituintes de (1) acima, (3) um grupo heterocíclico possuindo em torno de 1 a 13 átomos de carbono; (4) um resíduo glicosídeo; ou (5) um resíduo peptídeo. Dois ou mais dos grupos R1, R2, R3, R4, R5 e R6 podem estar combinados a qualquer forma das listadas partes cíclicas listadas acima (I e II). Os substituintes podem estar arranjados na forma cíclica ou acíclica, tão longo o farmacóforo furanona e a-metileno-y- lactona retenham as atividades descritas nesta patente. O átomo ou grupamento representado por “X” pode ser oxigênio (O), enxofre (S), nitrogênio (N), iminas (NR’) ou hidrazonas (NR’-NR’), onde R’ é um grupo alquil, alquenil ou alquinil de 1-5 carbonos.
[0030] Agentes ativos da presente invenção têm como grupos ativos a a- metileno-y-lactona, a furanona e ésteres conjugados e são preferencialmente substâncias contendo a estrutura do furanoeliangolido III, representada abaixo:
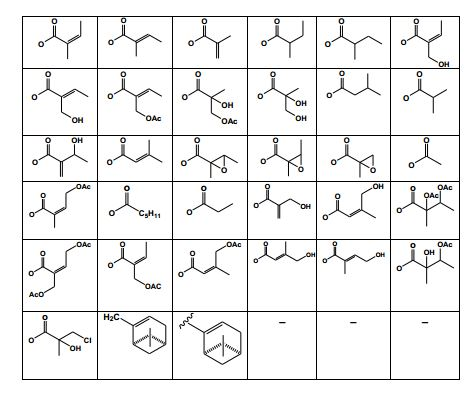
[0031] Assim sendo, podem ser considerados substâncias ativas desta invenção quaisquer umas das estruturas químicas contendo a estrutura química III, quando cada ou qualquer dos R7, R8, R9, R10 e R11 é (1) um substituinte selecionado a partir de grupos consistindo de hidrogênio (-H), halogênio (por exemplo, flúor, cloro ou bromo), hidroxila (-OH), alcoxila (OR’), acila (-COR’), carboxila (-CO2H), aldeídos (CHO), ésteres carboxílicos (- CO2R’), amida (CONR2’), amina (-NR2’), nitro (-NO2), nitroso (-NO), azo (-N=N- ), diazônio (-N2+), azida (-N3), hidrazina (-NR’-NR’2), ciano (-CN), isociano (CN- ), cianato (NCO-), isocianato (OCN-), tioéter (-SR’), tiol (-SH), sulfóxido (-SOR’), sulfona (-S(O)2R’), ácido sulfônico (HO3S-), ésteres sulfonila (R’O3S-), ácido sulfínico (HO2S-), ésteres sulfinila (R’O2S-), ácido sulfênico (HOS-), ésteres sulfenila (R’OS-), onde R’ é um grupo alquil, alquenil ou alquinil de 1 a 5 carbonos e os éteres seguintes: (-OCH2CH3, -OCH3), e também os seguintes ésteres: CO2CH3, CH3CH2CH2CH2CH2CH2CH2CO2-, CH3CH2CH2CH2CH2CH2CH2CH2CH2CO2-, CH3CH2CH2CH2CH2CH2CH2CH2CH2CH2CH2CO2-, CH3CH2CH2CH2CH2CH2CH2CH2CH2CH2CH2CH2CH2CO2-, CH3CH2CH2CH2CH2CH2CH2CH2CH2CH2CH2CH2CH2CH2CH2CO2-, CH3CH2CH2CH2CH2CH2CH2CH2CH2CH2CH2CH2CH2CH2CH2CH2CH2CO2-, CH3CH2CH2CH2CH2CH2CH2CH2CH=CHCH2CH2CH2CH2CH2CH2CH2CO2-, CH3CH2CH2CH2CH2CH2CH2CH2CH=CHCH2CH=CHCH2CH2CH2CH2CO2-, CH3CH2CH2CH2CH2CH=CHCH2CH=CHCH2CH=CHCH2CH2CH2CH2CO2-, CH3CH2CH2CH2CH2CH2CH2CH2CH=CHCH2CHOHCH2CH2CH2CH2CH2CO2-, CH3CH2CH2CH2CH2CH2CH2CH2CH2CH2CH2CH2CH2CH2CH2CH2CH2CH2CH2C O2-, CH3CH2CH2CH2CH2CH2CH2CH2CH2CH2CH2CH2CH2CH2CH2CH2CH2CH2CH2C H2CH2CO2-),
[0032] Para a estrutura química III, R7, R8, R9, R10 e R11 podem ser cada ou qualquer uma das opções dadas a seguir:
[0033] (2) um radical hidrocarboneto insaturado, alifático saturado, alicíclico ou aromático possuindo de 1 a 50 átomos de carbono, preferencialmente 1 a 30 átomos de carbono, e mais preferencialmente 1 a 20 átomos de carbono, que podem ser substituídos com um ou mais dos substituintes de (1) acima, (3) um grupo heterocíclico possuindo em torno de 1 a 20 átomos de carbono; (4) um resíduo glicosídeo; ou (5) um resíduo peptídeo. Dois ou mais dos grupos R1, R2, R3, R4, R5 e R6 podem estar combinados a qualquer forma das partes cíclicas listadas acima. Os substituintes podem estar arranjados na forma cíclica ou acíclica, na condição que a parte ativa da molécula centrada nos grupamentos α-metileno-y-lactona, furanona e ésteres conjugados mantenham as atividades biológicas descritas nesta patente. Para a estrutura química III, “X” pode ser oxigênio (O), enxofre (S), nitrogênio (N), iminas (NR’) ou hidrazonas (NR’-NR’), onde R’ é um grupo alquil, alquenil ou alquinil de 1-5 carbonos.
[0034] A presente invenção descreve o uso no tratamento da doença de Chagas experimental em camundongos e também em estudos in vitro em linhagens de células tumorais, de substância pertencente à classe das lactonas sesquiterpênicas do tipo furanoeliangolidos, descritos nesta invenção, particularmente a licnofolida (1), em um produto farmacêutico, formulado na forma de nanocápsulas poliméricas biodegradáveis, que aumentam a eficácia terapêutica dessas substâncias no tratamento de infecções por protozoários, particularmente Tripanossomíases e neoplasias. A atividade farmacológica exemplificada nesta invenção foi alcançada pelo uso da lactona sesquiterpênica do tipo furanoeliangolido, especialmente a licnofolida conforme estrutura química 1 e também com seus derivados, estruturas químicas de 2 a 77.
[0035] A nomenclatura da licnofolida (1), ou do licnofolídeo (1), conforme a IUPAC é: 2’-metil-2’-butenoato de 2, 3, 3a, 4, 5, 6, 7, 11a-octaidro-6,10-dimetil- 3-metileno-2,7-dioxo-6,9-epoxiciclodeca[b]-furan-4-ila [3aR*, 4S*(z), 6R*, 10Z, 11aR*].
[0036] Esta substância se apresenta na forma de cristais brancos, com ponto de fusão: 128-1290C. Recristalizado em etanol (Saúde-Guimarães, Transformações químicas, microbiológicas e atividades biológicas de lactonas sesquiterpênicas. Belo Horizonte. Tese (doutorado) UFMG,1998). Literatura: 1280C (Bohlmann, et al. Phytochemistry, 19(11), 2381-2385, 1980.) O Espectro no IV (KBr) v máx. (cm-1): 2900 (CH), 1770 (C=O de y-lactona), 1710 (C=O de cetona conjugada), 1660 (C=C), 1590 (C=C-OR, furanona), 1450, 1370, 1350, 1300, 1270, 1230, 1140, 1100, 1040, 1030, 950, 920, 880, 850, 820, 760 (Saúde-Guimarães, 1998). Por espectrometria de massas são obtidos: EMIE, m/z (int. rel.): 358 (M+, C20H22O6, 5), 295 (2), 275 (M - COC4H7, 6), 258 (M - C4H7CO2H, 2), 239 (2), 232 (10), 220 (5), 206 (5), 189 (5), 167 (10), 155 (5), 149 (34), 141 (5), 127 (7), 113 (12), 97 (20), 85 (41), 83 (C4H7CO+, 81), 71 (66), 57 (100). (Saúde-Guimarães, 1998).
[0037] RMN de 1H (CDCl3, 400 MHz):5,72 (s, H-2); 6,01-6,00 (m, J = 1,6; 2,8 Hz, H-5); 5,31-5,29 (m, H-6); 3,73-3,71 (m, H-7); 4,52 (ddd, J = 2,0; 2,4; 12,0 Hz, H-8); 2,30 (dd, J = 2,0; 14,0 Hz, H-9a); 2,49 (dd, J = 11,6; 13,6 Hz, H-9b); 6,21 (d, J = 2,8 Hz, H-13a); 5,44 (d, J = 2,8 Hz, H-13b); 1,53 (s, H-14); 2,08 (t, J = 2,0 Hz, H-15); 6,10 (qq, J = 1,6; 7,2 Hz, H-3’); 1,88 (dq, J = 1,4; 7,3 Hz); 1,78 (t, J = 1,6 Hz) (Saúde-Guimarães, 1998).
[0038] RMN de 13C (CDCl3, 75 MHz): 204,94 (C-1), 104,82 (C-2), 186,98 (C-3), 130,43 (C-4), 135,14 (C-5), 81,77 (C-6), 51,27 (C-7), 73,08 (C-8), 44,12 (C-9), 89,78 (C-10), 133,84 (C-11), 168,98 (C-12), 124,35 (C-13), 20,78 (C-14), 20,41 (C-15), 167,16 (C-1’), 126,50 (C-2’), 140,80 (C-3’), 20,11 (C-4’), 15,78 (C-5’) (Saúde-Guimarães, 1998).
[0039] A substância ativa compreende não apenas a substância química 1, mas também sesquiterpenóides com qualquer uma das estruturas químicas e substituintes específicos representados nas fórmulas 2 a 77.
[0040] Estão reivindicados nesta invenção substâncias químicas ativas pertencentes à classe dos sesquiterpenóides, especificamente a estrutura 1, como também qualquer estrutura química e substituintes específicos representados nas fórmulas de 2 a 77, quando cada ou qualquer dos R1, R2, R3 e R4 para (1) um substituinte selecionado a partir de grupos consistindo de hidrogênio (-H), halogênio (por exemplo flúor, cloro ou bromo), hidroxila (-OH), alcoxila (-OR’), acila (-COR’), carboxila (-CO2H), aldeídos (CHO), ésteres carboxílicos (-CO2R’), amida (-CONR2’), amina (-NR2’), nitro (-NO2), nitroso(- NO), azo (-N=N), diazônio (-N2+), azida (-N3), hidrazina (-NR’-NR’2), ciano (- CN), isociano (CN-), cianato (NCO-), isocianato (OCN-), tioéter (-SR’), tiol (- SH), sulfóxido (-SOR’), sulfona (-S(O)2R’), ácido sulfônico (HO3S-), ésteres sulfonila (R’O3S-), ácido sulfínico (HO2S-), ésteres sulfinila (R’O2S-), ácido sulfênico (HOS-), ésteres sulfenila (R’OS-), onde R’ é um grupo alquil, alquenil ou alquinil de 1 a 5 carbonos e os seguintes éteres: (-OCH2CH3, -OCH3), e também os seguintes ésteres: CO2CH3, CH3CH2CH2CH2CH2CH2CH2CO2-, CH3CH2CH2CH2CH2CH2CH2CH2CH2CO2-, CH3CH2CH2CH2CH2CH2CH2CH2CH2CH2CH2CO2-, CH3CH2CH2CH2CH2CH2CH2CH2CH2CH2CH2CH2CH2CO2-, CH3CH2CH2CH2CH2CH2CH2CH2CH2CH2CH2CH2CH2CH2CH2CO2-, CH3CH2CH2CH2CH2CH2CH2CH2CH2CH2CH2CH2CH2CH2CH2CH2CH2CO2-, CH3CH2CH2CH2CH2CH2CH2CH2CH=CHCH2CH2CH2CH2CH2CH2CH2CO2-, CH3CH2CH2CH2CH2CH2CH2CH2CH=CHCH2CH=CHCH2CH2CH2CH2CO2-, CH3CH2CH2CH2CH2CH=CHCH2CH=CHCH2CH=CHCH2CH2CH2CH2CO2-, CH3CH2CH2CH2CH2CH2CH2CH2CH=CHCH2CHOHCH2CH2CH2CH2CH2CO2- CH3CH2CH2CH2CH2CH2CH2CH2CH2CH2CH2CH2CH2CH2CH2CH2CH2CH2CH2C O2, CH3CH2CH2CH2CH2CH2CH2CH2CH2CH2CH2CH2CH2CH2CH2CH2CH2CH2CH2C H2CH2CO2-),
[0041] Para as estruturas químicas de 2 a 77, R1, R2, R3 e R4 podem ser cada ou qualquer uma das opções dadas a seguir:
[0042] (2) um radical hidrocarboneto insaturado, alifático saturado, alicíclico ou aromático possuindo de 1 a 50 átomos de carbono, preferencialmente 1 a 30 átomos de carbono, e mais preferencialmente 1 a 20 átomos de carbono, que podem ser substituídos com um ou mais dos substituintes de (1) acima, (3) um grupo heterocíclico possuindo em torno de 1 a 20 átomos de carbono; (4) um resíduo glicosídeo; ou (5) um resíduo peptídeo. Dois ou mais dos grupos R1, R2, R3 e R4, podem estar combinados a qualquer forma das partes cíclicas listadas acima. Os substituintes podem estar arranjados na forma cíclica ou acíclica, na condição que a parte ativa da molécula centrada nos grupamentos α-metileno-y-lactona, furanona e ésteres conjugados mantenham as atividades biológicas descritas nesta patente.
[0043] Para a preparação das nanocápsulas são utilizados polímeros geralmente na faixa de 0,2 a 4% (m/m) como determinado anteriormente pela patente EP 1531800 A2. Surfactantes hidrofílicos e lipofílicos também são utilizados, usualmente de 0,2 a 4%(m/m). Os óleos empregados podem ser de origem vegetal, sintético e mineral, devendo apresentar ausência de toxicidade, não serem capazes de degradar ou solubilizar o polímero e alta capacidade de dissolver a droga em questão (Legrand et al. S.T.P. Pharma Sci., 9, 411418,1999). As nanocápsulas podem ser classificados como convencionais ou estericamente estabilizados, também conhecidos como furtivos, pois escapam ao reconhecimento pela defesa imunológica do organismo. As convencionais possuem superfície mais hidrofóbica e por isso são facilmente reconhecidas pelo sistema de defesa do organismo, concentrando preferencialmente os fármacos encapsulados nos órgãos e células do sistema mononuclear fagocitário (SMF) (Mosqueira et al,. Biomaterials, 22, 2967-2979,2001). As nanocápsulas estericamente estabilizadas ou ditas furtivas possuem sua superfície modificada com cadeias de polietilenoglicol ligadas covalentemente à membrana hidrofóbica do polímero constituinte da parede da nanocápsula. Esta modificação superficial permite que quando injetadas por via intravenosa, estas sejam menos reconhecidas pelas células do SMF, o causa um prolongamento do tempo de residência dessas partículas na circulação sanguínea, as quais liberam lentamente, no compartimento plasmático, os princípios ativos encapsulados no seu interior. Tal liberação pode ser também prolongada em tecidos com alto fluxo sanguíneo (Mosqueira et al, Pharmaceutical Research, 2001). Esta é uma grande vantagem para o tratamento de infecções parasitárias onde os parasitas possuem um trânsito pelos vasos sanguíneos, como é o caso do Plasmodium spp. causador da malária, da doença de Chagas, da Tripanossomíase africana e nas metástases de tumores (Magalhães e Mosqueira, Adv. Drug. Deliver. Reviews, 2010). As nanocápsulas podem, através da regulagem tecnológica do tamanho nanométrico, serem direcionadas para alguns tecidos e liberarem de forma seletiva o fármaco em sítios de ação específicos. Essa é uma grande vantagem desta invenção, pois contribui para melhoria do regime de administração dos princípios ativos associados a essas nanocápsulas nesta invenção. Há também a conseqüente diminuição dos efeitos tóxicos obtidos pela alteração da biodistribuição da molécula ativa no organismo como já relatado por outros autores (Leite et al, Life Sciences, 80, 1327-1334, 2007). As nanocápsulas facilitam a penetração nas células hospedeiras de alguns parasitos, elevando a concentração intracelular dos ativos contra o agente infeccioso. Outra característica importante desses sistemas é a proteção das moléculas biologicamente ativas frente à degradação enzimática, química ou imunológica, levando a um aumento da resposta biológica com doses menores.
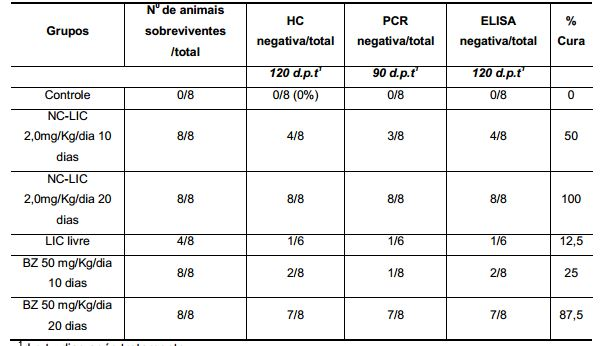
[0044] As nanocápsulas na presente invenção aumentaram a eficácia da licnofolida no tratamento da doença de Chagas experimental, como pode ser visto nas Tabelas sumarizadas 10, 11 e 12, mesmo em cepas de T. cruzi altamente resistentes a todos os compostos utilizados na quimioterapia atual, como o benzonidazol.
[0045] Além disso, esses compostos sesquiterpenóides do tipo furanoeliangolidos listados acima, foram testados em linhagens de células tumorais, especialmente a Licnofolida (1) no National Cancer Institute (NCI, Bethesda, Maryland, USA). Estes compostos foram submetidos a testes in vitro em 8 tipos de câncer e 60 linhagens de células tumorais humanas que determinaram o efeito antitumoral, expresso em CIC50 (concentração que inibe 50 % do crescimento das células tumorais em relação ao controle, em μg/mL), CIC100 (concentração que inibe 100 % do crescimento das células tumorais em relação ao controle, em μg/mL) e CL50 (concentração letal para 50 % das células tumorais em relação ao controle, em μg/mL).
[0046] O NCI considerou, para o composto (1), como concentrações mais significativas aquelas em que os valores de CL50 foram menores ou iguais a 4,3 μg/mL, de CIC100 menores ou iguais a 0,96 μg/mL e CIC50 menores ou iguais a 0,25 μg/mL (ver dados Tabela 13). O composto 1 apresentou valores de CL50 entre 0,3 e 5,0 μg/mL frente a 31 das 52 linhagens de células tumorais utilizadas no teste, valores de CIC100 entre 0,15 e 0,95 μg/mL contra 28 linhagens e valores de CIC50 entre 0,029 e 0,179 μg/mL frente a 24 linhagens (ver Tabela 13). Ressalta-se a atividade da licnofolida 1 em células tumorais de próstata, altamente resistentes à quimioterapia.
[0047] Os resultados dos testes para 1 são indicados na Tabela 13, onde foram listados apenas os valores de CL50, CIC100 e CIC50 nos quais as linhagens de células tumorais apresentaram maior sensibilidade.
[0048] As nanocápsulas fruto desta invenção associadas às lactonas sesquiterpênicas, descritas nesta presente invenção podem reduzir o crescimento de tumores e de parasitos da família Trypanosomatidae, reduzindo a freqüência e a concentração do fármaco administrada, reduzindo a toxicidade geral, e principalmente aumentando a adesão dos pacientes pela eficiência atribuída a essa nova terapia, especialmente eficaz conseguida com o uso da licnofolida nanoencapsulada.
[0049] Dentre as vantagens da presente invenção, que a difere da estratégia de quimioterapia atual e que devem ser ressaltados e enfatizados neste documento lista-se: 1) Os fármacos pertencentes à classe dos triazólicos, por exemplo, o pozaconazol e o ravuconazol que são atualmente, os candidatos mais prováveis para triagens clínicas em humanos devido as suas características farmacocinéticas e devido à capacidade de aumentar a sobrevida de animais infectados e de induzirem cura na fase aguda em cepas de T. cruzi com perfis de resistência intermediária ao benzonidazol, estes fármacos não induzem cura em cepas resistentes (cepa Colombiana), como aqui relatado para a licnofolida 1, ativa também em cepa resistentes ao benzonidazol (Tabela 10,11,12). Além disso, há baixa eficácia terapêutica em parasitos de multiplicação intracelular, a toxicidade ainda é um fator limitante para os derivados azólicos, devido aos esquemas terapêuticos empregados, geralmente, longos, além, dos elevados custos de produção destes compostos, o que torna o tratamento extremamente oneroso. A patente européia WO 03006012A1 descreve os compostos inibidores da biossíntese de ergosterol, especificamente inibidores da enzima lanosterol C-14α-demetilase, causando assim à depleção do ergosterol que é essencial a membrana plasmática e do complexo cinetoplasto-mitocôndria de fungos e protozoários, dentre eles o alvo do estudo aqui apresentado, o T. cruzi. 2) O tratamento aqui proposto nesta invenção com aplicação claramente descrita para doença de Chagas, tem fonte em planta típica da flora de Minas Gerais a Lychnophora trichocarpha, uma das regiões do Brasil com maior incidência de doença de Chagas, e onde o uso da única droga comercialmente disponível (o benzonidazol), apresenta várias limitações terapêuticas e limitada disponibilidade e foi descontinuado pelo laboratório de origem, Roche®; 3) Apesar de já existirem produtos naturais patenteados com atividade contra o T. cruzi (WO 04050092 A1, WO 04050092 B1, WO 03000272 A1) é a primeira vez na literatura que se faz menção de estudos de eficácia terapêutica in vivo das lactonas sesquiterpênicas furanoeliangolidas contra T. cruzi, sendo esta uma das novidades desta patente (inovação); 4) Além disso, esses compostos listados nesta invenção estão associados a formulações farmacêuticas bem descritas e exemplificadas neste documento e que são fruto de desenvolvimento tecnológico avançado, como o uso de nanocápsulas para determinarem maior eficácia terapêutica após administração em animais e em seres humanos; 5) Ressalta-se o fato diferencial em relação às terapias da doença de chagas e também do câncer em uso, o fato das lactonas sesquiterpênicas furanoeliangolidos possuírem alta potência e baixa solubilidade em água, com doses administradas muito baixas e que permitem o carreamento dessas lactonas nas nanocápsulas com potência antiprotozoário 50 vezes superior a droga padrão (benzonidazol), ou seja, esses compostos podem ser usados em doses 50 vezes inferiores ao benzonidazol e com eficácia de cura superior em todos os perfis de resistência das cepas ao benzonidazol; 6) Ressalta-se o fato dessas formulações serem aptas a serem utilizadas ou administradas por qualquer via de administração, devido à sua baixa toxicidade, sua dispersão em meio aquoso e em tamanho inferior a 1 micrômetro contendo excipientes inócuos e biodegradáveis, elas podem ser administradas por via parenteral (via intravenosa, intramuscular, intradérmica e subcutânea), oral, tópica, retal, inalatória, intranasal, transdérmica e bucal; 7) Essas lactonas sesquiterpênicas furanoeliangolidas são aplicáveis ao tratamento de doenças parasitárias, especialmente os parasitos pertencentes à família Trypanosomatidae (nas doenças de Chagas, leishmanioses diversas e na Tripanossomíase Africana) e nos tratamentos de tumores diversos, especialmente, câncer de próstata, câncer de mama, câncer de colo de útero, câncer renal, câncer de ovário, melanoma, câncer de cólon, leucemias, câncer de células não pequenas do pulmão dentre outros múltiplos tipos. Entretanto, diante de seus potenciais efeitos citotóxicos, foi demonstrado nesta invenção, que as nanocápsulas, protegeram todas as lactonas sesquiterpênicas da degradação, levando a um aumento significativo da resposta biológica, ou seja, da eficácia, controlando a velocidade de liberação e reduzindo seus efeitos adversos, principalmente pela alteração da biodistribuição da molécula no organismo (Figura 4).
[0050] As formulações de nanocápsulas utilizadas nesta invenção contendo lactonas sesquiterpênicas em seu interior foram preparadas baseando-se no método descrito por Fessi et al., Int. J. Pharm., 55, R1-R4, (1989), que se baseia na deposição interfacial do polímero pré-formada seguida do deslocamento do solvente miscível com a água. As nanocápsulas já foram objeto de uma patente norte americana US 5,049,322 como técnica de fácil execução e de fácil transposição para escala industrial, mas não estão reivindicadas no documento patentário anterior (US 5,049,322) a associação a das nanocápulas às lactonas sesquiterpênicas, listadas neste documento e o método de preparo destas formulações particulares.
[0051] Tem-se, portanto, nesta invenção a solicitação do diferenciada no pedido de patente em questão, de formulações de uso farmacêutico e também de uso animal de bioativos baseados em lactona sesquiterpênica furanoeliangolida nanoencapsulada, a licnofolida, desenvolvida para obtenção de melhor eficácia terapêutica em nanocápsulas que possuem grandes vantagens, tanto em sua utilização como em sua fabricação, agregando baixos custos para sua exequibilidade industrial e ainda pela disponibilidade das plantas fonte destes compostos estarem principalmente situadas no estado de Minas Gerais, na flora do Cerrado brasileiro onde se encontra as espécies Lychnophoras para isolamento dessas lactonas aqui descrita e relatadas, em área de aproximadamente 2,2 milhões de quilômetros quadrados, com enorme diversidade dessas espécies. Alia-se a todo o contexto, o interesse despertado em muitos setores da indústria, em particular do ramo farmacêutico para tratamento de doenças como o câncer, a doença de chagas e as Leishmanioses humana e animal. SUMÁRIO DA INVENÇÃO
[0052] A presente invenção revela um sistema de nanocápsulas, contendo lactonas sesquiterpênicas lipofílicas que se encontram absorvidas ao núcleo oleoso ou na matriz polimérica e mesmo na membrana polimérica das formulações, caracterizando um sistema de encapsulamento dos compostos químicos ativos citados nesta invenção. O referido sistema contém tensoativos hidrofílicos e lipofílicos, óleos e polímeros, além das substâncias ativas sesquiterpenóides furanoeliangolidos.
[0053] A atividade e eficácia in vitro e in vivo dos das referidas substâncias nas nanocápsulas desta invenção frente a várias linhagens celulares tumorais e em vários modelos animais infectados com cepas de T.cruzi com diferentes perfis de susceptibilidade ou resistência ao fármaco de referência (benzonidazol) é bastante evidente, como poderá ser observado nos exemplos desta invenção. É também uma concretização da presente invenção a preparação de solução de lactonas sesquiterpênicas não encapsuladas em formulações contendo solventes especiais aprovados para administração parenteral. A solução contém solventes orgânicos capazes de solubilizar completamente as lactonas, diluídas em solução glicosada ou água, para serem comparadas às formulações nanoencapsuladas. Foram ainda preparadas soluções de benzonidazol e suspensões de benzonidazol como fármaco de referência para o tratamento da doença de Chagas, utilizando os mesmos solventes orgânicos e excipientes farmacêuticos para sua solubilização com objetivo de comparar sua eficácia com as nanocápsulas objeto desta invenção. BREVE DESCRIÇÃO DAS FIGURAS
[0054] FIGURA 1: Representação esquemática da estrutura química da lactona sesquiterpênica, licnofolida, isolada de Lychnophora trichocarpha completamente absorvida ao núcleo oleoso da nanocápsula. Abaixo: a imagem das nanocápsulas em visão tridimensional obtida por microscopia de força atômica lado (A) imagem nanocápsula contendo licnofolida e lado (B) imagem de nanocápsula branca (sem licnofolida).
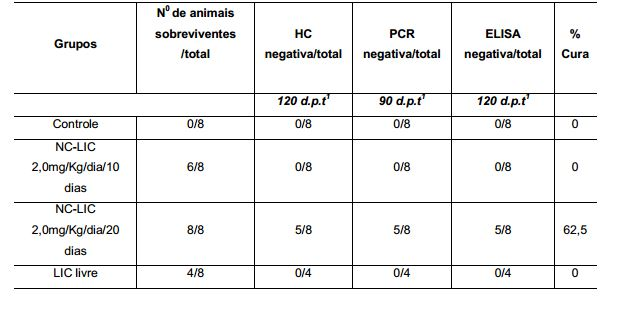
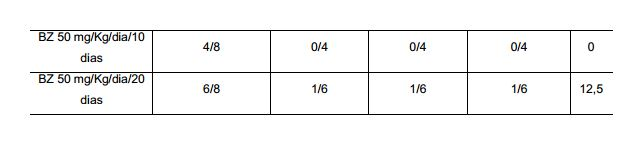
[0055] FIGURA 2: Cinética de liberação in vitro de licnofolida das nanocápsulas (NC-LIC) e dissolução de licnofolida livre (LIC livre) em PBS pH 7,4 contendo Tween® 80 na concentração de 0,5% a temperatura de 37±0,50C, onde observa-se um perfil de liberação mais lento a partir das nanocápsulas.
[0056] FIGURA 3: Curva de parasitemia média (A) e (B). Gráficos de sobrevida em camundongos infectados com 1,0 x 104 tripomastigotas sangüíneos da cepa CL de Trypanosoma.cruzi, tratados com diferentes formulações de BZ 50 mg/Kg/dia por 10 e 20 dias de tratamento (benzonidazol); NC-LIC 2,0 mg/Kg/dia por 10 e 20 dias de tratamento (nanocápsulas de licnofolida); LIC livre 2,0 mg/kg/dia por 10 dias de tratamento (licnofolida livre) administrados via intravenosa e controle.
[0057] Curva de parasitemia média (C) e (D) Gráficos de sobrevida de camundongos infectados com 1,0 x 104 tripomastigotas sangüíneos da cepa Y de Trypanosoma cruzi, tratados com diferentes formulações de BZ 50 mg/Kg/dia por 10 e 20 dias de tratamento (benzonidazol); NC-LIC 2,0 mg/Kg/dia por 10 e 20 dias de tratamento (nanocápsulas de licnofolida); LIC livre 2,0 mg/kg/dia por 10 dias de tratamento (licnofolida livre) administrados via intravenosa e controle.
[0058] Curva de parasitemia média (E) e (F) Gráficos de sobrevida de camundongos infectados com 1,0 x 104 tripomastigotas sangüíneos da cepa Colombiana de T. cruzi, tratados com diferentes formulações de BZ 50 mg/Kg/dia por 10 e 20 dias de tratamento (benzonidazol); NC-LIC 2,0 mg/Kg/dia por 10 e 20 dias de tratamento (nanocápsulas de licnofolida); LIC livre 2,0 mg/kg/dia por 10 dias de tratamento (licnofolida livre) administrados via intravenosa e controle.
[0059] Curva de parasitemia média (G) e (H) Gráficos de sobrevida de camundongos infectados com 1,0 x 104 tripomastigotas sangüíneos da cepa Y de T. cruzi, tratados com formulação de BZ 100 mg/Kg/dia por 20 dias (benzonidazol); NC-LIC 3,0 mg/Kg/dia por 20 dias (nanocápsulas de licnofolida); administradas via oral e controle.
[0060] FIGURA 4: Perfil de concentração plasmática de licnofolida após administração intravenosa de 200 μL de licnofolida em nanocápsulas de PCL na concentração de 2,0 mg/mL e de 200 μL de licnofolida livre na concentração de 2,0 mg/mL em camundongos sadios pesando 40,0 g ±2,0 g (média ± EPM). DESCRIÇÃO DETALHADA DA INVENÇÃO
[0061] As nanopartículas poliméricas presentes no referido sistema podem apresentar: (i) tamanho de partículas entre cerca de 50 nm a cerca de 1000 nm. (ii) índice de polidispersão entre cerca de 0,001 e cerca de 0,700, e (iii) potencial zeta em módulo entre -70 mV a +70mV, sendo os valores preferencialmente: de diâmetro médio inferior a 300 nm, polidispersão abaixo de cerca de 0,3 e potencial zeta de -50 mV.
[0062] De acordo com a presente invenção, os sistemas nanoestruturados podem compreender óleos de origem mineral, vegetal e sintético, sendo que os principais fatores para a escolha do óleo são: i) ausência de toxicidade, ii) incapacidade de degradar ou dissolver o polímero e iii) alta capacidade de dissolver o fármaco licnofolida.
[0063] O óleo utilizado na presente invenção é considerado como qualquer substância líquida insolúvel e imiscível com água, que seja imiscível com os polímeros biodegradáveis tal como: o óleo escolhido foi um triglicerídeo parcialmente sintético, estes podem ser selecionados ao grupo de triglicerídeos de cadeia média, tri, di ou monoacilgliceróis, e seus derivados, e ésteres de ácido graxo a partir de álcool de alto peso molecular (C8-C28).
[0064] Os óleos vegetais que podem ser usados são o de milho, óleo de coco, óleo de oliva, óleo de girassol, óleo de mamona, óleo de soja, óleo de canola, além de outros óleos vegetais e minerais.
[0065] De acordo com a presente invenção, o invólucro polimérico é selecionado do grupo consistindo de polímeros naturais ou sintéticos, biodegradáveis e/ou biocompatíveis, como por exemplo, poliésteres alifáticos sintéticos, PCL, PLA, PGA, PLGA e diblocos PCL-b-PEG, PLA-PEG, entre outros, os derivados acrílicos (poli(meta acrilato de alquila) e seus copolímeros, as poli-acrilamidas e poli-metarilamida, poli-cianocrilato de alquila, ou ainda poliuretanos e polissacarídeos, como por exemplo, a quitosana e derivados de celulose, os quais podem ser empregados ou isoladamente ou em misturas na faixa de 0,0001% e 50% de peso total, sendo preferencialmente cerca de 1%.
[0066] O polímero sintético é selecionado do grupo consistindo de poliestireno, poliésteres, polifosfazenos, polietilenoglicol (PEG), polivinilálcool (PVA), poliacrilamida, poliacrilatos, polivinilpirrolidonas (PVP), polialilamidas e seus copolímeros, polietilenos, Poliacrilicos, polimetacrilatos, polianidridos, polisiloxanos, polioxietilenos e seus copolímeros e/ou seus derivados e/ou copolímeros.
[0067] Os referidos poliésteres alifáticos sintéticos são selecionados do grupo consistindo de poli (ε-caprolactona) (PCL), poli (ácido glicólico) (PGA), poli (ácido lático) (PLA), poli (ácido láctico-co-ácido glicólico) (PLGA), poli (ácido hidroxibutírico) (PHB) e poli (ácido hidroxivalérico) (PHV), poli (cianocrilatos), poli (malonato de metilideno), poliésteres (exemplo: poli (ácido lático), poli (ácido glicólico), poli (ε-caprolactona)) e seus co-polímeros em bloco com o polietilenoglicol de variados pesos moleculares.
[0068] O sistema de nanopartículas poliméricas compreende ainda tensoativos, sendo selecionados do grupo consistindo de óleo de rícino etoxilado, os poloxamers (pluronics), Pluronic F68, todas as poloxaminas, todas as lecitinas de soja e de ovo, os polietilenoglicois succinato 1000, Steareth (Brij), Tween 20 (polissorbato 20), Tween 40 (polissorbato 40), Tween 60 (polissorbato 60), Tween 80 (polissorbato 80), lauril sulfato de sódio, Crillet 1, Crillet 4 HP, Crillet 4 NF, Cremophor RH40, Cremophor RH60, Cremophor EL, Etocas 30, Mkkol HCO-60, Labrasol, Accovov, MC-8, Gelucire 50/13, Gelucire 44/14, Myrj, polioxâmeros, Epikuron 170, lecitina e derivados, fosfolipídeos e derivados, Span (monoestearato de sorbitano), monoestearato de glicerol, Capmul MCM, Capmul MCM 8, Capmul MCM 10, Inwitor 988, Inwitor 742, Inwitor 308, Labrafil M-1944 CS, Labrafil-M 2125, Capryol 90, Laurogliol, Captex 200, ácidos graxos etoxilados, Plurol oleique, Crill 1, Crill 4, Maisine, Peceole, Arlacel P135, ácidos graxos etoxilados e mistura dos mesmos.
[0069] Os referidos tensoativos encontram-se presentes na faixa de cerca de 0,0001 a 50% do peso total, separadamente ou em combinação.
[0070] As formulações farmacêuticas utilizadas nesta invenção contendo lactonas sesquiterpênicas em seu interior foram preparadas pelo método descrito por Fessi et al., Nanocapsule formation by interfacial polymer deposition following solvent displacement. Int. J. Pharm., 55, R1-R4, 1989, baseado na deposição interfacial do polímero pré-formado seguida do deslocamento do solvente. As nanocápsulas já foram objeto de uma patente norte americana US 5,049,322 depositada em 1988.
[0071] A presente invenção provê ainda um processo que compreende a obtenção de um sistema de nanocápsulas poliméricas cujos invólucros compreendem material polimérico e cujos núcleos compreendem óleo sintético e lactonas sesquiterpênicas, pela deposição interfacial de polímeros pré- formados. As nanocápsulas contendo lactonas serão utilizados em concentração de 1,0 a 10,0 mg/mL .
[0072] Os solventes são selecionados do grupo consistindo de acetona, etanol, água, propilenoglicol, carbonato de propileno, clorofórmio, glicerina, diclorometano, metanol, acetato de etila, dimetilformamida, dimetilsufóxido, tetrahidrofurano, cetonas, álcoois, derivados halogenados.
[0073] Finalmente, é também uma concretização da invenção o tratamento da doença de Chagas em modelo experimental murino, combatendo o agente etiológico T. cruzi, tanto na fase aguda quanto crônica da infecção no modelo camundongo, tendo sido capaz de promover redução da carga parasitária, aumentar a sobrevida e promover cura parasitológica nas três diferentes cepas de sensibilidade diferentes à quimioterapia atual com benzonidazol.
[0074] Adiante, são mostrados alguns exemplos ilustrativos dos sistemas de nanopartículas poliméricas e de sua eficácia terapêutica frente às diferentes cepas de T. cruzi que apresentam diferentes perfis de susceptibilidade ao tratamento com benzonidazol, bem como os respectivos resultados de efeito terapêutico.
[0075] Como droga padrão para comparação, utilizou-se o fármaco benzonidazol, no qual foi formulado sob a forma farmacêutica de solução e suspensão. Os solventes são selecionados do grupo consistindo em álcool etílico, propilenoglicol, polietilenoglicol 400, polietilenoglicol 300, álcool benzílico, transcutol, dimetilformamida, dimetilsufóxido e tensoativos como polissorbatos (Tween) 40 e 80, dioctilsulfossuccinato de sódio, N-methyl-2- pyrrolidine. O benzonidazol será utilizado em concentração de 1,0 a 50,0 mg/mL nessas formulações de soluções onde o fármaco se encontra na forma não encapsulada.
[0076] Foi ainda formulada uma solução de lactonas sesquiterpênicas livres. Os solventes são selecionados do grupo consistindo em álcool etílico, propilenoglicol, polietilenoglicol 400, polietilenoglicol 300, álcool benzílico, dietilenoglicol monoetil éter (transcutol), dimetilformamida, dimetilsufóxido e tensoativos como polissorbatos (Tween) 40 e 80, dioctilsulfossuccinato de sódio, N-methyl-2- pyrrolidine. As lactonas serão utilizadas em concentração de 1,0 a 10,0 mg/mL.
[0077] Os exemplos listados têm mera finalidade de mostrar e ilustrar a realização prática da invenção e não tem o propósito de limitá-la. EXEMPLOS Exemplo 1: Isolamento das lactonas a partir de Lychnophoras 1.1 Planta
[0078] Lychnophora trichocarpha foi coletada em Ouro Preto, Minas Gerais, Brasil, em agosto de 2006. Uma exsicata (N0 20.635) está depositada no Herbário do Instituto de Ciências Exatas e Biológicas, UFOP, Ouro Preto, MG, Brasil. 1.1.1 Solventes, reagentes e preparação do extrato etanólico
[0079] As partes aéreas da planta foram secas em estufa a 40 ° C, durante uma semana, sendo reduzida a pó e depois de exaustivamente extraído com etanol à temperatura ambiente por duas semanas. O solvente foi removido sob vácuo, em rotavapor, sob 40°C para obenção do extrato etanólico bruto. 1.1.2 Isolamento e identificação de licnofolida
[0080] O extrato etanólico bruto foi analisado em coluna cromatográfica de gel de sílica Merck 60, 0,063-0,200 mm 0,063-0,400 milímetro partículas. A quantidade de sílica, o diâmetro da coluna e os eluentes utilizados foram de acordo com metodologia de Still, W.C. et al. Journal of Organic Chemistry, 43 (14), 2923-2925, 1978.
[0081] Os solventes foram utilizados seguindo a ordem crescente de polaridade utilizando hexano, acetato de etila, metanol. O processo de fracionamento cromatográfico foi monitorado por cromatografia em camada delgada (CCD) em placas analíticas de 0,25 mm de espessura. Para a fase estacionária de sílica Merck Kiesegel 60 G foi empregado. Os reveladores utilizados foram anisaldeido e solução ácida de sulfato sérico, ambos da Vetec Química Fina Ltda, Brasil.
[0082] O extrato etanólico (50,0g) foi submetido à filtração em coluna de cromatografia em sílica gel, eluído com hexano, acetato de etila e metanol produzindo as frações hexânica (H-0,3 g), acetato de etila (A-20,0 g), metanol (M-27,0g) . A fração A foi fracionada em coluna cromatográfica em sílica gel utilizando como solventes: hexano, acetato de etila (B) e metanol em ordem crescente de polaridade. Do fracionamento cromatográfico de B foi isolado um sólido branco que apresentou mancha branca na CCD quando aspergida com sulfato cérico, característica de lactona sesquiterpênica. O sólido foi pelo denominado como RLIC que apresentou faixa de fusão entre 119 e 121oC. Os espectros de RMN de RLIC foram obtidos em espectrômetro Bruker Avance DRx- 400 MHrz utilizando tertrametilsilano (TMS) como referência interna de CDCl3 (clorofómio deuterado) como solvente. Os deslocamentos químicos foram expressos em partes por milhão (ppm) e as constantes de acoplamento (J) expressas em Hertz. Os multipletos foram definidos com o valor de seu ponto médio. Os dados de RMN de 1H para a substância isolada pelo nosso grupo designada aqui RLIC corroboram com os dados da literatura para licnofolida descritos por Bohlmann, et al. Phytochemistry, 19(11), 2381-2385, 1980. O espectro de RMN de 13C confirma a presença do grupo furanona em LIC (Bohlmann et al., Phytochemistry, 19(11), 2381-2385, 1980; Vichnewski et al.Phytochemistry, 28 (5), 1441-1451,1989; Saúde et al., Fitoterapia, 69 (1), 9091, 1998).
[0083] Os dados de RMN de 1H e de RMN de 13C apresentados por RLIC estão de acordo com os dados descritos na literatura para a lactona sesquiterpênica licnofolida (Bohlmann, et al. Phytochemistry, 19(11), 2381-2385, 1980; Vichnewski et al. Phytochemistry, 28 (5), 1441-1451,1989). Portanto, ficou comprovada que a substância em estudo isolada (RLIC) era a licnofolida. Exemplo 2: Preparação e caracterização do sistema de nanocápsulas poliméricas 2.1 Preparações das nanocápsulas
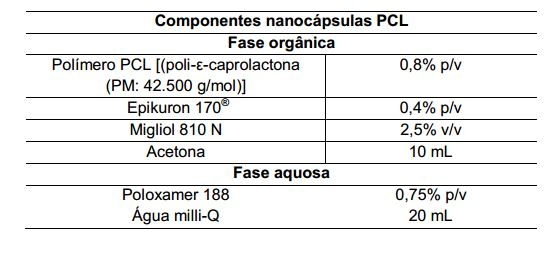
[0084] As nanocápsulas foram preparadas pelo método de deposição interfacial de um polímero pré-formado, no caso a poli-ε-caprolactona (PCL) e a nanocápsula de PLA-PEG, seguido da remoção do solvente como descrito por Fessi et al. (1989). Para fabricação da nanocápsula de PCL a fase orgânica foi constituída por 0,8% (p/v) de PCL [poli-ε-caprolactona, PM 42.500 g/mol, Sigma-Aldrich, (Brasil)]; e para produção da nanocápsula de PLA-PEG foi acrescentada na fase orgânica 0,6% (p/v) de PLA-PEG (PM 66.000 g/mol copolimerizado com PEG 5.000g/mol) e ainda 0,6% p/v do polímero PLA [poli- D,L-lático, Resomer®203, Boehringer Ingerlheim, (Alemanha)]; ainda como constituintes de fase orgânica foram incorporados os seguintes compostos: 0,4%(p/v), fosfatidilcolina de soja, «70% fosfatidilcolina,[Epikuron170®, (Lucas Meyer, França)]; 2,5%(v/v) triglicerídeos de cadeia média [Miglyol 810N (Hulls, Alemanha)] e diferentes concentrações das lactonas utilizadas de 1 a 10 mg/mL. As concentrações dos componentes da fase orgânica foram descritas em relação ao volume final da formulação. Os componentes da fase orgânica foram dissolvidos em 10 mL de acetona (Sigma-Aldrich, Brasil) em um agitador magnético (modelo PC-200, Corning, EUA) a 30°C, 250 rpm (rotações por minuto). A solução orgânica foi transferida para o interior da fase aquosa, contendo 0,75% p/v de poloxamer 188 [(Pluronic F68) PM 8400 g/mol (Aldrich, EUA)], no caso das nanocápsulas de PLA-PEG não se acrescenta poloxamer 188. A mistura foi então mantida 10 minutos sob agitação magnética (500 rpm). Finalmente a suspensão coloidal obtida foi concentrada em um rotavapor (Laborota 4000/4001 Heidolph Instruments, Alemanha) sendo seu volume final reduzido para 10 mL. (Tabelas 1 e 2)


Tabela 1: Composição de nanocápsulas de PCL contendo licnofolida Tabela 2: Composição de nanocápsulas de nanocápsulas de PLA-PEG contendo licnofolida
2.2 Caracterização físico-química do sistema de nanocápsulas poliméricas
[0085] A caracterização físico-química do sistema de nanocápsulas poliméricas é complexa de ser realizada em função de sua natureza coloidal e da complexidade de constituintes que compõem as formulações. No entanto, a determinação destes parâmetros é de extrema importância, pois avalia a estabilidade das preparações e permite determinar o perfil de distribuição das nanocápsulas, bem como sua interação com células do SFM (sistema fagocítico mononuclear). Análise de sua distribuição do tamanho das partículas, determinação do potencial zeta, eficiência e porcentagem de encapsulação e cinética de liberação da lactona a partir das nanopartículas são técnicas geralmente usadas nessa avaliação. 2.2.1 Determinação do tamanho e índice de polidispersão das nanocápsulas
[0086] A análise do tamanho médio das partículas e o índice de polidispersão (I.P) foram definidos utilizando o equipamento Nanosizer N5 Plus (Beckman Coulter, EUA) ambos os métodos determinados por espectroscopia de correlação de fótons (ECF).
[0087] Para realização das medidas de tamanho das partículas foram utilizados aproximadamente 10 μL das dispersões de nanocápsulas diluídas em 4990 μL de água recém-destilada e passada em filtro Millipore® 0,45 μm até se obter a contagem adequada de partículas. As medidas foram efetuadas a temperatura ambiente e ângulo de incidência do laser em relação à amostra de 900C. As determinações foram realizadas em sextuplicada e os valores obtidos correspondem à média ± desvio padrão.
[0088] O índice de polidispersão que também pode ser obtido nestas análises refere-se à distribuição de tamanho das nanopartículas. Índices menores que 0,3 indicam amostras monodispersas (Mosqueira et al. Biomaterials, 22, 29672979, 2001).
[0089] O tamanho médio das nanocápsulas variou entre 260 a 200 nm, sendo ainda observado aumento no tamanho das partículas para um carreamento de LIC em torno de 3 mg/mL (P<0,05). (Tabela 3)
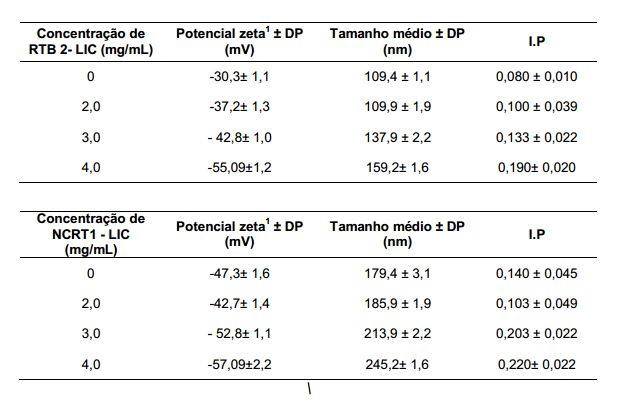
[0090] As formulações contendo de 0, 1, 2 e 3mg/mL de licnofolida mostraram- se monodispersas considerando-se o índice de polidispersão menor que 0,3. 2.2.2 Potencial Zeta
[0091] O potencial zeta foi feita em equipamento Zetasizer 3000HS (Malvern Instruments, Inglaterra). Para a realização das medidas do potencial zeta, 10μL das amostras foram diluídos em 9990μL de NaCl 1mM, previamente filtrados em filtro 0,45μm com o objetivo de se obter suspensões diluídas em soluções com forças iônicas constantes (1,2 ± 0,2 mS/cm2) aproximadamente 1,5 mL foram transferidos para cubetas de análise modelo DTS1060. Os valores obtidos correspondem à média ± desvio padrão.
[0092] O potencial zeta é um método muito utilizado para a caracterização de superfície das nanopartículas e é tal medida que fornece uma boa aproximação do potencial de superfície das partículas. Geralmente, para obter sistemas nanoparticulados fisicamente estáveis por períodos de tempo maiores o potencial zeta deve ser diferente de zero (Tabela 3).
[0093] O potencial zeta, o tamanho médio e o índice de polidispersão foram avaliados nas formulações de nanocápsulas contendo licnofolida em diferentes concentrações e nanocápsulas brancas. Observa-se pela Tabela 3 que: Os valores de potencial zeta não foram significativamente (P>0,05) alterados pela incorporação de licnofolida nas nanocápsulas, sendo que tanto as nanocápsulas brancas quanto as licnofolida encapsuladas possuem carga negativa, variando de -47,3 a -52,7 mV. Este resultado indica que nessa condição a maior parte da substância provavelmente encontra-se encapsulada, ou seja, absorvida no núcleo oleoso da nanocápsula. (Figura 1A). Tabela 3: Caracterização físico-química das formulações de nanocápsulas brancas e contendo diferentes concentrações de licnofolida incorporada ao núcleo oleoso

DP = Desvio padrão (n = 3); I.P = Índice de polidispersão (n=3). A análise estatística foi realizada através do teste t student entre as diferentes formulações, em relação às nanocápsulas brancas Concentração de RTB 2- LIC (mg/mL) Potencial zeta1 ± DP (mV) Tamanho médio ± DP (nm) I.P
2.2.3 Análise morfológica por microscopia de força atômica
[0094] A análise morfológica das nanocápsulas foi realizada por microscopia de varredura por sonda mecânica, utilizando-se a técnica de força atômica (MFA), à temperatura ambiente, nos equipamentos Multimode e Dimension 300, ambos monitorados por controlador Nanoscope IIIa (Digital Instruments, Santa Bárbara, EUA), do Centro Tecnológico de Minas Gerais (CETEC, MG). As imagens foram obtidas no modo de contato intermitente (tapping mode) segundo metodologia utilizada por Leite, et al., Microsc. Microanalysis, 11(3), 48-51, (2005), Assis, et al., Int. J. Pharm., 349,152-160, (2008).
[0095] As suspensões coloidais de nanocápsulas produzidas, contendo ou não licnofolida, foram também examinadas morfologicamente através da microscopia de força atômica (MFA). As imagens obtidas mostraram estruturas nanométricas arredondadas depositadas sobre as camadas de mica. (Figura 1B). De acordo com a Tabela 4, o tamanho médio observado apresentou-se significativamente maior que os valores obtidos pela MFA. O diâmetro médio das nanocápsulas brancas por MFA foi de164 ± 44 nm, e das nanocápsulas contendo 1,0 mg/mL de licnofolida foi de 156 ± 54 nm. Para se verificar a hipótese de achatamento das nanocápsulas sobre a mica, foi determinada a relação diâmetro/altura das nanopartículas pela MFA. O valor da relação obtida foi de 7,0 para as diferentes formulações analisadas, independente da presença de licnofolida e está de acordo com a hipótese de que as nanocápsulas possam se achatar na superfície da mica devido a sua estrutura fluida (Leite et al., Microsc. Microanalysis, 11(3), 48-51, 2005). Tabela 4: Medidas de tamanho médio das NC obtidas pelas técnicas de ECF e MFA
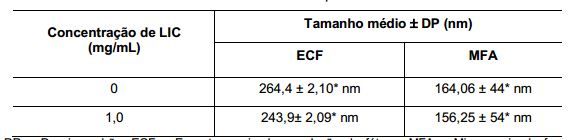
DP = Desvio padrão; ECF = Espectroscopia de correlação de fótons; MFA = Microscopia de força atômica. A análise estatística foi realizada através do teste t student entre as diferentes formulações e técnicas de análise, em relação às NC brancas. * Indica diferença estatística em relação às duas técnicas comparando com amostras pareadas.
[0096] Foram desenvolvidos e validados dois métodos analíticos para a quantificação de licnofolida em diferentes formulações de nanocápsulas. 1) Cromatografia Líquida de Alta Eficiência com detecção de arranjo de diodos (CLAE-DAD) e, 2) Espectrofotometria no ultravioleta (UV-espectrofotométrico). O sistema cromatográfico utilizado consistiu de uma bomba de módulo de separação Waters Alliance 2695, composto de injetor automático, forno de coluna e detector de arranjo de diodos Waters 2996. A separação foi realizada em uma coluna cromatográfica C18 Phenomenex®, modelo Gemini (150 x 4,6mm d.i.; 5 μm), pré-coluna C18 Phenomenex® modelo AJ0-4287 C18 (4 x 3,0 mm) com fase móvel constituída por uma mistura metanol: água (60:40 v/v) com eluição isocrática, fluxo de 0,8 mL/min., volume de injeção de 25 μL e temperatura de 25°C. O tempo de corrida foi 8,0 minutos, e detecção no arranjo diiodos em 265 nm. Foi utilizada água Milli-Q e metanol grau HPLC (Merck). Todos os solventes foram filtrados através de membrana Millipore® de acetato de celulose de 47 mm de diâmetro e poro de 0,22 μm desgaseificado em banho de ultrassom por 30 minutos. Para espectrofotometria foi utilizado o aparelho HeÀios α, Thermo Spectronic, com detecção no UV-Vis em 265 nm. O tempo de retenção obtido foi de 8,30 min. O método foi linear nas concentrações variando de 2,0 μg/ml a 25,0 μg/ml, com um coeficiente de correlação linear R2=0, 9999 (y=2801x + 3328) para CLAE-DAD, e nas concentrações de 5,0 μg/ml a 40,0 μg/ml com o coeficiente R2=0, 9999 (y = 0,0350x + 0,0150) para UV-espectrofotométrico. A precisão e exatidão dos métodos tiveram valores adequados (variação inferior a 2% nas amostras para análise). O limite de quantificação foi de 2,0 μg/ml para CLAE-DAD e 5,0 μg/ml para UV- espectrofotométrico. Já o limite de detecção foi de 0,25 μg/ml e 1,0 μg/ml para CLAE-DAD e espectrofotometria por UV, respectivamente, ressaltando a maior sensibilidade do método cromatográfico. Além disso, nenhum polímero constituinte das formulações de nanocápsulas apresentou picos interferentes no mesmo tempo de retenção das licnofolida.
[0097] A aplicabilidade no desenvolvimento e validação dos métodos para quantificação da licnofolida vem a seguir: na Determinação da porcentagem e eficiência de encapsulação e Determinação da cinética de liberação in vitro da LIC a partir das nanocápsulas.
[0098] A eficiência e o teor de encapsulação das licnofolida nas nanocápsulas foram quantificados pelos dois métodos de quantificação: CLAE-DAD e UV- espectrofotométrico. 2.2.4 Determinação da porcentagem e eficiência de encapsulação
[0099] Estes índices foram calculados pela diferença entre a quantidade de licnofolida na suspensão (medida por dissolução da suspensão total em solvente-acetonitrila) e a licnofolida livres no meio externo aquoso (ultrafiltrado) obtido por ultrafiltração-ultracentrigufação, utilizando-se uma microcentrífuga (Centrifuge 5415 D, Eppendorf). Na parte superior do filtro de AMICON (membranas MICROCON de 100.000Da, Millipore®), foram adicionados 400 μL de suspensão de nanocápsulas, centrifugados a 5900x g durante 15 minutos. Assim, 30 μL do ultrafiltrado contendo a licnofolida livre foram misturadas em 970 μL de acetonitrila, agitados no vórtex por 15 minutos, centrifugados a 5900 xg por 15 minutos e a quantidade de licnofolida no sobrenadante estimada por cromatografia líquida de alta eficiência com detector de arranjo de diiodos (CLAE-DAD) e espectrofotometria por ultravioleta (UV-espectrofotométrico).
[00100] A quantidade da licnofolida aderida à membrana de ultrafiltração foi estimada por remoção da membrana, seguida de sua lavagem em água MilliQ. A membrana foi adicionada a 1000 μL de acetonitrila agitada em vórtex por 15 minutos, centrifugada a 5900*g por 15 minutos e a quantidade de licnofolida determinada no sobrenadante.
[00101] Para a determinação da licnofolida total, 30 μL da suspensão de nanocápsulas foram solubilizados em 970 μL de acetonitrila, homogeneizados em vórtex e centrifugados a 5900 * g por 15 minutos. A quantidade de licnofolida no sobrenadante foi estimada por CLAE-DAD e espectrofotometria no UV. A eficiência e o teor de encapsulação das diferentes formulações de nanocápsulas preparadas foram determinados CLAE-DAD e UV- espectrofotométrico e as metodologias foram comparados utilizando-se o teste “t” de Student através do programa Prisma® 5.0. Os resultados se encontram na Tabela 5. Tabela 5: Eficiência e teor de encapsulação das nanocápsula contendo licnofolida
D=Desvio padrão (n = 3). Os valores de P entre os dois métodos: CLAE e UV foram determinados pelo teste t de Student. * Indica resultados semelhantes (P>0,05).
[00102] Os resultados apresentados na Tabela 5 mostram altos teores de encapsulação, para as nanocápsulas contendo 1 e 3 mg/mL de licnofolida. Não existe diferença significativa (P>0,05) entre os métodos avaliados para eficiência e percentagem de encapsulação. O valor alto obtido na porcentagem de encapsulação da licnofolida nas nanocápsulas reforça a hipótese de que a licnofolida encontram-se associadas às nanocápsulas em seu núcleo oleoso. 2.2.5 Determinação da cinética de liberação in vitro da LIC a partir das nanocápsulas
[00103] A avaliação do perfil de liberação in vitro da LIC a partir de nanocápsulas foi realizada de acordo com a técnica de diálise de equilíbrio direta, porém para realização do mesmo o teste de solubilidade da LIC foi previamente determinada. A solubilidade da LIC através PBS 7,4 foi determinada em 24 horas. Para esta 2,5 mg de LIC foi pesada com precisão e adicionado a um tubo Eppendorf ®. A este tubo foram adicionados 1 mL de PBS 7,4 e o sistema de agitação foi mantida a 37 ± 50C por 24 horas em um tubo de ultracentrífuga a 8000 rpm por 15 minutos. No final desse período ocorreu retirou-se, 100 μL do sobrenadante e diluiu volumetricamente com acetonitrila para obter um volume final de 2 mL. Esta solução foi vortexada por 15 minutos e novamente ultracentrifugadas em 8000 rpm. O sobrenadante foi quantificada por DAD- HPLC. A solubilidade da LIC em PBS 7,4 + Tween 80® de 0,5% foi determinado pelo mesmo método. Este experimento foi realizado em réplicas de seis (n = 6) e os resultados foram expressos como média e desvio padrão. Assim, na cinética de liberação in vitro da LIC da NC (1 mg/mL) procedeu conforme descrito abaixo: um total de 800 μL da suspensão de NC foram diretamente adicionados a três bolsas de diálise com poros de 12.000-14.000 Da. Estes sacos foram imersos em 40 mL de PBS contendo 7,4 0,50% Tween 80 ® (para melhorar a solubilidade do LIC), e foram mantidos em equilíbrio a 37 ± 0,5OC. Em intervalos de tempo pré-definido (0,25, 0,5, 1, 3, 6, 9, 12, 21, 24, 48 h) alíquotas de 1,0 mL foram retirados do ambiente externo e volumetricamente diluído em acetonitrila para obter um volume final de 2,0 mL. A solução final foi vortexada por 10 minutos e ultracentrifugada a 8000 rpm por 10 minutos. O sobrenadante foi analisado por HPLC-DAD.Condições Sink teórica (20% da concentração de saturação no meio de liberação após 100%) foram alcançados apenas no meio contendo 0,5% Tween ®. O experimento a liberação cinética foram realizados em banho-maria a 37 ± 0,50C com agitação (Banho Dubnoff mo.144, Fanem, Brasil).
[00104] Um sistema de transportador de sucesso não só deve ser capaz de incorporar uma carga elevada de droga, mas também devem ser capazes de lançamento que a uma taxa adequada em condições fisiológicas. O fato da baixa solubilidade de LIC em PBS 7,4 (33,0 ±1,3 μg/mL) e o alto limite de quantificação (LOQ) em CLAE-DAD, praticamente não foi possível quantificar o perfil de cinética de liberação LIC in vitro, sob condições aqui estabelecidas nas pequenas diluições de NC-LIC em PBS médio e usando o detector DAD. Em diluições mais elevadas, as concentrações LIC no meio externo foram abaixo do limite por CLAE-DAD. Portanto, a condição sink não pode ser obtida em PBS 7,4 e isto pode ser explicada pela baixa solubilidade em da LIC em água. A fim de melhorar a solubilidade LIC no meio de liberação e alcançar condição sink, 0,5% de Tween ®80 foi adicionado em PBS 7,4. A solubilidade máxima do LIC neste meio de liberação foi de aproximadamente 100,0 ± 3,5 μg/mL e uma diluição adequada do LIC-NC foi feito para garantir condições sink (concentração máxima possível de LIC em 20% da sua solubilidade). O estudo de liberação da licnofolida em função do tempo está apresentado na Figura 2.
[00105] Houve uma grande diferença no perfil de dissolução das licnofolida na sua forma livre e das licnofolida associadas às nanocápsulas. O perfil de dissolução indica que a substância é extremamente lipofílica e que sua dissolução e difusão através de membranas se fazem lentamente, pois a dissolução completa só acontece após 24 horas de contacto com o meio, mesmo em condições sink. No caso das nanocápsulas contendo a licnofolida observa-se um perfil de liberação da substância muito diferente do perfil de dissolução. Este perfil indica que o burst inicial é muito pequeno, quase negligenciável, indicando uma associação íntima entre a licnofolida e o vetor, e um encapsulamento efetivo no interior da vesícula polimérica. Observa-se um perfil trifásico para a liberação, com uma difusão extremamente baixa até 3h, seguida de uma fase de liberação de primeira ordem até 21 horas, tanto para a licnofolida pura quanto para a licnofolida associada às nanocápsulas, com diferença significativa na quantidade liberada (P<0,05). Após 21 horas a licnofolida associado às nanocápsulas foi liberado de maneira mais rápida para atingir uma liberação total em 48horas. Os dados evidenciam o caráter lipofílico da licnofolida, o que aumenta a afinidade pelo núcleo oleoso do carreador (Figura 2).
[00106] Por fim, a quantidade liberada da licnofolida a partir das nanocápsulas foi significativamente menor que a quantidade dissolvida do fármaco livre, embora, entre 3 e 21 horas a velocidade de liberação/dissolução tenha sido muito semelhante. Apesar da liberação da licnofolida das nanocápsulas para o meio externo ter sido lenta, ela foi completa, o que resultou na liberação de 97% desta substância em 48 h. Os resultados obtidos com a cinética de liberação da licnofolida encapsulada corroboram com a lipofilicidade da substância intimamente associada ao núcleo oleoso, sua alta porcentagem de encapsulação, bem como a baixa solubilidade em PBS. Exemplo 3: Preparo das soluções de licnofolida livre
[00107] Uma vez isolada, verificada a pureza e quimicamente identificada pelas técnicas de ponto de fusão e por análises espectrométricas de RMN de 1H e de 13C a licnofolida foi utilizado no preparo de soluções na concentração de 2,0 mg/mL, compostas de DMA (dimetilacetamida), PEG 300 (polietilenoglicol 300) e glicose 5% (p/v), denominadas de soluções de licnofolida livre. Para tal, 10 mg de licnofolida foi dissolvida, primeiramente em 400 μL de DMA, seguido da adição de 600 μL de PEG e acrescidos de solução de glicose a 5% até obtenção de um volume final de 5 mL. Durante o preparo das soluções de licnofolida, o mesmo foi primeiramente solubilizado em DMA e o PEG 300 adicionado à solução resultante. A solução de glicose 5% (p/v) foi incorporada lentamente à solução de DMA/PEG 300 sob leve agitação em vórtex (vórtex MS1, IKA Works, EUA). Antes da administração IV as nanocápsulas com e sem licnofolida bem como das soluções de benzonidazol e licnofolida livre, foram isotonizadas com glicose e filtradas em filtro Millipore 0,45 μm para garantir a administração de partículas inferiores a 5 μm, reduzindo assim a possibilidade de formação de êmbolos.
[00108] O derivado nitroimidazólico benzonidazol (BZ-Rochagan®,Suíca, Roche) foi utilizado para o tratamento dos animais infectados com cepas do T. cruzi. Sua produção está atualmente sob a supervisão da Organização Mundial de Saúde e a tecnologia para sua fabricação está sendo transferida ao Laboratório Farmacêutico de Pernambuco (LAFEPE), Brasil. O fármaco foi administrado por gavagem na forma de suspensão em goma arábica, utilizando uma dose diária de 100mg por Kg de peso corporal. Exemplo 4: Estudos da eficácia da LIC in vivo 4.1 Cepas de T.cruzi
[00109] A ação das diferentes formulações farmacêuticas foram estudadas nas seguintes cepas: 1) Cepa CL, isolada de vetor no RS e sensível ao tratamento pelo benzonidazol (Brener e Chiari, Journal of Parasitology, 51(6), 922926, 1965). 2) Cepa Y, isolada de uma paciente chagásica na fase aguda da infecção e proveniente do município de Marília, São Paulo considerada parcialmente resistente ao tratamento com benzonidazol (Filardi e Brener, Trans. R. Soc. Trop. Med. Hyg., 81, 755-759, 1987). 3) Cepa Colombiana isolada por meio de hemocultura de um caso humano da doença de Chagas na Colômbia e considerada resistente à quimioterapia com benzonidazol (Filardi, e Brener, Z.Trans. R. Soc. Trop. Med. Hyg., 81, 755-759, 1987). 4.2 Animais
[00110] Neste trabalho foram utilizados camundongos Swiss, fêmeas com idade entre 28 a 30 dias, provenientes do Centro de Ciência Animal da Universidade Federal de Ouro Preto. Durante os experimentos os animais foram mantidos neste Biotério segundo as diretrizes estabelecidas pelo Colégio Brasileiro de Experimentação Animal (COBEA), receberam água filtrada e foram alimentados com ração comercial balanceada “ad libidum”. Esse projeto foi aprovado pelo Comitê de Ética no Uso de Animais (CEUA) da Universidade Federal de Ouro Preto (UFOP), MG, Brasil, Protocolo No. 2085. 4.3 Infecções dos animais e protocolo de tratamento
[00111] Os testes in vivo foram feitos em grupos de oito camundongos. Os animais foram inoculados com as cepas CL, Y e Colombiana de T.cruzi por via intraperitonial com inóculo padronizado de 10.000 tripomastigotas sanguíneos obtidos no dia de pico de parasitemia e quantificado por contagem do parasito em preparações com 5 microlitros de sangue a fresco entre lâmina e lamínula segundo Brener,Z. Rev.Inst.Med.trop., 4, 389-396, 1962.
[00112] O tratamento da fase aguda foi feito em animais inoculados pela via intraperitonial (IP) com inóculo de 10.000 tripomastigotas sanguíneos.
[00113] O tratamento foi feito em três esquemas terapêuticos distintos: (1) Iniciado 24h após a inoculação. As formulações NC-LIC (nanocápsula de licnofolida), LIC livre (licnofolida livre), via intravenosa, 2,0 mg/Kg/dia por 10 dias consecutivos. O benzonidazol (BZ) foi administrado por via intravenosa 50 mg/Kg/dia por 10 dias consecutivos. Um grupo controle (animais infectados não tratados) foi mantido e avaliado em paralelo. (2) Iniciado no dia da confirmação da parasitemia para cada cepa (7° dia de infecção nos animais infectados com as cepas CL e Colombiana e 4° dia nos infectados com a cepa Y). Todos os animais tiveram a infecção por T. cruzi confirmada por exame de sangue a fresco. NC-LIC (nanocápsula de licnofolida) foi administrada por via intravenosa, 2,0 mg/Kg/dia por 20 dias consecutivos e benzonidazol (BZ) 50 mg/Kg/dia via intravenosa por 20 dias consecutivos. Um grupo controle (animais infectados não tratados) foi mantido e avaliado em paralelo. (3) Iniciado no 4° dia de infecção em animais infectados com a cepa Y. O tratamento com NC-LIC foi feito por via oral, 3,0 mg/Kg/dia por 20 dias consecutivos. O BZ 100 mg/Kg/dia foi utilizado via oral por 20 dias consecutivos. 4.4 Parâmetros avaliados nos animais tratados 4.4.1 Parasitemia (PAR)
[00114] A curva de parasitemia (PAR) representa a média diária de parasitos encontrados por exame de sangue a fresco nos animais expressa em número de tripomastigotas sanguíneos/0,01 ml de sangue.
[00115] A contagem da parasitemia foi feita diariamente ao longo da fase aguda da infecção, por meio do exame de sangue a fresco (ESF) coletado da veia caudal do animal de acordo com a metodologia de Brener, Z. Rev.Inst.Med.trop., 4, 389-396, 1962.
[00116] A contagem dos parasitos foi feita diariamente após a inoculação de todas as cepas. O ESF foi realizado até que não fossem mais observados parasitos no sangue periférico por pelo menos cinco dias consecutivos, ou até a morte dos camundongos. 4.4.2 Hemocultura
[00117] Em camundongos que apresentaram ESF negativo até o 30o dia pós tratamento (d.p.t) foi realizada a hemocultura (HC) sendo a coleta do sangue feita pela veia do plexo orbital. Para tal cerca de 500 microlitros de sangue foram coletados com heparina e cultivado em meio de cultura LIT, segundo a metodologia de Filardi e Brener. Trans. R. Soc. Trop. Med. Hyg., 81, 755-759, 1987.
[00118] O material foi mantido em estufa a 28oC, homogeneizado a cada 48h e uma gota do sedimento examinada no 30°, 60°, 90° e 120° dia. 4.4.3 Sobrevida dos animais
[00119] Foi avaliada diariamente a sobrevida dos animais durante a fase aguda da infecção registrada em percentagem cumulativa. 4.5 Análises estatísticas
[00120] A parasitemia (PAR) foi analisada pelo teste não-paramétrico de Kolmogorov-Sminnorv que compara a área sob a curva entre dois grupos experimentais. Em alguns casos, quando a variância foi heterogênea, foi utilizado o teste de Mann-Whitney.
[00121] A análise de sobrevida entre os grupos experimentais foi realizada pelo teste de Kaplan-Meier log rank. As diferenças foram consideradas como significativas quando o nível de significância foi menor ou igual a 0,05 (P< 0,05). Tratamentos por via intravenosa
[00122] As curvas de parasitemia dos animais infectados pela cepa CL de T.cruzi, sensível ao BZ e tratados com as diferentes formulações segundo o primeiro e segundo esquemas terapêuticos: estão representadas na Figura 3A.
[00123] Os resultados demonstraram que a formulação NC-LIC com maior duração de tratamento 2,0 mg/Kg/dia por 20 dias apresentou resultados semelhantes à BZ 50 mg/Kg/dia por 20 dias, reduzindo a parasitemia que se tornou subpatente durante e após o tratamento em 100% dos animais. NC-LIC na mesma dose 2,0 mg/Kg/dia porém com menor tempo de tratamento apenas 10 dias apresentou o segundo melhor resultado com parasitemia significantemente menor (P<0,05) em relação ao controle, porém semelhante em relação à BZ 50 mg/Kg/dia por 10 dias. Além disto, 5 animais (62,5% dos animais) tratados com NC-LIC 2,0 mg/Kg/dia por 10 dias apresentaram parasitemia subpatente durante e após o tratamento, mas 100% dos animais tratados por BZ 50 mg/Kg/dia por 10 dias apresentaram parasitemia subpatente. Animais tratados com LIC livre 2,0 mg/kg/dia por 10 dias apresentaram 12,5% dos animais com parasitemia subpatente.
[00124] A sobrevida dos animais infectados com a cepa CL e tratados durante a fase aguda da infecção está representada na Figura 3B. O grupo controle apresentou tempo médio de sobrevida de 24 dias. Nos animais tratados com as formulações NC-LIC (ambos os esquemas terapêuticos) e BZ correspondentes, 100% dos animais sobreviveram à fase aguda e apresentaram tempo médio de sobrevida superior a 6 meses pós-tratamento quando foram necropsiados para avaliações histopatológicas. Dos animais tratados com a formulação LIC livre, 6 (75%) também apresentaram igual sobrevida. Dois morreram na fase aguda sobrevivendo por um período de até 26 dias pós-tratamento. Não houve diferença estatística em relação à sobrevida entre os grupos de animais tratados com as formulações: NC-LIC, LIC livre e BZ (ambos os tratamentos).
[00125] As curvas de parasitemia dos animais infectados pela cepa Y de T.cruzi, parcialmente sensível ao BZ, estão representadas na Figura 3C.
[00126] A formulação NC-LIC 2,0 mg/Kg/dia por 20 dias apresentou redução da parasitemia semelhante à formulação BZ 50 mg/Kg/dia por 20 dias sendo100% e 87,5% dos animais com parasitemia subpatente, respectivamente. NC-LIC 2,0 mg/Kg/dia por 10 dias também apresentou redução de parasitemia dos animais, semelhante ao observado com BZ 50 mg/Kg/dia por 10 dias. Nestes 50% dos animais tratados com NC-LIC 2,0 mg/Kg/dia por 10 dias e 25% dos animais tratados com BZ 50 mg/Kg/dia por 10 dias apresentaram parasitemia subpatente. Nos animais tratados com LIC livre também foi observado que 12,5% apresentaram parasitemia subpatente.
[00127] A sobrevida dos animais infectados com a cepa Y e tratados está representada na Figura 3D. O grupo controle apresentou tempo médio de sobrevida de 17 dias. Nos animais tratados com as formulações: NC-LIC nos dois tempos de tratamento e BZ em ambas as formulações, 100% sobreviveram à fase aguda e apresentaram tempo sobrevida de até seis meses quando foram necropsiados para avaliações histopatológicas. Em animais tratados com a formulação LIC livre foi ainda observado que 4/8 (50%) deles também sobreviveram até 6 meses pós- tratamento.
[00128] As curvas de parasitemia dos animais infectados pela cepa Colombiana de T.cruzi, 100% resistente ao tratamento pelo BZ, estão representadas na Figura 3 E.
[00129] Os resultados demonstraram que o tratamento com as formulações NC- LIC 2,0 mg/Kg/dia nos dois tempos de duração (10 e 20 dias) apresentaram os melhores resultados com maior redução da parasitemia. Além disso, houve diferença significativa na redução da parasitemia (P<0,05) em relação aos respectivos tratamento com BZ 50 mg/Kg/dia (10 e 20 dias). Não houve diferenças significativas (P>0,05) entre o grupo controle em relação aos grupos tratados com a formulação BZ de ambos os tratamentos (10 e 20 dias). A formulação LIC livre 2,0 mg/Kg/dia por 10 dias reduziu a parasitemia. A parasitemia foi ainda significativamente maior em relação a ambos os tratamentos com NC-LIC e estatisticamente menor em relação aos tratamentos com BZ.
[00130] Apenas os animais tratados com NC-LIC 2,0 mg/Kg/dia por 20 dias apresentaram parasitemia subpatente o que foi observado em 5 dos 8 (62,5%) animais. Todos os demais tratamentos tornaram a parasitemia subpatente mais tardiamente e esta se deu antes nos animais tratados com NC-LIC (69 dias de infecção), seguida de LIC livre (70 dias) e BZ (80 dias). Todos os animais controle apresentaram parasitemia patente até a morte por consequência da infecção.
[00131] A sobrevida dos animais infectados com a cepa Colombiana e tratados está representada na Figura 3F. O grupo controle apresentou tempo médio de sobrevida de 49 dias, enquanto 100% dos animais tratados com NC-LIC 2,0 mg/Kg/dia por 20 dias sobreviveram ao tratamento. Animais tratados com as formulações NC-LIC 2,0 mg/Kg/dia por 10 dias e BZ 50 mg/Kg/dia por 20 dias apresentaram 75% de sobrevida por até seis meses, quando foram necropsiados para avaliação histopatológica. Em animais tratados com LIC livre e BZ 50 mg/Kg/dia por 10 dias, apenas metade (50%) apresentaram esta mesma sobrevida. Tratamentos por via oral
[00132] As curvas de parasitemia dos animais infectados pela cepa Y de T.cruzi, parcialmente sensível ao BZ, e tratados com as formulações NC-LIC e BZ, administrados via oral estão representadas na Figura 3G.
[00133] A formulação NC-LIC 3,0 mg/Kg/dia por 20 dias apresentou redução da parasitemia semelhante à formulação BZ 100 mg/Kg/dia por 20 dias. Nesta condição todos os animais tratados com BZ, apresentaram parasitemia subpatente durante e após o tratamento, o que foi verificado em apenas 4 dos 7 (57,2%) animais tratados com a formulação encapsulada. No entanto, vale ressaltar que a dose de BZ administrada foi cerca de 40 vezes superior à dose administrada de NC-LIC. Isto nos permite afirmar que a lactona sesquiterpênica licnofolida nanoencapsulada, mesmo não promovendo a cura em todos os camundongos infectados com uma cepa considerada parcialmente sensível (50%) ao BZ 100 mg/Kg/20dias pode ser considerada uma substância potente para o tratamento da doença de Chagas experimental e comparada ao tratamento com BZ. Cabe ainda ser estudado um aumento da dose, o que possivelmente promoverá um aumento de sua eficácia terapêutica e uma possível cura parasitológica em todos os animais tratados com NC-LIC. Segundo a literatura, as limitações do BZ administrados por via oral são bem descritas e relatadas. São graves efeitos colaterais existentes, que podem estar relacionados às propriedades farmacocinéticas desfavoráveis deste fármaco tais como: curta meia-vida e limitada penetração nos tecidos e células onde o parasita se multiplica, fatores estes que dificultam ou reduzem a sua eficácia terapêutica, especialmente na fase crônica da infecção.
[00134] A sobrevida dos animais infectados com a cepa Y e tratados com as formulações BZ e NC-LIC está representada na Figura 3H. O grupo controle apresentou tempo médio de sobrevida de 20 dias, enquanto 100% dos animais tratados com BZ 100 mg/Kg/dia por 20 dias e 87,5% dos animais tratados com as formulações NC-LIC 3,0 mg/Kg/dia por 20 dias, respectivamente sobreviveram até seis meses pós-tratamento quando foram necropsiados para avaliação histopatológica. Este resultado obtido com NC-LIC é muito animador se considerada a baixa dose da substância utilizada no tratamento, aliado ainda ao fato da lactona sesquiterpência estar totalmente associada ao núcleo de um vetor polimérico (NC) que confere à formulação redução de toxicidade da substância ativa e sua maior permanência na circulação sanguínea, agindo assim por mais tempo no parasito cuja eliminação de dá por exaustão do mesmo após suas sucessivas eliminações pelas células hospedeiras. 4.6 Controle de cura da infecção
[00135] O controle de cura da infecção foi feito por métodos parasitológicos (hemocultura, PCR), visando à detecção do T. cruzi ou seu DNA respectivamente, e sorológico convencional (ELISA) para pesquisa de anticorpos da classe IgG de imunoglobulinas anti-T.cruzi indicativos do contato ou presença do parasito no animal. 4.6.1 Avaliações Parasitológicas 4.6.1.1 Hemocultura (HC)
[00136] As HC foram realizadas no 30o após tratamento nos animais em que a parasitemia era subpatente ao exame a fresco (ESF), sendo realizada segundo a metodologia de Filardi e Brener, Trans. R. Soc. Trop. Med. Hyg., 81, 755-759, (1987). Como descrito anteriormente, a positividade de hemocultura na avaliação pós-tratamento é um claro indicador de fracasso terapêutico. 4.6.1.2 Reação em Cadeia da Polimerase (PCR)
[00137] A reação em cadeia da polimerase também foi realizada 30 e 90 dias após o tratamento, em amostras de DNA extraídas de sangue. A extração de DNA procedeu como descrito por Ávila et al., Parasitol., 48, 222-223, (1991). A PCR foi processada de acordo com o protocolo de Gomes et al., Am. J. Trop. Med. Hyg., 60, 205-210, (1999) modificada.
[00138] A positividade da PCR na avaliação pós-terapêutica é indicativa de fracasso terapêutica. Por outro lado, uma PCR negativa não indica com segurança cura parasitológica ou eficácia terapêutica, salvo se todos os demais parâmetros parasitológicos (ESF, hemocultura) e sorológicos (ELISA ou outros testes) forem também negativos. 4.6.2 Avaliação sorológica 4.6.2.1 Sorologia Convencional pela ELISA (ELISA)
[00139] A técnica de ELISA foi utilizada como controle de cura após tratamento para detecção de Imunoglobulina G total (IgG) de acordo com a metodologia estabelecida por Voller et al., Bull.World Health Organ. 53, 55-65, (1976). Para tal, as amostras de soro foram coletadas duas vezes: 90 e 120 dias após o término do tratamento e armazenadas. Os soros foram testados na diluição 1:80.
[00140] A negativação da sorologia por ELISA ou outros testes sorológicos é considerada o padrão ouro na avaliação da eficácia terapêutica, indicando assim cura parasitológica ou sucesso terapêutico. 4.7 Eficácia terapêutica
[00141] Seguindo o critério de cura clássico empregado para humanos (Consenso Brasileiro em Doença de Chagas, 2005) a susceptibilidade ou resistência aos tratamentos foi definida de acordo com os resultados obtidos pelo conjunto de métodos parasitológicos e sorológicos empregados na avaliação pós-tratamento.
[00142] Foram considerados curados aqueles animais que apresentaram simultaneamente negatividade em todos os testes parasitológicos (ESF, HC, PCR, parasitismo tecidual e sorológico (ELISA), atendendo assim o critério mais atual de cura parasitológica de infecção experimental com T.cruzi ou doença de Chagas humana). Tratamento via intravenosa
[00143] Os resultados obtidos com os animais infectados com a cepa CL de T.cruzi (Tabela 6) mostram que houve cura parasitológica em 100% (8/8) dos animais tratados com NC-LIC 2,0 mg/Kg/dia por 20 dias consecutivos; 62,5% (5/8) dos camundongos tratados com as formulações NC-LIC 2,0 mg/Kg/dia por 10 dias e em 0% dos tratados com LIC livre. Nos animais tratados com a formulação BZ 50 mg/Kg/dia 10 e 20 dias de tratamento a cura também aconteceu em 100% dos animais. Tabela 6: Resultados de sobrevida, hemocultura, PCR e ELISA em camundongos Swiss infectados pela cepa CL de Trypanosoma cruzi e tratados com diferentes formulações por via intravenosa
1d.p.t = dias pós tratamento
[00144] A porcentagem de sobrevida dos animais (Tabela 6) foi de 100% nos animais tratados com NC-LIC 2,0 mg/Kg/dia por 10 e 20 dias e nos tratados com BZ com ambas as formulações, 75% (6/8) dos animais tratados com LIC livre e de 0% nos animais do grupo controle não tratado.
[00145] Os resultados obtidos com os animais infectados com a cepa Y de T.cruzi (Tabela 7) mostram que houve cura parasitológica em 100% (8/8) dos animais tratados com NC-LIC 2,0 mg/Kg/dia por 20 dias; 50% (4/8) dos camundongos tratados com as formulações NC-LIC 2,0 mg/Kg/dia por 10 dias e em 12,5% dos tratados com LIC livre. Nos animais tratados com a formulação BZ 50 mg/Kg/dia por 20 dias a cura aconteceu 7 dos 8 (87,5%) animais tratados já nos tratados esta mesma dose de BZ 50 mg/Kg/dia por 10 dias a cura ocorreu em apenas 25% (2/8) dos animais.
[00146] Mais uma vez fica confirmado o grande potencial da substância licnofolida para o tratamento etiológico da doença de Chagas experimentalmente especialmente em formulações nanoencapsuladas, bem como em outras espécies de protozoários da família Trypanosomatidae, conforme reinvindicação nesta invenção. Tabela 7: Resultados de sobrevida, hemocultura, PCR e ELISA em camundongos Swiss infectados pela cepa Y de Trypanosoma cruzi e tratados com diferentes formulações via intravenosa
1d.p.t= dias após tratamento
[00147] A porcentagem de sobrevida dos animais (Tabela 7) foi de 100% nos animais tratados com NC-LIC 2,0 mg/Kg/dia por 10 e 20 dias consecutivos, de 50% (4/8) dos animais tratados com LIC livre. Nos animais tratados com BZ, ambos os tratamentos, a sobrevivência foi 100%, portanto igual aos dois esquemas de tratamentos com NC-LIC, enquanto no grupo controle não tratado não houve sobrevida (0%).
[00148] Dos animais infectados com a cepa Colombiana tratados com as diferentes formulações, foi verificado cura parasitológica em 62,5% (5/8) dos animais tratados com NC-LIC 2,0 mg/Kg/dia por 20 dias consecutivos. Este resultado foi estatisticamente superior em relação às demais formulações testadas em paralelo (Tabela 8).
[00149] Os animais tratados com as formulações NC-LIC 2,0 mg/Kg/dia por 10 dias e LIC livre não apresentaram cura parasitológica, pois apresentaram exames parasitológicos positivos (hemocultura e/ou PCR) indicativas de persistência de infecção ativa ou fracasso terapêutico. Os resultados do teste de ELISA foram compatíveis com os resultados dos testes parasitológicos também indicativos de fracasso terapêutico.
[00150] O tratamento com BZ 50 mg/Kg/dia por 10 dias não revelou cura em nenhum dos animais sobreviventes, já os animais tratados com BZ 100 mg/Kg/dia houve cura em apenas 12,5% (1/8) dos animais sobreviventes, respectivamente. Tabela 8: Resultados de sobrevida, hemocultura, PCR e ELISA em camundongos Swiss infectados pela cepa Colombiana de Trypanosoma cruzi e tratados com diferentes formulações via intravenosa
[00151] A Tabela 8 mostra que: houve sobrevida de 100% (8/8) e de 75% (6/8) dos animais tratados com a formulação NC-LIC 2,0 mg/Kg/dia por 20 dias e 10 dias consecutivos, respectivamente. Nos animais tratados com LIC livre o percentual de sobrevida foi de 50% (4/8). Nos animais tratados com BZ 50mg/kg/dia os índices de cura observados foram de 50 a 75% quando tratados por 10 e 20 dias, respectivamente. Nenhum animal do grupo controle sobreviveu à infecção.
[00152] De maneira especial os resultados obtidos com a cepa Colombina reforçam ainda mais o grande potencial das lactonas sesquiterpênicas para o tratamento etiológico da doença de Chagas experimental, pois camundongos infectados por esta cepa e tratados com o BZ em dosagem mais elevada (100mg/kg/dia por 20 dias) também pela via oral, o índice de cura é nulo (0%) e todos morrem durante a fase aguda da infecção, razão pela qual este cepa é considerada um protótipo de resistência ao tratamento convencional para a doença de Chagas por serem resistentes ao tratamento com o BZ ou o nifurtimox (Lampit® da Bayer), este último fármaco não disponibilizado no mercado. Tratamento via oral
[00153] Os animais tratados com as formulações por via oral (Tabela 9) mostram que houve cura parasitológica em 57,2% dos animais tratados com NC-LIC 3,0 mg/Kg/dia por 20 dias. Nos animais tratados com a formulação BZ 100 mg/Kg/dia a cura aconteceu 100% (8/8) dos animais. Tabela 9: Resultados de sobrevida, hemocultura, PCR e ELISA em camundongos Swiss infectados pela cepa Y de Trypanosoma cruzi e tratados com diferentes formulações por via oral
[00154] A porcentagem de sobrevida dos animais (tratados com NC-LIC 3,0 mg/Kg/dia por 20 dias foi de 87,5% (7/8). Nos animais tratados com BZ 100 mg/Kg/dia por 20 dias a sobrevivência foi 100% e a do grupo controle não tratado foi de 0% (Tabela 9).
[00155] As Tabelas 10, 11 e 12 apresentam o resumo dos principais parâmetros que permitem avaliar os resultados obtidos com o tratamento com as formulações NC-LIC, LIC livre e BZ, comparativamente.
*PMP=Pico Máximo de parasitemia (corresponde ao máximo de parasitemia e foi expresso em número de tripomastigotas sangüíneos/0.1mL de sangue, detectado pelo ESF segundo a metodologia de Brener (1962). # PI= Pós-infecção
[00156] Em resumo as Tabelas 10, 11 e 12, mostram que as formulações NC- LIC 2,0 mg/kg/dia, nas condições de tratamento via intravenosa (IV), iniciados no 4° ou 7° díade infecção, durante 20 dias consecutivos, apresentaram os melhores resultados que as formulações BZ (fármaco de referência) nas mesmas condições, para todas as cepas de T. cruzi sensível, parcialmente resistente e totalmente resistente ao BZ.
[00157] Em relação à cepa Colombiana (resistente ao BZ) em particular, foi verificado ainda que NC-LIC, 2,0 mg/kg/dia, via IV, iniciado no 7° dia de infecção e tratados por 20 dias consecutivos, foram melhores que BZ 100mg/kg/dia, via oral, por 20 dias consecutivos (tratamento clássico em modelo murino). Estudo in vitro em células tumorais
[00158] A tabela 13 resume os dados principais obtidos com atividade da Licnofolida em células tumorais. Tabela 13: Estudo da ação antitumoral in vitro para a licnofolida (1)
CIC50 (concentração que inibe 50 % do crescimento das células tumorais em relação ao controle, em ■ g/mL), CIC100 (concentração que inibe 100 % do crescimento das células tumorais em relação ao controle, em ■ g/mL) e CL50 (concentração letal para 50 % das células tumorais em relação ao controle, em - g/mL). Fonte: SAÚDE-GUIMARÃES, D.A. Transformações químicas, microbiológicas e atividades biológicas de lactonas sesquiterpênicas. Belo Horizonte. Tese (doutorado). UFMG, 1998. Exemplo 5: Estudo farmacocinético da LIC livre e encapsulada
[00159] A nanotecnologia tem sido bastante empregada no direcionamento de fármacos. Empregando uma formulação em escala nanométrica é possível aumentar o grau de dissolução de fármacos, resultando em um aumento de sua absorção e biodisponibilidade. Usando as nanopartículas para direcionamento de fármacos e melhorando a seletividade tecidual, isto pode ser alcançado com a captação seletiva do fármaco pelo tecido alvo, diminuindo também os efeitos adversos do fármaco. Frequentemente os perfis farmacocinéticos do fármaco livre e encapsulado em sistemas vetorizados são bem diferentes (Figura 4). Polímeros tais como o poli ácido lático (PLA), poli-ε- caprolactona (PCL), PLA-PEG são os mais comumente utilizados por serem biodegradáveis e biocompatíveis. Os sistemas nanoparticulares preparados a partir de tais polímeros permitem a encapsulação de agentes antiparasitários com vistas à liberação controlada dos mesmos ou ainda fornecer uma atividade terapêutica maior ou mais durável, quando comparada com aquela produzida pela administração do fármaco livre como foi observada neste estudo e demonstrado na figura 4 quando compara a licnofolida livre e a licnofolida nanoencapsulada. Além disso, tais sistemas possibilitam a administração intravenosa de moléculas hidrofóbicas, em elevadas concentrações, melhorando ainda a estabilidade proporcionada pela presença da barreira polimérica, frente à degradação química e enzimática em meio fisiológico Mosqueira et al., Biomaterials, 22, 2967-2979,2001)
[00160] Para a realização do estudo de farmacocinética, primeiramente fez-se necessário a validação para quantificação da licnofolida em matriz biológica, no aqui o plasma. Para a validação do método bioanalitico foram determinadas a especificidade, a recuperação, a linearidade, a precisão (inter e intradia), a exatidão, o limite de detecção, o limite de quantificação e a estabilidade, segundo os critérios de aceitação estabelecidos pelo Guia de Validação de Métodos Bioanalíticos do FDA (US Food and Drug Administration, 2001). O método bioanalítico aplicado aqui foi capaz de determinar as concentrações de licnofolida no plasma de camundongos após a administração intravenosa da substância livre e nanoencapsulada, utilizando polímeros de PCL. Um estudo de biodistribuição foi usado para investigar o efeito de doses crescentes de partículas no plasma de camundongos (Figura 4).
[00161] O estudo farmacocinético foi conduzido com licnofolida livre e nanoencapsulada, com polímeros de PCL na concentração de 2,0 mg/mL pela via intravenosa em camundongos sadios. Os perfis farmacocinéticos obtidos após a administração intravenosa de uma solução de licnofolida livre e licnofolida nanoencapsulada com polímeros de PCL são mostrados na Figura 4. A solução da LIC livre foi preparada numa concentração de 2,0 mg/mL sendo que foi injetada uma dose de 2,0 mg/Kg e um volume de 200μL. Durante o preparo das soluções de licnofolida, o mesmo foi primeiramente solubilizado em DMA e o PEG 300 adicionado à solução resultante. A solução de glicose 5% (p/v) foi incorporada lentamente à solução de DMA/PEG 300 sob leve agitação em vórtex (vórtex MS1, IKA Works, EUA). A suspensão coloidal de nanocápsulas contendo LIC também foi preparada numa concentração final de 2,0 mg/mL, sendo que foi injetada uma dose de 2,0 mg/Kg e um volume de 200μL. Antes da administração IV as nanocápsulas com e sem licnofolida, licnofolida livre, foram isotonizadas com glicose e filtradas em filtro Millipore 0,45 μm para garantir a administração de partículas inferiores a 5 μm, reduzindo assim a possibilidade de formação de êmbolos administrou-se 200 μL da solução de LIC livre e NC-LIC , respectivamente, por via intravenosa nos intervalos de tempos de 5min., 20min., 40min., 1h, 3h, 6h, 12h, após a injeção, o sangue (200 μL) foi coletado em tubos heparinizados, centrifugado por 15min a 800 x g separando-se 90 μL de plasma e adicionando 10 μL da solução de PI (no caso itraconazol). As amostras foram então extraídas, o solvente foi evaporado e o resíduo ressuspenso com 100 μL de fase móvel. Para determinar a quantidade total de LIC no plasma, foi assumido que o volume de plasma por camundongo é 4,9% do peso total (Auletta, Acute subchronic and chronic toxicology, in: M.J. Derelanko, M.A. Hollinger (Eds.), CRC Handbook of Toxicology, CRC Press, Boca Raton, 51-103, 1995). Os resultados foram expressos como média da porcentagem da dose administrada ± erro padrão (n=6 camundongos). Para comparar as diferentes formulações, a área sob a curva (AUC) (concentração x tempo) da LIC livre NC-LIC foi calculada pelo método trapezoidal durante o tempo experimental (AUC).
[00162] A concentração plasmática de licnofolida nanoencapsulada foi significativamente superior (p <0,001) do que as concentrações de licnofolida livre após 5, 20,40 e 60 min da injeção. Os perfis farmacocinéticos obtidos após a administração intravenosa da solução de LIC livre e de NC-LIC com nanocápsulas de PCL estão demonstrados da figura 4. A concentração plasmática da LIC em NC-PCL foi significativamente maior (p<0,001) que a sua concentração livre após 5,20,40,60 minutos de injeção, sugerindo uma rápida depuração plasmática da substância livre. Após 3 h, a concentração plasmática de LIC NC-PCL diminuiu acentuadamente até 6 h, porém, no tempo de 12 h ocorreu um aumento da concentração em relação à 6h, isto ocorreu provavelmente, pelo fato que a LIC foi liberada das NC ou as NC de PLC foram fagocitadas pelas células do SFM e se acumularam no fígado e baço dos animais, já que as NCs de PCL são nanocápsulas com superfície hidrofóbica, sendo vulneráveis a opsonização por proteínas plasmáticas que favorece a fagocitose destas nanopartículas e o acúmulo nos tecidos do sistema fagocítico mononuclear pouco tempo depois da sua administração endovenosa. A AUC da licnofolida nanoencapsulada foi 20 vezes superior em relação à AUC da substância administrada na forma livre, o que implica em maior tempo de residência no sangue após administração na forma nanoestruturada.
[00163] Portanto, as formas nanoparticuladas aumentaram a atividade da licnofolida, possivelmente devido ao aumento da biodisponibilidade do fármaco, com a associação da licnofolida com o núcleo oleoso da nanocápsula.