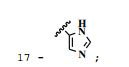Campo da Invenção
[0001] A presente invenção se refere a um conjunto de acil-hidrazonas derivadas da 3,4,5-trimetoxifenil- hidrazida, para o tratamento de doenças associadas à proliferação celular (como leucemias, tumores, inflamação e outras doenças proliferativas), dentre as quais, duas estruturas são consideradas possíveis protótipos de fármacos ou fármacos (compostos 02 e 07). Mais especificamente, a invenção se refere a compostos que têm atividade inibitória de proteínas quinase dependente de ciclina (CDKs) e da topoisomerase I, que podem ser consequentemente úteis no tratamento de leucemia linfóide aguda (LLA). A invenção se refere ainda à obtenção de acil- hidrazonas e oxadiazóis inéditos, a partir da 3,4,5- trimetoxifenil-hidrazida.
Antecedentes da Invenção
[0002] O câncer é uma doença caracterizada pela proliferação e propagação descontrolada pelo corpo de formas anormais das próprias células humanas. As células neoplásicas diferenciam-se das células normais pelo alto poder de invasão que possuem, pela perda de função, perda de diferenciação e pela capacidade de metástase, por possuírem menor adesão entre si (Rang, H. P.; Dale, M. M.; Ritter, J. M; Moore, P. K. Farmacologia. 5 ed. Rio de Janeiro: Elsevier, 2004. 703 p). A estimativa do Instituto Nacional do Câncer é do surgimento de 49 mil novos casos de câncer de mama, 47 mil de próstata, 27 mil de pulmão, 25 mil de cólon e reto, 23 mil de estômago e 19 mil de colo do útero em 2009 no Brasil (INCA, Instituto Nacional do Câncer. Disponível em: <http://www.inca.gov.br/estimativa/2006/index.asp?link=mapa .asp&ID=8> Acesso em: 23 fevereiro 2011), sendo a segunda maior causa de mortes no país.
[0003] As leucemias são um dos vários tipos de câncer, e ocorrem pela proliferação neoplásica de células hematopoiéticas linfóides ou mielóides, resultante da mutação de uma única célula-tronco, cuja prole forma um clone de células leucêmicas. Geralmente, ocorrem várias alterações genéticas para a transformação maligna, incluindo expressão inadequada de oncogenes e perda de função de genes supressores de tumor (Bain, B. J. Diagnóstico em leucemias. Rio de Janeiro: Elsevier, 2003, Cap. 1, 01-56), que podem estar associadas a fatores genéticos ou de risco (como tabagismo, exposição à radiação ou a produtos químicos como o benzeno) (INCA, Instituto Nacional do Câncer. Leucemia - prevenção, genética, outros fatores de risco. Disponível em: <http://www2.inca.gov.br/wps/wcm/connect/tiposdecancer/ site/home/leucemia/prevencao_genetica_outros_fatores_de_ris co>. Acesso em: 23 fevereiro 2011; e IARC (Internatonal Agency for Research on Cancer). World Cancer Report 2008. Disponível em: <http://www.iarc.fr/en/publications/pdfs- online/wcr/2008/index.php> e <http://www.iarc.fr/en/publications/pdfs- online/wcr/2008/wcr_2008.pdf>. Acesso em: 23 fevereiro 2011). As leucemias são subdivididas com base em quão rapidamente a doença evolui e torna-se grave, podendo ser crônicas ou agudas. As leucemias agudas se caracterizam por um defeito na maturação celular, o que ocasiona um desequilíbrio entre a proliferação e a maturação; uma vez que as células do clone leucêmico continuam a se proliferar, sem chegar aos estágios de maturação e morte, ocorre uma expansão contínua do clone leucêmico e predomínio das células imaturas (INCA 2009b, Bain, B. J. Diagnóstico em leucemias. Rio de Janeiro: Elsevier, 2003, Cap. 1, 01-56).
[0004] A leucemia linfóide aguda (LLA) é decorrente da proliferação descontrolada de células progenitoras linfóides imaturas na medula óssea, que resulta em um acúmulo muito rápido de células neoplásicas (Plasschaert, S.; Van Der Kolk, D.; De Bont, E.; Vellenga, E.; Kamps, W.; De Vries, E. Breast Cancer Resistance Protein (BCRP) in Acute Leukemia. Leukemia & Lymphoma, 2004, 45, 649-654). É responsável por 80% dos casos de leucemia aguda na infância (Laks, D.; Longhi, F., Bernardes, W. M.; Ramos, G. P. C. J Pediatr, 2003, 79, 149-158) e 50% de todas as malignidades hematopoiéticas (Downing, James R.; Shannon, Kevin M. Acute leukemia: A pediatric perspective. Cancer Cell, 2002, 2, 437-445). Em adultos, a LLA é relativamente rara, contabilizando 2-3% das malignidades hematopoiéticas (Downing, James R.; Shannon, Kevin M. Acute leukemia: A pediatric perspective. Cancer Cell, 2002, 2, 437-445); porém, o prognóstico é muito pior que para as crianças, pois acomete células-tronco multipotentes, originando uma leucemia muito mais agressiva (Greaves, M. F. Stem cell origins of leukaemia and curability. British Journal of Cancer, 1993, 67, 413-423).
[0005] As pesquisas do uso de compostos de fontes naturais como quimioterápicos é ampla. Um exemplo disso é o antineoplásico paclitaxel (TaxolTM), um dos mais importantes produtos antitumorais naturais já descobertos, reportado pela primeira vez em 1971 (Wani, M. C.; Taylor, H. L.; Wall, M. E.; Coggon, P.; McPhail, A. T. Plant antitumor agents. VI. Isolation and structure of taxol, a novel antileukemic and antitumor agent from Taxus brevifolia. Journal of the American Chemical Society, 1971, 93, 2325-2327), que somente depois de um longo período foi aprovado pelo FDA para o uso clínico (em 1992). Outro exemplo é a vincristina, um alcalóide utilizado na terapia de leucemia aguda e outros tipos de tumores, que atua da mesma forma que o paclitaxel, através da ligação à tubulina, interferindo na formação (polimerização) ou reorganização (despolimerização) dos microtúbulos (Goodman e Gilman. As bases farmacológicas da terapêutica,10aEd., editora Mc Graw Hill. 2006).
[0006] Os quimioterápicos mais usados na terapia atual das leucemias incluem daunorrubicina, doxorrubicina, dexametasona, vincristina, metotrexato e mercaptopurina (Plasschaert, S.; Van Der Kolk, D.; De Bont, E.; Vellenga, E.; Kamps, W.; De Vries, E. Breast Cancer Resistance Protein (BCRP) in Acute Leukemia. Leukemia & Lymphoma, 2004, 45, 649-654). Estes fármacos conferem benefício terapêutico, mas também significativa toxicidade ao organismo e às células normais, devido à sua atuação na indução de apoptose e inibição da proliferação celular (Herr, I.; Debatin, K. M. Cellular stress response and apoptosis in cancer therapy. Blood 2001, 98, 2603-2614; e Leszczyniecka, M.; Roberts, T.; Dent, P.; Grant, S.; Fisher, P. B. Differentiation therapy of human cancer: basic science and clinical applications. Pharmacology & Therapeutics, 2001, 90, 105-156). Os agentes antineoplásicos interferem também nos tecidos normais que possuem células de divisão rápida, podendo provocar muitos efeitos indesejáveis, como a redução da produção das células de defesa do organismo, a cicatrização deficiente de feridas, alopecia, lesão do epitélio gastrintestinal, esterilidade e teratogenicidade (Rang, H. P.; Dale, M. M.; Ritter, J. M; Moore, P. K. Farmacologia. 5 ed. Rio de Janeiro: Elsevier, 2004. 703 p).
[0007] Apesar dos recentes avanços na pesquisa contra o câncer, em 2008 foram registradas 5.686 mortes decorrentes de leucemia no Brasil, e para 2010 foram estimados 9.580 novos casos desta doença (INCA, Instituto Nacional do Câncer. Disponível em: <http://www.inca.gov.br/estimativa/2006/index.asp?link=mapa .asp&ID=8> Acesso em: 23 fevereiro 2011), o que motiva incessantemente a busca por novos fármacos para o tratamento de leucemias e outros tumores.
[0008] Em um recente estudo, Wang e colaboradores (Wang, Z.; Lu, Y.; Seibel, W.; Miller, D. D.; Li, W. Identifying Novel Molecular Structures for Advanced Melanoma by Ligand-Based Virtual Screening. Journal of Chemical Information and Modeling, 2009, 49, 1420-1427) avaliaram novos compostos antitumorais baseados na estrutura do composto LY-1-100, que tem comprovado mecanismo de ação e afinidade aos microtúbulos (sítio de ligação da colchicina), porém baixa seletividade.
[0009] Estruturas foram selecionadas através de screening virtual baseado no ligante (ligand-based), resultando em 14 novas moléculas com grande similaridade estrutural ao composto de partida (LY-1-100). Os resultados teóricos obtidos pelos pesquisadores revelaram que o anel trimetoxilado do composto LY-1-100 precisa ser mantido para proporcionar a atividade antitumoral, mas o anel triazol parece não ser importante, podendo ser substituído pela estrutura da N-metileno-hidrazina, o que gera a estrutura das acil-hidrazonas. Apesar dos resultados em células murinas B16F1 e A375 (in vitro) destes compostos não terem apresentado melhoria da potência e da seletividade do composto de partida, estes resultados foram muito promissores e identificaram alguns componentes estruturais importantes para a atividade anti-melanoma (Wang, Z.; Lu, Y.; Seibel, W.; Miller, D. D.; Li, W. Identifying Novel Molecular Structures for Advanced Melanoma by Ligand-Based Virtual Screening. Journal of Chemical Information and Modeling, 2009, 49, 1420-1427).

[0010] Neste sentido, as acil-hidrazonas surgem como uma classe interessante de compostos com atividade antitumoral. Assim, a presente invenção se refere à obtenção de acil-hidrazonas, especialmente derivadas da 3,4,5-trimetoxifenil-hidrazida, para o tratamento de doenças associadas à proliferação celular (como leucemias, tumores, inflamação e outras doenças proliferativas).
[0011] O pedido de patente PI 0112674-1 (Inibidores n-[5-[[[5-alquil-2-oxazolil]metil]tio]-2-tiazolil] carboxamida de quinases dependentes de ciclina) descreve compostos e seus enantiomorfos, diastereoisômeros, solvatos e sais farmaceuticamente aceitáveis como inibidores de proteína quinase, úteis no tratamento de doenças proliferativas, como por exemplo, câncer, inflamação e artrite. Os compostos sintetizados também podem ser de utilidade para o tratamento da doença de Alzheimer, alopécia induzida por quimioterapia e doença cardiovascular. Os compostos descritos no pedido PI 0112674-1 representam estruturas mais complexas que as apresentadas no decorrer da presente invenção.
[0012] Os compostos referenciados na publicação de pedido de patente n° US 20040138272 (1,4-Substituted cyclohexane derivatives) podem ser úteis na prevenção da proliferação celular em doenças malignas, por inibir Rho quinases, úteis na reparação do sistema nervoso central e periférico por indução do crescimento e regeneração dos axônios. O mecanismo de ação dos compostos da publicação US 20040138272 difere do proposto para as estruturas da presente invenção.
[0013] Inibidores de quinases dependentes de ciclina (cdks) úteis na modulação da progressão do ciclo celular são propostos no pedido de patente PI 0418095-0 (Inibidores de quinases dependentes de ciclina, composições e usos relacionados aos mesmos). Tais compostos seriam úteis para o tratamento de pacientes que apresentam distúrbios associados à proliferação celular excessiva. Os compostos descritos no PI 0418095-0 são acil-hidrazonas diferentes das propostas no decorrer da presente invenção, com processo de síntese mais complexo.
[0014] No pedido de patente PI 0508364-8 (Derivados de 4-benzimidazol-2-il-piridazin-3-ona) são descritos compostos e seus sais fisiologicamente tolerados, que apresentam ação como inibidores de quinases, em particular da quinase CDK2 (quinase 2 dependente de ciclina). Os compostos do PI 0508364-8 são diferentes dos propostos no decorrer da presente invenção, com síntese mais complexa.
[0015] A publicação de pedido de patente norte- americano n° US 20070066610 (Acylhydrazones as kinase modulators) descreve acil-hidrazonas como inibidores de tirosinas quinases, compreendendo c-Met, um receptor tirosina quinase que regula a proliferação celular, morfogênese e motilidade. As acil-hidrazonas descritas no documento US 20070066610 são diferentes das propostas no decorrer deste invento, com síntese mais complexa. Além disso, o alvo de ação dos compostos descritos se diferencia do proposto nesta invenção.
[0016] A publicação de pedido de patente US 20080194562 (Pyrazole Derivatives for The Inhibition Of Cdk's And Gsk's) se refere à síntese de pirazóis, compostos que inibem ou modulam a atividade de quinases dependentes de ciclina (CDK) e quinases glicogênio sintase (GSK), e ao seu uso no tratamento ou profilaxia de doença ou condição mediada por quinases. Também são descritas composições farmacêuticas contendo os compostos e intermediários químicos. Os compostos do documento US 20080194562 são diferentes dos propostos no decorrer do presente invento.
[0017] As acil-hidrazonas ainda são descritas na literatura por suas pronunciadas atividades inseticidas e estimulantes de crescimento de plantas (Robinson, B. Fischer indole synthesis. Chem. Rev., 1963, 4, 373-401); no tratamento da tuberculose (Vigorita, M. G.; Ottana, R.; Zappala, C.; Maccari, R.; Pizzimenti, F. C.; Gabbrielli, G. Halogenated isoniazid derivatives as possible antimycobacterial and anti-HIV agents - III. Farmaco, 1994, 49, 775-781); e como agentes bacteriológicos e bacteriostáticos (Samus, N. M.; Tsapkov, V. I.; Kuracheva, S. A.; Burdenko, T. A. Synthesis and antimicrobial activity of coordination compounds of 3d-elements with some hydrazones derived by using 5-nitro-2-furaldehyde. Khimiko- Farmatsevticheskii Zhurnal, 1994, 28, 41-44).
[0018] Desta forma, foi conduzido um estudo com acil- hidrazonas e seus derivados oxadiazóis obtidos pela estratégia de bioisosterismo não clássico, de fechamento de anel. Os oxadiazóis são uma importante classe de compostos heterocíclicos com uma ampla gama de atividades biológicas, tais como antiviral, antimicrobiana, antineoplásica, fungicida, inibição de tirosinase e catepsina K (Kumar, D.; Sundaree, S.; Johnson, E. O.; Shah, K. An efficient synthesis and biological study of novel indolyl-1,3,4- oxadiazoles as potent anticancer agents. Bioorganic & Medicinal Chemistry Letters, 2009, 19, 4492-4494). Além disso, são ótimos bioisósteros de amidas e ésteres, que podem contribuir substancialmente no aumento da atividade farmacológica, participando em ligações de hidrogênio como receptores (Guimaraes, C. R. W.; Boger, D. L.; Jorgensen, W. L. Elucidation of Fatty Acid Amide Hydrolase Inhibition by Potent α-Ketoheterocycle Derivatives from Monte Carlo Simulations. Journal of the American Chemical Society, 2005, 127, 17377-17384).
[0019] Assim, as acil-hidrazonas específicas e seus análogos, provenientes dos processos descritos detalhadamente neste invento a seguir, bem como seu uso no tratamento de leucemias, tumores e outras doenças proliferativas, como inflamação, é de grande interesse social e econômico.
Objetivos da Invenção
[0020] O objetivo da presente invenção é a obtenção de compostos sintéticos derivados da 3,4,5-trimetoxifenil- hidrazida (hidrazonas e oxadiazóis) e todos os seus compostos análogos e semelhantes, por meio de síntese simples, bem como o emprego destes compostos para o tratamento de doenças associadas à proliferação celular (como leucemias, especialmente leucemia linfóide aguda - LLA - tumores, inflamação e outras doenças proliferativas). A presente invenção também descreve os processos utilizados para a determinação da atividade biológica destes compostos.
Sumário da Invenção
[0021] A presente invenção se refere a uma classe de acil-hidrazonas, especialmente as derivadas da 3,4,5- trimetoxifenil-hidrazida, bem como a seus compostos análogos oxadiazóis e outros compostos análogos e semelhantes, e a aplicação farmacêutica de todos estes no tratamento de diferentes doenças associadas à proliferação celular, como leucemias, compreendendo leucemia linfóide aguda (LLA), tumores e inflamação. A presente invenção também descreve os processos utilizados para a determinação da atividade biológica de todos esses compostos.
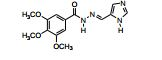
[0022] Foram obtidas acil-hidrazonas com atividade semelhante ao composto utilizado como padrão nos experimentos (colchicina). A maior seletividade dos compostos apresentados nesta invenção é uma característica importante relacionada com menores efeitos colaterais que os fármacos atualmente empregados na clínica. As acil- hidrazonas sintetizadas, mais especificamente os compostos 02 e 07, apresentaram importante atividade antileucêmica, o que indica 02 e 07 como candidatos a protótipos de fármacos, ou fármacos, para o tratamento de leucemias, especialmente leucemia linfóide aguda (LLA), tumores e outras doenças proliferativas, como inflamação.
[0023] A determinação do mecanismo de ação dos compostos mais ativos foi realizada pela utilização de microarranjos de DNA e testes subsequentes indicados através do chip, além dos estudos de seletividade em linfócitos humanos saudáveis.
Descrição Detalhada da Invenção
[0024] A presente invenção compreende a obtenção e mecanismo de ação de acil-hidrazonas sintéticas e seus compostos análogos e semelhantes, que podem ser úteis no tratamento de leucemias, especialmente leucemia linfóide aguda (LLA), tumores e outras doenças proliferativas, como inflamação.
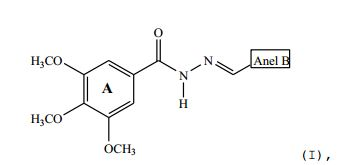
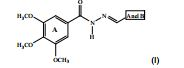
[0025] De acordo com um dos aspectos da presente invenção, é descrito um composto de estrutura (I):
fenil; 15 - 4-O(CH2)3CH3-fenil; 16 - 4-CH3-fenil;
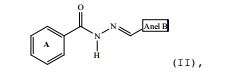
[0026] Um grupo complementar de compostos, de acordo com a presente invenção, compreende compostos com a estrutura (II):
em que o anel B representa: 30 - fenil; 31 - 3,4-OCH2O-fenil; 32 - 4-Br-fenil; 33 - 4-CH3-fenil; 34 - 1-naftil; e 35 - 2-naftil.
[0027] Adicionalmente, são também descritos os análogos sintéticos oxadiazóis, de acordo com a estrutura (III):
em que o anel B representa: 36 - 3-OCH3-4-OH-5-Br-fenil; 37 - 3-OCH3-4-OH-fenil; e 38 - 1-naftil.
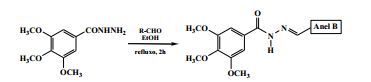
[0028] As acil-hidrazonas inéditas da presente invenção foram obtidas a partir da reação de condensação entre a 3,4,5-trimetoxifenil-hidrazida e diferentes aldeídos, usando etanol como solvente, em refluxo, conforme a reação:
em que o anel B representa:
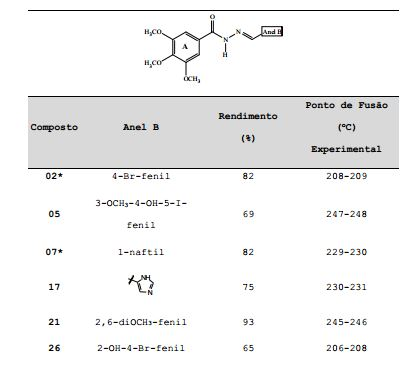
[0029] A Tabela 1 apresenta os rendimentos obtidos nas sínteses e os pontos de fusão experimental das 3,4,5- trimetoxifenil-hidrazonas inéditas. Tabela 1. Rendimentos e pontos de fusão das 3,4,5- trimetoxifenil-hidrazonas inéditas sintetizadas.
*Os compostos 02 e 07 não são inéditos, mas também são protegidos nesta invenção devido à sua pronunciada atividade antileucêmica, que será descrita no decorrer do presente invento.
[0030] A estrutura do composto 02 foi previamente publicada como intermediário de reação para obtenção de oxadiazóis (Mazzone, G.; Bonina, F.; Formica, F. Some aroylhydrazones of halobenzaldehydes and halo-substituted 2,5-diaryl-1,3,4-oxadiazoles. Farmaco, Edizione Scientifica, 1978, 33(12), 963-71) e o composto 07 foi previamente avaliado como inibidor da MAO (monoamino oxidase) (Mazzone, G.; Arrigo Reina, R. 3,4,5- Trimethoxybenzoyl hydrazides and their anti-MAO [monoamine oxidase] activity. Bollettino delle Sedute della Accademia Gioenia di Scienze Naturali in Catania, 1971, 10(8), 689 702). A caracterização química de ambos compostos (02 e 07) também foi previamente publicada por nosso grupo de pesquisas, em um trabalho que avaliou a atividade destes e de outros compostos como inibidores da cruzaína de Trypanosoma cruzi; entretanto, os compostos 02 e 07 não apresentaram atividade inibitória desta proteína (Borchhardt, Deise M.; Mascarello, Alessandra; Chiaradia, Louise Domeneghini; Nunes, Ricardo J.; Oliva, Glaucius; Yunes, Rosendo A.; Andricopulo, Adriano D. Biochemical evaluation of a series of synthetic chalcone and hydrazide derivatives as novel inhibitors of cruzain from Trypanosoma cruzi. Journal of the Brazilian Chemical Society, 2010, 21(1), 142-150).
[0031] As acil-hidrazonas complementares (preparadas para auxiliar na discussão dos testes biológicos) foram sintetizadas a partir da reação entre a fenil-hidrazida e diferentes aldeídos, pelo mesmo procedimento de preparação das hidrazonas descritas anteriormente, de acordo com a seguinte reação:
em que o anel B representa: 30 - fenil; 31 - 3,4-OCH2O-fenil; 32 - 4-Br-fenil; 33 - 4-CH3-fenil; 34 - 1-naftil; e 35 - 2-naftil.
[0032] Foram ainda preparados os análogos oxadiazóis sintéticos inéditos das 3,4,5-trimetoxifenil-hidrazonas, a partir da reação destas com anidrido acético, conforme a seguinte reação:
em que o anel B representa: 36 - 3-OCH3-4-OH-5-Br-fenil; 37 - 3-OCH3-4-OH-fenil; e 38 - 1-naftil.
[0033] A Tabela 2 apresenta os rendimentos obtidos nas sínteses e o ponto de fusão experimental dos oxadiazóis.Tabela 2. Rendimentos e ponto de fusão dos oxadiazóis inéditos sintetizados.
[0034] A presente invenção descreve também a determinação do mecanismo de ação das acil-hidrazonas sintetizadas e seus compostos análogos e semelhantes compreendendo oxadiazóis. A invenção se refere ainda ao uso de todos estes compostos como protótipos de fármacos, ou fármacos, para o tratamento de leucemias, especialmente leucemia linfóide aguda (LLA), tumores e outras doenças associadas à proliferação celular, como inflamação.
[0035] As acil-hidrazonas descritas na presente invenção atuam de maneira seletiva sobre células leucêmicas com atividade na ordem de nanomolar, quando comparadas à sua atividade em linfócitos humanos saudáveis, conforme será descrito a seguir na forma de exemplo. Fica evidenciada aqui a relevância dos resultados biológicos, inéditos para os compostos testados.
[0036] A presente invenção será agora descrita por meio de exemplos ilustrativos.
Exemplo 1. Procedimento geral para a preparação das 3,4,5-trimetoxifenil-hidrazonas 01-29
[0037] Para a síntese das 3,4,5-trimetoxifenil- hidrazonas 01-29, utilizou-se a metodologia descrita por Troeberg e col. (Troeberg, L.; Chen, X.; Flaherty, T. M.; Morty, R. E.; Cheng, M.; Hua, H.; Springer, C.; McKerrow, J. H.; Kenyon, G. L.; Lonsdale-Eccles, J. D.; Coetzer, T. H. T.; Cohen, F. E. Chalcone, acyl hydrazide, and related amides kill cultured Trypanosoma brucei brucei. Molecular Medicine, 2000, 6, 660-669). Em um balão de reação de 100 mL e 1 boca, colocou-se a 3,4,5-trimetoxifenil-hidrazida (2 mmol) preparada conforme descrito no exemplo 2, em um solvente orgânico: acetona, acetato de etila, éter etílico, etanol, metanol (20mL) e o aldeído apropriado (2 mmol). A mistura foi refluxada por 1 a 10 horas. Depois, a solução foi filtrada e o sólido recristalizado em um solvente orgânico.
Exemplo 2. Procedimento geral para obtenção da 3,4,5- trimetoxifenil-hidrazida, utilizada para obter as 3,4,5- trimetoxifenil-hidrazonas 01-29
[0038] Para a síntese da 3,4,5-trimetoxifenil- hidrazida usada na preparação das acil-hidrazonas 01-29, utilizou-se metodologia já descrita (Chida, A. S.; Vani, P. V. S. N.; Chandrasekharam, M.; Srinivasan, R.; Singh, A. K. Synthesis of 2,3-dimethoxy-5-methyl-1,4-benzoquinone: a key fragment in coenzyme-Q series. Synyhethic communications, 31, 657-660, 2001), realizada em duas etapas:
[0039] Obtenção do éster: Em um balão de reação de 1000 mL e 1 boca, colocou-se o ácido gálico (50g, 0,294mol), sulfato de dimetila (178,1g, 1,413mol), carbonato de potássio anidro (175,5g, 1,293mol) e TBAI (iodeto de tetra-n-butilamonio) (1g) em um solvente orgânico que pode ser etanol, éter etílico, acetato de etila, acetona, éter de petroleo (375mL) e foi refluxado por 1 a 12h. O sólido obtido foi filtrado e lavado com o mesmo solvente orgânico (3 x 50mL). O éster foi obtido em forma de sólido amorfo de cor creme, com rendimento de 78%; p.f.: 84 °C (lit. p.f.: 82-83 °C). RMN 1H (CDCI3) : 1,60 (s, 3H, CH3), 3,92 (s, 9H, OCH3), 7,33 (s, 2H, Ar).
[0040] Obtenção da hidrazida: Em um balão de reação de 1000 mL e 1 boca, colocou-se o éster obtido na primeira etapa (48g, 0,212mol), uma solução de hidrazina hidratada 99% (N2H4.H2O) (77,6g, 1,54mol) e um solvente orgânico que pode ser etanol, acetato de etila, diclorometano, acetona, metanol (200mL). A mistura foi refluxada por 1 a 5h e mantida apenas sob agitação magnética overnightà temperatura entre 0 e 50°C. O sólido obtido foi filtrado e recristalizado em metanol, obtendo a 3,4,5-trimetoxifenil- hidrazida na forma de cristais brancos, com rendimento de 85%; p.f.: 162-163 °C (lit. p.f.: 168 °C). RMN 1H (CDCl3): 3,80 (s, 3H, OCH3), 3,90 (s, 6H, OCH3), 7,18 (s, 2H, Ar), 9,55 (NH).
Exemplo 3. Procedimento geral para a preparação das fenil-hidrazonas 30-35
[0041] Para a síntese das fenil-hidrazonas 30-35, utilizou-se a metodologia descrita por Troeberg e col. (Troeberg, L.; Chen, X.; Flaherty, T. M.; Morty, R. E.; Cheng, M.; Hua, H.; Springer, C.; McKerrow, J. H.; Kenyon, G. L.; Lonsdale-Eccles, J. D.; Coetzer, T. H. T.; Cohen, F. E. Chalcone, acyl hydrazide, and related amides kill cultured Trypanosoma brucei brucei. Molecular Medicine, 2000, 6, 660-669), da mesma forma como descrito para a obtenção das 3,4,5-trimetoxifenil-hidrazonas.
Exemplo 4. Procedimento geral para a preparação dos oxadiazóis 36-38
[0042] Para a síntese dos oxadiazóis 36-38, utilizou- se a metodologia descrita por Jin e col. (Jin, L.; Chen, J.; Song, B.; Chen, Z.; Yang, S.; Li, Q.; Hu, D.; Xu, R. Bioorg Med Chem, 2006, 16, 5036-5040). Em um balão de reação de 100 mL e 1 boca, misturou-se a 3,4,5- trimetoxifenil-hidrazona correspondente (1 mmol) e anidrido acético (10mL). A mistura foi refluxada por 1 a 10 horas e após foi resfriada com adição de gelo picado e deixada a temperatura entre 0 e 60°C overnightpara precipitação do produto. O sólido obtido foi filtrado, lavado com água e recristalizado com solvente orgânico/água. DADOS ESPECTRAIS DE INFRAVERMELHO (IV) E RESSONÂNCIA MAGNÉTICA NUCLEAR (RMN) DE1H E DE13C DOS COMPOSTOS INÉDITOS δ ppm em relação ao TMS, Multiplicidade (J em Hz). Solvente CDCl3.
[0043] 05 - 1H NMR (DMSO-d6) δ 3.73 (s, 3H, p-OCH3), 3.86 (s, 6H, m-OCH3), 3.89 (s, 3H, m-OCH3), 7.22 (s, 2H, H2, H6), 7.34 (s, 1H, H6’), 7.60 (s, 1H, H2’), 8.31 (s, 1H, HC=N), 10.08 (1H, OH), 11.66 (s, 1H, NH). 13C NMR (DMSO-d6) δ 56.78 (m-OCH3), 60.81 (p-OCH3), 85.17 (C3’), 105.84 (C2, C6), 109.72 (C6’), 128.28 (C1), 129.31 (C1’), 130.75 (C2’), 141.03 (C4), 147.35 (C=N), 147.97 (C5’), 149.00 (C4’), 153.36 (C3, C5), 163.16 (C=O). IR Vmax/cm-1 3382 (N-H), 1636, 1228 (C=O), 1565 (C=N), 1290, 1045 (C-O), 2999, 2839, 1585, 1490, 1334, 1137, 997 (Ar) (KBr).
[0044] 17 - 1H NMR (DMSO-d6) δ 3.74 (s, 3H, p-OCH3), 3.88 (s, 6H, m-OCH3), 7.23 (s, 2H, H2, H6), 7.53 (s, 1H, H2’), 7.66 (m, 1H, H4’), 8.02 (s, 1H, NH5’), 8.43 (s, 1H, HC=N), 11.37 (s, 1H, NH). 13C NMR (DMSO-d6) δ 56.76 (m- OCH3), 60.81 (p-OCH3), 105.71 (C2, C6), 129.51 (C1), 131.94 (C1’), 132.16 (C2’), 135.37 (C4’), 145.45 (C=N), 153.35 (C3, C5), 162.29 (C=O). IR V^/cm-1 3212 (N-H), 1623, 1234 (C=O), 1580 (C=N), 1280, 1054 (C-O), 2994, 2941, 2838, 1503, 1456, 1411, 1344, 1125, 1006, 844 (Ar) (KBr).
[0045] 21 - 1H NMR (DMSO-de) δ 3.71 (s, 3H, p-OCH3), 3.79 (s, 6H, o-OCH3), 3.85 (s, 6H, m-OCH3), 6.72 (d, J = 8.0 Hz, 2H, H3’, H5’), 7.23 (s, 2H, H2, H6), 7.34 (t, J = 8.0 Hz, 1H, H4’), 8.60 (s, 1H, HC=N), 11.52 (s, 1H, NH). 13C NMR (DMSO-d6) δ 56.72 (m-OCH3), 56.78 (o-OCH3), 60.76 (p-OCH3), 105.08 (C3’, C5’), 105.76 (C2, C6), 111.75 (C1’), 129.37 (C1), 131.87 (C4’), 140.88 (C4), 143.92 (C=N), 153.31 (C3, C5), 159.38 (C2’, C6’), 162.81 (C=O). IR Vmax/cm-1 3186 (N-H), 1644, 1240 (C=O), 1586 (C=N), 1258, 1068 (C-O), 3002, 2928, 2838, 1502, 1473, 1417, 1378, 1342, 1121, 1007, 783 (Ar) (KBr).
[0046] 26 - 1H NMR (DMSO-d6) δ 3.42 (s, 3H, p-OCH3), 3.85 (s, 6H, m-OCH3), 6.89 (d, J = 8.6 Hz, 1H, H5’), 7.25 1416, 1352, 1335, 1189, 1011, 992, 951, 839, 760 (Ar) (KBr).
[0047] 29 - 1H NMR (DMSO-d6) δ 3.72 (s, 3H, p-OCH3), 3.85 (s, 6H, m-OCH3), 7.22 (s, 2H, H2, H6), 7.82 (d, J = 8.6 Hz, 1H, H5’), 8.02 (d, J = 8.6 Hz, 1H, H6’), 8.15 (s, 1H, H2’), 8.51 (s, 1H, HC=N), 11.96 (s, 1H, NH). 13C NMR (DMSO-d6) δ 56.65 (m-OCH3), 60.73 (p-OCH3), 105.96 (C2, C6), 126.29 (C2’), 127.98 (C1’), 128.88 (C1), 132.31 (CF3), 132.91 (C4’), 133.01 (C5’), 134.82 (C6’), 141.27 (C4), 145.61 (C=N), 153.40 (C3, C5), 163.50 (C=O). IR Vmax/cm-1 3182 (N-H), 1655, 1242 (C=O), 1587 (C=N), 1269, 1039 (C-O), 3008, 2938, 2838, 1506, 1480, 1417, 1336, 1316, 1173, 1121, 1006, 958, 666 (Ar) (KBr).
[0048] 36 - 1H NMR (DMSO-d6) δ 2.36 (s, 3H, CH3), 3.85 (s, 3H, p-OCH3), 3.91 (s, 3H, m-OCH3), 3.92 (s, 6H, m-OCH3), 6.00 (s, 1H, OH), 6.99 (s, 1H, H2’), 7.06 (s, 1H, H6’), 7.11 (s, 2H, H2, H6), 7.26 (s, 1H, HC-N). 13C NMR (DMSO-d6) δ 21.75 (CH3), 56.56 (m-OCH3), 61.25 (p-OCH3), 91.52 (C-N), 104.44 (C2, C6), 117.93 (C5’), 119.37 (C2’), 122.52 (C6’), 136.02 (C1’, C1), 139.24 (C4’), 141.49 (C4), 152.90 (C3’), 153.64 (C3, C5), 155.84 (C=N), 168.32 (C=O). IR V^/cm"1 1766, 1238 (C=O), 1667, 1582 (C=N), 1254, 1047 (C-O), 1177 (C-N), 3445 (OH), 1130, 621 (C-Br), 3004, 2941, 2838, 1466, 1416, 1366, 1306, 1190, 998, 858 (Ar) (KBr).
[0049] 37 - 1H NMR (DMSO-d6) δ 2.35 (s, 3H, CH3), 3.82 (s, 3H, p-OCH3), 3.90 (s, 3H, m-OCH3), 3.92 (s, 6H, m-OCH3), 6.97 (s, 1H, H2’), 7.06 (m, 1H, H5’), 7.10 (s, 2H, H2, H6), 7.25 (s, 1H, HC-N), 7.44 (m, 1H, H6’); o sinal correspondente ao grupo OH não é observado. 13C NMR (DMSO- d6) δ 21.54 (CH3), 56.27 (m-OCH3), 56.33 (m-OCH3), 61.02 (p-OCH3), 91.14 (C-N), 104.22 (C2, C6), 109.77 (C2’), 119.16 (C5’), 128.09 (C6’), 136.402 (C1’, C1), 141.24 (C4’), 141.96 (C4), 151.78 (C3’), 153.42 (C3, C5), 155.60 (C=N), 168.10 (C=O). IR Vmax/cm-1 1767, 1243 (C=O), 1665, 1581 (C=N), 1250, 1043 (C-O), 1177 (C-N), 3445 (OH), 1129, 644 (C-Br), 2967, 2945, 2838, 1507, 1466, 1417, 1365, 1036, 1287, 1197, 1083, 997, 958, 861, 699 (Ar) (KBr).
[0050] 38 - 1H NMR (DMSO-de) δ 2.47 (s, 3H, CH3), 3.87 (s, 9H, OCH3), 7.08 (s, 2H, H2, H6), 7.26 (s, 1H, HC-N), 7.26 (m, 1H, H2’), 7.48 (t, J = 8.0 Hz, 1H, H3’), 7.55 (t, J = 8.0 Hz, 1H, H7’, H8’), 7.62 (t, J = 8.0 Hz, 1H, H7’), 7.76 (m, 1H, H4’), 7.92 (d, J = 8.0 Hz, 1H, H6’), 8.22 (d, J = 8.0 Hz, 1H, H9’). 13C NMR (DMSO-d6) δ 21.57 (CH3), 56.28 (m-OCH3), 60.98 (p-OCH3), 91.17 (C-N), 104.27 (C2, C6), 119.61 (C1), 123.04 (C2’), 125.07 (C9’), 125.20 (C3’), 126.05 (C7’), 126.95 (C8’), 128.95 (C4’), 130.51 (C6’), 130.61 (C10’), 130.79 (C5’), 134.04 (C1’), 141.11 (C4), 153.29 (C3, C5), 155.83 (C=N), 168.26 (C=O). IR V^/cm-1 1731, 1243 (C=O), 1669, 1587 (C=N), 1254, 1039 (C-O), 1124 (C-N), 2997, 2941, 2827, 1509, 1465, 1416, 1369, 1332, 1191, 1006, 980, 847, 784, 699 (Ar) (KBr).
[0051] Diluição e armazenamento dos compostos: Todos os compostos foram ressuspendidos em DMSO na concentração estoque de 20 mM e armanezados a -20°C. Para os testes de citotoxicidade, as diluições a partir das soluções estoque foram feitas em meio de cultura (RPMI-1640 acrescido de 10% de soro fetal bovino, 100 UI/mL penicilina e 100 pg/mL estreptomicina), imediatamente antes de serem lançadas às células.
[0052] Ensaios in vitro da sensibilidade/resistência das células aos compostos pelo método do MTT: As linhagens celulares REH e Jurkat foram mantidas em meio RPMI-1640 com 10% de SFB (soro fetal bovino), 100 UI/mL penicilina, 100 pg/mL estreptomicina e incubadas a 37°C e 5% CO2. Os ensaios de citotoxicidade foram realizados de acordo com método descrito na literatura (Pieters, R.; et al. In vitro drug sensitivity of cells from children with leukemia using the MTT assay with improved culture conditions. Blood, 1990, 76, 2327-2336; Pieters, R.; et al. Relation of cellular drug resistance to long-term clinical outcome in childhood acute lymphoblastic leukaemia. Lancet, 1991, 338, 399-403 e Kaspers, G. J.; et al. In vitro cellular drug resistance and prognosis in newly diagnosed childhood acute lymphoblastic leukemia. Blood, 1997, 90, 2723). As células foram ressuspendidas na concentração de 3,75 x 105 células/mL em meio de cultura, como descrito. Oitenta microlitros desta suspensão foram semeados em placas de 96 poços de fundo arredondado, contendo 20 microlitros de diferentes concentrações do composto ou apenas veículo. Cada tratamento foi feito em triplicata. Após 48 horas de incubação a 37°C e 5% CO2, foram adicionados 20 μL de solução MTT (5 mg/mL de PBS 1x), seguindo-se nova incubação por 4h e 30min a 37°C e 5% CO2. Durante estas 4h e 30min, o MTT (de coloração amarelada) é metabolizado a sal de formazan (de coloração azulada) pelas células vivas. A seguir, foram acrescentados 100 μL de dodecil sulfato de sódio (DSS) 10% + 0,1M de HCl, para a dissolução dos cristais de formazan. Após nova incubação overnight, procedeu-se à leitura de absorbância a 570 nm. Os percentuais de células sobreviventes aos tratamentos foram calculados em relação ao número de células sobreviventes no meio sem adição dos compostos em questão (“controle negativo”).
[0053] c) Determinação da IC50: A IC50 é definida como a concentração de um composto na qual é obtida 50% da inibição máxima. Após a leitura da absorbância, foram construídas curvas de sobrevivência e obtidos os valores de IC50 com o auxílio do software GraphPad Prism.
RESULTADOS: AÇÃO DAS ACIL-HIDRAZONAS E OXADIAZÓIS SOBRE CÉLULAS LEUCÊMICAS DE LINHAGEM B E T (REH E JURKAT, RESPECTIVAMENTE)
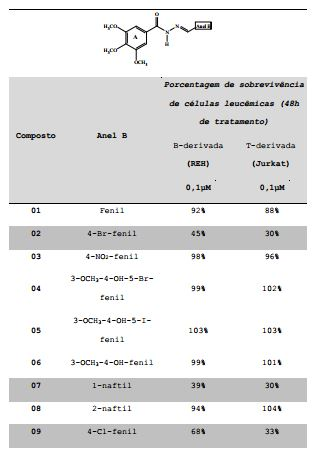
[0054] Foi estudado o efeito citotóxico dos compostos sintetizados em células leucêmicas humanas Jurkat e REH, por meio de ensaio de viabilidade celular (MTT), de acordo com os métodos descritos por Pieters e colaboradores (Pieters, R.; et al. In vitro drug sensitivity of cells from children with leukemia using the MTT assay with improved culture conditions. Blood, 1990, 76, 2327-2336; e Pieters, R.; et al. Relation of cellular drug resistance to long-term clinical outcome in childhood acute lymphoblastic leukaemia. Lancet, 1991, 338, 399-403) e Kaspers ecolaboradores (Kaspers, G. J.; et al. In vitro cellular drug resistance and prognosis in newly diagnosed childhood acute lymphoblastic leukemia. Blood, 1997 90, 2723). Os resultados obtidos estão apresentados na Tabela 3.Tabela 3. Screening dos compostos 01-29 na concentração de 0,1μM em células leucêmicas humanas Jurkat e REH (com destaque para os compostos mais ativos).
[0055] Para analisarmos a influência das metoxilas no anel A das acil-hidrazonas 01-29, foi testada a série de acil-hidrazonas complementares (30-35). Nenhum dos compostos desta série apresentou atividade frente às células no screening realizado, provando, assim, a necessidade do anel A trimetoxilado para a atividade antileucêmica.
[0056] Em uma terceira tentativa, na busca por compostos correlatos aos ativos, foram testados os 1,3,4- oxadiazóis (36-38) derivados cíclicos das 3,4,5- trimetoxifenil-hidrazonas. Do mesmo modo que para as acil- hidrazonas complementares (30-35), nenhum dos 1,3,4- oxadiazóis (36-38) apresentou-se ativo. A explicação destes resultados pode ser devido à rigidez dos anéis promovida pela ciclização, impedindo a interação da molécula com o alvo.
[0057] Assim, os compostos 02, 07, 09, 10, 11 e 16 foram selecionados para determinação da IC50; os valores estão apresentados na Tabela 4.Tabela 4. IC50 dos compostos mais ativos em células leucêmicas Jurkat e REH.
[0058] As acil-hidrazonas 02 e 07 apresentaram excelentes resultados, com IC50 de 33,7nM e 25,4nM para linhagem leucêmica REH e 31,4nM e 15,7nM para Jurkat.
Exemplo 6. Experimentos de determinação do mecanismo de ação dos compostos com microarranjos de DNA:
[0059] A linhagem celular Jurkat foi tratada ou não com a dose IC50 do composto 07, durante 6h, em meio RPMI- 1640, como descrito anteriormente. Ao final deste período, as células foram recuperadas por breve centrifugação e lisadas em solução de guanidina do kit RNeasy (Qiagen).
[0060] Purificação do RNA e preparo da sonda biotinilada: o RNA total foi extraído das células usando-se o kit RNeasy (Qiagen) seguindo instruções do fabricante. Um total de 5μg de RNA foi utilizado para preparar a sonda de RNA complementar biotinilado (Bio-cRNA), através de síntese de cDNA seguida de amplificação por transcrição in vitro, segundo recomendações da Affymetrix (www.affymetrix.com/ products/small_sample_prep_tn.html). Resumidamente, a síntese da primeira fita de cDNA foi sintetizada utilizando-se 500ng de T7-(dT)24 “primer” e 200U de SuperScript II por reação. A segunda fita foi sintetizada segundo recomendações SuperScript Choice System (Life Technologies). O cDNA sintetizado foi purificado através de precipitação por etanol, e então amplificado através de transcrição in vitro usando o kit Enzo BioArray HighYield RNA. O RNA biotinilado foi purificado através de mini coluna Rneasy. Vinte microgramas de cRNA biotinilado foram fragmentadas em MgCl2 a 95°C e guardadas a -80°C até o momento da hibridização.
[0061] Microarranjos de oligonucleotídeos e “arrays” de hibridização: um quarto do volume de sonda preparado ou 15μg foi hibridizado em microarranjos de oligonucleotídeos do genoma humano HG-U133A 2.0 (Affymetrix). As lavagens, revelação e scanning foram feitas seguindo recomendações do fabricante.
[0062] Análise dos dados: as imagens foram analisadas pelo Microarray Suite (MAS) 5.0 software (Affymetrix) e o DNA-Chip Analyzer (dCHIP) software (Li C, 2001). Os novos algorítmicos estatísticos no MAS 5.0 permitem valores adicionais de significância estatística (p value) e limites de confiança, que ajudam a elucidar a significância dos resultados. Uma limitação do MAS 5.0 é que ele não permite a comparação simultânea de mais de duas amostras. Comparações entre o grupo amostral foram feitas utilizando- se o software dCHIP. Imagens escaneadas foram normalizadas e foram calculados valores para um modelo de expressão basal. Os genes foram considerados diferentemente expressos quando: i) o Fold change entre a média do nível de expressão entre os dois grupos excedeu 2; ii) a diferença absoluta (absolute difference) entre a média do nível de expressão entre os dois grupos excedeu 50 (que corresponde a mais de 1000 no MAS software); e iii) o p value para o teste grupo A = grupo B foi <0,05. Os dados resultantes da análise de microarranjos foram interpretados com o CMap para identificação de possíveis mecanismos de ação correlatos à ação de compostos conhecidos (Lamb, J.; Crawford E. D. The Connectivity Map: Using Gene-Expression Signatures to Connect Small Molecules, Genes and Disease. Science, 2006, 313,1929-1935).
RESULTADOS: MECANISMO DE AÇÃO PROPOSTO ATRAVÉS DE ESTUDOS COM MICROARRANJOS DE DNA
[0063] No intuito de determinarmos o mecanismo de ação destes compostos, selecionamos o composto 07 para análise da expressão gênica, através de biochip de DNA. Estes ensaios geraram um conjunto de dados (algoritmos) que posteriormente foram interpretados através de Mapas de Conectividade (CMap) (www.broadinstitute.org/cmap).
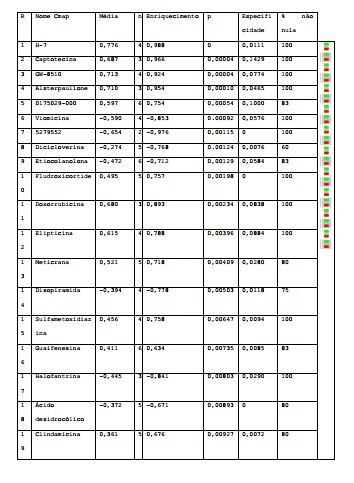
[0064] Os resultados obtidos na análise de microarranjo de expressão gênica, das células tratadas com o composto teste 07, foram analisados no CMap. Genes diferencialmente expressos nas células sob ação do composto 07 foram cruzados com uma série de listas de genes responsivos às mais diferentes drogas. Estas listas fazem parte do banco de dados do CMap. Foram identificados 13 compostos que apresentaram uma forte correlação positiva com a lista de genes obtida a partir das células tratadas com o composto 07 (p < 0,01). Os resultados da análise do composto 07 no CMap são apresentados na tabela 5a abaixo: Tabela 5a.
R = Ranking
[0065] Resultados obtidos para o composto 07 usando o CMap, com valores p< 0,01.
[0066] As quatro moléculas com maior correlação positiva com o composto 07 e menores valores p (p<0,0001) são apresentados na Tabela 5b, que apresenta também o alvo de ação já estudado para estas quatro estruturas. Os resultados obtidos indicam que o mecanismo de ação do composto 07 ocorre via inibição de proteínas quinase dependente de ciclina (CDKs) e da topoisomerase I.Tabela 5b: Compostos positivamente correlacionados com o composto 07.

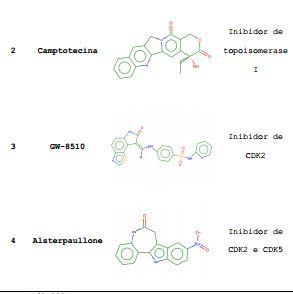
*(p<0,0001) Exemplo 7. Efeito do composto 07 sobre linfócitos humanos saudáveis e sobre a formação de colônias, avaliando sua seletividade: a) Ensaios in vitro da sensibilidade/resistência das células ao composto 07 pelo método do MTT: Para o teste do composto 07 contra linfócitos normais, sangue de doadores humanos foi separado em gradiente de ficoll e as células mononucleares foram cultivadas a 200.000 células/poço, em 80 microlitros de meio RPMI-1640 (contendo 10% de SFB, 100 UI/mL de penicilina e 100 pg/mL de estreptomicina) e 20 microlitros de solução de fitohemaglutinina (Cultilab) em cada poço, para estímulo dos linfócitos T. O composto 07 foi adicionado em diluições seriadas (50%) de 300 a 1 nM. A citotoxicidade foi avaliada após 48h pelo método do MTT, como descrito anteriormente. b) Experimentos de formação de colônias: A citotoxicidade do composto 07 também foi avaliada em ensaio de formação de colônias de células hematopoéticas. Utilizou-se kit “HSC-CFU complete with Epo” da Miltenyi (cat# 130-091-280), seguindo recomendações do fabricante. Células de medula óssea de doadores saudáveis foram cultivadas em meio semi-sólido de metilcelulose, acrescido de soro fetal bovino, soro albumina bovina, diferentes fatores de crescimento (GM-CSF, G-CSF, SCF, IL-3, IL-6, Epo) e do composto 07, cuja ação sobre a hematopoese foi avaliada. Após duas semanas de cultura a 37°C e 5% CO2, procedeu-se à contagem do número de colônias de granulócitos (CFU-G), macrófagos (CFU-M), granulócitos/macrófagos (CFU-GM), eritróide (BFU-E e CFU-E) e colônias mistas (CFU-GEMM).
[0067] O composto 07 foi testado em linfócitos T maduros e saudáveis, estimulados com fitohemaglutinina, para avaliar sua ação contra células normais. Como controle positivo usou-se a colchicina. Os resultados são apresentados na Tabela 6. Tabela 6. Porcentagem de sobrevivência de linfócitos saudáveis estimulados com fitohemaglutinina e tratados com diferentes concentrações do composto 07.
[0068] Em concentrações de 300 nM, o composto 07 apresentou toxicidade de 5% em relação ao controle, enquanto a colchicina, na mesma concentração, causou inibição de 37% na viabilidade dos linfócitos T. É importante salientar que a IC50 do composto 07 frente às células leucêmicas é de 15,7 nM para células da linhagem Jurkat e 25,4 nM para células da linhagem REH. Os resultados obtidos mostram que concentrações até 10 vezes maiores que a IC50 parecem não afetar os linfócitos normais induzidos por fitohemaglutinina. Isto indica que o composto 07 poderia atuar em células leucêmicas sem afetar a função imunológica normal dos pacientes.
[0069] Para averiguar o efeito do composto 07 na hematopoese, o mesmo foi testado em ensaio de formação de colônias de células da medula óssea, cultivadas em meio semi-sólido de metilcelulose acrescido de fatores de crescimento. Como mostra a Tabela 7, o composto 07, em concentração muito próxima a IC50 (20 nM), apresenta atividade inibitória sobre a formação de eritrócitos comparável a um inibidor da via Pi3K, usado no ensaio como controle positivo. O composto 07 também apresenta atividade inibitória contra granulócitos e macrófagos, porém em intensidade pequena quando comparada ao inibidor da Pi3K. Na concentração de 200 nM, correspondente a 10 vezes o IC50 médio em linhagens de LLA, o composto 07 inibiu completamente a hematopoese. Tabela 7. Formação de colônias de células de medula óssea (%) utilizando o composto 07.
• Para o controle são apresentados a média ± o desvio padrão de 3 repetições.
[0070] De acordo com a descrição da presente invenção, obtiveram-se cinco 3,4,5-trimetoxifenil- hidrazonas inéditas e três 1,3,4-oxadiazóis inéditos. Todas as acil-hidrazonas e oxadiazóis sintetizados e relatados no escopo da presente invenção foram avaliados em células leucêmicas das linhagens Jurkat e REH, e os compostos 02 e 07 apresentaram excelente atividade antileucêmica, semelhante ao composto padrão (colchicina). O mecanismo de ação do composto 07 foi determinado com a utilização de microarranjos de DNA, mostrando sua atividade inibitória de proteínas quinase dependente de ciclina (CDKs) e da topoisomerase I. Testes subsequentes mostraram a seletividade do composto 07 para células leucêmicas na ordem de 10 vezes, quando comparado à sua ação em linfócitos humanos saudáveis.
[0071] As 3,4,5-trimetoxifenil-hidrazonas, bem como seus compostos análogos e semelhantes, compreendendo os oxadiazóis, presentes nesta invenção, apresentam, portanto, grande potencial como protótipos de fármacos, pré-fármacos ou fármacos, para o tratamento de diferentes doenças associadas à proliferação celular, como leucemias, compreendendo leucemia linfóide aguda (LLA), tumores e inflamação.
[0072] A descrição da presente invenção foi apresentada com o propósito de ilustração e detalhamento para futuras aplicações. Entretanto, a presente descrição não tenciona limitar a invenção à forma aqui revelada e exemplificada: variações e modificações compatíveis com os descritos acima, e a habilidade ou conhecimento da técnica relevante, estão dentro do escopo da presente invenção. É intenção legítima que a presente invenção compreenda em seu escopo todas as modificações e variações da mesma, de acordo com a descrição do relatório e nas reivindicações anexas.