CN100361990C - 含吲哚基团的二阶非线性光学发色团及其制法 - Google Patents
含吲哚基团的二阶非线性光学发色团及其制法 Download PDFInfo
- Publication number
- CN100361990C CN100361990C CNB2005100193965A CN200510019396A CN100361990C CN 100361990 C CN100361990 C CN 100361990C CN B2005100193965 A CNB2005100193965 A CN B2005100193965A CN 200510019396 A CN200510019396 A CN 200510019396A CN 100361990 C CN100361990 C CN 100361990C
- Authority
- CN
- China
- Prior art keywords
- compound
- indoles
- nonlinear optical
- thiophene
- chromophore
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
- 230000003287 optical effect Effects 0.000 title claims abstract description 36
- 238000002360 preparation method Methods 0.000 title claims description 8
- 125000001041 indolyl group Chemical group 0.000 title abstract description 12
- 238000006243 chemical reaction Methods 0.000 claims abstract description 45
- -1 3-phenyl-isoxazolone Chemical compound 0.000 claims abstract description 37
- SIKJAQJRHWYJAI-UHFFFAOYSA-N Indole Chemical compound C1=CC=C2NC=CC2=C1 SIKJAQJRHWYJAI-UHFFFAOYSA-N 0.000 claims abstract description 32
- PZOUSPYUWWUPPK-UHFFFAOYSA-N indole Natural products CC1=CC=CC2=C1C=CN2 PZOUSPYUWWUPPK-UHFFFAOYSA-N 0.000 claims abstract description 20
- RKJUIXBNRJVNHR-UHFFFAOYSA-N indolenine Natural products C1=CC=C2CC=NC2=C1 RKJUIXBNRJVNHR-UHFFFAOYSA-N 0.000 claims abstract description 20
- RVBUGGBMJDPOST-UHFFFAOYSA-N 2-thiobarbituric acid Chemical compound O=C1CC(=O)NC(=S)N1 RVBUGGBMJDPOST-UHFFFAOYSA-N 0.000 claims abstract description 5
- 125000003118 aryl group Chemical group 0.000 claims abstract description 5
- 239000002994 raw material Substances 0.000 claims abstract description 4
- 229910052760 oxygen Inorganic materials 0.000 claims abstract description 3
- 239000001301 oxygen Substances 0.000 claims abstract description 3
- XHXFXVLFKHQFAL-UHFFFAOYSA-N phosphoryl trichloride Chemical compound ClP(Cl)(Cl)=O XHXFXVLFKHQFAL-UHFFFAOYSA-N 0.000 claims description 40
- YTPLMLYBLZKORZ-UHFFFAOYSA-N Thiophene Chemical compound C=1C=CSC=1 YTPLMLYBLZKORZ-UHFFFAOYSA-N 0.000 claims description 34
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 claims description 21
- MZRVEZGGRBJDDB-UHFFFAOYSA-N N-Butyllithium Chemical compound [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 claims description 20
- 229930192474 thiophene Natural products 0.000 claims description 17
- 150000002475 indoles Chemical class 0.000 claims description 12
- 229910052757 nitrogen Inorganic materials 0.000 claims description 11
- SBNOTUDDIXOFSN-UHFFFAOYSA-N 1h-indole-2-carbaldehyde Chemical compound C1=CC=C2NC(C=O)=CC2=C1 SBNOTUDDIXOFSN-UHFFFAOYSA-N 0.000 claims description 6
- 150000001335 aliphatic alkanes Chemical class 0.000 claims description 6
- 238000000034 method Methods 0.000 claims description 5
- 229910052799 carbon Inorganic materials 0.000 claims description 3
- 125000006575 electron-withdrawing group Chemical group 0.000 claims description 2
- 125000004433 nitrogen atom Chemical group N* 0.000 claims description 2
- LSSICPJTIPBTDD-UHFFFAOYSA-N 2-ethenyl-1h-indole Chemical class C1=CC=C2NC(C=C)=CC2=C1 LSSICPJTIPBTDD-UHFFFAOYSA-N 0.000 claims 5
- VGGSQFUCUMXWEO-UHFFFAOYSA-N Ethene Chemical compound C=C VGGSQFUCUMXWEO-UHFFFAOYSA-N 0.000 claims 3
- 239000005977 Ethylene Substances 0.000 claims 3
- CNUDBTRUORMMPA-UHFFFAOYSA-N formylthiophene Chemical compound O=CC1=CC=CS1 CNUDBTRUORMMPA-UHFFFAOYSA-N 0.000 claims 3
- JJNZXLAFIPKXIG-UHFFFAOYSA-N 2-Chlorobenzylidenemalononitrile Chemical compound ClC1=CC=CC=C1C=C(C#N)C#N JJNZXLAFIPKXIG-UHFFFAOYSA-N 0.000 claims 2
- WNRGVOZZLIIHHY-UHFFFAOYSA-N CC1=C(C=CC=C1)P(C1=CC=CC=C1)C1=CC=CC=C1.BrC=1OC=CC1 Chemical compound CC1=C(C=CC=C1)P(C1=CC=CC=C1)C1=CC=CC=C1.BrC=1OC=CC1 WNRGVOZZLIIHHY-UHFFFAOYSA-N 0.000 claims 2
- FWHWNDCFJTVQAK-UHFFFAOYSA-N CC1=C(C=CC=C1)P(C1=CC=CC=C1)C1=CC=CC=C1.BrC=1SC=CC1 Chemical compound CC1=C(C=CC=C1)P(C1=CC=CC=C1)C1=CC=CC=C1.BrC=1SC=CC1 FWHWNDCFJTVQAK-UHFFFAOYSA-N 0.000 claims 2
- 239000004215 Carbon black (E152) Substances 0.000 claims 2
- 229930195733 hydrocarbon Natural products 0.000 claims 2
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical group [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 claims 1
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical group [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 claims 1
- 238000004519 manufacturing process Methods 0.000 claims 1
- CUONGYYJJVDODC-UHFFFAOYSA-N malononitrile Chemical compound N#CCC#N CUONGYYJJVDODC-UHFFFAOYSA-N 0.000 abstract description 15
- FLMBQNOAWLPZPJ-UHFFFAOYSA-N 2-(3-cyano-4,5,5-trimethylfuran-2-ylidene)propanedinitrile Chemical compound CC1=C(C#N)C(=C(C#N)C#N)OC1(C)C FLMBQNOAWLPZPJ-UHFFFAOYSA-N 0.000 abstract description 13
- 239000000463 material Substances 0.000 abstract description 6
- 125000004430 oxygen atom Chemical group O* 0.000 abstract description 5
- 229910052717 sulfur Chemical group 0.000 abstract description 5
- 125000004434 sulfur atom Chemical group 0.000 abstract description 5
- 239000000370 acceptor Substances 0.000 abstract description 3
- 238000013500 data storage Methods 0.000 abstract description 3
- XQQBUAPQHNYYRS-UHFFFAOYSA-N 2-methylthiophene Chemical compound CC1=CC=CS1 XQQBUAPQHNYYRS-UHFFFAOYSA-N 0.000 abstract description 2
- QENGPZGAWFQWCZ-UHFFFAOYSA-N Methylthiophene Natural products CC=1C=CSC=1 QENGPZGAWFQWCZ-UHFFFAOYSA-N 0.000 abstract description 2
- 230000021615 conjugation Effects 0.000 abstract description 2
- 125000000524 functional group Chemical group 0.000 abstract description 2
- 150000002391 heterocyclic compounds Chemical class 0.000 abstract description 2
- 239000003153 chemical reaction reagent Substances 0.000 abstract 1
- 150000008282 halocarbons Chemical class 0.000 abstract 1
- 125000001183 hydrocarbyl group Chemical group 0.000 abstract 1
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 162
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 87
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 84
- 230000015572 biosynthetic process Effects 0.000 description 43
- 238000003786 synthesis reaction Methods 0.000 description 43
- 238000004440 column chromatography Methods 0.000 description 39
- 239000003480 eluent Substances 0.000 description 39
- 150000001875 compounds Chemical class 0.000 description 35
- 238000003756 stirring Methods 0.000 description 33
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 27
- 239000007787 solid Substances 0.000 description 25
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 24
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 19
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 18
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 18
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 18
- WSLDOOZREJYCGB-UHFFFAOYSA-N 1,2-Dichloroethane Chemical compound ClCCCl WSLDOOZREJYCGB-UHFFFAOYSA-N 0.000 description 12
- 239000003208 petroleum Substances 0.000 description 12
- QGVNJRROSLYGKF-UHFFFAOYSA-N thiobarbital Chemical compound CCC1(CC)C(=O)NC(=S)NC1=O QGVNJRROSLYGKF-UHFFFAOYSA-N 0.000 description 12
- LPNYRYFBWFDTMA-UHFFFAOYSA-N potassium tert-butoxide Chemical compound [K+].CC(C)(C)[O-] LPNYRYFBWFDTMA-UHFFFAOYSA-N 0.000 description 11
- 238000010992 reflux Methods 0.000 description 11
- GHYOCDFICYLMRF-UTIIJYGPSA-N (2S,3R)-N-[(2S)-3-(cyclopenten-1-yl)-1-[(2R)-2-methyloxiran-2-yl]-1-oxopropan-2-yl]-3-hydroxy-3-(4-methoxyphenyl)-2-[[(2S)-2-[(2-morpholin-4-ylacetyl)amino]propanoyl]amino]propanamide Chemical compound C1(=CCCC1)C[C@@H](C(=O)[C@@]1(OC1)C)NC([C@H]([C@@H](C1=CC=C(C=C1)OC)O)NC([C@H](C)NC(CN1CCOCC1)=O)=O)=O GHYOCDFICYLMRF-UTIIJYGPSA-N 0.000 description 10
- ONBQEOIKXPHGMB-VBSBHUPXSA-N 1-[2-[(2s,3r,4s,5r)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]oxy-4,6-dihydroxyphenyl]-3-(4-hydroxyphenyl)propan-1-one Chemical compound O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1OC1=CC(O)=CC(O)=C1C(=O)CCC1=CC=C(O)C=C1 ONBQEOIKXPHGMB-VBSBHUPXSA-N 0.000 description 10
- LNUFLCYMSVYYNW-ZPJMAFJPSA-N [(2r,3r,4s,5r,6r)-2-[(2r,3r,4s,5r,6r)-6-[(2r,3r,4s,5r,6r)-6-[(2r,3r,4s,5r,6r)-6-[[(3s,5s,8r,9s,10s,13r,14s,17r)-10,13-dimethyl-17-[(2r)-6-methylheptan-2-yl]-2,3,4,5,6,7,8,9,11,12,14,15,16,17-tetradecahydro-1h-cyclopenta[a]phenanthren-3-yl]oxy]-4,5-disulfo Chemical compound O([C@@H]1[C@@H](COS(O)(=O)=O)O[C@@H]([C@@H]([C@H]1OS(O)(=O)=O)OS(O)(=O)=O)O[C@@H]1[C@@H](COS(O)(=O)=O)O[C@@H]([C@@H]([C@H]1OS(O)(=O)=O)OS(O)(=O)=O)O[C@@H]1[C@@H](COS(O)(=O)=O)O[C@H]([C@@H]([C@H]1OS(O)(=O)=O)OS(O)(=O)=O)O[C@@H]1C[C@@H]2CC[C@H]3[C@@H]4CC[C@@H]([C@]4(CC[C@@H]3[C@@]2(C)CC1)C)[C@H](C)CCCC(C)C)[C@H]1O[C@H](COS(O)(=O)=O)[C@@H](OS(O)(=O)=O)[C@H](OS(O)(=O)=O)[C@H]1OS(O)(=O)=O LNUFLCYMSVYYNW-ZPJMAFJPSA-N 0.000 description 10
- 229940125797 compound 12 Drugs 0.000 description 10
- 229940126142 compound 16 Drugs 0.000 description 10
- 239000007788 liquid Substances 0.000 description 10
- 229940125773 compound 10 Drugs 0.000 description 9
- ZLVXBBHTMQJRSX-VMGNSXQWSA-N jdtic Chemical compound C1([C@]2(C)CCN(C[C@@H]2C)C[C@H](C(C)C)NC(=O)[C@@H]2NCC3=CC(O)=CC=C3C2)=CC=CC(O)=C1 ZLVXBBHTMQJRSX-VMGNSXQWSA-N 0.000 description 9
- 238000002844 melting Methods 0.000 description 8
- 230000008018 melting Effects 0.000 description 8
- VZGDMQKNWNREIO-UHFFFAOYSA-N tetrachloromethane Chemical compound ClC(Cl)(Cl)Cl VZGDMQKNWNREIO-UHFFFAOYSA-N 0.000 description 8
- 239000012295 chemical reaction liquid Substances 0.000 description 7
- SZUVGFMDDVSKSI-WIFOCOSTSA-N (1s,2s,3s,5r)-1-(carboxymethyl)-3,5-bis[(4-phenoxyphenyl)methyl-propylcarbamoyl]cyclopentane-1,2-dicarboxylic acid Chemical compound O=C([C@@H]1[C@@H]([C@](CC(O)=O)([C@H](C(=O)N(CCC)CC=2C=CC(OC=3C=CC=CC=3)=CC=2)C1)C(O)=O)C(O)=O)N(CCC)CC(C=C1)=CC=C1OC1=CC=CC=C1 SZUVGFMDDVSKSI-WIFOCOSTSA-N 0.000 description 6
- DOMRUOQXWPPSQH-UHFFFAOYSA-N 1-butylindole-3-carbaldehyde Chemical compound C1=CC=C2N(CCCC)C=C(C=O)C2=C1 DOMRUOQXWPPSQH-UHFFFAOYSA-N 0.000 description 6
- 229940126543 compound 14 Drugs 0.000 description 6
- 229940126214 compound 3 Drugs 0.000 description 6
- 239000012263 liquid product Substances 0.000 description 6
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical group CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 6
- 239000000843 powder Substances 0.000 description 6
- 238000001308 synthesis method Methods 0.000 description 6
- PSGFPTUMYLIVDX-UHFFFAOYSA-N 1-butylindole Chemical compound C1=CC=C2N(CCCC)C=CC2=C1 PSGFPTUMYLIVDX-UHFFFAOYSA-N 0.000 description 5
- OQXMGKDLNQYGLH-UHFFFAOYSA-N 1-hexylindole-3-carbaldehyde Chemical compound C1=CC=C2N(CCCCCC)C=C(C=O)C2=C1 OQXMGKDLNQYGLH-UHFFFAOYSA-N 0.000 description 5
- 238000005516 engineering process Methods 0.000 description 5
- 239000000047 product Substances 0.000 description 5
- UNILWMWFPHPYOR-KXEYIPSPSA-M 1-[6-[2-[3-[3-[3-[2-[2-[3-[[2-[2-[[(2r)-1-[[2-[[(2r)-1-[3-[2-[2-[3-[[2-(2-amino-2-oxoethoxy)acetyl]amino]propoxy]ethoxy]ethoxy]propylamino]-3-hydroxy-1-oxopropan-2-yl]amino]-2-oxoethyl]amino]-3-[(2r)-2,3-di(hexadecanoyloxy)propyl]sulfanyl-1-oxopropan-2-yl Chemical compound O=C1C(SCCC(=O)NCCCOCCOCCOCCCNC(=O)COCC(=O)N[C@@H](CSC[C@@H](COC(=O)CCCCCCCCCCCCCCC)OC(=O)CCCCCCCCCCCCCCC)C(=O)NCC(=O)N[C@H](CO)C(=O)NCCCOCCOCCOCCCNC(=O)COCC(N)=O)CC(=O)N1CCNC(=O)CCCCCN\1C2=CC=C(S([O-])(=O)=O)C=C2CC/1=C/C=C/C=C/C1=[N+](CC)C2=CC=C(S([O-])(=O)=O)C=C2C1 UNILWMWFPHPYOR-KXEYIPSPSA-M 0.000 description 4
- OXCITQLDOUGVRZ-UHFFFAOYSA-N 1-benzylindole-3-carbaldehyde Chemical compound C12=CC=CC=C2C(C=O)=CN1CC1=CC=CC=C1 OXCITQLDOUGVRZ-UHFFFAOYSA-N 0.000 description 4
- OJRUSAPKCPIVBY-KQYNXXCUSA-N C1=NC2=C(N=C(N=C2N1[C@H]3[C@@H]([C@@H]([C@H](O3)COP(=O)(CP(=O)(O)O)O)O)O)I)N Chemical compound C1=NC2=C(N=C(N=C2N1[C@H]3[C@@H]([C@@H]([C@H](O3)COP(=O)(CP(=O)(O)O)O)O)O)I)N OJRUSAPKCPIVBY-KQYNXXCUSA-N 0.000 description 4
- 229940125758 compound 15 Drugs 0.000 description 4
- 229940125782 compound 2 Drugs 0.000 description 4
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical class [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 3
- 238000009835 boiling Methods 0.000 description 3
- 239000003795 chemical substances by application Substances 0.000 description 3
- 229940125904 compound 1 Drugs 0.000 description 3
- 238000001816 cooling Methods 0.000 description 3
- 239000012153 distilled water Substances 0.000 description 3
- 238000000605 extraction Methods 0.000 description 3
- 239000012046 mixed solvent Substances 0.000 description 3
- 239000000203 mixture Substances 0.000 description 3
- AOSZTAHDEDLTLQ-AZKQZHLXSA-N (1S,2S,4R,8S,9S,11S,12R,13S,19S)-6-[(3-chlorophenyl)methyl]-12,19-difluoro-11-hydroxy-8-(2-hydroxyacetyl)-9,13-dimethyl-6-azapentacyclo[10.8.0.02,9.04,8.013,18]icosa-14,17-dien-16-one Chemical compound C([C@@H]1C[C@H]2[C@H]3[C@]([C@]4(C=CC(=O)C=C4[C@@H](F)C3)C)(F)[C@@H](O)C[C@@]2([C@@]1(C1)C(=O)CO)C)N1CC1=CC=CC(Cl)=C1 AOSZTAHDEDLTLQ-AZKQZHLXSA-N 0.000 description 2
- MPPPKRYCTPRNTB-UHFFFAOYSA-N 1-bromobutane Chemical compound CCCCBr MPPPKRYCTPRNTB-UHFFFAOYSA-N 0.000 description 2
- MNDIARAMWBIKFW-UHFFFAOYSA-N 1-bromohexane Chemical compound CCCCCCBr MNDIARAMWBIKFW-UHFFFAOYSA-N 0.000 description 2
- NOKPEQGJKFRMEL-UHFFFAOYSA-N 1-hexylindole Chemical compound C1=CC=C2N(CCCCCC)C=CC2=C1 NOKPEQGJKFRMEL-UHFFFAOYSA-N 0.000 description 2
- 229940126657 Compound 17 Drugs 0.000 description 2
- ATHHXGZTWNVVOU-UHFFFAOYSA-N N-methylformamide Chemical compound CNC=O ATHHXGZTWNVVOU-UHFFFAOYSA-N 0.000 description 2
- 150000001299 aldehydes Chemical class 0.000 description 2
- KCXMKQUNVWSEMD-UHFFFAOYSA-N benzyl chloride Chemical compound ClCC1=CC=CC=C1 KCXMKQUNVWSEMD-UHFFFAOYSA-N 0.000 description 2
- 229940073608 benzyl chloride Drugs 0.000 description 2
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 2
- 230000005540 biological transmission Effects 0.000 description 2
- 238000004891 communication Methods 0.000 description 2
- 238000013461 design Methods 0.000 description 2
- 238000004377 microelectronic Methods 0.000 description 2
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 2
- 125000001280 n-hexyl group Chemical group C(CCCCC)* 0.000 description 2
- 238000012545 processing Methods 0.000 description 2
- 239000002904 solvent Substances 0.000 description 2
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 2
- QFLWZFQWSBQYPS-AWRAUJHKSA-N (3S)-3-[[(2S)-2-[[(2S)-2-[5-[(3aS,6aR)-2-oxo-1,3,3a,4,6,6a-hexahydrothieno[3,4-d]imidazol-4-yl]pentanoylamino]-3-methylbutanoyl]amino]-3-(4-hydroxyphenyl)propanoyl]amino]-4-[1-bis(4-chlorophenoxy)phosphorylbutylamino]-4-oxobutanoic acid Chemical compound CCCC(NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](Cc1ccc(O)cc1)NC(=O)[C@@H](NC(=O)CCCCC1SC[C@@H]2NC(=O)N[C@H]12)C(C)C)P(=O)(Oc1ccc(Cl)cc1)Oc1ccc(Cl)cc1 QFLWZFQWSBQYPS-AWRAUJHKSA-N 0.000 description 1
- NJZQOCCEDXRQJM-UHFFFAOYSA-N 1-benzylindole Chemical compound C1=CC2=CC=CC=C2N1CC1=CC=CC=C1 NJZQOCCEDXRQJM-UHFFFAOYSA-N 0.000 description 1
- XAEBTCPOZVEMHR-UHFFFAOYSA-N 2-methylpropan-2-ol;potassium Chemical compound [K].CC(C)(C)O XAEBTCPOZVEMHR-UHFFFAOYSA-N 0.000 description 1
- FAWOZGBIUUKDMP-UHFFFAOYSA-N C(#N)C1OC(C(=C1)C)(C)C Chemical group C(#N)C1OC(C(=C1)C)(C)C FAWOZGBIUUKDMP-UHFFFAOYSA-N 0.000 description 1
- 239000007832 Na2SO4 Substances 0.000 description 1
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 1
- 238000010521 absorption reaction Methods 0.000 description 1
- 150000001448 anilines Chemical class 0.000 description 1
- 239000000969 carrier Substances 0.000 description 1
- 229940125898 compound 5 Drugs 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- 238000001914 filtration Methods 0.000 description 1
- 238000010438 heat treatment Methods 0.000 description 1
- 150000002430 hydrocarbons Chemical group 0.000 description 1
- 239000012535 impurity Substances 0.000 description 1
- 230000010365 information processing Effects 0.000 description 1
- 229910010272 inorganic material Inorganic materials 0.000 description 1
- 239000011147 inorganic material Substances 0.000 description 1
- 230000005693 optoelectronics Effects 0.000 description 1
- 229920000620 organic polymer Polymers 0.000 description 1
- 239000002861 polymer material Substances 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 239000011734 sodium Substances 0.000 description 1
- 229910052938 sodium sulfate Inorganic materials 0.000 description 1
- 235000011152 sodium sulphate Nutrition 0.000 description 1
- 238000005406 washing Methods 0.000 description 1
Landscapes
- Plural Heterocyclic Compounds (AREA)
- Indole Compounds (AREA)
Abstract
本发明涉及含吲哚基团的二阶非线性光学发色团,具有以下通式:式中R为各种烃基、芳香基团或其它杂环化合物功能基团。X为氧或者硫原子,A为各种拉电子的受体,如丙二腈、硫代巴比妥酸、3-苯基-异噁唑酮、2-二氰基亚甲基-3-氰基-4,5,5-三甲基-2,5-二氢呋喃等。本发明通过一系列化学反应,可以吲哚、卤代烃、各种拉电子受体、溴代呋喃甲基三苯基膦或2-甲基噻吩及其它一些试剂为原料,制备含吲哚基团的二阶非线性光学发色团。含吲哚基团的二阶非线性光学发色团作为二阶非线性光学材料可以在远程通讯、数据存储、相位共轭等方面得到实际应用。
Description
技术领域
本发明涉及含吲哚基团的二阶非线性光学发色团及其制备方法。
背景技术
信息技术是依靠电子学和微电子学技术发展的,但由于电子本身的物理极限——包括电子作为信息载体的传输速度极限(电子传输速度的最大值为600km/s)和元件(功能单元)由于电子电荷的相互干扰而形成的密度极限——“摩尔定律”并不能青春长在,电子信息技术的载体量级只能为吉位(Gb,即109bits)。另一方面,光子的速度远比电子的快,其频率比无线电(如微波)的频率高得多,以光子代替电子来传输信息,可以大大加快信息的处理速度,增加处理信息的容量,能够克服微电子技术的瓶颈,更准确、更高效、更远距离传输信息。对于全光信息技术的发展,非线性光学(NLO)是不可缺少的关键学科,它在高速光通讯、光信息处理和光电子学等实用领域具有极为重要的作用,非线性光学材料在这些领域中的应用前景得到越来越广泛的重视。
与无机材料相比,有机高分子非线性光学材料以其超快响应速度、较大的非线性光学响应、高光损伤阀值、优异的可加工性和低介电常数等优点而得到广泛的重视。为了达到实用化的要求,这些材料不仅要有大的非线性光学响应,而且要同时满足器件化对其透明性、热稳定性和可加工生等方面要求,由于高分子材料的非线性光学响应均首先取决于其中的非线性光学活性基团(常称发色团)分子的非线性光学特性,所以设计合成兼具大的分子一阶超极化率(β)和良好的光学透明性的二阶非线性光学发色团分子长期以来一直是最具挑战性的课题之一。
以下为与本发明相关的参考文献:Moemer,W.E.;Jepsen,A.G.;Thompson,C.L.Annu.Rev.Mater.Sci.1997,32,585./叶成;“有机固体的非线性光学特性”,《有机固体》第五章,主编:朱道本,王佛松,上海科学出版社,1999;p.181./Hua,J.;Luo,J.;Qin,J.;Shen,Y.;Zhang,Y.;Lu,Z.J. Mater.Chem.2002,12,863.
发明内容
本发明的目的就在于提供含吲哚基团的二阶非线性光学发色团及其制法。该类发色团兼具大的分子一阶超极化率(β)、良好的光学透明性以及高的热稳定性,可作为新型二阶非线性光学材料在远程通讯、数据存储、相位共轭等方面得到实际应用。
本发明提供的技术方案是:一类含吲哚基团的二阶非线性光学发色团,具有以下通式:

式中R为各种烃基、芳香基团或其它杂环化合物功能基团。X为氧或者硫原子,A为各种拉电子的受体,如丙二腈、硫代巴比妥酸、3-苯基-异噁唑酮、2-二氰基亚甲基-3-氰基-4,5,5-三甲基-2,5-二氢呋喃等吸电子基团。
上述R为C1-C9的烷烃基或C7-C11芳香基团。
本发明还提供了上述二阶非线性光学发色团的制法,以吲哚和C1-C9的卤代烷烃或C7-C11的卤代芳烃为原料制备取代吲哚A,其中吲哚和C1-C9的卤代烷烃或C7-C11的卤代芳烃的摩尔比例为1∶1.0~2.0,取代吲哚A为N原子上连有C1-C9的烷烃基或C7-C11芳香基团的吲哚;取代吲哚A与N,N-二甲基甲酰胺和三氯氧磷反应,制备吲哚醛B,其中取代吲哚A、N,N-二甲基甲酰胺、三氯氧磷的摩尔比为1∶1.1~1.9∶1.1~1.9;然后吲哚醛B与溴代呋喃甲基三苯基膦或溴代噻吩甲基三苯基膦反应,制备乙烯基吲哚呋喃C或乙烯基吲哚噻吩D,其中吲哚醛B与溴代呋喃甲基三苯基膦或者溴代噻吩甲基三苯基膦的摩尔比为1∶1.0~2.0;乙烯基吲哚呋喃C与N,N-二甲基甲酰胺和三氯氧磷反应,制备乙烯基吲哚呋喃醛E,其中乙烯基吲哚呋喃C、N,N-二甲基甲酰胺、三氯氧磷的摩尔比为1∶1.1~1.9∶2.0~3.7;或者乙烯基吲哚噻吩D与正丁基锂和N,N-二甲基甲酰胺反应制备乙烯基吲哚噻吩醛F,其中乙烯基吲哚噻吩D与正丁基锂和N,N-二甲基甲酰胺的摩尔比为1∶1.1~1.5∶1.0~1.4;最后以乙烯基吲哚呋喃醛E或者乙烯基吲哚噻吩醛F为原料,与丙二腈、硫代巴比妥酸、3-苯基-异噁唑酮或2-二氰基亚甲基-3-氰基-4,5,5-三甲基-2,5-二氢呋喃反应,制得含吲哚基团的二阶非线性光学发色团,其中乙烯基吲哚呋喃醛E或者乙烯基吲哚噻吩醛F与各种拉电子基团之间的摩尔比为1∶1~1.3。。
本发明的含吲哚基团的二阶非线性光学发色团可以作为二阶非线性光学材料在远程通讯、数据存储、相位共轭等方面的实际应用。
本发明的优点在于:
1.本发明含吲哚基团的二阶非线性光学发色团具有非常好的光学透明性,与苯胺类似物相比,其最大吸收波长紫移了20-30纳米,这种现象非常罕见。
2.本发明含吲哚基团的二阶非线性光学发色团具有很高的二阶非线性光学性能(例1和例2中八个发色团的一阶超极化率依次为1162,1540,412,615,1068,1108,685,597×10-30esu)。
3.本发明描述了含吲哚基团的二阶非线性光学发色团的制备,丰富了二阶非线性光学发色团研究的内容,从一定程度上拓展了二阶非线性光学发色团成化学的方法和设计思路。
具体实施方式
实施例1:
R为正己基,X为氧原子时,合成路线如下:

上述Ia、Ib、Ic、Id化合物中的A=分别为
合成方法为:
N-己基吲哚(化合物1)的合成
在250mL圆底烧瓶中,放入氢氧化钾固体和N,N-二甲基甲酰胺(为氢氧化钾质量的3-10倍),搅拌20分钟后加入吲哚,搅拌40分钟。用恒压漏斗加入1-溴-正己烷在N,N-二甲基甲酰胺中的溶液(质量百分比浓度为20%-50%之间)。然后升温至50℃,搅拌反应2-20小时。将反应液倒入适量水中,氯仿萃取,旋除氯仿后,用油泵减压蒸馏,得浅黄色液体,即为产物N-己基吲哚。其中,氢氧化钾、吲哚、1-溴-正己烷的摩尔比为5∶1∶1.2。
3-甲酰基-N-己基吲哚(化合物2)的合成
称取化合物1,用1,2-二氯乙烷溶解在100mL烧瓶中(质量百分比浓度为3%-10%之间),冰浴冷至0℃,搅拌。加入N,N-二甲基甲酰胺,然后加入三氯氧磷。加热回流反应2小时,冷却后将反应液倒入水中,用氯仿萃取。旋除氯仿,以氯仿为淋洗剂进行柱层析,得到红棕色液体产物3-甲酰基-N-己基吲哚。化合物1、N,N-二甲基甲酰胺、三氯氧磷的摩尔比为1∶1.3∶1.3。
2-(N-正己基-3-乙烯基吲哚)呋喃(化合物3)的合成
称取溴代呋喃甲基三苯基膦置于Schlenk管中,氮气保护下,加入无水四氢呋喃(为溴代呋喃甲基三苯基膦质量的5-10倍),加入叔丁醇钾,再加入3-甲酰基-N-己基吲哚(化合物2)。反应20-30小时。将反应液倒入蒸馏水中,氯仿萃取,旋除氯仿,用配比为1∶1(体积比)的氯仿和石油醚为淋洗剂进行柱层析,得到亮黄绿色液体2-(N-正己基-3-乙烯基吲哚)呋喃。其中,溴代呋喃甲基三苯基膦、叔丁醇钾和3-甲酰基-N-己基吲哚的摩尔比为1∶1.5∶1。
2-(N-正己基-3-乙烯基吲哚)-5-甲酰基呋喃(化合物4)的合成
在100mL二口烧瓶中,加入化合物3,用1,2-二氯乙烷溶解(质量百分比浓度为3%-10%之间)。将三氯氧磷和N,N-二甲基甲酰胺的混合液加到上述二口烧瓶中。室温搅拌2h后,将反应液倒入碳酸钠饱和溶液中,用1,2-二氯乙烷萃取。旋除1,2-二氯乙烷,用氯仿作淋洗剂进行柱层析,得到橙红色固体2-(N-正己基-3-乙烯基吲哚)-5-甲酰基呋喃。其中,化合物3、三氯氧磷和N,N-二甲基甲酰胺的摩尔比为1∶1.4∶2.4。
发色团Ia的合成
称取上述化合物4,用无水乙醇溶于100mL烧瓶中(质量百分比浓度为0.1%-1%之间),加入丙二腈,搅拌使其溶解。室温反应2h,将反应液在旋转蒸发仪上除去乙醇,然后进行柱层析,淋洗剂为1∶1(体积比)的氯仿和石油醚,得到红色固体发色团Ia。其中,化合物4和丙二腈的摩尔比为1∶1。熔点:95-96℃。IR:2218cm-1(C≡N)。UV-Vis(氯仿,λmax):526纳米。
发色团Ib的合成
称取上述化合物4,用无水乙醇溶于100mL烧瓶中(质量百分比浓度为0.1%-1%之间),加入硫代巴比妥,加热回流3小时。旋除乙醇,用配比为1∶1(体积比)的氯仿和石油醚(沸点60℃-90℃)为淋洗剂进行柱层析,得到蓝紫色固体发色团Ib。其中,化合物4和硫代巴比妥的摩尔比为1∶1。熔点:198-200℃。IR:1665cm-1(C=O)。UV-Vis(氯仿,λmax):590纳米。
发色团Ic的合成
称取上述化合物4,用无水乙醇溶于100mL烧瓶中(质量百分比浓度为0.1%-1%之间),加入3-苯基-异噁唑酮,搅拌使其溶解。室温搅拌2小时,旋除乙醇,用氯仿作柱层析淋洗剂,得到蓝紫色固体发色团Ic。其中,化合物4和3-苯基-异噁唑酮的摩尔比为1∶1.3。熔点:118-120℃。IR:1665cm-1(C=O)。UV-Vis(氯仿,λmax):566纳米。
发色团Id的合成
称取化合物4,用无水乙醇溶于100mL烧瓶中(质量百分比浓度为0.1%-1%之间),向其中加入2-二氰基亚甲基-3-氰基-4,5,5-三甲基-2,5-二氢呋喃,加热回流3小时。停止反应,旋除乙醇,以氯仿∶乙酸乙酯=20∶3(体积比)为淋洗剂进行柱层析,得到深蓝紫色固体粉末发色团Id。其中,化合物4和2-二氰基亚甲基-3-氰基-4,5,5-三甲基-2,5-二氢呋喃的摩尔比为1∶1.2。熔点:202-204℃。IR:2223cm-1(C≡N)。UV-Vis(氯仿,λmax):650纳米。
实施例2:
R为正己基,X为硫原子时,合成路线如下:
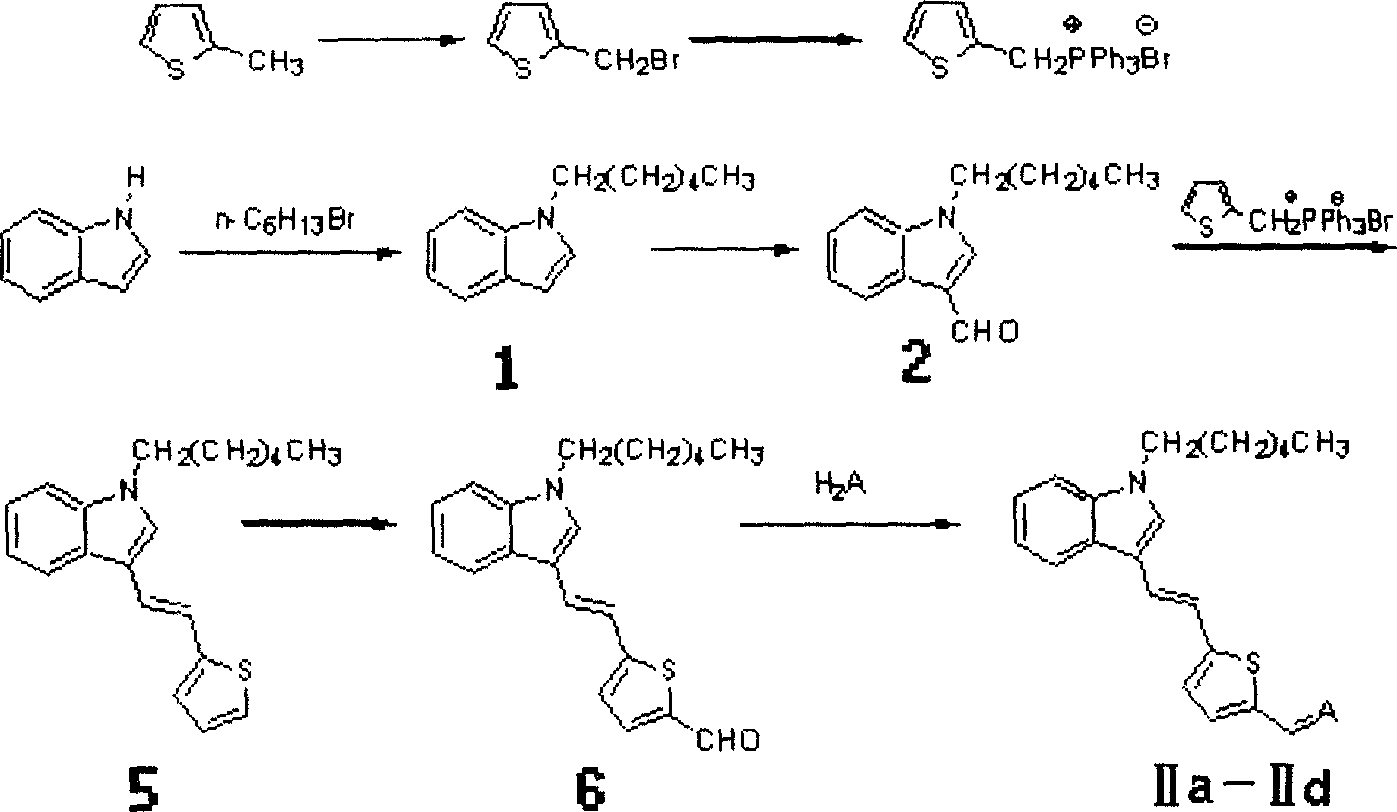
上述IIa、IIb、IIc、IId化合物中的A=分别为

合成方法为:
溴代噻吩甲基三苯基膦的制备
称取2-甲基噻吩(5.01g,50mmol),加入250ml圆底烧瓶中,然后加入四氯化碳150ml,NBS粉末(10.7g,60mmol),BPO粉末0.1g,加热至CCl4回流2h,反应液由浅黄色溶液逐渐变为较深黄色。将反应液过滤并用CCl4洗涤滤渣,收集所得CCl4溶液,旋除CCl4得到黄色油状液体,向其中加入水除去溶于水的杂质,用CHCl3萃取,加入无水Na2SO4干燥过夜。滤去Na2SO4,将所得黄色溶液转入-250ml烧瓶中,加入PPh3(10.7g,50mmol)及CHCl3100ml,加热搅拌,至CHCl3回流,反应2h后停止加热。回流过程中瓶壁出现大量白色固体,溶液颜色变浅。过滤并用少量CHCl3洗涤产物,得到白色固体粉末状产物溴代噻吩甲基三苯基膦。
3-甲酰基-N-己基吲哚(化合物2)的合成如实施例1中所述。
2-(N-正己基-3-乙烯基吲哚)噻吩(化合物5)的合成
于Schlenk管中加入正己基吲哚醛2和溴代噻吩甲基三苯基膦。氮气保护下向Schlek管中加入无水四氢呋喃(正己基吲哚醛2质量的10倍),然后加入叔丁醇钾,室温搅拌反应24h后将反应液倒入水中,用CHCl3为萃取剂萃取,旋除CHCl3,用配比为1∶1(体积比)的氯仿和石油醚为淋洗剂进行柱层析,得到橙红色油状液体产物2-(N-正己基-3-乙烯基吲哚)噻吩。其中,化合物2、溴代噻吩甲基三苯基膦和叔丁醇钾的摩尔比为1∶1∶2。
2-(N-正己基-3-乙烯基吲哚)-5-甲酰基噻吩(化合物6)的合成
向一干燥的Schlenk管中加入2-(N-正己基-3-乙烯基吲哚)噻吩以及干燥四氢呋喃(2-(N-正己基-3-乙烯基吲哚)噻吩质量的50倍),在N2保护下于-78℃液氮浴中加入正丁基锂溶液(溶剂为正己烷,浓度为2.5mol/L),搅拌1.5小时。然后将反应液提至空气中,加入N,N-二甲基甲酰胺,室温搅拌过夜。向反应液中加入水,搅拌后用CHCl3萃取。以CHCl3为淋洗剂进行柱层析,得到粘稠状棕色固体2-(N-正己基-3-乙烯基吲哚)-5-甲酰基噻吩。其中,2-(N-正己基-3-乙烯基吲哚)噻吩、正丁基锂和N,N-二甲基甲酰胺的摩尔比为1∶1.2∶1.1。
发色团IIa的合成
在一100ml圆底烧瓶中加入化合物6,再加入乙醇溶解。称取丙二腈加入反应溶液,升温至80℃,搅拌4小时。旋除乙醇,以CHCl3为淋洗剂进行柱层析,得到紫黑色微晶发色团IIa。其中,化合物6和丙二腈的摩尔比为1∶1。熔点:115-117℃。IR:2218cm-1(C≡N)。UV-Vis(氯仿,λmax):524纳米。
发色团IIb的合成
在一100ml圆底烧瓶中加入化合物6,再加入乙醇溶解。称取硫代巴比妥加入反应溶液,升温至80℃,搅拌4小时。旋除乙醇,以CHCl3为淋洗剂进行柱层析,得到蓝紫色微晶发色团IIb。其中,化合物6和硫代巴比妥的摩尔比为1∶1.3。熔点:191-193℃。IR:1653cm-1(C=O)。UV-Vis(氯仿,λmax):586纳米。
发色团IIc的合成
在一100ml圆底烧瓶中加入化合物6,再加入乙醇溶解。称取3-苯基异噁唑酮加入反应溶液,升温至80℃,搅拌4小时。旋除乙醇,以CHCl3为淋洗剂进行柱层析,得到蓝黑色微晶发色团IIc。其中,化合物6和3-苯基异噁唑酮的摩尔比为1∶1.2。熔点:139-140℃。IR:1733cm-1(C=O)。UV-Vis(氯仿,λmax):561纳米。
发色团IId的合成
在一100ml圆底烧瓶中加入化合物6,再加入乙醇溶解。称取2-二氰基亚甲基-3-氰基-4,5,5-三甲基-2,5-二氢呋喃加入反应溶液,升温至80℃,搅拌4小时。旋除乙醇,以CHCl3∶乙酸乙酯=20∶3(体积比)的混合溶剂为淋洗剂进行柱层析,得到蓝黑色微晶发色团IId。其中,化合物6和2-二氰基亚甲基-3-氰基-4,5,5-三甲基-2,5-二氢呋喃的摩尔比为1∶1。熔点:178-180℃。IR:2225cm-1(C=N)。UV-Vis(氯仿,λmax):638纳米。
实施例3:
R为正丁基,X为氧原子时,合成路线如下:

上述IIIa、IIIb、IIIc、IIId化合物中的A=分别为
合成方法为:
N-丁基吲哚(化合物7)的合成
在250mL圆底烧瓶中,放入氢氧化钾固体和N,N-二甲基甲酰胺(为氢氧化钾质量的3-10倍),搅拌20分钟后加入吲哚,搅拌40分钟。用恒压滴液漏斗加入1-溴-正丁烷溶液(溶剂为N,N-二甲基甲酰胺,质量百分比浓度为20%-50%之间)。然后升温至50℃,搅拌反应2-20小时。将反应液倒入水中,氯仿萃取,旋除氯仿后,用油泵减压蒸馏,得浅黄色液体,即为产物N-丁基吲哚。其中,氢氧化钾、吲哚、1-溴-正丁烷的摩尔比为2∶1∶1.2。
3-甲酰基-N-丁基吲哚(化合物8)的合成
称取化合物7,用1,2-二氯乙烷溶解在100mL烧瓶中(质量百分比浓度为3%-10%之间),冰浴冷至0℃,搅拌。加入N,N-二甲基甲酰胺,然后加入三氯氧磷。加热回流反应2小时,冷却后将反应液倒入盛有水中,用氯仿萃取。旋除氯仿,以氯仿为淋洗剂进行柱层析,得到红棕色液体产物。化合物7、N,N-二甲基甲酰胺、三氯氧磷的摩尔比为1∶1.3∶1.5。2-(N-正丁基-3-乙烯基吲哚)呋喃(化合物9)的合成
称取溴代呋喃甲基三苯基膦,置于Schlenk管中,氮气保护下,加入无水四氢呋喃(为溴代呋喃甲基三苯基膦质量的5-10倍),加入适量叔丁醇钾,在加入3-甲酰基-N-丁基吲哚。反应20-30小时。将反应液倒入蒸馏水中,氯仿萃取,旋除氯仿,用配比为1∶1(体积比)的氯仿和石油醚为淋洗剂进行柱层析,得到亮黄绿色液体2-(N-正丁基-3-乙烯基吲哚)呋喃。其中,溴代呋喃甲基三苯基膦、叔丁醇钾和3-甲酰基-N-丁基吲哚的摩尔比为1∶1.5∶1。
2-(N-正丁基-3-乙烯基吲哚)-5-甲酰基呋喃(化合物10)的合成
在100mL二口烧瓶中,加入化合物9,用1,2-二氯乙烷溶解(质量百分比浓度为3%-10%之间)。将三氯氧磷和N,N-二甲基甲酰胺的混合液加到上述二口烧瓶中。室温搅拌2小时后,将反应液倒入碳酸钠饱和溶液中,用1,2-二氯乙烷萃取。旋除1,2-二氯乙烷,用氯仿作淋洗剂进行柱层析,得到橙红色固体2-(N-正丁基-3-乙烯基吲哚)-5-甲酰基呋喃。其中,化合物9、三氯氧磷和N,N-二甲基甲酰胺的摩尔比为1∶1.4∶2.4。
发色团IIIa的合成
称取化合物10,用无水乙醇溶于100mL烧瓶中(质量百分比浓度为0.1%-1%之间),加入丙二腈,搅拌使其溶解。室温反应2小时,将反应液在旋转蒸发仪上除去乙醇,然后进行柱层析,淋洗剂为1∶1(体积比)的氯仿和石油醚,得到红色固体发色团IIIa。其中,化合物10和丙二腈的摩尔比为1∶1。IR:2219cm-1(C≡N)。UV-Vis(氯仿,λmax):527纳米。
发色团IIIb的合成
称取化合物10,用无水乙醇溶于100mL烧瓶中(质量百分比浓度为0.1%-1%之间),加入硫代巴比妥,加热回流3小时。旋除乙醇,用配比为1∶1(体积比)的氯仿和石油醚(沸点60-90℃)为淋洗剂进行柱层析,得到蓝紫色固体发色团IIIb。其中,化合物10和硫代巴比妥的摩尔比为1∶1.3。IR:1663cm-1(C=O)。UV-Vis(氯仿,λmax):5910纳米。
发色团IIIc的合成
称取化合物10,用无水乙醇溶于100mL烧瓶中(质量百分比浓度为0.1%-1%之间),加入3-苯基-异噁唑酮,搅拌使其溶解。室温搅拌2小时,旋除乙醇,用氯仿作柱层析淋洗剂,得到蓝紫色固体发色团IIIc。其中,化合物10和3-苯基-异噁唑酮的摩尔比为1∶1.2。IR:1667cm-1(C=0)。UV-Vis(氯仿,λmax):565纳米。
发色团IIId的合成
称取化合物10,用无水乙醇溶于100mL烧瓶中(质量百分比浓度为0.1%-1%之间),向其中加入2-二氰基亚甲基-3-氰基-4,5,5-三甲基-2,5-二氢呋喃,加热回流3小时。停止反应,旋除乙醇,以氯仿∶乙酸乙酯=20∶3(体积比)为淋洗剂进行柱层析,得到深蓝紫色固体粉末发色团IIId。其中,化合物10和2-二氰基亚甲基-3-氰基-4,5,5-三甲基-2,5-二氢呋喃的摩尔比为1∶1。IR:2224cm-1(C≡N)。UV-Vis(氯仿,λmax):652纳米。
实施例4:
R为正丁基,X为硫原子时,合成路线如下:

上述IVa、IVb、IVc、IVd化合物中的A=分别为
合成方法为:
3-甲酰基-N-丁基吲哚(化合物8)的合成如实施例3中所述。
2-(N-正丁基-3-乙烯基吲哚)噻吩(化合物11)的合成
于Schlenk管中加入化合物8和溴代噻吩甲基三苯基膦。氮气保护下向Schlek管中加入无水四氢呋喃(化合物8质量的10倍),然后加入叔丁醇钾,室温搅拌反应24小时后将反应液倒入水中,用CHCl3为萃取剂萃,旋除CHCl3,用配比为1∶1(体积比)的氯仿和石油醚为淋洗剂进行柱层析,得到橙红色油状液体产物2-(N-正丁基-3-乙烯基吲哚)噻吩。其中,化合物8、溴代噻吩甲基三苯基膦和叔丁醇钾的摩尔比为1∶1∶2。
2-(N-正丁基-3-乙烯基吲哚)-5-甲酰基噻吩(化合物12)的合成
向一干燥的Schlenk管中加入2-(N-正丁基-3-乙烯基吲哚)噻吩以及干燥四氢呋喃(2-(N-正丁基-3-乙烯基吲哚)噻吩质量的50倍),在N2保护下于-78℃液氮浴中加入正丁基锂(正己烷溶液,2.5mol/L),搅拌1.5小时。然后将反应液提至空气中,加入N,N-二甲基甲酰胺,室温搅拌过夜。向反应液中加入水,搅拌后用CHCl3萃取。以CHCl3为淋洗剂进行柱层析,得到粘稠状棕色固体化合物12。其中,2-(N-正丁基-3-乙烯基吲哚)噻吩、正丁基锂和N,N-二甲基甲酰胺的摩尔比为1∶1.2∶1.1。
发色团IVa的合成
在一100ml圆底烧瓶中加入化合物12,再加入乙醇溶解。称取丙二腈加入反应溶液,升温至80℃,搅拌4小时。旋除乙醇,以CHCl3为淋洗剂进行柱层析,得到紫黑色微晶发色团IVa。其中,化合物12和丙二腈的摩尔比为1∶1。IR:2217cm-1(C≡N)。UV-Vis(氯仿,λmax):523纳米。
发色团IVb的合成
在一100ml圆底烧瓶中加入化合物12,再加入乙醇溶解。称取硫代巴比妥加入反应溶液,升温至80℃,搅拌4小时。旋除乙醇,以CHCl3为淋洗剂进行柱层析,得到蓝紫色微晶发色团IVb。其中,化合物12和硫代巴比妥的摩尔比为1∶1.3。IR:1651cm-1(C=O)。UV-Vis(氯仿,λmax):585纳米
发色团IVc的合成
在一100ml圆底烧瓶中加入化合物12,再加入乙醇溶解。称取3-苯基异噁唑酮加入反应溶液,升温至80℃,搅拌4小时。旋除乙醇,以CHCl3为淋洗剂进行柱层析,得到蓝黑色微晶发色团IVc。其中,化合物12和3-苯基异噁唑酮的摩尔比为1∶1.1。IR:1731cm-1(C=O)。UV-Vis(氯仿,λmax):560纳米。
发色团IVd的合成
在-100ml圆底烧瓶中加入化合物12,再加入乙醇溶解。称取2-二氰基亚甲基-3-氰基-4,5,5-三甲基-2,5-二氢呋喃加入反应溶液,升温至80℃,搅拌4小时。旋除乙醇,以CHCl3∶乙酸乙酯=20∶3(体积比)的混合溶剂为淋洗剂进行柱层析,得到蓝黑色微晶发色团IVd。其中,化合物12和2-二氰基亚甲基-3-氰基-4,5,5-三甲基-2,5-二氢呋喃的摩尔比为1∶1。IR:2227cm-1(C≡N)。UV-Vis(氯仿,λmax):639纳米。
实施例5:
R为苄基,X为氧原子时,合成路线如下:

上述Va、Vb、Vc、Vd化合物中的A=分别为
合成方法为:
N-苄基吲哚(化合物13)的合成
在250mL圆底烧瓶中,放入氢氧化钾固体和N,N-二甲基甲酰胺(为氢氧化钾质量的3-10倍),搅拌20分钟后加入吲哚,搅拌40分钟。用恒压滴液漏斗加入苄氯在N,N-二甲基甲酰胺中的溶液(质量百分比浓度为20%-50%之间)。然后升温至50℃,搅拌反应2-20小时。将反应液倒入水中,氯仿萃取,旋除氯仿后,用油泵减压蒸馏,得浅黄色液体,即为产物化合物13。其中,氢氧化钾、吲哚、苄氯的摩尔比为4∶1∶1.2。
3-甲酰基-N-苄基吲哚(化合物14)的合成
称取化合物13,用1,2-二氯乙烷溶解在100mL烧瓶中(质量百分比浓度为3%-10%之间),冰浴冷至0℃,搅拌。加入N,N-二甲基甲酰胺,然后加入三氯氧磷。加热回流反应2小时,冷却后将反应液倒入盛有水中,用氯仿萃取。旋除氯仿,以氯仿为淋洗剂进行柱层析,得到红棕色液体产物化合物14。化合物13、N,N-二甲基甲酰胺、三氯氧磷的摩尔比为1∶13∶13。
2-(N-苄基-3-乙烯基吲哚)呋喃(化合物15)的合成
称取溴代呋喃甲基三苯基膦,置于Schlenk管中,氮气保护下,加入无水四氢呋喃(为溴代呋喃甲基三苯基膦质量的5-10倍),加入叔丁醇钾,再加入3-甲酰基-N-苄基吲哚。反应20-30小时。将反应液倒入蒸馏水中,氯仿萃取,旋除氯仿,用配比为1∶1(体积比)的氯仿和石油醚为淋洗剂进行柱层析,得到亮黄绿色液体化合物15。其中,溴代呋喃甲基三苯基膦、叔丁醇钾和3-甲酰基-N-苄基吲哚的摩尔比为1∶1.5∶1。
2-(N-苄基-3-乙烯基吲哚)-5-甲酰基呋喃(化合物16)的合成
在100mL二口烧瓶中,加入化合物15,用1,2-二氯乙烷溶解(质量百分比浓度为3%-10%之间)。将三氯氧磷和N,N-二甲基甲酰胺的混合液加到上述二口烧瓶中。室温搅拌2小时后,将反应液倒入碳酸钠饱和溶液中,用1,2-二氯乙烷萃取。旋除1,2-二氯乙烷,用氯仿作淋洗剂进行柱层析,得到橙红色固体化合物16。其中,化合物15、三氯氧磷和N,N-二甲基甲酰胺的摩尔比为1∶1.4∶2.4。
发色团Va的合成
称取化合物16,用无水乙醇溶于100mL烧瓶中(质量百分比浓度为0.1%-1%之间),加入丙二腈,搅拌使其溶解。室温反应2小时,将反应液在旋转蒸发仪上除去乙醇,然后进行柱层析,淋洗剂为1∶1(体积比)的氯仿和石油醚,得到红色固体发色团Va。其中,化合物16和丙二腈的摩尔比为1∶1.3。IR:2219cm-1(C≡N)。UV-Vis(氯仿,λmax):527纳米。发色团Vb的合成
称取化合物16,用无水乙醇溶于100mL烧瓶中(质量百分比浓度为0.1%-1%之间),加入硫代巴比妥,加热回流3小时。旋除乙醇,用配比为1∶1(体积比)的氯仿和石油醚(沸点60-90℃)为淋洗剂进行柱层析,得到蓝紫色固体发色团Vb。其中,化合物16和硫代巴比妥的摩尔比为1∶1.3。IR:1664cm-1(C=O)。UV-Vis(氯仿,λmax):591纳米。
发色团Vc的合成
称取化合物16,用无水乙醇溶于100mL烧瓶中(质量百分比浓度为0.1%-1%之间),加入3-苯基-异噁唑酮,搅拌使其溶解。室温搅拌2小时,旋除乙醇,用氯仿作柱层析淋洗剂,得到蓝紫色固体发色团Vc。其中,化合物16和3-苯基-异噁唑酮的摩尔比为1∶1。IR:1665cm-1(C=O)。UV-Vis(氯仿,λmax):566纳米。
发色团Vd的合成
称取化合物16,用无水乙醇溶于100mL烧瓶中(质量百分比浓度为0.1%-1%之间),向其中加入适量2-二氰基亚甲基-3-氰基-4,5,5-三甲基-2,5-二氢呋喃,加热回流3小时。停止反应,旋除乙醇,以氯仿∶乙酸乙酯=20∶3(体积比)为淋洗剂进行柱层析,得到深蓝紫色固体粉末发色团Vd。其中,化合物16和2-二氰基亚甲基-3-氰基-4,5,5-三甲基-2,5-二氢呋喃的摩尔比为1∶1.3。IR:2221cm-1(C≡N)。UV-Vis(氯仿,λmax):651纳米。
实施例6:
R为苄基,X为硫原子时,合成路线如下:

上述VIa、VIb、VIc、VId化合物中的A=分别为
合成方法为:
3-甲酰基-N-苄基吲哚(化合物14)的合成如实施例5中所述。
2-(N-苄基-3-乙烯基吲哚)噻吩(化合物17)的合成
于Schlenk管中加入化合物14和溴代噻吩甲基三苯基膦。氮气保护下向Schlek管中加入无水四氢呋喃(化合物14质量的10倍),然后加入叔丁醇钾,室温搅拌反应24小时后将反应液倒入水中,用CHCl3为萃取剂萃,旋除CHCl3,用配比为1∶1(体积比)的氯仿和石油醚为淋洗剂进行柱层析,得到橙红色油状液体产物化合物17。其中,化合物14、溴代噻吩甲基三苯基膦和叔丁醇钾的摩尔比为1∶1∶2。
2-(N-苄基-3-乙烯基吲哚)-5-甲酰基噻吩(化合物18)的合成
向一于燥的Schlenk管中加入2-(N-苄基-3-乙烯基吲哚)噻吩以及干燥四氢呋喃(2-(N-苄基-3-乙烯基吲哚)噻吩质量的50倍),在N2保护下于-78℃液氮浴中加入正丁基锂(正己烷溶液,2.5mol/L),搅拌1.5小时。然后将反应液提至空气中,加入N,N-二甲基甲酰胺,室温搅拌过夜。向反应液中加入水,搅拌后用CHCl3萃取。以CHCl3为淋洗剂进行柱层析,得到粘稠状棕色固体化合物18。其中,2-(N-苄基-3-乙烯基吲哚)噻吩、正丁基锂和N,N-二甲基甲酰胺的摩尔比为1∶1.2∶1.1。
发色团VIa的合成
在一100ml圆底烧瓶中加入化合物18,再加入乙醇溶解。称取丙二腈加入反应溶液,升温至80℃,搅拌4小时。旋除乙醇,以CHCl3为淋洗剂进行柱层析,得到紫黑色微晶发色团VIa。其中,化合物18和丙二腈的摩尔比为1∶1。IR:2220cm-1(C≡N)。UV-Vis(氯仿,λmax):524纳米。
发色团VIb的合成
在一100ml圆底烧瓶中加入化合物18,再加入乙醇溶解。称取硫代巴比妥加入反应溶液,升温至80℃,搅拌4小时。旋除乙醇,以CHCl3为淋洗剂进行柱层析,得到蓝紫色微晶发色团VIb。其中,化合物18和硫代巴比妥的摩尔比为1∶1.3。IR:1655cm-1(C=O)。UV-Vis(氯仿,λmax):586纳米
发色团VIc的合成
在一100ml圆底烧瓶中加入化合物18,再加入乙醇溶解。称取3-苯基异噁唑酮加入反应溶液,升温至80℃,搅拌4小时。旋除乙醇,以CHCl3为淋洗剂进行柱层析,得到蓝黑色微晶发色团VIc。其中,化合物18和3-苯基异噁唑酮的摩尔比为1∶1。IR:1730cm-1(C=O)。UV-Vis(氯仿,λmax):562纳米。
发色团VId的合成
在一100ml圆底烧瓶中加入化合物18,再加入乙醇溶解。称取2-二氰基亚甲基-3-氰基-4,5,5-三甲基-2,5-二氢呋喃加入反应溶液,升温至80℃,搅拌4小时。旋除乙醇,以CHCl3∶乙酸乙酯=20∶3(体积比)的混合溶剂为淋洗剂进行柱层析,得到蓝黑色微晶发色团VId。其中,化合物18和2-二氰基亚甲基-3-氰基-4,5,5-三甲基-2,5-二氢呋喃的摩尔比为1∶1.2。IR:2222cm-1(C≡N)。UV-Vis(氯仿,λmax):636纳米。
Claims (2)
1.含吲哚基团的二阶非线性光学发色团,其结构通式为:

式中R为C1-C9的烷烃基或C7-C11芳香基团;X为氧或者硫原子,A选自下述吸电子基团:丙二腈、硫代巴比妥酸、3-苯基-异噁唑酮、2-二氰基亚甲基-3-氰基-4,5,5-三甲基-2,5-二氢呋喃。
2.权利要求1所述含吲哚基团的二阶非线性光学发色团的制法,其特征在于:以吲哚和C1-C9的卤代烷烃或C7-C11的卤代芳烃为原料制备取代吲哚A,其中吲哚和C1-C9的卤代烷烃或C7-C11的卤代芳烃的摩尔比例为1∶1.0~2.0,取代吲哚A为N原子上连有C1-C9的烷烃基或C7-C11芳香基团的吲哚;取代吲哚A与N,N-二甲基甲酰胺和三氯氧磷反应,制备吲哚醛B,其中取代吲哚A、N,N-二甲基甲酰胺、三氯氧磷的摩尔比为1∶1.1~1.9∶1.1~1.9;然后吲哚醛B与溴代呋喃甲基三苯基膦或溴代噻吩甲基三苯基膦反应,制备乙烯基吲哚呋喃C或乙烯基吲哚噻吩D,其中吲哚醛B与溴代呋喃甲基三苯基膦或者溴代噻吩甲基三苯基膦的摩尔比为1∶1.0~2.0;乙烯基吲哚呋喃C与N,N-二甲基甲酰胺和三氯氧磷反应,制备乙烯基吲哚呋喃醛E,其中乙烯基吲哚呋喃C、N,N-二甲基甲酰胺、三氯氧磷的摩尔比为1∶1.1~1.9∶2.0~3.7;或者乙烯基吲哚噻吩D与正丁基锂和N,N-二甲基甲酰胺反应制备乙烯基吲哚噻吩醛F,其中乙烯基吲哚噻吩D与正丁基锂和N,N-二甲基甲酰胺的摩尔比为1∶1.1~1.5∶1.0~1.4;最后以乙烯基吲哚呋喃醛E或者乙烯基吲哚噻吩醛F为原料,与丙二腈、硫代巴比妥酸、3-苯基-异噁唑酮或2-二氰基亚甲基-3-氰基-4,5,5-三甲基-2,5-二氢呋喃反应,制得权利要求1所述含吲哚基团的二阶非线性光学发色团,其中乙烯基吲哚呋喃醛E或者乙烯基吲哚噻吩醛F与各种拉电子基团之间的摩尔比为1∶1~1.3。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CNB2005100193965A CN100361990C (zh) | 2005-09-06 | 2005-09-06 | 含吲哚基团的二阶非线性光学发色团及其制法 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CNB2005100193965A CN100361990C (zh) | 2005-09-06 | 2005-09-06 | 含吲哚基团的二阶非线性光学发色团及其制法 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN1740171A CN1740171A (zh) | 2006-03-01 |
| CN100361990C true CN100361990C (zh) | 2008-01-16 |
Family
ID=36092716
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CNB2005100193965A Expired - Fee Related CN100361990C (zh) | 2005-09-06 | 2005-09-06 | 含吲哚基团的二阶非线性光学发色团及其制法 |
Country Status (1)
| Country | Link |
|---|---|
| CN (1) | CN100361990C (zh) |
Families Citing this family (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN100386321C (zh) * | 2006-04-21 | 2008-05-07 | 武汉大学 | 含三苯胺基团y型二阶非线性光学发色团及其制法和用途 |
| CN101602759B (zh) * | 2009-07-07 | 2012-08-22 | 武汉大学 | 一类含吡咯基团的化合物及其制备方法和用途 |
| CN103709197B (zh) * | 2014-01-17 | 2015-11-04 | 四川大学 | 取代的水杨醛-tcf衍生物及其制备方法和用途 |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6348596B1 (en) * | 1998-01-23 | 2002-02-19 | Pe Corporation (Ny) | Non-fluorescent asymmetric cyanine dye compounds useful for quenching reporter dyes |
-
2005
- 2005-09-06 CN CNB2005100193965A patent/CN100361990C/zh not_active Expired - Fee Related
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6348596B1 (en) * | 1998-01-23 | 2002-02-19 | Pe Corporation (Ny) | Non-fluorescent asymmetric cyanine dye compounds useful for quenching reporter dyes |
Non-Patent Citations (1)
| Title |
|---|
| Synthesis and Propertes of PhotorefractivePolymersConttaining Idole-Based MultifunctionalChromophoire as aPendant Group. H.Moon et al.Macromolecules,Vol.33 No.14. 2000 * |
Also Published As
| Publication number | Publication date |
|---|---|
| CN1740171A (zh) | 2006-03-01 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| Xiong et al. | Tetrathio and tetraseleno [8] circulenes: synthesis, structures, and properties | |
| Shimogawa et al. | 4, 7‐Bis [3‐(dimesitylboryl) thien‐2‐yl] benzothiadiazole: Solvato‐, Thermo‐, and Mechanochromism Based on the Reversible Formation of an Intramolecular B− N Bond | |
| Huang et al. | Dibenzoannelated tetrathienoacene: synthesis, characterization, and applications in organic field-effect transistors | |
| Zhou et al. | Effects of cyano groups on the properties of thiazole-based β-ketoiminate boron complexes: aggregation-induced emission and mechanofluorochromism | |
| Zhao et al. | Mechanofluorochromism of difluoroboron β-ketoiminate boron complexes functionalized with benzoxazole and benzothiazole | |
| CN104098555B (zh) | 咪唑‑噻吩芳杂环超短波长光致变色二芳基乙烯化合物的合成方法及应用 | |
| Traskovskis et al. | Modular approach to obtaining organic glasses from low-molecular weight dyes using 1, 1, 1-triphenylpentane auxiliary groups: Nonlinear optical properties | |
| Zheng et al. | Syntheses and photochromism of new isomeric diarylethenes bearing an indole moiety | |
| Koeckelberghs et al. | Synthesis and properties of chiral helical chromophore-functionalised polybinaphthalenes for second-order nonlinear optical applications | |
| CN100361990C (zh) | 含吲哚基团的二阶非线性光学发色团及其制法 | |
| Zheng et al. | A fluorescent sensor for detection of grinding force and fluoride ion based on acylhydrazone derivative | |
| CN101239976B (zh) | 含萘酰亚胺单元的二噻吩乙烯类化合物 | |
| Cui et al. | Synthesis and nonlinear optical properties of a series of azo chromophore functionalized alkoxysilanes | |
| CN111057087B (zh) | 一种非对称噻吩[7]螺烯同分异构体及其制备方法和应用 | |
| Yuan et al. | Multistimuli-responsive dual-state emissive imidazo [1, 2-α] pyridine as imaging probe for lipid droplets | |
| CN116854652A (zh) | 一种自交联有机电光分子玻璃材料及其制备方法和应用 | |
| Tian et al. | Thieno [3, 2‐b] thiophene‐Based Discotic Liquid Crystal Mesogens: Rational Synthesis, Physical Properties and Self‐Assembly | |
| CN100386321C (zh) | 含三苯胺基团y型二阶非线性光学发色团及其制法和用途 | |
| CN105367592A (zh) | 一种中位烷基噻吩取代及3,5位给电子基取代氟化硼络合二吡咯甲川衍生物及其制备方法 | |
| CN101602743A (zh) | 光致变色噻唑六元环混联型不对称全氟环戊烯化合物及制备方法和应用 | |
| CN105968130B (zh) | 一种中位含咔唑及桥联基团的双中心氟化硼络合二吡咯甲川衍生物及其制备方法 | |
| CN101845041B (zh) | 光致变色苯并呋喃噻吩混联型全氟环戊烯化合物及合成方法和应用 | |
| Qu et al. | High contrast stimuli-responsive fluorescence emitters based on carbazole-cyanostilbene derivatives with aggregation induced emission properties | |
| CN108503669A (zh) | 一种高效二芳基乙烯光控开关分子及其制备方法 | |
| CN109232623B (zh) | 一种硼氮杂菲及其衍生物的合成方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C14 | Grant of patent or utility model | ||
| GR01 | Patent grant | ||
| C19 | Lapse of patent right due to non-payment of the annual fee | ||
| CF01 | Termination of patent right due to non-payment of annual fee |