CN101190899B - 一种合成1,2-二氢喹唑啉-4(3h)-酮类化合物的方法 - Google Patents
一种合成1,2-二氢喹唑啉-4(3h)-酮类化合物的方法 Download PDFInfo
- Publication number
- CN101190899B CN101190899B CN2007103042449A CN200710304244A CN101190899B CN 101190899 B CN101190899 B CN 101190899B CN 2007103042449 A CN2007103042449 A CN 2007103042449A CN 200710304244 A CN200710304244 A CN 200710304244A CN 101190899 B CN101190899 B CN 101190899B
- Authority
- CN
- China
- Prior art keywords
- dihydroquinazoline
- reaction
- synthetic
- ketone compounds
- ketone
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
- 0 *C(*)(Nc1ccccc11)NC1=O Chemical compound *C(*)(Nc1ccccc11)NC1=O 0.000 description 1
- HLCPWBZNUKCSBN-UHFFFAOYSA-N Nc1c(C#N)cccc1 Chemical compound Nc1c(C#N)cccc1 HLCPWBZNUKCSBN-UHFFFAOYSA-N 0.000 description 1
Landscapes
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Nitrogen And Oxygen As The Only Ring Hetero Atoms (AREA)
- Plural Heterocyclic Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
本发明提供了一种1,2-二氢喹唑啉-4(3H)-酮杂环化合物的合成新方法,反映通式为: 式中:R为取代基,可以为F,Cl,Br,I,NO2,NO,烷基,酮基,烷氧基和胺基等基团;取代基的数量和位置不限;R1、R2为H、烷基、环烷基、芳烃基等。催化剂可以是无水氯化锌、无水三氯化铝、氯化铜、氯化亚铜、甲基苯磺酸、对甲基苯磺酸等;反应的实施可常规加热,也可微波促进;纯化采用重结晶或柱色谱分离手段。本发明原料易得,工艺简单,反应条件温和;反应应用范围广,可用不同底物合成多种1,2-二氢喹唑啉-4(3H)-酮类化合物。
式中:R为取代基,可以为F,Cl,Br,I,NO2,NO,烷基,酮基,烷氧基和胺基等基团;取代基的数量和位置不限;R1、R2为H、烷基、环烷基、芳烃基等。催化剂可以是无水氯化锌、无水三氯化铝、氯化铜、氯化亚铜、甲基苯磺酸、对甲基苯磺酸等;反应的实施可常规加热,也可微波促进;纯化采用重结晶或柱色谱分离手段。本发明原料易得,工艺简单,反应条件温和;反应应用范围广,可用不同底物合成多种1,2-二氢喹唑啉-4(3H)-酮类化合物。
Description
技术领域
本发明涉及一种经由邻氨基芳香腈与酮反应制备1,2-二氢喹唑啉-4(3H)-酮(英文名1,2-dihydroquinazolin-4(3H)-one)杂环化合物的合成方法。
背景技术
1,2-二氢喹唑啉-4(3H)-酮杂环化合物的是一类具有良好生理活性的含氮杂环化合物,在抗肿瘤(Bioconjugate Chem.,2002,13:357-364;J.Med.Chem.,2007,50:663-673;Bioconjugate Chem.,2007,18:754-764)、抗炎症(J.Med.Chem.,1971,14:714-717;J.Org.Chem.,1993,58:741-743;Chem.Res.Toxicol.,2000,13:775-784,J.Org.Chem.,2000,65:2773-2777)、抗高血压(Bioorg.Med.Chem.,2003,11:2439-2444)、治疗哮喘(J.Org.Chem.,2001,66:997-1001)、杀菌[(J.Chem.Eng.Data,1986,31:501-502;J.Chem.Inf.Model,2005,45:634-644;J.Med.Chem.,1998,41:1855-1868;J.Chem.Inf.Model,2005,45:634-644)、治疗关节炎(J.Med.Chem.,1968,11:1208-1213;J.Med.Chem.,1985,28:568-576)、除草剂(Eur.Pat.Appl.EP58822,1982)和植物生长调节剂(US Pat.4431440,1981)等方面显示出良好的生物活性。
已知的1,2-二氢喹唑啉-4(3H)-酮类化合物的合成方法主要有:①以邻氨基苯甲酸或酯和甲酰氨经由Niementowski反应合成2,3-二氢喹唑啉-4(3H)-酮(J.Heterocyclic Chem.,1971,8:699-672),反应的催化剂可以是氯化亚锡(J.Org.Chem.,2005,70:6491-6493)、对甲苯磺酸(J.Med.Chem.,2000,43:4479-4487)等,该方法步骤少,但反应温度高,不易控制。②催化下以邻硝基或叠氮基苯甲酰胺与羰基化合物反应制得,反应的催化剂可以是碘化钐(J.Chem.Res.,2002,604-605;J.Heterocyclic Chem.,2002,39:1271-1272)、四氯化钛和锌(J.Heterocyclic Chem.,2005,42:173-183;中国化学,2004,22:743-746)、氯化钛和钐(J.Chem.Res.,2003,671-673)组成的低价钛混合物和[Ru3(CO)12]及其它过渡金属配合物(J.Org.Chem.,1993,58:310-312;J.Org.Chem.,2000,65:2773-2777;Synthesis,1991:1009-1010)等,这种方法反应条件温和,产率较高,但催化剂较贵、制备不易,催化活性难以保证。③在对甲苯磺酸催化下,由靛红酸酐、醛和胺进行三组分缩合反应制得(Synthesis,2006,344-348)。④也可以采用固相法合成法经由邻氨基苯甲酸或酯和甲酰胺合成制得(高等学校化学学报,2004,25:462-465),该技术可用于大组合库的合成,产品纯度高,但缺点是反应步骤多,总产率低(TetrahedronLett.,2002,43:939-942)。
综上所述,在1,2-二氢喹唑啉-4(3H)-酮的合成方法中,或者催化剂的价格过于昂贵、活性无保障;或者操作复杂;或者产率太低,这些不足对于它们的合成特别是工业生产都有不便。
发明内容
本发明涉及一种合成1,2-二氢喹唑啉-4(3H)-酮杂环化合物的新方法,即采用邻氨基芳香氰化物与醛或酮反应,生成目标化合物。反应通式为:

其中R为取代基,可以为F,Cl,Br,I,NO2,NO,烷基,酮基,烷氧基或胺基;该取代基的数量和位置不限。R1、R2为H、烷基、环烷基、芳烃基。
反应介质为苯、甲苯、二甲苯、硝基苯、氯苯、环丁砜、二甲亚砜、N,N-二甲基甲酰胺、N,N-二甲基乙酰胺、二氧六环、四氢呋喃以及卤代烃类极性溶剂;对于常温下为液态的羰基化合物,除了可以使用前述的反应介质外,自身也可以作为反应介质。
反应物1、2的物质的量的比为1∶1~1∶99。
反应的催化剂可以是无水氯化锌、无水三氯化铝、氯化铜、氯化亚铜等路易斯酸类,也可以是盐酸、硫酸等质子酸或吡啶、哌啶、碳酸钠等碱类催化剂。
路易斯酸类催化剂的用量为,但不限于反应物1的物质的量的1~1.5倍;质子酸或碱类催化剂的用量为反应物1的物质的量的1%~100%。
参与反应的邻氨基芳香氰化物可以在芳环上含有不同的取代基团,包括各种拉电子(硝基、卤素、羰基等)基团和推电子基团(烷基、烷氧基或胺基)。
醛可以是脂肪醛,也可以是芳香醛。
酮可以是脂肪酮,也可以是芳香酮。
所述的反应可以采用,但不限于常压油浴加热或微波加热方法;在常压油浴中的反应时间一般为1-10小时,产物的收率为10-99%;在微波促进条件下(Biotage合成仪),反应时间一般为6-50分钟,产率为10-99%。
产物的分离与提纯方法为:首先将反应物分散到少量水中,用碱中和除去催化剂,过滤后所得固体用有机溶剂溶出,而滤液进行萃取;将固体中溶出物和萃取液合并浓缩,得到粗产物,然后对所得的粗产品进行重结晶或者柱层析纯化,即得到纯的目标化合物。
除去催化剂后过滤所得的固体所用有机溶剂可以是,但不限于甲醇、乙醇、丙酮、四氢呋喃、乙酸乙酯、乙腈。
滤液的萃取剂可以是,但不限于乙酸乙酯、二氯甲烷、氯仿、乙醚。
粗产物的重结晶溶剂可以是,但不限于甲醇、乙醇、异丙醇、丙酮、乙腈、四氢呋喃、二氧六环、乙酸乙酯、二氯甲烷、氯仿、苯、甲苯、硝基苯。
柱层析采用硅胶柱或氧化铝柱,展开剂为,但不限于乙酸乙酯/石油醚(1∶1~1∶3,体积比)、甲醇/氯仿(1∶20~1∶50,体积比)、二氯甲烷、丙酮。
对于本发明的合成过程,可按参与反应的羰基化合物类型分别阐释为:
1)邻氨基芳香氰化物与醛的反应。反应通式中R1或R2为H。
将摩尔配比为1∶1~1∶99的邻氨基芳香氰化物与醛的混合液在100~160℃的反应温度下搅拌回流1-10小时,反应液冷却,分散于水中,搅拌下用氢氧化钠调节pH值到12-13,过滤,滤液和滤饼分别经过萃取和抽提后得到粗产物,再经重结晶或柱分离得到纯品。其中,此类反应的邻氨基芳香氰化物可以是,但不限于5-硝基-2-氨基苯甲腈、5-氯-2-氨基苯甲腈、5-溴-2-氨基苯甲腈、2-氨基苯甲腈。参加反应的醛可以是脂肪醛也可以是芳香醛。当醛为芳香类醛时,可以是、但不限于苯甲醛、3-甲氧基苯甲醛、茴香醛、邻硝基苯甲醛、间硝基苯甲醛、对硝基苯甲醛、水杨醛、对氯苯甲醛、吡啶甲醛、2-呋喃甲醛。
2)邻氨基芳香氰化物与酮的反应。反应通式中R1和R2皆不为H。
反应条件和参加反应的邻氨基芳香氰化物如1中所述。参加反应的酮可以是、但不限于丙酮、丁酮、戊酮、己酮、环戊酮、环己酮、环庚酮、甲基环己酮、对叔丁基环己酮、苯乙酮、各种取代苯乙酮。
本发明优点在于:原料易得,工艺简单,反应条件温和。应用范围广泛,可用不同的底物一步合成多种1,2-二氢喹唑啉-4(3H)-酮化合物。
具体实施方式
实施例1
在50mL的容器中加入苯甲醛4mL,DMF 10mL,搅拌下加入5-硝基邻氨基苯甲腈(1.0g,6mmol),再加入无水ZnCl2(0.8g,6mmol),加热回流1h。反应毕,混合液冷却,倒入20mL冷水中,析出的固体过滤。将所得固体分散于水中,搅拌下用20%的氢氧化钠溶液调节溶液的pH值到12~13,抽滤,得粗产品。粗产品用硅胶(200-300目)分离,洗脱液是乙酸乙酯和石油醚(1∶2,体积比)的混合溶剂,得到6-硝基-2-苯基-1,2-二氢喹唑啉-4(3H)-酮(I),收率80%,Mp 264~265℃。5-硝基-2-氨基苯甲腈与苯甲醛反应式为:

产物(I)的波谱数据为:IR(KBr):3385,3166,1690,1618,1530,1329,1140cm-1;1H NMR(DMSO-d6)δH:6.02(1H,t,J=1.7Hz,CH),6.83(1H,d,J=8.8Hz,ArH),7.39(1H,t,J=7.2Hz,ArH),7.42(2H,t,J=7.2,8.0Hz,ArH),7.49(2H,d,J=8.0Hz,ArH),8.11(1H,dd,J=2.8,8.8Hz,ArH),8.44(1H,d,J=2.8Hz,ArH),8.57(1H,s,NH),8.75(1H,s,NH);13C NMR(DMSO-d6)δC:66.30,112.64,114.24,124.18,126.54(2C),128.65(2C),128.88,128.97,137.12,141.09,152.13,161.28;MS(ESI):m/z(%)=270.1(100)[M+H]+;C14H11N3O3:calcd.C 62.45,H 4.12,N 15.61;found C 62.28,H 3.80,N 15.25。
实施例2
用2-氨基苯甲腈(0.75g,6mmol)代替5-硝基邻氨基苯甲腈,其它同实例1。得到目标化合物(II),产率71%,Mp 224~226℃。2-氨基苯甲腈与苯甲醛反应式为:

产物(II)的波谱数据为:IR(KBr):3370,3177,1678,1608,1472cm-1;1H NMR(DMSO-d6)δH:5.71(1H,t,J=1.7Hz,CH),6.77-6.79(4H,m,J=8.0Hz,ArH),7.11(1H,s,NH),7.25(1H,d,J=8.0Hz,ArH),7.31(1H,t,J=8.0Hz,ArH),7.78(3H,t,J=8.0Hz,ArH),8.28(1H,s,NH);13C NMR(DMSO-d6)δC:65.64,113.82,114.30,124.81,127.45(2C),128.80(2C),128.88,129.33,133.38,140.56,147.23,163.28;MS(ESI):m/z(%)=225.1(100)[M+H]+;C14H12N2O:calcd.C 74.98,H 5.39,N 12.49;found C 74.67,H 5.21,N 12.63。
实施例3
用3-甲氧基苯甲醛代替苯甲醛,其它同实例2。粗品用乙醇重结晶可得到纯的目标物(III),产率为78%,Mp 223~224℃。2-氨基苯甲腈与3-甲氧基苯甲醛反应式为:
产物(III)的波谱数据为:IR(KBr):3267,3162,1671,1612cm-1;1H NMR(DMSO-d6)δH:3.84(3H,s,CH3),5.67(1H,s,CH),6.52(2H,dd,J=2.0,7.6Hz,ArH),6.82-6.86(1H,m,J=8.0Hz,ArH),6.95(1H,s,NH),7.02(1H,d,J=8.0Hz,ArH),7.10-7.11(1H,m,J=8.0Hz,ArH),7.26(1H,dd,J=8.0Hz,ArH),7.36(2H,dd,J=2.0,7.6Hz,ArH),8.18(1H,s,NH);13C NMR(DMSO-d6)δC:55.43,65.36,114.56,114.65,117.12 117.34,117.58,128.12(2C),128.68,132.72,133.75,140.56,147.52,163.11;MS(ESI):m/z(%)=255.1(100)[M+H]+;C15H14N2O2:calcd.C 70.85,H 5.55,N 11.02;found C 70.88,H 5.15,N 10.76。
实施例4
用茴香醛(3.0ML)代替苯甲醛,其他同实例1,目标物(IV)产率分别为82%,Mp 242~243℃。5-硝基-2-氨基苯甲腈与茴香醛反应式为:
目标物(IV)的波谱数据:IR(KBr):3385,3162,1660,1620,1512,1309cm-1;1H NMR(DMSO-d6)δH:3.75(3H,s,CH3),5.97(1H,s,CH),6.82(1H,d,J=8.8Hz,ArH),6.98(2H,d,J=8.4Hz,ArH),7.39(2H,d,J=8.4Hz,ArH),8.11(1H,dd,J=2.8,8.8Hz,ArH),8.43(1H,d,J=2.8Hz,ArH),8.50(1H,s,NH),8.68(1H,s,NH);13C NMR(DMSO-d6)δC:55.19,65.91,112.62,113.90(2C),114.20,124.16,127.94(2C),128.93,132.94,136.99,152.19,159.67,161.36;MS(ESI):m/z(%)=300.1(100)[M+H]+;C15H13N3O4:calcd.C 60.19,H 4.38,N 14.04;foundC 60.05,H 4.40,N 14.11。
实施例5
用对硝基苯甲醛(0.9g,6mmoL)代替苯甲醛,其他同实例2,粗产品用柱层析分离后得到纯的目标物(V),其产率67%,Mp 198~200℃。2-氨基苯甲腈与对硝基苯甲醛反应式为:

目标物(V)的波谱数据:IR(KBr):3389,3282,1647,1615,1520,1349cm-1;1H NMR(DMSO-d6)δH:5.91(1H,s,CH),6.68(1H,t,J=7.6Hz,ArH),6.76(1H,d,J=8.0Hz,ArH),7.26(1H,t,J=7.6Hz,ArH),7.33(1H,s,NH),7.60(1H,d,J=8.0Hz,ArH),7.73(2H,d,J=8.4Hz,ArH),8.25(2H,d,J=8.4Hz,ArH),8.51(1H,s,NH);13CNMR(DMSO-d6)δC:65.76,115.02,115.38,117.92,124.02(2C),127.86,128.48(2C),134.00,147.68,147.91,149.81,163.71;MS(ESI):m/z(%)=270.1(100)[M+H]+;C14H11N3O3:calcd.C 62.45,H 4.12,N 15.61;found C 62.21,H 4.51,N 15.36。
实施例6
50ml的容器中加入已干燥的环戊酮15ml,搅拌下加入5-硝基邻氨基苯甲腈(1.0g,0.006mol),再加入无水ZnCl2(0.8g,0.006mol),搅拌下回流反应1h。反应液冷却,倒入15ml冷水中,析出的固体过滤。将所得固体分散于水中,搅拌下用20%的氢氧化钠溶液调节溶液的pH值到12~13,抽滤,得粗产品。粗产品用硅胶柱分离,洗脱液是乙酸乙酯和石油醚(1∶2,体积比)的混合溶剂,得到目标产物(VI),产率70%,Mp 281~283℃。5-硝基-2-氨基苯甲腈与环戊酮反应式为:

目标物(VI)的波谱数据:IR(KBr):3319,3180,2912,1672,1619,1534,1310cm-1;1H NMR(DMSO-d6)δH:1.67~1.88(8H,m,C4H8),6.82(1H,d,J=8.0Hz,ArH),8.10(1H,dd,J=2.4,8.0Hz,ArH),8.30(1H,s,NH),8.42(1H,d,J=2.4Hz,ArH),8.56(1H,s,NH);13C NMR(DMSO-d6)δC:21.90(2C),40.15(2C),77.32,112.31,114.14,124.11,128.62,136.68,151.63,161.21;MS(ESI):m/z(%)=248.2(100)[M+H]+;C12H13N3O3:calcd.C 58.29,H 5.30,N 17.00;found C 58.33,H 5.31,N 17.08。
实施例7
用环己酮(10.0mL)代替环戊酮,其他同实例6,得到目标化合物(VII),产率为75%,Mp 297~299℃。5-硝基-2-氨基苯甲腈与环己酮反应式为:

产物(VII)的波谱数据为:IR(KBr):3359,3188,2935,1672,1618,1529,1313cm-1;1H NMR(DMSO-d6)δH:1.05-1.80(10H,m,C5H10),6.94(1H,d,J=7.7Hz,ArH),8.08(1H,s,NH),8.10(1H,dd,J=2.8,7.7Hz,ArH),8.39(1H,d,J=2.8Hz,ArH),8.44(1H,s,NH);13C NMR(DMSO-d6)δC:21.13(2C),24.74,38.36(2C),69.19,112.88,114.99,124.57,129.26,137.27,151.85,161.66;MS(ESI):m/z(%)=262.2(100)[M+H]+;C13H15N3O3:calcd.C 59.76,H 5.78,N 16.08;found C 59.73,H 5.79,N 16.09。
实施例8
用丙酮(10.0mL)代替环己酮,2-氨基苯甲腈代替5-硝基邻氨基苯甲腈,其它同实例6。粗产品经柱分离得到化合物(VIII),产率为65%,Mp 190~191℃。2-氨基苯甲腈与丙酮反应式为:

产物(VIII)的波谱数据为:IR(KBr):3325,3166,1629,1606,1511cm-1;1H NMR(DMSO-d6,400MHz)δH:1.38(6H,s,CH3),6.59(1H,d,J=7.6Hz,ArH),6.62(1H,s,NH),6.64(1H,d,J=1.6Hz,ArH),7.20-7.23(1H,m,J=1.6,1.2,7.6Hz,ArH),7.57(1H,dd,J=1.2,7.6Hz,ArH),7.87(1H,s,NH);13C NMR(DMSO-d6,100MHz)δC:28.90(2C),66.73,113.78,114.13,116.31,127.06,133.05,146.96,162.92;MS(ESI):m/z(%)=177.1(100)[M+H]+;C10H12N2O:calcd.C 68.16,H 6.86,N 15.90;found C 68.50,H 6.98,N 15.74。
实施例9
用丁酮(10.0mL)代替丙酮,其它同实例8。粗产品经柱分离得到化合物(IX),产率为67%,Mp 153~155℃。2-氨基苯甲腈与对丁酮反应式为:

产物(IX)的波谱数据为:IR(KBr):3340,2913,1631,1609,1540cm-1;1H NMR(DMSO-d6,400MHz)δH:0.72(3H,t,J=6.8Hz,CH3),1.15(3H,s,CH3),1.70-1.76(2H,m,CH2),6.16(1H,s,NH),6.55-6.59(1H,m,J=8.0Hz,ArH),6.63(1H,d,J=8.0Hz,ArH),7.18-7.22(1H,m,J=1.2,8.0Hz,ArH),7.61(1H,dd,J=1.2,8.0Hz,ArH),8.11(1H,s,NH);13C NMR(DMSO-d6,100MHz)δC:8.35,27.37,34.88,71.59,112.35,115.51,118.16,128.72,136.16,148.57,167.68;MS(ESI):m/z(%)=191.1(100)[M+H]+;C11H14N2O:calcd.C 69.45,H 7.42,N 14.73;found C 69.23,H 7.71,N 14.58。
实施例10
用丁酮(10.0mL)代替环戊酮,其它同实例6。粗产品经柱分离得到化合物(X),产率为80%,Mp 283~285℃。5-硝基-2-氨基苯甲腈与丁酮反应式为:
产物(IX)的波谱数据为:IR(KBr):3322,3177,2919,1671,1618,1534,1306,1149cm-1;1H NMR(DMSO-d6)δH:0.88(3H,t,J=7.2Hz,CH3),1.43(3H,s,CH3),1.64-1.73(2H,m,J=7.2Hz,CH2),6.75(1H,d,J=8.8Hz,ArH),8.08(1H,dd,J=2.7,8.8Hz,ArH),8.17(1H,s,NH),8.36(1H,s,NH),8.40(1H,d,J=2.7Hz,ArH);13C NMR(DMSO-d6)δC:7.93,28.57,34.99,70.29,111.42,113.75,124.12,128.90,136.39,151.78,161.01;MS(ESI):m/z(%)=236.1(100)[M+H]+;C11H13N3O3:calcd.C 56.16,H 5.57,N 17.86;found C 55.98,H 5.67,N 17.48。
实施例11
用苯乙酮(10.0mL)代替环戊酮,其它同实例6。粗产品经柱分离得到化合物(XI),产率为78%,Mp>300℃。5-硝基-2-氨基苯甲腈与苯乙酮反应式为:
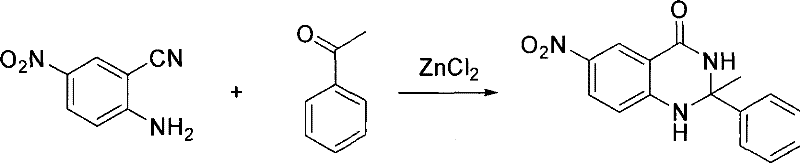
产物(XI)的波谱数据为:IR(KBr):3352,3188,1674,1617,1535,1310,1150cm-1;1H NMR(DMSO-d6,400MHz)δH:1.74(3H,s,CH3),6.89(1H,d,J=8.8Hz,ArH),7.24(1H,t,J=7.2Hz,ArH),7.34(2H,t,J=7.2,8.0Hz,ArH),7.47(2H,d,J=8.0Hz,ArH),8.10(1H,dd,J=2.7,8.8Hz,ArH),8.33(1H,d,J=2.7Hz,ArH),9.02(1H,s,NH),9.22(1H,s,NH);13C NMR(DMSO-d6,100MHz)δC:30.14,70.50,113.00,114.44,124.05,124.79(2C),127.61,128.33(2C),128.93,137.22,146.64,151.55,161.82;MS(ESI):m/z(%)=284.1(100)[M+H]+;C15H13N3O3:calcd.C 63.59,H 4.62,N 14.83;found C 63.42,H 4.75,N 14.96。
实施例12
用2-氨基苯甲腈代替5-硝基邻氨基苯甲腈,用苯乙酮(10.0mL)代替环己酮,其它同实例8。粗产品经柱分离得到化合物(XII),产率为62%,Mp232~234℃。2-氨基苯甲腈与苯乙酮反应式为:

产物(XII)的波谱数据为:IR(KBr):3389,3181,1663,1613cm-1;1H NMR(DMSO-d6,400MHz)δH:1.79(3H,s,CH3),6.63-6.67(1H,m,J=0.8,8.0Hz,ArH),6.83(1H,t,J=0.8,8.0Hz,ArH),6.89(1H,s,NH),7.21-7.23(2H,m,ArH),7.28-7.32(2H,m,ArH),7.61-7.68(3H,m,ArH),7.93(1H,s,NH);13C NMR(DMSO-d6,100MHz)δC:31.14,71.54,115.36,116.67,118.26,126.09(2C),128.00,128.45,128.87(2C),134.05,147.95,148.23,164.90;MS(ESI):m/z(%)=239.1(100)[M+H]+;C15H14N2O:calcd.C 75.61,H 5.92,N 11.76;found C 75.28,H 6.11,N 11.43。
Claims (14)
1.一种合成1,2-二氢喹唑啉-4(3H)-酮类化合物的方法,其特征在于:以邻氨基芳香氰化物与醛或酮反应,生成1,2-二氢喹唑啉-4(3H)-酮类杂环化合物,反应通式为:

其中R为取代基,为F,Cl,Br,I,NO2,烷基,烷氧基或胺基;该取代基位置不限;
R1、R2为H、烷基、环烷基、芳烃基。
2.如权利要求1所述的一种合成1,2-二氢喹唑啉-4(3H)-酮类化合物的方法,其特征在于:反应介质为苯、甲苯、二甲苯、硝基苯、氯苯、环丁砜、二甲亚砜、N,N-二甲基甲酰胺、N,N-二甲基乙酰胺、二氧六环、四氢呋喃或卤代烃极性溶剂;对于常温下为液态的羰基化合物,该羰基化合物是指反应原料2的醛或酮,除了可以使用前述的反应介质外,自身也可以作为反应介质。
3.如权利要求1所述的一种合成1,2-二氢喹唑啉-4(3H)-酮类化合物的方法,其特征在于:反应物1、2的物质的量的比为1∶1~1∶99。
4.如权利要求1所述的一种合成1,2-二氢喹唑啉-4(3H)-酮类化合物的方法,其特征在于:反应的催化剂是路易斯酸,质子酸或碱性催化剂。
5.如权利要求4所述的一种合成1,2-二氢喹唑啉-4(3H)-酮类化合物的方法,其特征在于:路易斯酸催化剂是无水氯化锌、无水三氯化铝、氯化铜或氯化亚铜,质子酸催化剂是盐酸或硫酸,碱性催化剂是吡啶、哌啶或碳酸钠。
6.如权利要求4或5所述的一种合成1,2-二氢喹唑啉-4(3H)-酮类化合物的方法,其特征在于:路易斯酸催化剂的用量为反应物1的物质的量的1~1.5倍;质子酸或碱性催化剂的用量为反应物1的物质的量的1%~100%。
7.如权利要求1所述的一种合成1,2-二氢喹唑啉-4(3H)-酮类化合物的方法,其特征在于:醛是脂肪醛或芳香醛。
8.如权利要求1所述的一种合成1,2-二氢喹唑啉-4(3H)-酮类化合物的方法,其特征在于:酮是脂肪酮或芳香酮。
9.如权利要求1所述的一种合成1,2-二氢喹唑啉-4(3H)-酮类化合物的方法,其特征在于:所述的反应采用常压油浴加热或微波加热方法;在常压油浴中的反应时间为1-10小时,产物的收率为10-99%;在微波促进条件下,反应时间为6-50分钟,产率为10-99%。
10.如权利要求1所述的一种合成1,2-二氢喹唑啉-4(3H)-酮类化合物的方法,其特征在于产物的分离与提纯方法为:首先将反应产物分散到少量水中,用碱中和除去催化剂,过滤后所得固体用有机溶剂溶出,而滤液进行萃取;将固体中溶出物和萃取液合并浓缩,得到粗产物,然后对所得的粗产品进行重结晶或者柱层析纯化,即得到纯的目标化合物。
11.如权利要求10所述的一种合成1,2-二氢喹唑啉-4(3H)-酮类化合物的方法,其特征在于:除去催化剂后过滤所得的固体所用有机溶剂是甲醇、乙醇、丙酮、四氢呋喃、乙酸乙酯或乙腈。
12.如权利要求10所述的一种合成1,2-二氢喹唑啉-4(3H)-酮类化合物的方法,其特征在于:滤液的萃取剂是乙酸乙酯、二氯甲烷、氯仿或乙醚。
13.如权利要求10所述的一种合成1,2-二氢喹唑啉-4(3H)-酮类化合物的方法,其特征在于:粗产物的重结晶溶剂是甲醇、乙醇、异丙醇、丙酮、乙腈、四氢呋喃、二氧六环、乙酸乙酯、二氯甲烷、氯仿、苯、甲苯或硝基苯。
14.如权利要求10所述的一种合成1,2-二氢喹唑啉-4(3H)-酮类化合物的方法,其特征在于:柱层析采用硅胶柱或氧化铝柱,展开剂为体积比为1∶1~1∶3的乙酸乙酯/石油醚、体积比为1∶20~1∶50的甲醇/氯仿、二氯甲烷或丙酮。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN2007103042449A CN101190899B (zh) | 2007-12-26 | 2007-12-26 | 一种合成1,2-二氢喹唑啉-4(3h)-酮类化合物的方法 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN2007103042449A CN101190899B (zh) | 2007-12-26 | 2007-12-26 | 一种合成1,2-二氢喹唑啉-4(3h)-酮类化合物的方法 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN101190899A CN101190899A (zh) | 2008-06-04 |
| CN101190899B true CN101190899B (zh) | 2012-07-18 |
Family
ID=39486116
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN2007103042449A Expired - Fee Related CN101190899B (zh) | 2007-12-26 | 2007-12-26 | 一种合成1,2-二氢喹唑啉-4(3h)-酮类化合物的方法 |
Country Status (1)
| Country | Link |
|---|---|
| CN (1) | CN101190899B (zh) |
Families Citing this family (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN102675223B (zh) * | 2012-04-28 | 2014-08-27 | 清华大学 | 一种多取代喹唑啉及杂环并嘧啶衍生物的制备方法 |
| CN102816164A (zh) * | 2012-08-31 | 2012-12-12 | 北京理工大学 | 一种合成7-氨基-2,3-二氢嘧啶[4,5-d]嘧啶-4(1H)-酮的方法 |
| CN103524432B (zh) * | 2013-10-23 | 2016-06-08 | 贵州大学 | 一种制备2,3-二氢喹唑啉-4(3h)-酮类衍生物的方法 |
| CN104987343A (zh) * | 2015-07-13 | 2015-10-21 | 北京理工大学 | 一种合成双噻吩并嘧啶酮衍生物的方法 |
| CN112457260B (zh) * | 2020-12-08 | 2023-01-17 | 江苏食品药品职业技术学院 | N-杂环芳基喹唑啉-4-胺化合物及其制备方法 |
| CN112645887B (zh) * | 2020-12-21 | 2023-01-13 | 淮阴工学院 | 一种喹唑啉酮衍生物的制备方法 |
| CN115160235B (zh) * | 2022-08-10 | 2023-12-15 | 宜宾学院 | 一种2,3-二氢喹唑啉-4(1h)-酮类化合物及其超声合成方法 |
| CN115181139B (zh) * | 2022-09-09 | 2023-02-10 | 深圳市小分子新药创新中心有限公司 | 利用氨基苯甲腈类化合物合成On-DNA二氢喹唑啉酮化合物的方法 |
| CN115978796B (zh) * | 2022-12-26 | 2025-03-07 | 烟台宁远药业有限公司 | 取代含氮喹唑啉酮类化合物的合成方法 |
-
2007
- 2007-12-26 CN CN2007103042449A patent/CN101190899B/zh not_active Expired - Fee Related
Non-Patent Citations (6)
| Title |
|---|
| Axel couture et al.,.An Expeditious Synthesis of 2-Aryl- and 2-Alkylquinazolin-4(3H)-ones.Synthesis.1991,1009-1010. * |
| Mann-Jen Hour et al.,.6-Alkylamino- and 2,3-Dihydro-3¢-methoxy-2-phenyl-4-quinazolinones andRelated Compounds: Their Synthesis, Cytotoxicity, andInhibition of TubulinPolymerization.J. Med. Chem.43.2000,434479-4487. * |
| Michael L.et al.,.Decoration of the Aromatic Ring ofDihydrocodeinone (Hydrocodone) and14-Hydroxydihydrocodeinone (Oxycodone).J.Org.Chem.,70.2005,706492-6495. * |
| Motohiro Akazome et al.,.Transition-Metal Complex-Catalyzed Reductive N-Heterocyclization: Synthesis of4(3R)-Quinazolinone Derivatives from N-(2- Nitrobenzoy1)amides.J. Org. Chem.,58.1993,58310-312. * |
| Richard Pater.2-Aryl-4(3H)quinazolinones.Journal of Heterocyclic Chemistry8 5.1971,8(5),699-702. |
| Richard Pater.2-Aryl-4(3H)quinazolinones.Journal of Heterocyclic Chemistry8 5.1971,8(5),699-702. * |
Also Published As
| Publication number | Publication date |
|---|---|
| CN101190899A (zh) | 2008-06-04 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN101190899B (zh) | 一种合成1,2-二氢喹唑啉-4(3h)-酮类化合物的方法 | |
| KR102684954B1 (ko) | 아미노피리미딘 유도체의 합성에 유용한 신규의 중간체, 이의 제조방법 및 이를 이용한 아미노피리미딘 유도체의 제조방법 | |
| CN109053625B (zh) | 一种取代苯并噻唑c2烷基化衍生物的制备方法 | |
| CN116354959B (zh) | 一种N-N桥连噻唑单元的β-咔啉衍生物及其制备方法、应用 | |
| CN102584841B (zh) | 一种喹啉香豆素衍生物及其制备方法及用途 | |
| CN109970679A (zh) | 丹皮酚噻唑衍生物及其制备方法和应用 | |
| CN111777564A (zh) | 一种在水相中光催化醇氧化合成喹唑啉酮化合物的方法 | |
| CN101195626A (zh) | 一种合成吡唑并[3,4-d]嘧啶-4(5H)-酮类化合物的方法 | |
| CN110256451A (zh) | 一种苯并呋喃并[2,3-b]喹啉衍生物的合成方法 | |
| CN104016929B (zh) | 一种合成喹唑啉-4(3h)-酮的方法 | |
| CN102127076A (zh) | 一种合成2,3-二氢吡啶并[2,3-d]嘧啶-4-(3H)-酮的方法 | |
| CN108033913A (zh) | 一种二氢吡唑啉衍生物及其制备方法和应用 | |
| CN106008305A (zh) | 一种五取代2-氨基吡咯衍生物的合成方法 | |
| Sharma et al. | Eco-friendly reactions in PEG-400: a highly efficient and green approach for stereoselective access to multisubstituted 3, 4-dihydro-2 (1 H)-quinazolines using 2-aminobenzylamines | |
| JPH0116837B2 (zh) | ||
| Zhai et al. | Syntheses and antiproliferative activities of novel diarylthiosemicarbazide derivatives | |
| CN106478526A (zh) | 一种喹唑啉酮席夫碱类化合物的合成方法 | |
| CN101580492A (zh) | 微波制备杀菌剂三芳基-2-吡唑啉衍生物的方法 | |
| CN106083649B (zh) | 一种3,5‑二芳基‑2,6,6‑三氰基‑1‑亚氨基‑2,4‑环己二烯衍生物的合成方法 | |
| CN105837522A (zh) | 一种1,5-苯并二氮卓酮衍生物的制备方法 | |
| CN104193669A (zh) | 一类阿比朵尔类似物或其盐、其制备方法及应用 | |
| CN112300181B (zh) | 二酮骨架化合物及其制备方法与应用 | |
| CN109776388B (zh) | 一种具有c2季碳中心的吲哚啉衍生物的合成方法 | |
| Fang et al. | Synthesis of 6, 7-Dihydrodibenzo [b, j] phenanthroline Derivatives by Pfitzinger Condensation of Isatin and Cyclic Diketones | |
| CN108822058B (zh) | 一种苯并噻嗪类化合物的制备方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C14 | Grant of patent or utility model | ||
| GR01 | Patent grant | ||
| C17 | Cessation of patent right | ||
| CF01 | Termination of patent right due to non-payment of annual fee |
Granted publication date: 20120718 Termination date: 20121226 |