CN1017151B - Processes for preparing naphthothiazepine derivative - Google Patents
Processes for preparing naphthothiazepine derivativeInfo
- Publication number
- CN1017151B CN1017151B CN88104589A CN88104589A CN1017151B CN 1017151 B CN1017151 B CN 1017151B CN 88104589 A CN88104589 A CN 88104589A CN 88104589 A CN88104589 A CN 88104589A CN 1017151 B CN1017151 B CN 1017151B
- Authority
- CN
- China
- Prior art keywords
- compound
- salt
- group
- pharmaceutically acceptable
- hydrogen atom
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D281/00—Heterocyclic compounds containing rings of more than six members having one nitrogen atom and one sulfur atom as the only ring hetero atoms
- C07D281/02—Seven-membered rings
- C07D281/04—Seven-membered rings having the hetero atoms in positions 1 and 4
- C07D281/08—Seven-membered rings having the hetero atoms in positions 1 and 4 condensed with carbocyclic rings or ring systems
- C07D281/10—Seven-membered rings having the hetero atoms in positions 1 and 4 condensed with carbocyclic rings or ring systems condensed with one six-membered ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D281/00—Heterocyclic compounds containing rings of more than six members having one nitrogen atom and one sulfur atom as the only ring hetero atoms
- C07D281/02—Seven-membered rings
- C07D281/04—Seven-membered rings having the hetero atoms in positions 1 and 4
- C07D281/08—Seven-membered rings having the hetero atoms in positions 1 and 4 condensed with carbocyclic rings or ring systems
-
- H—ELECTRICITY
- H10—SEMICONDUCTOR DEVICES; ELECTRIC SOLID-STATE DEVICES NOT OTHERWISE PROVIDED FOR
- H10W—GENERIC PACKAGES, INTERCONNECTIONS, CONNECTORS OR OTHER CONSTRUCTIONAL DETAILS OF DEVICES COVERED BY CLASS H10
- H10W72/00—Interconnections or connectors in packages
- H10W72/701—Tape-automated bond [TAB] connectors
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen- Or Sulfur-Containing Heterocyclic Ring Compounds With Rings Of Six Or More Members (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Nitrogen And Oxygen As The Only Ring Hetero Atoms (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Lubricants (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
Abstract
Description
本发明涉及一种新颖的萘并硫杂氮杂
![]()
![]()

的萘并硫杂氮杂
![]()
![]()
美国专利4,652,561揭示萘并硫杂氮杂
![]()
![]()
![]()
![]()
与所述的已知的化合物相比,本发明的新颖的萘并硫杂氮杂
![]()
![]()
本发明的化合物(Ⅰ)的实例包括那些化合物,其中R1为一种1至6个碳原子的烷氧基基团,R2为氢原子或一种2至6个碳原子的链烷酰基团,且R3和R4为氢原子或一种1至6个碳原子的烷基基 团。其中,较好的实例包括那些结构式(Ⅰ)的化合物,其中R1为一种1至4个碳原子的烷氧基基团,R2为氢原子或一种2至4个碳原子的链烷酰基团,R3为一种1至4个碳原子的烷基基团,且R4为氢原子或一种1至4个碳原子的烷基基团。Examples of the compound (I) of the present invention include those compounds wherein R 1 is an alkoxy group of 1 to 6 carbon atoms, R 2 is a hydrogen atom or an alkanoyl group of 2 to 6 carbon atoms group, and R3 and R4 are a hydrogen atom or an alkyl group of 1 to 6 carbon atoms. Among them, preferred examples include those compounds of formula (I), wherein R 1 is an alkoxy group of 1 to 4 carbon atoms, R 2 is a hydrogen atom or a chain of 2 to 4 carbon atoms An alkanoyl group, R 3 is an alkyl group of 1 to 4 carbon atoms, and R 4 is a hydrogen atom or an alkyl group of 1 to 4 carbon atoms.
根据本发明,化合物(Ⅰ)可通过一种结构式为:According to the present invention, compound (I) can be represented by a structural formula:
(Ⅱ) (II)
的化合物或其一种盐,其中R1和R2如上定义,和一种结构式为:A compound or a salt thereof, wherein R1 and R2 are as defined above, and a structural formula is:

的一种化合物或其一种盐,其中R3和R4如上定义,且X1为一种卤素原子的缩合而制备的。A compound or a salt thereof, wherein R 3 and R 4 are as defined above, and X 1 is prepared by condensation of a halogen atom.
本发明的化合物(Ⅰ),其中R2为一种低级链烷酰基团也可通过一种结构式为:The compound (I) of the present invention, wherein R 2 is a lower alkanoyl group can also be represented by a structural formula:
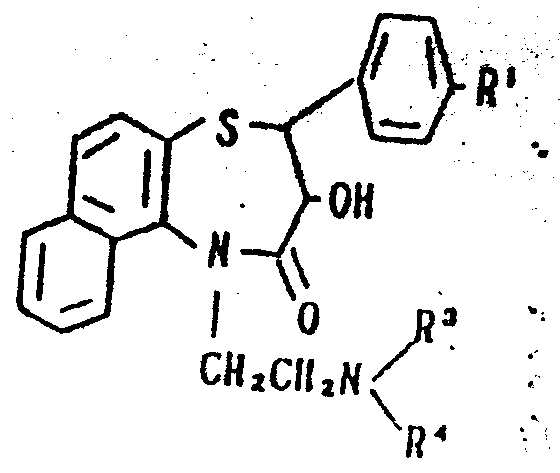
的化合物或其一种盐,其中R1、R3和R4如上定义,和一种结构式为:A compound or a salt thereof, wherein R 1 , R 3 and R 4 are as defined above, and a structural formula is:
的化合物或其一种活性衍生物,其中R2′为一种低级烷基基团的缩合而制备的。or a reactive derivative thereof, wherein R 2' is prepared by condensation of a lower alkyl group.
另一方面,本发明的化合物(Ⅰ),其中R3为一种低级烷基基团,且R4为氢原子,可通过一种结构式为:On the other hand, the compound (I) of the present invention, wherein R 3 is a lower alkyl group, and R 4 is a hydrogen atom, can be obtained by a structural formula:

的化合物或其一种盐,其中R3′和R4′均为一种低级烷基基团,且R1和R2如上定义,与一种卤代甲酸的反应活性衍生物的反应,然后用水,一种低级链烷醇或它们的一种混合物处理该产物或用在一种低级链烷酸中的锌处理该产物而制备的。A compound or a salt thereof, wherein R 3' and R 4' are both a lower alkyl group, and R 1 and R 2 are as defined above, reacted with a reactive derivative of haloformic acid, and then Prepared by treating the product with water, a lower alkanol or a mixture thereof or with zinc in a lower alkanoic acid.
如果需要,起始化合物(Ⅱ)可以以一种碱金属盐(例如,钠盐或钾盐)的形式用于上述反应,且,如果需要,起始化合物(Ⅲ)和化合物(Ⅰ-A)和(Ⅰ-B)可以以一种酸加成盐如一种无机酸加成盐(例如,氢氯化物或氢溴化物)或一种有机酸加成盐(例如,马来酸盐,延胡索酸盐或甲磺酸盐)的形式用于上述反应。If necessary, the starting compound (II) can be used in the above reaction in the form of an alkali metal salt (for example, sodium salt or potassium salt), and, if necessary, the starting compound (III) and compound (I-A) and (I-B) may be present as an acid addition salt such as an inorganic acid addition salt (for example, hydrochloride or hydrobromide) or an organic acid addition salt (for example, maleate, fumarate or mesylate) for the above reaction.
化合物(Ⅱ)或其一种盐与化合物(Ⅲ)或其一种盐的缩合可在一种碱性试剂存在或不存在下,在一种适合的溶剂中进行。当化合物(Ⅱ)以游离的形式被应用时,所述的缩合最好在一种碱性试剂存在下进行。适合的该碱性试剂的实例包括一种碱金属氢氧化物(例如,氢氧化钠,氢氧化钾),一种碱金属碳酸盐(例如,碳酸钠,碳酸钾)和一种碱金属氢化物(例如,氢化钠)等。较好地为在20至100℃,特别是25至70℃下进行该缩合。The condensation of compound (II) or a salt thereof with compound (III) or a salt thereof can be carried out in a suitable solvent in the presence or absence of a basic reagent. When the compound (II) is used in free form, said condensation is preferably carried out in the presence of a basic reagent. Examples of suitable alkaline reagents include an alkali metal hydroxide (e.g., sodium hydroxide, potassium hydroxide), an alkali metal carbonate (e.g., sodium carbonate, potassium carbonate) and an alkali metal hydrogenation substances (for example, sodium hydride), etc. It is preferred to carry out the condensation at 20 to 100°C, especially 25 to 70°C.
化合物(Ⅰ-A)或其一种盐与化合物(Ⅳ)的缩合可在一种缩合剂 存在下,在一种适合的溶剂中进行。适合的缩合剂的实例包括二环己基碳(化)二亚胺,N,N-二异丙基碳(化)二亚胺和N,N′-二-对-甲苯基碳二亚胺等。较好地为在0至30℃下进行该反应。The condensation of compound (I-A) or a salt thereof with compound (IV) can be carried out in a condensing agent in the presence of a suitable solvent. Examples of suitable condensing agents include dicyclohexylcarbo(3)diimide, N,N-diisopropylcarbo(3)diimide, and N,N'-di-p-tolylcarbodiimide, etc. . It is preferred to carry out the reaction at 0 to 30°C.
另一方面,化合物(Ⅰ-A)与化合物(Ⅳ)的反应活性衍生物的缩合可在一种酸性接受体存在或不存在下,在一种适合的溶剂中进行。适合的活性衍生物的实例包括相应的酰基卤(例如,氯化物,溴化物)酸酐,混合酸酐(例如,乙氧羰基酯,异丙氧基羰基酯,异丁氧基羰基酯)和活性酯(例如,对硝基苯基酯2,4,5-三氯苯基酯2,4,6-三氯苯基酯,N-羟基丁二酰亚胺酯,1-苯并三唑酯)等。适合的酸性接受体的实例包括吡啶、三乙胺、一种碱金属碳酸盐、一种碱金属碳酸氢盐和一种碱金属氢氧化物等。较好地为在0至100℃温度下进行该反应。On the other hand, the condensation of the compound (I-A) with the reactive derivative of the compound (IV) can be carried out in a suitable solvent in the presence or absence of an acidic acceptor. Examples of suitable reactive derivatives include the corresponding acid halides (e.g. chlorides, bromides) anhydrides, mixed anhydrides (e.g. ethoxycarbonyl esters, isopropoxycarbonyl esters, isobutoxycarbonyl esters) and active esters (e.g. p-nitrophenyl ester 2,4,5-trichlorophenyl ester 2,4,6-trichlorophenyl ester, N-hydroxysuccinimide ester, 1-benzotriazole ester) wait. Examples of suitable acid acceptors include pyridine, triethylamine, an alkali metal carbonate, an alkali metal bicarbonate, an alkali metal hydroxide, and the like. It is preferred to carry out the reaction at a temperature of 0 to 100°C.
化合物(Ⅰ-B)与一种卤代甲酸的活性衍生物的反应可在一种碱存在或不存在下,在一种适合的溶剂中进行。该卤代甲酸的活性衍生物的实例包括相应的酰基卤(例如,二氟化碳,二氯化碳,二溴化碳和二碘化碳),和卤代甲酸一种单-、双-或三卤代-低级烷基酯(例如,氯代甲酸三氯甲酯,氯代甲酸α-氯代乙基酯,氯代甲酸β,β,β-三氯乙基酯)。一种碱的实例包括三乙胺、吡啶、-甲基吡啶2,6-二甲基吡啶,四甲基脲,六甲基磷酰三胺和喹啉等。较好地为在0℃至140℃下进行。The reaction of compound (I-B) with a reactive derivative of haloformic acid can be carried out in a suitable solvent in the presence or absence of a base. Examples of reactive derivatives of the haloformic acid include the corresponding acid halides (e.g., carbon difluoride, carbon dichloride, carbon dibromide, and carbon diiodide), and haloformic acid mono-, bis- Or trihalo-lower alkyl esters (for example, trichloromethyl chloroformate, α-chloroethyl chloroformate, β,β,β-trichloroethyl chloroformate). Examples of a base include triethylamine, pyridine, -picoline, 2,6-lutidine, tetramethylurea, hexamethylphosphoric triamide, quinoline and the like. It is preferably carried out at 0°C to 140°C.
化合物(Ⅰ),其中R3为一种低级烷基基团,且R4为氢原子,可通过用水,一种低级链烷醇(例如,甲醇)或它们的一种混合物处理因此所得到的产物而获得。较好地为在20至120℃下进行。Compound (I), wherein R3 is a lower alkyl group, and R4 is a hydrogen atom, can be obtained by treating with water, a lower alkanol (for example, methanol) or a mixture thereof product obtained. It is preferably carried out at 20 to 120°C.
当氯代甲酸α-氯代乙基酯或氯代甲酸β,β,β-三氯代乙基酯被用作卤代甲酸的卤代烷基酯时,化合物(Ⅰ),其中R3为一种低级烷基基团,且R4为氢原子也可通过在一种低级链烷酸如乙酸中,用锌(例如,锌粉末)处理因此所得到的产物而获得。When α-chloroethyl chloroformate or β,β,β-trichloroethyl chloroformate is used as haloalkyl ester of haloformic acid, compound (I), wherein R3 is a Lower alkyl groups with R4 being a hydrogen atom can also be obtained by treating the product thus obtained with zinc (for example, zinc powder) in a lower alkanoic acid such as acetic acid.
较好地用于上述反应的适合溶剂的实例为苯,二噁烷,二氯甲烷,氯仿,丙酮,乙腈,乙酸乙酯和二甲基甲酰胺等。Examples of suitable solvents preferably used in the above reaction are benzene, dioxane, methylene chloride, chloroform, acetone, acetonitrile, ethyl acetate, dimethylformamide and the like.
本发明的化合物(Ⅰ),由于在萘并硫杂氮杂
![]()
![]()
本发明的因此所得到的化合物(Ⅰ)可以以游离的形式或以其药物学上可接受的酸加成盐的形式用于药物应用。化合物(Ⅰ)的盐的实施包括无机酸加成盐如氢氧化物,氢溴化物,氢碘化物,高氯酸盐,硫酸盐或磷酸盐,和有机酸加成盐和草酸盐,马来酸盐,延胡索酸盐,酒石酸盐,甲磺酸盐或琥珀酸盐。这些盐可以,例如,通过惯用的方法用一种酸处理化合物(Ⅰ)而容易地获得。The thus obtained compound (I) of the present invention can be used in pharmaceutical applications in the free form or in the form of a pharmaceutically acceptable acid addition salt thereof. Examples of salts of compound (I) include inorganic acid addition salts such as hydroxide, hydrobromide, hydroiodide, perchlorate, sulfate or phosphate, and organic acid addition salts and oxalate, horse tonate, fumarate, tartrate, mesylate or succinate. These salts can be easily obtained, for example, by treating the compound (I) with an acid in a conventional manner.
因为有效的降血压效应,和大脑血管舒张效应及冠状血管舒张效应,本发明的化合物(Ⅰ)和一种药学上可接受的酸加成盐用于治疗和预防低血压的形成,大脑疾病如脑血管痉挛,大脑局部缺血和大脑梗塞,及心脏疾病如必绞痛和心肌梗塞(形成)。Because of the effective hypotensive effect, and cerebral vasodilation effect and coronary vasodilation effect, the compound (I) of the present invention and a pharmaceutically acceptable acid addition salt are used for the treatment and prevention of hypotension, brain diseases such as Cerebral vasospasm, cerebral ischemia and cerebral infarction, and heart disorders such as angina and myocardial infarction (formation).
进一步讲,尽管已知的血管扩张剂如Verapamil已知通过抑制房室传导来延迟房室传导时间,且有时由于这种传导干扰而诱发心律失常,但本发明的化合物(Ⅰ)和其一种盐的特征在于它们具有较少不利的副作用如对房室传导的抑制较少或对心率的降低较少。Furthermore, although known vasodilators such as Verapamil are known to delay atrioventricular conduction time by inhibiting atrioventricular conduction, and sometimes induce arrhythmia due to this conduction disturbance, the compound (I) of the present invention and one of its Salts are characterized in that they have fewer adverse side effects such as less inhibition of atrioventricular conduction or less reduction in heart rate.
更进一步讲,本发明的化合物和其一种盐具有一种有效的血小板聚集抑制作用,且对治疗大脑疾病如脑血管痉挛,大脑局部缺血和大脑梗塞非常有用。Furthermore, the compound of the present invention and a salt thereof have a potent inhibitory effect on platelet aggregation and are very useful for the treatment of cerebral diseases such as cerebral vasospasm, cerebral ischemia and cerebral infarction.
本发明的化合物(Ⅰ)或其一种药物学上可接受的酸加成盐可以以一种药物制剂的形式运用,它可含有适合于口服或非经肠道施用 的药物赋形剂。适合的赋形剂的实例包括淀粉,乳糖,葡萄糖,磷酸钾,玉米淀粉,阿拉伯胶,硬脂酸和其它惯用的药物赋形剂。该药物制剂可以以固体剂量形式如片剂,丸剂,胶囊和栓剂等,或以液体剂量形式如溶液,悬浮液和乳剂等运用。进一步讲,当非经肠道施用时,药物制剂可以以注射剂的形式运用。The compound (I) of the present invention or a pharmaceutically acceptable acid addition salt thereof can be used in the form of a pharmaceutical preparation containing a compound suitable for oral or parenteral administration. pharmaceutical excipients. Examples of suitable excipients include starch, lactose, glucose, potassium phosphate, corn starch, acacia, stearic acid and other customary pharmaceutical excipients. The pharmaceutical preparations can be administered in solid dosage forms such as tablets, pills, capsules and suppositories, etc., or in liquid dosage forms such as solutions, suspensions and emulsions. Further, when administered parenterally, the pharmaceutical preparations can be administered in the form of injections.
本发明的化合物(Ⅰ)或其一种药物学上可接受的盐的每日剂量可根据施用途径,病人的年龄,体重或条件及待治疗疾病的严重性而变化。然而,总之,化合物(Ⅰ)或其一种盐的较好的每日剂量通常在0.05至10毫克/千克范围内,特别是在口服情况下约为0.5至10毫克/千克,或在非径肠道施用(例如,静脉注射)情况下为0.05至2毫克/千克。The daily dose of the compound (I) of the present invention or a pharmaceutically acceptable salt thereof may vary depending on the route of administration, the age, body weight or condition of the patient and the severity of the disease to be treated. However, in general, a preferred daily dose of compound (I) or a salt thereof is usually in the range of 0.05 to 10 mg/kg, especially about 0.5 to 10 mg/kg in the case of oral administration, or In the case of enteral administration (eg, intravenous injection) 0.05 to 2 mg/kg.
伴随着,本发明的起始化合物(Ⅱ)可,例如,通过2-氨基萘-2-硫醇(thiol)和一种结构式为:Concomitantly, the starting compound (II) of the present invention can, for example, be obtained by 2-aminonaphthalene-2-thiol (thiol) and a structural formula:

的缩水甘油酸酯,其中R为一种酯残余物,且R1如上定义缩合而制备。Glycidyl esters, wherein R is an ester residue, and R is prepared by condensation as defined above.
实验1Experiment 1
降血压活性hypotensive activity
(方法)(method)
将一种溶解或悬浮在水中的测试化合物以剂量30毫克/千克给在前一夜禁食的自发的低血压病鼠(SHR)口服,并用尾部体积描离(扫描)技术(实验室和临床医学杂志,卷78(1971),957页)每隔一段时间测量该鼠的收缩血压。测试化合物的降血压活性以“血压降低(△毫米汞柱)”的措词来估计,它通过下列公式来计算。A test compound dissolved or suspended in water was administered orally at a dose of 30 mg/kg to overnight fasted spontaneously hypotensive rats (SHR) and tail plethysmography (scanning) technique (Laboratory and Clinical Medicine Journal, Vol. 78 (1971), p. 957) The systolic blood pressure of the rat was measured at regular intervals. The hypotensive activity of the test compound was estimated in terms of "lowering blood pressure (ΔmmHg)", which was calculated by the following formula.
血压的减少(△毫米汞柱)=Reduction in blood pressure (△ mmHg) =
(结果)(result)
该结果如下列表1所示。The results are shown in Table 1 below.
表1Table 1
血压的降低(△毫米汞柱)Decrease in blood pressure (△ mm Hg)
服药后的时间time after medication
1小时 2小时 5小时1 hour 2 hours 5 hours
1.测试化合物*58.3±8.8 51.3±7.7 34.0±6.21. Test compound * 58.3±8.8 51.3±7.7 34.0±6.2
*):该测试化合物的化学名称:*): Chemical name of the test compound:
(±)-顺式-2-(4-甲氧基苯基)-3-乙酰氧基-5-〔2-(二甲基氨基)乙基〕-2,3-二氢-萘并〔2,1-b〕-1,5-硫杂氮杂
![]()
![]()
实施例1Example 1
(1)在100℃下加热一种7.86克1-氨基萘-2-硫醇,12.16克3-(4-甲氧基苯基)缩水甘油酸甲(基)酯(methyl 3-(4-methoxyphenyl)glycidate)和80毫升甲苯的混合物。在反应完全之后,浓缩该反应混合物。将二异丙醚加至含油残余物中。沉淀的结晶通过过滤收集,并从甲醇中重结晶而给出6.49克(38%)(±)-苏-3-(1-氨-2-萘基)硫代-3-(4-甲氧基苯基)-2-羟基丙酸甲(基)酯。(1) A 7.86 g 1-aminonaphthalene-2-thiol, 12.16 g 3-(4-methoxyphenyl) glycidic acid methyl (base) ester (methyl 3-(4- methoxyphenyl) glycidate) and 80 ml of toluene. After the reaction was complete, the reaction mixture was concentrated. Diisopropyl ether was added to the oily residue. The precipitated crystals were collected by filtration and recrystallized from methanol to give 6.49 g (38%) of (±)-threo-3-(1-amino-2-naphthyl)thio-3-(4-methoxy phenyl)-2-hydroxypropanoic acid methyl (yl) ester.
熔点:131.5-135℃Melting point: 131.5-135°C
质谱(Mass)(m/e):383(M+)Mass Spectrum (Mass) (m/e): 383 (M + )
IR(医用润滑油(Nujol,厘米-1):3500,3420,3120,1730IR (medical lubricant (Nujol, cm -1 ): 3500, 3420, 3120, 1730
(2)在室温下,将一种在2.60克5%的一种氢氧化钠水溶液中的4.0克上面所得到的产物,10毫升乙醇和2毫升四氢呋喃的溶液
搅拌2小时,用10%的盐酸酸化(pH2-3),并用氯仿萃取。萃取液混合,用水洗涤,干燥并浓缩。残余物从乙醇中重结晶而给出3.08(±)-苏-3-(1-氨基-2-萘基)硫代-3-(4-甲氧基苯基)-2-羟基丙酸)熔点:164-165℃(分解)。回流一种在140毫升二甲苯中的2.8克上面所得到的产物的悬浮液7小时。冷却后,沉淀的结晶从一种氯仿和乙醇的混合物中重结晶而给出1.0克(±)-顺式-2-(4-甲氧基苯基)-3-羟基-2,3-二氢-萘并〔2,1-b〕-1,5-硫杂氮杂
![]()
![]()
熔点:263-267℃Melting point: 263-267°C
Mass(m/e):351(M+)Mass (m/e): 351 (M + )
IR(Nujol,厘米-1:3520,3200,3100,3080,3060,1660IR (Nujol, cm -1 : 3520, 3200, 3100, 3080, 3060, 1660
(3)在135毫升丙酮中回流一种3.52克上面所得到的产物,1.17克2-(二甲基氨基)乙基氯盐酸化物和3.24克碳酸钾的混合物。反应完全之后,滤去无机物料,并在减压下浓缩滤液。该残余物溶于氯仿中,用水洗涤,干燥,并浓缩至无水。通过硅胶柱色谱分析〔溶剂:一种氯仿和甲醇(30∶1)的混合物〕纯化因此所得到的残余物而给出2.01克(±)-顺式-2-(4-甲氧基苯基)-3-羟基-5-〔2-(二甲氧基)乙基〕2,3-二氢-萘并〔2,1-b〕-1,5-硫杂氮杂
![]()
![]()
该产物用延胡索酸处理,并从乙醇中重结晶而给出相应的延胡索酸盐。The product is treated with fumaric acid and recrystallized from ethanol to give the corresponding fumarate salt.
熔点:216-218℃(分解)Melting point: 216-218°C (decomposition)
Mass(m/e):422(M+)Mass (m/e): 422 (M + )
IR(Nujol,厘米-1):3460,3050,1730,1660IR (Nujol, cm -1 ): 3460, 3050, 1730, 1660
实施例2Example 2
将2.32克(±)-顺式-2-(4-甲氧基苯基)-3-羟基-5-〔2-(二甲基氨基)乙基〕-2,3-二氢-萘并〔2,1-b〕-1,5-硫杂氮杂
-4(5H)-酮加至70毫升乙酸-乙酸酐(1∶1)中,并回流该混合物。反应完全之后,该混合物在减压下浓缩。将该残余物溶于少量乙醇中,并将1.1当
量延胡索酸加至该溶液中。沉淀的结晶从乙醇中重结晶而给出2.0克(±)-顺式-2-(4-甲氧基苯基)-3-乙酰氧基-5-〔2-(二甲基氨基)乙基〕-2,3-二氢-萘并〔2,1-b〕-1,5-硫杂氮杂
![]()
![]()
熔点:216-218℃Melting point: 216-218°C
Mass(m/e):464(M+)Mass (m/e): 464 (M + )
IR(Nujol,厘米-1):3060,3040,2600-2300,1740,1690IR (Nujol, cm -1 ): 3060, 3040, 2600-2300, 1740, 1690
实施例3Example 3
将一种碳酸钾的水溶液加至788克(±)-顺式-2-(4-甲氧基苯基)-3-乙酰氧基-5-〔2-(二甲基氨基)乙基〕-2,3-二氢-萘并〔2,1-b〕-1,5-硫杂氮杂
![]()
![]()
产率:82%Yield: 82%
熔点:199-202℃(分解)Melting point: 199-202°C (decomposition)
IR(Nujol,厘米-1):3060,3040,1740,1680,1660IR (Nujol, cm -1 ): 3060, 3040, 1740, 1680, 1660
Claims (5)
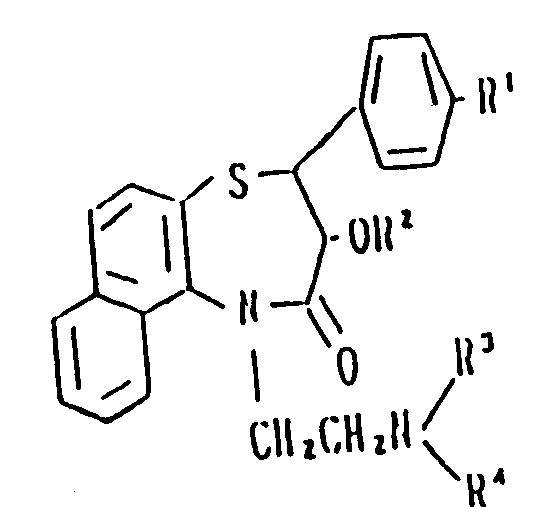
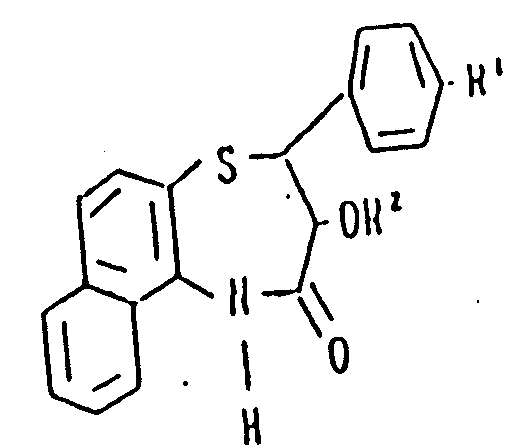

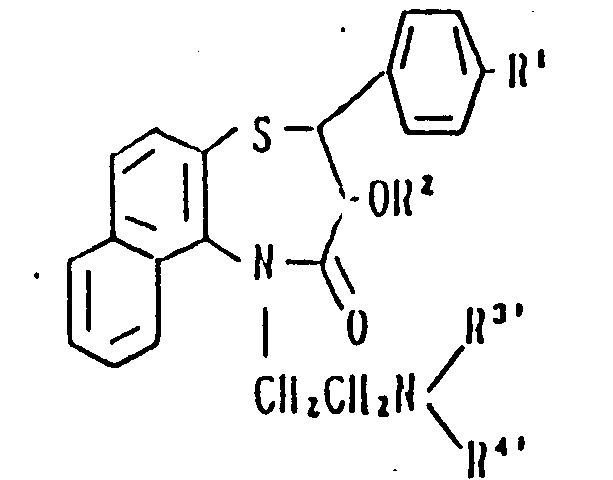
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP193131/87 | 1987-07-31 | ||
| JP19313187 | 1987-07-31 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN1031082A CN1031082A (en) | 1989-02-15 |
| CN1017151B true CN1017151B (en) | 1992-06-24 |
Family
ID=16302787
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN88104589A Expired CN1017151B (en) | 1987-07-31 | 1988-07-23 | Processes for preparing naphthothiazepine derivative |
Country Status (13)
| Country | Link |
|---|---|
| EP (1) | EP0302379B1 (en) |
| KR (1) | KR940003290B1 (en) |
| CN (1) | CN1017151B (en) |
| AT (1) | ATE95177T1 (en) |
| AU (1) | AU600588B2 (en) |
| CA (1) | CA1337652C (en) |
| DE (1) | DE3884518T2 (en) |
| DK (1) | DK424888A (en) |
| ES (1) | ES2059451T3 (en) |
| FI (1) | FI85976C (en) |
| HU (1) | HU200757B (en) |
| IE (1) | IE61939B1 (en) |
| IL (1) | IL87107A (en) |
Families Citing this family (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4652561A (en) * | 1986-02-26 | 1987-03-24 | Hoffmann-La Roche Inc. | Naphtho[1,2-b]-1,4-thiazepinones |
| US4808580A (en) * | 1987-12-17 | 1989-02-28 | Hoffmann-La Roche Inc. | Naphtho[1,2-b][1,4]thiazepin-4(5H)-ones and use thereof in treatment of ischemia and blood pressure lowering |
| CN108743585B (en) * | 2018-08-03 | 2021-03-23 | 杨威 | Small molecule compound with immunoregulation and antiviral effects |
| CN115260122B (en) * | 2022-08-31 | 2024-05-28 | 陕西科技大学 | A naphthothiazole derivative and a synthesis method thereof |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| AU547874B2 (en) * | 1980-03-27 | 1985-11-07 | Peter King Gerakiteys | Toy |
| US4590188A (en) * | 1984-02-18 | 1986-05-20 | Tanabe Seiyaku Co., Ltd. | 1,5-benzothiazepine derivatives and their pharmaceutical use |
| GB2167063A (en) * | 1984-11-17 | 1986-05-21 | Tanabe Seiyaku Co | 6 or 9-chloro-1, 5-benzothiazepine derivatives |
| US4652561A (en) * | 1986-02-26 | 1987-03-24 | Hoffmann-La Roche Inc. | Naphtho[1,2-b]-1,4-thiazepinones |
| US4808580A (en) * | 1987-12-17 | 1989-02-28 | Hoffmann-La Roche Inc. | Naphtho[1,2-b][1,4]thiazepin-4(5H)-ones and use thereof in treatment of ischemia and blood pressure lowering |
-
1988
- 1988-07-13 IE IE214288A patent/IE61939B1/en not_active IP Right Cessation
- 1988-07-14 IL IL87107A patent/IL87107A/en not_active IP Right Cessation
- 1988-07-23 CN CN88104589A patent/CN1017151B/en not_active Expired
- 1988-07-26 FI FI883507A patent/FI85976C/en not_active IP Right Cessation
- 1988-07-27 ES ES88112134T patent/ES2059451T3/en not_active Expired - Lifetime
- 1988-07-27 EP EP88112134A patent/EP0302379B1/en not_active Expired - Lifetime
- 1988-07-27 AT AT88112134T patent/ATE95177T1/en active
- 1988-07-27 DE DE88112134T patent/DE3884518T2/en not_active Expired - Fee Related
- 1988-07-28 AU AU20141/88A patent/AU600588B2/en not_active Ceased
- 1988-07-29 CA CA000573506A patent/CA1337652C/en not_active Expired - Fee Related
- 1988-07-29 HU HU884041A patent/HU200757B/en not_active IP Right Cessation
- 1988-07-29 KR KR1019880009590A patent/KR940003290B1/en not_active Expired - Fee Related
- 1988-07-29 DK DK424888A patent/DK424888A/en not_active Application Discontinuation
Also Published As
| Publication number | Publication date |
|---|---|
| DK424888D0 (en) | 1988-07-29 |
| FI883507A0 (en) | 1988-07-26 |
| KR890002064A (en) | 1989-04-07 |
| IL87107A0 (en) | 1988-12-30 |
| KR940003290B1 (en) | 1994-04-20 |
| ES2059451T3 (en) | 1994-11-16 |
| AU2014188A (en) | 1989-02-02 |
| IE61939B1 (en) | 1994-11-30 |
| DE3884518D1 (en) | 1993-11-04 |
| HU200757B (en) | 1990-08-28 |
| CN1031082A (en) | 1989-02-15 |
| EP0302379B1 (en) | 1993-09-29 |
| FI85976B (en) | 1992-03-13 |
| EP0302379A1 (en) | 1989-02-08 |
| IL87107A (en) | 1992-12-01 |
| IE882142L (en) | 1989-01-31 |
| FI883507L (en) | 1989-02-01 |
| ATE95177T1 (en) | 1993-10-15 |
| FI85976C (en) | 1992-06-25 |
| DE3884518T2 (en) | 1994-03-24 |
| HUT47923A (en) | 1989-04-28 |
| CA1337652C (en) | 1995-11-28 |
| DK424888A (en) | 1989-02-01 |
| AU600588B2 (en) | 1990-08-16 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JPH07503724A (en) | Blood lipid-lowering benzothiazepine compounds | |
| US4585768A (en) | 1,5-benzothiazepine derivatives and processes for preparing the same | |
| EP0234561A2 (en) | 2,3-Dihydro-2-phenyl-5-aminoalkyl-naphtho [1,2-b]-1,4-thiazepin-4(5H)-one derivatives | |
| JPH0427213B2 (en) | ||
| JPS6313994B2 (en) | ||
| CN1017151B (en) | Processes for preparing naphthothiazepine derivative | |
| JPS61122281A (en) | 1,5-benzothiazepine derivative and preparation thereof | |
| KR900005680B1 (en) | Process for preparing 1,5-benzo thiazepine derivatives | |
| CN1061341A (en) | Thiazoloisodihydroazindanone derivatives useful as antiviral agents) | |
| CN1024798C (en) | Process for preparing novel basic substituted 5-halo-thienoisothiazol-3 (2H) -one 1, 1-dioxides | |
| JPH0374661B2 (en) | ||
| EP0413300A1 (en) | Pyrido [3,4-b][1,4] benzoxazepines, a process for their preparation and their use as medicaments | |
| EP0146155A1 (en) | Ether of N-propanolamine derivative | |
| JPH05506654A (en) | 1,5-Benzothiazepinone derivatives, their production methods and pharmaceutical uses | |
| JPS63275572A (en) | 1,5-benzothiazepine derivative | |
| CN1031840A (en) | Benzofuran derivative and preparation method thereof | |
| KR910002879B1 (en) | Method for preparing 1,5-benzothiazepine derivative | |
| CN1030570C (en) | 1, the preparation method of 5-benzothiazepane derivatives | |
| JPH0649076A (en) | Substituted phenylquinazoline derivative | |
| JPH0637480B2 (en) | Naphthothiazepine derivative | |
| EP0093643A1 (en) | Dextrogyrating isomers of 1-aza-bicyclo(2,2,2)octane derivatives, their preparation and pharmaceutical preparations containing them | |
| JPH0421667B2 (en) | ||
| JPH01110679A (en) | 1,5-benzothiazepine derivative | |
| CN1052722C (en) | Tricyclic carboxylate | |
| EP0291270A2 (en) | Improvements in or relating to ergoline derivatives |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C13 | Decision | ||
| GR02 | Examined patent application | ||
| C14 | Grant of patent or utility model | ||
| GR01 | Patent grant | ||
| C19 | Lapse of patent right due to non-payment of the annual fee | ||
| CF01 | Termination of patent right due to non-payment of annual fee |