CN102002017A - 一种制备非布索坦中间体的方法 - Google Patents
一种制备非布索坦中间体的方法 Download PDFInfo
- Publication number
- CN102002017A CN102002017A CN 201010534098 CN201010534098A CN102002017A CN 102002017 A CN102002017 A CN 102002017A CN 201010534098 CN201010534098 CN 201010534098 CN 201010534098 A CN201010534098 A CN 201010534098A CN 102002017 A CN102002017 A CN 102002017A
- Authority
- CN
- China
- Prior art keywords
- acid
- methylthiazol
- ethyl ester
- hydroxy phenyl
- formic acid
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
- 238000000034 method Methods 0.000 title claims abstract description 12
- 229960005101 febuxostat Drugs 0.000 title abstract description 12
- BQSJTQLCZDPROO-UHFFFAOYSA-N febuxostat Chemical compound C1=C(C#N)C(OCC(C)C)=CC=C1C1=NC(C)=C(C(O)=O)S1 BQSJTQLCZDPROO-UHFFFAOYSA-N 0.000 title abstract description 12
- VKYKSIONXSXAKP-UHFFFAOYSA-N hexamethylenetetramine Chemical compound C1N(C2)CN3CN1CN2C3 VKYKSIONXSXAKP-UHFFFAOYSA-N 0.000 claims abstract description 12
- 239000002253 acid Substances 0.000 claims abstract description 11
- 239000007810 chemical reaction solvent Substances 0.000 claims abstract description 9
- 125000004203 4-hydroxyphenyl group Chemical group [H]OC1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims abstract description 7
- 229920000137 polyphosphoric acid Polymers 0.000 claims description 22
- 238000006243 chemical reaction Methods 0.000 claims description 16
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 claims description 12
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 claims description 12
- WISQBJLUORKXNY-UHFFFAOYSA-N ethyl 4-methyl-1,3-thiazole-5-carboxylate Chemical compound CCOC(=O)C=1SC=NC=1C WISQBJLUORKXNY-UHFFFAOYSA-N 0.000 claims description 6
- 150000001875 compounds Chemical class 0.000 claims description 5
- 239000000758 substrate Substances 0.000 claims description 4
- 238000002360 preparation method Methods 0.000 claims description 3
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 claims description 2
- 239000012046 mixed solvent Substances 0.000 claims description 2
- 229910017604 nitric acid Inorganic materials 0.000 claims description 2
- -1 heterocyclic aldehyde Chemical class 0.000 abstract description 3
- 239000012295 chemical reaction liquid Substances 0.000 abstract 1
- 238000010438 heat treatment Methods 0.000 abstract 1
- LOCYSKNNFCGDTR-UHFFFAOYSA-N ethyl 2-(4-hydroxyphenyl)-4-methyl-1,3-thiazole-5-carboxylate Chemical compound CC1=C(C(=O)OCC)SC(C=2C=CC(O)=CC=2)=N1 LOCYSKNNFCGDTR-UHFFFAOYSA-N 0.000 description 7
- LEHOTFFKMJEONL-UHFFFAOYSA-N Uric Acid Chemical compound N1C(=O)NC(=O)C2=C1NC(=O)N2 LEHOTFFKMJEONL-UHFFFAOYSA-N 0.000 description 6
- TVWHNULVHGKJHS-UHFFFAOYSA-N Uric acid Natural products N1C(=O)NC(=O)C2NC(=O)NC21 TVWHNULVHGKJHS-UHFFFAOYSA-N 0.000 description 5
- 229940116269 uric acid Drugs 0.000 description 5
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 3
- OGAZOYHQFBSRMC-UHFFFAOYSA-N ethyl 2-[3-cyano-4-(2-methylpropoxy)phenyl]-4-methyl-1,3-thiazole-5-carboxylate Chemical compound CC1=C(C(=O)OCC)SC(C=2C=C(C(OCC(C)C)=CC=2)C#N)=N1 OGAZOYHQFBSRMC-UHFFFAOYSA-N 0.000 description 3
- LRFVTYWOQMYALW-UHFFFAOYSA-N 9H-xanthine Chemical compound O=C1NC(=O)NC2=C1NC=N2 LRFVTYWOQMYALW-UHFFFAOYSA-N 0.000 description 2
- 201000005569 Gout Diseases 0.000 description 2
- 201000001431 Hyperuricemia Diseases 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- 239000003814 drug Substances 0.000 description 2
- 230000000694 effects Effects 0.000 description 2
- 230000007062 hydrolysis Effects 0.000 description 2
- 238000006460 hydrolysis reaction Methods 0.000 description 2
- 239000012535 impurity Substances 0.000 description 2
- 230000004144 purine metabolism Effects 0.000 description 2
- 239000002904 solvent Substances 0.000 description 2
- QBNJPSHRAWSBDW-UHFFFAOYSA-N 2-methylpropane;hydrobromide Chemical compound Br.CC(C)C QBNJPSHRAWSBDW-UHFFFAOYSA-N 0.000 description 1
- FMHRQJJWJQGSDR-UHFFFAOYSA-N 3-cyano-4-(2-methylpropoxy)benzenecarbothioamide Chemical compound CC(C)COC1=CC=C(C(N)=S)C=C1C#N FMHRQJJWJQGSDR-UHFFFAOYSA-N 0.000 description 1
- ZGWGSEUMABQEMD-UHFFFAOYSA-N 4-methyl-1,3-thiazole-5-carboxylic acid Chemical compound CC=1N=CSC=1C(O)=O ZGWGSEUMABQEMD-UHFFFAOYSA-N 0.000 description 1
- WTDHULULXKLSOZ-UHFFFAOYSA-N Hydroxylamine hydrochloride Chemical compound Cl.ON WTDHULULXKLSOZ-UHFFFAOYSA-N 0.000 description 1
- 229940123769 Xanthine oxidase inhibitor Drugs 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- 229960002708 antigout preparations Drugs 0.000 description 1
- UHOVQNZJYSORNB-UHFFFAOYSA-N benzene Substances C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 1
- 238000006555 catalytic reaction Methods 0.000 description 1
- 230000001684 chronic effect Effects 0.000 description 1
- DOBRDRYODQBAMW-UHFFFAOYSA-N copper(i) cyanide Chemical compound [Cu+].N#[C-] DOBRDRYODQBAMW-UHFFFAOYSA-N 0.000 description 1
- 230000008021 deposition Effects 0.000 description 1
- 238000006193 diazotization reaction Methods 0.000 description 1
- 201000010099 disease Diseases 0.000 description 1
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 1
- 229940079593 drug Drugs 0.000 description 1
- 238000005516 engineering process Methods 0.000 description 1
- 238000010931 ester hydrolysis Methods 0.000 description 1
- 150000002148 esters Chemical class 0.000 description 1
- RQDZYELGFUZPQX-UHFFFAOYSA-N ethyl 2-[3-amino-4-(2-methylpropoxy)phenyl]-4-methyl-1,3-thiazole-5-carboxylate Chemical compound CC1=C(C(=O)OCC)SC(C=2C=C(N)C(OCC(C)C)=CC=2)=N1 RQDZYELGFUZPQX-UHFFFAOYSA-N 0.000 description 1
- 125000000623 heterocyclic group Chemical group 0.000 description 1
- 238000005984 hydrogenation reaction Methods 0.000 description 1
- 238000001727 in vivo Methods 0.000 description 1
- 210000003734 kidney Anatomy 0.000 description 1
- UKVIEHSSVKSQBA-UHFFFAOYSA-N methane;palladium Chemical compound C.[Pd] UKVIEHSSVKSQBA-UHFFFAOYSA-N 0.000 description 1
- 230000008506 pathogenesis Effects 0.000 description 1
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 1
- 150000003212 purines Chemical class 0.000 description 1
- 239000002994 raw material Substances 0.000 description 1
- 238000007363 ring formation reaction Methods 0.000 description 1
- 230000002194 synthesizing effect Effects 0.000 description 1
- 238000010189 synthetic method Methods 0.000 description 1
- YUKQRDCYNOVPGJ-UHFFFAOYSA-N thioacetamide Chemical compound CC(N)=S YUKQRDCYNOVPGJ-UHFFFAOYSA-N 0.000 description 1
- DLFVBJFMPXGRIB-UHFFFAOYSA-N thioacetamide Natural products CC(N)=O DLFVBJFMPXGRIB-UHFFFAOYSA-N 0.000 description 1
- 229940075420 xanthine Drugs 0.000 description 1
- 239000003064 xanthine oxidase inhibitor Substances 0.000 description 1
Landscapes
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Thiazole And Isothizaole Compounds (AREA)
Abstract
本发明涉及一种制备非布索坦中间体的方法,包括将2-[4-羟基苯基]-4-甲基噻唑-5-甲酸乙酯溶于混合酸反应溶剂中,加入一定量的乌洛托品,在一定温度下加热反应1-36小时,反应液处理得到相应的杂环醛。
Description
技术领域
本发明属于药物化学领域,涉及非布索坦中间体2-[3-醛基-4-羟基苯基]-4-甲基噻唑-5-甲酸乙酯的制备。
背景技术
痛风现已成为当今世界尤其是中老年男性的常见病,致病原因在于体内产生尿酸过多、肾脏清除能力下降,导致尿酸在体内不断积蓄,形成高尿酸血症。体内尿酸的生成与嘌呤代谢有关,在嘌呤代谢的最后阶段,黄嘌呤在黄嘌呤氧化酶(XO)的作用下生成尿酸,抑制XO的活性可以有效的减少尿酸的生成,因此新型抗痛风药物的研制一直是药物研究的热点。。
非布索坦,化学名为2-[3-氰基-4-异丁氧基苯基]-4-甲基噻唑-5-甲酸,为新一代非嘌呤类选择性黄嘌呤氧化酶抑制剂,临床上用于治疗发生尿酸盐沉积的慢性高尿酸血症(包括曾经或现在出现痛风或痛风性关节炎)。
非布索坦的合成,报道较多的路线可归纳为三条:
(1)在文献Heterocycles,47(2),857-864中以4-异丁氧基-1,3-苯二腈为原料,与硫代乙酰胺反应得到3-氰基-4-异丁氧基硫代苯甲酰胺,后者再与2-氯乙酰乙酸乙酯环合,所得的2-(3-氰基-4-异丁氧基苯基)-4-甲基噻唑-5-甲酸乙酯在碱性条件下水解得到非布索坦。

(2)在中国医药工业杂志2009,40(10),726-728中2-(3-甲酰基-4-羟基苯基)-4-甲基噻唑-5-甲酸乙酯在甲醇中用盐酸羟胺氰化,得到的2-(3-氰基-4-羟基苯基)-4-甲基噻唑-5-甲酸乙酯与溴代异丁烷反应制得2-(3-氰基-4-异丁氧基苯基)-4-甲基噻唑-5-甲酸乙酯,后者水解后得到非布索坦。

(3)文献US5614520中,2-(3-硝基-4-异丁氧基苯基)-4-甲基噻唑-5-甲酸乙酯在钯碳催化下加氢,得到的2-(3-氨基-4-异丁氧基苯基)-4-甲基噻唑-5-甲酸乙酯,先重氮化再与氰化亚铜反应,得2-(3-氰基-4-异丁氧基苯基)-4-甲基噻唑-5-甲酸乙酯,后者水解得到非布索坦。

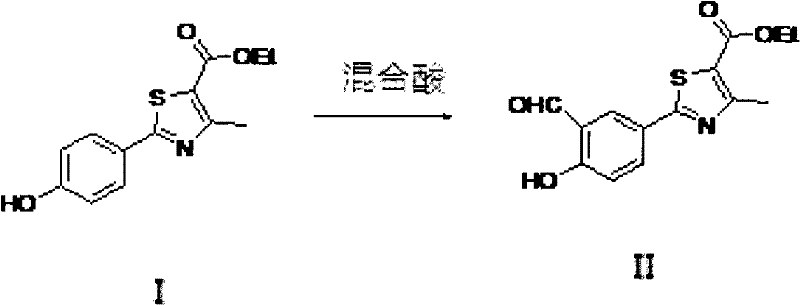
2-[3-醛基-4-羟基苯基]-4-甲基噻唑-5-甲酸乙酯是合成非布索坦的重要中间体,结构如下

已有的合成文献中,II式的合成方法是通过Duff-Bills反应,使用乌洛托品在苯环的三位上醛,反应溶剂有两种,
(1)在JP1045733中,使用PPA(多聚磷酸)作为溶剂,收率70%,
(2)在CN101412699中,使用三氟乙酸作为溶剂,收率95%,
使用PPA(多聚磷酸)做为反应溶剂,优点在于价格便宜,但反应产生杂质多,处理困难,产品收率低。
使用三氟乙酸做为反应溶剂,反应产生杂质少,产品收率高,但三氟乙酸价格高,难于回收,会增加合成成本。
我们在合成非布索坦过程发现,如果使用多聚磷酸与其它强酸的混合酸作为反应溶剂,既能获得高收率(92%),又能减少后处理的困难,降低合成成本,是一种合成非布索坦的中间体的好方法,该方法反应条件温和,操作简单,便于工业化。
发明内容
本发明提供一种结构II的化合物2-[3-醛基-4-羟基苯基]-4-甲基噻唑-5-甲酸乙酯的制备方法,包括以下步骤:
将结构I的2-[4-羟基苯基]-4-甲基噻唑-5-甲酸乙酯溶于混合酸反应溶剂中,加入乌洛托品,反应1-36小时,得到结构II的化合物。

其中所述混合酸反应溶剂选自:多聚磷酸与浓盐酸、多聚磷酸与浓硫酸、多聚磷酸与硝酸或多聚磷酸与三氟乙酸的混合溶剂,混合酸中多聚磷酸与其它酸的体积比例为4∶1-1∶1。
其中加入乌洛托品的量为底物2-[4-羟基苯基]-4-甲基噻唑-5-甲酸乙酯摩尔量的0.5-10倍。
反应温度40-120℃。
混合酸的加入量为底物2-[4-羟基苯基]-4-甲基噻唑-5-甲酸乙酯投料量的5-10倍。
本发明方法的优点在于:操作简单,纯度高,收率高,反应条件温和。
具体实施方式
以下通过实施例进一步说明本发明,但不作为对本发明的限制。
实施例1:
26.3g 2-[4-羟基苯基]-4-甲基噻唑-5-甲酸乙酯溶于100ml多聚磷酸和40ml浓盐酸中,加入13g乌洛托品,加热至80℃,保持12小时,反应液冷至室温,处理得到产品,收率90%。
实施例2:
26.3g 2-[4-羟基苯基]-4-甲基噻唑-5-甲酸乙酯溶于100ml多聚磷酸和50ml浓硫酸中,加入12g乌洛托品,加热至80℃,保持12小时,反应液冷至室温,处理得到产品,收率93%。
实施例3:
26.3g 2-[4-羟基苯基]-4-甲基噻唑-5-甲酸乙酯溶于100ml多聚磷酸和40ml磷酸中,加入10g乌洛托品,加热至120℃,保持12小时,反应液冷至室温,处理得到产品,收率70%。
实施例4:
26.3g 2-[4-羟基苯基]-4-甲基噻唑-5-甲酸乙酯溶于150ml多聚磷酸和25ml三氟乙酸中,加入13g乌洛托品,加热至90℃,保持6小时,反应液冷至室温,处理得到产品,收率93%。
实施例5:
26.3g 2-[4-羟基苯基]-4-甲基噻唑-5-甲酸乙酯溶于100ml多聚磷酸和40ml浓盐酸中,加入13g乌洛托品,加热至60℃,保持24小时,反应液冷至室温,处理得到产品,收率79%。
实施例6:
26.3g 2-[4-羟基苯基]-4-甲基噻唑-5-甲酸乙酯溶于100ml多聚磷酸和40ml浓盐酸中,加入13g乌洛托品,加热至100℃,保持3小时,反应液冷至室温,处理得到产品,收率80%。
实施例7:
26.3g 2-[4-羟基苯基]-4-甲基噻唑-5-甲酸乙酯溶于100ml多聚磷酸和40ml浓盐酸中,加入15g乌洛托品,加热至80℃,保持12小时,反应液冷至室温,处理得到产品,收率93%。
Claims (6)
1.一种结构式II化合物的制备方法,其特征在于,包括以下步骤:将结构I的化合物2-[4-羟基苯基]-4-甲基噻唑-5-甲酸乙酯溶于混合酸反应溶剂中,加入乌洛托品,反应1-36小时,得到结构II的化合物。
2.根据权利要求1的方法,其特征在于,所述混合酸反应溶剂选自:多聚磷酸与浓盐酸、多聚磷酸与浓硫酸、多聚磷酸与硝酸或多聚磷酸与三氟乙酸的混合溶剂。
3.根据权利要求1的方法,其特征在于,加入乌洛托品的量为底物2-[4-羟基苯基]-4-甲基噻唑-5-甲酸乙酯摩尔量的0.5-10倍。
4.根据权利要求1的方法,其特征在于,反应温度40-120℃。
5.根据权利要求1的方法,其特征在于,混合酸的加入量为底物2-[4-羟基苯基]-4-甲基噻唑-5-甲酸乙酯投料量的5-10倍。
6.根据权利要求2的方法,其特征在于,混合酸中多聚磷酸与其它酸的体积比例为4∶1-1∶1。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201010534098A CN102002017B (zh) | 2010-11-02 | 2010-11-02 | 一种制备非布索坦中间体的方法 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201010534098A CN102002017B (zh) | 2010-11-02 | 2010-11-02 | 一种制备非布索坦中间体的方法 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN102002017A true CN102002017A (zh) | 2011-04-06 |
| CN102002017B CN102002017B (zh) | 2012-09-26 |
Family
ID=43809751
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN201010534098A Active CN102002017B (zh) | 2010-11-02 | 2010-11-02 | 一种制备非布索坦中间体的方法 |
Country Status (1)
| Country | Link |
|---|---|
| CN (1) | CN102002017B (zh) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN103910695A (zh) * | 2014-04-24 | 2014-07-09 | 重庆科瑞制药(集团)有限公司 | 一种非布索坦的合成方法 |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH1045733A (ja) * | 1996-08-01 | 1998-02-17 | Teijin Ltd | 2−(4−アルコキシ−3−シアノフェニル)チアゾール誘導体の製造法 |
| CN101412699A (zh) * | 2007-10-19 | 2009-04-22 | 上海医药工业研究院 | 2-(3-甲醛基-4-羟基苯基)-4-甲基-5-噻唑甲酸乙酯的制备方法 |
| CN102086169A (zh) * | 2009-12-04 | 2011-06-08 | 重庆医药工业研究院有限责任公司 | 一种非布司他的中间体的制备方法 |
-
2010
- 2010-11-02 CN CN201010534098A patent/CN102002017B/zh active Active
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH1045733A (ja) * | 1996-08-01 | 1998-02-17 | Teijin Ltd | 2−(4−アルコキシ−3−シアノフェニル)チアゾール誘導体の製造法 |
| CN101412699A (zh) * | 2007-10-19 | 2009-04-22 | 上海医药工业研究院 | 2-(3-甲醛基-4-羟基苯基)-4-甲基-5-噻唑甲酸乙酯的制备方法 |
| CN102086169A (zh) * | 2009-12-04 | 2011-06-08 | 重庆医药工业研究院有限责任公司 | 一种非布司他的中间体的制备方法 |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN103910695A (zh) * | 2014-04-24 | 2014-07-09 | 重庆科瑞制药(集团)有限公司 | 一种非布索坦的合成方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| CN102002017B (zh) | 2012-09-26 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| García-Báez et al. | Benzothiazoles from condensation of o-aminothiophenoles with carboxylic acids and their derivatives: A review | |
| CN103012311A (zh) | 一种高纯度非布司他的制备方法 | |
| CN103739500B (zh) | 一种盐酸西那卡塞的合成与精制方法 | |
| CN103073519A (zh) | 一种制备右旋普拉克索盐酸盐的方法 | |
| CN102002017B (zh) | 一种制备非布索坦中间体的方法 | |
| CN104496936B (zh) | 一种盐酸普拉克索的制备方法 | |
| CN105566162B (zh) | 利匹韦林中间体的制备工艺 | |
| CN101723915B (zh) | 一种制备非布索坦中间体的方法 | |
| CN102936230A (zh) | 一种非布索坦的新工艺制法 | |
| CN103265497B (zh) | 一种替尼类抗肿瘤药合成所需中间体4-氯-6-氨基-7-羟基喹唑啉及其制备方法 | |
| CN103408507A (zh) | 一种2-氨基-1,3,4 噻二唑类化合物的制备方法 | |
| CN106748713B (zh) | 一种基于uio-66的固体酸催化酯交换反应合成(r)-2-氯丙酸的方法 | |
| CN103788010A (zh) | 非布索坦中间体及其制备方法 | |
| CN103739545B (zh) | 一种简便的维生素b6的制备方法 | |
| CN110229117A (zh) | 一种非布司他的制备新方法 | |
| CN114773270A (zh) | 一种二丙酸咪唑苯脲的生产制备方法 | |
| CN102911123A (zh) | 2-氯三氟甲基嘧啶类化合物的制备方法 | |
| CN103896784B (zh) | 一种芬戈莫德中间体硝基还原为氨基的方法 | |
| CN103508975A (zh) | 一种普拉克索的制备方法 | |
| CN102391170B (zh) | 一种n,n-二烯丙基-5-甲氧基色胺盐酸盐的制备方法 | |
| CN110698426B (zh) | 叔丁醇钾高效催化制备1,3-苯并噻唑衍生物的方法 | |
| CN102875399B (zh) | 一种d-缬氨酸的制备方法 | |
| CN107162920A (zh) | 一种r-1-氨基-2-丙醇的制备方法 | |
| CN108530438B (zh) | 一种苯并噻唑-噁唑型α-葡萄糖苷酶抑制剂及其制备方法和应用 | |
| CN101987837B (zh) | 1,2,4-噻二唑肟基乙酸系列化合物的制备方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C53 | Correction of patent for invention or patent application | ||
| CB02 | Change of applicant information |
Address after: 100021 Beijing city Chaoyang District West business center boziwan Jinhai rich 402 (A) 21 storey building Applicant after: China Resources Saike Pharmaceutical Co., Ltd. Address before: 100021 Beijing city Chaoyang District West business center boziwan Jinhai rich 402 (A) 21 storey building Applicant before: Saike Pharmaceutical Co., Ltd., Beijing |
|
| COR | Change of bibliographic data |
Free format text: CORRECT: APPLICANT; FROM: BEIJING SAIKE PHARMACEUTICAL CO., LTD. TO: CHINA RESOURCES SAIKE PHARMACEUTICAL CO., LTD. |
|
| C14 | Grant of patent or utility model | ||
| GR01 | Patent grant |