CN102796005A - N,n'-二异丙基乙二胺的合成方法 - Google Patents
N,n'-二异丙基乙二胺的合成方法 Download PDFInfo
- Publication number
- CN102796005A CN102796005A CN2012103092646A CN201210309264A CN102796005A CN 102796005 A CN102796005 A CN 102796005A CN 2012103092646 A CN2012103092646 A CN 2012103092646A CN 201210309264 A CN201210309264 A CN 201210309264A CN 102796005 A CN102796005 A CN 102796005A
- Authority
- CN
- China
- Prior art keywords
- isopropylamine
- reaction
- raw material
- diisopropyl ethylenediamine
- ethylene dichloride
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
- 229910052757 nitrogen Inorganic materials 0.000 title abstract description 23
- 238000010189 synthetic method Methods 0.000 title abstract 2
- JJWLVOIRVHMVIS-UHFFFAOYSA-N isopropylamine Chemical compound CC(C)N JJWLVOIRVHMVIS-UHFFFAOYSA-N 0.000 claims abstract description 56
- 239000002994 raw material Substances 0.000 claims abstract description 44
- 230000001105 regulatory effect Effects 0.000 claims abstract description 3
- 238000006243 chemical reaction Methods 0.000 claims description 66
- WSLDOOZREJYCGB-UHFFFAOYSA-N 1,2-Dichloroethane Chemical compound ClCCCl WSLDOOZREJYCGB-UHFFFAOYSA-N 0.000 claims description 36
- 239000000243 solution Substances 0.000 claims description 36
- 238000000034 method Methods 0.000 claims description 32
- 150000001875 compounds Chemical class 0.000 claims description 23
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 claims description 21
- 239000000203 mixture Substances 0.000 claims description 4
- 238000004821 distillation Methods 0.000 claims description 3
- 230000035484 reaction time Effects 0.000 claims description 3
- 239000012047 saturated solution Substances 0.000 claims description 2
- 239000003054 catalyst Substances 0.000 abstract description 4
- SCYULBFZEHDVBN-UHFFFAOYSA-N 1,1-Dichloroethane Chemical compound CC(Cl)Cl SCYULBFZEHDVBN-UHFFFAOYSA-N 0.000 abstract 2
- 239000012295 chemical reaction liquid Substances 0.000 abstract 2
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 32
- 239000012074 organic phase Substances 0.000 description 17
- 238000001514 detection method Methods 0.000 description 16
- 238000006073 displacement reaction Methods 0.000 description 16
- MFIGJRRHGZYPDD-UHFFFAOYSA-N n,n'-di(propan-2-yl)ethane-1,2-diamine Chemical group CC(C)NCCNC(C)C MFIGJRRHGZYPDD-UHFFFAOYSA-N 0.000 description 16
- 230000003595 spectral effect Effects 0.000 description 16
- 238000003756 stirring Methods 0.000 description 16
- 238000010792 warming Methods 0.000 description 16
- 238000002360 preparation method Methods 0.000 description 6
- 238000012423 maintenance Methods 0.000 description 5
- 230000000052 comparative effect Effects 0.000 description 4
- 238000005516 engineering process Methods 0.000 description 4
- 238000004519 manufacturing process Methods 0.000 description 4
- PAAZPARNPHGIKF-UHFFFAOYSA-N 1,2-dibromoethane Chemical compound BrCCBr PAAZPARNPHGIKF-UHFFFAOYSA-N 0.000 description 3
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 3
- 230000008569 process Effects 0.000 description 3
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 2
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical class [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 2
- 230000007812 deficiency Effects 0.000 description 2
- LEQAOMBKQFMDFZ-UHFFFAOYSA-N glyoxal Chemical compound O=CC=O LEQAOMBKQFMDFZ-UHFFFAOYSA-N 0.000 description 2
- 239000001257 hydrogen Substances 0.000 description 2
- 229910052739 hydrogen Inorganic materials 0.000 description 2
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 2
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 2
- 230000006872 improvement Effects 0.000 description 2
- 239000007788 liquid Substances 0.000 description 2
- 229910052763 palladium Inorganic materials 0.000 description 2
- 239000010970 precious metal Substances 0.000 description 2
- 239000000126 substance Substances 0.000 description 2
- 239000002699 waste material Substances 0.000 description 2
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 2
- NAMYKGVDVNBCFQ-UHFFFAOYSA-N 2-bromopropane Chemical compound CC(C)Br NAMYKGVDVNBCFQ-UHFFFAOYSA-N 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 1
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 1
- NSOXQYCFHDMMGV-UHFFFAOYSA-N Tetrakis(2-hydroxypropyl)ethylenediamine Chemical compound CC(O)CN(CC(C)O)CCN(CC(C)O)CC(C)O NSOXQYCFHDMMGV-UHFFFAOYSA-N 0.000 description 1
- 239000002253 acid Substances 0.000 description 1
- 230000029936 alkylation Effects 0.000 description 1
- 238000005804 alkylation reaction Methods 0.000 description 1
- 230000008901 benefit Effects 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 230000015572 biosynthetic process Effects 0.000 description 1
- 238000009835 boiling Methods 0.000 description 1
- 238000010531 catalytic reduction reaction Methods 0.000 description 1
- 150000004985 diamines Chemical class 0.000 description 1
- RAABOESOVLLHRU-UHFFFAOYSA-N diazene Chemical compound N=N RAABOESOVLLHRU-UHFFFAOYSA-N 0.000 description 1
- 229910000071 diazene Inorganic materials 0.000 description 1
- 230000007613 environmental effect Effects 0.000 description 1
- 239000002360 explosive Substances 0.000 description 1
- 239000000446 fuel Substances 0.000 description 1
- 229940015043 glyoxal Drugs 0.000 description 1
- -1 glyoxaline ion Chemical class 0.000 description 1
- 238000009776 industrial production Methods 0.000 description 1
- 150000002500 ions Chemical class 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 238000001819 mass spectrum Methods 0.000 description 1
- 239000000463 material Substances 0.000 description 1
- 229910052697 platinum Inorganic materials 0.000 description 1
- 238000007781 pre-processing Methods 0.000 description 1
- 150000003233 pyrroles Chemical class 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 238000006722 reduction reaction Methods 0.000 description 1
- 229910052707 ruthenium Inorganic materials 0.000 description 1
- 235000002639 sodium chloride Nutrition 0.000 description 1
- 239000007787 solid Substances 0.000 description 1
- 239000002904 solvent Substances 0.000 description 1
- 238000003786 synthesis reaction Methods 0.000 description 1
- 230000009466 transformation Effects 0.000 description 1
Landscapes
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
本发明公开了一种N,N'-二异丙基乙二胺的合成方法,采用MIx作为催化剂,以异丙胺和二氯乙烷为原料在高压釜中进行反应,反应温度为60~120℃,反应压力为0.4~1.3MPa,反应时间为2~6小时,二氯乙烷和异丙胺的摩尔比为1:8~12,所述催化剂占原料总重的0.05%~0.15%;所得的反应液调节pH=12.5~13.5,然后于35~45℃下蒸馏20~40分钟,再经过分液和精馏后,得到N,N'-二异丙基乙二胺。
Description
技术领域
本发明涉及一种有机化合物的合成方法,特别是一种化工中间体—N,N'-二异丙基乙二胺的合成方法。
背景技术
N,N'-二异丙基乙二胺,其分子式为C8H20N2,分子量为144.26,其结构式如S-1所示,沸点为169.5~170.3℃,有着广泛的工业用途,常用作缚酸剂,也是一种重要的有机化工中间体,用于合成双阳离子中心离子液体,以及合成咪唑类离子液体等。

S-1
综合文献报道,根据原料的不同,目前N,N'-二异丙基乙二胺的制备主要采用以下方法:
1、Yoo, Bok Ryul 等在美国专利US20110160454中报道,以异丙胺和二溴乙烷为原料,常温常压回流反应12h,得到产品N,N'-二异丙基乙二胺,收率为68%,反应式如S-2所示。该方法原料价格相对较高,收率偏低,经济效益差,且反应时间很久,生产效率低。
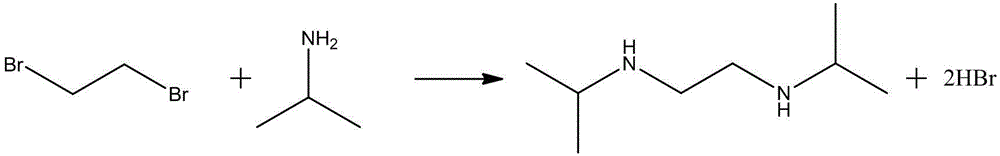
S-2
2、Klemn, Dieter等在Direct alkylation of diamines with isopropyl bromide, Journal fuel Praktische Chemie(Leipzig), 321(4), 680-2; 1979中报道以乙二胺和2-溴丙烷为原料生产N,N'-二异丙基乙二胺,收率为63%,反应式如S-3所示。与美国专利US20110160454中报道的二溴乙烷法相比,该方法原料成本依然较高,而且转化率更低,经济效益差,同样不适合大规模工业化生产。

S-3
3、Tom Dieck等在Glyoxal as synthone. III. A simple synthesis ofaminopyrroles and of dihydropyrrolo [3,2-b] pyrroles, Chemische Berichte, 122(1), 129-31;1989中报道在乙醇溶剂中,以钯为催化剂用氢气对N,N'-二异丙基乙二亚胺还原生成N,N'-二异丙基乙二胺,收率为73%,反应式如S-4所示。该方法虽然收率较上述方法较高,但是原料来源困难,且所用催化剂为贵金属,价格昂贵且,回收困难,成本投入高;此外还原过程需要在氢气环境下,反应过程易燃易爆,危险系数高,实现工业化生产难度大。

S-4
发明内容
本发明所要解决的技术问题是提供一种成本低、收率高、安全性好、环境友好、三废排放少的N,N'-二异丙基乙二胺的合成方法。
为了解决上述技术问题,本发明提供一种N,N'-二异丙基乙二胺的合成方法,采用MIx作为催化剂,以异丙胺和二氯乙烷为原料在高压釜中进行反应,反应温度为60~120℃,反应压力为0.4~1.3MPa,反应时间为2~6小时,二氯乙烷和异丙胺的摩尔比为1:8~12,催化剂占原料总重的0.05%~0.15%;
所得的反应液调节(利用强碱进行调节)pH=12.5~13.5,然后于35~45℃下蒸馏20~40分钟(从而蒸去大部分的异丙胺,较佳为40℃下蒸馏30分钟),再经过分液和精馏后,得到N,N'-二异丙基乙二胺。
作为本发明的N,N'-二异丙基乙二胺的合成方法的改进: MIx为NaI、KI、CaI2、MgI2、CuI、ZnI2、LiI、AlI3、AgI、CdI2、TiI4或FeI3。
作为本发明的N,N'-二异丙基乙二胺的合成方法的进一步改进:以强碱饱和溶液的形式调节所得的反应液pH=12.5~13.5(较佳为13),强碱为NaOH或KOH。
本发明的合成N,N'-二异丙基乙二胺的反应方程式如下式S-5:
S-5
本发明的N,N'-二异丙基乙二胺的合成方法,具有如下优点:
(1)与二溴乙烷工艺相比,采用碘化物为催化剂,产品收率高(最高可以达到91.3%),大大提高了原料利用率,缩短了反应流程,降低了成本。
(2)与催化还原工艺相比,以碘化物为催化剂,相对于Pt、Pd、Ru等贵金属可显著降低成本、且不需要催化剂制备等预处理过程,在降低了成本的基础上简化了操作过程,增强了反应可操作性。
(3)该反应原料来源广,反应条件要求不高,设备操作要求低且后处理简单,适合于工业化生产,大大降低了三废处理负担,减少对环境和人类危害。
附图说明
下面结合附图对本发明的具体实施方式作进一步详细说明。
图1是本发明所得的N,N'-二异丙基乙二胺的质谱图。
具体实施方式
实施例1、一种N,N'-二异丙基乙二胺的合成方法,以异丙胺和二氯乙烷为起始原料,依次进行以下步骤:
在带搅拌测温装置的高压反应釜中加入26g二氯乙烷(0.26mol),154g异丙胺(2.6mol),KI 0.18g(即占原料总重的0.1%),合上釜盖。用氮气检漏并置换数次空气后升温至100℃,压力为0.7MPa,保持上述温度反应6h后,结束反应。
向所得的反应液中加入饱和NaOH溶液直到pH=13,然后40℃下蒸馏半小时,从而蒸去大部分异丙胺,分出有机相(位于上层)进行常压间歇精馏,收集169-171℃馏分,得产品为N,N'-二异丙基乙二胺34.2g,产品纯度98.7%,收率91.3%。所得产品经质谱表征结构正确。
实施例2、一种N,N'-二异丙基乙二胺的合成方法,以异丙胺和二氯乙烷为起始原料,依次进行以下步骤:
在带搅拌测温装置的高压反应釜中加入26g二氯乙烷(0.26mol),123g异丙胺(2.1mol),NaI 0.18g(即占原料总重的0.12%),合上釜盖。用氮气检漏并置换数次空气后升温至100℃,压力为0.65MPa,保持上述温度反应6h后结束反应。
向反应液中加入饱和NaOH溶液直到pH=13,然后40℃下蒸馏半小时,从而蒸去大部分异丙胺,分出有机相(上层)进行常压间歇精馏,收集169-171℃馏分,得产品为N,N'-二异丙基乙二胺32.3g,产品纯度98.3%,收率86.1%。所得产品经质谱表征结构正确。
实施例3、一种N,N'-二异丙基乙二胺的合成方法,以异丙胺和二氯乙烷为起始原料,依次进行以下步骤:
在带搅拌测温装置的高压反应釜中加入26g二氯乙烷(0.26mol),154g异丙胺(2.6mol),CaI2 0.09g(即占原料总重的0.05%),合上釜盖。用氮气检漏并置换数次空气后升温至120℃,压力为1.25MPa,保持上述温度反应4h后结束反应。
向反应液中加入饱和NaOH溶液直到pH=13,然后40℃下蒸馏半小时,从而蒸去大部分异丙胺,分出有机相(上层)进行常压间歇精馏,收集169-171℃馏分,得产品为N,N'-二异丙基乙二胺32.0g,产品纯度97.9%,收率85.4%。所得产品经质谱表征结构正确。
实施例4、一种N,N'-二异丙基乙二胺的合成方法,以异丙胺和二氯乙烷为起始原料,依次进行以下步骤:
在带搅拌测温装置的高压反应釜中加入26g二氯乙烷(0.26mol),184g异丙胺(3.1mol),MgI2 0.21g(即占原料总重的0.1%),合上釜盖。用氮气检漏并置换数次空气后升温至60℃,压力为0.45MPa,保持上述温度反应4h后结束反应。
向反应液中加入饱和NaOH溶液直到pH=13,然后40℃下蒸馏半小时,从而蒸去大部分异丙胺,分出有机相(上层)进行常压间歇精馏,收集169-171℃馏分,得产品为N,N'-二异丙基乙二胺29.2g,产品纯度98.0%,收率78.0%。所得产品经质谱表征结构正确。
实施例5、一种N,N'-二异丙基乙二胺的合成方法,以异丙胺和二氯乙烷为起始原料,依次进行以下步骤:
在带搅拌测温装置的高压反应釜中加入26g二氯乙烷(0.26mol),154g异丙胺(2.6mol),CuI 0.18g(即占原料总重的0.1%),合上釜盖。用氮气检漏并置换数次空气后升温至80℃,压力为0.55MPa,保持上述温度反应2h后结束反应。
向反应液中加入饱和NaOH溶液直到pH=13,然后40℃下蒸馏半小时,从而蒸去大部分异丙胺,分出有机相(上层)进行常压间歇精馏,收集169-171℃馏分,得产品为N,N'-二异丙基乙二胺29.9g,产品纯度98.2%,收率79.9%。所得产品经质谱表征结构正确。
实施例6、一种N,N'-二异丙基乙二胺的合成方法,以异丙胺和二氯乙烷为起始原料,依次进行以下步骤:
在带搅拌测温装置的高压反应釜中加入26g二氯乙烷(0.26mol),184g异丙胺(3.1mol), ZnI2 0.31g(即占原料总重的0.15%),合上釜盖。用氮气检漏并置换数次空气后升温至80℃,压力为0.58MPa,保持上述温度反应4h后结束反应。
向反应液中加入饱和KOH溶液直到pH=13,然后40℃下蒸馏半小时,从而蒸去大部分异丙胺,分出有机相(上层)进行常压间歇精馏,收集169-171℃馏分,得产品为N,N'-二异丙基乙二胺31.3g,产品纯度98.5%,收率83.6%。所得产品经质谱表征结构正确。
实施例7、一种N,N'-二异丙基乙二胺的合成方法,以异丙胺和二氯乙烷为起始原料,依次进行以下步骤:
在带搅拌测温装置的高压反应釜中加入26g二氯乙烷(0.26mol),184g异丙胺(3.1mol),FeI30.11g(即占原料总重的0.05%),合上釜盖。用氮气检漏并置换数次空气后升温至120℃,压力为1.3MPa,保持上述温度反应2h后结束反应。
向反应液中加入饱和NaOH溶液直到pH=13,然后40℃下蒸馏半小时,从而蒸去大部分异丙胺,分出有机相(上层)进行常压间歇精馏,收集169-171℃馏分,得产品为N,N'-二异丙基乙二胺30.9g,产品纯度98.3%,收率82.4%。所得产品经质谱表征结构正确。
实施例8、一种N,N'-二异丙基乙二胺的合成方法,以异丙胺和二氯乙烷为起始原料,依次进行以下步骤:
在带搅拌测温装置的高压反应釜中加入26g二氯乙烷(0.26mol),123g异丙胺(2.1mol),AlI3 0.22g(即占原料总重的0.15%),合上釜盖。用氮气检漏并置换数次空气后升温至60℃,压力为0.4MPa,保持上述温度反应2h后结束反应。
向反应液中加入饱和NaOH溶液直到pH=13,然后40℃下蒸馏半小时,从而蒸去大部分异丙胺,分出有机相(上层)进行常压间歇精馏,收集169-171℃馏分,得产品为N,N'-二异丙基乙二胺27.0g,产品纯度97.7%,收率72.1%。所得产品经质谱表征结构正确。
实施例9、一种N,N'-二异丙基乙二胺的合成方法,以异丙胺和二氯乙烷为起始原料,依次进行以下步骤:
在带搅拌测温装置的高压反应釜中加入26g二氯乙烷(0.26mol),123g异丙胺(2.1mol),LiI 0.22g(即占原料总重的0.15%),合上釜盖。用氮气检漏并置换数次空气后升温至100℃,压力为0.67MPa,保持上述温度反应6h后结束反应。
向反应液中加入饱和NaOH溶液直到pH=13,然后40℃下蒸馏半小时,蒸去大部分异丙胺,分出有机相(上层)进行常压间歇精馏,收集169-171℃馏分,得产品为N,N'-二异丙基乙二胺32.3g,产品纯度98.7%,收率86.3%。所得产品经质谱表征结构正确。
实施例10、一种N,N'-二异丙基乙二胺的合成方法,以异丙胺和二氯乙烷为起始原料,依 次进行以下步骤:
在带搅拌测温装置的高压反应釜中加入26g二氯乙烷(0.26mol),184g异丙胺(3.1mol),CdI2 0.11g(即占原料总重的0.05%),合上釜盖。用氮气检漏并置换数次空气后升温至120℃,压力为1.3MPa,保持上述温度反应2h后结束反应。
向反应液中加入饱和NaOH溶液直到pH=13,然后40℃下蒸馏半小时,从而蒸去大部分异丙胺,分出有机相(上层)进行常压间歇精馏,收集169-171℃馏分,得产品为N,N'-二异丙基乙二胺30.8g,产品纯度98.2%,收率82.4%。所得产品经质谱表征结构正确。
实施例11、一种N,N'-二异丙基乙二胺的合成方法,以异丙胺和二氯乙烷为起始原料,依次进行以下步骤:
在带搅拌测温装置的高压反应釜中加入26g二氯乙烷(0.26mol),154g异丙胺(2.6mol),TiI4 0.18g(即占原料总重的0.1%),合上釜盖。用氮气检漏并置换数次空气后升温至60℃,压力为0.42MPa,保持上述温度反应4h后结束反应。
向反应液中加入饱和NaOH溶液直到pH=13,然后40℃下蒸馏半小时,从而蒸去大部分异丙胺,分出有机相(上层)进行常压间歇精馏,收集169-171℃馏分,得产品为N,N'-二异丙基乙二胺29.6g,产品纯度98.1%,收率79.0%。所得产品经质谱表征结构正确。
实施例12、一种N,N'-二异丙基乙二胺的合成方法,以异丙胺和二氯乙烷为起始原料,依次进行以下步骤:
在带搅拌测温装置的高压反应釜中加入26g二氯乙烷(0.26mol),123g异丙胺(2.1mol),AgI 0.22g(即占原料总重的0.15%),合上釜盖。用氮气检漏并置换数次空气后升温至80℃,压力为0.54MPa,保持上述温度反应6h后结束反应。
向反应液中加入饱和NaOH溶液直到pH=13,然后40℃下蒸馏半小时,蒸去大部分异丙胺,分出有机相(上层)进行常压间歇精馏,收集169-171℃馏分,得产品为N,N'-二异丙基乙二胺30.9g,产品纯度98.4%,收率82.5%。所得产品经质谱表征结构正确。
对比例1、一种N,N'-二异丙基乙二胺的制备方法,以异丙胺和二氯乙烷为起始原料,依次进行以下步骤:
在带搅拌测温装置的高压反应釜中加入26g二氯乙烷(0.26mol),154g异丙胺(2.6mol),NH4I 0.18g(即占原料总重的0.1%),合上釜盖。用氮气检漏并置换数次空气后升温至100℃,压力为0.7MPa,保持上述温度反应6h后,结束反应。
向所得的反应液中加入饱和NaOH溶液直到pH=13,然后40℃下蒸馏半小时,从而蒸去大部分异丙胺,分出有机相(上层)进行常压间歇精馏,收集169-171℃馏分,得产品为N,N'- 二异丙基乙二胺34.2g,产品纯度98.0%,收率51.7%。所得产品经质谱表征结构正确。
对比例2、一种N,N'-二异丙基乙二胺的制备方法,以异丙胺和二氯乙烷为起始原料,依次进行以下步骤:
在带搅拌测温装置的高压反应釜中加入26g二氯乙烷(0.26mol),154g异丙胺(2.6mol),合上釜盖。用氮气检漏并置换数次空气后升温至100℃,压力为0.7MPa,保持上述温度反应6h后,结束反应。
向所得的反应液中加入饱和NaOH溶液直到pH=13,然后40℃下蒸馏半小时,从而蒸去大部分异丙胺,分出有机相(上层)进行常压间歇精馏,收集169-171℃馏分,得产品为N,N'-二异丙基乙二胺4.8g,产品纯度98.7%,收率12.7%。所得产品经质谱表征结构正确。
对比例3、一种N,N'-二异丙基乙二胺的制备方法,以异丙胺和二氯乙烷为起始原料,依次进行以下步骤:
在带搅拌测温装置的高压反应釜中加入26g二氯乙烷(0.26mol),154g异丙胺(2.6mol),KI 0.05g(即占原料总重的0.027%),合上釜盖。用氮气检漏并置换数次空气后升温至100℃,压力为0.7MPa,保持上述温度反应6h后,结束反应。
向所得的反应液中加入饱和NaOH溶液直到pH=13,然后40℃下蒸馏半小时,从而蒸去大部分异丙胺,分出有机相(上层)进行常压间歇精馏,收集169-171℃馏分,得产品为N,N'-二异丙基乙二胺16.6g,产品纯度98.7%,收率44.2%。所得产品经质谱表征结构正确。
对比例4、一种N,N'-二异丙基乙二胺的制备方法,以异丙胺和二氯乙烷为起始原料,依次进行以下步骤:
在带搅拌测温装置的高压反应釜中加入26g二氯乙烷(0.26mol),154g异丙胺(2.6mol),KI 0.18g(即占原料总重的0.1%),合上釜盖。用氮气检漏并置换数次空气后升温至100℃,压力为0.7MPa,保持上述温度反应6h后,结束反应。
向所得的反应液中加入饱和NaOH溶液直到pH=13,然后转移入分液漏斗,分出有机相(上层)进行常压间歇精馏,由于水与异丙胺相互溶解度很好,有机相中还有大量溶解有饱和氯化钠的水,导致精馏时有大量固体氯化钠析出,精馏收率低。收集169-171℃馏分,得产品为N,N'-二异丙基乙二胺10.4g,产品纯度98.7%,收率27.8%。所得产品经质谱表征结构正确。
最后,还需要注意的是,以上列举的仅是本发明的若干个具体实施例。显然,本发明不限于以上实施例,还可以有许多变形。本领域的普通技术人员能从本发明公开的内容直接导出或联想到的所有变形,均应认为是本发明的保护范围。
Claims (3)
1.N,N'-二异丙基乙二胺的合成方法,其特征是:采用MIx作为催化剂,以异丙胺和二氯乙烷为原料在高压釜中进行反应,反应温度为60~120℃,反应压力为0.4~1.3MPa,反应时间为2~6小时,二氯乙烷和异丙胺的摩尔比为1:8~12,所述催化剂占原料总重的0.05%~0.15%;
所得的反应液调节pH=12.5~13.5,然后于35~45℃下蒸馏20~40分钟,再经过分液和精馏后,得到N,N'-二异丙基乙二胺。
2.根据权利要求1所述的N,N'-二异丙基乙二胺的合成方法,其特征是:所述MIx为NaI、KI、CaI2、MgI2、CuI、ZnI2、LiI、AlI3、AgI、CdI2、TiI4或FeI3。
3.根据权利要求2所述的N,N'-二异丙基乙二胺的合成方法,其特征是:以强碱饱和溶液的形式调节所得的反应液pH=12.5~13.5,所述强碱为NaOH或KOH。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201210309264.6A CN102796005B (zh) | 2012-08-28 | 2012-08-28 | N,n'-二异丙基乙二胺的合成方法 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201210309264.6A CN102796005B (zh) | 2012-08-28 | 2012-08-28 | N,n'-二异丙基乙二胺的合成方法 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN102796005A true CN102796005A (zh) | 2012-11-28 |
| CN102796005B CN102796005B (zh) | 2014-07-16 |
Family
ID=47195346
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN201210309264.6A Active CN102796005B (zh) | 2012-08-28 | 2012-08-28 | N,n'-二异丙基乙二胺的合成方法 |
Country Status (1)
| Country | Link |
|---|---|
| CN (1) | CN102796005B (zh) |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN101759571A (zh) * | 2010-01-28 | 2010-06-30 | 浙江大学 | N,n-二异丙基乙胺的制备方法 |
| CN101928222A (zh) * | 2010-07-08 | 2010-12-29 | 浙江大学 | 一种n,n,n’,n’-四异丙基乙二胺的合成方法 |
| US20110060167A1 (en) * | 2009-07-28 | 2011-03-10 | Bayer Cropscience Ag | Process for Preparing 2,2-Difluoroethylamine |
| CN102050687A (zh) * | 2010-11-30 | 2011-05-11 | 中山大学 | 一种水相体系中以氨水为氨源制备芳香伯胺的方法 |
| WO2011081277A2 (en) * | 2009-12-31 | 2011-07-07 | Korea Institute Of Science And Technology | Saturated n-heterocyclic carbene-ligand metal complex derivatives, preparing method thereof, and preparing method of silane compound by hydrosilylation reaction using the same as catalyst |
-
2012
- 2012-08-28 CN CN201210309264.6A patent/CN102796005B/zh active Active
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20110060167A1 (en) * | 2009-07-28 | 2011-03-10 | Bayer Cropscience Ag | Process for Preparing 2,2-Difluoroethylamine |
| WO2011081277A2 (en) * | 2009-12-31 | 2011-07-07 | Korea Institute Of Science And Technology | Saturated n-heterocyclic carbene-ligand metal complex derivatives, preparing method thereof, and preparing method of silane compound by hydrosilylation reaction using the same as catalyst |
| CN101759571A (zh) * | 2010-01-28 | 2010-06-30 | 浙江大学 | N,n-二异丙基乙胺的制备方法 |
| CN101928222A (zh) * | 2010-07-08 | 2010-12-29 | 浙江大学 | 一种n,n,n’,n’-四异丙基乙二胺的合成方法 |
| CN102050687A (zh) * | 2010-11-30 | 2011-05-11 | 中山大学 | 一种水相体系中以氨水为氨源制备芳香伯胺的方法 |
Non-Patent Citations (3)
| Title |
|---|
| 《Tetrahedron》 20031231 Michael K. Denk等 Reaction of 1,2-dibromoethane with primary amines: formation of N,N'-disubstituted ethylenediamines RNH-CH2CH2-NHR and homologous polyamines RNH-[CH2CH2NR]n-H 第7565-7570页 1-3 第59卷, * |
| MICHAEL K. DENK等: "Reaction of 1,2-dibromoethane with primary amines: formation of N,N’-disubstituted ethylenediamines RNH–CH2CH2–NHR and homologous polyamines RNH–[CH2CH2NR]n–H", 《TETRAHEDRON》, vol. 59, 31 December 2003 (2003-12-31), pages 7565 - 7570 * |
| 陈新志等: "N,N-二异丙基乙基胺的合成", 《精细化工》, vol. 20, no. 1, 31 January 2003 (2003-01-31), pages 60 - 64 * |
Also Published As
| Publication number | Publication date |
|---|---|
| CN102796005B (zh) | 2014-07-16 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| Zhao et al. | Eco-friendly acetylcholine-carboxylate bio-ionic liquids for controllable N-methylation and N-formylation using ambient CO 2 at low temperatures | |
| CN104496943A (zh) | 一种制备二羟甲基四氢呋喃的方法 | |
| CN110914249B (zh) | 2,5-双(氨基甲基)呋喃的制造方法 | |
| CN101811942B (zh) | 邻苯二甲醚的合成方法 | |
| CN103319313B (zh) | 氧芴开环制备邻苯基苯酚的方法 | |
| KR20100136430A (ko) | 방향족 아민 제조 방법 | |
| CN102827136A (zh) | 二氧化碳与环氧化合物环加成制备环状碳酸酯的方法 | |
| CN102432590A (zh) | 一种噻吩乙胺的合成方法 | |
| CN105198710A (zh) | 一种间叔丁基苯酚的合成方法 | |
| CN106748763B (zh) | 两釜联合相转移催化合成苯甲酸甲酯的方法 | |
| CN108947758A (zh) | 一种催化二苯并呋喃开环制备联苯的方法 | |
| CN101928222B (zh) | 一种n,n,n’,n’-四异丙基乙二胺的合成方法 | |
| CN102796005B (zh) | N,n'-二异丙基乙二胺的合成方法 | |
| CN108503608B (zh) | 一种1,4-二甲基哌嗪的制备方法 | |
| CN108976105B (zh) | 一种低分子量壬基环己醇聚氧乙烯醚的制备方法 | |
| CN112010730A (zh) | 一种二苯基甲烷的绿色制备方法 | |
| CN108752217B (zh) | 一种度鲁特韦关键中间体2,4-二氟苄胺的合成方法 | |
| CN104628626A (zh) | 一种2,2,6,6-四甲基-4-哌啶醇的制备方法 | |
| CN102304102B (zh) | 一种1-甲基哌嗪的制备方法 | |
| CN102600847B (zh) | 一种合成甲酸甲酯的催化剂及制备方法和应用 | |
| CN113666829B (zh) | 4-氟-n-异丙基苯胺及氟噻草胺的制备方法 | |
| TWI522335B (zh) | 用於藉由氫化1,1-二氟-2-硝基乙烷製備2,2-二氟乙基胺之方法 | |
| CN106631991B (zh) | 一种n-丁基-2,2,6,6-四甲基-4-哌啶胺的简便合成方法 | |
| CN105481702B (zh) | 一锅法合成间氨基苯乙醚的方法 | |
| CN106631819B (zh) | 一种1,2-环己二胺的制备方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C14 | Grant of patent or utility model | ||
| GR01 | Patent grant | ||
| CP01 | Change in the name or title of a patent holder | ||
| CP01 | Change in the name or title of a patent holder |
Address after: 311604 No.588, Nanfeng Road, Meicheng Town, Jiande City, Hangzhou City, Zhejiang Province Patentee after: Jiande Jianye Resource Recycling Technology Co.,Ltd. Address before: 311604 No.588, Nanfeng Road, Meicheng Town, Jiande City, Hangzhou City, Zhejiang Province Patentee before: HANGZHOU XINDE ENVIRONMENTAL PROTECTION TECHNOLOGY Co.,Ltd. |