CN1031264C - 1-3-二氢-4-吡啶酰基-2h-咪唑-2-酮类化合物的制备 - Google Patents
1-3-二氢-4-吡啶酰基-2h-咪唑-2-酮类化合物的制备 Download PDFInfo
- Publication number
- CN1031264C CN1031264C CN 91103558 CN91103558A CN1031264C CN 1031264 C CN1031264 C CN 1031264C CN 91103558 CN91103558 CN 91103558 CN 91103558 A CN91103558 A CN 91103558A CN 1031264 C CN1031264 C CN 1031264C
- Authority
- CN
- China
- Prior art keywords
- ketone
- pyridyl
- acid
- hydroxyl
- imidazoles
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Lifetime
Links
- 238000002360 preparation method Methods 0.000 title description 9
- CLFOXTYGCQWJOE-UHFFFAOYSA-N 4-(1,2,3,4-tetrahydropyridine-4-carbonyl)imidazol-2-one Chemical class N=1C(=O)N=CC=1C(=O)C1CCNC=C1 CLFOXTYGCQWJOE-UHFFFAOYSA-N 0.000 title 1
- 238000000034 method Methods 0.000 claims abstract description 39
- 238000006243 chemical reaction Methods 0.000 claims abstract description 22
- -1 alkaline earth metal cyanate Chemical class 0.000 claims abstract description 16
- 125000000339 4-pyridyl group Chemical group N1=C([H])C([H])=C([*])C([H])=C1[H] 0.000 claims abstract description 13
- 229910052739 hydrogen Inorganic materials 0.000 claims abstract description 13
- 239000001257 hydrogen Substances 0.000 claims abstract description 13
- 125000003349 3-pyridyl group Chemical group N1=C([H])C([*])=C([H])C([H])=C1[H] 0.000 claims abstract description 9
- 125000000217 alkyl group Chemical group 0.000 claims abstract description 6
- 125000004432 carbon atom Chemical group C* 0.000 claims abstract description 6
- 229910052784 alkaline earth metal Inorganic materials 0.000 claims abstract description 4
- KXDHJXZQYSOELW-UHFFFAOYSA-N Carbamic acid Chemical compound NC(O)=O KXDHJXZQYSOELW-UHFFFAOYSA-N 0.000 claims description 10
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 10
- 229910052799 carbon Inorganic materials 0.000 claims description 5
- 229910052728 basic metal Inorganic materials 0.000 claims description 3
- 150000003818 basic metals Chemical class 0.000 claims description 3
- GKKCIDNWFBPDBW-UHFFFAOYSA-M potassium cyanate Chemical class [K]OC#N GKKCIDNWFBPDBW-UHFFFAOYSA-M 0.000 claims description 3
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 claims description 2
- 125000004105 2-pyridyl group Chemical group N1=C([*])C([H])=C([H])C([H])=C1[H] 0.000 abstract description 6
- IBTSIFIGTARYRB-UHFFFAOYSA-N 1,3-dihydroxyurea Chemical compound ONC(=O)NO IBTSIFIGTARYRB-UHFFFAOYSA-N 0.000 abstract 1
- 229910052783 alkali metal Inorganic materials 0.000 abstract 1
- 150000001340 alkali metals Chemical class 0.000 abstract 1
- 125000004435 hydrogen atom Chemical group [H]* 0.000 abstract 1
- 238000011084 recovery Methods 0.000 abstract 1
- OFBQJSOFQDEBGM-UHFFFAOYSA-N Pentane Chemical compound CCCCC OFBQJSOFQDEBGM-UHFFFAOYSA-N 0.000 description 49
- 239000002904 solvent Substances 0.000 description 23
- 150000001875 compounds Chemical class 0.000 description 21
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 18
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 18
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 18
- 239000002253 acid Substances 0.000 description 17
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 17
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 14
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 13
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 13
- JUJWROOIHBZHMG-UHFFFAOYSA-N pyridine Substances C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 12
- 230000002829 reductive effect Effects 0.000 description 12
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 10
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 10
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 9
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 9
- 235000019253 formic acid Nutrition 0.000 description 9
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 9
- 150000003839 salts Chemical class 0.000 description 9
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 8
- 239000000047 product Substances 0.000 description 8
- 239000000243 solution Substances 0.000 description 8
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 8
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 6
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 6
- JRNVZBWKYDBUCA-UHFFFAOYSA-N N-chlorosuccinimide Chemical compound ClN1C(=O)CCC1=O JRNVZBWKYDBUCA-UHFFFAOYSA-N 0.000 description 6
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 6
- 235000019441 ethanol Nutrition 0.000 description 6
- 239000000376 reactant Substances 0.000 description 6
- 239000007787 solid Substances 0.000 description 6
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 5
- XLJMAIOERFSOGZ-UHFFFAOYSA-N anhydrous cyanic acid Natural products OC#N XLJMAIOERFSOGZ-UHFFFAOYSA-N 0.000 description 5
- 239000003795 chemical substances by application Substances 0.000 description 5
- XLJMAIOERFSOGZ-UHFFFAOYSA-M cyanate Chemical compound [O-]C#N XLJMAIOERFSOGZ-UHFFFAOYSA-M 0.000 description 5
- 239000000203 mixture Substances 0.000 description 5
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 4
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 4
- XEEYBQQBJWHFJM-UHFFFAOYSA-N Iron Chemical compound [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 description 4
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 4
- ATUOYWHBWRKTHZ-UHFFFAOYSA-N Propane Chemical compound CCC ATUOYWHBWRKTHZ-UHFFFAOYSA-N 0.000 description 4
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 4
- 239000003054 catalyst Substances 0.000 description 4
- 238000001816 cooling Methods 0.000 description 4
- UKVIEHSSVKSQBA-UHFFFAOYSA-N methane;palladium Chemical compound C.[Pd] UKVIEHSSVKSQBA-UHFFFAOYSA-N 0.000 description 4
- 150000007524 organic acids Chemical class 0.000 description 4
- 230000003647 oxidation Effects 0.000 description 4
- 238000007254 oxidation reaction Methods 0.000 description 4
- 239000011541 reaction mixture Substances 0.000 description 4
- 229910052708 sodium Inorganic materials 0.000 description 4
- 239000011734 sodium Substances 0.000 description 4
- 238000003756 stirring Methods 0.000 description 4
- 229910052725 zinc Inorganic materials 0.000 description 4
- 239000011701 zinc Substances 0.000 description 4
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 3
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 3
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 3
- 239000004642 Polyimide Substances 0.000 description 3
- 239000003513 alkali Substances 0.000 description 3
- 239000007864 aqueous solution Substances 0.000 description 3
- 239000001273 butane Substances 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- 238000001914 filtration Methods 0.000 description 3
- 125000004029 hydroxymethyl group Chemical group [H]OC([H])([H])* 0.000 description 3
- 229910052500 inorganic mineral Inorganic materials 0.000 description 3
- 239000011707 mineral Substances 0.000 description 3
- 235000010755 mineral Nutrition 0.000 description 3
- IJDNQMDRQITEOD-UHFFFAOYSA-N n-butane Chemical compound CCCC IJDNQMDRQITEOD-UHFFFAOYSA-N 0.000 description 3
- 229920001721 polyimide Polymers 0.000 description 3
- 239000001294 propane Substances 0.000 description 3
- 230000035484 reaction time Effects 0.000 description 3
- 238000006722 reduction reaction Methods 0.000 description 3
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 2
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 2
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 2
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 2
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 2
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 2
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 2
- PCLIMKBDDGJMGD-UHFFFAOYSA-N N-bromosuccinimide Chemical compound BrN1C(=O)CCC1=O PCLIMKBDDGJMGD-UHFFFAOYSA-N 0.000 description 2
- YIKSCQDJHCMVMK-UHFFFAOYSA-N Oxamide Chemical compound NC(=O)C(N)=O YIKSCQDJHCMVMK-UHFFFAOYSA-N 0.000 description 2
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 2
- KFSLWBXXFJQRDL-UHFFFAOYSA-N Peracetic acid Chemical compound CC(=O)OO KFSLWBXXFJQRDL-UHFFFAOYSA-N 0.000 description 2
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 2
- LCTONWCANYUPML-UHFFFAOYSA-N Pyruvic acid Chemical compound CC(=O)C(O)=O LCTONWCANYUPML-UHFFFAOYSA-N 0.000 description 2
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 2
- ATJFFYVFTNAWJD-UHFFFAOYSA-N Tin Chemical compound [Sn] ATJFFYVFTNAWJD-UHFFFAOYSA-N 0.000 description 2
- 229910052782 aluminium Inorganic materials 0.000 description 2
- 239000004411 aluminium Substances 0.000 description 2
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 2
- VSCWAEJMTAWNJL-UHFFFAOYSA-K aluminium trichloride Chemical compound Cl[Al](Cl)Cl VSCWAEJMTAWNJL-UHFFFAOYSA-K 0.000 description 2
- 239000011575 calcium Substances 0.000 description 2
- 229910052791 calcium Inorganic materials 0.000 description 2
- 238000001035 drying Methods 0.000 description 2
- JFCQEDHGNNZCLN-UHFFFAOYSA-N glutaric acid Chemical compound OC(=O)CCCC(O)=O JFCQEDHGNNZCLN-UHFFFAOYSA-N 0.000 description 2
- 150000002431 hydrogen Chemical class 0.000 description 2
- 229910052742 iron Inorganic materials 0.000 description 2
- JVTAAEKCZFNVCJ-UHFFFAOYSA-N lactic acid Chemical compound CC(O)C(O)=O JVTAAEKCZFNVCJ-UHFFFAOYSA-N 0.000 description 2
- 229910052744 lithium Inorganic materials 0.000 description 2
- 229910052751 metal Inorganic materials 0.000 description 2
- 239000002184 metal Substances 0.000 description 2
- 150000007522 mineralic acids Chemical class 0.000 description 2
- 238000002156 mixing Methods 0.000 description 2
- 235000011007 phosphoric acid Nutrition 0.000 description 2
- BASFCYQUMIYNBI-UHFFFAOYSA-N platinum Chemical compound [Pt] BASFCYQUMIYNBI-UHFFFAOYSA-N 0.000 description 2
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 2
- 238000001953 recrystallisation Methods 0.000 description 2
- 238000007086 side reaction Methods 0.000 description 2
- 239000012265 solid product Substances 0.000 description 2
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 2
- 239000011135 tin Substances 0.000 description 2
- 229910052718 tin Inorganic materials 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- NQBWNECTZUOWID-UHFFFAOYSA-N (E)-cinnamyl (E)-cinnamate Natural products C=1C=CC=CC=1C=CC(=O)OCC=CC1=CC=CC=C1 NQBWNECTZUOWID-UHFFFAOYSA-N 0.000 description 1
- UWYVPFMHMJIBHE-OWOJBTEDSA-N (e)-2-hydroxybut-2-enedioic acid Chemical compound OC(=O)\C=C(\O)C(O)=O UWYVPFMHMJIBHE-OWOJBTEDSA-N 0.000 description 1
- KEQGZUUPPQEDPF-UHFFFAOYSA-N 1,3-dichloro-5,5-dimethylimidazolidine-2,4-dione Chemical compound CC1(C)N(Cl)C(=O)N(Cl)C1=O KEQGZUUPPQEDPF-UHFFFAOYSA-N 0.000 description 1
- HTSGKJQDMSTCGS-UHFFFAOYSA-N 1,4-bis(4-chlorophenyl)-2-(4-methylphenyl)sulfonylbutane-1,4-dione Chemical compound C1=CC(C)=CC=C1S(=O)(=O)C(C(=O)C=1C=CC(Cl)=CC=1)CC(=O)C1=CC=C(Cl)C=C1 HTSGKJQDMSTCGS-UHFFFAOYSA-N 0.000 description 1
- CQVKMVQRSNNAGO-UHFFFAOYSA-N 2-[4-formyl-3-methyl-n-(2-methylsulfonyloxyethyl)anilino]ethyl methanesulfonate Chemical compound CC1=CC(N(CCOS(C)(=O)=O)CCOS(C)(=O)=O)=CC=C1C=O CQVKMVQRSNNAGO-UHFFFAOYSA-N 0.000 description 1
- JHWIEAWILPSRMU-UHFFFAOYSA-N 2-methyl-3-pyrimidin-4-ylpropanoic acid Chemical compound OC(=O)C(C)CC1=CC=NC=N1 JHWIEAWILPSRMU-UHFFFAOYSA-N 0.000 description 1
- PKRSYEPBQPFNRB-UHFFFAOYSA-N 2-phenoxybenzoic acid Chemical compound OC(=O)C1=CC=CC=C1OC1=CC=CC=C1 PKRSYEPBQPFNRB-UHFFFAOYSA-N 0.000 description 1
- JHUUPUMBZGWODW-UHFFFAOYSA-N 3,6-dihydro-1,2-dioxine Chemical compound C1OOCC=C1 JHUUPUMBZGWODW-UHFFFAOYSA-N 0.000 description 1
- AQZMINLSVARCSL-UHFFFAOYSA-N 4-chloro-3,6-dioxocyclohexa-1,4-diene-1,2-dicarbonitrile Chemical class ClC1=CC(=O)C(C#N)=C(C#N)C1=O AQZMINLSVARCSL-UHFFFAOYSA-N 0.000 description 1
- FJKROLUGYXJWQN-UHFFFAOYSA-N 4-hydroxybenzoic acid Chemical compound OC(=O)C1=CC=C(O)C=C1 FJKROLUGYXJWQN-UHFFFAOYSA-N 0.000 description 1
- 229910000497 Amalgam Inorganic materials 0.000 description 1
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 1
- 239000004135 Bone phosphate Substances 0.000 description 1
- GKRYOTUQPZKDFB-UHFFFAOYSA-N C1=CC=CC=C1C(=O)OO.[Cl] Chemical compound C1=CC=CC=C1C(=O)OO.[Cl] GKRYOTUQPZKDFB-UHFFFAOYSA-N 0.000 description 1
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 1
- 239000012027 Collins reagent Substances 0.000 description 1
- FKLJPTJMIBLJAV-UHFFFAOYSA-N Compound IV Chemical compound O1N=C(C)C=C1CCCCCCCOC1=CC=C(C=2OCCN=2)C=C1 FKLJPTJMIBLJAV-UHFFFAOYSA-N 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- 206010019280 Heart failures Diseases 0.000 description 1
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 1
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 description 1
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 1
- PXHVJJICTQNCMI-UHFFFAOYSA-N Nickel Chemical compound [Ni] PXHVJJICTQNCMI-UHFFFAOYSA-N 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- WCUXLLCKKVVCTQ-UHFFFAOYSA-M Potassium chloride Chemical compound [Cl-].[K+] WCUXLLCKKVVCTQ-UHFFFAOYSA-M 0.000 description 1
- 239000007868 Raney catalyst Substances 0.000 description 1
- 229910000564 Raney nickel Inorganic materials 0.000 description 1
- KJTLSVCANCCWHF-UHFFFAOYSA-N Ruthenium Chemical compound [Ru] KJTLSVCANCCWHF-UHFFFAOYSA-N 0.000 description 1
- VMHLLURERBWHNL-UHFFFAOYSA-M Sodium acetate Chemical compound [Na+].CC([O-])=O VMHLLURERBWHNL-UHFFFAOYSA-M 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-N Succinic acid Natural products OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 1
- 239000000654 additive Substances 0.000 description 1
- 230000000996 additive effect Effects 0.000 description 1
- 150000008431 aliphatic amides Chemical class 0.000 description 1
- 150000001342 alkaline earth metals Chemical class 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- 229910021529 ammonia Inorganic materials 0.000 description 1
- 239000003125 aqueous solvent Substances 0.000 description 1
- 230000009286 beneficial effect Effects 0.000 description 1
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 1
- CUBCNYWQJHBXIY-UHFFFAOYSA-N benzoic acid;2-hydroxybenzoic acid Chemical compound OC(=O)C1=CC=CC=C1.OC(=O)C1=CC=CC=C1O CUBCNYWQJHBXIY-UHFFFAOYSA-N 0.000 description 1
- KDYFGRWQOYBRFD-NUQCWPJISA-N butanedioic acid Chemical compound O[14C](=O)CC[14C](O)=O KDYFGRWQOYBRFD-NUQCWPJISA-N 0.000 description 1
- 235000011089 carbon dioxide Nutrition 0.000 description 1
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 1
- 230000003177 cardiotonic effect Effects 0.000 description 1
- 238000010531 catalytic reduction reaction Methods 0.000 description 1
- 239000007795 chemical reaction product Substances 0.000 description 1
- 239000003153 chemical reaction reagent Substances 0.000 description 1
- KRVSOGSZCMJSLX-UHFFFAOYSA-L chromic acid Substances O[Cr](O)(=O)=O KRVSOGSZCMJSLX-UHFFFAOYSA-L 0.000 description 1
- VTHIKKVKIVQWHV-UHFFFAOYSA-N chromium(6+) oxygen(2-) pyridine Chemical compound [O-2].[O-2].[O-2].[Cr+6].C1=CC=NC=C1 VTHIKKVKIVQWHV-UHFFFAOYSA-N 0.000 description 1
- NQBWNECTZUOWID-QSYVVUFSSA-N cinnamyl cinnamate Chemical compound C=1C=CC=CC=1\C=C/C(=O)OC\C=C\C1=CC=CC=C1 NQBWNECTZUOWID-QSYVVUFSSA-N 0.000 description 1
- 229940125904 compound 1 Drugs 0.000 description 1
- 235000019628 coolness Nutrition 0.000 description 1
- 239000006184 cosolvent Substances 0.000 description 1
- 239000013078 crystal Substances 0.000 description 1
- 230000006837 decompression Effects 0.000 description 1
- 125000005594 diketone group Chemical group 0.000 description 1
- 238000004090 dissolution Methods 0.000 description 1
- 230000008030 elimination Effects 0.000 description 1
- 238000003379 elimination reaction Methods 0.000 description 1
- 150000002170 ethers Chemical class 0.000 description 1
- HCPOCMMGKBZWSJ-UHFFFAOYSA-N ethyl 3-hydrazinyl-3-oxopropanoate Chemical compound CCOC(=O)CC(=O)NN HCPOCMMGKBZWSJ-UHFFFAOYSA-N 0.000 description 1
- 239000000706 filtrate Substances 0.000 description 1
- RNGGTCZLJZVJDO-UHFFFAOYSA-N formic acid;zinc Chemical compound [Zn].OC=O RNGGTCZLJZVJDO-UHFFFAOYSA-N 0.000 description 1
- 239000001530 fumaric acid Substances 0.000 description 1
- AWJWCTOOIBYHON-UHFFFAOYSA-N furo[3,4-b]pyrazine-5,7-dione Chemical compound C1=CN=C2C(=O)OC(=O)C2=N1 AWJWCTOOIBYHON-UHFFFAOYSA-N 0.000 description 1
- 239000007789 gas Substances 0.000 description 1
- 238000010438 heat treatment Methods 0.000 description 1
- 229910000042 hydrogen bromide Inorganic materials 0.000 description 1
- IXCSERBJSXMMFS-UHFFFAOYSA-N hydrogen chloride Substances Cl.Cl IXCSERBJSXMMFS-UHFFFAOYSA-N 0.000 description 1
- 229910000041 hydrogen chloride Inorganic materials 0.000 description 1
- 238000005984 hydrogenation reaction Methods 0.000 description 1
- PHTQWCKDNZKARW-UHFFFAOYSA-N isoamylol Chemical compound CC(C)CCO PHTQWCKDNZKARW-UHFFFAOYSA-N 0.000 description 1
- 125000000468 ketone group Chemical group 0.000 description 1
- 150000002576 ketones Chemical class 0.000 description 1
- 239000004310 lactic acid Substances 0.000 description 1
- 235000014655 lactic acid Nutrition 0.000 description 1
- 239000012280 lithium aluminium hydride Substances 0.000 description 1
- 239000011777 magnesium Substances 0.000 description 1
- 229910052749 magnesium Inorganic materials 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- 238000004519 manufacturing process Methods 0.000 description 1
- 229910052987 metal hydride Inorganic materials 0.000 description 1
- 150000004681 metal hydrides Chemical class 0.000 description 1
- 150000002739 metals Chemical class 0.000 description 1
- 150000002762 monocarboxylic acid derivatives Chemical class 0.000 description 1
- WWECJGLXBSQKRF-UHFFFAOYSA-N n,n-dimethylformamide;methanol Chemical compound OC.CN(C)C=O WWECJGLXBSQKRF-UHFFFAOYSA-N 0.000 description 1
- HSPSCWZIJWKZKD-UHFFFAOYSA-N n-chloroacetamide Chemical compound CC(=O)NCl HSPSCWZIJWKZKD-UHFFFAOYSA-N 0.000 description 1
- 238000007034 nitrosation reaction Methods 0.000 description 1
- 238000006053 organic reaction Methods 0.000 description 1
- 239000003960 organic solvent Substances 0.000 description 1
- 150000002923 oximes Chemical class 0.000 description 1
- MUMZUERVLWJKNR-UHFFFAOYSA-N oxoplatinum Chemical compound [Pt]=O MUMZUERVLWJKNR-UHFFFAOYSA-N 0.000 description 1
- 229910003445 palladium oxide Inorganic materials 0.000 description 1
- SHTYJBQEEQBWSX-UHFFFAOYSA-N pentane;dihydrochloride Chemical compound Cl.Cl.CCCCC SHTYJBQEEQBWSX-UHFFFAOYSA-N 0.000 description 1
- SIOXPEMLGUPBBT-UHFFFAOYSA-N picolinic acid Chemical compound OC(=O)C1=CC=CC=N1 SIOXPEMLGUPBBT-UHFFFAOYSA-N 0.000 description 1
- 229910052697 platinum Inorganic materials 0.000 description 1
- 229910003446 platinum oxide Inorganic materials 0.000 description 1
- 238000000247 postprecipitation Methods 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- CHKVPAROMQMJNQ-UHFFFAOYSA-M potassium bisulfate Chemical compound [K+].OS([O-])(=O)=O CHKVPAROMQMJNQ-UHFFFAOYSA-M 0.000 description 1
- 239000001103 potassium chloride Substances 0.000 description 1
- 235000011164 potassium chloride Nutrition 0.000 description 1
- 239000012286 potassium permanganate Substances 0.000 description 1
- NPRDHMWYZHSAHR-UHFFFAOYSA-N pyridine;trioxochromium Chemical compound O=[Cr](=O)=O.C1=CC=NC=C1.C1=CC=NC=C1 NPRDHMWYZHSAHR-UHFFFAOYSA-N 0.000 description 1
- 229940107700 pyruvic acid Drugs 0.000 description 1
- 239000002994 raw material Substances 0.000 description 1
- 239000010948 rhodium Substances 0.000 description 1
- MHOVAHRLVXNVSD-UHFFFAOYSA-N rhodium atom Chemical compound [Rh] MHOVAHRLVXNVSD-UHFFFAOYSA-N 0.000 description 1
- 229910003450 rhodium oxide Inorganic materials 0.000 description 1
- 238000007363 ring formation reaction Methods 0.000 description 1
- 229910001925 ruthenium oxide Inorganic materials 0.000 description 1
- 238000000926 separation method Methods 0.000 description 1
- 239000001632 sodium acetate Substances 0.000 description 1
- 235000017281 sodium acetate Nutrition 0.000 description 1
- 235000017557 sodium bicarbonate Nutrition 0.000 description 1
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 1
- 229910000033 sodium borohydride Inorganic materials 0.000 description 1
- 239000012279 sodium borohydride Substances 0.000 description 1
- HRZFUMHJMZEROT-UHFFFAOYSA-L sodium disulfite Chemical compound [Na+].[Na+].[O-]S(=O)S([O-])(=O)=O HRZFUMHJMZEROT-UHFFFAOYSA-L 0.000 description 1
- 229940001584 sodium metabisulfite Drugs 0.000 description 1
- 235000010262 sodium metabisulphite Nutrition 0.000 description 1
- 159000000000 sodium salts Chemical class 0.000 description 1
- 239000011949 solid catalyst Substances 0.000 description 1
- 229940095064 tartrate Drugs 0.000 description 1
- UGNWTBMOAKPKBL-UHFFFAOYSA-N tetrachloro-1,4-benzoquinone Chemical compound ClC1=C(Cl)C(=O)C(Cl)=C(Cl)C1=O UGNWTBMOAKPKBL-UHFFFAOYSA-N 0.000 description 1
- 229910052723 transition metal Inorganic materials 0.000 description 1
Landscapes
- Plural Heterocyclic Compounds (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
一种制备通式为的羟基甲基-咪唑-2-酮的方法,包括通式为的羟基氨基酮与碱金属或碱土金属氰酸盐的反应和回收产物,两个通式中的Ar均为2-,3-或4-吡啶基,R1均为氢或1-4个碳原子的烷基。
Description
本发明涉及生产通式I的1,3-二氢-4-吡啶酰基-2H-咪唑-2-酮类化合物及其药用盐的方法。 式中R1是氢或1-4个碳原子的烷基,Ar是一个2-,3-或4-吡啶基。这些化合物,特别是4-乙基-1,3-二氢-5-(4-吡啶酰基)-2H-咪唑-2-酮具有显著的强心作用,是治疗心脏衰竭的有效药剂。
式中R1是氢或1-4个碳原子的烷基,Ar是一个2-,3-或4-吡啶基。这些化合物,特别是4-乙基-1,3-二氢-5-(4-吡啶酰基)-2H-咪唑-2-酮具有显著的强心作用,是治疗心脏衰竭的有效药剂。
在现有技术中已有几种制备这些化合物的方法。其中之一是,在路易士酸催化剂特别是在氯化铝存在下,咪唑-2-酮与吡啶酰氯或吡啶酰溴或者呲啶羧酸或吡啶酸酐反应。此方法存在一些严重的问题,包括与固体铝络合物混合时的极度困难,以及主要由于反应产物难以从反应罐的固体中分离而导致产率很低。
现有技术的另一方法,例如本公司的专利申请EP59948中所披露,如反应示意式I所示,结构为II的二酮肟还原成结构为III的氨基二酮,然后再与氰酸盐反应得到化合物I。
反应示意式I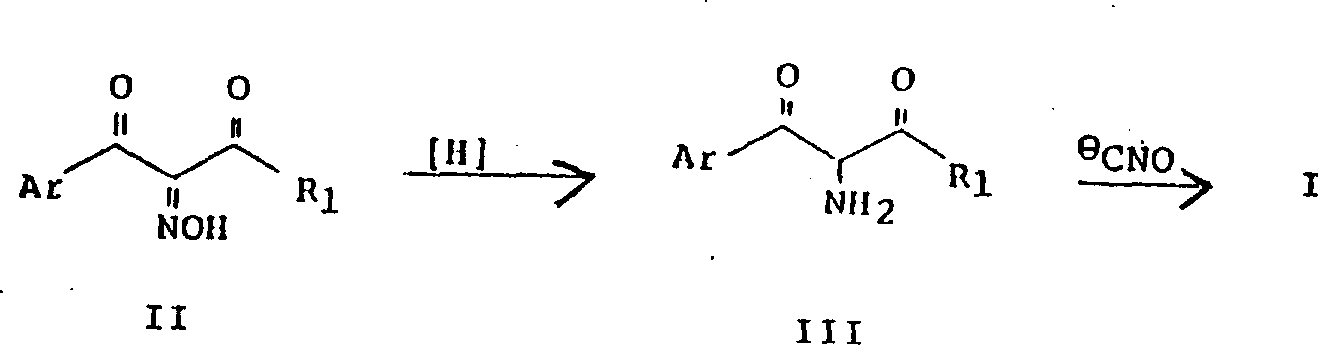 应用这一方法时,也遇到了一些困难。特别是,由于在化合物II中靠近吡啶环的酮基对氢化作用活泼,因此它与肟基同时被还原,得到羟基氨基酮IV。
应用这一方法时,也遇到了一些困难。特别是,由于在化合物II中靠近吡啶环的酮基对氢化作用活泼,因此它与肟基同时被还原,得到羟基氨基酮IV。 这个副反应导致必须从氢化反应混合物中除去生成的化合物VI,致使所要化合物I的总产率降低。
这个副反应导致必须从氢化反应混合物中除去生成的化合物VI,致使所要化合物I的总产率降低。
申请人发现,通过下面示意式II的反应途径,可以提高所需产物I的产率。
反应示意式II
此改进的反应流程,无需除去上一示意式中副反应所生成的羟基氨基酮IV。在反应示意式II中羟基氨基酮VI与氰酸根离子反应生成环化产物,其产率比前面提到的氨基二酮III与氰酸根离子反应生成环化产物的产率高得多。
申请人意外地发现,尽管示意式II的方法多了一步在示意式I中没有的反应,即需要把结构为V的醇氧化成需要的吡啶酰基咪唑-2-酮,但当应用示意式II时,化合物II以高得多的总产率转化成吡啶酰基咪唑-2-酮I。
根据本发明反应示意式II所阐明的原理,吡啶酰基咪唑-2-酮类化合物I,可由二酮肟类化合物II经三步反应而制得。更具体地说本发明的方法包括用适当的还原剂将二酮肟II还原成羟基氨基酮IV,以氰酸盐环化后再氧化成所需要的吡啶酰基咪唑-2-酮I。
申请人发现了用二酮肟II相继通过还原,环化和氧化三步来制备吡啶酰基咪唑-2-酮I的方法。由于产率高于其它已知方法,所以本发明的方法更适合于化合物I的大批量生产。
文中所说的“1-4个碳原子的烷基”,是指甲基、乙基、丙基、异丙基、正-丁基,或异丁基。
原料二酮肟II容易用任何已知的适当方法制得,例如,将相应的二酮VI亚硝化,其中R1为氢或1-4个碳原子的烷基,Ar是2-,3-,或4-吡啶基。适宜的亚硝化反应,可参看O.Tousler在“Organic Reaction”V VIIP.327-337中所叙的方法。
将二酮肟II还原成羟基氨基酮IV,可通过使用适当的还原剂采用已知方法来达到。申请人使用的还原剂是,a)氢气,用乙酸,接着用稀酸处理过的10%钯碳作为催化剂,或者b)金属锌和甲酸或乙酸。显然还有无数种其它适宜还原剂能将二酮肟II转化成烃基氨基酮VI。由于作为游离碱分离时,羟基氨基酮不稳定,所以化合物VI以酸的加成盐形式来分离是较得当的。能生成适当盐的无机酸有盐酸、氢溴酸、硫酸和磷酸,以及一些酸式金属盐,例如正磷酸~氢钠和硫酸氢钾。能生成适当盐的有机酸有一元羧酸、二元羧酸、三元羧酸。例如乙酸、乙醇酸、乳酸、丙酮酸、丙二酸、丁二酸、戊二酸、反丁烯二酸、苹果酸、酒石酸、柠檬酸、抗坏血酸、顺丁烯二酸、羟基顺丁烯二酸、苯甲酸、羟基苯甲酸、苯乙酸、肉桂酸、水杨酸、2-苯氧基苯甲酸和磺酸类化合物,如甲磺酸和2-羟基乙磺酸。生成的一酸盐或二酸盐可以水合形式或实际上无水形式存在。一般来说,这些化合物的酸加成盐是晶体,溶于水和各种亲水性有机溶剂中,与其游离碱形式相比,实际上更稳定。
还原二酮肟II的适宜方法包括用金属氢化物还原,如用氢化铝锂或硼氢化钠;应用氢气和金属催化剂,如阮内镍、铂、钯、铑、钌和氧化铂催化还原;应用锂、钠、钾、钙、锌、镁、锡或铁在液氨或低分子量脂肪胺中或者钠、铝、或锌汞齐、锌、锡或铁在羟基溶剂中或在含水无机酸或有机酸如甲酸、乙酸或盐酸存在下的溶解金属法还原。
申请人已经通过在甲酸中加入锌粉的方法将二酮肟II还原制备了羟基氨基酮化合物IV。把要还原的二酮肟溶解在适当的非活性溶剂中,如乙醇、异丙醇、正丁醇、异戊醇、水、无机酸水溶液,如盐酸、硫酸、或有机酸,如乙酸、甲磺酸、最好是甲酸。然后加酸,如盐酸或甲磺酸,最好是加甲磺酸到溶解了的反应物中。将所得的溶液慢慢加到金属还原物(最好是锌粉)与甲酸混合的桨状物中,搅拌直至反应完全,一般需用5分钟到10小时,最好是1-2小时。反应时间主要取决于反应物、溶剂和温度。温度可以从0℃到150℃,最好是25℃-80℃。产物可以游离碱形式从反应混合物中分离,但最好是用现有技术常用的方法,以酸加成盐的形式分离。例如,加含10%甲醇的异丙醇到浓的残留物中,羟基氨基酮就从溶液中沉淀出,然后过滤分离。
另外,申请人利用氢气和钯碳催化剂,最好是10%钯碳催化剂,还原二酮肟II,制备了羟基氨基酮类化合物IV。将被还原的二酮肟溶解在适当的溶剂中,加入少量催化剂,最好是小于被还原化合物重量的10%,反应一直进行到消耗掉3个当量的氢气。反应时间取决于被还原的化合物,氢气的压力,溶剂和温度。氢气压力可以是1-10个大气压,最好是一个大气压。温度可以是0℃-50℃,最好是25℃左右。适宜的溶剂包括任何非活性溶剂,如乙酸乙酯、乙醇、水、最好是乙酸。反应完全后,将盐酸或其它适当的无机酸加到反应混合物中,然后,滤除固体催化剂。羟基氨基酮或其酸加成盐可以通过适当方法回收。例如,简单的溶剂除去法。
普通工作人员就可通过任何适当的方法,将羟基氨基酮IV与氰酸根离子反应生成环化成羟基甲基咪唑-2-酮V。羟基氨基酮与1-5个克当量,最好是约两个克当量的氰酸盐反应5分钟到约24小时。反应时间的长短取决于反应物、溶剂和温度。温度可以从-78℃到100℃左右,最好在0℃-50℃左右。此反应的适宜溶剂是任何非活性溶剂,例如水,或能溶于水的溶剂,如乙酸(有机酸),甲醇或乙醇(醇),或者乙醚、四氢呋喃、1,4-二氧六环等醚类。最好是任一用水混合的非水溶剂。最可取的溶剂是水。任何氰酸根源都可以用于此环化反应中。申请人用的是氰酸钾,但是,任何简单碱金属或碱土金属如锂、钠或钙的氰酸盐,过渡族金属的氰酸盐也可应用。
此反应的产物或它的酸加成盐可以通过已知方法分离。例如,转化成相应的钠盐或钾盐,然后用二氧化碳或无机酸如稀盐酸使之再沉淀。反应示意式II的最后一步是将羟基咪唑-2-酮V氧化成所需的吡啶酰基咪唑-2-酮I。这可通过熟悉这一技术领域的人所知道的合适方法来完成。用于该方法的适宜氧化剂有二氧化锰,在乙酸或丙酮中的酸式铬酸水溶液;在乙酸中的重铬酸钠;三氧化铬吡啶络合物,如Sarrett试剂或Collins试剂;高锰酸钾与硫酸和乙酸水溶液;40%过乙酸;间-氯过苯甲酸;四氯苯醌,2,3-二氯-5,6-二氰基-1,4-苯醌和N-卤代酰亚胺,最好是N-氯代丁二酰亚胺。
申请人选用与以下N-卤代酰亚胺反应来氧化羟基甲基咪唑-2-酮V:1,3-二溴-5,5-二甲基乙内酰脲,1,3-二氯-5,5-二甲基乙内酰脲,N-氯代乙酰胺,N-溴代丁二酰亚胺,最好是N-氯代丁二酰亚胺。氧化可通过以下步骤进行。将被氧化的化合物溶于适当的溶剂中,加入1-5个克当量的N-卤代酰亚胺,最好是1克当量左右。反应温度大约从-78到80℃,反应完全约需要1/2小时到48小时,但这取决于所用的反应物、溶剂和其它反应条件。适宜的溶剂包括任何非活性溶剂,例如,二甲基乙酰胺,甲醇,二甲基甲酰胺,最好是二甲基甲酰胺-甲醇共溶剂。所得产物I可通过通常熟知的适当方法来分离,例如,先沉淀后重结晶的方法。
申请人已通过二氧化锰反应将羟基甲基咪唑-2-酮V氧化。把被氧化的化合物溶于适当的溶剂中,加1个克当量或更多一点的二氧化锰,最好是2或个克当量。反应15分钟到10个小时,最好是大约1-2小时,这主要取决于反应物、溶剂和温度。温度可以是0℃-150℃,最好是大约25℃-80℃。适宜的溶剂有戊烷、氯仿、二氯甲烷、苯、丙酮,最好是乙酸。得到的吡啶酰基咪唑-2-酮可用通常熟知的方法从反应混合物中分离。例如,申请人就是通过过滤和除去溶剂的方法来分离产物。
下面的一些具体例子进一步清楚地说明本发明中所述的方法并陈述发明人为了实施其发明而设想的最佳方案。然而,并不是要用来解释本发明的权利要求范围。
例1
1-(4-吡啶基)-1-羟基-2-氨基-3-酮戊烷的制备
在1000ml乙酸中溶解23.0g(0.11mol)1-(4-吡啶基)-1,3-二酮-2-肟基戊烷。加入1.0g10%钯碳,通入氢气,直至消耗3个克当量氢气。用18.5ml 12N盐酸酸化此混合物,过滤,蒸掉溶剂,得到1-(4-吡啶基)-1-羟基-2-氨基-3-酮戊烷的二盐酸盐;m.p.225℃。
用上面例1的方法,但1-(4-吡啶基)-1,3-二酮-2-肟基戊烷用下面化合物代替:
1-(2-吡啶基)-1,3-二酮-2-肟基戊烷;
1-(4-吡啶基)-1,3-二酮-2-肟基丁烷;
1-(3-吡啶基)-1,3-二酮-2-肟基丙烷;
1-(4-吡啶基)-1,3-二酮-4-甲基-2-肟基戊烷;或
1-(2-吡啶基)-1,3-二酮-2-肟基庚烷,
则分别得到:
1-(2-吡啶基)-1-羟基-2-氨基-3-酮戊烷;
1-(4-吡啶基)-1-羟基-2-氨基-3-酮丁烷;
1-(3-吡啶基)-1-羟基-2-氨基-3-酮丙烷;
1-(4-吡啶基)-1-羟基-2-氨基-4-甲基-3-酮戊烷;或
1-(2-吡啶基)-1-羟基-2-氨基-3-酮庚烷。
例2
1-(4-吡啶基)-1-羟基-2-氨基-3-酮戊烷的制备
在20ml乙酸中加热(50℃)溶解1.0g 1-(4-吡啶基)-1,3-二酮-2-肟基戊烷。用干燥氯化氢气酸化,慢慢加入锌粉,搅拌1小时,冷却。加入干燥乙醚,1-(4-吡啶基)-1-羟基-2-氨基-3-酮戊烷则从溶液中析出,得粗固体产物,可在下一步骤中使用,而无需提纯。
应用上述方法,但以如下化合物代替1-(4-吡啶基)-1,3-二酮-2-肟基戊烷;
1-(3-吡啶基)-1,3-二酮-4-甲基-2-肟基己烷;
1-(4-吡啶基)-1,3-二酮-2-肟基庚烷;
1-(3-吡啶基)-1,3-二酮-4-甲基-2-肟基戊烷,
则分别得到:
1-(3-吡啶基)-1-羟基-2-氨基-4-甲基-3-酮己烷;
1-(4-吡啶基)-1-羟基-2-氨基-3-酮戊烷;
1-(3-吡啶基)-1-羟基-2-氨基-4-甲基-3-酮戊烷。
例3
1-(4-吡啶基)-1-羟基-2-氨基-3-酮戊烷的制备
在37.8kg 88%甲酸中溶解7.5kg(91%纯度,36.37mol)1-(4-吡啶基)-1,3-二酮-2-肟基戊烷和7.0kg甲磺酸。将得到的溶液慢慢加到8.3kg锌粉和3 5.kg甲酸的混合浆中。通过适当冷却和控制加入速度,使反应温度保持在60℃左右。此混合物于55℃搅拌2小时,然后,冷却到20℃,滤去固体甲酸锌。在甲酸滤液中加入3.6kg甲磺酸。70℃减压(40mm Hg)除去甲酸。残余物中加入5.9kg甲醇和53.3kg异丙醇的混合溶液,20℃下搅拌4小时。用离心方法收集固体物质,以12.5kg含10%甲醇的异丙醇溶液洗涤,干燥后,得到11.0kg 1-(4-吡啶基)-1-羟基-2-氨基-3-酮戊烷的二甲磺酸盐,产率为87%。
例4
4-乙基-1,3-二氢-5-[羟基(4-吡啶基)-甲基]-2H-咪唑-2-酮的制备
在100ml水中溶解29.0g(0.11mol)1-(4-吡啶基)-1-羟基-2-氨基-3-酮戊烷二盐酸盐和17.9g(0.22mol)氰酸钾。此溶液于50℃温热10分钟,然后室温放置10小时,冷却,收集析出的固体,得到4-乙基-1,3-二氢-5-(羟基(4-吡啶基)-甲基1-2H-咪唑-2-酮。m.p.234-36℃
按照上面例4所述方法,但1-(4-吡啶基)-1-羟基-2-氨基-3-酮戊烷用以下化合物代替:
1-(2-吡啶基)-1-羟基-2-氨基-3-酮戊烷;
1-(4-吡啶基)-1-羟基-2-氨基-3-酮丁烷;
1-(3-吡啶基)-1-羟基-2-氨基-3-酮丙烷;
1-(4-吡啶基)-1-羟基-2-氨基-4-甲基-3-酮戊烷;
1-(2-吡啶基)-1-羟基-2-氨基-3-酮庚烷。
1-(3-吡啶基)-1-羟基-2-氨基-4-甲基-3-酮己烷;
1-(4-吡啶基)-1-羟基-2-氨基-3-酮庚烷。
1-(3-吡啶基)-1-羟基-2-氨基-4-甲基-3-酮戊烷,
分别得到:
1,3-二氢-4-乙基-5-[羟基(2-吡啶基)甲基]-2H-咪唑-2-酮;
1,3-二氢-4-[羟基(4-吡啶基)甲基]-5-甲基-2H-咪唑-2-酮;
1,3-二氢-4-[羟基(3-吡啶基)甲基]-2H-咪唑-2-酮;
1,3-二氢-4-[羟基(4-吡啶基)甲基]-5-(1-甲基)乙基-2H-咪唑-2-酮;
4-丁基-1,3-二氢-5-[羟基(2-吡啶)甲基]-2H-咪唑-2-酮;
1,3-二氢-4-[羟基(3-吡啶基)甲基]-5-(1-甲基)-丙基-2H-咪唑-2-酮;
4-丁基-1,3-二氢-5-[羟基(4-吡啶基)甲基]-2H-咪唑-2-酮;
1,3-二氢-4-[羟基-(3-吡啶基)甲基]-5-(1-甲基)-乙基-2H-咪唑-2-酮。
例5
1,3-二氢-4-乙基-5-(4-吡啶酰基)-2H-咪唑-2-酮的制备
在25ml乙酸中溶解2.15g(0.009mol)化合物1,并加热至50℃。慢慢加入0.55g(0.006mol)二氧化锰继续加热搅拌30分钟。过滤,蒸去溶剂。将残余物溶于稀(10%)盐酸中,用碳酸氢钠调节至PH4。析出固体1,3-二氢-4-乙基-5-)4-吡啶酰基)-2H-咪唑-2-酮,产物用乙醇重结晶方法提纯,m.p.264℃。
应用上述例4的方法,但1,3-二氢-4-乙基-5-[羟基(4-吡啶基)甲基]-2H-咪唑-2-酮,以如下化合物代替:
1,3-二氢-4-乙基-5-[羟基(2-吡啶基)甲基]-2H-咪唑-2-酮;
1,3-二氢-4-[羟基(4-吡啶基)甲基]-5-甲基-2H-咪唑-2-酮;
1,3-二氢-4-[羟基(3-吡啶基)甲基]-2H-咪唑-2-酮;
1,3-二氢-4-[羟基(4-吡啶基)甲基-5-(1-甲基)乙基-2H-咪唑-2-酮;
4-丁基-1,3-二氢-5-[羟基(2-吡啶基)甲基]-2H-咪唑-2-酮;
1,3-二氢-4-[羟基(3-吡啶基)甲基]-5-(1-甲基)-丙基-2H-咪唑-2-酮;
4-丁基-1,3-二氢-5-[羟基(4-吡啶基)甲基]-2H-咪唑-2-酮;
1,3-二氢-4-[羟基(3-吡啶基)甲基]-5-(1-甲基)-乙基-2H-咪唑-2-酮,
分别得到:
1,3-二氢-4-乙基-5-(2-吡啶酰基)-2H-咪唑-2-酮;
1,3-二氢-4-甲基-5-(4-吡啶酰基)-2H-咪唑-2-酮;
1,3-二氢-4-(3-吡啶酰基)-2H-咪唑-2-酮;
1,3-二氢-4-(1-甲基)乙基-5-(4-吡啶酰基)-2H-咪唑-2-酮;
4-丁基-1,3-二氢-5-(2-吡啶酰基)-2H-咪唑-2-酮;
1,3-二氢-4-(1-甲基)丙基-5-(3-吡啶酰基)-2H-咪唑-2-酮;
4-丁基-1,3-二氢-5-(4-吡啶酰基)-2 H-咪唑-2-酮;
1,3-二氢-4-(1-甲基)乙基-5-(3-吡啶酰基)-2H-咪唑-2-酮。
例6
1,3-二氢-4-乙基-5-(4-吡啶酰基)-2H-咪唑-2-酮的制备。
将2.8kg N-氯代丁二酰亚胺和17.0kg二甲基甲酰胺配成溶液,于0℃,用2小时以上时间,加到由4.6kg 4-乙基-1,3-二氢-5-[羟基(4-吡啶基)甲基]-2H-咪唑-2-酮与2.9kg甲醇和14.0kg二甲基甲酰胺所组成的浆状物中。得到的混合物,于0℃搅拌5小时,然后,于60℃温热3小时。在反应混合物中加入1.7kg乙酸钠和由0.39kg偏亚硫酸氢钠与5.5kg水配成的溶液,于25℃搅拌4小时。真空蒸馏(80℃,24mm Hg),除去21kg溶剂。搅拌下在浓溶液中加入17.5kg水,于-4℃冷却12小时。离心,收集固体产物,干燥后,得到3.3kg 1,3-二氢-4-乙基-5-(4-吡啶酰基)-2H-咪唑-2-酮(产率72%,纯度99%)。
Claims (4)
1.一种制备通式为
 的羟基甲基-咪唑-2-酮的方法,包括通式为
的羟基氨基酮与碱金属或碱土金属氰酸盐的反应和回收产物,两个通式中Ar均为2-,3-或4-吡啶基,R1均为氢或1-4个碳原子的烷基。
的羟基甲基-咪唑-2-酮的方法,包括通式为
的羟基氨基酮与碱金属或碱土金属氰酸盐的反应和回收产物,两个通式中Ar均为2-,3-或4-吡啶基,R1均为氢或1-4个碳原子的烷基。
2.在权利要求1规定的方法中,碱金属或碱土金属氰酸盐是氰酸钾。
3.在权利要求1规定的方法中,Ar是4-吡啶基,R1是1-4个碳原子的烷基。
4.在权利要求3规定的方法中,R1是乙基。
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN 85106722 CN1019197B (zh) | 1984-07-30 | 1985-09-06 | 1-3-二氢-4-吡啶酰基-2h-咪唑-2-酮类化合物的制备 |
Related Parent Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN8510672.2 Division | 1985-09-06 | ||
| CN 85106722 Division CN1019197B (zh) | 1984-07-30 | 1985-09-06 | 1-3-二氢-4-吡啶酰基-2h-咪唑-2-酮类化合物的制备 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN1057261A CN1057261A (zh) | 1991-12-25 |
| CN1031264C true CN1031264C (zh) | 1996-03-13 |
Family
ID=4795253
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN 91103558 Expired - Lifetime CN1031264C (zh) | 1985-09-06 | 1985-09-06 | 1-3-二氢-4-吡啶酰基-2h-咪唑-2-酮类化合物的制备 |
| CN 91103557 Pending CN1055737A (zh) | 1985-09-06 | 1985-09-06 | 1-3-二氢-4-吡啶酰基-2h-咪唑-2-酮类化合物的制备 |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN 91103557 Pending CN1055737A (zh) | 1985-09-06 | 1985-09-06 | 1-3-二氢-4-吡啶酰基-2h-咪唑-2-酮类化合物的制备 |
Country Status (1)
| Country | Link |
|---|---|
| CN (2) | CN1031264C (zh) |
-
1985
- 1985-09-06 CN CN 91103558 patent/CN1031264C/zh not_active Expired - Lifetime
- 1985-09-06 CN CN 91103557 patent/CN1055737A/zh active Pending
Also Published As
| Publication number | Publication date |
|---|---|
| CN1055737A (zh) | 1991-10-30 |
| CN1057261A (zh) | 1991-12-25 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US20100280237A1 (en) | Preparation of metal mesoporphyrin compounds | |
| JP2000516934A (ja) | 4―アセトキシアゼチジノンの立体選択的な製造方法 | |
| JP4309839B2 (ja) | 金属メソポルフィリンハライド化合物の調製 | |
| CN100436426C (zh) | 1,7'-二甲基-2'-丙基-2,5'-双-1h-苯并咪唑的制备和纯化方法 | |
| CN113831280A (zh) | 一种啶酰菌胺的制备方法 | |
| CN1679586A (zh) | 含哌嗪环化合物的新型合成和结晶方法 | |
| CN1031264C (zh) | 1-3-二氢-4-吡啶酰基-2h-咪唑-2-酮类化合物的制备 | |
| CN106431885B (zh) | 顺丁烯二酸酐混合溶剂臭氧化合成乙醛酸的方法 | |
| CN1019197B (zh) | 1-3-二氢-4-吡啶酰基-2h-咪唑-2-酮类化合物的制备 | |
| CN115197119B (zh) | 一种6,6-二甲基-3-氮杂双环[3.1.0]己烷-2,4-二酮的制备方法 | |
| CN117964467A (zh) | 一种制备4,4,4-三氟-1-(4-甲苯基)-1,3-丁二酮的方法 | |
| CN108409561B (zh) | 一种5-氨基酮戊酸盐酸盐及中间体的制备方法 | |
| JP3282372B2 (ja) | ピペロナールの製法 | |
| CN109096105B (zh) | 烯基活泼亚甲基化合物的还原方法及还原产物 | |
| JP2001278867A (ja) | 環式酸の製造 | |
| CN114890942A (zh) | 3-溴-6-氯吡啶-2-甲酸及其制备方法 | |
| US20210198176A1 (en) | Reduction method and reduction product of alkenyl active methylene compound | |
| JP2012020970A (ja) | {2−アミノ−1,4−ジヒドロ−6−メチル−4−(3−ニトロフェニル)−3,5−ピリジンジカルボン酸3−(1−ジフェニルメチルアゼチジン−3−イル)エステル5−イソプロピルエステル}の製造方法 | |
| KR910006636B1 (ko) | I,3-디하이드로-4-피리도일-2h-이미다졸-2-온의 제조방법 | |
| CN114920764A (zh) | 一种三仲丁基硼氢化锂的制备及其在制备抗菌剂中的应用 | |
| EP1757597B1 (en) | Method for producing 2-oxo-1-phenyl-3-oxabicyclo[3.1.0]hexane | |
| US4803278A (en) | Preparation of 1,3-dihydro-4-pyridoyl-2H-imidazol-2-ones | |
| CN112142728B (zh) | 一种咪草烟中间体及其合成方法和应用 | |
| JP4796776B2 (ja) | 4,4’−ジカルボキシ−2,2’−ビピリジンの製造方法 | |
| CN121800781A (zh) | 一种非奈利酮中间体的制备方法及其应用 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C14 | Grant of patent or utility model | ||
| GR01 | Patent grant | ||
| C17 | Cessation of patent right | ||
| CX01 | Expiry of patent term |