CN1042425C - 3-羟基-4-羟甲基-2-亚甲基环戊基嘌呤和嘧啶 - Google Patents
3-羟基-4-羟甲基-2-亚甲基环戊基嘌呤和嘧啶 Download PDFInfo
- Publication number
- CN1042425C CN1042425C CN94107231A CN94107231A CN1042425C CN 1042425 C CN1042425 C CN 1042425C CN 94107231 A CN94107231 A CN 94107231A CN 94107231 A CN94107231 A CN 94107231A CN 1042425 C CN1042425 C CN 1042425C
- Authority
- CN
- China
- Prior art keywords
- formula
- compound
- compounds
- methyl
- hydrogen
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
- ZBJCOKSYDWTPOC-UHFFFAOYSA-N 5-(hydroxymethyl)-2-methylidene-3-(7h-purin-2-yl)cyclopentan-1-ol Chemical class C=C1C(O)C(CO)CC1C1=NC=C(NC=N2)C2=N1 ZBJCOKSYDWTPOC-UHFFFAOYSA-N 0.000 title 1
- 150000003230 pyrimidines Chemical class 0.000 title 1
- 150000001875 compounds Chemical class 0.000 claims abstract description 310
- 150000003839 salts Chemical class 0.000 claims abstract description 17
- OKKJLVBELUTLKV-UHFFFAOYSA-N methanol Natural products OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 claims description 91
- 239000001257 hydrogen Substances 0.000 claims description 62
- 229910052739 hydrogen Inorganic materials 0.000 claims description 62
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 49
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 25
- 238000002360 preparation method Methods 0.000 claims description 25
- 150000002431 hydrogen Chemical class 0.000 claims description 24
- -1 iodine, hydrogen Chemical class 0.000 claims description 23
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 claims description 22
- 229910052794 bromium Inorganic materials 0.000 claims description 22
- 239000000460 chlorine Substances 0.000 claims description 19
- 229910052801 chlorine Inorganic materials 0.000 claims description 19
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 claims description 19
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 claims description 18
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 claims description 17
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 claims description 15
- 125000004777 2-fluoroethyl group Chemical group [H]C([H])(F)C([H])([H])* 0.000 claims description 14
- 125000001340 2-chloroethyl group Chemical group [H]C([H])(Cl)C([H])([H])* 0.000 claims description 13
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 claims description 12
- 125000000217 alkyl group Chemical group 0.000 claims description 12
- 229910052731 fluorine Inorganic materials 0.000 claims description 12
- 239000011737 fluorine Substances 0.000 claims description 12
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 claims description 10
- HSFWRNGVRCDJHI-UHFFFAOYSA-N alpha-acetylene Natural products C#C HSFWRNGVRCDJHI-UHFFFAOYSA-N 0.000 claims description 9
- 125000002534 ethynyl group Chemical group [H]C#C* 0.000 claims description 9
- 230000000840 anti-viral effect Effects 0.000 claims description 4
- 125000003118 aryl group Chemical group 0.000 claims description 4
- RGSFGYAAUTVSQA-UHFFFAOYSA-N pentamethylene Natural products C1CCCC1 RGSFGYAAUTVSQA-UHFFFAOYSA-N 0.000 claims description 3
- UYLWKSJTHLRFBX-UHFFFAOYSA-N purin-6-one Chemical compound O=C1N=CN=C2N=CN=C12 UYLWKSJTHLRFBX-UHFFFAOYSA-N 0.000 claims description 3
- 239000008194 pharmaceutical composition Substances 0.000 claims 1
- 125000000547 substituted alkyl group Chemical group 0.000 claims 1
- 230000002155 anti-virotic effect Effects 0.000 abstract 1
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 99
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 87
- 239000000243 solution Substances 0.000 description 67
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 63
- 238000000034 method Methods 0.000 description 51
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 43
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 39
- 239000000203 mixture Substances 0.000 description 37
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 36
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 34
- 238000006243 chemical reaction Methods 0.000 description 34
- 239000000047 product Substances 0.000 description 34
- 238000003756 stirring Methods 0.000 description 32
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 28
- 239000012044 organic layer Substances 0.000 description 27
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 26
- 239000007787 solid Substances 0.000 description 25
- 125000006239 protecting group Chemical group 0.000 description 24
- 125000000051 benzyloxy group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])O* 0.000 description 23
- 238000003818 flash chromatography Methods 0.000 description 23
- 239000003921 oil Substances 0.000 description 23
- 235000019198 oils Nutrition 0.000 description 23
- 239000011541 reaction mixture Substances 0.000 description 22
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 21
- 239000010410 layer Substances 0.000 description 21
- 239000000377 silicon dioxide Substances 0.000 description 20
- FAQYAMRNWDIXMY-UHFFFAOYSA-N trichloroborane Chemical compound ClB(Cl)Cl FAQYAMRNWDIXMY-UHFFFAOYSA-N 0.000 description 20
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical class [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 18
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 18
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 18
- 125000002221 trityl group Chemical group [H]C1=C([H])C([H])=C([H])C([H])=C1C([*])(C1=C(C(=C(C(=C1[H])[H])[H])[H])[H])C1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 description 18
- 229910052786 argon Inorganic materials 0.000 description 17
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 16
- 239000000463 material Substances 0.000 description 16
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 15
- 239000011630 iodine Substances 0.000 description 15
- 229910052740 iodine Inorganic materials 0.000 description 15
- 239000000376 reactant Substances 0.000 description 15
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 14
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 12
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 12
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 12
- 229910052938 sodium sulfate Inorganic materials 0.000 description 12
- 235000011152 sodium sulphate Nutrition 0.000 description 12
- 239000000725 suspension Substances 0.000 description 12
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 12
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 11
- OAYLNYINCPYISS-UHFFFAOYSA-N ethyl acetate;hexane Chemical compound CCCCCC.CCOC(C)=O OAYLNYINCPYISS-UHFFFAOYSA-N 0.000 description 11
- 239000012266 salt solution Substances 0.000 description 11
- XJDNKRIXUMDJCW-UHFFFAOYSA-J titanium tetrachloride Chemical compound Cl[Ti](Cl)(Cl)Cl XJDNKRIXUMDJCW-UHFFFAOYSA-J 0.000 description 11
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 10
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 10
- 239000012043 crude product Substances 0.000 description 10
- 238000005822 methylenation reaction Methods 0.000 description 10
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 10
- 239000012312 sodium hydride Substances 0.000 description 10
- 229910000104 sodium hydride Inorganic materials 0.000 description 10
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 9
- 241000700605 Viruses Species 0.000 description 9
- 229910052799 carbon Inorganic materials 0.000 description 9
- FJBFPHVGVWTDIP-UHFFFAOYSA-N dibromomethane Chemical compound BrCBr FJBFPHVGVWTDIP-UHFFFAOYSA-N 0.000 description 9
- HOFWRLQWIZPYEH-UHFFFAOYSA-J dibromomethane;tetrachlorotitanium;zinc Chemical compound [Zn].BrCBr.Cl[Ti](Cl)(Cl)Cl HOFWRLQWIZPYEH-UHFFFAOYSA-J 0.000 description 9
- 238000003810 ethyl acetate extraction Methods 0.000 description 9
- 229910052725 zinc Inorganic materials 0.000 description 9
- 239000011701 zinc Substances 0.000 description 9
- 0 CN(CC(*)C(N)=N1)C1=O Chemical compound CN(CC(*)C(N)=N1)C1=O 0.000 description 8
- WQDUMFSSJAZKTM-UHFFFAOYSA-N Sodium methoxide Chemical compound [Na+].[O-]C WQDUMFSSJAZKTM-UHFFFAOYSA-N 0.000 description 8
- 239000006260 foam Substances 0.000 description 8
- XYDYWTJEGDZLTH-UHFFFAOYSA-N methylenetriphenylphosphorane Chemical compound C=1C=CC=CC=1P(C=1C=CC=CC=1)(=C)C1=CC=CC=C1 XYDYWTJEGDZLTH-UHFFFAOYSA-N 0.000 description 8
- 230000003647 oxidation Effects 0.000 description 8
- 238000007254 oxidation reaction Methods 0.000 description 8
- 239000002002 slurry Substances 0.000 description 8
- APSBXTVYXVQYAB-UHFFFAOYSA-M sodium docusate Chemical compound [Na+].CCCCC(CC)COC(=O)CC(S([O-])(=O)=O)C(=O)OCC(CC)CCCC APSBXTVYXVQYAB-UHFFFAOYSA-M 0.000 description 8
- KZNICNPSHKQLFF-UHFFFAOYSA-N succinimide Chemical compound O=C1CCC(=O)N1 KZNICNPSHKQLFF-UHFFFAOYSA-N 0.000 description 8
- 238000005406 washing Methods 0.000 description 8
- WEVYAHXRMPXWCK-UHFFFAOYSA-N acetonitrile Substances CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 7
- 239000002253 acid Substances 0.000 description 7
- 125000002252 acyl group Chemical group 0.000 description 7
- 238000004458 analytical method Methods 0.000 description 7
- 150000001721 carbon Chemical group 0.000 description 7
- 239000002904 solvent Substances 0.000 description 7
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 6
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 6
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 6
- 241000700588 Human alphaherpesvirus 1 Species 0.000 description 6
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 6
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 6
- DKGAVHZHDRPRBM-UHFFFAOYSA-N Tert-Butanol Chemical compound CC(C)(C)O DKGAVHZHDRPRBM-UHFFFAOYSA-N 0.000 description 6
- 239000007864 aqueous solution Substances 0.000 description 6
- 239000003480 eluent Substances 0.000 description 6
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 6
- 229910052500 inorganic mineral Inorganic materials 0.000 description 6
- 235000010755 mineral Nutrition 0.000 description 6
- 239000011707 mineral Substances 0.000 description 6
- CTSLXHKWHWQRSH-UHFFFAOYSA-N oxalyl chloride Chemical compound ClC(=O)C(Cl)=O CTSLXHKWHWQRSH-UHFFFAOYSA-N 0.000 description 6
- 239000000843 powder Substances 0.000 description 6
- PODWXQQNRWNDGD-UHFFFAOYSA-L sodium thiosulfate pentahydrate Chemical compound O.O.O.O.O.[Na+].[Na+].[O-]S([S-])(=O)=O PODWXQQNRWNDGD-UHFFFAOYSA-L 0.000 description 6
- QSSXJPIWXQTSIX-UHFFFAOYSA-N 1-bromo-2-methylbenzene Chemical compound CC1=CC=CC=C1Br QSSXJPIWXQTSIX-UHFFFAOYSA-N 0.000 description 5
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 5
- 239000004593 Epoxy Substances 0.000 description 5
- 241000701074 Human alphaherpesvirus 2 Species 0.000 description 5
- 241000701085 Human alphaherpesvirus 3 Species 0.000 description 5
- PBCJIPOGFJYBJE-UHFFFAOYSA-N acetonitrile;hydrate Chemical compound O.CC#N PBCJIPOGFJYBJE-UHFFFAOYSA-N 0.000 description 5
- 239000013553 cell monolayer Substances 0.000 description 5
- 238000010790 dilution Methods 0.000 description 5
- 239000012895 dilution Substances 0.000 description 5
- SIAPCJWMELPYOE-UHFFFAOYSA-N lithium hydride Chemical compound [LiH] SIAPCJWMELPYOE-UHFFFAOYSA-N 0.000 description 5
- 229910000103 lithium hydride Inorganic materials 0.000 description 5
- 239000003880 polar aprotic solvent Substances 0.000 description 5
- 238000012545 processing Methods 0.000 description 5
- 239000011347 resin Substances 0.000 description 5
- 229920005989 resin Polymers 0.000 description 5
- 231100000419 toxicity Toxicity 0.000 description 5
- 230000001988 toxicity Effects 0.000 description 5
- OMBVEVHRIQULKW-DNQXCXABSA-M (3r,5r)-7-[3-(4-fluorophenyl)-8-oxo-7-phenyl-1-propan-2-yl-5,6-dihydro-4h-pyrrolo[2,3-c]azepin-2-yl]-3,5-dihydroxyheptanoate Chemical compound O=C1C=2N(C(C)C)C(CC[C@@H](O)C[C@@H](O)CC([O-])=O)=C(C=3C=CC(F)=CC=3)C=2CCCN1C1=CC=CC=C1 OMBVEVHRIQULKW-DNQXCXABSA-M 0.000 description 4
- PYRKKGOKRMZEIT-UHFFFAOYSA-N 2-[6-(2-cyclopropylethoxy)-9-(2-hydroxy-2-methylpropyl)-1h-phenanthro[9,10-d]imidazol-2-yl]-5-fluorobenzene-1,3-dicarbonitrile Chemical compound C1=C2C3=CC(CC(C)(O)C)=CC=C3C=3NC(C=4C(=CC(F)=CC=4C#N)C#N)=NC=3C2=CC=C1OCCC1CC1 PYRKKGOKRMZEIT-UHFFFAOYSA-N 0.000 description 4
- MHAJPDPJQMAIIY-UHFFFAOYSA-N Hydrogen peroxide Chemical compound OO MHAJPDPJQMAIIY-UHFFFAOYSA-N 0.000 description 4
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 4
- 239000003513 alkali Substances 0.000 description 4
- 150000003863 ammonium salts Chemical class 0.000 description 4
- 229940126540 compound 41 Drugs 0.000 description 4
- 238000001035 drying Methods 0.000 description 4
- 238000010438 heat treatment Methods 0.000 description 4
- 150000007523 nucleic acids Chemical class 0.000 description 4
- 102000039446 nucleic acids Human genes 0.000 description 4
- 108020004707 nucleic acids Proteins 0.000 description 4
- 229960002317 succinimide Drugs 0.000 description 4
- JBWKIWSBJXDJDT-UHFFFAOYSA-N triphenylmethyl chloride Chemical compound C=1C=CC=CC=1C(C=1C=CC=CC=1)(Cl)C1=CC=CC=C1 JBWKIWSBJXDJDT-UHFFFAOYSA-N 0.000 description 4
- SZUVGFMDDVSKSI-WIFOCOSTSA-N (1s,2s,3s,5r)-1-(carboxymethyl)-3,5-bis[(4-phenoxyphenyl)methyl-propylcarbamoyl]cyclopentane-1,2-dicarboxylic acid Chemical compound O=C([C@@H]1[C@@H]([C@](CC(O)=O)([C@H](C(=O)N(CCC)CC=2C=CC(OC=3C=CC=CC=3)=CC=2)C1)C(O)=O)C(O)=O)N(CCC)CC(C=C1)=CC=C1OC1=CC=CC=C1 SZUVGFMDDVSKSI-WIFOCOSTSA-N 0.000 description 3
- NGNBDVOYPDDBFK-UHFFFAOYSA-N 2-[2,4-di(pentan-2-yl)phenoxy]acetyl chloride Chemical compound CCCC(C)C1=CC=C(OCC(Cl)=O)C(C(C)CCC)=C1 NGNBDVOYPDDBFK-UHFFFAOYSA-N 0.000 description 3
- 125000004172 4-methoxyphenyl group Chemical group [H]C1=C([H])C(OC([H])([H])[H])=C([H])C([H])=C1* 0.000 description 3
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 3
- 241000701024 Human betaherpesvirus 5 Species 0.000 description 3
- CZPWVGJYEJSRLH-UHFFFAOYSA-N Pyrimidine Chemical compound C1=CN=CN=C1 CZPWVGJYEJSRLH-UHFFFAOYSA-N 0.000 description 3
- WREOTYWODABZMH-DTZQCDIJSA-N [[(2r,3s,4r,5r)-3,4-dihydroxy-5-[2-oxo-4-(2-phenylethoxyamino)pyrimidin-1-yl]oxolan-2-yl]methoxy-hydroxyphosphoryl] phosphono hydrogen phosphate Chemical compound O[C@@H]1[C@H](O)[C@@H](COP(O)(=O)OP(O)(=O)OP(O)(O)=O)O[C@H]1N(C=C\1)C(=O)NC/1=N\OCCC1=CC=CC=C1 WREOTYWODABZMH-DTZQCDIJSA-N 0.000 description 3
- 229910021529 ammonia Inorganic materials 0.000 description 3
- HQABUPZFAYXKJW-UHFFFAOYSA-N butan-1-amine Chemical compound CCCCN HQABUPZFAYXKJW-UHFFFAOYSA-N 0.000 description 3
- RCTYPNKXASFOBE-UHFFFAOYSA-M chloromercury Chemical compound [Hg]Cl RCTYPNKXASFOBE-UHFFFAOYSA-M 0.000 description 3
- 239000012230 colorless oil Substances 0.000 description 3
- 229940126543 compound 14 Drugs 0.000 description 3
- 229940125758 compound 15 Drugs 0.000 description 3
- 238000000354 decomposition reaction Methods 0.000 description 3
- WBKFWQBXFREOFH-UHFFFAOYSA-N dichloromethane;ethyl acetate Chemical compound ClCCl.CCOC(C)=O WBKFWQBXFREOFH-UHFFFAOYSA-N 0.000 description 3
- IJKVHSBPTUYDLN-UHFFFAOYSA-N dihydroxy(oxo)silane Chemical compound O[Si](O)=O IJKVHSBPTUYDLN-UHFFFAOYSA-N 0.000 description 3
- 238000007865 diluting Methods 0.000 description 3
- 125000005982 diphenylmethyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])(*)C1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 description 3
- 239000006185 dispersion Substances 0.000 description 3
- 208000015181 infectious disease Diseases 0.000 description 3
- PNDPGZBMCMUPRI-UHFFFAOYSA-N iodine Chemical compound II PNDPGZBMCMUPRI-UHFFFAOYSA-N 0.000 description 3
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 3
- 238000006386 neutralization reaction Methods 0.000 description 3
- 239000002777 nucleoside Substances 0.000 description 3
- 125000003835 nucleoside group Chemical group 0.000 description 3
- 125000001181 organosilyl group Chemical group [SiH3]* 0.000 description 3
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Substances [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 3
- 235000015320 potassium carbonate Nutrition 0.000 description 3
- NTTOTNSKUYCDAV-UHFFFAOYSA-N potassium hydride Chemical compound [KH] NTTOTNSKUYCDAV-UHFFFAOYSA-N 0.000 description 3
- 229910000105 potassium hydride Inorganic materials 0.000 description 3
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 3
- 238000001953 recrystallisation Methods 0.000 description 3
- 239000000741 silica gel Substances 0.000 description 3
- 229910002027 silica gel Inorganic materials 0.000 description 3
- 229960001866 silicon dioxide Drugs 0.000 description 3
- 229910052708 sodium Inorganic materials 0.000 description 3
- 239000011734 sodium Substances 0.000 description 3
- HXJUTPCZVOIRIF-UHFFFAOYSA-N sulfolane Chemical compound O=S1(=O)CCCC1 HXJUTPCZVOIRIF-UHFFFAOYSA-N 0.000 description 3
- DZLFLBLQUQXARW-UHFFFAOYSA-N tetrabutylammonium Chemical compound CCCC[N+](CCCC)(CCCC)CCCC DZLFLBLQUQXARW-UHFFFAOYSA-N 0.000 description 3
- WSLDOOZREJYCGB-UHFFFAOYSA-N 1,2-Dichloroethane Chemical compound ClCCCl WSLDOOZREJYCGB-UHFFFAOYSA-N 0.000 description 2
- VWCUMTCXBIRRSG-UHFFFAOYSA-N 1-[chloro(diphenyl)methyl]-2-methoxybenzene Chemical compound COC1=CC=CC=C1C(Cl)(C=1C=CC=CC=1)C1=CC=CC=C1 VWCUMTCXBIRRSG-UHFFFAOYSA-N 0.000 description 2
- OVSKIKFHRZPJSS-UHFFFAOYSA-N 2,4-D Chemical class OC(=O)COC1=CC=C(Cl)C=C1Cl OVSKIKFHRZPJSS-UHFFFAOYSA-N 0.000 description 2
- GOJUJUVQIVIZAV-UHFFFAOYSA-N 2-amino-4,6-dichloropyrimidine-5-carbaldehyde Chemical group NC1=NC(Cl)=C(C=O)C(Cl)=N1 GOJUJUVQIVIZAV-UHFFFAOYSA-N 0.000 description 2
- CFTPMBWYBDKGSA-UHFFFAOYSA-N 2-cyclopentyl-7h-purine Chemical compound C1CCCC1C1=NC=C(NC=N2)C2=N1 CFTPMBWYBDKGSA-UHFFFAOYSA-N 0.000 description 2
- 125000000954 2-hydroxyethyl group Chemical group [H]C([*])([H])C([H])([H])O[H] 0.000 description 2
- NHQDETIJWKXCTC-UHFFFAOYSA-N 3-chloroperbenzoic acid Chemical compound OOC(=O)C1=CC=CC(Cl)=C1 NHQDETIJWKXCTC-UHFFFAOYSA-N 0.000 description 2
- YYROPELSRYBVMQ-UHFFFAOYSA-N 4-toluenesulfonyl chloride Chemical compound CC1=CC=C(S(Cl)(=O)=O)C=C1 YYROPELSRYBVMQ-UHFFFAOYSA-N 0.000 description 2
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 2
- 241000701022 Cytomegalovirus Species 0.000 description 2
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 2
- BAPJBEWLBFYGME-UHFFFAOYSA-N Methyl acrylate Chemical compound COC(=O)C=C BAPJBEWLBFYGME-UHFFFAOYSA-N 0.000 description 2
- WCUXLLCKKVVCTQ-UHFFFAOYSA-M Potassium chloride Chemical compound [Cl-].[K+] WCUXLLCKKVVCTQ-UHFFFAOYSA-M 0.000 description 2
- KDCGOANMDULRCW-UHFFFAOYSA-N Purine Natural products N1=CNC2=NC=NC2=C1 KDCGOANMDULRCW-UHFFFAOYSA-N 0.000 description 2
- UIIMBOGNXHQVGW-DEQYMQKBSA-M Sodium bicarbonate-14C Chemical class [Na+].O[14C]([O-])=O UIIMBOGNXHQVGW-DEQYMQKBSA-M 0.000 description 2
- ISAKRJDGNUQOIC-UHFFFAOYSA-N Uracil Chemical compound O=C1C=CNC(=O)N1 ISAKRJDGNUQOIC-UHFFFAOYSA-N 0.000 description 2
- 229910052783 alkali metal Inorganic materials 0.000 description 2
- 150000001447 alkali salts Chemical class 0.000 description 2
- 238000013459 approach Methods 0.000 description 2
- CBHOOMGKXCMKIR-UHFFFAOYSA-N azane;methanol Chemical compound N.OC CBHOOMGKXCMKIR-UHFFFAOYSA-N 0.000 description 2
- 230000001580 bacterial effect Effects 0.000 description 2
- 230000015572 biosynthetic process Effects 0.000 description 2
- 239000011575 calcium Substances 0.000 description 2
- 229910052791 calcium Inorganic materials 0.000 description 2
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 2
- 238000004113 cell culture Methods 0.000 description 2
- 239000003153 chemical reaction reagent Substances 0.000 description 2
- 125000001309 chloro group Chemical group Cl* 0.000 description 2
- 239000012141 concentrate Substances 0.000 description 2
- 125000004093 cyano group Chemical group *C#N 0.000 description 2
- ZSWFCLXCOIISFI-UHFFFAOYSA-N cyclopentadiene Chemical compound C1C=CC=C1 ZSWFCLXCOIISFI-UHFFFAOYSA-N 0.000 description 2
- XCIXKGXIYUWCLL-UHFFFAOYSA-N cyclopentanol Chemical compound OC1CCCC1 XCIXKGXIYUWCLL-UHFFFAOYSA-N 0.000 description 2
- 125000002433 cyclopentenyl group Chemical class C1(=CCCC1)* 0.000 description 2
- FHHZOYXKOICLGH-UHFFFAOYSA-N dichloromethane;ethanol Chemical compound CCO.ClCCl FHHZOYXKOICLGH-UHFFFAOYSA-N 0.000 description 2
- CSJLBAMHHLJAAS-UHFFFAOYSA-N diethylaminosulfur trifluoride Chemical compound CCN(CC)S(F)(F)F CSJLBAMHHLJAAS-UHFFFAOYSA-N 0.000 description 2
- 230000008034 disappearance Effects 0.000 description 2
- 230000000694 effects Effects 0.000 description 2
- 238000010828 elution Methods 0.000 description 2
- 238000000605 extraction Methods 0.000 description 2
- 239000000706 filtrate Substances 0.000 description 2
- 238000001914 filtration Methods 0.000 description 2
- 229910052736 halogen Inorganic materials 0.000 description 2
- 150000002367 halogens Chemical class 0.000 description 2
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 2
- 230000007062 hydrolysis Effects 0.000 description 2
- 238000006460 hydrolysis reaction Methods 0.000 description 2
- 239000012442 inert solvent Substances 0.000 description 2
- 239000007788 liquid Substances 0.000 description 2
- 159000000003 magnesium salts Chemical class 0.000 description 2
- 230000014759 maintenance of location Effects 0.000 description 2
- LULAYUGMBFYYEX-UHFFFAOYSA-N metachloroperbenzoic acid Natural products OC(=O)C1=CC=CC(Cl)=C1 LULAYUGMBFYYEX-UHFFFAOYSA-N 0.000 description 2
- GBMDVOWEEQVZKZ-UHFFFAOYSA-N methanol;hydrate Chemical compound O.OC GBMDVOWEEQVZKZ-UHFFFAOYSA-N 0.000 description 2
- OKDQKPLMQBXTNH-UHFFFAOYSA-N n,n-dimethyl-2h-pyridin-1-amine Chemical compound CN(C)N1CC=CC=C1 OKDQKPLMQBXTNH-UHFFFAOYSA-N 0.000 description 2
- 239000002773 nucleotide Substances 0.000 description 2
- 125000003729 nucleotide group Chemical group 0.000 description 2
- 239000002674 ointment Substances 0.000 description 2
- 150000007530 organic bases Chemical class 0.000 description 2
- 239000003960 organic solvent Substances 0.000 description 2
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 2
- 239000001103 potassium chloride Substances 0.000 description 2
- 235000011164 potassium chloride Nutrition 0.000 description 2
- 235000017557 sodium bicarbonate Nutrition 0.000 description 2
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 2
- 238000003860 storage Methods 0.000 description 2
- YBBRCQOCSYXUOC-UHFFFAOYSA-N sulfuryl dichloride Chemical compound ClS(Cl)(=O)=O YBBRCQOCSYXUOC-UHFFFAOYSA-N 0.000 description 2
- 150000005621 tetraalkylammonium salts Chemical class 0.000 description 2
- VPAYJEUHKVESSD-UHFFFAOYSA-N trifluoroiodomethane Chemical compound FC(F)(F)I VPAYJEUHKVESSD-UHFFFAOYSA-N 0.000 description 2
- 238000007738 vacuum evaporation Methods 0.000 description 2
- 230000009385 viral infection Effects 0.000 description 2
- YQOLEILXOBUDMU-KRWDZBQOSA-N (4R)-5-[(6-bromo-3-methyl-2-pyrrolidin-1-ylquinoline-4-carbonyl)amino]-4-(2-chlorophenyl)pentanoic acid Chemical compound CC1=C(C2=C(C=CC(=C2)Br)N=C1N3CCCC3)C(=O)NC[C@H](CCC(=O)O)C4=CC=CC=C4Cl YQOLEILXOBUDMU-KRWDZBQOSA-N 0.000 description 1
- OTEKOJQFKOIXMU-UHFFFAOYSA-N 1,4-bis(trichloromethyl)benzene Chemical compound ClC(Cl)(Cl)C1=CC=C(C(Cl)(Cl)Cl)C=C1 OTEKOJQFKOIXMU-UHFFFAOYSA-N 0.000 description 1
- FXOOVOMGQUVLCK-UHFFFAOYSA-N 1-methyl-2-phenylmethoxy-6-oxabicyclo[3.1.0]hexane Chemical compound CC12OC1CCC2OCC1=CC=CC=C1 FXOOVOMGQUVLCK-UHFFFAOYSA-N 0.000 description 1
- 238000001644 13C nuclear magnetic resonance spectroscopy Methods 0.000 description 1
- SNTWKPAKVQFCCF-UHFFFAOYSA-N 2,3-dihydro-1h-triazole Chemical compound N1NC=CN1 SNTWKPAKVQFCCF-UHFFFAOYSA-N 0.000 description 1
- 125000002103 4,4'-dimethoxytriphenylmethyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C(*)(C1=C([H])C([H])=C(OC([H])([H])[H])C([H])=C1[H])C1=C([H])C([H])=C(OC([H])([H])[H])C([H])=C1[H] 0.000 description 1
- 101710169336 5'-deoxyadenosine deaminase Proteins 0.000 description 1
- GJEZBVHHZQAEDB-UHFFFAOYSA-N 6-oxabicyclo[3.1.0]hexane Chemical compound C1CCC2OC21 GJEZBVHHZQAEDB-UHFFFAOYSA-N 0.000 description 1
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 1
- 102000055025 Adenosine deaminases Human genes 0.000 description 1
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 1
- 241000271566 Aves Species 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 1
- OJIKWFOITYJUSO-UHFFFAOYSA-N CC(C)[SiH2]O[SiH3] Chemical compound CC(C)[SiH2]O[SiH3] OJIKWFOITYJUSO-UHFFFAOYSA-N 0.000 description 1
- RGSMQOKAAHVNLU-UHFFFAOYSA-N CN(C1N=C(N)N2)C=NC1C2=O Chemical compound CN(C1N=C(N)N2)C=NC1C2=O RGSMQOKAAHVNLU-UHFFFAOYSA-N 0.000 description 1
- KVWHYMBTDGAOHJ-UHFFFAOYSA-N CN1C=NC2C(N)=NCNC12 Chemical compound CN1C=NC2C(N)=NCNC12 KVWHYMBTDGAOHJ-UHFFFAOYSA-N 0.000 description 1
- DNDBMQQNPZKZMA-UHFFFAOYSA-N CN1C=NC2C=NC(N)=NC12 Chemical compound CN1C=NC2C=NC(N)=NC12 DNDBMQQNPZKZMA-UHFFFAOYSA-N 0.000 description 1
- VJSKGBHQAIZCTA-UHFFFAOYSA-N CN1[I]=Nc2c1ncnc2Cl Chemical compound CN1[I]=Nc2c1ncnc2Cl VJSKGBHQAIZCTA-UHFFFAOYSA-N 0.000 description 1
- BOUGRLIGKDAWCB-UHFFFAOYSA-N C[n]1c(N=C(NC2=O)N=C=O)c2nc1 Chemical compound C[n]1c(N=C(NC2=O)N=C=O)c2nc1 BOUGRLIGKDAWCB-UHFFFAOYSA-N 0.000 description 1
- PESGUQRDJASXOR-UHFFFAOYSA-N C[n]1c(N=CNC2=O)c2nc1 Chemical compound C[n]1c(N=CNC2=O)c2nc1 PESGUQRDJASXOR-UHFFFAOYSA-N 0.000 description 1
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 description 1
- 229910021595 Copper(I) iodide Inorganic materials 0.000 description 1
- XDTMQSROBMDMFD-UHFFFAOYSA-N Cyclohexane Chemical compound C1CCCCC1 XDTMQSROBMDMFD-UHFFFAOYSA-N 0.000 description 1
- 206010011831 Cytomegalovirus infection Diseases 0.000 description 1
- 206010013786 Dry skin Diseases 0.000 description 1
- 241000282326 Felis catus Species 0.000 description 1
- KRHYYFGTRYWZRS-UHFFFAOYSA-M Fluoride anion Chemical compound [F-] KRHYYFGTRYWZRS-UHFFFAOYSA-M 0.000 description 1
- 241000287828 Gallus gallus Species 0.000 description 1
- 241000700721 Hepatitis B virus Species 0.000 description 1
- 241000701044 Human gammaherpesvirus 4 Species 0.000 description 1
- 238000000023 Kugelrohr distillation Methods 0.000 description 1
- 241000124008 Mammalia Species 0.000 description 1
- 241001465754 Metazoa Species 0.000 description 1
- CERQOIWHTDAKMF-UHFFFAOYSA-M Methacrylate Chemical compound CC(=C)C([O-])=O CERQOIWHTDAKMF-UHFFFAOYSA-M 0.000 description 1
- XYEOALKITRFCJJ-UHFFFAOYSA-N NOCc1ccccc1 Chemical compound NOCc1ccccc1 XYEOALKITRFCJJ-UHFFFAOYSA-N 0.000 description 1
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 1
- IOVCWXUNBOPUCH-UHFFFAOYSA-N Nitrous acid Chemical compound ON=O IOVCWXUNBOPUCH-UHFFFAOYSA-N 0.000 description 1
- KRWMERLEINMZFT-UHFFFAOYSA-N O6-benzylguanine Chemical compound C=12NC=NC2=NC(N)=NC=1OCC1=CC=CC=C1 KRWMERLEINMZFT-UHFFFAOYSA-N 0.000 description 1
- WVDDGKGOMKODPV-UHFFFAOYSA-N OCc1ccccc1 Chemical compound OCc1ccccc1 WVDDGKGOMKODPV-UHFFFAOYSA-N 0.000 description 1
- 235000019502 Orange oil Nutrition 0.000 description 1
- 241001631646 Papillomaviridae Species 0.000 description 1
- 229930040373 Paraformaldehyde Natural products 0.000 description 1
- 102000004160 Phosphoric Monoester Hydrolases Human genes 0.000 description 1
- 108090000608 Phosphoric Monoester Hydrolases Proteins 0.000 description 1
- 241000700584 Simplexvirus Species 0.000 description 1
- 241000701093 Suid alphaherpesvirus 1 Species 0.000 description 1
- 241000700618 Vaccinia virus Species 0.000 description 1
- 241000726445 Viroids Species 0.000 description 1
- MVAGMBJXIBABNI-UHFFFAOYSA-N [Na].C=1C=CC=CC=1[Se]C1=CC=CC=C1 Chemical compound [Na].C=1C=CC=CC=1[Se]C1=CC=CC=C1 MVAGMBJXIBABNI-UHFFFAOYSA-N 0.000 description 1
- 238000005903 acid hydrolysis reaction Methods 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 230000004913 activation Effects 0.000 description 1
- 125000004423 acyloxy group Chemical group 0.000 description 1
- 239000000443 aerosol Substances 0.000 description 1
- 238000013019 agitation Methods 0.000 description 1
- 230000001476 alcoholic effect Effects 0.000 description 1
- 125000003545 alkoxy group Chemical group 0.000 description 1
- 125000003282 alkyl amino group Chemical group 0.000 description 1
- 150000008052 alkyl sulfonates Chemical class 0.000 description 1
- 125000003368 amide group Chemical group 0.000 description 1
- VZTDIZULWFCMLS-UHFFFAOYSA-N ammonium formate Chemical compound [NH4+].[O-]C=O VZTDIZULWFCMLS-UHFFFAOYSA-N 0.000 description 1
- 239000000908 ammonium hydroxide Substances 0.000 description 1
- 239000003443 antiviral agent Substances 0.000 description 1
- 239000011260 aqueous acid Substances 0.000 description 1
- 238000010533 azeotropic distillation Methods 0.000 description 1
- 239000002585 base Substances 0.000 description 1
- 239000004305 biphenyl Substances 0.000 description 1
- 235000010290 biphenyl Nutrition 0.000 description 1
- 125000006267 biphenyl group Chemical group 0.000 description 1
- 230000037396 body weight Effects 0.000 description 1
- 230000031709 bromination Effects 0.000 description 1
- 238000005893 bromination reaction Methods 0.000 description 1
- RJCQBQGAPKAMLL-UHFFFAOYSA-N bromotrifluoromethane Chemical compound FC(F)(F)Br RJCQBQGAPKAMLL-UHFFFAOYSA-N 0.000 description 1
- 229940045348 brown mixture Drugs 0.000 description 1
- SNCZNSNPXMPCGN-UHFFFAOYSA-N butanediamide Chemical compound NC(=O)CCC(N)=O SNCZNSNPXMPCGN-UHFFFAOYSA-N 0.000 description 1
- 230000003197 catalytic effect Effects 0.000 description 1
- 230000008859 change Effects 0.000 description 1
- 150000001805 chlorine compounds Chemical class 0.000 description 1
- MVPPADPHJFYWMZ-UHFFFAOYSA-N chlorobenzene Substances ClC1=CC=CC=C1 MVPPADPHJFYWMZ-UHFFFAOYSA-N 0.000 description 1
- SIHHLZPXQLFPMC-UHFFFAOYSA-N chloroform;methanol;hydrate Chemical compound O.OC.ClC(Cl)Cl SIHHLZPXQLFPMC-UHFFFAOYSA-N 0.000 description 1
- 238000004587 chromatography analysis Methods 0.000 description 1
- 238000005352 clarification Methods 0.000 description 1
- 229940125773 compound 10 Drugs 0.000 description 1
- 229940125844 compound 46 Drugs 0.000 description 1
- 229940125898 compound 5 Drugs 0.000 description 1
- 238000001816 cooling Methods 0.000 description 1
- 229910052802 copper Inorganic materials 0.000 description 1
- 239000010949 copper Substances 0.000 description 1
- LSXDOTMGLUJQCM-UHFFFAOYSA-M copper(i) iodide Chemical compound I[Cu] LSXDOTMGLUJQCM-UHFFFAOYSA-M 0.000 description 1
- 239000013078 crystal Substances 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- 238000010586 diagram Methods 0.000 description 1
- 125000003963 dichloro group Chemical group Cl* 0.000 description 1
- NKIZJHWBAYUHPE-UHFFFAOYSA-N dichloromethane;trichloroborane Chemical compound ClCCl.ClB(Cl)Cl NKIZJHWBAYUHPE-UHFFFAOYSA-N 0.000 description 1
- 229940079593 drug Drugs 0.000 description 1
- 239000003814 drug Substances 0.000 description 1
- 239000000975 dye Substances 0.000 description 1
- 238000006735 epoxidation reaction Methods 0.000 description 1
- DQYBDCGIPTYXML-UHFFFAOYSA-N ethoxyethane;hydrate Chemical compound O.CCOCC DQYBDCGIPTYXML-UHFFFAOYSA-N 0.000 description 1
- UREBWPXBXRYXRJ-UHFFFAOYSA-N ethyl acetate;methanol Chemical compound OC.CCOC(C)=O UREBWPXBXRYXRJ-UHFFFAOYSA-N 0.000 description 1
- 238000011156 evaluation Methods 0.000 description 1
- 238000001704 evaporation Methods 0.000 description 1
- 230000008020 evaporation Effects 0.000 description 1
- 230000003203 everyday effect Effects 0.000 description 1
- 239000003889 eye drop Substances 0.000 description 1
- 229940012356 eye drops Drugs 0.000 description 1
- 239000000499 gel Substances 0.000 description 1
- 230000026030 halogenation Effects 0.000 description 1
- 238000005658 halogenation reaction Methods 0.000 description 1
- 125000000623 heterocyclic group Chemical group 0.000 description 1
- 238000004128 high performance liquid chromatography Methods 0.000 description 1
- 238000007327 hydrogenolysis reaction Methods 0.000 description 1
- 230000006872 improvement Effects 0.000 description 1
- 150000007529 inorganic bases Chemical class 0.000 description 1
- 238000007912 intraperitoneal administration Methods 0.000 description 1
- ZLVXBBHTMQJRSX-VMGNSXQWSA-N jdtic Chemical compound C1([C@]2(C)CCN(C[C@@H]2C)C[C@H](C(C)C)NC(=O)[C@@H]2NCC3=CC(O)=CC=C3C2)=CC=CC(O)=C1 ZLVXBBHTMQJRSX-VMGNSXQWSA-N 0.000 description 1
- 125000000468 ketone group Chemical group 0.000 description 1
- 150000002576 ketones Chemical class 0.000 description 1
- 239000006210 lotion Substances 0.000 description 1
- 239000002808 molecular sieve Substances 0.000 description 1
- 208000005871 monkeypox Diseases 0.000 description 1
- 229910017604 nitric acid Inorganic materials 0.000 description 1
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 description 1
- 239000012299 nitrogen atmosphere Substances 0.000 description 1
- 239000004533 oil dispersion Substances 0.000 description 1
- 239000010502 orange oil Substances 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 239000012074 organic phase Substances 0.000 description 1
- 229910052763 palladium Inorganic materials 0.000 description 1
- 229920002866 paraformaldehyde Polymers 0.000 description 1
- XCRBXWCUXJNEFX-UHFFFAOYSA-N peroxybenzoic acid Chemical compound OOC(=O)C1=CC=CC=C1 XCRBXWCUXJNEFX-UHFFFAOYSA-N 0.000 description 1
- ZUOUZKKEUPVFJK-UHFFFAOYSA-N phenylbenzene Natural products C1=CC=CC=C1C1=CC=CC=C1 ZUOUZKKEUPVFJK-UHFFFAOYSA-N 0.000 description 1
- ORQWTLCYLDRDHK-UHFFFAOYSA-N phenylselanylbenzene Chemical compound C=1C=CC=CC=1[Se]C1=CC=CC=C1 ORQWTLCYLDRDHK-UHFFFAOYSA-N 0.000 description 1
- 150000003016 phosphoric acids Chemical class 0.000 description 1
- 230000008569 process Effects 0.000 description 1
- 238000004537 pulping Methods 0.000 description 1
- 125000000714 pyrimidinyl group Chemical group 0.000 description 1
- 238000010791 quenching Methods 0.000 description 1
- 230000000171 quenching effect Effects 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 238000010992 reflux Methods 0.000 description 1
- 230000000452 restraining effect Effects 0.000 description 1
- URGAHOPLAPQHLN-UHFFFAOYSA-N sodium aluminosilicate Chemical compound [Na+].[Al+3].[O-][Si]([O-])=O.[O-][Si]([O-])=O URGAHOPLAPQHLN-UHFFFAOYSA-N 0.000 description 1
- 229910000033 sodium borohydride Inorganic materials 0.000 description 1
- 239000012279 sodium borohydride Substances 0.000 description 1
- 239000001488 sodium phosphate Substances 0.000 description 1
- 229910000162 sodium phosphate Inorganic materials 0.000 description 1
- VPQBLCVGUWPDHV-UHFFFAOYSA-N sodium selenide Chemical compound [Na+].[Na+].[Se-2] VPQBLCVGUWPDHV-UHFFFAOYSA-N 0.000 description 1
- AKHNMLFCWUSKQB-UHFFFAOYSA-L sodium thiosulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=S AKHNMLFCWUSKQB-UHFFFAOYSA-L 0.000 description 1
- 235000019345 sodium thiosulphate Nutrition 0.000 description 1
- 238000003786 synthesis reaction Methods 0.000 description 1
- 238000012360 testing method Methods 0.000 description 1
- 150000005622 tetraalkylammonium hydroxides Chemical class 0.000 description 1
- DPKBAXPHAYBPRL-UHFFFAOYSA-M tetrabutylazanium;iodide Chemical compound [I-].CCCC[N+](CCCC)(CCCC)CCCC DPKBAXPHAYBPRL-UHFFFAOYSA-M 0.000 description 1
- ISXOBTBCNRIIQO-UHFFFAOYSA-N tetrahydrothiophene 1-oxide Chemical compound O=S1CCCC1 ISXOBTBCNRIIQO-UHFFFAOYSA-N 0.000 description 1
- 125000000383 tetramethylene group Chemical group [H]C([H])([*:1])C([H])([H])C([H])([H])C([H])([H])[*:2] 0.000 description 1
- 210000001541 thymus gland Anatomy 0.000 description 1
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 1
- 230000000699 topical effect Effects 0.000 description 1
- 125000005208 trialkylammonium group Chemical group 0.000 description 1
- CWMFRHBXRUITQE-UHFFFAOYSA-N trimethylsilylacetylene Chemical group C[Si](C)(C)C#C CWMFRHBXRUITQE-UHFFFAOYSA-N 0.000 description 1
- RYFMWSXOAZQYPI-UHFFFAOYSA-K trisodium phosphate Chemical compound [Na+].[Na+].[Na+].[O-]P([O-])([O-])=O RYFMWSXOAZQYPI-UHFFFAOYSA-K 0.000 description 1
- 241000701161 unidentified adenovirus Species 0.000 description 1
- 241001529453 unidentified herpesvirus Species 0.000 description 1
- 241000712461 unidentified influenza virus Species 0.000 description 1
- 229920002554 vinyl polymer Polymers 0.000 description 1
- 239000011782 vitamin Substances 0.000 description 1
- 235000013343 vitamin Nutrition 0.000 description 1
- 229940088594 vitamin Drugs 0.000 description 1
- 229930003231 vitamin Natural products 0.000 description 1
- 150000003722 vitamin derivatives Chemical class 0.000 description 1
- 238000010792 warming Methods 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D239/00—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings
- C07D239/02—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings
- C07D239/24—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members
- C07D239/28—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, directly attached to ring carbon atoms
- C07D239/46—Two or more oxygen, sulphur or nitrogen atoms
- C07D239/52—Two oxygen atoms
- C07D239/54—Two oxygen atoms as doubly bound oxygen atoms or as unsubstituted hydroxy radicals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/20—Antivirals for DNA viruses
- A61P31/22—Antivirals for DNA viruses for herpes viruses
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D239/00—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings
- C07D239/02—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings
- C07D239/24—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members
- C07D239/28—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, directly attached to ring carbon atoms
- C07D239/46—Two or more oxygen, sulphur or nitrogen atoms
- C07D239/52—Two oxygen atoms
- C07D239/54—Two oxygen atoms as doubly bound oxygen atoms or as unsubstituted hydroxy radicals
- C07D239/545—Two oxygen atoms as doubly bound oxygen atoms or as unsubstituted hydroxy radicals with other hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, directly attached to ring carbon atoms
- C07D239/553—Two oxygen atoms as doubly bound oxygen atoms or as unsubstituted hydroxy radicals with other hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, directly attached to ring carbon atoms with halogen atoms or nitro radicals directly attached to ring carbon atoms, e.g. fluorouracil
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D473/00—Heterocyclic compounds containing purine ring systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
Landscapes
- Organic Chemistry (AREA)
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Virology (AREA)
- Pharmacology & Pharmacy (AREA)
- Communicable Diseases (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Oncology (AREA)
- Animal Behavior & Ethology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Engineering & Computer Science (AREA)
- Biotechnology (AREA)
- Molecular Biology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
Abstract
下式的化合物或其可药用盐具有抗病毒活性。
Description
Zahler等人在EP431,754中公开了下式的4-羟基-3-羟甲基-2-亚甲基环戊基嘌呤和嘧啶其中R1是取代的嘌呤基或嘧啶基。
式1化合物及其可药用盐具有抗病毒活性。 在式1中和整个说明书中,各符号定义如下:R1是
在式1中和整个说明书中,各符号定义如下:R1是


R2是氟、氯、溴、碘、氢、甲基、三氟甲基、乙基、正丙基、2-氟乙基、2-氯乙基、乙炔基或 R3是氯、溴、碘、氢、甲基或三氟甲基。R4是烷基。R5是氢、烷基、取代的烷基或芳基。R6和R7独立地选自氢、-PO3H2和
R3是氯、溴、碘、氢、甲基或三氟甲基。R4是烷基。R5是氢、烷基、取代的烷基或芳基。R6和R7独立地选自氢、-PO3H2和
 其中R1是下式的式1化合物是优选的:
其中R1是下式的式1化合物是优选的:
术语“烷基”是指直链和支链烷基。优选含1-10个碳原子的烷基。术语“取代的烷基”是指含一个或多个、优选含一个取代基的烷基。优选的取代基是卤素、氨基、叠氮基、羟基、氰基、三烷基铵(其中每个烷基含1-6个碳原子)、1-6个碳原子的烷氧基、芳基和羧基。术语“芳基”是指苯基和被一个、两个或三个(优选一个)取代基取代的苯基。优选的取代基是1-6个碳原子的烷基、1-6个碳原子烷氧基、卤素、三氟甲基、氨基、1-6个碳原子的烷氨基、每个烷基有1-6个碳原子的二烷基氨基、硝基、氰基、2-11个碳原子的链烷酰氧基、羧基、氨基甲酰基和羟基。
式1化合物及其可药用盐是抗病毒剂,它们可用于治疗哺乳动物如驯养动物(例如狗、猫和马等)和人以及鸟类(如鸡和火鸡)和病毒感染。其中R1为下式的式1化合物对下列一种或多种病毒是有效的:单纯性疱疹病毒1和2、水痘一带状疱疹病毒和巨细胞病毒。  据信它们还有抗其他DNA病毒的活性。除上述那些病毒之外的DNA病毒的例子包括其他疱疹病毒(例如EB病毒、假性狂犬病病毒和人疱疹病毒6等)、痘病毒(例如牛痘病毒、猴痘和肌瘤)、乳多空病毒(例如乳头瘤病毒)、肝炎B病毒和腺病毒。据信所有其他式1化合物对下列一种或多种病毒有活性:单纯性疱疹病毒1和2、水痘-带状疱疹病毒、巨细胞病毒和上述其他DNA病毒。
据信它们还有抗其他DNA病毒的活性。除上述那些病毒之外的DNA病毒的例子包括其他疱疹病毒(例如EB病毒、假性狂犬病病毒和人疱疹病毒6等)、痘病毒(例如牛痘病毒、猴痘和肌瘤)、乳多空病毒(例如乳头瘤病毒)、肝炎B病毒和腺病毒。据信所有其他式1化合物对下列一种或多种病毒有活性:单纯性疱疹病毒1和2、水痘-带状疱疹病毒、巨细胞病毒和上述其他DNA病毒。
本发明化合物可以经非肠道(例如通过静脉内、腹膜内或肌内注射)、口或局部施用。
这些化合物可以以治疗感染有效量经口或非肠道施用。当然,所用剂量取决于感染的程度,但是通常能的剂量范围是约1.0-50mg/kg体重。所需剂量可以以合适的时间间隔分为每日几次施用。
对于眼或其他外部组织(如口和皮肤)感染,可以将组合物分以油膏、软膏、气雾剂、凝胶、粉末、洗剂、悬浮液或溶液(例如以滴眼剂)的形式局部施用于患者身体的被感染部位。当然,该化合物在赋形剂中的浓度将取决于感染的程度,但是通常的浓度范围是约0.1-7%(重量)。
可以从式2化合物制备本发明化合物。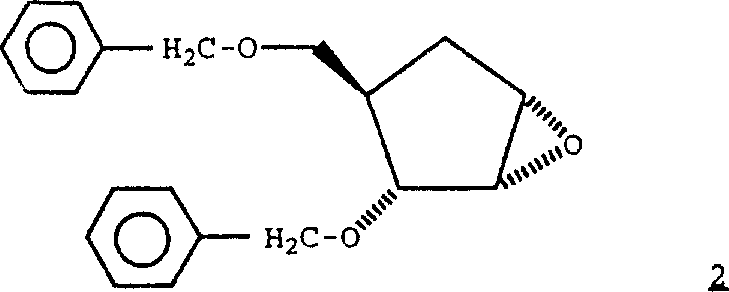 用苄基溴和氢化钠保护已知的式3化合物中的羟基,得到式2化合物。
用苄基溴和氢化钠保护已知的式3化合物中的羟基,得到式2化合物。
在Paulsen等人的Chem.Ber.,Vol.114,p.346-358(1981)和Hutchinson等人的J.Heterocyclic Chem..Vol.26,p.451-452(1989)中描述了式3的环氧化合物。采用这些改良的方法,可以通过下述方法制备式3的环氧化合物:在乙酸中将式4的环戊二烯与多聚甲醛和催化量的对甲苯磺酸反应, 然后用在甲醇中的甲醇钠处理,得到式5化合物。
然后用在甲醇中的甲醇钠处理,得到式5化合物。 用间氯过苯甲酸处理化合物5,得到式3的环氧化合物。
用间氯过苯甲酸处理化合物5,得到式3的环氧化合物。
或者,可以通过下述方法制备被保护的式2环氧化合物。用氢化钠处理式6化合物, 然后用苄基溴处理,得到式7化合物。
然后用苄基溴处理,得到式7化合物。
在含水二噁烷中用N-溴琥珀酰亚胺与化合物7反应,然后用碳酸钾甲醇液处理所得的溴化醇,得到式8化合物。在乙醇中用二苯硒化钠(sodium phenylselenide)处理化合物8,然后将所得二苯硒与过氧化氢反应,得到式9化合物。 在缓冲的二氯甲烷中用间氯过苯甲酸使化合物9环氧化,得到式10化合物。用氢化钠处理化合物10,然后用苄基溴处理,得到被保护的式2环氧化合物。在Branner-Jorgensen等人的Acta Chem.Scand.,Vol.20,p 2191-2194(1966);Maercker等人的Chem.Ber.,Vol.106,p.773-797(1973);和Chapman等人的J.Amer.Chem Soc.,Vol100,p.4878-4884(1978)文献中描述了式6化合物。
在缓冲的二氯甲烷中用间氯过苯甲酸使化合物9环氧化,得到式10化合物。用氢化钠处理化合物10,然后用苄基溴处理,得到被保护的式2环氧化合物。在Branner-Jorgensen等人的Acta Chem.Scand.,Vol.20,p 2191-2194(1966);Maercker等人的Chem.Ber.,Vol.106,p.773-797(1973);和Chapman等人的J.Amer.Chem Soc.,Vol100,p.4878-4884(1978)文献中描述了式6化合物。
其中R1是且R6和R7是氢的式1化合物可通过下述反应制备。在碱如氢化锂、氢化钠或氢化钾存在下,在极性非质子溶剂如二甲基甲酰胺、二甲基亚砜或环丁砜(四亚甲基亚砜)中,将式2化合物与式11化合物反应,
由此得到相应的式12化合物。
保护式12化合物中的氨基(-NH2)得到式13化合物,
其中保护基P1可以是三苯甲基或取代的三苯甲基(例如4-单甲氧基三苯基或4,4'-二甲氧基三苯甲基)。在三乙胺存在下(以及任意的在4-N,N-二甲氨基吡啶存在下),在二氯甲烷中,通过用三苯甲基氯或取代的三苯甲基氯处理式12化合物完成该保护作用。将式13化合物中的羟基氧化,得到式14化合物。
可以通过本领域公知的方法进行该氧化作用(例如在二氯甲烷中的草酰氯/二甲基亚砜,或在二氯甲烷中的1,1,1-三乙酸基-1,1-二氢-1,2-benziodoxol-3(1H)-酮和叔丁醇)。在二氯甲烷/四氢呋喃或亚甲基三苯基正膦中,用亚甲基化试剂如锌/四氯化钛/二溴甲烷处理式14化合物,得到式15化合物。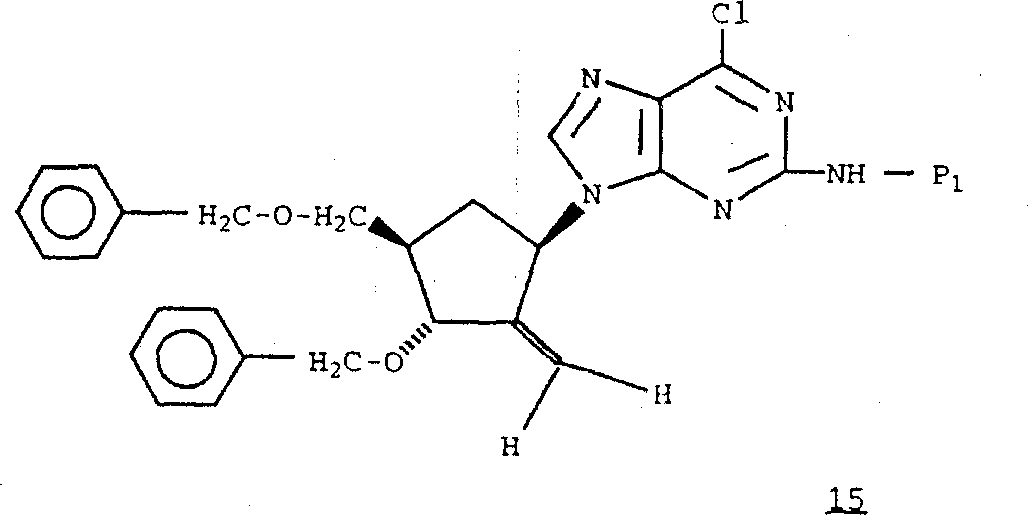
除去式15化合物的保护基,得到其中R1是 且R6和R7是氢的式1化合物。通过用无机酸含水的醇溶液(例如盐酸的甲醇水溶液)或含水乙酸进行处理可以除去保护基P1。随后,用三氯化硼处理可以除去苄基保护基。或者,P1和苄基保护基均可以通过用三氯化硼处理得以去除。
且R6和R7是氢的式1化合物。通过用无机酸含水的醇溶液(例如盐酸的甲醇水溶液)或含水乙酸进行处理可以除去保护基P1。随后,用三氯化硼处理可以除去苄基保护基。或者,P1和苄基保护基均可以通过用三氯化硼处理得以去除。
可以如下制备其中R1是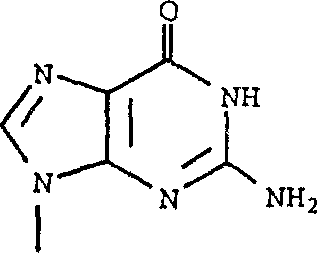 且R6和R7是氢的式1化合物:
且R6和R7是氢的式1化合物:
按照与制备式12化合物中所采用的类似方法,将式2化合物与式16化合物反应,
得到式17化合物。
按照与制备式13化合物中所采用的类似以方法,将式17化合物中的氨基(-NH2)保护,得到式18化合物。
在与制备式14化合物中所采用的相似条件下,氧化式18化合物中的羟基,得到式19化合物。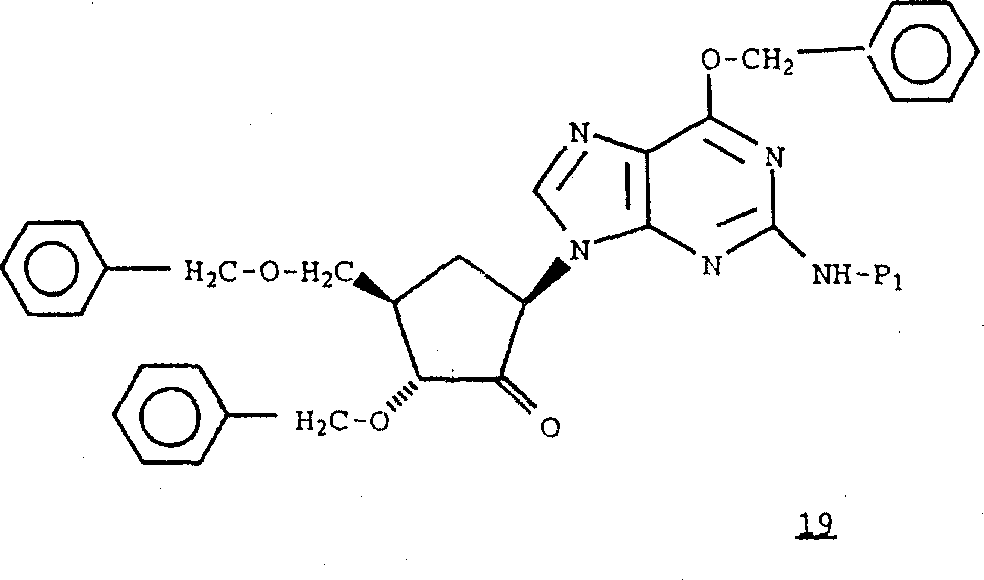
在二氯甲烷中,用锌/四氯化钛/二溴甲烷使式19化合物中的酮基亚甲基化,得到式20化合物。
最后,从式20化合物上除去保护基,得到式1化合物,其中R1是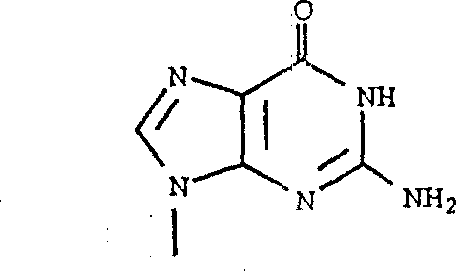 且R6和R7是氢。通过用无机酸的含水醇溶液(例如盐酸的甲醇水溶液)处理,可以除去P1和嘌呤O-苄基保护基。然后用三氯化硼处理,除去羟苄基保护基。或者可以用三氯化硼进行处理,除去所有的保护基。
且R6和R7是氢。通过用无机酸的含水醇溶液(例如盐酸的甲醇水溶液)处理,可以除去P1和嘌呤O-苄基保护基。然后用三氯化硼处理,除去羟苄基保护基。或者可以用三氯化硼进行处理,除去所有的保护基。
另外,也可以通过用酸水溶液(例如盐酸水溶液)水解氯基,由R1是 且R6和R7是氢的式1化合物制备上述的化合物。
且R6和R7是氢的式1化合物制备上述的化合物。
其中R1是 且R6和R7是氢的式1化合物可以从式12化合物制备。将该化合物氢解(例如用甲酸铵和Pd/C的甲醇液;PdOH/C和环己烷的乙醇液;或Pd/C、氢和乙醇),使得氯基还原并除去苄基保护基,从而得到式21化合物。
且R6和R7是氢的式1化合物可以从式12化合物制备。将该化合物氢解(例如用甲酸铵和Pd/C的甲醇液;PdOH/C和环己烷的乙醇液;或Pd/C、氢和乙醇),使得氯基还原并除去苄基保护基,从而得到式21化合物。
在吡啶中,将式21化合物与1,3-二氯-1,1,3,3-四异丙基二硅氧烷反应,得到式22化合物。
在三乙胺以及任意的在4-N,N-二甲氨基吡啶存在下,通过与三苯甲基氯或取代的三苯甲基氯反应,将式22化合物中的氨基(-NH2)保护,得到式23化合物, 其中P1是三苯甲基或取代的三苯甲基。在分别与制备式14和15化合物中所采用的相似条件下,将式23化合物相继氧化和亚甲基化,然后除去保护基,得到式1化合物,其中R1是
其中P1是三苯甲基或取代的三苯甲基。在分别与制备式14和15化合物中所采用的相似条件下,将式23化合物相继氧化和亚甲基化,然后除去保护基,得到式1化合物,其中R1是 且R6和R7是氢。首先可以通过用氟离子(例如氟化四丁基铵)处理除去甲硅烷基保护基,然后通过用无机酸的含水醇溶液(例如盐酸的甲醇水溶液)或含水乙酸处理除去三苯甲基保护基P1。或者,首先除去三苯甲基保护基P1,然后除去甲硅烷基保护基。
且R6和R7是氢。首先可以通过用氟离子(例如氟化四丁基铵)处理除去甲硅烷基保护基,然后通过用无机酸的含水醇溶液(例如盐酸的甲醇水溶液)或含水乙酸处理除去三苯甲基保护基P1。或者,首先除去三苯甲基保护基P1,然后除去甲硅烷基保护基。
或者,在与制备式12化合物中所采用的相似条件下,将式24化合物与式2化合物反应, (式24中P1是三苯甲基或取代的三苯甲基),得到式25化合物。
(式24中P1是三苯甲基或取代的三苯甲基),得到式25化合物。
在分别与制备化合物14和15中所采用的相似条件下,相继地将式25化合物氧化和亚甲基化,然后除去保护基,得到式1化合物,其中R1是 且R6和R7是氢。
且R6和R7是氢。
或者,将式26化合物与式2化合物反应, 得到式27化合物。
得到式27化合物。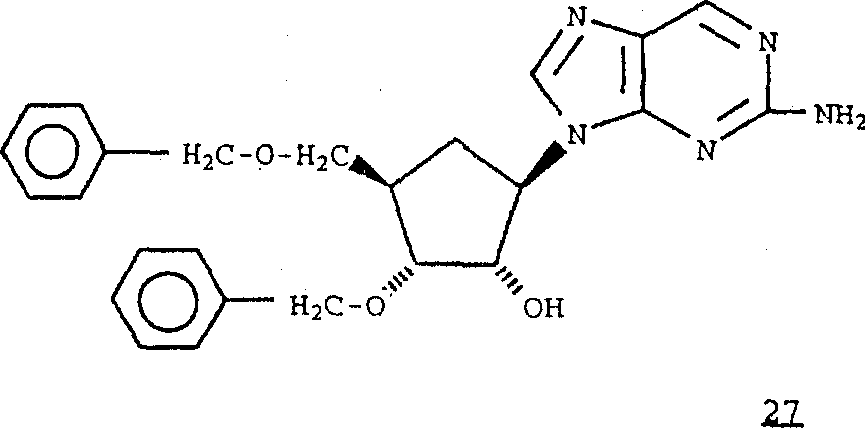 用三苯甲基或取代的三苯甲基保护氨基(-NH2),得到式25化合物,然后可将式25化合物转化为式1化合物(如上所述),其中R1是
用三苯甲基或取代的三苯甲基保护氨基(-NH2),得到式25化合物,然后可将式25化合物转化为式1化合物(如上所述),其中R1是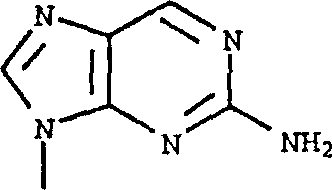 且R6和R7是氢。
且R6和R7是氢。
按照本领域已知的方法,可以从式15化合物制备式1化合物,其中R1是 且R6和R7是氢。因此,例如,当其中P1是三苯甲基的式15化合物用热的氨甲醇液处理时,氯被氨基取代。然后可以按照本领域已知的方法除去保护基。
且R6和R7是氢。因此,例如,当其中P1是三苯甲基的式15化合物用热的氨甲醇液处理时,氯被氨基取代。然后可以按照本领域已知的方法除去保护基。
或者,可以按照本领域已知的方法,用其中R1是 且R6和R7是氢的式1化合物,制备所述的式1化合物。[参见例如:J.C.Martin等人,J.Med.Chem.Vol.28,358(1985)]。通过用醇钠处理式12化合物,可以制备其中R1是
且R6和R7是氢的式1化合物,制备所述的式1化合物。[参见例如:J.C.Martin等人,J.Med.Chem.Vol.28,358(1985)]。通过用醇钠处理式12化合物,可以制备其中R1是 且R6和R7是氢的式1化合物,由此得到了式28化合物。
且R6和R7是氢的式1化合物,由此得到了式28化合物。 保护氨基、氧化并进行亚甲基化(按照与制备化合物13、14和15中所采用的相似方法),然后除去保护基,得到式1化合物,其中R1是
保护氨基、氧化并进行亚甲基化(按照与制备化合物13、14和15中所采用的相似方法),然后除去保护基,得到式1化合物,其中R1是 且R6和R7是氢。或者,按照本领域已知的方法,可以从其中R1是
且R6和R7是氢。或者,按照本领域已知的方法,可以从其中R1是 且R6和R7是氢的式1化合物制备该式1化合物。参见文献:例如J.F.Gerster,et al.,J.Amer.Chem.Soc.,87,3752(1965);K.K.ogilvie,et al.,Can.J.Chem.62,2702(1984);M.R.Harnden,et al.,J.Med.Chem.,30,1636(1987)。
且R6和R7是氢的式1化合物制备该式1化合物。参见文献:例如J.F.Gerster,et al.,J.Amer.Chem.Soc.,87,3752(1965);K.K.ogilvie,et al.,Can.J.Chem.62,2702(1984);M.R.Harnden,et al.,J.Med.Chem.,30,1636(1987)。
或者,利用与制备化合物12、13、14和15中所采用的相似方法,由式29化合物和式2化合物制备该式1化合物,然后除去保护基。式29化合物可以按照本领域已知的方法由式11化合物制备[参见:例如W.A.Bowles,et al.,J.Med.Chem.,6,471(1963);M.MacCoss,etal.,Tetrahedron Lett.,26,1815(1985)]。
按照与制备化合物17、18、19和20中所采用的相似方法,可以由式2化合物和式30化合物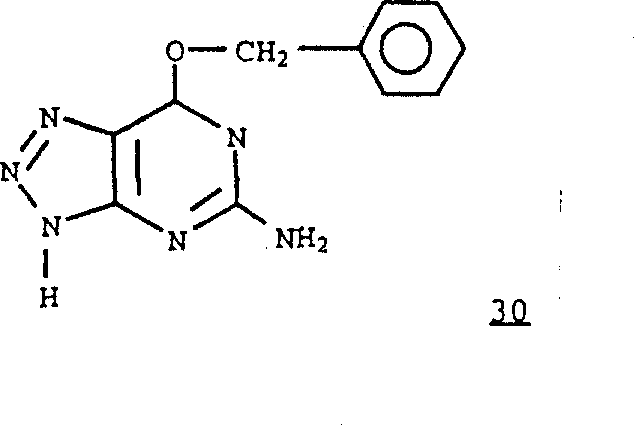 制备其中R1是
制备其中R1是 且R6和R7是氢的式1化合物,然后除去保护基。
且R6和R7是氢的式1化合物,然后除去保护基。
按照与制备式12化合物中所采用的相似方法,通过式31化合物 与式2化合物反应,可以制备其中R1是
与式2化合物反应,可以制备其中R1是 且R6和R7是氢的式1化合物,得到式32化合物。
且R6和R7是氢的式1化合物,得到式32化合物。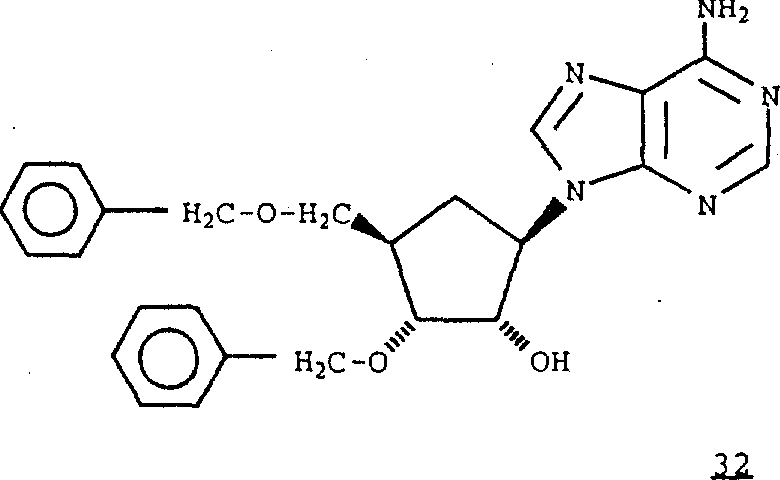
用酰基P2(例如乙酰基)选择性保护式32化合物中的氨基(-NH2),得到式33化合物,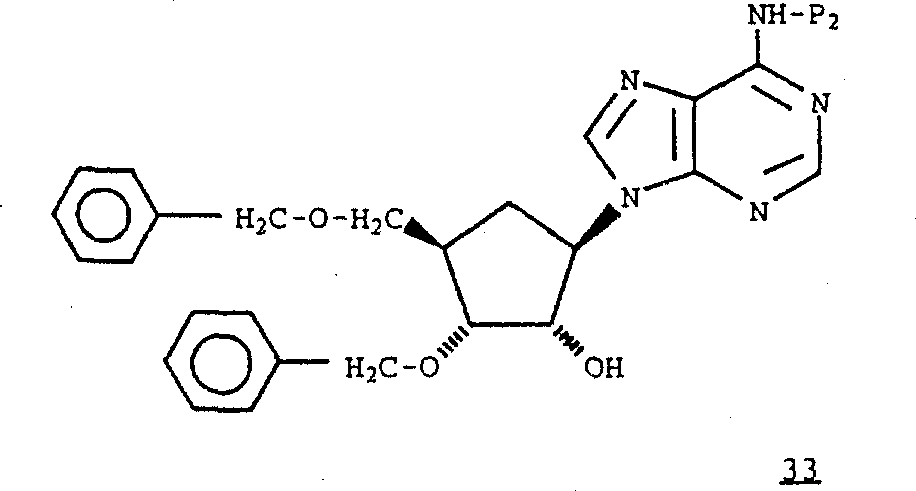 [参见例如G.S.Ti等人,J.Amer.Chem.Soc.,104,1316(1982)]。按照与制备化合物14和15中所采用的相似方法,将式33化合物氧化,然后用锌/四氯化钛/二溴甲烷或亚甲基三苯基正膦处理,得到式34化合物。
[参见例如G.S.Ti等人,J.Amer.Chem.Soc.,104,1316(1982)]。按照与制备化合物14和15中所采用的相似方法,将式33化合物氧化,然后用锌/四氯化钛/二溴甲烷或亚甲基三苯基正膦处理,得到式34化合物。
从该化合物中除去保护基,得到其中R1是 且R6和R7是氢的式1化合物。通过用甲醇钠的甲醇液或氨的甲醇液处理,可以除去氨基保护基P2。
且R6和R7是氢的式1化合物。通过用甲醇钠的甲醇液或氨的甲醇液处理,可以除去氨基保护基P2。
或者,在与制备式12化合物中所采用的相似条件下,通过式35化合物 与式2化合物反应,可以制备该式1化合物。按照与制备化合物14和15所采用的相似方法,相继地进行氧化和亚甲基化,可得到式36化合物。
与式2化合物反应,可以制备该式1化合物。按照与制备化合物14和15所采用的相似方法,相继地进行氧化和亚甲基化,可得到式36化合物。
用在热的醇(如甲醇或乙醇)中的氨处理式36化合物,然后除去保护基,得到其中R1是 且R6和R7是氢的式1化合物。或者,可以按照本领域已知的方法,由其中R1是
且R6和R7是氢的式1化合物。或者,可以按照本领域已知的方法,由其中R1是 且R6和R7是氢的式1化合物,制备该式1化合物[参见:例如J.C.Martin等人,J.Med.Chem.,28,358(1985)]。
且R6和R7是氢的式1化合物,制备该式1化合物[参见:例如J.C.Martin等人,J.Med.Chem.,28,358(1985)]。
用三氯化硼从式36化合物中除去保护基可以制备其中R1是 且R6和R7是氢的式1化合物。
且R6和R7是氢的式1化合物。
按照已知的方法,可以由其中R1是 且R6和R7是氢的式1化合物制备其中R1是且R6和R7是氢的式1化合物,参见文献:例如J.A.Montgomery等人,“Synthesic Procedures in Nucleic Acid Chemistry”,Vol.l,W.W.Zorbach和R.S.Tipson,Eds.,IntersciencePublishers(John Wiley and Sons),N.Y.p.205,1968。
且R6和R7是氢的式1化合物制备其中R1是且R6和R7是氢的式1化合物,参见文献:例如J.A.Montgomery等人,“Synthesic Procedures in Nucleic Acid Chemistry”,Vol.l,W.W.Zorbach和R.S.Tipson,Eds.,IntersciencePublishers(John Wiley and Sons),N.Y.p.205,1968。
通过酸水解(例如热盐酸水溶液)或碱水解(例如氢氧化钠的水甲醇溶液),可以由其中R1是 且R6和R7是氢的式1化合物制备其中R1是且R6和R7是氢的式1化合物。
且R6和R7是氢的式1化合物制备其中R1是且R6和R7是氢的式1化合物。
或者,按照本领域已知的方法,通过用亚硝酸的腺苷脱氨酶处理其中R1是 且R6和R7是氢的式1化合物,可以制备该式1化合物[参见:例如M.J.Robins,et al.,J.Med.Chem.,27,1486(1984);K.K.Ogilvie et al.,Can.J.Chem.,62 241(1984)];I.Iwai,etal.,in“Synthetic Procedures in Nucleic Acid Chemistry”,Vol.l,W.W.Zorbach and R.S.Tipson,Eds.,IntersciencePublishers(John Wiley and Sons),N.Y.p.135,1968;R.E.Holmes et al.,J.Amer.Chem.Soc.,86,1242(1964)。
且R6和R7是氢的式1化合物,可以制备该式1化合物[参见:例如M.J.Robins,et al.,J.Med.Chem.,27,1486(1984);K.K.Ogilvie et al.,Can.J.Chem.,62 241(1984)];I.Iwai,etal.,in“Synthetic Procedures in Nucleic Acid Chemistry”,Vol.l,W.W.Zorbach and R.S.Tipson,Eds.,IntersciencePublishers(John Wiley and Sons),N.Y.p.135,1968;R.E.Holmes et al.,J.Amer.Chem.Soc.,86,1242(1964)。
在碱如氢化锂、氢化钠或氢化钾存在下,在极性非质子溶剂(如二甲基甲酰胺、二甲基亚砜或环丁砜)中,将相应的式38化合物 与式2化合物反应,可以制备式37化合物,
与式2化合物反应,可以制备式37化合物, 其中R2是氢、氟、甲基、乙基、正丙基、2-氯乙基或2-氟乙基;从而可制得式39化合物。
其中R2是氢、氟、甲基、乙基、正丙基、2-氯乙基或2-氟乙基;从而可制得式39化合物。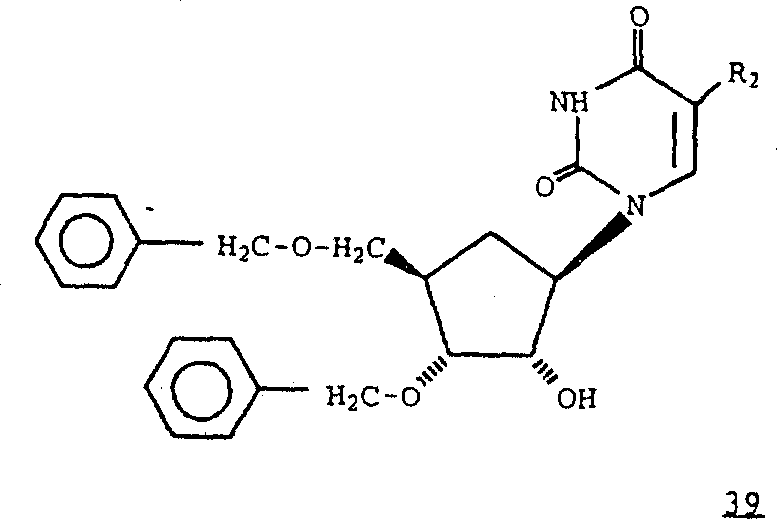
还可以在极性非质子溶剂如二甲基甲酰胺、二甲基亚砜或环丁砜中,通过式38化合物的单四烷基铵盐(例如四丁基铵盐)与式2化合物反应,制备式39化合物。式38化合物的单四烷基铵盐可以在极性非质子溶剂如二甲基甲酰胺中、通过式38化合物与氢氧化四烷基铵(例如氢氧化四丁基铵)的水溶液反应进行制备。
采用本领域已知的方法氧化式39化合物中的羟基(例如在二氯甲烷中的草酰氯/二甲基亚砜/三乙胺或在二氯甲烷中的1,1,1-三乙酸基-1,1-二氢-1,2-benziodoxol-3(1H)-酮和叔丁醇),可得到式40化合物。
用亚甲基化试剂如在二氯甲烷/四氢呋喃中的锌/四氯化钛/二溴甲烷或亚甲基三苯基正膦处理式40化合物,得到式41化合物。
从式41化合物除去保护基,得到相应的式37化合物。通过用三氯化硼处理可以除去苄基。
其中R2是2-氯乙基或2-氟乙基的式38化合物可以按本领域已知的方法制备[参见:H.Griengl等人,J.Med.,30,1199(1987);J.Med.Chem.,28,1679(1985)]。
其中R2是氟的式39化合物还可以按本领域已知的方法,通过用次氟酸三氟甲酯进行氟化,由其中R2是氢的相应的化合物39制备。例如参见:M.J.Robins.等人,J.Amer.Chem.Soc.,93,5277(1971);Chem.Communs.,18(1972);T.S.Lin等人,J.Med,Chem.,26,1691(1983)。
其中R2是2-氯乙基和2-氟乙基的式37化合物也可以从式42化合物制备其中P3是三苯甲基、取代的三苯甲基或甲硅烷基保护基。除去P3保护基,得到其中R2是2-羟乙基的式41化合物。用三苯膦/四氯化碳处理该化合物,然后用三氯化硼除去苄基保护基,得到其中R2是2-氯乙基的式37化合物。同样地,用三苯膦/N-溴琥珀酰亚胺或三苯膦/N-溴琥珀酰胺/四丁基碘化铵代替三苯膦/四氯化碳进行处理[例如参见H.Griengl等人的J.Med.Chem.,28,1679(1985)],可分别得到其中P是苄基且R2是溴乙基或2-碘乙基的式41化合物。然后用氟离子处理,除去苄基保护基,得到其中R2是2-氟乙基的式37化合物。或者,用二乙氨基三氟化硫处理其中R2是2-羟乙基的式41化合物,然后除去苄基保护基,得到其中R2是2-氟乙基的式37化合物。
按照与制备化合物39、40和41(其中R2例如是氢、甲基或乙基)中所采用的相似方法,可以由式43化合物和式2化合物制备式42化合物。式43化合物可以按照本领域已知的方法从相应的游离醇制备。
可以从相应的式41化合物制备式44化合物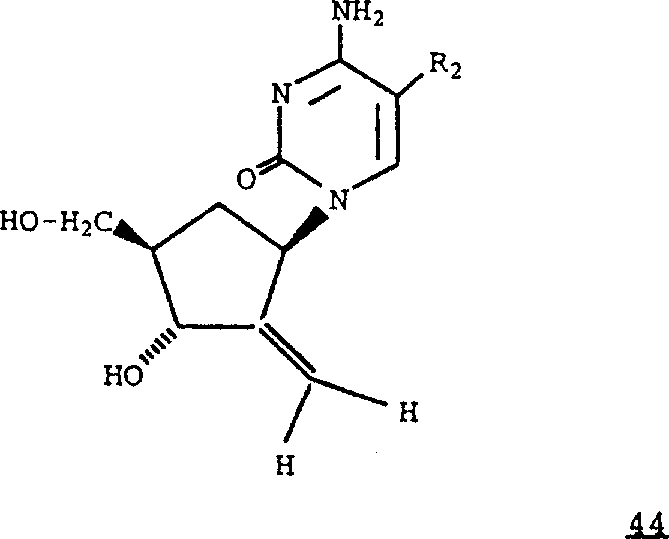 其中R2是氢、氟、甲基、乙基、正丙基、2-氯乙基或2-氟乙基。在溶剂如吡啶中,用例如二氯磷酸4-氯苯酯和1,2,4-三唑处理该化合物,并在溶液(如二噁烷)中用氢氧化铵与所得中间体反应,得到相应的式45化合物,参见文献:例如T.S.Lin等人,J.Med.Chem.,26,1691(1983);P.Herdewijn等人,J.Med.Chem.,28,550(1985)。除去式45化合物的保护基,得到相应的式44化合物。用三氯化硼处理可以除去苄基保护基。或者,可以在惰性溶剂(例如1,2-二氯乙烷或二噁烷)中,在碱(如碳酸钾)存在下,将其中R2是氢、氟、甲基、乙基、正丙基、2-氯乙基或2-氟乙基的式41化合物与磺酰氯(例如对甲苯磺酰氯)反应(其他的磺酰氯和溶剂可参见例如EP204264),由此得到相应的式46化合物。
其中R2是氢、氟、甲基、乙基、正丙基、2-氯乙基或2-氟乙基。在溶剂如吡啶中,用例如二氯磷酸4-氯苯酯和1,2,4-三唑处理该化合物,并在溶液(如二噁烷)中用氢氧化铵与所得中间体反应,得到相应的式45化合物,参见文献:例如T.S.Lin等人,J.Med.Chem.,26,1691(1983);P.Herdewijn等人,J.Med.Chem.,28,550(1985)。除去式45化合物的保护基,得到相应的式44化合物。用三氯化硼处理可以除去苄基保护基。或者,可以在惰性溶剂(例如1,2-二氯乙烷或二噁烷)中,在碱(如碳酸钾)存在下,将其中R2是氢、氟、甲基、乙基、正丙基、2-氯乙基或2-氟乙基的式41化合物与磺酰氯(例如对甲苯磺酰氯)反应(其他的磺酰氯和溶剂可参见例如EP204264),由此得到相应的式46化合物。 在惰性溶剂(例如1,2-二氯乙烷或二噁烷)中用氨气处理化合物46,得到相应的式45化合物。除去式45化合物的保护基,得到相应的式44化合物。
在惰性溶剂(例如1,2-二氯乙烷或二噁烷)中用氨气处理化合物46,得到相应的式45化合物。除去式45化合物的保护基,得到相应的式44化合物。
或者,可以按照本领域已知的方法,从相应的式37化合物制备其中R2是氟、氢、甲基、乙基、正丙基、2-氯乙基或2-氟乙基的式44化合物。参见例如T.S.Lin等人,J.Med.Chem.,26,1691(1983);P.Herdewijn等人,J.Med.Chem.,28,550(1985);EP360,018。
或者,可以在极性非质子溶剂如二甲基甲酰胺、二甲基亚砜或环丁烷中,在氢化锂、氢化钠或氢化钾存在下,通过相应的式47化合物 与式2化合物反应,制备其中R2是氟、氢、甲基、乙基、正丙基、2-氯乙基或2-氟乙基的式44化合物,从而得到式48化合物。
与式2化合物反应,制备其中R2是氟、氢、甲基、乙基、正丙基、2-氯乙基或2-氟乙基的式44化合物,从而得到式48化合物。
用酰基R2(例如乙酰基)选择性保护式48化合物中的氨基,得到式49化合物, [参见:例如G.S.Ti等人,J.Amer.Chem.Soc.,104,1316(1982)]。氧化式49化合物,然后用锌/四氯化钛/二溴甲烷进行亚甲基化,随后或同时除去酰基保护基P2,得到式45化合物。除去该化合物的保护基,得到其中R2是氟、氢、甲基、乙基、正丙基、2-氯乙基或2-氟乙基的式44化合物。通过在二氯甲烷中用三氯化硼处理可以除去苄基。
[参见:例如G.S.Ti等人,J.Amer.Chem.Soc.,104,1316(1982)]。氧化式49化合物,然后用锌/四氯化钛/二溴甲烷进行亚甲基化,随后或同时除去酰基保护基P2,得到式45化合物。除去该化合物的保护基,得到其中R2是氟、氢、甲基、乙基、正丙基、2-氯乙基或2-氟乙基的式44化合物。通过在二氯甲烷中用三氯化硼处理可以除去苄基。
或者,氧化式49化合物,并用亚甲基三苯基正膦处理,得到式50化合物, 除去该化合物的保护基,得到其中R2是氟、氢、甲基、乙基、正丙基、2-氯乙基或2-氟乙基的式44化合物。使用甲醇钠的甲醇液或氨甲醇液可以除去酰基保护基P2。
除去该化合物的保护基,得到其中R2是氟、氢、甲基、乙基、正丙基、2-氯乙基或2-氟乙基的式44化合物。使用甲醇钠的甲醇液或氨甲醇液可以除去酰基保护基P2。
或者,可以采用本领域已知的方法,从相应的式39化合物制备其中R2是氢、氟、甲基、乙基、正丙基、2-氯乙基或2-氟乙基的式48化合物。参见例如T.S.Lin等人,J.Med.Chem.,26,1691(1983);P.Herdewijn等人,J.Med.Chem.,28,550(1985);EP360018和EP204264。
或者,可以采用本领域已知的方法,通过用次氟酸三氟甲酯进行氟化,由其中R2是氢的相应的式48或49化合物制备其中R2是氟的式48或49化合物。例如参见M.J.Robins等人,J.Amer.Chem.Soc.,93,5277(1971)和Chem.Communs.,18(1972);T.S.Lin等人,J.Med.Chem.,26,1691(1983)。
其中R2是氯、溴或碘的式37化合物可以由其中R2是氢的式39化合物制备。按本领域已知的方法将该化合物卤化,得到相应的其中R2是氯、溴或碘的式39化合物,参见文献:例如P.Herdwijn et al.,J.Med.Chem,28,550(1985);M.J.Robins et al.,J,Oro.Chem.,48,1854(1983);T.S.Lin et al.,J.Med.Chem,26,598(1983);T.Ueda et al.,Nucleotides and Nucleosides,4,401(1985);"Synthetic Procedures in Nucleic AcidChemistry",Vol.l,W.W.Zorback and R.S.Tipson,Eds.,John Wiley and Sons,NY,p.491,(1968).将其中R2是氯、溴或碘的式39化合物氧化,然后用锌/四氯化钛/二溴甲烷或用亚甲基三苯基正膦进行亚甲基化,得到相应的其中R2是氯、溴或碘的式41化合物(参见例如EP360018)。通过用三氯化硼处理除去苄基保护基,得到其中R2是氯、溴或碘的式37化合物。
采用本领域已知的方法以及与将化合物41转化为化合物44(其中例如R2是氢、甲基或乙基)中所采用的相似方法,可以由相应的式41式化合物制备其中R2是氯、溴或碘的式44化合物。
或者,可以按照本领域已知的方法,从相应的式37化合物制备其中R2是氯、溴或碘的式44化合物。参见:例如T.S.Lin等人,J.Med.Chem.,26,1691(1983);P.Herdewijn等人,J,Med.Chem.,28,550(1985):EP 360018和EP 204264。
通过用三氟甲基碘和铜处理,然后用三氯化硼除去苄基保护基,可以由其中R2是碘的式41化合物制备其中R2是三氟甲基的式37化合物。参见例如Y.Kobayashi等人,J.Chem.Soc,Perkin 1,2755(1980);T.S.Lin等人,J.Med.Chem.,26,1691(1983)。
其中R2是三氟甲基的式37化合物可以按下述方法由其中R2是碘的式39化合物制备:可将其中R2是碘的式39化合物转化为式51化合物,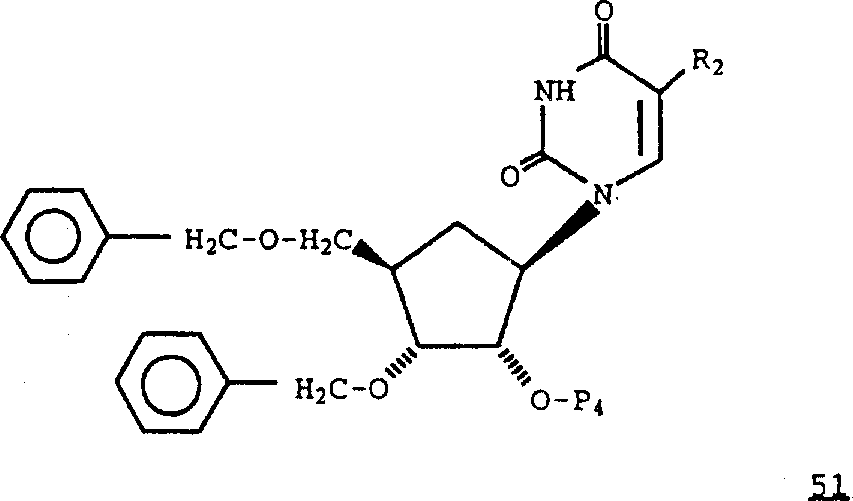 其中R2是碘,保护基P4是三苯甲基、取代的三苯甲基或酰基(例如乙酰基)。按本领域已知的方法,用三氟甲基碘和酮处理式51化合物(参见例如Y.Kobayashi等人,J.Chem.Soc.Perkin,2755(1980);T.S.Lin等人,J.Med.Chem.,26,1691(1983)),然后除去P4保护基,得到其中R2是三氟甲基的式39化合物。氧化其中R2是三氟甲基的式39化合物,然后用锌/四氯化钛/二溴甲烷或用亚甲基三苯基正膦处理,得到其中R2是三氟甲基的式41化合物。通过用三氯化硼处理除去苄基保护基,得到其中R2是三氟甲基的式37化合物。
其中R2是碘,保护基P4是三苯甲基、取代的三苯甲基或酰基(例如乙酰基)。按本领域已知的方法,用三氟甲基碘和酮处理式51化合物(参见例如Y.Kobayashi等人,J.Chem.Soc.Perkin,2755(1980);T.S.Lin等人,J.Med.Chem.,26,1691(1983)),然后除去P4保护基,得到其中R2是三氟甲基的式39化合物。氧化其中R2是三氟甲基的式39化合物,然后用锌/四氯化钛/二溴甲烷或用亚甲基三苯基正膦处理,得到其中R2是三氟甲基的式41化合物。通过用三氯化硼处理除去苄基保护基,得到其中R2是三氟甲基的式37化合物。
采用本领域已知的方法,以及与将化合物41转化为化合物44(其中例如R2是氢、甲基或乙基)中所采用的相似方法,可以由相应的式41化合物制备其中R2是三氟甲基的式44化合物。
或者,按本领域已知的方法,可以从相应的式37化合物制备其中R2是三氟甲基的式44化合物。参见例如T.S.Lin等人,J.Med.Chem.,26,1691(1983);P.Herdewijn等人,J.Med,Chem.,28,550(1985);EP 360018和EP 204264。其中R2是 且R3是氯、溴、碘、氢、甲基或三氟甲基的式37化合物可以由其中R2是碘或-HgCl的式39化合物制备。通过有机钯中间体使R2是碘或-HgCl的式39化合物反应,得到其中R2是且R3是氯、溴、碘、氢、甲基或三氟甲基的式39化合物。其中R2是HgCl的式39化合物可以由其中R2是氢的式39化合物制备。参见文献:例如E.DeClercq et al.,Pharmac.Ther.,26,1(1984);M.E.Perlman et al.,J.Med.Chem.,28,741(1985);P,Herdewijnet al.,J.Med.,Chem.,28,550(1985);D.E.Bergstrom etal.,J.Med.Chem.,27,279(1984)。
且R3是氯、溴、碘、氢、甲基或三氟甲基的式37化合物可以由其中R2是碘或-HgCl的式39化合物制备。通过有机钯中间体使R2是碘或-HgCl的式39化合物反应,得到其中R2是且R3是氯、溴、碘、氢、甲基或三氟甲基的式39化合物。其中R2是HgCl的式39化合物可以由其中R2是氢的式39化合物制备。参见文献:例如E.DeClercq et al.,Pharmac.Ther.,26,1(1984);M.E.Perlman et al.,J.Med.Chem.,28,741(1985);P,Herdewijnet al.,J.Med.,Chem.,28,550(1985);D.E.Bergstrom etal.,J.Med.Chem.,27,279(1984)。
将其中R2是 且R3是氯、溴、碘、氢、甲基或三氟甲基的式39化合物氧化,然后用锌/四氯化钛/二溴甲烷或用亚甲基三苯基正膦处理,得到相应的式41化合物。用三氯化硼除去该化合物的苄基保护基,得到其中R2是且R3是氯、溴、碘、氢、甲基或三氟甲基的式37化合物。
且R3是氯、溴、碘、氢、甲基或三氟甲基的式39化合物氧化,然后用锌/四氯化钛/二溴甲烷或用亚甲基三苯基正膦处理,得到相应的式41化合物。用三氯化硼除去该化合物的苄基保护基,得到其中R2是且R3是氯、溴、碘、氢、甲基或三氟甲基的式37化合物。
按照本领域已知的方法,可以从由相应的式37化合物制备其中R2是 且R3是氯、溴、碘、氢、甲基或三氟甲基的式44化合物。参见例如T.S.Lin等人,J.Med.Chem.,26,1691(1983);P.Herdewijn等人,J.Med.Chem.,28,550(1985);EP 360018和EP 204264。
且R3是氯、溴、碘、氢、甲基或三氟甲基的式44化合物。参见例如T.S.Lin等人,J.Med.Chem.,26,1691(1983);P.Herdewijn等人,J.Med.Chem.,28,550(1985);EP 360018和EP 204264。
或者,采用本领域已知的方法以及在将化合物41转化为化合物44(其中例如R2是氢、甲基或乙基)中所用的类似方法,可以由相应的式41化合物制备其中R2是 且R3是氯、溴、碘、氢、甲基或三氟甲基的式44化合物。
且R3是氯、溴、碘、氢、甲基或三氟甲基的式44化合物。
其中R2是乙炔基的式37化合物可以由其中R2是碘且保护基P4是酰基(例如乙酰基)、三苯甲基或取代的三苯甲基的式51化合物制备。在三乙胺中用三甲基甲硅烷基乙炔/二(三苯膦)氯化钯(Ⅱ)/碘化酮(Ⅰ)处理其中R2是碘且P4是酰基的式51化合物,然后,按照本领域已知的方法用在甲醇中的甲醇钠处理[参见例如E.DeClercq等人,J.Med.Chem.,26,661(1983)],得到其中R2是乙炔基的式39化合物。或者,相似地处理其中R2是碘且P4是三苯甲基或取代的三苯甲基的式51化合物,然后用例如乙酸水溶液或无机酸醇水溶液除去三苯甲基或取代的三苯甲基保护基P4,得到其中R2是乙炔基的式39化合物。氧化式39化合物,然后用锌/四氯化钛/二溴甲烷或用亚甲基三苯基正膦进行亚甲基化,得到其中R2是乙炔基的式41化合物,随后用三氯化硼除去苄基保护基,得到其中R2是乙炔基的式37化合物。
采用本领域已知的方法以及与将化合物41转化为化合物44(其中例如R2是氢、甲基或乙基)中所用的相似的方法,可以从相应的式41化合物制备其中R2是乙炔基的式44化合物。
或者,可以按照本领域已知的方法,从相应的式37化合物制备其中R2是乙炔基的式44化合物。参见例如:T.S.Lin等人,J.Med.Chem.,26,1691(1983);P.Herdewijn等人,J.Med.Chem.,28,550(1985);EP 360018和EP 204264。
其中R1是的式1化合物可以按照本领域已知的方法,由其中R1是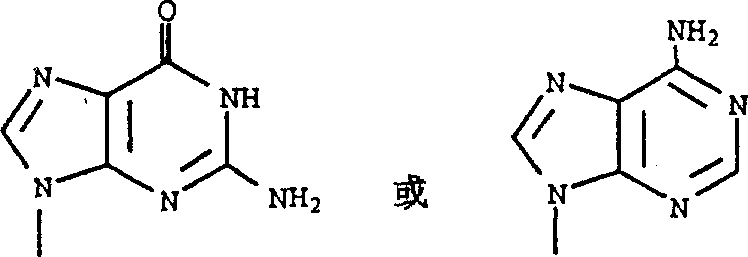 的相应的式1化合物制备。
的相应的式1化合物制备。
其中R6和R7之中一个或两个是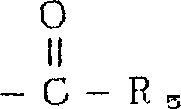 的式1化合物可以按照本领域已知的方法,由相应的其中R6和R7为氢的式1化合物制备。
的式1化合物可以按照本领域已知的方法,由相应的其中R6和R7为氢的式1化合物制备。
酰化方法的例子可参见下列文献:"Synthetic Procedures in Nucleic Acid Chemistry",Vol.1,W.W.Zorbach and R.S.Tipson,Eds.,John Wileyand Sons,1968;"Nucleic Acid Chemistry,"Part 1,L.B.Townsend and R.S.Tipson,Eds.,John Wiley andSons,1978;Y,Ishido,et al.,Nucleosides andNucleotides.5,159(1986);J.C.Martin,et al.,J.Pharm.Sci.,76,180(1987);A Matsuda,et al.,Synthesis,385(1986).其中R1是 的式1化合物可以按照本领域已知的方法,由相应的其中R1是
的式1化合物可以按照本领域已知的方法,由相应的其中R1是 的式1化合物制备。参见文献:例如A.Holy and J.Zemlicka,Collect.Czech.Chem.Commun.,32,3159(1967);K.K.Ogilvie,et al.,Nucleosides and Nucleotides,4,507(1985);M.H.Caruthers,et al.,J.Amer.Chem.Soc.,108,2040(1986)。
的式1化合物制备。参见文献:例如A.Holy and J.Zemlicka,Collect.Czech.Chem.Commun.,32,3159(1967);K.K.Ogilvie,et al.,Nucleosides and Nucleotides,4,507(1985);M.H.Caruthers,et al.,J.Amer.Chem.Soc.,108,2040(1986)。
其中R6和/或R7是-PO3H2的式1化合物可以按照本领域已知的方法,由相应的其中R6和R7是氢的式1化合物制备。参见例如H.Schallet,et al.,J.Amer.Chem.Soc.,85,3821(1963);J.Beres,et al.,J.Med.Chem.,29,494(1986);R.Noyori,et al.,Tet.Lett.,28,2259(1987);W.Pfeiderer,et al.,Helv.Chim.Acta.,70,1286(1987);“Nucleic AcidChemistry”,Part 2,L.B.Townsend and R.S.Tipson,Eds.,John Wiley and Sons,1978。
除非另外指明,本发明化合物以及制备本发明化合物的中间体的立体化学是相对的,而不是绝对的。在本发明化合物图示中,通过R1与-CH2OR6取代基为顺式而与直接连接于环戊环上的-OR7取代基为反式来表示。其中R1是下列基团

 的式1化合物可以与无机酸或有机酸形成酸加成盐。所述盐的实例是:例如卤化物(如氯化物和溴化物)、烷基磺酸盐、硫酸盐、磷酸盐和羧酸盐。
的式1化合物可以与无机酸或有机酸形成酸加成盐。所述盐的实例是:例如卤化物(如氯化物和溴化物)、烷基磺酸盐、硫酸盐、磷酸盐和羧酸盐。
其中R1是下列基团的式1化合物可以与无机或有机碱形成碱性盐。所述盐的实例是:例如碱金属盐(如钠和钾盐)、碱土金属盐(例如钙和镁盐)、铵盐和取代的铵盐。
其中R6和/或R7是-PO3H2的式1化合物可以与无机碱或有机碱形成碱性盐。所述盐的实例是:例如碱金属盐(如钠和钾盐)、碱土金属盐(如钙和镁盐)、铵盐和取代的铵盐。
下列实施例是本发明的具体实施方案。所有温度均为摄氏度。
实施例1(1α,3β,4α)-2-氨基-1,9-二氢-9-[3-羟基-4-(羟甲基)-2-亚甲基环戊基]-6H-嘌呤-6-酮a)3-[(苯甲氧基)甲基]环戊烯
将2-环戊烯-1-甲基[O.L.Chapman等人,J.Amer,Chem.Soc.,100,4878(1978);A.Maercker和R.Geuss,Chem.Ber.,106,773(1973)](14.5g,0.147mol)经3°A分子筛干燥,将其溶于无水二甲基甲酰胺(80ml)中,并干0℃、在搅拌下经套管转入于无水二甲基甲酰胺(60ml)中的氢化钠(7.06g,0.176mol,60%油分散液)悬浮液中,然后滴加苄基溴(10.9ml,0.09mol),并使反应混合物温热至室温过夜。用冷却的水(150ml)终止反应,并用乙酸乙酯(200ml)萃取水层三次。合并的有机层用盐水洗涤,用硫酸钠干燥,过滤并真空浓缩,得到残余物。该残余物经闪式色谱(Merck硅胶-60,230-400目,4.5升)纯化,得到无色油状标题化合物(19.1g)。b)2-[(苯甲氧基)甲基]-6-氧杂双环[3.1.0]己烷
在0℃,将3-[(苯甲氧基)甲基]-环戊烯(0.495g,2.63mmol)在对二噁烷(3.37ml)中的溶液加至N-溴琥珀酰亚胺(0.562g,3.16mmol)在水(3.37ml)中的悬浮液中。5分钟后,使反应混合物温热至室温并在此温度搅拌3小时。然后将反应混合物真空浓缩并在水和乙醚之间分配。用乙醚萃取水层,并用水和盐水洗涤合并的有机层,然后用硫酸钠干燥。真空除去挥发性物质,剩下黄色油(0.389g),不需进一步纯化即将该残余物用于下一反应。
在氮气氛下,将上述残余物(0.389g)溶于甲醇(4.3ml)中,并加入碳酸钾(0.473g,3.42mmol)。4小时后,将反应混合物真空浓缩,并将残余物在二氯甲烷和水之间分配。用二氯甲烷萃取水层,用硫酸镁干燥合并的有机层,并真空浓缩,得到黄色油。残余物经闪式色谱(Merck硅胶-60,230-400目,50ml)纯化,用1%丙酮/己烷洗脱,得到无色油状标题化合物(0.254g)。c)(反式)-5-[(苯甲氢基)甲基]-2-环戊烯-1-醇
小心地将硼氢化钠加至在脱气无水乙醇(150ml)中的二苯基二硒(8.43g,0.027mol)中,直到无色溶液形成,就地制备苯硒化钠。在室温向快速搅拌着的2-[(苯甲氧基)甲基]-6-氧杂双环[3.1.0]己烷(5.0g,0.02mol)在脱气无水乙醇(40ml)中的溶液中滴加该溶液(90ml)。将反应混合物在90℃回流30分钟,然后冷却至室温并用四氢呋喃(60ml)稀释。稀释后,将反应物再冷却至0℃,并滴加30%过氧化氢(25ml)。加完过氧化氢后,使反应物温热至室温过夜,然后加热回流2.5小时。将反应物冷却至室温并用乙醚(250ml)稀释。除去水层,并用饱和碳酸氢钠和水洗涤有机层,有机层用硫酸钠干燥,过滤并真空浓缩,得到黄色油。该油经闪式色谱(Merck硅胶-60,230-400目,1.5升)纯化,用30%乙酸乙酯-己烷洗脱,得到油状标题化合物(2.35g)。d)(1α,2β,3α,5α)-3-[(苯甲氧基)甲基]-6-氧杂双环[3.1.0]己-2-醇
在0℃。向搅拌着的步骤(c)产物(2.35g,0.01mol)和磷酸钠(8.09g,0.06mol)在二氯甲烷(60ml)中的悬浮液中加入3-氯过苯甲酸(4.90g,0.03mol)。在于室温搅拌过夜后,用饱和碳酸氢钠和硫代硫酸钠稀释反应物。除去有机溶剂,用乙酸乙酯(75ml)洗涤水层两次。用盐水洗涤合并的有机层,用硫酸钠干燥并浓缩,得到黄色油。该油经闪式色谱(Merck硅胶-60,230-400目)纯化,用15%乙酸乙酯-己烷洗脱,然后用50%乙酸乙酯-己烷洗脱,得到无色油状标题化合物(1.83g)。e)(1α,2β,3α,5α)-2-(苯甲氧基)-3-[(苯甲氧基)甲基]-6-氧杂双环[3.1.0]己烷
向步骤(d)产物(3.6g,16.34mmol)在二甲基甲酰胺(17ml)中的溶液中加入氢化钠(0.80g,19.61mmol,60%油分散体)。然后将该溶液冷却至0℃,并加入苄基溴(2.33ml,19.61mmol)。在室温3天后,用水稀释反应物,并用乙酸乙酯(200ml)萃取水层三次。用硫酸钠干燥合并的有机层,并真空浓缩,得到黄色油。该油经闪式色谱(Merck硅胶-60,230-400目,800ml)纯化,用15%乙酸乙酯-己烷洗脱,得到黄色油状标题化合物(4.6g)。f)(1α,2α,3β,5β)-5-[2-氨基-(苯甲氧基)-9H-嘌呤-9-基]-2-(苯甲氧基)-3-[(苯甲氧基)-甲基]环戊醇
在70℃,向搅拌着的步骤(e)产物(1.5g,4.83mmol)和6-(苯甲氧基)-9H-嘌呤-2-胺(2.91g,12.08mmol)在二甲基甲酰胺(23ml)中的溶液中加入氢化锂(0.023g,2.9mmol)。然后将反应混合物加热至112℃3天。冷却后,将反应混合物经真空Kugelrohr蒸馏浓缩,得到黄色油状固体残余物,该残余物经闪式色谱(Merck硅胶-60,230-400目)纯化,用1%甲醇-二氯甲烷洗脱,得到黄色油状标题化合物(1.25g)。g)(1α,2α,3β,5β)-5-[2-[[(4-甲氧苯基)二苯甲基]氨基]-6-(苯甲氧基)-9H-嘌呤-9-基]-2-(苯甲氧基)-3-(苯甲氧基)-甲基]环戊醇
在0℃,向步骤(f)产物(2.19g,3.98mmol)和三乙胺(0.55ml,3.98mmol)在二氯甲烷(50ml)中的溶液中加入甲氧苯基二苯基氯甲烷(1.23g,3.98mmol)。使反应物温热至室温过夜后,加入第二份甲氧苯基二苯基氯甲烷(0.37g,0.11mmol)。2小时后,用饱和碳酸氢钠终止反应,分出有机层,用乙酸乙酯(50ml)萃取水层,用硫酸钠干燥合并的有机层,并真空浓缩,得到油。该油经闪式色谱(Merck硅胶-60,230-400目,500ml)纯化,用50%乙酸乙酯-己烷洗脱,得到无色固体标题化合物(2.26g)。h)(2α,3β,5β)-5-[2-[[(4-甲氧苯基)二苯甲基]氨基]-6-(苯甲氧基)-9H-嘌呤-9-基]-2-苯甲氧基-3-[(苯甲氧基)甲基]环戊酮
在-78℃和氩气氛下,向草酰氯(0.15ml,1.72mmol)的二氯甲烷(4ml)溶液中滴加二甲基亚砜(0.25ml,3.53mmol)。在-78℃搅拌15分钟后,滴加步骤(g)产物(0.484g,0.587mmol)的二氯甲烷(2ml)溶液。30分钟后,加入三乙胺(0.57ml,4.09mmol),并使反应混合物在0℃保持3分钟,然后用饱和碳酸氢钠和盐水终止反应。用乙酸乙酯萃取水层三次。合并的有机层用硫酸钠干燥并真空蒸发,得到淡橙色泡沫状标题化合物(0.525g),在用于下一反应前将该产物与无水甲苯共沸三次。i)锌-四氯化钛-二溴甲烷复合物
按照L.Lombardo,Tetrahedron Lett,23,4293(1982)的方法制备该复合物,在-45°至-49°和氩气氛下,向搅拌着的锌粉(9.8g,0.15mol,用5%盐酸洗涤三次,然后用水洗涤,最后用乙醚洗涤使其活化)在无水凹氢呋喃(100ml)中的混合物中缓缓滴加四氯化钛(3.85ml,0.035mol)。加完四氯化钛后,在-45°-49°搅拌该混合物1小时,在0℃搅拌30分钟,然后在5℃搅拌4天。此浆料于-20℃贮存,在使用前温热至室温。j)(1α,3β,4α)-N-[(4-甲氧苯基)二苯甲基]-9-[2-亚甲基-3-(苯甲氧基)-4-[(苯甲氧基)甲基]环戊基]-9H-嘌呤-2胺
在氩气氛下,向搅拌着的步骤(h)产物(0.395g,少于0.48mmol)的二氯甲烷(3ml)溶液中加入步骤(i)的锌-四氯化钛-二溴甲烷复合物在四氢呋喃(4.3ml,约1.29mmol)中的浆料。将此黑色反应混合物在室温搅拌2小时,然后加入第二份锌-四氯化钛-二溴甲烷复合物(0.9ml,约0.27mmol)。1小时后,向快速搅拌着的乙酸乙酯和饱和碳酸氢钠的混合物中小心地加入该混合物,并再搅拌15分钟。用乙酸乙酯萃取水层三次,并用硫酸钠干燥合并的有机层。真空蒸发掉挥发性物质后,将该粗品残余物与从步骤(h)产物(43.9mg,0.053mmol)开始的相似反应的残余物合并。合并的残余物经闪式色谱(Mallinkrodt Silic AR,100-200目硅胶,60A型,3×14cm)纯化,用乙酸乙酯-己烷(1∶2,然后1∶1)洗脱,得到半纯化的残余物(218mg)。该混合物经闪式色谱(Merck硅胶-60,230-400目,2cm×18.5cm)纯化,用7%乙酸乙酯-二氯甲烷洗脱,得到纯的油状泡沫标题化合物(99mg)。k)(1α,3β,4α)-2-氨基-1,9-二氢-9-[3-羟基-4-(羟甲基)-2-亚甲基环戊基]-6H-嘌呤-6-酮
在-78℃和氩气氛下,用几分钟向步骤(j)产物(282mg,0.344mmol)的二氯甲烷(2.5ml)溶液中滴加三氯化硼(5.2ml,1M,在二氯甲烷中)的二氯甲烷溶液。在-78℃将所得导橙棕色混合物搅拌1小时,温热到-10℃(冰盐浴)5分钟,然后在-78℃加入第二份三氯化硼(1.5ml,1.5mmol)。在-78℃30分钟以及在-10℃10分钟后,将反应混合物从冰盐浴中移去,直到形成红色均匀的溶液。将反应混合物再冷却到-78℃,加入第三份三氯化硼(1.5ml,1.5mmol),在-78℃保持反应15分钟,在-10℃保持10分钟,并在用甲醇小心地终止反应之前再冷却到-78℃。在室温搅拌几分钟后,真空浓缩挥发性物质,将橙色油与甲醇一起蒸发三次,得到橙色玻璃状物。向该残余物中加入水,使该浆料pH为7。将所得混合物部分真空浓缩,并将此含水悬浮液在CHP-20P树脂柱(2×10cm)上纯化,首先用水洗脱,然后用3%乙腈-水洗脱,得到无色固体标题化合物(38.4mg)。质子NMR(270MHz,DMSO-d6)δ:10.51(br s,1H),7.60(s,1H),6.35(br s,2H),5.15(d,J=6.4Hz,1H),5.07-5.14(m,2H),4.66(s,1H),4.51(t,J=5.3Hz,1H),4.21-4.26(m,1H),3.58-3.65(m,1H),3.37-3.45(m,1H),2.14-2.21(m,1H),1.64-1.85(m,2H);碳NMR(270MHz,DMSO-d6)δ:156.8,154.1,153.3,151.2,135.6,116.4,109.6,72.9,61.6,53.7,47.2,32.1;熔点=146-158℃(分解)。
实施例2(1α,2β,4α)-4-(6-氨基-9H-嘌呤-9-基)-2-羟基-3-亚甲基环戊烷甲醇a)(1α,2α,3β,5β)-5-(6-氨基-9H-嘌呤-9-基)-2-(苯甲氧基)-3-[(苯甲氧基)甲基]-环戊醇
在室温和氩气氛下,向腺嘌呤(4.3g,31.8mmol)在无水二甲基甲酰胺(70ml)中的悬浮液中加入一部分氢化钠分散体(0.32g,8mmol,60%,在油中)。1小时后,加入一部分实施例1(e)产物(3.3g,10.6mmol)在无水二甲基甲酰胺(30ml)中的溶液,并将此悬浮液在130℃加热48小时。冷却至室温后,除去溶剂(30℃,高真空),得到油状固体残余物。该残余物用氯仿稀释,并过滤分出固体。将滤液蒸发至干,并将残余物与甲苯一起蒸发几次。该半固体残余物用二氯甲烷稀释,并再次滤出不溶物,用温热的乙酸乙酯萃取合并的固体。合并的有机相经真空浓缩,得到接近白色的油状固体残余物(6.4g)。该物质经闪式色谱(Merck硅胶-60,230-400目,2000ml)纯化,用5%甲醇-氯仿洗脱,得到白色粉末标题化合物(2.7g)。b)(1α,2β,3β,4α)-N-[9-[2-羟基-3-(苯甲氧基)-4-[(苯甲氧基)甲基]环戊基]-9H-嘌呤-6-基]-苯甲酰胺
在室温和氩气氛下,向步骤(a)产物(800mg)的无水吡啶(20ml)溶液中加入苄酰氯(0.836ml,7.2mmol)。1小时后,在室温真空除去溶剂,将半固体残余物溶于乙酸乙酯中,用饱和碳酸氢钠洗涤并用盐水洗涤两次。用硫酸镁干燥后,除去溶剂,得到橙色油状粗品标题化合物。
将含有标题化合物的该粗品残余物溶于乙醇(20ml)、对二噁烷(201ml)和1N氢氧化钠(10ml)中,并在室温搅拌该溶液1小时。用1NHCl将pH调至7.0(从13.5),并将该溶液蒸发至干。将残余物溶于乙酸乙酯中,用盐水洗涤该溶液3次,用硫酸镁干燥,除去溶剂,得到黄色油(1.41g)。该物质经闪式色谱(Merck硅胶-60,230-400目,150ml)纯化,用100%乙酸乙酯洗脱,得到白色泡沫状标题化合物(759mg)。c)(1α,3β,4α)-N-[9-[2-氧代-3-(苯甲氧基)-4-[(苯甲氧基)甲基]环戊基]-9H-嘌呤-6-基]苯甲酰胺
在室温和氩气氛下,向1,1,1-三乙酸基-1,1-二氢-1,2-benziodoxol-3(1H)-酮(1.75g,4.13mmol)的无水二氯甲烷(20ml)溶液中加入一部分纯叔丁醇(0.427ml,4.54mmol)。15分钟后,加入步骤(b)产物(755mg,1.37mmol)的二氯甲烷(5ml)溶液。30分钟后,在5℃将反应物倒入硫代硫酸钠(4.5g,28mmol)在饱和碳酸氢钠(20ml)和乙酸乙酯(100ml)中的混合物中。剧烈搅拌该混合物,直到得到澄清的有机层。分离有机层,用乙酸乙酯洗涤水层。合并的有机层用盐水洗涤两次,用硫酸镁干燥,真空除去溶剂,得到白色泡沫状标题化合物(707mg)。该物质不稳定,并且不需进一步纯化即直接使用。d)(1α,3β,4α)-N-[9-[2-亚甲基-3-(苯甲氧基)-4-[(苯甲氧基)甲基]环戊基]-9H-嘌呤-6-基]-苯甲酰胺
在室温和氩气氛下,向步骤(c)产物(400mg,0.73mmol)的二氯甲烷(10ml)溶液中加入-部分锌-四氯化钛-二溴甲烷复合物[如实施例1(i)所述制备]在四氢呋喃(50ml,约15mmol)中的浆料。继续搅拌30分钟,然后将此黑色反应物倒入剧烈搅拌着的饱和碳酸氢钠(100ml)和乙酸乙酯(100ml)的混合物中。继续搅拌,直到黑色消失(30分钟)。分离澄清的无色有机层,并用盐水洗涤两次,用硫酸镁干燥,真空除去溶剂,得到橙色油状残余物(378mg)。
将该物质与362mg相同反应的粗产物合并,并将此物质经闪式色谱(Merck硅胶-60,230-400目,500ml)纯化,最初用100%乙酸乙酯洗脱,得到不纯的黄色泡沫状标题化合物(270mg)。该物质经闪式色谱(Merck硅胶-60,230-400目,500ml)纯化,用60%乙酸乙酯-己烷洗脱,然后用70%乙酸乙酯-己烷洗脱,得到纯的自色泡沫状标题化合物(105mg)。e)(1α,3β,4α)-[9-[2-亚甲基-3-(苯甲氧基)-4-[(苯甲氧基)甲基]环戊基]-9H-嘌呤-6-胺
向步骤(d)产物(105mg,0.19mmol)的乙醇(1ml)溶液中加入1N氢氧化钠(0.5ml),并在室温搅拌所得橙色溶液过夜。另外加入1N氢氧化钠(1ml),然后加入足够量的可溶解加入氢氧化钠所得油的对二噁烷。继续搅拌1.5天。用1N HCl调节pH至7.0(从12.5)。除去有机溶剂,并用乙酸乙酯萃取该浑浊的含水残余物三次,合并的提取液用盐水洗涤两次,用硫酸镁干燥,并除去溶剂,得到粘稠的油(80mg)。将该物质与140mg不纯的前面获得的产物混合,并经闪式色谱(Merck硅胶-60,230-400目,280ml)纯化,用5%甲醇-乙酸乙酯洗脱,得到纯的白色泡沫状标题化合物(123mg)。f)(1α,2β,4α)-4-(6-氨基-9H-嘌呤-9-基)-2-羟基-3-亚甲基环戊烷甲醇
在室温和氩气氛下,向步骤(e)产物(150mg,0.34mmol)的无水二氯甲烷(3ml)溶液中滴加三氯化硼(1ml,1M,在二氯甲烷中)。在-78℃继续搅拌1小时,然后将反应物短暂地温热至0℃,然后再冷却至-78℃。滴加甲醇(0.5ml)使反应终止,然后使其温热至室温。除去溶剂,并用水稀释该残余物。用1NNaOH调节pH至7.0,将浑浊的悬浮液在CHP-20P树脂柱(95ml)上进行色谱。开始用水洗脱,然后用5%乙腈-水继续洗脱,得到澄清的、淡黄色玻璃状物(51mg)。该物质在WatersAssoc C-18 Prep-Pak Cartridge(25×10cm)上经半制备HPLC进一步纯化。用5%乙腈-水洗脱,得到玻璃状物,将其用甲醇研制,得到白色粉末(35mg)。该物质从甲醇中重结晶,得到纯的白色粉末状标题化合物(20mg)。质子NMR(270MHz,DMSO-d6):δ:8.15(s,1H),8.12(s,1H),7.21(s,2H),5.39(m,1H),5.26(d,1H),5.18(s,1H),4.71(s,1H),4.60(t,1H),4.38(m,1H),3.70(m,1H),3.50(m,1H),2.30(m,1H),1.98(m,1H),1.90(m,1H);熔点:218-219℃(在90℃于高真空干燥24小时)。
C12H15N5O2·0.12CH3 OH的分析:
计算值:C,54.96;H,5.89;N,26.40
实测值:C,55.01;H,5.48;N,26.00。
实施例3(1α,3β,4α)-1-[3-羟基-4-(羟甲基)-2-亚甲基环戊基]-5-甲基-2,4-(1H,3H)-嘧啶二酮a)(1α,2β,3β,4α)-1-[2-羟基-3-(苯甲氧基)-4-[(苯甲氧基)甲基]环戊基]-5-甲基-2,4(1H,3H)-嘧啶二酮
在室温和氩气氛下,向胸腺嘧啶(3.4g,27mmol)在无水二甲基甲酰胺(100ml)中的悬浮液中,加入一部分氢化钠分散体(0.29g,7.2mmol,60%,在油中)。将该混合物在60℃加热30分钟,得到乳状反应混合物向其中加入实施例1(e)产物(2.8g,9mmol)的二甲基甲酰胺(50ml)溶液。在140℃继续加热4天。冷却至室温后,除去溶剂(30℃,高真空),用氯仿稀释该油状固体残余物,并滤除不溶物。将滤液蒸发至干,并将该黑色油状残余物与甲苯一起蒸发。所得黑色油经闪式色谱(Merck硅胶-60,230-400目,2.2升)纯化,用80%乙酸乙酯-己烷洗脱,然后用100%乙酸乙酯洗脱,得到白色泡沫状标题化合物(1.5g)。b)(1α,3β,4α)-5-甲基-1-[2-氧代-3-(苯甲氧基)-4-[(苯甲氧基)甲基]环戊基]-2,4-(1H,3H)-嘧啶二酮
在室温和氩气氛下,向1,1,1-三乙酸基-1,1-二氢-1,2-benziodoxol-3(1H)-酮(1.46g,3.45mmol)的无水二氯甲烷(20ml)溶液中加入-部分叔丁醇(0.358ml,3.8mmol),然后加入步骤(a)产物(500mg,1.15mmol)的二氯甲烷(10ml)溶液。将所得溶液再搅拌45分钟。将反应混合物倒入充分搅拌的10%硫代硫酸钠(15ml)、饱和碳酸氢钠(15ml)、盐水(15ml)和乙酸乙酯(150ml)的混合物中。继续搅拌,直到获得澄清的溶液,分离水层并用乙酸乙酯洗涤。合并的有机层用盐水洗涤两次,用硫酸镁干燥,并真空除去溶剂,得到白色固体标题化合物(519mg)。该物质不需进一步纯化即立即使用。c)(1α,3β,4α)-5-甲基-1-[2-亚甲基-3-(苯甲氧基)-4-[(苯甲氧基)甲基]环戊基]-2,4-(1H,3H)-嘧啶二酮
在室温和氩气氛下,向步骤(b)产物(500mg,少于1.15mmol)的无水二氯甲烷(15ml)溶液中加入一部分如实施例1(i)所述制备的锌-四氯化钛-二溴甲烷复合物在四氢呋喃中的浆料(15ml,约4.5mmol)。将黑色反应混合物搅拌45分钟,然后倒入剧烈搅拌着的饱和碳酸氢钠(50ml)和乙酸乙酯(100ml)的混合物中。继续剧烈搅拌,直到黑色消失(约45分钟)。分离澄清的无色有机层,并用乙酸乙酯(50ml)洗涤水层两次。合并的有机层用盐水洗涤两次,用硫酸镁干燥,并真空除去溶剂,得到粘稠的泡沫状残余物(470mg)该物质经闪式色谱(Merck硅胶-60,230-400目,200ml)纯化,用50%乙酸乙酯-己烷洗脱,得到白色泡沫状标题化合物(158mg)。d)(1α,3β,4α)-1-[3-羟基-4-(羟甲基)-2-亚甲基环戊基]-5-甲基-2,4(1H,3H)-嘧啶二酮
在-78℃和氩气氛下,向步骤(c)产物(280mg,0.65mmol)的二氯甲烷(5ml)溶液中滴加三氯化硼(1.95ml,1M,在二氯甲烷中)。在-78℃ 15分钟后,滴加甲醇(0.5ml)终止反应,并使所得无色澄清的溶液温热至室温。除去溶剂,并将残余物与甲醇一起蒸馏两次。将残余物溶于水中,用1N氢氧化钠调节pH至7.0(从约2)。将此含水悬浮液在CHP-20P树脂(70ml上进行色谱。开始用水洗脱后,继续用3%乙腈-水洗脱,得到粗品白色固体(1α,3β,4α)-1-[3-羟基-4-(羟甲基)-2-亚甲基-环戊基]-5-甲基-2,4(1H,3H)-嘧啶二酮。该物质与74mg从相似反应得到的粗品(1α,3β,4α)-1-[3-羟基-4-(羟甲基)-2-亚甲基-环戊基]-5-甲基-2,4(1H,3H)-嘧啶二酮[从步骤(c)产物(160mg,0.37mmol)开始]合并,并从温热的甲醇-乙腈中重结晶,得到白色粉末状标题化合物(155mg)。质子NMR(270MHz,DMSO-d6):δ:11.27,(br s,1H),7.30(d,J=1.2Hz,1H),5.28(m,1H),5.23(d,J=6.5Hz,1H),5.18(d,1H),4.87(d,1H),4.55(m,1H),4.19(m,1H),3.66(m,1H),3.44(m,1H),2.06(m,1H),1.76(m,1H),1.49(q,J=11.7Hz 1H);熔点:212-213℃(在90℃高真空下干燥24小时)。
将标题化合物的分析样品从温热的乙腈中重结晶,得到白色晶体,m.p.213-214℃(在90℃于高真空下干燥24小时)。C12H16N2O4的分析:
计算值:C,57.13;H,6.39;N,11.10
实测值:C,56.91;H,6.40;N,11.10。
实施例4(1α,3β,4α)-1-[3-羟基-4-(羟甲基)-2-亚甲基环戊基]-5-碘-2,4(1H,3H)-嘧啶二酮a)尿嘧啶单四丁基铵盐
在室温向无水二甲基甲酰胺(14ml)中的尿嘧啶(1g,8.93mmol)中加入氢氧化四丁基铵水溶液(2.7ml,42%溶液,4.32mmol)。将反应混合物搅拌1小时,真空除去挥发性物质,并将残余物与无水二甲基甲酰胺一起蒸发4次,得到白色粉末状标题化合物(1.97g)。b)(1α,2β,3β,4α)-1-[2-羟基-3-(苯甲氧基)-4-[(苯甲氧基)甲基]环戊基]-2,4(1H,3H)-嘧啶二酮
在二甲基甲酰胺(60ml)中将实施例1(e)(3.0g,9.7mmol)产物和尿嘧啶单四丁基铵盐(18g,0.038mol)合并,并加热至110℃48小时。然后将该混合物冷却至室温,用水(100ml)稀释,并用乙酸乙酯(100ml)萃取水层3次。合并的有机层用硫酸钠干燥,过滤,并真空浓缩,得到米色固体。将固体残余物溶于乙酸乙酯-甲醇中,并预先吸附到Baker's硅胶(60-200目)上,然后将其装上硅胶柱(Merck硅胶-60,230-400目,1升)。用100%乙酸乙酯洗脱,得到油状标题化合物(2.5g)。
或者,还可以通过下述方法制备标题化合物:在室温氩气氛下,将氢化锂(1mg,0.125mmol)加至实施例1(e)产物(130mg,0.419mmol)和尿嘧啶(141mg,1.26mmol)在无水二甲基甲酰胺(1.95ml)中的混合物中。将该混合物在140℃加热24小时,然后真空浓缩,得到深棕色固体。该物质经闪式色谱(Merck硅胶-60,230-400目,100ml)纯化,用乙酸乙酯洗脱,得到油状标题化合物(78mg)。c)(1α,2β,3β,4α)-1-[2-羟基-3-(苯甲氧基)-4-[(苯甲氧基)甲基]环戊基]-5-碘-2,4(1H,3H)-嘧啶二酮
在室温,向步骤(b)产物(1.0g,2.4mmol)的对二噁烷(24ml)溶液中加入硝酸(0.5N,9.48ml)和碘(1.19g,4.7mmol)。在90℃加热该混合物2小时,将反应物冷却至室温并用水稀释。用碳酸氢钠中和反应混合物,并用二氯甲烷(15ml)萃取4次。浓缩合并的有机层,得到红色残余物,将其与氯仿-水-甲醇的混合物一起蒸发4次,得到淡黄色固体。该固体残余物经闪式色谱(Merck硅胶-60,230-400目)纯化,用50%乙酸乙酯-己烷洗脱,得到黄色泡沫状标题化合物(1.1g)。d)(1α,3β,4α)-5-碘-1-[2-氧代-3-(苯甲氧基)-4-[(苯甲氧基)甲基]环戊基]-2,4(1H,3H)-嘧啶二酮
在室温和氩气氛下,向快速搅拌着的1,1,1-三乙酸基-1,1-二氢-benziodoxol-3(1H)-酮(0.695g,1.6mmol)在二氯甲烷(3.5ml)中的悬浮液中加入叔丁醇(0.05ml,0.5mmol)。将该混合物在室温保持15分钟,然后加入步骤(c)产物(0.50g,0.91mmol)的二氯甲烷(4ml)溶液。30分钟后,将该反应物加至搅拌着的盐水(5ml)、10%硫代硫酸钠水溶液(5ml)和饱和碳酸氢钠(5ml)的混合物中。在室温搅拌30分钟后,用乙酸乙酯稀释该混合物,并用乙酸乙酯萃取水层两次。真空浓缩合并的有机层,得到不纯的白色固体标题化合物(0.498g)。e)(1α,3β,4α)-5-碘-1-[2-亚甲基-3-(苯甲氧基)-4-[(苯甲氧基)甲基]环戊基]-2,4(1H,3H)-嘧啶二酮
将步骤(d)产物经无水甲苯的短途蒸馏共沸干燥,并将所得残余物溶于二氯甲烷(3ml)中。在室温,向此二氯甲烷溶液中加入如实施例1(i)所述制备的锌-四氯化钛-二溴甲烷复合物在四氢呋喃中的浆料(8.4ml,约2.5mmol)。6小时后,向搅拌着的乙酸乙酯和饱和碳酸氢钠的混合物中加入该反应物。1小时后,用乙酸乙酯(50ml)萃取水层2次,并真空浓缩合并的有机层,得到黄色残余物。该残余物经闪式色谱(Merck硅胶-60,230-400目)纯化,用2%甲醇-二氯甲烷洗脱,得到米色固体标题化合物(0.165g)。f)(1α,3β,4α)-1-[3-羟基-4-(羟甲基)-2-亚甲基环戊基]-5-碘-2,4(1H,3H)-嘧啶二酮
在-78℃,向搅拌着的步骤(e)产物(0.165g,0.30mmol)的二氯甲烷(6ml)溶液中滴加三氯化硼的二氯甲烷溶液(3ml,3.0mmol)。10分钟后,将反应混合物从CO2/丙酮浴上移开15分钟,得到均匀的溶液。将所得溶液再冷却至-78℃,并再搅拌2小时。在0℃滴加甲醇使反应终止,将其温热至室温,并真空浓缩。将所得残余物与甲醇一起蒸发三次,得到红色油。将该油溶于甲醇中,用水稀释并用0.1N氢氧化钾中和。然后真空除去挥发性物质,并将所得残余物在水中匀浆,经CHP-20P树脂色谱纯化,用水洗脱,然后用2%甲醇-水洗脱,得到无色固体环题化合物(58mg)。质子NMR(270MHz,DMSO-d6):δ:11.55(br s,1H),7.84(s,1H),5.09-5.18(m,3H),4.86(s,1H),4.43-4.45(m,1H),4.13-4.16(m,1H),3.4-3.6(m,2H),1.9-2.05(m,1H),1.65-1.73(m,1H),1.48-1.53(m,1H);熔点(分解):232℃。
实施例5[1α(E),3β,4α]-5-[2-溴乙烯基]-1-[3-羟基-4-(羟甲基)-2-亚甲基环戊基]-2,4(1H,3H)-嘧啶二酮a)[1α(E),2β,3β,4α]-3-[1,2,3,4-四氢-1-[2-羟基-3-(苯甲氧基)-4-[(苯甲氧基)-甲基]环戊基]-2,4-二氧代-5-嘧啶基]-2-丙烯酸甲酯
在无水二氯甲烷(17ml,脱氧的)中将三乙胺(1.34ml,9.61mmol)、三苯膦(0.15g,0.57mmol)和乙酸钯(Ⅱ)(0.05g)合并,然后加热至70℃,得到红色溶液。15分钟后,加入如实施例4(c)所述制备的(1α,2β,3β,4α)-1-[2-羟基-3-(苯甲氧基)-4-[(苯甲氧基)-甲基]环戊基]-5-碘-2,4(1H,3H)-嘧啶二酮(1.5g,2.74mmol)的二甲基甲酰胺(10ml,脱氧的)溶液,然后加入丙烯酸甲酯(0.08ml,9.65mmol)。8小时后,将反应物冷却至室温并真空浓缩,得到黄色油,将此残余物与用实施例4(c)产物(2.1g,2.39mmol)开始的相似反应得到的粗产物合并。合并的残余物经闪式色谱(Merck硅胶-60,230-400目)纯化,用2%甲醇-二氯甲烷洗脱,得到黄色泡沫状标题化合物(2.1g)。b)[1α(E),2β,3β,4α]-3-[1,2,3,4-四氢-1-[2-羟基-3-(苯甲氧基)-4-[(苯甲氧基)-甲基]环戊基]-2,4-二氧代-5-嘧啶基]-2-丙烯酸
在室温,向步骤(a)产物(1.62g,3.26mmol)的四氢呋喃(23ml)溶液中加入2N氢氧化钾(16.3ml)。在室温搅拌该反应物6小时,然后加入第二份2N氢氧化钾(4ml)。1小时后,将反应混合物冷却到0℃,并用3NHCl将pH调至2。然后用乙酸乙酯稀释该溶液,并用乙酸乙酯萃取水层3次。合并的有机层用硫酸钠干燥,并真空浓缩,得到橙色固体标题化合物(1.6g)。不需进一步纯化即使用该物质。c)[1α(E),2β,3β,4α]-5-(2-溴乙烯基)-1-[2-羟基-3-(苯甲氧基)-4-[(苯甲氧基)-甲基]环戊基]-2,4(1H,3H)-嘧啶二酮
在室温,向步骤(b)产物(1.6g,3.26mmol,与二甲基甲酰胺共沸蒸馏干燥)和碳酸氢钾(1.05g,10.43mmol)在二甲基甲酰胺(18ml)中的混合物中加入0.5M N-溴琥珀酰亚胺的二甲基甲酰胺(6.1ml,3.05mmol)溶液,2小时后,真空浓缩反应混合物,将其溶于二氯甲烷中,并用稀硫代硫酸钠、0.1N碳酸氢钾和水洗涤。有机层用硫酸钠干燥并真空浓缩,得到黄色油。该油经闪式色谱(Merck硅胶-60,230-400目)纯化,用1%乙醇-二氯甲烷洗脱,然后用2%乙醇-二氯甲烷洗脱,得到米色固体标题化合物(0.75g)。d)[1α(E),3β,4α]-5-(2-溴乙烯基)-1-[2-氧代-3-(苯甲氧基)-4-[(苯甲氧基)甲基]环戊基]-2,4(1H,3H)-嘧啶二酮
在室温,向搅拌着的1,1,1-三乙酸基-1,1-二氢-1,2-benziodoxol-3(1H)-酮(0.59g,1.38mmol)在二氯甲烷(3ml)中的悬浮液中加入叔丁醇(0.043ml)。15分钟后,加入步骤(c)产物(0.40g,0.77mmol)的二氯甲烷(3ml)溶液。再过2小时后,将反应物加至剧烈搅拌着的盐水(10ml)、10%硫代硫酸钠(10ml)和泡和碳酸氢钠(10ml)的混合物中,1小时后,用乙酸乙酯稀释反应物,并用乙酸乙酯萃取水层两次。真空浓缩合并的有机层,得到白色固体粗品标题化合物。将该残余物与用步骤(c)产物(0.348g,0.66mmol)开始的相似反应的粗品标题化合物合并。将合并的残余物再溶于二氯甲烷中,并加至剧烈搅拌着的盐水(20ml)、10%硫代硫酸钠(20ml)和饱和碳酸氢钠(20ml)的混合物中。1小时后,用乙酸乙酯稀释反应物,并用乙酸乙酯萃取水层3次。真空浓缩合并的有机层,得到0.75g黄色固体粗品标题化合物。不需进一步纯化即使用该物质。e)[1α(E),3β,4α]-5-(2-溴乙烯基)-1-[2-亚甲基-3-(苯甲氧基)-4-[(苯甲氧基)-甲基]环戊基]-2,4(1H,3H)-嘧啶二酮
在室温和氩气氛下,向步骤(d)产物(约0.75g,约1.43mmol)的二氯甲烷(14ml)溶液中加入如实施例1(i)所述制备的锌-四氯化钛-二溴甲烷复合物在四氢呋喃(18ml,约5.5mmol)中的浆料。45分钟后,向该反应物中加入第二份锌-四氯化钛-二溴甲烷复合物(9ml,约2.79mmol),并将该混合物于室温再搅拌1小时,然后在-40℃贮存过夜。将反应混合物温热至室温后,加入第三份锌-四氯化钛-二溴甲烷复合物(9ml,约2.79mmol;总共36ml,约11.1mmol)。1小时后,将反应混合物加至快速搅拌着的乙酸乙酯和饱和碳酸氢钠溶液中。在室温搅拌该混合物1小时,然后用乙酸乙酯稀释。用乙酸乙酯萃取水层3次,将合并的有机层用硫酸钠干燥,过滤,并真空浓缩,得到无色油,所得残余物经闪式色谱(Merck硅胶-60,230-400目)纯化,用10%乙酸乙酯-二氯甲烷洗脱,得到无色泡沫(0.168g)。该物质经闪式色谱(Merck硅胶-60,230-400目)进一步纯化,用10%乙酸乙酯-二氯甲烷洗脱,得到纯的无色泡沫状标题化合物(0.1g)。f)[1α(E),3β,4α]-5-(2-溴乙烯基)-1-[3-羟基-4-(羟甲基)-2-亚甲基环戊基]-2,4(1H,3H)-嘧啶二酮
在-78℃和氩气氛下,向搅拌着的步骤(e)产物(0.10g,0.19mmol)的二氯甲烷(3.5ml)溶液中滴加三氯化硼(1.9ml,1M二氯甲烷溶液)。10分钟后,将该溶液温热至室温,直到形成均匀的溶液(约15分钟),然后再冷却至-78℃。再过1.5小时后,在0℃滴加甲醇使反应混合物骤冷。真空除去挥发性物质,并将橙色油状残余物与甲醇一起蒸发三次,将所得残余物溶于水中并用氢氧化钾中和,然后浓缩成浆。将此浆料装CHP-20P树脂柱,开始用水洗脱,然后用乙腈-水(1.3,5,7,9,15,20,30,然后40%乙腈)梯度洗脱,得到无色固体标题化合物(0.024g)。质子NMR(270MHZ,DMSO-DB):δ11.50(br s,1H),7.20(d,J=13.6Hz,DMSO-d6):δ:11.50(br s,1H),7.20(d,J=13.6Hz,1H),6.83(d,J=13.6Hz,1H),5.14-5.23(m,2H),5.12(d,J=6.8Hz,1H),4.86(s,1H),4.4-4.5(m,1H),4.13-4.16(m,1H),3.6-3.65(m,1H),3.3-3.4(m,1H),1.95-2.05(m,1H),1.65-1.75(m,1H),1.40-1.50(m,1H);熔点(分解):190℃。
实施例6
在离体细胞培养中病毒感染的治疗
在细胞培养体系中进行分析,以测定有效防止几类病毒感染的化合物浓度。按下面的描述进行分析,结果示于表1中。缩写:
HSV-1(单纯性疱疹病毒1型,Schooler株),HSV-2(单纯性疱疹病毒2型,186株),VZV(水痘带状疱疹病毒,ELLEN株),HCMV(人巨细胞病毒,AD169株)。细胞培养测定:
HSV-1、HSV-2、HCMV和VZV抗病毒测定:将病毒吸附于6孔培养板(Costar,Cambridgl,MA)的WI-38细胞培养单层上1小时,然后加入含倍数稀释的试验化合物的维持培养基。在37℃培养HSV-1和HSV-24天后,培养HCMV和VZV5-7天后,固定并染色各单层,评价菌斑发展的抑制作用。从与病毒对照组比至少减少50%菌斑的药物浓度测定ID50值。
表1 对下列病毒的ID50(μM)
对下列病毒的ID50(μM)
 1在361μM和分析条件下对各细胞单层有明显毒性。2在383μM和分析条件下对各细胞单层有明显毒性。3在396μM和分析条件下对各细胞单层有明显毒性。4在275μM和分析条件下对各细胞单层有明显毒性。5在291μM和分析条件下对各细胞单层有明显毒性。6未测定。
1在361μM和分析条件下对各细胞单层有明显毒性。2在383μM和分析条件下对各细胞单层有明显毒性。3在396μM和分析条件下对各细胞单层有明显毒性。4在275μM和分析条件下对各细胞单层有明显毒性。5在291μM和分析条件下对各细胞单层有明显毒性。6未测定。
Claims (11)
1.下式化合物或其可药用盐:其中R1是
 其中R2是氟、氯、溴、碘、氢、甲基、三氟甲基、乙基、正丙基、2-氟乙基、2-氯乙基、乙炔基或其中R3是氯、溴、碘、氢、甲基或三氟甲基:R4是烷基;R5是氢、烷基、取代烷基或芳基;并且R6和R7独立地为氢、-PO3H2
其中R2是氟、氯、溴、碘、氢、甲基、三氟甲基、乙基、正丙基、2-氟乙基、2-氯乙基、乙炔基或其中R3是氯、溴、碘、氢、甲基或三氟甲基:R4是烷基;R5是氢、烷基、取代烷基或芳基;并且R6和R7独立地为氢、-PO3H2 
2.按照权利要求1的化合物,其中R1是

3.按照权利要求2的化合物,其中R6和R7是氢。
4.按照权利要求3的化合物,其中R1是
5.按照权利要求1的化合物,它是(1α,3β,4α)-2-氨基-1,9-二氢-9-[3-羟基-4-羟甲基)-2-亚甲基环戊基]-6H-嘌呤-6-酮。
6.按照权利要求1的化合物,它是(1α,3β,4α)-4-(6-氨基-9 H-嘌呤-9-基)-2-羟基-3-亚甲基环戊烷甲醇。
7.按照权利要求1的化合物,它是(1α,3β,4α)-1-[3-羟基-4-(羟甲基)-2-亚甲基环戊基]-5-甲基-2,4(1H,3H)-嘧啶二酮。
8.按照权利要求1的化合物,它是(1α,3β,4α)-1-[3-羟基-4-(羟甲基)-2-亚甲基环戊基]-5-碘-2,4(1H,3H)-嘧啶二酮。
9.按照权利要求1的化合物,它是[1α(E),3β,4α]-5-[2-(溴乙烯基)-1-[3-羟基-4-(羟甲基)-2-亚甲基环戊基]-2,4-(1H,3H)-嘧啶二酮。
10.权利要求1的化合物或其可药用的盐在制备抗病毒药物中的应用。
11.-种抗病毒药物组合物,它包括治疗有效量的权利要求1的化合物或其可药用的盐及可药用的载体。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US8171893A | 1993-06-25 | 1993-06-25 | |
| US081718 | 1993-06-25 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN1106403A CN1106403A (zh) | 1995-08-09 |
| CN1042425C true CN1042425C (zh) | 1999-03-10 |
Family
ID=22165948
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN94107231A Expired - Fee Related CN1042425C (zh) | 1993-06-25 | 1994-06-24 | 3-羟基-4-羟甲基-2-亚甲基环戊基嘌呤和嘧啶 |
Country Status (5)
| Country | Link |
|---|---|
| EP (1) | EP0630897A3 (zh) |
| JP (1) | JPH0725856A (zh) |
| CN (1) | CN1042425C (zh) |
| AU (1) | AU673259B2 (zh) |
| CA (1) | CA2126363A1 (zh) |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1997037993A1 (en) * | 1996-04-09 | 1997-10-16 | Yamasa Corporation | 9-(2-DEOXY-2-FLUORO-4-THIO-β-D-ARABINOFURANOSYL)PURINE DERIVATIVES |
| AU2003300884B2 (en) * | 2002-12-11 | 2010-01-21 | Bristol-Myers Squibb Holdings Ireland Unlimited Company | Process and intermediates for synthesis entecavir |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0304889A2 (en) * | 1987-08-26 | 1989-03-01 | Merrell Pharmaceuticals Inc. | Novel aristeromycin/adenosine derivatives |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4754026A (en) | 1985-06-04 | 1988-06-28 | Takeda Chemical Industries, Ltd. | Conversion of uracil derivatives to cytosine derivatives |
| GB8926623D0 (en) | 1989-11-24 | 1990-01-17 | Glaxo Group Ltd | Chemical compounds |
| GB9007569D0 (en) * | 1990-04-04 | 1990-05-30 | Nycomed As | Carbo-nucleoside derivatives |
| US5206244A (en) * | 1990-10-18 | 1993-04-27 | E. R. Squibb & Sons, Inc. | Hydroxymethyl (methylenecyclopentyl) purines and pyrimidines |
| GB9204015D0 (en) * | 1992-02-25 | 1992-04-08 | Wellcome Found | Therapeutic nucleosides |
-
1994
- 1994-06-13 EP EP94109034A patent/EP0630897A3/en not_active Ceased
- 1994-06-21 CA CA002126363A patent/CA2126363A1/en not_active Abandoned
- 1994-06-23 AU AU65932/94A patent/AU673259B2/en not_active Ceased
- 1994-06-24 CN CN94107231A patent/CN1042425C/zh not_active Expired - Fee Related
- 1994-06-24 JP JP6142960A patent/JPH0725856A/ja active Pending
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0304889A2 (en) * | 1987-08-26 | 1989-03-01 | Merrell Pharmaceuticals Inc. | Novel aristeromycin/adenosine derivatives |
Also Published As
| Publication number | Publication date |
|---|---|
| AU673259B2 (en) | 1996-10-31 |
| JPH0725856A (ja) | 1995-01-27 |
| AU6593294A (en) | 1995-01-05 |
| CA2126363A1 (en) | 1994-12-26 |
| EP0630897A2 (en) | 1994-12-28 |
| CN1106403A (zh) | 1995-08-09 |
| EP0630897A3 (en) | 1995-03-01 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP2994117B2 (ja) | ヒドロキシメチル(メチレンシクロペンチル)プリン類およびピリミジン類 | |
| CN1031054C (zh) | 制备双(羟甲基)环丁基嘌呤和嘧啶的方法 | |
| AU596400B2 (en) | Carbocyclic purine nucleosides, their production and use | |
| CN1388123A (zh) | 氯代嘧啶中间体 | |
| RU2001125421A (ru) | Способ получения трет-бутил(е)-(6-[2-[4-(4-фторфенил)-6-изопропил-2- [метил(метилсульфонил)амино]пиримидин-5-ил]винил](4r,6s)-2,2- диметил[1,3]диоксан-4-ил]ацетата | |
| HU203236B (en) | Process for producing purinyl and pyrimidinyl cyclobutanes | |
| KR20060017848A (ko) | 로수바스타틴 칼슘염의 향상된 제조 | |
| CN1046288C (zh) | 嘌呤的不饱和膦酸酯衍生物 | |
| US8394956B2 (en) | Process for preparing pyrimidine propenaldehyde | |
| KR100232619B1 (ko) | 사이클로프로판 유도체, 이의 제조방법 및 이를 함유하는 약제학적 조성물 | |
| JP2002539125A (ja) | 抗ウイルス剤としての2−ヒドロキシメチルシクロプロピリデンメチルプリンおよび−ピリミジン類 | |
| CN1042425C (zh) | 3-羟基-4-羟甲基-2-亚甲基环戊基嘌呤和嘧啶 | |
| JPH06271574A (ja) | 抗ウイルス性テトラヒドロピラン化合物 | |
| KR100535450B1 (ko) | 피리미딘유도체의제조방법 | |
| EP0219838A2 (en) | Carbocyclic purine nucleosides, their production and use | |
| EP1210347B1 (en) | Carbocyclic nucleosides and process for obtaining such | |
| IE901291L (en) | Purinyl and pyrimidinyl tetrahydrofurans | |
| CN1021820C (zh) | N9-取代的腺嘌呤n1-取代的胞嘧啶的制备方法 | |
| JPH0625061B2 (ja) | 抗ウイルス性化合物 | |
| CN1049502A (zh) | 制备抗病毒化合物的方法 | |
| Sokolov et al. | Reaction of 6-amino-1, 3-dimethyluracil with hexafluoroacetone and ethyl trifluoropyruvate benzoylimines | |
| JPH07126282A (ja) | 新規なチオヌクレオシド誘導体 | |
| EP0504868B1 (en) | Method of manufacturing 2'-3'-dideoxy-2',3'.didehydronucleosides | |
| CA1339647C (en) | Bis-(hydroxymethyl) cyclobutyl purines and pyrimidines | |
| Pomeisl et al. | Pyrimidine 1-[2-(phosphonomethoxy) propyl] derivatives: Their syntheses and utilization as potent inhibitors of thymidine phosphorylase (PD-ECGF) from SD-lymphoma |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C14 | Grant of patent or utility model | ||
| GR01 | Patent grant | ||
| C19 | Lapse of patent right due to non-payment of the annual fee | ||
| CF01 | Termination of patent right due to non-payment of annual fee |