CN1102829A - 紫杉酚、紫杉酚类似物及其中间体的合成及它的组合物 - Google Patents
紫杉酚、紫杉酚类似物及其中间体的合成及它的组合物 Download PDFInfo
- Publication number
- CN1102829A CN1102829A CN94102253A CN94102253A CN1102829A CN 1102829 A CN1102829 A CN 1102829A CN 94102253 A CN94102253 A CN 94102253A CN 94102253 A CN94102253 A CN 94102253A CN 1102829 A CN1102829 A CN 1102829A
- Authority
- CN
- China
- Prior art keywords
- taxol
- hydrogen
- phch
- aryl
- alkyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D305/00—Heterocyclic compounds containing four-membered rings having one oxygen atom as the only ring hetero atoms
- C07D305/14—Heterocyclic compounds containing four-membered rings having one oxygen atom as the only ring hetero atoms condensed with carbocyclic rings or ring systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C271/00—Derivatives of carbamic acids, i.e. compounds containing any of the groups, the nitrogen atom not being part of nitro or nitroso groups
- C07C271/06—Esters of carbamic acids
- C07C271/08—Esters of carbamic acids having oxygen atoms of carbamate groups bound to acyclic carbon atoms
- C07C271/10—Esters of carbamic acids having oxygen atoms of carbamate groups bound to acyclic carbon atoms with the nitrogen atoms of the carbamate groups bound to hydrogen atoms or to acyclic carbon atoms
- C07C271/22—Esters of carbamic acids having oxygen atoms of carbamate groups bound to acyclic carbon atoms with the nitrogen atoms of the carbamate groups bound to hydrogen atoms or to acyclic carbon atoms to carbon atoms of hydrocarbon radicals substituted by carboxyl groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F7/00—Compounds containing elements of Groups 4 or 14 of the Periodic Table
- C07F7/02—Silicon compounds
- C07F7/08—Compounds having one or more C—Si linkages
- C07F7/18—Compounds having one or more C—Si linkages as well as one or more C—O—Si linkages
- C07F7/1804—Compounds having Si-O-C linkages
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/07—Optical isomers
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/55—Design of synthesis routes, e.g. reducing the use of auxiliary or protecting groups
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Epoxy Compounds (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Preparation Of Compounds By Using Micro-Organisms (AREA)
Abstract
本发明描述了一种合成紫杉酚,紫杉酚类似物及
其中间体的有效方法。该法包括将紫杉酚A-环侧
链连接到浆果赤霉素Ⅲ并合成具有可变A-环侧链
结构的紫杉酚和紫杉酚类似物。将带有N-苯甲酰
氧羰基的O-保护的异丝氨酸和3-苯基异丝氨酸在
7-O保护的浆果赤霉素Ⅲ上快速高效地酯化,接着
去保护一乙酰化以制备紫杉酚,Celphalomannine和
各种类似物,包括光亲核性标记的替代物。
Description
本发明涉及紫杉酚,紫杉酚类似物及其中间体的合成产品。
紫杉酚为天然存在的紫杉烷类二萜化合物,具有显著的临床活性,可作为具有相当潜力的抗肿瘤剂而进一步研究。用紫杉酚广泛地治疗各种疾病受限于目前药物的缺乏。紫杉酚在它的天然来源即生长缓慢的太平洋紫杉的树皮中的低含量使得通过分离得到紫杉酚的长期前景暗淡。紫杉酚具有如下所示的通式和相应的编号系统:

在解决紫杉酚供应问题中,有机化学家提出的是通过在象浆果赤霉素Ⅲ的天然来源的物质在C-13位连接A-环侧链来部分合成该药,或具有临床活性的类似物。紫杉酚及其类似物的制备是已知的。例如,Denis等于1990年5月8日颁布的美国专利4,924,011,公开了通过(2R,3S)侧链酚与紫杉烷衍生物缩合,接着除去各个羟基保护基来制备紫杉酚的方法。Colin等于1990年5月8日颁布的美国专利4,924,012公开了由一种酸与浆果赤霉素Ⅲ或10-去乙酰基浆果赤霉素Ⅲ衍生物缩合,随后由氢除去保护基来制备浆果赤霉素和10-去乙酰基-浆果赤霉素Ⅲ衍生物的方法。TAXOTERER(在Rhone-Pou-lenc-Sante注册)及其相关化合物的几种合成方法已在Journal of Organic Chemistry:1986,51,46;1990,55,1957;1991,56,1681;1991,56,6939;1992,57,4320;1992,57,6387;及1993,58,255公开;另外Holton于1991年5月14日颁布的美国专利5,015,744也描述了这样的合成。
紫杉酚的最直接的部分合成需要合适地使用手性的,非外消旋的侧链和衍生物,充足的天然来源的浆果赤霉素Ⅲ或密切相关的二萜类物质,并需要一种连接二者的有效方法。特别有利的是下式所示的浆果赤霉素Ⅲ和10-去乙酰基浆果赤霉素Ⅲ:

但是,由于浆果赤霉素Ⅲ的C-13位羟基位于半球形紫杉烷骨架的凹区而存在位阻,这二者的酚化很困难。例如,Greene和Gueritte-Voegelein报导在紫杉酚部分合成中,100小时后仅有50%转化。J.Am.Chem.Soc,1988,110,5917。Holten的专利提供了一种更有效的合成方法。
仍然需要更有效的方案在紫杉烷骨架例如,浆果赤霉素Ⅲ上连接紫杉酚A-环侧链并合成紫杉酚,紫杉酚类似物及其具有可变A-环侧链结构的中间体。
因此,本发明总的目的是提供一种新的,有用的并且有效的方案,在紫杉烷骨架上连接紫杉酚A-环侧链以合成紫杉酚,紫杉酚类似物及其具有可变A-环侧链结构的中间体,该方案克服以前文献中的缺陷。
本发明的另一个目的是在浆果赤霉素Ⅲ上连接紫杉酚A-环侧链以合成紫杉酚,紫杉酚类似物其具有可变A-环侧链结构的中间体。
本发明的另一目的为提供一种快速高效的方法,将7-0-保护的浆果赤霉素Ⅲ的C-13羟基用具有N-苄氧羰基的O-保护的异丝氨酸和3-苯基异丝氨酸酯化。
本发明的另一目的是提供一种前述的去保护-酰化的步骤,提供紫杉酚,cephalomanmine,10-乙酰基TAXOTERE
 和各种类似物,包括光亲和性标记的替代物。
和各种类似物,包括光亲和性标记的替代物。
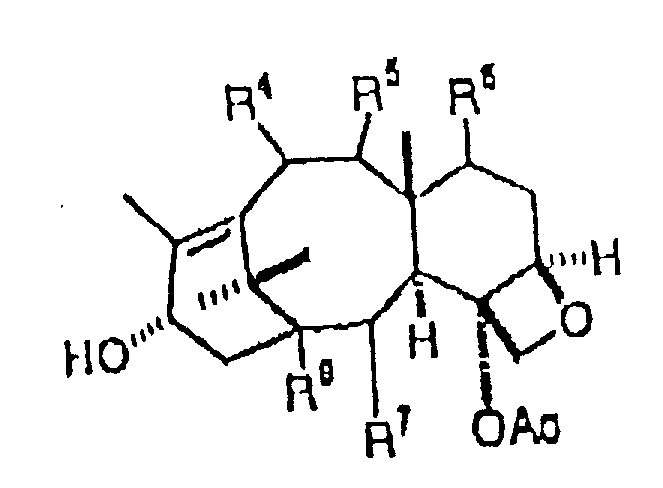
作为它的广泛的形式,本发明提供一种用于制备紫杉酚,紫杉酚类似物及其中间体的实用的化学方法。该方法包括将下面通式所示化合物:

与具有下面通式结构的紫杉烷
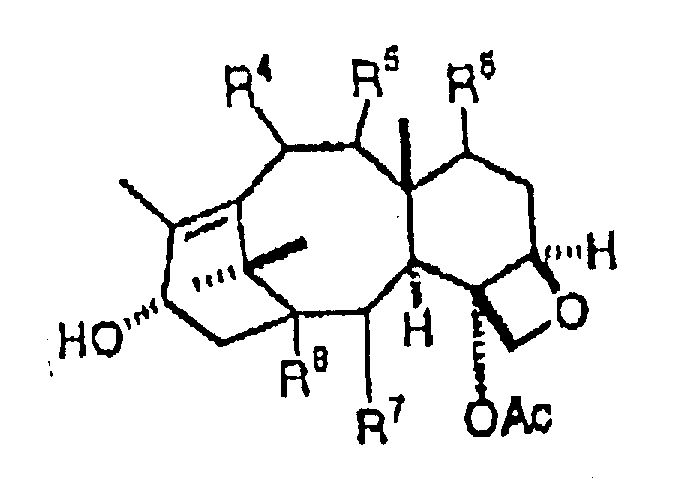
缩合得到具有下面通式结构的中间体:

其中:R1=烷基,烯基,芳基或phCH2
R3=氢,烷基,烯基,芳基或Ph
R4=氢,氧基或乙酰氧基
R5=氢,氧基或羰基
R6=氢,氧基或乙酰氧基
R7=氢,氧基,苯甲酰氧基或芳酰氧基
R8=氢或羟基
P1=羟基保护基。
按总的方法,各种取代,特别是其中R1为PhCH2,R3为Ph,R4为AcO,R5为双键O,R6为OH,R7为PhCO2和R8为OH的,为所期望的。该总的方法可接着进行将中间体的R1O和P1复原的步骤,以得到如下通式结构的化合物:

其中R2=氢,烷基,烯基,芳基,氧,氮,硫或Ph基而且,各种优选的取代也是所期望的。反应可在芳香性溶剂,活化剂以及可能的缩合剂中进行。优选的反应温度为70°-75℃。
本发明涉及有效地制备紫杉酚,紫杉酚类似物及其中间体的化学方法。这可通过在浆果赤霉素Ⅲ上连接紫杉酚A-环侧链来实现。这样便合成了紫杉酚,紫杉酚类似物及其具有可变A-环侧链结构的中间体。本发明进一步涉及快速有效的方法,将7-0保护的浆果赤霉素Ⅲ的C-13位羟基用具有N-苄氧羰基的O-保护的异丝氨酸和3-苯基异丝氨酸酯化。
首先我们报告与Greene,Gueritte-Voegelein方案不同的除去A-环侧链取代基的紫杉酚类似物的部分合成。Swindell等人,J.Med.Chem.1991,34,1176,Greene,Gueritte-Voegelein的方案可概括为:

按照通式反应:

其中P1为羟基保护基,例如,(三氯)乙氧甲基,甲氧甲基,1-乙氧甲基,苄氧甲基,2-(三甲硅烷基)乙氧甲基,四氢吡喃基和烯丙氧甲基;P2为羟基保护基,例如,3,3,3-(三氯)乙氧羰基,三烷基甲硅烷基,烯丙氧羰基和苄氧羰基,可在下表1中概述我们的发现:
表1乳酸,苯基乳酸和异丝氨酸侧链酯化
R 酸当量偶合剂 时间 产物 产率(%)
H(8)a6 DPC 24 h 12 53a
ph(9)a6 DPC 48 h 13 37b
phCONH(10)a6 DPC 6h 14 87
phCH2OCHONH(11)c6 DCC 15m 15 97
2.2 DCC 30m 100
a见Swindell等,J.Med.Chem,1991,34,1176。b包括随后2′和7-羟基去保护的产率。c我们的研究工作。
在进行这项研究工作中我们的一个目标是发现具有可通过常规的酯化方法比紫杉酚侧链更有效地被接在浆果赤霉素Ⅲ上的结构简单的侧链的有生理活性的紫杉酚类似物。我们发现乳酸和苯基乳酸类似物(12和13)在侧链复杂性和侧链连接比率和效率间的直接联系。但是我们惊奇地发现N-(苯甲酰基)异丝氨酸侧链酸10,可证明其空间需要如苯基乳酸9,可以明显高的比率和效率酯化7-0-保护的浆果赤霉素Ⅲ7,Magri等,J.Org.Chem,1986,51,3239。我们提出最初由苯甲酰胺羰基氧分子内酰化形成的如下式的二氢噁嗪中介物:
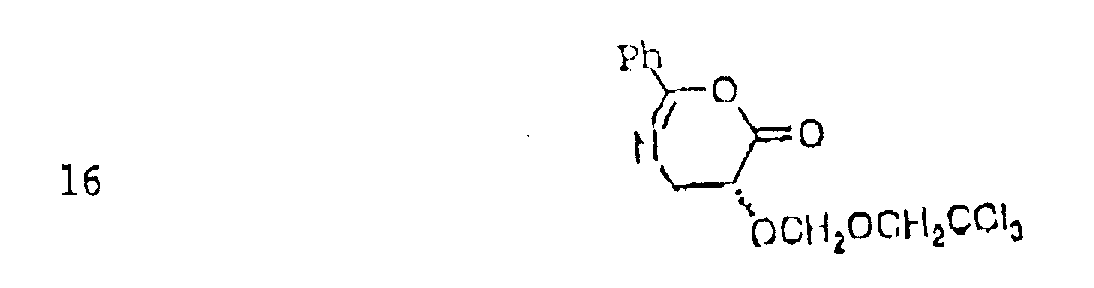
我们提出:(Ⅰ)由于空间需要的限制,环状乙酰化试剂16对浆果赤霉素ⅢC-13位于羟基具有特别的反应性;(Ⅱ)比16更复杂的二氢噁嗪类似物可对紫杉酚连接问题提供普通的解决。
由于在紫杉酚结构活性曲线中的A-环侧链的中心位置并由于通过微管连接位置的识别,侧链上用光官能度进行紫杉酚类似物光亲和性标记在紫杉酚-微管相互作用的化学的详细研究中是很有价值的。因此我们确定追踪出乎期望的苄基氨基甲酸乙酯-保护的侧链酸11的复杂情况,在该情况下,侧链连接后的N-去保护-酰化步骤将对下一步光不稳定的官能度和放射性标记的结合提供光标记试剂。
当7,11,二环已基碳化二亚胺(DCC),和4-(二甲氨基)吡啶(DMAP)在甲苯中的混合物被加热浓缩时,比较收集于表1中的酯化反应,侧链连接此用苯甲酰胺10时更迅速,产率更高。在假设有下式所示二氢噁嗪:
介入并且它的形成为限速无中间体过程,可发现在酯化中11的较高反应性为它的氨基甲酸乙酯羰基氧的较高亲和性的结果。这个特征使过量使用的侧链酸的用量有相当大的减少。
锌诱导的三氯乙氧基的O-保护基的除去得到紫杉酚类似物:
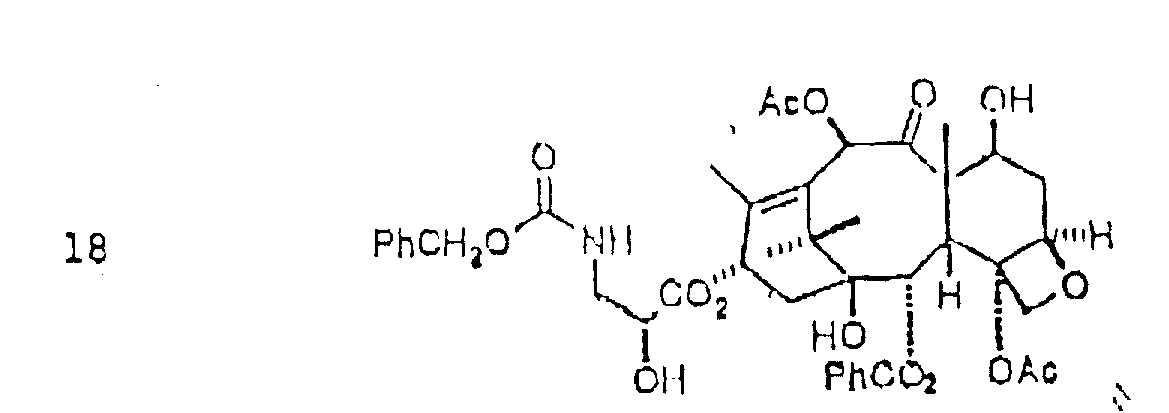
随后在含三氟乙酸(TEA)的溶液中氢解除去苄基氨基甲酸乙酯,按下列反应:

选择性酰化氨基,得到如表2所示紫杉酚类似物:
表2化合物18的去保护-酰化
RCO 产物 总产率(%)
4-叠氮苯甲酰基 19 79a
75b
4-(三氟甲基)苯甲酰基 20 52a
2-萘甲酰基 21 64a
已酰基 22 64a
新戊酰基 23 79a
环己羰基 24 50a
1-金刚烷基羰基 25 70a
t-丁氧羰基 26 89c
a用酰氯制备。b用过量氨和4-叠氮基苯甲酸N-羟基琥珀酰亚胺酯制备。c用碳酸二叔丁酯制备。苄基保护基在本文中很有用。因为已知紫杉桥头烯要抵抗氢化作用。Baxter等,J.Chem.Soc,1962,2964。
在制备上述类似物中方便地提高了侧链11的连接比率的同时,在更复杂的N-乙酰基-3-苯基异丝氨酸侧链的浆果赤霉素ⅢC-13羟基的酯化中的相似的提高对紫杉酚的特性具有更显著的影响。我们按下列方案研究3-苯基异丝氨酸侧链的酯化:

其结果概括于下列表3:
表3 3-苯基异丝氨酸侧链的酯化
R 酸当量 偶合剂 时间 产物 产率(%)
ph (27) 10 DPC 36 h 29 50 (96a)
6 DPC 48 h 0
phCH2O (28) 10 DPC 36 h 30 50 (78a)
6 DCC 1 h 95
3.9 DCC 2 h 78 (84a)
a按回收的7计算。
令人遗憾的是Greene,Gueritte-Voegelein紫杉酚部分合成的低速率的酯化以及我们对侧链酸27的相似观察暗示出二氢恶嗪不能由N-苯甲酰基和3-苯基取代基结合的侧链酸迅速得到。更近一步。当DPC作为羧基活化剂时,由Greene(Denis等,J.Org.Chem,1990,55,1957)和Jacobsen(Deng等,J.Org.Chem,1992,57,4320)紫杉酚侧链合成制得的苄基氨基甲酸乙酯酸28的酯化的速率没有提高。(而且,可比较总结于表3的酯化)。但是与DCC结合的28却导致了上述证明的高速率和产率。通过DCC未能导致27有效连接可证明这是由于亲核性更大的氨基甲酸乙酯氧和特别有效的羧基活化剂DCC的结合,可能导致就地生成下式化合物:

如适当,式31的形成必须是限速的,因为在与28的酯化中未发现中间体。至今在我们已经研究过的侧链连接中,由Greene和Gueritte-Voegelein为紫杉酚侧链酯化提出的DPC,当侧链氮连着苯甲酰基时为最有效的试剂,而在进行含苄基氨基甲酸乙酯的侧链的快速酯化时,DCC是最有效的。
锌处理30得到33。一系列氢解-乙酰化实验得到紫杉酚,cephalomannine,10-乙酰基TAXOTERE
 以及列于表4的其余类似物,其反应归纳为下面反应式:
以及列于表4的其余类似物,其反应归纳为下面反应式:

(其中X为任意离去基团,例如,-C1,OCO2(CH3)3或
而i-PrOH为异丙醇)
表4 33的去保护-乙酰化
RCO
产率(%) 产物 总的
tigloyl cephalomannine 81a
t-丁氧羰基 10-乙酰基TaxotereR94b
4-叠氮基苯甲酰基 34 71a
79c
4-(三氟甲基)苯甲酰基 35 86a
4-溴苯甲酰基 36 76a
4-(乙酰基)苯甲酰基 37 67a
4-(苯甲酸基)苯甲酰基 38 66a
邻羟苄基 39 51d
a由酰氯制备。b用碳酸二叔丁酯制备。c用过量氨和4-叠氮基苯甲酸N-羟基琥珀酰亚胺酯制备。d用邻羟基苯甲酸N-羟基琥珀酰亚胺酯制备。
33氢解后所得到的氨基三醇在c-2′位特别易于0-酰化。例如,在cephalomannine的制备中,乙酰化发生在使用顺式-乙-甲基-2-丁烯酸的N-羟基琥珀酰亚胺酯时也会发生显著的0-酰化反应。更进一步,在三乙胺-二氯甲烷溶液中使用酰氯,即使在低温下,对所有类似物均无的选择性。令人惊奇地,影响氨基乙酰化选择性的最佳条件包括在吡啶溶液中的酰氯与在室温下的DMAP催化剂。
当令人满意地实现了紫杉酚的部分合成时,光亲和性标记的叠氮化物类似物34的制备强调了这种方法的实用性。在我们的工作中由于叠氮化合物部分的热不稳定性使Greene,Gueritte-Voegelein的酯化反应不能用于由连有N-对-叠氮基苯甲酰基的O-保护的侧链酸制备34。是通过上面的化学描述,可通过适于制备放射性标记的物质的方法而以优良的产率得到34。
在这项工作中研究了几项羟基保护方案。任何用于紫杉酚和紫杉酚类似物部分合成的就地活化羧基要求这些保护基对酸稳定。我们工作中未达到这个标准的有对异丝氨酸侧链的C-2′羟基保护的三乙基甲硅烷基和对3-苯基异丝氨酸侧链的这个羟基保护的乙氧基乙基。总的说来,我们发现在3-苯基异丝氨酸侧链酸中的侧链羟基保护基的酸稳定性比在相似的异丝氨酸中所需的要大。同样地,对于异丝氨酸紫杉酚类似物来说酯化后除去酚不稳定羟基保护基更为困难。因为这个原因,乙氧乙基不适于后者的侧链类型。在侧链羟基保护基中复杂的或防止酯化的是三异丙基甲硅烷基和三氯乙氧羰基。苄氧甲基对于异丝氨酸侧链相当合适,但不能从更具障碍的连于3-苯基异丝氨酸侧链的C-2′羟基上除去。从这个实验中我们已知的三氯乙氧甲基保护基和已知的三氯乙氧羰基对C-2′和C-7羟基是较好的保护基。这些基团对酯化反应时的侧链羰酸是稳定的,并且在与18,33,紫杉酚,及相关的复杂紫杉烷17的官能团相适应的单一还原反应时都被除去。
一种制备紫杉酚及类似物的简单有效的方法已被得到,它基于对O-保护的异丝氨酸和3-苯基异丝氨酸侧链酸的快速高产率的酯化反应,即将N-苄氧羰基官能基引入我们确信有类似17的二氢恶嗪和就地形成的下式化合物:

介入。酯化产物去保护和侧链氨基的酰化提供了目标化合物。从浆果赤霉素Ⅲ总共需要5步。
在这项工作之前,在浆果赤霉素ⅢC-13位羟基上连接紫杉酚侧链最有效的方法需要独立地形成β-内酰胺和二氢恶嗪酰化试剂。按本法从合适地保护的侧链酸就地生成二氢恶嗪,已报导的几种紫杉酚侧链的不对称合成方法对于紫杉酚及相关药物的部分合成变得特别有用。
本发明制备紫杉酚,紫杉酚类似物及其中间体的化学方法概括为下面通式化合物:
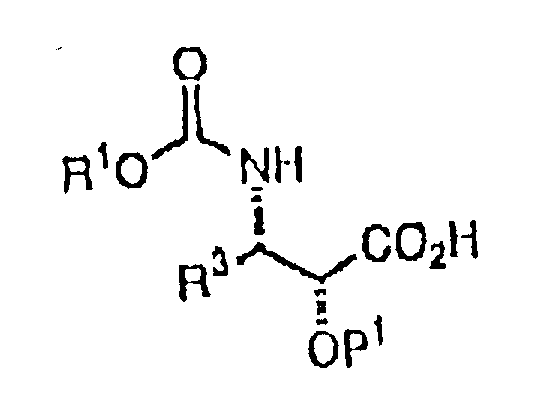
与下面通式结构的紫杉烷:
缩合,得到下面通式结构的中间体:

其中,R1=烷基,烯基或芳基或PhCH2
R3=氢,烷基,烯基,芳基或Ph
R4=氢,氧基或乙酰氧基
R5=氢,氧基或羰基
R6=氢,氧基或乙酰氧基
R7=氢,氧基,苯甲酰氧基或芳酰氧基
R8=氢或羟基
P1=羟基保护基
这个总方法中,某些更具体的反应包括其中:(Ⅰ)R1为PhCH2,R3为Ph而P1为CH2OCH2CCl3;(Ⅱ)R1为PhCH2,R3为Ph,R4为AcO,R5为双键O,R6为OCO2CH2cCl3,R7为PhCO2,R6为OH及P1为CH2OCH2CCl3;(Ⅲ)R1为PhCH2,R3为Ph,R4为AcO,R5为双键O,R6为OHR7为PhCO2,R8为OH而P1为H;以及(Ⅳ)R3为Ph,R4为AcO,R5为双键O,R6为OP2,R7为PhCO2,R8为OH而其中P2为羟基保护基。
上述总方法后接着进行取代R1O和P1的步骤,得到如下通式结构的化合物:

其中R2=氢,烷基,芳基,氧,氮,硫或Ph基。
此外,上述总方法可以按下列方法更有效地进行:(Ⅰ)R1为烷基,烯基或芳基,R3为Ph,R4为AcO,R5为双键O,R6为OP2,R7为PhCO2,R8为OH,P2为羟基保护基,并包括用Ph取代R1O以及用氢取代P1和P2以制备紫杉酚的步骤;(Ⅱ)R1为PhCH2,P1为CH2OCH2CCl3,P2为CO2CH2CCl3以及通过PhCH2OC=0与PhC=0交换来完成的用Ph取代R1O的步骤;(Ⅲ)R1为烷基,烯基或芳基,R3为氢,烷基,烯基或芳基,R4为AcO,R5为双键O,R6为OP2,R7为PhCO2,R8为OH,P2为羟基保护基,并包括用ph取代R1O及用氢取代P1和P2以制备紫杉酚类似物的步骤;以及(Ⅳ)R1为PhCh2O,R3为Ph,R4为AcO,R5为双键O,R6为OP2,R7为PhCO2,R8为OH及P2为羟基保护基以及在芳香溶剂和活化剂存在下进行的3-苯基异丝氨酸侧链的酯化缩合。
在最后一种情况(Ⅳ)中,P1可为CH2OCH2CCl3而P2可为CO2CH2CCl3。这里活化剂选用二烷氨基吡啶,如4-(二甲氨基)吡啶。有机溶剂选用苯,甲苯,二甲苯,乙苯,异丙苯和氯苯。反应最好在大约70-75℃的温度下进行。酯化反应在缩合剂存在下进行,该缩合剂可选自二环已基碳化二亚胺和二-2-吡啶基碳酸酯。总起来反应(Ⅳ)可在选自二环已基碳化二亚胺和二-吡啶基碳酸酯及活化剂4-(二甲氨基)吡啶的存在下进行。
另外,制备紫杉酚类似物的化学方法也可通过由下列步骤组成的方法来概述:
将10-去乙酰浆果赤霉素Ⅲ转化为:

然后通过与

最终转化成下式的紫杉酚类似物:

其中:R1=烷基,烯基,或芳基
R2=氢,烷基,烯基,芳基,氧,氮,或硫基
R3=氢,烷基,烯基或芳基
P1,P2和P3=CO2CH2Cl3或其他羟基保护基
按Magri等,J.Org.Chem.1986,51,3239所述,通过由Dr。Kenneth Snade,Natioal Cancer Institute,National Institues of Health提供的紫杉酚-Cephalomannine混合物的降解而制备浆果赤霉素Ⅲ。按照此方法它被转化为浆果赤霉素Ⅲ7-三氯乙基碳酸酯(7)。需要无水条件和惰性气氛的反应在火焰干燥的玻璃仪器在氮气中使用无水溶剂进行。CDCl3溶液的1H和13CNMR谱分别使用四甲基硅烷(1Hδ=0ppm),氯仿(1Hδ=1.27ppm),或CDECl3,(13Cδ=77ppm)作为内标而在300MHZ和75MHZ被记录。对于13CNMR谱,质子取代的程度用辅助1FDEPT的实验指定。高效液相色谱(HPLC)用Waters牌的仪器在3.9mm×15cm Novapak牌D18柱,或在7.8×30cm NovapakC13柱上进行,在254纳米(nm)处使用UV波检测。另外色谱用硅胶进行。在Mass Spectrometry Laboratory,Qepartment of Chemistry,University of Pennsylvania,Philadelphia,PA,USA进行高分辨质谱的测定。
以标准方法制备简单的异丝氨酸侧链,包括:(Ⅰ)(2R)-N-苄氧羰基-O-三氯乙氧甲基-异丝氨酸(化合物11);(Ⅱ)浆果赤霉素Ⅲ酯15(由化合物11生成的化合物15);及(Ⅲ)浆果赤霉素Ⅲ酯18(由化合物15生成的化合物18)。
43 X=N345 R1=PhCH2O1R2=H1R3-El
44 X=NH248 R1=PhCH2O1R2=CH2OCH2CCl3,R3-El
制备化合物28,(2R,3S)-N-苄氧羰基-O-(三氯乙氧甲基)-3-苯基异丝氨酸,这里将3.12g(16.3mmol)(2R,3R)-乙基3-苯基甘氨酸酯,13.5m甲酸甲酯,和5.28g(81.5mmol)叠氮化钠在81m18∶1 95%乙醇-水中的溶液在50℃加热45小时。将反应混合物用水和乙醚分配,用乙醚萃取水层,将有机层合并,干燥(MgSO4),过滤并浓缩。通过柱层析纯化残留物,用乙酚乙酯-已烷洗脱,得到2.91g(94%)的无色油状物43。
1H NMR δ 1.28(3H,t,J=7.1,CH3),3.18K(1H,d,J=6.7,OH)4.27(2H,q,J=7.1,CH2),4.37(1H,dd,J=3.1,6.7,CHOH),4.84(1H,d,J=3.1,PhCH),7.4-7.5(5H,m,ph);13CNMR δ 171.84,135.52(;四重峰),128.77,128.71,127.81,73.87,67.14,(CH),62.37(CH2),14.02(CH3);IR(CHCl3)3158,2932,2100,1731,1302,1110 cm-1
C11H13N3O3元素分析计算值:C,56.15;H,5.57;N,17.87.
实测值:C,56.33;H,5.79;N,17.65.
将1.88g43(8.00mmol)和188mg10%pd/炭在80ml乙酸乙酯中的混合物在氢气中(35Psi)摇动14小时。过滤并浓缩得到白色粉末状氨基醇44的粗品,mp69-71℃,它无需纯化便可使用。
1H NMR δ 1.25(3H,t,J=7.2,CH3),2.24(3H,bs,OH and NH2),4.23(2H,m,CH2)4.27(2H,pp d,J=1.2,CH),7.3-7.4(m,5H,Ph).
0℃下在1.98g(9.48mmol)44和2.98g(10.43mmol)碳酸钠的57ml 1∶1乙醚-水悬浮液中加入1.35ml(9.48mmol)氯甲酸苄酯。将该混合物搅拌30分钟,蒸发溶剂,用二氯甲烷和水分配残留物。将有机层干燥(硫酸钠)并浓缩得到氨基甲酸乙酯45(由43的产率91%);mp103-105℃(二氯甲烷-已烷)。
1H NMR δ 1.25(3H,t,J=6.9,CH3),3.30(1H,bs,OH),4.25(2H,q,J=6.9,CH2CH3),4.44(1H,app s,CHOH),5.03(1H,1/2 AB q,J=11.9,CH2Ph),5.07(1H,1/2 AB q,J=11.9,CH2Ph),5.28(1H,app d,J=9.5,PhCH),5.76(1H,bd,J=9.5,NH),7.3-7.4(10H,m,Ph):13C NMR δ 172.66,155.61,138.90,136.24(四重峰),128.54(双峰)128.42,128.02,127.76,126.70,73.41,66.93,(CH),62.49,56.42(CH2),13.98(CH3);IR CHCl3)3520,3440,1730,1260,1095 cm-1.
C19H21NO3元素分析计算值:C,66.45;H,6.17;N,4.08.实测值:C,66.29;H,6.28;N,3.98.
-68℃搅拌下在6.78g(19.8mmol)氨基甲酸乙酯45的9ml无水THF溶液中滴加n-BuLi(1.6M已烷溶液,12.4ml,19.8mmol)。5分钟后加入3.89ml(2.97mmol)三氯乙氧甲基氯和2.65g(19.8mmol)LiI。随后,使混合物温热到室温超过1小时。然后将反应混合物倒入1N盐酸中,用二氯甲烷萃取,将有机层干燥(MgSO4)并浓缩。通过柱层析纯化残留物,用乙酸乙酯-已烷洗脱,得到8.22g(82%)无色油状物46。
H NMR δ 1.23(3H,t,J=7.2,CH3),3.16(1H,1/2 AB q,J=11.7,CH2CCl3),3.63(1H,1/2 AB q,J=11.7,CH2CCl3),4.20(2H,m,CH2CH3),4.52(1H,bs,CHCO2Et),4.70(1H 1/2 AB q,J=7.3,OCH2O),4.85(1H,1/2 AB q,J=7.3,OCH2O),5.04(1H,1/2 AB q,J=11.9,CH2Ph),5.08(1H 1/2 AB q,J=11.9,CH2Ph),5.41(1H,app bd,J-9.2,PhCH),5.80(1H,d,J=9.2,NH),7.28-7.34(10H,m,Ph):13C NMR δ 168.92,155.24,138.38,135.98(q 四重峰 s),128.31,128.14,127.77,127.72,127.60,126.12,95.86,94.39,79.02(CH),77.00,66.65,61.43,56.05(CH2),13.77(CH3);IR(CHCl3)3440,2912,1760,1290,1095cm-1.
C22H24Cl3NO3元素分析计算值:C,52.48;H,4.81;N,2.78.实测值:C,52.33 H,4.82;N,2.68.
室温下将494mg(0.98mmol)46和205mg(4.88mmol)氢氧化锂-水合物在16.3ml3∶1甲醇水中的混合物搅拌2小时,蒸发溶剂并用二氯甲烷和1N盐酸分配残留物,将有机相干燥(硫酸钠)并浓缩得到458mg(96%)28。为元素分析,将酸28转化成它的二环已基铵盐,mp133.5-136℃(乙醚)。
,mp 133.5-136.0℃(乙醚).1H NMR δ 3.17(1H,1/2 AB q,J=11.7,OCH2CCl3),3.65(1H,1/2 AB q,J=11.7,OCH2CCl3),4.60(1H,m,CHOC2H),4.73(1H,1/2 AB q,J=7.3,OCH2O),4.89(1H,1/2 AB q,J=7.3,OCH2O),5.08(1H,1/2 AB q,J=11.9,PhCH2O),5.17(1H,1/2 AB q,J=11.9,PhCH2O),5.51(1H,app bd,J=9.0 PhCH),5.95(1H,d,J=9.0,NH),7.2-7.5(10H,m,Ph);13C NMR δ 171.94,156.21,138.48,135.95,96.14(q 四重峰 s),128.75,128.52,128.16,128.09,126.40,76.33,56.28(CH),94.88,79.51,67.51(CH2);IR(CHCl3)3460,3100,2990,2910,1760,1720,1360,1290,1140,1090,1060,1010 cm-1.
C34H43Cl3N2O6元素分析计算值:(28 二环已基胺):C,58.41;H,6.59;N,4.26,实测值:C,58.65;H,6.51;N,4.17.
浆果赤霉素Ⅲ酯30:将80mg(0.105mmol)7,301mg(9.629mmol)28,36mg(0.210mmol)4-(二甲氨基)吡啶,和130mg(0.629mmol)二环已基碳化二亚胺在5.25ml甲苯中的混合物在75℃搅拌1小时。将反应混合物冷却,过滤,并将滤液用0.5N盐酸、水洗涤并干燥(硫酸钠)。浓缩并通过柱层析纯化残留物,用乙酸乙酯-已烷洗脱,得到120mg(94%)无色,无定形固体30。
1H NMR δ 1.10(3H,s,C-17),1.17(3H,s,C-16),1.77(3H,s,C-19)1.90(3H,s,C-18),1.9-2.4(3H,m,1/2 C-6,C-14),2.10(3H,s,OAc),2.39(3H,s,OAc),2.5-2.6(1H,m,1/2 C-6),3.23(1H,1/2 AB q,J=11.4,OCH2CCl3),3.63(1H,1/2 AB q,J=11.4,OCH2CCl3),3.87(1H,d,J=7.1,C-3),4.13(1H,1/2 AB q,J=8.3 C-20),4.26(1H,1/2 ABq,J=8.3,C-20),4.58(1H,1/2 AB q,J=12.0,OCH2CCl3),4.71(1H,1/2 AB q,J=7.3 OCH2O),4.82(1H,1/2 AB q,J=7.3,OCH2O),4.85-5.0(4H,m,C-5,PhCH2O,1/2 OCH2CCl3),5.42(1H,br d,J=8.5 C-2′),5.51(1H,dd,J=7.1,10.7,C-7),5.61(1H,d,J=7.1,C-2),5.68(1H,br d,J=9.1,C-3′),6.22(1H,t,J=6.7,C-13),6.28(1H,s,C-10),7.1-7.4(11H,m,Ph,NH),7.45(2H,app t,J=7.4,OBz),7.55(1H,app t,J=7.4,OBz),8.06(2H,app d,J=7.4,OBz).
用7,10当量的28,10当量的二-2-吡啶基碳酸酯,和3.3当量的4-(二甲氨基)吡啶,在75℃甲苯中进行相似的反应36小时,得到50%产率的30(按回收的7计算为78%)。
浆果赤霉素Ⅲ酯33:对120mg(0.0996mmol)30和97mg(1.49mmol)Zn粉在1.2ml1∶1乙酸-甲醇中的混合物声处理30分钟。另加锌65mg(0.996mmol)并对其继续声处理,共声处理5小时。倾析溶剂,用甲醇冲洗锌和锌盐,将有机相合并,用水稀释,并用二氯甲烷萃取。将二氯甲烷层用水洗涤,干燥(硫酸钠),并浓缩。层析残留物,用乙酸乙酯-已烷洗脱,得到57-75mg(65-85%)33。

33的去保护-乙酰化得到紫杉酚,Cephalomannine,和34-39:将242mg(0.274mmol)33,42μL三氯乙酸,和43mg钯炭黑在4ML异丙醇中的混合物在氢气(35Psi)中摇动1.5小时。过滤并浓缩滤液得到相应的三氟乙酸铵盐,它无需纯化便可使用。室温搅拌下在20mg(0.023mmol)这种物质和2mg4-(二甲氨基)吡啶在1.3ML吡啶的混合物中滴加0.028mmol乙酰化试剂。30分钟后,另加0.0184mmol乙酰化试剂,如果需要,继续搅拌30分钟。然后将混合物用二氯甲烷稀释,用1N盐酸,水洗涤,并干燥(硫酸钠)有机相。浓缩并通过柱层析纯化残留物,用乙酸乙酯-已烷洗脱,将得到的产物层析,用乙酸乙酯-已烷洗脱,得到色谱及谱纯的产物。
紫杉酚:以90%的产率由苯甲酰氯制备,得到色谱及光谱与真实样品一致的产物。
Cephalomannine:以81%的产率由甘氨酰氯制备,得到色谱及光谱与可p真实样品一致的产物。HRMS(阴离子CI;甲烷试剂气)计算值(M)831.3466,实测值831.3389。
叠氮基紫杉酚34:以71%的产率由4-叠氮基苯甲酰氯制备。
1H NMR δ 1.13(3H,s,C-17),1.23(3H,s,C-16),1.67(3H,s,C-19),1.78(cH,s,C-18),1.9-2.1(3H,m,1/2 C-6,C-14),2.22(3H,s,OAc),2.37(3H,s,OAc),2.45-2.55(1H,m,1/2 C-16),3.79(1H,d,J=6.9,C-3),4.18(1H,1/2 AB q,J=8.2,C-20),4.28(1H,1/2 AB q,J=8.2,C-20),4.38(1H,dd,J=6.8,11.0,C-7),4.75-4.8(1H,m,C-2′),4.92(1H,d,J=7.6,C-5),5.66(1H,d,J=7.0,C-2),5.76(1H,dd,J=2.4,8.8,C-3′),6.22(1H,t,J=8.2,C-13),6.27(1H,s,C-10),6.98(1H,app d,J=8.8,NH),7.01(2H,app d,J=8.6,N3Ar),7.3-7.5(7H,m,ph,OBz),7.61(1H,app t,J=7.4,OBz),7.73(2H,d,J=8.6,N3Ar),8.12(2H,app d,J=7.4,OBz);13CNMR δ 203.55,172.76,171.17,170.36,167.03,165.96,143.83,141.87,137.94,133.20,130.03,129.15,81.15,79.04,58.58,43.16(四重峰),133.70,130.19,129.00,128.88,128.70,128.44,128.35,127.03,119.07,84.38,75.54,74.96,73.18,72.27,72.14,55.08,45.67(CH),76.48,35.63(双峰)(CH2),26.84,22.58,21.77,20.80,14.79,9.55(CH3);UV[MeOH,波长 nm(
 )]230(23 000),270(20 000);HRFABMS计算值(M+Na)917.3221,实测值
)]230(23 000),270(20 000);HRFABMS计算值(M+Na)917.3221,实测值
 917.3236.
917.3236.
三氟甲基紫杉酚35:以86%的产率由4-(三氯甲基)苯甲酰氯制备。
1H NMR δ 1.19(3H,s,C-17),1.28(3H,s,C-16),1.71(3H,s,C-19),1.81(3H,s,C-18),1.8-2.4(3H,m,1/2 C-6,C-14),2.28(3H,s,OAc),2.44(3H,s,OAc),2.5-2.6(1H,m,1/2 C-6),3.43(1H,d,J=4.7,OH),3.79(1H,d,J=7.5,C-3),4.19(1H,1/2 AB q,J=8.5,C-20),4.31(1H,1/2 AB q,J=8.5,C-20),4.35-4.45(1H,m,C-7),4.79(1H,dd,J=2.4,4.7,C-2′),4.94(1H,d,J=8.0,C-5),5.67(1H,d,J=7.0,C-2),5.79(1H,dd,J=2.4,9.0,C-3′),6.23(1H,t,J=7.0,C-13),6.27(1H,s,C-10),7.03(1H,d,J=9.0,NH),7.2-7.7(10H,m,Ar),7.84(2H,app d,J=8.2,Ar),8.13(2H,app d,J=8.2,Ar);13C NMR δ 203.51,172.62,171.25,170.34,167.05,165.00,141.77,137.60,136.92,133.37,129.15,128.72,128.46,81.22,79.09,58.62,43.18(四重峰),133.77,130.20,129.11,128.72,128.54,127.52,127.01,125.79,84.37,75.51,74.89,72.92,72.47,72.19,55.02,45.66,(CH),77.91,35.57(双峰)(CH2),26.88,22.63,21.75,20.84,14.84,9.54(CH3);UV[MeOh,波长 nm(ε)]230(29 000);HRFABMS(计算值:(M+Na)944.3081,实测值:944.3022.
溴代紫杉酚36:以76%的产率由4-溴苯甲酰氯制备。
1H NMR δ 1.14(3H,s,C-17),1.24(3H,s,C-16),1.68(3H,s,C-19),1.78(3H,s,C-18),1.9-2.4(3H,m,1/2 C-6,C-14),2.24(3H,s,OAc),2.38(3H,s,OAc),2.5-2.6(1H,m,1/2 C-6),3.79(1H,d,J=6.9,C-3),4.19(1H,1/2 AB q,J=8.3,C-20),4.31(1H,1/2 AB q,J=8.3,C-20),4.40(1H,dd,J=7.0,11.1,C-7),4.78(1H,d,J=2.5,C-2′),4.94(1H,d,J=7.9,C-5),5.67(1H,d,J=6.9,C-2),5.77(1H,dd,J=2.5,8.7,C-3′),6.22(1H,t,J=8.6,C-13),6.26(1H,s,C-10),6.97(1H,d,J=8.7,NH),7.3-7.6(12H,m Ar),8.13(2H,d,J=7.3,Ar);13C NMR δ 203.57,172.62,171.28,170.35,167.01,165.98,141.86,137.70,133.19,132.33,128.46,126.74,81.13,79.04,58.59,43.13(四重峰),133.77,131.92,130.19,129.08,128.72,128.62,126.99,84.35,75.51,74.85,73.00,72.39,72.19,54.94,45.57,(CH),76.58,35.56,(双峰)(CH2),26.85,22.62,21.75,20.86,14.86,9.53(CH2);HRFABMS计算值(M[78Br]+H)932.2493,实测值
 .932.2577.
.932.2577.
乙酰基紫杉酚37:以67%的产率由4-(乙酰基)苯甲酰氯制备。
1H NMR δ 1.14(3H,s,C-17),1.24(3H,s,C-16),1.68(3H,s,C-19),1.77(3H,s,C-18),1.9-2.4(3H,m,1/2 C-6,C-14),2.22(3H,s,OAc),2.38(3H,s,OAc),2.5-2.6(1H,m,1/2 C-6),2.61(3H,s,Ac),3.79(1H,d,J=7.3,C-3),4.19(1H,1/2 AB q,J=8.6,C-20),4.31(1H,1/2 AB q,J=8.6,C-20),4.37(1H,dd,J=6.6,10.4,C-7),4.79(1H,d,J=2.1,C-2′),4.94(1H,d,J=7.0,C-5),5.67(1H,d,J=7.3,C-2),5.76(1H,dd,J=2.1,7.1,C-3′),6.23(1H,t,J=7.9,C-13),6.26(1H,s,C-10),7.05(1H,d,J=7.1,NH)7.3-7.65(8H,m,Ar),7.82(2H,app d,J=8.3,Ar),7.97(2H,app d,J=8.3,Ar),8.13(2H,d,J=7.3,Ar);13C NMR δ 203.56,197.33,172.61,171.28,170.36,167.00,166.02,141.80,139.48,137.65,137.41,133.22,128.50,81.14,79.02,58.57,43.13,(四重峰),133.77,130.02,129.18,129.08,128.72,128.66,127.37,127.01,84.35,75.51,74.85,73.01,72.39,72.18,55.02,45.60,(CH),77.20,35.58,(双峰)(CH2),26.84,22.63,21.74,20.87,14.86,14.18,9.54,(CH3);UV[MeOH,波长 nm(ε)]236(37 000);HRFABMS计算值(M+H)896.3493,实测值:896.3536。
苯甲酰基紫杉酚38:以66%的产率由4-(苯甲酰基)苯甲酰氯制备。
1H NMR δ 1.14(3H,s,C-16),1.23(3H,s,C-17),1.68(3H,s,C-19),1.79(3H,s,C-18),2.23(3H,s,OAc),2.3-2.4(2H,m,C-14),2.38(3H,s,OAc),2.4-2.6(2H,m,C-6),3.67(1H,d,J=5.2,OH),3.79(1H,d,J=7.0,C-3),4.18(1H,1/2 AB q,J=8.4,C-20),4.30(1H,1/2 AB q,J=8.4,C-20),4.35-4.42(1H,m,C-7),4.80(1H,dd,J=5.2,2.5,C-2′),4.94(1H,d,J=7.8,C-5),5.66(1H,d,J=7.0,C-2),5.80(1H,dd,J=8.9,2.6,C-3′),6.24(1H,t,J=7.5,C-13),6.27(1H,s,C-10),7.18(1H,bd,J=8.9,NH),7.3-7.6(5H,m,ph),7.61(2H,t,J=7.2,OBz),7.7-7.9(5H,m,OBz,Ar),8.12(2H,d,J=7.2,OBz);13C NMR δ 203.56,195.86,172.65,171.25,170.35,166.96,166.20,141.81,140.44,137.70,136.77,133.18,81.12,78.99,77.42,77.00,58.53,43.13,(四重峰),133.74,132.99,130.17,130.13,130.07,129.05,128.70,128.44,127.04,127.02,84.34,77.20,75.50,74.85,73.03,72.32,72.14,55.06,45.61,(CH),76.57,35.57,(双峰)(CH2),26.83,22.61,21.75,20.85,14.84,9.54,(CH3):UV[MeOH,波长 nm(ε)]232(28 000),258(27 000);HRFABMS计算值,(M+H)958.3650,实测值:958.3591。
羟基紫杉酚39:以51%的产率由邻羟基苯甲酸N-羟基琥珀酰亚胺酯制备。
1H NMR δ 1.14(3H,s,C-17),1.22(3H,s,C-16),1.68(3H,s,C-19),1.76(3H,s,C-18)2.23(3H,s,OAc),2.3-2.4(2H,m,C-14),2.35(3H,s,OAc),2.4-2.6(2H,m,C-6),3.78(1H,d,J=7.0,C-3),4.19(1H,1/2 AB q,J=8.4,C-20),4.29(1H,1/2 AB q,J=8.4,C-20),4.37(1H,dd,J=10.4,6.6,C-7),4.80(1H,d,J=2.5,C-2′),4.93(1H,d,J=8.2,C-5),5.65(1H,d,J=7.0,C-2),5.79(1H,dd,J=8.6,2.5,C-3′),6.22(1H,bt,J=9.6,C-13),6.26(1H,s,C-10),6.86(1H,t,J=7.6,Ar),6.93(1H,bd,J=8.6,NH),7.3-7.5(10H,m,Ar,OBz),7.63(1H,t,J=7.4,OBz),8.09(2H,d,J=7.4,OBz);13C NMR δ 203.50,172.27,171.27,170.40,169.49,166.90,161.51,141.57,137.51,133.33,129.02,113.68,81.23,78.85,58.57,43.08,(四重峰),134.72,133.83,130.03,129.07,128.71,128.51,126.93,125.62,118.91,118.70,84.30,75.51,74.77,73.01,72.32,72.18,54.32,45.66,(CH),76.47,35.61,(双峰)(CH2),26.81,22.61,21.56,20.85,14.90,9.51(CH3);UV[MeOH,波长 nm(ε)]232(28 000),300(5 000);萤光光谱 MeOH,波长 (300激发 nm]342,410;HRFABMS计算值,(M+H)870.3337,实测值:870.3354.
33的去保护-乙酰化得到10-乙酰基TAXOTERER和34:除了在室温下乙酰化外按与上述相似的方法将33氢解去保护(16小时)及乙酰化。
10-乙酰基TAXOTERER:以94%的产率由碳酸二叔丁酯制备,得到光谱数据与文献中一致的物质12。HRFABMS计算值(M+H)850.3650,实测值850.3687。
叠氮基紫杉酚34:以79%的产率由4-叠氮基苯甲酸N-羟基琥珀酰亚胺酯和过量铵盐制备。
因此,按前述,本发明特别关注结构如下的一些中间体和紫杉酚类似物:


无需作进一步说明,上述内容将充分说明我们的发明,以致于其他人可通过应用现在或将来的知识,使其适应各种用途。
Claims (19)
1、一种用于制备紫杉酚,紫杉酚类似物及其中间体的化学方法,其中包括将下面通式的化合物:

与下面通式结构的紫杉烷:

缩合得到下面通式结构的中间体:

其中:R1=烷基,烯基或芳基或PhCH2
R3=氢,烷基,烯基,芳基或Ph
R4=氢,氧基或乙酰氧基
R5=氢,氧基或羰基
R6=氢,氧基或乙酰氧基
R7=氢,氧基,苯甲酰氧基或芳酰氧基
R8=氢或羟基
P1=羟基保护基。
2、按照权利要求1的方法,其中R1为PhCh2,R3为Ph而P1为CH2OCH2CCl3。
3、按照权利要求2的方法,其中R4为AcO,R5为双键O,R6为OCO2CH2CCl3,R7为PhCO2,而R8为OH。
4、按照权利要求1的方法,其中R1为PhCH2,R3为Ph,R
 为AcO,R5为双键O,R6为OH,R7为PhCO2,R8为OH而P1为H。
为AcO,R5为双键O,R6为OH,R7为PhCO2,R8为OH而P1为H。
5、按照权利要求1的方法,其中R3为Ph,R4为AcO,R5为双键O,R6为OP2,R7为PhCO2,R8为OH而其中P2为羟基保护基。
6、按照权利要求1的方法,包括取代R1O和P1得到下列通式结构的化合物的步骤:

其中R2=氢,烷基,芳基,氧,氮,硫或Ph基
7、按照权利要求1的方法,其中R1为烷基,烯基或芳基,R3为Ph,R4为AcO,R5为双键O,R6为OP2,R7为PhCO2,R8为OH,而P2为羟基保护基,并包括用Ph取代R1O及用氢取代P1和P2以制备紫杉酚的步骤。
8、按照权利要求7的方法,其中R1为PhCH2,P1为CH2OCH2CCl3而P2为CO2CH2CCl3并包括通过将PhCH2OC=0与PhC=O交换而使Ph取代R1O的步骤。
9、按照权利要求1的方法,其中R1为烷基,烯基或芳基,R3是氢,烷基,烯基或芳基,R4为AcO,R5为双键O,R6为OP2,R7为PhCO2,R8为OH而P2为羟基保护基,并包括用Ph取代R1O及用氢取代P1和P2以制备紫杉酚的步骤。
10、按照权利要求1的方法,其中R1为PhCH2O,R3为Ph,R4为AcO,R5为双键O,R6为OP2,R7为PhCO2,R8为OH而P2为羟基保护基,并且其中的缩合是在芳香溶剂和活化剂存在下,由3-苯基异丝氨酸侧链酯化完成的。
11、权利要求10的方法,其中P1为CH2OCH2CCl3而P2为CO2CH2CCl3。
12、权利要求11的方法,其中活化剂选自二烷氨基吡啶。
13、权利要求12的方法,其中二烷氨基吡啶为4-(二甲氨基)吡啶。
14、权利要求12的方法,其中有机溶剂选自苯,甲苯,二甲苯,乙苯,异丙苯和氯苯。
15、权利要求12的方法,其中反应在大约70-75℃的温度下进行。
16、权利要求12的方法,其中酯化在缩合剂存在下进行。
17、权利要求16的方法,其中缩合剂选自二环已基碳化二亚胺和二-2-吡啶基碳酸酯。
18、权利要求12的方法,其中反应在选自二环已基碳化二亚胺和二-2-吡啶基碳酸酯的缩合剂和4-(二甲氨基)吡啶作为活化剂存在下进行。
19、按权利要求1-18的任何一项的方法制备的化合物。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US1509593A | 1993-02-05 | 1993-02-05 | |
| US08/015,095 | 1993-02-05 |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN00103725A Division CN1284501A (zh) | 1993-02-05 | 2000-03-01 | 用于合成紫杉酚、紫杉酚类似物及其中间体的化合物 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| CN1102829A true CN1102829A (zh) | 1995-05-24 |
Family
ID=21769500
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN94102253A Pending CN1102829A (zh) | 1993-02-05 | 1994-02-05 | 紫杉酚、紫杉酚类似物及其中间体的合成及它的组合物 |
| CN00103725A Pending CN1284501A (zh) | 1993-02-05 | 2000-03-01 | 用于合成紫杉酚、紫杉酚类似物及其中间体的化合物 |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN00103725A Pending CN1284501A (zh) | 1993-02-05 | 2000-03-01 | 用于合成紫杉酚、紫杉酚类似物及其中间体的化合物 |
Country Status (12)
| Country | Link |
|---|---|
| US (6) | US5770745A (zh) |
| EP (2) | EP0682660B1 (zh) |
| JP (1) | JP3031715B2 (zh) |
| CN (2) | CN1102829A (zh) |
| AT (1) | ATE253563T1 (zh) |
| AU (1) | AU695530B2 (zh) |
| BR (1) | BR9405687C1 (zh) |
| CA (1) | CA2155304C (zh) |
| DE (1) | DE69433297T2 (zh) |
| IL (1) | IL108581A0 (zh) |
| NZ (1) | NZ262030A (zh) |
| WO (1) | WO1994018186A1 (zh) |
Families Citing this family (44)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5973160A (en) * | 1992-12-23 | 1999-10-26 | Poss; Michael A. | Methods for the preparation of novel sidechain-bearing taxanes |
| US5684175A (en) * | 1993-02-05 | 1997-11-04 | Napro Biotherapeutics, Inc. | C-2' hydroxyl-benzyl protected, N-carbamate protected (2R, 3S)- 3-phenylisoserine and production process therefor |
| ATE253563T1 (de) | 1993-02-05 | 2003-11-15 | Bryn Mawr College | Synthese von taxol, seinen analogen und intermediaten mit variablen a-ring seitenketten |
| US5442065A (en) * | 1993-09-09 | 1995-08-15 | Board Of Regents, The University Of Texas System | Synthesis of tetrahydroquinoline enediyne core analogs of dynemicin |
| US5677470A (en) * | 1994-06-28 | 1997-10-14 | Tanabe Seiyaku Co., Ltd. | Baccatin derivatives and processes for preparing the same |
| CA2162759A1 (en) * | 1994-11-17 | 1996-05-18 | Kenji Tsujihara | Baccatin derivatives and processes for preparing the same |
| EP0868422A1 (de) * | 1996-09-24 | 1998-10-07 | Marigen S.A. | Ultramikroemulsionen aus spontan dispergierbaren konzentraten mit antitumoral und antiviral wirksamen estern von baccatin-iii-verbindungen |
| US5750737A (en) * | 1996-09-25 | 1998-05-12 | Sisti; Nicholas J. | Method for paclitaxel synthesis |
| CA2188190A1 (en) * | 1996-10-18 | 1998-04-18 | Sarala Balachandran | The semi-synthesis of a protected bacatin iii compound |
| US6150537A (en) | 1997-12-12 | 2000-11-21 | Emory University | Methods for the esterification of alcohols and compounds useful therefor as potential anticancer agents |
| US6020507A (en) | 1998-03-02 | 2000-02-01 | Bristol-Myers Squibb Company | Synthesis of paclitaxel from baccatin III by protection of the 7-hydroxyl of baccatin III using a strong base and an electrophile |
| KR20010043230A (ko) | 1998-05-01 | 2001-05-25 | 패트리샤 에이, 필리어 | C-7, c-10 디-cbz 박카틴 iii로부터 파클리탁셀을제조하는 방법 및 이를 제조하는데 사용하기 위한 중간체 |
| FI981717A7 (fi) | 1998-08-07 | 2000-02-08 | Borealis As | Katalysaattorikomponentti, joka käsittää magnesiumia, titaania, halogeenia ja elektronidonorin, sen valmistus ja käyttö |
| US6025516A (en) * | 1998-10-14 | 2000-02-15 | Chiragene, Inc. | Resolution of 2-hydroxy-3-amino-3-phenylpropionamide and its conversion to C-13 sidechain of taxanes |
| EP1183250B1 (en) * | 1999-05-28 | 2006-10-11 | Bristol-Myers Squibb Company | Semi-synthesis of paclitaxel using dialkyldichlorosilanes |
| US6136999A (en) * | 1999-06-21 | 2000-10-24 | Napro Biotherapeutics, Inc. | C-2 hydroxyl protected-n-acyl (2R,3S)-3-phenylisoserine substituted phenyl activated esters and method for production thereof |
| US6143902A (en) * | 1999-06-21 | 2000-11-07 | Napro Biotherapeutics, Inc. | C-2 hydroxyl protected-N-acyl (2R,3S)-3-phenylisoserine N-imido activated esters and method for production thereof |
| AU775373B2 (en) | 1999-10-01 | 2004-07-29 | Immunogen, Inc. | Compositions and methods for treating cancer using immunoconjugates and chemotherapeutic agents |
| IL145637A0 (en) * | 2000-02-02 | 2002-06-30 | Univ Florida State Res Found | C7 carbonate substituted taxanes as antitumor agents |
| AU778991B2 (en) * | 2000-02-02 | 2004-12-23 | Florida State University Research Foundation, Inc. | C7 carbamoyloxy substituted taxanes as antitumor agents |
| US6452024B1 (en) | 2000-02-22 | 2002-09-17 | Chaichem Pharmaceuticals International | Process for extraction and purification of paclitaxel from natural sources |
| US6362217B2 (en) * | 2000-03-17 | 2002-03-26 | Bristol-Myers Squibb Company | Taxane anticancer agents |
| US6358996B1 (en) | 2000-06-09 | 2002-03-19 | Napro Biotherapeutics, Inc. | Stable isotope labeling of paclitaxel |
| CA2410632A1 (en) | 2000-06-22 | 2001-12-27 | David S. Garvey | Nitrosated and nitrosylated taxanes, compositions and methods of use |
| DE60126103T2 (de) * | 2000-08-18 | 2007-11-15 | Nikon Corp. | Haltevorrichtung für optisches Element |
| HUP0302599A3 (en) | 2000-09-22 | 2005-05-30 | Bristol Myers Squibb Co | Method for reducing toxicity of combined chemotherapic compositions |
| EP1383754B1 (en) * | 2001-03-23 | 2011-02-23 | ScinoPharm Taiwan, Ltd. | Process for making taxane derivatives |
| CA2441484A1 (en) * | 2001-03-23 | 2002-10-03 | Napro Biotherapeutics, Inc. | Molecular conjugates for use in treatment of cancer |
| US6452025B1 (en) * | 2001-04-25 | 2002-09-17 | Napro Biotherapeutics, Inc. | Three-step conversion of protected taxane ester to paclitaxel |
| US6479679B1 (en) | 2001-04-25 | 2002-11-12 | Napro Biotherapeutics, Inc. | Two-step conversion of protected taxane ester to paclitaxel |
| US6653501B2 (en) | 2001-06-27 | 2003-11-25 | Napro Biotherapeutics, Inc. | Chiral resolution method for producing compounds useful in the synthesis of taxanes |
| US7173145B2 (en) * | 2001-11-29 | 2007-02-06 | University Of Maryland, College Park | Process for extraction and purification of lutein, zeaxanthin and rare carotenoids from marigold flowers and plants |
| ITMI20020782A1 (it) * | 2002-04-12 | 2003-10-13 | Indena Spa | Processo semisintetico per la preparazione di n-debenzoilpaclitaxel |
| US7981928B2 (en) | 2002-09-05 | 2011-07-19 | Nanodynamics, Inc. | Chemotherapy method using x-rays |
| JP2006513152A (ja) * | 2002-10-09 | 2006-04-20 | ファイトジェン ライフ サイエンシズ インコーポレイテッド | 新規タキサンならびにその使用および調製に関する方法 |
| US6759539B1 (en) | 2003-02-27 | 2004-07-06 | Chaichem Pharmaceuticals International | Process for isolation and purification of paclitaxel from natural sources |
| US7202370B2 (en) | 2003-10-27 | 2007-04-10 | Conor Medsystems, Inc. | Semi-synthesis of taxane intermediates from 9-dihydro-13-acetylbaccatin III |
| EP1875178A4 (en) * | 2005-04-12 | 2010-05-26 | X Rite Inc | SYSTEMS AND METHOD FOR VALIDATING A SECURITY FEATURE OF AN OBJECT |
| CN101370798A (zh) * | 2005-12-21 | 2009-02-18 | 挂毯药品公司 | 用于形成紫杉烷的新化合物和方法及其应用 |
| JP2009531446A (ja) * | 2006-03-27 | 2009-09-03 | タペストリー ファーマシューティカルズ インコーポレーテッド | タキサン誘導体の合成のためのコンバージェントプロセス |
| GB0701523D0 (en) | 2007-01-26 | 2007-03-07 | Chatham Biotec Ltd | Semi-synthetic process for the preparation of taxane derivatives |
| KR101096282B1 (ko) | 2009-04-24 | 2011-12-20 | 주식회사 삼양제넥스 | 탁산 유도체를 제조하는 방법 |
| WO2017055522A1 (en) | 2015-09-29 | 2017-04-06 | Academisch Medisch Centrum | Stabilized env proteins of hiv |
| EP3672983A1 (en) | 2017-08-26 | 2020-07-01 | Academisch Medisch Centrum | Improved hiv envelope glycoprotein immunogens |
Family Cites Families (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| FR2601675B1 (fr) * | 1986-07-17 | 1988-09-23 | Rhone Poulenc Sante | Derives du taxol, leur preparation et les compositions pharmaceutiques qui les contiennent |
| FR2601676B1 (fr) * | 1986-07-17 | 1988-09-23 | Rhone Poulenc Sante | Procede de preparation du taxol et du desacetyl-10 taxol |
| FR2629819B1 (fr) * | 1988-04-06 | 1990-11-16 | Rhone Poulenc Sante | Procede de preparation de derives de la baccatine iii et de la desacetyl-10 baccatine iii |
| FR2629818B1 (fr) * | 1988-04-06 | 1990-11-16 | Centre Nat Rech Scient | Procede de preparation du taxol |
| US5175315A (en) * | 1989-05-31 | 1992-12-29 | Florida State University | Method for preparation of taxol using β-lactam |
| US5015744A (en) * | 1989-11-14 | 1991-05-14 | Florida State University | Method for preparation of taxol using an oxazinone |
| US5136060A (en) * | 1989-11-14 | 1992-08-04 | Florida State University | Method for preparation of taxol using an oxazinone |
| FR2658513B1 (fr) * | 1990-02-21 | 1994-02-04 | Rhone Poulenc Sante | Procede de preparation de l'acide cis-beta-phenylglycidique-(2r,3r). |
| FR2680506B1 (fr) * | 1991-08-19 | 1994-09-02 | Rhone Poulenc Rorer Sa | Procede de preparation de derives de la beta-phenylisoserine et leur utilisation. |
| FR2687150B1 (fr) * | 1992-02-07 | 1995-04-28 | Rhone Poulenc Rorer Sa | Procede de preparation de derives du taxane. |
| FR2687151B1 (fr) | 1992-02-07 | 1994-03-25 | Rhone Poulenc Rorer Sa | Nouveaux derives de la baccatine iii et de la desacetyl-10 baccatine iii, leur preparation et les compositions pharmaceutiques qui les contiennent. |
| US5470866A (en) * | 1992-08-18 | 1995-11-28 | Virginia Polytechnic Institute And State University | Method for the conversion of cephalomannine to taxol and for the preparation of n-acyl analogs of taxol |
| US5319112A (en) * | 1992-08-18 | 1994-06-07 | Virgnia Tech Intellectual Properties, Inc. | Method for the conversion of cephalomannine to taxol and for the preparation of N-acyl analogs of taxol |
| FR2696460B1 (fr) | 1992-10-05 | 1994-11-25 | Rhone Poulenc Rorer Sa | Procédé de préparation de dérivés du taxane. |
| ATE253563T1 (de) | 1993-02-05 | 2003-11-15 | Bryn Mawr College | Synthese von taxol, seinen analogen und intermediaten mit variablen a-ring seitenketten |
| US5684175A (en) * | 1993-02-05 | 1997-11-04 | Napro Biotherapeutics, Inc. | C-2' hydroxyl-benzyl protected, N-carbamate protected (2R, 3S)- 3-phenylisoserine and production process therefor |
| FR2703353B1 (fr) * | 1993-03-29 | 1995-05-05 | Rhone Poulenc Rorer Sa | Procédé de préparation de dérivés de la beta-phénylisosérine. |
| US5679807A (en) | 1995-01-30 | 1997-10-21 | Hauser, Inc. | Preparation of taxol and docetaxel through primary amines |
-
1994
- 1994-02-04 AT AT94907973T patent/ATE253563T1/de not_active IP Right Cessation
- 1994-02-04 JP JP6518249A patent/JP3031715B2/ja not_active Expired - Fee Related
- 1994-02-04 EP EP94907973A patent/EP0682660B1/en not_active Expired - Lifetime
- 1994-02-04 EP EP02014256A patent/EP1260507A1/en not_active Withdrawn
- 1994-02-04 AU AU61336/94A patent/AU695530B2/en not_active Expired
- 1994-02-04 NZ NZ262030A patent/NZ262030A/en unknown
- 1994-02-04 BR BR9405687-0A patent/BR9405687C1/pt not_active Application Discontinuation
- 1994-02-04 DE DE69433297T patent/DE69433297T2/de not_active Expired - Lifetime
- 1994-02-04 CA CA2155304A patent/CA2155304C/en not_active Expired - Lifetime
- 1994-02-04 WO PCT/US1994/001293 patent/WO1994018186A1/en not_active Ceased
- 1994-02-05 CN CN94102253A patent/CN1102829A/zh active Pending
- 1994-02-07 IL IL10858194A patent/IL108581A0/xx unknown
- 1994-12-15 US US08/357,507 patent/US5770745A/en not_active Expired - Lifetime
-
1995
- 1995-06-07 US US08/483,083 patent/US5939566A/en not_active Expired - Lifetime
-
1998
- 1998-02-09 US US09/020,742 patent/US6262281B1/en not_active Expired - Fee Related
-
1999
- 1999-02-19 US US09/253,325 patent/US6072060A/en not_active Expired - Fee Related
-
2000
- 2000-03-01 CN CN00103725A patent/CN1284501A/zh active Pending
- 2000-04-11 US US09/547,327 patent/US6307088B1/en not_active Expired - Fee Related
-
2001
- 2001-05-22 US US09/863,889 patent/US6509484B2/en not_active Expired - Fee Related
Also Published As
| Publication number | Publication date |
|---|---|
| CA2155304A1 (en) | 1994-08-18 |
| CN1284501A (zh) | 2001-02-21 |
| DE69433297T2 (de) | 2004-10-21 |
| EP0682660B1 (en) | 2003-11-05 |
| US6072060A (en) | 2000-06-06 |
| NZ262030A (en) | 1997-10-24 |
| EP0682660A1 (en) | 1995-11-22 |
| US6307088B1 (en) | 2001-10-23 |
| BR9405687C1 (pt) | 2001-08-07 |
| ATE253563T1 (de) | 2003-11-15 |
| US5939566A (en) | 1999-08-17 |
| JP3031715B2 (ja) | 2000-04-10 |
| CA2155304C (en) | 2010-07-20 |
| US6262281B1 (en) | 2001-07-17 |
| BR9405687A (pt) | 1995-11-21 |
| US5770745A (en) | 1998-06-23 |
| AU695530B2 (en) | 1998-08-13 |
| EP0682660A4 (zh) | 1995-12-20 |
| US6509484B2 (en) | 2003-01-21 |
| AU6133694A (en) | 1994-08-29 |
| DE69433297D1 (de) | 2003-12-11 |
| EP1260507A1 (en) | 2002-11-27 |
| WO1994018186A1 (en) | 1994-08-18 |
| JPH08506587A (ja) | 1996-07-16 |
| US20020052517A1 (en) | 2002-05-02 |
| IL108581A0 (en) | 1994-05-30 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN1102829A (zh) | 紫杉酚、紫杉酚类似物及其中间体的合成及它的组合物 | |
| CN1067682C (zh) | 抗肿瘤化合物、药物组合物、其制备方法以及其用于治疗的方法 | |
| CN1108653A (zh) | 2-脱苯甲酰基-2-酰基紫杉醇衍生物及其制备方法 | |
| CN101048394A (zh) | 太平洋紫杉醇向多烯紫杉醇的半合成转化 | |
| CN1075718A (zh) | 金属醇盐 | |
| CN1239476A (zh) | 选择性衍生紫杉烷的方法 | |
| CN1080256C (zh) | 新的β-内酰胺类及制备紫杉烷的方法 | |
| CN1234679C (zh) | 3,3-二芳基丙基胺衍生物的简化合成法 | |
| CN1198805C (zh) | 制备光学活性的半酯的方法 | |
| CN1200731A (zh) | 半合成塔三烷用的中间体及其制备方法 | |
| CN1239493C (zh) | 3,6-二烷基-5,6-二氢-4-羟基-吡喃-2-酮的合成方法 | |
| CN1942460A (zh) | 紫杉烷衍生物的一锅合成及其向太平洋紫杉醇和多烯紫杉醇的转化 | |
| CN1703409A (zh) | 新紫杉烷和用途及制备方法 | |
| CN100349867C (zh) | 制备r,r(或s,s)构型的格隆铵-立体异构体的方法 | |
| CN1143641A (zh) | 利用噁唑烷中间体制备塔三烷的新方法 | |
| CN101035778A (zh) | 紫杉烷中间体的半合成及其向太平洋紫杉醇和多烯紫杉醇的转化 | |
| CN1244569C (zh) | C-10位紫杉烷衍生物及其药物组合物 | |
| CN1090625C (zh) | 选择性保护浆果赤霉素衍生物的方法及其在合成紫杉烷中的应用 | |
| CN1784378A (zh) | 4-羟基异亮氨酸及其衍生物的合成方法 | |
| CN1216039A (zh) | 制备紫杉酚的方法 | |
| US7605278B2 (en) | Methods and compositions for converting taxane amides to paclitaxel or other taxanes | |
| CN1547567A (zh) | 制备用于合成紫杉烷的化合物的手性分离方法 | |
| CN1048982C (zh) | 用金属烷氧化物和β-内酰胺半合成紫杉烷衍生物的方法 | |
| HK1035190A (zh) | 用於合成紫杉酚、紫杉酚类似物及其中间体的化合物 | |
| CN1942458A (zh) | 紫杉烷中间体和氮丙啶类似物的半合成及其向太平洋紫杉醇和多烯紫杉醇的转化 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C02 | Deemed withdrawal of patent application after publication (patent law 2001) | ||
| WD01 | Invention patent application deemed withdrawn after publication |