CN1114106A - 苄胺衍生物 - Google Patents
苄胺衍生物 Download PDFInfo
- Publication number
- CN1114106A CN1114106A CN94190643A CN94190643A CN1114106A CN 1114106 A CN1114106 A CN 1114106A CN 94190643 A CN94190643 A CN 94190643A CN 94190643 A CN94190643 A CN 94190643A CN 1114106 A CN1114106 A CN 1114106A
- Authority
- CN
- China
- Prior art keywords
- benzylamine derivative
- compound
- defined above
- salt
- reaction
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C217/00—Compounds containing amino and etherified hydroxy groups bound to the same carbon skeleton
- C07C217/54—Compounds containing amino and etherified hydroxy groups bound to the same carbon skeleton having etherified hydroxy groups bound to carbon atoms of at least one six-membered aromatic ring and amino groups bound to acyclic carbon atoms or to carbon atoms of rings other than six-membered aromatic rings of the same carbon skeleton
- C07C217/56—Compounds containing amino and etherified hydroxy groups bound to the same carbon skeleton having etherified hydroxy groups bound to carbon atoms of at least one six-membered aromatic ring and amino groups bound to acyclic carbon atoms or to carbon atoms of rings other than six-membered aromatic rings of the same carbon skeleton with amino groups linked to the six-membered aromatic ring, or to the condensed ring system containing that ring, by carbon chains not further substituted by singly-bound oxygen atoms
- C07C217/58—Compounds containing amino and etherified hydroxy groups bound to the same carbon skeleton having etherified hydroxy groups bound to carbon atoms of at least one six-membered aromatic ring and amino groups bound to acyclic carbon atoms or to carbon atoms of rings other than six-membered aromatic rings of the same carbon skeleton with amino groups linked to the six-membered aromatic ring, or to the condensed ring system containing that ring, by carbon chains not further substituted by singly-bound oxygen atoms with amino groups and the six-membered aromatic ring, or the condensed ring system containing that ring, bound to the same carbon atom of the carbon chain
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/20—Hypnotics; Sedatives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/24—Antidepressants
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2601/00—Systems containing only non-condensed rings
- C07C2601/02—Systems containing only non-condensed rings with a three-membered ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2601/00—Systems containing only non-condensed rings
- C07C2601/04—Systems containing only non-condensed rings with a four-membered ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2601/00—Systems containing only non-condensed rings
- C07C2601/12—Systems containing only non-condensed rings with a six-membered ring
- C07C2601/14—The ring being saturated
Landscapes
- Organic Chemistry (AREA)
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Neurosurgery (AREA)
- Life Sciences & Earth Sciences (AREA)
- Neurology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Veterinary Medicine (AREA)
- Pharmacology & Pharmacy (AREA)
- Biomedical Technology (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Anesthesiology (AREA)
- Pain & Pain Management (AREA)
- Psychiatry (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
Abstract
本发明的目标是提供一种具有抗抑郁和抗焦虑
活性的苄胺衍生物或其盐。本发明的苄胺衍生物或
其盐可用上列通式表示,式中各基团定义详见说明
书。
Description
本发明涉及新颖的苄胺衍生物及其盐。
由于当今人类社会的结构和机制日趋复杂化,因而,人们所承受的各种压力不断增加。在这样一种变化多端的社会中,精神失常病人的人数,特别是抑郁及焦虑型精神病人的人数不断增加,并产生了严重的社会问题。
与以往的抑郁病不同,现在的抑郁病人大多相对轻微且趋于转为慢性的。抑郁病通常很难与精神病区别且容易转为慢性的。据报导,大多数慢性的抑郁病人同时患有精神病,且近些年来,抑郁病人的数目越来越多。(参见:Japanese Journal of Clinical Psychiatry,Vol.21,No.4,pp.691-695,1992)。
如同社会的结构和机制变得复杂一样,精神失常的特征也趋于复杂化。下面两个事实是近来精神失常特征方面的主要变化。
(1)在精神病中,表现为抑郁症的精神病人数增加,且很难将其与抑郁病人区别开来。
(2)在被诊断为精神病的病人中,包括有轻微的抑郁病。特别是在被诊断为焦虑型及恐惧型精神病人中,有抑郁病症的比例相当高(参见Japanese Journal of Clinical Psychiatry,Vol.21,No.4,pp.691-695,1992).
根据临床研究(参见:“Anxious Depression”,Racagni G,Smeraldi编,Raven Press,N.Y.,1987),分析被诊断有严重抑郁症病人的症状时发现:中等程度的焦虑(72%),心理上的焦虑(62%),物理上的焦虑(自主神经系统及肌肉系统)(42%),恐惧心理(29%),恐惧病症(19%)及强迫综合症(1.2%)。抑郁病人中包括有焦虑症状的比例很高。
同上所述,在精神失常病人中表现出复杂且多样化的症状,目前在许多精神失常病例的治疗中,一般是根据症状,采用抗抑郁药、抗焦虑药或其组合物。在某些病例中,上述治疗是有效的,但在多数情况下效果欠佳。常规的抗抑郁药仅对内源性抑郁症有效,而对性格引发的抑郁症或高度神经性抑郁症无效。(参见:Journal of Neuropsychopharmacology,Vol.9,No.4,pp.279-285,1987)。给表面反映为焦虑症,而实际为抑郁型的病人服用抗焦虑药,在某些情况下会拖延抑郁症。(参见Japanese Journal of Clinical Psychiatry,Vol.21,No.4,pp.691-695,1992)。基于临床治疗部门而不是精神病学部门轻率的诊断,而给病人服用抗抑郁药,会忽略自杀的心理作用,并且会导致抑郁症状的延长(参见:Journal of Neuropsychopharmacology,Vol.9,No.4,pp.279-285,1987)。
常规的抗抑郁剂存在如下问题:(1)它们不能即刻起作用,需至少连续服用2周,(2)有不良的副作用,如抗胆碱效应等和(3)在某些病例中无效(有效率:65%)(参见Journal of Neuropsychopharmacology,Vol.11,No.10,pp.753-761,1989)。常规的抗焦虑剂存在如下的问题:(1)有副作用,例如过度的镇静,困倦,肌内松弛等,(2)常出现严重的不良症状如,依赖性,戒断症状,记忆损害等(参见Journal of Neuropsychopharmacology,Vol.11,No.9,pp.709-719,1989)。
给病人单独或结合服用上述抗抑郁药和抗焦虑药(这些药物有许多副作用和不良反应且据说对精神失常的核心的病症无效)会在精神病人的治疗中产生自相矛盾的结果。因此,如能开发一种针对精神病根本病因,而且同时具有抗郁活性和抗焦虑活性的药物,将会解决上述与治疗精神失常有联系的诸问题。
与本发明中的苄胺衍生物结构类似的化合物在下列文献中被公开:PCT International Applications WO 90/14 334(出版日期:1990年11月29日),WO 93/07113(出版日期:1993年4月15日),WO 91/09594(出版日期:1991年7月11日)及WO 93/00313(出版日期:1993年1月7日),特别是在WO 91/09594和WO 93/00313中公开了具有下列通式的化合物:

其中:
Ar是芳基或杂芳基,其中芳基或杂芳基可被如下基团取代:氢,卤素如氯,氟,溴或碘,CF3,C1-C6烷氧基,C2-C6二烷氧甲基,C1-C6烷基,氰基,C3-C15二烷氨烷基,羧基,羧酰胺基,C1-C6卤代烷基,C1-C6卤代烷硫基,烯丙基,芳烷基,C3-C6环烷基,芳酰基,芳烷氧基,C2-C6酰基,芳基,取代的芳基,杂芳基,取代的杂芳基,一个芳基与一个取代的苯环稠合,一个取代的芳基与一个苯环稠合,一个杂芳基与一个苯环稠合,一个取代的杂芳基与一个苯环稠合,C3-C6杂环烷基,一个C3-C6杂环烷基与一个苯环稠合,C1-C6烷硫基,C1-C6烷基磺酰基,C1-C6卤代烷基磺酰基,C1-C6烷基亚硫酰基,C1-C6卤代烷基亚硫酰基,芳硫基,C1-C6卤代烷氧基,氨基,C1-C6烷氨基,C5-C15二烷基氨基,羟基,氨甲羰基,C1-C6N-烷基氨甲酰基,C5-C15N,N-二烷基氨甲酰基,硝基或C2-C15二烷基氨磺酰基;
R为氢或C1-C6烷基;
R1为氢,C1-C6烷基,羟基,氨基,C1-C6烷氨基及-0(双键氧);或
R和R1一起构成吗啉环;
n为0-5;
W为-(CH2)P-或-HH-,其中P为1-3;
X为-(CH2)q-其中q为1-6,-(CH2)r-C≡C-(CH2)r-其中r为0-3,-(CH2)r-CH=CH-(CH2)r-,-(CH2)r-
-(CH2)r-,-(CH2)r-Y-(CH2)r-其中Y为O或S,或C1-C6烷基(其中Z为氢):且
Z为氢,芳基,芳基取代的羧酸基,杂芳基或环烷基,其中芳基,杂芳基和环烷基可被如下基团所取代:氢,卤素如氯,氟,溴或碘,CF3,C1-C6烷氧基,C2-C6二烷氧甲基,C1-C6烷基,氰基,C3-C15二烷基氨基烷基,羧基,羧酰胺基,C1-C6卤代烷基,C1-C6卤代烷硫基,烯丙基,芳烷基;C3-C6环烷基,芳羰基,芳烷氧基,C2-C6羧酰基,芳基,取代的芳基,杂芳基,取代的杂芳基,芳基与取代的苯环稠合,取代的芳基与苯环稠合,杂芳基与苯环稠合,取代的杂芳基与苯环稠合,C3-C6杂环烷基,一个C3-C6杂环烷基与苯环稠合,C1-C6烷硫基,C1-C6烷磺酰基,C1-C6卤代烷磺酰基,C1-C6烷基亚硫酰基,C1-C6卤代烷基亚硫酰基,芳硫基,C1-C6卤代烷氧基,氨基,C1-C6烷基氨基,C2-C15二烷氨基,羟基,氨甲酰基,C1-C6N-烷氨甲酰基,C2-C15N,N-二烷基氨甲酰基,硝基,C2-C15二烷基氨磺酰基或原亚甲二氧基。
根据所述的两篇文献,具上述通式的化合物可用作σ受体配体,且可作为治疗精神分裂症或其它精神病的药物,或用于治疗中枢神经系统疾病,药物滥用,胃肠道疾病,高血压,偏头痛,扁桃体周炎,抑郁症,等等。
因此,上述文献公开了与本发明中的苄胺衍生物化学结构类似的化合物,但并未公开本文中的苄胺衍生物。
基于上述情况,本发明者作了深入的研究,发现上述通式(1)所示的苄胺衍生物及其盐,具有抗抑郁活性和抗焦虑活性,并且作为出色的抗抑郁药和抗焦虑药是有效的。在此发现的基础上完成了本发明。本发明中的苄胺化合物和其盐的特点是它们能有效地改善意识紊乱,以及能用于治疗强迫和强制性的神经机能病。
本发明的苄胺衍生物是任何文献中均未报导过的新颖化合物,且可用下列通式(1)来表示:
其中R1是低级烷基;R2是环烷基;且R3是卤原子。
本发明中的由通式(1)所表示的苄胺衍生物及其盐具有抗抑郁活性和抗焦虑活性,而且是医疗上十分有效的抗抑郁药和抗焦虑药。
本发明中的苄胺衍生物或其盐具有活化中枢神经系统的效应,且能改善意识紊乱,它们可用于治疗如下病症:头部神经损伤,脑溢血,脑栓塞,蜘蛛膜下溢血,药物中毒,大气缺氧,缺氧造成的意外,大脑手术或心脏分流手术后造成的意识紊乱,及上述疾病的后遗症例如,精神发育迟缓,反应能力下降,语言障碍,识别紊乱,学习能力丧失,运动能力低下,厌食以及情感紊乱等等,也可用于改善如下疾病,例如老年痴呆性抑郁症,精神错乱,语言障碍,识别紊乱,学习能力丧失,运动迟缓,反应能力低下,老年性健忘症等等。进一步地本发明化合物具有σ受体激动活性,且可用于治疗抑郁症,焦虑性精神病,压抑造成的精神病(身心病),厌食症,垂体机能下降,高催乳素病,脑血管性痴呆,多动症,痴呆健忘症,怕金森病等等。本发明化合物也可作为抗抑郁和抗焦虑药。
按照本发明中的苄胺衍生物及其盐,当经口服给药时表现出抗抑郁和抗焦虑的活性,能有效活化中枢神经系统,有效地改善意识紊乱,且具有σ受体激动作用。
在当今复杂的社会中,许多人由于环境压力而患有精神失常病(例如,强迫和强制性的神经机能病)。
本化合物可用于治疗此病。
优选实施例的详细描述:
本发明中具有通式(Ⅰ)的苄胺衍生物及其盐中所采用的基团特指如下基团:
R1所表示的低级烷基包括直链或支链的1-6个碳原子的烷基,例如,甲基,乙基,丙基,异丙基,丁基,叔丁基,戊基,己基等。
R2所表示的环烷基系指3-8碳原子的环烷基,例如环丙基,环戊基,环丁基,环己基,环庚基,环辛基等。
R3所表示的卤素包括氟,氯,溴,碘原子等。
在本发明的化合物中,较可取的化合物为R1是甲基或乙基,R2为环丙基,或环丁基,且R3为氯原子的化合物。特别可取的是R1为甲基,R2为环丙基,R3为氯原子的化合物。
可用不同的方法来制备本发明中具备通式(1)的苄胺衍生物,较可取的有如下几种方法:
[反应式-1]

其中R1,R2和R3同上述定义。
在反应式1中,化合物(2)与化合物(3)的反应没有溶剂或有合适的溶剂下进行,加或不加脱水剂。溶剂的例子有,醇类如甲醇,乙醇,异丙醇等,芳烃类如苯,甲苯,二甲苯等;卤代烃如二氯甲烷,二氯乙烷,氯仿,四氯化碳等;非质子极性溶剂,如二甲基甲酰胺,二甲基乙酰胺,N-甲基吡咯烷酮等;以及它们的混合剂。所提到的脱水剂可以是,例如,溶剂干燥时用的吸水剂,如分子筛等等;矿物酸,如盐酸,硫酸,三氟化硼等;有机酸,如对甲苯磺酸等。该反应通常在室温至150℃间进行,较可取的是在室温至100℃,该反应通常在5分钟至10小时内完成。式(3)化合物的用量无严格限制,但通常至少与式(2)化合物等量,较可取的是每摩尔式(2)化合物用1-2摩尔的式(3)化合物。当采用吸水剂作为脱水剂时,用量一般为大大过量,当用酸时,常用催化量已足。得到下列通式(A)所表示的化合物

(其中,R1,R2和R3同上述定义),无须分离即可将式(A)化合物进行还原。
在式(A)化合物的还原反应中,可采用多种方法。例如,催化氢化通式(9)化合物时所采用的条件(后面将提到)。较可取的还原方法是采用氢化物还原剂。该还原剂包括,氢化铝锂,硼氢化钠及二硼烷。对每摩尔式(2)化合物,还原剂的用量最少要1摩尔,较可取的为1-10摩尔。该还原反应一般是在合适的溶剂中进行的,例如水,低级醇(如:甲醇;乙醇或异丙醇),醚(如,四氢呋喃,乙醚或甘醇二甲醚)等等。还原温度一般在-60至50℃之间,较可取的是在-30℃至室温下进行大约10分钟至5小时。当采用氢化铝锂或二硼烷时,最好采用无水溶剂,如乙醚,四氢呋喃,甘醇二甲醚等等。
[反应式-2]


其中R1,R2和R3同上述定义;X为卤原子,且R4和R5分别为氢原子或低级烷基。
化合物(4)与化合物(5)的反应,以及化合物(7)与化合物(10)的反应,一般是在合适的惰性溶剂中,在碱性化合物存在或不存在下进行的。所述惰性溶剂包括:例如,芳烃,如苯,甲苯,二甲苯等;醚如四氢呋喃,二噁烷,二甘醇二甲醚等;低级醇如甲醇,乙醇,异丙醇,丁醇等;乙酸;乙酸乙酸;丙酮;乙腈;二甲基亚砜;二甲基甲酰胺;六甲基磷酸三酰胺;及它们的混合溶剂。所述的碱性化合物有:例如,碳酸钠,碳酸钾,碳酸氢钠,碳酸氢钾等;金属氢氧化物如氢氧化钠,氢氧化钾等;氢化钠;钾;钠;铵化钠;金属醇化物,如甲醇钠,乙醇钠等;及有机碱,如吡啶,N-乙基二异丙基胺,二甲氨基吡啶,三乙胺,1,5-二氮杂双环[4.3.0]壬烯-5(DBN),1,8-二氮杂双环[5.4.0]十一烯-7(DBU),1,4-二氮杂双环[2.2.2]辛烷(DABCO)等。化合物(4)与(5)或化合物(7)与(10)之间的比例无严格限制,且可在很宽的范围内进行选择;然而,较可取的为:当后者为1摩尔时,前者为1-5摩尔。该反应一般在0-200℃间反应,较可取的为0-170℃,且一般在约30分钟至30小时内完成反应。偶尔也可向反应体系中加入卤代碱金属(如碘化钠或碘化钾)。
化合物(4)与化合物(6)的反应在没有溶剂或有适当溶剂的条件下,在还原剂存在下进行。溶剂的实施例有水;醇,如甲醇,乙醇,异丙醇等;乙腈;甲酸;乙酸;醚,如二噁烷,乙醚,二甘醇二甲醚,四氢呋喃等;芳烃,如苯,甲苯,二甲苯等;以及它们的混合剂。还原剂的实施例有甲酸;脂肪酸的碱金属盐,如甲酸钠等;氢化物还原剂,如硼氢化钠,氰基硼氢化钠,氢化铝锂等等;以及催化还原剂,如钯黑,钯碳,氧化钯,铂黑,Raney镍等等。
当采用甲酸作还原剂时,反应最好在室温至200℃间进行,较可取的是在大约50-150℃,在1-10小时左右完成。所需的甲酸量为大大过量于化合物(4)。
当采用氢化物还原剂时,反应一般在约-30℃至100℃间进行,较可取的为0-70℃,在大约30分钟至15小时内完成。还原剂用量为每摩尔化合物(4)需用1-20摩尔还原剂,较可取的为1-6摩尔。当采用氢化铝锂作为氢化物还原剂时,较可取的是采用醚(如,乙醚,二噁烷,四氢呋喃,或二甘醇二甲醚)或芳烃(如苯,甲苯或二甲苯)作为溶剂。
当采用催化还原剂时,反应需在氢气中进行,氢气压力通常约为正常大气压至20大气压,较可取的为正常大气压至10个大气压左右,且有氢供体存在,如甲酸,甲酸铵,环己烷,水合肼等,反应温度一般为约-30℃-100℃,较可取的为0-60℃,在1-12小时内完成。催化还原剂用量一般需要0.1-40%(重量比,相对于化合物(4)),较可取的为1-20%。
化合物(6)用量一般至少与化合物(4)等量,较可取的为大大过量于化合物(4)。
按常规的酰胺键形成反应的方法使化合物(7)与化合物(8)进行反应。该反应可按多种已知方法进行,例如,(a)混合酸酐方法,例如,将羧酸(8)与卤代羧酸酯反应形成混合酸酐,而后将该酸酐与胺(7)进行反应;(b)活性酯方法,例如,将羧酸(8)转化为活性酯,如对硝基苯酯,N-羟基琥珀酰亚胺酯,1-羟基苯并三唑酯等,将该活性酯与胺(7)反应;(c)碳化二亚胺方法,其包括将羧酸(8)与胺(7)进行缩合反应,在反应中加入活性剂,如二环己基碳化二亚胺,羰基二咪唑等;(d)其他方法,包括:例如,将羧酸(8)转化为酸酐(采用脱水剂,如乙酐等)再与胺(7)反应,将羧酸(8)与低级醇的酯与胺(7)进行反应;将羧酸(8)的卤化物,如酰卤与胺(7)进行反应。还可采用如下方法:例如,将羧酸(8)用磷化物,如三苯膦,氯磷酸二乙酯等进行活化,再将所生成的产物与胺(7)进行反应;使用光气,氯甲酸三氯甲酯等将羧酸(8)转化为N-羧氨基酸酐,再将该酸酐与胺(7)进行反应。还可采用乙炔化合物,如三甲基甲硅烷基乙氧基乙炔等将羧酸(8)活化,再用活化产物与胺(7)反应。
在混合酸酐步骤(a)中,可用普通的Schotten-Baumann反应来获得混合酸酐。使酸酐与胺(7)进行反应,不用分离即可制得通式(9)化合物。Schotten-Baumann反应在有碱化合物存在下进行。碱化物是通常用于Schotten-Baumann反应中的化合物,包括有机碱,例如三乙胺,三甲胺,吡啶,N,N-二甲基苯胺,N-甲基吗啉,4-二甲基氨基吡啶,DBN,DBU,DABCO等等,无机碱,例如K2CO3,Na2CO3,KBH4,NaBH4等等。反应是在-20~100℃之间,较好的是在0-50℃下反应5分钟至10小时,较好地是约5分钟至2小时。生成的混合酸酐与胺(7)在-20~150℃下,较好的是10-50℃下反应5分钟至10小时,更好的是5分钟至5小时。混合酸酐步骤(a)可以在合适的溶剂存在下或无任何溶剂存在下进行。溶剂可以是常规混合酸酐步骤中用的溶剂,可以卤代烃为例,例如CH2Cl2,CHCl3,ClCH2CH2Cl等等;芳香烃,如苯,甲苯,二甲苯等等;醚类,如乙醚,二噁烷,二异丙醚,THF,二甲氧基乙烷等等;酯类,如乙酸甲酯,乙酸乙酯等等;非质子性极性溶剂,如1,1,3,3-四甲基尿素,N,N-二甲基甲酰胺,二甲亚砜,六甲基磷酰三胺等,以及它们的混合溶剂。用于混合酸酐步骤(a)的烷基卤代羧酸酯可举例如下,氯代甲酸甲酯,溴代甲酸甲酯,氯代甲酸乙酯,溴代甲酸乙酯和氯代甲酸异丁酯。所用的烷基卤代羧酸酯的量通常是每摩尔胺(7)用1摩尔,较好的是1-1.5摩尔。羧酸(8)的用量通常是每摩尔胺(7)至少1摩尔,较好的是约1-1.5摩尔。
活性酯步骤(b),例如,当使用N-羟基琥珀酰亚胺酯时,通常是在合适的溶剂中(其对反应不产生副作用),于有碱化合物存在或无碱化合物存在下进行。进入此反应系统,要加缩合剂,如二环己基碳化二亚胺,羰基二咪唑,1-乙基-3-(3’-二甲基氨基丙基)碳化二亚胺等等。作为碱性化合物,可以使用上述Schotten-Baumann反应中所用的任何碱性化物;可以进一步使用羧酸的碱金属盐(例如乙酸钠,苯甲酸钠,甲酸钠,乙酸钾,苯甲酸锂以及乙酸铈),碱金属卤化物(例如氟化钾和氯化铈)等。特定的溶剂的例子有卤代烃,如二氯甲烷,氯仿,二氯乙烷等,芳烃如苯,甲苯,二甲苯等;醚如乙醚,二噁烷,THF,二甲氧乙烷等,酯,如乙酸甲酯,乙酸乙酯等;非质子极性溶剂,如N,N-二甲基甲酰胺,二甲亚砜,六甲基磷酰三胺等;以及它们的混合溶剂。反应在0~150℃下,较好的是10-100℃下进行5-30小时,N-羟基琥珀酰亚胺酯的量至少为每摩尔胺(7)需用1摩尔,较好的是1-2摩尔。
在有磷化物作缩合剂的存在下,使胺(7)与羧酸(8)进行反应,也可以制得化合物(9),这类缩合剂有三苯基磷,三苯基膦-2,2’-二吡啶基二硫化物,氯磷酸二乙酯,二苯基次膦酰氯,N-苯基胺基氯磷酸苯酯,氰基磷酸二乙酯,双(2-氧代-3-噁唑烷基)膦氯化物等等。作为碱性化合物,可使用的范围很广,例如在上述Schotten-Baumann反应中所用的碱性化合物,NaOH,KOH等。此反应所用的溶剂包括在混合酸酐步骤(a)中所用的那些溶剂,吡啶,丙酮,乙腈和上述两种或多种混合溶剂。反应通常在-20-150℃下,较好的在0-100℃下进行5分钟-30小时。所需的缩合剂和羧酸(8)各自的量至少为每摩尔胺(7)1摩尔,较好的是约1-2摩尔。
在有缩合剂存在下,使胺(7)和羧酸(8)进行反应,也可以获得化合物(9)。反应是在有或无催化剂存在下,于合适的溶剂中进行。溶剂的实例有卤代烃,如CH2Cl2,二氯乙烷,CHCl3,CCl4等,乙腈;二甲基甲酰胺。催化剂的例子有有机碱,如二甲基氨基吡啶,4-哌啶子基吡啶等;盐类如吡啶鎓甲苯磺酸盐等,樟脑磺酸以及氧化汞。缩合剂的例子有乙炔化合物,如三甲基甲硅烷基乙氧基乙炔等。通常缩合剂的用量每摩尔胺(7)至少1-10摩尔,较好的是2-6摩尔。羧酸(8)的用量为每摩尔胺(7)至少1摩尔,较好的是1-2摩尔。反应通常在0-150℃下,较好的是在室温至100℃下进行1-10小时。
当采用其中之一的步骤(d)时(其包括使羧酸(8)的卤代化物,即羧酸卤化物与胺(7)进行反应),反应是在有脱氢卤化剂存在下,于合适的溶剂中进行,通常用碱性化合物作脱氢卤化剂,可选择的碱性化合物范围很广,例如,在Schotten-Baumann反应中所用的碱性化合物,NaOH,KOH,NaH和KH。所用溶剂包括在混合酸酐步骤(a)中所用的那些溶剂,醇类,如甲醇,乙醇,丙醇,丁醇,3-甲氧-1-丁醇,乙基溶纤剂,甲基溶纤剂等;吡啶,丙酮;乙腈,所述溶剂中两或多种混合溶剂等等。胺(7)与羧酰基卤代物的比例不加以特别限制,可以在一个较宽的范围内选择,但每摩尔前者,至少要用1摩尔后者,较好的是1-5摩尔后者。反应通常在-20~180℃下,较好的是在0-150℃下进行5分钟~30小时。
通常羧酸(8)与卤化剂在有或无溶剂存在的条件下,可以反应生成上述的羧酸卤化剂。所用的溶剂可以是任何不对反应产生影响的溶剂,包括芳烃,如苯,甲苯,二甲苯等;卤代烃,如CHCl3,CH2Cl2,CCl4等;醚类,如二噁烷,THF,乙醚等;二甲基甲酰胺;二甲亚砜。卤化剂可以是普通的可使羧基基团上的羟基转化成卤素的卤化剂,如亚硫酰氯,氯化氧磷,溴化氧磷,PCl5和PBr5。对羧酸(8)和卤化剂的比例并不加以特别限制,可以适当选择;然而当在无溶剂下进行反应时,后者的量要大大过量于前者,当有溶剂存在下进行反应时,每1摩尔前者至少要用1摩尔后者,较好的是2-4摩尔后者。对反应的温度和时间也不加特别限制,通常是室温至100℃,较好的是50-80℃下进行30分钟~6小时。
要使化合物(9)转化成化合物(1)可用各种方法。例如(1)使化合物(9)在有催化剂存在下,于合适的溶剂中进行催化氢化,溶剂包括有,水,乙酸;醇类,如甲醇,乙醇,异丙醇等;烃类,如己烷,环己烷等;醚类,如二甘醇二甲醚,二噁烷,THF,乙醚等;酯类,如乙酸乙酯,乙酸甲酯等;非质子极性溶剂如二甲基甲酰胺等,及混合溶剂。作为催化剂,可以选择Pd,钯黑,Pd-C,铂,铂的氧化物,亚铬酸铜(copper chromite),和Raney镍。所用催化剂的量通常为0.02-1倍化合物(9)的量。反应通常在-20-100℃下,较好的是0-70℃下进行,所需的氢气压力为1-10大气压,反应时间通常为0.5~20小时。
使化合物(9)转化成化合物(1),通常采用氢化物还原剂进行还原反应。氢化物还原剂包括AlLiH4,NaBH4和乙硼烷,每摩尔化合物(9)至少要用1摩尔,较好的是1-10摩尔氢化剂。反应通常在合适的溶剂中进行,如水,低级醇(例如甲醇,乙醇或异丙醇),醚类(例如THF,乙醚或二甘醇二甲醚),乙酸等,反应温度为约0-200℃,较好的在0-170℃下进行10分钟~10小时。当使用AlLiH4或乙硼烷作还原剂时,最好采用无水溶剂,如乙醚,THF,二甘醇二甲醚等。
[反应式-3]
其中,R1,R2和R3的定义同前。
羧酸化合物(11)与胺化合物(3)的反应,可按与胺化合物(7)和羧酸化合物(8)在反应式-2中相同的条件进行,胺化合物(3)的用量取决于羧酸化合物(11)的每摩尔质量。
化合物(12)的还原反应,可按与反应式-2中化合物(9)的还原反应相同的条件进行。化合物(12)可按下列反应式-4所示的方法获得:
[反应式-4]

其中R3和X的定义同前;R6和R7分别为R1或-CH2R2(R1和R2的定义同前),附带条件是当R6为R1时,R7为-CH2R2,且R6为一CH2R2时,R7为R1。
化合物(11)与化合物(13)的反应可按与反应式-3中化合物(11)与化合物(3)相同的反应条件进行。化合物(14)与化合物(15)的反应可按与反应式-2中化合物(4)与化合物(5)相同的反应条件进行。
反应式-2中起始物(4)或(7)可用下列反应式-5所示的方法来制备:
[反应式-5]

其中R3和R6的定义同前。
在反应式-5中,化合物(16)(其中R6为R1)为化合物(7),化合物(16)(其中R6为-CH2R2)为化合物(4)。化合物(2)与化合物(13)的反应可按与反应式-1中化合物(2)与化合物(3)相同的反应条件进行。
反应式-1中的起始原料(2)和反应式-3或反应式-4中的起始原料(11)可按下列反应式-6所述的方法获得:
[反应式-6]

其中R3和X的定义同前。
化合物(17)与化合物(19)的反应以及化合物(18)与化合物(19)的反应可按与反应式-2中化合物(4)与化合物(5)相同的反应条件进行。
其中式(1)表示的本发明化合物带有碱性基团,每一个碱性基团均可容易地与药理学上可接受的酸反应生成酸加成盐。所述的酸可以是无机酸,如盐酸,硫酸,磷酸,氢溴酸等,也可以是有机酸,如草酸,乙酸,琥珀酸,丙二酸,甲磺酸,马来酸,富马酸,苹果酸,酒石酸,柠檬酸,苯甲酸等。
每个通过上述反应制得的所要的化合物均可以用常规的分离方法进行分离和纯化。分离方法有溶剂提取,稀释,重结晶,柱层析以及制备薄层层析。
通式(Ⅰ)中的每个化合物均以其普通的药物制剂形式使用。制备药物制剂采用稀释剂或赋形剂,如填充剂,增量剂,粘合剂,湿润剂,崩解剂,表面活性剂,润滑剂等。根据治疗的目的不同,药物制剂可以制成各种形式,典型的剂型有片剂,丸剂,粉剂,溶液,悬浮液,乳化液,颗粒,胶囊,栓剂,注射剂(例如溶液或悬浮液)等。在制备片剂时,可采用本领域中熟知的各种载体,如赋形剂,如乳糖,白蔗糖,NaCl,葡萄糖,尿素,淀粉,CaCO3,高岭土,结晶纤维素,硅酸,等等,粘合剂如水,乙醇,丙醇,单糖浆,乳糖溶液,淀粉溶液,明胶溶液,羧甲基纤维素,紫胶片,甲基纤维素,K3PO4,聚乙烯吡咯烷酮等;崩解剂如干淀粉,藻酸钠,粉末状琼脂,粉末状昆布多糖,NaHCO3,CaCO3,聚氧乙烯山梨糖醇酐脂肪酸酯,十二烷基硫酸钠,硬脂酸单甘油酯,淀粉,乳糖等;崩解抑制剂,如白蔗糖,硬脂精,可可脂,氢化油等;吸收促进剂,如季铵盐类;十二烷基硫酸钠等;湿润剂,如甘油,淀粉等;吸附剂,如淀粉乳糖,高岭土,膨润土,胶体硅酸等等;润滑剂,如精细滑石,硬脂酸盐,硼酸粉末,聚乙二醇等等。片子可采用常用的包衣形式,如糖-包衣片,明胶包衣片,肠包衣片或膜包衣片,或以双层药片或多层药片的形式存在。在制备丸剂时,可加入各种本领域内熟知的载体,如赋形剂,如葡萄糖,乳糖,淀粉,可可脂,硬化的蔬菜油,高岭土,滑石等;粘合剂如粉末状的阿拉伯胶,粉末状黄蓍胶,明胶,乙醇等;崩解剂有昆布多糖,琼脂等。在制备栓剂时,可采用本领域中熟知的载体,如聚乙二醇,可可脂,高级醇酯,明胶和半合成甘油酯。在制备注射液(溶液,乳化剂或悬浮剂)时,需对溶液或悬浮液进行无菌处理,并最好使之与血液等渗。在制备溶液,乳化液,悬浮液时,可采用本领域熟知的所有稀释剂,如水,乙醇,丙二醇,乙氧基化异十八烷醇,聚氧基-异十八烷醇和聚氧乙烯基脱水山梨醇-脂肪酸酯。在这种情况下,药物制剂可以含有NaCl,葡萄糖或甘油,其量足可以使之等渗,且可以进一步含有常用的增溶剂,缓冲剂,润滑剂等。如有所需,药物制剂还可以进一步含有增色剂,防腐剂,香料,香味剂,甜味剂和其它药物。
对含于药物制剂中的本发明通式(1)化合物或其盐的用量不加以特别限制,可以在一个较宽的范围内进行选择,但其通常占药物制剂重量的1-70%。
对本发明药物制剂的给药方法也不加以限制,其取决于剂型,患者的年龄,性别以及其他条件,病人的疾病状况等。例如,片剂,丸剂,溶液,悬浮液,乳化剂,颗粒或胶囊经口服给药。注射液采用单独静脉给药或与普通的辅助剂如葡萄糖,氨基酸等混合静脉给药,或者,在需要的情况下单独进行肌肉注射,真皮内注射,皮下注射或腹膜内注射。栓剂用于直肠给药。
根据给药的方法,患者的年龄,性别和其它条件以及患病的程度,恰当地选择药物制剂的量,但是就活性成份来讲,通常是每公斤体重每天约0.2~200mg活性成份即通式(1)化合物或其盐。
在下文中,通常制备实例,参考实例,实例和药理学实验结果对本发明作了详细的描述。
制备实例1
4-(4-氯苄氧基)-N-环丙基甲基-N-甲基苄胺
5mg
淀粉 132mg
硬脂酸镁 18mg
乳糖 45mg
总量 200mg
含有上述组份的药片可用常规方法制备。
制备实例2
2-(4-氯苄氧基)-N-环丙基-甲基-N-甲苄胺
150mg
Aricel(注册商标,ASAHI化学工业有限公司产品) 40g
玉米淀粉 30g
硬脂酸镁 2g
羟丙基甲基纤维素 10g
聚乙二醇6000 3g
蓖麻油 40g
甲醇 40g
将本发明化合物,Avicel,玉米淀粉和硬脂酸镁混合并粉碎,然后用压片机(R:10mm)将其压成片子。然后用含羟丙基甲基纤维素,聚乙二醇6000,蓖麻油和甲醇的膜包衣剂对其进行包衣,生成了膜包衣片。
参考实例1
向1.2g3-羟苯甲醛的50mlN,N-二甲基甲酰胺溶液中加入2.4g4-氯代氯苄和2.1gK2CO3。混合物在60℃下搅拌3小时,冷却后,用乙酸乙酯-水处理反应物,分出乙酸乙酯层,水洗,无水MgSO4干燥,真空蒸馏除去溶剂,残渣经硅胶柱层析纯化(洗脱液:正己烷/乙酸乙酯=10/1)得1.9g3-(4-氯苄氧)苯甲醛。
无色针状
1H-NMR(CDCl3)δ ppm:
5.10(2H,s),7.21-7.25(1H,m),7.38(4H,s),
7.43-7.49(3H,m),9.98(1H,s)
参考实例2
向10g4-羟苯甲酸于100mlN,N-二甲基甲酰胺的溶液中加入29.1g4-氯代氯苄和25gK2CO3,混合物在60℃下搅拌3小时,冷却后,用乙酸乙酯-水处理混合物,分出乙酸乙酯层,水洗,无水MgSO4干燥,真空蒸除溶剂。残渣用正己烷洗涤,滤集后悬浮于50ml乙醇中。向其中加入100ml 2N NaOH溶液,混合物于室温下搅拌4天,用浓HCl中和。滤集生成的结晶,氯仿重结晶,得16.7g4-(4-氯苄氧)苯甲酸。
无色鳞状物
1H-NMR(DMSO-d6)δ ppm:
5.14(2H,s),6.95(2H,d,J=8.5 Hz),7.47
(2H,d,J=11 Hz),7.52(2H,d,J=11 Hz),7.87
(2H,d,J=8.5 Hz)
参考实例3
用与参考实例2相同的方法,以2-羟基苯甲酸为起始物,可以制得2-(4-氯苄氧基)苯甲酸。
黄色棱柱状
1H-NMR(CDCl3)δ ppm:
5.26(2H,s),7.07-7.17(2H,m),7.39(4H,s),
7.51-7.58(1H,m),8.16(1H,dd,J=2.0 Hz,
J=8.0 Hz)
参考实例4
以3-羟苯甲醛作起始原料,用与参考实例1相同的方法可以制得下列化合物。
·3-(2-氯苄氧基)苯甲醛
无色颗粒状
1H-NMR(CDCl3)δ ppm:
5.21(2H,s),7.24-7.31(3H,m),7.39-7.57
(5H,m),9.98(1H,s)
·3-(3-氯苄氧基)苯甲醛
无色油状物
1H-NMR(CDCl3)δ ppm:
5.10(2H,s),7.22-7.33(4H,m),7.43-7.52
(4H,m),9.98(1H,s)
·3-(4-氟苄氧基)苯甲醛
粉色粉末
1H-NMR(CDCl3)δ ppm:
5.08(2H,s),7.09(2H,dd,J=8.5 Hz,J=8.5
Hz),7.22-7.26(1H,m),7.39-7.50(5H,m),
9.98(1H,s)
·3-(4-溴苄氧基)苯甲醛
白色粉末
1H-NMR(CDCl3)δ ppm:
5.07(2H,s),7.20-7.26(1H,m),7.30-7.34
(2H,m),7.42-7.55(5H,m),9.97(1H,s)
参考实例5
在室温下,将62g3-(4-氯苄氧基)苯甲醛和20g环丙甲胺的750ml甲醇溶液一起搅拌15分钟,然后于冰浴冷却下,慢慢加入12g硼氢化钠。生成的混合物于同样温度下再搅拌1小时。将混合物溶于1升氯仿中,水洗,无水MgSO4干燥,真空蒸除溶剂,得81g3-(4-氯苄氧基)-N-环丙甲基苄胺。
黄色油状物
1H-NMR(CDCl3)δ ppm:
0.06-0.12(2H,m),0.44-0.51(2H,m),0.92-
1.00(1H,m),1.55(1H,m),2.47(2H,d,J=7
Hz),3.79(2H,s),5.04(2H,s),6.81-6.96
(3H,m),7.24(1H,dd,J=8 Hz,J=8 Hz),7.36
(4H,s)
参考实例6
以合适的起始原料并采用与参考实例5相同的方法可以制得3-(4-氯苄氧基)-N-甲基苄基胺。
黄色油状物
1H-NMR(CDCl3)δ ppm:
1.40(1H,bs),2.45(3H,s),3.73(2H,s),
5.03(2H,s),6.82-6.95(3H,m),7.24(1H,dd,
J=8.0 Hz,J=8.0 Hz),7.36(4H,s)
参考实例7
在冰浴冷却下,向2.1g3-(4-氯苄氧基)-N-甲基苄胺和3.4ml三乙胺的50mlCH2Cl2溶液中滴加1.0ml环丁烷羰基氯。混合物于室温下搅拌1小时。反应混合物用水洗涤,无水MgSO4干燥,真空蒸除溶剂,得3.1g3-(4-氯苄氧基)-N-环丁烷羰基-N-甲基苄基胺。
黄色油状物。
1H-NMR(CDCl3)δ ppm:
1.81-2.53(6H,m),2.80,2.90(total 3H,s),
3.20-3.48(1H,m),4.40,4.54(total 2H,s),
5.01(2H,s),6.70-6.93(3H,m),7.19-7.33
(1H,m),7.35(4H,s)
参考实例8
将80ml亚硫酰氯溶于5g4-(4-氯苄氧基)苯甲酸中。加热回流1小时,真空蒸除亚硫酰氯,将残渣溶于30mlCH2Cl2中,于冰浴下滴加入2.2g环丙甲胺和10ml三乙胺的100mlCH2Cl2溶液中。混合物于室温下搅拌1小时,用2N HCl和水洗涤,无水MgSO4干燥,蒸除溶剂,得4.2g4-(4-氯苄氧基)-N-环丙甲基苯甲酰胺。
白色粉末。
1H-NMR(CDCl3)δ ppm:
0.23-0.30(2H,m),0.51-0.59(2H,m),1.00-
1.15(1H,m),3.30(2H,dd,J=5.5 Hz,J=7.0
Hz),5.07(2H,s),6.09-6.21(1H,m),6.95-
7.00(2H,m),7.36(4H,s),7.72-7.77(2H,m)
参考实例9
以2-(4-氯苄氧基)苯甲酸为起始原料,采用与参考实例8相同的方法,得2-(4-氯苄氧基)-N-环丙甲基苯甲酰胺。
白色粉末
1H-NMR(CDCl3)δ ppm:
0.02-0.08(2H,m),0.29-0.37(2H,m),0.77-
0.85(1H,m),3.22(2H,dd,J=5.0 Hz,J=7.0
Hz),5.14(2H,s),7.03-7.15(2H,m),7.38-
7.48(1H,m),7.41(4H,s),7.72-7.98(1H,m),
8.24(1H,dd,J=2.0 Hz,J=8.0 Hz)
参考实例10
将1.5g4-(4-氯苄氧基)-N-环丙甲苯甲酰胺溶于30mlN,N-二甲基甲酰胺中,向其中加入0.25gNaH。混合物于室温下搅拌30分钟,然后于60℃下搅拌30分钟。冰浴冷却,加入0.5mlCH3I,室温下放置过夜,混合物用乙酸乙酯-水处理,分出乙酸乙酯层,水洗,无水MgSO4干燥,蒸除溶剂。残渣经硅胶柱层析纯化(洗脱液CH2Cl2/丙酮=50/1),得1.4g4-(4-氯苄氧基)-N-环丙甲基-N-甲基苯甲酰胺。
无色油状物
1H-NMR(CDCl3)δ ppm:
0.00-0.40(2H,m),0.51-0.63(2H,m),0.88-
1.11(1H,m),2.95-3.50(5H,m),5.05(2H,s),
6.93-6.97(2H,m),7.26-7.40(2H,m),7.37
(4H,s)
参考实例11
以2-(4-氯苄氧基)-N-环丙甲基苯甲酰胺为起始原料,用与参考实例10相同的方法可以制得2-(4-氯苄氧基)-N-环丙甲基-N-甲基苯甲酰胺。
浅黄色油状物
1H-NMR(CDCl3)δ ppm:
-0.05-0.10,0.23-0.37,0.37-0.55(total 4H,
m),0.75-0.92,0.92-1.08(total 1H,m),2.88-
3.25(5H,m),5.07-5.14(2H,m),6.89-7.14
(2H,m),7.25-7.45(6H,m)
参考实例12
以合适的起始原料,用与参考实例5相同的方法,可以制得下列化合物。
·3-(2-氯苄氧基)-N-环丙甲基苄胺,
无色油状物
1H-NMR(CDCl3)δ ppm:
0.07-0.13(2H,m),0.44-0.51(2H,m),0.92-
1.10(1H,m),1.35-1.61(1H,m),2.48(2H,d,
J=7.0 Hz),3.80(2H,s),5.17(2H,s),6.85-
6.99(3H,m),7.25-7.32(3H,m),7.38-7.42
(1H,m),7.55-7.59(1H,m)
·3-(3-氯苄氧基)-N-环丙基甲基苄基胺
无色油状物
1H-NMR(CDCl3)δ ppm:
0.07-0.12(2H,m),0.44-0.51(2H,m),0.50-
1.08(1H,m),1.35-1.52(1H,m),2.48(2H,d,
J=7.0 Hz),3.80(2H,s),5.04(2H,s),6.82-
6.97(3H,m),7.21-7.30(4H,m),7.44-7.45
(1H,m)
·3-(4-氟苄氧基)-N-环丙甲基苄胺
无色油状物
1H-NMR(CDCl3)δ ppm:
0.07-0.13(2H,m),0.44-0.51(2H,m),0.92-
1.08(1H,m),1.40-1.70(1H,m),2.47(2H,d,
J=7.0 Hz),3.80(2H,s),5.03(2H,s),6.83-
6.97(3H,m),7.04-7.10(2H,m),7.26(1H,dd,
J=8.0 Hz,J=8.0 Hz),7.38-7.44(2H,m)
·3-(4-溴苄氧基)-N-环丙甲基苄胺
无色油状物
1H-NMR(CDCl3)δ ppm:
0.06-0.12(2H,m),0.44-0.51(2H,m),0.91-
1.06(1H,m),1.34-1.60(1H,m),2.47(2H,d,
J=7.0 Hz),3.79(2H,s),5.02(2H,s),6.81-
6.95(3H,m),7.20-7.33(3H,m),7.49-7.52
(2H,m)
参考实例13
以合适的起始原料,用与参考实例8相同的方法,可以制备下列化合物。
·3-(4-氯代苄氧基)-N-环己基甲基苯甲酰胺
白色粉末
1H-NMR(CDCl3)δ ppm:
0.95-1.11(2H,m),1.11-1.36(3H,m),1.56-
1.90(6H,m),3.29(2H,dd,J=6.5 Hz,J=6.5
Hz),5.06(2H,s),6.18-6.29(1H,m),7.04-
7.09(1H,m),7.26-7.43(3H,m),7.36(4H,s)
·3-(4-氯代苄氧基)-N-环丙基甲基-N-丙基苯甲酰胺
白色粉末
1H-NMR(CDCl3)δ ppm:
-0.01-1.82(10H,m),3.00-3.63(4H,m),5.05
(2H,s),6.91-6.99(3H,m),7.30(1H,dd,
J=8.0 Hz,J=8.0 Hz),7.35(4H,s)
参考实例14
以3-(4-氯代苄氧基)-N-环己基甲基苯甲酰胺为起始原料,用与参考实例10相同的方法,可以制得3-(4-氯苄氧基)-N-环己基甲基-N-甲基苯甲酰胺。
无色油状物
1H-NMR(CDCl3)δ ppm:
0.55-0.72,0.95-1.37,1.52-1.85(total 11H,
m),2.89,3.04(total 3H,s),3.06,3.37
(total 2H,s),5.05(2H,s),6.92-6.99(3H,
m),7.30(1H,dd,J=8.5 Hz,J=8.5 Hz),7.36
(4H,s)
实例1
将80g3-(4-氯苄氧基)-N-环丙基甲基苄胺溶于150ml甲酸和150ml37%甲醛组成的混合物中,加热回流3小时,混合物冷却至室温,加入150ml浓HCl,真空蒸除溶剂,残渣用氯仿-NaOH溶液处理,分出氯仿层,水洗,无水Na2SO4干燥,真空下蒸除溶剂,得86g3-(4-氯代苄氧基)-N-环丙甲基-N-甲基苄胺,其为棕色油状物。
将86g中的3g转变成盐酸盐,然后用乙酸乙酯重结晶,得2.1g3-(4-氯苄氧基)-N-环丙甲基-N-甲基苄胺盐酸盐。
无色针状物
熔点:119.0-121.0℃。
用合适的起始原料,并以与实例1相同的方法,可以制得表1中实例2-10化合物。


实例11
将0.5gAlLiH4加到1.4g4-(4-氯苄氧基)-N-环丙基甲基-N-甲基苯甲酰胺的50mlTHF溶液中。加热回流3小时,冷却后,加2ml水。混合物于室温下搅拌14小时,过滤,滤液用水、乙醚提取,分出乙醚层,水洗,无水MgSO4干燥,真空下蒸除溶剂。生成的残渣转化成草酸盐。用乙醇对草酸盐重结晶,得1.3g4-(4-氯苄氧基)-N-环丙甲基-N-甲基苄胺草酸盐。
白色粉末。
熔点:154-154.5℃
以合适的起始原料,用与实例11相同的方法,可以制得实例1和3-10中的化合物。
实例12
在冰浴冷却下,将12g硼氢化钠慢慢加到62g4-(4-氯苄氧基)苯甲醛和24gN-环丙基甲基-N-甲胺的750ml甲醇溶液中,混合物在相同温度下搅拌1小时。将生成的混合物溶于1升氯仿中。加水洗涤,无水MgSO4干燥,真空蒸除溶剂,残渣转化成草酸盐,生成的草酸盐用乙醇重结晶,得84g4-(4-氯苄氧基)-N-环丙甲基-N-甲基苄胺草酸盐。
白色粉末
熔点:154~154.5℃
用合适的起始原料,并以与实例12相同的方法,得实例1和3-10中的化合物。
实例13
将0.92gAlLiH4加入2.8g3-(4-氯苄氧基)-N-环丁烷羰基-N-甲基苄胺的50mlTHF溶液中,加热回流3小时,冷却后,加入2ml水,混合物搅拌14小时,过滤,滤液用水-乙醚处理,分出乙醚层,水洗,无水MgSO4干燥,真空蒸除溶剂。残渣转化成马来酸盐,并用乙醇重结晶,得1.9g3-(4-氯苄氧基)-N-环丁基甲基-N-甲基苄基胺马来酸盐。
白色粉末
熔点:102.5~103.0℃
用合适的起始原料,并以与实例13相同的方法可以制得实例1-3及5-10中的化合物。
药理学实验
(1)强制性游泳实验
参照文献Nature Vol.266,pp.730-732(1977)以及European Journal of Pharmacology,Vol.47,pp.379-391(1978)中所描述的方法,并加以改良来实施本实验。将自来水(温度约25℃)灌入一个圆筒形水浴(内径:29.5cm,高度25cm)中,该水浴是用透明的丙烯树脂制成的,水深11.3cm。强迫7-8周大小的雄性小鼠(体重约30-35g)的游泳6分钟,然后测量所游泳的活动量,测量是借助红外光束以及能够检测光束的传感器自动记数,作为评判标准,其可显示出小鼠的抑制状态。较大的数字表示被测试化合物有较高的抗抑制活性。
每个小鼠在游泳前禁食16-18小时,并在实验前1小时口服给予被测试化合物和溶剂。溶剂是含5%阿拉伯胶的生理盐水,被测试化合物悬浮于或溶于溶剂中。对照组的每只小鼠仅给予溶剂,即含5%阿拉伯胶的生理盐水。
结果列于表2。在表2中,以对照组的游泳量为100%来计量实验组小鼠的游泳活动量的值(%)
表2
实验化合物 给药量(mg/kg) 泳值(%)
实例1化合物 1 120
实例2化合物 3 110
实例3化合物 30 118
(2)架高十字迷宫试验
根据Pharmacology Biochemistry&Behavior,Vol.24,pp.525-529(1986)和Psychopharmacology,Vol.92,pp.180-185(1987)中所描述的方法来实施本实验。实验仪器是用丙烯树脂为材料的迷宫,其由一个平台(5×5cm),两个开放的臂(每个25×5cm且没有墙围着臂)以及两个封闭的臂(每个25×5cm,三面被透明的15cm高的丙烯树脂墙围起来)组成,其中两个开放的臂从平台相反的两边延伸出去,两个封闭的臂从平台余下的相反的两边延伸出去(两个开放臂和两个封闭臂经平台形成一个交叉形状)。迷宫的平台,开放臂,封闭臂被置于室内距地面38cm高的位置上。将已禁食16-18小时,4-5周令,体重20-24g的雄性小鼠置于迷宫的平台上,脸朝着任意一侧的开放臂,计时5分钟,观察小鼠在迷宫中的运动,记录进入开放臂和进入封闭臂的时间,以“进入开放臂的频率”作为受试化合物抗焦虑活性的评判标准,其定义如下:
进入开放臂的频率(%)=(进入开放臂的时间)-(进入开放臂的时间+进入封闭臂的时间)×100
较高的频率意味着所试化合物具有较高的抗焦虑活性。
在将小鼠置于迷宫前1小时时,口服给予每只小鼠悬浮于或溶于含5%阿拉伯胶的生理盐水中的受试化合物。对照组小鼠仅给予生理盐水。
结果列于表3,表3中的频率值(%)是以对照组值为100%时计算的。
表3
受试化合物 给药量(mg/kg) 频率(%)
实例1化合物 1 262
实例2化合物 3 175
实例3化合物 3 128
(3)对由于暴力造成的头损伤引起的昏迷,有改善意识紊乱的功效的评价
根据文献Journal of Japan Accident Medical Association,Vol.25,P.202(1977)-和Jounal of Clinical and Experimental Medicine(IGAKUNO AYUMI),Vol.102,pp.867-869(1977)中所描述的方法来实施本实验。将4-5周令,体重约20-29g,禁食18-20小时的雄性小鼠的头,固定在一个泡沫状的聚苯乙烯制的枕头上。沿着透明的塑料管用丙烯树脂制成的圆筒形棒,对每只小鼠的头颅顶部进行撞击。然后观测以下两个项目,对由此引起的小鼠的意识丧失进行测定:撞击后从昏迷到正确反射恢复的时间(此时间称为RR时间)以及从所述的昏迷至自发运动恢复所需时间(此时间称为SM时间)。将受试化合物悬浮于或溶于含5%阿拉伯胶的生理盐水中,然后在小鼠受到麻醉1小时前口服给予每只小鼠。对照组的小鼠仅给予含5%阿拉伯胶的生理盐水。通过给予小鼠/受试化合物的RR或SM时间与对照组小鼠的RR或SM时间的比率(%)来评价每个受试化合物对意识丧失的改善作用。现有的每个受试化合物对意识丧失的改善均显示出显著的功效。
(4)在控制条件下小鼠运动量实验
(a)实验动物
ddy-株雄性小鼠,7-8周令,体重30-35g之间。
(b)实验仪器
透明的由丙烯树脂制成的实验箱,体积为30×20×29cm,带有格栅地面;光电测试器,用来测定小鼠的运动量;撞击发生器用于对小鼠进行电击。
(C)实验步骤
实验第一天,将小鼠置于上述仪器中,从地面格栅处对其进行电击(AC160V×3A,200mse,0.1HZ)6分钟。第二天将小鼠放回同样的仪器中(但不予电击),用上述光电测试器测定小鼠的运动量(这些小鼠属于控制条件组)。对照组的小鼠进行同样的实验,所不同的是它们第一天不接受电击。
将每种受试化合物悬浮于含5%阿拉伯胶的生理盐水中,或溶于生理盐水中。在实验第二天进行小鼠运动量测定的30分钟~1小时前,每只小鼠口服给予上述悬浮液或溶液,将条件控制组的参数与单单给予溶剂的组(对照组)的参数进行比较。
在这项实验中,“参数”是小鼠第二天的运动量(通过光电测试器记数)。
在上述实验中,当将已经过电击的小鼠放回同一位置时,会记忆起电击并产生一种紧张感,这种紧张感实际上降低了它们的活动量。经测试的本发明化合物可以有效地恢复其运动量。
Claims (24)
1、由通式(1)表示的苄胺衍生物或其盐:
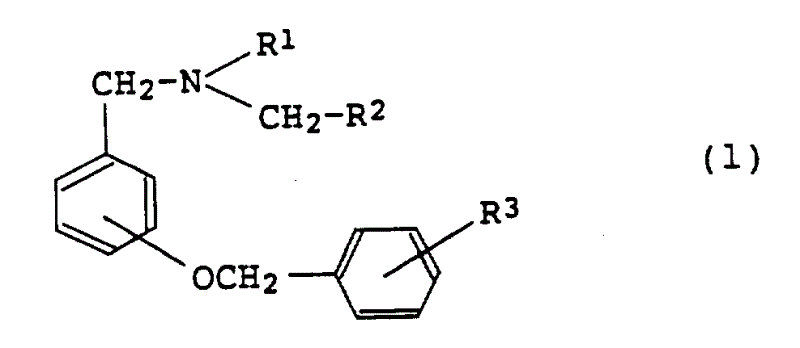
其中R1为低级烷基;
R2为环烷基;且
R3为卤素。
2、权利要求1中的苄胺衍生物或其盐,其中R1为甲基或乙基。
3、权利要求2中的苄胺衍生物或其盐,其中R1为甲基。
4、权利要求1中的苄胺衍生物或其盐,其中R2为环丙基或环丁基。
5、权利要求1中的苄胺衍生物或其盐,其中R2为环丙基。
6、权利要求1中的苄胺衍生物或其盐,其中R3为氯原子。
7、权利要求1中的苄胺衍生物或其盐,其中R1为甲基或乙基,R2为环丙基或环丁基;且R3为氯原子。
8、权利要求6或7中的苄胺衍生物,其中R3取代于苯环的4-位上。
9、3-(4-氯苄氧基)-N-环丙基-N-甲基苄胺。
10、4-(4-氯苄氧基)-N-环丙基-N-甲基苄胺。
11、2-(4-氯苄氧基)-N-环丙基-N-甲基苄胺。
12、含有权利要求1的苄胺衍生物或其盐,作为活性成分的抗抑郁、抗焦虑剂。
13、含有权利要求1中的苄胺衍生物或其盐,作为活性成分的中枢神经系统活化剂。
14、含有权利要求1中的苄胺衍生物或其盐,作为活性成分的知觉紊乱改善剂。
15、含有权利要求1中的苄胺衍生物或其盐,作为活性成分的σ-受体激动剂。
16、按照权利要求12的抗抑郁、抗焦虑剂,其中的活性成分是权利要求9,10或11中的苄胺衍生物。
17、按照权利要求13的中枢神经系统活化剂,其中活性成分是权利要求9,10或11中的苄胺衍生物。
18、按照权利要求14的知觉紊乱改善剂,其中的活性成分是权利要求9,10或11中的苄胺衍生物。
19、按照权利要求15的σ-受体激动剂,其中的活性成分是权利要求9,10或11中的苄胺衍生物。
20、制备苄胺衍生物(1)的方法,

其中R1是低级烷基;R2是环烷基;R3是卤原子;
将苄氧基苯甲醛(2)(其中R3同上述定义)与

胺化合物(3)(其中R1和R2同上述定义)进行反应,

得到shiff氏碱化合物A:
其中R1,R2和R3同上述定义;不经分离,即将化合物(A)进行还原。制得所需的苄胺衍生物(1)。
21、制备苄胺衍生物(1)的方法:

其中R1,R2和R3同上述定义,将苄氧基苄胺化合物(4):

(其中R2和R3同上述定义)与卤代烷(5)反应,
R1-X (5)
(其中R1同上述定义,且X为卤原子),或与醛或酮(6)进行反应:
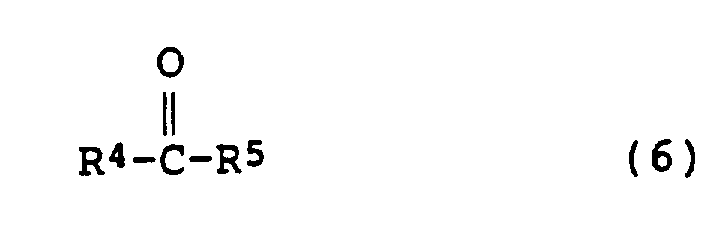
(其中R4和R5分别为氢原子或低级烷基),制得所需的苄胺衍生物(1)。
22、制备苄胺衍生物(1)的方法:

(其中,R1,R2和R3同上述定义);将苄氧基苄胺化合物(9):

(其中R1,R2和R3同上述定义);进行还原反应制得所需的苄胺衍生物(1)。
23、制备苄胺衍生物(1)的方法:

(其中R1,R2和R3同上述定义);将苄氧基苄胺化合物(7):
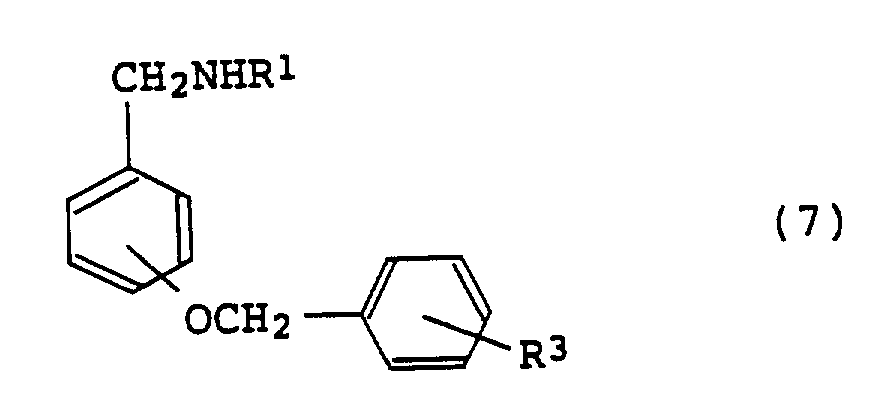
(其中R1和R3同上述定义);与卤代环烷甲基(10):
R2-CH2-X (10)
(其中R2和X同上述定义)进行反应,得到所需的苄胺衍生物(1)
24、制备苄胺衍生物(1)的方法:
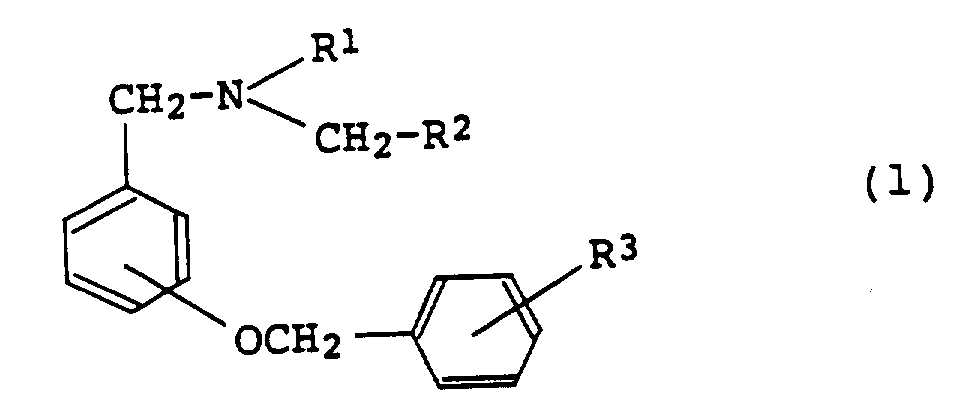
(其中R1,R2和R3同上述定义);通过还原苄氧基苯甲酰胺(12):

(其中R1,R2和R3同上述定义);制得所需的苄胺衍生物(1)。
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP214147/93 | 1993-08-30 | ||
| JP21414793 | 1993-08-30 | ||
| JP4400394 | 1994-03-15 | ||
| JP44003/94 | 1994-03-15 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| CN1114106A true CN1114106A (zh) | 1995-12-27 |
Family
ID=26383844
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN94190643A Pending CN1114106A (zh) | 1993-08-30 | 1994-08-11 | 苄胺衍生物 |
Country Status (9)
| Country | Link |
|---|---|
| US (1) | US5550292A (zh) |
| EP (1) | EP0666840A1 (zh) |
| KR (1) | KR950704235A (zh) |
| CN (1) | CN1114106A (zh) |
| AU (1) | AU667280B2 (zh) |
| CA (1) | CA2147356A1 (zh) |
| PH (1) | PH30825A (zh) |
| TW (1) | TW260661B (zh) |
| WO (1) | WO1995006630A1 (zh) |
Families Citing this family (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5980941A (en) * | 1997-08-20 | 1999-11-09 | Fuisz Technologies Ltd. | Self-binding shearform compositions |
| US5840334A (en) * | 1997-08-20 | 1998-11-24 | Fuisz Technologies Ltd. | Self-binding shearform compositions |
| US5986141A (en) * | 1998-09-29 | 1999-11-16 | Eastman Chemical Company | Process for the production of cyclopropanemethylamine |
| EP1634598A1 (en) * | 2004-09-07 | 2006-03-15 | Laboratorios Del Dr. Esteve, S.A. | Use of piperazine derivatives and analogues for the manufacture of a medicament for the prophylaxis and/or treatment of disorders of food ingestion |
Family Cites Families (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4687865A (en) * | 1986-06-04 | 1987-08-18 | E. R. Squibb & Sons, Inc. | Process for preparing 7-oxabicycloheptane amino-alcohol intermediates useful in making thromboxane A2 receptor antagonists |
| CA1327795C (en) * | 1987-08-14 | 1994-03-15 | Jules Freedman | Antidepressants which are aryloxy inadanamines |
| IL94466A (en) * | 1989-05-25 | 1995-01-24 | Erba Carlo Spa | Pharmaceutical preparations containing the history of A-amino carboxamide N-phenylalkyl are converted into such new compounds and their preparation |
| CA2071897A1 (en) * | 1989-12-28 | 1991-06-29 | Richard A. Glennon | Sigma receptor ligands and the use thereof |
| AU676993B2 (en) * | 1991-06-27 | 1997-04-10 | Virginia Commonwealth University | Sigma receptor ligands and the use thereof |
| DE4132013A1 (de) * | 1991-09-26 | 1993-04-01 | Basf Ag | Neue phenylbenzylamine und diese enthaltende arzneimittel |
| ES2125906T3 (es) * | 1991-10-04 | 1999-03-16 | Taisho Pharmaceutical Co Ltd | Derivado de alcoxifenilalquilamina. |
| DE4216941A1 (de) * | 1992-05-22 | 1993-11-25 | Basf Ag | Para-substituierte Benzylamine und diese enthaltende therapeutische Mittel |
| DE4327365A1 (de) * | 1993-08-14 | 1995-02-16 | Boehringer Mannheim Gmbh | Verwendung von Phenolen und Phenolderivaten als Arzneimittel mit fibrinogensenkender Wirkung |
-
1994
- 1994-08-11 WO PCT/JP1994/001332 patent/WO1995006630A1/en not_active Ceased
- 1994-08-11 AU AU73505/94A patent/AU667280B2/en not_active Ceased
- 1994-08-11 CA CA002147356A patent/CA2147356A1/en not_active Abandoned
- 1994-08-11 EP EP94922362A patent/EP0666840A1/en not_active Ceased
- 1994-08-11 KR KR1019950701681A patent/KR950704235A/ko not_active Ceased
- 1994-08-11 CN CN94190643A patent/CN1114106A/zh active Pending
- 1994-08-11 US US08/428,114 patent/US5550292A/en not_active Expired - Fee Related
- 1994-08-16 TW TW083107505A patent/TW260661B/zh active
- 1994-08-30 PH PH48875A patent/PH30825A/en unknown
Also Published As
| Publication number | Publication date |
|---|---|
| WO1995006630A1 (en) | 1995-03-09 |
| EP0666840A1 (en) | 1995-08-16 |
| TW260661B (zh) | 1995-10-21 |
| AU667280B2 (en) | 1996-03-14 |
| KR950704235A (ko) | 1995-11-17 |
| AU7350594A (en) | 1995-03-22 |
| PH30825A (en) | 1997-10-17 |
| US5550292A (en) | 1996-08-27 |
| CA2147356A1 (en) | 1995-03-09 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN1688544A (zh) | 作为蕈毒碱受体拮抗剂的含氟代和磺酰氨基的3,6-二取代的氮杂双环(3.1.0)己烷衍生物 | |
| CN1255117A (zh) | 兴奋性氨基酸受体调节剂 | |
| CN1890208A (zh) | 类鸦片受体拮抗剂 | |
| CN1859904A (zh) | 用于治疗阿尔茨海默氏病的苄醚和苄氨基β-分泌酶抑制剂 | |
| CN1168720C (zh) | 芳基链烷酰基哒嗪化合物 | |
| CN1147248A (zh) | N-(3-吡咯烷基)苯甲酰胺衍生物 | |
| CN1032438A (zh) | 新的取代的n-(1-烷基-3-羟基-4-哌啶基)苯甲酰胺 | |
| CN1007727B (zh) | 制备四氢化萘衍生物的方法 | |
| CN1205704A (zh) | 2,7-取代的八氢-吡咯并[1,2-a]吡嗪衍生物 | |
| HK1045684A1 (zh) | 联苯基衍生物 | |
| CN1216856C (zh) | 具有亚环烷基链的新环状化合物,其制备方法和含有它们的药物组合物 | |
| CN1114106A (zh) | 苄胺衍生物 | |
| CN1723204A (zh) | 苯并噁唑衍生物及其作为腺苷受体配体的用途 | |
| CN1172478A (zh) | 喹诺酮衍生物抗血栓形成药 | |
| CN1871013A (zh) | 作为mao-b抑制剂的苯并氮杂䓬衍生物 | |
| CN1345317A (zh) | (1-苯甲酰甲基-3-苯基-3-哌啶基乙基)哌啶衍生物,制备它们的方法以及包含它们的药物组合物 | |
| CN1023479C (zh) | 乙二胺的单酰胺衍生物的制备方法 | |
| CN1478074A (zh) | 环己基(烷基)-丙醇胺及其制备方法和含有它们的药物组合物 | |
| CN101039912A (zh) | 吡啶衍生物和它们的制备方法及其治疗应用 | |
| CN1023599C (zh) | 氯苯胺衍生物的制备方法 | |
| CN1062526A (zh) | 烷氨基烷基胺和醚化合物及其制造方法和中间产物以及含这些化合物的药剂 | |
| CN1230173A (zh) | 环丁烯-3,4-二酮的取代的n-芳甲基氨基衍生物 | |
| CN1147482C (zh) | 取代的(二氢)苯并噁嗪和(二氢)苯并噻嗪化合物,其制法和药物组合物 | |
| CN88101939A (zh) | 对白三烯拮抗剂或有关方面的改进 | |
| CN1466573A (zh) | 5-苯基苄胺化合物、其制备方法以及其合成中间体 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C01 | Deemed withdrawal of patent application (patent law 1993) | ||
| WD01 | Invention patent application deemed withdrawn after publication |