Preparation method of indoleamine 2, 3-dioxygenase inhibitor
The application is a divisional application, and the Chinese application number of the parent case is as follows: 201880057640.2, International application number PCT/CN2018/124110, International application date 12/27 of 2018.
The present invention claims priority from chinese patent applications CN201711478307.2 and CN201810754253.6, and the contents of the specification, drawings and claims of this priority document are incorporated in their entirety into the specification of the present invention and are included as a part of the original description of the present invention. Applicants further claim that applicants have the right to amend the description and claims of this invention based on this priority document.
Technical Field
The invention relates to the field of medicines, and in particular relates to a preparation method of an indoleamine 2, 3-dioxygenase inhibitor.
Background
Tryptophan (TRP) is an α -amino acid used in protein biosynthesis. It contains an alpha-amino group, an alpha-carboxylic acid group and a side chain indole. It is essential in humans, who cannot synthesize it, but must be obtained from the diet. Tryptophan is also a precursor in the synthesis of the neurotransmitter 5-hydroxytryptamine (serotonin) and the hormone N-acetyl-5-methoxytryptamine (melatonin). The heme-dependent enzyme indoleamine 2, 3-dioxygenase (also called IDO, or IDO1) is a metabolic enzyme outside the liver responsible for the conversion of tryptophan to N-formyl-kynurenine, which is the first step in the tryptophan metabolism process and is also the rate-limiting step of the overall process. N-formyl-kynurenine is a precursor of the diverse bioactive molecules kynurenine (kynurenine, or Kyn) that has immunomodulatory functions (Schwarcz et al, Nat Rev neurosci.2012; 13(7): 465).
Indoleamine 2, 3-dioxygenase (IDO) is widely expressed in solid tumors (Uyttenhove et al, natmed. 2003; 10:1269), and is also expressed in both primary and metastatic cancer cells. IDO is induced in tumors by pro-inflammatory factors, including type I and type II interferons produced by infiltrating lymphocytes (Tnani and Bayard, Biochim biophysis acta 1999; 1451(l): 59; Mellor and Munn, Nat Rev Immunol 2004; 4(10): 762; Munn, Front biosci.2012; 4:734) and transforming growth factor-beta (TGF-beta) (Pallotta et al, nature immunol.2011; 12(9): 870). In recent years, there has been increasing evidence that IDO plays a major role in immune cell regulation as an inducible enzyme. Decreased tryptophan levels and increased kynurenines suppress immune effector cells and promote adaptive immune suppression by inducing and maintaining regulatory T cells (Tregs; Munn, Front biosci.2012; 4: 734); the concentration of tryptophan in the immune system is positively correlated with T cells. In the tumor immune microenvironment, activated or overexpressed IDO leads to tryptophan depletion, which in turn leads to T cell death, immune system inactivation, and ultimately to the development of tumor immune tolerance and immune escape. The existing research shows that the immune balance disorder caused by IDO is deeply involved in the generation and the progression of tumors. Therefore, IDO receptor has become an important target for immunotherapy such as tumor. IDO is associated with, in addition to tumors, viral infections, depression, organ transplant rejection or autoimmune diseases (Johnson and Munn, Immunol Invest 2012; 41(6-7): 765). Therefore, agents targeting IDO are also of great value for the treatment of the above mentioned diseases. In conclusion, it is necessary to develop an IDO inhibitor having activity and selectivity to effectively treat diseases due to harmful substances in the kynurenine pathway by modulating the kynurenine pathway and maintaining tryptophan levels in the body, either as a single agent or a combination therapy.
Numerous published preclinical data also further confirm the role of IDO in antitumor immune responses. IDO inhibitors may be used to activate T cells, thereby increasing T cell activation when the T cells are suppressed by viruses such as pregnancy, malignancy, or HIV. Forced induction of IDO in cancer cells has proven to be a survival advantage (Uyttenhove et al, Nat Med. 2003; 10: 1269). Another in vivo study showed that IDO inhibitors reduce lymphocyte dependence by reducing kynurenine levels in tumor growth (Liu et al, blood. 2010; 115(17): 3520). Preclinical studies have also shown that IDO inhibitors have synergistic effects if combined with other tumor drugs, such as radiation therapy, chemotherapy, or vaccines. (Koblish et al, Molcancer ther. 2010; 9(2):489, Hou et al, Cancer Res.2007; 67(2): 792; Sharma et al, blood.2009; 113(24):6102).
Research on IDO inhibitor antitumor drugs has currently made significant progress worldwide, such as INCB024360, NLG919 and BMS-986205 all entering the clinic. However, the INCB024360 has the problem of toxic and side effects, so that the dosage (50mg bid or 100mg bid) of the existing clinical research is about 30 percent of the optimal dosage (300mg bid or 600mg bid), and the clinical activity is greatly limited; meanwhile, the toxic group of INCB024360 is a pharmacophore, and the INCB024360 and derivatives thereof have the problem of high toxicity. The safety of NLG919 is good, but the bioactivity of NLG919 is poor. BMS-986205 has already entered the clinic at present, but the clinical data are limited, and based on BMS-986205, the research of novel compounds with high biological activity and high safety is carried out, which has very important practical significance for finding novel IDO tumor immunotherapy drugs with better clinical therapeutic activity, such as possible tumor cure, rather than just tumor inhibition.
Disclosure of Invention
The invention provides a compound shown as a formula I,
wherein
Represents: - (O) b,
Or
A represents-C (O) -, -S (O)2-or-s (o) -;
wherein each R is1Each is independentSelected from hydrogen atom, halogen, hydroxyl, nitro, cyano, sulfonic acid group and C1-6Alkyl radical, C3-6Cycloalkyl radical, C2-6Alkenyl radical, C2-6Alkynyl, C1-6Alkoxy, halo C1-C6Alkyl, halo C1-C6Alkoxy, halo C1-C6Cycloalkyl radical, C1-6Alkylthio radical, C1-6Alkylcarbonyl group, C1-6Alkoxycarbonyl, di (C)1-6Alkyl) amino C2-6Alkoxycarbonyl, amino, C1-6Alkylamino radical, di (C)1-6Alkyl) amino, carbamoyl, C1-6Alkylcarbamoyl, di (C)1-6Alkyl) carbamoyl, di (C)1-6Alkyl) amino C2-6Alkylcarbamoyl, sulfamoyl, C1-6Alkylsulfamoyl, di (C)1-6Alkyl) sulfamoyl, di (C)1-6Alkyl) amino C2-6Alkylsulfamoyl, C1-6Alkylsulfonyl radical, C1-6Alkylsulfinyl, di (C)1-6Alkyl) phosphono, hydroxy C1-6Alkyl, hydroxy carbonyl C1-6Alkyl radical, C1-6Alkoxy radical C1-6Alkyl radical, C1-6Alkylsulfonyl radical C1-6Alkyl radical, C1-6Alkylsulfinyl C1-6Alkyl, di (C)1-6Alkyl) phosphono C1-6Alkyl, hydroxy C2-6Alkoxy radical, C1-6Alkoxy radical C2-6Alkoxy, amino C1-6Alkyl radical, C1-6Alkylamino radical C1-6Alkyl, di (C)1-6Alkyl) amino C1-6Alkyl, di (C)1-6Alkyl) aminoacetyl, amino C2-6Alkoxy radical, C1-6Alkylamino radical C2-6Alkoxy, di (C)1-6Alkyl) amino C2-6Alkoxy, hydroxy C2-6Alkylamino radical, C1-6Alkoxy radical C2-6Alkylamino radical, amino radical C2-6Alkylamino radical, C1-6Alkylamino radical C2-6Alkylamino radical, di (C)1-6Alkyl) amino C2-6An alkylamino group; or adjacent R1Mutually cyclized to form a 3-8 membered ring, and the ring contains 0, 1, 2 and 3 heteroatoms;
Cy1selected from 5-15 membered bridged ring group, 5-15 membered spiro ring group, 5-15 membered bridged heterocyclic group, or 5-15 membered spiro heterocyclic group substituted by any substituent group: halogen, hydroxy, C1-6Alkyl, amino, halo C1-6Alkyl, mercapto, C1-6Alkyl mercapto group, C1-6Alkylamino radical, di (C)1-6Alkyl) amino, cyano;
Ra、Rb、R2each independently selected from hydrogen and C1-C6Alkyl or C3-6A cycloalkyl group;
Cy2is C containing one or more substituents5-C10Aryl radical, C5-C10Heteroaryl group, C5-C10Cycloalkyl radical, C5-C10A heterocycloalkyl group; the substituent can be selected from halogen, hydroxyl, nitro, cyano, sulfonic acid group and C1-6Alkyl radical, C2-6Alkenyl radical, C2-6Alkynyl, C3-6Cycloalkyl radical, C1-6Alkoxy, halo C1-C6Alkyl, halo C1-C6Alkoxy radical, C1-6Alkylthio radical, C1-6Alkylcarbonyl group, C1-6Alkylcarbonyloxy, C1-6Alkoxycarbonyl, di (C)1-6Alkyl) amino C2-6Alkoxycarbonyl, amino, C1-6Alkylamino radical, di (C)1-6Alkyl) amino, carbamoyl, C1-6Alkylcarbamoyl, di (C)1-6Alkyl) carbamoyl, di (C)1-6Alkyl) amino C2-6Alkylcarbamoyl, sulfamoyl, C1-6Alkylsulfamoyl, di (C)1-6Alkyl) sulfamoyl, di (C)1-6Alkyl) amino C2-6Alkylsulfamoyl, C1-6Alkylsulfonyl radical, C1-6Alkylsulfinyl, di (C)1-6Alkyl) phosphono, hydroxy C1-6Alkyl, hydroxy carbonyl C1-6Alkyl radical, C1-6Alkoxy radical C1-6Alkyl radical, C1-6Alkylsulfonyl radical C1-6Alkyl radical, C1-6Alkylsulfinyl C1-6Alkyl, di (C)1-6Alkyl) phosphono C1-6Alkyl, hydroxy C2-6Alkoxy radical, C1-6Alkoxy radical C2-6Alkoxy, amino C1-6Alkyl radical, C1-6Alkylamino radical C1-6Alkyl, di (C)1-6Alkyl) amino C1-6Alkyl, di (C)1-6Alkyl) aminoacetyl, amino C2-6Alkoxy radical, C1-6Alkylamino radical C2-6Alkoxy, di (C)1-6Alkyl) amino C2-6Alkoxy, hydroxy C2-6Alkylamino radical, C1-6Alkoxy radical C2-6Alkylamino radical, amino radical C2-6Alkylamino radical, C1-6Alkylamino radical C2-6Alkylamino radical, di (C)1-6Alkyl) amino C2-6Alkylamino, -S (O) C1-6An alkyl group; or when two substituents are adjacent, can form a 3-8 membered ring, which 3-8 membered ring may contain 0, 1, 2, 3O, S, N atoms; m and n are 0, 1, 2,3 and 4.
In another embodiment of the present invention, Cy is1Selected from 8-12 membered spiro ring group or 8-12 membered spiro ring group, 8-12 membered bridged heterocyclic group, or 8-12 membered spiro heterocyclic group substituted by substituent groups: halogen, hydroxy, C1-6Alkyl, amino, halo C1-6Alkyl, mercapto, C1-6Alkyl mercapto group, C1-6Alkylamino radical, di (C)1-6Alkyl) amino, cyano. In another embodiment of the present invention, Cy is1Selected from the following groups:
the above groups may be selected from halogen, hydroxy, C1-6Alkyl, amino, halo C1-6Alkyl, mercapto, C1-6Alkyl mercapto group, C1-6Alkylamino radical, di (C)1-6Alkyl) amino, cyano.
In another embodiment of the present invention, Cy is1Selected from the following groups:
the above groups may be selected from halogen, hydroxy, C1-6Alkyl, amino, halo C1-6Alkyl, mercapto, C1-6Alkyl mercapto group, C1-6Alkylamino radical, di (C)1-6Alkyl) amino, cyano.
The invention provides a compound shown as a formula (II),
wherein R is1、R2、Ra、Rb、Cy2M, n, A are as defined for formula I; x is selected from (CR)cRd)oWherein is optionally CRcRdMay be substituted by O or NReReplacing; y is selected from CRfOr N; wherein R isc、Rd、Re、RfEach independently selected from hydrogen or C1-6An alkyl group; o is selected from 0, 1, 2,3, 4, 5.
The invention provides a compound shown as a formula (III),
wherein W and Q are selected from CRcRdOr NReOr O; A. r1、R2、Ra、Rb、Rc、 Rd、Re、Cy2M, n and Y are as defined above for formula (II).
The invention also provides a compound shown as a formula (IV),
wherein R is1、R2、Ra、Rb、Cy2M, n, X, Y, A are as defined above for formula (II); z is selected from (CR)g)pWherein CR is arbitrarygMay be replaced by N; rgEach independently selected from hydrogen or C1-6An alkyl group; p is selected from 0, 1, 2,3, 4, 5.
In the preferred technical scheme of the invention, the structure of the compound has the formula (V):
wherein R is1、R2、Rc、Rd、Rf、Cy2M and A are as defined above for formula (II).
In the preferred technical scheme of the invention, the structure of the compound has the formula (VI):
wherein R is1、R2、Rc、Rd、Cy2M and A are as defined above for formula (II).
In a preferred embodiment of the present invention, the compound has a structure represented by formula (VII):
wherein R is1、R2、Re、Rf、Cy2M and A are as defined above for formula (II).
In a preferred technical scheme of the invention, the compound has a structure as shown in a formula (VIII):
wherein R is1、R2、Rc、Rd、Rf、Cy2M and A are as defined above for formula (II).
In a preferred technical scheme of the invention, the compound has a structure as shown in a formula (IX):
wherein R is1、R2、Rc、Rd、Re、Rf、Cy2M and A are as defined above for formula (II).
In the technical scheme of the invention, the air conditioner is provided with a fan,
is shown to,
Or
In the technical scheme of the invention, the air conditioner is provided with a fan,
preferably, it is
In the technical scheme of the invention, A is selected from-C (O) -or S (O)2-。
The invention also provides a preparation method of the compound with the structure of the formula (X):
in the route I reaction:
wherein the base used in step (1) is selected from inorganic bases or organic bases, including but not limited to: sodium hydride, calcium hydride, sodium amide, sodium methoxide, sodium ethoxide, potassium hydroxide, sodium hydroxide, lithium aluminum hydride, tert-butyl lithium, tert-butyl potassium, potassium tert-butoxide, lithium diisopropylamide, barium hydroxide, or any combination thereof;
wherein the organic solvent used in step (1) includes but is not limited to: 1, 4-dioxane, N-dimethylformamide, dichloromethane, chloroform, DMSO, DMF, THF, acetone, methanol, ethanol, or any combination thereof;
wherein the ylide used in step (2) is selected from the group consisting of a sulfur ylide and a phosphorus ylide;
wherein the Grignard reagent used in step (3) is selected from CH3MgCl、CH3MgBr、 C2H5MgCl、C2H5MgBr、i-PrMgCl、i-PrMgBr,PhCH2MgCl、 PhCH2MgBr, or any combination thereof.
The invention also provides a preparation method of the compound with the structure of the formula (XI):
in the route II reaction:
wherein the catalyst used in step (1) is selected from methyl titanate, ethyl titanate, n-propyl titanate, isopropyl titanate, butyl titanate or any combination thereof;
wherein the base used in step (2) is selected from inorganic bases or organic bases, including but not limited to: sodium hydride, calcium hydride, sodium amide, sodium methoxide, sodium ethoxide, potassium hydroxide, sodium hydroxide, lithium aluminum hydride, tert-butyl lithium, tert-butyl potassium, potassium tert-butoxide, lithium diisopropylamide, barium hydroxide, or any combination thereof;
wherein the organic solvent used in step (2) includes but is not limited to: 1, 4-dioxane, N-dimethylformamide, dichloromethane, chloroform, DMSO, DMF, THF, acetone, methanol, ethanol, or any combination thereof;
wherein the ylide used in step (2) is selected from the group consisting of a sulfur ylide and a phosphorus ylide;
wherein the hydrolysis of step (3) is carried out under acidic conditions, said acid being selected from, but not limited to, hydrochloric acid, sulfuric acid, hydrobromic acid, oxalic acid, citric acid, formic acid, acetic acid or any combination thereof;
wherein the organic solvent used in step (4) includes but is not limited to: 1, 4-dioxane, N-dimethylformamide, dichloromethane, chloroform, DMSO, DMF, THF, acetone, methanol, ethanol, or any combination thereof.
The invention also provides a preparation method of the compound with the structure of the formula (XII):
in path III:
wherein the catalyst used in step (1) is selected from methyl titanate, ethyl titanate, n-propyl titanate, isopropyl titanate, butyl titanate or any combination thereof;
wherein the step (2) is carried out under the action of non-nucleophilic strong base selected from the group consisting of but not limited to lithium diisopropylamide, lithium diethylamide, lithium isopropylcyclohexylamide, lithium dicyclohexylamide, lithium 2,2,6, 6-tetramethylpiperidino and lithium hexamethyldisilazide;
wherein the hydrolysis reaction of step (3) is carried out under acidic conditions, and the acid is selected from, but not limited to, hydrochloric acid, sulfuric acid, hydrobromic acid, oxalic acid, citric acid, formic acid, acetic acid, or any combination thereof;
wherein the Grignard reagent used in step (4) is selected from CH3MgCl、CH3MgBr、 C2H5MgCl、C2H5MgBr、i-PrMgCl、i-PrMgBr,PhCH2MgCl、 PhCH2MgBr or any combination thereof;
wherein, when the alkylation reaction in the step (5) is performed, the alkylation reaction reagent is selected from halogenated alkyl, the reaction is performed under the condition of Lewis acid as a catalyst, and the Lewis acid is preferably AlCl3、FeCl2、CuCl2。
The invention also provides a preparation method of the compound with the structure of the formula (XIII):
in path IV
Wherein the base used in step (1) and step (3) is selected from inorganic bases or organic bases, including but not limited to: sodium hydride, calcium hydride, sodium amide, sodium methoxide, sodium ethoxide, potassium hydroxide, sodium hydroxide, lithium aluminum hydride, tert-butyl lithium, tert-butyl potassium, potassium tert-butoxide, lithium diisopropylamide, barium hydroxide, or any combination thereof;
wherein the organic solvent used in steps (1) to (3) includes but is not limited to: 1, 4-dioxane, N-dimethylformamide, dichloromethane, chloroform, DMSO, DMF, THF, acetone, methanol, ethanol, or any combination thereof;
wherein the oxidant used in step (2) is selected from but not limited to m-chloroperoxybenzoic acid, CrO3、KMnO4、MnO2、NaCr2O7、HIO4、PbAc4、OsO4Hydrogen peroxide or any combination thereof;
wherein the hydrolysis reaction of step (4) is carried out under acidic conditions, and the acid is selected from, but not limited to, hydrochloric acid, sulfuric acid, hydrobromic acid, oxalic acid, citric acid, formic acid, acetic acid, or any combination thereof;
wherein the Grignard reagent used in step (5) is selected from CH3MgCl、CH3MgBr、 C2H5MgCl、C2H5MgBr、i-PrMgCl、i-PrMgBr,PhCH2MgCl、 PhCH2MgBr, or any combination thereof.
The invention also provides a preparation method of the compound with the structure of the formula (XIV):
in path V:
wherein step (1) is carried out in the presence of an alkali metal fluoride selected from, but not limited to, LiF, Na, or an alkaline earth metal fluorideF、KF、MgF2、 CaF2;
Wherein the reduction reaction in the step (2) can be palladium carbon catalytic hydrogenation reduction or Na/liquid ammonia reduction;
wherein the Grignard reagent used in step (3) is selected from CH3MgCl、CH3MgBr、 C2H5MgCl、C2H5MgBr、i-PrMgCl、i-PrMgBr,PhCH2MgCl、 PhCH2MgBr or any combination thereof;
wherein, when the alkylation reaction in the step (4) is carried out, the alkylation reaction reagent is selected from halogenated alkyl, the reaction is carried out under the condition of Lewis acid as a catalyst, and the Lewis acid is preferably AlCl3、FeCl2、CuCl2。
Detailed Description
The present invention relates to a method of inhibiting indoleamine 2, 3-dioxygenase comprising exposing a compound or pharmaceutical composition described herein to an indoleamine 2, 3-dioxygenase.
Design and reaction examples
The compound of the present invention can be synthesized by known procedures with reference to the following descriptions. All solvents and reagents purchased were used directly without treatment. All synthesized compounds can be analytically validated by, but not limited to, the following methods: LCMS (liquid chromatography mass spectrometry) and NMR (nuclear magnetic resonance). Nuclear Magnetic Resonance (NMR) was measured by Bruker AVANCE-500 NMR spectrometer using deuterated dimethyl sulfoxide (d)6-DMSO), deuterated chloroform (CDCl)3) Tetramethylsilane (TMS) was used as an internal standard. The following abbreviations represent various types of split peaks: singlet(s), doublet (d), triplet (t), multiplet (m), broad (br). Mass Spectrometry (MS) was measured using a Thermo Fisher-MSQ Plus LC Mass spectrometer. General synthetic analysis and examples are described below:
example 1
N- (4-chlorophenylyl) -6- (6-fluoroquinolin-4-yl) spiro [2.5] octane-1-carboxamide N- (4-chlorophenyl) -6- (6-fluoroquinolin-4-yl) spiro [2.5] octane-1-carboxamide
The first step is as follows: 1, 4-cyclohexanedione monoethylene glycol ketal (10.0g,64.03mmol) was dissolved in 250 mL of methyl tert-butyl ether and N-phenylbis (trifluoromethanesulfonyl) imide (22.9g,64.03 mmol) was added. The reaction mixture was cooled to-78 ℃ and sodium bis (trimethylsilyl) amide (2mol/L tetrahydrofuran solution) (32mL,64.03mmol) was added dropwise to the reaction mixture under a nitrogen atmosphere. After the completion of the dropwise addition, the reaction mixture was stirred at that temperature for 60 minutes, then allowed to warm to room temperature and stirred overnight until the reaction material was completely consumed by TLC. The reaction solution was quenched with 3mL of an aqueous potassium hydrogen sulfate solution, filtered to remove solids, and the filtrate was concentrated. To the residue was added 3mL of methyl t-butyl ether, and the organic layer was washed three times with 45mL of sodium hydroxide (5%) solution and once with 50mL of saturated brine. The organic layer was dried over anhydrous sodium sulfate, filtered, and concentrated to give compound 1a (17.23g) as an orange oily liquid in 93% yield.1H NMR(500MHz,CDCl3) 5.66(J=4.0Hz,1H),4.01–3.96(m,4H),2.56–2.52(m,2H),2.42–2.40 (m,2H),1.90(t,J=6.5Hz,2H).
The second step is that: compound 1a (13g,45.1mmol) was dissolved in 100mL dioxane, and pinacol diboron (14.9g,58.64mmol), potassium acetate (13.3g,135.3mmol) and Pd (dppf) Cl were added sequentially2(1.65g,2.26 mmol). The reaction mixture was refluxed overnight under nitrogen atmosphere. Then the reaction solvent dioxane was evaporated to dryness, ethyl acetate was added, celite was filtered, the filtrate was concentrated and then separated by flash column chromatography to give compound 1b (7.6g) as a pale yellow solid in 63% yield.1HNMR(500MHz,CDCl3)6.48–6.45(m,1H),3.98(s,4H), 2.40–2.34(m,4H),1.73(t,J=6.5Hz,2H),1.25(s,12H).
The third step: compound 1b (5.7g,21.48mmol) was dissolved in 60mL/15mL dioxane/water and 4-chloro-6-fluoroquinoline (3.0g,16.53mmol), potassium carbonate (6.8g,49.56 mmol) and Pd (PPh3)4(954mg,0.83mmol) were added sequentially. The reaction mixture was refluxed overnight under nitrogen atmosphere. Then concentrating the reaction solution, extracting with ethyl acetate, and concentrating the organic phaseAfter the condensation, compound 1c (2.42g) was isolated by flash column chromatography as a pale yellow liquid in 51% yield. MS (ESI) M/z 286.1(M + H)+.1H NMR(500MHz,CDCl3)8.81(d,J=4.5 Hz,1H),8.15(dd,J=9.0,5.5Hz,1H),7.65(dd,J=10.0,2.5Hz,1H), 7.49(td,J=9.0,2.5Hz,1H),7.26(d,J=4.5Hz,1H),5.77(t,J=3.5Hz, 1H),4.08–40.6(m,4H),2.65–2.60(m,2H),2.56–2.53(m,2H),2.00(t,J=6.5Hz,2H).
The fourth step: compound 1c (2.42g,8.49mmol) was dissolved in 45mL of isopropanol and 10% palladium on carbon (300mg) was added. The reaction mixture was heated to 55 ℃ under hydrogen atmosphere for overnight reaction. The palladium on carbon was then filtered off with celite and the filtrate was concentrated to give crude compound 1d (2.04g) as a slurry in 84% yield, which was used directly in the next reaction. MS (ESI) M/z288.1(M + H)+.
The fifth step: compound 1d (2.04g,7.11mmol) was dissolved in 36mL of acetone, and 9mL of 4mol/L hydrochloric acid was added. The reaction mixture was heated to 45 ℃ and reacted overnight. The solvent was then evaporated off, the aqueous solution was neutralized with 6mol/L sodium hydroxide to pH 9 and the aqueous phase was extracted with ethyl acetate. The organic phases were combined and washed with saturated brine, dried over anhydrous sodium sulfate, filtered and concentrated. The residue was isolated by flash column chromatography to give compound 1e (1.17g) as a pale yellow solid in 67% yield. MS (ESI) M/z 244.3(M + H)+.1H NMR(500MHz,CDCl3)8.85(d,J=4.5 Hz,1H),8.22(dd,J=9.0,5.5Hz,1H),7.74(dd,J=10.0,2.5Hz,1H), 7.57–7.50(m,1H),7.33(d,J=4.5Hz,1H),3.74–3.66(m,1H), 2.72–2.58(m,4H),2.41–2.34(m,2H),2.11–2.00(m,2H).
And a sixth step: triethyl phosphonoacetate (968mg,4.32mmol) was dissolved in 16mL of ultra dry tetrahydrofuran and sodium tert-butoxide (415mg,4.32mmol) was added at 0 ℃ in an ice bath. After 10 minutes, a solution of compound 1e (1g,4.12mmol) in tetrahydrofuran (4mL) was added to the reaction. After 2 hours of reaction, quench with water. The aqueous solution was extracted three times with 20mL of ethyl acetate, the organic phases were combined, washed with 20mL of saturated brine, dried over anhydrous sodium sulfate, filtered and concentrated. The residue was isolated by flash column chromatography to give compound 1f (1.18g) as a white solid in 92% yield. MS (ESI) M/z 314.0(M + H)+.1H NMR(500MHz,CDCl3)8.81 (d,J=4.5Hz,1H),8.17(dd,J=9.0,5.5Hz,1H),7.72(dd,J=10.0,2.5 Hz,1H),7.53–7.47(m,1H),7.28(d,J=4.5Hz,1H),5.75(s,1H),4.19 (q,J=7.0Hz,2H),3.52–3.42(m,1H),2.54–2.48(m,2H),2.26–2.11(m, 4H),1.80–1.68(m,2H),1.30(t,J=7.0Hz,3H).
The seventh step: NaH (383mg,9.57mmol) was added to 15mL of dimethyl sulfoxide, and trimethyl sulfoxide iodide (2.11g,9.57mmol) was added to the suspension. The mixture was stirred at room temperature for 1.5 hours. Then, a solution of compound 1f (1.0g,3.19mmol) in dimethyl sulfoxide (5mL) was added to the reaction solution. The reaction was stirred at room temperature overnight. Then quenched with water, extracted with ethyl acetate, and isolated by flash column chromatography to give 1g (820mg) of the compound as a colorless oily liquid in 78% yield. MS (ESI) M/z 328.1(M + H)+.1HNMR(500MHz, CDCl3)8.83(d,J=4.5Hz,1H),8.24(dd,J=9.0,5.5Hz,1H),7.71 (dd,J=10.0,2.5Hz,1H),7.55–7.49(m,1H),7.35(d,J=4.5Hz,1H), 4.19(q,J=7.0Hz,2H),3.32–3.24(m,1H),2.17(td,J=13.0,3.5Hz, 1H),2.07–1.90(m,4H),1.87–1.78(m,1H),1.58(dd,J=8.0,5.5Hz, 1H),1.46–1.37(m,1H),1.30(t,J=7.0Hz,3H),1.28–1.24(m,2H), 1.16–1.11(m,1H),1.00(dd,J=8.0,4.5Hz,1H).
Eighth step: 4-chloroaniline (94mg,0.73mmol) was dissolved in 5mL tetrahydrofuran and a 2mol/L solution of isopropyl magnesium chloride in tetrahydrofuran (0.4mL,0.73 mmol) was added at 0 deg.C in an ice bath. The mixture was stirred at room temperature for 5 minutes, and a solution of compound 1g (60mg,0.18mmol) in tetrahydrofuran (2mL) was added to the mixture. The reaction was stirred at room temperature overnight, then quenched with saturated ammonium chloride solution and the aqueous phase extracted with ethyl acetate. The combined organic phases were dried over anhydrous sodium sulfate, filtered and concentrated. The residue was purified by reverse phase preparative chromatography to give compound 1(16.04mg) as a white solid in 21% yield. MS (ESI) M/z 408.9(M + H)+.1H NMR(500MHz,d6-DMSO)10.37(s,1H),8.81(s,1H),8.11–8.05(m, 1H),8.02(d,J=11.0Hz,1H),7.69–7.63(m,3H),7.38–7.31(m,3H), 3.48–3.40(m,1H),2.20(t,J=12.0Hz,1H),1.97–1.84(m,4H),1.78(d, J=12.5Hz,1H),1.72(t,J=6.5Hz,1H),1.35–1.26(m,1H),1.17–1.08 (m,2H),0.96–0.90(m,1H).
Example 2
N-(4-fluorophenyl)-6-(6-fluoroquinolin-4-yl)spiro[2.5]octane-1-carboxamide
N- (4-fluorophenyl) -6- (6-fluoroquinolin-4-yl) spiro [2.5] octane-1-carboxamide
Synthesis of Compound 2 Compound 1 was synthesized by substituting 4-fluoroaniline for 4-chloroaniline in example 1. Compound 2(16.97mg) was obtained as a white solid in 22% yield. MS (ESI) M/z 393.3 (M + H)+.1H NMR(500MHz,d6-DMSO)10.28(s,1H),8.81(s,1H), 8.10–8.05(m,1H),8.03(d,J=11.0Hz,1H),7.69–7.60(m,3H),7.37(s, 1H),7.13(t,J=8.0Hz,2H),3.49–3.40(m,1H),2.20(t,J=12.0Hz,1H), 1.98–1.85(m,4H),1.78(d,J=11.0Hz,1H),1.72(t,J=6.5Hz,1H), 1.37–1.28(m,1H),1.17–1.07(m,2H),0.94–0.89(m,1H).
Example 3
N-(4-chlorobenzyl)-6-(6-fluoroquinolin-4-yl)spiro[2.5]octane-1-carboxamide
N- (4-chlorobenzyl) -6- (6-fluoroquinolin-4-yl) spiro [2.5] octane-1-carboxamide
Synthesis of compound 3 starting from intermediate 1g in example 1, prepared via the following steps:
the first step is as follows: compound 1g (200mg,0.61mmol) was dissolved in 10mL of ethanol, and 4mL of 2mol/L sodium hydroxide solution was added. The reaction solution was heated to 50 ℃ and reacted for 2 hours. After the reaction solution was cooled to room temperature, it was neutralized with a 4mol/L hydrochloric acid solution to pH 1. The aqueous phase was extracted with ethyl acetate. The combined organic phases were dried over anhydrous sodium sulfate, filtered and concentrated. The residue was separated by preparative thin layer chromatography to give Compound 3a (150mg), whiteA colored solid, yield 83%. MS (ESI) M/z 300.0(M + H)+.1H NMR(500MHz,d6-DMSO)12.02(br, 1H),8.83(d,J=4.5Hz,1H),8.10(dd,J=9.0,5.5Hz,1H),8.03(dd,J= 10.0,2.5Hz,1H),7.71–7.64(m,1H),7.38(d,J=4.5Hz,1H),3.48–3.41 (m,1H),2.21–2.13(m,1H),2.01–1.80(m,4H),1.75–1.65(m,1H),1.51 (dd,J=8.0,5.5Hz,1H),1.38–1.32(m,1H),1.11–1.05(m,1H), 1.04–0.99(m,1H),0.95(dd,J=7.5,4.0Hz,1H).
The second step is that: compound 3a (40mg,0.13mmol) was dissolved in 5mLN, N-dimethylformamide, diisopropylethylamine (52mg,0.39mmol) and HATU (61mg,0.16 mmol) were added in this order, and the mixture was stirred at room temperature for 30 minutes. 4-chlorobenzylamine (57mg,0.39mmol) was then added, and the reaction was stirred at room temperature for an additional 2 hours. The reaction solution was quenched by adding 20mL of water, and the aqueous phase was extracted with ethyl acetate. The combined organic phases were dried over anhydrous sodium sulfate, filtered and concentrated. The residue was purified by reverse phase preparative chromatography to give compound 3(6.34mg) as a white solid in 11% yield. MS (ESI) M/z 423.4(M + H)+.1H NMR(500MHz,d6-DMSO) 8.81(s,1H),8.68(s,1H),8.12–8.07(m,1H),8.00(d,J=11.0Hz,1H), 7.67(t,J=8.5Hz,1H),7.36–7.31(m,4H),7.14(s,1H),4.44(dd,J=15.0,6.5Hz,1H),4.18(dd,J=15.0Hz,5.0Hz,1H),3.42–3.34(m,1H), 2.13(t,J=12.5Hz,1H),1.86–1.74(m,4H),1.63(d,J=12.5Hz, 1H),1.54–1.48(m,1H),1.25–1.15(m,1H),1.08–0.96(m,2H),0.82–0.76(m,1H).
Example 4
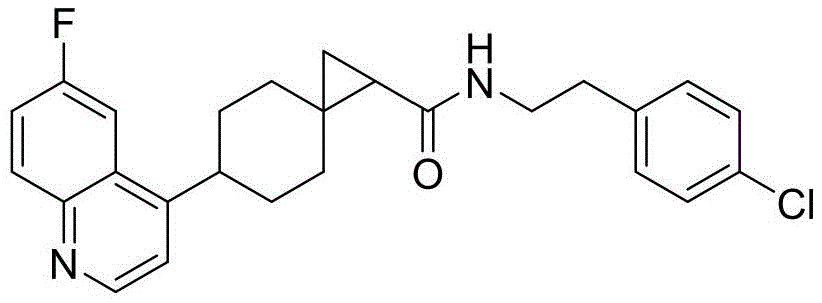
N-(4-chlorophenethyl)-6-(6-fluoroquinolin-4-yl)spiro[2.5]octane-1-carboxamide
N- (4-Chlorobenzoethyl) -6- (6-fluoroquinolin-4-yl) spiro [2.5] octane-1-carboxamide
Synthesis of Compound 4 As compound 3, 4-chlorophenylethylamine was used in place of 4-chlorobenzylamine in example 3. Compound 4(17.15mg) was obtained as a white solid in 29% yield. MS (ESI) M/z437.4(M + H)+.1HNMR(500MHz,d6-DMSO)8.84(d,J=4.5Hz,1H), 8.19(t,J=5.5Hz,1H),8.12–8.07(m,1H),7.99(d,J=11.0Hz,1H), 7.67(t,J=8.5Hz,1H),7.27(d,J=4.1Hz,1H),7.25–7.21(m,4H), 3.49–3.42(m,1H),3.30–3.22(m,2H),2.74(t,J=6.5Hz,2H),2.11(t,J =12.5Hz,1H),1.85–1.75(m,4H),1.67(d,J=12.5Hz,1H),1.46–1.41 (m,1H),1.26–1.14(m,1H),1.03–0.95(m,2H),0.75–0.70(m,1H).
Example 5
6-(6-fluoroquinolin-4-yl)-N-(4-(trifluoromethyl)phenyl)spiro[2.5]octane-1-carboxamide
6- (6-Fluoroquinolin-4-yl) -N- (4- (trifluoromethyl) phenyl) spiro [2.5] octane-1-carboxamide
Synthesis of compound 5 starting from intermediate 3a in example 3, was prepared via the following steps:
compound 3a (40mg,0.13mmol) was dissolved in 5mL of ethyl acetate, and pyridine (32mg,0.39mmol) and tripropyl phosphoric anhydride (127mg,0.33mmol) were added in this order, followed by stirring at room temperature for 10 minutes. 4-Trifluoromethylaniline (65mg,0.39mmol) was then added and the reaction was continued to stir at room temperature overnight. 2mL of 2mol/L sodium hydroxide solution was added to the reaction solution, followed by addition of 20mL of water for dilution, and the aqueous phase was extracted with ethyl acetate. The combined organic phases were dried over anhydrous sodium sulfate, filtered and concentrated. The residue was purified by reverse phase preparative chromatography to give compound 5(2.65 mg) as a white solid in 5% yield. MS (ESI) M/z437.4(M + H)+.1HNMR(500 MHz,d6-DMSO)10.63(s,1H),8.80(d,J=4.5Hz,1H),8.10–8.05(m, 1H),8.03(d,J=11.0Hz,1H),7.84(d,J=8.0Hz,2H),7.69–7.63(m, 3H),7.36(s,1H),3.48–3.41(m,1H),2.25–2.17(m,1H),1.99–1.85(m, 4H),1.80–1.74(m,2H),1.35–1.25(m,1H),1.21–1.09(m,2H), 0.99–0.95(m,1H).
Example 6
N-(4-cyanophenyl)-6-(6-fluoroquinolin-4-yl)spiro[2.5]octane-1-carboxamide
N- (4-cyanophenyl) -6- (6-fluoroquinolin-4-yl) spiro [2.5] octane-1-carboxamide
Synthesis of Compound 6 Compound 5 was prepared by substituting 4-aminobenzonitrile for 4-trifluoromethylaniline in example 5. Compound 6(28.36mg) was obtained as a white solid in 53% yield. MS (ESI) M/z 400.4(M + H)+.1H NMR(500MHz,d6-DMSO)10.70(s,1H),8.80 (d,J=4.5Hz,1H),8.10–8.06(m,1H),8.03(d,J=11.0Hz,1H),7.82(d, J=8.5Hz,2H),7.75(d,J=8.5Hz,2H),7.66(t,J=9.0Hz,1H),7.35(d, J=3.5Hz,1H),3.48–3.40(m,1H),2.25–2.17(m,1H),1.99–1.83(m, 4H),1.81–1.74(m,2H),1.33–1.23(m,1H),1.21–1.10(m,2H), 1.02–0.96(m,1H).
Example 7
N-(6-chloropyridin-3-yl)-6-(6-fluoroquinolin-4-yl)spiro[2.5]octane-1-c arboxamide
N- (6-Chloropyridin-3-yl) -6- (6-fluoroquinolin-4-yl) spiro [2.5] octane-1-carboxamide
Synthesis of Compound 7 Compound 5 was synthesized by substituting 3-amino-6-chloropyridine for 4-trifluoromethylaniline in example 5. Compound 7(7.07mg) was obtained as a white solid in 13% yield. MS (ESI) M/z 410.4(M + H)+.1H NMR(500MHz,d6-DMSO)10.62(s, 1H),8.81(s,1H),8.63(s,1H),8.15–8.06(m,2H),8.03(d,J=11.0Hz, 1H),7.66(t,J=8.5Hz,1H),7.45(d,J=8.0Hz,1H),7.38(s,1H), 3.48–3.41(m,1H),2.25–2.16(m,1H),1.99–1.82(m,4H),1.82–1.72(m, 2H),1.35–1.26(m,1H),1.20–1.09(m,2H),1.00–0.95(m,1H).
Example 8
N-(5-chloropyridin-2-yl)-6-(6-fluoroquinolin-4-yl)spiro[2.5]octane-1-c arboxamide
N- (5-Chloropyridin-2-yl) -6- (6-fluoroquinolin-4-yl) spiro [2.5] octane-1-carboxamide
Synthesis of Compound 8 Compound 5 was synthesized by substituting 2-amino-5-chloropyridine for 4-trifluoromethylaniline in example 5. Compound 8(19.54mg) was obtained as a white solid in 36% yield. MS (ESI) M/z 410.4(M + H)+.1H NMR(500MHz,d6-DMSO)11.04(s, 1H),8.80(s,1H),8.38(s,1H),8.13(d,J=8.5Hz,1H),8.07(t,J=8.0 Hz,1H),8.02(d,J=11.0Hz,1H),7.85(d,J=9.0Hz,1H),7.66(t,J= 8.0Hz,1H),7.34(s,1H),3.47–3.40(m,1H),2.19(t,J=12.5Hz,1H), 2.02–1.84(m,5H),1.78(d,J=11.0Hz,1H),1.33–1.22(m,1H), 1.21–1.15(m,1H),1.08(d,J=12.5Hz,1H),0.98–0.92(m,1H).
Example 9
N-(4-chlorophenyl)-6-(6-fluoro-7-methylquinolin-4-yl)spiro[2.5]octane-1-carboxamide
N- (4-chlorophenyl) -6- (6-fluoro-7-methylquinolin-4-yl) -spiro [2.5] octane-1-carboxamide
Synthesis of compound 9 starting from intermediate 1g in example 1, was prepared via the following steps:
the first step is as follows: n-butyllithium (2.5M,0.5mL,1.22mmol) was added dropwise to a solution of diisopropylamine (123mg,1.22mmol) in tetrahydrofuran (5mL) at-78 ℃. A solution of compound (1g, 200mg,0.61mmol) in tetrahydrofuran (2mL) was added dropwise thereto. The reaction was stirred at-78 ℃ for 1 hour. Then, a solution of iodomethane (173mg,1.22mmol) in tetrahydrofuran (2mL) was added dropwise to the reaction mixture, and the reaction was maintained at-78 ℃ for half an hour, then warmed to room temperature, and stirred overnight. Quench with saturated ammonium chloride solution and extract the aqueous phase with ethyl acetate. The organic phases are combined and washed with waterDried over sodium sulfate, filtered, and concentrated. The residue was separated by preparative thin layer chromatography to give compound 9a (24mg) as a colorless oily liquid in a yield of 12%. MS (ESI) M/z 342.4 (M + H)+.
The second step is that: 4-chloroaniline (36mg,0.28mmol) was dissolved in tetrahydrofuran (2mL) and a 2.0mol/L solution of isopropyl magnesium chloride in tetrahydrofuran (0.2mL, 0.28mmol) was added at 0 deg.C in an ice bath. The mixture was stirred at room temperature for 5 minutes, and a solution of compound 9a (24mg,0.07 mmol) in tetrahydrofuran (1mL) was added to the mixture. The reaction was stirred at room temperature overnight, then quenched with saturated ammonium chloride solution and the aqueous phase extracted with ethyl acetate. The combined organic phases were dried over anhydrous sodium sulfate, filtered and concentrated. The residue was purified by reverse phase preparative chromatography to give compound 9(12.01mg) as a white solid in 41% yield. MS (ESI) M/z 423.4(M + H)+.1H NMR(500MHz,d6-DMSO)10.37(s,1H),8.75(s,1H), 7.99–7.91(m,2H),7.80–7.56(m,3H),7.34(d,J=8.0Hz,2H),7.28 (s,1H),3.44–3.37(m,1H),2.44(s,3H),2.23–2.14(m,1H), 1.98–1.84(m,4H),1.78–1.69(m,2H),1.17–1.07(m,2H),0.95–0.92 (m,1H)。