CN112079779B - Synthetic method of pentazocine - Google Patents
Synthetic method of pentazocine Download PDFInfo
- Publication number
- CN112079779B CN112079779B CN202010848205.0A CN202010848205A CN112079779B CN 112079779 B CN112079779 B CN 112079779B CN 202010848205 A CN202010848205 A CN 202010848205A CN 112079779 B CN112079779 B CN 112079779B
- Authority
- CN
- China
- Prior art keywords
- pentazocine
- bicarbonate
- compound
- quaternary ammonium
- synthesizing
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- VOKSWYLNZZRQPF-GDIGMMSISA-N pentazocine Chemical compound C1C2=CC=C(O)C=C2[C@@]2(C)[C@@H](C)[C@@H]1N(CC=C(C)C)CC2 VOKSWYLNZZRQPF-GDIGMMSISA-N 0.000 title claims abstract description 93
- 229960005301 pentazocine Drugs 0.000 title claims abstract description 92
- 238000010189 synthetic method Methods 0.000 title description 2
- 238000006243 chemical reaction Methods 0.000 claims abstract description 25
- 238000000034 method Methods 0.000 claims abstract description 21
- 238000001308 synthesis method Methods 0.000 claims abstract description 18
- 238000003756 stirring Methods 0.000 claims abstract description 17
- BVKZGUZCCUSVTD-UHFFFAOYSA-M Bicarbonate Chemical compound OC([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-M 0.000 claims abstract description 14
- 230000015572 biosynthetic process Effects 0.000 claims abstract description 14
- 238000003786 synthesis reaction Methods 0.000 claims abstract description 14
- 239000002253 acid Substances 0.000 claims abstract description 11
- -1 quaternary ammonium ions Chemical class 0.000 claims abstract description 10
- 239000011230 binding agent Substances 0.000 claims abstract description 9
- 230000008569 process Effects 0.000 claims abstract description 6
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-dimethylformamide Substances CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 claims description 82
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 claims description 27
- 238000001816 cooling Methods 0.000 claims description 15
- 239000002904 solvent Substances 0.000 claims description 13
- JMMWKPVZQRWMSS-UHFFFAOYSA-N isopropanol acetate Natural products CC(C)OC(C)=O JMMWKPVZQRWMSS-UHFFFAOYSA-N 0.000 claims description 9
- 229940011051 isopropyl acetate Drugs 0.000 claims description 9
- GWYFCOCPABKNJV-UHFFFAOYSA-N isovaleric acid Chemical compound CC(C)CC(O)=O GWYFCOCPABKNJV-UHFFFAOYSA-N 0.000 claims description 9
- 239000000047 product Substances 0.000 claims description 9
- 125000001453 quaternary ammonium group Chemical group 0.000 claims description 9
- 229940125904 compound 1 Drugs 0.000 claims description 8
- 229940125782 compound 2 Drugs 0.000 claims description 8
- 239000005457 ice water Substances 0.000 claims description 8
- 230000002194 synthesizing effect Effects 0.000 claims description 8
- VFHDWENBWYCAIB-UHFFFAOYSA-M hydrogen carbonate;tetramethylazanium Chemical compound OC([O-])=O.C[N+](C)(C)C VFHDWENBWYCAIB-UHFFFAOYSA-M 0.000 claims description 7
- 239000012043 crude product Substances 0.000 claims description 5
- 238000004042 decolorization Methods 0.000 claims description 3
- XUBMPLUQNSSFHO-UHFFFAOYSA-M hydrogen carbonate;tetraethylazanium Chemical compound OC([O-])=O.CC[N+](CC)(CC)CC XUBMPLUQNSSFHO-UHFFFAOYSA-M 0.000 claims description 3
- BVKZGUZCCUSVTD-UHFFFAOYSA-N carbonic acid Chemical class OC(O)=O BVKZGUZCCUSVTD-UHFFFAOYSA-N 0.000 claims description 2
- 238000007670 refining Methods 0.000 claims description 2
- 125000000217 alkyl group Chemical group 0.000 claims 1
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 abstract description 24
- 238000002347 injection Methods 0.000 abstract description 19
- 239000007924 injection Substances 0.000 abstract description 19
- 238000010438 heat treatment Methods 0.000 abstract description 12
- 235000017557 sodium bicarbonate Nutrition 0.000 abstract description 12
- 229910000030 sodium bicarbonate Inorganic materials 0.000 abstract description 12
- 238000004090 dissolution Methods 0.000 abstract description 2
- 230000035484 reaction time Effects 0.000 abstract description 2
- BZLVMXJERCGZMT-UHFFFAOYSA-N Methyl tert-butyl ether Chemical compound COC(C)(C)C BZLVMXJERCGZMT-UHFFFAOYSA-N 0.000 description 26
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 18
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 14
- LOYZVRIHVZEDMW-UHFFFAOYSA-N 1-bromo-3-methylbut-2-ene Chemical compound CC(C)=CCBr LOYZVRIHVZEDMW-UHFFFAOYSA-N 0.000 description 8
- JVTAAEKCZFNVCJ-UHFFFAOYSA-N lactic acid Chemical compound CC(O)C(O)=O JVTAAEKCZFNVCJ-UHFFFAOYSA-N 0.000 description 8
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 7
- 230000000052 comparative effect Effects 0.000 description 7
- 239000007787 solid Substances 0.000 description 7
- 230000036592 analgesia Effects 0.000 description 6
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Chemical compound O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 6
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 5
- 230000000202 analgesic effect Effects 0.000 description 5
- 230000002980 postoperative effect Effects 0.000 description 5
- 206010002091 Anaesthesia Diseases 0.000 description 4
- 230000037005 anaesthesia Effects 0.000 description 4
- 238000001035 drying Methods 0.000 description 4
- 239000004310 lactic acid Substances 0.000 description 4
- 235000014655 lactic acid Nutrition 0.000 description 4
- 238000002156 mixing Methods 0.000 description 4
- BQJCRHHNABKAKU-KBQPJGBKSA-N morphine Chemical compound O([C@H]1[C@H](C=C[C@H]23)O)C4=C5[C@@]12CCN(C)[C@@H]3CC5=CC=C4O BQJCRHHNABKAKU-KBQPJGBKSA-N 0.000 description 4
- 238000005406 washing Methods 0.000 description 4
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 3
- 230000000694 effects Effects 0.000 description 3
- 238000002474 experimental method Methods 0.000 description 3
- 229940124636 opioid drug Drugs 0.000 description 3
- 239000000843 powder Substances 0.000 description 3
- 239000011780 sodium chloride Substances 0.000 description 3
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical class O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 3
- 238000003860 storage Methods 0.000 description 3
- 239000008215 water for injection Substances 0.000 description 3
- 206010067484 Adverse reaction Diseases 0.000 description 2
- 229910000013 Ammonium bicarbonate Inorganic materials 0.000 description 2
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 2
- 208000004454 Hyperalgesia Diseases 0.000 description 2
- 208000035154 Hyperesthesia Diseases 0.000 description 2
- XADCESSVHJOZHK-UHFFFAOYSA-N Meperidine Chemical compound C=1C=CC=CC=1C1(C(=O)OCC)CCN(C)CC1 XADCESSVHJOZHK-UHFFFAOYSA-N 0.000 description 2
- WQDUMFSSJAZKTM-UHFFFAOYSA-N Sodium methoxide Chemical compound [Na+].[O-]C WQDUMFSSJAZKTM-UHFFFAOYSA-N 0.000 description 2
- 230000001154 acute effect Effects 0.000 description 2
- 230000006838 adverse reaction Effects 0.000 description 2
- 235000012538 ammonium bicarbonate Nutrition 0.000 description 2
- 239000001099 ammonium carbonate Substances 0.000 description 2
- 230000009286 beneficial effect Effects 0.000 description 2
- 239000003153 chemical reaction reagent Substances 0.000 description 2
- 239000013078 crystal Substances 0.000 description 2
- 125000000753 cycloalkyl group Chemical group 0.000 description 2
- 238000000354 decomposition reaction Methods 0.000 description 2
- 238000011049 filling Methods 0.000 description 2
- 238000002695 general anesthesia Methods 0.000 description 2
- 238000004128 high performance liquid chromatography Methods 0.000 description 2
- 230000006872 improvement Effects 0.000 description 2
- 238000010255 intramuscular injection Methods 0.000 description 2
- 239000007927 intramuscular injection Substances 0.000 description 2
- 229960005181 morphine Drugs 0.000 description 2
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 2
- 238000002360 preparation method Methods 0.000 description 2
- 238000001953 recrystallisation Methods 0.000 description 2
- 208000001953 Hypotension Diseases 0.000 description 1
- 206010028980 Neoplasm Diseases 0.000 description 1
- 102000003840 Opioid Receptors Human genes 0.000 description 1
- 108090000137 Opioid Receptors Proteins 0.000 description 1
- 229940121954 Opioid receptor agonist Drugs 0.000 description 1
- 229940123257 Opioid receptor antagonist Drugs 0.000 description 1
- ZTVQQQVZCWLTDF-UHFFFAOYSA-N Remifentanil Chemical compound C1CN(CCC(=O)OC)CCC1(C(=O)OC)N(C(=O)CC)C1=CC=CC=C1 ZTVQQQVZCWLTDF-UHFFFAOYSA-N 0.000 description 1
- 208000004756 Respiratory Insufficiency Diseases 0.000 description 1
- 206010038678 Respiratory depression Diseases 0.000 description 1
- UIIMBOGNXHQVGW-DEQYMQKBSA-M Sodium bicarbonate-14C Chemical compound [Na+].O[14C]([O-])=O UIIMBOGNXHQVGW-DEQYMQKBSA-M 0.000 description 1
- 206010047700 Vomiting Diseases 0.000 description 1
- 206010000210 abortion Diseases 0.000 description 1
- 231100000176 abortion Toxicity 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 230000001270 agonistic effect Effects 0.000 description 1
- 230000003321 amplification Effects 0.000 description 1
- 239000003708 ampul Substances 0.000 description 1
- 229940035674 anesthetics Drugs 0.000 description 1
- 230000003042 antagnostic effect Effects 0.000 description 1
- 239000008346 aqueous phase Substances 0.000 description 1
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 1
- 201000011510 cancer Diseases 0.000 description 1
- 230000008859 change Effects 0.000 description 1
- 150000001875 compounds Chemical class 0.000 description 1
- 230000003247 decreasing effect Effects 0.000 description 1
- 206010013663 drug dependence Diseases 0.000 description 1
- 230000008451 emotion Effects 0.000 description 1
- 238000002674 endoscopic surgery Methods 0.000 description 1
- 238000005265 energy consumption Methods 0.000 description 1
- NPUKDXXFDDZOKR-LLVKDONJSA-N etomidate Chemical compound CCOC(=O)C1=CN=CN1[C@H](C)C1=CC=CC=C1 NPUKDXXFDDZOKR-LLVKDONJSA-N 0.000 description 1
- 229960001690 etomidate Drugs 0.000 description 1
- 238000001914 filtration Methods 0.000 description 1
- 238000002575 gastroscopy Methods 0.000 description 1
- 239000003193 general anesthetic agent Substances 0.000 description 1
- 230000000004 hemodynamic effect Effects 0.000 description 1
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 1
- 208000021822 hypotensive Diseases 0.000 description 1
- 230000001077 hypotensive effect Effects 0.000 description 1
- 230000006698 induction Effects 0.000 description 1
- 238000010253 intravenous injection Methods 0.000 description 1
- 238000011031 large-scale manufacturing process Methods 0.000 description 1
- 230000007774 longterm Effects 0.000 description 1
- 238000004519 manufacturing process Methods 0.000 description 1
- 239000000463 material Substances 0.000 description 1
- 239000012528 membrane Substances 0.000 description 1
- 239000000203 mixture Substances 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 230000003472 neutralizing effect Effects 0.000 description 1
- 229910052757 nitrogen Inorganic materials 0.000 description 1
- 238000003199 nucleic acid amplification method Methods 0.000 description 1
- 239000003402 opiate agonist Substances 0.000 description 1
- 239000003401 opiate antagonist Substances 0.000 description 1
- 230000003204 osmotic effect Effects 0.000 description 1
- 239000001301 oxygen Substances 0.000 description 1
- 229910052760 oxygen Inorganic materials 0.000 description 1
- 238000004806 packaging method and process Methods 0.000 description 1
- 238000007911 parenteral administration Methods 0.000 description 1
- 239000008194 pharmaceutical composition Substances 0.000 description 1
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N phenol group Chemical group C1(=CC=CC=C1)O ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 1
- 229910000027 potassium carbonate Inorganic materials 0.000 description 1
- 230000036632 reaction speed Effects 0.000 description 1
- 229960003394 remifentanil Drugs 0.000 description 1
- 230000029058 respiratory gaseous exchange Effects 0.000 description 1
- 230000004044 response Effects 0.000 description 1
- 238000007789 sealing Methods 0.000 description 1
- 239000007858 starting material Substances 0.000 description 1
- 230000001954 sterilising effect Effects 0.000 description 1
- 238000010254 subcutaneous injection Methods 0.000 description 1
- 239000007929 subcutaneous injection Substances 0.000 description 1
- 208000011117 substance-related disease Diseases 0.000 description 1
- 238000006467 substitution reaction Methods 0.000 description 1
- 238000005303 weighing Methods 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D221/00—Heterocyclic compounds containing six-membered rings having one nitrogen atom as the only ring hetero atom, not provided for by groups C07D211/00 - C07D219/00
- C07D221/02—Heterocyclic compounds containing six-membered rings having one nitrogen atom as the only ring hetero atom, not provided for by groups C07D211/00 - C07D219/00 condensed with carbocyclic rings or ring systems
- C07D221/22—Bridged ring systems
- C07D221/26—Benzomorphans
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Other In-Based Heterocyclic Compounds (AREA)
Abstract
The invention provides a synthesis method of pentazocine, in the synthesis process, bicarbonate of quaternary ammonium ions is adopted to replace sodium bicarbonate to be used as an acid-binding agent, acid generated in the reaction is neutralized, heating or stirring in the reaction process is not needed to accelerate the dissolution of the acid-binding agent, and the reaction time can be shortened. The pentazocine synthesized by the synthesis method has high yield and stable yield. The prepared pentazocine injection has stable property and is not easy to decompose.
Description
Technical Field
The invention relates to the technical field of pharmaceutical chemistry, in particular to a synthesis method of pentazocine.
Background
Pentazocine was successfully marketed in 1967 by the wensber group of stirling, uk. The pentazocine injection is also called analgesic, and is an opioid receptor agonistic/antagonistic analgesic. The active component of the product is pentazocine, and the auxiliary materials are sodium chloride and lactic acid. The pentazocine compound etomidate is used for painless gastroscopy of the old, has satisfactory anesthesia effect, stable hemodynamics, less adverse reaction and safe operation, and is one of very reliable anesthesia methods.
Pentazocine is the first clinically applied opioid receptor agonist/antagonist analgesic, and can provide similar analgesic effects to opioid drugs including morphine, dolantin and the like. The parenteral administration has rapid and strong analgesic effect, and the acting time is shorter than that of morphine and dolantin; central depression is mild, especially in respiratory depression and nausea and vomiting, compared to other opioid drugs; there was no hypotensive response; drug dependence is less than other opioid drugs; the emotion is not influenced; the half-life period is moderate, the suitable half-life period is suitable for various operations, and the postoperative effect is rapidly eliminated; can be administered by intramuscular injection, subcutaneous injection, intravenous injection, pump-in, etc.; 30mg of pentazocine (diluted to 5ml by 0.9% normal saline) is given 15-20min before the operation is finished, the intramuscular injection of the pentazocine can effectively relieve the postoperative acute hyperalgesia of remifentanil general anesthesia patients, does not prolong the tracheal catheter removal time after general anesthesia, has no obvious adverse reaction when used for preventing and treating the postoperative acute hyperalgesia, and has no obvious influence on respiration and revival time.
The pentazocine is suitable for analgesia of various surgical anesthetics, and can be used for anesthesia induction, intraoperative anesthesia, postoperative analgesia and the like. Can be used for postoperative analgesia of various operating departments, analgesia of various endoscopic surgeries, analgesia of painless induced abortion, analgesia of cancer patients and the like.
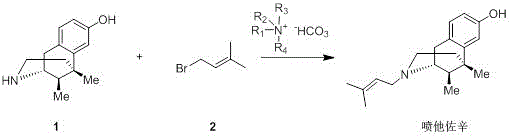
In the prior art, the synthesis method and route of pentazocine are as follows:

wherein, the compound 1 is an important intermediate for synthesizing pentazocine, and reacts with 1-bromo-3-methyl-2-butene (compound 2) to generate the pentazocine. In the reaction, it is usually necessary to add basic sodium bicarbonate in order to neutralize the hydrobromic acid formed in the reaction. Whether hydrobromic acid can react with sodium bicarbonate in sufficient quantity in time is a key point for determining the reaction speed and the reaction completeness. However, it is difficult to achieve this in production because sodium bicarbonate has very low solubility in DMF and even when heated, the solubility is not great. In order to achieve the purpose, measures such as increasing the stirring speed, improving the mixing effect, increasing the solvent amount, increasing the reaction temperature and the like are often adopted, but the effect is still poor after amplification, the reproducibility is poor, the yield is high and low, and the industrial large-scale production is not facilitated.
CN111217749A relates to deuterated pentazocine, its preparation method, pharmaceutical composition and use, wherein sodium bicarbonate is added in step 4 of the synthesis process. Dissolving the starting material 2 in DMF, adding sodium bicarbonate, stirring and heating to 105-110 ℃, and dripping the DMF solvent of the intermediate 1. In the reaction process, in order to dissolve the sodium bicarbonate in the DMF solvent and exert the action of neutralizing the acid of the reaction, the stirring is needed, and the temperature is raised to 105-110 ℃, so that the operation is complicated.
Disclosure of Invention
In view of the above, the present invention aims to provide a synthesis method of pentazocine, so as to solve the problems of poor solubility and low yield of sodium bicarbonate adopted in the synthesis process in the prior art.
In order to achieve the purpose, the technical scheme of the invention is realized as follows:
the synthesis method of pentazocine provided by the invention comprises the following steps:

the pentazocine is generated by the reaction of a compound 1 and a compound 2, and bicarbonate of quaternary ammonium ions is adopted as an acid-binding agent in the synthesis process, wherein the structural formula of the bicarbonate of the quaternary ammonium ions is shown in the specification Wherein R is1、R2、R3And R4Same or different, each independently selected from C1-C10Alkyl radical, C3-C7A cycloalkyl group.
Wherein R is1、R2、R3And R4Same or different, each independently selected from C1-C10Alkyl radical, C3-C7A cycloalkyl group.
Further, the bicarbonate of the quaternary ammonium ion is at least one of tetramethylammonium bicarbonate and tetraethylammonium bicarbonate.
Further, the synthesis method of pentazocine provided by the invention specifically comprises the following steps:
s1, dissolving a compound 1 in a DMF solvent, adding bicarbonate of quaternary ammonium ions, and dropwise adding a compound 2 under the cooling of ice water;
s2, after the compound 2 is dropwise added, controlling the temperature, stirring and cooling to room temperature to obtain a crude product of pentazocine;
s3, refining the crude product of pentazocine to prepare a pure product of pentazocine.
Further, in step S1, the molar ratio of the compound 1 to the bicarbonate of the quaternary ammonium ion is 1: 1.1-1.5.
Further, in step S2, the control temperature is 0 to 180 ℃.
Further, in step S2, the control temperature is 25 to 50 ℃.
Further, in step S2, the stirring time is 1-2 h.
Further, in step S3, the crude product of pentazocine is extracted, washed, dried, concentrated, decolorized and recrystallized to obtain a pure product of pentazocine.
Further, in step S3, isopropyl acetate and n-heptane are used for decoloring.
Compared with the prior art, the synthesis method of pentazocine has the following advantages:
(1) the hydrogen carbonate of quaternary ammonium ions is used as an acid-binding agent, so that hydrobromic acid generated in the reaction process can be neutralized in time, the reaction is smoothly carried out, and the problem of incomplete reaction is avoided;
(2) the quaternary ammonium bicarbonate has good solubility in DMF solvent, and also has good solubility at room temperature or even lower temperature, and the synthesis of pentazocine can be carried out at low temperature without adopting additional means to improve the solubility, simplify the operation, reduce the energy consumption and shorten the reaction time;
(3) by adopting the synthesis method of pentazocine, disubstituted products are not easy to generate, and the yield can be improved;
(4) the pentazocine synthesized by the synthesis method has stable property and is not easy to decompose after being prepared into the pentazocine injection.
Detailed Description
It should be noted that the embodiments and features of the embodiments may be combined with each other without conflict.
In the invention, the synthesis method of pentazocine in the prior art is improved, and the hydrogen carbonate of quaternary ammonium ions is used as an acid-binding agent to replace sodium hydrogen carbonate to neutralize hydrobromic acid generated in the reaction process.
Since sodium bicarbonate has a low solubility in DMF solvent, the improvement in solubility is small even if the heating temperature is increased, the stirring speed is increased, or the content of DMF solvent is increased. The inventors have made a number of experiments to try to select other alkaline reagents with good solubility in DMF solvent. Alkaline reagents such as sodium hydroxide, anhydrous potassium carbonate and sodium methoxide are selected in sequence, although the solubility can be improved, the oxygen on the phenolic hydroxyl group of the synthesized pentazocine is easy to react to generate a disubstituted product, which is not beneficial to the synthesis of the pentazocine.
The inventor unexpectedly finds that by adopting quaternary ammonium bicarbonate with the alkalinity almost the same as that of sodium bicarbonate as an acid-binding agent to neutralize hydrobromic acid generated in the reaction process, the problem of low solubility of the acid-binding agent in a DMF solvent can be solved, other substitution products cannot be generated, and the purity of finally generated pentazocine is ensured.
Specifically, the synthesis method of pentazocine of the invention is as follows:

wherein the bicarbonate of quaternary ammonium ion has a structural formula R1、R2、R3And R4Same or different, each independently selected from C1-C10Alkyl radical, C3-C7A cycloalkyl group. Due to R1、R2、R3And R4The groups can be selected more frequently, and are not listed in detail due to space limitation.
R1、R2、R3And R4Same or different, each independently selected from C1-C10Alkyl radical, C3-C7A cycloalkyl group. Due to R1、R2、R3And R4The groups can be selected more frequently, and are not listed in detail due to space limitation.
The present invention will be described in detail with reference to specific examples.
Example 1 synthesis of pentazocine
Dissolving 5, 9-dimethyl-2' -hydroxy-6, 7-benzomorphane, namely compound 1(217g, 1.0mol), in 2.17L of Dimethylformamide (DMF), adding tetramethylammonium bicarbonate (162g, 1.2mol), dropwise adding 1-bromo-3-methyl-2-butene, namely compound 2(182g, 1.1mol, 90% content) under ice water cooling, maintaining the temperature at 0-5 ℃ after adding, stirring for reaction for 2 hours, adding 4L of methyl tert-butyl ether, adding 2L of saturated saline solution, layering, extracting the lower layer once with 2L of methyl tert-butyl ether, combining ether layers, washing once with 1L of saturated saline solution, drying with anhydrous magnesium sulfate, concentrating to obtain a light yellow solid, decoloring and recrystallizing with isopropyl acetate and n-heptane to obtain 222g of white powdery pentazocine, the yield thereof was found to be 78%.
The preparation method of the pentazocine injection comprises the following steps:
| composition (I) | Dosage of |
| Pentazocine | 30.0g |
| Lactic acid | 45.0g |
| Sodium chloride | Proper amount of |
| Water for injection | Adding to 1000ml |
Make 1000 pieces
Weighing the prepared pentazocine according to the formula amount in the table above, placing the pentazocine in a container, adding 800ml of water for injection, adding lactic acid, heating to 50-60 ℃, stirring for 30min to dissolve, then adding a proper amount of sodium chloride to adjust the osmotic pressure ratio to 1, adjusting the pH value to 4.0-5.0 by using the lactic acid, adding the water for injection to a scale, then filtering by using a 0.22 mu m microporous filter membrane, charging nitrogen, filling into 1ml of ampoule bottles, filling 1ml of each bottle, sealing, sterilizing by flowing steam at 121 ℃ for 30min, cooling, inspecting by a lamp, packaging and warehousing the finished product.
Example 2 synthesis of pentazocine
Dissolving 5, 9-dimethyl-2' -hydroxy-6, 7-benzomorphane (217g, 1.0mol) in 2.17L of Dimethylformamide (DMF), adding tetramethylammonium bicarbonate (135g, 1mol), adding 1-bromo-3-methyl-2-butene (182g, 1.1mol, 90% content) dropwise under ice water cooling, after the addition, heating to 25 ℃, stirring at room temperature for 1 hour, adding 4L of methyl tert-butyl ether, adding 2L of saturated saline solution, layering, extracting the lower layer with 2L of methyl tert-butyl ether, combining ether layers, washing with 1L of saturated saline solution, drying with anhydrous magnesium sulfate, concentrating to obtain a pale yellow solid, decolorizing with isopropyl acetate and n-heptane, and recrystallizing to obtain 239g of white powdery crystalline pentazocine, 84%.
The pentazocine prepared in example 2 was prepared into an injection by the method for preparing the pentazocine injection in example 1.
Example 3 synthesis of pentazocine
Dissolving 5, 9-dimethyl-2' -hydroxy-6, 7-benzomorphane (217g, 1.0mol) in dimethyl formamide (DMF)2.17L, adding tetramethylammonium bicarbonate (202.5g, 1.5mol), adding 1-bromo-3-methyl-2-butene (182g, 1.1mol, 90% content) dropwise under ice water cooling, heating to 40 deg.C after adding, reacting and stirring for 2 hr, cooling to room temperature, adding 4L of methyl tert-butyl ether, adding 2L of saturated saline solution, layering, extracting the lower layer with 2L of methyl tert-butyl ether, mixing the ether layers, washed once with saturated brine 1L, dried over anhydrous magnesium sulfate, concentrated to give a pale yellow solid, the isopropyl acetate and n-heptane were decolorized and recrystallized to yield pentazocine 245g as a white powder in 86% yield.
The pentazocine prepared in example 3 was prepared into an injection by the method for preparing the pentazocine injection in example 1.
Example 4 synthesis of pentazocine
Dissolving 5, 9-dimethyl-2' -hydroxy-6, 7-benzomorphane (217g, 1.0mol) in dimethyl formamide (DMF)2.17L, adding tetramethylammonium bicarbonate (162g, 1.2mol), adding 1-bromo-3-methyl-2-butene (182g, 1.1mol, 90% content) dropwise under ice water cooling, heating to 80 deg.C after adding, reacting and stirring for 2 hr, cooling to room temperature, adding 4L of methyl tert-butyl ether, adding 2L of saturated saline solution, layering, extracting the lower layer with 2L of methyl tert-butyl ether, mixing the ether layers, washed once with saturated brine 1L, dried over anhydrous magnesium sulfate, concentrated to give a pale yellow solid, decolorization and recrystallization with isopropyl acetate and n-heptane gave 231g of pentazocine as a white powder in 81% yield.
The pentazocine prepared in example 4 was prepared into an injection by the method for preparing the pentazocine injection in example 1.
Example 5 Synthesis of pentazocine
Dissolving 5, 9-dimethyl-2' -hydroxy-6, 7-benzomorphane (217g, 1.0mol) in dimethyl formamide (DMF)2.17L, adding tetramethylammonium bicarbonate (162g, 1.2mol), adding 1-bromo-3-methyl-2-butene (182g, 1.1mol, 90% content) dropwise under ice water cooling, heating to 180 deg.C after adding, reacting and stirring for 1.5 hr, cooling to room temperature, adding 4L of methyl tert-butyl ether, adding 2L of saturated saline solution, layering, extracting the lower layer with 2L of methyl tert-butyl ether, mixing the ether layers, washed once with saturated brine 1L, dried over anhydrous magnesium sulfate, concentrated to give a pale yellow solid, decolorization and recrystallization with isopropyl acetate and n-heptane gave 211g of pentazocine as a white powder in 74% yield.
The pentazocine prepared in example 5 was prepared into an injection by the method for preparing the pentazocine injection in example 1.
Example 6 Synthesis of pentazocine
Dissolving 5, 9-dimethyl-2' -hydroxy-6, 7-benzomorphane (217g, 1.0mol) in 2.17L of Dimethylformamide (DMF), adding tetraethylammonium bicarbonate (234g, 1.2mol), dropwise adding 1-bromo-3-methyl-2-butene (182g, 1.1mol, 90% content) under cooling with ice water, after the addition, heating to 50 ℃, stirring for reaction for 1 hour, adding methyl tert-butyl ether (2L), adding saturated saline solution (4L), layering, extracting the lower layer with 2L of methyl tert-butyl ether, combining ether layers, washing with saturated saline solution (1L), drying with anhydrous magnesium sulfate, concentrating to obtain a pale yellow solid, decoloring and recrystallizing with isopropyl acetate and n-heptane to obtain white powdery crystals of pentazocine (234 g), with a yield of 82%.
The pentazocine prepared in example 6 was prepared into an injection by the method for preparing the pentazocine injection in example 1.
Comparative example 1
5, 9-dimethyl-2' -hydroxy-6, 7-benzomorphan (217g, 1.0mol) was dissolved in 3L Dimethylformamide (DMF), sodium bicarbonate (152g, 1.8mol) was added, 1-bromo-3-methyl-2-butene (200g, 1.2mol, 90% content) was added, heating was carried out, and the temperature was maintained at 110 ℃ for reaction for 4.5 hours. And after the reaction is finished, cooling to room temperature. Adding 6L of saturated saline solution and 3L of methyl tertiary butyl ether, stirring vigorously, standing for layering, extracting the lower aqueous phase once with 3L of methyl tertiary butyl ether, combining ether layers, washing once with 1L of saturated saline solution, drying with anhydrous magnesium sulfate, concentrating to obtain a light yellow solid, decoloring and recrystallizing with isopropyl acetate and n-heptane to obtain 148g of white powdery crystal pentazocine, wherein the yield is 52%.
The pentazocine prepared in comparative example 1 was prepared into an injection by the method for preparing the pentazocine injection of example 1.
In the synthesis process of pentazocine, by adopting the method of the application of the embodiment 1-6, after the bicarbonate of the quaternary ammonium ion is added, the temperature is controlled to be 0-180 ℃, the reaction is stirred for 1-2 hours, and the reaction can be normally carried out. Even at low temperatures, the bicarbonate salt of the quaternary ammonium ion can be dissolved in DMF solvent. The yield of the finally prepared pure pentazocine product is as high as 74-86%.
In the method for synthesizing pentazocine in the prior art, i.e., in comparative example 1, in order to dissolve sodium bicarbonate in DMF solvent, heating to 110 ℃ is required, and the reaction is carried out for 4.5h while maintaining the temperature, so that the yield is only 52%.
Therefore, the synthesis method of pentazocine can reduce the reaction temperature, does not need heating and other means to increase the dissolution of the acid-binding agent in the DMF solvent, and is beneficial to simplifying the reaction operation. And the reaction is complete, and the yield is much higher than that of comparative example 1.
Example 7 stability experiment
Examining the stability of the injection of pentazocine prepared in examples 1-6 and comparative example 1, samples were taken after leaving each sample at room temperature for 0h, 24h, 7d, 14d and 30d, respectively, and the content of pentazocine in each sample was analyzed by HPLC; each sample was stored in 80 ℃ hot water at constant temperature and sampled at 30min, 1h, 5h and 24h, respectively, and analyzed by HPLC. The results are shown in Table 1.
TABLE 1


For injections, it is important to maintain the contents during long-term storage because of the variety of storage conditions. As can be seen from the data of the stability experiment in table 1, the injection prepared by using the pentazocine synthesized in the embodiments 1-6 of the present invention has almost no change or little decrease in the content of the pentazocine when stored at normal temperature, even after being stored at normal temperature for 30 days, the content is still as high as 99.4-99.7%, and the decomposition does not occur with the increase of the storage time. In contrast, in the sample of comparative example 1, the content of pentazocine was 97.0% when left to stand at normal temperature for 24 hours, and was reduced to 93.3% when left to stand for 30 days for a long period. The samples of examples 1-6 were stored in hot water at 80 ℃ for 24h with a content of 98.6% to 99.0% and a smaller drop. The sample of comparative example 1, on the other hand, was placed in hot water at 80 ℃ and the content decreased continuously from 97.1% to 91.0% after 24 hours of placement, showing that decomposition occurred, indicating that pentazocine stability was poor. By adopting the synthesis method of pentazocine, the pentazocine is stable both in room temperature placement and high temperature placement, and can not be decomposed even being heated.
The above description is only for the purpose of illustrating the preferred embodiments of the present invention and is not to be construed as limiting the invention, and any modifications, equivalents, improvements and the like that fall within the spirit and principle of the present invention are intended to be included therein.
Claims (9)
1. The synthesis method of pentazocine is characterized by comprising the following steps:
the pentazocine is generated by the reaction of a compound 1 and a compound 2, and bicarbonate of quaternary ammonium ions is adopted as an acid-binding agent in the synthesis process, wherein the structural formula of the bicarbonate of the quaternary ammonium ions is shown in the specification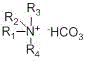 Wherein R is1、R2、R3And R4Same or different, each independently selected from C1-C10An alkyl group.
Wherein R is1、R2、R3And R4Same or different, each independently selected from C1-C10An alkyl group.
2. The method for synthesizing pentazocine according to claim 1, wherein the bicarbonate of the quaternary ammonium ion is at least one of tetramethylammonium bicarbonate and tetraethylammonium bicarbonate.
3. The synthesis method of pentazocine according to claim 1, characterized in that the synthesis method specifically comprises:
s1, dissolving a compound 1 in a DMF solvent, adding bicarbonate of quaternary ammonium ions, and dropwise adding a compound 2 under the cooling of ice water;
s2, after the compound 2 is dropwise added, controlling the temperature, stirring and cooling to room temperature to obtain a crude product of pentazocine;
s3, refining the crude product of pentazocine to prepare a pure product of pentazocine.
4. The method for synthesizing pentazocine according to claim 3, wherein in step S1, the molar ratio of Compound 1 to the bicarbonate salt of the quaternary ammonium ion is 1: 1.1-1.5.
5. The method for synthesizing pentazocine according to claim 3, wherein in step S2, the control temperature is 0-180 ℃.
6. The method for synthesizing pentazocine according to claim 5, wherein in step S2, the control temperature is 25-50 ℃.
7. The method for synthesizing pentazocine according to claim 3, characterized in that in step S2, the stirring time is 1-2 h.
8. The method for synthesizing pentazocine according to claim 3, characterized in that, in step S3, the crude pentazocine is extracted, washed, dried, concentrated, decolorized and recrystallized to obtain pure pentazocine.
9. The synthesis method of pentazocine according to claim 8, characterized in that the decolorization is carried out using isopropyl acetate and n-heptane.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202010848205.0A CN112079779B (en) | 2020-08-21 | 2020-08-21 | Synthetic method of pentazocine |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202010848205.0A CN112079779B (en) | 2020-08-21 | 2020-08-21 | Synthetic method of pentazocine |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN112079779A CN112079779A (en) | 2020-12-15 |
| CN112079779B true CN112079779B (en) | 2022-03-18 |
Family
ID=73729481
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN202010848205.0A Active CN112079779B (en) | 2020-08-21 | 2020-08-21 | Synthetic method of pentazocine |
Country Status (1)
| Country | Link |
|---|---|
| CN (1) | CN112079779B (en) |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN113845477B (en) * | 2021-10-28 | 2023-06-02 | 华润双鹤药业股份有限公司沧州分公司 | Refining method of pentazocine |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN101714660A (en) * | 2009-11-25 | 2010-05-26 | 中国海洋石油总公司 | Method for removing free acids in non-aqueous electrolyte |
| CN111217749A (en) * | 2020-03-13 | 2020-06-02 | 安徽省逸欣铭医药科技有限公司 | Deuterated pentazocine, preparation method, medical composition and application |
| CN111269124A (en) * | 2020-02-11 | 2020-06-12 | 浙江理工大学 | A kind of reactive ultraviolet absorber and its preparation method and application |
| CN111349111A (en) * | 2020-03-23 | 2020-06-30 | 安徽省逸欣铭医药科技有限公司 | Pentazocine prodrug, preparation method and application thereof |
-
2020
- 2020-08-21 CN CN202010848205.0A patent/CN112079779B/en active Active
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN101714660A (en) * | 2009-11-25 | 2010-05-26 | 中国海洋石油总公司 | Method for removing free acids in non-aqueous electrolyte |
| CN111269124A (en) * | 2020-02-11 | 2020-06-12 | 浙江理工大学 | A kind of reactive ultraviolet absorber and its preparation method and application |
| CN111217749A (en) * | 2020-03-13 | 2020-06-02 | 安徽省逸欣铭医药科技有限公司 | Deuterated pentazocine, preparation method, medical composition and application |
| CN111349111A (en) * | 2020-03-23 | 2020-06-30 | 安徽省逸欣铭医药科技有限公司 | Pentazocine prodrug, preparation method and application thereof |
Non-Patent Citations (2)
| Title |
|---|
| Conjugated Thiophene-Containing Polymer Zwitterions: Direct Synthesis and Thin Film Electronic Properties;Todd Emrick et al.;《Macromolecules》;20121231;第46卷(第2期);第344-351页 * |
| Enantiocontrolled Synthesis of (-)-9-epi-Pentazocine and (-)-Aphanorphine;Xiaobao Yang et al.;《Org. Lett. 》;20081231;第10卷(第12期);第2457-2460页 * |
Also Published As
| Publication number | Publication date |
|---|---|
| CN112079779A (en) | 2020-12-15 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| NO171765B (en) | PROCEDURE FOR PREPARING SOLUTIONS OF Lactic Acid Salts of PIPERAZINYLKINOLONE AND PIPERAZINYLAZAKINOLONCARBOXYL ACIDS | |
| HRP20010132A2 (en) | CRYSTALLINE FORMS OF EtO<->2<P>C-CH<->2<P>-(R)Cgl-Aze-Pab-OH | |
| US4673666A (en) | 2-amino-3-ethoxycarbonylamino-6-(p-fluoro-benzylamino)-pyridine gluconate and pharmaceutical preparations containing it | |
| CN112079779B (en) | Synthetic method of pentazocine | |
| AU2008220800B2 (en) | Acid addition salts, hydrates and polymorphs of 5-(2,4-dihydroxy-5-isopropyl-phenyl)-4-(4-morpholin-4-ylmethyl-phenyl)-isoxazole-3-carboxylic acid ethylamide and formulations comprising these forms | |
| EP1485393B1 (en) | 9-dexo-9a-aza-9a-methyl-9a-homoerythromycin a derivatives | |
| AU2003266940C1 (en) | Novel raloxifene acid addition salts and/or solvates thereof, improved method for purification of said raloxifene acid addition salts and/or solvates thereof and pharmaceutical compositions comprising these | |
| RU2594732C2 (en) | Hydrate of cyclopeptide compound as well as preparation method and use thereof | |
| US6936591B2 (en) | Amorphous 9-deoxo-9a-aza-9a-methyl-9a-homoerythromycin A, process for preparing the same, and uses thereof | |
| KR20020020765A (en) | A Substantially Crystalline Form of Melagatran | |
| EP3981759A1 (en) | New crystal form of treprostinil sodium salt and preparation method therefor | |
| IE50319B1 (en) | Fosfomycin salt | |
| AU726662B2 (en) | Solid compound, its preparation and use in medicine | |
| JPS61148173A (en) | Novel amine and its salt | |
| GB2101587A (en) | Methylenedioxybenzene derivatives. | |
| CN115227656B (en) | Preparation method of omeprazole sodium for injection | |
| RU2173994C1 (en) | Fluconazole-containing medicinal agent and method of synthesis of fluconazole | |
| US5556879A (en) | Aqueous spectinomycin borate solutions | |
| NO117505B (en) | ||
| GRIMAUX | THE CONVERSION OF MORPHIA INTO CODEIA. | |
| JPH0569107B2 (en) | ||
| KR810000431B1 (en) | Process for preparing substituted 2-(2-hydroxyethyl)-tetrahydro-1,4-oxazines | |
| HK40067809A (en) | New crystal form of treprostinil sodium salt and preparation method therefor | |
| MXPA01002044A (en) | CRYSTALLINE FORMS OF EtO2 | |
| JPS6023362A (en) | Preparation of dl-cysteine of single crystal |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PB01 | Publication | ||
| PB01 | Publication | ||
| SE01 | Entry into force of request for substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| GR01 | Patent grant | ||
| GR01 | Patent grant |