CN112341402A - Preparation method and application of pyrimidine compound capable of inhibiting bacteria in medical care process - Google Patents
Preparation method and application of pyrimidine compound capable of inhibiting bacteria in medical care process Download PDFInfo
- Publication number
- CN112341402A CN112341402A CN202011441230.3A CN202011441230A CN112341402A CN 112341402 A CN112341402 A CN 112341402A CN 202011441230 A CN202011441230 A CN 202011441230A CN 112341402 A CN112341402 A CN 112341402A
- Authority
- CN
- China
- Prior art keywords
- reaction
- hydroxybenzene
- pyrimidinone
- hydroxyphenyl
- dichloromethane
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
- -1 pyrimidine compound Chemical class 0.000 title claims abstract description 32
- 238000000034 method Methods 0.000 title claims abstract description 31
- 230000008569 process Effects 0.000 title claims abstract description 20
- 238000002360 preparation method Methods 0.000 title claims abstract description 15
- 230000002401 inhibitory effect Effects 0.000 title abstract description 8
- 241000894006 Bacteria Species 0.000 title description 13
- 238000006243 chemical reaction Methods 0.000 claims abstract description 74
- LUJMEECXHPYQOF-UHFFFAOYSA-N 3-hydroxyacetophenone Chemical compound CC(=O)C1=CC=CC(O)=C1 LUJMEECXHPYQOF-UHFFFAOYSA-N 0.000 claims abstract description 16
- FXLOVSHXALFLKQ-UHFFFAOYSA-N p-tolualdehyde Chemical compound CC1=CC=C(C=O)C=C1 FXLOVSHXALFLKQ-UHFFFAOYSA-N 0.000 claims abstract description 16
- 150000003230 pyrimidines Chemical class 0.000 claims abstract description 12
- 239000001431 2-methylbenzaldehyde Substances 0.000 claims abstract description 8
- 230000000844 anti-bacterial effect Effects 0.000 claims abstract description 7
- 150000001875 compounds Chemical class 0.000 claims abstract description 6
- GVSNQMFKEPBIOY-UHFFFAOYSA-N 4-methyl-2h-triazole Chemical compound CC=1C=NNN=1 GVSNQMFKEPBIOY-UHFFFAOYSA-N 0.000 claims abstract description 3
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims abstract description 3
- 125000002568 propynyl group Chemical group [*]C#CC([H])([H])[H] 0.000 claims abstract 2
- 150000003673 urethanes Chemical class 0.000 claims abstract 2
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 claims description 117
- 238000003756 stirring Methods 0.000 claims description 43
- 125000004208 3-hydroxyphenyl group Chemical group [H]OC1=C([H])C([H])=C([H])C(*)=C1[H] 0.000 claims description 33
- 239000012074 organic phase Substances 0.000 claims description 28
- 239000003054 catalyst Substances 0.000 claims description 27
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 claims description 26
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 claims description 24
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 claims description 24
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 claims description 20
- 238000001914 filtration Methods 0.000 claims description 20
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical compound ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 claims description 20
- 230000007935 neutral effect Effects 0.000 claims description 19
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 claims description 18
- 125000003944 tolyl group Chemical group 0.000 claims description 18
- 238000010898 silica gel chromatography Methods 0.000 claims description 16
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 claims description 14
- DKGAVHZHDRPRBM-UHFFFAOYSA-N Tert-Butanol Chemical compound CC(C)(C)O DKGAVHZHDRPRBM-UHFFFAOYSA-N 0.000 claims description 14
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 claims description 12
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Natural products CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 claims description 11
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Chemical compound NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 claims description 11
- 239000004202 carbamide Substances 0.000 claims description 11
- 239000012295 chemical reaction liquid Substances 0.000 claims description 11
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 claims description 10
- SEDZOYHHAIAQIW-UHFFFAOYSA-N trimethylsilyl azide Chemical compound C[Si](C)(C)N=[N+]=[N-] SEDZOYHHAIAQIW-UHFFFAOYSA-N 0.000 claims description 9
- WQDUMFSSJAZKTM-UHFFFAOYSA-N Sodium methoxide Chemical group [Na+].[O-]C WQDUMFSSJAZKTM-UHFFFAOYSA-N 0.000 claims description 8
- WGLPBDUCMAPZCE-UHFFFAOYSA-N chromium trioxide Inorganic materials O=[Cr](=O)=O WGLPBDUCMAPZCE-UHFFFAOYSA-N 0.000 claims description 8
- 150000002576 ketones Chemical class 0.000 claims description 8
- 239000012046 mixed solvent Substances 0.000 claims description 8
- 239000012141 concentrate Substances 0.000 claims description 7
- 230000009471 action Effects 0.000 claims description 6
- YORCIIVHUBAYBQ-UHFFFAOYSA-N propargyl bromide Chemical compound BrCC#C YORCIIVHUBAYBQ-UHFFFAOYSA-N 0.000 claims description 6
- 238000010992 reflux Methods 0.000 claims description 6
- 229910021591 Copper(I) chloride Inorganic materials 0.000 claims description 5
- 229910021595 Copper(I) iodide Inorganic materials 0.000 claims description 5
- MHAJPDPJQMAIIY-UHFFFAOYSA-N Hydrogen peroxide Chemical compound OO MHAJPDPJQMAIIY-UHFFFAOYSA-N 0.000 claims description 5
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical class [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 claims description 5
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 claims description 5
- 229910000024 caesium carbonate Inorganic materials 0.000 claims description 5
- OXBLHERUFWYNTN-UHFFFAOYSA-M copper(I) chloride Chemical group [Cu]Cl OXBLHERUFWYNTN-UHFFFAOYSA-M 0.000 claims description 5
- JZCCFEFSEZPSOG-UHFFFAOYSA-L copper(II) sulfate pentahydrate Chemical compound O.O.O.O.O.[Cu+2].[O-]S([O-])(=O)=O JZCCFEFSEZPSOG-UHFFFAOYSA-L 0.000 claims description 5
- LSXDOTMGLUJQCM-UHFFFAOYSA-M copper(i) iodide Chemical compound I[Cu] LSXDOTMGLUJQCM-UHFFFAOYSA-M 0.000 claims description 5
- 229940045803 cuprous chloride Drugs 0.000 claims description 5
- 239000000706 filtrate Substances 0.000 claims description 5
- 150000004675 formic acid derivatives Chemical class 0.000 claims description 5
- WJCXADMLESSGRI-UHFFFAOYSA-N phenyl selenohypochlorite Chemical compound Cl[Se]C1=CC=CC=C1 WJCXADMLESSGRI-UHFFFAOYSA-N 0.000 claims description 5
- 235000010378 sodium ascorbate Nutrition 0.000 claims description 5
- PPASLZSBLFJQEF-RKJRWTFHSA-M sodium ascorbate Substances [Na+].OC[C@@H](O)[C@H]1OC(=O)C(O)=C1[O-] PPASLZSBLFJQEF-RKJRWTFHSA-M 0.000 claims description 5
- 229960005055 sodium ascorbate Drugs 0.000 claims description 5
- PPASLZSBLFJQEF-RXSVEWSESA-M sodium-L-ascorbate Chemical compound [Na+].OC[C@H](O)[C@H]1OC(=O)C(O)=C1[O-] PPASLZSBLFJQEF-RXSVEWSESA-M 0.000 claims description 5
- 239000008346 aqueous phase Substances 0.000 claims description 4
- 239000007789 gas Substances 0.000 claims description 4
- 239000012948 isocyanate Substances 0.000 claims description 4
- 239000002904 solvent Substances 0.000 claims description 4
- XJDNKRIXUMDJCW-UHFFFAOYSA-J titanium tetrachloride Chemical compound Cl[Ti](Cl)(Cl)Cl XJDNKRIXUMDJCW-UHFFFAOYSA-J 0.000 claims description 4
- YNHIGQDRGKUECZ-UHFFFAOYSA-L bis(triphenylphosphine)palladium(ii) dichloride Chemical compound [Cl-].[Cl-].[Pd+2].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 YNHIGQDRGKUECZ-UHFFFAOYSA-L 0.000 claims description 3
- 238000007363 ring formation reaction Methods 0.000 claims description 2
- 238000000926 separation method Methods 0.000 claims description 2
- DUNKXUFBGCUVQW-UHFFFAOYSA-J zirconium tetrachloride Chemical compound Cl[Zr](Cl)(Cl)Cl DUNKXUFBGCUVQW-UHFFFAOYSA-J 0.000 claims description 2
- YCIPQJTZJGUXND-UHFFFAOYSA-N Aglaia odorata Alkaloid Natural products C1=CC(OC)=CC=C1C1(C(C=2C(=O)N3CCCC3=NC=22)C=3C=CC=CC=3)C2(O)C2=C(OC)C=C(OC)C=C2O1 YCIPQJTZJGUXND-UHFFFAOYSA-N 0.000 claims 12
- VTGOHKSTWXHQJK-UHFFFAOYSA-N pyrimidin-2-ol Chemical compound OC1=NC=CC=N1 VTGOHKSTWXHQJK-UHFFFAOYSA-N 0.000 claims 12
- 150000007514 bases Chemical class 0.000 claims 3
- CKJNUZNMWOVDFN-UHFFFAOYSA-N methanone Chemical compound O=[CH-] CKJNUZNMWOVDFN-UHFFFAOYSA-N 0.000 claims 2
- 230000029936 alkylation Effects 0.000 claims 1
- 238000005804 alkylation reaction Methods 0.000 claims 1
- 229910001902 chlorine oxide Inorganic materials 0.000 claims 1
- MAYPHUUCLRDEAZ-UHFFFAOYSA-N chlorine peroxide Chemical compound ClOOCl MAYPHUUCLRDEAZ-UHFFFAOYSA-N 0.000 claims 1
- 229940117975 chromium trioxide Drugs 0.000 claims 1
- GAMDZJFZMJECOS-UHFFFAOYSA-N chromium(6+);oxygen(2-) Chemical compound [O-2].[O-2].[O-2].[Cr+6] GAMDZJFZMJECOS-UHFFFAOYSA-N 0.000 claims 1
- RCJVRSBWZCNNQT-UHFFFAOYSA-N dichloridooxygen Chemical compound ClOCl RCJVRSBWZCNNQT-UHFFFAOYSA-N 0.000 claims 1
- ROSKZJGILXBSFM-UHFFFAOYSA-N pyrimidin-2-ylmethanamine Chemical class NCC1=NC=CC=N1 ROSKZJGILXBSFM-UHFFFAOYSA-N 0.000 claims 1
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 claims 1
- 229920002554 vinyl polymer Polymers 0.000 claims 1
- 239000003814 drug Substances 0.000 abstract description 19
- 229940079593 drug Drugs 0.000 abstract description 15
- 238000012360 testing method Methods 0.000 abstract description 6
- 241000588724 Escherichia coli Species 0.000 abstract description 4
- 229940124350 antibacterial drug Drugs 0.000 abstract description 4
- 238000003786 synthesis reaction Methods 0.000 abstract description 4
- JGSARLDLIJGVTE-MBNYWOFBSA-N Penicillin G Chemical compound N([C@H]1[C@H]2SC([C@@H](N2C1=O)C(O)=O)(C)C)C(=O)CC1=CC=CC=C1 JGSARLDLIJGVTE-MBNYWOFBSA-N 0.000 abstract description 3
- 230000015572 biosynthetic process Effects 0.000 abstract description 3
- 229920001817 Agar Polymers 0.000 abstract description 2
- 239000008272 agar Substances 0.000 abstract description 2
- 230000003385 bacteriostatic effect Effects 0.000 abstract description 2
- 238000009792 diffusion process Methods 0.000 abstract description 2
- 238000001308 synthesis method Methods 0.000 abstract description 2
- 229940118531 bicillin Drugs 0.000 abstract 1
- 239000007858 starting material Substances 0.000 abstract 1
- 239000000243 solution Substances 0.000 description 41
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 20
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 13
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 12
- 238000001035 drying Methods 0.000 description 11
- 238000005160 1H NMR spectroscopy Methods 0.000 description 10
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 9
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 8
- 238000001704 evaporation Methods 0.000 description 8
- 229910000423 chromium oxide Inorganic materials 0.000 description 7
- 238000000921 elemental analysis Methods 0.000 description 7
- 230000005764 inhibitory process Effects 0.000 description 7
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 6
- KAESVJOAVNADME-UHFFFAOYSA-N Pyrrole Chemical compound C=1C=CNC=1 KAESVJOAVNADME-UHFFFAOYSA-N 0.000 description 6
- 239000003242 anti bacterial agent Substances 0.000 description 6
- 229940088710 antibiotic agent Drugs 0.000 description 6
- 230000000694 effects Effects 0.000 description 6
- 239000002994 raw material Substances 0.000 description 6
- 241000191967 Staphylococcus aureus Species 0.000 description 5
- 238000010438 heat treatment Methods 0.000 description 5
- WSFSSNUMVMOOMR-BJUDXGSMSA-N methanone Chemical compound O=[11CH2] WSFSSNUMVMOOMR-BJUDXGSMSA-N 0.000 description 5
- 238000012544 monitoring process Methods 0.000 description 5
- 238000004809 thin layer chromatography Methods 0.000 description 5
- 239000000203 mixture Substances 0.000 description 4
- 229910052757 nitrogen Inorganic materials 0.000 description 4
- 238000005406 washing Methods 0.000 description 4
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 3
- 108090000695 Cytokines Proteins 0.000 description 3
- 102000004127 Cytokines Human genes 0.000 description 3
- 206010059866 Drug resistance Diseases 0.000 description 3
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 3
- 230000003115 biocidal effect Effects 0.000 description 3
- 238000012258 culturing Methods 0.000 description 3
- 238000007599 discharging Methods 0.000 description 3
- 239000001963 growth medium Substances 0.000 description 3
- UQPUONNXJVWHRM-UHFFFAOYSA-N palladium;triphenylphosphane Chemical compound [Pd].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 UQPUONNXJVWHRM-UHFFFAOYSA-N 0.000 description 3
- 239000012071 phase Substances 0.000 description 3
- DQFBYFPFKXHELB-UHFFFAOYSA-N Chalcone Natural products C=1C=CC=CC=1C(=O)C=CC1=CC=CC=C1 DQFBYFPFKXHELB-UHFFFAOYSA-N 0.000 description 2
- QUSNBJAOOMFDIB-UHFFFAOYSA-N Ethylamine Chemical compound CCN QUSNBJAOOMFDIB-UHFFFAOYSA-N 0.000 description 2
- 241000192125 Firmicutes Species 0.000 description 2
- 201000009906 Meningitis Diseases 0.000 description 2
- 229930182555 Penicillin Natural products 0.000 description 2
- PXIPVTKHYLBLMZ-UHFFFAOYSA-N Sodium azide Chemical compound [Na+].[N-]=[N+]=[N-] PXIPVTKHYLBLMZ-UHFFFAOYSA-N 0.000 description 2
- 150000001299 aldehydes Chemical class 0.000 description 2
- 210000004027 cell Anatomy 0.000 description 2
- 230000004663 cell proliferation Effects 0.000 description 2
- 235000005513 chalcones Nutrition 0.000 description 2
- 238000002474 experimental method Methods 0.000 description 2
- 238000011010 flushing procedure Methods 0.000 description 2
- 230000036541 health Effects 0.000 description 2
- 208000015181 infectious disease Diseases 0.000 description 2
- UWYHMGVUTGAWSP-JKIFEVAISA-N oxacillin Chemical compound N([C@@H]1C(N2[C@H](C(C)(C)S[C@@H]21)C(O)=O)=O)C(=O)C1=C(C)ON=C1C1=CC=CC=C1 UWYHMGVUTGAWSP-JKIFEVAISA-N 0.000 description 2
- 229960001019 oxacillin Drugs 0.000 description 2
- 229940049954 penicillin Drugs 0.000 description 2
- DQFBYFPFKXHELB-VAWYXSNFSA-N trans-chalcone Chemical compound C=1C=CC=CC=1C(=O)\C=C\C1=CC=CC=C1 DQFBYFPFKXHELB-VAWYXSNFSA-N 0.000 description 2
- 208000023275 Autoimmune disease Diseases 0.000 description 1
- 208000035473 Communicable disease Diseases 0.000 description 1
- 101000934996 Homo sapiens Tyrosine-protein kinase JAK3 Proteins 0.000 description 1
- 102000042838 JAK family Human genes 0.000 description 1
- 108091082332 JAK family Proteins 0.000 description 1
- 241000588653 Neisseria Species 0.000 description 1
- 229940124158 Protease/peptidase inhibitor Drugs 0.000 description 1
- 201000004681 Psoriasis Diseases 0.000 description 1
- CZPWVGJYEJSRLH-UHFFFAOYSA-N Pyrimidine Chemical compound C1=CN=CN=C1 CZPWVGJYEJSRLH-UHFFFAOYSA-N 0.000 description 1
- 241000194017 Streptococcus Species 0.000 description 1
- 241000193998 Streptococcus pneumoniae Species 0.000 description 1
- 102100025387 Tyrosine-protein kinase JAK3 Human genes 0.000 description 1
- AKZWRTCWNXHHFR-PDIZUQLASA-N [(3S)-oxolan-3-yl] N-[(2S,3S)-4-[(5S)-5-benzyl-3-[(2R)-2-carbamoyloxy-2,3-dihydro-1H-inden-1-yl]-4-oxo-3H-pyrrol-5-yl]-3-hydroxy-1-phenylbutan-2-yl]carbamate Chemical compound NC(=O)O[C@@H]1Cc2ccccc2C1C1C=N[C@](C[C@H](O)[C@H](Cc2ccccc2)NC(=O)O[C@H]2CCOC2)(Cc2ccccc2)C1=O AKZWRTCWNXHHFR-PDIZUQLASA-N 0.000 description 1
- 238000002835 absorbance Methods 0.000 description 1
- 229940121359 adenosine receptor antagonist Drugs 0.000 description 1
- 150000001336 alkenes Chemical class 0.000 description 1
- HXBPYFMVGFDZFT-UHFFFAOYSA-N allyl isocyanate Chemical compound C=CCN=C=O HXBPYFMVGFDZFT-UHFFFAOYSA-N 0.000 description 1
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 1
- 230000009286 beneficial effect Effects 0.000 description 1
- 230000008499 blood brain barrier function Effects 0.000 description 1
- 210000001218 blood-brain barrier Anatomy 0.000 description 1
- 229940082638 cardiac stimulant phosphodiesterase inhibitors Drugs 0.000 description 1
- 239000006285 cell suspension Substances 0.000 description 1
- 210000001175 cerebrospinal fluid Anatomy 0.000 description 1
- 238000005660 chlorination reaction Methods 0.000 description 1
- 206010009887 colitis Diseases 0.000 description 1
- 229910000366 copper(II) sulfate Inorganic materials 0.000 description 1
- 238000010227 cup method (microbiological evaluation) Methods 0.000 description 1
- 239000002875 cyclin dependent kinase inhibitor Substances 0.000 description 1
- 229940043378 cyclin-dependent kinase inhibitor Drugs 0.000 description 1
- WEPUZBYKXNKSDH-UHFFFAOYSA-N cyclopentanecarbonyl chloride Chemical compound ClC(=O)C1CCCC1 WEPUZBYKXNKSDH-UHFFFAOYSA-N 0.000 description 1
- 231100000135 cytotoxicity Toxicity 0.000 description 1
- 230000003013 cytotoxicity Effects 0.000 description 1
- 238000013461 design Methods 0.000 description 1
- 238000001514 detection method Methods 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- 206010012601 diabetes mellitus Diseases 0.000 description 1
- 201000010099 disease Diseases 0.000 description 1
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 1
- 238000005516 engineering process Methods 0.000 description 1
- 230000003203 everyday effect Effects 0.000 description 1
- 230000014509 gene expression Effects 0.000 description 1
- 230000002949 hemolytic effect Effects 0.000 description 1
- 150000002391 heterocyclic compounds Chemical class 0.000 description 1
- 238000002347 injection Methods 0.000 description 1
- 239000007924 injection Substances 0.000 description 1
- 239000007788 liquid Substances 0.000 description 1
- 231100000053 low toxicity Toxicity 0.000 description 1
- 238000001819 mass spectrum Methods 0.000 description 1
- 230000007246 mechanism Effects 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 239000002547 new drug Substances 0.000 description 1
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 1
- JRZJOMJEPLMPRA-UHFFFAOYSA-N olefin Natural products CCCCCCCC=C JRZJOMJEPLMPRA-UHFFFAOYSA-N 0.000 description 1
- 238000005580 one pot reaction Methods 0.000 description 1
- 210000000056 organ Anatomy 0.000 description 1
- 238000006053 organic reaction Methods 0.000 description 1
- 239000003960 organic solvent Substances 0.000 description 1
- 230000008520 organization Effects 0.000 description 1
- 239000001301 oxygen Substances 0.000 description 1
- 229910052760 oxygen Inorganic materials 0.000 description 1
- 239000000137 peptide hydrolase inhibitor Substances 0.000 description 1
- 239000012466 permeate Substances 0.000 description 1
- 239000002571 phosphodiesterase inhibitor Substances 0.000 description 1
- 230000026731 phosphorylation Effects 0.000 description 1
- 238000006366 phosphorylation reaction Methods 0.000 description 1
- 239000013641 positive control Substances 0.000 description 1
- 230000003389 potentiating effect Effects 0.000 description 1
- 108090000623 proteins and genes Proteins 0.000 description 1
- 238000000746 purification Methods 0.000 description 1
- 239000000296 purinergic P1 receptor antagonist Substances 0.000 description 1
- 229940083082 pyrimidine derivative acting on arteriolar smooth muscle Drugs 0.000 description 1
- 125000000714 pyrimidinyl group Chemical group 0.000 description 1
- 239000012429 reaction media Substances 0.000 description 1
- 230000035484 reaction time Effects 0.000 description 1
- 108020003175 receptors Proteins 0.000 description 1
- 102000005962 receptors Human genes 0.000 description 1
- 230000008261 resistance mechanism Effects 0.000 description 1
- 201000003068 rheumatic fever Diseases 0.000 description 1
- 159000000000 sodium salts Chemical class 0.000 description 1
- 238000001179 sorption measurement Methods 0.000 description 1
- 238000001228 spectrum Methods 0.000 description 1
- 229940031000 streptococcus pneumoniae Drugs 0.000 description 1
- 238000006467 substitution reaction Methods 0.000 description 1
- SEEPANYCNGTZFQ-UHFFFAOYSA-N sulfadiazine Chemical compound C1=CC(N)=CC=C1S(=O)(=O)NC1=NC=CC=N1 SEEPANYCNGTZFQ-UHFFFAOYSA-N 0.000 description 1
- 229960004306 sulfadiazine Drugs 0.000 description 1
- 239000006228 supernatant Substances 0.000 description 1
- 230000002194 synthesizing effect Effects 0.000 description 1
- 231100000331 toxic Toxicity 0.000 description 1
- 230000002588 toxic effect Effects 0.000 description 1
- 238000012546 transfer Methods 0.000 description 1
- 238000002054 transplantation Methods 0.000 description 1
- 230000035899 viability Effects 0.000 description 1
Images

Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D263/00—Heterocyclic compounds containing 1,3-oxazole or hydrogenated 1,3-oxazole rings
- C07D263/02—Heterocyclic compounds containing 1,3-oxazole or hydrogenated 1,3-oxazole rings not condensed with other rings
- C07D263/30—Heterocyclic compounds containing 1,3-oxazole or hydrogenated 1,3-oxazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D263/34—Heterocyclic compounds containing 1,3-oxazole or hydrogenated 1,3-oxazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D263/36—One oxygen atom
- C07D263/38—One oxygen atom attached in position 2
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02A—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE
- Y02A50/00—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE in human health protection, e.g. against extreme weather
- Y02A50/30—Against vector-borne diseases, e.g. mosquito-borne, fly-borne, tick-borne or waterborne diseases whose impact is exacerbated by climate change
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Communicable Diseases (AREA)
- Pharmacology & Pharmacy (AREA)
- Oncology (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Nitrogen And Oxygen As The Only Ring Hetero Atoms (AREA)
Abstract
本发明公开了一种可用于医疗护理过程中抑菌的嘧啶类化合物的制备方法和应用,属于抑菌药物的合成技术领域。本发明的技术方案要点为:该嘧啶类化合物具有结构

The invention discloses a preparation method and application of a pyrimidine compound that can be used for bacteriostasis in the medical care process, and belongs to the technical field of the synthesis of bacteriostatic drugs. The main point of the technical solution of the present invention is: the pyrimidine compound has a structure
Description
Technical Field
The invention belongs to the technical field of synthesis of antibacterial drugs, and particularly relates to a preparation method and application of a pyrimidine compound for bacteriostasis in a medical care process.
Background
Since the discovery of penicillin in 1928, antibiotics become common medicines for treating various diseases in clinic. While the variety and the quantity of antibiotics are updated, the selection difficulty of the drugs and the situations of excessive drugs, abuse and the like are increased under the influence of various human and objective factors, and both gram-positive bacteria and gram-negative bacteria have serious drug resistance. In addition, the bacteria obtain exogenous drug-resistant genes through horizontal transfer, and the generation of drug-resistant strains is accelerated. In 2006 Science was reported that a staphylococcus aureus strain stored in the laboratory in 1930 was sensitive to currently clinically used antibiotics, while a staphylococcus aureus strain isolated from a patient was resistant to almost all antibiotics, and this resistance exhibited multiple resistance mechanisms to different classes of antibiotics in the same bacterium. According to the statistics of the world health organization, about ten thousand patients die from infectious diseases every day in the world, and the human health and social development are seriously threatened, so that the high attention of people is attracted. Among them, the resistance problem of multi-resistant bacteria is particularly prominent, which brings great difficulty to clinical treatment. In the face of the vicious circle of 'drug resistance-new drug development-drug resistance', and the existing drugs are still difficult to effectively control the infection of novel drug-resistant bacteria, pharmaceutical chemists are struggling to develop novel drug-resistant bacteria resistant drugs, design and screen novel antibacterial drugs with brand new structures, unique action mechanisms or new action targets, or hybrid drugs with other drugs.
The pyrimidine compound is an important nitrogen-containing and oxygen-containing heterocyclic compound and is important in organic synthesis and medicine, for example, the pyrrolopyrimidine compound plays an important role due to high efficiency, low toxicity and multi-orientation substitution on pyrrole and pyrimidine rings. For example, they are useful as adenosine receptor antagonists, cyclin-dependent kinase inhibitors, phosphodiesterase inhibitors and the like in medicine. There are reports in the literature that a pyrrole [2,3-d ] pyrimidine derivative with a potent protease inhibitor action can effectively and selectively inhibit JAK3, and can block cytokine signals and cytokine-induced gene expression, while having no inhibitory action on JAK enzyme family members related to other cytokines and receptor phosphorylation, and can be used for organ transplantation and the treatment of various autoimmune diseases; furthermore, the pyrrole [2,3-d ] pyrimidine derivatives are also effective in the treatment of rheumatic arthritis, psoriasis, colitis, diabetes and the like. For example, classical antibacterial drugs, sulfadiazine, have wide antibacterial spectrum, have inhibition effect on most gram-positive bacteria and gram-negative bacteria, have stronger inhibition effect on meningococcus, streptococcus pneumoniae, gonococcus and hemolytic streptococcus, and can permeate into cerebrospinal fluid through a blood brain barrier. The traditional Chinese medicine composition is mainly used for treating epidemic cerebrospinal meningitis in clinic, is a first-choice medicine for treating epidemic cerebrospinal meningitis, and can also be used for treating other infections caused by the sensitive bacteria. It is also often made into water soluble sodium salt for injection.
The invention takes 3-hydroxyacetophenone and 4-methylbenzaldehyde as initial raw materials, obtains a pyrimidine compound with a novel structure through five steps of reactions, and performs antibacterial activity tests on escherichia coli and staphylococcus aureus.
Disclosure of Invention
The invention aims to provide a preparation method and application of a pyrimidine compound for bacteriostasis in a medical care process.
The invention adopts the following technical scheme for solving the technical problems, and the pyrimidine compound for inhibiting bacteria in the medical care process is characterized by having the following structure:

The invention adopts the following technical scheme for solving the technical problems, and the preparation method of the pyrimidine compound for bacteriostasis in the medical care process is characterized by comprising the following specific steps:
(1) condensing 3-hydroxyacetophenone and 4-methylbenzaldehyde under the action of sodium methoxide to obtain 1- (3-hydroxyphenyl) -3- (tolyl) -2-alkenyl-1-ketone;
(2) obtaining (3-hydroxyphenyl) (3- (tolyl) chloroethylene-2-yl) ketone by the 1- (3-hydroxyphenyl) -3- (tolyl) -2-alkenyl-1-ketone under the action of a catalyst;
(3) cyclizing ((3-hydroxyphenyl) (3- (tolyl) chloroethylene-2-yl) ketone oxide with urea to obtain 4- (3-hydroxyphenyl) -6- (4-methylbenzene) pyrimidinone;
(4) reacting 4- (3-hydroxyphenyl) -6- (4-methylbenzene) pyrimidone with 3-bromopropyne to obtain N-propynyl-4- (3-hydroxyphenyl) -6- (4-methylbenzene) pyrimidone;
(5) reacting the N-propynyl-4- (3-hydroxyphenyl) -6- (4-methylbenzene) pyrimidone with TMS azide to obtain N-methyltriazole-4- (3-hydroxyphenyl) -6- (4-methylbenzene) pyrimidone;
(6) and reacting the N-propynyl-4- (3-hydroxybenzene) -6- (4-methylbenzene) pyrimidone with an isocyanate compound to obtain the aminomethyl pyrimidine compound.
Further limiting, the specific process of step (1) is as follows: adding a certain amount of 3-hydroxyacetophenone into a certain amount of toluene, adding a certain amount of 4-methylbenzaldehyde and sodium methoxide, slowly heating to reflux, pouring the reaction liquid into water after reacting for a period of time, adjusting the pH of the reaction liquid to be neutral by using dilute hydrochloric acid, extracting for multiple times by using dichloromethane, merging organic phases, and separating and purifying by using a silica gel column chromatography to obtain the 1- (3-hydroxyphenyl) -3- (tolyl) -2-alkenyl-1-ketone.
Further limiting, the specific process of step (2) is as follows: adding a certain amount of 1- (3-hydroxyphenyl) -3- (tolyl) -2-alkenyl-1-ketone, a certain amount of catalyst 1, a certain amount of catalyst 2 and a certain amount of catalyst 3 into thionyl chloride and tetrahydrofuran, adding triphenylphosphine palladium chloride, stirring and reacting at 10 ℃ for a period of time, slowly dropwise adding a hydrogen peroxide solution, filtering the reaction solution after dropwise adding, evaporating unreacted thionyl chloride under vacuum conditions, adjusting the pH value to be neutral by using a saturated sodium bicarbonate solution after filtering, extracting for multiple times by using dichloromethane, combining organic phases, concentrating, and separating by using a silica gel column chromatography to obtain (3-hydroxyphenyl) (3- (tolyl) chloroethylene-2-yl) methanone; the molar ratio of the 1- (3-hydroxyphenyl) -3- (tolyl) -2-alkenyl-1-ketone to the catalyst 1 to the catalyst 2 to the catalyst 3 is 1: 0.1: 0.01-0.02: 0.01; the catalyst 1 is phenyl selenium chloride; the catalyst 2 is chromium oxide; the catalyst 3 is titanium chloride.
Further limiting, the specific process of step (3) is as follows: adding a certain amount of (3-hydroxyphenyl) (3- (tolyl) chloroethylene-2-yl) ketone oxide, urea and cesium carbonate into N, N-dimethylformamide, heating to 100 ℃, after the reaction is finished, evaporating in vacuum to remove the N, N-dimethylformamide, then adding ethyl acetate into the concentrate, stirring, adding dilute hydrochloric acid to adjust the pH of the reaction solution to be neutral, discharging a large amount of gas in the process of dropwise adding hydrochloric acid, separating out an organic phase, drying with anhydrous magnesium sulfate, filtering, concentrating the reaction solution, and separating by silica gel column chromatography to obtain the 4- (3-hydroxyphenyl) -6- (4-methylbenzene) pyrimidinone.
Further limiting, the specific process of step (4) is as follows: adding a certain amount of 4- (3-hydroxyphenyl) -6- (4-methylbenzene) pyrimidone and triethylamine into dichloromethane, then adding dichloromethane solution dissolved with 3-bromopropyne, refluxing and stirring for reaction for a period of time after dropwise addition is finished, adding water, adjusting the pH of a reaction solution to be neutral by using dilute hydrochloric acid, separating out an organic phase, extracting a water phase by using dichloromethane for multiple times, combining the organic phases, drying by using anhydrous magnesium sulfate, and concentrating to obtain the N-propynyl-4- (3-hydroxyphenyl) -6- (4-methylbenzene) pyrimidone.
Further limiting, the specific process of step (5) is as follows: adding a certain amount of N-propynyl-4- (3-hydroxybenzene) -6- (4-methylbenzene) pyrimidone and TMS azide into a mixed solvent, uniformly stirring, adding a catalyst, reacting at the temperature of about 80 ℃, adding dichloromethane into a reaction solution after stirring reaction is finished, filtering the reaction solution, concentrating the filtrate, adding dichloromethane, washing with water for multiple times, and finally concentrating and separating by silica gel column chromatography to obtain a triazole compound; the catalyst is cuprous chloride or cuprous iodide or sodium ascorbate and copper sulfate pentahydrate; the molar ratio of the N-propynyl-4- (3-hydroxybenzene) -6- (4-methylbenzene) pyrimidone to the cuprous chloride or cuprous iodide is 1: 0.1; the mass ratio of the N-propynyl-4- (3-hydroxybenzene) -6- (4-methylbenzene) pyrimidone to the sodium ascorbate to the copper sulfate pentahydrate is 1: 0.1: 0.1; the molar ratio of the N-propynyl-4- (3-hydroxybenzene) -6- (4-methylbenzene) pyrimidone to the TMS azide is 1: 1.1; the mixed solvent is a mixed solvent of tert-butyl alcohol, water and tetrahydrofuran.
Further limiting, the specific process of step (6) is as follows: adding a certain amount of N-propynyl-4- (3-hydroxybenzene) -6- (4-methylbenzene) pyrimidone and triethylamine into dichloromethane, stirring at room temperature for a period of time, adding a dichloromethane solution in which an isocyanate compound is dissolved, continuously stirring at room temperature, adding into water after the reaction is finished, adjusting the pH of a reaction solution to be neutral by using dilute hydrochloric acid, stirring at room temperature, separating an organic phase, extracting the reaction solution by using dichloromethane for multiple times by using an aqueous phase, combining the organic phases, drying by using anhydrous magnesium sulfate, and concentrating to obtain the target compound.
The invention has the following beneficial effects: firstly, in terms of synthesis, the following two synthesis methods are available for pyrimidine compounds: 1, synthesizing chalcone and urea; 2, aldehyde, ketone and urea are subjected to a three-component one-pot method; the former method firstly prepares aldehyde and arone into chalcone, then reacts with urea, the reaction time is long, the reaction yield is low, and the post-treatment is relatively complex because a large amount of concentrated hydrochloric acid is used as a catalyst in the reaction; the latter method needs expensive and inaccessible catalyst and volatile and toxic organic solvent as reaction medium, and does not accord with the trend of greening organic reaction; the preparation method creatively adopts a positioning olefin chlorination method, and expensive catalysts are not used in the cyclization process of urea. In the aspect of activity, an antibacterial activity test is carried out by an oxford cup method to find that the target compound has good antibacterial action.
Drawings
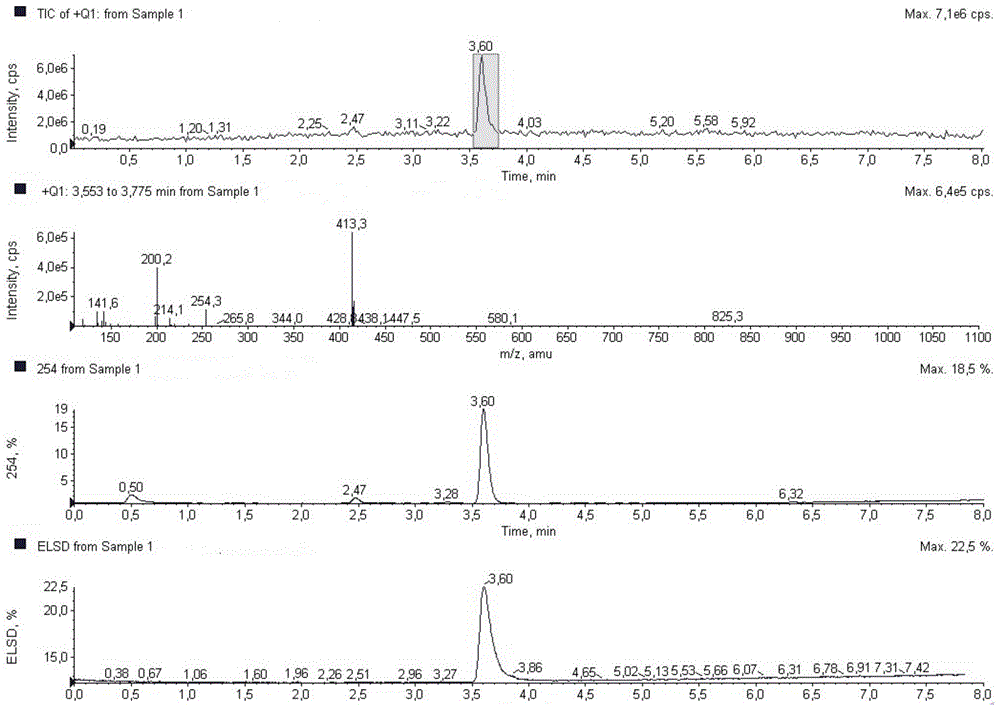
FIG. 1 conventional mass spectra of formate compounds
Detailed Description
The present invention is described in further detail below with reference to examples, but it should not be construed that the scope of the above subject matter of the present invention is limited to the following examples, and that all the technologies realized based on the above subject matter of the present invention belong to the scope of the present invention.
Example 1

Adding 13.5g of 3-hydroxyacetophenone into 150mL of toluene in a reaction bottle with a water separator, stirring and dissolving, then adding 12g of liquid 4-methylbenzaldehyde and 11g of sodium methoxide, slowly heating to reflux, discharging water generated in the reaction process through the water separator in the reflux reaction process, after reacting for 3h, monitoring the complete reaction of raw materials by TLC (thin layer chromatography), evaporating and removing 50mL of toluene under reduced pressure under a vacuum condition, then pouring the reaction liquid into 200mL of water, adjusting the pH of the reaction liquid to be neutral by using dilute hydrochloric acid, then extracting for 3 times by using 100mL of dichloromethane, combining organic phases, and then carrying out silica gel column chromatography separation and purification to obtain 18.1g of 1- (3-hydroxyphenyl) -3- (tolyl) -2-alkenyl-1-ketone; LC-MS (ESI) M/z 239[ M + H]+。
Example 2

Adding 24g of 1- (3-hydroxyphenyl) -3- (tolyl) -2-alkenyl-1-ketone, 1.9g of phenyl selenium chloride, 0.3g of chromium oxide and 0.2g of titanium chloride into 200mL of thionyl chloride and 200mL of tetrahydrofuran in a reaction device with stirring, adding 0.7g of palladium triphenylphosphine chloride, stirring and reacting at 10 ℃ for 6.5h to obtain a light green solution, slowly dropwise adding 150mL of hydrogen peroxide solution (30%), filtering the reaction solution after dropwise adding, evaporating unreacted thionyl chloride under vacuum conditions, adjusting the pH to be neutral by using saturated sodium bicarbonate solution for filtering, adding activated carbon, stirring and adsorbing the color generated by the chromium oxide in the solution, filtering the reaction solution again, extracting with 100mL of dichloromethane for multiple times, combining organic phases, drying with anhydrous magnesium sulfate, concentrating, the concentrate was subjected to silica gel column chromatography to give 11.9g of (3-hydroxyphenyl) (3- (tolyl) chloroethylene-2-yl) methanone; LC-MS (ESI) M/z273[ M + H]+;1H NMR(400MHz,DMSO-d6):δ9.19(s,1H),7.83-7.79(m,2H),7.72(s,1H),7.49(t,J1=8.0Hz,J2=4.0Hz,2H),7.31-7.28(m,2H),7.09(dd,J1=8.0Hz,J2=8.0Hz,2H),2.34(s,3H)。
Example 3

Adding 24g of 1- (3-hydroxyphenyl) -3- (tolyl) -2-alkenyl-1-ketone, 1.9g of phenyl selenium chloride, 0.15g of chromium oxide and 0.2g of titanium chloride into 200mL of thionyl chloride and 200mL of tetrahydrofuran in a reaction device with stirring, adding 0.7g of palladium triphenylphosphine chloride, stirring and reacting at 10 ℃ for 9h to obtain a light green solution, slowly dropwise adding 150mL of hydrogen peroxide solution (30%), filtering the reaction solution after dropwise adding, evaporating unreacted thionyl chloride under vacuum condition, filtering the solution by using saturated sodium bicarbonate solution to adjust the pH to be neutral, adding activated carbon, stirring and adsorbing the color generated by the chromium oxide in the solution, filtering the reaction solution again, and then drying the reaction solutionThen, the mixture was extracted with 100mL of methylene chloride for several times, the organic phases were combined, dried over anhydrous magnesium sulfate, concentrated, and separated by silica gel column chromatography to obtain 19.3g of (3-hydroxyphenyl) (3- (tolyl) oxyethylene-2-yl) methanone; LC-MS (ESI) M/z273[ M + H]+;1H NMR(400MHz,DMSO-d6):δ9.19(s,1H),7.83-7.79(m,2H),7.72(s,1H),7.49(t,J1=8.0Hz,J2=4.0Hz,2H),7.31-7.28(m,2H),7.09(dd,J1=8.0Hz,J2=8.0Hz,2H),2.34(s,3H)。
Example 4

Adding 24g of 1- (3-hydroxyphenyl) -3- (tolyl) -2-alkenyl-1-ketone, 1.9g of phenyl selenium chloride, 0.15g of chromium oxide and 0.24g of zirconium chloride into 200mL of thionyl chloride and 200mL of tetrahydrofuran in a reaction device with stirring, adding 0.7g of palladium triphenylphosphine chloride, stirring and reacting at 10 ℃ for 24 hours, slowly dropwise adding 150mL of hydrogen peroxide solution (30%), filtering the reaction solution after dropwise adding, evaporating unreacted thionyl chloride under vacuum conditions, filtering, adjusting the pH to be neutral by using a saturated sodium bicarbonate solution, adding activated carbon, stirring the color generated by the chromium oxide in the adsorption solution, filtering the reaction solution again, extracting with 100mL of dichloromethane for multiple times, combining organic phases, drying with anhydrous magnesium sulfate, concentrating, and separating by silica gel column chromatography to obtain (3-hydroxyphenyl) (3- (tolyl) chloroethylene-2-oxide -yl) methanone 23.5 g; LC-MS (ESI) M/z273[ M + H]+;1H NMR(400MHz,DMSO-d6):δ9.19(s,1H),7.83-7.79(m,2H),7.72(s,1H),7.49(t,J1=8.0Hz,J2=4.0Hz,2H),7.31-7.28(m,2H),7.09(dd,J1=8.0Hz,J2=8.0Hz,2H),2.34(s,3H)。
Example 5

(3-hydroxyphenyl) in a reaction flask with stirrerAdding 27g of 3- (tolyl) chloroethylene-2-yl) ketone oxide, 6g of urea and 16g of cesium carbonate into 250mL of N, N-dimethylformamide, heating to 100 ℃, reacting for 15h, monitoring the reaction completion of raw materials by TLC, evaporating a solvent N, N-dimethylformamide in vacuum, adding ethyl acetate into a concentrate, stirring, adding diluted hydrochloric acid to adjust the pH of the reaction solution to be neutral, releasing a large amount of gas in the process of dropwise adding hydrochloric acid, controlling the stirring rate to prevent flushing, separating an organic phase, drying with anhydrous magnesium sulfate, filtering, concentrating the reaction solution, and separating by silica gel column chromatography to obtain 14.7g of 4- (3-hydroxybenzene) -6- (4-methylbenzene) pyrimidinone; LC-MS (ESI) M/z 279[ M + H]+;1H NMR(400MHz,DMSO-d6):δ10.29(s,1H),8.71(s,1H),7.62-7.60(m,1H),7.55(d,J=8.0Hz,1H),7.35-7.30(m,3H),7.24(t,J1=4.0Hz,J24.0Hz,1H),7.04-6.99(m,2H),5.81(s,1H),2.36(s, 3H); calculated value of elemental analysis [ C17H14N2O2]C, 73.37; h, 5.07; n,10.07, found C, 73.31; h, 5.09; and N, 10.03.
Example 6

In a reaction flask equipped with a stirrer, 27g of (3-hydroxyphenyl) (3- (tolyl) oxido-chloroethyl-2-yl) methanone and 6.5g of sodium azide were added to 300mL of acetonitrile, and the mixture was heated to 50 ℃ first, reacted for 3 hours, concentrated, then adding 200mL of N, N-dimethylformamide, 8g of cesium carbonate and 6g of urea, heating to 70 ℃, reacting for 2h, evaporating N, N-dimethylformamide in vacuum, then adding ethyl acetate into the concentrate, stirring, adding dilute hydrochloric acid to adjust the pH of the reaction solution to be neutral, discharging a large amount of gas in the process of dropwise adding hydrochloric acid, controlling the stirring speed to prevent flushing, then separating out an organic phase, drying the organic phase by using anhydrous magnesium sulfate, filtering the organic phase, concentrating the reaction solution, and recrystallizing the reaction solution by using methanol to obtain 25.1g of 4- (3-hydroxybenzene) -6- (4-methylbenzene) pyrimidone; LC-MS (ESI) M/z 279[ M + H]+;1H NMR(400MHz,DMSO-d6):δ10.29(s,1H),8.71(s,1H),7.62-7.60(m,1H),7.55(d,J=8.0Hz,1H),7.35-7.30(m,3H),7.24(t,J1=4.0Hz,J24.0Hz,1H),7.04-6.99(m,2H),5.81(s,1H),2.35(s, 3H); calculated value of elemental analysis [ C17H14N2O2]C, 73.37; h, 5.07; n,10.07, found C, 73.31; h, 5.09; and N, 10.03.
Example 7

Under the protection of nitrogen, adding 28g of 4- (3-hydroxyphenyl) -6- (4-methylbenzene) pyrimidinone and 20g of triethylamine into 200mL of dichloromethane, stirring at room temperature for 30min, then adding 50mL of dichloromethane dissolved with 13.5g of 3-bromopropyne, refluxing and stirring for reaction for 1h after dropwise addition, adding 150mL of water, adjusting the pH of a reaction solution to be neutral by using diluted hydrochloric acid, separating out an organic phase, extracting an aqueous phase for 3 times by using 20mL of dichloromethane, combining the organic phases, drying by using anhydrous magnesium sulfate, and concentrating to obtain 21.9g of N-propynyl-4- (3-hydroxyphenyl) -6- (4-methylbenzene) pyrimidinone; LC-MS (ESI) M/z 317[ M + H]+。
Example 8

Under the protection of nitrogen, adding 3.2g of N-propynyl-4- (3-hydroxybenzene) -6- (4-methylbenzene) pyrimidone, 1.2g of TMS azide, 20mL of tert-butyl alcohol, 20mL of water and 20mL of tetrahydrofuran into a reaction bottle, uniformly stirring, then adding 0.19g of cuprous iodide, stirring to react for 21h at the reaction temperature of about 80 ℃, monitoring the complete reaction of raw materials by TLC, adding 100mL of dichloromethane into the reaction liquid, filtering the reaction liquid, adding 50mL of dichloromethane into the filtrate after concentrating, washing for multiple times by using 10mL of water, and finally concentrating and separating by using a silica gel column chromatography to obtain 1.4g of triazole compound; LC-MS (ESI) M/z360[ M + H]+;1H NMR(400MHz,DMSO-d6):δ11.71(s,1H),9.12(s,1H),7.79(d,J=12.0Hz,1H),7.61(s,1H),7.53(d,J=8.0Hz,1H),7.39-7.35(m,2H),7.33(d,J=8.0Hz,1H),7.22-7.19(m,1H),7.06-7.01(m,2H),5.83(s,1H),4.37(s2H),2.35(s, 3H); calculated value of elemental analysis [ C20H17N5O2]C, 66.84; h, 4.77; n,19.49, found C, 66.76; h, 4.75; n, 19.43.
Example 9

Under the protection of nitrogen, adding 3.2g of N-propynyl-4- (3-hydroxybenzene) -6- (4-methylbenzene) pyrimidine, 1.2g of TMS azide, 20mL of tert-butyl alcohol, 20mL of water and 20mL of tetrahydrofuran into a reaction bottle, uniformly stirring, then adding 0.1g of cuprous chloride, stirring to react for 19 hours at the reaction temperature of about 80 ℃, monitoring the complete reaction of raw materials by TLC, adding 100mL of dichloromethane into the reaction liquid, filtering the reaction liquid, concentrating the filtrate, adding 50mL of dichloromethane, washing for multiple times by using 10mL of water, and finally concentrating and separating by using a silica gel column chromatography to obtain 1.9g of triazole compound; LC-MS (ESI) M/z360[ M + H]+;1H NMR(400MHz,DMSO-d6) δ 11.71(s,1H),9.12(s,1H),7.79(d, J ═ 12.0Hz,1H),7.61(s,1H),7.53(d, J ═ 8.0Hz,1H),7.39-7.35(m,2H),7.33(d, J ═ 8.0Hz,1H),7.22-7.19(m,1H),7.06-7.01(m,2H),5.83(s,1H),4.37(s,2H),2.35(s, 3H); calculated value of elemental analysis [ C20H17N5O2]C, 66.84; h, 4.77; n,19.49, found C, 66.76; h, 4.75; n, 19.43.
Example 10

Adding 3.2g of N-propynyl-4- (3-hydroxybenzene) -6- (4-methylbenzene) pyrimidine, 1.2g of TMS azide, 50mL of tert-butyl alcohol, 50mL of water and 50mL of tetrahydrofuran into a reaction bottle under the protection of nitrogen, uniformly stirring, adding 0.32g of sodium ascorbate and 0.32g of blue vitriol after uniformly stirring, stirring for reacting for 24 hours when the reaction temperature is about 80 ℃, monitoring the reaction completion of raw materials by TLC, adding 100mL of dichloromethane into the reaction liquid, filtering the reaction liquid, adding 50mL of dichloromethane after concentrating the filtrate, washing with 10mL of water for multiple times, and finally filtering the reaction liquidThen concentrating and separating by silica gel column chromatography to obtain 3.1g of triazole compounds; LC-MS (ESI) M/z360[ M + H]+;1H NMR(400MHz,DMSO-d6) δ 11.71(s,1H),9.12(s,1H),7.79(d, J ═ 12.0Hz,1H),7.61(s,1H),7.53(d, J ═ 8.0Hz,1H),7.39-7.35(m,2H),7.33(d, J ═ 8.0Hz,1H),7.22-7.19(m,1H),7.06-7.01(m,2H),5.83(s,1H),4.37(s,2H),2.35(s, 3H); calculated value of elemental analysis [ C20H17N5O2]C, 66.84; h, 4.77; n,19.49, found C, 66.76; h, 4.75; n, 19.43.
Example 11

Adding 32g of N-propynyl-4- (3-hydroxybenzene) -6- (4-methylbenzene) pyrimidine, 10g of triethylamine and 300mL of dichloromethane into a four-neck flask, stirring at room temperature for 30min, then adding 70mL of dichloromethane solution dissolved with 10g of allyl isocyanate, continuing stirring at room temperature for 8h for reaction, then adding into 500mL of water, adjusting the pH of the reaction solution to be neutral by using dilute hydrochloric acid, stirring at room temperature for 30min, separating out an organic phase, extracting the reaction solution for 5 times by using 200mL of dichloromethane for a water phase, combining the organic phases, drying by using anhydrous magnesium sulfate, and concentrating to obtain 31.6g of carbamate compound; LC-MS (ESI) M/z400[ M + H]+;1H NMR(400MHz,DMSO-d6) δ 8.68(s,1H),7.82(d, J ═ 4.0Hz,1H),7.75(s,1H),7.59 to 7.51(m,3H),7.48(d, J ═ 8.0Hz,1H),7.19(d, J ═ 12.0Hz,2H),5.85(s,1H),5.81(d, J ═ 8.0Hz,1H),5.07(d, J ═ 8.0Hz,2H),4.19 to 4.15(m,2H),3.72 to 3.69(m,2H),3.15 to 3.13(m,1H),2.38(s, 3H); calculated value of elemental analysis [ C24H21N3O3]C, 72.17; h, 5.30; n,10.52, found C, 72.09; h, 5.33; n, 10.58.
Example 12

Adding 32g of N-propynyl-4- (3-hydroxybenzene) -6- (4-methylbenzene) pyrimidine and three10g of ethylamine and 300mL of dichloromethane, stirring at room temperature for 30min, then adding 70mL of dichloromethane solution in which 13.5g of cyclopentyl formyl chloride is dissolved, continuing to stir at room temperature for reaction for 1h, then adding into 300mL of water, adjusting the pH of the reaction solution to be neutral by using dilute hydrochloric acid, stirring at room temperature for 30min, separating out an organic phase, extracting the reaction solution by using 200mL of dichloromethane for 5 times for a water phase, combining the organic phases, drying by using anhydrous magnesium sulfate, and concentrating to obtain 35.1g of formate compounds; LC-MS (ESI) M/z413[ M + H]+;1H NMR(400MHz,DMSO-d6):δ7.96-7.94(m,1H),7.68(s,1H),7.38-7.34(m,2H),7.32-7.29(m,2H),7.15(d,J=12.0Hz,2H),5.88(s,1H),3.74-3.70(m,2H),3.17-3.15(m,1H),2.40(s,3H),2.25(dd,J1=8.0Hz,J28.0Hz,1H),1.89-1.85(m,2H),1.83-1.78(m,2H),1.71-1.65(m, 4H); calculated value of elemental analysis [ C26H24N2O3]C, 75.71; h, 5.86; n,6.79, found C, 75.62; h, 5.89; n, 6.74.
Example 13
And (3) testing antibacterial activity: testing the bacteriostatic activity of the pyrimidine compounds on escherichia coli and staphylococcus aureus by an oxford cup agar diffusion method; preparing a dimethyl sulfoxide solution with the concentration of 1mg/mL of pyrimidine compounds, taking the dimethyl sulfoxide solution of 1mg/mL of penicillin as a positive control, and taking a solvent dimethyl sulfoxide as a blank control; each sample is cultured for 24h at 37 ℃ in a repeated way for 5 times, in the culture process, on one hand, the test bacteria start to grow, on the other hand, the antibiotics are diffused in a spherical shape, and the closer to the cup, the higher the antibiotic concentration is, and the farther from the cup, the smaller the antibiotic concentration is. As the concentration of the antibiotic is reduced, a minimum inhibitory concentration zone exists, bacteria cannot grow in the zone range and are in a transparent circle, namely an 'inhibitory zone', and the inhibitory diameter is taken as the average value.


As can be seen from the table above, the obtained pyrimidine target compounds have better inhibition effect on escherichia coli than that of oxacillin, and the formate compounds have the best effect, but have weaker inhibition effect on staphylococcus aureus than that of oxacillin.
Example 14
Cytotoxicity experiments: we prepared the human normal cell 293T with viability into a single cell suspension of 50000cells/mL using the corresponding culture medium, and then inoculated it into a 96-well plate at 100. mu.L per well. The 96-well plate was placed at 37 ℃ in 5% CO2Culturing in an incubator for 24 h. The resulting formate compound (compound of example 12) was formulated to the desired concentration: 0.16. mu. mol/L, 0.8. mu. mol/L, 4.0. mu. mol/L, 20.0. mu. mol/L, 100. mu. mol/L. From CO2The 96-well plate is taken out from the incubator, 100 mu L of drug-containing culture medium is added into each well, and 3 multiple wells are simultaneously arranged for each concentration of drug. As a blank well, an equal volume of the corresponding culture medium was added. Placing it at 37 ℃ and 5% CO2Culturing for 72h in an incubator. Each drug was tested in triplicate with the same batch of cells at different passage numbers. After 72 hours, 20. mu.L of MTT solution (5 mg/mL) was added to each well in the dark, and CO addition was continued2Culturing for 4h in an incubator, absorbing supernatant by using a pipette gun, adding 150 mu L DMSO into each hole, placing a shaking table for 5min to mix uniformly, measuring the absorbance OD value of the mixture at the wavelength of 562nm by using a microplate reader, and calculating the cell proliferation inhibition rate by the following method: inhibition rate of cell proliferation [ OD ]Control-ODExperiment of]/ODControlX is 100%; the detection shows that the formate compounds have the inhibition rate IC on 293T cells50It was 105.2. mu. mol/L.
The foregoing embodiments illustrate the principles, principal features and advantages of the invention, and it will be understood by those skilled in the art that the invention is not limited to the foregoing embodiments, which are merely illustrative of the principles of the invention, and that various changes and modifications may be made therein without departing from the scope of the principles of the invention.
Claims (9)

Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN2020104009250 | 2020-05-13 | ||
| CN202010400925.0A CN111559985A (en) | 2020-05-13 | 2020-05-13 | Oxazolone compounds with bactericidal effect and preparation method thereof |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN112341402A true CN112341402A (en) | 2021-02-09 |
| CN112341402B CN112341402B (en) | 2021-05-14 |
Family
ID=72070954
Family Applications (3)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN202010400925.0A Pending CN111559985A (en) | 2020-05-13 | 2020-05-13 | Oxazolone compounds with bactericidal effect and preparation method thereof |
| CN202011442396.7A Expired - Fee Related CN112321523B (en) | 2020-05-13 | 2020-12-08 | A kind of preparation method and application of oxazolone compound which can be used for antibacterial in patient care process |
| CN202011441230.3A Expired - Fee Related CN112341402B (en) | 2020-05-13 | 2020-12-08 | A kind of preparation method and application of pyrimidine compound that can be used for antibacterial in medical care process |
Family Applications Before (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN202010400925.0A Pending CN111559985A (en) | 2020-05-13 | 2020-05-13 | Oxazolone compounds with bactericidal effect and preparation method thereof |
| CN202011442396.7A Expired - Fee Related CN112321523B (en) | 2020-05-13 | 2020-12-08 | A kind of preparation method and application of oxazolone compound which can be used for antibacterial in patient care process |
Country Status (1)
| Country | Link |
|---|---|
| CN (3) | CN111559985A (en) |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN112574215B (en) * | 2020-12-15 | 2021-08-10 | 河南科技大学第一附属医院 | Preparation method and application of benzoxazole compound for hospital disinfection |
| CN115010677B (en) * | 2022-07-15 | 2024-05-03 | 扬州市普林斯医药科技有限公司 | Preparation method of 4-phenyl-2 (3H) -oxazolone |
Citations (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4663329A (en) * | 1984-05-04 | 1987-05-05 | Sanofi | Thienopyridinone derivatives and anti-bacterial compositions containing them |
| WO2005019213A1 (en) * | 2003-08-22 | 2005-03-03 | Pharmacia & Upjohn Company Llc | N-aryl-2-cyanooxazolidinones and their derivatives |
| WO2007048065A2 (en) * | 2005-10-21 | 2007-04-26 | Exelixis, Inc. | Pyrimidinones as casein kinase ii (ck2) modulators |
| WO2008133955A1 (en) * | 2007-04-25 | 2008-11-06 | Exelixis, Inc. | 6-phenylpyrimidinones as pim modulators |
| CN101671336A (en) * | 2009-09-23 | 2010-03-17 | 辽宁利锋科技开发有限公司 | Aromatic heterocyclic pyrimidine derivative and analogue, preparation method and application thereof |
| CN105229012A (en) * | 2013-01-23 | 2016-01-06 | 恩塔西斯治疗有限公司 | Be used for the treatment of the Compounds and methods for of bacteriological infection |
| WO2016049586A2 (en) * | 2014-09-25 | 2016-03-31 | University Of Notre Dame Du Lac | Non-beta lactam antibiotics |
| CN108349968A (en) * | 2015-07-28 | 2018-07-31 | 维奥梅生物科学私人有限公司 | Antimicrobial Therapeutics and Preventives |
| CN109776545A (en) * | 2018-12-24 | 2019-05-21 | 赵言元 | A kind of triazolo pyrimidine antibacterial agent and its composition pesticide |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| BRPI0407252A (en) * | 2003-02-07 | 2006-01-31 | Warner Lambert Co | Antibacterial agents. |
| CN110372624A (en) * | 2019-08-13 | 2019-10-25 | 北京海美桐医药科技有限公司 | A kind of Oxazolidinone derivative, preparation method and application |
-
2020
- 2020-05-13 CN CN202010400925.0A patent/CN111559985A/en active Pending
- 2020-12-08 CN CN202011442396.7A patent/CN112321523B/en not_active Expired - Fee Related
- 2020-12-08 CN CN202011441230.3A patent/CN112341402B/en not_active Expired - Fee Related
Patent Citations (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4663329A (en) * | 1984-05-04 | 1987-05-05 | Sanofi | Thienopyridinone derivatives and anti-bacterial compositions containing them |
| WO2005019213A1 (en) * | 2003-08-22 | 2005-03-03 | Pharmacia & Upjohn Company Llc | N-aryl-2-cyanooxazolidinones and their derivatives |
| WO2007048065A2 (en) * | 2005-10-21 | 2007-04-26 | Exelixis, Inc. | Pyrimidinones as casein kinase ii (ck2) modulators |
| WO2008133955A1 (en) * | 2007-04-25 | 2008-11-06 | Exelixis, Inc. | 6-phenylpyrimidinones as pim modulators |
| CN101671336A (en) * | 2009-09-23 | 2010-03-17 | 辽宁利锋科技开发有限公司 | Aromatic heterocyclic pyrimidine derivative and analogue, preparation method and application thereof |
| CN105229012A (en) * | 2013-01-23 | 2016-01-06 | 恩塔西斯治疗有限公司 | Be used for the treatment of the Compounds and methods for of bacteriological infection |
| WO2016049586A2 (en) * | 2014-09-25 | 2016-03-31 | University Of Notre Dame Du Lac | Non-beta lactam antibiotics |
| CN108349968A (en) * | 2015-07-28 | 2018-07-31 | 维奥梅生物科学私人有限公司 | Antimicrobial Therapeutics and Preventives |
| CN109776545A (en) * | 2018-12-24 | 2019-05-21 | 赵言元 | A kind of triazolo pyrimidine antibacterial agent and its composition pesticide |
Non-Patent Citations (4)
| Title |
|---|
| SAURABH GARG等: "Investigation of C-5 alkynyl (alkynyloxy or hydroxymethyl) and/or N-3 propynyl substituted pyrimidine nucleoside analogs as a new class of antimicrobial agents", 《BIOORGANIC & MEDICINAL CHEMISTRY》 * |
| SHIWANI SHUKLA等: "4,6-Diaryl/heteroarylpyrimidin-2(1H)-ones as a New Class of Xanthine Oxidase Inhibitors", 《ARCH. PHARM. CHEM. LIFE SCI.》 * |
| 唐传球等: "3种含1,2,4-三氮唑噻吩并嘧啶酮衍生物的合成及抗菌活性", 《长江大学学报(自科版)》 * |
| 喻建波: "新型嘧啶酮及四嗪衍生物的合成与性质", 《中国优秀硕士学位论文全文数据库 工程科技Ⅰ辑》 * |
Also Published As
| Publication number | Publication date |
|---|---|
| CN111559985A (en) | 2020-08-21 |
| CN112321523A (en) | 2021-02-05 |
| CN112341402B (en) | 2021-05-14 |
| CN112321523B (en) | 2021-04-30 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US20230056365A1 (en) | PROCESS FOR MAKING CRYSTALLINE 2-(3-(4-(7H-PYRROLO[2,3-d]PYRIMIDIN-4-YL)-1H-PYRAZOL-1-YL)-1-(CYCLOPROPYLSULFONYL)AZETIDIN-3-YL)ACETONITRILE | |
| KR20180101643A (en) | Crystalline forms of a macrolide, and uses therefor | |
| CN112341402B (en) | A kind of preparation method and application of pyrimidine compound that can be used for antibacterial in medical care process | |
| CN114507158B (en) | Pleuromutilin alpha-cyano cinnamic acid ester compounds with drug-resistant bacteria resisting activity and preparation method and application thereof | |
| CN111646937B (en) | Propenone derivative of N-acetyl ciprofloxacin and preparation method and application thereof | |
| CN102746309B (en) | 1-N-ethyl-4-N-2'-substituted acylhydrazine-1H-pyrazol [3, 4-d] miazines derivative as well as preparation method and application thereof | |
| CN103360370A (en) | Synthesis of nitroimidazole derivatives and application of nitroimidazole derivatives in field of antibacterial drugs | |
| CN114097804B (en) | Use of a diaminopyrimidine compound in preventing and controlling agricultural pathogens | |
| CN112321580B (en) | Oxazole linked triazole medicine molecule for sterilization and disinfection and preparation method and application thereof | |
| CN112824391A (en) | Propylene ketone derivative of gatifloxacin and preparation method and application thereof | |
| CN112574215B (en) | Preparation method and application of benzoxazole compound for hospital disinfection | |
| CN109251196A (en) | Amino benzo [d] azepine * base quinazoline compounds and its preparation method and application | |
| CN112824396B (en) | A kind of propenone derivative of N-acetyllomefloxacin and its preparation method and application | |
| CN115636815A (en) | Preparation method and application of a novel pomalidomide-linked urea compound | |
| WO2022105845A1 (en) | Glucoside derivative, and preparation method therefor and application thereof | |
| CN109111396B (en) | Quinoline aromatic vinyl derivative and preparation method and application thereof | |
| CN112645940A (en) | 1,3, 4-oxadiazole-norfloxacin heterozygote and preparation method and application thereof | |
| CN110143963B (en) | Pyridazine compound with sterilization and disinfection activity and preparation method and application thereof | |
| CN112661766A (en) | Preparation method and application of benzopyran compound for hospital disinfection | |
| CN118440080B (en) | Triphenylamine cationic compound and preparation method and application thereof | |
| CN114573502B (en) | Pleuromutilin aromatic heterocyclic acrylate compound, and synthetic method and application thereof | |
| CN119528934B (en) | 2-Pyridone thiophene compound, preparation method, use and anti-tuberculosis drug thereof | |
| CN115417857B (en) | Piperidine alkaloid in Chinese medicinal Alangium chinense and extraction purification and semisynthesis method and application thereof | |
| CN111646938B (en) | Propenone derivative of pefloxacin, and preparation method and application thereof | |
| CN112824388A (en) | Acrylic ketone derivative of norfloxacin, and preparation method and application thereof |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PB01 | Publication | ||
| PB01 | Publication | ||
| SE01 | Entry into force of request for substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| GR01 | Patent grant | ||
| GR01 | Patent grant | ||
| CF01 | Termination of patent right due to non-payment of annual fee | ||
| CF01 | Termination of patent right due to non-payment of annual fee |
Granted publication date: 20210514 |