CN112403473A - Synthetic method for preparing reforming catalyst through MOFs - Google Patents
Synthetic method for preparing reforming catalyst through MOFs Download PDFInfo
- Publication number
- CN112403473A CN112403473A CN202011229347.5A CN202011229347A CN112403473A CN 112403473 A CN112403473 A CN 112403473A CN 202011229347 A CN202011229347 A CN 202011229347A CN 112403473 A CN112403473 A CN 112403473A
- Authority
- CN
- China
- Prior art keywords
- precursor
- mofs
- catalyst
- reaction
- auxiliary agent
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
Classifications
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/70—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of the iron group metals or copper
- B01J23/76—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of the iron group metals or copper combined with metals, oxides or hydroxides provided for in groups B01J23/02 - B01J23/36
- B01J23/80—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of the iron group metals or copper combined with metals, oxides or hydroxides provided for in groups B01J23/02 - B01J23/36 with zinc, cadmium or mercury
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/002—Mixed oxides other than spinels, e.g. perovskite
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/70—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of the iron group metals or copper
- B01J23/76—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of the iron group metals or copper combined with metals, oxides or hydroxides provided for in groups B01J23/02 - B01J23/36
- B01J23/78—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of the iron group metals or copper combined with metals, oxides or hydroxides provided for in groups B01J23/02 - B01J23/36 with alkali- or alkaline earth metals
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/70—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of the iron group metals or copper
- B01J23/76—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of the iron group metals or copper combined with metals, oxides or hydroxides provided for in groups B01J23/02 - B01J23/36
- B01J23/83—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of the iron group metals or copper combined with metals, oxides or hydroxides provided for in groups B01J23/02 - B01J23/36 with rare earths or actinides
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J35/00—Catalysts, in general, characterised by their form or physical properties
- B01J35/20—Catalysts, in general, characterised by their form or physical properties characterised by their non-solid state
- B01J35/23—Catalysts, in general, characterised by their form or physical properties characterised by their non-solid state in a colloidal state
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J35/00—Catalysts, in general, characterised by their form or physical properties
- B01J35/30—Catalysts, in general, characterised by their form or physical properties characterised by their physical properties
- B01J35/391—Physical properties of the active metal ingredient
- B01J35/393—Metal or metal oxide crystallite size
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J35/00—Catalysts, in general, characterised by their form or physical properties
- B01J35/60—Catalysts, in general, characterised by their form or physical properties characterised by their surface properties or porosity
- B01J35/61—Surface area
- B01J35/615—100-500 m2/g
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J37/00—Processes, in general, for preparing catalysts; Processes, in general, for activation of catalysts
- B01J37/02—Impregnation, coating or precipitation
- B01J37/03—Precipitation; Co-precipitation
- B01J37/031—Precipitation
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J37/00—Processes, in general, for preparing catalysts; Processes, in general, for activation of catalysts
- B01J37/08—Heat treatment
- B01J37/082—Decomposition and pyrolysis
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J37/00—Processes, in general, for preparing catalysts; Processes, in general, for activation of catalysts
- B01J37/08—Heat treatment
- B01J37/082—Decomposition and pyrolysis
- B01J37/086—Decomposition of an organometallic compound, a metal complex or a metal salt of a carboxylic acid
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J37/00—Processes, in general, for preparing catalysts; Processes, in general, for activation of catalysts
- B01J37/08—Heat treatment
- B01J37/10—Heat treatment in the presence of water, e.g. steam
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01B—NON-METALLIC ELEMENTS; COMPOUNDS THEREOF; METALLOIDS OR COMPOUNDS THEREOF NOT COVERED BY SUBCLASS C01C
- C01B3/00—Hydrogen; Gaseous mixtures containing hydrogen; Separation of hydrogen from mixtures containing it; Purification of hydrogen; Reversible storage of hydrogen
- C01B3/02—Production of hydrogen; Production of gaseous mixtures containing hydrogen
- C01B3/32—Production of hydrogen; Production of gaseous mixtures containing hydrogen by reaction of gaseous or liquid organic compounds with gasifying agents, e.g. water, carbon dioxide or air
- C01B3/34—Production of hydrogen; Production of gaseous mixtures containing hydrogen by reaction of gaseous or liquid organic compounds with gasifying agents, e.g. water, carbon dioxide or air by reaction of hydrocarbons with gasifying agents
- C01B3/38—Production of hydrogen; Production of gaseous mixtures containing hydrogen by reaction of gaseous or liquid organic compounds with gasifying agents, e.g. water, carbon dioxide or air by reaction of hydrocarbons with gasifying agents using catalysts
- C01B3/40—Production of hydrogen; Production of gaseous mixtures containing hydrogen by reaction of gaseous or liquid organic compounds with gasifying agents, e.g. water, carbon dioxide or air by reaction of hydrocarbons with gasifying agents using catalysts characterised by the catalyst
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J2523/00—Constitutive chemical elements of heterogeneous catalysts
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01B—NON-METALLIC ELEMENTS; COMPOUNDS THEREOF; METALLOIDS OR COMPOUNDS THEREOF NOT COVERED BY SUBCLASS C01C
- C01B2203/00—Integrated processes for the production of hydrogen or synthesis gas
- C01B2203/02—Processes for making hydrogen or synthesis gas
- C01B2203/0205—Processes for making hydrogen or synthesis gas containing a reforming step
- C01B2203/0227—Processes for making hydrogen or synthesis gas containing a reforming step containing a catalytic reforming step
- C01B2203/0238—Processes for making hydrogen or synthesis gas containing a reforming step containing a catalytic reforming step the reforming step being a carbon dioxide reforming step
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01B—NON-METALLIC ELEMENTS; COMPOUNDS THEREOF; METALLOIDS OR COMPOUNDS THEREOF NOT COVERED BY SUBCLASS C01C
- C01B2203/00—Integrated processes for the production of hydrogen or synthesis gas
- C01B2203/10—Catalysts for performing the hydrogen forming reactions
- C01B2203/1041—Composition of the catalyst
- C01B2203/1047—Group VIII metal catalysts
- C01B2203/1052—Nickel or cobalt catalysts
- C01B2203/1058—Nickel catalysts
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/52—Improvements relating to the production of bulk chemicals using catalysts, e.g. selective catalysts
Landscapes
- Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Materials Engineering (AREA)
- Physics & Mathematics (AREA)
- Thermal Sciences (AREA)
- Inorganic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Combustion & Propulsion (AREA)
- Catalysts (AREA)
Abstract
The invention discloses a synthesis method for preparing a reforming catalyst by MOFs, which is characterized in that an auxiliary agent precursor M1 and an organic ligand are simultaneously added into an organic solvent, so that the auxiliary agent precursor M1 is fully dissolved to form a transparent clear solution; transfer to reactionHeating the mixture in a kettle for reaction, naturally cooling the mixture to room temperature after the reaction is finished to obtain turbid liquid, and centrifuging, washing and drying the turbid liquid to obtain an M1-MOFs precursor; mixing M1-MOFs precursor and Ni (NO)3)2·6H2Dissolving O and an auxiliary agent precursor M2 in deionized water, adding ammonia water, reacting, and cooling to room temperature; and drying the obtained filter cake in an oven, and roasting in a muffle furnace to obtain the oxidation precursor catalyst. The catalyst has good threshold effect during the coprecipitation reaction by synthesizing MOFs precursors, thereby controlling the dispersion degree and size of Ni, prolonging the service life of the catalyst in the reforming reaction process and ensuring higher catalytic efficiency.
Description
Technical Field
The invention belongs to the technical field of petrochemical industry, and relates to a synthesis method and application of a reforming catalyst obtained by synthesizing MOFs structures.
Background
With the development of industry, the emission of carbon dioxide in human society is increasing year by year. The excessively high carbon dioxide content in the atmosphere has a great negative effect on the climate and ecological balance, and methane, as a clean energy source, has a high application value, but is also a "greenhouse gas" having adverse effects on the environment. Therefore, how to utilize and reduce the emission of carbon dioxide/methane has been a focus of attention. The synthesis gas prepared by reforming methane and carbon dioxide has good economic and environmental protection values while eliminating two greenhouse gases.
Catalysts for catalytic reforming of methane and carbon dioxide can be classified into two types: noble metals and non-noble metals. Among them, the noble metals Rh, Ru and Ir have the best catalytic activity. Although the noble metal catalyst has good catalytic activity and carbon deposit resistance, the industrial application of the noble metal catalyst is poor in economic benefit due to limited resources and high price. On the contrary, although the catalytic activity and the carbon deposit resistance of the non-noble metal are not as good as those of noble metals, the non-noble metal is low in price and rich in resources, so that the main research content is still the non-noble metal catalyst. Wherein the catalytic activity sequence of the non-noble metal is Ni & gt Co & gt Cu & gt Fe. Therefore, the nickel-based catalyst has been the focus of research on the carbon dioxide reforming reaction of methane.
Patent CN106391020A discloses a method for preparing a methane carbon dioxide reforming catalyst prepared by using a carbon material as a carrier to load active metal. The process, by subcritical H2Carbon material as carrier of methane-carbon dioxide reforming catalyst prepared from O-CO modified lignite, and reforming catalyst prepared by using carbon material as carrier to load active metal. The catalyst can be used at lower temperature, saves energy, but has the defects of complex synthesis process and difficult control of conditions, and is difficult to apply on a large scale.
Patent CN102240566B discloses a method for preparing a methane carbon dioxide reforming catalyst. The method comprises the steps of immersing semicoke in hydrogen peroxide for modification, impregnating with an auxiliary agent precursor, and roasting with nitrogen to obtain a catalyst precursor. The catalyst prepared by the method has high activity, but hydrogen peroxide is used in the synthesis process, so that the catalyst has certain danger; air is insulated during roasting, and higher cost is needed, so that large-scale preparation is not facilitated.
The synthesis gas prepared by reforming methane and carbon dioxide is an important component of C1 chemical research, and is an effective path for methane conversion and carbon dioxide utilization. The VIII transition metals (Ni, Co, Fe, Cu, etc.) are favored for their low cost and high activity, especially for Ni-based catalysts, which exhibit superior performance. However, the Ni-based catalyst has a serious carbon deposit phenomenon, and partial sintering of metal particles occurs during the reaction at a high temperature, and the catalyst is deactivated by the combined action of the two. Therefore, the control of the size of Ni metal particles on the surface of the catalyst and the improvement of the dispersion degree not only solve the problem of carbon deposition, but also solve the key of the problem of catalyst sintering.
Disclosure of Invention
The technical problem to be solved by the invention is as follows: how to control the size of the Ni metal particles on the catalyst surface.
In order to solve the above technical problems, the present invention provides a synthesis method for preparing a reforming catalyst by MOFs, comprising the steps of:
step 1): slowly adding the assistant precursor M1 and the organic ligand into the organic solvent under the condition of vigorous stirring simultaneously to fully dissolve the assistant precursor M1 to form a transparent clear solution; the assistant precursor M1 is Zn (NO)3)2·5H2O、Fe(NO3)3·9H2O and Mg (NO)3)2·6H2At least one of O; the organic ligand is terephthalic acid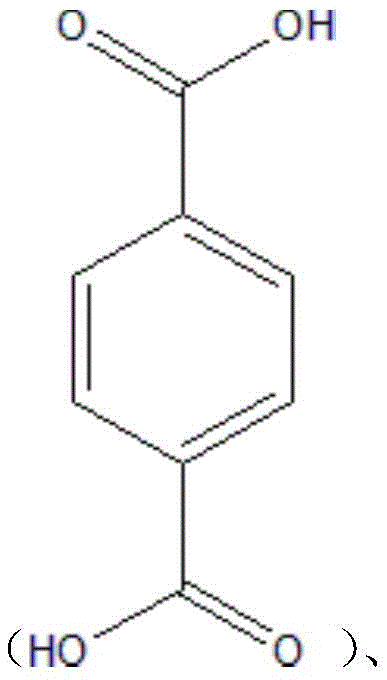 Isophthalic acid
Isophthalic acid Trimesic acid
Trimesic acid Or an organic compound containing one of these three types of substructures;
Or an organic compound containing one of these three types of substructures;
step 2): transferring the solution obtained in the step 1) into a reaction kettle, heating for reaction, naturally cooling to room temperature after the reaction is finished to obtain turbid liquid, and centrifuging, washing and drying the turbid liquid to obtain an M1-MOFs precursor;
step 3): mixing M1-MOFs precursor and Ni (NO)3)2·6H2Dissolving O and an auxiliary agent precursor M2 in deionized water, slowly adding ammonia water to control the pH to be 9-10, violently stirring for reaction, cooling to room temperature, filtering, and washing with deionized water until the filtrate is neutral to obtain a filter cake; the assistant precursor M2 is Ce (NO)3)3·6H2O、Al(NO3)3·9H2O and Zr (NO)3)4·5H2At least one of O;
step 4): and drying the obtained filter cake in an oven, and roasting in a muffle furnace to obtain the oxidation precursor catalyst.
Preferably, the molar ratio of the organic ligand to the auxiliary agent precursor M1 in the step 1) is 1: (0.1-2), preferably 1: (0.5 to 1).
Preferably, the organic solvent in step 1) is N, N-dimethylformamide.
Preferably, the amount of the organic solvent used in step 1) is 200mL of the organic solvent per 0.1mol of the promoter precursor M1.
Preferably, the reaction temperature in the step 2) is 120 ℃ and the reaction time is 24 h.
Preferably, the reaction temperature in the step 3) is 90 ℃ and the reaction time is 24 h.
Preferably, M2 in the filter cake obtained in the step 3)a+And Ni2+And M1b+The molar ratio of the sum of the moles of (a) is 0.25 to 2.5: 1; ni2+And M1b+The molar ratio of (a) to (b) is 0.2-2: 1.
preferably, the drying temperature in the step 4) is 80 ℃, and the time is 12 hours; the roasting temperature is 750 ℃ and the roasting time is 5 h.
Preferably, the oxidation precursor catalyst obtained in step 4) comprises an active component, an assistant precursor M1, an assistant precursor M2, and an organic ligand, wherein the active component is Ni, the assistant precursor M1 is at least one of Zn, Mg, and Fe, and the assistant precursor M2 is at least one of Ce, Al, and Zr.
The invention provides a synthesis method for preparing a reforming catalyst by forming an assistant precursor M1 and an organic ligand into a metal organic framework material (M1-MOFs), then carrying out coprecipitation reaction on the metal organic framework material, Ni and another assistant precursor M2, and finally washing, drying and roasting the obtained product. The catalyst effectively synthesizes MOFs precursor, and has good threshold limiting effect during coprecipitation reaction, thereby controlling the dispersion degree and particle size of Ni, prolonging the service life of the catalyst in the reforming reaction process, and simultaneously ensuring higher catalytic efficiency.
Compared with the prior art, the invention has the following advantages:
(1) the MOFs structure has a certain size of pore diameter, the dispersion degree and size of Ni in the pore diameter can be effectively controlled during synthesis, and the particle size of the finally synthesized oxidation precursor catalyst is smaller than that of a common preparation method, so that better catalytic activity and stability are obtained.
(2) The coprecipitation preparation method is simple and easy to operate and low in cost.
Detailed Description
In order to make the invention more comprehensible, preferred embodiments are described in detail below.
The evaluation procedure for the reforming reaction using the catalyst prepared in the example was as follows:
mixing the oxidation precursor catalyst (0.1g, 80-100 mesh) with analytically pure quartz sand (0.9g, 80-100 mesh), and adding into H2/N2Atmosphere (volume percent)50% each, flow rate 120mL/min), 700 ℃ pre-reduction for 1 h. After completion of reduction, at H2/N2The reaction temperature is raised to 850 ℃ under the atmosphere (50 percent by volume), and after the temperature is raised to 850 ℃, the reaction is switched to CO2/CH4The mixed gas (molar ratio 1.2:1, GHSV: 40000 mL/g.h) undergoes a reforming reaction. After the reaction was stable, the composition of the product was measured on-line by gas chromatography.
Example 1
Weighing Zn (NO)3)2·5H2Dissolving O (0.1mol, 42.93g) in 200mL DMF, adding terephthalic acid (0.02mol, 3.32g) under the condition of vigorous stirring, transferring the solution to a stainless steel reaction kettle with a polytetrafluoroethylene lining after the dissolution is finished, placing the reaction kettle in a constant-temperature oven, reacting for 24h at 120 ℃, cooling, centrifuging and collecting the product, and washing the product with DMF for several times to obtain Zn-MOFs. Prepared Zn-MOFs (0.1mol) and Ni (NO)3)2·6H2O(0.2mol,58.16g)、Al(NO3)3·9H2Dissolving O (0.075mol, 28.14g) in 500mL of deionized water, slowly dropwise adding 50mL of ammonia water under the condition of vigorous stirring, reacting at 90 ℃ for 24 hours under vigorous stirring, cooling to room temperature, filtering, washing with deionized water until the filtrate is neutral to obtain a filter cake, drying at 80 ℃ for 12 hours, and roasting at 750 ℃ for 5 hours to obtain the oxidized precursor catalyst.
Calculating to obtain CO2Conversion 82.01%, CH4Conversion of 87.58%, H in the output2The ratio of/CO is 0.82, the reaction lasts for 300h, CO2、CH4Conversion and H2the/CO ratio remains substantially unchanged.
Example 2
Weighing Mg (NO)3)2·6H2Dissolving O (0.1mol, 25.64g) in 200mL of DMF, adding 2, 5-dihydroxyterephthalic acid (0.15mol, 29.72g) under the condition of vigorous stirring, transferring the solution to a stainless steel reaction kettle with a polytetrafluoroethylene lining after the dissolution is finished, placing the reaction kettle in a constant-temperature oven, reacting for 24 hours at 120 ℃, cooling, centrifuging and collecting the product, and washing the product with DMF for several times to obtain Mg-MOFs. Mixing prepared Mg-MOFs (0.1mol) and Ni (NO)3)2·6H2O(0.05mol,14.54g)、Zr(NO3)4·5H2Dissolving O (0.225mol, 93.48g) in 500mL of deionized water, slowly dropwise adding 65mL of ammonia water under the condition of vigorous stirring, reacting for 24h under vigorous stirring at 90 ℃, cooling to room temperature, filtering, washing with deionized water until the filtrate is neutral to obtain a filter cake, drying for 12h at 80 ℃, and roasting for 5h at 750 ℃ to obtain the oxidation precursor catalyst.
Calculating to obtain CO2Conversion 82.15%, CH4Conversion of 91.62%, H in the output2The ratio of/CO is 0.85, the reaction lasts for 300h, CO2、CH4Conversion and H2the/CO ratio remains substantially unchanged.
Example 3
Weighing Mg (NO)3)2·6H2O (0.1mol, 25.64g) is dissolved in 200mL DMF, 2, 5-dihydroxyterephthalic acid (0.1mol, 19.81g) is added under the condition of vigorous stirring, the solution is transferred to a stainless steel reaction kettle which is lined with polytetrafluoroethylene after the dissolution is finished, the reaction kettle is placed in a constant-temperature oven to react for 24 hours at 120 ℃, and after cooling, the product is collected by centrifugation and washed with DMF for several times to obtain Mg-MOFS. Mixing prepared Mg-MOFS (0.1mol) and Ni (NO)3)2·6H2O(0.1mol,29.08g)、Al(NO3)3·9H2Dissolving O (0.2mol, 75.02g) in 500mL of deionized water, slowly dropwise adding 55mL of ammonia water under the condition of vigorous stirring, reacting at 90 ℃ for 24h under vigorous stirring, cooling to room temperature, filtering, washing with deionized water until the filtrate is neutral to obtain a filter cake, drying at 80 ℃ for 12h, and roasting at 750 ℃ for 5h to obtain the oxidation precursor catalyst.
Calculating to obtain CO2Conversion 88.64%, CH4Conversion of 95.21%, H in the output2The ratio of/CO is 0.89, the reaction lasts for 300h, CO2、CH4Conversion and H2the/CO ratio remains substantially unchanged.
Example 4
Weighing Mg (NO)3)2·6H2O (0.1mol, 25.64g) was dissolved in 200mL of DMF, 2, 5-dihydroxyterephthalic acid (0.2mol, 39.62g) was added with vigorous stirring, and after completion of the dissolution, the mixture was transferred to a column packed with DMFAnd (3) putting the reaction kettle in a stainless steel reaction kettle with a polytetrafluoroethylene lining, reacting for 24 hours at 120 ℃ in a constant-temperature oven, cooling, centrifuging and collecting a product, and washing with DMF (dimethyl formamide) for several times to obtain Mg-MOFs. Mixing prepared Mg-MOFs (0.1mol) and Ni (NO)3)2·6H2O(0.02mol,5.82g)、Ce(NO3)3·6H2Dissolving O (0.3mol, 130.27g) in 500mL of deionized water, slowly dropwise adding 65mL of ammonia water under the condition of vigorous stirring, reacting for 24h under vigorous stirring at 90 ℃, cooling to room temperature, filtering, washing with deionized water until the filtrate is neutral to obtain a filter cake, drying for 12h at 80 ℃, and roasting for 5h at 750 ℃ to obtain the oxidation precursor state catalyst.
Calculating to obtain CO2Conversion 82.25%, CH4Conversion of 86.54%, H in the output2The ratio of/CO is 0.81, the reaction lasts for 300h, CO2、CH4Conversion and H2the/CO ratio remains substantially unchanged.
Example 5
Weighing Fe (NO)3)3·9H2Dissolving O (0.1mol, 40.4g) in 200mL DMF, adding 1,3, 5-trimesic acid (0.08mol, 16.81g) under the condition of vigorous stirring, transferring the solution to a stainless steel reaction kettle with a polytetrafluoroethylene lining after the dissolution is finished, placing the reaction kettle in a constant-temperature oven, reacting for 24 hours at 120 ℃, cooling, centrifuging and collecting products, and washing the products with DMF for several times to obtain Fe-MOFs. Mixing prepared Mg-MOFs (0.1mol) and Ni (NO)3)2·6H2O(0.15mol,43.62g)、Al(NO3)3·9H2Dissolving O (0.125mol, 46.90g) in 500mL of deionized water, slowly dropwise adding 55mL of ammonia water under the condition of vigorous stirring, reacting for 24h under vigorous stirring at 90 ℃, cooling to room temperature, filtering, washing with deionized water until the filtrate is neutral to obtain a filter cake, drying for 12h at 80 ℃, and roasting for 5h at 750 ℃ to obtain the oxidation precursor catalyst.
Calculating to obtain CO2Conversion 86.11%, CH4Conversion rate of 92.84%, H in the output2The ratio of/CO is 0.86, the reaction lasts for 300h, CO2、CH4Conversion and H2the/CO ratio remains substantially unchanged.
Example 6
Weighing Zn (NO)3)2·5H2Dissolving O (0.1mol, 42.93g) in 200mL DMF, adding isophthalic acid (0.15mol, 24.92g) under the condition of vigorous stirring, transferring the solution to a stainless steel reaction kettle with a polytetrafluoroethylene lining after the dissolution is finished, placing the reaction kettle in a constant-temperature oven, reacting for 24h at 120 ℃, cooling, centrifuging and collecting the product, and washing the product with DMF for several times to obtain Zn-MOFs. Prepared Zn-MOFs (0.1mol) and Ni (NO)3)2·6H2O(0.1mol,29.08g)、Al(NO3)3·9H2Dissolving O (0.4mol, 150.05g) in 500mL of deionized water, slowly dropwise adding 75mL of ammonia water under the condition of vigorous stirring, reacting for 24h under vigorous stirring at 90 ℃, cooling to room temperature, filtering, washing with deionized water until the filtrate is neutral to obtain a filter cake, drying for 12h at 80 ℃, and roasting for 5h at 750 ℃ to obtain the oxidation precursor state catalyst.
Calculating to obtain CO2Conversion 83.89%, CH4Conversion rate 89.01%, H in the output2The ratio of/CO is 0.83, the reaction lasts for 300h, CO2、CH4Conversion and H2the/CO ratio remains substantially unchanged.
Comparative example 1
Nickel nitrate hexahydrate (0.1mol, 29.08g) and Mg (NO) were weighed3)2·6H2O(0.1mol,25.64g),Al(NO3)3·9H2Dissolving O (0.1mol, 75.02g) in 500mL deionized water, slowly dropwise adding 55mL ammonia water under the condition of vigorous stirring, reacting for 24h under vigorous stirring at 90 ℃, cooling to room temperature, filtering, washing with deionized water until the filtrate is neutral to obtain a filter cake, drying for 12h at 80 ℃, and roasting for 5h at 750 ℃ to obtain the oxidation precursor state catalyst.
Calculating to obtain CO2Conversion 71.26%, CH4Conversion of 74.61%, H in the output2The ratio of/CO is 0.74, the obvious inactivation phenomenon appears after the reaction is carried out for 300 hours, and the carbon deposition is serious in thermogravimetric analysis of the catalyst.
Table 1 shows a comparison of the performance of the catalysts of examples 1 to 6 and comparative example 1. As can be seen from the experimental procedure for catalyst synthesis, examples 1-6 were all prepared firstAnd when M1-MOFs is obtained, the obtained material is subjected to coprecipitation reaction with Ni and an auxiliary agent precursor M2, and then the obtained product is washed, dried and roasted to obtain the oxide precursor catalyst. The comparative example is the preparation of the catalyst directly by coprecipitation. From the data in the table, it can be clearly found that the average particle size of examples 1-6 is significantly smaller than that of the comparative example, while the difference trend of the specific surface area is opposite, and the smaller particle size and the larger specific surface area can effectively prevent the generation of carbon, improve the carbon deposition resistance of the catalyst, and ensure the high stability and good activity of the catalyst. The catalysts of examples 1-6 and comparative example 1 were used for methane carbon dioxide reforming reaction, and example 3 had the highest CO2And CH4Conversion rate and CO after 300h operation2And CH4The conversion rate is basically kept unchanged, and the catalyst activity and stability are good. The activity of the catalyst of other examples is slightly lower than that of the catalyst of example 3 at first, but the catalyst of other examples also basically maintains stable after running for 300 hours, and has good stability and higher catalytic activity. The initial reactivity of the catalysts of example 3 and comparative example was similar, but the CO of the comparative example was similar2And CH4The conversion rate is rapidly reduced along with the progress of the reforming reaction, and after the reaction for 300 hours, CO is obtained2And CH4The conversion was much lower than that of example 32And CH4The conversion rate is also greatly lower than that of CO in other examples2And CH4And (4) conversion rate. Meanwhile, the carbon deposition of the catalyst bed is found to be serious, which proves that the carbon deposition resistance of the catalyst in the comparison example is poor, and the activity and the stability of the catalyst are far inferior to those of the catalyst prepared by M1-MOFs.
Therefore, the synthesis method can be seen that the M1-MOFs can effectively control the dispersion degree of metal Ni during coprecipitation by utilizing the threshold effect, and then the metal Ni is dried and roasted to obtain the oxidized precursor catalyst, so that the particle size of the Ni catalyst is effectively reduced, the specific surface area is increased, and the high CO content is obtained2And CH4The reforming catalyst with high conversion rate and high stability has simple and easy operation in the synthesis process, and has large-scale preparation and application prospects.
Table 1 comparison of the performance of examples 1-6 with the catalyst of comparative example 1 over 300h of operation

Claims (9)
1. A synthesis process for the preparation of reforming catalysts by means of MOFs, characterized in that it comprises the following steps:
step 1): slowly adding the assistant precursor M1 and the organic ligand into the organic solvent under the condition of vigorous stirring simultaneously to fully dissolve the assistant precursor M1 to form a transparent clear solution; the assistant precursor M1 is Zn (NO)3)2·5H2O、Fe(NO3)3·9H2O and Mg (NO)3)2·6H2At least one of O; the organic ligand is terephthalic acid, isophthalic acid, trimesic acid or an organic compound containing one of the three sub-structures;
step 2): transferring the solution obtained in the step 1) into a reaction kettle, heating for reaction, naturally cooling to room temperature after the reaction is finished to obtain turbid liquid, and centrifuging, washing and drying the turbid liquid to obtain an M1-MOFs precursor;
step 3): mixing M1-MOFs precursor and Ni (NO)3)2·6H2Dissolving O and an auxiliary agent precursor M2 in deionized water, slowly adding ammonia water to control the pH to be 9-10, violently stirring for reaction, cooling to room temperature, filtering, and washing with deionized water until the filtrate is neutral to obtain a filter cake; the assistant precursor M2 is Ce (NO)3)3·6H2O、Al(NO3)3·9H2O and Zr (NO)3)4·5H2At least one of O;
step 4): and drying the obtained filter cake in an oven, and roasting in a muffle furnace to obtain the oxidation precursor catalyst.
2. A synthesis process for the preparation of reforming catalysts via MOFs according to claim 1, wherein the molar ratio of organic ligand and promoter precursor M1 in step 1) is 1: (0.1-2).
3. A synthesis process for the preparation of reforming catalysts via MOFs according to claim 1, characterized in that the organic solvent in step 1) is N, N-dimethylformamide.
4. A synthesis process for the preparation of reforming catalysts via MOFs according to claim 1, wherein the amount of organic solvent used in step 1) is 200mL of organic solvent per 0.1mol of promoter precursor M1.
5. A synthesis process for the preparation of reforming catalysts via MOFs according to claim 1, wherein the reaction in step 2) is carried out at 120 ℃ for 24 h.
6. A synthesis process for the preparation of reforming catalysts via MOFs according to claim 1, wherein the reaction in step 3) is carried out at a temperature of 90 ℃ for a time of 24 h.
7. Synthesis process for reforming catalysts via MOFs according to claim 1, wherein M2 is present in the filter cake obtained in step 3)a+And Ni2+And M1b+The molar ratio of the sum of the moles of (a) is 0.25 to 2.5: 1; ni2+And M1b+The molar ratio of (a) to (b) is 0.2-2: 1.
8. a synthesis process for the preparation of reforming catalysts via MOFs according to claim 1, characterized in that the drying in step 4) is carried out at a temperature of 80 ℃ for a time of 12 h; the roasting temperature is 750 ℃ and the roasting time is 5 h.
9. The synthesis method of preparing a reforming catalyst through MOFs according to claim 1, wherein the oxidation precursor state catalyst obtained in the step 4) comprises an active component, an auxiliary agent precursor M1, an auxiliary agent precursor M2 and an organic ligand, wherein the active component is Ni, the auxiliary agent precursor M1 is at least one of Zn, Mg and Fe, and the auxiliary agent precursor M2 is at least one of Ce, Al and Zr.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202011229347.5A CN112403473B (en) | 2020-11-06 | 2020-11-06 | Synthesis method for preparing reforming catalyst through MOFs |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202011229347.5A CN112403473B (en) | 2020-11-06 | 2020-11-06 | Synthesis method for preparing reforming catalyst through MOFs |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN112403473A true CN112403473A (en) | 2021-02-26 |
| CN112403473B CN112403473B (en) | 2022-12-30 |
Family
ID=74780453
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN202011229347.5A Active CN112403473B (en) | 2020-11-06 | 2020-11-06 | Synthesis method for preparing reforming catalyst through MOFs |
Country Status (1)
| Country | Link |
|---|---|
| CN (1) | CN112403473B (en) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN116850998A (en) * | 2023-06-15 | 2023-10-10 | 内蒙古科技大学 | Preparation method and application of Fe-Zr based catalyst |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2008129024A1 (en) * | 2007-04-24 | 2008-10-30 | Basf Se | Porous organometallic framework materials loaded with catalyst metal components |
| CN106582655A (en) * | 2016-11-29 | 2017-04-26 | 太原理工大学 | Method for preparing high-dispersion easy-reduction loaded nickel-aluminum catalyst |
| CN106902829A (en) * | 2017-04-01 | 2017-06-30 | 太原理工大学 | A kind of load type double-metal reforming catalyst and its preparation method and application |
| CN108283939A (en) * | 2018-01-12 | 2018-07-17 | 湘潭大学 | A kind of catalysis of phenol hydroxylating solid catalyst and the preparation method and application thereof |
| CN108704647A (en) * | 2018-06-15 | 2018-10-26 | 华东理工大学 | A kind of anti-carbon type methane dry gas reforms cladded type Raney nickel and preparation method |
-
2020
- 2020-11-06 CN CN202011229347.5A patent/CN112403473B/en active Active
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2008129024A1 (en) * | 2007-04-24 | 2008-10-30 | Basf Se | Porous organometallic framework materials loaded with catalyst metal components |
| CN106582655A (en) * | 2016-11-29 | 2017-04-26 | 太原理工大学 | Method for preparing high-dispersion easy-reduction loaded nickel-aluminum catalyst |
| CN106902829A (en) * | 2017-04-01 | 2017-06-30 | 太原理工大学 | A kind of load type double-metal reforming catalyst and its preparation method and application |
| CN108283939A (en) * | 2018-01-12 | 2018-07-17 | 湘潭大学 | A kind of catalysis of phenol hydroxylating solid catalyst and the preparation method and application thereof |
| CN108704647A (en) * | 2018-06-15 | 2018-10-26 | 华东理工大学 | A kind of anti-carbon type methane dry gas reforms cladded type Raney nickel and preparation method |
Non-Patent Citations (2)
| Title |
|---|
| LEILA KARAM ET AL.: ""Porous Nickel-Alumina Derived from Metal-Organic Framework (MIL-53): A New Approach to Achieve Active and Stable Catalysts in Methane Dry Reforming"", 《CHEMCATCHEM》 * |
| 陈勇强: "《配位聚合物的结构性能及应用研究》", 30 September 2019, 中国原子能出版社 * |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN116850998A (en) * | 2023-06-15 | 2023-10-10 | 内蒙古科技大学 | Preparation method and application of Fe-Zr based catalyst |
Also Published As
| Publication number | Publication date |
|---|---|
| CN112403473B (en) | 2022-12-30 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN112755996A (en) | Catalyst for synthesizing methanol by carbon dioxide hydrogenation, preparation method and application | |
| CN106824165A (en) | The preparation method of CeO 2 supporting high-dispersion nano catalyst | |
| CN108311154A (en) | One kind being used for CO2The modification of the novel nickel-base catalyst of methanation and preparation method | |
| CN107051433A (en) | The preparation method of zinc oxide supported palladium/platinum catalyst and the application in CO catalysis oxidations | |
| CN110433815A (en) | A kind of carbon dioxide methanation nickel-based catalyst and its preparation method and application | |
| CN114272950A (en) | CH (physical channel)4、CO2Catalyst for reforming preparation of synthesis gas and preparation method and application thereof | |
| CN115591547B (en) | Hydroxyl anchored monoatomic catalyst and preparation method and application thereof | |
| CN113694929B (en) | Supported single-atom copper-based metal oxide catalyst, and preparation method and application thereof | |
| CN103831111A (en) | Catalyst for low-temperature CO catalytic oxidation and preparation method of catalyst | |
| CN107597119A (en) | Anti-carbon type cobalt-based low temperature methane carbon dioxide reformation catalyst and preparation method thereof | |
| CN116078393A (en) | Transition metal supported high-entropy oxide low-temperature methane dry reforming catalyst and preparation method and application thereof | |
| CN102527382A (en) | Metal-supported cerium-based core-shell structure catalyst and preparation method thereof | |
| CN112619654A (en) | Catalyst for preparing synthesis gas by reforming methane and carbon dioxide and preparation method thereof | |
| CN113019394B (en) | Ni-Pt/CeO2 catalyst for hydrogen production by decomposition of ammonia and its preparation method and application | |
| CN112403473B (en) | Synthesis method for preparing reforming catalyst through MOFs | |
| CN114602496B (en) | Nanometer carbon-loaded platinum-iron bimetallic catalyst, preparation method thereof and application thereof in CO selective oxidation reaction in hydrogen-rich atmosphere | |
| CN115970742A (en) | A low-temperature oxidation of CH4 coupling CO2 catalyst for direct production of oxides and its preparation method and application | |
| CN114308063A (en) | PtCo/Co3O4-x-Al2O3Multi-interface structure catalyst and preparation method and application thereof | |
| CN115999554B (en) | A methanol catalyst using a metal organic framework as a precursor and a preparation method thereof | |
| CN116393136A (en) | Preparation method and application of cobalt-based catalyst for synthesizing higher alcohol by hydrogenation of carbon dioxide | |
| CN114308061B (en) | NiAu Bimetallic Alloy Nanocatalyst and Its Synthesis and Application | |
| CN104841448B (en) | Organic silicon waste contact body borne nickel-based methanation catalyst and preparation method therefor | |
| CN116983974A (en) | A ZnO-In2O3 solid solution hollow tube catalyst, preparation method and application | |
| CN110882726A (en) | Cu-doped MOF-5 catalyst and preparation method and application thereof | |
| CN118976497B (en) | A CH4/CO2 dry reforming CoNi bimetallic catalyst, preparation method and application |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PB01 | Publication | ||
| PB01 | Publication | ||
| SE01 | Entry into force of request for substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| GR01 | Patent grant | ||
| GR01 | Patent grant |