CN114014792A - 一种7-(4-溴苯甲酰基)-1,3-二氢-2h-吲哚-2-酮的制备方法 - Google Patents
一种7-(4-溴苯甲酰基)-1,3-二氢-2h-吲哚-2-酮的制备方法 Download PDFInfo
- Publication number
- CN114014792A CN114014792A CN202111535125.0A CN202111535125A CN114014792A CN 114014792 A CN114014792 A CN 114014792A CN 202111535125 A CN202111535125 A CN 202111535125A CN 114014792 A CN114014792 A CN 114014792A
- Authority
- CN
- China
- Prior art keywords
- bromobenzoyl
- dihydro
- indol
- indole
- acid
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
- 238000002360 preparation method Methods 0.000 title claims abstract description 30
- JDSPXIPYMUGSIJ-UHFFFAOYSA-N 7-(4-bromobenzoyl)-1,3-dihydroindol-2-one Chemical compound C1=CC(Br)=CC=C1C(=O)C1=CC=CC2=C1NC(=O)C2 JDSPXIPYMUGSIJ-UHFFFAOYSA-N 0.000 title claims description 14
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 claims abstract description 58
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 claims abstract description 33
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 claims abstract description 29
- RNXKQFMCSXIADL-UHFFFAOYSA-N (4-bromophenyl)-(1h-indol-7-yl)methanone Chemical compound C1=CC(Br)=CC=C1C(=O)C1=CC=CC2=C1NC=C2 RNXKQFMCSXIADL-UHFFFAOYSA-N 0.000 claims abstract description 23
- 125000002672 4-bromobenzoyl group Chemical group BrC1=CC=C(C(=O)*)C=C1 0.000 claims abstract description 17
- 239000002253 acid Substances 0.000 claims abstract description 16
- 238000000034 method Methods 0.000 claims abstract description 14
- 230000002140 halogenating effect Effects 0.000 claims abstract description 12
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 claims abstract description 11
- 238000006722 reduction reaction Methods 0.000 claims abstract description 6
- 230000009471 action Effects 0.000 claims abstract description 3
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 claims description 30
- -1 3, 3-dichloro-7- (4-bromobenzoyl) -1, 3-dihydro-2H-indol-2-one Chemical compound 0.000 claims description 26
- JRNVZBWKYDBUCA-UHFFFAOYSA-N N-chlorosuccinimide Chemical group ClN1C(=O)CCC1=O JRNVZBWKYDBUCA-UHFFFAOYSA-N 0.000 claims description 26
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 claims description 22
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 claims description 10
- PCLIMKBDDGJMGD-UHFFFAOYSA-N N-bromosuccinimide Chemical compound BrN1C(=O)CCC1=O PCLIMKBDDGJMGD-UHFFFAOYSA-N 0.000 claims description 8
- 239000003795 chemical substances by application Substances 0.000 claims description 7
- LQZMLBORDGWNPD-UHFFFAOYSA-N N-iodosuccinimide Chemical compound IN1C(=O)CCC1=O LQZMLBORDGWNPD-UHFFFAOYSA-N 0.000 claims description 6
- 239000003153 chemical reaction reagent Substances 0.000 claims description 4
- 238000005660 chlorination reaction Methods 0.000 claims description 3
- 238000005658 halogenation reaction Methods 0.000 claims description 3
- 230000008569 process Effects 0.000 claims description 3
- 229910052794 bromium Inorganic materials 0.000 claims description 2
- 229910052801 chlorine Inorganic materials 0.000 claims description 2
- 229910052731 fluorine Inorganic materials 0.000 claims description 2
- 229910052736 halogen Inorganic materials 0.000 claims description 2
- 125000005843 halogen group Chemical group 0.000 claims description 2
- 229910052740 iodine Inorganic materials 0.000 claims description 2
- 238000006243 chemical reaction Methods 0.000 abstract description 10
- 238000009776 industrial production Methods 0.000 abstract description 5
- 239000002994 raw material Substances 0.000 abstract description 3
- 239000007787 solid Substances 0.000 description 14
- 238000001914 filtration Methods 0.000 description 12
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 9
- 239000012065 filter cake Substances 0.000 description 9
- 239000008213 purified water Substances 0.000 description 9
- 238000001035 drying Methods 0.000 description 8
- 238000005303 weighing Methods 0.000 description 8
- 239000000706 filtrate Substances 0.000 description 5
- 239000002904 solvent Substances 0.000 description 5
- 238000003756 stirring Methods 0.000 description 5
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 4
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 3
- 238000005160 1H NMR spectroscopy Methods 0.000 description 2
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 2
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 2
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 2
- 238000002425 crystallisation Methods 0.000 description 2
- 230000008025 crystallization Effects 0.000 description 2
- 230000007547 defect Effects 0.000 description 2
- 238000004128 high performance liquid chromatography Methods 0.000 description 2
- 229910052739 hydrogen Inorganic materials 0.000 description 2
- 239000001257 hydrogen Substances 0.000 description 2
- 230000007062 hydrolysis Effects 0.000 description 2
- 238000006460 hydrolysis reaction Methods 0.000 description 2
- NUJOXMJBOLGQSY-UHFFFAOYSA-N manganese dioxide Chemical compound O=[Mn]=O NUJOXMJBOLGQSY-UHFFFAOYSA-N 0.000 description 2
- 238000001819 mass spectrum Methods 0.000 description 2
- 238000007363 ring formation reaction Methods 0.000 description 2
- 238000001228 spectrum Methods 0.000 description 2
- 238000000967 suction filtration Methods 0.000 description 2
- 230000002194 synthesizing effect Effects 0.000 description 2
- WIISOMGJWLLMDG-UHFFFAOYSA-N (2-aminophenyl)-(4-bromophenyl)methanone Chemical compound NC1=CC=CC=C1C(=O)C1=CC=C(Br)C=C1 WIISOMGJWLLMDG-UHFFFAOYSA-N 0.000 description 1
- JVSFQJZRHXAUGT-UHFFFAOYSA-N 2,2-dimethylpropanoyl chloride Chemical compound CC(C)(C)C(Cl)=O JVSFQJZRHXAUGT-UHFFFAOYSA-N 0.000 description 1
- HQSCPPCMBMFJJN-UHFFFAOYSA-N 4-bromobenzonitrile Chemical compound BrC1=CC=C(C#N)C=C1 HQSCPPCMBMFJJN-UHFFFAOYSA-N 0.000 description 1
- MDIAKIHKBBNYHF-UHFFFAOYSA-N Ethyl 2-(methylthio)acetate Chemical compound CCOC(=O)CSC MDIAKIHKBBNYHF-UHFFFAOYSA-N 0.000 description 1
- SIKJAQJRHWYJAI-UHFFFAOYSA-N Indole Chemical compound C1=CC=C2NC=CC2=C1 SIKJAQJRHWYJAI-UHFFFAOYSA-N 0.000 description 1
- 239000007868 Raney catalyst Substances 0.000 description 1
- NPXOKRUENSOPAO-UHFFFAOYSA-N Raney nickel Chemical compound [Al].[Ni] NPXOKRUENSOPAO-UHFFFAOYSA-N 0.000 description 1
- 229910000564 Raney nickel Inorganic materials 0.000 description 1
- ATJFFYVFTNAWJD-UHFFFAOYSA-N Tin Chemical compound [Sn] ATJFFYVFTNAWJD-UHFFFAOYSA-N 0.000 description 1
- VSCWAEJMTAWNJL-UHFFFAOYSA-K aluminium trichloride Chemical compound Cl[Al](Cl)Cl VSCWAEJMTAWNJL-UHFFFAOYSA-K 0.000 description 1
- 150000001408 amides Chemical class 0.000 description 1
- 229960002716 bromfenac sodium Drugs 0.000 description 1
- HZFGMQJYAFHESD-UHFFFAOYSA-M bromfenac sodium Chemical compound [Na+].NC1=C(CC([O-])=O)C=CC=C1C(=O)C1=CC=C(Br)C=C1 HZFGMQJYAFHESD-UHFFFAOYSA-M 0.000 description 1
- 239000003054 catalyst Substances 0.000 description 1
- 238000006555 catalytic reaction Methods 0.000 description 1
- 239000012320 chlorinating reagent Substances 0.000 description 1
- 229940126214 compound 3 Drugs 0.000 description 1
- 150000001875 compounds Chemical class 0.000 description 1
- 230000000694 effects Effects 0.000 description 1
- 239000003889 eye drop Substances 0.000 description 1
- 229940012356 eye drops Drugs 0.000 description 1
- ANSXAPJVJOKRDJ-UHFFFAOYSA-N furo[3,4-f][2]benzofuran-1,3,5,7-tetrone Chemical compound C1=C2C(=O)OC(=O)C2=CC2=C1C(=O)OC2=O ANSXAPJVJOKRDJ-UHFFFAOYSA-N 0.000 description 1
- 230000026030 halogenation Effects 0.000 description 1
- 125000000623 heterocyclic group Chemical group 0.000 description 1
- 208000027866 inflammatory disease Diseases 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 230000003647 oxidation Effects 0.000 description 1
- 238000007254 oxidation reaction Methods 0.000 description 1
- 229920000642 polymer Polymers 0.000 description 1
- 238000000746 purification Methods 0.000 description 1
- 238000011403 purification operation Methods 0.000 description 1
- 230000035484 reaction time Effects 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 238000002636 symptomatic treatment Methods 0.000 description 1
- FAQYAMRNWDIXMY-UHFFFAOYSA-N trichloroborane Chemical compound ClB(Cl)Cl FAQYAMRNWDIXMY-UHFFFAOYSA-N 0.000 description 1
Images






Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D209/00—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D209/02—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom condensed with one carbocyclic ring
- C07D209/04—Indoles; Hydrogenated indoles
- C07D209/30—Indoles; Hydrogenated indoles with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, directly attached to carbon atoms of the hetero ring
- C07D209/32—Oxygen atoms
- C07D209/34—Oxygen atoms in position 2
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/55—Design of synthesis routes, e.g. reducing the use of auxiliary or protecting groups
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Indole Compounds (AREA)
Abstract
本发明涉及药物化学领域,具体涉及一种7‑(4‑溴苯甲酰基)‑1,3‑二氢‑2H‑吲哚‑2‑酮的制备方法。所述的制备方法包括如下步骤:(1)7‑(4‑溴苯甲酰基)吲哚在酸存在下,在四氢呋喃中卤代反应制得3,3‑二卤代‑7‑(4‑溴苯甲酰基)‑1,3‑二氢‑2H‑吲哚‑2‑酮;(2)3,3‑二卤代‑7‑(4‑溴苯甲酰基)‑1,3‑二氢‑2H‑吲哚‑2‑酮,在四氢呋喃中,在醋酸和锌粉作用下进行还原反应制得7‑(4‑溴苯甲酰基)‑1,3‑二氢‑2H‑吲哚‑2‑酮。本方法具有原料易得,反应条件温和,操作简单,收率高,纯度高,适用于工业化生产的特点。
Description
技术领域
本发明涉及药物化学领域,具体涉及一种7-(4-溴苯甲酰基)-1,3-二氢-2H-吲哚-2-酮的制备方法。
背景技术
7-(4-溴苯甲酰基)-1,3-二氢-2H-吲哚-2-酮是合成溴芬酸钠的重要中间体,溴芬酸钠由日本千寿制药株式会社开发为滴眼液上市,于2000年3月获得日本PMDA批准上市,用于外眼部及前眼部的炎症性疾病对症治疗。

目前7-(4-溴苯甲酰基)-1,3-二氢-2H-吲哚-2-酮的制备方法主要包括以下两种:
方法一,美国专利US 4126635和Journal of Medicinal Chemistry,1990 33(8),2296-2304报道了以2-氨基-4'-溴二苯甲酮和2-甲巯基乙酸乙酯为原料,用特戊酰氯催化下环合,再经雷尼镍或锡还原,该路线成环反应条件需要-70℃,工艺较复杂。
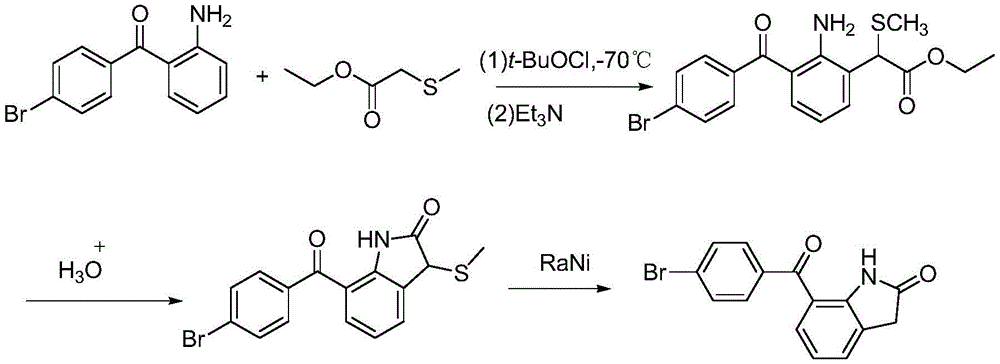
方法二,欧洲专利EP0221753、Journal of Heterocyclic Chemistry,17(8),1663-4,1980和Journal of Medicinal Chemistry 27(11),1379-1388,1984报道了以对溴苯甲腈和吲哚啉为原料,用三氯化硼和三氯化铝为催化剂,进行Houben-Hoesch反应,再经活性二氧化锰氧化、NBS或N-氯代丁二酰亚胺卤化、磷酸酸水解制备。该路线中缺点是磷酸水解时反应温度高时间长,有红色聚合物导致分离纯化困难。

现有技术中,7-(4-溴苯甲酰基)-1,3-二氢-2H-吲哚-2-酮的制备方法存在着各种不同的缺陷,寻找一种反应条件温和,纯化操作简便,易于工业化生产的工艺路线显然格外重要。
发明内容
本发明的目的在于克服现有技术的不足,提供一种7-(4-溴苯甲酰基)-1,3-二氢-2H-吲哚-2-酮的制备方法,该方法具有原料易得,反应条件温和,操作简单,收率高,纯度高,适用于工业化生产的特点。
本发明通过如下技术方案实现:
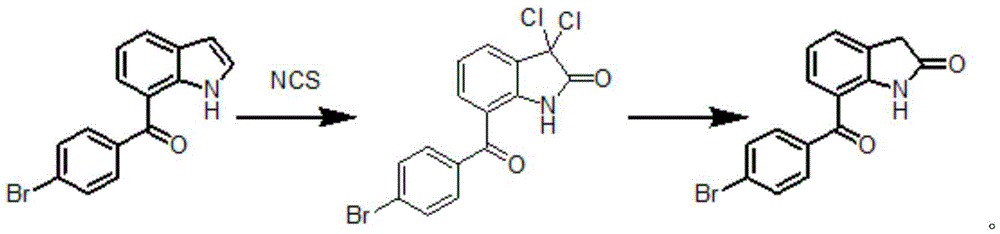
7-(4-溴苯甲酰基)吲哚进行卤代反应制得α-双卤代酰胺即3,3-二卤代-7-(4-溴苯甲酰基)-1,3-二氢-2H-吲哚-2-酮,3,3-二卤代-7-(4-溴苯甲酰基)-1,3-二氢-2H-吲哚-2-酮进行还原反应制得7-(4-溴苯甲酰基)-1,3-二氢-2H-吲哚-2-酮。

其中,R为卤素,选自F、Cl、Br、I。
进一步地,本发明包括如下步骤:
(1)7-(4-溴苯甲酰基)吲哚在酸存在下,在四氢呋喃中卤代反应制得3,3-二卤代-7-(4-溴苯甲酰基)-1,3-二氢-2H-吲哚-2-酮;
(2)3,3-二卤代-7-(4-溴苯甲酰基)-1,3-二氢-2H-吲哚-2-酮,在四氢呋喃中,在醋酸和锌粉作用下进行还原反应制得7-(4-溴苯甲酰基)-1,3-二氢-2H-吲哚-2-酮。
步骤(1)中,所述的酸为硫酸或盐酸,优选为浓硫酸或浓盐酸。
步骤(1)中,所述的卤代反应的卤代试剂为N-氯代丁二酰亚胺、N-溴代丁二酰亚胺、或N-碘代丁二酰亚胺。
优选地卤代试剂为N-氯代丁二酰亚胺。
步骤(1)中,7-(4-溴苯甲酰基)吲哚:卤代试剂:酸:水:四氢呋喃的质量比为1:0.5-2.5:0.1-3:0-5:7-20;
进一步地,为了保障反应的安全,本发明优选7-(4-溴苯甲酰基)吲哚:卤代试剂:酸:水:四氢呋喃的质量比为1:1.4-2:0.2-1.0:0.5-2:9-11;
步骤(2)中,3,3-二卤代-7-(4-溴苯甲酰基)-1,3-二氢-2H-吲哚-2-酮:醋酸:锌粉的摩尔比为1:1-4:2-4。
优选的反应流程如下:
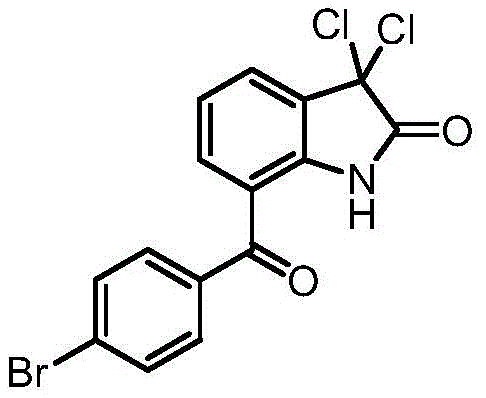
本发明还涉及化合物3,3-二氯代-7-(4-溴苯甲酰基)-1,3-二氢-2H-吲哚-2-酮及其制备方法。
所述的3,3-二氯代-7-(4-溴苯甲酰基)-1,3-二氢-2H-吲哚-2-酮结构如下:
其制备方法如下:
7-(4-溴苯甲酰基)吲哚在酸存在下,在四氢呋喃中氯代反应制得3,3-二氯代-7-(4-溴苯甲酰基)-1,3-二氢-2H-吲哚-2-酮:
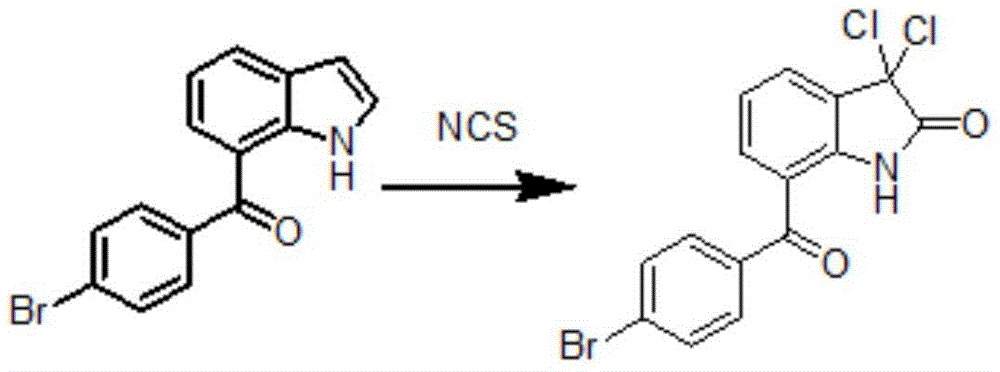
其中,所述的酸为硫酸或盐酸,优选为浓硫酸或浓盐酸。
所述的氯化反应的氯代试剂为N-氯代丁二酰亚胺。
7-(4-溴苯甲酰基)吲哚:氯代试剂:酸:水:四氢呋喃的质量比为1:0.5-2.5:0.1-3:0-5:7-20;
优选地,7-(4-溴苯甲酰基)吲哚:卤代试剂:酸:水:四氢呋喃的质量比为1:1.4-2:0.2-1.0:0.5-2:9-11。
本发明提供了7-(4-溴苯甲酰基)-1,3-二氢-2H-吲哚-2-酮的制备方法,以3,3-二卤代-7-(4-溴苯甲酰基)-1,3-二氢-2H-吲哚-2-酮为关键中间体合成7-(4-溴苯甲酰基)-1,3-二氢-2H-吲哚-2-酮新的制备方法,该方法具有原料易得,反应条件温和可控,操作简单,适合工业化生产,同时提供了新化合物3,3-二氯代-7-(4-溴苯甲酰基)-1,3-二氢-2H-吲哚-2-酮及其制备方法,且以3,3-二氯代-7-(4-溴苯甲酰基)-1,3-二氢-2H-吲哚-2-酮作为中间体制备7-(4-溴苯甲酰基)-1,3-二氢-2H-吲哚-2-酮时,不仅成本低,而且收率高,操作简单,更适合工业化生产。
附图说明
图1为3,3-二氯-7-(4-溴苯甲酰基)-1,3-二氢-2H-吲哚-2-酮的HPLC图谱;
图2为3,3-二氯-7-(4-溴苯甲酰基)-1,3-二氢-2H-吲哚-2-酮的氢谱;
图3为3,3-二氯-7-(4-溴苯甲酰基)-1,3-二氢-2H-吲哚-2-酮的质谱;
图4为7-(4-溴苯甲酰基)-1,3-二氢-2H-吲哚-2-酮的HPLC图谱;
图5为7-(4-溴苯甲酰基)-1,3-二氢-2H-吲哚-2-酮的氢谱;
图6为7-(4-溴苯甲酰基)-1,3-二氢-2H-吲哚-2-酮的质谱。
具体实施方式
下面将通过实施例对本发明作进一步的描述,这些描述并不是对本发明内容作进一步限定,对本发明的技术特征所做的等同替换或相应的改进,仍属于本发明的保护范围之内。
实施例1 3,3-二氯-7-(4-溴苯甲酰基)-1,3-二氢-2H-吲哚-2-酮的制备:
称取四氢呋喃9kg、7-(4-溴苯甲酰基)吲哚1.0kg和36-38%盐酸0.1kg,水0.1kg,加入N-氯代丁二酰亚胺1.5kg,反应4小时后,加纯化水析晶抽滤,滤饼50℃干燥得浅黄固体1.16kg,收率90%。1H-NMR(DMSO-D6,600MHz)7.28(t,1H,J=7.8Hz),7.56(dd,1H,J=7.9Hz),7.71(d,2H,J=6.7Hz),7.80(d,2H,J=6.7Hz),7.96(d,1H,J=7.4Hz),11.41(s,1H)。HRMS(ESI,neg)[M-H]=384。
实施例2 3,3-二氯-7-(4-溴苯甲酰基)-1,3-二氢-2H-吲哚-2-酮的制备:
称取四氢呋喃9kg、7-(4-溴苯甲酰基)吲哚1.0kg和36-38%盐酸0.5kg,加入N-氯代丁二酰亚胺1.0kg,反应4小时后,加纯化水析晶抽滤,滤饼50℃干燥得浅黄固体0.9kg,收率70%。
实施例3 3,3-二氯-7-(4-溴苯甲酰基)-1,3-二氢-2H-吲哚-2-酮的制备:
称取四氢呋喃10kg、7-(4-溴苯甲酰基)吲哚1.0kg和36-38%盐酸1.0kg,水2.0kg,加入N-氯代丁二酰亚胺2.0kg,反应4小时后,加纯化水析晶抽滤,滤饼50℃干燥得浅黄固体1.2kg,收率94%。
实施例4 3,3-二氯-7-(4-溴苯甲酰基)-1,3-二氢-2H-吲哚-2-酮的制备:
称取四氢呋喃7kg、7-(4-溴苯甲酰基)吲哚1.0kg和36-38%盐酸0.2kg,加入N-氯代丁二酰亚胺0.5kg,反应4小时后,加纯化水析晶抽滤,滤饼50℃干燥得浅黄固体1.15kg,收率70%。
实施例5 3,3-二氯-7-(4-溴苯甲酰基)-1,3-二氢-2H-吲哚-2-酮的制备:
称取四氢呋喃10kg、7-(4-溴苯甲酰基)吲哚1.0kg和36-38%盐酸3.0kg、水5.0kg,加入N-氯代丁二酰亚胺1.4kg,反应4小时后,加纯化水析晶抽滤,滤饼50℃干燥得浅黄固体1.21kg,收率95%。
实施例6 3,3-二氯-7-(4-溴苯甲酰基)-1,3-二氢-2H-吲哚-2-酮的制备:
称取四氢呋喃20kg、7-(4-溴苯甲酰基)吲哚1.0kg和36-38%盐酸0.6kg、水1.0kg,加入N-氯代丁二酰亚胺1.5kg,反应4小时后,加纯化水析晶抽滤,滤饼50℃干燥得浅黄固体1.2kg,收率94%。
实施例7 3,3-二氯-7-(4-溴苯甲酰基)-1,3-二氢-2H-吲哚-2-酮的制备:
称取四氢呋喃10kg、7-(4-溴苯甲酰基)吲哚1.0kg和36-38%盐酸1.0kg,加入N-氯代丁二酰亚胺1.4kg,反应4小时后,加纯化水析晶抽滤,滤饼50℃干燥得浅黄固体1.2kg,收率94%。
实施例8 3,3-二溴-7-(4-溴苯甲酰基)-1,3-二氢-2H-吲哚-2-酮的制备:
称取四氢呋喃9kg、7-(4-溴苯甲酰基)吲哚1.0kg和36-38%盐酸0.2kg、水0.5kg,加入N-溴代丁二酰亚胺1.8kg,反应5小时后,加纯化水析晶抽滤,滤饼50℃干燥得浅黄固体1.46kg,收率93%。
实施例9 3,3-二碘-7-(4-溴苯甲酰基)-1,3-二氢-2H-吲哚-2-酮的制备:
称取四氢呋喃12kg、7-(4-溴苯甲酰基)吲哚1.0kg和36-38%盐酸0.5kg、水0.5kg,加入N-碘代丁二酰亚胺2.25kg,反应6小时后,加纯化水析晶抽滤,滤饼50℃干燥得浅黄固体1.8kg,收率95%。
实施例10 7-(4-溴苯甲酰基)-1,3-二氢-2H-吲哚-2-酮的制备:
实施例1制备的3,3-二氯-7-(4-溴苯甲酰基)-1,3-二氢-2H-吲哚-2-酮1kg,四氢呋喃15kg和醋酸0.5kg(3.2equiv.)溶解,搅拌下加入锌粉0.675kg(4equv.),40℃反应2小时滤除不溶物后,所得滤液浓缩回收溶剂后加乙酸乙酯打浆得类白色固体0.6kg,收率73%。1H-NMR(DMSO-D6,600MHz)3.59(s,2H),7.05(t,1H,J=7.5Hz),7.33(d,1H,J=8Hz),7.48(d,1H,J=7.3Hz),7.66(d,2H,J=6.7Hz),7.78(d,2H,J=6.7Hz),10.36(s,1H)。HRMS(ESI,neg)[M+H]=316,318。
实施例11 7-(4-溴苯甲酰基)-1,3-二氢-2H-吲哚-2-酮的制备:
实施例1制备的3,3-二氯-7-(4-溴苯甲酰基)-1,3-二氢-2H-吲哚-2-酮1kg,四氢呋喃13kg和醋酸0.156(1equiv.)kg溶解,搅拌下加入锌粉0.7kg(4.1equiv.),35℃反应2小时滤除不溶物后,所得滤液浓缩回收溶剂后加乙醇打浆得类白色固体0.66kg,收率80%。
实施例12 7-(4-溴苯甲酰基)-1,3-二氢-2H-吲哚-2-酮的制备:
实施例1制备的3,3-二氯-7-(4-溴苯甲酰基)-1,3-二氢-2H-吲哚-2-酮1kg,四氢呋喃10kg和醋酸0.624kg(4equiv.)溶解,搅拌下加入锌粉0.34kg(2.0equiv.),30℃反应2小时滤除不溶物后,所得滤液浓缩回收溶剂后加甲醇打浆得类白色固体0.66kg,收率80%。
实施例13 7-(4-溴苯甲酰基)-1,3-二氢-2H-吲哚-2-酮的制备:
实施例1制备的3,3-二溴-7-(4-溴苯甲酰基)-1,3-二氢-2H-吲哚-2-酮1kg,四氢呋喃13kg和醋酸0.3kg(1.9equiv.)溶解,搅拌下加入锌粉0.6kg(3.5equiv.),40℃反应2小时滤除不溶物后,所得滤液浓缩回收溶剂后加乙酸乙酯打浆得类白色固体0.62kg,收率75%。
实施例14 7-(4-溴苯甲酰基)-1,3-二氢-2H-吲哚-2-酮的制备:
实施例1制备的3,3-二溴-7-(4-溴苯甲酰基)-1,3-二氢-2H-吲哚-2-酮1kg,四氢呋喃13kg和醋酸0.8kg(5.1equiv.)溶解,搅拌下加入锌粉1.0kg(6equiv.),40℃反应2小时滤除不溶物后,所得滤液浓缩回收溶剂后加乙酸乙酯打浆得类白色固体0.56kg,收率68%。
步骤(1)中卤代试剂种类、7-(4-溴苯甲酰基)吲哚:卤代试剂:酸:水:四氢呋喃的质量比3,3-二氯-7-(4-溴苯甲酰基)-1,3-二氢-2H-吲哚-2-酮收率的影响
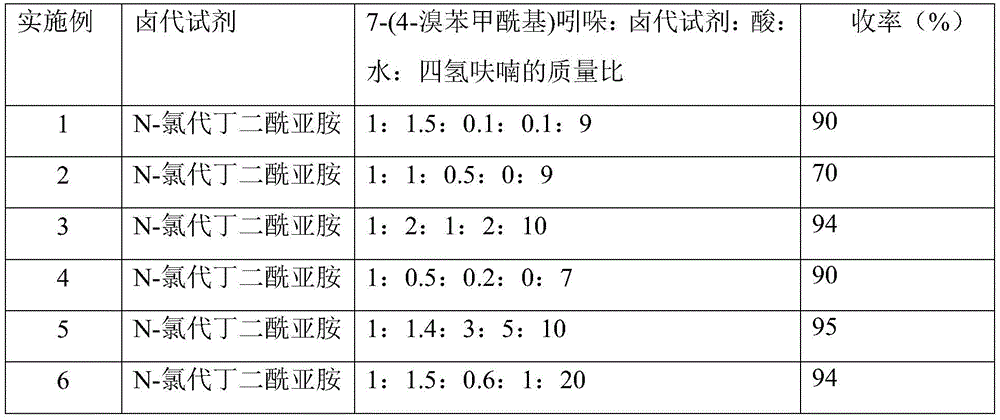

步骤(2)中3,3-二卤代-7-(4-溴苯甲酰基)-1,3-二氢-2H-吲哚-2-酮:醋酸:锌粉的摩尔比对7-(4-溴苯甲酰基)-1,3-二氢-2H-吲哚-2-酮的收率的影响

Claims (10)
1.7-(4-溴苯甲酰基)-1,3-二氢-2H-吲哚-2-酮的制备方法,其特征在于,7-(4-溴苯甲酰基)吲哚进行卤代反应制得3,3-二卤代-7-(4-溴苯甲酰基)-1,3-二氢-2H-吲哚-2-酮,3,3-二卤代-7-(4-溴苯甲酰基)-1,3-二氢-2H-吲哚-2-酮进行还原反应制得7-(4-溴苯甲酰基)-1,3-二氢-2H-吲哚-2-酮;

其中,R为卤素,选自F、Cl、Br、I。
2.如权利要求1所述的制备方法,其特征在于,包括如下步骤:
(1)7-(4-溴苯甲酰基)吲哚在酸存在下,在四氢呋喃中卤代反应制得3,3-二卤代-7-(4-溴苯甲酰基)-1,3-二氢-2H-吲哚-2-酮;
(2)3,3-二卤代-7-(4-溴苯甲酰基)-1,3-二氢-2H-吲哚-2-酮,在四氢呋喃中,在醋酸和锌粉作用下进行还原反应制得7-(4-溴苯甲酰基)-1,3-二氢-2H-吲哚-2-酮。
3.如权利要求2所述的制备方法,其特征在于,步骤(1)中所述的酸为硫酸或盐酸,所述的卤代试剂为N-氯代丁二酰亚胺、N-溴代丁二酰亚胺、或N-碘代丁二酰亚胺。
4.如权利要求2所述的制备方法,其特征在于,步骤(1)中,7-(4-溴苯甲酰基)吲哚:卤代试剂:酸:水:四氢呋喃的质量比为1:0.5-2.5:0.1-3:0-5:7-20,优选为1:1.4-2:0.2-1.0:0.5-2:9-11。
5.如权利要求2所述的制备方法,其特征在于,步骤(2)中,3,3-二卤代-7-(4-溴苯甲酰基)-1,3-二氢-2H-吲哚-2-酮:醋酸:锌粉的摩尔比为1:1-4:2-4。
6.如下结构的3,3-二氯代-7-(4-溴苯甲酰基)-1,3-二氢-2H-吲哚-2-酮:

7.权利要求6所述的3,3-二氯代-7-(4-溴苯甲酰基)-1,3-二氢-2H-吲哚-2-酮的制备方法,其特征在于,7-(4-溴苯甲酰基)吲哚在酸存在下,在四氢呋喃中经N-氯代丁二酰亚胺氯代反应制得3,3-二氯代-7-(4-溴苯甲酰基)-1,3-二氢-2H-吲哚-2-酮;

8.如权利要求7所述的制备方法,其特征在于,所述的酸为硫酸或盐酸。
9.如权利要求7所述的制备方法,其特征在于,7-(4-溴苯甲酰基)吲哚:氯代试剂:酸:水:四氢呋喃的质量比为1:0.5-2.5:0.1-3:0-5:7-20,优选为1:1.4-2:0.2-1.0:0.5-2:9-11。
10.权利要求6所述的3,3-二氯代-7-(4-溴苯甲酰基)-1,3-二氢-2H-吲哚-2-酮在制备7-(4-溴苯甲酰基)-1,3-二氢-2H-吲哚-2-酮中的应用。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202111535125.0A CN114014792B (zh) | 2021-12-15 | 2021-12-15 | 一种7-(4-溴苯甲酰基)-1,3-二氢-2h-吲哚-2-酮的制备方法 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202111535125.0A CN114014792B (zh) | 2021-12-15 | 2021-12-15 | 一种7-(4-溴苯甲酰基)-1,3-二氢-2h-吲哚-2-酮的制备方法 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN114014792A true CN114014792A (zh) | 2022-02-08 |
| CN114014792B CN114014792B (zh) | 2024-01-26 |
Family
ID=80068837
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN202111535125.0A Active CN114014792B (zh) | 2021-12-15 | 2021-12-15 | 一种7-(4-溴苯甲酰基)-1,3-二氢-2h-吲哚-2-酮的制备方法 |
Country Status (1)
| Country | Link |
|---|---|
| CN (1) | CN114014792B (zh) |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN101563319A (zh) * | 2006-12-19 | 2009-10-21 | 弗·哈夫曼-拉罗切有限公司 | 杂芳基吡咯烷基和哌啶基甲酮衍生物 |
| CN102066320A (zh) * | 2008-06-18 | 2011-05-18 | 弗·哈夫曼-拉罗切有限公司 | 作为mri的芳基甲酮 |
| CN106278988A (zh) * | 2016-08-12 | 2017-01-04 | 合肥久诺医药科技有限公司 | 一种溴芬酸钠杂质标准品的合成方法 |
| CN108569975A (zh) * | 2018-04-16 | 2018-09-25 | 扬子江药业集团有限公司 | 一种溴芬酸钠倍半水合物的制备方法 |
| CN110172036A (zh) * | 2018-02-19 | 2019-08-27 | 齐鲁制药有限公司 | 一种溴芬酸钠中间体的制备方法 |
-
2021
- 2021-12-15 CN CN202111535125.0A patent/CN114014792B/zh active Active
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN101563319A (zh) * | 2006-12-19 | 2009-10-21 | 弗·哈夫曼-拉罗切有限公司 | 杂芳基吡咯烷基和哌啶基甲酮衍生物 |
| CN102066320A (zh) * | 2008-06-18 | 2011-05-18 | 弗·哈夫曼-拉罗切有限公司 | 作为mri的芳基甲酮 |
| CN106278988A (zh) * | 2016-08-12 | 2017-01-04 | 合肥久诺医药科技有限公司 | 一种溴芬酸钠杂质标准品的合成方法 |
| CN110172036A (zh) * | 2018-02-19 | 2019-08-27 | 齐鲁制药有限公司 | 一种溴芬酸钠中间体的制备方法 |
| CN108569975A (zh) * | 2018-04-16 | 2018-09-25 | 扬子江药业集团有限公司 | 一种溴芬酸钠倍半水合物的制备方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| CN114014792B (zh) | 2024-01-26 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN107793418A (zh) | 一种枸橼酸托法替布的工业化生产方法 | |
| JP2024009896A (ja) | 一酸化窒素を供与するプロスタグランジン類似体の製造方法 | |
| CA1043791A (en) | Process for preparing imidazolinylaminoquinoxalines | |
| JPH0148911B2 (zh) | ||
| CN107955002A (zh) | 阿哌沙班及其中间体的制备方法 | |
| CN114014792A (zh) | 一种7-(4-溴苯甲酰基)-1,3-二氢-2h-吲哚-2-酮的制备方法 | |
| CN110981800A (zh) | 一种乐伐替尼的制备方法 | |
| US2797218A (en) | Cinnoline derivatives | |
| CN105646327A (zh) | 2-全氟烷基吲哚衍生物及其合成方法 | |
| US12503441B2 (en) | Process for preparing diphenylureido-dihalokynurenic acids and tosylate addition salts thereof | |
| JP2019507156A (ja) | 4−アルコキシ−3−ヒドロキシピコリン酸を製造する方法 | |
| CN104892485A (zh) | 2-全氟烷基吲哚衍生物及其合成方法 | |
| JPH0116837B2 (zh) | ||
| KR101485418B1 (ko) | 고순도 미르타자핀의 제조방법 | |
| CN114591238A (zh) | 一种异喹啉类药物中间体的合成方法 | |
| CN113429333B (zh) | 一种加雷沙星中间体的合成方法 | |
| CN110407702A (zh) | 一种艾曲波帕关键中间体3’-氨基-2’-羟基联苯-3-羧酸的制备方法 | |
| JPH07121931B2 (ja) | ベンゾ〔b〕フラン誘導体 | |
| EP4227297A1 (en) | Process for preparing diphenylureido-dihalokynurenic acids and tosylate addition salts thereof | |
| CN112552261B (zh) | 左氧氟沙星及其中间体的制备方法 | |
| CN118359560B (zh) | 一种亚甲蓝的制备方法 | |
| Mitchell et al. | Nitration and bromination of isocytosine-6-acetic acid. Corrections | |
| US20060183738A1 (en) | Crystalline forms of nevirapine | |
| CN110407735B (zh) | 一种3,4,5,6-四氟-n-甲基邻苯二甲酰亚胺的合成工艺 | |
| JP6917612B2 (ja) | ペリレンテトラカルボン酸二無水物の臭素一置換体の製造方法及び精製方法。 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PB01 | Publication | ||
| PB01 | Publication | ||
| SE01 | Entry into force of request for substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| GR01 | Patent grant | ||
| GR01 | Patent grant |