CN1229367C - 呋喃并4a羟甲基多氢萘化合物、合成方法及其用途 - Google Patents
呋喃并4a羟甲基多氢萘化合物、合成方法及其用途 Download PDFInfo
- Publication number
- CN1229367C CN1229367C CN 02151114 CN02151114A CN1229367C CN 1229367 C CN1229367 C CN 1229367C CN 02151114 CN02151114 CN 02151114 CN 02151114 A CN02151114 A CN 02151114A CN 1229367 C CN1229367 C CN 1229367C
- Authority
- CN
- China
- Prior art keywords
- compound
- acid
- polyhydronaphthalene
- hydroxymethyl
- furo
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
Landscapes
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
本发明涉及一种呋喃并4a羟甲基多氢萘化合物、合成方法和用途。该化合物具有如下的结构式:右式(I),其中R1、R2或R3=H或C1-C3,或R2+R3=甾体,表示为单键或双键,所述的甾体为右式(II)或右式(III),R4=H,C1-C4或甲氧甲基,右式(IV)或右式(V)n=0-2。本发明不仅合成方法简便,适合工业化生产,而且能用于合成结构式为右式(VI)的海洋天然产物(±)spiniferin-1及其类似物。
Description
技术领域
本发明涉及多种呋喃并4a羟甲基多氢萘化合物、合成方法及其用途。可在酸催化条件下及用氟代磺酰氟诱发的碳正离子重排反应方法(参见CN 97106576.4)合成这类化合物,上述化合物可高效的合成了海洋天然产物spiniferin-1及其类似物。
背景技术
天然spiniferin-1是意大利G.Cimino教授从那不勒斯湾的一种海绵生物pleraplysilla中分离得到的一种呋喃倍半萜(Tetra.Lett.1975(45),3727)。1980年,J.A.Marshall小组通过合成spiniferin-1的二氢衍生物(±)dihydrospiniferin-1确定其结构如下(参见J.Amer.Chem.Soc.1980(102),4274):


(±)spiniferin-1 (±)dihydrospiniferin-1
众所周知,自然界中存在大量含有苯并呋喃结构的天然产物,这些天然产物具有广泛的生理活性(Comprehensive Hetercycles Chemistry II)。环庚三烯是苯的高碳同系物,具有环庚三烯并呋喃结构的spiniferin-1可能具有与苯并呋喃类化合物相似的生理作用。

benzofuran analog
此结构特点的化合物尽管仅有spiniferin-1,但是它代表了一类新型结构的有机分子。它的结构不稳定,容易分解,相对而言,其双氢衍生物dihydrospiniferin-1较为稳定。虽然spiniferin-1的确切生理活性还未能被研究,但是一些含有苯并呋喃结构单元的有机分子已被用作药物的事实,足以使我们关注spiniferin-1的潜在用途。
spiniferin-1研究进展缓慢的原因在于获得样品困难。1983年,美国教授J.A.Marshall研究了spiniferin-1及其双氢衍生物的合成。但是合成路线冗长,产率很低,未能解决为进一步研究提供足够样品的问题。
在研究氟代磺酰氟试剂与19-羟基甾体反应中,田伟生等人已经发现了一个有趣的碳正离子引发的串联重排反应(CN 97106576.4)

田伟生等人还使用这个已经发现的串联重排反应,合成了(±)dihydrospiniferin-1(CN 02145067.6)。
(±)dihydrospiniferin-1
但是在合成(±)spiniferin-1及其类似物的过程中,使用同样的策略时,未能得到目标产物,主要的问题在于臭氧化反应条件过于剧烈,对分子其它部位的官能团有影响。因此,仍在不断探索新的(±)spiniferin-1及其类似物和能在温和的条件下,高效,简捷地合成(±)spiniferin-1及其类似物的方法。
发明内容
本发明目的是提供一系列呋喃并4a羟甲基多氢萘化合物;
本发明的另一目的是提供一种上述呋喃并4a羟甲基多氢萘化合物的合成方法。即使用酸催化的环化反应合成了呋喃并4a羟甲基多氢萘化合物;
本发明还提供一种上述呋喃并4a羟甲基多氢萘化合物的用途,可使用此化合物合成海洋天然产物spiniferin-1及其多种类似物。
本发明的呋喃并4a羟甲基多氢萘化合物具有如下的结构式:
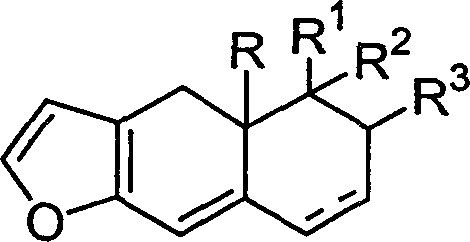
其中,R=OH,OTHP或COOR6,
表示为单键或双键,R1、R2或R3=H或C1-C3,或R2+R3=甾体,如R1=R2=R3=H;R1=R2=C1-C3,R3=H;或R1=H或C1-C3,R2+R3=甾体。所述的甾体为
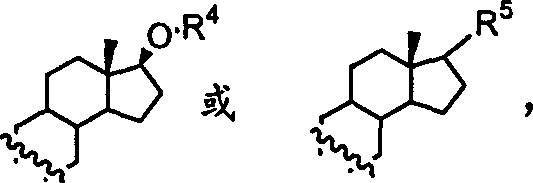 ,其中R4=H、C1-C4或甲氧甲基,,n=0-2,R6=H,C1-C4,THP为四氢吡喃。
,其中R4=H、C1-C4或甲氧甲基,,n=0-2,R6=H,C1-C4,THP为四氢吡喃。
本发明的化合物可以以下述结构式的化合物为例:
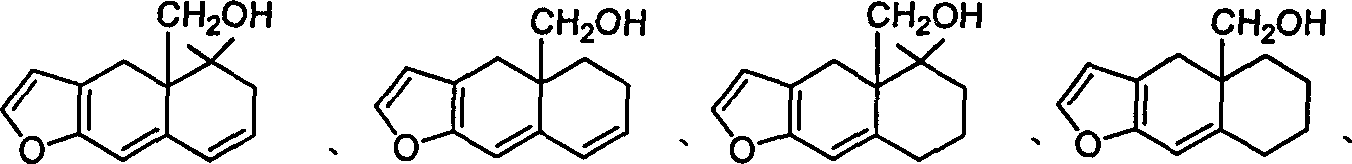

其中R4=H,C1-C4或甲氧甲基,
或
n=0-2。
本发明的呋喃并4a羟甲基多氢萘化合物4的合成方法,使用α,β不饱和酮为原料,THPOCH2CHO为加成片断,两步反应合成了呋喃并4a羟甲基多氢萘化合物。反应式如下:
1 2
3 4,
其中-OPG为羟基的潜在官能团,可以为酯基或四氢吡喃保护的羟基。
化合物3去保护的方法分别为还原或酸水解成羟基。
所述的化合物1、2和3的结构式如下:


化合物1, 化合物2, 化合物3,
其中
 为-COOMe,-COOEt,CH2OTHP,THP即四氢吡喃保护基,上述化合物也可以进一步分别描述为:
为-COOMe,-COOEt,CH2OTHP,THP即四氢吡喃保护基,上述化合物也可以进一步分别描述为:
化合物1:
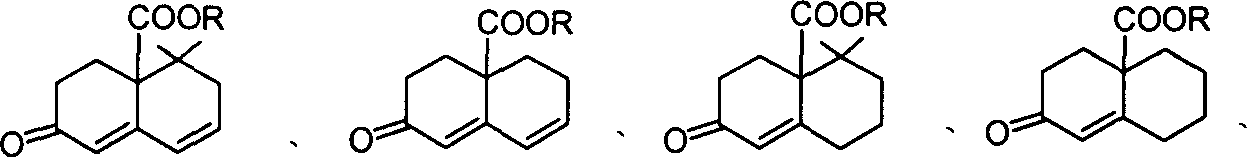
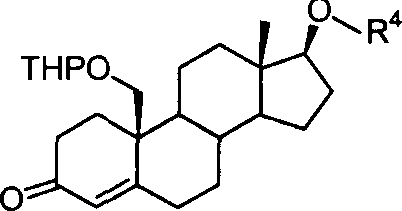

化合物2:

化合物3
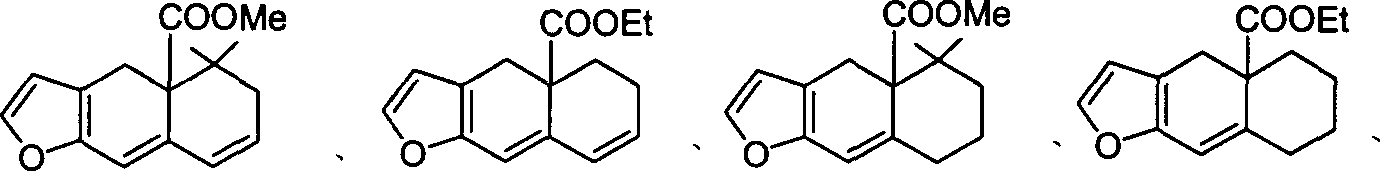
或

上述化合物,2,3中,R4=H,C1-C4或甲氧甲基,
或
本发明的方法中,加成片断THPOCH2CHO可按文献方法制备(J.Amer.Chem.Soc.1995(117),634)。
本发明的方法用下述1)和2);1)、2)和3)或1)、2)和4)三种方法合成,具体如下:
1)在非质子极性溶剂中和-40-78℃温度下,化合物1与强碱反应0.5-3小时,得到的中间体继续与ZnCl2反应,加入THPOCH2CHO得到化合物2,产率90-100%;其中,所述的强碱是二异丙胺锂或MN(SiMe3)2等,M=Li,Na,K,化合物、强碱、ZnCl2和THPOCH2CHO的摩尔比为1∶1-10∶1-4∶1-4;
2)在惰性溶剂中,化合物2与酸在0℃到回流温度下反应0.5-10小时,得到化合物3,产率70-90%,所述的化合物2与酸的摩尔比为1∶0.1-0.5,所述的酸可以是无机酸或有机酸;所述的无机酸可以是氢卤酸,硫酸,磷酸等,所述的有机酸可以是甲酸,乙酸,对甲苯磺酸,樟脑磺酸或对甲苯磺酸吡啶盐等。
3)在惰性溶剂中,化合物3与LiALH4在0℃到回流温度下反应0.5-2小时,得到化合物4,产率70-90%,所述的化合物3与LiALH4的摩尔比为1∶1-5。
4)在惰性溶剂中,化合物3与酸在0℃到回流温度下反应0.5-5小时,得到化合物4,产率70-90%,所述的化合物3与酸的摩尔比为1∶0.1-0.5所述的酸可以是无机酸或有机酸;所述的无机酸可以是氢卤酸,硫酸,磷酸等,所述的有机酸可以是甲酸,乙酸,对甲苯磺酸,樟脑磺酸或对甲苯磺酸吡啶盐等。
上述的惰性溶剂可以是乙醚、四氢呋喃、石油醚、正己烷、苯、甲苯,醇或卤代烃等。
所述的非质子极性溶剂可以是乙醚、四氢呋喃、二甲基亚砜或二甲基甲酰胺等。
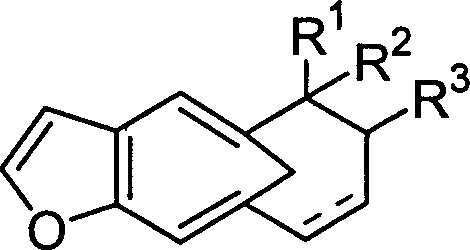
本发明提供的方法合成了一系列呋喃并4a羟甲基多氢萘化合物,结构为:
其中R1、R2或R3=H或C1-C3,或R2+R3=甾体,
表示为单键或双键,所述的甾体为
 其中R1=H,C1-C4或甲氧甲基,
其中R1=H,C1-C4或甲氧甲基, 或
或

使用专利方法(CN 02145067.6),在氟代磺酰氟试剂的诱导下发生重排反应,
可以高效地转化为结构式为
(±)spiniferin-1的海洋天然产物spiniferin-1及其类似物。该海洋天然产物(±)spiniferin-1及其类似物的结构如下:
其中R1、R2或R3=H或C1-C3,或R2+R3=甾体,
表示为单键或双键,其中甾体为
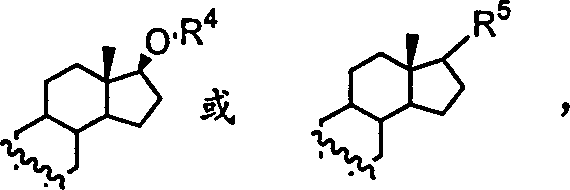 ,其中R4=H,C1-C4或甲氧甲基,或
,其中R4=H,C1-C4或甲氧甲基,或
本发明的发明主要优点有:
1、使用易制备的α,β不饱和酮为原料,使用ZnCl2催化的羟醛缩合反应得到加成产物,在酸催化下高效地构建了呋喃环。控制反应条件可以很高的产率得到呋喃并4a羟甲基多氢萘化合物。
2、呋喃环的合成,增强了2位烯键的电子密度,使氟代磺酰氟试剂的诱导下发生重排反应速度大大加快,无需换用更强的碱室温下即可反应。简化了试验操作。
3、呋喃环的合成相当于保护了α,β不饱和酮的羰基。简化了spiniferin-1类似物的合成。以(±)dihydrospiniferin-1的合成为例,进一步简化为4步(文献为13步,专利(CN 02145067.6)为6步),各步反应产率中等到良好。没有使用昂贵的试剂,均可在市场购得。是一条具有工业化价值的合成路线。
具体实施方式
通过下述实施例将有助于理解本发明,但并不限制本发明的内容。
实施例1化合物2a的合成

0.26ml(2mmol)二异丙胺溶于1ml四氢呋喃,冷却至-78℃,滴加1.0ml 2M(2mmol)的n-BuLi。加毕搅拌15分钟。
-78℃下缓慢向其中滴加220mg(1mmol)化合物1a的2ml四氢呋喃溶液。-78℃下搅拌反应2小时后,加入2mmol(2eq)ZnCl2,搅拌5min,加入2mmol(2eq)THPOCH2CHO加毕,反应1小时。饱和NH4Cl溶液淬灭反应。二氯甲烷提取。合并有机相,水洗,饱和NaCl溶液洗,Na2SO4干燥。抽去溶剂,过柱得320mg化合物2a(产率90%)。
化合物2a:C20H30O6(366.45)
1H-NMR(CDCl3,300MHz):δ5.94(m,1H),4.61(m,1H),3.5-4.3(m,8H),1.3-1.8(m,17H)1.28(m,3H)。
实施例2化合物3a的合成

200mg(0.8mmol)化合物2a溶于4ml THF,加入20mg对甲苯磺酸,60℃下搅拌3hr。抽去溶剂,残留物过柱得130mg(77%)无色油状物3a。
化合物3a:C15H18O3(246.30)
1H-NMR(CDCl3 300MHz):δ7.17(d,1H,J=1.8Hz),6.24(s,1H),6.19(d,1H,J=1.8Hz),4.0-4.2(m,2H),3.20(d,1H,J=16.5Hz),2.59(d,1H,J=16.2Hz),),1.3-2.5(m,8H),1.19(t,3H,J=7.2Hz)。
实施例3化合物4a的合成
将100mg(0.4mmol)化合物3a溶于2mlTHF。室温滴入40mg(2.5eq)LiAlH4的5mlTHF悬浮液中,反应0.5hr。少量水淬灭反应,过滤,蒸去溶剂,残留物经柱层析分离得88mg化合物4a(100%)。
化合物4a:C13H16O2(204.26)
m.p.92-93℃
1H-NMR(CDCl3 300MHz)δ7.17(d,1H,J=1.8Hz),6.19(d,1H,J=1.8Hz),6.15(d,1H,J=0.9Hz),3.46(m,2H),2.89(d,1H,J=16.8Hz),2.36(d,1H,J=16.5Hz),),1.2-2.3(m,8H)。
实施例4化合物5a的合成

90mg4a(0.44mmol)溶于3mlTHF,0℃加入150mg(2eq)DBU和300mg(2eq)RfSO2F,搅拌0.5hr,走板原料完全转化。反应液抽干,过柱,得65mg无色粘液5a(80%)。
化合物5a:C25H38O4(402.57)
1H-NMR(CDCl3 300MHz)δ7.34(d,1H,J=1.8Hz),6.57(d,1H,J=2.1Hz),3.09(d,1H,J=10.8Hz),2.55(m,2H),2.35(m,2H),1.86(m,2H),1.24(d,1H,J=11.2Hz),1.18(m,2H)。
实施例5化合物2b的合成
-78℃下缓慢向1.2eq(1.2mmol)LiN(SiMe3)2的2mlTHF溶液中滴加400mg(1mmol)化合物1b的2ml四氢呋喃溶液。-78℃下搅拌反应2小时后,加入1mmol(leq)ZnCl2,搅拌5min,加入1mmol(leq)THPOCH2CHO加毕,反应1小时。饱和NH4Cl溶液淬灭反应。二氯甲烷提取。合并有机相,水洗,饱和NaCl溶液洗,Na2SO4干燥。抽去溶剂,过柱得540mg化合物2b(产率100%)。
化合物2b:C30H50O7(546.74)
1H-NMR(CDCl3,300MHz):δ5.90(d,1H),4.61(m,1H),3.34(s,3H),3.22(t,2H,J=8.1Hz)0.79(s,3H)。
实施例6化合物4b的合成
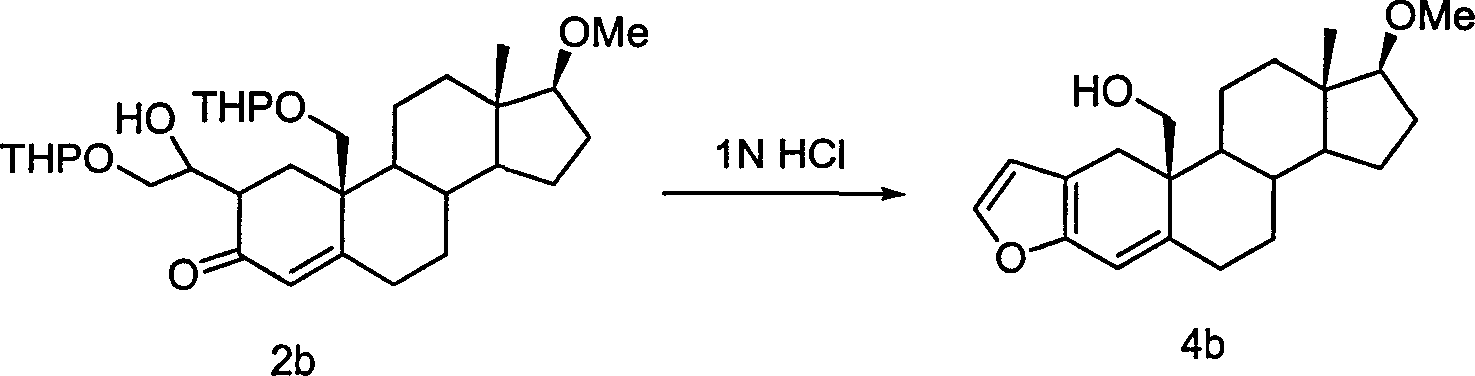
270mg(0.5mmol)化合物2b溶于2ml THF,加入0.1ml 1N HCl,室温下搅拌1hr。抽去溶剂,残留物过柱得150mg(88%)无色油状物4b。
化合物4b:C22H30O3(342.47)
1H-NMR(CDCl3 300MHz):δ7.16(d,1H,J=2.1Hz),6.25(d,1H,J=1.5Hz),6.18(s,1H),3.87,(m,1H,3.66,(m,1H),3.34(s,3H),3.23(t,1H,J=8.1Hz),3.01(d,1H,J=16.8Hz),2.71(d,1H,J=16.8Hz),0.80(s,3H)。
实施例7化合物5b的合成
70mg4b(0.2mmol)溶于2mlTHF,0℃加入30mg(leq)DBU和60mg(leq)RfSO2F,搅拌0.5hr,走板原料完全转化。反应液抽干,过柱,得60mg无色粘液5b(92%)。
化合物5b:C22H28O2(324.46)
1H-NMR(CDCl3 300MHz)δ7.32(d,1H,J=2.1Hz),6.55(d,1H,J=2.1Hz),6.27(s,1H),6.27(s,1H),6.22(s,3H),3.29(d,1H,J=10.2Hz),3.27(t,1H,J=8.1Hz),1.14(d,1H,J=10.5Hz),0.92(s,1H)。
实施例8化合物2c的合成

0.26ml(2mmol)二异丙胺溶于1ml四氢呋喃,冷却至-78℃,滴加1.0ml 2M(2mmol)的n-BuLi。加毕搅拌15分钟。
-78℃下缓慢向其中滴加220mg(1mmol)化合物1c的2ml四氢呋喃溶液。-40℃下搅拌反应2小时后,加入1mmol(leq)ZnCl2,搅拌5min,加入1mmol(leq)THPOCH2CHO加毕,反应1小时。饱和NH4Cl溶液淬灭反应。二氯甲烷提取。合并有机相,水洗,饱和NaCl溶液洗,Na2SO4干燥。抽去溶剂,过柱得310mg化合物2c(产率85%)。
化合物2a:C20H28O6(364.43)
1H-NMR(CDCl3,300MHz):δ6.29(s,2H),5.88(d,1H,J=1.8Hz),4.63(m,1H),4.19(m,2H),1.24(m,3H)
实施例9化合物5c的合成
200mg(0.55mmol)化合物2c溶于4ml THF,加入20mg对甲苯磺酸,60℃下搅拌3hr。抽去溶剂,残留物230mg3c直接用于下步反应。
将230mg粗品溶于10mlTHF。室温滴入100mg(2.5eq)LiAlH4的5mlTHF悬浮液中,反应0.5hr。少量水淬灭反应,过滤,蒸去溶剂,残留物200mg 4c直接用于下步反应。
200mg粗品溶于5mlTHF,0℃加入300mg(2eq)DBU和600mg(2eq)RfSO2F,搅拌0.5hr,走板原料完全转化。反应液抽干,过柱,得80mg无色粘液5a(三步79%)。
化合物5c:C13H12O(184.23)
1H-NMR(CDCl3 300MHz)δ7.39(d,1H,J=2.1Hz),6.61(d,1H,J=1.8Hz),6.3-6.4(d,4H),3.61(d,1H,J=13.5Hz),1.03(d,1H,J=9.9Hz)
Claims (7)
1,一种呋喃并4a羟甲基多氢萘化合物,具有如下的结构式:
其中R1、R2或R3=H或C1-C3,或R2+R3=甾体,
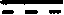 表示为单键或双键,甾体为
表示为单键或双键,甾体为
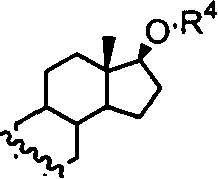 或
R4=H,C1-C4或甲氧甲基,或
n=0-2。
或
R4=H,C1-C4或甲氧甲基,或
n=0-2。
2,如权利要求1所述的呋喃并4a羟甲基多氢萘化合物,其特征是具有如下的结构式:
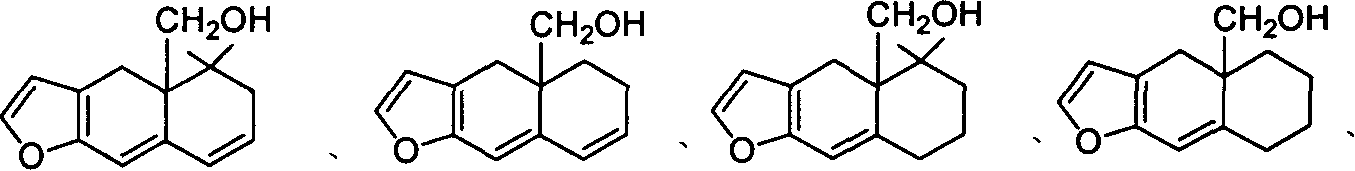
或
其中R4=H,C1-C4或甲氧甲基,
 或
或
 ,n=0-2。
,n=0-2。
3,一种如权利要求1所述的呋喃并4a羟甲基多氢萘化合物的合成方法,其特征是用下述1)和2);1)、2)和3)或1)、2)和4)三种方法合成:
1)在非质子极性溶剂中和-40-78℃温度下,化合物1与强碱反应0.5-3小时,得到的中间体继续与ZnCl2反应,加入THPOCH2CHO,THP即四氢吡喃保护基,得到化合物2;其中,所述的强碱是二异丙胺锂或MN(SiMe3)2,M=Li,Na,K,化合物、强碱、ZnCl2和THPOCH2CHO的摩尔比为l∶l-10∶1-4∶1-4;
2)在惰性溶剂中,化合物2与酸在0℃到回流温度下反应0.5-10小时,得到化合物3,所述的化合物2与酸的摩尔比为1∶0.1-0.5;
3)在惰性溶剂中,化合物3与LiALH4在0℃到回流温度下反应0.5-2小时,得到化合物4,所述的化合物3与LiALH4的摩尔比为1∶1-5;
4)在惰性溶剂中,化合物3与有机或无机酸在0℃到回流温度下反应0.5-5小时,得到化合物4,所述的化合物3与有机或无机酸的摩尔比为1∶0.1-0.5;
其中,化合物1,2,3和4的结构式如下:


化合物1, 化合物2, 化合物3, 化合物4,
上述化合物中R1R2和R3如权利要求1所述,其中
 为-COOMe,-COOEt,-CH2OTHP,THP即四氢吡喃基。
为-COOMe,-COOEt,-CH2OTHP,THP即四氢吡喃基。
4,一种如权利要求3所述的呋喃并4a羟甲基多氢萘化合物的合成方法,其特征是所述的非质子极性溶剂是乙醚、四氢呋喃、二甲基亚砜或二甲基甲酰胺。
5,一种如权利要求3所述的呋喃并4a羟甲基多氢萘化合物的合成方法,其特征是所述的惰性溶剂是乙醚、四氢呋喃、石油醚、正己烷、苯、甲苯,醇或卤代烃。
6,一种如权利要求3所述的呋喃并4a羟甲基多氢萘化合物的合成方法,其特征是所述的无机酸可以是氢卤酸,硫酸,磷酸,所述的有机酸可以是甲酸,乙酸,对甲苯磺酸,樟脑磺酸或对甲苯磺酸吡啶盐。
7,一种如权利要求1所述的呋喃并4a羟甲基多氢萘化合物的用途,其特征是用于合成结构通式如下的海洋天然产物:
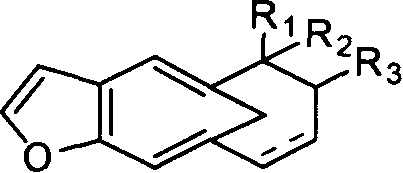
其中R1、R2、R3和
 如权利要求1所述。
如权利要求1所述。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN 02151114 CN1229367C (zh) | 2002-12-06 | 2002-12-06 | 呋喃并4a羟甲基多氢萘化合物、合成方法及其用途 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN 02151114 CN1229367C (zh) | 2002-12-06 | 2002-12-06 | 呋喃并4a羟甲基多氢萘化合物、合成方法及其用途 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN1421443A CN1421443A (zh) | 2003-06-04 |
| CN1229367C true CN1229367C (zh) | 2005-11-30 |
Family
ID=4751919
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN 02151114 Expired - Fee Related CN1229367C (zh) | 2002-12-06 | 2002-12-06 | 呋喃并4a羟甲基多氢萘化合物、合成方法及其用途 |
Country Status (1)
| Country | Link |
|---|---|
| CN (1) | CN1229367C (zh) |
-
2002
- 2002-12-06 CN CN 02151114 patent/CN1229367C/zh not_active Expired - Fee Related
Also Published As
| Publication number | Publication date |
|---|---|
| CN1421443A (zh) | 2003-06-04 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN100338051C (zh) | 西酞普兰的制备方法 | |
| CN1206207C (zh) | 氰酞氟苯胺的制备方法 | |
| CN1078203C (zh) | 全氟不饱和腈化合物及其制造方法 | |
| CN1229367C (zh) | 呋喃并4a羟甲基多氢萘化合物、合成方法及其用途 | |
| CN1095842C (zh) | 抗病毒有效的环丙烷衍生物的制备方法及所用中间体 | |
| CN1681800A (zh) | 制备前列腺素衍生物及其原料的方法 | |
| CN1496345A (zh) | 链状低聚乳酸酯 | |
| CN1379771A (zh) | 环状乳酸齐聚物的制备方法 | |
| CN1304346C (zh) | 光学活性2,3-联烯醇和联烯醇酯、合成方法及其用途 | |
| CN1239492C (zh) | 具有1,6-亚甲基-[10]-轮烯并呋喃和甾体基本结构的药物及其用途 | |
| CN1296342C (zh) | 炔化合物的制造方法 | |
| CN1033274C (zh) | 甾体青蒿素 | |
| CN1179933C (zh) | 用于制备烯烃聚合催化剂的γ-酰氧基取代醚化合物 | |
| CN101037425A (zh) | 2-呋喃甲酰基-3-芳基-4-乙氧基甲酰基-5-甲基-反式-2,3-二氢呋喃及其制备方法 | |
| CN1033084C (zh) | 取代的乙烯基苯的生产方法 | |
| CN100335454C (zh) | 一种二苯乙炔衍生物及其制备方法与应用 | |
| CN1876644A (zh) | 一种(4R-cis)-6-甲酰基-2,2-二甲基-1,3-二氧己环-4-乙酸叔丁酯的合成方法 | |
| CN1660806A (zh) | 2-三氟甲基吲哚的合成方法 | |
| CN1164589C (zh) | 旋光的吡咯并氮杂环庚三烯衍生物的制备方法 | |
| CN1898228A (zh) | 3-(4-四氢吡喃)-3-氧代丙酸烷基酯化合物以及4-酰基四氢吡喃的制备方法 | |
| CN1199962C (zh) | 一种抗肿瘤药物及其用途 | |
| CN1220674C (zh) | 一种左旋多巴甲酯盐酸盐的纯化方法 | |
| CN1252039C (zh) | 2-羟基-5-酰胺基苯酮类化合物的化学合成方法 | |
| CN1032001C (zh) | 氨基酮类的制备方法 | |
| CN1660766A (zh) | 带手性甲基侧链的ω-羟基酸衍生物 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C14 | Grant of patent or utility model | ||
| GR01 | Patent grant | ||
| C17 | Cessation of patent right | ||
| CF01 | Termination of patent right due to non-payment of annual fee |
Granted publication date: 20051130 Termination date: 20111206 |