CN1272111A - 三环后叶加压素激动剂 - Google Patents
三环后叶加压素激动剂 Download PDFInfo
- Publication number
- CN1272111A CN1272111A CN98809641A CN98809641A CN1272111A CN 1272111 A CN1272111 A CN 1272111A CN 98809641 A CN98809641 A CN 98809641A CN 98809641 A CN98809641 A CN 98809641A CN 1272111 A CN1272111 A CN 1272111A
- Authority
- CN
- China
- Prior art keywords
- benzodiazepine
- pyrrolo
- phenyl
- ketone
- compound
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/02—Drugs for disorders of the urinary system of urine or of the urinary tract, e.g. urine acidifiers
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/10—Drugs for disorders of the endocrine system of the posterior pituitary hormones, e.g. oxytocin, ADH
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/04—Antihaemorrhagics; Procoagulants; Haemostatic agents; Antifibrinolytic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/12—Antidiuretics, e.g. drugs for diabetes insipidus
Landscapes
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Veterinary Medicine (AREA)
- Animal Behavior & Ethology (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Engineering & Computer Science (AREA)
- Public Health (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Health & Medical Sciences (AREA)
- Diabetes (AREA)
- Hematology (AREA)
- Urology & Nephrology (AREA)
- Endocrinology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
Abstract
本发明涉及选自通式(Ⅰ)的新化合物或其药物学上可接受的盐、酯或前药形式,其中D、E和G是N或CH,这些化合物作为后叶加压素激动剂用于治疗尿崩症、夜间遗尿、夜尿症、尿失禁、出血和血凝固紊乱或不能暂时延迟排尿等疾患,以及涉及药物组合物和治疗方法。
Description
本发明涉及新的具有后叶加压素激动剂活性的化合物,以及使用该化合物的治疗方法和药物组合物。
发明背景
后叶加压素(抗利尿激素,ADH),一种九氨基酸肽激素和神经递质,是在大脑的丘脑下部中合成,并通过视上核垂体束传递至用于储存它们的垂体后叶中。当由大脑渗压感受器感觉到血浆克分子渗透浓度增加或通过压力感受器和体积感受器检测血量或血压降低时,后叶加压素被释放到血循环中并激活血管上的加压素V1a受体,引起血管收缩血压升高,并激活肾单位中的后叶加压素V2受体,使之主要保留水分和较小浓度的电解质,从而增大血量(Cervoni P.和Chan P.S.,“利尿剂”In Kirk-Othmer,“化学技术百科全书”,Wiley,volume8,398-432,1993)。早在1895年,人们就知道脑垂体中后叶加压素的存在(Oliver,H.和Schaefer,“生理学杂志”(London),18,277-279,1895)。du Vigneaud和同事在1954年完成了后叶加压素的结构测定和全部合成(du Vigneaud,V.,Gish.D.T.和Katsoyannis,“美国化学会志”,76:4751-4752,1954)。
后叶加压素V1a受体是通过磷脂酰肌醇途径介导的。后叶加压素V1a受体的激活引起血管平滑肌收缩、使得血压升高。后叶加压素V2受体是由激活腺苷酸环化酶体系和细胞内cAMP的浓度升高介导的。通过后叶加压素或后叶加压素类(肽或非肽)化合物激活后叶加压素V2受体,增加肾收集管的水渗透力并使得大量游离水重吸收。最后结果形成浓缩尿并排出,并伴随尿量减少和尿的同渗重量摩尔浓度增加。
通过在肾收集管位置处浓缩尿,后叶加压素在保存水方面起到重要作用。受体中没有后叶加压素时,肾收集管几乎不渗透水,因此在通过肾小球过滤、经过邻近的肾曲小管、汉勒氏袢和远侧的肾曲小管后形成的低渗流体将作为稀释尿液排泄出。然而,在脱水、体液流失或失血过程中,从大脑中释放出后叶加压素并激活肾收集管中的后叶加压素V2受体,使得该管对水的渗透力加大;因此重吸收水并排泄浓缩尿液。在患有中枢性或神经性尿崩症的病人和动物身上,大脑中后叶加压素的合成欠缺,因此他们不会产生或产生很少的后叶加压素,但是他们肾中的后叶加压素受体是正常的。由于他们不能浓缩尿液,会产生健康人十倍的尿量,而且对后叶加压素和后叶加压素激动剂的作用非常敏感。后叶加压素和去氨加压素(天然后叶加压素的肽类似物)可用于中枢性尿崩症的病人。当需要时,后叶加压素V2激动剂也可用于治疗夜间遗尿、夜尿症、尿失禁和提供给接受者暂时延迟排尿的能力。
通过激活其V1a受体,后叶加压素起着使血管收缩的作用使得血压升高。后叶加压素V1a受体激动剂将抵消该效果。后叶加压素和后叶加压素激动剂释放出VIII因子和维勒布兰德因子,这样它们可用于治疗出血症如血友病。后叶加压素和后叶加压素类激动剂也释放出组织型纤维蛋白溶原活化剂(t-PA)至血循环中,这样它们可用于溶解如心肌梗塞和其他血栓栓塞症的病人身上的血块(Jackson,E.K.,“使肾保留水分的后叶加压素和其它药剂”In:Goodman’s和Gilman’s,“治疗药物基础”第九版,Eds.Hardman,Limbird,Molinoff,Ruddon和Gilman,McGraw-hill,New York,pp.715-731,1996,Lethagen.S.,“血液学年报”69期、173-180(1994);Cash,J.D.等人,“不列颠血液学杂志”,27,363-364,1974;Dayid,J-L.,“调节肽”,45,311-317,1993和Burggraaf.J.等人,“临床学”86期497-503(1994)。
下面的现有技术文献中描述了肽的后叶加压素拮抗剂:M.Manning等人。“医药化学杂志”35,382(1992);M.Manning等人,“医药化学杂志”35,3895(1992);H.Gavras和B.Lammek,美国专利5070187(1991);M.Manning和W.H.Sawyer,美国专利5055448(1991);F.E.Ali,美国专利4766108(1998);R.R.Ruffolo等人,“药物最新报道和展望”,4(4),217(1991,5月)。P.D.Williams等人报道过有效的六肽催产素拮抗剂[“医药化学杂志”,35,3905(1992)],与V1和V2受体结合也显示出较弱的后叶加压素对抗活性。肽后叶加压素拮抗剂缺点在于缺少口服活性,且许多这样的肽不是选择过的拮抗剂,因为它们也显示部分激动剂活性。
最近已经有人公开了非肽的后叶加压素对抗剂。Albright等人在美国专利5516774(1996年5月14)中描述了三环二氮杂用作后叶加压素和催产素拮抗剂;J.P.0801460-A(1996年3月26)中公开了四氢苯并二氮杂衍生物用作后叶加压素拮抗剂;Ogawa等人在WO 9534540-A中公开了苯并杂环衍生物用作后叶加压素和催产素对抗剂,以及用作后叶加压素激动剂;Albright等人在美国专利5512563(1996年4月30)中公开了三环苯并氮杂衍生物用作后叶加压素拮抗剂;Venkatesan等人在美国专利5521173(1996年5月28)中公开了三环苯并氮杂衍生物用作后叶加压素和催产素拮抗剂。
如上所述,去氨加压素(1-脱氨-8-D-精氨酸后叶加压素)(Huguenin and Boissonnas,Helv.Chim.Acta,49,695(1996))是一种后叶加压素激动剂。该化合物是具有可变生物利用率的合成肽。鼻内途径服药较难忍受,而用于治疗夜间遗尿的口服药剂需要比鼻内服药多10-20倍的剂量。
本发明的化合物为非肽并具有较好的口服生物利用率。它们是特定的后叶加压素V2激动剂并且没有V1a激动剂效果,因此不会升高血压。相反,现有技术(Ogawa,H.等人的WO9534540-A)的化合物是后叶加压素/催产素拮抗剂。
发明概述
本发明涉及选自通式(I)的新化合物或其药物学上可接受的盐、酯或前药形式: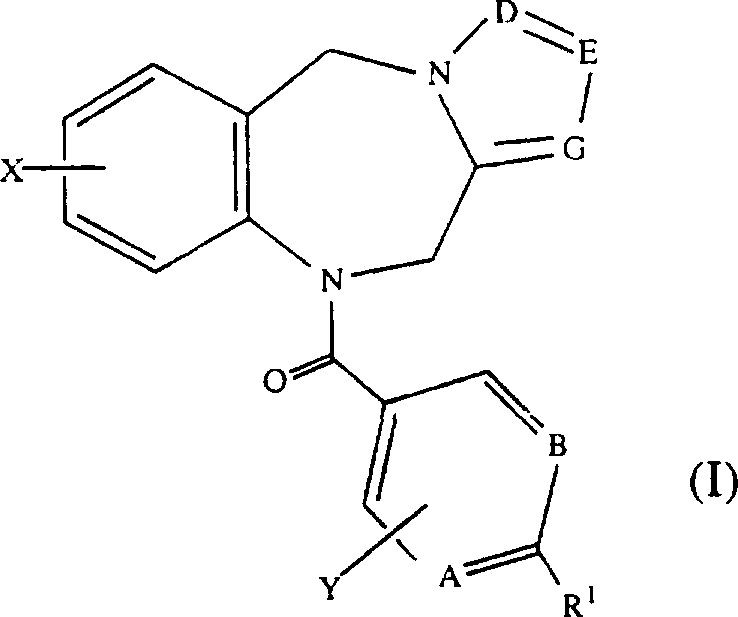
其中
A、B、E和G分别各自是CH或氮;
D独自是C-W或氮;
R1是2-7个碳原子的链烷酰基,选自CN、COOH、CONH2、
的基团,或选自如下基团:
 或
或
R2、R3和R5分别各自是氢,1-6个碳原子的直链烷基,3-7个碳原子的支链烷基,3-7个碳原子的环烷基或1-6个碳原子的全氟烷基;
R4是氢,1-6个碳原子的直链烷基,3-7个碳原子的支链烷基,3-7个碳原子的环烷基,2-7个碳原子的烷氧基烷基,或选自2-7个碳原子的链烷酰基、3-7个碳原子的链烯酰基、3-7个碳原子的环烷酰基、芳酰基或芳烷酰基的酰基取代基;
X和Y分别各自是氢,1-6个碳原子的直链烷基,3-7个碳原子的支链烷基,3-7个碳原子的环烷基,1-6个碳原子的全氟烷基,2-7个碳原子的烷氧基烷基,卤素(包括氯、溴、氟和碘),1-6个碳原子的烷氧基,羟基,CF3或2-6个碳原子的全氟烷基;
W是氢,卤素(优选氯、溴或碘),烷基,2-7个碳原子的烷氧基烷基,1-6个碳原子的羟基烷基或CH2NR6R7;
R6和R7分别各自是氢,1-6个碳原子的直链烷基,3-7个碳原子的支链烷基;或R6和R7与CH2NR6R7中的氮原子一起形成五元或六元环,该环任意含有一个或多个其他杂原子,例如(但不局限于)如下基团:  或
或
R8是1-6个碳原子的直链烷基;
R9分别是氢,三甲基甲硅烷基或1-6个碳原子的直链烷基;
E和G优选为CH;D优选为N或C-W,其中W是氢、烷基、CH2NR6R7或卤素,更优选其中W是氢、甲基、CH2NMe2或溴。
R2、R3和R5各自优选为氢,1-6个碳原子的直链烷基,3-7个碳原子的环烷基或1-6个碳原子的全氟烷基,更优选为氢或1-6个碳原子的直链烷基,最优选为氢或甲基。
R4优选是氢,1-6个碳原子的直链烷基或酰基取代基,更优选是氢、甲基、乙基、正丙基、正丁基、甲氧基甲基、乙酰基、环丙基羰基、正丙基羰基、2-噻吩基羰基、2-甲基、5-氟苯基羰基、2-甲基苯基羰基、2-氯-4-氟苯基羰基、2,4-二氟苯基羰基或2,4-二氟苄基羰基。
X和Y各自优选是氢,1-6个碳原子的全氟烷基,卤素,1-6个碳原子的烷氧基或羟基,更优选是氢、三氟甲基、氯、溴、氟、甲氧基或羟基。最优选X和Y中至少一个是氢。
R6和R7优选均为甲基。
R1的优选基团包括CN、CONH2、乙酰基或如下基团中的一种:
-基团a,其中R2、R3和R5分别各自是氢或1-6个碳原子的直链基团,更优选其中烷基是甲基;
-基团a,其中R2、R3和R5中的两个是氢,第三个是3-7个碳原子的环烷基或1-6个碳原子的全氟烷基,更优选其中第三个是环丙基或三氟甲基;
-基团b,c,d或i,其中R2是氢或1-6个碳原子的直链烷基,更优选其中烷基为甲基;
-基团f,其中R2是氢和/或R4是氢,1-6个碳原子的直链烷基或选自2-7个碳原子的链烷酰基、3-7个碳原子的链烯酰基、3-7个碳原子的环烷酰基、芳酰基或芳烷酰基的酰基取代基;更优选其中R4是氢、甲基、乙基、正丙基、正丁基、甲氧基甲基或乙酰基、环丙基羰基、正丙基羰基、2-噻吩基羰基、2-甲基、5-氟苯基羰基、2-甲基苯基羰基、2-氯-4-氟苯基羰基、2,4-二氟苯基羰基或2,4-二氟苄基羰基;
-基团f或g,其中R4是氢和/或R2是1-6个碳原子的直链烷基,更优选其中烷基是甲基;
-基团k或h,其中R2是甲基;
-基团m,其中R2是氢。
本发明更优选的化合物为下式化合物或其药物学上可接受的盐:
其中
A和B分别是CH或氮;
D是C-W或氮;
R1是2-7个碳原子的链烷酰基或选自如下基团:
 或
或

R2、R3和R5分别各自是氢,1-6个碳原子的直链烷基,3-7个碳原子的支链烷基,3-7个碳原子的环烷基或1-6个碳原子的全氟烷基;
R4、X、Y、W、R6、R7和R8定义同上;
这里使用的作为基团或基团一部分的术语烷基,如烷氧基,包括直链或支链的烷基,如甲基、乙基、丙基、异丙基、正丁基、仲丁基、异丁基、叔丁基、戊基、己基和庚基。这里使用的术语环烷基包括饱和和不饱和的环基,如环丙基、环丁基、环戊基、环己基、环庚基、环丙烯基、环丁烯基、环戊烯基、环己烯基和环庚烯基。优选饱和的环烷基。
用于上述化合物定义和本文所指的芳酰基,除非另有说明,均包括可以分别被一个或多个取代基取代的苯甲酰基和萘甲酰基,所述取代基选自氢、卤素、氰基、1-6个碳原子的直链烷基、3-7个碳原子的支链烷基、1-6个碳原子的烷氧基、CF3或苯基(该苯基本身可以被任意取代)。这里提到的杂芳酰基是指与五元杂环的碳原子直接连接的羰基,其中杂环含有一个或两个选自氮、氧或硫的杂原子,例如2-噻吩羰基。杂芳酰基的杂环还可以包括,(但不局限于)其中杂芳基部分是呋喃、吡咯、2H-吡咯、咪唑、吡唑、异噻唑、异噁唑、噻吩、吡唑啉、咪唑烷或吡唑烷的基团。这里的杂芳基可以单独被一个或多个取代基取代,所述取代基选自氢、卤素、氰基、1-6个碳原子的直链烷基或3-7个碳原子的支链烷基。
这里所述的芳基烷酰基,是指与1-6个碳原子的烷基直接连接的羰基,其中烷基末端被芳基取代,例如苯乙酸。芳基可以单独被一个或多个取代基取代,所述取代基选自氢、卤素、氰基、1-6个碳原子的直链烷基、3-7个碳原子的支链烷基、1-6个碳原子的烷氧基、CF3或苯基或取代的苯基,其中苯基中的取代基选自卤素、氰基、1-6个碳原子的直链烷基、3-7个碳原子的支链烷基、1-6个碳原子的烷氧基或CF3。
如上文所定义。
这里提到的卤素,除非另有说明,可以选自氟、氯、溴或碘。
本领域实施人员可以理解,当R1、R2、R3、R4、R5、R6、R7、X或Y含有不对称碳原子时,定义的式(I)化合物包括所有可能的具有如下所述活性的其立体异构体和混合物。具体讲,包括任何旋光异构体和非对映异构体;以及外消旋体和拆分出的对映异构纯的R和S立体异构体;以及具有所述活性的其它R和S立体异构体的混合物和其药物上可接受的盐。按照常规的分离技术可以获得纯的旋光异构体。同样可以理解的式(I)化合物中R1、R2、R3、R4、R5、R6、R7、X或Y的定义包括了所有可能的具有所述活性的区域异构体(regioisomers),及其混合物。按照本领域技术人员熟知的常规分离方法可以获得纯的该区域异构体。
同样优选的本发明化合物是如下一类:
a)下式化合物:
其中A、B、W、R1、R2、R3、R4、R5、R6、R7、R8、R9、X和Y定义同上;
b)下式化合物: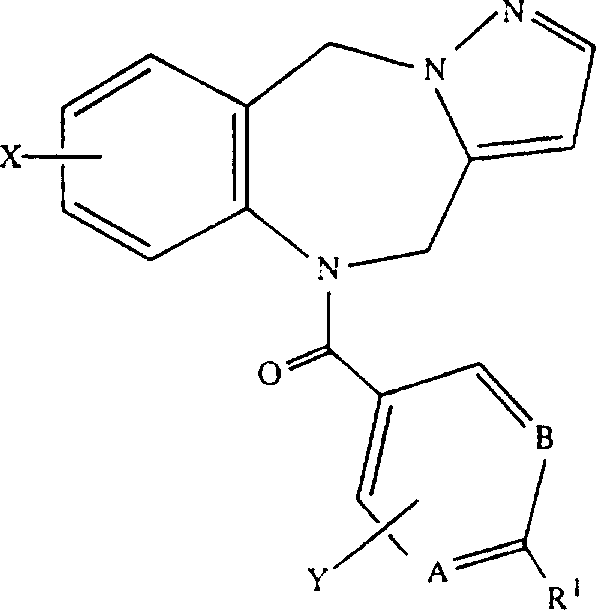
其中A、B、R1、R2、R3、R4、R5、R9、X和Y定义同上;及
c)下式化合物:
其中A、B、R1、R2、R3、R4、R5、R9、X和Y定义同上。
可以理解上述亚类化合物a)-c)还包括如下述亚类化合物,其中:
A和B分别是CH或氮;
R1是2-7个碳原子的链烷酰基或选自如下基团
R2、R3和R5分别是氢、1-6个碳原子的直链烷基、3-7个碳原子的支链烷基、3-7个碳原子的环烷基或1-6个碳原子的全氟烷基;
R4、X、Y、W、R6、R7和R8定义同上;
或其药物上可接受的盐。
上述化合物a)中,特别优选的是其中W是H,A和B分别是CH,R1是2-7个碳原子的链烷酰基或选自上面列出的基团(a),(b),(e),(f),(g),(h),(i)或(k)。
药物上可接受的盐包括由如下有机酸和无机酸衍生出的那些盐:柠檬酸、乳酸、乙酸、酒石酸、琥珀酸、马来酸、丙二酸、盐酸、氢溴酸、磷酸、硝酸、硫酸、甲磺酸和已知类似可接受的酸。
本发明还提供了治疗需要后叶加压素激动剂活性的疾病、症状或紊乱的方法,该方法包括给需要这种治疗的人或其它哺乳动物服用有效量的本发明化合物或药物组合物。这些治疗方法包括那些用于希望释放出VIII因子和维勒布兰德因子至循环体系中,释放出组织型纤维蛋白溶原活化剂(t-PA)至血循环中,或使肾保持水和尿的浓度从而治疗疾病、症状和紊乱的方法。这种治疗方法包括,但不局限于治疗人或其它哺乳动物的尿崩症、夜间遗尿、夜尿症、尿失禁或出血和血凝固紊乱。
这里的方法包括促进人或其它哺乳动物暂时延迟排尿,这也可以被理解为无论何时需要,对不能暂时延迟排尿的情况进行控制或治疗。这种方法可以被理解为包括不同于和不包括称为夜间遗尿症症状治疗在内的促进暂时延迟排尿治疗法。
相应地,本发明提供了用于治疗上述疾病、症状或紊乱的药物组合物,药物组合物包括一个或多个本发明的化合物或其药物上可接受的盐,并与药物上可接受的载体相结合。
该组合物优选适用于口服。然而,它们也适合其它服用方式,例如,用于患有心力衰竭病人的肠胃外用药。
为了达到连续服药,本发明的组合物优选单位剂量形式。合适的单位剂量形式包括片剂、胶囊剂和装在小药囊或小瓶中的粉末。这样的单位剂量可以含有0.1-1000mg本发明的化合物,优选含有2-50mg。更优选的单位剂量含有5-25mg的本发明化合物。本发明化合物的口服剂量范围可以是0.01-100mg/Kg或优选为0.1-10mg/Kg。该组合物一天可以服用1-6次,更通常为一天1-4次。
本发明的组合物可以用常用的载体和赋形剂配制,如填料、崩解剂、粘合剂、润滑剂、调味剂等。它们可以按照常用的方法进行配制,例如按照类似于用于配制已知抗高血压药剂、利尿剂和β-抑制剂的方法。
本发明还提供了用于制备本发明化合物的方法。
发明方法
本发明的化合物可以按照如下列出的一般方法中的一种进行制备。
按照反应路线I所示的方法,在-40℃至50℃的温度下,在碱,如吡啶或三烷基胺如三乙胺存在下、在非质子传递有机溶剂,如二氯甲烷或四氢呋喃中,用合适取代的乙酰芳酰基(杂芳酰基)卤,优选式(2)的芳酰(杂芳酰)氯处理式(1)的三环苯并二氮杂制得酰化的衍生物式(3)。按照Lin等人在“杂环化学杂志”14,345(1977)中的方法,在0℃-溶剂回流温度范围内,在非质子传递有机溶剂,如二氯甲烷中,用二烷基酰胺二烷基缩醛(4)处理(3)制得烯酮(5)。在室温-溶剂回流温度的范围内,在乙酸中用羟胺或取代的肼(6)处理(5)制得目标化合物式(I),其中A、B、D、E、G、X、Y、R2和R4定义如上,R1是选自上述定义的杂环基团(f),(g)或(j)。
在室温-溶剂回流温度的范围内用亚硫酰氯处理相应的羧酸,或在0℃-40℃的温度范围内、在催化量的二甲基甲酰胺存在下、在非质子传递溶剂如二氯甲烷或四氢呋喃中用草酰氯处理相应的羧酸,可以方便地制备优选的反应路线I中的取代的乙酰基芳酰基(杂芳酰基)氯(2)。
优选的二烷基酰胺二烷基乙缩醛可以购买到,或是本领域已知的,或者可以按照文献中的类似方法方便地制备出(参见Kantlehner,W.Chem.Ber.,105,1340(1972))。
优选的式(1)三环苯并二氮杂是10,11-二氢-5H-吡咯并[2,1-c][1,4]苯并二氮杂(Albright et al.,美国专利5536718,1996年7月16公开),10,11-二氢-5H-吡唑[5,1-c][1,4]苯并二氮杂(Cecchi,L.等人的“杂环化学杂志”,20,871(1983))和10,11-二氢-5H-四唑[5,1-c][1,4]苯并二氮杂(Klaubert,D.H.,“杂环化学杂志”,22,333(1985))。
反应路线I 下面反应路线II中描述了另一种制备中间体式(3)的方法。反应路线II
下面反应路线II中描述了另一种制备中间体式(3)的方法。反应路线II
这样,在-40℃至50℃的温度下,在有机碱,如吡啶,或三烷基胺,如三乙胺存在下、在非质子传递有机溶剂如二氯甲烷或四氢呋喃中,用合适取代的溴代芳酰基(杂芳酰基)卤,优选是式(8)的芳酰(杂芳酰)氯处理式(1)的三环苯并二氮杂制得乙酰化中间体式(9)。随后主要按照Martinez等人,“医药化学杂志”,35,620(1992)中的方法,在室温-100℃的温度范围内,在密封的耐压管中,以有机碱,如三乙胺作为溶剂,在吡啶和催化剂,如氯化二(三苯基膦)钯(II)和碘化亚铜存在下,将中间体(9)与单取代的、用三甲基甲硅烷基或1-6个碳原子的直链烷基等封端的乙炔偶合。然后主要按照Reed等人在“有机化学杂志”,52,3491(1987)中的方法,在室温下,在非质子传递有机溶剂,如硫酸汞(II)饱和的四氢呋喃中,用1%的硫酸水合得到的乙炔中间体式(10),制得所需的酰基化合物式(3),其中A、B、D、E、G、X和Y定义如上,R9是氢或1-6个碳原子的直链烷基。另外,在醚溶剂,如四氢呋喃中,用氟化四丁基铵处理化合物9(其中R9是三甲基甲硅烷基),得到化合物(10),其中R9是氢。
在室温-溶剂回流温度的范围内用亚硫酰氯,或在0℃-40℃的温度范围内、在催化量的二甲基甲酰胺存在下、在非质子传递溶剂如二氯甲烷或四氢呋喃中用草酰氯处理合适取代的芳基(杂芳基)羧酸式(7),可以方便地制备优选的反应路线II中式(8)的酰化剂。
反应路线II中保护的乙炔中间体可以市购,或是本领域已知的,或者可以按照文献中的类似方法容易地制备出。
如反应路线III中所示,在室温-溶剂回流温度的范围内,在催化量的氯化二(三苯基膦)钯(II)存在下,在非质子传递有机溶剂如甲苯中,将反应路线II中式(9)溴代芳基(杂芳基)化合物与(α-乙氧基乙烯基)三烷基锡、优选(α-乙氧基乙烯基)三丁基锡进行Stille偶合反应,也可以制备反应路线I中间体乙酰基化合物(3),即主要按照Kosugi等人在“日本化学会公告”,(Bull.Chem.Soc.Jpn.),60,767(1987)中所述方法。
反应路线III
也可以按照Cabri等人,在“四面体通讯”,32,1753(1991)中的方法,通过用芳基卤中间体式(9)与乙烯基烷基醚,如乙烯基丁基醚进行钯催化的芳基化反应,完成乙酰基化合物(3)的制备。
反应路线III中(α-烷氧基乙烯基)三烷基锡中间体可以购买到,或是本领域已知的,或者可以按照文献中的类似方法容易地制备出。
当反应路线I中的R4是氢时,杂环氮原子可以按照反应路线IV中列出的反应进行烷基化或酰化。
反应路线IV
这样,通过在0℃-80℃的温度范围内,在非质子传递有机溶剂,如二甲基甲酰胺或四氢呋喃中,用强碱,如氢化钠或氢化钾和烷基化剂,如烷基卤、优选烷基氯(溴或碘)处理,将吡唑化合物式(I,R4是H)烷基化,制得化合物式(I,R1=(f)或(g)),其中A、B、D、E、G、X、Y和R2定义如上,R4是烷基或酰基部分。此外,通过在-40℃至室温的范围内,在胺碱,如吡啶或三烷基胺、优选三乙胺存在下,在非质子传递有机溶剂,如二氯甲烷或四氢呋喃中,或者如果使用吡啶作为碱时不加溶剂,用羧酰卤、优选羧酰氯或羧酸酐处理,将化合物(I)酰化,制得化合物(I),其中A、B、D、E、G、X、Y和R2定义如上,R4是烷基或酰基部分。化合物式(I,R4是H)的烷基化或酰化产生区域异构体混合物,其中R2是氢,R1分别选自上面定义的杂环基团(f)或(g)的杂环部分且如下所示。
按照反应路线V中列出的一般方法可以制备出反应路线I中的通式(I)化合物,其中A和B是碳,R2是H,R1是选自上面定义的杂环基团(g)的杂环部分。
这样,在室温-80℃的温度范围内,在催化剂如氯化二(三苯基膦)钯(II)和碘化亚铜(I)存在下,在有机碱,如三乙胺的溶剂中用二烷基氨基丙炔、优选1-二甲基氨基丙炔偶合合适取代的卤代芳基(杂芳基)羧酸酯,优选其溴代(或碘代)甲基酯式(11),主要按照Alami等人在,“四面体通讯”,34,6403(1993)和Sanogashira等人在,“四面体通讯”,4467(1975)中的方法,制得取代的乙炔中间体通式(12)。接着在低于室温的温度下,在非质子传递有机溶剂如二氯甲烷中,采用任一种标准的氧化方法(Albini,A.,“合成”,263(1993))用氧化剂或用二环氧乙烷试剂(Murray,R.W.,“化学综述”,1187(1989))处理,将中间体(12)转化为其N-氧化物。中间体N-氧化物未经分离,通过处理、优选在羟基溶剂中加热就地重排形成通式(13)的烯酮(enone),其中所述的羟基溶剂包括含有水、任意的C1-C8直链或支链烷基醇、乙二醇、聚乙二醇、1,2-丙二醇、聚丙二醇、丙三醇、2-甲氧基乙醇、2-乙氧基乙醇、2,2,2-三氟乙醇、苄醇、酚或本领域技术人员已知的含有一个或多个游离羟基(-OH)取代基的任意相当溶剂中的一种溶剂或这些溶剂的混合物。
含有一种或多种助溶剂,以及一种或多种溶剂的溶剂体系,也可以用于将N-氧化物重排至所需烯氨酮(enaminone)的方法中。这里提到的助溶剂可以定义为稀释的主溶剂,可以选自:烃,如戊烷、己烷或庚烷;芳烃,如苯、甲苯或二甲苯;醚,如乙醚、四氢呋喃、二噁烷或二甲氧基乙烷;氯代烃,如二氯甲烷、氯仿、二氯乙烷或四氯乙烷;或其他常用的溶剂,如乙酸乙酯、N,N-二甲基甲酰胺、N,N-二甲基乙酰胺、乙腈、二甲亚砜或丙酮等。
通过在约室温-溶剂回流的温度下,将胺的N-氧化物导入合适的羟基溶剂中并优选搅拌,可以完成胺的N-氧化物至烯胺酮的转化。其他情况下,在可接受的催化剂如钯(II)催化剂或铜(I)催化剂存在下,在室温-溶剂回流的温度下可以将胺的N-氧化物导入合适的羟基溶剂中,并优选搅拌。
该方法提供了一种新的在羟基溶剂中由炔丙基胺或其N-氧化物合成烯胺酮(enamineone)的方法,其中溶剂影响了反应的最终结果。这种新的烯胺酮(enaminone)的合成方法成为已知方法的一种方便的替代方法,此外扩大了可转化为烯胺酮产物的原料范围。
尽管目前还没有精确地确定炔丙基胺N-氧化物转化为烯胺酮(enaminone)产物的确切机理,但可能类似于两种已知的方法;炔丙基胺N-氧化物的热[2,3]-σ迁移重排(Craig等人“四面体通讯”,4025,1979;Hallstrom等人“四面体通讯”,667,1980;Khuthier,A-H等人“化学会志化学通讯”,9,1979)和特定异噁唑至烯胺酮(enaminone)的转化(Liguori等人“四面体”,44,1255(1988))。
在室温-回流的温度范围内,在乙酸中,用取代的肼(6)处理(13),产生可变比例的通式(14)和(15)区域异构体化合物的混合物。通过色谱法和/或结晶法分离出较多量的异构体式(14),接着水解成所需的羧酸式(16)。
然后通过类似上文描述的那些方法将中间体(16)转化为酰化剂,优选是酰氯(溴或碘)或式(17)的混合酐。然后按照上文描述的任一种方法,使用酰化剂(17)将式(1)三环苯并二氮杂进行酰化制得目标化合物式(I),其中A和B是CH,D、E、G、X、Y和R4定义如上,R2是氢,R1是选自杂环基团(g)的杂环部分,且如下所示。
同样,在室温-溶剂回流温度的范围内,在乙酸中用未取代的肼(6,R4是H)处理(13)制得式(18)中间体吡唑酯。在此情况下,杂环氮可以按照反应路线VI中所示的方法被烷基化或酰化,制得式(I)化合物,其中R2是氢,R1是选自上述定义的杂环基团(f)的杂环部分。
反应路线VI
这样,通过在0℃-80℃的温度范围内,在非质子传递溶剂,如二甲基甲酰胺或四氢呋喃中,用强碱,如氢化钠或氢化钾和烷基化剂,如烷基卤、优选烷基氯(溴或碘)处理,将中间体酯式(18)烷基化制得可变比例的通式(14)和(15)区域异构体的混合物。通过色谱法和/或结晶法分离出较多量的异构体(15),接着水解成所需的羧酸式(19),然后通过类似上文描述的那些方法转化为酰化剂,优选是酰氯或混合酐。然后使用酰化剂式(20)将式(1)三环苯并二氮杂进行酰化制得目标化合物式(I),其中A、B、D、E、G、X、Y和R4定义如上,R2是氢,R1是选自上述定义的杂环基团(f)的杂环部分。
按照反应路线VII中列出的方法可以制备通式(I)化合物,其中R1是选自上述定义的杂环基团(h)的杂环部分。
反应路线VII
(23)J=酰化部分
首先在室温-溶剂回流温度的范围内,在乙酸中用肼处理合适取代的丙二醛式(21),然后在室温-溶剂回流温度的范围内,在碱性水溶液中用高锰酸钾氧化中间体吡唑,制得羧酸中间体式(22)。通过类似上文描述的那些方法将酸(22)转化为酰化剂,优选是酰氯(溴或碘)或混合的酐。最后将酰化剂式(23)与式(1)的三环苯并二氮杂进行反应制得通式(I)化合物,其中A、B、D、E、G、X、Y和R4定义如上,R1是选自上述定义的杂环基团(h)的杂环部分。
当反应路线VII中的R4是氢时,可以按照上文中列出的方法将杂环氮烷基化或酰化。
优选的丙二醛式(21)和反应路线VII中的肼可以购买到,或是本领域已知的,或者可以按照文献中用于制备已知化合物的类似方法容易地制备出,例如Knorr等人,在“有机化学杂志”,49,1288(1984)和Coppola等人,在杂环化学杂志,51(1974)中所述。
反应路线VIII中列出了另一种制备反应路线VII中的式(22)中间体羧酸的方法,其中Y定义如上,R4是氢以外的其它基团。
反应路线VIII
在室温-150℃的温度范围内,在催化剂,如四(三苯基膦)钯(O)和碘化亚铜(I)存在下,在有机非质子传递溶剂,如二甲基甲酰胺中,将有机锡试剂式(25)与合适取代的芳基(杂芳基)卤,优选式(28)的溴化物或碘化物进行Stille偶合反应,主要按照类似于Farina等人在“有机化学杂志”,59,5905(1994)中描述的那些方法。在室温-溶剂回流温度的范围内,用氢氧化钠或氢氧化锂在含水的醇或四氢呋喃中碱性水解得到的式(26)的酯,制得所需的羧酸式(22)。
反过来,按照类似于Martina等人在“合成”,8,613(1991)中的那些方法,在-40℃至室温的温度范围内,在非质子传递有机溶剂如乙醚中,在金属化试剂如烷基锂,例如正丁基锂、仲丁基锂或叔丁基锂存在下,通过用三烷基锡卤化物、优选氯化三丁基锡(或溴化三丁基锡)金属化4-溴-N-烷基吡唑式(24),可以方便地制备式(25)的有机锡试剂,其中R优选是烷基。
通过在0℃-80℃的温度范围内,在非质子传递有机溶剂如二甲基甲酰胺或四氢呋喃中,在强碱如氢化锂、氢化钠或氢化钾存在下,用烷基卤、优选烷基氯(溴或碘)将4-溴吡唑烷基化,可以方便地制备优选的N-烷基取代的4-溴吡唑式(24)。另外,可以用上述烷基化试剂和强碱如氢氧化锂、氢氧化钠或氢氧化钾,在相转移催化剂(Jones,R.A.Aldrichimica Acta,9(3),35,1976)如氯化苄基二甲基十四烷基铵或氯化苄基三甲基铵存在下,进行4-溴吡唑的烷基化反应。
优选的芳基(杂芳基)碘化物式(28)可以通过如下方法方便地制备:将相应的取代的苯胺式(27)重氮化,随后将相应的重氮盐与碘或碘化钾在酸性含水的介质中反应,主要按照Street等人在“医药化学杂志”,36,1529(1993)和Coffen等人在“有机化学杂质”,49,296(1984)中的方法进行。
反应路线IX中列出了另一种制备通式(I)化合物的方法。
反应路线IX
在-40℃至溶剂回流温度下,在碱,如三乙胺或二异丙基乙胺存在下、在非质子传递有机溶剂,如二氯甲烷或四氢呋喃中,用合适取代的卤代芳酰基(杂芳酰基)卤,优选氟代芳酰或氟代(或氯代)杂芳酰氯式(29)处理式(1)的三环苯并二氮杂,制得酰化的中间体式(30)。
另外,酰基化部分可以是上述羧酸的混合酐,例如按照Inanaga等人,“日本化学会志公告”(Bull.Chem.Soc.Jpn.),52,1989(1979)中的方法,使用2,4,6-三氯苯甲酰氯在溶剂,如二氯甲烷中反应而制备出的混合酐。在0℃-溶剂的回流温度范围内,在有机碱,如4-二甲基氨基吡啶存在下,在溶剂,如二氯甲烷中,用式(1)三环苯并二氮杂处理所述的通式(29)的混合酐,制得反应路线IX中的中间体酰基化衍生物(30)。
然后在室温-溶剂回流温度的范围内,在极性非质子传递有机溶剂,如二甲基甲酰胺或四氢呋喃中,用式(31)的合适取代的杂环锂、钠或钾盐处理式(30)化合物,制得通式(I)化合物,其中A、B、D、E、G、X、Y、R2、R3和R5定义如上,R1选自上述定义的杂环基团(a),(b),(c),(d),(l),(n)或(o)。
式(30)中间体与式(31)的中间体盐缩合产生可变比例的通式(I)的区域异构体,该异构体通过色谱法和/或结晶法分离。
优选取代的氟代芳酰基和氟代(或氯代)杂芳酰基氯式(29)可以购买到,或是本领域已知的,或者可以按照文献中用于制备已知化合物的类似方法容易地制备出。
在-40℃至室温的范围内,在非质子传递有机溶剂,如二甲基甲酰胺或四氢呋喃中,用强碱,如氢化锂、氢化钠、氢化钾或金属醇化物处理所述的杂环,制备杂环式(31)的锂、钠或钾盐。
此外,反应路线IX中所述的通式(I)化合物可以按照反应路线X中列出的方法进行制备。
反应路线X
这样,使用本领域已知的方法,将合适取代的氟代芳基或氟代(或氯代)杂芳基羧酸式(32)进行酯化,例如在催化量的二甲基甲酰胺存在下,在醇溶剂,如甲醇中用草酰氯(或亚硫酰氯)处理;或者通过在室温-回流温度的范围内,在酸催化剂,如对甲苯磺酸存在下与甲醇进行缩合。
在室温-150℃的温度范围内,在极性非质子传递有机溶剂,如二甲基甲酰胺中,将制得的式(33)的酯,与合适取代的杂环式(31)的锂、钠或钾盐反应,制得式(34)的中间体酯。(33)与(31)的缩合产生可变比例的式(34)的区域异构体,该异构体通过色谱法和/或结晶法分离。
随后将式(34)的中间体酯与碱溶液,如氢氧化钠或氢氧化锂的甲醇溶液或四氢呋喃溶液进行水解,得到式(35)的羧酸。
然后使用上文中描述的任意方法将中间体羧酸(35)转化为酰化剂,优选通式(36)的酰基氯或混合酐。
随后按照上文描述的任意方法将式(1)的三环苯并二氮杂与式(36)的中间体酰化剂进行反应,制得反应路线IX中的目标化合物式(I)。
此外,反应路线X中所述的式(35)的取代羧酸可以按照反应路线XI中列出的方法进行制备。
反应路线XI
这样,在室温-150℃的温度范围内,在极性非质子传递溶剂,如二甲基甲酰胺中,将式(37)的氟代芳基或氟代(氯代)杂芳基腈与取代的杂环式(31)的理、钠或钾盐反应,制得通式(38)的中间体。(37)与(31)的反应产生可变比例的式(38)的区域异构体,该异构体通过色谱法和/或结晶法分离。中间体式(38,Y≠CF3)腈的水解优选在室温-60℃的温度范围内,与无机酸,如硫酸进行。
此外,腈(38)的水解可以通过在乙醇中,在强碱,如氢氧化钠存在下进行,其中含有或不含相转移催化剂(Jones,R.A.Aldrichimica,9(3),35,1976),如氯化苄基二甲基十四烷基铵。
然后按照类似上文描述的那些方法将制得的羧酸式(35)转化为反应路线IX中目标化合物式(I)。
此外,反应路线X中取代的羧酸式(35),可以按照反应路线XII中描述的方法进行制备,即用碱性过氧化氢在二甲亚砜中连续处理式(38)的腈,其中A和B是CH,R1不是2-7个碳原子的链烷酰基、炔基(b)或(d),主要按照Katritzky等在“合成”,949(1989)中所述方法,随后水解制得式(38)的酰胺,优选按照Hales等人,“四面体”,51,7403(1995)中所述方法,用稀硫酸和硝酸钠进行处理。
反应路线XII
其中R1不是(b)或(d)
反应路线XIII中列出了制备反应路线X中的中间体,取代的羧酸式(35)的优选方法,其中R1是选自上述定义的R1杂环基团(a)的杂环部分。
反应路线XIII
将合适取代的式(40)的苯胺重氮化,然后按照Street等人在“医药化学杂志”,36,1529(1993)中的方法,在浓盐酸中用氯化锡(II)还原制得的重氮盐式(41),得到中间体肼的盐酸盐式(42)。随后在室温-100℃的温度范围内,在溶剂,如含水的甲醇中,将(42)与醛衍生物式(47,其中R2定义如上,R3和R5是H,P是二烷基乙缩醛),如乙酰基乙醛二甲基乙缩醛或酮式(47,其中R2、R3和R5定义如上,P=O或(O-烷基)2)进行缩合,结晶后得到目标中间体酯式(34,R1是(a),R5是H),然后再按照上文反应路线X列出的方法将其转化为化合物式(I)。
当Y是OCH3时,按照反应路线XIV列出的方法,可以方便地将反应路线I中的通式(I)化合物去甲基化。
反应路线XIV
这样,将化合物(I)(其中Y是OCH3)与三溴化硼在有机溶剂,如二氯甲烷中反应,制得式(I)相应的酚,其中Y是OH,而A、B、D、E、G、X、R2和R3定义如上,R1选自上述定义的杂环基团(a)且如下所示。
按照反应路线XV制备其中R1含有3个杂原子的化合物。
反应路线XV
这样,在-40℃至80℃的温度范围内,在非质子传递有机溶剂,如二氯甲烷或四氢呋喃中,在碱存在下,用合适取代的氰基芳酰基(杂芳酰基)卤、优选芳酰(杂芳酰)氯式(43)处理式(1)的三环苯并二氮杂,制得中间体腈式(46,反应路线XVI),随后在室温-50℃的温度范围内,通过用无机酸,如硫酸将其水解为通式(44)的酰胺中间体。在0℃-80℃的温度范围内,在非质子传递有机溶剂,如二氯甲烷或四氢呋喃中,用二烷基酰胺二烷基缩醛式(4)处理酰胺(44),制得中间体式(45)。在室温-回流温度的范围内,在乙酸中用羟胺或肼式(6)处理(45),制得所需的目标化合物式(I),其中A、B、D、E、G、X、Y、R2和R4定义如上,R1是选自上述定义的杂环基团(e),(i)或(k)。
反应路线XVI中列出了另一种用于制备中间体酰胺式(44)(参见反应路线XV)的优选方法,其中A和B是CH,D不是CH。该方法包括用碱性过氧化氢在二甲亚砜中处理腈式(46),主要按照Katritzky等人在“合成”,949(1989)中所述的方法。
反应路线XVI
反应路线XVII中列出了用于制备通式(I)化合物的优选方法,其中R1含有4个杂原子,R4是氢。
反应路线XVII
在室温-溶剂回流温度的范围内,在非质子传递有机溶剂,如二甲基甲酰胺中,用叠氮化钠和氯化铵处理反应路线XVI中腈中间体式(46),制得目标化合物式(I),其中A、B、D、E、G、X和Y定义如上,R4是氢,R1是选自上述定义的杂环基团(m)的杂环部分。
如反应路线XVIII中所示,通式(I)化合物可以进行Mannich缩合反应,其中D是CW,W是氢。
反应路线XVIII
这样,在室温-回流的温度范围内,将化合物式(I,D是CH)与含水的甲醛或低聚甲醛、取代的胺式(47)和冰醋酸,在醇溶剂,如甲醇中进行反应,制得通式(I)相应的Mannich碱,其中A、B、E、G、X、Y、R2、R3、R5、R6和R7定义如上;D是CW;W是二烷基氨基烷基残基、优选为二甲基氨基甲基残基,R1是选自上述定义的基团(a),(c),(e),(f),(g),(h),(i),(j),(k),(l),(m),(n)和(o)的杂环基。
同样,如反应路线XIX所示,通式(I)化合物(其中D是CH)可以进行卤化反应。
反应路线XIX
这样,在-80℃至室温的温度范围内,在极性非质子传递有机溶剂,如二氯甲烷中,将(I,D是CH)与N-卤代丁二酰亚胺,如N-氯(溴或碘)代丁二酰亚胺反应,制得通式(I)相应的卤代衍生物,其中A、B、E、G、X、R2、R3和R5定义如上,D是CW,W是卤素如氯(溴或碘),R1是选自上述定义的基团(a),(c),(e),(f),(g),(h),(i),(j),(k),(l),(m),(n)和(o)的杂环基。
按照下面方法测试本发明主题化合物的生物活性。
试验化合物对正常神志清醒喂过水的大鼠作为后叶加压素V2激动剂的效果
给350-500克体重的雄性或雌性的正常血压的Sprague-Dawley鼠(Charles River,Laboratories,inc.,Kingston,NY)提供标准啮齿动物的食物(Purina Rodent lab.Chow 5001)和任意的水。在实验当天,将鼠单独放置在装有分离粪便和尿的装置及收集尿的容器的代谢测定笼中。将试验化合物或参照剂按照10mg/Kg、体积为10ml/Kg的剂量给鼠口服。使用的赋形剂为含20%二甲亚砜(DMSO)的2.5%事先煮沸的玉米淀粉。服用试验化合物30分钟后,用喂食针给鼠喂入30ml/Kg的水至胃中。在实验过程中不提供水或食物。服用试验化合物后收集尿4小时。4小时后测量尿的体积。使用Fiske One-Ten渗透压力计(Fiske Associates,Norwood,MA,02062)或先进的3C2型CRYOMATIC渗透压力计(Advanced Instruments,Norwood,MA)测量尿的渗透性。使用Beckman SYNCHRON EL-ISE电解质系统分析器中的离子特异性电极测量Na+、K+和Cl-。尿的渗透性应当成比例地增加。在筛选实验中,每种化合物分别使用两只鼠。如果两只鼠的尿体积之差大于50%,则要使用第三只鼠。
试验化合物对正常神志清醒的患有中枢性尿崩症的纯合brattleboro鼠作为后叶加压素V2激动剂的效果
给250-350克体重的雄性或雌性的纯合Brattleboro鼠(HarlanSprague Dawley,Inc.,Indianapolis,IN)提供标准啮齿动物的食物(Purina Rodent lab.Chow 5001)和任意的水。在实验当天,将鼠单独放置在装有分离粪便和尿的装置及收集尿的容器的代谢测定笼中。将试验化合物或参照剂按照1-10mg/Kg、体积为10ml/Kg的剂量给鼠口服。使用的赋形剂为含20%二甲亚砜(DMSO)的2.5%事先煮沸的玉米淀粉。在实验过程中给鼠提供任意量的水。服用试验化合物后收集尿6小时。6小时后测量尿的体积。使用Fiske One-Ten渗透压力计(Fiske Associates,Norwood,MA,02062)或先进的3C2型CRYOMATIC渗透压力计(Advanced Instruments,Norwood,MA)测量尿的渗透性。使用Beckman SYNCHRON EL-ISE电解质系统分析器中的离子特异性电极测量Na+、K+和Cl-。这种动物模型主要用于估价活性化合物作用的效力和持续时间。该实验的结果见表I。
表1 实施例# 尿体积 渗透性 鼠型
(% 减少)a (% 增加)b2 80%(1mg/kg) 306%(1mg/kg) CD3 58% 240% CD4 57% 225% CD5 56% 231% CD6 58% 270% CD7 13% 137% CD9A 70% 325% CD9B 21% 168% CD11 70% 285% CD12 69% 330% CD13 50% 229% CD14 86% 406% CD15 47% 38% CD16 88% 400% CD18 52% 214% CD20 25%(1mg/kg) 152%(lmg/kg) CD21 49% 181% CD22 80% 322% CD24 47% 159% CD25 87% 979% CD26 54% 279% CD27 76% 183% CD28 75% 37% CD29 66% 305% CD30 81% 334% BB31 72% 298% CD32 77% 373% CD33 68% 362% CD34 76% 407% BB35 63% 308% CD36 66% 164% BB37 71% 370% CD38 66% 256% BB39 69% 253% CD40 46% 183% CD41 69% 240% CD49 74% 221% BB50 53% 223% CD 实施例# 尿体积 渗透性 鼠型
(% 减少)a (% 增加)b51 72% CD52 66% 261% CD55 80% 164% CD57 77% 288% CD58 49% 324% CD59 80% 607% CD60 54% 165% CD61 59% 245% CD62 22% 150% CD63 27% 214% CD64 79% 349% CD71 84% 264% CD77 13% 90% CD78 21% 115% CD79 38% 123% CD81 82% 490% CD83 85% 442% CD84 56% 291% CD85 76% 436% CD86 5% 86% CD87 71% 214% CD88 68% 226% CD90 61% 413% CD91 22% 69% CD92 69% 454% CD95 68% 300% CD97 3% 106% CD99 43% 205% CD100 24% 248% CD101 76% 376% CD107 31% 125% CD108 30% 145% CD109 21% 95% CD115 66% 229% CD116 66% 256% CD117 68% 311% CD120A 66% 269% CD120B 67% 272% CD121 22% 155% CD123 88% 663% CD
a除非另有说明均按10mg/Kg给药后,与对照试样相比,尿体积减少百分比。
b除非另有说明,均按10mg/Kg给药后与对照试样相比渗透性。
c使用的鼠型:Sprague-Dawley(CD)或Brattleboro(BB)。
下面给出的实施例用于举例说明,但不限制本发明的范围。
实施例1
(4-氯-2-三氟甲基-苯基)-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮
将草酰氯(2.0g)加入到含有4-氟-2-三氟甲基苯甲酸(2.0g)的二氯甲烷(25mL)悬浮液中。加入2滴二甲基甲酰胺,室温下搅拌混合物18小时。将得到的溶液蒸发至干制得粗酰氯。将粗酰氯再溶解于二氯甲烷中并过滤。蒸发该物质得到液体,然后再溶解于己烷中,过滤并蒸发得到酰氯,为浅黄色粘稠液体,无需进一步纯化直接使用。
将含有酰氯(2.26g)的二氯甲烷(25mL)分批加入到含有10,11-二氢-5H-吡咯并[2,1-c][1,4]苯并二氮杂(1.66g)、二氯甲烷(10mL)和二异丙基乙胺(1.30g)的混合物中,冰浴冷却。室温下保持18小时后,反应混合物用水和饱和碳酸氢钠溶液洗涤。二氯甲烷溶液用无水硫酸钠干燥并通过水合硅酸镁钠短柱过滤,再用几倍体积的二氯甲烷洗脱。在电热板上浓缩合并的有机相并逐渐加入己烷直至开始结晶。冷却后,过滤收集晶体得到2.57g标题化合物,熔点为154-155℃。
实施例2
[4-(3-甲基-吡唑-1-基)-2-三氟甲基-苯基]-(5H,11H-吡咯并[2,1-c]-[1,4]苯并二氮杂-10-基)-甲酮
将油(0.15g)中60%氢化钠用己烷洗涤,并加入二甲基甲酰胺(25mL),随后加入3-甲基吡唑(0.25g)。氢气停止放出后,加入(4-氟-2-三氟甲基-苯基)(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮(1.0g)。在沙浴中110℃下加热反应混合物15小时。将反应混合物倒入冰中并加入饱和盐水溶液。过滤收集沉淀。将粗反应产物溶解于二氯甲烷中并通过水合硅酸镁钠短柱过滤,再用几倍体积的二氯甲烷洗脱。在电热板上浓缩合并的有机相并逐渐加入己烷。冷却后,过滤收集晶体得到0.77g粗产物。再通过水合硅酸镁钠短柱过滤来进一步纯化,然后加入己烷,制得标题化合物,为结晶固体(0.66g),熔点为194-195℃。
实施例3
[4-(4-甲基-吡唑-1-基)-2-三氟甲基-苯基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮
按照实施例2中的方法,使用(4-氟-2-三氟甲基-苯基)(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮(0.8g)、含有60%氢化钠的油(0.15g)、4-甲基吡唑(0.20g)和二甲基甲酰胺(25mL),得到产物(0.47g),为无色无定形固体,MS,m/z:437.3(M+H)+,873.2(2M+H)+。
实施例4
(4-吡唑-1-基-2-三氟甲基-苯基)-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮
按照实施例2中的方法,使用(4-氟-2-三氟甲基-苯基)(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮(1.0g)、含有60%氢化钠的油(0.20g)、吡唑(0.25g)和二甲基甲酰胺(35mL)。得到产物(0.62g),为无色无定形固体,MS,m/z:423.2(M+H)+,445.2(M+Na)+,845.3(2M+H)+。
实施例5
[4-(3-环丙基-吡唑-1-基)-2-三氟甲基-苯基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮
按照实施例2中的方法,使用(4-氟-2-三氟甲基-苯基)(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮(1.42g)、含有60%氢化钠的油(0.20g)、3-环丙基吡唑(0.43g)和二甲基甲酰胺(50mL),得到产物(1.22g),为结晶固体,熔点为163-164℃。
实施例6
[4-(4-甲基-咪唑-1-基)-2-三氟甲基-苯基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮
按照实施例2中的方法,使用(4-氟-2-三氟甲基-苯基)(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮(1.0g)、含有60%氢化钠的油(0.20g)、4-甲基咪唑(0.25g)和二甲基甲酰胺(25mL),得到标题化合物(0.66g),为无定形固体,MS,m/z:437.2(M+H)+,873.2(2M+H)+。
实施例7
(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-(4-[1,2,4]三唑-1-基-2-三氟甲基苯基)-甲酮
按照实施例2中的方法,使用(4-氟-2-三氟甲基-苯基)(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮(1.0g)、含有60%氢化钠的油(0.20g)、1,2,4-三唑(0.20g)和二甲基甲酰胺(25mL),得到标题化合物(0.36g),为无色无定形固体,MS,m/z:424.2(M+H)+,847.3(2M+H)+。
实施例8
(2-氯-4-氟苯基)-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮
将草酰氯(2.60g)加入到含有2-氯-4-氟苯甲酸(3.44g)的二氯甲烷(50mL)的悬浮液中。加入2滴二甲基甲酰胺,室温下搅拌混合物18小时。将得到的溶液蒸发制得粗2-氯-4-氟苯甲酰氯,为粘稠油状物(3.72g)。
将含有粗2-氯-4-氟苯甲酰氯(3.68g)的二氯甲烷(25mL)分批加入到含有10,11-二氢-5H-吡咯并[2,1-c][1,4]苯并二氮杂(2.76g)、二异丙基乙胺(2.47g)和二氯甲烷(50mL)的搅拌且冰冷却的溶液中。室温下保持18小时后,反应混合物用水和饱和碳酸氢钠溶液洗涤。二氯甲烷溶液用无水硫酸钠干燥并通过水合硅酸镁钠短柱过滤,再用几倍体积二氯甲烷洗脱。在电热板上浓缩合并的有机相,并逐渐加入己烷直至开始结晶。冷却后,过滤收集晶体得到标题化合物(3.85g),熔点为110-112℃。
实施例9
[2-氯-4-(3-甲基-吡唑-1-基)-苯基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮(异构体A)
和
[2-氯-4-(5-甲基-吡唑-1-基)-苯基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮(异构体B)
方法1:向含有60%氢化钠的油(0.3g,用己烷去脂)的二甲基甲酰胺(25mL)中加入3-甲基吡唑(0.55g)。当氢气停止放出后,加入(2-氟-4-氟苯基)(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮(1.70g)。在沙浴中(内部温度为125℃)加热反应混合物18小时。然后将反应混合物倒入冰中,再用饱和盐水溶液稀释。过滤回收沉淀出的固体。将粗产物溶解于二氯甲烷中,无水硫酸钠干燥,然后通过水合硅酸镁钠短柱过滤,再用几倍体积二氯甲烷洗脱。在电热板上回流合并的洗脱液并逐渐加入己烷,直至观察到溶液浑浊。冷却后,得到无定形固体。将该物质通过第二根水合硅酸镁钠柱,真空下蒸发溶剂,得到区域异构体9A和9B约9∶1的混合物,为无定形玻璃(1.11g),MS,m/z:403.2(M+H)+。
方法2:在0℃下和氮气中,向预冷却和搅拌的含有己烷洗涤的60%氢化钠(3.00g)的无水二甲基甲酰胺(250mL)的悬浮液中滴加3-甲基吡唑(5.50g)。将混合物加热至室温。停止放出气体后,加入(2-氟-4-氟苯基)(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲基酮固体(17.0g),混合物加热至130℃1小时。将反应混合物倒入冰水中,过滤收集沉淀并风干。将沉淀溶解于二氯甲烷中,无水硫酸钠干燥,通过硅胶短柱过滤,用乙酸乙酯洗脱。真空下蒸发合并的滤液得到残余的泡沫状物(18.5g)。通过低压硅胶柱色谱法纯化和分离区域异构体,乙酸乙酯-己烷(10∶90-25∶75)混合物梯度洗脱,制得两种纯化的区域异构体:
异构体A,[2-氯-4-(3-甲基-吡唑-1-基)-苯基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮(13.5g),为无色无定形固体;MS(EI),m/z:402(M)+。将试样(0.5g)在乙醚中结晶,随后在乙醇中重结晶得到区域异构体A(0.275g),为无色结晶固体,熔点为141-143℃;
异构体B,[2-氯-4-(5-甲基-吡唑-1-基)-苯基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮(1.93g),为无色无定形固体。将试样在乙醚中结晶,随后在甲醇中重结晶得到区域异构体B,为无色针状(1.4g),熔点为160-163℃;MS(EI),m/z:402(M)+,MS(+FAB),m/z:403(M+H)+。
实施例10
[2-氯-4-(3-甲基-吡唑-1-基)-苯基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮
步骤a)2-氯-4-(3-甲基吡唑-1-基)苄腈:向冷却(0℃)的含有氢化钠(油中60%;2.0g)的二甲基甲酰胺(50mL)的悬浮液中,分批加入3-甲基吡唑(3.39g)。氢气停止放出后,加入2-氯-4-氟苄腈(5.17g)并使混合物在室温下搅拌18小时。将混合物倒入冰中并用盐水洗涤,过滤收集得到的沉淀。将粗产物溶解于二氯甲烷中,通过无水硅酸镁钠柱过滤,并加入己烷使之结晶。在乙醇中重结晶得到4.42g产物,熔点为148-150℃。
步骤b)2-氯-4-(3-甲基吡唑-1-基)苯甲酰胺:在冰浴中冷却含有步骤a)的2-氯-4(3-甲基吡唑-1-基)苄腈(4.35g)的含碳酸钾(0.40g)的二甲亚砜(20mL)悬浮液。加入过氧化氢(30%,2.4mL)并在1小时内将混合物加热至室温。过滤回收得到的沉淀并在乙醇中重结晶制得2.44g细针状产物,熔点为159-160℃;MS,m/z:235.9(M+H)+。
步骤c)2-氯-4-(3-甲基吡唑-1-基)苯甲酸:在冰浴中冷却含有步骤b)的2-氯-4-(3-甲基吡唑-1-基)苯甲酰胺(1.09g)的75%硫酸水溶液(25mL)并加入硝酸钠(1.73g)。在1小时内将混合物加热至室温并倒入冰中。过滤收集沉淀并直接用于下一步。
步骤d)[2-氯-4-(3-甲基-吡唑-1-基)-苯基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮:将含有步骤c)的2-氯-4-(3-甲基吡唑-1-基)苯甲酸(0.69g)、二氯甲烷(25mL)、草酰氯(1.0g)和1滴二甲基甲酰胺的混合物在室温下搅拌18小时。浓缩混合物并溶解于二氯甲烷(25mL)中,加入到含有10,11-二氢-5H-吡咯并[2,1-c][1,4]苯并二氮杂(0.51g)的含二异丙基乙胺(0.76g)的二氯甲烷(25mL)混合物中。混合物在室温下搅拌18小时并用饱和碳酸氢钠水溶液洗涤。有机层用无水硫酸钠干燥并通过无水硅酸镁钠短柱过滤。浓缩溶液并将得到的物质在乙醚中结晶,制得0.67g产物,熔点为137-138℃:MS,m/z:403.2(M+H)+,805.8(2M+H)+。
实施例11
[2-氯-4-(4-甲基-吡唑-1-基)-苯基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮
按照实施例9的方法1,使用(2-氯-4-氟-苯基)-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮(1.0g)、60%氢化钠的油(0.3g,用己烷去脂)、4-甲基吡唑(0.48g)和二甲基甲酰胺(25mL),制得标题化合物(0.74g),为无定形固体,MS,m/z:403.2(M+H)+,425.2(M+Na)+,805.3(2M+H)+。
实施例12
[2-氯-4-(4-甲基-咪唑-1-基)-苯基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10基)-甲酮
按照实施例9的方法1,使用(2-氯-4-氟-苯基)-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮(1.0g)、60%氢化钠的油(0.3g,用己烷去脂)、4-甲基咪唑(0.48g)和二甲基甲酰胺(25mL),制得标题化合物(0.38g),为无定形固体,MS,m/z:403.3(M+H)+。
实施例13
[2-氯-4-(3-三氟甲基-吡唑-1-基)-苯基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮
按照实施例9的方法1,使用(2-氯-4-氟-苯基)-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮(0.8g)、60%氢化钠的油(0.25g,用己烷去脂)、3-三氟甲基吡唑(0.61g)和二甲基甲酰胺(25mL),制得标题化合物(0.74g),为无定形固体,MS,m/z:457.2(M+H)+。
实施例14
[2-氯-4-(1,2,4-三唑-1-基)-苯基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10基)-甲酮
按照实施例9的方法1,使用(2-氯-4-氟-苯基)-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮(1.7g)、60%氢化钠的油(0.5g,用己烷去脂)、1,2,4-三唑(0.70g)和二甲基甲酰胺(50mL),制得标题化合物(0.51g),为无定形固体,MS,m/z:309.3(M+H)+,779.3(2M+H)+。
实施例15
(2-氯-4-吡咯-1-基-苯基)-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮
按照实施例9的方法1,使用(2-氯-4-氟-苯基)-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮(1.7g)、60%氢化钠的油(0.3g,用己烷去脂)、吡咯(0.42g)和二甲基甲酰胺(25mL),制得标题化合物(0.60g),为无定形固体,MS,m/z:388.2(M+H)+。
实施例16
(2-氯-4-吡唑-1-基-苯基)-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮
按照实施例9的方法1,使用(2-氯-4-氟-苯基)-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮(1.0g)、60%氢化钠的油(0.2g,用己烷去脂)、吡唑(0.20g)和二甲基甲酰胺(25mL),制得标题化合物(0.60g),为无定形固体,MS,m/z:389.2(M+H)+,777.1(2M+H)+。
实施例17
[2-氯-4-(1H-咪唑-1-基)-苯基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮
按照实施例9的方法1,使用(2-氯-4-氟-苯基)-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮(2.0g)、60%氢化钠的油(0.50g,用己烷去脂)、咪唑(0.50g)和二甲基甲酰胺(25mL),制得标题化合物(0.57g),为黄褐色无定形固体,MS,m/z:389(M+H)+。
实施例18
[2-氯-4-(3-甲基吡唑-1-基)-苯基]-(3-甲基-5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮
步骤a)1-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-2,2,2-三氟乙基酮:向含有10,11-二氢-5H-吡咯并[2,1-c][1,4]苯并二氮杂(5.62g)和二异丙基乙胺(4.0g)的二氯甲烷(75mL)的冰冷却的溶液中,滴加含有三氟乙酐(7.0g)的二氯甲烷。混合物在室温下搅拌18小时,用水和饱和碳酸氢钠水溶液洗涤。有机相用无水硫酸钠干燥并通过无水硅酸镁钠短柱过滤。在电热板上浓缩合并的有机相,并逐渐加入己烷直至开始结晶。冷却后,过滤收集晶体得到细针状的产物7.70g,熔点为134-135℃,MS,m/z:281(M+H)+。
步骤b)1-(3-二甲基氨基甲基-5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-2,2,2-三氟乙酮:将含有步骤a)的1-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-2,2,2-三氟乙酮(2.80g)、双-二甲基氨基甲烷(2.04g)、低聚甲醛(2.70g)和乙酸(1.20g)与四氢呋喃(50mL)和甲醇(50mL)的混合物在室温下搅拌18小时。真空下浓缩混合物,加入水,用二氯甲烷萃取含水的混合物。合并的萃取液用无水硫酸钠干燥并通过无水硅酸镁钠短柱过滤。真空下浓缩该溶液,并将残余物在己烷中结晶,得到2.05g产物,为无色固体,熔点为109-110℃,MS,m/z:338.3(M+H)+。
步骤c)碘化三甲基-(10-三氟乙酰基-10,11-二氢-5H-吡咯并[2,1-c][1,4]苯并二氮杂-3-基-甲基)铵:将含有步骤b)的1-(3-二甲基氨基甲基5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-2,2,2-三氟乙酮(1.83g)和碘甲烷(1.0g)的二氯甲烷(20mL)的混合物在室温下搅拌18小时。加入乙醚,过滤收集得到的沉淀,制得2.54g产物,为无色固体,熔点为140-155℃(分解)。
步骤d)10,11-二氢-3-甲基-5H-吡咯并[2,1-c][1,4]苯并二氮杂:将硼氢化钠(2.6g)分两批加入到含有步骤c)的碘化三甲基-(10-三氟乙酰基-10,11-二氢-5H-吡咯并[2,1-c][1,4]苯并二氮杂-3-基-甲基)铵(2.60g)的乙醇回流混合物中。4小时后,真空下浓缩混合物。将水加入到残余物中并用二氯甲烷萃取该混合物。合并的萃取液用无水硫酸钠干燥,并通过无水硅酸镁钠短柱过滤。真空下浓缩该溶液并将残余物在己烷中结晶,得到1.14g产物,熔点为150-151℃,MS,m/z:199.1(M+H)+。
步骤e)[2-氯-4-(3-甲基吡唑-1-基)-苯基]-(3-甲基-5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮:将含有实施例10步骤c)的2-氯-4-(3-甲基吡唑-1-基)苯甲酸(0.18g)、草酰氯(0.18g)和一滴二甲基甲酰胺的二氯甲烷(10mL)的混合物在室温下搅拌18小时。真空下浓缩混合物,并将残余物再溶解于二氯甲烷中,再浓缩得到2-氯-4-(3-甲基-吡唑-1-基)-苯甲酰氯。将含有酰氯的二氯甲烷(25mL)浆状物滴加到含有10,11-二氢-3-甲基-5H-吡咯并[2,1-c][1,4]苯并二氮杂(0.12g)和二异丙基乙胺(0.10g)的二氯甲烷(25mL)溶液中。混合物在室温下搅拌18小时并用饱和碳酸氢钠水溶液洗涤。有机相用无水硫酸钠干燥并通过无水硅酸镁钠短柱过滤。真空下浓缩该溶液,并用乙醚研碎制得0.115g产物,为无色晶体,熔点为178-180℃;MS,m/z:417.3(M+H)+,833.3(2M+H)+。
实施例19
(2-氯-4-三氟甲基-嘧啶-5-基)-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮
将2-氯-4-(三氟甲基)嘧啶-5-碳酰氯(2.57g)逐渐加入到含有10,11-二氢-5H-吡咯并[2,1-c][1,4]苯并二氮杂(1.84g)和二异丙基乙胺(1.37g)的二氯甲烷(50mL)的冰冷却溶液中。室温下搅拌18小时后,反应混合物用水和饱和碳酸氢钠溶液洗涤。二氯甲烷溶液用无水硫酸钠干燥并通过水合硅酸镁钠短柱过滤,再用几倍体积二氯甲烷洗脱。在电热板上浓缩合并的有机相,并逐渐加入己烷直至开始结晶。冷却后,过滤收集晶体得到标题化合物(3.22g),熔点为221-223℃。
实施例20
[2-(3-甲基-吡唑-1-基)-4-三氟甲基-嘧啶-5-基)-(5H,11H-吡咯并[2.1-c][1.4]苯并二氮杂-10-基)-甲酮
向含有60%氢化钠的油(0.15g,用己烷去脂)的二甲基甲酰胺(25mL)中加入3-甲基吡唑(0.25g)。当氢气停止放出后,加入(2-氯-4-三氟甲基-嘧啶-5-基)-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮(0.98g)。在沙浴中(内部温度为110℃)加热反应混合物18小时。然后将反应混合物倒入冰中,再用饱和盐水溶液稀释。过滤沉淀,再溶解于二氯甲烷中,无水硫酸钠干燥。通过水合硅酸镁钠短柱过滤进行纯化,再用几倍体积二氯甲烷洗脱。在电热板上浓缩合并的洗脱液并逐渐加入己烷直至开始结晶。冷却后,过滤收集固体,制得标题化合物(0.54g),为无色晶体,熔点为202-204℃。
实施例21
[2-(4-甲基-吡唑-1-基)-4-三氟甲基-嘧啶-5-基)-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮
按照实施例9的方法1,使用(2-氯-4-三氟甲基-嘧啶-5-基)-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮(0.98g)、60%氢化钠的油(0.15g)、4-甲基吡唑(0.42g)和二甲基甲酰胺(25mL),制得标题化合物(0.73g),为结晶固体,熔点为214-217℃。
实施例22
1-[4-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-羰基)-苯基]-乙酮
在氩气中将4-乙酰基苯甲酸(5.0g)和亚硫酰氯(10mL)在蒸汽浴上加热0.75小时,减压下除去挥发物。加入甲苯并再次除去挥发物得到橙红色油状的粗酰氯。该化合物易于固化,可以直接用于进一步转化。
将含有酰氯(4.56g)的二氯甲烷(25mL)分批加入到含有10,11-二氢-5H-吡咯并[2,1-c][1,4]苯并二氮杂(3.68g)和二异丙基乙胺(3.25g)的二氯甲烷(100mL)的冰冷却的溶液中。室温下搅拌18小时后,反应混合物用水和饱和碳酸氢钠溶液洗涤。二氯甲烷溶液用无水硫酸钠干燥并通过水合硅酸镁钠短柱过滤,再用几倍体积二氯甲烷洗脱。在电热板上浓缩合并的有机相,并逐渐加入己烷直至开始结晶。冷却后,过滤收集晶体,得到标题化合物(1.75g),熔点为135-137℃。
实施例23
3-二甲基氨基-1-[4-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-羰基)-苯基]-2-丙烯-1-酮
将含有1-[4-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-羰基)-苯基]-乙酮(1.40g)、叔丁氧基-双-二甲基氨基甲烷(5.0mL)和二氯甲烷(10mL)的反应混合物搅拌18小时。过滤橙红色的沉淀制得标题化合物(1.22g),熔点为203-205℃。通过浓缩从反应混合物中分离出另外的产物(0.18g)。
实施例24
[4-(1H-吡唑-3-基)-苯基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮
将含有3-二甲基氨基-1-[4-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-羰基)-苯基]-2-丙烯-1-酮(1.0g)、无水肼(0.20g)和冰乙酸(20mL)的反应混合物回流7小时,并蒸发至干。将粗残余物溶解于二氯甲烷中,用饱和碳酸氢钠溶液洗涤,无水硫酸钠干燥。将该溶液通过水合硅酸镁钠短柱过滤,用几倍体积二氯甲烷洗脱。在电热板上浓缩合并的有机相并逐渐加入己烷直至开始结晶。冷却后,过滤收集晶体。重复上述柱过滤法制得标题化合物(0.65g),熔点为219-221℃。
实施例25
[4-(1-甲基-1H-吡唑-3-基)-苯基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮
向含有60%氢化钠的油(0.35g,用己烷去脂)和二甲基甲酰胺(20mL)的混合物中加入[4-(1H-吡唑-3-基)-苯基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮(0.98g),几分钟后再加入碘甲烷(0.50g)。反应混合物在室温下搅拌18小时,然后倒入水中并用二氯甲烷萃取。干燥后,将有机层通过水合硅酸镁钠短柱过滤,用几倍体积二氯甲烷洗脱。在电热板上浓缩合并的有机相并逐渐加入己烷直至开始结晶。冷却后,过滤收集晶体制得标题化合物(0.70g),熔点为194-195℃。
实施例26
[4-(1-乙基-1H-吡唑-3基)-苯基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮
按照实施例25的方法,使用[4-(1H-吡唑-3-基)-苯基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮(1.0g)、60%氢化钠的油(0.27g)、二甲基甲酰胺(25mL)和碘乙烷(0.87g),制得标题化合物(0.69g),为结晶固体,熔点为180-183℃。
实施例27
[4-(1-丙基-1H-吡唑-3-基)-苯基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮
按照实施例25的方法,使用[4-(1H-吡唑-3-基)-苯基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲基酮(0.98g)、60%氢化钠的油(0.30g)、二甲基甲酰胺(25mL)和1-碘丙烷(0.60g),制得标题化合物(0.32g),为结晶固体,熔点为159-161℃。
实施例28
[4-(1-丁基-1H-吡唑-3-基)-苯基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮
按照实施例25的方法,使用[4-(1H-吡唑-3-基)-苯基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲基酮(0.98g)、60%氢化钠的油(0.30g)、二甲基甲酰胺(25mL)和1-碘丁烷(0.60g),制得标题化合物(0.32g),为结晶固体,熔点为122-123℃。
实施例29
[4-(1-甲氧基甲基-1H-吡唑-3-基)-苯基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮
按照实施例25的方法,使用[4-(1H-吡唑-3-基)-苯基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮(1.0g)、60%氢化钠的油(0.15g)、二甲基甲酰胺(25mL)和碘代甲基甲基醚(0.50g),制得标题化合物(0.26g),为无定形固体,MS,m/z:399.2(M+H)+,797.2(2M+H)+。
实施例30
1-{3-[4-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-羰基)-苯基]-吡唑-1-基}-乙酮
向含有[4-(1H-吡唑-3-基)-苯基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮(0.50g)的无水吡啶(10mL)溶液中加入乙酐(0.20g)。室温下搅拌18小时后,将反应混合物倒入水中并通过过滤收集沉淀。将粗产物溶解于二氯甲烷中,用无水硫酸钠干燥。将该溶液通过水合硅酸镁钠短柱过滤,用几倍体积二氯甲烷洗脱。在电热板上浓缩合并的洗脱液,并逐渐加入己烷直至开始结晶。冷却后,过滤收集晶体,制得标题化合物(0.46g),熔点为192-194℃。
实施例31
1-{3-[4-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-羰基)-苯基]-吡唑-1-基}-丙-1-酮
按照实施例30的方法,使用[4-(1H-吡唑-3-基)-苯基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮(0.16g)的无水吡啶(10mL)和丙酸酐(0.10g),制得标题化合物(0.17g),为结晶固体,熔点为150-152℃。
实施例32
[4-(1-环丙烷羰基-1H-吡唑-3-基)-苯基]-(5H,11H-吡咯并[2.1-c][1,4]苯并二氮杂-10-基)-甲酮
向含有[4-(1H吡唑-3-基)-苯基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮(1.0g)的无水吡啶(10mL)溶液中加入环丙烷碳酰氯(0.44g)。室温下搅拌18小时后,将反应混合物倒入水中,并过滤收集沉淀。将粗产物溶解于二氯甲烷中,以无水硫酸钠干燥。将该溶液通过水合硅酸镁钠短柱过滤,用几倍体积二氯甲烷洗脱。在电热板上浓缩合并的洗脱液,并逐渐加入己烷直至开始结晶。冷却后,过滤收集晶体,制得标题化合物(0.88g),为结晶固体,熔点为197-199℃。
实施例33
1-{3-[4-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-羰基)-苯基]-吡唑-1-基}-丁-1-酮
按照实施例32的方法,使用[4-(1H-吡唑-3-基)-苯基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮(0.71g)的无水吡啶(10mL)和丁酰氯(0.32g)液,制得标题化合物(0.54g),为固体,熔点为105-110℃;MS,m/z:424(M)+。
实施例34
(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-羰基)-{4-[1-(噻吩-2-羰基)-1H-吡唑-3-基]苯基}-甲酮
按照实施例32的方法,使用[4-(1H-吡唑-3-基)-苯基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲基酮(0.5g)的无水吡啶(10mL)和噻吩-2-碳酰氯(0.25g)液,制得标题化合物(0.41g),为结晶固体,熔点为195-197℃;MS,m/z:464(M)+。
实施例35
{4-[1-(5-氟-2-甲基-苯甲酰基)-1H-吡唑-3-基]-苯基}-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮
按照实施例32的方法,使用[4-(1H-吡唑-3-基)-苯基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮(0.35g)的无水吡啶(10mL)和2-甲基-5-氟苯甲酰氯(0.22g)液,制得标题化合物(0.11g),为无定形浅黄色固体,MS,m/z:490(M)+。
实施例36
{4-[1-(2-甲基-苯甲酰基)-1H-吡唑-3-基]-苯基}-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮
按照实施例32的方法,使用[4-(1H-吡唑-3-基)-苯基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮(0.71g)的无水吡啶(20mL)和邻-甲苯酰氯(0.39g)液,制得标题化合物(0.59g),为结晶固体,熔点为170-173℃;MS,m/z:472(M)+。
实施例37
{4-[1-(2-氯-4-氟-苯甲酰基)-1H-吡唑-3-基]-苯基}-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮
将2-氯-4-氟苯甲酰氯(0.82g)分批加入含有[4-(1H-吡唑-3-基)-苯基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮(1.0g)和二异丙基胺(0.55g)的二氯甲烷(25mL)的用冰浴冷却的溶液中。反应混合物在室温下搅拌过夜。反应混合物用水和饱和碳酸氢钠溶液洗涤,用无水硫酸钠干燥。将二氯甲烷溶液通过水合硅酸镁钠短柱过滤,再用几倍体积二氯甲烷洗脱。蒸发洗脱液至干得到1.06g固体产物,熔点为150-157℃;MS,m/z:510(M)+。
实施例38
{4-[1-(2,4-二氯-苯甲酰基)-1H-吡唑-3-基]-苯基}-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮
按照实施例32的方法,使用[4-(1H-吡唑-3-基)-苯基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮(0.71g)的无水吡啶(20mL)和2,4-二氯苯甲酰氯(0.52g)液,制得标题化合物(0.66g),为结晶固体,熔点为180-182℃;MS,m/z:528(M)+。
实施例39
2-(2,4-二氯-苯基)-1-{3-[4-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-羰基)-苯基]-吡唑-1-基}-乙酮
按照实施例32的方法,使用[4-(1H-吡唑-3-基)-苯基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲基酮(0.71g)的无水吡啶(25mL)和2,4-二氯苯乙酰氯(0.56g)液,制得标题化合物(0.20g),为结晶固体,熔点为130-140℃,再固化,熔点为180-182℃。
实施例40
{4-[1-(二苯基-2-羰基)-1H-吡唑-3-基]-苯基}-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮
按照实施例32的方法,使用[4-(1H-吡唑-3-基)-苯基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮(0.71g)的无水吡啶(20mL)和2-二苯基碳酰氯(0.65g)液,制得标题化合物(0.49g),为无定形固体,MS,m/z:534(M)+。
实施例41
{4-[1-(4’-三氟甲基-二苯基-2-羰基)-1H-吡唑-3-基]-苯基}-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮
按照实施例32的方法,使用[4-(1H-吡唑-3-基)-苯基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮(0.71g)的无水吡啶(20mL)和4’-三氟甲基-2-二苯基碳酰氯(0.71g),制得标题化合物(0.59g),为无定形固体,MS,m/z:602(M)+。
实施例42
3-二甲基氨基-1-[4-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-羰基)-苯基]-2-丁烯-1-酮
在惰性气氛中将含有1-[4-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-羰基)-苯基]-乙酮(2.0g)和二甲基乙酰胺二甲基乙缩醛(15mL)的混合物回流15小时,减压下除去挥发物。将粗产固体溶解于二氯甲烷中,通过水合硅酸镁钠短柱过滤,然后用几倍体积二氯甲烷洗脱。浓缩合并的洗脱液,并逐渐加入己烷直至开始结晶。过滤冷却的溶液回收标题化合物(1.03g),为结晶固体,熔点为183-185℃。
实施例43
[4-(5-甲基-1H-吡唑-3-基)-苯基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮
将无水肼加入到含有3-二甲基氨基-1-[4-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-羰基)-苯基]-2-丁烯-1-酮(0.50g)的冰醋酸(25mL)溶液中。反应混合物回流18小时然后真空浓缩。用二氯甲烷萃取出固体并用饱和碳酸氢钠溶液洗涤。二氯甲烷溶液用无水硫酸钠干燥,并通过水合硅酸镁钠短柱过滤,再用几倍体积二氯甲烷洗脱。在蒸汽浴上浓缩合并的有机相,并逐渐加入己烷得到浑浊溶液。冷却后,过滤回收无定形固体,制得产物(0.33g),MS,m/z:368(M)+。
实施例44
4-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-羰基)-苄腈
将4-氰基苯甲酸(5.0g)和亚硫酰氯(5.0mL)在蒸汽浴上加热1小时,减压下除去所有挥发物。加入己烷,过滤回收粗结晶的酰氯(5.30g),无需进一步分离直接使用。
向含有10,11-二氢-5H-吡咯并[2,1-c][1,4]苯并二氮杂(3.68g)、二氯甲烷(100mL)和二异丙基乙胺(2.80g)的反应混合物中加入4-氰基苯甲酰氯(2.97g)。反应混合物在室温下保持18小时后,用水和饱和碳酸氢钠水溶液洗涤。二氯甲烷溶液用无水硫酸钠干燥并通过无水硅酸镁钠短柱过滤,再用几倍体积二氯甲烷洗脱。在电热板上浓缩合并的有机相,并逐渐加入己烷直至开始结晶。冷却后,过滤收集晶体得到标题化合物(5.05g),熔点为184-186℃。
实施例45
4-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-羰基)-苯甲酰胺
将实施例44中的4-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-羰基)-苄腈(0.5g)加入到浓硫酸(5mL)中,混合物在室温下搅拌18小时得到嫩黄色的溶液。将溶液倒入水中并加入浓氨水使之呈碱性。过滤得到的固体,溶解于二氯甲烷中,并通过无水硅酸镁钠短柱过滤,再用几倍体积二氯甲烷洗脱。在电热板上浓缩合并的有机相,并逐渐加入己烷直至开始结晶。冷却后,过滤收集晶体得到标题化合物(5.05g),熔点为226-228℃。
实施例46
N-(二甲基氨基亚甲基)-4-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-羰基)-苯甲酰胺
将含有实施例45的4-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-羰基)-苯甲酰胺(1.25g)和二甲基甲酰胺二甲基缩醛(20mL)的混合物回流4小时,真空下除去挥发物得到固体。将固体溶解于二氯甲烷中,通过水合硅酸镁钠短柱过滤,再用几倍体积二氯甲烷洗脱。在蒸汽浴上浓缩合并的有机相,并逐渐加入己烷直至开始结晶。冷却后,过滤收集晶体,制得标题化合物(1.40g),熔点为232-234℃。
实施例47
N-(1-二甲基氨基亚乙基)-4-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-羰基)-苯甲酰胺
将含有实施例45的4-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-羰基)-苯甲酰胺(1.24g)和二甲基乙酰胺二甲基缩醛(5.0mL)的混合物在蒸汽浴上加热4小时。冷却18小时后,沉淀出结晶固体,通过过滤回收。将该固体用己烷洗涤制得产物(1.54g)。熔点为210-212℃;MS,m/z:400(M)+。
实施例48
(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-[4-(2H-[1,2,4]三唑-3-基)-苯基]-甲酮
将含有实施例46中的N-(二甲基氨基亚甲基)-4-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-羰基)-苯甲酰胺(1.0g)、冰醋酸(15mL)和无水肼(0.16g)的混合物回流15小时,真空除去挥发物。加入饱和碳酸氢钠溶液并过滤回收得到的固体。将固体回流4小时并在真空中除去挥发物得到固体。将固体溶解于二氯甲烷中,通过水合硅酸镁钠短柱过滤,再用几倍体积二氯甲烷洗脱。在蒸汽浴上浓缩合并的有机相,并逐渐加入己烷直至开始结晶。冷却后,过滤收集晶体制得标题化合物(0.39g),熔点为225-227℃;MS,m/z:355(M)+。
实施例49
[4-(2-甲基-2H-[1,2,4]三唑-3-基)-苯基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮
按照实施例48的同样方法,使用实施例46中的N-(二甲基氨基亚甲基)-4-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-羰基)-苯甲酰胺(1.56g)的冰醋酸(75mL)和甲基肼(0.32g)液,制得标题化合物(0.10g),为固体,熔点为155-158℃;MS,m/z:369(M)+。
实施例50
[4-(5-甲基-2H-[1,2,4]三唑-3-基)-苯基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮
按照实施例48的同样方法,使用实施例47中的N-(1-二甲基氨基亚乙基)-4-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-羰基)-苯甲酰胺(1.00g)的冰醋酸(75mL)和无水肼(0.25g)液,制得标题化合物(0.20g),为无定形固体,MS,m/z:369(M)+。
实施例51
[4-(2,5-二甲基-2H-[1,2,4]三唑-3-基)-苯基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮
按照实施例48的方法,使用实施例47中的N-(1-二甲基氨基亚乙基)-4-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-羰基)-苯甲酰胺(1.18g)的冰醋酸(75mL)和甲基肼(0.30g)液,制得标题化合物(0.33g),为固体,熔点为193-195℃;MS,m/z:383(M)+。
实施例52
[4-(3-甲基-[1,2,4]氧二唑-5-基)-苯基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮
将含羟胺盐酸盐(0.40g)和乙酸钾(1.0g)的实施例47的N-(1-二甲基氨基亚乙基)-4-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-羰基)-苯甲酰胺(1.15g)的冰醋酸(50mL)溶液回流2小时。减压下除去所有挥发物并加入饱和碳酸氢钠水溶液。混合物用二氯甲烷萃取,萃取液用无水硫酸钠干燥。将该溶液通过水合硅酸镁钠短柱过滤,再用几倍体积二氯甲烷洗脱。在蒸汽浴上浓缩合并的有机相,并逐渐加入己烷直至开始结晶。冷却后,过滤收集晶体制得标题化合物(0.38g),熔点为177-179℃;MS,m/z:371.3(M)+,741.3(2M)+。
实施例53
1-甲基-4-(4-甲基苯基)-1H-吡唑
将含有2-(4-甲基苯基)-丙二醛(3.05g)、无水乙醇(40mL)和甲基肼(1.09g)的混合物在室温下搅拌18小时,室温下除去挥发物。加入水并用二氯甲烷萃取混合物。无水硫酸钠干燥后,将该溶液通过水合硅酸镁钠短柱过滤,再用几倍体积二氯甲烷洗脱。在蒸汽浴上浓缩合并的有机相,并逐渐加入己烷直至开始结晶。冷却后,过滤收集晶体,制得标题化合物(2.91g),熔点为107-108℃。
实施例54
4-(1-甲基-1H-吡唑-4基)-苯甲酸
将含有1-甲基-4-(4-甲基苯基)-1H-吡唑(1.70g)、高锰酸钾(9.70g)和1N氢氧化钠(100mL)的混合物回流18小时。将悬浮液通过硅藻土过滤并冷却。水溶液用二氯甲烷萃取然后丢弃二氯甲烷液。水溶液酸化至pH5.5。得到的沉淀很难过滤,因此用二氯甲烷萃取。蒸发溶剂后,将得到的固体在丙酮中重结晶,制得标题化合物(0.60g),熔点为274-275℃;MS,m/z:202(M)+。
实施例55
[4-(1-甲基-1H-吡唑-4-基)-苯基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮
将草酰氯(0.30g)加入到含有4-(1-甲基-1H-吡唑-4-基)-苯甲酸(0.46g)的二氯甲烷(25mL)的悬浮液中。加入2滴二甲基甲酰胺,室温下搅拌混合物18小时。将得到的溶液蒸发至干,制得粗酰氯(0.57g),无需进一步纯化直接使用。
将酰氯加入到含有10,11-二氢-5H-吡咯并[2,1-c][1,4]苯并二氮杂(0.37g)和二异丙基乙胺(0.58g)的二氯甲烷(50mL)的溶液中。室温下保持18小时后,反应混合物用水和饱和碳酸氢钠溶液洗涤。二氯甲烷溶液用无水硫酸钠干燥并通过水合硅酸镁钠短柱过滤,再用几倍体积二氯甲烷洗脱。在电热板上浓缩合并的有机相并逐渐加入己烷直至开始结晶。冷却后,过滤收集晶体,得到标题化合物(0.38g),熔点为200-201℃;MS,m/z:368(M)+。
实施例56
6-(1-甲基-1H-吡唑-4-基)-吡啶-3-羧酸
将含有6-(1-甲酰基-2-羟基乙烯基)吡啶-3-羧酸(1.93g)(Eastman Chemicals)的无水乙醇(50mL)和甲基肼(0.50g)的悬浮液在室温下搅拌18小时。过滤反应混合物得到产物(1.30g)。蒸发滤液得到固体,在乙酸乙酯中重结晶,制得标题化合物的分析样品(0.55g)。熔点为262-264℃。
实施例57
[6-(1-甲基-1H-吡唑-4-基)-吡啶-3-基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮
将含有6-(1-甲基-1H-吡唑-4-基)-吡啶-3-羧酸
(0.48g)的亚硫酰氯(5.0mL)的悬浮液在室温下搅拌2小时。减压下除去挥发物得到6-(1-甲基-1H-吡唑-4-基)-吡啶-3-碳酰氯固体,无需进一步纯化直接使用。
将含有10,11-二氢-5H-吡咯并[2,1-c][1,4]苯并二氮杂(0.37g)和二异丙基乙胺(0.61g)的二氯甲烷(25mL)溶液冷却到0℃,分批加入含有6-(1-甲基-1H-吡唑-4-基)-吡啶-3-碳酰氯的二氯甲烷(25mL)溶液。室温下保持18小时后,反应混合物用水和饱和碳酸氢钠溶液洗涤。二氯甲烷溶液用无水硫酸钠干燥并通过水合硅酸镁钠短柱过滤,再用几倍体积二氯甲烷洗脱。在电热板上浓缩合并的有机相,并逐渐加入己烷直至开始结晶。冷却后,过滤收集晶体得到标题化合物(0.31g),熔点为173-175℃;MS,m/z:370.3(M+H)+。
实施例58
[4-(吡唑-1基)-苯基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮
将草酰氯(1.04g)和1滴二甲基甲酰胺加入到含有4-(吡唑-1-基)-苯甲酸(1.56g)的二氯甲烷(25mL)的悬浮液中。室温下搅拌混合物18小时得到一透明溶液。减压下除去挥发物得到4-(吡唑-1-基)苯甲酰氯,为浅黄色固体(1.58g),无需进一步纯化直接使用。
将4-(吡唑-1-基)苯甲酰氯(0.75g)加入到含有10,11-二氢-5H-吡咯并[2,1-c][1,4]苯并二氮杂(0.61g)和二异丙基乙胺(0.47g)的二氯甲烷(25mL)溶液中。室温下保持18小时后,反应混合物用水和饱和碳酸氢钠溶液洗涤。二氯甲烷溶液用无水硫酸钠干燥并通过水合硅酸镁钠短柱过滤,再用几倍体积二氯甲烷洗脱。在电热板上浓缩合并的有机相,并逐渐加入己烷直至开始结晶。冷却后,过滤收集晶体得到标题化合物(0.90g),熔点为179-181℃。
实施例59
[4-(3-甲基-吡唑-1-基)-苯基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮
将草酰氯(1.16g)和1滴二甲基甲酰胺加入到含有4-(3-甲基吡唑-1-基)-苯甲酸(1.84g)的二氯甲烷(25mL)的悬浮液中。室温下搅拌混合物18小时,减压下除去挥发物。加入二氯甲烷,过滤溶液,减压下蒸发溶剂,得到4-(3-甲基吡唑-1-基)苯甲酰氯,为黄色油(1.76g),无需进一步纯化直接使用。
将4-(3-甲基吡唑-1-基)苯甲酰氯加入到含有10,11-二氢-5H-吡咯并[2,1-c][1,4]苯并二氮杂(0.55g)和二异丙基乙胺(0.44g)的二氯甲烷(25mL)冰冷却溶液中。室温下搅拌18小时后,反应混合物用水和饱和碳酸氢钠溶液洗涤。二氯甲烷溶液用无水硫酸钠干燥,并通过水合硅酸镁钠短柱过滤,再用几倍体积二氯甲烷洗脱。在电热板上浓缩合并的有机相,并逐渐加入己烷直至开始结晶。冷却后,得到标题化合物(0.90g),为无定形固体,MS,m/z:369(M+H)+。
实施例60
[4-(4-甲基-吡唑-1-基)-苯基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮
将草酰氯(0.50g)和1滴二甲基甲酰胺加入到含有4-(4-甲基吡唑-1-基)-苯甲酸(0.75g)的二氯甲烷(15mL)的悬浮液中。室温下搅拌混合物18小时,减压下除去挥发物。将残余物溶解于己烷中,通过硅藻土过滤。真空下蒸发溶剂得到4-(4-甲基吡唑-1-基)苯甲酰氯(0.77g),无需进一步纯化直接使用。
将4-(4-甲基吡唑-1-基)苯甲酰氯(0.72g)加入到含有10,11-二氢-5H-吡咯并[2,1-c][1,4]苯并二氮杂(0.60g)和二异丙基乙胺(0.48g)的二氯甲烷(25mL)冰冷却溶液中。室温下搅拌18小时后,反应混合物用水和饱和碳酸氢钠溶液洗涤。二氯甲烷溶液用无水硫酸钠干燥,并通过水合硅酸镁钠短柱过滤,再用几倍体积二氯甲烷洗脱。在电热板上浓缩合并的有机相,逐渐加入己烷直至开始结晶。冷却后,过滤收集晶体得到标题化合物(0.75g),熔点为179-181℃,MS,m/z:369(M+H)+。
实施例61
[4-(3,5-二甲基-吡唑-1-基)-苯基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮
将草酰氯(1.0g)和1滴二甲基甲酰胺加入到含有4-(3,5-二甲基吡唑-1-基)-苯甲酸(1.34g)的二氯甲烷(25mL)悬浮液中。室温下搅拌混合物18小时,减压下除去挥发物。将残余物溶解于己烷中,通过硅藻土过滤。真空下蒸发溶剂,得到4-(3,5-二甲基吡唑-1-基)苯甲酰氯(0.80g),无需进一步纯化直接使用。
将4-(3,5-二甲基吡唑-1-基)苯甲酰氯(0.75g)加入到含有10,11-二氢-5H-吡咯并[2,1-c][1,4]苯并二氮杂(0.55g)和二异丙基乙胺(0.42g)的二氯甲烷(25mL)的冰冷却溶液中。室温下搅拌18小时后,反应混合物用水和饱和碳酸氢钠溶液洗涤。二氯甲烷溶液用无水硫酸钠干燥,并通过水合硅酸镁钠短柱过滤,再用几倍体积二氯甲烷洗脱。在电热板上浓缩合并的有机相,并逐渐加入己烷直至开始结晶。冷却后,得到标题化合物(0.79g),为无定形固体,MS,m/z:383(M+H)+。
实施例62
(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-[4-(3-三氟甲基-吡唑-1-基)-苯基]-甲酮
将含有4-(3-三氟甲基吡唑-1-基)-苯甲酸(1.45g)的亚硫酰氯(5.0mL)的悬浮液在回流下加热3小时。减压下除去挥发物,将残余物溶解于二氯甲烷中,通过硅藻土过滤。真空下蒸发溶剂,得到4-(3-三氟甲基吡唑-1-基)苯甲酰氯(1.45g),无需进一步纯化直接使用。
将4-(3-三氟甲基吡唑-1-基)苯甲酰氯(1.40g)加入到含有10,11-二氢-5H-吡咯并[2,1-c][1,4]苯并二氮杂(0.88g)和二异丙基乙胺(0.66g)的二氯甲烷(50mL)溶液中。室温下搅拌18小时后,反应混合物用水和饱和碳酸氢钠溶液洗涤。二氯甲烷溶液用无水硫酸钠干燥,并通过水合硅酸镁钠短柱过滤,再用几倍体积二氯甲烷洗脱。在电热板上浓缩合并的有机相,并逐渐加入己烷直至开始结晶。冷却后,过滤收集晶体,得到标题化合物(1.70g),熔点为166-167℃,MS,m/z:423.3(M+H)+,845.4(2M+H)+。
实施例63
[4-(咪唑-1-基)-苯基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮
在氩气中将含有4-(咪唑-1-基)-苯甲酸(0.90g)的亚硫酰氯(2.0mL)的悬浮液在蒸汽浴中加热1小时。减压下蒸发去挥发物得到残余物,加入己烷使之结晶得到4-(咪唑-1-基)苯甲酰氯的盐酸盐(1.17g),熔点为242-247℃。
将4-(咪唑-1-基)苯甲酰氯盐酸盐(1.12g)加入到含有10,11-二氢-5H-吡咯并[2,1-c][1,4]苯并二氮杂(0.75g)、二异丙基乙胺(1.20g)和4-二甲基氨基吡啶(0.1g)的二氯甲烷(50mL)的溶液中。室温下搅拌18小时后,反应混合物用水和饱和碳酸氢钠溶液洗涤。二氯甲烷溶液用无水硫酸钠干燥,并通过水合硅酸镁钠短柱过滤,再用几倍体积二氯甲烷洗脱。在电热板上浓缩合并的有机相,并逐渐加入己烷直至开始结晶。冷却后,过滤收集晶体得到标题化合物(0.57g),熔点为171-172℃;MS,m/z:354(M+H)+。
实施例64
[4-(4-甲基-咪唑-1-基)-苯基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮
将草酰氯(0.50g)和1滴二甲基甲酰胺加入到含有4-(4-甲基咪唑-1-基)-苯甲酸(0.80g)的二氯甲烷(25mL)悬浮液中。混合物在室温下搅拌18小时,减压下除去挥发物,得到4-(4-甲基咪唑-1-基)苯甲酰氯(1.02g),无需进一步纯化直接使用。
将4-(4-甲基咪唑-1-基)苯甲酰氯(0.99g)加入到含有10,11-二氢-5H-吡咯并[2,1-c][1,4]苯并二氮杂(0.64g)和二异丙基乙胺(0.60g)的二氯甲烷(25mL)冰冷却溶液中。室温下搅拌18小时后,反应混合物用水和饱和碳酸氢钠溶液洗涤。二氯甲烷溶液用无水硫酸钠干燥并通过水合硅酸镁钠短柱过滤,再用几倍体积二氯甲烷洗脱。在电热板上浓缩合并的有机相并逐渐加入己烷直至开始结晶。冷却后,得到标题化合物(0.52g)固体,熔点为140-145℃,MS,m/z:369(M+H)+。
实施例65
4-溴-2-氯-苯甲酸甲酯
将亚硫酰氯(1.64mL)滴加到含有4-溴-2-氯苯甲酸(6.92g)的甲醇悬浮液中,加热至60℃2小时。真空下除去溶剂,将残余物再溶解于乙酸乙酯中,用0.5N氢氧化钠(2x)、水和盐水依次洗涤。有机相用无水硫酸钠干燥,真空除去溶剂,制得标题化合物(7.8g)。1HNMR(300MHz),(DMSO-d6)δ:3.87(s,3H),7.68-7.9(m,3H)。
实施例66
2-氯-4-(3-二甲基氨基-丙炔-1-基)苯甲酸甲酯
向含有4-溴-2-氯苯甲酸甲酯(18.69g)的三乙胺(110mL)的搅拌溶液中,加入1-二甲基氨基-2-丙炔(12.1mL)、氯化二(三苯基膦)钯(II)(1.26g)和碘化亚铜(I)(0.136g)。混合物缓慢加热至60℃,在该温度下保持1小时。反应冷却至室温,通过硅藻土过滤,收集的固体用乙酸乙酯洗涤。真空下除去溶剂,将得到的残余物再溶解于乙酸乙酯中,用水洗涤(3x)。用无水硫酸钠干燥合并的有机萃取液,真空下除去溶剂得到粗产物。将粗产物通过硅胶(225g)柱色谱法纯化,用40%乙酸乙酯/己烷洗脱。真空下除去溶剂后,制得标题化合物,为粘稠油(17.7g),MS(FAB),m/z:252(M+H)+。
实施例67
2-氯-4-(3-二甲基氨基-2-丙烯-1-酮-1-基)苯甲酸甲酯
以保持反应混合物为-20℃的加入速率,将3-氯过苯甲酸(10.76g)逐渐加入到含有2-氯-4-(3-二甲基氨基-丙炔-1-基)-苯甲酸甲酯(15.07g)的二氯甲烷(40mL)的溶液中。搅拌混合物10-15分钟。将得到的N-氧化物通过活性I级碱性氧化铝(215g)色谱柱纯化,用10%甲醇/二氯甲烷洗脱。在12-18℃下真空蒸发溶剂。将得到的残余物溶解于甲醇(100mL)中,并在60-65℃及搅拌下加热18小时。真空除去溶剂后,将产物通过硅胶(190g)色谱柱纯化,用70%乙酸乙酯/己烷洗脱。用含有一些己烷的乙醚研碎,得到标题化合物固体(5.68g),熔点为92-96℃。
实施例68
2-氯-4-(1H-吡唑-3-基)-苯甲酸甲酯
向含有2-氯-4-(3-二甲基氨基-2-丙烯-酮-1-基)-苯甲酸甲酯(13.67g)的乙醇(53mL)悬浮液中,加入肼的单盐酸盐(7.0g)。在油浴中75-80℃下加热混合物1小时。真空下除去溶剂。将得到的残余物溶解于乙酸乙酯中,用水和盐水洗涤,无水硫酸钠干燥,真空除去溶剂,制得粗固体的标题化合物(12g)。纯化的样品的熔点为130-131℃。
实施例69
2-氯-4-(1-甲基-1H-吡唑-3-基)-苯甲酸甲酯
在氮气中向含有己烷洗涤的氢化钠(3.05g,60%分散液)的二甲基甲酰胺(6mL)的悬浮液中,用15分钟时间加入含有2-氯-4-(1H-吡唑-3-基)苯甲酸甲酯(12.0g)的二甲基甲酰胺(30mL)溶液。混合物在室温下搅拌30分钟。用15分钟滴加碘甲烷(9.5mL)。混合物在室温下搅拌45分钟。再加入碘甲烷(5.16mL),反应再搅拌75分钟。用少量水稀释反应物并真空浓缩。将残余物用水(500mL)稀释并用少量乙酸乙酯萃取(5x)。真空浓缩合并的有机相得到粗产物(13.48g)。将粗产物通过硅胶(195g)色谱柱纯化,用15%乙酸乙酯/己烷洗脱,得到纯的1-甲基区域异构体(4.29g),然后是1-甲基和2-甲基区异域构体的混合物(4.6g)。用己烷研磨异构体混合物三次,再得到一些纯的1-甲基区域异构体的样品(2.55g),熔点为66.5-67℃;MS(+FAB),m/z:251(M+H)+。
实施例70
2-氯-4-(1-甲基-1H-吡唑-3-基)-苯甲酸
向含有2-氯-4-(1-甲基-1H-吡唑-3-基)-苯甲酸甲酯(6.85g)的甲醇(32mL)溶液中,加入2.5N氢氧化钠溶液(15.3mL)。反应加热至50℃1小时。真空除去溶剂,将残余物溶解于水(250mL)中,冰浴中冷却,用2N盐酸(24mL)酸化。过滤得到的沉淀,干燥制得无色固体(6.3g),熔点为232-233℃;MS(+FAB),m/z:236(M+H)+。
实施例71
[2-氯-4-(1-甲基-1H-吡唑-3-基)-苯基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮
在氮气中,将粉碎好的2-氯-4-(1-甲基-1H-吡唑-3-基)-苯甲酸(6.3g)和二甲基甲酰胺(2.16mL)悬浮在四氢呋喃(70mL)和二氯甲烷(15mL)的混合物中。滴加含有草酰氯(2.43mL)的二氯甲烷(5ml)溶液,反应物搅拌1小时。得到的2-氯-4-(1-甲基-1H-吡唑-3-基)-苯甲酰氯悬浮液无需进一步纯化直接使用。
向含有10,11-二氢-5H-吡咯并[2,1-c][1,4]苯并二氮杂(4.93g)的二氯甲烷(15mL)悬浮液中,加入二异丙基乙胺(7mL)。在正向氮气流中,用15分钟逐渐加入新制备好的酰氯悬浮液。微温的反应混合物在氮气中搅拌50分钟。搅拌1小时后,真空浓缩混合物。将残余物溶解于二氯甲烷中,用水、5%碳酸氢钠和水洗涤。有机相用盐水洗涤,以无水硫酸钠干燥,真空除去溶剂得到粗产物(10.95g)。将粗产物通过硅胶(200g)色谱柱纯化,柱中装载25%乙酸乙酯/己烷。用25-30%乙酸乙酯/己烷洗脱极性较小的杂质。产物用30-40乙酸乙酯/己烷洗脱得到纯样品(7.42g);用晶体种晶后,用含有一些己烷的乙醚研磨24小时。过滤得到标题化合物,为结晶固体(6.88g),熔点为148.5-150℃;MS,m/z:402(M)+。
实施例72
2-氯-4-(2-甲基-1H-吡唑-3-基)-苯甲酸甲酯
按照实施例68中描述的同样方法制备标题化合物,使用2-氯-4-(3-二甲基氨基-2-丙烯-1-酮)-苯甲酸甲酯(0.8g)和甲基肼(0.319mL)。通过硅胶柱色谱分离出较多量的2-甲基区域异构体,1H NMR(300MHz),(DMSO-d6)δ:3.87(s,3H),3.89(s,3H),6.58(d,1H),7.5(d,1H),7.62-7.93(m,3H)。
实施例73
2-氯-4-(2-甲基-1H-吡唑-3-基)-苯甲酸
按照实施例70中描述的同样方法制备标题化合物,使用2-氯-4-(2-甲基-1H-吡唑-3-基)-苯甲酸甲酯(0.464g)和2.5N氢氧化钠(1.04mL)
1HNMR(300MHz),(DMSOd6)δ:3.89(s,3H),6.56(d,1H),7.49(d,1H),7.59-7.90(m,3H)。
实施例74
[2-氯-4-(2-甲基-1H-吡唑-3-基)-苯基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮
按照实施例71中同样方法制备标题化合物,使用2-氯-4-(2-甲基-1H-吡唑-3-基)-苯甲酸(3.98g)制得相应的酰氯,与10,11-二氢-5H-吡咯并[2,1-c][1,4]苯并二氮杂(0.293g)酰化,制得泡沫状的标题化合物,熔点为78-79℃;MS(EI),m/z:402(M)+。
实施例75
2-氯-4-氰基苯甲酸甲酯
将2-氯-4-氨基苯甲酸甲酯(13.95g)悬浮在水(65mL)和浓盐酸(15.7mL)的混合物中。室温下搅拌10分钟后,将悬浮液冷却至0℃。用20分钟时间逐渐加入硝酸钠(5.71g)的水(37mL)溶液,保持反应温度为0℃。在0℃下搅拌35分钟后,通过加入固体碳酸钠(3.16g)部分中和反应混合物,得到重氮盐的冷溶液。
用45-50分钟的时间,向事先冷却好的含有氰化铜(I)(8.4g)和氰化钠(9.19g)的水(112mL)溶液中,逐渐加入上述重氮盐溶液。在添加过程中,重氮盐溶液保持0℃。得到的混合物在室温下搅拌18小时。过滤沉淀,风干并溶解于乙酸乙酯(250mL)中,过滤除去不溶物质。以无水硫酸镁干燥有机相,真空下除去溶剂得到粗产物,为褐色固体(13.2g)。将粗产物通过硅胶(250g)柱色谱法纯化,用5-10%乙酸乙酯/己烷洗脱,制得标题化合物固体(10.9g)。熔点为90-92℃;MS(EI),m/z:195(M)+。
实施例76
2-氯-4氰基苯甲酸
向含有2-氯-4-氰基苯甲酸甲酯(24.3g)的甲醇(150mL)搅拌溶液中,加入2.5N氢氧化钠溶液(54.5mL)。室温下搅拌45分钟后,真空除去溶剂。将残余物溶解于水中,冰浴冷却,用2N盐酸(14mL)酸化。将过滤得到的沉淀真空干燥,制得标题化合物固体(22.55g),熔点为154-158℃。
实施例77
3-氯-4-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-羰基)-苄腈
0℃下,向含有2-氯-4-氰基苯甲酸(9.1g)的二氯甲烷(40ml)和二甲基甲酰胺(3.88ml)混合物的冷却的悬浮液中,滴加含有草酰氯(4.6mL)的二氯甲烷(10mL)溶液。用1小时时间将搅拌反应物加热至室温。浑浊的2-氯-4-氰基苯甲酰氯溶液无需进一步分离直接使用。
在氮气中,向含有10,11-二氢-5H-吡咯并[2,1-c][1,4]苯并二氮杂(7.32g)和二异丙基乙胺(13.6ml)的二氯甲烷(35mL)搅拌悬浮液中,加入浑浊的2-氯-4-氰基苯甲酰氯溶液。反应混合物在室温下保持1小时后,用二氯甲烷稀释,用水、5%的碳酸氢钠和50%的饱和盐水依次洗涤。以无水硫酸钠干燥后,真空除去溶剂,得到粗产物(18.0g)。将粗产物通过硅胶(250g)柱色谱法纯化,用20%乙酸乙酯/己烷、然后用25%乙酸乙酯/己烷进行洗脱,制得标题化合物(13.56g),为淡黄色泡沫状物;MS(EI),m/z:347(M)+。
实施例78
3-氯-4-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-羰基)-苯甲酸
向含有3-氯-4-(5H,11H-咯并[2,1-c][1,4]苯并二氮杂-10-羰基)-苄腈(90.72g)的乙醇悬浮液中,加入10N氢氧化钠溶液(1.02mL),混合物在回流下加热2小时。真空除去溶剂,将残余物溶解于水(250mL)中,用2N盐酸(4.7mL)酸化。得到的沉淀用乙酸乙酯萃取,以无水硫酸钠干燥有机相。真空除去溶剂后,泡沫状物用乙醚研磨18小时,过滤得到粗产物(0.69g)。将粗产物通过用含有活性炭的甲醇处理进行纯化。在甲醇/乙醚中结晶得到标题化合物,为纯化的固体(0.29g),熔点为198-199℃;MS(EI),m/z:366(M)+。
实施例79
3-氯-4-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-羰基)-苯甲酰胺
将浓硫酸(70mL)加入到3-氯-4-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-羰基)-苄腈(12.85g)中。混合物在60℃下搅拌3小时,然后在室温下搅拌18小时。将反应混合物倒入冰中,并在0℃下用30%氢氧化铵(184ml)中和。得到的悬浮液用乙酸乙酯萃取。过滤含水的混合物,再用乙酸乙酯萃取。以无水硫酸钠干燥合并的有机相,并在真空下除去溶剂。将残余物用乙醚(50-60ml)和少量乙酸乙酯的混合物研磨。过滤沉淀得到标题化合物为结晶固体(10.44g),熔点为211-212℃;MS(EI),m/z:365(M)+。
实施例80
N-(1-二甲基氨基亚乙基)-3-氯-4-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-羰基)-苯甲酰胺
将含有3-氯-4-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-羰基)-苯甲酰胺(5.48g)和二甲基乙酰胺二甲基缩醛(10.97ml)的悬浮液在90℃下加热20分钟。减压下除去过量的反应物,得到的标题化合物无需进一步纯化直接使用,MS(EI),m/z:434(M)+。
实施例81
[2-氯-4-(5-甲基-2H-[1,2,4]三唑-3-基)苯基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-羰基)-甲酮
向含有N-(1-二甲基氨基亚乙基)-3-氯-4-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-羰基)-苯甲酰胺(3.01g)的乙酸(4ml)溶液中,加入含有无水肼(0.435ml)的乙酸(4ml)溶液。反应混合物在85-90℃下搅拌45分钟。真空下除去乙酸后,反应混合物用水(35-40ml)稀释,用碳酸氢钠水溶液中和至pH7.0,再用乙酸乙酯萃取。有机萃取液用盐水洗涤,以无水硫酸钠干燥,真空除去溶剂得到粗产物(2.68g)。将粗产物通过硅胶(45g)柱色谱法纯化,用70%乙酸乙酯/己烷洗脱,制得纯化的产物(2.5g),用乙醚研磨后制得标题化合物固体(2g),熔点为211-212℃;MS(EI),m/z:403(M)+。
实施例82
N-(二甲基氨基亚甲基)-3-氯-4-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-羰基)-苯甲酰胺
按照实施例80中描述的同样方法制备标题化合物,使用3-氯-4-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-羰基)-苯甲酰胺(1.83g)和二甲基甲酰胺二甲基缩醛(5.3ml),MS(EI),m/z:420(M)+。
实施例83
[2-氯-4-(2H-1,2,4三唑-3-基)苯基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮
按照实施例81中描述的同样方法制备标题化合物,使用N-(二甲基氨基亚甲基)-3-氯-4-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-羰基)-苯甲酰胺(2.53g)和无水肼(0.38ml),熔点为174-177℃;MS(EI),m/z:389(M)+。
实施例84
[2-氯-4-(2-甲基-2H-[1,2,4]三唑-3-基)苯基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-羰基)-甲酮
按照实施例48中描述的同样方法制备标题化合物,使用N-(二甲基氨基亚甲基)-3-氯-4-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-羰基)-苯甲酰胺(0.572g)和甲基肼(0.149ml),熔点为141-143℃;MS(EI):403(M)+。
实施例85
4-[(2,5-二甲基-2H-[1,2,4]三唑-3-基)-2-氯-苯基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-羰基)-甲酮
按照实施例48中描述的同样方法制备标题化合物,使用N-(1-二甲基氨基亚乙基)-3-氯-4-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-羰基)-苯甲酰胺(0.51g)和甲基肼(0.125ml),熔点为197-199℃;MS(EI):417(M)+。
实施例86
[2-氯-4-(1H-四唑-5-基)苯基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮
向含有3-氯-4-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-羰基)-苄腈(0.348g)的二甲基甲酰胺(2ml)溶液中,加入叠氮化钠(0.078g)和氯化铵(0.065g)。混合物加热至100℃18小时。
真空除去大部分二甲基甲酰胺。将残余物溶解与水(约8ml)中,用2.5N氢氧化钠(0.6ml)碱化至pH为9.0,用乙酸乙酯萃取。含水的萃取液用2N盐酸(1.1ml)酸化,再用乙酸乙酯萃取,以无水硫酸钠干燥,真空除去溶剂得到油状的粗产物(0.350g)。将该油状的粗产物用乙醚研磨,通过酸处理过的硅胶过滤,用40%乙酸乙酯/己烷洗脱得到较纯的样品。该样品再用乙醚研磨,过滤得到样品(0.88g)。熔点为218-220℃。MS(+FAB):391(M+H)+。
实施例87
[2-氯-4-(3-甲基-吡唑-1-基)-苯基]-(3-二甲基氨基甲基-5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮
向含有[2-氯-4-(3-甲基-吡唑-1-基)-苯基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮(1.61g)、N,N,N’,N’-四甲基二氨基甲烷(0.82g)和冰醋酸(0.48g)的甲醇(25ml)搅拌溶液中,加入37%的甲醛水溶液(4ml)。将混合物加热至40℃持续10分钟。室温下搅拌1小时后,真空浓缩反应物,再溶解于二氯甲烷中,依次用碳酸氢钠水溶液和水萃取(4x)。有机相用无水硫酸钠干燥,通过硅胶塞过滤,用乙酸乙酯洗脱。真空蒸发溶剂得到油状物,用己烷研磨制得0.36g标题化合物,为无色粉末,熔点为100-102℃;MS(+FAB),m/z:482(M+Na)+,460(M+H)+。
实施例88
(3-溴-5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-[2-氯-4-(3-甲基-吡唑-1-基)-苯基]-甲酮
在-78℃下,10分钟内向含有[2-氯-4-(3-甲基-吡唑-1-基)-苯基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮(1.61g)的二氯甲烷(25ml)的预冷却搅拌溶液中,加入固体N-溴丁二酰亚胺(0.712g)。30分钟内将反应升温至-40℃。混合物用二氯甲烷稀释,依次用饱和碳酸氢钠水溶液(2×100ml)和水(100ml)萃取。有机相用无水硫酸钠干燥,通过硅胶塞过滤,真空蒸发得到残余物。在乙醚中结晶,制得1.47g标题化合物,为无色固体,熔点为148-149℃(分解);MS(EI),m/z:480(M)+。
实施例89
(4-溴-2-氯苯基)-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮
将二甲基甲酰胺(1滴)加入到含有4-溴-2-氯苯甲酸(2.20g)的无水四氢呋喃(20mL)溶液中。加入草酰氯(1.46g),将混合物加热至回流。将得到的溶液冷却至室温,然后蒸发至干,得到粗4-溴-2-氯苯甲酰氯,为金色粘稠液体,无需进一步纯化直接使用。
将4-溴-2-氯苯甲酰氯(2.42g)的二氯甲烷(20ml)溶液滴加到含有10,11-二氢-5H-吡咯并[2,1-c][1,4]苯并二氮杂(1.44g)和三乙胺(0.95g)的二氯甲烷(40mL)冰浴冷却的混合物中。除去冰浴,搅拌22小时后,反应混合物依次用水、饱和碳酸氢钠溶液、0.5N盐酸和水洗涤。二氯甲烷溶液用无水硫酸钠干燥,过滤,然后真空蒸发至干得到黄白色泡沫状物。通过硅胶快速色谱法纯化,用己烷-乙酸乙酯(2∶1)洗脱,制得白色泡沫状物(3.02g),熔点为77-80℃,MS,m/z:400(M)+。
实施例90
[2-溴-4-(3-甲基-吡唑-1-基)-苯基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮
步骤a)4-氟-2-溴苯甲酰氯:将二甲基甲酰胺(2滴)加入到含有4-氟-2-溴苯甲酸(4.91g)的无水四氢呋喃(55mL)溶液中。加入草酰氯(3.41g),将混合物加热至回流。将得到的溶液冷却至室温,真空蒸发得到粗酰氯,为金色粘稠液体,无需进一步纯化直接使用。
步骤b)(4-氟-2-溴苯基)-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮:将步骤a)的4-氟-2-溴苯甲酰氯(5.32g)的二氯甲烷(35ml)溶液,滴加到含有10,11-二氢-5H-吡咯并[2,1-c][1,4]苯并二氮杂(3.44g)和三乙胺(2.27g)的二氯甲烷(80mL)冰浴冷却的溶液中。除去冰浴,搅拌16小时后,反应混合物依次用水、饱和碳酸氢钠溶液和饱和氯化钠溶液洗涤。二氯甲烷溶液用无水硫酸钠干燥,过滤,然后真空蒸发得到浅紫色泡沫状物。通过硅胶快速色谱法纯化,用己烷-乙酸乙酯(1∶1)洗脱,制得中间体(4-氟-2-溴苯基)-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮褐色泡沫状物(6.91g),MS,m/z:384(M)+。该物质无需进一步纯化直接用于下一步。
步骤c)[2-溴-4-(3-甲基吡唑-1-基)-苯基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮:将60%氢化钠的油(0.20g)用己烷洗涤,然后悬浮在二甲基甲酰胺(15ml)中。向该悬浮液中加入3-甲基吡唑(0.41g)。当氢气停止放出后,加入步骤b)的(4-氟-2-溴苯基)-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮(1.74g)。反应混合物加热至130℃6小时。反应混合物冷却至室温后,倒入50%的饱和氯化钠水溶液中,并用乙酸乙酯萃取。乙酸乙酯溶液用无水硫酸镁干燥,过滤,真空蒸发得到褐色油状物。通过硅胶快速色谱法纯化,用己烷-乙酸乙酯(1∶1)洗脱得到无色固体(0.75g)。在乙醇中重结晶得到黄白色结晶固体(0.53g),熔点为141-142.5℃,MS,m/z:446(M)+。
实施例91
(2,4-二氟-苯基)-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮
步骤a)2,4-二氟苯甲酰氯:在氮气中,滴加草酰氯(2.4ml),处理含有几滴二甲基甲酰胺的2,4-二氟苯甲酸(3.6g)的二氯甲烷(40mL)悬浮液。停止放出气体后,将反应混合物再回流15分钟。将该溶液真空蒸发至干,残余物无需进一步纯化直接使用。
步骤b)(2,4-二氟苯基)-(5H,11H-二氢-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮:在氮气中,向含有步骤a)中的粗2,4-二氟苯甲酰氯的二氯甲烷溶液中加入固体10,11-二氢-5H-吡咯并[2,1-c][1,4]苯并二氮杂胺(2.0g)和二异丙基乙胺(3.4ml)。反应混合物变为橙黄色。室温下搅拌10分钟后,反应混合物用水、1N盐酸、1N氢氧化钠和盐水洗涤。有机相用无水硫酸钠干燥并蒸发至干,得到褐色固体。将该粗产物通过硅胶柱色谱(Merck-60)纯化,用20%的乙酸乙酯-己烷洗脱制得2.9g白色泡沫状的标题化合物。MS(EI,m/z):324(M)+。
实施例92
[2-氟-4-(3-甲基-吡唑-1-基)-苯基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮
在室温下,氮气中通过滴加3-甲基吡唑(0.62ml)处理含有用己烷洗涤过的60%氢化钠(0.31g)的无水二甲基甲酰胺悬浮液。连续搅拌直至停止放出气体(10分钟)。一次性加入实施例91步骤b)中的(2,4-二氟苯基)-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮(2.5g),继续搅拌直至溶液澄清。将该混合物放置在预加热至130℃的油浴中加热1小时。冷却后混合物分为水层和乙酸乙酯层。用无水硫酸钠干燥有机萃取液,并蒸发至干。将该粗产物通过快速硅胶色谱柱(Merck-60)纯化,用20%乙酸乙酯-己烷洗脱,制得0.82g泡沫状的标题产物,通过超声处理在乙醇/己烷中结晶,熔点为192-193℃。MS(EI)m/z:386(M)+。
实施例93
4-(3-甲基-吡唑-1-基)-2-三氟甲基-苯甲酸甲酯
步骤a)4-氟-2-三氟甲基苯甲酸甲酯:在氮气中,滴加草酰氯(11.3ml)处理含有4-氟-2-三氟甲基苯甲酸(25.6g)和几滴二甲基甲酰胺的二氯甲烷(250ml)悬浮液。停止放出气体后,将反应混合物再回流15分钟。冷却反应并加入甲醇(50ml)。搅拌2小时后,真空浓缩反应物,残余物分成乙酸乙酯层和水层。有机相用饱和碳酸氢钠洗涤,无水硫酸钠干燥,蒸发至干得到18.0g标题化合物,为金色油状物。MS,(EI)m/z:222(M)+。
水层用2N盐酸酸化得到无色固体,过滤收集得到7.5g原料苯甲酸。
步骤b)4-(3-甲基-吡唑-1-基)-2-三氟甲基-苯甲酸甲酯:在室温下,氮气中通过滴加3-甲基吡唑(7.75ml)的二氯甲烷(50ml)溶液处理含有用己烷洗涤过的60%氢化钠(3.85g)的无水二甲基甲酰胺(150ml)悬浮液。连续搅拌直至停止放出气体(10分钟)。向该澄清溶液中滴加含有步骤a)中的4-氟-2-三氟甲基苯甲酸甲酯(17.8g)的二甲基甲酰胺(50ml)溶液。搅拌30分钟后,室温下将反应物用饱和氯化铵骤冷,并用乙酸乙酯萃取。用无水硫酸钠干燥有机萃取液(3x),并蒸发至干。将该粗产物通过快速硅胶色谱柱(Merck-60)纯化,用二氯甲烷-己烷梯度液洗脱(50%-75%),制得13.6g标题产物,为无色固体,熔点为59-61℃。MS(EI)m/z:284(M)+。
实施例94
4-(3-甲基-吡唑-1-基)-2-三氟甲基-苯甲酸
将实施例93步骤b)中的4-(3-甲基-吡唑-1-基)-2-三氟甲基-苯甲酸甲酯(1.19g)溶解于甲醇(10ml)中,并加入2.5N的氢氧化钠(3.3ml)溶液。反应在回流下加热90分钟,冷却至室温并真空浓缩至干。残余物分成乙酸乙酯层和1N盐酸层。以无水硫酸钠干燥合并的有机萃取液并真空浓缩,得到1.14g标题化合物,为无色固体。MS(FAB)m/z:271(M+H)+。
实施例95
[4-(3-甲基-吡唑-1-基)-2-三氟甲基苯基]-(5H,11H-吡咯并[5,1-c][1,4]苯并二氮杂-10-基)-甲酮
将含有实施例94中的4-(3-甲基-吡唑-1-基)-2-三氟甲基-苯甲酸(0.26g)的四氢呋喃(5ml)溶液,用二甲基甲酰胺(0.020ml)处理,然后用草酰氯(0.090ml)处理。溶液在室温下搅拌直至气体停止放出,然后将溶液加热回流10分钟。将样品冷却至室温,浓缩为固体,该固体溶解于四氢呋喃(25mL)中。将该溶液加入到含有(5H-10,11-二氢-吡咯并[5,1-c][1,4]苯并二氮杂(0.143g)和三乙胺(0.150ml)的四氢呋喃(20ml)溶液中。室温下将该溶液搅拌过夜。形成沉淀。将该样品用二氯甲烷稀释,溶解沉淀,然后真空浓缩样品至起始体积的1/3。该样品分成二氯甲烷层和饱和氯化铵水层。用二氯甲烷萃取该样品,合并有机层,用无水硫酸钠干燥,过滤,浓缩为油状物。将该油通过快速硅胶色谱柱,使用40%乙酸乙酯/己烷-100%乙酸乙酯进行梯度洗脱,得到泡沫状的标题化合物(0.30g)。将一部分该物质在丙酮/己烷中重结晶得到重质板状晶体,熔点为100-102℃,MS m/z:437(M)+。
实施例96
2-氯-4-(3-甲基-1H-吡唑-1-基)-苯甲酸甲酯和2-氯-4-(5-甲基-1H-吡唑-1-基)-苯甲酸甲酯
在搅拌下一次加入3-甲基吡唑(0.85ml),处理含有己烷洗涤过的氢化钾(0.424g)的二甲基甲酰胺(5ml)悬浮液。停止放出气体后,向该澄清溶液中加入2-氯-4-氟苯甲酸甲酯(2.0g,10.6),并在130℃下加热15分钟。反应混合物冷却至室温并分成乙酸乙酯层和盐水层。有机相用水和盐水洗涤并用无水硫酸钠干燥。真空除去溶剂得到2.2g黄色油状物。(注:通过粗产物的NMR谱图分析检测到酯水解20%)。通过快速硅胶(merck 60)色谱法,从其他异构体(如下所述)中分离出所需的区域异构体2-氯-4-(3-甲基-1H-吡唑-1-基)-苯甲酸甲酯,用二氯甲烷-己烷(2∶1)洗脱,得到1.55g标题化合物,为无色固体。MS(EI)m/z:250-252(M)+。
5-区域异构体,即2-氯-4-(5-甲基-1H-吡唑-1-基)-苯甲酸甲酯,再用二氯甲烷-己烷2∶1洗脱从上述硅胶(Merck 60)快速色谱柱中分离出来,得到0.20g产物,为无色固体。MS(EI),m/z:250/252(M)+。
实施例97
2-氯-4-(3-甲基-1H-吡唑-1-基)-苯甲酸
室温下,将含有实施例96中的2-氯-4-(3-甲基-1H-吡唑-1-基)-苯甲酸甲酯(1.42g)和6ml 1M含水氢氧化锂的四氢呋喃(20ml)的溶液搅拌18小时。反应混合物分成乙酸乙酯层和1N盐酸层。有机层用水和盐水洗涤并用无水硫酸钠干燥。真空蒸发溶剂,得到1.05g标题化合物,为无色固体。熔点为192-193℃。MS(EI),m/z:236/238(M)+。
实施例98
(2,6-二氯吡啶-3-基)(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮
室温下,将含有2,6-二氯烟酸(3.84g)、草酰氯(2.0g)和1滴二甲基甲酰胺的二氯甲烷(25ml)溶液搅拌18小时。真空浓缩溶液,得到3.50g 2,6-二氯烟酰氯,将含有酰氯的二氯甲烷(25ml)分批加入到含有10,11-二氢-5H-吡咯并[2,1-c][1,4]苯并二氮杂(2.15g)和二异丙基乙胺(2.03g)的二氯甲烷(50ml)的冰冷却溶液中。混合物在室温下搅拌18小时,并用饱和碳酸氢钠水溶液洗涤。有机相用无水硫酸钠干燥,并通过无水硅酸镁钠短柱过滤。在电热板上浓缩合并的有机相,并逐渐加入己烷直至开始结晶。冷却后,过滤收集晶体,得到2.65g标题化合物,为无定形固体,熔点为115-130℃;MS,m/z:358.1(M+H)+。
实施例99
(2-氯-6-吡唑-1-基-吡啶-3-基)(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮
向含有60%氢化钠的油(0.1g)的二甲基甲酰胺(25ml)悬浮液中滴加吡唑(0.15g)。停止放出氢气后,加入(2,6-二氯吡啶-3-基)(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮(0.67g)。在沙浴中110℃下加热反应混合物18小时。将混合物倒入冰中并用盐水稀释,用二氯甲烷萃取。以无水硫酸钠干燥合并的萃取液,并通过无水硅酸镁钠短柱过滤。真空浓缩该溶液,用乙醚研磨得到0.18g标题化合物,为无色固体。熔点为133-135℃。MSm/z:390.8(M+H)+,779.1(2M+H)+。
实施例100
[2-氯-6-(3-甲基吡唑-1-基)-吡啶-3-基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮
向含有60%氢化钠的油(0.1g)的二甲基甲酰胺(25ml)悬浮液中滴加3-甲基吡唑(0.15g)。停止放出氢气后,加入(2,6-二氯吡啶-3-基)(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮(0.67g),在沙浴中110℃下加热反应混合物18小时。将混合物倒入冰中并用盐水稀释,用二氯甲烷萃取。无水硫酸钠干燥合并的萃取液,并通过无水硅酸镁钠短柱过滤。将粗产物通过制备型hplc(Dynamax c60二氧化硅筒)柱纯化,用含40%乙酸乙酯的己烷洗脱得到0.21g无色晶体。熔点为171-172℃。MSm/z:404.2(M+H)+,807.1(2M+H)+。
实施例101
[2-氯-6-(4-甲基吡唑-1-基)-吡啶-3-基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮
向含有60%氢化钠的油(0.1g)的二甲基甲酰胺(25ml)悬浮液中滴加3-甲基吡唑(0.45g)。停止放出氢气后,加入(2,6-二氯吡啶-3-基)(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮(1.79g),在沙浴中110℃下加热反应混合物18小时。将混合物倒入冰中并用盐水稀释,用二氯甲烷萃取。无水硫酸钠干燥合并的萃取液,并通过无水硅酸镁钠短柱过滤。将粗产物通过制备型hplc(Dynamax c60二氧化硅筒)柱纯化,用含40%乙酸乙酯的己烷洗脱得到0.26g无色晶体。熔点为155-156℃。MSm/z:404.2(M+H)+,807.0(2M+H)+。
实施例102
[2-氯-4-(3-甲基-1,2,4三唑-1-基)-苯基](5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮
向含有60%氢化钠的油(0.3g)的二甲基甲酰胺(50ml)悬浮液中滴加3-甲基-1,2,4-三唑(0.45g)。停止放出氢气后,加入2-氯-4-氟苯基-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮(1.70g),在沙浴中110℃下加热反应混合物18小时。将混合物倒入冰中并用盐水稀释,用二氯甲烷萃取。以无水硫酸钠干燥合并的萃取液,并通过无水硅酸镁钠短柱过滤。真空浓缩该溶液,用乙醚研磨残余物,得到1.25g标题化合物,为无色晶体。熔点为191-193℃。MS m/z:404.1(M+H)+。
实施例103
[4-(3-甲基-1,2,4-三唑-1-基)-2-三氟甲基-苯基](5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮
向含有60%氢化钠的油(0.3g)的二甲基甲酰胺(50ml)悬浮液中滴加3-甲基-1,2,4-三唑(0.45g)。停止放出氢气后,加入4-氟-2-三氟甲基-苯基-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮(1.76g),在沙浴中110℃下加热反应混合物18小时。将混合物倒入冰中并用盐水稀释,用二氯甲烷萃取。无水硫酸钠干燥合并的萃取液,并通过无水硅酸镁钠短柱过滤。真空浓缩该溶液,用乙醚研磨残余物,得到0.81g标题化合物,为无色晶体。熔点为148-150℃。MS m/z:438.2(M+H)+,875.8(2M+H)+。
实施例104
4-肼基-2-甲氧基苯甲酸甲酯盐酸盐(1∶1),水合物(2∶1)
以保持反应温度低于0℃所需的速率,用预冷却的硝酸钠(8.5g)的水(45ml)溶液处理含有4-氨基-2-甲氧基苯甲酸甲酯(21.74g)的浓盐酸(110ml)搅拌悬浮液。添加完后,将反应混合物在-2℃下搅拌10分钟。在-10℃下,将浑浊的橙色溶液滴加到含有氯化锡(II)二水合物(101g)的浓盐酸(67ml)的剧烈搅拌预冷却的溶液中。控制添加速率,以保持反应温度低于-5℃。加完后,将乳油色的悬浮液加热至室温并过滤出固体。固体用乙醚洗涤,无水硫酸钠干燥,制得52g粗产物。将粗产物(20g)在2.5N氢氧化钠水溶液和二氯甲烷之间分配。有机相通过硅藻土过滤,盐水洗涤并用无水硫酸镁干燥。过滤并真空蒸发溶剂得到乳油色的固体(7.1g),用含有1当量氯化氢的乙醚处理得到标题化合物,为盐酸盐,熔点为76-79℃,MS,m/z:197(M+H)+。
实施例105
2-甲氧基-4-(3-甲基-吡唑-1-基)-苯甲酸甲酯
将乙酰基乙醛二甲基缩醛(0.53g),加入到含有实施例104中的4-肼基-2-甲氧基苯甲酸甲酯盐酸盐(0.88g)和一滴浓盐酸的1∶1水/甲醇(10ml)混合物的搅拌溶液中。将反应加热至90℃5分钟。真空浓缩反应物,并在1N氢氧化钠(10ml)和乙酸乙酯(50ml)间分配。移出有机相并用盐水洗涤,无水硫酸镁干燥并过滤。真空蒸发溶剂,得到褐色油状物,与前面所得部分(0.54g)合并,在二异丙醚中重结晶3次,得到2-甲氧基-4-(3-甲基-吡唑-1-基)-苯甲酸甲酯(0.5g),熔点为167-169℃,MS,m/z:246(M)+。
实施例106
2-甲氧基-4-(3-甲基-吡唑-1-基)-苯甲酸
在室温下,用1N氢氧化锂(2.13ml)处理含有实施例105中的2-甲氧基-4-(3-甲基-吡唑-1-基)-苯甲酸甲酯(0.5g)的四氢呋喃(2.5ml)溶液。14小时后,真空除去溶剂,在0℃下加入1N盐酸沉淀出标题化合物。真空干燥后,得到0.42g标题化合物固体。MS,m/z:232(M)+。
实施例107
[2-甲氧基-4-(3-甲基-吡唑-1-基)-苯基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮
将草酰氯(0.17ml)加入到含有实施例106中的2-甲氧基-4-(3-甲基-吡唑-1-基)-苯甲酸(0.41g)和二甲基甲酰胺(0.004ml)的无水四氢呋喃(10ml)的搅拌溶液中。反应在35℃下加热10分钟。真空蒸发得到的溶液,制得粗2-甲氧基-4-(3-甲基-吡唑-1-基)-苯甲酸碳酰氯。与二氯甲烷共蒸发后,将酰氯溶解于二氯甲烷(10ml)中,并加入10,11-二氢-5H-吡咯并[2,1-c][1,4]苯并二氮杂(0.31g)。加入二异丙基乙胺(0.37ml),反应在室温下搅拌2小时。反应物用二氯甲烷稀释并用水、然后用1N盐酸洗涤。有机相用盐水洗涤,无水硫酸镁干燥并真空浓缩至干。将固体残余物通过快速硅胶色谱柱纯化,用己烷/乙酸乙酯(2/1)洗脱制得0.35g标题化合物,为无色固体,熔点为92-94℃。
实施例108
(3-二甲基氨基甲基-5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-[2-甲氧基4-(3-甲基-吡唑-1-基)-苯基]-甲酮
向含有实施例107中[2-甲氧基-4-(3-甲基-吡唑-1-基)-苯基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮(0.57g)的热甲醇(10ml)的搅拌溶液中,加入N,N,N’,N’-四甲基二氨基甲烷(0.392ml)和乙酸(0.164ml)。加入37%福尔马林水溶液(2.9ml)后,反应搅拌15分钟。混合物在真空中浓缩,并在二氯甲烷和碳酸氢钠之间分配。分离出有机相,用水洗涤并用无水硫酸镁干燥。过滤该溶液,真空除去溶剂。将残余物通过快速二氧化硅色谱柱纯化,用氯仿/甲醇(50/1)洗脱制得固体。将该固体在丙酮中重结晶,得到标题化合物,为无色固体,熔点为196-198℃。
实施例109
[2-羟基-4-(3-甲基-吡唑-1-基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮
将实施例107中的[2-甲氧基-4-(3-甲基-吡唑-1-基)-苯基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮(0.82g)溶解于二氯甲烷(20ml)中,并冷却至-78℃。加入三溴化硼(6.2ml),反应物在0℃下搅拌5分钟。加入氢氧化铵(15ml)并用二氯甲烷萃取。有机相用盐水洗涤,并用无水硫酸镁干燥。过滤除去固体并在真空中除去溶剂。将残余物通过快速加压的二氧化硅色谱柱纯化,用己烷/乙酸乙酯洗脱(3/1然后2/1)制得0.19g标题化合物,为无色固体,熔点为134-136℃。
实施例110
2-氯-4-碘-苯甲酸甲酯
将4-氨基-2-甲氧基-苯甲酸甲酯(22.97g)冷却到内部温度为-10℃,在浓盐酸(110ml)中搅拌成悬浮液。以保持反应温度低于0℃的速率,向该混合物中加入预冷却的硝酸钠(98.71g)的水(45ml)溶液。0℃下搅拌25分钟后,以保持反应温度低于-4℃的速率,向反应物中加入碘化钾(24.44g)和碘(18.37g)的水(50ml)溶液。在添加过程中加入乙酸乙酯(100ml),深色混合物在0℃下搅拌1小时。有机层用乙酸乙酯稀释,并用饱和硫代硫酸钠溶液洗涤。得到的橙色溶液用盐水洗涤,并用无水硫酸镁干燥。真空蒸发溶剂得到油状物,通过硅胶抽吸过滤进行纯化,使用己烷/乙酸乙酯(50/1)洗脱。将制得的纯化的油冷却固化,得到33.71g标题化合物。MS,m/z:296(M)+。
实施例111
4-溴-1-甲基-1H-吡唑
向含有事先洗涤(四氢呋喃)过的60%氢化钠的油(11.67g)的四氢呋喃(200ml)悬浮液中,滴加含有4-溴吡唑(39.77g)的四氢呋喃(50ml)溶液。该溶液在室温下搅拌2小时。以保持反应温度略微上升的速率,加入含有过量碘甲烷(33ml)的四氢呋喃(50ml)。该反应再搅拌2小时。真空除去溶剂,将残余物在乙醚中搅拌。通过抽吸过滤移除沉淀并用乙醚洗涤。真空蒸发合并的有机相,制得42.22g油状的标题化合物。MS,m/z:160(M)+。
实施例112
1-甲基-4-三丁基甲锡烷基-1H-吡唑
在氩气中,向含有1.6M正丁基锂的己烷(100ml)的无水乙醚(100ml)的预冷却(内部温度<-10℃)溶液中,以保持该温度的速率,加入含有实施例111中4-溴-1-甲基-1H-吡唑(23.42g)的乙醚(50ml)溶液。反应物再搅拌20分钟,然后加入氯化三丁基锡(43.4ml)的乙醚液(50ml)。反应温度升高至20℃。反应物用乙醚稀释,通过抽吸过滤除去不溶物质。真空蒸发溶剂得到56g油状的标题化合物。MS,m/z:373[M+H]+。使用Kugelrohr仪器,在高真空及170℃下,通过蒸馏从油中除去残余量的锡残余物。
实施例113
2-氯-4-(1-甲基-1H-吡唑-4-基)-苯甲酸甲酯
将含有实施例110中的2-氯-4-碘-苯甲酸甲酯(25.4g)吡唑、1-甲基-4-三丁基甲锡烷基-1H-(31.77g)、四(三苯基膦)钯(O)(1.8g)和催化碘化铜(I)的用氩气脱气的二甲基甲酰胺溶液(70ml),在80℃下加热7小时。真空除去溶剂并将残余物吸附在硅胶上。通过硅胶垫抽吸过滤进行纯化,依次用己烷、然后用己烷/乙酸乙酯(2/1)进行洗脱,蒸发溶剂后制得固体残余物,在二异丙醚中重结晶,制得7.82g标题化合物。MS,m/z 250(M)+。
实施例114
2-氯-4-(1-甲基-1H-吡唑-4-基)-苯甲酸
向含有实施例113中的2-氯-4-(1-甲基-1H-吡唑-4-基)-苯甲酸甲酯(6.25g)的甲醇(80ml)溶液中加入1N氢氧化钠(30ml)。反应在回流下加热1小时。真空下将溶剂的体积降低3/4,在0℃下用2N盐酸处理残余物。过滤沉淀并真空干燥,制得5.84g标题化合物,MS,m/z:237[M+H]+。
实施例115
[2-氯-4-(1-甲基-1H-吡唑-4-基)-苯基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮
将草酰氯(0.49ml)加入到含有实施例114中的2-氯-4-(1-甲基-1H-吡唑-4-基)-苯甲酸(0.41g)和二甲基甲酰胺(0.012ml)的无水四氢呋喃(20ml)的溶液中。反应在35℃下加热10分钟。真空蒸发所得溶液,制得粗2-氯-4-(1-甲基-1H-吡唑-4-基)-苯甲酸碳酰氯。与无水二氯甲烷共蒸发后,将酰氯溶解于二氯甲烷(20ml)中,然后加入10,11-二氢-5H-吡咯并[2,1-c][1,4]苯并二氮杂(0.888g)和二异丙基乙胺(1.06ml)。得到的溶液在室温下搅拌2小时。反应物用二氯甲烷稀释并用水、然后用1N盐酸洗涤。有机相用盐水洗涤,无水硫酸镁干燥。过滤二氯甲烷并真空浓缩至干。将残余物通过快速硅胶加压色谱柱纯化,用己烷/乙酸乙酯(2/1)洗脱,制得1.4g标题化合物,为无色固体,熔点为105-109℃。
实施例116
[2-氯-4-(3-甲基-吡唑-1-基)-苯基]-(4H,10H-吡唑并[5,1-c][1,4]苯并二氮杂-5-基)-甲酮
向含有实施例18步骤e)中的2-氯-4-(3-甲基-吡唑-1-基)-苯甲酰氯(0.214g)的二氯甲烷(10ml)溶液中,加入5H-10,11-二氢-吡咯并[5,1-c][1,4]苯并二氮杂(0.153g)和二异丙基乙胺(0.173ml)。反应在室温下搅拌2小时。反应物用二氯甲烷稀释,并用水、然后用1N盐酸洗涤。有机相用盐水洗涤,无水硫酸镁干燥。过滤二氯甲烷溶液并真空浓缩至干。将残余物通过快速硅胶加压色谱柱纯化,用己烷/乙酸乙酯(1/1)洗脱制得0.3g标题化合物,为无色固体,熔点为187-188℃。
实施例117
[2-氯-4-(3-甲基-吡唑-1-基)-苯基]-(5,10-二氢-4H-四唑并[5,1-c][1,4]苯并二氮杂-5-基)-甲酮
向含有实施例18步骤e)中的2-氯-4-(3-甲基-吡唑-1-基)-苯甲酰氯(0.18g)的二氯甲烷(10ml)溶液中,加入10,11-二氢-5H-四唑[5,1-c][1,4]苯并二氮杂(0.13g)和二异丙基乙胺(0.145ml)。反应在室温下搅拌2小时。反应物用二氯甲烷稀释,并用水、然后用1N盐酸洗涤。有机相用盐水洗涤,无水硫酸镁干燥。过滤二氯甲烷溶液并真空浓缩至干。将残余物通过快速硅胶色谱柱纯化,用己烷/乙酸乙酯(1/1)洗脱,制得0.14g标题化合物,为无色固体,熔点为110-114℃。
实施例118
1-[4-(4H,10H-吡唑并[5,1-c][1,4]苯并二氮杂-5-羰基)-苯基]-乙酮
将含有5,10-二氢-4H-吡唑并[5,1-c][1,4]苯并二氮杂(0.555g)、4-乙酰基苯甲酰氯(0.657g)和N,N-二异丙基乙胺(0.464g)的二氯甲烷(15mL)的混合物在室温下搅拌4小时。将混合物倒入水中并用二氯甲烷萃取。二氯甲烷萃取液用饱和碳酸氢钠、水和盐水洗涤,无水硫酸钠干燥。萃取液通过水合硅酸镁的薄垫过滤,过滤器垫用二氯甲烷洗涤。真空浓缩滤液,得到1.53g黄色固体。固体用乙酸乙酯研磨,得到0.747g玻璃状的标题化合物,熔点为201-210℃。蒸发研磨后的母液,并将残余物(0.30g)通过薄层硅胶板(200目)进行色谱分离,使用己烷-乙酸乙酯(1∶1)作为溶剂。乙酸乙酯研磨固体并与0.747g先前分离出的产物合并。从二氯甲烷-己烷的混合物中沉淀出合并的固体,制得0.73g玻璃状的产物。
实施例119
1-[4-(4H,10H-吡唑并[5,1-c][1,4]苯并二氮杂-5-羰基)-苯基]-3-(二甲基氨基)-丙-2-烯-1-酮
将含有1-[4-(4H,10H-吡唑并[5,1-c][1,4]苯并二氮杂-5-羰基)-苯基]-乙酮(0.73g)、叔-丁氧基双-[二甲基氨基]甲烷(0.964g)的二氯甲烷(10ml)混合物,在室温下搅拌2天。真空浓缩混合物,将残余物在二氯甲烷-己烷中结晶,得到0.65g标题化合物,为黄色晶体,熔点为225-230℃。
实施例120
[4-(1-甲基-1H-吡唑-3-基)-苯基]-(4H,10H-吡唑并[5,1-c][1,4]苯并二氮杂-5-基)-甲酮(异构体A)
和
[4-(2-甲基-1H-吡唑-3-基)-苯基]-(4H,10H-吡唑并[5.1-c][1,4]苯并二氮杂-5-基)-甲酮(异构体B)
将含有1-[4-(4H,10H-吡唑并[5,1-c][1,4]苯并二氮杂-5-羰基)-苯基]-3-(二甲基氨基)-丙-2-烯-1-酮(0.83g)、肼(0.198g)和乙酸(0.336g)的10ml乙醇混合物回流4小时。真空除去挥发物,并将残余物溶解于二氯甲烷中。该溶液用水、1N碳酸氢钠、水和盐水洗涤,以无水硫酸钠干燥。将该溶液通过水合硅酸镁的薄垫过滤,过滤器垫用乙酸乙酯洗涤。真空浓缩滤液得到0.56g浅黄色固体。该固体通过厚层硅胶板(200微米)进行色谱分离,使用乙酸乙酯作为溶剂,制得0.35g白色固体A和B的混合物(1∶4)。在乙酸乙酯中多级结晶制得89mg晶体,熔点为155-156℃,为A和B的混合物(9∶1),以及65mg玻璃状的A和B的混合物(1∶6)。
实施例121
1-[4-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-羰基)3-氯苯基]-乙酮
步骤a)在20mlCarrius试管中,将三乙胺(8.80ml)加入到含有(4-溴-2-氯苯基)-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮(2.37g)的吡啶(1.80ml)溶液中。得到的溶液用氮气净化25分钟,然后加入(三甲基甲硅烷基)乙炔(1.67ml)、氯化二(三苯基膦)钯(II)(0.08g)和碘化铜(0.01g)。该试管装满用氮气净化的三乙胺,密封并在油浴中90℃下加热80小时。将溶液冷却至室温,真空蒸发溶剂,残余物分成二氯甲烷层和水层。二氯甲烷萃取液用无水硫酸钠干燥,过滤,真空蒸发至褐色泡沫状。通过快速硅胶色谱柱,用己烷-乙酸乙酯(1∶1)洗脱,制得黄白色泡沫状的中间体乙炔(2.11g),MS,m/z:418(M)+。该物质无需进一步纯化直接用于下一步。
步骤b)将含有1%硫酸的四氢呋喃溶液,用硫酸汞(II)饱和。将中间体乙炔(1.00g)的四氢呋喃(5ml)溶液与30ml上述硫酸汞(II)的四氢呋喃溶液一起搅拌50小时。另外再加入一定量的硫酸汞(II)(0.01g)和水0.3ml。搅拌120小时后,将反应混合物倒入水中,并用二氯甲烷萃取。二氯甲烷溶液依次用饱和碳酸氢钠水溶液和水洗涤。以无水硫酸镁干燥二氯甲烷溶液,过滤蒸发至干,制得褐色固体。通过快速硅胶色谱柱纯化,用己烷-乙酸乙酯(1∶1)洗脱得到白色固体(0.30g),熔点为98-100℃,MS m/z:364(M)+。
实施例122
1-[4-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-羰基)3-氯苯基]-乙酮
将三丁基(乙氧基乙烯基)锡(1.17g)加入到含有(4-溴-2-氯苯基)-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮(1.24g)的甲苯(10ml)溶液中。得到的溶液用氮气净化10分钟,然后加入氯化二(三苯基膦)钯(II)(0.11g)。反应混合物加热回流24小时。该溶液冷却至室温,加入5%盐酸水溶液(10ml)。搅拌1小时后,混合物通过硅藻土垫过滤。滤液中加入乙醚(5ml),得到的混合物用乙酸乙酯萃取。有机层用无水硫酸镁干燥,过滤真空蒸发得到褐色玻璃状物质。通过快速硅胶色谱柱纯化,用己烷-乙酸乙酯(1∶1)洗脱,得到白色固体(0.30g),MS m/z:364(M)+。
实施例123
[2-氯-4-(3-甲基-4-乙炔基-苯基)-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮
在室温下,用1M氟化四丁基铵的四氢呋喃溶液处理实施例121步骤A中的中间体乙炔,除去溶剂得到84%产率的标题化合物,为橙黄色固体,熔点为84-86℃,MS,m/z:346(M)+。
Claims (20)
1.式(I)化合物和其药物学上可接受的盐:其中
A、B、E和G分别各自是CH或氮;
D独自是C-W或氮;
R1是2-7个碳原子的链烷酰基,选自CN、COOH、CONH2、
的基团,或选自如下基团:
 或
或

R2、R3和R5分别各自是氢,1-6个碳原子的直链烷基,3-7个碳原子的支链烷基,3-7个碳原子的环烷基或1-6个碳原子的全氟烷基;
R4是氢,1-6个碳原子的直链烷基,3-7个碳原子的支链烷基,3-7个碳原子的环烷基,2-7个碳原子的烷氧基烷基,或选自2-7个碳原子的链烷酰基、3-7个碳原子的链烯酰基、3-7个碳原子的环烷酰基、芳酰基或芳烷酰基的酰基取代基;
X和Y分别各自是氢,1-6个碳原子的直链烷基,3-7个碳原子的支链烷基,3-7个碳原子的环烷基,1-6个碳原子的全氟烷基,2-7个碳原子的烷氧基烷基,卤素(包括氯、溴、氟和碘),1-6个碳原子的烷氧基,羟基,CF3或2-6个碳原子的全氟烷基;
W是氢,卤素(优选氯、溴或碘),烷基,2-7个碳原子的烷氧基烷基,1-6个碳原子的羟基烷基或CH2NR6R7;
R6和R7分别各自是氢,1-6个碳原子的直链烷基,3-7个碳原子的支链烷基;或R6和R7与CH2NR6R7中的氮原子一起形成五元或六元环,该环任意含有一个或多个其他杂原子;
R8是1-6个碳原子的直链烷基;
R9分别各自是氢,三甲基甲硅烷基或1-6个碳原子的直链烷基。
2.根据权利要求1的化合物,其中R6和R7与CH2NR6R7中的氮原子一起,形成选自如下基团的五元或六元环:
 或
或
 或所述化合的药物学上可接受的盐。
或所述化合的药物学上可接受的盐。
3.根据权利要求1的式(I)化合物:其中
A、B、E和G分别各自是CH或氮;
D独自是C-W或氮;
R1是2-7个碳原子的链烷酰基或选自如下基团: 或

R2、R3和R5分别各自是氢,1-6个碳原子的直链烷基,3-7个碳原子的支链烷基,3-7个碳原子的环烷基或1-6个碳原子的全氟烷基;
R4、X、Y、W、R6、R7和R8如权利要求1中所定义;
或所述化合物药物学上可接受的盐。
4.根据权利要求3的化合物,其中R6和R7与CH2NR6R7中的氮原子一起形成选自如下基团的五元或六元环:
 或
或
或所述化合物药物学上可接受的盐。
5.下式化合物: 其中:
其中:
A和B分别各自是CH或N;
D是C-W或N;
R1是2-7个碳原子的链烷酰基或选自如下基团; 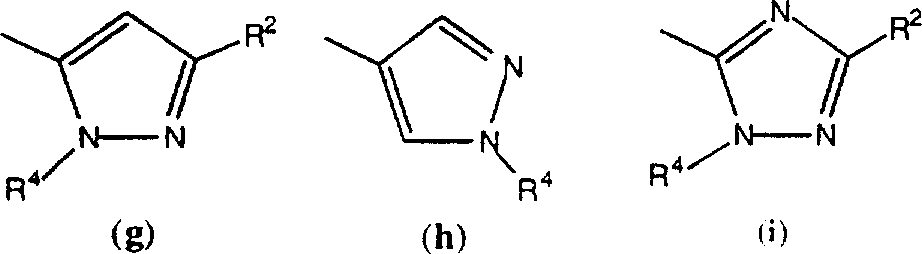 或
或

R2、R3和R5分别各自是氢、1-6个碳原子的直链烷基、3-7个碳原子的支链烷基、3-7个碳原子的环烷基或1-6个碳原子的全氟烷基;
R4、X、Y、W、R6、R7和R8如权利要求1中所定义;
或所述化合物药物学上可接受的盐。
6.根据权利要求5的化合物,其中R6和R7与CH2NR6R7中的氮原子一起形成选自如下基团的五元或六元环:
或所述化合物药物学上可接受的盐。
7.下式化合物: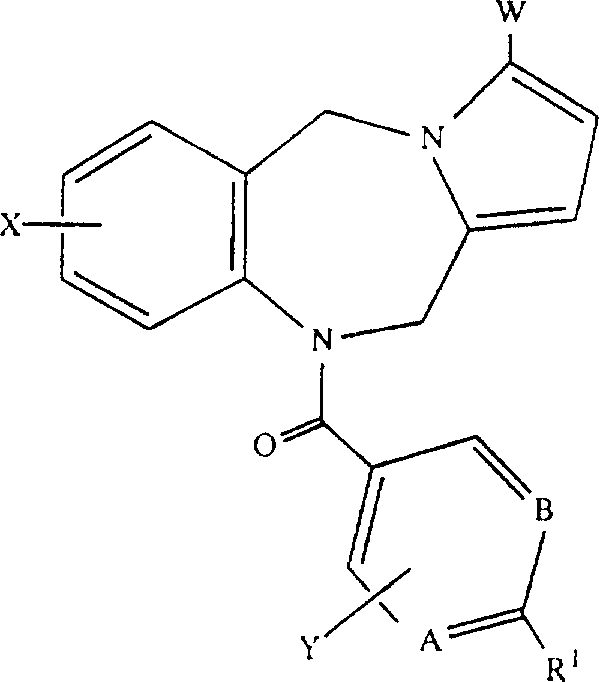
其中A、B、W、R1、R2、R3、R4、R5、R6、R7、R8、R9、X和Y如权利要求1中所定义;
或所述化合物药物学上可接受的盐。
8.根据权利要求7的化合物,其中R6和R7与CH2NR6R7中的氮原子一起形成选自如下基团的五元或六元环: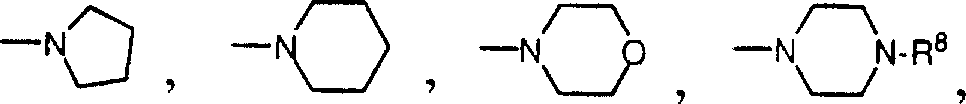 或
或

或所述化合物药物学上可接受的盐。
9.根据权利要求8的化合物,其中
W是H;
A和B各自是CH;
R1、R2、R3、R4、R5、R8、R9、X和Y如权利要求1中所定义;
或所述化合物药物学上可接受的盐。
10.下式化合物:
其中A、B、R1、R2、R3、R4、R5、R9、X和Y如权利要求1中所定义;
或所述化合物药物学上可接受的盐。
11.下式化合物:
其中A、B、R1、R2、R3、R4、R5、X和Y如权利要求1中所定义;
或所述化合物药物学上可接受的盐。
12.根据权利要求1的化合物,选自
[4-(3-甲基-吡唑-1-基)-2-三氟甲基-苯基]-(5H,11H-吡咯并[2,1-c]-[1,4]苯并二氮杂-10-基)-甲酮,
[4-(4-甲基-吡唑-1-基)-2-三氟甲基-苯基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮,
(4-吡唑-1-基-2-三氟甲基-苯基)-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮,
[4-(3-环丙基-吡唑-1-基)-2-三氟甲基-苯基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮,
[4-(4-甲基-咪唑-1-基)-2-三氟甲基-苯基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮,
(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-(4-[1,2,4]三唑-1-基-2-三氟甲基苯基)-甲酮,
[2-氯-4-(3-甲基-吡唑-1-基)-苯基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮,
[2-氯-4-(5-甲基-吡唑-1-基)-苯基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮,
[2-氯-4-(4-甲基-吡唑-1-基)-苯基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮,
[2-氯-4-(4-甲基-咪唑-1-基)-苯基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10基)-甲酮,
[2-氯-4-(3-三氟甲基-吡唑-1-基)-苯基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮,
[2-氯-4-(1,2,4-三唑-1-基)-苯基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮,
(2-氯-4-吡咯-1-基-苯基)-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮,
(2-氯-4-吡唑-1-基-苯基)-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮,
[2-氯-4-(1H-咪唑-1-基)-苯基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮,
[2-氯-4-(3-甲基吡唑-1-基)-苯基]-(3-甲基-5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮,
[2-(3-甲基-吡唑-1-基)-4-三氟甲基-嘧啶-5-基)-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮,
[2-(4-甲基-吡唑-1-基)-4-三氟甲基-嘧啶-5-基)-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮,
1-[4-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-羰基)-苯基]-乙酮,
[4-(1H-吡唑-3-基)-苯基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮,
[4-(1-甲基-1H-吡唑-3-基)-苯基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮,
[4-(1-乙基-1H-吡唑-3-基)-苯基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮,
[4-(1-丙基-1H-吡唑-3-基)-苯基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮,
[4-(1-丁基-1H-吡唑-3-基)-苯基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮,
[4-(1-甲氧基甲基-1H-吡唑-3-基)-苯基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮,
1-{3-[4-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-羰基)-苯基]-吡唑-1-基}-乙酮,
1-{3-[4-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-羰基)-苯基]-吡唑-1-基}-丙-1-酮,
[4-(1-环丙烷羰基-1H-吡唑-3-基)-苯基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮,
1-{3-[4-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-羰基)-苯基]-吡唑-1-基}-丁-1-酮,
(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-羰基)-{4-[1-(噻吩-2-羰基)-1H-吡唑-3-基]苯基}-甲酮,
{4-[1-(5-氟-2-甲基-苯甲酰基)-1H-吡唑-3-基]-苯基}-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮,
{4-[1-(2-甲基-苯甲酰基)-1H-吡唑-3-基]-苯基}-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮,
{4-[1-(2-氯-4-氟-苯甲酰基)-1H-吡唑-3-基]-苯基}-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮,
{4-[1-(2,4-二氯-苯甲酰基)-1H-吡唑-3-基]-苯基}-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮,
2-(2,4-二氯-苯基)-1-{3-[4-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-羰基)-苯基]-吡唑-1-基}-乙酮,
{4-[1-(二苯基-2-羰基)-1H-吡唑-3-基]-苯基}-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮,
{4-[1-(4’-三氟甲基-二苯基-2-羰基)-1H-吡唑-3-基]-苯基}-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮,
[4-(5-甲基-1H-吡唑-3-基)-苯基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮,
(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-[4-(2H-[1,2,4]三唑-3-基)-苯基]-甲酮,
[4-(2-甲基-2H-[1,2,4]三唑-3-基)-苯基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮,
[4-(5-甲基-2H-[1,2,4]三唑-3-基)-苯基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮,
[4-(2,5-二甲基-2H-[1,2,4]三唑-3-基)-苯基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮,
[4-(3-甲基-[1,2,4]噁二唑-5-基)-苯基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮,
[4-(1-甲基-1H-吡唑-4-基)-苯基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮,
[6-(1-甲基-1H-吡唑-4-基)-吡啶-3-基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮,
[4-(吡唑-1-基)-苯基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮,
[4-(3-甲基-吡唑-1-基)-苯基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮,
[4-(4-甲基-吡唑-1-基)-苯基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮,
[4-(3,5-二甲基-吡唑-1-基)-苯基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10基)-甲酮,
(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-[4-(3-三氟甲基-吡唑-1-基)-苯基]-甲酮,
[4-(咪唑-1-基)-苯基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮,
[4-(4-甲基-咪唑-1-基)-苯基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮,
[2-氯-4-(1-甲基-1H-吡唑-3-基)-苯基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮,
[2-氯-4-(2-甲基-1H-吡唑-3-基)-苯基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮,
3-氯-4-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-羰基)-苄腈,
3-氯-4-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-羰基)-苯甲酸,
3-氯-4-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-羰基)-苯甲酰胺,
[2-氯-4-(5-甲基-2H-[1,2,4]三唑-3-基)苯基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-羰基)-甲酮,
[2-氯-4-(2H-1,2,4三唑-3-基)苯基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮,
[2-氯-4-(2-甲基-2H-[1,2,4]三唑-3-基)苯基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-羰基)-甲酮,
4-[(2,5-二甲基-2H-[1,2,4]三唑-3-基)-2-氯-苯基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-羰基)-甲酮,
[2-氯-4-(1H-四唑-5-基)苯基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮,
[2-氯-4-(3-甲基-吡唑-1-基)-苯基]-(3-二甲基氨基甲基-5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮,
(3-溴-5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-[2-氯-4-(3-甲基-吡唑-1-基)-苯基]-甲酮,
[2-溴-4-(3-甲基-吡唑-1-基)-苯基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮,
(2,4-二氟-苯基)-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮,
[2-氟-4-(3-甲基-吡唑-1-基)-苯基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮,
[4-(3-甲基吡唑-1-基)-2-三氟甲基苯基]-(5H,11H-吡唑并[5,1-c][1,4]苯并二氮杂-10-基)-甲酮,
(2-氯-6-吡唑-1-基-吡啶-3-基)(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮,
[2-氯-6-(3-甲基吡唑-1-基)-吡啶-3-基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮,
[2-氯-6-(4-甲基吡唑-1-基)-吡啶-3-基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮,
[2-氯-4-(3-甲基-1,2,4-三唑-1-基)-苯基](5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮,
[4-(3-甲基-1,2,4-三唑-1-基)-2-三氟甲基-苯基](5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮,
[2-甲氧基-4-(3-甲基-吡唑-1-基)-苯基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮,
(3-二甲基氨基甲基-5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-[2-甲氧基-4-(3-甲基-吡唑-1-基)-苯基]-甲酮,
[2-羟基-4-(3-甲基-吡唑-1-基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮,
[2-氯-4-(1-甲基-1H-吡唑-4-基)-苯基]-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮,
[2-氯-4-(3-甲基-吡唑-1-基)-苯基]-(4H,10H-吡唑并[5,1-c][1,4]苯并二氮杂-5-基)-甲酮,
[2-氯-4-(3-甲基-吡唑-1-基)-苯基]-(5,10-二氢-4H-四唑并[5,1-c][1,4]苯并二氮杂-5-基)-甲酮,
1-[4-(4H,10H-吡唑并[5,1-c][1,4]苯并二氮杂-5-羰基)-苯基]-乙酮,
[4-(1-甲基-1H-吡唑-3-基)-苯基]-(4H,10H-吡唑并[5,1-c][1,4]苯并二氮杂-5-基)-甲酮,
[4-(2-甲基-1H-吡唑-3-基)-苯基]-(4H,10H-吡唑并[5,1-c][1,4]苯并二氮杂-5-基)-甲酮,
1-[4-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-羰基)3-氯苯基]-乙酮,或
[2-氯-4-(3-甲基-4-乙炔基-苯基)-(5H,11H-吡咯并[2,1-c][1,4]苯并二氮杂-10-基)-甲酮。
13.一种用于治疗需要后叶加压素激动剂活性的哺乳动物疾病或症状的药物组合物,该药物组合物包括有效量的权利要求1-12中任一项的化合物,或其药物上可接受的盐、酯或前药形式,以及合适的药物载体。
14.根据权利要求13的药物组合物,其中所述需要后叶加压素激动剂活性的哺乳动物疾病或症状指尿崩症、夜间遗尿、夜尿症、尿失禁、出血和血凝固紊乱或不能暂时延迟排尿。
15.一种治疗需要后叶加压素激动剂活性的哺乳动物疾病或症状的方法,该方法包括给需要这种治疗的哺乳动物服用有效量的权利要求1-12中任一项的化合物或其药物学上可接受的盐、酯或前药形式,以及合适的药物载体。
16.根据权利要求15的方法,其中需要后叶加压素激动剂活性的哺乳动物疾病或症状指尿崩症、夜间遗尿、夜尿症、尿失禁、出血和血凝固紊乱或不能暂时延迟排尿。
17.权利要求1-12中任一项的化合物作为药物的用途。
18.权利要求1-12中任一项的化合物用于制备治疗尿崩症、夜间遗尿、夜尿症、尿失禁、出血和血凝固紊乱或不能暂时延迟排尿药物方面的用途。
19.制备式I化合物的方法,包括:
a)将式5化合物其中A、B、D、E、G、X、Y和R2分别各自如权利要求1所定义,
与羟胺或适当取代的式R4-NHNH2肼反应,其中R4如权利要求1中所定义,由此制得式I的目标化合物,其中R1是选自权利要求1中所定义的f,g和j的杂环基;
b)将式1化合物
其中D、E、G和X分别如权利要求1所定义,
与下式化合物反应,
其中A、B和Y分别如权利要求1所定义,J是酰化基,R10是烷基,由此
制得式I化合物,其中R1是链烷酰基;
c)将下式化合物
其中A、B、D、E、G、X和Y如权利要求1所定义,
与化合物HC=C-R9反应,其中R9如权利要求1所定义,由此
制得式I化合物,其中R1是-C=C-R9,其中R9如权利要求1所定义;
d)将下式化合物
其中A、B、D、E、G、X和Y分别如权利要求1所定义,
转化为相应的式I化合物,其中R1是链烷酰基;
e)将式1化合物
其中D、E、G和X如权利要求1所定义,
与式9的酰化剂反应,
其中A和B是碳,J是酰化基,R1是选自权利要求1中定义的g杂环基,而R2是氢,由此制得式I的目标化合物,其中A和B是碳,R1是选自权利要求1中定义的g杂环基,而R2是氢;
f)将式1化合物
其中D、E、G和X如权利要求1所定义,
与式9的酰化剂反应,
其中A和B如权利要求1所定义,J是酰化基,R1是选自权利要求1中所定义的f杂环基,而R2是氢,由此制得式I的目标化合物,其中R1是选自权利要求1中所定义的f杂环基,R2是氢;
g)将下式化合物
其中A、B、D、E、G、X和Y分别如权利要求1所定义,
与合适的式R1H化合物反应,其中R1是选自权利要求1中所定义的a,b,c,d,l,n和o杂环基,由此制得式I的目标化合物,其中R1是选自权利要求1中所定义的a,b,c,d,l,n和o的杂环基,
h)将式1化合物
其中D、E、G和X如权利要求1所定义,
与式9的酰化剂反应,
其中A和B如权利要求1所定义,J是酰化基,R1是选自权利要求1中所定义的a,b,c,d,l,n和o的杂环基;由此
制得式I的目标化合物,其中R1是选自权利要求1中所定义的a,b,c,d,l,n和o的杂环基;
i)将式45化合物
其中A、B、D、E、G、X、Y和R2分别各自如权利要求1所定义,
转化为相应的式I化合物,其中R1选自权利要求1中定义的e,i和k的杂环基;
j)将式I化合物
其中D、E、G和X如权利要求1所定义,
与式43化合物反应
其中A、B和Y如权利要求1所定义,J是酰化剂,由此
制得式I的目标化合物,其中R1是CN或CONH2;
k)将式I游离酸化合物转化为相应的药物上可接受的盐;或
l)将一种式I化合物转化为另一种化合物。
20.根据权利要求19中的方法,其中步骤l包括:
i)将式I化合物,其中R1是基团-C≡CR9,R9如权利要求1所定义,转化为相应的化合物,其中R1是链烷酰基;
ii)将式I化合物,其中R1是权利要求1中定义的杂环基,其包括的R2或R4是氢,进行烷基化或酰基化,制得相应的式I化合物,其中R2或R4是氢以外的其它基团;
iii)将Y是烷氧基的式I化合物,转化为相应的Y是羟基的式I化合物;
iv)将R1是CN的式I化合物,转化为相应的其中R1是权利要求1中定义的杂环基团m的式I化合物;
v)将式I化合物,其中R1是选自权利要求1中定义的a,c,e,f,g,h,i,j,k,l,m,n和o的杂环基,D是CW,而W是氢,转化为相应的式I化合物,其中D是CW,而W是卤素或基团CH2NR6R7;或
vi)将R1是CN的式I化合物,转化为R1是CONH2的式I化合物。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US90336997A | 1997-07-30 | 1997-07-30 | |
| US08/903369 | 1997-07-30 | ||
| US08/903,369 | 1997-07-30 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN1272111A true CN1272111A (zh) | 2000-11-01 |
| CN1183134C CN1183134C (zh) | 2005-01-05 |
Family
ID=25417392
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CNB988096412A Expired - Fee Related CN1183134C (zh) | 1997-07-30 | 1998-07-24 | 三环后叶加压素激动剂 |
Country Status (22)
| Country | Link |
|---|---|
| EP (1) | EP1000062B1 (zh) |
| JP (1) | JP4338305B2 (zh) |
| KR (1) | KR100580805B1 (zh) |
| CN (1) | CN1183134C (zh) |
| AR (1) | AR016557A1 (zh) |
| AT (1) | ATE277050T1 (zh) |
| AU (1) | AU756959B2 (zh) |
| BR (1) | BR9811585A (zh) |
| CA (1) | CA2297406C (zh) |
| DE (1) | DE69826487T2 (zh) |
| DK (1) | DK1000062T3 (zh) |
| ES (1) | ES2229525T3 (zh) |
| HU (1) | HUP0002480A3 (zh) |
| IL (1) | IL133788A0 (zh) |
| NO (1) | NO315273B1 (zh) |
| NZ (1) | NZ502449A (zh) |
| PT (1) | PT1000062E (zh) |
| RU (1) | RU2213094C2 (zh) |
| SI (1) | SI1000062T1 (zh) |
| TW (1) | TW502035B (zh) |
| WO (1) | WO1999006409A1 (zh) |
| ZA (1) | ZA986784B (zh) |
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN102884065A (zh) * | 2010-05-10 | 2013-01-16 | 霍夫曼-拉罗奇有限公司 | 杂芳基-环己基-四氮杂苯并[e]薁 |
| US9376424B2 (en) | 2010-07-01 | 2016-06-28 | Azevan Pharmaceuticals, Inc. | Methods for treating post traumatic stress disorder |
| CN105712985A (zh) * | 2001-10-12 | 2016-06-29 | 阿泽范药品公司 | β-内酰胺后叶加压素V1a拮抗剂 |
| US9597314B2 (en) | 2005-03-22 | 2017-03-21 | Azevan Pharmaceuticals, Inc. | Beta-lactamylalkanoic acids for treating premenstrual disorders |
| US9802925B2 (en) | 2014-03-28 | 2017-10-31 | Azevan Pharmaceuticals, Inc. | Compositions and methods for treating neurodegenerative diseases |
| US11628160B2 (en) | 2017-09-15 | 2023-04-18 | Azevan Pharmaceuticals, Inc. | Compositions and methods for treating brain injury |
Families Citing this family (27)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7138393B2 (en) * | 1998-07-24 | 2006-11-21 | Wyeth | Biologically active vasopressin agonist metabolites |
| ME00129B (me) * | 1999-01-19 | 2010-10-10 | Ortho Mcneil Janssen Pharmaceuticals Inc | Triciklicnj benzodiazepini kao antagonisti receptora vazopresina |
| JP2003510280A (ja) * | 1999-09-27 | 2003-03-18 | アメリカン・サイアナミド・カンパニー | バソプレッシンアゴニスト処方および製法 |
| US6831079B1 (en) * | 1999-09-27 | 2004-12-14 | American Cyanamid Company | Vasopressin agonist formulation and process |
| GB0000079D0 (en) | 2000-01-05 | 2000-02-23 | Ferring Bv | Novel antidiuretic agents |
| GB0015601D0 (en) | 2000-06-26 | 2000-08-16 | Ferring Bv | Novel antidiuretic agents |
| US6977254B2 (en) | 2001-04-12 | 2005-12-20 | Wyeth | Hydroxy cyclohexenyl phenyl carboxamides tocolytic oxytocin receptor antagonists |
| US6900200B2 (en) | 2001-04-12 | 2005-05-31 | Wyeth | Tricyclic hydroxy carboxamides and derivatives thereof tocolytic oxytocin receptor antagonists |
| US6903087B2 (en) | 2001-04-12 | 2005-06-07 | Wyeth | Pyrido cyclohexenyl phenyl carboxamides tocolytic oxytocin receptor antagonists |
| US7326700B2 (en) | 2001-04-12 | 2008-02-05 | Wyeth | Cyclohexenyl phenyl carboxamides tocolytic oxytocin receptor antagonists |
| US7109193B2 (en) | 2001-04-12 | 2006-09-19 | Wyeth | Tricyclic diazepines tocolytic oxytocin receptor antagonists |
| US7202239B2 (en) | 2001-04-12 | 2007-04-10 | Wyeth | Cyclohexylphenyl carboxamides tocolytic oxytocin receptor antagonists |
| US7022699B2 (en) | 2001-04-12 | 2006-04-04 | Wyeth | Cyclohexenyl phenyl diazepines vasopressin and oxytocin receptor modulators |
| US7064120B2 (en) | 2001-04-12 | 2006-06-20 | Wyeth | Tricyclic pyridyl carboxamides and derivatives thereof tocolytic oxytocin receptor antagonists |
| RU2338747C2 (ru) * | 2003-03-31 | 2008-11-20 | Каунсил Оф Сайентифик Энд Индастриал Рисерч | ДИМЕРЫ ПИРРОЛО [2, 1-c][1, 4] БЕНЗОДИАЗЕПИНА В КАЧЕСТВЕ ПРОТИВООПУХОЛЕВЫХ СРЕДСТВ И СПОСОБ ИХ ПОЛУЧЕНИЯ |
| SI1619185T1 (sl) | 2003-04-28 | 2011-09-30 | Astellas Pharma Inc | 4,4-difluoro-1,2,3,4-tetrahidro-5H-1-benzazepinski derivat ali njegova sol |
| GB0406917D0 (en) | 2004-03-26 | 2004-04-28 | Photocure Asa | Compounds |
| EP1627876A1 (en) * | 2004-08-20 | 2006-02-22 | Ferring B.V. | Heterocyclic condensed compounds useful as antidiuretic agents |
| CA2578370A1 (en) * | 2004-08-25 | 2006-03-02 | Pfizer Inc. | Triazolobenzodiazepines and their use as vasopressin antagonists |
| PT2356123E (pt) | 2008-11-13 | 2012-10-31 | Hoffmann La Roche | Espiro-5,6-di-hidro-4h-2,3,5,10b-tetra-azabenzo[ e]azulenos |
| EP2308866A1 (de) | 2009-10-09 | 2011-04-13 | Bayer CropScience AG | Phenylpyri(mi)dinylpyrazole und ihre Verwendung als Fungizide |
| US8420633B2 (en) * | 2010-03-31 | 2013-04-16 | Hoffmann-La Roche Inc. | Aryl-cyclohexyl-tetraazabenzo[e]azulenes |
| GB201005623D0 (en) | 2010-04-01 | 2010-05-19 | Vantia Ltd | New polymorph |
| US8461151B2 (en) * | 2010-04-13 | 2013-06-11 | Hoffmann-La Roche Inc. | Aryl-/heteroaryl-cyclohexenyl-tetraazabenzo[e]azulenes |
| US8481528B2 (en) * | 2010-04-26 | 2013-07-09 | Hoffmann-La Roche Inc. | Heterobiaryl-cyclohexyl-tetraazabenzo[e]azulenes |
| AR094929A1 (es) | 2013-02-28 | 2015-09-09 | Bristol Myers Squibb Co | Derivados de fenilpirazol como inhibidores potentes de rock1 y rock2 |
| CN105102448B (zh) | 2013-02-28 | 2018-03-06 | 百时美施贵宝公司 | 作为rock1和rock2抑制剂的苯基吡唑衍生物 |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5516774A (en) * | 1993-07-29 | 1996-05-14 | American Cyanamid Company | Tricyclic diazepine vasopressin antagonists and oxytocin antagonists |
| US5521173A (en) * | 1995-01-17 | 1996-05-28 | American Home Products Corporation | Tricyclic benzazepine vasopressin antagonists |
| TW359669B (en) * | 1995-12-15 | 1999-06-01 | Otsuka Pharma Co Ltd | Benzazepine derivatives |
-
1998
- 1998-07-24 BR BR9811585-5A patent/BR9811585A/pt not_active IP Right Cessation
- 1998-07-24 DK DK98938017T patent/DK1000062T3/da active
- 1998-07-24 NZ NZ502449A patent/NZ502449A/xx unknown
- 1998-07-24 WO PCT/US1998/015495 patent/WO1999006409A1/en not_active Ceased
- 1998-07-24 CN CNB988096412A patent/CN1183134C/zh not_active Expired - Fee Related
- 1998-07-24 DE DE69826487T patent/DE69826487T2/de not_active Expired - Fee Related
- 1998-07-24 AU AU86633/98A patent/AU756959B2/en not_active Ceased
- 1998-07-24 RU RU2000104865/04A patent/RU2213094C2/ru not_active IP Right Cessation
- 1998-07-24 JP JP2000505167A patent/JP4338305B2/ja not_active Expired - Fee Related
- 1998-07-24 SI SI9830693T patent/SI1000062T1/xx unknown
- 1998-07-24 PT PT98938017T patent/PT1000062E/pt unknown
- 1998-07-24 EP EP98938017A patent/EP1000062B1/en not_active Expired - Lifetime
- 1998-07-24 IL IL13378898A patent/IL133788A0/xx unknown
- 1998-07-24 KR KR1020007000977A patent/KR100580805B1/ko not_active Expired - Fee Related
- 1998-07-24 AT AT98938017T patent/ATE277050T1/de not_active IP Right Cessation
- 1998-07-24 HU HU0002480A patent/HUP0002480A3/hu unknown
- 1998-07-24 ES ES98938017T patent/ES2229525T3/es not_active Expired - Lifetime
- 1998-07-24 CA CA002297406A patent/CA2297406C/en not_active Expired - Fee Related
- 1998-07-28 TW TW087112353A patent/TW502035B/zh not_active IP Right Cessation
- 1998-07-29 ZA ZA9806784A patent/ZA986784B/xx unknown
- 1998-07-29 AR ARP980103736A patent/AR016557A1/es unknown
-
2000
- 2000-01-18 NO NO20000242A patent/NO315273B1/no unknown
Cited By (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN105712985A (zh) * | 2001-10-12 | 2016-06-29 | 阿泽范药品公司 | β-内酰胺后叶加压素V1a拮抗剂 |
| US9597314B2 (en) | 2005-03-22 | 2017-03-21 | Azevan Pharmaceuticals, Inc. | Beta-lactamylalkanoic acids for treating premenstrual disorders |
| CN102884065A (zh) * | 2010-05-10 | 2013-01-16 | 霍夫曼-拉罗奇有限公司 | 杂芳基-环己基-四氮杂苯并[e]薁 |
| CN102884065B (zh) * | 2010-05-10 | 2015-11-25 | 霍夫曼-拉罗奇有限公司 | 杂芳基-环己基-四氮杂苯并[e]薁 |
| US9376424B2 (en) | 2010-07-01 | 2016-06-28 | Azevan Pharmaceuticals, Inc. | Methods for treating post traumatic stress disorder |
| US9987265B2 (en) | 2010-07-01 | 2018-06-05 | Azevan Pharmaceuticals, Inc. | Methods for treating post traumatic stress disorder |
| US10953001B2 (en) | 2010-07-01 | 2021-03-23 | Azevan Pharmaceuticals, Inc. | Methods for treating post traumatic stress disorder |
| US9802925B2 (en) | 2014-03-28 | 2017-10-31 | Azevan Pharmaceuticals, Inc. | Compositions and methods for treating neurodegenerative diseases |
| US10364236B2 (en) | 2014-03-28 | 2019-07-30 | Azevan Pharmaceuticals, Inc. | Compositions and methods for treating neurodegenerative diseases |
| US11319306B2 (en) | 2014-03-28 | 2022-05-03 | Azevan Pharmaceuticals, Inc. | Compositions and methods for treating neurodegenerative diseases |
| US11628160B2 (en) | 2017-09-15 | 2023-04-18 | Azevan Pharmaceuticals, Inc. | Compositions and methods for treating brain injury |
Also Published As
| Publication number | Publication date |
|---|---|
| CA2297406C (en) | 2009-02-24 |
| RU2213094C2 (ru) | 2003-09-27 |
| EP1000062B1 (en) | 2004-09-22 |
| ES2229525T3 (es) | 2005-04-16 |
| SI1000062T1 (en) | 2004-12-31 |
| DE69826487T2 (de) | 2005-12-01 |
| BR9811585A (pt) | 2000-09-26 |
| AU756959B2 (en) | 2003-01-30 |
| CN1183134C (zh) | 2005-01-05 |
| DE69826487D1 (de) | 2004-10-28 |
| AU8663398A (en) | 1999-02-22 |
| PT1000062E (pt) | 2004-12-31 |
| ZA986784B (en) | 2000-06-22 |
| AR016557A1 (es) | 2001-07-25 |
| IL133788A0 (en) | 2001-04-30 |
| ATE277050T1 (de) | 2004-10-15 |
| EP1000062A1 (en) | 2000-05-17 |
| NZ502449A (en) | 2002-08-28 |
| JP4338305B2 (ja) | 2009-10-07 |
| DK1000062T3 (da) | 2004-11-22 |
| HUP0002480A3 (en) | 2002-09-30 |
| JP2001512125A (ja) | 2001-08-21 |
| TW502035B (en) | 2002-09-11 |
| HUP0002480A2 (hu) | 2000-11-28 |
| NO20000242D0 (no) | 2000-01-18 |
| NO20000242L (no) | 2000-03-06 |
| KR20010022398A (ko) | 2001-03-15 |
| CA2297406A1 (en) | 1999-02-11 |
| KR100580805B1 (ko) | 2006-05-17 |
| WO1999006409A1 (en) | 1999-02-11 |
| NO315273B1 (no) | 2003-08-11 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN1272111A (zh) | 三环后叶加压素激动剂 | |
| CN1052725C (zh) | 三环加压素拮抗剂 | |
| CN1039908C (zh) | 三环二氮杂䓬加压素拮抗剂和催产素拮抗剂 | |
| CN1065244C (zh) | 三环苯并氮杂䓬加压素拮抗剂 | |
| CN1273469C (zh) | 作为保胎用催产素受体拮抗剂的三环二氮杂䓬 | |
| CN1177848C (zh) | 可用作lh激动剂的二环杂芳族化合物 | |
| CN1061658C (zh) | 三环苯并吖庚因加压素拮抗剂 | |
| CN1313450C (zh) | 速激肽受体拮抗剂 | |
| CN1646502A (zh) | 用作速激肽受体拮抗剂的三唑衍生物 | |
| CN1926139A (zh) | 稠合吡唑衍生物 | |
| CN1827603A (zh) | 作用于大麻受体的新型吡唑类似物 | |
| CN1099755A (zh) | 环状衍生物、含该衍生物的药物组合物及制备方法 | |
| CN1615300A (zh) | 苯并咪唑衍生物以及它们作为gnrh拮抗剂的用途 | |
| CN1708492A (zh) | 经选择cgrp拮抗剂、其制法及作为药物的用途 | |
| CN1231666A (zh) | 三环苯并氮杂䓬血管加压素拮抗剂 | |
| CN1529697A (zh) | 用作血管紧张素ⅱ激动剂的三环化合物 | |
| CN1072219C (zh) | 二氮杂䓬酮、其生产和用途 | |
| CN1768061A (zh) | 作为大麻素受体配体的吡唑并[1,5-a][1,3,5]三嗪衍生物 | |
| CN1976916A (zh) | 选择的cgrp-拮抗剂,其制备方法以及它们作为药物的用途 | |
| CN1105993A (zh) | 取代的唑类化合物的制备方法 | |
| CN1708493A (zh) | 选择的cgrp拮抗剂、其制法及作为药物的用途 | |
| CN1040748C (zh) | 苯并吖庚因衍生物及其药物组合物和中间体 | |
| CN1798738A (zh) | 作为钠通道阻滞剂的联芳基取代吡唑 | |
| CN1603314A (zh) | 作为cdk2抑制剂的吡唑并苯并二氮䓬 | |
| CN1761670A (zh) | 新化合物 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C14 | Grant of patent or utility model | ||
| GR01 | Patent grant | ||
| C17 | Cessation of patent right | ||
| CF01 | Termination of patent right due to non-payment of annual fee |
Granted publication date: 20050105 Termination date: 20090824 |