CN1280302C - 对抗真菌活性有增强作用的大环内酯衍生物 - Google Patents
对抗真菌活性有增强作用的大环内酯衍生物 Download PDFInfo
- Publication number
- CN1280302C CN1280302C CNB028081358A CN02808135A CN1280302C CN 1280302 C CN1280302 C CN 1280302C CN B028081358 A CNB028081358 A CN B028081358A CN 02808135 A CN02808135 A CN 02808135A CN 1280302 C CN1280302 C CN 1280302C
- Authority
- CN
- China
- Prior art keywords
- mmol
- activity
- antifungal agents
- methanol
- crude product
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H17/00—Compounds containing heterocyclic radicals directly attached to hetero atoms of saccharide radicals
- C07H17/04—Heterocyclic radicals containing only oxygen as ring hetero atoms
- C07H17/08—Hetero rings containing eight or more ring members, e.g. erythromycins
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7042—Compounds having saccharide radicals and heterocyclic rings
- A61K31/7048—Compounds having saccharide radicals and heterocyclic rings having oxygen as a ring hetero atom, e.g. leucoglucosan, hesperidin, erythromycin, nystatin, digitoxin or digoxin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/10—Antimycotics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Animal Behavior & Ethology (AREA)
- Molecular Biology (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Engineering & Computer Science (AREA)
- Epidemiology (AREA)
- Genetics & Genomics (AREA)
- Biotechnology (AREA)
- Biochemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Communicable Diseases (AREA)
- Oncology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Saccharide Compounds (AREA)
Abstract
本发明涉及对唑类抗真菌剂的活性具有增强作用、在低浓度下和短期内对真菌感染起作用而且可降低耐药性微生物的出现率的物质。所述物质之一是式[I]所示化合物:其中R1为Ac时,R2和R3分别为Ac,R4为Me;R1为H时,R2和R3分别为Ac,R4为Me;R1为H时,R2和R3分别为Ac,R4为H;R1为Bz1时,R2和R3分别为Bz1,R4为Me;R1为Ac时,R2和R3分别为Pr,R4为Me;R1为Ac时,R2和R3分别为Hex,R4为Me;R1为Ac时,R2和R3分别为Bz1,R4为Me;R1为H时,R2和R3分别为Pr,R4为Me;R1为H时,R2和R3分别为Hex,R4为Me;R1为H时,R2和R3分别为Bz1,R4为Me;R1为H时,R2为H,R3为Bz1,R4为Me;R1为H时,R2和R3分别为Hex,R4为H;或R1为H时,R2和R3分别为Hex,R4为Et。
Description
技术领域
本发明涉及增强抗真菌剂用于真菌感染的效果的物质。更具体地,本发明涉及对抗真菌活性有增强作用的新大环内酯衍生物,与用于伴有免疫减弱病症的真菌感染如HIV感染和血液病的化学疗法的唑类抗真菌剂组合可增强抗真菌效果。
背景技术
用于治疗真菌感染的已知唑类(azole)化合物的例子是1-[2-(2,4-二氯苄氧基)-2-(2,4-二氯苯基)乙基]咪唑(非商标名称(generic name):咪康唑,Sigma Inc.,U.S.)、2,4-二氟-α,α-双(1H-1,2,4-三唑-1-基甲基)苄醇(非商标名称:氟康唑,ICNPharmaceuticals Inc.,U.S.)、和(±)-1-仲丁基-4-[p-[4-[p-[[(2R,4S)-2-(2,4-二氯苯基)-2-(1H-1,2,4-三唑-1-基乙基)-1,3-二氧戊环-4-基]甲氧基]苯基]-1-哌嗪基]苯基]-Δ2-1,2,4-三唑啉-5-酮(非商标名称:伊曲康唑,Kyowa Hakko Co.,Japan)。
与用于真菌感染的多烯类抗真菌剂即1R,3S,5R,6R,9R,11R,15S,16R,17R,18S,19E,21E,23E,25E,27E,31E,33R,35S,36S,37S-33-(3-氨基-3,6-二脱氧-β-D-吡喃甘露糖基氧基)-1,3,5,6,9,11,17,37-八羟基-15,16,18-三甲基-13-氧-14,39-二氧双环(doxoabicyclo)[33.3.1]三十九碳-19,21,23,25,27,29,31-七烯-36-羧酸(非商标名称:两性霉素B)相比,唑类化合物很安全,而经常使用(Anaissie E.J.et al.,Clinical Infectious Diseases,23,964-972,1996)。
最近,因长期或经常使用唑类抗真菌剂导致出现耐药性菌株的问题,需要开发耐药性微生物的出现率低而且很安全的药物。但主要以杀真菌或抑制真菌作用为目标进行抗真菌剂的开发,从增强抗真菌剂活性的观点尚未创造出新药剂。
发明概述和目的
在伴有免疫减弱病症的疾病如HIV感染和血液病中,产生缺乏免疫力的病症,结果真菌感染的发病率随机会感染增加。许多伴有免疫减弱病症的疾病病例很严重,需要长期治疗。为此,需要长期进行真菌感染的化学疗法,经常使用唑类抗真菌剂容易诱导耐药性。
已提出的对唑类抗真菌剂的耐药性机理是:例如,白色念珠菌中P450 14-α-脱甲基酶(目标酶)的过度表达,和因氨基酸突变导致与药物的亲和力减小(Vanden Bossche,H.et al.Antimicrob.Agents and Chemoth.,36,2602-2610,1992;Sanglard,D.etal.ibid.42,241-253,1998);多种药物排泄转运蛋白如MSF(主要促进蛋白超家族)和ABC(ATP结合盒)的作用使细胞内药物浓度下降(Fling,M.E.et al.,Molecular Genetics and Genomics,227,318-329,1991;Sanglard,D.et al.,Microbiology,143,405-416,1997)。
酿酒酵母的脂类代谢作用中涉及MDR(多耐药性)基因PDR16和PDR17,在缺少这些基因的情况下,微生物可能对唑类化合物很敏感(H.Bart van den Hazel et al.J.Biol.Chem.,274,1934-1941,1999)。
因此,预计能提高唑类抗真菌剂活性的药物可减小药物剂量和缩短给药期,从而可降低耐药性微生物的出现率。同时,预计有不同骨架结构的两类药物组合使用或用于对唑类化合物具有耐药性菌株的此类药物组合使用可克服对唑类抗真菌剂的耐药性。因此,认为提供对唑类抗真菌剂的活性有增强作用的药物可用于预防和治疗因深部大片霉菌病等引起的真菌感染和吡咯耐药性真菌感染。
在此情况下,我们广泛地研究了各种大环内酯衍生物增强唑类抗真菌剂活性的作用,发现有与用于治疗真菌病的已知唑类抗真菌剂不同的新骨架结构的大环内酯衍生物对唑类抗真菌剂活性的增强作用,从而完成本发明。
本发明的目的是提供对抗真菌剂的活性具有增强作用、在低浓度下和短期内对真菌感染起作用而且可降低耐药性微生物的出现率的新大环内酯衍生物。
本发明还提供式[I]所示对抗真菌剂的活性具有增强作用的新大环内酯衍生物:

其中R1为Ac时,R2和R3分别为Ac,R4为Me;
当R1为H时,R2和R3分别为Ac,R4为Me;
当R1为H时,R2和R3分别为Ac,R4为H;
当R1为苯甲酰基时,R2和R3分别为苯甲酰基,R4为Me;
当R1为Ac时,R2和R3分别为丙酰基,R4为Me;
当R1为Ac时,R2和R3分别为己酰基,R4为Me;
当R1为Ac时,R2和R3分别为苯甲酰基,R4为Me;
当R1为H时,R2和R3分别为丙酰基,R4为Me;
当R1为H时,R2和R3分别为己酰基,R4为Me;
当R1为H时,R2和R3分别为苯甲酰基,R4为Me;
当R1为H时,R2为H,R3为苯甲酰基,R4为Me;
当R1为H时,R2和R3分别为己酰基,R4为H;或
当R1为H时,R2和R3分别为己酰基,R4为Et。
上式[I]所示化合物具有增强唑类抗真菌剂抗白色念珠菌和黑曲霉(二者均包括耐药性菌株)的活性的作用。本发明提供对抗真菌剂抗例如白色念珠菌如白色念珠菌ATCC 64548或白色念珠菌ATCC 64550和黑曲霉如黑曲霉ATCC 6275的活性有增强作用的新大环内酯衍生物。
本发明还提供式[II]所示对抗真菌剂的活性具有增强作用的新大环内酯衍生物:
其中R1为Ac时,R2为苯磺酰基;R1为Ac时,R2为苄基磺酰基;或R1为H时,R2为苄基磺酰基。
上式[II]所示化合物具有增强唑类抗真菌剂抗白色念珠菌和黑曲霉(二者均包括耐药性菌株)的活性的作用。
本发明提供对抗真菌剂抗例如白色念珠菌如白色念珠菌ATCC64550和黑曲霉如黑曲霉ATCC 6275的活性有增强作用的新大环内酯衍生物。本发明还提供式[III]所示对抗真菌剂的活性具有增强作用的新大环内酯衍生物:

其中R1是


上式[III]所示化合物具有增强唑类抗真菌剂抗白色念珠菌和黑曲霉(二者均包括耐药性菌株)的活性的作用。本发明提供对抗真菌剂抗例如白色念珠菌如白色念珠菌ATCC 64548和黑曲霉如黑曲霉ATCC6275的活性有增强作用的新大环内酯衍生物。
本发明还提供式[IV]所示对抗真菌剂的活性具有增强作用的新大环内酯衍生物:
其中R1为H或Me。
上式[IV]所示化合物具有增强唑类抗真菌剂抗白色念珠菌和黑曲霉(二者均包括耐药性菌株)的活性的作用。本发明提供对抗真菌剂抗例如白色念珠菌如白色念珠菌ATCC 64548和黑曲霉如黑曲霉ATCC6275的活性有增强作用的新大环内酯衍生物。
本发明还提供式[V]所示对抗真菌剂的活性具有增强作用的新大环内酯衍生物:

其中R1为H或Me。
上式[V]所示化合物具有增强唑类抗真菌剂抗白色念珠菌和黑曲霉(二者均包括耐药性菌株)的活性的作用。本发明提供对抗真菌剂抗例如白色念珠菌如白色念珠菌ATCC 64548和黑曲霉如黑曲霉ATCC6275的活性有增强作用的新大环内酯衍生物。
本发明还提供式[VI]所示对抗真菌剂的活性具有增强作用的新大环内酯衍生物:

上式[VI]所示化合物具有增强唑类抗真菌剂抗白色念珠菌和黑曲霉(二者均包括耐药性菌株)的活性的作用。本发明提供对抗真菌剂抗例如白色念珠菌如白色念珠菌ATCC 64548和黑曲霉如黑曲霉ATCC6275的活性有增强作用的新大环内酯衍生物。
本发明还提供式[VII]所示对抗真菌剂的活性具有增强作用的新大环内酯衍生物:
上式[VII]所示化合物具有增强唑类抗真菌剂抗白色念珠菌和黑曲霉(二者均包括耐药性菌株)的活性的作用。本发明提供对抗真菌剂抗例如白色念珠菌如白色念珠菌ATCC 64548和黑曲霉如黑曲霉ATCC6275的活性有增强作用的新大环内酯衍生物。
本发明还提供式[VIII]所示对抗真菌剂的活性具有增强作用的新大环内酯衍生物:

上式[VIII]所示化合物具有增强唑类抗真菌剂抗白色念珠菌的活性的作用。本发明提供对抗真菌剂抗例如白色念珠菌如白色念珠菌ATCC 64548的活性有增强作用的新大环内酯衍生物。
本发明还提供式[IX]所示对抗真菌剂的活性具有增强作用的新大环内酯衍生物:

上式[IX]所示化合物具有增强唑类抗真菌剂抗白色念珠菌的活性的作用。本发明提供对抗真菌剂抗例如白色念珠菌如白色念珠菌ATCC 64548的活性有增强作用的新大环内酯衍生物。
本发明还提供式[X]所示对抗真菌剂的活性具有增强作用的新大环内酯衍生物:

上式[X]所示化合物具有增强唑类抗真菌剂抗白色念珠菌和黑曲霉(二者均包括耐药性菌株)的活性的作用。本发明提供对抗真菌剂抗例如白色念珠菌如白色念珠菌ATCC 64548和黑曲霉如黑曲霉ATCC6275的活性有增强作用的新大环内酯衍生物。
本发明还提供式[XI]所示对抗真菌剂的活性具有增强作用的新大环内酯衍生物:
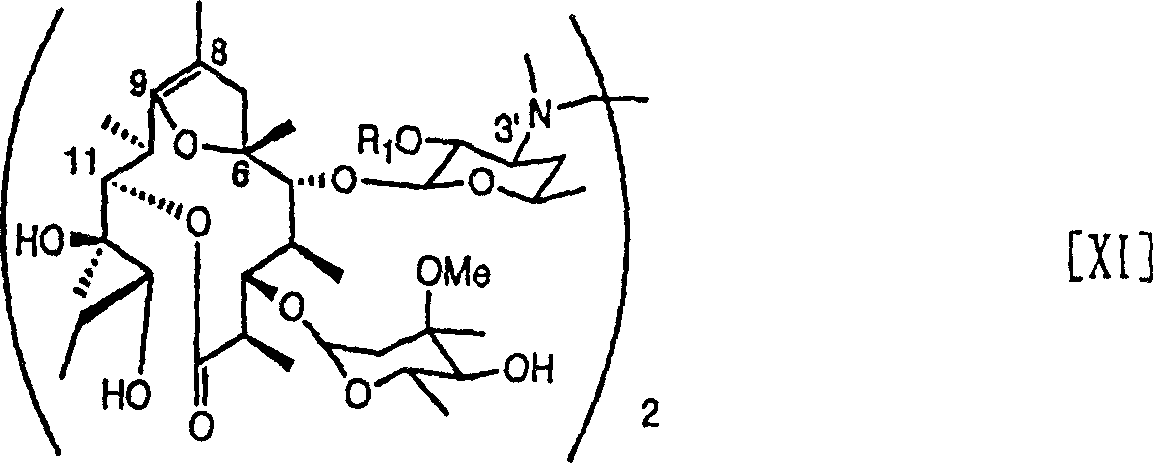
上式[XI]所示化合物具有增强唑类抗真菌剂抗白色念珠菌的活性的作用。本发明提供对抗真菌剂抗例如白色念珠菌如白色念珠菌ATCC 64548的活性有增强作用的新大环内酯衍生物。
本发明提供选自式[I]至[XI]所示化合物的任一化合物用于生产增强抗真菌剂预防或治疗由HIV感染或血液病引起的伴有免疫减弱病症的真菌感染的活性的药物的用途。本发明还提供选自式[I]至[XI]所示化合物的任一化合物用于生产增强抗真菌剂预防或治疗由HIV感染或血液病引起的伴有免疫减弱病症的真菌感染的活性的药物的物质。
以下列出本发明式[I]所示大环内酯衍生物的化合物号、式中的R1、R2、R3和R4、及实施例号。
| 化合物号 | R 1 | R 2 | R 3 | R 4 | 实施例号 |
| EM719EM755EM756EM770EM771EM772EM773EM776EM777EM778EM779EM852EM853 | AcHHBzlAcAcAcHHHHHH | AcAcAcBzlPrHexBzlPrHexBzlHHexHex | AcAcAcBzlPrHexBzlp rHexBzlBzlHexHex | MeMeHMeMeMeMeMeMeMeMeHEt | 12345106711891213 |
以下列出本发明式[II]所示大环内酯衍生物的化合物号、式中的R1和R2、及实施例号。
| 化合物号 | R 1 | R 2 | 实施例号 |
| EM774EM775EM780 | AcAcH | SO2PhSO2BnSO2Bn | 141516 |
以下列出本发明式[III]所示大环内酯衍生物的化合物号、式中的R1、及实施例号。
以下列出本发明式[IV]所示大环内酯衍生物的化合物号、式中的R1、及实施例号。
| 化合物号 | R 1 | 实施例号 |
| EM752EM753 | HMe | 2021 |
以下列出本发明式[V]所示大环内酯衍生物的化合物号、式中的R1、及实施例号。
| 化合物号 | R 1 | 实施例号 |
| EM757EM758 | HMe | 2223 |
以下列出本发明式[VI]所示大环内酯衍生物的化合物号、及实施例号。
| 化合物号 | R 1 | 实施例号 |
| EM759 | 24 |
以下列出本发明式[VII]所示大环内酯衍生物的化合物号、及实施例号。
| 化合物号 | 实施例号 |
| EM760 | 25 |
以下列出本发明式[VIII]所示大环内酯衍生物的化合物号、及实施例号。
| 化合物号 | 实施例号 |
| EM761 | 26 |
以下列出本发明式[IX]所示大环内酯衍生物的化合物号、及实施例号。
| 化合物号 | 实施例号 |
| EM764 | 27 |
以下列出本发明式[X]所示大环内酯衍生物的化合物号、及实施例号。
| 化合物号 | 实施例号 |
| EM765 | 28 |
以下列出本发明式[XI]所示大环内酯衍生物的化合物号、及实施例号。
| 化合物号 | 实施例号 |
| EM741 | 29 |
优选实施方案详述
用实施例详细解释本发明,但不能解释本发明限于这些实施例。
参考例1
8,9-脱水-假红霉素(pseudoerythromycin)A 6,9-半缩酮(EM701)的合成
EM701的合成详细地公开在WO02/14338A1中。将红霉素的冰醋酸溶液在室温下搅拌2小时,缓慢加入碳酸氢钠水溶液中和。将该反应混合物用氯仿萃取。通过添加无水硫酸钠使有机层脱水,过滤除去硫酸钠得到粗产品。所述粗产品通过硅胶柱色谱法用氯仿-甲醇-氨水(10∶0.5∶0.01→10∶1∶0.05)纯化得到红霉素A烯醇醚。向红霉素A烯醇醚的甲醇溶液中加入碳酸钾,回流2小时。除去溶剂后,使残余物溶于碳酸氢钠水溶液,将所述混合物用氯仿萃取。用硫酸钠使提取物脱水,过滤和蒸馏。所得粗产品通过硅胶柱色谱法用氯仿-甲醇-氨水(10∶0.5∶0.01→10∶1∶0.05)纯化得到EM701(白色粉末)。
实施例1
2’,4”,13-三-0-乙酰基-8,9-脱水-假红霉素A 6,9-半缩酮(EM719)的合成
向EM701(165.4mg,0.231mmol)的吡啶(2.3ml)溶液中加入乙酸酐(327.0μl),将该混合物在室温下搅拌96小时。向混合物中加纯水,用二氯甲烷萃取。有机层用饱和盐水洗涤,经无水硫酸钠干燥,过滤,除去溶剂得到粗产品。所述粗产品通过硅胶柱色谱法用氯仿-甲醇-氨水(10∶0.5∶0.01→10∶1∶0.05)纯化得到EM719(118.5mg,60%,白色粉末)。
EM719:M.p.:107-109℃;
IR(KBr)ν:3467.4,2973.7,2935.1,2879.2,1700.9,1637.3,1457.9,1380.8,1265.1,1166.7,1126.2,1079.9,1037.5,1016.3cm-1;
HRMS(FAB)m/z:C35H61NO12Na[M+Na]+的计算值为710.4091,实测值为710.4060。
实施例2
4”,13-二-O-乙酰基-8,9-脱水-假红霉素A 6,9-半缩酮(EM755)的合成
向EM701(165.4mg,0.231mmol)的吡啶(2.3ml)溶液中加入乙酸酐(327μl),将该混合物在室温下搅拌96小时。向混合物中加纯水,用二氯甲烷萃取。有机层用饱和盐水洗涤,经无水硫酸钠干燥,过滤,除去溶剂得到粗产品。所述粗产品通过硅胶柱色谱法用氯仿-甲醇-氨水(10∶0.5∶0.01→10∶1∶0.05)纯化得到EM755(62.6mg,34%,白色粉末)。
EM755:M.p.:108-110℃;
IR(KBr)ν:3455.8,2975.6,2937.1,1735.6,1629.6,1457.9,1378.9,1243.9,1168.7,1078.0,1043.3,1016.3cm-1;
HRMS(FAB)m/z:C41H70NO14[M+H]+的计算值为800.4795,实测值为800.4784。
实施例3
4”,13-二-O-乙酰基-脱(3’-N-甲基)-8,9-脱水-假红霉素A 6,9-半缩酮(EM756)的合成
在室温下向搅拌的EM719(50.3mg,0.0598mmol)的甲醇(4.8ml)和水(1.2ml)溶液中相继加入乙酸钠(24.5mg,0.299mmol)和碘(15.2mg,0.0598mmol)。将反应混合物在50℃下搅拌3小时。反应过程中,滴加1N的氢氧化钠水溶液将溶液的pH调至8.0-9.0。将反应混合物用水稀释,用二氯甲烷萃取。萃取液经无水硫酸钠干燥,真空浓缩得到粗产品。所得粗产品通过薄层色谱法用氯仿-甲醇-氨水(20∶1∶0.1)纯化得到EM756(49.0mg,72%,白色粉末)。
EM756:M.p.:119-121℃;
IR(KBr)ν:3450.0,2975.6,2939.0,1735.6,1457.9,1376.9,1241.9,1126.2,1093.4,1041.4,1016.3cm-1;
HRMS(FAB)m/z:C40H68NO14[M+H]+的计算值为786.4639,实测值为786.4649。
实施例4
2’,4”,13-三-O-苯甲酰基-8,9-脱水-假红霉素A 6,9-半缩酮(EM770)的合成
在室温下向溶于吡啶(1.0ml)的EM701(33.8mg,0.0472mmol)中滴加苯甲酸酐(133.6μl,0.708mmol),然后向该溶液中加入DMAP(痕量)。将该反应混合物在室温下搅拌5小时,然后在60℃下搅拌23小时。将混合物倒入水中使反应终止,用二氯甲烷萃取。萃取液用饱和盐水洗涤,经无水硫酸钠干燥,过滤,除去溶剂得到粗产品。所述粗产品通过薄层色谱法用氯仿-甲醇-氨水(15∶1∶0.1)纯化得到EM770(27.6mg,57%,白色粉末)。
EM770:M.p.:119-121℃;
IR(KBr)ν:3374.8,2975.6,2937.1,1724.0,1672.0,1602.6,1452.1,1378.9,1338.4,1268.9,1170.6,1097.3,1070.3,1016.3,cm-1;
HRMS(FAB)m/z:C58H78NO15[M+H]+的计算值为1028.5371,实测值为1028.5353。
参考例2
2’-O-乙酰基-8,9-脱水-假红霉素A 6,9-半缩酮(EM718)的合成EM718的合成描述在Kazuo Tsuzuki,Toshiaki Sunazuka,Shogo Marui,Hajime Toyoda,Satoshi Omura,Nobuhiro Inatomi,and Zen Itoh,Chem.Pharm.Bull.37(10),2687-2700,1989中。使EM701(693mg,1.0mmol)溶于丙酮(10ml),加入无水乙酸(918μl,9.7mmol),在室温下搅拌40分钟。将反应混合物用水(50ml)稀释,用氯仿(50ml)萃取。通过加无水硫酸钠使氯仿层脱水,除去溶剂得到粗产品。所述粗产品通过硅胶柱色谱法用氯仿-甲醇-氨水(10∶0.5∶0.1)纯化得到EM718(586mg,80%,无定型白色固体)。
实施例5
2’-O-乙酰基-4”,13-二-O-丙酰基-8,9-脱水-假红霉素A 6,9-半缩酮(EM771)的合成
在室温下向EM718(34.0mg,0.0449mmol)的吡啶(1.0ml)溶液中滴加丙酸酐(86.3μl,0.673mmol),然后向溶液中加入DMAP(痕量)。将该混合物在室温下搅拌18小时。将混合物倒入水中使反应终止,用二氯甲烷萃取。萃取液用饱和盐水洗涤,经无水硫酸钠干燥,过滤,除去溶剂得到粗产品。所述粗产品通过薄层色谱法用氯仿-甲醇-氨水(15∶1∶0.1)纯化得到EM771(27.6mg,71%,白色粉末)。
EM771:M.p.:96-98℃;
IR(KBr)ν:3521.4,3446.2,2977.6,2940.9,2883.1,1745.3,1463.7,1375.0,1344.1,1240.0,1170.6,1066.4,1016.3cm-1;
HRMS(FAB)m/z:C45H75NO15Na[M+Na]+的计算值为892.5034,实测值为892.5037。
实施例6
2’-O-乙酰基-4”,13-二-O-苯甲酰基-8,9-脱水-假红霉素A 6,9-半缩酮(EM773)的合成
在室温下向EM718(64.0mg,0.0845mmol)的吡啶(1.7ml)溶液中滴加苯甲酸酐(159.4μl,0.845mmol),然后向溶液中加入DMAP(痕量)。将该反应混合物在室温下搅拌120小时。将混合物倒入水中使反应终止,用二氯甲烷萃取。萃取液用饱和盐水洗涤,经无水硫酸钠干燥,过滤,除去溶剂得到粗产品。所述粗产品通过薄层色谱法用氯仿-甲醇-氨水(15∶1∶0.1)纯化得到EM773(34.1mg,42%,白色粉末)和副产物(20.6mg),该副产物用于制备EM779。
EM773:M.p.:108-110℃;
IR(KBr)ν:3473.2,2975.6,2939.0,2881.1,1745.3,1375.0,1338.4,1268.9,1168.7,1110.8,1070.3,1016.3cm-1;
HRMS(FAB)m/z:C53H75NO15Na[M+Na]+的计算值为988.5034,实测值为988.5030。
实施例7
4”,13-二-O-丙酰基-8,9-脱水-假红霉素A 6,9-半缩酮(EM776)的合成
使EM771(27.6mg,0.0317mmol)溶于甲醇(1.0ml),在室温下搅拌48小时。在真空中除去甲醇之后,残余物通过薄层色谱法用氯仿-甲醇-氨水(15∶1∶0.1)纯化得到EM776(25.4mg,97%,白色粉末)。
EM776:M.p.:138-140℃;
IR(KBr)ν:3430.7,2977.6,2933.2,1739.5,1621.8,1589.1,1456.0,1419.4,1384.6,1344.1,1315.2,1243.9,1164.8,1124.3,1074.2,1016.3cm-1;
HRMS(FAB)m/z:C43H73NO14Na[M+Na]+的计算值为850.4929,实测值为850.4928。
实施例8
4”,13-二-O-苯甲酰基-8,9-脱水-假红霉素A 6,9-半缩酮(EM778)的合成
使EM773(34.1mmg,0.0353mmol)溶于甲醇(1.0ml),在室温下搅拌96小时。在真空中除去甲醇之后,残余物通过薄层色谱法用氯仿-甲醇-氨水(15∶1∶0.1)纯化得到EM778(32.0mg,98%,白色粉末)。
EM778:M.p.:113-116℃;
IR(KBr)ν:3486.7,2977.6,2939.0,2879.2,1724.0,1602.6,1585.2,1452.1,1382.7,1334.5,1268.9,1170.6,1112.7,1070.-3,1018.2cm-1;
HRMS(FAB)m/z:C51H74NO14[M+H]+的计算值为924.5109,实测值为924.5120。
实施例9
13-O-苯甲酰基-8,9-脱水-假红霉素A 6,9-半缩酮(EM779)的合成
使制备EM773中所得副产物混合物(20.6mg)溶于甲醇(1.0ml),在室温下搅拌96小时。在真空中除去甲醇之后,残余物通过薄层色谱法用氯仿-甲醇-氨水(15∶1∶0.1)纯化得到EM779(18.6mg,90%,白色粉末)。
EM779:M.p.:125-127℃;
IR(KBr)ν:3467.4,3436.5,2971.8,2879.2,1722.1,1631.5,1454.1,1380.8,1317.1,1270.9,1166.7,1110.8,1074.2,1016.3,937.2cm-1;
HRMS(FAB)m/z:C44H70NO13[M+H]+的计算值为820.4847,实测值为820.4859。
实施例10
2’-O-乙酰基-4”,13-二-O-己酰基-8,9-脱水-假红霉素A 6,9-半缩酮(EM772)的合成
在室温下向EM718(36.5mg,0.0482mmol)的吡啶(1.0ml)溶液中滴加己酸酐(167.3μl,0.723mmol),然后向溶液中加入DMAP(痕量)。将该混合物在室温下搅拌18小时。将混合物倒入水中使反应终止,用二氯甲烷萃取。萃取液用饱和盐水洗涤,经无水硫酸钠干燥,过滤,蒸馏出溶剂得到粗产品。所述粗产品通过薄层色谱法用氯仿-甲醇-氨水(15∶1∶0.1)纯化得到EM772(32.5mg,71%,白色粉末)。
EM772:M.p.:90-91℃;
IR(KBr)ν:3519.5,2971.8,2937.1,2861.8,2831.0,2782.8,1743.3,1457.9,1375.0,1340.3,1241.9,1168.7,1095.4,1058.7,1016.3cm-1;
HRMS(FAB)m/z:C58H77NO15Na[M+Na]+的计算值为1050.5191,实测值为1050.5210。
实施例11
4”,13-二-O-己酰基-8,9-脱水-假红霉素A 6,9-半缩酮(EM777)的合成
使EM772(32.5mg,0.0341mmol)溶于甲醇(1.0ml),在室温下搅拌48小时。在真空中除去甲醇之后,残余物通过薄层色谱法用氯仿-甲醇-氨水(15∶1∶0.1)纯化得到EM777(31.1mg,99%,白色粉末)。
EM777:M.p.:94-96℃;
IR(KBr)ν:3648.4,3465.5,2962.1,2933.2,2861.8,1737.5,1629.6,1456.0,1382.7,1243.9,1166.7,1110.8,1074.2,1016.3cm-1;
HRMS(FAB)m/z:C49H85NO14Na[M+Na]+的计算值为934.5868,实测值为934.5867。
实施例12
4”,13-二-O-己酰基-3′-脱-N-甲基-8,9-脱水-假红霉素A 6,9-半缩酮(EM852)的合成
使EM777(108.4mg,0.1191mmol)溶于80%甲醇(1.0ml),通过分成小份逐渐加入乙酸钠(48.8mg,0.595mmol)和碘(30.2mg,0.119mmol),然后在47℃下搅拌75分钟。搅拌过程中,连续地加1N氢氧化钠水溶液以调至pH8-9。通过TLC确定反应终止后,将反应混合物用氨水(5ml)-水(10ml)稀释,用氯仿萃取该混合物。通过加无水硫酸钠使有机层脱水,滤出硫酸钠,蒸馏出溶剂得到粗产品。所述粗产品通过薄层色谱法用氯仿-甲醇-氨水(50∶1∶0.1)纯化得到EM852(88.1mg,83%,白色粉末)。
EM852:M.p.:94-96℃;
HRMS(FAB)m/z:C48H83NO14Na[M+Na]+的计算值为920.5711,实测值为920.5743。
实施例13
4”,13-二-O-己酰基-脱(3’-N-甲基)-N-乙基-8,9-脱水-假红霉素A 6,9-半缩酮(EM853)的合成
向EM777(33.2mg,0.037mmol)和N,N-二异丙胺(32.2μl,0.185mmol)的氯仿(1.2ml)溶液中加入乙基碘(14.8μl,0.185mmol),在50℃下搅拌3小时。通过TLC确定反应终止后,将反应混合物用水(10ml)稀释,用氯仿萃取。通过加无水硫酸钠使有机层脱水,滤出硫酸钠,蒸馏出溶剂得到粗产品。除去甲醇后,残余物通过薄层色谱法用氯仿-甲醇-氨水(100∶1∶0.1)纯化得到EM853(24.0mg,70%,白色粉末)。
EM853:M.p.:94-96℃;
HRMS(FAB)m/z:C50H87NO14Na[M+Na]+的计算值为948.6024,实测值为948.6024。
实施例14
2’-O-乙酰基-4”-O-苯磺酰基-12,13-环氧-8,9-脱水-假红霉素A6,9-半缩酮(EM774)的合成
在室温下向溶于吡啶(1.7ml)的EM718(62.5mg,0.0825mmol)中滴加苯磺酰氯(105.3μl,0.825mmol),然后向溶液中加入DMAP(痕量)。将该反应混合物在室温下搅拌48小时。将混合物倒入水中使反应终止,用二氯甲烷萃取。萃取液用饱和盐水洗涤,经无水硫酸钠干燥,过滤,蒸馏出溶剂得到粗产品。所述粗产品通过薄层色谱法用氯仿-甲醇-氨水(15∶1∶0.1)纯化得到EM774(42.0mg,589%,白色粉末)。
EM774:M.p.:94-96℃;
IR(KBr)ν:2973.7,2939.0,2881.1,2859.9,2832.9,2782.8,1743.3,1420.2,1371.1,1346.1,1243.9,1187.9,1126.2,1095.4,1051.1,1016.3cm-1;
HRMS(FAB)m/z:C45H70NO14S[M+H]+的计算值为880.4517,实测值为880.4545。
实施例15
2’-O-乙酰基-4”-O-苄磺酰基-12,13-环氧-8,9-脱水-假红霉素A6,9-半缩酮(EM775)的合成
在室温下向溶于吡啶(1.7ml)的EM718(62.5mg,0.0825mmol)中滴加苯磺酰氯(157.3μl,0.825mmol),然后向溶液中加入DMAP(痕量)。将该反应混合物在室温下搅拌48小时。将混合物倒入水中使反应终止,用二氯甲烷萃取。萃取液用饱和盐水洗涤,经无水硫酸钠干燥,过滤,蒸馏出溶剂得到粗产品。所述粗产品通过薄层色谱法用氯仿-甲醇-氨水(15∶1∶0.1)纯化得到EM775(57.2mg,78%,白色粉末)。
EM775:M.p.:103-106℃;
IR(KBr)ν:2967.9,2931.3,2831.0,1749.1,1731.8,1633.4,1456.0,1369.2,1340.3,1240.0,1170.6,1130.1,1095.4,1056.8,1002.8,966.2cm-1;
HRMS(FAB)m/z:C46H72NO14S[M+H]+的计算值为894.4674,实测值为894.4673。
实施例16
4”-O-苄磺酰基-12,13-环氧-8,9-脱水-假红霉素A 6,9-半缩酮(EM780)的合成
使EM775(11.5mg,0.0129mmol)溶于甲醇(4.0ml)和二氯甲烷(2ml),然后在室温下搅拌48小时。在真空中除去甲醇后,残余物通过薄层色谱法用氯仿-甲醇-氨水(15∶1∶0.1)纯化得到EM780(10.1mg,92%,白色粉末)。
EM780:M.p.:197-199℃;
IR(KBr)ν:3463.5,2971.8,2939.0,2886.9,1735.6,1457.9,1384.6,1340.3,1247.7,1172.5,1093.4,1051.0,1016.3,962.3,916.0,887.1cm-1;
HRMS(FAB)m/z:C44H70NO13S[M+H]+的计算值为852.4568,实测值为852.4567。
参考例3
9(E)-肟红霉素A[E9(E)-肟]的合成
9-肟红霉素A的合成描述在Richard S.Egan,Leslie A.Freiberig,和Wolliam H.Washbum,J.Org.Chem.39(17),2492-2494(1974)中。
使红霉素A(15g,21mmol)溶于甲醇(225ml)。向其中加入三乙胺(9.6ml)和盐酸羟胺(6.3mg,91mmol),回流96小时。使反应混合物冷却,用水(2.6L)稀释,用氯仿(2.6L)萃取。通过加无水硫酸钠使氯仿层脱水,蒸馏出溶剂得到粗产品。所述粗产品通过硅胶柱色谱法用氯仿-甲醇-氨水(10∶0.5∶0.1)纯化得到标题化合物(12.3mg,84%,无定型白色固体)。
实施例17
9(E)-4-吗啉代-2-丁烯基肟-红霉素A(EM762)的合成
使EM(9)-肟(96.2mg,0.129mmol)溶于DMF(1.3ml)和乙醚(2.6ml)。将氢化钠(60%)(7.1mg,0.193mmol)和1,4-溴-2-丁烯(32.9mg,0.154mmol)相继加入该溶液中。在室温下搅拌1小时后,将哌嗪(4.4mg,0.0514mmol)和N,N-二异丙基乙胺(67.2μl,0.386mmol)加入该反应混合物中。将反应混合物在室温下搅拌12小时,然后倾入水中使反应终止,用二氯甲烷萃取。有机层用饱和盐水洗涤,经无水硫酸钠干燥,过滤,蒸馏出溶剂得到粗产品。所述粗产品通过薄层色谱法用氯仿-甲醇-氨水(20∶1∶0.1)纯化得到EM762(38.9mg,34%,白色粉末)。
EM762:M.p.:124-126℃;
IR(KBr)ν:3425.0,2973.7,2939.0,2879.2,2829.1,1735.6,1633.4,1459.8,1378.9,1344.1,1282.4,1168.7,1112.7,1085.7,1052.9,1000.9cm-1;
HRMS(FAB)m/z:C45H82N4O13Na[M+Na]+的计算值为909.5767,实测值为909.5774。
实施例18
9(E)-4-(氨基乙氨基)-2-丁烯基肟-红霉素A(EM763)的合成
使EM9(E)-肟(51.2mg,0.0684mmol)溶于DMF(0.6ml)和乙醚(1.4ml)。将氢化钠(60%)(4.2mg,0.103mmol)和1,4-溴-2-丁烯(17.6mg,0.0820mmol)相继加入该溶液中。在室温下搅拌1小时后,将乙二胺(2.4μl,0.0342mmol)和N,N-二异丙基乙胺(35.8μl,0.206mmol)加入该反应混合物中。将反应混合物在室温下搅拌16小时,然后倾入水中使反应终止,用二氯甲烷萃取。有机层用饱和盐水洗涤,经无水硫酸钠干燥,过滤,蒸馏出溶剂得到粗产品。所述粗产品通过薄层色谱法用氯仿-甲醇-氨水(20∶1∶0.1)纯化得到EM763(22.6mg,38%,白色粉末)。
EM763:M.p.:105-108℃;
IR(KBr)v:3446.2,3430.7,2973.7,2939.0,2877.3,1735.6,1625.7,1457.9,1378.9,1346.1,1284.4,1168.7,1110.8,1085.7,1052.9,1012.4cm-1;
HRMS(FAB)m/z:C43H81N4O13[M+H]+的计算值为861,实测值为861。
实施例19
9(E)-3-丁烯基肟-红霉素A(EM769)的合成
使EM9(E)-肟(92.3mg,0.123mmol)溶于DMF(3.1ml)和乙醚(6.1ml)。将氢化钠(60%)(7.4mg,0.185mmol)和1,4-溴-2-丁烯(31.7mg,0.148mmol)相继加入该溶液中。在室温下搅拌1小时后,将肼(31.0μl,0.9872mmol)加入该反应混合物中。将反应混合物在室温下搅拌18小时,然后倾入水中使反应终止,用二氯甲烷萃取。有机层用饱和盐水洗涤,经无水硫酸钠干燥,过滤,蒸馏出溶剂得到粗产品。所述粗产品通过薄层色谱法用氯仿-甲醇-氨水(15∶1∶0.1)纯化得到EM769(34.0mg,34%,白色粉末)。
EM769:M.p.:102-104℃;
IR(KBr)ν:3453.9,3430.7,2973.7,2939.0,2877.3,1737.5,1641.1,1457.9,1380.8,1344.1,1282.4,1168.7,1112.7,1081.9,1052.9,1000.9cm-1;
HRMS(FAB)m/z:C41H74N2O13Na[M+Na]+的计算值为825.5089,实测值为825.5056。
实施例20
3’-脱-N-甲基-3’-N-{2-氨基-(3’-脱-N-甲基-红霉素A)}乙基-红霉素A(EM752)的合成
在室温下向溶于DMF(1.6ml)的脱-N-甲基红霉素A(34.5mg,0.0479mmol)中滴加N,N-二异丙基乙胺(166.9μl,0.958mmol)和1-溴-2-氯乙烷(79.8μl,0.958mmol)。将该反应混合物在室温下搅拌120小时。将该溶液用水稀释,用二氯甲烷萃取。有机层用饱和盐水洗涤,经无水硫酸钠干燥。溶剂蒸发后,残余物通过薄层色谱法用氯仿-甲醇-氨水(15∶1∶0.1)纯化得到EM752(21.0mg,60%,白色粉末)。
EM752:M.p.:164-167℃
IR(KBr)ν:3455.8,2971.6,2937.1,1729.8,1693.2,1457.9,1378.9,1344.1,1284.4,1168.7,1108.9,1079.9,1054.9,1012.4cm-1;
HRMS(FAB)m/z:C74H133N2O26[M+H]+的计算值为1465,实测值为1465。
实施例21
3’-脱-N-甲基-3’-N-{2-氨基-(3’-脱-N-甲基-克拉霉素A)}乙基-克拉霉素(clarythromycin)(EM753)的合成
在室温下向溶于DMF(1.6ml)的脱-N-甲基克拉霉素(93.2mg,0.127mmol)中滴加N,N-二异丙基乙胺(442.4μl,2.540mmol)和1-溴-2-氯乙烷(211.4μl,2.540mmol)。将该反应混合物在室温下搅拌120小时。将该溶液用水稀释,用二氯甲烷萃取。有机层用饱和盐水洗涤,经无水硫酸钠干燥。溶剂蒸发后,残余物通过薄层色谱法用氯仿-甲醇-氨水(20∶1∶0.1)纯化得到EM753(74.2mg,78%,白色粉末)。
EM753:M.p.:188-191℃;
IR(KBr)ν:3461.6,2971.8,2937.1,1733.7,1691.3,1629.6,1459.8,1405.9,1378.9,1346.1,1286.3,1245.8,1168.7,1110.8,1083.8,1054.9cm-1:
HRMS(FAB)m/z:C76H137N2O26[M+H]+的计算值为1493,实测值为1493。
实施例22
3’-脱-N-甲基-3’-N-{4-氨基-(3’-脱-N-甲基-红霉素A)}-2(E)-丁烯基-红霉素A(EM757)的合成
在室温下向溶于二氯甲烷(2.6ml)的脱-N-甲基红霉素A(56.5mg,0.0785mmol)中加入1,4-溴-2-丁烯(8.4mg,0.0393mmol)和N,N-二异丙基乙胺(41.0μl,0.236mmol)。将该反应混合物在室温下搅拌120小时。将混合物倒入水中使反应终止,用二氯甲烷萃取。萃取液用饱和盐水洗涤,经无水硫酸钠干燥,过滤,蒸馏出溶剂得到粗产品。所述粗产品通过薄层色谱法用氯仿-甲醇-氨水(20∶1∶0.1)纯化得到EM757(35.1mg,60%,白色粉末)。
EM757:M.p.:151-154℃;
IR(KBr)ν:3519.5,3473.2,2973.7,2940.9,2879.2,1714.4,1637.3,1459.8,1376.9,1348.0,1284.4,1270.9,1240.0,1193.7,1170.6,1108.9,1085.7,1052.9,1008.6cm-1;
HRMS(FAB)m/z:C76H135N2O26[M+H]+的计算值为1491,实测值为1491。
实施例23
3’-脱-N-甲基-3’-N-{4-氨基-(3’-脱-N-甲基-克拉霉素A)}-2(E)-丁烯基-克拉霉素A(EM758)的合成
在室温下向脱-N-甲基-克拉霉素(39.8mg,0.0542mmol)和N,N-二异丙基乙胺(28.3μl,0.163mmol)的二氯甲烷(1.8ml)中加入1,4-溴-2-丁烯(5.8mg,0.0271mmol)。将该反应混合物在室温下搅拌120小时。将混合物倒入水中,用二氯甲烷萃取。萃取液用饱和盐水洗涤,经无水硫酸钠干燥,过滤,蒸馏出溶剂得到粗产品。所述粗产品通过薄层色谱法用氯仿-甲醇-氨水(20∶1∶0.1)纯化得到EM758(26.3mg,66%,白色粉末)。
EM758:M.p.:167-169℃;
IR(KBr)ν:3619.7,3453.9,3426.9,2973.7,2939.0,1735.6,1689.3,1457.9,1407.8,1378.9,1346.1,1286.3,1170.6,1110.8,1083.8,1052.9,1010.5cm-1;
HRMS(FAB)m/z:C78H139N2O26[M+H]+的计算值为1519,实测值为1519。
实施例24
9(E)-4-{9(E)-肟-红霉素A}-2-丁烯基肟-红霉素A(759)的合成
使EM9(E)-肟(99.8mg,0.133mmol)溶于DMF(1.33ml)和乙醚(1.33ml)。将氢化钠(60%)(8.0mg,0.200mmol)和1,4-溴-2-丁烯(14.3mg,0.0667mmol)相继加入该溶液中。在室温下搅拌1小时后,将反应混合物倾入水中使反应终止,用二氯甲烷萃取。有机层用饱和盐水洗涤,经无水硫酸钠干燥,过滤,蒸馏出溶剂得到粗产品。所述粗产品通过薄层色谱法用氯仿-甲醇-氨水(20∶1∶0.1)纯化得到EM759(64.0mg,62%,白色粉末)。
EM759:M.p.:138-140℃;
IR(KBr)ν:3448.1,3423.0,2973.7,2939.0,1735.6,1623.8,1457.9,1405.9,1380.8,1344.1,1280.5,1168.7,1112.7,1085.7,1052.9,1000.9cm-1;
HRMS(FAB)m/z:C78H141N4O28[M+H]+的计算值为1549,实测值为1549。
实施例25
9(E)-4-哌嗪-{N-4-(9(E)-肟-红霉素A-2(E)丁烯基)-2-丁烯基肟-红霉素A(EM760)的合成
使EM9(E)-肟(96.2mg,0.129mmol)溶于DMF(1.3ml)和乙醚(2.6ml)。将氢化钠(60%)(7.1mg,0.193mmol)和1,4-溴-2-丁烯(32.9mg,0.154mmol)相继加入该溶液中。在室温下搅拌1小时后,将哌嗪(4.4mg,0.0514mmol)和N,N-二异丙基乙胺(67.2μl,0.386mmol)加入反应混合物中。将反应混合物在室温下再搅拌12小时,然后倒入水中使反应终止,用二氯甲烷萃取。有机层用饱和盐水洗涤,经无水硫酸钠干燥,过滤,蒸馏出溶剂得到粗产品。所述粗产品通过薄层色谱法用氯仿-甲醇-氨水(20∶1∶0.1)纯化得到EM760(41.7mg,38%,白色粉末)。
EM760:M.p.:152-155℃;
IR(KBr)ν:3452.0,3423.0,2973.7,2937.1,2879.2,1733.7,1633.4,1456.0,1378.9,1346.1,1282.4,1168.7,1110.8,1085.7,1052.9,1002.8cm-1;
HRMS(FAB)m/z:C86H156N6O26[M+H]+的计算值为1688,实测值为1688。
实施例26
9(E)-4-氨基乙氨基-{N-4-(9(E)-肟-红霉素A)-2-丁烯基}-2-丁烯基肟-红霉素A(EM761)的合成
使EM9(E)-肟(51.2mg,0.0684mmol)溶于DMF(0.6ml)和乙醚(1.4ml)。将氢化钠(60%)(4.2mg,0.103mmol)和1,4-溴-2-丁烯(17.6mg,0.0820mmol)相继加入该溶液中。在室温下搅拌1小时后,将乙二胺(2.4μl,0.0342mmol)和N,N-二异丙基乙胺(35.8μl,0.206mmol)加入反应混合物中。将反应混合物在室温下搅拌16小时,然后倾入水中使反应终止,用二氯甲烷萃取。有机层用饱和盐水洗涤,经无水硫酸钠干燥,过滤,蒸馏出溶剂得到粗产品。所述粗产品通过薄层色谱法用氯仿-甲醇-氨水(20∶1∶0.1)纯化得到EM761(14.2mg,25%,白色粉末)。
EM761:M.p.:140-143℃;
IR(KBr)ν:3467.4,3428.8,3423.0,2973.7,2939.0,2877.3,1733.7,1627.6,1459.8,1378.9,1348.0,1282.4,1168.7,1112.7,1085.7,1052.9,1012.4cm-1;
HRMS(FAB)m/z:C84H152N6O26Na[M+H]+的计算值为1684,实测值为1684。
实施例27
9(E)-4-{9(E)-肟-红霉素A}-丁基肟-红霉素A(EM764)的合成
向钯/活性炭(4.0mg)的乙醇(2.6ml)混合物中加入EM759(19.9mg,0.0129mmol)。将该反应混合物在室温和H2(1atm)下搅拌48小时。将混合物用硅藻土过滤,用乙醇洗涤。蒸馏出溶剂后,残余物通过薄层色谱法用氯仿-甲醇-氨水(15∶1∶0.1)纯化得到EM764(16.5mg,83%,白色粉末)。
EM764:M.p.:150-153℃;
IR(KBr)ν:3455.8,3436.5,2973.7,2939.0,2877.3,1737.5,1627.6,1459.8,1380.8,1344.1,1280.5,1168.7,1085.7,1052.9,1012.4cm-1;
HRMS(FAB)m/z:C78H143N4O26[M+H]+的计算值为1551,实测值为1551。
参考例4
3’-脱-N-甲基红霉素A(EM798)的合成
EM798的合成描述在L.A.Freiberg和JP-A-47-9129中。在室温下向搅拌的红霉素A(267.0mg,0.375mmol)的甲醇(58.0ml)和水(12.0ml)溶液中相继加入乙酸钠(146.4mg,1.785mmol)和碘(99.6mg,0.392mmol)。将反应混合物在50℃下搅拌3.5小时。反应过程中,滴加1N氢氧化钠水溶液将溶液的pH调至8.0-9.0。使反应混合物真空浓缩除去甲醇。残余物溶于饱和盐水,用二氯甲烷萃取。萃取液经无水硫酸钠干燥,蒸馏出溶剂得到粗产品。所得粗产品通过柱色谱法在硅胶上用氯仿-甲醇-氨水(10∶0.5∶0.1)洗脱纯化得到EM798(221.0mg,84%,白色无定型固体)。
实施例28
3’-脱-N-甲基-3’-N-{4-9(E)-肟-红霉素A}-2(E)-丁烯基-红霉素A(EM765)的合成
使EM9(E)-肟(68.6mg,0.0967mmol)溶于DMF(1.0ml)和乙醚(1.9ml)。将氢化钠(60%)(5.5mg,0.137mmol)和1,4-溴-2-丁烯(23.5mg,0.110mmol)相继加入该溶液中。在室温下搅拌1小时后,将EM798(66.0mg,0.0967mmol)和N,N-二异丙基胺(47.9μl,0.275mmol)加入反应混合物中。将反应混合物在室温下搅拌18小时,然后倒入水中使反应终止,用二氯甲烷萃取。有机层用饱和盐水洗涤,经无水硫酸钠干燥,过滤,蒸馏出溶剂得到粗产品。所述粗产品通过薄层色谱法用氯仿-甲醇-氨水(20∶1∶0.1)纯化得到EM765(37.6mg,25%,白色粉末)。
EM765:M.p.:148-151℃;
IR(KBr)ν:3448.1,3436.5,2973.7,2939.0,2879.2,1733.7,1635.3,1459.8,1378.9,1346.1,1284.4,1168.7,1110.8,1085.7,1052.9,1012.4cm-1;
HRMS(FAB)m/z:C77H138N3O26[M+H]+的计算值为1520,实测值为1520。
实施例29
3’-脱-N-甲基-3’-N-{2-氨基-(3’-脱-N-甲基-脱水-假红霉素A6,9-半缩酮)}-乙基-脱水-假红霉素A 6,9-半缩酮(EM741)的合成
在室温下向溶于DMF(3.4ml)的EM703(72.4mg,0.103mmol)中滴加N,N-二异丙基乙胺(359.5μl,2.064mmol)和1-溴-2-氯乙烷(171.8μl,2.064mmol)。将反应混合物在室温下搅拌48小时。将该溶液用水稀释,用二氯甲烷萃取。有机层用饱和盐水洗涤,经无水硫酸钠干燥。蒸馏出溶剂后,残余物通过薄层色谱法用氯仿-甲醇-氨水(15∶1∶0.1)纯化得到EM741(38.7mg,53%,白色粉末)。回收10.2mg EM703(14%)。
EM741:M.p.:149-152℃;
IR(KBr)ν:3675.7,2971.8,2935.1,2879.2,1708.6,1631.5,1457.9,1378.9,1263.1,1166.7,1112.7,1074.2,1049.1,1039.4,1016.3cm-1;
HRMS(FAB)m/z:C74H129N2O24[M+H]+的计算值为1429,实测值为1429。
生物学性质
下面详细地解释对唑类抗真菌剂的活性的增强作用。
所采用的测试微生物是白色念珠菌ATCC 64548、抗氟康唑的白色念珠菌ATCC 64550和黑曲霉ATCC 6275。念珠菌属在Waksman培养基(葡萄糖2.0%,胨0.5%,干酵母0.3%,肉膏0.5%,NaCl 0.5%和CaCO3 0.3%,pH 7.0)中于27℃培养40小时,然后将其0.1%接种在GY琼脂培养基(葡萄糖1.0%,酵母膏0.5%和琼脂0.8%,pH 6.0)之上。将0.2%黑曲霉ATCC 6275的孢子悬浮液接种在GY琼脂培养基之上。
所用唑类抗真菌剂是咪康唑(Sigma Inc.,U.S.)和氟康唑(ICNPharmaceuticals Inc.,U.S.),以如下对各测试微生物的生长无影响的浓度加入。
白色念珠菌ATCC 64548:咪康唑0.03μg/ml或氟康唑0.5μg/ml。
白色念珠菌ATCC 64550:咪康唑0.15μg/ml或氟康唑30μg/ml。
黑曲霉ATCC 6275:咪康唑0.1μg/ml或氟康唑10μg/ml。
以上培养基中,与咪康唑一起加入GY培养基(对照)中的培养基指定为GYM,与氟康唑一起加入的培养基指定为GYF。用纸碟法(厚:8mm,ADVANTEC MFS INC.,Japan)评定活性。培养念珠菌属24小时和黑曲霉ATCC 6275菌48小时后以mm为单位表示抑制环的直径。用A、B、C、D和E(透明<A<B<C<D<E<不透明)评定抑制环的透明度。各透明度在下面说明,结果示于表1中。
A:测试微生物生长抑制率大于或等于95%。
B:测试微生物生长抑制率大于或等于75%而且小于或等于95%。
C:测试微生物生长抑制率大于或等于55%而且小于或等于75%。
D:测试微生物生长抑制率大于或等于35%而且小于或等于55%。
E:测试微生物生长抑制率小于或等于35%。
N.T.:未试验。
表1
| 化合物试样 | 抑制活性(抑制环mm) | |||||||||
| 白色念珠菌ATCC 64548 | 白色念珠菌ATCC 64550 | 黑曲霉ATCC 6275 | ||||||||
| 号 | (μg/8mm碟) | GY | GYM | GYF | GY | GYM | GYF | GY | GYM | GYF |
| 719 | 1050100 | --- | 9E10E13E | --- | --- | --- | --- | --- | --- | --- |
| 755 | 1050100 | --- | --17D | --- | --- | --10E | --- | --- | -16EN.T. | --- |
| 756 | 1050100 | --- | --15D | --- | --- | --10E | --- | -10EN.T. | -14EN.T. | -14EN.T. |
| 770 | 105010 | --- | 11D14CN.T. | --11D | --N.T. | -13AN.T. | -13CN.T. | -11EN.T. | -12EN.T. | 11E18EN.T. |
| 771 | 1050100 | --- | -12DN.T. | --N.T. | --N.T. | -10AN.T. | -10EN.T. | --N.T. | -23EN.T. | 14E15EN.T. |
| 772 | 1050100 | --N.T. | 14B12BN.T. | 9D10DN.T. | --N.T. | -12AN.T. | 11E14BN.T. | -12EN.T. | 12E13AN.T. | 10D10DN.T. |
| 773 | 1050100 | --N.T. | 13C16AN.T. | -12EN.T. | --N.T. | 10C14AN.T. | 11E15AN.T. | --N.T. | 13E15BN.T. | 10E15EN.T. |
| 776 | 1050100 | --- | -11DN.T. | --15E | --N.T. | -11EN.T. | -9EN.T. | --N.T. | -20CN.T. | 16E16EN.T. |
| 777 | 1050100 | --N.T. | 11D14AN.T. | -11DN.T. | --N.T. | -14AN.T. | 11D14A.N.T. | -9EN.T. | 11E13BN.T. | 11E17DN.T. |
| 778 | 1050100 | --N.T. | 12B16AN.T. | -9DN.T. | --N.T. | -14BN.T. | 10E13BN.T. | -10EN.T. | 14E16AN.T. | 12E13DN.T. |
| 779 | 1050100 | --N.T. | 10E15AN.T. | -9DN.T. | --N.T. | -11DN.T. | -10EN.T. | -11EN.T. | -17EN.T. | 11E13DN.T. |
| 852 | 1050100 | --- | -9A10A | --- | --- | 9C10B12B | 9C11A12A | -9E10E | -8C9A | -8D9D |
| 853 | 1050100 | --- | 10B14A15A | 9E14B16B | --- | -15A16A | 11D14A15A | 10E12D13D | 10E15B16B | 9E13B14B |
| 774 | 1050100 | --N.T. | --N.T. | --N.T. | --N.T. | 14A17AN.T. | 13A17AN.T. | --N.T. | 13E18DN.T | 14E17EN.T. |
| 775 | 1050100 | --N.T. | -10EN.T. | --N.T. | --- | --- | --- | --N.T. | -16DN.T. | 16E16EN.T. |
| 780 | 1050100 | --N.T. | --N.T. | --N.T. | --N.T. | -12CN.T. | -14CN.T | -12EN.T | 14E14EN.T | 11E12EN.T. |
| 762 | 1050100 | --- | -13DN.T. | --- | --- | --- | --- | --- | --- | 14E15EN.T. |
| 763 | 1050 | -- | -16C | -10D | -- | -- | -- | -- | -- | 12E15E |
| 100 | N.T. | N.T. | N.T. | - | - | - | - | - | N.T. | |
| 769 | 1050100 | --- | --14E | --- | --- | --- | --- | -10EN.T. | --- | 11E16EN.T. |
| 752 | 1050100 | --- | --12E | --- | --- | --- | --- | --- | --- | -17EN.T. |
| 753 | 1050100 | --- | 17C22CN.T. | --- | --- | --- | --- | --- | --13E | --- |
| 757 | 1050100 | --- | --18D | --17E | --- | --- | --- | -10E- | --- | --- |
| 758 | 1050100 | --- | 11E12DN.T. | --15E | --- | --- | --- | --- | --- | --- |
| 759 | 1050100 | --- | --13D | --- | --- | --- | --- | --- | --- | --(-) |
| 760 | 1050100 | --- | 10E14DN.T. | --- | --- | --- | --- | --- | --- | 15E16EN.T. |
| 761 | 1050100 | --- | --14E | --- | --- | --- | --- | --- | --- | --- |
| 764 | 1050100 | --- | -9EN.T. | --- | --- | --- | --- | --- | --- | --- |
| 765 | 1050100 | --- | -14EN.T. | --- | --- | --- | --- | --- | --- | --14E | 14E21EN.T. |
| 741 | 1050100 | --- | --14D | --- | --- | --- | --- | --- | --- | --- | --- |
工业实用性
如上文所解释,由于本发明物质有增强唑类抗真菌剂抗白色念珠菌和黑曲霉(二者均包括耐药性菌株)的活性的作用,它们具有在低浓度下和短期内抗真菌感染的作用,可用于降低耐药性微生物的出现率。此外,预计有不同骨架结构的两类药物的组合使用或用于对于唑类化合物具有耐药性菌株的这类药物的组合使用可克服对唑类抗真菌剂的耐药性。
Claims (6)
1.式[I]所示对抗真菌剂的活性具有增强作用的大环内酯衍生物:

其中R1为H时,R2和R3分别为Ac,R4为Me;R1为H时,R2和R3分别为Ac,R4为H;R1为苯甲酰基时,R2和R3分别为苯甲酰基,R4为Me;R1为Ac时,R2和R3分别为丙酰基,R4为Me;R1为Ac时,R2和R3分别为己酰基,R4为Me;R1为Ac时,R2和R3分别为苯甲酰基,R4为Me;R1为H时,R2和R3分别为丙酰基,R4为Me;R1为H时,R2和R3分别为己酰基,R4为Me;R1为H时,R2和R3分别为苯甲酰基,R4为Me;R1为H时,R2为H,R3为苯甲酰基,R4为Me;R1为H时,R2和R3分别为己酰基,R4为H;或R1为H时,R2和R3分别为己酰基,R4为Et。
2.[II]所示对抗真菌剂的活性具有增强作用的大环内酯衍生物:

其中R1为Ac时,R2为苯磺酰基;R1为Ac时,R2为苄基磺酰基;或R1为H时,R2为苄基磺酰基。
3.式[XI]所示对抗真菌剂的活性具有增强作用的大环内酯衍生物:
其中,R1为H。
4.权利要求1所述的大环内酯衍生物用于制备增强抗真菌剂预防或治疗真菌感染的活性的药物的用途,所述的真菌感染为伴有由HIV感染或血液病引起的免疫减弱病症的真菌感染。
5.权利要求2所述的大环内酯衍生物用于制备增强抗真菌剂预防或治疗真菌感染的活性的药物的用途,所述的真菌感染为伴有由HIV感染或血液病引起的免疫减弱病症的真菌感染。
6.权利要求3所述的大环内酯衍生物用于制备增强抗真菌剂预防或治疗真菌感染的活性的药物的用途,所述的真菌感染为伴有由HIV感染或血液病引起的免疫减弱病症的真菌感染。
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| PCT/JP2002/011213 WO2004039823A1 (ja) | 2002-10-29 | 2002-10-29 | 抗真菌活性増強作用を有する新規マクロライド誘導体 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN1561342A CN1561342A (zh) | 2005-01-05 |
| CN1280302C true CN1280302C (zh) | 2006-10-18 |
Family
ID=32260007
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CNB028081358A Expired - Fee Related CN1280302C (zh) | 2002-10-29 | 2002-10-29 | 对抗真菌活性有增强作用的大环内酯衍生物 |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US7521428B2 (zh) |
| EP (1) | EP1559719A4 (zh) |
| JP (1) | JP4440108B2 (zh) |
| CN (1) | CN1280302C (zh) |
| AU (1) | AU2002368309A1 (zh) |
| WO (1) | WO2004039823A1 (zh) |
Families Citing this family (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN101326192B (zh) * | 2005-10-14 | 2012-06-20 | 社团法人北里研究所 | 新型二氢伪红霉素衍生物 |
| JP2009518396A (ja) | 2005-12-08 | 2009-05-07 | ファイザー・インク | エリスロマイシン化合物の3’−ジメチルアミノ基を脱メチルする方法 |
| CN101597311B (zh) * | 2009-07-02 | 2014-05-21 | 沈阳药科大学 | 红霉素衍生物及其用途 |
| JP5843188B2 (ja) * | 2011-02-28 | 2016-01-13 | 学校法人北里研究所 | 抗真菌剤の活性増強作用を有する新規物質およびその製造方法と用途 |
| CN103536613B (zh) * | 2013-10-14 | 2015-07-29 | 中国科学院微生物研究所 | 一种抗真菌的药物组合物 |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA1321581C (en) | 1987-05-26 | 1993-08-24 | Herbert A. Kirst | Ring-contracted macrolides |
| US4920102A (en) | 1988-04-18 | 1990-04-24 | Eli Lilly And Company | Method for treating gastrointestinal disorders |
| JP2000198795A (ja) * | 1996-10-31 | 2000-07-18 | Taisho Pharmaceut Co Ltd | エリスロマイシンa誘導体 |
| KR100283990B1 (ko) | 1998-12-29 | 2001-03-02 | 민경윤 | 에리스로마이신 a 유도체 및 그의 제조방법 |
-
2002
- 2002-10-29 WO PCT/JP2002/011213 patent/WO2004039823A1/ja not_active Ceased
- 2002-10-29 JP JP2004547983A patent/JP4440108B2/ja not_active Expired - Fee Related
- 2002-10-29 EP EP02807125A patent/EP1559719A4/en not_active Withdrawn
- 2002-10-29 AU AU2002368309A patent/AU2002368309A1/en not_active Abandoned
- 2002-10-29 CN CNB028081358A patent/CN1280302C/zh not_active Expired - Fee Related
- 2002-10-29 US US10/472,044 patent/US7521428B2/en not_active Expired - Fee Related
Also Published As
| Publication number | Publication date |
|---|---|
| WO2004039823A1 (ja) | 2004-05-13 |
| AU2002368309A1 (en) | 2004-05-25 |
| US7521428B2 (en) | 2009-04-21 |
| JPWO2004039823A1 (ja) | 2006-03-02 |
| US20050176655A1 (en) | 2005-08-11 |
| CN1561342A (zh) | 2005-01-05 |
| EP1559719A1 (en) | 2005-08-03 |
| JP4440108B2 (ja) | 2010-03-24 |
| EP1559719A4 (en) | 2006-12-20 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN1310907C (zh) | 杂环化合物和以其为有效成分的抗肿瘤药 | |
| CN1088709C (zh) | 胺类化合物及其酸加成盐 | |
| CN1768072A (zh) | 糖脂衍生物及其制造方法,以及其合成中间体及其制造方法 | |
| CN1137275A (zh) | 具有免疫抑制活性的o-芳基、o-烷基、o-烯基和o-炔基大环内酯物 | |
| CN1699343A (zh) | 盐酸多奈哌齐的多晶型物及其制备方法 | |
| CN1053787A (zh) | 三唑类抗真菌剂 | |
| CN1155585C (zh) | 3,5-取代噁唑烷酮衍生物及其制备方法和应用 | |
| CN1681801A (zh) | 喹啉基丙基哌啶衍生物以及其作为抗微生物剂的应用 | |
| CN1151165C (zh) | 具有抗炎活性的大环内酯 | |
| CN1240707C (zh) | 2-卤代-6-0-取代的酮内酯衍生物 | |
| CN1290707A (zh) | 核苷5’-硫代磷酰氨基酸酯化合物 | |
| CN1271363A (zh) | 具有抗菌活性的3’-n-修饰的6-o-取代的红霉素酮基内酯衍生物 | |
| CN1922138A (zh) | 芳氧基烷基氨基甲酸酯类衍生物,它们的制备方法与治疗用途 | |
| CN1687088A (zh) | N-取代苯并噻唑基-1-取代苯基-O,O-二烷基-α-氨基膦酸酯类衍生物及制备方法和用途 | |
| CN1059336A (zh) | 抗真菌的三唑衍生物及其制备和用途 | |
| CN1280302C (zh) | 对抗真菌活性有增强作用的大环内酯衍生物 | |
| CN1054980A (zh) | 具有旋光活性的8-甲氧基喹诺酮羧酸衍生物,它们的制备方法以及它们的中间体 | |
| CN1960965A (zh) | 氨基-烷氧基-庚酸烷基酯的合成 | |
| CN1894211A (zh) | 借助费歇尔-芬克型合成和随后的酰化制备4,5-二烷基-3-酰基-吡咯-2-羧酸衍生物 | |
| CN1910177A (zh) | 取代的喹啉及其作为分枝杆菌抑制剂的用途 | |
| CN1040877C (zh) | 红霉素的新衍生物、它们的制备方法和作为药物的应用 | |
| CN1049662A (zh) | 香豆素衍生物及其制备和在治疗脑血管失调中的应用 | |
| CN1633502A (zh) | 制备(3r,5s)-(e)-7-[2-环丙基-4-(4-氟苯基)-喹啉-3-基]-3,5-二羟基庚-6-烯酸酯的方法 | |
| CN1308342C (zh) | 核糖取代的芳族化合物,其制备方法和作为药物的应用 | |
| CN88101987A (zh) | 喹啉羧酸衍生物和含有它们的抗菌剂 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C14 | Grant of patent or utility model | ||
| GR01 | Patent grant | ||
| C19 | Lapse of patent right due to non-payment of the annual fee | ||
| CF01 | Termination of patent right due to non-payment of annual fee |