CN1617848A - 结晶文拉法辛碱、新的盐酸文拉法辛多晶型物及它们的制备方法 - Google Patents
结晶文拉法辛碱、新的盐酸文拉法辛多晶型物及它们的制备方法 Download PDFInfo
- Publication number
- CN1617848A CN1617848A CNA028276485A CN02827648A CN1617848A CN 1617848 A CN1617848 A CN 1617848A CN A028276485 A CNA028276485 A CN A028276485A CN 02827648 A CN02827648 A CN 02827648A CN 1617848 A CN1617848 A CN 1617848A
- Authority
- CN
- China
- Prior art keywords
- venlafaxine
- crystalline
- purity
- base
- venlafaxine hydrochloride
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Images

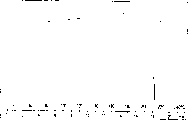


Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C217/00—Compounds containing amino and etherified hydroxy groups bound to the same carbon skeleton
- C07C217/54—Compounds containing amino and etherified hydroxy groups bound to the same carbon skeleton having etherified hydroxy groups bound to carbon atoms of at least one six-membered aromatic ring and amino groups bound to acyclic carbon atoms or to carbon atoms of rings other than six-membered aromatic rings of the same carbon skeleton
- C07C217/74—Compounds containing amino and etherified hydroxy groups bound to the same carbon skeleton having etherified hydroxy groups bound to carbon atoms of at least one six-membered aromatic ring and amino groups bound to acyclic carbon atoms or to carbon atoms of rings other than six-membered aromatic rings of the same carbon skeleton with rings other than six-membered aromatic rings being part of the carbon skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C217/00—Compounds containing amino and etherified hydroxy groups bound to the same carbon skeleton
- C07C217/54—Compounds containing amino and etherified hydroxy groups bound to the same carbon skeleton having etherified hydroxy groups bound to carbon atoms of at least one six-membered aromatic ring and amino groups bound to acyclic carbon atoms or to carbon atoms of rings other than six-membered aromatic rings of the same carbon skeleton
- C07C217/64—Compounds containing amino and etherified hydroxy groups bound to the same carbon skeleton having etherified hydroxy groups bound to carbon atoms of at least one six-membered aromatic ring and amino groups bound to acyclic carbon atoms or to carbon atoms of rings other than six-membered aromatic rings of the same carbon skeleton with amino groups linked to the six-membered aromatic ring, or to the condensed ring system containing that ring, by carbon chains further substituted by singly-bound oxygen atoms
- C07C217/66—Compounds containing amino and etherified hydroxy groups bound to the same carbon skeleton having etherified hydroxy groups bound to carbon atoms of at least one six-membered aromatic ring and amino groups bound to acyclic carbon atoms or to carbon atoms of rings other than six-membered aromatic rings of the same carbon skeleton with amino groups linked to the six-membered aromatic ring, or to the condensed ring system containing that ring, by carbon chains further substituted by singly-bound oxygen atoms with singly-bound oxygen atoms and six-membered aromatic rings bound to the same carbon atom of the carbon chain
- C07C217/70—Compounds containing amino and etherified hydroxy groups bound to the same carbon skeleton having etherified hydroxy groups bound to carbon atoms of at least one six-membered aromatic ring and amino groups bound to acyclic carbon atoms or to carbon atoms of rings other than six-membered aromatic rings of the same carbon skeleton with amino groups linked to the six-membered aromatic ring, or to the condensed ring system containing that ring, by carbon chains further substituted by singly-bound oxygen atoms with singly-bound oxygen atoms and six-membered aromatic rings bound to the same carbon atom of the carbon chain linked by carbon chains having two carbon atoms between the amino groups and the six-membered aromatic ring or the condensed ring system containing that ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C213/00—Preparation of compounds containing amino and hydroxy, amino and etherified hydroxy or amino and esterified hydroxy groups bound to the same carbon skeleton
- C07C213/10—Separation; Purification; Stabilisation; Use of additives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2601/00—Systems containing only non-condensed rings
- C07C2601/12—Systems containing only non-condensed rings with a six-membered ring
- C07C2601/14—The ring being saturated
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
本发明涉及新的基本上是纯的文拉法辛及其制备方法。本发明也涉及新的盐酸文拉法辛溶剂合物及其制备方法。而且,本发明提供了一种由文拉法辛制备盐酸文拉法辛的新方法,该方法包括以下步骤:i)制备文拉法辛的丙酮混合物;ii)将混合物暴露在氯化氢气体中。
Description
相关申请的交叉参考
本申请是由Ben-Zion Dolitzky、Judith Aronhime、Shlomit Weizel和Gennady Nisnevish (Doc.No.01662/54902)在2001年10月19日提交的名称为“结晶文拉法辛碱、新的盐酸文拉法辛多晶型物及它们的制备方法”的美国专利申请系列第10/045,510号的部分继续申请,该申请要求在2000年10月19日提交的临时申请系列第60/241,577号、在2000年12月29日提交的临时申请系列第60/258,861号、在2001年3月26日提交的临时申请系列第60/278,721号和在2001年5月21日提交的临时申请系列第60/292,469号的优先权。这些申请的内容通过引用其整体而结合到本文中。
发明背景
文拉法辛是第一种抗抑郁药物,其化学名为(±)-1-[2-(二甲氨基)-1-(4-乙氧基苯基)-乙基]-环己醇,具有下式I的结构。文拉法辛通过抑制去甲肾上腺素和5-羟色胺的再摄取起作用,是三环类抗抑郁药物和选择性再摄取抑制剂的替代产品。

美国专利第4,535,186号(`186专利)描述了通过中间体文拉法辛碱制备盐酸文拉法辛的方法。通过引用`186专利的整体而将其结合到本文中。但是,`186专利并没有描述如此得到的文拉法辛是否是固体。
欧洲专利申请EP 0 797 991 A1提及盐酸文拉法辛的某些多晶型物的存在。
Summary Basis of Approval of New Drug Application(新药申请批准概要)第20-151号(盐酸文拉法辛片)和第20-699号(文拉法辛缓释胶囊)中提及三种盐酸文拉法辛多晶型物。
我们现在发现一种新的分离文拉法辛固体的方法。分离出的文拉法辛为白色晶体,高效液相色谱(HPLC)测定其纯度为99.3%或以上。
我们发现结晶文拉法辛可以按照新方法,由盐酸文拉法辛通过N,N-二去甲基文拉法辛甲基化制备得到。
我们发现两种新的盐酸文拉法辛的多晶型物(命名为晶型I和晶型II)和两种新的溶剂合物(命名为晶型III和晶型IV)。
我们发现通过文拉法辛碱和氯化氢气体(HCl)在丙酮或异丙醇中制备盐酸文拉法辛的方法。我们还发现该方法在制备盐酸文拉法辛晶型I和晶型II中的用途。
发明概要
本发明的一方面涉及基本上是纯的文拉法辛。
本发明的另一方面涉及基本上是纯的盐酸文拉法辛。
本发明的另一方面提供了一种由盐酸文拉法辛制备文拉法辛碱的方法。
本发明的另一方面提供了一种通过N,N-二去甲基文拉法辛的烷基化反应制备文拉法辛碱的方法。
本发明的另一方面提供了一种结晶文拉法辛碱,所述文拉法辛碱为白色晶体,纯度约为97%。
本发明的另一方面提供了一种结晶文拉法辛碱,所述文拉法辛碱为白色晶体,纯度约为98%。
本发明的另一方面提供了一种结晶文拉法辛碱,所述文拉法辛碱为白色晶体,纯度约为99%。
本发明的另一方面提供了一种结晶文拉法辛碱,所述文拉法辛碱为白色晶体,纯度约为99.3%。
本发明的另一方面提供了一种结晶文拉法辛碱,所述文拉法辛碱为白色晶体,纯度约为99.5%。
本发明的一方面涉及一种由固体文拉法辛制备基本上是纯的盐酸文拉法辛的方法。
本发明的另一方面涉及两种新的分别命名为晶型I和晶型II的盐酸文拉法辛的多晶型物以及分别命名为晶型III和晶型IV的盐酸文拉法辛的溶剂合物。
本发明的另一方面提供了一种通过将所述化合物溶于水中并加入DMF(二甲基甲酰胺)或MEK(丁酮)使之沉淀制备无水晶型I的方法。
本发明的另一方面提供了一种通过将所述化合物溶于质子溶剂如水、乙醇或甲醇中并加入非质子溶剂如丙酮、乙酸乙酯、异丙醚或叔丁基甲基醚(MTBE)使之沉淀制备晶型III的溶剂合物的方法。
本发明的另一方面提供了一种通过将所述化合物溶于氯仿中并加入正己烷或甲苯使之沉淀制备晶型III的溶剂合物的方法。
本发明的另一方面提供了一种通过将所述化合物在无水乙醇或异丙醇中结晶制备晶型III的溶剂合物的方法。
本发明的另一方面提供了一种通过在非质子溶剂如乙酸乙酯、异丙醚或正己烷中研磨化合物制备晶型III的溶剂合物的方法。
本发明的另一方面提供了一种通过将所述化合物在DMF(二甲基甲酰胺)和DMSO(二甲基亚砜)中结晶、或将所述化合物溶于水中并加入DMSO使之沉淀制备晶型IV的溶剂合物的方法。
本发明的另一方面也提供了一种由文拉法辛碱制备盐酸文拉法辛的方法。
本发明的另一方面提供了一种制备盐酸文拉法辛的方法,所述方法包括以下步骤:形成文拉法辛、优选文拉法辛碱的丙酮混合物并将所述混合物暴露在氯化氢气体(HCl)中。
本发明的另一方面提供了一种制备盐酸文拉法辛的方法,所述方法包括以下步骤:形成文拉法辛、优选文拉法辛碱的异丙醇混合物,并加入盐酸(HCl)、优选氯化氢气体直至pH约为5-8,优选pH约为6-7.5,最优选pH约为7。
本发明的另一方面提供了一种制备盐酸文拉法辛的方法,所述方法包括将文拉法辛/丙酮的均匀溶液暴露在氯化氢气体(HCl)中。
本发明的另一方面提供了一种制备盐酸文拉法辛的方法,所述方法包括将文拉法辛/异丙醇的均匀溶液暴露在氯化氢气体(HCl)中。
本发明的另一方面提供了一种制备文拉法辛均匀溶液的方法,其中在所述溶液中文拉法辛基本不溶或微溶,溶剂优选为丙酮或异丙醇。
本发明的另一方面提供了制备文拉法辛晶型I和晶型II的方法。
本发明的另一方面提供了一种制备盐酸文拉法辛的方法,所述方法包括以下步骤:1)制备文拉法辛、优选文拉法辛碱和丙酮的混合物(或均匀溶液),和2)将所述混合物暴露在氯化氢气体(HCl)中。
本发明的另一方面提供了一种制备盐酸文拉法辛的方法,所述方法包括以下步骤:1)制备文拉法辛的异丙醇混合物;和2)将所述混合物暴露在一定pH范围的氯化氢气体中,其中所述pH范围约为5至8,优选pH范围约为6-7.5,最优选pH约为7。
本发明的另一方面提供了盐酸文拉法辛,其中所述盐酸文拉法辛为白色晶体,纯度约为99.92%。
本发明的另一方面提供了一种制备盐酸文拉法辛晶型1的方法,所述方法包括在丙酮中研磨盐酸文拉法辛,然后在搅拌下减压干燥并使盐酸文拉法辛结晶。
本发明的另一方面提供了盐酸文拉法辛晶型I,所述盐酸文拉法辛晶型I通过包括以下步骤的方法制备:在丙酮中研磨盐酸文拉法辛,然后在搅拌下减压干燥并使盐酸文拉法辛结晶。
本发明的另一方面提供了一种制备盐酸文拉法辛晶型I的方法,所述方法包括在异丙醇中研磨盐酸文拉法辛,然后在搅拌下减压干燥并使盐酸文拉法辛结晶。
本发明的另一方面提供了盐酸文拉法辛晶型I,所述盐酸文拉法辛晶型I通过包括以下步骤的方法制备:在异丙醇中研磨盐酸文拉法辛,然后在搅拌下减压干燥并使盐酸文拉法辛结晶。
本发明的另一方面提供了盐酸文拉法辛晶型I,所述盐酸文拉法辛晶型I为白色晶体,纯度约为99.95%。
本发明的另一方面提供了一种制备盐酸文拉法辛晶型II的方法,所述方法包括在丙酮或异丙醇中研磨盐酸文拉法辛,然后置于托盘中减压干燥并使盐酸文拉法辛结晶。
本发明的另一方面提供了盐酸文拉法辛晶型II,所述盐酸文拉法辛晶型II通过包括以下步骤的方法制备:在丙酮或异丙醇中研磨盐酸文拉法辛,然后置于托盘中减压干燥并使盐酸文拉法辛结晶。
本发明的另一方面提供了盐酸文拉法辛晶型II,所述盐酸文拉法辛晶型II为白色晶体,纯度约为99.95%。
附图简单描述
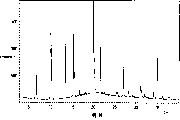
图1表示盐酸文拉法辛晶型I的差示扫描量热法(DSC)曲线。
图2表示盐酸文拉法辛晶型I的X-射线粉末衍射图谱(PXRD)。
图3表示盐酸文拉法辛晶型II的DSC曲线。
图4表示盐酸文拉法辛晶型II的PXRD图谱。
图5表示盐酸文拉法辛晶型III的DSC曲线。
图6表示盐酸文拉法辛晶型III的PXRD图谱。
图7表示盐酸文拉法辛晶型IV的DSC曲线。
图8表示盐酸文拉法辛晶型IV的PXRD图谱。
图9表示结晶文拉法辛碱的PXRD图谱。
图10表示在氯化氢气体(HCl)和丙酮存在下,由文拉法辛碱制备盐酸文拉法辛的方法示意图。
发明详细描述
在本文中使用下述缩写术语:“DMF”是指二甲基甲酰胺;“MEK”是指丁酮;“MTBE”是指叔丁基甲醚;“DMSO”是指二甲亚砜;“DSC”是指差示扫描量热法;“PXRD”是指X-射线粉末衍射图谱;“IPA”是指异丙醇;“HCl”是指盐酸。
1)文拉法辛游离碱
本发明涉及基本上是纯的文拉法辛,所述文拉法辛可令人惊讶地以游离碱形式获得。所得的文拉法辛游离碱以固体结晶形态存在。
基本上是纯的文拉法辛通过将氢氧化钠加入盐酸文拉法辛的水溶液中制得。另一种优选的碱溶液是氢氧化钾。所得混合物用有机溶剂萃取。萃取可用乙酸乙酯、庚烷、正己烷及其混合物来实施。萃取溶剂优选为乙酸乙酯。优选用无水硫酸钠干燥合并的有机层并蒸发。然后残余物在庚烷或己烷中结晶。
过滤如此得到的结晶,用冷的庚烷或己烷洗涤,干燥得到文拉法辛固体,纯度为99.3%或更高。所述文拉法辛固体的纯度一般高于97%,优选高于98%,最优选高于99%。
文拉法辛固体进一步与盐酸反应并结晶得到基本上是纯的盐酸文拉法辛。
通过下面的实施列进一步说明本发明,但并不因此将本发明限制在所述的实施例范围中。
实施例1
在冰水浴中,将32%的氢氧化钠水溶液(10.0克,80.0毫摩尔)加入至搅拌的盐酸文拉法辛(20.0克,63.7毫摩尔)的水(100毫升)溶液中。混合物在冰水浴中搅拌约30分钟,用乙酸乙酯(3×30毫升)萃取。合并的有机层用无水硫酸钠干燥,过滤,约50-60℃下减压蒸发(水浴)。残余物溶于沸腾的己烷(50毫升)中并在冰箱中冷却(-18℃)。
过滤如此得到的结晶,用冷的己烷(20毫升)洗涤,减压干燥得到15.5克(87.7%)文拉法辛,为白色晶体,HPLC测定纯度约为99.3%,熔点为78.3-79.5℃。
实施例2
由N,N-二去甲基盐酸文拉法辛制备结晶文拉法辛游离碱
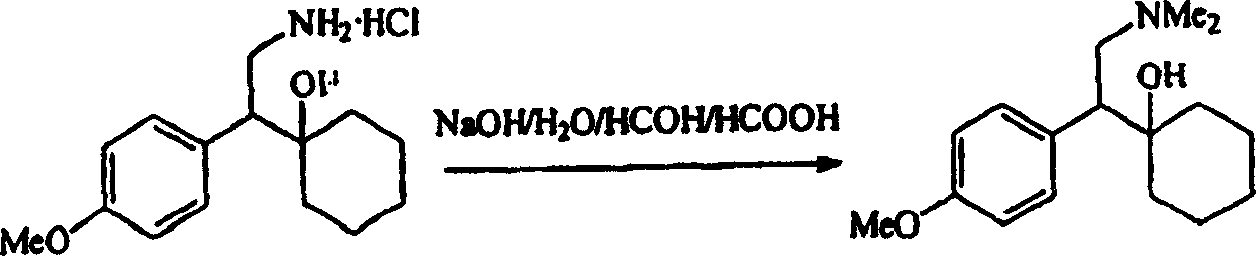
室温下,将32%的氢氧化钠水溶液(2.75克,0.022摩尔)加入至搅拌的N,N-二去甲基盐酸文拉法辛(5.72克,0.02摩尔)的水(13毫升)溶液中。将88.5%的甲酸水溶液(4.16克,0.08摩尔)和35.8%的甲醛水溶液(3.7克,0.044摩尔)加入至该乳状液中。所得混合物在回流条件下搅拌8小时,冷却至室温,用32%的氢氧化钠水溶液调节pH至约11,用庚烷(100毫升)萃取。
有机萃取液用水(20毫升)洗涤,硫酸钠干燥,蒸发掉两体积的溶剂,过滤得到结晶文拉法辛碱。
实施例3
文拉法辛碱的制备
室温下,将N,N-二去甲基文拉法辛(20毫摩尔)加入至水(480毫升)、甲酸(88.5%,5.2克,~100毫摩尔)、甲醛(35.8%,5克,62毫摩尔)中。所得混合物在回流条件下搅拌21小时,冷却至室温。用32%的氢氧化钠水溶液调节pH至约11,同样可用氢氧化钾调节pH至约11。调节pH后混合物用甲苯(50毫升×5)萃取。
合并的有机相用水(50毫升)洗涤,硫酸钠干燥,蒸发至干,得到结晶文拉法辛碱(5.4克,98%)。HPLC测定纯度约为99.5%。所得物可在己烷、戊烷、石油醚等中结晶。结晶文拉法辛碱的熔点范围为78.3-79.5℃。
II)盐酸文拉法辛
本发明提供了一种纯化盐酸文拉法辛的方法,所述方法包括碱化盐酸文拉法辛。
本发明提供了一种纯化盐酸文拉法辛的方法,所述方法进一步包括使文拉法辛结晶。
本发明提供了一种纯化盐酸文拉法辛的方法,所述方法进一步包括使如此制备的文拉法辛与盐酸反应并结晶重新得到纯度更高的盐酸文拉法辛。盐酸文拉法辛的纯度一般高于约97%,优选高于98%最优选高于约99%。
按照美国专利第4,535,186号描述的方法制得盐酸文拉法辛,该专利通过引用结合到本文中。
III)盐酸文拉法辛的新溶剂合物和多晶型物
盐酸文拉法辛晶型I
本发明的一方面涉及一种命名为晶型I的盐酸文拉法辛的新多晶型物。该晶型的特征为在2θ为约10.2、15.5、20.3、21.7±0.2°处有特强X射线衍射峰,和在2θ为6.7、13.5、18.2、19.8、22.6、25.6、28.1、35.1±0.2°处有中等强度的X射线衍射峰。
由于发生熔化,晶型I的DSC热分析图在210-213℃包含一吸热峰。
盐酸文拉法辛晶型II
本发明另一方面涉及一种命名为晶型II的盐酸文拉法辛的新多晶型物。该晶型特征为在2θ为约12.8、20.5、21.3±0.2°处有特强X射线衍射峰,和在2θ为6.8、8.5、10.3、13.6、15.6、16.5、19.8、19.9、21.9、25.2、28.7、31.2、31.7、35.3±0.2°处有中等强度的X射线衍射峰。
由于发生熔化,晶型II的DSC热分析图在210-213℃包含一吸热峰,在约219-222℃经常可以观察到因相变化产生的峰。该变化的发生程度的可能不同并可能伴随发生升华现象。
盐酸文拉法辛晶型III
本发明另一方面涉及一种命名为晶型III的盐酸文拉法辛的新溶剂合物晶型。该晶型特征为在2θ为约7.4、14.9、26.5±0.2°处有特强X射线衍射峰,和在2θ为约12.9、16.4、17.5、18.6、18.9、20.5、21.4、38.2±0.2°处有中等强度的X射线衍射峰。
由于去溶剂化作用,晶型III的DSC热分析图包含一宽的吸热峰,并且由于发生熔化,在大约180-200℃包含一小的吸热峰和在大约212℃包含一吸热峰。
该溶剂合物可以包含水、甲醇、乙醇或己烷。含有甲醇或乙醇的化合物的干燥失重值范围约为5.6%-6.0%,含有异丙醇的化合物的干燥失重约为4.6%,和含有己烷的化合物的干燥失重约为5.5%。
这些数值表明每分子盐酸文拉法辛含约1/2分子的甲醇或乙醇和1/4分子的异丙醇的化学计量组成。这些数据表明存在乙醇或甲醇的半溶剂合物和异丙醇的1/4溶剂合物。
盐酸文拉法辛晶型IV
本发明另一方面涉及一种命名为晶型IV的盐酸文拉法辛的新溶剂合物晶型。该晶型特征为在2θ为约10.3、20.3±0.2°处有特强X射线衍射峰,和在2θ为6.8、13.5、15.6、21.8、27.2、35.2±0.2°处有中等强度的X射线衍射峰。
由于去溶剂化作用,晶型IV的DSC热分析图包含一宽的吸热峰,并且由于发生熔化,在大约212℃包含一吸热峰。
所述该溶剂合物晶型可包含DMSO或DMF。在DMSO中结晶的化合物的干燥失重经TGA测定约为41%,在DMF中结晶的化合物的干燥失重经TGA测定约为33%。这些数值(约44%和33%)相应于每分子盐酸文拉法辛三分子DMSO和两分子DMF的化学组成。由此我们可以推知该溶剂化的晶型IV可以是DMSO的三溶剂合物和DMF的二溶剂合物。
IV)结晶盐酸文拉法辛的多晶型物的制备
本发明公开了盐酸文拉法辛的不同多晶型物的制备方法。
在干燥过程观察到,溶剂合物转化得到新的多晶型物(命名为晶型I和晶型II)。
结晶过程观察到生成新的溶剂合物(命名为晶型III和晶型IV)。
观察到溶剂合物III和IV在干燥过程会导致生成晶型I、晶型II或两种晶型的混合物。通过使用旋转蒸发器,其中干燥条件包括减压、粉末连续旋转、适度加热(约60℃),主要得到晶型I,但在一些情况下也得到晶型I或晶型I和晶型II的混合物。通过在静态烘箱(static oven)中干燥溶剂合物(约160℃下1/2小时),晶型III转化为晶型II,晶型IV转化为晶型I。
还观察到,晶型III可以和不同的溶剂例如乙醇、甲醇或异丙醇形成溶剂合物。
还观察到,晶型IV可以和DMF和DMSO形成溶剂合物。
发现了一种可生成晶型III的新溶剂合物的方法。在该方法中,将盐酸文拉法辛溶于质子溶剂(即具有羟基(-OH)的溶剂,例如水、乙醇或甲醇)中,并加入非质子溶剂(即不含羟基(-OH)的溶剂,例如丙酮、乙酸乙酯、异丙醚或叔丁基甲醚(MTBE))生成晶型III的溶剂合物。在旋转蒸发器中约60℃下进一步减压(~10毫巴)干燥样品约45分钟,得到新的晶型I多晶型物。
发现了一种制备晶型III的新溶剂合物的方法,所述方法包括将盐酸文拉法辛溶于氯仿中,并向所得溶液中加入DMF和DMSO。在旋转蒸发器中约60℃下进一步减压(~10毫巴)干燥样品约45分钟,得到新的晶型I多晶型物。
在乙醇、异丙醇、氯仿中直接结晶也生成晶型III,在旋转蒸发器中约60℃下进一步减压(~10毫巴)干燥样品约45分钟,得到新的多晶型物晶型I或晶型I和晶型II的混合物。
在DMF和DMSO中直接结晶生成晶型IV的新溶剂合物,在旋转蒸发器中约60℃下进一步减压(~10毫巴)干燥样品约45分钟,得到新的晶型II多晶型物或晶型I和晶型II的混合物。
发现了一种制备新的晶型I多晶型物的方法,所述方法包括将盐酸文拉法辛溶于水中,并将MEK或DMF加入所述溶液中。
发现了一种制备新的晶型II多晶型物的方法,所述方法包括将盐酸文拉法辛溶于甲醇中,并以溶剂∶反溶剂约为3∶30的比例将乙酸乙酯加入所述溶液中。
发现了一种方法,其中将盐酸文拉法辛溶于异丙醇中,并将所得溶液暴露在一定pH范围内的氯化氢气体中,pH范围约为5-8,优选pH范围约为6-7.5,最优选pH值约为7。
方法
X-射线粉末衍射法(PXRD)
X-射线衍射仪,飞利浦TW1830发生器
PW3020测角仪
PW3710 MPD控制
X-射线管,铜靶阳极
单色仪,正比计数器
发散狭缝1°(Divergence Slits),接受狭缝0.2毫米,发散狭缝1°
功率:40千伏,30毫安
扫描速度:2°/分钟 步长:0.05°
热重分析(TGA)
岛津DTG-50
样品重量:7-15毫克
温度范围:最高至185℃
加热速率:10℃/分钟
差示扫描量热法(DSC)
Mettler Toledo DSC821e
样品重量:3-5毫克
温度范围:30-250℃
加热速率:10℃/分钟
坩锅孔数:3
实施例4
用溶剂/反溶剂制备晶型III和晶型I
比例:0.7毫升水:9.7毫升丙酮:3克盐酸文拉法辛
将盐酸文拉法辛在回流下溶入水中。加入丙酮。将形成的悬浮液再回流10分钟,并在室温下暴露过夜。然后过滤悬浮液,用约2毫升相同的溶剂混合物洗涤。结晶得到的固体为晶型III。在旋转蒸发器中约60℃下进一步减压(~10毫巴)干燥约45分钟得到晶型I。
实施例5
用溶剂/反溶剂制备晶型III和晶型I
比例:3毫升甲醇:9.5毫升乙酸乙酯:2.5克盐酸文拉法辛
比例:3.8毫升甲醇:2毫升异丙醚:3克盐酸文拉法辛
比例:3.5毫升甲醇:2毫升MTBE:3.1克盐酸文拉法辛
将盐酸文拉法辛在回流下溶入甲醇中。加入乙酸乙酯、异丙醚或MTBE。将形成的悬浮液再回流10分钟,在室温下暴露过夜。然后过滤悬浮液,用2毫升相同的溶剂混合物洗涤。结晶得到的固体为晶型III。在旋转蒸发器中约60℃下进一步减压(~10毫巴)干燥约45分钟得到晶型I。
实施例6
用溶剂/反溶剂制备晶型III和晶型I/II
比例:12毫升氯仿∶5毫升己烷∶2.5克盐酸文拉法辛
比例:6毫升乙醇∶9毫升乙酸乙酯∶3克盐酸文拉法辛
比例:12毫升氯仿∶5毫升甲苯∶2.6克盐酸文拉法辛
将盐酸文拉法辛在回流下溶入溶剂中。加入反溶剂。将形成的悬浮液再回流10分钟,在室温下暴露过夜。然后过滤悬浮液,用2毫升相同的溶剂混合物洗涤。结晶得到的固体为晶型III。在旋转蒸发器中约60℃下进一步减压(~10毫巴)干燥约45分钟得到晶型II或晶型I,或两种晶型的混合物。
实施例7
直接结晶制备晶型III和晶型I/晶型II
将盐酸文拉法辛(2克)在回流下溶于乙醇(8毫升)或异丙醇(10毫升)中,将所得溶液在室温下静置过夜。将结晶的物质过滤,用2毫升相同的溶剂洗涤。结晶得到的固体为晶型III。在旋转蒸发器中约60℃下进一步减压(~10毫巴)干燥约45分钟得到晶型II或晶型I,或两种晶型的混合物。
实施例8
直接结晶制备晶型IV和晶型I/II
将盐酸文拉法辛(2克)在回流下溶于DMF或DMSO(8毫升)中,所得溶液在室温下静置过夜。将结晶的物质过滤,用2毫升相同的溶剂洗涤。结晶得到的固体为晶型III。在旋转蒸发器中约60℃下进一步减压(~10毫巴)干燥约45分钟得到晶型II或晶型I,或两种晶型的混合物。
实施例9
用溶剂/反溶剂制备晶型I
比例:0.5毫升水∶13毫升DMF∶3克盐酸文拉法辛
比例:0.5毫升水∶13毫升DMSO∶3.1克盐酸文拉法辛
将盐酸文拉法辛在回流下溶入水中。加入反溶剂。将形成的悬浮液再回流10分钟,在室温下暴露过夜。然后过滤悬浮液,用2毫升相同的溶剂混合物洗涤。结晶得到的固体为晶型I。在旋转蒸发器中约60℃下进一步减压(~10毫巴)干燥约45分钟得到晶型I。
实施例10
用溶剂/反溶剂制备晶型II
比例:10毫升甲醇∶30毫升乙酸乙酯∶3克盐酸文拉法辛
在约0-5℃下将盐酸文拉法辛溶入甲醇中。加入反溶剂。搅拌形成的悬浮液30分钟。然后过滤悬浮液,用2毫升相同的溶剂混合物洗涤。结晶得到的固体为晶型II。在旋转蒸发器中约60℃下进一步减压(~10毫巴)干燥约45分钟得到晶型II。
实施例11
在静态烘箱中加热晶型III制备晶型II
将晶型III的样品在静态烘箱中约160℃下放置约1/2小时,得到的多晶型物是晶型II。
实施例12
在静态烘箱中加热晶型IV制备晶型I
将晶型IV样品在静态烘箱中约160℃下放置约1/2小时,得到的多晶型物是晶型I。
实施例13
通过研磨晶型I制备晶型III
将盐酸文拉法辛晶型I样品(2克)在异丙醚、己烷或乙酸乙酯(8毫升)中在回流条件下研磨约1小时,或在室温条件下研磨过夜。固体中包含溶剂化的晶型III。
V)在丙酮中由文拉法辛碱和HCl气体制备盐酸文拉法辛
本发明提供一种制备盐酸文拉法辛的方法,该方法包括将文拉法辛碱暴露在氯化氢气体中(HCl)。
由文拉法辛碱制备盐酸文拉法辛的示意方法见图10。
实施例14
制备盐酸文拉法辛粗产物
由文拉法辛碱制备盐酸文拉法辛所需要的反应物和溶剂见表1总结。
表1:反应物和溶剂
1.文拉法辛碱 27.7克 100毫摩尔 1.0当量
2.HCl,气体
3.丙酮 846克
产物(即盐酸文拉法辛)的理论收率是约31.34克(即100毫摩尔)。
向装有机械搅拌器、温度计、pH电极和聚四氟乙烯长管的1升的双夹套反应器中装入文拉法辛碱(约27.7克)和丙酮(约526克)。在室温下搅拌混合物约20分钟直至获得均匀溶液。
在约10℃、剧烈搅拌下,用氯化氢气体酸化溶液至溶液pH约为2.0。所得悬浮液在约10℃下搅拌约2小时。
将沉淀出的晶体过滤,用冷的丙酮(约120克)洗涤,在约50℃(水浴)下减压干燥至恒重,得到约29.57克(约94.4%)盐酸文拉法辛的白色晶体,HPLC测定纯度约为99.92%。
实施例15
制备盐酸文拉法辛(晶型I)
在约60℃下,将盐酸文拉法辛粗品(约15.0克)在丙酮(约60.0克)中研磨约1小时和在0℃下研磨约1小时,过滤,用冷的丙酮(约120克)洗涤,在搅拌及约50℃(水浴)下减压干燥至恒重,得到约14.8克(约93.2%)盐酸文拉法辛的白色晶体,HPLC测定纯度约为99.95%。
实施例16
制备盐酸文拉法辛(晶型II)
在约60℃下,将盐酸文拉法辛粗品(约15.0克)在丙酮(约60.0克)中研磨约1小时和在0℃下研磨约1小时,过滤,用冷的丙酮(约120克)洗涤,置于托盘中在约50℃(水浴)下减压干燥至恒重,得到约14.8克(约93.2%)盐酸文拉法辛的白色晶体,HPLC测定纯度为99.95%。
实施例17
制备盐酸文拉法辛(晶型I)
将文拉法辛碱(1kg)溶于异丙醇(6升)中。约20℃下通过通入盐酸(气体)鼓泡直至pH范围达到约为5至8,优先pH范围约为6至7.5,最优选pH值约为7。加热反应混合物至成为澄清溶液,然后逐渐冷却至10℃。将沉淀过滤,用异丙醇洗涤并真空干燥。
本发明并不仅限于本文描述的具体实施方案的范围。事实上,本领域技术人员通过阅读本发明说明书和附图会认识到可以对本发明作出各种修改。这些修改属于专利要求的范围。
Claims (27)
1.一种白色晶型的结晶文拉法辛碱,所述文拉法辛碱的纯度至少为约97%。
2.一种白色晶型的结晶文拉法辛碱,所述文拉法辛碱的纯度至少为约98%。
3.一种白色晶型的结晶文拉法辛碱,所述文拉法辛碱的纯度至少为约99%。
4.一种白色晶型的结晶文拉法辛碱,所述文拉法辛碱的纯度至少为约99.3%。
5.一种白色晶型的结晶文拉法辛碱,所述文拉法辛碱的纯度至少为约99.5%。
6.一种制备纯度至少为约97%的结晶文拉法辛碱的方法,所述方法包括以下步骤:1)在第一种有机溶剂中制备N,N-二去甲基文拉法辛的混合物;2)将选自一组碱的碱溶液加入至所述混合物中调节pH至碱性;和3)用第二种有机溶剂萃取所述文拉法辛碱。
7.权利要求6的方法,其中所述纯度至少约为98%。
8.权利要求7的方法,其中所述纯度至少约为99%。
9.权利要求8的方法,其中所述纯度至少约为99.3%。
10.权利要求9的方法,其中所述纯度至少约为99.5%。
11.权利要求6-10中任一项的方法,其中所述碱溶液选自氢氧化钠和氢氧化钾。
12.权利要求6-10中任一项的方法,其中所述第一种有机溶剂为甲酸和甲醛。
13.权利要求6-10中任一项的方法,其中所述第二种有机溶剂选自甲苯和庚烷。
14.权利要求6-10中任一项的方法,所述方法还包括将第二种有机溶剂干燥至无水。
15.权利要求14的方法,其中所述干燥通过加热或真空实现。
16.权利要求6-10中任一项的方法,所述方法还包括步骤4)将文拉法辛碱从选自己烷、戊烷和石油醚的溶剂中结晶。
17.权利要求6-10中任一项的方法制备的结晶文拉法辛碱,所述结晶文拉法辛碱的纯度至少约为97%。
18.权利要求6-10中任一项的方法制备的结晶文拉法辛碱,所述结晶文拉法辛碱的纯度至少约为98%。
19.权利要求6-10中任一项的方法制备的结晶文拉法辛碱,所述结晶文拉法辛碱的纯度至少约为99%。
20.权利要求6-10中任一项的方法制备的结晶文拉法辛碱,所述结晶文拉法辛碱的纯度至少约为99.3%。
21.权利要求6-10中任一项的方法制备的结晶文拉法辛碱,所述结晶文拉法辛碱的纯度至少约为99.5%。
22.一种制备盐酸文拉法辛晶型I的方法,所述方法包括以下步骤:
1)制备文拉法辛的异丙醇混合物;和
2)加入盐酸直至pH范围约为5-8。
23.权利要求22的方法,其中所述pH范围约为6-7.5。
24.权利要求22的方法,其中所述pH约为7。
25.权利要求22的方法,其中所述盐酸是氯化氢气体。
26.权利要求22的方法,其中所述文拉法辛是文拉法辛碱。
27.权利要求22的方法,其中所述混合物是文拉法辛的均相溶液。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US10/000,428 | 2001-11-30 | ||
| US10/000,428 US20020183553A1 (en) | 2000-10-19 | 2001-11-30 | Crystalline venlafaxine base and novel polymorphs of venlafaxine hydrochloride, processes for preparing thereof |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CNA2007101970607A Division CN101219962A (zh) | 2001-11-30 | 2002-11-20 | 结晶文拉法辛碱、新的盐酸文拉法辛多晶型物及它们的制备方法 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| CN1617848A true CN1617848A (zh) | 2005-05-18 |
Family
ID=21691493
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CNA028276485A Pending CN1617848A (zh) | 2001-11-30 | 2002-11-20 | 结晶文拉法辛碱、新的盐酸文拉法辛多晶型物及它们的制备方法 |
| CNA2007101970607A Pending CN101219962A (zh) | 2001-11-30 | 2002-11-20 | 结晶文拉法辛碱、新的盐酸文拉法辛多晶型物及它们的制备方法 |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CNA2007101970607A Pending CN101219962A (zh) | 2001-11-30 | 2002-11-20 | 结晶文拉法辛碱、新的盐酸文拉法辛多晶型物及它们的制备方法 |
Country Status (16)
| Country | Link |
|---|---|
| US (4) | US20020183553A1 (zh) |
| EP (1) | EP1474379A4 (zh) |
| JP (1) | JP2005511668A (zh) |
| KR (1) | KR20040068163A (zh) |
| CN (2) | CN1617848A (zh) |
| AU (1) | AU2002365874A1 (zh) |
| CA (1) | CA2468728A1 (zh) |
| HR (1) | HRP20040565A2 (zh) |
| HU (1) | HUP0700037A2 (zh) |
| IL (1) | IL162171A0 (zh) |
| IS (1) | IS7289A (zh) |
| MX (1) | MXPA04005193A (zh) |
| NO (1) | NO20042732L (zh) |
| PL (1) | PL374273A1 (zh) |
| WO (1) | WO2003048082A2 (zh) |
| ZA (1) | ZA200404080B (zh) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN101279926A (zh) * | 2008-05-22 | 2008-10-08 | 杭州盛美医药科技开发有限公司 | 盐酸文拉法辛晶型FormⅠ的制备方法 |
Families Citing this family (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR20030059206A (ko) * | 2000-10-19 | 2003-07-07 | 테바 파마슈티컬 인더스트리즈 리미티드 | 결정질 벤라팍신 염기 및 벤라팍신 히드로클로라이드의신규한 다형태, 이의 제조 방법 |
| AU2002212340B2 (en) * | 2000-10-31 | 2006-12-14 | Sandoz Ag | Crystalline forms of venlafaxine hydrochloride |
| CN1630631A (zh) * | 2001-12-05 | 2005-06-22 | 惠氏公司 | 新的文拉法星盐酸盐多晶型物及其制备方法 |
| UA77234C2 (en) * | 2001-12-05 | 2006-11-15 | Wyeth Corp | Monohydrate of venlafaxine hydrochloride and methods for its preparation (variants) |
| US20030190352A1 (en) * | 2002-03-28 | 2003-10-09 | Synthon Bv | Compositions of venlafaxine base |
| DE10359154A1 (de) * | 2003-12-16 | 2005-07-28 | Krka Tovarna Zdravil, D.D. | Verfahren zur Herstellung von Venlafaxin und Venlafaxinhydrochlorid der Form I |
| KR20080056311A (ko) * | 2005-10-19 | 2008-06-20 | 테바 파마슈티컬 인더스트리즈 리미티드 | 고순도1-[2-디메틸아미노-(4-메톡시페닐)에틸)시클로헥산올염산염의 제조 방법 |
| PT1954669E (pt) * | 2005-12-01 | 2015-10-23 | Auspex Pharmaceuticals Inc | Feniletilaminas substituídas com actividade serotoninérgica e/ou norepinefrinérgica |
| SI2125698T1 (sl) * | 2007-03-15 | 2016-12-30 | Auspex Pharmaceuticals, Inc. | Devterirani d9-venlafaksin |
| EP4345100A3 (en) | 2012-09-18 | 2024-04-10 | Auspex Pharmaceuticals, Inc. | Formulations pharmacokinetics of deuterated benzoquinoline inhibitors of vesicular monoamine transporter 2 |
| MY180626A (en) | 2013-11-15 | 2020-12-03 | Akebia Therapeutics Inc | Solid forms of {[5-(3-chlorophenyl)-3-hydroxypyridine-2-carbonyl]amino}acetic acid, compositions, and uses thereof |
| AU2016229949B2 (en) | 2015-03-06 | 2021-04-08 | Auspex Pharmaceuticals Llc | Methods for the treatment of abnormal involuntary movement disorders |
Family Cites Families (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| IL63968A (en) * | 1980-10-01 | 1985-10-31 | Glaxo Group Ltd | Form 2 ranitidine hydrochloride,its preparation and pharmaceutical compositions containing it |
| US4535186A (en) * | 1983-04-19 | 1985-08-13 | American Home Products Corporation | 2-Phenyl-2-(1-hydroxycycloalkyl or 1-hydroxycycloalk-2-enyl)ethylamine derivatives |
| US4611078A (en) * | 1983-10-26 | 1986-09-09 | American Home Products Corporation | Substituted phenylacetonitriles |
| US6197828B1 (en) * | 1998-12-01 | 2001-03-06 | Sepracor, Inc. | Derivatives of (+)-venlafaxine and methods of preparing and using the same |
| KR20030059206A (ko) * | 2000-10-19 | 2003-07-07 | 테바 파마슈티컬 인더스트리즈 리미티드 | 결정질 벤라팍신 염기 및 벤라팍신 히드로클로라이드의신규한 다형태, 이의 제조 방법 |
| AU2002212340B2 (en) | 2000-10-31 | 2006-12-14 | Sandoz Ag | Crystalline forms of venlafaxine hydrochloride |
| WO2002046140A1 (en) | 2000-12-07 | 2002-06-13 | Dr. Reddy's Laboratories Ltd. | Novel crystalline polymorphic forms of venlafaxine hydrochloride and a process for their preparation |
| DE60136263D1 (de) * | 2001-04-10 | 2008-12-04 | Alembic Ltd | Zwischenprodukt zur Herstellung von 1-Ä2-Dimethylamino-1-(4-methoxyphenyl)-ethylÜ-cyclohexanol |
| CN1630631A (zh) * | 2001-12-05 | 2005-06-22 | 惠氏公司 | 新的文拉法星盐酸盐多晶型物及其制备方法 |
| WO2003050074A1 (en) * | 2001-12-13 | 2003-06-19 | Cadila Healthcare Limited | Manufacture of venlafaxine hydrochloride and crystalline polymorphs thereof |
-
2001
- 2001-11-30 US US10/000,428 patent/US20020183553A1/en not_active Abandoned
-
2002
- 2002-11-20 CA CA002468728A patent/CA2468728A1/en not_active Abandoned
- 2002-11-20 CN CNA028276485A patent/CN1617848A/zh active Pending
- 2002-11-20 PL PL02374273A patent/PL374273A1/xx not_active Application Discontinuation
- 2002-11-20 ZA ZA200404080A patent/ZA200404080B/en unknown
- 2002-11-20 CN CNA2007101970607A patent/CN101219962A/zh active Pending
- 2002-11-20 EP EP02791279A patent/EP1474379A4/en not_active Withdrawn
- 2002-11-20 KR KR10-2004-7008012A patent/KR20040068163A/ko not_active Ceased
- 2002-11-20 HU HU0700037A patent/HUP0700037A2/hu unknown
- 2002-11-20 AU AU2002365874A patent/AU2002365874A1/en not_active Abandoned
- 2002-11-20 HR HR20040565A patent/HRP20040565A2/xx not_active Application Discontinuation
- 2002-11-20 IL IL16217102A patent/IL162171A0/xx unknown
- 2002-11-20 WO PCT/US2002/037268 patent/WO2003048082A2/en not_active Ceased
- 2002-11-20 JP JP2003549277A patent/JP2005511668A/ja active Pending
- 2002-11-20 MX MXPA04005193A patent/MXPA04005193A/es active IP Right Grant
-
2004
- 2004-05-27 IS IS7289A patent/IS7289A/is unknown
- 2004-06-08 US US10/863,958 patent/US20040220278A1/en not_active Abandoned
- 2004-06-29 NO NO20042732A patent/NO20042732L/no not_active Application Discontinuation
- 2004-10-07 US US10/961,337 patent/US6924393B2/en not_active Expired - Fee Related
-
2008
- 2008-03-13 US US12/075,875 patent/US20080167498A1/en not_active Abandoned
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN101279926A (zh) * | 2008-05-22 | 2008-10-08 | 杭州盛美医药科技开发有限公司 | 盐酸文拉法辛晶型FormⅠ的制备方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| US20040220278A1 (en) | 2004-11-04 |
| US6924393B2 (en) | 2005-08-02 |
| US20050049304A1 (en) | 2005-03-03 |
| EP1474379A4 (en) | 2006-06-07 |
| IL162171A0 (en) | 2005-11-20 |
| WO2003048082A2 (en) | 2003-06-12 |
| ZA200404080B (en) | 2007-12-27 |
| US20080167498A1 (en) | 2008-07-10 |
| NO20042732L (no) | 2004-06-29 |
| AU2002365874A1 (en) | 2003-06-17 |
| JP2005511668A (ja) | 2005-04-28 |
| WO2003048082A3 (en) | 2004-08-26 |
| IS7289A (is) | 2004-05-27 |
| EP1474379A2 (en) | 2004-11-10 |
| MXPA04005193A (es) | 2005-02-22 |
| PL374273A1 (en) | 2005-10-03 |
| US20020183553A1 (en) | 2002-12-05 |
| KR20040068163A (ko) | 2004-07-30 |
| CA2468728A1 (en) | 2003-06-12 |
| HRP20040565A2 (en) | 2005-08-31 |
| CN101219962A (zh) | 2008-07-16 |
| HUP0700037A2 (en) | 2007-05-02 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN1245974C (zh) | 卡维地洛 | |
| CN1769262A (zh) | 苯甲酰基衍生物和其盐酸盐及其制备方法 | |
| CN1617848A (zh) | 结晶文拉法辛碱、新的盐酸文拉法辛多晶型物及它们的制备方法 | |
| CN1589256A (zh) | 盐酸舍曲林的新多晶型物和含有它们的组合物,制备盐酸舍曲林多晶型物和无定形物的新方法 | |
| CN1620420A (zh) | 结晶文拉法辛碱和新的文拉法辛盐酸盐多晶型物及其制备方法 | |
| CN1083430C (zh) | 咔唑酮衍生物 | |
| CN1155882A (zh) | 萘酚衍生物及其制备方法 | |
| CN1349522A (zh) | 2,2-二甲基-1,3-二氧杂环己烷中间体的盐及其制备方法 | |
| CN1095830C (zh) | 制备烷氧基亚氨基乙酰胺衍生物的方法 | |
| CN1030611C (zh) | 形式ii地红霉素的制备方法 | |
| CN101058543A (zh) | 制备胺衍生物的方法 | |
| CN1461301A (zh) | 苯并[b]噻吩衍生物及其制备方法 | |
| CN1247556C (zh) | 二苯并硫氮䓬衍生物的制备方法 | |
| CN1309718C (zh) | R-硫辛酸的氨基丁三醇盐的新的变性体及其制备方法 | |
| CN1227252C (zh) | 制备6-甲基-2-(4-甲基-苯基)-咪唑并[1,2-a]吡啶-3-(N,N-二甲基-乙酰胺)和中间体的方法 | |
| CN1266131C (zh) | 制备杂环茚类似物的方法 | |
| CN1178934C (zh) | 苯并呋喃衍生物 | |
| CN1195739C (zh) | 2-氨磺酰苯甲酸衍生物 | |
| CN1993369A (zh) | 特比萘芬及其衍生物的合成方法 | |
| CN101056853A (zh) | 异吲哚衍生物的制备方法 | |
| WO2010078250A1 (en) | Synthesis of (2-amino)-tetrahydrocarbazole-propanoic acid | |
| CN1875018A (zh) | 制备噻唑烷二酮的方法 | |
| CN1087337A (zh) | 取代的喹啉-2-基甲氧基苯乙酸衍生物 | |
| CN1325381A (zh) | 酮亚胺的制备方法 | |
| CN1360574A (zh) | 制备三环氨基醇衍生物的方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| AD01 | Patent right deemed abandoned | ||
| C20 | Patent right or utility model deemed to be abandoned or is abandoned |