CN1993376A - 制备雌酮和雌二醇的2-取代-衍生物的方法 - Google Patents
制备雌酮和雌二醇的2-取代-衍生物的方法 Download PDFInfo
- Publication number
- CN1993376A CN1993376A CNA2005800261640A CN200580026164A CN1993376A CN 1993376 A CN1993376 A CN 1993376A CN A2005800261640 A CNA2005800261640 A CN A2005800261640A CN 200580026164 A CN200580026164 A CN 200580026164A CN 1993376 A CN1993376 A CN 1993376A
- Authority
- CN
- China
- Prior art keywords
- compound
- general formula
- alkyl
- group
- carbon atom
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Images

Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07J—STEROIDS
- C07J75/00—Processes for the preparation of steroids in general
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07J—STEROIDS
- C07J75/00—Processes for the preparation of steroids in general
- C07J75/005—Preparation of steroids by cyclization of non-steroid compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
- A61P15/08—Drugs for genital or sexual disorders; Contraceptives for gonadal disorders or for enhancing fertility, e.g. inducers of ovulation or of spermatogenesis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/02—Drugs for dermatological disorders for treating wounds, ulcers, burns, scars, keloids, or the like
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Animal Behavior & Ethology (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Immunology (AREA)
- Rheumatology (AREA)
- Reproductive Health (AREA)
- Heart & Thoracic Surgery (AREA)
- Cardiology (AREA)
- Dermatology (AREA)
- Vascular Medicine (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Physical Education & Sports Medicine (AREA)
- Urology & Nephrology (AREA)
- Gynecology & Obstetrics (AREA)
- Pregnancy & Childbirth (AREA)
- Pain & Pain Management (AREA)
- Endocrinology (AREA)
- Steroid Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Heterocyclic Compounds That Contain Two Or More Ring Oxygen Atoms (AREA)
- Pyrane Compounds (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
本发明提供了制备雌酮和雌二醇的2-取代-衍生物的方法,包含i)如下制备通式(II)化合物,使通式(I)化合物在一个或多个步骤中反应得到通式(II)化合物,其中R1是C1-C10烷基、链链烯基或芳基;-Cl、-Br、-I、-F;-CN;-OH;或者-OR2、-O(CO)R2或-R2-OH基团,其中R2是包含1至6个碳原子的烷基或亚烷基;并且原子9与10之间的键合是单键或双键;ii)芳构化通式II化合物为通式III化合物,其中R1和R2具有上述含义;和iii)任选地,还原通式III化合物为通式IV化合物,其中R1和R2具有上述含义。进一步地,本发明提供了几种在上述方法中可作为中间体的新颖的化合物和制备这些新颖化合物的方法。本发明还提供了基本上不含其它雌激素中间体的2-烷氧基-甾酮,2-烷氧基-雌二醇或其混合物。
Description
技术领域
本发明涉及制备雌酮和雌二醇的2-取代-衍生物的方法。
发明背景
17-酮基甾类或17-羟基甾类、例如雌酮和雌二醇的治疗价值是熟知的。除了甾类本身以外,还已发现雌酮和雌二醇的衍生物具有治疗价值。在这方面,尤其需要提到雌酮和雌二醇的2-烷氧基-衍生物,例如2-甲氧基-雌二醇。
2-甲氧基雌二醇、即1,3,5(10)-雌三烯-2,3,17b-三醇-2-甲基-醚(2-ME2)是雌二醇的内源性代谢产物。2-ME2具有较低的雌激素活性,但是已经发现具有重要的其他生物学作用,例如抗癌活性,如下文所述。
美国专利5,504,074、5,66,143和5,892,069描述了使用2-ME2治疗哺乳动物以异常细胞有丝分裂为特征的疾病的方法。另外,WO-A-02/42319描述了治疗以异常血管生成为特征的疾病状态。
不期望的细胞有丝分裂是很多疾病的特征,包括但不限于癌症、动脉粥样硬化、实体肿瘤增殖、血管机能障碍、子宫内膜异位、视网膜病、关节病和异常伤口愈合。另外,细胞有丝分裂在多种生物功能中都是重要的,包括但不限于正常的胚胎发育、黄体生成、子宫内膜的环状增殖、伤口愈合和炎性及免疫应答。
美国专利5,521,168描述了2-ME2降低眼内压的用途。2-ME2也抑制雌激素-诱导的垂体肿瘤血管生成,抑制Fisher 344大鼠中的肿瘤生长,如Banerjee,S.K.et al.,Proc.Amer.Assoc.CancerRes.39,March 1998所报道的。
2-ME2在人类中的任何治疗用途要求2-ME2具有较高的纯度水平。确切而言,由于2-ME2的作用被雌二醇和其他雌激素代谢产物所抵消,理想的是2-ME2制备物基本上不含这类污染物。可以从污染性雌二醇、雌酮和2-羟基雌二醇看到的作用包括雌激素作用,例如女性化、子宫内膜增生、子宫与乳腺癌的危险增加、对性器官的发育作用、白细胞生成的抑制和对造血细胞的作用。另外,已知4-羟基雌二醇、4-甲氧基雌二醇和雌二醇至少是受怀疑的致癌物。
这些发现提示我们寻找制备雌酮和雌二醇的2-烷氧基衍生物、例如2-甲氧基雌二醇的新合成工艺。
制备2-ME2的方法是本领域已知的。例如,由J.Fishman发表在J.Am.Chem.Soc.,5 March 1958,1213-1216页中的、题为“2-甲氧基雌激素的合成”的文章描述了从雌二醇开始制备2-甲氧基-雌二醇。美国专利6,051,726也描述了从雌二醇开始制备2-烷氧基雌二醇。不过,这类从雌二醇开始的方法的缺点是具有最终产物2-ME2将被不期望的雌激素活性化合物所污染的危险,例如起始化合物雌二醇和/或任何雌激素中间体。正如上文所解释的,这类雌激素杂质是非常不可取的。
EP-A-0776904涉及完全不同的技术领域,即3-酮基-5(10),9(11)-性二烯(gonadiene)-衍生物的烷基缩酮的制备。在其实施例中,描述了从雌-4,9-二烯-3,17-二酮开始制备这类性二烯-衍生物。雌-4,9-二烯-3,17-二酮化合物是这样制备的,缩合(+)5α-羟基-7aβ-甲基-2,3,3aα,4,5,6,7,7a-八氢-1H-茚-1-酮-4α-(3-丙酸)-内酯与2-戊酮-新戊基乙缩醛(acetale)-5-氯化镁;氧化5α-羟基;第一个环(B)的环闭合;缩酮裂解;和第二个环(A)的环闭合。EP-A-0776904既没有描述也没有提示雌-4,9-二烯-3,17-二酮的2-取代衍生物的制备。
此外,H.Ali et al,J.Chem.Soc.Perkin Trans.1,1991,page 2485-2491描述了使用具有适合定位的双键的19-降甾类、继之以在C2或C4位选择性引入官能团、随后芳构化A-环为2-或4-取代-雌二醇的途径的可能性。没有给出根据这种途径的这种2-取代作用的具体实例。
2-取代雌二醇的其他制备参见P.W.Le Quesne et al,Steroids,vol.53/6,June 1989,page 649-661;L.R.Axelrod et al,Chem.&Ind.,November 1959,page 1454-1455;and M.Mihailovic,Tetrahedron,vol.33,1977,page 235-237。
附图
下列附图旨在阐述本发明:
图1:制备2-(3-氯-1-甲氧基-丙基)-2,5,5-三甲基-[1,3]二烷的反应方案。
图2:制备2-甲氧基-雌二醇的反应方案。
发明概述
现在已经有利地发现制备雌酮和雌二醇的2-烷氧基-衍生物的新途径。另外,这种方法也能够用于制备雌二醇和雌酮的其他衍生物。
在新发现的途径中,在合成的最后一步进行A-环的芳构化。结果,所制备的最终产物本质上不含雌激素中间体。新发现的途径能够有利地开始于谷内酯(sitolactone)或其衍生物。谷内酯相对便宜,使这样一种途径在经济上比例如J.Fishman所述途径更具吸引力。
因此,本发明提供制备雌酮和雌二醇的2-取代-衍生物的方法,包含
i)如下制备通式(II)化合物,使通式(I)化合物
在一个或多个步骤中反应得到通式(II)化合物

其中R1是C1-C10烷基、链链烯基或芳基;-Cl、-Br、-I、-F;-CN;-OH;或者-OR2、-O(CO)R2或-R2-OH基团,其中R2是具有1-6个C原子的烷基或亚烷基;并且原子9与10之间的键合是单键或双键;
ii)芳构化通式(II)化合物为通式III化合物

其中R1和R2具有上述含义;和
iii)任选地,还原通式III化合物为通式IV化合物

其中R1和R2具有上述含义。
此外,本发明提供若干新颖的化合物,它们可以是上述方法中的中间体,和制备这些新颖化合物的方法。
此外,正如上文所解释的,本发明的方法能够用于制备本质上不含雌激素中间体的2-烷氧基-雌酮或2-烷氧基-雌二醇或其混合物。
本发明的详细说明
正如上文所指出的,本发明提供制备雌酮、雌酮-衍生物、雌二醇和/或雌二醇-衍生物的方法。不过,该方法尤其有利于制备雌酮和/或雌二醇的2-烷氧基衍生物。因此在具体实施方式中,本发明提供如上所述的方法,用于制备雌酮和/或雌二醇的2-烷氧基衍生物,也就是在2位被烷氧基取代的雌酮和/或雌二醇。而且,本发明提供这样一种方法,用于制备2-甲氧基-雌酮和/或2-甲氧基-雌二醇。正如上文所解释的,这样一种方法尤其是有利的,因为最终产物中的任何雌激素中间体最少。
式II、III和IV中的R1可以是具有1至10个碳原子的直链或支链烷基,例如甲基、乙基、丙基、异丙基、丁基、异丁基、叔丁基、戊基、异戊基、叔戊基或新戊基、己基、庚基、辛基、壬基或癸基。在具体实施方式中,若R1是烷基,R1是甲基或乙基。若R1是链链烯基,实例包括乙链链烯基、丙链链烯基、异丙链链烯基、丁链链烯基和戊链链烯基。若R1是芳基,实例包括苯基、苄基和甲苯基。
在具体的发明实施方式中,式II、III和IV中的R1是-OH;或者-OR2基团,其中R2是具有1至6个碳原子的烷基。在进一步的实施方式中,R1是C1-C6-烷氧基,即-OR2基团,其中R2是具有1至6个碳原子的直链或支链烷基。这类烷氧基的实例包括甲氧基-、乙氧基-、丙氧基-、异丙氧基-、丁氧基-、戊氧基-、己氧基-。在更进一步的实施方式中,式II、III和IV中的R1是甲氧基或乙氧基,在更进一步的实施方式中,R1是甲氧基。
当R1是烷氧基时,本发明提供制备2-烷氧基-雌酮和/或2-烷氧基-雌二醇的方法。通过还原按照该方法步骤ii)制备的2-烷氧基-雌酮,可以得到2-烷氧基-雌二醇。
式II中原子9与10之间的键合可以是单键或双键。不过在本发明的具体实施方式中,原子9与10之间的键合是双键。
步骤ii)中通式II化合物向通式III化合物的芳构化可以按照技术人员已知适合于该方法的任何方式进行。这类芳构化方法的实例包括使用芳构化剂,例如叔戊醇钾和乙酸酐;并且使用酶,例如芳构化酶。
在具体实施方式中,芳构化是用戊醇钾进行的。
在另一种具体实施方式中,芳构化是用锂的乙二胺溶液进行的。
当式II中原子9与10之间的键合是双键时,芳构化能够导致通式III化合物的两种异构体,即9-α-H-和9-β-H-异构体。其中,天然存在于人体中的9-α-H-异构体的制备是优选的。有利地,其中原子9与10之间的键合是双键的通式II化合物用锂的乙二胺溶液芳构化,导致包含大多数9-α-H-异构体的异构体混合物。
在进一步的实施方式中,反应是在适合的溶剂中进行的。适合的溶剂的实例包括二乙胺、四氢呋喃;及其混合物。反应期间的温度可以各不相同。在一种实施方式中,反应是在0至60℃范围内的温度下进行的。在具体实施方式中,反应是在室温(20-25℃)下进行的。
在更进一步的实施方式中,混合物随后用碱溶液淬灭,经由酸-碱萃取从混合物中除去残留的原料。
步骤iii)中通式III化合物向通式IV化合物的还原可以按照技术人员已知适合于这种目的的任何方式进行。在一种实施方式中,还原是借助还原剂进行的。适合的还原剂的实例包括NaBH4和LiAlH4。在具体实施方式中,使用NaBH4作为还原剂。在进一步的实施方式中,反应是在适合的溶剂中进行的。适合的溶剂的实例包括链烷醇,例如甲醇和乙醇;二乙胺;四氢呋喃;及其混合物。反应期间的温度可以各不相同。在一种实施方式中,反应是在0至70℃范围内的温度下进行的。在具体实施方式中,反应是在室温(20-25℃)下进行的。
有些通式II化合物被认为是新颖的。因此,本发明也提供通式II化合物
其中R1和R2是如上所指定的;并且原子9与10之间的键合是单键或双键。
在具体实施方式中,R1是C1-C6-烷氧基,即-OR2基团,其中R2是具有1至6个碳原子的直链或支链烷基。这类烷氧基的实例包括甲氧基-、乙氧基-、丙氧基-、异丙氧基-、丁氧基-、戊氧基-、己氧基-。在更进一步的实施方式中,式II、III和IV中的R1是甲氧基或乙氧基,在进一步的实施方式中,R1是甲氧基。
有利地,通式II化合物是从式I化合物制备的,也被称为谷内酯((4aS-(4aα,6aα,9aβ,9bα))-十氢-6a-甲基环戊二烯并(cyclopenta)(f)(1)苯并吡喃-3,7-二酮)。
在具体实施方式中,通式II化合物是借助这样一种方法制备的,它包含下列步骤;
a)使式I化合物

与通式V化合物反应,

其中R1和R2具有上述含义;R3和R4独立地是包含1至6个碳原子的烷基;X选自Cl或Br;
制得通式(VI)化合物

其中R1、R2、R3和R4具有上述含义;
b)氧化通式(VI)化合物的羟基,生成通式(VII)化合物

其中R1、R2、R3和R4具有上述含义;
c)环闭合通式VII化合物的B-环,制得通式VIII化合物

其中R1、R2、R3和R4具有上述含义;
d)使式VIII化合物在一个或多个步骤中反应为通式(II)化合物。
在具体实施方式中,步骤a)中的偶联反应是借助格氏反应进行的,其中通式(V)化合物首先被转化为通式(Va)化合物。

适合于这种反应的溶剂包括二乙醚、四氢呋喃及其混合物。
在具体实施方式中,使用粉末状的镁,它是用二溴乙烷活化的,然后加入通式V化合物,以制备通式Va化合物。后者是逐批加入到包含通式I化合物的溶液或悬浮液中的,生成通式VI化合物。在进一步的实施方式中,反应是在回流温度和大气压(1巴)下进行的。优选地,所得产物经过庚烷与乙酸乙酯1∶1混合物结晶纯化。
步骤b)中通式(VI)化合物的羟基氧化生成通式VII化合物可以借助技术人员已知适合于这种目的的任何氧化作用进行。适合的氧化方法的实例包括用氧化剂处理,例如铬酸、氧化铬/吡啶、氯/吡啶、重铬酸钙、吡啶铬酸盐和N-溴代琥珀酰亚胺;和Oppenauer氧化。在一种具体实施方式中,氧化是在丙酮中用铬酸进行的。在第二种实施方式中,氧化是用四正丙基过钌酸铵(TPAP)和N-甲基吗啉N-氧化物(NMO)进行的。作为替代溶剂,可以使用二氯甲烷。
步骤c)中通式VII化合物B-环的环闭合制备通式(VIII)化合物可以按照技术人员已知适合于这种目的的任何方式进行。B-环被理解为构成最终产物雌酮或雌二醇衍生物中B-环的那些碳原子。在一种具体实施方式中,环闭合步骤是在碱性条件下借助羟醛缩合作用进行的,这种缩合作用在进一步的实施方式中继之以羟醛产物的脱水。适合使用的碱的实例包括叔丁醇钾、叔戊醇钾、氢氧化钠、氢氧化钾。技术人员将进一步认识到,可以使用很多其他的碱。在进一步的实施方式中,反应是在适合的溶剂中进行的。适合的溶剂的实例包括甲苯;链烷醇,例如甲醇、乙醇和异丙醇;四氢呋喃;及其混合物。在具体实施方式中,使用甲醇、甲苯与水的混合物作为溶剂。反应期间的温度可以各不相同。在一种实施方式中,反应是在0至80℃范围内的温度下进行的。在具体实施方式中,反应是在室温(20-25℃)下进行的。在另一种具体实施方式中,反应是在55至75℃范围内的温度下进行的。在进一步的实施方式中,所得通式VIII化合物经过适合的溶剂结晶纯化,例如醇。在具体实施方式中,通式VIII化合物经过异丙醇结晶纯化。
按照步骤d)从通式VIII化合物制备通式II化合物可以按照技术人员已知适合于这种目的的任何方式进行。在一种具体实施方式中,步骤d)的反应包含
d1)通式VIII化合物的水解,其中原子9与10之间的键合是双键,得到通式IX化合物
其中R1和R2具有上述含义;
d2)通式IX化合物A-环的环闭合,得到通式II化合物,其中原子9与10之间的带点键合是双键。
在替代实施方式中,步骤d)的反应包含
d3)使通式VIII化合物原子9与10之间的双键的氢化,得到通式X化合物

d4)通式X化合物的水解,得到通式XI化合物
d5)通式XI化合物A-环的环闭合,得到通式II化合物,其中原子9与10之间的带点键合是单键。
A-环被理解为构成最终产物雌酮或雌二醇衍生物A-环的那些碳原子。
步骤d1)的水解可以按照技术人员已知适合于这种目的的任何方式进行。在一种实施方式中,水解是在酸性条件下进行的。适合的水解剂的实例包括氢卤化物、磷酸、磺酸和硫酸,例如盐酸、硫酸、对-甲苯磺酸和磷酸。在一种实施方式中,使用硫酸(H2SO4)作为水解剂。可以使用技术人员已知适合于这种目的的任何溶剂作为反应期间的溶剂。适合的溶剂的实例包括C1-C6烷醇,例如甲醇、乙醇、丙醇和异丙醇。在一种实施方式中,使用乙醇作为溶剂。反应期间的温度可以各不相同。在一种实施方式中,反应是在0至40℃范围内的温度下进行的,在具体实施方式中,反应是在室温(20-25℃)下进行的。
步骤d2)的A-环环闭合可以按照技术人员已知适合于这种目的的任何方式进行。在一种具体实施方式中,环闭合步骤是在碱性条件下借助羟醛缩合进行的,这种缩合作用在进一步的实施方式中继之以羟醛产物的脱水。适合使用的碱的实例包括叔丁醇钾、叔戊醇钾、氢氧化钠、氢氧化钾。技术人员将进一步认识到,可以使用很多其他的碱。在进一步的实施方式中,反应是在适合的溶剂中进行的。适合的溶剂的实例包括甲苯;链烷醇,例如甲醇、乙醇和异丙醇;四氢呋喃;及其混合物。反应期间的温度可以各不相同。在一种实施方式中,反应是在0至80℃范围内的温度下进行的。在具体实施方式中,反应是在室温(20-25℃)下进行的。
步骤d3)的氢化可以按照技术人员已知适合于这种目的的任何方式进行。在一种实施方式中,使用Pd/C和氢进行氢化。在进一步的实施方式中,反应是在适合的溶剂中进行的。适合的溶剂的实例包括四氢呋喃;链烷醇,例如甲醇、乙醇和异丙醇;及其混合物。在具体实施方式中,使用乙醇作为溶剂。反应期间的温度可以各不相同。在一种实施方式中,反应是在0至80℃范围内的温度下进行的。在具体实施方式中,反应是在25至60℃范围内的温度下进行的。
步骤d4)的水解可以按照技术人员已知适合于这种目的的任何方式进行。在一种实施方式中,水解是在酸性条件下进行的。适合的水解剂的实例包括氢卤化物、磷酸、磺酸和硫酸,例如盐酸、硫酸、对-甲苯磺酸和磷酸。在一种实施方式中,使用硫酸(H2SO4)作为水解剂。可以使用技术人员已知适合于这种目的的任何溶剂作为反应期间的溶剂。适合的溶剂的实例包括C1-C6烷醇,例如甲醇、乙醇、丙醇和异丙醇。在一种实施方式中,使用乙醇作为溶剂。反应期间的温度可以各不相同。在一种实施方式中,反应是在0至40℃范围内的温度下进行的,在具体实施方式中,反应是在室温(20-25℃)下进行的。
步骤d5)的A-环环闭合可以按照技术人员已知适合于这种目的的任何方式进行。在一种具体实施方式中,环闭合步骤是在碱性条件下借助羟醛缩合进行的,这种缩合作用在进一步的实施方式中继之以羟醛产物的脱水。适合使用的碱的实例包括叔丁醇钾、叔戊醇钾、氢氧化钠、氢氧化钾。技术人员将进一步认识到,可以使用很多其他的碱。在进一步的实施方式中,反应是在适合的溶剂中进行的。适合的溶剂的实例包括甲苯;链烷醇,例如甲醇、乙醇和异丙醇;四氢呋喃;及其混合物。反应期间的温度可以各不相同。在一种实施方式中,反应是在0至80℃范围内的温度下进行的。在具体实施方式中,反应是在室温(20-25℃)下进行的。
大量上文提到的中间体被视为新颖的。因此,本发明也提供通式V化合物
和通式Va的其Mg活化副本(counterpart)
其中R1、R2、R3和R4具有上述含义;X选自Cl或Br。在进一步的实施方式中,R3和R4独立地是甲基或乙基。在具体实施方式中,R3和R4是甲基,并且R1是甲氧基。适合的根据通式V的化合物的实例例如包括2-(3-氯-1-甲氧基-丙基)-2,5,5-三甲基-[1,3]二烷和2-(3-溴-1-甲氧基-丙基)-2,5,5-三甲基-[1,3]二烷。适合的根据通式Va的化合物的实例例如包括3-甲氧基-2-戊酮-新戊基乙缩醛-5-氯化镁和3-甲氧基-2-戊酮-新戊基乙缩醛-5-溴化镁。
通式(V)化合物例如可以这样制备,卤化适当取代的丙烯化合物;用腈基团取代一个卤原子;借助格氏反应加成甲基,再转化为5-氯-戊烷-2-酮化合物,在其3位被适当的基团取代;并与新戊基二醇在酸性条件下反应,得到通式(V)化合物。2-(3-氯-1-甲氧基-丙基)-2,5,5-三甲基-[1,3]二烷的制备如图1所述。
此外,本发明提供通式(VI)化合物

其中R1、R2、R3和R4具有上述含义。
进而,本发明提供通式VII化合物
其中R1、R2、R3和R4具有上述含义。
进而,本发明提供通式VIII化合物
其中R1、R2、R3和R4具有上述含义。
进而,本发明提供通式IX化合物

其中R1和R2具有上述含义。
进而,本发明提供通式X化合物
其中R1、R2、R3和R4具有上述含义。
进而,本发明提供通式XI化合物
其中R1和R2具有上述含义。
下列非限制性实施例和图2反应方案进一步阐述发明。
实施例1
步骤A:(3aα,4α,5α,7aα)5-羟基-7a-甲基-4[7-(2,5,5-三甲基-1,3-二烷-2-基)-5-甲氧基-3-氧代己基]-1H-茚-1-酮的制备
将50目镁粉(10g)悬浮在干燥四氢呋喃(70ml)中。将悬浮液加热至50℃。加入二溴乙烷(500μl)后,发生剧烈的反应。加入初始部分的2-(3-氯-1-甲氧基-丙基)-2,5,5-三甲基-[1,3]二烷(2g)。在回流下搅拌1h后,加入2-(3-氯-1-甲氧基-丙基)-2,5,5-三甲基-[1,3]二烷(35g)。在回流下搅拌20h后,将反应混合物冷却至20℃。使未反应的镁沉积下来,用注射器中吸取上层,在-35℃下加入到(4aS-(4aα,6aα,9aα,9bα))-十氢-6a-甲基环戊二烯并(f)(1)苯并吡喃-3,7-二酮(谷内酯,28g)的四氢呋喃(110ml)悬浮液中。在-30℃下搅拌4h后,在1h内升高温度至20℃。在40min内向反应混合物滴加氯化铵溶液(10g,200ml水)。将悬浮液搅拌1h,经过Dicalite滤器过滤。滤器用二氯甲烷洗涤(2×100ml)。在30℃真空中蒸馏除去溶剂后,残余物用二氯甲烷萃取(5×100ml)。合并有机层,在30℃真空中浓缩。干燥后,得到51.6g粗的(3aα,4α,5α,7aα)5-羟基-7a-甲基-4[7-(2,5,5-三甲基-1,3-二烷-2-基)-5-甲氧基-3-氧代己基]-1H-茚-1-酮。
实施例2
步骤B:(3aα,4β,5α,7aβ)-5-氧代-7a-甲基-4[6-(2,5,5-三甲基-1,3-二烷-2-基)-5-甲氧基-3-氧代己基]-1H-茚-1-酮的制备,羟基的氧化
在20℃下,向(3aα,4α,5α,7aα)5-羟基-7a-甲基-4[7-(2,5,5-三甲基-1,3-二烷-2-基)-5-甲氧基-3-氧代己基]-1H-茚-1-酮(85g)的二氯甲烷(450ml)溶液加入N-甲基吗啉(35g)和四丙基过钌酸铵(2g)。在20℃下搅拌20h,加入二氧化硅(50g)。将反应混合物经过Dicalite滤器和二氧化硅过滤,滤器用二氯甲烷(500ml)洗涤。在30℃下蒸发滤液至干,得到75.36g粗的(3aα,4β,5α,7aβ)-5-氧代-7a-甲基-4[6-(2,5,5-三甲基-1,3-二烷-2-基)-5-甲氧基-3-氧代己基]-1H-茚-1-酮,含有约10%原料。
实施例3
步骤C:(3aα,9aα,9bβ)-4,5,8,9,9a,9b-六氢-3a-甲基-6-(2-(2,5,5-三甲基-1,3-二烷-2-基)-1-甲氧基-乙基)-1H-苯并(e)茚-3,7(2H,3aH)-二酮的制备,B-环的生成
在20℃下,将叔丁醇钾(6.4g)悬浮在甲苯(100ml)和2-丙醇(32ml)中。在30min内向叔丁醇钾/甲苯/2-丙醇混合物中加入(3aα,4α,5α-7aα)-5-氧代-7a-甲基-4[6-(2,5,5-三甲基-1,3-二烷-2-基)-5-甲氧基-3-氧代己基]-1H-茚-1-酮(80g)的甲苯(400ml)溶液。将反应混合物在20℃下搅拌2h后,反应混合物用乙酸(8ml)酸化至pH=4。加入水(300ml)并在20℃下搅拌15min后,分离各层。水层用甲苯萃取(3×100ml),合并有机层,用氢氧化钠(5g)的水(100ml)溶液萃取。在50℃真空中蒸发有机层至干。在50℃下将残余物溶于2-丙醇(300ml),将溶液冷却至10℃。在10℃下搅拌2h后,得到第一批白色晶体(7.8g)。蒸发母液至干,溶于庚烷(50ml)。将溶液在70℃下搅拌30min,然后冷却至20℃。在20℃下搅拌悬浮液达50h后,得到第二批白色晶体(11.5g)。
实施例4:
步骤B和C:(3aα,9aα,9bβ)-4,5,8,9,9a,9b-六氢-3a-甲基-6-(2-(2,5,5-三甲基-1,3-二烷-2-基)-1-甲氧基-乙基)-1H-苯并(e)茚-3,7(2H,3aH)-二酮的制备;合并的氧化和羟醛缩合
将(3aα,4α,5α,7aα)5-羟基-7a-甲基-4[7-(2,5,5-三甲基-1,3-二烷-2-基)-5-甲氧基-3-氧代己基]-1H-茚-1-酮(147g)在甲苯(690ml)与吡啶(162ml)混合物中的溶液冷却至-2℃。在2h内向反应混合物引入氯气(42g)。在-2℃下搅拌反应混合物达2h后,在10℃下将反应混合物倒入亚硫酸钠(93g)与碳酸钠(78g)的水(750ml)溶液中。将混合物在20℃下搅拌30min,分离各层。水层用甲苯萃取(4×150ml)。合并有机层,在50℃真空中蒸发至干。将残余物溶于甲苯(660ml)。向甲苯溶液加入氢氧化钾(127.5g)的水(185ml)与甲醇(430ml)溶液。在65℃下搅拌45min后,将反应混合物冷却至20℃。分离各层,有机层用50%甲醇洗涤(aq)(2×175ml)。合并的甲醇/水萃取液用二氯甲烷洗涤(4×100ml)。合并有机层,在40℃真空中蒸发至干,在50℃下将残余物悬浮在2-丙醇(250ml)中。在50℃下搅拌15min后,将悬液在10℃下搅拌1h,滤出晶体,用2-丙醇洗涤(2×10ml)。干燥后,得到58.04g产物。
实施例5
步骤D:2-甲氧基-(+)4,5-开环(seco)-雌(estra)-9-烯-3,5,17-三酮的制备,缩酮的水解
在20℃下,向(3aα,9aα,9bα)-4,5,8,9,9a,9b-六氢-3a-甲基-6-(2,5,5-三甲基-1,3-二烷-2-基)-1-甲氧基-乙基)-1H-苯并(e)茚-3,7(2H,3aH)-二酮(16.2g)的乙醇(200ml)悬浮液加入硫酸(2ml)(pH=2)。将反应混合物在40℃下搅拌2h。向反应混合物加入乙酸钠(3.5g)的水(100ml)溶液。将反应混合物冷却至20℃。加入二氯甲烷(100ml)和水(100ml)后,将混合物在20℃下搅拌30min,分离各层。水层用二氯甲烷萃取(3×50ml)。合并二氯甲烷萃取液,在50℃下蒸发至干,得到16.7g粗产物。
实施例6
步骤E:2-甲氧基-雌-4,9-二烯-3,17-二酮的制备和纯化,A-环的生成
在20℃下,将2-甲氧基-(+)4,5-开环(seco)-雌-9-烯-3,5,17-三酮(10g)的四氢呋喃(50ml)溶液在30min内加入到叔丁醇钾(750mg)的四氢呋喃(50ml)悬液中。在20℃下搅拌7h后,将反应混合物用4.0N硫酸中和至pH=6。在35℃真空中浓缩反应混合物。将残余物溶于二氯甲烷(100ml),用水萃取(2×50ml)。在35℃下蒸发有机层至干,得到8.1g粗产物。
实施例7
步骤F:2-甲氧基-雌酮的制备,芳构化
在100℃下,将锂(0.58g)缓慢加入到乙二胺(100ml)中。在100℃下搅拌混合物达30min后,冷却至20℃。在20℃下,在30min内向该试剂中加入2-甲氧基-雌-4,9-二烯-3,17-二酮(5g)的乙二胺(20ml)溶液。在20℃下搅拌3h后,将反应混合物倒入水(250ml)中。用硫酸酸化至pH=7。在20℃下搅拌12h后,得到浅褐色晶体,滤出。粗产物(6g)经过二氧化硅过滤以除去无机盐,得到1.7g产物。所形成的9α-H异构体与所形成的9α-H异构体之比为约7∶1。
实施例8
步骤F:2-甲氧基-雌酮的制备,芳构化
在20℃下,将2-甲氧基-雌-4,9-二烯-3,17-二酮(500mg,1.66mmol)溶于四氢呋喃(12.5ml)。然后加入1.7M叔戊醇钾的甲苯溶液(5ml甲苯,8.5mmol叔戊醇钾)。用25ml 1M氢氧化钠(NaOH)溶液淬灭反应。分离水层,用2M硫酸调节pH至4。用三份二氯甲烷(15ml)萃取后,蒸发有机层至干,得到200mg粗产物。第一有机层(四氢呋喃/甲苯)似乎含有可观数量的产物。蒸发至干后,得到250mg产物。所形成的9α-H异构体与所生成的9α-H异构体之比为约1∶1。
实施例9
步骤G:2-甲氧基-雌二醇的制备,还原
在20℃下,向2-甲氧基雌酮(2g)的四氢呋喃(15ml)溶液加入33%氢氧化钠(200μl)和水(2ml)。向反应混合物中缓慢加入硼氢化钠(0.25g)。将反应混合物在20℃下搅拌1h,用乙酸中和至pH=7。搅拌30min后,分离各层,水层用二氯甲烷萃取(2×10ml)。合并有机层,在35℃下蒸发至干。残余物(2.1g)经过二氧化硅(200g)色谱处理,用甲苯与乙酸乙酯的混合物(甲苯与乙酸乙酯的体积比为9∶1)洗脱。从丙酮和乙醇中结晶,得到400mg 2-甲氧基雌二醇。
Claims (17)
1.制备雌酮和雌二醇的2-取代-衍生物的方法,包含
i)如下制备通式(II)化合物,使通式(I)化合物
在一个或多个步骤中反应得到通式(II)化合物

其中R1是C1-C10烷基、链链烯基或芳基;-Cl、-Br、-I、-F;-CN;-OH;或者-OR2、-O(CO)R2或-R2-OH基团,其中R2是包含1至6个碳原子的烷基或亚烷基;原子9与10之间的键合是单键或双键;
ii)芳构化通式(II)化合物为通式(III)化合物
其中R1和R2具有上述含义;和
iii)任选地,还原通式(III)化合物为通式(IV)化合物

其中R1和R2具有上述含义。
2.根据权利要求1的方法,其中在通式(II)、(III)和(IV)化合物中,R1是-OR2基团,其中R2是具有1至6个碳原子的烷基。
3.根据权利要求1或2的方法,其中在通式(II)化合物中,原子9与10之间的键合是双键。
4.根据权利要求1-3任意一项的方法,其中步骤ii)中的芳构化是借助锂的乙二胺溶液进行的。
5.根据权利要求1-4任意一项的方法,其中步骤iii)中的还原是使用NaBH4作为还原剂进行的。
6.通式(II)化合物
其中R1是C1-C10烷基、链链烯基或芳基;-Cl、-Br、-I、-F;-CN;-OH;或者-OR2、-O(CO)R2或-R2-OH基团,其中R2是包含1至6个碳原子的烷基或亚烷基;并且原子9与10之间的键合是单键或双键。
7.制备通式(II)化合物的方法,

其中R1是C1-C10烷基、链链烯基或芳基;-Cl、-Br、-I、-F;-CN;-OH;或者-OR2、-O(CO)R2或-R2-OH基团,其中R2是包含1至6个碳原子的烷基或亚烷基;并且原子9与10之间的键合是单键或双键;
包含下列步骤:
a)使式(I)化合物
与通式(V)化合物反应,
其中R1和R2具有上述含义;R3和R4独立地是包含1至6个碳原子的烷基;X选自Cl或Br;
制得通式(VI)化合物
其中R1、R2、R3和R4具有上述含义;
b)氧化通式(VI)化合物的羟基,生成通式(VII)化合物
其中R1、R2、R3和R4具有上述含义;
c)环闭合通式(VII)化合物的B-环,制得通式(VIII)化合物
其中R1、R2、R3和R4具有上述含义;
d)使式(VIII)化合物在一个或多个步骤中反应为通式(II)化合物。
8.根据权利要求7的方法,其中步骤d)的反应包含
d1)通式(VIII)化合物的水解,其中原子9与10之间的键合是双键,得到通式(IX)化合物
其中R1是C1-C10烷基、链链烯基或芳基;-Cl、-Br、-I、-F;-CN;-OH;或者-OR2、-O(CO)R2或-R2-OH基团,其中R2是包含1至6个碳原子的烷基或亚烷基;
d2)通式(IX)化合物A-环的环闭合,得到通式(II)化合物,其中原子9与10之间的键合是双键。
9.根据权利要求7的方法,其中步骤d)的反应包含
d3)通式(VIII)化合物原子9与10之间的双键的氢化,得到通式X化合物
d4)通式(X)化合物的水解,得到通式(XI)化合物
d5)通式(XI)化合物A-环的环闭合,得到通式(II)化合物,其中原子9与10之间的键合是单键。
10.通式(V)化合物
其中R1是C1-C10烷基、链链烯基或芳基;-Cl、-Br、-I、-F;-CN;-OH;或者-OR2、-O(CO)R2或-R2-OH基团,其中R2是包含1至6个碳原子的烷基或亚烷基;R3和R4独立地是包含1至6个碳原子的烷基;并且X选自Cl或Br。
11.通式(VI)化合物
其中R1是C1-C10烷基、链链烯基或芳基;-Cl、-Br、-I、-F;-CN;-OH;或者-OR2、-O(CO)R2或-R2-OH基团,其中R2是包含1至6个碳原子的烷基或亚烷基;R3和R4独立地是包含1至6个碳原子的烷基。
12.通式(VII)化合物

其中R1是C1-C10烷基、链链烯基或芳基;-Cl、-Br、-I、-F;-CN;-OH;或者-OR2、-O(CO)R2或-R2-OH基团,其中R2是包含1至6个碳原子的烷基或亚烷基;R3和R4独立地是包含1至6个碳原子的烷基。
13.通式(VIII)化合物
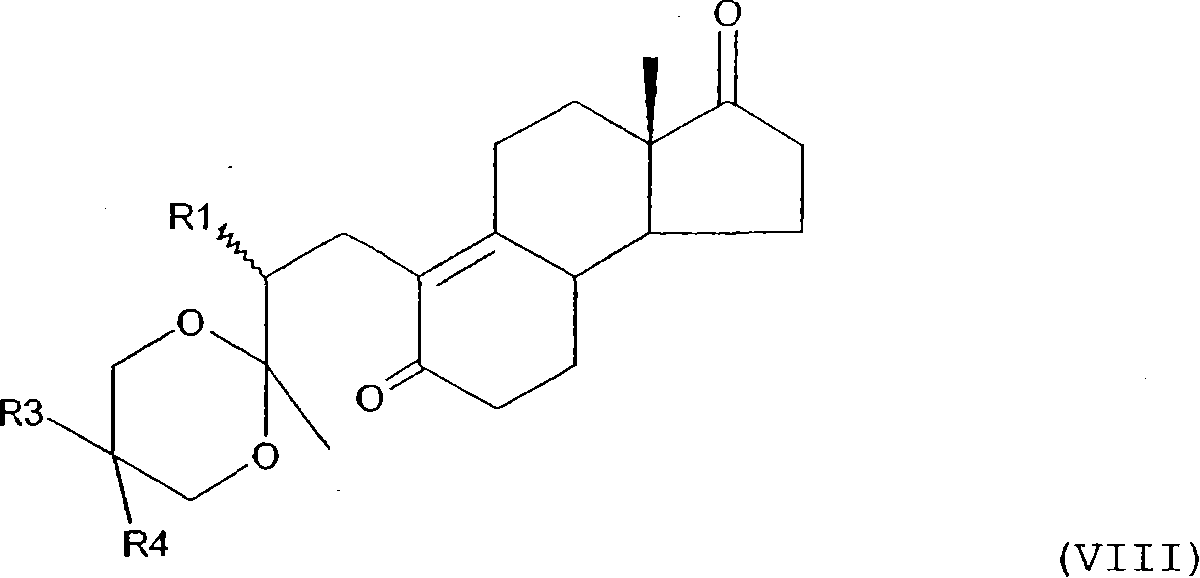
其中R1是C1-C10烷基、链链烯基或芳基;-Cl、-Br、-I、-F;-CN;-OH;或者-OR2、-O(CO)R2或-R2-OH基团,其中R2是包含1至6个碳原子的烷基或亚烷基;R3和R4独立地是包含1至6个碳原子的烷基。
14.通式(IX)化合物
其中R1是C1-C10烷基、链链烯基或芳基;-Cl、-Br、-I、-F;-CN;-OH;或者-OR2、-O(CO)R2或-R2-OH基团,其中R2是包含1至6个碳原子的烷基或亚烷基。
15.通式(X)化合物

其中R1是C1-C10烷基、链链烯基或芳基;-Cl、-Br、-I、-F;-CN;-OH;或者-OR2、-O(CO)R2或-R2-OH基团,其中R2是包含1至6个碳原子的烷基或亚烷基;R3和R4独立地是包含1至6个碳原子的烷基。
16.通式(XI)化合物
其中R1是C1-C10烷基、链链烯基或芳基;-Cl、-Br、-I、-F;-CN;-OH;或者-OR2、-O(CO)R2或-R2-OH基团,其中R2是包含1至6个碳原子的烷基或亚烷基。
17.本质上不含雌激素中间体的2-烷氧基-雌酮、2-烷氧基-雌二醇或其混合物。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP04103742.5 | 2004-08-04 | ||
| EP04103742 | 2004-08-04 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| CN1993376A true CN1993376A (zh) | 2007-07-04 |
Family
ID=34929415
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CNA2005800261640A Pending CN1993376A (zh) | 2004-08-04 | 2005-08-01 | 制备雌酮和雌二醇的2-取代-衍生物的方法 |
Country Status (20)
| Country | Link |
|---|---|
| US (2) | US7910756B2 (zh) |
| EP (1) | EP1776378B1 (zh) |
| JP (1) | JP5075626B2 (zh) |
| KR (1) | KR20070040386A (zh) |
| CN (1) | CN1993376A (zh) |
| AR (1) | AR050195A1 (zh) |
| AT (1) | ATE447580T1 (zh) |
| AU (1) | AU2005268768B2 (zh) |
| BR (1) | BRPI0514096A (zh) |
| CA (1) | CA2573133C (zh) |
| DE (1) | DE602005017500D1 (zh) |
| EC (1) | ECSP077228A (zh) |
| ES (1) | ES2334244T3 (zh) |
| IL (1) | IL180381A0 (zh) |
| MX (1) | MX2007001340A (zh) |
| NO (1) | NO20070747L (zh) |
| RU (1) | RU2007107914A (zh) |
| TW (1) | TW200611909A (zh) |
| WO (1) | WO2006013196A1 (zh) |
| ZA (1) | ZA200700483B (zh) |
Cited By (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN104497088A (zh) * | 2014-12-30 | 2015-04-08 | 湖南新合新生物医药有限公司 | 19-去甲-4-雄甾烯-3,17-二酮的制备方法 |
| CN104592339A (zh) * | 2014-12-30 | 2015-05-06 | 湖南新合新生物医药有限公司 | 雌甾-4,9-二烯-3,17-二酮的制备方法 |
| CN108003210A (zh) * | 2018-01-24 | 2018-05-08 | 四川理工学院 | 一种雌甾-4,9-二烯-3,17-二酮的制备方法 |
| CN108997463A (zh) * | 2018-08-09 | 2018-12-14 | 四川理工学院 | 一种雌甾-4, 9-二烯-3,17-二酮制备方法 |
| CN112409434A (zh) * | 2020-11-27 | 2021-02-26 | 厦门欧瑞捷生物科技有限公司 | 一种去氢孕酮的合成方法 |
| CN113861158A (zh) * | 2021-11-22 | 2021-12-31 | 湖南新合新生物医药有限公司 | 一种合成甲基双烯双酮关键中间体的方法 |
| CN114853802A (zh) * | 2022-03-22 | 2022-08-05 | 河北康泰药业有限公司 | 一种谷内酯衍生物及其制备方法、药物组合物和应用 |
Families Citing this family (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CZ300376B6 (cs) | 2008-05-05 | 2009-05-06 | Prírodovedecká Fakulta Uk | Ligandy estrogenových receptoru alfa a beta, zpusob jejich prípravy a farmaceutické prostredky, které je obsahují |
| WO2012020417A1 (en) | 2010-08-10 | 2012-02-16 | Reliance Life Sciences Pvt. Ltd. | Process for the preparation of estradiol and its derivatives |
| EP2742352B1 (en) * | 2011-08-12 | 2018-02-28 | Siemens Healthcare Diagnostics Inc. | Detection of sex steroids |
| ITUB20155260A1 (it) * | 2015-10-30 | 2017-04-30 | Ind Chimica Srl | PROCESSO PER LA PREPARAZIONE DI 17?-idrossi-des-A-androst-9,10-en-5-one |
Family Cites Families (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US2997488A (en) * | 1959-06-20 | 1961-08-22 | Organon | 2-methyl-19-nor-steroids |
| FR1468608A (fr) * | 1960-08-04 | 1967-02-10 | Upjohn Co | 2-alpha-fluoro stéroïdes de la série de la 19-norandrostène et leur procédé d'obtention |
| CH409944A (fr) | 1961-04-14 | 1966-03-31 | Roussel Uclaf | Procédé de préparation de dérivés de la 19-nor-testostérone |
| FR1368721A (fr) * | 1961-04-14 | 1964-08-07 | Roussel Uclaf | Dérivés de la 19-nor testostérone et procédé de préparation |
| US3519714A (en) * | 1966-03-15 | 1970-07-07 | Herchel Smith | Synthesis of gona-1,3,5(10)-trienes |
| US4400524A (en) * | 1981-07-28 | 1983-08-23 | The Upjohn Company | Grignard reagents prepared from 5-halopentan-2-one propylene ketals |
| DE3208432A1 (de) | 1982-03-09 | 1983-09-15 | The Upjohn Co., 49001 Kalamazoo, Mich. | Verfahren zur herstellung von 19-norandrostendion, zwischenprodukt des 19-norandrostendions und verfahren zu seiner herstellung |
| US5504074A (en) * | 1993-08-06 | 1996-04-02 | Children's Medical Center Corporation | Estrogenic compounds as anti-angiogenic agents |
| US5521168A (en) * | 1994-10-13 | 1996-05-28 | Alcon Laboratories, Inc. | Estrogen metabolites for lowering intraocular pressure |
| US5998639A (en) * | 1995-11-06 | 1999-12-07 | Akzo Nobel, N.V. | Sulfatation of estrogen mixtures |
| IL119649A (en) | 1995-11-30 | 2002-03-10 | Akzo Nobel Nv | Preparation of Cyclic Kills of History - 3 keto- (5) 10, (-9) 11 steroidadians |
| US6051726A (en) * | 1997-03-13 | 2000-04-18 | Pharm-Eco Laboratories, Inc. | Synthesis of 2-alkoxyestradiols |
| US7087592B1 (en) * | 1999-08-23 | 2006-08-08 | Entre Med, Inc. | Compositions comprising purified 2-methoxyestradiol and methods of producing same |
| AU7572600A (en) | 1999-08-23 | 2001-03-19 | Entremed, Inc | Methods of obtaining 2-methoxyestradiol of high purity |
| EP1343803A2 (en) | 2000-11-27 | 2003-09-17 | EntreMed, Inc. | 2-substituted estrogens as antiangiogenic agents |
-
2005
- 2005-07-14 TW TW094123942A patent/TW200611909A/zh unknown
- 2005-08-01 JP JP2007524335A patent/JP5075626B2/ja not_active Expired - Fee Related
- 2005-08-01 US US11/659,147 patent/US7910756B2/en not_active Expired - Fee Related
- 2005-08-01 EP EP05767940A patent/EP1776378B1/en not_active Expired - Lifetime
- 2005-08-01 AT AT05767940T patent/ATE447580T1/de not_active IP Right Cessation
- 2005-08-01 WO PCT/EP2005/053732 patent/WO2006013196A1/en not_active Ceased
- 2005-08-01 KR KR1020077002976A patent/KR20070040386A/ko not_active Ceased
- 2005-08-01 AU AU2005268768A patent/AU2005268768B2/en not_active Ceased
- 2005-08-01 ES ES05767940T patent/ES2334244T3/es not_active Expired - Lifetime
- 2005-08-01 CA CA2573133A patent/CA2573133C/en not_active Expired - Fee Related
- 2005-08-01 DE DE602005017500T patent/DE602005017500D1/de not_active Expired - Lifetime
- 2005-08-01 CN CNA2005800261640A patent/CN1993376A/zh active Pending
- 2005-08-01 MX MX2007001340A patent/MX2007001340A/es active IP Right Grant
- 2005-08-01 RU RU2007107914/04A patent/RU2007107914A/ru not_active Application Discontinuation
- 2005-08-01 BR BRPI0514096-0A patent/BRPI0514096A/pt not_active IP Right Cessation
- 2005-08-04 AR ARP050103233A patent/AR050195A1/es unknown
-
2006
- 2006-12-27 IL IL180381A patent/IL180381A0/en unknown
-
2007
- 2007-01-16 ZA ZA200700483A patent/ZA200700483B/xx unknown
- 2007-02-05 EC EC2007007228A patent/ECSP077228A/es unknown
- 2007-02-08 NO NO20070747A patent/NO20070747L/no not_active Application Discontinuation
-
2011
- 2011-02-22 US US13/031,975 patent/US20110152544A1/en not_active Abandoned
Cited By (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN104497088A (zh) * | 2014-12-30 | 2015-04-08 | 湖南新合新生物医药有限公司 | 19-去甲-4-雄甾烯-3,17-二酮的制备方法 |
| CN104592339A (zh) * | 2014-12-30 | 2015-05-06 | 湖南新合新生物医药有限公司 | 雌甾-4,9-二烯-3,17-二酮的制备方法 |
| CN108003210A (zh) * | 2018-01-24 | 2018-05-08 | 四川理工学院 | 一种雌甾-4,9-二烯-3,17-二酮的制备方法 |
| CN108997463A (zh) * | 2018-08-09 | 2018-12-14 | 四川理工学院 | 一种雌甾-4, 9-二烯-3,17-二酮制备方法 |
| CN112409434A (zh) * | 2020-11-27 | 2021-02-26 | 厦门欧瑞捷生物科技有限公司 | 一种去氢孕酮的合成方法 |
| CN112409434B (zh) * | 2020-11-27 | 2021-10-26 | 厦门欧瑞捷生物科技有限公司 | 一种去氢孕酮的合成方法 |
| CN113861158A (zh) * | 2021-11-22 | 2021-12-31 | 湖南新合新生物医药有限公司 | 一种合成甲基双烯双酮关键中间体的方法 |
| CN113861158B (zh) * | 2021-11-22 | 2023-02-10 | 湖南新合新生物医药有限公司 | 一种合成甲基双烯双酮关键中间体的方法 |
| CN114853802A (zh) * | 2022-03-22 | 2022-08-05 | 河北康泰药业有限公司 | 一种谷内酯衍生物及其制备方法、药物组合物和应用 |
| CN114853802B (zh) * | 2022-03-22 | 2023-11-28 | 河北康泰药业有限公司 | 一种谷内酯衍生物及其制备方法、药物组合物和应用 |
Also Published As
| Publication number | Publication date |
|---|---|
| CA2573133A1 (en) | 2006-02-09 |
| ZA200700483B (en) | 2008-09-25 |
| MX2007001340A (es) | 2007-03-27 |
| DE602005017500D1 (de) | 2009-12-17 |
| JP2008509109A (ja) | 2008-03-27 |
| AU2005268768B2 (en) | 2011-10-13 |
| RU2007107914A (ru) | 2008-09-10 |
| US20090012319A1 (en) | 2009-01-08 |
| NO20070747L (no) | 2007-04-23 |
| IL180381A0 (en) | 2007-06-03 |
| BRPI0514096A (pt) | 2008-05-27 |
| US7910756B2 (en) | 2011-03-22 |
| CA2573133C (en) | 2012-07-10 |
| WO2006013196A1 (en) | 2006-02-09 |
| EP1776378B1 (en) | 2009-11-04 |
| TW200611909A (en) | 2006-04-16 |
| US20110152544A1 (en) | 2011-06-23 |
| ATE447580T1 (de) | 2009-11-15 |
| KR20070040386A (ko) | 2007-04-16 |
| EP1776378A1 (en) | 2007-04-25 |
| JP5075626B2 (ja) | 2012-11-21 |
| ES2334244T3 (es) | 2010-03-08 |
| AU2005268768A1 (en) | 2006-02-09 |
| AR050195A1 (es) | 2006-10-04 |
| ECSP077228A (es) | 2007-03-29 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US20110152544A1 (en) | Process for the preparation 2-substituted derivatives of estrone and estradiol | |
| CN100343269C (zh) | 通过雌酮衍生的甾体化合物合成雌四醇 | |
| US20120214987A1 (en) | Methods and compounds for preparing 3alpha-oxygen substituted steroids | |
| CN1257181C (zh) | 雄激素甾类化合物以及制备和使用它们的方法 | |
| CN86104664A (zh) | 取代的雄-1,4-二烯-3,17-二酮类化合物的制法 | |
| CN1360589A (zh) | 口服活性雄激素 | |
| CN87105307A (zh) | 11β-(4-异丙烯基苯基)-4,9-雌二烯,其制法及含此化合物的药剂 | |
| CN103781795A (zh) | 用于制备雌四醇的方法 | |
| JPS6220998B2 (zh) | ||
| JP2643943B2 (ja) | 新規な6−もしくは7−メチレンアンドロスタ−1,4−ジエン−3,17−ジオン誘導体およびその製造方法 | |
| CN1402735A (zh) | 新颖的雄激素 | |
| FI87791C (fi) | Foerfarande foer framstaellning av 17 -(cyklopropyloxi)androst-5-en-3 -ol och naerstaoende foereningar, som aer anvaendbara som c17-20 lyas inhibitorer | |
| CN1104439C (zh) | 苯基取代的4-氮杂甾族氟衍生物、其制备方法和用途及含有它们的药物组合物 | |
| CN1372565A (zh) | 在11位带有烃取代基的非芳香族雌激素甾族化合物 | |
| CN101863947A (zh) | 一种地诺孕素的合成方法 | |
| CN87105736A (zh) | 14,17β-桥亚乙基-14β-雌三烯和雌四烯的制备方法和含有它们的药物制剂 | |
| CN86107158A (zh) | 1-甲基-15α-烷基-雄甾-1,4-二烯3,17-二酮,及其制法和含有该类化合物的药物的制法 | |
| EP0338065B1 (fr) | Derives de la 19-nor progesterone, leur preparation et leur utilisation | |
| CN1205219C (zh) | 不饱和14,15-环丙烷并-雄烷,其制备方法以及包含该化合物的药物组合物 | |
| CN1226304C (zh) | 作为雄激素的亚甲基类固醇 | |
| Perni et al. | A synthetic approach to (+/-)-aristomakine | |
| US3717663A (en) | 9-alpha-methyl steroids | |
| Biggerstaff et al. | 17-(5-Substituted-2-thienyl) and 17-(5-substituted-2-thienylidene) derivatives of selected 17-oxo steroids | |
| US3655652A (en) | Aromatically-unsaturated 9alpha-methyl steroids | |
| IE73240B1 (en) | Steroids and therapeutic compositions containing same |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C02 | Deemed withdrawal of patent application after publication (patent law 2001) | ||
| WD01 | Invention patent application deemed withdrawn after publication |