CN87107343A - 23位上酮基的甾族化合物,其制备方法及用其制备20酮孕甾烷 - Google Patents
23位上酮基的甾族化合物,其制备方法及用其制备20酮孕甾烷 Download PDFInfo
- Publication number
- CN87107343A CN87107343A CN87107343.9A CN87107343A CN87107343A CN 87107343 A CN87107343 A CN 87107343A CN 87107343 A CN87107343 A CN 87107343A CN 87107343 A CN87107343 A CN 87107343A
- Authority
- CN
- China
- Prior art keywords
- product
- formula
- group
- carbon atoms
- acid
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07J—STEROIDS
- C07J9/00—Normal steroids containing carbon, hydrogen, halogen or oxygen substituted in position 17 beta by a chain of more than two carbon atoms, e.g. cholane, cholestane, coprostane
- C07J9/005—Normal steroids containing carbon, hydrogen, halogen or oxygen substituted in position 17 beta by a chain of more than two carbon atoms, e.g. cholane, cholestane, coprostane containing a carboxylic function directly attached or attached by a chain containing only carbon atoms to the cyclopenta[a]hydrophenanthrene skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07J—STEROIDS
- C07J31/00—Normal steroids containing one or more sulfur atoms not belonging to a hetero ring
- C07J31/006—Normal steroids containing one or more sulfur atoms not belonging to a hetero ring not covered by C07J31/003
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07J—STEROIDS
- C07J43/00—Normal steroids having a nitrogen-containing hetero ring spiro-condensed or not condensed with the cyclopenta(a)hydrophenanthrene skeleton
- C07J43/003—Normal steroids having a nitrogen-containing hetero ring spiro-condensed or not condensed with the cyclopenta(a)hydrophenanthrene skeleton not condensed
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Steroid Compounds (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
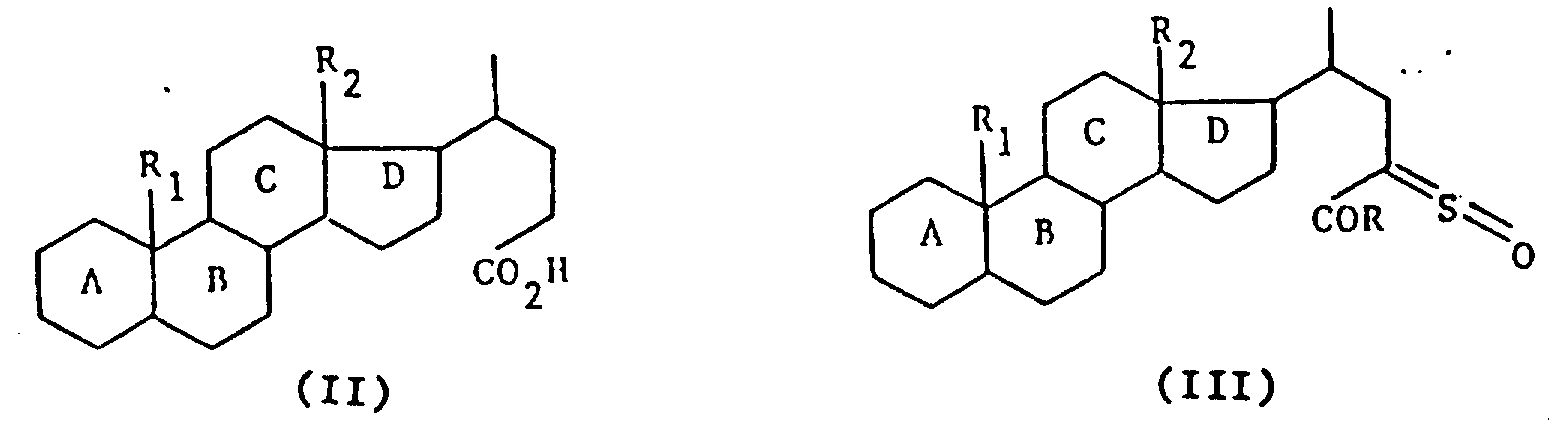
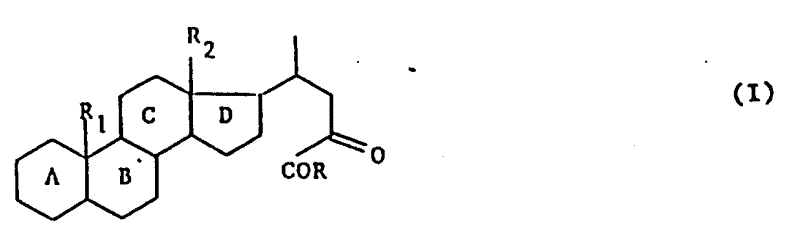
A.本发明的目的是提供制备式(I)化合物的方法。式中R1是氢或甲基,R2是甲基或乙基,A,B,C和D环可任意含有1个或多个双键,并且任意由1个或多个官能团取代或者1个或多个原子或基团取代,R是卤素,羟基,烷氧基,芳烷硫基或
Description
本发明涉及包括23位上的酮基的新的甾族化合物产物及其制备方法、它们在制备20酮孕甾烷类产物中的应用和用于此目的中间体。
本发明的任务在于提供通式(Ⅰ)的产物:
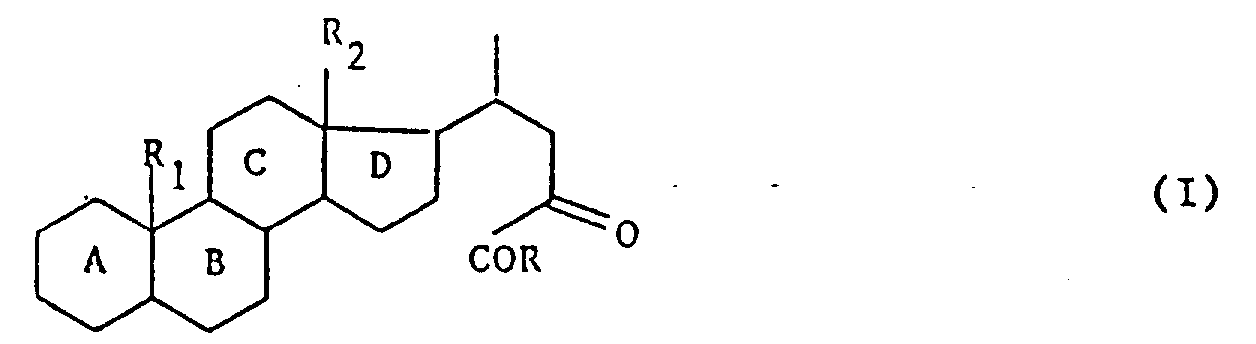
式中,R1代表氢原子或甲基,R2代表甲基或乙基,母核A,B,C,D可以含1个或多个双键并且可以由下列基团取代:1个或多个羟基或可以被保护的酮官能团,1个或多个卤原子,含有1~4个碳原子的1个或多个烷基或烷氧基,或含有2~4个碳原子的1个或多个链烯基或炔基,R代表卤原子、羟基、含有1~6个碳原子的烷氧基、含有7~15个碳原子的芳烷氧基、
 基,其中R3和R4可以相同,也可不同,代表氢原子、含有1~6个碳原子的烷基、含有7~15个碳原子的芳烷基,或R3、R4与之键合的氮原子一起形成杂环,该环可以包括选自氮原子和氧原子的另一个杂原子,或者R代表含有1~6个碳原子的烷硫基,或最多可含15个碳原子的芳硫基或芳烷硫基。
基,其中R3和R4可以相同,也可不同,代表氢原子、含有1~6个碳原子的烷基、含有7~15个碳原子的芳烷基,或R3、R4与之键合的氮原子一起形成杂环,该环可以包括选自氮原子和氧原子的另一个杂原子,或者R代表含有1~6个碳原子的烷硫基,或最多可含15个碳原子的芳硫基或芳烷硫基。
当环A,B,C和D含1个或多个双键时,双键的最佳键位是在1(2),4(5),5(6)或9(11)位上,或共轭双键系统在3(4)和5(6)或4(5)和6(7)位上,或3个双键芳族系统上1,3,5位上,或3个双键系统在1(2),4(5),6(7)位上。但是最好使用不含双键的产物。
当环A,B,C和D由1个或多个羟基官能团取代时,则1个或多个羟基官能团在3,6,7,11和/或12位上为最佳。
当环A,B,C和D由1个或多个酮官能团取代时,酮官能团在3,7,11或12位上为最佳1。
当环A,B,C和D由1个或多个卤原子取代时,举例来说,则氟、氯或溴原子在6或9α位上为最佳。
当环A,B,C和D由1个或多个烷氧基取代时,甲基或乙基在2,6,7或16α或16β位上为最佳1。
当环A,B,C和D由1个或多个烷氧基取代时,甲氧基或乙氧基在3或11β位上为最佳。
当环A,B,C和D由1个或多个链烯基取代时,例如乙烯基或烯丙基则在11β位上为最佳。
当环A,B,C和D由1个或多个炔基取代时,例如乙炔基在11β位上最佳。
羟基可以按文献中已知的常规方法加以保护。例如能够用:丙酮化合物类,环碳酸酯,原酸酯,环亚硫酸酯,与四氢吡喃基、三苯甲基或苄基、酰基如乙酰、琥珀酰或甲酰生成醚。
酮基也可用常用保护基予以保护,例如缩酮,尤指乙二醇缩酮,硫缩醛,半硫缩醛,烯醇醚,烯醇乙酸酯,烯胺和肟。
然而缩酮,尤其是乙二醇缩酮用于保护酮基为最佳。当式Ⅰ的产物在3位上含酮基时,对酮基保护尤为理想。
R代表卤原子,尤以氯或溴原子为佳;
R也可代表烷氧基,尤以甲氧基或乙氧基为佳,但也可以是丙氧基,异丙氧基,丁氧基,仲丁氧基,叔丁氧基,戊氧基或己氧基;R也可代表苄氧基或苯乙氧基。
相同或不相同的R3和R4代表氢原子或甲基,乙基,丙基,异丙基,丁基,仲丁基,叔丁基,戊基,己基或苄基,或R3、R4与之键合的氮原子共同形成吗啉基,哌啶基或吡咯烷基。R也可代表甲硫基或乙硫基或由如上所述的烷基或烷氧基衍生的烷硫基。R还可以代表苯硫或苄硫基。
本发明的化合物尤指如上定义的通式Ⅰ的产物,其中R和R分别为甲基和母核A,B,C和D在3位上可以含保护的羟基官能团,在6,7,11和12位上可以含选自保护的羟基官能团的1个或多个其他官能团,在7,11,和12位上可以含保护的酮官能团,R代表羟基,最多可含4个碳原子的烷氧基,或
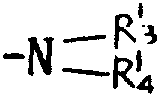 基,其中R1 3和R1 4代表氢原子,最多可含4个碳原子的烷基,或R1 3、R1 4与之键合的氮原子共同形成哌啶子基,吡啶烷子基或吗啉子基,尤其是如上定义的通式Ⅰ的产物,其环A,B,C和D在3位上可以含保护的羟基,在12位上可以含选自保护的羟基官能团的1个或多个其他官能团,以及在11或12位上可以含保护的酮官能团,R代表羟基,甲氧基或乙氧基,或吗啉子基。
基,其中R1 3和R1 4代表氢原子,最多可含4个碳原子的烷基,或R1 3、R1 4与之键合的氮原子共同形成哌啶子基,吡啶烷子基或吗啉子基,尤其是如上定义的通式Ⅰ的产物,其环A,B,C和D在3位上可以含保护的羟基,在12位上可以含选自保护的羟基官能团的1个或多个其他官能团,以及在11或12位上可以含保护的酮官能团,R代表羟基,甲氧基或乙氧基,或吗啉子基。
在最佳产物中,特别有利是如上定义的通式Ⅰ的产物的亚族产物,其中母核A,B,C和D在其3位上含保护的羟基,在6,7或12位上含选自保护的羟基官能团的1个或多个其他官能团,在其7,11或12位上含保护的酮官能团。
在最后这一族中,可以例举的产物包括以环A,B,C D为骨架,由天然的或半人工含成的胆汁烷酸衍生的产物。这些产物列表于下:
式中,R1代表羟基,甲氧基,乙氧基或吗啉子基,R6,R7和R12含有下列意义:
R6R7R12
H OHα OHα
H OHβ OHα
H H H
H H OHα
H OHα H
OHα H H
H OHβ H
OHα OHα H
OHβ OHα H
OHβ OHβ H
H H OHα
H OHα OHα
在这些产物中,对一个或多个羟基也能加以保护,特别是在3位上的羟基。最佳保护基是乙酰基或甲酰基。
在包括1个或多个酮官能团的这些产物中,下列产物为最佳:
式中,R1代表羟基,甲氧基,乙氧基或吗啉子基,并且在3,7,11和12位上的取代基具有下列含义:
-3保护酮
-3OH α,7酮基,12OH α
-3OH α,11酮基
-3OH α,7OH α,12酮基
-3OH α,7酮基
-3OH α,7OH β,12酮基
-3OH,11酮基,12OH
-3OH,11酮基。
如前所述,对羟基当然也可加以保护。同样也适用于7位或12位上的酮基。酮基的最佳保护基为环状缩酮或非环状缩酮。
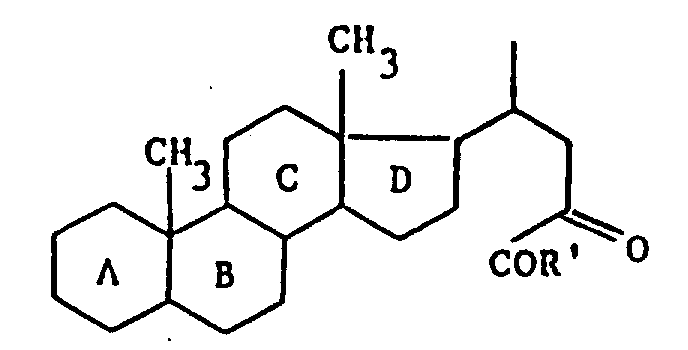
最佳产物将在实施例中详述,尤其是下列产物:
3α,5β-4-(3-乙酰氧基-11,23,24-三氧-胆烷-24-基)吗啉。
3α,5β-11,23-二氧-3-羟基-胆烷-24-酸。
3α-羟基-11,23-二氧-5β-胆烷-24酸甲酯
3α,5β-3-乙酰氧基-11,23-二氧-胆烷-24-酸。
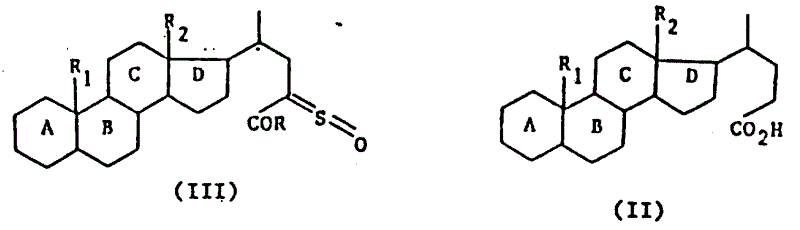
本发明的任务还在于提供制备如上定义的通式Ⅰ的产物的方法,其特征在于通式(Ⅱ)的产物先用一种试剂进行处理形成酸性卤化物,继之用叔碱,亚链酰氯处理,最后可用水、链烷醇、芳链烷醇、含
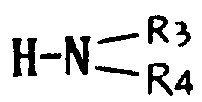 的伯胺或仲胺(R3和R4的定义同上)、烷硫醇、芳硫醇或芳烷硫醇处理,得到式(Ⅲ)产物:
的伯胺或仲胺(R3和R4的定义同上)、烷硫醇、芳硫醇或芳烷硫醇处理,得到式(Ⅲ)产物:
式中A,B,C,D,R,R1和R2的含意同上,再将式Ⅲ的产物进行下列处理:
或用含水酸或氧化剂,
或先后分别用卤化试剂和碱性水解剂,
亦可用链烷醇、芳链烷醇、含
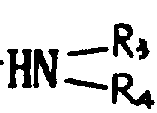 的伯胺或仲胺(式中R3和R4的定义同上)、烷硫醇、芳硫醇或芳烷硫醇,处理含R为烃基的式Ⅰ得到的产物,
的伯胺或仲胺(式中R3和R4的定义同上)、烷硫醇、芳硫醇或芳烷硫醇,处理含R为烃基的式Ⅰ得到的产物,
或用形成酸性卤化物的试剂处理含R为羟基的式Ⅰ得到的产物。
在实施上述方法的最佳途径中,最好用形成酸性卤化物的试剂,这种试剂选自亚硫酰氯、草酰氯或草酰溴、尤以亚硫酰氯为佳。
所用的叔碱选自三乙胺、甲基乙基吡啶,吡啶,二氮二环辛烷,二氮二环壬烯,二氮二环十一碳烯,尤以三乙胺或吡啶为佳。
所用最佳的链烷醇或芳链烷醇是甲醇,乙醇,或苄醇。
能用的伯胺或仲胺选自甲胺或乙胺,二乙胺,吗啉,哌啶,吡咯烷,以吗啉为最佳。当然,相应的硫醇也可使用。
可以用的烷硫醇、芳硫醇或芳烷硫醇,最好选目甲硫醇、乙硫醇和硫苄醇。
用于优先将式Ⅲ产物转化为式Ⅰ产物的含水酸是硫酸,也可用另一种无机酸或有机酸,如盐酸或乙酸。
使用氧化剂时,可用高锰酸钾、过氧化氢、臭氧、过硼酸盐或过硫酸盐。
一般说来,含水酸或氧化剂对式Ⅱ产物的作用导致R为羟基的式Ⅰ产物的形成。如有需要并且期望得到式Ⅰ的其他产物时,也可以进行制备,例如用链烷醇如甲醇分别或同时与之作用,即可得到R为羟基的式Ⅰ产物,或用伯胺或仲胺如吗啉与之作用,即可得到
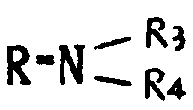 的式Ⅰ产物,还可采用相应的硫醇。最佳的方法是在存在期望得到式Ⅰ衍生物的链烷醇或伯胺或仲胺的条件下,进行酸水解或氧化反应。
的式Ⅰ产物,还可采用相应的硫醇。最佳的方法是在存在期望得到式Ⅰ衍生物的链烷醇或伯胺或仲胺的条件下,进行酸水解或氧化反应。
当然,也可以将选自上面所列的酸性卤化物的形成剂与R=OH的式Ⅰ产物相作用。
上述用式Ⅲ产物制备产物的反应最好是在溶剂或与水略微相溶或根本不相溶的溶剂(如二氯甲烷和三氯甲烷)的混合物中进行。
当然,环A,B,C和D上的官能团的常规保护或去保护反应,在含成开始时就能在式Ⅱ产物或用式(Ⅰ)得到的产物上进行。例如,为了得到A环上含有自由羟基的相应产物,可将在A环3位上含有由酰基(如乙酰基或甲酰基)保护的羟基的式(Ⅰ)产物进行常规的皂化反应。操作按常规方法,通过溶于溶剂中,如甲醇、二氯甲烷、水或它们的混合物的碱作用,这些碱例如有:氢氧化钠,氢氧化钾或钾的碳酸盐。
反之,为了保护例如3位上的自由羟基,也可将式(Ⅰ)产物通过保护基衍生物如乙酐的作用达到。
本发明的任务还在将如上定义的式(Ⅰ)产物用于制备式(Ⅵ)产物:
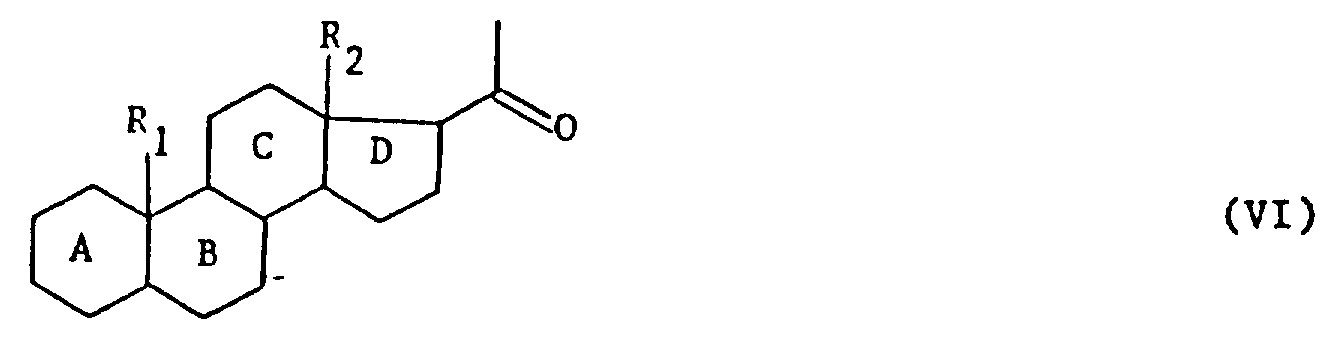
式中,A,B,C,D,R1和R2的定义同上,其特征在于用强氧化剂处理式(Ⅰ)产物,可得到式(Ⅳ)产物:
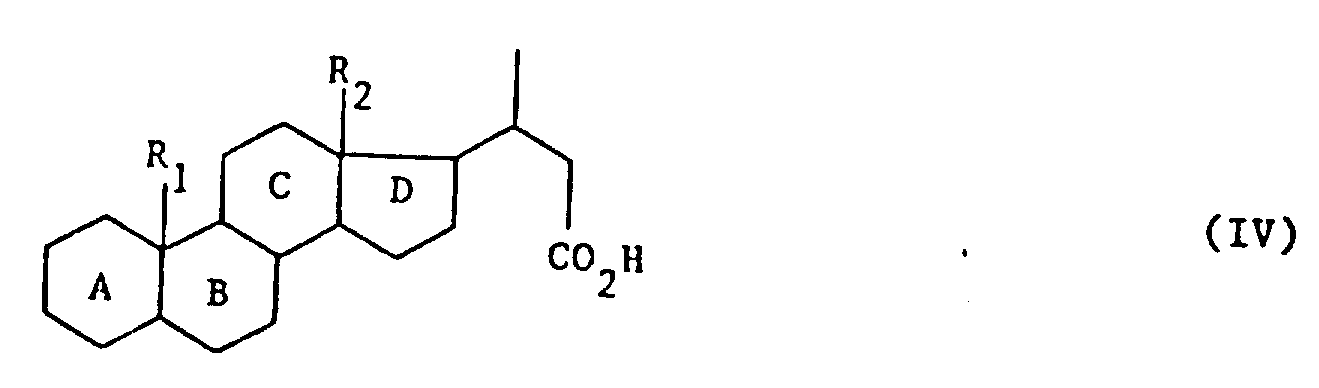
式中,A,B,C,D,R1和R2的定义同上,式(Ⅳ)产物先后用酸性卤化物形成剂、叔碱、亚硫酰氯和卤化剂处理,最后可用水、链烷醇、芳链烷醇、含
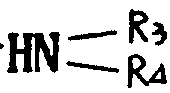 的伯胺或仲胺(式中R3和R4的定义同上)、烷硫醇、芳硫醇或芳烷硫醇处理,得到式(Ⅴ)产物:
的伯胺或仲胺(式中R3和R4的定义同上)、烷硫醇、芳硫醇或芳烷硫醇处理,得到式(Ⅴ)产物:
式中,A,B,C,D,R1,R2的定义同上,Hal代表卤原子,式(Ⅴ)产物先用卤化氢剂,再用氧化切割剂(oxidizing cutting agent)处理,得到所需的式(Ⅵ)产物。
所用强氧化剂可选自琼斯试剂(铬酸和硫酸水溶液)、乙酸高铅、过氧化氢、重铬酸钾。
在式(Ⅳ)产物上所用酸性卤化物形成剂可从由上所述中选择,尤以亚硫酰氯为佳。其他试剂也可从上所述中选择。
所用卤化试剂可以是卤素,如溴,也可以是卤化试剂,如磺酰氯。
用于在式(Ⅴ)产物上的脱卤化氢剂最好是强碱剂,例如三通B,(
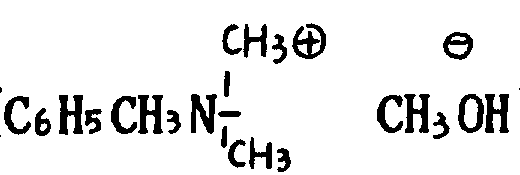 ),碱金属醇化物,如乙醇钠或乙醇钾,叔丁醇钾,氨基钠或氨基钾1。还可预计到可利用碱如氢氧化钠或氢氧化钾在链烷醇如甲醇、乙醇或甘醇二甲醚中回流。最后,应予注意的还有碱性树脂,如离子交换树脂(amberlitc)。
),碱金属醇化物,如乙醇钠或乙醇钾,叔丁醇钾,氨基钠或氨基钾1。还可预计到可利用碱如氢氧化钠或氢氧化钾在链烷醇如甲醇、乙醇或甘醇二甲醚中回流。最后,应予注意的还有碱性树脂,如离子交换树脂(amberlitc)。
在合成最终阶段用于获得式(Ⅵ)产物的氧化切割剂,选自臭氧和氧化剂,如氧化钌或氧化锰。
根据申请人的测定,脱卤化氢剂对式(Ⅴ)产物的作用,从而产生式(Ⅴ1)的产物:
该产物在氧化切割以后,生成式(Ⅵ)产物。
当然,在将式(Ⅰ)产物用于制备式(Ⅵ)产物时,环A,B,C,和D上进行保护和去保护官能团的常规反应可以用于式(Ⅰ)的原料产物,也可以用于合成的中间产物。
特别是在对式(Ⅴ)中间产物进行脱囟化氢反应,导致酰基保护基如乙酰基或甲酰基皂化的情况下,含有自由羟基的产物在存在吡啶下由乙酐再酰基化。
上述诸方法也属本发明的任务,其特征在于所用的酸性卤化物形成剂是亚硫酰氯。
由于在任何情况下,形成亚磺基官能团的反应都包括使用的亚硫酰氯,所以在以酸性卤化物形成剂为亚硫酰氯的最佳条件下,上述试剂即a)酸性囟化物形成剂的作用,b)叔碱,c)亚硫酰氯结合在一起,相当于在叔碱存在下对亚硫酰氯式(Ⅱ)产物的作用。
最后,如上定义的式(Ⅳ)和(Ⅴ)产物也是本发明的任务,这类产物是一种新的工业产品并且是一种利用式(Ⅰ)产物所必需的值得注意的工业产品。
对胆汁烷酸类的许多天然产物和以这些天然产物为原料通过常规方法能够制备的产物而言,在制备式(Ⅰ)产物工艺开始阶段所用的式(Ⅱ)产物是一种已知产物。
式(Ⅵ)产物是孕甾酮系产物,这些产物具有含人感兴趣的药理特征。此外,这些产物可用作构成去氧可的松链:
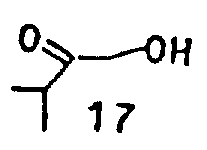 或17位上其他链的基本原料。
或17位上其他链的基本原料。
以下实施例用于说明本发明而不是限制本发明。
实施例1:3α,5β-11,23-二氧-3-羟基-胆烷-24-酸
步骤A:3α,5β-3-乙酰氧基-11-氧-胆烷-24-酸。
将200g3α-羟基-11-氧-3β胆烷-24-酸和400cm3乙酐混合,加热至45℃,一次性加入2g对甲苯磺酸和20cm3乙酸。5分后温度升至63℃,在60℃保持1小时,然后温度降至55℃。约1小时后,在55℃下加入400cm3蒸馏水,冷却至10℃后,对生成的沉淀物进行分离、洗涤和减压干燥。得到211g所需产品,熔点225℃(纯度约99%)。
将106克所得产品溶于二氯甲烷,进行硅过滤,用二氯甲烷和乙酸乙酯混合物(9/1)洗脱,得到105克纯产品,熔点225℃。
检验:
红外光谱(三氯甲烷)Cm-1

核磁共振谱(CDCl3)pmm
18位上CH3的H 0.62
21位上CH3的H 0.88~0.93
19位上CH3的H 1.2
ACO的H 2.03
3位上的H 4.72
COOH的H 8.1
步骤B:3α,5β-4-(3-乙酰氧基-11,23,24-三氧-胆烷-24-基)吗啉
在惰性气氛下,将68克3α,5β-3-乙酰氧基-11-氧-胆烷-24-酸、250cm3二氯甲烷和0.35cm3N,N-二甲基甲酰胺混合并在二氯甲烷中回流约15分,加入12.8cm3亚硫酰氯。再回流45分钟,继之通过减压蒸馏浓缩干燥。将250cm3二氯甲烷加入结晶酰基氯并在-15℃下再加12.8cm3亚硫酰氯。在-25℃和大约1小时30分钟内,加入46.5cm3三乙胺和46.5cm3二氯甲烷混合物,得到的悬浮液搅拌30分钟。维持温度在-25℃下并保持大约30分钟下,加入35.5cm3吗啉和50cm3二氯甲烷混合物,搅拌30分钟,然后在大约10分钟内,加入350cm3水,同时允许温度升至0℃。加4.7cm3乙酸,并在2~5℃和大约1小时30分钟内,加49.6g高锰酸钾,混合物用240cm3水稀释,在2~5℃下搅拌1小时。在5~10℃和大约30分钟内,加入43g亚硫酸氢钠,同时加浓硫酸(12cm3)的冰水(150cm3)溶液。滗析后,二氯甲烷相用水洗涤,脱水,在20℃和均匀搅拌下,大约1小时30分钟内先后加入5g硫酸镁和60g铝CBT1。再在室温下搅拌1小时30分钟,过滤后通过减压蒸馏将滤液浓缩干燥。将80cm3乙酸乙酯加入残渣,然后通过减压蒸馏浓缩干燥,去除残留的二氯甲烷,将100cm3乙醇加入残留物,在40℃左右温度下将其搅拌溶解,溶液冷却至0℃开始结晶,静置16小时后,分离得到57.6g所需产品,熔点122~123℃。
将母液浓缩干燥,得到22g残留物,径滴定测定含有83.5%所需产品。
检验:红外光谱(三氯甲烷)cm-1。

1715
1704
核磁共振谱(CDCL3),ppm
18位上CH3的H 0.67
21位上CH3的H 0.9~1.0
19位上CH3的H 1.17
ACO的H 2.0
3位上的H 4.7
吗啉中的H 3.4~3.8
步骤C:3α,5β-11,23-二氧-3-羟基-胆烷-24酸。
在惰性气氛下,将前一步骤得到的0.5g产品,5cm3的5%甲醇水溶液和0.75%氢氧化钠(锭中)混合在一起,在室温下放置24小时。溶液沉清后加入2N盐酸水溶液,至pH呈酸性。用乙酸乙酯提取。提取液脱水后经减压蒸馏浓缩干燥,得到0.4g所需产品。
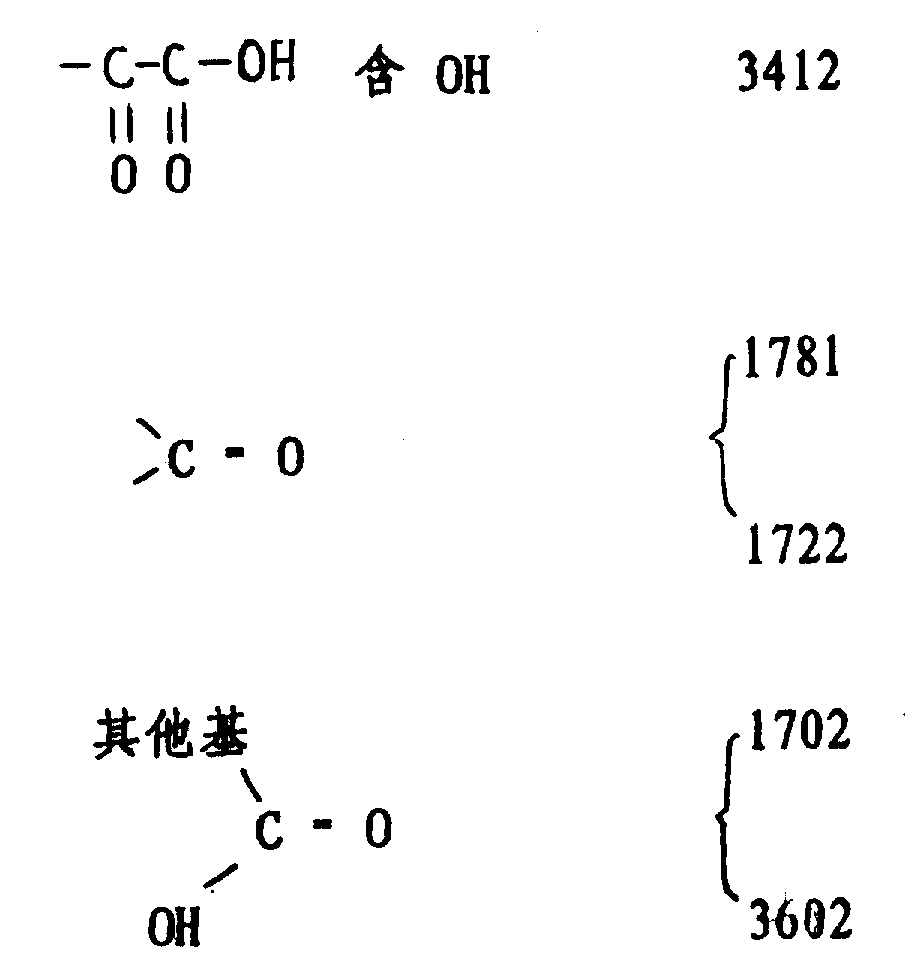
检验:红外光谱(三氯甲烷)cm-1。
3羟基OH 3605 区域C:O1781
酸的3种形式 3410 ep 1750
1781 1700
3510(单体) 1710
1703
实施例2:3α,5β-11,23-二氧-3-羟基-胆烷-24-酸。
步骤A:3α-甲酰氧基-11-氧-23-亚硫酰基-5β-胆烷-24-酸。
将83.7g3α-甲酰氧基-11-氧-23-亚硫酰基-5β胆烷-24-酸、840cm3二氯甲烷和168cm3吡啶混合,冷却至10℃,大约5分钟,同时允许温度升至20℃,加32cm3亚硫酰。在20℃下搅拌1小时后,在大约5分内加入84cm3水并在20℃下搅拌15分钟。然后将反应混合物注入盐酸冰水溶液,搅拌后滗析,用二氯甲烷提取。提取液用活性炭处理,再经减压蒸馏浓缩干燥。得到98.5g所需的粗制品,Rf.=0.45,用三氯甲烷、异丙醇和乙酸混合物(85∶14∶1)洗脱。
检验:
元素分析:C25H36O6S(464.60)
计算值:C%64.82,H%7.81,S%6.90
实际值:64.8,7.7,7.00
紫外光谱(乙醇)
吸收峰值:282nm,E1 1=157,ε=6400
77%锍。
步骤B:3α-羟基-11,23-二氧-5β-胆烷-24-酸的甲基醚。
在惰性气氛下,将步骤A得到的2g3α-甲酰氧基-11-氧-23-亚硫酰基-5-β胆烷-24-酸粗制品、20cm3甲醇和0.4cm3浓硫酸混合,回流加热2小时,然后注入水和冰混合物中,用乙酸乙酯提取。提取液经水洗涤,通过减压蒸馏干燥浓缩。残渣经硅层析并用二氯甲烷和乙酸乙酯混合物(9∶1)洗脱,得到0.64g所需产品,再由二氯甲烷和异丙醚混合物结晶,纯化得到的所需产品,熔点约75℃。(Rf.=0.32,用二氯甲烷和乙酸乙酯混合物(85∶15)洗脱)。
红外光谱(三氯甲烷)
不存在甲酸而存在OH,酮并不共轭而且酯带很宽。
18位上甲基的H:0.67
20位上甲基的H:0.9~1.0
19位上甲基的H:1.14
H :3.67
COOCH3的H :3.9
步骤C:3α,5β-11,23-二氧-3-羟基-胆烷-24-酸。
在惰性气氛下,将前一步骤所得的产品5g、5Cm3甲醇、1.3cm3水和35毫克氢氧化钠(锭中)混合,在20℃下搅拌20小时。然后将反应混合物注入N盐酸和冰混合物中,搅拌后生成的沉淀物经分离和水洗涤后,溶于二氯甲烷,经减压蒸馏浓缩干燥,得到0.43g所需产品。
检验:
红外光谱(三氯甲烷)cm-1。
实施例3:3α-羟基-11,23-二氧-5β-胆烷-24-酸。
步骤A:3α-甲酰氧基-11-氧-22,23-二溴-5β-胆烷-24-酸。
在惰性气氛下,将20.9g3α-甲酰氧基-11-氧-5β-胆烷-24-酸、200cm3二氯甲烷和32cm3吡啶混合。在大约5分钟内在5℃下同时允许温度升至20℃,加入8cm3亚硫酰氯,在20℃下搅拌1小时,然后冷却至10℃。在大约5分钟内,加入8cm3溴并在20℃下搅拌1小时。将反应混合物注入水和冰混合物中,搅拌,滗析,再用二氯甲烷提取。提取液经脱水,用含少量铝的活性炭处理,过滤和减压蒸馏浓缩干燥。将40cm3甲酸加入残留物,加热至沸点保持5分钟后,经减压蒸馏去除甲酸。再缓慢加入40cm3异丙醚,冷却后分离得到24.6g所需产品。
熔点248℃,Rf.=0.40,用三氯甲烷、异丙醇和乙酸混合物(85∶14∶1)洗脱。
分析:C25H36O5Br2,576,38
计算值:C%52.09,H%6.30,Br%27.73
实际值:52.0,6.30,27.4
步骤B:3α-羟基-11,23-二氧-5β-胆烷-24-酸。
在惰性气氛下,将前一步骤得到的产品46.6g和930cm3的N氢氧化钠混合,悬浮液在100℃下加热4小时,冷却后加冰,再加100cm3盐酸,然后浓缩并用乙酸乙酯提取。提取液经脱水,用含少量铝的活性炭处理,然后过滤,滤液经减压蒸馏浓缩干燥,再将二氯甲烷加入残渣进行结晶。分离得到25.4g所需产品,熔点约155℃。
产品经硅层析,用三氯甲烷、异丙醇和乙酸混合物(80∶18.5∶1.5)洗脱。所需的部分经浓缩,再加乙酸乙酯,用水洗涤后脱水,并经减压蒸馏浓缩干燥,由丙酮和石油醚(沸点60~80℃)混合物结晶。得到所需产品,熔点130℃,Rf.=0.35,用三氯甲烷、异丙醇和乙酸混合物(78∶20∶2)洗脱。
红外光谱(三氯甲烷)
存在OH,11-酮基和-C-COOH
核磁共振谱(CDCl3)ppm.
18位上甲基的H:0.68
20位上甲基的H:0.9~1.0
19位上甲基的H:1.18
游离H :4.4
实施例4:3α,5β-3-乙酰氧基-11,23-二氧-胆烷-24-酸。
在惰性气氛下,将141mg3α,5β-11,23-二氧-3-羟基-胆烷-24-酸、0.3cm3乙酐和3mg对甲苯磺酸混合,在20℃下搅拌2小时,然后加2cm3水,再搅拌15分钟。用乙酸乙酯提取后,提取液用水洗涤、脱水并经减压蒸馏浓缩干燥。残渣经硅层析并用二氯甲烷、异丙醇和乙酸混合物(87∶12.5∶0.5)洗脱,得到80mg所需产物。
红外光谱(三氯甲烷)cm-1。
存在
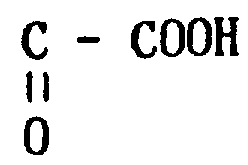
OH 酸 3420
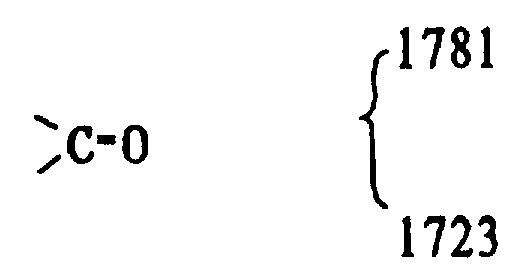
11酮基 1723
实施例5:3α-乙酰氧基-11,20-二氧-5β-孕甾烷
步骤A:3α,5β-3-乙酰氧基-11-氧-24-降胆烷-23-酸。
在惰性气氛下,将893mg3α,5β-3-乙酰氧基-11-,23-二氧-胆烷-24-酸和6cm3乙酸混合,在15℃和大约20分钟内,加4.5cm3琼斯氧化溶液(制备方法:267g CrO3和230cm H2SO4,加水至1000cm3),在15℃搅拌5分钟。然后将反应混合物注入冰水,用二氯甲烷提取。提取液先后用0.1M的硫代硫酸钠水溶液和水洗涤,脱水并经减压蒸馏浓缩干燥,残渣用硅层析并用二氯甲烷和丙酮混合物(9∶1)洗脱,得到680mg所需产物,熔点110~120℃,不很纯。
红外光谱(三氯甲烷)
存在乙酸酯、11-氧和酸。
核磁共振谱(CDCl3)ppm.
18位上甲基的H:0.66
21位上甲基的H:0.97~1.02
19位上甲基的H:1.18
ACO的H :2.0
H3:4.68
步骤B:4-3α,5β-3-乙酰氧基-22,22-二溴-11-氧-24-降胆烷-23-基-吗啉
在惰性气氛中,将4.1g前一步骤所得产品、41cm3二氯甲烷和6.35cm3吡啶混合,在0~5℃下加1.57cm3亚硫酰氯后,随即加1.6cm3溴,在20℃搅拌1小时。在0~5℃和大约15分钟内,加8.5cm3吗啉,搅拌1小时,同时允许温度回升至20℃。然后将反应混合物注入400cm3的2N冰盐酸中并用三氯甲烷提取。提取液用水洗涤、脱水,通过减压蒸馏浓缩干燥。残渣(6.5g)经硅层析并用环己烷和乙酸乙酯混合物(8∶2)洗脱,得到2.075g所需产品。
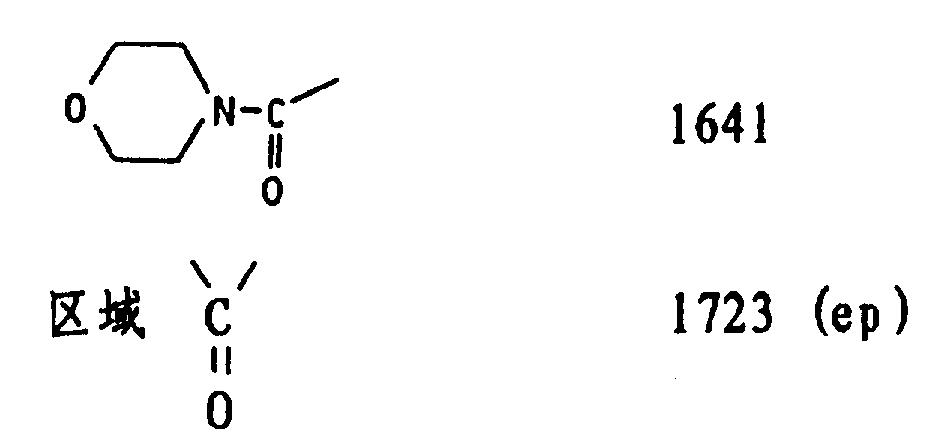
检验:
红外光谱(三氯甲烷)cm-1。
OAC 1724
1364
C-O配合物 1267-1251-1235
11-氧 1704
酰胺 1645
核磁共振谱(CDCl3)ppm
18位上甲基的H:0.74
19位上甲基的H:1.16
21位上甲基的H:1.34~1.42
ACO的H :1.8
吗啉的H :3.76
H3:4.7
分析:C29H43Br2NO5645.48
计算值:C%53.96,H%6.71,N%2.17,Br%24.76
实际值:53.9,6.7,2.1,24.6
步骤C:3α-乙氧基-11,20-二氧-5β-孕甾烷。
1.脱溴水合反应
在惰性气氛下,将250mg前一步骤得到的产品,2.5cm3甲醇和2.5cm3三通β(或氢氧化苄基三甲基铵)的40%水溶液混合,回流加热1小时。冷却后,将反应混合物注入冰水中并用二氯甲烷提取。提取液用水洗涤、脱水并经减压蒸馏浓缩干燥,得到的120mg所需产品,用于下步反应。
2.乙酰化反应
在惰性气氛下,将上述得到的产品、1.2cm3吡啶和0.48cm3乙酐混合,接触20小时,然后全部注入冰水,30分钟后用二氯甲烷提取。提取液用水洗涤、脱水并经减压蒸馏浓缩干燥,得到的170mg所需乙酰化产品用于下步反应。
3.臭氧分解反应
在惰性气氛下,将所得产品、2.5cm31,2-二氯乙烷和1cm3乙酸混合,在-5℃下臭氧化氧流经15分钟。过量的臭氧用通入氮气去除,然将反应混合物徐徐注入过量的碳酸氢钠水溶液。用二氯甲烷提取,提取液用水洗涤、脱水并经减压蒸馏浓缩干燥,得到115mg所需产品(3α-乙酰氧基-11,20-二氧-5β-孕甾烷)。
粗制品经硅层析并用环己烷和乙酸乙酯混合物(8∶2)洗脱,得到16mg所需的纯产品,经红外光谱测定,其与真实样品相同。
实施例6:3α-乙酰氧基-11,20-二氧-5β孕甾烷,从3α,5β-3-乙酰氧基-11-氧-24-降胆烷-23-酸起始制备
1.溴化反应
在惰性气氛下,将实施例5中步骤A所得的3.2g3α,5β-3-乙酰氧基-11氧-24降胆烷-23-酸(滴定率为85%)、32cm3二氯甲烷和6.1cm3吡啶混合,在-50℃和大约10分钟内,滴加1.2cm3亚硫酰氯,然后在-10℃和大约10分钟内,加入0.6cm3溴。在20℃下搅拌2小时30分钟后,在-5℃和大约20分钟内加入8cm3二乙胺。然后在20℃下搅拌1小时,再将反应混合物注入2N冰盐酸,搅拌15分钟。用二氯甲烷提取,提取液用水洗涤、脱水、活性炭处理和减压蒸馏干燥。得到5.1克所需粗溴化产品用于下步反应。
2.三通B(脱溴水合)处理
在惰性气氛下,将1.5g上述得到的粗溴化产品、15cm3甲醇和12cm3三通B(氢氧化苄基三甲基铵)的40%水溶液混合,回流加热1小时30分钟,冷却后注入水中并用二氯甲烷提取。提取液用氯化钠和水溶液、0.5M磷酸二氢钠水溶液和氯化钠饱和水溶液洗涤,然后脱水并通过减压蒸馏浓缩,得到的837mg所需粗制品用于下步反应。
3.乙酰化反应
在惰性气氛下,将837mg上述产品、3cm3吡啶和1.5cm3乙酐混合,在20℃下搅拌20小时。加入几毫升水,搅搏1小时后,将反应混合物注入氯化钠饱和水溶液中,用二氯甲烷提取。提取液用饱和氯化钠水溶液洗涤、脱水,并经减压蒸馏浓缩干燥,得到954mg所需乙酰化粗制品用于下步反应。
4.臭氧化反应
将上一步得到的954mg乙酰化粗制品、15cm3二氧甲烷和8cm3乙酸混合,在0℃下臭氧化氧流经1小时。然后将反应混合物注入水中并用二氯甲烷提取。提取液用氯化钠饱和水溶液洗涤,脱水,并经减压蒸馏浓缩干燥,得到992mg粗臭氧化产品。粗产品经硅层析并用环己烷和乙酸乙酯混合物(75∶25)洗脱,得到106mg所需3α-乙酰氧基-11,20-二氧-5β孕甾烷。
红外光谱(三氯甲烷):
经测定,得到的产品与3α-乙酰氧基-11,20-二氧-5β-孕甾烷的真实样品相同。
对照实施例:3α,5β-4-(3-乙酰氧基-11,23,24-三氧-胆烷-24-基)吗啉
在惰性气氛下,将68克3α,5β-3-乙酰氧基-11-氧-胆烷-24-酸、250cm3二氯甲烷和0.35cm3N,N-二甲基甲酰胺混合并在二氯甲烷中回流约15分,加入12.8cm3亚硫酰氯。再回流45分钟,继之通过减压蒸馏浓缩干燥。将250cm3二氯甲烷加入结晶酰基氯并在-15℃下再加12.8cm3亚硫酰氯。在-25℃和大约1小时30分钟内,加入46.5cm3三乙胺和46.5cm3二氯甲烷混合物,得到的悬浮液搅拌30分钟。维持温度在-25℃下并保持大约30分钟下,加入35.5cm3吗啉和50cm3二氯甲烷混合物,搅拌30分钟,然后在大约10分钟内,加入350cm3水,同时允许温度升至0℃。加4.7cm3乙酸,并在2~5℃和大约1小时30分钟内,加49.6g高锰酸钾,混合物用240cm3水稀释,在2~5℃下搅拌1小时。在5~10℃和大约30分钟内,加入43g亚硫酸氢钠,同时加浓硫酸(12cm3)的冰水(150cm3)溶液。滗析后,二氯甲烷相用水洗涤,脱水,在20℃和均匀搅拌下,大约1小时30分钟内先后加入5g硫酸镁和60g铝CBT1。再在室温下搅拌1小时30分钟,过滤后通过减压蒸馏将滤液浓缩干燥。将80cm3乙酸乙酯加入残渣,然后通过减压蒸馏浓缩干燥,去除残留的二氯甲烷,将100cm3乙醇加入残留物,在40℃左右温度下将其搅拌溶解,溶液冷却至0℃开始结晶,静置16小时后,分离得到57.6g所需产品,熔点122~123℃。
将母液浓缩干燥,得到22g残留物,径滴定测定含有83.5%所需产品。
检验:红外光谱(三氯甲烷)cm-1。
1715
1704
核磁共振谱(CDCL3),ppm
18位上CH3的H 0.67
21位上CH3的H 0.9~1.0
19位上CH3的H 1.17
ACO的H 2.0
3位上的H 4.7
吗啉中的H 3.4~3.8
Claims (6)
1、制备通式(Ⅰ)产物的方法,
式中,R1代表氢原子或甲基,R2代表甲基或乙基,母核A,B,C,D可以含1个或多个双键并且可以由下列基团取代:1个或多个羟基或可以被保护的酮官能团,1个或多个点原子,含有1~4个碳原子的1个或多个烷基或烷氧基,或含有2~4个碳原子的一个或多个链稀基或炔基,R代表卤原子、羟基、含有1~6个碳原子的烷氧基、含有7~15个碳原子的芳烷氧基、
 基,其中R3和R4可以相同,也可不同,代表氢原子、含有1~6个碳原子的烷基、含有7~15个碳原子的芳烷基,或R3、R4与之键合的氮原子一起形成杂环,该环可以包括选自氮原子和氧原子的另一个杂原子,或者R代表含有1~6个碳原子的烷硫基,或最多可含15个碳原子的芳硫基或芳烷硫基。
基,其中R3和R4可以相同,也可不同,代表氢原子、含有1~6个碳原子的烷基、含有7~15个碳原子的芳烷基,或R3、R4与之键合的氮原子一起形成杂环,该环可以包括选自氮原子和氧原子的另一个杂原子,或者R代表含有1~6个碳原子的烷硫基,或最多可含15个碳原子的芳硫基或芳烷硫基。
该方法特征在于通式(Ⅱ)的产物先用形成酸性卤化物的试剂进行处理,然后再用叔碱、亚硫酰氯处理,最后可用水、链烷醇、芳链烷醇含
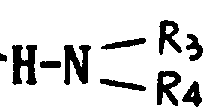 的伯胺或仲胺(R3和R4的定义同上)、烷硫醇、芳硫醇或芳烷硫醇处理,得到式(Ⅲ)产物:
的伯胺或仲胺(R3和R4的定义同上)、烷硫醇、芳硫醇或芳烷硫醇处理,得到式(Ⅲ)产物:
式中A,B,C,D,R,R1和R2的含意同上,再将式(Ⅲ)的产物进行下列处理:
或用含水酸或氧化剂,
或先后分别用卤化试剂和碱性水解剂,
亦可用链烷醇、芳链烷醇、含
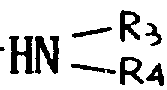 的伯胺或仲胺(式中R3和R4的定义同上)、烷硫醇、芳硫醇或芳烷硫醇,处理含R为烃基的式Ⅰ得到的产物,
的伯胺或仲胺(式中R3和R4的定义同上)、烷硫醇、芳硫醇或芳烷硫醇,处理含R为烃基的式Ⅰ得到的产物,
或用形成酸性卤化物的试剂处理含R为羟基的式Ⅰ得到的产物。
2、式(Ⅰ)产品在制备式(Ⅵ)产品中的应用,
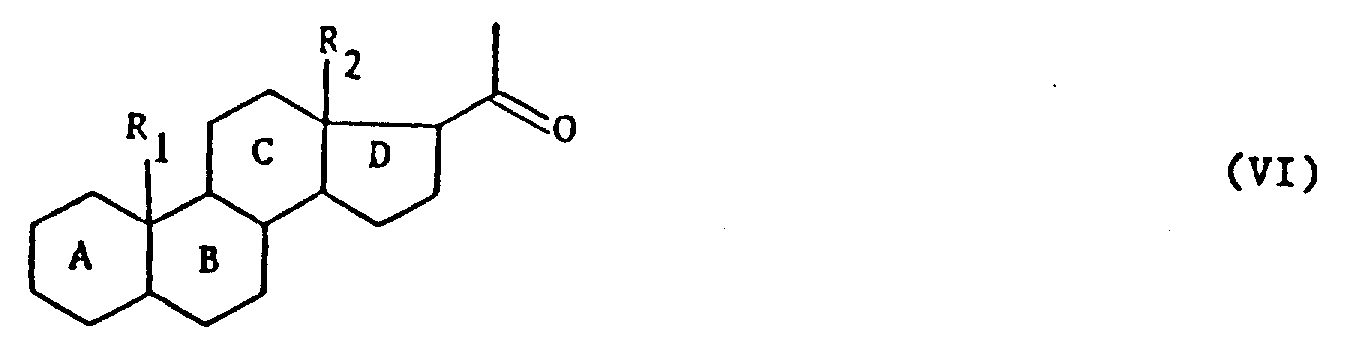
式中,A,B,C,D,R1和R2的定义同上,其特征在于用强氧化剂处理式(Ⅰ)产物,可得到式(Ⅳ)产物:
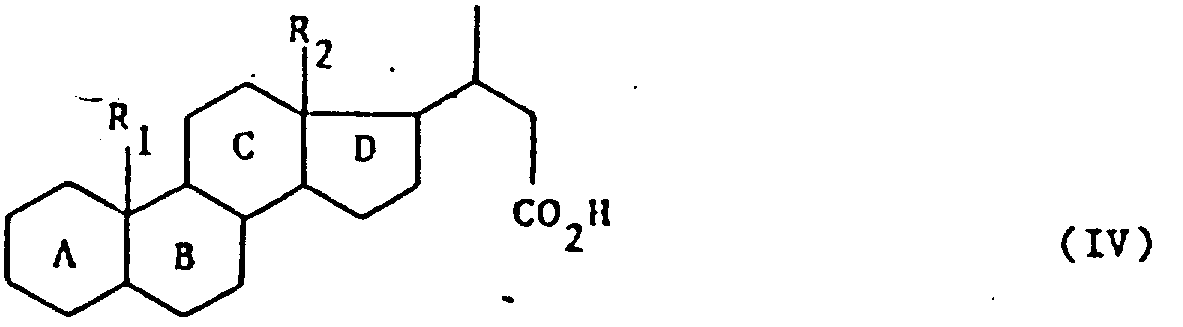
式中,A,B,C,D,R1和R2的定义同上,式(Ⅳ)产物先后用酸性卤化物形成剂、叔碱、亚硫酰氯和卤化剂处理,最后可用水、链烷醇、芳链烷醇、含
的伯胺或仲胺(式中R3和R4的定义同上)、烷硫醇、芳硫醇或芳烷硫醇处理,得到式(Ⅴ)产物:
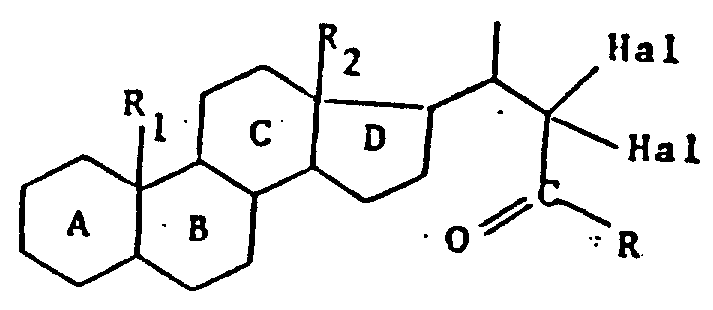
式中,A,B,C,D,R1,R2的定义同上,Hal代表卤原子,式(Ⅴ)产物先用脱卤化氢剂,再用氧化切割剂处理,得到所需的式(Ⅵ)产物。
3、按权利要求1或2之一的方法,其特征在于用于形成酸性卤化物的试剂是亚硫酰氯。
4、按权利要求1所述的方法,其中制得的通式(Ⅰ)的产品,其中R和R分别为甲基和母核A,B,C和D在其3位上可以含保护的羟基官能团,在6,7,11和12位上可以含选自保护的羟基官能团的1个或多个其他官能团,在7,11和12位上可以保护的酮官能团,R代表羟基,最多可含4个碳原子的烷氧基,或
 基,其中R1 3和R1 4代表氢原子,最多可含4个碳原子的烷基,或R1 3、R1 4与之键合的氮原子共同形成哌啶子基,吡咯烷子基或吗啉子基。
基,其中R1 3和R1 4代表氢原子,最多可含4个碳原子的烷基,或R1 3、R1 4与之键合的氮原子共同形成哌啶子基,吡咯烷子基或吗啉子基。
5、按权利要求1所述的方法,所制得的通式(Ⅰ)的产品,其中母核A,B,C和D在3位上可以含保护的羟基,在12位上可以含选自保护的羟基官能团的1个或多其他官能团,以及在11或12位上可以含保护的酮官能团,R代表羟基,甲氧基或乙氧基,或吗啉子基。
6、按权利要求1所述的方法,其中制得的通式(Ⅰ)的产物,符合下列各式:
3 α,5β-4-(3-乙酰氧基-11,23,24-三氧-胆烷-24-基)吗啉。
3 α,5β-11,23-二氧-3-羟基-胆烷-24-酸。
3 α羟基-11,23-二氧-5β-胆烷-24-酸甲酯。
3 α,5β-3-乙酰氧基-11,23-二氧-胆烷-24-酸。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| FR8617051A FR2607815B1 (fr) | 1986-12-05 | 1986-12-05 | Nouveaux produits steroides comportant, en position 23, un radical cetonique, leur procede de preparation, leur application a la preparation de produits de la serie des 20-cetopregnanes et des intermediaires de cette association |
| FR8617051 | 1986-12-05 | ||
| FR86-17051 | 1986-12-05 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN87107343A true CN87107343A (zh) | 1988-06-15 |
| CN1031575C CN1031575C (zh) | 1996-04-17 |
Family
ID=9341614
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN87107343A Expired - Fee Related CN1031575C (zh) | 1986-12-05 | 1987-12-05 | 23位上酮基的甾族化合物的制备方法 |
Country Status (15)
| Country | Link |
|---|---|
| US (2) | US4927921A (zh) |
| EP (1) | EP0275729B1 (zh) |
| JP (1) | JP2660413B2 (zh) |
| KR (1) | KR960013444B1 (zh) |
| CN (1) | CN1031575C (zh) |
| AT (1) | ATE63315T1 (zh) |
| CA (2) | CA1299170C (zh) |
| DE (1) | DE3769964D1 (zh) |
| ES (1) | ES2022422B3 (zh) |
| FI (1) | FI86306C (zh) |
| FR (1) | FR2607815B1 (zh) |
| GR (1) | GR3002414T3 (zh) |
| HU (1) | HU198082B (zh) |
| MX (1) | MX173419B (zh) |
| PT (1) | PT86284B (zh) |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE4432708A1 (de) * | 1994-09-14 | 1996-03-21 | Hoechst Ag | Modifizierte Gallensäuren, Verfahren zu ihrer Herstellung und ihre Verwendung |
Family Cites Families (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US2180095A (en) * | 1935-03-09 | 1939-11-14 | Schering Corp | Valuable degradation products of sterols and a method of producing the same |
| US2445006A (en) * | 1942-09-26 | 1948-07-13 | Schering Corp | Steroid keto hydroxy acids and their polybasic acid esters and method of preparing same |
| US2495735A (en) * | 1947-02-19 | 1950-01-31 | Research Corp | Process for preparing lower alkyl esters of 3-hydroxy-11-keto-12-bromonorcholanic acid |
| US2705232A (en) * | 1953-02-20 | 1955-03-29 | Searle & Co | Ternorcholanylthiazoles |
| DE1568499C3 (de) * | 1966-03-10 | 1975-04-17 | Farbwerke Hoechst Ag, Vormals Meister Lucius & Bruening, 6000 Frankfurt | Verfahren zur Herstellung von ungesättigten Lactonen der Steroidreihe |
| US3906095A (en) * | 1972-08-30 | 1975-09-16 | Schering Ag | Novel pregnanoic acid derivatives |
| GB2028333B (en) * | 1978-07-03 | 1982-10-27 | Radiochemical Centre Ltd | Selenium and tellurium derivatives of bile acids |
| US4502991A (en) * | 1983-08-18 | 1985-03-05 | Wisconsin Alumni Research Foundation | 23,23-Difluoro-1α,25-dihydroxy-vitamin D3 |
| US4500460A (en) * | 1983-08-18 | 1985-02-19 | Wisconsin Alumni Research Foundation | 23,23-Difluoro-25-hydroxy-vitamin D3 and process for preparing same |
-
1986
- 1986-12-05 FR FR8617051A patent/FR2607815B1/fr not_active Expired
-
1987
- 1987-12-03 EP EP87402740A patent/EP0275729B1/fr not_active Expired - Lifetime
- 1987-12-03 ES ES87402740T patent/ES2022422B3/es not_active Expired - Lifetime
- 1987-12-03 PT PT86284A patent/PT86284B/pt not_active IP Right Cessation
- 1987-12-03 AT AT87402740T patent/ATE63315T1/de not_active IP Right Cessation
- 1987-12-03 DE DE8787402740T patent/DE3769964D1/de not_active Expired - Fee Related
- 1987-12-04 JP JP62306057A patent/JP2660413B2/ja not_active Expired - Fee Related
- 1987-12-04 US US07/128,997 patent/US4927921A/en not_active Expired - Lifetime
- 1987-12-04 MX MX009632A patent/MX173419B/es unknown
- 1987-12-04 FI FI875349A patent/FI86306C/fi not_active IP Right Cessation
- 1987-12-04 HU HU875457A patent/HU198082B/hu not_active IP Right Cessation
- 1987-12-04 CA CA000553529A patent/CA1299170C/fr not_active Expired - Fee Related
- 1987-12-05 KR KR1019870013913A patent/KR960013444B1/ko not_active Expired - Fee Related
- 1987-12-05 CN CN87107343A patent/CN1031575C/zh not_active Expired - Fee Related
-
1990
- 1990-03-05 US US07/488,674 patent/US5095130A/en not_active Expired - Fee Related
- 1990-12-07 CA CA000615953A patent/CA1305131C/fr not_active Expired - Fee Related
-
1991
- 1991-08-02 GR GR90401168T patent/GR3002414T3/el unknown
Also Published As
| Publication number | Publication date |
|---|---|
| MX9632A (es) | 1993-09-01 |
| FR2607815B1 (fr) | 1988-12-30 |
| CN1031575C (zh) | 1996-04-17 |
| CA1299170C (fr) | 1992-04-21 |
| DE3769964D1 (de) | 1991-06-13 |
| FI875349A7 (fi) | 1988-06-06 |
| JP2660413B2 (ja) | 1997-10-08 |
| US4927921A (en) | 1990-05-22 |
| FI86306B (fi) | 1992-04-30 |
| PT86284A (fr) | 1988-01-01 |
| EP0275729B1 (fr) | 1991-05-08 |
| ATE63315T1 (de) | 1991-05-15 |
| FR2607815A1 (fr) | 1988-06-10 |
| FI86306C (fi) | 1992-08-10 |
| CA1305131C (fr) | 1992-07-14 |
| EP0275729A1 (fr) | 1988-07-27 |
| FI875349A0 (fi) | 1987-12-04 |
| ES2022422B3 (es) | 1991-12-01 |
| GR3002414T3 (en) | 1992-12-30 |
| HU198082B (en) | 1989-07-28 |
| MX173419B (es) | 1994-03-03 |
| HUT46036A (en) | 1988-09-28 |
| KR880007558A (ko) | 1988-08-27 |
| JPS63154697A (ja) | 1988-06-27 |
| US5095130A (en) | 1992-03-10 |
| KR960013444B1 (ko) | 1996-10-05 |
| PT86284B (pt) | 1990-11-07 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN1104426C (zh) | 紫杉醇和三尖杉宁碱的分离和纯化 | |
| CN1036342C (zh) | 9,11-脱氢甾类化合物及9-α-羟基-16,17-脱氢甾类化合物的制备方法 | |
| CN1041054C (zh) | 含有甾类5-α-还原酶抑制剂的药物组合物制备方法 | |
| CN1187365C (zh) | 9α-羟基-17-亚甲基甾类在制备皮质甾类中的用途 | |
| CN1990499A (zh) | 环丙孕酮乙酸酯的改进合成方法 | |
| CN1046502C (zh) | 22-硫代维生素d3衍生物 | |
| CN1049499A (zh) | 具有粘液调节和抗局部缺血性质的2,3-二氢-5-氧-4,6,7-三甲基苯并呋喃的2-(rs)-衍生物及其作为抗氧化药剂的用途 | |
| CN1653081A (zh) | 甾族化合物的c-17螺甾内酯化和6,7氧化 | |
| CN1067044C (zh) | 新的三环衍生物和它们的制备方法 | |
| CN87107343A (zh) | 23位上酮基的甾族化合物,其制备方法及用其制备20酮孕甾烷 | |
| CN101061133A (zh) | 制备17-羟基-6β,7β;15β,16β-双亚甲基-3-氧代-17α-孕甾-4-烯-21-羧酸γ-内酯的工业方法和用于该方法的关键中间体 | |
| CN1043426C (zh) | 孕甾-4,9(11),16-三烯-3,20-二酮类甾族化合物的制法 | |
| CN1918168A (zh) | 制备3-o-保护吗啡酮和3-o-保护吗啡酮二烯醇羧酸酯的方法 | |
| CN1031576C (zh) | 23位上酮基的甾族化合物的制备方法 | |
| CN1096790A (zh) | 新的17,20-环氧孕甾烷衍生物及其制法和用于制备可的松衍生物及其中间体 | |
| CN1780849A (zh) | 生产7-甲异炔诺酮的方法 | |
| CN85101840A (zh) | 制造16-苯氧基-和16-取代苯氧基-前列腺素三烯酸衍生物及它们的立体异构体的方法和中间体 | |
| CN1042030C (zh) | 取代氧芴的制备方法 | |
| CN1795201A (zh) | 没药固酮和没药固醇的制备方法 | |
| CN1032134C (zh) | 2,2-二甲基-5-(2,5-二甲基苯氧基)戊酸的制备方法 | |
| CN87102944A (zh) | 新麦角烯(Ergolene)衍生物、其药物组合物及其制备方法 | |
| CN100343274C (zh) | 一类14α,17α-氧桥甾体化合物、合成方法及其用途 | |
| CN1191263C (zh) | 3-脱氧-去碳霉糖太乐菌素衍生物和它们的制备方法 | |
| CN1069734A (zh) | 11.19-环合孕烯酮类似物的合成方法 | |
| HK1085485A (zh) | 甾族化合物的c-17螺甾內酯化和6,7氧化 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C14 | Grant of patent or utility model | ||
| GR01 | Patent grant | ||
| C15 | Extension of patent right duration from 15 to 20 years for appl. with date before 31.12.1992 and still valid on 11.12.2001 (patent law change 1993) | ||
| OR01 | Other related matters | ||
| C19 | Lapse of patent right due to non-payment of the annual fee | ||
| CF01 | Termination of patent right due to non-payment of annual fee |