DE10328844A1 - Alkoxysilanterminierte Prepolymere - Google Patents
Alkoxysilanterminierte Prepolymere Download PDFInfo
- Publication number
- DE10328844A1 DE10328844A1 DE10328844A DE10328844A DE10328844A1 DE 10328844 A1 DE10328844 A1 DE 10328844A1 DE 10328844 A DE10328844 A DE 10328844A DE 10328844 A DE10328844 A DE 10328844A DE 10328844 A1 DE10328844 A1 DE 10328844A1
- Authority
- DE
- Germany
- Prior art keywords
- prepolymers
- molecular weight
- prepolymer
- dollar
- isocyanate
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Withdrawn
Links
- 150000003077 polyols Chemical class 0.000 claims abstract description 55
- 229920005862 polyol Polymers 0.000 claims abstract description 36
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 claims abstract description 30
- -1 alkyl radical Chemical class 0.000 claims abstract description 24
- IQPQWNKOIGAROB-UHFFFAOYSA-N isocyanate group Chemical group [N-]=C=O IQPQWNKOIGAROB-UHFFFAOYSA-N 0.000 claims abstract description 16
- 229920001228 polyisocyanate Polymers 0.000 claims abstract description 14
- 125000004432 carbon atom Chemical group C* 0.000 claims abstract description 13
- 239000005056 polyisocyanate Substances 0.000 claims abstract description 13
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims abstract description 7
- 125000003342 alkenyl group Chemical group 0.000 claims abstract description 3
- 150000005840 aryl radicals Chemical class 0.000 claims abstract description 3
- 125000000753 cycloalkyl group Chemical group 0.000 claims abstract description 3
- 239000000203 mixture Substances 0.000 claims description 49
- 239000000945 filler Substances 0.000 claims description 19
- WERYXYBDKMZEQL-UHFFFAOYSA-N butane-1,4-diol Chemical compound OCCCCO WERYXYBDKMZEQL-UHFFFAOYSA-N 0.000 claims description 18
- 229920000642 polymer Polymers 0.000 claims description 18
- 150000004756 silanes Chemical class 0.000 claims description 12
- VTYYLEPIZMXCLO-UHFFFAOYSA-L Calcium carbonate Chemical compound [Ca+2].[O-]C([O-])=O VTYYLEPIZMXCLO-UHFFFAOYSA-L 0.000 claims description 10
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical compound OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 claims description 10
- NIMLQBUJDJZYEJ-UHFFFAOYSA-N isophorone diisocyanate Chemical compound CC1(C)CC(N=C=O)CC(C)(CN=C=O)C1 NIMLQBUJDJZYEJ-UHFFFAOYSA-N 0.000 claims description 10
- 150000001298 alcohols Chemical class 0.000 claims description 9
- 229920000570 polyether Polymers 0.000 claims description 9
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 claims description 8
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 claims description 6
- 229910000019 calcium carbonate Inorganic materials 0.000 claims description 5
- 229920000728 polyester Polymers 0.000 claims description 5
- PUPZLCDOIYMWBV-UHFFFAOYSA-N (+/-)-1,3-Butanediol Chemical compound CC(O)CCO PUPZLCDOIYMWBV-UHFFFAOYSA-N 0.000 claims description 4
- 239000005057 Hexamethylene diisocyanate Substances 0.000 claims description 4
- 125000004122 cyclic group Chemical class 0.000 claims description 4
- RRAMGCGOFNQTLD-UHFFFAOYSA-N hexamethylene diisocyanate Chemical compound O=C=NCCCCCCN=C=O RRAMGCGOFNQTLD-UHFFFAOYSA-N 0.000 claims description 4
- 229910052739 hydrogen Inorganic materials 0.000 claims description 4
- 239000001257 hydrogen Substances 0.000 claims description 4
- DVKJHBMWWAPEIU-UHFFFAOYSA-N toluene 2,4-diisocyanate Chemical compound CC1=CC=C(N=C=O)C=C1N=C=O DVKJHBMWWAPEIU-UHFFFAOYSA-N 0.000 claims description 4
- 239000006229 carbon black Substances 0.000 claims description 3
- 125000004435 hydrogen atom Chemical class [H]* 0.000 claims description 3
- 125000005647 linker group Chemical group 0.000 claims description 3
- 229920000058 polyacrylate Polymers 0.000 claims description 3
- 229920000098 polyolefin Polymers 0.000 claims description 3
- 229920001296 polysiloxane Polymers 0.000 claims description 3
- 229920001290 polyvinyl ester Polymers 0.000 claims description 3
- 229920002554 vinyl polymer Polymers 0.000 claims description 3
- DNIAPMSPPWPWGF-VKHMYHEASA-N (+)-propylene glycol Chemical compound C[C@H](O)CO DNIAPMSPPWPWGF-VKHMYHEASA-N 0.000 claims description 2
- YPFDHNVEDLHUCE-UHFFFAOYSA-N 1,3-propanediol Substances OCCCO YPFDHNVEDLHUCE-UHFFFAOYSA-N 0.000 claims description 2
- 239000004952 Polyamide Substances 0.000 claims description 2
- 239000004793 Polystyrene Substances 0.000 claims description 2
- LNWBFIVSTXCJJG-UHFFFAOYSA-N [diisocyanato(phenyl)methyl]benzene Chemical compound C=1C=CC=CC=1C(N=C=O)(N=C=O)C1=CC=CC=C1 LNWBFIVSTXCJJG-UHFFFAOYSA-N 0.000 claims description 2
- OHJMTUPIZMNBFR-UHFFFAOYSA-N biuret Chemical compound NC(=O)NC(N)=O OHJMTUPIZMNBFR-UHFFFAOYSA-N 0.000 claims description 2
- 150000004679 hydroxides Chemical class 0.000 claims description 2
- WGCNASOHLSPBMP-UHFFFAOYSA-N hydroxyacetaldehyde Natural products OCC=O WGCNASOHLSPBMP-UHFFFAOYSA-N 0.000 claims description 2
- ZFSLODLOARCGLH-UHFFFAOYSA-N isocyanuric acid Chemical compound OC1=NC(O)=NC(O)=N1 ZFSLODLOARCGLH-UHFFFAOYSA-N 0.000 claims description 2
- 150000002734 metacrylic acid derivatives Chemical class 0.000 claims description 2
- 239000003960 organic solvent Substances 0.000 claims description 2
- 229920002647 polyamide Polymers 0.000 claims description 2
- 229920000515 polycarbonate Polymers 0.000 claims description 2
- 239000004417 polycarbonate Substances 0.000 claims description 2
- 229920002223 polystyrene Polymers 0.000 claims description 2
- 229920000166 polytrimethylene carbonate Polymers 0.000 claims description 2
- 239000000377 silicon dioxide Substances 0.000 claims description 2
- AAAQKTZKLRYKHR-UHFFFAOYSA-N triphenylmethane Chemical compound C1=CC=CC=C1C(C=1C=CC=CC=1)C1=CC=CC=C1 AAAQKTZKLRYKHR-UHFFFAOYSA-N 0.000 claims description 2
- UFWIBTONFRDIAS-UHFFFAOYSA-N Naphthalene Chemical compound C1=CC=CC2=CC=CC=C21 UFWIBTONFRDIAS-UHFFFAOYSA-N 0.000 claims 2
- 238000002360 preparation method Methods 0.000 description 29
- 239000003054 catalyst Substances 0.000 description 24
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 21
- 238000006243 chemical reaction Methods 0.000 description 18
- BLRPTPMANUNPDV-UHFFFAOYSA-N Silane Chemical compound [SiH4] BLRPTPMANUNPDV-UHFFFAOYSA-N 0.000 description 16
- 230000000052 comparative effect Effects 0.000 description 15
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 14
- 230000015572 biosynthetic process Effects 0.000 description 13
- 239000002904 solvent Substances 0.000 description 13
- 229910000077 silane Inorganic materials 0.000 description 12
- 125000005370 alkoxysilyl group Chemical group 0.000 description 11
- 229920002959 polymer blend Polymers 0.000 description 10
- 229920001451 polypropylene glycol Polymers 0.000 description 9
- 238000003756 stirring Methods 0.000 description 9
- JOYRKODLDBILNP-UHFFFAOYSA-N urethane group Chemical group NC(=O)OCC JOYRKODLDBILNP-UHFFFAOYSA-N 0.000 description 9
- 239000005058 Isophorone diisocyanate Substances 0.000 description 8
- 239000000853 adhesive Substances 0.000 description 8
- 230000001070 adhesive effect Effects 0.000 description 8
- 150000001412 amines Chemical class 0.000 description 8
- 238000010438 heat treatment Methods 0.000 description 8
- 229910052757 nitrogen Inorganic materials 0.000 description 8
- IBMUMCCOQRVIMN-UHFFFAOYSA-N trimethoxy(methoxymethyl)silane Chemical compound COC[Si](OC)(OC)OC IBMUMCCOQRVIMN-UHFFFAOYSA-N 0.000 description 8
- 230000008901 benefit Effects 0.000 description 7
- 238000004519 manufacturing process Methods 0.000 description 7
- 230000009257 reactivity Effects 0.000 description 7
- 238000003786 synthesis reaction Methods 0.000 description 7
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 6
- RTAQQCXQSZGOHL-UHFFFAOYSA-N Titanium Chemical compound [Ti] RTAQQCXQSZGOHL-UHFFFAOYSA-N 0.000 description 6
- UKLDJPRMSDWDSL-UHFFFAOYSA-L [dibutyl(dodecanoyloxy)stannyl] dodecanoate Chemical compound CCCCCCCCCCCC(=O)O[Sn](CCCC)(CCCC)OC(=O)CCCCCCCCCCC UKLDJPRMSDWDSL-UHFFFAOYSA-L 0.000 description 6
- 150000001875 compounds Chemical class 0.000 description 6
- 238000001816 cooling Methods 0.000 description 6
- 239000012975 dibutyltin dilaurate Substances 0.000 description 6
- 239000012948 isocyanate Substances 0.000 description 6
- DWYWQJWQNQLGLB-UHFFFAOYSA-N n-(dimethoxymethylsilylmethyl)cyclohexanamine Chemical compound COC(OC)[SiH2]CNC1CCCCC1 DWYWQJWQNQLGLB-UHFFFAOYSA-N 0.000 description 6
- 239000010936 titanium Substances 0.000 description 6
- 229910052719 titanium Inorganic materials 0.000 description 6
- 238000004566 IR spectroscopy Methods 0.000 description 5
- 229920001730 Moisture cure polyurethane Polymers 0.000 description 5
- 239000004721 Polyphenylene oxide Substances 0.000 description 5
- 238000010348 incorporation Methods 0.000 description 5
- 150000002513 isocyanates Chemical class 0.000 description 5
- 239000000047 product Substances 0.000 description 5
- XSQUKJJJFZCRTK-UHFFFAOYSA-N urea group Chemical group NC(=O)N XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 5
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 5
- XDTMQSROBMDMFD-UHFFFAOYSA-N Cyclohexane Chemical compound C1CCCCC1 XDTMQSROBMDMFD-UHFFFAOYSA-N 0.000 description 4
- WCUXLLCKKVVCTQ-UHFFFAOYSA-M Potassium chloride Chemical compound [Cl-].[K+] WCUXLLCKKVVCTQ-UHFFFAOYSA-M 0.000 description 4
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 4
- 238000004132 cross linking Methods 0.000 description 4
- 238000004821 distillation Methods 0.000 description 4
- 230000006872 improvement Effects 0.000 description 4
- 150000003254 radicals Chemical class 0.000 description 4
- 239000000565 sealant Substances 0.000 description 4
- SJECZPVISLOESU-UHFFFAOYSA-N 3-trimethoxysilylpropan-1-amine Chemical compound CO[Si](OC)(OC)CCCN SJECZPVISLOESU-UHFFFAOYSA-N 0.000 description 3
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 3
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 3
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 3
- ATJFFYVFTNAWJD-UHFFFAOYSA-N Tin Chemical compound [Sn] ATJFFYVFTNAWJD-UHFFFAOYSA-N 0.000 description 3
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 3
- 229920004482 WACKER® Polymers 0.000 description 3
- 239000002318 adhesion promoter Substances 0.000 description 3
- 125000003545 alkoxy group Chemical group 0.000 description 3
- 125000005442 diisocyanate group Chemical group 0.000 description 3
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 3
- 229910001385 heavy metal Inorganic materials 0.000 description 3
- 125000005842 heteroatom Chemical group 0.000 description 3
- 238000002156 mixing Methods 0.000 description 3
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 3
- 229920003023 plastic Polymers 0.000 description 3
- 239000004033 plastic Substances 0.000 description 3
- FZHAPNGMFPVSLP-UHFFFAOYSA-N silanamine Chemical class [SiH3]N FZHAPNGMFPVSLP-UHFFFAOYSA-N 0.000 description 3
- 239000000243 solution Substances 0.000 description 3
- 239000000725 suspension Substances 0.000 description 3
- 239000012974 tin catalyst Substances 0.000 description 3
- GQHTUMJGOHRCHB-UHFFFAOYSA-N 2,3,4,6,7,8,9,10-octahydropyrimido[1,2-a]azepine Chemical compound C1CCCCN2CCCN=C21 GQHTUMJGOHRCHB-UHFFFAOYSA-N 0.000 description 2
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 2
- 229920000089 Cyclic olefin copolymer Polymers 0.000 description 2
- XEEYBQQBJWHFJM-UHFFFAOYSA-N Iron Chemical compound [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 description 2
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- WQDUMFSSJAZKTM-UHFFFAOYSA-N Sodium methoxide Chemical compound [Na+].[O-]C WQDUMFSSJAZKTM-UHFFFAOYSA-N 0.000 description 2
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 2
- 239000002253 acid Substances 0.000 description 2
- TZCXTZWJZNENPQ-UHFFFAOYSA-L barium sulfate Chemical compound [Ba+2].[O-]S([O-])(=O)=O TZCXTZWJZNENPQ-UHFFFAOYSA-L 0.000 description 2
- 238000009835 boiling Methods 0.000 description 2
- FPOSCXQHGOVVPD-UHFFFAOYSA-N chloromethyl(trimethoxy)silane Chemical compound CO[Si](CCl)(OC)OC FPOSCXQHGOVVPD-UHFFFAOYSA-N 0.000 description 2
- 238000003776 cleavage reaction Methods 0.000 description 2
- PAFZNILMFXTMIY-UHFFFAOYSA-N cyclohexylamine Chemical compound NC1CCCCC1 PAFZNILMFXTMIY-UHFFFAOYSA-N 0.000 description 2
- 229910021485 fumed silica Inorganic materials 0.000 description 2
- 239000011521 glass Substances 0.000 description 2
- 239000000463 material Substances 0.000 description 2
- 229910052751 metal Inorganic materials 0.000 description 2
- 239000002184 metal Substances 0.000 description 2
- 150000002739 metals Chemical class 0.000 description 2
- 238000000034 method Methods 0.000 description 2
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 2
- 239000001103 potassium chloride Substances 0.000 description 2
- 235000011164 potassium chloride Nutrition 0.000 description 2
- 239000000843 powder Substances 0.000 description 2
- 239000002516 radical scavenger Substances 0.000 description 2
- 239000012763 reinforcing filler Substances 0.000 description 2
- 230000007017 scission Effects 0.000 description 2
- 239000011780 sodium chloride Substances 0.000 description 2
- 239000007787 solid Substances 0.000 description 2
- 239000004071 soot Substances 0.000 description 2
- 238000003860 storage Methods 0.000 description 2
- 229920003048 styrene butadiene rubber Polymers 0.000 description 2
- 239000000758 substrate Substances 0.000 description 2
- SGUVLZREKBPKCE-UHFFFAOYSA-N 1,5-diazabicyclo[4.3.0]-non-5-ene Chemical compound C1CCN=C2CCCN21 SGUVLZREKBPKCE-UHFFFAOYSA-N 0.000 description 1
- WWBITQUCWSFVNB-UHFFFAOYSA-N 3-silylpropan-1-amine Chemical class NCCC[SiH3] WWBITQUCWSFVNB-UHFFFAOYSA-N 0.000 description 1
- HAAZMOAXEMIBAJ-UHFFFAOYSA-N 4-chloro-2-methylquinazoline Chemical compound C1=CC=CC2=NC(C)=NC(Cl)=C21 HAAZMOAXEMIBAJ-UHFFFAOYSA-N 0.000 description 1
- HVCNXQOWACZAFN-UHFFFAOYSA-N 4-ethylmorpholine Chemical compound CCN1CCOCC1 HVCNXQOWACZAFN-UHFFFAOYSA-N 0.000 description 1
- 229910052582 BN Inorganic materials 0.000 description 1
- PZNSFCLAULLKQX-UHFFFAOYSA-N Boron nitride Chemical compound N#B PZNSFCLAULLKQX-UHFFFAOYSA-N 0.000 description 1
- DKPFZGUDAPQIHT-UHFFFAOYSA-N Butyl acetate Natural products CCCCOC(C)=O DKPFZGUDAPQIHT-UHFFFAOYSA-N 0.000 description 1
- BUUXTCACASYRFP-UHFFFAOYSA-N C1(CCCCC1)NC[SiH](OC)OC Chemical compound C1(CCCCC1)NC[SiH](OC)OC BUUXTCACASYRFP-UHFFFAOYSA-N 0.000 description 1
- 239000004215 Carbon black (E152) Substances 0.000 description 1
- 239000004971 Cross linker Substances 0.000 description 1
- JYFHYPJRHGVZDY-UHFFFAOYSA-N Dibutyl phosphate Chemical compound CCCCOP(O)(=O)OCCCC JYFHYPJRHGVZDY-UHFFFAOYSA-N 0.000 description 1
- MYMOFIZGZYHOMD-UHFFFAOYSA-N Dioxygen Chemical compound O=O MYMOFIZGZYHOMD-UHFFFAOYSA-N 0.000 description 1
- 239000004593 Epoxy Substances 0.000 description 1
- VTLYFUHAOXGGBS-UHFFFAOYSA-N Fe3+ Chemical class [Fe+3] VTLYFUHAOXGGBS-UHFFFAOYSA-N 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- 239000005909 Kieselgur Substances 0.000 description 1
- 239000002841 Lewis acid Substances 0.000 description 1
- 239000002879 Lewis base Substances 0.000 description 1
- BZLVMXJERCGZMT-UHFFFAOYSA-N Methyl tert-butyl ether Chemical compound COC(C)(C)C BZLVMXJERCGZMT-UHFFFAOYSA-N 0.000 description 1
- SVYKKECYCPFKGB-UHFFFAOYSA-N N,N-dimethylcyclohexylamine Chemical compound CN(C)C1CCCCC1 SVYKKECYCPFKGB-UHFFFAOYSA-N 0.000 description 1
- PHIIOKFICBAPOS-UHFFFAOYSA-N NCCNCCC[SiH3] Chemical class NCCNCCC[SiH3] PHIIOKFICBAPOS-UHFFFAOYSA-N 0.000 description 1
- 239000005062 Polybutadiene Substances 0.000 description 1
- 239000004698 Polyethylene Substances 0.000 description 1
- 229910052581 Si3N4 Inorganic materials 0.000 description 1
- XUIMIQQOPSSXEZ-UHFFFAOYSA-N Silicon Chemical group [Si] XUIMIQQOPSSXEZ-UHFFFAOYSA-N 0.000 description 1
- 229910002808 Si–O–Si Inorganic materials 0.000 description 1
- 239000004809 Teflon Substances 0.000 description 1
- 229920006362 Teflon® Polymers 0.000 description 1
- LCKIEQZJEYYRIY-UHFFFAOYSA-N Titanium ion Chemical compound [Ti+4] LCKIEQZJEYYRIY-UHFFFAOYSA-N 0.000 description 1
- ISKQADXMHQSTHK-UHFFFAOYSA-N [4-(aminomethyl)phenyl]methanamine Chemical compound NCC1=CC=C(CN)C=C1 ISKQADXMHQSTHK-UHFFFAOYSA-N 0.000 description 1
- NBJODVYWAQLZOC-UHFFFAOYSA-L [dibutyl(octanoyloxy)stannyl] octanoate Chemical compound CCCCCCCC(=O)O[Sn](CCCC)(CCCC)OC(=O)CCCCCCC NBJODVYWAQLZOC-UHFFFAOYSA-L 0.000 description 1
- XQBCVRSTVUHIGH-UHFFFAOYSA-L [dodecanoyloxy(dioctyl)stannyl] dodecanoate Chemical compound CCCCCCCCCCCC(=O)O[Sn](CCCCCCCC)(CCCCCCCC)OC(=O)CCCCCCCCCCC XQBCVRSTVUHIGH-UHFFFAOYSA-L 0.000 description 1
- 125000005595 acetylacetonate group Chemical group 0.000 description 1
- 239000006230 acetylene black Substances 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 239000000654 additive Substances 0.000 description 1
- 230000002411 adverse Effects 0.000 description 1
- 125000001931 aliphatic group Chemical group 0.000 description 1
- CSDREXVUYHZDNP-UHFFFAOYSA-N alumanylidynesilicon Chemical compound [Al].[Si] CSDREXVUYHZDNP-UHFFFAOYSA-N 0.000 description 1
- 229910052782 aluminium Inorganic materials 0.000 description 1
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- 239000003963 antioxidant agent Substances 0.000 description 1
- 125000003118 aryl group Chemical group 0.000 description 1
- 239000010425 asbestos Substances 0.000 description 1
- PASDCCFISLVPSO-UHFFFAOYSA-N benzoyl chloride Chemical compound ClC(=O)C1=CC=CC=C1 PASDCCFISLVPSO-UHFFFAOYSA-N 0.000 description 1
- CDQSJQSWAWPGKG-UHFFFAOYSA-N butane-1,1-diol Chemical compound CCCC(O)O CDQSJQSWAWPGKG-UHFFFAOYSA-N 0.000 description 1
- 229910052918 calcium silicate Inorganic materials 0.000 description 1
- 239000000378 calcium silicate Substances 0.000 description 1
- OYACROKNLOSFPA-UHFFFAOYSA-N calcium;dioxido(oxo)silane Chemical compound [Ca+2].[O-][Si]([O-])=O OYACROKNLOSFPA-UHFFFAOYSA-N 0.000 description 1
- 239000004202 carbamide Substances 0.000 description 1
- 239000000919 ceramic Substances 0.000 description 1
- ZXZMFKUGAPMMCJ-UHFFFAOYSA-N chloromethyl-dimethoxy-methylsilane Chemical compound CO[Si](C)(CCl)OC ZXZMFKUGAPMMCJ-UHFFFAOYSA-N 0.000 description 1
- 238000004140 cleaning Methods 0.000 description 1
- 239000003426 co-catalyst Substances 0.000 description 1
- 238000000576 coating method Methods 0.000 description 1
- 239000003086 colorant Substances 0.000 description 1
- 238000013329 compounding Methods 0.000 description 1
- 238000010276 construction Methods 0.000 description 1
- 239000012045 crude solution Substances 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 230000006735 deficit Effects 0.000 description 1
- AYOHIQLKSOJJQH-UHFFFAOYSA-N dibutyltin Chemical compound CCCC[Sn]CCCC AYOHIQLKSOJJQH-UHFFFAOYSA-N 0.000 description 1
- 150000005690 diesters Chemical class 0.000 description 1
- 239000003085 diluting agent Substances 0.000 description 1
- 150000002009 diols Chemical class 0.000 description 1
- 239000006185 dispersion Substances 0.000 description 1
- 229920001971 elastomer Polymers 0.000 description 1
- 239000000806 elastomer Substances 0.000 description 1
- 150000002148 esters Chemical class 0.000 description 1
- 238000006266 etherification reaction Methods 0.000 description 1
- 150000002170 ethers Chemical class 0.000 description 1
- 125000001301 ethoxy group Chemical group [H]C([H])([H])C([H])([H])O* 0.000 description 1
- 230000002349 favourable effect Effects 0.000 description 1
- 239000000835 fiber Substances 0.000 description 1
- 239000012765 fibrous filler Substances 0.000 description 1
- 239000012065 filter cake Substances 0.000 description 1
- 239000003063 flame retardant Substances 0.000 description 1
- 239000000417 fungicide Substances 0.000 description 1
- 239000006232 furnace black Substances 0.000 description 1
- 239000010440 gypsum Substances 0.000 description 1
- 229910052602 gypsum Inorganic materials 0.000 description 1
- 125000005843 halogen group Chemical group 0.000 description 1
- FUZZWVXGSFPDMH-UHFFFAOYSA-N hexanoic acid Chemical compound CCCCCC(O)=O FUZZWVXGSFPDMH-UHFFFAOYSA-N 0.000 description 1
- 229930195733 hydrocarbon Natural products 0.000 description 1
- 150000002430 hydrocarbons Chemical group 0.000 description 1
- 230000002209 hydrophobic effect Effects 0.000 description 1
- UWNADWZGEHDQAB-UHFFFAOYSA-N i-Pr2C2H4i-Pr2 Natural products CC(C)CCC(C)C UWNADWZGEHDQAB-UHFFFAOYSA-N 0.000 description 1
- 229910052500 inorganic mineral Inorganic materials 0.000 description 1
- 229910052742 iron Inorganic materials 0.000 description 1
- 235000013980 iron oxide Nutrition 0.000 description 1
- AQBLLJNPHDIAPN-LNTINUHCSA-K iron(3+);(z)-4-oxopent-2-en-2-olate Chemical compound [Fe+3].C\C([O-])=C\C(C)=O.C\C([O-])=C\C(C)=O.C\C([O-])=C\C(C)=O AQBLLJNPHDIAPN-LNTINUHCSA-K 0.000 description 1
- 150000007517 lewis acids Chemical class 0.000 description 1
- 150000007527 lewis bases Chemical class 0.000 description 1
- 239000004611 light stabiliser Substances 0.000 description 1
- 150000002736 metal compounds Chemical class 0.000 description 1
- 229910044991 metal oxide Inorganic materials 0.000 description 1
- 150000004706 metal oxides Chemical class 0.000 description 1
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 1
- VOFZZZOHJMJYKC-UHFFFAOYSA-N methyl n-(trimethoxysilylmethyl)carbamate Chemical compound COC(=O)NC[Si](OC)(OC)OC VOFZZZOHJMJYKC-UHFFFAOYSA-N 0.000 description 1
- BFXIKLCIZHOAAZ-UHFFFAOYSA-N methyltrimethoxysilane Chemical compound CO[Si](C)(OC)OC BFXIKLCIZHOAAZ-UHFFFAOYSA-N 0.000 description 1
- 239000011707 mineral Substances 0.000 description 1
- 235000010755 mineral Nutrition 0.000 description 1
- QRANWKHEGLJBQC-UHFFFAOYSA-N n-(trimethoxysilylmethyl)cyclohexanamine Chemical compound CO[Si](OC)(OC)CNC1CCCCC1 QRANWKHEGLJBQC-UHFFFAOYSA-N 0.000 description 1
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 230000006855 networking Effects 0.000 description 1
- 238000006386 neutralization reaction Methods 0.000 description 1
- 229910017464 nitrogen compound Inorganic materials 0.000 description 1
- 150000002830 nitrogen compounds Chemical class 0.000 description 1
- 230000003287 optical effect Effects 0.000 description 1
- 150000001282 organosilanes Chemical class 0.000 description 1
- 125000005375 organosiloxane group Chemical group 0.000 description 1
- 150000002923 oximes Chemical class 0.000 description 1
- 229910052760 oxygen Inorganic materials 0.000 description 1
- 239000001301 oxygen Substances 0.000 description 1
- UKODFQOELJFMII-UHFFFAOYSA-N pentamethyldiethylenetriamine Chemical compound CN(C)CCN(C)CCN(C)C UKODFQOELJFMII-UHFFFAOYSA-N 0.000 description 1
- 239000000546 pharmaceutical excipient Substances 0.000 description 1
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 1
- 239000000049 pigment Substances 0.000 description 1
- 239000004014 plasticizer Substances 0.000 description 1
- 239000002574 poison Substances 0.000 description 1
- 231100000614 poison Toxicity 0.000 description 1
- 229920002857 polybutadiene Polymers 0.000 description 1
- 229920005906 polyester polyol Polymers 0.000 description 1
- 229920000573 polyethylene Polymers 0.000 description 1
- 239000004814 polyurethane Substances 0.000 description 1
- 229920002635 polyurethane Polymers 0.000 description 1
- GKKCIDNWFBPDBW-UHFFFAOYSA-M potassium cyanate Chemical compound [K]OC#N GKKCIDNWFBPDBW-UHFFFAOYSA-M 0.000 description 1
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 1
- MHZDONKZSXBOGL-UHFFFAOYSA-N propyl dihydrogen phosphate Chemical compound CCCOP(O)(O)=O MHZDONKZSXBOGL-UHFFFAOYSA-N 0.000 description 1
- 239000010453 quartz Substances 0.000 description 1
- 239000002994 raw material Substances 0.000 description 1
- 239000000376 reactant Substances 0.000 description 1
- 239000011541 reaction mixture Substances 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 229910052895 riebeckite Inorganic materials 0.000 description 1
- LMHHRCOWPQNFTF-UHFFFAOYSA-N s-propan-2-yl azepane-1-carbothioate Chemical compound CC(C)SC(=O)N1CCCCCC1 LMHHRCOWPQNFTF-UHFFFAOYSA-N 0.000 description 1
- 150000003839 salts Chemical class 0.000 description 1
- 229910052710 silicon Inorganic materials 0.000 description 1
- HBMJWWWQQXIZIP-UHFFFAOYSA-N silicon carbide Chemical compound [Si+]#[C-] HBMJWWWQQXIZIP-UHFFFAOYSA-N 0.000 description 1
- 229910010271 silicon carbide Inorganic materials 0.000 description 1
- HQVNEWCFYHHQES-UHFFFAOYSA-N silicon nitride Chemical compound N12[Si]34N5[Si]62N3[Si]51N64 HQVNEWCFYHHQES-UHFFFAOYSA-N 0.000 description 1
- 125000003808 silyl group Chemical group [H][Si]([H])([H])[*] 0.000 description 1
- 238000010583 slow cooling Methods 0.000 description 1
- 239000011734 sodium Substances 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 239000011877 solvent mixture Substances 0.000 description 1
- 239000003381 stabilizer Substances 0.000 description 1
- 239000007858 starting material Substances 0.000 description 1
- 125000001424 substituent group Chemical group 0.000 description 1
- 229910052717 sulfur Inorganic materials 0.000 description 1
- 125000004434 sulfur atom Chemical group 0.000 description 1
- DKGAVHZHDRPRBM-UHFFFAOYSA-N tert-butyl alcohol Substances CC(C)(C)O DKGAVHZHDRPRBM-UHFFFAOYSA-N 0.000 description 1
- 229920001169 thermoplastic Polymers 0.000 description 1
- 229920001187 thermosetting polymer Polymers 0.000 description 1
- 239000004416 thermosoftening plastic Substances 0.000 description 1
- 239000013008 thixotropic agent Substances 0.000 description 1
- 150000003606 tin compounds Chemical class 0.000 description 1
- 231100000331 toxic Toxicity 0.000 description 1
- 230000002588 toxic effect Effects 0.000 description 1
- STCOOQWBFONSKY-UHFFFAOYSA-N tributyl phosphate Chemical compound CCCCOP(=O)(OCCCC)OCCCC STCOOQWBFONSKY-UHFFFAOYSA-N 0.000 description 1
- IMFACGCPASFAPR-UHFFFAOYSA-N tributylamine Chemical compound CCCCN(CCCC)CCCC IMFACGCPASFAPR-UHFFFAOYSA-N 0.000 description 1
- PIILXFBHQILWPS-UHFFFAOYSA-N tributyltin Chemical class CCCC[Sn](CCCC)CCCC PIILXFBHQILWPS-UHFFFAOYSA-N 0.000 description 1
- IMNIMPAHZVJRPE-UHFFFAOYSA-N triethylenediamine Chemical compound C1CN2CCN1CC2 IMNIMPAHZVJRPE-UHFFFAOYSA-N 0.000 description 1
- ZNOCGWVLWPVKAO-UHFFFAOYSA-N trimethoxy(phenyl)silane Chemical compound CO[Si](OC)(OC)C1=CC=CC=C1 ZNOCGWVLWPVKAO-UHFFFAOYSA-N 0.000 description 1
- 238000009827 uniform distribution Methods 0.000 description 1
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 1
- 238000010626 work up procedure Methods 0.000 description 1
- 239000010457 zeolite Substances 0.000 description 1
- 235000014692 zinc oxide Nutrition 0.000 description 1
- RNWHGQJWIACOKP-UHFFFAOYSA-N zinc;oxygen(2-) Chemical class [O-2].[Zn+2] RNWHGQJWIACOKP-UHFFFAOYSA-N 0.000 description 1
- GFQYVLUOOAAOGM-UHFFFAOYSA-N zirconium(iv) silicate Chemical compound [Zr+4].[O-][Si]([O-])([O-])[O-] GFQYVLUOOAAOGM-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09J—ADHESIVES; NON-MECHANICAL ASPECTS OF ADHESIVE PROCESSES IN GENERAL; ADHESIVE PROCESSES NOT PROVIDED FOR ELSEWHERE; USE OF MATERIALS AS ADHESIVES
- C09J175/00—Adhesives based on polyureas or polyurethanes; Adhesives based on derivatives of such polymers
- C09J175/04—Polyurethanes
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G18/00—Polymeric products of isocyanates or isothiocyanates
- C08G18/06—Polymeric products of isocyanates or isothiocyanates with compounds having active hydrogen
- C08G18/08—Processes
- C08G18/10—Prepolymer processes involving reaction of isocyanates or isothiocyanates with compounds having active hydrogen in a first reaction step
- C08G18/12—Prepolymer processes involving reaction of isocyanates or isothiocyanates with compounds having active hydrogen in a first reaction step using two or more compounds having active hydrogen in the first polymerisation step
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G18/00—Polymeric products of isocyanates or isothiocyanates
- C08G18/06—Polymeric products of isocyanates or isothiocyanates with compounds having active hydrogen
- C08G18/28—Polymeric products of isocyanates or isothiocyanates with compounds having active hydrogen characterised by the compounds used containing active hydrogen
- C08G18/65—Low-molecular-weight compounds having active hydrogen with high-molecular-weight compounds having active hydrogen
- C08G18/66—Compounds of groups C08G18/42, C08G18/48, or C08G18/52
- C08G18/6666—Compounds of group C08G18/48 or C08G18/52
- C08G18/667—Compounds of group C08G18/48 or C08G18/52 with compounds of group C08G18/32 or polyamines of C08G18/38
- C08G18/6674—Compounds of group C08G18/48 or C08G18/52 with compounds of group C08G18/32 or polyamines of C08G18/38 with compounds of group C08G18/3203
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Polyurethanes Or Polyureas (AREA)
- Compositions Of Macromolecular Compounds (AREA)
Abstract
Gegenstand der Erfindung sind Prepolymere (A) mit Endgruppen der allgemeinen Formel (1) DOLLAR A -SiR·1·¶a¶(OR·3·)¶3-a¶, DOLLAR A wobei DOLLAR A R·1· einen gegebenenfalls halogensubstituierten Alkyl-, Cycloalkyl-, Alkenyl- oder Arylrest mit 1-10 Kohlenstoffatomen, DOLLAR A R·2· einen Alkylrest mit 1-6 Kohlenstoffatomen oder einen omega-Oxaalkyl-alkylrest mit insgesamt 2-10 Kohlenstoffatomen, DOLLAR A a eine Zahl von 0 bis 2 bedeuten, DOLLAR A wobei die Prepolymere (A) erhältlich sind durch Umsetzung von DOLLAR A 1) Polyol (A1) mit einem mittleren Molekulargewicht Mn von 1000 bis 25000, DOLLAR A 2) niedermolekularem Alkohol (A2) mit mindestens zwei Hydroxylgruppen pro Molekül und einem Molekulargewicht von 62 bis 300, DOLLAR A 3) Di- oder Polyisocyanat (A3) und DOLLAR A 4) Alkoxysilan (A4), welches über eine Isocyanatgruppe oder über eine isocyanatreaktive Gruppe verfügen, DOLLAR A wobei der niedermolekulare Alkohol (A2) und das Polyol (A1) in einem molaren Verhältnis von 0,3 : 1 bis 7 : 1 eingesetzt werden.
Description
- Die Erfindung betrifft alkoxysilanterminierte Prepolymere und Prepolymere enthaltende Massen.
- Prepolymersysteme, die über reaktive Alkoxysilylgruppen verfügen, sind seit langem bekannt und werden vielfach zur Herstellung von elastischen Dicht- und Klebstoffen im Industrie- und Baubereich verwendet. In Gegenwart von Luftfeuchtigkeit und geeigneten Katalysatoren sind diese alkoxysilantermierten Prepolymere bereits bei Raumtemperatur in der Lage, unter Abspaltung der Alkoxygruppen und Ausbildung einer Si-O-Si-Bindung miteinander zu kondensieren. Somit lassen sich diese Prepolymere u.a. als einkomponentige Systeme verwenden, welche den Vorteil einer einfachen Handhabung besitzen, da keine zweite Komponente zudosiert und eingemischt werden muß.
- Ein weiterer Vorteil von alkoxysilanterminierten Prepolymeren besteht in der Tatsache, daß bei der Härtung weder Säuren noch Oxime oder Amine freigesetzt werden. Anders als bei Kleb- oder Dichtstoffen auf Isocyanatbasis entsteht auch kein CO2, das als gasförmige Komponente zu einer Blasenbildung führen kann. Anders als isocyanatbasierende Systeme sind alkoxysilanterminierte Prepolymermischungen auch toxikologisch in jedem Falle unbedenklich. Je nach Gehalt an Alkoxysilangruppen und deren Aufbau bilden sich bei der Härtung dieses Prepolymertyps hauptsächlich langkettige Polymere (Thermoplaste), relativ weitmaschige dreidimensionale Netzwerke (Elastomere) oder aber hochvernetzte Systeme (Duroplaste).
- Alkoxysilanterminierte Prepolymere können aus unterschiedlichen Bausteinen aufgebaut sein. Üblicherweise besitzen diese Prepolymere ein organisches Rückgrat, d.h. sie sind beispielsweise aus Polyurethanen, Polyethern, Polyestern, Polyacrylaten, Polyvinylestern, Ethylen-Olefincopolymeren, Styrol-Butadiencopolymeren oder Polyolefinen aufgebaut, beschrieben u.a. in
EP 0 372 561 ,EP 0 269 819 , WO 00/37533,US 6,207,766 undUS 3,971,751 . Daneben sind aber auch Systeme weit verbreitet, deren Rückgrat ganz oder zumindest zum Teil aus Organosiloxanen besteht, beschrieben u.a. in WO 96/34030 undUS 5,254,657 ). - Bei einem besonders vorteilhaften Herstellverfahren werden alkoxysilanterminierte Prepolymere durch Umsatz von Polyolen, z.B. von Polyester- oder Polyetherpolyolen, mit einem γ-Isocyanatopropyl-alkoxysilan umgesetzt. Alternativ können hier auch OH-terminierte Prepolymere, hergestellt aus einem Polyol und einem Unterschuß eines Di- oder Polyisocyanates, mit einem γ-Isocyanatopropyl-alkoxysilan zu alkoxysilanterminierten Prepolymeren umgesetzt werden. Derartige Systeme sind beispielsweise in
EP 0 931 800 ,EP 0 070 475 oderUS 5,068,304 beschrieben. - Bei einem zweiten besonders vorteilhaften Herstellungsverfahren für alkoxysilanterminierte Prepolymere wird von Polyolen, z.B. von Polyether- oder Polyesterpolyolen, ausgegangen, die in einem ersten Reaktionsschritt mit einem Überschuß eines Di- oder Polyisocyanates umgesetzt werden. Anschließend werden die dabei erhaltenen isocyanatterminierten Prepolymere mit einem γ-aminopropylfunktionellen Alkoxysilan zu dem gewünschten alkoxysilanterminierten Prepolymer umgesetzt. Derartige Systeme sind beispielsweise in
EP 1 256 595 ,EP 0 569 360 oderEP 0 082 528 oderDE 198 49 817 beschrieben. - In
DE 198 49 817 wird zudem festgestellt, daß bei der Herstellung der dabei als Zwischenprodukt dienenden isocyanatterminierten Prepolymere gegebenenfalls auch untergeordnete Mengen an niedermolekularen 2- und 3-wertigen Alkoholen mitverwendet werden können. Allerdings führen diese untergeordneten Mengen eines Alkohols zu keinerlei Eigenschaftsverbesserungen von den resultierenden Prepolymeren oder deren Aushärtungsprodukten. - Nachteilig an den beschriebenen Systemen ist deren nur mäßige Reaktivität gegenüber Feuchtigkeit, sowohl in Form von Luftfeuchtigkeit als auch in Form von gegebenenfalls zugesetztem Wasser. Um auch bei Raumtemperatur eine hinreichende Härtungsgeschwindigkeit zu erreichen, ist daher der Zusatz eines Katalysators unbedingt erforderlich. Das ist vor allem deshalb problematisch, da die in der Regel als Katalysatoren eingesetzten zinnorganischen Verbindungen toxikologisch bedenklich sind. Zudem enthalten die Zinnkatalysatoren oftmals auch noch Spuren hochtoxischer Tributylzinnderivate.
- Besonders problematisch ist die relativ geringe Reaktivität der alkoxysilanterminierten Prepolymersysteme, wenn keine Methoxysilylterminierungen sondern die nochmals unreaktiveren Ethoxysilylterminierungen verwendet werden. Gerade ethoxysilylterminierte Prepolymere wären jedoch in vielen Fällen besonders vorteilhaft, weil bei ihrer Aushärtung lediglich Ethanol als Spaltprodukt freigesetzt wird.
- Um dieses Problem zu umgehen, wurde bereits nach zinnfreien Katalysatoren gesucht. Denkbar sind hier vor allem titanhaltige Katalysatoren, z.B. Titantetraisopropoxylat oder Bis-(acetylacetonato)-diisobutytitanat (beschrieben u.a. in
EP 0 885 933 ). Allerdings besitzen diese Titankatalysatoren den Nachteil, daß sie nicht gemeinsam mit zahlreichen stickstoffhaltigen Verbindungen eingesetzt werden können, da letztere hier als Katalysatorgifte wirken. Die Verwendung von stickstoffhaltigen Verbindungen, z.B. als Haftvermittler, wäre in vielen Fällen jedoch wünschenswert. Zudem dienen Stickstoffverbindungen, z.B. Aminosilane, in vielen Fällen als Edukte bei der Herstellung der silanterminierten Prepolymere. - Einen großen Vorteil können daher alkoxysilanterminierte Prepolymersysteme darstellen, wie sie beispielsweise in
DE 101 42 050 beschrieben sind. Diese Prepolymere zeichnen sich dadurch aus, daß sie Alkoxysilylgruppen enthalten, die nur durch einen Methylspacer von einem elektronegativen Heteroatom mit mindestens einem freien Elektronenpaar getrennt sind, d.h. von einem Sauerstoff-, Stickstoff- oder Schwefelatom. Dadurch besitzen diese Prepolymere eine extrem hohe Reaktivität gegenüber (Luft-)Feuchtigkeit, so daß sie zu Prepolymer abmischungen verarbeitet werden können, die auch mit wenig oder sogar ohne Katalysatoren auskommen können, die Titan, Zinn oder weitere (Schwer-)Metalle enthalten, und dennoch bei Raumtemperatur mit hinreichend kurzen Klebfreizeiten bzw. mit hinreichend hoher Geschwindigkeit aushärten. - In
DE 101 42 050 wird zudem festgestellt, daß zur Herstellung von alkoxysilanterminierten Prepolymeren prinzipiell auch monomere Alkohole/Amine mit mindestens zwei OH-/NH-Funktionen in Kombination mit Di- oder Polyisocyanaten sowie organofunktionellen Silanen eingesetzt werden können. Allerdings werden hier keinerlei Alkohol-/Amingehalte oder aber Mengenverhältnisse zwischen dem monomeren Alkoholen/Aminen und weiteren Prepolymerbausteinen beschrieben, die zu einer Eigenschaftsverbesserung des Prepolymers führen. Auch wird nicht beschrieben, daß der Einsatz von monomeren Alkoholen/Aminen bei der Prepolymersynthese überhaupt geeignet sein könnte, um Eigenschaften von silanterminierten Prepolymeren oder deren Aushärtungsprodukten zu verbessern. - Alle alkoxysilanterminierten Prepolymere des Standes der Technik weisen jedoch den Nachteil auf, daß sie bei einer Aushärtung nur zu Massen mit einer mäßigen Reißfestigkeit und/oder Reißdehnung führen. Ausnahme stellen hier lediglich Systeme mit einem hohen Gehalt an Harnstoffeinheiten im Prepolymer dar, wie sie beispielsweise in
DE 21 55 258 beschrieben werden. Dieser hohe Gehalt an Harnstoffeinheiten führt jedoch dazu, daß diese Prepolymere bereits im unvernetzten Zustand fest sind und nur in stark verdünnten Lösungen mit einem Feststoffgehalt « 50 % handhabbar sind. Derartige Prepolymerlösungen sind für die meisten Anwendungen völlig ungeeignet. - Silanvernetzende Abmischungen, die zu Massen mit hoher Reißfestigkeit und Bruchdehnung aushärten, werden vor allem bei Klebstoffanwendungen, u.a. in der Automobilindustrie, gefordert.
- Ein Ansatz zur Verbesserung der Reißfestigkeit von alkoxysilanvernetzenden Klebstoffen kann der Einsatz von optimierten Füllstoffgemischen darstellen, die in das alkoxysilanterminierte Polymer eingearbeitet werden. Ein derartiges Verfahren wird in
EP 1 256 595 beschrieben. Hier werden einem alkoxysilanterminierten Prepolymer eine bestimmte Rußsorte sowie feinteiliges beschichtetes Calciumcarbonat zugemischt. Mit diesem System konnten zwar hervorragende Reißdehnungen von 4,0-5,9 MPa erreicht werden, die erzielbaren Bruchdehnungen hingegen waren mit 250-300 jedoch noch nicht befriedigend. Zudem lassen sich mit derartigen rußgefüllten Massen lediglich schwarze Klebstoffe herstellen. Andere Farben, obgleich oftmals erwünscht, sind nicht möglich. Außerdem kann es, z.B. wenn man aus optischen Gründen transparente Massen erhalten will, wünschenswert sein, ganz auf Füllstoffe zu verzichten. - Es bestand die Aufgabe, Massen aus Basis silanterminierter Prepolymere mit verbesserter Reißfestigkeit und Bruchdehnung bereitzustellen, die die vorgenannten Nachteile nicht aufweisen.
- Gegenstand der Erfindung sind Prepolymere (A) mit Endgruppen der allgemeinen Formel [1]
R1 einen gegebenenfalls halogensubstituierten Alkyl-, Cycloalkyl-, Alkenyl- oder Arylrest mit 1-10 Kohlenstoffatomen,
R2 einen Alkylrest mit 1-6 Kohlenstoffatomen oder einen ω-Oxaalkyl-alkylrest mit insgesamt 2-10 Kohlenstoffatomen,
a eine Zahl von 0 bis 2 bedeuten,
wobei die Prepolymere (A) erhältlich sind durch Umsetzung von - 1) Polyol (A1) mit einem mittleren Molekulargewicht Mn von 1000 bis 25000,
- 2) niedermolekularem Alkohol (A2) mit mindestens zwei Hydroxylgruppen pro Molekül und einem Molekulargewicht von 62 bis 300,
- 3) Di- oder Polyisocyanat (A3) und
- 4) Alkoxysilan (A4), welches über eine Isocyanatgruppe oder über eine isocyanatreaktive Gruppe verfügen,
- Bei der Herstellung der alkoxysilanvernetzenden Prepolymere (A) Werden neben Di- oder Polyisocyanaten und organofunktionellen Silanen eine bestimmte Mischung aus langkettigen Polyolen (A1) und niedermolekularen Alkoholen (A2) verwendet. Die so hergestellten Prepolymere (A) weisen nach der Vernetzung unabhängig von gegebenenfalls verwendeten Füllstoffen eine erheblich verbesserte Reißfestigkeit sowie eine erheblich verbesserte Bruchdehnung auf. Auch Massen (M), die die silanterminierten Prepolymere (A) enthalten, zeigen die verbesserte Reißfestigkeit und Bruchdehnung.
- Die Prepolymere (A) sind bevorzugt isocyanatfrei.
- Bevorzugt wird dabei ein molares Verhältnis des niedermolekularen Alkohols (A2) zum Polyol (A1) von 0,5:1 bis 5:1, wobei ein Verhältnis dieser beiden Komponenten von 0,7:1 bis 3:1 besonders bevorzugt wird. Sowohl beim niedermolekularem Alkohol (A2) als auch beim Polyol (A1) werden Verbindungen mit zwei OH-Gruppen bevorzugt, die bei der Prepolymersynthese zu linearen und unverzweigten Prepolymeren (A) führen.
- Die Wirkungsweise der Kombination eines niedermolekularen Alkohols (A2) und eines Polyols (A1) während der Prepolymersynthese besteht zum einen darin, daß der Einsatz des Alkohols (A2) in der Prepolymersynthese durch dessen Reaktion mit den Isocyanatgruppen des Di- oder Polyisocyanates (A3) oder mit einem gegebenfalls vorhandenen isocyanatfunktionellen Silan (A4) in der resultierenden Polymerkette zu einer erhöhten Dichte an Urethaneinheiten führt. Diese verbessert die mechanischen Eigenschaften der Prepolymere (A) und Prepolymere (A) enthaltenden Massen (M) nach deren Aushärtung.
- Vor allem aber führt der Einsatz des niedermolekularen Alkohols (A2) in Kombination mit einem oder mehreren langkettigen Polyolen (A1) dazu, daß Prepolymerketten gebildet werden, in denen die Urethaneinheiten nicht gleichmäßig verteilt sind. So bilden sich durch den Einbau eines Polyolmoleküls (A1) in die Prepolymerkette stets ein langer urethangruppenfreier Kettenabschnitt, während der Einbau des niedermolekularen Alkohols (A2) stets zu (mindestens) zwei Urethaneinheiten führt, die nur durch einen sehr kurzen, aus wenigen Kohlenstoffatomen bestehenden Kettenabschnitt getrennt sind. Werden Polyol (A1) und niedermolekularer Alkohol (A2) in dem erfindungsgemäßen relativen Verhältnis zueinander eingesetzt, so wirkt sich diese ungleichmäßige Anordnung der Urethaneinheiten im Polymer ungewöhnlich positiv auf die Reißfestigkeit der ausgehärteten Masse (M) aus. So lassen sich mit den Prepolymeren (A) Massen (M) mit deutlich besserer Reißfestigkeit herstellen, als dies mit herkömmlichen Prepolymeren mit einer relativ gleichmäßigen Verteilung der Urethaneinheiten in der Prepolymerkette möglich ist. Dies gilt auch dann, wenn die Prepolymere (A) und nicht erfindungsgemäßen Prepolymere in ihren übrigen Merkmalen wie mittlere Kettenlänge, Urethan-, Harnstoff- und Silylgruppendichte übereinstimmen und beide Polymere aus demselben Polyoltyp (z.B. Polypropylenglykol), Isocyanat- und Silantypen aufgebaut sind.
- In einer bevorzugten Ausführung der Erfindung verfügen die alkoxysilanterminierten Polymere (A) über Endgruppen der allgemeinen Formel [2]
A eine zweibindige Bindegruppe ausgewählt aus -O-, -S-, -(R3)N-, -O-CO-N(R3)-, -N(R3)-CO-O-, -NH-CO-NH-, -N(R4)-CO-NH-, -NH-CO-N(R4)-, -N(R4)-CO-N(R4)-,
R3 Wasserstoff, einen gegebenenfalls halogensubstituierten cyclischen, linearen oder verzweigten C1- bis C18-Alkyl- oder Alkenylrest oder einen C6- bis C18-Arylrest,
R4 einen gegebenenfalls halogensubstituierten cyclischen, linearen oder verzweigten C1- bis C18-Alkyl- oder Alkenylrest oder einen C6- bis C18-Arylrest bedeuten
und R1, R2 und a die bei der allgemeinen Formel [1] angegebenen Bedeutungen aufweisen. - Die Polymere (A) mit Endgruppen der allgemeinen Formel [2] zeichnen sich dadurch aus, daß sie Alkoxysilylgruppen enthalten, die nur durch einen Methylspacer von einem elektronegativen Heteroatom mit mindestens einem freien Elektronenpaar getrennt sind. Dadurch besitzen diese Polymere eine extrem hohe Reaktivität gegenüber (Luft-) Feuchtigkeit, so daß sie zu Polymerabmischungen (M) verarbeitet werden können, die auch mit wenig oder sogar ohne Zinnkatalysator, bevorzugt ohne Zinn- oder Titankatalysator, besonders bevorzugt ganz ohne schwermetallhaltigen Katalysator bei Raumtemperatur mit hinreichend kurzen Klebfreizeiten bzw. mit hinreichend hoher Geschwindigkeit aushärten.
- Als Reste R1 werden Methyl-, Ethyl- oder Phenylgruppen bevorzugt. Bei den Resten R2 handelt es sich bevorzugt um Methyl- oder Ethylgruppen und als Rest R3 wird Wasserstoff bevorzugt, während es sich bei den Resten R4 bevorzugt um Alkylreste mit 1-4 Kohlenstoffatomen, Cyclohexyl- und Phenylreste handelt.
- Besonders bevorzugt sind alkoxysilylterminierte Polymere (A), deren vernetzbare Alkoxysilylgruppen durch einen Methylspacer von einer Bindegruppe wie Urethan- oder Harnstoffgruppen getrennt sind, d.h. Polymere (A) der allgemeinen Formel [2], bei denen A ausgewählt wird aus den Gruppen -O-CO-N(R3)-, -N(R3)-CO-O-, -N(R4)-CO-NH-, und -NH-CO-N(R4)-.
- Besonders günstige Eigenschaften weisen dabei Prepolymere (A) auf, die mit Alkoxysilylgruppen der allgemeinen Formel [2] terminiert sind, wenn diese Alkoxysilylgruppen zu mindestens 50 aus Dialkoxysilylgruppen (Formel [2] mit a = 1) bestehen. So kann ein zu hoher Gehalt an Monoalkoxysilylgruppen in den Prepolymeren (A) zu einem Verlust an Reißfestigkeit führen, während hohe Gehalte an Trialkoxysilylgruppen zu einer Verringerung der Reißdehnung führen können, ohne die Reißfestigkeit signifikant zu steigern. Die entsprechenden Prepolymere (A), deren Alkoxysilylgruppen zu mindestens 50 aus Dialkoxysilylgruppen der allgemeinen Formel [2] mit a = 1 bestehen, werden daher bevorzugt. Besonders bevorzugt werden Prepolymere (A) mit einem Anteil an Dialkoxysilylgruppen der allgemeinen Formel [2] von mindestens 70 %, wobei Prepolymere (A), die ausschließlich Dialkoxysilylgruppen der allgemeinen Formel [2] enthalten, nicht nur besonders bevorzugt sondern auch logistisch einfach zugänglich sind, da zur ihrer Herstellung lediglich ein Silantyp (A4) benötigt wird.
- Die Hauptketten der alkoxysilanterminierten Polymere (A) können verzweigt oder unverzweigt sein, wobei unverzweigte oder nur schwach verzweigte Hauptketten bevorzugt werden. Die mittleren Kettenlängen können beliebig, entsprechend der jeweils gewünschten Eigenschaften sowohl der unvernetzten Mischung als auch der ausgehärteten Masse, angepaßt werden.
- Als Polyole (A1) für die Herstellung der Prepolymere (A) können prinzipiell sämtliche Polyole mit einem mittleren Molekulargewicht Mn von 1000 bis 25000 eingesetzt werden. Dabei kann es sich beispielsweise um hydroxylfunktionelle Polyether, Polyester, Polyacrylate und -methacrylate, Polycarbonate, Polystyrole, Polysiloxane, Polyamide, Polyvinylester, Polyvinylhydroxide oder Polyolefine wie z.B. Polyethylen, Polybutadien, Ethylen-Olefincopolymere oder Styrol-Butadiencopolymere handeln.
- Bevorzugt werden Polyole (A1) mit einem Molekulargewicht Mn von 2000 bis 25000, besonders bevorzugt von 4000 bis 20000 eingesetzt. Besonders geeignete Polyole (A1) sind aromatische und/oder aliphatische Polyesherpolyole und Polyetherpolyole, wie sie in der Literatur vielfach beschrieben sind. Die als Polyole (A1) eingesetzten Polyether und/oder Polyester können dabei sowohl linear als auch verzweigt sein, wobei jedoch unverzweigte, lineare Polyole bevorzugt werden. Zudem können Polyole (A1) auch Substituenten wie z.B. Halogenatome besitzen.
- Ebenso können als Polyole (A1) auch hydroxyalkyl- oder aminoalkylterminierte Polysiloxane der allgemeinen Formel [3]
R5 einen Kohlenwasserstoffrest mit 1 bis 12 Kohlenstoffatomen, bevorzugt Methylreste,
R6 eine verzweigte oder unverzweigte Kohlenwasserstoffkette mit 1-12 Kohlenstoffatomen, bevorzugt n-Propyl,
n eine Zahl von 1 bis 3000, bevorzugt eine Zahl von 10 bis 1000 und
Z eine OH- oder NHR3-Gruppe
bedeuten und R3 die bei der allgemeinen Formel [2] angegebenen Bedeutungen aufweist. - Selbstverständlich ist auch der Einsatz beliebiger Mischungen der verschiedenen Polyoltypen möglich. Besonders bevorzugt werden jedoch lineare Polyetherpolyole, insbesondere Polypropylenglycole als Polyole (A1) verwendet.
- Als niedermolekulare Alkohole mit mindestens zwei Hydroxylgruppen pro Molekül (A2) eignen sich prinzipiell sämtliche entsprechende Verbindungen mit einem Molekulargewicht von 32 bis 300. Bevorzugt werden hier jedoch niedermolekulare Diole eingesetzt, wie z.B. Glycol, 1,3-Propandiol, 1,3-Butandiol, 1,4-Butandiol, sämtliche regioisomere Penta- und Hexadiole sowie Etylenglykol oder Propylenglykol. Einen besonders bevorzugten niedermolekularen Alkohol (A2) stellt 1,4-Butandiol dar.
- Als Di- oder Polyisocyanate (A3) für die Herstellung der Prepolymere (A) können prinzipiell sämtliche gebräuchlichen Isocyanate eingesetzt werden, wie sie in der Literatur vielfach beschrieben sind. Gängige Diisocyanate (A3) sind beispielsweise Diisocyanatodiphenylmethan (MDI), sowohl in Form von rohem oder technischem MDI als auch in Form reiner 4,4' bzw. 2,4' Isomeren oder deren Mischungen, Tolylendiisocyanat (TDI) in Form seiner verschiedenen Regioisomere, Diisocyanatonaphthalin (NDI), Isophorondiisocyanat (IPDI), perhydriertes MDI (H-MDI) oder auch von Hexamethylendiisocyanat (HDI). Beispiele für Polyisocyanate (A3) sind polymeres MDI (P-MDI), Triphenylmethantriisocanat oder Isocyanurat- oder Biurettriisocyanate. Sämtliche Di- und/oder Polyisocyanate (A3) können einzeln oder auch in Mischungen eingesetzt werden. Bevorzugt werden jedoch ausschließlich Diisocyanate eingesetzt. Falls die UV-Stabilität der Prepolymere (A) oder der aus diesen Prepolymeren hergestellten ausgehärteten Materialien auf Grund der jeweiligen Anwendung von Bedeutung ist, werden bevorzugt aliphatische Isocyanate als Komponente (A3) verwendet.
- Als Alkoxysilane (A4) für die Herstellung der Prepolymere (A) können prinzipiell sämtliche Alkoxysilane eingesetzt werden, die entweder über eine Isocyanatfunktion oder über eine isocyanatreaktive Gruppe verfügen. Die Alkoxysilane dienen zum Einbau der Alkoxysilylterminierungen in die Prepolymere (A). Als Alkoxysilane (A4) werden dabei vorzugsweise Verbindungen eingesetzt, welche ausgewählt werden aus Silanen der allgemeinen Formeln [4] und [5]wobei

B1 eine OH-, SH-, NH2- oder eine HR4N-Gruppe bedeutet und
R1, R2, R4 und a die bei den allgemeinen Formeln [1] und [2] angegebenen Bedeutungen aufweisen. - Die isocyanatreaktive Gruppe B1 in der allgemeinen Formel [5] stellt dabei bevorzugt eine HR4N-Gruppe dar.
- Dabei können einzelne Silane (A4) sowie auch Mischungen verschiedener Silane (A4) eingesetzt werden. Die entsprechenden Silane lassen sich durch eine Reaktion aus Chlormethyltrialkoxysilan, Chlormethyldialkoxymethylsilan oder Chlordimethylalkoxymethylsilan mit einem Amin der allgemeinen Formel NH2R4, d.h. aus sehr einfachen und preiswerten Edukten, problemlos in nur einem Reaktionsschritt herstellen.
- Die Herstellung der Prepolymere (A) erfolgt durch ein einfaches Zusammengeben der beschriebenen Komponenten, wobei gegebenenfalls noch ein Katalysator zugegeben und/oder bei erhöhter Temperatur gearbeitet werden kann. Dabei reagieren die Isocyanatgruppen der Di- und/oder Polyisocyanate (A3) sowie – falls vorhanden – die Isocyanatgruppen des Silans der allgemeinen Formel [4] mit den OH- bzw. NH-Funktionen der zugegebenen Polyole (A1) und niedermolekularen Alkohole (A2) sowie – falls vorhanden – mit den OH- bzw. NH-Funktionen der Silane der allgemeinen Formel [5]. Ob der relativ hohen Exothermie dieser Reaktionen kann es dabei vorteilhaft sein, die einzelnen Komponenten sukzessive zuzugeben, um die freiwerdende Wärmemenge besser kontrollieren zu können. Die Zugabereihenfolge und -geschwindigkeit der einzelnen Komponenten kann dabei beliebig gestaltet werden. Auch können die verschiedenen Rohstoffe sowohl einzeln als auch in Mischungen vorgelegt bzw. zugegeben werden. Auch eine kontinuierliche Prepolymerherstellung, z.B. in einem Röhrenreaktor, ist möglich.
- Die Konzentrationen aller an sämtlichen Reaktionsschritten beteiligten Isocyanatgruppen und aller isocyanatreaktiven Gruppen sowie die Reaktionsbedingungen sind dabei bevorzugt so gewählt; daß im Laufe der Prepolymersynthese sämtliche Isocyanatgruppen abreagieren. Das fertige Prepolymer (A) ist somit isocyanatfrei. In einer bevorzugten Ausführungsform der Erfindung sind die Konzentrationsverhältnisse sowie die Reaktionsbedingungen so gewählt, daß nahezu sämtliche Kettenenden (> 80 der Kettenenden, besonders bevorzugt > 90 % der Kettenenden) der Prepolymere (A) mit Alkoxysilylgruppen terminiert sind.
- Bei einem bevorzugten Herstellungsverfahren wird die Isocyanatkomponente (A3) in einem ersten Reaktionsschritt mit der Polyolkomponente (A1) sowie mit der Alkoholkomponente (A2) umgesetzt, wobei – entsprechend der eingesetzten Mengenverhältnissen – ein hydroxyl- oder isocyanatterminiertes Prepolymer erhalten wird. Die Komponenten (A1) und (A2) können dabei nacheinander oder auch als Mischung eingesetzt werden. In einem zweiten Reaktionsschritt werden diese hydroxyl- oder isocyanatterminierten Prepolymere dann mit einem Silan der allgemeinen Formel [4] oder [5] umgesetzt, wobei die Konzentrationen so gewählt sind, daß sämtliche Isocyanatgruppen abreagieren. Es resultiert das silanterminierte Prepolymer (A). Eine gesonderte Reinigung oder sonstige Aufarbeitung des Prepolymers (A) ist dabei nicht erforderlich.
- Bei diesem Herstellungsverfahren werden bevorzugt Aminosilane der allgemeinen Formel [4] mit B1 = HR4N- als Silane (A4) verwendet und mit einem isocyanatterminierten Prepolymer umgesetzt. In einer besonders bevorzugten Ausführungsform der Erfindung wird das Silan dabei im Überschuß eingesetzt. Der Überschuß beträgt bevorzugt 20-400 %, besonders bevorzugt 50- 200 %. Das überschüssige Silan kann dem Prepolymer zu jedem beliebigen Zeitpunkt zugesetzt werden, bevorzugt wird der Silanüberschuß jedoch bereits während der Synthese der Prepolymere (A) zugegeben.
- Wenn bei der Herstellung der Prepolymere (A) ein Überschuß eines Silans (A4) der allgemeinen Formel [5] eingesetzt wird, können mit den Prepolymeren (A) Massen (M) mit besonders hoher Reißfestigkeit hergestellt werden.
- Die bei der Herstellung der Prepolymere (A) auftretenden Reaktionen zwischen Isocyanatgruppen und isocyanatreaktiven Gruppen können gegebenenfalls durch einen Katalysator beschleunigt werden. Bevorzugt werden dabei dieselben Katalysatoren eingesetzt, die unten auch als Härtungskatalysatoren (C) aufgeführt sind. Gegebenenfalls ist es sogar möglich, daß die Herstellung der Prepolymere (A) durch dieselben Katalysatoren katalysiert wird, die später bei der Aushärtung der fertigen Prepolymerabmischungen auch als Härtungskatalysator (C) dient. Dies hat den Vorteil, daß der Härtungskatalysator (C) bereits in dem Prepolymer (A) enthalten ist und bei der Compoundierung der fertigen Prepolymerabmischung (M) nicht mehr gesondert zugegeben werden muß. Selbstverständlich können dabei anstelle eines Katalysators auch Kombinationen mehrerer Katalysatoren eingesetzt werden.
- Um bei Raumtempratur eine schnelle Aushärtung der Massen (M) zu erreichen, kann gegebenenfalls ein Härtungskatalysator (C) zugesetzt werden. Wie bereits erwähnt kommen hier u.a. die zu diesem Zwecke üblicherweise verwendeten organischen Zinnverbindungen, wie z.B. Dibutylzinndilaurat, Dioctylzinndilaurat, Dibutylzinndiacetylacetonat, Dibutylzinndiacetat oder Dibutylzinndioctoat etc., in Frage. Des weiteren können auch Titanate, z.B. Titan(IV)isopropylat, Eisen(III)-Verbindungen, z.B. Eisen(III)-acetylacetonat, oder auch Amine, z.B. Triethylamin, Tributylamin, 1,4-Diazabicyclo[2,2,2]octan, 1,8-Diazabicyclo[5.4.0]undec-7-en, 1,5-Diazabicyclo[4.3.0]non-5-en, N,N-Bis-(N,N-dimethyl-2-amino ethyl)-methylamin, N,N-Dimethylcyclohexylamin, N,N-Dimethylphenlyamin, N-Ethylmorpholinin etc., eingesetzt werden. Auch organische oder anorganische Brönstedsäuren wie Essigsäure, Trifluoressigsäure oder Benzoylchlorid, Salzsäure, Phosphorsäure deren Mono- und/oder Diester, wie z.B. Butylphosphat, (Iso-) Propylphosphat, Dibutylphosphat etc., sind als Katalysatoren [C] geeignet. Daneben können hier aber auch zahlreiche weitere organische und anorganische Schwermetallverbindungen sowie organische und anorganische Lewissäuren oder -basen eingesetzt werden. Zudem kann die Vernetzungsgeschwindigkeit auch durch die Kombination verschiedener Katalysatoren bzw. von Katalysatoren mit verschiedenen Cokatalysatoren weiter gesteigert bzw. genau auf den jeweiligen Bedarf hin abgestimmt werden. Dabei werden Abmischungen (M) deutlich bevorzugt, die Prepolymere (A) mit hochreaktiven Alkoxysilylgruppen der allgemeinen Formel [2] enthalten, und somit keine schwermetallhaltigen Katalysatoren (C) benötigen, um auch bei Raumtemperatur ausreichend kurze Härtungszeiten zu erreichen.
- Der Einsatz von Prepolymeren (A) mit Silantermini der allgemeinen Formel [2] hat zudem den besonderen Vorteil, daß sich so auch Prepolymere (A) herstellen lassen, welche ausschließlich Ethoxysilylgruppen enthalten, d.h. Silylgruppen der allgemeinen Formel [2] mit R2 = Ethyl. Diese Massen (M) sind gegenüber Feuchtigkeit so reaktiv, daß sie auch ohne Zinnkatalysatoren mit hinreichend hoher Geschwindigkeit aushärten, obgleich Ethoxysilylgruppen generell weniger reaktiv sind als die entsprechenden Methoxysilylgruppen. So sind auch mit ethoxysilanterminierten Polymeren (A) zinnfreie Systeme möglich. Derartige Polymerabmischungen (M), die ausschließlich ethoxysilanterminierte Polymere (A) enthalten, besitzen den Vorteil, daß sie beim Härten lediglich Ethanol als Spaltprodukt freisetzen. Sie stellen eine bevorzugte Ausführungsform dieser Erfindung dar.
- Die Prepolymere (A) werden bevorzugt in Abmischungen (M) eingesetzt, die zudem noch niedermolekulare Alkoxysilane (D) enthalten. Diese Alkoxysilane (D) können dabei mehrere Funktionen übernehmen. So können sie beispielsweise als Wasserfänger dienen, d.h. sie sollen eventuell vorhandene Feuchtigkeitsspuren abfangen und so die Lagerstabilität der entsprechenden silanvernetzenden Massen (M) erhöhen. Selbstverständlich müssen diese zumindest eine vergleichbar hohe Reaktivität gegenüber Feuchtigkeitsspuren besitzen wie das Prepolymer (A). Geeignet als Wasserfänger sind daher vor allen hochreaktive Alkoxysilane (D) der allgemeinen Formel [6]wobei

B2 eine R4O-CO-NH-, R4R3N-CO-NH-, OH-, OR4-, SH-, SR4-, NH2- ,NHR4-, oder N(R4)2-Gruppe bedeutet und
R1, R2, R3, R4 und a die bei den allgemeinen Formeln [1] und [2] angegebenen Bedeutungen aufweisen. Ein besonders bevorzugter Wasserfänger ist dabei das Carbamatosilan, bei dem
B2 eine R4O-CO-NH-Gruppe darstellt. - Des weiteren können die niedermolekularen Alkoxysilane (D) auch als Vernetzer und/oder Reaktivverdünner dienen. Zu diesem Zweck sind prinzipiell sämtliche Silane geeignet, die über reaktive Alkoxysilylgruppen verfügen, über die sie während der Aushärtung der Polymerabmischung mit in das entstehende dreidimensionale Netzwerk eingebaut werden können. Die Alkoxysilane (D) können dabei zu einer Steigerung der Netzwerkdichte und damit zur Verbesserung der mechanischen Eigenschaften, z.B. der Reißfestigkeit, der ausgehärteten Masse (M) beitragen. Zudem können sie auch die Viskosität der entsprechenden Prepolymerabmischungen senken. Als Alkoxysilane (D) eignen sich in dieser Funktion beispielsweise Alkoxymethyltrialkoxysilane und Alkoxymethyldialkoxyalkylsilane. Als Alkoxygruppen werden dabei Methoxy- und Ethoxygruppen bevorzugt. Zudem können auch die preisgünstigen Alkyltrimethoxysilane, wie Methyltrimethoxysilan sowie Vinyl- oder Phenyltrimethoxysilan, sowie deren Teilhydrolysate geeignet sein.
- Auch können die niedermolekularen Alkoxysilane (D) als Haftvermitteler dienen. Hier können vor allem Alkoxysilane eingesetzt werden, die über Aminofunktionen oder Epoxyfunktionen verfügen. Als Beispiele seien γ-Aminopropyltrialkoxysilane, γ-[N-Aminoethylamino]-propyltrialkoxysilane, γ-Glycidoxy-propyltrialkoxysilane sowie sämtliche Silane der allgemeinen Formel [6], bei denen B2 für eine stickstoffhaltige Gruppe steht, genannt.
- Schließlich können die niedermolekularen Alkoxysilane (D) sogar als Härtungskatalysatoren oder -cokatalysatoren dienen. Zu diesem Zweck eignen sich vor allem sämtliche basischen Aminosilane, wie z.B. sämtliche Aminopropylsilane, N-Aminoethylaminopropylsilane sowie auch sämtliche Silane der allgemeinen Formel [6] soweit es sich bei B2 um eine NH2-, NHR4-, N(R4)2-Gruppe handelt.
- Die Alkoxysilane (D) können den Prepolymeren (A) zu jedem beliebigen Zeitpunkt zugegeben werden. Soweit sie über keine NCO-reaktiven Gruppen verfügen, können sie sogar bereits während der Synthese der Prepolymere (A) zugesetzt werden. Dabei können, bezogen auf 100 Gewichtsteile Prepolymer (A), bis zu 100 Gewichtsteile, vorzugsweise 1 bis 40 Gewichtsteile eines niedermolekularen Alkoxysilanes (D) zugesetzt werden.
- Des weiteren werden Abmischungen aus den alkoxysilantermierten Prepolymeren (A) üblicherweise Füllstoffe (E) zugegeben. Die Füllstoffe (E) führen dabei zu einer erheblichen Eigenschaftsverbesserung der resultierenden Abmischungen (M). Vor allem die Reißfestigkeit wie auch die Bruchdehnung können durch den Einsatz von geeigneten Füllstoffen erheblich gesteigert werden.
- Als Füllstoffe (E) eigenen sich dabei sämtliche Materialien, wie sie im Stand der Technik vielfach beschrieben sind. Beispiele für Füllstoffe sind nicht verstärkende Füllstoffe, also Füllstoffe mit einer BET-Oberfläche von bis zu 50 m2/g, wie Quarz, Diatomeenerde, Calciumsilikat, Zirkoniumsilikat, Zeolithe, Calciumcarbonat, Metalloxidpulver, wie Aluminium-, Titan-, Eisen-, oder Zinkoxide bzw. deren Mischoxide, Bariumsulfat, Calciumcarbonat, Gips, Siliciumnitrid, Siliciumcarbid, Bornitrid, Glas- und Kunststoffpulver; verstärkende Füllstoffe, also Füllstoffe mit einer BET-Oberfläche von mindestens 50 m2/g, wie pyrogen hergestellte Kieselsäure, gefällte Kieselsäure, Ruß, wie Furnace- und Acetylenruß und Silicium-Aluminium-Mischoxide großer BET-Oberfläche; faserförmige Füllstoffe, wie Asbest sowie Kunststoffasern. Die genannten Füllstoffe können hydrophobiert sein, beispielsweise durch die Behandlung mit Organosilanen bzw. -siloxanen oder durch Verätherung von Hydroxylgruppen zu Alkoxygruppen. Es kann eine Art von Füllstoff (E), es kann auch ein Gemisch von mindestens zwei Füllstoffen (E) eingesetzt werden.
- Die Füllstoffe (E) werden bevorzugt in einer Konzentration von 0-90 Gew.-% bezogen auf die fertige Abmischung eingesetzt, wobei Konzentrationen von 30-70 Gew.-% besonders bevorzugt sind. In einer bevorzugten Anwendung werden Füllstoffkombinationen (E) eingesetzt, die neben Calciumcarbonat noch pyrogene Kieselsäure und/oder Ruß enthalten.
- Auch Massen (M), welche keine Füllstoffe (E) enthalten, sind bevorzugt. So besitzen die Prepolymere (A) nach der Aushärtung bereits eine relativ hohe Reißdehnung, so daß sie auch ungefüllte Massen (M) ermöglichen. Vorteile ungefüllter Systeme sind deutlich niedrigere Viskosität sowie Transparenz.
- Die Abmischungen (M) enthaltend die Prepolymere (A) können zudem auch noch geringe Mengen eines organischen Lösungsmittels (F) enthalten. Dieses Lösungsmittel dient dabei der Erniedrigung der Viskosität der unvernetzten Massen (M). Als Lösungsmittel (F) kommen prinzipiell sämtliche Lösungsmittel sowie Lösemittelmischungen in Betracht. Als Lösungsmittel (F) werden bevorzugt Verbindungen eingesetzt, die über ein Dipolmoment verfügen. Besonders bevorzugte Lösungsmittel verfügen über ein Heteroatom mit freien Elektronenpaaren, die Wasserstoffbrückenbindungen eingehen können. Bevorzugte Beispiele für solche Lösungsmittel sind Ether wie z.B. t-Butylmethylether, Ester, wie z.B. Ethylacetat oder Butylacetat sowie Alkohole, wie z.B. Methanol, Ethanol , n-Butanol oder – besonders bevorzugt – t-Butanol. Die Lösungsmittel (F) werden bevorzugt in einer Konzentrationen von 0-20 Vol.-% bezogen auf die fertige Prepolymermischung incl. aller Füllstoffe (E) eingesetzt, wobei Lösungsmittelkonzentrationen von 0-5 Vol.-% besonders bevorzugt sind.
- Die Polymerabmischungen (M) können als weitere Komponenten an sich bekannte Hilfsstoffe, wie von den Komponenten (D) abweichende Wasserfänger und/oder Reaktivverdünner sowie Haftvermittler, Weichmacher, Thixotropiermittel, Fungizide, Flammschutzmittel, Pigmente etc. enthalten. Auch Lichtschutzmittel, Antioxidantien, Radikalfänger sowie weitere Stabilisatoren können den Massen (M) zugesetzt werden. Zur Erzeugung der jeweils gewünschten Eigenschaftsprofile, sowohl der unvernetzten Polymerabmischungen (M) als auch der ausgehärteten Massen (M), sind derartige Zusätze bevorzugt.
- Für die Polymerabmischungen (M) existieren zahllose verschiedene Anwendungen im Bereich der Kleb-, Dicht- und Fugendichtstoffe, Oberflächenbeschichtungen sowie auch bei der Herstellung von Formteilen. Auf Grund ihrer verbesserten Reißfestigkeit sind die Massen (M) besonders für Klebstoffanwendungen geeignet. Der Einsatz der Prepolymere (A) und Polymerabmischungen (M) in Klebstoffen ist daher bevorzugt. Dabei sind sie für zahllose unterschiedliche Untergründe, wie z.B. mineralische Untergründe, Metalle, Kunststoffe, Glas, Keramik etc., geeignet.
- Die Polymerabmischungen (M) können dabei sowohl in reiner Form als auch in Form von Lösungen oder Dispersionen zum Einsatz kommen.
- Alle vorstehenden Symbole der vorstehenden Formeln weisen ihre Bedeutungen jeweils unabhängig voneinander auf. In allen Formeln ist das Siliciumatom vierwertig.
- Soweit nicht anders angegeben sind alle Mengen- und Prozentangaben auf das Gewicht bezogen, alle Drücke 0,10 MPa (abs.) und alle Temperaturen 20°C.
- Als Maß für die Reaktivitäten der Polymerabmischungen (M) – bzw. für die Reaktivitäten der nicht erfindungsgemäßen Polymerabmischungen in den Vergleichsbeispielen – sind jeweils die Hautbildungszeiten angegeben. Unter Hautbildungszeiten ist dabei diejenige Zeitspanne zu verstehen, die nach dem Ausbringen des Prepolymers an die Luft vergeht, bis die Polymeroberfläche soweit gehärtet ist, daß nach einer Berührung dieser Oberfläche mit einem Bleistift die Polymermasse nicht mehr an diesem haften bleibt.
- Beispiel 1:
- Herstellung von N-Cyclohexyl-aminomethyl-dimethoxysilan:
- 1486,5 g (15 mol) Cyclohexylamin und 600 g Cyclohexan als Lösungsmittel werden in einem 4 Liter 4-Hals-Kolben komplett vorgelegt und anschließend mit Stickstoff inertisiert. Man heizt auf eine Temperatur von 85 °C auf und tropft 773,4 g (5 mol) Chlormethyl-methyldimethoxysilan über 2 h zu (Temperatur < 95 °C) und rührt für weitere 2 Stunden bei 95 °C. Nach einer Zugabe von ca. 300 g des Silans fällt vermehrt Cyclohexylamin-hydrochlorid als Salz aus, jedoch bleibt die Suspension bis zum Dosierende gut rührbar. Die Suspension wird über Nacht stehen lassen und dann mit ca. 300 ml Cyclohexan versetzt. Im Teilvakuum wird das überschüssige Amin und das Lösungsmittel Cyclohexan bei 60 – 70 °C destillativ entfernt. Der Rückstand wird abgekühlt und mit weiteren 300 ml Cyclohexan versetzt, um das Hydrochlorid vollständig zu fällen. Die Suspension wird filtriert und das Lösungsmittel wiederum im Teilvakuum bei 60-70 °C entfernt. Der Rückstand wird destillativ gereinigt (106-108 °C bei 15 mbar). Es wird eine Ausbeute von 761 g, d.h. 70% der Theorie erreicht, bei einer Produktreinheit von ca. 99,5 %.
- Beispiel 2:
- Herstellung von Methoxymethyltrimethoxysilan (MeO-TMO):
- Zu 315 ml Methanol werden unter leichter Rührung 68 g (1,26 mol) Natriummethanolat zugegeben. Nachdem sich das Natriummethanolat bei 65 °C vollständig gelöst hat, werden 205 g (1,2 mol) Chlormethyltrimethoxysilan innerhalb von 2 h bei einer Temperatur von 45 – 50 °C zugetropft. Bei der leicht exothermen Neutralisation fällt NaCl aus. Danach wird unter langsamen Abkühlen auf 25 °C 1 Stunde nachgerührt. NaCl wird über eine Fritte por. 3 filtriert und mit wenig Methanol nachgewaschen.
- Im Teilvakuum wird das Lösungsmittel Methanol bei 60 °C entfernt. Der Rückstand wird destillativ gereinigt (78-93 °C bei 90 mbar). Es wird eine Ausbeute von 140 g, d.h. 70 % der Theorie erreicht.
- Beispiel 3:
- Herstellung von Trimethoxysilylmethylcarbaminsäuremethylester (C-TMO):
- 61,3 g (7,56 mol) extra fein gemahlenes Kaliumisocyanat werden in einem l Liter 4-Hals-Kolben eingewogen. Anschließend werden 404 g (0,51 l, 12,6 mol) Methanol, 184,0 g (0,196 l) Dimethylformamid sowie 100,7 g (0,59 mol) Chlormethyltrimethoxysilan eingefüllt. Unter Rühren wurde die Reaktionsmischung zum Sieden erhitzt und insgesamt 10 h unter Rückfluß gehalten, wobei die Siedetemperatur von 100 °C auf 128 °C ansteigt und dann stabil bleibt. Nach dem Abkühlen auf Raumtemperatur wird das gebildete Kaliumchlorid über eine Nutsche abgetrennt und der Filterkuchen mit 1,1 l Methanol gewaschen. Im Rotationsverdampfer werden die Lösungsmittel Methanol und Dimethylformamid entfernt. Die verbliebenen Mengen an Kaliumchlorid werden abgetrennt. Die Rohlösung wird destillativ gereinigt (Kopftemperatur 79-85 °C bei 3 mbar). Insgesamt konnten 60,4 g (53 % d. Th. [114 g]) C-TMO erhalten werden.
- Beispiel 4:
- Herstellung eines Prepolymers (A):
- In einem 250 ml Reaktionsgefäß mit Rühr-, Kühl und Heizmöglichkeiten werden 152 g (16 mmol) eines Polypropylenglykols mit einem mittleren Molekulargewicht von 9500 g/mol (Acclaim 12200 der Fa. Bayer) vorgelegt und 30 Minuten bei 80 °C im Vakuum entwässert. Anschließend wird die Heizung entfernt und unter Stickstoff 2,16 g (24 mmol) 1,4-Butandiol, 12,43 g (56 mmol) Isophorondiisocyanat und 80 mg Dibutylzinndilaurat (entspricht einem Zinngehalt von 100ppm) zugegeben. Es wird für 60 Minuten bei 80 °C gerührt. Das erhaltene NCO-terminierte Polyurethanprepolymer wird danach auf 75 °C abgekühlt und mit 11,13 g (51,2 mmol) N-Cyclohexylaminomethyldimethoxymethylsilan versetzt und 60 min bei 80 °C gerührt. In der resultierenden Prepolymermischung lassen sich IR-spektroskopisch keine Isocyanatgruppen mehr nachweisen. Man erhält ein leicht trübes Prepolymer, daß sich bei 20 °C mit einer Viskosität von 370 Pas problemlos gießen und weiterverarbeiten läßt.
- Herstellung von Abmischungen mit Prepolymer (A):
- Allgemeine Vorschrift (Die konkreten Mengenangaben für die einzelnen Komponenten können der Tabelle 1 entnommen werden. Sind einzelnen Komponenten dabei nicht vorhanden, wird auf die jeweiligen Einarbeitungsschritte verzichtet):
Zu dem oben beschriebenen Prepolymer wird Carbamatomethyltrimethoxysilan (C-TMO – hergestellt nach Beispiel 3) zugegeben und 15 Sekunden lang bei 27000 rpm in einem Speedmixer (DAC 150 FV der Firma Hausschild) gemischt. Dann werden Kreide (BLR 3 der Firma Omya), HDK V 15 (Wacker Chemie GmbH, Germany) und Methoxymethyltrimethoxysilan (MeO-TMO hergestellt nach Beispiel 2) zugegeben und 2 mal 20 Sekunden bei einer Umdrehung von 30000 rpm gemischt. Schließlich wird Aminopropyltrimethoxysilan (A-TMO – Silquest A1110® der Firma Crompton) hinzugegeben und ebenfalls 20 Sekunden bei einer Umdrehung von 30000 rpm gemischt. Tabelle 1:
- Vergleichsbeispiel 1:
- Dieses Vergleichsbeispiel bezieht sich auf das Beispiel 4 Jedoch werden hier anstelle einer Mischung aus 1,4-Butandiol und einem Polypropylenglycol mit der Masse 9500 ein Polypropylenglycol mit der Masse 4000 eingesetzt. Dabei sind die Konzentrationsverhältnisse so gewählt, daß die Prepolymere aus Beispiel 4 und Vergleichsbeispiel 1 weitgehend dieselben mittleren Molekülmassen, Urethan- und Harnstoffgruppendichte sowie denselben Silangruppengehalt aufweisen:
- Herstellung eines nicht erfindungsgemäßen Prepolymers:
- In einem 250 ml Reaktionsgefäß mit Rühr-, Kühl und Heizmöglichkeiten werden 160 g (40 mmol) eines Polypropylenglykols mit einem mittleren Molekulargewicht von 4000 g/mol vorgelegt und 30 Minuten bei 80 °C im Vakuum entwässert. Anschließend wird die Heizung entfernt und unter Stickstoff 12,43 g (56 mmol) Isophorondiisocyanat und 80 mg Dibutylzinndilaurat zugegeben. Es wird für 60 Minuten bei 80 °C gerührt. Das erhaltene NCO-terminierte Polyurethanprepolymer wird danach auf 75 °C abgekühlt und mit 11,13 g (51,2 mmol) N-Cyclohexylaminomethyldimethoxymethylsilan versetzt und 60 min bei 80 °C gerührt. In der resultierenden Prepolymermischung lassen sich IR-spektroskopisch keine Isocyanatgruppen mehr nachweisen. Man erhält ein leicht trübes Prepolymer, daß sich bei 20 °C mit einer Viskosität von 155 Pas problemlos gießen und weiterverarbeiten läßt.
- Herstellung nicht erfindungsgemäßer Prepolymerabmischungen:
- Allgemeine Vorschrift (Die konkreten Mengenangaben für die einzelnen Komponenten können der Tabelle 2 entnommen werden. Sind einzelnen Komponenten dabei nicht vorhanden, wird auf die jeweiligen Einarbeitungsschritte verzichtet):
Zu dem oben beschriebenen Prepolymer wird Carbamatomethyltrimethoxysilan (C-TMO hergestellt nach Beispiel 3) zugegeben und 15 Sekunden lang bei 27000 rpm in einem Speedmixer (DAC 150 FV der Firma Hausschild) gemischt. Dann werden Kreide (BLR 3 der Firma Omya), HDK V 15 (Wacker Chemie GmbH, Germany) und Methoxymethyltrimethoxysilan (MeO-TMO hergestellt nach Beispiel 2) und 2 mal 20 Sekunden bei einer Umdrehung von 30000 rpm gemischt. Schließlich wird Aminopropyltrimethoxysilan (A-TMO Silquest A1110® der Firma Crompton) hinzugegeben und ebenfalls 20 Sekunden bei einer Umdrehung von 30000 rpm gemischt. Tabelle 2:
- Vergleichsbeispiel 2
- Dieses Vergleichsbeispiel bezieht sich auf das Beispiel 4. Jedoch wird hier auf den Einsatz von 1,4-Butandiol verzichtet und die Menge des einzusetzenden Isophorondiisocyanates wird entsprechend reduziert.
- Herstellung eines nicht erfindungsgemäßen Prepolymers:
- In einem 250 ml Reaktionsgefäß mit Rühr-, Kühl und Heizmöglichkeiten werden 152 g (16 mmol) eines Polypropylenglykols mit einem mittleren Molekulargewicht von 9500 g/mol (Acclaim® 12200 der Fa. Bayer) vorgelegt und 30 Minuten bei 80 °C im Vakuum entwässert. Anschließend wird die Heizung entfernt und unter Stickstoff 7,1 g (32 mmol) Isophorondiisocyanat und 80 mg Dibutylzinndilaurat zugegeben. Nun werden 60 Minuten bei 80 °C gerührt. Das erhaltene NCO-terminierte Polyurethanprepolymer wird danach auf 75 °C abgekühlt und mit 11,13 g (51,2 mmol) N-Cyclohexylaminomethyldimethoxymethylsilan versetzt und 60 min bei 80 °C gerührt. In der resultierenden Prepolymermischung lassen sich IR-spektroskopisch keine Isocyanatgruppen mehr nachweisen. Man erhält ein leicht trübes Prepolymer, daß sich bei 20 °C mit einer Viskosität von 77 Pas problemlos gießen und weiterverarbeiten läßt.
- Herstellung nicht erfindungsgemäßer Prepolymerabmischungen
- Allgemeine Vorschrift (Die konkreten Mengenangaben für die einzelnen Komponenten können der Tabelle 3 entnommen werden. Sind einzelnen Komponenten dabei nicht vorhanden, wird auf die jeweiligen Einarbeitungsschritte verzichtet):
Zu dem oben beschriebenen Prepolymer wird Carbamatomethyltrimethoxysilan (C-TMO hergestellt nach Beispiel 3) zugegeben und 15 Sekunden lang bei 27000 rpm in einem Speedmixer (DAC 150 FV der Firma Hausschild) gemischt. Dann werden Kreide (BLR 3 der Firma Omya), HDK V 15 (Wacker Chemie GmbH, Germany) und Methoxymethyltrimethoxysilan (MeO-TMO hergestellt nach Beispiel 2) zugegeben und 2 mal 20 Sekunden bei einer Umdrehung von 30000 rpm gemischt. Schließlich wird Aminopropyltrimethoxysilan (A-TMO Silquest A1110® der Firma Crompton) hinzugegeben und ebenfalls 20 Sekunden bei einer Umdrehung von 30000 rpm gemischt. Tabelle 3:
- Beispiel 5:
- Eigenschaften der ausgehärteten Prepolymerabmischungen
- Die fertige Prepolymerabmischung wird mit Hilfe eines Rakels in eine 2 mm hohe Teflonform verstrichen, wobei die Durchhärtegeschwindikeit ca. 2 mm am Tag beträgt. Nach zweiwöchiger Lagerung werden S1-Prüfkörper ausgestanzt, deren Zugeigenschaften nach EN ISO 527-2 an der Z010 der Firma Zwick vermessen werden. Die dabei bestimmten Eigenschaften der jeweiligen Prepolymerabmischungen sind in Tabelle 4 aufgelistet. Dabei sind die Abmischungen von
- • Beispiel 4.1, Vergleichsbeispiel 1.1 und Vergleichsbsp. 2.1;
- • Beispiel 4.2, Vergleichsbeispiel 1.2 und Vergleichsbsp. 2.2;
- • Beispiel 4.3, Vergleichsbeispiel 1.3 und Vergleichsbsp. 2.3;
- Beispiel 6:
- Mit diesem Beispiel soll gezeigt werden, daß die Prepolymere (A) auch zur Herstellung von nicht gefüllten Prepolymerabmischungen geeignet sind, die zu Massen mit einer für derartige Systeme aussergewöhnlich hohen Reißfestigkeit geeinnet sind.
- Herstellung eines Prepolymers (A):
- In einem 250 ml Reaktionsgefäß mit Rühr-, Kühl und Heizmöglichkeiten werden 152 g (16 mmol) eines Polypropylenglykols mit einem mittleren Molekulargewicht von 9500 g/mol (Acclaim® 12200 der Fa. Bayer) vorgelegt und 30 Minuten bei 80 °C im Vakuum entwässert. Anschließend wird die Heizung entfernt und unter Stickstoff 2,88g (32 mmol) 1,4-Butandiol, 14,21 g (64 mmol) Isophorondiisocyanat und 80 mg Dibutylzinndilaurat zugegeben. Es wird für 60 Minuten bei 80 °C gerührt. Das erhaltene NCO-terminierte Polyurethanprepolymer wird danach auf 75 °C abgekühlt und mit 13,91 g (64 mmol) N-Cyclohexylaminomethyldimethoxymethylsilan versetzt und 60 min bei 80 °C gerührt. In der resultierenden Prepolymermischung lassen sich IR-spektroskopisch keine Isocyanatgruppen mehr nachweisen. Man erhält ein leicht trübes Prepolymer, daß sich bei 20 °C mit einer Viskosität von 620 Pas problemlos gießen und weiterverarbeiten läßt.
- Herstellung einer Abmischung mit Prepolymer (A):
- Dieses Prepolymer wird entspechend Beispiel 4 zu einer Prepolymerabmischung verarbeitet. Dabei wird die in Tabelle 5 dargestellte Rezeptur verwendet. Tabelle 5:

- Diese besitzt eine Hautbildungszeit von 2 Stunden, eine Bruchdehung von 812 %, eine Reißfestigkeit von 2,4 MPa und ein 100 %-Modul von 0,3 MPa.
- Vergleichsbeispiel 3:
- Dieses Vergleichsbeispiel bezieht sich auf das Beispiel 6. Jedoch wird hier auf den Einsatz von 1,4-Butandiol verzichtet und die Menge des einzusetzenden Isophorondiisocyanates wird entsprechend reduziert.
- Herstellung eines nicht erfindungsgemäßen Prepolymers:
- Die Herstellung des Polymers erfolgt genauso wie in Beispiel 6 beschrieben, mit dem Unterschied, daß auf den Zusatz von Butandiol verzichtet wurde, und daß 7,1 g (32 mmol) anstelle von 14,21 g (64 mmol) Isophorondiisocyanat eingesetzt werden.
- Herstellung einer nicht erfindungsgemäßen Prepolymerabmischung
- Aus diesem Prepolymer wird entspechend Beispiel 4 zu einer Prepolymerabmischung verarbeitet. Dabei wird die Tabelle 6 dargestellte Rezeptur verwendet. Tabelle 6:
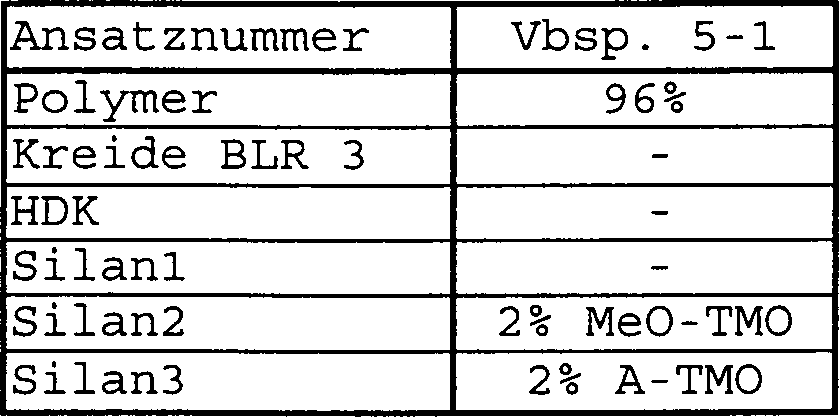
- Beispiel 7:
- Wie in Bsp. 5 beschrieben werden Probenkörper hergestellt und vermessen. Jedoch werden dabei die in Beispiel 6 und Vergleichsbeispiel 3 hergestellten Prepolymere eingesetzt. Die dabei bestimmten Eigenschaften der jeweiligen Prepolymerabmischungen sind in Tabelle 7 aufgelistet. Tabelle 7:
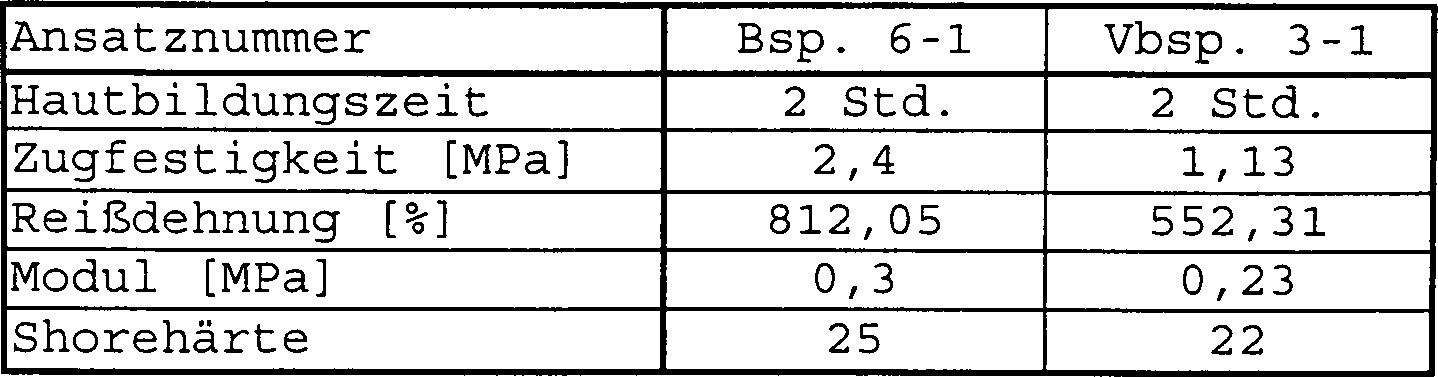
- Beispiel 8:
- Dieses Beispiel dient dazu die Leistungsfähigkeit der Prepolymere (A) noch weiter zu belegen.
- Herstellung eines Prepolymers (A):
- In einem 250 ml Reaktionsgefäß mit Rühr-, Kühl und Heizmöglichkeiten werden 152 g (16 mmol) eines Polypropylenglykols mit einem mittleren Molekulargewicht von 9500 g/mol (Acclaim® 12200 der Fa. Bayer) vorgelegt und 30 Minuten bei 80 °C im Vakuum entwässert. Anschließend wird die Heizung entfernt und bei 60 °C 2,16 g (24 mmol) 1,4-Butandiol., 12,43 g (56 mmol) Isophorondiisocyanat und 80 mg Dibutylzinndilaurat unter Stickstoff zugegeben. Es wird für 60 Minuten bei 80 °C gerührt. Das erhaltene NCO-terminierte Polyurethanprepolymer wird danach auf 60 °C abgekühlt und mit 13,91g (64 mmol) N-Cyclohexylaminomethyldimethoxymethylsilan versetzt und 60 Minuten bei 80 °C gerührt. In der resultierenden Prepolymermischung lassen sich IR-spektroskopisch keine Isocyanatgruppen mehr nachweisen. Man erhält ein leicht trübes Prepolymer, daß sich bei 20 °C mit einer Viskosität von 505 Pas problemlos gießen und weiterverarbeiten läßt.
- Herstellung einer Abmischung mit Prepolymer (A):
- Dieses Prepolymer wird entspechend Beispiel 4 zu einer Prepolymerabmischung verarbeitet. Dabei wird die in Tabelle 8 dargestellte Rezeptur verwendet. Tabelle 8:

- Wie in Bsp. 5 beschrieben werden aus dieser Abmischung Probenkörper hergestellt und vermessen. Diese besitzten eine Hautbildungszeit von 15 Minuten, eine Reißfestigkeit von 5,4 MPa, eine Bruchdehung von 667 % und ein 100 %-Modul von 1,8 MPa.
- Beispiel 9:
- Dieses Beispiel dient dazu die Leistungsfähigkeit der Prepolymere (A) noch weiter zu belegen.
- Herstellung eines Prepolymers (A):
- Das Prepolymer wird hergestellt wie in Beispiel 4 beschrieben, mit der Ausnahme, daß statt 11,13g (51,2 mmol) N-Cyclohexylaminomethyldimethoxymethylsilan 7,42g (34,1 mmol) und zusätzlich 4,64g (17,1 mmol) N-Cyclohexylaminomethyltrimethoxysilan verwendet werden.
- Herstellung einer Abmischung mit Prepolymer (A):
- Dieses Prepolymer wird entspechend Beispiel 4 zu einer Prepolymerabmischung verarbeitet. Dabei wird die in Tabelle 9 dargestellte Rezeptur verwendet. Tabelle 9:

- Wie in Bsp. 5 beschrieben werden aus dieser Abmischung Probenkörper hergestellt und vermessen. Diese besitzten eine Hautbildungszeit von 5 Minuten, eine Bruchdehung von 502 %, eine Reißfestigkeit von 4,2 MPa und ein 100 %-Modul von 1,71 MPa.
- Beispiel 10:
- Dieses Beispiel dient dazu die Leistungsfähigkeit der Prepolymere noch weiter zu belegen.
- Herstellung eines Prepolymers (A):
- Das Prepolymer wird hergestellt wie in Beispiel 8 beschrieben. Es werden entsprechend Probenkörper hergestellt, bei denen das Prepolymer vor der Abmischung mit Triveron® versetzt wird.
- Herstellung einer Abmischung mit Prepolymer (A):
- Dieses Prepolymer wird mit 5 Gew% Triveron® versetzt und entspechend Beispiel 4 zu einer Prepolymerabmischung verarbeitet. Dabei wird die in Tabelle 10 dargestellte Rezeptur verwendet. Tabelle 10:

- Wie in Bsp. 5 beschrieben werden aus dieser Abmischung Probenkörper hergestellt und vermessen. Diese besitzten eine Hautbildungszeit von 5 Minuten, eine Bruchdehung von 633 %, eine Reißfestigkeit von 5,74 MPa und ein 100 %-Modul von 2,09 MPa.

Claims (11)
- Prepolymere (A) mit Endgruppen der allgemeinen Formel [1]
- Prepolymere (A) nach Anspruch 1, welche isocyanatfrei sind.
- Prepolymere (A) nach Anspruch 1 oder 2, bei denen die alkoxysilanterminierten Polymere (A) über Endgruppen der allgemeinen Formel [2]
- Prepolymere (A) nach Anspruch 1 bis 3, bei denen die Polyole (A1) ausgewählt werden aus hydroxylfunktionellen Polyethern, Polyestern, Polyacrylaten und -methacrylaten, Polycarbonaten, Polystyrolen, Polysiloxanen, Polyamiden, Polyvinylestern, Polyvinylhydroxiden und Polyolefinen.
- Prepolymere (A) nach Anspruch 1 bis 4, bei denen die niedermolekularen Alkohole (A2) ausgewählt werden aus Glycol, 1,3-Propandiol, 1,3- Butandiol, 1,4-Butandiol, regioisomeren Penta- und Hexadiolen, Etylenglykol und Propylenglykol.
- Prepolymere (A) nach Anspruch 1 bis 5, bei denen die Di- oder Polyisocyanate (A3) ausgewählt werden aus Diisocyanatodiphenylmethan (MDI), Tolylendiisocyanat (TDI), Diisocyanatonaphthalin (NDI), Isophorondiisocyanat (IPDI), perhydriertes MDI (H-MDI), Hexamethylendiisocyanat (HDI), polymerem MDI (P-MDI), Triphenylmethantriisocanat, Isocyanurat- und Biuret-triisocyanaten.
- Prepolymere (A) nach Anspruch 1 bis 6, bei denen die Alkoxysilane (A4) ausgewählt werden aus Silanen der allgemeinen Formeln [4] und [5]wobei B1 eine OH-, SH-, NH2- oder eine HR4N-Gruppe bedeutet und R1, R2, R4 und a die bei den allgemeinen Formeln [1] und [2] gemäss Anspruch 1 und 3 angegebenen Bedeutungen aufweisen.
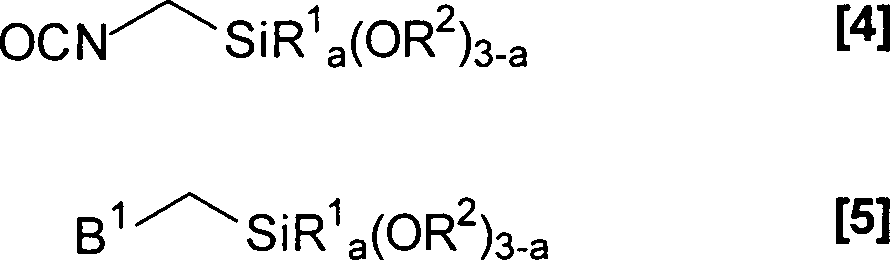
- Prepolymere (A) gemäss nach Anspruch 1 bis 7 enthaltende Massen (M).
- Massen (M) nach Anspruch 8, welche Füllstoffe (E) enthalten, die ausgewählt werden aus Calciumcarbonat, Kieselsäure und Ruß.
- Massen (M) nach Anspruch 8, welche keine Füllstoffe (E) enthalten.
- Massen (M) nach den Ansprüchen 8 bis 10, welche 0-20 Vol.-% eines organischen Lösungsmittels (F) enthalten.
Priority Applications (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DE10328844A DE10328844A1 (de) | 2003-06-26 | 2003-06-26 | Alkoxysilanterminierte Prepolymere |
| US10/595,010 US20070100111A1 (en) | 2003-06-26 | 2004-06-03 | Alkoxysilane terminated prepolymers |
| PCT/EP2004/006010 WO2005000931A1 (de) | 2003-06-26 | 2004-06-03 | Alkoxysilanterminierte prepolymere |
| CNA2004800179228A CN1813014A (zh) | 2003-06-26 | 2004-06-03 | 烷氧基硅烷封端的预聚物 |
| EP04735887A EP1636283A1 (de) | 2003-06-26 | 2004-06-03 | Alkoxysilanterminierte prepolymere |
| JP2006515826A JP2007513203A (ja) | 2003-06-26 | 2004-06-03 | アルコキシシラン−末端プレポリマー |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DE10328844A DE10328844A1 (de) | 2003-06-26 | 2003-06-26 | Alkoxysilanterminierte Prepolymere |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| DE10328844A1 true DE10328844A1 (de) | 2005-02-03 |
Family
ID=33546674
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| DE10328844A Withdrawn DE10328844A1 (de) | 2003-06-26 | 2003-06-26 | Alkoxysilanterminierte Prepolymere |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US20070100111A1 (de) |
| EP (1) | EP1636283A1 (de) |
| JP (1) | JP2007513203A (de) |
| CN (1) | CN1813014A (de) |
| DE (1) | DE10328844A1 (de) |
| WO (1) | WO2005000931A1 (de) |
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE102006059473A1 (de) * | 2006-12-14 | 2008-06-19 | Henkel Kgaa | Silylgruppen enthaltende Mischung von Prepolymeren und deren Verwendung |
| DE102007012908A1 (de) | 2007-03-19 | 2008-09-25 | Momentive Performance Materials Gmbh | Neue Polyamid-Polysiloxan-Verbindungen |
| DE102007058344A1 (de) * | 2007-12-03 | 2009-06-04 | Henkel Ag & Co. Kgaa | Härtbare Zusammensetzungen enthaltend silylierte Polyurethane |
| DE102009025944A1 (de) * | 2009-06-10 | 2010-12-23 | Kömmerling Chemische Fabrik GmbH | Feuchtehärtbarer Kleb- bzw. Dichtstoff auf Polyurethanbasis |
| WO2011069971A1 (de) * | 2009-12-09 | 2011-06-16 | Bayer Materialscience Ag | Polyurethan-prepolymere |
| CN115427510A (zh) * | 2020-04-16 | 2022-12-02 | 信越化学工业株式会社 | 室温固化性有机聚硅氧烷组合物及物品 |
Families Citing this family (42)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE102004059379A1 (de) * | 2004-12-09 | 2006-06-22 | Consortium für elektrochemische Industrie GmbH | Alkoxysilanterminierte Prepolymere |
| DE102004059378A1 (de) * | 2004-12-09 | 2006-06-22 | Consortium für elektrochemische Industrie GmbH | Alpha-Aminomethyl-alkoxysilane mit hoher Reaktivität und verbesserter Stabilität |
| ATE491753T1 (de) † | 2006-02-16 | 2011-01-15 | Kaneka Corp | Härtbare zusammensetzung |
| DE102006036438A1 (de) | 2006-08-04 | 2008-02-14 | Fischerwerke Artur Fischer Gmbh & Co. Kg | Verwendung von Kunstharzen beim Befestigen von Schrauben und ähnlichen Verankerungsmitteln, entsprechende Verfahren und Kunstharze |
| US8153261B2 (en) | 2006-09-01 | 2012-04-10 | Momentive Performance Materials Inc. | Solid polymeric substrate having adherent resin component derived from curable silylated polyurethane composition |
| DE102006054155A1 (de) | 2006-11-16 | 2008-05-21 | Wacker Chemie Ag | Schäumbare Mischungen enthaltend alkoxysilanterminierte Prepolymere |
| US7863398B2 (en) * | 2007-03-27 | 2011-01-04 | Momentive Performance Materials Inc. | Process for making hydrolyzable silylated polymers |
| FR2916969B1 (fr) * | 2007-06-05 | 2009-10-02 | Oreal | Kit comprenant des composes x et y fonctionnalises alcoxysilane. |
| DE102008018861A1 (de) | 2008-04-15 | 2009-12-17 | Fischerwerke Gmbh & Co. Kg | Verwendung definierter Kunstharze beim Befestigen von Schrauben und ähnlichen Verankerungsmitteln, entsprechende Verfahren und Kunstharze |
| KR20110036730A (ko) * | 2008-07-28 | 2011-04-08 | 아사히 가라스 가부시키가이샤 | 점착체, 점착 시트 및 그 용도 |
| DE102008043218A1 (de) | 2008-09-24 | 2010-04-01 | Evonik Goldschmidt Gmbh | Polymere Werkstoffe sowie daraus bestehende Kleber- und Beschichtungsmittel auf Basis multialkoxysilylfunktioneller Präpolymerer |
| DE102009022628A1 (de) | 2008-12-05 | 2010-06-10 | Evonik Goldschmidt Gmbh | Verfahren zur Modifizierung von Oberflächen |
| DE102009001489A1 (de) * | 2009-03-11 | 2010-09-16 | Wacker Chemie Ag | Verfahren zur kontinuierlichen Herstellung von silanterminierten Prepolymeren |
| DE102009022631A1 (de) | 2009-05-25 | 2010-12-16 | Evonik Goldschmidt Gmbh | Härtbare Silylgruppen enthaltende Zusammensetzungen und deren Verwendung |
| EP2267052A1 (de) | 2009-05-27 | 2010-12-29 | Sika Technology AG | Feuchtigkeitshärtende Zusammensetzung mit verbesserter Anfangsfestigkeit |
| EP2267051A1 (de) * | 2009-05-27 | 2010-12-29 | Sika Technology AG | Silanfunktionelle Polyester in feuchtigkeitshärtenden Zusammensetzungen auf Basis silanfunktioneller Polymere |
| DE102009029200A1 (de) * | 2009-09-04 | 2011-03-17 | Wacker Chemie Ag | Isocyanatfreie silanvernetzende Zusammensetzungen |
| DE102009046190A1 (de) * | 2009-10-30 | 2011-05-05 | Henkel Ag & Co. Kgaa | Kaschierklebstoff mit Silanvernetzung |
| DE102009057600A1 (de) * | 2009-12-09 | 2011-06-16 | Bayer Materialscience Ag | Polyurethan-Prepolymere |
| DE102009057597A1 (de) * | 2009-12-09 | 2011-06-16 | Bayer Materialscience Ag | Polyrethan-Prepolymere |
| DE102010000881A1 (de) * | 2010-01-14 | 2011-07-21 | Henkel AG & Co. KGaA, 40589 | 1K- Kaschierklebstoff mit Silanvernetzung |
| DE102010030096A1 (de) | 2010-06-15 | 2011-12-15 | Wacker Chemie Ag | Silanvernetzende Zusammensetzungen |
| EP2586784B8 (de) | 2010-06-22 | 2016-01-13 | Kaneka Corporation | Verfahren zur herstellung von alkoxyhydrosilan |
| WO2012020560A1 (ja) | 2010-08-10 | 2012-02-16 | 株式会社カネカ | 硬化性組成物 |
| JP2012214755A (ja) * | 2011-03-31 | 2012-11-08 | Kaneka Corp | 硬化性組成物 |
| JP2012233040A (ja) * | 2011-04-28 | 2012-11-29 | Shin-Etsu Chemical Co Ltd | 室温硬化性オルガノポリシロキサン組成物 |
| DE102011087603A1 (de) | 2011-12-01 | 2013-06-06 | Wacker Chemie Ag | Vernetzbare Massen auf Basis von organyloxysilanterminierten Polyurethanen |
| DE102011087604A1 (de) | 2011-12-01 | 2013-06-06 | Wacker Chemie Ag | Vernetzbare Massen auf Basis von organyloxysilanterminierten Polyurethanen |
| DE102012201734A1 (de) | 2012-02-06 | 2013-08-08 | Wacker Chemie Ag | Massen auf Basis von organyloxysilanterminierten Polymeren |
| CN103351461B (zh) * | 2013-06-26 | 2015-11-11 | 佛山市顺德区德美瓦克有机硅有限公司 | 一种封闭型异氰酸酯改性聚醚有机硅及其制备方法 |
| US20160326344A1 (en) | 2013-12-26 | 2016-11-10 | Kaneka Corporation | Curable composition and cured product thereof |
| EP2952533A1 (de) * | 2014-06-04 | 2015-12-09 | Sika Technology AG | Zinn- und Phthalat-freier Dichtstoff auf Basis von silanterminierten Polymeren |
| FR3027903B1 (fr) * | 2014-10-29 | 2016-11-25 | Oreal | Polymere a groupes alcoxysilane et utilisation en cosmetique |
| RU2020124123A (ru) * | 2017-12-22 | 2022-01-25 | ХЕНКЕЛЬ АйПи ЭНД ХОЛДИНГ ГМБХ | Полиуретановый полимер с концевыми силановыми группами для поперечной сшивки для адгезива с высокой прочностью на разрыв |
| EP3505548A1 (de) * | 2017-12-28 | 2019-07-03 | Covestro Deutschland AG | Alkoxysilanmodifizierte polyharnstoffverbindungen auf basis einer mischung von dialkoxy- und trialkoxysilanen |
| JP7513016B2 (ja) * | 2019-03-18 | 2024-07-09 | 信越化学工業株式会社 | 室温硬化性樹脂組成物、コーティング剤、接着剤及びシーリング剤、並びに物品 |
| CN110437791B (zh) * | 2019-09-06 | 2022-07-05 | 陕西杨凌磐基新材料科技有限公司 | 用于铁路无砟轨道的单组分嵌缝防水密封胶及其制备方法 |
| JP7809058B2 (ja) * | 2020-08-04 | 2026-01-30 | 信越化学工業株式会社 | 鎖長延長剤とそれを用いた室温硬化性樹脂組成物、コーティング剤、接着剤及びシーリング剤、並びに物品 |
| CA3197281A1 (en) | 2020-11-04 | 2022-05-12 | Lingtao Yu | Improved adhesive formulations including at least one silyl modified polymer |
| JP7491258B2 (ja) * | 2021-04-15 | 2024-05-28 | 信越化学工業株式会社 | 精製(オルガノオキシメチル)オルガノオキシシランの製造方法 |
| US20240043601A1 (en) * | 2022-07-27 | 2024-02-08 | The Government Of The United States Of America, As Represented By The Secretary Of The Navy | Cross-linked organosilicon networks that degrade with fluoride salts |
| CN119552347B (zh) * | 2024-11-19 | 2025-07-22 | 江苏瑞洋安泰新材料科技有限公司 | 一种高活性湿固化含有机硅聚合物组分及其制备工艺 |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE19849817A1 (de) * | 1998-10-29 | 2000-05-04 | Bayer Ag | Alkoxysilan-Endgruppen aufweisende Polyurethanprepolymere, ein Verfahren zu ihrer Herstellung sowie ihre Verwendung zur Herstellung von Dichtstoffen |
| WO2002066532A1 (de) * | 2001-02-20 | 2002-08-29 | Consortium für elektrochemische Industrie GmbH | Isocyanatfrie schäumbare mischungen mit hoher härtungsgeschwindigkeit |
Family Cites Families (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3971751A (en) * | 1975-06-09 | 1976-07-27 | Kanegafuchi Kagaku Kogyo Kabushiki Kaisha | Vulcanizable silylether terminated polymer |
| US4567228A (en) * | 1984-05-21 | 1986-01-28 | Ppg Industries, Inc. | Aqueous dispersion, internally silylated and dispersed polyurethane resins, and surfaces containing same |
| DE3629237A1 (de) * | 1986-08-28 | 1988-03-03 | Henkel Kgaa | Alkoxysilanterminierte, feuchtigkeitshaertende polyurethane sowie ihre verwendung fuer klebe- und dichtungsmassen |
| DE3636974A1 (de) * | 1986-10-30 | 1988-05-05 | Bayer Ag | Poly-(ether-urethan-harnstoff)polyadditionsprodukte, ihre herstellung, abmischung enthaltend diese sowie ihre verwendung als abformmassen |
| DE3827464A1 (de) * | 1988-08-12 | 1990-02-22 | Henkel Kgaa | Alkoxysilanterminierte, feuchtigkeitsvernetzende schmelzkleber sowie ihre verwendung als klebe- und dichtmassen |
| US5068304A (en) * | 1988-12-09 | 1991-11-26 | Asahi Glass Company, Ltd. | Moisture-curable resin composition |
| US5254657A (en) * | 1991-05-30 | 1993-10-19 | Shin-Etsu Chemical Co., Ltd. | RTV silicone rubber compositions and cured products thereof |
| EP0918062B1 (de) * | 1997-04-21 | 2004-02-18 | Asahi Glass Company Ltd. | Bei raumtemperatur härtende zusammensetzungen |
| US6319311B1 (en) * | 1998-04-24 | 2001-11-20 | Osi Specialties, Inc. | Powder coatings employing silyl carbamates |
| ATE394437T1 (de) * | 1998-12-11 | 2008-05-15 | Henkel Kgaa | Verwendung von dispersionen silylterminierter polymerer als dichtmassen |
| JP4099451B2 (ja) * | 2001-08-28 | 2008-06-11 | ワッカー ケミー アクチエンゲゼルシャフト | アルコキシシラン末端のポリマーを含有する速硬性1成分混合物 |
| DE10204523A1 (de) * | 2002-02-05 | 2003-08-07 | Bayer Ag | Alkoxysilan- und OH-Endgruppen aufweisende Polyurethanprepolymere mit erniedrigter Funktionalität, ein Verfahren zu ihrer Herstellung sowie ihre Verwendung |
-
2003
- 2003-06-26 DE DE10328844A patent/DE10328844A1/de not_active Withdrawn
-
2004
- 2004-06-03 JP JP2006515826A patent/JP2007513203A/ja active Pending
- 2004-06-03 EP EP04735887A patent/EP1636283A1/de not_active Withdrawn
- 2004-06-03 WO PCT/EP2004/006010 patent/WO2005000931A1/de not_active Ceased
- 2004-06-03 CN CNA2004800179228A patent/CN1813014A/zh active Pending
- 2004-06-03 US US10/595,010 patent/US20070100111A1/en not_active Abandoned
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE19849817A1 (de) * | 1998-10-29 | 2000-05-04 | Bayer Ag | Alkoxysilan-Endgruppen aufweisende Polyurethanprepolymere, ein Verfahren zu ihrer Herstellung sowie ihre Verwendung zur Herstellung von Dichtstoffen |
| WO2002066532A1 (de) * | 2001-02-20 | 2002-08-29 | Consortium für elektrochemische Industrie GmbH | Isocyanatfrie schäumbare mischungen mit hoher härtungsgeschwindigkeit |
Cited By (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE102006059473A1 (de) * | 2006-12-14 | 2008-06-19 | Henkel Kgaa | Silylgruppen enthaltende Mischung von Prepolymeren und deren Verwendung |
| DE102007012908A1 (de) | 2007-03-19 | 2008-09-25 | Momentive Performance Materials Gmbh | Neue Polyamid-Polysiloxan-Verbindungen |
| DE102007058344A1 (de) * | 2007-12-03 | 2009-06-04 | Henkel Ag & Co. Kgaa | Härtbare Zusammensetzungen enthaltend silylierte Polyurethane |
| DE102009025944A1 (de) * | 2009-06-10 | 2010-12-23 | Kömmerling Chemische Fabrik GmbH | Feuchtehärtbarer Kleb- bzw. Dichtstoff auf Polyurethanbasis |
| EP2270060A1 (de) | 2009-06-10 | 2011-01-05 | Kömmerling Chemische Fabrik GmbH & Co. | Feuchtehärtbarer Polyurethanklebstoff |
| WO2011069971A1 (de) * | 2009-12-09 | 2011-06-16 | Bayer Materialscience Ag | Polyurethan-prepolymere |
| CN115427510A (zh) * | 2020-04-16 | 2022-12-02 | 信越化学工业株式会社 | 室温固化性有机聚硅氧烷组合物及物品 |
| CN115427510B (zh) * | 2020-04-16 | 2023-11-28 | 信越化学工业株式会社 | 室温固化性有机聚硅氧烷组合物及物品 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP1636283A1 (de) | 2006-03-22 |
| WO2005000931A1 (de) | 2005-01-06 |
| US20070100111A1 (en) | 2007-05-03 |
| CN1813014A (zh) | 2006-08-02 |
| JP2007513203A (ja) | 2007-05-24 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| DE10328844A1 (de) | Alkoxysilanterminierte Prepolymere | |
| EP1641854B1 (de) | Alkoxysilanterminierte prepolymere | |
| EP1421129B1 (de) | Einkomponentige alkoxysilanterminierte polymere enthaltende schnell härtende abmischungen | |
| EP1465936B1 (de) | Aloxysilanterminierte polymere enthaltende vernetzbare polymerabmischungen | |
| EP1529071B1 (de) | Polymermassen auf basis alkoxysilanterminierter polymere mit regulierbarer härtungsgeschwindigkeit | |
| EP1824904B1 (de) | Alkoxysilanterminierte prepolymere | |
| EP2582738B1 (de) | Silanvernetzende zusammensetzungen | |
| EP2744842B1 (de) | Vernetzbare massen auf basis von organyloxysilanterminierten polymeren | |
| EP0261409B1 (de) | Verfarhen zur herstellung von Alkoxysilanterminierten, feuchtigkeitsärtenden Polyurethanen sowie ihre Verwendung für Klebe- und Dichtungsmassen | |
| EP1619215B1 (de) | Niedrigviskose Polyurethan-Prepolymere auf Basis von 2,4'-MDI | |
| EP2473545B1 (de) | Isocyanatfreie silanvernetzende zusammensetzungen | |
| EP1124872A1 (de) | Alkoxysilan-endgruppen aufweisende polyurethanprepolymere, ein verfahren zu ihrer herstellung sowie ihre verwendung zur herstellung von dichtstoffen | |
| EP1414909A1 (de) | Alkoxyvernetzende einkomponentige feuchtigkeitshärtende massen | |
| EP1196469A2 (de) | Spezielle aminosilane enthaltende, kondensationsvernetzende polyurethanmassen, ein verfahren zu ihrer herstellung sowie ihre verwendung | |
| EP1680457A1 (de) | a-ALKOXYSILANE SOWIE IHRE ANWENDUNG IN ALKOXYSILANTERMINIERTE N PREPOLYMEREN | |
| DE10108038C1 (de) | Isocyanatfreie schäumbare Mischungen | |
| WO2025247739A1 (de) | REAKTIVE HEIßSCHMELZKLEBSTOFFZUSAMMENSETZUNGEN BASIEREND AUF ALPHA-SILAN-TERMINIERTEN ORGANISCHEN POLYMEREN |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| OP8 | Request for examination as to paragraph 44 patent law | ||
| 8127 | New person/name/address of the applicant |
Owner name: WACKER CHEMIE AG, 81737 MUENCHEN, DE |
|
| 8139 | Disposal/non-payment of the annual fee |