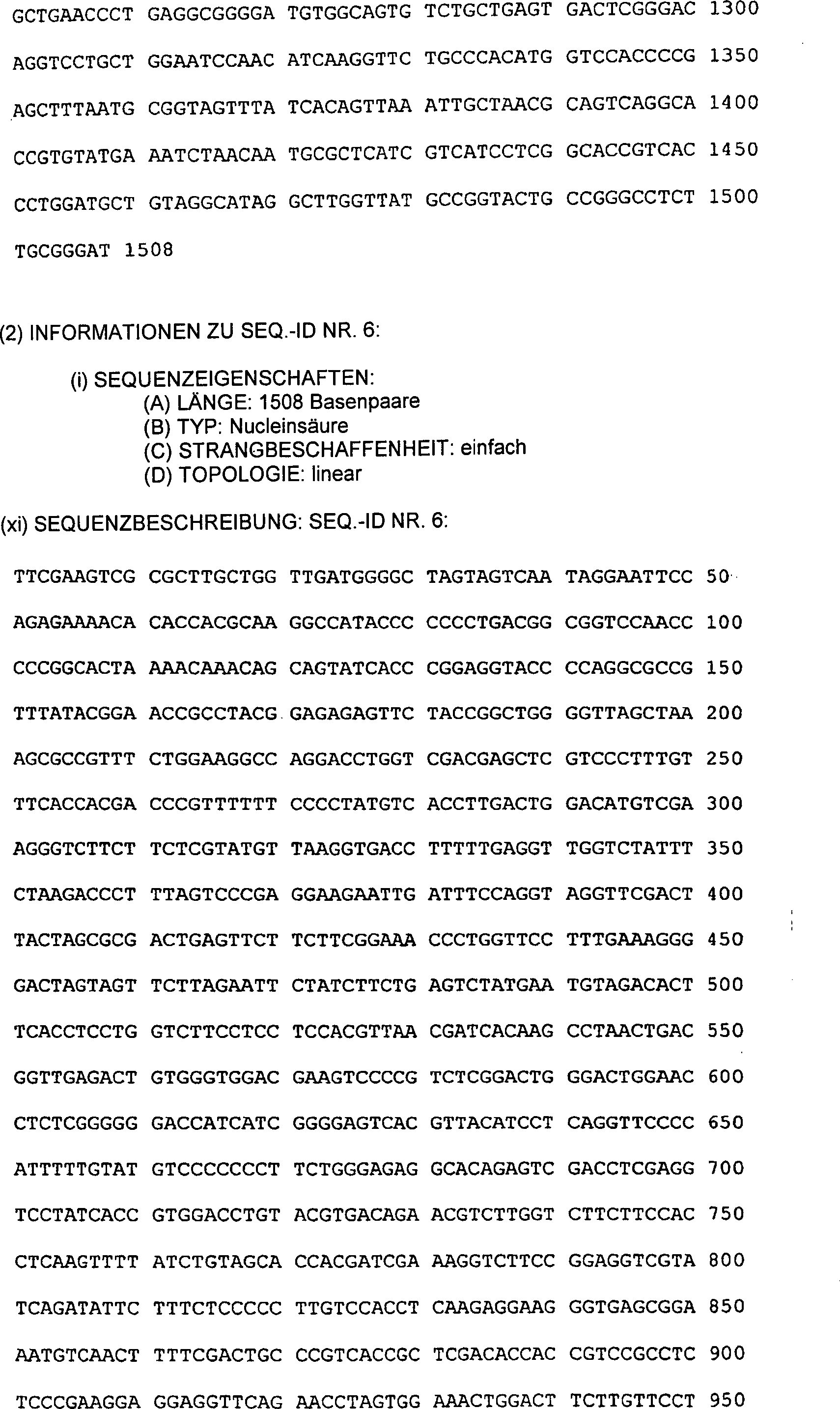
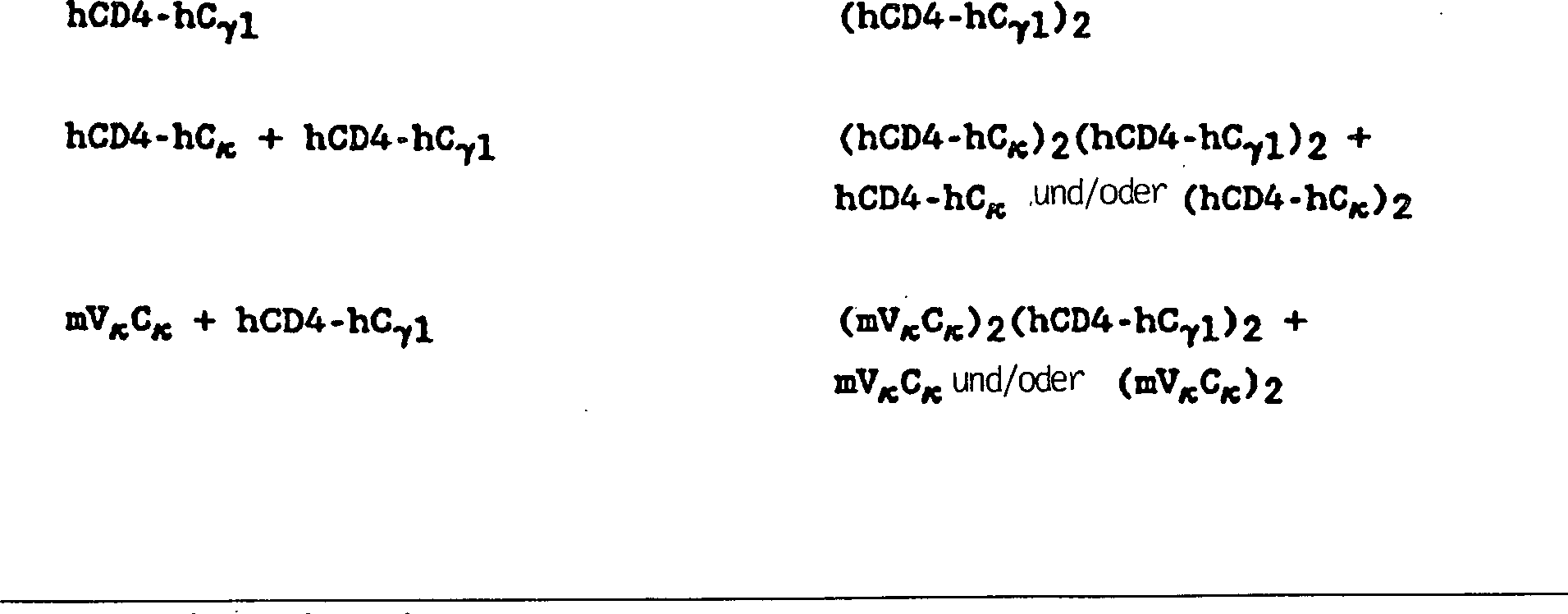-
Hintergrund
der Erfindung
-
Diese
Anmeldung betrifft Zusammensetzungen für antivirale oder immunmodulatorische
Therapieverfahren. Im Besonderen betrifft sie Zusammensetzungen,
die bei der Behandlung von Infektionen mit dem „Human Immunodeficiency Virus" (HIV) nützlich sind.
-
Die
immunologische Hauptanomalie, die aus einer Infektion mit HIV resultiert,
ist die progressive Verarmung und funktionelle Schädigung der
das CD4-Zelloberflächen-Glykoprotein exprimierenden
T-Lymphozyten (H. Lane et al., Ann. Rev. Immunol. 3, 477 [1985]).
CD4 ist ein nicht polymorphes Glykoprotein mit einer Homologie mit
der Immunglobulingenüberfamilie
(P. Maddon et al., Cell 42, 93 [1985]). Zusammen mit dem CD8-Oberflächen-Antigen
definiert CD4 zwei verschiedene Untergruppen reifer peripherer T-Zellen
(E. Reinherz et al., Cell 19, 821 [1980]), die sich jeweils durch
ihre Fähigkeit,
mit nominellen Antigen-Targets wechselzuwirken, im Kontext der Klasse-I-
bzw. Klasse-II-Haupthistokompatibilitätskomplex-(MHC-) Antigene unterscheiden
(S. Swain, Proc. Natl. Acad. Sci. 78, 7101 [1981]; E. Engleman et
al., J. Immunol. 127, 2124 [1981]; H. Spitz et al., J. Immunol.
129; 1563 [1982]; W. Biddison et al., J. Exp. Med. 156, 1065 [1982],
und D. Wilde et al., J. Immunol. 131, 2178 [1983]). Größtenteils
weisen CD4-T-Zellen den Helfer/Induktor-T-Zellen-Phänotyp auf
(E. Reinherz, s.o.), obwohl als cytotoxische/Suppressor-T-Zellen
identifizierte CD4-T-Zellen ebenso identifiziert wurden (Y. Thomas
et al., J. Exp. Med. 154, 459 [1981]; S. Meuer et al., Proc. Natl.
Acad. Sci. USA 79, 4395 [1982], und A. Krensky et al., Proc. Natl.
Acad. Sci. USA 79, 2365 [1982]). Der Verlust von CD4-Helfer/Induktor-T-Zellfunktionen
liegt wahrscheinlich den tiefgreifenden Defekten der zellulären und
humoralen Immunität
zugrunde, die zu den opportunistischen Infektionen und Malignomen
führen,
die für
das Erworbene Immundefektsyndrom (AIDS) charakteristisch sind (H.
Lane, s.o.).
-
Studien über eine
HIV-I-Infektion fraktionierter CD4- und CD8-T-Zellen von normalen
Spendern und AIDS-Patienten haben gezeigt, dass die Verarmung der
CD4-T-Zellen das Resultat der Fähigkeit
von HIV-I ist, diese T-Lymphozyten-Untergruppe selektiv zu infizieren,
in diesen zu replizieren und sie schließlich zu zerstören (D.
Klatzmann et al., Science 225, 59 [1984]). Auf die Möglichkeit,
dass CD4 selbst eine essentielle Komponente des zellulären Rezeptors
für HIV-I
ist, wurde erstmals durch die Beobachtung, dass monoklonale, gegen
CD4 gerichtete Antikörper
eine HIV-I-Infektion und Synzytiainduktion blockieren, hingewiesen
(A. Dalgleish et al., Nature 312, 767 [London] [1984]; J. McDougal
et al., J. Immunol. 135, 3151 [1985]). Diese Hypothese wurde durch
die Demonstration der Bildung eines Molekularkomplexes zwischen
CD4 und gp120, dem Haupt-Hüllglykoprotein
von HIV-I, bestätigt
(J. McDougal et al., Science 231, 382 [1986]) sowie durch die Entdeckung,
dass HIV-I-Tropismus nach der stabilen Expression einer CD4-cDNA
auf normale, nichtpermissive menschliche Zellen übertragen werden kann (P. Maddon
et al., Cell 47, 333 [1986]). Weiters scheinen die neurotropen Eigenschaften
von HIV-I, die sich im häufigen
Auftreten von Dysfunktionen des Zentralnervensystems bei HIV-I-infizierten
Individuen widerspiegeln (W. Snider et al., Ann. Neurol. 14, 403
[1983]), und die Fähigkeit, HIV-I
im Gewebe des Gehirns sowie in der Zerebrospinalflüssigkeit
von AIDS-Patienten nachzuweisen (G. Shaw et al., Science 227, 177
[1985]; L. Epstein, AIDS Res. 1, 447 [1985]; S. Koenig, Science
233, 1089 [1986]; D. Ho et al., N. Engl. J. Med. 313, 1498 [1985];
J. Levy et al., Lancet II, 586 [1985]), ihre Erklärung in der
Expression von CD4 in Neuronen-, Glia- und Monozyten-/Makrophagen-Zellen
zu haben (P. Maddon, Cell 47, 444 [1986]; I. Funke et al., J. Exp.
Med. 165, 1230 [1986]; B. Tourvieille et al., Science 234, 610 [1986]).
-
Zusätzlich zur
Bestimmung der Anfälligkeit
für eine
HIV-I-Infektion scheint das Auftreten von zytopathischen Wirkungen
im infizierten Wirt CD4 zu involvieren. Von Antikörpern für CD4 wurde
herausgefunden, dass sie in vitro die Fusion uninfizierter CD4-T-Zellen
mit HIV-I-infizierten Zellen hemmten; weiters sterben die großen vielkernigen
Zellen, die durch dieses Vorkommnis entstehen, kurz nach ihrer Entstehung,
was zu einer Verarmung der Population von CD4-Zellen führt (J.
Lifson et al., Science 232, 1123 [1986]). Die Bildung von Synzytien
erfordert ebenso eine gp120-Expression
und kann durch Co-Kultivieren von CD4-positiven Zelllinien mit Zelllinien,
die das HIV-I-env-Gen in Abwesenheit anderer viraler struktureller
oder regulatorischer Proteine exprimieren, ausgelöst werden
(J. Sodroski et al., Nature 322, 470 [1986]; J. Lifson et al., Nature
323, 725 [1986]). Beim Vermitteln zwischen der initialen Infektion
durch HIV-I und einem schlussendlichen Zelltod stellt die Wechselwirkung
zwischen gp120 und CD4 einen von mehreren kritischen Eintrittspunkten
im viralen Lebenszyklus dar, der sich für therapeutisches Einschreiten
eignet (H. Mitsuya et al., Nature 325, 773 [1987]).
-
Die
bekannte Sequenz des CD4-Vorläufers
prognostiziert ein hydrophobes Signalpeptid, eine extrazelluläre Region
von etwa 370 Aminosäuren,
einen hochgradig hydrophoben Abschnitt mit einer signifikanten Identität mit der
membrandurchdringenden Domäne
der Klasse-II-MHC-β-Kette
sowie eine stark geladene intrazelluläre Sequenz von 40 Resten (P.
Madden, Cell 42, 93 [1985]). Die extrazelluläre Domäne von CD4 besteht aus vier
zusammenhängenden
Regionen, die jede eine Aminosäuren-
und strukturelle Ähnlichkeit
mit den variablen und den Verbindungs-(V-J-) Domänen der Immunglobulin-Leichtketten
aufweisen sowie aus verwandten Regionen in anderen Mitgliedern der
Immunglobulingenüberfamilie
(eine Unterklasse, die hierin durch den Begriff "Adhesone" definiert wird). Diese strukturell ähnlichen
Regionen von CD4 werden die V1-, V2-, V3- und V4-Domänen genannt
(in 3 mit 1-4 nummeriert).
-
Eine
erfolgreiche Strategie in der Entwicklung von Arzneimitteln für die Behandlung
vieler rezeptorvermittelter Anomalien war die Identifikation von
Antagonisten, welche die Bindung des natürlichen Liganden blockieren.
Da das CD4-Adheson normalerweise an die Erkennungsstellen der HIV-Hülle bindet,
scheint es ein Kandidat für
das therapeutische Maskieren dieser HIV-Stellen zu sein, wodurch
die virale Infektiosität
blockiert wird. CD4 voller Länge
und andere Adhesone sind jedoch Zellmembranproteine, die in der
Lipiddoppelschicht der Zellen verankert sind. Die Gegenwart von
Membrankomponenten ist vom Standpunkt der Herstellung und der Reinigung
unerwünscht.
Zusätzlich
wäre es
wünschenswert,
Adhesone in einer Form zu produzieren, die in der Zirkulation stabiler
ist, da diese normalerweise nur auf Zelloberflächen vorhanden sind. Weiters
kann lösliches
CD4-Adheson, sogar in trunkierter Form, (im Allgemeinen als CD4T
bekannt) als Therapeutikum nicht optimal wirksam sein, da es eine
relativ kurze biologische Halbwertszeit aufweist, an HIV nicht besser
als Zell oberflächen-CD4
bindet, die Plazentabarriere oder andere biologische Barrieren nicht überschreiten
kann und da es die HIV-Erkennungsstellen lediglich maskiert, ohne
selbst eine Funktion zum Töten
von infizierten Zellen oder zum Töten von Viren in sich zu tragen.
-
Dementsprechend
ist es ein Ziel dieser Erfindung, lösliche, sekretierte Adhesone
zu produzieren. Ein weiteres Ziel ist es, CD4-Derivate herzustellen,
die bei der Behandlung von AIDS und verwandten Erkrankungen nützlich sind
und im Wesentlichen vom extrem Ausmaß der genetischen Variation,
die unter verschiedenen HIV-I-Isolaten
und ihren jeweiligen env-Polypeptiden beobachtet wurde, unbeeinflusst
bleiben (J. Coffin, Cell 46, 1 [1986]). Wiederum ein anderes Ziel
ist es, an andere Polypeptide fusionierte Adhesone herzustellen, um
Moleküle
mit neuen Funktionalitäten,
wie z.B. jene, die oben stehend für therapeutische Zwecke beschrieben
wurden, oder aber diagnostische Reagenzien für den In-Vitro-Test für Adhesone
oder ihre Liganden bereitzustellen. Im Besonderen ist es ein Ziel,
Moleküle
herzustellen, um Toxine oder Effektormoleküle (z.B. die Fc-Domäne eines
Immunglobulins) zu Zellen zu dirigieren, die Rezeptoren für die Adhesone
in sich tragen, z.B. HIV-gp120 im Fall von CD4, sowie zur Verwendung
bei der Vereinfachung der Reinigung der Adhesone. Ein weiteres Ziel
ist es, stabile hochgereinigte Adheson-Präparate herzustellen.
-
Zusammenfassung
-
Gemäß der vorliegenden
Erfindung wird ein Immunglobulinschwerkettendimer, in dem die extrazelluläre Domänensequenz
eines Zelloberflächenmitglieds
der Immunglobulingenüberfamilie,
deren Mitglied der Genüberfamilie
kein Klasse-I- oder Klasse-II-Haupthistokompatibilitäts-Antigen,
ein Immunglobulin oder eine T-Zellen-Rezeptor-α-, -β-, -γ- oder -δ-Kette ist, anstelle der variablen
Region einer Immunglobulin-Schwerkette substituiert wird, wobei
das Dimer ein Polypeptid umfasst, das die extrazelluläre Domäne umfasst,
die an eine konstante Region einer Immunglobulin-Schwerkette fusioniert ist, und wobei
in diesem Dimer die extrazelluläre Domäne einen
Liganden des Mitglieds der Immunglobulingenüberfamilie bindet, zur Verwendung
in einem therapeutischen Behandlungsverfahren eines Patienten bereitgestellt.
-
In
einer bevorzugten Ausführungsform
wird ein Polypeptid, das eine gp120-bindende Domäne des CD4-Adhesons umfasst,
an seinem C-Terminus an eine konstante Domäne eines Immunglobulins fusioniert.
-
Die
hierin bereitgestellten CD4-Adheson-Varianten werden gereinigt und
in pharmakologisch akzeptablen Trägermitteln zur Verabreichung
an Patienten formuliert, die eine antivirale, neuromodulatorische
oder immunmodulatorische Therapie benötigen, insbesondere Patienten,
die mit HIV infiziert sind, sowie zur Verwendung in der Modulierung
der Zelladhäsion.
-
Kurzbeschreibung
der Zeichnungen
-
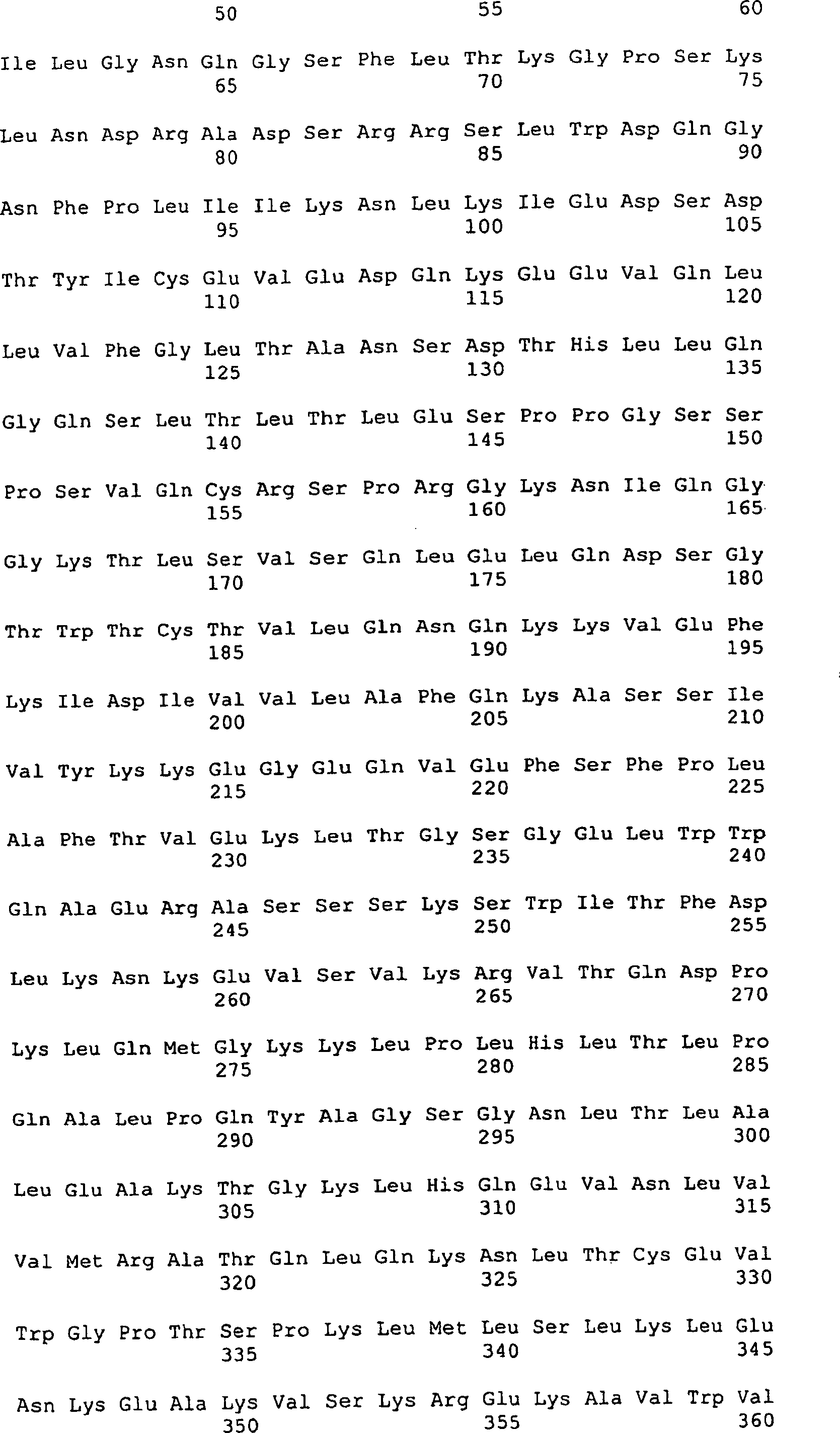
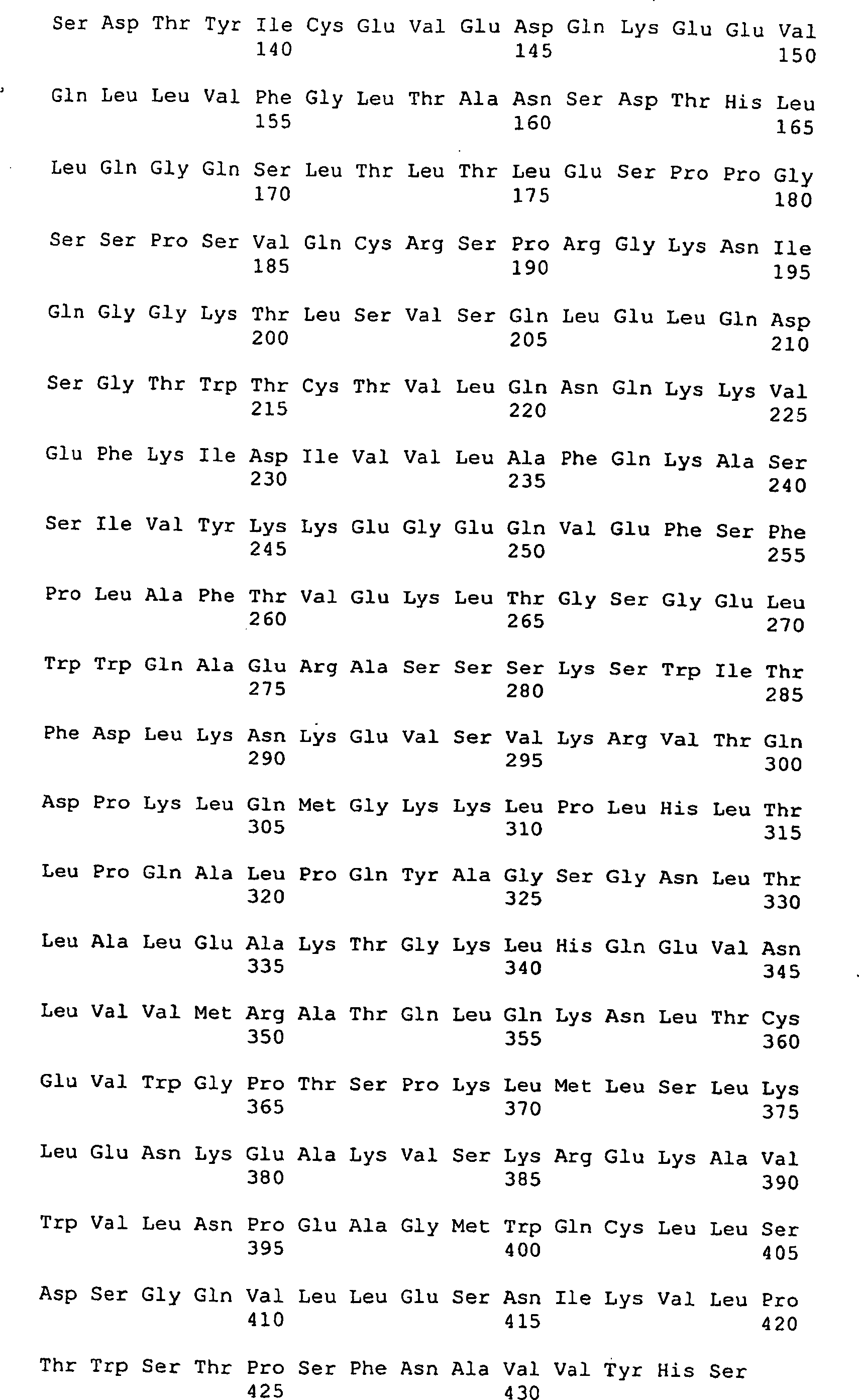
Die 1a-1c stellen
die Aminosäure-
und Nucleotidsequenz einer sekretierten Form des CD4-Adhesons dar.
Die Signalbearbeitungsstelle ist mit einem Pfeil markiert.
-
Die 2a-2c stellen
die Aminosäure-
und Nucleotidsequenz einer Fusion des Herpes-gD-Leaders und die
27 N-terminalen Reste bis zum mutmaßlichen reifen N-Terminus von CD4T
dar.
-
3 stellt
die strukturellen Elemente des nativen und löslichen CD4-Adhesons, die native
Human-IgG1-(γ1-)Schwerkette
und zwei Schwerketten-CD4-Chimären
als Beispiel dar.
-
Die 4a-4b zeigen eine Karte des gebundenen Human-IgG1-(γ1-)Kettenfragments, das bei der Herstellung
von CD4-Fusionen verwendet wird. Insertionsstellen sind mit γ1 und Fc
gekennzeichnet.
-
5 zeigt
eine Karte des menschlichen κ-Leichtkettenfragments,
das für
CD4-Fusionen an
dem von VκJκ (leicht,
variabel und bindend) und Cκ (leicht,
konstant) flankierten Pfeil nützlich
ist.
-
Ausführliche
Beschreibung
-
Adhesone
sind Zelloberflächen-Polypeptide
mit einer extrazellulären
Domäne,
welche die Sequenz eines Mitglieds der Immunglobulingenüberfamilie
aufweist, jedoch unter Ausschluss hochgradig polymorpher Mitglieder
dieser Überfamilie,
die aus der Gruppe der Klasse-I- und Klasse-II-Haupthistokompatibilitäts-Antigene,
Immunglobuline und T-Zelllen-Rezeptor-α-, -β-, -γ- oder -δ-Kette ausgewählt wurden.
Beispiele für
Adhesone umfassen CD1, CD2, CD4, CD8, CD28, die γ-, δ- und ε-Ketten von CD3, OX-2, Thy-1,
die intrazellulären
oder neuralen Zelladhäsionsmoleküle (I-CAM
oder N-CAM), das lymphozytenfunktionsassoziierte Antigen-3 (LFA-3),
das neurocytoplasmatische Protein (NCP-3), den poly-Ig-Rezeptor,
das myelinassoziierte Glykoprotein (MAG), den Hochaffinitäts-IgE-Rezeptor,
das Hauptglykoprotein von peripherem Myelin (Po), den Rezeptor des
aus Blutplättchen
gewonnenen Wachstumsfaktors, den Rezeptor des das Koloniewachstum
stimulierenden Faktors 1, den Makrophagen-Fc-Rezeptor, Fc-Gamma-Rezeptoren
und das karzinoembryonale Antigen. Bevorzugte Adhesone sind CD4,
CD8 und der Hochaffinitäts-IgE-Fc-Rezeptor.
-
Hierin
werden Aminosäuresequenzvarianten
von Adhesonen beschrieben. Aminosäuresequenzvarianten von Adhesonen
werden mit dem Gedanken an verschiedene Ziele hergestellt, dazu
gehören
unter anderem eine Erhöhung
der Affinität
des Adhesons für
seinen Bindungspartner, eine erleichterte Stabilität, Reinigung
und Herstellung des Adhesons, eine Erhöhung seiner Plasmahalbwertszeit,
eine Verbesserung der therapeutischen Wirksamkeit, wie oben im Abschnitt "Hintergrund" beschrieben, das
Einführen
zusätzlicher
Funktionalitäten
sowie eine Verringerung des Schweregrads oder des Auftretens von
Nebenwirkungen während
der therapeutischen Verwendung des Adhesons. Aminosäuresequenzvarianten
von Adhesonen sind einer oder einer Kombination der folgenden Klassen
zuzuordnen: Insertionsvarianten, Substitutionsvarianten oder Deletionsvarianten.
-
Insertionsaminosäuresequenzvarianten
sind jene, bei denen ein oder mehrere Aminosäurerest(e), der/die dem Adheson
fremd ist/sind, an einer vorweg festgelegten Stelle in das die C-
oder N-Termini umfassende Adheson eingeführt wird/werden. Solche Varianten
werden als Fusionen des Adhesons und eines anderen Polypeptids bezeichnet.
Diese anderen Polypeptide enthalten andere Sequenzen als die, die
normalerweise in dem Adheson an der insertierten Position zu finden
sind. Es werden mehrere Gruppen von Fusionen beschrieben. Immunologisch
aktive Adheson-Fusionen
umfassen ein Adheson und ein Polypeptid, das ein nichtadhesonartiges
Epitop umfasst. Das nichtadhesonartige Epitop ist ein immunologisch
kompetentes Polyeptid, d.h. ein beliebiges Polypeptid, das fähig ist,
eine Immunantwort in dem Tier auszulösen, dem die Fusion verabreicht
werden soll, oder das fähig
ist, sich durch einen Antikörper,
der gegen das nichtadhesonartige Polypeptid hervorgebracht wurde,
binden zu lassen. Typische nichtadhesonartige Epitope sind jene,
die von Allergenen, Autoimmun-Epitopen oder anderen wirksamen Immunogenen
oder Antigenen in sich getragen werden, die von bereits existierenden
Antikörpern
im Fusionsrezipienten erkannt werden, unter anderem bakterielle
Polypeptide, wie etwa trpLE, β-Galactosidase, virale
Polypeptide, wie etwa Herpes-gD-Protein, und dergleichen. Immunogene
Fusionen werden in vitro durch Vernetzung oder durch mit DNA, die
für ein
immunogenes Polypeptid kodiert, transformierter rekombinanter Zellkultur
hergestellt. Vorzugsweise ist die immunogene Fusion eine, in der
die immunogene Sequenz an das Adheson-Antigen oder ein Fragment
davon durch eine/mehrere Peptidbindung(en) bindet oder in dieses
insertiert wird. Diese Produkte bestehen daher aus einer linearen
Polypeptidkette, die Adheson-Epitope und zumindest ein dem Adheson
fremdes Epitop enthält.
Solche Fusionen sind in rekombinanten Wirtszellen oder unter Verwendung
bifunktioneller Vernetzungsmittel leicht herzustellen. Die Verwendung
eines Vernetzungsmittels, um das Adheson an das immunogene Polypeptid
zu fusionieren, ist als lineare Fusion nicht erwünscht, da die vernetzten Produkte
in einer strukturell homogenen Form nicht so leicht zu synthetisieren
sind.
-
Diese
immunogenen Insertionen sind besonders nützlich, wenn sie in einen pharmakologisch
annehmbaren Träger
formuliert und einem Subjekt verabreicht werden, um Antikörper gegen
das Adheson zu erzeugen, die wiederum in der Diagnostik oder bei
der Reinigung von Adhesonen durch per se bekannte Immunaffinitätsverfahren
nützlich
sind. Alternativ dazu werden bei der Reinigung von Adhesonen Bindungspartner für das fusionierte
nichtadhesonartige Polypeptid, z.B. Antikörper, Rezeptoren oder Liganden,
verwendet, um die Fusion aus unreinen Beimengungen zu adsorbieren,
wonach die Fusion eluiert wird und das Adheson, falls gewünscht, aus
der Fusion gewonnen wird, z.B. durch enzymatische Spaltung.
-
Andere
Fusionen, die ebenso immunologisch aktiv sein können oder auch nicht, umfassen
Fusionen der Adhesonsequenz mit einer Signalsequenz, die mit dem
Adheson heterolog ist, Fusionen transmembranmodifizierter CD4-Adhesone,
z.B. an Polypeptide mit einer verbesserten Plasmahalbwertszeit (normalerweise > etwa 20 Stunden),
wie etwa Immunglobulinketten oder Fragmente davon, sowie Fusionen
mit cytotoxischen Funktionalitäten.
Signalsequenzfusionen werden verwendet, um die Sekretion des Adhesons
schneller zu steuern. Das heterologe Signal ersetzt das native Adhesonsignal,
und wenn die resultierende Fusion erkannt, d.h. von der Wirtszelle
bearbeitet und gespalten wird, wird das Adheson sekretiert. Signale
werden basierend auf der intendierten Wirtszelle ausgewählt und
können
bakterielle Hefe-, Säugetier-
und virale Sequenzen umfassen. Das Herpes-gD-Glykoprotein-Signal
ist zur Verwendung in Säugetierexpressionssystemen
geeignet.
-
Plasmaproteine,
die eine verlängerte
Plasmahalbwertszeit haben, die länger
als die von transmembranmodifiziertem CD4 ist, umfassen Serumalbumin,
Immunglobuline, Apolipoproteine und Transferrin. Vorzugsweise ist
die Adheson-Plasma-Proteinfusion in dem Tier, in dem sie verwendet
wird, nicht signifikant immunogen, und das Plasmaprotein führt in Patienten
durch seine normale biologische Aktivität nicht zu unerwünschten
Nebenwirkungen.
-
In
einer speziellen Ausführungsform
wird die extrazelluläre
Domäne
des Adhesons mit einer konstanten Region einer Immunglobulinsequenz
verbunden. Die daraus resultierenden Produkte werden hierin als
Immunoadhesone bezeichnet. Immunglobuline und gewisse Varianten
davon sind bekannt, und es wurden viele in rekombinanter Zellkultur
hergestellt. Siehe z.B. U.S.-Patent 4.745.055;
EP 256.654 ; Faulkner et al., Nature 298,
286 (1982);
EP 120.694 ;
EP 125.023 ; Morrison, J.
Immun. 123, 793 (1979); Köhler
et al., P.N.A.S. USA 77, 2197 (1980); Raso et al., Cancer Res. 41, 2073
(1981); Morrison et al., Ann. Rev. Immunol. 2, 239 (1984); Morrison,
Science 229, 1202 (1985); Morrison et al., P.N.A.S. USA 81, 6851
(1984);
EP 255.694 ;
EP 266.663 ; und WO 88/03559.
Neuangeordnete Immunglobulinketten sind ebenso bekannt. Siehe z.B.
U.S.-Patent 4.444.878, WO 88/03565 und
EP
68.763 und die darin zitierten Verweise.
-
Normalerweise
sind die Domänen
von Adhesonen, die homolog zu Immunglobulinen und extrazellulär in ihrem
nativen Umfeld sind, C-terminal an den N-Terminus der konstanten
Region der Immunglobuline anstelle der variablen Region(en) dieser
fusioniert, wodurch zumindest funktionell aktive Gelenk-CH2- und CH3-Domänen der
konstanten Region einer Immunglobulin-Schwerkette beibehalten werden.
Dies wird normalerweise durch Konstruieren der geeigneten DNA-Sequenz
und durch deren Expression in Rekombinationszellkultur erreicht.
Immunglobuline und andere Polypeptide mit erhöhter Plasmahalbwertszeit werden
auf dieselbe Art und Weise an die extrazellulären oder ligandenbindenden
Domänen
anderer Adhesone fusioniert.
-
Die
Grenzdomänen
für die
CD4-V-artigen Regionen (V1-V4) sind etwa 100-109, etwa 175-184,
etwa 289-298 bzw. etwa 360-369 (basierend auf der Vorläufer-CD4-Aminosäuresequenz,
in der das initiierende Met-25 ist;
1a). CD4-Sequenzen,
die eine beliebige der CD4-V-Domänen
enthalten, werden an die Immunglobulinsequenz fusioniert. Vorzugsweise
wird die V1-Domäne
von CD4 oder CD4, dem die Transmembran- und zytoplasmatischen Domänen fehlen,
an ihren C-Termini an die konstante Region des Immunglobulins fusioniert.
Die genaue Stelle, an der die Fusion durchgeführt wird, ist nicht entscheidend;
die hierin erwähnten Grenzdomänen dienen
lediglich der Führung,
es können
andere Stellen, die an die V-Regionen angrenzen oder sich in ihnen
befinden, ausgewählt
werden, um die Sekretions- oder Bindungscharakteristika der CD4
zu optimieren. Die optimale Stelle wird durch Routineexperimente
bestimmt werden. Im Allgemeinen wurde herausgefunden, dass die Fusionen
intrazellulär
exprimiert werden, man fand jedoch ein großes Ausmaß an Variation beim Ausmaß der Sekretion
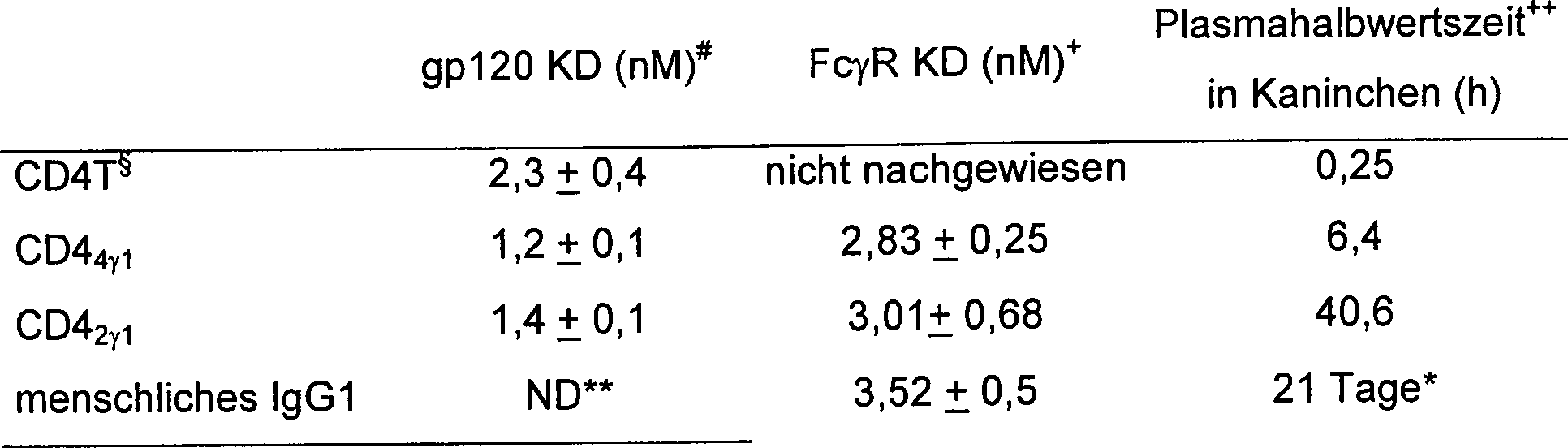
der Fusionen aus rekombinanten Wirten. Die folgende Tabelle zeigt
z.B. die verschiedenen Immunglobulinfusionen, die durch das Verfahren
dieser Erfindung erhalten wurden. In allen Beispielen für CD4- Immunadhesone wurde
das CD4-Signal verwendet, um die Sekretion aus 293-Zellen zu leiten.
Ein klein geschriebenes m zeigt einen murinen Ursprung an, während ein
klein geschriebenes h für
einen menschlichen Ursprung steht. V und C sind Abkürzungen
für variable
bzw. konstante Domänen
von Immunglobulin. Die numerischen tiefgestellten Indices zeigen
die Anzahl der eingeklammerten Einheiten an, die im kreierten Dimer
zu finden sind. Es ist klar, dass von den Ketten der Multimere angenommen
wird, dass sie auf dieselbe Art und Weise wie native Immunglobuline über Disulfidbindung
gebunden sind. Die CD4-Immunadhesone enthielten typischerweise entweder
die ersten 366 N-terminalen Reste von CD4 (CD4
4)
oder die ersten 180 N-terminalen
Reste von CD4 (CD4
2), gebunden an seinem
C-Terminus an die konstante Region (γ1) der κ-(Leicht-) Kette oder der igG1-Schwerkette. Tabelle
1
- *
ND = nicht nachgewiesen
-
Aus
dieser Tabelle geht interessanterweise hervor, dass das menschliche
CD4-Schwerketten-Immunadheson
als ein Dimer sekretiert wurde, während die analoge murine Konstruktion
nicht nachgewiesen wurde (was jedoch nicht die extrazelluläre Akkumulation
des Proteins ausschließt).
Die Fähigkeit
der hCD4-hCγ1-Transformanten,
ein Schwerkettendimer zu produzieren, kam unerwartet, da vorausgegangene Arbeiten
nahe gelegt hatten, Immunglobulin-Schwerketten würden nicht sekretiert, es sei
denn, die Wirte werden mit sowohl für Schwer- als auch für Leichtketten
kodierender Nucleinsäure
co-transformiert (Valle et al., Nature 241, 338 [1981]). Gemäß dieser
Erfindung werden CD4-IgG-Immunadheson-Chimären leicht sekretiert, worin
das CD4-Epitop in Schwerkettendimeren, Leichtkettenmonomeren oder – dimeren
und Schwer- und Leichtkettenheterotetrameren vorhanden ist, worin
das CD4-Epitop in einer an eine oder mehrere Leicht- oder Schwerketten
fusionierten Form, umfassend Heterotetramere, in denen bis zu und
einschließlich
alle vier Analoga der variablen Region von CD4 abstammen, vorhanden
ist. Wo eine variable nicht-CD4-Leicht-Schwerketten-Domäne vorhanden
ist, wird ein heterofunktioneller Antikörper bereitgestellt.
-
Geeignete
Begleit-Immunglobulin-Kombinationsstellen und Fusionspartner werden
aus IgG-1-, -2-, -3- oder -4-Subtypen, IgA, IgE, IgD oder IgM, jedoch
vorzugsweise IgG-1, erhalten.
-
Eine
bevorzugte Ausführungsform
ist eine Fusion eines N-terminalen Abschnitts von CD4, der die Bindungsstelle
für das
gp120-Hüllprotein
von HIV enthält,
an den C-terminalen
Fc-Abschnitt eines Antikörpers, der
die Effektorfunktionen des Immunglobulins G1 enthält. Von
dieser Art gibt es zwei bevorzugte Ausführungsformen; in einer ist
die gesamte konstante Region der schweren Kette an einen Abschnitt
von CD4 fusioniert; in einer anderen ist eine Sequenz, die in der
Gelenkregion unmittelbar stromauf der Papain-Spaltstelle beginnt, die
IgG-Fc chemisch definiert (Rest 216, wobei 114 als erster Rest der
konstanten Region der schweren Kette angenommen wird [Kobat et al.,
Sequences of Proteins of Immunological Interest, 4. Auflage (1987)],
oder analoge Stellen anderer Immunglobuline), an einen Abschnitt
von CD4 fusioniert. Diese Ausführungsformen
werden in den Beispielen beschrieben.
-
Im
Besonderen wird von jenen Varianten, in denen die variable Region
einer Immunglobulinkette durch eine oder mehrere immunglobulinähnliche
Domänen
eines Adhesons substituiert sind, angenommen, sie hätten in
vivo eine verbesserte Plasmahalbwertszeit. Diese Chimären sind
auf eine ähnliche
Art und Weise wie chimäre
Antikörper
aufgebaut, in denen die variable Domäne eines Antikörpers einer
Spezies für
eine variable Domäne
einer anderen Spezies substituiert wird. Siehe z.B
EP 0 125 023 ; Munro, Nature 312 (13.
Dezember 1984); Neuberger et al., Nature 312 (13. Dezember 1984);
Sharon et al., Nature 309 (24. Mai 1984); Morrison et al., Proc.
Natl. Acad. Sci. USA 81, 6851-6855 (1984); Morrison et al., Science
229, 1202-1207 (1985), und Boulianne et al., Nature 312, 643-646
(13. Dezember 1984). Die für
die immunglobulinähnliche(n)
Domäne(n) des
Adhesons kodierende DNA wird von einem Restriktionsenzym am oder
proximal zum 3'-Ende
der für
die immunglobulinähnliche(n)
Domäne(n)
kodierenden DNA sowie an einem Punkt an oder in der Nähe der für das N-Terminal-Ende
des reifen Adheson-Polypeptids kodierenden DNA (wo die Verwendung
eines anderen Leaders erwogen wird) oder an der oder proximal zu
der für
den N-terminal kodierenden Region für das Adheson (wo das native
Adhesonsignal verwendet wird) gespalten. Dieses DNA-Fragment wird
dann leicht in für
eine konstante Region einer Immunglobulin-Leicht- oder -Schwerkette
kodierende DNA insertiert und, falls notwendig, durch Deletionsmutagenese
zugeschnitten. Vorzugs weise handelt es sich dabei um ein menschliches
Immunglobulin, falls die Variante zur In-vivo-Therapie von Menschen
gedacht ist. DNA, die für
konstante Regionen einer Immunglobulin-Leicht- oder -Schwerkette
kodiert, ist bekannt oder leicht aus cDNA-Bibliotheken erhältlich oder
wird synthetisiert. Siehe z.B. Adams et al., Biochemistry 19, 2711-2719
(1980); Gough et al., Biochemistry 19, 2702-2710 (1980); Dolby et
al., P.N.A.S. USA 77, 6027-6031 (1980); Rice et al., P.N.A.S. USA 79,
7862-7865 (1982); Falkner et al., Nature 298, 286-288 (1982), und
Morrison et al., Ann. Rev. Immunol. 2, 239-256 (1984).
-
Für die chimäre(n) Kette(n)
des Immunglobulins oder Immunadhesons kodierende DNA wird zur Expression
in eine Wirtszelle transfiziert. Falls die Wirtszelle vor der Transfektion
ein Immunglobulin produziert, so muss nur mit dem an Leicht- oder
Schwerketten fusionierten Adheson transfiziert werden, um einen
Heteroantikörper
herzustellen. Die zuvor erwähnten
Immunglobuline, die einen oder mehrere Arme aufweisen, die Adhesondomänen tragen,
und einen oder mehrere Arme aufweisen, die variable Begleiterregionen
tragen, führen
zu einer dualen Spezifität
für den
Adheson-Liganden und für
ein Antigen. Sie werden durch die oben beschriebenen Rekombinationsverfahren
oder durch In-vitro-Verfahren hergestellt. In letzterem Fall werden z.B.
F(ab')2-Fragmente
einer Adheson-Fusion und ein Immunglobulin hergestellt, die F(ab')2-Fragmente werden
durch Reduktion unter milden Reduktionsbedingungen in Fab'-Fragmente konvertiert
und dann jeweils in Gegenwart des anderen unter sauren Bedingungen
gemäß per se
bekannter Verfahren reoxidiert. Siehe auch U.S.-Patent 4.444.878.
-
Zusätzlich sind
Verfahren zur Herstellung intakter Heteroantikörper aus Immunglobulinen mit
verschiedenen Spezifitäten
bekannt. Diese Verfahren werden zur In-vitro-Herstellung von heterochimären Antikörpern durch
einfaches Substituieren eines der zuvor verwendeten Immunglobuline
durch die Immunadhesonketten verwendet.
-
In
einem alternativen Verfahren zur Herstellung eines heterofunktionellen
Antikörpers
werden Wirtszellen, die eine Adheson-Immunglobulinfusion produzieren,
z.B. transfizierte Myelome, ebenso mit B-Zellen oder Hybridomen
fusioniert, die Antikörper sekretieren,
welche die gewünschte
Begleitspezifität
für ein
Antigen aufweisen. Heterobifunktionelle Antikörper werden aus dem Kulturmedium
solcher Hybridome gewonnen und sind daher etwas leichter zu produzieren
als durch herkömmliche
In-vitro-Anwendungsverfahren
(
EP 68.763 ).
-
Eine
weitere Gruppe an Fusionen sind jene, in denen ein Adheson mit einer
toxischen Substanz, z.B. einem Polpypeptid, wie etwa Ricin (umfassend
eine deglykosylierte Ricin-A-Kette), Diphtherietoxin A oder ein nichtpeptidylartiges
Zytotoxin, konjugiert ist. Wenn es sich bei dem Toxin um ein Polypeptid
handelt, ist es zweckdienlich, das Polypeptid durch herkömmliche
In-vitro-Proteinvernetzungsmittel mit dem Adheson oder seiner transmembrandeletierten
Variante zu vernetzen (für
geeignete Verfahren zur Bindung einer Ricin-A-Kette oder einer deglykosylierten
A-Kette an CD4 siehe z.B. Duncan et al., Analy. Biochem. 132, 68-73
[1983]; Thorpe et al., Cancer Res. 47, 5924 [1987], und Ghotie et
al., Cancer Res. 48, 2610 [1988]) oder aber dies durch Rekombinationssynthese
als Fusion zu tun (siehe z.B. U.S.-Patent 4.765.382). Alternativ
dazu werden in Fällen,
in denen Begleitantikörper
variable Immunglobulindomänen
von Anti-Ricin-Antikörpern
sind, solche Immunglobulin-Heteroantikörper verwendet, um, gemäß dem allgemeinen
Verfahren von Raso et al., Cancer Research 41, 2073 (1981), Ricin
an HIV-infizierte Zellen zu liefern.
-
Eine
weitere Klasse von Adhesonvarianten sind Deletionsvarianten. Deletionen
sind durch das Entfernen von einem oder mehreren Aminosäurerest(en)
aus einer Adhesonsequenz charakterisiert. Typischerweise sind die
Transmembran- und die zytoplasmatischen Domänen von Adhesonen deletiert.
Im Fall von CD4 sind zumindest die Reste 368 bis 395 (die Transmembranregion)
und normalerweise ebenso 396-433
(die zytoplasmatische Domäne)
deletiert, um sekretierte Formen dieses Adhesons zu erhalten. Ebenso
ist anzumerken, dass die Aminosäurereste
den Nummern folgen, die für
reifes CD4 gegeben werden, wie dies z.B. in den 1a-1c dargestellt
ist. CD4T-Moleküle
werden daher im Allgemeinen ungefähr in der Nähe der Reste 366-368 enden
oder an einer beliebigen anderen geeigneten Stelle, die sich N-terminal dazu befindet,
die die gp120-Bindungsfähigkeit
der CD4-Variante erhält.
-
Substitutionsvarianten
sind jene, in denen zumindest ein Rest in der Adhesonsequenz entfernt
wurde und ein anderer Rest stattdessen insertiert wurde. Der native
N-terminale Rest für
reifes CD4 ist, wie nun bekannt ist, Lysin. Die in
1 gezeigte
Sequenz mit einem N-terminalen Asparagin ist daher eine Aminosäuresequenzvariante
des nativen reifen CD4. In der unten stehenden Tabelle 2 werden
Substitutionen beschrieben, die im Allgemeinen zu einer Feinmodulation
der Eigenschaften des CD-Antigens führen. Tabelle
2
-
Substantielle
Veränderungen
der Funktion oder der immunologischen Identität werden durch Auswählen von
Substitutionen erreicht, die weniger konservativ als die in Tabelle
2 angeführten
sind, d.h. durch Auswählen
von Resten, die sich auf signifikantere Art und Weise in ihrer Wirkung
auf den Erhalt von (a) der Struktur der Polypeptidhauptkette im
Bereich der Substitution, z.B. als Faltblatt oder Helixkonformation,
(b) der Ladung oder der Hydrophobizität des Moleküls an der Targetstelle oder
(c) der Sperrigkeit der Seitenkette unterscheiden. Die Substitutionen,
von denen im Allgemeinen erwartet wird, dass sie die größten Veränderungen
der Adhesoneigenschaften hervorrufen, sind jene, in denen (a) ein
hydrophiler Rest, z.B. Seryl oder Threonyl, für (oder durch) einen hydrophoben
Rest, z.B. Leucyl, Isoleucyl, Phenylalanyl, Valyl oder Alanyl, substituiert
wird; (b) ein Cysteinyl oder Prolyl für (oder durch) einen beliebigen
anderen Rest substituiert wird; (c) ein Rest mit einer elektropositiven
Seitenkette, z.B. Lysyl, Arginyl oder Histidyl, für (oder
durch) einen elektronegativen Rest, z.B. Glutamyl oder Aspartyl,
substituiert wird oder (d) ein Rest mit einer sperrigen Seitenkette,
z.B. Phenylalanyl, für
(oder durch) einen Rest ohne Seitenkette, z.B. Glycyl, substituiert
wird.
-
Eine
bevorzugte Klasse von Substitutions- oder Deletionsvarianten sind
jene, die die Transmembranregion des Adhesons involvieren. Die Transmembranregion
des Adhesons ist eine hochgradig hydrophobe oder lipophile Domäne, die
die richtige Größe aufweist,
um die Lipiddoppelschicht der Zellmembran zu umspannen. Es wird
angenommen, sie würde
das Adehson in der Zellmembran verankern.
-
Eine
Deletion oder Substitution der Transmembrandomäne erleichtert die Gewinnung
und stellt durch die Reduktion ihrer Zell- oder Membranlipidaffinität und die
Verbesserung ihrer Wasserlöslichkeit
eine lösliche Form
des Adhesons bereit. Falls die Transmembran- und die zytoplasmatischen
Domänen
deletiert werden, wird die Einführung
von potentiell immunogenen Epitopen vermieden, entweder durch die
Exposition von andernfalls intrazellulären Polypeptiden, die vom Körper als
fremdartig erkannt werden könnten,
oder durch Einführung
heterologer Polypeptide, die potentiell immunogen sind. Ein Hauptvorteil
des transmembrandeletierten Adhesons ist die Tatsache, dass es in
das Kulturmedium rekombinanter Wirte sekretiert wird. Diese Variante
ist wasserlöslich
und besitzt keine nennenswerte Affinität zu Zellmembranlipiden, was
ihre Gewinnung aus der Rekombinationszellkultur bedeutend vereinfacht.
-
Aus
der vorhergehenden Diskussion wird klar und deutlich ersichtlich,
dass Substitutionen, Deletionen, Insertionen oder eine beliebige
Kombination dieser eingeführt
werden, um ein Endkonstrukt zu erhalten. Als allgemeine Behauptung
werden alle Varianten keine funktionelle Transmembrandomäne und vorzugsweise
keine funktionelle zytoplasmatische Sequenz aufweisen. Dies wird
im Allgemeinen durch Deletion der relevanten Domäne erreicht, obwohl adäquate Insertions-
oder Substitutionsmutagene zu diesem Zweck ebenso wirksam sein können. Die
Transmembrandomäne
wird z.B. durch eine beliebige Aminosäuresequenz, z.B. eine Zufalls-
oder Homopolynucleinsequenz von etwa 5 bis 50 Serin-, Threonin-,
Lysin-, Arginin-, Glutamin-, Asparaginsäure- und ähnlichen hydrophilen Resten,
die insgesamt ein hydrophiles Hydropathie-Diagramm aufweisen, ersetzt,
sodass sie in das Kulturmedium rekombinanter Wirte sekretiert wird.
Diese Variante sollte ebenso als Adhesonvariante angesehen werden.
-
Diese
Varianten werden normalerweise durch stellenspezifische Mutagenese
von Nucleotiden in der für
das Adheson kodierenden DNA hergestellt, wodurch für die Variante
kodierende DNA produziert wird und wonach die DNA in Rekombinationszellkultur
exprimiert wird. Variantenadhesone werden jedoch auch durch In-vitro-Synthese hergestellt.
Natürlich
dürfen
Variationen in der für
die Variantenadhesone kodierenden DNA die Sequenz nicht außerhalb
des Leserasters platzieren und erzeugen vorzugsweise keine komplementären Regionen,
die sekundäre
mRNA-Strukturen
produzieren könnten,
die für
die Expression schädlich
sind (
EP 75.444A ).
Die CD4-Varianten weisen normalerweise dieselbe gp120-Bindungsaktivität wie der
natürlich
vorkommende Prototyp auf, obwohl Varianten ebenso ausgewählt werden,
um die Merkmale des CD4-Adhesons wie oben angegeben zu modifizieren.
-
Während die
Stelle zur Einführung
einer Aminosäuresequenzvariation
im Vorhinein festgelegt ist, muss die Mutation per se nicht im Vorhinein
festgelegt sein. Um z.B. die Leistung einer Mutation an einer bestimmten
Stelle zu optimieren, kann eine Zu fallsmutagenese am Target-Codon
oder der Target-Region durchgeführt
werden, und die exprimierten Adhesonvarianten können auf die optimale Kombination
der gewünschten
Aktivitäten
gescreent werden. Verfahren zur Herstellung von Substitutionsmutationen
an im Vorhinein festgelegten Stellen in DNA mit einer bekannten
Sequenz sind wohlbekannt, z.B. M13-Primer-Mutagenese.
-
Adhesonvarianten,
die nicht fähig
sind, HIV-gp120 zu binden, sind trotzdem als Immunogene zur Produktion
von Antikörpern
gegen das Adheson oder als Immuntest-Set-Komponenten von Nutzen (markiert
als kompetitives Reagens für
den gp120-Test oder
unmarkiert als Standard für
einen Adhesontest), solange zumindest ein Adheson-Epitop aktiv bleibt.
-
Die
für Adhesone
kodierende DNA wird durch bekannte Verfahren erhalten. Siehe Williams,
Immunol. Today 8, 298-303 (1987), und die darin enthaltenen Zitate.
Im Allgemeinen werden Prakaryoten zum Klonieren von DNA-Sequenzen
von CD4-Varianten
verwendet. So sind z.B. der E.-coli-Stamm SR101 (zum Vermehren von
m13-Phagen, einem λ-resistenten
Stamm von JM 101; Messing et al., Nucl. Acids. Res. 9(2), 309-321 [1981])
und der E.-coli-K12-Stamm 294 (ATCC Nr. 31446) besonders nützlich.
Andere Mikrobenstämme,
die verwendet werden können,
umfassen E. coli B, UM101 und E.coli X1776
(ATCC Nr. 31537). Diese Beispiele dienen der Veranschaulichung und
sollen keine Einschränkung
darstellen.
-
Für die Variantenadhesone
kodierende DNA wird zur Expression in Vektoren eingeführt, die
Promotoren und Kontrollsequenzen enthalten, die von mit der beabsichtigten
Wirtszelle kompatiblen Spezies abstammen. Der Vektor trägt normalerweise,
muss dies jedoch nicht tun, eine Replikationsstelle sowie eine oder
mehrere Markersequenzen in sich, die fähig sind, in transformierten
Zellen für
eine phänotypische
Selektion zu sorgen. E. coli wird z.B. typischerweise unter Verwendung
eines Derivats von pBR322, einem von einer E.-coli-Spezies abstammenden
Plasmid, transformiert (Bolivar et al., Gene 2, 95 [1977]). pBR322
enthält
Gene für Ampicillin-
und Tetracyclinresistenz und stellt daher ein einfaches Mittel zur
Identifikation transfor mierter Zellen bereit. Das pBR322-Plasmid
oder ein anderes Mikrobenplasmid muss ebenso Promotoren und andere
Kontrollelemente, die in rekombinanten DNA-Konstruktionen häufig verwendet werden, umfassen
oder es muss modifiziert werden, um diese zu enthalten.
-
Für die Verwendung
mit prokaryotischen Wirten geeignete Promotoren umfassen veranschaulichenderweise
die β-Lactamase-
und Lactose-Promotor-Systeme (Chang et al., Nature 275, 615 [1978];
und Goeddel et al., Nature 281, 544 [1979]), alkalische Phosphatase,
das Tryptophan-(trp-) Promotor-System (Goeddel, Nucleic Acids Res.
8, 4057 [1980], und EPO Appln. Publ. Nr. 36.776) und Hybridpromotoren,
wie etwa den tac-Promotor (H. de Boer et al., Proc. Natl. Acad.
Sci. USA 80, 21-25 [1983]). Andere funktionelle bakterielle Promotoren
sind jedoch ebenso geeignet. Ihre Nucleotidsequenzen sind im Allgemeinen
bekannt, wodurch ein Fachmann sie unter Verwendung von Linkern oder
Apatoren operabel an für
die Adhesonvariante kodierende DNA ligieren kann, um jegliche erforderliche
Restriktionsstelle bereitzustellen (Siebenlist et al., Cell 20,
269 [1980]). Promotoren zur Verwendung in bakteriellen Systemen
umfassen ebenso eine Shine-Dalgarno-(S.D.-) Sequenz, die an die
für das
Antigen kodierende DNA operabel gebunden ist.
-
Zusätzlich zu
Prokaryoten sind eukaryotische Mikroben, wie z.B. Hefekulturen,
ebenso als Klonierungs- oder Expressionswirte nützlich. Saccharomyces cerevisiae
oder Gemeine Bäckerhefe
ist der am häufigsten
verwendete eukaryotische Mikroorganismus, obwohl eine Reihe anderer
Stämme
leicht erhältlich
sind. Zur Expression in Saccharomyces wird z.B. das Plasmid YRp7
(Stinchcomb et al., Nature 282, 39 [1979]; Kingsman et al., Gene
7, 141 [1979]; Tschemper et al., Gene 10, 157 [1980]) häufig verwendet.
Dieses Plasmid umfasst bereits das trp1-Gen, das einen Selektionsmarker
für einen
mutierten Hefestamm bereitstellt, dem die Fähigkeit, in Tryptophan zu wachsen,
fehlt, z.B. ATCC Nr. 44076 oder PEP4-1 (Jones, Genetics 85, 12 [977]). Die
Gegenwart der trp1-Läsion
als Merkmal des Hefewirtszellengenoms stellt dann ein wirksames
Mittel der Selektion durch Wachstum in Abwesenheit von Tryptophan
bereit.
-
Geeignete
Promotorsequenzen zur Verwendung mit Hefewirten umfassen die Promotoren
für 3-Phosphoglyceratkinase
(Hitzeman et al., J. Biol. Chem. 255, 2073 [1980]) oder andere glykolytische
Enzyme (Hess et al., J. Adv. Enzyme Reg. 7, 149 [1968]; und Holland,
Biochemistry 17, 4900 [1978]), wie z.B. Enolase, Glyceraldehyd-3-phosphatdehydrogenase,
Hexokinase, Pyruvatdecarboxylase, Phosphofructokinase, Glucose-6-phosphatisomerase,
3-Phosphoglyceratmutase, Pyruvatkinase, Triosephosphatisomerase,
Phosphoglucoseisomerase und Glucokinase.
-
Weitere
Hefepromotoren, die induzierbare Promotoren mit dem zusätzlichen
Vorteil einer von Wachstumsbedingungen kontrollierten Transkription
sind, umfassen die Promotorregionen für Alkoholdehydrogenase 2, Isocytochrom
C, saure Phosphatase, abbauende Enzyme, die mit dem Stickstoffstoffwechsel
assoziiert sind, Metallothionein, Glyceraldehyd-3-phosphatdehydrogenase
sowie Enzyme, die für
die Maltose- und
Galactoseverwertung verantwortlich sind. Geeignete Vektoren und
Promotoren für
die Verwendung in der Hefeexpression werden weiter in R. Hitzeman
et al., EP-Veröffentlichung
Nr. 73.657A, beschrieben. Hefe-Enhancer werden ebenso mit Hefepromotoren
vorteilhaft verwendet.
-
Promotoren,
um die Transkription aus Vektoren in Säugetierwirtszellen zu kontrollieren,
können
aus verschiedenen Quellen erhalten werden, z.B. den Genomen von
Viren, wie z.B.: Polyoma, Simian-Virus 40 (SV40), Adenovirus, Retroviren,
Hepatitis-B-Virus
und insbesondere Zytomegalievirus, oder aus heterologen Säugetierpromotoren,
z.B. dem β-Actin-Promotor.
Die frühen
und späten
Promotoren des SV40-Virus sind leicht als ein SV40-Restriktionsfragment
zu erhalten, das auch den viralen SV40-Replikationsstartpunkt umfasst.
Fiers et al., Nature 273, 113 (1978). Der unmittelbare frühe Promotor
des menschlichen Zytomegalievirus ist leicht als ein HindIII-E-Restriktionsfragment
zu erhalten. P.J. Greenaway et al., Gene 18, 355-360 (1982). Natürlich sind
auch Promotoren aus der Wirtszelle oder verwandten Spezies hierin
von Nutzen.
-
Die
DNA-Transkription in höheren
Eukaryoten wird durch Insertieren einer Enhancer-Sequenz in den Vektor verstärkt. Enhancer
sind cis-wirkende DNA-Elemente, norma lerweise von etwa 10 bis 300
bp, die die Transkriptionsinitiationsfähigkeit eines Promotors verstärken. Enhancer
sind relativ orientierungs- und positionsunabhängig, da sie 5' (L. Laimins et al.,
Proc. Natl. Acad. Sci 78, 993 [1981]) und 3' (M.L. Lusky et al., Mol. Cell Bio.
3, 1108 [1983]) zu der Transkriptionseinheit gefunden wurden, in
einem Intron (J.L. Banerji et al., Cell 33, 729 [1983]) sowie in
der kodierenden Sequenz selbst (T.F. Osborne et a., Mol. Cell Bio.
4, 1293 [1984]). Viele Enhancer-Sequenzen von Säugetiergenen sind nun bekannt
(Globin, Elastase, Albumin, α-Fötoprotein und
Insulin). Typischerweise wird jedoch ein Enhancer aus einem eukaryotischen
Zellvirus verwendet. Beispiele umfassen den SV40-Enhancer auf der
späten
Seite des Replikationsstartpunktes (bp 100-270), den Enhancer des
frühen
Promotors des Zytomegalievirus, den Polyoma-Enhancer auf der späten Seite
des Replikationsstartpunkts und Adenovirus-Enhancer.
-
Expressionsvektoren,
die in eukaryotischen Wirtszellen (Hefe-, Pilz-, Insekten-, Pflanzen-,
Tier-, menschliche oder kernhaltige Zellen) verwendet werden, können ebenso
Sequenzen umfassen, die für
die Termination der Transkription erforderlich sind und die die
mRNA-Expression beeinflussen können.
Diese Regionen werden als polyadenylierte Segmente im untranslatierten
Teil der für
das Adheson kodierenden mRNA transkribiert.
-
Expressionsvektorsysteme
umfassen im Allgemeinen ein Selektionsgen, auch selektierbarer Marker genannt.
Beispiele für
geeignete selektierbare Marker für
Säugetierzellen
sind Dihydrofolatreduktase (DHFR), Thymidinkinase oder Neomycin.
Werden diese selektierbaren Marker erfolgreich in eine Säugetierwirtszelle transferiert,
so kann die transformierte Säugetierwirtszelle überleben,
wenn sie Selektionsdruck ausgesetzt wird. Es gibt zwei weit verbreitete,
verschiedene Kategorien von Selektionsegimen. Die erste Kategorie
basiert auf dem Stoffwechsel einer Zelle und der Verwendung einer
mutierten Zelllinie, der die Fähigkeit
fehlt, unabhängig
von einem angereichertem Medium zu wachsen. Zwei Beispiele sind
CHO-DHFR--Zellen sowie Maus-LTK--Zellen.
Diesen Zellen fehlt die Fähigkeit,
ohne den Zusatz solcher Nährstoffe
wie Thymidin oder Hypoxanthin zu wachsen. Da diesen Zellen gewisse,
für einen
vollständigen
Nucleotidsyntheseweg notwendige Gene fehlen, können sie nicht überleben,
wenn die fehlenden Nucleotide nicht in einem angereicherten Medium
bereitgestellt werden. Eine Alternative zum Anreichern des Mediums
ist die Einführung
eines intakten DHFR- oder TK-Gens in jene Zellen, denen die entsprechenden
Gene fehlen, wodurch ihre Wachstumsbedingungen verändert werden.
Einzelne Zellen, die nicht mit dem DHFR- oder TK-Gen transformiert
wurden, sind nicht fähig,
in einem nichtangereicherten Medium zu überleben.
-
Die
zweite Kategorie ist die der dominanten Selektion, die sich auf
ein Selektionsschema bezieht, das in jedem beliebigen Zelltyp verwendet
wird und nicht die Verwendung einer mutierten Zelllinie erforderlich macht.
Diese Schemen verwenden typischerweise ein Arzneimittel, um das
Wachstum einer Wirtszelle zum Stillstand zu bringen. Jene Zellen,
die ein neues Gen aufweisen, würden
ein Protein exprimieren, das eine Arzneimittelresistenz verleiht,
und würden
die Selektion überleben.
In Beispielen für
diese dominante Selektion werden die Wirkstoffe Neomycin, P. Southern & P. Berg, J. Molec.
Appl. Genet. 1, 327 (1982), Mycophenolsäure, R.C. Mulligan & P. Berg, Science
209, 1422 (1980), oder Hygromycin, B. Sugden et al., Mol. Cell.
Biol. 5, 410-413 (1985), verwendet. Die drei oben angeführten Beispiele
verwenden bakterielle Gene unter eukaryotischer Kontrolle, um eine
Resistenz gegen den jeweiligen Wirkstoff G418 oder Neomycin (Geneticin),
Xgpt (Mycophenolsäure)
bzw. Hygromycin zu verleihen.
-
"Amplifikation" bezieht sich auf
die Verstärkung
oder Replikation einer isolierten Region in der chomosomalen DNA
einer Zelle. Die Amplifikation wird unter Verwendung eines Selektionsmittels,
z.B. Methotrexat (MTX), das DHFR deaktiviert, erreicht. Die Amplifikation
oder die Herstellung von aufeinander folgenden Kopien des DHFR-Gens führt angesichts
größerer Mengen
von MTX zu einer Produktion von größeren Mengen von DHFR. Der
Amplifikationsdruck wird trotz der Gegenwart endogener DHFR durch
Zusatz von noch größeren Mengen
an MTX zu dem Medium angewandt. Die Amplifikation eines gewünschten
Gens kann durch Co-Transfektion einer Säugetierwirtszelle mit einem
Plasmid mit einer für
ein gewünschtes
Protein kodierenden DNA und dem DHFR- oder Amplifikationsgen, das
die Co-Integration ermöglicht,
erreicht werden. Es wird sichergestellt, dass die Zelle mehr DHFR
benötigt,
ei ne Anforderung, der durch Replikation des Selektionsgens nachgekommen
wird, indem nur nach Zellen selektiert wird, die in Gegenwart von
ansteigenden MTX-Konzentrationen
wachsen können.
So lange das für
ein gewünschtes
heterologes Protein kodierende Gen mit dem Selektionsgen cointegriert
hat, führt
die Replikation dieses Gens zur Replikation des für das gewünschte Protein
kodierenden Gens. Das Ergebnis ist, dass mehr Kopien des Gens, d.h.
ein amplifiziertes Gen, das für
das gewünschte
heterologe Protein kodiert, mehr des gewünschten heterologen Proteins
exprimieren.
-
Bevorzugte
Wirtszellen zur Expression der CD-Antigenvarianten dieser Erfindung
sind Säugetierzelllinien,
wobei die Beispiele folgende umfassen: Affennierenlinie CV1, transformiert
durch SV40 (COS-7, ATCC CRL 1651); menschliche embryonale Nierenlinie
(293, F.L. Graham et al., J. Gen Virol 36, 59 [1977], und 293S-Zellen
[293-Subklone, die aufgrund von besserem Suspensionswachstum ausgewählt wurden]);
Babyhamster-Nierenzellen (BHK, ATCC CCL 10); Chinahamster-Eierstockzellen-DHFR
(CHO, Urlaub & Chasin, Proc.
Natl. Acad. Sci. (USA) 77, 4216 [1980]); Maus-Sertoli-Zellen (TM4,
J.P. Mather, Biol. Reprod. 23, 243-251 [1980]); Affennierenzellen
(CV1 ATCC CCL 70); AGMK-Zellen (VERO-76, ATCC CRL-1587); menschliche
Zervixkarzinomzellen (HELA, ATCC CCL 2); Hundenierenzellen (MDCK,
ATCC 34); Büffelratten-Leberzellen
(BRL 3A, ATCC CRL 1442), menschliche Lungenzellen (W138, ATCC CCL
75); menschliche Leberzellen (Hep G2, HB 8065); Maus-Mammatumor
(MMT 060562, ATCC CCL51-Zellen); und TRI-Zellen (J.P. Mather et
al., Annals N.Y. Acad. Sci. 383, 44-68 [1982]).
-
"Transformation" bedeutet das Einführen von
DNA in einen Organismus, sodass die DNA entweder als extrachomosomales
Element oder durch chromosomale Integration replizierbar ist. Ein
Verfahren, das zur Transformation der Wirtszellen geeignet ist,
ist jenes von F. Graham und A. van der Eb, Virology 52, 456-457 (1973).
Es können
jedoch auch andere Verfahren zum Einführen von DNA in Zellen verwendet
werden, z.B. durch Kerninjektion oder durch Protoplastenfusion.
Falls prokaryotische Zellen oder Zellen, die substantielle Zellwände enthalten,
als Wirte verwendet werden, so ist Calciumbehandlung unter Verwendung
von Calciumchlorid, wie von F.N. Cohen et al., Proc. Natl. Acad.
Sci. (USA) 69, 2110 (1972), beschrieben, das bevorzugte Transfektionsverfahren.
-
Die
Konstruktion geeigneter Vektoren, die die gewünschten kodierenden und Kontrollsequenzen
enthalten, erfolgt unter Anwendung von Standard- und Manipulations-Ligationsverfahren.
Isolierte Plasmide oder DNA-Fragmente werden gespalten, nach Maß zusammengeschnitten
und in der gewünschten
Form erneut ligiert, um die geforderten Plasmide zu bilden. Geeignete
Verfahren sind für
die hierin beschriebene Konstruktion wohlbekannt. Siehe z.B. T.
Maniatis et al., Molecular Cloning, 133-134, Cold Spring Harbor
[1982]; Current Protocols in Molecular Biology, Greene Publishing
Associates & Wiley-Interscience,
Ausubel et al. (Hrsg.) [1987].
-
Korrekte
Plasmidsequenzen werden durch Transformieren des E.-coli-K12-Stamms
294 (ATCC 31446) mit Ligationsgemischen bestätigt, erfolgreiche Transformanten
werden durch Ampicillin- oder Tetracyclin-Resistenz ausgewählt, wo
dies angebracht ist, danach werden Plasmide der Transformaten hergestellt und
durch Restriktionsenzymverdau analysiert und/oder durch das Verfahren
von Messing et al., Nucleic Acids Res. 9, 309 (1981), oder das Verfahren
von Maxam et al., Methods in Enzymology 65, 499 (1980), sequenziert.
-
Die
Wirtszellen werden mit den Expressionsvektoren transformiert. Danach
werden sie in geeignetem Kulturmedium, z.B. umfassend Substanzen
zur Induktion von Promotoren, zur Selektion von Transformanten oder
zur Amplifikation von Genen, kultiviert. Die Kulturbedingungen,
wie z.B. Temperatur, pH und dergleichen, sind jene, die zuvor für die zur
Expression ausgewählte
Wirtszelle verwendet wurden, und sind dem durchschnittlichen Fachmann
bekannt.
-
Die
sekretierten Adhesonvarianten werden gewonnen und von den Kulturüberständen oder
Lysaten rekombinanter Wirte gereinigt. Typischerweise werden die Überstände durch
Ultrafiltration eingeengt, mit einer Ligandaffinitäts- oder
Immunaffinitätsmatrix
kontaktiert, sodass die Adhesonvariante adsorbiert wird, und aus der
Mat rix eluiert. Gegebenenfalls wird das Adheson durch Ionenaustauschchromatographie
gereinigt.
-
Überraschenderweise
war die Reinigung von löslichem
CD4-Adheson aus dem Kulturmedium unerwartet schwierig. Trotz der
Deletion der hydrophoben Transmembranregion des Antigens wies das
Antigen eine starke Tendenz auf, Aggregate zu bilden, die aus der
Suspension leicht durch Zentrifugieren bei 1000 × g zu entfernen waren und
die Oberflächen,
wie z.B. Ultrafiltrationsmembranen, schnell beschichteten. Dies scheint
das Resultat der Reduktion der Albuminkonzentration oder der Konzentration
anderer Serumproteine (die normalerweise im Präparat im Rohzustand vorhanden
sind) auf ein bestimmtes Niveau zu sein, unter dem das trunkierte
Antigen nicht mehr löslich
bleibt. Dieses Phänomen
scheint durch eine Exposition des CD4-Adhesons gegenüber niedrigen
pH-Werten (< etwa
pH 4) verstärkt
zu werden. Als ein Ergebnis sollten Trennungsverfahren (besonders
jene, die eine Säureelution
verwenden, wie z.B. Immunaffinität)
modifiziert werden, sodass das Eluat neutral gehalten wird oder
sofort wieder neutral gemacht wird. Weiters sollte ein Tensid, z.B.
ein Detergens wie etwa Tween 80, mit dem Antigen während des
Trennungsverfahrens inkludiert werden. Das gereinigte Endprodukt
wird mit einem vorher festgelegten Protein, wie z.B. Albumin, und/oder
einem Detergens stabilisiert.
-
Das
gereinigte Adheson wird in herkömmliche,
pharmakologisch annehmbare Arzneimittelträger formuliert.
-
Es
wird Patienten mit einer HIV-Infektion in einer Dosis verabreicht,
durch die eine höhere
Konzentration als etwa 100 ng lösliches
CD4-Adheson/ml Plasma beibehalten werden kann. Für CD4-Adhesonvarianten mit
unterschiedlichem Molekulargewicht werden anfänglich etwa 2 Picomol löslicher
Rezeptor pro ml Plasma klinisch evaluiert, um ein stöchiometrisches
Gleichgewicht mit nativem (membrangebundenem) und löslichem Rezeptor
zu schaffen. Die normale Dosis von löslichem CD4 liegt bei 100 μg/kg Körpergewicht
des Patienten pro Tag.
-
Die
therapeutischen CD4-Varianten werden mit anderen Therapien und Agenzien
zur Behandlung von AIDS, unter anderem AZT, neutralisierende Antikörper und
Immuncytotoxine, gp120-Fragmente und Vakzinen, verwendet.
-
Um
das Verständnis
der folgenden Beispiele zu erleichtern, werden einige, häufig verwendete
Verfahren und/oder Begriffe beschrieben.
-
"Plasmide" werden durch ein
klein geschriebenes p mit vorangehenden und/oder darauf folgenden Großbuchstaben
und/oder Nummern bezeichnet. Die hierin genannten Startplasmide
sind entweder im Handel erhältlich, öffentlich
auf unangeschränkter
Basis erhältlich
oder können
gemäß veröffentlichter
Verfahren aus erhältlichen
Plasmiden konstruiert werden. Zusätzlich sind nach dem Stand
der Technik Plasmide bekannt, die mit den beschriebenen äquivalent
und dem durchschnittlichen Fachmann bekannt sind.
-
"Verdau" von DNA bezieht
sich auf die katalytische Spaltung von DNA mit einem Restriktionsenzym, das
lediglich an bestimmten Sequenzen in der DNA agiert. Die verschiedenen
hierin verwendeten Restriktionsenzyme sind im Handel erhältlich,
und ihre Reaktionsbedingungen, Co-Faktoren und anderen Bedingungen wurden
so angewandt, wie dies dem durchschnittlichen Fachmann nach dem
Stand der Technik bekannt wäre. Für Analysezwecke
wird typischerweise 1 μg
Plasmid- oder DNA-Fragment
mit etwa 2 Enzymeinheiten in etwa 20 μl Pufferlösung verwendet. Um DNA-Fragmente
für die
Plasmidkonstruktion zu isolieren, werden typischerweise 5 bis 50 μg DNA mit
20 bis 250 Enzymeinheiten in einer größeren Menge verdaut. Geeignete
Puffer- und Substratmengen für
bestimmte Restriktionsenzyme werden von Hersteller spezifiziert.
Normalerweise werden Inkubationszeiten von etwa 1 Stunde bei 37 °C angewandt,
können
jedoch gemäß den Anweisungen des
Herstellers variieren. Nach dem Verdau wird die Reaktion direkt
auf einem Polyacrylamidgel der Elektrophorese unterzogen, um das
gewünschte
Fragment zu isolieren.
-
"Gewinnung" oder "Isolierung" eines bestimmten
DNA-Fragments aus einem Restriktionsverdau bedeutet eine Trennung
des Verdaus auf Polyacrylamid- oder Agarosegel durch Elektrophorese,
Identifikation des Fragments von Interesse durch Vergleich seiner
Mobilität
in Gegenüberstellung
mit jener der Marker-DNA-Fragmente mit bekanntem Molekulargewicht,
Entfernen der Gelsektion, die das gewünschte Fragment enthält, und
Trennung des Gels von der DNA. Dieses Verfahren ist allgemein bekannt
(R. Lawn et al., Nucleic Acids Res. 9, 6103-6114 [1981], und D.
Goeddel et al., Nucleic Acids Res. 8, 4057 [1980]).
-
"Dephosphorylierung" bezieht such auf
die Entfernung der terminalen 5'-Phosphate
durch Behandlung mit bakterieller alkalischer Phosphatase (BAP).
Dieses Verfahren verhindert, dass die zwei durch Restriktionsverdau
gespaltene Enden eines DNA-Fragments "zirkularisieren" oder eine geschlossene
Schleife bilden, die die Insertion eines anderen DNA-Fragments an
der Restriktionsstelle verhindern würde. Verfahren und Reagenzien
zur Dephosphorylierung sowie andere rekombinante Manipulationen
sind die herkömmlichen.
Reaktionen unter Verwendung von BAP werden in 50 mM Tris bei 68 °C durchgeführt, um
die Aktivität
jeglicher Exonucleasen zu unterdrücken, die in den Enzympräparaten
vorhanden sein können.
Die Reaktionen wurden 1 Stunde lang laufen gelassen. Nach der Reaktion
wird das DNA-Fragment gelgereinigt.
-
"Ligation" bezieht sich auf
den Prozess der Bildung von Phosphodiesterbindungen zwischen zwei
doppelsträngigen
Nucleinsäurefragmenten
(T. Maniatis et al., id. auf 146). Falls nicht anders bereitgestellt,
kann die Ligation unter Verwendung bekannter Puffer und Bedingungen
mit 10 Einheiten T4-DNA-Ligase ("Ligase") pro 0,5 μg an ungefähr äquimolaren
Mengen der zu ligierenden DNA-Fragmente durchgeführt werden.
-
"Auffüllen" oder "Abstumpfen" bezieht sich auf
die Verfahren, durch die das einzelsträngige Ende im Kohäsionsterminus
einer durch ein Restriktionsenzym gespaltenen Nucleinsäure in einen
Doppelstrang konvertiert wird. Dadurch wird der Kohäsionsterminus
eliminiert und ein stumpfes Ende gebildet. Dieses Verfahren ist
ein viel seitig verwendbares Werkzeug, um ein restriktionsgeschnittenes
Ende, das mit den von lediglich einem oder einigen anderen Restriktionsenzymen
geschaffenen Enden kohäsiv
sein kann, in einen Terminus zu konvertieren, der mit jeder stumpfschneidenden
Restriktions-Endonuclease oder anderen gefüllten Kohäsionstermini kompatibel ist.
Typischerweise wird das Abstumpfen durch Inkubieren von 2-15 μg der Target-DNA in
10 mM MgCl2, 1 mM Dithiothreit, 50 mM NaCl,
10 Mm Tris-Puffer (pH 7,5) bei etwa 37 °C in Gegenwart von 8 Einheiten
des Klenow-Fragments von DNA-Polymerase
I und 250 μM
von jedem der vier Desoxynucleosidtriphosphate erreicht. Die Inkubation
ist im Allgemeinen nach 30 Minuten Phenol- und Chloroformextraktion
und Ethanolfällung
beendet.
-
Die
folgenden Beispiele veranschaulichen lediglich die besten, zur Zeit
zur praktischen Umsetzung der Erfindung in Betracht gezogenen Ausführungsart,
sollten aber nicht als Einschränkung
der Erfindung interpretiert werden.
-
Beispiel 1
-
Konstruktion von Vektoren
zur Expression von nativem CD4 und sekretierten Derivaten
-
Abschnitt 1
-
Das
für die
rekombinante Synthese von menschlichem CD4 verwendete Plasmid war
pSVeCD4DHFR. Das Plasmid wurde wie folgt konstruiert:
λCD4P1, das
die meisten der kodierenden Sequenzen von menschlichem CD4 enthielt
(aus einer menschlichen Plazenta-cDNA-Bibliothek unter Verwendung
von Oligonucleotid-Sonden auf Basis der veröffentlichten Sequenz erhalten
[Maddon et al. 1985]), wurde mit EcoRI verdaut, um das cDNA-Insert
herzustellen. Dieses Fragment wurde mittels Polyacrylamid-Gelelektrophorese
gewonnen (Fragment 1).
-
pUC18
wurde mit EcoRI verdaut, und das Einzelfragment wurde mittels Polyacrylamid-Gelelektrophorese
(Fragment 2) gewonnen. Fragment 1 wurde an Fragment 2 ligiert, und
das Ligationsgemisch wurde in den E.-coli-Stamm 294 transformiert.
Die transformierte Kultur wurde auf Ampicillin-Mediumplatten ausplattiert, und
es wurden die resistenten Kolonien ausgewählt. Plasmid-DNA wurde aus
Transformanten hergestellt und mittels Restriktionsanalyse auf die
Gegenwart der korrekten DNA-Fragmente
untersucht. Dieses Plasmid wird als pUCCD4 bezeichnet.
-
pSVeE'DHFR (Muesing et
al., Cell 48, 691-701 [1987]) wurde mit KpnI und BamHI verdaut und
mit E.-coli-DNA-Polymerase-I (Klenow-Fragment) und den vier dNTPs
abgestumpft. Fragment 3, das die pML-Ampr-Region,
den frühen
SV40-Promotor, den HIV-LTR und das Maus-DHFR-Gen enthielt, wurde
mittels Gelelektrophorese gewonnen, ligiert, und das Ligationsgemisch
wurde in den E.-coli-Stamm 294 transformiert. Die transformierte
Kultur wurde auf Ampicillin-Mediumplatten ausplattiert, und danach
wurden die resistenten Kolonien auswählt. Aus Transformanten wird
Plasmid-DNA hergestellt und mittels Restriktionsanalyse auf die Gegenwart
der BamHI-Restriktionsstelle und die Abwesenheit der KpnI-Restriktionsstelle
untersucht. Dieses Plasmid wird pSVeΔBKDHFR genannt und ermöglicht nach
der Transfektion in eine geeignete Zelllinie die Insertion von EcoRI-BamHI-Fragmenten
nach dem frühen
SV40-Promotor sowie eine Transkription unter seiner Kontrolle.
-
Synthetische
Oligonucleotide (Adaptoren 1-8, unten stehend) wurden so behandelt,
dass sie sich nun vom 76 bp 5' zum
Initiationscodon der CD4-Translation bis zur RsaI-Restriktionsstelle
bei 121 bp 3' zum
Initiator erstrecken, wobei die Sequenz AATT am 5'-Ende des kodierenden
Strangs ein Ende generiert, das an ein Eco-RI-Restriktionsfragment ligieren konnte.
Diese Oligonucleotide wurden ligiert, und das 204-bp-Fragment, das
die gesamte Sequenz enthielt, wurde mittels Gelelektrophorese gewonnen
(Fragment 4).
-
-
-
pUCCD4
wurde mit RsaI und SstI verdaut, und das 401-bp-Fragment, das einen
Teil der CD4-Kodiersequenz enthielt, wurde mittels Gelelektrophorese
gewonnen (Fragment 5). pUC18 wurde mit EcoRI und SstI verdaut, und
das Fragment, das den Hauptteil des Plasmids umfasste, wurde mittels
Gelelektrophorese gewonnen (Fragment 6). Die Fragmente 4 und 5 wurden
an Fragment 6 ligiert, und das Ligationsgemisch wurde in den E.-coli-Stamm
294 transformiert. Die transformierte Kultur wurde auf Ampicillin-Mediumplatten
ausplattiert, und es wurden die resistenten Kolonien ausgewählt. Aus
Transformanten wurde Plasmid-DNA hergestellt und mittels Restriktionsanalyse
auf die Gegenwart des korrekten Fragments untersucht. Die Sequenz
der insertierten synthetischen DNA wurde durch Herausschneiden der
605-bp-EcoRI-SstI-Fragmente
aus mehreren Transformanten und Ligieren dieser an M13mp19, das
mit den selben Enzymen verdaut wurde, überprüft. Nach der Transformation
in den E.-coli-Stamm JM101 wurde einzelsträngige DNA hergestellt und sequenziert. Ein
Plasmid, das die korrekte Sequenz enthielt, wurde ausgewählt und
wird als pCD4int bezeichnet.
-
pCD4int
wurde mit EcoRI und SstI verdaut, und das Fragment 7, das das 5'-Ende der CD4-Kodierregion
enthielt, wurde mittels Gelelektrophorese gewonnen. pUCCD4 wurde
mit SstI und BamHI verdaut, und das 1139-bp-Fragment, das den Rest
der CD4-Kodierregion enthielt (Fragment 8), wurde mittels Gelelektrophorese
gewonnen.
-
pSVeΔBKDHFR wurde
mit EcoRI und BamHI verdaut, und Fragment 9, das den Hauptteil des
Plasmids umfasste, wurde isoliert. Die Fragmente 7, 8 und 9 wurden
ligiert, und das Ligationsgemisch wurde in den E.-coli-Stamm 294
transformiert. Die transformierte Kultur wurde auf Ampicillin-Mediumplatten
ausplattiert, und es wurden die resistenten Kolonien ausgewählt. Aus
Transformanten wurde Plasmid-DNA hergestellt und mittels Restriktionsanalyse
auf die Gegenwart des korrekten Fragments untersucht. Dieses Plasmid
wird als pSVeCD4DHFR bezeichnet und wurde für die Steuerung der Synthese
rekombinanter intakter CD4 verwendet.
-
Abschnitt 2
-
Es
wurde ein Plasmid konstruiert, um die Synthese eines CD4-Derivats
zu steuern, dem die mutmaßliche
Transmembrandomäne
und der Großteil
der mutmaßlichen
zytoplasmatischen Domäne
fehlt (Madden et al.). Dies wurde mit der Intention getan, eine
sekretierte Form von CD4 zu schaffen, basierend auf der Annahme,
dass diese Domänen
das CD4-Glykoprotein an der Zellmembran verankern und dass ihre
Deletion zu der Sekretion des Produkts führen würde. Dieses Plasmid wird als
pSVeCD4ΔNIaDHFR
bezeichnet und wurde wie folgt konstruiert:
pUCCD4 wurde mit
SstI und TaqI verdaut, und das 531-bp-Fragment wurde gewonnen (Fragment
10). pUCCD4 wurde mit NlaIII und TaqI verdaut, und das 112-bp-Fragment wurde gewonnen
(Fragment 11). pUCCD4 wurde mit BamHI und NIaIII verdaut, und das
301-bp-Fragment wurde gewonnen (Fragment 12). pCD4int wurde mit
SstI und BamHI verdaut, und Fragment 13, das den Hauptteil des Plasmids
umfasst, wurde gewonnen. Die Fragmente 10, 11 und 12 wurden zusammen
mit Fragment 13 ligiert, und das Ligationsgemisch wurde in den E.-coli-Stamm
294 transformiert. Die transformierte Kultur wurde auf Ampicillin-Mediumplatten ausplattiert,
und es wurden die resistenten Kolonien ausgewählt. Aus Transformanten wurde
Plasmid-DNA hergestellt
und mittels Restriktionsanalyse auf die Gegenwart des korrekten
Fragments untersucht. Plasmid-DNA aus mehreren Transformanten wurde
sequenziert, um sicherzustellen, dass das 195-bp-NIaIII-Fragment
deletiert wurde und dass der korrekte Leseraster wiederhergestellt
wurde. Das daraus resultierende Plasmid wird als pCD4ΔNIa bezeichnet.
-
pCD4ΔNIa wurde
mit EcoRI und BamHI verdaut, und das 1541-bp-Fragment, das die Sequenz
eines CD4-Derivats enthielt, dem die Transmembran- und die zytoplasmati schen
Domänen
fehlten, wurde gewonnen (Fragment 14) und an Fragment 9 ligiert,
und das Ligationsgemisch wurde dann in den E.-coli-Stamm 294 transformiert.
Die transformierte Kultur wurde auf Ampicillin-Mediumplatten ausplattiert,
und es wurden resistente Kolonien ausgewählt. Aus Transformanten wurde
Plasmid-DNA hergestellt und mittels Restriktionsanalyse auf die
Gegenwart des korrekten Fragments untersucht. Dieses Plasmid wird
als pSVeCD4ΔNIaDHFR
bezeichnet.
-
Sowohl
pSVeCD4DHFR als auch pSVeCD4ΔNIaDHFR
wurden mittels desselben Verfahrens, das verwendet wurde, um Zelllinien
zu schaffen, die HIV-I-Polypeptide stabil exprimieren, in CHO-Zellen
transfiziert (Muesing, Smith & Capon,
Cell 48, 6910701 [1987]). Diese Zellen wurden mittels Radioimmunfällung wie
unten stehend beschrieben auf ihre Produktion getestet. Während in
den anfänglichen
Experimenten keine Produkte nachgewiesen wurden, zeigten darauf
folgende Experimente, dass das oben beschriebene kodierende Segment
tatsächlich
die Synthese einer löslichen
CD4-Adhesonvariante sowohl in CHO- als auch in 293-Zellen steuern
konnte.
-
Abschnitt 3
-
Ein
anderes Expressionssystem wurde anfänglich für die Synthese und die Expression
einer CD4-Variante verwendet, der die zytoplasmatischen und Transmembrandomänen vollständig fehlten.
Dieses System verwendet den Zytomegalieviruspromotor und kann in
kultivierten Zellen menschlichen Ursprungs verwendet werden. Das
erste zur Verwendung in diesem System konstruierte Plasmid enthielt
die gesamte kodierende Region für
CD4 und war als Kontrolle in den folgenden Studien gedacht. Es wird
als pRKCD4 bezeichnet und wurde wie folgt konstruiert:
pSVeCD4DHFR
wurde mit EcoRI und BamHI verdaut, und das Fragment 15, das die
gesamte CD4-kodierende Region enthielt, wurde isoliert. pRK5 (U.S.S.N.
97.472, eingereicht am 11. September 1987) wurde mit EcoRI und BamHI
verdaut, und das Fragment 16, das den Hauptteil des mittels Gelelektrophorese
gewonnenen Plasmids enthielt, wurde an Fragment 15 ligiert, und
das Ligationsgemisch wurde in den E.-coli-Stamm 294 transformiert.
Die transformierte Kultur wurde auf Ampicillin- Mediumplatten ausplattiert, und es wurden
resistente Kolonien ausgewählt.
Aus Transformanten wurde Plasmid-DNA hergestellt und mittels Restriktionsanalyse
auf die Gegenwart des korrekten Fragments untersucht. Dieses Plasmid
wird als pRKCD4 bezeichnet.
-
Abschnitt 4
-
Das
nächste
konstruierte Plasmid wurde geschaffen, um die Expression des oben
angeführten
(Abschnitt 3) sekretierten Derivats von CD4 zu steuern. Die kodierende
Region von CD4 wurde nach dem Aminosäurerest 368 des reifen CD4
an eine Sequenz von pBR322 fusioniert, die für 9 weitere Reste vor einem
Translationsterminationscodon kodiert. Dadurch werden die mutmaßlichen
Transmembran- und zytoplasmatischen Domänen von CD4, von denen angenommen
wird, sie würden
CD4 an der Zelloberfläche
verankern, entfernt. Dieses Plasmid wird als pRKCD4T bezeichnet
(und produziert ein Protein, das CD4T genannt wird) und wurde wie
folgt konstruiert:
pSVeCD4DHFR wurde mit HpaII verdaut, mit
dem Klenow-Fragment und den vier dNTPs abgestumpft und mit BstEII
verdaut. Das 382-bp-Fragment (Fragment 17), das einen Teil der CD4-Kodiersequenz
enthielt, wurde mittels Gelelektrophorese gewonnen. pSVeCD4DHFR
wurde mit EcoRI und BstEII verdaut, und das 874-bp-Fragment (Fragment
18) wurde gewonnen. pBR322 wurde mit HindIII verdaut, mit dem Klenow-Fragment
und den vier dNTPs abgestumpft und mit EcoRI verdaut. Fragment 19,
das den Hauptteil des Plasmids enthielt, wurde isoliert und an die
Fragmente 17 und 18 ligiert, und das Ligationsgemisch wurde in den
E.-coli-Stamm 294 transformiert. Die transformierte Kultur wurde
auf Ampicillin-Mediumplatten ausplattiert, und es wurden resistente
Kolonien ausgewählt.
Aus Transformanten wurde Plasmid-DNA hergestellt und mittels Restriktionsanalyse
auf die Gegenwart des korrekten Fragments untersucht. Dieses Plasmid
wird als pCD4Tint bezeichnet.
-
pRK5
wurde mit EcoRI und SmaI verdaut, und das Fragment 20, das den Hauptteil
des Plasmids enthielt, wurde isoliert. pCD4Tint wurde mit EcoRI
und EcoRV verdaut, und das 1410-bp-Fragment, das die CD4-Kodiersequenz
bis zu der HpaII-Stelle bei 1176 bp 3' zum Initiationscodon enthielt, sowie
das 154-bp-HindIII-EcoRV-Fragment von pBR322 wurden gewonnen (Fragment
21). Die Fragmente 20 und 21 wurden ligiert, und das Ligationsgemisch
wurde in den E.-coli-Stamm 294 transformiert. Die transformierte
Kultur wurde auf Ampicillin-Mediumplatten ausplattiert, und es wurden
resistente Kolonien ausgewählt.
Aus Transformanten wurde Plasmid-DNA hergestellt und mittels Restriktionsanalyse
auf die Gegenwart des korrekten Fragments untersucht. Dieses Plasmid
wird als pRKCD4T bezeichnet.
-
Abschnitt 5a
-
Um
eine sekretierte Form von CD4 zu schaffen, die mit einem auf das
Typ-I-Glykoprotein-D
des Herpes-Virus gerichteten Antikörper gereinigt werden konnte,
wurde ein Plasmid konstruiert, um ein Derivat von CD4T zu exprimieren,
in dem die für
das reife, aufbereitete CD4T-Polypeptid kodierende Region an eine
Sequenz fusioniert war, die für
das Signalpeptid und die ersten 27 Reste des reifen Typ-I-gD-Glykoproteins des Herpes-Simplex-Virus
kodiert. Dieses Plasmid wird als pRKGDCD4T bezeichnet und wurde
wie folgt konstruiert:
pgDTrunc.DHFR wurde mit EcoRI und PvuII
verdaut, und das Fragment, das die kodierende Region für das Signalpeptid
und die ersten 27 Reste des reifen HSV-I-gD-Glykoproteins enthielt, wurde isoliert
(Fragment 22). pRKCD4T wurde mit EcoRI und BstEII verdaut, und das
Fragment 23, das das 3'-Ende
der CD4-Kodiersequenz und die pRK5-Region enthielt, wurde isoliert.
-
Die
synthetischen Oligonucleotide GD (Adaptoren 1-2, unten), die die
kodierende Sequenz von CD4 vom Codon für den aminoterminalen Rest
des reifen CD4 bis zur Rsa-Stelle bei 121 bp 3' zu der Translationsinitiation sowie
die Sequenz CTGCTCGAG am 5'-Ende
des kodierenden Strangs enthielten, wurden hergestellt (Fragment
24). pRKCD4 wurde mit RsaI und BstEII verdaut, und das 665-bp-Fragment, das einen
Teil der kodierenden Region für
CD4 enthielt, wurde gewonnen (Fragment 25) und an Fragment 24 ligiert.
Nach dem Verdau mit BstEII wurde, um sicherzustellen, dass nur monomere
Fragmente vorhanden waren, das 724-bp-Fragment, das beide Sequenzen enthielt,
mittels Gelelektrophorese gewonnen (Fragment 26).
-
Die
Fragmente 22, 23 und 26 wurden ligiert, und das Ligationsgemisch
wurde in den E.-coli-Stamm 294 transformiert. Die transformierte
Kultur wurde auf Ampicillin-Mediumplatten
ausplattiert, und es wurden resistente Kolonien ausgewählt. Aus
Transformanten wurde Plasmid-DNA hergestellt und mittels Restriktionsanalyse
auf die Gegenwart des korrekten Fragments untersucht. Die Sequenz
einiger Transformanten wurde untersucht, um sicherzustellen, dass
das synthetische Insert korrekt war und dass der Leseraster erhalten blieb.
Dieses Plasmid wird als pRKGDCD4T bezeichnet.
-
Diese
von pRK5 abstammenden Plasmide wurden für eine stabile Expression gemäß Muesing
et al., Cell 48, 691 (1987), vorzugsweise in 293S-Zellen transfiziert,
jedoch mit der Ausnahme, dass zusätzlich zu dem Plasmid von Interesse
ein das Neomycin-Resistenzgen
pRSV neo exprimierendes Plasmid (Gorman et al., Science 221, 553-555 (1985)) co-transfiziert
wurde. 293-Zellen werden ebenfalls zufriedenstellend als Wirtszellen
verwendet. 2 Tage nach der Transfektion wurden die Zellen mit 0,5
mg/ml G418 (Genticinsulfat; Gibco) zur Selektion von stabilen Zelllinien
in ein Standardmedium (1:1 F12/DME, ergänzt mit L-Glutamin, Penicillin-Streptomycin
und 10 FBS) passagiert, anstatt in ein methotrexathältiges Medium,
wie dies von Muesing et al. gezeigt wurde. Zellen wurden mittels
Radioimmunfällung
auf die Produktion von CD4 oder CD4-Analoga getestet. In Bindungsstudien
(Abschnitt 5c) wurden konditionierte Überstände dieser Zellen in dem 1:1-F12/DME-Medium
verwendet. Materialien, die in Infektiositätstests (Abschnitt 5b) verwendet
wurden, wurden wie unten stehend in Abschnitt 8 beschrieben erhalten.
gDCD4-Adaptor
1:
CTGCTCGAGCAGGGAAACAAAGTGGTGCTGGGCAAAAAAGGGGATACAGTGGAACTGAC
gDCD4-Adaptor
2:
pACAGGTCAGTTCCACTGTATCCCCTTTTTTGCCCAGCACCACTTTGTTTCCCTGCTCGA
-
Abschnitt 5b
-
Im
Folgenden wird eine Studie über
die Neutralisierung der HIV-I-Infektiosität durch lösliche CD4-Analoga dargelegt.
Darauf folgte eine Modifikation des Neutralisierungsverfahrens von
Robert-Guroff et al., Nature 316, 72 (1985). Gleiche Volumina an
Inhibitorüberstand
und Virus (60 Mikroliter) wurden bei 4 °C 1 Stunde lang inkubiert, dann
wurde dasselbe Volumen H9 (Gallo et al., Science 224, 500 (1984))
bei 5 × 106/ml
hinzugefügt, und
die Inkubation wurde bei 37 °C
1 Stunde lang fortgesetzt. Nach der Absorption wurden 2,5 × 105 Zellen in 150 Mikroliter in 2 ml Inkubationsmedium
transferiert. Nach 4 Tagen bei 37 °C wurden die Kulturen 1:2 mit
frischem Medium geteilt und für
3 weitere Tage inkubiert. Die Kulturen wurden geerntet, die Aktivität der reversen Transkriptase
wurde gemessen (Groopman et al., AIDS Research and Human Retroviruses
3,71 (1987)), und die Immunfluoreszenzreaktivität mit HIV-1-positivem Serum wurde wie beschrieben
nachgewiesen (Poiesz et al., Proc. Acad. Nat. Sci. USA 77, 7415
(1980)).
-
Inhibitorüberstände wurden
aus konfluenten Plattenkulturen von 293S/CDT4-, 293S/gDCD4T-Zellen oder
nicht transfizierten 293S-Zellen erhalten, und zwar durch Ersetzen
der Wachstumsmedium-Inkubationsmedien sowie durch Ernten der Überstände 24 Stunden
später.
Der Inhibitorüberstand
ersetzte einen Teil des Inkubationsmediums oder das gesamte Inkubationsmedium
während
der ersten drei Kulturtage, wie dies in der zweiten Spalte von Tabelle
3 angegeben ist. Die Challenge-Dosis
des Virus lag bei 100 TCID
50 (Groopman et al.,
s.o.) des HIV-1-Stamms HTLV-IIIB,
der in H9-Zellen kultiviert wurde, die im selben System getestet
wurden. Das Inkubationsmedium bestand aus einem RPMI-1640-Medium,
das 2 mM L-Glutamin, 100 Einheiten/ml Penicillin, 100 Mikrogramm/ml
Streptomycin, 2 Mikrogramm/ml Polybren und 20 % fötales Kälberserum
(M.A. Bioproducts) enthielt. Tabelle
3
-
Beide
Formen des löslichen
CD4 brachten bei einer Inkubation mit virusinfizierten Zellen ohne
vorherige Verdünnung
das Wachstum von HIV-1 beinahe zum Stillstand (Tabelle 2). Bei einer
Verdünnung
von 1:4 waren die löslichen
CD4-Präparate
lediglich teilweise wirksam bei der Inhibierung des Viruswachstums,
die Menge an fluoreszenz-positiven Zellen und reverser Transkriptase
war jedoch immer noch signifikant geringer als bei Kulturen, die
mock-transfizierte Zellüberstände erhielten
(Tabelle 2). Da es beim Viruswachstum keinen signifikanten Unterschied
zwischen verdünnten
und unverdünnten
Kontrollüberständen gegeben
hat und keiner der Überstände das
Wachstum der nicht infizierten H9-Zellen (Daten nicht dargestellt)
beeinflusst hat, wurde angenommen, die in diesen Überständen vorhandenen
löslichen
CD4-Proteine wären
für die
Neutralisierung der HIV-1-Infektion der H9-Zellen verantwortlich.
-
Abschnitt 5c
-
Um
die Affinitätskonstante
für Wechselwirkungen
zwischen gp120 und CD4 oder CD4-Varianten zu bestimmen, wurde eine
Sättigungs-Bindungsanalyse
mit löslichem
CD4 (s.o.) durchgeführt,
und ein Detergens solubilisierte intaktes CD4 (Lasky et al., Cell
50, 957 [1987]) unter Verwendung von radioiodiertem gp120, das mit
Lactoperoxidase markiert war. Die Bindungsreaktionen bestanden aus 125I-gp120 (3 ng bis 670 ng, 2,9 nCi/ng),
das 1 Stunde lang bei 0 °C
mit Zelllysaten, die intaktes CD4 enthiel ten (Lasky et al., s.o.),
oder mit Zellüberständen, die
unmarkiertes CD4T oder gDCD4T enthielten, das wie in Abschnitt 5a
beschrieben hergestellt wurde, inkubiert wurde. Die Reaktionen (0,2
ml) besaßen
eine Endzusammensetzung von 0,5X McDougal-Lysepuffer (McDLB) (1 × McDLB
enthält
0,5 % Nonidet NP-40, 0,2 % Na-Desoxycholat,
0,12 M NaCl, 0,02 M Tris-HCl, pH 8,0) und wurden zweifach durchgeführt, beide
in Gegenwart oder in Abwesenheit von 50 Mikrogramm unmarkiertem
gereinigtem gp120 (74facher oder größerer Überschuss). Nach der Inkubation
wurde das gebundene gp120 mittels Immunfällung quantifiziert und in
einem Gammazähler
gezählt.
Für die
Immunfällung
wurden Bindungsreaktionslösungen
mit 5 Mikrolitern normalem Kaninchenserum 1 Stunde lang bei 0 °C präabsorbiert
und mit 40 Mikrolitern Pansorbin (10 % (Gew./Vol.), Calbiochem)
30 Minuten lang bei 0 °C geklärt. Die
Proben wurden dann über
Nacht bei 0 °C
mit 2 Mikrolitern normalem Serum oder 5 Mikrolitern (0,25 Mikrogramm)
monoklonalem OKT4-Antikörper
(Ortho) inkubiert, gefolgt vom Sammeln der Immunkomplexe mit 10
Mikrolitern Pansorbin. Die Präzipitate
wurden zwei Mal in 1X McDLB und einmal in Wasser gewaschen, danach
durch Eluieren bei 100 °C
2 Minuten lang in Probenpuffer (0,12 M Tris-HCl, pH 6,8, 4 % SDS, 0,7
M Mercaptoethanol, 20 % Glycerin und 0,1 % Bromphenolblau) eluiert.
Die CD4-Moleküle wurden
durch gp120 sättigungsgebunden
und ergaben eine einfache Massenwirkungsbindungskurve.
-
Die Überstände von
mock-transfizierten Zellen ergaben eine Menge an spezifisch gebundenem gp120,
die unter 1 % jenes Wertes lag, der für lösliches CD4 enthaltende Überstände gefunden
wurde. Die Scatchard-Analyse ergab eine einzige Klasse an Bindungsstellen
auf jedem Molekül,
mit erkennbaren Dissoziationskonstanten (Kd) von 1,3 × 10-9 M, 0,83 × 10-9 M
und 0,72 × 10-9 M für
intaktes CD4, CD4T bzw. gDCD4T. Die Werte, die für eine CD4-gp120-Bindung in
einer Lösung
erhalten wurden, sind mit der zuvor für eine gp120-Bindung an CD4
auf ganzen Zellen (Kd=4,0 × 10-9, M. Lasky, Cell, s.o.) gemessenen Affinität vergleichbar.
-
Abschnitt 6
-
Um
sekretierte Derivate von CD4 zu produzieren, die frei von fremden
Aminosäureresten
sind, wurden zwei Plasmide für
die Expression in 293-Zellen konstruiert. Die Plasmide enthalten
CD4-Gene, die trunkiert wurden ohne das Hinzufügen von zusätzlichen Resten und pRKCD4ΔNIa und pRKCD4TP
genannt werden (und Proteine, die CD4TP und CD4ΔNIa genannt werden, produzieren),
und wurden wie folgt konstruiert:
Das Fragment 14, das das
CD4-Gen mit dem deletierten 195-bp-NIaIII-Restriktionsfragment enthielt,
wurde an Fragment 16 ligiert, das mit EcoRI und BamHI verdautes
pRK5 ist. Das Ligationsgemisch wurde in den E.-coli-Stamm 294 transformiert,
die transformierte Kultur wurde auf Ampicillin-Mediumplatten ausplattiert,
und es wurden resistente Kolonien ausgewählt. Aus Transformanten wurde
Plasmid-DNA hergestellt und mittels Restriktionsanalyse auf die
Gegenwart des korrekten Fragments untersucht. Das resultierende
Plasmid wird als pRKCD4ΔNIa
bezeichnet.
-
Synthetische
DNA (5' CGT GAT
AGA AGC TTT CTA GAG 3')
wurde so bearbeitet, dass sie an die HpaII-Stelle bei 1176 bp bindet
und bei einer solchen Bindung die Translation nach dem Aminosäurerest
368 des reifen CD4 (Fragment 27) zu Ende bringen würde. Das
andere Ende dieses Fragments wurde so geschaffen, dass es an BamHI-Restriktionsfragmente
ligiert. pUCCD4 wurde mit BstEII und HpaII verdaut, und das 382-bp-Fragment,
das einen Teil des CD4-Gens enthielt, wurde gewonnen (Fragment 28).
Die Fragmente 27 und 28 wurden ligiert und danach mit BstEII verdaut,
um dimerisierte Fragmente zu Monomeren zu reduzieren, und das resultierende
401-bp-Fragment wurde gewonnen (Fragment 29).
-
pRKCD4
wurde mit BstII und BamHI verdaut, und das Fragment, das den Großteil des
Plasmids (Fragment 30) umfasste, wurde isoliert und an Fragment
29 ligiert. Das Ligationsgemisch wurde in den E.-coli-Stamm 294
transformiert, die transformierte Kultur wurde auf Ampicillin-Mediumplatten
ausplattiert, und es wurden resistente Kolonien ausgewählt. Aus
Transformanten wurde Plasmid-DNA hergestellt und mittels Restriktionsanalyse
auf die Gegenwart des korrekten Fragments untersucht. Das resultierende
Plasmid wird als pRKCD4TP bezeichnet. Beide Plasmide werden in 293-Zellen transfiziert,
um, wie oben beschrieben, stabile, Varianten-CD4-exprimierende Zelllinien
zu generieren.
-
Abschnitt 7
-
Es
wurden zwei Plasmide konstruiert, um die Expression von sekretiertem
CD4, dem fremde Aminosäurereste
fehlten, in CHO-Zellen zu steuern. Diese werden als pSVeCD4ΔNIaSVDHFR
und pSVeCD4TPSVDHFR bezeichnet (und kodieren für Proteine, die die Hauptsequenz
von CD4ΔNIa
und CD4TP aufweisen) und wurden wie folgt konstruiert:
pE348HBV.E400D22
wurde mit PvuI und EcoRI verdaut, und das Fragment, das den frühen Promotor
von SV40 und einen Teil des β-Lactamase-Gens
enthielt, wurde gewonnen (Fragment 31). pE348HBV.E400D22 wurde mit
PvuI und BamHI verdaut, und das große Fragment, das den Rest des β-Lactamase-Gens
sowie den frühen
Promotor von SV40 und das DHFR-Gen enthielt, wurde isoliert (Fragment
32).
-
Die
Fragmente 31 und 32 wurden zusammen mit dem Fragment 14 ligiert
und in den E.-coli-Stamm 294 transformiert. Die transformierte Kultur
wurde auf Ampicillin-Mediumplatten
ausplattiert, und es wurden resistente Kolonien ausgewählt. Aus
Transformanten wurde Plasmid-DNA hergestellt und mittels Restriktionsanalyse
auf die Gegenwart des korrekten Fragments untersucht.
-
Das
resultierende Plasmid wird als pSVECD4ΔNIaSVDHFR bezeichnet. Dieses
Plasmid enthält
dasselbe DNA-Fragment, das für
das lösliche
CD4-Molekül
kodiert, das im oben genannten Plasmid pSVeCD4ΔNIaDHFR zu finden ist (Abschnitt
2).
-
pRKCD4TP
wurde mit EcoRI und BamHI verdaut, und das Fragment, das die trunkierte
kodierende Region von CD4 enthielt, wurde isoliert und an die Fragmente
31 und 32 ligiert. Das Ligationsgemisch wurde in den E.-coli-Stamm
294 transformiert, die transformierte Kultur wurde auf Ampicillin-Mediumplatten
ausplattiert, und es wurden resistente Kolonien ausgewählt. Aus
Transformanten wurde Plasmid-DNA hergestellt und mittels Restriktionsanalyse
auf die Gegenwart des korrekten Fragments untersucht. Das resultierende
Plasmid wird als pSVeCD4TPSVDHFR bezeichnet. Beide dieser Plasmide
werden in CHO-Zellen transfiziert, und amplifizierte Transfektanten
werden durch Methotrexat unter Verwendung herkömmlicher Verfahren ausgewählt.
-
Beispiel 2
-
Fusionen
der V-Region des CD4-Gens, das zu der variablen Region von Immunglobulin-Genen
homolog ist (siehe Maddon et al. (1985)), mit der konstanten (C-)
Region von menschlichen Immunglobulin-κ- und -γ2-Ketten werden wie folgt konstruiert:
Synthetische
DNA wird, basierend auf der von Morin et al., Proc. Natl. Acad.
Sci. 82, 7025-7029, publizierten Sequenz so behandelt, dass sie
für die
C-Region einer menschlichen κ-Kette
(Reste 109-214) kodiert, mit der Addition am 5'-Ende des kodierenden Strangs der Sequenz
GGGG, was eine Ligation dieses Fragments an die BspMI-Stelle am
Ende der mutmaßlichen
V-artigen Region von CD4 ermöglicht.
Am 3'-Ende der kodierenden
Region wird ein Translationsstopcodon sowie eine Sequenz, die eine
Ligation dieses Endes an BamHI-Restriktionsfragmente ermöglicht,
addiert. Die synthetische DNA wird in 8 Fragmenten hergestellt,
4 für jeden Strang,
70-90 Basen lang. Diesen wird dann die Möglichkeit zum Annealing gegeben,
und sie werden vor der Isolation auf einem Polyacrylamidgel ligiert
(Fragment 33).
-
pRKCD4
wird mit EcoRI und BspMI verdaut, und das 478-bp-Fragment, das die
für die
mutmaßliche V-artige
Domäne
von CD4 kodiert, wird gewonnen (Fragment 34). Die Fragmente 33 und
34 werden mit Fragment 16 (aus dem Expressionsvektor pRK5) zusammen
ligiert. Das Ligationsgemisch wird in den E.-coli-Stamm 294 transformiert,
die transformierte Kultur wurde auf Ampicillin-Mediumplatten ausplattiert, und
es wurden resistente Kolonien ausgewählt. Aus Transformanten wird
Plasmid-DNA hergestellt
und mittels Restriktionsanalyse auf die Gegenwart des korrekten
Fragments untersucht. Das resultierende Plasmid wird als pRKCD4Ck
bezeichnet.
-
Ein
Plasmid, das für
eine Fusion der V-artigen Domäne
von CD4 mit der menschlichen Immunglobulin-Cγ2-Region kodiert, wird auf eine ähnliche
Art und Weise konstruiert und wird als pRKCD4Cγ2 bezeichnet. Beide dieser Plasmide
werden in 293-Zellen,
Myelomzellen oder andere kompetente Zellen transfiziert, um Zelllinien
zu erhalten, die, wie oben beschrieben, unterschiedliche CD4-Moleküle exprimieren.
-
Beispiel 3
-
Das
durch das Verfahren aus Beispiel 1 sekretierte gDCD4T wurde vom
Zellkulturfluid, das entweder 10 % FBS (Fetales Rinderserum) oder
kein zugesetztes FBS enthielt, gereinigt. Das konditionierte Zellkulturfluid
wurde erst durch Ultrafiltration eingeengt und dann mittels Immunaffinitätschromatographie
gereinigt. Die Immunaffinitätssäule wurde
durch Binden monoklonaler muriner Antikörper 5B6 (deren Epitop sich
auf dem HSV-1-gD-Teil des gDCD4T-Moleküls befindet) an glycerylbeschichtetes
Glas mit definierter Porengröße mittels
des Verfahrens von Roy et al. (1984) hergestellt. Das konzentrierte
Zellkulturfluid wird direkt auf die Säule aufgebracht, und die verunreinigenden
Proteine werden mit einem neutralen pH-Puffer weggewaschen. Die Säule wird
dann mit neutralem Puffer, der Tetramethylammoniumchlorid enthält, gewaschen,
gefolgt von einem neutralen Puffer, der Tween 80 enthält. Das
gebundene gDCD4T wird aus der Säule
mit einem Puffer bei einem pH von 3, der Tween 80 enthält (0,1
% (Gew./Vol.)), eluiert und wird sofort nach dem Eluieren neutralisiert.
Das eluierte, neutralisierte gDCD4T wird dann durch Ultrafiltration
konzentriert und dialysiert/diafiltriert, um den Puffer gegen eine
physiologische Salzlösung,
die Tween 80 zu etwa 0,1 % (Gew./Vol.) enthält, auszutauschen.
-
Falls
das Detergens nicht vorhanden ist, bildet das gDCD4T Aggregate,
wie dies durch die Fähigkeit der
Zentrifugierung bei etwa 10.000 × g 2 Minuten lang gezeigt wird,
um das gDCD4T aus der Lösung
zu entfernen. Eine Inkubation von gDCD4T bei 4 °C in 0,1 M Natriumacetat, 0,5
M NaCl und 0,25 M Tris bei einem pH von 7 zusammen mit BSA, Tween
80 oder Glycerin als Kandidatenstabilisatoren zeigte, dass gDCD4T
in Abwesenheit eines Stabilisators über einen Zeitraum von 12 Tagen
nach und nach bis zu dem Punkt aggregierte, an dem lediglich etwa
60-70 % des Proteins löslich
waren. Die Verwendung von 0,1 % (Gew./Vol.) Tween 80 oder 0,5 mg/ml
BSA stellte jedoch sicher, dass etwa 100 % bzw. 80 % des gDCD4T über diesen Zeitraum
löslich
blieben. Überraschenderweise
war Glycerin als Stabilisator nicht wirksam und ergab sogar schlechtere
Resultate als die Kontrollgruppe – nach 8 Tagen waren etwa 80
% des gDCD4T aggregiert, wenn dieses in Gegenwart von Glycerin aufgewahrt
wurde.
-
Beispiel 4
-
Es
wurden Plasmide konstruiert, um die Expression von Proteinen zu
steuern, die verschiedene Längen
der extrazellulären
aminoterminalen Domäne
von CD4 enthielten, die an die konstante Region von menschlichem
Immunglobulin γ1
fusioniert war. Diese Plasmide werden als pRKCD42γ1,
pRKCD4e4γ1, pRKCD42γ1,
pRKCD4e2γ1, pRKCD41γ1 und
pRKCD4e1γ1 bezeichnet.
-
Das
Plasmid pRKCD44γ1 enthält den Teil
des CD4-Gens vom Initiationscodon bis zur Fusionsstelle nach dem
Codon für
den Serin-Rest 366 des reifen CD4-Polypeptids, unmittelbar gefolgt
von der für
die konstante Region des menschlichen Immunglobulins γ1 kodierenden
Sequenz, beginnend beim Codon für
den Serin-Rest 114 des reifen menschlichen Immunglobulins γ1 (Kabat
et al.).
-
Das
Plasmid pRKCD4e4γ1 enthält den Teil
des CD4-Gens vom Initiationscodon bis zur Fusionsstelle nach dem
Codon für
den Lysin-Rest 360 des reifen CD4-Polypeptids, unmittelbar gefolgt von
der für
die konstante Region des menschlichen Immunglobulins γ1 kodierenden
Sequenz, beginnend beim Codon für
den Serin-Rest 114 des reifen menschlichen Immunglobulins γ1 (Kabat
et al.).
-
Das
Plasmid pRKCD42γ1 enthält den Teil
des CD4-Gens vom Initiationscodon bis zur Fusionsstelle nach dem
Codon für
den Glutamin-Rest 180 des reifen CD4-Polypeptids, unmittelbar gefolgt von
der für
die konstante Region des menschlichen Immunglobulins γ1 kodierenden
Sequenz, beginnend beim Codon für
den Serin-Rest 114 des reifen menschlichen Immunglobulins γ1 (Kabat
et al.).
-
Das
Plasmid pRKCD4e2γ1 enthält den Teil
des CD4-Gens vom Initiationscodon bis zur Fusionsstelle nach dem
Codon für
den Leucin-Rest 177 des reifen CD4-Polypeptids, unmittelbar gefolgt von
der für
die konstante Region des menschlichen Immunglobulins γ1 kodierenden
Sequenz, beginnend beim Codon für
den Serin-Rest 114 des reifen menschlichen Immunglobulins γ1 (Kabat
et al.).
-
Das
Plasmid pRKCD41γ1 enthält den Teil
des CD4-Gens vom Initiationscodon bis zur Fusionsstelle nach dem
Codon für
den Asparaginsäure-Rest
105 des reifen CD4-Polypeptids,
unmittelbar gefolgt von der für die
konstante Region des menschlichen Immunglobulins γ1 kodierenden
Sequenz, beginnend beim Codon für den
Serin-Rest 114 des reifen menschlichen Immunglobulins γ1 (Kabat
et al.).
-
Das
Plasmid pRKCD4e1γ1 enthält den Teil
des CD4-Gens vom Initiationscodon bis zur Fusionsstelle nach dem
Codon für
den Leucin-Rest 100 des reifen CD4-Polypeptids, unmittelbar gefolgt von
der für
die konstante Region des menschlichen Immunglobulins γ1 kodierenden
Sequenz, beginnend beim Codon für
den Serin-Rest 114 des reifen menschlichen Immunglobulins γ1 (Kabat
et al.).
-
Die
Konstruktion dieser Plasmide erforderte die vorausgehende Konstruktion
des Plasmids pRKCD4TP/γ1.
Dieses wurde wie folgt konstruiert:
Ein cDNA-Klon, der für das menschliche
Immunglobulin γ1
kodiert, wurde aus einer menschlichen Milz-cDNA-Bibliothek (Clontech
Laboratories, Inc.) unter Verwendung von Oligonucleotiden erhalten,
die auf der veröffentlichten
Sequenz basierten (Ellison et al., Nucl. Acids Res. 10, 4071-4079
[1982]), und ein EcoRI-EagI-Fragment (die EcoRI-Stelle wurde von
einem Linker beigesteuert; siehe 4a, b),
das einen Teil der variablen und die gesamte konstante Region enthielt,
wurde erhalten. Dieses Fragment wurde mit einem Klenow-Fragment
abgestumpft und mittels Gelelektrophorese (Fragment a1) gewonnen.
-
Das
Plasmid pRKCD4TP-kk, das für
eine Substitutionsvariante eines löslichen CD4 (Reste 1-368) kodiert,
das einen Lysin-Rest anstelle eines Asparagins an Position 1 des
reifen Polypeptids trägt,
wurde mittels ortspezifischer Mutagenese aus dem Plasmid pRKCD4TP
konstruiert. Ein synthetisches Oligonucleotid wurde als ein Primer
für eine
Mutagenesereaktion hergestellt, um die gewünschte kodierende Sequenz zu
erhalten. Diese wurde als ein 51-mer synthetisiert, das zwei stille
Mutationen aus der natürlichen
Sequenz zusätzlich
zu der Substitutionsmutation sowie 21 Basen auf jeder Seite der
mutierten Codons enthielt:
5'-CCC TTT TTT GCC CAG CAC CAC CTT CTT
GCC CTG-
AGT GGC TGC TGG GAG GAG-3'
-
Das
Plasmid pRKCD4TP wurde in den E.-coli-Stamm SR101 transformiert,
und die transformierten Kolonien wurden auf Ampicillin-Mediumplatten
ausplattiert. Es wurden resistente Kolonien ausgewählt und
in Gegenwart von m13K07-Helfer-Bakteriophagen
kultiviert, um sekretierte, enkapsidierte, einzelsträngige Matrizen
von pRKCD4TP zu erhalten. Die einzelsträngige Plasmid-DNA wurde isoliert
und mit den oben beschriebenen synthetischen Oligonucleotiden als
Primer als Matrize für
Mutagenesereaktionen verwendet. Die Mutagenesereaktion wurde in
E. coli SR101 transformiert, und die transformierte Kultur wurde
auf Ampicillin-Mediumplatten ausplattiert. Die Transformanten wurden
mittels Koloniehybridisierung (siehe Grunstein-Hogness) unter Verwendung des folgenden
16-mers als Sonde auf die Gegenwart der geeigneten Sequenz gescreent.
5'-C CAC CTT CTT GCC
CTG-3'
-
Die
ausgewählten
Hybridisierungsbedingungen waren stringent genug, sodass die Sonde
lediglich das korrekt fusionierte Produkt nachweist. Als positiv
identifizierte Kolonien wurden ausgewählt, und die Plasmid-DNA wurde
isoliert und in den E.-coli-Stamm
SR101 transformiert. Die transformierten Kulturen wurden auf Ampicillin-Mediumplatten ausplattiert,
und es wurden resistente Kolonien ausgewählt und in Gegenwart von m13K07-Bakteriophagen
kultiviert. Matrizen wurden wie oben beschrieben hergestellt und
mittels Sequenzierung gescreent.
-
Das
Plasmid pRKCD4TP-kk wurde mit Xbal verdaut und mit dem Klenow-Enzym
behandelt, und das Fragment a2, das das linearisierte Plasmid enthielt,
wurde mittels Gelelektrophorese gewonnen und mit dem Fragment a1
ligiert. Das Ligationsgemisch wurde in den E.-coli-Stamm 294 transformiert,
die transformierte Kultur wurde auf Ampicillin-Mediumplatten ausplattiert,
und es wurden resistente Kolonien ausgewählt. Aus den Transformanten
wurde Plasmid-DNA hergestellt und mittels Restriktionsanalyse auf
die Gegenwart des korrekten Fragments in der korrekten Ausrichtung
(d.h. die kodierende Region des Immunglobulins in der gleichen Ausrichtung
wie die kodierende Region von CD4 sowie am 3'-Ende der kodierenden Region von CD4) untersucht.
Dieses Plasmid wird als pRKCD4TP/γ1
bezeichnet.
-
Synthetische
Oligonucleotide wurden als Primer für Deletionsmutagenesereaktionen
hergestellt, um die geeigneten kodierenden Sequenzen von IgG1 und
CD4 wie oben beschrieben zu fusionieren. Diese wurden als 48-mere,
die 24 Nucleotide auf jeder Seite der gewünschten Fusionsstelle umfassen
(d.h. entsprechend den 8 COOH-terminalen
Resten der gewünschten
CD4-Gruppierung sowie den 8 NH2-terminalen
Resten der gewünschten
Immunglobulin-Gruppierung), synthetisiert. Das Plasmid pRKCD4TP/γ1 wurde in
den E.-coli-Stamm SR101 transformiert, und die transformierten Kulturen
wurden auf Ampicillin-Mediumplatten ausplattiert. Es wurden resistente
Kolonien ausgewählt
und in Gegenwart von m13K07-Helfer-Bakteriophagen kultiviert, um
sekretierte, enkapsidierte, einzelsträngige Matrizen von pRKCD4TP/γ1 zu erhalten.
Die einzelsträngige
Plasmid-DNA wurde isoliert und mit den oben beschriebenen synthetischen
Oligonucleotiden als Primer als Matrize für Mutagenesereaktionen verwendet.
Die Mutagenesereaktionen wurden in E. coli SR101 transformiert,
und die transformierte Kultur wurde auf Ampicillin-Mediumplatten
ausplattiert.
-
Die
Transformanten wurden mittels Koloniehybridisierung (siehe Grunstein-Hogness)
unter Verwendung von 16-meren als Sonden auf die Gegenwart der geeigneten
Fusionsstelle gescreent. Die 16-mere umfassen 8 Basen an jeder Seite
der Fusionsstelle, und die ausgewählten Hybridisierungsbedingungen
waren stringent genug, sodass die Sonden lediglich das korrekt fusionierte
Produkt nachweisen. Als positiv identifizierte Kolonien wurden ausgewählt, und
die Plasmid-DNA wurde isoliert und in den E.-coli-Stamm SR101 transformiert.
Die transformierten Kulturen wurden auf Ampicillin-Mediumplatten
ausplattiert, und es wurden resistente Kolonien ausgewählt und
in Gegenwart von m13K07-Bakteriophagen kultiviert. Matrizen wurden
wie oben beschrieben hergestellt und mittels Sequenzierung gescreent.
-
Die
Plasmide wurden unter Verwendung von Standardverfahren in 293-Zellen
transfiziert und wie oben beschrieben auf Expression und Produktion
getestet.
-
-
Es
wurden ebenfalls Plasmide konstruiert, um die Expression von Fusionsproteinen
zu steuern, die verschiedene Längen
der aminoterminalen extrazellulären
Domäne
von CD4 enthielten, die an den trunkierten Teil der konstanten Region
des menschlichen Immunglobulins γ1
fusioniert sind, und lediglich die Gelenkregion und die konstanten
Domänen
CH2 und CH3 umfassen.
-
Synthetische
Oligonucleotide wurden als Primer für Mutagenesereaktionen hergestellt,
um die Immunglobulinsequenz von Ser114 bis Cys215 (inklusive) zu
deletieren (Kabat et al.). Diese wurden als 48-mere, die 24 Nucleotide
auf jeder Seite der ge wünschten
Fusionsstelle umfassen (d.h. entsprechend den 8 COOH-terminalen
Resten der gewünschten
CD4-Gruppierung sowie den 8 NH2-terminalen
Resten der gewünschten
Immunglobulin-Gruppierung), synthetisiert. Die Plasmide pRKCD44γ1, pRKCD42γ1 und
pRKCD41γ1 wurden getrennt voneinander in den E.-coli-Stamm
SR101 transformiert, und die transformierte Kultur wurde auf Ampicillin-Mediumplatten ausplattiert.
Es wurden resistente Kolonien ausgewählt und in Gegenwart von m13K07-Helfer-Bakteriophagen
kultiviert, um sekretierte, enkapsidierte, einzelsträngige Matrizen
dieser Plasmide zu erhalten. Die einzelsträngige Plasmid-DNA wurde isoliert
und mit den oben beschriebenen synthetischen Oligonucleotiden als
Primer als Matrize für
Mutagenesereaktionen verwendet. Die Mutagenesereaktionen wurden
in E. coli SR101 transformiert, und die transformierte Kultur wurde
auf Ampicillin-Mediumplatten ausplattiert. Die Transformanten wurden
mittels Koloniehybridisierung (Grunstein-Hogness) unter Verwendung von
16-meren als Sonden auf die Gegenwart der geeigneten Fusionsstelle
gescreent. Diese 16-mere umfassen 8 Basen an jeder Seite der Fusionsstelle,
und die ausgewählten
Hybridisierungsbedingungen waren stringent genug, sodass die Sonden
lediglich das korrekt fusionierte Produkt nachweisen. Als positiv
identifizierte Kolonien wurden ausgewählt, und die Plasmid-DNA wurde
isoliert und in den E.-coli-Stamm SR101 transformiert. Die transformierten
Kulturen wurden auf Ampicillin-Mediumplatten ausplattiert, und es
wurden resistente Kolonien ausgewählt und in Gegenwart von m13K07-Bakteriophagen
kultiviert. Matrizen wurden wie oben beschrieben hergestellt und
mittels Sequenzierung gescreent.
-
Das
vom Plasmid pRKCD44γ1 abstammende
Plasmid wird als pRKCD44Fc1 bezeichnet,
jenes, das vom Plasmid pRKCD42γ1 abstammt, wird als pRKCD42Fc1 bezeichnet und jenes, das vom Plasmid
pRKCD41γ1 abstammt,
wird als pRKCD41Fc1 bezeichnet.
-
pRKCD42Fc1, pRKCD41Fc1 und
pRKCD44Fc1 werden auf dieselbe Art und Weise
wie oben beschrieben kultiviert, und die CH1-deletierten CD4-Immunadhesone
werden wie hierin anderorts beschrieben gewonnen.
-
Leichtkettenfusionen
-
Es
wurden Plasmide konstruiert, um die Expression von Proteinen zu
steuern, die verschiedene Längen
der aminoterminalen extrazellulären
Domäne
von CD4 enthielten, die an die konstante Region des menschlichen
Immunglobulins κ fusioniert
ist. Diese Plasmide werden als pRKCD44κ und pRKCD4e4κ bezeichnet.
-
Das
Plasmid pRKCD44κ enthält den Abschnitt des CD4-Gens
vom Initiationscodon bis zur Fusionsstelle nach dem Codon für den Serin-Rest
366 des reifen CD4-Polypeptids,
unmittelbar gefolgt von der Sequenz für die konstante Region des
menschlichen Immunglobulins κ,
beginnend beim Codon für
den Threonin-Rest 109 des reifen menschlichen Immunglobulins κ (Kabat et
al.).
-
Das
Plasmid pRKCD4e4κ enthält den Teil des CD4-Gens vom
Initiationscodon bis zur Fusionsstelle nach dem Codon für den Lysin-Rest
360 des reifen CD4-Polypeptids, unmittelbar gefolgt von der Sequenz
für die
konstante Region des menschlichen Immunglobulins κ, beginnend
beim Codon für
den Threonin-Rest 109 des reifen menschlichen Immunglobulins κ (Kabat et
al.).
-
Diese
Plasmide wurden analog zu den oben beschriebenen Plasmiden pRKCD44γ1 und pRKCD4e4γ1 konstruiert,
jedoch mit folgender Ausnahme:
Die kodierende Sequenz des menschlichen
Immunglobulins κ (5)
wurde aus einer menschlichen Milz-cDNA-Bibliothek (Clontech Laboratories,
Inc.) unter Verwendung von Oligonucleotiden erhalten, die auf der
veröffentlichten
Sequenz basierten (P.A. Hieter et al., Cell 22, 197-207 [1980]),
und ein EcoRI-BspMI-Fragment, das einen Teil der variablen Region
und die gesamte konstante Region enthielt, wurde erhalten (siehe 5). Dieses
Fragment wurde mit einem Klenow-Fragment und den vier dNTPs abgestumpft.
Dieses Fragment wurde anstelle des Fragments a1 verwendet und wurde
benutzt, um das Plasmid pRKCD4TP/hκ zu erhalten.
-
Expression
in CHO-Zellen
-
Plasmide
wurden oder werden konstruiert, um die Expression der oben beschriebenen
Immunadhesone in CHO-Zellen zu steuern. Sie werden als pSVeCD44γ1SVDHFR, pSVeCD42γ1SVDHFR, pSVeCD41γ1SVDHFR, pSVeCD4e4γ1SVDHFR,
pSVeCD4e2γ1SVDHFR, pSVeCD4e1γ1SVDHFR, pSVeCD44Fc1SVDHFR, pSVeCD42Fc1SVDHFR,
pSVeCD41Fc1SVDHFR, pSVeCD44κSVDHFR und pSVeCD42κSVDHFR
bezeichnet.
-
Fragment
31 wurde wie oben beschrieben hergestellt. Fragment 32a wurde durch
den Verdau des Plasmids pE348HBV.E400 D22 mit BamHI, Abstumpfen
mit dem Klenow-Fragment und den vier dNTPs und darauf folgendem
Verdau mit PvuI und Isolation des großen Fragments hergestellt,
das den Rest des β-Lactamase-Gens
und den frühen
Promotor von SV40 sowie das DHFR-Gen enthielt. Die Plasmide pRKCD44γ1, pRKCD42γ1,
pRKCD41γ1, pRKCD4e4γ1,
pRKCD4e2γ1, pRKCD4e1γ1,
pRKCD44Fc1, pRKCD42Fc1,
pRKCD41Fc1, pRKCD44κ und
pRKCD42κ wurden
getrennt voneinander mit HindIII verdaut, mit dem Klenow-Fragment
und den vier dNTPs abgestumpft, dann mit EcoRI verdaut, und die
für das
CD4-Ig-Fusionsprotein kodierenden Fragmente wurden isoliert. Die
resultierenden DNA-Fragmente wurden mit den Fragmenten 31 und 32a
zusammen ligiert und in den E.-coli-Stamm 294 transformiert. Kolonien
wurden ausgewählt
und wie oben auf die Gegenwart des korrekten Plasmids untersucht,
dann in CHO-Zellen transfiziert und durch Methotrexatselektion unter
Verwendung konventioneller Verfahren amplifiziert.
-
Beispiel 5
-
Kultivierung, Reinigung
und Formulierung von CD4-Varianten
-
Für lösliche CD4-Adhesone,
wie z.B. CD4T, CD4TP, oder lösliche
CD4-Immunadhesone
kodierende Plasmide wurden in CHO-DP7 (eine proinsulintransformierte,
autokrine Wirtszelle, die von CHO abstammt; U.S.S.N. 97.472) calci umphosphat-transfiziert,
und die Transformanten wurden in selektivem Medium (1:1 HAM F12/DMEM
GHT-, das 1–10 % diafiltriertes oder dialysiertes
Rinderserum enthielt) kultiviert. Andere geeignete Wirtszellen sind
CHO-Zellen oder menschliche embryonale 293S-Nierenzellen. Die Transformanten wurden
durch Methotrexatselektion im selben Medium, das jedoch 500 nm Methotrexat
enthielt, amplifiziert. Es wurde ein Subklon ausgewählt, der
fähig war,
CD4TP, CD4tp 500 b zu sekretieren. CD4tp 500 b wird in einem DMEM/HAM-F12-Medium
bei etwa 37 °C
kultiviert, bis das CD4TP in der Kultur akkumuliert, wonach das
Medium von den Zellen und von unlöslichen Stoffen durch Zentrifugieren
getrennt wird.
-
Kulturfluid
von CD4TP-Transformanten wurde konzentriert und diafiltriert, um
die Ionenstärke
zu reduzieren. Das Konzentrat wurde durch eine große Menge
an Q-Sepharose-Anionenaustauschharz
(zuvor mit 25 mM NaCl, pH 8,5, äquilibriert)
laufen gelassen, um verunreinigende Stoffe aus der Kulturflüssigkeit
zu adsorbieren. Der isoelektrische Punkt von CD4TP liegt bei etwa
9,5, was eine Unterscheidung zwischen den trunkierten Formen von
CD4 und den meisten Verunreinigungen aufgrund von unterschiedlicher
Adsorption auf einem Kationenaustauschharz, wie z.B. Carboxymethyl-
oder Sulfonylsepharose, bzw. einem Anionenaustauschharz, wie z.B.
quaternäre
Ammoniumsepharose, ermöglicht.
Zusätzlich
wird, da hochgradig elektropositive Domänen im extrazellulären Segment
von CD4 vorhanden sind, jede CD4-haltige Variante auf dieselbe Art
und Weise wie CD4TP gereinigt. Die nicht adsorbierte Kulturflüssigkeit
aus dem Anionenaustauschharz involvierenden Schritt wurde dann durch
ein Kationenaustauschharz (zuvor mit 25 mM NaCl, pH 8,5, äquilibriert) laufen
gelassen, wodurch CD4TP an das Harz adsorbiert wurde. Das CD4TP
wurde mit einem NaCl-Gradienten bei einem pH von 8,5 eluiert, wobei
diese CD4-Variante
bei etwa 0,2 M NaCl eluiert. Ammoniumsulfat wurde zu dem Eluat bis
zu einer Konzentration von 1,7 M hinzugefügt, und die Lösung wurde
durch eine Säule aus
hydrophobem Wechselwirkungschromatographieharz (Phenyl- oder Butylsepharose)
laufen gelassen. Das CD4TP wurde aus der hydrophoben Wechselwirkungssäule mit
einem Gradienten aus Ammoniumsulfat eluiert, wobei das CD4TP bei
etwa 0,7 M Ammoniumsulfat austrat. Das Eluat wurde konzentriert,
und der Puffer wurde auf einer G-25-Säule unter Verwendung von phosphatgepufferter
Salzlösung,
die 0,02 % (Gew./Vol.) Tween 20 oder Tween 80 enthielt, ausgetauscht.
Das CD4TP war löslich
und stabil in dieser Lösung,
die sterilfiltriert war und als wässrige Formulierung in Phiolen
gefüllt
wurde. Andere polymere, nichtionische Tenside werden geeigneterweise
mit den CD4-Formulierungen verwendet, unter anderem Pluronic-Block-Copolymere oder
Polyethylenglykol.
-
Es
ist ebenso möglich,
eine Immunaffinitätsreinigung
von löslichem
CD4 durchzuführen,
wobei das CD4 an einen immobilisierten Antikörper gegen CD4 adsorbiert wird.
Dieses Verfahren leidet unter dem Nachteil, dass eine Elution des
löslichen
CD4 unter sauren Bedingungen zu einer Proteinaggregation führt, die
lediglich bei relativ hohen Tensidmengen weitgehend zurückgeht.
Das vorhergehende Verfahren ermöglicht
die Verwendung von viel geringeren Tensidmengen, etwa von 0,01 bis
0,10 (Gew./Vol.) Tensid.
-
Das
angewandte Verfahren für
die Reinigung von CD4-Fusionen mit Immunglobulin-Schwerketten bestand in der Konzentration
rekombinanter Überstände mittels
Ultrafiltration und darauf folgender Adsorption der Fusion an harz-immobilisiertem
Staphylokokken-Protein A. Die Fusion wurde mit 0,1 M Citratpuffer
mit einem pH von 3 ohne Salz oder Detergens eluiert. Dieses Präparat wird
in Tris-Puffer bei einem pH von 7,5 gepuffert. Die Immunglobulin-Fusionen
mit V1-V4 von CD4 werden gegebenenfalls durch das oben für nicht
fusionierte CD4-Varianten beschriebene Verfahren weiter gereinigt.
CD4-Immunglobulin-Fusionen mit V1-V4 von CD4 können ebenfalls durch das oben
beschriebene Verfahren gereinigt werden, jedoch mit der Ausnahme,
dass nicht erwartet wird, dass der isoelektrische Punkt dieser Molekülklasse
so alkalisch ist wie jener der Arten, die alle vier V-Regionen von
CD4 enthalten.
-
Beispiel 6
-
Es
wurden die Merkmale mehrerer Adhesonvarianten bestimmt. Wie in Tabelle
4 gezeigt wird, zeigen die Immunadhesone CD4
4γ
1 und
CD4
2γ
1 eine verbesserte Plasmahalbwertszeit in
Kaninchen, gekoppelt mit hochaffiner gp120-Bindung und einer Affi nität zum Fcγ-Rezeptor
(bestimmt durch U937-Zellen), die mit der des Hauptteils des menschlichen
IgG1 vergleichbar ist. Tabelle
4
- * in Menschen bestimmt
- + KD wurde durch das Verfahren von Anderson et al., J. Immunol.
125, 2735-2741 (1980), bestimmt.
- # durch das Verfahren von Smith et al., Science 238, 1704-07
(1987), bestimmt.
- § nur
Reste 1-368
- ++ Die Adhesonvariante wurde Kaninchen intravenös injiziert,
und es wurden in regelmäßigen Abständen Blutproben
genommen und auf die Gegenwart der Adhesonvariante getestet.
- ** nicht durchgeführt
SEQUENZPROTOKOLL