DE60005183T2 - Polymerisierbare sauere verbindungen und verfahren zur herstellung - Google Patents
Polymerisierbare sauere verbindungen und verfahren zur herstellung Download PDFInfo
- Publication number
- DE60005183T2 DE60005183T2 DE60005183T DE60005183T DE60005183T2 DE 60005183 T2 DE60005183 T2 DE 60005183T2 DE 60005183 T DE60005183 T DE 60005183T DE 60005183 T DE60005183 T DE 60005183T DE 60005183 T2 DE60005183 T2 DE 60005183T2
- Authority
- DE
- Germany
- Prior art keywords
- independently
- alkyl
- formula
- reaction
- alkyl group
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Lifetime
Links
- 150000001875 compounds Chemical class 0.000 title claims description 38
- 239000002253 acid Substances 0.000 title claims description 24
- 238000004519 manufacturing process Methods 0.000 title description 3
- 238000006243 chemical reaction Methods 0.000 claims description 31
- 239000003054 catalyst Substances 0.000 claims description 19
- 125000004400 (C1-C12) alkyl group Chemical group 0.000 claims description 18
- 125000000217 alkyl group Chemical group 0.000 claims description 16
- XBDQKXXYIPTUBI-UHFFFAOYSA-N dimethylselenoniopropionate Natural products CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 claims description 10
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 9
- 238000000034 method Methods 0.000 claims description 9
- 239000007795 chemical reaction product Substances 0.000 claims description 8
- 239000012948 isocyanate Substances 0.000 claims description 8
- 150000002513 isocyanates Chemical class 0.000 claims description 8
- 125000001424 substituent group Chemical group 0.000 claims description 8
- 239000000047 product Substances 0.000 claims description 6
- IUVKMZGDUIUOCP-BTNSXGMBSA-N quinbolone Chemical compound O([C@H]1CC[C@H]2[C@H]3[C@@H]([C@]4(C=CC(=O)C=C4CC3)C)CC[C@@]21C)C1=CCCC1 IUVKMZGDUIUOCP-BTNSXGMBSA-N 0.000 claims description 6
- 229920002939 poly(N,N-dimethylacrylamides) Polymers 0.000 claims description 5
- 235000019260 propionic acid Nutrition 0.000 claims description 5
- AHLWZBVXSWOPPL-RGYGYFBISA-N 20-deoxy-20-oxophorbol 12-myristate 13-acetate Chemical compound C([C@]1(O)C(=O)C(C)=C[C@H]1[C@@]1(O)[C@H](C)[C@H]2OC(=O)CCCCCCCCCCCCC)C(C=O)=C[C@H]1[C@H]1[C@]2(OC(C)=O)C1(C)C AHLWZBVXSWOPPL-RGYGYFBISA-N 0.000 claims description 4
- 241001602688 Pama Species 0.000 claims description 4
- 230000003197 catalytic effect Effects 0.000 claims description 4
- 239000007809 chemical reaction catalyst Substances 0.000 claims description 4
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Chemical compound NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 claims description 3
- 150000001408 amides Chemical class 0.000 claims description 3
- 239000004202 carbamide Substances 0.000 claims description 3
- 238000002360 preparation method Methods 0.000 claims description 3
- 150000003839 salts Chemical class 0.000 claims description 2
- 150000003512 tertiary amines Chemical class 0.000 claims description 2
- IUTCEZPPWBHGIX-UHFFFAOYSA-N tin(2+) Chemical compound [Sn+2] IUTCEZPPWBHGIX-UHFFFAOYSA-N 0.000 claims description 2
- SYRHIZPPCHMRIT-UHFFFAOYSA-N tin(4+) Chemical compound [Sn+4] SYRHIZPPCHMRIT-UHFFFAOYSA-N 0.000 claims description 2
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 18
- 239000000178 monomer Substances 0.000 description 17
- 239000000463 material Substances 0.000 description 12
- 239000000203 mixture Substances 0.000 description 8
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 6
- 230000002378 acidificating effect Effects 0.000 description 6
- 239000000243 solution Substances 0.000 description 6
- 239000007858 starting material Substances 0.000 description 6
- SFUUDZYXHNYCTM-UHFFFAOYSA-N 2-methylprop-2-enamide;prop-2-enamide Chemical compound NC(=O)C=C.CC(=C)C(N)=O SFUUDZYXHNYCTM-UHFFFAOYSA-N 0.000 description 5
- RBQRWNWVPQDTJJ-UHFFFAOYSA-N methacryloyloxyethyl isocyanate Chemical compound CC(=C)C(=O)OCCN=C=O RBQRWNWVPQDTJJ-UHFFFAOYSA-N 0.000 description 5
- 239000002904 solvent Substances 0.000 description 5
- NLZUEZXRPGMBCV-UHFFFAOYSA-N Butylhydroxytoluene Chemical compound CC1=CC(C(C)(C)C)=C(O)C(C(C)(C)C)=C1 NLZUEZXRPGMBCV-UHFFFAOYSA-N 0.000 description 4
- UKLDJPRMSDWDSL-UHFFFAOYSA-L [dibutyl(dodecanoyloxy)stannyl] dodecanoate Chemical compound CCCCCCCCCCCC(=O)O[Sn](CCCC)(CCCC)OC(=O)CCCCCCCCCCC UKLDJPRMSDWDSL-UHFFFAOYSA-L 0.000 description 4
- 235000010354 butylated hydroxytoluene Nutrition 0.000 description 4
- 239000005548 dental material Substances 0.000 description 4
- 239000012975 dibutyltin dilaurate Substances 0.000 description 4
- PTBDIHRZYDMNKB-UHFFFAOYSA-N 2,2-Bis(hydroxymethyl)propionic acid Chemical compound OCC(C)(CO)C(O)=O PTBDIHRZYDMNKB-UHFFFAOYSA-N 0.000 description 3
- 150000007513 acids Chemical class 0.000 description 3
- 239000004568 cement Substances 0.000 description 3
- 125000001261 isocyanato group Chemical group *N=C=O 0.000 description 3
- 229910052751 metal Inorganic materials 0.000 description 3
- 239000002184 metal Substances 0.000 description 3
- 239000011347 resin Substances 0.000 description 3
- 229920005989 resin Polymers 0.000 description 3
- 239000003381 stabilizer Substances 0.000 description 3
- 238000003756 stirring Methods 0.000 description 3
- GQHTUMJGOHRCHB-UHFFFAOYSA-N 2,3,4,6,7,8,9,10-octahydropyrimido[1,2-a]azepine Chemical compound C1CCCCN2CCCN=C21 GQHTUMJGOHRCHB-UHFFFAOYSA-N 0.000 description 2
- 0 CC(C)**(NC(OC(C)(C)CC(C(O)O)N)=O)=C Chemical compound CC(C)**(NC(OC(C)(C)CC(C(O)O)N)=O)=C 0.000 description 2
- JOYRKODLDBILNP-UHFFFAOYSA-N Ethyl urethane Chemical compound CCOC(N)=O JOYRKODLDBILNP-UHFFFAOYSA-N 0.000 description 2
- -1 ZnF 2 Inorganic materials 0.000 description 2
- 239000000853 adhesive Substances 0.000 description 2
- 230000001070 adhesive effect Effects 0.000 description 2
- 239000002521 compomer Substances 0.000 description 2
- 239000002131 composite material Substances 0.000 description 2
- 239000003479 dental cement Substances 0.000 description 2
- PWEVMPIIOJUPRI-UHFFFAOYSA-N dimethyltin Chemical compound C[Sn]C PWEVMPIIOJUPRI-UHFFFAOYSA-N 0.000 description 2
- 238000009826 distribution Methods 0.000 description 2
- 239000012467 final product Substances 0.000 description 2
- 230000009969 flowable effect Effects 0.000 description 2
- 150000002221 fluorine Chemical class 0.000 description 2
- 238000009472 formulation Methods 0.000 description 2
- 238000007306 functionalization reaction Methods 0.000 description 2
- 239000011521 glass Substances 0.000 description 2
- 239000003178 glass ionomer cement Substances 0.000 description 2
- 150000001261 hydroxy acids Chemical class 0.000 description 2
- 239000007788 liquid Substances 0.000 description 2
- 239000002244 precipitate Substances 0.000 description 2
- 239000007787 solid Substances 0.000 description 2
- 238000004381 surface treatment Methods 0.000 description 2
- IMNIMPAHZVJRPE-UHFFFAOYSA-N triethylenediamine Chemical compound C1CN2CCN1CC2 IMNIMPAHZVJRPE-UHFFFAOYSA-N 0.000 description 2
- 238000003828 vacuum filtration Methods 0.000 description 2
- AHDSRXYHVZECER-UHFFFAOYSA-N 2,4,6-tris[(dimethylamino)methyl]phenol Chemical compound CN(C)CC1=CC(CN(C)C)=C(O)C(CN(C)C)=C1 AHDSRXYHVZECER-UHFFFAOYSA-N 0.000 description 1
- GTEXIOINCJRBIO-UHFFFAOYSA-N 2-[2-(dimethylamino)ethoxy]-n,n-dimethylethanamine Chemical compound CN(C)CCOCCN(C)C GTEXIOINCJRBIO-UHFFFAOYSA-N 0.000 description 1
- CMCOFAYLDYIEBR-UHFFFAOYSA-L 2-[carboxymethylsulfanyl(dioctyl)stannyl]sulfanylacetic acid Chemical compound [O-]C(=O)CS.[O-]C(=O)CS.CCCCCCCC[Sn+2]CCCCCCCC CMCOFAYLDYIEBR-UHFFFAOYSA-L 0.000 description 1
- IGMYAFRPSRRXNA-UHFFFAOYSA-L 2-[dibutyl(carboxymethylsulfanyl)stannyl]sulfanylacetic acid Chemical compound [O-]C(=O)CS.[O-]C(=O)CS.CCCC[Sn+2]CCCC IGMYAFRPSRRXNA-UHFFFAOYSA-L 0.000 description 1
- VPCAFPAKZIJBRH-UHFFFAOYSA-N 2-methylprop-2-enoic acid;oxiran-2-ylmethyl 2-methylprop-2-enoate Chemical group CC(=C)C(O)=O.CC(=C)C(=O)OCC1CO1 VPCAFPAKZIJBRH-UHFFFAOYSA-N 0.000 description 1
- ZRWNRAJCPNLYAK-UHFFFAOYSA-N 4-bromobenzamide Chemical compound NC(=O)C1=CC=C(Br)C=C1 ZRWNRAJCPNLYAK-UHFFFAOYSA-N 0.000 description 1
- HVCNXQOWACZAFN-UHFFFAOYSA-N 4-ethylmorpholine Chemical compound CCN1CCOCC1 HVCNXQOWACZAFN-UHFFFAOYSA-N 0.000 description 1
- 229910016569 AlF 3 Inorganic materials 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- IAYPIBMASNFSPL-UHFFFAOYSA-N Ethylene oxide Chemical group C1CO1 IAYPIBMASNFSPL-UHFFFAOYSA-N 0.000 description 1
- KRHYYFGTRYWZRS-UHFFFAOYSA-M Fluoride anion Chemical compound [F-] KRHYYFGTRYWZRS-UHFFFAOYSA-M 0.000 description 1
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 1
- CERQOIWHTDAKMF-UHFFFAOYSA-N Methacrylic acid Chemical compound CC(=C)C(O)=O CERQOIWHTDAKMF-UHFFFAOYSA-N 0.000 description 1
- UEEJHVSXFDXPFK-UHFFFAOYSA-N N-dimethylaminoethanol Chemical compound CN(C)CCO UEEJHVSXFDXPFK-UHFFFAOYSA-N 0.000 description 1
- 229910008449 SnF 2 Inorganic materials 0.000 description 1
- ISKQADXMHQSTHK-UHFFFAOYSA-N [4-(aminomethyl)phenyl]methanamine Chemical compound NCC1=CC=C(CN)C=C1 ISKQADXMHQSTHK-UHFFFAOYSA-N 0.000 description 1
- KYDGMZSIZYYJJJ-UHFFFAOYSA-L [dimethyl-(2-sulfanylacetyl)oxystannyl] 2-sulfanylacetate Chemical compound C[Sn+2]C.[O-]C(=O)CS.[O-]C(=O)CS KYDGMZSIZYYJJJ-UHFFFAOYSA-L 0.000 description 1
- XQBCVRSTVUHIGH-UHFFFAOYSA-L [dodecanoyloxy(dioctyl)stannyl] dodecanoate Chemical compound CCCCCCCCCCCC(=O)O[Sn](CCCCCCCC)(CCCCCCCC)OC(=O)CCCCCCCCCCC XQBCVRSTVUHIGH-UHFFFAOYSA-L 0.000 description 1
- 239000000654 additive Substances 0.000 description 1
- 150000001298 alcohols Chemical class 0.000 description 1
- 125000003118 aryl group Chemical group 0.000 description 1
- 239000002585 base Substances 0.000 description 1
- NSPSPMKCKIPQBH-UHFFFAOYSA-K bismuth;7,7-dimethyloctanoate Chemical compound [Bi+3].CC(C)(C)CCCCCC([O-])=O.CC(C)(C)CCCCCC([O-])=O.CC(C)(C)CCCCCC([O-])=O NSPSPMKCKIPQBH-UHFFFAOYSA-K 0.000 description 1
- 239000006227 byproduct Substances 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- 239000011575 calcium Substances 0.000 description 1
- 150000001732 carboxylic acid derivatives Chemical group 0.000 description 1
- 239000013522 chelant Substances 0.000 description 1
- JQZRVMZHTADUSY-UHFFFAOYSA-L di(octanoyloxy)tin Chemical compound [Sn+2].CCCCCCCC([O-])=O.CCCCCCCC([O-])=O JQZRVMZHTADUSY-UHFFFAOYSA-L 0.000 description 1
- AYOHIQLKSOJJQH-UHFFFAOYSA-N dibutyltin Chemical compound CCCC[Sn]CCCC AYOHIQLKSOJJQH-UHFFFAOYSA-N 0.000 description 1
- HGQSXVKHVMGQRG-UHFFFAOYSA-N dioctyltin Chemical compound CCCCCCCC[Sn]CCCCCCCC HGQSXVKHVMGQRG-UHFFFAOYSA-N 0.000 description 1
- 125000003700 epoxy group Chemical group 0.000 description 1
- 238000005755 formation reaction Methods 0.000 description 1
- 125000000524 functional group Chemical group 0.000 description 1
- 229910052739 hydrogen Inorganic materials 0.000 description 1
- 239000001257 hydrogen Substances 0.000 description 1
- 238000010348 incorporation Methods 0.000 description 1
- 239000000543 intermediate Substances 0.000 description 1
- 229920000554 ionomer Polymers 0.000 description 1
- 229910001512 metal fluoride Inorganic materials 0.000 description 1
- 229910021645 metal ion Inorganic materials 0.000 description 1
- VHRYZQNGTZXDNX-UHFFFAOYSA-N methacryloyl chloride Chemical compound CC(=C)C(Cl)=O VHRYZQNGTZXDNX-UHFFFAOYSA-N 0.000 description 1
- CYCFYXLDTSNTGP-UHFFFAOYSA-L octadecanoate;tin(2+) Chemical compound [Sn+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O CYCFYXLDTSNTGP-UHFFFAOYSA-L 0.000 description 1
- 230000020477 pH reduction Effects 0.000 description 1
- OOCYPIXCHKROMD-UHFFFAOYSA-M phenyl(propanoyloxy)mercury Chemical compound CCC(=O)O[Hg]C1=CC=CC=C1 OOCYPIXCHKROMD-UHFFFAOYSA-M 0.000 description 1
- 238000006116 polymerization reaction Methods 0.000 description 1
- 239000004814 polyurethane Substances 0.000 description 1
- 229920002635 polyurethane Polymers 0.000 description 1
- ZUFQCVZBBNZMKD-UHFFFAOYSA-M potassium 2-ethylhexanoate Chemical compound [K+].CCCCC(CC)C([O-])=O ZUFQCVZBBNZMKD-UHFFFAOYSA-M 0.000 description 1
- 239000000843 powder Substances 0.000 description 1
- 239000000376 reactant Substances 0.000 description 1
- 239000003829 resin cement Substances 0.000 description 1
- 125000006850 spacer group Chemical group 0.000 description 1
- 239000000126 substance Substances 0.000 description 1
- ILWRPSCZWQJDMK-UHFFFAOYSA-N triethylazanium;chloride Chemical compound Cl.CCN(CC)CC ILWRPSCZWQJDMK-UHFFFAOYSA-N 0.000 description 1
- HVYVMSPIJIWUNA-UHFFFAOYSA-N triphenylstibine Chemical compound C1=CC=CC=C1[Sb](C=1C=CC=CC=1)C1=CC=CC=C1 HVYVMSPIJIWUNA-UHFFFAOYSA-N 0.000 description 1
- 238000005406 washing Methods 0.000 description 1
- XOOUIPVCVHRTMJ-UHFFFAOYSA-L zinc stearate Chemical compound [Zn+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O XOOUIPVCVHRTMJ-UHFFFAOYSA-L 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C271/00—Derivatives of carbamic acids, i.e. compounds containing any of the groups, the nitrogen atom not being part of nitro or nitroso groups
- C07C271/06—Esters of carbamic acids
- C07C271/08—Esters of carbamic acids having oxygen atoms of carbamate groups bound to acyclic carbon atoms
- C07C271/10—Esters of carbamic acids having oxygen atoms of carbamate groups bound to acyclic carbon atoms with the nitrogen atoms of the carbamate groups bound to hydrogen atoms or to acyclic carbon atoms
- C07C271/16—Esters of carbamic acids having oxygen atoms of carbamate groups bound to acyclic carbon atoms with the nitrogen atoms of the carbamate groups bound to hydrogen atoms or to acyclic carbon atoms to carbon atoms of hydrocarbon radicals substituted by singly-bound oxygen atoms
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Polyurethanes Or Polyureas (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
Description
- Die vorliegende Erfindung betrifft neue polymerisierbare Urethansäuren. Genauer gesagt betrifft die vorliegende Erfindung multifunktionelle Verbindungen, die durch eine hochgradig selektive Reaktion erzeugt werden können.
- Polymerisierbare saure Materialien sind zuvor in Harzverbindungssystemen und in Hybridzementsystemen verwendet worden. Beispielsweise werden polymerisierbare saure Materialien in U.S. Pat. Nr. 5,130,347 zur Verwendung in Glasionomerzementen beschrieben. Diese Verbindungen werden in U.S. Pat. Nr. 5,525,648 auch zur Verwendung in Dentalklebstoffen beschrieben.
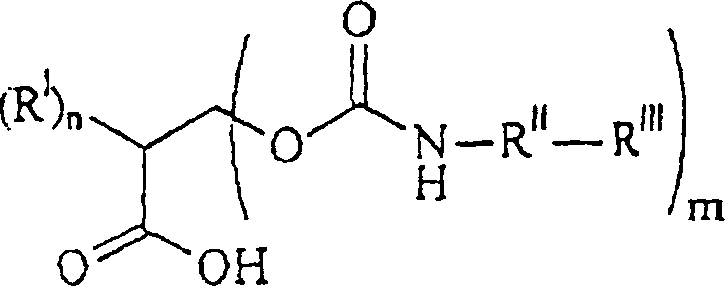
- Neue polymerisierbare erfindungsgemäße Urethansäureverbindungen haben die Formel:wobei

n gleich 0, 1 oder 2 ist,
m gleich 1, 2 oder 3 ist,
und n + m = 3 ist;
RI unabhängig voneinander ein C1–12-Alkylrest, ein Rest -O-C1–12-alkyl, C1–12-alkyl-OHist;
RV ein Alkylrest oder ein Rest -OH ist,
mit der Maßgabe, dass, falls m = 1 ist, ein RI ein Rest -CH2OH ist;
RII unabhängig voneinander wobei p gleich 1 bis 12 ist,
wobei p gleich 1 bis 12 ist,
oder jede Kombination davon ist;
RIII unabhängig voneinander-acrylamid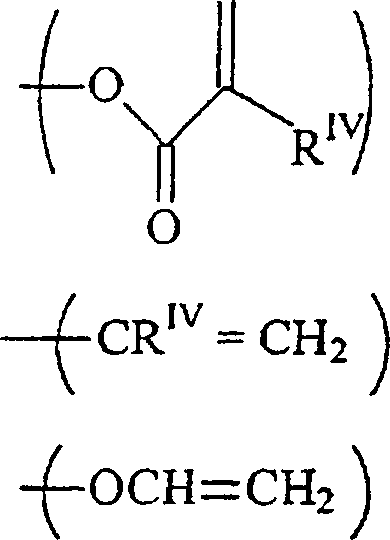
-methacrylamid ist;
RIV unabhängig voneinander H oder ein C1–12-Alkylrest ist. - Neue Herstellungsmethoden für diese Verbindungen werden auch beschrieben.
- Die erfindungsgemäßen polymerisierbaren Urethansäuren haben mehrere Vorteile im Vergleich zu den bislang bekannten sauren Monomeren. Materialien, insbesondere Dentalmaterialien, umfassend diese polymerisierbaren sauren Monomere, zeigen hohe Festigkeit und Zähigkeit des gehärteten (vernetzten) Materials. Ferner ist das erfindungsgemäße polymerisierbare saure Monomer verhältnismäßig wenig viskos im Vergleich zu den bislang hergestellten polymerisierbaren sauren Monomeren. Die vorliegende Erfindung ermöglicht deshalb die Formulierung von Materialien, wie Dentalmaterialien, mit einem höheren Prozentsatz an polymerisierbarem saurem Monomer im Material als zuvor bei einer gewünschten Viskosität oder Konsistenz möglich. Anders als die Monomeren, die zur Verwendung nach dem Stand der Technik für Zementsysteme empfohlen werden, die mehrere Säurereste enthalten, haben die erfindungsgemäßen polymerisierbaren Urethansäuren lediglich einen Carbonsäurerest. Bei Formulierungen vom Glasionomer- oder Compomer-Typ wurde gefunden, dass es zur Verbesserung der Stabilität der Paste beiträgt, wenn sie lediglich einen Säurerest tragen. Ferner können die erfindungsgemäßen polymerisierbaren Säuremonomeren anders als viele andere polyfunktionellen Monomere, die oft ein Gemisch aus verschiedenen Komponenten sind, mit hoher Reinheit hergestellt werden. Hohe Reinheit trägt sehr stark dazu bei, ein konsistentes Herstellungsverfahren zu erreichen und konsistente Endprodukte herzustellen.
- Die erfindungsgemäßen polymerisierbaren Säuremonomer sind nützliche Komponenten in verschiedenen Dentalmaterialien, bei denen eine Säurefunktionalität erwünscht ist, wie Compomere, Verbundstoffe, fließfähige Verbundstoffe, Glasionomere, Harzzemente und Dentalklebstoffe. Diese Monomere finden auch in weiteren industriellen oder biomedizinischen Klebstoffprodukten Anwendung.
- Bestimmte erfindungsgemäße Moleküle stellen polymerisierbare chelatisierende Monomere bereit. Die β-Hydroxycarbonsäureeinheit an diesem Molekül kann Metallionen, z. B. Calcium, chelatisieren, so dass sich ein sechsgliedriger Ring bildet. Die erfindungsgemäßen β-Hydroxycarbonsäureverbindungen stellen also eine überraschend gute Wirksamkeit bei der Oberflächenbehandlung bereit, verglichen mit Materialien, die nicht die β-Hydroxycarbonsäurekombination von Funktionalitäten enthalten, während sie polymerisierbar sind, wodurch ein Material bereitgestellt wird, das nach der Polymerisation des Harzes insgesamt keine freien Säuremonomere enthält. Dieses Material ist deshalb zur Verwendung in Klebstoffen hochgradig erwünscht und verringert im Wesentlichen oder beseitigt die Notwendigkeit für vorherige Oberflächenbehandlung vor dem Binden an die Oberfläche. Die erfindungsgemäßen β-Hydroxycarbonsäureverbindungen sind auch hochgradig reaktiv gegenüber säurereaktivem Glas, was rasche Zementreaktionen erleichtert.
- Die erfindungsgemäßen polymerisierbaren Säuremonomere sind auch als Zwischenprodukte für andere nützliche Materialien verwendbar. Beispielsweise können die erfindungsgemäßen sauren polymerisierbaren Monomere mit einem Metallfluorid, wie ZnF2, SnF2, AlF3, umgesetzt werden, wodurch nützliche Metallfluorkomplexe hergestellt werden. Diese Metallfluorkomplexe können dann als Zusatzstoffe in Dentalmaterialien zur Fluoridfreisetzung verwendet werden.
- In einer anderen Ausführungsform weisen erfindungsgemäße polymerisierbare Säuremonomere mindestens eine (und im Falle der hydroxyfunktionellen Verbindungen zwei) aktive Wasserstoffstelle auf, die weiter reagieren kann, wodurch polymerisierbare Materialien mit einzigartiger chemischer Struktur zur Einbringung in polymerisierbare Harze bereitgestellt werden.
- Beispielsweise kann die weitere Funktionalisierung der Hydroxylgruppen an den erfindungsgemäßen hydroxyfunktionellen polymerisierbaren sauren Monomeren in alternativen Umwandlungswegen durchgeführt werden. Beispielsweise können die Hydroxylgruppen direkt durch Umsetzung mit Methacrylsäure oder Methacrylchlorid; oder durch Umsetzen mit den Epoxidgruppen in Glycidylmethacrylat zu Methacrylatgruppen umgewandelt werden. Gleichermaßen können die erfindungsgemäßen Monomeren auch zur Herstellung von Verbindungen mit verschiedenen Spacern (z. B. aromatischen Resten, Ethylenoxideinheiten, mehr als zwei Urethanverknüpfungen) zwischen verschiedenen funktionellen Gruppen und der Propionsäureeinheit verwendbar sein.
- Von den vorstehend beschriebenen Verbindungen besitzt eine bevorzugte Klasse von Verbindungen die Formel:n gleich 2 ist,

und m gleich 1 ist;
ein RI ein Rest -CH2OH ist,
und der andere Rest RI ein C1–12-Alkylrest, ein Rest -O-C1–12-alkyl oderist;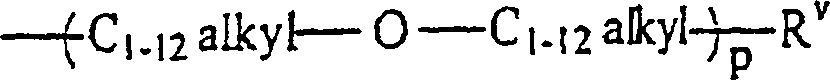
RV ein C1–12-Alkylrest ist;
RII unabhängig voneinanderwobei p gleich 1 bis 12 ist,
oder jede Kombination davon ist;
RIII unabhängig voneinander-acrylamid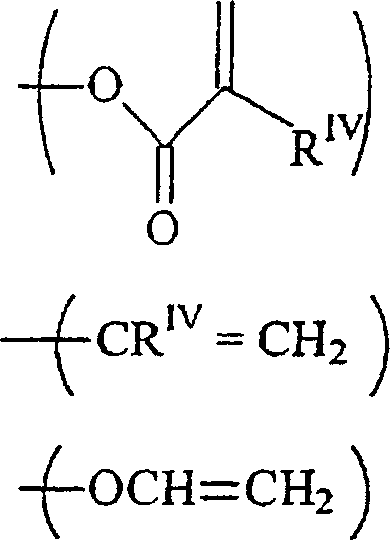
-methacrylamid ist;
RIV unabhängig voneinander H oder ein C1–12-Alkylrest ist. - Eine weitere bevorzugte Klasse von Verbindungen besitzt die Formel:wobei

n gleich 1 ist,
und m gleich 2 ist;
RI ein C1–12-Alkylrest, ein Rest -O-C1–12-alkyl oderist;
RV ein C1–12-Alkylrest ist;
RII unabhängig voneinanderwobei p gleich 1 bis 12 ist,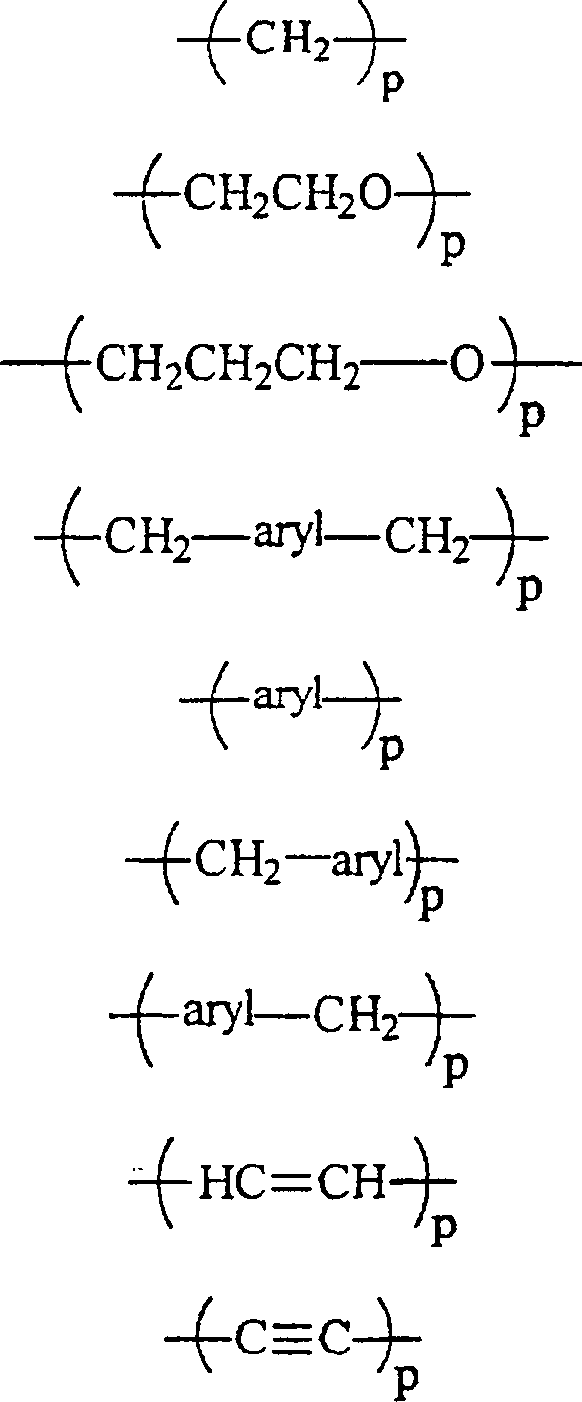
oder jede Kombination davon ist;
RIII unabhängig voneinander-acrylamid
-methacrylamid ist;
RIV unabhängig voneinander H oder ein C1–12-Alkylrest ist. - Eine besonders bevorzugte Klasse von erfindungsgemäßen Verbindungen ist
wobei
n = 1;
m = 2;
RI = CH3. -

- Eine weitere besonders bevorzugte Klasse sind die Verbindungen, bei denen n = 2, m = 1, ein RI ein Rest CH2-OH und der andere RI ein Rest -CH3 ist, RII gleich
( CH2CH2 )
ist und RIII gleichist.
- Eine weitere besonders bevorzugte Klasse von Verbindungen ist die, bei der n gleich 2 ist und ein RI ein Rest CH2-OH ist.
- Am stärksten bevorzugt wird die polymerisierbare Carbonsäureverbindung aus 2,2-Di(N-methacryloxyethylcarbamoylmethyl)propionsäure („PDMA") und 2-Hydroxymethyl-2-[(N-methacryloxyethyl)carbamoylmethyl]propionsäure („PAMA") ausgewählt.
- Ein weiterer Gesichtspunkt der vorliegenden Erfindung stellt ein Verfahren bereit zur Herstellung von Verbindungen der Formel:wobei
n gleich 2 ist,
und m gleich 1 ist;
ein RI ein Rest -CH2OH ist,
und der andere Rest RI ein C1–12-Alkylrest, ein Rest -O-C1–12-alkyl oderist;
RV ein C1–12-Alkylrest ist;
RII unabhängig voneinander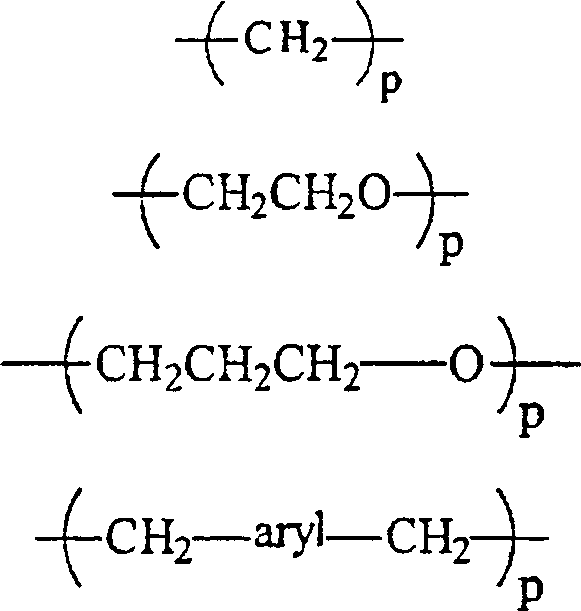 wobei p gleich 1 bis 12 ist,
wobei p gleich 1 bis 12 ist,
oder jede Kombination davon ist;
RIII unabhängig voneinander-acrylamid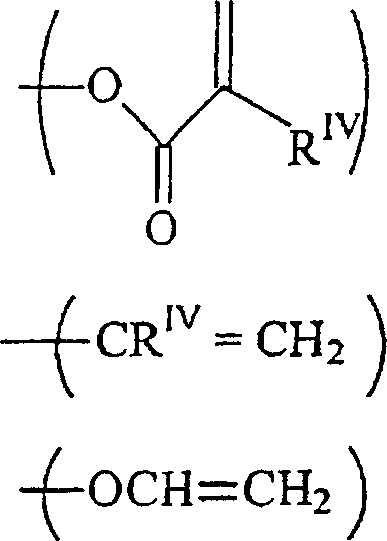
-methacrylamid ist;
RIV unabhängig voneinander H oder ein C1–12-Alkylrest ist. - Bei diesem Verfahren wird eine Verbindung der Formelin der die Substituenten die vorstehende angegebene Bedeutung haben, mit einer Verbindung der Formel
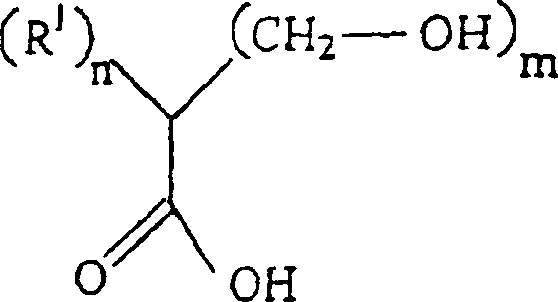 umgesetzt, in der die Substituenten die vorstehende angegebene Bedeutung haben.
umgesetzt, in der die Substituenten die vorstehende angegebene Bedeutung haben.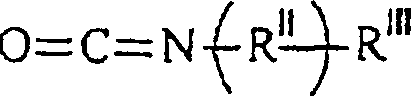
- Diese Verbindungen werden in Gegenwart einer katalytischen Menge eines nicht-basischen Katalysators bei einer Temperatur zwischen 40 und 80 °C für eine Zeitdauer größer als 15 Stunden umgesetzt.
- Überraschenderweise wurde gefunden, dass diese Reaktion, wenn sie wie vorstehend beschrieben durchgeführt wird, eine hohe Ausbeute an der gewünschten mono-hydroxyfunktionellen polymerisierbaren Säure ergibt. Dies ist hinsichtlich der ansonsten schnellen Reaktion von Isocyanato- mit primären Hydroxylverbindungen unerwartet, wie in Saunders und Frisch, Polyurethanes Chemistry and Technology, (1962, John Wiley and Sons), S. 73–75, 80–81, offenbart. Es wäre zu erwarten gewesen, dass alle Hydroxylverbindungen unter den vorliegenden aggressiven Bedingungen reagieren. Diese Ausbeute ist weit höher als die statistisch erwartete Ausbeute, wobei über 80% des Reaktionsprodukts das gewünschte Material ist und weniger als 20% des Reaktionsprodukts die bis-polymerisierbare Verbindung (erzeugt durch Reaktion der Isocyanat- mit beiden Hydroxylfunktionalitäten) und das amidofunktionelle Produkt (erzeugt durch Reaktion der Isocyanat- mit der Säurefunktionalität) sind. Stärker bevorzugt erzeugt die Reaktion einen Überschuss von 85% des Reaktionsprodukts, das die gewünschte mono-hydroxyfunktionelle polymerisierbare Säure ist. Dieses Ergebnis steht in überraschendem Gegensatz zur erwarteten Verteilung der Reaktionsprodukte, von der zu erwarten wäre, dass sie eine statistische Verteilung der Reaktion des Isocyanats mit Alkoholen ist, was zu etwa 66% mono-hydroxyfunktioneller Verbindung und 33% bis-polymerisierbarer Verbindung führt. Ferner hätte man erwartet, dass jede amido-funktionelle Verbindung lediglich so wirkt, dass sie die Ausbeute an gewünschtem Produkt anteilmäßig verringert.
- Weil keine Base als Katalysator eingesetzt wird, stellt die erfindungsgemäße Reaktion den bedeutenden Vorteil bereit, dass das Salz der Säure nicht gebildet wird, das vor der Verwendung im letztendlichen System, in dem eine Säure gewünscht ist, wieder zurück in die Säure umgewandelt werden müsste.
- Bevorzugte nicht-basische Katalysatoren zur Verwendung in der vorliegenden Erfindung schließen Zinn(IV)-Katalysatoren ein, wie Dibutylzinndilaurat, Dibutylzinndiacetat, Dibutylzinndimercaptid, Dibutylzinndithioglycolat, Dimethylzinndilaurat, Dimethylzinndimaleat, Dimethylzinndimercaptid, Dimethylzinndithioglycolat, Dioctylzinndilaurat, Dioctylzinndimercaptid und Dioctylzinndithioglycolat. Weitere Zinn(II)-Katalysatoren schließen Zinn(II)octoat und Zinn(II)stearat ein. Weitere Urethanmetallkatalysatoren schließen Bismutneodecanoat, Phenylquecksilberpropionat, Kaliumoctoat und Zinkstearat ein.
- Vorzugsweise wird diese Reaktion so durchgeführt, dass das Molverhältnis vom Ausgangsmaterial Hydroxysäure zum Ausgangsmaterial Isocyanat größer als 1:1 ist. Diese Reaktionsbedingung stellt ein Endprodukt bereit, das ein Gemisch von mono- und di-polymerisierbaren funktionellen Verbindungen mit einer vorherrschenden Menge an mono-polymerisierbarer funktioneller Verbindung ist.
- In einer weiteren bevorzugten Reaktion wird die Reaktion so durchgeführt, dass das Molverhältnis vom Ausgangsmaterial Hydroxysäure zum Ausgangsmaterial Isocyanat größer als etwa 2:1 ist. Diese Reaktionsbedingung stellt ein Endprodukt bereit, das ein Gemisch von mono- und di-polymerisierbaren funktionellen Verbindungen mit einem überraschend hohen Überwiegen der mono-polymerisierbaren funktionellen Verbindung ist.
- Ein weiterer überraschender Gesichtspunkt der vorliegenden Erfindung ist das Verfahren zur Herstellung von Verbindungen der Formel:wobei

n gleich 1 ist,
und m gleich 2 ist;
RI ein C1–12-Alkylrest, ein Rest -O-C1–12-alkyl oderist;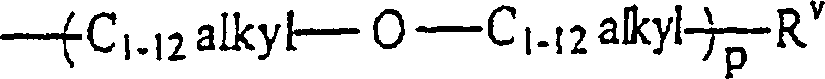
RV ein C1–12-Alkylrest ist;
RII unabhängig voneinander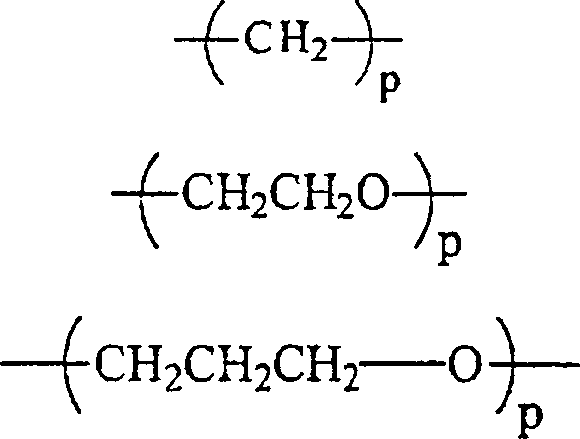 wobei p gleich 1 bis 12 ist,
wobei p gleich 1 bis 12 ist,
oder jede Kombination davon ist;
RIII unabhängig voneinander-acrylamid
-methacrylamid ist;
RIV unabhängig voneinander H oder ein C1–12-Alkylrest ist. - Diese Verbindung wird bereitgestellt, indem ein Teil einer Verbindung der Formelin der die Substituenten die vorstehende angegebene Bedeutung haben, mit zwei Teilen einer Verbindung der Formel
 umgesetzt wird, in der die Substituenten die vorstehende angegebene Bedeutung haben.
umgesetzt wird, in der die Substituenten die vorstehende angegebene Bedeutung haben.
- Diese Reaktanten werden in Gegenwart eines Hydroxyl/Isocyanato-Reaktionskatalysators bei einer Temperatur zwischen 22 und 80 °C für eine Zeitdauer größer als 15 Stunden umgesetzt, wenn lediglich nicht-basischer Katalysator verwendet wird. Der Hydroxyl/Isocyanato-Reaktionskatalysator für diese Reaktion kann ein organischer basischer Katalysator, ein nicht-basischer Katalysator oder Kombinationen davon sein. Vorzugsweise ist der Katalysator ein nicht-basischer Katalysator.
- Unter diesen Reaktionsbedingungen wäre zu erwarten, dass entweder die Säurefunktionalität durch die harten Reaktionsbedingungen zerstört wird oder das Isocyanat mit der Säure zu einem Amid oder einem Harnstoff reagiert. Unter Verwendung unterschiedlicher Ausgangsmaterialien wurde zuvor beobachtet, dass solche Amid- oder Harnstoffbildungsreaktionen unter weniger aggressiven Bedingungen eine ziemlich rasche Reaktion sind. Ein Beispiel für eine solche Reaktion bei Zimmertemperatur wird in U.S. Pat. Nr. 5,260,483 offenbart.
- Bevorzugte organische basische Katalysatoren sind tertiäre Aminkatalysatoren, einschließlich Triethylamin; Triethylendiamin; Bis(dimethylaminoethyl)ether; Tris(dimethylaminomethyl)phenol; N,N'-Dimorpholinodiethylether; N,N'-Dimethylcyclohexylamin; Pentamethyl-N,N'-dipropylentriamin; 1,8-Diazabicyclo-[5.4.0]-undec-7-en, N,N'-Dimethylethanolamin; und N-Ethylmorpholin.
- Die folgenden Beispiele werden zum Zweck der Veranschaulichung der vorliegenden Erfindung bereitgestellt und sollen nicht die breitesten Konzepte der vorliegenden Erfindung begrenzen. Sofern nicht anders angegeben beziehen sich alle Teile und Prozentsätze auf das Gewicht und alle Molekulargewichte sind Gewichtsmittel des Molekulargewichts.
- BEISPIELE
- Beispiel 1
- 2,2-Di(N-methacryloxyethylcarbamoylmethyl)propionsäure (PDMA) wird synthesisiert, indem 2,2-Bis(hydroxymethyl)propionsäure (BHMPA) und zwei Äquivalente Isocyanatoethylmethacrylat (IEM) wie folgt umgesetzt werden:
2,2-Bis(hydroxymethyl)propionsäure (BHMPA, 225,21 g, 1,679 mol), geringe Mengen an Stabilisator(en), wie 2,6-Di-tert-butyl-4-methylphenol (BHT, 1,6781 g, 7,615 mmol) und/oder Triphenylantimon (TPS, 1,3463 g, 3,813 mmol), und eine katalytische Menge Dibutylzinndilaurat (2,4396 g, 3,863 mmol) und trockenes THF oder andere geeignete Lösungsmittel wurden zuerst in den Reaktor gegeben. Nachdem die Lösung eine kurze Weile gerührt worden war, wurde IEM (592,64 g, 3,823 mol) zugegeben. Der Ansatz wurde unter ständigem Rühren etwa 30 Stunden auf 65 °C erhitzt. Das Lösungsmittel wurde abgezogen, nachdem die Umwandlung beendet war. Das Endprodukt PDMA war eine farblose, viskose Flüssigkeit. - Beispiel 2
- In einer anderen Ausführungsform kann die vorstehende Reaktion unter Verwendung von Triethylamin als basischem Katalysator durchgeführt werden, um die Reaktion zu beschleunigen. 0,075 bis 0,15 Äquivalente Triethylamin sind typischerweise erforderlich. Diese Reaktion wurde wie folgt durchgeführt:
2,2-Bis(hydroxymethyl)propionsäure (14,9946 g, 0,112 mol), geringe Mengen des Stabilisators 2,6-Di-tert-butyl-4-methylphenol (0,1012 g, 0,456 mmol) [ein alternativer Stabilisator, wie Triphenylantimon (0,0831 g, 0,235 mmol) kann verwendet werden] und eine katalytische Menge Dibutylzinndilaurat (0,1450 g, 0,230 mmol), Triethylamin (1,132 g, 0,0112 mol) und trockenes THF wurden zuerst in den Reaktor gegeben. Nachdem die Lösung eine kurze Weile gerührt worden war, wurde IEM (35,55 g, 0,229 mol) zugegeben. Der Ansatz wurde unter ständigem Rühren 8 Stunden auf 65 °C erhitzt. Nach dem Funktionalisierungsschritt wurde das Triethylamin entfernt, indem mit 37 gew.%iger wässr. HCl (1,218 g, 0,0124 mol) bei niedriger Temperatur wieder angesäuert wurde. Ein weißer Feststoff, Triethylaminhydrochlorid, fiel aus. Nach dem Abfiltrieren des weißen Niederschlags wurde dann, während die Lösung noch kalt war, das Lösungsmittel abgezogen. Wieder Ansäuern ist nicht notwendig, wenn das Vorliegen einer geringen Menge an Triethylamin in einer gegebenen Anwendung toleriert werden kann. - Beispiel 3
- Der Reaktor wurde zuerst mit einem Überschuss an BHMPA (139,94 g, 1,043 mol), 2,6-Di-tert-butyl-4-methylphenol (0,2322 g, 1,054 mmol), Triphenylantimon (0,1891 g, 0,536 mmol) und Dibutylzinndilaurat (0,6801 g, 1,077 mmol) beschickt. Das Ausgangsmaterial BHMPA war in THF bei Zimmertemperatur lediglich gering löslich. IEM wurde allmählich in das vorstehende Gemisch zugetropft (80,94 g, 0,522 mol). Die Reaktion wurde unter ständigem Rühren 24 Stunden bei 60 °C durchgeführt. Am Ende der Reaktion setzte sich das meiste des unumgesetzten BHMPA als weißes festes Pulver ab, nachdem die Lösung abgekühlt war. Unumgesetztes BHMPA wurde mittels Vakuumfiltration abfiltriert und dann wurde das Lösungsmittel abgezogen. Das zurückgewonnene BHMPA kann in zukünftigen Reaktionen eingesetzt werden.
- Nach der Entfernung des Lösungsmittels wurde das Produkt auf Grund des langsamen Ausfallens von restlichem BHMPA leicht trübe. Genügend Diethylether wurde zugegeben, um das Produkt zu lösen, und dann konnte die Lösung ungestört über Nacht stehen bleiben (ungefähr 18 Stunden), um so das meiste des in Lösung verbliebenen BHMPA auszufällen. Der weiße Niederschlag wurde mittels Vakuumfiltration abfiltriert und Diethylether wurde abgezogen.
- Das resultierende Produkt PAMA war eine farblose, fließfähige Flüssigkeit. Die Reinheit von PAMA im Endprodukt betrug ungefähr 80% nach dem Molverhältnis, wobei PDMA das Haupt-Nebenprodukt (ungefähr 17%) neben geringen Mengen an verbliebenem BHMPA (ungefähr 3%) war.
Claims (13)
- Verbindung der Formelwobei n gleich 0, 1 oder 2 ist; m gleich 1, 2 oder 3 ist, und n + m = 3 ist; RI unabhängig voneinander ein C1–12-Alkylrest, ein Rest -O-C1–12-alkyl, C1–12-alkyl-OH oder
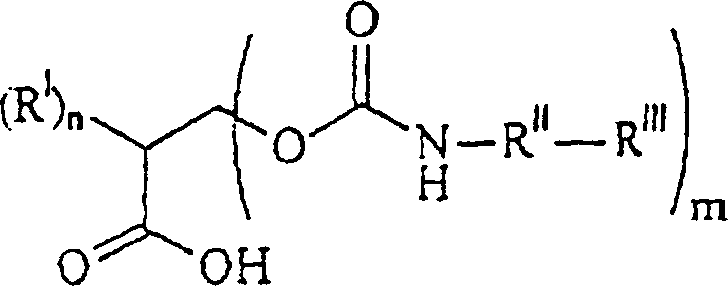 ist; RV ein C1–12-Alkylrest oder ein Rest -OH ist, mit der Maßgabe, dass, falls m = 1 ist, ein RI ein Rest -CH2OH ist; RII unabhängig voneinander
ist; RV ein C1–12-Alkylrest oder ein Rest -OH ist, mit der Maßgabe, dass, falls m = 1 ist, ein RI ein Rest -CH2OH ist; RII unabhängig voneinander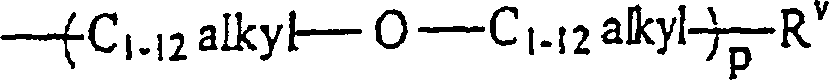
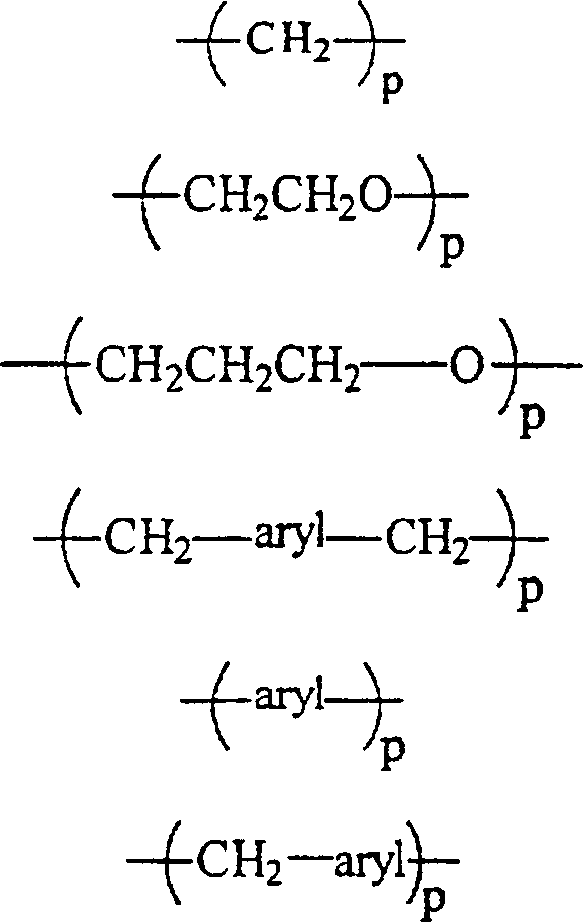 wobei p gleich 1 bis 12 ist, oder jede Kombination davon ist; RIII unabhängig voneinander
wobei p gleich 1 bis 12 ist, oder jede Kombination davon ist; RIII unabhängig voneinander -acrylamid -methacrylamid ist; RIV unabhängig voneinander H oder ein C1–12-Alkylrest ist; oder ein Salz davon.
-acrylamid -methacrylamid ist; RIV unabhängig voneinander H oder ein C1–12-Alkylrest ist; oder ein Salz davon.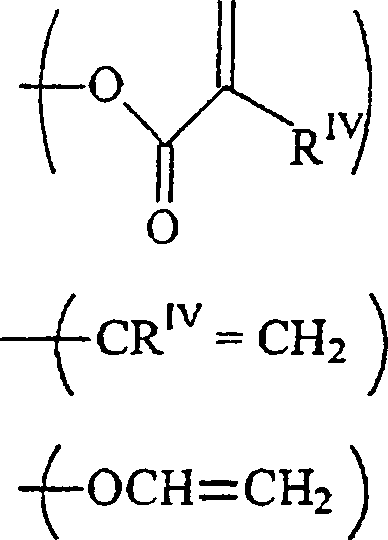
- Verbindung gemäß Anspruch 1, welche die Formel aufweist:wobei n gleich 2 ist, und m gleich 1 ist; ein RI ein Rest -CH2OH ist, und der andere Rest RI ein C1–12-Alkylrest, ein Rest -O-C1–12-alkyl oder
 ist; RV ein C1–12-Alkylrest ist; RII unabhängig voneinander
ist; RV ein C1–12-Alkylrest ist; RII unabhängig voneinander wobei p gleich 1 bis 12 ist, oder jede Kombination davon ist; RIII unabhängig voneinander
wobei p gleich 1 bis 12 ist, oder jede Kombination davon ist; RIII unabhängig voneinander -acrylamid -methacrylamid ist; RI Vunabhängig voneinander H oder ein C1–12-Alkylrest ist.
-acrylamid -methacrylamid ist; RI Vunabhängig voneinander H oder ein C1–12-Alkylrest ist.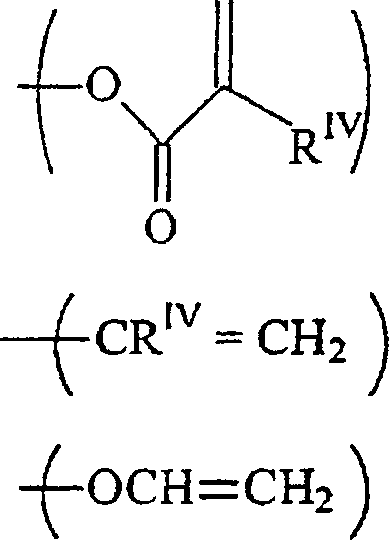
- Verbindung gemäß Anspruch 1, welche die Formel aufweist:wobei n gleich 1 ist, und m gleich 2 ist; RI ein C1–12-Alkylrest, ein Rest -O-C1–12-alkyl oder
 ist; RV ein C1–12-Alkylrest ist; RII unabhängig voneinander
ist; RV ein C1–12-Alkylrest ist; RII unabhängig voneinander wobei p gleich 1 bis 12 ist, oder jede Kombination davon ist; RIII unabhängig voneinander
wobei p gleich 1 bis 12 ist, oder jede Kombination davon ist; RIII unabhängig voneinander -acrylamid -methacrylamid ist; RIV unabhängig voneinander H oder ein C1–12-Alkylrest ist.
-acrylamid -methacrylamid ist; RIV unabhängig voneinander H oder ein C1–12-Alkylrest ist.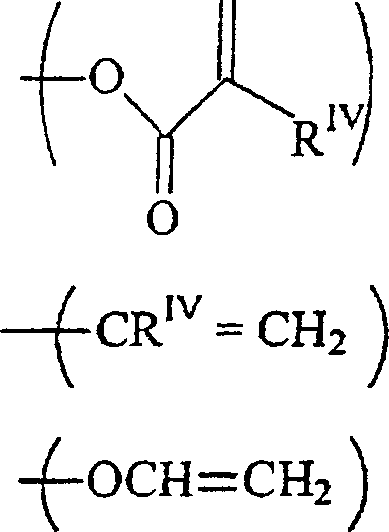
- Verbindung gemäß Anspruch 1, wobei n = 1; m = 2; RI = CH3;

- Verbindung gemäß Anspruch 1, wobei n = 2, m = 1, ein RI ein Rest -CH2-OH ist und der andere Rest RI ein Rest -CH3 ist; RII gleich ( CH2CH2 ) ist und RIII gleichist.

- Verbindung gemäß Anspruch 1, wobei n gleich 2 ist und ein RI ein Rest -CH2-OH ist.
- Verbindung gemäß Anspruch 1, welche 2,2-Di(N-methacryloxyethylcarbamoyl- methyl)propionsäure (PDMA) ist.
- Verbindung gemäß Anspruch 1, welche 2-Hydroxymethyl-2-[(N-methacryloxyethyl)-carbamoylmethyl]propionsäure (PAMA) ist.
- Verfahren zur Herstellung von Verbindungen der Formel:wobei n gleich 2 ist, und m gleich 1 ist; ein RI ein Rest -CH2OH ist, und der andere Rest RI ein C1–12-Alkylrest, ein Rest-O-C1–12-alkyl oder
 ist; RV ein C1–12-Alkylrest ist; RII unabhängig voneinander
ist; RV ein C1–12-Alkylrest ist; RII unabhängig voneinander
 wobei p gleich 1 bis 12 ist, oder jede Kombination davon ist; RIII abhängig voneinander
wobei p gleich 1 bis 12 ist, oder jede Kombination davon ist; RIII abhängig voneinander -acrylamid -methacrylamid ist; RIV unabhängig voneinander H oder ein C1–12-Alkylrest ist; wobei das Verfahren das Umsetzen einer Verbindung der Formel
-acrylamid -methacrylamid ist; RIV unabhängig voneinander H oder ein C1–12-Alkylrest ist; wobei das Verfahren das Umsetzen einer Verbindung der Formel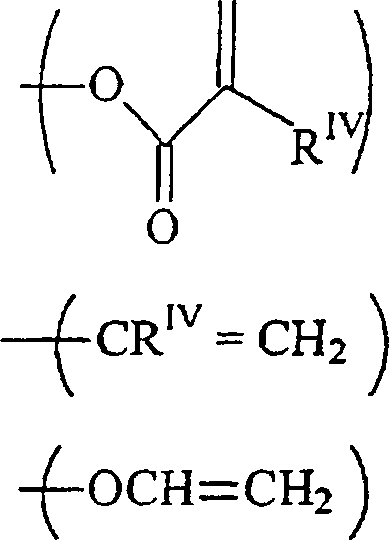 wobei die Substituenten die vorstehend angegebene Bedeutung haben, mit einer Verbindung der Formel
wobei die Substituenten die vorstehend angegebene Bedeutung haben, mit einer Verbindung der Formel umfasst, wobei die Substituenten die vorstehende angegebene Bedeutung haben, die Umsetzung in Gegenwart einer katalytischen Menge eines nicht-basischen Katalysators bei einer Temperatur zwischen 22 und 80 °C für eine Zeitdauer größer als 15 Stunden durchgeführt wird, wobei die Umsetzung einen Überschuss von 80% der theoretischen Ausbeute der mono-Hydroxy-funktionalisierten polymerisierbaren Säure erzeugt.
umfasst, wobei die Substituenten die vorstehende angegebene Bedeutung haben, die Umsetzung in Gegenwart einer katalytischen Menge eines nicht-basischen Katalysators bei einer Temperatur zwischen 22 und 80 °C für eine Zeitdauer größer als 15 Stunden durchgeführt wird, wobei die Umsetzung einen Überschuss von 80% der theoretischen Ausbeute der mono-Hydroxy-funktionalisierten polymerisierbaren Säure erzeugt.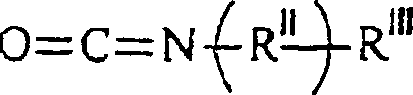
- Verfahren gemäß Anspruch 9, wobei der nicht-basische Katalysator aus Zinn(IV)- und Zinn(II)-Katalysatoren ausgewählt ist.
- Verfahren zur Herstellung von Verbindungen der Formel:wobei n gleich 1 ist, und m gleich 2 ist; RI ein C1–12-Alkylrest, ein Rest -O-C1–12-alkyl oder
 ist; RV ein C1–12-Alkylrest ist; RII unabhängig voneinander
ist; RV ein C1–12-Alkylrest ist; RII unabhängig voneinander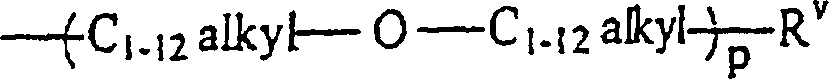 wobei p gleich 1 bis 12 ist, oder jede Kombination davon ist; RIII abhängig voneinander
wobei p gleich 1 bis 12 ist, oder jede Kombination davon ist; RIII abhängig voneinander -acrylamid -methacrylamid ist; RIV unabhängig voneinander H oder ein C1–12-Alkylrest ist, umfassend das Umsetzen einer Verbindung der Formel
-acrylamid -methacrylamid ist; RIV unabhängig voneinander H oder ein C1–12-Alkylrest ist, umfassend das Umsetzen einer Verbindung der Formel wobei die Substituenten die vorstehend angegebene Bedeutung haben, mit einer Verbindung der Formel
wobei die Substituenten die vorstehend angegebene Bedeutung haben, mit einer Verbindung der Formel wobei die Substituenten die vorstehend angegebene Bedeutung haben, bei einer Temperatur zwischen 22 und 80 °C für eine Zeitdauer größer als 15 Stunden, wobei die Umsetzung weniger als 5% Amid- und Harnstoffreaktionsprodukte der theoretischen Ausbeute der Produkte erzeugt.
wobei die Substituenten die vorstehend angegebene Bedeutung haben, bei einer Temperatur zwischen 22 und 80 °C für eine Zeitdauer größer als 15 Stunden, wobei die Umsetzung weniger als 5% Amid- und Harnstoffreaktionsprodukte der theoretischen Ausbeute der Produkte erzeugt.
- Verfahren gemäß Anspruch 11, wobei die Umsetzung in Gegenwart eines Hydroxyl/Isocyanat-Reaktionskatalysators stattfindet.
- Verfahren gemäß Anspruch 12, wobei der Hydroxyl/Isocyanat-Reaktionskatalysator ein organischer, basischer Katalysator ist, welcher aus tertiären Aminkatalysatoren ausgewählt ist.
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US09/314,489 US6143919A (en) | 1999-05-18 | 1999-05-18 | Polymerizable acidic compounds and methods of preparation |
| US314489 | 1999-05-18 | ||
| PCT/US2000/008319 WO2000069944A1 (en) | 1999-05-18 | 2000-03-29 | Polymerizable acidic compounds and methods of preparation |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| DE60005183D1 DE60005183D1 (de) | 2003-10-16 |
| DE60005183T2 true DE60005183T2 (de) | 2004-07-01 |
Family
ID=23220167
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| DE60005183T Expired - Lifetime DE60005183T2 (de) | 1999-05-18 | 2000-03-29 | Polymerisierbare sauere verbindungen und verfahren zur herstellung |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US6143919A (de) |
| EP (1) | EP1185573B1 (de) |
| JP (1) | JP2002544290A (de) |
| AU (1) | AU4042700A (de) |
| CA (1) | CA2371572A1 (de) |
| DE (1) | DE60005183T2 (de) |
| WO (1) | WO2000069944A1 (de) |
Families Citing this family (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2008137948A (ja) * | 2006-12-01 | 2008-06-19 | Showa Denko Kk | 重合性単量体組成物および重合防止方法 |
| US8735463B2 (en) | 2007-05-31 | 2014-05-27 | Creighton University | Self-healing dental composites and related methods |
| JP6781062B2 (ja) * | 2017-01-31 | 2020-11-04 | 株式会社松風 | 高い機械的強度をもつ歯科用硬化性組成物 |
Family Cites Families (44)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB1139430A (en) * | 1966-12-30 | 1969-01-08 | Nat Res Dev | Improvements relating to surgical cements |
| US3814717A (en) * | 1970-12-04 | 1974-06-04 | Dental Materials Section Labor | Poly(carboxylic acid)-fluoroalumino-silicate glass surgical cement |
| US4209434A (en) * | 1972-04-18 | 1980-06-24 | National Research Development Corporation | Dental cement containing poly(carboxylic acid), chelating agent and glass cement powder |
| GB1532954A (en) * | 1974-10-24 | 1978-11-22 | Nat Res Dev | Poly-(carboxylate)cements |
| US4144324A (en) * | 1976-10-08 | 1979-03-13 | Colgate Palmolive Company | Oral compositions for calculus retardation |
| US4208401A (en) * | 1977-08-19 | 1980-06-17 | Colgate-Palmolive Company | Quaternary ammonium alkylene diphosphonate anti-calculus agents |
| DE2929121A1 (de) * | 1979-07-18 | 1981-02-12 | Espe Pharm Praep | Calciumaluminiumfluorosilikatglas- pulver und seine verwendung |
| DE2932823A1 (de) * | 1979-08-13 | 1981-03-12 | Espe Pharm Praep | Anmischkomponente fuer glasionomerzemente |
| US4695251A (en) * | 1980-04-07 | 1987-09-22 | Minnesota Mining And Manufacturing Company | Orthodontic bracket adhesive and abrasive for removal thereof |
| DE3016289A1 (de) * | 1980-04-28 | 1981-10-29 | Henkel KGaA, 4000 Düsseldorf | Verfahren zur herstellung von omega -amino-1-hydroxyalkyliden-1,1-bis-phosphonsaeuren |
| US4394494A (en) * | 1980-07-04 | 1983-07-19 | Lion Corporation | Dental filling material |
| JPS57106688A (en) * | 1980-12-20 | 1982-07-02 | Lion Corp | 1-methacryloxyethane-1,1-diphosphonic acid |
| US4503169A (en) * | 1984-04-19 | 1985-03-05 | Minnesota Mining And Manufacturing Company | Radiopaque, low visual opacity dental composites containing non-vitreous microparticles |
| GB8419489D0 (en) * | 1984-07-31 | 1984-09-05 | Leo Pharm Prod Ltd | Chemical compounds |
| US4639338A (en) * | 1984-08-06 | 1987-01-27 | Ciba-Geigy Corporation | Preparation of crystalline disodium 3-amino-1-hydroxypropane-1,1-diphosphonate pentahydrate |
| EP0243890A3 (de) * | 1986-04-30 | 1988-08-03 | EASTMAN KODAK COMPANY (a New Jersey corporation) | Diolmonomere und deren Polymere |
| US5270431A (en) * | 1987-07-23 | 1993-12-14 | Basf Aktiengesellschaft | Preparation of oligomeric or polymeric radiation-reactive intermediates for solvent-structured layers |
| US4877603A (en) * | 1987-12-18 | 1989-10-31 | The Procter & Gamble Company | Oral compositions |
| AU618772B2 (en) * | 1987-12-30 | 1992-01-09 | Minnesota Mining And Manufacturing Company | Photocurable ionomer cement systems |
| US4877401A (en) * | 1988-03-09 | 1989-10-31 | University Of Utah | Method of preventing tooth decay by laser beam irradiation and chemical treatment |
| US5260483A (en) * | 1988-03-29 | 1993-11-09 | Drexel University | Preparation of n-aryl amides from isocyanates |
| US5258067A (en) * | 1988-06-25 | 1993-11-02 | Bayer Aktiengesellschaft | Liquid for conditioning tooth or bone substance |
| TW198039B (de) * | 1988-11-28 | 1993-01-11 | Ciba Geigy Ag | |
| US5015180A (en) * | 1989-03-01 | 1991-05-14 | Minnesota Mining And Manufacturing Company | Dental article containing light-curable paste |
| US5204426A (en) * | 1989-04-27 | 1993-04-20 | National Research Development Corporation | Command-curable composition |
| EP0419043B1 (de) * | 1989-08-24 | 1995-10-11 | Mitsubishi Rayon Co., Ltd. | Hitzehärtbare flüssige (meth)acrylate Harze und Zusammenfassungen, die diesen enthalten |
| US5019651A (en) * | 1990-06-20 | 1991-05-28 | Merck & Co., Inc. | Process for preparing 4-amino-1-hydroxybutylidene-1,1-bisphosphonic acid (ABP) or salts thereof |
| US5096699A (en) * | 1990-12-20 | 1992-03-17 | Colgate-Palmolive Company | Anticalculus oral compositions |
| US5208009A (en) * | 1990-12-20 | 1993-05-04 | Colgate-Palmolive Company | Anticalculus oral compositions |
| US5218070A (en) * | 1991-02-11 | 1993-06-08 | Dentsply G.M.B.H. | Dental/medical composition and use |
| US5332429A (en) * | 1991-05-31 | 1994-07-26 | Minnesota Mining And Manufacturing Company | Method for treating fluoroaluminosilicate glass |
| US5354199A (en) * | 1991-08-02 | 1994-10-11 | Minnesota Mining And Manufacturing Company | Adhesive for packaged orthodontic appliance |
| JP2686684B2 (ja) * | 1991-08-27 | 1997-12-08 | 寅雄 大塚 | 水酸化アパタイトを充填し生体内溶解性繊維により編機されたチューブ網編成物 |
| US5270365A (en) * | 1991-12-17 | 1993-12-14 | Merck & Co., Inc. | Prevention and treatment of periodontal disease with alendronate |
| US5525648A (en) * | 1991-12-31 | 1996-06-11 | Minnesota Mining And Manufacturing Company | Method for adhering to hard tissue |
| JPH06298779A (ja) * | 1993-04-15 | 1994-10-25 | Hoechst Japan Ltd | ヘテロ環イミノビスメチレンビスホスホン酸誘導体 |
| US5451401A (en) * | 1993-09-29 | 1995-09-19 | The Procter & Gamble Company | Diphosphonic acid esters as tartar control agents |
| US5547379A (en) * | 1994-10-06 | 1996-08-20 | Hasel; Robert W. | Method of restoring a tooth |
| US5652227A (en) * | 1995-01-30 | 1997-07-29 | Teronen; Olli Pekka | Inhibition of the degradation of connective tissue matrix protein components in mammals |
| JP4213207B2 (ja) * | 1995-11-17 | 2009-01-21 | スリーエム カンパニー | フッ化物を遊離する組成物 |
| US6391286B1 (en) * | 1995-11-17 | 2002-05-21 | 3M Innovative Properties Company | Use of metallofluorocomplexes for dental compositions |
| US6126922A (en) * | 1995-11-17 | 2000-10-03 | 3M Innovative Properties Company | Fluorid-releasing compositions and compositions with improved rheology |
| EP0971678B9 (de) * | 1997-04-02 | 2010-07-07 | Dentsply International Inc. | Dental-verbundwerkstoff und verfahren zur herstellung von restaurationen |
| US6506816B1 (en) * | 1997-07-17 | 2003-01-14 | 3M Innovative Properties Company | Dental resin cements having improved handling properties |
-
1999
- 1999-05-18 US US09/314,489 patent/US6143919A/en not_active Expired - Lifetime
-
2000
- 2000-03-29 EP EP00919805A patent/EP1185573B1/de not_active Expired - Lifetime
- 2000-03-29 DE DE60005183T patent/DE60005183T2/de not_active Expired - Lifetime
- 2000-03-29 JP JP2000618359A patent/JP2002544290A/ja not_active Withdrawn
- 2000-03-29 WO PCT/US2000/008319 patent/WO2000069944A1/en not_active Ceased
- 2000-03-29 CA CA002371572A patent/CA2371572A1/en not_active Abandoned
- 2000-03-29 AU AU40427/00A patent/AU4042700A/en not_active Abandoned
Also Published As
| Publication number | Publication date |
|---|---|
| DE60005183D1 (de) | 2003-10-16 |
| EP1185573A1 (de) | 2002-03-13 |
| AU4042700A (en) | 2000-12-05 |
| WO2000069944A1 (en) | 2000-11-23 |
| EP1185573B1 (de) | 2003-09-10 |
| JP2002544290A (ja) | 2002-12-24 |
| CA2371572A1 (en) | 2000-11-23 |
| US6143919A (en) | 2000-11-07 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| DE2230728A1 (de) | Verfahren zur stereoregulären Polymerisation von alpha-Olefinen | |
| DE2427661A1 (de) | Verfahren zur herstellung eines polyoxyalkylens und das dabei erhaltene produkt | |
| DE69022232T2 (de) | Verfahren zur herstellung von polyethern aus oxetanen. | |
| EP0346788B1 (de) | Strahlungsempfindliche, ethylenisch ungesättigte, copolymerisierbare Verbindungen und Verfahren zu deren Herstellung | |
| EP1213292B1 (de) | Oligomere Silasesquioxane, deren Herstellung und deren Verwendung zur Synthese von nicht vollständig kondensierten Silasesquioxanen, von Katalysatoren und deren Ausgangsverbindungen sowie von Polymeren | |
| DE4006097A1 (de) | Ethylenisch ungesaettigte, fluorhaltige urethanderivate und verfahren zu ihrer herstellung | |
| DE60005183T2 (de) | Polymerisierbare sauere verbindungen und verfahren zur herstellung | |
| DE3025227A1 (de) | Verfahren zur herstellung von ungesaettigten urethan-monoisocyanatverbindungen | |
| DE3850230T2 (de) | Verfahren zur Herstellung von Alkylthioethylaminsalzen. | |
| EP1794212B1 (de) | Verfahren zur herstellung von polymeren hydroxylalkylterminierten polysulfiden | |
| DE1157602B (de) | Verfahren zur Herstellung organischer Isothiocyanate | |
| EP0244747B1 (de) | Ethylenisch ungesättigte Phosphinsäure- oder Thiophosphinsäureisocyanate bzw. -isothiocyanate und Verfahren zu ihrer Herstellung | |
| EP0001996B1 (de) | Verfahren zur herstellung von N,N-Bis-(2-hydroxyalkyl)-aminomethan-phosphonsäure-dimethylester und ihre Verwendung als flammhemmende Mittel für Kunststoffe | |
| CH666479A5 (de) | Herstellung von polyolen aus polycarbonsaeuren. | |
| DE69420331T2 (de) | Verfahren zur Herstellung von 3-Methyl-1-phenylphospholenoxyd | |
| DE1249274B (de) | Verfahren zur Herstellung von alkoholi sehe Hydroxylgruppen und Phosphor enthaltenden Polyathern, Zus z Änm C | |
| DE1418961C (de) | Verfahren zur Herstellung von Anlagerungsverbindungen aus Fluor enthaltenden Olefinen und Stickstoffmonoxid | |
| EP0681567B1 (de) | Diacylperoxidester | |
| DE2360248C3 (de) | Verfahren zur Herstellung von Estern von Thiolcarbaminsäuren | |
| DE60111642T2 (de) | Verwendung von zinnderivaten als transcarbamatierungskatalysatoren,carabamatezusammenstellungen und transcarbamatierungsverfahren | |
| EP0045390A2 (de) | Monoalkylfluorozinn-Verbindungen | |
| EP1203780A1 (de) | Isocyanate mit Aminogruppen | |
| DE19523169C1 (de) | Verfahren zur Herstellung von Acryloylcarbamaten oder -harnstoffen | |
| DE2658955A1 (de) | Verfahren zur herstellung eines vernetzerkomponentengemisches fuer dispersions-anstrichstoffe und -kleber | |
| EP0220485A1 (de) | Sulfonsäureestergruppen aufweisenden Polyhydroxylverbindungen und ihre Verwendung als Polyolkomponente in Giessharzen |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 8364 | No opposition during term of opposition |