-
Hintergrund der Erfindung
-
Die
p38 Kinase ist eine durch Mitogen aktivierte Proteinkinase (MAP),
die zur Serin/Threoninkinasesuperfamilie gehört. Diese Kinase wird durch
extrazellulären
Stress, wie Hitze, UV Licht und osmotischen Stress, wie auch entzündliche
Stimuli, wie Lipopolysaccharid aktiviert. Bei einer Aktivierung
phosphoryliert die p38 Kinase intrazelluläre Proteinsubstrate, die die
Biosynthese der proinflammatorischen Cytokine Tumornekrosefaktor α (TNF-α) und Interleukin
1β (IL-1β) reguliert.
Diese Cytokine sind bei der Pathologie von mehreren chronischen
entzündlichen
Störungen
(Lee et al., Ann. N.Y. Acad. Sci., 696, 149-170 (1993), Muller-Ladner, Curr.
Opin. Rheumatol., 8, 210-220 (1996)), kardiovaskulären Störungen und
Störungen
des zentralen Nervensystems (Salituro et al., Current Medicinal
Chemistry, 6, 807-823
(1999)) und Autoimmunstörungen
(Pargellis et al., Nature Structural Biology, 9 (4), 268-272 (2002))
beteiligt.
-
Es
wurden mehrere Verbindungen innerhalb der Strukturplattformen der
Pyridinylimidazole (WO 96 21 452 A, WO 96 40 143 A, WO 97 25 045
A,
US 5 656 644 A ,
US 5 686 455 A ,
US 5 717 100 A ,
WO 97 12 876 A, WO 98 21 957 A, WO 98 47 892 A, WO 99 903 837 A,
WO 99 01 449 A, WO 00 61 576 A, WO 00 10 563 A, WO 01 72 737 A,
Revesz et al., Bioorganic and Med. Chem. Lett. 10, 1261-1264 (2000))
und der Pyrimidinylimidazole (WO 97 25 048 A, WO 99 01 452 A, WO
97 25 046 A, WO 99 32 121 A, WO 99 01 131 A, WO 99 01 130 A, WO
99 01 136 A, WO 98 07 452 A, WO 97 47 618 A, WO 98 56 788 A, WO
98 57 996 A) als Inhibitoren der p38 Kinase oder als Cytokininhibitoren
identifiziert. Die WO 02 072 576 A (vom 19. September 2002) identifiziert
ebenfalls eine Reihe an Verbindungen, die als p38 Kinaseinhibitoren
identifiziert werden. Von selektiven Inhibitoren der p38 Kinase
ist bekannt, dass sie die Expression von TNF-α und IL-1β unterdrücken (McKenna et al., J. Med.
Chem 45 (11), 2173-2184 (2002)). Es wurde eine antiinflammatorische
Aktivität
für Verbindungen
innerhalb der Pyrimidinylimidazolstrukturplattform berichtet (Lantos
et al., J. Med. Chem, 27, 72-75 (1984)) und es ist eine Vielzahl
an Inhibitoren der p38 Kinase zur Behandlung einer Vielzahl an Störungen in
Untersuchung (Boehm und Adams, Exp. Opin. Ther. Patents, 10 (1),
25-37 (2000)). Es bleibt ein Bedarf in diesem Behandlungsgebiet
für Verbindungen,
die Cytokin-suppressiv sind, das heißt Verbindungen, die zur Hemmung
der p38 Kinase fähig
sind.
-
Die
vorliegende Erfindung liefert neue Inhibitoren der p38 Kinase, die
zur Behandlung von Zuständen brauchbar
sind, welche aus einer übermäßigen Cytokinbildung
resultieren.
-
Kurze Zusammenfassung
der Erfindung
-
Die
vorliegende Erfindung liefert Verbindungen der Formel I
worin
W steht für
X für N(R
4) oder S steht,
R
0 ist
- (a) ausgewählt
aus der Gruppe, die besteht aus Wasserstoff, C1-C6 Alkyl, Cyano, C1-C4 Alkylen-R11, 3-Hydroxyprop-2-yl,
(1-Phenyl)-2-hydroxyeth-1-yl, (1-Cyclohexyl)-3-hydroxyprop-2-yl,
4-Methoxybenzyl, 1,4-Dioxoaspiro[4,5]dec-8-yl, Tetrahydropyran,
2,2,6,6-Tetramethylpiperidin-4-yl und Cyclohexan-1-on-4-yl,
- (b) Phenyl, das optional mit einem Substituenten substituiert
ist, der aus der Gruppe ausgewählt
ist, welche besteht aus Nitro und Amino,
- (c) Piperidin-4-yl, das optional mit einem Substituenten substituiert
ist, der aus der Gruppe ausgewählt
ist, welche besteht aus C1-C4 Alkyl,
C1-C4 Alkoxycarbonyl
und Benzyl oder
- (d) C3-C6 Cycloalkyl,
das optional mit einem Substituenten substituiert ist, der aus der
Gruppe ausgewählt ist,
welche besteht aus C1-C4 Alkoxycarbonylamino,
Amino, Hydroxy und C1-C4 Alkylen-OH,
R1 ist
- (a) ausgewählt
aus der Gruppe, die besteht aus Wasserstoff, C1-C6 Alkyl, C2-C4 Alkinyl, Halogen, Amino, Azido, Formyl,
1-(C1-C4 Alkoxycarbonyl)ethen-2-yl,
1-(C1-C4 Alkoxycarbonyl)ethyl,
1-(C1-C4 Carboxy)ethyl, (C1-C4 Alkylen)benzyloxy,
Trifluormethyl, Trimethylsilylethinyl, But-3-in-1-ol, C3-C6 Cycloalkyl, Tetrahydropyran-4-yl, Hydroxymethyl,
2-(Piperidin-1-yl)methyl, N,N',N'-[Trimethyl]-2-(aminoethylamino) methyl,
(Morpholin-4-yl)methyl, Dimethylaminomethyl, N-[2-(Piperidin-1-yl)eth-1-yl]aminomethyl,
N',N'-Dimethyl-2-(aminoethylamino)methyl,
Pyridinyl, Thiazolyl, Triazolyl, Benzo(1,3)dioxolan-5-yl und Imidazol-2-yl,
- (b) Phenyl, das optional mit einem bis drei Substituenten substituiert
ist, die unabhängig
aus der Gruppe ausgewählt
sind, die besteht aus C1-C4 Alkyl,
Halogen, Nitro, Amino, C1-C4 Alkoxy,
Trifluormethyl, Trifluormethoxy, Trifluormethylsulfanyl, Methylsulfonyl,
Methylsulfonamidyl, Pyrrolidin-1-yl, Morpholin-4-yl, 4-(C1-C4 Alkyl)piperazin-1-yl, -NR6R7 und C1-C4 Alkoxy, das optional mit einem Substituenten
substituiert ist, der aus der Gruppe ausgewählt ist, welche besteht aus
Piperidin-1-yl, Pyrrolidin-1-yl, Morpholin-4-yl, Azepin-4-yl und
Di(C1-C4 alkyl)amino,
- (c) Thienyl, das optional mit einem Substituenten substituiert
ist, der aus der Gruppe ausgewählt
ist, die besteht aus Halogen, Nitro, Amino und C1-C4 Alkyl oder
- (d) Piperidin-4-yl, das optional an der Position 1 aus der Gruppe
substituiert ist, die aus C1-C4 Alkyl,
C1-C4 Alkoxycarbonyl,
Benzyloxycarbonyl und (C1-C4 Alkylen)-R8 besteht.
-
Alternativ
dazu können
R und R unter Bildung einer vollkommen gesättigten C
3-C
4 Kohlenstoffkette oder einer vollkommen
ungesättigten
C
3-C
4 Kohlenstoffkette
zusammengenommen werden, die optional mit Halogen oder C
1-C
4 Alkyl substituiert
ist,
R
2 steht für Wasserstoff, C
1-C
4 Alkyl oder Benzyl,
R
3 steht
für Thienyl
oder Phenyl, das wahlweise mit einem bis zwei Substituenten substituiert
ist, die unabhängig
aus der Gruppe ausgewählt
sind, welche besteht aus Halogen, C
1-C
4 Alkyl, C
1-C
4 Alkoxy und Trifluormethyl,
R
4 steht für
Wasserstoff, (C
1-C
4 Alkyl)sulfonyl
oder (C
3-C
6 Cycloalkyl)sulfonyl
oder (C
1-C
4 Alkyl)
2-N-sulfonyl,
R
5 steht für
Halogen, Wasserstoff oder -NR
9R
10,
R
6 und R
7 sind jeweils
einzeln aus Wasserstoff, Carbonyl oder C
1-C
4 Alkyl ausgewählt, mit der Maßgabe, dass zumindest
eines von R
6 und R
7 für Wasserstoff
steht,
R
8 steht für Hydroxy, Trifluormethyl,
Dimethylamino, Phenyl, Pyridinyl oder 1-Methylimidazol-2-yl,
R
9 steht jeweils unabhängig für Wasserstoff oder C
1-C
4 Alkyl,
R
10 steht für Wasserstoff, C
1-C
4 Alkyl oder Benzyl,
R
11 steht
für C
1-C
4 Alkoxy, Hydroxy,
C
1-C
4 Alkoxycarbonyl,
C
1-C
4 Alkoxycarbonylamino,
C
3-C
6 Cycloalkyl,
Phenyl, das optional mit einem oder zwei Substituenten substituiert
ist, die unabhängig
aus der Gruppe ausgewählt sind,
die aus C
1-C
4 Alkoxy
und Halogen besteht, für
Morpholin-4-yl oder Pyridinyl, mit der Maßgabe, dass wenn W für
steht, dann folgendes gilt
- (a) zumindest eines von R0 und
R1 für
Wasserstoff oder C1-C6 Alkyl
steht, oder
- (b) R und R unter Bildung einer vollkommen gesättigten
C3-C4 Kohlenstoffkette
oder einer vollkommen ungesättigten
C3-C4 Kohlenstoffkette
zusammen genommen werden können,
die optional mit Halogen oder C1-C4 Alkyl substituiert ist,
und ebenfalls
mit der Maßgabe,
dass wenn X für
S steht, W dann steht für oder ein pharmazeutisch annehmbares
Salz oder ein pharmazeutisch annehmbares Solvat hiervon.
-
Die
vorliegende Erfindung liefert ein Verfahren zur Hemmung der p38
Kinase bei einem Säuger,
das die Verabreichung einer wirksamen Menge einer Verbindung der
Formel I oder eines pharmazeutisch annehmbaren Salzes oder eines
pharmazeutisch annehmbaren Solvats hiervon an einen Säuger umfasst,
der einer solchen Behandlung bedarf.
-
Die
vorliegende Erfindung liefert auch ein Verfahren zur Unterdrückung der
Bildung des Tumornekrosefaktors α (TNF-α) bei einem
Säuger,
das die Verabreichung einer wirksamen Menge einer Verbindung der Formel
I oder eines pharmazeutisch annehmbaren Salzes hiervon oder eines
pharmazeutisch annehmbaren Solvats hiervon an einen Säuger umfasst,
der einer solchen Behandlung bedarf.
-
Die
vorliegende Erfindung liefert auch ein Verfahren zur Suppression
der Bildung von Interleukin-1β (IL-1β) bei einem
Säuger,
das die Verabreichung einer wirksamen Menge einer Verbindung der
Formel I oder eines pharmazeutisch annehmbaren Salzes oder eines
pharmazeutisch annehmbaren Solvats hiervon an einen Säuger umfasst,
der einer solchen Behandlung bedarf.
-
Die
vorliegende Erfindung liefert ferner ein Verfahren zur Behandlung
von Zuständen,
die aus einer überschüssigen Cytokinbildung
resultieren, bei einem Säuger,
das die Verabreichung einer Cytokinunterdrückenden Menge einer Verbindung
der Formel I oder eines pharmazeutisch annehmbaren Salzes oder eines pharmazeutisch
annehmbaren Solvats hiervon an einen Säuger umfasst, der einer solchen
Behandlung bedarf.
-
Die
vorliegende Erfindung liefert auch ein Verfahren zur Hemmung des
Wachstums eines empfindlichen Neoplasmas bei einem Säuger, das
die Verabreichung einer p38 hemmenden Menge einer Verbindung der
Formel I oder eines pharmazeutisch annehmbaren Salzes oder eines
pharmazeutisch annehmbaren Solvats hiervon an einen Säuger umfasst,
der einer solchen Behandlung bedarf.
-
Die
vorliegende Erfindung liefert auch ein Verfahren zur Hemmung der
Metastasierung bei einem Säuger,
das die Verabreichung einer p38 hemmenden Menge einer Verbindung
der Formel I oder eines pharmazeutisch annehmbaren Salzes oder eines
pharmazeutisch annehmbaren Solvats hiervon an einen Säuger umfasst,
der einer solchen Behandlung bedarf.
-
Die
vorliegende Erfindung liefert auch ein Verfahren zur Behandlung
der rheumatoiden Arthritis bei einem Säuger, das die Verabreichung
einer p38 hemmenden Menge einer Verbindung der Formel I oder eines pharmazeutisch
annehmbaren Salzes oder eines pharmazeutisch annehmbaren Solvats
hiervon an einen Säuger
umfasst, der einer solchen Behandlung bedarf.
-
Die
vorliegende Erfindung liefert auch eine pharmazeutische Formulierung,
die eine Verbindung der Formel I oder ein pharmazeutisch annehmbares
Salz oder ein pharmazeutisch annehmbares Solvat hiervon in Kombination
mit einem pharmazeutisch annehmbaren Träger, Verdünnungsmittel oder Hilfsstoff
umfasst.
-
Die
Erfindung liefert auch die Verwendung einer Verbindung der Formel
I oder eines pharmazeutisch annehmbaren Salzes oder eines pharmazeutisch
annehmbaren Solvats hiervon zur Herstellung eines Arzneimittels
zur Hemmung der p38 Kinase. Zusätzlich
liefert die Erfindung eine Verbindung der Formel I oder ein pharmazeutisch
annehmbares Salz oder ein pharmazeutisch annehmbares Solvat hiervon
zur Verwendung bei der Hemmung der p38 Kinase bei Säugern. Ferner
liefert die Erfindung eine pharmazeutische Zusammensetzung, die
zur Hemmung der p38 Kinase angepasst ist, welche eine Verbindung
der Formel I oder ein pharmazeutisch annehmbares Salz hiervon oder
ein pharmazeutisch annehmbares Solvat hiervon in Kombination mit
einem oder mehreren pharmazeutisch annehmbaren Hilfsstoffen, Trägern oder
Verdünnungsmitteln
umfasst.
-
Die
Erfindung liefert auch die Verwendung einer Verbindung der Formel
I oder eines pharmazeutisch annehmbaren Salzes oder pharmazeutisch
annehmbaren Solvats hiervon zur Herstellung eines Arzneimittels zur
Unterdrückung
der Bildung des Tumornekrosefaktors α (TNF-α). Zusätzlich liefert die Erfindung
eine Verbindung der Formel I oder eines pharmazeutisch annehmbaren
Salzes oder eines pharmazeutisch annehmbaren Solvats hiervon zur
Verwendung bei der Unterdrückung
der Bildung des Tumornekrosefaktors α (TNF-α) bei Säugern. Ferner liefert die Erfindung
eine pharmazeutische Zusammensetzung, die zur Unterdrückung der Bildung
des Tumronekrosefaktors α (TNF-α) angepasst
ist, die eine Verbindung der Formel I oder ein pharmazeutisch annehmbares
Salz oder ein pharmazeutisch annehmbares Solvat hiervon in Kombination
mit einem oder mehreren pharmazeutisch annehmbaren Hilfsstoffen,
Trägern
oder Verdünnungsmitteln
umfasst.
-
Die
Erfindung liefert auch die Verwendung einer Verbindung der Formel
I oder eines pharmazeutisch annehmbaren Salzes oder eines pharmazeutisch
annehmbaren Solvats hiervon zur Herstellung eines Arzneimittels
zur Unterdrückung
der Bildung von Interleukin 1β (IL-1β). Zusätzlich liefert
die Erfindung eine Verbindung der Formel I oder ein pharmazeutisch
annehmbares Salz oder ein pharmazeutisch annehmbares Solvat hiervon
zur Verwendung bei der Unterdrückung
der Bildung von Interleukin 1β (IL-1β) bei Säugern. Ferner liefert die Erfindung
eine pharmazeutische Zusammensetzung, die zur Unterdrückung der
Bildung von Interleukin 1β (IL-1β) angepasst
ist, die eine Verbindung der Formel I oder ein pharmazeutisch annehmbares
Salz oder ein pharmazeutisch annehmbares Solvat hiervon in Kombination
mit einem oder mehreren pharmazeutisch annehmbaren Hilfsstoffen,
Trägern
oder Verdünnungsmitteln
umfasst.
-
Die
Erfindung liefert auch die Verwendung einer Verbindung der Formel
I oder eines pharmazeutisch annehmbaren Salzes oder eines pharmazeutisch
annehmbaren Solvats hiervon zur Herstellung eines Arzneimittels
zur Behandlung von Zuständen,
die aus einer übermäßigen Cytokinbildung
resultieren. Zusätzlich
liefert die Erfindung eine Verbindung der Formel I oder ein pharmazeutisch
annehmbares Salz oder ein pharmazeutisch annehmbares Solvat hiervon
zur Verwendung bei der Behandlung von Zuständen, die aus einer übermäßigen Cytokinbildung
resultieren, bei Säugern.
Ferner liefert die Erfindung eine pharmazeutische Zusammensetzung,
die zur Behandlung von Zuständen
angepasst ist, welche aus einer übermäßigen Cytokinbildung resultieren,
die eine Verbindung der Formel I oder ein phar mazeutisch annehmbares
Salz oder ein pharmazeutisch annehmbares Solvat hiervon in Kombination
mit einem oder mehreren pharmazeutisch annehmbaren Hilfsstoffen,
Trägern
oder Verdünnungsmitteln
umfasst.
-
Die
Erfindung liefert auch die Verwendung einer Verbindung der Formel
I oder eines pharmazeutisch annehmbaren Salzes oder eines pharmazeutisch
annehmbaren Solvats hiervon zur Herstellung eines Arzneimittels
zur Wachstumshemmung eines empfindlichen Neoplasmas. Zusätzlich liefert
die Erfindung eine Verbindung der Formel I oder ein pharmazeutisch
annehmbares Salz oder ein pharmazeutisch annehmbares Solvat hiervon
zur Verwendung bei der Wachstumshemmung eines empfindlichen Neoplasmas
bei Säugern.
Ferner liefert die Erfindung eine pharmazeutische Zusammensetzung,
die zur Wachstumshemmung eines empfindlichen Neoplasmas angepasst
ist, die eine Verbindung der Formel I oder ein pharmazeutisch annehmbares Salz
oder ein pharmazeutisch annehmbares Solvat hiervon in Kombination
mit einem oder mehreren pharmazeutisch annehmbaren Hilfsstoffen,
Trägern
oder Verdünnungsmitteln
umfasst.
-
Die
Erfindung liefert auch die Verwendung einer Verbindung der Formel
I oder eines pharmazeutisch annehmbaren Salzes oder eines pharmazeutisch
annehmbaren Solvats hiervon zur Herstellung eines Arzneimittels
zur Hemmung der Metastasierung. Zusätzlich liefert die Erfindung
eine Verbindung der Formel I oder ein pharmazeutisch annehmbares
Salz oder ein pharmazeutisch annehmbares Solvat hiervon zur Verwendung
bei der Hemmung der Metastasierung bei Säugern. Ferner liefert die Erfindung
eine pharmazeutische Zusammensetzung, die zur Hemmung der Metastasierung
angepasst ist, die eine Verbindung der Formel I oder ein pharmazeutisch
annehmbares Salz oder ein pharmazeutisch annehmbares Solvat hiervon
in Kombination mit einem oder mehreren pharmazeutisch annehmbaren
Hilfsstoffen, Trägern
oder Verdünnungsmitteln umfasst.
-
Die
Erfindung liefert auch die Verwendung einer Verbindung der Formel
I oder eines pharmazeutisch annehmbaren Salzes oder eines pharmazeutisch
annehmbaren Solvats hiervon zur Herstellung eines Arzneimittels
zur Behandlung der rheumatoiden Arthritis. Zusätzlich liefert die Erfindung
eine Verbindung der Formel I oder ein pharmazeutisch annehmbares
Salz oder ein pharmazeutisch annehmbares Solvat hiervon zur Verwendung
bei der Behandlung der rheumatoiden Arthritis bei Säugern. Ferner
liefert die Erfindung eine pharmazeutische Zusammensetzung, die
zur Behandlung der rheumatoiden Arthritis angepasst ist, die eine
Verbindung der Formel I oder ein pharmazeutisch annehmbares Salz
oder ein pharmazeutisch annehmbares Solvat hiervon in Kombination
mit einem oder mehreren pharmazeutisch annehmbaren Hilfsstoffen,
Trägern
oder Verdünnungsmitteln
umfasst.
-
Detaillierte Beschreibung
der Erfindung
-
Die
allgemeinen chemischen Ausdrücke,
die in den obigen Formeln verwendet werden, haben ihre allgemeinen
Bedeutungen. Beispielsweise umfasst der Ausdruck "C1-C6 Alkyl" Methyl-,
Ethyl-, Propyl-, Isopropyl-, Butyl-, Isobutyl-, sek-Butyl-, tert-Butyl-,
Pentyl- und Hexylreste. Der Ausdruck "C1-C4 Alkyl" ist
innerhalb der Bedeutung von "C1-C6 Alkyl" enthalten und umfasst
Methyl-, Ethyl-, Propyl-, Isopropyl-, Butyl-, Isobutyl-, sek-Butyl-
und tert-Butylreste. Der Ausdruck "C1-C4 Alkoxy" soll
eine C1-C4 Alkylgruppe
bezeichnen, die an das Ausgangsmolekül über ein Sauerstoffatom gebunden
ist und umfasst die Gruppen Methoxy, Ethoxy, Isopropoxy und dergleichen.
Der Ausdruck "C3-C6 Cycloalkyl" umfasst Cyclopropyl-,
Cyclobutyl-, Cyclopentyl- und Cyclohexylreste. Der Ausdruck "Halogen" umfasst Fluor-,
Chlor-, Brom- und Iod.
-
Der
Ausdruck "C1-C4 Alkylen-R8" wird
verwendet, um eine lineare oder verzweigte Alkylenkette zu bezeichnen,
die an einem beliebigen Kohlenstoffatom mit der Variablen R8 substituiert ist und umfasst beispielsweise
lineare oder verzweigte Alkylketten, Benzyl- und α-Methylbenzylreste. Ähnlich soll
der Ausdruck "C1-C4 Alkylen-R12" eine
lineare oder verzweigte Alkylenkette bezeichnen, die an einem beliebigen
Kohlenstoffatom mit der Variablen R12 substituiert
ist und umfasst beispielsweise lineare oder verzweigte Alkylketten,
Benzyl- und α-Methylbenzylreste.
-
Der
Ausdruck "C1-C4 Alkylen-OH" soll eine lineare
oder verzweigte Alkylenkette bezeichnen, die an einem beliebigen
Kohlenstoffatom mit einer Hydroxygruppe substituiert ist.
-
Der
Ausdruck "1,4-Dioxaspiro[4.5]dec-8-yl" soll die folgende
Formel aufweisen:
-
Der
Ausdruck "vollkommen
gesättigte
C3-C4 Kohlenstoffkette" soll eine Kette
mit 3 oder 4 Methylengruppen bezeichnen. Der Ausdruck "vollkommen unsubstituierte
C3-C4 Kohlenstoffkette" soll eine Kette
mit 3 Kohlenstoffatomen, die eine Kohlenstoff-Kohlenstoff-Doppelbindung
enthält
oder eine Kette mit 4 Kohlenstoffen bezeichnen, die zwei Kohlenstoff-Kohlenstoff-Doppelbindungen
enthält.
-
Der
Ausdruck "p-38 Kinase" soll die p-38α und/oder
p38β Kinaseisoformen
umfassen.
-
Der
Ausdruck "Unterdrückung der
Bildung von TNF-α (IL-1β, Cytokin)" wird verwendet,
um überschüssige in
vivo Mengen an TNF-α,
IL-1β oder
einem anderen Cytokin in einem Säuger
auf normale oder sub-normale Mengen zu verringern. Dies kann durch
die Hemmung der in vivo Freisetzung von TNF-α, IL-1β oder einem anderen Cytokin
durch alle Zellen, einschließlich
Makrophagen, durch Herunterregulierung der überschüssigen in vivo Mengen von TNF-α, IL-1β oder einem
anderen Cytokin bei einem Säuger
auf normale oder sub-normale Mengen auf Genomebene, durch Hemmung
der Synthese von TNF-α,
IL-1β oder
einem anderen Cytokin als posttranslationales Ereignis oder durch
eine Herabregulierung von TNF-α,
IL-1β oder
einem anderen Cytokin auf Transkriptions- oder Translationsebene
erreicht werden.
-
Der
Fachmann erkennt, dass bestimmte Verbindungen der Formel I zumindest
ein chirales Zentrum enthalten. Die vorliegende Erfindung umfasst
alle einzelnen Enantiomere oder Diastereomere wie auch Gemische
aus Enantiomeren und Diastereomeren dieser Verbindungen einschließlich Razemate.
Es ist bevorzugt, dass die Verbindungen der Formel I, die zumindest
ein chirales Zentrum enthalten, als einzelne Enantiomere oder Diastereomere
existieren. Die einzelnen Enantiomere oder Diastereomere können ausgehend
von chiralen Reagenzien oder durch stereoselektive oder stereospezifische
Synthesetechniken hergestellt werden. Alternativ dazu können die
einzelnen Enatiomere oder Diastereomere aus Gemischen durch chirale
Standardchromatographie oder Standardkristallisationstechniken isoliert
werden. Der Fachmann erkennt auch, dass die Verbindungen der vorliegenden
Erfindung als Tautomere existieren können. Die vorliegende Erfindung
umfasst alle tautomeren Formen.
-
Ferner
können
bestimmte Verbindungen der Formel Ials geometrische cis- und trans-Isomere
existieren. Die vorliegende Erfindung umfasst alle einzelnen geometrischen
Isomere, wie auch Gemische der geometrischen Isomere dieser Verbindungen.
Es ist bevorzugt, dass die Verbindungen der Formel I als einzelne geometrische
Isomere existieren. Die einzelnen Isomere können selektiv durch Verfahren
hergestellt werden, die dem Fachmann bekannt sind, oder die Gemische
der Isomere können
durch Standardchromatograpie- oder Standardkristallisationstechniken
getrennt werden.
-
Der
geschulte Leser versteht, dass die meisten oder alle Verbindungen
der vorliegenden Erfindung zur Bildung von Salzen fähig sind.
In allen Fällen
sind die pharmazeutisch annehmbaren Salze aller Verbindungen in
deren Namen enthalten. Die Verbindungen der vorliegenden Erfindung
sind Amine und reagieren demnach mit vielen anorganischen und organischen
Säuren
unter Bildung von pharmazeutisch annehmbaren Säureadditionssalzen. Bevorzugte
pharmazeutisch annehmbare Salze sind jene, die mit Chlorwasserstoffsäure, Maleinsäure oder
Methansulfonsäure
gebildet werden.
-
Der
geschulte Leser versteht ebenfalls, dass pharmazeutisch annehmbare
Solvate der Verbindungen der Formel I als Teil der Erfindung betrachtet
werden und durch herkömmliche
Verfahren hergestellt werden können,
wie Auflösen
der Verbindungen der Formel I in Lösemitteln, wie Wasser, Methanol,
Ethanol, usw. und Umkristallisierung unter Verwendung unterschiedlicher
Kristallisationstechniken.
-
Während alle
Verbindungen der Formel I brauchbare Inhibitoren der p38 Kinase
sind, sind bestimmte Verbindungsklassen bevorzugt. Die folgenden
Absätze
beschreiben solche bevorzugten Klassen:
- a)
R0 steht für Wasserstoff,
- b) R0 steht für Methyl,
- c) R0 steht für Cyclopropyl,
- d) R0 steht für Cyclohexyl,
- e) R1 steht für Phenyl, das optional mit
einem oder zwei Substituenten substituiert ist, die individuell
aus der Gruppe ausgewählt
sind, die aus Halogen und Trifluormethyl besteht,
- f) R1 steht für Phenyl, das mit Chlor substituiert
ist,
- g) R1 steht für Phenyl, das mit zwei Chloratomen
substituiert ist,
- h) R1 steht für Phenyl, das mit zwei Fluoratomen
substituiert ist,
- i) R1 steht für Phenyl, das mit Fluor und
Chlor substituiert ist,
- j) R1 steht für Phenyl, das mit Trifluormethyl
substituiert ist,
- k) R1 steht für 4-Chlorphenyl,
- l) R1 steht für 2,6-Dichlorphenyl,
- m) R1 steht für 2,6-Difluorphenyl,
- n) R1 steht für 2-Chlor-6-fluorphenyl,
- o) R1 steht für 2-Fluor-6-trifluormethyl,
- p) R1 steht für Methyl,
- q) R1 steht für Ethyl,
- r) R1 steht für tert-Butyl,
- s) R1 steht für Isopropyl,
- t) R1 steht für 2,2-Dimethylpropyl,
- u) R1 steht für Cycylopropyl,
- v) R1 steht für Cyclohexyl,
- w) R1 steht für Wasserstoff,
- x) R2 steht für Wasserstoff,
- y) R3 steht für Phenyl,
- z) R3 steht für Phenyl, das mit einem Fluor
substituiert ist,
- aa) R3 steht für Phenyl, das mit zwei Fluoratomen
substituiert ist,
- bb) R3 steht für 4-Fluorphenyl,
- cc) R3 steht für 2,4-Difluorphenyl,
- dd) R4 steht für C1-C4 Alkylsulfonyl,
- ee) R4 steht für Isopropylsulfonyl,
- ff) R4 steht für tert-Butylsulfonyl,
- gg) R4 steht für C3-C6 Cycloalkylsulfonyl,
- hh) R4 steht für Cyclopentylsulfonyl,
- ii) R4 steht für Cyclohexylsulfonyl,
- jj) R4 steht für Dimethylaminosulfonyl,
- kk) R5 steht für NR9R10,
- ll) R5 steht für -NH2,
- mm) R5 steht für Wasserstoff,
- nn) W steht für
- oo) W steht für
- pp) W steht für
- qq) W steht für
- rr) X steht für
N(R4),
- ss) X steht für
N-Isopropylsulfonyl, R5 steht für NH2, W steht für R3 steht
für Phenyl
und R1 steht für Phenyl, das wahlweise mit
einem oder zwei Halogenatomen oder C1-C4 Alkyl
substitiuiert,
- tt) Die Verbindung der Formel I ist eine freie Base,
- uu) Die Verbindung der Formel I ist ein Solvat,
- vv) Die Verbindung der Formel I ist ein pharmazeutisch annehmbares
Salz,
- ww) Die Verbindung der Formel I ist das Hydrochloridsalz,
- xx) Die Verbindung der Formel I ist das Napadysilatsalz,
- yy) Die Verbindung der Formel I ist das Dimaleatsalz,
- zz) Die Verbindung der Formel I ist das Methansulfonatsalz.
-
Bevorzugte
Ausführungsformen
der vorliegenden Erfindung umfassen alle Kombinationen von a) bis zz).
-
Die
Verbindungen der Formel I sind Inhibitoren der p38 Kinase. Daher
liefert die Erfindung auch ein Verfahren zur Hemmung der p38 Kinase
bei einem Säuger,
das die Verabreichung einer p38 Kinasehemmenden Menge einer Verbindung
der Formel I umfasst. Es ist bevorzugt dass der durch die Verabreichung
der Verbindung der Formel I zu behandelnde Säuger ein Mensch ist.
-
Als
Inhibitoren der p38 Kinase sind die erfindungsgemäßen Verbindungen
zur Suppression der Bildung der proinflammatorischen Cytokine, des
Tumornekrosefaktors α (TNF-α) und des
Interleukins 1β (IL-1β) und daher
zur Behandlung von Störungen
brauchbar, die aus einer exzessiven Cytokinbildung resultieren.
Die vorliegenden Verbindungen sind daher bei der Behandlung von
entzündlichen
Störungen,
einschließlich
Ekzem, atoper Dermatitis, rheumatoider Arthritis, Osteoarthritis,
entzündlicher
Darmerkrankung und toxischem Schocksyndrom brauchbar. Die Verbindungen
der vorliegenden Erfindung dürften
auch bei der Behandlung von kardiovaskulären Störungen, wie akutem Myokardinfarkt,
chronischem Herzversagen, Atherosklerose, viraler Myokarditis, kardialer
Allotransplantatabstoßung
und Sepsisassoziierter kardialer Dysfunktion brauchbar sein. Ferner
sind die Verbindungen der vorliegenden Erfindung auch zur Behandlung
der zentralen Nervensystemstörungen,
wie Meningokokkenmeningitis, Alzheimerscher Erkrankung, Parkinsonscher
Erkrankung und multipler Sklerose brauchbar.
-
Die
meisten soliden Tumoren nehmen an Masse durch die Proliferation
von malignen Zellen und Stromazellen, einschließlich Endothelzellen zu. Um
einen Tumor auf mehr als 2-3 Zentimeter Durchmesser wachsen zu lassen,
muss er Gefäße bilden,
ein Prozess, der als Angiogenese bekannt ist. Von einer Unterdrückung der
durch Tumoren induzierten Angiogenese durch Angiostatin und Endostatin
wurde berichtet, dass sie zu einer Antitumoraktivität führt (O'Reilly et al., Cell.
88, 277-285 (1997)). Vom selektiven p38 Kinaseinhibitor SB22025
wurde gezeigt, dass er die Angiogenese hemmt (J.R. Jackson, et al.,
J. Pharmacol. Exp. Therapeutics, 284, 687 (1998)). Da die Angiogenese
eine kritische Komponente der Massenexpansion der meisten soliden
Tumoren ist, stellt die Entwicklung von p38 Kinaseinhibitoren zur
Hemmung dieses Prozesses einen vielversprechenden Ansatz für die Antitumortherapie
dar. Diesem Ansatz zur Antitumortherapie könnten die toxischen Nebenwirkungen
oder Arzneimittelresistenz induzierenden Eigenschaften der herkömmlichen
Chemotherapie fehlen (Judah Folkman, Endogenous Inhibitors of Angiogenesis,
The Harvey Lectures, Reihe 92, Seiten 65-82, Wiley-Liss Inc. (1998)).
-
Als
Inhibitoren der p38 Kinase sind die erfindungsgemäßen Verbindungen
daher auch zur Hemmung des Wachstums von sensitiven Neoplasmen brauchbar.
R.M. Schultz, Potential von p38 MAP Kinaseinhibitoren der Behandlung
von Krebs. In: E. Jucker (Herausgeber), Progress in Drug Research,
60, 59-92 (2003). Ein sensitives Neoplasma wird als Neoplasma definiert,
das von der p38 Kinase für
dessen Überleben,
Wachstum oder Metastase abhängt.
Empfindliche Neoplasmen, einschließlich Tumoren des Gehirns,
des Genitourinaltrakts, des lymphatischen Systems, des Magens, des
Larynx und der Lunge (
US
5 717 100 A ). Vorzugsweise umfasst der Ausdruck "empfindliche Neoplasmen", wie er in der vorliegenden
Beschreibung verwendet wird, die humanen Krebsarten, einschließlich kleinzelliges
Lungencarzi nom (A. Greeberg et al., Am. J. Respir. Cell Mol. Biol.,
26, 558 (2002)), Brustcarzinom (J. Chen et al., J. Biol. Chem. 276,
47901 (2001), B. Salh et al., Int. J. Cancer, 98, 148 (2002) und
S. Xiong et al., Cancer Res., 61, 1727 (2001), Magencarzinom (Y.D.
Jung et al., Proc. Am. Assoc. Cancer Res., 43, 9 (2002)), Colorektalcarzinome
(S. Xiong et al., Cancer Res., 61, 1727 (2001)) und maligne Melanome
(C. Denkert et al., Clin. Exp. Metastasis, 19, 79 (2002)).
-
Die
Hemmung der Angiogenese durch die Suppression von TNF-α wurde als
brauchbar bei der Hemmung oder Prävention der Metastase beschrieben
(
US 6 414 150 A ,
US 6 335 336 A ).
Ferner ist die Suppression von TNF-α zur Behandlung der Prävention
von Kachexie indiziert, einem Zerstörungssyndrom, das bei etwa
der Hälfte
aller Krebspatienten vorkommt (T. Yoneda et al., J. Clin. Invest.,
87, 977 (1991)).
-
Ferner
kann die Hemmung der p38 Kinase durch die Behandlung von bestimmten
viralen Zuständen wirksam
sein, wie Influenza (K. Kujime et al., J. Immunology, 164, 3222-3228
(2000)), Rhinovirus (S. Griego et al., J. Immunology, 165, 5211-5220
(2000)), und HIV (L. Shapiro et al., Proc. Natl. Acad. Sci. USA,
95, 7422-7426 (1998)).
-
Verbindungen
der Formel I, worin W für
Imidazol (i) oder (ii) steht und R5 für -NH2 steht, können hergestellt werden, wie
dies im folgenden Schema dargestellt wird, worin "TBS" als tert-Butyldimethylsilyl
definiert ist und alle anderen Variablen wie vorher definiert sind.
Im folgenden Schema ist nur W = Imidazol (i) dargestellt. Dies soll
den Umfang der Erfindung in keiner Weise beschränken.
-
-
Ein
Gemisch des α-Ketosilylethers
(a) wird mit einem geeigneten Aldehyd in Gegenwart von Kupfer-(II)-acetat
und Ammoniumacetat in einem geeigneten Lösemittel, typischerweise Essigsäure erhitzt.
Die Säure
wird neutralisiert und das gewünschte
Imidazol (Ia) wird durch Standardextraktions- und Standardchromatographietechniken
isoliert.
-
Der
erforderliche α-Ketosilylether
(a) kann hergestellt werden, wie dies im folgenden Schema beschrieben
ist, worin "TBS" für tert-Butyldimethylsilyl
steht, R5 für -NH2 steht
und alle anderen Variablen wie vorher definiert sind.
-
-
Eine
geeignete α-Hydroxysäure (b)
wird in das entsprechende Weinrebamid (c) unter Standardbedingungen
umgewandelt. Kurz gesagt wird die α-Hydroxysäure (b) in den entsprechenden
Methylester umgewandelt und der Ester wird dann mit N-Methyl-O-methylhydoxylaminhydrochlorid
in Gegenwart von Trimethylaluminium in einem geeigneten Lösemittel
umgesetzt. Das α-Hydroxyamid
(c) wird dann mit tert-Butyldimethylsilyltriflat
in Gegenwart einer Base unter Standardbedingungen unter Bildung
des α-Silyletheramids (d)
behandelt (Tius et al. (Tetrahedron, 56, 3339-3351 (2000)). Die
Verbindung (d) wird dann mit 6-Iodbenzamid oder 6-Iodbenzothiazol
(e) in Gegenwart von Isopropylmagnesiumchlorid unter Bildung der
gewünschten
Verbindung (a) gekuppelt. Das erforderliche Iodbenzimidazol (e)
kann aus 2-Aminobenzimidazol
hergestellt werden, wie dies von Mitchell et al beschrieben ist
(Journal of Organic Chemistry, 63, 5050-5058 (1998)).
-
Alternativ
dazu kann 3,4-Dinitrobenzoesäure
(f) in das entsprechende Weinrebamid (g) durch Umwandlung der Benzoesäure in das
entsprechende Benzoylhalogenid, vorzugsweise durch Umsetzung mit
Oxalylchlorid und anschließender
Umsetzung des Benzoylchlorids mit N-Methyl-O-methylhydroxylamin
in Gegenwart einer geeigneten Base, typischerweise Pyridin unter
Bildung des entsprechenden Amids umgewandelt werden. Das Amid (g)
wird dann katalytischen Hydrierungsbedingungen unter Bildung des
entsprechenden Diamins unterzogen, das dann mit einem geeigneten
Sulfonylhalogenid in Gegenwart einer Base, typischerweise Pyridin
unter Bildung des entsprechenden Sulfonamids (h) behandelt wird.
Dieses Sulfonamid wird zuerst mit einer Base und dann mit Cyanogenbromid
in einem geeigneten Lösemittel
unter Bildung des Aminobenzimidazols (j) umgesetzt. Das Aminobenzimidazol
(j) wird mit dem aus dem Silylether (k) erzeugten Anion und tert-Butyllithium
unter Bildung des gewünschten
Zwischenprodukts (a) umgesetzt. Der erforderliche Silylether kann
aus dem entsprechenden Alkohol unter Standardbedingungen hergestellt
werden (siehe Greene et al., Protective Groups in Organic Synthesis,
Herausgeber John Wiley and Sons, 1981).
-
-
Alternativ
dazu können
Verbindungen der Formel I, worin W für Imidazol (i) steht, hergestellt
werden, wie dies im folgenden Schema erläutert ist, worin R5 für -NH2 steht, R2 für Wasserstoff
steht und alle anderen Variablen wie vorher definiert sind.
-
-
Das
Diketon (I) wird mit Ammoniumacetat und einem geeigneten Aldehyd
in einem geeigneten Lösemittel,
vorzugsweise Essigsäure,
unter Bildung des entsprechenden Imidazolylbenzimidazols oder Imidazolylbenzothiazols
umgesetzt. Die erforderlichen Diketone (I) können wie im folgenden Schema
beschrieben hergestellt werden, worin alle Variablen wie vorher
definiert sind.
-
-
Das
geeignete Alkinyl wird mit 6-Iodbenzimidazol oder 6-Iodbenzothiazol
(e) gekuppelt und dann unter Bildung der gewünschten Diketonverbindung (I)
durch das Verfahren von Khan et al. oxidiert (JOC, 17, 1063-1065
(1952)).
-
Alternativ
dazu kann die gewünschte
Diketonverbindung ausgehend von α-Ketosilylether
(a) hergestellt werden, wobei die Silylgruppe gefolgt von einer
Oxidation hydrolysiert wird.
-
Die
Verbindungen der Formel I, worin W für Imidazol (ii) steht, können hergestellt
werden, wie dies im folgenden Schema beschrieben ist, worin R5 für
-NH2 steht und alle anderen Variablen wie
vorher definiert sind.
-
-
Das
Heteroaryliodid (e) wird mit Phenyllithium, gefolgt von tert-Butyllithium
bei niedriger Temperatur umgesetzt. Die Dianion wird mit Dimethylformamid
abgefangen und das entsprechende Aldehyd (m) wird unter Standardbedingungen
isoliert. Das Aldehyd wird dann mit einem geeigneten Amin (n) in
einem geeigneten Lösemittel,
typischerweise Dimethylformamid, unter Bildung des entsprechenden
Imins (o) umgesetzt. Dieses Imin wird dann mit einem geeignet substituierten
p-Toluolsulfonylisocyanat (p) in Methanol mit einem primären Alkylamin
bei Rückfluss
unter Bildung der gewünschten
Verbindung (Ib) umgesetzt. Die erforderlichen Amine (n) sind entweder
im Handel erhältlich
oder können
durch dem Fachmann gut bekannte Verfahren hergestellt werden. Die
erforderlichen p-Toluolsulfonylmethylisocyanate (p) können hergestellt
werden, wie dies in dem folgenden Schema beschrieben ist, worin
alle Variablen wie vorher definiert sind.
-
-
Ein
Gemisch aus p-Toluolsulfinsäure,
Formamid und einem geeigneten Aldehyd wird vereinigt und zusammen
in Gegenwart einer geeigneten Säure
unter Bildung des N-Formyl-p-toluolsulfonylmethylamins
(q) erhitzt. Das Zwischenprodukt (q) wird mit einem geeigneten Dehydrierungsmittel,
typischerweise Phosphoroxychlorid unter Bildung des Isocyanids (p)
umgesetzt. Die erforderlichen Aldehyde sind entweder im Handel erhältlich oder
können
durch in der Technik gut bekannte Standardverfahren hergestellt
werden.
-
Die
Verbindungen der Formel I, worin W für Thiazol (vii) steht, können hergestellt
werden, wie dies im folgenden Schema gezeigt ist, worin R5 für
-NH2 steht und alle anderen Variablen wie
vorher definiert sind.
-
-
Ein
geeignetes α-Halogenketon
(r) wird mit einem geeigneten Thioamid (s) in einem geeigneten Lösemittel
unter Bildung des Thiazols (t) umgesetzt. Dieses Thiazol wird dann
mit n-Butyllithium behandelt und das entstehende Anion wird mit
Tributylzinnchlorid unter Bildung des Zinnderivats (u) umgesetzt.
Dieses Zwischenprodukt wird mit 1-(Isopropylsulfonyl)-2-amino-6-iodbenzimidazol
oder 2-Amino-6-iodbenzothiazol
(e) in Gegenwart eines Palladiumkatalysators unter Standardbedingungen
unter Bildung der gewünschten
Verbindung (Ic) gekuppelt.
-
Die
erforderlichen α-Halogenketone
sind entweder im Handel erhältlich
oder können
durch Standardbedingungen aus der entsprechenden Carbonylverbindung
hergestellt werden, wie dies beispielsweise beschrieben ist von
House (H.O. House, Modern Synthetic Reactions, W.A. Benjamin, Inc.
Menlo Park, California (1972), Seiten 459-478) und Larock (R.C.
Larock, Comprehensive Organic Transformations, VCH Publishers, Inc.
New York, New York (1989), Seiten 369-471, 755). Die erforderlichen
Thioamide sind entweder im Handel erhältlich oder können durch
Standardverfahren, die dem Fachmann bekannt sind, hergestellt werden,
beispielsweise durch Behandlung eines geeigneten Amids mit [2,4-Bis-(4-methoxyphenyl)-1,3-dithia-2,4-diphosphetan-2,4-disulfid]
(Lawesson's Reagent).
-
Die
Verbindungen der Formel I, worin W für das Pyrazol (v) steht, können hergestellt
werden, wie dies im folgenden Schema beschrieben ist, worin R5 für
-NH2 steht und alle anderen Variablen wie
vorher definiert sind.
-
-
Das
geeignete Pyrazol-5-on wird mit einem geeigneten Bromierungsmittel,
wie Phosphoroxybromid, unter Bildung des geeigneten 5-Brom-1H-pyrazols
bromiert. Das Pyrazol wird dann mit der geeigneten Benzimidazol-6-borsäure umgesetzt,
um beim geeigneten Pyrazolylbenzimidazol anzukommen.
-
Alternativ
dazu können
Verbindungen, worin W = (iv) steht, hergestellt werden, wie dies
im folgenden Schema erläutert
ist, worin R5 für -NN2 steht
und alle anderen Variablen wie vorher definiert sind.
-
-
Ein
geeignetes Alkinyl wird mit einer geeigneten Chloridverbindung gekuppelt
und dann unter Bildung des Ketons oxidiert. Das Keton wird mit wasserfreiem
Hydrazin und einem geeigneten Aldehyd in einem geeigneten Lösemittel
unter Bildung des Pyrazols umgesetzt. Die Nitrogruppe wird reduziert
und der Ringschluss wird bewirkt, um beim geeigneten Pyrazolylbenzamidazol
anzukommen.
-
Die
Verbindungen der Formel I, worin W für das Pyrazolon (vi) steht,
können
hergestellt werden, wie dies im folgenden Schema beschrieben ist,
worin R5 für -NH2 steht
und alle anderen Variablen wie vorher definiert sind.
-
-
Die
Dianion von 6-Iodbenzimidazol (c) wird durch sequenzielle Behandlung
mit Phenyllithium, gefolgt von tert-Butyllithium bei niedriger Temperatur
hergestellt. Das Anion wird mit Dimethylformamid abgefangen und
das entsprechende Aldehyd (m) wird unter Standardbedingungen isoliert.
Dieses Aldehyd wird dann mit einem geeigneten Ethylacetat, gefolgt
von einer Oxidation unter Bildung des Diketobenzamidazols umgesetzt. Das
Diketobenzimidazol wird mit der geeigneten Hydrazinverbindung umgesetzt
und in Gegenwart von Toluol unter Bildung des gewünschten
Pyrazolonbenzimidazols am Rückfluss
erhitzt.
-
Die
Verbindungen der Formel I, worin W für ein Imidazol (viii) steht
und X für
N(R4) steht, können hergestellt werden, wie
dies im folgenden Schema gezeigt ist, worin alle Variablen wie vorher
definiert sind. Es sollte bemerkt werden, dass das am Imidazol gebundene
2-Phenyl unsubstituiert gezeigt ist, aber auch substituiert sein
kann.
-
-
Paramethoxybenzylmethylamin
wird mit einem geeigenten Sulfonylchlorid gekuppelt. Das Amin wird dann
geschützt
und die entstehende Verbindung wird mit 2-Phenylimidazol-3-yl gekuppelt.
Das Amin wird von den Schutzgruppen befreit und das benzimidazol
wird wie in Scehma III konstruiert.
-
Verbindungen
der Formel I, worin W für
ein Triazol (ix) steht, können
hergestellt werden, wie dies im folgenden Schema gezeigt ist, worin
R5 für
-NH2 steht und 11 Variablen wie vorher definiert
sind.
-
-
Das
Dibrombenzol wird gefolgt von der Aminierung nitriert. Das entstehende
Nitroanilin wird mit einem geeigneten Phenylacetylen unter Bildung
des Diphenylacetylens gekuppelt. Das Sulfonamid wird durch die Zugabe
eines geeigneten Sulfonylhalogenids gebildet und das Triazol wird
durch die Zugabe einer Azidquelle, typischerweise Natriumazid gebildet.
-
Viele
der erfindungsgemäßen Verbindungen
sind nicht nur Inhibitoren von p38 Kinase, aber sind auch brauchbare
Zwischenprodukte zur Herstellung von zusätzlichen Verbindungen der vorliegenden
Erfindung. Beispielsweise können
primäre
und sekundäre
Amine acyliert, alkyliert oder mit Carbonsäuren oder Aminosäuren unter
Standardpeptidkupplungsbedingungen gekuppelt werden. Ferner können Esterreste
zu den entsprechenden Alkoholen reduziert oder unter Standardbedingungen
zu Amiden umgewandelt werden. Alkohole können durch mehrere Nukleophile
unter Bildung der erfindungsgemäßen Verbindungen
aktiviert und verdrängt
werden.
-
Präparation 1
-
1-Isopropylsulfonyl-2-amino-6-formylbenzimidazol
-
Phenyllithium
(750 ml, 1,8 M in Cyclohexan/Ether, 70/30) wird über 1 Stunde zu einer Lösung aus 1-Isopropylsulfonyl-2-amino-6-iodbenzimidazol
(150 g, 0,41 mol) in Tetrahydrofuran (5,6 l) bei –76°C gegeben. Es
wird für
15 Minuten gerührt
und dann wird tert-Butyllithium (750 ml, 1,7 M in Pentan) zugegeben.
Nach 1 Stunde wird Dimethylformamid (250 ml) langsam über eine
Stunde zugegeben und dann auf 0°C
erwärmt.
Es wird durch Gießen
des Reaktionsgemisches in ein Gemisch aus kaltem gesättigtem
wässrigem
Ammoniumchlorid (500 g, 5 l) und konzentrierter Chlorwasserstoffsäure (300
ml) gestoppt. Die Phasen werden getrennt und die organische Phase
wird mit Wasser gewaschen und dann unter verringertem Druck konzentriert.
Der Rückstand
wird in Methanol (500 ml) aufgeschlämmt, der gelbe Niederschlag
wird filtriert und unter Bildung von 85 g (76%) der Titelverbindung
getrocknet.
1H NMR (DMSO-d6): δ 9,89 (s,
1H), 7,97 (s, 1H), 7,76 (d, 1H), 7,43 (s, 2H), 7,37 (d, 1H), 3,98
(m, 1H), 1,35 (m, 6H).
-
Präparation 2
-
α-(p-Toluolsulfonyl)benzylisocyanid
-
Schritt A: N-[Formyl]-α-(p-toluolsulfonyl)benzylamin
-
Verfahren A
-
Konzentrierte
Chlorwasserstoffsäure
(3 ml) wird tropfenweise zu einer Lösung aus p-Toluolsulfinsäurenatriumsalz
in Wasser (20 ml) und tert-Butylmethylether (10 ml) gegeben. Es
wird für
10 Minuten gerührt
und dann werden die Phasen getrennt. Die organische Phase wird mit
gesättigtem
wässrigem
Natriumchlorid gewaschen, über
Natriumsulfat getrocknet und unter verringertem Druck unter Bildung
von 5 g an p-Toluolsulfinsäure
konzentriert. Diese Säure
wird mit Benzaldehyd (4,75 g, 44,8 mmol), Formamid (4,9 g, 0,11
mol) und Camphersulfonsäure
(0,86 g, 3,7 mmol) vereinigt und für 18 Stunden auf 60°C erhitzt.
Die Reaktion wird aus der Hitze entfernt und der weiße Feststoff
wird in 3 : 1 Hexan : Methanol aufgeschlämmt. Die Aufschlämmung wird
unter Bildung von 7,6 g (82%) des gewünschten Produkts als weißer Feststoff
filtriert.
1H NMR (DMSO-d6): δ 9,75 (d,
1H), 7,98 (s, 1H), 7,69 (d, 2H), 7,53 (d, 2H), 7,39 (m, 5H), 6,36
(d, 1H), 2,38 (s, 1H).
-
Verfahren
B
-
Eine
Lösung
aus p-Toluolsulfinsäurenatriumsalz
(6,0 g, 33,7 mmol) in H2O (20 ml) und tert-Butylmethylether
(10 ml) wird tropfenweise mit konzentriertem HCl (3 ml) behandelt
und für
10 Minuten gerührt.
Die Lösung
wird in einem Trenntrichter getrennt und die organische Phase wird
mit gesättigtem
wässrigem
Natriumchlorid gewaschen. Die organische Phase wird über Na2SO4 getrocknet,
filtriert und das Lösemittel
wird unter Bildung von 5,2 g (quantitativ) an p-Toluolsulfinsäure entfernt.
Die Säure
wird mit Benzaldehyd (2,4 g, 22,5 mmol), Formamid (3,8 g, 84,2 mmol)
und Trimethylsilylchlorid (TMSCI) (4,0 g, 37,0 mmol) in 30 ml einer
1 : 1 Lösung
aus Toluol/Acetonitril vereinigt. Die Reaktion wird auf 50°C erhitzt
und für
5 Stunden gerührt.
Die Reaktion wird gekühlt
und in H2O (100 ml) und tert-Butylmethylether
(TBME) (30 ml) verdünnt.
Die Lösung
wird in einem Eisbad gekühlt
und dann unter Bildung von 4,5 g (70%) des gewünschten Produkts filtriert.
Der Feststoff wird unter Vakuum über
Nacht unter Entfernung des restlichen Wassers getrocknet.
1H NMR (DMSO-d6):
9,75 (d, 1H), 7,98 (s, 1H), 7,69 (d, 2H), 7,53 (d, 2H), 7,39 (m,
5H), 6,36 (d, 1H), 2,38 (s, 1H).
-
Schritt B: Dehydratation
-
Eine
Lösung
aus N-[Formyl]-α-(p-toluolsulfonyl)benzylamin
(7,0 g, 0,024 mol) in Dimethoxyethan (200 ml) wird auf –10°C gekühlt. Phosphoroxychlorid
(5,6 ml, 0,56 mmol) wird gefolgt von der tropfenweisen Zugabe von
Triethylamin (16,8 ml, 0,12 mol) in Dimethoxyethan (10 ml) zugegeben,
während
die Reaktionstemperatur unter –5°C gehalten
wird. Das Reaktionsgemisch wird schrittweise über 1 Stunde erwärmt, Wasser
wird zugegeben und es wird mit Ethylacetat extrahiert. Die Phasen
werden getrennt, die organische Phase wird mit gesättigtem
wässrigem
Natriumbicarbonat gewaschen, über
Natriumsulfat getrocknet und unter verringertem Druck unter Bildung
von 6,5 g der Titelverbindung konzentriert. MS (ES–)
m/z = 270,1 (M–H)–.
-
Die
Verbindungen der Präparationen
3-4 werden im wesentlichen wie in Präparation 2 beschrieben mittels
des Verfahrens A aus dem ersten Schritt, hergestellt.
-
-
Die
Verbindungen der Präparationen
5-8 werden im wesentlichen wie in Präparation 2 beschrieben mittels
des Verfahrens B aus dem ersten Schritt, hergestellt.
-
-
Präparation 9
-
N-[1-(Ethoxycarbonyl)piperidin-4-yl]((1-isopropylsulfonyl-2-amino-6-formyl)-benzimidazol)imin
-
1-Isopropylsulfonyl-2-amino-6-formylbenzimidazol
(10 g, 3,7 mmol) und 1-(Ethoxycarbonyl)-4-aminopiperidin (0,64 g, 3,7 mmol) in
Dimethylformamid (5 ml) werden vereinigt und bei Raumtemperatur über Nacht gerührt. Das
Reaktionsgemisch wird mit Ethylacetat (50 ml) verdünnt und
nacheinander mit Wasser (2 × 10 ml)
und gesättigtem
wässrigem
Natriumchlorid (2 × 10
ml) gewaschen. Die verbleibende organische Phase wird über Natriumsulfat
getrocknet und unter verringertem Druck unter Bildung von 1,5 g
(95%) der Titelverbindung konzentriert. MS (ES–):
m/z = 422,2 (M–H)–.
-
Die
Verbindungen der Präparationen
10-41 werden im wesentlichen wie in Präparation 9 beschrieben, hergestellt.
In einigen Beispielen ist die Zugabe von einem Äquivalent an Triethylamin nötig, um
die Umwandlung in das Iminderivat zu vervollständigen.
-
-
-
-
Präparation 42
-
N-[Methyl]-N-[methoxy]-1-(isopropylsulfonyl)-2-aminobenzimidazol-6-carboxamid
-
A. N-[Methyl]-N-[methoxy]-3,4-dinitrobenzamid
-
Ein
Gemisch aus 3,4-Dinitrobenzoesäure
(195 g, 0,92 mol), 1,3 l trockenem Dichlormethan und 2 ml Dimethylformamid
wird auf –12°C unter einer
Stickstoffatmosphäre
gekühlt.
Oxalylchlorid (134 mg, 1,54 mol) wird tropfenweise mittels eines
Zugabetrichters über
35 Minuten zugegeben und das Reaktionsgemisch wird bei Raumtemperatur
unter einer Stickstoffatmosphäre über Nacht
gerührt.
Das überschüssige Oxalylchlorid wird
aus dem Reaktionsgemisch durch wiederholte Cyclen der Konzentration
einer Dichlormethanlösung
des Reaktonsgemisches unter verringertem Druck entfernt. Ein Gemisch
des Rückstands
in 1 l Dichlormethan wird unter einer Stickstoffatmosphäre auf –5°C gekühlt und
N,O-Dimethylhydroxylaminhydrochlorid
(98,7 g, 1,01 mol) wird zugegeben, gefolgt von der vorsichtigen
portionsweisen Zugabe von 209 ml (2,62 mol) an trockenem Pyridin.
Das Gemisch wird bei Raumtemperatur für 4 Stunden gerührt und
dann unter verringertem Druck konzentriert. Der Rückstand
wird in 500 ml Dichlormethan suspendiert und unter verringertem
Druck zweimal konzentriert. Der Rückstand wird in 500 ml Dichlormethan
suspendiert und filtriert. Das Filtrat wird bei –13°C für 3 Tage gelagert, der Feststoff
wird filtriert und mit kaltem Dichlormethan gewaschen. Das Filtrat
wird mit Wasser (40 ml) gewaschen und bei –13°C unter Bildung eines zweiten
Kristallisats gelagert. Die vereinigten Kristallisate werden unter
verringertem Druck unter Bildung von 182 g (77%) der gewünschten
Verbindung getrocknet.
-
B. N-[Methyl]-N-[methoxy]-3,4-diaminobenzamid
-
18,0
g an 10 Gewichtsprozent Pd/C Katalysator werden zu einer Lösung aus
N-[Methyl]-N-[methoxy]-3,4-dinitrobenzamid
(182 g, 0,712 mol) in 900 ml Tetrahydrofuran und 900 ml Ethanol
unter einer Stickstoffatmosphäre
gegeben. Es wird bei Raumtemperatur für 6 Stunden und unter 60 psi
hydriert. Das Gemisch wird durch Celite® filtriert
und das Filtrat wird unter verringertem Druck konzentriert. Der
Rückstand
wird in 500 ml Dichlormethan suspendiert, unter verringertem Druck
konzentriert und der Rückstand
wird unter verringertem Druck unter Bildung von 135 g (97%) der
gewünschten
Verbindung konzentriert.
-
C. N-[Methyl]-N-[methoxy]-3-(isopropylsulfonyl)amino-4-aminobenzamid
-
Trockenes
Pyridin (234 ml, 2,94 mol) wird zu einer kalten (0°C) Lösung aus
N-[Methyl9-N-[methoxy]-3,4-diaminobenzamid
(135 g, 0,69 mol) in 1 Liter trockenem Dichlormethan unter einer
Stickstoffatmosphäre
gegeben. Isopropylsulfonylchlorid (85,4 ml, 0,76 mol) wird bei 0°C über 30 Minuten
zugegeben, das Gemisch wird bei 0°C
für weitere
30 Minuten und dann bei Raumtemperatur über Nacht gerührt. Das
Reaktionsgemisch wird unter verringertem Druck konzentriert und
der Rückstand
wird mit Diethylether (1 l) und 5 N Chlorwasserstoffsäure (1 l)
aufgeteilt. Die Phasen werden getrennt, die Diethyletherphase wird
verworfen, Ethylacetat (1,5 l) wird zu der wässrigen Phase gegeben und gerührt, während festes
Natriumcarbonat bis pH 6,5 zugegeben wird. Die wässrige Phase wird mittels Ethylacetat
extrahiert, die vereinigten organischen Phasen werden gewaschen, über Magnesiumsulfat
getrocknet und unter verringertem Druck konzentriert. Der Rückstand
wird unter Elution mit einem Gradienten an Ethylacetat : Hexan 65
: 3 bis 100% Ethylacetat unter Bildung einer 40% Ausbeute der gewünschten
Verbindung einer Silicagelchromatographie unterzogen.
-
D. Imidazolringbildung
-
Es
wird 5 N Natriumhydroxid (55 ml) über 1 Stunde zu einer Suspension
aus N-[Methyl]-N-[methoxy]-3-(isopropylsulfonyl)amino-4-aminobenzamid
(83 g, 0,28 mol) in 550 ml Isopropylalkohol und 28 ml Wasser gegeben.
Das Reaktionsgemisch wird für
eine weitere Stunde gerührt
und dann auf 3°C
gekühlt.
Cyanogenbromid (29,0 g, 0,27 mol) wird portionsweise zugegeben und
bei Raumtemperatur über
Nacht und am Rückfluss
für 5 Stunden
gerührt
und dann bei Raumtemperatur über
Nacht. Ethylacetat (1,5 l) wird zugegeben, kräftig gerührt und dann wird die entstehende
Suspension filtriert. Das Filtrat wird mit gesättigtem wässrigem Natrtiumchlorid gewaschen, über Magnesiumsulfat
getrocknet und dann auf etwa 1/4 Volumen konzentriert. Die Suspension
wird filtriert und der Feststoff wird mit kaltem Ethylacetat gewaschen.
Das Filtrat wird unter verringertem Druck konzentriert und der Rückstand
wird aus Ethylacetat unter Bildung eines zweiten Kristallisats kristallisiert.
Die vereinigten Kristallisats ergeben 60% Ausbeute der Titelverbindung.
MS (FD+): m/z = 326 (M+H)+.
-
Präparation 43
-
N-[Methyl]-N-[methoxy]-2-(tert-butyldimethylsilyl)oxy-2-(4-fluorphenyl)acetamid
-
A. Methyl-p-fluormandelat
-
Kaliumcarbonat
(12 g, 87 mmol) gefolgt von Iodmethan (7,37 ml, 118 mmol) wird zu
einer 0°C
Lösung aus
p-Fluormandelsäure
(79 mmol, 13,4 g) in 160 ml trockenem Dimethylformamid unter einer Stickstoffatmosphäre gegeben.
Das entstehende Gemisch wird bei 0°C für 1 Stunde und bei Raumtemperatur über Nacht gerührt. Das
Reaktionsgemisch wird über
Eis gegossen, mit Wasser und Ethylacetat verdünnt und die wässrige Phase
wird dreimal mit Ethylacetat extrahiert. Die vereinigten organischen
Phasen werden mit kaltem Wasser und gesättigtem wässrigem Natriumchlorid vereinigt, über Natriumsulfat
getrocknet und unter verringertem Druck unter Bildung von 12,7 g
(87%) der gewünschten
Verbindung als hellgelbes Öl
konzentriert.
-
B. N-[Methyl]-N-[methoxy]-2-hydroxy-2-(4-fluorphenyl)acetamid
-
Ein
Gemisch aus N-Methyl-O-methylhydroxylaminhydrochlorid (118 mmol)
und Toluol (125 ml) wird auf –5°C gekühlt. Trimethylaluminium
(2 M in Heptan, 59,2 ml, 118 mmol) wird langsam zu dem Gemisch über 20 Minuten
gegeben, während
die Reaktionstemperatur von –1
bis 8°C
gehalten wird. Nach etwa 5 Minuten wird das Gemisch langsam auf
Raumtemperatur erwärmt
und für
1,5 Stunden gerührt.
Eine Lösung
aus Methyl-p-fluormandelat (11,1 g, 60 mmol) in 75 ml Toluol wird über 30 min
ohne externes Kühlen
zugegeben. Die Reaktion wird auf 0°C gekühlt und mit 10% Chlorwasserstoffsäure gestoppt.
Es wird mit Ethylacetat (4 × 250 ml)
extrahiert. Die vereinigten Ethylacetatphasen werden nacheinander
mit Wasser und gesättigtem
wässrigem
Natriumchlorid gewaschen, über
Natriumsulfat getrocknet und unter verringertem Druck unter Bildung
von 12,1 g (82%) der gewünschten
Verbindung konzentriert.
-
C. O-Silylierung
-
Triethylamin
(17,2 ml, 123 mmol) wird gefolgt von tert-Butyldimethylsilyltriflat
(20,8 ml, 90 mmol) zu einer 0°C
Lösung
aus N-[Methyl]-N-[methoxy]-2-hydroxy-2-(4-fluorphenyl)acetamid (12,1
g, 62 mmol) in 180 ml Dichlormethan unter einer Stickstoffatmosphäre gegeben.
Das Reaktionsgemisch wird bei 0°C
für 1 Stunde und
bei Raumtemperatur für
4 Stunden gerührt.
Eine gesättigte
wässrige
Lösung
aus Ammoniumchlorid wird zugegeben und mit Diethylether verdünnt. Die
organische Phase wird nacheinander mit Wasser und gesättigtem
Natriumchlorid gewaschen, über
Natriumsulfat getrocknet und unter verringertem Druck konzentriert.
Der Rückstand
wird durch Silicagelchromatographie unter Elution mit 9 : 1 Hexan
: Ethylacetat unter Bildung der Titelverbindung als gelbes Öl mit 55%
Ausbeute gereinigt.
-
Präparation 44
-
N-[Methyl]-N-[methoxy]-2-(tert-butyldimethylsilyl)oxy-2-(4-(trifluormethyl)phenyl)acetamid
-
Ausgehend
von p-(Trifluormethyl)mandelsäure
wird die Titelverbindung im wesentlichen wie in Präparation
43 beschrieben, hergestellt.
-
Präparation 45
-
1-Isopropylsulfonyl-2-amino-6-(α-((tert-butyldimethylsilyl)oxy)-α-(phenyl)acetyl)-benzimidazol
-
Isopropylmagnesiumchlorid
(2,0 M in THF, 235 ml, 470 mmol) wird über 15 Minuten zu einer Lösung aus
1-Isopropylsulfonyl-2-amino-6-iodbenzimidazol (42,9 g, 118 mmol)
in Tetrahydrofuran (850 ml) bei –70°C unter einer Stickstoffatmosphäre gegeben.
Es wird für
1 Stunde bei 0°C
gerührt
und dann wird eine Lösung aus
N-[Methyl]-N-[methoxy]-2-(tert-butyldimethylsilyl)oxy-2-(phenyl)acetamid
(90,0 g, 294 mmol) (Tius et al. Tetrahedron, 56, 3339-3351 (2000))
in Tetrahydrofuran (150 ml) mittels einer Kanüle zugegeben. Die entstehende
Aufschlämmung
wird bei 0-5°C
für 1 Stunde
und dann bei Raumtemperatur für
1,5 Stunden gerührt. Das
Gemisch wird auf 10°C
gekühlt
und dann wird gesättigtes
wässriges
Ammoniumchlorid zugegeben. Das Gemisch wird für 15 Minuten gerührt und
die Phasen werden getrennt. Die wässrige Phase wird mit Ethylacetat (400
ml) extrahiert, die vereinigten organischen Phasen werden über Natriumsulfat
getrocknet und unter verringertem Druck konzentriert. Der Rückstand
wird durch Silicagelchromatographie unter Elution mit 1-10% Acetonitril
in Dichlormethan, das 0,5% Triethylamin enthält, unter Bildung von 50% Ausbeute
der Titelverbindung als weißer
Feststoff gereinigt. MS (ES+): m/z = 488,1
(M+H)+
-
Die
Verbindungen der Präparationen
46-47 werden im wesentlichen wie in Präparation 45 beschrieben, hergestellt.
-
-
Präparation 48
-
Alternative Synthese von
1-Isopropylsulfonyl-2-amino-6-(α-((tert-butyldimethylsilyl)oxy)-α-(phenyl)acetyl)benzimidazol
-
Tert-Butyllithium
(1,5 M Lösung,
5,8 ml, 8,65 mmol) wird langsam zu einer Lösung aus O-(tert-Butyldimethyl)silylbenzylalkohol
(1,9 g, 8,54 mmol) in 40 ml wasserfreiem Tetrahydrofuran bei –78°C unter einer
Stickstoffatmosphäre
gegeben. Die Lösung
wird für
3,5 Stunden gerührt
wobei sich die Reaktion auf –25°C erwärmen kann.
Es wird auf –35°C gekühlt und
eine Lösung
aus N-[Methyl]-N-[methoxy]-1-isopropylsulfonyl-2-aminobenzimidazol-6-carboxamid
(0,7 g, 2,13 mmol) in 24 ml wasserfreiem Tetrahydrofuran wird zugegeben.
Die Reaktion wird für
1 Stunde gerührt
während
sie sich langsam auf 0°C
erwärmen
kann. Gesättigtes
wässriges Ammoniumchlorid
wird zugegeben und mit Ethylacetat verdünnt. Die wässrige Phase wird mit Ethylacetat
extrahiert, die vereinigten organischen Phasen werden nacheinander
mit Wasser und gesättigtem
wässrigem
Natriumchlorid gewaschen, über
Natriumsulfat getrocknet und unter verringertem Druck konzentriert.
Der Rückstand
wird durch Silicagelchromatographie unter Elution mit 5 : 1 Dichlormethan
: Acetonitril unter Bildung von 730 mg (70%) der Titelverbindung
gereinigt. MS (ES+): m/z = 488,1 (M+H)+.
-
Präparation 49
-
1-Isopropylsulfonyl-2-amino-6-(α-((tert-butyldimethylsilyl)oxy)-α-(3-(trifluormethyl)phenyl)acetyl)benzimidazol
-
Ausgehend
von O-(tert-Butyldimethyl)silyl-3-(trifluormethyl)benzylalkohol
wird die Titelverbindung im wesentlichen wie in Präparation
48 (83% Ausbeute) beschrieben, hergestellt. MS (ES+):
m/z = 556,2 (M+H)+.
-
Präparation 50
-
1-Isopropylsulfonyl-2-amino-6-(α-hydroxy)-α-(4-fluorphenyl)acetyl)benzimidazol
-
Fluorwasserstoffsäure (48%
wässrig,
14,6 ml) wird zu einer Suspension aus 1-Isopropylsulfonyl-2-amino-6-(α-((tert-butyldimethylsilyl)oxy)-α-(4-phenyl)acetyl)benzimidazol
(7,3 g, 14,4 mmol) in Acetonitril (60 ml) bei Raumtemperatur gegeben.
Die klare Lösung
wird für
2,5 Stunden bei Raumtemperatur gerührt. Der entstehende weiße Feststoff
wird durch Filtration gesammelt und mit zusätzlichem Acetonitril (25 ml)
gewaschen. Der weiße
Feststoff wird unter Vakuum unter Bildung von 5,49 g (97%) der Titelverbindung
getrocknet. MS (ES+): m/z = 392 (M+H)+.
-
Präparation 51
-
1-Isopropylsulfonyl-2-amino-6-(2-(4-fluorphenyl)ethan-1,2-dion)benzimidazol
-
1-Hydroxy-1,2-benzoiodoxol-3(1H)-on-1-oxid
(IBX) (2,83 g, 10,11 mmol) wird zu einer Lösung aus 1-Isopropylsulfonyl-2-amino-6-(α-hydroxy)-α-(4-fluorphenyl)acetyl)benzimidazol
(3,16 g, 8,08 mmol) in wasserfreiem Dimethylsulfoxid (25 ml) gegeben.
Nach 2 Stunden wird Natriumthiosulfat (gesättigte wässrige Lösung, 50 ml) zugegeben und
bei Raumtemperatur für
5 Minuten gerührt.
Wasser wird zugegeben und mit Ethylacetat extrahiert. Die organische
Phase wird mit wässrigem
gesättigtem
Natriumchlorid gewaschen und getrocknet (MgSO4).
Nach der Eindampfung des Lösemittels
wird das rohe Produkt durch Säulenchromatographie
unter Bildung von 2,08 g (74% Ausbeute) der Titelverbindung als
blassgelber Feststoff gereinigt. MS (ES+):
m/z = 390 (M+H)+.
-
Präparation 52
-
1-Isopropylsulfonyl-2-amino-6-(phenylethinyl)benzimidazol
-
Unter
einer Stickstoffatmosphäre
werden 1-Isopropylsulfonyl-2-amino-6-iod-benzimidazol (400 g, 1,095
mol), Bis(triphenylphosphin)palladium(II)acetat (32,8 g, 0,044 mol)
und Cul (41,7 g, 0,219 mol) gefolgt von DMSO (8,4 l) und Triethylamin
(317 g, 3,133 mol) vereinigt. Das Rühren des entstehenden Gemisches
bei 20 bis 25°C
für 15
Minuten, gefolgt von der Zugabe von Phenylacetylen (168 g, 1,645
mol) über
30 Minuten ergibt einen Temperaturanstieg auf 34°C. Die Reaktion wird langsam
auf 20-25°C
gekühlt
und nach 15 Stunden wird ein Aliquot der Reaktion in Wasser gestoppt
und mit CH3CN zur HPLC Analyse verdünnt. Das
HPLC Chromatogramm (Säule:
Zorbax SB-C8, 4,6 × 250
mm, 5 Micron UV = 218 nm, Gradienten Acetonitril/Puffer) zeigt den
vollständigen
Verbrauch des Iodids mit der Bildung von zwei neuen weniger polaren
Produkten (gewünschtes
Produkt und Phenylacetylendimer). Wasser (4 l) wird zu der Reaktion über 1 Stunde
gegeben, wobei sich die Temperatur auf 40°C erhöht und sich ein dunkler Niederschlag
bildet (hauptsächlich
Katalysator). Die Reaktion wird durch eine dünne Phase auf Celite® (24
cm Durchmesser) filtriert wobei der Katalysatorabfallkuchen nicht
gewaschen wird. Das Filtrat wird in den Reaktor zurückgegeben
und zusätzliche
4 l Wasser werden über
1 Stunde unter Bildung einer Endtemperatur von 45°C und einer
gelben Aufschlämmung
zugegeben. Das Gemisch kann sich über Nacht auf 20 bis 25°C unter Rühren abkühlen. Die
Feststoffe werden durch Vakuumfiltration gesammelt und mit Wasser
(4 l) gewaschen. Die Feststoffe werden bei 60°C unter Vakuum auf ein konstantes
Gewicht getrocknet. Ausbeute: 319 g (86%) des hellgelben granulären Feststoffs.
MS (ES+) m/z = 340,2 (M+H)+.
-
Präparation 53
-
1-Cyclopentylsulfonyl-2-amino-6-(phenylethinyl)benzimidazol
-
Ausgehend
von 1-Cyclopentylsulfonyl-2-amino-6-iodbenzimidazol wird die Titelverbindung
im wesentlichen wie in Präparation
52 beschrieben (44% Ausbeute) hergestellt. MS (APC+):
m/z = 366,2 (M+H)+.
-
Präparation 54
-
1-Isopropylsulfonyl-2-amino-6-(2-phenylethan-1,2-dion)benzimidazol
-
Wasser
(4 l), NaHCO3 (30 g, 0,357 mol) und MgSO4 (145 g, 1,205 mol) werden bei 20-25°C vereinigt und
bis zur Homogenität
(exotherm bis 30,5°C)
gerührt.
Aceton (4 l) wird unter Bildung eines trüben Gemisches zu der Reaktion
gegeben, gefolgt von 1-Isopropylsulfonyl-2-amino-6-(phenylethinyl)benzimidazol
(200 g, 0,590 mol). Die entstehende Aufschlämmung wird mit KMnO4 (360 g, 2,278 mol) unter Bildung einer
schrittweisen Exothermie bis 40°C über 1 Stunde
behandelt. Nach einer weiteren Stunde (35°C) wird ein Aliquot des Reaktionsgemisches
mit CH3CN zur HPLC Analyse verdünnt. Das
HPLC Chromatogramm zeigt den vollständigen Verbrauch des Ausgangsmaterials
mit einer reinen Bildung eines leicht stärker polaren Produkts. Na2SO3 (400 g) wird
zu dem Reaktionsgemisch gefolgt von EtOAc (3 l) gegeben. Es wird
20% H2SO4 in Wasser (300
ml) zu dem Reaktionsgemisch über
25 Minuten (Temperaturbereich 30 bis 40°C. Eine große Menge an festem MnO2 wird gebildet) gegeben. Die Phasen können sich
trennen und ein Aliquot der oberen organischen Phase zur HPLC Analyse
(HPLC Chromatogramm zeigt ein reines Produkt) wird gesammelt. Die
organische Phase wird abgetrennt und durch Filtration durch Celite® gereinigt.
Die wässrige
Phase (schwarz und dick) wird mit EtOAc (2 l) rückextrahiert und die entstehende
organische Phase wird durch Filtration durch Celite® geklärt. EtOAc
(1 l) wird zum Waschen der Celite® verwendet.
Die organischen Phasen werden vereinigt und bei 40°C unter Vakuum
konzentriert (7 l werden entfernt), wobei sich zwei Phasen bilden.
EtOAc (2 l) wird zu dem Gemisch gegeben, gefolgt von Wasser (0,5
l) und NaCl (30 g). Die Phasen werden getrennt und die wässrige Phase
wird mit EtOAc (0,5 l) rückextrahiert.
Die organischen Phasen werden vereinigt, mit MgSO4 getrocknet und
EtOAc (2 l) wird zum Waschen des MgSO4 zugegeben.
Das Lösemittel
wird unter Vakuum bei 40°C
unter Bildung von 189 g der gelben Feststoffe entfernt. Die Feststoffe
werden unter Vakuum bei 60°C
bis zu einem konstanten Gewicht unter Bildung eines Endgewichts
von 182 g (83%) getrocknet. Das analytisch reine Endprodukt wird
mittels Umkristallisation aus EtOAc erhalten. MS (ES+)
m/z = 372,2 (M+H)+.
-
Präparation 55
-
1-Cyclopentylsulfonyl-2-amino-6-(2-phenylethan-1,2-dion)benzimidazol
-
Ausgehend
von 1-Cyclopentylsulfonyl-2-amino-6-(phenylethinyl)benzimidazol
wird die Titelverbindung im wesentlichen wie in Präparation
54 beschrieben, als gelber Feststoff mit 1,29 g (99% Ausbeute) hergestellt.
MS (APC+): m/z = 398,8 (M+H)+.
-
Präparation 56
-
1-Isopropylsulfonyl-2-aminobenzimidazol-6-borsäure
-
Ein
5 l Bodenkolben, der mit einem Trockeneis/Acetonbad, einer Stickstoffatmosphäre, einem
mechanischen Rührer,
einem Thermoelement und einem Zugabetrichter mit einem Septum mit
1-Isopropylsulfonyl-2-amino-6-iodbenzimidazol
(125 g, 342 mmol) und THF (1,2 l) ausgestattet ist, wird auf –77°C gekühlt (es entsteht
eine Aufschlämmung).
Zu dieser Aufschlämmung
wird PhLi (1,8 M in c-Hexan/Ether, 599 ml, 1078 mmol) über 25 Minuten
gegeben. wobei die Temperatur unter –67°C gehalten wird. Nach dem Rühren der
entstehenden grünen
Aufschlämmung
für 20
Minuten unter Kühlen
auf –77°C wird t-BuLi
(1,7 M in Pentan, 503 ml, 856 mmol) über 25 Minuten zugegeben, wobei
die Temperatur der Reaktion unter –67°C gehalten wird. Das Gemisch
wird bei –75°C für 40 Minuten
gerührt
und dann wird Triisopropylborat (276 ml, 1198 mmol) über 15 Minuten
zugegeben, wobei die Temperatur unter –65°C gehalten wird. Die entstehende
Aufschlämmung wird
langsam über
2 Stunden auf 0°C
gekühlt,
dann wird ein Gemisch aus konzentriertem HCl (wässrig) und Wasser (400 ml)
zugegeben, bis pH 2 erreicht ist. Das Gemisch wird für 1 Stunde
gerührt
und dann mit 5 N NaOH auf pH > 12
eingestellt, während
die Temperatur < 10°C beträgt. Wasser
(1 l) wird zugegeben und die Phasen werden getrennt. Die wässrige Phase
wird mit EtOAc (300 ml) gewaschen und dann mit konzentriertem HCl
(wässrig)
auf pH 6 eingestellt. Das Gemisch wird mit EtOAc (2 × 700 ml)
extrahiert und die vereinigten Extrakte (Na2SO4) werden getrocknet, filtriert und im Vakuum
zu einer orangen Paste (116 g) konzentriert. Die Paste wird in warmem
(–60°C) 2-Propanol
(360 ml) gelöst
und dann Wasser (1,4 l) zugegeben. Die Lösung wird für 3 Stunden auf 0°C gekühlt. Der
entstehende Feststoff wird durch Filtration gesammelt, mit Wasser
gewaschen und dann unter konstantem Gewicht unter Bildung der Titelverbindung
(34,9 g, 36% Ausbeute) als hellbraune Kristalle luftgetrocknet.
Es wird ein zweites Kristallisat aus dem Filtrat (4,95 g, 41% Gesamtausbeute)
erhalten. MS (ES+): m/z = 284 (M+H)+.
-
Präparation 57
-
2,4-(Dibrom)-5-(phenyl)imidazol
-
Zu
einer Suspension aus 4-Phenylimidazol (51,0 g, 354 mol) in AcOH
(450 ml) wird tropfenweise Brom (39,9 ml, 778 mmol) bei Raumtemperatur
unter Stickstoff bei einer derartigen Geschwindigkeit so gegeben, dass
die innere Temperatur < 30°C beträgt, wobei
ein Eisbad zur Kühlung
verwendet wird. Das Gemisch wird bei Raumtemperatur für 22 Stunden
gerührt
(zu welcher Zeit die Analyse gemäß LC-MS
anzeigt, dass das Monobromzwischenprodukt noch vorhanden ist). Zusätzliches
Brom (12 ml) wird zugegeben und die Reaktion wird für 24 Stunden
gerührt
und dann mit Wasser (1600 ml) verdünnt. Das Gemisch wird mit Ether
(800 dann 400 ml) verdünnt
und die vereinigten organischen Phasen werden mit NaHCO3 (5%,
1000 ml), Wasser (2 × 1000
ml) und dann NaHCO3 (500 ml) gewaschen.
Die organische Phase (Na2SO4)
wird getrocknet, filtriert und im Vakuum teilweise konzentriert.
Hexan (500 ml) wird zugegeben und das Gemisch wird wieder teilweise
im Vakuum zu etwa 600-700 ml konzentriert. Der Feststoff wird durch
Filtration gesammelt und unter Bildung der Titelverbindungen (70,9
g, 66% Ausbeute) als blassgelbes Pulver luftgetrocknet. MS (ES+): m/z = 301 (M+H)+.
-
Präparation 58
-
1-(Trimethylsilylethoxymethyl)-2,5-(dibrom)-4-(phenyl)imidazol
-
Zu
einer Lösung
aus 2,4-(Dibrom)-5-(phenyl)imidazol (73,86 g, 244,6 mmol) in trockenem
THF (800 ml) die unter Stickstoff auf 1°C gekühlt ist, wird portionsweise
NaH (60% ungewaschen in Mineralöl,
11,25 g, 281 mmol) gegeben, wobei die Reaktionstemperatur bei < 10°C gehalten
wird. Das Gemisch wird auf –7°C gekühlt und
dann wird Silylethoxymethylchlorid (SEMCI) (42,0 g, 252 mmol) tropfenweise
(2°C) zugegeben. Das
Gemisch wird für
2,5 Stunden (10°C)
gerührt
und dann mit Wasser (1400 ml) verdünnt und mit Ether (2 × 600 ml)
extrahiert. Die vereinigten organischen Phasen werden getrocknet
(Na2SO4), filtriert
und im Vakuum konzentriert. Das rohe Material wird durch ein Kissen
aus Silicagel (700 g) filtriert, mit Hexan (3 l), dann 10% (3 l)
und 20% (3 l) EtOAc/Hexan unter Bildung des Materials eluiert, das
aus heißem
Isopropylalkohol (500 ml) umkristallisiert ist, der über 2 Stunden
auf Raumtemperatur und dann für
30 Minuten auf 5°C
gekühlt
wird. Der Feststoff wird durch Filtration gesammelt, mit kaltem
Isopropylalkohol gewaschen und dann unter Bildung von 54,2 g (51%
Ausbeute) als weiße
Kristalle luftgetrocknet. MS (ES+): m/z
= 431 (M+H)+.
-
Präparation 59
-
1-(Trimethylsilylethoxymethyl)-2-(formyl)-5-(brom)-4-(phenyl)imidazol
-
Zu
einer Lösung
aus 1-(Trimethylsilylethoxy)-2,5-(dibrom)-4-(phenyl)imidazol (60,3
g, 140 mmol) in trockenem THF (600 ml), die unter Stickstoff auf –11°C gekühlt ist,
wird tropfenweise eine Lösung
aus Isopropylmagnesiumchlorid (2 M THF, 87,2 ml, 174 mmol) gegeben,
wobei sich eine Exothermie auf 1°C
entwickelt. Das Gemisch wird bei dieser Temperatur für 30 Minuten
gerührt
und dann wird trockenes DMF (32,4 ml, 419 mmol) (8°C) zugegeben
und für
45 Minuten gerührt.
Eine gesättigte
wässrige
Lösung
aus NH4Cl (500 ml) und Wasser (100 ml) wird
zugegeben, die Phasen werden getrennt und die wässrige Phase wird mit Ethylacetat (300
ml) extrahiert. Die vereinigten organischen Phasen werden getrocknet
(Na2SO4), filtriert
und im Vakuum konzentriert und der Rückstand wird durch Chromatographie
(Biotage 75 lang, eluiert mit 5% EtOAc/Hexan) unter Bildung von
33,6 g (63% Ausbeute) als gelbes Öl gereinigt. MS (ES+): m/z = 323 (M+H)+.
-
Präparation 60
-
1-Isopropylsulfonyl-2-amino-6-(1-(trimethylsilylethoxymethyl)-2-(formyl)-4-(phenyl)imidazol-5-yl)benzimidazol
-
Zu
einer Lösung
aus 1-(Trimethylsilylethoxymethyl)-2-(formyl)-5-(brom)-4-(phenyl)-imidazol
(532 mg, 1,39 mmol) in trockenem Toluol (9 ml), das vorher mit einem
Stickstoffstrom gespült
wurde, wird PdCl2 (PPh3)2 (49 mg, 0,0697 mmol) in einer Portion gegeben.
Nach 5 Minuten wird eine Suspension aus 1-Isopropylsulfonyl-2-amino-6-borsäurebenzimidazol
(472 mg, 1,67 mmol) in Ethanol (6 ml) des vorher mit einem Stickstoffstrom
gespült
wurde, gefolgt von Natriumcarbonat (2 M in Wasser, 3,4 ml, 6,95
mmol) zugegeben. Das Gemisch wird für 2,5 Stunden bei 90°C gerührt, auf
Raumtemperatur gekühlt
und mit Wasser (20 ml) verdünnt. Das
Gemisch wird mit EtOAc (3 × 30
ml) extrahiert und die vereinigten organischen Phasen werden mit
gesättigtem
wässrigem
Natriumchlorid (30 ml) gewaschen, getrocknet (MgSO4)
und im Vakuum konzentriert. Der Rückstand wird durch Blitzchromatographie
(SiO2, Eluent Hexan/EtOAc 1 : 1 bis 1 :
3) unter Bildung eines braunen Feststoffs, 384 mg, 51% Ausbeute,
gereinigt. MS (ES+): m/z = 540,2 (M+H)+.
-
Präparation 61
-
1-Benzyl-2,4,5-tribromimidazol
-
Es
werden 2,4,5-Tribromimidazol (15,29 g, 0,050 mol), Cäsiumcarbonat
(18,0 g, 0,055 mol), Benzylbromid (6,3 ml, 0,053 mol) und Dimethylformamid
(100 ml) vereinigt und bei 20°C
für 18
Stunden gerührt.
Die Feststoffe werden filtriert und unter verringertem Druck konzentriert.
Es wird in Dichlor methan suspendiert, durch ein 2 cm Silicagelkissen
filtriert und mit Dichlormethan (1 l) gewaschen. Die Filtrate werden
unter verringertem Druck konzentriert. Sie werden in Dichlormethan
(15 ml) rückgelöst, 4 M
HCl in Dioxan (12,5 ml, 0,050 mol) wird zugegeben, auf –20°C gekühlt und
die Feststoffe werden filtriert. Das Filtrat wird konzentriert,
in Ethylether gelöst,
1 M HCl/Ether (20 ml) und Hexan (40 ml) werden zugegeben, auf –20°C gekühlt und
filtriert. Die Feststoffe werden vereinigt und unter verringertem
Druck unter Bildung von 19,6 g (90%, 0,045 mol) der Titelverbindung
getrocknet. MS (ES+): m/z = 393,0 (M+H)+.
-
Beispiel 62
-
1-Benzyl-4,5-dibrom-2-(2,4-difluorphenyl)imidazol
-
Ein
Gemisch aus 1-Benzyl-2,4,5-tribromimidazol (1,043 g, 2,42 mmol),
2,4-Difluorphenylborsäure (0,682
g, 4,32 mmol), Palladiumacetat (0,027 g, 0,12 mmol), R(+)-2,2'-Bis(di-p-tolylphosphino)-1,1'-binaphthyl (0,098 g, 0,14 mmol), 2 M
Natriumcarbonat (3,6 ml, 4,83 mmol), Methanol (3,6 ml) und Toluol
(36 ml) wird für 18
Stunden am Rückfluss
erhitzt. Es wird auf Umgebungstemperatur gekühlt und mit Ethylacetat verdünnt. Es wird
mit gesättigtem
Natriumcarbonat und gesättigtem
Natriumchlorid gewaschen, mit Magnesiumsulfat getrocknet und der
Rückstand
wird auf Silicagel unter Elution mit Hexan/Ethylacetatgemischen
unter Bildung von 1-Benzyl-4,5-dibrom-2-(2,4-difluorphenyl)-1H-imidazol
(0,39 g) gereinigt. MS (ES+): m/z = 461,1
(M+H)+.
-
Präparation 63
-
1-Benzyl-4-brom-5-(3,5-difluorphenyl)-2-(2,4-difluorphenyl)imidazol
-
Ein
Gemisch aus 1-Benzyl-4,5-dibrom-2-(2,4-difluorphenyl)imidazol (0,386
g, 0,90 mmol), 3,5-Difluorphenyloborsäure (0,15
g, 0,945 mmol), trans-Dichlorbis(triphenylphosphin)palladium(II)
(0,065 g, 0,09 mmol), 2 M Natriumcarbonat (0,90 ml), Methanol (1,5
ml) und Toluol (10 ml) wird für
18 Stunden am Rückfluss
erhitzt und dann auf Umgebungstemperatur gekühlt. Es wird mit Ethylacetat
verdünnt
und mit gesättigtem
Natriumbicarbonat, gesättigtem
Natriumchlorid gewaschen, mit Magnesiumsulfat getrocknet, filtriert
und die Lösemittel werden
unter verringertem Druck entfernt. Eine Reinigung des Rückstands
auf Silicagel unter Elution mit Hexan/Ethylacetatgemischen ergibt
1-Benzyl-4-brom-5-(3,5-difluorphenyl)-2-(2,4-difluorphenyl)-1H-imidazol (0,19
g). MS (ES+): m/z = 461,0 (M+H)+.
-
Präparation 64
-
1-Isopropylsulfonyl-2-amino-6-(1-(benzyl)-5-(3,5-difluorphenyl)-2-(2,4-difluorphenyl)-imidazol-4-ylbenzimidazol
-
Ein
Gemisch aus 1-Benzyl-4-brom-5-(3,5-difluorphenyl)-2-(2,4-difluorphenyl)imidazol
(0,061 g, 0,13 mmol), 1-Isopropylsulfonyl-2-amino-benzimidazol-6-borsäure (0,113
g, 0,40 mmol) (beschrieben in Präparation
56), trans-Dichlorbis(triphenylphosphin)palladium (II) (0,009 g,
0,013 mmol), 2 M Natriumcarbonat (0,20 ml) und 1,2-Dimethoxyethan
(5 ml) wird für
18 Stunden am Rückfluss
erhitzt. Es wird auf Umgebungstemperatur gekühlt, mit Ethylacetat verdünnt und
mit gesättigtem
Natriumbicarbonat, gesättigtem
Natriumchlorid gewaschen, mit Magnesiumsulfat getrocknet, filtriert
und die Lösemittel
werden unter verringertem Druck entfernt. Eine Reinigung aus Silicagel
mit Dichlormethan/Methanolgemisch ergibt die Titelverbindung (0,084
g). MS (ES+): m/z = 620,1 (M+H)+.
-
Präparation 65
-
1-Benzyl-4,5-dibrom-2-methylimidazol
-
Zu
einer Lösung
aus 4,5-Dibrom-2-methylimidazol (3,0 g, 12,5 mmol) in 25 ml DMF
werden Natriumcarbonat (2,0 g, 18,8 mmol) und Benzylbromid (1,64
ml, 13,8 mmol) gegeben. Die Lösung
wird bei Raumtemperatur für
72 Stunden gerührt.
Die Reaktion wird in Wasser gegossen und mit EtOAc extrahiert. Die
organische Phase wird mit Wasser und gesättigtem wässrigem Natriumchlorid gewaschen
und über
Natriumsulfat getrocknet. Das Gemisch wird filtriert und zu 3,8
g rohem Öl
konzentriert. Das Öl
wird durch Radialchromatographie unter Elution mit 40% EtOAc in
Hexan gereinigt. Die geeigneten Fraktionen werden zu 3,1 g (75%)
der Titelverbindung als farbloses Öl konzentriert. MS (ES+): m/z = 331,0 (M+H)+.
-
Präparation 66
-
1-Benzyl-4-brom-5-(2,4-difluorphenyl)-2-methylimidazol
-
Zu
einer Lösung
aus 1-(Benzyl)-4,5-(dibrom)-2-(methyl)imidazol (1,71 g, 5,18 mmol)
in 8 ml DME wird 2,4-Difluorphenylborsäure (0,90 g, 5,70 mmol) gegeben.
Natriumcarbonat (1,65 g, 15,5 mmol) gelöst in 1 ml Wasser und trans-Dichlorbis(triphenylphosphin)palladium
(II) (1,09 g, 1,55 mmol) wird zugegeben. Das Gemisch wird unter
Rühren
unter Stickstoff auf 100°C
erhitzt. Nach 3 Stunden wird die Lösung gekühlt und über ein Kissen aus Celite® und
Natriumsulfat filtriert. Das rohe Produkt wird durch Radialchromatographie
unter Elution mit 35% EtOAc, 1% CH2Cl2 in Hexan unter Bildung von 1,12 g (60%)
der Titelverbindung als gelbes Öl gereinigt.
Diese Verbindung wird direkt ohne weitere Reinigung verwendet. MS
(ES+): m/z = 364,0 (M+H)+.
-
Präparation 67
-
1-Isopropylsulfonyl-2-amino-6-(1-(benzyl)-5-(2,4-difluorphenyl)-2-(methyl)imidazol-4-yl)benzimidazol
-
Zu
einer Lösung
aus 1-(Benzyl)-4-(brom)-5-(2,4-difluorphenyl)-2-(methyl)imidazol
(1,12 g, 3,08 mmol) in 8 ml DME wird 1-Isopropylsulfonyl-2-aminobenzimidazol-6-borsäure (2,62
g, 9,25 mmol) (beschrieben in Präparation
58) gegeben. Natriumcarbonat (0,98 g, 9,25 mmol), das in Wasser
(1 ml) gelöst
ist, und trans-Dichlorbis(triphenylphosphin)palladium (II) (0,65
g, 0,93 mmol) werden zugegeben. Das Gemisch wird unter Rühren unter
Stickstoff auf 100°C
erhitzt. Nach 3 Stunden wird die Lösung gekühlt und über ein Kissen aus Celite und
Natriumsulfat filtriert. Der Rückstand
wird durch Radialchromatographie unter Elution mit 3% Methanol in
CH2Cl2 mit einem
Gradienten bis 10% Methanol gereinigt. Eine zweite Reinigung durch
Radialchromatographie unter Elution mit 4% Methanol in CH2Cl2 mit einem Gradienten
bis 6% Methanol wird unter Bildung von 390 mg (24%) der Titelverbindung
als gelber Feststoff ausgeführt.
MS (ES+): m/z = 522,0 (M+H)+.
-
Präparation 68
-
5-Brom-3-methyl-1-phenylpyrazol
-
Zu
einer Lösung
aus 3-Methyl-1-phenyl-2-pyrazol-5-on (2,0 g, 11,5 mmol) in 5 ml
wasserfreiem Acetonitril wird Phosphoroxybromid (9,9 g, 34,5 mmol)
gegeben. Das Gemisch wird am Rückfluss
für 3 Stunden unter
Stickstoff erhitzt. Die Reaktion wird auf 0°C gekühlt und mit Ethylacetat und
Wasser verdünnt.
Die organische Phase wird über
Magnesiumsulfat getrocknet, filtriert und zu einem rohen gelben Öl gegeben,
das ein 1 : 1 Gemisch der mono- bis dibromierten Produkte enthält. Eine
Reinigung des Rückstands
durch Radialchromatographie unter Elution mit Methylenchlorid ergibt
677 mg (25%) eines farblosen Öls.
Es werden 925 mg des weniger polar dibromierten Produkts als weißer Feststoff
isoliert. MS (ES+): m/z = 238,0 (M+H)+.
-
Präparation 69
-
5-Brom-3-tert-butyl-1-phenylpyrazol
-
Zu
einer Lösung
aus 3-tert-Butyl-1-phenyl-2-pyrazol-5-on (1,0 g, 4,6 mmol) in 6
ml wasserfreiem Acetonitril wird Phosphoroxybromid (2,0 g, 6,9 mmol)
gegeben. Das Gemisch wird für
3 Stunden unter Stickstoff am Rückfluss
erhitzt. Die Reaktion wird auf 0°C
gekühlt
und mit Ethylacetat und Wasser verdünnt. Die organische Phase wird über Magnesiumsulfat
getrocknet, filtriert und zu einem rohen farblosen Öl konzentriert.
Die Titelverbindung wird durch Radialchromatographie unter Elution
mit 70 : 30 Methylenchlorid : Hexan unter Bildung von 1,1 g (86%)
als farbloses Öl
gereinigt. MS (ES+): m/z = 280,0 (M+H)+.
-
Präparation 70
-
2-Phenylimidazo[1,2-a]pyridin
-
Es
wird eine Lösung
aus 2-Bromacetophenon (1 g, 5,024 mmol) und 2-Aminopyridin (591
mg, 6,280 mmol) in Ethanol (4 ml), die NaHCO3 (658
mg, 7,837 mmol) enthält,
bei Raumtemperatur für
6 Stunden gerührt. Das
Gemisch wird mit Wasser (15 ml) verdünnt und mit Ether (3 × 20 ml)
extrahiert. Die vereinigten organischen Phasen werden mit gesättigtem
wässrigem
Natriumchlorid (25 ml) gewaschen, getrocknet (MgSO4)
und im Vakuum konzentriert. Eine Reinigung des Rückstands durch Blitzchromatographie
(SiO2, Eluent : Hexan/EtOAc 4 : 1) ergibt
einen weißen
Feststoff mit 79% Ausbeute. MS (ES+): m/z
= 195,1 (M+H)+.
-
Präparation 71
-
8-Methyl-2-phenylimidazo[1,2-a]pyridin
-
Ausgehend
von 2-Amino-3-methylpyridin wird die Titelverbindung im wesentlichen
wie in Präparation 70
beschrieben, mit 73% Ausbeute hergestellt. MS (ES+):
m/z = 195,1 (M+H)+.
-
Präparation 72
-
3-Iod-2-phenylimidazo[1,2-a]pyridin
-
Eine
Lösung
aus 2-Phenylimidazo[1,2-a]pyridin (50 mg, 0,257 mmol) in Acetonitril
(1,25 ml) wird mit N-Iodsuccinimid (NIS) (69 mg, 0,309 mmol) bei
Raumtemperatur für
5 Stunden behandelt. Das Gemisch wird mit Ether (10 ml) verdünnt, mit
einer gesättigten
wässrigen
Lösung
aus NaHCO3 (15 ml) und NaHSO3 (40%, 15
ml) gewaschen, getrocknet (Na2SO4) und im Vakuum konzentriert. Es wird ein
gelber Feststoff mit 80 mg, 97% Ausbeute erhalten. MS (ES+): m/z = 321,0 (M+H)+.
-
Präparation 73
-
3-Iod-8-methyl-2-phenylimidazo[1,2-a]pyridin
-
Ausgehend
von 8-Methyl-2-phenylimidazo[1,2-a]pyridin wird die Titelverbindung
im wesentlichen wie in Präparation
72 beschrieben, mit 65% Ausbeute hergestellt. MS (ES+):
m/z = 335,0 (M+H)+.
-
Präparation 74
-
2-Phenyl-6,7-dihydro-5H-pyrrolo[1,2-a]imidazol
-
Es
wird 2-Iminopyrrolidinhydrochlorid (6,98 g, 57,8 mmol) gemäß Callahan,
et al., J. Med. Chem. 2002, 45, 999-1001) hergestellt und 2-Bromacetophenon
(3,8 g, 19,3 mmol) wird mit Na2CO3 (8,2 g, 77,2 mmol) in trockenem DMF (25
ml) vereinigt und bei 80°C
für 18
Stunden erhitzt. Dann wird das Gemisch auf Raumtemperatur gekühlt, Wasser
(60 ml) wird zugegeben und mit EtOAc (3 × 100 ml) extrahiert. Die vereinigten
organischen Phasen werden im Vakuum konzentriert, der Rückstand
wird mit Ether (100 ml) verdünnt
und mit gekühltem
Wasser (3 × 80
ml) gewaschen. Die organische Phase wird im Vakuum unter Bildung
eines weißen Feststoffs
mit 3,2 g, 89% Ausbeute, konzentriert. MS (ES+):
m/z = 185,1 (M+H)+.
-
Präparation 75
-
2-(4-Fluorphenyl)-6,7-dihydro-5H-pyrrolo[1,2-a]imidazol
-
Ausgehend
von 2-Iminopyrrolidinhydrochlorid und 2-Fluoracetophenon wird die
Titelverbindung im wesentlichen wie in Präparation 74 beschrieben, mit
99% Ausbeute hergestellt.
-
Präparation 76
-
3-Brom-2-phenyl-6,7-dihydro-5H-pyrrolo[1,2-a]imidazol
-
Zu
einer Lösung
aus 2-Phenyl-6,7-dihydro-5H-pyrrolo[1,2-a]imidazol (3,38 g, 18,3
mmol) in trockenem CH2Cl2 (113
ml) wird langsam Brom (1,0 ml, 20,2 mmol) gegeben und das Gemisch
wird bei Raumtemperatur für
1,5 Stunden gerührt.
Eine gesättigte
wässrige
Lösung
aus NaHCO3 (100 ml) wird zugegeben und mit CH2Cl2 (3 × 100 ml)
extrahiert. Die vereinigten organischen Phasen werden mit NaHSO3 (40%, 30 ml) gewaschen, getrocknet (MgSO4) und im Vakuum konzentriert. Der Rückstand
wird durch Blitzchromatographie (SiO2, Eluent:
CH2Cl2 zu CH2Cl2/MeOH 20 : 1)
unter Bildung eines roten Feststoffs mit 3,54 g, 73% Ausbeute, gereinigt.
MS (ES+): m/z = 263,0 (M+H)+.
-
Präparation 77
-
3-Brom-2-(4-fluorphenyl)-6,7-dihydro-5H-pyrrolo[1,2-a]imidazol
-
Ausgehend
von 2-(4-Fluorphenyl)-6,7-dihydro-5H-pyrrolo[1,2-a]imidazol wird
die Titelverbindung im wesentlichen wie in Präparation 76 beschrieben, mit
95% Ausbeute hergestellt.
-
Präparation 78
-
2-Phenyl-5,6,7,8-tetrahydroimidazo[1,2-a]pyridin
-
2-Iminopiperidinhydrochlorid
(4,9 g, 36,4 mmol, 3 Äquivalente)
und 2-Bromacetophenon (2,4 g, 12,1 mmol) werden mit Na2CO3 (5,1 g, 48,4 mmol) in trockenem DMF (15
ml) vereinigt und das Gemisch wird bei 80°C für 16 Stunden erhitzt. Dann
wird das Gemisch auf Raumtemperatur gekühlt, Wasser (200 ml) wird zugegeben
und mit Ether (3 × 100
ml) extrahiert. Die vereinigten organischen Phasen werden mit Wasser
(2 × 50
ml) gewaschen und die wässrige
Phase wird mit Ether (2 × 50
ml) rückextrahiert.
Die vereinigten organischen Phasen werden mit gesättigtem
wässrigem
Natriumchlorid (100 ml) gewaschen, getrocknet (MgSO4) und
im Vakuum konzentriert. Der Rückstand
wird durch Blitzchromatographie (SiO2 :
Eluent: CH2Cl2 bis
CH2Cl2 : MeOH 50
: 1) unter Bildung eines braunen Feststoffs mit 1,7 g, 71% Ausbeute,
gereinigt. MS (ES+): m/z = 199,1 (M+H)+.
-
Präparation 79
-
3-Brom-2-phenyl-5,6,7,8-tetrahydroimidazo[1,2-a]pyridin
-
Zu
einer Lösung
aus 2-Phenyl-5,6,7,8-tetrahydroimidazo[1,2-a]pyridin (1,7 g, 8,9
mmol) in trockenem CH2Cl2 (55
ml) wird langsam Brom (0,5 ml, 9,8 mmol) gegeben und das Gemisch
wird bei Raumtemperatur für 3
Stunden gerührt.
Eine gesättigte
wässrige
Lösung
aus NaHCO3 (100 ml) wird dann zugegeben
und mit CH2Cl2 (3 × 100 ml)
extrahiert. Die vereinigten organischen Phasen werden mit NaHSO3 (40%, 30 ml), gefolgt von gesättigtem
wässrigem
Natriumchlorid (50 ml) gewaschen, getrocknet (MgSO4)
und im Vakuum konzentriert. Der Rückstand wird durch Blitzchromatographie
(SiO2, Eluent: CH2Cl2 bis CH2Cl2/MeOH 20 : 1) unter Bildung eines braunen
Feststoffs mit 2,0 g, 82% Ausbeute, gereinigt. MS (ES+):
m/z = 277,0 (M+H)+.
-
Präparation 80
-
3-Ethoxycarbonyl-2-phenyl-5,6-dihydro-4H-pyrrolo[1,2-b]pyrazol
-
Zu
einer Lösung
aus 1-Aminopyrrolidin-2-onhydrochlorid (3,8 g, 27,8 mmol) (WO 02/094833)
wird Ethylbenzoylacetat (4,3 ml, 25 mmol) gefolgt von Pyridin (10
ml) bei Raumtemperatur unter Stickstoff gegeben. Das Gemisch wird
bei Raumtemperatur für
20 Stunden gerührt
und dann mit Wasser (50 ml) verdünnt
und mit Toluol (2 × 50
ml) extrahiert. Die vereinigten organischen Phasen (MgSO4) werden getrocknet und im Vakuum unter
Bildung eines braunen Öls
mit 6,23 g, 82% Ausbeute, konzentriert und mit NaOEt (frisch hergestellt,
3,1 g, 45,4 mmol, 2 Äquivalente)
in Toluol bei 120°C
für 8 Stunden
behandelt. Das Gemisch wird auf Raumtemperatur gekühlt und
Wasser (100 ml) und konzentrierte HCl werden zugegeben, bis ein
pH von 4 erreicht ist. Das Gemisch wird mit EtOAc (3 × 200 ml)
extrahiert und getrocknet (MgSO4) und die
vereinigten organischen Phasen werden im Vakuum konzentriert. Der
Rückstand
wird in ein Biotagesystem (Eluent: CH2Cl2/MeOH 40 : 1) unter Bildung eines gelben
Feststoffs, 1,8 g, 36% Ausbeute, gereinigt. MS (ES+):
m/z = 257,1 (M+H)+.
-
Präparation 81
-
3-Brom-2-phenyl-5,6-dihydro-4H-pyrrolo[1,2-b]pyrazol
-
A. 3-Carboxy-2-phenyl-5,6-dihydro-4H-pyrrolo[1,2-b]pyrazol
-
Zu
einer Lösung
aus 3-Ethoxycarbonyl-2-phenyl-5,6-dihydro-4H-pyrrolo[1,2-b]pyrazol
(2,1 g, 8,2 mmol) in MeOH (20 ml) wird eine Lösung aus NaOH (15%, 40 ml)
gefolgt von Acetonitril (5 ml) gegeben und bei 50°C für 3 Stunden
gerührt.
Das Gemisch wird auf Raumtemperatur gekühlt und HCl (10%) wird zugegeben,
bis ein pH von 3 erreicht ist. Das Gemisch wird mit EtOAc (3 × 100 ml)
extrahiert und mit gesättigtem wässrigem
Natriumchlorid (100 ml) gewaschen, getrocknet (MgSO4)
und im Vakuum unter Bildung eines gelben Feststoffs mit 1,82 g 97%
Ausbeute, konzentriert. MS (ES+): m/z =
229,1 (M+H)+.
-
B. 3-Brom-2-phenyl-5,6-dihydro-4H-pyrrolo[1,2-b]pyrazol
-
Zu
einer Lösung
aus 3-Carboxy-2-phenyl-5,6-dihydro-4H-pyrrolo[1,2-b]pyrazol (820
mg, 3,59 mmol) in trockenem DMF (15 ml), die vorher entgast wurde,
wird N-Bromsuccinimid (NBS) (702 mg, 3,95 mmol) gegeben und bei
Raumtemperatur für
18 Stunden gerührt.
Dann wird sie mit EtOAc (80 ml) verdünnt und mit gekühltem Wasser
(5 × 10
ml) gewaschen. Die organische Phase (MgSO4)
wird getrocknet und im Vakuum zu einem gelben Feststoff mit 1,0
g, konzentriert. MS (ES+): m/z = 263,0 (M+H)+.
-
Präparation 82
-
1-(4-Methoxybenzyl)-4-phenylimidazol
-
Zu
einer Lösung
aus 4-Phenylimidazol (7,49 g, 51,9 mmol) in DMF (30 ml), die unter
Stickstoff auf 0°C gekühlt ist,
wird portionsweise NaH (95%, 2,56 g, 101 mmol) gegeben. Nach 10
Minuten wird p-Methoxybenzylchlorid
(PMBCI) (7,50 ml, 55,3 mmol) tropfenweise zugegeben. Es wird für 5 Stunden
gerührt,
mit MeOH (5 ml) gestoppt, mit Wasser (100 ml) verdünnt und
mit CH2Cl2 (2 × 100 ml)
verdünnt.
Die vereinigten organischen Phasen (Na2SO4) werden getrocknet und im Vakuum zu einem
Volumen von 80 ml unter Bildung von 12,2 g (89% Ausbeute) nach der
Kristallisation konzentriert. MS (ES+):
m/z = 265 (M+H)+.
-
Präparation 83
-
1-(4-Methoxybenzyl)-2-formyl-4-phenylimidazol
-
Zu
einer Lösung
aus 1-(4-Methoxybenzyl)-4-phenylimidazol (2,00 g, 7,56 mmol) in
THF (20 ml), die unter Stickstoff auf –78°C gekühlt ist, wird tropfenweise
eine Lösung
aus n-Butyllithium (1,6 M Hexan, 5,60 ml, 8,96 mmol) gegeben. Das
Gemisch wird bei dieser Temperatur für 10 Minuten gerührt und
dann wird DMF (1,43 ml, 18,5 mmol) tropfenweise zugegeben und das
Rühren
wird für
30 Minuten fortgesetzt. Eine gesättigte wässrige Lösung aus
NH4Cl (50 ml) wird zugegeben, die Temperatur
wird auf Raumtemperatur erhöht
und die wässrige
Phase wird mit CH2Cl2 (150
ml) extrahiert, getrocknet (Na2SO4) und im Vakuum konzentriert. Der Rückstand
wird durch Blitzchromatographie (SiO2, Hexan/EtOAc
9 : 1) unter Bildung von 1,75 g (79% Ausbeute) gereinigt. MS (ES+): m/z = 293 (M+H)+.
-
Präparation 84
-
1-(4-Methoxybenzyl)-2-hydroxymethyl-4-phenylimidazol
-
Eine
Lösung
aus 1-(4-Methoxybenzyl)-2-formyl-4-phenylimidazol (1,75 g, 5,99
mmol) in CH2Cl2/MeOH (1:1,
60 ml) wird mit NaBH4 (480 mg, 12,7 mmol)
bei Raumtemperatur für
15 Minuten behandelt. Sie wird mit H2O (50
ml) gestoppt, mit CH2Cl2 (2 × 100 ml)
extrahiert, getrocknet (Na2SO4)
und im Vakuum konzentriert. Der Rückstand wird durch Blitzchromatographie
(SiO2, CH2Cl2/MeOH 20 : 1 → 7 : 1) unter Bildung von 1,69
g (96% Ausbeute) gereinigt. MS (ES+): m/z
= 295 (M+H)+.
-
Präparation 85
-
1-(4-Methoxybenzyl)-2-tert-butyldimethylsilyloxymethyl-4-phenylimidazol
-
Die
Lösung
aus 1-(4-Methoxybenzyl)-2hydroxymethyl-4-phenylimidazol (1,68 g,
5,71 mmol) in CH2Cl2 (35
ml) mit N,N-Diisopropylethylamin (3,2 ml, 18,4 mmol) und tert-Butyldimethylsilylchlorid
(TBSCI) (1,46 g, 9,69 mmol) wird bei Raumtemperatur für 60 Stunden
behandelt. Es wird mit H2O (40 ml) gestoppt,
mit CH2Cl2 (100
ml) extrahiert, getrocknet (Na2SO4) und im Vakuum konzentriert. Man reinigt den
Rückstand
durch Blitzchromatographie (SiO2, Hexan/EtOAc
20 : 1 → 9
: 1) unter Bildung von 2,09 g (90% Ausbeute gereinigt. MS(ES+): m/z = 409 (M+H)+.
-
Präparation 86
-
1-(4-Methoxybenzyl)-2-tert-butyldimethylsilyloxymethyl-4-phenyl-5-iodimidazol
-
Eine
Lösung
aus 1-(4-Methoxybenzyl)-2-tert-biutyldimethylsilyloxymethyl-4-phenylimidazol
(2,08 g, 5,09 mmol) in MeCN (25 ml) mit N-Iodsuccinimid (NIS) (1,35
g, 6,00 mmol) wird bei Raumtemperatur für 24 Stunden behandelt. Das
gemisch wird mit Et2O (100 ml) verdünnt, mit
gesättigten
NaHCO3 (25 ml) gewaschen, getrocknet (Na2SO4) und im Vakuum
konzentriert. Der Rückstand
wird durch Blitzchromatographie (SiO2, Hexan/EtOAc
20 : 1 → 9
: 1) unter Bildung von 2,30 g (85% Ausbeute) gereinigt. MS (ES+): m/z 535 (M+H)+.
-
Präparation 87
-
1-(Benzyloxycarbonyl)-4-formylpiperidin
-
Diisobutylaluminiumhydrid
(1 M in Toluol, 30 ml, 30 mmol) wird über 5 Minuten zu einer Lösung aus 1-(Benzyloxycarbonyl)-4-(ethoxycarbonyl)piperidin
(7 g, 24 mmol) in wasserfreiem Dichlormethan (150 ml) bei –78°C unter einer
Stickstoffatmosphäre
gegeben. Das Reaktionsgemisch wird bei dieser Temperatur für 30 Minuten
gerührt
und dann werden 10% wässriges
Natriumtartrat (100 ml) gefolgt von Dichlormethan zugegeben. Das
Reaktionsgemisch wird bei Raumtemperatur über Nacht gerührt. Die
Phasen werden getrennt und die wässrige
Phase wird mit Dichlormethan extrahiert. Die vereinigten organischen
Phasen werden nacheinander mit 10% wässrigem Natriumtartrat, Wasser
und gesättigtem
wässrigem
Natriumchlorid gewaschen. Sie werden über Natriumsulfat getrocknet
und unter verringertem Druck konzentriert. Der Rückstand wird unter Elution mit
3 : 1 Hexan : Ethylacetat unter Bildung von 3,15 g (53%) der Titelverbindung
einer Silicagelchromatographie unterzogen.
-
Präparation 88
-
1-(tert-Butoxycarbonyl)-4-formylpiperidin
-
Ausgehend
von 1-(tert-Butoxycarbonyl)-4-(ethoxycarbonyl)piperidin wird die
Titelverbindung im wesentlichen wie in Präparation 51 beschrieben, hergestellt.
-
Präparation 89
-
4-Formyltetrahydropyran
-
Ausgehend
von 4-(Methoxycarbonyl)tetrahydropyran wird die Titelverbindung
im wesentlichen wie in Präparation
87 beschrieben, hergestellt.
-
Präparation 90
-
4-(N-tert-Butoxycarbonyl-N-ethylamino)benzaldehyd
-
A. N-Boc-Schutzgruppenanbringung
-
Zu
einer Lösung
aus 4-Aminobenzoesäuremethylester
(4 g, 26,4 mmol), (Boc)2O (8,67 g, 39,6
mmol) und DMAP (0,322 mg, 2,64 mmol) in 50 ml Acetonitril wird Triethylamin
(7,345 ml, 52,8 ml) tropfenweise gegeben und über Nacht bei Raumtemperatur
gerührt.
Der Niederschlag wird in Ethylacetat gelöst und mit 3% HCl behandelt,
mit Ethylacetat extrahiert und die organischen Phasen werden mit einer
gesättigten
Lösung
aus Natriumbicarbonat und gesättigtem
wässrigem
Natriumchlorid gewaschen, über
Natriumsulfat getrocknet und unter Bildung von 5,3 g des Produkts
konzentriert. Ausbeute: 80%.
1H NMR
(CDCl3, 300 MHz): 8,07 (d, 2H, J = 8,8),
7,53 (d, 2H, J = 8,8), 6,78 (s, 1H), 4,00 (s, 3H), 1,64 (s, 9H).
-
B. N-Alkylierung
-
Zu
einer Suspension aus 60% Natriumhydrid (595 mg, 1,1 Äquivalente)
in trockenem DMF und unter Argonatmosphäre werden langsam 4-tert-Butoxycarbonylaminobenzoesäuremethylester
(3,24 g, 12,9 mmol) und Ethyliodid (1,206 ml, 1,1 mmol) mittels
einer Spritze gegeben und über
Nacht gerührt.
Ethylacetat wird zugegeben und die entstehende Lösung wird mit gesättigtem
Ammoniumchlorid und gesättigtem
wässrigem
Natriumchlorid gewaschen, über
Natriumsulfat getrocknet und konzentriert. Eine Reinigung des Rückstands durch
Chromatographie unter Elution mit Hexan/16% Ethylacetat ergibt 2,85
g des reinen Produkts. Ausbeute: 79%.
1H
NMR (CDCl3, 300 MHz): 8,11 (dd, J = 8,5,
2,0 Hz, 2H), 7,40 (dd, J = 8,5, 2,0 Hz, 2H), 4,02 (s, 3H), 3,84
(q, J = 6,9 Hz, 2H), 1,56 (s, 9H), 1,28 (t, J = 6,9 Hz, 3H).
-
C. Esterreduktion
-
Zu
einer Lösung
aus 4-(N-tert-Butoxycarbonyl-N-ethylamino)benzoesäuremethylester
(2,85 g, 10,2 mmol) in 65 ml trockenem Toluol unter Argonatmosphäre und bei –78°C wird tropfenweise
Diisobutylaluminiumhydrid (DIBAL-H) 1 M in Toluol (12,77 ml, 12,77
mmol) gegeben. Das Gemisch wird für 1 Stunde gerührt. Natriumtartrat
wird zugegeben, das Bad wird entfernt und das Gemisch wird über Nacht
gerührt.
Die organische Phase wird mit Ethylacetat extrahiert und die organischen
Phasen werden mit gesättigtem
wässrigem
Natriumchlorid gewaschen, über
Natriumsulfat getrocknet und konzentriert. Der Rückstand wird durch Chromatographie
unter Elution mit Hexan/Acetat (16%) unter Bildung von 1,24 g des
reinen Alkohols gereinigt. Ausbeute: 54%.
1H
NMR (CDCl3, 300 MHz): 7,45 (d, J = 8,5 Hz,
2H), 7,30 (d, J = 8,5 Hz, 2H), 4,80 (d, J = 6,1 Hz, 2H), 3,78 (q, J
= 7,3 Hz, 2H), 1,78 (t, J = 6,1 Hz, 1H), 1,55 (s, 9H), 1,25 (t,
J = 7,3 Hz, 3H).
-
D. Alkoholoxidation
-
4-(N-tert-Butoxycarbonyl-N-ethylamino)benzylalkohol
(1,24 g, 5,52 mmol) wird in 50 ml Acetonitril gelöst. Zu dieser
Lösung
wird Mangandioxid gegeben und das Gemisch wird für zwei Tage bei Raumtemperatur gerührt. Der
Rückstand
wird durch Celite® filtriert und das Lösemittel
wird unter Bildung von 632 mg an 4-(N-tert-Butoxycarbonyl-N-ethylamino)benzaldehyd
konzentriert. Ausbeute: 52%.
1H NMR
(CDCl3, 300 MHz): 9,95 (s, 1H), 7,83 (d,
J = 8,7 Hz, 2H), 7,39 (d, J = 8,7 Hz, 2H), 3,74 (q, J = 7,0 Hz, 2H),
1,45 (s, 9H), 1,18 (t, J = 7,0 Hz, 3H).
-
Präparation 91
-
2,6-Difluor-4-(pyrrolidin-1-yl-ethoxy)benzaldehyd
-
A. 1-(3,5-Difluorphenoxymethyl)pyrrolidin
-
Pyrrolidin-1-yl-ethanolhydrochlorid
(1,5 g) wird zu einem Gemisch aus 3,5-Difluorphenol (1,0 g), Cäsiumcarbonat
(8,78 g), Natriumiodid (1,15 g) in trockenem Dimethylformamid (20
ml) gegeben. Es wird bei Raumtemperatur über Nacht gerührt. Es
wird unter verringertem Druck konzentriert und der Rückstand
wird durch Chromatographie unter Elution mit Ethylacetat : Hexan
(1 : 1) unter Bildung eines Öls
(0,8 g, 50%) gereinigt.
-
B. 2,6-Difluor-4-(pyrrolidin-1-yl-ethoxy)benzaldehyd
-
Ein
Gemisch aus 1-(3,5-Difluorphenoxymethyl)pyrrolidin (0,1 g) und trockenem
Tetrahydrofuran (5 ml) wird unter einer Stickstoffatmosphäre auf –78°C gekühlt. Butyllithium
(0,29 ml, 1,6 M in Tetrahydrofuran) und N,N,N',N'-Tetramethylethylendiamin
(0,5 ml) werden zugegeben und für
30 Minuten gerührt.
Dann wird Dimethylformamid (0,07 ml) zugegeben und bei Raumtemperatur
für 60
Minuten gerührt.
Es wird durch Gießen
des Reaktionsgemisches in ein Gemisch aus kaltem gesättigtem
wässrigem
Ammoniumchlorid und Ethylacetat gestoppt. Die Phasen werden getrennt,
die organische Phase wird mit Wasser gewaschen und dann unter verringertem
Druck unter Bildung der Titelverbindung als Öl (0,10 g, 94%) konzentriert.
-
Präparation 92
-
2-Fluor-4-nitrobenzaldehyd
-
Ein
Bortetrahydrofurankomplex (16,21 ml, 1 M) wird zu einem Gemisch
aus 2-Fluor-4-nitrobenzoesäure (1,2
g) und Tetrahydrofuran (10 ml) bei 0°C gegeben. Es wird für 3 Stunden
auf 80°C
erhitzt. Dann wird auf Raumtemperatur gekühlt und 1 N wässrige Chlorwasserstoffsäure (20
ml) wird zugegeben und es wird mit Ethylacetat extrahiert. Es wird
mit gesättigtem
wässrigem
Natriumbicarbonat gewaschen und die verbleibende organische Phase
wird über
Natriumsulfat getrocknet und unter verringertem Druck unter Bildung
eines Öls (0,92
g, 83%) konzentriert. (2-Fluor-4-nitrophenyl)methanol (0,91 g) wird
zu einem Gemisch aus Mangandioxid (1,1 g) in 20 ml Dichlormethan
gegeben. Es wird bei Raumtemperatur über Nacht gerührt und über Celite® filtriert.
Es wird unter verringertem Druck unter Bildung der Titelverbindung
als Öl
(0,27 g, 30%) konzentriert.
-
Präparation 93
-
4-Formyl-N-methylbenzolsulfonamid
-
A. 4-Hydroxymethyl-N-methylbenzolsulfonamid
-
Methylamin
(4,5 ml, 2 M in Dichlormethan) wird zu einem Gemisch aus 4-Chlorsulfonylbenzoesäure (0,5
g) und Dichlormethan (20 ml) gegeben. Es wird bei Raumtemperatur über Nacht
gerührt.
Es wird durch Gießen
des Reaktionsgemisches in ein Gemisch aus gesättigtem wässrigem Ammoniumchlorid und
Ethylacetat gestoppt. Die Phasen werden getrennt, die organische
Phase wird mit Wasser gewaschen und die verbleibende Phase wird über Natriumsulfat
getrocknet und dann unter verringertem Druck unter Bildung eines Öls (0,40
g, 82%) konzentriert. Ein Borantetrahydrofurankomplex (7,44 ml,
1 M) wird zu einem Gemisch aus 4-Methylsulfamoylbenzoesäure (0,4
g) und Tetrahydrofuran (10 ml) bei 0°C gegeben. Es wird für 3 Stunden
auf 80°C
erhitzt. Dann wird bei Raumtemperatur gekühlt und 1 N wässrige Chlorwasserstoffsäure (20
ml) wird zugegeben und mit Ethylacetat extrahiert. Es wird mit gesättigtem
wässrigem
Natriumbicarbonat gewaschen und die verbleibende organische Phase
wird über
Natriumsulfat getrocknet und unter verringertem Druck unter Bildung
eines Öls
(0,28 g, 75%) konzentriert.
-
B. 4-Formyl-N-methylbenzolsulfonamid
-
4-Hydroxymethyl-N-methylbenzolsulfonamid
(0,28 g) wird zu einem Gemisch aus Mangandioxid (2,8 g) in 20 ml
Dichlormethan gegeben. Es wird bei Raumtemperatur über Nacht
gerührt
und über
Celite® filtriert. Es
wird unter verringertem Druck unter Bildung der Titelverbindung
als Öl
(0,11 g, 40%) konzentriert.
-
Präparation 94
-
2-Fluor-4-methylaminobezaldehyd
-
Diazomethan
(22,3 ml, 2 M in Ether) wird zu einem Gemisch aus 2-Fluor-4-nitrobenzoesäure (4,14
g) und Ether (50 ml) gegeben. Es wird bei Raumtemperatur über Nacht
gerührt.
Dann wird unter verringertem Druck unter Bildung eines Öls (4,08
g, 92%) konzentriert.
-
Das
Gemisch aus 2-Fluor-4-nitrobenzoesäuremethylester (0,85 g), Ammoniumformiat
(1,1 g) und Pd/C (10%, 0,32 g) in Ethanol (20 ml) wird bei 95°C für 6 Stunden
am Rückfluss
erhitzt. Es wird über
Celite® filtriert
und unter Bildung eines grauen Feststoffs (0,64 g, 89%) konzentriert.
-
Formaldehyd
(0,15 g) wird zu einem Gemisch aus 2-Fluor-4-aminobenzosäuremethylester
(0,62 g), Essigsäure
(2,1 ml) und 1,2-Dichlorethan (20 ml) gegeben. Es wird bei Raumtemperatur über Nacht
gerührt. Natriumtriacetoxyborhydrid
(1,15 g) wird zugegeben und bei Raumtemperatur für 2 Stunden gerührt. Dann wird
durch Gießen
des Reaktionsgemisches in ein Gemisch aus gesättigtem wässrigem Natriumbicarbonat und
Ethylacetat gestoppt. Die Phasen werden getrennt, die organische
Phase wird mit Wasser gewaschen und die verbleibende Phase wird über Natriumsulfat
getrocknet und dann unter verringertem Druck konzentriert. Der Rückstand
wird durch Chromatographie unter Elution mit Ethylacetat : Hexan
(1 : 4) unter Bildung von 2-Fluor-4-methylaminobenzoesäuremethylester
(0,10 g, 15%) als Öl
gereinigt.
-
Diisobutylaluminiumhydrid
(1,0 ml, 1 M in Toluol) wird tropfenweise zu einem Gemisch aus 2-Fluor-4-methylaminobenzolsäuremethylester
(0,09 g) und trockenem Toluol (7 ml) bei –78°C unter einer Stickstoffatmosphäre gegeben.
Das Gemisch wird für
2 Stunden bei Raumtemperatur gerührt.
Es wird durch Gießen
des Reaktionsgemisches in ein Gemisch aus gesättigtem wässrigem Natriumtartrat (20
ml) und Ethylacetat (20 ml) gestoppt. Die Phasen werden getrennt,
die organische Phase wird mit Wasser gewaschen und die verbleibende
Phase wird über
Natriumsulfat getrocknet und dann unter verringertem Druck unter
Bildung eines Öls
(0,07 g, 88%) konzentriert.
-
Tetrapropylammoniumperruthenat
(0,35 g) wird zu einem Gemisch aus (2-Fluor-4-methylaminophenyl)methanol
(0,18 g), 4-Methylmorpholin-N-oxid (0,38 g) und frisch aktivierten
pulverisierten Molekularsieben (0,30 g) in trockenem Dichlormethan
(10 ml) bei Raumtemperatur unter einer Stickstoffatmosphäre gegeben. Es
wird für
30 Minuten gerührt
und dann wird weiteres Dichlormethan zugegeben und durch ein Florisil-Celite® Kissen
filtriert. Eine Konzentration unter verringertem Druck ergibt die
Titelverbindung (0,25 g, 76%) als Öl.
-
Präparation 95
-
1-Isopropylsulfonyl-2-chlor-6-iodbenzimidazol
-
1-Isopropylsulfonyl-2-amino-6-iodbenzimidazol
(0,20 g, 0,55 mmol) wird in drei Portionen über 5 Minuten zu einer Suspension
aus Kupfer(II)chlorid (0,089 mg, 0,66 mmol) und tert-Butylnitrit
(0,085 g, 0,1 ml, 0,82 mmol) in 2 ml Acetonitril bei 65°C gegeben.
Nach 30 Minuten wird die entstehende grüne Lösung in Wasser gegossen und
die wässrige
Phase wird mit Ethylacetat (3 × 25
ml) extrahiert. Die vereinigten organischen Phasen werden nacheinander
mit Wasser (2 × 15
ml) und gesättigtem
wässrigem
Natriumchlorid (15 ml) gewaschen, über Natriumsulfat getrocknet,
filtriert und unter verringertem Druck unter Bildung der Titelverbindung (0,18
g, 85%) konzentriert.
-
Präparation 96
-
2-(tert-Butyldimethylsilyloxy)-1-[2-chlor-3-(isopropylsulfonyl)-3H-benzimidazol-5-yl]-2-phenylethanon
-
Unter
einer Stickstoffatmosphäre
wird tert-Butylnitrit (2,44 ml, 18,45 mmol, 1,5 Äquivalente) tropfenweise zu
einer gerührten
Suspension aus CuCl2 (2,20 g, 14,76 mmol)
in 43 ml CH3CN gegeben. Das entstehende
Gemisch wird bei 65°C
erhitzt und langsam wird 1-[2-Amino-3-(propan-2-sulfonyl)-3H-benzoimidazol-5-yl]-2-(tert-butyldimethylsilyloxy)-2-phenylethanon
(6 g, 12,3 mmol), 1 Äquivalent)
portionsweise über
einen Gesamtzeitraum von 10 Minuten zugegeben. Das Gemisch wird
für 30
Minuten gerührt,
auf Raumtemperatur gekühlt, über 200
ml Wasser gegossen und mit 200 ml EtOAc verdünnt. Die Phasen werden getrennt
und die organische wird mit weiterem EtOAc (dreimal) rückextrahiert.
Die vereinigten organischen Phasen werden über MgSO4 getrocknet,
filtriert und im Vakuum unter Bildung des rohen Produkts konzentriert,
das durch Chromatographie mittels CH2Cl2 als Eluent gereinigt wird. 42% Ausbeute.
MS (ES+): m/z = 507,1 (M+H)+.
-
Präparation 97
-
1-[2-Benzylamino-3-(propan-2-sulfonyl)-3H-benzimidazol-5-yl]-2-(tert-butyldimethylsilyloxy)-2-phenylethanon
-
Zu
einer gerührten
Lösung
des anfänglichen
2-(tert-Butyldimethylsilyloxy)-1-[2-chlor-3-(propan-2-sulfonyl)-3H-benzimidazol-5-yl]-2-phenylethanons
(588 mg, 1,16 mmol, 1 Äquivalent)
in 2 ml trockenem THF wird Benzylamin (0,4 ml, 3,48 mmol, 3 Äquivalente)
tropfenweise gegeben. Die entstehende gelbe Lösung wird bei 40°C für 30 Minuten
erhitzt, auf Raumtemperatur gekühlt
und Wasser und CH2Cl2 werden
zugegeben. Die Phasen werden getrennt und die organische wird mit
weiterem CH2Cl2 (dreimal)
extrahiert. Die vereinigten organischen Phasen werden über MgSO4 getrocknet, filtriert und im Vakuum unter
Bildung eines rohen Produkts konzentriert. Eine Reinigung durch
Chromatographie mittels CH2Cl2 Hexan
(3 : 1) ergibt die Titelverbindung (80% Ausbeute). MS (ES+): m/z = 578,2 (M+H)+.
-
Präparation 98
-
2-(tert-Butyldimethylsilyloxy)-1-[2-ethylamino-3-(propan-2-sulfonyl)-3H-benzoimidazol-5-yl]-2-phenylethanon
-
Die
Herstellung erfolgt wie in der vorherigen Präparation (Präparation
97) beschrieben, ausgehend von 2-(tert-Butyldimethylsilyloxy)-1-[2-chlor-3-(propan-2-sulfonyl)-3H-benzimidazol-5-yl]-2-phenylethanon (760
mg, 1,50 mmol) und Ethylamin (0,25 ml, 4,50 mmol, 3 Äquivalente)
in 2 ml THF. Ausbeute: 84%. MS (ES+): m/z
= 516,3 (M+H)+.
-
Präparation 99
-
1-Isopropylsulfonyl-2-amino-6-(2-ethoxycarbonyl)-1-hydroxy-2-phenylethyl)benzimidazol
-
Zu
einer gerührten
Lösung
aus Ethylphenylacetat (18,5 mmol, 5 Äquivalente) in trockenem THF
(20 ml) wird Lithiumhexamethyldisilazan (LHMDS) (0,5 M in THF, 22,2
mmol, 6 Äquivalente)
bei –78°C gegeben. Das
Gemisch wird unter Stickstoff für
30 Minuten gerührt
und dann zu einer Lösung
aus 1-Isopropylsulfonyl-2-amino-6-formylbenzimidazol
(Präparation
1,1 g, 3,7 mmol) in THF (60 ml) gegeben. Die Reaktion wird bei –78°C gerührt, gefolgt
von TLC. Dann werden gesättigtes
NH4Cl (20 ml) und Ethylacetat (100 ml) zugegeben.
Die Phasen werden getrennt und die wässrige Phase wird mit Ethylacetat
(3 × 50
ml) extrahiert. Die vereinigten organischen Phasen (Na2SO4) werden getrocknet und im Vakuum konzentriert.
Das rohe Produkt wird mit Hexan gewaschen und der verbleibende Feststoff
wird durch Dichlormethan unter Bildung der entsprechen Verbindung
(0,75 g, 62% Ausbeute) gewaschen.
-
Präparation 100
-
1-Isopropylsulfonyl-2-amino-6-(α-((1-hydroxy)-α-(phenyl)acetyl)benzimidazol
-
Zu
einer gerührten
Lösung
aus IBX (3,48 mmol) in DMSO (5 ml) wird 1-Isopropylsulfonyl-2-amino-6-(2-ethoxycarbonyl)-1-hydroxy-2-phenylethyl)benzimidazol
(2,32 mmol, 1,5 Äquivalente)
bei Raumtemperatur gegeben und das Gemisch wird über Nacht (16 Stunden) gerührt. Dann
wird eine gesättigte
Lösung
aus NaCl (50 ml) gefolgt von Ethylacetat (100 ml) zugegeben. Die
Phasen werden getrennt und der Extrakt der wässrigen Phase wird mit Ethylacetat
(3 × 50
ml) extrahiert. Die organische Phase wird mit Na2S2O3 (25 ml) gewaschen
und die vereinigten organischen Phasen (Na2SO4) werden getrocknet. Eine Chromatographie
mittels Hexan : Ethylacetat 1 : 4 ergibt die entsprechende Verbindung
(90% Ausbeute).
-
Präparation 101
-
N-[Isopropylsulfonyl]-4-methoxybenzylamin
-
4-Methoxybenzylamin
(11,7 ml, 89,5 mmol) wird zu einer Lösung aus Isopropylsulfonylchlorid
(5,0 ml, 44,74 mmol) in Dichlormethan (100 ml) gegeben und für 18 Stunden
bei Umgebungstemperatur gerührt.
Die Lösung
wird mit 1 N Chlorwasserstoffsäure
(3 × 100
ml) gewaschen, mit Magnesiumsulfat getrocknet und konzentriert.
Eine Reinigung auf Silicagel mittels Hexan/Ethylacetatgemischen
ergibt 4,73 g (43%) der Titelverbindung als weißen Feststoff. MS (ES–):
m/z = 242,1 (M–H)–.
-
Präparation 102
-
N-[Isopropylsulfonyl]-N-[5-fluor-2-nitrophenyl]-4-methoxybenzylamin
-
Natriumhydrid
(60% in Mineralöl,
9,85 g, 21,2 mmol) wird in Tetrahydrofuran (100 ml) suspendiert. 1-N-[Isopropylsulfonyl)]-4-methoxybenzylamin
(4,73 g, 19,45 mmol) wird in Tetrahydrofuran (400 ml) gegeben und
auf 70°C
erhitzt. Es wird für
10 Minuten gerührt,
dann wird 2,4-Difluornitrobenzol (3,2 ml, 29,2 mmol) in einer Portion
zugegeben und für
18 Stunden bei 70°C
gerührt.
Es wird auf Umgebungstemperatur gekühlt und die Lösemittel
werden unter verringertem Druck entfernt. Es wird in Ethylacetat
gelöst
und zweimal mit wässrigem
1 N HCl, dreimal mit gesättigtem
Natriumhydrogencarbonat, gesättigtem
Natriumcarbonat, 1 N wässrigem
HCl gewaschen, über
Magnesiumsulfat getrocknet, filtriert und die Lösemittel werden unter verringertem Druck
entfernt. Eine Reinigung auf Silicagel mit Dichlormethan/Hexan ergibt
5,395 g (72%) der Titelverbindung.
-
Präparation 103
-
N-[Isopropylsulfonyl]-N-[5-(2-phenylimidazol-1-yl)-2-nitrophenyl]-4-methoxybenzylamin
-
Es
werden N-[Isopropylsulfonyl]-N-[5-fluor-2-nitrophenyl-4-methoxybenzylamin
(0,36 g, 0,94 mmol) und 2-Phenylimidazol (0,20 g, 1,41 mmol) in
N,N-Dimethylformamid (4 ml) gelöst.
Natriumhydrid (60% in Mineralöl,
0,056 g, 1,41 mmol) wird zugegeben und für 30 Minuten auf 90°C erhitzt.
Es wird auf Umgebungstemperatur gekühlt und die Lösemittel
werden unter verringertem Druck entfernt. Eine Reinigung auf Silicagel
mit Dichlormethan/Ethylacetatgemischen ergibt 0,31 g (65%) der Titelverbindung.
-
Präparation 104
-
N-[Isopropylsulfonyl]-N-[5-(2-phenylimidazol-1-yl)-2-nitrophenyl]amin
-
Ein
Kolben der N-[Isopropylsulfonyl]-N-[5-(2-phenylimidazol-1-yl)-2-nitrophenyl]-4-methoxybenzylamin
(0,30 g, 0,58 mmol) enthält,
wird in einem Eisbad gekühlt
und eiskalte Trifluoressigsäure
(5 ml) wird zugegeben und dann auf Umgebungstemperatur erwärmt und
für 18
Stunden gerührt.
Das Lösemittel
wird unter verringertem Druck entfernt. Es wird in Dichlormethan
gelöst
und mit gesättigtem
Natriumhydrogencarbonat gewaschen, mit Magnesiumsulfat getrocknet,
filtriert und auf Silicagel mit Dichlormethan/Ethylacetatgemischen
unter Bildung von 0,21 g (92%) an 1-Isopropylsulfonyl-[2-nitro-5-(2-phenylimidazol-1-yl)phenyl]amid
gereinigt. MS (ES+): m/z = 387,1 (M+H)+.
-
Präparation 105
-
N-[Isopropylsulfonyl]-N-[2-amino-5-(2-phenylimidazol-1-yl)phenyl]amid
-
N-[Isopropylsulfonyl]-N-[5-(2-phenylimidazol-1-yl)-2-nitrophenyl]amin
(0,20 g, 0,52 mmol), 10% Palladium auf Kohle (0,02 g), Methanol
(10 ml) und Hydrogenat werden unter einem Ballon aus Wasserstoff
für 2 Stunden
zugegeben. Es wird filtriert und die Lösemittel werden unter verringertem
Druck entfernt. Eine Reinigung auf Silicagel mit Dichlormethan/Ethylacetatgemischen
ergibt 0,12 g (63%) der Titelverbindung. MS (ES+): m/z
= 357,1 (M+H)+.
-
Präparation 106
-
2-Aminobenzothiazol-6-carboxaldehyd
-
Unter
einer Stickstoffatmosphäre
wird langsam Phenyllithium (5 ml, 9,16 mmol, 2,1 Äquivalente
aus einer 1,8 M Lösung
in Cyclohexan/Ether, 70/30) zu einer kalten (–78°C) Lösung aus kommerziell erhältlichem 2-Brombenzothiazol
in 30 ml trockenem THF gegeben. Nachdem die Zugabe vollständig ist,
wird das Gemisch für
5 Minuten gerührt
und t-Butyllithium (5,6 ml, 9,16 mmol, 2,1 Äquivalente aus einer 1,7 M
Lösung
in Pentan) wird zugegeben. Das Gemisch verändert sich während einer
Stunde von einer cremeweißen
Farbe zu einer grünen
Farbe. An diesem Punkt wird schnell Formylpiperidin (2,4 ml, 21,8
mmol, 5 Äquivalente)
tropfenweise zugegeben und das Gemisch wird langsam auf 0°C (> 1 Stunde) erwärmt. Die
Reaktion wird in einem Gemisch aus kaltem gesättigem NH4Cl
gestoppt und mit EtOAc extrahiert. Die Phasen werden getrennt und
die organische wird mit weiterem EtOAc (dreimal) rückextrahiert.
Die vereinigten organischen Phasen werden über MgSO4 getrocknet,
filtriert und im Vakuum unter Bildung des Aldehyds (50% Ausbeute)
konzentriert.
-
Präparation 107
-
2,4-Dibromnitrobenzol
-
1,3-Dibrombenzol
(25 g) wird über
rauchender Schwefelsäure
(20 ml) zugegeben. Es wird für
5 Stunden gerührt
und dann in Eiswasser gegossen, mit Ethylacetat extrahiert, mit
gesättigtem
wässrigem
Natriumchlorid gewaschen, über
Magnesiumsulfat getrocknet und unter verringertem Druck konzentriert.
Der Rückstand
wird mit Methanol unter Bildung des gewünschten Produkts als hellgelber
Feststoff (26,8 g, 90%) kristallisiert. MS (ES+):
281,90 (M+H)+.
-
Präparation 108
-
(5-Brom-2-nitrophenyl)amin
-
In
ein verschlossenes Reaktionsröhrchen
werden vorsichtig 6 ml einer wässrigen
Lösung
aus Ammoniak (32%) zu einer Lösung
aus 2,4-Dibromnitrobenzol (1,6 g) in 2 ml DMSO gegeben. Es wird
für 18
Stunden bei 100°C
gerührt
und auf Raumtemperatur gekühlt.
Das heterogene Gemisch wird in Wasser gegossen und die Titelverbindung
wird als gelber Feststoff (1,1 g, 92%) filtriert.
-
Präparation 109
-
3-Amino-4-nitro-1-phenylethinylphenyl
-
Pd(OAc)2 (31 mg), Cul (43 mg) und PPh3 (48
mg) werden zu einem Gemisch aus 5-Brom-2-nitrophenylamin (0,5 g) in 6 ml Et3N gegeben. Stickstoff wird für 5 Minuten
durchgeblasen und Phenylacetylen (0,37 ml) wird zugegeben. Das Gemisch
wird bei 90°C
für 1 Stunde
gerührt
und auf Raumtemperatur gekühlt.
Triethylamin wird im Vakuum entfernt und der Rückstand wird zwischen gesättigtem
wässrigem
Natriumchlorid und EtOAc aufgeteilt. Die Phasen werden getrennt,
die organische wird über
MgSO4 getrocknet, filtriert und eingedampft.
Der Rückstand
wird durch Chromatographie unter Elution mit einem 8 : 1 Gemisch
aus Hexan : EtOAc unter Bildung der Titelverbindung (0,49 g, 90%)
als gelber Feststoff gereinigt.
-
Präparation 110
-
3-Isopropylsulfonamidyl-4-nitro-1-phenylethinylphenyl
-
Es
werden Isopropylsulfonylchlorid (0,93 ml) und 1,8-Diazabicyclo(5,4,0)undec-7-en
(DBU) (2,48 ml) zu einer Lösung
aus 3-Amino-4-nitro-1-phenylethinylphenyl (1 g) in 40 ml CH2Cl2 gegeben. Es
wird für
17 Stunden bei 45°C
gerührt
und auf Raumtemperatur gekühlt.
Dann wird mit weiterem CH2Cl2 verdünnt und
mit wässriger
10% HCl Lösung
und Wasser gewaschen. Die organische Phase wird über Magnesiumsulfat getrocknet und
unter verringertem Druck konzentriert. Der Rückstand wird durch Silicagelchromatographie
unter Elution mit 1 : 1 Hexan : CH2Cl2 unter Bildung der Titelverbindung (0,76
g, 53%) als gelber Feststoff gereinigt.
-
Präparation 111
-
3-Isopropylsulfonamidyl-4-nitro-1-(phenylethanon)phenyl
-
Es
werden 30 ml einer wässrigen
Lösung
aus HgO (624 mg) in 100 ml an 4% H2SO4 zu einer Lösung aus 3-Ispropylsulfonamidyl-4-nitro-1-phenylethinylphenyl
(1,48 g) in 40 ml Methanol gegeben. Es wird bei 100°C für 17 Stunden
gerührt
und auf Raumtemperatur gekühlt.
Das Gemisch wird mit einer gesättigten
wässrigen
Lösung
aus NaHCO3 neutralisiert und mit CH2Cl2 (zweimal) extrahiert.
Die Phasen werden getrennt, die organische Phase wird über Magnesiumsulfat
getrocknet und unter verringertem Druck konzentriert. Der Rückstand
wird durch Silicagelchromatographie unter Elution mit 1 : 2 Hexan
CH2Cl2 unter Bildung
der Titelverbindung (1,2 g, 79%) als gelber Feststoff gereinigt.
MS (ES–):
m/z = 361,3 (M–H)–.
-
Präparation 112
-
3-Isopropylsulfonamidyl-4-nitro-1-(5-phenyl)pyrazol-4-yl
-
Dimethylformamid
(DMF)-Dimethylacetal (0,9 ml) wird zu einer gerührten Lösung aus 3-Isopropylsulfonamidyl-4-nitro-1-(phenylethanon)phenyl
(0,5 g) in 1,5 ml trockenem DMF gegeben. Das Gemisch wird bei 80°C für 1 h und
45 min erhitzt, auf Raumtemperatur gekühlt und die Lösemittel
werden im Vakuum entfernt. Der Rückstand
wird in 8 ml EtOH gelöst
und 0,5 ml wasserfreies Hydrazin werden zugegeben. Es wird für 17 Stunden
gerührt
und unter verringertem Druck konzentriert. Eine Reinigung des Rückstands
durch Silicagelchromatographie unter Elution mit 5% CH2Cl2 : MeOH ergibt die Titelverbindung (0,5
g, 86%) als roten Feststoff. MS (ES+) m/z
= 387,4 (M+H)+.
-
Präparation 113
-
3-Isopropylsulfonamidyl-4-amino-1-(5-phenyl)pyrazol-4-yl-phenyl
-
Es
werden 10 Gewichtsprozent an Pd/C Katalysator (250 mg) zu einer
gerührten
Suspension aus Nitroanilin (0,5 g) in 10 ml EtOH gegeben. Die Suspension
wird mit Wasserstoff für
5 Minuten gewaschen und das Gemisch wird unter einer Wasserstoffatmosphäre (1 atm)
für 3 Stunden
gerührt.
Es wird unter verringertem Druck konzentriert und der Rückstand
wird unter Elution mit 5% CH2Cl2 :
MeOH unter Bildung der Titelverbindung (0,3 g, 70%) als Feststoff
einer Silicagelchromatographie unterzogen. MS (ES+):
m/z = 357,1 (M+H)+.
-
Präparation 114
-
3-Isopropylsulfonamidyl-4-nitro-1-(5-phenyl)-1,2,3-triazol-4-yl
-
Natriumazid
(0,033 g) wird zu einer Lösung
aus 2-Isopropylsulfonamidyl-4-nitro-1-phenylethinyl (0,173 g) in
5 ml Dimethoxyethan gegeben. Es wird am Rückfluss (80°C) für 2 Stunden erhitzt und dann
auf Raumtemperatur gekühlt.
Es werden 10 ml an 1 N Chlorwasserstoffsäure zugegeben und mit Ethylacetat
(20 ml) extrahiert und mit gesättigtem
wässrigem
Natriumchlorid (2 × 10
ml) gewaschen. Die verbleibende organische Phase wird über Natriumsulfat
getrocknet und unter verringertem Druck konzentriert. Der Rückstand
wird durch Chromatographie unter Elution mit Ethylacetat : Hexan
(1 : 1) unter Bildung der Titelverbindung als gelber Feststoff (0,10
g, 50%) gereinigt. MS (ES+): m/z = 388,42
M+H)+.
-
Präparation 115
-
3-Isopropylsulfonamidyl-4-amino-1-(5-phenyl)-1,2,3-triazol-4-yl
-
Zinn(II)chloriddihydrat
(0,350 g) wird zu einem Gemisch aus 3-Isopropylsulfonamidyl-4-nitro-1-(5-phenyl)-1,2,3-triazol-4-yl
(0,10 g), Ethanol (5 ml) und Ethylacetat (10 ml) gegeben. Das Gemisch
wird für
3 Stunden bei 70°C
erhitzt. Das Gemisch wird in Eiswasser gegossen. Gesättigtes
wässriges
Natriumbicarbonat wird zugegeben und mit Ethylacetat extrahiert.
Die Phasen werden getrennt, über
Natriumsulfat getrocknet und unter verringertem Druck konzentriert.
Der Rückstand
wird unter Elution mit 1 : 1 Ethylacetat : Hexan unter Bildung der
Titelverbindung als weißer
Feststoff (0,084 g, 92%) einer Silicagelchromatographie unterzogen.
MS (ES+): m/z = 358,4 (M+H)+.
-
Präparation 116
-
1-Dimethylaminosulfonyl-2-aminobenzimidazol
-
Zu
einer Lösung
aus Natriumhydroxid (1,8 g) in Wasser (9 ml) wird Acetonitril (44
ml) gegeben. Zu dieser Lösung
werden 2-Aminobenzimidazol (3,0 g, 22,5 mmol) und Dimethylsulfamoylchlorid
(2,4 ml, 22,5 mmol) gegeben. Das Reaktionsgemisch wird bei Raumtemperatur über Nacht
gerührt.
Dann wird das Gemisch bei 0°C
gekühlt
und das Produkt wird aus der Lösung
kristallisiert. Das Produkt wird filtriert und im Vakuum unter Bildung
von 5,0 g (90% Ausbeute) der Titelverbindung als weißer Feststoff
getrocknet. MS (ES+): m/z = 241,1 (M+H)+.
-
Präparation 117
-
1-Dimethylaminosulfonyl-2-amino-6-iodbenzimidazol
-
Dimethylaminosulfonyl-2-aminobenzimidazol
(1,0 g, 4,2 mmol) wird zu Essigsäure
(10 ml) unter Bildung einer Lösung
gegeben. Zu dieser Lösung
wird N-Iodsuccinimid (0,945 g, 4,2 mmol) gegeben und das Reaktionsgemisch
wird bei 55°C über Nacht
erhitzt. Das Gemisch wird bei 0°C
gekühlt
und Wasser wird zugegeben. Das Produkt wird aus der Lösung kristallisiert.
Der Feststoff wird filtriert und im Vakuum unter Bildung von 1,5
g (99%) der Titelverbindung getrocknet. MS (ES+):
m/z = 366,9 (M+H)+.
-
Präparation 118
-
1-Dimethylaminosulfonyl-2-amino-6-(phenylethinyl)benzimidazol
-
Ausgehend
von 1-Dimethylaminosulfonyl-2-amino-6-iodbenzimidazol wird die Titelverbindung
im wesentlichen wie in Präparation
52 beschrieben hergestellt (88% Ausbeute). MS (ES+):
m/z = 341,0 (M+H)+.
-
Präparation 119
-
1-Dimethylaminosulfonyl-2-amino-6-(2-phenylethan-1,2-dionel)benzimidazol
-
Ausgehend
von 1-Dimethylaminosulfonyl-2-amino-6-(phenylethinyl)benzimidazol
wird die Titelverbindung im wesentlichen wie in Präparation
54 beschrieben hergestellt (72% Ausbeute). MS (ES+):
m/z = 373,0 (M+H)+.
-
Beispiel 1
-
1-Isopropylsulfonyl-2-amino-6-(2-(thien-2-yl)-5-(phenyl)imidazol-4-yl)benzimidazolmethansulfonat
-
Thiophen-2-carboxaldehyd
(3,16 ml, 33,8 mmol) wird zu einem gerührten Gemisch aus 1-Isopropylsulfonyl-2-amino-6-(α-((tert-butyldimethylsilyl)oxy)-α-(phenyl)acetyl)benzimidazol
(15,0 g, 30,8 mmol), Kupfer(II)acetat (11,2 g, 61,5 mmol) und Ammoniumacetat
(23,7 g, 308 mmol) in Essigsäure
(300 ml) gegeben. Das Gemisch wird bei 95-100°C erhitzt und für 2 Stunden
kräftig
gerührt.
Das Gemisch wird auf 15°C
gekühlt, in
ein Gemisch aus 750 ml gesättigtem
wässrigem
Ammoniumchlorid und 250 ml konzentriertem Ammoniumhydroxid gegossen.
Das Gemisch wird mit Ammoniumhydroxid, das auf 5°C vorgekühlt ist, auf pH 10 eingestellt
und dann werden 4 l an 4 : 1 Ethylacetat : Methanol zugegeben. Die
Phasen werden getrennt und die organische Phase wird mit gesättigtem
wässrigem
Ammoniumchlorid (500 ml) gewaschen, über Magnesiumsulfat getrocknet
und unter verringertem Druck konzentriert. Der Rückstand wird unter Elution
mit Dichlormethan, das 15-50% Acetonitril und 0,5% Triethylamin
enthält,
einer Silicagelchromatographie unterzogen. Das gewonnene Material
wird in Ethanol (75 ml) suspendiert, auf 55-60°C für 30 Minuten erwärmt, für 30 Minuten auf –10°C gekühlt, filtriert
und nacheinander mit kaltem Ethanol gefolgt von Diethylether gewaschen.
Es wird unter verringertem Druck unter Bildung der Titelverbindung
mit 40% Ausbeute als dunkelgelbes Pulver getrocknet. MS (ES+): m/z = 464,1 (M+H)+.
-
Zu
einer homogenen Lösung
aus 1-Isopropylsulfonyl-2-amino-6-(2-(thien-2-yl)-5-(phenyl)imidazol-4-yl)benzimidazol
(0,4 g, 0,86 mmol) in 10% Methanol/Dichlormethan (10 ml) wird eine
1 N Lösung
der Methansulfonsäure
in demselben Reaktionsgemisch (10% Methanol/Dichlormethan) (0,86
ml, 1 Äquivalent)
gegeben. Das Gemisch wird bei Raumtemperatur für 2 Stunden gerührt, die
Lösemittel
werden unter verringertem Druck entfernt und der Rückstand
wird mit Diethylether (80 ml) für
3 Stunden gerührt.
Der Feststoff wird filtriert und unter Vakuum unter Bildung eines
blasscremefarbenen Feststoffs getrocknet (97% Ausbeute). MS (ES+): m/z = 464,1 (M+H)+.
-
Die
Verbindungen der Beispiele 2-62 können im wesentlichen wie in
Beispiel 1 beschrieben als entweder freie Base oder Methansulfonatsalz
hergestellt werden. Das Dimethansulfonatsalz kann auch mittels im wesentlichen
demselben Verfahren, wie in Beispiel 1 beschrieben, mit 2,0 Äquivalenten
Methansulfonsäure, hergestellt
werden.
-
-
-
-
-
Beispiel 63
-
1-Isopropylsulfonyl-2-amino-6-(2-(2-fluor-4-aminophenyl)-5-(phenyl)imidazol-4-yl)benzimidazol
-
Ein
Gemisch aus 1-Isopropylsulfonyl-2-amino-6-(2-(2-fluor-4-nitrophenyl)-5-(phenyl)imidazol-4-yl)benzimidazol (0,59
g), Ammoniumformiat (0,29 g) und Pd/C 10%, 0,12 g) in Ethanol (20
ml) wird bei 95°C
für 6 Stunden
am Rückfluss
erhitzt. Es wird über
Celite® filtriert
und das Filtrat wird unter verringertem Druck unter Bildung der
Titelverbindung (0,41 g, 75%) konzentriert. MS (ES+):
m/z = 491,2 (M+H)+.
-
Beispiel 64
-
1-Isopropylsulfonyl-2-amino-6-(2-((4-ethylamino)phenyl)-5-(phenyl)imidazol-4-yl)benzimidazol
-
Zu
einer Lösung
aus 1-Isopropylsulfonyl-2-amino-6-(α-((tert-butyldimethylsilyl)oxy)-α-(phenyl)acetyl)benzimidazol
(1,16 g, 2,37 mmol) in 20 ml Eisessig werden Kupfer(II)acetat (861
mg, 4,74 mmol), Ammoniumacetat (2,92 g, 28,44 mmol) und 4-(N-tert-Butoxycarbonyl-N-ethylamino)benzaldehyd
(630 mg, 2,84 mmol) gegeben. Das Reaktionsgemisch wird bei 80°C für 3 Stunden
gerührt.
Das Lösemittel
wird konzentriert und der Rückstand
wird in Ethylacetat/Methanol (20%) gelöst, in 3 : 1 gesättigte wässrige Ammoniumchloridlösung/Ammoniak
30% gegossen, in 3 : 1 gesättigter
wässriger
Ammoniumchloridlösung/Ammoniak
30% gegossen und mit Ethylacetat extrahiert, die organischen Phasen
werden mit gesättigtem
Ammoniumchlorid, gesättigtem
wässrigem
Natriumchlorid gewaschen, über
Natriumsulfat getrocknet und konzentriert. Es werden 1,43 g des
Rückstands
in 40 ml Dichlormethan gelöst
und 1,82 ml Trifluoressigsäure
(TFA) werden zugegeben. Das Gemisch wird bei Raumtemperatur unter
Argonatmosphäre
für 3 Stunden
gerührt.
Dann wird das Lösemittel
konzentriert, der Rückstand
wird in Ethylacetat/Methanol (20%) gelöst und mit gesättigtem
Natriumbicarbonat und gesättigtem
wässrigem
Natriumchlorid gewaschen, über
Natriumsulfat getrocknet und konzentriert. Der erhaltene Feststoff
wird durch Chromatographie unter Elution mit Dichlormethan/Methanol
(2%) unter Bildung von 273 mg der Titelverbindung gereinigt. Ausbeute:
27%. MS (ES+): m/z = 501,2 (M+H)+.
-
Beispiel 65
-
1-Isopropylsulfonyl-2-amino-6-(2-(4-(2-(piperidin-1-yl)ethoxy)phenyl)-5-(phenyl)imidazol-4-yl)benzimidazoltrifluoracetat
-
Zu
einer Lösung
aus 4-(2-Piperidin-1-yl)ethoxybenzaldehyd (0,42 g, 1,8 mmol) in
Essigsäure
(18 ml) (G. Kaliappa, Tetrahedron Letter, 34(35), 5631-5634, (1993))
wird 1-(Isopropylsulfonyl)-2-amino-6-((α-((tert-butyldimethylsilyl)oxy)-α-(phenyl)acetyl)benzimidazol
(0,88 g, 1,8 mmol), Ammoniumacetat (1,39 g, 18 mmol) Kupfer(II)acetat
(0,65 g, 3,6 mmol) unter Stickstoff gegeben. Das entstehende Reaktionsgemisch
wird bei 100°C
erhitzt und für
2 Stunden gerührt.
Es wird auf Raumtemperatur gekühlt
und dann wird die Essigsäure
konzentriert. Der Rückstand
wird in Methanol gelöst,
die Methanollösung
wird durch eine SCX Säule
gegeben, mit Methanol und dann 2 N Ammoniak in Methanol gewaschen.
Das Lösemittel
wird konzentriert. Der Rückstand
wird durch HPLC unter Bildung der Titelverbindung als Trifluoressigsäuresalz
(0,17 g, 23%) gereinigt. MS (ES+) m/z =
585,3 (M+H)+.
-
Die
Verbindungen der Beispiele 66-75 werden im wesentlichen wie in Beispiel
65 beschrieben, hergestellt.
-
-
Beispiel 76
-
1-Isopropylsulfonyl-2-amino-6-(2-(2,6-difluorphenyl)-5-(phenyl)oxazol-4-yl)benzimidazol
-
Zu
einer Lösung
aus 1-Isopropylsulfonyl-2-amino-6-(α-((tert-butyldimethylsilyl)oxy)-α-(phenyl)acetyl)benzimidazol
(5,84 g, 12,0 mmol) in Essigsäure
(Eisessig, 100 ml) werden Ammoniumacetat (9,26 g, 120 mmol) und
Kupfer(I)acetat (4,31 g, 23,7 mmol), gefolgt von 2,6-Difluorbenzaldehyd
(1,41 ml, 13,1 mmol) gegeben und das Gemisch wird bei 100°C für 4 Stunden
gerührt.
Das Gemisch wird auf 0°C
gekühlt
und langsam über
ein Gemisch aus NH4Cl/NH4OH
3 : 1, pH = 9 gegossen. Das Gemisch wird mit H2O
(200 ml) verdünnt, mit
EtOAc (3 × 200
ml) extrahiert, mit gesättigtem
wässrigem
NaCl (150 ml) gewaschen, getrocknet (Na2SO4) und im Vakuum konzentriert. Der Rückstand
wird in einem Biotagesystem (Eluent: CH2Cl2/MeCN 5 : 1) und dann durch HPLC unter Bildung
eines gelben Feststoffs, 675 mg, 11% Ausbeute, gereinigt. MS (ES+): m/z = 495,0 (M+H)+.
-
Beispiel 77
-
1-Isopropylsulfonyl-2-amino-6-(2-(2,6-difluorphenyl)-5-(phenyl)imidazol-4-yl)benzimidazol
und 1- Isopropylsulfonyl-2-amino-6-(2-(2,6-difluorphenyl)-5-(phenyl)imidazol-4-yl)benzimidazolethanolat
-
Unter
einer Stickstoffatmosphäre
werden 1-Isopropylsulfonyl-2-amino-6-(diketo(2-phenyl)ethyl)benzimidazol
(225 g, 0,606 mol), NH4OAc (700,4 g, 9,08
mol), Butanol (4,5 l) und 2,6-Difluorbenzaldehyd (172,2 g, 1,212
mol) vereinigt. Das entstehende Gemisch wird unter Bildung einer
gelben Aufschlämmung
auf 55°C
erhitzt. Das Reaktionsgemisch wird auf 15°C gekühlt und Wasser (2,5 l) wird
zugegeben. Nach dem Mischen für 15
Minuten werden die Phasen getrennt und die organische Phase wird
mit Wasser (2,5 l) rückextrahiert.
Die organische Phase wird abgetrennt und unter Vakuum bei 55°C unter Bildung
von 513 g eines schwarzen Öls konzentriert.
Das Öl
wird in MeOH (505 ml) bei 5°C
gelöst
und dann wird Methyl-tert-butylether (MTBE) (2,25 l) über 1,5
Stunden zu dem Gemisch gegeben. Die entstehende Aufschlämmung wird
langsam auf 22°C
gekühlt
und über
Nacht gerührt.
Die hellbraunen Feststoffe werden filtriert und mit MTBE (750 ml)
gewaschen. Die Feststoffe werden für 1,5 Stunden unter Bildung
von 239 g des Feststoffs einem Hausvakuum unterzogen. Die Feststoffe
werden in EtOAc (3,85 l) bei 50°C
gelöst
und Silicagel (250 g) wird zu dem Gemisch gegeben. Nach 0,5 Stunden
wird das Gemisch auf 35°C
gekühlt,
im Vakuum über
Silicagel (250 g nass mit EtOAc) filtriert und mit EtOAc (6 l) durchgewaschen.
Das entstehende Gemisch wird unter Vakuum bei 50°C unter Bildung eines hellbraunen
Feststoffs konzentriert (233,6 g, 78%, HPLC > 99 Flächenprozent, 1H NMR zeigt daß EtOAc im Feststoff eingeschlossen
ist. DSC Beginn 106,65°C,
Maximum 116,79°C,
18,67 J/g).
-
Die
Ethanolatverbindung wird folgendermaßen hergestellt: Es werden
395 g der Verbindung in EtOH (2 l) bei 53°C gelöst und zur Trockne im Vakuum
destilliert (dieses Verfahren kann zur Entfernung des restlichen
EtOAc aus der Verbindung wiederholt werden und kann gemäß 1H NMR bestätigt werden). Die entstehenden
Feststoffe werden in EtOH (1,185 l) bei 50°C für wenige Minuten aufgeschlämmt und
langsam über Nacht
auf 22°C
gekühlt.
Die entstehende Aufschlämmung
wird auf –3
bis 0°C
gekühlt
und für
1 Stunde gehalten. Die entstehenden Kristalle werden filtriert,
mit EtOH (395 ml, 0°C)
gewaschen und unter Vakuum bei 50°C zu
einem konstanten Gewicht getrocknet. Das Ethanolsolvat (324 g, 84%)
wird als hellpinkfarbener Feststoff isoliert. Die HPLC zeigt dass
das Produkt eine Reinheit von 99,5 Flächenprozent aufweist. MS (ES+) m/z = 492,3 (M+H)+.
-
Beispiel 78
-
1-Isopropylsulfonyl-2-amino-6-(2-(2,6-difluorphenyl)-5-(phenyl)imidazol-4-yl)benzimidazol-1,5-naphthalindisulfonat
-
Es
werden 247 mg der freien Base ausgewogen und in 4 ml an 95% EtOH
suspendiert. Davon getrennt werden 181 mg an 1,5-Naphthalindisulfonsäuretetrahydrat
in 2 ml an 95% EtOH gelöst.
Die saure Lösung
wird zu der Suspension in 0,2 ml Anteilen über einen 5 Minutenintervall
unter Mischen zwischen den Zugaben gegeben. Die Lösung mit
den Kristallen angeimpft, die aus einem kürzlichen Lauf mit Salzselektionssieb
erhalten werden und innerhalb weniger Minuten zeigt sich ein Niederschlag.
Das Gefäß kann für 2 Stunden
unter gelegentlichem Schütteln
stehen bleiben. Der Niederschlag wird filtriert und unter Bildung
von 325 mg an nicht ganz weißen
Kristallen luftgetrocknet.
-
Impfkristalle
für das
Napadisylat werden auf folgende Weise hergestellt:
Eine Salzscreenmatrix
aus 24 Gegenionen und 4 Lösemitteln
wird mittels Library DesignTM Software,
die von Symyx Technologies, Inc. entwickelt wurde, aufgebaut. Die
Lösung
der freien Base und die Lösungen
der Gegenionen werden mittels einer CavroTM Flüssigkeitsvorrichtung
zugegeben. Die Flüssigkeiten
werden dann mittels einer GenvacTM Vakuumzentrifuge
entfernt. Die Feststoffe werden dann auf eine Symyx automatisierte Kristallisationsausstattung
gegeben, die: 1) Lösemittel
in die Vertiefungen gibt, 2) erhitzt, 3) filtriert und 4) das Filtrat
zugibt, um die Kristallisationsstationen zu trennen (Eindampfung,
Ausfällung
und Kühlen).
Bei diesen getrennten Verfahrensschritten zeigt sich, dass kleine
Mengen (< 2 mg)
des kristallinen Materials an Napadisylatsalz auf den Glasplatten
vorhanden sind. Die Lösemittelsysteme,
die das kristalline Material ergeben, bestehen aus 95% Ethanol und
Ethylacetat.
-
Beispiel 79
-
1-Cyclopentylsulfonyl-2-amino-6-(2-(2,6-difluorphenyl)-5-(phenyl)imidazol-4-yl)benzimidazol
-
Ausgehend
von 1-Cyclopentylsulfonyl-2-amino-6-(diketo-(2-phenylethyl))benzimidazol
(1,29 g) werden im wesentlichen wie in Beispiel 77 beschrieben,
0,46 g (28% Ausbeute) als gelber Feststoff erhalten. MS (APC+), m/z = 520,2 (M+H)+.
Das Dimethansulfonat wird aus 0,23 g der freien Base unter Bildung
von 0,25 g (78% Ausbeute) als weißer Feststoff erhalten. MS
(Ionenfalle/ES+): m/z = 520,1 (M+H)+.
-
Die
Verbindungen der Beispiele 80-84 werden im wesentlichen wie in Beispiel
77 beschrieben als freie Base hergestellt. Das Methansulfonat oder
Dimethansulfonatsalz kann auch unter Verwendung derselben Verfahren
von Beispiel 1 und 1,0 oder 2,0 Äquivalenten
der Methansulfonsäure
hergestellt werden.
-
-
-
Beispiel 85
-
1-Isopropylsulfonyl-2-amino-6-(2-(isopropyl)-5-(4-fluorphenyl)imidazol-4-yl)benzimidazolmethansulfonat
-
Isobutyraldehyd
(0,25 ml, 3,3 mmol) wird zu einer gerührten Lösung aus 1-(6-(1-Isopropylsulfonyl)-2-aminobenzimidazol)-2-(4-fluor)phenylethan-1,2-dion
(0,5 g, 1,28 mmol) und Ammoniumacetat (1,48 g, 19,2 mmol) in n-BuOH
(10 ml) gegeben. Das Gemisch wird bei 80-90°C erhitzt und für 2 Stunden
in einem verschlossenen Röhrchen
kräftig
gerührt.
Das Gemisch wird auf Raumtemperatur gekühlt und unter verringertem
Druck konzentriert. Ethylacetat wird zugegeben und mit Wasser gewaschen.
Die Phasen werden getrennt und die organische Phase wird mit gesättigtem
wässrigem
Natriumchlorid (50 ml) gewaschen, über Magnesiumsulfat getrocknet
und unter verringertem Druck konzentriert. Der Rückstand wird unter Elution
mit 40 : 1 bis 20 : 1 Dichlormethan : Methanol unter Bildung von
1-Isopropylsulfonyl-2-amino-6-(2-(isopropyl)-5-(4-fluorphenyl)imidazol-4-yl)benzimidazol
einer Silicagelchromatographie (Biotage) unterzogen. MS (ES+): m/z = 442,1 (M+H)+.
-
Das
erhaltene 1-Isopropylsulfonyl-2-amino-6-(2-(isopropyl)-5-(4-fluorphenyl)imidazol-4-yl)benzimidazol
wird in Diethylether suspendiert, filtriert und mit kaltem Diethylether
gewaschen. Es wird unter verringertem Druck, unter Bildung der Titelverbindung
mit 59% Ausbeute als weißes
Pulver getrocknet. MS (ES+): m/z = 442,1
(M+H)+.
-
Methansulfonsäure (0,68
ml, 1 M in Dichlormethan : Methanol 95 : 5 frisch hergestellt) wird
sofort zu einer Lösung
aus 1-Isopropylsulfonyl-2-amino-6-(2-(isopropyl)-5-(4-fluorphenyl)imidazol-4-yl)benzimidazol (0,299
g, 0,68 mmol) in Dichlormethan : Methanol 10 ml, 95 : 5) gegeben.
Nach 5 Minuten wird das Lösemittel durch
Stickstoffdampf entfernt und unter Vakuum endgetrocknet. Das rohe
Material wird mit Diethylether (20 ml) über Nacht behandelt. Das Salz
wird durch Filtration gesammelt und mit zusätzlichem Diethylether (20 ml) unter
Bildung von 0,353 mg (97% Ausbeute) der Titelverbindung gewaschen.
MS (ES+): m/z = 442,1 (M+H)+.
-
Beispiel 86
-
1-Isopropylsulfonyl-2-amino-6-(2-(isopropyl)-5-(phenyl)imidazol-4-yl)benzimidazolmethansulfonat
-
Isobutyraldehyd
(0,73 ml, 8,1 mmol) wird zu einem gerührten Gemisch aus 1-Isopropylsulfonyl-2-amino-6-(diketo-(2-phenyl)ethyl))benzimidazol
(1,5 g, 4,04 mmol) gegeben und Ammoniumacetat (3 g, 40,4 mmol) in
Essigsäure
(20 ml) wird gegeben. Das Gemisch wird bei 80-90°C erhitzt und für 2 Stunden
in einem verschlossenen Röhrchen
kräftig
gerührt.
Das Gemisch wird auf Raumtemperatur gekühlt und unter verringertem Druck
konzentriert. Eis wird zugegeben und das Gemisch wird für 15 Minuten
kräftig
gerührt.
Ein vorgekühltes (Eisbad)
Gemisch 4/1 (V/V) aus gesättigtem
wässrigem
Ammoniumchlorid/konzentriertem Ammoniumhydroxid (50 ml) wird zugegeben.
Es werden 125 ml an 4 : 1 Ethylacetat : Methanol zugegeben. Die
Phasen werden getrennt und die organische Phase wird mit gesättigtem
wässri gem
Ammoniumchlorid (50 ml) gewaschen, über Magnesiumsulfat getrocknet
und unter verringertem Druck konzentriert. Der Rückstand wird unter Elution mit
25 : 1 Dichlormethan : Methanol unter Bildung von 1-Isopropylsulfonyl-2-amino-6-(2-(isopropyl)-5-(phenyl)imidazol-4-yl)benzimidazol
einer Silicagelchromatographie unterzogen. MS (ES+):
m/z = 424,1 (M+H)+.
-
Das
erhaltene 1-Isopropylsulfonyl-2-amino-6-(2-(isopropyl)-5-(phenyl)imidazol-4-yl)benzimidazol
wird in Diethylether suspendiert, filtriert und mit kaltem Diethylether
gewaschen. Es wird unter verringertem Druck unter Bildung der freien
Base mit 28% als weißes
Pulver getrocknet. MS (ES+): m/z = 424,1
(M+H)+.
-
Methansulfonsäure (0,2
ml, 1 M in CH2Cl2 :
MeOH 95 : 5 kurz vorher hergestellt) wird auf einmal zu einer Lösung aus
1-Isopropylsulfonyl-2-amino-6-(2-(isopropyl)-5-(phenyl)imidazol-4-yl)benzimidazol
(90 mg, 0,2 mmol) in CH2Cl2 :
MeOH 8 : 10 ml, 95 : 5) gegeben. Nach 5 Minuten wird das Lösemittel
durch Stickstoffdampf entfernt und unter Vakuum endgetrocknet. Das
rohe Material wird mit Diethylether (20 ml) über Nacht behandelt. Das Salz
wird durch Filtration gesammelt und mit zusätzlichem Diethylether unter
Bildung von 88 mg (81% Ausbeute) der Titelverbindung gewaschen.
-
Die
Verbindungen der Beispiele 87-93 werden im wesentlichen wie in den
Beispielen 85 und 86 beschrieben, hergestellt.
-
-
Beispiel 94
-
1-Isopropylsulfonyl-2-amino-6-(2-(formyl)-5-(phenyl)imidazol-4-yl)benzimidazol
-
Zu
einer Lösung
aus 1-Isopropylsulfonyl-2-amino-6-(1-(trimethylsilylethoxymethyl)-2-(formyl)-5-(phenyl)imidazol-4-yl)benzimidazol
(100 mg, 0,185 mmol) in Ethanol (4 ml) wird HCl (37%, 4 ml) gegeben
und bei Raumtemperatur für
4 Stunden gerührt.
Das Gemisch wird mit Wasser (20 ml) verdünnt und mit EtOAc (3 × 10 ml)
gewaschen. NaOH (15%) wird zu der wässrigen Phase gegeben, bis
der pH = 6,7 ist und das Gemisch wird mit EtOAc (3 × 20 ml)
extrahiert. Die vereinigten organischen Phasen werden getrocknet
(MgSO4) und im Vakuum konzentriert. Der
Rückstand
wird in einer Festphasenextraktionskartusche (SPE) (OasisTM) und dann mittels HPLC unter Bildung eines
weißen
Feststoffs, 8 mg, 10% Ausbeute, gereinigt. MS (ES+):
m/z = 410 (M+H)+.
-
Beispiel 95
-
1-Isopropylsulfonyl-2-amino-6-(2-(2-(piperidin-1-yl)methyl)-5-(phenyl)imidazol-4-yl)benzimidazol
-
A. Reduktive Aminierung
-
Zu
einer Lösung
aus 1-Isopropylsulfonyl-2-amino-6-(1-trimethylsilylethoxymethyl)-2-(formyl)-5-(phenyl)imidazol-4-yl)benzimidazol
(200 mg, 0,37 mmol) und Piperidin (0,037 ml, 0,37 mmol) in 1,2-Dichlorethan (5 ml)
wird Natriumtriacetoxyborhydrid (110 mg, 0,53 mmol) in einer Portion
gegeben und bei Raumtemperatur für
5 Stunden gerührt.
Dann wird Wasser (50 ml) zugegeben und mit EtOAc (3 × 50 ml)
extrahiert. Die vereinigten organischen Phasen werden mit gesättigtem
wässrigem
Natriumchlorid (50 ml) gewaschen, getrocknet (MgSO4)
und im Vakuum unter Bildung eines gelben Feststoffs, 1-Isopropylsulfonyl-2-amino-6-(1-trimethylsilylethoxymethyl)-2-((2-piperidin-1-yl)methyl)-5-(phenyl)imidazol-4-yl)benzimidazol,
251 mg, konzentriert. MS (ES+) m/z = 609,2
(M+H)+.
-
B. Schutzgruppenabspaltung
-
Zu
einer Lösung
aus 1-Isopropylsulfonyl-2-amino-6-(1-trimethylsilylethoxymethyl-2-((2-piperidin-1-yl)methyl)-5-(phenyl)imidazol-4-yl)benzimidazol
(0,37 mmol) in Acetonitril (0,1 M) wird Fluorwasserstoffsäure (HF)
(48%, 53,7 mmol) gegeben und bei 50°C für 3 Stunden gerührt. Dann
wird das Gemisch aus Raumtemperatur gekühlt und eine gesättigte wässrige Lösung aus
NaHCO3 wird zugegeben, bis der pH = 8 beträgt. Das
Gemisch wird mit EtOAc (3 × 20
ml) extrahiert und die vereinigten organischen Phasen werden getrocknet (MgSO4) und im Vakuum konzentriert. Der Rückstand
wird durch eine SCX Säule
gereinigt und dann durch HPLC unter Bildung eines gelben Feststoffs,
46 mg, 28% Ausbeute, gereinigt. MS (ES+):
m/z = 479,2 (M+H)+.
-
Die
Herstellung des Methansulfonatsalzes der Titelverbindung wird wie
vorher beschrieben, ausgeführt
(89% Ausbeute). MS (ES+): m/z = 479,2 (M+H)+.
-
Die
Verbindungen der Beispiele 96-98 können im wesentlichen wie in
Beispiel 95 beschrieben, hergestellt werden.
-
-
Beispiel 99
-
1-Isopropylsulfonyl-2-amino-6-(2-(N-[2-(piperidin-1-yl)eth-1-yl]aminomethyl)-5-(phenyl)imidazol-4-yl)benzimidazol
-
A. Reduktive Aminierung
-
Zu
einer Lösung
aus 1-Isopropylsulfonyl-2-amino-6-(1-(trimethylsilylethoxymethyl)-2-(formyl)-5-(phenyl)imidazol-4-yl)benzimidazol
(200 mg, 0,37 mmol) in Methanol (8 ml) werden 4 Å Molekularsiebe und 2-(N-Piperidinyl)ethylamin
(0,37 mmol, 1 Äquivalent)
gegeben und über
Nacht gerührt.
Dann wird Natriumborhydrid (70 mg, 1,85 mmol, 5 Äquivalente) in zwei Portionen
zugegeben und bei Raumtemperatur für 1 Stunde gerührt. Das
Gemisch wird mit Methanol (20 ml) verdünnt und durch ein Kissen aus
Celite® und
SiO2 filtriert. Die Filtrate werden im Vakuum
konzentriert und der Rückstand
wird durch eine SCX Säule
unter Bildung eines gelben Feststoffs mit 97% Ausbeute gereinigt.
MS (ES+): m/z = 652,3 (M+H)+.
-
B. Schutzgruppenabspaltung
-
Die
Ausführung
erfolgt gemäß dem Verfahren
für Beispiel
95. MS (ES+): m/z = 522 (M+H)+.
Die Verbindung in Beispiel 100 kann im wesentlichen wie in Beispiel
99 beschrieben, hergestellt werden.
-
-
Beispiele 101 und 102
-
1-Isopropylsulfonyl-2-amino-6-(2-(1-(ethoxycarbonyl)ethen-2-yl)-5-(phenyl)imidazol-4-yl)benzimidazol
und 1-Isopropylsulfonyl-6-(2-(2-carboxyethyl)-5-(phenyl)imidazol-4-yl)benzimidazol
-
A. Horner-Emmonds Kondensation
-
Zu
einer Lösung
aus Triethylphosphonoacetat (0,081 ml, 0,407 mmol) in trockenem
THF (0,8 ml) wird tropfenweise eine Lösung aus Kaliumbis(trimethylsilyl)amid
(KHMDS) (0,5 M in Toluol, 0,85 ml) bei –78°C unter Stickstoff gegeben.
Das Gemisch wird für
30 Minuten gerührt
und das Bad wird entfernt. Nach 5 Minuten wird das Gemisch auf –78°C während der
Zugabe einer Lösung
aus 1-Isopropylsulfonyl-2-amino-6-(1-(trimethylsilylethoxymethyl)-2-(formyl)-5-(phenyl)imidazol-4-yl)benzimidazol
(100 mg, 0,185 mmol) in trockenem THF (1 ml) gekühlt und langsam auf 0°C für 2 Stunden
erwärmt.
Dann wird eine gesättigte
wässrige
Lösung
aus NH4Cl (10 ml) zugegeben und das Gemisch
wird mit EtOAc (3 × 15
ml) extrahiert. Die vereinigten organischen Phasen werden getrocknet
(MgSO4) und im Vakuum konzentriert. Der
Rückstand
wird durch Blitzchromatographie (SiO2, Eluent:
Hexan/EtOAc 1 : 2) unter Bildung eines gelben Feststoffs, 97 mg,
86% Ausbeute, gereinigt. MS (ES+): m/z =
610 (M+H)+.
-
B. Reduktion
-
Zu
einer Lösung
aus 1-Isopropylsulfonyl-2-amino-6-(1-(trimethylsilylethoxymethyl)-2-(2-(ethoxycarbonyl)ethen-2-yl)-5-(phenyl)imidazol-4-yl)benzimidazol
(97 mg, 0,159 mmol) in MeOH (5 ml) wird Pd/C (20 Gewichtsprozent,
19,4 mg) gegeben. Es wird Wasserstoff bei Raumtemperatur für 5 Minuten
durch geblasen. Dann wird bei Raumtemperatur für 20 Stunden unter Wasserstoff
gerührt
und durch ein Kissen aus Celite® filtriert.
Die Filtrate werden im Vakuum unter Bildung eines gelben Feststoffs
konzentriert (91 mg, 93% Ausbeute). MS (ES+):
m/z = 612 (M+H)+.
-
C. Schutzgruppenabspaltung
-
Zu
einer Lösung
aus 1-Isopropylsulfonyl-2-amino-6-(1-trimethylsilylethoxymethyl)-2-(1-(ethoxycarbonyl)ethyl)-5-(phenyl)imidazol-4-yl)benzimidazol
(91 mg, 0,14 mmol) in Ethanol (3 ml) wird HCl (37%, 3 ml) gegeben
und bei Raumtemperatur für
24 Stunden gerührt.
Dann wird das Gemisch mit Wasser (10 ml) verdünnt und eine Lösung aus
NaOH (15%) wird zugegeben, bis der pH = 6–7 beträgt. Das Gemisch wird mit EtOAc
(3 × 30
ml) extrahiert, getrocknet (MgSO4) und die
vereinigten organischen Phasen werden im Vakuum konzentriert. Der
Rückstand
wird durch HPLC unter Bildung von 1-Isopropylsulfonyl-2-amino-6-(1-(trimethylsilylethoxymethyl)-2-(ethoxycarbonylethyl)-5-(phenyl)imidazol-4-yl)benzimidazol
als weißer
Feststoff, 11 mg, 16% Ausbeute, gereinigt. FIA MS (ES+):
m/z = 482 (M+H)+. 1-Isopropylsulfonyl-6-(2-(2-carboxyethyl)-5-(phenyl)imidazol-4-yl)benzimidazol
wird als weißer
Feststoff, 3 mg, 5% Ausbeute, ebenfalls erhalten. MS (ES+): m/z = 454 (M+H)+.
-
Die
Verbindungen in den Beispielen 103-108 können im wesentlichen wie in
Beispiel 102 beschrieben hergestellt werden.
-
-
Beispiel 109
-
1-Isopropylsulfonyl-2-amino-6-(1-(4-methoxybenzyl)-2-(hydroxymethyl)-4-(phenyl)imidazol-5-yl)benzimidazol
-
Zu
einer Lösung
aus 1-(4-Methoxybenzyl)-2-tert-butyldimethylsilyloxymethyl-4-(phenyl)-5-iodimidazol (50
mg, 0,0935 mmol) in trockenem Toluol (0,8 ml), die vorher mit einem
Stickstoffstrom gespült
wurde, wird PdCl2(PPh3)2 (4,5 mg, 0,00641 mmol) in einer Portion
gegeben. Nach 5 Minuten wird eine Suspension aus 1-Isopropylsulfonyl-2-aminobenzimidazol-6-borsäure (40
mg, 0,141 mmol) in Ethanol (0,8 ml), die vorher mit einem Stickstoffstrom
gespült
wurde, gefolgt von Natriumcarbonat (2 M in Wasser, 0,32 ml, 0,640
mmol) zugegeben. Das Gemisch wird für 2,5 Stunden bei 100°C gerührt, auf
Raumtemperatur gekühlt,
mit 10% HCl (5 ml) verdünnt,
mit EtOAc (15 ml) extrahiert, getrocknet (Na2SO4) und im Vakuum konzentriert. Der Rückstand wird
durch Blitzchromatographie (SiO2, CH2Cl2/MeOH 19 : 1 → 5 : 1)
gereinigt, das erhaltene Material wird in MeCN (0,5 ml) gelöst und mit
HF (48% in H2O, 0,12 ml, 3,31 mmol) bei
Raumtemperatur für
4 Stunden behandelt. Das Gemisch wird mit EtOAc (15 ml) verdünnt, mit
gesättigtem
NaHCO3 (5 ml) gewaschen, getrocknet (Na2SO4) und im Vakuum
konzentriert. Der Rückstand
wird durch Blitzchromatographie (SiO2, CH2Cl2/MeCN/MeOH 85
: 15 : 5) unter Bildung von 18 mg (36% Ausbeute) gereinigt. MS (ES+): m/z = 532 (M+H)+.
-
Beispiel 110
-
1-Isopropylsulfonyl-2-amino-6-(1-(1-(tert-butoxycarbonyl)piperidin-4-yl)-4-(phenyl)imidazol-5-yl)benzimidazol
-
tert-Butylamin
(0,125 g, 1,7 mmol) wird zu einer Lösung aus N-[1-(Ethoxycarbonyl)piperidin-4-yl](1-isopropylsulfonyl-2-amino-6-formylbenzimidazol)imin
(0,36 g, 0,86 mmol) und α-(p-Toluolsulfonyl)benzylisocyanid
(0,463 g, 1,7 mmol) in Methanol (10 ml) gegeben. Das Reaktionsgemisch
wird auf Rückfluss
erhitzt und über
Nacht gerührt.
Es wird auf Raumtemperatur gekühlt,
unter verringertem Druck konzentriert und der Rückstand wird zwischen Dichlormethan
(50 ml) und Wasser (50 ml) aufgeteilt. Die Phasen werden getrennt,
die organische Phase wird mit gesättigtem wässrigem Natriumchlorid (2 × 15 ml)
gewaschen, über
Natriumsulfat getrocknet und unter verringertem Druck konzentriert.
Der Rückstand
wird unter Elution mit 9 : 1 Ethylacetat : Hexan unter Bildung von
0,130 g (28%) der Titelverbindung einer Silicagelchromatographie unterzogen.
MS (ES+): m/z = 537,2 (M+H)+.
-
Die
Verbindungen der Beispiele 111-171 können im wesentlichen wie in
Beispiel 110 beschrieben, hergestellt werden.
-
-
-
-
-
Beispiel 172
-
1-Isopropylsulfonyl-2-amino-6-(1-(cyclopropyl)-4-(2-fluorphenyl))imidazol-5-yl)benzimidazolmethansulfonat
-
Methansulfonsäure (0,029
ml, 0,455 mmol) wird zu einer Lösung
aus 1-Isopropylsulfonyl-2-amino-6-(1-(cyclopropyl)-4-(2-fluorphenyl)imidazol-5-yl)benzimidazol
(0,200 g, 0,455 mmol), die gemäß dem Verfahren
von Beispiel 110 in 10% Methanol/Dichlormethan (3 ml) hergestellt
wurde, gegeben. Die Lösemittel
werden unter verringertem Druck zu 1-Isopropylsulfonyl-2-amino-6-(1-(cyclopropyl)-4-(2-fluorphenyl)imidazol-5-yl)benzimidazolmethansulfonat
(0,25 g) entfernt. MS (ES+): m/z = 440,1
(M+H)+.
-
Beispiel 173
-
1-Isopropylsulfonyl-2-amino-6-(5-(3-fluorphenyl)imidazol-4-yl)benzimidazol
-
1-Isopropylsulfonyl-2-amino-6-formylbenzimidazol
(0,3 g, 1,1 mmol) in THF (5 ml) wird mit Ammoniumhydroxid (0,39
ml, 3,4 mmol) behandelt und für
3 Stunden bei Raumtemperatur gerührt.
Die Reaktion wird mit 3-Fluorphenyltolylsulfonomethylisocyanid (0,23
g, 0,8 mmol) und Piperazin (0,096 g, 1,1 mmol) behandelt. Die Reaktion
wird für
5 Stunden gerührt
und dann in EtOAc verdünnt.
Die organische Phase wird mit Wasser und gesättigtem wässrigem Natriumchlorid gewaschen, über Natriumsulfat
getrocknet, filtriert und das Lösemittel
wird im Vakuum entfernt. Der Rückstand
wird in kaltem EtOAc aufgeschlämmt
und unter Bildung von 0,195 g (44%) des gewünschten Produkts filtriert.
MS (ES+) m/z = 400,2 (M+H)+.
-
Die
Verbindungen der Beispiele 174-176 können im wesentlichen wie in
Beispiel 173 beschrieben, hergestellt werden.
-
-
Beispiel 177
-
1-Isopropylsulfonyl-2-amino-6-(1-p-nitrophenyl-4-(phenyl)imidazol-5-yl)benzimidazol
-
Zu
dem Reaktionsröhrchen
werden p-Nitroanilin (0,138 g, 1 mmol), 1-Isopropylsulfonyl-2-amino-6-formylbenzimidazol
(Präparation
1, 0,267 g, 1 mmol), Essigsäure
(10 μl)
und Toluol (2 ml) gegeben. Das Reaktionsgefäß wird verschlossen und die
Suspension wird für
24 Stunden auf 120°C
erhitzt. Das rohe Imin wird mit CHCl3 gewaschen
und im Vakuum unter Bildung von 300 mg eines gelben Pulvers getrocknet.
MS (ES)+: m/z = 388,1 (M+H)+.
-
NaH
(65 mg, 60% Mineralöl)
wird in Dimethoxyethan (DME) (2 ml) bei Raumtemperatur suspendiert. α-(p-Toluolsulfonyl)benzylisocyanid
(Präparation
2, 0,135 g, 0,5 mmol) wird zu der Suspension gegeben, für 5 Minuten
gerührt
und das Imin (0,116 g, 0,3 mmol) wird zugegeben. Nach 2 Stunden
werden Wasser und EtOAc in die Reaktion gegeben und der Inhalt wird
für 1-2
Minuten schnell gerührt,
in einen Trenntrichter überführt, die
organische Phase wird mit Wasser und dann mit gesättigtem
wässrigem
Natriumchlorid gewaschen. Die organische Phase wird über Na2SO4 getrocknet,
filtriert und unter Bildung des gewünschten Produkts konzentriert.
Es wird durch Chromatographietrennung über einer kleinen Silicasäule mittels
3 : 2 MeCl2/MeCN + 10% MeOH unter Bildung
von 138 mg des gewünschten
Produkts gereinigt. MS (ES+): m/z = 503,3
(M+H)+.
-
Beispiel 178
-
1-Isopropylsulfonyl-2-amino-6-(1-(4-amino)-4-(phenyl)imidazol-5-yl)benzimidazol
-
Zu
0,025 g (0,05 mmol) an 1-Isopropylsulfonyl-2-amino-6-(1-p-nitrophenyl-4-(phenyl)imidazol-5-yl)benzimidazol in
EtOH (1 ml) bei Raumtemperatur wird SnCl2dihydrat
(50 mg, 0,22 mmol) gegeben. Nach 3 Stunden wird die rohe Reaktion
mit H2O (1 ml) behandelt, weitere 25 Minuten
gerührt
und dann wird Na2CO3 zugegeben,
um den pH auf etwa 10 zu bringen. Das Reaktionsgemisch wird in einen
Trenntrichter überführt und
mit EtOAc und Wasser verdünnt.
Die wässrige
Phase wird dreimal mit EtOAc rückextrahiert,
die vereinigten Phasen werden mit gesättigtem wässrigem Natriumchlorid gewaschen, über Na2SO4 getrocknet, filtriert
und im Vakuum unter Bildung des rohen Produkts konzentriert. Es
wird durch Chromatographie mittels präparativer TLC (Silica, 20 × 20 cm × 1 mm,
20 : 1 MeCl2/MeOH) unter Bildung von 6 mg
des gewünschten Produkts
gereinigt. MS (ES+): m/z = 473,2 (M+H)+.
-
Beispiel 179
-
1-Isopropylsulfonyl-2-amino-6-(2-(methyl)-4-(phenyl)thiazol-5-yl)benzimidazol
-
A. 2-Methyl-4-phenylthiazol
-
Thioacetamid
(0,09 g, 1,2 mmol) wird zu einer Lösung aus 2-Bromacetophenon
(0,2 g, 1,0 mmol) in 30 ml Dioxan gegeben. Diese wird bei Raumtemperatur
für 4 Stunden
gerührt,
mit Ethylacetat verdünnt
und nacheinander mit wässrigem
Natriumcarbonat (3 × 15
ml) und Wasser (3 × 20
ml) gewaschen. Die organische Phase wird über Natriumsulfat getrocknet,
unter verringertem Druck konzentriert und der Rückstand wird unter Elution
mit Hexan, das 10% Ethylacetat enthält unter Bildung der gewünschten
Verbindung mit 78% Ausbeute, einer Silicagelchromatographie unterzogen.
MS (ES+): m/z = 176,1 (M+H)+.
-
B. 2-Methyl-4-phenyl-5-(tributylstannyl)thiazol
-
n-Butyllithium
(0,46 ml, 0,74 mmol, 1,6 M in Tetrahydrofuran) wird zu einer Lösung aus
2-Methyl-4-phenylthiazol
(0,13 g, 0,74 mmol) in Tetrahydrofuran (7 ml), die auf –78°C gekühlt ist,
unter einer Stickstoffatmosphäre
gegeben. Es wird bei –78°c für 45 Minuten
gerührt,
Tributylzinnchlorid (0,2 ml, 0,74 mmol) wird zugegeben und es wird
für 2 Stunden
gerührt,
wobei sich das Reaktionsgemisch auf Raumtemperatur erwärmt. Wässriges
Ammoniumchlorid wird zugegeben und dann wird das Gemisch zwischen
Ethylacetat und Wasser aufgeteilt. Die wässrige Phase wird mit Ethylacetat
extrahiert. Die vereinigten organischen Phasen werden nacheinander
mit Wasser und gesättigtem
wässrigem
Natriumchlorid gewaschen, über
Natriumsulfat getrocknet und unter verringertem Druck konzentriert.
Der Rückstand
wird unter Elution mit Hexan, das 10% Ethylacetat enthält unter
Bildung der gewünschten
Verbindung mit 68% Ausbeute einer Silicagelchromatographie unterzogen. MS
(ES+): m/z = 466,1 (M+H)+.
-
C. Kupplung
-
Ein
Gemisch aus 2-Methyl-4-phenyl-5-(tributylstannyl)thiazol (0,1 g,
0,21 mmol), 1-Isopropylsulfonyl-2-amino-6-iodbenzimidazol (0,07
g, 0,21 mmol) und (Acetonitril)palladium(II)chlorid (0,021 mmol)
in trockenem Dimethylformamid (2 ml) wird bei 100°C für 4 Stunden
unter einer Stickstoffatmosphäre
gerührt.
Es wird auf Raumtemperatur gekühlt
und mit Ethylacetat (20 ml) verdünnt.
Dann wird nacheinander mit Wasser (3 × 5 ml) und gesättigtem
wässrigem
Natriumchlorid (3 × 5
ml) gewaschen, über
Natriumsul fat getrocknet und unter verringertem Druck konzentriert.
Der Rückstand
wird unter Elution mit 7 : 1 Dichlormethan : Acetonitril unter Bildung
der Titelverbindung als weißer
Feststoff mit 2% Ausbeute einer Silicagelchromatgraphie unterzogen. MS
(ES+): m/z = 413,0 (M+H)+.
-
Beispiel 180
-
1-Isopropylsulfonyl-2-amino-6-(2,4-diphenylthiazol-5-yl)benzimidazol
-
Ausgehend
von Thiobenzamid und 2-Bromacetophenenon wird die Titelverbindung
im wesentlichen wie in Beispiel 179 beschrieben, mit 12% Ausbeute
hergestellt. MS (ES+): m/z = 474,8 (M+H)+.
-
Beispiel 181
-
2-Amino-6-(2-(2,6-difluorphenyl)-5-(phenyl)imidazol-4-yl)benzimidazol
-
Es
wird ein Gemisch aus 1-Isopropylsulfonyl-2-amino-6-(2-(2,6-difluorphenyl)-5-(phenyl)imidazol-4-yl)benzimidazol
(0,07 g, 0,142 mmol) und 1,42 ml an 1 N Natriumhydroxid in 1 : 1
Wasser : Acetonitril bei 60°C
für 1 Stunde
gerührt.
Das Gemisch wird bei Raumtemperatur gekühlt und mit Wasser und Ethylacetat verdünnt. Die
wässrige
Phase wird mit Ethylacetat (dreimal) extrahiert. Die vereinigten
organischen Phasen werden nacheinander mit Wasser und gesättigem wässrigem
Natriumchlorid gewaschen, über
Natriumsulfat getrocknet und unter verringertem Druck konzentriert.
Der Rückstand
wird unter Elution mit 2 : 1 Dichlormethan : Methanol unter Bildung
der Titelverbindung mit 93% Ausbeute einer Silicagelchromatographie
unterzogen. MS (ES+): m/z = 388,0 (M+H)+.
-
Die
Verbindungen der Beispiele 182-183 werden im wesentlichen wie in
Beispiel 181 beschrieben, hergestellt.
-
-
Beispiel 184
-
1-Isopropylsulfonyl-2-amino-6-(2-(4-aminophenyl)-5-(phenyl)imidazol-4-yl)benzimidazol
-
Eine
Suspension aus 1-Isopropylsulfonyl-2-amino-6-(2-(4-nitrophenyl)-5-(phenyl)imidazol-4-yl)benzimidazol (0,149
g, 0,3 mmol) und 10% Palladium auf Kohle (0,03 mmol) in Methanol
wird mit Wasserstoff für
10 Minuten gespült
und dann unter einer Wasserstoffatmosphäre für 6 Stunden gerührt. Dann
wird durch ein Bett aus Celite® filtriert und der Rückstand
wird unter verringertem Druck konzentriert. Der Rückstand
wird unter Elution mit 4% Methanol in Dichlormethan unter Bildung
der Titelverbindung mit 28% Ausbeute einer Silicagelchromatographie
unterzogen. MS (ES+): m/z = 473,0 (M+H)+.
-
Beispiel 185
-
1-Isopropylsulfonyl-2-amino-6-(2-(2-aminothien-5-yl)-5-(phenyl)imidazol-4-yl)benzimidazol
-
Ausgehend
von 1-Isopropylsulfonyl-2-amino-6-(2-(2-nitrothien-5-yl)-5-(phenyl)imidazol-4-yl)benzimidazol wird
die Titelverbindung im wesentlichen wie in Beispiel 184 beschrieben,
mit 22% Ausbeute hergestellt. MS (ES+):
m/z = 479,0 (M+H)+.
-
Beispiele 186 und 187
-
1-Isopropylsulfonyl-2-amino-6-(1-(methyl)-2-(2,6-difluorphenyl)-5-(phenyl)imidazol-4-yl)benzimidazol
und 1-Isopropylsulfonyl-2-amino-6-(1-methyl)-2-(2,6-difluorphenyl)-4-(phenyl)imidazol-5-yl)benzimidazol
-
Cäsiumcarbonat
(0,134 mmol) und Methyliodid (0,134 mmol) werden zu einer Lösung aus
1-Isopropylsulfonyl-2-amino-6-(2-(2,6-difluorphenyl)-5-(phenyl)imidazol-4-ylbenzimidazol
(0,12 mmol) in trockenem Dimethylformamid (2,5 ml) bei 0°C gegeben.
Das Gemisch wird bei Raumtemperatur für 16 Stunden gerührt, mit Wasser
und Ethylacetat verdünnt
und die Phasen werden getrennt. Die wässrige Phase wird mit Ethylacetat (dreimal)
extrahiert. Die vereinigten organischen Phasen werden nacheinander
mit kaltem Wasser (fünfmal) und
gesättigtem
wässrigem
Natriumchlorid gewaschen, über
Natriumsulfat getrocknet und unter verringertem Druck konzentriert.
Der Rückstand
wird einer Silicagelchromatographie unterzogen. Das Isomergemisch
wird in Dichlormethan gelöst
und dieses Gemisch wird unter Elution mit Dichlormethan, das 4%
Methanol (100 ml/min) enthält
unter Bildung von 1-Isopropylsulfonyl-2-amino-6-(1-methyl)-2-(2,6-difluorphenyl)-5-(phenyl)imidazol-4-yl)benzimidazol
(11,1 Minuten) mit 22% Ausbeute, MS (ES+):
m/z = 508,2 (M+H)+ und 1-Isopropylsulfonyl-2-amino-6-(1-methyl)-2-(2,6-difluorphenyl)-4-(phenyl)imidazol-5-yl)benzimidazol
(16,1 min) mit 33% Ausbeute, MS (ES+): m/z
= 508,2 (M+H)+, einer präparativen HPLC (Kromasil Si60,
7 μm, 20 × 250 mm ID)
unterzogen.
-
Beispiele 188 und 189
-
1-Isopropylsulfonyl-2-amino-6-(1-methyl)-2-(thien-2-yl)-5-(phenyl)imidazol-4-yl)benzimidazol
und 1-Isopropylsulfonyl-2-amino-6-(1-(methyl)-2-(thien-2-yl)-4-(phenyl)imidazol-5-yl)benzimidazol
-
Ausgehend
von 1-Isopropylsulfonyl-2-amino-6-(2-(thien-2-yl)-5-(phenyl)imidazol-4-yl)benzimidazol und
1-Isopropylsulfonyl-2-amino-6-(1-methyl)-2-(thien-2-yl)-5-(phenyl)imidazol-4-yl)benzimidazol
und 1-Isopropylsulfonyl-2-amino-6-((1-methyl-2-(thien-2-yl)-4-phenyl)imidazol-5-yl)benzimidazol
wird die Titelverbindung im wesentlichen wie in Beispiel 187 beschrieben,
hergestellt. MS (ES+): m/z 478,2 (M+H)+.
-
Beispiel 190
-
1-Isopropylsulfonyl-2-amino-6-(2-(piperidin-4-yl)-5-(phenyl)imidazol-4-yl)benzimidazol
-
Ein
Gemisch aus 1-Isopropylsulfonyl-2-amino-6-(2-(1-(benzyloxycarbonyl)piperidin-4-yl)-5-(phenyl)imidazol-4-yl)benzimidazol
(0,74 g, 1,24 mmol), Ammoniumformiat (4,94 mmol) und 10% Palladium
auf Kohle (0,12 mmol) in 30 ml absolutem Ethanol wird für 5 Stunden
am Rückfluss
kräftig
gerührt.
Das Reaktionsgemisch wird auf Raumtemperatur gekühlt, durch ein Bett aus Celite® filtriert
und das Filtrat wird unter verringertem Druck konzentriert. Der
Rückstand
wird unter Elution mit 19 : 1 Dichlormethan : Methanol unter Bildung der
Titelverbindung einer Silicagelchromatographie unterzogen. MS (ES+): m/z = 465,2 (M+H)+.
-
Beispiel 191
-
1-Isopropylsulfonyl-2-amino-6-(2-(piperidin-4-yl)-5-(4-fluorphenyl)imidazol-4-yl)benzimidazolditrifluoracetat
-
Chlorwasserstoff
in Ethylacetat wird zu einer Lösung
aus 1-Isopropylsulfonyl-2-amino-6-((2-1-(tert-butoxycarbonyl)piperidin-4-yl)-5-(4-fluorphenyl)imidazol-4-yl)benzimidazol
(0,05 mmol) in Ethylacetat (2 ml) gegeben und das Gemisch wird bei
Raumtemperatur über
Nacht gerührt.
Die entstehende Suspension wird filtriert und mit Diethylether unter
Bildung der Titelverbindung mit 25% Ausbeute gewaschen. Der Feststoff
wird in 1 : 1 Dimethylsulfoxid : Acetonitril gelöst und unter Elution mit einem
Gradienten aus Wasser + 0,05% Trifluoressigsäure : Acetonitril + 0,05% Trifluoressigsäure aus
90 : 10 bis 45 : 55 in 15 Minuten (9 ml/min) unter Bildung der Titelverbindung
(25%, 9,33 min) einer HPLC (YMC C18, 5 mm, 20 × 50 mm ID) unterzogen. MS
(ES+): m/z = 483,2 (M+H)+.
-
Beispiel 192
-
1-Isopropylsulfonyl-2-amino-6-(1-(piperidin-4-yl)-4-(phenyl)imidazol-5-yl)benzimidazol
-
Ein
Gemisch aus 1-Isopropylsulfonyl-2-amino-6-(1-(1-ethoxycarbonyl)piperidin-4-yl)-4-(phenyl)imidazol-5-yl)benzimidazol
(55 mg) in konzentrierter Chlorwasserstoffsäure wird für 24 Stunden am Rückfluss
erhitzt. Das Gemisch wird bei Raumtemperatur gekühlt, 5 N Natriumhydroxid wird
zugegeben, bis das Gemisch basisch ist und es wird mit Dichlormethan
extrahiert. Die organische Phase wird unter verringertem Druck konzentriert
und der Rückstand
wird unter Elution mit Dichlormethan gefolgt von 1 : 1 Dichlormethan
: Methanol unter Bildung von 20 mg (43%) der Titelverbindung einer
Silicagelchromatographie unterzogen. MS (ES+):
m/z = 465,0 (M+H)+.
-
Beispiel 193
-
1-Isopropylsulfonyl-2-amino-6-(1-(piperidin-4-yl)-4-(4-fluorphenyl)imidazol-5-yl)benzimidazol
-
Ausgehend
von 1-Isopropylsulfonyl-2-amino-6-(1-(1-(ethoxycarbonyl)piperidin-4-yl)-4-(4-fluorphenyl)imidazol-5-yl)benzimidazol
wird die Titelverbindung im wesentlichen wie in Beispiel 192 beschrieben,
hergestellt. MS (ES+): m/z = 483,0 (M+H)+.
-
Beispiel 194
-
1-Isopropylsulfonyl-2-amino-6-(2-(1-ethyl)piperidin-4-yl)-5-(phenyl)imidazol-4-yl)benzimidazol
-
Ein
Gemisch aus 1-Isopropylsulfonyl-2-amino-6-(2-(piperidin-4-yl)-5-(phenyl)imidazol-4-yl)benzimidazol (0,574
g, 1,23 mmol), Triethylamin (3,1 mmol) und Iodethan (1,54 mmol)
in wasserfreiem Dimethylformamid (5 ml) wird bei Raumtemperatur
für 6 Stunden
gerührt.
Das Gemisch wird zwischen Ethylacetat und Wasser aufgeteilt. Die
wässrige
Phase wird mit Ethylacetat (dreimal) extrahiert. Die vereinigten
organischen Phasen werden mit kaltem Wasser (fünfmal) und gesättigtem
wässrigem
Natriumchlorid gewaschen, über
Natriumsulfat getrocknet und unter verringertem Druck konzentriert.
Der Rückstand
wird unter Bildung der Titelverbindung mit 76% Ausbeute einer Silicagelchromatographie
unterzogen. MS (ES+): m/z = 493,2 (M+H)+.
-
Die
Verbindungen der Beispiele 195-203 können im wesentlichen wie in
Beispiel 194 beschrieben, hergestellt werden.
-
-
Beispiel 204
-
1-Isopropylsulfonyl-2-amino-6-(1-(1-(methyl)piperidin-4-yl)-4-(phenyl)imidazol-5-yl)benzimidazol
-
Wässriges
Formaldehyd (37 Gewichtsprozent, 0,15 mmol) wird zu einer Lösung aus
1-Isopropylsulfonyl-2-amino-6-(1-(piperidin-4-yl)-4-(phenyl)mimidazol-5-yl)benzimidazol
(75 mg) in Methanol (10 ml) gegeben. Das Gemisch wird bei Raumtemperatur
für 30
Minuten gerührt
und dann auf 0°C
gekühlt.
Essigsäure
(0,3 ml) gefolgt von Natriumcyanoborhydrid (18 mg) wird zugegeben
und über
Nacht bei Raumtemperatur gerührt.
Das Gemisch wird unter verringertem Druck konzentriert und dann
wird der Rückstand
in Ethylacetat gelöst.
Die Lösung
wird zweimal mit 1 N Natriumhydroxid gewaschen und die organische
Phase wird unter verringertem Druck unter Bildung von 60 mg (79%)
der Titelverbindung konzentriert. MS (ES+):
m/z = 479,6 (M+H)+.
-
Die
Verbindungen der Beispiele 205-206 können im wesentlichen wie in
Beispiel 204 beschrieben, hergestellt werden.
-
-
Beispiel 207
-
1-Isopropylsulfonyl-2-amino-6-((1-(cyclohexan-1-on-4-yl)-4-(phenyl)imidazol-5-yl)benzimidazol
-
Eine
Lösung
aus 1-Isopropylsulfonyl-2-amino-6-(1-(1,4-dioxaspiro[4.5]dec-8-yl)-4-(phenyl)imidazol-5-yl)benzimidazol
(0,05 g, 0,1 mmol) in 3 N Chlorwasserstoffsäure (5 ml) wird für 3 Tage
bei Raumtemperatur gerührt.
5 N Natriumhydroxid wird zur Neutralisierung des Gemisches zugegeben
und mit Dichlormethan (3 × 10
ml) extrahiert. Die vereinigten organischen Phasen werden über Natriumsulfat
getrocknet und unter verringertem Druck unter Bildung von 0,038
g (83%) der Titelverbindung konzentriert. MS (ES+):
m/z = 478,2 (M+H)+.
-
Beispiel 208
-
1-Isopropylsulfonyl-2-amino-6-(1-(4-hydroxycyclohex-1-yl)-4-(phenyl)imidazol-5-yl)benzimidazol
-
Eine
Lösung
aus Natriumborhydrid (0,28 g) in Methanol (5 ml) wird zu einer Lösung aus
1-Isopropylsulfonyl-2-amino-6-(1-(cyclohexan-1-on-4-yl)-4-(phenyl)imidazol-5-yl)benzimidazol
(0,35 g, 0,7 mmol) in 10 ml Tetrahydrofuran und 10 ml Methanol gegeben.
Das Reaktionsgemisch wird für
30 Minuten bei Raumtemperatur gerührt, mit Wasser (50 ml) verdünnt und
mit Ethylacetat (2 × 15
ml) extrahiert. Die vereinigten organischen Extrakte werden mit
gesättigtem
wässrigem
Natriumchlorid gewaschen, über
Natriumsulfat getrocknet und unter verringertem Druck unter Bildung
von 0,345 g (98%) der Titelverbindung konzentriert. MS (ES+): m/z = 480,2 (M+H)+.
-
Beispiel 209
-
1-Isopropylsulfonyl-2-amino-6-(1-(4-aminocyclohex-1-yl)-4-(phenyl)imidazol-5-yl)benzimidazolhydrochlorid
-
Eine
Lösung
aus 1-Isopropylsulfonyl-2-amino-6-(1-(4-(N-[tert-butoxycarbonyl]amino)cyclohex-1-yl)-4-(phenyl)imidazol-5-yl)benzimidazol
(0,14 g, 0,2 mmol) in 1 M Chlorwasserstoff wird in Essigsäure (5 ml)
bei Raumtemperatur für
1 Stunde gerührt.
Die Suspension wird filtriert und der Feststoff wird unter Bildung
von 0,017 g (15%) der Titelverbindung getrocknet. MS (ES+): m/z = 479,0 (M+H)+.
-
Beispiel 210
-
1-Isopropylsulfonyl-2-aminobenzyl-6-(4-(phenyl)imidazol-5-yl)benzimidazol
-
Ausgehend
von 1-Isopropylsulfonyl-2-aminobenzyl-6-(α-((tert-butyldimethylsilyl)oxy)-α-(phenyl)acetyl)benzimidazol
wird die Titelverbindung im wesentlichen wie in Beispiel 1 beschrieben,
hergestellt. Ausbeute 12%. MS (ES+): m/z
= 472,1 (M+H)+.
-
Beispiel 211
-
2-Aminobenzyl-6-(4-(phenyl)imidazol-5-yl)benzimidazol
-
Ausgehend
von 1-Isopropylsulfonyl-2-aminobenzyl-6-(4-(phenyl)imidazol-5-yl)benzimidazol
wird die Titelverbindung im wesentlichen wie in Beispiel 181 beschrieben,
hergestellt. Ausbeute 91%. MS (ES+): m/z
= 366,1 (M+H)+.
-
Beispiele 212 und 213
-
1-Isopropylsulfonyl-2-aminoethyl-6-(2-(2,6-(difluor)phenyl)-5-(phenyl)imidazol-4-yl)benzimidazol
und 2-Aminoethyl-6-(2-(2,6-(difluor)phenyl)-5-(phenyl)imidazol-4-yl)benzimidazol
-
Ausgehend
von 1-Isopropylsulfonyl-2-aminoethyl-6-(α-((tert-butyldimethylsilyl)oxy)-α-(phenyl)acetyl))benzimidazol
wird die Titelverbindung im wesentlichen wie in Beispiel 1 beschrieben,
hergestellt. Ausbeute 42%. MS (ES+): m/z
= 522,1 (M+H)+. Gemeinsam mit dem gewünschten
Produkt wird das zu 12% desulfonylierte Produkt 2-Aminoethyl-6-(2-(2,6-(difluor)phenyl)-5-(phenyl)imidazol-4-yl)benzimidazol
isoliert. MS (ES+): m/z = 416,2 (M+H)+
-
Beispiel 214
-
1-Isopropylsulfonyl-6-(1-(4-(methoxy)benzyl)-4-(phenyl)imidazol-5-yl)benzimidazol
-
A. 1-Isopropylsulfonyl-2-chlor-6-(1-(4-methoxybenzyl)-4-(phenyl)imidazol-5-yl)benzimidazol
-
Ausgehend
von 1-Isopropylsulfonyl-2-amino-6-(1-(4-methoxybenzyl)-4-(phenyl)imidazol-5-yl)benzimidazol
wird die Titelverbindung im wesentlichen wie in Präparation
100 beschrieben, hergestellt. Ausbeute: 14%. MS (ES+):
m/z = 521,2 (M+H)+.
-
B. Reduktion
-
Zu
einer gerührten
Suspension des anfänglichen
2-Chlorbenzimidazolderivats (30 mg, 0,057 mmol, 1 Äquivalent)
in 3 ml MeOH wird Et3N (0,04 ml) tropfenweise
gefolgt von der Zugaber von 10 Gewichtsprozent Pd/C Katalysator
(100 Molprozent) gegeben. Die Suspension wird mit Wasserstoff für 5 Minuten
gespült.
Das Gemisch wird unter Wasserstoffatmosphäre (1 atm) über Nacht gerührt. Der
Katalysator wird durch ein Kissen aus Celite® filtriert
und mehrere Male mit EtOAc gewaschen. Die Lösemittel werden im Vakuum konzentriert und
der Rückstand
wird durch Silicagelchromatographie unter Elution mit 3% CH2Cl2 : MeOH unter
Bildung der Titelverbindung (44% Ausbeute) gereinigt. MS (ES): m/z
= 487,1 (M+H)+.
-
Beispiel 215
-
1-Isopropylsulfonyl-2-amino-6-(1-(cyclopropyl)-2-(brom)-4-(2-fluorphenyl)imidazol-5-yl)benzimidazol
-
Brom
(0,34 ml, 6,545 mmol) wird tropfenweise über 5 Minuten zu einer Lösung aus
1-Isopropylsulfonyl-2-amino-6-(1-(cyclopropyl)-4-(2-fluorphenyl)imidazol-5-yl)benzimidazol
(2,61 g, 5,95 mmol) in Chloroform (0,7 l) gegeben und für 18 Stunden
gerührt.
Es wird mit Wasser, gesättigtem
Bicarbonat gewaschen, mit Magnesiumsulfat getrocknet, filtriert
und die Lösemittel
werden unter verringertem Druck entfernt. Der Rückstand wird auf Silicagel
unter Elution mit Ethylacetat/Dichlormethangemischen unter Bildung
der Titelverbindung (2,41 g) gereinigt. MS (ES+):
m/z = 518,0 (M+H)+.
-
Beispiel 216
-
1-Isopropylsulfonyl-2-amino-6-(1-(cyclopropyl)-2-(4-chlorphenyl)-4-(2-fluorphenyl)imidazol-5-yl)benzimidazolmethansulfonat
-
1-Isopropylsulfonyl-2-amino-6-(1-(cyclopropyl)-2-(brom)-4-(2-fluorphenyl)imidazol-5-yl)benzimidazol (0,214
g, 0,41 mmol), 4-Chlorphenylborsäure
(0,077 g, 0,50 mmol), Tetrakis(triphenylphosphin)palladium (0,234
g, 0,021 mmol), 2 M Natriumcarbonat (0,62 ml), Methanol (2,0 ml)
und Toluol (20 ml) werden am Rückfluss
für 3 Stunden
erhitzt. Es wird auf Umgebungstemperatur gekühlt, mit Ethylacetat verdünnt, mit
Wasser, gesättigtem
Natriumcarbonat und gesättigtem
Natriumchlorid gewaschen, mit Magnesiumsulfat getrocknet, filtriert
und die Feststoffe werden unter verringertem Druck entfernt. Der
Rückstand
wird auf Silicagel unter Elution mit Dichlormethan/Methanolgemischen
unter Bildung von 0,196 g der freien Base gereinigt. Es wird mit Methansulfonsäure wie
vorher beschrieben, unter Bildung der Titelverbindung (0,232 g)
behandelt. MS (ES+): m/z = 550,2 (M+H)+.
-
Beispiel 217
-
1-Isopropylsulfonyl-2-amino-6-(1-(cyclopropyl)-2-(2,4-difluorphenyl)-4-(2-fluorphenyl))imidazol-5-yl)benzimidazolmethansulfonat
-
Ausgehend
von 1-Isopropylsulfonyl-2-amino-6-(1-(cyclopropyl)-2-(brom)-4-(2-fluorphenyl)imidazol-5-yl)benzimidazol
wird die Titelverbindung im wesentlichen wie in Beispiel 216 beschrieben
(0,179 g) hergestellt. MS (ES+): m/z = 552,2
(M+H)+.
-
Beispiel 218
-
1-Isopropylsulfonyl-2-amino-6-(1-(cyclopropyl)-2-(trimethylsilylethinyl)-4-(2-fluorphenyl)imidazol-5-yl)benzimidazol
-
1-Isopropylsulfonyl-2-amino-6-(1-(cyclopropyl)-2-(brom)-4-(2-fluorphenyl)imidazol-5-yl)benzimidazol (0,332
g, 0,64 mmol), Trimethylsilylacetylen (0,36 ml, 2,56 mmol), Tetrakis(triphenylphosphin)palladium
(0,150 g, 0,128 mmol), Triethylamin (2 ml) und Dimethylformamid
(4 ml) werden vereinigt und für
18 Stunden auf 70°C erhitzt.
Es wird auf Umgebungstemperatur gekühlt und die Lösemittel
werden unter verringertem Druck entfernt. Der Rückstand wird auf Silicagel
unter Elution mit Acetonitril/Dichlormethangemischen unter Bildung
der Titelverbindung (0,295 g) gereinigt. MS (ES+):
m/z = 536,2 (M+H)+.
-
Beispiel 219
-
1-Isopropylsulfonyl-2-amino-6-(1-cyclopropyl)-(2-but-3-in-1-ol)-4-(2-fluorphenyl)imidazol-5-yl)benzimidazolmethansulfonat
-
Ausgehend
von 1-Isopropylsulfonyl-2-amino-6-(1-(cyclopropyl)-2-(brom)-4-(2-fluorphenyl)imidazol-5-yl)benzimidazol
wird die freie Base im wesentlichen wie in Beispiel 218 beschrieben,
hergestellt. Die Methansulfonsäure
wird wie vorher beschrieben unter Bildung der Titelverbindung (0,096
g) behandelt. MS (ES+): m/z = 508,4 (M+H)+.
-
Beispiel 220
-
1-Isopropylsulfonyl-2-amino-6-(1-(cyclopropyl)-2-(ethinyl)-4-(2-fluorphenyl)imidazol-5-yl)benzimidazolmethansulfonat
-
1
M Tetra-n-butylammoniumfluorid (0,15 ml, 0,15 mmol) wird zu einer
Lösung
aus 1-Isopropylsulfonyl-2-amino-6-(1-cyclopropyl)-2-(trimethylsilylethinyl)-4-(2-fluorphenyl)imidazol-5-yl)benzimidazol
(0,054 g, 0,101 mmol) in Tetrahydrofuran (4 ml) gegeben und für 5 Minuten
gerührt.
Es wird mit Ethylacetat verdünnt, mit
Wasser, 50% gesättigtem
Natriumchlorid gewaschen, mit Magnesiumsulfat getrocknet, filtriert
und die Lösemittel
werden unter verringertem Druck entfernt. Der Rückstand wird auf Silicagel
unter Elution mit Hexan/Ethylacetatgemischen unter Bildung der freien
Base (0,037 g) gereinigt. Es wird mit Methansulfonsäure wie
vorher beschrieben unter Bildung der Titelverbindung (0,045 g) behandelt.
MS (ES+): m/z = 464,1 (M+H)+.
-
Beispiel 221
-
1-Isopropylsulfonyl-2-amino-6-(1-(cyclopropyl)-2-(ethyl)-4-(2-fluorphenyl)imidazol-5-yl)benzimidazolmethansulfonat
-
Es
werden 10% Palladium auf Kohle (0,010 g) zu einer Lösung aus
1-Isopropylsulfonyl-2-amino-6-(1-(cyclopropyl)-2-(ethinyl)-4-(2-fluorphenyl))imidazol-5-yl)benzimidazol
(0,074 g, 0,16 mmol) in Methanol (15 ml) gegeben und unter 1 Atmosphäre an Wasserstoffgas
für 1,5
Stunden hydriert. Es wird filtriert und die Lösemittel werden unter verringertem
Druck entfernt. Der Rückstand
wird auf Silicagel gereinigt. Es wird mit Methansulfonsäure wie
vorher beschrieben unter Bildung der Titelverbindung (0,087 g) behandelt.
MS (ES+): m/z = 468,1 (M+H)+.
-
Beispiel 222
-
1-Isopropylsulfonyl-2-amino-6-(1-(cyclopropyl)-2-(1,2,3-triazol-4-yl)-4-(2-fluorphenyl)imidazol-5-yl)benzimidazolmethansulfonat
-
1-Isopropylsulfonyl-2-amino-6-(1-(cyclopropyl)-2-(ethinyl)-4-(2-fluorphenyl)imidazol-5-yl)benzimidazol (0,114
g, 0,247 mmol), Trimethylsilylazid (0,165 ml, 1,23 mmol) und 1,4-Dioxan
(2 ml) werden vereinigt und unter Mikrowellenbestrahlung bei 140°C für 1 Stunde
erhitzt. Es wird auf Umgebungstemperatur gekühlt, Methanol (5 ml) wird zugegeben
und die Lösemittel
werden unter verringertem Druck entfernt. Der Rückstand wird auf Silicagel
unter Elution mit Dichlormethan/Methanolgemischen gereinigt. Es
wird mit Methansulfonsäure
wie vorher beschrieben unter Bildung der Titelverbindung (0,088
g) behandelt. MS (ES+): m/z = 507,1 (M+H)+.
-
Beispiel 223
-
1-Isopropylsulfonyl-2-amino-6-(1-cyclopropyl)-2-azido-4-(2-fluorphenyl)imidazol-5-yl)benzimidazol
-
Es
wird 1,9 M Phenyllithium (0,42 ml, 0,80 mmol) tropfenweise mit einer
Lösung
aus 1-Isopropylsulfonyl-2-amino-6-(1-(cyclopropyl)-2-(brom)-4-(2-fluorphenyl))imidazol-5-yl)benzimidazol
(0,103 g, 0,20 mmol) in Tetrahydrofuran (3 ml) bei –78°C vereinigt
und für
20 Minuten gerührt.
1,7 M tert-Butyllithium
(0,28 ml, 0,48 mmol) wird tropfenweise über 5 Minuten zugegeben und
für 60
Minuten gerührt.
p-Toluolsulfonylazid (0,064 g, 0,323 mmol) in Tetrahydrofuran (0,4
ml) wird tropfenweise über
2 Minuten zugegeben und bei –78°C für 1 Stunde
gerührt.
Es wird auf 0°C
erwärmt,
mit Ethylacetat ver dünnt
und mit gesättigtem
Natriumchlorid gewaschen. Das wässrige
Natriumchlorid wird neutralisiert, mit wässriger 1 N Chlorwasserstoffsäure gewaschen
und mit Ethylacetat rückextrahiert.
Die organischen Phasen werden vereinigt und mit Magnesiumsulfat
getrocknet, filtriert und die Lösemittel
werden unter verringertem Druck entfernt. Der Rückstand wird auf Silicagel
unter Elution mit Acetonitril/Dichlormethangemischen unter Bildung
der Titelverbindung (0,035 g) gereinigt. MS (ES+): m/z
= 481,1 (M+H)+.
-
Beispiel 224
-
1-Isopropylsulfonyl-2-amino-6-(1-(cyclopropyl)-2-(amino)-4-(2-fluorphenyl)imidazol-5-yl)benzimidazolmethansulfonat
-
Ausgehend
von 1-Isopropylsulfonyl-2-amino-6-(1-cyclopropyl)-2-(nitro)-4-(2-fluorphenyl))imidazol-5-yl)benzimidazol
wird die Titelverbindung im wesentlichen wie in Beispiel 221 (0,031
g) beschrieben hergestellt. MS (ES+): m/z
= 455,1 (M+H)+.
-
Beispiel 225
-
1-Isopropylsulfonyl-2-amino-6-(1-cyclohexyl-2-(brom)-4)-2-fluorphenyl))imidazol-5-yl)benzimidazol
-
Ausgehend
von 1-Isopropylsulfonyl-2-amino-6-(1-cyclohexylphenyl)imidazol-5-yl)benzimidazol
wird die Titelverbindung im wesentlichen wie in Beispiel 215 beschrieben,
hergestellt. MS (ES+): m/z = 543,9 (M+H)+.
-
Beispiel 226
-
1-Isopropylsulfonyl-2-amino-6-(1-cyclohexyl)-2-(ethinyl)-4-(phenyl)imidazol-5-yl)benzimidazol
-
Ausgehend
von 1-Isopropylsulfonyl-2-amino-6-(1-(cyclohexyl)-2-(brom)-4-(phenyl)imidazol-5-yl)benzimidazol wird
die Titelverbindung im wesentlichen wie in Beispiel 218 und 220
beschrieben, hergestellt. MS (ES+): m/z
= 488,1 (M+H)+.
-
Beispiel 227
-
1-Isopropylsulfonyl-2-amino-6-(2-(2,4-difluorphenyl)-5-(3,5-difluorphenyl)imidazol-4-yl)benzimidazol
-
Ein
Gemisch aus 1-Isopropylsulfonyl-2-amino-6-(1-(benzyl)-2-(2,4-difluorphenyl)-5-(3,5-dilfluorphenyl)imidazol-4-yl)benzimidazol
(0,084 g, 0,778 mmol), Palladiumschwarz (0,084 g) und Methanol (50
ml) wird bei 50°C
unter Wasserstoffgas (55 psi) für
18 Stunden hydriert. Die Reaktion wird auf Umgebungstemperatur gekühlt, filtriert
und das Lösemittel
wird unter verringertem Druck entfernt. Der Rückstand wird auf Silicagel
unter Elution mit Dichlormethan/Methanolgemischen unter Bildung
der Titelverbindung (0,025 g) gereinigt. MS (ES+):
m/z = 530,0 (M+H)+.
-
Die
freie Base der Beispiele 228-231 wird im wesentlichen wie in Präparation
66-67 und Beispiel 227 beschrieben, hergestellt. Die Methansulfonatsalze
werden durch Lösen
der freien Base in 10% Methanol/Dichlormethan unter Zugabe von 1,0 Äquivalenten
Methansulfonsäure
gelöst
und die Lösemittel
werden unter verringertem Druck unter Bildung der Methansulfonatsalze
entfernt.
-
-
Die
freie Base der Beispiele 232-235 wird im wesentlichen wie in Präparation
67 und Beispiel 227 beschrieben, hergestellt. Die Methansulfonatsalze
werden durch Lösen
der freien Base in 10% Methanol/Dichlormethan unter Zugabe von 1,0 Äquivalenten
Methansulfonsäure
hergestellt und die Lösemittel
werden unter verringertem Druck unter Bildung der Monomethansulfonatsalze
zugegeben.
-
-
Beispiel 236
-
1-Isopropylsulfonyl-2-amino-6-(2-(methyl)-5-(2,4-difluorphenyl)imidazol-4-yl)benzimidazolmethansulfonat
-
Zu
einer Lösung
aus 1-Isopropylsulfonyl-2-amino-6-(1-(benzyl)-2-(methyl)-5-(2,4-difluorphenyl)imidazol-4-yl)benzimidazol
(390 mg, 0,75 mmol) in 100 ml Methanol werden Palladiumschwarz (390
mg) und Wasserstoffgas bei 50°C über Nacht
bei 60 psi gegeben. Die Lösung
wird filtriert und durch Radialchromatographie unter Elution mit
8% Methanol in Methylenchlorid unter Bildung von 152 mg (47%) der
Titelverbindung als nicht ganz weißer Feststoff gereinigt. MS
(ES+): m/z = 432,0 (M+H)+.
-
Zu
einer Lösung
aus 1-Isopropylsulfonyl-2-amino-6-(2-(methyl)-5-(2,4-difluorphenyl)imidazol-4-yl)benzimidazol (152
mg, 0,35 mmol) in 5 ml Methylenchlorid mit 3% Methanol wird ein Äquivalent
an Methansulfonsäure
(23 μl)
gegeben und unter Bildung von 186 mg (100%) der Titelverbindung
als hellbrauner Feststoff getrocknet. MS (ES+):
m/z = 432,0 (M+H)+.
-
Die
Verbindung von Beispiel 237 kann im wesentlichen wie in Beispiel
236 beschrieben, hergestellt werden.
-
-
Beispiel 238
-
1-Isopropylsulfonyl-2-amino-6-(3-(methyl)-1-(phenyl)pyrazol-S-yl)benzimidazol
-
Zu
einer Lösung
aus 5-(Brom-3-(methyl)-1-(phenyl)pyrazol (264 mg, 1,11 mmol) in
6 ml DME wird 1-Isopropylsulfonyl-2-aminobenzimidazol-6-borsäure (694
mg, 2,45 mmol) gegeben. Natriumcarbonat (354 mg, 3,34 mmol) wird
in 0,5 ml Wasser gelöst
und trans-Dichlorbis(triphenylphosphin)palladium(II) (234 mg, 0,33
mmol) wird zugegeben. Das Gemisch wird unter Rühren unter Stickstoff auf 100°C erhitzt.
Nach 2 Stunden wird sie gekühlt
und über
ein Kissen aus Celite® und Natriumsulfat filtriert.
Das rohe Produkt wird durch Radialchromatographie unter Elution
mit 4% Methanol in CH2Cl2 unter
Bildung von 201 mg (46%) der Titelverbindung gereinigt. MS (ES+): m/z = 396,0 (M+H)+.
-
Die
Verbindungen der Beispiele 239-240 werden im wesentlichen wie in
Beispiel 238 beschrieben, hergestellt.
-
-
Beispiel 241
-
1-Isopropylsulfonyl-2-amino-6-(2,4-(diphenyl)-1,2-dihydropyrazol-3-on-5-yl)benzimidazol
-
Zu
einer gerührten
Lösung
aus 1-Isopropylsulfonyl-2-amino-6-(α-((1-hydroxy)-α-(phenyl)acetyl)benzimidazol
(0,25 mmol, 1 Äquivalent)
in trockenem Toluol (3 ml) wird Phenylhydrazin (0,27 mmol, 1,1 Äquivalente) gegeben
und das Gemisch wird am Rückfluss
unter Stickstoff gefolgt von einer TLC erhitzt. Das Reaktionsgemisch
wird konzentriert und mittels Dichlormethan : Methanol 30 : 1 unter
Bildung der Titelverbindung (24% Ausbeute) konzentriert. MS (ACP+): m/z = 474,2 (M+H)+.
-
Die
Verbindungen der Beispiele 242-245 können im wesentlichen wie in
Beispiel 241 beschrieben, hergestellt werden.
-
-
-
Beispiel 246
-
1-Isopropylsulfonyl-2-amino-6-(1-(benzyl)-2-(methyl)-5-(2,4-difluorphenyl)imidazol-4-yl)benzimidazol
-
Zu
einer Lösung
aus 1-(Benzyl)-4-(brom)-5-(2,4-difluorphenyl)-2-(methyl)imidazol
(1,12 g, 3,08 mmol) in 8 ml DME wird 1-Isopropylsulfonyl-2-aminobenzimidazol-6-borsäure (2,62
g, 0,25 mmol) gegeben. Natriumcarbonat (0,98 g, 9,25 mmol) gelöst in 1
ml Wasser und trans-Dichlorbis(triphenylphosphin)palladium(II) (0,65 g,
0,93 mmol) werden zugegeben. Das Gemisch wird unter Rühren unter
Stickstoff auf 100°C
erhitzt. Nach 3 Stunden wird die Lösung gekühlt und über ein Kissen aus Celite® und
Natriumsulfat filtriert. Der Rückstand
wird durch Radialchromatographie unter Elution mit 3% Methanol in
CH2Cl2 mit einem
Gradienten aus 10% Methanol gereinigt. Eine zweite Reinigung durch
Radialchromatographie wird unter Elution mit 4% Methanol in CH2Cl2 mit einem Gradienten
aus 6% Methanol unter Bildung von 390 mg (24%) der Titelverbindung
als gelber Feststoff durchgeführt.
MS (ES+): m/z = 522,0 (M+H)+.
-
Beispiel 247
-
2-Amino-6-(5-phenylimidazol-4-yl)benzothiazol
-
Ausgehend
von 2-Aminobenzothiazol-6-carboxaldehyd wird die Titelverbindung
im wesentlichen wie in Beispiel 182 beschrieben, hergestellt. Ausbeute
(16%). MS (ES+): m/z = 293,0 (M+H)+.
-
Beispiel 248
-
2-Amino-6-(1-(cyclohexyl)-4-(phenyl)imidazol-5-yl)benzothiazol
-
Ausgehend
von 2-Aminobenzothiazol-6-carboxaldehyd und Cyclohexylamin wird
die Titelverbindung im wesentlichen wie in Beispiel 247 beschrieben,
hergestellt. Ausbeute (68%). MS (ES+): m/z
= 375,1 (M+H)+.
-
Allgemeines Verfahren
A. Sandmeyer Reaktion:
-
Ein Äquivalent
Benzothiazol wird in drei Portionen über 5 Minuten zu einer Suspension
aus einem Äquivalent
Kupfer(II)chlorid und 1,5 Äquivalenten
tert-Butylnitrit in 1 ml Acetonitril bei 65°C gegeben. 0,1 ml Ethylendiamin
wird zugegeben, das Reaktionsgemisch wird in Wasser gegossen und
mit Ethylacetat (3 × 25 ml)
extrahiert. Die vereinigten organischen Phasen werden nacheinander
mit Wasser (2 × 15
ml) gefolgt von gesättigtem
wässrigem
Natriumchlorid (15 ml) gewaschen, über Natriumsulfat getrocknet
und unter verringertem Druck konzentriert. Der Rückstand wird durch Silicagelchromatographie
unter Elution mit einem Gradienten aus Hexan, der 25-50% Ethylacetat
enthält,
gereinigt.
-
Beispiel 249
-
2-Chlor-6-(1-(cyclohexyl)-4-(phenyl)imidazol-5-yl)benzothiazol
-
Ausgehend
von 2-Amino-6-(1-(cyclohexyl)-4-(phenyl)imidazol-5-yl)benzothiazol
wird die Titelverbindung im wesentlichen wie in dem Allgemeinen
Verfahren A: Sandmeyer Reaktion, beschrieben hergestellt. Ausbeute
(24%). MS (ES+): m/z = 394,1 (M+H)+.
-
Allgemeines Verfahren
B
-
Ein Äquivalent
des Amins wird zu der Lösung
aus drei Äquivalenten
an Benzothiazol in 1 ml Tetrahydrofuran gegeben. Nach 24 Stunden
wird das Reaktionsgemisch unter verringertem Druck konzentriert.
Der Rückstand
wird durch Silicagelchromatographie unter Elution mit Hexan, das
25% Ethylacetat enthält,
gereinigt.
-
Beispiel 250
-
2-Aminoethyl-6-(1-(cyclohexyl)-4-(phenyl)imidazol-5-yl)benzothiazol
-
Ausgehend
von 2-Chlor-6-(1-(cyclohexyl)-4-((phenyl)imidazol-5-yl)benzothiazol
und Ethylamin wird die Titelverbindung im wesentlichen wie im allgemeinen
Verfahren B beschrieben, hergestellt. MS (ES+):
m/z = 403,1 (M+H)+.
-
Beispiel 251
-
1-Isopropylsulfonyl-2-amino-6-(5-(phenyl)-1,2,3-(triazol-4-yl))benzimidazol
-
Ein
Gemisch aus 3-Isopropylsulfonamidyl-4-amino-1-(5-phenyl)-1,2,3-triazol-4-yl)
(0,060 g), Lithiummethoxid (0,013 g) und Cyanogenbromid (0,053 g)
in Dichlormethan (5 ml) wird unter Stickstoff für 48 Stunden vereinigt und
gerührt.
Der Rückstand
wird durch eine Oasis® HLB Extraktionskartusche
60 μM (LP)
unter Bildung eines weißen
Feststoffs (0,050 g, 30%) gereinigt. MS (ES+):
m/z = 382,44 (M+H)+.
-
Beispiel 252
-
1-Isopropylsulfonyl-2-amino-6-(5-(phenyl)pyrazol-4-yl)benzimidazol
-
Lithiummethoxid
(21 mg) und Cyanogenbromid (89 mg) werden zu einer Lösung aus
3-Isopropylsulfonamidyl-4-amino-1-(5-phenyl)pyrazol-4-yl-phenyl
in 4,5 ml CH2Cl2 gegeben.
Es wird unter Stickstoff für
18 Stunden gerührt.
Es wird mit 2 ml THF verdünnt
und 0,25 ml MeOH werden zum Lösen
der Feststoffe zugegeben und mit 5% NaHCO3 gewaschen.
Die wässrige
Phase wird mit einer 9 : 1 Lösung
aus CH2Cl2 : MeOH rückextrahiert.
Die Phasen werden getrennt und die organische Phase wird über Magnesiumsulfat
getrocknet und unter verringertem Druck konzentriert. Der Rückstand
wird unter Elution mit einem Gradienten von 2-3,5% CH2Cl2 : MeOH unter Bildung eines weißen Feststoffs
(34 mg, 31%) einer Silicagelchromatographie unterzogen. MS (ES+): m/z = 382,5 (M+H)+.
-
Beispiel 253
-
1-Isopropylsulfonyl-2-amino-6-(2-(2,6-difluorphenyl)-5-(phenyl)imidazol-4-yl)benzimidazoldimaleat
-
Es
werden 2,50 g (5,0 mmol) in 15 ml Methanol gelöst und 1,20 g (10,0 mmol) Maleinsäure werden zugegeben.
Das Reaktionsgemisch wird bei 40°C
unter Rühren
für 10
Minuten erhitzt. Das Reaktionsgemisch wird filtriert. Das Filtrat
wird auf 40-4°C
erhitzt, während
35 ml Ethylacetat zugegeben wer den. Das Gemisch wird unter kontinuierlichem
Rühren
auf Raumtemperatur gekühlt.
Das ausgefallene Produkt bildet sich langsam. Man sammelt das feste
Produkt durch Filtration, wäscht
mit Ethylacetat und trocknet bei 50°C in einem Vakuumofen, 3,25
g, 92,8%.
1H NMR (500 MHz, CD3OD) δ 7,775
(m, 1H), 7,62 (s, 1H), 7,54 (d, J = 7,0 Hz, 2H), 7,48 (s, 1H), 7,46
(d, J = 7,00 Hz, 2H), 7,38 (d, J = 7,00 Hz, 1H), 7,38 (dd, J = 7,00
Hz, 2H), 6,29 (s, 4H), 3,60 (m, 1H), 1,26 (d, J = 7,00 Hz, 6H).
-
Die
Fähigkeit
der erfindungsgemäßen Verbindungen
zur Hemmung der p38 Kinase wird durch Standardtests gezeigt, die
dem Fachmann gut bekannt sind und ist kurz in den folgenden Abschnitten
beschrieben.
-
Hemmung der p38 Kinase
-
Standardlösungspräparationen
-
Die
Kinasepufferlösung
wird durch Kombination von 2,5 ml an 1 M Tris-HCl (pH 7,5), 0,1
ml an 1 M Dithiothreit, 1,0 ml an 1 M Magnesiumchlorid und 300 μl 1% Triton
X-100 und einer Verdünnung
auf 100 ml mit Wasser hergestellt. 84 ml dieser Kinasepufferlösung werden
mit 16 ml Dimethylsulfoxid unter Bildung der 16% DMSO Lösung kombiniert.
-
Die
200 μM ATP
Lösung
wird durch Zugabe von 102,6 μl
an 10 mM wässrigem
ATP, 25 μl
an 33P-ATP und 163,5 μl an 4 mM wässrigem epidermalen Wachstumsfaktorpeptid
661-668 (Biomol, Katalognummer P-121) in 5 ml Kinasepufferlösungen hergestellt.
-
Die
p38 Kinaseenzymlösung
wird durch Lösen
von 9,5 μl
an konzentrierter Enzymlösung
(250 ng an p38 Enzym/μl
Kinasepufferlösung)
in 1536 μl
Kinasepufferlösung
hergestellt.
-
Probenherstellung
-
Eine
80 μM Lösung jeder
Testverbindung und der Kontrollverbindung werden durch Lösen von
2 μl einer
10 mM Stammlösung
der jeweiligen Verbindungen in Dimethylsulfoxid in 248 ml der 16%
DMSO Lösung in
einer Costar Mikrotiterplatte mit 96 Vertiefungen hergestellt. Die
Platte wird auf den automatisierten Tecan Genesis Flüssigkeitsroboter
für 1 :
3 Verdünnungen
gestellt.
-
Test
-
10 μl seriell
verdünnte
Verbindung werden mit einem automatisierten Beckman Multimek Flüssigkeitsrobotor
für 96
Vertiefungen in die Platte gegeben. 20 μl an 200 μM ATP Lösung werden mit einem Titertek
Multidrop 8-Kanalflüssigkeitsrobotor
zugegeben. 10 μl
an p38 Kinaseenzymlösung
wird mittels des Multimek zur Testplatte gegeben. Das Gemisch kann
für 40
Minuten bei 30°C
reagieren und dann wird die Reaktion durch die Zugabe von 60 μl eines frisch
hergestellten 5% Eisessig mit Multidrop gestoppt. 80 μl dieser
Lösung
werden in eine "MAPH" Platte mittels des
Multimek gegeben. Die Platten können
sich für
30 Minuten auf Raumtemperatur einstellen und dann auf einem Titertek
MAP Extraktionsgerät
mit frisch hergestelltem 0,5% Eisessig gewaschen/abgesaugt (1 × 300 μl, 2 × 200 μl). Die Vertiefungen
werden geblottet und es werden 100 μl MicroScint 20 Scintillationsflüssigkeit
(Packard Bioscience) mit dem Multidrop zugegeben. Die Platten können für 30 Minuten
stehen und werden auf einem PE/Wallac Microbeta Trilux Scintillationszähler auf
das 33P-Isotop gezählt.
-
Alle
beispielhaft dargestellten Verbindungen werden anfänglich mit
10 Konzentrationen getestet (20 μM – 1 nM mittels
1 : 3 Reihenverdünnungen).
Verbindungen mit HK50 Werten von weniger
als 25 nM werden bei der Ausgangskonzentration von 2 μM bis 0,1
nM (1 : 3 Reihenverdünnungen)
erneut getestet. Die HK50 Werte werden für jede Verbindung
mittels einer nicht-linearen Regression berechnet (IDBS Activity
Base Software). Alle beispielhaft dargestellten Verbindungen werden
getestet, wie dies im wesentlichen oben beschrieben ist und hemmen
das p38 Kinaseenzym mit einer HK50 von mindestens
5 μM.
-
Hemmung von THF-α in vitro
-
Peritoneale Mausmakrophagen
-
1
ml Thioglycolatmedium (5,0 g Hefeextrakt, 15,0 Casiton oder Trypticase,
5,0 g Dextrose, 2,5 g Natriumchlorid, 0,75 g L-Cystein, 0,5 g Natriumthioglycolat,
1,0 mg Resazurin und 0,75 g Agar in 1,0 l destilliertem Wasser)
wird in den Peritonealraum jeder weiblichen Balb/C Maus injiziert.
Am Tag 4 oder 5 nach der Injektion werden die Mäuse getötet und dann mit 4 ml Dulbecco's phosphat-gepufferter
Kochsalzlösung
(BioWhittaker) injiziert und die Peritonealmakrophagen werden mit
einer Spritze entnommen.
-
Cytokinbildung
-
Peritoneale
Mausmakrophagen werden mit einem Hämocytometer gezählt und
auf 2,5 × 106 Zellen/ml Vollmedium eingestellt (RPMI-1640
Medium, das 10% fetales Rinderserum enthält). Die Zellen werden in Flachbodenplatten
mit 96 Vertiefungen mit 5 × 105 Zellen pro 200 μl pro Vertiefung ausplattiert
und können
sich absetzen und am Boden der Vertiefung für mindestens 3 Stunden haften.
Die Zellen werden mit der Testverbindung oder dem Standard p38 Kinaseinhibitor
mittels einer Reihe an 8 Konzentrationen für 1 Stunde bei 37°C (20 μl/Vertiefung)
vorbehandelt. Die Zellen werden dann mit einem Gemisch aus 50 ng/ml
Lipopolysaccharid (LPS) und 10 E/ml Interferon γ für 18 Stunden bei 37°C behandelt
(20 μl/Vertiefung).
Das konditionierte Medium wird geerntet und auf eine TNF-α Bildung
mittels des Luminex-Verfahrens getestet.
-
TNF-α/Luminexdetektionstest zur Hemmung
von TNF-α in
vitro (Bio Rad Bio Plex Kit – Katalog
Nr. 171-G12221)
-
Der
lyophilisierte, vorgemischte TNF-α Standard
(1 Standardröhrchen/zwei
Platten mit 96 Vertiefungen) wird mit 50 ml sterilem Wasser rekonstituiert
(500 000 pg/ml). Die Proben werden für 5 Sekunden gevortext, für 30 Minuten
auf Eis inkubiert und vor der Verwendung für 5 Sekunden gevortext. Ein
Satz aus zwölf
1,5 ml Röhrchen
wird mit Nr. 1 bis Nr. 12 beschriftet und dann werden die im folgenden
gezeigten Mengen an Zellmedien zu den geeigneten Röhrchen gegeben
(Standardkonzentration wie folgt: 50 000, 25 000, 12 500, 6 250,
3 125, 1 562,5, 781,3, 390,6, 195,3, 97,7, 48,8 und 24,4 pg/ml).
Die vorgemischten Anti-Cytokin konjugierten Kügelchen werden stark für 30 Sekunden
gevortext (25 ×).
Die Anti-Cytokin konjugierten Kügelchen werden
auf eine 1 × Konzentration
mittels 1 × Bio-Plex
Testpuffer verdünnt.
Für jede
Platte werden 240 μl
der vorgemischten Kügelchen
zu 5760 μl
Bio-Plex Testpuffer gegeben. Eine Milliporeplatte mit 96 Vertiefungen wird
mit 100 μl/Vertiefung
an Blockierungspuffer blockiert. Der Blockierungspuffer wird durch
ein Millipore Filtrationssystem filtriert und dann getrocknet. Es
werden zwei Waschschritte auf der Filterplatte mit 100 μl/Vertiefung
an Bio-Plex Testpuffer ausgeführt
und getrocknet. Die 1 × Anti-Cytokin-konjugierten
Kügelchen
werden für
15 Sekunden gevortext und es werden 50 μl zu jeder Vertiefung gegeben.
Es wird erneut durchfiltriert und getrocknet. Es werden zwei Waschschrite
auf den Platten mit 100 μl/Vertiefung
an Bioplexwaschpuffer ausgeführt.
Es wird erneut durchfiltriert und getrocknet. 50 μl Probe oder
Standard wird zu jeder Probenvertiefung gegeben. Dies wird für 60 Sekunden
bei Raumtemperatur auf einem Schüttler
bei der Einstellung 6 inkubiert, der vor Licht geschützt ist,
und dann für
30 Minuten bei der Einstellung 3 und dann über Nacht in den Kühlschrank
gestellt. Es werden 3 Waschschritte mit dem Bio-Plex Waschpuffer
ausgeführt
und dann filtriert und getrocknet. Der Cytokinantikörper wird
für jede
Platte hergestellt (~10 Minuten vor der Verwendung) und 60 μl der vorgemischten
Cytokindetektionsantikörperstammlösung wird
zu 5940 μl
Bio-Plex Detection Antibody Diluent gegeben. 50 μl des Cytokindetektionsantikörpers werden
zugegeben und bei Raumtemperatur für 60 Sekunden bei der Einstellung
6 und dann für
30 Minuten bei der Einstellung 3 auf einem Schüttler inkubiert, der vor Licht
geschützt
ist. Es werden 3 Waschschritte mit dem Bio-Plex Waschpuffer ausgeführt. Dies
wird durchfiltriert und getrocknet. Strept-PE (~10 Minuten vor der
Verwendung) wird für
jede Platte hergestellt und 60 μl bis
5940 μl
Bio-Plex Testpuffer werden zugegeben. 50 μl Streptavidin-PE werden zu
jeder Vertiefung gegeben und für
60 Sekunden bei Raumtemperatur bei Einstellung 6 und dann für 10 Minuten
bei Einstellung 3 auf einem Schüttler
inkubiert, der vor Licht geschützt
ist. Es werden 3 Waschschritte mit Bio-Plex Waschpuffer ausgeführt. Dies
wird durchfiltriert. Die Kügelchen
werden in 100 μl/Vertiefung
an Bio Plex Testpuffer resuspendiert. Standards und Proben werden
auf einem Luminexgerät
ausgelesen. Diese Intensitätsauswertungen
werden dann auf Picogramm/Milliliter Einheiten basierend auf einer
Standardkurve mit 12 Punkten umgewandelt, die zweifach mittels einer
logischen Regressionsmethode mit 4 Parametern erzeugt wird (Bio
Plex Manager, Versionen 2.0 und 3.0, Bio Rad) und es wird die HK50
berechnet.
-
Die
Verbindungen der Beispiele 1, 6, 16 und 17 werden getestet, wie
dies im wesentlichen oben beschrieben ist und unterdrücken TNF-α in vitro
mit einer HK50 von weniger als 100 nM.
-
Hemmung von TNF-α in vivo
-
Die
Verbindungen werden p.o. (100, 30, 10 und 3 mg/kg) an weibliche
Balb/c Mäuse
(5 Mäuse/Dosis) verabreicht.
Nach 2 Stunden wird Lipopolysaccharid (LPS, E. coli Serotyp 0111:B4,
5 mg/kg) i.v. in die Schwanzvene jeder Maus verabreicht. Eine Stunde
nach der LPS Verabreichung werden die Mäuse durch CO2 Inhalation
getötet
und das Blut wird über
eine Herzpunktion gewonnen. Das Serum wird auf TNF-α Spiegel durch
das Luminexverfahren analysiert.
-
TNF-α/Luminexdetektionstest zur Hemmung
von TNF-α in
vivo (Bio-Rad Bio-Plex Kit Katalog Nr. 171-G12221)
-
Es
wird der lyophilisierte vorgemischte TNF-α Standard (1 Standardröhrchen/zwei
Platten mit 96 Vertiefungen) mit 50 μl sterilem Wasser (500 000 pg/ml)
rekonstituiert. Man vortext sanft für 5 Sekunden, inkubiert für 30 Minuten
auf Eis und vortext für
5 Sekunden vor der Verwendung. Ein Satz aus zwölf 1,5 ml Röhrchen wird mit Nr. 1 bis Nr.
12 beschriftet und dann werden die im folgenden gezeigten Mengen
an Mausserumstandardverdünnung
zu den geeigneten Röhrchen
gegeben (Standardkonzentration wie folgt: 50 000, 25 000, 12 500,
6 250, 3 125, 1 562,5, 781,3, 390,6, 195,3, 97,7, 48,8 und 24,4
pg/ml). Die vorgemischten Anti-Cytokin konjugierten Kügelchen
werden stark für
30 Sekunden gevortext (25 ×).
Die Anti-Cytokin konjugierten Kügelchen
werden auf eine 1 × Konzentration
mittels 1 × Bio-Plex
Testpuffer verdünnt.
Für jede
Platte werden 240 μl
der vorgemischten Kügelchen
zu 5760 μl
Bio-Plex Testpuffer gegeben. Eine Milliporeplatte mit 96 Vertiefungen
wird mit 100 μl/Vertiefung
an Blockierungspuffer blockiert. Der Blockierungspuffer wird durch
ein Millipore Filtrationssystem filtriert und dann getrocknet. Es
werden zwei Waschschritte auf der Filterplatte mit 100 μl/Vertiefung
an Bio-Plex Testpuffer ausgeführt
und getrocknet. Es werden 1 × Anti-Cytokin
konjugierte Kügelchen für 15 Sekunden
gevortext und 50 μl
werden zu jeder Vertiefung gegeben. Es wird erneut durchfiltriert
und getrocknet. Es werden 25 μl
Serumprobe und 25 μl
Verdünnungsmittel
(Bio Rad) oder 50 μl
Standard zu jeder Probenvertiefung gegeben. Es wird bei Raumtemperatur
für 60
Sekunden bei der Einstellung 6 und dann für 30 Minuten bei der Einstellung
3 auf einem Schüttler
inkubiert, der vor Licht geschützt
ist, und über
Nacht in den Kühlschrank
gestellt. Es werden 3 Waschschritte mit Bio Plex Waschpuffer ausgeführt. Es
wird durchfiltriert und getrocknet. Es wird Cytokindetektionsantikörper (~10
Minuten vor der Verwendung) für
jede Platte hergestellt und man gibt 60 μl der vorgemischten Cytokindetektionsantikörperstammlösung zu
5940 μl Bio-Plex
Detection Antibody Diluent. Es werden 50 μl Cytokindetektionsantikörper zugegeben
und für
60 Sekunden bei Raumtemperatur bei einer Einstellung von 6 und dann
für 30
Minuten bei einer Einstellung von 3 auf einem Schüttler inkubiert,
der vor Licht geschützt
ist. Man führt
3 Waschschritte mit Bio-Plex Waschpuffer aus. Man filtriert und
trocknet. Strept-PE (–10
Minuten vor der Verwendung) wird für jede Platte hergestellt und 60 μl bis 5940 μl Bio-Plex
Testpuffer werden zugegeben. 50 μl
Streptavidin-PE werden zu jeder Vertiefung gegeben und für 60 Sekunden
bei Raumtemperatur bei Einstellung 6 und dann für 10 Minuten bei Einstellung
3 auf einem Schüttler
inkubiert, der vor Licht geschützt
ist. Es werden 3 Waschschritte mit Bio-Plex Waschpuffer ausgeführt. Dies
wird durchfiltriert. Die Kügelchen
werden in 100 μl/Vertiefung
an Bio Plex Testpuffer resuspendiert. Standards und Proben werden
auf einem Luminexgerät
ausgelesen. Diese Intensitätsauswertungen werden
dann auf Picogramm/Milliliter Einheiten basierend auf einer Standardkurve
mit 12 Punkten umgewandelt, die zweifach mittels einer logischen
Regressionsmethode mit 4 Parametern erzeugt wird (Bio Plex Manager
2.0, Bio Rad) und es wird die HK50 berechnet.
-
Die
Verbindungen der Beispiele 1, 6 und 16 werden getestet, wie dies
im wesentlichen oben beschrieben ist und unterdrücken TNF-α in vivo mit einer HK50 von weniger als 100 mg/kg.
-
Wirkung TNF-α, das durch
intraartikuläres
LPS induziert wird
-
Die
intraartikuläre
Injektion von LPS in die Rattengelenke induziert die Synthese von
TNF-α, das
in der Synovialflüssigkeit
gemessen werden kann. Es sind innerhalb von 2 Stunden hohe Mengen
an TNF-α detektierbar.
Da das Gelenk die Stelle ist, an der sich die Arthritis entwickelt,
kann dieses Modell schnell bestimmen, ob eine oral verabreichte
Verbindung einen Effekt auf die Entzündungsreaktion im Synovium
hat.
-
Sechs
weibliche Lewis-Ratten (150-200 g) werden in jede Behandlungsgruppe
gegeben. Den Tieren werden Träger
(1% Natriumcarboxymethylcellulose – 0,25% Tween 80) oder die
Verbindung von Beispiel 77 (freie Base) (1 mg/kg, 3 mg/kg, 10 mg/kg
und 30 mg/kg) oral verabreicht. Eine Stunde später werden 10 μl LPS (10 μg) intraartikulär in das
rechte Gelenk jeder Ratte verabreicht, während das linke Gelenk 10 μl Kochsalzlösung erhält. Nach
2 Stunden wird jedes Gelenk mit 100 ml Kochsalzlösung gewaschen. Die Lavage
wird gewonnen und bei –80°C gelagert.
- Gruppe Nr. 1: Träger
(1% Na-CMC – 0,25%
Tween 80, 1 ml, PO)
- Gruppe Nr. 2: Beispiel 77 (freie Base) (1 mg/kg, 1 ml PO)
- Gruppe Nr. 3: Beispiel 77 (freie Base) (3 mg/kg, 1 ml, PO)
- Gruppe Nr. 4: Beispiel 77 (freie Base) (10 mg/kg, 1 ml PO)
- Gruppe Nr. 5: Beispiel 77 (freie Base) (30 mg/kg, 1 ml, PO)
-
TNF-α wird mit
einem im Handel erhältlichen
ELISA Kit (R&D,
RTA00) gemessen. Die Behandlung mit der Verbindung von Beispiel
77 ruft eine Dosis-abhängige
Hemmung der TNF-α Synthese
hervor, wie dies in der Synoviallavageflüssigkeit gemessen wird. Die
TMED50 beträgt
10 mg/kg für
Beispiel 77 in diesem Test.
-
B16F10 Melanomziel (MAPKAP-K2
Phosphorylierung) und B16F10 Melanommetastaseeffizienzmodell Hemmung
der B16F10 Melanomlungenmetastasen
-
Die
B16F10 Melanomzelllinie erhält
man von der American Type Culture Collection, Rockville, MD. Die Zellen
werden in RPMI-1640 Medium kultiviert, das mit 10% fetalem Kälberserum
supplementiert ist. Die Zellen werden in vitro angezogen und werden
während
ihrer exponentiellen Wachstumsphase durch sanfte Trypsinisierung
geerntet, zweimal in Medium gewaschen und in serumfreiem RPMI-1640
Medium resuspendiert. Die Anzahl an monodispers lebensfähigen Zellen
wird mittels eines Hämocytometers
bestimmt und auf 1 × 106 Zellen/ml eingestellt. Die Tumorzellen
werden intravenös
in die Schwanzvene von normalen C57B16 Mäusen mit einem Inokulumvolumen
von 0,2 ml injiziert, worin 200 000 Zellen enthalten sind. Die Mäuse werden
mit Testverbindung oder Trägerkontrolle
ausgehend vom Tag 1 vor der i.v. Tumorinokulation behandelt. Die
Verbindung von Beispiel 77 (freie Base) wird als Suspensionsformulierung
in 1% Na-CMC/0,25% Polysorbat 80 hergestellt und die Sonde wird
in einem Injektionsvolumen von 1% Körpergewicht ultrabeschallt
(beispielsweise wird die 30 mg/kg Dosismenge mit 3 mg/ml hergestellt
und 0,2 cm3 pro 20 g Maus verabreicht).
Die Mäuse werden
dreimal am Tag oral mit der Verbindung mit 30, 10 und 3 mg/kg (90,
30 und 9 mg/kg/Tag) von den Tagen –1 bis 16 nach der Tumorzellbeimpfung
behandelt. Die Kontrollmäuse
erhalten den Träger
alleine auf identische Weise. Am Tag 16 werden die Mäuse getötet und
die Lungen werden geerntet und in 3% Paraformaldehyd fixiert. Die
Lungenläsionen
werden durch eine manuelle Zählung
unter dem Sektionsmikroskop quantifiziert.
-
Die
Testverbindung wird im B16F10 Melanomlungenmetastasemodell oral
mit 3, 10 und 30 mg/kg dreimal am Tag von 1 Tag vorher bis 16 Tage
nach der i.v. Tumorbeimpfung getestet (Gesamtdosis 9, 30 und 90 mg/kg/Tag).
Die Lungen werden am Tag 16 geerntet und die Lungenläsionen werden
quantifiziert. Die Testverbindung verursacht jeweils 54%, 73% und
95% Hemmung der Lungenmetastasenbildung für die 3, 10 und 30 mg/kg Dosismengen
(p < 0,001 bei
allen 3 Dosismengen).
-
B16F10 Zielstudien (phosphorylierte
MAPKAPK-2)
-
Die
B16F10 Melanomzelllinie erhält
man von der American Type Culture Collection, Rockville, MD. Die Zellen
werden in RPMI-1640 Medium kultiviert, das mit 10% fetalem Kälberserum
supplementiert ist. Die in vitro angezogenen Zellen werden während ihrer
exponentiellen Wachstumsphase durch sanfte Trypsinierung geerntet,
zweimal in Medium gewaschen und in serumfreiem RPMI 1640 Medium
resuspendiert. Die Anzahl an lebensfähigen Zellen wird mittels eines
Hämocytometers
bestimmt und auf 1 × 107/ml eingestellt. Die Tumorzellen werden
subkutan in normale C57B16 Mäuse
injiziert. Das Inoku lumvolumen pro Maus beträgt 0,2 ml (2 000 000 Zellen).
Wenn die Tumoren 300 bis 500 mg erreichen, werden die Mäuse für Zielhemmstudien entweder
zu einer fixierten Zeit (2,5 Stunden) nach der p.o. Behandlung mit
der Verbindung oder für
pharmakodynamische Studien verwendet, worin die Tumoren zu verschiedenen
Zeitpunkten (beispielsweise 3, 6, 9, 12, 15 und 18 Stunden) nach
der p.o. Behandlung mit der Verbindung gewonnen werden.
-
Proteinextraktion und
Immunblotanalyse
-
Die
wie oben beschrieben gewonnen Tumoren werden unmittelbar in flüssigem Stickstoff
schockgefroren und bei –80°C gelagert.
Die Tumorgewebe werden auf Eis mittels eines Daunce Homogenisiergeräts in einem
Extraktionspuffer homogenisiert (25 mM Tris pH 7,5, worin die folgenden
Proteaseinhibitoren enthalten sind: 10 μg/ml Leupeptin, 10 μg/ml Sojabohnen
Tryp-Chymotrypsininhibitor, 10 mg/ml N-Tosyl-L-Phenylalaninchlormethylketon,
10 μg/ml
Aprotinin, N-α-p-Tosyl-L-Argininmethylester,
7 mM Benzamidin, 0,3 mM Phenylmethylsulfonylfluorid und zwei Tabletten
vollständiger
Proteaseinhibitorcocktail von Roche, gefolgt von Phosphataseinhibitoren:
60 mM β-Glycerophosphat,
1 mM Natriumvanadat, 10 mM Natriumfluorid, 20 mM p-Nitrophenylphosphat,
1 μM Okadainsäure, 1 μM Mikrocystin,
2,5 mM Natriumpyrophosphat und 1 mM Dithiothreit, 15 mM EDTA, 5
mM EGTA, 1% Triton × 100
und 150 mM Natriumchlorid). Die Gewebelysate werden durch Zentrifugation
in einer gekühlten
Mirkozentrifuge bei 14 000 Upm und bei 1°C für 20 Minuten geklärt. Die Überstände werden
in frische Mikrofugenröhrchen überführt, die
auf Eis vorgekühlt
sind und in flüssigem
Stickstoff oder Trockeneis schockgefroren. Nach einem schnellen
Auftauen bis etwa 80% Vollständigkeit
in lauwarmem Wasser werden die Proben bis zum vollständigen Auftauen
auf Eis gestellt. Die Proben werden erneut bei 14 000 Upm und bei
1°C für 15 Minuten
zentrifugiert. Der Überstand
wird in frisch vorgekühlte
Mikrofugenröhrchen überführt und
die Proteinkonzentrationen werden mittels Bio-Rad Proteintestreagenzien
mittels Rinderserumalbumin als Proteinstandard gemessen.
-
Die
Proteinextrakte werden mit dem Extraktionspuffer ausgeglichen. Es
wird ein gleiches Volumen an 2 × SDS
Probenpuffer zu den Proteinextrakten gegeben und in einem Wasserbad
für 5 Minuten
gekocht. Es werden 100 μg
Proteinextrakt pro Probe für
die Elektrophorese auf einem 4-20% Gradienten SDS-PAGE Gel verwendet
und auf Nitrozellulosemembranen (NC Membranen) überführt. Die NC Membranen werden
in 5% BSA in TBST (20 mM Tris pH 7,5, 500 mM Natriumchlorid, 0,05%
Tween 20 und 0,02% Natriumazid) für mindestens 1 Stunde blockiert.
Die Membranen werden dann in primärem Antikörper bei 1 : 1 000 mit 5% BSA
in TBST über
Nacht in einem Schüttler
mit 80 Upm bei 4°C
inkubiert. Die Membranen werden 4 × jeweils 10 Minuten mit TBST
gewaschen. Die Membranen werden dann für 40 Minuten mit einem sekundären Antikörper-HRP
(Meerrettichperoxidase) Konjugat mit einer 1 : 10 000 Verdünnung in
3% Magermilch in TBST inkubiert und erneut viermal mit TBST für jeweils
10 Minuten gewaschen. Die Immunblots werden dann durch verstärkte Chemoluminszenz
(ECL, Amersham) gemäß den Anleitungen
des Herstellers sichtbar gemacht. Alle primären Antikörper werden von Cell Signaling
bezogen und sekundäre
Antikörper
HRP Konjugate werden von Amersham bezogen. Gele, Membranen und das
Gerät,
die zur Elektrophorese und zum Western Blot verwendet werden, werden
von Invitrogen bezogen. Proteinbanden von Interesse werden von Filmen
mittels Kodak Image Station 1000 quantifiziert.
-
Die
Verbindung von Beispiel 77 zeigt ausgezeichnete Dosis-abhängige Aktivitäten gegen
p38 MAPK in Tumoren, die 2,5 Stunden nach der Dosierung geerntet
werden, wie dies als Dosis-abhängige
Hemmung der MAPKAPK-2 Phosphorylierung beobachtet wird. MAPKAPK-2
ist ein physiologisches Substrat der p38 MAPK. Eine beträchtliche
Hemmung der MAPKAPK-2 Phosphorylierung durch die Verbindung von
Beispiel 77 wird in Dosen bis zu 0,3 mg/kg beobachtet und die ANOVA
TMED 50 wird mit 3 mg/kg bestimmt. Die Verbindung von Beispiel 77
verringert den Phosphorylierungsgrad von MAPKAPK-2 in den Tumoren
um > 80% (P < 0,001) bei Dosen
von 30 mg/kg. Im Zeitverlauf der Studie wird die Verbindung von
Beispiel 77 mit 30 mg/kg verabreicht und die Tumoren werden zu verschiedenen
Zeitintervallen nach einer Dosierung für bis zu 18 Stunden herausgeschnitten.
Eine Western Blot Analyse der MAPKAPK-2 Phosphorylierung zeigt,
dass die Verbindung von Beispiel 77 die p38 MAPK Aktivität für bis zu
9 Stunden hemmt. 12 Stunden nach der Dosierung geht das Maß der MAPKAPK-2
Phosphorylierung in den behandelten Tumoren auf ein ähnliches
Maß wie
in der Trägerkontrolle
zurück.
-
Die
Verbindung von Beispiel 77 wird im B16F10 Melanom Lungenmetastasemodell
oral mit 3, 10 und 30 mg/kg dreimal am Tag von 1 Tag vor bis 16
Tage nach der i.v. Tumorinjektion getestet (Gesamtdosis 9, 30 und
90 mg/kg/Tag). Die Lungen werden am Tag 16 entnommen und die Lungenläsionen werden
quantifiziert. Die Verbindung von Beispiel 77 verursacht jeweils
54%, 73% und 95% Hemmung der Lungenmetastasenbildung jeweils für die 3,
10 und 30 mg/kg Dosismengen (p < 0,001
bei allen drei Dosismengen).
-
P815 Tumormodell
-
Weibliche
(6-8 Wochen alte) DBA/2 Mäuse
(Taconic) werden subkutan in die hintere Flankenregion am Tag 0
mit P815 Zellen (0,5 × 106 Zellen in 200 μl RPMI 1640) implantiert. P815
Tumorzellen werden von der ATCC bezogen und in RPMI 1640 Medium,
das mit Glutamin und 10% Rinderserumalbumin supplementiert ist,
bei 37°C
und in einem 5% CO2 Zellkulturinkubator
inkubiert. Tumortragende Tiere werden durch die orale Verabreichung
der Verbindung von Beispiel 77 (freie Base) bei unterschiedlichen
Dosen oder Trägern
mit einer Häufigkeit
von dreimal am Tag beginnend am Tag der Implantierung behandelt.
Das Tumorwachstum wird alle 2 Tage durch Messung der senkrechten
Durchmesser gemessen. Das in Milligramm (mg) ausgedrückte Tumorvolumen
wird als Produkt des größten Durchmessers
(a) und dessen Senkrechte (b) gemäß der Formel [Tumorvolumen
= a × b2 × 0,536]
bestimmt.
-
In vivo Zielhemmungsstudie
im P815 Mastocytommodell
-
Die
in vivo Zielhemmung wird durch Messen des Effekts der Inhibitorbehandlung
auf die Phosphorylierung von MAPKAP-K2 bestimmt, das in P815 Tumorgeweben
exprimiert wird. Tumoren in DBA/2 Mäusen erhalten eine subkutane
Implantierung mit P815 Zellen und können ohne Behandlung bis auf
eine Größe von 300-500
mg wachsen. Den Tumor-tragenden Mäusen wird dann eine orale Verabreichung
der Verbindung von Beispiel 77 (freie Base) oder eines Trägers gegeben.
Um die zeitabhängige
Zielhemmung durch die Verbindung von Beispiel 77 (freie Base) zu
untersuchen, werden die Tumoren aus CO2 getöteten Tieren
zu den angegebenen Zeiten (3 Stunden, 6 Stunden, 12 Stunden und
18 Stunden) nach der Dosierung der Verbindung mit 30 mg/kg entnommen.
Die Dosis-abhängige
Zielhemmung durch die Verbindung von Beispiel 77 (freie Base) wird
durch die Entnahme der Tumoren 3 Stunden nach der oralen Gabe von
verschiedenen Dosen der Verbindung von Beispiel 77 (freie Base)
oder des Trägers untersucht.
Die entnommenen Tumoren werden unmittelbar auf Trockeneis schockgefroren,
pulverisiert, homogenisiert und in gekühltem Lysepuffer lysiert, der
Proteinase- und Phosphataseinhibitoren enthält. Nach der Zentrifugation
zur Entfernung des Zelldebris werden die Überstände, die 100 μg Gesamtproteine
enthalten, in 2 × Tris-Glycin
Auftragepuffer resuspendiert und einer Natriumdodecylsulfat-Polyacrylamidgelelektrophorese
(10% Tris-Glycin) unter reduzierenden Bedingungen unterzogen. Die
Proteine werden anschließend
auf eine PDVF Membran geblottet und werden dann in 5% Milch PBS,
worin 0,1% Tween 20 enthalten sind, für 1 Stunde bei Raumtemperatur
blockiert. Die Membran wird dann mit primärem Antikörper (anti-Phospho-MAPKAP-K2,
Cell Signalliing) bei 4°C über Nacht,
gefolgt von einer Inkubation mit sekundärem Antikörper (anti-Kaninchen HRP-konjugiertes
IgG) bei Raumtemperatur für
1 Stunde inkubiert.
-
Das
Phospho-MAPKAP-K2 Expressionsniveau wird durch das Phospho-Image
Detektionssystem visualisiert, nachdem die verstärkte Chemilumineszenz (ECL)
Detektion verwendet wurde, um das Vorkommen der Proteine auf den
PVDF Blots zu reflektieren. Das Expressionsniveau der Phospho-p38
MAP Kinase und der Gesamt p38 MAP Kinase wird auch durch ein ähnliches
Westernblotverfahren verfolgt.
-
Zeit- und dosisabhängige Hemmung
der MAPKAP-K2 Phsophorylierung druch die Verbindung von Beispiel
77 (freie Base) bei P815 Tumoren in vivo
-
Die
Wirkung der oralen Verabreichung einer Einzeldosis der Verbindung
von Beispiel 77 (freie Base) auf die MAPKAP-K2 Phosphorylierung
wird in DBA/2 Mäusen
evaluiert, die P815 Tumoren tragen. Die Verabreichung von Beispiel
77 (freie Base) mit 30 mg/kg verursacht eine zeitabhängige Hemmung
der MAPKAP-K2 Phosphorylierung. Der hemmende Effekt der Verbindung
tritt schon 3 Stunden nach der Dosierung der Tiere auf und erstreckt
sich über
12 Stunden. Es ist auch ersichtlich, dass die Phosphorylierung von
MAPKAP-K2 18 Stunden nach der Dosierung vollständig wiederhergestellt ist.
Wenn den P815 Tumor tragenden Tieren die Verbindung von Beispiel
77 (freie Base) in verschiedenen Dosen (3, 10 und 30 mg/kg) verabreicht
wird und ihre Tumoren zur Messung der Proteinexpression 3 Stunden
nach der Dosierung entnommen werden, wird eine signifikante Dosis-abhängige Hemmung
der MAPKAP-K2 Phosphorylierung gezeigt. In den Tierversuchen verändert die
Verbindung von Beispiel 77 (freie Base) weder die p28 MAP Kinasephosphorylierung
noch die p38 MAP Kinaseexpression in vivo.
-
Hemmung des P815 Tumorwachstums
in vivo
-
Die
in vivo Aktivität
der Verbindung von Beispiel 77 (freie Base) wird auf das Wachstum
von P815 Tumoren untersucht, die subkutan in DBA/2 Mäuse transplantiert
werden. Eine orale Verabreichung der Verbindung von Beispiel 77
(freie Base) dreimal am Tag mit Dosen zwischen 0,1 und 30 mg/kg
führt zu
einer Dosis-abhängigen
Hemmung des P815 Tumorwachstums mit einer 67% Hemmung des Tumorvolumens
bei 30 mg/kg (p < 0,01,
drei unabhängige
Studien), 55% Hemmung bei 3 mg/kg (p < 0,05, 2 unabhängige Studien) und 30 bis 14%
Hemmung bei geringeren Dosen.
-
Kollagen-induziertes Arthritiswirksamkeitsmodell
der Ratte
-
Weibliche
Lewis Ratten (etwa 190 g, Charles River Labs) werden mit Rinderkollagen
vom Typ II (2 mg/ml) immunisiert, das mit einem gleichen Volumen
Adjuvans (Aluminiumhydroxid) emulgiert ist. Die Ratten werden mit
etwa 0,3 mg der Emulsion intradermal am Rücken nahe der Schwanzbasis
immuni siert. Alle Tiere werden 7 Tage später gemäß demselben Protokoll immunisiert.
Die Ratten beginnen eine Arthritis (gekennzeichnet durch Schwellung
und Rötung
eines oder beider Knöchel)
12 bis 14 Tage nach der ersten Immunisierung zu entwickeln. Die
Ratten werden gleichmäßig in 5
Behandlungsgruppen bei den ersten Anzeichen von Arthritis eingeteilt
und die Behandlung wird initiiert, wobei jede Ratte zweimal am Tag
für 14
Tage die Dosis erhält.
-
Behandlungsgruppen:
-
- Gruppe 1: Träger
(1% Na-Carboxymethylcellulose + 0,25% Tween 80) 1 ml, PO, zweimal
täglich
für 14
Tage
- Gruppe 2: Beispiel 77 (freie Base), 5 mg/kg, 1 ml, PO, zweimal
täglich
für 14
Tage
- Gruppe 3: Beispiel 77 (freie Base), 15 mg/kg, 1 ml, PO, zweimal
täglich
für 14
Tage
- Gruppe 4: Beispiel 77 (freie Base), 30 mg/kg, 1 ml, PO, zweimal
täglich
für 14
Tage
- Gruppe 5: Prednisolon 10 mg/kg, 1 ml, PO, zweimal täglich für 14 Tage
-
Der
Gelenkdurchmesser wird mit Schublehren 5 Tage pro Woche gemessen
und aufgezeichnet. Die Daten werden als Fläche unter der Kurve (AUC) exprimiert,
die von den gesammelten Entzündungsbewertungen
erzeugt wird und es wird eine statistische Analyse ausgeführt. Bei
allen Dosen der Verbindung von Beispiel 77 zeigt sich eine signifikante
Verringerung im Gelenkdurchmesser mit einer maximalen Verringerung
von 46% bei 30 mg/kg (p < 0,0001).
Prednisolon verringert die Entzündung
auf das Niveau vor der Arthritis.
-
Am
Ende der Studie werden die Tiere getötet und die Gelenke werden
einer histologischen Evaluierung unterzogen. Die Histologiebewertung
basiert auf einer 0 bis 5 Skala für jeden der 4 Parameter (Entzündung, Pannus,
Knorpelschädigung,
Knochenresorption). Bei Tieren, die mit der Verbindung von Beispiel
77 behandelt wurden, wird eine Dosis-abhängige Hemmung der Gelenkspathologie
beobachtet. Es wird eine signifikante Reduktion in den Gesamtgelenksbewertungen
bei Tieren erhalten, die mit der Verbindung von Beispiel 77 mit
15 und 30 mg/kg behandelt wurden (jeweils p < 0,03 und p = 0,0031).
-
Die
orale Verabreichung der erfindungsgemäßen Verbindungen ist bevorzugt.
Jedoch ist die orale Verabreichung nicht der einzige Weg oder der
einzig bevorzugte Weg. Beispielsweise kann eine transdermale Verabreichung
für Patienten
sehr erwünscht
sein, die bezüglich
der Einnahme von oralen Arzneimitteln vergesslich oder verärgert sind
und der intravenöse
Weg kann aus Komfort oder zur Vermeidung von Komplikationen, die
mit einer oralen Verabreichung verbunden sind, bevorzugt sein. Die
Verbindungen der Formel I können
auch unter bestimmten Umständen
durch den perkutanen, intramuskulären, intranasalen oder intrarektalen
Weg verabreicht werden. Der Verabreichungsweg kann auf jede Weise
variiert werden, eingeschränkt durch
die physikalischen Eigenschaften der Arzneimittel, den Komfort des
Patienten und des Pflegepersonals und anderer relevanter Umstände (Remington's Pharmaceutical
Sciences, 18. Ausgabe, Mack Publishing Co. (1990)).
-
Die
pharmazeutischen Zusammensetzungen werden auf eine in der pharmazeutischen
Technik gut bekannte Weise hergestellt. Der Träger oder der Hilfsstoff kann
ein festes, halbfestes oder flüssiges
Material sein, das als Vehikel oder Medium für den Wirkstoff dient. Geeignete
Träger
oder Hilfsstoffe sind in der Technik gut bekannt. Die pharmazeutische
Zusammensetzung kann zur oralen, inhalativen, parenteralen oder
topischen Verwendung angepasst werden und kann in Form von Tabletten,
Kapseln, Aero solen, Inhalationsmitteln, Zäpfchen, Lösungen, Suspensionen oder dergleichen
an den Patienten verabreicht werden.
-
Die
erfindungsgemäßen Verbindungen
können
oral verabreicht werden, beispielsweise mit einem inerten Verdünnungsmittel
für Kapseln
oder zu Tabletten verpresst werden. Zum Zweck der oralen therapeutischen
Verabreichung können
die Verbindungen mit Hilfsstoffen eingearbeitet werden und in Form
von Tabletten, Dragees, Kapseln, Elixieren, Suspensionen, Sirupen,
Obladen, Kaugummis und dergleichen verwendet werden. Diese Präparationen
sollten mindestens 4% der erfindungsgemäßen Verbindung, dem Wirkstoff
enthalten, aber dies kann in Abhängigkeit
der bestimmten Form variieren und kann zwischen 4% bis etwa 70% des
Gewichts der Einheit betragen. Die Menge der in den Zusammensetzungen
vorhandenen Verbindung ist so groß, dass eine geeignete Dosierung
erhalten wird. Bevorzugte Zusammensetzungen und Präparationen gemäß der vorliegenden
Erfindung können
durch den Fachmann bestimmt werden.
-
Die
Tabletten, Pillen, Kapseln, Dragees und dergleichen können auch
einen oder mehrere der folgenden Hilfsstoffe enthalten: Bindemittel,
wie Povidon, Hydroxypropylcellulose, mikrokristalline Cellulose
oder Gelatine, Hilfsstoffe oder Verdünnungsmittel wie: Stärke, Lactose,
mikrokristalline Cellulose oder Dicalciumphosphat, Zerfallshilfsstoffe,
wie: Croscarmellose, Crospovidon, Natriumstärkeglycolat, Maisstärke und
dergleichen, Schmiermittel, wie: Magnesiumstearat, Stearinsäure, Talkum
oder hydriertes Pflanzenöl:
Gleitmittel, wie kolloidales Siliziumdioxid, Netzmittel, wie: Natriumlaurylsulfat
und Polysorbat 80 und Süßstoffe,
wie Saccharose, Aspartam oder Saccharin können zugegeben werden oder
ein Geschmacksstoff, wie: Pfefferminze, Methylsalicylat oder Orangenaroma.
Wenn die Dosierungsform eine Kapsel ist, kann sie zusätzlich zu
Materialien des obigen Typs einen flüssigen Träger, wie Polyethylenglycol
oder ein fettes Öl
enthalten. Andere Einheitsdosierungsformen können andere verschiedene Materialien
enthalten, die die physikalische Form der Dosierungseinheit modifizieren,
wie beispielsweise als Beschichtungen. Daher können Tabletten oder Pillen
mit Zucker, Hydroxypropylmethylcellulose, Polymethacrylaten oder
anderen Beschichtungsmitteln beschichtet werden. Sirupe können zusätzlich zu
den vorliegenden Verbindungen Saccharose als Süßstoff und bestimmte Konservierungsstoffe,
Farbstoffe und Färbungen
und Geschmacksstoffe enthalten. Materialien, die zur Herstellung
dieser verschiedenen Zusammensetzungen verwendet werden, sollten
pharmazeutisch rein und in den verwendeten Mengen nicht toxisch
sein.
-
Die
Verbindungen der Formel I sind im allgemeinen über einen weiten Dosierungsbereich
wirksam. Beispielsweise fallen Tagesdosierungen normalerweise in
den Bereich von etwa 0,0001 bis etwa 30 mg/kg Körpergewicht. In einigen Fällen können Dosierungsmengen,
die unter der unteren Grenze des vorher erwähnten Bereichs liegen, passender
sein, während
in anderen Fällen
höhere
Dosen ohne eine Verursachung von schädlichen Nebenwirkungen verwendet
werden können
und daher soll der obige Dosierungsbereich den Umfang der Erfindung
in keiner Weise beschränken.
Es ist verständlich,
dass die Menge an tatsächlich
verabreichter Verbindung vom Arzt in Anbetracht der relevanten Umstände bestimmt
wird, einschließlich
des zu behandelnden Zustands, des gewählten Verabreichungswegs, der
im einzelnen verabreichten Verbindung oder Verbindungen, dem Alter,
dem Gewicht und der Reaktion des individuellen Patienten und der
Schwere der Symptome des Patienten.

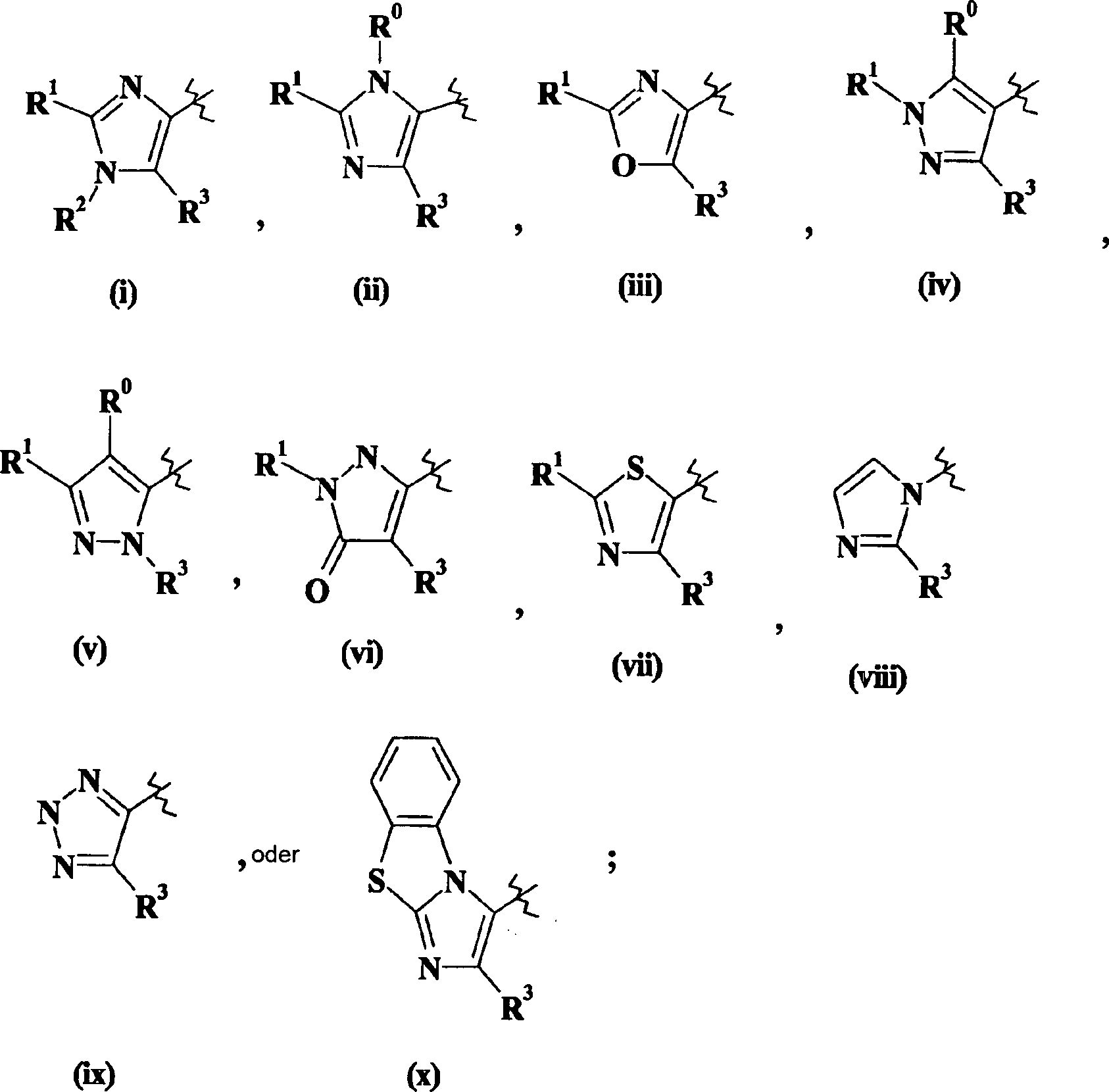







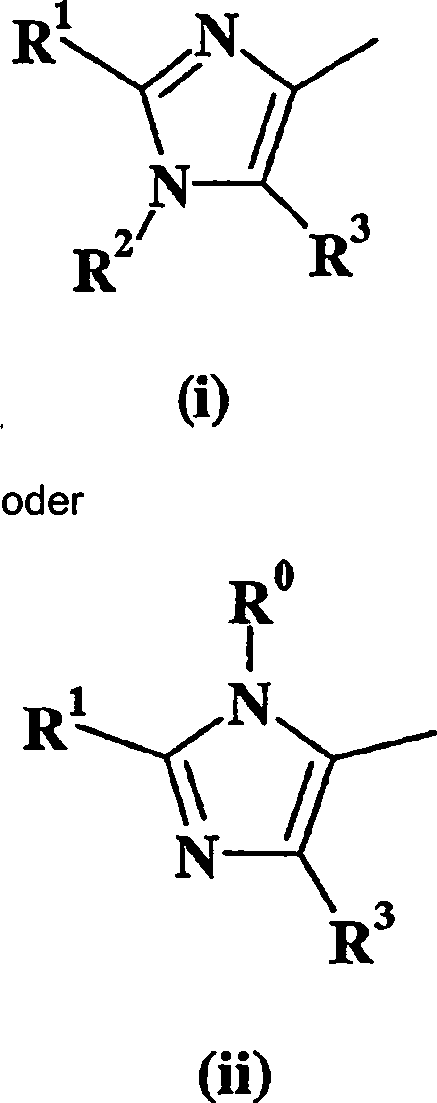


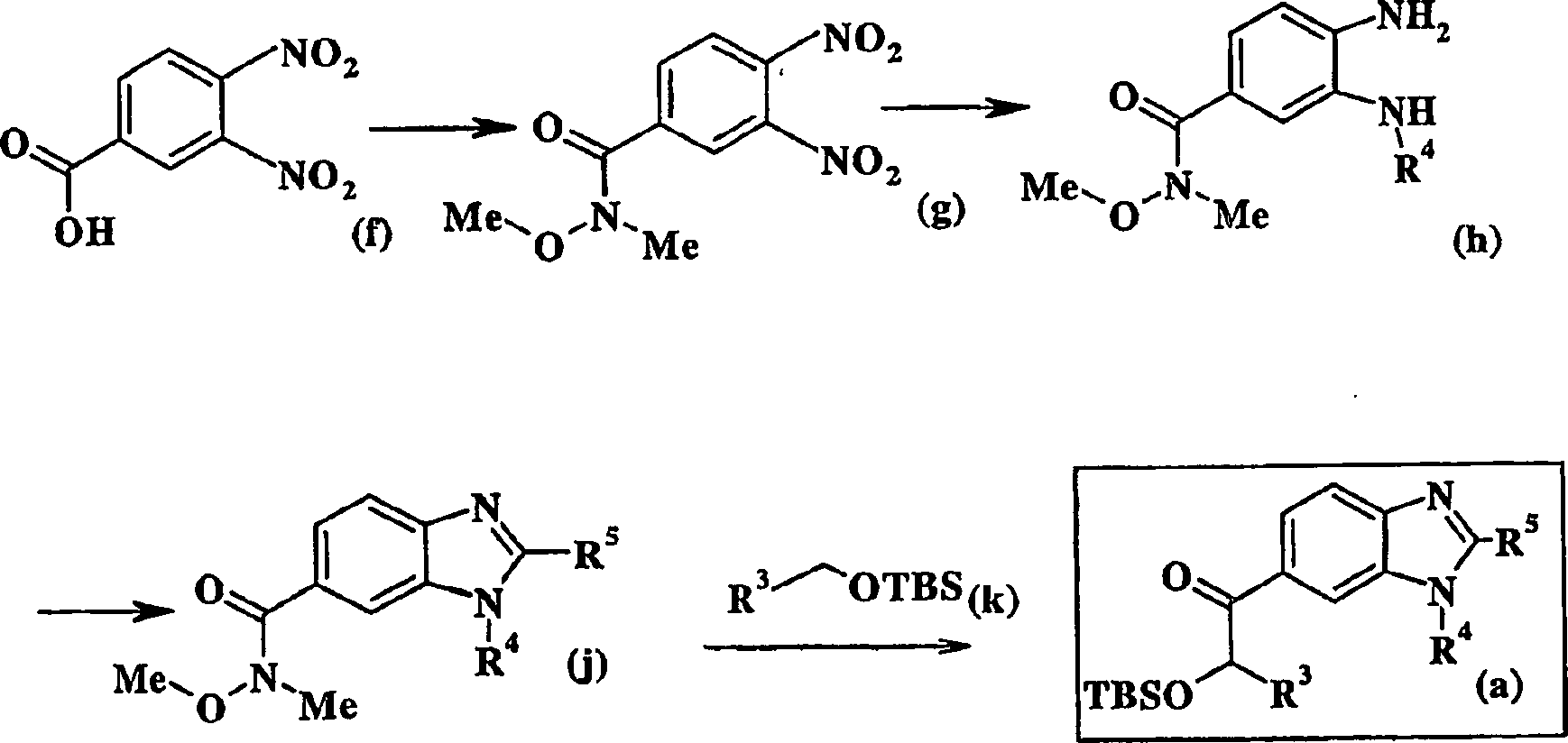







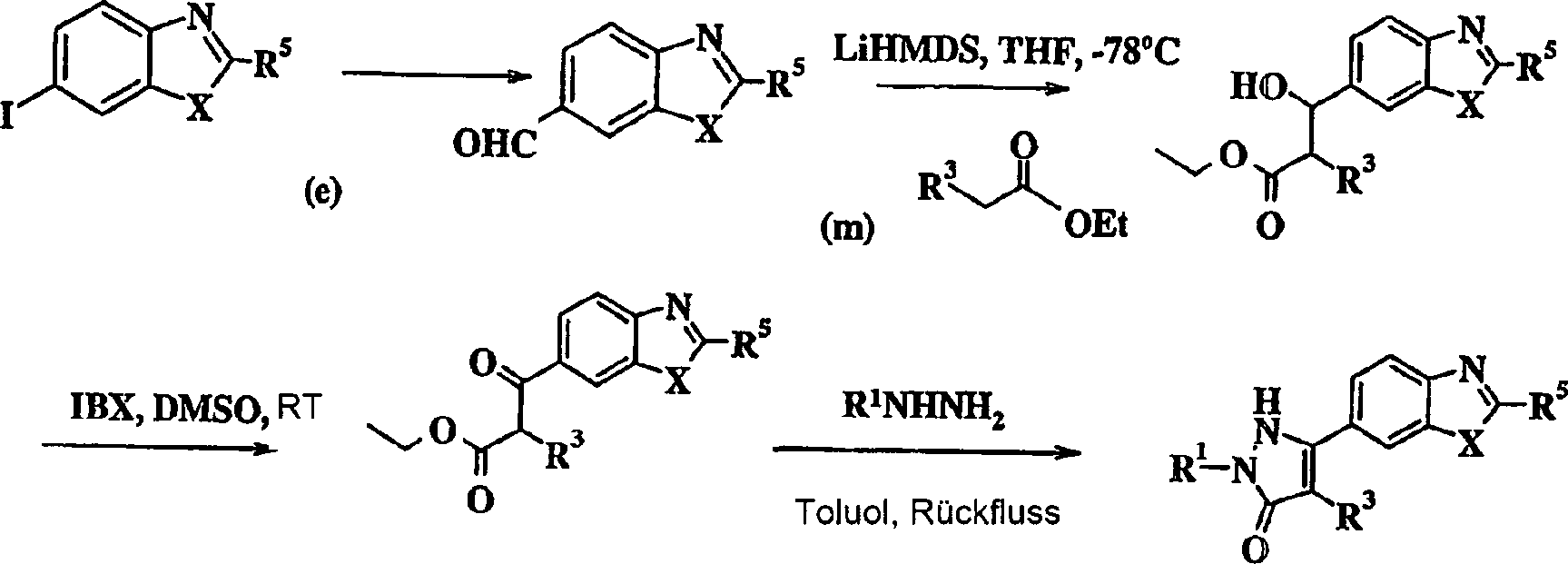






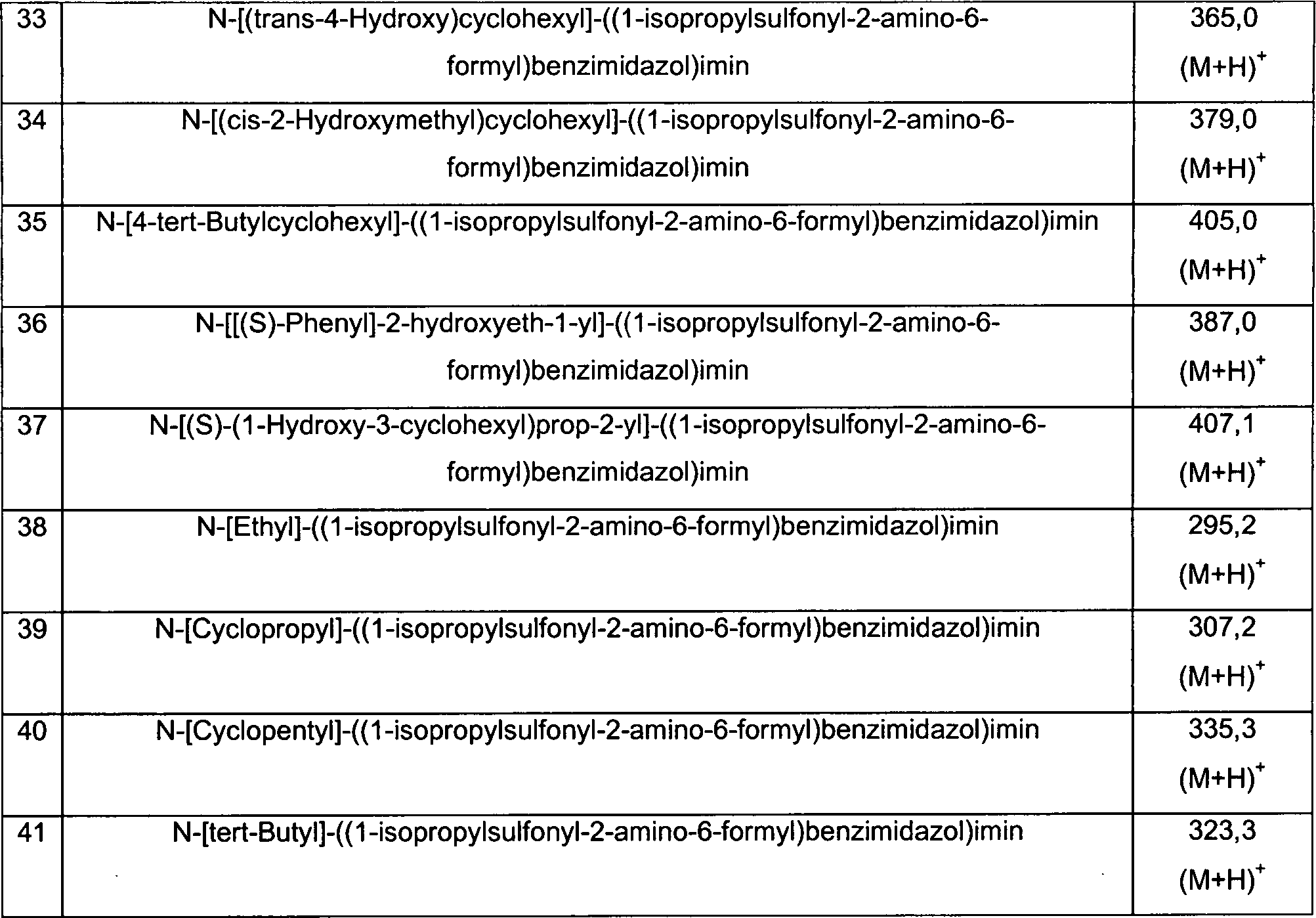

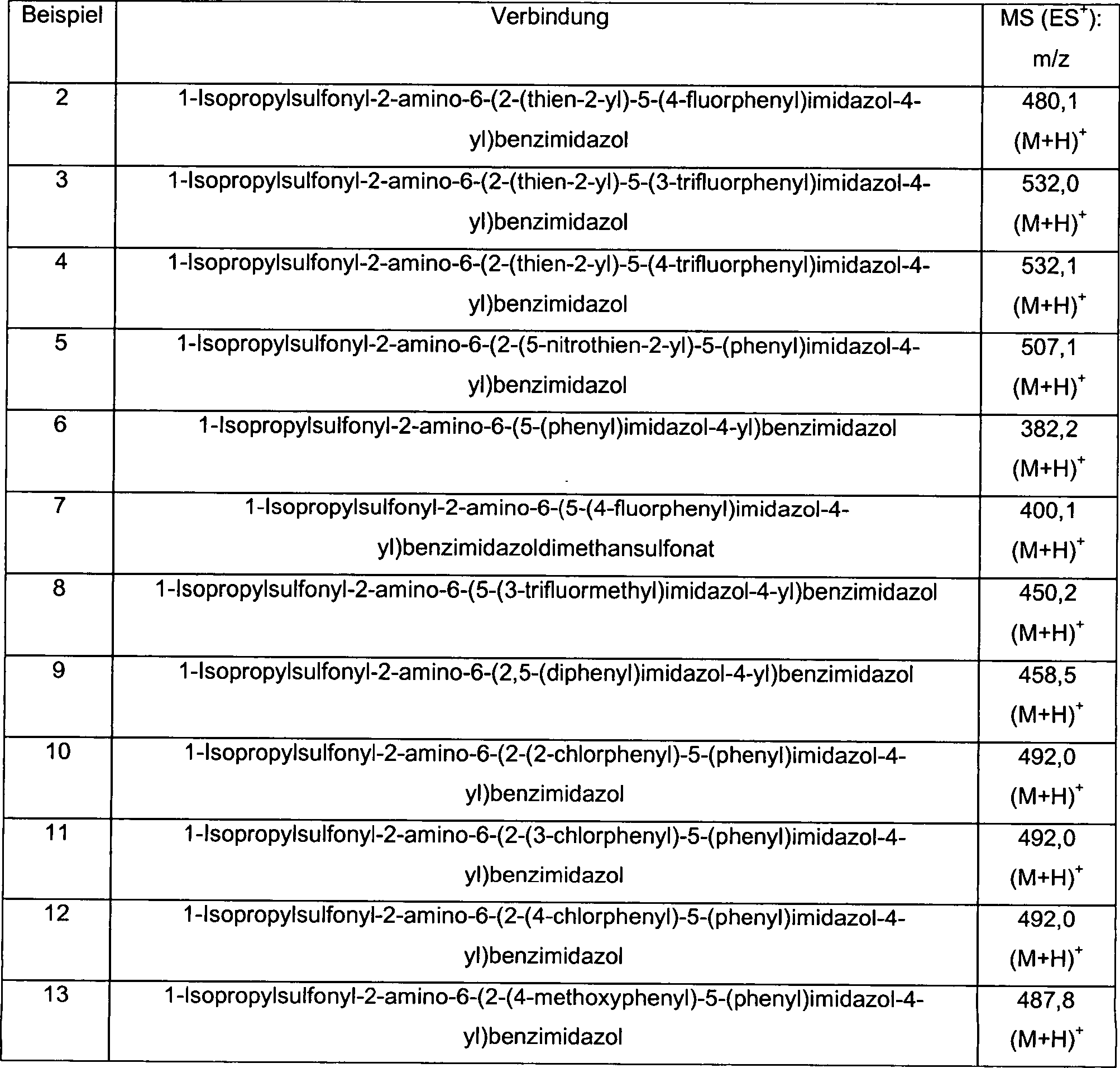



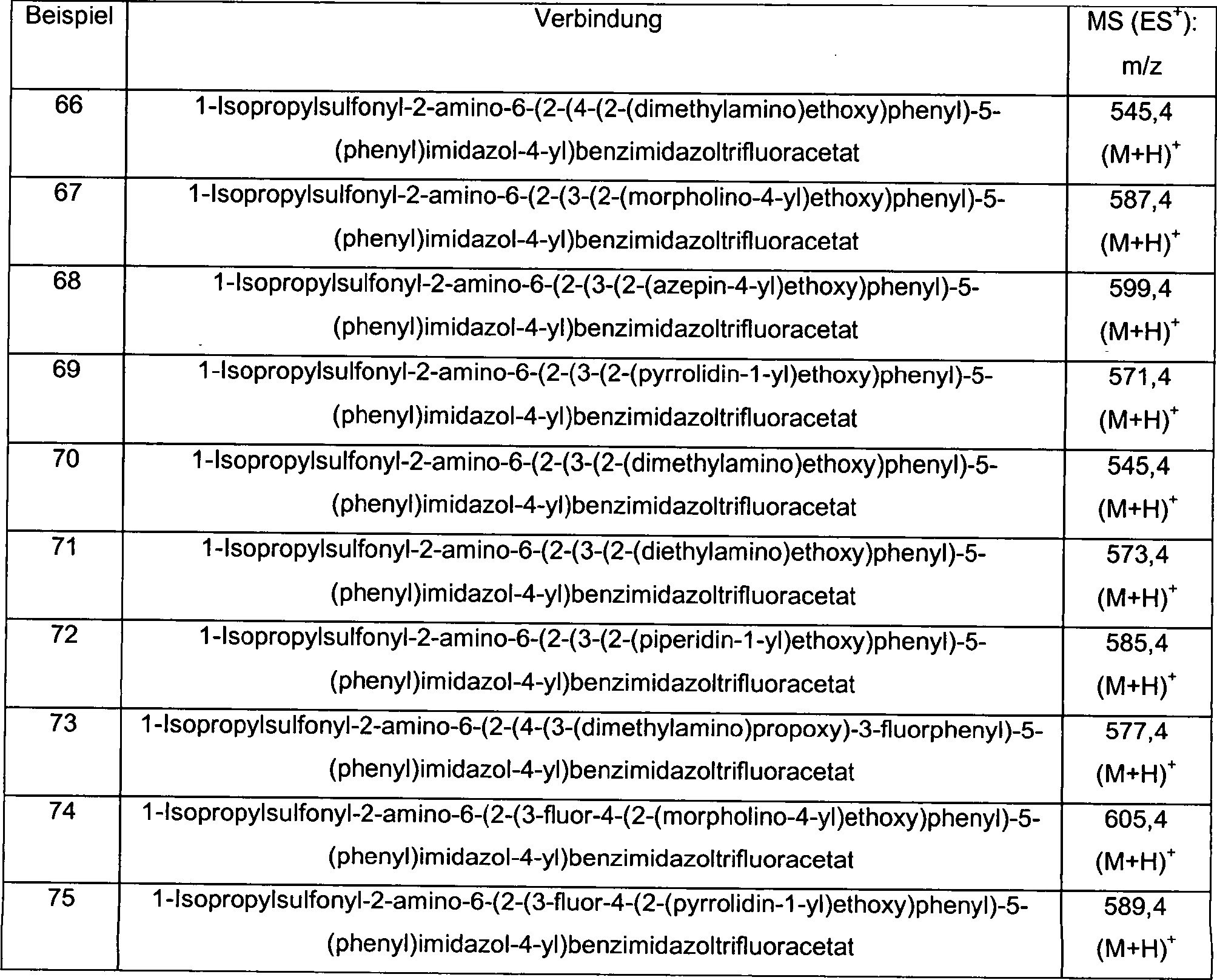
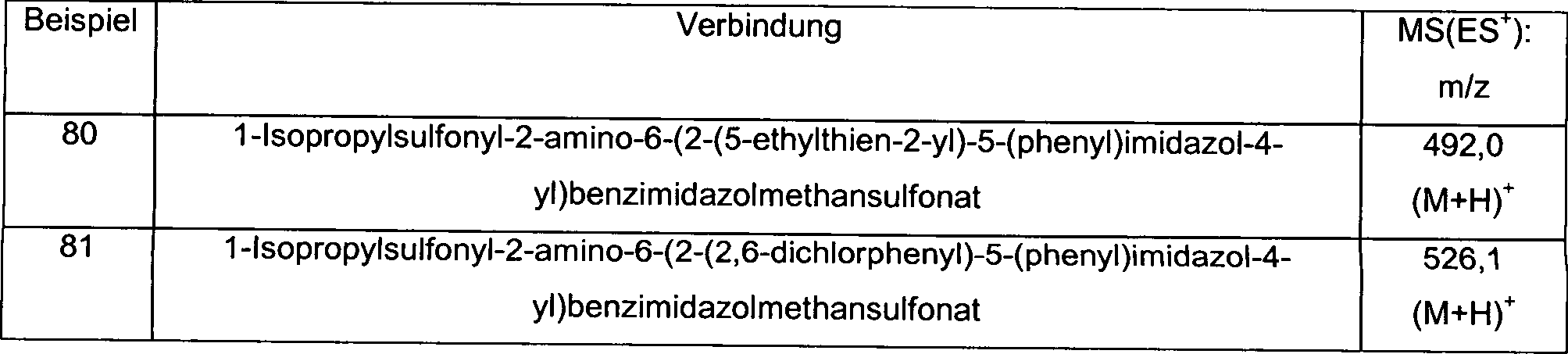



















 X für N(R4) oder S steht, R0 ist (a) ausgewählt aus der Gruppe, die besteht aus Wasserstoff, C1-C6 Alkyl, Cyano, C1-C4 Alkylen-R11, 3-Hydroxyprop-2-yl, (1-Phenyl)-2-hydroxyeth-1-yl, (1-Cyclohexyl)-3-hydroxyprop-2-yl, 4-Methoxybenzyl, 1,4-Dioxoaspiro[4,5]dec-8-yl, Tetrahydropyran, 2,2,6,6-Tetramethylpiperidin-4-yl und Cyclohexan-1-on-4-yl, (b) Phenyl, das optional mit einem Substituenten substituiert ist, der aus der Gruppe ausgewählt ist, welche besteht aus Nitro und Amino, (c) Piperidin-4-yl, das optional mit einem Substituenten substituiert ist, der aus der Gruppe ausgewählt ist, welche besteht aus C1-C4 Alkyl, C1-C4 Alkoxycarbonyl und Benzyl oder (d) C3-C6 Cycloalkyl, das optional mit einem Substituenten substituiert ist, der aus der Gruppe ausgewählt ist, welche besteht aus C1-C4 Alkoxycarbonylamino, Amino, Hydroxy und C1-C4 Alkylen-OH, R1 ist (a) ausgewählt aus der Gruppe, die besteht aus Wasserstoff, C1-C6 Alkyl, C2-C4 Alkinyl, Halogen, Amino, Azido, Formyl, 1-(C1-C4 Alkoxycarbonyl)ethen-2-yl, 1-(C1-C4 Alkoxycarbonyl)ethyl, 1-(C1-C4 Carboxy)ethyl, (C1-C4 Alkylen)benzyloxy, Trifluormethyl, Trimethylsilylethinyl, But-3-in-1-ol, C3-C6 Cycloalkyl, Tetrahydropyran-4-yl, Hydroxymethyl, 2-(Piperidin-1-yl)methyl, N,N',N'-[Trimethyl]-2-(aminoethylamino)methyl, (Morpholin-4-yl)methyl, Dimethylaminomethyl, N-[2-(Piperidin-1-yl)eth-1-yl]aminomethyl, N',N'-Dimethyl-2-(aminoethylamino)methyl, Pyridinyl, Thiazolyl, Triazolyl, Benzo(1,3)dioxolan-5-yl und Imidazol-2-yl, (b) Phenyl, das optional mit einem bis drei Substituenten substituiert ist, die unabhängig aus der Gruppe ausgewählt sind, die besteht aus C1-C4 Alkyl, Halogen, Nitro, Amino, C1-C4 Alkoxy, Trifluormethyl, Trifluormethoxy, Trifluormethylsulfanyl, Methylsulfonyl, Methylsulfonamidyl, Pyrrolidin-1-yl, Morpholin-4-yl, 4-(C1-C4 Alkyl)piperazin-1-yl, -NR6R7 und C1-C4 Alkoxy, das optional mit einem Substituenten substituiert ist, der aus der Gruppe ausgewählt ist, welche besteht aus Piperidin-1-yl, Pyrrolidin-1-yl, Morpholin-4-yl, Azepin-4-yl und Di(C1-C4 alkyl)amino, (c) Thienyl, das optional mit einem Substituenten substituiert ist, der aus der Gruppe ausgewählt ist, die besteht aus Halogen, Nitro, Amino und C1-C4 Alkyl oder (d) Piperidin-4-yl, das optional an der Position 1 aus der Gruppe substituiert ist, die aus C1-C4 Alkyl, C1-C4 Alkoxycarbonyl, Benzyloxycarbonyl und (C1-C4 Alkylen)-R8 besteht, wobei alternativ dazu R0 und R1 unter Bildung einer vollkommen gesättigten C3-C4 Kohlenstoffkette oder einer vollkommen ungesättigten C3-C4 Kohlenstoffkette zusammengenommen werden können, die optional mit Halogen oder C1-C4 Alkyl substituiert ist, R2 für Wasserstoff, C1-C4 Alkyl oder Benzyl steht, R3 für Thienyl oder Phenyl steht, das wahlweise mit einem bis zwei Substituenten substituiert ist, die unabhängig aus der Gruppe ausgewählt sind, welche besteht aus Halogen, C1-C4 Alkyl, C1-C4 Alkoxy und Trifluormethyl, R4 für Wasserstoff, (C1-C4 Alkyl)sulfonyl oder (C3-C6 Cycloalkyl)sulfonyl oder (C1-C4 Alkyl)2-N-sulfonyl steht, R5 für Halogen, Wasserstoff oder -NR9R10 steht, R6 und R7 jeweils einzeln aus Wasserstoff, Carbonyl oder C1-C4 Alkyl ausgewählt sind, mit der Maßgabe, dass zumindest eines von R6 und R7 für Wasserstoff steht, R8 für Hydroxy, Trifluormethyl, Dimethylamino, Phenyl, Pyridinyl oder 1-Methylimidazol-2-yl steht, R9 jeweils unabhängig für Wasserstoff oder C1-C4 Alkyl steht, R10 für Wasserstoff, C1-C4 Alkyl oder Benzyl steht, R11 für C1-C4 Alkoxy, Hydroxy, C1-C4 Alkoxycarbonyl, C1-C4 Alkoxycarbonylamino, C3-C6 Cycloalkyl, Phenyl, das optional mit einem oder zwei Substituenten substituiert ist, die unabhängig aus der Gruppe ausgewählt sind, die aus C1-C4 Alkoxy und Halogen besteht, für Morpholin-4-yl oder Pyridinyl steht, mit der Maßgabe, dass wenn W für
X für N(R4) oder S steht, R0 ist (a) ausgewählt aus der Gruppe, die besteht aus Wasserstoff, C1-C6 Alkyl, Cyano, C1-C4 Alkylen-R11, 3-Hydroxyprop-2-yl, (1-Phenyl)-2-hydroxyeth-1-yl, (1-Cyclohexyl)-3-hydroxyprop-2-yl, 4-Methoxybenzyl, 1,4-Dioxoaspiro[4,5]dec-8-yl, Tetrahydropyran, 2,2,6,6-Tetramethylpiperidin-4-yl und Cyclohexan-1-on-4-yl, (b) Phenyl, das optional mit einem Substituenten substituiert ist, der aus der Gruppe ausgewählt ist, welche besteht aus Nitro und Amino, (c) Piperidin-4-yl, das optional mit einem Substituenten substituiert ist, der aus der Gruppe ausgewählt ist, welche besteht aus C1-C4 Alkyl, C1-C4 Alkoxycarbonyl und Benzyl oder (d) C3-C6 Cycloalkyl, das optional mit einem Substituenten substituiert ist, der aus der Gruppe ausgewählt ist, welche besteht aus C1-C4 Alkoxycarbonylamino, Amino, Hydroxy und C1-C4 Alkylen-OH, R1 ist (a) ausgewählt aus der Gruppe, die besteht aus Wasserstoff, C1-C6 Alkyl, C2-C4 Alkinyl, Halogen, Amino, Azido, Formyl, 1-(C1-C4 Alkoxycarbonyl)ethen-2-yl, 1-(C1-C4 Alkoxycarbonyl)ethyl, 1-(C1-C4 Carboxy)ethyl, (C1-C4 Alkylen)benzyloxy, Trifluormethyl, Trimethylsilylethinyl, But-3-in-1-ol, C3-C6 Cycloalkyl, Tetrahydropyran-4-yl, Hydroxymethyl, 2-(Piperidin-1-yl)methyl, N,N',N'-[Trimethyl]-2-(aminoethylamino)methyl, (Morpholin-4-yl)methyl, Dimethylaminomethyl, N-[2-(Piperidin-1-yl)eth-1-yl]aminomethyl, N',N'-Dimethyl-2-(aminoethylamino)methyl, Pyridinyl, Thiazolyl, Triazolyl, Benzo(1,3)dioxolan-5-yl und Imidazol-2-yl, (b) Phenyl, das optional mit einem bis drei Substituenten substituiert ist, die unabhängig aus der Gruppe ausgewählt sind, die besteht aus C1-C4 Alkyl, Halogen, Nitro, Amino, C1-C4 Alkoxy, Trifluormethyl, Trifluormethoxy, Trifluormethylsulfanyl, Methylsulfonyl, Methylsulfonamidyl, Pyrrolidin-1-yl, Morpholin-4-yl, 4-(C1-C4 Alkyl)piperazin-1-yl, -NR6R7 und C1-C4 Alkoxy, das optional mit einem Substituenten substituiert ist, der aus der Gruppe ausgewählt ist, welche besteht aus Piperidin-1-yl, Pyrrolidin-1-yl, Morpholin-4-yl, Azepin-4-yl und Di(C1-C4 alkyl)amino, (c) Thienyl, das optional mit einem Substituenten substituiert ist, der aus der Gruppe ausgewählt ist, die besteht aus Halogen, Nitro, Amino und C1-C4 Alkyl oder (d) Piperidin-4-yl, das optional an der Position 1 aus der Gruppe substituiert ist, die aus C1-C4 Alkyl, C1-C4 Alkoxycarbonyl, Benzyloxycarbonyl und (C1-C4 Alkylen)-R8 besteht, wobei alternativ dazu R0 und R1 unter Bildung einer vollkommen gesättigten C3-C4 Kohlenstoffkette oder einer vollkommen ungesättigten C3-C4 Kohlenstoffkette zusammengenommen werden können, die optional mit Halogen oder C1-C4 Alkyl substituiert ist, R2 für Wasserstoff, C1-C4 Alkyl oder Benzyl steht, R3 für Thienyl oder Phenyl steht, das wahlweise mit einem bis zwei Substituenten substituiert ist, die unabhängig aus der Gruppe ausgewählt sind, welche besteht aus Halogen, C1-C4 Alkyl, C1-C4 Alkoxy und Trifluormethyl, R4 für Wasserstoff, (C1-C4 Alkyl)sulfonyl oder (C3-C6 Cycloalkyl)sulfonyl oder (C1-C4 Alkyl)2-N-sulfonyl steht, R5 für Halogen, Wasserstoff oder -NR9R10 steht, R6 und R7 jeweils einzeln aus Wasserstoff, Carbonyl oder C1-C4 Alkyl ausgewählt sind, mit der Maßgabe, dass zumindest eines von R6 und R7 für Wasserstoff steht, R8 für Hydroxy, Trifluormethyl, Dimethylamino, Phenyl, Pyridinyl oder 1-Methylimidazol-2-yl steht, R9 jeweils unabhängig für Wasserstoff oder C1-C4 Alkyl steht, R10 für Wasserstoff, C1-C4 Alkyl oder Benzyl steht, R11 für C1-C4 Alkoxy, Hydroxy, C1-C4 Alkoxycarbonyl, C1-C4 Alkoxycarbonylamino, C3-C6 Cycloalkyl, Phenyl, das optional mit einem oder zwei Substituenten substituiert ist, die unabhängig aus der Gruppe ausgewählt sind, die aus C1-C4 Alkoxy und Halogen besteht, für Morpholin-4-yl oder Pyridinyl steht, mit der Maßgabe, dass wenn W für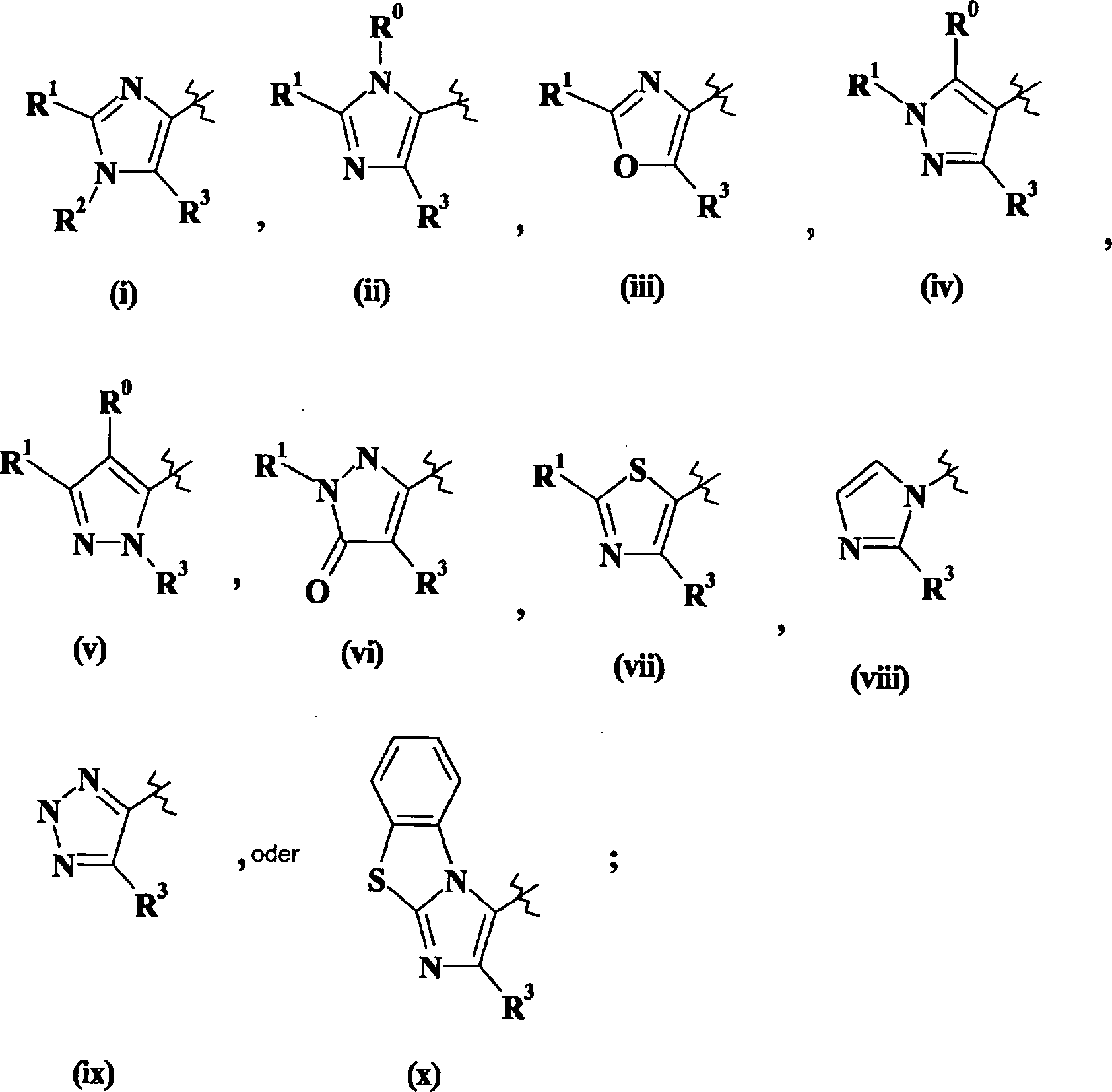 steht, dann folgendes gilt (a) zumindest eines von R0 und R1 für Wasserstoff oder C1-C6 Alkyl steht, oder (b) R0 und R1 unter Bildung einer vollkommen gesättigten C3-C4 Kohlenstoffkette oder einer vollkommen ungesättigten C3-C4 Kohlenstoffkette zusammen genommen werden können, die optional mit Halogen oder C1-C4 Alkyl substituiert ist, und ebenfalls mit der Maßgabe, dass wenn X für S steht, W dann steht für
steht, dann folgendes gilt (a) zumindest eines von R0 und R1 für Wasserstoff oder C1-C6 Alkyl steht, oder (b) R0 und R1 unter Bildung einer vollkommen gesättigten C3-C4 Kohlenstoffkette oder einer vollkommen ungesättigten C3-C4 Kohlenstoffkette zusammen genommen werden können, die optional mit Halogen oder C1-C4 Alkyl substituiert ist, und ebenfalls mit der Maßgabe, dass wenn X für S steht, W dann steht für oder ein pharmazeutisch annehmbares Salz oder ein pharmazeutisch annehmbares Solvat hiervon.
oder ein pharmazeutisch annehmbares Salz oder ein pharmazeutisch annehmbares Solvat hiervon.
