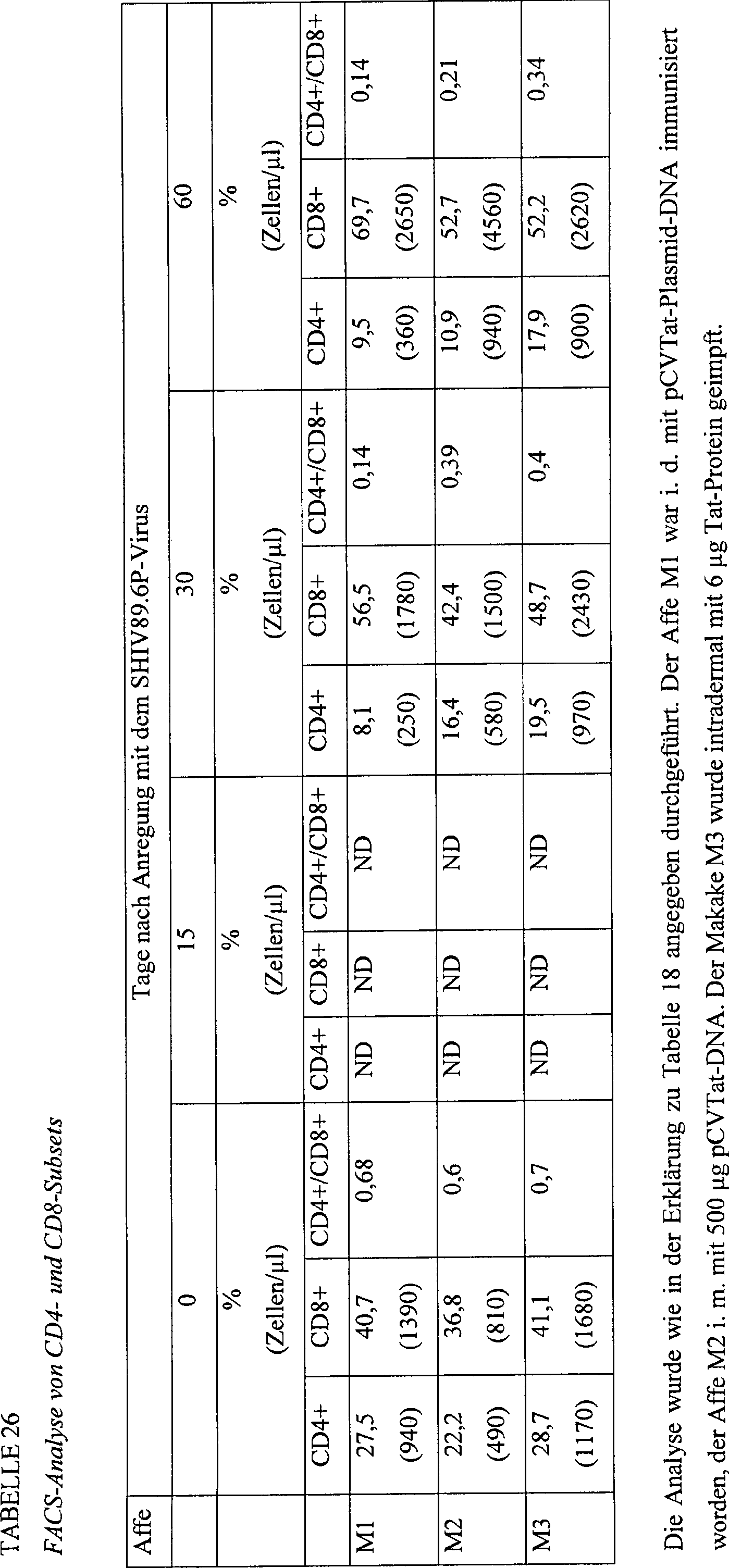
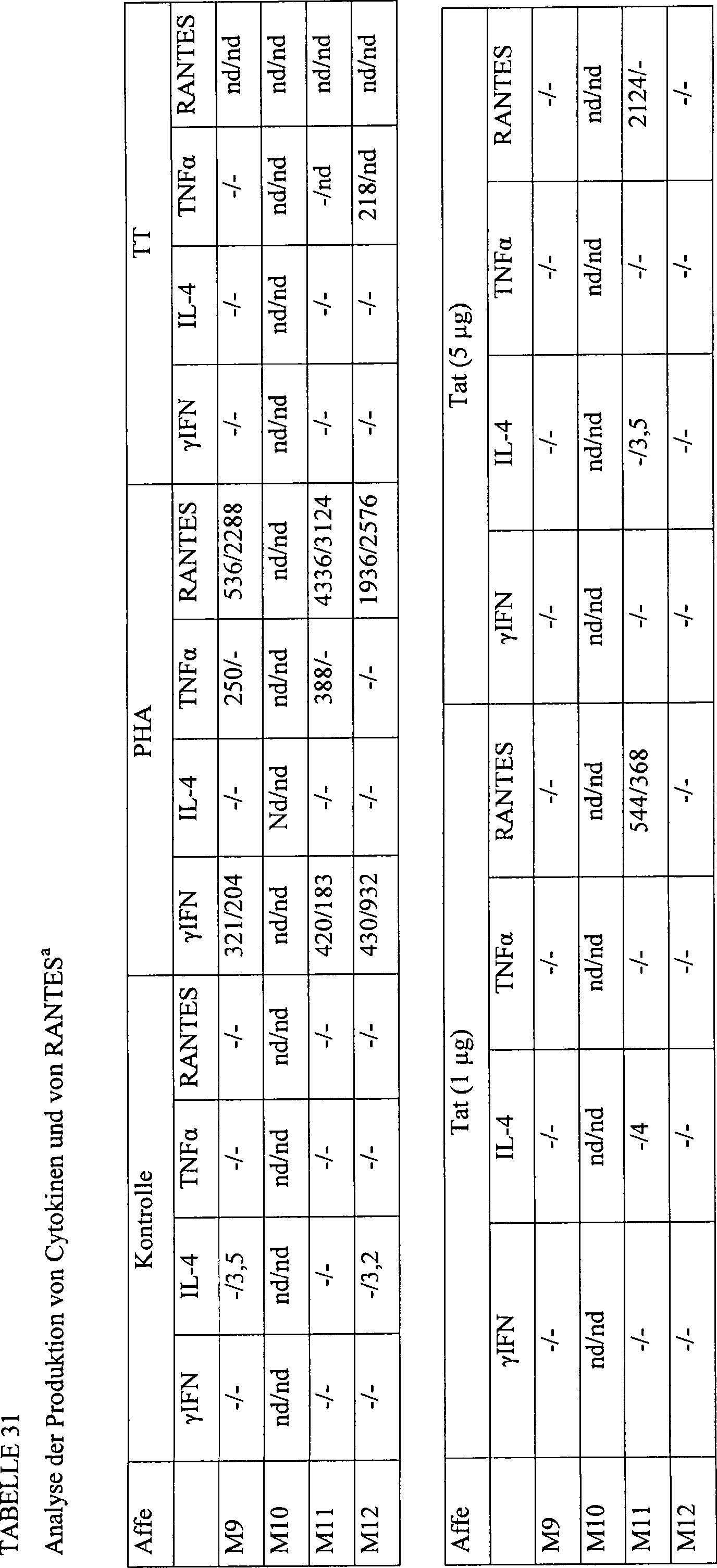
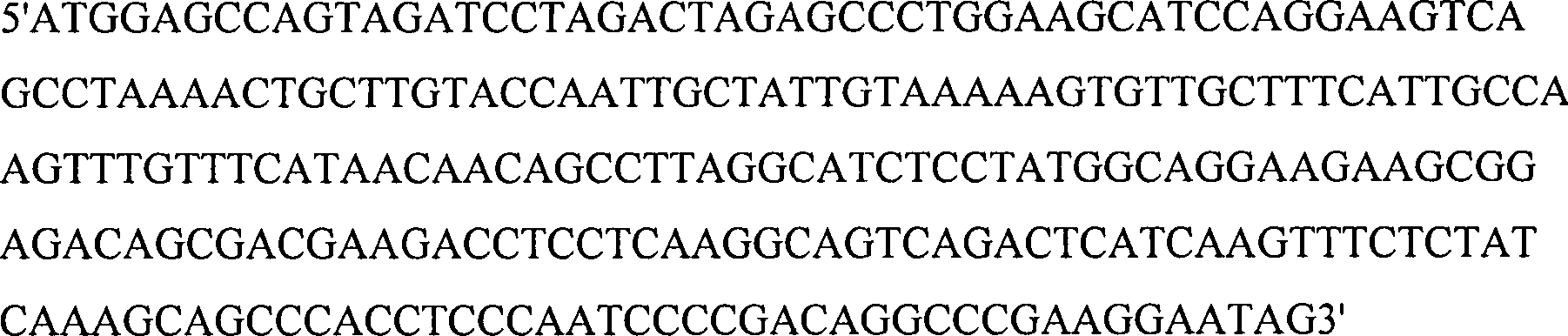-
Gebiet der Erfindung
-
Die
vorliegende Erfindung bezieht sich auf einen prophylaktischen und/oder
therapeutischen Impfstoff gegen HIV, gegen AIDS und gegen Tumore
und Syndrome, die mit einer HIV-Infektion
zusammenhängen,
welcher als Immunogene HIV Tat, alleine oder assoziiert mit Proteinen,
Peptide oder DNA anderer viraler Produkte (Nef, Rev, Gag) oder Cytokine,
welche die antivirale Immunantwort potenzieren, verwendet.
-
Die
Erfindung bezieht sich auch auf die Immunisierung von Tat, wie sie
in den Ansprüchen
definiert ist, mittels mukosaler Immunisierung oder ex-vivo-Immunisierung
von peripheren Blutzellen, die durch Co-Stimulation mit monoklonalen
Anti-CD3- und Anti-CD28-Antikörpern erweitert
sind, und auf die Zuführung
der oben erwähnten
Immunogene unter Verwendung von Erythrozyten oder Nanopartikeln.
-
Hintergrund der Erfindung
-
AIDS
(erworbenes Immunschwächesyndrom)
wird durch HIV (menschliches Immunschwächevirus) verursacht und ist
durch Immunschwäche,
Tumore, wie das Kaposi-Sarkom
(KS) und B-Zellenlymphome, opportunistische Infektionen und Störungen des
zentralen Nervensystems gekennzeichnet. Da AIDS weltweit verbreitet
ist und eine hohe Sterblichkeitsrate aufweist, ist eines der wichtigsten
Ziele des Gesundheitswesens, einen prophylaktischen und/oder therapeutischen
Impfstoff gegen HIV oder AIDS zu entwickeln. Die meisten der früheren und
aktuellen Strategien benutzten die Virushülle oder ihre Untereinheiten
als Immunogene, aufgrund der hohen Variabilität der Virushülle jedoch
mit unbefriedigenden Ergebnissen (Ref. 162, 112 – in gesamten vorliegenden
Beschreibung sind verschiedene Literaturstellen in Klammern angegeben,
um den Stand der Technik, dem die vorliegende Erfindung angehört, näher zu beschreiben.
Vollständige
bibliographische Angaben für
jedes Zitat finden sich am Ende der Beschreibung, unmittelbar vor
den Ansprüchen).
Daher ist man allgemein der Meinung, dass es als Alternative zur
sterilisierenden Immunität
ausreichen könnte,
die Progression einer Infektion und den Ausbruch der Krankheit zu
blockieren. Darüber
hinaus können
Immunschutzantworten unter Verwendung von DNA-Regionen von HIV als
Immunogenen (Ref. 91, 17) erhalten werden. Aufgrund der veröffentlichten
Versuchsdaten geht die Erfinderin davon aus, dass es notwendig ist,
einen Impfstoff zu produzieren, der andere virale Produkte als env
verwendet. Insbesondere müssen
die als Immunogene zu verwendenden viralen Proteine unter HIV-Isolaten besser konserviert
sein, eine wirksame Immunantwort, sowohl humoral als auch zellulär, induzieren
können
und eine lebenswichtige Funktion für das Virus haben. Solche Produkte
müssen
im Modell von nicht-menschlichen Primaten getestet werden (da deren
Immunsystem dem von Menschen im Vergleich zu phylogenetisch entfernteren
Lebewesen ähnlicher
ist), bei denen sich nach einer Virusinfektion AIDS entwickeln kann.
-
Das
HIV-1-Tat-Protein hat alle Merkmale, um ein gutes Immunogen für Impfzwecke
zu sein: es ist konserviert, immunogen und in den frühen Phasen
der viralen Infektion essentiell. Darüber hinaus spielt Tat nicht nur
bei der viralen Replikation, Transmission und Progression der Infektion
eine Schlüsselrolle,
sondern auch bei Ausbruch und Progression von AIDS-assoziierten Tumoren,
beispielsweise KS, dem häufigsten
AIDS-assoziierten Tumor, und anderen Syndromen und Symptomen, die
sich nach einer HIV-Infektion entwickeln.
-
Tat
ist ein Protein mit je nach Virusstamm 86-102 Aminosäuren, das
von zwei Exonen kodiert wird. Tat wird bald nach der Infektion produziert,
lokalisiert im Kern und transaktiviert die Expression aller viralen
Gene durch Wechselwirkung mit dem „Tatreaktiven Element" (TAR), das in den
LTR (Ref. 25) vorliegt. Tat spielt auch eine Rolle für die HIV-Virulenz
(Ref. 63, 113, 60, 84). Das Produkt des ersten Exons (Aminosäuren 1-72)
ist unter verschiedenen viralen Isolaten konserviert (Ref. 112)
und reicht für
die Transaktivierung der HIV-1-Produkte aus (Ref. 25). Es enthält 4 Domänen. Die
saure Domäne
(Aminosäuren
1-21) ist für
die Tat-Wechselwirkung mit zellulären Proteinen wichtig; die
Cystein-reiche Region (Aminosäuren
22-37) stellt die Transaktivierungsdomäne dar. Diese Region ist unter
den primären
Isolaten (Ref. 108) von Cystein 22 am besten konserviert und hebt
mit einem Glycin die Fähigkeit
von Tat auf, die HIV-LTR zu transaktivieren (Ref. 166), wobei die Kerndomäne (Aminosäuren 38-48)
ebenfalls konserviert und für
die Funktion wichtig ist. Eine Substitution von Lysin 41 mit einem
Threonin deaktiviert die Transaktivierungsaktivität von Tat
gegenüber
den HIV-LTR (Ref. 70); die basische Domäne (Aminosäuren 49-57), die reich an Arginin
und Lysin ist, ist für
die Kernlokalisierung von Tat notwendig und bindet ihr RNA-Ziel
(TAR) spezifisch (Ref. 25). Darüber
hinaus ist die basische Region für
die Bindung von extrazellulärem
Tat an Heparin und an Heparansulfatproteoglykane (HSPG) verantwortlich (Ref.
26). Mutationen in der basischen Region heben solche Wechselwirkungen
auf. Der carboxyterminale Abschnitt von Tat ist für die LTR-Transaktivierung
nicht notwendig, enthält
jedoch eine Arginin-Glycin-Asparaginsäure-Sequenz
(RGD), die üblicherweise
in den extrazellulären
Matrixproteinen (ECM) vorliegt und für die Bindung von Tat an die
Integrin-Rezeptoren α5β1 und αv β3 verantwortlich
ist. Diese Wechselwirkungen vermitteln die Tat-Effekte gegenüber AIDS-assoziierten Tumoren
und gegenüber
dem Immun-, Gefäß- und Nervensystem
(Ref. 11, 42, 170, 25). Während
der akuten Infektion von T-Zellen mit HIV-1 oder nach Transfektion
des tat-Gens in COS-1-Zellen wird das Tat-Protein ohne Zelltod in
der extrazellulären
Umgebung freigesetzt (Ref. 40, 41, 25). Die Tat-Freisetzung aus
infizierten Zellen erfolgt auch in vivo, da extrazelluläres Tat
im Serum infizierter Personen (Ref. 164) und in AIDS-KS-Läsionen (Ref.
42) vorhanden ist. Nach der Freisetzung bleibt das Protein teilweise
löslich
und bindet teilweise an die HSPG der ECM. An die HSPG gebundenes
Tat kann durch Zugabe von Heparin in löslicher Form zurückgewonnen
werden. Die Bindung mit Heparin ist durch die basische Tat-Region
bedingt; sie verhindert die Effekte von extrazellulärem Tat
und schützt
das Protein vor Oxidation. Dieses Merkmal wurde von uns eingesetzt,
um Tat mit einer hohen biologischen Aktivität zu reinigen (Ref. 26). Extrazelluläres Tat
kann von Zellen internalisiert werden, kann in den Kern migrieren
und eine Virusgenexpression transaktivieren (Ref. 49, 98, 100, 41).
Die Internalisierung von Tat erfolgt durch Endozytose, die durch
die Bindung der RGD-Region von Tat an α5β1 und αvβ3 (Ref.
10, 42, Ensoli et al., unveröffentlichte
Daten) und/oder durch die basische Region, welche an HSPG bindet,
vermittelt ist.
-
Tat
kann die virale Replikation und die Virusübertragung auch durch indirekte
Mechanismen unter Beteiligung der Modulation der Expression zellulärer Gene,
die eine Schlüsselrolle
bei der Steuerung des Zellüberlebens
und hinsichtlich der Expression inflammatorischer Cytokine (IC)
mit einem Effekt auf die virale Replikation spielen, aktivieren
(Ref. 25).
-
Neben
seiner Bedeutung bei der viralen Replikation spielt Tat eine wichtige
Rolle in der AIDS-Pathogenese. Tat ist in der Lage, das Überleben
und die Proliferation infizierter und nicht-infizierter Zellen zu
modulieren, indem es eine Aktivierung oder Unterdrückung von
Cytokinen, wie IL-2 (Ref. 123, 163, 31) oder von Genen mit einer
Schlüsselrolle
im Zellzyklus (Ref. 145, 169, 164, 173) bewirkt. Die anti- oder
proapoptotischen Effekte von Tat hängen von einer Reihe von Faktoren
ab, wie dem Zelltyp, der Tatsache, dass Tat von der Zelle exprimiert
oder der Zelle zugegeben wird, und von seiner Konzentration (Ref.
40, 41,171).
-
Tat
ist der Faktor, der für
die verstärkte
Häufigkeit
und Aggressivität
des KS in HIV-infizierten
Personen verantwortlich ist (Ref. 43, 33). Das KS ist ein Tumor
vaskulären
Ursprungs und die häufigste
Neoplasie in HIV-infizierten Individuen. Tat induziert KS-Zellen
und durch IC aktivierte Endothelzellen dazu, zu migrieren, Kollagenase
vom Typ IV zu exprimieren, in die ECM einzudringen und zu proliferieren,
wobei diese Mechanismen für
die Angiogenese und Tumorinvasion notwendig sind (Ref. 40, 41, 42,
2, 46). Solche Effekte von Tat werden durch IC induziert, da sie
die Expression der Tat-Rezeptoren α5β1 und αvβ3 stimulieren
(Ref. 10). Tat ahmt den Effekt von ECM-Proteinen, wie Fibronektin
und Vitronektin nach, und sowohl die RGD-Region als auch die basische
Region sind für
die Wirkungen von extrazellulärem
Tat auf KS-Zellen, auf die Angiogenese und auf die Progression von
KS notwendig. Die Fähigkeit
von extrazellulärem
Tat, in vivo an seine Rezeptoren in den AIDS-KS-Läsionen zu
binden (Ref. 40), stützt
die Überlegung,
dass Tat am Ausbruch und am Aufrechterhalten des AIDS-assoziierten
KS beteiligt ist. Darüber
hinaus entwickeln Mäuse,
die für
das tat-Gen transgen sind, abhängig
vom Expressionsgrad des Transgens das KS oder andere Phänotypen
(Ref. 160, 34).
-
Es
wurde nahegelegt, dass Tat eine Rolle bei den hyperproliferativen
Phänomenen
und bei der Pathogenese der B-Lymphome spielt, die häufig bei
seropositiven Personen und bei tat-transgenen Mäusen beobachtet werden (Ref.
157), aufgrund von Mechanismen unter Beteiligung der Verstärkung der
bcl-2- und Cytokin-Expression (Ref. 122). Andere Hinweise bestätigen eine
wahrscheinliche Rolle von Tat in der Onkogenese (Ref. 72).
-
Tat
kann auch die Expression viraler Promotoren, wie derer von Herpesviren
und anderen Viren, aktivieren, die AIDS in Individuen reaktivieren,
was Ausbruch und Progression opportunistischer Infektionen fördert (Ref.
25).
-
Tat
scheint auch in der Lage zu sein, neurotoxische Effekte sowohl direkt
(durch die basischen Regionen und die RGD-Regionen) und indirekt,
durch Induktion von IC, die eine toxische Wirkung auf die Neuronen des
zentralen Nervensystems oder auf die Blut-Hirn-Schranke haben, auszuüben (Ref.
25). Bezüglich
der Immunantwort auf Tat legt eine Reihe von Studien nahe, dass
Anti-Tat-Antikörper
eine schützende
Rolle bei der Steuerung der Evolution der Erkrankung in vivo spielen
(Ref. 130, 135, 136, 149, 127). Darüber hinaus unterdrücken Anti-Tat-Antikörper in
vitro nicht nur die Internalisierung, die transzelluläre Aktivierung
von Tat und von Virusinfektionen (Ref. 41, 127), sondern sie inhibieren
auch die Proliferation und die Tat-induzierte Migration von KS-Zellen
sowie die Bildung von KS-artigen
Läsionen
in Mäusen
(Ref. 40, 41, 42). Schließlich
zeigen unsere vorläufigen
Ergebnisse, dass Anti-Tat-Antikörper
in AIDS-KS-Personen fehlen, was nahelegt, dass diese Personen die
Aktivität
von extrazellulärem
Tat nicht blockieren können.
-
Die
Entwicklung einer zellvermittelten Anti-Tat-Reaktion in der Anfangsphase
der Infektion ist wichtig für
die Steuerung der Infektion selbst (Ref. 161, 133, 59), und es besteht
eine inverse Korrelation zwischen dem Vorliegen von spezifischem
Anti-Tat-CTL und der Progression der Erkrankung (Ref. 156). Diese
Ergebnisse wurden in Studien mit Makaken erhalten, die mit SIVmac
inokuliert waren (Ref. 91, 158). Darüber hinaus zeigten jüngste Daten
in Mäusen
unterschiedlicher Spezien, in die Tat entweder als Plasmid oder
als Protein inokuliert wurde, dass es möglich ist, sowohl eine humorale
als auch eine zelluläre
Antwort auf das Protein zu induzieren (Ref. 61). Jedoch wurde zwischen
verschiedenen Mausspezien eine Variabilität beobachtet, und diese Ergebnisse
wurden mit den gleichen Immunogenen in nicht-menschlichen Primaten
nicht reproduziert (Ref. 124). Die mangelnde Reproduzierbarkeit
im nicht-menschlichen Primaten-Modell der Ergebnisse aus den Impfstoffversuchen,
die bei Mäusen
durchgeführt
wurden, ist häufig
und möglicherweise
durch das unterschiedliche Immunsystem dieser beiden Spezien bedingt,
die mit dem gleichen Immunogen unterschiedliche Immunantworten hervorrufen
können,
wie dies am HIV-Env-Protein
gezeigt wurde. Somit müssen
für Menschen
in Frage kommende Impfstoffe in nicht-menschlichen Primaten, und nicht nur
in niederen Spezien getestet werden.
-
Die
Erfinderin geht davon aus, dass andere virale Proteine oder Teile
davon mit Tat assoziiert werden könnten, um eine spezifische
Immunantwort gegen HIV zu verstärken,
und auch bei der Impfung gegen den Ausbruch von Tumoren und anderen
Pathologien und Symptomen, die mit HIV-Infektion assoziiert sind,
von Nutzen sein könnten.
Diese Produkte sind die Nef-, Rev- und Gag-Proteine von HIV.
-
Nef
ist ein weiteres virales Regulatorprotein, das für die Entwicklung einer Krankheit
wichtig ist (Ref. 3, 48, 58). Nef wird früh nach der Infektion produziert
und in die extrazelluläre
Umgebung freigesetzt (Ensoli, unveröffentlichte Daten). Im SIVmac/Makaken-System korreliert
das Vorliegen von Nef mit einer hohen viralen Replikation und mit
der Progression zu AIDS (Ref. 71). Nef ist variabler als Tat (Ref.
112). Nef ist ein immunogenes Protein (Ref. 53, 32, 35, 151) und
in der Lage, CTL zu induzieren (Ref. 16, 36). Insbesondere wurde eine
immundominante Region von Nef identifiziert (Region 73-144), die
von den CTLs in den meisten HIV-infizierten Patienten erkannt wird.
-
Rev
ist ein virales Regulatorprotein, das bei einer Infektion früh produziert
(Ref. 51, 119) und in die extrazelluläre Umgebung zugeführt wird
(Ensoli et al., unveröffentlichte
Daten). Rev ist essenziell für
die HIV-Replikation und für
die Progression der Erkrankung und wird von zwei Exonen kodiert,
welche die Tat-Codierungsregionen teilweise überlappen. Rev ist ein Kernprotein
(Ref. 44), das für
die Expression der viralen Boten-RNAs benötigt wird, welche für die späten Proteine
kodieren (Ref. 97). Rev ist ein hoch konserviertes Protein unter
den verschiedenen viralen Isolaten von HIV-1 (Ref. 111) und ist
immunogen. Tatsächlich
induziert es die Produktion spezifischer Antikörper, die gegen die beiden
funktionalen Domänen
des Proteins (Ref. 120) gerichtet sind, während der natürlichen
Infektion beim Menschen (Ref. 131) und nach Inokulation in Mäusen (Ref.
61). Niedrigere Spiegel von Anti-Rev-Antikörpern in
den Seren infizierter Individuen scheinen mit der Progression zu
AIDS zu korrelieren (Ref. 131). Rev kann CTL sowohl beim Menschen
als auch beim Affen induzieren (Ref. 156, 158), und es wurde berichtet,
dass eine spezifische Anti-Rev-CTL-Antwort im frühen Infektionsstadium mit der
Progression der Erkrankung invers korreliert (Ref. 156, 158).
-
Ein
weiteres virales Ziel ist das gag-Gen, das im späten Infektionsstadium exprimiert
wird und für
eine Gruppe von hoch immunogenen Strukturproteinen des Kapsids kodiert
(Ref. 18, 147). Die Anti-Gag-Antikörpertiter sind während der
asymptomatischen Infektionsphase hoch und stabil und erreichen sehr
niedrige Werte, wenn die Infektion zu voll entwickeltem AIDS fortschreitet,
in Kombination mit dem Abfall der CD4+-Lymphocyten und dem Vorliegen
des Virus im peripheren Blut (Ref. 174, 73). Gag-Proteine induzieren
bei einer Infektion sowohl beim Menschen als auch bei Primaten früh eine CTL-Aktivität (Ref.
103, 168), und ihr Vorliegen steht in deutlichem Zusammenhang mit
der Steuerung der anfänglichen
Virämie
und mit der Progression der Erkrankung (Ref. 175, 6, 134, 167, 92).
Schließlich
enthalten die p17- und p24-Proteine immundominante Epitope, die
in unterschiedlichen HIV-1- und HIV-2-Isolaten gehalten und von CTL erkannt
werden (Ref. 89, 19, 114, 155, 115).
-
Die
Erfinderin geht davon aus, dass Cytokine oder Teile davon, wie IL-12
und IL-15 oder andere immun-modulierende Cytokine, wie IFNα oder IFNβ, oder andere
Proteine, welche die immunogene Wirkung von Tat verstärken, als
Adjuvanzien zur Formulierung des Anti-Tat-Impfstoffs verwendet werden können. IL-12
ist ein starkes immunregulatorisches Cytokin, das von Antigen-präsentierenden
Zellen (APC), wie B-Zellen und dendritischen Zellen, produziert
wird (Ref. 154). IL-12 wird bald nach einer HIV-Infektion produziert
und hat eine entzündungsfördernde
Wirkung, die NK-Zellen und T-Lymphocyten zur Produktion von IFNγ induziert, welches
Phagocyten aktiviert und die Induktion von Th1-Lymphocyten fördert. IL-12
spielt eine grundlegende Rolle für
die Resistenz gegen eine Reihe von Infektionen, die durch Bakterien,
Pilze und Viren verursacht werden, und zeigt eine hohe Antitumor-Aktivität. Mehrere
Indizien legen nahe, dass Viren, welche eine Immunsuppression induzieren,
wie HIV- und Masern-Viren, auch durch Mechanismen wirken, welche
die IL-12-Produktion unterdrücken
(Ref. 57, 50, 144).
-
IL-15
ist ein pleiotropes Cytokin, das von nicht-lymphoiden Geweben, von
aktivierten Monocyten/Makrophagen und von dendritischen Zellen (DC)
exprimiert wird (Ref. 125, 66). IL-15 spielt eine wichtige Rolle
bei der Regulierung der NK-Aktivität, bei der Proliferation von
T-Lymphocyten und bei der CTL-Aktivität (Ref. 67, 24). IL-15 induziert
die Expression von CTLs gegen HIV-Antigene in Abwesenheit von IL-2
und funktionalen CD4+ T-Lymphocyten
(Ref. 68, 1). Darüber
hinaus induziert IL-15, ähnlich
wie IL-2, die Expansion von Lymphocyten mit cytotoxischer Aktivität („Lymphokin-aktivierter
Killer", LAK) und
stimuliert die Produktion von IFNγ in
PBMCs von seropositiven Patienten (Ref. 93). IL-15 aktiviert Monocyten,
um Chemokine zu produzieren, die eine Rolle beim Einsetzen entzündlicher
Prozesse spielen (Ref. 8).
-
Jüngste Studien
haben gezeigt, dass die Co-Stimulation von CD4+-Lymphocyten mit
paramagnetischen Kügelchen,
die mit monoklonalen Anti-CD3- und Anti-CD28-Antikörpern beschichtet
sind, eine logarithmische und polyklonale Expansion von Lymphocyten
HIV-infizierter
Personen bestimmt (Ref. 82), ohne eine Virusreplikation und -transmission
zu aktivieren. Eine solche antivirale Aktivität ist eine Folge sowohl der
negativen Modulation der Expression von CCR5, des Co-Rezeptors von
HIV-1-monozytotropen Stämmen
(Ref. 23), und in geringerem Maße
der hohen Spiegel von Chemokinen, die durch die Co-Stimulation mit
monoklonalen Anti-CD3- und Anti-CD28-Antikörpern induziert werden (Ref.
132). Die Erfinderin geht davon aus, dass durch die Möglichkeit,
autologe Lymphocyten von HIV- infizierten
Personen in Abwesenheit von viraler Replikation/Transmission zu
expandieren, eine wirksame ex vivo-Immunisierung erhalten werden
kann, die in den Beispielen beschrieben ist und äußerst hilfreich bei der Entwicklung
eines Anti-Tat-Impfstoffs sein kann.
-
Innerhalb
der unterschiedlichen Systeme, die auf die Herstellung wirksamer
Antivirus- und Antitumor-Impfstoffe gerichtet sind, geht die Erfinderin
davon aus, dass die Verwendung von dendritischen Zellen ein Schlüssel zur
Induktion einer Immunantwort auf Tat sein könnte. Dies ist dadurch bedingt,
dass diese Zellen am wirksamsten das Antigen präsentieren und als einzige in
der Lage sind, naive Lymphocyten in Abwesenheit von Adjuvanzien
zu stimulieren (Ref. 150). Die Verwendung dendritischer Zellen ersetzt
die Funktion mehrerer Adjuvanzien, die in der Induktion einer nicht-spezifischen
Immunantwort (natürliche
Immunität)
besteht, welche wiederum eine starke primäre, spezifische Antwort in
Gegenwart des Antigens erzeugt.
-
Da
die Übertragung
der HIV-Infektion vorwiegend auf mukosaler Ebene (genital und rektal
beim Erwachsenen, oral beim Neugeborenen) erfolgt, geht die Erfinderin
davon aus, dass die Induktion schützender Immunität auf mukosaler
Ebene ein vorrangiges Ziel ist. Zahlreiche Studien zeigten vor kurzem
die Möglichkeit, eine
mukosale Immunisierung lokal und systemisch zu induzieren. Insbesondere
der nasale und der orale Weg haben sich als die wirksamsten Wege
zur Induktion einer wirksamen mukosalen Immunantwort, selbst an
entfernten Orten, wie der Genitalschleimhaut, gezeigt (Ref. 138,
118). Insbesondere geht die Erfinderin davon aus, dass die Verwendung
von S. Gordonii- und Lactobacillus-Bakterien, die so modifiziert
sind, dass sie die oben erwähnten
viralen Antigene exprimieren, eine zulässige Strategie zum Induzieren
oder Potenzieren einer spezifischen Immunantwort auf mukosaler Ebene
beim Affen und beim Menschen sein könnte. Diese Bakterien sind
tatsächlich
in der Lage, die Mund- und Vaginalschleimhaut der Maus zu kolonisieren
und eine spezifische, lokale und systemische Antikörperantwort
gegen heterologe Antigene, die auf der Oberfläche rekombinanter Bakterien
exprimiert werden, zu induzieren (Ref. 116, 104, 106, 121, 117,
139, 105, 107). Schließlich wirken
diese Bakterien als lebende Vektoren und können eine lang anhaltende Stimulation
des Immunsystems induzieren. Darüber
hinaus geht die Erfinderin davon aus, dass nicht-replizierende und
nicht-pathogene, rekombinante virale Vektoren, wie Herpes simplex
Typ 1-Viren (HSV-1) (Ref. 99), dazu verwendet werden können, virale
Proteine zur systemischen (intradermalen) und mukosalen (oraler,
vaginaler und nasaler Weg) Immunisierung zu exprimieren. Tatsächlich können diese
Vektoren große exogene
Sequenzen aufnehmen (Ref. 52, 64), wie mehrere HIV-Gene (regulatorisch,
akzessorisch und strukturell). Zudem können Herpesvektoren auch auf
oralem, nasalem oder vaginalem Wege verabreicht werden (Ref. 176,
75).
-
Die
Erfinderin geht davon aus, dass Tat (entweder als Protein oder DNA),
alleine oder in Kombination mit den anderen oben beschriebenen Immunogenen,
auch unter Verwendung neuer Zuführungssysteme,
wie Erythrozyten oder Nanopartikel, inokuliert werden kann. Insbesondere
geht die Erfinderin davon aus, dass es möglich ist, Antigene abzugeben,
die an die Membran autologer Erythrozyten gebunden sind (Ref. 95,
96). Da diese Erythrozyten von Makrophagen, professionellen Antigen-präsentierenden
Zellen, nach nur 120 Tagen aus dem Blut entfernt werden, kann dieses
Merkmal für
Impfstoffzwecke genutzt werden. Schließlich besteht eine weitere
Zuführungsstrategie
in der Verwendung von Nanopartikeln, die Proteine und DNA tragen
können (Ref.
27, 172). Nanosphären
sind polymere, kolloidale Partikel vielfältiger chemischer Zusammensetzung,
die von 10-1000 nm variieren können.
Unterschiedliche Substanzen (Oligonukleotide, Arzneimittel, Proteine,
Peptide, DNA) können
auf ihre Oberfläche
geladen oder in dem Partikel absorbiert und in das Cytoplasma oder den
Kern der Zellen zugeführt
werden, von wo sie langsam freigesetzt werden. Dies ermöglicht die
Nutzung sehr kleiner Mengen der abzugebenden Substanz.
-
Aufgrund
der oben beschriebenen Ergebnisse geht die Erfinderin davon aus,
dass die Immunisierung mit Tat, alleine oder in Kombination mit
anderen viralen Produkten oder immun-modulierenden Cytokinen oder Teilen
davon, gegebenenfalls in Gegenwart von Adjuvanzien, die virale Replikation
in Personen, die nach der Impfung exponiert werden, und in den infizierten
Personen blockieren könnte,
wodurch die Infektion in einer Abbruchphase gehalten würde, die
vom Immunsystem leichter kontrolliert werden kann. Daher geht die
Erfinderin davon aus, dass ein Impfstoff auf Tat-Basis sowohl humoral
als auch zellulär
eine Immunantwort induzieren können
sollte, die ausreicht, um die Replikation oder die Transmission
des Virus zu blockieren oder zu verringern, und daher in der Lage
sein sollte, die Virusreplikation zu kontrollieren und eine produktive
Infektion, eine Progression zur Erkrankung und der Ausbruch von
Tumoren und anderen mit AIDS assoziierten Syndromen und Symptomen
zu blockieren. Daher ist es möglich,
den Anti-Tat-Impfstoff sowohl für
präventive
als auch für
therapeutische Zwecke zu verwenden. Tatsächlich könnte eine humorale Antwort
gegen Tat die Effekte von extrazellulärem Tat, welches die Infektion
vermindert und eingrenzt, neutralisieren, während die zellinduzierte Antwort
gegen Tat sowie gegen andere virale Proteine, die in der Impfstoff-Formulierung
enthalten sind, die mit dem Virus infizierten Zellen zerstören und
damit zur Kontrolle der Infektion führen könnte. Dies gibt dem Immunsystem
die notwendige Zeit, eine vollständige
Antwort auf alle viralen Komponenten des infizierenden Virus, ohne
irreversible Schäden
aufgrund der viralen Replikation, zu entwickeln.
-
Die
Verwendung von Tat als Immunogen ist vorbeschrieben (WO95/31999).
Allerdings ist die Verwendung eines biologisch inaktiven Proteins
offenbart; zudem findet sich in derselben Patentanmeldung kein Hinweis
auf die biologische Aktivität
des „nativen" Tat-Proteins.
-
Die
WO9415634 bezieht sich auf
synthetische Oligopeptide, die homolog zu den Signalsequenzen von
Tat- und Rev-Proteinen sind, und zur Behandlung von HIV-Infektionen
verwendet werden können.
Es finden sich jedoch weder Hinweise zur Herstellung noch Versuche,
welche die Wirksamkeit der offenbarten Proteine zeigen.
-
Hinkula
et al., Vaccine, Vol. 15, 8, 874-878 (1997) offenbart Impfversuche
an Mäusen,
ausgehend von Impfstoffen, die Tat enthalten, wobei jedoch die dortigen
Ergebnisse, die bei niederen Spezien erhalten wurden, nicht direkt
auf nicht-menschliche Primaten übertragbar
sind.
-
Zudem
gibt es insofern eine starke technische Voreingenommenheit gegen
die Verwendung eines biologisch aktiven Tat-Proteins, als angenommen
wird, dass es die virale Replikation bei infizierten Personen verstärkt und/oder
bei seronegativen oder seropositiven Individuen eine Immunsuppression
ergibt (A. Tonelli: Aids, un vaccino per seerare. "La Repubblica", S. 10, 24. Okt.
1998).
-
Wie
aus dem Obigen ersichtlich ist, wurde trotz der unternommenen Anstrengungen
noch kein wirksamer Anti-HIV-Impfstoff auf Basis von Tat entwickelt.
-
Zusammenfassung der Erfindung
-
Ein
Ziel der vorliegenden Erfindung ist es, ein Tat-Protein zur Verwendung
als Impfstoff bereitzustellen, wie dies in den Ansprüchen definiert
ist.
-
Ein
weiteres Ziel der Erfindung ist ein Proteinimpfstoff, der beim Menschen
prophylaktisch oder therapeutisch gegen AIDS, AIDS-assoziierte Tumore
und HIV-assoziierte Syndrome und Symptome verwendet werden soll
und aus rekombinantem Wildtyp-Tat-Protein besteht (Sequenz ID 1),
das wie beschrieben exprimiert und gereinigt und alleine, oder mit
einem T-Helfer-Tetanustoxoidepitop
oder anderen T-Helfer-Epitopen konjugiert, verabreicht wird.
-
Noch
ein weiteres Ziel der Erfindung ist ein Impfstoff der oben beschriebenen
Art in Kombination mit rekombinanten HIV-Nef-, -Rev- und/oder -Gag-Proteinen
oder Peptiden von Nef, Rev und Gag, die als Tat/Nef-, Tat/Rev-,
Tat/Gag-Fusionsproteine oder als Teile dieser Proteine verabreicht
werden.
-
Ein
weiteres Ziel der Erfindung ist ein Impfstoff der oben beschriebenen
Art in Kombination mit rekombinanten Proteinen von immun-modulierenden
Cytokinen, wie IL-12, IL-15 oder anderen Molekülen oder Teilen davon, die
in der Lage sind, die antivirale Immunantwort zu verstärken, oder
ein Impfstoff, der durch Tat/IL-12, Tat/IL-15 oder Tat/andere Fusionsproteine
oder Teile davon gebildet und in der Lage ist, die antivirale Immunantwort
zu verstärken.
-
Ein
weiteres Ziel der Erfindung ist ein Anti-Tat-Impfstoff als Protein,
alleine oder wie oben beschrieben kombiniert, zur Immunisierung
mit autologen dendritischen Zellen durch ex vivo-Behandlung.
-
Noch
ein weiteres Ziel der Erfindung ist ein Anti-Tat-Impfstoff als Protein,
alleine oder wie oben beschrieben kombiniert, zur mukosalen Immunisierung
(nasal, oral, vaginal oder rektal).
-
Wieder
ein weiteres Ziel der Erfindung ist ein Anti-Tat-Impfstoff als Protein,
alleine oder wie oben beschrieben kombiniert, zur ex vivo-Immunisierung
von peripheren Blutzellen aus infizierten Personen, die durch Co-Stimulation
mit monoklonalen Anti-CD3- und Anti-CD28-Antikörpern erweitert sind, welche
mit paramagnetischen Kügelchen
konjugiert sind und in den Wirt reinfundiert werden.
-
Ein
weiteres Ziel der Erfindung ist ein Anti-Tat-Impfstoff als Protein,
wie oben beschrieben, kombiniert mit Inhibitoren der viralen Replikation.
-
Noch
ein weiteres Ziel der Erfindung ist ein Anti-Tat-Impfstoff der bereits
beschriebenen Art, in Kombination mit Adjuvanzien, welche die Immunantwort
verstärken.
-
Wieder
ein weiteres Ziel der Erfindung ist ein Anti-Tat-Impfstoff, alleine
oder in Kombination, wie bereits beschrieben, verabreicht mittels
spezifischer Zuführungssysteme,
wie Nanopartikel, Herpesvektoren, rote Blutzellen, Bakterien oder
irgendein anderes Zuführungssystem,
durch das der oben beschriebene Impfstoff in all seinen Kombinationen
verabreicht werden kann.
-
Weitere
Ziele sind aus der detaillierten Beschreibung der Erfindung ersichtlich.
-
Kurze Beschreibung der Figuren
-
1A. Inhibierung der Aufnahme von 10 ng/ml rhodaminiertem
Tat-Protein durch Vorinkubation von Cytokin-aktivierten Endothelzellen
mit Anti-Integrin-Antikörpern.
-
1A. Bild A, Zellen, vorinkubiert mit Puffer, inkubiert
mit BSA.
-
1A. Bild B, Zellen, vorinkubiert mit Puffer, inkubiert
mit Tat.
-
1A. Bild C, Zellen, vorinkubiert mit den monoklonalen
Antikörpern
CDw49e und CD29, inkubiert mit Tat.
-
1A. Bild D, Zellen, vorinkubiert mit den monoklonalen
Antikörpern
CD51 und CD61, inkubiert mit Tat.
-
1A. Bild E, Zellen, vorinkubiert mit Anti-Humanfaktor
VIII-Antikörpern
(Kontroll-Antikörper), inkubiert
mit Tat.
-
1B. Fähigkeit
von gereinigtem Tat-cys22 (Tat22)-Protein, um die Transaktivierungsaktivität von Wildtyp-Tat-Protein
zu konkurrieren, überwacht
durch CAT-Assays.
-
2A. Anti-Tat-spezifische IgG-Produktion
in mit dem Tat-Protein geimpften Affen, bestimmt durch immunenzymatischen
Assay (ELISA). Es wurden Ergebnisse von zwei Affen erhalten, die
subkutan inokuliert waren mit 10 oder 100 μg rekombinantem Tat-Protein,
resuspendiert in 250 μl
autologem Serum und 250 μl RIBI.
-
2B. Anti-Tat-spezifische IgG-Produktion
bei mit dem Tat-Protein geimpften Affen, bestimmt durch immunenzymatischen
Assay (ELISA). Ergebnisse für
den Kontrollaffen (M3).
-
3.
Titration von Anti-Tat-Antikörpern
in Plasma von Affen, die mit 100 (M1) und 10 (M2) μg rekombinantem
Tat-Protein inokuliert wurden, wie dies in 2A und 2B beschrieben ist.
-
4A. Kartierung der Tat-Epitope, die von
Anti-Tat IgG aus Affen, denen 100 (M1) und 10 (M2) μg rekombinantes
Tat-Protein injiziert worden waren, wie in 2A und 2B beschrieben, erkannt wurden. Gezeigt
sind die durchschnittlichen Ergebnisse von Plasma, das für jedes
zweifach getestete Peptid 1:50 verdünnt war.
-
4B. Kartierung gemäß 4A.
Gezeigt sind die Antikörpertiter
im Plasma, ausgedrückt
als Kehrwert der höchsten
Verdünnung,
bei welcher der Test noch positiv war.
-
5.
Analyse der spezifischen humoralen Anti-Tat-IgM-Antwort in mit Tat-Protein
inokulierten Affen, bestimmt mittels ELISA.
-
6.
Analyse der spezifischen Anti-Tat IgG-Produktion in mit Tat-Protein
inokulierten Affen, getestet mittels ELISA.
-
7.
Titration von Anti-Tat-Antikörpern
im Plasma der mit rekombinantem Tat (10 μg) in Gegenwart von RIBI (MI-3)
oder Alaun (M4-6) inokulierten Affen, wie in 6 beschrieben.
-
8A. Epitope von Tat, die von Anti-Tat
IgG aus Affen erkannt werden, welche wie in 6 beschrieben
inokuliert sind. Die Ergebnisse beziehen sich auf Proben mit einer
Verdünnung
von 1:50 und sind der Mittelwert aus zwei Vertiefungen.
-
8B. Epitope von Tat gemäß 8A. Die Ergebnisse beziehen sich auf die
Plasmatitration, die in 8A gezeigt
ist, und sind als die höchste
reziproke Plasmaverdünnung
ausgedrückt,
bei welcher der Test noch positiv war.
-
9.
Analyse von Tat-spezifischen CTL.
-
10. Analyse der Antwort einer verzögerten Hypersensibilität gegenüber Tat
mittels Hauttest.
-
11A. Humorale IgG-Antwort auf Tat in mit
Tat-DNA geimpften Affen. Gezeigt sind die Ergebnisse, die von zwei
Affen erhalten wurden, welche mit 200 (M1) und 500 (M2) μg pCV-Tat-Plasmid
geimpft waren.
-
11B. Humorale IgG-Antwort auf Tat in mit Tat-DNA
geimpften Affen. Ergebnisse für
den Kontrollaffen (M3).
-
12. Titration von Anti-Tat-Antikörpern in
Plasma des Affen M2, der i. d. mit 200 μg pCV-Tat inokuliert war.
-
13. Analyse der Anti-Tat IgG-Produktion in drei
Affen (M9 bis M11), die mit 1 mg pCV-Tat inokuliert waren, und in
einem Kontrollaffen (M12), inokuliert mit 1 mg des Kontrollvektors
pCV-0.
-
14. Kinetik der proliferativen Antwort von PBMC
aus Macaca fascicularis auf die Co-Stimulation mit monoklonalen Anti-CD3-
und Anti-CD28-Antikörpern
auf paramagnetischen Kügelchen
(Anti-CD3/28-Kügelchen).
-
15A. Antiviraler Effekt der Co-Stimulation
mit Anti-CD3/28-Kügelchen
auf PBMC von Macaca fascicularis. Affe MK 193.
-
15B. Antiviraler Effekt der Co-Stimulation
mit Anti-CD3/28-Kügelchen
auf PBMC von Macaca fascicularis. Affe MK D91.
-
15C. Antiviraler Effekt der Co-Stimulation
mit Anti-CD3/28-Kügelchen
auf PBMC von Macaca fascicularis. Affe MK 9301.
-
15D. Antiviraler Effekt der Co-Stimulation
mit Anti-CD3/28-Kügelchen
auf PBMC von Macaca fascicularis. Affe MK 9401.
-
16A. Funktionale Charakterisierung dendritischer
Zellen (DC), erhalten aus peripherem Blut von Affen. 3H-Thymidin-Aufnahme
von allogener gemischter Leukozytenkultur (AMLR) an Tag 4.
-
16B. Funktionale Charakterisierung dendritischer
Zellen, erhalten aus peripherem Blut von Affen. APCs, wie DC und
M∅, die wie in 16A angegeben
erhalten worden waren, wurden mit T-Lymphocyten eines anderen Affen
angeregt.
-
Detaillierte Beschreibung
der Erfindung
-
Die
vorliegende Erfindung bezieht sich auf Tat, wie in den Ansprüchen definiert,
als Wirkprinzip für
einen prophylaktischen und/oder therapeutischen Impfstoff gegen
HIV-Infektion, gegen
die Progression zu AIDS und gegen die Entwicklung von Tumoren und
anderen Syndromen und Symptomen in HIV-infizierten Personen. Tat
oder Wildtyp-Tat liegt in seiner aktiven Form oder, korrekter gesagt,
in seiner biologisch aktiven Form (wie hier nachfolgend erläutert) als
rekombinantes Protein vor. Insbesondere bezieht sich die Erfindung
auf einen Impfstoff, der auf HIV-1 Tat als Immunogen beruht, welches
als rekombinantes Protein, alleine oder in Kombination mit anderen
Genen oder viralen Genprodukten (Nef, Rev, Gag) oder Teilen davon,
oder in Kombination mit verschiedenen immun-modulierenden Cytokinen
(IL-12, IL-15) oder mit dem Gen, das für ein immun-modulierendes Cytokin
oder einen Teil davon kodiert, inokuliert wird. Tat, Nef, Rev, Gag
und die immun-modulierenden Cytokine werden sowohl als Mischung
rekombinanter Proteine, Peptide oder Fusionsproteine (Tat/Nef, Tat/Rev,
Tat/Gag, Tat/IL-12, Tat/IL-15) wie auch als Plasmid-DNA verabreicht.
-
In
der vorliegenden Beschreibung sind „Wildtyp-Tat" und „Tat in
seiner aktiven Form" als
Synonyme für „biologisch
aktives Tat" zu
betrachten.
-
„Biologisch
aktives Tat” soll
sich auf das Protein beziehen, das bei pikomolaren bis nanomolaren
Konzentrationen (von 10 ng/ml oder weniger bis 1 μg/ml, vorzugsweise
0,1 ng/ml bis 100 ng/ml) in der Lage ist:
- (i)
in die Kerne aktivierter Endothelzellen oder dendritischer Zellen
einzudringen und sich dort zu lokalisieren, wie in Beispiel 1A bestimmt;
- (ii) die Proliferation, Migration und Invasion des Proteins
von Kaposi-Sarkom(KS)-Zellen und Cytokin-aktivierten Endothelzellen
zu aktivieren (Ref. 40,2);
- (iii) die Virusreplikation zu aktivieren, wenn es zu infizierten
Zellen gegeben wird, bestimmt durch a) die Rettung von Tat-defizienten
Proviren in HLM-1-Zellen nach Zugabe von exogenem Protein (Ref:
41); b) die Transaktivierung der HIV-1-Genexpression in mit einem
HIV-1-Promotor-Reporterplasmid-Protein transfizierten Zellen (Ref.
41);
- (iv) in Mäusen
die Entwicklung von KS-artigen Läsionen
in Gegenwart angiogener Faktoren oder inflammatorischer Cytokine
zu induzieren (Ref. 42).
-
Die
Erfinderin erachtet es als grundlegend für biologisch aktives Tat, dass
einer der Punkte (i) oder (ii) überprüft wird;
vorzugsweise sollten beide überprüft werden,
noch bevorzugter sollten Punkt (i) oder Punkt (ii) oder beide in
Kombination mit Punkt (iii) a) und/oder (iii) b) überprüft werden.
Die besten Ergebnisse werden erhalten, wenn alle Punkte (i) bis
(iv) überprüft werden.
Ein Tat-Protein mit diesen Merkmalen ist in der Lage, in vivo eine
cytotoxische und antivirale Immunantwort zu induzieren. Tatsächlich ist
ein biologisch aktives Tat mit den oben erwähnten Merkmalen in der Lage,
spezifische Zelloberflächenrezeptoren
zu binden, und wird über
diese Rezeptoren aufgenommen. Die Tat-Aufnahme ist essentiell für die Induktion
einer cytotoxischen Antwort.
-
Frühere oder
noch andauernde Studien, die sich mit der Entwicklung eines auf
Tat basierenden Impfstoffs befassen, nutzten kein biologisch aktives
Tat-Protein mit den oben erwähnten
Merkmalen.
-
Ein
Verfahren zum Erhalten und Handhaben von biologisch aktivem Tat
gemäß der vorliegenden
Erfindung ist in Beispiel 1 beschrieben.
-
Ebenso
ist ein Immunisierungsverfahren beschrieben, das autologe dendritische
Zellen einsetzt, die ex vivo mit rekombinantem Tat-Protein oder
Peptiden davon, allein oder mit einer Mischung von rekombinanten Proteinen
oder Peptiden (Tat, Nef, Rev, Gag), oder mit dem Tat-Protein und
einem oder mehreren immun-modulierenden Cytokin(en) oder Teilen
davon behandelt oder mit eukaryontischen Vektoren transduziert sind, welche
das tat-Gen allein oder in Kombination mit viralen Genen, welche
für Nef,
Gag oder Rev kodieren, oder tat und das Gen, das für ein immun-modulierendes
Cytokin oder einen Teil davon kodiert, enthalten.
-
Strategien
zum Induzieren einer Immunantwort auf mukosaler Ebene sind ebenfalls
beschrieben.
-
Tat
wird alleine oder in Kombination mit viralen Proteinen und/oder
Cytokinen auf mukosaler Ebene inokuliert, um die lokale Immunantwort
zu verstärken
und zu induzieren. Das HIV-1 Tat-Protein wird auch zur ex vivo-Immunisierung
von CD4+- und CD8+-Lymphocyten
eingesetzt, die aus dem peripheren Blut infizierter Personen isoliert
sind. Dann werden die Tat-antigenspezifischen Zellen in vitro durch
Co-Stimulation mit monoklonalen Antikörpern, die gegen CD3 und CD28
gerichtet sind, erweitert und reinfundiert. Schließlich ist
auch die Verwendung von in den Beispielen angegebenen Tat-Mutanten
beschrieben, die als Immunogene alternativ zum Tat-Wildtyp zu verwenden
sind.
-
Die
Tat-Mutanten liegen vor in i) der Cysteinregion (cys22) und ii)
der Kernregion (lys41), iii) der in der RGD-Sequenz deletierten
Mutante; iv) der bei Lysin 41 und in der RGD deletierten Doppelmutante.
Alternativ zur Verwendung von Tat-Mutanten oder Tat-Peptiden (Wildtyp
oder mutiert als Protein) im Falle einer therapeutischen Impfung
werden Inhibitoren der viralen Replikation zusammen mit dem Immunogen
eingesetzt.
-
In
diesem Zusammenhang sollen „Inhibitoren
der viralen Replikation" alle
derzeit bekannten Moleküle oder
solche sein, die später
noch entdeckt werden (Nukleosid- und Nicht-Nukleosidinhibitoren von reverser Transkriptase,
Proteaseinhibitoren, Antisense-RNA und im allgemeinen alle Moleküle, welche
in der Lage sind, die HIV-Genexpression zu blockieren) und in der
Lage sind, die HIV-Replikation zu verringern oder zu blockieren.
Wie zuvor erwähnt,
sind unterschiedliche Immunisierungsverfahren beschrieben, die Tat-Protein,
-Peptide und Tat-DNA in Verbindung mit anderen viralen Genen oder
Proteinen oder einem Teil davon oder immun-modulierende Cytokine
oder Gene, welche für
immun-modulierende Cytokine kodieren, oder einen Teil davon einsetzen.
Ein „Teil
davon" sollen Segmente
von Genen oder Proteinen der oben beschriebenen Art sein, deren Wirksamkeit
zur Induktion derselben immunogenen Effekte wie beim ganzen Gen
oder Protein bewiesen ist. Da zudem die Wirksamkeit von Adjuvanzien
bei Impfstoffstrategien bekannt ist, bezieht sich die vorliegende Erfindung
auf die Verwendung bekannter später
noch zu entdeckender Adjuvanzien, die zusammen mit Tat (Protein
oder DNA) und mit Kombinationen von Tat und anderen Genen oder viralen
oder zellulären
Proteinen verabreicht werden.
-
Entsprechend
wird die Wirksamkeit unterschiedlicher Zuführungssysteme für Tat (Protein
oder DNA) und Kombinationen von Tat und anderen Genen oder viralen
oder zellulären
Proteinen zur Induktion sowohl einer systemischen als auch einer
lokalen Immunantwort auf Tat (mukosale Immunisierung) angenommen.
-
Von
der Erfinderin erhaltene (unveröffentlichte)
Ergebnisse zeigen, dass nur das Tat-Protein in seiner biologisch
aktiven Form in der Lage ist, spezifische zelluläre Rezeptoren zu binden und
in die Zelle einzudringen. Dieses Merkmal liegt der Immunantwort
akzessorischer Zellen und allgemeiner der Immunzellen zugrunde,
und laut der Erfinderin ist es von grundlegender Bedeutung für die Induktion
einer deutlich stärkeren
Immunantwort, als sie das deaktivierte Protein hervorrufen kann.
Im Gegensatz zur Verwendung von deaktiviertem Tat als Immunogen,
wie sie von einigen Wissenschaftlern vorgeschlagen wurde, beabsichtigt
die Erfinderin folglich, HIV-1 Tat in seiner biologisch aktiven
Form einzusetzen, um eine sehr starke Immunantwort gegen HIV zu
induzieren, die eine Infektion oder die Entwicklung der Erkrankung
verhindern und wirksame therapeutische Strategien in HIV-1-infizierten
Individuen zulassen kann. Laut der Erfinderin kann der Impfstoff
auf systemischem (intramuskulärem,
d. h. subkutanem usw.) oder lokalem (mukosalem) Wege zugeführt werden.
Der letztgenannte Weg ist bevorzugt, wenn Bakterien (siehe unten)
als Zuführungssysteme
eingesetzt werden.
-
Der
Impfstoff kann wie folgt produziert werden. Tat kann gemäß Beispiel
1 hergestellt, lyophilisiert und gelagert werden. Zum Zeitpunkt
der Verwendung kann es in einem biologisch verträglichen Fluid, wie Serum, Plasma
oder Fraktionen davon, resuspendiert werden.
-
Transformierte
Zellen, umfassend einen Tat-exprimierenden Vektor, oder Teile davon,
wie zuvor beschrieben, und Zellen, die kultiviert werden, um Tat-Protein
zu exprimieren, und für
die Verwendung isoliert werden, sind alle im Umfang des vorliegenden
Patentes enthalten.
-
Alle
Tat-Varianten (einschließlich
aller Arten und Unterarten von HIV-Stämmen) mit analoger oder größerer Aktivität als die
oben beschriebene sind in dieser Erfindung enthalten.
-
Die
vorliegende Erfindung wird nun anhand ihrer veranschaulichenden
und nicht einschränkenden, spezifischen
Beispiele beschrieben, in denen auf die beigefügten Zeichnungen Bezug genommen
wird.
-
Detaillierte Beschreibung
der Figuren
-
1A. Inhibierung der Aufnahme von 10 ng/ml rhodaminiertem
Tat-Protein durch Vorinkubation Cytokin-aktivierter Endothelzellen
mit Anti-Integrin-Antikörpern.
Cytokin-aktivierte
Humanumbilikalvenen(HUVE)-Zellen, die wie in der Erklärung zu
Tabelle 2A beschrieben behandelt wurden, wurden in serumfreiem Medium
vorinkubiert, enthaltend Puffer oder Antikörper, und dann 15 Minuten bei
37°C mit
10 ng/ml rhodaminiertem Tat oder rhodaminiertem BSA inkubiert.
-
Bild
A, Zellen, vorinkubiert mit Puffer, inkubiert mit BSA.
-
Bild
B, Zellen, vorinkubiert mit Puffer, inkubiert mit Tat.
-
Bild
C, Zellen, vorinkubiert mit den monoklonalen Antikörpern CDw49e
(anti-α5)
und CD29 (anti-β1), inkubiert
mit Tat.
-
Bild
D, Zellen, vorinkubiert mit den monoklonalen Antikörpern CD51
(anti-αv)
und CD61 (anti-β3),
inkubiert mit Tat.
-
Bild
E, Zellen, vorinkubiert mit Antihuman-Faktor VIII-Antikörpern (Kontroll-Antikörper), inkubiert
mit Tat.
-
1B. Gezeigt ist die Fähigkeit von gereinigtem Tat-cys22
(Tat22)-Protein, mit der Transaktivierungsaktivität von Wildtyp-Tat-Protein
zu konkurrieren, die mittels CAT-Assays überwacht wird. H3T1-Zellen, enthaltend
das HIV-1 LTR-CAT-Reportergen (Ref. 148), wurden mit Wildtyp-Tat-Protein
(100 ng), alleine oder in Gegenwart eines molaren Überschusses
von Tat-cys22-Protein (1 μg)
inkubiert. Die HIV-1 LTR-Transaktivierungsaktivität von Tat
und die Fähigkeit
des Tat-cys22-Proteins mit Wildtyp-Tat zu konkurrieren, wurden 48 h
nach Transfektion durch Bestimmen der CAT-Aktivität in Cytoplasmaextrakten
(entsprechend 200 μg
Protein) wie beschrieben (Ref. 41) bestimmt. Die Prozentsätze (%)
der Acetylierung von 14C-Chloramphenicol
sind angegeben.
-
2.
Anti-Tat-spezifische IgG-Produktion in mit dem Tat-Protein geimpften
Affen, bestimmt durch immun-enzymatischen Assay (ELISA). (A) zeigt
die Ergebnisse, die in zwei Affen erhalten wurden, welche mit 10 oder
100 μg rekombinantem
Tat-Protein, resuspendiert in 250 μl autologem Serum und 250 μl RIBI, subkutan inokuliert
waren; (B) zeigt die Ergebnisse für den Kontrollaffen (M3). Die
Affen wurden zum Zeitpunkt 0 und nach 2, 5, 10, 15, 22, 27, 32 und
37 Wochen inokuliert. Auch die Anti-Tat-Antikörper wurden in Woche 41 im Affen
M2, der mit 10 μg
Tat-Protein inokuliert war, und für den Affen M3 bestimmt.
-
Das
Vorliegen der Anti-Tat-Antikörper
im Plasma geimpfter Tiere wurde mittels ELISA ausgewertet, das wie
folgt hergestellt und charakterisiert wurde: Das Tat-Protein wurde
12 h bei 4°C
in PVC-Platten mit 96 Vertiefungen adsorbiert (100 ng/Vertiefung
in 200 μl
Carbonatpuffer, 0,05 M, pH 9,6). Nach 3 Wäschen mit PBS 1 × ohne Ca++ und Mg++ (PBS-A),
enthaltend Tween 20 (0,05%), wurde Plasma 1:50 verdünnt in 200 μl Carbonatpuffer
zugegeben (zweifach), und die Platten wurden 90' bei 37°C inkubiert. Dann wurden die
Vertiefungen mit PBS-A 1 ×/Tween
0,05% gewaschen, gefolgt von 90-minütiger Zugabe von 100 μl des sekundären Antikörpers (1:1000
verdünnt
in PBS-A 1 ×/Tween
0,1%/BSA 1%), konjugiert mit Meerrettichperoxidase, bei Raumtemperatur.
Nach 5 Wäschen
der Vertiefungen wurden 100 μl
Substrat (ABTS 1 mM, Amersham) 30-45' bei Raumtemperatur zugegeben. Es wurde
eine Ablesung am Spektrophotometer (405 nm) durchgeführt. Jeder
ELISA umfasste ein polyklonales Anti-Tat-Kaninchenserum (positive
Kontrolle) in einer Verdünnung
von 1:200 bis 1:6400 und das präimmune
Plasma (negative Kontrolle) in einer Verdünnung von 1:50. Der Cutoff-Wert
wurde als Mittelwert der optischen Dichten (O. D.) von negativem
Affenplasma +3 Standardabweichungen (S. D.), erhalten in allen Experimenten
mit dem präimmunen
Plasma, berechnet. Die gezeigten Ergebnisse sind der Mittelwert
aus zwei Vertiefungen. > 2,7
zeigt an, dass die optischen Dichtewerte außerhalb der Skala lagen.
-
3.
Titration von Anti-Tat-Antikörpern
im Plasma von Affen, inokuliert mit 100 (M1) und 10 (M2) μg rekombinantem
Tat-Protein, beschrieben in 2.
-
Ein
Elisa wurde, wie in 2 beschrieben, durchgeführt und
Plasma wurde (zweifach) in skalaren Verdünnungen von 1:50 bis 1:25.600
getestet.
-
Die
Werte auf der Ordinate stellen die Umkehrfunktion der höchsten Plasmaverdünnung dar,
bei welcher der Test noch positiv war. Der Cutoff-Wert wurde für jede Verdünnung berechnet
und entsprach der durchschnittlichen O. D. des präimmunen
Plasmas aller Affen in allen Experimenten + 3 S. D.
-
4.
Kartierung der Tat-Epitope, die erkannt werden vom Anti-Tat IgG
von Affen, denen 100 (M1) und 10 (M2) μg rekombinantes Tat-Protein
injiziert wurden, wie dies in 2 beschrieben
ist. Für
die Epitopkartierung wurden Elisa unter Verwendung von 8 synthetischen
Peptiden durchgeführt,
die den Tat-Aminosäuren
(aa) 1-20, 21-40, 36-50, 46-60, 52-72, 56-70, 65-80, 73-86 entsprachen.
Einhundert Mikroliter jedes Peptids (10 μg/ml in PBS-A/0,1% BSA) wurden
12 Stunden bei 4°C
an eine PVC-Platte mit 96 Vertiefungen adsorbiert. Dann wurden die
Platten gewaschen und 2 Stunden bei 37° mit 100 μl PBS-A/3% BSA inkubiert. Nach der
Inkubation wurden die Platten mit PBS-A/0,05% Tween 20 gewaschen,
und dann wurden 50 μl
Plasma, verdünnt
in PBS-A und 3% BSA, in jede Vertiefung gegeben. Anschließend wurden
die Elisa wie in 2 beschrieben fortgesetzt.
Plasma wurde in Woche 37 nach der primären Immunisierung erhalten.
Die für
jedes Peptid und für
jede Plasmaverdünnung
berechneten Cutoff-Werte entsprechen der durchschnittlichen O. D. des
präimmunen
Plasmas aller Experimente + 3 S. D. (A) zeigt die durchschnittlichen
Ergebnisse von 1:50 verdünntem
Plasma für
jedes zweifach getestete Peptid; (B) zeigt die Antikörpertiter
des in (A) gezeigten Plasmas, ausgedrückt als Kehrwert der höchsten Verdünnung, bei
welcher der Test noch positiv war.
-
5.
Analyse der spezifischen Anti-Tat IgM-Antwort in Affen, inokuliert
mit Tat, und bestimmt mittels ELISA. Drei Affen (M1-3) wurden subkutan
mit 10 μg
rekombinantem Tat-Protein
inokuliert, das in 250 μl
autologem Serum und 250 μl
RIBI resuspendiert war; 3 Affen (M4-6) wurden subkutan inokuliert
mit 10 μg
rekombinantem Tat-Protein, resuspendiert in 250 μl autologem Serum und 250 μl Alaun;
2 Kontrollaffen wurden subkutan inokuliert mit RIBI (250 μl und 250 μl autologes
Serum) (M7) oder mit Alaun (250 μl
und 250 μl
autologes Serum) (M8). Die Affen wurden zum Zeitpunkt 0 und nach
2, 6, 11 und 15 Wochen inokuliert. Das Vorliegen von Antikörpern wurde
nach 2, 6, 11 und 15 Wochen untersucht. Das ELISA-Verfahren ist
in 2 beschrieben. In diesem Fall wurde das Plasma
der Tiere (zweifach) bei einer Verdünnung von 1:100 getestet und
ein IgM-Ziege-Anti-Affe-Serum
(1:1000 verdünnt),
konjugiert mit Meerrettichperoxidase, als sekundärer Antikörper verwendet.
-
Der
Cutoff-Wert wurde als Durchschnitt (+2 S. D.) der O. D.-Werte des
präimmunen
Plasmas berechnet. Die Ergebnisse sind der Durchschnitt der O. D.-Werte
(bei 405 nm) von zwei Vertiefungen, subtrahiert vom Cutoff-Wert
(ΔO. D.
405).
-
6.
Analyse der spezifischen Anti-Tat IgG-Produktion in Affen, inokuliert
mit Tat, getestet mittels ELISA. Drei Affen (M1-3) wurden mit 10 μg rekombinantem
Tat-Protein, resuspendiert in 250 μl autologem Serum und 250 μl RIBI, inokuliert;
3 Affen (M4-6) wurden mit 10 μg
rekombinantem Tat-Protein, resuspendiert in 250 μl autologem Serum und 250 μl Alaun,
inokuliert; zwei Kontrollaffen wurden mit RIBI (250 μl und 250 μl autologes
Serum) (M7) oder mit Alaun (250 μl
und 250 μl
autologes Serum) inokuliert (M8). Die Affen wurden zum Zeitpunkt
0 und nach 2, 6, 11, 15, 21, 28 und 32 Wochen inokuliert. In Woche
36 wurden die Affen M1 bis M6 mit 16 μg Tat-Protein, resuspendiert
in 200 μl
ISCOM und 300 μl
PBS, inokuliert. Zudem wurden die Antikörper in Woche 40 und 44 evaluiert.
Das ELISA-Verfahren
und die Bestimmung des Cutoff-Wertes sind in 2 beschrieben.
Die gezeigten Ergebnisse beziehen sich auf 1:50 verdünnte Proben. > 2,7 zeigt an, dass der
O. D.-Wert außerhalb
der Skala lag.
-
7.
Titration von Anti-Tat-Antikörpern
in Plasma der Affen, die mit rekombinantem Tat (10 μg) in Gegenwart
von RIBI (M1-3) oder Alaun (M4-6), wie in 6 beschrieben,
inokuliert wurden. Die Ergebnisse sind für jedes Plasma als Kehrwert
der höchsten
Serumverdünnung
gezeigt, bei welcher der Test noch positiv war.
-
8.
Epitope von Tat, erkannt von Anti-Tat-IgG aus Affen, die mit rekombinantem
Tat-Protein (10 μg) in Gegenwart
von RIBI (M1 bis M3) oder Alaun (M4 bis M6), wie in 6 beschrieben,
inokuliert wurden. Plasma wurde in Woche 21 nach der Primärimmunisierung
erhalten. Das Elisa-Verfahren und die Cutoff-Bestimmung sind in 4 beschrieben.
Die Ergebnisse in (A) beziehen sich auf Proben in einer Verdünnung von
1:50 und sind der Durchschnitt von zwei Vertiefungen. Die Ergebnisse
in (B) beziehen sich auf die Titration von Plasma, gezeigt in (A),
und sind als die höchste
reziproke Verdünnung
von Plasma ausgedrückt,
bei welcher der Test noch positiv war.
-
9.
Analyse von Tat-spezifischen CTL. Der Assay wurde wie in Tabelle
5 beschrieben durchgeführt. Gezeigt
ist ein Beispiel in der 36. Woche für den Affen M1, dem 10 μg Tat und
RIBI subkutan injiziert worden waren, wie dies in 6 beschrieben
ist. Die Quadrate (Kontrolle) entsprechen den mit ungepulsten BLCL-Zielzellen
inkubierten Zellen; die Rhomben entsprechen den Zellen, die mit
den BLCL-Zielzellen, gepulst mit Tat (1 μg/250.000 Zellen), inkubiert
wurden.
-
10. Analyse der verzögerten Hypersensibilitätsreaktion
auf Tat mittels Hauttest. Tat-Protein
(5,1 und 0,2 μg),
resuspendiert in 150 μl
PBS, enthaltend 0,1% BSA, oder Puffer, in dem Tat resuspendiert
war, wurde intradermal (i. d.) in einem rasierten Bereich auf dem
Rücken
des Tieres inokuliert. Der Bereich wurde zum Zeitpunkt 0 und nach
24, 48 und 72 Stunden photographiert. Die Kontrollaffen wurden nur
mit Puffer inokuliert. Gezeigt ist ein Beispiel des Affen M2 (Woche
15), inokuliert mit 10 μg
Tat und RIBI, wie in 6 beschrieben. Die positive
Reaktion auf Tat wurde 48 Stunden nach dem Hauttest deutlich.
-
11. Humorale IgG-Antwort auf Tat in einem Affen
(M1), der mit 200 μg
des pCV-Tat-Plasmids,
resuspendiert in 150 μl
PBS-A, an zwei Stellen nahe den axillären Lymphknoten i.d. inokuliert
war; einem Affen (M2) wurden 500 μg
pCV-Tat, resuspendiert in 250 μl
PBS-A, intramuskulär
in zwei Stellen am Rücken
injiziert; der Kontrollaffe (M3) wurde nicht mit Tat-DNA inokuliert, sondern
erhielt als Kontrolle der Spezifität wiederholte Hauttests mit
Tat. Den Affen wurde pCV-Tat zum Zeitpunkt 0 und nach 5, 10, 15,
22, 27, 32 und 37 Wochen injiziert. Schließlich wurden die Affen nach
42 Wochen mit rekombinantem Tat-Protein (16 μg), resuspendiert in 200 μl ISCOMs
und 300 μl
PBS, geboostet. Antikörper
wurden in den Wochen 2, 5, 10, 15, 22, 27, 32, 37, 42, 48 und 58
evaluiert. Die Anti-Tat-Antikörperantwort
in Plasma (1:50 verdünnt)
wurde mittels Elisa analysiert, wie in 2 beschrieben.
Die Ergebnisse sind die durchschnittlichen O.D. von zweifachen Vertiefungen. (A)
zeigt die Ergebnisse, die mit den zwei Affen erhalten wurden, welche
mit 200 (M1) und 500 (M2) μg pCV-Tat-Plasmid
geimpft waren. (B) zeigt die Ergebnisse des Kontrollaffen (M3).
-
12. Titration von Anti-Tat-Antikörpern in
Plasma des mit 200 μg
pCV-Tat i. d. inokulierten Affen M2. Der Elisa ist in 2 beschrieben.
Die Ergebnisse an der Ordinate sind als Kehrwert der höchsten Verdünnung ausgedrückt, bei
welcher der Test noch positiv war.
-
13. Analyse der Anti-Tat IgG-Produktion in drei
Affen (M9 bis M11), inokuliert mit 1 mg pCV-Tat, und in einem Kontrollaffen
(M12), inokuliert mit 1 mg des Kontrollvektors pCV-0. DNA wurde
in 1 ml PBS-A resuspendiert und intramuskulär in zwei Stellen am Rücken injiziert.
Die Affen wurden zum Zeitpunkt 0 und nach 6, 11, 15, 21, 28 und
32 Wochen inokuliert. In der 36. Woche erhielten die Affen M9 bis
M11 einen Boost mit 16 μg
rekombinantem Tat-Protein, resuspendiert in 200 μl ISCOMs und 300 μl PBS. Das
Vorliegen von Anti-Tat-Antikörpern
wurde in den Wochen 2, 6, 11, 15, 21, 28, 32, 36, 40 und 44 evaluiert.
Elisa und Cutoff-Bestimmung sind in 2 beschrieben.
-
14. Kinetik der proliferativen Antwort von PBMC
aus Macaca fascicularis auf die Co-Stimulation mit monoklonalen Anti-CD3-
und Anti-CD28-Antikörpern
auf paramagnetischen Kügelchen
(Anti-CD3/28-Kügelchen).
Aus den PBMC wurde die CD8-positive Subpopulation mittels immunmagnetischer
Verfahren depletiert. Danach wurde die Hälfte der Anti-CD8-depletierten
Lymphocyten ab Tag 3 mit PHA und IL-2 (40 U/ml) stimuliert; den übrigen Teil
ließ man
an den Anti-CD3/28-beschichtete Kügelchen-Antikörpern haften,
wodurch eine CD8-depletierte und CD3/28-positive Lymphocytenpopulation
erhalten wurde. Dieser Zellfraktion wurde IL-2 (40 U/ml) ab Tag
10 der Kultur zugegeben. Alle 2-3 Tage wurden die Zellen gezählt und
ihre Lebensfähigkeit
bestimmt. Das Verhältnis
Kügelchen:Zellen
wurde konstant gehalten. Es ist die Anzahl der Zellen zu verschiedenen
Zeitpunkten angegeben.
-
15. Antiviraler Effekt der Co-Stimulation mit
Anti-CD3/28-Kügelchen
auf PBMC von Macaca fascicularis. Die CD8-depletierten und CD8-depletierten
CD3+/CD28+-Lymphocyten,
erhalten aus 4 Affen (15A bis 15D) durch die in 14 beschriebenen
Verfahren, wurden stimuliert, wie dies in Beispiel 7 beschrieben
ist. Die beiden Fraktionen wurden am Tag 0 mit 0,1 M.O.I. SIVmac251/63M
in vitro infiziert.
-
Die
Stimulation wurde mit PHA und IL-2, zugegeben seit Tag 3, und mit
den Anti-CD3/28-Kügelchen ohne
Zugabe von exogenem IL-2 durchgeführt. Die virale Produktion
wurde durch Bestimmung der p27-Spiegel (ng/ml) in den Zellüberständen an
den Tagen 6 und 12 nach der Infektion evaluiert, wie dies in Beispiel
7 beschrieben ist (hellgrau: PHA+L–2;
dunkelgrau: Anti-CD3/28-Kügelchen
auf PBMC CD8–/CD3+/CD28+).
-
16. Funktionale Charakterisierung dendritischer
Zellen (DC), erhalten aus dem peripheren Blut von Affen. (A) 3H-Thymidininaufnahme am Tag 4 der allogenen,
gemischten Leukozytenkultur (AMLR), zum Vergleich der Antigen-präsentierenden
Funktion (APC, bestimmt als Induktion der Proliferation von allogenen T-Zellen)
von DC und Makrophagen (M⌀),
erhalten aus PBMC von Macaca fascicularis nach Auftrennung auf Percoll-Gradient
und Adhäsion
an Kunststoff. Nicht-adhärente
Zellen wurden entfernt, und adhärente
Zellen wurden durch Zugabe von GMCSF (200 ng/ml) und IL-4 (200 Einheiten/ml)
alle 3 Tage zur Reifung zu DC induziert. Alle 3 Tage wurde die Hälfte des
Kulturmediums (RPMI, 10% FCS) entfernt und durch frisches Medium ersetzt.
Nach 6-7 Tagen wurde eine morphologische Veränderung der Cytokin-induzierten
Zellen beobachtet, die einen typischen DC-Phänotypen (Adhäsionsverlust,
Clusterbildung, Finger) erwarben, was auch durch Bestimmung typischer
Membranmarker verifiziert wurde (Daten nicht gezeigt). Die Monocyten
waren nicht Cytokin-induziert und wurden in dem gleichen Medium
kultiviert, das alle 3 Tage ersetzt wurde. Die Zellen behielten die
Monocyten-Makrophagen-Merkmale, wie die Adhäsion, bei. Am Tag 7 wurden
beide Zellpopulationen mit T-Lymphocyten eines menschlichen Blutspenders
angeregt, durch Ficoll- und Percoll-Gradient und durch Adhäsion gereinigt
und dann eingefroren. Zellproliferationsassays wurden in einer Platte
mit 48 Vertiefungen durchgeführt.
Fünfhunderttausend
T-Lymphocyten wurden mit 5000 DC oder M⌀ stimuliert (Verhältnis T:APC =
100:1). Die Kultur wurde 4 Tage aufrecht erhalten, und festgelegte
Aliquots der Zellsuspension wurden dreifach in Platten mit 96 Vertiefungen übertragen.
Dann wurde 1 μCi 3H-Thymidin über 16 Stunden zugegeben, und
die Counts pro Minute (cpm) des integrierten Präkursors wurden mit einem β-Zähler bestimmt.
-
(B)
APCs, wie DC und M⌀,
erhalten wie in 16A angegeben, wurden
mit T-Lymphocyten
eines anderen Affen angeregt, die, wie oben für den menschlichen Spender
angegeben, erhalten wurden. Die größere Fähigkeit, das Antigen zu präsentieren,
ist ein typisches Merkmal der DC im Vergleich zu M⌀. APCs
wurden in skalaren Konzentrationen zu T-Lymphocyten gegeben, um
die proliferativen Antworten zu evaluieren, die bei unterschiedlichen
Verhältnissen
von T:APCs (DC oder M⌀)
erhalten wurden.
-
Die
folgenden Beispiele sind als veranschaulichend aufzufassen und nicht
als den Umfang der Erfindung einschränkend auszulegen.
-
Beispiel 1. Expression, Reinigung und
Charakterisierung von Wildtyp-Tat-Protein (IIIB-Isolat), mutierten Tat-Proteinen
und Wildtyp-Tat-Peptiden.
-
Früher begegnete
man zahlreichen Schwierigkeiten beim Reinigen und Aufrechterhalten
der biologischen Aktivität
des Tat-Proteins aufgrund der Leichtigkeit, mit der es oxidiert,
aggregiert und seine Aktivität
verliert. Dies ist bedingt durch die hohen Mengen an Cysteinresten,
die intra- und intermolekulare Bindungen bilden können und
somit die Struktur des nativen Proteins (Ref. 159, 41) modifizieren.
Die cDNA oder das tat-Gen (Seq. 1, Beispiel 2), das in den pL-syn-Vektor
kloniert wurde, bereitgestellt von Dr. J. F. DeLamarter und B. Allet (Glaxo
Institute for Molecular Biology S. A., Genf, Schweiz), wurde für die Expression
des Proteins in E. Coli verwendet.
-
Um
eine wirksame Immunisierung mit Tat für Impfstoffzwecke zu erreichen,
betrachtet es die Erfinderin als fundamental, ein biologisch aktives
Tat-Protein zu erhalten, wie dies im Abschnitt „Detaillierte Beschreibung der
Erfindung" beschrieben
ist. Daher beschreiben die Verfahren zur Herstellung und Reinigung
von Tat, die in diesem Beispiel und in den nächsten Beispielen 1B, 2 und
3 beschrieben sind, notwendige Prozeduren und Kontrollen, um ein
biologisch aktives Tat-Protein zu erhalten, das ein wirksames Immunogen
zum Schutz vor HIV-Infektion, AIDS oder vor der Entwicklung von
HIV-bedingten Erkrankungen ist.
-
Ein
erstes Verfahren, das wir einsetzten, um ein aktives Protein zu
erhalten, basierte auf aufeinanderfolgenden Schritten einer Hochdruckflüssigkeitschromatographie
sowie Flüssigkeits-
und Ionenaustauschchromatographie (Ref: 15, 41). Das mit diesen
Verfahren erhaltene Protein hat eine Reinheit von mehr als 95% und
ist aktiv (Ref. 41, 42).
-
Jedoch
wurde aufgrund der Proteinoxidation, die das Hauptproblem der kommerziellen
Tat-Präparate ist,
keine gute Reproduzierbarkeit von Charge zu Charge erhalten. Aufgrund
unserer Beobachtungen, dass die basische Region des Tat-Proteins
eine starke Affinität
gegenüber
Heparin aufweist und dass Heparin dessen Oxidation verhindert, setzten
wir die Heparin-Affinitätschromatographie
ein und definierten ein neues Tat-Reinigungsprotokoll, wie von Chang
et al. (Ref. 26) beschrieben. Zellen (10 g Gewicht) von E. coli,
die Tat exprimieren, wurden in 40 ml Lysepuffer (Dinatriumphosphat
20 mM, pH 7,8; Glycerin bei 2,5%; PMSF 0,2 mM; DTT 5 mM; Mannitol
50 mM; Ascorbinsäure
10 mM; NaCI 500 mM) unter Verwendung eines Ultrasonic Liquid Processor
(Model XL2020, Heat System Inc.) mit drei Entladungen von jeweils
20 Sek. beschallt. Das Lysat wurde 30 Min. bei 12.000 g zentrifugiert,
und der Überstand
wurde eine Stunde bei Raumtemperatur mit 2 ml Heparinsepharoseharz,
das mit dem Lysepuffer vorgewaschen war, inkubiert. Das Harz wurde
auf eine Glassäule geladen
mit dem Lysepuffer gewaschen, bis das Protein in dem Waschmedium
undetektierbar war. Das gebundene Material wurde mit Lysepuffer
eluiert, der 2 M NaCI enthielt, und das Eluat wurde in Fraktionen
von 1 ml gesammelt. Die Homogenität des eluierten Proteins wurde
mittels Gelelektrophorese (SDS-PAGE) analysiert. Das gereinigte
Protein wurde bei –70°C lyophilisiert
gelagert und vor Verwendung in einem entgasten Puffer resuspendiert.
-
Die
biologische Aktivität
von gereinigtem Tat-Protein wurde gemäß dem obigen Protokoll evaluiert durch
einen „Rescue"-Assay viraler Infektion
in HLM-1-Zellen, die von HeLCD4+-Zellen
abgeleitet waren, die Proviren enthielten, denen das tat-Gen fehlte,
und die von Sadaie et al. (Ref. 140) erhalten und beschrieben worden
waren. Der "Rescue"-Assay viraler Infektion,
beschrieben von Ensoli et al. (Ref. 41), bestand darin, die fehlende
Tat-Expression in HLM-1 Zellen (2 × 105)
durch Zugabe von exogenem Tat-Protein (2 μg/ml) zu komplementieren und
die virale Replikation durch die Bestimmung des p24-Antigens, das
im Kulturmedium 48 Stunden nach der Zugabe des exogenen Tat-Proteins
freigesetzt wurde, mit handelsüblichen
Kits zu evaluieren. Die Ergebnisse der von Chang et al. (Ref. 26)
beschriebenen "Rescue"-Experimente zeigen,
dass das mit diesem Verfahren gereinigte Tat-Protein aktiv ist und dieses Reinigungsverfahren
im Vergleich zu den vorbeschriebenen Verfahren (Ref. 40, 41, 42)
sowohl hinsichtlich der Reinheit als auch der biologischen Aktivität von Tat
besser, einfacher und kostengünstiger
ist.
-
Unterschiedliche
Präparate
von rekombinantem Tat, die wie oben beschrieben gereinigt waren,
wurden in Gegenwart von Freund-Adjuvans gemäß üblichen Protokollen in Mäuse und
Kaninchen inokuliert (Ref. 4). Die Ergebnisse der durch die Immunisierung
induzierten Antikörperantwort
sind in Tabelle 1 gezeigt. TABELLE 1. Analyse der Antwort Anti-Tat-spezifischer
Antikörper
in Seren von Mäusen
und Kaninchen, immunisiert mit dem rekombinanten Tat-Protein.
| Anti-Tat-Antikörper | OD-ELISA/Tat | Western
Blot |
| 1:500 | 1:1000 | 1:2000 |
| Kaninchen
Maus | 0,651
0,502 | 0,400
0,240 | 0,175
0,150 | +
+ |
-
Das
in E. coli produzierte, rekombinante Tat-Protein wurde eingesetzt,
um Mäuse
und Kaninchen gemäß üblichen
Immunisierungsprotokollen zu immunisieren (Ref. 4). Die Seren der
immunisierten Tiere wurden mittels ELISA auf das Vorliegen von Anti-Tat-Antikörpern unter
Verwendung von drei Serumverdünnungen (1:500
bis 1:2000) analysiert. Die Ergebnisse sind der Mittelwert der Messwerte
von zwei Kaninchen und drei Mäusen
bei 405 nm. Darüber
hinaus wurden die Seren mittels Western Blot mit dem rekombinanten
Tat-Protein (100
ng) getestet.
-
Die
Ergebnisse von Tabelle 1 zeigen, dass das von uns hergestellte,
rekombinante Tat in der Lage war, in beiden Tierspezien eine Antikörperantwort
zu induzieren, wie dies mit ELISA und Western Blot unter Verwendung
des rekombinanten Tat-Proteins getestet wurde. Diese Antikörper waren
in der Lage, die Internalisierung und die biologischen Aktivitäten von
Tat zu inhibieren (Ref. 40, 41, 42). Der pL-syn-Vektor und das Reinigungsprotokoll
des Tat-Proteins
werden eingesetzt, um die in Beispiel 2 beschriebenen Mutanten von
Tat zu exprimieren und zu reinigen. Die biologische Aktivität der mutierten
und gereinigten Tat-Proteine
wird mittels „Rescue"-Assays der viralen
Infektion in HLM-1 Zellen, Assays der Proliferation von KS-Zellen
und in vivo in Mäusen
bestimmt, wie dies oben für
das Wildtyp-Tat-Protein
beschrieben wurde. Darüber
hinaus werden die mutierten Tat-Proteine in Gegenwart von Wildtyp-Tat
(in Reihenkonzentrationen) getestet, um den negativen, transdominanten
Effekt auf die nicht-virale Replikation zu verifizieren. Der pL-syn-Vektor
und das Reinigungsprotokoll werden verwendet, um Fusionsproteine
dieses Typs zu exprimieren und zu reinigen: Tat (Wildtyp oder Mutanten
davon)/IL-12 oder Tat (Wildtyp oder Mutanten davon)/IL-15 oder Teile
desselben oder Tat (Wildtyp oder Mutanten davon)/andere Moleküle (oder
Teile davon), die in der Lage sind, die Immunantwort auf Tat alleine
oder in Verbindung mit anderen viralen Produkten zu verstärken. Rekombinante
Fusionsmoleküle
werden unter Verwendung der in den Beispielen 2 und 3 beschriebenen
Sequenzen und Primer hergestellt. Als Alternative werden synthetische
Peptide, die Regionen von Tat oder von anderen viralen Produkten
oder von Cytokinen entsprechen, welche in Kombination mit Tat zu
verwenden sind, als Immunogene eingesetzt. Die Peptidsequenzen von
Tat sind:

-
Die
Tat-Mutantenpeptide enthalten die gleichen Aminosäuresubstitutionen
der mutierten Tat-Proteine, die
in Beispiel 2 beschrieben sind. Die Peptide werden in Kombination
mit dem Peptid, welches das universelle T-Hilfsepitop des Tetanoustoxoids
darstellt, oder mit anderen Peptiden, die T-Hilfsepitope darstellen,
verwendet (Ref. 77).
-
Beispiel 1A. Aufnahme pikomolarer Konzentrationen
(10 bis 100 ng/ml) von biologisch aktivem Tat durch aktivierte Endothelzellen
wird von Integrin-Rezeptoren vermittelt.
-
Wenn
normale Endothelzellen in vitro mit inflammatorischen Cytokinen
aktiviert werden, sprechen sie auf die Effekte von extrazellulärem Tat
an, was durch die Induktion der α5β1- und αvβ3-Integrine
bedingt ist (Ref. 9, 10). Gleichermaßen erhöhen inflammatorische Cytokine
(IC) oder bFGF die Integrinexpression auf Endothelzellen in vivo,
was zu einem synergistischen KS-fördernden Effekt führt, wenn
ein biologisch aktives Tat in Mäuse
gleichzeitig mit oder nach bFGF inokuliert wird (Ref. 42). Zudem
erwerben IC-aktivierte Endothelzellen eine APC-Funktion.
-
In
diesem Beispiel ist gezeigt, dass mit IC aktivierte Endothelzellen
rhodaminierte, biologisch aktives Tat-Protein effizienter aufnehmen,
und dass dies durch die Integrinrezeptoren vermittelt wird.
-
Wegen
der Schwierigkeit, die Internalisierung sehr niedriger Konzentrationen
von kaltem Tat zu beobachten, wurde das Protein mit Rhodamin markiert
(Ref. 98). Das rhodaminierte Tat zeigte noch immer eine Aktivierung
der KS-Zellproliferation im gleichen Konzentrationsbereich wie unmarkiertes
Tat, was darauf hindeutet, dass der Markierungsvorgang dessen biologische
Funktion nicht beeinträchtigte.
Tat-Aufnahmeexperimente
wurden wie folgt durchgeführt:
humane Umbilikalvenen (HUVE)-Zellen
wurden 5 Tage mit IC gezüchtet
und behandelt, wie beschrieben (Ref. 9, 46). Dann wurden die Zellen
trypsiniert, auf Objektträgern
mit 8 Vertiefungen (Nunc Inc., Naperville, IL) bei 0,5 × 105 Zellen pro Vertiefung plattiert und 18
Stunden in Medium, das 15% fötales
Rinderserum (FBS) enthält,
in Gegenwart von IC inkubiert. Serumfreie (SF, RPMI, 1% BSA, 0,1%
Antibiotika, Fungizon) Medien wurden zugegeben und die Träger 2 h
bei 4°C
vorinkubiert. Frisches Medium, enthaltend rhodaminiertes Tat in
Reihenverdünnung,
wurde den Zellen zugegeben, die Zellen über den angegebenen Zeitraum
bei 37°C
inkubiert wurden. Negative Kontrollen waren rhodaminiertes BSA in
dem gleichen Puffer wie Tat. Die Zellen wurden in eiskaltem Aceton-Methanol
(1:1) fixiert und die Aufnahme und Lokalisierung von Tat mittels
Fluoreszenzmikroskopie visualisiert und photographiert. Die Ergebnisse
wurden durch Vergleichen der Fluoreszenz von Proben mit der negativen
Kontrolle evaluiert und bezüglich
der Aufnahmemenge ohne vorherige Kenntnis des Probencodes von 0
bis ++++ bewertet.
-
Um
die Wege zu untersuchen, über
die Tat von aktivierten Endothelzellen aufgenommen wird, wurden Experimente
unter Verwendung von aktivierten HUVE-Zellen mit einem großen Bereich
von Konzentrationen an exogenemen Tat durchgeführt, wie sie zuvor zum Induzieren
des HUVE- oder KS-Zellwachstums (10-50 ng/ml) oder zur HIV-1-Transaktivierung
durch Zugabe des Proteins zu Zellen, die den HIV-1-Promoter oder
das Provirus tragen (0,5 bis 1 μg/ml),
verwendet worden waren.
-
In
diesen Experimenten wurden zur Übereinstimmung
mit Aufnahmeinhibierungsexperimenten (siehe unten) Zellen 2 Stunden
bei 4°C
mit Medium vorinkubiert, dem fötales
Kälberserum
fehlte. Diese Vorinkubation hat keinen Einfluss auf die nachfolgende
Aufnahme von rhodaminiertem Tat.
-
Mit
rhodaminiertem Tat begann die Aufnahme und Translokation des Proteins
am Kern oder an den Nucleoli aktivierter HUVE-Zellen innerhalb von
15 Minuten der Inkubation mit nur 10 ng/ml rhodaminiertem Tat sichtbar
zu werden. Die Dichte des aufgenommenen Tat in den Zellen nahm dosis-
und zeitabhängig
zu. Rhodaminiertes BSA oder rhodaminierter Puffer zeigten keine
Signale und wurden routinemäßig als
negative Kontrollen verwendet.
-
Um
zu bestimmen, ob die Aufnahme von Tat in aktivierten HUVE-Zellen
durch die gleichen Integrine vermittelt wurde, wie sie auf KS-Zellen
exprimiert zu finden sind, wurden Inhibierungsexperimente durch
Vorinkubieren IC-aktivierter Endothelzellen mit kaltem Tat (Kompetitor),
den physiologischen Liganden für
diese Rezeptoren, wie Fibronektin (FN) oder Vitronektin (VN), oder
durch Vorinkubieren der Zellen mit monoklonalen Antikörpern, die
gegen die RGD-Bindungsregionen der α
5β
1-
und α
vβ
3-Rezeptoren gerichtet sind, durchgeführt. Über das
experimentelle Vorgehen wird kurz berichtet. Nach dem Plattieren
auf Trägern
mit 8 Vertiefungen wurden HUVE-Zellen 18 h mit Medium, das 15% FBS
enthielt, und dann mit SF-Medium, enthaltend unmarkiertes Tat (kalter
Kompetitor) (Tabelle 1A), FN, VN (Tabelle 1B) oder monoklonale Antikörper, die
gegen die RGD-Bindungssequenz der FN- oder VN-Rezeptoren (α
5β
1 bzw. α
vβ
3)
gerichtet sind, oder monoklonale Antikörper, die gegen den menschlichen
Faktor VIII gerichtet sind (Kontroll-Antikörper)(
1A),
2 h bei 4°C
inkubiert. Dann wurden die Zellen über die angegebenen Zeiträume mit
rhodaminiertem Tat inkubiert. Die Kontrolle bestand aus Zellen,
die 2h bei 4°C
nur mit SF-Medium behandelt und mit rhodaminiertem BSA inkubiert
wurden. Die Zellen wurden fixiert, visualisiert und photographiert,
und die Ergebnisse wurden wie oben angegeben bewertet. TABELLE 1A Inhibierung der Aufnahme von 100 ng/ml
und 1 μg/ml
rhodaminiertem Tat durch Cytokin-aktivierte
HUVE mittels Vorinkubation der Zellen mit 1 μg/ml unmarkiertem Tat.
a | Vorinkubation | Rhodaminiertes
Tat | Aufnahme
von Tat |
| Serumfreies
Medium | 100
ng/ml | +++ |
| 1 μg/ml unmarkiertes
Tat | 100
ng/ml | +/– |
| Serumfreies
Medium | 1 μg/ml | ++++ |
| 1 μg/ml unmarkiertes
Tat | 1 μg/ml | +/– |
- aHUVE-Zellen wurden
wie zuvor beschrieben kultiviert (Ref. 40). IC wurden aus mit humanem
T-lymphotrophem Virus Typ II (HTLV-II) transformierten CD4+ T-Zellen oder Phytohämagglutinin-stimulierten T-Zellen
erhalten und die Überstände verwendet
(1:8), um die HUVE-Zellen (Passage 8-14) 5 Tage zu aktivieren, wie
dies zuvor beschrieben wurde (Ref. 9, 46). Dieser Überstand
enthält
Interleukin-1α (IL-1α) und -β (IL-1β), Tumornekrosefaktor-α (TNF-α) und -β (TNF-β) sowie Interferon-γ (IFN-γ) (Ref. 9).
-
Das
Tat-Protein wurde an Lysinresten im Wesentlichen wie beschrieben
rhodaminiert (Ref. 98). Kurz gesagt, wurden 50 μg rekombinantes Tat (2 mg/ml)
durch Zugabe von 2,5 μl
1M Na2CO3 auf pH
9,0 gebracht. 2,5 μl
von 1 mg/ml TRITC in Dimethylsulfoxid (DMSO) wurden zugegeben, und
man ließ die
Reaktion 8 h bei 4°C
ablaufen. Nicht umgesetztes TRITC wurde durch Zugabe von 2,5 μl 0,5M NH4CI gequencht, der pH wurde unter Verwendung
von 1 M HCl auf 7,0 gesenkt, und das rhodaminierte Tat wurde mit
zwei Wechseln von 50 mM Tris-HCI, pH 7,0, 1 mM Dithiothreitol (DTT),
dialysiert, um das abgeschreckte TRITC abzuziehen. In gleicher Weise
rhodaminierte(s) BSA oder PBS wurde als negative Kontrolle verwendet.
Rhodaminiertes Tat wurde auf die AIDS-KS-Zellwachstumsaktivität getestet, wie beschrieben,
um sicher zu stellen, dass die biologische Aktivität erhalten
blieb (Ref. 40).
-
HUVE-Zellen
wurden 2 Stunden mit serumfreiem Medium oder 1 μg/ml unmarkiertem Tat in serumfreiem
Medium vorinkubiert, dann 60 Minuten mit 100 ng/ml oder 1 μg/ml rhodaminiertem
Tat inkubiert, und die Tat-Aufnahme wurde mittels Fluoreszenzmikroskopie
visualisiert. Die negativen Kontrollen (+/– Aufnahme) waren Vorinkubation
mit serumfreiem Medium, gefolgt von Inkubation mit rhodaminiertem
BSA. TABELLE 1B Inhibierung der Aufnahme von 10 ng/ml
rhodaminiertem Tat durch Cytokin-aktivierte HUVE mittels Vorinkubation
der Zellen mit einem Überschuss
von FN oder VN.
a | Vorinkubation | Aufnahme
von rhodaminiertem Tat |
| Serumfreies
Medium | ++++ |
| 100
ng/ml FN | +/– |
| 100
ng/ml VN | +/– |
- aHUVE-Zellen wurden
2 Stunden mit serumfreiem Medium oder FN oder VN in serumfreiem
Medium vorinkubiert, dann 60 Minuten mit 10 ng/ml rhodaminiertem
Tat inkubiert, und die Tat-Aufnahme wurde mittels Fluoreszenzmikroskopie
visualisiert. Die negativen Kontrollen (+/– Aufnahme) waren die Vorinkubation
mit serumfreiem Medium, gefolgt von Inkubation mit rhodaminiertem
BSA.
-
Die
Aufnahme von Tat wurde mit kaltem Tat (Tabelle 1A), mit FN oder
VN (Tabelle 1B) oder durch vorherige Behandlung der Zellen mit monoklonalen
Antikörpern,
die gegen die RGD-Bindungsregionen sowohl des FN-Rezeptors α5β1 als
auch des VN-Rezeptors αvβ3 gerichtet waren, inhibiert (1A). Die Intensität der Fluoreszenz in den Zellen
wurde auf Werte verringert, die bei der negativen Kontrolle zu sehen
sind, und vor der Inkubation der Zellen mit gegen den Humanfaktor
VIII gerichteten monoklonalen Antikörpern, verwendet als negative
Kontrolle, wurde keine Inhibierung beobachtet, was darauf hinweist,
dass die Inhibierung spezifisch war (1A).
-
Die
Aufnahme und Kernlokalisierung von 100 ng/ml Tat wurde inhibiert
durch Vorinkubation der Zellen mit den monoklonalen Antikörpern, die
gegen die RGD-Bindungsregion
des α5ß1-Rezeptors und des αvβ3-Rezeptors
gerichtet waren. In beiden Fällen
war die Inhibierung jedoch nicht vollständig. Diese Ergebnisse zeigen,
dass die Aufnahme pikomolarer Konzentrationen von Tat durch die
gleichen Integrine vermittelt wird, die an der Zelladhäsion an
Tat beteiligt sind (Ref. 10). Bei einer höheren Konzentration von extrazellulärem Tat (wie
z. B. ≥ 100
ng/ml) ist ein nicht von Integrinen vermittelter Weg für die Aufnahme
eines Teils des Proteins verantwortlich.
-
Im
Gegensatz zu diesen Ergebnissen wurde gezeigt, dass die Aufnahme
von iodiertem Tat mit Lymphocyten- und Epithelzelllinien linear
und abhängig
von der Tat-Konzentration im Medium erfolgte und keine oder nur
geringe Konkurrenz von einem Überschuss
an kaltem Tat bekam, was auf eine fehlende Rezeptorbeteiligung hinweist
(Ref. 98). Allerdings betrug der Konzentrationsbereich von Tat in
dem Medium bei dieser Studie etwa 1-100 μg/ml (Ref. 98), was deutlich
höher ist
als die Konzentrationen, die benötigt
werden, um die Aufnahme von Tat durch Zellen, die auf seine biologische
Aktivität
ansprechen, wie aktivierte primäre
Endothelzellen, zu beobachten. Zudem kann die Iodierung von Tat
dessen Struktur und dessen Aufnahme durch die Zellen erschweren,
und von diesen Autoren wurden keine Ergebnisse für die biologische Aktivität von iodiertem Tat
gezeigt.
-
Diese
Ergebnisse, die noch unveröffentlicht
sind, zeigen, dass die Aufnahme von Tat auf mindestens zwei Wegen
erfolgt, die von der Konzentration des Proteins abhängen. Bei
niedrigen Tat-Konzentrationen (10-100 ng/ml) wird die Tat-Aufnahme
von den α5β1- und αvβ3-Rezeptoren durch die Wechselwirkung mit
der RGD-Sequenz des Proteins vermittelt, während bei einer höheren Konzentration
an extrazellulärem
Tat ein Integrin-unabhängiger
Weg wichtiger ist. Die Integrin-vermittelte Aufnahme pikomolarer
Konzentrationen von Tat durch IC-aktivierte Endothelzellen weist
auf ein vollständig
aktives Protein hin, das in Antigene präsentierende Zellen, wie aktivierte
Endothelzellen und dendritische Zellen, welche die Immunantwort
initiieren, eindringen kann.
-
Beispiel 2. Konstruktion und Charakterisierung
von mutierten tat-Genen (nicht beansprucht)
-
Wir
produzierten 19 Mutanten in unterschiedlichen Tat-Regionen mittels
ortsspezifischer Mutagenese oder mittels Deletion. Die Sequenz jeder
mutierten DNA wurde durch Sequenzierung kontrolliert. Die cDNAs der
mutierten tat-Gene wurden in die Pst1-Stelle des pCV0-Vektors kloniert,
wie dies in Beispiel 3 beschrieben ist. Jede Mutante wurde, wie
von Ensoli et al. beschrieben (Ref. 41), in COS-1-Zellen oder in
die Jurkat T-Zelllinie mit dem HIV-1 LTR-CAT-Plasmid cotransfiziert,
in dem das CAT-Reportergen von den HIV-1-LTR gesteuert wird. Die
unveröffentlichten
Ergebnisse dieser Experimente sind in Tabelle 2 angegeben. TABELLE 2 Effekt von Tat-Mutanten auf die HIV-1
LTR-CAT-Transaktivierung und blockierender Effekt (negative Transdominante)
gegenüber
der Tat-Wildtyp-Aktivität
| MUTANTEN | Transaktivierende
Aktivitäta | Transdominante Aktivitätb (%
Inhibierung) Mittelwert |
| Mittelwert
(Faktor) | (Min-Max-Werte) |
| CYS
22 | 0,09 | (0,021-0,22) | 21 |
| THR
23 | 0,36 | (0,16-1) | |
| THR
23A | 0,30 | (0,16-0,78) | |
| ASN
24 | 0,34 | (0,34-0,82) | |
| ASN
24A | 0,42 | (0,45-0,95) | |
| TYR
26 | 0,14 | (0,08-0,19) | |
| LYS
28/29 | 0,52 | (0,19-1,04) | |
| CYS
30 | 0,30 | (0,045-0,65) | |
| CYS
31 | 0,60 | (0,27-1,09) | |
| PHE
32 | 0,31 | (0,077-0,097) | |
| LYS
33 | 0,04 | (0,0027-0,068) | 46 |
| GLU
35 | 0,31 | (0,19-0,43) | |
| PHE
38 | 0,05 | (0,043-0,057) | 98 |
| LYS
41 | 0,04 | (0,025-0,061) | 97 |
| TYR
47 | 0,58 | (0,31-0,8) | |
| 57
A | 0,35 | (0,26-0,44) | |
| TAT-RGD | 0,94 | (0,73-1,15) | |
| TAT-KGE | 1,11 | (0,67-1,49) | |
| TAT-Wildtyp | 1 | 1 | |
- aDie Ergebnisse
sind als Inkrement der Aktivierung der CAT-Aktivitätswerte
angegeben, die durch Wildtyp Tat (Faktor = 1) induziert werden. bDie Ergebnisse sind als prozentuale (%)
Inhibierung der Wildtyp-Tat-Aktivität ausgedrückt.
-
Aus
den in Tabelle 2 angegebenen Ergebnissen ist ersichtlich, dass bei
der Mehrheit der Mutanten der transaktivierende Effekt der HIV-1
LTR verringert war oder fehlte, mit Ausnahme der RGD-Mutante, die
eine ähnliche
Aktivität
wie Wildtyp Tat aufwies. Wir wählten
die 4 Mutanten (cys22, lys33, phe38, Iys41) mit der niedrigsten
(nahezu Null) transaktivierenden Aktivität und bestimmten den negativen,
transdominanten Effekt auf die transaktivierende Aktivität von Wildtyp-Tat.
Zu diesem Zweck wurden COS-1 Zellen mit jedem Vektor, der eine Tat-Mutante
enthielt, und dem pCV-Tat-Vektor (in einem Molverhältnis von
10:1) in Gegenwart des HIV-1 LTR-CAT-Vektors cotransfiziert. Wie
in Tabelle 2 gezeigt, inhibierten die lys41- und phe38-Mutanten
die Tat-Aktivität
nahezu vollständig,
während
die lys33- und cys22-Mutanten die Tat-Aktivität nur teilweise inhibierten.
Allerdings konkurrierte das cys22-rekombinante Protein (beschrieben
im folgenden Beispiel 3) mit dem Wildtyp-Tat-Protein um die Transaktivierung
von HIV-1 LTR-CAT
(1B). Gewählt
wurden eine Mutante in der Cysteinregion (cys22), eine in der Kernregion
(lys41), eine mit deletierter RGD-Sequenz (RGDΔ) und eine Doppelmutante, welche
die Mutation bei Iys41 und die Deletion der RGD-Sequenz (Iys41-RGDΔ) enthielt.
-
Nachfolgend
sind die Sequenz des tat-Inserts und die für die Impfung ausgewählten Mutanten
angegeben. Dabei wird eine Reihe von tat-Mutanten beschrieben, hergestellt
durch 1) Substitution einer Base, um eine Aminosäuresubstitution zu erhalten,
und 2) Deletion einer Base, um eine Deletion der entsprechenden Aminosäuren zu
erhalten. Die Substitutionen und Deletionen wurden durch ortsgerichtete
Mutagenese erhalten. Die Sequenzen des Wildtyp-tat-Gens und der tat-Gen-Mutanten,
die nachfolgend angegeben sind, wurden in den pCV0-Plasmidvektor wie
oben beschrieben eingesetzt.
-
Mit
Seq. 1 ist die HIV-1 tat-Gensequenz des BH-10-Klons und seines abgeleiteten
Proteins gemeint. Mit Seq. 2 ist die cys22-Mutantensequenz (und
ihr abgeleitetes Protein) gemeint, dargestellt durch eine Substitution
des Thymin(T)-Nukleotids in Position 64, beginnend vom 5'-Ende mit dem Guanin(G)-Nukleotid.
Diese Substitution erzeugt in der abgeleiteten Aminosäuresequenz
eine Substitution eines Cysteins (C im Einbuchstabencode) in Position
22 am aminoterminalen Ende, mit einem Glycin (G im Einbuchstabencode).
Mit Seq. 3 ist die lys41-Mutantensequenz (und ihr abgeleitetes Protein)
gemeint, dargestellt durch eine Substitution des Adenin(A)-Nukleotids
in Position 122 vom 5'-Ende
mit dem Cytosin(C)-Nukleotid.
Diese Substitution erzeugt in der abgeleiteten Aminosäuresequenz
eine Substitution eines Lysins (K im Einbuchstabencode) in Position 41
vom aminoterminalen Ende mit einem Threonin (T im Einbuchstabencode).
Mit Seq. 4 ist eine Sequenz der RGD-Mutante (und ihres abgeleiteten Proteins)
gemeint, dargestellt durch die Deletion der Nukleotidsequenz CGAGGGGAC
von Nukleotid 232 bis Nukleotid 240, beginnend vom 5'-Ende des Wildtyp-tat-Gens. Dies ergibt eine
Deletion der Aminosäuren
Arginin-Glycidin-Asparaginsäure (RGD
im Einbuchstabencode) in den Positionen 78-80 vom aminoterminalen
Ende. Mit Seq. 5 ist eine Sequenz der lys41-RGDΔ-Doppelmutante (und ihres abgeleiteten
Proteins) gemeint, hervorgegangen aus einer Kombination der oben
beschriebenen Mutanten.
-
Wildtyp-tat-Nukleotidsequenz
(Seq. 1) (Sequenz ID Nr. 1)
-
-
Aminosäuresequenz
(Sequenz ID Nr. 2)
-
Cys22-Mutanten-Nukleotidsequenz
(Seq. 2) (Sequenz ID Nr. 3)
-
Aminosäuresequenz
(Sequenz ID Nr. 4)
-
Lys41-Nukleotidsequenz
(Seq. 3) (Sequenz ID Nr. 5)
-
Aminosäuresequenz
(Sequenz ID Nr. 6)
-
RGDΔ-Mutanten-Nukleotidsequenz
(Seq. 4) (Sequenz ID Nr. 7)
-
Aminosäuresequenz
(Sequenz ID Nr. 8)
-
Lys41-RGDΔ-Mutanten-Nukleotidsequenz
(Seq. 5) (Sequenz ID Nr. 9)
-
Aminosäuresequenz
(Sequenz ID Nr. 10)
-
Beispiel 3. Konstruktion und Charakterisierung
der DNA-Immunogene.
-
Die
DNA-Moleküle
für die
Inokulierung von Tieren werden in den 6,4 kb pCV0-Plasmidvektor eingesetzt
(Ref. 5). Dieses Plasmid umfasst zwei SV40-Replikationsursprünge, den
späten
Hauptpromoter des Adenovirus (AdMLP) und die Spleiss-Sequenzen des
Adenovirus und der Mäuse-Immunglobulingene,
die cDNA des Mäuse-Dihydrofolatreduktasegens
(dhfr) und das SV40-Polyadenilierungssignal. Die Stelle für das Pst1-Restriktionsenzym
liegt an der 3'-Position
des AdMLP und stellt die Stelle dar, in die das interessierende exogene
Gen kloniert wird. Die tat-Gen-cDNA (261 Basenpaare) (Seq. 1, Beispiel
2) von HIV-1 wurde vom HIV-1 BH10-Klon abgeleitet (Ref. 126) und
kodierte ein 86 Aminosäuren
langes Protein. Der pCV-Tat-Vektor (Ref. 5) wurde erhalten durch
Klonieren der tat-cDNA in die pCV0 Pst1-Stelle, gesteuert durch
den AdMLP. Die Wahl dieses Vektors basiert darauf, dass der AdMLP
eine höhere
Expression und Freisetzung von Tat gegenüber anderen eukaryontischen
Promotern, wie z. B. dem Promoter der Immediate Early-Region des
Cytomegalovirus (CMV), induzierte, wie von Ensoli et al. (Ref. 41)
gezeigt und in Tabelle 3 angegeben. TABELLE 3. Expression, subzelluläre Lokalisierung,
Freisetzung und Aktivität
von Tat in COS-1-Zellen, transfiziert mit pCV-Tat und CMV-Tat
a.
| Vektoren | Tat-Expression | Tatb-Gehalt | Tat-Aktivität |
| Positive
Zellen | Kernc | (%) | Cytoplasma | Gesamt | Intrazell.
(%) | Extrazell.
(%) | Intrazell.d
(Faktor) | Extrazell.c
(cpm) |
| PCV-Tat
CMV-Tat
Kontrolle | 5-10
3-5
0 | ++
++
– | ++
+
– | 25
14,6
0 | 63,5
92,2
0 | 36,5
7,8
0 | 50
72
1 | 2.478
2.254
1.400 |
- aCOS-1-Zellen (5 × 106) wurden mittels Elektroporation mit 30 μg pCV-Tat,
CMV-Tat oder einer Kontroll-DNA transfiziert. 48 Stunden nach Transfektion
wurde die Tat-Expression mittels Immunhistochemie mit monoklonalen
Anti-Tat-Antikörpern
(die angegebenen Werte sind Mittelwerte der Prozentwerte positiver
Zellen) und durch Lokalisierung von Kern- und Cytoplasma-Tat evaluiert.
Das Vorliegen von intra- und extrazellulärem Tat wurde mittels Radioimmunpräzipitation
auf den Zellextrakten (500 μl)
und in den Kulturmedien (4 ml) und anschließende densitometrische Ablesung
(Gelscan XL; Pharmacia) der präzipitierten
Tat-Banden analysiert.
Die Aktivität
von intrazellulärem
Tat wurde auf Zellextrakten von COS-1-Zellen gemessen, cotransfiziert mit
Tat-Expressionsvektoren oder dem Kontrollvektor und mit dem LTR-CAT-HIV-1-Plasmid;
die extrazelluläre
Tat-Aktivität
hinsichtlich der AIDS-KS-Zellenproliferation
(bestimmt mittels 3H-Thymidinaufnahme-Assay)
wurde im Kulturmedium (1:2 und 1:4 verdünnt) der Zellen bestimmt, die
mit Tat exprimierenden Plasmiden oder mit dem Kontrollplasmid transfiziert
waren. Die Ergebnisse stellen den Durchschnitt aus fünf unabhängigen Experimenten
dar.
- bDensitometrische Analyse der immunpräzipitierten
Tat-Proteinbande. Die Werte sind in einem willkürlichen Maßstab ausgedrückt, wobei
der gesamte detektierte Mindestwert (intra- und extrazelluläres Tat) bei 10 liegt.
- c–:
negativ; +: 50% Tat-positive Zellen; ++: 50-100% Tat-positive Zellen.
- dCAT-Aktivität nach 20 Minuten Inkubation
gegenüber
dem Kontrollvektor, dessen Aktivierungswert als 1 betrachtet wird.
- eDas AIDS-KS-Zellwachstum wurde mit
einem 3[H]-Thymidinaufnahme-Assay (Standardabweichung
SD: 12%) bestimmt. Die Überstände der
mit der Kontroll-DNA transfizierten Zellen wiesen eine 3[H]-Thymidinaufnahme
von 1.400 cpm (SD: 11,5%) auf.
-
Das
Kulturmedium, das von aktivierten T-Lymphocyten (positive Kontrolle)
stammte, wies eine 3[H]-Thymidinaufnahme
von 2.400 cpm (SD: 10%) auf.
-
Tabelle
3 zeigt, dass in den pCV-Tat-transfizierten Zellen im Vergleich
zu den CMV-Tat-transfizierten Zellen
der Prozentsatz Tat-positiver Zellen und der Gesamtgehalt an Tat
höher sind,
die Menge an freigesetztem Tat deutlich höher ist und mit dem Gesamtgehalt
und dem Cytoplasmagehalt von Tat zusammenhängt, und die biologische Aktivität von extrazellulärem Tat
hinsichtlich des AIDS-KS-Zellwachstums daher stärker ist. Diese Ergebnisse
zeigen, dass der pCV-Tat-Vektor für ein biologisch aktives Protein
kodiert, hohe Expressionsniveaus des tat-Gens induziert und aus
den Zellen deutlich höhere
Tat-Mengen freisetzen kann als der CMV-Tat-Vektor.
-
Der
pCV0-Vektor wird auch zur Expression der HIV-1-nef-, -rev- und -gag-Gene
und der Gene, die für IL-12-
und IL-15-Cytokine kodieren, eingesetzt. Die cDNAs der nef- (618
Basenpaare, Stamm NL43) (Ref. 112) rev- (348 Basenpaare, Stamm NL43)
(Ref. 95) und der gag-Gene (1500 Basenpaare, Stamm NL43) (Ref. 95), oder
die cDNAs der IL-12- (Ref. 165) oder IL-15-Gene (Ref. 56) werden
durch die Technik der Polymerasekettenreaktion (PCR) amplifiziert,
wobei spezifische Primer verwendet werden, die komplementär zu den
ersten 15 Nukleotiden der 5'-Region
(Vorwärtsprimer)
(Seq. P1, P3, P5, P7, P9) oder zu den letzten 15 Nukleotiden der
3'-Region des Gens
(Rückwärtsprimer)
(Seq. P2, P4, P6, P8, P10) sind. Darüber hinaus umfasst jeder Primer,
ob vorwärts
oder rückwärts gerichtet,
die Sequenz für
das Restriktions-Pst1-Enzym, um das Klonieren des amplifizierten
Produktes in den pCV0-Vektor
zu gestatten. Nach dem Klonieren wird die Sequenz der insertierten
Gene durch DNA-Sequenzierung
kontrolliert. Der pCV0-Vektor wird auch zur Tat-Coexpression mit anderen
viralen Genen von fHIV-1 (rev, nef oder gag) oder mit den IL-12-
oder IL-15-Cytokinkodierenden Genen verwendet. Zu diesem Zweck wird
die cDNA des HIV-1-tat-Gens mit 261 Basenpaaren (Seq. 1, Beispiel 2)
mittels PCR mit einem Vorwärtsprimer,
der die Sequenz für
das Pst1-Restriktionsenzym (Seq. P11) umfasst, und mit einem Rückwärtsprimer,
der komplementär
zu den letzten 15 Nukleotiden des tat-Gens (Seq. P12) ist, amplifiziert.
Die viralen Gene (nef, rev oder gag) oder die Gene, welche für die IL-12-
oder IL-15-Cytokine kodieren, werden durch einen Vorwärtsprimer,
der auch eine Sequenz von 15 Basen umfasst, die komplementär zur tat-3'-Region ist, wodurch
das Gen in einem Rahmen mit dem tat-Gen (Seq. P13, P14, P15, P16,
P17) liegt, und durch einen Rückwärtsprimer,
der die Sequenz für
das Pst1-Restriktionsenzym (Seq. P2, P4, P6, P8, P10) umfasst, amplifiziert.
Anschließend wird
eine dritte PCR-Reaktion durchgeführt, bei der die DNA-Matritze durch
die amplifizierten Produkte des tat-Gens und des Gens von Interesse
dargestellt wird. Der Vorwärtsprimer
ist durch den Primer dargestellt, der eingesetzt wird, um tat (Seq.
P11) zu amplifizieren, und der Rückwärtsprimer
durch den, der eingesetzt wird, um das interessierende Gen zu amplifizieren
(Seq. P2, P4, P6, P8, P10). Das amplifizierte tat/interessierende
Gen wird mit Agarosegel gereinigt, mit Pst1 verdaut und in pCV0 kloniert.
-
Nach
dem Klonieren wird die Sequenz der insertierten Gene durch DNA-Sequenzierung
kontrolliert, während
die Proteinexpression anhand der Transfektion bestimmt wird, wie
dies oben beschrieben ist (Ref. 41).
-
Die
Sequenzen der oben erwähnten
Primer lauten:
-
Beispiel 4. Inokulierung eines Anti-Tat-Proteinimpfstoffs
in gesunde Macaca fascicularis: Evaluation von Sicherheit, Verträglichkeit,
spezifischer Immunantwort und Wirksamkeit des Schutzes gegen Virusanregung
-
Die
Verträglichkeit,
die Sicherheit und die Fähigkeit,
eine spezifische Immunantwort (humoral und zellulär) hervorzurufen,
und die Schutzwirkung gegen eine Virenanregung eines rekombinanten
Tat-Proteinimpfstoffs, hergestellt durch das beschriebene Verfahren
und gereinigt durch Heparinaffinitätssäulen, wurde im Versuchsmodell
mit Cynomolgus-Affen (Macaca fascicularis) beurteilt. Um eine breite
Immunantwort zu induzieren, verwendeten wir Aluminiumphosphat (Alaun),
das in zahlreichen Modellen getestet wurde, als einziges zur Verwendung
beim Menschen zugelassen ist. Als teilchenförmige Adjuvanzien verwendeten
wir RIBI (zur Gruppe der Emulgatoren gehörig oder aus Monophosphoryl-Lipid
A, dimycolischem Trehasol und dem Skelett der Bakterienwand des
Calmette-Guerin-Bazillus zusammengesetzt) (Ref. 7,109).
-
Im
ersten Pilotversuch evaluierten wir die Verträglichkeit, die Sicherheit und
die Fähigkeit
zum Hervorrufen einer spezifischen Immunantwort (humoral und zellulär). Dabei
wurden 3 Affen gemäß folgendem
Schema inokuliert: Affe 1 (M1) wurde mit dem rekombinanten Tat-Protein (100 μg), resuspendiert
in 250 μl
autologem Serum und 250 μI
RIBI, auf subkutanem Wege an einer Stelle inokuliert; Affe 2 (M2)
wurde mit dem rekombinanten Tat-Protein (10 μg), resuspendiert in 250 μl autologem
Serum und 250 μl
RIBI, auf subkutanem Wege an einer Stelle inokuliert; und Affe 3
(M3) war der nicht-inokulierte Kontrollaffe. Allen Affen wurden
an den Tagen –42
und –35
vor der ersten Impfstoffinokulation zehn ml Blut entnommen, um die
Grundparameter zu bestimmen. Serum- und Plasmaproben wurden bei –20°C oder –80°C eingefroren
und später
dazu verwendet, das Protein-Inokulum zu resuspendieren. Die Affen
1 und 2 wurden zum Zeitpunkt 0 und nach 2, 5, 10, 15, 22, 27, 32
und 37 Wochen inokuliert. Der Immunisierungsplan wurde in Woche
37 für
den Affen M1 und in Woche 41 für
den Affen M2 unterbrochen. Die Tiere wurden sakrifiziert, um die
immunologischen Parameter in mehreren Organen und Geweben (Milz
und Lymphknoten) zu untersuchen, wie die Evaluation des Vorliegens
einer proliferativen Antwort auf Tat, und der CAF- und CTL-Aktivität gegen
Tat. Die CAF-Aktivität
ist die antivirale Aktivität,
die durch CD8+-Lymphocyten weder MHC-restriktiv noch cytolytisch
vermittelt wird. An den selben Tagen der Inokulierung des Immunogens
wurden jedem Tier 10 ml Blut entnommen, um Labortests (chemisch-physikalische
Analysen, Elektrolyte, Leukozyten, Plättchenzählungen und Hämoglobinquantifizierung),
die Evaluierung der immunologischen Parameter, wie des Vorliegens
spezifischer Immunglobuline (IgM, IgG, IgA), der Spiegel der Cytokine
vom Th1-Typ (IL-2,
IFNγ) und
vom Th2-Typ (IL-4, IL-10), der Produktion von Chemokinen (RANTES,
M1P-1α und
M1P-1β),
des lymphocytischen Phänotyps
(CD4, CD8, CD3, CD14, CD20, CD25, CD56, HLA-DR, CD45RA), der proliferativen
Antwort auf Tat, des Vorliegens spezifischer cytotoxischer Aktivität (CTL),
des Vorliegens antiviraler Aktivität (CAF), und des Vorliegens
antiviraler Gesamtaktivität
(TAA), vermittelt durch PBMC und durch autologes Serum, durchzuführen. Zudem
wurden, um das Vorliegen einer zellvermittelten Immunantwort in
vivo zu evaluieren, alle geimpften Affen und der Kontrollaffe einem
Tat-Hauttest unterzogen.
-
Die
Ergebnisse dieses Exprimentes sind wie folgt: Es waren keine Änderungen
der chemisch-physikalischen,
hämatologischen
und verhaltensbezogenen Parameter zu beobachten. Bei den geimpften
Affen und dem Kontrollaffen wurden keine Anzeichen von Entzündung und
Gefäßneubildung
an den Inokulationsstellen detektiert. Diese Ergebnisse zeigen,
dass das Tat-Protein
von den Tieren gut toleriert wurde und in den verabreichten Dosen
sowie auf dem gegebenen Inokulationsweg nicht toxisch war. Bei den
Affen M1 und M2 wurde das Vorliegen von Antikörpern des IgG-Typs, der für Tat spezifisch
ist, in Woche 5 nach der ersten Inokulierung detektiert. In Woche
37 waren Anti-Tat-IgG bis zu einer Plasmaverdünnung von 1:6400 in beiden
Affen und in Woche 41 bis zu einer Plasmaverdünnung von 1:12.800 im Affen
M2 detektierbar. Die Ergebnisse sind in den
2 und
3 gezeigt.
Im Kontrollaffen M3 wurden Anti-Tat-Antikörper mit niedrigen Titern detektiert,
die wahrscheinlich durch die wiederholten Inokulierungen einer geringen
Menge Tat hervorgerufen wurden, die diesem Affen injiziert wurde,
um die Spezifität
der Hauttestreaktionen zu kontrollieren. In den Affen M1 und M2
waren die Anti-Tat-Antikörper
mit einem Titer von 1:3200 hauptsächlich gegen die aminoterminale Region
(aa 1-20) von Tat gerichtet (
4). Im
mit 10 μg
Tat geimpften Affen M2 wurden auch gegen aa 36-50 und 46-60 von
Tat gerichtete Antikörper
mit Titern von 1:50 bzw. 1:100 detektiert (
4). Die
Fähigkeit
des Serums der Affen, Tat zu neutralisieren, wurde mittels in vitro-Assays
bestimmt, welche die Inhibierung der Rettung der HIV-1-Replikation
in HLM-1 Zellen nach Zugabe von exogenem Tat-Protein maßen, wie
zuvor beschrieben (Ref. 41). Diese Assays zeigten, dass Plasma von
den Affen M1 und M2 in Woche 27 nach der ersten Inokulierung die
von exogenem Tat induzierte Virusreplikation blockierten, wie dies
durch Quantifizierung des p24-Antigens in den Kulturüberstanden
bestimmt wurde. Dagegen blockierte präimmunes Plasma derselben Affen
die Tat-Aktivität
nicht (Tabelle 4). TABELLE 4 Neutralisierende Aktivität von Affenplasma
hinsichtlich der Rettung der durch extrazelluläres Tat induzierten Virusreplikation
a | Proben | Inhibierung
(%) |
| Tat
(30 ng/ml) + Präimmun
M1 | 0 |
| Tat
(30 ng/ml) + Präimmun
M2 | 0 |
| Tat
(39 ng/ml) + Immun M1 | 79,12 |
| Tat
(30 ng/ml) + Immun M2 | 100 |
- aDie neutralisierende
Aktivität
des Plasmas wurde in HLM-1-Zellen (HeLa-CD4+-Zellen,
enthaltend eine integrierte Kopie eines HIV-1-Provirus, dem das
tat-Gen fehlt) bestimmt. Die HLM-1 Zellen wurden bei 6 × 105 Zellen/Vertiefungen in Platten mit 24 Vertiefungen
angeimpft und 16 Stunden bei 37°C
inkubiert. Die Zellen wurden zweimal mit PBS, enthaltend 0,1% Rinderserumalbumin
(BSA), gewaschen und mit frischem Medium (0,3 ml) in Gegenwart von
rekombinantem Tat-Protein und einem gleichen Volumen des Tierplasmas,
entnommen zum Zeitpunkt 0 (präimmunes
Plasma) oder in Woche 27 (immunes Plasma), 48 Stunden kultiviert.
Die negativen Kontrollen wurden durch Zellen dargestellt, die nur
mit dem gepoolten präimmunen
Plasma, mit dem gepoolten immunen Plasma oder mit PBS, enthaltend
0,1% BSA (PBS + 0,1% BSA), ohne Tat, behandelt wurden. Bei allen
Kontrollproben wurden keine Auswirkungen auf die Rettung der Virusreplikation
beobachtet. Jedes Plasma wurde zweifach getestet. Das Vorliegen
eines von den Zellen freigesetzten Virus wurde durch Quantifizierung
des p24 Gag-Antigens unter Verwendung eines handelsüblichen
p24-Antigen-Einfang-ELISA-Kits (NEN-Dupont) getestet. Die Ergebnisse
sind als Prozentsatz der Inhibierung der Virus-Rettung [für jedes
Plasma als Durchschnittswert von p24 (pg/ml) in zwei Vertiefungen
bestimmt] durch das immune Plasma im Vergleich zum präimmunen
Plasma (0% Inhibierung) ausgedrückt.
Die Affen M1 und M2 wurden mit dem rekombinanten Tat-Protein (100 μg bzw. 10 μg) geimpft,
das in 250 μl
autologem Serum und 250 μl
RIBI resuspendiert war und auf subkutanem Wege in eine einzige Stelle
injiziert wurde.
-
Die
Ergebnisse zeigen das Vorliegen einer proliferativen Antwort auf
Tat in Woche 22 (Tabelle 5), die bei mit dem rekombinanten Tat-Protein
geimpften Affen höher
war als beim Affen M2, der bei jedem Boost (Auffrischungsimpfung)
10 μg rekombinantes
Tat-Protein erhielt. Tabelle 5 Proliferative Antwort auf Tat
a | Affe | Stimulus | Wochen ab
Primärimmunisierung |
| | | 15 | 22 | 27 | 32 | 37 |
| M1 | PHA
TT
Tat | 15,3
1,2
0,8 | 13,9
4,7
2,4 | 19,9
2,1
1,1 | 40,6
3,8
1,3 | 3,2
2
0,6 |
| M2 | PHA
TT
Tat | 8,1
2
0,9 | 11,6
3,8
3 | 17,1
1,7
1,4 | 16,8
1
1,2 | 1,7
0,6
0,6 |
| M3 | PHA
TT
Tat | 5,1
7,2
2,1 | 19,9
6,2
1,4 | 18,2
5,5
1,3 | 6,6
2,8
0,7 | 8,1
5,6
0,9 |
- aPBMCs, isoliert
durch den Ficoll-Dichtegradienten, wurden in einer Konzentration
von 2 × 105 Zellen/Vertiefung dreifach in eine flachbödige Platte
mit 96 Vertiefungen plattiert, in mit 10% fötalem Kälberserum (FCS) ergänztem RPMI-1640
kultiviert, und mit Tat (1 oder 5 μg/ml), PHA (4 μg/ml) oder
Tetanustoxoid (TT), gegen das die Affen geimpft waren, stimuliert.
-
Unstimulierte
Kontrollen wurden in RPMI, 10% FCS-Medium, inkubiert. Der Anstieg
der Zellproliferation wurde am Tag 5 mittels 3[H]-Thymidinaufnahme
bestimmt, wie zuvor beschrieben (Ref. 39, 22). Die Ergebnisse sind
als Stimulationsindex ausgedrückt
und wurden wie folgt berechnet: Durchschnitt der Testprobe (cpm)/Durchschnitt
der Kontrolle (cpm). Werte größer 2,5
galten als positiv. Die Affen M1 und M2 wurden mit 100 μg oder 10 μg rekombinantem
Tat, resuspendiert in 250 μl
autologem Serum und 250 μl
RIBI, subkutan immunisiert. M3 stellt einen Kontrollaffen dar.
-
Wie
in Tabelle 6 gezeigt, wurde in den Affen M1 und M2, die mit rekombinantem
Tat immunisiert waren, keine cytotoxische Aktivität gegen
Tat detektiert. TABELLE 6 Analyse der cytotoxischen Aktivität gegen
Tat (CTL)
a | Affe | Woche | Verhältnis Ziel:Effektor | CTL-Aktivität |
| | | 1:50 | 1:25 | 1:12,5 | 1:6,25 | 1:3,125 | Medien | |
| M1 | 41 | ND | ND | ND | ND | ND | ND | ND |
| M2 | 41* | 0 | 0 | 0 | 0 | 0 | 0 | – |
| M3 | 41 | 0 | 0 | 0 | 0 | 0 | 0 | – |
- aPBMCs, isoliert
mittels Ficoll-Dichtezentrifugation, wurden in einer Konzentration
von 1 × 107 Zellen/ml in RPMI 1640, ergänzt mit
10% hitzeinaktiviertem FCS, resuspendiert und in einer Platte mit
24 Vertiefungen (500 μl
pro Vertiefung) 12 Stunden bei 37°C
in Gegenwart von 1 μg
Tat angeimpft. Einen Tag später
wurden die ohne Tat inkubierten Zellen bei 1500 UpM zentrifugiert
und in 50 μl
RPMI 1640, ergänzt
mit 10% FCS, resuspendiert, 3 Stunden bei 37°C mit 1 μg Tat inkubiert, gewaschen,
in 500 μl
frischem Medium resuspendiert und in die Vertiefung gegeben, welche
die zuvor stimulierten PBMCs enthielt. Am Tag 2 wurden die Zellen
mit 1 ml Medium, enthaltend IL-2 (2 IU/ml), verdünnt und 14 Tage kultiviert.
Autologe B-Lymphocyten, die aus jedem Affen vor dem Impfungsprotokoll
isoliert worden waren, wurden als Zielzellen (BLCL) verwendet. Zu
diesem Zweck wurden mittels Ficoll-Dichtezentrifugation isolierte PBMCs
am Tag 35 in einer Konzentration von 3 × 105 Zellen/Vertiefung
in einer Platte mit 96 Vertiefungen angeimpft und 2 oder 3 Wochen
in Gegenwart von 50% eines Mediums kultiviert, das aus einer Zelllinie
gesammelt wurde, die Papioviren produziert, wie zuvor beschrieben
(Ref. 28). Zehn B-Zelllinien, die für jedes Tier erhalten worden
waren, wurden erweitert und eingefroren. Zum Testen der Toxizität wurde
der Delta Cytotoxic Test (Wallac, Turku, Finnland), basierend auf
der zeitaufgelösten
Fluoreszenz, verwendet (Ref. 12, 13, 14). Zu diesem Zweck wurden
BLCL in einer Konzentration von 1 × 106 Zellen/200 μl RPMI1640,
ergänzt
mit 10% FCS, das 4 μg
Tat enthielt, 12 Stunden bei 37°C kultiviert.
Als Kontrolle wurde ein anderer Aliquot von autologen BLCL mit dem
gleichen Medium, ohne Tat, inkubiert. Die BLCL wurden in 1 ml RPMI
1640, ergänzt
mit 10% FCS, das 5 μl
eines fluoreszenzverstärkenden Liganden
enthielt, gewaschen und resuspendiert sowie 15 Min. bei 37°C gemäß den Anweisungen
des Herstellers inkubiert. Nach 5-maligem Waschen wurden die BLCL
in einer Konzentration von 5 × 104 Zellen/ml resuspendiert und umgehend zentrifugiert,
um den Überstand
zu ernten, der zum Messen des Hintergrundpegels verwendet wurde.
PBMCs (Effektoren) wurden zweifach in einer Konzentration von 2,5 × 104 Zellen/100 μl in Medium, das IL-2 enthielt
und entsprechend verdünnt
war, in einer Platte mit 96 Vertiefungen angeimpft. In jede Vertiefung
wurden 5 × 103 Zielzellen/100 μl (mit oder ohne Tat kultiviert)
gegeben. Die Ziel:Effektor-Verhältnisse
betrugen 1:50, 1:25, 1:12,5, 1:6,25, 1:3,125. PBMCs und Zielzellen
(Tat-gepulst oder ungepulst) wurden 2 Stunden bei 37°C inkubiert
mit i) 20 μl
5% Triton, um die maximale Freisetzung zu bestimmen, ii) 100 μl Wachstumsmedium,
um die spontane Freisetzung zu detektieren, iii) 200 μl Überstand
der Zielzellen, um den Hintergrundpegel zu detektieren. Am Ende
des Inkubationszeitraums wurden die Platten zentrifugiert, 20 μl jedes Überstandes
auf eine neue Platte übertragen
und in Gegenwart von 200 μl
einer in dem Kit enthaltenen Europiumlösung inkubiert. Die Fluoreszenz
wurde nach 20-minütiger
Inkubation mit einem zeitaufgelösten Floureszenzmessgerät (Victor,
Wallac, Turku, Finnland) bestimmt. Die spezifische CTL-Aktivität wurde
wie folgt bestimmt: % spezifische Freisetzung = [(Durchschnitt von
Probendetektion – Hintergrund) – (spontane Freisetzung – Hintergrund)]/[(maximale
Freisetzung – Hintergrund) – (spontane
Freisetzung – Hintergrund)] × 100. Der
Test galt als positiv, wenn die Tat-spezifische Freisetzung bei
den meisten getesteten Effektor:Ziel-Verhältnissen größer als 4% war. 4% ist ein
willkürlicher
Wert, der aufgrund früherer
Kontrollexperimente bestimmt wurde. ND: nicht bestimmt. Der Affe
M2 wurde mit 10 μg
rekombinantem Tat, resuspendiert in 250 μl autologem Serum und 250 μl RIBI, subkutan
immunisiert. M3 stellt einen Kontrollaffen dar.
- ND: nicht durchgeführt.
- *PBMCs wurden aus peripheren Lymphknoten isoliert, nachdem M2
sakrifiziert worden war. Zudem zeigen die Ergebnisse in den Wochen
22, 27 und 37 das Vorliegen einer löslichen antiviralen Aktivität, vermittelt
durch CD8+ T-Lymphocyten, gemessen als die Fähigkeit von Zellüberständen der
CD8+ T-Lymphocyten von Affen, eine akute Infektion des chimären Virus
SHIV 89.6P in CEMx174-Zellen zu inhibieren, oder die Reaktivierung einer
chronischen HIV-1-Infektion in OM-10-1-Zellen zu steuern (Tabelle
7). Die CAF-Aktivität
war meist eher in den geimpften Affen als in den Kontroll-Tieren
zu beobachten.
TABELLE 7 Analyse des Vorliegen einer löslichen,
antiviralen Aktivität,
vermittelt durch CD8+ T-Lymphocyten
(CAF)a | Affen-ID | Woche
nach Primärimmunisierung | % Inhibierung
der viralen Replikation |
| | | Akute
Infektion | Chronische
Infektion |
| M1 | 22 | 89,5 | ND |
| | 27 | 62 | 61,7 |
| | 37 | ND | ND |
| M2 | 22 | 44 | ND |
| | 27 | 54 | 27 |
| | 37 | 48 | 53 |
| M3 | 22 | 24 | ND |
- aPBMC von Affen,
die mit 100 μg
(M1) und mit 10 μg
(M2) rekombinantem Tat-Protein geimpft waren, und eines Kontrollaffen
(M3), der nicht geimpft war, aber wiederholte Hauttests mit Tat
durchlief, wurden mittels Ficoll-Dichtegradient isoliert. Mit CD8+
T-Lymphocyten angereicherte
Kulturen wurden aus PBMC mittels Anti-CD8-Magnetkügelchen
(Dynabeads, Dynal, Norwegen) gemäß den Anweisungen
des Herstellers isoliert. Die Reinheit der Kulturen wurde mittels
FACS-Analyse unter Verwendung einer Reihe von Antikörpern kontrolliert,
die gegen spezifische Zellmarker (CD3, CD4, CD8) gerichtet waren.
Mit CD8+ angereicherte Kulturen wurden (zweifach) bei 5 × 105 Zellen/500 μl pro Vertiefung in Platten
mit 48 Vertiefungen angeimpft, die zuvor 12 Stunden bei 4°C mit einem
monoklonalen Anti-CD3-Antikörper
beschichtet worden waren (2,5 μg/ml,
BioSource International, Camarillo, CA), und in RPMI 1640, enthaltend
10% fötales
Rinderserum und IL-2 (20 U/ml), kultiviert. Zwei Wochen lang wurden
alle drei Tage 250 μl
Medium gesammelt und durch ein gleiches Volumen an frischem Medium
ersetzt. Die Zellüberstände wurden
zentrifugiert, filtriert (0,45 μm)
und bei –80°C gelagert.
Von allen Zeitpunkten stammende Zellüberstände, mit Ausnahme des ersten,
wurden gepoolt, und das Vorliegen antiviraler Aktivität wurde
als ihre Fähigkeit
getestet, die virale Replikation in zwei Systemen zu inhibieren,
repräsentiert
durch eine akute bzw. eine chronische Infektion. Für das akute
Infektionssystem wurde die CEM x 174-Zelllinie verwendet, die von
der menschlichen B-Zelllinie
721.174 stammt, fusioniert mit der menschlichen T-Zelllinie CEM
(Ref. 143). Die Zellen (2 × 105) wurden 2 Stunden bei 37°C in Polypropylenröhrchen mit
oder ohne 200 μl
von CD8+-Überständen inkubiert,
die wie oben beschrieben hergestellt waren. Die Zellen wurden 3-mal
mit frischem Medium gewaschen, bei 2 × 104 Zellen
pro Vertiefung in Platten mit 96 Vertiefungen angeimpft und in 200 μl mit (behandelte
Zellen) oder ohne (unbehandelte Zellen) unterschiedliche(n) Volumina
(50 μl,
5 μl und
0,5 μl)
von Kulturüberständen, die
von CD8+ T-Lymphocyten des mit dem Impfstoff geimpften Affen oder
des Kontrollaffen stammten, inkubiert. Nach der Infektion wurden
Aliquots der Kulturüberstände alle
drei Tage gesammelt und durch ein gleiches Volumen Komplettmedium
ersetzt, das zuvor mit dem CD8+-Kulturüberstand von geimpften Affen
und vom Kontrollaffen versetzt worden war. Die in der Tabelle gezeigten
Ergebnisse entsprechen Tag 7 nach der Infektion und sind als Prozentsatz
(%) der Inhibierung der viralen Replikation von Zellen, die mit
CD8+-Kulturüberständen behandelt
wurden, welche aus geimpften Affen stammen, im Vergleich zu unbehandelten
Zellen ausgedrückt.
Die virale Replikation wurde durch Messen der RT-Werte in der beschriebenen
Weise (Ref. 54) oder der p27 Gag-Werte mittels ELISA in den Zellüberständen bestimmt,
die zu jedem Zeitpunkt gesammelt wurden. Für das chronische Infektionssystem
wurde die OM-10-1-Zelllinie verwendet (Ref. 20, 21), die eine menschliche
T-Lymphocytenlinie darstellt, welche chronisch mit HIV-1 infiziert
ist. Die Zellen wurden (zweifach) bei 5 × 104 Zellen/200 μl pro Vertiefung
in Platten mit 96 Vertiefungen in Gegenwart von Anti-TNFβ-Antikörpern (40 μg/ml), mit
oder ohne unterschiedliche(n) Volumina (50 μl, 5 μl, 0,5 μl) von Zellüberständen aus CD8+ T-Lymphocyten, die
von geimpften Affen oder vom Kontrollaffen stammten, angeimpft.
Die Zellen wurden zur Proliferation durch PMA (10–7 M)
aktiviert. Nach 24 Stunden wurden Aliquots des Kulturmediums gesammelt,
um die virale Replikation durch Messen der RT- oder p24 Gag-Spiegel mittels ELISA
zu bestimmen. Die Ergebnisse sind als % der Inhibierung der Reaktivierung der
Infektion in behandelten Zellen im Vergleich zu unbehandelten Zellen
dargestellt. Die Ergebnisse akuter und chronischer Infektion, die
in der Tabelle gezeigt sind, beziehen sich auf Zellen, die mit 5 μl Überstand,
der aus CD8+-Zellkulturen
stammte, behandelt wurden. ND: nicht durchgeführt.
-
Eine
Analyse der verzögerten
Hypersensibilität
(DTH) durch einen Hauttest zeigte, dass sowohl die geimpften Affen
(M1 und M2) als auch der Kontrollaffe (M3) negativ waren (Tabelle
8). TABELLE 8 Tat-Hauttest
a | Wochen
nach Primärimmunisierung | Affen |
| | M1 | M2 | M3 |
| 10 | – | – | – |
| 15 | – | – | – |
| 22 | – | – | – |
| 27 | – | – | – |
| 32 | – | – | – |
| 37 | – | – | – |
- aTat (1 und 5 μg) in 150 μl PBS-0,1%
BSA oder Puffer alleine wurden auf intradermalem Wege in einem zuvor rasierten
dorsalen Bereich der geimpften Affen und des Kontrollaffen (Kontrolle
der Spezifität
der Antwort) in den Wochen 10, 15, 22, 27, 32 und 37 nach der Erstimmunisierung
inokuliert. Die Affen M1 und M2 wurden mit rekombinantem Tat-Protein
(100 μg
bzw. 10 μg)
in 250 μl
autologem Serum und 250 μl
RIBI geimpft, das auf subkutanem Wege in eine einzige Stelle injiziert
wurde.
-
Affe
M3 ist ein Kontrollaffe, der nicht geimpft wurde. Das Auftreten
eines knotigen Erythems nach 48-72 Stunden legte eine verzögerte Hypersensibilitätsreaktion
(DTH) nahe: ++, ∅ ≥ 5
mm; +, ∅ ≥ 1-4
mm; +/–,
Erythem ohne Verhärtung; –, ∅ < 1 mm.
-
Die
Ergebnisse dieses Pilotexperimentes deuten darauf hin, dass rekombinantes
Tat-Protein, hergestellt und gereinigt gemäß einem von uns beschriebenen
Protokoll, in den Dosen von 100 und 10 μg, verabreicht auf subkutanem
Wege, nicht toxisch war. Zudem rief das Tat-Protein eine spezifische und breite
Immunantwort mit antiviralen Aktivitäten, sowohl humoral als auch
zellvermittelt, hervor. Eine stärkere
und spezifische Anti-Tat-Immunantwort wurde im Affen M2 beobachtet,
der mit 10 μg
rekombinantem Protein geimpft war. Darüber hinaus zeigte das RIBI-Adjuvans
keinerlei auffälliges
Anzeichen von Toxizität
in den geimpften Affen.
-
Ausgehend
von diesen Ergebnissen, wurde ein zweites Pilotexperiment entworfen,
um die Effekte einer Immunisierung mit 10 μg Tat, kombiniert mit RIBI oder
Alaun-Adjuvanzien, zu bestimmen. Die Affen erhielten gemäß folgendem
Schema Injektionen auf subkutanem Wege in eine einzige Stelle: Affen
M1-3: 10 μg
rekombinantes Tat-Protein in 250 μl
autologem Serum und 250 μl
RIBI. Affen M4-6: 10 μg
rekombinantes Tat-Protein in 250 μl
autologem Serum und 250 μl
Alaun. Affe M7: 250 μl
RIBI und 250 μl
autologes Serum (Kontrollaffe). Affe M8: 250 μl Alaun und 250 μl autologes
Serum (Kontrollaffe). Am Tag –9
vor der Erstimmunisierung wurden jedem Affen zehn ml Blut entnommen,
um die im vorhergehenden Pilotexperiment beschriebenen Untersuchungen
durchzuführen
und die Grundparameter jedes Tiers zu bestimmen. Die Affen wurden zum
Zeitpunkt 0 und nach 2, 6, 11, 15, 21, 28 und 32 Wochen inokuliert.
In Woche 36 erhielten die Affen M1-6 den letzten Boost mit rekombinantem
Tat-Protein (16 μg)
in 200 μl
ISCOM (immunstimulierender Komplex) und 300 μl PBS. ISCOM ist ein Adjuvans,
bestehend aus Quil-A-Saponin, Cholesterin und Phospholipiden, das
die humorale und zellvermittelte Immunantwort verstärkt (Ref.
109, 90). Den Affen M7 und M8 wurden zu den gleichen Zeitpunkten
nur Adjuvanzien injiziert. Zu jedem Impfungszeitpunkt und in den
Wochen 40, 44 und 50 wurden den Tieren 10 ml Blut entnommen, um
die im vorhergehenden Pilotexperiment beschriebenen klinischen und
immunologischen Parameter zu analysieren. Zudem wurden Urinproben
und Vaginalabstriche gesammelt, um das Vorliegen von Tat-spezifischem,
sekretorischem IgA zu analysieren. Zum Evaluieren der Schutzwirkung
der Tat-Immunisierung gegen die Infektion wurden die geimpften Affen
und der Kontrollaffe mit dem chimären „Simian/Human-Immunschwächevirus" (SHIV), Stamm 89.6P,
enthaltend des HIV-1-tat-Gen, das zuvor in Macaca fascicularis gezüchtet und
titriert worden war (Ref. 128, 129, 69). Nach der Anregung wurden
die Tiere auf virologische Parameter, wie Plasma p27-Antigenämie und
Plasma- und Zellen-Virenlast überwacht (im
ersten Monat alle zwei Wochen, über
die nächsten
drei Monate alle vier Wochen und über bis zu 6-12 Monate alle
8 Wochen). Um zu bestätigen,
dass eine Infektion erfolgt war, wurden auch Anti-SIV-Antikörper mit einem
handelsüblichen
Kit gesucht, der zur Detektion von Anti-HIV-2-Antikörpern dient
und auch Anti-SIV-Antikörper
erkennt (Elavia Ac-Ab-Ak II-Kit, Diagnostic Pasteur, Paris, Frankreich).
-
Die
Ergebnisse der zweiten Pilotexperimente sind derzeit folgende: Es
waren keine Veränderungen der
chemisch-physikalischen, hämatologischen
und verhaltensbezogenen Parameter zu beobachten. Die Affen zeigten
keinerlei Anzeichen von Entzündung
oder Gefäßneubildung
an der Inokulationsstelle. Zu beobachten war eine spezifische Antikörperantwort
(IgM, IgG). In Woche 15 erreichten die Anti-Tat-Antikörper(IgG)-Titer
hohe Spiegel im Bereich von 1:6400 bis 1:25600 (
5-
7).
Die Antikörper
reagierten im Wesentlichen mit der aminoterminalen Region (aa 1-20)
von Tat, mit Titern im Bereich von 1:1600 to 1:3200 (
8),
wie dies in Woche 22 gezeigt wurde. Darüber hinaus wurden auch gegen
aa 46-60 von Tat gerichtete Antikörper mit Titern im Bereich
von 1:100 bis 1:200 detektiert (
8). Die
Fähigkeit
des Plasmas der Affen, die Tat-Aktivität zu neutralisieren, wurde
getestet durch einen Assay der Inhibierung der viralen Rettung in
HLM-1 Zellen, inkubiert mit seriellen Mengen von exogenem Tat, wie
dies zuvor im ersten Pilotexperiment beschrieben wurde. Die Ergebnisse
dieser Experimente zeigten, dass immunes Plasma (1:2 verdünnt) der
Affen M1-6 in Woche 15 die virale Replikation, induziert durch 30
ng/ml exogenes Tat, blockierte, wie dies durch die Messung von in das
Kulturmedium freigesetztem p24-Antigen bestimmt wurde. Dagegen blockierte
präimmunes
Plasma der Affen M1-6 oder Plasma der Kontrollaffen (M7, M8) die
Tat-Aktivität
nicht (Tabelle 9). Darüber
hinaus blockierte immunes Plasma (1:2 verdünnt) der Affen M1-6, das in
Woche 21 entnommen worden war, die Virusreplikation, die durch 60
ng/ml, 120 ng/ml, 240 ng/ml und 500 ng/ml exogenes Tat induziert
worden war. Insbesondere bestimmte dieses Plasma eine 10-fache Verringerung
der Virusreplikation, die durch sehr hohe Dosen von extrazellulärem Tat
(240 ng/ml und 500 ng/ml) induziert war (Tabelle 9). TABELLE 9 Neutralisierende Aktivität von immunem
Plasma gegenüber
der Rettung der Virusreplikation, induziert durch extrazelluläres Tat
a | Proben | Inhibierung
(%) |
| Tat
(30 ng/ml) + Präimmun
M1 | 0 |
| Tat
(30 ng/ml) + Präimmun
M2 | 0 |
| Tat
(30 ng/ml) + Präimmun
M3 | 0 |
| Tat
(30 ng/ml) + Präimmun
M4 | 0 |
| Tat
(30 ng/ml) + Präimmun
M5 | 0 |
| Tat
(30 ng/ml) + Präimmun
M6 | 0 |
| Tat
(30 ng/ml) + Immun M1 (Woche 15) | 89,8 |
| Tat
(30 ng/ml) + Immun M2 (Woche 15) | 78,7 |
| Tat
(30 ng/ml) + Immun M3 (Woche 15) | 100 |
| Tat
(30 ng/ml) + Immun M4 (Woche 15) | 100 |
| Tat
(30 ng/ml) + Immun M5 (Woche 15) | 70,8 |
| Tat
(30 ng/ml) + Immun M6 (Woche 15) | 94,2 |
| Tat
(60 ng/ml) + Präimmun
M1 | 0 |
| Tat
(60 ng/ml) + Präimmun
M2 | 0 |
| Tat
(60 ng/ml) + Präimmun
M3 | 0 |
| Tat
(60 ng/ml) + Präimmun
M4 | 0 |
| Tat
(60 ng/ml) + Präimmun
M5 | 0 |
| Tat
(60 ng/ml) + Präimmun
M6 | 0 |
| Tat
(60 ng/ml) + Immun M1 (Woche 21) | 96,3 |
| Tat
(60 ng/ml) + Immun M2 (Woche 21) | 100 |
| Tat
(60 ng/ml) + Immun M3 (Woche 21) | 100 |
| Tat
(60 ng/ml) + Immun M4 (Woche 21) | 98,7 |
| Tat
(60 ng/ml) + Immun M5 (Woche 21) | 99 |
| Tat
(60 ng/ml) + Immun M6 (Woche 21) | 98,8 |
| Tat
(120 ng/ml) + Präimmun
M1-6, gepoolt | 0 |
| Tat
(120 ng/ml) + Immun M1 (Woche 21) | 59,2 |
| Tat
(120 ng/ml) + Immun M2 (Woche 21) | 90,4 |
| Tat
(120 ng/ml) + Immun M3 (Woche 21) | 96,8 |
| Tat
(120 ng/ml) + Immun M4 (Woche 21) | 98,3 |
| Tat
(120 ng/ml) + Immun M5 (Woche 21) | 100 |
| Tat
(120 ng/ml) + Immun M6 (Woche 21) | 97,8 |
| Tat
(240 ng/ml) + Präimmun
M1-6, gepoolt | 0 |
| Tat
(240 ng/ml) + Immun M1 (Woche 21) | 26,1 |
| Tat
(240 ng/ml) + Immun M2 (Woche 21) | 49,4 |
| Tat
(240 ng/ml) + Immun M3 (Woche 21) | 70,3 |
| Tat
(240 ng/ml) + Immun M4 (Woche 21) | 91,2 |
| Tat
(240 ng/ml) + Immun M5 (Woche 21) | 94,5 |
| Tat
(240 ng/ml) + Immun M6 (Woche 21) | 86 |
| Tat
(500 ng/ml) + Präimmun
M1-6, gepoolt | 0 |
| Tat
(500 ng/ml) + Immun M1 (Woche 21) | 32,7 |
| Tat
(500 ng/ml) + Immun M2 (Woche 21) | 38,9 |
| Tat
(500 ng/ml) + Immun M3 (Woche 21) | 57,5 |
| Tat
(500 ng/ml) + Immun M4 (Woche 21) | 89,4 |
| Tat
(500 ng/ml) + Immun M5 (Woche 21) | 72 |
| Tat
(500 ng/ml) + Immun M6 (Woche 21) | 71,8 |
- aDie Fähigkeit
von Anti-Tat-Plasma, die Tat-Aktivität zu neutralisieren, wurde
in HLM-1-Zellen
bestimmt, wie dies in der Erklärung
zu Tabelle 4 beschrieben ist. Rekombinantes Tat-Protein (30 ng/ml, 60 ng/ml, 120 ng/ml, 240
ng/ml und 500 ng/ml) wurde alleine oder zusammen mit einem gleichen
Volumen an präimmunem
Affen-Plasma oder in Woche 15 oder 21 (immunes Plasma) zugegeben.
Die Affen M1-3 wurden mit 10 μg
Tat in 250 μl
autologem Serum und 250 μl
RIBI geimpft; die Affen M4-6 wurden mit 10 μg Tat in 250 μl autologem Serum
und 250 μl
Alaun geimpft; zwei Kontrollaffen wurde RIBI (250 μl und 250 μl autologes
Serum) (M7) oder Alaun (250 μl
und 250 μl
autologes Serum) (M8) injiziert. Die Ergebnisse sind wie in der
Erklärung
zu Tabelle 4 beschrieben dargestellt.
-
Die
Fähigkeit
von Affenplasma, die Aktivität
von extrazellulärem
Tat, das von den Zellen während
einer akuten Infektion freigesetzt wird, zu neutralisieren, wurde
in CEM x 174 Zellen getestet, die mit dem chimären Virus SHIV 89.6P infiziert
waren. Am Tag 7 nach der Infektion war eine Virusreplikation in
50% der mit SHIV infizierten und mit dem präimmunen Plasma der Affen M1-6
kultivierten Kontrollzellen zu beobachten. Umgekehrt wurde keine
Virusreplikation in infizierten Zellen detektiert, die in Gegenwart
des immunen Plasmas der Affen M1-6 gezüchtet wurden, das in Woche
44 entnommen worden war (Tabelle 10). TABELLE 10 Neutralisierende Aktivität von immunem
Plasma gegenüber
der Übertragung
einer Virusinfektion
a | Probe | p27
(pg/ml) |
| SHIV
+ Präimmun
M1 | Neg |
| SHIV
+ Präimmun
M2 | Neg |
| SHIV
+ Präimmun
M3 | 1,080 |
| SHIV
+ Präimmun
M4 | 0,602 |
| SHIV
+ Präimmun
M5 | 1,169 |
| SHIV
+ Präimmun
M6 | Neg |
| SHIV
+ Immun M1 | Neg |
| SHIV
+ Immun M2 | Neg |
| SHIV
+ Immun M3 | Neg |
| SHIV
+ Immun M4 | Neg |
| SHIV
+ Immun M5 | Neg |
| SHIV
+ Immun M6 | Neg |
- aCEM x 174 Zellen
(3 × 104 Zellen/150 μl) in Platten mit 96 Vertiefungen
wurden 2 Stunden bei 37°C
in RPMI 1640, enthaltend 10% FCS, mit dem chimären SHIV 89.6P-Virus (5 × 10–5 TCID50/Zelle) infiziert. Die Zellen wurden zweimal
mit RPMI 1640 gewaschen und in 150 μl Komplettmedium resuspendiert,
das mit 5% des präimmunen
oder immunen Affenplasmas (Woche 44) von mit rekombinantem Tat (10 μg) und RIBI
(M1-3) oder Alaun (M4-6) geimpften Tieren versetzt war. Das Tierplasma
wurde zuvor 30 Min. bei 56° erwärmt und
mittels ELISA analysiert, um die Anti-Tat-Antikörpertiter zu kontrollieren.
Jedes Serum wurde zweifach getestet. An den Tagen 3, 5 und 7 nach
der Infektion wurden 120 μl
Kulturmedium gesammelt und durch ein gleiches Volumen an frischem
Medium ersetzt, das 5% präimmunes
oder immunes Plasma der Affen M1-6 enthielt. Die Fähigkeit
des Plasmas, extrazelluläres
Tat zu neutralisieren, das während
einer akuten Infektion freigesetzt wird, und die Übertragung
der Infektion in vitro zu kontrollieren, wurde durch Detektieren
von viralem Gag p27 im Kulturmedium mittels ELISA (Coulter International,
Miami, FL) bestimmt. Die Ergebnisse, die als p27-Werte (pg/ml) dargestellt
sind, entsprechen dem Mittelwert von zwei Vertiefungen für jedes
Serum am Tag 7 nach der Infektion.
-
Zudem
wurde ab Woche 11 eine proliferative Antwort auf Tat beobachtet
(Tabelle 11). TABELLE 11 Proliferative Antwort auf Tat
a | Affe | Stimulus | Wochen
ab Primärimmunisierung |
| | | 0 | 11 | 15 | 21 | 28 | 32 | 36 | 40 | 44 | 50 |
| M1 | PHA
TT
Tat | 16,96
11,69
1,12 | 10,50
1,96
1,55 | 15,27
3,01
0,52 | 33,8
1,2
1,7 | 7,2
1,2
0,8 | 51,5
1,3
0,8 | 64,3
0,93
0,6 | 36,05
1,4
0,7 | 24
10,05
9,27 | 65,7
7,2
4,7 |
| M2 | PHA
TT
Tat | 31,27
1,12
1,08 | 25,75
1,8
3,65 | 21,28
0,57
6,22 | 87,1
1,7
14,14 | 25,7
1,15
3,5 | 56
1,6
1,8 | 38,2
4,95
4,1 | 40,3
1,2
1,9 | 29,03
1,51
7,67 | 26
2,9
13,2 |
| M3 | PHA
TT
Tat | 22,42
11,43
1,65 | 7,89
0,95
2,69 | 16,88
1,71
18,82 | 36,3
1,25
23,51 | 148,5
1,2
12,03 | 42
1,1
0,9 | 78,9
1
1,3 | 27
1
0,5 | 53,71
1,81
23,85 | ND
ND
ND |
| M4 | PHA
TT
Tat | 3,88
2,85
1,29 | 20,77
4,49
3,01 | 15,22
9,07
3,24 | 83,7
6,9
7,9 | 18,6
15,8
10,1 | 35
3,7
2,6 | 38,2
3,8
1,5 | 45,2
5
3,9 | 57,47
19,77
33,61 | 15,8
6,6
4,7 |
| M5 | PHA
TT
Tat | 6,25
2,31
1,80 | 5,74
1,07
0,66 | 16,74
4,84
1,76 | 72,2
3,9
3,6 | 7,45
0,9
2,22 | 41
0,83
0,8 | 56,5
1,4
1,14 | 32,9
1,24
1,3 | 33,85
10,22
1,33 | 12
1,95
1,4 |
| M6 | PHA
TT
Tat | 11,96
4,14
1,37 | 17,94
1,71
1,06 | 2,77
0,13
0,11 | 29,4
1,7
2,95 | 7,3
10,34
9,3 | 25
1,3
1,13 | 8,3
1,8
1,3 | 6,85
1,1
1 | 18,01
2,49
5,8 | 5,2
0,9
0,3 |
| M7 | PHA
TT
Tat | 21,65
0,97
1,78 | 20,30
0,80
0,68 | 37,93
0,88
0,73 | 17,6
1
1 | 17,9
0,6
0,42 | 75
1,04
0,9 | 12,9
0,6
0,5 | 34,8
0,4
0,8 | 41,81
1,11
1,07 | 27,5
1,1
0,4 |
| M8 | PHA
TT
Tat | 26,51
1,20
1,12 | 67,09
10,78
0,00 | 16,38
0,20
0,21 | 14,9
1,6
1,03 | 17,2
0,62
0,57 | 28,2
0,8
0,6 | 18,95
1,2
0,5 | 20,6
0,9
0,9 | 28,61
1,11
1,04 | 13,6
2,1
1 |
- aPeripherblut-Lymphocyten
wurden isoliert, mit PHA (4 μg/ml),
mit dem Tetanustoxoid (TT) (10 μg/ml)
und mit Tat (5 μg/ml)
aktiviert und einem Assay unterzogen, wie dies in der Erklärung zu
Tabelle 5 beschrieben ist. Die Affen M1-3 wurden mit 10 μg rekombinantem
Tat-Protein in 250 μl
autologem Serum und 250 μl
RIBI inokuliert; die Affen M4-6 wurden mit 10 μg rekombinantem Tat in 250 μl autologem
Serum und 250 μl
Alaun inokuliert; zwei Kontrollaffen wurden mit RIBI (250 μl und 250 μl autologes
Serum) (M7) und mit Alaun (250 μl
und 250 μl autologes
Serum) (M8) inokuliert. ND: nicht durchgeführt.
-
Eine
starke cytotoxische T-Zellantwort (CTL) wurde in einem Affen, der
mit dem Tat-Protein und RIBI geimpft war (M1), und in zwei Affen,
die mit dem Tat-Protein und Alaun geimpft waren (M4 und M5), detektiert, während eine
schwächere
CTL-Antwort im Affen M6, der mit Tat und Alaun immunisiert war (
9 und
Tabelle 12), beobachtet wurde. TABELLE 12 Analyse der CTL-Antwort
| Affe | Woche | Ziel:Effektor-Verhältnis | CTL-Aktivität |
| | | 1:50 | 1:25 | 1:12,5 | 1:6,25 | 1:3,125 | Durchschnitt | |
| M1 | 28 | 5,9 | 4,7 | 4,1 | 7,9 | 5,3 | 5,5 | + |
| | 36 | ND | 14,4 | 8,8 | 4,9 | 6,7 | 8,7 | + |
| M2 | 28 | ND | ND | ND | ND | ND | ND | ND |
| | 36 | ND | ND | ND | ND | ND | ND | ND |
| M3 | 28 | 0 | 0 | 0 | 0 | 0 | 0 | – |
| | 36 | ND | 0 | 0,6 | 0,5 | 2,0 | 0,7 | – |
| M4 | 28 | 0 | 0 | 1,1 | 1,1 | 2,6 | 0,9 | – |
| | 36 | ND | 2,7 | 8,3 | 15 | 1,9 | 6,9 | + |
| M5 | 28 | 4,9 | 3,9 | 4,7 | 5,5 | 1,7 | 4,1 | + |
| | 36 | 0 | 1 | 0 | 0 | 0 | 0,2 | – |
| M6 | 28 | 0 | 2,6 | 1,1 | 7,2 | 7,2 | 3,6 | ± |
| | 36 | ND | 0 | 0 | 0 | 0 | 0 | – |
| M7 | 36 | 0 | 0 | 0 | 0 | 0 | 0 | – |
| M8 | 36 | 0 | 0 | 0 | 0 | 0 | 0 | – |
-
- aDer Assay wurde durchgeführt, wie
in Tabelle 6 beschrieben. Die Affen M1-3 wurden mit 10 μg rekombinantem Tat
in 250 μl
autologem Serum und 250 μl
RIBI immunisiert; die Affen M4-6 wurden mit 10 μg rekombinantem Tat in 250 μl autologem
Serum und 250 μl
Alaun inokuliert; zwei Kontrollaffen wurden mit RIBI (250 μl und 250 μl autologes
Serum) (M7) und Alaun (250 μl
und 250 μl
autologes Serum) (M8) inokuliert. ND: nicht durchgeführt.
-
In
Woche 44 wurde das Vorliegen der antiviralen Gesamtaktivität (TAA)
bestimmt. Die TAA wurde als Fähigkeit
der PBMC von Affen, die mit in Gegenwart von autologem Serum kultiviertem,
rekombinantem Tat-Protein geimpft waren, zur Resistenz gegen eine
SHIV 89.6P-Infektion gemessen (Tabelle 13). TABELLE 13 Analyse des Vorliegen der antiviralen
Gesamtaktivität
(TAA)
a | Affen-ID | Tage nach
Infektion |
| | 7 | 17 |
| | Minimale
infektiöse
Dosis (TCID50/Zelle) | Minimale
infektiöse
Dosis (TCID50/Zelle) |
| M1 | 10–2 | 10–2 |
| M2 | 10–4 | 10–4 |
| M3 | 10–3 | 10–3 |
| M4 | 10–2 | 10–2 |
| M5 | 10–2 | 10–2 |
| M6 | 10–3 | 10–3 |
| M7 | 10–3 | 10–3 |
| M8 | 10–4 | 10–3 |
- aPBMC wurden in
Woche 44 von mit dem rekombinanten Tat-Protein (10 μg) und RIBI
(M1-3) oder Alaun (M4-6) geimpften Affen und von mit RIBI (M7) oder
Alaun (M8) inokulierten Kontrollaffen gesammelt. Die PBMC, gereinigt
mittels Ficoll-Gradient und dreifach bei 5 × 105/200 μl pro Vertiefung
in Platten mit 48 Vertiefungen angeimpft, wurden in RPMI 1640, enthaltend
10% FCS und 5% autologes Plasma, das zuvor 30 Min. bei 56°C erwärmt worden
war, in Gegenwart eines monoklonalen Anti-CD3-Antikörpers (5
ng/ml) und von IL-2 (2 U/ml) 48-72 Stunden bei 37°C gezüchtet. Die
Zellen wurden 2 Stunden bei 37°C
mit Reihenverdünnungen des
chimären
Virus SHIV 89.6P (10–2, 10–3,
10–4,
10–5 TCID50/Zelle) infiziert,
3-mal mit PBS-A gewaschen und in 50% konditioniertem Medium sowie
50% frischem Medium bei 5 × 105 Zellen/ml/Vertiefung resuspendiert. An
den Tagen 3, 7, 10, 14 und 17 nach der Infektion wurden Aliquots
des Kulturmediums gesammelt und durch gleiche Volumina an frischem
Medium ersetzt. Die Virusreplikation wurde in Zellüberständen mittels
p27 Gag ELISA (Coulter International, Miami, FL) bestimmt. Die Ergebnisse
sind als minimale infektiöse
Dosis von SHIV (TCID50/Zelle) an den Tagen 7 und 17 nach der Infektion
angegeben, die in der Lage war, Affen-Lymphocyten zu infizieren.
-
Die
Ergebnisse zeigen das Vorliegen löslicher viraler Aktivität, vermittelt
durch CD8+ T-Lymphocyten (CAF)
(Tabelle 14). In den geimpften Affen war im Vergleich zu den Kontrolltieren
ein Gesamtanstieg der CAF-Aktivität zu beobachten. TABELLE
14 Analyse
des Vorliegen löslicher
antiviraler Aktivität,
vermittelt durch CD8+ T-Lymphocyten CAF)
a | Affen-ID | Wochen
nach Primärimmunisierung | % Inhibierung
der viralen Replikation |
| | | Akute
Infektion | Chronische
Infektion |
| M1 | 0 | 8 | 30 |
| | 32 | 53 | 53 |
| M2 | 0 | 36 | 0 |
| | 32 | 60 | 27 |
| M3 | 0 | 0 | 37 |
| | 32 | 55 | 29 |
| M4 | 0 | 45 | 0 |
| | 32 | 85 | 66 |
| M5 | 0 | 41 | 0 |
| | 32 | ND | ND |
| M6 | 0 | 49 | 18 |
| | 32 | 34 | 41,4 |
| M7 | 0 | 39 | 39 |
| | 32 | 71 | 44 |
| M8 | 0 | 37 | 0 |
| | 32 | 76 | 26,8 |
- aAnalyse des Vorliegens
löslicher
antiviraler Aktivität,
vermittelt durch CD8+ T-Lymphocyten
(CAF) von mit rekombinantem Tat-Protein (10 μg) und RIBI (M1-3) oder Alaun
(M4-6) geimpften Affen und von mit RIBI (M7) oder Alaun (M8) inokulierten
Kontrollaffen. Eine akute Infektion wurde bei CEM x 174 Zellen getestet,
die mit SHIV 89.6P infiziert waren. Der Assay wurde wie in Tabelle
7 beschrieben durchgeführt,
und die Ergebnisse beziehen sich auf Tag 7 nach der Infektion. Das
Vorliegen von CAF beim chronischen Infektionssystem wurde in der
U1-Zelllinie getestet (Ref. 47), bei der es sich um eine promonocytische,
menschliche Zelllinie handelt, die chronisch mit HIV-1 infiziert
ist. U1-Zellen, die bei 1 × 104 Zellen/200 μl pro Vertiefung in Platten
mit 96 Vertiefungen angeimpft worden waren, wurden, um eine Reaktivierung
der HIV-1-Infektion zu induzieren, mit PMA (10–8 M)
und mit oder ohne unterschiedliche(n) Volumina (50 μl, 5 μI, 0,5 μI) von Kulturüberständen aus
CD8+ T-Lymphocyten, die von geimpften Affen und von Kontrollaffen
stammten, inkubiert. Drei Tage nach der PMA-Behandlung wurde das
Vorliegen von HIV-1 im Kulturmedium mittels RT-Assay oder p24 Gag
ELISA bestimmt. Die Ergebnisse sind als % Inhibierung der HIV-1-Replikation
in CD8+ T-Zellüberständen gegenüber unbehandelten
Zellen gezeigt. Die Ergebnisse der Inhibierung akuter und chronischer
Infektion beziehen sich auf Zellen, die mit 5 μl CD8+-Überständen behandelt wurden.
-
Die
Produktion von Cytokinen (γIFN,
IL-4, TNFα)
und des RANTES-Chemokins aus PBMC von Affen, die mit Tat und RIBI
(M1-3) oder Tat und Alaun (M46) geimpft waren, und der Kontrollaffen
M7 und M8 wurde ebenfalls bestimmt (Tabelle 15).
- aAnalyse der Produktion von Cytokinen und
Chemokinen nach 48 und 96 Stunden Kultur (48/96) von PBMC aus mit
10 μg Tat
und RIBI (M1-3) oder Alaun (M4-6) geimpften Affen. Die Kontrollaffen
(M7 und M8) wurden mit RIBI bzw. Alaun-Adjuvanzien inokuliert. In
Woche 44 entnommene und mittels Ficoll-Gradient gereinigte PBMC
wurden bei 1 × 106 Zellen/ml pro Vertiefung in Platten mit
24 Vertiefungen angeimpft und in RPMI 1640, enthaltend 10% FCS,
gezüchtet.
Die PBMC waren unstimuliert (Kontrolle), um die spontane Freisetzung
von Cytokinen und Chemokinen zu evaluieren, oder wurden mit PHA
(2 μg/ml),
mit dem Tetanustoxoid (TT, 5 μg/ml) oder
mit Tat (1 oder 5 μg/ml)
stimuliert. Aliquots von Kulturüberstanden
wurden 48 und 96 Stunden nach der Stimulation gesammelt, um das
Vorliegen von Cytokinen und Chemokinen mittels handelsüblicher
ELISA-Kits von BioSource International (Camarillo, CA, USA) für den Assay
der Cytokinproduktion und von R & D
Systems (Abdigdon, Oxon, UK) zum Evaluieren der RANTES-Produktion
zu bestimmen. Die Ergebnisse sind als pg/ml nach 48 bzw. 96 Stunden
Kultur (48/96) gezeigt. Die Cutoff-Werte waren (pg/ml): γ1FN: 31,2;
IL-4: 3,12; TNFα: 15,6;
RANTES: 6,25. (–):
Die Werte waren niedriger als entsprechende Cutoff-Werte. Nd: nicht
durchgeführt.
-
Darüber hinaus
zeigten fünf
mit dem rekombinanten Protein geimpfte Affen (M2-6) in Woche 15
eine positive Reaktion auf den Tat-Hauttest mit einer starken, verzögerten Hypersensibilitätsreaktion
(Tabelle 16 und
19). Bei den Affen
4 und 5 war die Hauttestreaktion in den folgenden Wochen sogar noch
stärker
(Tabelle 16). TABELLE 16 Tat-Hauttest
a | Affe | Wochen nach
Primärimmunisierung |
| | 11 | 15 | 21 | 28 | 32 | 36 | 44 |
| M1 | – | – | – | – | – | – | – |
| M2 | – | + | + | +/– | +/– | + | +/– |
| M3 | +/– | + | +/– | +/– | – | – | – |
| M4 | – | + | ++ | ++ | ++ | ++ | ++ |
| M5 | +/– | + | ++ | + | ++ | ++ | + |
| M6 | – | + | +/– | +/– | – | – | – |
| M7 | ND | ND | ND | ND | ND | ND | ND |
| M8 | ND | ND | ND | ND | ND | ND | ND |
- aTat (1 und 5 μg) in 150 μl PBS-0,1%
BSA oder der Puffer alleine wurden auf intradermalem Wege in einem rasierten
Bereich des Rückens
von geimpften Affen inokuliert. Die Kontrolltiere wurden in den
Wochen 11, 15, 21, 28, 32, 36 und 44 nach der Erstimmunisierung
nicht inokuliert (ND, nicht durchgeführt). Die Affen M1-3 wurden
mit 10 μg
rekombinantem Tat-Protein in 250 μl
autologem Serum und 250 μl
RIBI geimpft; die Affen M4-6 wurden mit 10 μg rekombinantem Tat-Protein
in 250 μl
autologem Serum und 250 μl
Alaun geimpft; zwei Kontrollaffen wurden mit RIBI (250 μl und 250 μl autologes
Serum) (M7) oder Alaun (250 μl
und 250 μl
autologes Serum) (M8) inokuliert. Das Vorliegen eines erythematösen Knötchens nach
48-72 Stunden legte eine verzögerte
Hypersensibilitätsreaktion
(DTH) nahe: ++, ⌀ ≥ 5 mm; +, ⌀ ≥ 1-4 mm; +/+,
Erythem ohne Verhärtung; –, ⌀ < 1 mm.
-
Die
Ergebnisse nach der Anregung zeigen, dass 4/6 (67%) der geimpften
Affen vor einer Infektion mit 10 MID50 von
SHIV 89.6P geschützt
waren, wie durch die Ergebnisse der virologischen Assays gezeigt
(Tabelle 17). Insbesondere wurde kein p27 Gag-Antigen im Plasma
der Affen M1, M2, M4 und M6 detektiert und keine provirale DNA mittels
PCR in Lymphocyten dieser Affen gefunden, und die Cyto-Virämie war
negativ. Die Affen M3 und M5 waren infiziert, wie sich durch das
Vorliegen des p27 Gag-Antigens im Plasma durch Detektion von proviraler
DNA in den Zellen und durch eine positive Cytomegalie-Virämie zeigte
(Tabelle 17). Beide Kontrollen (M7 und M8) stellten sich anhand
der gleichen virologischen Assays als infiziert heraus. Um die Infektiösität der viralen
Dosis, die zur Anregung verwendet wurde, weiter zu kontrollieren,
wurde ein weiterer naiver Affe (M13) zu den Kontrolltieren hinzugenommen
und mit 2,85 MID50 von SHIV 89.6P (was einer
Virusdosis entspricht, die 3,5-mal niedriger als die zur Anregung
der Tiere im Protokoll verwendete Dosis ist) infiziert. Der Affe
M13 stellte sich anhand aller virologischen Assays als infiziert
heraus. Um zu bestätigen,
dass die Tiere dem Virus ausgesetzt waren, wurde das Vorliegen von
Antikörpern
gegen SIV-Antigene, kodiert durch das chimäre SHIV 89.6P-Virus (Gag, Pol,
RT, Nef), analyisert, wie in diesem Beispiel bereits beschrieben.
Das Vorliegen von Anti-SIV-Antikörpern
in den Affen, die bezüglich
der virologischen Parameter negativ waren (M1, M2, M4 und M6), bestätigt, dass
diese Tiere dem Virus ausgesetzt waren, und zeigt, dass in diesen
Affen eine abortive SHIV-Infektion stattgefunden hatte. Die Affen,
die niedrige Anti-SIV-Antikörper-Titer
zeigten, wurden gemäß folgendem
Verfahren auf die in vitro-Produktion von spezifischen antiviralen
IgG (IVAP) (Ref. 177,178) untersucht: PBMC (2 × 106/Vertiefung)
wurden in Platten mit 24 Vertiefungen angeimpft und mit PWM (2 μg/ml, Sigma,
St. Louis, USA) stimuliert. Nach 7-tägiger Inkubation (bei 37°C in Gegenwart
von 5% CO2 und 95% Feuchtigkeit) wurden
die Kulturüberstände gesammelt,
um sie mit einem handelsüblichen
Elisa-Kit zur Detektion von HIV-1- und HIV-2-Antikörpern (Abbott,
HIV-1/HIV-2 EIA Third Generation Plus) auf Anti-HIV-Antikörperproduktion
zu testen. Alle angeregten Affen stellten sich bezüglich der
Produktion von Anti-HIV Env-Antikörpern als
positiv heraus, da HIV-1 Env in SHIV 89.6P vorliegt.
-
-
Analyse
der virologischen Parameter nach Anregung von Affen, die mit 10 μg rekombinantem
Tat-Protein und RIBI (MI-3) oder Alaun (M4-6) geimpft waren. Die
Kontrollaffen (M7 und M8) wurden mit RIBI bzw. Alaun inokuliert.
Affe M13 war ein naives Tier, das mit 2,85 MID50 von
SHIV 89.6P infiziert war.
- aDie Plasmaantigenämie wurde
mittels p27 Gag ELISA (Innogenetics, Belgien) evaluiert und ist
in p27-Werten (pg/ml) ausgedrückt.
Neg: der Wert war niedriger als der entsprechende Cutoff-Wert (18
pg/ml).
- bDNA wurde mit Vollblut unter Verwendung
des Q1Aamp-Blutkits (Angingen GmbH und Qiagen Inc., Hilden, Deutschland)
gereinigt. Die Qualität
der DNA wurde mittels PCR-Amplifikation
des β-Globingens
kontrolliert, wie zuvor beschrieben (Ref. 141). Das Vorliegen von
proviraler DNA wurde mittels semiquantitativer PCR-Amplifikation
von SIV gag analysiert. Eine PCR wurde mit 1 μg Zell-DNA unter Verwendung
der Primer SG1096Ngag (entsprechend den Nukleotiden 1096-1119 des
S1Vmac251-Genoms: 5'TTAGGCTACGACCCGGCGGAAAGA3') (Sequenz ID Nr.
35) und SG1592CgagD (Kartierung an den Nukleotiden 1569-1592 des
S1Vmac251-Genoms: 5'ATAGGGGGTGCAGCCTTCTGACAG3') (Sequenz ID Nr.
36), die ein 496 Basenpaare umfassendes Fragment des SHIV gag-Gens
amplifizieren, wie beschrieben durchgeführt (Ref. 153). Um die Kopienzahl
proviraler DNA zu quantifizieren, wurde in jedem Experiment eine
Standardkurve unter Verwendung des Plasmids pCMRII-Δgag (das
eine 100 Basenpaare umfassende Deletion im gag-Gen von S1Vmac251
enthielt) als Matritzen-DNA und der oben beschriebenen Primer, die
ein 396 Basenpaare umfassendes DNA-Fragment amplifizieren, hergestellt.
Die PCR-Produkte wurden mittels Elektophorese analysiert und durch
densitometrische Analyse (Ultrascan LX Enhancer Laser, LKB, Bromma,
Schweden) quantifiziert. Die Beziehung zwischen den OD-Werten und
der Zahl der Moleküle
des Δgag-Plasmids wurde mittels
linearer Regressionsanalyse (Statgraphics, Manugistics, Inc., Cambridge,
MA) korreliert. Die OD-Werte waren bei bis zu 1000 Molekülen linear
(Korrelationskoeffizient = 0,954 ± 0,026). Die Kopienzahl von
SHIV-proviraler DNA/μg
Zell-DNA wurde durch Interpolation der OD-Werte jeder Probe mit
der Standardkurve bestimmt. Die Empfindlichkeit des Assays betrug
1 Provirus-Kopie/μg
DNA.
- cDie Cytomegalie-Virämie wurde in Co-Kultivierungsassays
bestimmt. Zu diesem Zweck wurden 1 × 104 CEM x
174 Zellen in Gegenwart von Reihenverdünnungen von zu untersuchenden,
CD-8-depletierten PBMC (insgesamt 12 Verdünnungen, von 1 × 106 bis 3,9 × 103 Zellen
pro Vertiefung) in Platten mit 96 Vertiefungen kultiviert. An den
Tagen 3, 7 und 10 nach der Infektion wurden 150 μl entnommen, um das Vorliegen
von p27 Gag mittels ELISA (Innogenetics, Belgien) zu testen, und
durch ein gleiches Volumen frisches Medium ersetzt. Die Ergebnisse
wurden anhand der Formel von Reed und Muench analysiert, um die
Anzahl produktiv infizierter PBMC pro Million der gesamten Zellen
zu bestimmen.
- dDas Vorliegen von Antikörpern gegen
SHIV wurde mit Reihenverdünnungen
von Tierplasma bestimmt, die mit dem Elavia Ac-Ab Ak II-Kit (Diagnostic
Pasteur, Paris, Frankreich) gemäß den Anweisungen
des Herstellers zweifach getestet wurden. Gezeigt ist die höchste Verdünnung, bei
der die Plasmawerte höher
als der Cutoff-Wert waren.
- eBeim Affen M13 wurde statt einer Cytomegalie-Virämie eine
Virusisolierung durchgeführt.
Zu diesem Zweck wurden PBMC (3 × 106) von Affen, die mit unterschiedlichen Dosen
von SHIV 89.6P, gereinigt mittels Ficoll, infiziert waren, mit CEM
x 174 Zellen (1 × 106) in 1 ml Medium kultiviert, das 10% FCS
enthielt. Nach 24 Stunden wurden die Zellen auf 1 × 106/ml verdünnt
und drei Tage kultiviert. Dann wurden zwei ml Medium gesammelt und
die Zellen bei 3 × 105/m1 in 7 ml wieder angeimpft. Der Überschuss
der Zellen wurde verworfen. Dieser Vorgang wurde zweimal pro Woche über 4 Wochen
wiederholt. Das Vorliegen von Viren wurde mittels p27 Gag ELISA
(Innogenetics, Belgien) und anschließend mittels RT-Assay bestimmt.
Die Virusisolierung galt als positiv (+), wenn beide Assays (p27
und RT) in 3 aufeinanderfolgenden Proben positiv waren. Andernfalls
wurde die Virusisolierung als negativ (–) betrachtet.
- fEine qualitative DNA-PCR wurde für den Affen
M13 durchgeführt.
Die virologischen Daten überschneiden
sich mit der absoluten Anzahl CD4-Lymphocyten, die sich bei den
infizierten Affen (M3, M5, M7, M8) als drastisch verringert und
bei den Virus-negativen
Tieren (M1, M2, M4, M6) als hoch und stabil herausstellte, wie dies
in Tabelle 18 gezeigt ist.

- aFACS-Analyse von CD4+- und CD8+-Lymphocyten
von Affen, die mit 10 μg
rekombinantem Tat-Protein und RIBI (M1-3) oder Alaun (M4-6) geimpft
waren. Die Kontrollaffen (M7 und M8) wurden nur mit RIBI- bzw. Alaun-Adjuvanzien
inokuliert. Der Affe M13 war ein naives Tier, das mit 2,85 MID50 SHIV 89.6P infiziert war. Eine Analyse
wurde mit einem Fluoreszenz-aktivierten Zellsorter (FACS), wie beschrieben
(Ref. 137), unter Verwendung markierter monoklonaler Antikörper (Anti-CD4-FITC,
BioSource; Anti-CD8-PerCp, Becton-Dickinson) durchgeführt. ND:
nicht durchgeführt.
-
Die
Ergebnisse vor der Anregung zeigen, dass Tat als Immunogen sowie
RIBI und Alaun als Adjuvanzien (oder ISCOM, das im letzten Boost
als Adjuvans verwendet wurde) von den Tieren gut toleriert wurden und
nicht-toxisch waren, was die Ergebnisse der Sicherheit und Verträglichkeit
der Immunisierung mit Tat bestätigt,
die im ersten Pilotexperiment erhalten wurden. Zudem bestätigen diese
Daten die Beobachtungen des ersten Pilotexperimentes und liefern
zusätzliche
Beweise für
die Tatsache, dass das rekombinante Tat-Protein eine starke humorale
und zelluläre,
Tat-spezifische Antwort mit antiviralen Effekten in vitro und in
vivo hervorruft. Die Ergebnisse nach der Anregung (4/6 geschützte Affen)
bestätigen
die Erwartung der in vitro-Ergebnisse und deuten darauf hin, dass
ein Anti-Tat-Impfstoff einen Schutz gegen die Infektion und damit
gegen die Erkrankung induziert. Die Nachfolgeuntersuchung der beiden
geimpften und infizierten Affen wird die Effekte der Impfung auf
die Progression der Erkrankung klären.
-
Beispiel 5. Inokulierung eines Anti-Tat-DNA-Impfstoffes
in Macaca fascicularis: Analyse von Sicherheit, Verträglichkeit,
spezifischer Immunantwort und Wirksamkeit des Schutzes gegen virale
Anregung (nicht beansprucht).
-
Vorgeschlagen
wird die direkte Inokulierung von DNA des Plasmids pCV-Tat, enthaltend
die cDNA des tat-Gens, und des Plasmids pCV0 als Kontroll-DNA. Die
den Tieren zu verabreichenden Plasmid-DNAs werden in E. Coli (Stamm
DH5) gemäß üblichen
Verfahren (Ref. 110) und gemäß von der „European
Agency for the evaluation of medical products; Human Medicine Evaluation
Unit" (Technical
Report Series Nr. 17 Januar 1997) etablierten Protokollen amplifiziert,
mit zwei CsCI-Gradienten gereinigt und 48-72 Stunden mit 100 Volumina
PBS dialysiert. Dann werden die DNAs mittels Restriktionsenzymverdau überprüft. Die
Funktionalität der
Plasmid-DNA wird mittels Transfektion von 5-10 μg DNA unter Anwendung von Calciumphosphat-Techniken
(Ref. 110) in H3T1-Zellen (1 × 10
6), die eine integrierte Kopie des Reporter-Plasmids
HIV-1 LTR-CAT enthalten, und 48 Stunden später mittels Analyse der CAT-Aktivität (Ref.
55) kontrolliert. Die Verträglichkeit,
die Sicherheit, die Fähigkeit
zum Hervorrufen einer spezifischen Immunantwort (sowohl humoral
als auch zellulär), und
die Wirksamkeit des Schutzes gegen die Virusanregung nach der Immunisierung
mit pCV-Tat-Plasmid-DNA wurden in Cynomolgus-Affen (Macaca fascicularis)
evaluiert. In einem ersten Pilotexperiment wurden drei Affen gemäß folgendem
Schema immunisiert: Affe M1 wurde mit 200 μg pCV-Tat in 300 μl PBS auf i.d.
Wege in 2 Stellen am Rücken
nahe den axillären
Lymphknoten inokuliert (150 μI/Stelle);
Affe M2 wurde mit 500 μg
pCV-Tat in 500 μl
PBS auf i.m. Wege in 2 Stellen am Rücken inokuliert (250 μI/Stelle).
An den Tagen 1 oder 5 vor der i.m. Inokulation wurden 250 μl physiologische
Lösung,
enthaltend 0,5% Bupivacain und 0,1% Methylparaben, in die zwei zuvor
markierten Stellen injiziert, wo Plasmid-DNA inokuliert werden musste.
Dies wurde durchgeführt,
um die Aufnahme und Expression von DNA im Muskel zu erhöhen (Ref.
37, 45). Der Affe M3 wurde nicht inokuliert, sondern als Kontrolltier
verwendet. Ab Woche 10 wurde dieser Affe jedoch mit 6 μg (5 + 1 μg) Tat i.
d. als Kontrolle für
Hauttests inokuliert. Allen Affen wurden 42 und 35 Tage vor der
ersten Inokulation zehn ml Blut zur Analyse von Grundparametern
entnommen. Die Affen wurden zum Zeitpunkt 0 und nach 5, 10, 15,
22, 27, 32 und 37 Wochen inokuliert. Schließlich erhielten die Tiere in
Woche 42 den letzten Boost mit rekombinantem Tat-Protein (16 μg) in 200 μl ISCOM und
300 μl PBS.
Die Tiere wurden täglich
hinsichtlich klinischer Parameter beobachtet, wie dies in Beispiel
4 beschrieben ist. Darüber
hinaus wurden am selben Tag 10 ml Blut entnommen, wie dies in Beispiel
4 beschrieben ist. Die Schutzwirkung der Impfung wurde nach Anregung
der Affen mit 10 MID
50 SHIV89.6P bestimmt,
das in Woche 65 auf intravenösem
Wege injiziert wurde. Die noch andauernde Nachuntersuchung nach
der Anregung wurde wie in Beispiel 4 beschrieben durchgeführt. Die
Ergebnisse dieses Experimentes lauten wie folgt: Bei zwei geimpften
Affen und beim Kontrollaffen wurden keine Veränderungen klinischer, hämatologischer
und verhaltensbezogener Parameter beobachtet. Auch wurden weder
Anzeichen einer Entzündung
noch eine Gefäßneubildung
an der Injektionsstelle beobachtet. Diese Ergebnisse zeigen, dass
die pCV-Tat-DNA von den Tieren gut toleriert wurde und bei den Dosierungen
und Inokulationswegen, die bei diesem Experiment verwendet wurden,
nicht-toxisch war. Der Affe M1, der auf i. d. Wege mit 200 μg DNA geimpft
wurde, entwickelte ab Woche 32 Tat-spezifische IgG-Antikörper (
11). Die Antikörper-Titer
(von Woche 32 bis Woche 58) lagen in einem Bereich von 1:100 bis 1:800
(
12). In Woche 37 zeigte eine Epitopkartierungsanalyse
(durchgeführt,
wie in der Erklärung
zu
4 beschrieben), dass diese Antikörper gegen
spezifische Regionen von Tat gerichtet waren, mit einer Kartierungsposition
bei aa 1-20, aa 46-60 und aa 65-80 und Titern von 1:200, 1:100 bzw.
1:50 (Daten nicht gezeigt). Im Affen M2, der auf i. m. Wege mit
500 μg DNA
geimpft war, wurden über
den gesamten Zeitraum der Studie kaum Anti-Tat-Antikörper detektiert
(mit einem Titer von 1:50, nicht gezeigt). Die Ergebnisse sind in
11 gezeigt. Die Fähigkeit von Plasma des Affen
M1, der auf i. d. Wege mit 200 μg
DNA geimpft war, die Tat-Aktivität
zu neutralisieren, wurde getestet, indem ein Assay der Inhibierung
der Rettung der viralen Replikation in HLM1-Zellen, inkubiert mit
exogenem Tat-Protein, durchgeführt
wurde, wie dies in Beispiel 4 beschrieben ist. Dieser Assay zeigte,
dass das Plasma des Affen M1, 1:2 verdünnt und in Woche 37 erhalten,
die mit 30 ng/ml exogenem Tat induzierte virale Replikation verringerte.
Dagegen wurde extrazelluläres
Tat durch das Plasma desselben Affen, das zum Zeitpunkt 0 (präimmun) erhalten
wurde, nicht blockiert (Tabelle 19). TABELLE 19 Neutralisierende Aktivität von Plasma
gegenüber
der Rettung einer mit extrazellulärem Tat induzierten, viralen Infektion
a | Proben
Tat
+ M1 präimmun
Tat
+ M1 immun | Inhibierung
0
51 |
- aDie Fähigkeit
von Anti-Tat-Antikörpern,
die Tat-Aktivität
zu neutralisieren, wurde in HLM1-Zellen durch Zugabe von 30 ng/ml
rekombinantem Tat-Protein bestimmt, das zuvor mit einem gleichen
Volumen an Plasma inkubiert wurde, welches zum Zeitpunkt 0 (präimmun) oder
in Woche 37 (immun) vom Affen M1 erhalten wurde, der auf i. d. Wege
mit 200 μg
pCV-Tat-Plasmid-DNA geimpft war. Der Assay wurde durchgeführt und
die Ergebnisse sind ausgedrückt,
wie dies in Tabelle 4 beschrieben ist.
-
Die
in Tabelle 20 angegebenen Ergebnisse zeigen das Vorliegen einer
proliferativen Antwort auf Tat in Woche 42 im Affen M1, der i. d.
mit 200 μg
DNA immunisiert war, während
diese Art einer zellulären
Antwort im Affen M2 nicht detektiert wurde. TABELLE 20 Proliferative Antwort auf Tat
a | Affe | Stimulus | Wochen
nach Erstimmunisierung |
| | | 15 | 22 | 27 | 32 | 37 | 42 | 48 | 58 |
| M1 | PHA
TT
Tat | 32,9
0,8
0,9 | 45
2,7
1,7 | 89,3
1,5
1,2 | 40,5
1,3
1,1 | 3,1
0,6
1,1 | 13,3
9
5,9 | ND
1,2
1 | 13,1
1,6
1 |
| M2 | PHA
TT
Tat | 11,7
0,9
0,8 | 18,5
1,8
1,4 | 21,8
0,8
0,9 | 32,2
1,1
1,1 | 1,1
1
1,1 | 6,2
1,5
1,3 | 7
1,1
1,1 | 18,9
1
1 |
| M3 | PHA
TT
Tat | 5,1
7,2
2,1 | 19,9
6,2
1,4 | 18,2
5,5
2,2 | 6,6
2,8
0,7 | 8,1
5,6
1,5 | 77,8
36,8
2,8 | ND
1
0,8 | 2,1
2,1
0,9 |
- aPBMC wurden isoliert,
mit PHA (4 μg/ml),
Tetanustoxoid (TT) und Tat (1 oder 5 μg/ml) stimuliert und getestet, wie
in Tabelle 5 beschrieben. Die Affen wurden auf i. d. Wege mit 200 μg (M1) pCV-Tat
oder auf i. m. Wege mit 500 μg
(M2) pCV-Tat geimpft. Der Affe (M3) wurde nicht geimpft, sondern
ab Woche 10 mit 6 μg
(5 + 1 μg)
Tat als Kontrolle für
Hauttests i. d. inokuliert. ND: nicht durchgeführt.
-
Die
cytotoxische Anti-Tat-Aktivität
(CTL) wurde im Affen M1 in den Wochen 42 und 48 sowie im Affen M2
in Woche 48 detektiert. Ferner wurde in Woche 48 eine positive CTL-Antwort im Affen
M3 detektiert, der seit Woche 10 mit 6 μg Tat als Kontrolle für Hauttests
inokuliert wurde (Tabelle 21). TABELLE 21 Analyse der Tat-spezifischen cytotoxischen
Aktivität
(CTL)
a | Affe | Woche | Ziel:Effektor-Verhältnis | CTL-Aktivität |
| 1:50 | 1:25 | 1:12,5 | 1:6,25 | 1:3,125 | Medien |
| M1 | 42 | 27,4 | 27,8 | 17,1 | 9,8 | 3,9 | 17,2 | + |
| | 48 | ND | ND | 21,3 | 0 | 11,7 | 11 | + |
| M2 | 42 | 1,2 | 5,9 | 2,4 | 1 | 0 | 2,1 | – |
| | 48 | ND | ND | ND | 57 | 25,1 | 41 | + |
| M3 | 42 | 0 | 0 | 0 | 1,2 | 0 | 0,6 | – |
| | 48 | ND | 12,4 | 4,2 | 0 | 0 | 0 | + |
- aDer Assay wurde
wie in Tabelle 6 beschrieben durchgeführt. Die Affen wurden mit 200 μg (M1) pCV-Tat
auf i.d. Wege oder mit 500 μg
(M2) pCV-Tat auf i.m. Wege geimpft. Der Affe (M3) wurde nicht geimpft,
sondern ab Woche 10 mit 6 μg
(5 + 1 μg)
Tat als Kontrolle für
Hauttests i.d. inokuliert. ND: nicht durchgeführt.
-
Die
in Tabelle 22 angegebenen Ergebnisse zeigen in Woche 52 das Vorliegen
der antiviralen Gesamtaktivität
(TAA) in den beiden mit 200 bzw. 500 μg DNA geimpften Affen. TABELLE 22 Analyse der antiviralen Gesamtaktivität (TAA)
a | Affe | Tage nach
Infektion |
| | 7 | 17 |
| | Minimale
infektiöse
Dosis (TCID50/Zelle) | Minimale
infektiöse
Dosis (TCID50/Zelle) |
| M1 | 10–4 | 10–4 |
| M2 | 10–4 | 10–4 |
| M3 | 10–8 | 10–8 |
- aDer Assay wurde
wie in Tabelle 13 beschrieben durchgeführt. Die Affen wurden mit 200 μg (M1) pCV-Tat
auf i. d. Wege oder mit 500 μg
pCV-Tat auf i. m. Wege inokuliert. Der Affe (M3) wurde nicht inokuliert,
erhielt aber ab Woche 10 i. d. 6 μg
(5 +1 μg)
Tat als Kontrolle für
Hauttests. PBMC wurden in Woche 52 nach der Erstimmunisierung gesammelt
und mit SHIV 89.6P (10–2, 10–4,
10–6,
10–8 TCID50/Zelle) infiziert. Die Ergebnisse sind dargestellt
als minimale infektiöse
Dosis von SHIV (TCID50/Zelle), die noch
in der Lage war, die Zellen zu infizieren.
-
Die
in Tabelle 23 gezeigten Ergebnisse deuten auf das Vorliegen von
löslicher
antiviraler Aktivität (CAF),
vermittelt durch CD8+ T-Lymphocyten, in den Wochen 22 und 27 bei
beiden geimpften Affen hin. Die Aktivität war im Kontrollaffen niedriger. TABELLE 23 Analyse der CD8+-Zellen-vermittelten,
löslichen
antiviralen Aktivität
(CAF)
a | Affe | Wochen
nach Erstimmunisierung | % Inhibierung
der viralen Replikation |
| | | Akute
Infektion | Chronische
Infektion |
| M1 | 22 | 62 | 27 |
| | 27 | 56 | 25 |
| M2 | 22 | 74 | ND |
| | 27 | 28 | ND |
| M3 | 22 | 24 | ND |
| | 27 | 37 | 22 |
- aAnalyse des Vorliegens
von löslicher
antiviraler Aktivität,
erzeugt durch CD8+ T-Lymphocyten
(CAF), die von mit 200 μg
(M1) und 500 μg
(M2) pCV-Tat inokulierten Affen und vom Affen M3 stammten. Die antivirale
Aktivität
wurde bei einer akuten und einer chronischen Infektion in CEM x
174 Zellen, die mit SHIV 89.6P infiziert waren, und in OM-10-1-Zellen, die
chronisch mit HIV-1 infiziert waren, wie in Tabelle 7 beschrieben,
getestet. Die Ergebnisse sind dargestellt als Prozentsatz (%) der
Inhibierung viraler Replikation in Zellen, die mit Überständen von
CD8+ T-Lymphocyten behandelt wurden, im Vergleich zu unbehandelten
Zellen. Die in der Tabelle gezeigten Ergebnisse der akuten und der
chronischen Infektion beziehen sich auf Proben, die mit 5 μl der CD8+-Kulturüberstände behandelt
wurden. ND: nicht durchgeführt.
-
Die
in Tabelle 24 angegebenen Ergebnisse zeigen, dass der Affe M1, inokuliert
mit 200 μg
DNA auf i. d. Wege, in Woche 22 einen positiven Tat-Hauttest hatte. TABELLE 24 Tat-Hauttest
a | Wochen
nach Immunisierung | Affe |
| | M1 | M2 | M3 |
| 10 | – | – | – |
| 15 | – | – | – |
| 22 | + | – | – |
| 27 | – | – | – |
| 32 | – | – | – |
| 37 | – | – | – |
| 42 | – | – | – |
| 48 | – | – | – |
| 52 | – | – | – |
| 58 | – | – | – |
- aTat (1 und 5 μg) in 150 μl PBS-0,1%
BSA oder der Puffer alleine (Kontrolle) wurden in den Wochen 10,
15, 22, 27, 32, 37, 42, 48, 52 und 58 nach der Erstimmunisierung
i. d. in einen zuvor trichotomisierten Bereich am oberen Rücken der
geimpften Tiere und in den Kontrollaffen (Kontrolle für die Spezifität der Antwort)
inokuliert. Der Affe M1 wurde i.d. mit 200 μg DNA des Plasmids pCV-Tat inokuliert,
während
der Makake M2 500 μg
des gleichen Plasmids i. m. erhielt. Der Affe M3 (Kontrolle) wurde
nicht geimpft, erhielt aber ab Woche 10 i. d. 6 μg (5 + 1 μg) Tat als Kontrolle für Hauttests.
Das Auftreten eines erythematösen
Knötchens
48 bis 72 Stunden später deutete
auf das Vorliegen einer verzögerten
Hypersensibilität
(DTH) hin: ++, ∅ ≥ 5
mm; + ≥ 1-4
mm; ±,
Erythem ohne Verhärtung; –, ∅ < 1 mm.
-
Diese
Ergebnisse zeigen, dass das Plasmid pCVTat (pCVTat-DNA) in den gegebenen
Dosierungen sowohl intradermal als auch intramuskulär gut toleriert
wurde und sicher war. Darüber
hinaus zeigen diese Ergebnisse, dass die Immunisierung mit der pCVTat-DNA
sowohl eine humorale (wenn auch eine geringere, als die durch die
Immunisierung mit dem rekombinanten Tat-Protein induzierte) als
auch eine zelluläre
Anti-Tat-Immunantwort mit antiviralen Effekten induziert. Bezüglich der
Schutzwirksamkeit nach Anregung (durchgeführt in Woche 65 nach der Erstimmunisierung)
deuten die virologischen Daten, einschließlich Antigenämie- und
Cytomegalie-Virämiemessungen,
und die Bestimmung der Anzahl proviraler DNA-Kopien (DNA-PCR) in
PBMCs darauf hin, dass der Affe M2, der i. m. mit Tat-DNA immunisiert
wurde, sich bei Anregung mit 10 MID50 von SHIV-89.6P
als geschützt
herausstellte, während
der Makake M1, der i. d. mit einer niedrigeren Dosis Tat-DNA (200 μg) immunisiert
wurde, sich als infiziert herausstellte, was nahelegt, dass hinsichtlich
der Immunisierung mit DNA der i. m. Weg wirksamer als die i. d.
Inokulierung ist. Der Kontrollaffe M3 erwies sich ebenfalls als resistent
gegen die Infektion. Wie zuvor beschrieben, erhielt dieser Affe
jedoch im Gegensatz zu den Kontrollen der anderen experimentellen
Protokolle wiederholte Hauttests für Tat, um die Testspezifität zu kontrollieren (Tabelle
24), und Anti-Tat-Antikörper
wurden, wenn auch mit niedrigeren Titern (1:100), ab Woche 32 seit
Beginn der Immunisierung detektiert (Daten nicht gezeigt). Darüber hinaus
zeigte die proliferative Antwort auf Tat in diesem Affen eine schwache
und sporadische Reaktivität
gegenüber
dem Antigen (Tabelle 20). Schließlich zeigte der Affe M3 das
Vorliegen spezifischer Anti-Tat-CTLs (Tabelle 21). Obwohl sie nur
vorläufig
sind, deuten diese Daten darauf hin, dass die wiederholte i. d.
Injektion von 6 μg
Tat zur Immunisierung des Tiers und zum Schutz gegen Anregung hätte führen können. Daher
soll der Affe M3 als i. d. mit dem Tat-Protein geimpft betrachtet
und als solcher untersucht werden.
-
-
Die
FACS-Evaluation des Prozentsatzes und der absoluten Anzahl der CD4-
und CD8-Lymphocyten bestätigten die
virologischen Daten mit einer klaren Verringerung (etwa 4-fach)
der CD4-Lymphocyten im infizierten Affen bereits bei der ersten
Analyse nach Anregung (Tag 30), was später (Tag 60) bestätigt wurde
(Tabelle 26).
-
-
Ausgehend
von diesen Ergebnissen, wurde ein zweites Experiment entwickelt,
in dem die Effekte der Immunisierung mit der pCVTat-DNA in 3 Affen
(M9-M11) im Vergleich zum Kontrollaffen (M12), der die pCV0-DNA
erhielt, evaluiert wurden. Alle Tiere wurden i. m. in 2 Stellen
am Rücken
mit insgesamt 1 mg pCVTat (M9-M11) oder pCV0 (M12) inokuliert. Entweder
1 oder 5 Tage vor der Impfung wurden 250 μl Kochsalzlösung, enthaltend 0,5% Bupivacain
und 0,1% Methylparaben, in die beiden markierten Stellen inokuliert,
in die anschließend
das Plasmid injiziert worden wäre.
Die Makaken wurden zum Zeitpunkt 0 und in den Wochen 6, 11, 15,
21, 28 und 32 geimpft. Ein abschließender Booster wurde in Woche
36 mit dem rekombinanten Tat-Protein (16 μg), resuspendiert in 200 μI ISCOM und
300 μI PBS,
durchgeführt.
Die Tiere wurden jeden Tag hinsichtlich klinischer Parameter kontrolliert,
wie dies in Beispiel 4 beschrieben ist. Darüber hinaus wurden 9 Tage vor
der Erstimmunisierung und bei jeder Immunisierung 10 ml Blut entnommen,
wie dies in Beispiel 4 beschrieben ist. Um die Schutzwirkungen der
Impfung zu evaluieren, wurden die Affen in Woche 50 nach Beginn
der Immunisierung mittels intravenöser (i. v.) Injektion von 10
MID50 SHIV89.6P angeregt. Die Nachfolgeuntersuchung nach
der Anregung dauert noch an und wird wie in Beispiel 4 beschrieben
durchgeführt.
-
Die
Ergebnisse dieses Experimentes sind die folgenden: Sowohl in den
geimpften Tieren als auch in den Kontrolltieren waren keine Veränderungen
hinsichtlich Verhalten, klinischer Parameter und Blutchemie festzustellen.
An den Injektionsstellen wurden weder Anzeichen für eine Entzündung noch
Gefäßneubildungen detektiert.
Diese Ergebnisse bestätigen,
dass 1 mg der Plasmid pCVTat-DNA, i. m. injiziert, gut toleriert
wurde und nicht-toxisch war. Anti-Tat IgG wurden ab Woche 15 (
13) mit Titern in einem Bereich von 1:50 bis 1:100
detektiert (Daten nicht gezeigt). Darüber hinaus wurde eine proliferative
Antwort auf Tat bei einem Affen (M11) bereits in Woche 2 detektiert
(Tabelle 27). TABELLE 27 Proliferative Antwort auf Tat
a | Affe | Stimulus | Wochen
ab Primärimmunisierung |
| | | 2 | 6 | 11 | 15 | 21 | 28 | 32 | 36 | 40 | 44 | 50 |
| M9 | PHA
TT
Tat | 8,9
2,9
0,4 | 9,2
1,7
0,5 | 17,1
0,9
0,6 | 58,2
1
1,5 | 18
1,8
1,6 | 47,1
0,7
0,9 | 43,4
1,1
1 | 3,1
0,8
0,7 | 72,6
1
1,1 | 64,6
7
7 | 7
2,7
1,9 |
| M10 | PHA
TT
Tat | 8,5
2,4
1 | 18
0,3
0,3 | 19,8
0,8
0,7 | ND
ND
ND | 10,1
1,1
1,1 | 2,2
0,6
0,5 | 14,7
1
1 | 15,2
0,9
0,9 | 4,4
0,6
0,7 | 8,4
6,4
4,2 | ND
ND
ND |
| M11 | PHA
TT
Tat | 25,7
4,2
5,1 | 43,3
1,9
0,8 | 1,3
1,6 | 12,1
0,9
0,7 | 27,8
1,1
1,1 | 3,4
3,6
1,1 | 21,3
1,2
1,2 | 14,1
0,8
0,7 | 15,9
0,3
0,7 | 25,8
1,8
3 | ND
ND
ND |
| M12 | PHA
TT
Tat | 28,7
3,2
3,2 | 30,9
1,6
1,4 | 41
0,9
0,8 | 50,7
5,2
1,3 | 30,8
1,6
1 | 7,6
1,6
1,6 | 43
1,3
1 | 22,6
1,1
0,8 | 34,6
1
1 | 19,9
0,7
1,6 | 55,1
3,1
1,3 |
- aPBMC wurden isoliert,
mit PHA (4 μg/ml)
oder Tetanustoxoid (TT, 10 μg/ml
oder Tat (1 und 5 μg/ml)
stimuliert und einem Assay unterzogen, wie in Tabelle 5 beschrieben.
Den Affen wurden 1 mg pCVTat (M9-M11) oder pCV0 (M12, Kontrolle)
i. m. injiziert. ND: nicht bestimmt.
-
Anti-Tat-CTLs
wurden in Woche 32 nach der Immunisierung detektiert (Tabelle 28). TABELLE 28 Analyse der cytotoxischen Anti-Tat-Aktivität (CTLS)
a | Affe | Woche | Ziel:Effektor-Verhältnis | CTL-Aktivität |
| 1:50 | 1:25 | 1:12,5 | 1:6,25 | 1:3,125 | Medien |
| M9 | 32 | 0 | 0 | 0 | 0 | 0 | 0 | – |
| | 50 | 4,2 | 0 | 0 | 0 | 0,9 | 1 | – |
| M10 | 32 | 0 | 0 | 9,9 | 2,7 | 0 | 2,5 | – |
| | 50 | 3,5 | 0 | 2,3 | 0 | 0 | 1,1 | – |
| M11 | 32 | 0 | 10,5 | 8,9 | 3,5 | 0,9 | 4,7 | + |
| | 50 | 0 | 0 | 0 | 3,8 | 0,3 | 0,8 | – |
| M12 | 32 | 0 | 0 | 0 | 0 | 0 | 0 | – |
| | 50 | 0 | 0 | 0 | 0 | 0 | 0 | – |
- aDer Assay wurde
wie in Tabelle 6 beschrieben durchgeführt. Den Makaken wurde 1 mg
pCVTat (M9-M11) oder pCV0 (M12, Kontrolle) i.m. injiziert.
-
PBMCs,
die vom Affen M11 in Woche 44 erhalten wurden, erwiesen sich als
resistent gegen in vitro-Infektion mit Reihenverdünnungen
des chimären
SHIV-89.6P-Virus in einem zuvor beschriebenen Assay, der das Vorliegen
der antiviralen Gesamtaktivität
(TAA) detektiert. Tatsächlich
wird die TAA evaluiert als die Fähigkeit
von PBMCs aus Affen, die mit pCVTat-DNA geimpft sind, die in Gegenwart von
autologem Serum gezüchtet
wurden, der Infektion mit Virus-Reihenverdünnungen zu widerstehen. (Tabelle
29) TABELLE 29 Analyse der antiviralen Gesamtaktivität (TAA)
| Affe | Tage nach
Infektion |
| | 7 | 17 |
| | Minimale
infektiöse
Dosis (TCID50/Zelle) | Minimale
infektiöse
Dosis (TCID50/Zelle) |
| M9 | 10–2 | > 10–2** |
| M10 | 10–3 | 10–2 |
| M11 | > 10–2* | > 10–2* |
| M12 | 10–2 | > 10–2** |
- aDer Assay wurde
wie in Tabelle 13 beschrieben durchgeführt. Den Makaken wurde 1 mg
pCVTat (M9-M11) oder pCV0 (M12, Kontrolle) i.m. injiziert. PBMCs
wurden in Woche 44 nach der Erstimmunisierung entnommen und in vitro
mit 10–2,
10–3,
10–4,
10–5 TCID50 von SHIV-89.6P infiziert. Die Ergebnisse
sind als die minimale infektiöse
Dosis des SHIV (TCID50/Zelle) angegeben,
die noch in der Lage ist, die Zellen zu infizieren.
- *Keine Kultur stellte sich bei der höchsten SHIV-Konzentration,
die im Assay verwendet wurde (10–2 TCID50/Zelle), als infiziert heraus. **Die Kulturen
wurden am Tag 17 nach der Infektion negativ.
-
Die
in Tabelle 30 angegebenen Ergebnisse zeigen das Vorliegen der durch
die CD8+ T-Lymphocyten vermittelten,
löslichen
antiviralen Aktivität
(CAF) in den geimpften Affen und in dem Kontrollaffen (M12), dem der
leere Vektor (pCV0) injiziert worden war. TABELLE 30 Analyse der durch die CD8+ T-Lymphocyten
vermittelten, löslichen
antiviralen Aktivität
(CAF)
a | Affe | Wochen nach Primärimmunisierung | % Inhibierung
der viralen Replikation |
| Akute
Infektion | Chronische
Infektion |
| M9 | 0 | 21 | 14,6 |
| | 36 | 77 | 2,6 |
| M10 | 0 | 40 | 13,8 |
| | 36 | 67 | 25 |
| M11 | 0 | 49 | 19 |
| | 36 | 42 | 14 |
| M12 | 0 | 65 | 23 |
| | 36 | 62 | 14 |
- aAnalyse des Vorliegens
der durch die CD8+ T-Lymphocyten vermittelten, löslichen antiviralen Aktivität (CAF). PBMCs
wurden von den drei Affen (M9-M11), denen 1 mg pCVTat injiziert
worden war, und von dem Kontrollaffen (M12) erhalten, der mit 1
mg pCV0 inokuliert war. Der Assay für die akute Infektion wurde
in CEM x 174 Zellen, infiziert mit SHIV-89.6P, wie in Tabelle 14
beschrieben durchgeführt.
Der Assay für
die chronische Infektion wurde in U1-Zellen, die chronisch mit HIV-1
infiziert waren, wie in Tabelle 14 beschrieben durchgeführt. Die
Ergebnisse sind als Prozentsatz (%) der Inhibierung der viralen
Replikation in Zellen, die in Gegenwart oder in Abwesenheit (Kontrolle)
von 5 μl
der Überstände von
CD8+ T-Zellen kultiviert wurden, ausgedrückt.
-
Die
Produktion von Cytokinen (γIFN,
IL-4, TNFα)
und des Chemokins RANTES wurde in Woche 44 in PBMCs sowohl der geimpften
Affen als auch des Kontrollaffen evaluiert (Tabelle 31).
- aDer Assay wurde wie in Tabelle 15 beschrieben
durchgeführt.
Den Makaken wurde 1 mg pCVTat (M9-M11) oder pCV0 (M12, Kontrolle)
i.m. injiziert. PBMCs wurden in Woche 44 nach der Erstimmunisierung
entnommen. Die Ergebnisse sind gezeigt als pg/ml von Cytokinen und
RANTES, detektiert nach 48 bzw. 96 Stunden (48/96).
- (–):
Die Werte lagen unter dem Cutoff-Wert. Die Cutoff-Werte (pg/ml)
betrugen: γIFN:
31,2; IL-4: 3,12; TNF-α: 15,6;
RANTES: 62,5. ND: nicht durchgeführt.
TABELLE 32 Die Ergebnisse zeigen das Vorliegen einer
schwachen Reaktivität
auf die Hauttests mit Tat bei einem Affen (M9) in Woche 11 (Tabelle
32) Tat-Hauttesta | Wochen nach
Primärimmunisierung |
| Affe | 11 | 15 | 21 | 28 | 32 | 36 | 44 |
| M9 | +/– | – | – | – | – | – | – |
| M10 | – | – | – | – | – | – | – |
| M11 | – | – | – | – | – | – | – |
| M12 | ND | ND | ND | ND | ND | ND | ND |
- aTat (1 und 5 μg) in 150 μl PBS-A,
0,1% BSA oder der Puffer alleine (Kontrolle) wurden i. d. in einen
zuvor trichotomisierten Bereich am oberen Rücken der geimpften Affen, aber
nicht in die Kontrollaffen, in den Wochen 11, 15, 21, 28, 32, 36,
und 44 nach der Erstimmunisierung inokuliert. Den Makaken wurde
1 mg pCVTat (M9-M11) oder pCV0 (M12, Kontrolle) i.m. injiziert.
Das Auftreten eines erythematösen
Knötchens
48 bis 72 Stunden später
deutete auf das Vorliegen einer verzögerten Hypersensibilität (DTH)
hin: ++, ⌀ ≥ 5 mm; + ⌀ 1-4 mm; ±, Erythem
ohne Verhärtung; –, ⌀ < 1 mm.
-
Die
Ergebnisse nach Anregung deuten darauf hin, dass alle geimpften
Tiere gegen die Infektion mit 10 MID50 von
SHIV-89.6P geschützt
waren, wie sich durch die virologischen Tests (Plasmaantigenämie, Bestimmung
der Kopienzahl proviraler DNA, Cytomegalie-Virämie)
zeigte, die alle negativ waren (Tabelle 33). Darüber hinaus deutete das Vorliegen
von Anti-SIV-Antikörpern
im Affen M11 auf die Exposition mit dem Virus oder eine abortive
Infektion hin. Im Gegensatz dazu wurden sie in den übrigen Affen
nicht detektiert, weshalb wir beschlossen, den in vitro-Antikörperproduktionsassay
(IVAP) sowie die lymphoproliferative Antwort auf SIV-Antigene durchzuführen. Diese
Assays dauern noch an, und vorläufige
Daten zeigen das Vorliegen von Anti-HIV Env-Antikörpern in
allen DNA-inokulierten
Affen. Die Makaken werden mit einer höheren Dosis des Virus inokuliert
werden, da sich auch das Kontrolltier M12 als gegen die Infektion
resistent erwies.
-
Dieser
Affe war mit dem leeren Vektor pCV0 geimpft worden. Jüngste Daten
aus der Literatur zeigten die Adjuvansrolle, die von bestimmten
DNA-Sequenzen gespielt wird, welche in Bakterien deutlich häufiger als in
eukaryontischen Zellen sind, und die ähnlich wie bei LPS und Mannose
einen starken Stimulus für
die natürliche
Immunität
darstellen (Ref. 179). Somit ist denkbar, dass der beim Affen M12
beobachtete Schutz möglicherweise
durch die Induktion einer nicht-spezifischen antiviralen Immunität durch
diese bakteriellen Sequenzen bedingt ist, wie die Produktion von
IFNα, IFNβ, IL-12 und
IL18, die bekanntlich immun-modulierende und antivirale Funktionen
induziert. Dies wird durch das Vorliegen von TAA- (Tabelle 29) und
CAF- (Tabelle 30) antiviralen Aktivitäten bei fehlender Anti-Tat-spezifischer, humoraler
und zellulärer
Immunität
in diesem Makaken deutlich nahegelegt. Tatsächlich messen diese Assays
auch nicht-antigenspezifische antivirale Aktivitäten. Der naive Affe M13, inokuliert
mit einer 3,5-mal niedrigeren Virusdosis als sie dem Makaken M12
injiziert wurde, erwies sich als infiziert. Diese Ergebnisse bestätigen, dass
die 10 MID50-Anregungsdosis, mit welcher der Affe M12
inokuliert wurde, infektiös
war (Tabelle 33). Auf Grundlage dieses Ergebnisses plant die Erfinderin
die Verwendung des pCV0-Vektors oder von Teilen davon als Adjuvans.
-
-
Eine
FACS-Analyse der CD4- und CD8-Subsets (Tabelle 34) bestätigte die
virologischen Daten.
-
Tatsächlich wurde
ein signifikanter Rückgang
des Prozentsatzes und der absoluten Anzahl der CD4-Lymphocyten an
den Tagen 15 und 60 nach Anregung lediglich beim naiven Affen M13
beobachtet, der sich als infiziert erwies, wie das positive Ergebnis
der Plasmaantigenämie,
der proviralen DNA und der Virusisolierung zeigt (Tabelle 33).
-
-
Diese
Ergebnisse zeigen, dass die Impfung mit dem pCVTat-Plasmid gut toleriert
wurde und nicht-toxisch war, und bestätigen die Ergebnisse bezüglich Sicherheit
und Verträglichkeit
der DNA-Impfung, die in der ersten Pilotstudie erhalten wurden.
Zudem liefern diese Daten den Nachweis, dass das pCVTat-DNA-Plasmid eine
spezifische humorale (wenn auch schwächere, als die durch das Tat-Protein
induzierte) und zelluläre
Immunantwort mit antiviralen Effekten induziert, die zum Teil durch
bestimmte, im pCV0-Vektor vorhandene DNA-Sequenzen bedingt sein
kann, die als Adjuvanzien fungieren könnten.
-
Es
werden Immunisierungsprotokolle evaluiert, die Kombinationen der
für andere
HIV-1- und Cytokingene
kodierenden DNA enthalten, wie in Beispiel 3 beschrieben. In diesen
Experimenten soll SHIV, enthaltend die tat-, rev- und nef-Gene von
HIV, verwendet werden (Ref. 146, 85, 142, 65, 94, 129).
-
Die
pCV0- und pCVTat-Plasmide werden in die Tiere unter Verwendung anderer
Zuführungssysteme inokuliert,
welche die Immunisierungswirksamkeit verbessern können, wie
Liposome, Nanopartikel, Erythrozyten, Zuführung mittels Genpistole, oder
Tat-DNA wird unter Verwendung von Herpesvektoren zugeführt, wie
in den vorausschauenden Beispielen 9 und 10 beschrieben.
-
Beispiel 6: Therapeutischer Impfstoff
-
Ein
Impfprotokoll, das sowohl auf Tat-Protein als auch auf Tat-DNA beruht,
wurde erstellt, um die Sicherheit und Toxizität eines Anti-Tat-Impfstoffs
bei bereits infizierten Individuen zu evaluieren.
-
Das
Experiment wurde an Affen durchgeführt, die mit niedriger werdenden
Dosen von SHIV89.6P und mit der Immunschwächeerkrankung (AIDS) infiziert
wurden. Das zur Infektion verwendete virale Ausgangsmaterial wurde
aus Milz und Lymphknoten eines 14 Tage zuvor infizierten Cynomolgusaffen
erhalten. Lymphocyten, gereinigt durch mechanische Auftrennung,
wurden in zwei Aliquots (jeweils 1,5 × 106 Zellen/ml)
aufgeteilt. Aus einem Aliquot wurden mittels immunmagnetischer Kügelchen
(Dynal, Norwegen) CD8+ T-Zellen depletiert. Beide Kulturen wurden
drei Tage mit PHA (1 μg/ml)
stimuliert und in der Konzentration von 1 × 106 Zellen/ml
in Gegenwart von 50 U/ml IL-2 angeimpft. Die virale Replikation
wurde durch das Vorliegen reverser Transkriptase (RT) im nach drei
Tagen geernteten Kulturmedium detektiert. Vor dem Testen wurde der Überstand
geklärt
und bei 100.000 UpM 11 Minuten bei +4°C ultrazentrifugiert (Beckman
TL-100-Ultrazentrifuge) und das Pellet lysiert. Dreißig μl der Suspension
wurden zu dem Reaktionsgemisch (IRIS HCl 1M, pH 8; MgCl2, 0,5
M; KCl, 1M; Poly A 1 mg/ml; Oligo-dT 12-18 100 μg/ml; DTT 0,02 M; 1,2 3[H]-Methylthymidintriphosphat 1 mCi/ml)
gegeben und 60 Minuten bei 37°C
inkubiert. Die Reaktion wurde durch Zugabe von 500 μl 0,1 M Na-Pyrophosphat,
pH5, und 600 μl
20%iger Trichloressigsäure
(TCA) beendet, und die Probe wurde auf einen 0,45 μm-Filter
(Millipore) getupft und dann mit einem β-Zähler nach Zugabe von 5 ml eines
Szintillationscocktails (Filter Count, Packard) gelesen.
-
Kulturmedien
mit mehr als 20,000 cpm wurden zentrifugiert und mit 10% menschlichem
Serum AB ergänzt.
Das Virus wurde durch Ultrazentrifugation bei 30.000 UpM (90 Minuten
bei 4°C)
konzentriert, in RPMI 1640, enthaltend 10% menschliches Serum (AB-Gruppe),
resuspendiert und dann in kleinen Aliquots in flüssigem Stickstoff gelagert.
Das virale Ausgangsmaterial wurde in vitro auf den menschlichen
Zelllinien CEMx174 und C8166 (3 × 103 TCID50/Zelle) und in vivo auf Cynomolgus-Affen
(3,17 ≥ 105,69 MID50/ml) titriert.
-
Ein
erstes Pilotexperiment wurde an 7 Affen durchgeführt, die mit wie oben hergestelltem
SHIV89.6P i. v. infiziert wurden. Jeder Affe erhielt 1 ml SHIV,
verdünnt
in Kochsalzpuffer, ergänzt
mit 2% menschlichem Serum (AB, Rh–), gemäß folgendem Protokoll: Ein
Affe (IM1) wurde mit 1:500 viraler Verdünnung inokuliert; zwei Affen
(IM2, IM3) erhielten die Verdünnung
1:5.000; zwei Affen (IM4, IM5) wurden mit 1:50.000 inokuliert; der
Affe IM6 erhielt die Verdünnung
1:500.000; der letzte Affe (IM7) erhielt die Verdünnung 1:5.000.000.
Jedem Affen wurde am Tag 7 vor der Infektion mit SHIV zur Bestimmung
der Grundparameter Blut entnommen. Serum und Plasmaproben wurden
bei –20°C oder –80°C eingefroren
und dann dazu verwendet, das Proteininokulum zu resuspendieren.
Zum Zeitpunkt 0 wurden alle Affen mit SHIV89.6P inokuliert. Die
Affen wurden täglich überprüft. Zudem
wurde ihnen am Tag 0 und nach 2 und 4 Wochen Blut entnommen und
wurden 10 ml Blut für hämatochemische
Bestimmungen (chemisch-klinische Analyse, Elektrolyten, weiße Zellen
und Plättchenzählungen,
Hämoglobin)
und virologische und immunologische Analyse (d. h. Plasma p27 Ag-Bestimmung
und virale Last in Plasma und Zellen) verwendet. In Woche 4 nach
der Infektion waren 6 Affen (IM1-6) infiziert. Der Affe IM7, der
die geringste virale Verdünnung
erhielt (1:5.000.000) war SHIV-negativ (Tabelle 35). TABELLE 35 Detektion des Vorliegens von SHIV89.6P
in Affen, infiziert mit viralen Reihenverdünnungen
| Affe | SHIV 89.6P-Verdünnung | Wochen nach
Infektion |
| | | 0 | | 2 | | 4 | |
| | | Virusisolierunga | p27 (pg/ml)b | Virusisolierunga | p27 (pg/ml)b | Virusisolierunga | p27 (pg/ml)b |
| IM1 | 1:500 | ND | ND | + | > 450 | + | 47 |
| IM2 | 1:5.000 | ND | ND | + | > 450 | + | 161,8 |
| IM3 | 1:5.000 | ND | ND | + | > 450 | + | 6,67 |
| IM4 | 1:50.000 | ND | ND | – | < 20 | + | > 450 |
| IM5 | 1:50.000 | ND | ND | – | > 450 | + | 166,7 |
| IM6 | 1:500.000 | ND | ND | – | > 450 | + | 0 |
| IM7 | 1:5.000.000 | ND | ND | – | 0 | – | 0 |
- aVirusisolierung
und bPlasma p27 Ag (pg/ml) wurden wie in
der Erklärung
zu Tabelle 17 beschrieben durchgeführt. Die Affen wurden i. v.
mit Reihenverdünnungen
des Virusausgangsmaterials inokuliert, wie im Text beschrieben.
-
7
Wochen nach der Infektion wurden alle Tiere, die ernste Immunschwächesymptome
zeigten, sowohl mit dem Tat-Protein als auch mit der DNA des Plasmids
pCVTat gemäß folgendem
Protokoll geimpft. Die Affen IM1, IM3, IM5 und IM6 erhielten das
Tat-Protein (20 g), gelöst
in 250 μl
PBS-A, ergänzt
mit 0,1% BSA und 20% autologem Plasma und dann zu 250 μl Alaun-Adjuvans
gegeben. Das Proteininokulum wurde subkutan an einer einzigen Stelle
am oberen Rücken
des Affen durchgeführt,
wobei das Plasmid pCVTat (1 mg), resuspendiert in 1 ml PBS-A, an
einer anderen Stelle i.m. in den Rücken injiziert wurde. Den Affen
IM2 und IM4 (Kontrollen) wurden 250 μl Alaun und 250 μl PBS-A,
0,1% BSA 20% autologes Plasma, s.c. in eine einzige Stelle am oberen
Rücken
injiziert, und pCV-0 (1 mg), resuspendiert in 1 ml PBS-A, i. m.
in eine einzige Stelle im oberen Rücken injiziert, die sich von
der vorhergehenden unterschied. Der uninfizierte Affe IM7 wurde
nicht geimpft. Das Impfschema bestand aus einem Zeitpunkt 0, entsprechend
7 Wochen nach der SHIV-Infektion, und 1, 4, 5, 10, 11, 13, 14, 17,
18 Wochen danach. Um die Wirkungen dieser Impfung auf die Progression
der Erkrankung zu evaluieren, wurde jeder Makake täglich auf
das Vorliegen oder auf Anzeichen der Erkrankung untersucht, und
zum Zeitpunkt 0 sowie nach 3, 8, 12, 16 und 21 Wochen wurden 10
ml Blut für
Labortests (chemisch-klinische Analyse, Elektrolyte, weiße Zellen
und Plättchenzählungen,
Hämoglobin),
zur Evaluation des immunologischen Status (Vorliegen von spezifischen
Immunglobulinen, Bestimmung von Th1- und Th2-Cytokinen, Chemokinproduktion), zur
Charakterisierung von Lymphocyten mittels FACS-Analyse (CD4, CD8, CD28, CD40, CD86,
CD20, CD2, CD26 und CD20), und schließlich zur Evaluation virologischer
Parameter (provirale DNA-Detektion durch semi-quantitative PCR,
Plasmaviruslast durch kompetitive RT-PCR, Plasma p27 Gag-Antigen
mittels ELISA und Vorliegen von Anti-SHIV Ab, wie zuvor beschrieben)
entnommen. Andere Boosts sollen auf Basis der immunologischen, virologischen
und klinischen Ergebnisse durchgeführt werden.
-
Nach
dem letzten Inokulum soll eine Überprüfung monatlich
und bei Auftreten klinischer Modifikationen durchgeführt werden.
PBMC, Seren, Plasma und Urinproben sollen zu jedem Zeitpunkt für zukünftige Tests
eingefroren werden, wie zuvor beschrieben.
-
Nun
werden die bereits verfügbaren
Ergebnisse dieses Experimentes beschrieben, die in Woche 8 nach
Immunisierung erhalten wurden. Sowohl in den geimpften, asymptomatischen
Affen als auch in den Kontrollaffen wurden keinerlei Anzeichen einer
Entzündung
und Neoangiogenese an den Inokulationsstellen oder allgemeine Erkrankungssymptome
beobachtet. In den bereits symptomatischen Affen waren keine Modifikationen
des klinischen Status sichtbar. Zudem wurde keine Aktivierung der
viralen Replikation detektiert. Zusammen genommen, deuten diese
Ergebnisse auf das Fehlen von Toxizität oder erhöhter viraler Replikation in
den mit einem (einer) biologisch aktiven Tat-Protein oder -DNA geimpften
Affen hin (Tabelle 36). TABELLE 36 Analyse virologischer Parameter
| Affe | Wochen ab
Beginn der Impfung |
| | 0 | 3 | 8 |
| | p27
(pg/ml) | DNA
PCR
Kopien/μg | p27
(pg/ml) | DNA
PCR
Kopien/μg | p27
(pg/ml) | DNA
PCR
Kopien/μg |
| IM1 | 12,3 | 68 | 17,3 | 52 | 141 | 41 |
| IM3 | 0 | 61 | 0 | 48 | 0 | 71 |
| IM5 | 97,1 | 20 | 21,7 | 15 | 23,6 | 95 |
| IM6 | 0 | 43 | 0 | 55 | 0 | 24 |
| IM2 | 21,2 | ND | 36,6 | 53 | 27,4 | 78 |
| IM4 | 81 | 195 | 22 | 288 | 15,4 | 135 |
| IM7 | ND | ND | ND | ND | 0 | > 1 |
-
Die
Tests wurden wie in Tabelle 17 beschrieben durchgeführt. Den
Affen IM1, IM3, IM5 und IM6 wurden Tat-Protein (20 μg) und Alaun-Adjuvans
s. c. und pCVTat (1 mg) i. m. injiziert. Den Affen IM2 und IM4 (infizierte
Kontrollen) wurden Alaun-Adjuvans s. c. und pCV0 (1 mg) i. m. injiziert.
IM7 war ein nicht-infizierter, naiver Affe.
-
FACS-Analysen
zeigen, dass keine Modifikationen der CD4+- und CD8+-T-Lymphocyten
nach Impfung zu beobachten waren (Tabelle 37).
-
-
Diese
Daten bestätigen,
dass sowohl das Tat-Protein als auch das pCVTat-Plasmid bei den
verwendeten Dosierungen und Inokulationswegen gut toleriert wurden
und keinen toxischen Effekt in den geimpften Affen hatten, und zudem
erhöhten
sie weder die virale Replikation noch die CD4 T-Zellenverringerung
in infizierten Tieren.
-
Beispiel 7. Co-Stimulation von gereinigten
CD4+-Lymphocyten aus SIV-infizierten Affen mit Anti-CD3/28-beschichteten
Kügelchen
führt zu
einer logarithmischen Expansion der Zellzahl ohne signifikante virale
Replikation und Transmission
-
Aus
mononukleären
Peripherblutzellen wurde die CD8+-Zellpopulation unter Verwendung
von immunmagnetischen Anti-CD8-Kügelchen
(Dynal, Oslo; Dynabeads M-450 CD8) depletiert. Der Reinigungsgrad
wurde mittels FACS-Analyse evaluiert und als akzeptabel betrachtet,
wenn er höher
als 95% war. Die CD8-depletierten Zellen (als CD8-PBMC bezeichnet)
wurden gezüchtet
in Gegenwart von PHA (2 μg/ml)
und IL-2 (40 U/ml) oder immunmagnetischen Kügelchen, die zuvor mit zwei
monoklonalen Antikörpern
gegen die Antigene CD3 (Klon FN18, BioSource) und CD28 (Klon 9.3)
beschichtet worden waren (Anti-CD3/28-Kügelchen). Um die Bindung der
Anti-CD3/28-Kügelchen
mit Zielzellen zu verbessern, wurde die Inkubation auf einer Drehscheibenanordnung
durchgeführt.
Dann wurden die gebundenen Zellen (als CD8-CD3+CD28+ bezeichnet) mit
einem Magneten selektiert und in Kultur angeimpft. Dreimal pro Woche
wurden die Zellkonzentrationen auf den Anfangsspiegel eingestellt
und IL-2 zugegeben, wo dies indiziert war; zudem legen Ergebnisse
hinsichtlich der mit Anti-CD3/28-Kügelchen stimulierten Zellen
nahe, dass das Dauerstimulationsregime, gekoppelt mit einer ständigen Kontrolle
des Verhältnisses
Kügelchen:Zelle,
welches zu jedem Zeitpunkt eingestellt wird, hochwirksam bei der
Induktion der proliferativen Antwort ist. Unsere früheren Studien
haben gezeigt, dass in Abwesenheit von exogenem IL-2 die CD8-CD3+CD28+-Zellpopulation
besser proliferiert als mit Anti-CD3/28-Kügelchen stimulierte CD8-PBMC.
Darüber
hinaus erhöht
die Zugabe von exogenem IL-2 (40 U/ml, dreimal pro Woche) die Proliferationskinetik
signifikant, sowohl bezüglich
der Zellenzahl als auch der Wirkungsdauer (14).
-
Um
die antivirale Aktivität
dieser Stimulation zu evaluieren, wurden gereinigte CD8-CD3+CD28+-Zellen
von 4 nicht-infizierten Affen am Tag 0 mit 0,1 M.O.I. von SIV infiziert
und dann unter kontinuierlicher Stimulation kultiviert. CD8-PBMC,
stimuliert mit PHA und IL-2, waren die Kontrolle des Experimentes.
Die virale Infektion wurde durch Detektion von p27 Gag-Antigen im
Kulturüberstand
mittels eines handelsüblichen
ELISA (Coulter, Hialeah, FL) verfolgt. Die p27 Gag-Antigenspiegel
(ng/ml) wurden an den Tagen 6 und 12 nach der Infektion gemessen.
Wie in 15 gezeigt, gibt es zwischen
den beiden Stimulationsregimes einen signifikanten Unterschied hinsichtlich
der Infektion. Tatsächlich
war am Tag 6 nach der Infektion das p27-Antigen in den CD3/28 Kügelchen-stimulierten
Kulturen 40% bis 87% geringer als in mit PHA plus IL-2 stimulierten
Kulturen, und am Tag 12 war dieser Unterschied in 2 von 4 Affen
noch stärker.
Dies legt eine Verminderung der Anfälligkeit für eine virale Infektion nahe.
In nur einem Fall (MK 9401) beobachteten wir eine Virusausbreitung
bei beiden Stimulationsregimes. Die hier beschriebenen Ergebnisse
zeigen, dass Macaca fascicularis ein gutes Modell für die ex
vivo-Expansion von Lymphocyten-Subpopulationen durch Anti-CD3/28
Kügelchen-Co-Stimulation ohne
virale Replikation ist. Dies stellt das Grundprinzip für den therapeutischen
Impfstoff dar, den wir vorschlagen, ausgehend von der Expansion
und Reinfusion autologer, antiviraler spezifischer Lymphocyten in HIV-infizierten
Individuen.
-
Beispiel 8. Verwendung dendritischer Zellen
zur Impfung (nicht beansprucht)
-
Die
dendritischen Zellen (DC) und Makrophagen sind in geringerem Maße in der
Lage, wirksam Antigene gegen die T-Lymphocyten zu präsentieren,
und induzieren in diesem Zell-Subset
eine Proliferation oder einen Erwerb spezifischer cytotoxischer
Aktivitäten.
Diese Zellen werden als „Antigen-präsentierende
Zellen" (APCs) bezeichnet
und können
die Immunantwort starten. Somit können DC bei ex vivo-Immunisierungsprotokollen
eingesetzt werden. Daher wurden DC-Vorläufer aus dem peripheren Blut
von Macaca fascicularis durch in vitro-Kultivierung adhärenter Zellen
nach sieben Tagen GM-CSF- und IL-2-Stimulation isoliert. Alternativ
wurden CD34+-Zellen mit immunmagnetischen Kügelchen gereinigt und dann
14 Tage in vitro mit GM-CSF und TNF-α kultiviert. Um zu bestätigen, dass
DC isoliert wurden, führte
man eine morphologische Analyse und Phänotyp-Charakterisierung (FACS-Analyse und Immunhistochemie)
durch. Die Funktionsanalyse beruhte auf der einzigartigen Fähigkeit
von DC, die Proliferation allogener Lymphocyten zu induzieren.
-
Die
erhaltenen Ergebnisse bestätigen
voll und ganz die Wirksamkeit der Reinigung und der funktionalen
Charakterisierung von DC. Im Einzelnen wurden, um DC-Vorläufer zu
isolieren, PBMCs, erhalten durch Ficoll-Dichtegradientenzentrifugation,
erneut auf einem diskontinuierlichen Percoll-Gradienten (50% und
42,5%) stratifiziert. Die Zellfraktion, die nach 30-minütiger Zentrifugation
bei 500 g zwischen zwei Gradienten vorlag, bestand hauptsächlich aus
Monocyten (wie durch FACS-Analyse bestätigt, Daten nicht gezeigt).
Diese Zellen wurden bei 4°C
gehalten, um eine Zelladhäsion
an die Kunststoffröhrchen
zu vermeiden, wurden dann gesammelt, gewaschen, gezählt und
bei 37°C
in Kultur angeimpft. Am Tag danach wurden nicht-adhärente Zellen
mit 4 sanften Waschungen weggewaschen. Um eine Differenzierung zu
DC zu induzieren, wurde zu den adhärenten Zellen Komplettmedium
gegeben, das mit GM-CSF (200 ng/ml, Leucomax, Sandoz, Mailand, Italien)
und IL-4 (200 U/ml, Pepro tech, London, England) ergänzt war.
Als Kontrolle wurde ein Komplettmedium ohne Cytokine zugegeben,
um die normale Differenzierung von Monocyten in der Makrophagenlinie
zu induzieren. Zweimal pro Woche wurde die Hälfte des Überstandes durch frisches Medium
ersetzt, das mit dem am Tag 0 eingesetzten identisch war. Die Reifung
von DC in den mit Cytokinen behandelten Vertiefungen wurde anhand typischer
morphologischer Veränderungen,
wie Clusterbildung, Adhäsionsverlust
und Entwicklung von Ablegerzellen, detektiert. Die ohne Cytokine
kultivierten Monocyten-/Makrophagen-adhärenten
Zellen wurden durch EDTA-Behandlung (0,5 mM in PBS-A) abgelöst, zweimal
gewaschen, gezählt
und bei unterschiedlichen Konzentrationen, abhängig vom durchgeführten Experiment,
in frischem Medium resuspendiert.
-
Für die allogenen
gemischten Leukozytenreaktionen (AMLR) wurden die erhaltenen APCs
(DC oder Makrophagen) mit einer festgelegten Menge allogener T-Lymphocyten,
gereinigt mittels Ficoll- und Percoll-Gradienten und Adhäsion, getestet
und dann eingefroren. Die AMLR wurde in Platten mit 48 Vertiefungen mit
0,5 × 106 T-Lymphocyten und Reihenverdünnungen
von APCs durchgeführt.
Am Tag 4 der Kultur wurde eine festgelegte Menge der Zellsuspension
in einer Platte mit 96 Vertiefungen dreifach angeimpft. Ein μCi von 3H-Tymidin wurde in jede Vertiefung gegeben
und die Platte dann 16 Stunden bei 37°C inkubiert. Am Ende der Inkubation
wurde die Menge an von den Zellen aufgenommenem 3H-Tymidin mit einem β-Zähler gemessen und
in Counts pro Minute (cpm) ausgedrückt. Die Ergebnisse deuten
darauf hin, dass die erhaltenen DC potente APC sind, wie sich durch
die höhere
Induktion der Proliferation in allogenen menschlichen Lymphocyten im
Vergleich zur Makrophagenstimulation und durch die Fähigkeit
zur Induktion einer T-Lymphocytenproliferation
in Affen bei allen verwendeten Konzentrationen zeigt (16B).
-
Zur
Verwendung für
die Impfung werden DC in einer Konzentration von 1 × 105 Zellen/100 μl in RPMI 1640, ergänzt mit
5% autologem Serum, 10 mM Hepes-Puffer, 100 U/ml Penicillin- Streptomycin, 0,5
mg/ml Amphotericin B und 0,03% Glutamin resuspendiert, und dann
2 Stunden bei 37°C
in Gegenwart von Tat-Protein oder Tat-Peptiden oder einer Kombination
aus Tat, Rev, Nef, Gag und/oder Cytokinen inkubiert. Diese behandelten
DC werden dann innerhalb 2-4 Wochen nach der Erstinjektion zweimal
oder öfter
intravenös
inokuliert. Alternativ dazu werden die DC mit tat-Gen-enthaltenden
Vektoren alleine oder in Verbindung mit anderen, oben erwähnten Vektoren
transduziert und dann intravenös
injiziert.
-
Vorausschauendes Beispiel 9
-
Die
beschriebenen Immunogene werden dazu eingesetzt werden, eine spezifische
Immunantwort auf mukosaler Ebene zu induzieren und/oder zu potenzieren.
Einer der Ansätze
basiert auf der Verwendung von Bakterien (S. Gordonii und Lactobacillus)
die „manipuliert" wurden, um die oben
erwähnten
viralen Antigene zu exprimieren. Diese Bakterien kolonisieren die
Mund- und Vaginalschleimhaut von Mäusen und induzieren eine spezifische,
sowohl lokale als auch systemische Antikörperantwort gegen heterologe
Antigene, die an der Oberfläche
von rekombinanten Bakterien exprimiert werden (Ref. 116, 104, 106,
121, 117, 139, 105, 107). Diese Bakterien können als lebende Vektoren für Impfstoffe
dienen und diesen Vorteil nutzen, um eine lang anhaltende Stimulation
des Immunsystems zu verursachen. Zudem werden wir die Möglichkeit
evaluieren, virale Antigene und Moleküle, die an der Immunantwort
beteiligt sind, wie die B-Untereinheit des temperaturempfindlichen
Toxins von E. Coli oder Cytokine, an der bakteriellen Oberfläche zu co-exprimieren.
Die Herstellung der rekombinanten Stämme von S. Gordonii wird wie
zuvor beschrieben durchgeführt
werden (Ref. 116), kurz gesagt: (i) chromosomale Integration rekombinanter
DNA-Moleküle;
(ii) transkriptionale Fusionen mit starken chromosomalen Promotoren;
(iii) transkriptionale Fusionen mit dem Gen, das für das Protein
M6, ein Oberflächenprotein
von Streptococcus, kodiert. Die rekombinanten Stämme von S. Gordonii werden
eingesetzt werden, um die Vaginalschleimhaut der Affen zu kolonisieren.
Es wurde gezeigt, dass die rekombinanten Stämme von S. Gordonii, welche
die V3-Region von gp120 von HIV-1 und das E7-Protein von HPV-16
exprimieren, die Vaginalschleimhaut der Maus nach einem einzigen
Inokulum dauerhaft kolonisieren, was eine antigenspezifische Antikörperantwort
sowohl lokal als auch systemisch induziert. Die systemische Antwort
besteht vorwiegend aus IgG2a-Antikörpern, was eine Antwort vom
Th1-Typ nahelegt (Ref. 105, 106). Wir werden menschliche vaginale
Stämme
von Lactobacillus auswählen,
die in der Lage sind, die Vaginalschleimhaut der Affen zu kolonisieren.
Danach wird ein bereits entwickeltes genetisches System eingesetzt
werden, das die Expression heterologer Antigene an der Oberfläche von
Lactobacillus gestattet (Rush, 1997). Diese Strategie beruht auf: (i)
dem Klonieren genetischer Fusionen (emm6/heterologes Gen) in Insertionsvektoren,
die Homologien zum konjugativen Transposon Tn916 tragen; (ii) der
Transformation der Vektoren in Bakterienstämmen, die als Zwischenwirt
dienen (Bacillus subtilis); (iii) der konjugativen Mobilisierung
der rekombinanten Transposons aus B. subtilis zu Lactobacillus.
Die rekombinanten Stämme
von Lactobacillus werden zum Kolonisieren der Vaginalschleimhaut
der Affen eingesetzt werden.
-
Vaginale
Proben werden unter Einsatz spezieller absorbierender Filter erhalten
werden (Ref. 38, 105, 106). Die Kolonisierung wird durch Plattieren
der vaginalen Proben auf selektiven Platten evaluiert werden, und
die Expression von HIV-Antigenen in vivo wird mittels Immunfluoreszenz
an Vaginalabstrichen überprüft werden
(Ref. 105). Mit bereits standardisierten Verfahren (Ref. 38) werden
die Vaginalabstriche eingesetzt werden für i) den Papanicolau-Test im
Falle einer vaginalen Impfung; ii) das Vorliegen von Impfstoffantigenen in
den Zellen; iii) die Phänotypcharakterisierung
von Zellen mittels Durchflusscytometrie-Analyse (CD1, CD2, CD4, CD5, CD8, CD11c,
CD14, CD20, CD28, CD40, CD25, HLA-DR); iv) die Evaluation der Cytokinproduktion (IL-2,
IFNγ, TNFα, IL-4, IL-10,
IL-15, semiquantitative RT-PCR), die Bestimmung des Vorliegens von
Cytokinen und β-Chemokinen in den
mukosalen Fluiden mittels Elisa-Assays; v) die Dosierung der gesamten
und der spezifischen Immunglobuline (IgA und IgG) im mukosalen Fluid
mittels Elisa [Di Fabio et al., Vaccine 15: 1 (1997)]. Einen Monat
nach dem letzten Inokulum des Immunogens werden die Affen intravenös oder auf
mukosalem Wege mit dem SHIV 89.6P infiziert werden. Die Nachuntersuchung
der Affen wird wie im Beispiel 4 beschrieben durchgeführt werden.
Blutproben werden genommen werden, um die routinemäßigen Laboruntersuchungen,
die Evaluierung immunologischer Parameter, sowohl humoral als auch
zellulär,
wie in Beispiel 4 beschrieben durchzuführen. Die Erfinderin geht davon
aus, dass dieses Verfahren erfolgreich eingesetzt werden kann, um
eine spezifische Immunisierung von Affen auf vaginalem Wege zu induzieren.
Alternativ dazu kann die mukosale Immunität durch Verabreichung der Proteinimmunogene,
wie oben beschrieben, direkt auf mukosalem Weg in Gegenwart von
Adjuvanzien, wie dem thermosensitiven Toxin von E. Coil und dem
Choleratoxin, oder unter Verwendung anderer bakterieller und nicht-bakterieller
Zuführungssysteme,
wie Cytofectine und Liposome, oder durch Inokulation über andere
Wege induziert werden, welche die wirksamste und den besten Schutz
bietende Immunantwort induzieren können (Ref. 83, 81, 62).
-
Zudem
geht die Erfinderin davon aus, dass rekombinante Herpesvektoren,
welche die oben beschriebenen viralen Proteine exprimieren, hervorragende
Systeme zum Induzieren einer wirksamen mukosalen Immunantwort sein
können.
Rekombinante virale Vektoren des Herpes Simplex Typ 1-Virus (HSV-1)
werden eingesetzt, um virale Proteine zur Induktion einer systemischen
Antwort (durch kutane Immunisierung, i. d.) und einer mukosalen
Antwort (auf oralem, vaginalem oder nasalem Wege) zu exprimieren.
Nicht-pathogene, nicht-replikative Herpes-Vektoren werden eingesetzt
(Ref. 99) wegen ihrer Fähigkeit,
große
exogene Sequenzen zu enthalten, ohne die Wirksamkeit der Infektion
zu beeinträchtigen
(Ref. 52, 64). Daher werden Vektoren konstruiert, die mehr als ein
HIV-Gen (akzessorisch, regulatorisch und strukturell) enthalten.
Die mukosale Immunität
könnte
durch einen Oral-, Vaginal- oder Nasalimpfstoff induziert werden.
Die Herpes-Vektoren können in
diesen Impfmethoden verwendet werden, da HSV-1 direkt auf mucosalem
Wege verabreicht werden kann (Ref. 176, 75). Die rekombinanten Viren
werden unter Nutzung eines zweistufigen Verfahrens konstruiert,
das die Insertion exogener Sequenzen in das virale Genom erleichtert.
Der erste Schritt erfordert die Insertion einer Expressionskassette
mit einem Reportergen (β-Galactosidase, LacZ),
kloniert in die Restriktionsstelle PacI, die im HSV-1-Genom nicht
vorhanden ist, flankiert von der gewünschten Zielsequenz von HSV-1,
unter Anwendung des üblichen
Vorgehens für
die homologe Rekombination, um das HSV-1-Gen zu unterbrechen. Das rekombinante
Virus wird durch Bildung von Plaques mit einem blauen Phänotyp mittels „X-Gal-Färbung" ausgewählt. Der
Verdau der viralen DNA mit Pac1 setzt das Markergen frei und erzeugt
zwei große
Fragmente viraler DNA, die nicht in der Lage sind, infektiöse Viruspartikel
zu produzieren. Der zweite Schritt besteht aus einer Cotransfektion
der viralen DNA, verdaut mit dem gleichen Plasmid, das zum Erzeugen
der Deletion verwendet wurde, wobei das Reportergen durch das gewünschte Gen
substituiert wird. Die rekombinanten Viren werden durch die Auswahl
von Plaques mit einem weißen
Phänotyp
nach „X-Gal-Färbung" identifiziert. Diese Rekombination
führt zur
Elimination von Pac1-Stellen, wodurch dieses Verfahren viele Gene
an unterschiedlichen Orten des HSV-1-Genoms insertieren kann (Ref.
74). Durch Kreuzen der unterschiedlichen Vektoren, welche die einzelnen
Gene enthalten, könnten
wir möglicherweise
all die verschiedenen genetischen Kombinationen erzeugen. Der Vektor,
der alle gewünschten
Gene enthält,
wird durch Screening mit unterschiedlichen Markern, Phänotypen
und selektives Wachstum auf kompetenten Zellen isoliert. Alle Kombinationen
werden durch Alternieren von DNA-Transfektionen und viralen Rekombinationen
erzeugt werden.
-
Vektoren,
welche die einzelnen Gene tat, rev, nef oder gag enthalten, werden
konstruiert werden, indem als Grundvektor der verwendet wird, der
die Mutationen in den Genen 4-/22/27/41 enthält und im Vergleich mit den
anderen nicht-replikativen HSV1-Vektoren besser für die niedrige
Toxizität
und die starke Expression ist. Konstitutive Promotoren werden, wie
die von HCMV (Immediate Early Promoter des menschlichen Cytomegalovirus)
oder ICP0 lep (Immediate Early Promoter von infiziertem Zellprotein)
und die Moloney-Mäuseleukämievirus
LTR verwendet werden, um die Expression der oben erwähnten Gene
zu induzieren. Nicht-replikative HSV-1-Vektoren, die HIV-Proteine
in unterschiedlichen Kombinationen exprimieren, werden konstruiert
werden. Die Produktion dieser Viren, die mehr unterschiedliche Gene
enthalten, wird durch genetische Kreuzung der Vektoren, welche die
unter dem vorhergehenden Punkt beschriebenen, einzelnen Gene enthalten,
erhalten werden. Zweifach-, Dreifach- und Vierfachvektoren werden
erzeugt werden. Die Vektoren werden in die Affen i.d. oder auf mucosalem
(oralem, vaginalem oder nasalem) Wege inokuliert werden, mit besonderem
Augenmerk auf der letztgenannten Art der Verabreichung (Ref. 176,
101, 102). Das Impfschema besteht aus Mehrfach-Inokula zu unterschiedlichen
Zeitpunkten, die im Verhältnis
zum Immunogen oder zur Kombination von Immunogenen bestimmt werden
müssen.
Während
der Immunisierung werden die Tiere zur Evaluation hämatochemischer
und immunologischer Parameter, wie in Beispiel 4 beschrieben, überwacht
werden. Mit bereits standardisierten Methoden werden Vaginalproben
erhalten, die wie zuvor in diesem Beispiel beschrieben untersucht
werden.
-
Vorausschauendes Beispiel 10
-
Zuführungssysteme
-
Tat
(Protein und/oder DNA) wird alleine oder in Kombination (wie oben
beschrieben) unter Verwendung neuer Zuführungssysteme, wie Erythrozyten
oder Nanopartikel, inokuliert werden. Das Zuführungssystem unter Verwendung
von Erythrozyten basiert auf der Möglichkeit, das Antigen an autologe
Erythrozyten gebunden abzugeben. Tatsächlich werden Erythrozyten
am Ende ihrer Lebensspanne (beim Menschen etwa 120 Tage) durch die
Makrophagen, die bekanntlich die Funktion professioneller Antigen-präsentierender
Zellen haben, aus dem Kreislauf entfernt. Diese Eigenschaft lässt sich
für Impfstoffstrategien
nutzen. Somit werden Antigene an die Erythrozyten mit einer bestimmten
Technik gebunden (Ref. 95, 96), welche die Bewahrung der immunogenen
Eigenschaften des Antigens erlaubt (Ref: 29, 30). Durch diese Prozedur
kann eine Biotinilierung von Erythrozyten ohne signifikante Modifikation
ihrer Eigenschaften und Lebensspanne durchgeführt werden (Ref. 95). Eine
Phagocytose alter Erythrozyten durch Makrophagenzellen wird eine
Immunantwort auslösen. Eine
Antikörper-Opsonisierung
von Erythrozyten, die das Antigen tragen, unterstützt die
Antigenentfernung aus dem Kreislauf. Die Hauptvorteile dieser Methodologie
sind: 1) Bedarf an einer geringen Menge des Antigens, um eine humorale
und zelluläre
Immunantwort zu induzieren, 2) lang andauernde Immunisierung aufgrund
der anhaltenden Gegenwart von Antigenen, die von den Erythrozyten
an der Peripherie getragen werden, 3) Adjuvansfunktionen, die vom
System selbst bereitgestellt werden.
-
Tatsächlich wurde
in Tierstudien gezeigt, dass die Verabreichung an die Membran autologer
Erythrozyten gebundener Antigene eine ähnliche oder stärkere Immunantwort
als die Immunantwort induziert, die mit dem gleichen Antigen, verabreicht
mit Freund-Adjuvans (Ref. 29), erhalten wird. Diese Eigenschaften
sind sehr nützlich
zur Entwicklung eines Anti-HIV-Impfstoffs,
insbesondere wenn er benötigt
wird, um die Immunogenität
des Antigens und die Antigenverfügbarkeit
zu erhöhen,
und wenn eine geringe Anzahl Immunisierungen erforderlich ist. Zudem
kann diese Strategie eingesetzt werden, wenn keine Adjuvanzien im
Impfprotokoll enthalten sind. Tatsächlich wurde im Mausmodell
gezeigt, dass durch autologe Erythrozyten verabreichte Antigene ähnliche
oder stärkere
Immunantworten induzieren, als sie mit dem gleichen Antigen erhalten
werden, wenn es mit Freund-Adjuvans verabreicht wird, das als das
stärkste
im Handel erhältliche
Adjuvans bekannt ist (Ref. 29), obwohl es aufgrund beträchtlicher
Nebenwirkungen nicht für
menschliche Studien zugelassen ist. Daher wird der Adjuvanseffekt
von Erythrozyten, die das Tat-Protein alleine oder in Kombination
mit anderen zuvor beschriebenen Immunogenen tragen, in nicht-menschlichen
Primaten analysiert werden. Es wird ein Vergleich zwischen diesen
Daten und den bei Verabreichung des Tat-Proteins in Gegenwart von
Alaun, RIBI oder ISCOM erhaltenen durchgeführt werden.
-
Die
Verwendung von Nanopartikeln kann eine zusätzliche Zuführungsstrategie darstellen.
Funktionale Nanopartikel stellen ein wichtiges System für den Transport
und die Freisetzung von Proteinen und DNA dar (Ref. 27, 172). Die
Nanosphären
sind kolloidale Polymerpartikel unterschiedlicher chemischer Zusammensetzung
mit einem großen
Durchmesserbereich von 10 bis 1000 nm. Es ist möglich, unterschiedliche Arten
von Substanzen an der Oberfläche
oder im Inneren der Nanosphären
zu adsorbieren (Oligonukleotide, Arzneimittel, Proteine, Peptide,
DNA), die dann zum Cytoplasma oder zum Kern von Zellen verbracht
werden, wo sie langsam freigesetzt werden. Zudem muss aufgrund der
Charakteristiken von Nanosphären
nur eine kleine Menge des Immunogens zugeführt werden. Nanopartikel sind
ein gute Zuführungssystem,
insbesondere für Moleküle mit geringer
Stabilität
in der extrazellulären
Umgebung, oder wenn die Zuführung
gegen eine spezifische Zielzelle gerichtet ist.
-
Die
Erfinderin geht davon aus, dass Nanosphären verwendet werden können, um
die oben beschriebenen viralen Antigene abzugeben. Es ist möglich, drei
Arten von Nanosphären
herzustellen und zu charakterisieren, die für die Zuführung und kontrollierte Freisetzung
von DNA (Nanosphären
Typ 1 und 2) und Proteinen (Nanosphären Typ 3) vorgesehen sind.
-
Für die DNA-Zuführung sind
zwei Arten von Nanosphären
(Nanosphären
Typ 1 und 2) verfügbar.
Der erste Typ Nanosphären
(Nanosphären
Typ 1) weist einen dreischichtigen Aufbau mit einer äußeren Schicht aus
Polyoxyethylenglykol (PEG) auf. Jüngste Berichte, die auf Studien
von Tarnsystemen beruhen (Ref. 180, 78), zeigen, dass PEG Nanosphären für Kupferzellen
unsichtbar macht. Im Gegensatz dazu besteht die weiter innen gelegene
Schicht aus Monomeren mit die Oberflächenspannung beeinflussenden
Merkmalen, die quaternäre
Ammoniumgruppen enthalten, welche die DNA durch einen Ionenaustausch-Mechanismus
und einen inneren Kern aus Methylmethacrylat als Monomer reversibel
adsorbieren. Diese Nanosphären
sind erhältlich durch
Polymerisation in Mikroemulsion unter Beteiligung der Polymerisation
eines Vinyl- oder Vinylidenmonomers in Gegenwart einer Mischung
von die Oberflächenspannung
beeinflussenden Reagenzien. Diese Reagenzien sind somit in der Lage,
das Monomer zu polymerisieren. Eines davon weist eine quaternäre Ammoniumgruppe
auf, die mit Oligonukleotiden interagiert, und das andere hat eine
lange PEG-Kette.
-
Der
zweite Typ eines DNA-Zuführungssystems
besteht aus funktionalen Nano- und Mikrosphären (Nanosphären Typ
2) mit Hydrogelmerkmalen. Diese Nanosphären sollten in Gegenwart von
DNA hergestellt werden, um diese im Zuführungssystem einzuschließen. Kern-Hülle-Nanosphären werden
benötigt,
um Proteine zuzuführen
(Nanosphären
Typ 3). Sie bestehen aus einem inneren Kern aus Polymethylmethacrylat
und einer äußeren Hülle aus
wasserlöslichem,
statistischem Copolymer von Acrylsäure und Methylmethacrylat,
das bekanntlich eine hochgradige Affinität gegenüber Proteinen hat (Ref. 79,
80). Dieses Copolymer ist im Handel erhältlich (EUDRAGIT) und wird
mit unterschiedlichen prozentualen Anteilen der beiden Co-Monomere
erhalten. Das Herstellverfahren, das zur Herstellung dieses zweiten
Typs von Nanosphären
führt,
beinhaltet die Polymerisation in Dispersion. Die Synthese umfasst
die Radikalpolymerisation eines Vinyl- oder Vinylidenmonomers in
Gegenwart von EUDRAGIT mit sterischen Stabilisierungsfunktionen.
Nach der Nanosphären-Keimbildung
lagert sich das EUDRAGIT außerhalb
der Partikel an. Durch Ändern
der Konzentration des Radikalinitiators, des Verhältnisses
zwischen dem Monomer und EUDRAGIT sowie der Reaktionszeit, werden
somit zahlreiche Nanosphären-Proben mit unterschiedlichen
morphologischen und chemischen Merkmalen erhalten.
-
Damit
kann evaluiert werden, ob die Zuführung von Tat-Protein oder
Tat-DNA durch Nanopartikel, alleine oder in Kombination mit den
oben erwähnten
Immunogenen (Protein oder DNA), eine Immunantwort gegen HIV induziert.
Insbesondere werden die humoralen oder zellvermittelten Immunantworten
evaluiert und mit denen verglichen werden, die mit den nicht zugeführten Immunogenen
im Affenmodell erhalten wurden.
-
Die
Erfinderin geht davon aus, dass die aus diesen Studien abgeleiteten
Informationen für
die Entwicklung eines Anti-HIV-Impfstoffes nützlich sein können. Zudem
werden die aus diesem Versuchsprotokoll abgeleiteten Informationen
auch auf andere Impfstoffstudien, insbesondere auf solche, die sich
mit rekombinanten Proteinen oder Peptiden geringer Immunogenität befassen, übertragen
werden. Die Möglichkeit,
einen Impfstoff zur einmaligen Verabreichung zu entwickeln, wird
enorme Vorteile hinsichtlich der Effizienz des Impfstoffes und eine
Senkung der Durchführungskosten
von Impfstoffprogrammen mit sich bringen.
-
Literaturstellen
-
- 1. Agostini et al., Blood 90: 1115 (1997)
- 2. Albini et al., Proc. Natl. Acad. Sci. USA 92: 4838 (1995)
- 3. Allan et al., Science 230: 813 (1985)
- 4. Antibodies-A laboratory manual, Hrsg. Hariow E., Lane D.,
Cold Spring Harbor Laboratory (1988)
- 5. Arya et al., Science 229: 69 (1985)
- 6. Aryoshi et al., AIDS 9: 555 (1995)
- 7. Audibert et al., Immunol. Today 14: 281 (193)
- 8. Badolato et al., Blood 90: 2804 (1997)
- 9. Barillari et al., J. Immunol. 149: 3727 (1992)
- 10. Barillari et al., Proc. Natl. Acad. Sci. USA 90: 7941 (1993)
- 11. Barillari et al., Proc. Natl. Acad. Sci. USA 149: 3727 (1993)
- 12. Blomberg et al., J.Immunol. Methods 160: 27-34 (1993)
- 13. Blomberg et al., J.Immunol. Methods 168: 267-273 (1994)
- 14. Blomberg et al., J.Immunol. Methods 193: 199-206 (1996)
- 15. Bohan et al., Gene Expr. 2: 391 (1992)
- 16. Bourgault et al., J. Virol. 66: 75 (1992)
- 17. Boyer et al., Nature Med. 3: 526 (1997)
- 18. Bruisten et al., J. Infect. Dis. 166: 620 (1992)
- 19. Buseyne et al., J. Virol. 67: 694 (1993)
- 20. Butera et al., J. Virol. 65: 4645 (1991)
- 21. Butera et al., J. Virol. 68: 2726 (1994)
- 22. Cafaro et al., AIDS Res. Hum. Retrov. 7: 204 (1991)
- 23. Carrol et al, Science. 276: 273-276, (1997)
- 24. Carson et al., J. Clin. Invest. 99: 937 (1997)
- 25. Chang et al., J. Biomed. Sci. 2: 189 (1995)
- 26. Chang et al., AIDS 11: 1421 (1997)
- 27. Chavany et al., Phar. Res. 9: 441 (1994)
- 28. Chef et al., J. Immunol. 149: 4060 (1992)
- 29. Chiarantini et al., Vaccine 15: 276 (1997)
- 30. Chiarantini eat., Clin. Diag. Lab.Immunol. 5: 235 (1998)
- 31. Chirmule et al., J. Virol. 69: 492 (1995)
- 32. Choppin et al., J. Immunol. 147: 569 (1991)
- 33. Corallin et al., Cancer Res. 53: 1 (1993)
- 34. Corallin et al., Cancer Res, 53: 5569 (1993)
- 35. Culman et al., J. Immunol. 146: 1560 (1991)
- 36. Couillin et al., J. Exp. Med. 180: 1129 (1994)
- 37. Danko et al., Vaccine 12: 1499 (1994)
- 38. Di Fabio et al., Vaccine 15: 1 (1997)
- 39. Ensoli et al., IV International Conference on AIDS, Stockholm,
1: 241 (1988)
- 40. Ensoli et al., Nature 345: 84 (1990)
- 41. Ensoli et al., J. Virol. 67: 277 (1993)
- 42. Ensoli et al., Nature 371: 674 (1994)
- 43. Ensoli et al., AIDS Updates, Hrsg. V. De Vita, Jr., Hellman
S., Rosenberg S. A., Lippincott J. B., Philadelphia; 7: 1 (1994)
- 44. Felber et al., Proc. Natl. Acad. Sci. 86: 1495 (1989)
- 45. Fine et al., Ann. Plast. Surg. 20: 6 (1988)
- 46. Fiorelli et al., J. Clin. Invest. 95: 1723 (1995)
- 47. Folks et al., Science 238: 800 (1987)
- 48. Franchini et al., Virology 155: 593 (1986)
- 49. Frankel et al., Cell 55: 1189 (1988)
- 50. Fugier-Vivier et al, J. Exp. Med. 186: 813 (1997)
- 51. Gait et al., Trends Biochem. Sci. 18: 255 (1993)
- 52. Glorioso et al., Ann. Rev. Microbiol. 49: 675 (1995)
- 53. Gobert et al., Virology 176: 458 (1990)
- 54. Goletti et al., J. Virol. 69: 2540 (1995)
- 55. Gorman et al., Mol. Cell. Biol. 2: 1044 (1982)
- 56. Grabstein et al., Science 264: 965 (1994)
- 57. Grosjean et al, J. Exp. Med. 186: 801 (1997)
- 58. Guy et al., Nature 330: 266 (1987)
- 59. Harrer et al., AIDS Res. Hum. Retrov. 12: 585 (1996)
- 60. Harrich et al., EMBO J. 16: 6 (1997).
- 61. Hinkula et al., J. Virol. 71: 5528 (1997)
- 62. Honenbang et al., Infect. Immun. 62: 15 (1994)
- 63. Huang et al., EMBO J. 13: 2886 (1994)
- 64. Huard et al. Gene Ther. 2: 385 (1995)
- 65. Igarashi et al., AIDS Res. Hum. Retrov. 10: 1021 (1994)
- 66. Jonuleit et al., J. Immunol. 158: 2610 (1997)
- 67. Jullien et al., J. Immunol. 158: 800 (1997)
- 68. Kanai et al., J.Immunol. 157: 3681 (1996)
- 69. Karlosson et al., J. Virol. 71: 4218 (1997)
- 70. Kashanchi et al., J. Virol. 70: 5503 (1996)
- 71. Kestler et al., Science 248: 1109 (1991)
- 72. Kim et al., Oncogene 7: 1525 (1992)
- 73. Koup et al., J. Virol. 68: 4650 (1994)
- 74. Krisky et al., Gene Ther. 4: 1120 (1997)
- 75. Kuklin et al., J. Virol. 240: 245 (1998)
- 76. Landes, Austin, S.107 (1997)
- 77. Lanzavecchia, Science 260: 937 (1993)
- 78. Lasic et al., Chemical Reviews 95: 2601 (1995)
- 79. Laus et al., Polymer 37: 343 (1996)
- 80. Laus et al., Polymers for Adv. Techn. 7: 548 (1996)
- 81. Lehnen et al., Vaccine Research 1: 319 (1992)
- 82. Levine et al., Science. 272: 1939-1943 (1996)
- 83. Lewis et al., Vaccine Press, Hrsg. Robinson, Farrar, Wiblin;
Human Press, Totowa, New Jersey (1996)
- 84. Li et al., Proc. Natal. Acad. Sci. USA 94: 8116 (1997)
- 85. Li et al., J. AIDS 5: 639 (1992)
- 86. Li et al., Science 268: 229 (1995)
- 87. Li et al., Proc. Natl. Acad. Sci. USA 94: 8116 (1997)
- 88. Lippincott J. B., Stockholm, Schweden, 31. Mai – 3. Juni
(1997)
- 89. Littaua et al., J. Virol. 65: 40 (1991)
- 90. Lvgren et al., Vaccine 14: 753 (1996)
- 91. Lu et al., J. Virol. 70: 3978 (1996)
- 92. Lubaki et al., J. Infect. Dis. 175: 1360 (1997)
- 93. Lucey et al., Clin. Diagn. Lab. Immunol. 4: 43 (1997)
- 94. Luciw et al., Proc. Natal. Acad. Sci. 92: 7490 (1995)
- 95. Magnani et al., Biotech. Appl. Biochem. 16: 188 (1992)
- 96. Magnani et al., Biotech. Appl. Biochem. 20: 335 (1994)
- 97. Malim et al., Nature 338: 254 (1989)
- 98. Mann et al., EMBO J. 10: 1733 (1991)
- 99. Marconi et al., Proc. Natl. Acad. Sci 93: 11319 (1996)
- 100. Marcuzzi et al., J. Virol. 66: 4228 (1992)
- 101. McLean et al., J. Infect. Dis. 66: 341 (1994)
- 102. McLean et al., Vaccine 14: 987 (1996)
- 103. Mcfarland et al., J. Inf. Dis. 170: 766 (1994)
- 104. Medaglini et al., Proc. Natl. Acad. Sci. USA 92: 6868 (1995)
- 105. Medaglini et al., Biotech. Annu. Rev. 3: 297 (1997)
- 106. Medaglini et al., Vaccine 15: 1330 (1997)
- 107. Medaglini et al., Am. J. Reprod. Immunol. 39: 199 (1998)
- 108. Meyerhans et al., Cell 58: 901 (1989)
- 109. Morein et al., AIDS Res. Hum. Retrov. 510: 5109 (1994)
- 110. Molecular cloning-A laboratory manual; Hrsg. Maniais T.,
Fritsch E. F., Sambrook J., Cold Spring Harbor Laboratory, Cold
Spring Harbor, New York (1992)
- 111. Myers et al., Human Retroviruses and AIDS: A compilation
and analysis of nucleic acid and amino acid sequences, Los Alamos
Laboratory, Los Alamos, NM, S. 1 (1993)
- 112. Myers et al., Human Retroviruses and AIDS. Theoretical
Biology and Biophysics Group. Los Alamos, NH (1995)
- 113. Neuvet et al., J. Virol. 70: 5572 (1996)
- 114. Nietfield et al., J. Immunol. 154: 2189 (1995)
- 115. Nixon et al., Nature 336: 484 (1988)
- 116. Oggioni et al., Vaccine 13: 775 (1995)
- 117. Oggioni, et al., Gene 169: 85 (1996)
- 118. O'Hagan
et al., Novel Delivery Systems for Oral Vaccines, Hrsg. O'Hagan, D. T. CRC
Press Boca Raton, FL, S. 176 (1994)
- 119. Parslow, Human Retroviruses, Ed. B. R. Cullen, IRLpress,
Oxford, England, S. 101 (1993)
- 120. Pilkington et al., Mol. Immunol. 33: 439 (1996)
- 121. Pozzi et al., in „Gram-positive
bacteria as vaccine vehicles for mucosal immunization", Hrsg. Pozzi G. & Wells, J. M.-Landes,
Austin, S. 35 (1997)
- 122. Puri et al., Cancer Res., 52: 3787 (1992)
- 123. Puri et al., AIDS Res. 11: 31 (1995)
- 124. Quesada-Rolander et al., ABS 6-S1, 2nd European Conference
on Experimental AIDS Research, Stockholm, Schweden, 31. Mai – 3. Juni
(1997)
- 125. Quinn et al., Biochem. Biophys. Res. Commun. 239: 6 (1997)
- 126. Ratner et al., Nature 313: 277 (1985)
- 127. Re et al., J. Acquir. Immun. Defic. Syndr. 10: 408 (1995)
- 128. Reimann et al., J. Virol. 70: 3189 (1996)
- 129. Reimann et al., J. Virol. 70: 6922 (1996)
- 130. Reiss et al., J. Med Virol. 30: 163 (1990)
- 131. Reiss et al., AIDS Res. Hum. Retrov. 5: 621 (1989)
- 132. Riley et al, J. Immunol. 158: 5545-5553, (1997)
- 133. Rinaldo et al., AIDS Res. Hum. Retrov. 11: 481 (1995)
- 134. Rinaldo et al., J. Virol., 69: 5838 (1995)
- 135. Rodman et al., Proc. Natl. Acad. Sci. USA 90: 7719 (1993)
- 136. Rodman et al., J. Exp. Med. 175: 1247 (1992)
- 137. Rosenberg et al., Vint. Immunol. 9 (5): 703 (1997)
- 138. Rosenthal et al., Seminars in Immunology 9: 303 (1997)
- 139. Rush, et al., in „Gram-positive
bacteria as vaccine vehicles for mucosal immunization", Hrsg. Pozzi G. & Wells, J. M.-Landes,
Austin, S.107 (1997)
- 140. Sadaie et al., New Biol. 2: 479 (1990)
- 141. Saiki et al., Science 230: 1350 (1985)
- 142. Sakuragi et al., J. Gen. Virol. 73: 2983 (1992)
- 143. Salter et al., Immunogenetics 21: 235 (1985)
- 144. Schnorr et al., Proc. Natl. Acad. Sci. USA. 94: 5326 (1997)
- 145. Sharma et al., Biochem. Biophys. Res. Co. 208: 704 (1995)
- 146. Shibata et al., J. Virol. 65: 314 (1991)
- 147. Sipsas et al., J. Clin. Invest. 99: 752 (1997)
- 148. Sodroski et al., Science 227: 171, (1985)
- 149. Steinaa et al., Arch. Virol. 139: 263 (1994)
- 150. Steinman R. M., Exp. Hematol. 24: 859 (1996)
- 151. Tahtinen et al., Virology 187: 156 (1992)
- 152. Theoretical Biology and Biophysics, Los Alamos, NH, (1995)
- 153. Titti et al., Cell. Pharmacol. AIDS 3: 123 (1996)
- 154. Trinchieri, Curr. Opin. Hematol. 4: 59 (1997)
- 155. van Baalen et al., J.Gen. Virol., 77: 1659 (1996)
- 156. van Baalen et al., J.Gen. Virol. 78: 1913 (1997)
- 157. Vellutini et al., AIDS Res. Hum. Retrov. 11: 21 (1995)
- 158. Venet et al., J. Immunol. 148: 2899 (1992)
- 159. Viscidi et al., Science 246: 1606 (1989)
- 160. Vogel et al., Nature 335: 601 (1988)
- 161. Voss et al., Virology 208: 770 (1995)
- 162. Wain-Hobson, Curr. Opin. Genet. Dev. 3: 878 (1993)
- 163. Westendorp et al., J. Virol. 68: 4177 (1994)
- 164. Westendorp et al., Nature 375: 497 (1995)
- 165. Wolf et al., J.Immunol. 146: 3074 (1991)
- 166. Yang et al., J. Virol. 70: 4576 (1996)
- 167. Yang et al., J. Virol. 70: 5799 (1996)
- 168. Yasutomi et al., J. Virol. 70: 678 (1996)
- 169. Zauli et al., Blood 86: 3823 (1995)
- 170. Zauli et al., Blood 80: 3036 (1996)
- 171. Zauli et al., J. Immunol. 157: 2216 (1996)
- 172. Zobel et al., Antisense Nucleic Acid Drug Dev. 7: 483 (1997)
- 173. Gibellini et al., Blood 89: 1654 (1997)
- 174. Bauer et al., J. Infect. Dis. 165: 419 (1992)
- 175. Klein et al., J. Exp. Med. 181: 1365 (1995)
- 176. Bowen et al., Res. Virol. 143: 269 (1992)
- 177. Zamarchi et al., AIDS Res. Human Retrov. 9: 1139 (1993)
- 178. Fiore et al., AIDS 5: 1034 (19912)
- 179. Roman et al., Nature Med. 3: 849 (1997)
- 180. Allen et al., Biochim. Biophis. Acta 1237: 99 (1995)
-
-
-
-
-
-
-