-
Gebiet der Erfindung
-
Die
vorliegende Erfindung betrifft chemische Verfahren zur Herstellung
von Verbindungen, welche bei der Behandlung von verschiedenen medizinischen
Störungen
nützlich
sind, wobei solche Anwendungen die Verwendung als Antifibrillantia
und Antiarrhythmika einschließen,
aber nicht darauf begrenzt sind. Die Verfahren dieser Erfindung
sind zur Herstellung von 1,3-disubstituierten 4-oxocyclischen Harnstoffen,
insbesondere 1-[[[5-(4-Chlorphenyl)-2-furanyl]methylen]amino]-3-[4-(4-methyl-1-piperazinyl)butyl]-2,4-imidazolidindion,
und Salzen hiervon nützlich.
-
Hintergrund
der Erfindung
-
Die
vorliegende Erfindung betrifft ein Verfahren zur Herstellung von
1,3-disubstituierten 4-oxocyclischen Harnstoffen, insbesondere 1-[[[5-(4-Chlorphenyl)-2-furanyl]-methylen]amino]-3-[4-(4-methyl-1-piperazinyl)butyl]-2,4-imidazolidindion,
oder Salzen hiervon, wobei das Endprodukt in reiner Form und hoher
Ausbeute erhalten wird.
-
1-[[[5-(4-Chlorphenyl)-2-furanyl]methylen]amino]-3-[4-(4-methyl-1-piperazinyl)butyl]-2,4-imidazolidindion
(Azimilid) ist in US-Patent Nr. 5,462,940 (1995) an Norwich Eaton
Pharmaceuticals, Inc., offenbart. In US-Patent Nr. 5,462,940, erteilt
an Yu et al. am 31. Oktober 1995, werden zwei allgemeine Verfahren
für diesen Verbindungstyp
offenbart. Jedes beschreibt eine Reihe von Reaktionen, welche die
Isolierung von drei bis fünf Zwischenverbindungen
beinhalten. Die Nachteile von beiden Verfahren sind die Verwendung
von leicht entzündlichem
und feuchtigkeitsempfindlichem Natriumhydrid, potentiell explosiven
DMF/Natriumhydrid-Mischungen, überschüssigen Lösungsmittelvolumina,
Natriumiodid und mehreren Isolierungsschritten. Weitere Nachteile
eines Verfahrens sind die Verwendung einer Amin-Schutzgruppe und
die Erfordernis einer Hydrierungsreaktion um sie zu entfernen.
-
Auf
dem Fachgebiet ist bekannt, daß sichere,
ertragsreichere, wirtschaftlichere Verfahren zur Herstellung von
Azimilid vorteilhaft wären.
Besonders, vorteilhaft wären
eine Verringerung der Anzahl an Syntheseschritten, ein erhöhter Reaktionsdurchsatz
(höhere
Reaktionskonzentrationen), die Ausschließung einer Hydrierungsreaktion,
die Elimination einer Amin-Schutzgruppe, höhere Gesamtausbeuten, die Möglichkeit
einer großtechnischen
Herstellung und bessere Endproduktisolierungen. Es ist überraschend
entdeckt worden, daß die
Nachteile der in der Literatur beschriebenen Synthesen für diese
Verbindungen durch die Ausführung
der Reaktionsabfolge in einer milden Base, wie Kaliumcarbonat, zur
Alkylierung, die Elimination der Verwendung von Natriumiodid, um
die Alkylierung der Amineinheit zu erleichtern, und die Verwendung
von Lösungsmitteln wie
Methylsulfoxid (DMSO) und N-Methylpyrrolidon (NMP), um wesentlich
höhere
Reaktionskonzentrationen, eine höhere
Produktausbeute und Reinheit zu ermöglichen, überwunden werden können.
-
Der
Gegenstand dieses Patents ist ein Verfahren zur Herstellung von
1,3-disubstituierten 4-oxocyclischen Harnstoffen, wodurch die 1,3-disubstituierten
4-oxocyclischen Harnstoffe geeigneterweise in hohen Ausbeuten, ohne
Isolierung von Zwischenverbindungen, synthetisiert werden, indem
zuerst der entsprechende 1-substituierte 4-oxocyclische Harnstoff
mit einer Kohlenstoffkette, welche bis zu zwei Abgangsgruppen enthält, alkyliert
wird, um ein Addukt zu bilden, welches ohne Isolierung verwendet
wird, um ein Amin zur Bildung eines 1,3-disubstituierten 4-oxocyclischen
Harnstoffs zu alkylieren, der schließlich mit einer Säure umgesetzt
wird, um das gewünschte
Salz zu bilden. Das vorliegende Verfahren ermöglicht die Herstellung von 1,3-disubstituierten
4-oxocyclischen Harnstoffen unter Reaktionsbedingungen, welche die
Erfordernis für
einen Hydrierungsschritt und die Verwendung einer Amin-Schutzgruppe
eliminieren. Dieses Verfahren ermöglicht eine bessere Ausbeute
und Produktreinheit, einen höheren
Durchsatz und stellt zusätzlich
eine einfache Synthese für
die Herstellung dieser Molekülklassen
bereit.
-
Insbesondere
stellen die bevorzugten Verfahren der vorliegenden Erfindung eine
neue Methodik bereit, welche für
die maßstäbliche Vergrößerung und
die Herstellung von Azimilid besonders geeignet ist.
-
Zusammenfassung der Erfindung
-
Die
vorliegende Erfindung stellt ein Verfahren zur Herstellung von 1,3-disubstituierten
4-oxocyclischen Harnstoffen der allgemeinen Formel:
wie in Anspruch 1 angegeben,
bereit.
-
Dieses
Verfahren wird zur Herstellung von Azimilid besonders bevorzugt.
Der bei der Herstellung von Azimilid verwendete 1-substituierte
4-oxocyclische Harnstoff ist 1-[[[5-(4-Chlorphenyl)-2-furanyl]methylen]amino]-2,4-imidazolidindion.
-
Definitionen und Verwendung
von Begriffen
-
Das
Folgende ist eine Aufstellung von Definitionen für hierin verwendete Begriffe:
So
wie hierin verwendet, bedeutet "Säure" eine anorganische
oder organische Säure.
Eine anorganische Säure
ist eine Mineralsäure,
wie Schwefel-, Salpeter-, Salz- und Phosphorsäure. Eine organische Säure ist
eine organische Carbonsäure,
wie Ameisensäure, Essigsäure, Chloressigsäure, Dichloressigsäure, Propionsäure, Benzoesäure, Maleinsäure, Fumarsäure, Bernsteinsäure und
Weinsäure.
So
wie hierin verwendet, bedeutet "Addukt" eine) chemisches)
Reaktionszwischenverbindung oder -produkt, welche(s) eine neu eingebaute
funktionelle Gruppe enthält.
So
wie hierin verwendet, bedeutet "Base" ein basisches Reagens,
welches zu einem Reaktionsgemisch zugegeben wird, um die Alkylierung
eines Stickstoffs unter Verwendung eines Alkylierungsmittels zu
erleichtern. Basen schließen
Stickstoffbasen und anorganische Basen wie N,N-Diisopropylethylamin,
Triethylamin, Trimethylamin, 4-Dimethylaminopyridin, Pyridin, Natriumhydrid,
Kaliumhydrid, Kaliumcarbonat, Natriumcarbonat, Kaliumbicarbonat
und Natriumbicarbonat ein.
So wie hierin verwendet, ist ein "Halogen" ein Chlor-, Brom-,
Fluor oder Iodatomrest. Brom und Chlor sind bevorzugte Halogene.
So
wie hierin verwendet, ist ein "heterocyclischer
Ring" ein gesättigter,
ungesättigter
oder aromatischer Ringrest, bestehend aus Kohlenstoffatomen und
einem oder mehreren Heteroatomen in dem Ring. Heterocyclische Ringe
sind monocyclisch oder sind kondensierte, verbrückte oder spiro-polycyclische
Ringsysteme. Monocyclische Ringe enthalten 3 bis 9 Atome, vorzugsweise
4 bis 7 Atome und am meisten bevorzugt 5 oder 6 Atome. Polycyclische
Ringe enthalten 7 bis 17 Atome, vorzugsweise 7 bis 14 Atome und
am meisten bevorzugt 9 oder 10 Atome.
So wie hierin verwendet,
bedeutet "Abgangsgruppe" irgendein substituiertes
oder unsubstituiertes Alkyl- oder Arylsulfonat oder Halogenid. Bevorzugte
Substituenten sind Halogene.
So wie hierin verwendet, ist "Methylen" ein -CH2-Rest.
So
wie hierin verwendet, ist ein "polares
aprotisches Lösungsmittel" ein Lösungsmittel,
welches die Eigenschaft einer starken Polarität aufweist, aber dennoch die
Fähigkeit
besitzt, ein Proton abzugeben. Bevorzugte polare aprotische Lösungsmittel
schließen
N,N-Dimethylformamid (DMF), N,N-Dimethylacetamid (DMAC), N-Methylpyrrolidon
(NMP) und Methylsulfoxid (DMSO) ein.
Wie oben definiert und
wie hierin verwendet, können
die Substituentengruppen selbst substituiert sein. Eine solche Substitution
kann mit einem oder mehreren Substituenten erfolgen. Solche Substituenten
schließen
die bei Hansch C. und Leo A., Substituent Constants for Correlation
Analysis in Chemistry and Biology (1979), aufgeführten ein. Bevorzugte Substituenten
schließen
(zum Beispiel) Alkyl, Alkenyl, Alkoxy, Hydroxy, Oxo, Amino, Aminoalkyl
(z.B. Aminomethyl, etc.), Cyano, Halogen, Alkoxy, Alkoxyacyl (z.B.
Carboethoxy, etc.), Thiol, Aryl, Cycloalkyl, Heteroaryl, Heterocycloalkyl
(z.B. Piperidinyl, Morpholinyl, Pyrrolidinyl, etc.), Imino, Thioxo,
Hydroxyalkyl, Aryloxy, Arylalkyl und Kombinationen hiervon ein.
So
wie hierin verwendet, verweist "Volumina" auf die Liter eines
angegebenen Lösungsmittels
pro Kilogramm des Ausgangsmaterials.
-
Ausführliche
Beschreibung der Erfindung
-
Die
vorliegende Erfindung betrifft Verfahren zur Herstellung von 1,3-disubstituierten
4-oxocyclischen Harnstoffen, einschließlich, aber nicht begrenzt
auf, Azimilid und anderen pharmazeutisch annehmbaren Salzen hiervon,
welche in hohen Ausbeuten, mit einer hohen Produktreinheit, in einem
hohen Durchsatz und mit einer einfachen Synthese erhalten werden
können.
Die Erfindung schließt
ein aufeinanderfolgendes Verfahren ein, umfassend das Umsetzen eines
1-substituierten 4-oxocyclischen Harnstoffs mit einem Kohlenstoffkettenreagens,
enthaltend zwei Abgangsgruppen, in einem polaren aprotischen Lösungsmittel
in Gegenwart einer milden Base, das weitere Umsetzen mit einem Amin,
das Ausfällen
von Salzen mit einem Co-Lösungsmittel, das
Filtrieren und schließlich
das Zugeben einer Säure
und das Gewinnen eines 1,3-disubstituierten 4-oxocyclischen Harnstoffs
oder anderen Salzen hiervon.
-
Die
erste Alkylierung erfolgt bei Temperaturen von 40°C bis 120°C, vorzugsweise
bei etwa 60°C
bis 75°C.
Die Base, welche verwendet werden kann, ist aus solchen gewählt, welche
einfach filtrierbare und anderweitig entfernbare Salze haben. Genauer
schließen
bevorzugte Basen N,N-Diisopropylethylamin, Triethylamin, Trimethylamin,
4-Dimethylaminopyridin, Pyridin, Kaliumcarbonat, Natriumcarbonat,
Kaliumbicarbonat und Natriumbicarbonat ein. Die stärker bevorzugten
Basen sind Kaliumcarbonat, Natriumcarbonat, Kaliumbicarbonat und
Natriumbicarbonat. Die am meisten bevorzugte Base ist Kaliumcarbonat;
im allgemeinen 0,8 bis 4,0 Äquivalente,
vorzugsweise 1,2 bis 2 Äquivalente
pro Mol Imidazolidindion. Bevorzugte Kohlenstoffkettenreagenzien
sind gewählt
aus der Gruppe, enthaltend Halogengruppen, einschließlich, aber
nicht begrenzt auf, 1-Brom-4-chlorbutan, 1,4-Dichlor- oder 1,4-Dibrombutan;
wobei 1-Brom-4-chlorbutan mehr bevorzugt wird. Den Fachleuten ist
bekannt, daß Butylalkohole,
Butylsulfonylate und Tetrahydrofuran auch als Kohlenstoffkettenreagenzien
verwendet werden. Im allgemeinen werden 0,8 bis 2,5 Äquivalente,
vorzugsweise 1 bis 1,2 Äquivalente
pro Mol Imidazolidindion verwendet. Die Lösungsmittel, welche verwendet
werden, sind DMF, DMAC, DMSO und NMP, bevorzugt NMP. Im allgemeinen
werden 2 bis 20 Volumina, vorzugsweise 2,5 bis 5 Volumina NMP verwendet.
-
Bevorzugte
1-substituierte 4-oxocyclische Harnstoffe sind gewählt aus
der Gruppe, bestehend aus: 1-[[[5-(4-Chlorphenyl)-2-furanyl]methylen]amino]-2,4-imidazolidindion;
1-[[[5-(4-Methansulfonamidophenyl)-2-furanyl]methylen]amino]-2,4-imidazolidindion;
1-[[[5-(4-Fluorphenyl)-2-furanyl]methylen]amino]-2,4-imidazolidindion;
1-[[[5-(4-Nitrophenyl)-2-oxazolidinyl]methylen]amino]-2,4-imidazolidindion;
1-[[[5-(4-Methylphenyl)-2-furanyl]methylen]amino]-2,4-imidazolidindion;
und 1-[[[5-(3,4-Dimethoxyphenyl)-2-furanyl]methylen]amino]-2,4-imidazolidindion.
Bei der Herstellung von Azimilid ist der verwendete 1-substituierte
4-oxocyclische Harnstoff 1-[[[5-(4-Chlorphenyl)-2-furanyl]-methylen]amino]-2,4-imidazolidindion.
-
Die
zweite Alkylierung erfolgt bei Temperaturen von 50°C bis 120°C, vorzugsweise
bei etwa 75°C
bis 95°C.
Bevorzugte Amine für
diesen Schritt sind gewählt
aus der Gruppe, bestehend aus Dimethylamin; Diethylamin; N,N-Bis-(2-hydroxyethyl)amin;
Isopropylamin; N-Benzyl-N-methylamin; N-(2-Hydroxyethyl)-N-methylamin;
N-Methylpiperazin; Morpholin; 4-Hydroxypiperidin; und N-Methyl-N-phenylamin.
Das zur Herstellung von Azimilid verwendete Amin ist N-Methylpiperazin.
Im allgemeinen werden 0,8 bis 5 Äquivalente,
vorzugsweise 1,2 bis 3 Äquivalente
Amin pro Mol Imidazolidindion zugegeben.
-
Nach
der zweiten Alkylierung wird das Reaktionsgemisch im allgemeinen
auf –10°C bis 50°C, vorzugsweise
5°C bis
35°C gekühlt. Das
zum Ausfällen
der Salze verwendete Co-Lösungsmittel
ist entweder Aceton, Methanol, Ethanol oder Mischungen hiervon,
bevorzugt Aceton. Im allgemeinen werden 0 bis 20 Volumina, vorzugsweise
6 bis 10 Volumina verwendet. Die unlöslichen Salze werden durch
Filtration gesammelt und mit dem Co-Lösungsmittel gewaschen.
-
Wasser
wird zu dem Reaktionsgemisch zugegeben, um die Salzbildung vorzubereiten.
Im allgemeinen werden 0 bis 5 Volumina, vorzugsweise 0,5 bis 2,8
Volumina Wasser verwendet. Die Säure,
welche zur Bildung des gewünschten
Salzes verwendet wird, ist Salzsäure.
-
Im
allgemeinen wird der pH in dem Bereich von pH 3 bis 7, vorzugsweise
pH 4,5 bis 5, für
eine Kristallisationskeimbildung kontrolliert, gefolgt durch die
weitere Zugabe von Säure
bis pH 0–3,
um das Azimilid auszufällen,
welches durch Filtration in einer Ausbeute von 80 bis 90% gewonnen
wird.
-
Das
im Einklang mit dem erfindungsgemäßen Verfahren hergestellte
Azimilid ist zur Behandlung von verschiedenen medizinischen Störungen nützlich,
wobei solche Anwendungen die Verwendung als Antifibrillantia und
Antiarrhythmika einschließen,
aber nicht darauf begrenzt sind. Den Fachleuten ist auch bekannt, daß verschiedene
Säuren
in den letzten Schritten des Verfahrens zugegeben werden können, um
verschiedene Salzformen zu bilden, welche die Isolierung und Handhabung
erleichtern können.
Andere pharmazeutisch annehmbare Salze, wie zum Beispiel Sulfat
und Hydrobromid, können
im Einklang mit dem erfindungsgemäßen Verfahren hergestellt werden
und sind im Schutzumfang der Erfindung eingeschlossen.
-
Das
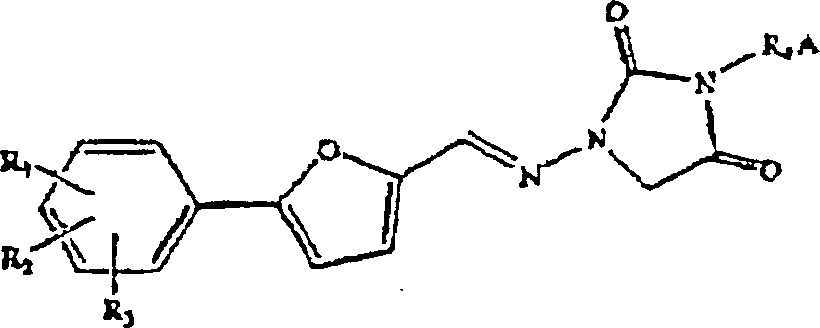
Verfahren wird durch das folgende allgemeine Schema veranschaulicht:
worin:
R
1, R
2 und R
3 unabhängig
gewählt
sind aus H, Cl, F, Br, NH
2, NO
2,
COOH, CH
3SO
2NH,
SO
3H, OH, Alkoxy, Alkyl, Alkoxycarbonyl,
Hydroxyalkyl, Carboxyalkyl und Acyloxy;
R
4 gewählt ist
aus einem substituierten oder unsubstituierten Alkyl, Alkenyl, Alkinyl,
Alkylacyl und Heteroalkyl;
A eine substituierte oder unsubstituierte,
geradkettige oder verzweigte Alkyl- oder Alkenylaminogruppe, umfassend
1-7 Kohlenstoffatome, darstellt; oder A ein substituierter oder
unsubstituierter, gesättigter
oder ungesättigter
Heterocyclus mit 5, 6 oder 7 Ringatomen ist, enthaltend mindestens
einen Stickstoff, und R
4 an diesen Stickstoff
gebunden ist;
X und Y unabhängig
Abgangsgruppen sind, vorzugsweise verschiedene Abgangsgruppen, gewählt aus
Halogeniden und substituierten oder unsubstituierten Alkyl- oder
Arylsulfonaten, und Mischungen hiervon;
wobei der 1,3-disubstituierte
4-oxocyclische Harnstoff ohne Isolierung von Zwischenverbindungen
hergestellt wird und die Herstellung die Schritte umfaßt:
(Ia)
Umsetzen eines 1-substituierten 4-oxocyclischen Harnstoffs mit einer
Kohlenstoffkette, enthaltend mindestens zwei Abgangsgruppen, in
Gegenwart einer milden Base und eines Lösungsmittels, um ein Addukt
zu bilden, welches mindestens eine Abgangsgruppe enthält; und
(Ib)
Kondensieren des Addukts mit einem Amin unter Bildung eines 1,3-disubstituierten
4-oxocyclischen Harnstoffs; und
(II) Gewinnen des 1,3-disubstituierten
4-oxocyclischen Harnstoffs.
-
Die
folgenden nichtbegrenzenden Beispiele veranschaulichen die Verfahren
der vorliegenden Erfindung:
-
Beispiel 1
-
Verwendung von Dimethylformamid
(DMF) als Reaktionslösungsmittel
zur Herstellung von Azimilid
-
Ein
12 1-Dreihalskolben, ausgestattet mit einem Thermometer, mechanischen
Rührwerk,
Heizmantel, Rückflußkondensator
und Zugabetrichter, wird mit DMF (4,77 1) gefüllt und auf 50°C erhitzt.
1-[[[5-(4-Chlorphenyl)-2-furanyl]methylen]amino]-2,4-imidazolidindion
(597 g) wird zugegeben, und das Erhitzen wird fortgesetzt. Wenn
das Auflösen
beendet ist, wird Kaliumcarbonat (276 g) in den Kolben eingebracht,
und das Erhitzen wird bis 85°C
fortgesetzt. Nach 10 Minuten wird 1-Brom-4-chlorbutan (370 g) zugegeben,
und das Erhitzen wird bis etwa 100°C fortgesetzt. Nach 35 Minuten
wird N-Methylpiperazin (465 g) zugegeben, und man läßt die Mischung
für 1 Stunde
bei 100°C
rühren.
Das Reaktionsgemisch wird auf etwa 10°C gekühlt und filtriert, um unlösliche Stoffe
zu entfernen. Das DMF wird unter vermindertem Druck bei 65–68°C entfernt
und durch absoluten Ethanol (3,6 1) ersetzt. Die Mischung wird erhitzt,
um die freie Base zu lösen,
und filtriert, um unlösliche
Stoffe zu entfernen. Das Produkt wird unter Zugabe von 418 g konzentrierter
Salzsäure
aus Ethanol (6,0 l insgesamt) ausgefällt und anschließend abfiltriert,
wobei 680 g der Verbindung erhalten werden.
-
Beispiel 2
-
Verwendung von Methylsulfoxid
(DMSO) als Reaktionslösungsmittel
zur Herstellung von Azimilid
-
Ein
500 ml-Dreihalskolben, ausgestattet mit einem Thermometer, mechanischen
Rührwerk,
Heizmantel, Rückflußkondensator
und Zugabetrichter, wird mit DMSO (200 ml) und 1-[[[5-(4-Chlorphenyl)-2-furanyl]methylen]amino]-2,4-imidazolidindion
(20 g) gefüllt.
Nach dem Auflösen
werden Kaliumcarbonat (15,5 g) und 1-Brom-4-chlorbutan (13,6 g)
zugegeben, und die Mischung wird über 30 Minuten auf 70°C erhitzt.
N-Methylpiperazin (19,8 g) wird zu der Mischung über 15 Minuten zugegeben, während das
Gemisch auf 90°C
erhitzt wird. Nach insgesamt 2 Stunden und 15 Minuten wird das Reaktionsgemisch
auf etwa 30°C
gekühlt,
und Methanol (200 ml) wird zugegeben. Die Mischung wird auf Raumtemperatur
gekühlt
und filtriert, um unlösliche Stoffe
zu entfernen. Das Filtrat wird mit konzentrierter Salzsäure auf
pH 1-2 angesäuert.
Die Mischung wird 15°C
gekühlt
und filtriert, wobei 30,4 g der Verbindung erhalten werden.
-
Beispiel 3
-
Verwendung von N,N-Dimethylacetamid
(DMAC) als Reaktionslösungsmittel
zur Herstellung von Azimilid
-
Ein
2 1-Dreihalskolben, ausgestattet mit einem Thermometer, mechanischen
Rührwerk,
Heizmantel, Rückflußkondensator
und Zugabetrichter, wird mit DMAC (200 ml), 1-[[[5-(4-Chlorphenyl)-2-furanyl]methylen]amino]-2,4-imidazolidindion
(100 g), 1-Brom-4-chlorbutan (59 g) und Kaliumcarbonat (73 g) gefüllt. Die
Mischung wird für
etwa 100 Minuten unter Erhitzen auf 70°C gerührt. N-Methylpiperazin (59,5
g) wird zugegeben, und die Mischung wird für weitere 3 Stunden unter Erhitzen
auf 86°C
gerührt.
Das Reaktionsgemisch wird auf 20°C
gekühlt,
und Aceton (900 ml) wird zugegeben. Die Mischung wird filtriert,
um unlösliche
Stoffe zu entfernen. Das Filtrat wird mit konzentrierter Salzsäure auf
pH 1-2 angesäuert,
auf 15°C
gekühlt
und filtriert, wobei 122,7 g der Verbindung erhalten werden.
-
Beispiel 4
-
Verwendung von N-Methylpyrrolidon
(NMP) als Reaktionslösungsmittel
zur Herstellung von Azimilid
-
Ein
5 1-Dreihalskolben, ausgestattet mit einem Thermometer, mechanischen
Rührwerk,
Heizmantel, Rückflußkondensator
und Zugabetrichter, wird mit NMP (1,2 1), 1-[[[5-(4-Chlorphenyl)-2-furanyl]methylen]amino]-2,4-imidazolidindion
(300 g), 1-Brom-4-chlorbutan
(187 g) und Kaliumcarbonat (219 g) gefüllt. Die Mischung wird für etwa 1
Stunde unter Erhitzen auf 70°C
gerührt.
N-Methylpiperazin (149 g) wird zugegeben, und die Mischung wird
für etwa
150 Minuten unter Erhitzen auf 90°C
gerührt.
Das Reaktionsgemisch wird auf 20°C
gekühlt,
und Aceton (2,4 1) wird zugegeben. Die Mischung wird filtriert,
um unlösliche
Stoffe zu entfernen. Wasser (0,42 1) wird zu dem Filtrat zugegeben,
und die Mischung wird auf 30°C
bis 35°C
erwärmt.
Die Mischung wird mit konzentrierter Salzsäure auf pH 4,5 bis 5 angesäuert, mit
Produkt beimpft, für
1 Stunde gerührt und
dann mit konzentrierter Salzsäure
weiter angesäuert
auf pH 0 bis 3. Die Mischung wird auf 10°C gekühlt und filtriert, wobei 382,8
g der Verbindung erhalten werden.