EP1613304B1 - Dpp-iv-hemmer - Google Patents
Dpp-iv-hemmer Download PDFInfo
- Publication number
- EP1613304B1 EP1613304B1 EP04803694A EP04803694A EP1613304B1 EP 1613304 B1 EP1613304 B1 EP 1613304B1 EP 04803694 A EP04803694 A EP 04803694A EP 04803694 A EP04803694 A EP 04803694A EP 1613304 B1 EP1613304 B1 EP 1613304B1
- Authority
- EP
- European Patent Office
- Prior art keywords
- alkyl
- optionally substituted
- group
- compound according
- independently
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Lifetime
Links
- 0 *N[C@@](CC(N1CC(COc2ccccc2)C1)=O)Cc(cccc1)c1F Chemical compound *N[C@@](CC(N1CC(COc2ccccc2)C1)=O)Cc(cccc1)c1F 0.000 description 6
- DTQVDTLACAAQTR-UHFFFAOYSA-N OC(C(F)(F)F)=O Chemical compound OC(C(F)(F)F)=O DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 3
- DFMYSQPIUFIOJZ-GOSISDBHSA-N N[C@@H](CC(N1CC(CNC(c2ccccc2)=O)C1)=O)Cc(cccc1)c1F Chemical compound N[C@@H](CC(N1CC(CNC(c2ccccc2)=O)C1)=O)Cc(cccc1)c1F DFMYSQPIUFIOJZ-GOSISDBHSA-N 0.000 description 2
- VUELKBGRNAWTPV-SJORKVTESA-N N[C@@H](CC(N1[C@H](CNC(c2ccccn2)=O)CCC1)=O)Cc(cccc1)c1F Chemical compound N[C@@H](CC(N1[C@H](CNC(c2ccccn2)=O)CCC1)=O)Cc(cccc1)c1F VUELKBGRNAWTPV-SJORKVTESA-N 0.000 description 2
- NCADHSLPNSTDMJ-UHFFFAOYSA-N CC(C)(C)OC(N(C1)CC1C(O)=O)=O Chemical compound CC(C)(C)OC(N(C1)CC1C(O)=O)=O NCADHSLPNSTDMJ-UHFFFAOYSA-N 0.000 description 1
- SOGXYCNKQQJEED-QMMMGPOBSA-N CC(C)(C)OC(N1[C@H](CN)CCC1)=O Chemical compound CC(C)(C)OC(N1[C@H](CN)CCC1)=O SOGXYCNKQQJEED-QMMMGPOBSA-N 0.000 description 1
- MCOPOJXBKPXZCR-QMMMGPOBSA-N CC(C)(C)OC(N1[C@H](CNC(C(F)(F)F)=O)CCC1)=O Chemical compound CC(C)(C)OC(N1[C@H](CNC(C(F)(F)F)=O)CCC1)=O MCOPOJXBKPXZCR-QMMMGPOBSA-N 0.000 description 1
- OXAHRXMFRIOLTN-AWEZNQCLSA-N CC(C)(C)OC(N1[C@H](CNC(c2ccccc2)=O)CCC1)=O Chemical compound CC(C)(C)OC(N1[C@H](CNC(c2ccccc2)=O)CCC1)=O OXAHRXMFRIOLTN-AWEZNQCLSA-N 0.000 description 1
- YSDGNANNAZSIBB-JTQLQIEISA-N CC(C)(C)OC(N1[C@H](CNS(C2CC2)(=O)=O)CCC1)=O Chemical compound CC(C)(C)OC(N1[C@H](CNS(C2CC2)(=O)=O)CCC1)=O YSDGNANNAZSIBB-JTQLQIEISA-N 0.000 description 1
- XQPQTMLCJXCGNU-FIWHBWSRSA-N CC(C)(C)OC(N[C@@H](CC(N1C(CNc2nc(-c3ncccc3)n[o]2)CCC1)=O)Cc1ccccc1F)=O Chemical compound CC(C)(C)OC(N[C@@H](CC(N1C(CNc2nc(-c3ncccc3)n[o]2)CCC1)=O)Cc1ccccc1F)=O XQPQTMLCJXCGNU-FIWHBWSRSA-N 0.000 description 1
- ITUPANPGWUIOSR-ABLWVSNPSA-N CC(C)C(C)OC(N1[C@H](CN(C)S(C2CC2)(=O)=O)CCC1)=O Chemical compound CC(C)C(C)OC(N1[C@H](CN(C)S(C2CC2)(=O)=O)CCC1)=O ITUPANPGWUIOSR-ABLWVSNPSA-N 0.000 description 1
- AFSTUBSMTQRBAA-AAFJCEBUSA-N CN(CC(CCC1)N1C(C[C@@H](Cc(cc(c(F)c1)F)c1F)N)=O)c1cc(C(F)(F)F)ccn1 Chemical compound CN(CC(CCC1)N1C(C[C@@H](Cc(cc(c(F)c1)F)c1F)N)=O)c1cc(C(F)(F)F)ccn1 AFSTUBSMTQRBAA-AAFJCEBUSA-N 0.000 description 1
- NDUKSSFKHVGCNM-CABCVRRESA-N CN(C[C@H](CCC1)N1C(C[C@@H](Cc(c(F)c1)cc(F)c1F)N)=O)C(C1CC1)=O Chemical compound CN(C[C@H](CCC1)N1C(C[C@@H](Cc(c(F)c1)cc(F)c1F)N)=O)C(C1CC1)=O NDUKSSFKHVGCNM-CABCVRRESA-N 0.000 description 1
- UUBDZSXHODIDHL-VIFPVBQESA-N CN(C[C@H]1NCCC1)C(C1CC1)=O Chemical compound CN(C[C@H]1NCCC1)C(C1CC1)=O UUBDZSXHODIDHL-VIFPVBQESA-N 0.000 description 1
- LEWXABZAAARZNR-MSOLQXFVSA-N CS(c(cccc1)c1C(NC[C@H](CCC1)N1C(C[C@@H](Cc1ccccc1F)N)=O)=O)(=O)=O Chemical compound CS(c(cccc1)c1C(NC[C@H](CCC1)N1C(C[C@@H](Cc1ccccc1F)N)=O)=O)(=O)=O LEWXABZAAARZNR-MSOLQXFVSA-N 0.000 description 1
- QRGBOABBMKYMLG-UXHICEINSA-N CS(c1cc(C(NC[C@H](CCC2)N2C(C[C@@H](Cc2cccc(Cl)c2)N)=O)=O)ccc1)(=O)=O Chemical compound CS(c1cc(C(NC[C@H](CCC2)N2C(C[C@@H](Cc2cccc(Cl)c2)N)=O)=O)ccc1)(=O)=O QRGBOABBMKYMLG-UXHICEINSA-N 0.000 description 1
- BUKAZCMVMUYNMT-JTQLQIEISA-N C[C@@H](CCCC(C(OC(C)(C)C)=O)=C)CN Chemical compound C[C@@H](CCCC(C(OC(C)(C)C)=O)=C)CN BUKAZCMVMUYNMT-JTQLQIEISA-N 0.000 description 1
- FRDPDLLWDHVFLS-PMRGXDQWSA-N C[C@H](CNC(c1ccccn1)=O)C(CC1)CCC1C(CC(Cc1ccccc1F)N)=O Chemical compound C[C@H](CNC(c1ccccn1)=O)C(CC1)CCC1C(CC(Cc1ccccc1F)N)=O FRDPDLLWDHVFLS-PMRGXDQWSA-N 0.000 description 1
- OFYKGOYPNBYZKV-RTWAWAEBSA-N Cc1ccccc1C[C@H](CC(N1[C@H](CNc2nc(-c3cc(F)ccc3)n[o]2)CCC1)=O)N Chemical compound Cc1ccccc1C[C@H](CC(N1[C@H](CNc2nc(-c3cc(F)ccc3)n[o]2)CCC1)=O)N OFYKGOYPNBYZKV-RTWAWAEBSA-N 0.000 description 1
- BTAMWUHAAIDZPC-UHFFFAOYSA-N ClC(c1nc(-c2ccccn2)n[o]1)(Cl)Cl Chemical compound ClC(c1nc(-c2ccccn2)n[o]1)(Cl)Cl BTAMWUHAAIDZPC-UHFFFAOYSA-N 0.000 description 1
- DXTNOORZGCJSRV-UHFFFAOYSA-N Fc1cccc(-c2n[o]c(NCC3NCCC3)n2)c1 Chemical compound Fc1cccc(-c2n[o]c(NCC3NCCC3)n2)c1 DXTNOORZGCJSRV-UHFFFAOYSA-N 0.000 description 1
- YXRBUJMLHVRPCD-UHFFFAOYSA-N NC(CC(N1C(CNc2nc(-c3cc(Cl)ccc3)n[o]2)CCC1)=O)Cc(cccc1)c1F Chemical compound NC(CC(N1C(CNc2nc(-c3cc(Cl)ccc3)n[o]2)CCC1)=O)Cc(cccc1)c1F YXRBUJMLHVRPCD-UHFFFAOYSA-N 0.000 description 1
- SUOPSKUNEUIZLU-ZVAWYAOSSA-N NC(CC(N1[C@H](CNC(c2cccnc2)=O)CCC1)=O)Cc(cccc1)c1F Chemical compound NC(CC(N1[C@H](CNC(c2cccnc2)=O)CCC1)=O)Cc(cccc1)c1F SUOPSKUNEUIZLU-ZVAWYAOSSA-N 0.000 description 1
- GUKXSTARZIHVLU-DJNXLDHESA-N NC(CC(N1[C@H](CNS(c(cc(cc2)Cl)c2Cl)(=O)=O)CCC1)=O)Cc(cccc1)c1F Chemical compound NC(CC(N1[C@H](CNS(c(cc(cc2)Cl)c2Cl)(=O)=O)CCC1)=O)Cc(cccc1)c1F GUKXSTARZIHVLU-DJNXLDHESA-N 0.000 description 1
- YJKFKJDPCJBSJP-ZVAWYAOSSA-N NC(CC(N1[C@H](CNc(nc2)ccc2C#N)CCC1)=O)Cc(cccc1)c1F Chemical compound NC(CC(N1[C@H](CNc(nc2)ccc2C#N)CCC1)=O)Cc(cccc1)c1F YJKFKJDPCJBSJP-ZVAWYAOSSA-N 0.000 description 1
- FYCRVTDSDFOKTD-GGYWPGCISA-N NC(CC(N1[C@H](COc2cccc(F)c2)CCC1)=O)Cc1cccc(Cl)c1 Chemical compound NC(CC(N1[C@H](COc2cccc(F)c2)CCC1)=O)Cc1cccc(Cl)c1 FYCRVTDSDFOKTD-GGYWPGCISA-N 0.000 description 1
- BHRMMYVOEWKURV-ZVAWYAOSSA-N NC(CC(N1[C@H](COc2cccc(F)c2)CCC1)=O)Cc1ccccc1F Chemical compound NC(CC(N1[C@H](COc2cccc(F)c2)CCC1)=O)Cc1ccccc1F BHRMMYVOEWKURV-ZVAWYAOSSA-N 0.000 description 1
- YLNNGMZPGMFNOR-ZVAWYAOSSA-N NC(CC(N1[C@H](COc2ccccc2)CCC1)=O)Cc(cccc1)c1F Chemical compound NC(CC(N1[C@H](COc2ccccc2)CCC1)=O)Cc(cccc1)c1F YLNNGMZPGMFNOR-ZVAWYAOSSA-N 0.000 description 1
- OSNIFVNVWAVGFL-OLZOCXBDSA-N NC[C@H](CCC1)N1C(C[C@@H](Cc(cccc1)c1F)N)=O Chemical compound NC[C@H](CCC1)N1C(C[C@@H](Cc(cccc1)c1F)N)=O OSNIFVNVWAVGFL-OLZOCXBDSA-N 0.000 description 1
- QIVJQINZGCWSNW-PCRWRXJSSA-N N[C@@H](CC(N(CCC1)/C1=C/NC(C1CC1)=O)=O)Cc(cc(c(F)c1)F)c1F Chemical compound N[C@@H](CC(N(CCC1)/C1=C/NC(C1CC1)=O)=O)Cc(cc(c(F)c1)F)c1F QIVJQINZGCWSNW-PCRWRXJSSA-N 0.000 description 1
- SSOHKGQVPWCIKU-RNCFNFMXSA-N N[C@@H](CC(N(CCC1)[C@@H]1[IH]NC(C(C(F)(F)F)(F)F)=O)=O)Cc(cc(c(F)c1)F)c1F Chemical compound N[C@@H](CC(N(CCC1)[C@@H]1[IH]NC(C(C(F)(F)F)(F)F)=O)=O)Cc(cc(c(F)c1)F)c1F SSOHKGQVPWCIKU-RNCFNFMXSA-N 0.000 description 1
- IGLNCVBGTJJFNA-TZHYSIJRSA-N N[C@@H](CC(N1C(CNC(C2CC2)=O)CCC1)=O)Cc1cc(Cl)ccc1 Chemical compound N[C@@H](CC(N1C(CNC(C2CC2)=O)CCC1)=O)Cc1cc(Cl)ccc1 IGLNCVBGTJJFNA-TZHYSIJRSA-N 0.000 description 1
- JTHGYFUJXMFRGK-CQSZACIVSA-N N[C@@H](CC(N1C(COCC2CC2)=CCC1)=O)Cc(cc(c(F)c1)F)c1F Chemical compound N[C@@H](CC(N1C(COCC2CC2)=CCC1)=O)Cc(cc(c(F)c1)F)c1F JTHGYFUJXMFRGK-CQSZACIVSA-N 0.000 description 1
- SUOPSKUNEUIZLU-MSOLQXFVSA-N N[C@@H](CC(N1[C@H](CNC(c2cccnc2)=O)CCC1)=O)Cc(cccc1)c1F Chemical compound N[C@@H](CC(N1[C@H](CNC(c2cccnc2)=O)CCC1)=O)Cc(cccc1)c1F SUOPSKUNEUIZLU-MSOLQXFVSA-N 0.000 description 1
- JQTKCIIZYREHAN-MJGOQNOKSA-N N[C@@H](CC(N1[C@H](CNc2ccccc2)CCC1)=O)Cc(cccc1)c1F Chemical compound N[C@@H](CC(N1[C@H](CNc2ccccc2)CCC1)=O)Cc(cccc1)c1F JQTKCIIZYREHAN-MJGOQNOKSA-N 0.000 description 1
- VNKLHOHFAWRPIT-CVEARBPZSA-N N[C@@H](CC(N1[C@H](CNc2cnccn2)CCC1)=O)Cc(cccc1)c1F Chemical compound N[C@@H](CC(N1[C@H](CNc2cnccn2)CCC1)=O)Cc(cccc1)c1F VNKLHOHFAWRPIT-CVEARBPZSA-N 0.000 description 1
- KGBUFUOVZRROTR-UXHICEINSA-N N[C@@H](CC(N1[C@H](COCc2ccccc2)CCC1)=O)Cc(cccc1)c1F Chemical compound N[C@@H](CC(N1[C@H](COCc2ccccc2)CCC1)=O)Cc(cccc1)c1F KGBUFUOVZRROTR-UXHICEINSA-N 0.000 description 1
- QAEDZJGFFMLHHQ-UHFFFAOYSA-N O=C(C(F)(F)F)OC(C(F)(F)F)=O Chemical compound O=C(C(F)(F)F)OC(C(F)(F)F)=O QAEDZJGFFMLHHQ-UHFFFAOYSA-N 0.000 description 1
- GFZWHAAOIVMHOI-UHFFFAOYSA-N OC(C1CNC1)=O Chemical compound OC(C1CNC1)=O GFZWHAAOIVMHOI-UHFFFAOYSA-N 0.000 description 1
- WPYMKLBDIGXBTP-UHFFFAOYSA-N OC(c1ccccc1)=O Chemical compound OC(c1ccccc1)=O WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/04—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D211/06—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D211/08—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms
- C07D211/18—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms with substituted hydrocarbon radicals attached to ring carbon atoms
- C07D211/26—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms with substituted hydrocarbon radicals attached to ring carbon atoms with hydrocarbon radicals, substituted by nitrogen atoms
- C07D211/28—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms with substituted hydrocarbon radicals attached to ring carbon atoms with hydrocarbon radicals, substituted by nitrogen atoms to which a second hetero atom is attached
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D207/00—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D207/02—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D207/04—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D207/08—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon radicals, substituted by hetero atoms, attached to ring carbon atoms
- C07D207/09—Radicals substituted by nitrogen atoms, not forming part of a nitro radical
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/397—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having four-membered rings, e.g. azetidine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/02—Stomatological preparations, e.g. drugs for caries, aphtae, periodontitis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/18—Drugs for disorders of the alimentary tract or the digestive system for pancreatic disorders, e.g. pancreatic enzymes
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/08—Drugs for disorders of the urinary system of the prostate
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/08—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease
- A61P19/10—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease for osteoporosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/22—Anxiolytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/24—Antidepressants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/06—Antihyperlipidemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
- A61P31/18—Antivirals for RNA viruses for HIV
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/10—Drugs for disorders of the endocrine system of the posterior pituitary hormones, e.g. oxytocin, ADH
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/12—Antihypertensives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D205/00—Heterocyclic compounds containing four-membered rings with one nitrogen atom as the only ring hetero atom
- C07D205/02—Heterocyclic compounds containing four-membered rings with one nitrogen atom as the only ring hetero atom not condensed with other rings
- C07D205/04—Heterocyclic compounds containing four-membered rings with one nitrogen atom as the only ring hetero atom not condensed with other rings having no double bonds between ring members or between ring members and non-ring members
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D207/00—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D207/02—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D207/04—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D207/08—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon radicals, substituted by hetero atoms, attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/04—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D211/06—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D211/08—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms
- C07D211/18—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms with substituted hydrocarbon radicals attached to ring carbon atoms
- C07D211/26—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms with substituted hydrocarbon radicals attached to ring carbon atoms with hydrocarbon radicals, substituted by nitrogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/04—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D211/06—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D211/08—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms
- C07D211/18—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms with substituted hydrocarbon radicals attached to ring carbon atoms
- C07D211/34—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms with substituted hydrocarbon radicals attached to ring carbon atoms with hydrocarbon radicals, substituted by carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
Definitions
- the present invention relates to a novel class of dipeptidyl peptidase inhibitors, including pharmaceutically acceptable salts and prodrugs thereof, which are useful as therapeutic compounds, particularly in the treatment of Type 2 diabetes mellitus, often referred to as non-insulin dependent diabetes mellitus (NIDDM), and of conditions that are often associated with this disease, such as obesity and lipid disorders.
- NIDDM non-insulin dependent diabetes mellitus
- the invention also relates to a process for the preparation of such inhibitors.
- Diabetes refers to a disease process derived from multiple causative factors and characterized by elevated levels of plasma glucose or hyperglycemia in the fasting state or after administration of glucose during an oral glucose tolerance test
- Persistent or uncontrolled hyperglycemia is associated with increased and premature morbidity and mortality.
- Often abnormal glucose homeostasis is associated both directly and indirectly with alterations of the lipid, lipoprotein and apolipoprotein metabolism and other metabolic and hemodynamic disease. Therefore patients with Type 2 diabetes mellitus are at an increased risk of macrovascular and microvascular complications, including coronary heart disease, stroke, peripheral vascular disease, hypertension, nephropathy, neuropathy, and retinopathy. Therefore, therapeutical control of glucose homeostasis, lipid metabolism and hypertension are critically important in the clinical management and treatment of diabetes mellitus.
- Type 1 diabetes mellitus IDDM
- Type 2 diabetes mellitus NIDDM
- Type 3 diabetes mellitus NIDDM
- Type 3 diabetes mellitus NIDDM
- Type 2 diabetes mellitus NIDDM
- NIDDM noninsulin dependent, diabetes mellitus
- these patients develop a resistance to the insulin stimulating effect on glucose and lipid metabolism in the main insulin-sensitive tissues, namely the muscle, liver and adipose tissues. Further, the plasma insulin levels, while elevated, are insufficient to overcome the pronounced insulin resistance.
- Insulin resistance is not primarily due to a diminished number of insulin receptors but to a post-insulin receptor binding defect that is not yet understood. This resistance to insulin responsiveness results in insufficient insulin activation of glucose uptake, oxidation and storage in muscle, and inadequate insulin repression of lipolysis in adipose tissue and of glucose production and secretion in the liver.
- Type 2 diabetes which have not changed substantially in many years, have recognized limitations. While physical exercise and reductions in dietary intake of calories will dramatically improve the diabetic condition, compliance with this treatment is very poor because of well-entrenched sedentary lifestyles and excess food consumption, especially of foods containing high amounts of saturated fat.
- sulfonylureas e.g., tolbutamide and glipizide
- meglitinide which stimulate the pancreatic ⁇ -cells to secrete more insulin, and/or by injection of insulin when sulfonylureas or meglitinide become ineffective, can result in insulin concentrations high enough to stimulate the very insulin-resistant tissues.
- sulfonylureas or meglitinide sulfonylureas or meglitinide
- the biguanides increase insulin sensitivity resulting in some correction of hyperglycemia.
- the two biguanides, phenformin and metformin can induce lactic acidosis and nausea/diarrhoea.
- Metformin has fewer side effects than phenformin and is often prescribed for the treatment of Type 2 diabetes.
- the glitazones are a recently described class of compounds with potential for ameliorating many symptoms of Type 2 diabetes. These agents substantially increase insulin sensitivity in muscle, liver and adipose tissue in several animal models of Type 2 diabetes, resulting in partial or complete correction of the elevated plasma levels of glucose without occurrence of hypoglycemia.
- the glitazones that are currently marketed are agonists of the peroxisome proliferator activated receptor (PPAR), primarily the PPAR-gamma subtype.
- PPAR-gamma agonism is generally believed to be responsible for the improved insulin sensitization that is observed with the glitazones.
- Newer PPAR agonists that are being tested for treatment of Type 2 diabetes are agonists of the alpha, gamma or delta subtype, or a combination of these, and in many cases are chemically different from the glitazones ( i.e ., they are not thiazolidinediones). Serious side effects (e . g ., liver toxicity) have occurred with some of the glitazones, such as troglitazone.
- alpha-glucosidase inhibitors e.g., acarbose
- PTP-1B protein tyrosine phosphatase-IB
- DPP-IV dipeptidyl peptidase-IV
- WO-A-97/40832 WO-A-98/19998
- WO-A-03/180 WO-A-03/181 .
- the usefulness of DPP-IV inhibitors in the treatment of Type 2 diabetes is based on the fact that DPP-IV in vivo readily inactivates glucagon like peptide-1 (GLP-1) and gastric inhibitory peptide (GIP).
- GLP-1 and GIP are incretins and are produced when food is consumed. The incretins stimulate production of insulin.
- DPP-IV Inhibition of DPP-IV leads to decreased inactivation of the incretins, and this in turn results in increased effectiveness of the incretins in stimulating production of insulin by the pancreas. DPP-IV inhibition therefore results in an increased level of serum insulin.
- DPP-IV inhibition since the incretins are produced by the body only when food is consumed, DPP-IV inhibition is not expected to increase the level of insulin at inappropriate times, such as between meals, which can lead to excessively low blood sugar (hypoglycemia). Inhibition of DPP-IV is therefore expected to increase insulin without increasing the risk of hypoglycemia, which is a dangerous side effect associated with the use of insulin secretagogues.
- DPP-IV inhibitors may also have other therapeutic utilities, as discussed elsewhere in this application.
- DPP-IV inhibitors have not been studied extensively to date, especially for utilities other than diabetes. New compounds are needed so that improved DPP-IV inhibitors can be found for the treatment of diabetes and potentially other disease and conditions.
- the object of the present invention is to provide a new class of DPP-IV inhibitors which may be effective in the treatment of Type 2 diabetes and other DPP-IV modulated diseases.
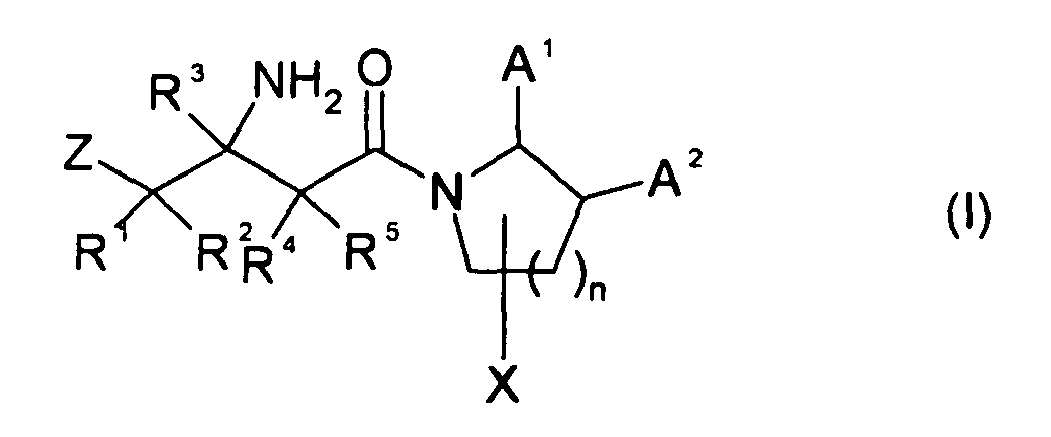
- the present invention provides novel compounds of formula (I): or a pharmaceutically acceptable salt thereof, wherein Z is selected from the group consisting of phenyl; naphthyl; C 3-7 cycloalkyl; heterocycle; and heterobicycle; wherein Z is optionally substituted with one, or independently from each other, more of
- Preferred compounds of formula (I) or (Ia) are those compounds in which one or more of the residues contained therein have the meanings given below, with all combinations of preferred substituent definitions being a subject of the present invention. With respect to all preferred compounds of the formulas (I) or (Ia) the present invention also includes all tautomeric and stereoisomeric forms and mixtures thereof in all ratios, and their pharmaceutically acceptable salts.
- the substituents R 1 - R 5 , Z, X, n, A 1 and A 2 of the formula (I) or (Ia) independently from each other have the following meaning.
- one or more of the substituents R 1 - R 5 , Z, X, n, A 1 and A 2 can have the preferred or more preferred meanings given below.
- Z is preferably is phenyl or heterocycle and Z is optionally substituted independently from each other with 1, 2 or 3, more preferably up to 2 or 3, of Cl, F, CN, CH 3 or OCH 3 . In one embodiment Z is substituted with up to 3 F.
- R 1 , R 2 , R 4 , R 5 are independently from each other selected from the group consisting of H, F, OH CH 3 , OCH 3 .
- R 3 is preferably H.
- X is preferably H, F or CH 3 .
- n is 1. In other embodiments, n is 0.
- a 1 is R 6 and A 2 is H, F or CH 3 .
- n is preferably 1.
- a 2 is preferably R 6 .
- a 2 is preferably H, F or CH 3 .
- R 6 is preferably -CH 2 -Y-T.
- Y is preferably -O-, -N(R 9 )- or -S(O) 2 -, more preferably -O- or -N(R 9 )-.
- R 9 is selected from the group consisting of H, CH 3 , COOH, COOCH 3 , C(O)NH 2 , C(O)N(CH 3 ) 2 , and S(O) 2 CH 3 , more preferably H, CH 3 , most preferably H.
- T is T 1 -T 2 or T 2 , wherein T 1 is selected from the group consisting of
- T 1 selected from the group consisting of -C(O)-; -CH 2 -; -S(O) 2 -; and -C(O)NH-.
- T is T 1 -T 2 .
- T 1 -T 2 is preferably a group as defined below.
- T 1 -T 2 is preferably CH 2 -phenyl, whereby phenyl may be substituted with 1-3, preferably 1 or 2, substituents selected from halogen, CN, O-C 1-4 alkyl, C 1-4 alkyl or S(O) 2 CH 3 , preferably F, CI, O-Me, Me or S(O) 2 CH 3 .
- T 1 -T 2 is preferably CH 2 -C 3-7 cycloalkyl, more preferably cyclopropyl or cyclobutyl, more preferably cyclopropyl, whereby cycloalkyl may be substituted with 1 or 2, preferably 1, of halogen; CN; OH; NH 2 COOH; C(O)NH 2 ; or S(O) 2 NH 2 , more preferably COOH or C(O)NH 2 .
- T 1 -T 2 is preferably C 1-4 alkyl, preferably methyl, ethyl or propyl, most preferably methyl.
- T 1 - T 2 is preferably C(O)-phenyl, whereby phenyl may be substituted with 1-3, preferably 1 or 2, substituents selected from halogen, CN, O-C 1-4 alkyl, C 1-4 alkyl or S(O) 2 CH 3 , preferably F, Cl, O-Me, Me or S(O) 2 CH 3 .
- T 1 -T 2 is preferably C(O)-C 3-7 cycloalkyl, more preferably cyclopropyl or cyclobutyl, more preferably cyclopropyl, whereby cycloalkyl may be substituted with 1-3, preferably 1 or 2, substituents selected from halogen, CN, O-C 1-4 alkyl, C 1-4 alkyl, whereby alkyl may be further substituted with 1 to 3 F; more preferably cycloalkyl may be substituted with 1 C 1-4 alkyl substituted with 1 to 3 F.
- T 1 -T 2 is preferably C(O)-heterocycle, whereby heterocycle may be substituted with 1-3, preferably 1 or 2, substituents selected from halogen, CN, O-C 1-4 alkyl, C 1-4 alkyl or S(O) 2 CH 3 ; preferably, the heterocycle is aromatic, more preferably containing 1 or 2 heteroatoms selected from N and O most preferably N.
- T 1 -T 2 is preferably S(O) 2 -phenyl, whereby phenyl may be substituted with 1-3, preferably 1 or 2, substituents selected from halogen, CN, O-C 1-4 alkyl, C 1-4 alkyl or S(O) 2 CH 3 , preferably F, Cl, O-Me, Me or S(O) 2 CH 3 .
- T 1 -T 2 is preferably S(O) 2 -C 3-7 cycloalkyl, more preferably cyclopropyl or cyclobutyl, more preferably cyclopropyl, whereby cycloalkyl may be substituted with 1-3, preferably 1 or 2, substituents selected from halogen, CN, O-C 1-4 alkyl, C 1-4 alkyl, whereby alkyl may be further substituted with 1 to 3 F; more preferably cycloalkyl may be substituted with 1 C 1-4 alkyl substituted with 1 to 3 F.
- T 1 - T 2 is preferably S(O) 2 -C 1-4 alkyl, preferably S(O) 2 CH 3 .
- T 1 -T 2 is preferably C(O)-NH-phenyl, whereby phenyl may be substituted with 1-3, preferably 1 or 2, substituents selected from halogen, CN, O-C 1-4 alkyl, C 1-4 alkyl or S(O) 2 CH 3 .
- T is T 2
- T 2 is preferably a group as defined below.
- T 2 is preferably H.
- T 2 is preferably phenyl, whereby phenyl may be substituted with 1-3, preferably 1 or 2, substituents selected from halogen, CN, O-C 1 - 4 alkyl, C 1 - 4 alkyl or S(O) 2 CH 3 , preferably F, Cl, O-Me, Me or S(O) 2 CH 3 .
- T 2 is preferably heterocycle, whereby heterocycle may be substituted with 1-3, preferably 1 or 2, substituents selected from halogen, CN, phenyl, heterocycle, O-C 1-4 alkyl, C 1-4 alkyl or S(O) 2 CH 3 ; preferably, the heterocycle is aromatic, more preferably containing 1, 2 or 3 heteroatoms selected from N and 0, most preferably N.
- the heterocycle is preferably aromatic, more preferably containing 1, 2 or 3 heteroatoms selected from N and O, most preferably N, and the phenyl or heterocycle may be further substituted by 1 or 2 F or S(O) 2 CH 3 .
- T 2 is preferably CF 3 .
- T 2 is preferably phenyl or heterocycle.
- R 6 is -CH 2 -N(R 36 )-T, wherein R 36 is H, S(O) 2 CH 3 or S(O) 2 -C 3-7 cycloalkyl, most preferably H.
- R 6 is -CH 2 -O-T.
- R 9 contains the group R 9 , the following is preferred in embodiments:
- R 9 and T may form together a 3 to 7 membered cyclic group containing 1 N, preferably a 5 or 6 membered cyclic group.
- the present invention provides prodrug compounds of the compounds of the invention as described above.
- Prodrug compound means a derivative that is converted into a compound according to the present invention by a reaction with an enzyme, gastric acid or the like under a physiological condition in the living body, e.g. by oxidation, reduction, hydrolysis or the like, each of which is carried out enzymatically.
- Examples of the prodrug are compounds, wherein the amino group in a compound of the present invention is acylated, alkylated or phosphorylated to form, e.g., eicosanoylamino, alanylamino, pivaloyloxymethylamino. These compounds can be produced from compounds of the present invention according to well-known methods.
- tautomerism like e.g. keto-enol tautomerism, of compounds of general formula (I) or (Ia) or their prodrugs
- the individual forms like e.g. the keto and enol form, are claimed separately and together as mixtures in any ratio.
- stereoisomers like e.g. enantiomers, cis/trans isomers, conformers and the like.
- isomers can be separated by methods well known in the art, e.g. by liquid chromatography.
- enantiomers by using e.g. chiral stationary phases.
- enantiomers may be isolated by converting them into diastereomers, i.e.
- any enantiomer of a compound of formula (I) or (Ia) may be obtained from stereoselective synthesis using optically pure starting materials.
- the invention also comprises their corresponding pharmaceutically or toxicologically acceptable salts, in particular their pharmaceutically utilizable salts.
- the compounds of the formula (I) or (la) which contain acidic groups can be present on these groups and can be used according to the invention, for example, as alkali metal salts, alkaline earth metal salts or as ammonium salts. More precise examples of such salts include sodium salts, potassium salts, calcium salts, magnesium salts or salts with ammonia or organic amines such as, for example, ethylamine, ethanolamine, triethanolamine or amino acids.
- Compounds of the formula (I) or (la) which contain one or more basic groups, i.e. groups which can be protonated, can be present and can be used according to the invention in the form of their addition salts with inorganic or organic acids.
- acids examples include hydrogen chloride, hydrogen bromide, phosphoric acid, sulfuric acid, nitric acid, methanesulfonic acid, p-toluenesulfonic acid, naphthalenedisulfonic, acids, oxalic acid, acetic acid, tartaric acid, lactic acid, salicylic acid, benzoic acid, formic acid, propionic acid, pivalic acid, diethylacetic acid, malonic acid, succinic acid, pimelic acid, fumaric acid, maleic acid, malic acid, sulfaminic acid, phenylpropionic acid, gluconic acid, ascorbic acid, isonicotinic acid, citric acid, adipic acid, and other acids known to the person skilled in the art.
- the invention also includes, in addition to the salt forms mentioned, inner salts or betaines (zwitterions).
- inner salts or betaines zwitterions.
- the respective salts according to the formula (I) or (la) can be obtained by customary methods which are known to the person skilled in the art like, for example by contacting these with an organic or inorganic acid or base in a solvent or dispersant, or by anion exchange or cation exchange with other salts.
- the present invention also includes all salts of the compounds of the formula (I) or (Ia) which, owing to low physiological compatibility, are not directly suitable for use in pharmaceuticals but which can be used, for example, as intermediates for chemical reactions or for the preparation of pharmaceutically acceptable salts.
- DPP-IV is a cell surface protein that has been implicated in a wide range of biological functions. It has a broad tissue distribution (intestine, kidney, liver, pancreas, placenta, thymus, spleen, epithelial cells, vascular endothelium, lymphoid and myeloid cells, serum), and distinct tissue and cell-type expression levels. DPP-IV is identical to the T cell activation marker CD26, and it can cleave a number of immunoregulatory, endocrine, and neurological peptides in vitro. This has suggested a potential role for this peptidase in a variety of disease processes.
- the present invention provides compounds of formula (I) or (Ia) or their prodrugs or pharmaceutically acceptable salt thereof for use as a medicament.
- the present invention provides the use of compounds of formula (I) or (Ia) or their prodrugs or a pharmaceutically acceptable salt thereof for the manufacture of a medicament for the treatment or prophylaxis of non-insulin dependent (Type II) diabetes mellitus; hyperglycemia; obesity; insulin resistance; lipid disorders; dyslipidemia; hyperlipidemia; hypertriglyceridemia; hypercholestrerolemia; low HDL; high LDL; atherosclerosis; growth hormone deficiency; diseases related to the immune response; HIV infection; neutropenia; neuronal disorders; anxiety; depression; tumor metastasis; benign prostatic hypertrophy; gingivitis; hypertension; osteoporosis; diseases related to sperm motility; low glucose tolerance; insulin resistance; ist sequelae; vascular restenosis; irritable bowel syndrome; inflammatory bowel disease; including Crohn's disease and ulcerative colitis; other inflammatory conditions; pancreatitis; abdominal obesity; neurodegenerative disease; retinopathy, nephropathy
- the present invention provides pharmaceutical compositions comprising a compound of formula (I) or (Ia), or a prodrug compound thereof, or a pharmaceutically acceptable salt thereof as active ingredient together with a pharmaceutically acceptable carrier.
- “Pharmaceutical composition” means one or more active ingredients, and one or more inert ingredients that make up the carrier, as well as any product which results, directly or indirectly, from combination, complexation or aggregation of any two or more of the ingredients, or from dissociation of one or more of the ingredients, or from other types of reactions or interactions of one or more of the ingredients. Accordingly, the pharmaceutical compositions of the present invention encompass any composition made by admixing a compound of the present invention and a pharmaceutically acceptable carrier.
- a pharmaceutical composition of the present invention may additionally comprise one or more other compounds as active ingredients like one or more additional compounds of formula (I) or (Ia), or a prodrug compound or other DPP-IV inhibitors.
- Other active ingredients are disclosed in WO-A-03/181 under the paragraph "Combination Therapy”.
- other active ingredients may be insulin sensitizers; PPAR agonists; biguanides; protein tyrosinephosphatase-IB (PTP-1 B) inhibitors; insulin and insulin mimetics; sulfonylureas and other insulin secretagogues; a-glucosidase inhibitors; glucagon receptor antagonists; GLP-1, GLP-1 mimetics, and GLP-1 receptor agonists; GIP, GIP mimetics, and GIP receptor agonists; PACAP, PACAP mimetics, and PACAP receptor 3 agonists; cholesterol lowering agents; HMG-CoA reductase inhibitors; sequestrants; nicotinyl alcohol; nicotinic acid or a salt thereof; PPARa agonists; PPARoly dual agonists; inhibitors of cholesterol absorption; acyl CoA : cholesterol acyltransferase inhibitors; anti-oxidants; PPARo agonists; antiobesity
- pharmaceutically acceptable salts refers to salts prepared from pharmaceutically acceptable non-toxic bases or acids, including inorganic bases or acids and organic bases or acids.
- the compounds of formula (I) or (Ia) can be combined as the active ingredient in intimate admixture with a pharmaceutical carrier according to conventional pharmaceutical compounding techniques.
- the carrier may take a wide variety of forms depending on the form of preparation desired for administration, e . g ., oral or parenteral (including intravenous).
- any of the usual pharmaceutical media may be employed, such as, for example, water, glycols, oils, alcohols, flavoring agents, preservatives, coloring agents and the like in the case of oral liquid preparations, such as, for example, suspensions, elixirs and solutions; or carriers such as starches, sugars, microcrystalline cellulose, diluents, granulating agents, lubricants, binders, disintegrating agents and the like in the case of oral solid preparations such as, for example, powders, hard and soft capsules and tablets, with the solid oral preparations being preferred over the liquid preparations.
- oral liquid preparations such as, for example, suspensions, elixirs and solutions
- carriers such as starches, sugars, microcrystalline cellulose, diluents, granulating agents, lubricants, binders, disintegrating agents and the like in the case of oral solid preparations such as, for example, powders, hard and soft capsules and tablets, with the solid oral preparations being preferred over the liquid preparation
- tablets and capsules represent the most advantageous oral dosage unit form in which case solid pharmaceutical carriers are obviously employed. If desired, tablets may be coated by standard aqueous or nonaqueous techniques. Such compositions and preparations should contain at least 0.1 percent of active compound. The percentage of active compound in these compositions may, of course, be varied and may conveniently be between about 2 percent to about 60 percent of the weight of the unit. The amount of active compound in such therapeutically useful compositions is such that an effective dosage will be obtained.
- the active compounds can also be administered intranasally as, for example, liquid drops or spray.
- the tablets, pills, capsules, and the like may also contain a binder such as gum tragacanth, acacia, com starch or gelatin; excipients such as dicalcium phosphate; a disintegrating agent such as com starch, potato starch, alginic acid; a lubricant such as magnesium stearate; and a sweetening agent such as sucrose, lactose or saccharin.
- a dosage unit form is a capsule, it may contain, in addition to materials of the above type, a liquid carrier such as a fatty oil.
- tablets may be coated with shellac, sugar or both.
- a syrup or elixir may contain, in addition to the active ingredient, sucrose as a sweetening agent, methyl and propylparabens as preservatives, a dye and a flavoring such as cherry or orange flavor.
- Compounds of formula (I) or (Ia) may also be administered parenterally. Solutions or suspensions of these active compounds can be prepared in water suitably mixed with a surfactant such as hydroxy-propylcellulose. Dispersions can also be prepared in glycerol, liquid polyethylene glycols and mixtures thereof in oils. Under ordinary conditions of storage and use, these preparations contain a preservative to prevent the growth of microorganisms.
- the pharmaceutical forms suitable for injectable use include sterile aqueous solutions or dispersions and sterile powders for the extemporaneous preparation of sterile injectable solutions or dispersions.
- the form must be sterile and must be fluid to the extent that easy syringability exists. It must be stable under the conditions of manufacture and storage and must be preserved against the contaminating action of microorganisms such as bacteria and fungi.
- the carrier can be a solvent or dispersion medium containing, for example, water, ethanol, polyol (e.g., glycerol, propylene glycol and liquid polyethylene glycol), suitable mixtures thereof, and vegetable oils.
- Any suitable route of administration may be employed for providing a mammal, especially a human, with an effective dose of a compound of the present invention.
- oral, rectal, topical, parenteral, ocular, pulmonary, nasal, and the like may be employed.
- Dosage forms include tablets, troches, dispersions, suspensions, solutions, capsules, creams, ointments, aerosols, and the like.
- compounds of formula (I) or (la) are administered orally.
- the effective dosage of active ingredient employed may vary depending on the particular compound employed, the mode of administration, the condition being treated and the severity of the condition being treated. Such dosage may be ascertained readily by a person skilled in the art.
- the compounds of the present invention are administered at a daily dosage of from about 0.1 milligram to about 100 milligram per kilogram of animal body weight, preferably given as a single daily dose or in divided doses two to six times a day, or in sustained release form.
- the total daily dosage is from about 1.0 milligrams to about 1000 milligrams, preferably from about 1 milligrams to about 50 milligrams. In the case of a 70 kg adult human, the total daily dose will generally be from about 7 milligrams to about 350 milligrams. This dosage regimen may be adjusted to provide the optimal therapeutic response.
- the compounds of formula (I) of the present invention can be prepared from beta amino acid intermediates such as those of formula (IV) and substituted amine intermediates such as those of formula (III), using standard peptide coupling conditions.
- the preparation of these intermediates is described in the following schemes.
- Available starting materials may be amines having the formula (III).
- Enantiomerically pure beta amino acids having the formula (IV) may be commercially available, known in the literature or may be conveniently synthesized using one of the methods already published and reviewed in e.g., Cole, Tetrahedron, 32, 9517 (1994 ), Juaristi et al., Aldrichimica Acta, 27, 3, 1994 , or Juaristi, Enantioselective Synthesis of ⁇ -Amino Acids, Ed. Wiley-VCH, New York, 1997 .
- 3-amino-4-(2,4,5-trifluoro-phenyl)-butyric acid may be synthesized by a variety of methods as reported in the patent applications WO 2004069162 , WO 2004064778 , WO 2004037169 , WO 2004032836 and in the articles JACS, 126, 3048 (2004 ) and JACS, 126, 9918 (2004 ).
- compounds having the formula (I) wherein the variables have the above described meanings may be prepared using standard peptide coupling conditions, reagents and protective groups.
- EDC 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride
- HOBt 1-hydroxybenzotriazole
- a base triethylamine or diisopropylethylamine
- solvents such as methylene chloride or N,N-dimethylformamide.
- Scheme H outlines a procedure for using the amines formed according to Schemes A through G to synthesize compounds that are embodiments of the invention.
- the protective group may be removed with, for example, diethylamine in dichloromethane in the case of 9-fluorenylmethoxycarbonyl or using acidic conditions (such as trifluoroacetic acid in dichloromethane or hydrochloric acid in dioxane) in the case of tert-butoxycarbonyl, as described in Protective Groups in Organic Synthesis 3rd ed., Ed. Wiley-VCH, New York; 1999 .
- acidic conditions such as trifluoroacetic acid in dichloromethane or hydrochloric acid in dioxane
- tert-butoxycarbonyl as described in Protective Groups in Organic Synthesis 3rd ed., Ed. Wiley-VCH, New York; 1999 .
- flash chromatography on silica gel may be suitable for the free amines whereas the use of preparative HPLC leads to the isolation of the corresponding trifluoroacetic acid or formate salts.
- step 1 A solution of 20.0 mg (0.07 mmol) of (2S) -(benzoylamino-methyl)-pyrrolidine-1-carboxylic acid tert -butyl ester (step 1) in 1.0 mL of dichloromethane and 0.5 mL of trifluoroacetic acid is stirred at room temperature for 30 minutes and then evaporated under reduced pressure to give the title compound.
- Boc-protected compound (Step 1): 1.19 (m, 2H), 1.21 (m, 2H), 1.40 (s, 9H), 1.60-1.85.(m, 4H), 2.99-3.22 (m, 3H), 3.38-3.52 (m, 1 H), 3.73-3.90 (m, 1H). 7.91 (bs, 1H). 6 LC/MS (11) rt 1.30 m/z 183 (M+H) + . 8 LC/MS (II) rt 1.58 m/z 191 (M+H) +
- step 1 A solution of 300 mg (1.03 mmol) of (2S) -benzyloxymethyl-pyrrolidine-1-carboxylic acid tert- butyl ester (step 1) in 1.5 mL of dichloromethane and 1.5 mL of trifluoroacetic acid is stirred at room temperature for 1 h and then evaporated under reduced pressure. The crude mixture is diluted in 5 mL of dichloromethane and stirred for 1 h with 1.43 g (4.12 mmol) of (polystyrylmethyl)trimethylammonium bicarbonate, then filtered and evaporated under reduced pressure to give the title compound.
- 24 Boc-protected compound (step 1) LC/MS (II) rt 3.90, m/z 332 (M+H-CH 3 ) + .
- step 1 To a solution of 45 mg (0.15 mmol) of 2-(cyclopropanesulfonylamino-methyl)-pyrrolidine-1-carboxylic acid tert-butyl ester (step 1, example 18) in 1 mL of tetrahydrofuran, 7.1 mg (0.30 mmol) of sodium hydride in 0.5 mL THF is added and the reaction is stirred 5 minutes. 14 ⁇ l (0.22 mmol) of methyliodide are added slowly and reaction is stirred overnight.
- step 1 A solution of 253 mg (approx. 0.37 mmol) of (2S)-[(3-phenyl-ureido)-methyl]-pyrrolidine-1-carboxylic acid tert -butyl ester (step 1) in 0.5 mL of trifluoroacetic acid and 1.0 mL of dichloromethane is stirred at room temperature for 2 h and then evaporated , under reduced pressure.
- the crude mixture is dissolved in 2 mL of a 1M ammonia solution in methanol, concentrated under reduced pressure and then purified using flash chromatography (aluminium oxide, eluent: 0% to 10% methanol in dichloromethane containing 0.1% of ammonia) to give the title compound.
- step 2 To a solution of 94 mg (0.5 mmol) Boc protected hydroxymethyl azetidine (step 2) in 5 mL of THF was added 354 mg (0.5 mmol) fluorous triphenyl phosphine and 47 mg (0.5 mmol) phenol. The mixture was cooled down to 0 °C and 405 mg (0.5 mmol) fluorous diethyl azodicarboxylate (DEAD) was added and allowed for warm up to room temperature. The reaction was stirred for 3 days, evaporated to dryness over 1 g of alumina. Alumina containing the reaction product was placed over fluorous silica cartridge and washed with methanol:water 4:1 eluent (4 x 1 mL).
- DEAD diethyl azodicarboxylate
- step 3 A solution of 34.0 mg (0.13 mmol) of 3-phenoxymethyl-azetidine-1-carboxylic acid tert-butyl ester (step 3) in 300 ⁇ L of trifluoroacetic acid and 300 ⁇ L of dichloromethane is stirred at room temperature for 30 minutes and then evaporated under reduced pressure to give the title compound.
- step 2 73 mg (0.35 mmol) of 3-azidomethyl-azetidine-1-carboxylic acid tert-butyl ester (step 2) dissolved in 20 mL methanol, 1 mL ammonia (2M in MeOH) and Pd/C (5% with 50% water) added and the mixture stirred at 1 atm H 2 for 1 h. Filtration over Celite and evaporation of the solvent affords the crude amine that is taken directly to the next step. LC/MS (IV) rt 1.75, m/z 172 (M+H- CH 3 ) + .
- step 3 39 mg (0.21 mmol) of 3-aminomethyl-azetidine-1-carboxylic acid tert-butyl ester (step 3) and 32 ⁇ l (0.25 mmol) triethylamine are dissolved in dichloromethane and 17 ⁇ l (0.23 mmol) of benzenesulfonylchloride added at 0°C .
- the reaction mixture is subsequently stirred for 1h and diluted with dichloromethane.
- the organic layer is washed with 5% citric acid, saturated sodium bicarbonate solution and brine and dried over sodium sulphate.
- step 3 Obtained from the product of step 3 and 3-tert butoxycarbonylamino-4-(2-fluorophenyl)-butyric acid according to the procedure described for step 1 in example 32.
- step 1 The product of step 1 is dissolved In 1 mL of 4N hydrochloric acid in dioxane. The solution is stirred for 1 hour at room temperature and the solvent is evaporated under reduced pressure. The crude material Is redissolved in methanol and the solvent Is evaporated under reduced pressure to give the title compound. LC/MS (II) rt 2.09, m/z 382 (M+H) + .
- the solution is cooled to room temperature, washed successively with a 5 % citric acid solution, saturated sodium hydrogen carbonate solution, water and brine, dried over magnesium sulphate, filtered and the solvent is removed under reduced pressure.
- the residue is subjected to preparative thin layer chromatography on silica gel (eluent: dichloroethane/ethanol 5:1) to afford the title compound which is taken directly into the next step without further characterisation.
- Example 101 Obtained from (2S)-[(3-Pyridin-2-yl-[1,2,4]oxadiazol-5-ylamino)-methyl]-pyrrolidine-1-carboxylic acid tert -butyl ester (Example 101, Step 1), and synthesised according to the procedure for Example 100, Step 2.
- Example 101 Obtained from (3-pyridin-2-yl-[1,2,4]oxadiazol-5-yl)-pyrrolidin-(2S)-ylmethylamine di-hydrochloride (Example 101, Step 2) and (3R)-tert-butoxycarbonylamino-4-(2-fluorophenyl)-butyric acid, and synthesised according to Example 100, Step 3.
- Example 103 Obtained from [3-(3-fluorophenyl)-[1,2,4]oxadiazol-5-yl]-pyrrolidin-(2S)-ylmethylamine hydrochloride (Example 103, Step 2) and (3R)-tert-butoxycarbonylamino-4-(2-fluorophenyl)-butyric acid, and synthesised according to Example 100, Step 3.
- Example 104 Obtained from (2S)- ⁇ [3-(4-methanesulphonylphenyl)-[1,2,4]oxadiazol-5-ylamino]-methyl ⁇ -pyrrolidine-1-carboxylic acid tert- butyl ester (Example 104, Step 1), and synthesised according to the procedure for Example 100, Step 2.
- DPP-IV peptidase activity was monitored with a continuous fluorimetric assay.
- This assay is based on the cleavage of the substrate Gly-Pro-AMC (Bachem) by DPP-IV, releasing free AMC.
- the assay is carried out in 96-well microtiterplates. In a total volume of 100 ⁇ L, compounds are preincubated with 50 pM DPP-IV employing a buffer containing 10mM Hepes, 150mM NaCl, 0.005% Tween 20 (pH 7.4).
- the reaction is started by the addition of 16 ⁇ M substrate and the fluorescence of liberated AMC is detected for 10 minutes at 25 °C with a fluorescence reader (BMG-Fluostar; BMG-Technologies) using an excitation wavelength of 370 nm and an emission wavelength of 450 nm.
- the final concentration of DMSO is 1 %.
- the inhibitory potential of the compounds were determined.
- DPP-IV activity assays were carried out with human and porcine DPP-IV (see below); both enzymes showed comparable activities-include.
- Soluble human DPP-IV lacking the transmembrane anchor (Gly31-Pro766) was expressed in a recombinant YEAST-strain as Pre-Pro-alpha-mating fusion.
- the secreted product (rhuDPP-IV-Gly31-Pro766) was purified from fermentation broth (>90% purity) and used for inhouse screening.
- IC 50 values for inhibition of DPP-IV peptidase activity determined in assays as described above.
- the IC 50 values were grouped in 3 classes: a ⁇ 100 nM; b ⁇ 101 nM and ⁇ 1001 nM; c ⁇ 1001 nM ⁇ 2000 nM.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmacology & Pharmacy (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Diabetes (AREA)
- Hematology (AREA)
- Obesity (AREA)
- Urology & Nephrology (AREA)
- Physical Education & Sports Medicine (AREA)
- Neurosurgery (AREA)
- Neurology (AREA)
- Endocrinology (AREA)
- Biomedical Technology (AREA)
- Cardiology (AREA)
- Heart & Thoracic Surgery (AREA)
- Immunology (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Virology (AREA)
- Rheumatology (AREA)
- Communicable Diseases (AREA)
- Tropical Medicine & Parasitology (AREA)
- Oncology (AREA)
- Molecular Biology (AREA)
- Pain & Pain Management (AREA)
- Psychiatry (AREA)
- Emergency Medicine (AREA)
- Ophthalmology & Optometry (AREA)
- Reproductive Health (AREA)
- Vascular Medicine (AREA)
Claims (24)
- Eine Verbindung der Formel (I)
Z ausgewählt ist aus der Gruppe bestehend aus
Phenyl;
Naphthyl;
C3-7-Cycloalkyl;
Heterocyclus; und
Heterobicyclus;worin Z optional substituiert ist mit einem oder, unabhängig voneinander, mehreren ausHalogen;CN;OH;=O worin der Ring zumindest teilweise gesättigt ist;C1-6-Alkyl, optional mit einem oder mehreren F substituiert; undO-C1-6-Alkyl, optional mit einem oder mehreren F substituiert;R1, R2, R4, R5 unabhängig voneinander ausgewählt sind aus der Gruppe bestehend aus
H;
F;
OH;
C1-6-Alkyl, optional mit einem oder mehreren F substituiert; und
O-C1-6-Alkyl, optional mit einem oder mehreren F substituiert;
und/oder R1 und R2 optional zusammen ein C3-7Cycloalkyl bilden, welches optional mit einem oder mehreren F substituiert ist;
und/oder R2 und R3 optional zusammen ein C3-7-Cycloalkyl bilden, welches optional mit einem oder mehreren F substituiert ist;
und/oder R3 und R4 optional zusammen ein C3-7-Cycloalkyl bilden, welches optional mit einem oder mehreren F substituiert ist;
und/oder R4 und R5 optional zusammen ein C3-7Cycloalkyl bilden, welches optional mit einem oder mehreren F substituiert ist;
R3 H oder C1-6-Alkyl ist;
X ausgewählt ist aus der Gruppe bestehend aus
H;
F; und
C1-6-Alkyl, optional mit einem oder mehreren F substituiert;
n ist 0 oder 1;
A1, A2 unabhängig voneinander ausgewählt sind aus der Gruppe bestehend aus
H;
Halogen;
C1-6-Alkyl, optional mit einem oder mehreren F substituiert; und
R6; mit der Maßgabe, dass eines von A1 und A2 R6 ist;
R6 -C(R7R8)-Y- T ist;
R7, R8 unabhängig voneinander ausgewählt sind aus der Gruppe bestehend aus
H;
F; und
C1-6-Alkyl, optional mit einem oder mehreren F substituiert;
und/oder R7 und R8 optional zusammen ein C3-7Cycloalkyl bilden, welches optional mit einem oder mehreren F substituiert ist;
Y ausgewählt ist aus der Gruppe bestehend aus
-O-;
-C1-6-Alkyl-O-;
-N(R9)-;
-C1-6-Alkyl-N(R9)-
-S-;
-C1-6-Alkyl-S-;
-S(O)-;
-C1-6-Alkyl-S(O)-;
-S(O)2-; und
-C1-6-Alkyl-S(O)2-;worin jedes C1-6-Alkyl optional mit einem oder mehreren F substituiert ist;R9, T unabhängig voneinander T1- T2 oder T2 sind;
T1 ausgewählt ist aus der Gruppe bestehend aus
-C1-6-Alkyl-;
-C1-6-Alkyl-O-;
-C1-6-Alkyl-N(R10)-;
-C(O)-;
-C(O)-C1-6-Alkyl-;
-C(O)-C1-6-Alkyl-O-;
-C(O)-C1-6-Alkyl-N(R10)-,
-C(O)O-;
-C(O)O-C1-6-Alkyl-;
-C(O)O-C1-6-Alkyl-O-;
-C(O)O-C1-6-Alkyl-N(R10)-;
-C(O)N(R10)-;
-C(O)N(R10)-C1-6-Alkyl-;
-C(O)N(R10)-C1-6-Alkyl-O-;
-C(O)N(R10)-C1-6-Alkyl-N(R11)-;
-S(O)2-;
-S(O)2-C1-6-Alkyl-;
-S(O)2-C1-6-Alkyl-O-; und
-S(O)2-C1-6-Alkyl-N(R10)-;worin jedes C1-6-Alkyl optional mit einem oder mehreren F substituiert ist;R10, R11 unabhängig voneinander H oder C1-6-Alkyl, optional mit einem oder mehreren F substituiert, sind;
T2 ausgewählt ist aus der Gruppe bestehend aus
H;
CF3;
Phenyl;
Naphthyl;worin Phenyl und Naphthyl optional substituiert sind mit einem oder, unabhängig voneinander, mehreren ausHalogen;CN;R12;COOH;OH;C(O)NH2;S(0)2NH2;COOT3;OT3;C(O)NHT3;S(O)2NHT3; oderT3;C3-7-Cycloakyl;
Heterocyclus; und
Heterobicyclus;worin C3-7-Cycloalkyl, Heterocyclus und Heterobicyclus optional mit einem oder, unabhängig voneinander, mehreren ausHalogen;CN;R13;OH;=O, worin der Ring zumindest teilweise gesättigt ist;NH2COOH;C(O)NH2;S(O)2NH2;COOT3;OT3;C(O)NHT3;S(O)2NHT3;NHT3; oderT3;worin, wenn R9 T1- T2 ist und C1-6-Alkyl darstellt und T T1 - T2 ist und C1-6-Alkyl darstellt, dann können R9 und T zusammen eine 3- bis 7-gliedrige zyklische Gruppe enthaltend 1 N bilden;
R12 ausgewählt ist aus der Gruppe bestehend aus
C1-6-Alkyl;
O-C1-6-Alkyl;
COO-C1-6-Alkyl;
OC(O)-C1-6-Alkyl;
C(O)N(R15)-C1-6-Alkyl;
S(O)2N(R17)-C1-6-Alkyl;
S(O)-C1-6-Alkyl;
S(O)2-C1-6-Alkyl; und
N(R18)S(O)2-C1-6-Alkyl;worin jedes C1-6-Alkyl optional mit einem oder, unabhängig voneinander, mehreren aus F, COOR19, C(O)N(R20R21), S(O)2N(R22R23), OR24,N(R25R26), T3, O-T3 oder N(R27)-T3 substituiert ist;R13 ausgewählt ist aus der Gruppe bestehend aus
C1-6-Alkyl;
O-C1-6-Alkyl;
N(R14)-C1-6-Alkyl;
COO-C1-6-Alkyl;
OC(O)-C1-6-Alkyl;
C(O)N(R15)-C1-6-Alkyl;
N(R16)-C(O)-C1-6-Alkyl;
S(O)2N(R17)-C1-6-Alkyl;
S(O)-C1-6-Alkyl;
S(O)2-C1-6-Alkyl; und
-N(R18)S(O)2-C1-6-Alkyl;worin jedes C1-6-Alkyl optional mit einem oder, unabhängig voneinander, mehreren aus F, COOR19, C(O)N(R20R21), S(O)2N(R22R23), OR24, N(R25R26), T3, O-T3 oder N(R27)-T3 substituiert ist;R14, R15, R16, R17, R18, R19, R20, R21, R22, R23, R24, R25, R26, R27 unabhängig voneinander H oder C1-6-Alkyl sind;
T3 ausgewählt ist aus der Gruppe bestehend aus
Phenyl;
Naphthyl;worin Phenyl und Naphthyl optional mit einem oder, unabhängig voneinander, mehreren substituiert sind ausHalogen;CN;COOH;OH;C(0)NH2;S(0)2NH2;C1-6-Alkyl;O-C1-6-Alkyl;COO-C1-6-Alkyl;OC(O)-C1-6-Alkyl;C(O)N(R28)-C1-6-Alkyl;S(O)2N(R29)-C1-6-Alkyl;S(O)2-C1-6-Alkyl; oderN(R30)S(O)2-C1-6-Alkyl;Heterocyclus;
Heterobicyclus; und
C3-7Cycloalkyl;worin C3-7-Cycloalkyl, Heterocylus und Heterobicyclus optional substituiert sind mit einem oder mehreren ausHalogen,CN;OH;=O, worin der Ring zumindest teilweise gesättigt ist;NH2COOH;C(O)NH2;S(O)2NH2;C1-6-Alkyl;O-C1-6-Alkyl;N(R31)-C1-6-Alkyl;COO-C1-6-Alkyl;OC(O)-C1-6-Alkyl;C(O)N(R32)-C1-6-Alkyl;N(R33)-C(O)-C1-6-Alkyl;S(O)2N(R34)-C1-6-Alkyl;S(O)2-C1-6-Alkyl; oder-N(R35)S(O)2-C1-6-Alkyl. - Verbindung gemäß Anspruch 1 nach Formel (la)

- Verbindung gemäß Anspruch 1 oder 2, worin Z Phenyl oder Heterocyclus ist und Z optional unabhängig voneinander mit bis zu 2 aus Cl, F, CN, CH3 oder OCH3 substituiert ist.
- Verbindung gemäß einem der vorangehenden Ansprüche, worin R1, R2, R4, R5 unabhängig voneinander ausgewählt sind aus der Gruppe bestehend aus H, F, OH, CH3, OCH3.
- Verbindung gemäß einem der vorangehenden Ansprüche, worin R3 H ist.
- Verbindung gemäß einem der vorangehenden Ansprüche, worin X H, F oder CH3 ist.
- Verbindung gemäß einem der vorangehenden Ansprüche, worin n 1 ist.
- Verbindung gemäß einem der vorangehenden Ansprüche, worin A1 R6 ist und A2 H, F oder CH3 ist.
- Verbindung gemäß einem der vorangehenden Ansprüche, worin R6 -CH2-X-T ist.
- Verbindung gemäß einem der vorangehenden Ansprüche, worin Y -O-, -N(R9)- oder -S(O)2- ist.
- Verbindung gemäß einem der vorangehenden Ansprüche, worin R9 ausgewählt ist aus der Gruppe bestehend aus H, CH3, COOH, COOCH3, C(O)NH2, C(O)N(CH3)2 und S(O)2CH3 .
- Verbindung gemäß einem der vorangehenden Ansprüche, worin T T1-T2 oder T2 ist und worin T1 ausgewählt ist aus der Gruppe bestehend aus-CH2-;-C(O)-;-C(O)-CH2-;-C(O)O-;-C(O)O-CH2-;-C(O)NH-;-C(O)NH-CH2-;-S(O)2-; und-S(O)2-CH2-.
- Verbindung gemäß Anspruch 12, worin T T1-T2 oder T2 ist und worin T1 ausgewählt ist aus der Gruppe bestehend aus -C(O)-; -CH2-; -S(O)2-; and -C(O)NH-.
- Verbindung gemäß einem der vorangehenden Ansprüche, worin R6-CH2-N(R36)-T ist und worin R36 H oder S(O)2CH3 ist.
- Verbindung gemäß einem der vorangehenden Ansprüche, worin T2 Phenyl oder Heterocyclus ist.
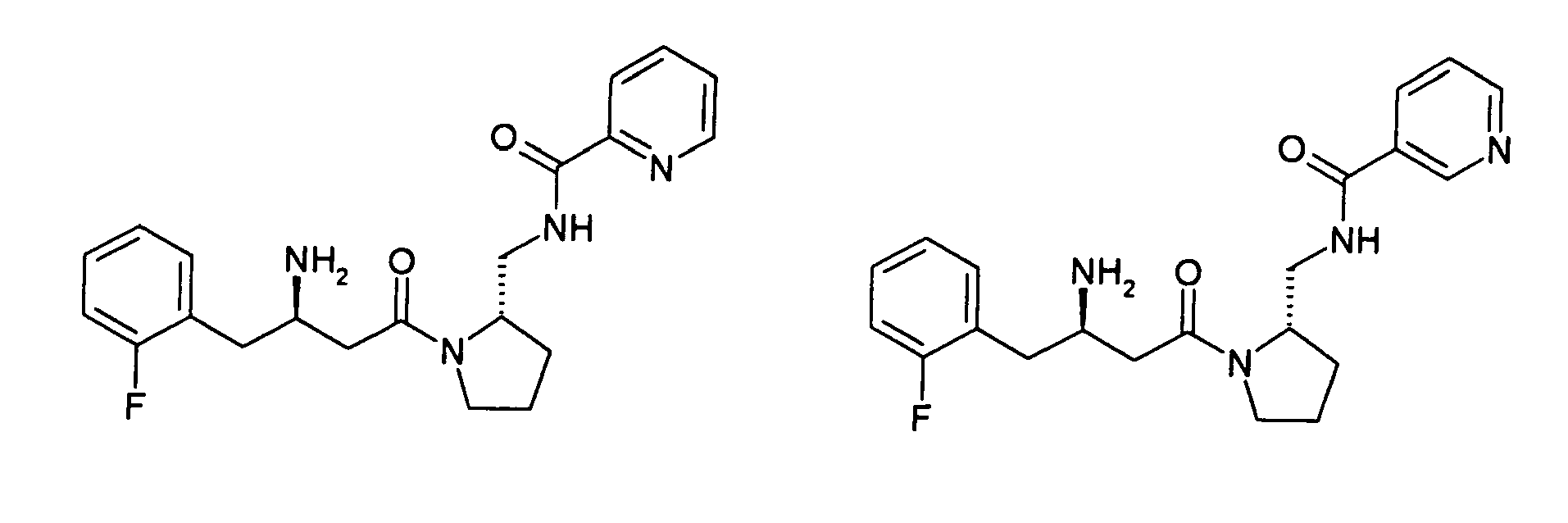
- Verbindung gemäß Anspruch 1 ausgewählt aus der Gruppe bestehend aus




























- Eine Prodrug-Verbindung einer Verbindung gemäß einem der Ansprüche 1 bis 16, worin die Aminogruppe der Formel (I) acyliert, alkyliert oder phosphoryliert ist.
- Eine pharmazeutische Zusammensetzung umfassend eine Verbindung oder ein pharmazeutisch akzeptables Salz davon gemäß einem der Ansprüche 1 bis 17 zusammen mit einem pharmazeutisch akzeptablen Träger.
- Pharmazeutische Zusammensetzung gemäß Anspruch 18, umfassend eine oder mehrere zusätzliche Verbindungen oder pharmazeutisch akzeptable Salze davon, ausgewählt aus der Gruppe bestehend aus einer weiteren Verbindung gemäß einem der Ansprüche 1 bis 17; einem weiteren DPP-IV-Inhibitor; Insulin-Sensibilisatoren; PPAR-Agonisten; Biguanide; Protein-Tyrosinphosphatase-IB (PTP-1 B)-Inhibitoren; Insulin und Insulin-Mimetika; Sulfonylharnstoffe und weitere Insulin-Sekretationsstimulanzien; a-Glucosidase-Inhibitoren; Glukagon-Rezeptor-Antagonisten; GLP-1, GLP-1-Mimetika und GLP-1-Rezeptor-Agonisten; GIP, GIP-Mimetika und GIP-Rezeptor-Agonisten; PACAP, PACAP-Mimetika und PACAP-Rezeptor-3-Agonisten; Cholesterin-senkende Mittel; HMG-CoA-Reduktase-Inhibitoren; Komplexbildner; Nikotinalkohol; Nikotinsäure oder ein Salz davon; PPARa-Agonisten; PPARoly-Dualagonisten; Inhibitoren der Cholesterinabsorption; Acyl-CoA : Cholesterinacyltransferaseinhibitoren; Antioxidantien; PPARo-Agonisten; Verbindungen gegen Fettleibigkeit; ein Inhibitor des Ileal-Gallensäuretransporters; und antientzündliche Mittel.
- Verbindung oder ein pharmazeutisch akzeptables Salz davon gemäß einem der Ansprüche 1 bis 17 zur Verwendung als ein Arzneimittel.
- Verwendung einer Verbindung oder eines pharmazeutisch akzeptablen Salzes davon nach einem der Ansprüche 1 bis 17 für die Herstellung eines Arzneimittels zur Behandlung oder Prophylaxe von nicht-Insulin-abhängigen (Typ II) Diabetes mellitus; Hyperglykämie; Fettleibigkeit; Insulinresistenz; Lipid-Störungen; Dyslipidämie; Hyperlipidämie, Hypertriglyceridämie; Hypercholesterinämie; niedriges HDL; hohes LDL; Artherosklerose; Wachstumshormondefizienz; Krankheiten verbunden mit einer Immunantwort; HIV-Infektion; Neutropenie; neuronale Krankheiten; Angstzustände; Depression; Tumormetastasen; gutartige Prostata-Hypertrophy; Gingivitis; Bluthochdruck; Osteoporose; Krankheiten verbunden mit Spermabeweglichkeit; niedrige Glukose-Toleranz; Insulinresistenz; ist Sequelae; vaskuläre Restinose; Reizdarm-Syndrom; entzündliche Darmkrankheit; beinhaltend Crohn's Krankheit und Dickdarmentzündung; weitere entzündliche Bedingungen; Pankreatitis; Bierbauch; neurodegenerative Krankheiten; Retinopathy; Nephropathy; Neuropathy; Syndrom X; polycystisches Ovarien-Syndrom; Typ n Diabetes; oder Wachstumshormon-Defizienz.
- Verfahren zur Herstellung einer Verbindung gemäß einem der Ansprüche 1 bis 17, umfassend die Schritte von• Kuppeln einer aminogeschützten Beta-Aminosäure der Formel (IVa)

 • Entfernen der Schutzgruppe (PG).
• Entfernen der Schutzgruppe (PG). - Verfahren gemäß Anspruch 22, worin die Kupplungsreagenzien 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimidhydrochlorid (EDC) in Kombination mit 1-Hydroxybenzotriazol (HOBt) und einer Base (Triethylamin oder Diisopropylethylamin) oder O-(7-azabenzotriazol-1-yl)-N,N,N',N'-tetramethyluroniumhexafluorphosphat (HATU) sind in der Gegenwart einer Base und in die Schutzgruppe 9-Fluorenylmethoxycarbonyl oder tert-Butoxycarbonyl ist.
- Verfahren gemäß Anspruch 22 oder 23, worin die Schutzgruppe unter Verwendung von Diethylamin in Dichlormethan im Falle von 9-Fluorenylmethoxycarbonyl entfernt wird oder unter Verwendung von sauren Bedingungen im Falle von tert-Butoxycarbonyl.
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP04803694A EP1613304B1 (de) | 2003-12-09 | 2004-12-09 | Dpp-iv-hemmer |
| SI200430469T SI1613304T1 (sl) | 2003-12-09 | 2004-12-09 | Dpp-iv zaviralci |
| PL04803694T PL1613304T3 (pl) | 2003-12-09 | 2004-12-09 | Inhibitory DPP-IV |
| CY20071101404T CY1106969T1 (el) | 2003-12-09 | 2007-10-30 | Αναστολεις dpp-iv |
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP03028211A EP1541143A1 (de) | 2003-12-09 | 2003-12-09 | Dpp-iv inhibitoren |
| EP04803694A EP1613304B1 (de) | 2003-12-09 | 2004-12-09 | Dpp-iv-hemmer |
| PCT/EP2004/014040 WO2005056003A1 (en) | 2003-12-09 | 2004-12-09 | Dpp-iv inhibitors |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| EP1613304A1 EP1613304A1 (de) | 2006-01-11 |
| EP1613304B1 true EP1613304B1 (de) | 2007-09-12 |
Family
ID=34486147
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| EP03028211A Withdrawn EP1541143A1 (de) | 2003-12-09 | 2003-12-09 | Dpp-iv inhibitoren |
| EP04803694A Expired - Lifetime EP1613304B1 (de) | 2003-12-09 | 2004-12-09 | Dpp-iv-hemmer |
Family Applications Before (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| EP03028211A Withdrawn EP1541143A1 (de) | 2003-12-09 | 2003-12-09 | Dpp-iv inhibitoren |
Country Status (23)
| Country | Link |
|---|---|
| US (1) | US20080027035A1 (de) |
| EP (2) | EP1541143A1 (de) |
| JP (1) | JP2007513910A (de) |
| KR (1) | KR100845397B1 (de) |
| CN (1) | CN1905868A (de) |
| AT (1) | ATE372767T1 (de) |
| AU (1) | AU2004296553B2 (de) |
| BR (1) | BRPI0417458A (de) |
| CA (1) | CA2548742A1 (de) |
| CY (1) | CY1106969T1 (de) |
| DE (1) | DE602004008895T2 (de) |
| DK (1) | DK1613304T3 (de) |
| ES (1) | ES2291966T3 (de) |
| HR (1) | HRP20070560T3 (de) |
| IL (1) | IL176062A0 (de) |
| MX (1) | MXPA06006652A (de) |
| NO (1) | NO20062644L (de) |
| PL (1) | PL1613304T3 (de) |
| PT (1) | PT1613304E (de) |
| RU (1) | RU2006119302A (de) |
| SI (1) | SI1613304T1 (de) |
| WO (1) | WO2005056003A1 (de) |
| ZA (1) | ZA200604719B (de) |
Families Citing this family (39)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN1723196A (zh) | 2001-06-27 | 2006-01-18 | 史密丝克莱恩比彻姆公司 | 作为二肽酶抑制剂的氟代吡咯烷 |
| EP1589969A4 (de) | 2003-01-17 | 2008-08-13 | Merck & Co Inc | 3-amino-4-phenylbutansäurederivateals dipeptidylpeptidase-hemmer zur behandlung oder vorbeugung von diabetes |
| CA2513684A1 (en) | 2003-01-31 | 2004-08-19 | Merck & Co., Inc. | 3-amino-4-phenylbutanoic acid derivatives as dipeptidyl peptidase inhibitors for the treatment or prevention of diabetes |
| US7560455B2 (en) | 2003-05-14 | 2009-07-14 | Merck & Co., Inc. | 3-Amino-4-phenylbutanoic acid derivatives as dipeptidyl peptidase inhibitors for the treatment or prevention of diabetes |
| CN1798556A (zh) | 2003-06-06 | 2006-07-05 | 麦克公司 | 作为治疗或者预防糖尿病的二肽基肽酶抑制剂的稠合吲哚 |
| CN1809544A (zh) | 2003-06-17 | 2006-07-26 | 麦克公司 | 作为二肽基肽酶抑制剂用于治疗或预防糖尿病的环己基甘氨酸衍生物 |
| US7259160B2 (en) | 2003-07-31 | 2007-08-21 | Merck & Co., Inc. | Hexahydrodiazepinones as dipeptidyl peptidase-IV inhibitors for the treatment or prevention of diabetes |
| WO2005108382A1 (en) | 2004-05-04 | 2005-11-17 | Merck & Co., Inc. | 1,2,4-oxadiazole derivatives as dipeptidyl peptidase-iv inhibitors for the treatment or prevention of diabetes |
| WO2005116029A1 (en) | 2004-05-18 | 2005-12-08 | Merck & Co., Inc. | Cyclohexylalanine derivatives as dipeptidyl peptidase-iv inhibitors for the treatment or prevention of diabetes |
| WO2006098342A1 (en) * | 2005-03-16 | 2006-09-21 | Astellas Pharma Inc. | Piperazinyl compounds |
| EP1943215A2 (de) * | 2005-10-31 | 2008-07-16 | Brystol-Myers Squibb Company | Inhibitoren von dipeptidylpeptidase iv auf pyrrolidinyl-beta-aminoamidbasis und verfahren |
| GB0526291D0 (en) | 2005-12-23 | 2006-02-01 | Prosidion Ltd | Therapeutic method |
| US20090156465A1 (en) | 2005-12-30 | 2009-06-18 | Sattigeri Jitendra A | Derivatives of beta-amino acid as dipeptidyl peptidase-iv inhibitors |
| AU2007235876A1 (en) | 2006-04-12 | 2007-10-18 | Probiodrug Ag | Enzyme inhibitors |
| WO2008028662A1 (en) * | 2006-09-07 | 2008-03-13 | Santhera Pharmaceuticals (Schweiz) Ag | N-[1-(3-amino-4-phenyl-butyryl)-4-hydroxy-pyrrolidin-2-ylmethyl}-propionamide and related compounds as dpp-iv inhibitors for the treatment of type 2 diabetes mellitus |
| EP1905759A1 (de) * | 2006-09-07 | 2008-04-02 | Santhera Pharmaceuticals (Schweiz) AG | N-[1-(3-amino-4-phenyl-butyryl)-4-hydroxy-pyrrolidin-2-ylmethyl}-propionamid und verwandte verbindungen als dpp-iv inhibitoren zur behandlung von typ 2 diabetes mellitus |
| US8278345B2 (en) | 2006-11-09 | 2012-10-02 | Probiodrug Ag | Inhibitors of glutaminyl cyclase |
| ATE554085T1 (de) | 2006-11-30 | 2012-05-15 | Probiodrug Ag | Neue inhibitoren von glutaminylcyclase |
| KR20080071476A (ko) * | 2007-01-30 | 2008-08-04 | 주식회사 엘지생명과학 | 신규한 디펩티딜 펩티데이즈 iv(dpp-iv) 저해제 |
| EP2142514B1 (de) | 2007-04-18 | 2014-12-24 | Probiodrug AG | Thioharnstoffderivate als glutaminylcyclaseinhibitoren |
| EP2142519B1 (de) * | 2007-04-19 | 2014-09-24 | Dong-A Pharm.Co., Ltd. | Dpp-iv-inhibitor mit beta-aminogruppe, verfahren zu dessen herstellung und pharmazeutische zusammensetzung, die diesen enthält, zur prävention und behandlung von diabetes oder obesitas |
| US7860904B2 (en) * | 2007-04-24 | 2010-12-28 | Microsoft Corporation | Standalone execution of incomplete data flows |
| EP2019099A1 (de) * | 2007-07-02 | 2009-01-28 | Santhera Pharmaceuticals (Schweiz) AG | DPP-IV-Hemmer |
| WO2009003681A1 (en) * | 2007-07-02 | 2009-01-08 | Santhera Pharmaceuticals (Schweiz) Ag | Dpp-iv inhibitors |
| CN102838589B (zh) * | 2008-09-23 | 2014-03-26 | 成都地奥制药集团有限公司 | 经甲磺酰化的制备n取代的吡咯烷衍生物的方法 |
| CN101823987B (zh) * | 2009-03-06 | 2014-06-04 | 中国科学院上海药物研究所 | 一类新的dpp-ⅳ抑制剂及其制备方法和用途 |
| UA107360C2 (en) | 2009-08-05 | 2014-12-25 | Biogen Idec Inc | Bicyclic aryl sphingosine 1-phosphate analogs |
| JP5934645B2 (ja) | 2009-09-11 | 2016-06-15 | プロビオドルグ エージー | グルタミニルシクラーゼ阻害剤としてのヘテロ環式誘導体 |
| JP6026284B2 (ja) | 2010-03-03 | 2016-11-16 | プロビオドルグ エージー | グルタミニルシクラーゼの阻害剤 |
| NZ602312A (en) | 2010-03-10 | 2014-02-28 | Probiodrug Ag | Heterocyclic inhibitors of glutaminyl cyclase (qc, ec 2.3.2.5) |
| WO2011131748A2 (en) | 2010-04-21 | 2011-10-27 | Probiodrug Ag | Novel inhibitors |
| EA034451B1 (ru) | 2011-02-07 | 2020-02-10 | Байоджен Ма Инк. | Модуляторы s1p |
| EP2686313B1 (de) | 2011-03-16 | 2016-02-03 | Probiodrug AG | Benzimidazolderivate als glutaminylcyclase-hemmer |
| WO2012170702A1 (en) | 2011-06-08 | 2012-12-13 | Arena Pharmaceuticals, Inc. | Modulators of the gpr119 receptor and the treatment of disorders related thereto |
| US20160350201A1 (en) * | 2015-05-27 | 2016-12-01 | International Business Machines Corporation | Etl data flow design assistance through progressive context matching |
| EP3316877B1 (de) | 2015-07-02 | 2019-10-30 | Horizon Orphan LLC | Ado-resistente cysteamin-analoga und verwendungen davon |
| CN105362268A (zh) * | 2015-12-15 | 2016-03-02 | 上海壹志医药科技有限公司 | 脱亚甲基小檗碱的药物用途 |
| CN106526047A (zh) * | 2016-11-15 | 2017-03-22 | 迪沙药业集团有限公司 | R‑琥珀酸曲格列汀光学纯度的测定方法 |
| ES2812698T3 (es) | 2017-09-29 | 2021-03-18 | Probiodrug Ag | Inhibidores de glutaminil ciclasa |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6573287B2 (en) * | 2001-04-12 | 2003-06-03 | Bristo-Myers Squibb Company | 2,1-oxazoline and 1,2-pyrazoline-based inhibitors of dipeptidyl peptidase IV and method |
| ES2257555T3 (es) * | 2001-06-20 | 2006-08-01 | MERCK & CO., INC. | Inhibidores de dipeptidilpeptidasa para el tratamiento de la diabetes. |
| CA2450579A1 (en) * | 2001-06-20 | 2003-01-03 | Merck & Co., Inc. | Dipeptidyl peptidase inhibitors for the treatment of diabetes |
| DE10340800A1 (de) * | 2002-12-20 | 2004-07-01 | Merck Patent Gmbh | Funktionalisierung unreaktiver Substrate |
-
2003
- 2003-12-09 EP EP03028211A patent/EP1541143A1/de not_active Withdrawn
-
2004
- 2004-12-09 HR HR20070560T patent/HRP20070560T3/xx unknown
- 2004-12-09 MX MXPA06006652A patent/MXPA06006652A/es not_active Application Discontinuation
- 2004-12-09 US US10/582,054 patent/US20080027035A1/en not_active Abandoned
- 2004-12-09 WO PCT/EP2004/014040 patent/WO2005056003A1/en not_active Ceased
- 2004-12-09 PL PL04803694T patent/PL1613304T3/pl unknown
- 2004-12-09 AT AT04803694T patent/ATE372767T1/de not_active IP Right Cessation
- 2004-12-09 ES ES04803694T patent/ES2291966T3/es not_active Expired - Lifetime
- 2004-12-09 DE DE602004008895T patent/DE602004008895T2/de not_active Expired - Fee Related
- 2004-12-09 DK DK04803694T patent/DK1613304T3/da active
- 2004-12-09 KR KR1020067013752A patent/KR100845397B1/ko not_active Expired - Fee Related
- 2004-12-09 EP EP04803694A patent/EP1613304B1/de not_active Expired - Lifetime
- 2004-12-09 CN CNA2004800404476A patent/CN1905868A/zh active Pending
- 2004-12-09 CA CA002548742A patent/CA2548742A1/en not_active Abandoned
- 2004-12-09 PT PT04803694T patent/PT1613304E/pt unknown
- 2004-12-09 RU RU2006119302/04A patent/RU2006119302A/ru not_active Application Discontinuation
- 2004-12-09 AU AU2004296553A patent/AU2004296553B2/en not_active Ceased
- 2004-12-09 JP JP2006543480A patent/JP2007513910A/ja not_active Withdrawn
- 2004-12-09 BR BRPI0417458-5A patent/BRPI0417458A/pt not_active IP Right Cessation
- 2004-12-09 SI SI200430469T patent/SI1613304T1/sl unknown
-
2006
- 2006-05-31 IL IL176062A patent/IL176062A0/en unknown
- 2006-06-08 NO NO20062644A patent/NO20062644L/no not_active Application Discontinuation
- 2006-06-08 ZA ZA200604719A patent/ZA200604719B/xx unknown
-
2007
- 2007-10-30 CY CY20071101404T patent/CY1106969T1/el unknown
Also Published As
| Publication number | Publication date |
|---|---|
| MXPA06006652A (es) | 2007-02-20 |
| PT1613304E (pt) | 2007-11-14 |
| HRP20070560T3 (hr) | 2008-01-31 |
| SI1613304T1 (sl) | 2007-12-31 |
| PL1613304T3 (pl) | 2008-02-29 |
| IL176062A0 (en) | 2006-10-05 |
| DE602004008895D1 (en) | 2007-10-25 |
| HK1087022A1 (en) | 2006-10-06 |
| AU2004296553B2 (en) | 2008-09-04 |
| CN1905868A (zh) | 2007-01-31 |
| EP1613304A1 (de) | 2006-01-11 |
| WO2005056003A1 (en) | 2005-06-23 |
| BRPI0417458A (pt) | 2007-03-13 |
| CA2548742A1 (en) | 2005-06-23 |
| DK1613304T3 (da) | 2008-01-21 |
| ATE372767T1 (de) | 2007-09-15 |
| DE602004008895T2 (de) | 2008-06-19 |
| US20080027035A1 (en) | 2008-01-31 |
| ES2291966T3 (es) | 2008-03-01 |
| CY1106969T1 (el) | 2012-09-26 |
| KR20060108756A (ko) | 2006-10-18 |
| AU2004296553A1 (en) | 2005-06-23 |
| RU2006119302A (ru) | 2008-01-20 |
| EP1541143A1 (de) | 2005-06-15 |
| KR100845397B1 (ko) | 2008-07-09 |
| ZA200604719B (en) | 2007-12-27 |
| NO20062644L (no) | 2006-09-07 |
| JP2007513910A (ja) | 2007-05-31 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP1613304B1 (de) | Dpp-iv-hemmer | |
| AU2005229330B2 (en) | DPP-IV inhibitors | |
| US20080275086A1 (en) | Dpp-Iv Inhibitors | |
| US20080064728A1 (en) | Heterocyclic Compounds Useful as Dpp-Iv Inhibitors | |
| EP1604989A1 (de) | DPP-IV-Hemmer | |
| US20080176838A1 (en) | Dpp-IV Inhibitors | |
| EP1541148A1 (de) | Dpp-iv inhibitoren | |
| US20070265301A1 (en) | Dpp-IV Inhibitors | |
| WO2009003681A1 (en) | Dpp-iv inhibitors | |
| EP1791817A1 (de) | Dpp-iv-hemmer | |
| EP2019099A1 (de) | DPP-IV-Hemmer | |
| HK1087022B (en) | Dpp-iv inhibitors | |
| EP1905759A1 (de) | N-[1-(3-amino-4-phenyl-butyryl)-4-hydroxy-pyrrolidin-2-ylmethyl}-propionamid und verwandte verbindungen als dpp-iv inhibitoren zur behandlung von typ 2 diabetes mellitus | |
| KR20070030884A (ko) | 2형 진성 당뇨병의 치료를 위한 다이펩티딜 펩티다제iv(dpp-iv) 억제제로서의1-[(3r)-아미노-(4-2-플루오로-페닐)-뷰틸]-피롤리딘-(2r)-카복실산-벤질 아민 유도체 및 관련 화합물 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PUAI | Public reference made under article 153(3) epc to a published international application that has entered the european phase |
Free format text: ORIGINAL CODE: 0009012 |
|
| 17P | Request for examination filed |
Effective date: 20051024 |
|
| AK | Designated contracting states |
Kind code of ref document: A1 Designated state(s): AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IS IT LI LT LU MC NL PL PT RO SE SI SK TR |
|
| AX | Request for extension of the european patent |
Extension state: AL BA HR LV MK YU |
|
| RAP1 | Party data changed (applicant data changed or rights of an application transferred) |
Owner name: SANTHERA PHARMACEUTICALS (SCHWEIZ) GMBH |
|
| RAP1 | Party data changed (applicant data changed or rights of an application transferred) |
Owner name: SANTHERA PHARMACEUTICALS (SCHWEIZ) GMBH |
|
| REG | Reference to a national code |
Ref country code: HK Ref legal event code: DE Ref document number: 1087022 Country of ref document: HK |
|
| 17Q | First examination report despatched |
Effective date: 20051215 |
|
| RAP1 | Party data changed (applicant data changed or rights of an application transferred) |
Owner name: SANTHERA PHARMACEUTICALS (SCHWEIZ) AG |
|
| 17Q | First examination report despatched |
Effective date: 20051215 |
|
| GRAP | Despatch of communication of intention to grant a patent |
Free format text: ORIGINAL CODE: EPIDOSNIGR1 |
|
| GRAS | Grant fee paid |
Free format text: ORIGINAL CODE: EPIDOSNIGR3 |
|
| GRAA | (expected) grant |
Free format text: ORIGINAL CODE: 0009210 |
|
| AK | Designated contracting states |
Kind code of ref document: B1 Designated state(s): AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IS IT LI LT LU MC NL PL PT RO SE SI SK TR |
|
| AX | Request for extension of the european patent |
Extension state: AL BA HR LV MK YU |
|
| REG | Reference to a national code |
Ref country code: GB Ref legal event code: FG4D |
|
| REG | Reference to a national code |
Ref country code: CH Ref legal event code: EP |
|
| REG | Reference to a national code |
Ref country code: CH Ref legal event code: NV Representative=s name: BOVARD AG PATENTANWAELTE |
|
| REF | Corresponds to: |
Ref document number: 602004008895 Country of ref document: DE Date of ref document: 20071025 Kind code of ref document: P |
|
| REG | Reference to a national code |
Ref country code: IE Ref legal event code: FG4D |
|
| REG | Reference to a national code |
Ref country code: RO Ref legal event code: EPE Ref country code: SE Ref legal event code: TRGR |
|
| REG | Reference to a national code |
Ref country code: PT Ref legal event code: SC4A Free format text: AVAILABILITY OF NATIONAL TRANSLATION Effective date: 20071031 |
|
| REG | Reference to a national code |
Ref country code: GR Ref legal event code: EP Ref document number: 20070403081 Country of ref document: GR |
|
| REG | Reference to a national code |
Ref country code: HR Ref legal event code: TUEP Ref document number: P20070560 Country of ref document: HR |
|
| REG | Reference to a national code |
Ref country code: HR Ref legal event code: ODRP Ref document number: P20070560 Country of ref document: HR Payment date: 20071213 Year of fee payment: 4 |
|
| REG | Reference to a national code |
Ref country code: EE Ref legal event code: FG4A Ref document number: E001447 Country of ref document: EE Effective date: 20071030 |
|
| REG | Reference to a national code |
Ref country code: DK Ref legal event code: T3 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: CZ Payment date: 20071204 Year of fee payment: 4 Ref country code: DK Payment date: 20071220 Year of fee payment: 4 Ref country code: EE Payment date: 20071204 Year of fee payment: 4 Ref country code: ES Payment date: 20071227 Year of fee payment: 4 Ref country code: LU Payment date: 20071221 Year of fee payment: 4 Ref country code: MC Payment date: 20071220 Year of fee payment: 4 |
|
| REG | Reference to a national code |
Ref country code: HR Ref legal event code: T1PR Ref document number: P20070560 Country of ref document: HR |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: AT Payment date: 20071219 Year of fee payment: 4 Ref country code: FI Payment date: 20071221 Year of fee payment: 4 Ref country code: BG Payment date: 20071220 Year of fee payment: 4 Ref country code: PL Payment date: 20071207 Year of fee payment: 4 Ref country code: SK Payment date: 20071203 Year of fee payment: 4 |
|
| REG | Reference to a national code |
Ref country code: PL Ref legal event code: T3 |
|
| REG | Reference to a national code |
Ref country code: ES Ref legal event code: FG2A Ref document number: 2291966 Country of ref document: ES Kind code of ref document: T3 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: BE Payment date: 20071221 Year of fee payment: 4 Ref country code: SE Payment date: 20071219 Year of fee payment: 4 Ref country code: LT Payment date: 20071204 Year of fee payment: 4 |
|
| ET | Fr: translation filed | ||
| REG | Reference to a national code |
Ref country code: HK Ref legal event code: GR Ref document number: 1087022 Country of ref document: HK |
|
| REG | Reference to a national code |
Ref country code: HU Ref legal event code: AG4A Ref document number: E002744 Country of ref document: HU |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: CY Payment date: 20071204 Year of fee payment: 4 Ref country code: DE Payment date: 20080130 Year of fee payment: 4 Ref country code: HU Payment date: 20071221 Year of fee payment: 4 Ref country code: IS Payment date: 20080117 Year of fee payment: 4 Ref country code: PT Payment date: 20071204 Year of fee payment: 4 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: RO Payment date: 20071210 Year of fee payment: 4 Ref country code: TR Payment date: 20071210 Year of fee payment: 4 |
|
| PLBE | No opposition filed within time limit |
Free format text: ORIGINAL CODE: 0009261 |
|
| STAA | Information on the status of an ep patent application or granted ep patent |
Free format text: STATUS: NO OPPOSITION FILED WITHIN TIME LIMIT |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: FR Payment date: 20071214 Year of fee payment: 4 |
|
| 26N | No opposition filed |
Effective date: 20080613 |
|
| REG | Reference to a national code |
Ref country code: HR Ref legal event code: PBON Ref document number: P20070560 Country of ref document: HR Effective date: 20081210 |
|
| REG | Reference to a national code |
Ref country code: PT Ref legal event code: MM4A Free format text: LAPSE DUE TO NON-PAYMENT OF FEES Effective date: 20090609 |
|
| BERE | Be: lapsed |
Owner name: SANTHERA PHARMACEUTICALS (SCHWEIZ) A.G. Effective date: 20081231 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: SI Payment date: 20071204 Year of fee payment: 4 |
|
| LTLA | Lt: lapse of european patent or patent extension |
Effective date: 20081209 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: FI Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20081209 Ref country code: MC Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20081231 Ref country code: LT Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20081209 |
|
| REG | Reference to a national code |
Ref country code: CH Ref legal event code: PL |
|
| REG | Reference to a national code |
Ref country code: DK Ref legal event code: EBP |
|
| REG | Reference to a national code |
Ref country code: EE Ref legal event code: MM4A Ref document number: E001447 Country of ref document: EE Effective date: 20081231 |
|
| EUG | Se: european patent has lapsed | ||
| GBPC | Gb: european patent ceased through non-payment of renewal fee |
Effective date: 20081209 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: PT Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20090609 Ref country code: AT Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20081209 Ref country code: CZ Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20081209 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: IE Payment date: 20071216 Year of fee payment: 4 |
|
| NLV4 | Nl: lapsed or anulled due to non-payment of the annual fee |
Effective date: 20090701 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: BE Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20081231 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: IE Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20081209 Ref country code: LI Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20081231 Ref country code: DE Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20090701 Ref country code: EE Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20081231 Ref country code: CY Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20081209 Ref country code: CH Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20081231 |
|
| REG | Reference to a national code |
Ref country code: SI Ref legal event code: KO00 Effective date: 20090717 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: HU Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20081210 Ref country code: SK Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20081209 Ref country code: GB Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20081209 Ref country code: NL Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20090701 Ref country code: SI Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20081210 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: RO Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20081209 Ref country code: DK Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20090105 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: GR Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20090703 Ref country code: PL Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20081209 |
|
| REG | Reference to a national code |
Ref country code: ES Ref legal event code: FD2A Effective date: 20081210 |
|
| REG | Reference to a national code |
Ref country code: PL Ref legal event code: LAPE |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: ES Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20081210 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: IS Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20081231 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: LU Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20081209 Ref country code: SE Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20081210 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: BG Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20070912 |
|
| REG | Reference to a national code |
Ref country code: FR Ref legal event code: ST Effective date: 20110722 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: TR Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20100916 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: FR Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20081231 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: IT Payment date: 20071231 Year of fee payment: 4 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: TR Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20081209 |