EP3768668B1 - Shp2-phosphatase-inhibitoren und verfahren zur verwendung davon - Google Patents
Shp2-phosphatase-inhibitoren und verfahren zur verwendung davon Download PDFInfo
- Publication number
- EP3768668B1 EP3768668B1 EP19715690.4A EP19715690A EP3768668B1 EP 3768668 B1 EP3768668 B1 EP 3768668B1 EP 19715690 A EP19715690 A EP 19715690A EP 3768668 B1 EP3768668 B1 EP 3768668B1
- Authority
- EP
- European Patent Office
- Prior art keywords
- mmol
- compound
- mixture
- tert
- stirred
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Images



Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/433—Thidiazoles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
- A61K31/4523—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems
- A61K31/454—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems containing a five-membered ring with nitrogen as a ring hetero atom, e.g. pimozide, domperidone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/4965—Non-condensed pyrazines
- A61K31/497—Non-condensed pyrazines containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/4985—Pyrazines or piperazines ortho- or peri-condensed with heterocyclic ring systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/506—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/519—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/5375—1,4-Oxazines, e.g. morpholine
- A61K31/5377—1,4-Oxazines, e.g. morpholine not condensed and containing further heterocyclic rings, e.g. timolol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/04—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D211/06—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D211/08—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms
- C07D211/18—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms with substituted hydrocarbon radicals attached to ring carbon atoms
- C07D211/30—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms with substituted hydrocarbon radicals attached to ring carbon atoms with hydrocarbon radicals, substituted by doubly bound oxygen or sulfur atoms or by two oxygen or sulfur atoms singly bound to the same carbon atom
- C07D211/32—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms with substituted hydrocarbon radicals attached to ring carbon atoms with hydrocarbon radicals, substituted by doubly bound oxygen or sulfur atoms or by two oxygen or sulfur atoms singly bound to the same carbon atom by oxygen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/62—Oxygen or sulfur atoms
- C07D213/70—Sulfur atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D221/00—Heterocyclic compounds containing six-membered rings having one nitrogen atom as the only ring hetero atom, not provided for by groups C07D211/00 - C07D219/00
- C07D221/02—Heterocyclic compounds containing six-membered rings having one nitrogen atom as the only ring hetero atom, not provided for by groups C07D211/00 - C07D219/00 condensed with carbocyclic rings or ring systems
- C07D221/20—Spiro-condensed ring systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D241/00—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings
- C07D241/02—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings not condensed with other rings
- C07D241/10—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings not condensed with other rings having three double bonds between ring members or between ring members and non-ring members
- C07D241/14—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings not condensed with other rings having three double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D241/18—Oxygen or sulfur atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/06—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/14—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/06—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/22—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed systems contains four or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/12—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains three hetero rings
- C07D487/20—Spiro-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/02—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains two hetero rings
- C07D491/10—Spiro-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/02—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains two hetero rings
- C07D491/10—Spiro-condensed systems
- C07D491/107—Spiro-condensed systems with only one oxygen atom as ring hetero atom in the oxygen-containing ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/12—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains three hetero rings
- C07D491/14—Ortho-condensed systems
- C07D491/147—Ortho-condensed systems the condensed system containing one ring with oxygen as ring hetero atom and two rings with nitrogen as ring hetero atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/12—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains three hetero rings
- C07D491/20—Spiro-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D495/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having sulfur atoms as the only ring hetero atoms
- C07D495/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having sulfur atoms as the only ring hetero atoms in which the condensed system contains two hetero rings
- C07D495/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D498/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D498/12—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms in which the condensed system contains three hetero rings
- C07D498/20—Spiro-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D498/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D498/22—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms in which the condensed system contains four or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D513/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for in groups C07D463/00, C07D477/00 or C07D499/00 - C07D507/00
- C07D513/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for in groups C07D463/00, C07D477/00 or C07D499/00 - C07D507/00 in which the condensed system contains two hetero rings
- C07D513/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D519/00—Heterocyclic compounds containing more than one system of two or more relevant hetero rings condensed among themselves or condensed with a common carbocyclic ring system not provided for in groups C07D453/00 or C07D455/00
Definitions
- Src homology region 2 (SH2)-containing protein tyrosine phosphatase 2 is a protein tyrosine phosphatase encoded by the PTPN11 gene.
- SHP2 contains two Src homology 2 (SH2) NH2-terminal domains and a C-terminal protein-tyrosine phosphatase domain. It is ubiquitously expressed in various tissues and cell types.
- SHP2 plays an important role in diverse signaling pathways to regulate cellular biological processes and is involved in the signaling pathways of a variety of growth factors and cytokines. Within a single signaling pathway, SHP2 can play both positive (signal enhancing) and negative (signal diminishing) roles in intracellular signaling processes.
- SHP2 is believed to function by dephosphorylating its associated signaling molecules, thereby attenuating the local signaling flow.
- the main effect of SHP2 action in most signaling pathways is to enhance signal transduction.
- SHP2 is a positive regulator of the ERK/MAPK signaling pathway, playing a key role in regulating cellular proliferation and survival.
- SHP2 is normally auto-inhibited due to intramolecular interactions between its N-terminal SH2 (N-SH2) domain and its catalytic (PTP) domain, which blocks access to the catalytic site.
- N-SH2 N-terminal SH2
- PTP catalytic
- Activating proteins that interact with the SH2 domains induce a conformational change that reverses this inhibition and allows substrate access to the catalytic site.
- Mutations in the PTPN11 gene that affect the N-SH2 or PTP domain residues involved in basal inhibition of SHP2 result in more readily activatable forms of SHP2 protein, which can lead to unregulated or increased SHP2 activity.
- Such activated mutants of SHP2 have been associated with developmental disorders such as Noonan syndrome, where nearly all mutated forms of SHP2 demonstrate increased PTP activity.
- WO2018172984 and WO2017211303 relate to heterocyclic derivatives useful as SHP2 inhibitors.
- WO2018013597 relates to 2,5-disubstituted 3-methyl pyrazines and 2,5,6-trisubstituted 3-methyl pyrazines as allosteric SHP2 inhibitors.
- WO2016203406 relates to compounds and compositions for inhibiting the activity of SHP2.
- the presently claimed invention provides a compound selected from the group consisting of: and or a pharmaceutically acceptable salt thereof.
- the presently claimed invention provides a compound selected from the group consisting of: or a pharmaceutically acceptable salt thereof.
- the presently claimed invention provides a compound selected from the group consisting of: and or a pharmaceutically acceptable salt thereof.
- the presently claimed invention provides a compound having the formula: or a pharmaceutically acceptable salt thereof.
- the presently claimed invention provides a compound having the formula: or a pharmaceutically acceptable salt thereof.
- the presently claimed invention also provides, for example, a pharmaceutical composition
- a pharmaceutical composition comprising a compound as recited above, or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable carrier.
- the presently claimed invention further provides, for example, a compound or composition as recited above for use in the treatment of a disorder, the treatment comprising administering to a human subject in need thereof an effective amount of said compound, or a pharmaceutically acceptable salt thereof, or said composition.
- disorders include Noonan syndrome, neutropenia, diabetes, neuroblastoma, melanoma, acute myeloid leukemia, juvenile leukemia, juvenile myelomonocytic leukemia, breast cancer, lung cancer, and colorectal cancer.
- such treatment may include administration of a therapeutically effective amount of an antibody, an antibody-drug conjugate, an immunomodulator, or a histone deacetylase inhibitor.
- the presently claimed invention further provides a compound or composition as recited above for use in the treatment of a SHP2-mediated cancer, the treatment comprising administering to a human subject in need thereof an effective amount of said compound, or a pharmaceutically acceptable salt thereof, or said composition.
- such treatment may include administration of a therapeutically effective amount of an antibody, an antibody-drug conjugate, an immunomodulator, or a histone deacetylase inhibitor.
- the presently claimed invention further provides a compound or composition as recited above for use in the treatment of a SHP2-mediated cancer, the treatment comprising administering to a human subject in need thereof an effective amount of said compound, or a pharmaceutically acceptable salt thereof, or said composition, wherein the SHP2-mediated cancer is chronic myelomonocytic leukemia, acute myeloid leukemia, breast cancer, non-small cell lung cancer (NSCLC), colorectal cancer (CRC), esophageal cancer, gastric cancer, squamous-cell carcinoma of the head and neck (SCCHN), or ovarian cancer.
- SHP2-mediated cancer is chronic myelomonocytic leukemia, acute myeloid leukemia, breast cancer, non-small cell lung cancer (NSCLC), colorectal cancer (CRC), esophageal cancer, gastric cancer, squamous-cell carcinoma of the head and neck (SCCHN), or ovarian cancer.
- the presently claimed invention further provides a compound or composition as recited above for use in the treatment of a SHP2-mediated cancer, the treatment comprising administering to a human subject in need thereof an effective amount of said compound, or a pharmaceutically acceptable salt thereof, or said composition, wherein the SHP2-mediated cancer is breast cancer and wherein the breast cancer is HER2-positive breast cancer, triple-negative breast cancer, ductal carcinoma, or invasive ductal carcinoma.
- the presently claimed invention further provides a compound or composition as recited above for use in the treatment of a SHP2-mediated cancer, the treatment comprising administering to a human subject in need thereof an effective amount of said compound, or a pharmaceutically acceptable salt thereof, or said composition, wherein the SHP2-mediated cancer is NSCLC.
- the presently claimed invention further provides a compound or composition as recited above for use in the treatment of a SHP2-mediated cancer, the treatment comprising administering to a human subject in need thereof an effective amount of said compound, or a pharmaceutically acceptable salt thereof, or said composition, wherein the SHP2-mediated cancer is CRC.
- the presently claimed invention further provides a compound or composition as recited above for use in the treatment of a SHP2-mediated Noonan syndrome, juvenile leukemia, or juvenile myelomonocytic leukemia (JMML) in a human subject having Noonan syndrome, juvenile leukemia, or juvenile myelomonocytic leukemia (JMML), the treatment comprising administering to the subject an effective amount of said compound, or a pharmaceutically acceptable salt thereof, or said composition.
- a SHP2-mediated Noonan syndrome, juvenile leukemia, or juvenile myelomonocytic leukemia (JMML) in a human subject having Noonan syndrome, juvenile leukemia, or juvenile myelomonocytic leukemia (JMML)
- the treatment comprising administering to the subject an effective amount of said compound, or a pharmaceutically acceptable salt thereof, or said composition.
- the presently claimed invention further provides a compound or composition as recited above for use in the treatment of SHP2-mediated Noonan syndrome, the treatment comprising administering to a human subject in need thereof an effective amount of said compound, or a pharmaceutically acceptable salt thereof, or said composition.
- the present disclosure is based, in part, on certain discoveries which are described more fully in the Examples section of the present application.
- the present disclosure is based, in part, on the discovery of compounds disclosed herein, and the SHP2 phosphatase inhibition exhibited by such compounds.
- references to methods of treatment herein are to be interpreted as references to compounds and pharmaceutical compositions of the presently claimed invention for use in those methods.
- Activating SHP2 mutations have been detected in juvenile myelomonocytic leukemia (e.g ., Q506P), chronic myelomonocytic leukemia (e.g ., Y63C), neuroblastoma ( e.g., T507K), melanoma ( e.g ., R138Q), acute myeloid leukemia (e.g ., G503V), breast cancer, lung cancer ( e.g ., E76V), colorectal cancer ( e.g., E76G).
- Q506P juvenile myelomonocytic leukemia
- chronic myelomonocytic leukemia e.g ., Y63C
- neuroblastoma e.g., T507K
- melanoma e.g ., R138Q
- acute myeloid leukemia e.g ., G503V
- breast cancer e.
- SHP2 phosphatase inhibitors are disclosed, e.g ., in WO 2015/107493 ; WO 2015/107494 ; WO 2015/107495 ; and J.G. Fortanet et al., in J. Med. Chem. 2016, DOI: 10.1021/acs.jmedchem.6b00680 ; and references cited therein.
- the effects of SHP2 phsophatase inhibition are described, e.g. , Y.-N. P. Chen et al., in Nature, 2016, doi: 10.1038/nature18621 ; J. Wang et al., in J. Clin. Invest. 2016, 126, 2077-2092 ; and references cited therein.
- the compounds and/or compositions of the disclosure may be effective in treating, reducing, and/or suppressing disorders related to SHP2 phosphatase activity such as, e.g., Noonan syndrome, Leopard Syndrome, diabetes, neuroblastoma, melanoma, juvenile leukemia, juvenile myelomonocytic leukemia (JMML), chronic myelomonocytic leukemia, acute myeloid leukemia, HER2-positive breast cancer, triple-negative breast cancer, ductal carcinoma of the breast, invasive ductal carcinoma of the breast, non-small cell lung cancer (including adenocarcinoma of the lung), colorectal cancer, esophageal cancer, gastric cancer, squamous-cell carcinoma of the head and neck (SCCHN), neutropenia (Kostmann's syndrome), and systemic lupus erythematosus.
- disorders related to SHP2 phosphatase activity such as, e.g., Noonan syndrome,
- the methods described herein may also include additionally administering a therapeutically effective amount of an antibody, an antibody-drug conjugate, an immunomodulator, or a histone deacetylase inhibitor.
- compounds of the disclosure may contain "optionally substituted” moieties.
- substituted whether preceded by the term “optionally” or not, means that one or more hydrogens of the designated moiety are replaced with a suitable substituent.
- an “optionally substituted” group may have a suitable substituent at each substitutable position of the group, and when more than one position in any given structure may be substituted with more than one substituent selected from a specified group, the substituent may be either the same or different at every position.
- Combinations of substituents envisioned by this disclosure are preferably those that result in the formation of stable or chemically feasible compounds.
- stable refers to compounds that are not substantially altered when subjected to conditions to allow for their production, detection, and, in certain embodiments, their recovery, purification, and use for one or more of the purposes disclosed herein.
- Suitable monovalent substituents on R° are independently halogen, -(CH 2 ) 0-2 R • , -(haloR • ), -(CH 2 ) 0-2 OH, -(CH 2 ) 0-2 OR • , -(CH 2 ) 0-2 CH(OR • ) 2 ; -O(haloR • ), -CN, -N 3 , -(CH 2 ) 0-2 C(O)R • , -(CH 2 ) 0-2 C(O)OH, -(CH 2 ) 0-2 C(O)OR • , -(CH 2 ) 0-2 SR • , -(CH 2 ) 0-2 SH, -(CH 2 ) 0-2 NH 2 , -(CH 2 ) 0-2 NHR • , -(CH 2 ) 0-2 NR • 2 ,
- Suitable divalent substituents that are bound to vicinal substitutable carbons of an "optionally substituted” group include: -O(CR* 2 ) 2-3 O-, wherein each independent occurrence of R* is selected from hydrogen, C 1-6 aliphatic which may be substituted as defined below, or an unsubstituted 5-6-membered saturated, partially unsaturated, or aryl ring having 0-4 heteroatoms independently selected from nitrogen, oxygen, or sulfur.
- Suitable substituents on the aliphatic group of R* include halogen, -R • , -(haloR • ), -OH, -OR • , -O(haloR • ), -CN, -C(O)OH, -C(O)OR • , NH 2 , -NHR • , -NR • 2 , or -NO 2 , wherein each R • is unsubstituted or where preceded by "halo" is substituted only with one or more halogens, and is independently C 1-4 aliphatic, -CH 2 Ph, -O(CH 2 ) 0-1 Ph, or a 5-6-membered saturated, partially unsaturated, or aryl ring having 0-4 heteroatoms independently selected from nitrogen, oxygen, or sulfur.
- Suitable substituents on a substitutable nitrogen of an "optionally substituted" group include -R ⁇ , -NR ⁇ 2 , -C(O)R ⁇ , -C(O)OR ⁇ , -C(O)C(O)R ⁇ , -C(O)CH 2 C(O)R ⁇ , -S(O) 2 R ⁇ , -S(O) 2 NR ⁇ 2 , -C(S)NR ⁇ 2 , -C(NH)NR ⁇ 2 , or -N(R ⁇ )S(O) 2 R ⁇ ; wherein each R ⁇ is independently hydrogen, C 1-6 aliphatic which may be substituted as defined below, unsubstituted -OPh, or an unsubstituted 5-6-membered saturated, partially unsaturated, or aryl ring having 0-4 heteroatoms independently selected from nitrogen, oxygen, or sulfur, or, notwithstanding the definition above, two independent occurrence
- Suitable substituents on the aliphatic group of R ⁇ are independently halogen, -R • , -(haloR • ), -OH, -OR • , -O(haloR • ), -CN, -C(O)OH, -C(O)OR • , NH 2 , -NHR • , -NR • 2 , or -NO 2 , wherein each R • is unsubstituted or where preceded by "halo" is substituted only with one or more halogens, and is independently C 1-4 aliphatic, -CH 2 Ph, -O(CH 2 ) 0-1 Ph, or a 5-6-membered saturated, partially unsaturated, or aryl ring having 0-4 heteroatoms independently selected from nitrogen, oxygen, or sulfur.
- isomeric molecules that have the same molecular formula but differ in positioning of atoms and/or functional groups in the space. All stereoisomers of the present compounds (e.g ., those which may exist due to asymmetric carbons on various substituents), including enantiomeric forms and diastereomeric forms, are contemplated within the scope of this disclosure.
- tautomer refers to one of two or more structural isomers which exist in equilibrium and which are readily converted from one isomeric form to another. It is understood that tautomers encompass valence tautomers and proton tautomers (also known as prototropic tautomers). Valence tautomers include interconversions by reorganization of some of the bonding electrons. Proton tautomers include interconversions via migration of a proton, such as keto-enol and imine-enamine isomerizations.
- isotopic substitution refers to the substitution of an atom with its isotope.
- isotope refers to an atom having the same atomic number as that of atoms dominant in nature but having a mass number (neutron number) different from the mass number of the atoms dominant in nature. It is understood that a compound with an isotopic substitution refers to a compound in which at least one atom contained therein is substituted with its isotope. Atoms that can be substituted with its isotope include, but are not limited to, hydrogen, carbon, and oxygen. Examples of the isotope of a hydrogen atom include 2 H (also represented as D) and 3 H. Examples of the isotope of a carbon atom include 13 C and 14 C. Examples of the isotope of an oxygen atom include 18 O.
- alkyl refers to a monovalent aliphatic hydrocarbon radical having a straight chain, branched chain, monocyclic moiety, or polycyclic moiety or combinations thereof, wherein the radical is optionally substituted at one or more carbons of the straight chain, branched chain, monocyclic moiety, or polycyclic moiety or combinations thereof with one or more substituents at each carbon, wherein the one or more substituents are independently C 1 -C 10 alkyl.
- alkyl groups include methyl, ethyl, propyl, isopropyl, butyl, iso -butyl, sec -butyl, tert -butyl, pentyl, hexyl, heptyl, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, norbornyl, and the like.
- heteroaryl or “heteroaromatic group” as used herein refers to a monocyclic aromatic 5-6 membered ring system containing one or more heteroatoms, for example one to three heteroatoms, such as nitrogen, oxygen, and sulfur, or a 8-10 membered bicyclic unsaturated or partially unsaturated ring system containing one or more heteroatoms, for example one to three heteroatoms, such as nitrogen, oxygen, and sulfur.
- said heteroaryl ring may be linked to the adjacent radical though carbon or nitrogen.
- heteroaryl rings include but are not limited to furan, thiophene, pyrrole, thiazole, oxazole, isothiazole, isoxazole, imidazole, pyrazole, triazole, pyridine or pyrimidine, tetrahydroquinoline, etc.
- heterocyclyl or “heterocyclic group” are art-recognized and refer to saturated 4-10 membered monocyclic and bicyclic ring structures, including bridged or fused rings, and whose ring structures include one to three heteroatoms, such as nitrogen, oxygen, and sulfur. Where possible, heterocyclyl rings may be linked to the adjacent radical through carbon or nitrogen.
- salts derived from inorganic or organic acids including, e.g., hydrochloric, hydrobromic, sulfuric, nitric, perchloric, phosphoric, formic, acetic, lactic, maleic, fumaric, succinic, tartaric, glycolic, salicylic, citric, methanesulfonic, benzenesulfonic, benzoic, malonic, trifluroacetic, trichloroacetic, naphthalene-2 sulfonic and other acids; and salts derived from inorganic or organic bases including, e.g ., sodium, potassium, calcium, magnesium, zinc, ammonia, lysine, arginine, histidine, polyhydroxylated amines or tetrafluoroborate.
- Exemplary pharmaceutically acceptable salts are found, e.g., in Berge, et al. (J. Pharm. Sci. 1977, 66(1), 1 ; and Gould, P.L., Int. J. Pharmaceutics 1986, 33, 201-217 .
- Pharmaceutically acceptable salts are also intended to encompass hemi-salts, wherein the ratio of compound:acid is respectively 2: 1.
- Exemplary hemi-salts are those salts derived from acids comprising two carboxylic acid groups, such as malic acid, fumaric acid, maleic acid, succinic acid, tartaric acid, glutaric acid, oxalic acid, adipic acid and citric acid.
- exemplary hemi-salts are those salts derived from diprotic mineral acids such as sulfuric acid.
- Exemplary preferred hemi-salts include, but are not limited to, hemimaleate, hemifumarate, and hemisuccinate.
- the term “about” is used herein to mean approximately, roughly, around, or in the region of. When the term “about” is used in conjunction with a numerical range, it modifies that range by extending the boundaries above and below the numerical values set forth. In general, the term “about” is used herein to modify a numerical value above and below the stated value by a variance of 20 percent up or down (higher or lower).
- an “effective amount”, “sufficient amount” or “therapeutically effective amount” as used herein is an amount of a compound that is sufficient to effect beneficial or desired results, including clinical results.
- the effective amount may be sufficient, e.g ., to reduce or ameliorate the severity and/or duration of afflictions related to SHP2 phosphatase, or one or more symptoms thereof, prevent the advancement of conditions or symptoms related to afflictions related to SHP2 phosphatase, or enhance or otherwise improve the prophylactic or therapeutic effect(s) of another therapy.
- An effective amount also includes the amount of the compound that avoids or substantially attenuates undesirable side effects.
- treatment is an approach for obtaining beneficial or desired results, including clinical results.
- beneficial or desired clinical results may include, but are not limited to, alleviation or amelioration of one or more symptoms or conditions, diminution of extent of disease or affliction, a stabilized ( i.e ., not worsening) state of disease or affliction, preventing spread of disease or affliction, delay or slowing of disease or affliction progression, amelioration or palliation of the disease or affliction state and remission (whether partial or total), whether detectable or undetectable.
- Treatment can also mean prolonging survival as compared to expected survival if not receiving treatment.
- SHP2 phosphatase enzymes can be useful in applications that benefit from inhibition of SHP2 phosphatase enzymes.
- inhibition of SHP2 phosphatase may offer a therapeutic approach for the treatment of cancer.
- Inhibition of SHP2 phosphatase also has been found to ameliorate the pathogensis of systemic lupus erythematosus. (See, e.g., J. Wang et al., in J. Clin. Invest. 2016, 126, 2077-2092 ; and references cited therein.)
- compounds or compositions of the disclosure can be useful in suppressing tumor cell growth. In some embodiments, compounds or compositions of the disclosure can be useful in ameliorating the pathogenesis of systemic lupus erythematosus. In some embodiments, compounds or compositions of the disclosure can be useful in the treatment of various other disorders, including neurofibromatosis (e.g. neurofibromatosis type 1 (NF1), Noonan syndrome (NS)), diabetes, neuroblastoma, melanoma (see. Hill et al, Mol. Cancer Res. 2019, 17, 583-593 ), juvenile leukemia, juvenile myelomonocytic leukemia (JMML, see Yu et al, Mol. Cancer Ther.
- neurofibromatosis e.g. neurofibromatosis type 1 (NF1), Noonan syndrome (NS)
- NF1 neurofibromatosis type 1
- NS Noonan syndrome
- JMML juvenile myelomonocytic leukemia
- the disorder treated is a proliferative disorder.
- the disorder treated is a solid tumor.
- the disorder treated is a neurofibromatosis (e.g. neurofibromatosis type 1 (NF1), Noonan syndrome (NS)).
- the disorder treated is diabetes.
- the disorder treated is a neuroblastoma.
- the disorder treated is melanoma.
- the disorder treated is a hematological cancer.
- the disorder treated is a juvenile leukemia. In some embodiments, the disorder treated is a juvenile myelomonocytic leukemia (JMML). In some embodiments, the disorder treated is a chronic myelomonocytic leukemia. In some embodiments, the disorder treated is an acute myeloid leukemia. In some embodiments, the disorder treated is a breast cancer. In some embodiments, the disorder treated is a HER2-positive breast cancer. In some embodiments, the disorder treated is a triple-negative breast cancer. In some embodiments, the disorder treated is a ductal carcinoma of the breast. In some embodiments, the disorder treated is an invasive ductal carcinoma of the breast.
- JMML juvenile myelomonocytic leukemia
- the disorder treated is a chronic myelomonocytic leukemia.
- the disorder treated is an acute myeloid leukemia.
- the disorder treated is a breast cancer. In some embodiments, the disorder treated is a HER2-positive breast cancer
- the disorder treated is a non-small cell lung cancer (including adenocarcinoma of the lung). In some embodiments, the disorder treated is a colorectal cancer. In some embodiments, the disorder treated is an esophageal cancer. In some embodiments, the disorder treated is a gastric cancer. In some embodiments, the disorder treated is a squamous-cell carcinoma of the head and neck (SCCHN). In some embodiments, the disorder treated is a neutropenia (e.g. Kostmann's syndrome). In some embodiments, the disorder treated is an ovarian cancer. In some embodiments, the disorder treated is an FGFR1-amplified/mutant cancer (e.g.
- the disorder treated is an FGFR2-amplified cancer (e.g. FGFR2-amplified gastric cancer).
- the disorder treated is an FGFR2-fusion/mutant cancer (e.g. FGFR2-fusion/mutant cholangiocarcinoma).
- the disorder treated is or an FGFR3-fusion/mutant cancer (e.g. FGFR3-fusion or mutant bladder cancer).
- compounds or compositions of the disclosure can be used in combination with other treatments and/or cancer therapies.
- compounds or compositions of the disclosure can be used in combination with, but are not limited to, antibodies, antibody-drug conjugates, kinase inhibitors, immunomodulators, and histone deacetylase inhibitors.
- the compounds or compositions of the disclosure can also be used in combination with other treatments and/or cancer therapies as disclosed in WO 2015/107495 ; and references cited therein.
- the compounds disclosed herein can be used in the treatment of one or more of the diseases mentioned herein, alone or in combination with another therapeutic agent.
- a provided compound can be used in combination with one or more of the following agents, or a pharmaceutically acceptable salt thereof: BCR-ABL inhibitors: e.g.
- ALK inhibitors see Dardaei et al, 2018, Nat Med.; 24(4):512-517 : e.g. crizotinib, NVP-TAE684, ceritinib, alectinib, brigatinib, entrecinib, lorlatinib
- BRAF inhibitors see Prahallad et al, 2015, Cell Rep. 12, 1978-1985 ): e.g.
- FGFR inhibitors e.g. infigratinib, dovitinib, erdafitinib, BLU-554, AZD4547
- FLT3 inhibitors e.g. sunitinib, midostaurin, tanutinib, sorafenib, lestaurtinib, quizartinib, and crenolanib
- MEK Inhibitors see Fedele et al, 2018, BioRxiv 307876 ; Torres-Ayuso et al, 2018, Cancer Discov. 8, 1210-1212 ; and Wong et al, 2016, Oncotarget.
- VEGF receptor inhibitors e.g. bevacizumab, axitinib, aflibercept, brivanib, motesanib, pasireotide, sorafenib
- Tyrosine kinase inhibitors e.g.
- erlotinib linifanib, sunitinib, pazopanib
- Epidermal growth factor receptor (EGFR) inhibitors gefitnib, osimertinib, cetuximab, panitumumab
- HER2 receptor inhibitors e.g. trastuzumab, neratinib, lapatinib, lapatinib
- MET inhibitors e.g. crizotinib, cabozantinib
- CD20 antibodies e.g. rituximab, tositumomab, ofatumumab
- DNA Synthesis inhibitors e.g.
- capecitabine gemcitabine, nelarabine, hydroxycarbamide
- Antineoplastic agents e.g. oxaliplatin, cisplatin
- HER dimerization inhibitors e.g. pertuzumab
- Human Granulocyte colony-stimulating factor (G-CSF) modulators e.g. filgrastim
- Immunomodulators e.g. afutuzumab, lenalidomide, thalidomide, pomalidomide
- CD40 inhibitors e.g. dacetuzumab
- PARAs Pro-apoptotic receptor agonists
- HSP Heat Shock Protein
- tanespimycin (17-allylamino-17-desmethoxygeldanamycin
- Hedgehog antagonists e.g. vismodegib
- Proteasome inhibitors e.g. bortezomib
- PI3K inhibitors e.g. pictilisib, dactolisib, buparlisib, taselisib, idelalisib, duvelisib, umbralisib
- Phospholipase A2 inhibitors e.g. anagrelide
- BCL-2 inhibitors e.g.
- Aromatase inhibitors exemestane, letrozole, anastrozole, faslodex, tamoxifen; Topoisomerase I inhibitors: e.g. irinotecan, topotecan; Topoisomerase II inhibitors: e.g. etoposide, teniposide; mTOR inhibitors: e.g. temsirolimus, ridaforolimus, everolimus, sirolimus; Osteoclastic bone resorption inhibitors: e.g. zoledronic acid; CD33 Antibody Drug Conjugates: e.g.
- gemtuzumab ozogamicin CD22 Antibody Drug Conjugates: e.g. inotuzumab ozogamicin; CD20 Antibody Drug Conjugates: e.g. ibritumomab tiuxetan; Somatostain analogs: e.g. octreotide; Interleukin-11 (IL-11): e.g. oprelvekin; Synthetic erythropoietin: e.g. darbepoetin alfa; Receptor Activator for Nuclear Factor ⁇ B (RANK) inhibitors: e.g.
- RANK Nuclear Factor ⁇ B
- Thrombopoietin mimetic peptides e.g. romiplostim
- Cell growth stimulators e.g. palifermin
- Anti-Insulin-like Growth Factor-1 receptor (IGF-1R) antibodies e.g. figitumumab
- Anti-CSl antibodies e.g. elotuzumab
- CD52 antibodies e.g. alemtuzumab
- CTLA-4 inhibitors e.g. tremelimumab, ipilimumab
- PD1 inhibitors e.g. nivolumab, pembrolizumab
- an immunoadhesin e.g.
- pidilizumab pidilizumab, AMP-224; PDL1 inhibitors: e.g. MSB0010718C; YW243.55.S70, MPDL3280A; MEDI-4736, MSB-0010718C, or MDX-1105; LAG-3 inhibitors: e.g. BMS-986016; GITR agonists; GITR fusion proteins and anti-GITR antibodies; Histone deacetylase inhibitors (HDI): e.g. voninostat; Anti-CTLA4 antibodies: e.g. tremelimumab, ipilimumab; Alkylating agents: e.g.
- PDL1 inhibitors e.g. MSB0010718C; YW243.55.S70, MPDL3280A; MEDI-4736, MSB-0010718C, or MDX-1105
- LAG-3 inhibitors e.g. BMS-986016
- temozolomide dactinomycin, melphalan, altretamine carmustine, bendamustine, busulfan, carboplatin, lomustine, cisplatin, chlorambucil, cyclophosphamide, dacarbazine , altretamine, ifosfamide, procarbazine , mechlorethamine, mustine and mechloroethamine, streptozocin, thiotepa; Biologic response modifiers: e.g. bacillus calmette-guerin, denileukin diftitox; Anti-tumor antibiotics: e.g.
- Anti-microtubule agents e.g. estramustine; Cathepsin K inhibitors: e.g. odanacatib; Epothilone analogs: e.g. ixabepilone; TpoR agonists: e.g. eltrombopag; Anti-mitotic agents: e.g. docetaxel; Adrenal steroid inhibitors: e.g. aminoglutethimide; Anti-androgens: e.g.
- nilutamide nilutamide
- Androgen Receptor inhibitors e.g. enzalutamide, abiraterone acetate, orteronel, galeterone, and seviteronel, bicalutamide, flutamide; Androgens: e.g. fluoxymesterone
- CDK1 inhibitors e.g. alvocidib, palbociclib, ribociclib, trilaciclib, abemaciclib
- Gonadotropin-releasing hormone (GnRH) receptor agonists e.g. leuprolide or leuprolide acetate
- Taxane anti-neoplastic agents e.g.
- 5-HTla receptor agonists e.g. xaliproden
- HPV vaccines e.g. Cervarix ® sold by GlaxoSmithKline, Gardasil ® sold by Merck
- Iron Chelating agents e.g. deferasirox
- Anti-metabolites e.g. claribine, 5-fluorouracil, 6-thioguanine, pemetrexed, cytarabine, cytarabine liposomal, decitabine, hydroxyurea, fludarabine, floxuridine, cladribine, methotrexate, pentostatin
- Bisphosphonates e.g.
- Demethylating agents e.g. 5-azacitidine, decitabine
- Anti-tumor Plant Alkaloids e.g. paclitaxel proteinbound; vinblastine, vincristine, vinorelbine, paclitaxel
- Retinoids e.g. alitretinoin, tretinoin, isotretinoin, bexarotene
- Glucocorticosteroids e.g. hydrocortisone, dexamethasone, prednisolone, prednisone, methylprednisolone
- Cytokines e.g.
- interleukin-2 interleukin-2, interleukin-11 (oprevelkin), alpha interferon alfa (IFN-alpha); estrogen receptor downregulators: fulvestrant; Anti-estrogens: e.g. tamoxifen, toremifene; Selective estrogen receptor modulators (SERMs): e.g. raloxifene; Luteinizing hormone releasing hormone (LHRH) agonists: e.g. goserelin; Progesterones: e.g.
- cytotoxic agents arsenic trioxide, asparaginase (also known as L-asparaginase, Erwinia L-asparaginase; Anti-nausea drugs: e.g. NK-1 receptor antagonists (e.g. casopitant); Cytoprotective agents: e.g. amifostine, leucovorin; and Immune checkpoint inhibitors.
- NK-1 receptor antagonists e.g. casopitant
- Cytoprotective agents e.g. amifostine, leucovorin
- Immune checkpoint inhibitors refers to a group of molecules on the cell surface of CD4 and CD8 T cells.
- Immune checkpoint molecules include, but are not limited to, Programmed Death 1 (PD-1), Cytotoxic T-Lymphocyte Antigen 4 (CTLA-4), B7H1, B7H4, OX-40, CD 137, CD40, and LAG3.
- Immunotherapeutic agents which can act as immune checkpoint inhibitors useful in the methods of the present disclosure, include, but are not limited to, inhibitors of PD-L1, PD-L2, CTLA4, TIM3, LAG3, VISTA, BTLA, TIGIT, LAIR1, CD 160, 2B4 and/or TGFR beta.
- the compounds described herein can function as allosteric inhibitors and block the activation of SHP2 by targeting the auto-inhibited conformation of SHP2.
- the compounds described herein can also inhibit SHP2 function through incorporation into agents that catalyze the destruction of SHP2.
- the compounds can be incorporated into proteolysis targeting chimeras (PROTACs).
- a PROTAC is a bifunctional molecule, with one portion capable of engaging an E3 ubiquitin ligase, and the other portion having the ability to bind to a target protein meant for degradation by the cellular protein quality control machinery. Recruitment of the target protein to the specific E3 ligase results in its tagging for destruction (i.e., ubiquitination) and subsequent degradation by the proteasome. Any E3 ligase can be used.
- the portion of the PROTAC that engages the E3 ligase is connected to the portion of the PROTAC that engages the target protein via a linker which consists of a variable chain of atoms. Recruitment of SHP2 to the E3 ligase will thus result in the destruction of the SHP2 protein.
- the variable chain of atoms can include, for example, rings, heteroatoms, and/or repeating polymeric units. It can be rigid or flexible. It can be attached to the two portions described above using standard techniques.
- the compounds described herein can be linked to one end of a variable chain, while the other end of the variable chain can be bound to the E3 ligase. Recruitment of SHP2 to the ligase will thus result in the destruction of the SHP2 protein.
- compounds or compositions of the disclosure can be used in combination with an antibody. In some embodiments, compounds or compositions of the disclosure can be used in combination with an antibody-drug conjugate. In some embodiments, compounds or compositions of the disclosure can be used in combination with a kinase inhibitor. In some embodiments, compounds or compositions of the disclosure can be used in combination with an immunomodulator. In some embodiments, compounds or compositions of the disclosure can be used in combination with a histone deacetylase inhibitor.
- the present disclosure provides a method of treating a SHP2-mediated disorder comprising administering to a subject in need thereof a compound described herein, wherein the disorder is selected from those described in WO2019051084A1 .
- the present disclosure provides a method of treating a SHP2-mediated disorder comprising administering to a subject in need thereof a compound described herein together with an additional therapeutic agent, wherein the additional therapeutic agent is not a SHP2 inhibitor, and is selected from those described in WO2019051084A1 .
- a disclosed compound can be administered to a subject in need of treatment at dosages ranging from about 0.0001 mg to about 100 mg/kg body weight of the subject to be treated per day, such as from about 1.0 to 10 mg/kg.
- dosages ranging from about 0.0001 mg to about 100 mg/kg body weight of the subject to be treated per day, such as from about 1.0 to 10 mg/kg.
- additional variations are within the scope of the disclosure.
- a disclosed compound can be administered alone or in combination with pharmaceutically acceptable carriers, such as diluents, fillers, aqueous solution, and even organic solvents.
- pharmaceutically acceptable carriers such as diluents, fillers, aqueous solution, and even organic solvents.
- the compound and/or compositions of the disclosure can be administered as a tablet, powder, lozenge, syrup, injectable solution, and the like. Additional ingredients, such as flavoring, binder, excipients, and the like are within the scope of the disclosure.
- compositions can contain a disclosed compound and/or a pharmaceutically acceptable salt thereof at a concentration ranging from about 0.01 to about 90 wt%, about 0.01 to about 80 wt%, about 0.01 to about 70 wt%, about 0.01 to about 60 wt%, about 0.01 to about 50 wt%, about 0.01 to about 40 wt%, about 0.01 to about 30 wt%, about 0.01 to about 20 wt%, about 0.01 to about 2.0 wt%, about 0.01 to about 1 wt%, about 0.05 to about 0.5 wt%, about 1 to about 30wt%, or about 1 to about 20wt%.
- the composition can be formulated as a solution, suspension, ointment, or a capsule, and the like.
- the pharmaceutical composition can be prepared as an aqueous solution and can contain additional components, such as preservatives, buffers, tonicity agents, antioxidants, stabilizers, viscosity-modifying ingredients and the like.
- the present disclosure provides for the use of pharmaceutical compositions and/or medicaments comprised of a disclosed compound or a pharmaceutically acceptable salt thereof, in a method of treating a disease state, and/or condition caused by or related to SHP2 phosphatase.
- a disease state e.g., a chronic hemangioma
- a pharmaceutically acceptable salt thereof e.g., a pharmaceutically acceptable salt thereof.
- methods of treating subjects in need thereof e.g., subjects suffering from cancer (e.g.,leukemia, breast, lung and/or colorectal cancer) an effective amount of a disclosed compound, and optionally an effective amount of an additional compound (e.g., therapeutic agent) such as disclosed herein.
- cancer e.g.,leukemia, breast, lung and/or colorectal cancer
- an additional compound e.g., therapeutic agent
- the method of treatment comprises the steps of: i) identifying a subject in need of such treatment; (ii) providing a disclosed compound or a pharmaceutically acceptable salt thereof; and (iii) administering said disclosed compound in a therapeutically effective amount to treat, suppress and/or prevent the disease state or condition in a subject in need of such treatment.
- the method of treatment comprises the steps of: i) identifying a subject in need of such treatment; (ii) providing a composition comprising a disclosed compound or a pharmaceutically acceptable salt thereof; and (iii) administering said composition in a therapeutically effective amount to treat, suppress and/or prevent the disease state or condition in a subject in need of such treatment.
- the subject is an animal.
- Animals include all members of the animal kingdom, but are not limited to humans, mice, rats, cats, monkeys, dogs, horses, and swine.
- the subject is a human.
- the subject is a mouse, a rat, a cat, a monkey, a dog, a horse, or a pig.
- a compound or composition of the disclosure is administered orally, intravenously, by inhalation, intranasally, intraocularly, topically, subcutaneously, rectally, intravaginally, or intrathecally. In some embodiments, the compound or composition is administered orally. In some embodiments, the compound or composition is administered intravenously.
- the methods comprise administering to the subject an effective amount of a disclosed compound or a pharmaceutically acceptable salt thereof; or a composition comprising a disclosed compound or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable carrier.
- Pharmaceutically acceptable carriers are well-known to those skilled in the art, and include, e.g., adjuvants, diluents, excipients, fillers, lubricants and vehicles.
- the carrier is a diluent, adjuvant, excipient, or vehicle.
- the carrier is a diluent, adjuvant, or excipient.
- the carrier is a diluent or adjuvant.
- the carrier is an excipient.
- the pharmaceutically acceptable carrier is chemically inert toward the active compounds and is non-toxic under the conditions of use.
- Examples of pharmaceutically acceptable carriers may include, e.g., water or saline solution, polymers such as polyethylene glycol, carbohydrates and derivatives thereof, oils, fatty acids, or alcohols.
- oils as pharmaceutical carriers include oils of petroleum, animal, vegetable or synthetic origin, such as peanut oil, soybean oil, mineral oil, sesame oil and the like.
- the pharmaceutical carriers may also be saline, gum acacia, gelatin, starch paste, talc, keratin, colloidal silica, urea, and the like.
- auxiliary, stabilizing, thickening, lubricating and coloring agents may be used.
- suitable pharmaceutical carriers are described in e.g., Remington's: The Science and Practice of Pharmacy, 22nd Ed.
- the method of treatment, prevention and/or suppression of a condition related to SHP2 phosphatase comprises the steps of: i) identifying a subject in need of such treatment; (ii) providing a disclosed compound or a pharmaceutically acceptable salt thereof; or a composition comprising a disclosed compound or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable carrier; and (iii) administering said compound or composition in a therapeutically effective amount to treat, prevent and/or suppress the disease state or condition related to SHP2 phosphatase in a subject in need of such treatment.
- the compounds of the disclosure are formulated into pharmaceutical compositions for administration to subjects in a biologically compatible form suitable for administration in vivo.
- the present disclosure provides a pharmaceutical composition comprising a disclosed compound in admixture with a pharmaceutically acceptable diluent and/or carrier.
- the pharmaceutically-acceptable carrier is "acceptable" in the sense of being compatible with the other ingredients of the composition and not deleterious to the recipient thereof.
- the pharmaceutically-acceptable carriers employed herein may be selected from various organic or inorganic materials that are used as materials for pharmaceutical formulations and which are incorporated as analgesic agents, buffers, binders, disintegrants, diluents, emulsifiers, excipients, extenders, glidants, solubilizers, stabilizers, suspending agents, tonicity agents, vehicles and viscosity-increasing agents.
- Pharmaceutical additives such as antioxidants, aromatics, colorants, flavorimproving agents, preservatives, and sweeteners, may also be added.
- acceptable pharmaceutical carriers include carboxymethyl cellulose, crystalline cellulose, glycerin, gum arabic, lactose, magnesium stearate, methyl cellulose, powders, saline, sodium alginate, sucrose, starch, talc and water, among others.
- pharmaceutically acceptable means approved by a regulatory agency of the Federal or a state government or listed in the U.S. Pharmacopeia or other generally recognized pharmacopeia for use in animals, and more particularly in humans.
- Surfactants such as, e.g., detergents, are also suitable for use in the formulations.
- Specific examples of surfactants include polyvinylpyrrolidone, polyvinyl alcohols, copolymers of vinyl acetate and of vinylpyrrolidone, polyethylene glycols, benzyl alcohol, mannitol, glycerol, sorbitol or polyoxyethylenated esters of sorbitan; lecithin or sodium carboxymethylcellulose; or acrylic derivatives, such as methacrylates and others, anionic surfactants, such as alkaline stearates, in particular sodium, potassium or ammonium stearate; calcium stearate or triethanolamine stearate; alkyl sulfates, in particular sodium lauryl sufate and sodium cetyl sulfate; sodium dodecylbenzenesulphonate or sodium dioctyl sulphosuccinate; or fatty acids, in particular those derived from
- a disclosed compound and pharmaceutically acceptable carriers can be sterile.
- suitable pharmaceutical carriers may also include excipients such as starch, glucose, lactose, sucrose, gelatin, malt, rice, flour, chalk, silica gel, sodium stearate, glycerol monostearate, talc, sodium chloride, dried skim milk, glycerol, propylene, glycol, polyethylene glycol 300, water, ethanol, polysorbate 20, and the like.
- the present compositions may also contain minor amounts of wetting or emulsifying agents, or pH buffering agents.
- compositions of the present disclosure are prepared by methods well-known in the pharmaceutical arts.
- one or more accessory ingredients e.g., buffers, flavoring agents, surface active agents, and the like
- the choice of carrier is determined by the solubility and chemical nature of the compounds, chosen route of administration and standard pharmaceutical practice.
- the compounds and/or compositions of the present disclosure are administered to a human or animal subject by known procedures including oral administration, sublingual or buccal administration. In some embodiments, the compound and/or composition is administered orally.
- a formulation of the compounds of the disclosure may be presented in dosage forms such as capsules, tablets, powders, granules, or as a suspension or solution.
- Capsule formulations may be gelatin, soft-gel or solid. Tablets and capsule formulations may further contain one or more adjuvants, binders, diluents, disintegrants, excipients, fillers, or lubricants, each of which are known in the art.
- compositions may contain one or more optional agents such as, e.g ., sweetening agents such as fructose, aspartame or saccharin; flavoring agents such as peppermint, oil of wintergreen, or cherry; coloring agents; and preservative agents, to provide a pharmaceutically palatable preparation.
- optional agents such as, e.g ., sweetening agents such as fructose, aspartame or saccharin; flavoring agents such as peppermint, oil of wintergreen, or cherry; coloring agents; and preservative agents, to provide a pharmaceutically palatable preparation.
- the composition is in unit dose form such as a tablet, capsule or single-dose vial.
- Suitable unit doses i.e ., therapeutically effective amounts, may be determined during clinical trials designed appropriately for each of the conditions for which administration of a chosen compound is indicated and will, of course, vary depending on the desired clinical endpoint.
- the compounds of the disclosure are administered to the subject in a therapeutically effective amount, e.g., to reduce or ameliorate symptoms related to SHP2 phosphatase activity in the subject.
- a therapeutically effective amount e.g., to reduce or ameliorate symptoms related to SHP2 phosphatase activity in the subject.
- the methods comprise administration of a therapeutically effective dosage of the compounds of the disclosure.
- the therapeutically effective dosage is at least about 0.0001 mg/kg body weight, at least about 0.001 mg/kg body weight, at least about 0.01 mg/kg body weight, at least about 0.05 mg/kg body weight, at least about 0.1 mg/kg body weight, at least about 0.25 mg/kg body weight, at least about 0.3 mg/kg body weight, at least about 0.5 mg/kg body weight, at least about 0.75 mg/kg body weight, at least about 1 mg/kg body weight, at least about 2 mg/kg body weight, at least about 3 mg/kg body weight, at least about 4 mg/kg body weight, at least about 5 mg/kg body weight, at least about 6 mg/kg body weight, at least about 7 mg/kg body weight, at least about 8 mg/kg body weight, at least about 9 mg/kg body weight, at least about 10 mg/kg body weight, at least about 15 mg/kg body weight, at least about 20 mg/kg body weight,
- the therapeutically effective dosage is in the range of about 0.1 mg to about 10 mg/kg body weight, about 0.1 mg to about 6 mg/kg body weight, about 0.1 mg to about 4 mg /kg body weight, or about 0.1 mg to about 2 mg/kg body weight.
- the therapeutically effective dosage is in the range of about 1 to 500 mg, about 2 to 150 mg, about 2 to 120 mg, about 2 to 80 mg, about 2 to 40 mg, about 5 to 150 mg, about 5 to 120 mg, about 5 to 80 mg, about 10 to 150 mg, about 10 to 120 mg, about 10 to 80 mg, about 10 to 40 mg, about 20 to 150 mg, about 20 to 120 mg, about 20 to 80 mg, about 20 to 40 mg, about 40 to 150 mg, about 40 to 120 mg or about 40 to 80 mg.
- the methods comprise a single dosage or administration (e.g., as a single injection or deposition).
- the methods comprise administration once daily, twice daily, three times daily or four times daily to a subject in need thereof for a period of from about 2 to about 28 days, or from about 7 to about 10 days, or from about 7 to about 15 days, or longer.
- the methods comprise chronic administration.
- the methods comprise administration over the course of several weeks, months, years or decades.
- the methods comprise administration over the course of several weeks.
- the methods comprise administration over the course of several months.
- the methods comprise administration over the course of several years.
- the methods comprise administration over the course of several decades.
- the dosage administered can vary depending upon known factors such as the pharmacodynamic characteristics of the active ingredient and its mode and route of administration; time of administration of active ingredient; age, sex, health and weight of the recipient; nature and extent of symptoms; kind of concurrent treatment, frequency of treatment and the effect desired; and rate of excretion. These are all readily determined and may be used by the skilled artisan to adjust or titrate dosages and/or dosing regimens.
- suitable dose ranges for oral administration of the compounds of the disclosure are generally about 1 mg/day to about 1000 mg/day. In some embodiments, the oral dose is about 1 mg/day to about 800 mg/day. In some embodiments, the oral dose is about 1 mg/day to about 500 mg/day. In some embodiments, the oral dose is about 1 mg/day to about 250 mg/day. In some embodiments, the oral dose is about 1 mg/day to about 100 mg/day. In some embodiments, the oral dose is about 5 mg/day to about 50 mg/day. In some embodiments, the oral dose is about 5 mg/day.
- the oral dose is about 10 mg/day. In some embodiments, the oral dose is about 20 mg/day. In some embodiments, the oral dose is about 30 mg/day. In some embodiments, the oral dose is about 40 mg/day. In some embodiments, the oral dose is about 50 mg/day. In some embodiments, the oral dose is about 60 mg/day. In some embodiments, the oral dose is about 70 mg/day. In some embodiments, the oral dose is about 100 mg/day. It will be recognized that any of the dosages listed herein may constitute an upper or lower dosage range, and may be combined with any other dosage to constitute a dosage range comprising an upper and lower limit.
- any of the compounds and/or compositions of the disclosure may be provided in a kit comprising the compounds and/or compositions.
- the compound and/or composition of the disclosure is provided in a kit.
- the compounds described herein can be prepared in a number of ways based on the teachings contained herein and synthetic procedures known in the art.
- synthetic procedures known in the art.
- all proposed reaction conditions including choice of solvent, reaction atmosphere, reaction temperature, duration of the experiment and workup procedures, can be chosen to be the conditions standard for that reaction, unless otherwise indicated.
- the functionality present on various portions of the molecule should be compatible with the reagents and reactions proposed.
- Substituents not compatible with the reaction conditions will be apparent to one skilled in the art, and alternate methods are therefore indicated.
- the starting materials for the examples are either commercially available or are readily prepared by standard methods from known materials.
- an aryl compound such as a compound of Formula 1001 undergoes a cross-coupling reaction with a metalated or otherwise activated moiety to provide a compound of Formula 1002 .
- the compound of Formula 1002 then undergoes a substitution reaction with an amine such as Compound 1005, followed by removal of the protecting group to provide a compound of Formula (I).
- LG is I.
- LG is Cl.
- LG is OTf or OTs.
- a protected heteroaryl ether such as a compound of Formula 1003 , undergoes a cross-coupling reaction to provide a compound of Formula 1004 .
- the ether protecting group is subsequently removed and the resulting hydroxyl group activated to form a Q group, such as OSO 2 Me, OMs, OTs, OTf, and the like, to form a compound of Formula 1002 , which can then be carried forward to prepare compounds having the Formula (I).
- a Q group such as OSO 2 Me, OMs, OTs, OTf, and the like
- compounds of the disclosure can generally be prepared according to exemplary Scheme 2: wherein X, R 1 , R 6 , R 4 , U, V, B and D are as defined above, Q is independently a halogen, such as Cl, Br, I, and the like, or any other leaving group, such as OSO 2 Me, OMs, OTs, OTf, and the like. LG is a leaving group, such as Cl, Br, I, OTs, OTf, and the like, and P is a protecting group, such as 4-methoxybenzyl and the like. Alternative protecting groups that can be used are described, e.g ., in Greene et al., Protective Groups in Organic Synthesis (4th ed. 2006 ).
- an aryl compound such as a compound of Formula 1001 undergoes a undergoes a substitution reaction with an amine such as 1005 to provide a compound of Formula 1006 .
- the compound of Formula 1006 then undergoes a cross-coupling reaction with a metalated or otherwise activated moiety to provide a compound of Formula 1007.
- the compound of Formula 1007 can be deprotected to produce a compound of Formula (I).
- the compound of Formula 1007 can be left protected and functional groups on the R 1 moiety refunctionalized by methods known to those of ordinary skill in the art.
- the cross-coupling reaction is a Buchwald-Hartwig reaction. In some embodiments, the cross-coupling reaction is a Chan-Lam coupling reaction. In some embodiments, the cross-coupling reaction is an Ullmann reaction. In some embodiments, the cross-coupling reaction is a Suzuki reaction. In some embodiments, the cross-coupling reaction is a Stille reaction. In some embodiments, the cross-coupling reaction is a Negishi reaction. In some embodiments, the cross-coupling reaction is a Hiyama reaction. Other cross-coupling reactions may be employed as would be apparent to one of ordinary skill in the art.
- the protecting group is removed under acidic conditions, such as HBr in AcOH. Conditions for removal of the protecting group will depend on the nature of the protecting group. Conditions for the removal of various protecting groups can be found, e.g. , in Greene et al., Protective Groups in Organic Synthesis (4th ed. 2006 ).
- LCMS standard conditions were: Waters HPLC system equipped with an Alliance 2695 main module, Waters 996 diode array detector and ZQ micromass ESI-MS detector.
- Mobile phase A H 2 O (10.0 mM NH 4 HCO 2 )
- mobile phase B CH 3 CN.
- HPLC conditions were: XBridge C18 column, 4.6 ⁇ 30 mm, 3.5 ⁇ m, 0.0-0.2 min. isocratic (5% B), 0.2-2.0 min. gradient (5-100% B), 3.0-3.0 min. isocratic (100% B); flow rate: 3.0 mL/min; UV channel: 254 nm.
- Semi preparative HPLC A Gilson 215 system equipped with a Waters 996 diode array detector and a Waters 2525 pump.
- Semi preparative HPLC B Waters 2767 system equipped with a Waters 996 diode array detector, 2 X Waters 515 pumps, a Waters 2525 pump and a ZQ micromass ESI-MS detector.
- Semi preparative SFC Mettler Toledo Minigram SFC equipped with a Knauer K-2501 UV detector and an Alcott Model 1719 Autosampler.
- Step a In a 100 mL round-bottomed flask, 3,5-dichloropyrazine-2-carboxylic acid (3.65 g, 18.9 mmol) and NaHCO 3 (4.70 g, 22.7 mmol) were dissolved in dimethylformamide (38 mL). Iodomethane (7.14 mL, 113 mmol) was added dropwise and the resulting mixture stirred overnight at rt. The mixture was diluted with water (50 mL) and extracted with ethyl acetate (3 ⁇ 15 mL). The combined organics were washed with brine (4 ⁇ 10 mL), dried over anhydrous Na 2 SO 4 , filtered, and concentrated under reduced pressure.
- Step b Methyl 3,5-dichloropyrazine-2-carboxylate (5.0 g, 24.2 mmol) was dissolved in a 9:1 mixture of dry tetrahydrofuran (242 mL) and methanol (27 mL). The mixture was cooled to 1.5-2 °C with an ice/water bath and stirred at this temperature for 10 min. A 2 M solution of lithium borohydride in THF (13.3 mL, 26.6 mmol) was then added carefully keeping the temperature below 4-5 °C. After addition, the reaction mixture was stirred for an additional 10-15 min at 0-4 °C. Methanol (120 mL) was added to the flask and the mixture stirred for 15 min at rt.
- Step c (3,5-Dichloropyrazin-2-yl)methanol (4.3 g, 24 mmol) was dissolved in dichloromethane (100 mL) and MnO 2 (20.2 g, 240 mmol) was then added in one portion. The resulting dark heterogeneous mixture was stirred for 16 h at rt. After this time, the reaction mixture was sonicated for 5 min. and additional MnO 2 (4 g) was added to the reaction mixture. The resulting suspension was stirred for 2 h at rt. Then the mixture was filtered over a pad of celite, and the cake washed with dichloromethane.
- Step d 3,5-Dichloropyrazine-2-carbaldehyde (2.9 g, 16.4 mmol) was dissolved in N -methyl-2-pyrrolidone (16 mL), then hydrazine hydrate (0.78 mL, 49.2 mmol) was added dropwise. The resulting brown suspension was stirred at 65 °C for 40 min. After this time, additional hydrazine hydrate (0.4 mL) was added and the mixture stirred at 65 °C for 2 h. The mixture was cooled to rt, poured into 1 M HCl solution (100 mL), and ethyl acetate (400 mL) was added.
- Step e 6-Chloro-1 H -pyrazolo[3,4- b ]pyrazine was dissolved in acetonitrile (24 mL). N -iodosuccinimide (3.43 g, 14.5 mmol) and tetrafluoroboric acid solution (2.8 mL, 21.7 mmol, 48% in water) were successively added. The resulting brown/orange mixture was then stirred at reflux for 2 h. A beige/brown precipitate formed and the mixture cooled to room temperature, then placed into an ice/water bath for 5 min.
- Step f 6-Chloro-3-iodo-1 H -pyrazolo[3,4- b ]pyrazine (850 mg, 3 mmol) was dissolved in dichloromethane (15 mL). 3,4-Dihydro-2H-pyran (0.85 mL, 9.1 mmol) and p- toluenesulfonic acid monohydrate (176 mg, 0.91 mmol) were successively added to the flask. The resulting mixture was stirred at room temperature for 10 min. The mixture became homogeneous and darkish overtime. After this time, a saturated aqueous solution of NaHCO 3 (20 mL) was added to the flask and the biphasic mixture stirred for 10 min.
- Step a A solution of sodium hydride (213 mg, 5.34 mmol) in DMF (10 mL) was cooled to 0 °C, then 6-chloro-3-iodo-1H-pyrazolo[3,4-b]pyrazine (1 g, 3.56 mmol, synthesized via Steps a-e of Intermediate B) was added. The reaction mixture was allowed to warm to rt and the reaction was stirred for 2.25 hr. The reaction mixture was then cooled to 0-10°C and methyl carbonochloridate (817 ⁇ L, 10.6 mmol) was added and the reaction mixture was stirred for 20 min. On completion, water was added (20 mL), then the mixture was poured in water (60mL).
- Step a A round bottomed flask was charged with methyl 6-chloro-3-iodo-1H-pyrazolo[3,4-b]pyrazine-1-carboxylate (525 mg, 1.55 mmol, Intermediate B), 2-(acetyloxy)acetic acid (1.46 g, 12.4 mmol), silver nitrate (52.6 mg, 0.31 mmol), and acetonitrile (15 mL) and water (9 mL). To the mixture was added ammonium persulfate (2.82 g, 12.4 mmol), and the reaction was heated to 85 °C. After 2 h, the reaction was cooled to room temperature and poured into ethyl acetate and brine.
- Step b A reaction tube containing methyl 5-[(acetyloxy)methyl]-6-chloro-3-iodo-1H-pyrazolo[3,4-b]pyrazine-1-carboxylate (220 mg, 0.5358 mmol) in dichloromethane (4 mL) was charged with piperidine (52.9 ⁇ L, 0.5358 mmol) at room temperature. After 15 min, further piperidine (0.2 equiv) was added. After 15 min, 3,4-dihydro-2H-pyran (145 ⁇ L, 1.60 mmol) and 4-methylbenzene-1-sulfonic acid (92.2 mg, 0.5358 mmol) were added.
- Step a A mixture of 1,3-dihydrospiro[indene-2,4'-piperidin]-3-amine dihydrochloride (800.0 mg, 2.9 mmol, Intermediate E), 6-chloro-3-iodo-1-(oxan-2-yl)-1 H -pyrazolo[3,4- b ]pyrazine (1.05 g, 2.9 mmol, Intermediate A) and Et 3 N (2.0 mL, 14.4 mmol) in DMF (20 mL) was stirred at 60 °C for 1 hour. To the reaction mixture was added Boc 2 O (757.0 mg, 3.5 mmol) and the reaction was stirred at 60 °C for another 2 hours.
- Boc 2 O 757.0 mg, 3.5 mmol
- Step a To a vial with [6-chloro-3-iodo-1-(oxan-2-yl)-1H-pyrazolo[3,4-b]pyrazin-5-yl]methyl acetate (76 mg, 0.1740 mmol, Intermediate C) in DMF (0.13 mL, 1 mL) was added 1,3-dihydrospiro[indene-2,4'-piperidin]-3-amine ditrifluoroacetate (89.4 mg, 0.2088 mmol, Intermediate E) in 1 mL DMF and ethylbis(propan-2-yl)amine (121 ⁇ L, 0.696 mmol, Hunig's base).
- Step a Dissolved tert-butyl 3-oxo-1,3-dihydrospiro[indene-2,4'-piperidine]-1'-carboxylate (1 g, 3.31 mmol, Intermediate D) in 2-Me-THF (20 mL). Then tetratitanium-1-ylium tetraethanolate (4.50 mL, 13.2 mmol) was added followed by (R)-2-methylpropane-2-sulfinamide (721 mg, 5.95 mmol) and the reaction mixture was stirred at 90 °C for 16 h.
- reaction mixture was then cooled to 0 °C and lithium(1+) borohydride (79.2 mg, 3.64 mmol) was added portion-wise and the mixture was stirred for 0.5 h.
- the reaction was then quenched with methanol and concentrated in vacuo. EtOAc and water were added and the mixture was extracted with EtOAc, and the organic layer was dried over Na 2 SO 4 , filtered and concentrated.
- Step a Dissolved tert-butyl (3S)-3- ⁇ [(R)-2-methylpropane-2-sulfinyl]amino ⁇ -1,3-dihydrospiro[indene-2,4'-piperidine]-1'-carboxylate (500 mg, 1.22 mmol, Intermediate H) in MeOH (15 mL) and added hydrogen chloride (3.05 mL, 12.2 mmol), and the reaction mixture was stirred at rt for 16 h. Then 1 mL more 4N HCl was added and the reaction mixture was stirred at rt for 1 h, then heated to 60 °C for 2 h. The reaction mixture was then concentrated.
- Step a Dissolved (3S)-1,3-dihydrospiro[indene-2,4'-piperidin]-3-amine dihydrochloride (1.37 g, 5.01 mmol, Intermediate I) in DMF (15 mL), then added 6-chloro-3-iodo-1-(tetrahydro-2H-pyran-2-yl)-1H-pyrazolo[3,4-b]pyrazine (2 g, 7.13 mmol, Intermediate A) followed by ethylbis(propan-2-yl)amine (4.97 mL, 28.5 mmol).
- Step a Dissolved [6-chloro-3-iodo-1-(oxan-2-yl)-1H-pyrazolo[3,4-b]pyrazin-5-yl]methyl acetate (528 mg, 1.21 mmol, Intermediate C) in DMF (10 mL). Then added (3S)-1,3-dihydrospiro[indene-2,4'-piperidin]-3-amine dihydrochloride (335 mg, 1.21 mmol, Intermediate I) followed by ethylbis(propan-2-yl)amine (844 ⁇ L, 4.84 mmol) to the reaction and the mixture was stirred at 75 °C for 16 h.
- reaction mixture was then cooled to rt and di-tert-butyl dicarbonate (305 ⁇ L, 1.33 mmol) was added and the reaction was stirred at rt for 3 h.
- the reaction was then diluted wtih EtOAc and water. The layers were separated, then the organic layer was washed with brine, dried over Na 2 SO 4 , filtered and concentrated directly onto SiO 2 .
- Step a A solution of BnOH (30.8 g, 0.285 mol, 29.6 mL, 1.1 eq ) in THF (450 mL) was cooled to 0 °C. NaH (12.4 g, 0.311 mol, 60% oil dispersion, 1.2 eq ) was added slowly to the mixture at 0 °C. After addition, the mixture was stirred at 25°C for 1 hr. The resultant solution was added a solution of 3,5-dichloropyrazine-2-carbonitrile (45.0 g, 258.64 mmol, 1.0 eq ) in THF (450 mL) at -78 °C and the mixture was stirred at -78 °C for 0.5 hr.
- Step b To a solution of 5-(benzyloxy)-3-chloropyrazine-2-carbonitrile (20.0 g, 75.6 mmol, 1.0 eq ) in THF (200 mL) was added DIBAL-H (1.0 M, 227 mL, 3.0 eq ) at -78 °C under N 2 . The mixture was stirred at -78 °C for 1hr. The reaction was quenched by a solution of 10% aqueous HOAc (2.00 L) at -78 °C and extracted with EtOAc (1.50 L*3). The combined organic layer was adjusted pH to 8-9 with saturated aqueous of NaHCO 3 and separated.
- DIBAL-H 1.0 M, 227 mL, 3.0 eq
- Step c To a mixture of 5-(benzyloxy)-3-chloropyrazine-2-carbaldehyde (11.3 g, 45.4 mmol, 1.0 eq ) and NH 2 NH 2 ⁇ H 2 O (6.96 g, 137 mmol, 6.76 mL, 3.0 eq ) in EtOH (113 mL) was stirred at 25 °C. Then Et 3 N (23.0 g, 228 mmol, 31.6 mL, 5.00 eq ) was added to the mixture at 25 °C. The mixture was heated to 80 °C and stirred at 80 °C for 16 hrs. Then the reaction was concentrated to give a residue.
- Step d To a solution of 6-(benzyloxy)-1H-pyrazolo[3,4-b]pyrazine (9.50 g, 35.4 mmol, 1.0 eq ) in DMF (190 mL) was added NIS (10.4 g, 46.0 mmol, 1.3 eq ) at 25 °C. Then the mixture was heated to 80 °C and stirred for 1 h. The reaction was then cooled to 25 °C and then poured into ice-water (2.00 L). The mixture was extracted with EtOAc (2.00 L).
- Step e To a solution of 6-(benzyloxy)-3-iodo-1H-pyrazolo[3,4-b]pyrazine (12.1 g, 34.3 mmol, 1.0 eq ) in DCM (20.0 mL) was added DHP (8.65 g, 103 mmol, 9.40 mL, 3.0 eq ) and TsOH ⁇ H 2 O (1.96 g, 10.3 mmol, 0.3 eq ) . The mixture was stirred at 25 °C for 0.5 hr. Three batched in parallel were combined for work-up. The mixture was poured into saturated NaHCO 3 solution (250 mL) and then extracted with EtOAc (250 mL*2).
- Step f To a mixture of 6-(benzyloxy)-3-iodo-1-(tetrahydro-2H-pyran-2-yl)-1H-pyrazolo[3,4-b]pyrazine (12.9 g, 27.9 mmol, 1.00 eq ) and 1,2,3,4-tetrahydro-1,5-naphthyridine (3.74 g, 27.9 mmol, 1.00 eq ) in toluene (130 mL) was added RuPhos (2.60 g, 5.57 mmol, 0.2 eq ), Pd 2 (dba) 3 (766 mg, 836 umol, 0.03 eq ) and Cs 2 CO 3 (27.3 g, 83.6 mmol, 3.0 eq ) at 25 °C under N 2 .
- the mixture was heated to 100 °C and stirred at 100 °C for 20 hrs.
- the mixture was filtered and to the filtrate was added water (500 mL) and extracted with EtOAc (500 mL).
- the combined organic layer was washed with 0.5 M aq.HCl (200 mL).
- the aqueous layer was further extracted with DCM (200 mL*2).
- the combined organic layers (EtOAc and DCM) were washed with sat. NaHCO 3 (200 mL), brine (200 mL), dried over Na 2 SO 4 , filtered and concentrated.
- Step g To a solution of 1-(6-(benzyloxy)-1-(tetrahydro-2H-pyran-2-yl)-1H-pyrazolo[3,4-b]pyrazin-3-yl)-1,2,3,4-tetrahydro-1,5-naphthyridine (9.00 g, 20.4 mmol, 1.00 eq ) in MeOH (950 mL) was added Pd(OH) 2 /C (1.14 g, 4.07 mmol, 50% purity, 0.20 eq ) under N 2 . The suspension was degassed under vacuum and purged with H 2 several times. The mixture was stirred under H 2 (50 psi) at 25 °C for 30 hrs.
- Step h To a mixture of 3-(3,4-dihydro-1,5-naphthyridin-1(2H)-yl)-1-(tetrahydro-2H-pyran-2-yl)-1H-pyrazolo[3,4-b]pyrazin-6-ol (7.00 g, 19.9 mmol, 1.0 eq ) in DCM (70.0 mL) was added DIPEA (2.82 g, 21.9 mmol, 3.81 mL, 1.1 eq ) at 25 °C.
- Step a Dissolved tert-butyl 2-oxo-2,3-dihydrospiro[indene-1,4'-piperidine]-1'-carboxylate (500 mg, 1.65 mmol, CAS# 241819-85-2) and 1-phenylmethanamine (264 mg, 2.47 mmol) in DCE (10 mL). Then acetic acid (9.42 ⁇ L, 0.165 mmol) was added and the reaction mixture was stirred at rt for 1 hr. Next, sodium cyanoboranuide (155 mg, 2.47 mmol) was added and the reaction was stirred at rt for 48 h.
- Step b Dissolved tert-butyl 2-(benzylamino)-2,3-dihydrospiro[indene-1,4'-piperidine]-1'-carboxylate (242 mg, 0.616 mmol) and trifluoroacetic acid (70.2 mg, 0.616 mmol) in MeOH (5 mL). The reaction mixture was then cycled through a H-Cube at 3 bar hydrogen gas with a 10% Pd/C cartridge for 2 hr at rt.
- Step b A solution of 3-[(3-hydroxycyclobutyl)amino]-4-nitrobenzonitrile (5.60 g, 24.0 mmol) and Pd/C (1.00 g, 10%) in MeOH (100.0 mL) was stirred at 10 °C for 12 hours under H 2 (15 psi). The reaction mixture was then filtered and the filtrate was concentrated in vacuo to give 4-amino-3-[(3-hydroxycyclobutyl)amino]benzonitrile (5.00 g, quant. crude yield) as a yellow gum.
- LCMS m/z [M+H] + 203.9.
- Step c A solution of 4-amino-3-[(3-hydroxycyclobutyl)amino]benzonitrile (5.00 g, 24.6 mmol), 1,2-dibromoethane (18.40 g, 98.4 mmol), TBAB (31.70 g, 98.4 mmol) and TEA (13.7 mL, 98.4 mmol) was stirred at 60 °C for 12 hours. The solution was added into H 2 O (500.0 mL) and then extracted with EtOAc (500.0 mL ⁇ 2). The combined organic layers were washed with brine (500.0 mL), dried over anhydrous Na 2 SO 4 , filtered and concentrated in vacuo to give crude product as orange gum.
- Step d A solution of cis -3-hydroxycyclobutyl]-1,2,3,4-tetrahydroquinoxaline-6-carbonitrile (3.50 g, 15.2 mmol), TBSCl (2.96 g, 19.7 mmol) and imidazole (2.06 g, 30.4 mmol) in CH 2 Cl 2 (30.0 mL) was stirred at 40 °C for 0.5 hour. The mixture was concentrated in vacuo.
- Step a The mixture of 3-bromo-6-chloropyrazin-2-amine (600 mg, 2.87 mmol, 1.0 eq , CAS# 212779-21-0), (2,3-dichlorophenyl)boronic acid (547 mg, 2.87 mmol, 1.0 eq ), Pd(dppf)Cl 2 (210 mg, 287 ⁇ m ⁇ l, 0.1 eq ) and K 3 PO 4 (1.82 g, 8.61 mmol, 3.0 eq ) in dioxane (15 mL) and H 2 O (3 mL) was evacuated and refilled 3 times with N 2 gas, then stirred at 70 °C for 12 hours.
- Step a A round bottomed flask was charged with tert-butyl 4-cyanopiperidine-1-carboxylate (533 mg, 2.53 mmol) and THF (10 mL) before being cooled to -78°C for the addition of lithiobis(propan-2-yl)amine (2.90 mL, 2.90 mmol). After 45 min, a solution of 2-(bromomethyl)-1-fluoro-3-iodobenzene (954 mg, 3.03 mmol) in THF (2 mL) was added, and the reaction warmed to rt. After 45 min, the reaction was diluted with water and ethyl acetate.
- Step b A round bottomed flask was charged with tert-butyl 4-cyano-4-[(2-fluoro-6-iodophenyl)methyl]piperidine-1-carboxylate (860 mg, 1.93 mmol), Pd/P(tBu) 3 G2 (98.8 mg, 0.1930 mmol), DMF (15 mL), water (1.5 mL), and triethylamine (320 ⁇ L, 2.31 mmol). Nitrogen was bubbled through the mixture for 5 min, before the vial was sealed and heated to 130 °C.
- Step c A reaction tube was charged with tert-butyl 4-fluoro-1-oxo-1,3-dihydrospiro[indene-2,4'-piperidine]-1'-carboxylate (280 mg, 0.8767 mmol), (R)-2-methylpropane-2-sulfinamide (158 mg, 1.31 mmol), 2-MeTHF, and tetratitanium-1-ylium tetraethanolate (1.19 mL, 3.50 mmol). The vial was sealed and heated to 80 °C overnight. The mixture was cooled to rt and charged with boranium lithiumuide (28.5 mg, 1.31 mmol).
- Step d A reaction tube was charged with tert-butyl (3S)-7-fluoro-3- ⁇ [(R)-2-methylpropane-2-sulfinyl]amino ⁇ -1,3-dihydrospiro[indene-2,4'-piperidine]-1'-carboxylate (170 mg, 0.40 mmol) and methanol (3 mL), followed by hydrogen chloride (1 mL, 4.00 mmol). The reaction mixture was stirred at rt for 16 h. The solvent was then removed in vacuo, and the residue was triturated with methyl tertbutyl ether.
- Step a Dissolved (3 S)-4-fluoro-1,3-dihydrospiro[indene-2,4'-piperidin]-3-amine dihydrochloride (1.38 g, 4.70 mmol, Intermediate T) in DMF (12 mL). 6-chloro-3-iodo-1-(oxan-2-yl)-1H-pyrazolo[3,4-b]pyrazine (1.71 g, 4.70 mmol, Intermediate A) was then added followed by ethylbis(propan-2-yl)amine (3.27 mL, 18.8 mmol) and the reaction mixture was stirred at 75 °C for 2 h.
- reaction mixture ws then cooled to rt and di-tert-butyl dicarbonate (1.17 mL, 5.17 mmol) was added and the reaction mixture was stirred at rt for 2 h.
- the reaction mixture was then diluted with EtOAc and water. The layers were separated, then the organic layer was washed with brine, dried over Na 2 SO 4 , filtered and concentrated onto SiO 2 .
- Step a To a solution of 6-bromo-1,2,3,4-tetrahydro-1,5-naphthyridine (4.0 g, 18.7 mmol) in dioxane (50 mL) and H 2 O (5 mL) were added Cs 2 CO 3 (12.1 g, 37.4 mmol), 1-(oxan-2-yl)-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1 H -pyrazole (7.8 g, 28.0 mmol, CAS# 903550-26-5) and Pd(dppf)Cl 2 (684 mg, 935 mmol).
- Step a To the mixture of 6-chloro-1,2,3,4-tetrahydro-1,5-naphthyridine (2 g, 11.8 mmol) and 1-methyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1 H -pyrazole (2.93 g, 14.1 mmol) in dioxane (35.0 mL) and H 2 O (5.0 mL) were added Pd(dppf)Cl 2 (1.29 g, 1.77 mmol) and K 3 PO 4 (5.49 g, 25.9 mmol) under N 2 . The mixture was stirred at 100 °C under N 2 for 3 hrs.
- Step a 6-chloro-3-iodo-1-(oxan-2-yl)-1H-pyrazolo[3,4-b]pyrazine (150 mg, 0.4114 mmol, Intermediate A) in DMF (4 mL) was charged with ethylbis(propan-2-yl)amine (355 ⁇ L, 2.05 mmol) and (S)-6-fluoro-1,3-dihydrospiro[indene-2,4'-piperidin]-1-amine dihydrochloride (120 mg, 0.411 mmol, Intermediate Y) and the solution was heated to 75°C for 4 hr.
- the org layer was pre-absorbed on SiO 2 (2g) and purified on by column chromatography (12g column, 20-70%EA/hep) to give tert-butyl ((3S)-5-fluoro-1'-(3-iodo-1-(tetrahydro-2H-pyran-2-yl)-1H-pyrazolo[3,4-b]pyrazin-6-yl)-1,3-dihydrospiro[indene-2,4'-piperidin]-3-yl)carbamate (175 mg, 66% yield) as a white solid.
- LCMS m/z [M+H] + 649.2.
- Step a A vial was charged with 6-chloro-3-iodo-1-(oxan-2-yl)-1H-pyrazolo[3,4-b]pyrazine (2 g, 5.48 mmol, Intermediate A), XantPhos-Pd-G4 (263 mg, 0.274 mmol), 1,2,3,4-tetrahydro-1,5-naphthyridine (720 mg, 5.37 mmol), and Cs 2 CO 3 (3.54 g, 10.9 mmol) in PhMe (20 mL). The mixture was bubbled with nitrogen for 10 min, then the vial was sealed and heated to 60 °C for 48 h. The reaction mixture was cooled water and EA were added.
- Step a To the stirred mixture of propane-1,2-diamine (5.00 g, 67.4 mmol, 5.76 mL, 1.00 eq ) in EtOH (25.0 mL) was added diethyl 2-oxomalonate (11.7 g, 67.4 mmol, 10.4 mL, 1.00 eq ) dropwise at 0 °C. The mixture was warmed to 25 °C. The reaction was stirred at 25 °C for 2 h, then the reaction was stirred at 95 °C for 18 h.
- Step b To the stirred solution of ethyl 3-hydroxy-5-methylpyrazine-2-carboxylate (2.24 g, 12.3 mmol, 1.00 eq ) in DMF (11.2 mL) was added NBS (2.30 g, 12.9 mmol, 1.05 eq ) in one portion at 0 °C under N 2 , then the mixture was stirred at 20 °C for 2 h. The reaction mixture was poured into H 2 O (60.0 mL) where solid precipitate formed. The suspension was filtered and the solid filtrate was dried under reduced pressure to give ethyl 6-bromo-3-hydroxy-5-methylpyrazine-2-carboxylate (1.90 g, 59% yield) as a light yellow solid.
- Step c To the stirred mixture of ethyl 6-bromo-3-hydroxy-5-methylpyrazine-2-carboxylate (1.90 g, 7.28 mmol, 1.00 eq ) and K 2 CO 3 (4.02 g, 29.1 mmol, 4 eq ) in ACN (9.50 mL) and H 2 O (1.90 mL) was added (2,3-dichlorophenyl)boronic acid (1.39 g, 7.28 mmol, 1.00 eq ) and Pd(dppf)Cl 2 ⁇ CH 2 Cl 2 (594 mg, 728 umol, 0.10 eq ) under N 2 at 20 °C.
- Step d A solution of ethyl-6-(2,3-dichlorophenyl)-3-hydroxy-5-methylpyrazine-2-carboxylate (150.0 mg, 458.0 ⁇ m ⁇ l), TsCl (130.0 mg, 686.0 umol) and DIPEA (241.0 uL, 1.37 mmol) in CH 2 Cl 2 (3.0 mL) was stirred at 20 °C for 1 hour. The solution was poured into H 2 O (10.0 mL) and extracted with CH 2 Cl 2 (10.0 mL ⁇ 2).
- Step a A 250 mL round bottomed flask was charged with 3-chloro-4-iodopyridin-2-amine (1 g, 3.92 mmol), 9- ⁇ [5-(diphenylphosphanyl)-9,9-dimethyl-9H-xanthen-4-yl]diphenyl-4-phosphanyl ⁇ -O-methanesulfonyl-8-methyl-8-4-aza-9-palladatricyclo[8.4.0.0 2 ,7]tetradeca-1(14),2,4,6,10,12-hexaene-9,9-bis(ylium)-10-uid-9-olate(188 mg, 0.196 mmol), dioxane (30 mL), methyl 3-sulfanylpropanoate (476 ⁇ L, 4.31 mmol) and ethylbis(propan-2-yl)amine (1.36 mL, 7.84 mmol).
- Step b Methyl 3-[(2-amino-3-chloropyridin-4-yl)sulfanyl]propanoate (890 mg, 3.60 mmol), and ethoxysodium (1.40 mL, 3.78 mmol) were dissolved in THF (10 mL). The mixture was stirred at 25°C for 10 min. The mixture was diluted with DCM (10-15 mL) and stirred until nucleation occurred; after 5 min, large amount of solid formed in suspension. Additional DCM (86 mL) was added, the reaction was filtered and the filter cake washed with DCM and was air dried.
- Step a A mixture of 6-bromo-1,2,3,4-tetrahydro-1,5-naphthyridine (1 g, 4.69 mmol, CAS# 1219022-46-4), 2-(tributylstannyl)-1,3-oxazole (2.51 g, 7.03 mmol), Pd 2 (dba) 3 (429 mg, 469 ⁇ mol) and XPhos (447 mg, 938 ⁇ mol) in dioxane (30 mL) was stirred at 100 °C for 12 hours under N 2 atmosphere. After cooling to room temperature, KF (2 g) was added and the reaction mixture was stirred at 20 °C for 0.5 hour.
- Step c A mixture of 4-amino-3-[(1-cyanocyclopropyl)amino]benzonitrile (500.0 mg, 2.5 mmol), TBAB (3.2 g, 10.0 mmol), TEA (1.1 mL, 8.6 mmol) and 1,2-dibromoethane (1.5 mL, 17.4 mmol) was stirred at 60 °C for 24 hours. The mixture was poured into water (50 mL) and extracted with DCM (50 mL ⁇ 3). The organic layers were washed with brine and dried over anhydrous Na 2 SO 4 , filtered and the filtrate was concentrated in vacuo to give a residue.
- Step a The mixture of 1,3-dihydrospiro[indene-2,4'-piperidin]-3-amine (100 mg, 494 ⁇ mol, Intermediate E), Boc 2 O (322 mg, 1.48 mmol) and TEA (149 mg, 1.48 mmol) in DCM (3 mL) was stirred at 30 °C for 2 hours. Then the mixture was concentrated under reduced pressure.
- Step a A solution of tert-butyl 6-bromo-1-oxo-1,3-dihydrospiro[indene-2,4'-piperidine]-1'-carboxylate (1.5 g, 3.94 mmol, synthesized via Steps a-c of Intermediate AL), AcONH 4 (3.03 g, 39.4 mmol) and NaBH 3 CN (297 mg, 4.72 mmol) in EtOH (30 mL) was stirred at 80 °C for 1 h.
- Step b A solution of tert-butyl 1-amino-6-bromo-1,3-dihydrospiro[indene-2,4'-piperidine]-1'-carboxylate (1.5 g, 3.93 mmol), Boc 2 O (1.02 g, 4.71 mmol) and Et 3 N (1.61 mL, 11.7 mmol) in DCM (30 mL) was stirred at 20 °C for 1 h. The solution was added into H 2 O (100 mL) and then extracted with CH 2 Cl 2 (50 mL x 2).
- Step a A solution of tert-butyl 6-bromo-1-((tert-butoxycarbonyl)amino)-1,3-dihydrospiro[indene-2,4'-piperidine]-1'-carboxylate (300.0 mg, 623.0 ⁇ m ⁇ l), Pd(dppf)Cl 2 (45.6 mg, 62.3 umol) and TEA (256.0 uL, 1.86 mmol) in MeOH (20.0 mL) was stirred at 80 °C for 12 hours under CO (50 psi).
- Step c A solution of 1'-[( tert -butoxy)carbonyl]-1- ⁇ [( tert -butoxy)carbonyl]amino ⁇ -1,3-dihydrospiro[indene-2,4'-piperidine]-6-carboxylic acid (200.0 mg, 447.0 ⁇ m ⁇ l), dimethylamine hydrochloride (109.0 mg, 1.34 mmol), HATU (254.0 mg, 670.0 umol) and TEA (245.0 uL, 1.78 mmol) in DMF (5.0 mL) was stirred at 50 °C for 0.5 hour. The reaction mixture was poured into H 2 O (20.0 mL) and extracted with EtOAc (20.0 mL ⁇ 2).
- Step d A solution of tert -butyl 1- ⁇ [( tert -butoxy)carbonyl]amino ⁇ -6-(dimethylcarbamoyl)-1,3-dihydrospiro[indene-2,4'-piperidine]-1'-carboxylate (200.0 mg, 422.0 ⁇ mol) in HCl/MeOH (5.0 mL, 4 M) was stirred at 20 °C for 0.5 hour. The reaction mixture was concentrated under reduced pressure to give the product of 1-amino- N , N- dimethyl-1,3-dihydrospiro[indene-2,4'-piperidine]-6-carboxamide dihydrochloride (150.0 mg, quant. crude yield) as a yellow oil.
- LCMS m/z [M+H] + 274.1.
- Step a To a mixture of 2-chloro-3-fluoro-4-iodopyridine (900 mg, 3.5 mmol) and 2-ethylhexyl 3-sulfanylpropanoate (912 mg, 4.2 mmol) in dioxane (10 mL) were added Pd 2 (dba) 3 (319 mg, 0.3 mmol), XantPhos (403 mg, 0.7 mmol) and DIPEA (1.8 mL, 10.4 mmol). The reaction mixture was purged with N 2 for 3 min and stirred at 100 °C for 12 hours under N 2 .
- Step a A mixture of methyl 3,5-dichloropyrazine-2-carboxylate (2.0 g, 9.7 mmol, synthesized via Step a of Intermediate A), trimethyl-1,3,5,2,4,6-trioxatriborinane (2.4 g, 19.3 mmol), Pd(PPh 3 ) 4 (558 mg, 483 ⁇ mol) and Cs 2 CO 3 (6.3 g, 19.3 mmol) in dioxane (70 mL) was stirred at 110 °C for 12 hours under N 2 atmosphere.
- Step a To a solution of 1- tert -butyl-4-methyl piperidine-1,4-dicarboxylate (10.00 g, 41.10 mmol) in THF (150.0 mL) was added LDA (24.6 mL, 49.3 mmol, 2 M) at - 78 °C under N 2 . The mixture was stirred at - 78 °C for 1 hour. To the mixture was added 1-bromo-4-(bromomethyl)benzene (10.70 g, 43.10 mmol) in THF (50.0 mL) at -78 °C. The mixture was then stirred at 20 °C for 11 hours under N 2 .
- Boc 2 O (10.90 g, 50.2 mmol) at 20 °C.
- the mixture was stirred at 20 °C for 1 hour.
- the reaction mixture was filtered and the filtrate was extracted with CH 2 Cl 2 (100.0 mL ⁇ 2).
- the combined organic layers were washed with brine (200.0 mL), dried over anhydrous Na 2 SO 4 , filtered and concentrated to give a yellow residue.
- Step d To a solution of tert -butyl 6-bromo-1-oxo-1,3-dihydrospiro[indene-2,4'-piperidine]-1'-carboxylate (1.00 g, 2.62 mmol) and Ti(OEt) 4 (2.17 mL, 10.4 mmol) in 2-Me-THF (20.0 mL) was added ( R )-2-methylpropane-2-sulfinamide (635.0 mg, 5.24 mmol). The reaction mixture was stirred at 90 °C for 12 hours under N 2 .
- Step e To a solution of tert -butyl (1 E )-6-bromo-1- ⁇ [( R )-2-methylpropane-2-sulfinyl]imino ⁇ -1,3-dihydrospiro[indene-2,4'-piperidine]-1'-carboxylate (1.30 g, 2.68 mmol) in 2-Me-THF (20.0 mL) was added LiBH 4 (2.68 mL, 5.36 mmol) at 0 °C. The mixture was stirred at 20 °C for 1 hour. The reaction mixture was quenched with MeOH, then triturated with H 2 O (200.0 mL) and extracted with EtOAc (200.0 mL ⁇ 2).
- Step a A solution of tert -butyl (1 S )-6-bromo-1- ⁇ [( R )-2-methylpropane-2-sulfinyl]amino ⁇ -1,3-dihydrospiro[indene-2,4'-piperidine]-1'-carboxylate (550.0 mg, 1.13 mmol, Intermediate AL), Zn(CN) 2 (265.0 mg, 2.26 mmol) and XantPhos-Pd-G4 (108.0 mg, 113.0 umol) in DMF (20.0 mL) was stirred at 100 °C for 12 hours under N 2 .
- reaction mixture was poured into H 2 O (100.0 mL) and extracted with EtOAc (100.0 mL x 2). The combined organic layers were washed with brine (200.0 mL), dried over anhydrous Na 2 SO 4 , filtered and concentrated to give an orange residue.
- Step b A solution of tert -butyl (1 S )-6-cyano-1- ⁇ [( R )-2-methylpropane-2-sulfinyl]amino ⁇ -1,3-dihydrospiro[indene-2,4'-piperidine]-1'-carboxylate (450.0 mg, 1.04 mmol) in HCl/MeOH (15.0 mL, 4 M) was stirred at 20 °C for 0.5 hour. The reaction mixture was concentrated to give the product of ( S )-1-amino-1,3-dihydrospiro[indene-2,4'-piperidine]-6-carbonitrile dihydrochloride (450.0 mg, 70% purity, quant. crude yield) as a white solid.
- LCMS m/z [M+H] + 228.1.
- Step a To the mixture of 4-bromo-2-fluoro-1-nitrobenzene (25.0 g, 113.0 mmol) in MeOH (100.0 mL) and THF (50.0 mL) was added MeNH 2 (67.5 mL, 135.0 mmol, 2 M in THF) dropwise. The mixture was stirred at 10 °C for 12 hours. Then more MeNH 2 (60.0 mL, 2 M in THF) was added to the mixture and the mixture was stirred at 45 °C for 12 hours. The mixture was concentrated in vacuo to give residue. Water (200.0 mL) added to the mixture and the mixture was extracted with EtOAc (200.0 mL ⁇ 2).
- Step b To the mixture of 5-bromo- N -methyl-2-nitroaniline (10.0 g, 43.2 mmol) in MeOH (150.0 mL) was added sodium dithionite (67.5 g, 388.0 mmol) in H 2 O (60.0 mL) dropwise. The mixture was stirred at 60 °C for 12 hours. The mixture was then filtered and the filtrate was concentrated in vacuo. The residue was extracted with EtOAc (200.0 mL ⁇ 3), the organic layers were washed with H 2 O (100.0 mL) and brine (100.0 mL), then dried over anhydrous Na 2 SO 4 . The mixture was filtered and the filtrate was concentrated in vacuo to give the product of 5-bromo- N 1-methylbenzene-1,2-diamine (8.60 g, crude) as a brown oil.
- Step a To a solution of 2-fluorobenzaldehyde (4 g, 32.2 mmol) in DCM (20 mL) were added propane-1, 3-dithiol (3.5 mg, 32.2 mmol) and I 2 (244 mg, 966 umol). The mixture was stirred at 25 °C for 12 hours. The reaction mixture was poured into the solution of Na 2 S 2 O 3 (0.4 M, 180 mL) and 150 mL of NaOH solution was added. The organic phase was separated and the aqueous phase was extracted with CH 2 Cl 2 (200 mL).
- Step b To a mixture of 2-(2-fluorophenyl)-1,3-dithiane (4 g, 18.6 mmol) in THF (50 mL) was added LDA (18.6 mL, 37.2 mmol) at -78 °C slowly. The resulting mixture was stirred at -20 °C for 0.5 hour, then tert -butyl 4-oxopiperidine-1-carboxylate (3.7 g, 18.6 mmol) was added at -78 °C. The reaction mixture was stirred at -78 °C for 2 hours. The reaction mixture was then poured into saturated NH 4 Cl (50 mL) and extracted with EtOAc (80 mL ⁇ 3).
- Step c A mixture of tert -butyl 4-[2-(2-fluorophenyl)-1,3-dithian-2-yl]-4-hydroxypiperidine-1-carboxylate (2 g, 4.83 mmol) in DCM (20 mL) and H 2 O (5 mL) were added pyridine (2 mL), pyridine ⁇ HBr 3 (1.82 g, 5.79 mmol) and TBAB (158 mg, 483 umol). The mixture was stirred at 25 °C for 12 hours. The solution was poured into water (30 mL) and extracted with DCM (50 mL ⁇ 3).
- Step d To a solution of tert -butyl 4-(2-fluorobenzoyl)-4-hydroxypiperidine-1-carboxylate (600 mg, 1.85 mmol) in dioxane (5 mL) was added t -BuOK (207 mg, 1.85 mmol). The mixture was stirred at 70 °C for 2 hours. The mixture was concentrated under reduced pressure and diluted with water (20 mL), extracted by EtOAc (30 mL ⁇ 3). The combined organic layers were washed with brine (30.0 mL), dried over anhydrous Na 2 SO 4 , filtered and concentrated under reduced pressure to give a residue.
- Step a 6-methyl-1,6-naphthyridin-6-ium iodide (1.56 g, 5.73 mmol, CAS# 37960-58-0) was suspended in water (10 mL) and cooled to 0°C. The reaction was charged with sodium hydroxide (1.25 g, 31.5 mmol) in water (10 mL) and tripotassium hexakis(iminomethanide) iron (4.04 g, 12.3 mmol) in water (10 mL). The solution was stirred for 1 hr at 0 °C, then 16 hr at rt. The mixture was extracted with CHCl 3 , dried and pre-absorbed on SiO 2 (3g).
- Step a tert-butyl 4-cyanopiperidine-1-carboxylate (1.65 g, 7.84 mmol) in THF (10 mL) was cooled to -78 °C and charged with lithiobis(propan-2-yl)amine (9.01 mL, 9.01 mmol) (max temperature -65 °C on addition) and the reaction was stirred at -78 °C for 1.5 hr.
- Step b tert-butyl 4-[(2-bromopyridin-3-yl)methyl]-4-cyanopiperidine-1-carboxylate (910 mg, 2.39 mmol) in 2-MeTHF (15 mL) was cooled to 0 °C and charged with chloro(propan-2-yl)magnesium; chlorolithium(3.67 mL, 4.78 mmol) and the reaction was stirred at 0 °C for 30 min. Next, the reaction was cooled to -78 °C and charged with butyllithium (1.04 mL, 2.62 mmol) and the reaction was stirred for 1 hr at -78 °C. Then an additional 0.25 eq.
- Step c tert-butyl 7-imino-5,7-dihydrospiro[cyclopenta[b]pyridine-6,4'-piperidine]-1'-carboxylate (720 mg, 2.38 mmol) was dissolved in EA (15 mL) and run in a H-Cube for 90 min (5 bar, 40 °C). The solvent was then removed by rotary evaporation and the crude residue was purified by prep-HPLC (5-40% ACN/water/FA). tert-butyl 7-amino-5,7-dihydrospiro[cyclopenta[b]pyridine-6,4'-piperidine]-1'-carboxylate (208 mg, 29% yield).
- Step d tert-butyl 7-amino-5,7-dihydrospiro[cyclopenta[b]pyridine-6,4'-piperidine]-1'-carboxylate (208 mg, 0.6855 mmol) in MeOH (5 mL) was charged with hydrogen chloride (1.71 mL, 6.85 mmol) and the reaction was stirred at rt for 2.5 h. Then the reaction was heated to 50 °C for 5 h. The reaction was cooled to rt and stirred for 16 h.
- Step a 6-chloro-3-iodo-1-(oxan-2-yl)-1H-pyrazolo[3,4-b]pyrazine (244 mg, 0.6697 mmol, Intermediate A) in DMF (4 mL) was charged with ethylbis(propan-2-yl)amine (580 ⁇ L, 3.34 mmol) and 5,7-dihydrospiro[cyclopenta[b]pyridine-6,4'-piperidin]-7-amine dihydrochloride (185 mg, 0.6697 mmol, Intermediate AT) and the solution was heated to 80 °C for 4 h.
- Step a To a solution of 2,3-dichloro-4-iodopyridine (2 g, 7.30 mmol) and 2-ethylhexyl 3-sulfanylpropanoate (1.75 g, 8.01 mmol) in dioxane (20 mL) was added XantPhos (844 mg, 1.46 mmol), Pd 2 dba 3 (668 mg, 0.7300 mmol) and DIPEA (3.81 mL, 21.9 mmol). Then the mixture was stirred at 100 °C for 12 h under N 2 . Brine and EtOAc were then added to the reaction mixture, which was then extracted with EtOAc (3x).
- Step a To a solution of 2-ethylhexyl 3-[(2,3-dichloropyridin-4-yl)sulfanyl]propanoate (800 mg, 2.19 mmol, synthesized via Step a of Intermediate AV) and trimethylboroxine (411 mg, 3.28 mmol) in dioxane (0.3M, 7 mL) and water ( 4M, 0.5 mL) was added XphosG4 (376 mg, 0.438 mmol) and Pd 2 dba 3 (376 mg, 0.438 mmol). The mixture was degassed for 3 min then heated to 110 °C for 2 h. The mixture was then cooled to rt, and EtOAc and brine were added.
- 2-ethylhexyl 3-[(2,3-dichloropyridin-4-yl)sulfanyl]propanoate 800 mg, 2.19 mmol, synthesized via Step a of Intermediate AV
- Step a 6-chloro-3-iodo-1-(oxan-2-yl)-1H-pyrazolo[3,4-b]pyrazine (91.9 mg, 0.252 mmol, Intermediate A) in DMF (4 mL) was charged with ethylbis(propan-2-yl)amine (218 ⁇ L, 1.26 mmol) and (S)-7-fluoro-1,3-dihydrospiro[indene-2,4'-piperidin]-1-amine dihydrochloride (74 mg, 0.252 mmol, Intermediate AS) and the solution was heated to 80 °C for 3 hr.
- Step a Dissolved 6-chloro-3-iodo-1-(oxan-2-yl)-1H-pyrazolo[3,4-b]pyrazine (223 mg, 0.6118 mmol, Intermediate A) in DMF (5 mL).
- DMF 5 mL
- 5S -5,7-dihydrospiro[cyclopenta[b]pyridine-6,4'-piperidin]-5-amine dihydrochloride
- ethylbis(propan-2-yl)amine (425 ⁇ L, 2.44 mmol
- di-tert-butyl dicarbonate (153 ⁇ L, 0.6729 mmol) was added and the reaction was stirred at rt for 1.5 hr. The reaction mixture was then diluted with EtOAc and extracted with water. The layers were separated, and the organic layer was washed with brine, dried over Na 2 SO 4 , filtered and concentrated onto SiO 2 .
- Step a Dry DMSO (5 mL) was added to a 50 mL flask, which was then bubbled with N 2 gas and equipped with a thermocouple. To the solution was added sodium hydride (173 mg, 4.36 mmol, 60% in oil) in small portions while the temperature was monitored so as not to exceed 35 °C. Then trimethyl(oxo)- ⁇ 6 -sulfanylium iodide (959 mg, 4.36 mmol) was added in small portions while monitoring temperature. The suspension was then stirred at rt for 45 min.
- tert-butyl 1-oxo-8-azaspiro[4.5]dec-2-ene-8-carboxylate (1 g, 3.97 mmol, synthesized as described in PCT Int. Appl., 2016203406) was dissolved in 2.5 mL dry DMSO. This solution was then added dropwise to reaction mixture while stirring vigorously and monitoring temperature so as not to exceed 27 °C. The reaction mixture was then stirred at rt for 16 h. Then 10 mL of water was added dropwise and the solution was extracted with diethyl ether (2x 30 mL). The combined organic layer was dried over Na 2 SO 4 , filtered and concentrated in vacuo.
- Step b Dissolved hydrogen chloride (288 mg, 7.91 mmol) in EtOH (15 mL), then added tert-butyl 4-oxospiro[bicyclo[3.1.0]hexane-3,4 (210 mg, 0.791 mmol) followed by acetic acid amine (909 mg, 11.8 mmol) and NaCNBH 3 (54.6 mg, 0.870 mmol). The reaction mixture was then heated in a microwave at 130 °C for 1h. Additional NaCNBH 3 (54.6 mg, 0.870 mmol) was added and the mixture was stirred in a microwave at 130 °C for 1h more. The reaction mixture was then concentrated in vacuo and the residue was treated with NaOH (2N, 15 mL).
- Step a Spiro[bicyclo[3.1.0]hexane-3,4 (120 mg, 0.502 mmol, Intermediate BA) and 6-chloro-3-iodo-1-(oxan-2-yl)-1H-pyrazolo[3,4-b]pyrazine (182 mg, 0.502 mmol, Intermediate A) were placed into a round bottom flask and dissolved in DMF (2 mL). Then ethylbis(propan-2-yl)amine (435 ⁇ L, 2.50 mmol) was added and the reaction mixture was stirred at rt for 2 h.
- Step a The mixture of methyl 3,5-dichloropyrazine-2-carboxylate (1.0 g, 4.83 mmol, CAS# 330786-09-9), (3 S )-1,3-dihydrospiro[indene-2,4'-piperidin]-3-amine dihydrochloride (1.65 g, 4.83 mmol, Intermediate I) and CsF (3.66 g, 24.1 mmol) in DMF (15 mL) was stirred at 70 °C for 2 hours. Boc 2 O (1.57 g, 7.24 mmol) and TEA (1 mL) were then added to the mixture and the mixture was stirred at 20 °C for 1 hour.
- Step b The mixture of methyl 5-[(3 S )-3- ⁇ [( tert -butoxy)carbonyl]amino ⁇ -1,3-dihydrospiro[indene-2,4'-piperidin]-1'-yl]-3-chloropyrazine-2-carboxylate (1.5 g, 3.17 mmol), PMBNHNH 2 ⁇ 2HCl (927 mg, 4.12 mmol) and TEA (2.01 mL, 15.8 mmol) in EtOH (20 mL) was stirred at 80 °C for 10 hours.
- Step a A mixture of 3-bromo-6-chloropyrazin-2-amine (300 mg, 1.4 mmol, CAS# 212779-21-0), DMAP (87 mg, 0.7 mmol) and (Boc) 2 O (936 mg, 4.3 mmol) in DCM (15 mL) was stirred at 25 °C for 16 h. The reaction mixture was then washed with H 2 O (15 mL x 2) and brine (15 mL). The organic phase was dried over anhydrous Na 2 SO 4 , filtered and concentrated to give a residue.
- Step b tert -butyl N -(3-bromo-6-chloropyrazin-2-yl)- N -[( tert- butoxy)carbonyl]carbamate (270 mg, 0.7 mmol), (3 S )-1,3-dihydrospiro[indene-2,4'-piperidin]-3-amine (200 mg, 1.0 mmol, Intermediate I) and DIPEA (0.3 mL, 2.0mmol) were added into DMF (2 mL). The mixture was stirred at 85 °C for 12 h. The reaction mixture was then diluted with EtOAc (50 mL).
- Step a A mixture of 5-bromoquinoline (2.0 g, 9.61 mmol, 1.0 eq), Zn(CN) 2 (2.26 g, 19.23 mmol, 2.0 eq) and XantPhos-Pd-G4 (924.8 mg, 961.3 umol, 0.1 eq) in dioxane (20.0 mL) and H 2 O (2.0 mL) was stirred at 80 °C for 16 hours under N 2 . The reaction mixture was concentrated under reduced pressure.
- Step b To a solution of quinoline-5-carbonitrile (780.0 mg, 5.06 mmol, 1.0 eq) in MeOH (10.0 mL) were added Raney-Ni (300.0 mg, 5.11 mmol, 1.0 eq) and NH 3 ⁇ H 2 O (1.91 g, 2.10 mL, 28% solution). The reaction mixture was degassed and refilled with H 2 for three times. The reaction mixture was stirred at 15 °C for 16 hours under H 2 (15 psi). The reaction mixture was filtered through a pad of celite and washed with MeOH (5.0 mL x 4). The filtrate was concentrated under reduced pressure to give a green residue.
- Step c To a solution of 5-quinolylmethanamine (550.0 mg, 3.48 mmol, 1.0 eq) in DCM (7.0 mL) were added isopropyl carbonochloridate (852.1 mg, 6.95 mmol, 965.0 uL, 2.0 eq) and TEA (1.06 g, 10.43 mmol, 1.45 mL, 3.0 eq). The reaction mixture was stirred at 15 °C for 16 hours under N 2 . The reaction mixture was concentrated under reduced pressure to give a yellow residue.
- Step d To a solution of isopropyl- N -(5-quinolylmethyl)carbamate (830.0 mg, 3.40 mmol, 1.0 eq) and Cs 2 CO 3 (3.32 g, 10.19 mmol, 3.0 eq) in DMF (10.0 mL) was added a solution of MeI (578.7 mg, 4.08 mmol, 253.8 uL, 1.2 eq) in DMF (2.0 mL). The reaction mixture was stirred at 15 °C for 16 hours under N 2 . The reaction mixture was concentrated under reduced pressure. The residue was washed with water (70.0 mL) and extracted with EtOAc (50.0 mL x 3).
- Step e To a solution of isopropyl- N -methyl- N -(5-quinolylmethyl)carbamate (130.0 mg, 503.3 umol, 1.0 eq) in MeOH (3.0 mL) was added PtO 2 (20.0 mg, 88.08 umol, 1.75 eq). The reaction mixture was degassed and refilled with H 2 for three times. The reaction mixture was stirred at 30 °C for 16 hours under H 2 (15 psi). The reaction mixture was filtered through a pad of celite and washed with MeOH (5.0 mL x 3).
- Step a A mixture of 2-bromo-3-methylbenzoic acid (10.0 g, 46.5 mmol, CAS# 53663-39-1), DIPEA (38.2 mL, 232.0 mmol), HATU (22.9 g, 60.4 mmol) and DMF (80.0 mL) was stirred at 25 °C for 1 hour. Then NH 4 Cl (7.4 g, 139.0 mmol) was added, and the resulting mixture was stirred at 25 °C for 12 hours. The reaction mixture was concentrated to remove DMF. Then water (200.0 mL) was added into the residue.
- Step b To the reaction mixture of 2-bromo-3-methylbenzamide (8.5 g, 39.7 mmol) and TEA (8.2 mL, 59.5 mmol) in DCM (100.0 mL) was added TFAA (8.3 mL, 59.5 mmol) slowly at 0°C. The reaction mixture was stirred at 0 °C for 15 min. The reaction mixture was then quenched with H 2 O (20.0 mL) and extracted with DCM (50.0 mL). The combined organic layers were washed with brine (20.0 mL), dried over anhydrous Na 2 SO 4 , filtered and concentrated under reduced pressure to give a residue.
- Step d To a solution of tert -butyl 4-cyanopiperidine-1-carboxylate (1.9 g, 9.2 mmol) in THF (20.0 mL) at -78 °C was added LDA (6.9 mL, 13.8 mmol, 2 M in THF) dropwise and stirred at -78 °C for 1 hour. 2-bromo-3-(bromomethyl)benzonitrile (2.1 g, 7.7 mmol) was then added into the reaction and the reaction mixture was allowed stirring at -78 °C for 0.5 hour. The reaction mixture was then warmed to 20 °C. The reaction mixture was quenched with sat.
- Step e The mixture of tert-butyl 4-[(2-bromo-3-cyanophenyl)methyl] -4-cyanopiperidine-1-carboxylate (1.3 g, 3.3 mmol), P(t-Bu) 3- Pd-G4(387.0 mg, 0.7 mmol) and TEA (915.0 ⁇ L, 6.6 mmol) in DMF(13.5 mL) and H 2 O(1.5 mL) was stirred at 130 °C for 12 hours under N 2 atmosphere. The reaction mixture was quenched with water (80.0 mL), and extracted with EtOAc (100.0 mL x 2). The combined organic layers were dried over anhydrous Na 2 SO 4 , filtered and concentrated to give a residue.
- Step f A reaction mixture of tert -butyl 7-cyano-1-oxo-1,3-dihydrospiro[indene-2,4 (400.0 mg, 1.2 mmol), (R)-2-methylpropane-2-sulfinamide (591.0 mg, 4.9 mmol), Ti(OEt) 4 (1.7 g, 7.3 mmol) and 2-Me-THF (10.0 mL) was stirred at 90 °C for 12 hours under N 2 atmosphere. The addition of ( R )-2-methylpropane-2-sulfinamide (591.0 mg, 4.9 mmol) and Ti(OEt) 4 (1.7 g, 7.3 mmol) at 90 °C was repeated for 2 times in 24 hours. The crude solution was used directly in the next step.
- Step g NaBH 4 (13.7 mg, 0.4 mmol) was added into the crude solution of tert -butyl (1 Z )-7-cyano-1- ⁇ [( R )-2-methylpropane-2-sulfinyl]imino ⁇ -1,3-dihydrospiro[indene-2,4 (524.0 mg, 1.2 mmol) in 2-Me-THF(10.0 mL) at 0 °C. The reaction mixture was stirred at 0 °C for 0.5 hour. Then NaBH 4 (13.7 mg, 0.4 mmol) was added again, and the resulting mixture was stirred at 0 °C for 0.5 hour.
- Step a To a solution of quinolin-6-ol (1.00 g, 6.88 mmol) and TEA (2.84 mL, 20.6 mmol) in DCM (50.0 mL) was added acetyl chloride (1.07 g, 13.7 mmol) dropwise at 0 °C. The reaction mixture was stirred at 20 °C for 1 hour. The reaction mixture was poured into H 2 O (100.0 mL) and extracted with DCM (50.0 mL x 2). The combined organic layers were washed with brine (50.0 mL), dried over anhydrous Na 2 SO 4 , filtered and concentrated under reduced pressure to give an orange residue.
- Step b A solution of quinolin-6-yl acetate (1.20 g, 6.41 mmol) and PtO 2 (218.0 mg, 961.0 umol) in THF (50.0 mL) was stirred at 20 °C for 12 hours under H 2 (15 psi). The reaction mixture was filtered and the filtrate was concentrated under reduced pressure to give a residue, which was purified by flash silica gel chromatography (20 g column, ethyl acetate in petroleum ether from 0% to 20%) to give 1,2,3,4-tetrahydroquinolin-6-yl acetate (1.00 g, 82% yield) as a yellow oil.
- LCMS m/z [M+H] + 192.1.
- Step a To a solution of LiN(SO 2 F) 2 (2.74 g, 14.7 mmol) and PhI(OAc) 2 (3.54 g, 11.0 mmol) in DCE (30.0 mL) was added N -phenylacetamide (1.00 g, 7.39 mmol) in DCE (20.0 mL) dropwise under N 2 at 20 °C. The reaction mixture was stirred at 90 °C for 20 min.
- Step a NaBH 4 (327.0 mg, 8.6 mmol) was added in portions to the mixture of 2-bromo-6-methoxypyridine-3-carbaldehyde (3.75 g, 17.3 mmol, CAS# 1060810-41-4) in MeOH (120 mL) at 25 °C. The mixture was stirred at 25 °C for 5 min. The reaction was quenched with H 2 O (150 mL). The MeOH was removed under reduced pressure. The combined mixture was extracted with ethyl acetate (100 mL x 3). The combined organic layers were washed with brine (150 mL), dried over anhydrous Na 2 SO 4 , filtered and concentrated to give a residue.
- Step b The compound of (2-bromo-6-methoxypyridin-3-yl)methanol (4.50 g, 20.6 mmol) and CBr 4 (8.19 g, 24.7 mmol) were added in DCM (200 mL). PPh 3 (6.47 g, 24.7 mmol) in DCM (50 mL) was then added dropwise at 0 °C. The mixture was stirred at 0 °C for 0.5 h. The reaction was then quenched with brine (100 mL) and the partitioned layers were separated. The aqueous phase was extracted with DCM (100 mL x 2).
- Step a The compound of tert-butyl 4-cyanopiperidine-1-carboxylate (2.43 g, 11.6 mmol) was placed in THF (100 mL). The LDA (10.6 mL, 21.2 mmol, 2M in THF) was added dropwise into the mixture at 0 °C. The mixture was stirred at 0 °C for 0.5 hour. The 2-bromo-3-(bromomethyl)-6-methoxypyridine (3.0 g, 10.6 mmol, Intermediate BK) in THF (50 mL) was added dropwise into the mixture at 0 °C. The mixture was allowed to warm to 25 °C and stirred for 2 hours.
- Step b The compound of tert-butyl 4-[(2-bromo-6-methoxypyridin-3-yl)methyl]-4-cyanopiperidine-1-carboxylate (1.7 g, 4.1 mmol) was added in the 2-Me-THF (20 mL) and PhMe (20 mL).
- i -PrMgCl ⁇ LiCl (6.4 mL, 8.3 mmol, 1.3 M in THF)
- n-BuLi 1.7 mL, 4.1 mmol, 2.5 M in hexane ) were added at -78 °C.
- the reaction mixture was stirred at -78 °C for 1 hour.
- the mixture was slowly warmed to 25 °C and stirred for 15 h.
- Step c To a solution of tert -butyl 2-methoxy-7-oxo-5,7-dihydrospiro[cyclopenta[ b ]pyridine-6,4'-piperidine]-1'-carboxylate (160 mg, 0.5 mmol) and Ti(OEt) 4 (0.5 mL, 2.4 mmol) in 2-Me-THF (10 mL) was added ( R )-2-methylpropane-2-sulfinamide (116 mg, 1.0 mmol). The reaction mixture was stirred at 90 °C for 12 h under N 2 .
- Step d The compound of tert-butyl (7 Z )-2-methoxy-7- ⁇ [( R )-2-methylpropane-2-sulfinyl]imino ⁇ -5,7-dihydrospiro[cyclopenta[b]pyridine-6,4'-piperidine]-1'-carboxylate (70 mg, 160 ⁇ mol) in THF (2 mL) was added NaBH 4 (18 mg, 480 ⁇ mol) at 0 °C. The mixture was stirred at 25 °C for 1 h. The reaction mixture was quenched with MeOH. The solution was added into H 2 O (10 mL) and EtOAc (10 mL). The mixture was filtered and extracted with EtOAc (10 mL x 2).
- Step a A mixture of 3-bromo-6-chloropyridine-2-carboxylic acid (10.0 g, 42.2 mmol, CAS# 929000-66-8) in MeOH (100.0 mL)/SOCl 2 (10.0 mL) was stirred at 80 °C for 3 hours. The reaction mixture was concentrated in vacuo to give methyl 3-bromo-6-chloropyridine-2-carboxylate (10.4 g, 99% yield) as a yellow solid.
- Step b To the solution of methyl 3-bromo-6-chloropyridine-2-carboxylate (5.0 g, 19.9 mmol) and MeOH (1.0 mL, 25.8 mmol) in THF (15.0 mL, freshly dried over NaH) was added t-BuOK (29.8 mL, 29.8 mmol, 1 M in THF) slowly over 20 min at 0 °C under N 2 atmosphere. The reaction mixture was stirred at 0 °C for 5 min. The reaction mixture was quenched with ice-cold sat. NH 4 Cl solution (30.0 mL), and extracted with EtOAc (50.0 mL x 2) rapidly.
- Step d (3-bromo-6-methoxypyridin-2-yl)methanol (2.5 g, 11.4 mmol) and CBr 4 (4.5 g, 13.6 mmol) were added into DCM (30 mL). PPh 3 (3.6 g, 13.6 mmol) in DCM (10 mL) was added dropwise into the reaction mixture at 0 °C. The mixture was stirred at 0 °C for 0.5 hour. The reaction was concentrated to give a residue.
- Step a 4-Iodo-3-methylbenzonitrile (2.00 g, 8.20 mmol), BPO (199.0 mg, 822.0 ⁇ mol) and NBS (2.20 g, 12.30 mmol) were added in DCE (30.0 mL), and the reaction mixture was evacuated and refilled for 3 times with N 2 and stirred at 80 °C for 2 hours. Another batch of NBS (1.50 g, 8.44 mmol) was added and the mixture was stirred at 80 °C for another 12 hours.
- Step b To a solution of 5,6-dichloropyridin-2-amine (2.1 g, 12.8 mmol) in anhydrous THF (20 mL) was added NaHMDS (25.6 mL, 25.6 mmol) at 0 °C. The reaction mixture was stirred at this temperature for 30 mins, then the solution of (Boc) 2 O (2.9 g, 13.4 mmol) in anhydrous THF (10 mL) was added. The resulting mixture was stirred at 0 °C for 1.5 hours. The mixture was quenched with saturated NH 4 Cl and extracted with ethyl acetate (50 mL x 2).
- Step c To a mixture of tert-butyl (5,6-dichloropyridin-2-yl)carbamate (1.2 g, 4.6 mmol) in anhydrous THF (15 mL) at -70 °C was added LDA (5.7 mL, 11.4 mmol) under N 2 atmosphere. After stirring at this temperature for 2 hours, NCS (1.1 g, 8.2 mmol) in THF (5 mL) was added. The resulting mixture was stirred at -70 °C for 2 hours and 10 hours at 20 °C. The reaction mixture was diluted with H 2 O (40 mL) and extracted with ethyl acetate (45 mL x 2).
- Step a A mixture of ( R )- N -[(3 S )-1,3-dihydrospiro[indene-2,4'-piperidin]-3-yl]-2-methylpropane-2-sulfinamide (450 mg, 1.5 mmol, synthesized via Step a of Example 120), 2,5-dibromopyrazine (416 mg, 1.8 mmol) and TEA (1.0 mL, 7.3 mmol) in DMF (10 mL) was stirred at 80 °C for 2 hours. The reaction mixture was then diluted with ethyl acetate (30 mL), and washed with H 2 O (20 mL x 2).
- Step b A mixture of ( R )- N -[(3 S )-1'-(5-bromopyrazin-2-yl)-1,3-dihydrospiro[indene-2,4'-piperidin]-3-yl]-2-methylpropane-2-sulfinamide (550 mg, 1.2 mmol), 2-ethylhexyl 3-mercaptopropanoate (307 mg, 1.4 mmol), Pd 2 (dba) 3 (108 mg, 118 ⁇ mol), XantPhos (136 mg, 236 ⁇ mol) and TEA (0.5 mL, 3.5 mmol) in toluene (30 mL) was stirred at 100 °C for 12 hours under N 2 atmosphere.
- Step c To a mixture of 2-ethylhexyl 3-((5-((S)-1-((R)-1,1-dimethylethylsulfinamido)-1,3-dihydrospiro[indene-2,4'-piperidin]-1'-yl)pyrazin-2-yl)thio)propanoate (650 mg, 1.1 mmol) in anhydrous THF (3.0 mL) was added MeONa (116 mg, 2.2 mmol), the resulting mixture was stirred at 20 °C for 12 hours under N 2 atmosphere.
- Step a A mixture of 2,5-dibromopyrazine (287 mg, 1.2 mmol), 2,3-dichloropyridin-4-ol (300 mg, 1.8 mmol) and Cs 2 CO 3 (593 mg, 1.8 mmol) in DMF (5.0 mL) was stirred at 85 °C for 24 hours. The reaction mixture was concentrated under reduced pressure. The residue was diluted with water (30.0 mL) and extracted ethyl acetate (50.0 mL x 2). The combined organic layers were washed with H 2 O (30.0 mL) and brine (30.0 mL), dried over anhydrous Na 2 SO 4 , filtered and concentrated under reduced pressure to give a residue.
- Step a To a solution of 1- tert -butyl 4-methyl piperidine-1,4-dicarboxylate (10.0 g, 41.1 mmol) in THF (150.0 mL) was added LDA (24.6 mL, 49.3 mmol, 2 M) at -78 °C under N 2 . The mixture was stirred at -78 °C for 1 hour. Then the solution of 1-bromo-2-(bromomethyl)benzene (12.3 g, 49.3 mmol) in THF (50.0 mL) was added at -78 °C. The mixture was stirred at 20 °C for 11 hours under N 2 .
- Step d To a solution of tert-butyl 7-bromo-3-oxo-1,3-dihydrospiro[indene-2,4'-piperidine]-1'-carboxylate (1.1 g, 2.9 mmol) in EtOH (20.0 mL) were added NH 4 OAc (8.9 g, 115.0 mmol) and NaBH 3 CN (907.0 mg, 14.4 mmol) in portions (4 times). The mixture was stirred at 80 °C for 12 hours. The mixture was concentrated under reduced pressure to give a residue. The residue was extracted with EtOAc (100.0 mL x 2), and the combined organic phases were washed with 2N aqueous NaOH (50.0 mL x 2).
- Step a To a solution of tert -butyl 3-amino-7-bromo-1,3-dihydrospiro[indene-2,4'-piperidine]-1'-carboxylate (1.1 g, Intermediate BT) in DCM (10.0 mL) were added (Boc) 2 O (1.3 g, 5.8 mmol) and Et 3 N (1.2 mL, 8.6 mmol). The mixture was stirred at 20 °C for 1 hour.
- Step b To a solution of tert -butyl 7-bromo-3- ⁇ [( tert -butoxy)carbonyl]amino ⁇ -1,3-dihydrospiro[indene-2,4'-piperidine]-1'-carboxylate (600.0 mg, 1.2 mmol) in DMF (20.0 mL) were added Zn(CN) 2 (1.2 g, 9.9 mmol) and XantPhos-Pd-G4 (119.0 mg, 124.0 ⁇ mol). The mixture was stirred at 100 °C for 12 hours under N 2 . The combined mixture was filtered, extracted with EtOAc (50.0 mL x 3) and washed with brine (50.0 mL).
- Step a 1,6-naphthyridine (0.965 g, 7.41 mmol) was dissolved in MeOH (8.5 mL) and charged with iodomethane (921 ⁇ L, 14.8 mmol). The vial was sealed and heated to 65°C and stirred for 16hrs. The solvent was removed, the residue taken up in a small amount of MeOH (1-2 mL) and ethyla acetate was charged. The mixture was filtered, washed with EA and air dried to constant weight to afford 6-methyl-1,6-naphthyridin-6-ium iodide (1.56 g).
- Step b 6-methyl-1,6-naphthyridin-6-ium iodide (1.56 g, 5.73 mmol) was suspended in water (10 mL) and cooled to 0°C. The reaction was charged with NaOH (1.25 g, 31.5 mmol) in water (10 mL) and tripotassium hexakis(iminomethanide) iron (4.04 g, 12.3 mmol) in water (10 mL). The solution was stirred for 1hr at 0°C then overnight at rt. The mixture was extracted with CHCl 3 , dried and concentrated.
- Step a To a mixture of 2,3-difluorobenzaldehyde (10 g, 70.3 mmol) and N,N-dimethylpyridin-4-amine (103 mg, 0.8436 mmol) in ACN (100 mL) was added trimethylsilanecarbonitrile (7.87 g, 79.4 mmol), where a cold bath was used to offset the small exotherm during addition. The reaction was stirred at rt for 5 h. The reaction was concentrated to give 2-(2,3-difluorophenyl)-2-((trimethylsilyl)oxy)acetonitrile as a yellow oil.
- Step b 2-(2,3-difluorophenyl)-2-[(trimethylsilyl)oxy]acetonitrile (8.45 g, 35.0 mmol) in THF (65 mL) was cooled to -78 °C and charged with 1M LHMDS (38.5 mL, 38.5 mmol) not allowing the temperature to rise above -65 °C during the addition.
- reaction mixture was stirred at -78 °C for 1.5 h, then tert-butyl 4-oxopiperidine-1-carboxylate (7.67 g, 38.5 mmol) in THF (10 mL) was added, again not allowing the temperature to rise above -65 °C during the addition, and the reaction was stirred at -78 °C for 3 h.
- Hydrogen chloride (84.0 mL, 84.0 mmol) was then added to the reaction mixture and the solution was allowed to warm to rt and stirred 16 h. The organic layer was separated and the aqueous layer was back extracted with EA.
- Step c Tert-butyl 4-(2,3-difluorobenzoyl)-4-hydroxypiperidine-1-carboxylate (2.25 g, 6.59 mmol) and (tert-butoxy)potassium (7.24 mL, 7.24 mmol) were dissolved in THF (3 mL) and heated in a microwave at 70 °C for 1 hr. Water was then added and the mixture was extracted with EA. The combined organic layer was dried and concentrated to afford tert-butyl 7-fluoro-3-oxo-3H-spiro[benzofuran-2,4'-piperidine]-1'-carboxylate as a light yellow oil.
- LCMS m/z [M+H-100] + 222.2.
- Step d To a mixture of tert-butyl 7-fluoro-3-oxo-3H-spiro[1-benzofuran-2,4'-piperidine]-1'-carboxylate (4.2 g, 13.0 mmol) and (R)-2-methylpropane-2-sulfinamide (2.36 g, 19.5 mmol) in 2-MeTHF (5 mL) was added tetratitanium-1-ylium tetraethanolate (17.7 mL, 52.0 mmol). The vial was then sealed and heated to 95 °C for 16 h. The reaction mixture was then cooled and was diluted with 2-MeTHF (20mL) and further cooled to -10 °C.
- Step e tert-butyl (3R)-7-fluoro-3- ⁇ [(R)-2-methylpropane-2-sulfinyl]amino ⁇ -3H-spiro[1-benzofuran-2,4'-piperidine]-1'-carboxylate (1.54 g, 3.61 mmol) in MeOH (25 mL) was charged with hydrogen chloride (9.02 mL, 36.1 mmol) and the reaction mixture was stirred at rt for 5 h. The solvent was then removed and chased with MTBE to yield (R)-7-fluoro-3H-spiro[benzofuran-2,4'-piperidin]-3-amine dihydrochloride as a white solid.
- LCMS m/z [M+H] + 649.2.
- Step a A mixture of tert-butyl 3-oxo-3H-spiro[benzofuran-2,4'-piperidine]-1'-carboxylate (200 mg, 659 ⁇ mol, synthesized via Steps a-d of Intermediate AP), Ti(OEt) 4 (599 mg, 2.63 mmol) and(R)-2-methylpropane-2-sulfinamide (119 mg,988 umol) in 2-Me-THF (10 mL) was stirred at 80 °C for 12 hours under N 2 atmosphere.
- Step b To a mixture of tert-butyl (3Z)-3- ⁇ [(R)-2-methylpropane-2-sulfmyl]imino ⁇ -3H-spiro[1-benzofuran-2,4'-piperidine]-1'-carboxylate (5.5 g, crude) in THF (50 mL) was added borane lithium hydride (331 mg, 15.1 mmol) at 0 °C, then the resulting mixture was stirred at 25 °C for 2 hours. The reaction mixture was quenched with sat.NH 4 Cl, diluted with H 2 O (200 mL), then extracted with ethyl acetate (200 mL x 2).
- Step c Dissolved tert-butyl (3R)-3- ⁇ [(R)-2-methylpropane-2-sulfinyl]amino ⁇ -3H-spiro[1-benzofuran-2,4'-piperidine]-1'-carboxylate (1.15 g, 2.81 mmol) in 20 mL MeOH then added hydrogen chloride (7.00 mL, 28.0 mmol). The reaction mixture was stirred at 60 °C for 30 min. The reaction mixture was then concentrated to an oil. MTBE was added and the product precipitated. The mixture was filtered and the solid was washed with MTBE and dried to give (3R)-3H-spiro[1-benzofuran-2,4'-piperidin]-3-amine dihydrochloride (750 mg, 96% yield).
- tert-butyl (1 S )-4,6-difluoro-1- ⁇ [( R )-2-methylpropane-2-sulfinyl]amino ⁇ -1,3-dihydrospiro[indene-2,4'-piperidine]-1'-carboxylate was then deprotected as follows: the mixture of tert-butyl (1S)-4,6-difluoro-1- ⁇ [( R )-2-methylpropane-2-sulfinyl]amino ⁇ -1,3-dihydrospiro[indene-2,4'-piperidine]-1'-carboxylate (190 mg, 429 ⁇ mol) in DCM (5 mL) and TFA (1 mL) was stirred at 20 °C for 2 hours.
- Step a A resealable reaction vial was charged with 5-chloropyrazine-2-carboxylic acid (500 mg, 3.15 mmol) and sulfurooyl dichloride (5.73 mL, 78.7 mmol). The mixture was charged with DMF (3 drops) and heated to 80 °C for 3.5 h. The solvent was removed in vacuo and chased with toluene to give a yellow crystalline solid.
- Step a A disposable tube was charged with (S)-1,3-dihydrospiro[indene-2,4'-piperidin]-1-amine (203 mg, 0.738 mmol, Intermediate I) triethylamine (148 mg, 1.47 mmol), 5-chloro-1,3,4-thiadiazol-2-amine (100 mg, 0.738 mmol, CAS# 37566-40-8) and a stir bar.
- Step a A mixture of 2,5-dibromo-1,3-thiazole (106 mg, 0.436 mmol), (S)-1,3-dihydrospiro[indene-2,4'-piperidin]-1-amine dihydrochloride (120 mg, 0.436 mmol, Intermediate I) and triethylamine (121 ⁇ L, 0.873 mmol) in 2 mL DMF was heated at 100 °C for 20 hr. The reaction mixture was cooled to rt and di-tert-butyl dicarbonate (109 ⁇ L, 0.480 mmol) was added and the mixture was stirred at rt for 48 hr. The reaction mixture was then partitioned between EtOAc and water.
- Step a Dissolved 2,5-dichloropyrazine (50 mg, 0.336 mmol), dicaesium(1+) carbonate (436 mg, 1.34 mmol), and (3S)-1,3-dihydrospiro[indene-2,4'-piperidin]-3-amine dihydrochloride (92.3 mg, 0.336 mmol, Intermediate I) in DMF (2 mL). The reaction mixture was stirred at 80 °C for 3 h. The reaction mixture was then partitioned between EtOAc and water, and the aqueous layer was extracted 3 x with EtOAc. The organic layers were combined, dried over Na 2 SO 4 , filtered and concentrated in vacuo.
- Step a 2,5-dibromopyrazine (1.23 g, 5.19 mmol), (S)-1,3-dihydrospiro[indene-2,4'-piperidin]-1-amine dihydrochloride (1.3 g, 4.72 mmol, Intermediate I) and TEA (3.26 mL, 23.6 mmol) were added in DMF (20 mL). The reaction mixture was stirred at 85 °C for 12 hr.
- Step a The mixture of 5-bromo-2-chloropyrimidine (550 mg, 2.84 mmol), 3-chloro-4-(sodiosulfanyl)pyridin-2-amine (491 mg, 2.69 mmol, Intermediate AC) and Cs 2 CO 3 (1.85 g, 5.68 mmol) in DMF (5 mL) was stirred at 80 °C for 1 hour. The mixture was diluted with H 2 O (20 mL), then extracted with EtOAc (20 mL x 2). The organic layer was washed with brine (20 mL), dried over anhydrous Na 2 SO 4 , filtered and concentrated under reduced pressure.
- Step b The mixture of 4-[(5-bromopyrimidin-2-yl)sulfanyl]-3-chloropyridin-2-amine (200 mg, 629 ⁇ mol), Boc 2 O (164 mg, 754 ⁇ mol) and DMAP (115 mg, 943 ⁇ mol) in DCM (10 mL) was stirred at 25 °C for 12 hours. The mixture was concentrated under reduced pressure.
- Step a The mixture of ( R )- N -(( S )-1,3-dihydrospiro[indene-2,4'-piperidin]-1-yl)-2-methylpropane-2-sulfinamide (200 mg, 652 ⁇ mol, synthesized via Step a of Example 120), 5-bromo-2-chloropyrimidine (126 mg, 652 ⁇ mol), XantPhos-Pd-G4 (62.7 mg, 65.2 ⁇ mol) and Cs 2 CO 3 (423 mg, 1.30 mmol) in DMF (10 mL) was stirred at 80 °C for 10 hours under N 2 atmosphere.
- Step a A mixture of 2,4-dichloropyridine (1.00 g, 6.75 mmol) and aq. MeNH 2 (30.0 mL) in MeOH (10.0 mL) was stirred in sealed tube at 85 °C for 12 hours. The reaction mixture was poured into H 2 O (100.0 mL) and extracted with EtOAc (100.0 mL x 2). The combined organic layers were washed with brine (200.0 mL), dried over anhydrous Na 2 SO 4 , filtered and the filtrate was concentrated under reduced pressure to give a residue.
- Step a To a solution of tert-butyl 3-cyanoazetidine-1-carboxylate (3.60 g, 19.70 mmol, CAS# 142253-54-1) in THF (40 mL) was added LDA (11.8 mL, 23.60 mmol, 2.0 M) dropwise at - 78 °C under N 2 . The mixture was stirred at 0 °C for 15 min. Then to the mixture was added 1-bromo-2-(bromomethyl)benzene (5.40 g, 21.60 mmol) in THF (20 mL) at -78 °C. The mixture was stirred at 0 ⁇ 25 °C for 12 hours under N 2 .
- Step b A mixture of tert-butyl 3-[(2-bromophenyl)methyl]-3-cyanoazetidine-1-carboxylate (800.0 mg, 2.27 mmol), PdCl 2 (Amphos) (160.0 mg, 227.0 ⁇ mol, CAS# 887919-35-9) and TEA (918.0 mg, 9.08 mmol) in DMA/H 2 O (10 mL, 10/1) was stirred at 120 °C under N 2 for 12 hours. The reaction mixture was then poured into EtOAc (50 mL) and washed with water (30 mL x 3).
- Step c A mixture of tert-butyl 1'-oxo-1',3'-dihydrospiro[azetidine-3,2'-indene]-1-carboxylate (520.0 mg, 1.90 mmol), (R)-2-methylpropane-2-sulfinamide (460.0 mg, 3.80 mmol, CAS# 196929-78-9) and Ti(OEt) 4 (3.50 g, 15.20 mmol) in 2-Me-THF (10 mL) was stirred at 100 °C for 12 hours. The reaction mixture was used for next step directly.
- Step d To a mixture of (A)-tert-butyl 1'-((tert-butylsulfinyl)imino)-1',3'-dihydrospiro[azetidine-3,2'-indene]-1-carboxylate (1.90 mmol from Step c) in 2-Me-THF (10 mL) was added L-selectride (2.85 mmol, 2.85 mL, 1.0 M in THF, CAS# 38721-52-7) slowly at -78°C. After addition, the mixture was stirred at 0 °C for 1 hour.
- reaction mixture was then quenched with MeOH (10 mL), poured into EtOAc (500 mL) and H 2 O (5 mL), and stirred for 0.5 hour. The mixture was filtered through celite and washed with EtOAc (300 mL x 2).
- tert-butyl (1' S )-1'- ⁇ [( R )-2-methylpropane-2-sulfinyl]amino ⁇ -1',3'-dihydrospiro[azetidine-3,2'-indene]-1-carboxylate (300.0 mg, 42% yield, the faster eluting isomer) as a light yellow solid
- tert-butyl (1'R)-1'- ⁇ [(R)-2-methylpropane-2-sulfinyl]amino ⁇ -1',3'-dihydrospiro[azetidine-3,2'-indene]-1-carboxylate 280.0 mg, 39% yield, the slower eluting isomer) as a light yellow solid.
- Step e A solution of tert-butyl (1' S )-1'- ⁇ [( R )-2-methylpropane-2-sulfinyl]amino ⁇ -1',3'-dihydrospiro[azetidine-3,2'-indene]-1-carboxylate (300.0 mg, 792.0 ⁇ mol) in 2M HCl/MeOH (20 mL) was stirred at 25 °C for 1 hour. The reaction mixture was concentrated to give (1' S )-1',3'-dihydrospiro[azetidine-3,2'-inden]-1'-amine dihydrochloride (240.0 mg, 123% crude yield) as a light yellow solid.
- Step f A solution of tert -butyl (1' R )-1'- ⁇ [( R )-2-methylpropane-2-sulfinyl]amino ⁇ -1',3'-dihydrospiro[azetidine-3,2'-indene]-1-carboxylate (280.0 mg, 739.0 ⁇ mol) in 2M HCl/MeOH (20 mL) was stirred at 25 °C for 1 hour. The reaction mixture was concentrated to give (1'R)-1',3'-dihydrospiro[azetidine-3,2'-inden]-1'-amine dihydrochloride (220.0 mg, 120% crude yield) as a light yellow solid.
- Step a A mixture of 4-bromo-1-methyl-1 H -pyrazole-5-carboxylic acid (4.80 g, 23.4 mmol, CAS# 84547-84-2) in THF (40.00 mL) was added BH 3 /THF (93.60 mL, 1 M). The mixture was stirred at 80 °C for 12 hours under N 2 atmosphere. To the mixture was added EtOAc (200 mL). The mixture was washed with saturated NaHCO 3 (200 mL x 3). The organic layer was dried over anhydrous Na 2 SO 4 , filtered and concentrated to give a residue.
- Step b The compound of (4-bromo-1-methyl-1 H -pyrazol-5-yl)methanol (3.00 g, 15.7 mmol) and CBr 4 (6.23 g, 18.8 mmol) was added in DCM (100 mL). Then PPh 3 (4.93 g, 18.8 mmol) in DCM (50 mL) was added dropwise at 0 °C and the mixture was stirred at 0 °C for 0.5 h. The reaction was quenched with brine (100 mL) and the partitioned layers were separated. The aqueous phase was extracted with DCM (100 mL x 2).
- Step c The compound of tert-butyl 4-cyanopiperidine-1-carboxylate (2.96 g, 14.1 mmol) was placed in THF (100 mL). LDA (8.85mL, 17.7 mmol, 2M in THF) was added dropwise into the mixture at 0 °C and the mixture was stirred at 0 °C for 0.5 h. The mixture was then cooled to - 78 °C. Then 4-bromo-5-(bromomethyl)-1-methyl-1H-pyrazole (3 g, 11.8 mmol) in THF (50 mL) was added dropwise into the mixture at -78 °C and the mixture was stirred at -78 °C for 1h.
- Step d The compound of tert-butyl 4-[(4-bromo-1-methyl-1 H -pyrazol-5-yl)methyl]-4-cyanopiperidine-1-carboxylate (1.00 g, 2.6 mmol), PdCl 2 (AmPhos) (92.0 mg, 130 ⁇ mol) and TEA (1.43 mL, 10.4 mmol) were placed into DMA (50.00 mL) and H 2 O (1.00 mL). The reaction mixture was evacuated and refilled 3 times using N 2 . The reaction mixture was stirred at 120 °C for 12 hours.
- Step f NaBH 4 (48.0 mg, 1.27 mmol) was added in the mixture of tert -butyl (4 Z )-1-methyl-4- ⁇ [( R )-2-methylpropane-2-sulfinyl]imino ⁇ -4,6-dihydro-1 H -spiro[cyclopenta[c]pyrazole-5,4'-piperidine]-1'-carboxylate (260.0 mg, 636 ⁇ mol) in 2-Me-THF (5.00 ml) at 0 °C. The mixture was stirred at 25 °C for 12 h. The mixture was diluted with ethyl acetate (50 mL).
- Step a 1- tert -butyl 4-methyl piperidine-1,4-dicarboxylate (25.70 g, 106.0 mmol, CAS# 124443-68-1) was dissolved in THF (200 mL), and the reaction mixture was cooled to -78 °C. Then LDA (57.5 mL, 115.0 mmol) was added, and the reaction mixture was stirred at -78 °C for 2 hours. Then a solution of 2-(bromomethyl)-1,4-difluorobenzene (20.00 g, 96.6 mmol, CAS# 85117-99-3) in THF (100 mL) was added, and the reaction mixture was warmed to 20 °C and stirred for 2 hours.
- Step c 1-[( tert -butoxy)carbonyl]-4-[(2,5-difluorophenyl)methyl]piperidine-4-carboxylic acid (10.00 g, 28.1 mmol) was dissolved in 1,2-dichloroethane (200 mL) and the reaction mixture was cooled to 0 °C. Then SOCl 2 (4.1 mL, 56.2 mmol) was added, and the reaction mixture was warmed to 20 °C and stirred for 4 hours. AlCl 3 (5.60 g, 42.1 mmol) was then added, and the reaction mixture was stirred at 75 °C for 12 hours.
- the reaction mixture was concentrated under reduced pressure, diluted with EtOAc (500 mL), washed with H 2 O (300 mL x 3), brine (300 mL), dried over anhydrous Na 2 SO 4 , filtered and concentrated under reduced pressure.
- Step e Tert -butyl (1 Z )-4,7-difluoro-1- ⁇ [( R )-2-methylpropane-2-sulfinyl]imino ⁇ -1,3-dihydrospiro[indene-2,4'-piperidine]-1'-carboxylate (3.26 g, crude) in 2-Me-THF (40 mL) was cooled to 0 °C, then NaBH 4 (136.0 mg, 3.7 mmol) was added. The reaction mixture was warmed to 20 °C and stirred for 2 hours.
- Step g Tert- butyl (1 R )-4,7-difluoro-1- ⁇ [( R )-2-methylpropane-2-sulfinyl]amino ⁇ -1,3-dihydrospiro[indene-2,4'-piperidine]-1'-carboxylate (900.0 mg, 2.0 mmol) was added in 4N HCl/MeOH (20 mL), the reaction mixture was stirred at 25 °C for 2 hours.
- Step h Tert -butyl (1 S )-4,7-difluoro-1- ⁇ [( R )-2-methylpropane-2-sulfinyl]amino ⁇ -1,3-dihydrospiro[indene-2,4'-piperidine]-1'-carboxylate (300.0 mg, 677.0 ⁇ mol) was added in 4N HCl/MeOH (6 mL), the reaction mixture was stirred at 25 °C for 1 hour.
- Step a 6-chloro-3-iodo-1-(oxan-2-yl)-1 H -pyrazolo[3,4- b ]pyrazine (150.0 mg, 411.0 ⁇ mol, Intermediate A), (1 R )-4,7-difluoro-1,3-dihydrospiro[indene-2,4'-piperidin]-1-amine dihydrochloride (127.0 mg, 411.0 ⁇ mol, Intermediate CQ), TEA (283.0 ⁇ L,2.1 mmol) were added in DMF (6 mL), and the reaction mixture was stirred at 80 °C for 12 hours.
- Step a To a mixture of 2-chloro-3-iodopyridine (1.00 g, 4.17 mmol, CAS# 78607-36-0 ) and 2-ethylhexyl 3-mercaptopropanoate (1.13 g, 5.21 mmol, CAS# 50448-95-8 ) in dioxane (30 mL) were added Pd 2 (dba) 3 (317 mg, 347 ⁇ mol), XantPhos (401.0 mg, 695 ⁇ mol) and DIPEA (1.80 mL, 10.4 mmol). The reaction mixture was purged with N 2 for 3 min and stirred at 100 °C for 12 hours under N 2 protection. The reaction mixture was concentrated under reduced pressure.
- Step a To the mixture of ( R )- N -[(3 S )-1,3-dihydrospiro[indene-2,4'-piperidin]-3-yl]-2-methylpropane-2-sulfinamide (50.0 mg, 163.0 ⁇ mol, synthesized via Step a of Example 120) and 2-bromo-5-iodopyridine (46.2 mg, 163 ⁇ mol, CAS# 73290-22-9) in toluene (3.0 mL) were added XantPhos-Pd-G4 (15.6 mg, 16.3 ⁇ mol) and Cs 2 CO 3 (116.0 mg, 358.0 ⁇ mol) under N 2 .
- Step a To the reaction mixture of 2-bromoaniline (10.00 g, 58.1 mmol, CAS# 615-36-1 ) and tert-butyl 4-oxopiperidine-1-carboxylate (11.50 g, 58.1 mmol, CAS# 79099-07-3 ) in HOAc (80 mL) was added Me 3 SiCN (7.98 mL, 63.9 mmol, CAS# 7677-24-9 ) at 25 °C under N 2 atmosphere. The reaction mixture was stirred at 25 °C for 12 hours. The combined reaction mixture was poured into ice-cold NH 4 OH solution (500 mL, 28% solution), then extracted with EtOAc (300 mL ⁇ 2).
- Step b The mixture of tert -butyl 4-[(2-bromophenyl)amino]-4-cyanopiperidine-1-carboxylate (6.00 g, 15.7 mmol), PdCl 2 (Amphos) 2 (1.11 g, 1.57 mmol, CAS# 887919-35-9) and TEA (8.67 mL, 62.7 mmol) in DMA (120 mL) and H 2 O (2.4 mL) was stirred at 120 °C for 12 hours under N 2 atmosphere. The reaction mixture was diluted with water (120 mL), then extracted with EtOAc (100 mL ⁇ 3).
- Step c To the reaction mixture of tert -butyl 3-oxospiro[indoline-2,4'-piperidine]-1'-carboxylate (450.0 mg, 1.5 mmol) in THF (9 mL) was added NaHMDS (2.21 mL, 2.2 mmol, 1 M in THF ) under N 2 atmosphere. The reaction mixture was stirred at 0 °C for 0.5 hour. Then (MeO) 2 SO 2 (1.86 g, 14.8 mmol) was added and the resulting mixture was stirred at 0 °C for 0.5 hour. The combined reaction mixture was poured into saturated NaHCO 3 (40 mL) and extracted with EtOAc (50 mL ⁇ 2).
- Step d The reaction mixture of tert -butyl 1-methyl-3-oxo-1,3-dihydrospiro[indole-2,4'-piperidine]-1'-carboxylate (600.0 mg, 1.9 mmol), ( R )-2-methylpropane-2-sulfinamide (916.0 mg, 7.6 mmol) and Ti(OEt) 4 (6 mL) was stirred at 100 °C for 12 hours under N 2 atmosphere. The addition of ( R )-2-methylpropane-2-sulfinamide (916.0 mg, 7.6 mmol) at 100 °C was repeated one time. The resulting mixture was stirred at 100 °C for 20 hours.
- Step e To a mixture of tert -butyl (3 E )-1-methyl-3- ⁇ [( R )-2-methylpropane-2-sulfinyl]imino ⁇ -1,3-dihydrospiro[indole-2,4'-piperidine]-1'-carboxylate (190.0 mg, 0.4 mmol) in 2-Me-THF (4 mL) was added NaBH 4 (170.0 mg, 4.5 mmol) and MeOH (1 mL) at 25 °C. The mixture was stirred at 40 °C for 0.5 hour. The reaction mixture was quenched with MeOH (1 mL) and poured into the mixture of H 2 O (50 mL) and EtOAc (60 mL).
- Step a The compound of 4-chlorothieno[3,2-d]pyrimidine (800 mg, 4.68 mmol, CAS# 16269-66-2), NBS (1.05 g, 4.68 mmol) and HOAc (0.2 mL) were added in MeCN (20 mL). The mixture was stirred at 85 °C for 18 h. The mixture was then extracted with ethyl acetate (30 mL ⁇ 3). The combined organic layers were washed with brine (20 mL ⁇ 2), dried over anhydrous Na 2 SO 4 , filtered and concentrated to give a residue.
- Step b The compound of 7-bromo-4-chlorothieno[3,2-d]pyrimidine (210 mg, 841 ⁇ m ⁇ l), (S)-1,3-dihydrospiro[indene-2,4'-piperidin]-1-amine dihydrochloride (254 mg, 925 ⁇ mol, Intermediate I) and TEA (582 ⁇ L, 4.20 mmol) were placed into DMF (10 mL). The reaction mixture was evacuated and refilled 3 times using N 2 . The reaction mixture was stirred at 85 °C for 12 hours. The reaction mixture was concentrated and H 2 O (30 mL) was added and the mixture was extracted with ethyl acetate (100 mL).
- Step c The compound of (3 S )-1( S )-1'-(7-bromothieno[3,2-d]pyrimidin-4-yl)-1,3-dihydrospiro[indene-2,4'-piperidin]-1-amine (330 mg, 794 ⁇ mol), TEA (400 mg, 4.0 mmol) and (Boc) 2 O (519 mg, 2.4 mmol) were placed into DMF (10 mL). The reaction mixture was stirred at 25 °C for 2 hours. The reaction mixture was concentrated and H 2 O (20 mL) was added, then extracted with ethyl acetate (30 mL ⁇ 3).
- Step a To a solution of ( R )-3 H -spiro[benzofuran-2,4'-piperidin]-3-amine dihydrochloride (230 mg, 0.8 mmol, Intermediate CB) and [6-chloro-3-iodo-1-(oxan-2-yl)-1 H -pyrazolo[3,4-b]pyrazin-5-yl]methyl acetate (361 mg, 0.8 mmol, Intermediate C) in DMF (5.0 mL) was added TEA (573 ⁇ L, 4.1 mmol). The reaction was stirred at 70 °C for 12 hours.
- Step a A solution of 6-chloro-3-iodo-1-(oxan-2-yl)-1H-pyrazolo[3,4-b]pyrazine (12 g, 32.9 mmol, Intermediate A), ( R )- N -(( S )-1,3-dihydrospiro[indene-2,4'-piperidin]-1-yl)-2-methylpropane-2-sulfinamide (10.0 g, 32.9 mmol, synthesized via Step a of Example 120) and CsF (14.9 g, 98.6 mmol) in DMSO (150 mL) was stirred at 60 °C for 2 h.
- reaction mixture was poured into H 2 O (700 mL) and extracted with EtOAc (700 mL ⁇ 2). The combined organic layers were washed with brine (800 mL), dried over anhydrous Na2SO4, filtered and filtrate concentrated under reduced pressure to give an orange residue.
- Step a To a solution of (3 S )-1'-(5-bromopyrazin-2-yl)-1,3-dihydrospiro[indene-2,4'-piperidin]-3-amine (1.30 g, 3.6 mmol, Intermediate CI) in DMF (15 mL) was added Boc 2 O (1.65 mL, 7.22 mmol). The resulting mixture was stirred at 25 °C for 12 hours. The reaction mixture was diluted with water (30 mL), then extracted with EtOAc (50 mL ⁇ 2). The organic layers were dried over anhydrous Na 2 SO 4 , filtered and concentrated to give a residue.
- Step a To a mixture of 1-benzyl-3-iodo-7-[(4-methoxyphenyl)methyl]-5-methyl-1 H ,4 H ,5 H ,6 H ,7 H -pyrazolo[3,4- d ]pyrimidine-4,6-dione (2.00 g, 4.0 mmol, CAS# 2055938-41-3) and 2,3-dichlorobenzene-1-thiol (1.06 g, 6.0 mmol) in dioxane (20 mL) were added Pd 2 (dba) 3 (291 mg, 0.4 mmol), XantPhos (370.0 mg, 0.8 mmol) and DIPEA (1.4 mL, 8.0 mmol).
- Step c To a mixture of 1-benzyl-3-[(2,3-dichlorophenyl)sulfanyl]-5-methyl-1 H ,4 H ,5 H ,6 H ,7 H -pyrazolo[3,4- d ]pyrimidine-4,6-dione (300.0 mg, 0.7 mmol) and DIPEA (1.2 mL, 6.9 mmol) was added POCl 3 (1.7 mL, 18.5 mmol). The reaction mixture was stirred at 120 °C for 12 hours. The reaction mixture was concentrated under reduced pressure to give a residue which was diluted with ethyl acetate (50 mL). The mixture was added slowly into ice-cooled sat.
- Step a A solution of tert -butyl ((1 S )-1'-(3-iodo-1-(tetrahydro-2 H -pyran-2-yl)-1 H- pyrazolo[3,4- b ]pyrazin-6-yl)-1,3-dihydrospiro[indene-2,4'-piperidin]-1-yl)carbamate (500.0 mg, 792.0 ⁇ mol, Intermediate J), 10% Pd/C (100.0 mg) and TEA (220.0 ⁇ L, 1.58 mmol) in THF (15.0 ml) was stirred at 20 °C for 12 hours under H 2 (15 psi).
- Step b To a solution of tert -butyl N -[(3 S )-1'-[1-(oxan-2-yl)-1 H -pyrazolo[3,4- b ]pyrazin-6-yl]-1,3-dihydrospiro[indene-2,4'-piperidin]-3-yl]carbamate (380.0 mg, 753.0 ⁇ mol) in AcOH/ACN (10.0 mL/10.0 mL) was added NBS (134.0 mg, 753.0 ⁇ mol). The reaction mixture was stirred at 20 °C for 0.5 hour. The reaction mixture was then concentrated under reduced pressure.
- Step c A solution of tert -butyl N -[(3 S )-1'-[5-bromo-1-(oxan-2-yl)-1 H -pyrazolo[3,4- b ]pyrazin-6-yl]-1,3-dihydrospiro[indene-2,4'-piperidin]-3-yl]carbamate (330.0 mg, 565.0 ⁇ mol), trimethyl-1,3,5,2,4,6-trioxatriborinane (141.0 mg, 1.13 mmol, CAS# 823-96-1), Pd(dppf)Cl 2 (82.7 mg, 113.0 umol, CAS# 72287-26-4) and K 2 CO 3 (233.0 mg, 1.69 mmol) in dioxane/H 2 O (10.0 mL/2.0 mL) was stirred at 90 °C for 12 hours under N 2 .
- reaction mixture was poured into H 2 O (100.0 mL) and extracted with EtOAc (100.0 mL ⁇ 2). The combined organic layers were washed with brine (200.0 mL), dried over anhydrous Na 2 SO 4 , filtered and the filtrate was concentrated under reduced pressure to give a residue.
- Step d A solution of tert -butyl N -[(3 S )-1'-[5-methyl-1-(oxan-2-yl)-1 H -pyrazolo[3,4- b ]pyrazin-6-yl]-1,3-dihydrospiro[indene-2,4'-piperidin]-3-yl]carbamate (250.0 mg, 482.0 ⁇ m ⁇ l) in HCl/MeOH (15.0 mL, 4 M) was stirred at 20 °C for 1 hour. The reaction mixture was then concentrated, triturated with EtOAc and stirred for 20 min.
- Step e The compound of (3 S )-1'- ⁇ 5-methyl-1 H -pyrazolo[3,4- b ]pyrazin-6-yl ⁇ -1,3-dihydrospiro[indene-2,4'-piperidin]-3-amine hydrochloride (100.0 mg, 269.0 ⁇ m ⁇ l) and TEA (111 ⁇ L, 807.0 ⁇ m ⁇ l) was dissolved in DCM (10 ml). Then (Boc) 2 O (73.8 ⁇ L, 322.0 ⁇ m ⁇ l) in DCM (0.13 mL) was added. The mixture was stirred at 25 °C for 2 hours.
- Step f Tert -butyl N -[(3 S )-1'- ⁇ 5-methyl-1 H -pyrazolo[3,4- b ]pyrazin-6-yl ⁇ -1,3-dihydrospiro[indene-2,4'-piperidin]-3-yl]carbamate (70.0 mg, 161.0 ⁇ mol) and NIS (54.2 mg, 241.0 ⁇ m ⁇ l) were added in DMF (2 mL). The reaction mixture was stirred at 110 °C for 16 hours. The mixture was quenched with the mixture of sat. Na 2 SO 3 (10 mL) and sat. NaHCO 3 (10 mL) and stirred for 10 min where a lot of precipitate formed.
- Step a 4-Chloro-2 H -pyrazolo[3,4- d ]pyrimidine ( 1.00 g , 6.5 mmol , CAS# 5399-92-8) was dissolved in THF (33.0 mL). Then NaHMDS (10 mL, 1.0 M in THF) was added at 0 °C and the mixture was stirred for 5 min. MeI (1.03mL, 16.7 mmol) was added slowly over 5 min at 0 °C under N 2 gas protection. The reaction mixture was warmed to 25 °C and stirred for 1 h. The mixture was diluted with H 2 O (200 mL) and CH 2 Cl 2 (200 mL), then the partitioned layers were separated.
- Step a A mixture of ( S )- tert -butyl (1'-(5-bromopyrazin-2-yl)-1,3-dihydrospiro[indene-2,4'-piperidin]-1-yl)carbamate (590.0 mg, 1.3 mmol, Intermediate CZ), 2-ethylhexyl 3-mercaptopropanoate (334.0 mg, 1.5 mmol, CAS# 50448-95-8 ), Pd 2 (dba) 3 (117.0 mg, 0.13 mmol), XantPhos (148.0 mg, 0.26 mmol, CAS# 161265-03-8 ) and TEA (522 ⁇ L, 3.8 mmol) in toluene (15 mL) was stirred at 100 °C for 12 hours under N 2 atmosphere.
- Step a The compound of 1-[6-chloro-1-(oxan-2-yl)-1 H -pyrazolo[3,4- b ]pyrazin-3-yl]-1,2,3,4-tetrahydro-1,5-naphthyridine (4.00 g, 10.7 mmol, Intermediate AA), 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)cyclohex-3-ene-1-carbonitrile (2.72 g, 11.7 mmol, CAS# 1310384-20-3 ), Pd(dppf)Cl 2 (782.0 mg, 1.1 mmol) and Cs 2 CO 3 (6.97 g, 21.4 mmol) were placed into the solvent of dioxane (250 mL) and H 2 O (25 mL).
- reaction mixture was evacuated and refilled for 3 times using N 2 ; then the reaction mixture was stirred at 90 °C for 12 hours.
- the reaction mixture was then concentrated and H 2 O (200 mL) was added, then the mixture was extracted with ethyl acetate (300 mL ⁇ 3). The combined organic layers were washed with brine (500 mL), dried over anhydrous Na 2 SO 4 , filtered and concentrated to give a residue.
- Step b 4-[1-(Oxan-2-yl)-3-(1,2,3,4-tetrahydro-1,5-naphthyridin-1-yl)-1 H -pyrazolo[3,4- b ]pyrazin-6-yl]cyclohex-3-ene-1-carbonitrile (2.70 g, 6.1 mmol) and 1-bromo-2-(bromomethyl)benzene (1.67 g, 6.7 mmol) were dissolved in THF (150 mL). Then LDA (3.66 mL, 7.3 mmol, 2 M in THF) was added dropwise into the mixture at -10 °C.
- Step c 1-[(2-Bromophenyl)methyl]-4-[1-(oxan-2-yl)-3-(1,2,3,4-tetrahydro-1,5-naphthyridin-1-yl)-1 H -pyrazolo[3,4- b ]pyrazin-6-yl]cyclohex-3-ene-1-carbonitrile (700.0 mg, 1.1 mmol), PdCl 2 (AmPhos) 2 (23.1 mg, 32.6 ⁇ m ⁇ l) and TEA (631 ⁇ L, 4.6 mmol) were placed into DMA (25 mL) and H 2 O (0.5 mL). The reaction mixture was evacuated and refilled 3 times using N 2 .
- reaction mixture was stirred at 120 °C for 12 hours.
- the mixture was then diluted with ethyl acetate (100 mL).
- the mixture was washed with H 2 O (30 mL ⁇ 5), brine (50 mL ⁇ 2), dried over anhydrous Na 2 SO 4 , filtered and concentrated to give a residue.
- Step a To a solution of 4-chloro-3 H -imidazo[4,5- c ]pyridine (1 g, 6.51 mmol, CAS# 81053-66-9 ) in DMSO/THF (2.5 mL/25 mL) was added NaHMDS (9.76 mL, 9.76 mmol) at 20 °C under N 2 . The reaction mixture was stirred at 20 °C for 1 h. Then, to the reaction mixture was added MeI (1.21 mL, 19.5 mmol) at 20 °C. The reaction mixture was then stirred at 20 °C for 11 h. The reaction mixture was filtered and the filtrate was concentrated to give a residue.
- Step a A mixture of 5-bromo-1 H -pyrrolo[2,3- b ]pyridine (8.00 g, 40.6 mmol, CAS# 183208-35-7 ), cyclopropylboronic acid (8.01 g, 93.3 mmol) and K 2 CO 3 (16.60 g, 121.0 mmol) in toluene (100.0 mL) was stirred at 25 °C for 0.5 hours under N 2 atmosphere. Then, Pd 2 (dba) 3 (1.85 g, 2.0 mmol) and SPhos (1.66 g, 4.1 mmol) were added, and the resulting mixture was stirred at 100 °C for 11.5 hours under N 2 atmosphere.
- Step b To a stirring solution of 5-cyclopropyl-1 H -pyrrolo[2,3- b ]pyridine (2.10 g, 13.2 mmol) in t -BuOH (120.0 mL) was added pyridium tribromide (12.50 g, 39.5 mmol) portion-wise, then the resulting mixture was stirred at 25 °C for 12 hours. The reaction mixture was concentrated in vacuo to give a residue, which was then dissolved in ethyl acetate (200.0 mL) and washed with H 2 O (150.0 mL ⁇ 2).
- Step c To a mixture of 3,3-dibromo-5-cyclopropyl-1 H ,2 H ,3 H -pyrrolo[2,3- b ]pyridin-2-one (4.30 g, 12.9 mmol) in AcOH (20.0 mL) and MeOH (20.0 mL) was added Zn (4.21 g, 64.5 mmol), and the resulting mixture was stirred at 25 °C for 2 hours. The reaction mixture was then filtered and concentrated in vacuo to give a residue, which was then dissolved in ethyl acetate (100.0 mL), and washed with H 2 O (80.0 mL ⁇ 2).
- Step a A mixture of 2,5-dibromopyrazine (4.00 g, 16.8 mmol, CAS# 23229-26-7 ), (3 S )-1,3-dihydrospiro[indene-2,4'-piperidin]-3-amine dihydrochloride (5.06 g, 18.4 mmol, Intermediate I) and TEA (11.6 mL, 84.0 mmol) in DMF (50 mL) was stirred at 80 °C for 3 hours. The crude solution was then cooled to rt and used directly in the next step. LC-MS (ESI + ) m / z: 341.9, 343.9 (M-NH 2 ) + .
- Step b To the crude solution of ( S )-1'-(5-bromopyrazin-2-yl)-1,3-dihydrospiro[indene-2,4'-piperidin]-1-amine (6.03 g, 16.7 mmol) in DMF (50 mL) was added (Boc) 2 O (5.73 mL, 25.0 mmol). The resulting mixture was stirred at 25 °C for 12 hours. Then the mixture was diluted with EtOAc (500 mL). The mixture was next washed with H 2 O (100 mL ⁇ 5), brine (500 mL), dried over anhydrous Na 2 SO 4 , filtered and concentrated to give a residue.
- Step c ( S )- tert -butyl (1'-(5-bromopyrazin-2-yl)-1,3-dihydrospiro[indene-2,4'-piperidin]-1-yl)carbamate (3.00 g, 6.53 mmol), 2-ethylhexyl 3-sulfanylpropanoate (1.56 g, 7.18 mmol, CAS# 50448-95-8 ), XantPhos (752 g, 1.30 mmol), Pd 2 (dba) 3 (597.0 mg, 0.65 mmol), and DIPEA (3.40 mL, 19.5 mmol) were added into dioxane (60 mL).
- the reaction mixture was evacuated and refilled 3 times with N 2 .
- the solution was stirred at 100 °C for 12 h.
- the solvent was then removed under reduced pressure.
- the residue was triturated with DCM (100 mL) and H 2 O (100 mL), and the yellow solid was filtered off.
- the organic layer was separated and the aqueous layer was extracted with DCM (100 mL ⁇ 2).
- the combined organic layers were washed with brine (100 mL), dried over anhydrous Na 2 SO 4 , filtered and concentrated to give a brown residue.
- Step d The compound of 2-ethylhexyl 3-( ⁇ 5-[(3 S )-3- ⁇ [(tert-butoxy)carbonyl]amino ⁇ -1,3-dihydrospiro[indene-2,4'-piperidin]-1'-yl]pyrazin-2-yl ⁇ sulfanyl)propanoate (200.0 mg, 335 ⁇ m ⁇ l) was dissolved in THF (1.00 mL). The reaction mixture was cooled to -78 °C and t- BuOK (1.00 mL, 1 M in THF) was added dropwise over 10 min under N 2 . The reaction mixture was stirred at -78 °C for 20 min.
- the mixture was washed with H 2 O (20 mL) and brine (20 ml).
- the organic layer were dried over anhydrous Na 2 SO 4 , filtered and concentrated to give tert -butyl N -[(3 S )-1'-(5-sulfanylpyrazin-2-yl)-1,3-dihydrospiro[indene-2,4'-piperidin]-3-yl]carbamate (138.0 mg, 100% yield, crude) as a yellow oil.
- Step a A mixture of (R)-N-((S)-1'-(5-bromo-3-methylpyrazin-2-yl)-1,3-dihydrospiro[indene-2,4'-piperidin]-1-yl)-2-methylpropane-2-sulfinamide and (R)-N-((S)-1'-(5-bromo-6-methylpyrazin-2-yl)-1,3 -dihydrospiro[indene-2,4'-piperidin]-1 -yl)-2-methylpropane-2-sulfinamide (320.0 mg, 670.0 ⁇ mol, synthesized via Step a of Examples 83 and 84 ), 2-ethylhexyl 3-sulfanylpropanoate (292.0 mg, 1.3 mmol, CAS# 50448-95-8), XantPhos (77.5 mg, 134.0 ⁇ mol), DIPEA (344.0 ⁇ L, 2.0 mmol) and Pd 2
- Step b To a mixture of 2-ethylhexyl 3-((5-((S)-1-(((R)-tert-butylsulfinyl)amino)-1,3-dihydrospiro[indene-2,4'-piperidin]-1'-yl)-6-methylpyrazin-2-yl)thio)propanoate and 2-ethylhexyl 3-((5-((S)-1-(((R)-tert-butylsulfinyl)amino)-1,3-dihydrospiro[indene-2,4'-piperidin]-1'-yl)-3-methylpyrazin-2-yl)thio)propanoate (390.0 mg, 634.0 ⁇ m ⁇ l) in anhydrous THF (10.0 mL) was added MeONa (68.0 mg, 1.3 mmol), and the resulting mixture was stirred at 20 °C for 1 hour under N 2 atmosphere.
- Step a To a mixture of cyclopentanecarboxylic acid (3.00 g, 26.2 mmol) in SOCl 2 (10.0 mL) was added DMF (2 drops), and the resulting mixture was stirred at 50 °C for 3 hours under N 2 atmosphere. The mixture was concentrated in vacuo to give a residue, which was dissolved in DCM (30.0 mL) and then re-concentrated in vacuo to give cyclopentanecarbonyl chloride (3.40 g, 98% crude yield) as a colorless oil.
- Step b To a mixture of cyclopentanecarbonyl chloride (3.40 g, 25.6 mmol) in anhydrous THF (60.0 mL) at 0 °C was added TMSCHN 2 (38.4 mL, 76.8 mmol) slowly, then the mixture was stirred at this temperature for 0.5 hours. Then the resulting mixture was warmed to 20 °C and stirred for 11.5 hours. The mixture was concentrated in vacuo to give a residue, which was then dissolved in THF (60.0 mL) and HCl/1,4-dioxane (4 M, 19.2 mL) was added slowly at 0 °C. The resulting mixture was warmed to 20 °C and stirred for 2 hours.
- Step a Methyl 6-bromo-3-chloropyrazine-2-carboxylate (3 g, 11.9 mmol, CAS# 1256921-67-1 ), TEA (8.26 mL, 59.5 mmol) and ( S )-1,3-dihydrospiro[indene-2,4'-piperidin]-1-amine dihydrochloride (3.27 g, 11.9 mmol, Intermediate I) were placed into DMF (80 mL). The reaction mixture was evacuated and refilled 3 times using N 2 . The reaction mixture was then stirred at 50 °C for 2 hours. The mixture was used for the next step without further purification. LC-MS (ESI + ) m/z: 440.8 (M+Na) + .
- Step b Methyl 3-[(3 S )-3-amino-1,3-dihydrospiro[indene-2,4'-piperidin]-1'-yl]-6-bromopyrazine-2-carboxylate (4.96 g, 11.8 mmol), TEA (8.20 mL, 59.0 mmol) and (Boc) 2 O (7.72 g, 35.4 mmol) were placed into DMF (80 mL). The reaction mixture was stirred at 25 °C for 2 hours.
- Step b A mixture of 6-bromo-3-[(3 S )-3- ⁇ [( tert -butoxy)carbonyl]amino ⁇ -1,3-dihydrospiro[indene-2,4'-piperidin]-1'-yl]pyrazine-2-carboxylic acid (600.0 mg, 1.19 mmol) , DIPEA (1.83 g, 14.2 mmol), HATU (676 mg, 1.78 mmol) and DMF(10 mL) was stirred at 25 °C for 1 h. Then NH 4 Cl (631 mg, 11.8 mmol) was added, and the resulting mixture was stirred at 25 °C for 12 h.
- Step c To a reaction mixture of tert -butyl N -[(3 S )-1'-(5-bromo-3-carbamoylpyrazin-2-yl)-1,3-dihydrospiro[indene-2,4'-piperidin]-3-yl]carbamate (480 mg, 955 ⁇ mol) and TEA (197 ⁇ L, 1.43 mmol) in DCM (10.0 mL) was added TFFA (201 ⁇ L, 1.43 mmol) slowly at 0 °C. The reaction mixture was stirred at 0 °C for 15 min. Then the reaction mixture was quenched with H 2 O (20.0 mL) and extracted with DCM (50.0 mL).
- Step b To a solution of 2-chloro-5-iodo-3,4-dihydropyrimidin-4-one (400 mg, 1.55 mmol) in DMF (10 mL) was added LDA (1.55 mL, 2.0 M in THF) dropwise at 0 °C. The mixture was stirred at 0 °C for 5 min, then MeI (212.0 ⁇ L, 3.41 mmol) was added. The mixture was allowed to warm up to 15 °C and stirred at this temperature for 18 h. The mixture was then diluted with H 2 O (20 mL), and extracted with ethyl acetate (200 mL ⁇ 3).
- Step a 2-Chloro-5-iodo-3-methyl-3,4-dihydropyrimidin-4-one (234.0 mg, 865 ⁇ mol, Intermediate DP), DIPEA (766 ⁇ L, 4.32 mmol), and ( S )-1,3-dihydrospiro[indene-2,4'-piperidin]-1-amine dihydrochloride (283 mg, 1.03 mmol, Intermediate I) were placed into DMF (10 mL). The reaction mixture was evacuated and refilled 3 times using N 2 . The reaction mixture was stirred at 100 °C for 2 hours. The mixture cooled to rt and was used in the next step without further purification. LC-MS (ESI + ) m / z: 458.9 (M +H) + .
- Step b A mixture of 2-[(3 S )-3-amino-1,3-dihydrospiro[indene-2,4 (377.0 mg, 864 ⁇ mol), TEA (362 ⁇ L, 2.59 mmol) and (Boc) 2 O (295 ⁇ L, 1.29 mmol) in DMF (10 mL) was stirred at 15 °C for 2 hours.
- Step a A mixture of 1-benzyl-3-iodo-7-(4-methoxybenzyl)-5-methyl-1 H -pyrazolo[3,4- d ]pyrimidine-4,6(5 H ,7 H )-dione (1.0 g, 2.0 mmol, CAS# 2055938-41-3 , synthesis described in PCT Int. Appl. 2016203404 ) in TFA (10 mL) and TfOH (0.1 mL) was stirred at 70 °C for 15 hours. The mixture was then concentrated to give a residue which was triturated with saturated aqueous NH 4 Cl (100 mL), then extracted with EtOAc (100 mL ⁇ 2).
- Step b 1-benzyl-3-iodo-5-methyl-1H,4H,SH,6H,7H-pyrazolo[3,4-d]pyrimidine-4,6-dione (500 mg, 1.30 mmol) and DIPEA (681 ⁇ L, 3.90 mmol) were added in the POCl 3 (5 mL). The mixture was stirred at 120 °C for 12 h. The reaction mixture was then concentrated to give a residue which was diluted with EtOAc (20 mL). The mixture was added slowly into an ice and sat. aq. NaHCO 3 (30 mL). The partitioned layers were separated and the aqueous phase was extracted with ethyl acetate (20 mL ⁇ 3).
- Step a 1-Benzyl-6-chloro-3-iodo-5-methyl-1 H , 4 H , 5 H -pyrazolo[3,4- d ]pyrimidin-4-one (210.0 mg, 524 ⁇ mol, Intermediate DR), (3 S )-1,3-dihydrospiro[indene-2,4'-piperidin]-3-amine dihydrochloride (172.0 mg, 628 ⁇ mol, Intermediate I) and DIPEA (455 ⁇ L, 2.61 mmol) were added in DMSO (2.00 mL). The reaction mixture was stirred at 120 °C for 12 hours. The reaction mixture was cooled to rt and the crude solution was used into the next step without further purification. LC-MS (ESI + ) m / z: 550.0 (M-NH 2 ) + .
- Step b The (Boc) 2 O (178 ⁇ L, 783 ⁇ m ⁇ l) was added into a mixture of ( S )-6-(1-amino-1,3-dihydrospiro[indene-2,4'-piperidin]-1'-yl)-1-benzyl-3-iodo-5-methyl-1 H- pyrazolo[3,4- d ]pyrimidin-4(5 H )-one (296.0 mg, 522 ⁇ m ⁇ l) in DMSO (3 mL). The mixture was stirred at 30 °C for 2 hours. Then the mixture was diluted with EtOAc (30 mL).
- Step a To a solution of (R)-N-((S)-1,3-dihydrospiro[indene-2,4'-piperidin]-1-yl)-2-methylpropane-2-sulfinamide (1.00 g, 3.76 mmol, synthesized via Step a of Example 120 ) in DMF (10.0 mL) was added 2,5-dibromo-3,6-dimethylpyrazine (1.33 g 3.76 mmol, CAS# 121594-49-8 ) and TEA (1.90 g, 18.8 mmol, 2.62 mL), and the resulting mixture was stirred at 80 °C for 12 hrs.
- Step a To a solution of 2-bromo-5-iodo-3-methylpyridine (500 mg, 167.8 umol, CAS# 65550-78-9) in toluene (1.00 mL) was added (R)-N-((S)-1,3-dihydrospiro[indene-2,4'-piperidin]-1-yl)-2-methylpropane-2-sulfinamide (594.8 mg, 1.68 mmol, synthesized via Step a of Example 120), Cs 2 CO 3 (1.64 g, 5.03 mmol, 3.00 eq ) and Xantphos-Pd-G4 (161.5 mg, 167.8 umol, 0.10 eq ), and the resulting mixture was stirred at 80 °C for 12 hrs.
- the yellow residue was purified by prep-HPLC (column: Phenomenex luna C18 250*50mm*10 um;mobile phase: [water(0.225%FA)-ACN]; B%: 55%-85%, 10min) to give (R)-N-((S)-1'-(6-bromo-5-methylpyridin-3-yl)-1,3-dihydrospiro[indene-2,4'-piperidin]-1-yl)-2-methylpropane-2-sulfinamide (330 mg, 678 umol, 40 % yield) as a yellow solid.
- Step a To a solution of 2-bromo-5-chloro-3-methylpyrazine (400 mg, 1.93 mmol, CAS# 1260664-82-1) in DMF (2.00 mL) was added (R)-N-((S)-1,3-dihydrospiro[indene-2,4'-piperidin]-1-yl)-2-methylpropane-2-sulfinamide (683.36 mg, 1.93 mmol, synthesized via Step a of Example 120) and TEA (975.53 mg, 9.64 mmol, 1.34 mL), and the mixture was stirred at 80 °C for 3 h. The mixture was then adjusted to pH to 6-7 with FA.
- Step a To a solution of (S)-5-fluoro-1,3-dihydrospiro[indene-2,4'-piperidin]-1-amine (3.00 g, 10.2 mmol, Intermediate BX) in DMF (30.0 mL) was added NaOH (2.29 g, 57.3 mmol). The mixture was stirred at 25 °C for 5 min, then to the mixture was added Boc 2 O (6.70 g, 30.7 mmol). The mixture was stirred at 25 °C for 5 hr. The mixture was then poured into water (50.0 mL), and extracted with ethyl acetate (40.0 mL ⁇ 3).
- Step b To a solution of tert-butyl (S)-1-((tert-butoxycarbonyl)amino)-5-fluoro-1,3-dihydrospiro[indene-2,4'-piperidine]-1'-carboxylate (3.21 g, 7.59 mmol) in DCM (30.0 mL) was added ZnBr 2 (3.42 g, 15.2 mmol, 759 uL) and the mixture was stirred at 25 °C for 12 h. The mixture was then concentrated under reduced pressure to give the residue.
- the mixture was purified by Pre-HPLC (column: Phenomenex luna C18 250*50mm*10 um;mobile phase: [water(0.225%FA)-ACN];B%: 10%-25%, 13min) to give tert-butyl (S)-(5-fluoro-1,3-dihydrospiro[indene-2,4'-piperidin]-1-yl)carbamate (1.5 g, 4.68 mmol, 62% yield) as a white oil.
- Step a To a solution of 2-bromo-5-iodopyridine (1.33 g, 4.68 mmol), tert-butyl (S)-(5-fluoro-1,3-dihydrospiro[indene-2,4'-piperidin]-1-yl)carbamate (1.50 g, 4.68 mmol, Intermediate DX) and Cs 2 CO 3 (4.58 g, 14.04 mmol) in toluene (15.0 mL) was added XantPhos-Pd-G4 (451 mg, 468 umol), and the mixture was stirred at 80 °C for 12 h. The mixture was then filtered and the filtrate was concentrated under reduced pressure to give a residue.
- Step a To a solution of (S)-6-fluoro-1,3-dihydrospiro[indene-2,4'-piperidin]-1-amine (2.00 g, 6.82 mmol, Intermediate Y) in DMF (20.0 mL) was added Boc 2 O (4.47 g, 20.5 mmol, 4.70 mL) and NaOH (1.53 g, 38.2 mmol), and the mixture was stirred at 25 °C for 5 h. The mixture was poured into water (40.0 mL), and extracted with ethyl acetate (30.0 mL ⁇ 3).
- Step b To a solution of tert-butyl (S)-1-((tert-butoxycarbonyl)amino)-6-fluoro-1,3-dihydrospiro[indene-2,4'-piperidine]-1'-carboxylate (1.78 g, 4.23 mmol) in DCM (20.0 mL) was added ZnBr 2 (1.91 g, 8.47 mmol, 424 uL), and the mixture was stirred at 25 °C for 12 h. The mixture was concentrated under reduced pressure to give the residue.
- the mixture was purified by Pre-HPLC (column: Phenomenex luna C18 250*50mm*10 um; mobile phase: [water(0.225%FA)-ACN];B%: 10%-25%, 13min) to give tert-butyl (S)-(5-fluoro-1,3-dihydrospiro[indene-2,4'-piperidin]-3-yl)carbamate (1.1 g, 3.43 mmol, 81% yield) was obtained as a white oil.
- Step a To a solution of 2-bromo-5-iodopyridine (975 mg, 3.43 mmol), tert-butyl (S)-(5-fluoro-1,3-dihydrospiro[indene-2,4'-piperidin]-3-yl)carbamate (1.10 g, 3.43 mmol, Intermediate DZ) and Cs 2 CO 3 (3.36 g, 10.3 mmol) in toluene (15.0 mL) was added XantPhos-Pd-G4 (330 mg, 343 umol,). The mixture was stirred at 80 °C for 12 h.
- Step a Tert-butyl (R)-3-((tert-butoxycarbonyl)amino)-3H-spiro[benzofuran-2,4'-piperidine]-1'-carboxylate, synthesized via Boc protection of (R)-3H-spiro[benzofuran-2,4'-piperidin]-3-amine, Intermediate CB (under standard conditions with Boc 2 O) was mono-deprotected using ZnBr 2 , as described in Step b of Intermediate DZ to give tert-butyl (R)-(3H-spiro[benzofuran-2,4'-piperidin]-3-yl)carbamate.
- Step a To a solution of 2-bromo-5-iodopyridine (1.00 g, 3.52 mmol), tert-butyl (R)-(3H-spiro[benzofuran-2,4'-piperidin]-3-yl)carbamate (1.09 g, 3.52 mmol, Intermediate EB) and Cs 2 CO 3 (3.44 g, 10.6 mmol) in toluene (10.0 mL) was added XantPhos-Pd-G4 (339 mg, 352 umol). The mixture was stirred at 80 °C for 12 h. The mixture was then filtered and the filtrate was concentrated under reduced pressure to give a residue.
- Step a To a mixture of 2-bromo-5-chloropyrazine (46.0 g, 237 mmol) and ethyl 3-mercaptopropanoate (31.9 g, 237 mmol) in dioxane (460 mL) was added DIPEA (61.4 g, 475 mmol, 82.8 mL), Xantphos (13.7 g, 23.8 mmol) and Pd 2 (dba) 3 (10.8 g, 11.8 mmol) in one portion under N 2 . The mixture was stirred at 80 °C for 2 hours under N 2 . The mixture was then filtered and the filtrate was concentrated in vacuo.
- Step b To a solution of ethyl 3-((5-chloropyrazin-2-yl)thio)propanoate (18.0 g, 72.9 mmol) and (S)-1,3-dihydrospiro[indene-2,4'-piperidin]-1-amine, (20.1 g, 72.9 mmol, Intermediate I, 2HCl) in NMP (180 mL) was added Na 2 CO 3 (30.9 g, 291 mmol) at 25 °C. Then the mixture was heated to 130 °C and stirred for 1 h. The mixture was then cooled to 25 °C.
- Step a To a solution of quinolin-8-ylmethanol (1.70 g, 10.7 mmol) in DMF (65.0 mL) was added NaH (428 mg, 10.7 mmol, 60% dispersion in mineral oil) at 0 °C. Then the mixture was stirred at 0 °C for 1 h. Next, 2,5-dibromopyrazine (2.12 g, 8.91 mmol) was added to the mixture. Then the reaction mixture was stirred at 0 to 25°C for 6 h. The mixture was then poured into water (200 mL) and extracted with ethyl acetate (150 mL ⁇ 3).
- Step a To a solution of (S)-1,3-dihydrospiro[indene-2,4'-piperidin]-1-amine (3.00 g, 10.9 mmol, Intermediate I, 2HCl) in DMF (30.0 mL) was added NaOH (2.44 g, 61.0 mmol). The mixture was stirred at 25 °C for 5 min, then to the mixture was added Boc 2 O (7.14 g, 32.7 mmol, 7.51 mL). The mixture was stirred at 25°C for another 16 h. The mixture was then poured into water (100 mL) and extracted with ethyl acetate (50.0 mL ⁇ 3).
- Step b To a solution of tert-butyl (S)-1-((tert-butoxycarbonyl)amino)-1,3-dihydrospiro[indene-2,4'-piperidine]-1'-carboxylate (4.00 g, 9.94 mmol) in DCM (40.0 mL) was added ZnBr 2 (4.48 g, 19.9 mmol, 995 uL) and the mixture was stirred at 25 °C for 12 h. Then, additional ZnBr 2 (2.24 g, 9.94 mmol, 497 uL) was added to the mixture and the mixture was stirred at 25 °C for another 4 h. The mixture was then concentrated under reduced pressure to remove DCM.
- Step a Boron trifluoride-Et 2 O (103 mL, 402 mmol, 48% solution) was added dropwise to a solution of 2-fluorobenzaldehyde (200 g, 168 mL,1.61 mol) and propane-1,3-dithiol (176 g, 163 mL, 1.63 mol) in DCM (1.00 L) at 0 °C. The reaction was then stirred at 25 °C for 1 h. The reaction mixture was then poured into water (200 mL) and the organic phase was separated and the aqueous phase was extracted with dichloromethane (200 mL x2).
- Step b A mixture of 2-(2-fluorophenyl)-1,3-dithiane (100 g, 464 mmol) in THF (500 mL) was cooled to -50 ⁇ -40 °C for 30 min with stirring under N 2 atmosphere. To the mixture was added LDA (2.00 M, 278 mL) dropwise at -50 ⁇ -40 °C for 30 min. Then the mixture was warmed to -30 ⁇ -20°C for 1 hr.
- Step c A mixture of tert-butyl (1R,3r,5S)-3-(2-(2-fluorophenyl)-1,3-dithian-2-yl)-3-hydroxy-8-azabicyclo[3.2.1]octane-8-carboxylate (180 g, 409 mmol), Pyridine (66.1 mL, 818. mmol), Py ⁇ HBr 3 (26.4 g, 81.8 mmol) in water (90.0 mL) and DCM (900 mL) was added TBAB (261 g, 818 mmol) at 0 °C. The mixture was stirred at 25 °C for 2 hrs.
- Step d A mixture of tert-butyl (1R,3r,5S)-3-(2-fluorobenzoyl)-3-hydroxy-8-azabicyclo[3.2.1]octane-8-carboxylate (140 g, 400 mmol) in dioxane (700 mL) was added t-BuOK (67.4 g, 601 mmol) at 20 °C. The mixture was then stirred at 70 °C for 2 hrs. The reaction mixture was poured into water (1.50 L) and extracted with ethyl acetate (1.00 L ⁇ 2). The combined organic layers were washed with brine (1.00 L), dried over anhydrous Na 2 SO 4 , then filtered and evaporated under reduced pressure.
- Step e To a solution of tert-butyl (1'R,2r,5'S)-3-oxo-3H-8'-azaspiro[benzofuran-2,3'-bicyclo[3.2.1]octane]-8'-carboxylate (45.0 g, 135 mmol) in 2-Me-THF (90.0 mL) were added Ti(OEt) 4 (92.7 g, 406 mmol, 84.3 mL) and (R)-2-methylpropane-2-sulfinamide (32.8 g, 271 mmol) at 25 °C. Then the mixture was stirred at 90 °C for 96 hrs.
- the reaction mixture was then cooled to -5 ⁇ 0°C, and LiBH4 (3.25 g, 149 mmol, 1.10 eq) was added carefully portionwise keeping the temperature at -5 ⁇ 0°C over 1 hr, then the mixture was stirred at -5-0 °C for 2 hours.
- the solution was quenched by addition methanol (2.00 mL) at 0 ⁇ 10 °C and 2-[2-[bis(2-hydroxyethyl)amino]ethyl-(2-hydroxyethyl)amino]ethanol (50.0 g) was added, and the mixture was stirred for 1 hr at 20 °C.
- tert-butyl (1'R,2r,3R,5'S)-3-(((R)-tert-butylsulfinyl)amino)-3H-8'-azaspiro[benzofuran-2,3'-bicyclo[3.2.1]octane]-8'-carboxylate (28.0 g, 64.1 mmol, 47% yield) as a white solid.
- Step f To a solution of tert-butyl (1'R,2r,3R,5'S)-3-(((R)-tert-butylsulfinyl)amino)-3H-8'-azaspiro[benzofuran-2,3'-bicyclo[3.2.1]octane]-8'-carboxylate (10.0 g, 22.9 mmol) in MeOH (20.0 mL) was added HCl/MeOH (4.00 M, 50.0 mL). The mixture was stirred at 20 °C for 2 hrs. The reaction mixture was concentrated under reduced pressure to remove methanol to give a residue.
- Step a To a solution of 2-bromo-5-chloropyrazine (20.0 g, 103 mmol) and sodium 2-amino-3-chloropyridine-4-thiolate (18.9 g 103 mmol, Intermediate AC) in dioxane (80.0 mL) was added Pd 2 (dba) 3 (3.79 g, 4.14 mmol), Xantphos (2.99 g, 5.17 mmol) and DIPEA (26.7 g, 207 mmol, 36.0 mL) at 25 °C. Then the mixture was heated to 80 °C and stirred at 80°C for 3 h. The mixture was then cooled to 25 °C and filtered.
- Step a To a solution of ethyl 6-bromo-3-chloro-5-methylpyrazine-2-carboxylate (1.00 g, 3.58 mmol, CAS# 2091009-80-0) and K 2 CO 3 (494 mg, 3.58 mmol) in ACN (10.0 mL) and H 2 O (2.00 mL) was added (2,3-dichlorophenyl)boronic acid (887 mg, 4.65 mmol) and Pd(dppf)Cl 2 (262 mg, 358 umol) at 25 °C. The solution was heated to 80 °C and stirred for 2 h.
- Step a To the stirred solution of 2-chloro-6-methylisonicotinic acid (500 mg, 2.91 mmol, CAS# 25462-85-5), methanamine hydrochloride (984 mg, 14.6 mmol) and TEA (2.03 mL, 14.6 mmol,) in DMF (20.0 mL) was added HATU (2.22 g, 5.83 mmol). The reaction mixture was stirred at 15 °C for 2 h. The reaction mixture was then poured into H 2 O (50.0 mL) and extracted with ethyl acetate (30.0 mL ⁇ 3). The combined organic layers were washed with brine (30.0 mL ⁇ 3), dried over Na 2 SO 4 , filtered and concentrated.
- Step a To the stirred solution of 2-fluoro-4-iodo-3-methylpyridine (1.00 g, 4.22 mmol, CAS# 153034-80-1) and DIPEA (2.94 mL, 16.9 mmol) in NMP (5.00 mL) was added (S)-1,3-dihydrospiro[indene-2,4'-piperidin]-1-amine (1.39 g, 5.06 mmol, 2HCl, Intermediate I) at 20 °C. The mixture was stirred at 120 °C for 14 h. Then more DIPEA (2.00 mL) was added and the reaction mixture was stirred at 130 °C for 14 h.
- Boc 2 O (1.38 g, 6.33 mmol, 1.45 mL) was added at 15 °C and the reaction mixture was stirred at 15 °C for 4 h.
- the reaction mixture was then diluted with H 2 O (20.0 mL) and extracted with ethyl acetate (30.0 mL ⁇ 3). The combined organic layers were washed with brine (20.0 mL ⁇ 2), dried over Na 2 SO 4 , filtered and concentrated under reduced pressure to give a residue.
- Step a To a solution of 2-fluoro-4-iodo-6-methylpyridine (800 mg, 3.38 mmol, CAS# 884494-45-5 ) and (S)-1,3-dihydrospiro[indene-2,4'-piperidin]-1-amine (1.11 g, 4.05 mmol, Intermediate I, 2HCl) in NMP (10.0 mL) was added DIPEA (2.35 mL, 13.50 mmol) at 25 °C, then the mixture was stirred at 120 °C for 24 h.
- Step a To a solution of 1,3-dibromo-5-methylbenzene (450 mg, 1.80 mmol, CAS# 1611-92-3), tert-butyl (S)-(1,3-dihydrospiro[indene-2,4'-piperidin]-1-yl)carbamate (327 mg, 1.08 mmol, Intermediate EF) and Cs 2 CO 3 (1.76 g, 5.40 mmol) in dioxane (2 mL) was added Xantphos (208 mg, 360 umol) and Pd 2 (dba) 3 (165 mg, 180 umol) under N 2 , then the mixture was stirred at 100 °C for 4 h.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Public Health (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Epidemiology (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Diabetes (AREA)
- Hematology (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Emergency Medicine (AREA)
- Obesity (AREA)
- Endocrinology (AREA)
- Oncology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Steroid Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Pyridine Compounds (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Oxygen Or Sulfur (AREA)
Claims (27)
- Verbindung, ausgewählt aus der Gruppe bestehend aus:


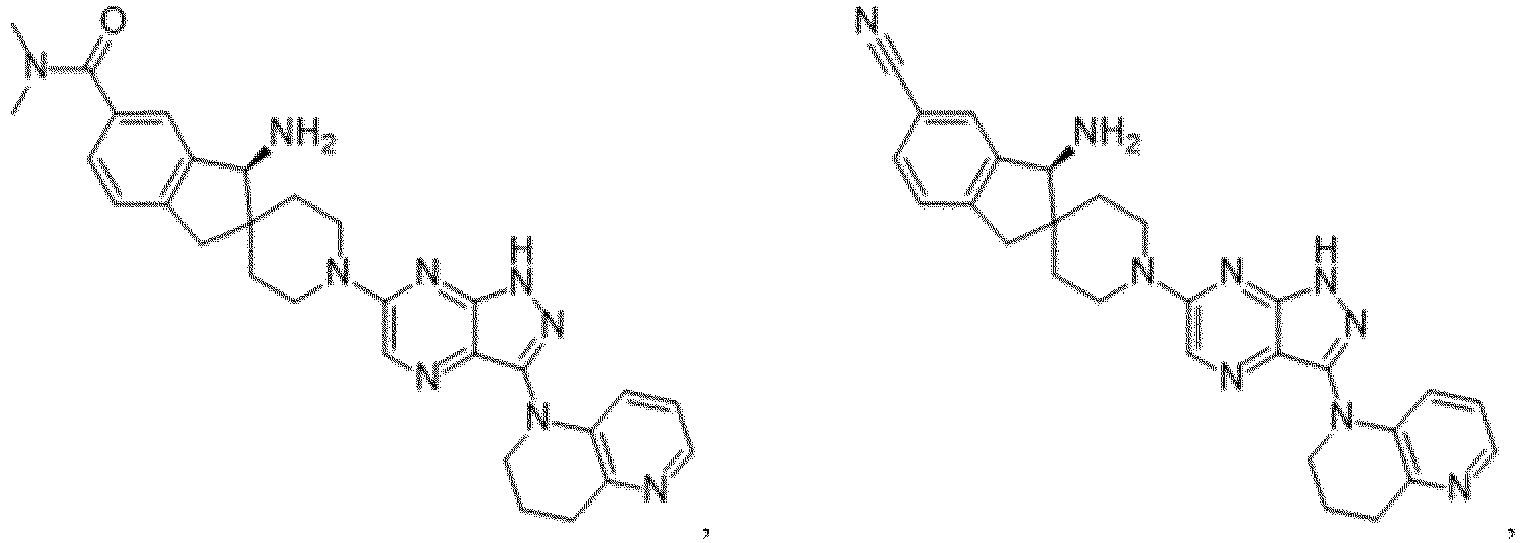




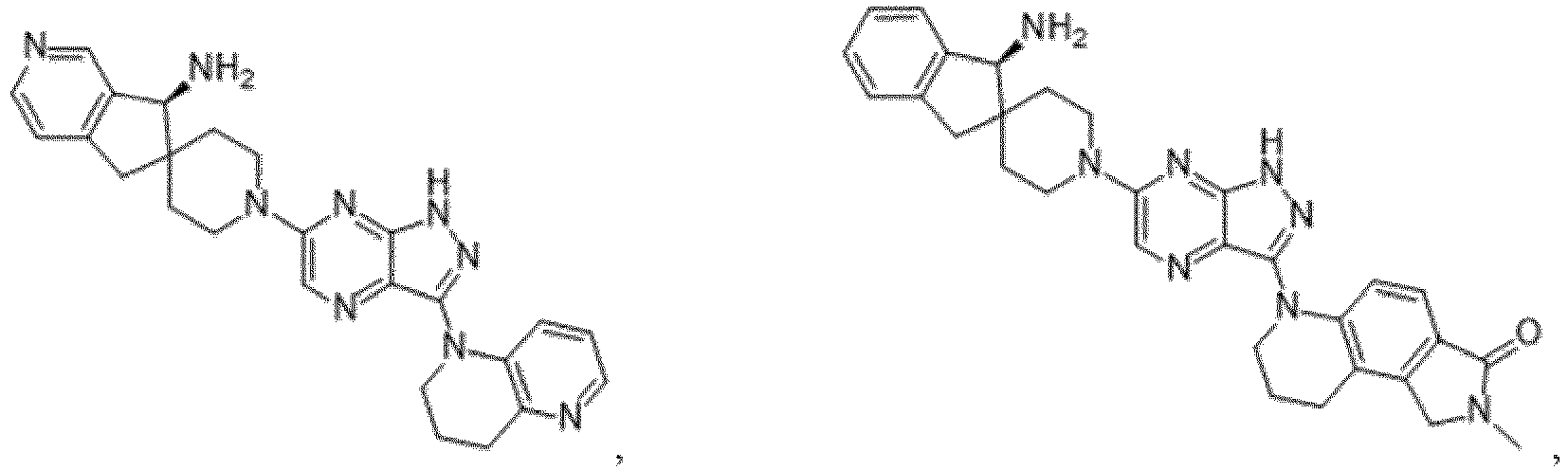







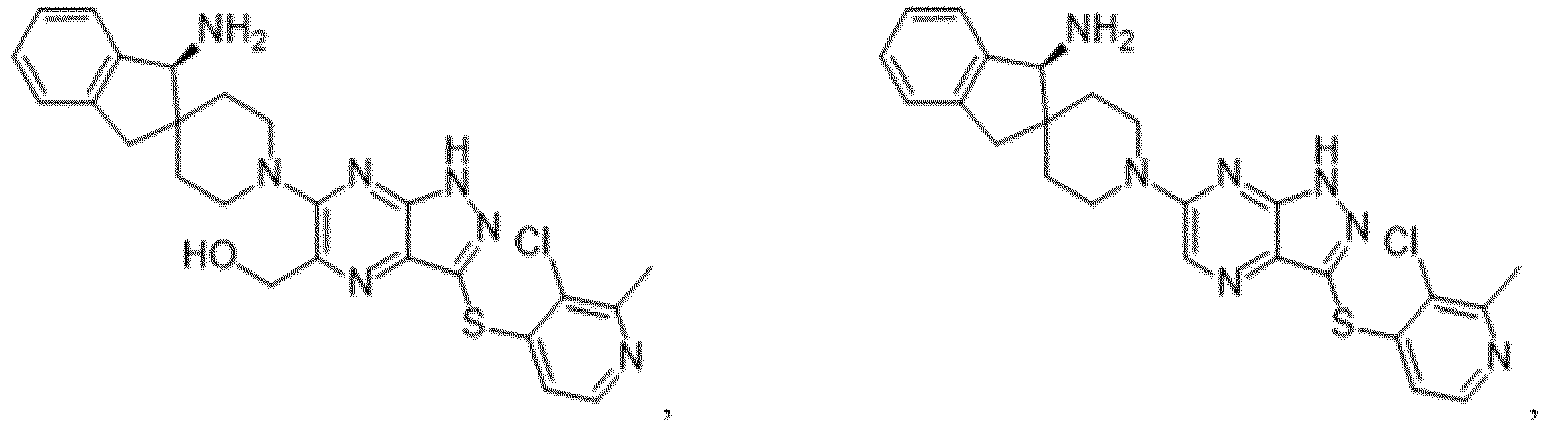
















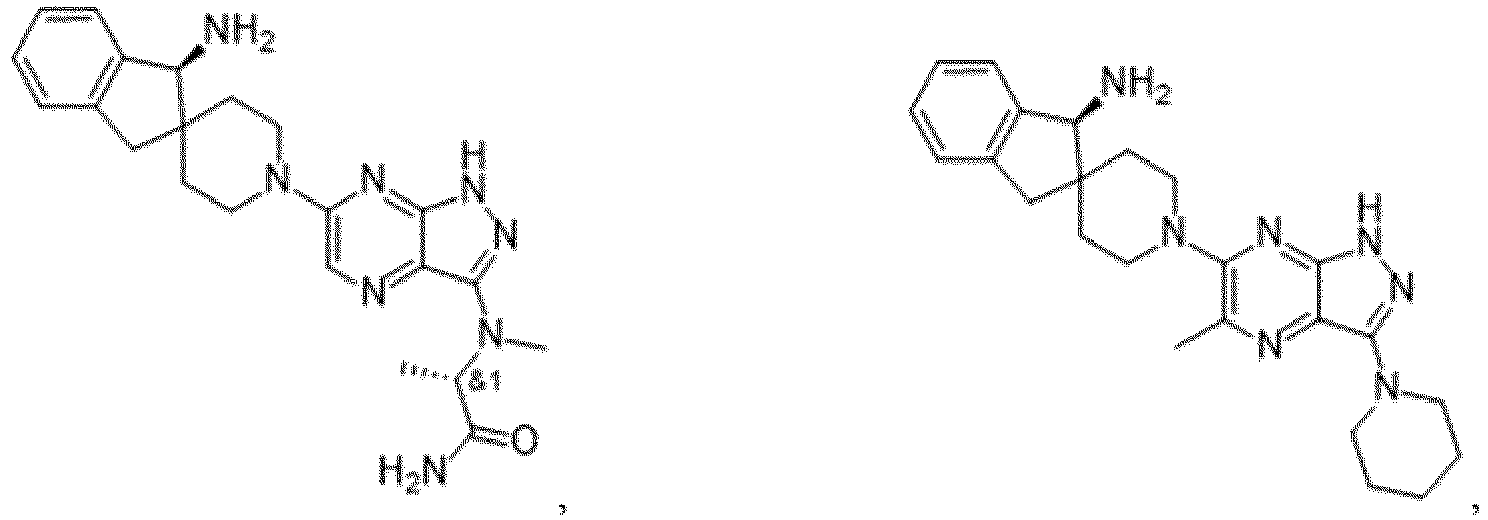













- Verbindung nach Anspruch 1, ausgewählt aus der Gruppe bestehend aus:

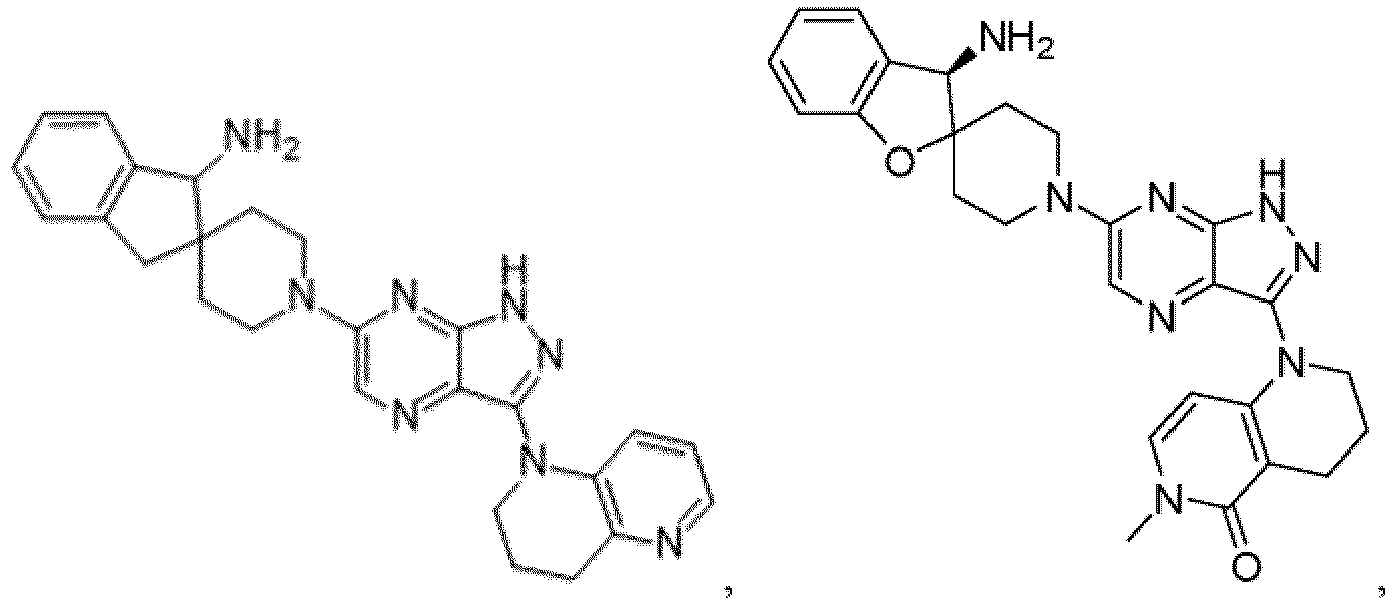




- Verbindung nach Anspruch 2, wobei die Verbindung ausgewählt ist aus der Gruppe bestehend aus:



- Pharmazeutische Zusammensetzung, umfassend eine Verbindung nach Anspruch 1, 2 oder 3 oder ein pharmazeutisch unbedenkliches Salz davon und einen pharmazeutisch unbedenklichen Träger.
- Verbindung nach Anspruch 2 mit der Formel:

- Pharmazeutische Zusammensetzung, umfassend eine Verbindung nach Anspruch 5 oder ein pharmazeutisch unbedenkliches Salz davon und einen pharmazeutisch unbedenklichen Träger.
- Verbindung nach Anspruch 2 mit der Formel:

- Pharmazeutische Zusammensetzung, umfassend eine Verbindung nach Anspruch 7 oder ein pharmazeutisch unbedenkliches Salz davon und einen pharmazeutisch unbedenklichen Träger.
- Verbindung nach Anspruch 1 oder Zusammensetzung nach Anspruch 4 zur Verwendung bei der Behandlung einer Störung, wobei die Behandlung die Verabreichung einer wirksamen Menge der Verbindung oder eines pharmazeutisch unbedenklichen Salzes davon oder der Zusammensetzung an ein dessen bedürftiges menschliches Subjekt umfasst.
- Verbindung nach Anspruch 1 oder Zusammensetzung nach Anspruch 4 zur Verwendung bei der Behandlung einer SHP2-vermittelten Krebserkrankung, wobei die Behandlung die Verabreichung einer wirksamen Menge der Verbindung oder eines pharmazeutisch unbedenklichen Salzes davon oder der Zusammensetzung an ein dessen bedürftiges menschliches Subjekt umfasst.
- Verbindung oder Zusammensetzung zur Verwendung nach Anspruch 10, wobei es sich bei der SHP2-vermittelten Krebserkrankung um chronische myelomonozytäre Leukämie, akute myeloische Leukämie, Brustkrebs, nichtkleinzelligen Lungenkrebs (Non-Small Cell Lung Cancer, NSCLC), kolorektalen Krebs (Colorectal Cancer, CRC), Speiseröhrenkrebs, Magenkrebs, Plattenepithelkarzinom des Kopfes und Halses (Squamous-Cell Carcinoma of the Head and Neck, SCCHN) oder Eierstockkrebs handelt.
- Verbindung oder Zusammensetzung zur Verwendung nach Anspruch 10, wobei es sich bei der SHP2-vermittelten Krebserkrankung um Brustkrebs handelt und wobei es sich bei dem Brustkrebs um HER2-positiven Brustkrebs, dreifach negativen Brustkrebs, duktales Karzinom oder invasives duktales Karzinom handelt.
- Verbindung oder Zusammensetzung zur Verwendung nach Anspruch 10, wobei es sich bei der SHP2-vermittelten Krebserkrankung um NSCLC handelt.
- Verbindung oder Zusammensetzung zur Verwendung nach Anspruch 10, wobei es sich bei der SHP2-vermittelten Krebserkrankung um CRC handelt.
- Verbindung oder Zusammensetzung zur Verwendung nach Anspruch 9 oder Anspruch 10, wobei die Behandlung weiterhin die Verabreichung einer wirksamen Menges eines Antikörpers, eines Antikörper-Arzneimittel-Konjugats, eines Immunmodulators oder eines Histondeacetylasehemmers umfasst.
- Verbindung oder Zusammensetzung zur Verwendung nach Anspruch 9 oder Anspruch 10, wobei die Verbindung die Formel:






- Verbindung oder Zusammensetzung zur Verwendung nach Anspruch 9 oder Anspruch 10, wobei die Verbindung die Formel:



- Verbindung oder Zusammensetzung zur Verwendung nach Anspruch 9 oder Anspruch 10, wobei die Verbindung die Formel:

- Verbindung oder Zusammensetzung zur Verwendung nach Anspruch 9 oder Anspruch 10, wobei die Verbindung die Formel:

- Verbindung nach Anspruch 1 oder Zusammensetzung nach Anspruch 4 zur Verwendung bei der Behandlung eines SHP2-vermittelten Noonan-Syndroms, einer juvenilen Leukämie oder einer juvenilen myelomonozytären Leukämie (JMML) bei einem menschlichen Subjekt mit Noonan-Syndrom, juveniler Leukämie oder juveniler myelomonozytärer Leukämie (JMML), wobei die Behandlung die Verabreichung einer wirksamen Menge der Verbindung oder eines pharmazeutisch unbedenklichen Salzes davon oder der Zusammensetzung an das Subjekt umfasst.
- Verbindung oder Zusammensetzung zur Verwendung nach Anspruch 20, wobei die Verbindung oder Zusammensetzung zur Verwendung bei der Behandlung von SHP2-vermitteltem Noonan-Syndrom bestimmt ist.
- Verbindung oder Zusammensetzung zur Verwendung nach Anspruch 20, wobei die Verbindung die Formel:






- Verbindung oder Zusammensetzung zur Verwendung nach Anspruch 20, wobei die Verbindung die Formel:

- Verbindung oder Zusammensetzung zur Verwendung nach Anspruch 20, wobei die Verbindung die Formel:

- Verbindung oder Zusammensetzung zur Verwendung nach Anspruch 11, wobei die Verbindung die Formel:






- Verbindung oder Zusammensetzung zur Verwendung nach Anspruch 11, wobei die Verbindung die Formel:

- Verbindung oder Zusammensetzung zur Verwendung nach Anspruch 11, wobei die Verbindung die Formel:

Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP24196397.4A EP4506035A3 (de) | 2018-03-21 | 2019-03-21 | Shp2-phosphatasehemmer und verfahren zur verwendung davon |
Applications Claiming Priority (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201862646083P | 2018-03-21 | 2018-03-21 | |
| US201862646099P | 2018-03-21 | 2018-03-21 | |
| US201862649834P | 2018-03-29 | 2018-03-29 | |
| US201862661902P | 2018-04-24 | 2018-04-24 | |
| US201862737819P | 2018-09-27 | 2018-09-27 | |
| PCT/US2019/023389 WO2019183367A1 (en) | 2018-03-21 | 2019-03-21 | Shp2 phosphatase inhibitors and methods of use thereof |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| EP24196397.4A Division EP4506035A3 (de) | 2018-03-21 | 2019-03-21 | Shp2-phosphatasehemmer und verfahren zur verwendung davon |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| EP3768668A1 EP3768668A1 (de) | 2021-01-27 |
| EP3768668B1 true EP3768668B1 (de) | 2024-08-28 |
Family
ID=66041715
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| EP19715690.4A Active EP3768668B1 (de) | 2018-03-21 | 2019-03-21 | Shp2-phosphatase-inhibitoren und verfahren zur verwendung davon |
| EP24196397.4A Pending EP4506035A3 (de) | 2018-03-21 | 2019-03-21 | Shp2-phosphatasehemmer und verfahren zur verwendung davon |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| EP24196397.4A Pending EP4506035A3 (de) | 2018-03-21 | 2019-03-21 | Shp2-phosphatasehemmer und verfahren zur verwendung davon |
Country Status (15)
| Country | Link |
|---|---|
| US (4) | US20220315586A1 (de) |
| EP (2) | EP3768668B1 (de) |
| JP (2) | JP7418395B2 (de) |
| KR (1) | KR102926301B1 (de) |
| CN (2) | CN112166110B (de) |
| AU (2) | AU2019240299B2 (de) |
| BR (1) | BR112020019385A2 (de) |
| CA (1) | CA3094690A1 (de) |
| CL (2) | CL2020002419A1 (de) |
| IL (2) | IL301106B2 (de) |
| MX (2) | MX2020009782A (de) |
| SG (1) | SG11202009245TA (de) |
| TW (1) | TWI879728B (de) |
| WO (1) | WO2019183367A1 (de) |
| ZA (1) | ZA202106851B (de) |
Families Citing this family (118)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US11466017B2 (en) | 2011-03-10 | 2022-10-11 | Board Of Regents, The University Of Texas System | Heterocyclic inhibitors of PTPN11 |
| JO3517B1 (ar) | 2014-01-17 | 2020-07-05 | Novartis Ag | ان-ازاسبيرو الكان حلقي كبديل مركبات اريل-ان مغايرة وتركيبات لتثبيط نشاط shp2 |
| IL294124B2 (en) | 2015-12-10 | 2024-02-01 | Ptc Therapeutics Inc | Methods for treating huntington's disease |
| WO2017210134A1 (en) | 2016-05-31 | 2017-12-07 | Board Of Regents, University Of Texas System | Heterocyclic inhibitors of ptpn11 |
| WO2017216706A1 (en) | 2016-06-14 | 2017-12-21 | Novartis Ag | Compounds and compositions for inhibiting the activity of shp2 |
| WO2018057884A1 (en) | 2016-09-22 | 2018-03-29 | Relay Therapeutics, Inc. | Shp2 phosphatase inhibitors and methods of use thereof |
| TW202500565A (zh) | 2016-10-24 | 2025-01-01 | 美商傳達治療有限公司 | Shp2磷酸酶抑制劑及其使用方法 |
| EP3630770B1 (de) | 2017-05-26 | 2024-08-28 | Relay Therapeutics, Inc. | Pyrazolo[3,4-b]pyrazin-derivate als shp2-phosphatase-inhibitoren |
| US11407753B2 (en) | 2017-06-05 | 2022-08-09 | Ptc Therapeutics, Inc. | Compounds for treating Huntington's disease |
| US11382918B2 (en) | 2017-06-28 | 2022-07-12 | Ptc Therapeutics, Inc. | Methods for treating Huntington's Disease |
| WO2019005993A1 (en) | 2017-06-28 | 2019-01-03 | Ptc Therapeutics, Inc. | METHODS OF TREATING HUNTINGTON'S DISEASE |
| EP3687997A1 (de) | 2017-09-29 | 2020-08-05 | Relay Therapeutics, Inc. | Pyrazolo[3,4-b]pyrazin-derivate als shp2-phosphatase-inhibitoren |
| AU2019240299B2 (en) | 2018-03-21 | 2023-06-22 | D.E. Shaw Research, Llc | SHP2 phosphatase inhibitors and methods of use thereof |
| US12138263B2 (en) | 2018-03-21 | 2024-11-12 | Relay Therapeutics, Inc. | Pyrazolo[3,4-b]pyrazine SHP2 phosphatase inhibitors and methods of use thereof |
| MX2020009957A (es) | 2018-03-27 | 2021-01-15 | Ptc Therapeutics Inc | Compuestos para el tratamiento de enfermedad de hungtinton. |
| MX2020011528A (es) * | 2018-05-02 | 2021-02-09 | Navire Pharma Inc | Inhibidores heterociclicos sustituidos de ptpn11. |
| PL3814357T3 (pl) * | 2018-06-27 | 2024-09-16 | Ptc Therapeutics, Inc. | Związki heterocykliczne i heteroarylowe do leczenia choroby huntingtona |
| EP3814345B8 (de) | 2018-06-27 | 2024-10-30 | PTC Therapeutics, Inc. | Heteroaryl verbindungen zur behandlung von morbus huntington |
| CN112601750B (zh) | 2018-08-10 | 2023-10-31 | 纳维尔制药有限公司 | Ptpn11(shp2)抑制剂 |
| CR20210175A (es) | 2018-09-18 | 2021-06-01 | Nikang Therapeutics Inc | Derivados de anillo tricíclico condensado como inhibidores de la fosfatasa de homología a src 2 (shp2) |
| CA3113689A1 (en) | 2018-09-25 | 2020-04-02 | Antabio Sas | Indane derivatives for use in the treatment of bacterial infection |
| US20210393623A1 (en) * | 2018-09-26 | 2021-12-23 | Jacobio Pharmaceuticals Co., Ltd. | Novel Heterocyclic Derivatives Useful as SHP2 Inhibitors |
| EP3860717A1 (de) | 2018-10-03 | 2021-08-11 | Gilead Sciences, Inc. | Imidozopyrimidinderivate |
| TW202028183A (zh) * | 2018-10-10 | 2020-08-01 | 大陸商江蘇豪森藥業集團有限公司 | 含氮雜芳類衍生物調節劑、其製備方法和應用 |
| BR112021005733A2 (pt) | 2018-10-17 | 2021-07-27 | Array Biopharma Inc. | inibidores de proteína tirosina fosfatase |
| CN117143079A (zh) | 2018-11-06 | 2023-12-01 | 上海奕拓医药科技有限责任公司 | 一种螺芳环化合物及其应用 |
| WO2020156243A1 (zh) * | 2019-01-31 | 2020-08-06 | 贝达药业股份有限公司 | Shp2抑制剂及其应用 |
| WO2020156242A1 (zh) * | 2019-01-31 | 2020-08-06 | 贝达药业股份有限公司 | Shp2抑制剂及其应用 |
| EP3917911A1 (de) * | 2019-01-31 | 2021-12-08 | Kyorin Pharmaceutical Co., Ltd. | 15-pgdh-inhibitoren |
| US11033547B2 (en) | 2019-03-07 | 2021-06-15 | Merck Patent Gmbh | Carboxamide-pyrimidine derivatives as SHP2 antagonists |
| CU20210080A7 (es) | 2019-04-02 | 2022-05-11 | Array Biopharma Inc | Triazinas sustituidas como inhibidores de proteína tirosina fosfatasa |
| JP7586834B2 (ja) | 2019-04-08 | 2024-11-19 | メルク パテント ゲゼルシャフト ミット ベシュレンクテル ハフツング | Shp2拮抗薬としてのピリミジノン誘導体 |
| WO2020231977A1 (en) | 2019-05-13 | 2020-11-19 | Ptc Therapeutics, Inc. | Compounds for treating huntington's disease |
| CN114008035A (zh) * | 2019-06-14 | 2022-02-01 | 北京盛诺基医药科技股份有限公司 | 一种shp2磷酸酶变构抑制剂 |
| CN111704611B (zh) * | 2019-07-25 | 2022-01-14 | 上海凌达生物医药有限公司 | 一类芳基螺环类shp2抑制剂化合物、制备方法和用途 |
| EP3772513A1 (de) | 2019-08-09 | 2021-02-10 | C.N.C.C.S. S.c.a.r.l. Collezione Nazionale Dei Composti Chimici e Centro Screening | Shp2-inhibitoren |
| GB201911928D0 (en) * | 2019-08-20 | 2019-10-02 | Otsuka Pharma Co Ltd | Pharmaceutical compounds |
| MX2022003454A (es) | 2019-09-24 | 2022-04-19 | Relay Therapeutics Inc | Inhibidores de fosfatasa shp2 y metodos para su fabricacion y uso. |
| US20240139193A1 (en) * | 2019-10-15 | 2024-05-02 | Amgen Inc. | Combination therapy of kras inhibitor and shp2 inhibitor for treatment of cancers |
| CA3156359A1 (en) | 2019-11-08 | 2021-05-14 | Adrian Liam Gill | Bicyclic heteroaryl compounds and uses thereof |
| WO2021088945A1 (zh) * | 2019-11-08 | 2021-05-14 | 南京圣和药业股份有限公司 | 作为shp2抑制剂的化合物及其应用 |
| CN117683033A (zh) * | 2019-12-19 | 2024-03-12 | 首药控股(北京)股份有限公司 | 取代的炔基杂环化合物 |
| WO2021121397A1 (zh) * | 2019-12-19 | 2021-06-24 | 首药控股(北京)股份有限公司 | 取代的炔基杂环化合物 |
| CA3164995A1 (en) | 2019-12-20 | 2021-06-24 | Erasca, Inc. | Tricyclic pyridones and pyrimidones |
| CN114846005B (zh) * | 2020-01-21 | 2024-04-02 | 贝达药业股份有限公司 | Shp2抑制剂及其应用 |
| WO2021148010A1 (zh) * | 2020-01-22 | 2021-07-29 | 南京明德新药研发有限公司 | 吡唑并杂芳环类化合物及其应用 |
| EP4110338A1 (de) | 2020-02-28 | 2023-01-04 | Novartis AG | Dreifache pharmazeutische kombination mit dabrafenib, einem erk-inhibitor und einem shp2-inhibitor |
| CN113493440B (zh) * | 2020-04-03 | 2024-08-23 | 上海翰森生物医药科技有限公司 | 含氮杂芳类衍生物的盐及其晶型 |
| TW202144334A (zh) * | 2020-04-03 | 2021-12-01 | 大陸商上海翰森生物醫藥科技有限公司 | 含氮雜芳類衍生物游離鹼的晶型 |
| CN115362149B (zh) * | 2020-04-26 | 2024-05-14 | 贝达药业股份有限公司 | Shp2抑制剂及其组合物和应用 |
| WO2021218755A1 (zh) * | 2020-04-30 | 2021-11-04 | 贝达药业股份有限公司 | Shp2抑制剂及其组合物和应用 |
| CA3181898A1 (en) * | 2020-06-11 | 2021-12-16 | Bang Fu | Shp2 inhibitors, compositions and uses thereof |
| IL299131A (en) | 2020-06-18 | 2023-02-01 | Revolution Medicines Inc | Methods for delaying, preventing and treating acquired resistance to RAS inhibitors |
| CN115515947B (zh) * | 2020-07-24 | 2024-02-06 | 贝达药业股份有限公司 | Shp2抑制剂及其组合物和应用 |
| US20250195521A1 (en) | 2020-09-03 | 2025-06-19 | Revolution Medicines, Inc. | Use of sos1 inhibitors to treat malignancies with shp2 mutations |
| CA3194067A1 (en) | 2020-09-15 | 2022-03-24 | Revolution Medicines, Inc. | Ras inhibitors |
| JP2023544450A (ja) | 2020-09-23 | 2023-10-23 | エラスカ・インコーポレイテッド | 三環式ピリドン及びピリミドン |
| CN116848121A (zh) * | 2020-12-18 | 2023-10-03 | 建新公司 | 制备shp2抑制剂的方法 |
| WO2022133345A1 (en) | 2020-12-18 | 2022-06-23 | Erasca, Inc. | Tricyclic pyridones and pyrimidones |
| TW202241907A (zh) * | 2020-12-18 | 2022-11-01 | 美商瑞佛路森醫藥公司 | 製備shp2抑制劑之方法 |
| WO2022156765A1 (zh) * | 2021-01-22 | 2022-07-28 | 南京明德新药研发有限公司 | 吡唑并吡嗪联三环类化合物及其应用 |
| EP4288436A4 (de) * | 2021-02-05 | 2025-01-22 | HUTCHMED Limited | Tricyclische verbindungen und verwendungen davon |
| US20240025924A1 (en) * | 2021-03-23 | 2024-01-25 | Shanghai Haiyan Pharmaceutical Technology Co., Ltd. | Heterocycle substituted ketone derivative, and composition and medicinal use thereof |
| WO2022226145A1 (en) * | 2021-04-21 | 2022-10-27 | Hospital Rhode Island | Method and composition for the prevention or treatment of osteoarthritis |
| KR20240017811A (ko) | 2021-05-05 | 2024-02-08 | 레볼루션 메디슨즈, 인크. | 암의 치료를 위한 ras 억제제 |
| TW202309052A (zh) | 2021-05-05 | 2023-03-01 | 美商銳新醫藥公司 | Ras抑制劑 |
| JP2024516450A (ja) | 2021-05-05 | 2024-04-15 | レボリューション メディシンズ インコーポレイテッド | 共有結合性ras阻害剤及びその使用 |
| CN115304612A (zh) * | 2021-05-08 | 2022-11-08 | 南京圣和药业股份有限公司 | 杂环类shp2抑制剂的晶型 |
| CN115304613A (zh) * | 2021-05-08 | 2022-11-08 | 南京圣和药业股份有限公司 | 杂环类shp2抑制剂的制备方法 |
| CN115960109B (zh) * | 2021-05-31 | 2024-06-25 | 药雅科技(上海)有限公司 | 稠环类shp2磷酸酶抑制剂的制备及其应用 |
| TW202244049A (zh) * | 2021-05-12 | 2022-11-16 | 大陸商藥雅科技(上海)有限公司 | Shp2磷酸酶抑制劑的製備及其應用 |
| CN115340545A (zh) * | 2021-05-14 | 2022-11-15 | 浙江海正药业股份有限公司 | 双环杂芳基类衍生物及其制备方法和用途 |
| WO2022243940A1 (en) | 2021-05-21 | 2022-11-24 | Peptone, Ltd. | Spacio-temporal determination of polypeptide structure |
| WO2022242767A1 (zh) * | 2021-05-21 | 2022-11-24 | 石药集团中奇制药技术(石家庄)有限公司 | 螺环类化合物及其用途 |
| WO2022259157A1 (en) | 2021-06-09 | 2022-12-15 | Novartis Ag | A triple pharmaceutical combination comprising dabrafenib, trametinib and a shp2 inhibitor |
| CN115466273A (zh) * | 2021-06-11 | 2022-12-13 | 首药控股(北京)股份有限公司 | 取代的炔基杂环化合物 |
| TW202317100A (zh) | 2021-06-23 | 2023-05-01 | 瑞士商諾華公司 | 包含kras g12c抑制劑的藥物組合及其用於治療癌症之用途 |
| WO2023280283A1 (zh) * | 2021-07-07 | 2023-01-12 | 浙江同源康医药股份有限公司 | 用作shp2抑制剂的化合物及其应用 |
| TW202319056A (zh) | 2021-07-09 | 2023-05-16 | 南韓商治納輔醫藥科技有限公司 | Shp2抑制劑及其用途 |
| KR20240055778A (ko) | 2021-09-01 | 2024-04-29 | 노파르티스 아게 | Tead 억제제를 포함하는 제약 조합물 및 암의 치료를 위한 이의 용도 |
| WO2023046198A1 (zh) * | 2021-09-27 | 2023-03-30 | 中国医药研究开发中心有限公司 | 磺胺酮类化合物及其制备方法和医药用途 |
| WO2023051648A1 (zh) * | 2021-09-28 | 2023-04-06 | 甘李药业股份有限公司 | 可用作shp2抑制剂的化合物及其制备方法和用途 |
| WO2023051717A1 (zh) * | 2021-09-29 | 2023-04-06 | 微境生物医药科技(上海)有限公司 | 作为shp2抑制剂的稠环化合物 |
| AR127308A1 (es) | 2021-10-08 | 2024-01-10 | Revolution Medicines Inc | Inhibidores ras |
| EP4417600A4 (de) * | 2021-10-14 | 2025-02-19 | Beijing Tide Pharmaceutical Co., Ltd. | Shp2-inhibitor, pharmazeutische zusammensetzung damit und verwendung davon |
| CN116063307B (zh) | 2021-10-29 | 2025-08-19 | 中国药科大学 | Shp2与cdk4/6双靶点抑制化合物合成及其制备方法与应用 |
| CN114213417B (zh) * | 2021-11-16 | 2023-08-22 | 郑州大学 | 吡唑并六元氮杂环类化合物及其合成方法和应用 |
| CN118369315A (zh) * | 2021-12-15 | 2024-07-19 | 贝达药业股份有限公司 | 吡唑并嘧啶酮类化合物及其盐的结晶 |
| JP2025510572A (ja) | 2022-03-08 | 2025-04-15 | レボリューション メディシンズ インコーポレイテッド | 免疫不応性肺癌を治療するための方法 |
| CN116731038B (zh) * | 2022-03-10 | 2025-11-21 | 苏州亚盛药业有限公司 | 含氮杂环化合物及其制备方法和应用 |
| CN117088887A (zh) * | 2022-05-20 | 2023-11-21 | 安徽中科拓苒药物科学研究有限公司 | Shp2抑制剂及其用途 |
| US11878958B2 (en) | 2022-05-25 | 2024-01-23 | Ikena Oncology, Inc. | MEK inhibitors and uses thereof |
| WO2023240263A1 (en) | 2022-06-10 | 2023-12-14 | Revolution Medicines, Inc. | Macrocyclic ras inhibitors |
| CN119923402A (zh) * | 2022-09-22 | 2025-05-02 | 上海海雁医药科技有限公司 | 杂环取代的甲酮类衍生物的固体形式及其应用 |
| CN118206550A (zh) * | 2022-12-15 | 2024-06-18 | 江苏威凯尔医药科技有限公司 | Shp2抑制剂及其应用 |
| TWI879384B (zh) * | 2023-01-16 | 2025-04-01 | 大陸商上海美悅生物科技發展有限公司 | 雜環取代的吡嗪類化合物及其製備方法和用途 |
| AU2024241633A1 (en) | 2023-03-30 | 2025-11-06 | Revolution Medicines, Inc. | Compositions for inducing ras gtp hydrolysis and uses thereof |
| CR20250420A (es) | 2023-04-07 | 2025-11-20 | Revolution Medicines Inc | Inhibidores macrocíclicos de ras |
| AR132338A1 (es) | 2023-04-07 | 2025-06-18 | Revolution Medicines Inc | Inhibidores de ras |
| CN121100123A (zh) | 2023-04-14 | 2025-12-09 | 锐新医药公司 | Ras抑制剂的结晶形式 |
| CN121464140A (zh) | 2023-04-14 | 2026-02-03 | 锐新医药公司 | Ras抑制剂的结晶形式、含有其的组合物及其使用方法 |
| TW202508595A (zh) | 2023-05-04 | 2025-03-01 | 美商銳新醫藥公司 | 用於ras相關疾病或病症之組合療法 |
| TW202504588A (zh) * | 2023-06-12 | 2025-02-01 | 美商雷密克斯醫療公司 | 用於調節剪接之化合物及方法 |
| US20250049810A1 (en) | 2023-08-07 | 2025-02-13 | Revolution Medicines, Inc. | Methods of treating a ras protein-related disease or disorder |
| AU2024360465A1 (en) | 2023-10-12 | 2026-04-09 | Revolution Medicines, Inc. | Macrocyclic ras inhibitors |
| WO2025165802A1 (en) | 2024-01-30 | 2025-08-07 | Genentech, Inc. | Processes for making 1h-pyrazolo [3, 4-b] pyrazine compounds |
| WO2025171296A1 (en) | 2024-02-09 | 2025-08-14 | Revolution Medicines, Inc. | Ras inhibitors |
| TW202547461A (zh) | 2024-05-17 | 2025-12-16 | 美商銳新醫藥公司 | Ras抑制劑 |
| WO2025255438A1 (en) | 2024-06-07 | 2025-12-11 | Revolution Medicines, Inc. | Methods of treating a ras protein-related disease or disorder |
| WO2025265060A1 (en) | 2024-06-21 | 2025-12-26 | Revolution Medicines, Inc. | Therapeutic compositions and methods for managing treatment-related effects |
| WO2026006747A1 (en) | 2024-06-28 | 2026-01-02 | Revolution Medicines, Inc. | Ras inhibitors |
| WO2026015825A1 (en) | 2024-07-12 | 2026-01-15 | Revolution Medicines, Inc. | Use of ras inhibitor for treating pancreatic cancer |
| WO2026015796A1 (en) | 2024-07-12 | 2026-01-15 | Revolution Medicines, Inc. | Methods of treating a ras related disease or disorder |
| WO2026015790A1 (en) | 2024-07-12 | 2026-01-15 | Revolution Medicines, Inc. | Methods of treating a ras related disease or disorder |
| WO2026015801A1 (en) | 2024-07-12 | 2026-01-15 | Revolution Medicines, Inc. | Methods of treating a ras related disease or disorder |
| WO2026050446A1 (en) | 2024-08-29 | 2026-03-05 | Revolution Medicines, Inc. | Ras inhibitors |
| WO2026072904A2 (en) | 2024-09-26 | 2026-04-02 | Revolution Medicines, Inc. | Compositions and methods for treating lung cancer |
| CN119059990B (zh) * | 2024-11-01 | 2025-02-07 | 安徽昊帆生物有限公司 | 1-氨基甲酸叔丁酯哌嗪的制备方法 |
Family Cites Families (59)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5466823A (en) * | 1993-11-30 | 1995-11-14 | G.D. Searle & Co. | Substituted pyrazolyl benzenesulfonamides |
| EP1622569B1 (de) | 2003-04-24 | 2015-12-02 | Incyte Corporation | Aza-spiro-alkan-derivate als hemmer von metallproteasen |
| US20060135571A1 (en) | 2003-06-13 | 2006-06-22 | Jaquith James B | Imidazo(2,1-b)-1,3,4-thiadiazole sulfoxides and sulfones |
| WO2010011666A2 (en) | 2008-07-21 | 2010-01-28 | University Of South Florida | Indoline scaffold shp-2 inhibitors and cancer treatment method |
| AU2009275544B2 (en) | 2008-07-29 | 2013-09-12 | Merck Patent Gmbh | Imidazothiadiazoles derivatives |
| WO2010097798A1 (en) | 2009-02-25 | 2010-09-02 | Urifer Ltd. | 6-phenyl-2-[((piperidin-4-ylmethyl)-piperazin-1yl) or piperazin 1-ylmethyl)-piperidin-1-yl)]-imidazo[2,1-b][1,3,4]thiadiazole derivatives and their use |
| WO2010121212A2 (en) | 2009-04-17 | 2010-10-21 | H. Lee Moffit Cancer Center And Research Institute, Inc. | Indoline scaffold shp-2 inhibitors and method of treating cancer |
| US20110256331A1 (en) | 2010-04-14 | 2011-10-20 | Dak Americas Llc | Ultra-high iv polyester for extrusion blow molding and method for its production |
| CN103570622B (zh) * | 2012-09-07 | 2015-02-04 | 北京京卫燕康药物研究所有限公司 | 塞来昔布的制备方法 |
| ES2699351T3 (es) | 2014-01-17 | 2019-02-08 | Novartis Ag | Derivados de 1-piridazin/triazin-3-il-piper(-azina)/idina/pirolidina y composiciones de las mismas para inhibir la actividad de SHP2 |
| WO2015107494A1 (en) | 2014-01-17 | 2015-07-23 | Novartis Ag | 1 -(triazin-3-yi_/pyridazin-3-yl)-piper(-azine)idine derivatives and compositions thereof for inhibiting the activity of shp2 |
| JO3517B1 (ar) | 2014-01-17 | 2020-07-05 | Novartis Ag | ان-ازاسبيرو الكان حلقي كبديل مركبات اريل-ان مغايرة وتركيبات لتثبيط نشاط shp2 |
| EP3310779B1 (de) | 2015-06-19 | 2019-05-08 | Novartis AG | Verbindungen und zusammensetzungen zur hemmung der aktivität von shp2 |
| US10287266B2 (en) | 2015-06-19 | 2019-05-14 | Novartis Ag | Compounds and compositions for inhibiting the activity of SHP2 |
| WO2016203405A1 (en) | 2015-06-19 | 2016-12-22 | Novartis Ag | Compounds and compositions for inhibiting the activity of shp2 |
| WO2017156397A1 (en) | 2016-03-11 | 2017-09-14 | Board Of Regents, The University Of Texas Sysytem | Heterocyclic inhibitors of ptpn11 |
| CN107286150B (zh) | 2016-04-11 | 2020-07-07 | 中国科学院上海有机化学研究所 | N-杂环类化合物、其中间体、制备方法、药物组合物和应用 |
| WO2017210134A1 (en) | 2016-05-31 | 2017-12-07 | Board Of Regents, University Of Texas System | Heterocyclic inhibitors of ptpn11 |
| MX388576B (es) | 2016-06-07 | 2025-03-20 | Jacobio Pharmaceuticals Co Ltd | Derivados heterociclicos novedosos utiles como inhibidores de shp2. |
| WO2017216706A1 (en) | 2016-06-14 | 2017-12-21 | Novartis Ag | Compounds and compositions for inhibiting the activity of shp2 |
| EP4302834A3 (de) | 2016-07-12 | 2024-07-17 | Revolution Medicines, Inc. | 2,5-disubstituierte 3-methyl pyrazine und 2,5,6-trisubstituierte 3-methylpyrazine als allosterische shp2-inhibitoren |
| WO2018013957A1 (en) * | 2016-07-14 | 2018-01-18 | Greco Chad | Hybrid formulation of responsive polymeric nanocarriers for therapeutic and diagnostic delivery |
| WO2018057884A1 (en) | 2016-09-22 | 2018-03-29 | Relay Therapeutics, Inc. | Shp2 phosphatase inhibitors and methods of use thereof |
| TW202500565A (zh) | 2016-10-24 | 2025-01-01 | 美商傳達治療有限公司 | Shp2磷酸酶抑制劑及其使用方法 |
| JP7240319B2 (ja) | 2017-01-23 | 2023-03-15 | レヴォリューション・メディスンズ,インコーポレイテッド | アロステリックshp2阻害剤としての二環式化合物 |
| KR20190110588A (ko) | 2017-01-23 | 2019-09-30 | 레볼루션 메디슨즈, 인크. | 알로스테릭 shp2 억제제로서의 피리딘 화합물 |
| EA202190196A1 (ru) * | 2017-03-23 | 2021-08-31 | Джакобио Фармасьютикалс Ко., Лтд. | Новые гетероциклические производные, применимые в качестве ингибиторов shp2 |
| EP3630770B1 (de) | 2017-05-26 | 2024-08-28 | Relay Therapeutics, Inc. | Pyrazolo[3,4-b]pyrazin-derivate als shp2-phosphatase-inhibitoren |
| CA3074690A1 (en) | 2017-09-07 | 2019-03-14 | Revolution Medicines, Inc. | Shp2 inhibitor compositions and methods for treating cancer |
| JP7447002B2 (ja) | 2017-09-11 | 2024-03-11 | クルーゾン・ファーマシューティカルズ・インコーポレイテッド | SHP2のオクタヒドロシクロペンタ[c]ピロールのアロステリック阻害剤 |
| EP3687997A1 (de) | 2017-09-29 | 2020-08-05 | Relay Therapeutics, Inc. | Pyrazolo[3,4-b]pyrazin-derivate als shp2-phosphatase-inhibitoren |
| TW201930292A (zh) | 2017-10-12 | 2019-08-01 | 美商銳新醫藥公司 | 作為變構shp2抑制劑的吡啶、吡嗪和三嗪化合物 |
| TWI697490B (zh) | 2017-12-06 | 2020-07-01 | 大陸商北京加科思新藥研發有限公司 | 用於作為shp2抑制劑之新穎雜環衍生物 |
| JP7361693B2 (ja) | 2017-12-15 | 2023-10-16 | レヴォリューション・メディスンズ,インコーポレイテッド | アロステリックshp2阻害剤としての多環式化合物 |
| JP7335882B2 (ja) | 2018-02-13 | 2023-08-30 | ブルーレイ セラピューティクス (シャンハイ) カンパニー,リミティド | ピリミジン縮合環式化合物及びその製造方法、並びに使用 |
| WO2019165073A1 (en) | 2018-02-21 | 2019-08-29 | Relay Therapeutics, Inc. | Shp2 phosphatase inhibitors and methods of use thereof |
| IL276232B2 (en) | 2018-03-02 | 2024-04-01 | Otsuka Pharma Co Ltd | Pyrazine derivatives, pharmaceutical compositions comprising them and their use for treating diseases |
| RU2020133727A (ru) | 2018-03-21 | 2022-04-21 | Сучжоу Пухе Биофарма Ко., Лтд. | Ингибиторы shp2 и их применение |
| AU2019240299B2 (en) * | 2018-03-21 | 2023-06-22 | D.E. Shaw Research, Llc | SHP2 phosphatase inhibitors and methods of use thereof |
| US12138263B2 (en) | 2018-03-21 | 2024-11-12 | Relay Therapeutics, Inc. | Pyrazolo[3,4-b]pyrazine SHP2 phosphatase inhibitors and methods of use thereof |
| MX2020010719A (es) | 2018-04-10 | 2020-11-06 | Revolution Medicines Inc | Composiciones de inhibidores de shp2, metodos para tratar el cancer y metodos para identificar a un sujeto con mutaciones en shp2. |
| MX2020011528A (es) | 2018-05-02 | 2021-02-09 | Navire Pharma Inc | Inhibidores heterociclicos sustituidos de ptpn11. |
| CN110143949A (zh) | 2018-05-09 | 2019-08-20 | 北京加科思新药研发有限公司 | 可用作shp2抑制剂的新型杂环衍生物 |
| EP3801613A1 (de) | 2018-06-04 | 2021-04-14 | Bayer Aktiengesellschaft | Inhibitoren von shp2 |
| MX2021000795A (es) | 2018-07-24 | 2021-04-12 | Taiho Pharmaceutical Co Ltd | Compuestos heterociclicos para inhibir la actividad de shp2. |
| CN112601750B (zh) | 2018-08-10 | 2023-10-31 | 纳维尔制药有限公司 | Ptpn11(shp2)抑制剂 |
| CR20210175A (es) | 2018-09-18 | 2021-06-01 | Nikang Therapeutics Inc | Derivados de anillo tricíclico condensado como inhibidores de la fosfatasa de homología a src 2 (shp2) |
| US20210393623A1 (en) | 2018-09-26 | 2021-12-23 | Jacobio Pharmaceuticals Co., Ltd. | Novel Heterocyclic Derivatives Useful as SHP2 Inhibitors |
| CN112867718B (zh) | 2018-09-29 | 2025-03-04 | 诺华股份有限公司 | 用于抑制shp2活性的化合物和组合物的制造 |
| IL305106B2 (en) | 2018-09-29 | 2025-08-01 | Novartis Ag | Process of manufacture of a compound for inhibiting the activity of shp2 |
| EP3860717A1 (de) | 2018-10-03 | 2021-08-11 | Gilead Sciences, Inc. | Imidozopyrimidinderivate |
| JP2022508651A (ja) | 2018-10-08 | 2022-01-19 | レヴォリューション・メディスンズ,インコーポレイテッド | 癌を処置するためのshp2阻害剤組成物および方法 |
| TW202028183A (zh) | 2018-10-10 | 2020-08-01 | 大陸商江蘇豪森藥業集團有限公司 | 含氮雜芳類衍生物調節劑、其製備方法和應用 |
| CN111295384B (zh) | 2018-10-10 | 2022-08-12 | 江苏豪森药业集团有限公司 | 双环类衍生物抑制剂、其制备方法和应用 |
| BR112021005733A2 (pt) | 2018-10-17 | 2021-07-27 | Array Biopharma Inc. | inibidores de proteína tirosina fosfatase |
| CN117143079A (zh) | 2018-11-06 | 2023-12-01 | 上海奕拓医药科技有限责任公司 | 一种螺芳环化合物及其应用 |
| JP2022506887A (ja) | 2018-11-07 | 2022-01-17 | シャンハイ リンジーン バイオファーマ カンパニー リミテッド | 窒素含有縮合複素環系shp2阻害剤化合物、製造方法及び使用 |
| CN111153899B (zh) | 2018-11-08 | 2023-12-01 | 四川科伦博泰生物医药股份有限公司 | 一种取代吡啶化合物、其制备方法和用途 |
| MX2022003454A (es) | 2019-09-24 | 2022-04-19 | Relay Therapeutics Inc | Inhibidores de fosfatasa shp2 y metodos para su fabricacion y uso. |
-
2019
- 2019-03-21 AU AU2019240299A patent/AU2019240299B2/en active Active
- 2019-03-21 BR BR112020019385-2A patent/BR112020019385A2/pt unknown
- 2019-03-21 CN CN201980034042.8A patent/CN112166110B/zh active Active
- 2019-03-21 EP EP19715690.4A patent/EP3768668B1/de active Active
- 2019-03-21 MX MX2020009782A patent/MX2020009782A/es unknown
- 2019-03-21 US US16/982,401 patent/US20220315586A1/en not_active Abandoned
- 2019-03-21 TW TW108109755A patent/TWI879728B/zh active
- 2019-03-21 IL IL301106A patent/IL301106B2/en unknown
- 2019-03-21 CA CA3094690A patent/CA3094690A1/en active Pending
- 2019-03-21 EP EP24196397.4A patent/EP4506035A3/de active Pending
- 2019-03-21 JP JP2021500497A patent/JP7418395B2/ja active Active
- 2019-03-21 KR KR1020207030055A patent/KR102926301B1/ko active Active
- 2019-03-21 WO PCT/US2019/023389 patent/WO2019183367A1/en not_active Ceased
- 2019-03-21 CN CN202310913246.7A patent/CN117263942A/zh active Pending
- 2019-03-21 SG SG11202009245TA patent/SG11202009245TA/en unknown
-
2020
- 2020-05-28 US US16/886,105 patent/US10934302B1/en active Active
- 2020-09-17 IL IL277434A patent/IL277434B2/en unknown
- 2020-09-18 MX MX2023001917A patent/MX2023001917A/es unknown
- 2020-09-21 CL CL2020002419A patent/CL2020002419A1/es unknown
- 2020-11-17 US US16/950,576 patent/US12084447B2/en active Active
-
2021
- 2021-09-17 ZA ZA2021/06851A patent/ZA202106851B/en unknown
-
2022
- 2022-03-25 CL CL2022000751A patent/CL2022000751A1/es unknown
- 2022-12-15 US US18/066,551 patent/US12331056B2/en active Active
-
2023
- 2023-09-22 AU AU2023233205A patent/AU2023233205A1/en not_active Abandoned
- 2023-09-22 JP JP2023158204A patent/JP2023166614A/ja active Pending
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP3768668B1 (de) | Shp2-phosphatase-inhibitoren und verfahren zur verwendung davon | |
| EP3630770B1 (de) | Pyrazolo[3,4-b]pyrazin-derivate als shp2-phosphatase-inhibitoren | |
| US12570672B2 (en) | Macrocyclic compounds and methods of use | |
| KR101659193B1 (ko) | Btk 활성의 억제제로서의 헤테로아릴 피리돈 및 아자-피리돈 화합물 | |
| WO2023091726A1 (en) | Inhibitors of cyclin‑dependent kinase 12 (cdk12) | |
| ES3060156T3 (en) | (s)-1-(5-((pyridin-3-yl)thio)pyrazin-2-yl)-4'h,6'h-spiro[piperidine-4,5'-pyrrolo[1,2-b]pyrazol]-4'-amine derivatives and similar compounds as shp2 inhibitors for the treatment of e.g. cancer | |
| CN118556059A (zh) | 用于治疗癌症的杂环化合物 | |
| CN119053600A (zh) | 用于治疗癌症的杂环化合物 | |
| RU2846525C2 (ru) | Ингибиторы shp2 фосфатазы и способы их применения | |
| HK40101612A (zh) | Shp2磷酸酶抑制剂及其使用方法 | |
| HK40044087B (zh) | Shp2磷酸酶抑制剂及其使用方法 | |
| HK40044087A (en) | Shp2 phosphatase inhibitors and methods of use thereof | |
| WO2026096330A1 (en) | Glp-1 receptor agonists and methods of use | |
| WO2025194054A1 (en) | Spirocyclic compounds as modulators of kras and uses thereof |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| STAA | Information on the status of an ep patent application or granted ep patent |
Free format text: STATUS: UNKNOWN |
|
| STAA | Information on the status of an ep patent application or granted ep patent |
Free format text: STATUS: THE INTERNATIONAL PUBLICATION HAS BEEN MADE |
|
| PUAI | Public reference made under article 153(3) epc to a published international application that has entered the european phase |
Free format text: ORIGINAL CODE: 0009012 |
|
| STAA | Information on the status of an ep patent application or granted ep patent |
Free format text: STATUS: REQUEST FOR EXAMINATION WAS MADE |
|
| 17P | Request for examination filed |
Effective date: 20201015 |
|
| AK | Designated contracting states |
Kind code of ref document: A1 Designated state(s): AL AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HR HU IE IS IT LI LT LU LV MC MK MT NL NO PL PT RO RS SE SI SK SM TR |
|
| AX | Request for extension of the european patent |
Extension state: BA ME |
|
| DAV | Request for validation of the european patent (deleted) | ||
| DAX | Request for extension of the european patent (deleted) | ||
| STAA | Information on the status of an ep patent application or granted ep patent |
Free format text: STATUS: EXAMINATION IS IN PROGRESS |
|
| 17Q | First examination report despatched |
Effective date: 20211130 |
|
| GRAP | Despatch of communication of intention to grant a patent |
Free format text: ORIGINAL CODE: EPIDOSNIGR1 |
|
| STAA | Information on the status of an ep patent application or granted ep patent |
Free format text: STATUS: GRANT OF PATENT IS INTENDED |
|
| RIC1 | Information provided on ipc code assigned before grant |
Ipc: A61P 35/00 20060101ALI20240305BHEP Ipc: C07D 519/00 20060101ALI20240305BHEP Ipc: C07D 491/20 20060101ALI20240305BHEP Ipc: C07D 491/107 20060101ALI20240305BHEP Ipc: C07D 491/10 20060101ALI20240305BHEP Ipc: C07D 487/20 20060101ALI20240305BHEP Ipc: C07D 487/04 20060101ALI20240305BHEP Ipc: C07D 471/04 20060101ALI20240305BHEP Ipc: C07D 417/06 20060101ALI20240305BHEP Ipc: C07D 417/04 20060101ALI20240305BHEP Ipc: C07D 241/18 20060101ALI20240305BHEP Ipc: C07D 401/12 20060101ALI20240305BHEP Ipc: C07D 401/06 20060101ALI20240305BHEP Ipc: C07D 401/04 20060101ALI20240305BHEP Ipc: C07D 221/20 20060101ALI20240305BHEP Ipc: C07D 405/14 20060101ALI20240305BHEP Ipc: C07D 401/14 20060101AFI20240305BHEP |
|
| INTG | Intention to grant announced |
Effective date: 20240320 |
|
| GRAS | Grant fee paid |
Free format text: ORIGINAL CODE: EPIDOSNIGR3 |
|
| GRAA | (expected) grant |
Free format text: ORIGINAL CODE: 0009210 |
|
| STAA | Information on the status of an ep patent application or granted ep patent |
Free format text: STATUS: THE PATENT HAS BEEN GRANTED |
|
| AK | Designated contracting states |
Kind code of ref document: B1 Designated state(s): AL AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HR HU IE IS IT LI LT LU LV MC MK MT NL NO PL PT RO RS SE SI SK SM TR |
|
| REG | Reference to a national code |
Ref country code: GB Ref legal event code: FG4D |
|
| REG | Reference to a national code |
Ref country code: CH Ref legal event code: EP |
|
| REG | Reference to a national code |
Ref country code: DE Ref legal event code: R096 Ref document number: 602019057838 Country of ref document: DE |
|
| REG | Reference to a national code |
Ref country code: IE Ref legal event code: FG4D |
|
| P01 | Opt-out of the competence of the unified patent court (upc) registered |
Free format text: CASE NUMBER: APP_50967/2024 Effective date: 20240909 |
|
| REG | Reference to a national code |
Ref country code: NL Ref legal event code: FP |
|
| REG | Reference to a national code |
Ref country code: LT Ref legal event code: MG9D |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: NO Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20241128 |
|
| REG | Reference to a national code |
Ref country code: AT Ref legal event code: MK05 Ref document number: 1717855 Country of ref document: AT Kind code of ref document: T Effective date: 20240828 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: GR Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20241129 Ref country code: PL Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20240828 Ref country code: PT Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20241230 Ref country code: FI Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20240828 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: BG Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20240828 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: LV Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20240828 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: IS Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20241228 Ref country code: AT Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20240828 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: HR Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20240828 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: RS Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20241128 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: RS Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20241128 Ref country code: PT Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20241230 Ref country code: PL Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20240828 Ref country code: NO Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20241128 Ref country code: LV Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20240828 Ref country code: IS Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20241228 Ref country code: HR Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20240828 Ref country code: GR Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20241129 Ref country code: FI Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20240828 Ref country code: BG Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20240828 Ref country code: AT Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20240828 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: SM Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20240828 Ref country code: DK Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20240828 Ref country code: RO Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20240828 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: ES Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20240828 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: EE Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20240828 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: CZ Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20240828 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: SK Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20240828 Ref country code: IT Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20240828 |
|
| REG | Reference to a national code |
Ref country code: DE Ref legal event code: R097 Ref document number: 602019057838 Country of ref document: DE |
|
| PLBE | No opposition filed within time limit |
Free format text: ORIGINAL CODE: 0009261 |
|
| STAA | Information on the status of an ep patent application or granted ep patent |
Free format text: STATUS: NO OPPOSITION FILED WITHIN TIME LIMIT |
|
| 26N | No opposition filed |
Effective date: 20250530 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: SE Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20240828 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: MC Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20240828 |
|
| REG | Reference to a national code |
Ref country code: CH Ref legal event code: H13 Free format text: ST27 STATUS EVENT CODE: U-0-0-H10-H13 (AS PROVIDED BY THE NATIONAL OFFICE) Effective date: 20251023 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: LU Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20250321 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: CH Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20250331 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: GB Payment date: 20260327 Year of fee payment: 8 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: DE Payment date: 20260327 Year of fee payment: 8 Ref country code: IE Payment date: 20260327 Year of fee payment: 8 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: BE Payment date: 20260327 Year of fee payment: 8 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: NL Payment date: 20260326 Year of fee payment: 8 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: FR Payment date: 20260325 Year of fee payment: 8 |