ES2700901A2 - Proceso para la preparación de 3ß-hidroxi-17-(1h-benzimidazol-1-il)androsta-5,16-dieno. - Google Patents
Proceso para la preparación de 3ß-hidroxi-17-(1h-benzimidazol-1-il)androsta-5,16-dieno. Download PDFInfo
- Publication number
- ES2700901A2 ES2700901A2 ES201990014A ES201990014A ES2700901A2 ES 2700901 A2 ES2700901 A2 ES 2700901A2 ES 201990014 A ES201990014 A ES 201990014A ES 201990014 A ES201990014 A ES 201990014A ES 2700901 A2 ES2700901 A2 ES 2700901A2
- Authority
- ES
- Spain
- Prior art keywords
- benzimidazol
- androsta
- hydroxy
- preparation
- compound
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
- 238000000034 method Methods 0.000 title claims abstract description 37
- PAFKTGFSEFKSQG-PAASFTFBSA-N Galeterone Chemical compound C1=NC2=CC=CC=C2N1C1=CC[C@H]2[C@H](CC=C3[C@@]4(CC[C@H](O)C3)C)[C@@H]4CC[C@@]21C PAFKTGFSEFKSQG-PAASFTFBSA-N 0.000 title claims abstract description 27
- 238000002360 preparation method Methods 0.000 title claims description 25
- 150000001875 compounds Chemical class 0.000 claims abstract description 33
- 229950003400 galeterone Drugs 0.000 claims abstract description 27
- MDHIWBNJNHUBJL-VMXHOPILSA-N (8s,9s,10r,13r,14s)-10,13-dimethyl-2,3,4,7,8,9,11,12,14,15-decahydro-1h-cyclopenta[a]phenanthrene Chemical compound C1CCC[C@]2(C)[C@H]3CC[C@](C)(C=CC4)[C@@H]4[C@@H]3CC=C21 MDHIWBNJNHUBJL-VMXHOPILSA-N 0.000 claims abstract description 26
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical group CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 claims description 42
- 238000006243 chemical reaction Methods 0.000 claims description 38
- -1 1H-benzimidazol-1-yl Chemical group 0.000 claims description 20
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 claims description 16
- 239000005725 8-Hydroxyquinoline Substances 0.000 claims description 13
- 229960003540 oxyquinoline Drugs 0.000 claims description 13
- MCJGNVYPOGVAJF-UHFFFAOYSA-N quinolin-8-ol Chemical compound C1=CN=C2C(O)=CC=CC2=C1 MCJGNVYPOGVAJF-UHFFFAOYSA-N 0.000 claims description 13
- 239000002904 solvent Substances 0.000 claims description 13
- 229910021595 Copper(I) iodide Inorganic materials 0.000 claims description 12
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 claims description 12
- LSXDOTMGLUJQCM-UHFFFAOYSA-M copper(i) iodide Chemical compound I[Cu] LSXDOTMGLUJQCM-UHFFFAOYSA-M 0.000 claims description 12
- HYZJCKYKOHLVJF-UHFFFAOYSA-N 1H-benzimidazole Chemical compound C1=CC=C2NC=NC2=C1 HYZJCKYKOHLVJF-UHFFFAOYSA-N 0.000 claims description 10
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 claims description 10
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 claims description 9
- 239000002253 acid Substances 0.000 claims description 8
- 239000003960 organic solvent Substances 0.000 claims description 8
- 229910000027 potassium carbonate Inorganic materials 0.000 claims description 7
- 239000011541 reaction mixture Substances 0.000 claims description 7
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical group [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 claims description 6
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 claims description 5
- GUVUOGQBMYCBQP-UHFFFAOYSA-N dmpu Chemical compound CN1CCCN(C)C1=O GUVUOGQBMYCBQP-UHFFFAOYSA-N 0.000 claims description 5
- 229940113088 dimethylacetamide Drugs 0.000 claims description 4
- 238000001914 filtration Methods 0.000 claims description 4
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 claims description 3
- 229910000024 caesium carbonate Inorganic materials 0.000 claims description 3
- 238000010992 reflux Methods 0.000 claims description 3
- 229910000029 sodium carbonate Inorganic materials 0.000 claims description 3
- 238000009835 boiling Methods 0.000 claims description 2
- 238000001556 precipitation Methods 0.000 claims description 2
- MFRIHAYPQRLWNB-UHFFFAOYSA-N sodium tert-butoxide Chemical compound [Na+].CC(C)(C)[O-] MFRIHAYPQRLWNB-UHFFFAOYSA-N 0.000 claims 2
- TUUSTVXFVGQGSB-TYVXZMKCSA-N C(C(=O)O)(=O)O.C(C(=O)O)(=O)O.C[C@@]12C=CC[C@H]1[C@@H]1CC=C3CCCC[C@]3(C)[C@H]1CC2 Chemical compound C(C(=O)O)(=O)O.C(C(=O)O)(=O)O.C[C@@]12C=CC[C@H]1[C@@H]1CC=C3CCCC[C@]3(C)[C@H]1CC2 TUUSTVXFVGQGSB-TYVXZMKCSA-N 0.000 claims 1
- GEVPUGOOGXGPIO-UHFFFAOYSA-N oxalic acid;dihydrate Chemical compound O.O.OC(=O)C(O)=O GEVPUGOOGXGPIO-UHFFFAOYSA-N 0.000 claims 1
- LPNYRYFBWFDTMA-UHFFFAOYSA-N potassium tert-butoxide Chemical compound [K+].CC(C)(C)[O-] LPNYRYFBWFDTMA-UHFFFAOYSA-N 0.000 claims 1
- 230000015572 biosynthetic process Effects 0.000 abstract description 14
- 238000003786 synthesis reaction Methods 0.000 abstract description 14
- 206010060862 Prostate cancer Diseases 0.000 abstract description 5
- 208000000236 Prostatic Neoplasms Diseases 0.000 abstract description 5
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 28
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 24
- 238000004809 thin layer chromatography Methods 0.000 description 17
- 239000000047 product Substances 0.000 description 14
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 12
- DKGAVHZHDRPRBM-UHFFFAOYSA-N Tert-Butanol Chemical compound CC(C)(C)O DKGAVHZHDRPRBM-UHFFFAOYSA-N 0.000 description 12
- 238000004128 high performance liquid chromatography Methods 0.000 description 11
- 239000007787 solid Substances 0.000 description 11
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 11
- 239000000203 mixture Substances 0.000 description 10
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 9
- 239000000243 solution Substances 0.000 description 8
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 7
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 6
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 6
- 238000000746 purification Methods 0.000 description 5
- 239000000741 silica gel Substances 0.000 description 5
- 229910002027 silica gel Inorganic materials 0.000 description 5
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 4
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical group [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 4
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 4
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 4
- 239000012074 organic phase Substances 0.000 description 4
- 235000006408 oxalic acid Nutrition 0.000 description 4
- IOLCXVTUBQKXJR-UHFFFAOYSA-M potassium bromide Chemical compound [K+].[Br-] IOLCXVTUBQKXJR-UHFFFAOYSA-M 0.000 description 4
- 239000000523 sample Substances 0.000 description 4
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 3
- WMFOQBRAJBCJND-UHFFFAOYSA-M Lithium hydroxide Chemical compound [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 description 3
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 3
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 3
- 229910052782 aluminium Inorganic materials 0.000 description 3
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 3
- 238000004458 analytical method Methods 0.000 description 3
- 229910052799 carbon Inorganic materials 0.000 description 3
- 239000003054 catalyst Substances 0.000 description 3
- 239000003795 chemical substances by application Substances 0.000 description 3
- 238000004587 chromatography analysis Methods 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- IJKVHSBPTUYDLN-UHFFFAOYSA-N dihydroxy(oxo)silane Chemical compound O[Si](O)=O IJKVHSBPTUYDLN-UHFFFAOYSA-N 0.000 description 3
- 238000003818 flash chromatography Methods 0.000 description 3
- 239000013067 intermediate product Substances 0.000 description 3
- 229910052751 metal Inorganic materials 0.000 description 3
- 239000002184 metal Substances 0.000 description 3
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 3
- 229910052757 nitrogen Inorganic materials 0.000 description 3
- 235000015320 potassium carbonate Nutrition 0.000 description 3
- 229920006395 saturated elastomer Polymers 0.000 description 3
- 235000011121 sodium hydroxide Nutrition 0.000 description 3
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 2
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 2
- 229910052684 Cerium Inorganic materials 0.000 description 2
- 229940124766 Cyp17 inhibitor Drugs 0.000 description 2
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 2
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 2
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 2
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- 150000007513 acids Chemical class 0.000 description 2
- 239000008186 active pharmaceutical agent Substances 0.000 description 2
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 2
- 230000002051 biphasic effect Effects 0.000 description 2
- GWXLDORMOJMVQZ-UHFFFAOYSA-N cerium Chemical compound [Ce] GWXLDORMOJMVQZ-UHFFFAOYSA-N 0.000 description 2
- 239000003153 chemical reaction reagent Substances 0.000 description 2
- XBDQKXXYIPTUBI-UHFFFAOYSA-N dimethylselenoniopropionate Natural products CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 2
- JVTAAEKCZFNVCJ-UHFFFAOYSA-N lactic acid Chemical compound CC(O)C(O)=O JVTAAEKCZFNVCJ-UHFFFAOYSA-N 0.000 description 2
- 150000007522 mineralic acids Chemical class 0.000 description 2
- 150000007524 organic acids Chemical class 0.000 description 2
- 239000013110 organic ligand Substances 0.000 description 2
- 230000003647 oxidation Effects 0.000 description 2
- 238000007254 oxidation reaction Methods 0.000 description 2
- 239000012071 phase Substances 0.000 description 2
- 235000011181 potassium carbonates Nutrition 0.000 description 2
- 238000000926 separation method Methods 0.000 description 2
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 2
- 235000017557 sodium bicarbonate Nutrition 0.000 description 2
- 239000011780 sodium chloride Substances 0.000 description 2
- 239000007858 starting material Substances 0.000 description 2
- CZDYPVPMEAXLPK-UHFFFAOYSA-N tetramethylsilane Chemical compound C[Si](C)(C)C CZDYPVPMEAXLPK-UHFFFAOYSA-N 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- 238000005406 washing Methods 0.000 description 2
- CJFMJEUEUPHGHU-UHFFFAOYSA-N (5-methoxy-1h-indol-2-yl)-[4-(2-methylbenzimidazol-1-yl)piperidin-1-yl]methanone Chemical compound CC1=NC2=CC=CC=C2N1C(CC1)CCN1C(=O)C1=CC2=CC(OC)=CC=C2N1 CJFMJEUEUPHGHU-UHFFFAOYSA-N 0.000 description 1
- SJJCQDRGABAVBB-UHFFFAOYSA-N 1-hydroxy-2-naphthoic acid Chemical compound C1=CC=CC2=C(O)C(C(=O)O)=CC=C21 SJJCQDRGABAVBB-UHFFFAOYSA-N 0.000 description 1
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 1
- 239000005711 Benzoic acid Substances 0.000 description 1
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 description 1
- XDTMQSROBMDMFD-UHFFFAOYSA-N Cyclohexane Chemical compound C1CCCCC1 XDTMQSROBMDMFD-UHFFFAOYSA-N 0.000 description 1
- 102000018832 Cytochromes Human genes 0.000 description 1
- 108010052832 Cytochromes Proteins 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- 238000001157 Fourier transform infrared spectrum Methods 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- CTQNGGLPUBDAKN-UHFFFAOYSA-N O-Xylene Chemical compound CC1=CC=CC=C1C CTQNGGLPUBDAKN-UHFFFAOYSA-N 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-N Succinic acid Natural products OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- 238000005874 Vilsmeier-Haack formylation reaction Methods 0.000 description 1
- 229960000853 abiraterone Drugs 0.000 description 1
- GZOSMCIZMLWJML-VJLLXTKPSA-N abiraterone Chemical compound C([C@H]1[C@H]2[C@@H]([C@]3(CC[C@H](O)CC3=CC2)C)CC[C@@]11C)C=C1C1=CC=CN=C1 GZOSMCIZMLWJML-VJLLXTKPSA-N 0.000 description 1
- 229960004103 abiraterone acetate Drugs 0.000 description 1
- UVIQSJCZCSLXRZ-UBUQANBQSA-N abiraterone acetate Chemical compound C([C@@H]1[C@]2(C)CC[C@@H]3[C@@]4(C)CC[C@@H](CC4=CC[C@H]31)OC(=O)C)C=C2C1=CC=CN=C1 UVIQSJCZCSLXRZ-UBUQANBQSA-N 0.000 description 1
- 239000004480 active ingredient Substances 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- 230000002280 anti-androgenic effect Effects 0.000 description 1
- 230000000259 anti-tumor effect Effects 0.000 description 1
- 239000000051 antiandrogen Substances 0.000 description 1
- 229940030495 antiandrogen sex hormone and modulator of the genital system Drugs 0.000 description 1
- 239000008346 aqueous phase Substances 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 1
- 235000010233 benzoic acid Nutrition 0.000 description 1
- 230000004071 biological effect Effects 0.000 description 1
- KDYFGRWQOYBRFD-NUQCWPJISA-N butanedioic acid Chemical compound O[14C](=O)CC[14C](O)=O KDYFGRWQOYBRFD-NUQCWPJISA-N 0.000 description 1
- 239000002775 capsule Substances 0.000 description 1
- 230000015556 catabolic process Effects 0.000 description 1
- VZDYWEUILIUIDF-UHFFFAOYSA-J cerium(4+);disulfate Chemical compound [Ce+4].[O-]S([O-])(=O)=O.[O-]S([O-])(=O)=O VZDYWEUILIUIDF-UHFFFAOYSA-J 0.000 description 1
- 229910000355 cerium(IV) sulfate Inorganic materials 0.000 description 1
- 239000007795 chemical reaction product Substances 0.000 description 1
- 238000011097 chromatography purification Methods 0.000 description 1
- 235000015165 citric acid Nutrition 0.000 description 1
- 238000001816 cooling Methods 0.000 description 1
- 229910052802 copper Inorganic materials 0.000 description 1
- 239000010949 copper Substances 0.000 description 1
- GBRBMTNGQBKBQE-UHFFFAOYSA-L copper;diiodide Chemical compound I[Cu]I GBRBMTNGQBKBQE-UHFFFAOYSA-L 0.000 description 1
- 239000012043 crude product Substances 0.000 description 1
- 238000006731 degradation reaction Methods 0.000 description 1
- 229910003460 diamond Inorganic materials 0.000 description 1
- 239000010432 diamond Substances 0.000 description 1
- 238000004821 distillation Methods 0.000 description 1
- 238000000921 elemental analysis Methods 0.000 description 1
- 230000008030 elimination Effects 0.000 description 1
- 238000003379 elimination reaction Methods 0.000 description 1
- 239000003480 eluent Substances 0.000 description 1
- 238000000105 evaporative light scattering detection Methods 0.000 description 1
- 239000012467 final product Substances 0.000 description 1
- 239000001530 fumaric acid Substances 0.000 description 1
- 235000011087 fumaric acid Nutrition 0.000 description 1
- 239000011521 glass Substances 0.000 description 1
- 239000001257 hydrogen Substances 0.000 description 1
- 229910052739 hydrogen Inorganic materials 0.000 description 1
- 125000004435 hydrogen atom Chemical class [H]* 0.000 description 1
- 238000000338 in vitro Methods 0.000 description 1
- 239000003112 inhibitor Substances 0.000 description 1
- JMMWKPVZQRWMSS-UHFFFAOYSA-N isopropanol acetate Natural products CC(C)OC(C)=O JMMWKPVZQRWMSS-UHFFFAOYSA-N 0.000 description 1
- 229940011051 isopropyl acetate Drugs 0.000 description 1
- GWYFCOCPABKNJV-UHFFFAOYSA-N isovaleric acid Chemical compound CC(C)CC(O)=O GWYFCOCPABKNJV-UHFFFAOYSA-N 0.000 description 1
- 239000004310 lactic acid Substances 0.000 description 1
- 235000014655 lactic acid Nutrition 0.000 description 1
- 230000014759 maintenance of location Effects 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- 239000012528 membrane Substances 0.000 description 1
- 150000002739 metals Chemical class 0.000 description 1
- 230000003472 neutralizing effect Effects 0.000 description 1
- 238000011275 oncology therapy Methods 0.000 description 1
- 239000001301 oxygen Substances 0.000 description 1
- 229910052760 oxygen Inorganic materials 0.000 description 1
- 239000008194 pharmaceutical composition Substances 0.000 description 1
- DHRLEVQXOMLTIM-UHFFFAOYSA-N phosphoric acid;trioxomolybdenum Chemical compound O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.OP(O)(O)=O DHRLEVQXOMLTIM-UHFFFAOYSA-N 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- 229960003975 potassium Drugs 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- 239000011736 potassium bicarbonate Substances 0.000 description 1
- 235000015497 potassium bicarbonate Nutrition 0.000 description 1
- 229910000028 potassium bicarbonate Inorganic materials 0.000 description 1
- TYJJADVDDVDEDZ-UHFFFAOYSA-M potassium hydrogencarbonate Chemical compound [K+].OC([O-])=O TYJJADVDDVDEDZ-UHFFFAOYSA-M 0.000 description 1
- 229940086066 potassium hydrogencarbonate Drugs 0.000 description 1
- 230000003389 potentiating effect Effects 0.000 description 1
- 239000000843 powder Substances 0.000 description 1
- 235000019260 propionic acid Nutrition 0.000 description 1
- IUVKMZGDUIUOCP-BTNSXGMBSA-N quinbolone Chemical compound O([C@H]1CC[C@H]2[C@H]3[C@@H]([C@]4(C=CC(=O)C=C4CC3)C)CC[C@@]21C)C1=CCCC1 IUVKMZGDUIUOCP-BTNSXGMBSA-N 0.000 description 1
- LISFMEBWQUVKPJ-UHFFFAOYSA-N quinolin-2-ol Chemical compound C1=CC=C2NC(=O)C=CC2=C1 LISFMEBWQUVKPJ-UHFFFAOYSA-N 0.000 description 1
- IQQDNMHUOLMLNJ-UHFFFAOYSA-N quinolin-3-ol Chemical compound C1=CC=CC2=CC(O)=CN=C21 IQQDNMHUOLMLNJ-UHFFFAOYSA-N 0.000 description 1
- PMZDQRJGMBOQBF-UHFFFAOYSA-N quinolin-4-ol Chemical compound C1=CC=C2C(O)=CC=NC2=C1 PMZDQRJGMBOQBF-UHFFFAOYSA-N 0.000 description 1
- OVYWMEWYEJLIER-UHFFFAOYSA-N quinolin-6-ol Chemical compound N1=CC=CC2=CC(O)=CC=C21 OVYWMEWYEJLIER-UHFFFAOYSA-N 0.000 description 1
- 230000035484 reaction time Effects 0.000 description 1
- 239000013074 reference sample Substances 0.000 description 1
- 150000003839 salts Chemical class 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 239000011734 sodium Substances 0.000 description 1
- 238000005063 solubilization Methods 0.000 description 1
- 230000007928 solubilization Effects 0.000 description 1
- 238000001228 spectrum Methods 0.000 description 1
- 230000003637 steroidlike Effects 0.000 description 1
- 150000003431 steroids Chemical class 0.000 description 1
- 239000000126 substance Substances 0.000 description 1
- 239000000725 suspension Substances 0.000 description 1
- 239000011975 tartaric acid Substances 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- 239000008096 xylene Substances 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07J—STEROIDS
- C07J1/00—Normal steroids containing carbon, hydrogen, halogen or oxygen, not substituted in position 17 beta by a carbon atom, e.g. estrane, androstane
- C07J1/0003—Androstane derivatives
- C07J1/0011—Androstane derivatives substituted in position 17 by a keto group
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07J—STEROIDS
- C07J13/00—Normal steroids containing carbon, hydrogen, halogen or oxygen having a carbon-to-carbon double bond from or to position 17
- C07J13/005—Normal steroids containing carbon, hydrogen, halogen or oxygen having a carbon-to-carbon double bond from or to position 17 with double bond in position 16 (17)
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07J—STEROIDS
- C07J43/00—Normal steroids having a nitrogen-containing hetero ring spiro-condensed or not condensed with the cyclopenta(a)hydrophenanthrene skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07J—STEROIDS
- C07J43/00—Normal steroids having a nitrogen-containing hetero ring spiro-condensed or not condensed with the cyclopenta(a)hydrophenanthrene skeleton
- C07J43/003—Normal steroids having a nitrogen-containing hetero ring spiro-condensed or not condensed with the cyclopenta(a)hydrophenanthrene skeleton not condensed
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07J—STEROIDS
- C07J75/00—Processes for the preparation of steroids in general
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
Abstract
Description




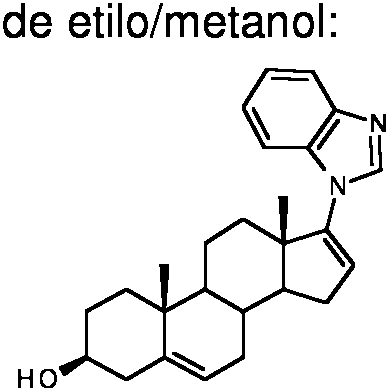






Claims (16)


Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| IT102016000083406A IT201600083406A1 (it) | 2016-08-08 | 2016-08-08 | PROCESSO PER LA PREPARAZIONE DI 3β-IDROSSI-17-(1H-BENZIMIDAZOL-1-IL)ANDROSTA-5,16-DIENE |
| IT102016000121375A IT201600121375A1 (it) | 2016-11-30 | 2016-11-30 | PROCESSO PER LA PREPARAZIONE DI 3ß-IDROSSI-17-(1H-BENZIMIDAZOL-1-IL)ANDROSTA-5,16-DIENE |
| PCT/EP2017/070124 WO2018029223A1 (en) | 2016-08-08 | 2017-08-08 | PROCESS FOR THE PREPARATION OF 3ß-HYDROXY-17-(1H-BENZIMIDAZOL-1-YL)ANDROSTA-5,16-DIENE |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| ES2700901A2 true ES2700901A2 (es) | 2019-02-19 |
| ES2700901R1 ES2700901R1 (es) | 2019-03-04 |
| ES2700901B2 ES2700901B2 (es) | 2019-08-06 |
Family
ID=59745873
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES201990014A Active ES2700901B2 (es) | 2016-08-08 | 2017-08-08 | Proceso para la preparación de 3ß-hidroxi-17-(1h-benzimidazol-1-il)androsta-5,16-dieno. |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US11390645B2 (es) |
| CA (1) | CA3063339C (es) |
| CH (1) | CH714173B1 (es) |
| ES (1) | ES2700901B2 (es) |
| RU (1) | RU2749134C1 (es) |
| WO (1) | WO2018029223A1 (es) |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP4608841A1 (en) * | 2022-10-25 | 2025-09-03 | University of Maryland, Baltimore | Salts of galeterone and salts of next generation galeterone analogs, and uses thereof |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| AU2006218711B2 (en) * | 2005-03-02 | 2010-11-11 | University Of Maryland, Baltimore | Novel C-17-heteroaryl steroidal CYP17 inhibitors/antiandrogens: synthesis, in vitro biological activities, pharmacokinetics and antitumor activity |
| CN105732759A (zh) * | 2015-01-29 | 2016-07-06 | 苏州晶云药物科技有限公司 | (3β)-17-(1H-苯并咪唑-1-基)雄甾-5,16-二烯-3-醇的盐及其制备方法 |
| CN112851741B (zh) * | 2016-02-19 | 2022-10-25 | 深圳市塔吉瑞生物医药有限公司 | 一种取代的甾体类化合物及其应用 |
-
2017
- 2017-08-08 CH CH00147/19A patent/CH714173B1/it unknown
- 2017-08-08 CA CA3063339A patent/CA3063339C/en active Active
- 2017-08-08 RU RU2019143855A patent/RU2749134C1/ru active
- 2017-08-08 WO PCT/EP2017/070124 patent/WO2018029223A1/en not_active Ceased
- 2017-08-08 US US16/323,317 patent/US11390645B2/en active Active
- 2017-08-08 ES ES201990014A patent/ES2700901B2/es active Active
Also Published As
| Publication number | Publication date |
|---|---|
| CH714173B1 (it) | 2021-04-15 |
| US20210284681A1 (en) | 2021-09-16 |
| RU2749134C1 (ru) | 2021-06-04 |
| CA3063339A1 (en) | 2018-02-15 |
| US11390645B2 (en) | 2022-07-19 |
| CA3063339C (en) | 2024-05-28 |
| ES2700901R1 (es) | 2019-03-04 |
| ES2700901B2 (es) | 2019-08-06 |
| WO2018029223A1 (en) | 2018-02-15 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN1315828C (zh) | 通过1h-咪唑并[4,5-c]喹啉-4-邻苯二甲酰亚胺类中间体制备1h-咪唑并[4,5-c]喹啉-4-胺类化合物 | |
| AU780989B2 (en) | 3alpha-hydroxy-3beta methoxymethyl-21-heterocycle substituted steroids with anesthetic activity | |
| JPS6212795A (ja) | 新規なプソラレン誘導体 | |
| JP2001511807A (ja) | 高親油性カンプトテシン誘導体 | |
| CN112961120B (zh) | 一种萘基脲类化合物、其制备方法及应用 | |
| WO2023160727A1 (zh) | 吲唑类化合物用于治疗银屑病的用途 | |
| CN102746306B (zh) | 别嘌醇类衍生物及其制备方法和用途 | |
| ES2700901A2 (es) | Proceso para la preparación de 3ß-hidroxi-17-(1h-benzimidazol-1-il)androsta-5,16-dieno. | |
| WO2018041260A1 (zh) | 一类溴结构域识别蛋白抑制剂及其制备方法和用途 | |
| Keyser et al. | Linear benzoguanine. Synthesis by two independent methods | |
| KR102606167B1 (ko) | 불소 함유 치환 벤조티오펜 화합물, 그의 약학적 조성물 및 응용 | |
| ES2309567T3 (es) | Derivados de imidazopiridinas como inhibidores de no-sintasa inducible. | |
| ES2741505T3 (es) | Procedimiento para la preparación de acetato de abiraterona y sus productos intermedios | |
| JPS5839684A (ja) | 10−置換カンプテシン誘導体の製造法 | |
| JP7638031B2 (ja) | ピロロトリアジン系化合物の塩形、その結晶形及びその製造方法 | |
| CN103288842B (zh) | 氟取代e环喜树碱类似物及其作为药物的用途 | |
| CN108947916A (zh) | 一种Perimidine醌类衍生物及其制备方法和应用 | |
| CN115785189B (zh) | 一种5α,8α-过氧化甾醇-17-苯基噻唑衍生物及其合成方法和应用 | |
| CN112300235B (zh) | 一种苯并咪唑衍生物bi321及其制备方法和应用 | |
| CN102603759B (zh) | 喜树碱e环类似物及其作为药物的用途 | |
| RU2426737C1 (ru) | 4-ГЕТЕРО-16α, 17α-ЦИКЛОГЕКСАНОПРЕГНАНЫ | |
| WO2019020068A1 (zh) | 甾体类衍生物fxr激动剂的结晶或无定型物、其制备方法和用途 | |
| JPH0616553A (ja) | 血行促進のために使用する医薬 | |
| CN104497086B (zh) | 雄甾-3β,5α,6β,19-四醇及其制备方法与应用 | |
| CN106928303B (zh) | 一种螺环化合物及其制备方法和应用 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| BA2A | Patent application published |
Ref document number: 2700901 Country of ref document: ES Kind code of ref document: A2 Effective date: 20190219 |
|
| EC2A | Search report published |
Ref document number: 2700901 Country of ref document: ES Kind code of ref document: R1 Effective date: 20190225 |
|
| FG2A | Definitive protection |
Ref document number: 2700901 Country of ref document: ES Kind code of ref document: B2 Effective date: 20190806 |