ES2702474T3 - Ciclohexilmetanosulfonamidas - Google Patents
Ciclohexilmetanosulfonamidas Download PDFInfo
- Publication number
- ES2702474T3 ES2702474T3 ES15713431T ES15713431T ES2702474T3 ES 2702474 T3 ES2702474 T3 ES 2702474T3 ES 15713431 T ES15713431 T ES 15713431T ES 15713431 T ES15713431 T ES 15713431T ES 2702474 T3 ES2702474 T3 ES 2702474T3
- Authority
- ES
- Spain
- Prior art keywords
- trans
- alkyl
- compound
- formula
- reaction mixture
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/12—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains three hetero rings
- C07D471/14—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/02—Nasal agents, e.g. decongestants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/06—Antiasthmatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/04—Antipruritics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/06—Antipsoriatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
- A61P27/14—Decongestants or antiallergics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/08—Antiallergic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D239/00—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings
- C07D239/02—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings
- C07D239/24—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members
- C07D239/28—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, directly attached to ring carbon atoms
- C07D239/32—One oxygen, sulfur or nitrogen atom
- C07D239/42—One nitrogen atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D239/00—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings
- C07D239/02—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings
- C07D239/24—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members
- C07D239/28—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, directly attached to ring carbon atoms
- C07D239/46—Two or more oxygen, sulphur or nitrogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D239/00—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings
- C07D239/02—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings
- C07D239/24—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members
- C07D239/28—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, directly attached to ring carbon atoms
- C07D239/46—Two or more oxygen, sulphur or nitrogen atoms
- C07D239/48—Two nitrogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D473/00—Heterocyclic compounds containing purine ring systems
- C07D473/26—Heterocyclic compounds containing purine ring systems with an oxygen, sulphur, or nitrogen atom directly attached in position 2 or 6, but not in both
- C07D473/32—Nitrogen atom
- C07D473/34—Nitrogen atom attached in position 6, e.g. adenine
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D495/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having sulfur atoms as the only ring hetero atoms
- C07D495/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having sulfur atoms as the only ring hetero atoms in which the condensed system contains two hetero rings
- C07D495/04—Ortho-condensed systems
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Veterinary Medicine (AREA)
- Pharmacology & Pharmacy (AREA)
- Public Health (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Immunology (AREA)
- Pulmonology (AREA)
- Diabetes (AREA)
- Dermatology (AREA)
- Neurosurgery (AREA)
- Neurology (AREA)
- Biomedical Technology (AREA)
- Rheumatology (AREA)
- Endocrinology (AREA)
- Hematology (AREA)
- Hospice & Palliative Care (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Pain & Pain Management (AREA)
- Transplantation (AREA)
- Oncology (AREA)
- Obesity (AREA)
- Psychiatry (AREA)
- Ophthalmology & Optometry (AREA)
- Physical Education & Sports Medicine (AREA)
- Emergency Medicine (AREA)
- Otolaryngology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Oxygen Or Sulfur (AREA)
Abstract
Un compuesto de fórmula I**Fórmula** en la que R es metilo; y en la que A consiste en la fórmula 2a**Fórmula** en la que Y se selecciona de entre el grupo que consiste en NH, N(alquilo C1-C2) u O, y R2 se selecciona de entre el grupo que consiste en ciano (CN), nitro (NO2), o NR'R", y R' y R" se seleccionan independientemente de entre el grupo que consiste en H o alquilo C1-C4; o en la que Y y R2 forman juntos**Fórmula** en la que R0 se selecciona de entre el grupo que consiste en H, alquilo C1-C4, hidroxi-alquilo C1-C4, alcoxi C1-C4- alquilo C1-C4 o NH2; o una sal del mismo aceptable desde el punto de vista veterinario.
Description
DESCRIPCIÓN
Ciclohexilmetanosulfonamidas
La presente invención se refiere a novedosas ciclohexilmetansulfonamidas sustituidas con heterociclilo que son inhibidoras de la quinasa Janus, conocidas también como inhibidores de JAK, y a su uso en el tratamiento de reacciones alérgicas que incluyen dermatitis alérgica, eczema, dermatitis atópica, prurito y otras afecciones pruríticas y también enfermedades inflamatorias.
Oclacitinib, una pirrolopirimidinaminociclohexilmetanosulfonamida, es un inhibidor de JAK, que está aprobado para el control del prurito asociado con la dermatitis alérgica y el control de la dermatitis atópica en perros. Sin embargo, la búsqueda de nuevas moléculas inhibidoras de JAK, más potentes, continúa. Sorprendentemente, se han encontrado nuevos inhibidores de JAK específicos que proporcionan una actividad mejorada en lo concerniente a enfermedades de la piel, en particular dermatitis atópica y prurito.
Por lo tanto, la presente invención, en un aspecto, se refiere a un compuesto de fórmula I

en la que R es metilo; y en la que A consiste en la fórmula 2a

en la que Y se selecciona de entre el grupo que consiste en NH, N (alquilo C1-C2) u O, y R2 se selecciona de entre el grupo que consiste en ciano (CN), nitro (NO2), o NR'R", y R' y R" se seleccionan independientemente de entre el grupo que consiste en H o alquilo C1-C4; o en la que Y y R2 juntos forman

en la que R0 se selecciona de entre el grupo que consiste en H, alquilo C1-C4 , hidroxi-alquilo C1-C4 , alcoxi C1-C4-alquilo C1-C4 o NH2 ; o una sal del mismo aceptable desde el punto de vista veterinario.
R0 como hidroxi-alquilo C1-C4 es preferentemente hidroximetilo o hidroxietilo, en particular hidroximetilo. R0 como alcoxi C1-C4-alquilo C1-C4 es preferentemente metoximetilo o etoximetilo, en particular metoximetilo. La variable R0 es preferentemente H, metilo hidroximetilo, metoximetilo o NH2 , más preferentemente H, metilo o NH2 , en particular, H. R' y R" son, cada uno independientemente del otro, preferentemente H, metilo o etilo. R2 es preferentemente ciano o nitro.
Una realización de la invención se refiere a un radical A de fórmula (2a), en la que R2 e Y tiene los significados definidos. Un radical A preferente es de fórmula (2a), en la que R2 es ciano o nitro e Y es NH, N(CH3) u O, preferentemente NH o N(CH3), en particular N(CH3).
Todavía un radical A adicional preferente es un radical de fórmula

Los compuestos de la presente invención pueden existir como uno o más estereoisómeros. Los diversos estereoisómeros incluyen enantiómeros, diastereómeros, atropisómeros e isómeros geométricos.
En el caso en el que los compuestos de la fórmula (I) tienen un átomo de carbono quiral, pueden tener una configuración (R) o (S). La presente invención abarca los compuestos de fórmula (I) con configuración tanto (S) como (R) en los átomos de carbono quirales particulares, lo que significa que la presente invención cubre los compuestos de la fórmula general (I) en los que los átomos de carbono en cuestión tienen, cada uno independientemente, una configuración (R); o tienen una configuración (S).
Si hay presentes una pluralidad de centros quirales en los compuestos de fórmula (I), es posible cualquier combinación deseada de las configuraciones de los centros quirales, lo que significa que (1) un centro quiral puede tener una configuración (R) y el otro centro quiral una configuración (S); (2) un centro quiral puede tener una configuración (R) y el otro centro quiral una configuración (R); y (3) un centro quiral puede tener una configuración (S) y el otro centro quiral una configuración (S).
Un experto en la técnica apreciará que un estereoisómero puede ser más activo y/o puede exhibir efectos beneficiosos cuando se enriquece con relación al otro o a los otros estereoisómeros o cuando se separa del otro o de los otros estereoisómeros. Además, el experto en la materia sabe cómo separar, enriquecer y/o preparar de manera selectiva dichos estereoisómeros. Los compuestos de la invención pueden estar presentes como una mezcla de estereoisómeros, estereoisómeros individuales o como una forma ópticamente activa.
Un experto en la técnica apreciará que no todos los anillos heterocíclicos que contienen nitrógeno pueden formar N-óxidos, ya que el nitrógeno requiere un par solitario disponible para la oxidación al óxido; un experto en la técnica reconocerá aquellos anillos heterocíclicos que contienen nitrógeno que pueden formar N-óxidos. Un experto en la técnica reconocerá también que las aminas terciarias pueden formar N-óxidos. Un experto en la técnica conoce muy bien los procedimientos sintéticos para la preparación de N-óxidos de anillos heterocíclicos y aminas terciarias, incluyendo la oxidación de anillos heterocíclicos y aminas terciarias con ácidos peroxi, tales como ácido peracético y m-cloroperbenzoico (MCPBA), peróxido de hidrógeno, hidroperóxidos de alquilo, tales como hidroperóxido de t-butilo, perborato de sodio y dioxiranos tales como dimetil dioxirano. Estos procedimientos para la preparación de N-óxidos se han descrito y revisado ampliamente en la literatura. La fabricación de S-óxidos adecuados puede realizarse de manera análoga usando, por ejemplo, el mismo tipo de oxidantes que los indicados anteriormente para los N-óxidos.
Un experto en la materia reconoce que, debido al medio ambiente y en condiciones fisiológicas, las sales de los compuestos químicos están en equilibrio con sus formas no salinas correspondientes, las sales comparten la utilidad biológica de las formas no salinas. De esta manera, son útiles una amplia diversidad de sales de los compuestos de fórmula (I) (es decir, son adecuadas para uso veterinario). Las sales de los compuestos de fórmula (I) incluyen sales de adición de ácido con ácidos inorgánicos u orgánicos tales como ácidos bromhídrico, clorhídrico, nítrico, fosfórico, sulfúrico, acético, butírico, fumárico, láctico, maleico, malónico, oxálico, propiónico, salicílico, tartárico, 4-toluensulfónico o valérico. Cuando un compuesto de fórmula (I) contiene un resto ácido, tal como un ácido carboxílico o fenol, las sales incluyen también aquellas formadas con bases orgánicas o inorgánicas, tales como piridina, trietilamina o amoniaco, o amidas, hidruros, hidróxidos o carbonatos de sodio, potasio, litio, calcio, magnesio o bario. Por consiguiente, la presente invención comprende compuestos seleccionados de fórmula (I), N-óxidos y sales de los mismos aceptables desde el punto de vista veterinario. Los compuestos de la presente invención pueden formar también sales internas.
Los compuestos de fórmula (I) pueden prepararse, por ejemplo, haciendo reaccionar un compuesto de fórmula A'-Hal (3),
con un compuesto de fórmula

en la que Y y R son tal como se han definido anteriormente, Hal es halógeno, por ejemplo, cloro, y A' es un radical de fórmula

en la que R2 es tal como se ha definido anteriormente. La reacción de sustitución nucleófila puede realizarse, por ejemplo, según se describe en los libros de texto de química orgánica. Por ejemplo, los compuestos de fórmula (3) y (4) se hacen reaccionar en un disolvente adecuado o mezcla de disolventes en presencia de una base. La elección del disolvente y de la base depende en gran medida de la naturaleza específica de los compuestos de fórmulas (3) y (4). La reacción puede tener lugar a temperatura ambiente o a temperatura elevada, por ejemplo, por encima de 100°C. En el caso de un compuesto de fórmula (3) que comprende un radical A' con un grupo NH, puede ser aconsejable proteger dicho grupo amina antes de realizar la reacción con el compuesto de fórmula (4). La protección del grupo amina y la desprotección posterior pueden realizarse de una manera conocida en sí misma.
De manera alternativa, los compuestos de fórmula (3) y (4) pueden acoplarse mediante aminación Buchwald-Hartwig catalizada por Pd, tal como se describe en los libros de texto de química orgánica.
Los compuestos de fórmula (3) son conocidos en sí mismos o pueden prepararse según procedimientos conocidos en sí mismos.
Los compuestos de fórmula (4) son igualmente conocidos, o pueden prepararse según procedimientos conocidos en sí mismos. Por ejemplo, el grupo HY de un compuesto de fórmula

en la que Y es tal como como se ha definido anteriormente, se protege primero de una manera conocida en sí misma, antes de reducir el grupo carboxilo para producir el correspondiente alcohol de fórmula
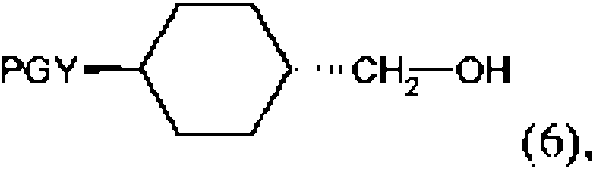
en la que PG es un grupo protector e Y es tal como se ha definido anteriormente. El alcohol de fórmula (6) se convierte a continuación en el compuesto correspondiente de fórmula (7)

en la que LG es un grupo saliente, por ejemplo, cloro, bromo, mesilato o tosilato, que a su vez se convierte en el correspondiente ácido sulfónico de fórmula (8)

usando sulfito de sodio o tioacetato de sodio, seguido de oxidación con peróxido de hidrógeno.
El compuesto de fórmula (8), después de haber sido convertido en el haluro metanosulfónico correspondiente, por ejemplo, mediante reacción con cloruro de tionilo, se hace reaccionar con una amina de fórmula
H2N-R (9),
en la que R es tal como como se ha definido anteriormente, para producir un compuesto de fórmula (4) en forma protegida, que finalmente se desprotege. Las etapas descritas anteriormente desde el compuesto de fórmula (5) al compuesto de fórmula (4) son reacciones bien conocidas que pueden realizarse tal como se describe en los libros de texto de química orgánica. Los ejemplos de trabajo ilustran adicionalmente las reacciones.
Como alternativa, los compuestos de fórmula (1) pueden sintetizarse de manera similar al Esquema II de la página 14 del documento WO2010/020905.
Además, un compuesto de fórmula (1), en la que A es un radical de fórmula

en la que R0 es tal como como se ha definido anteriormente, puede obtenerse preparando en primer lugar un compuesto de fórmula

en la que PG es un grupo protector, mediante un procedimiento como se ha descrito anteriormente, reduciendo el grupo nitro del compuesto de fórmula (1a) de una manera conocida en sí misma, por ejemplo, catalíticamente con H2/níquel Raney, y haciendo reaccionar la diamina resultante con un ácido haluro R0-C(O)Hal o con un trialquil orto-éster (AlkO)3-C-R0 , en la que Hal es Br o Cl, Alk es, por ejemplo, etilo, y R0 es tal como se ha descrito anteriormente, con el fin de producir un compuesto de fórmula

después de la desprotección de la amina, en donde R y R0 son como se describe anteriormente.
Los compuestos de la presente invención son inhibidores de Janus kinasa (JAK-i) con eficacia, por ejemplo, contra Janus kinasa-1 (JAK-I), Janus kinasa-2 (JAK-2), Janus kinasa-3 (JAK-3) y tirosina quinasa-2 quinasa (TYK-2), en particular JAK-I o JAK-3. Por consiguiente, son útiles como agentes terapéuticos para trasplantes de órganos, lupus, esclerosis múltiple, artritis reumatoide, psoriasis, diabetes tipo I y complicaciones de la diabetes, cáncer, asma, dermatitis atópica, trastornos autoinmunes de la tiroides, colitis ulcerosa, enfermedades inflamatorias del
intestino, enfermedad de Crohn, enfermedad de Alzheimer, leucemia, osteoartritis, control del prurito, enfermedad respiratoria crónica, queratoconjuntivitis y otras indicaciones en las que sería deseable una inmunosupresión/inmunomodulación.
En particular, ha resultado que los compuestos de la presente invención son agentes seguros y eficaces para controlar enfermedades de la piel, afecciones o trastornos que incluyen dermatitis atópica, eccema, psoriasis, esclerodermia, prurito y otras afecciones pruríticas, reacciones alérgicas que incluyen dermatitis alérgica en mamíferos incluyendo caballos, enfermedades alérgicas tales como hipersensibilidad a las mordeduras, eccema de verano y “sweet itch” (picazón o eccema de verano) en caballos.
Debido a que los compuestos de la presente invención son inhibidores de JAK con eficacia contra JAK-I y JAK-3, proporcionan resolución para el prurito y la inflamación crónicos que persistirían en la dermatitis atópica o disminuirían lentamente después de la eliminación del alérgeno o del agente causal, tal como pulgas en la dermatitis alérgica a las pulgas.
Los compuestos de la presente invención pueden administrarse en una forma farmacéuticamente aceptable, bien solos o bien en combinación con uno o más agentes adicionales que modulan el sistema inmunitario de un mamífero o con agentes antiinflamatorios. Los ejemplos son ciclosporina A aspirina, acetaminofeno, ibuprofeno, naproxeno, piroxicam y esteroides antiinflamatorios (por ejemplo, prednisolona o dexametasona). Estos agentes pueden administrarse como parte de las mismas formas de dosificación o formas de dosificación separadas, a través de las mismas vías de administración o vías de administración diferentes, y en los mismos esquemas de administración o en esquemas de administración diferentes según la práctica farmacéutica estándar conocida por un experto en la técnica.
En una realización, la invención proporciona uno o más compuestos descritos en la presente memoria para su uso en el tratamiento o la prevención de una enfermedad, afección o trastorno asociado con JAK en un sujeto, tal como un mamífero humano o no humano. La enfermedad, afección o trastorno asociado con JAK puede estar relacionado con JAK-I, JAK-2, JAK-3 y/o TYK-2. Los sujetos adecuados que pueden ser tratados incluyen animales domésticos o salvajes, animales de compañía, tales como perros, gatos, caballos y similares; ganado incluyendo vacas y otros rumiantes, cerdos, aves de corral, conejos y similares; primates, por ejemplo, monos; y roedores, tales como ratas, ratones, jerbos, cobayas y similares. En una realización, el compuesto se administra en una forma farmacéuticamente aceptable, opcionalmente en un vehículo farmacéuticamente aceptable.
En la presente memoria, se divulga un procedimiento de inhibición de una enzima JAK, incluyendo JAK-I, JAK-2, JAK-3 y/o Tyk-2, que incluye poner en contacto la enzima JAK con una cantidad no terapéutica o una cantidad terapéuticamente eficaz de una o más de los presentes compuestos. Dichos procedimientos pueden ocurrir in vivo o in vitro. El contacto in vitro puede implicar un ensayo de selección para determinar la eficacia de los uno o más compuestos contra una enzima seleccionada en diversas cantidades o concentraciones. El contacto in vivo con una cantidad terapéuticamente eficaz de los uno o más compuestos puede implicar el tratamiento de una enfermedad, trastorno o afección descrita o la profilaxis del rechazo de un trasplante de órganos en el animal en el que se produce el contacto. También puede determinarse o medirse el efecto de los uno o más compuestos sobre la enzima JAK y/o el animal huésped. Los procedimientos para determinar la actividad de JAK se muestran en la parte de Ejemplos a continuación.
En el uso terapéutico para el tratamiento de trastornos en un mamífero (es decir, seres humanos y animales), un compuesto de la presente invención o sus composiciones farmacéuticas pueden administrarse por vía oral, parenteral, tópica, rectal, transmucosal o intestinal. Las administraciones parenterales incluyen inyecciones indirectas para generar un efecto sistémico o inyecciones directas en el área afectada. Las administraciones tópicas incluyen el tratamiento de la piel u órganos fácilmente accesibles mediante aplicación local, por ejemplo, ojos u oídos. Incluye también el suministro transdérmico para generar un efecto sistémico. La administración rectal incluye la forma de supositorios. Las vías de administración preferentes son oral y parenteral.
Las composiciones farmacéuticas de la presente invención pueden fabricarse mediante procesos bien conocidos en la técnica, por ejemplo, por medio de procedimientos convencionales de mezclado, disolución, granulación, fabricación de grageas, levigación, emulsificación, encapsulación, atrapamiento, liofilización o secado por pulverización. Las composiciones farmacéuticas para su uso según la presente invención pueden formularse de manera convencional usando uno o más vehículos farmacéuticamente aceptables que comprenden excipientes y auxiliares, que facilitan el procesamiento del compuesto activo en preparaciones, que pueden usarse farmacéuticamente. La formulación apropiada depende de la vía de administración elegida. Los excipientes y los vehículos farmacéuticamente aceptables son generalmente conocidos por los expertos en la técnica y, de esta manera, se incluyen en la presente invención.
Las formulaciones de la invención pueden diseñarse de manera que sean de acción corta, de liberación rápida, de acción prolongada y de liberación sostenida. De esta manera, las formulaciones farmacéuticas pueden formularse también para una liberación controlada o para una liberación lenta.
Las composiciones farmacéuticas adecuadas para su uso en la presente invención incluyen composiciones en las que los ingredientes activos están contenidos en una cantidad suficiente para conseguir el propósito deseado, es decir, el control o el tratamiento de trastornos o enfermedades. Más específicamente, una cantidad terapéuticamente eficaz significa una cantidad de compuesto eficaz para prevenir, aliviar o mejorar los síntomas/signos de la enfermedad o prolongar la supervivencia del sujeto que está siendo tratado.
La cantidad de componente activo, que es el compuesto de la presente invención, en la composición farmacéutica y la forma de dosificación unitaria del mismo, puede variarse o ajustarse ampliamente dependiendo de la forma de administración, la potencia del compuesto particular y la concentración deseada. La determinación de una cantidad terapéuticamente eficaz está dentro de la capacidad de los expertos en la técnica. En general, la cantidad de componente activo estará comprendida entre el 0,01% y el 99% en peso de la composición.
Generalmente, una cantidad de dosificación terapéuticamente eficaz del componente activo estará comprendida en el intervalo de aproximadamente 0,01 a aproximadamente 100 mg/kg de peso corporal/día, preferentemente de aproximadamente 0,1 a aproximadamente 10 mg/kg de peso corporal/día, más preferentemente de aproximadamente 0,3 a 3 mg/kg de peso corporal/día, incluso más preferentemente de aproximadamente 0,3 a 1,5 mg/kg de peso corporal/día. Debe entenderse que las dosis pueden variar dependiendo de los requisitos de cada sujeto y de la gravedad de los trastornos o enfermedades tratados. La dosis deseada puede presentarse convenientemente en una dosis única o como dosis divididas administradas a intervalos apropiados, por ejemplo, como dos, tres, cuatro o más sub-dosis por día. La propia sub-dosis puede dividirse además, por ejemplo, en una serie de administraciones discretas con poca separación; tales como múltiples inhalaciones desde un insuflador o mediante la aplicación de una pluralidad de gotas en el ojo. Además, debe entenderse que la dosis inicial administrada puede incrementarse más allá del nivel superior anterior para conseguir rápidamente la concentración plasmática deseada. Por otra parte, la dosis inicial puede ser más pequeña que la óptima y la dosis diaria puede incrementarse progresivamente durante el curso del tratamiento, dependiendo de la situación particular. Si se desea, la dosis diaria puede dividirse también en múltiples dosis para la administración, por ejemplo, de dos a cuatro veces por día.
Los Ejemplos ilustran adicionalmente la invención.
Ejemplo 1
Este ejemplo ilustra la preparación de 1-[trans-4-(imidazo[4,5-d]pirrolo[2,3-b]piridin-1(6H)-il)ciclohexil]-N-metilmetanosulfonamida (Compuesto 22 en la Tabla 3)
Etapa A: Se colocaron trans-4-aminociclohexanocarboxilato de metilo (0,19 g), acetonitrilo (3 ml) y K2CO3 (0,407 g) en un matraz de fondo redondo con agitador magnético y baño de calentamiento. Se añadió bromuro de bencilo (0,29 ml) y la mezcla de reacción se agitó vigorosamente a una temperatura de 25 a 30°C durante 3 horas. La TLC (DCM/MeOH 8:1) reveló una conversión completa del material de partida y la presencia de amina monobencilada. El precipitado inorgánico se separó mediante filtración. El filtrado se evaporó. El residuo se purificó mediante cromatografía en gel de sílice (DCM/Hexano 1:1 a DCM/MeOH 10:1) para producir trans-4-(dibencilamino)ciclohexanocarboxilato de metilo como agujas blancas (0,33 g).
Etapa B: Se disolvió trans-4-(dibencilamino)ciclohexanocarboxilato de metilo (0,22 g) en THF anhidro (2,2 ml) y se enfrió a 0°C. Se añadió hidruro de litio y aluminio (0,125 g) en porciones durante aproximadamente 15 minutos. Cuando cesó la formación de espuma, la temperatura del lote se aumentó lentamente y la reacción se aceleró (exotérmica). Después de 30 minutos, se tomaron muestras para TLC (DCM) y no se detectó material de partida. La mezcla de reacción se inactivó con agua y NaOH al 10%. Se separaron las fases. La parte acuosa se extrajo con t-butil éter de metilo. Las fases orgánicas combinadas se lavaron con salmuera, se secaron sobre sulfato de sodio anhidro y se concentraron en vacío para dar [trans-4-(dibencilamino)ciclohexil]metanol como un aceite incoloro que se solidifica tras dejarlo en reposo (0,20 g). El producto crudo obtenido se usó sin purificación adicional.
Etapa C: Se disolvieron [trans-4-(dibencilamino)ciclohexil]metanol (7,74 g) y trifenilfosfina (9,84 g) en THF anhidro (60 ml). Se disolvió tetrabromometano (12,44 g) en THF (17,5 ml) y se añadió gota a gota a la mezcla de reacción. El matraz se enfrió con agua (~10°C) debido a la exotermia. Apareció el precipitado. Después de 1 hora, se tomaron muestras para TLC (DCM 100%) que reveló la reacción completa. El disolvente se evaporó y el residuo se purificó mediante cromatografía en gel de sílice (DCM 100%) Las fracciones que contenían el producto se
combinaron y se evaporaron. El residuo sólido se tomó con hexano (30 ml) y se enfrió a entre 2 y 4°C. El precipitado se separó mediante filtración, se enjuagó con hexano frío y se secó bajo vacío para dar N,N-dibenciltrans-4-(bromometil)cidohexanamina como un sólido blanco (9,2 g).
Etapa D: Se suspendió N,N-dibencil-trans-4-(bromometil)ciclohexanamina (5,0 g) en alcohol isopropílico (10 ml). Se disolvieron Na2SO3 (2,2 g) y KI (cat.) en agua (20 ml) y se añadieron a la suspensión de N,N-dibencil-4-(bromometil)ciclohexanamina en un reactor de presión. Se selló y se calentó a 130°C con buena agitación. El progreso de la reacción se controló mediante TLC (DCM 100%). Cuando se encontró que la reacción se había completado, los disolventes se evaporaron y se secaron mediante destilación azeotrópica con tolueno para dar ácido [trans-4-(dibencilamino)ciclohexil]metanosulfónico. El producto crudo obtenido se usó sin purificación adicional.
Etapa E: Se suspendió ácido trans-4-(dibencilamino)ciclohexil]metanosulfónico crudo (4,5 g) en cloroformo (75 ml) y se enfrió en un baño de hielo. Se añadió cloruro de tionilo (19,4 ml) gota a gota. La mezcla de reacción se agitó durante 20 minutos a temperatura ambiente, a continuación, se calentó a 65°C y se agitó a 65°C durante la noche. El disolvente y el exceso de cloruro de tionilo se evaporaron para dar cloruro de [trans-4-(dibencilamino)ciclohexil]metanosulfonilo. El producto crudo obtenido se usó sin purificación adicional.
Etapa F: Se suspendió cloruro de trans-4-(dibencilamino)ciclohexil]metanosulfonilo crudo (~11,5 mmol) en THF anhidro (45 ml) y se enfrió a 0°C. Se añadió trietilamina (2,4 ml) seguido de metilamina (2M en THF, 11,5 ml). La mezcla de reacción se agitó a 0°C durante 1 hora, se calentó a temperatura ambiente y se mantuvo durante 1 hora a temperatura ambiente. El disolvente se evaporó y el residuo se tomó con EtOAc (100 ml) y se lavó con NaHCO3 (sat.). La fase acuosa se lavó de nuevo con EtOAc (50 ml). Las fases orgánicas combinadas se lavaron con agua y salmuera (50 ml cada una), se secaron sobre sulfato de magnesio y se concentraron en vacío. El material bruto se purificó mediante cromatografía en gel de sílice (DCM/MeOH 8:1). Las fracciones que contenían el producto se combinaron, se concentraron en vacío y se trituraron con DCM/hexano (1:1). El precipitado se separó mediante filtración, se lavó con DCM/hexano frío (1:1) y se secó bajo vacío para producir 1-[trans-4-(dibencilamino)ciclohexil]-N-metil-metanosulfonamida como un sólido blanco (1,67 g).
Etapa G: Se disolvió (parcialmente) 1-[trans-4-(dibencilamino)ciclohexil]-N-metil-metanosulfonamida (1,6 g) en MeOH (16 ml). Se añadieron formiato de amonio (1,04 g) y Pd/C húmedo al 10% (0,3 g). La mezcla de reacción se calentó a reflujo con buena agitación. Se encontró una conversión muy lenta (TLC - DCM/MeOH 8:1). El disolvente se rellenó con THF (10 ml). Se añadió otra porción de formiato de amonio (1,04 g) seguido de Pd(OH)2/C al 10% (0,3 g). La mezcla de reacción se volvió a muestrear para TLC (DCM/MeOH 8:1) después de 1 hora, se observó una conversión completa. El catalizador de paladio se separó mediante filtración a través de un tapón de Celite. La torta del filtro se lavó con MeOH (2 x 20 ml). Los filtrados se combinaron y se evaporaron. El residuo sólido se tomó con metanol y se evaporó (para eliminar los volátiles aromáticos restantes y el formiato de amonio). Esta etapa se repitió dos veces para dar 1-(trans-4-aminociclohexil)-N-metil-metanosulfonamida como un sólido blanquecino (0,85 g).
Etapa H: A una suspensión de 4-cloro-7-azaindol (1,37 g), trietilamina (1,9 ml) y DMAP (0,11 g) en DCM (70 ml) se añadió a temperatura ambiente cloruro de bencenosulfonilo (1,3 ml). La mezcla de reacción se agitó a temperatura ambiente durante la noche. La mezcla de reacción se diluyó con DCM y se inactivó con una solución acuosa de HCl (1 M, 70 ml). La fase orgánica se separó y se extrajo con una solución saturada de NaHCO3 (70 ml), con agua y con una solución acuosa saturada de NaCl, se secó sobre Na2SO4 y se concentró en vacío para dar 1-(bencenosulfonil)-4-cloro-pirrolo[2,3-b]piridina como un sólido marrón (2,65 g). El producto crudo obtenido se usó sin purificación adicional.
Etapa I: Se añadió gota a gota nitrato de tetrabutilamonio (381 mg) disuelto en DCM (5 ml) a una solución de 1-(bencenosulfonil)-4-cloro-pirrolo[2,3-b]piridina (292 mg) en DCM (5 ml) bajo nitrógeno a -10°C. Se añadió gota a gota anhídrido trifluoroacético (180 pl), se agitó durante 30 minutos a la misma temperatura y a continuación durante 4 horas a temperatura ambiente. Se añadieron nitrato de tetrabutilamonio adicional (80 mg) y anhídrido trifluoroacético (40 pl) y la mezcla de reacción se agitó a temperatura ambiente durante la noche. Se añadieron nitrato de tetrabutilamonio adicional (380 mg) y anhídrido trifluoroacético (180 pl) y la mezcla de reacción se agitó a temperatura ambiente durante 3 horas. Después de diluir con DCM, la mezcla de reacción se inactivó con agua. La fase orgánica se separó y se extrajo 3 veces con agua y una vez con una solución acuosa saturada de NaCl, se secó sobre Na2SO4 y se concentró en vacío. El producto bruto se purificó mediante cromatografía en gel de sílice (EtOAc/heptano 1:5) para dar 1-(bencenosulfonil)-4-cloro-5-nitro-pirrolo[2,3-b]piridina como un sólido de color beige (111 mg).
Etapa J: Se suspendieron 1-(bencenosulfonil)-4-cloro-5-nitro-pirrolo[2,3-b]piridina (337 mg), 1-(trans-4-aminocidohexil)-N-metil-metanosulfonamida (ejemplo 1, etapa G, 206 mg) y carbonato de potasio (304 mg) en dioxan/agua 9:1 (10 ml). La suspensión resultante se calentó a 120°C en un horno de microondas. Después de 2 horas, se tomaron muestras de la mezcla de reacción para HPLC/MS, sin material de partida visible; se formó el producto. La mezcla de reacción se concentró en vacío. Se añadieron THF (15 ml), EtOAc (45 ml) y agua (30 ml) al residuo. La mezcla de reacción se agitó a temperatura ambiente durante 30 minutos. La fase acuosa se separó y se extrajo dos veces con una mezcla de THF/EtOAc 1:3. Las fases orgánicas combinadas se extrajeron con una solución acuosa saturada de NaCl, se secaron sobre Na2SO4 y se concentraron en vacío. El producto bruto se purificó mediante cromatografía en gel de sílice (EtOAc/heptano 1:1 a EtOAc 100%) para dar 1-[trans-4-[[1-(bencenosulfonil)-5-nitro-1H-pirrolo[2,3-b]piridin-4-il]amino]ciclohexil]-N-metil-metanosulfonamida como una espuma amarilla (276 mg).
Etapa K: Se disolvió 1-[trans-4-[[1-(bencenosulfonil)-5-nitro-1H-pirrolo[2,3-b]piridin-4-il]amino]ciclohexil]-N-metilmetanosulfonamida (254 mg) en THF (20 ml). Esta solución se hidrogenó durante 6 horas sobre un catalizador Raney-Nickel usando el reactor de flujo H-cube® (Modo Full H2, temperatura = temperatura ambiente, caudal = 1 ml/min). El THF se eliminó bajo vacío. El residuo se disolvió en EtOAc, se secó sobre sulfato de magnesio y se concentró en vacío para dar 1-[trans-4-[[5-amino-1-(bencenosulfonil)-1H-pirrolo[2,3-b]piridin-4-il]amino]ciclohexil]-N-metil-metanosulfonamida como una resina amarilla (222 mg). El producto crudo obtenido se usó sin purificación adicional.
Etapa L: Una mezcla de 1-[trans-4-[[5-amino-1-(bencenosulfonil)-1H-pirrolo[2,3-b]piridin-4-il]amino]ciclohexil]-N-metil-metanosulfonamida (95 mg), ortoformiato de trietilo (80 pl) y monohidrato de ácido p-toluensulfónico (4 mg) en tolueno (6 ml) se sometió a reflujo durante la noche.
Después de diluir con EtOAc, la mezcla de reacción se inactivó con una solución acuosa saturada de NaHCO3. La fase orgánica se separó y se extrajo una vez más con una solución acuosa saturada de NaHCO3 y con una solución acuosa saturada de NaCl, se secó sobre MgSO4 y se concentró en vacío. El producto bruto se purificó en una HPLC semipreparativa para dar 1-[trans-4-(6-(fenilsulfonil)-imidazo[4,5-d]pirrolo[2,3-b]piridin-1(6H)-il)ciclohexil]-N-metil-metanosulfonamida como una resina incolora (50 mg).
Etapa M: Una mezcla de 1-[trans-4-(6-(fenilsulfonil)-imidazo[4,5-d]pirrolo[2,3-b]piridin-1(6H)-il)ciclohexil]-N-metilmetanosulfonamida (68 mg) e hidróxido de litio (14 mg) en isopropanol/agua 1:1 (1 ml) se agitó a 40°C durante la noche. Después de 20 horas, la mezcla de reacción se calentó a 50°C y se agitó a 50°C durante un día. Se añadió hidróxido de litio adicional (14 mg). La mezcla de reacción se calentó a 60°C y se agitó a 60°C durante un día. Se añadió una solución acuosa de HCl al 37% (120 pl) hasta alcanzar un pH = 5 y a continuación se añadió una solución acuosa saturada de NaHCO3 (100 pl) hasta alcanzar un pH = 8. El isopropanol se evaporó. Se añadió EtOAc/THF (2-3 ml) al residuo y la suspensión resultante se agitó a temperatura ambiente. Se añadió una solución acuosa de carbonato de potasio (2 M, 2-3 ml) hasta alcanzar un pH = 12. La suspensión se agitó adicionalmente a temperatura ambiente y el precipitado se separó mediante filtración, se enjuagó con agua y se secó en vacío para dar 1-[trans-4-(imidazo[4,5-d]pirrolo[2,3-b]piridin-1(6H)-il)ciclohexil]-N-metil-metanosulfonamida (13 mg, compuesto 22 en la Tabla 3). MS (HPLC/MS): 348 (MH+). Tiempo de retención: 1,70 min.
Ejemplo 2
Este ejemplo ilustra la preparación de N-metil-1-[trans-4-[(5-nitro-1H-pirrolo[2,3-b]piridin-4-il)oxi]ciclohexil]metanosulfonamida (Compuesto 25 en la Tabla 3)
Etapa A: Se mezclaron juntos trans-4-hidroxiciclohexanocarboxilato de etilo (10 g), diisopropilamina (21 ml), bromuro de bencilo (10 ml) y yoduro de sodio (0,9 g) y se calentaron en un tubo sellado a 120°C durante la noche. Después de diluir con EtOAc, la mezcla de reacción se inactivó con agua. La fase acuosa se separó y se extrajo dos veces con EtOAc. Las fases orgánicas combinadas se secaron sobre MgSO4 anhidro y se concentraron en vacío. El producto bruto se purificó mediante cromatografía en gel de sílice (hexano/EtOAc 9:1) para dar trans-4-(benciloxi)ciclohexanocarboxilato de etilo como un aceite amarillo (16,0 g).
Etapa B: Se suspendió trans-4-(benciloxi)ciclohexanocarboxilato de etilo (16,0 g) en THF seco mientras se enfriaba en un baño de hielo. Se añadió hidruro de litio y aluminio (5,2 g) en porciones. Después de la adición, la mezcla de reacción se calentó a 60°C durante 4 horas. Después de ese tiempo, la mezcla de reacción se enfrió a 0°C y se añadió EtOAc (30 ml) seguido de agua (30 ml). Las sales inorgánicas resultantes se separaron mediante filtración a través de una almohadilla de Celite. Las fases se separaron. La parte acuosa se extrajo con EtOAc. Las fases orgánicas combinadas se secaron sobre MgSO4 anhidro y se concentraron en vacío para dar (trans-4
benciloxicidohexil)metanol como un sólido amarillo claro (14,2 g). El producto crudo obtenido se usó sin purificación adicional.
Etapa C: Se suspendió (trans-4-benciloxiciclohexil)metanol (14,2 g) en THF seco mientras se enfriaba en un baño de hielo. Se añadió trifenilfosfina (22,32 g). La solución resultante se agitó a 0°C durante 10 minutos, a continuación, se añadió tetrabromometano (28,22 g) en porciones y se dejó que la suspensión alcanzara la temperatura ambiente. Después de 24 horas de agitación, el precipitado blanco se filtró y se lavó con THF seguido de EtOAc. El filtrado se evaporó a presión reducida y se purificó mediante cromatografía en columna sobre gel de sílice (hexano/EtOAc 9:1) para dar trans-4-(bromometil)ciclohexil éter de bencilo como un sólido amarillo (17,0 g). Etapa D: Se disolvió trans-4-(bromometil)ciclohexil éter de bencilo (8,0 g) en isopropanol (100 ml) y se añadió sulfito de sodio (7,12 g) en agua (100 ml). A continuación, la mezcla de reacción se agitó vigorosamente mientras se calentaba a 100°C durante la noche. Después de enfriar a temperatura ambiente, la mezcla de reacción se concentró para dar un sólido blanco. Se añadió metanol y la mezcla se agitó a temperatura ambiente durante 3 horas, a continuación, el precipitado se filtró, se enjuagó con metanol, el filtrado se evaporó para obtener ácido (trans-4-benciloxiciclohexil)metanosulfónico como un sólido blanco (9,5 g).
Etapa E: Se suspendió ácido (trans-4-benciloxiciclohexil)metanosulfónico (1,0 g) en un cloroformo recién destilado (50 ml) mientras se enfriaba en un baño de hielo. Se añadió DMF seco (3-5 gotas). La solución resultante se agitó a 0°C durante 10 minutos, a continuación, se añadió gota a gota cloruro de tionilo (0,52 ml). La mezcla se agitó a esta temperatura durante 15 minutos, 30 minutos a temperatura ambiente y durante la noche a 45°C. Después de enfriar, el disolvente se evaporó; se añadió DCM seco y se evaporó para eliminar el cloruro de tionilo residual. Este procedimiento se repitió dos veces para dar cloruro de (trans-4-benciloxiciclohexil)metanosulfonilo en forma de un aceite amarillo (1,0 g).
Etapa F: Se suspendió cloruro de (trans-4-benciloxiciclohexil)metanosulfonilo (1,0 g) en DCM seco (50 ml) mientras se enfriaba en un baño de hielo, se añadió gota a gota metilamina (2M en THF, 5,0 ml). Posteriormente, se dejó que la mezcla de reacción alcanzara la temperatura ambiente y se agitó a esta temperatura durante la noche. Después de ese tiempo, el disolvente se evaporó y se purificó mediante cromatografía en columna sobre gel de sílice (hexano/EtOAc 9:1) para dar 1-(trans-4-benciloxiciclohexil)-N-metil-metanosulfonamida como un sólido amarillo claro (0,56 g).
Etapa G: Se suspendió 1-(trans-4-benciloxiciclohexilo)-N-metil-metanosulfonamida (3,7 g) en metanol, se añadió Pd(OH)2 (1,75 g). Posteriormente, la reacción se continuó en un aparato Parr durante 12 horas. A continuación, el catalizador se separó mediante filtración a través de una almohadilla de Celite. El filtrado se evaporó, se lavó con Et2O y se secó en vacío para dar 1-(trans-4-hidroxiciclohexil)-N-metil-metanosulfonamida como un sólido blanco (2,44 g).
Etapa H: Se añadió hidruro de sodio (60% en aceite mineral, 0,09 g) bajo nitrógeno a una solución de 1-(trans-4-hidroxiciclohexil)-N-metil-metanosulfonamida (0,40 g) en DMF (19 ml). Después de 30 minutos a temperatura ambiente, se añadió cloruro de 2-(trimetilsilil)etoximetilo durante 15 minutos a la mezcla de reacción. Después de 18 horas a temperatura ambiente, la mezcla de reacción se diluyó con EtOAc y se inactivó con una solución acuosa de fosfato de sodio (1 M). La fase acuosa se separó y se extrajo dos veces con EtOAc. Las fases orgánicas combinadas se lavaron con agua y dos veces con salmuera, se secaron sobre sulfato de magnesio anhidro y se concentraron en vacío para dar 1-(trans-4-hidroxiciclohexil)-N-metilo-N-(2-trimetilsililetoximetil)metanosulfonamida como un aceite amarillo (0,66 g). El producto crudo obtenido se usó sin purificación adicional.
Etapa I: Se añadió hidruro de sodio (60% en aceite mineral, 0,07 g) bajo nitrógeno a una solución de 1-(trans-4-hidroxiciclohexil)-N-metilo-N-(2-trimetilsililetoximetil)metanosulfonamida (0,49 g) en DMF (10 ml). Después de 30 minutos a temperatura ambiente, se añadió 1-(bencenosulfonil)-4-cloro-5-nitro-pirrolo[2,3-b]piridina (ejemplo 1, etapa I, 0,49 g) en DMF (5 ml) durante 15 minutos a la mezcla de reacción. Después de tres días a temperatura ambiente, se añadió EtOAc y la mezcla de reacción se vertió en agua. La fase acuosa se separó y se extrajo con EtOAc. Las fases orgánicas combinadas se lavaron dos veces con agua, dos veces con una solución acuosa de hidróxido de sodio (2 N) y dos veces con salmuera, se secaron sobre sulfato de magnesio anhidro y se concentraron en vacío. El producto bruto se purificó en una HPLC semipreparativa para dar N-metil-1-[trans-4-[(5-nitro-1 H-pirrolo[2,3-b]piridin-4-il)oxi]ciclohexil]-N-(2-trimetilsililetoximetil)metanosulfonamida como un sólido amarillo (0,12 g).
Etapa J: Una solución de N-metil-1 -[trans-4-[(5-nitro-1 H-pirrolo[2,3-b]piridin-4-il)oxi]ciclohexil]-N-(2-trimetilsililetoximetil)metanosulfonamida (120 mg) en AcOH (10 ml) y agua (5 ml) se agitó a 70°C durante 1 hora
bajo nitrógeno. La mezcla de reacción se enfrió a temperatura ambiente y se vertió en agua. Se añadió una solución acuosa de NaOH (4N) hasta que se alcanzó un pH = 7-8. La fase acuosa se extrajo dos veces con EtOAc. Las fases orgánicas combinadas se lavaron con agua, dos veces con una solución acuosa de hidróxido de sodio (2 N) y con salmuera, se secaron sobre sulfato de magnesio anhidro y se concentraron en vacío. El residuo se recristalizó en DCM/EtOAc (9:1) para dar N-metil-1-[trans-4-[(5-nitro-1H-pirrolo[2,3-b]piridin-4-il)oxi]ciclohexil]metanosulfonamida (50 mg, Compuesto 25 en la Tabla 3). MS (HPLC/MS): 369 (MH+). Tiempo de retención: 1,12 min.
Ejemplo 3
Este ejemplo ilustra la preparación de 1-[trans-4-[(5-ciano-1H-pirrolo[2,3-b]piridin-4-il)-metil-amino]ciclohexil]-N-metil-metanosulfonamida (Compuesto 26 en la Tabla 3).
Etapa A: Se añadió una solución de hidruro de bis(2-metoxietoxi)aluminio sódico (Red-Al®, 65% en tolueno, 183 ml) durante 60 minutos a una solución de ácido trans-4-(tert-butoxicarbonilamino)ciclohexanocarboxílico (24,3 g) en tolueno (250 ml) a 0°C. La mezcla de reacción se calentó a continuación a 130°C y se agitó a esta temperatura durante 1 hora. Después de enfriar a 0°C, se añadió gota a gota una solución acuosa saturada de sulfato de sodio (195 ml). La mezcla de reacción se filtró a continuación a través de un filtro Hyflo. La torta del filtro se enjuagó con DCM (150 ml) y agua (24 ml). La fase acuosa se separó y se extrajo dos veces con DCM (2 x 150 ml). Las fases orgánicas combinadas se secaron sobre sulfato de magnesio anhidro y se concentraron en vacío para dar [trans-4-(metilamino)ciclohexil]metanol como cristales de color blanco (12,5 g). El producto crudo obtenido se usó sin purificación adicional.
Etapa B: Se añadió gota a gota cloruro de benzoilo (8,8 ml) a una emulsión de hidrogenocarbonato de sodio (12,6 g) en agua (50 ml) y [trans-4-(metilamino)ciclohexil]metanol (10,9 g) en DCM (50 ml) a 0°C. Posteriormente, se dejó que la mezcla de reacción alcanzara la temperatura ambiente y se agitó a esta temperatura durante 3 horas. La mezcla de reacción se diluyó con agua (150 ml) y con DCM (200 ml). La fase orgánica se separó, se secó sobre sulfato de magnesio anhidro y se concentró en vacío para dar N-[trans-4-(hidroximetil)ciclohexil]-N-metilbenzamida en forma de cristales de color beige (17,2 g). El producto crudo obtenido se usó sin purificación adicional.
Etapa C: Se añadieron trietilamina (11 ml), DMAP (0,43 g) y cloruro de p-toluenosulfonilo (13,2 g) a una solución de N-[trans-4-(hidroximetil)ciclohexil]-N-metilbenzamida (17,0 g) en DCM (250 ml). Después de 3 horas a temperatura ambiente, la mezcla de reacción se inactivó con agua (150 ml). La fase orgánica se separó, se secó sobre sulfato de magnesio anhidro y se concentró en vacío. El producto bruto se purificó mediante cromatografía en gel de sílice (EtOAc/heptano 1:1) para dar 4-metilbencenosulfonato de [trans-4-[benzoil(metil)amino]ciclohexil]metilo como cristales de color blanco (19,5 g).
Etapa D: Se añadió tioacetato de potasio (6,3 g) a una suspensión de 4-metilbencenosulfonato de [trans-4-[benzoil(metil)amino]ciclohexil]metilo (19,5 g) en DMSO (66 ml) a temperatura ambiente. La mezcla de reacción se calentó a 55°C y se agitó a 55°C durante 3 horas. Después de enfriar a temperatura ambiente, la mezcla de reacción se diluyó con EtOAc (200 ml) y se inactivó con una solución acuosa de NaHCO3 (0,1 M, 300 ml). La fase acuosa se separó y se extrajo dos veces con EtOAc (2 x 200 ml). Las fases orgánicas combinadas se lavaron con agua y salmuera, se secaron sobre sulfato de magnesio anhidro y se concentraron en vacío. El residuo (17 g, cristales de color amarillo claro) se disolvió en ácido fórmico (73 ml) y la mezcla de reacción se calentó a 25-35°C. Se añadió una solución de peróxido de hidrógeno (30% en agua, 25 ml) durante 60 minutos a esta temperatura. Posteriormente, la mezcla de reacción se enfrió a temperatura ambiente y se agitó a temperatura ambiente durante 15 minutos. Después de enfriar a 0°C, la mezcla de reacción se inactivó con una solución acuosa de metabisulfito de sodio (33%, 27 ml). A continuación, se añadió una solución acuosa de NaOH (33%, 121 ml) a 0°C hasta que se alcanzó un pH = 5 y la mezcla de reacción se agitó a temperatura ambiente durante la noche. La mezcla de reacción se concentró en vacío. El residuo obtenido se recogió en agua (160 ml) y 2-propanol (40 ml) y se agitó a 45°C. Posteriormente, el 2-propanol se evaporó y se añadió una solución acuosa de HCl (2 M, 10 ml). La suspensión se agitó a 0°C y se filtró. El precipitado se secó en vacío sobre Sicapent® para dar ácido [trans-4-[benzoil(metil)amino]ciclohexil]metanosulfónico como un sólido blanco (31,0 g) usado en la siguiente etapa sin purificación adicional.
Etapa E: Se suspendió ácido [trans-4-[benzoil(metil)amino]ciclohexil]metanosulfónico (10,1 g) en DCM (40 ml) con la adición de unas pocas gotas de DMF. La mezcla de reacción se enfrió a 0°C y se añadió gota a gota cloruro de tionilo (4,7 ml) durante 10 minutos. La mezcla de reacción se sometió a continuación a reflujo durante 6 horas. Después de enfriar a 0°C, se añadió gota a gota cloruro de tionilo adicional (4,7 ml) durante 10 minutos. La mezcla de reacción se sometió a continuación a reflujo durante la noche. Después de ese tiempo, la mezcla se enfrió a
temperatura ambiente y se concentró casi hasta la sequedad en vacío. Se añadió tolueno seco al residuo y se eliminó a presión reducida para asegurar la eliminación de cualquier cloruro de tionilo sin reaccionar. El residuo se recogió en THF (40 ml) y se añadió metilamina (2 M en THF, 47 ml). La mezcla de reacción se agitó a temperatura ambiente durante la noche. La suspensión resultante se filtró y se enjuagó con THF. El filtrado se evaporó para dar una resina de color marrón claro (2,6 g). El precipitado se suspendió en DCM (20 ml) con la adición de unas pocas gotas de DMF. Se añadió gota a gota cloruro de tionilo (2,4 ml) durante 10 minutos. La mezcla de reacción se sometió a continuación a reflujo durante la noche. Después de 16 horas a reflujo, se añadió gota a gota cloruro de tionilo adicional (2,4 ml) durante 10 minutos. La mezcla de reacción se sometió a continuación a reflujo durante la noche. Después de ese tiempo, la mezcla se enfrió a temperatura ambiente y se concentró casi hasta la sequedad en vacío. Se añadió tolueno seco al residuo y se eliminó a presión reducida para asegurar la eliminación de cualquier cloruro de tionilo sin reaccionar. El residuo se recogió en THF (20 ml) y se añadió metilamina (2 M en THF, 24 ml). La mezcla de reacción se agitó a temperatura ambiente durante 4 horas, a continuación, se filtró y se enjuagó con THF. El filtrado se evaporó para dar una resina de color marrón claro (2,0 g). Las resinas de color marrón claro combinadas (4,6 g) se purificaron mediante cromatografía en gel de sílice (MeOH/DCM 1:49 a 1:19) para dar N-metil-N-[trans-4-(metilsulfamoilmetil)ciclohexil]benzamida como cristales de color beige (2,18 g).
Etapa F: La N-metil-N-[trans-4-(metilsulfamoilmetil)ciclohexil]benzamida (875 mg) se recogió en una solución acuosa de HCl (6 M, 10 ml) y la mezcla de reacción se calentó a reflujo durante dos días. Después de enfriar a temperatura ambiente, la suspensión resultante se filtró. El filtrado se concentró en vacío para dar clorhidrato de N-metil-1-[trans-4-(metilamino)ciclohexil]metanosulfonamida como cristales de color beige (710 mg). El producto crudo obtenido se usó sin purificación adicional.
Etapa G: Se suspendieron clorhidrato de N-metil-1-[trans-4-(metilamino)ciclohexil]metanosulfonamida (129 mg), 4-cloro-1 H-pirrolo[2,3-b]piridin-carbonitrilo (89 mg) y carbonato de potasio (225 mg) en agua/dioxano 1:9 (4,5 ml). La suspensión resultante se calentó a 160°C en un horno de microondas durante 8 horas. La mezcla de reacción se concentró en vacío. El residuo se recogió en THF (10 ml) y se añadió una mezcla de agua/solución acuosa saturada de NaCl 1:1 (20 ml). La mezcla se agitó a TA. La fase acuosa se separó y se extrajo dos veces con THF (2 x 10 ml). Las fases orgánicas combinadas se lavaron con salmuera, se secaron sobre sulfato de magnesio anhidro y se concentraron en vacío. El residuo se suspendió en acetonitrilo, se agitó a temperatura ambiente, se filtró y se enjuagó con acetonitrilo. El filtrado se concentró en vacío y se purificó en una HPLC semipreparativa para dar 1-[trans-4-[(5-ciano-1H-pirrolo[2,3-b]piridin-4-il)-metil-amino]ciclohexil]-N-metil-metanosulfonamida (22 mg, Compuesto 26 en la Tabla 3). MS (HPLC/MS): 362 (MH+). Tiempo de retención: 2,19 min.
Ejemplo 6
Este ejemplo ilustra la preparación de 1-[trans-4-((2-metoximetil)imidazo[4,5-d]pirrolo[2,3-b]piridin-1(6H)-il)ciclohexil]-N-metil-metanosulfonamida (Compuesto 35 en la Tabla 3).
Etapa A:
Una mezcla de 1-[trans-4-[[5-amino-1-(bencenosulfonil)-1H-pirrolo[2,3-b]piridin-4-il]amino]ciclohexil]-N-metilmetanosulfonamida (Ejemplo 1, etapa K) (212 mg), cloruro de metoxiacetilo (40 pl) y trietilamina (70 pl) en cloruro de metileno (5 ml) se agitó una hora a temperatura ambiente. La mezcla de reacción se concentró en vacío. El crudo se recogió en ácido acético (2 ml) y se calentó a 100°C durante 3 h en un microondas. Después de diluir con EtOAc, la mezcla de reacción se inactivó con una solución acuosa saturada de NaHCO3. La fase orgánica se separó y se extrajo una vez más con una solución acuosa saturada de NaHCO3 y con una solución acuosa saturada de NaCl, se secó sobre MgSO4 y se concentró en vacío. El producto bruto se purificó en una HPLC semipreparativa para dar 1-[trans-4-((2-metoximetil)-6-(fenilsulfonil)-imidazo[4,5-d]pirrolo[2,3-b]piridin-1(6H)-il)ciclohexil]-N-metil-metanosulfonamida como una resina incolora.
Etapa B:
La 1-[trans-4-((2-metoximetil)-6-(fenilsulfonil)-imidazo[4,5-d]pirrolo[2,3-b]piridin-1(6H)-il)ciclohexil]-N-metilmetano sulfonamida se desprotegió usando un procedimiento similar al descrito en el Ejemplo 1, Etapa I, para dar 1-[trans-4-((2-metoximetil)imidazo[4,5-d]pirrolo[2,3-b]piridin-1(6H)-il)ciclohexil]-N-metilmetanosulfonamida (26 mg, Compuesto 35 en la Tabla 3). MS (HPLC/MS): 392 (MH+). Tiempo de retención: 1,89 min.
El análisis de las muestras purificadas se realizó en cada caso usando un sistema Waters Autopurification (HPLC/MS) con una columna de fase inversa (Daisogel SP-120-ODS-AP 5pm, 150X3mm) de Bischoff, Leonberg, Alemania. Las muestras se caracterizaron por m/z y por tiempo de retención. Los tiempos de retención proporcionados anteriormente se refieren en cada caso al uso de un sistema disolvente que comprende dos
disolventes diferentes, disolvente A: H2O HCOOH al 0,01% y disolvente B: CH3CN HCOOH al 0,01%). Dichos dos disolventes A y B se emplean a un caudal de 2,00 ml/min con un gradiente dependiente del tiempo tal como se indica en la Tabla:
Procedimiento A: columna Daisogel SP-120-ODS-AP 5|jm, 150X3mm) de Bischoff, Leonberg, Alemania, caudal de 2,00 ml/min con un gradiente dependiente del tiempo tal como se indica en la Tabla 1:
Tabla 1:

Procedimiento B: columna Waters XTerra MS C18 5jm, 50X4,6mm (Waters), caudal de 3,00 ml/min con un gradiente dependiente del tiempo tal como se indica en la Tabla 2
Tabla 2:

Las sustancias nombradas en la Tabla 3 siguiente se prepararon de manera análoga a los procedimientos descritos anteriormente. Los siguientes datos físicos se obtuvieron según el procedimiento de caracterización HPLC/MS descrito anteriormente.
Tabla 3

(Cont.)

Las cuatro quinasas de la familia de quinasas JAK/TYK se usaron como proteínas de fusión GST recombinantes purificadas, que contenían los dominios de quinasa activos. GST-JAK1 (866-1154), GST-JAK3 (811-1124) y GST-TYK2 (888-1187) se expresaron y purificaron mediante cromatografía de afinidad en la unidad de biología EPK.
Los ensayos de quinasa se basaron en el ensayo de cambio de movilidad Caliper usando los sistemas LabChip 3000. Esta tecnología es similar a la electroforesis capilar y usa la separación mediada por carga de sustrato y producto en un chip microfluídico.
Todas las reacciones de quinasa se realizaron en placas de microtitulación de 384 pocillos en un volumen de reacción total de 18 pI. Las placas de ensayo se prepararon con 0,1 pI por pocillo de compuesto de ensayo en la concentración de ensayo apropiada, tal como se describe en la sección "Preparación de diluciones de compuesto". Las reacciones se iniciaron combinando 9 pI de la mezcla de sustrato (que consistía en péptido y ATP) con 9 pI de dilución de quinasa. Las reacciones se incubaron durante 60 minutos a 30°C y se detuvieron añadiendo 70 pI de tampón de parada (Hepes 100 mM, DMSO al 5%, reactivo de revestimiento al 0,1%, EDTA 10 mM, Brij 35 al 0,015%).
Se usaron péptidos sintéticos marcados con fluorescencia como sustratos en todas las reacciones. Se usó un péptido derivado de la secuencia de IRS-1 (péptido IRS-1, FITC-Ahx-KKSRGDYMTMQIG-NH2) para JAK1 y TYK2 y un péptido denominado JAK3tide (FITC-GGEEEEYFELVKKKK-NH2) para JAK3. Las condiciones de ensayo específicas se describen en la Tabla 4:
Tabla 4: Condiciones de ensayo de los ensayos de quinasa individuales

Las reacciones terminadas se transfirieron al lector Caliper LabChip 3000 y se midió la rotación de cada reacción determinando la relación sustrato/producto.
Preparación de diluciones de compuestos
Los compuestos de ensayo se disolvieron en DMSO (10 mM) y se transfirieron a tubos de matriz plana de fondo plano o en forma de V de 1,4 ml que transportaban un único chip de matriz 2D por centros de compuestos individuales. Los números de estos chips se vincularon distintivamente a los números de identificación de los compuestos individuales. Las soluciones madre se almacenaron a -20°C si no se usaron inmediatamente. Para el procedimiento de ensayo, los viales se descongelaron y se identificaron mediante un escáner mediante el cual se generó una hoja de trabajo que sirvió de guía para las etapas de trabajo posteriores Las diluciones de los
compuestos se realizaron en placas de 96 pocilios. Este formato permitió el ensayo de un máximo de 40 compuestos de ensayo individuales en 8 concentraciones (puntos únicos), incluyendo 4 compuestos de referencia. El protocolo de dilución incluía la producción de placas de predilución, placas maestras y placas de ensayo:
Placas de predilución: se usaron placas de 96 pocillos de polipropileno como placas de predilución. Se prepararon un total de 4 placas de predilución que incluían 10 compuestos de ensayo cada una en las posiciones de placa A1-A10, un compuesto estándar en A11 y un control con DMSO en A12. Todas las etapas de dilución se realizaron en un robot HamiltonSTAR.
Placas maestras: se transfirieron 100 |j I de diluciones de compuestos individuales que incluían el compuesto estándar y los controles de las 4 "placas de predilución" a una "placa maestra" de 384 pocillos que incluía las siguientes concentraciones 1'820, 564, 182, 54,6, 18,2, 5,46, 1,82 y 0,546 j M, respectivamente, en 90% de DMSO.
Placas de ensayo: A continuación, se prepararon placas de ensayo idénticas pipeteando 100 nl de diluciones de compuestos de las placas maestras en las "placas de ensayo" de 384 pocillos. A continuación, los compuestos se mezclaron con 9 j I de componentes de ensayo más 9 j I de enzima correspondiente a etapas de dilución de 1:181 que permitían una concentración final de 10, 3,0, 1,0, 0,3, 0,1, 0,03, 0,01 y 0,003 j M, respectivamente. La preparación de las placas maestras se realizó mediante el robot Matrix PlateMate Plus y la replicación de las placas de ensayo con el robot HummingBird.
En base a este estudio, un compuesto de la invención muestra una eficacia terapéutica especialmente contra trastornos dependientes de la proteína quinasa, especialmente enfermedades proliferativas mediadas por la actividad quinasa JAK/TYK.

Claims (9)
1. Un compuesto de fórmula I
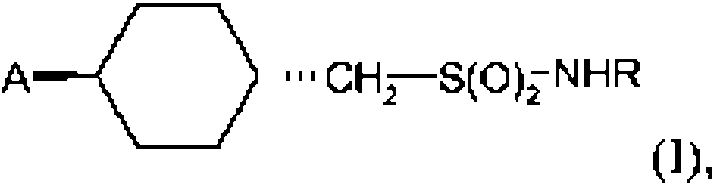
en la que R es metilo; y en la que A consiste en la fórmula 2a

en la que Y se selecciona de entre el grupo que consiste en NH, N(alquilo C1-C2) u O, y R2 se selecciona de entre el grupo que consiste en ciano (CN), nitro (NO2), o NR'R", y R' y R" se seleccionan independientemente de entre el grupo que consiste en H o alquilo C1-C4; o en la que Y y R2 forman juntos

en la que R0 se selecciona de entre el grupo que consiste en H, alquilo C1-C4 , hidroxi-alquilo C1-C4 , alcoxi C1-C4-alquilo C1-C4 o NH2 ; o una sal del mismo aceptable desde el punto de vista veterinario.
2. El compuesto según la reivindicación 1, en el que Y se selecciona de entre el grupo que consiste en NH, N(alquilo C1-C2) u O, y R2 se selecciona de entre el grupo que consiste en CN, NO2 , o NR'R", y R' y R" se seleccionan independientemente de entre el grupo que consiste en H o alquilo C1-C4 ; o una sal del mismo aceptable desde el punto de vista veterinario.
3. El compuesto según la reivindicación 1, en el que A consiste en la fórmula 2a'

en la que R0 se selecciona de entre el grupo que consiste en H, alquilo C1-C4 , hidroxi-alquilo C1-C4 , alcoxi C1-C4-alquilo C1-C4 o NH2 , o una sal del mismo aceptable desde el punto de vista veterinario.
4. un compuesto según la reivindicación 1, que es 1-[trans-4-((2-amino)imidazo[4,5-d]pirrolo[2,3-b]piridin-1(6H)-il)ciclohexil]-N-metilmetanosulfonamida y puede ser representado estructuralmente como

5. Una composición que comprende un compuesto de cualquiera de las reivindicaciones 1 a 4, y un vehículo aceptable desde el punto de vista veterinario.
6. Un compuesto según una cualquiera de las reivindicaciones 1 a 4, para su uso en el tratamiento de enfermedades inflamatorias o relacionadas con el sistema inmune.
7. Un compuesto según una cualquiera de las reivindicaciones 1 a 4, para su uso en el tratamiento de dermatitis atópica, eccema, psoriasis, escleroderma, prurito y otras afecciones pruríticas o dermatitis alérgica en un mamífero.
8. Un compuesto para su uso según la reivindicación 7, en el que el mamífero es un animal de compañía.
9. Un compuesto para su uso según la reivindicación 8, en el que el animal de compañía es un perro.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP14162391.8A EP2924026A1 (en) | 2014-03-28 | 2014-03-28 | Aminosulfonylmethylcyclohexanes as JAK inhibitors |
| PCT/EP2015/056430 WO2015144773A1 (en) | 2014-03-28 | 2015-03-25 | New compounds |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2702474T3 true ES2702474T3 (es) | 2019-03-01 |
Family
ID=50389928
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES15713431T Active ES2702474T3 (es) | 2014-03-28 | 2015-03-25 | Ciclohexilmetanosulfonamidas |
Country Status (10)
| Country | Link |
|---|---|
| US (2) | US10059709B2 (es) |
| EP (2) | EP2924026A1 (es) |
| JP (1) | JP6239139B2 (es) |
| AU (1) | AU2015238361B2 (es) |
| CA (1) | CA2940321C (es) |
| DK (1) | DK3122731T3 (es) |
| ES (1) | ES2702474T3 (es) |
| PL (1) | PL3122731T3 (es) |
| PT (1) | PT3122731T (es) |
| WO (1) | WO2015144773A1 (es) |
Families Citing this family (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2924026A1 (en) * | 2014-03-28 | 2015-09-30 | Novartis Tiergesundheit AG | Aminosulfonylmethylcyclohexanes as JAK inhibitors |
| WO2017004134A1 (en) * | 2015-06-29 | 2017-01-05 | Nimbus Iris, Inc. | Irak inhibitors and uses thereof |
| CN107365312B (zh) * | 2016-05-13 | 2021-10-01 | 瑞博(杭州)医药科技有限公司 | 一种制备Oclacitinib的新方法 |
| EP3528816A4 (en) * | 2016-10-21 | 2020-04-08 | Nimbus Lakshmi, Inc. | TYK2 INHIBITORS AND USES THEREOF |
| LT3555097T (lt) | 2016-12-16 | 2022-08-25 | Janssen Pharmaceutica Nv | Imidazo[4,5-d]pirolo[2,3-b]piridino junginiai kaip janus kinazės inhibitoriai |
| JOP20190144A1 (ar) | 2016-12-16 | 2019-06-16 | Janssen Pharmaceutica Nv | إيميدازو بيرولو بيريدين كمثبطات لعائلة jak الخاصة بإنزيمات الكيناز |
| EP3710431A4 (en) * | 2017-11-03 | 2021-07-07 | Aclaris Therapeutics, Inc. | SUBSTITUTED PYRROLOPYRIMIDINE-BASED JAK INHIBITORS AND THEIR MANUFACTURING AND USE PROCEDURES |
| TW202016110A (zh) | 2018-06-15 | 2020-05-01 | 比利時商健生藥品公司 | Jak激酶家族之小分子抑制劑 |
| WO2020033955A1 (en) | 2018-08-10 | 2020-02-13 | Aclaris Therapeutics, Inc. | Pyrrolopyrimidine itk inhibitors |
| EP3856742B1 (en) * | 2018-11-01 | 2024-10-02 | Lynk Pharmaceuticals Co. Ltd. | Tricyclic janus kinase 1 inhibitors, and compositions and methods thereof |
| KR102737181B1 (ko) * | 2019-03-14 | 2024-12-02 | 상하이 시너지 파마슈티컬 사이언시스 코., 엘티디. | Jak 억제제 및 그 제조방법과 의약분야에서의 응용 |
| WO2020223728A1 (en) * | 2019-05-02 | 2020-11-05 | Aclaris Therapeutics, Inc. | Substituted pyrrolopyridines as jak inhibitors |
| CN116925077A (zh) * | 2020-09-08 | 2023-10-24 | 一又生物(上海)有限公司 | 吡啶并环类化合物及其制备方法、中间体、组合物和应用 |
| BR112023018286A2 (pt) | 2021-03-11 | 2023-10-31 | Janssen Pharmaceutica Nv | Lorpucitinib para uso no tratamento de distúrbios mediados por jak |
| WO2025102268A1 (en) * | 2023-11-15 | 2025-05-22 | Lynk Pharmaceuticals Co. Ltd. | Solid state forms, pharmaceutical compositions, preparation methods and use thereof |
| TW202602438A (zh) * | 2024-03-21 | 2026-01-16 | 大陸商凌科藥業有限公司 | 藥物成分、劑型及其製備及使用方法 |
Family Cites Families (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| PT72878B (en) | 1980-04-24 | 1983-03-29 | Merck & Co Inc | Process for preparing mannich-base hydroxamic acid pro-drugs for the improved delivery of non-steroidal anti-inflammatory agents |
| AR054416A1 (es) * | 2004-12-22 | 2007-06-27 | Incyte Corp | Pirrolo [2,3-b]piridin-4-il-aminas y pirrolo [2,3-b]pirimidin-4-il-aminas como inhibidores de las quinasas janus. composiciones farmaceuticas. |
| WO2006127587A1 (en) * | 2005-05-20 | 2006-11-30 | Vertex Pharmaceuticals Incorporated | Pyrrolopyridines useful as inhibitors of protein kinase |
| WO2007007919A2 (en) * | 2005-07-14 | 2007-01-18 | Astellas Pharma Inc. | Heterocyclic janus kinase 3 inhibitors |
| JP5391587B2 (ja) * | 2008-06-18 | 2014-01-15 | 株式会社ニコン | 撮像装置 |
| US20110269740A1 (en) * | 2008-07-02 | 2011-11-03 | Ambit Biosciences Corporation | Jak kinase modulating compounds and methods of use thereof |
| JP4884570B2 (ja) | 2008-08-20 | 2012-02-29 | ファイザー・インク | ピロロ[2,3−d]ピリミジン化合物 |
| TWM370852U (en) * | 2009-07-14 | 2009-12-11 | Dragonstate Technology Co Ltd | Adapter module for combined electrical connector and electrical connector |
| RU2012107101A (ru) * | 2009-07-31 | 2013-09-10 | Байокрист Фармасьютикалз, Инк. | Производные пирроло[1,2-в] пиридазина как ингибиторы янус-киназы |
| CA2776028C (en) * | 2009-10-15 | 2015-12-01 | Pfizer Inc. | Pyrrolo[2,3-d]pyrimidine compounds |
| ES2461967T3 (es) * | 2009-12-18 | 2014-05-21 | Pfizer Inc. | Compuestos de pirrolo[2,3-d]pirimidina |
| CN102905888B (zh) | 2009-12-18 | 2016-03-09 | 塞特克技术公司 | 赋予复合制品制造及其材料中所用的材料导电性的方法 |
| EP2523957A1 (en) * | 2010-01-12 | 2012-11-21 | F. Hoffmann-La Roche AG | Tricyclic heterocyclic compounds, compositions and methods of use thereof |
| US20130317045A1 (en) * | 2010-09-01 | 2013-11-28 | Ambit Biosciences Corporation | Thienopyridine and thienopyrimidine compounds and methods of use thereof |
| WO2013007765A1 (en) | 2011-07-13 | 2013-01-17 | F. Hoffmann-La Roche Ag | Fused tricyclic compounds for use as inhibitors of janus kinases |
| AU2012295802B2 (en) | 2011-08-12 | 2017-03-30 | Nissan Chemical Industries, Ltd. | Tricyclic heterocyclic compounds and JAK inhibitors |
| EP2924026A1 (en) * | 2014-03-28 | 2015-09-30 | Novartis Tiergesundheit AG | Aminosulfonylmethylcyclohexanes as JAK inhibitors |
-
2014
- 2014-03-28 EP EP14162391.8A patent/EP2924026A1/en not_active Ceased
-
2015
- 2015-03-25 DK DK15713431.3T patent/DK3122731T3/en active
- 2015-03-25 US US15/119,800 patent/US10059709B2/en active Active
- 2015-03-25 AU AU2015238361A patent/AU2015238361B2/en active Active
- 2015-03-25 ES ES15713431T patent/ES2702474T3/es active Active
- 2015-03-25 PL PL15713431T patent/PL3122731T3/pl unknown
- 2015-03-25 JP JP2016554336A patent/JP6239139B2/ja active Active
- 2015-03-25 EP EP15713431.3A patent/EP3122731B1/en active Active
- 2015-03-25 WO PCT/EP2015/056430 patent/WO2015144773A1/en not_active Ceased
- 2015-03-25 CA CA2940321A patent/CA2940321C/en active Active
- 2015-03-25 PT PT15713431T patent/PT3122731T/pt unknown
-
2018
- 2018-08-06 US US16/055,560 patent/US10370375B2/en active Active
Also Published As
| Publication number | Publication date |
|---|---|
| PL3122731T3 (pl) | 2019-05-31 |
| DK3122731T3 (en) | 2018-12-10 |
| JP2017506656A (ja) | 2017-03-09 |
| AU2015238361B2 (en) | 2017-07-13 |
| US20170050965A1 (en) | 2017-02-23 |
| JP6239139B2 (ja) | 2017-11-29 |
| WO2015144773A1 (en) | 2015-10-01 |
| CA2940321A1 (en) | 2015-10-01 |
| EP3122731A1 (en) | 2017-02-01 |
| EP2924026A1 (en) | 2015-09-30 |
| EP3122731B1 (en) | 2018-10-10 |
| US10370375B2 (en) | 2019-08-06 |
| PT3122731T (pt) | 2018-11-30 |
| CA2940321C (en) | 2018-10-02 |
| AU2015238361A1 (en) | 2016-09-01 |
| US10059709B2 (en) | 2018-08-28 |
| US20180346465A1 (en) | 2018-12-06 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2702474T3 (es) | Ciclohexilmetanosulfonamidas | |
| AU2014400628B2 (en) | Aminopyridazinone compounds as protein kinase inhibitors | |
| TW201704231A (zh) | Jak抑制劑 | |
| JP7600119B2 (ja) | アデノシン受容体アンタゴニストとしてのチアゾロピリジン誘導体 | |
| TW201920123A (zh) | 作為腺苷受體拮抗劑之喹㗁啉衍生物 | |
| TWI905843B (zh) | 嘧啶胺類nuak抑制劑及其製備方法和用途 | |
| AU2017221193B2 (en) | Process for preparing 7H-pyrrolo (2, 3-d) pyrimidine compounds | |
| WO2022093552A9 (en) | Rev-erb agonists | |
| TW202342444A (zh) | 特定化學實體、組合物及方法 | |
| EP2825537A1 (en) | Dihydropyridopyrimidine and dihydronaphthyridine derivatives as tyrosine kinase inhibitors of especially vegf and pdgf | |
| CN114907350B (zh) | 一类含氮稠环类化合物、制备方法和用途 | |
| CN121487934A (zh) | 能够用于治疗smarca4缺失型癌症的作为smarca2抑制剂的1.6-萘啶化合物 | |
| HK40002231A (en) | Process for preparing 7h-pyrrolo [2,3-d]pyrimidine compounds | |
| HK40002231B (en) | Process for preparing 7h-pyrrolo [2,3-d]pyrimidine compounds |