ES2702891T3 - Método para producir (R)-1,1,3-Trimetil-4-aminoindano - Google Patents
Método para producir (R)-1,1,3-Trimetil-4-aminoindano Download PDFInfo
- Publication number
- ES2702891T3 ES2702891T3 ES14882034T ES14882034T ES2702891T3 ES 2702891 T3 ES2702891 T3 ES 2702891T3 ES 14882034 T ES14882034 T ES 14882034T ES 14882034 T ES14882034 T ES 14882034T ES 2702891 T3 ES2702891 T3 ES 2702891T3
- Authority
- ES
- Spain
- Prior art keywords
- trimethyl
- aminoindane
- parts
- methyl
- aminoindan
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 0 C[n]1nc(*)c(C(O)=O)c1* Chemical compound C[n]1nc(*)c(C(O)=O)c1* 0.000 description 3
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B57/00—Separation of optically-active compounds
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C209/00—Preparation of compounds containing amino groups bound to a carbon skeleton
- C07C209/82—Purification; Separation; Stabilisation; Use of additives
- C07C209/84—Purification
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C209/00—Preparation of compounds containing amino groups bound to a carbon skeleton
- C07C209/82—Purification; Separation; Stabilisation; Use of additives
- C07C209/86—Separation
- C07C209/88—Separation of optical isomers
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C209/00—Preparation of compounds containing amino groups bound to a carbon skeleton
- C07C209/82—Purification; Separation; Stabilisation; Use of additives
- C07C209/90—Stabilisation; Use of additives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C211/00—Compounds containing amino groups bound to a carbon skeleton
- C07C211/43—Compounds containing amino groups bound to a carbon skeleton having amino groups bound to carbon atoms of six-membered aromatic rings of the carbon skeleton
- C07C211/57—Compounds containing amino groups bound to a carbon skeleton having amino groups bound to carbon atoms of six-membered aromatic rings of the carbon skeleton having amino groups bound to carbon atoms of six-membered aromatic rings being part of condensed ring systems of the carbon skeleton
- C07C211/60—Compounds containing amino groups bound to a carbon skeleton having amino groups bound to carbon atoms of six-membered aromatic rings of the carbon skeleton having amino groups bound to carbon atoms of six-membered aromatic rings being part of condensed ring systems of the carbon skeleton containing a ring other than a six-membered aromatic ring forming part of at least one of the condensed ring systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D231/00—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings
- C07D231/02—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings
- C07D231/10—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D231/00—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings
- C07D231/02—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings
- C07D231/10—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D231/12—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D231/00—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings
- C07D231/02—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings
- C07D231/10—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D231/14—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D231/00—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings
- C07D231/02—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings
- C07D231/10—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D231/14—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D231/16—Halogen atoms or nitro radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/07—Optical isomers
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2602/00—Systems containing two condensed rings
- C07C2602/02—Systems containing two condensed rings the rings having only two atoms in common
- C07C2602/04—One of the condensed rings being a six-membered aromatic ring
- C07C2602/08—One of the condensed rings being a six-membered aromatic ring the other ring being five-membered, e.g. indane
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Indole Compounds (AREA)
Abstract
Un método para producir (R)-1,1,3-trimetil-4-aminoindano, que comprende las siguientes etapas (A), (B) y (C): (A): una etapa de resolución óptica del 1,1,3-trimetil-4-aminoindano con ácido D-tartárico para obtener (R)-1,1,3- trimetil-4-aminoindano y (S)-1,1,3-trimetil-4-aminoindano; (B): una etapa de racemización del (S)-1,1,3-trimetil-4-aminoindano obtenido en las etapas (A) o (C) para obtener 1,1,3-trimetil-4-aminoindano; y (C): una etapa de resolución óptica del 1,1,3-trimetil-4-aminoindano obtenido en la etapa (B) para obtener (R)- 1,1,3-trimetil-4-aminoindano y (S)-1,1,3-trimetil-4-aminoindano, en el que la etapa (B) es una etapa en la que se pone en contacto el (S)-1,1,3-trimetil-4-aminoindano obtenido en las etapas (A) o (C) con un catalizador de metal de transición para realizar la racemización.
Description
DESCRIPCIÓN
Método para producir (R)-1,1,3-Trimetil-4-aminoindano
Campo técnico
La presente invención se refiere a un método para producir (R)-1,1,3-trimetil-4-aminoindano.
Antecedentes de la técnica
El documento de patente 1 describe que el (R)-1,1,3-trimetil-4-aminoindano es útil como intermedio para la síntesis de la amida del ácido (R)-(-)-N-(1,1,3-trimetilindan-4-il)-1-metil-3-difluorometilpirazol-4-carboxílico que tiene un efecto de control sobre las enfermedades de las plantas.
Documento de la técnica anterior
Documento de patente
Documento de Patente 1: WO2011/162397
Sumario de la invención
Problemas a resolver por la invención
Ha habido una demanda de un método para producir (R)-1,1,3-trimetil-4-aminoindano con un alto rendimiento.
Medios para resolver los problemas
La presente invención incluye los siguientes aspectos de las invenciones.
[1] Un método para producir (R)-1,1,3-trimetil-4-aminoindano, que incluye las siguientes etapas (A), (B) y (C):
(A) : una etapa de resolución óptica del 1,1,3-trimetil-4-aminoindano con ácido D-tartárico para obtener (R)-1.1.3- trimetil-4-aminoindano y (S)-1,1,3-trimetil-4-aminoindano;
(B) : una etapa de racemización del (S)-1,1,3-trimetil-4-aminoindano obtenido en la etapa (A) o (C) para obtener 1.1.3- trimetil-4-aminoindano; y
(C) : una etapa de resolución óptica del 1,1,3-trimetil-4-aminoindano obtenido en la etapa (B) para obtener (R)-1.1.3- trimetil-4-aminoindano y (S)-1,1,3-trimetil-4-aminoindano, en el que la etapa (B) es una etapa en la que se pone en contacto el (S)-1,1,3-trimetil-4-aminoindano obtenido en la etapa (A) o (C) con un catalizador de metal de transición para realizar la racemización.
[2] El método de producción descrito en [1], en el que se repiten las etapas (B) y (C).
[3] El método de producción descrito en [1] o [2], en el que la etapa (C) es una etapa de resolución óptica del 1,1,3-trimetil-4-aminoindano obtenido en la etapa (B) y el 1,1,3-trimetil-4-aminoindano obtenido en una etapa diferente a la etapa (B) con ácido D-tartárico para obtener (R)-1,1,3-trimetil-4-aminoindano.
[4] El método de producción descrito en [1], que incluye las siguientes etapas (A), (B1), (D) y (E):
(A): una etapa de resolución óptica del 1,1,3-trimetil-4-aminoindano con ácido D-tartárico para obtener (R)-1.1.3- trimetil-4-aminoindano y (S)-1,1,3-trimetil-4-aminoindano;
(B'): una etapa de racemización del (S)-1,1,3-trimetil-4-aminoindano obtenido en la etapa (A) o (E) para obtener 1.1.3- trimetil-4-aminoindano;
(D) : una etapa de purificación del 1,1,3-trimetil-4-aminoindano obtenido en la etapa (B1); y
(E) : una etapa de resolución óptica del 1,1,3-trimetil-4-aminoindano obtenido en la etapa (D) para obtener (R)-1.1.3- trimetil-4-aminoindano y (S)-1,1,3-trimetil-4-aminoindano.
[5] El método de producción descrito en [4], en el que se repiten las etapas (B'), (D) y (E).
[6] El método de producción descrito en cualquiera de [1] a [5], en el que la etapa (B) o (B') es una etapa en la que se pone en contacto el (S)-1,1,3-trimetil-4-aminoindano obtenido en la etapa (A) con un catalizador de metal de transición para realizar la racemización.
[7] El método de producción descrito en una cualquiera de [1] a [6], en el que la etapa (A) es una etapa que incluye las siguientes etapas (A1), (A2), (A3) y (A4):
(A1): una etapa de mezclar 1,1,3-trimetil-4-aminoindano con ácido D-tartárico y metanol para obtener una mezcla que incluye un solvato de metanol de D-tartrato de (R)-1,1,3-trimetil-4-aminoindano y D-tartrato de (S)-1.1.3- trimetil-4-aminoindano;
(A2): una etapa de separación de una solución que contiene D-tartrato de (S)-1,1,3-trimetil-4-aminoindano y el
solvato de metanol de D-tartrato de (R)-1,1,3-trimetilo -4-aminoindano de la mezcla obtenida en la etapa (A1); (A3): una etapa de mezclar el solvato de metanol de D-tartrato de (R)-1,1,3-trimetil-4-aminoindano obtenido en la etapa (A2) y una solución acuosa de hidróxido de metal alcalino o solución acuosa de carbonato de metal alcalino para obtener (R)-1,1,3-trimetil-4-aminoindano; y
(A4): una etapa de mezclar la solución que incluye el D-tartrato de (S)-1,1,3-trimetil-4-aminoindano obtenido en la etapa (A2) y una solución acuosa de hidróxido de metal alcalino o solución acuosa de carbonato de metal alcalino para obtener (S)-1,1,3-trimetil-4-aminoindano.
[8] El método de producción descrito en [7], en el que el agua se mezcla con el sistema de reacción antes de la etapa (A2).
[9] El método de producción descrito en uno cualquiera de [4] a [8], en el que la etapa (D) es una etapa que incluye las siguientes etapas (D1), (D2), (D3) y (D4):
(D1): una etapa en la que se hace reaccionar el 1,1,3-trimetil-4-aminoindano obtenido en la etapa (B') con hidruro de hidrógeno en presencia de agua y un disolvente orgánico insoluble en agua;
(D2): una etapa de separar una capa disolviendo en la misma una salde haluro de hidrógeno de 1,1,3-trimetil-4-aminoindano incluida en la mezcla obtenida en la etapa (D1) de otra capa;
(D3): una etapa de precipitación de la sal de haluro de hidrógeno de 1,1,3-trimetil-4-aminoindano de la capa disolviendo en la misma la sal de haluro de hidrógeno de 1,1,3-trimetil-4-aminoindano obtenida en la etapa (D2); y
(D4): una etapa de extracción de la sal de haluro de hidrógeno de 1,1,3-trimetil-4-aminoindano obtenida en la etapa (D3) y hacer reaccionar la sal de haluro de hidrógeno obtenida de esta manera con una base.
[10] El método de producción descrito en [9], en el que el haluro de hidrógeno es cloruro de hidrógeno.
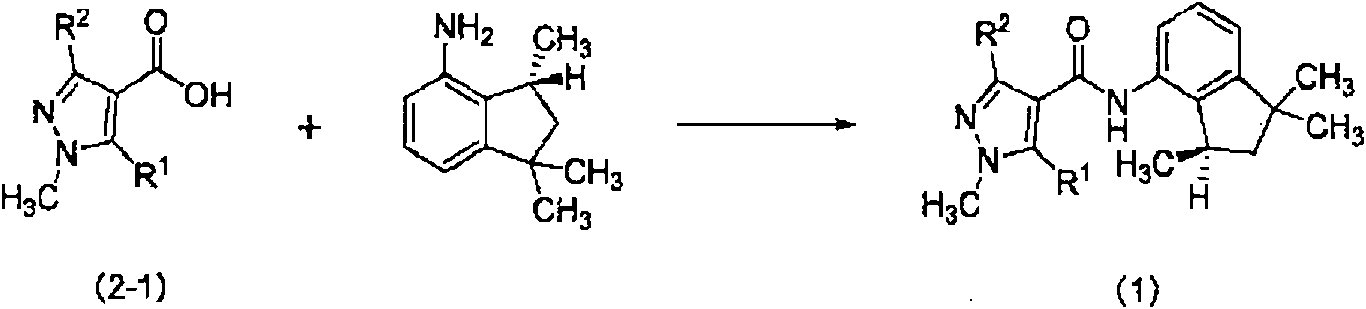
[11] Un método de producción de un compuesto representado por la siguiente fórmula (1):

en la que R1 y R2 cada uno representa independientemente un grupo alquilo que puede estar sustituido con un átomo de halógeno o un átomo de hidrógeno, que comprende además la etapa (F): una etapa en la que se hace reaccionar el (R)-1,1,3-trimetil-4-aminoindano obtenido en la etapa (C) con un compuesto representado por la fórmula (2) para obtener un compuesto representado por la fórmula (1):

en la que R1 y R2 tienen los mismos significados que antes, y R3 representa un átomo de halógeno, un grupo hidroxilo o un grupo alcoxi que puede estar sustituido con un átomo de halógeno.
[12] Un método para producir un compuesto representado por la siguiente fórmula (1):

en la que R1 y R2 cada uno representa independientemente un grupo alquilo que puede estar sustituido con un átomo de halógeno o un átomo de hidrógeno, que comprende además las siguientes etapas (G) y (H):
(G): una etapa para obtener un compuesto representado por la fórmula (4) a partir de un compuesto representado por la fórmula (3):

(en la fórmula, R1 y R2 tienen los 

(en la fórmula, R1 y R2 tienen los mismos significados que antes); y
(H): una etapa en la que se hace reaccionar el compuesto representado por la fórmula (4) obtenido en la etapa (G) con el (R)-1,1,3-trimetil-4-aminoindano obtenido en la etapa (C) en presencia de una base para obtener el compuesto representado por la fórmula (1).
[13] El método de producción descrito en [11] o [12], en el que R1 es un átomo de hidrógeno o un grupo metilo, y R2 es un grupo metilo, un grupo monofluorometilo, un grupo difluorometilo o un grupo trifluorometilo.
Efectos de la ventaja de la invención
De acuerdo con la presente invención, se puede obtener (R)-1,1,3-trimetil-4-aminoindano con un alto rendimiento.
Breve descripción de los dibujos
La Fig. 1 muestra un gráfico de XRD de la amida del ácido (R)-(-)-N-(1,1,3-trimetilindan-4-il)-1-metil-3-difluorometilpirazol-4-carboxílico en el Ejemplo 3.
La Fig. 2 muestra un gráfico de XRD de la amida del ácido (R)-(-)-N-(1,1,3-trimetilindan-4-il)-1-metil-3-difluorometilpirazol-4-carboxílico en el Ejemplo de referencia 1.
La Fig. 3 muestra un espectro de FT-Raman de la amida del ácido (R)-(-)-N-(1,1,3-trimetilindan-4-il)-1-metil-3-difluorometilpirazol-4-carboxílico en el Ejemplo de referencia. 1.
La Fig. 4 muestra un gráfico DSC/TGA de la amida del ácido (R)-(-)-N-(1,1,3-trimetilindan-4-il)-1-metil-3-difluorometilpirazol-4-carboxílico en el Ejemplo de referencia 1.
La Fig. 5 muestra un gráfico de XRD de un etanol/hidrato de la amida del ácido (R)-(-)-N-(1,1,3-trimetilindan-4-il)-1-metil-3-difluorometilpirazol-4-carboxílico en el Ejemplo de referencia 2.
La Fig. 6 muestra un espectro de FT-Raman de un etanol/hidrato de la amida del ácido (R)-(-)-N-(1,1,3-trimetilindan-4-il)-1-metil-3-difluorometilpirazol-4-carboxílico en el Ejemplo de referencia 2.
La Fig. 7 muestra un gráfico de DSC/TGA de un etanol/hidrato de la amida del ácido (R)-(-)-N-(1,1,3-trimetilindan-4-il)-1-metil-3-difluorometilpirazol-4-carboxílico en el Ejemplo de referencia 2.
La Fig. 8 muestra un gráfico de XRD de la amida del ácido (R)-(-)-N-(1,1,3-trimetilindan-4-il)-1-metil-3-difluorometilpirazol-4-carboxílico en el Ejemplo de referencia 4.
La Fig. 9 muestra un espectro de FT-Raman de la amida del ácido (R)-(-)-N-(1,1,3-trimetilindan-4-il)-1-metil-3-difluorometilpirazol-4-carboxílico en el Ejemplo de referencia 4.
La Fig. 10 muestra un gráfico de DSC/TGA de la amida del ácido (R)-(-)-N-(1,1,3-trimetilindan-4-il)-1-metil-3-difluorometilpirazol-4-carboxílico en el Ejemplo de referencia. 4.
La Fig. 11 muestra un gráfico de XRD de un hidrato de la amida del ácido (R)-(-)-N-(1,1,3-trimetilindan-4-il)-1-metil-3- difluorometilpirazol-4-carboxílico en el Ejemplo de referencia 5.
La Fig. 12 muestra un espectro de FT-Raman de un hidrato de la amida del ácido (R)-(-)-N-(1,1,3-trimetilindan-4-il)-1-metil-3-difluorometilpirazol-4-carboxílico en el Ejemplo de referencia 5.
La Fig. 13 muestra un gráfico de DSC/TGA de un hidrato de la amida del ácido (R)-(-)-N-(1,1,3-trimetilindan-4-il)-1-metil-3-difluorometilpirazol-4-carboxílico en el Ejemplo de referencia 5.
La Fig. 14 muestra gráfico de XRD de un 2-metoxietanol/hidrato de la amida del ácido (R)-(-)-N-(1,1,3-trimetilindan-4- il)-1-metil-3-difluorometilpirazol-4-carboxílico en el Ejemplo de referencia 7.
La Fig. 15 muestra un espectro de FT-Raman de un 2-metoxietanol/hidrato de la amida del ácido (R)-(-)-N-(1,1,3-trimetilindan-4-il)-1-metil-3-difluorometilpirazol-4-carboxílico en el Ejemplo de referencia 7.
La Fig. 16 muestra un gráfico de DSC/TGA de un 2-metoxietanol/hidrato de la amida del ácido (R)-(-)-N-(1,1,3-trimetilindan-4-il)-1-metil-3-difluorometilpirazol-4-carboxílico en el Ejemplo de referencia 7.
La Fig. 17 muestra un gráfico de XRD de un 1-propanol/ciclohexano/hidrato de la amida del ácido (R)-(-)-N-(1,1,3-trimetilindan-4-il)-1-metil-3-difluorometilpirazol-4-carboxílico en el Ejemplo de referencia 9.
La Fig. 18 muestra un espectro de RT-Raman de un 1-propanol/ciclohexano/hidrato de la amida del ácido (R)-(-)
N-(1,1,3-trimetilindan-4-il)-1-metil-3-difluorometilpirazol-4-carboxílico en el Ejemplo de referencia 9.
La Fig. 19 muestra un gráfico de DSC/TGA de un 1-propanol/ciclohexano/hidrato de la amida del ácido (R)-(-)-N-(1,1,3-trimetilindan-4-il)-1-metil-3-difluorometilpirazol-4-carboxílico en el Ejemplo de referencia 9.
La Fig. 20 muestra un gráfico de XRD de un tetrahidrofurano/hidrato de la amida del ácido (R)-(-)-N-(1,1,3-trimetilindan-4-il)-1-metil-3-difluorometilpirazol-4-carboxílico en el Ejemplo de referencia 11.
La Fig. 21 muestra un espectro de FT-Raman de un tetrahidrofurano/hidrato de la amida del ácido (R)-(-)-N-(1,1,3-trimetilindan-4-il)-1-metil-3-difluorometilpirazol-4-carboxílico en el Ejemplo de referencia 11.
La Fig. 22 muestra un gráfico de DSC/TGA de un tetrahidrofurano/hidrato de la amida del ácido (R)-(-)-N-(1,1,3-trimetilindan-4-il)-1-metil-3-difluorometilpirazol-4-carboxílico en el Ejemplo de referencia 11.
La Fig. 23 muestra un gráfico de XRD de un solvato de dimetilsulfóxido de la amida del ácido (R)-(-)-N-(1,1,3-trimetilindan-4-il)-1-metil-3-difluorometilpirazol-4-carboxílico en Ejemplo de referencia 13.
La Fig. 24 muestra un espectro de FT-Raman de un solvato de dimetilsulfóxido de la amida del ácido (R)-(-)-N-(1,1,3-trimetilindan-4-il)-1-metil-3-difluorometilpirazol-4-carboxílico en el Ejemplo de referencia 13.
La Fig. 25 muestra un gráfico de DSC/TGA de un solvato de dimetilsulfóxido de la amida del ácido (R)-(-)-N-(1,1,3-trimetilindan-4-il)-1-metil-3-difluorometilpirazol-4-carboxílico en el Ejemplo de referencia 13.
La Fig. 26 muestra un gráfico de XRD de un solvato de xileno de la amida del ácido (R)-(-)-N-(1,1,3-trimetilindan-4-il)-1-metil-3-difluorometilpirazol-4-carboxílico en Ejemplo de referencia 14.
La Fig.27 muestra un gráfico de TG/DTA de un solvato de xileno de la amida del ácido (R)-(-)-N-(1,1,3-trimetilindan-4-il)-1-metil-3-difluorometilpirazol-4-carboxílico en el Ejemplo de referencia 14.
Descripción de las realizaciones
La presente invención se refiere a un método para producir (R)-1,1,3-trimetil-4-aminoindano, que incluye las etapas (A), (B) y (C).
<Etapa (A)>
El 1,1,3-trimetil-4-aminoindano está representado por la siguiente fórmula. Los ejemplos del 1,1,3-trimetil-4-aminoindano incluyen 1,1,3-trimetil-4-aminoindano que tiene generalmente una pureza óptica de aproximadamente 0 % ee a 25 % ee, y el 1,1,3-trimetil-4-aminoindano tiene preferiblemente una pureza óptica de aproximadamente 0 % ee a 10 % ee, y más preferiblemente tiene una pureza óptica de aproximadamente 0 % ee a 5 % ee.

El 1,1,3-trimetil-4-aminoindano se puede producir de acuerdo con el método descrito en J. Chem. Soc. (C), 514 (1966), por ejemplo.
El 1,1,3-trimetil-4-aminoindano puede obtenerse hidrogenando un compuesto representado por la fórmula (6) para obtener un compuesto representado por la fórmula (7); y haciendo reaccionar el compuesto representado por la fórmula (7) con un ácido:

El 1,1,3-trimetil-4-aminoindano puede obtenerse haciendo reaccionar un compuesto representado por la fórmula (6) con un reactivo de protección tal como ácido acético anhidro para obtener un compuesto representado por la fórmula
(6) que tiene su átomo de nitrógeno protegido con un grupo protector; hidrogenar el compuesto representado por la fórmula (6) que tiene su átomo de nitrógeno protegido con un grupo protector para obtener un compuesto representado por la fórmula (7) que tiene su átomo de nitrógeno protegido con un grupo protector representado por la fórmula (7); y haciendo reaccionar el compuesto representado por la fórmula (7) que tiene su átomo de nitrógeno protegido con un grupo protector representado por la fórmula (7) con un ácido.
Como ácido, se prefiere el ácido sulfúrico. La concentración del ácido sulfúrico es generalmente del 90 % en peso al 98 % en peso, y en cuanto al rendimiento, la concentración es preferiblemente del 92 % en peso al 97 % en peso. La reacción del compuesto representado por la fórmula (7) con un ácido se lleva a cabo sin un disolvente y la temperatura de reacción es generalmente de 20 °C a 80 °C.
Una vez completada la reacción, la mezcla de reacción obtenida y el agua se mezclan, la mezcla obtenida se neutraliza con un álcali y se extrae con un disolvente orgánico insoluble en agua, como tolueno, obteniendo así una solución que contiene 1,1,3-trimetil-4-aminoindano. La pureza del 1,1,3-trimetil-4-aminoindano es generalmente del 60 % al 97 %. El compuesto representado por la fórmula (6) se puede obtener por despolimerización de un oligómero del compuesto representado por la fórmula (6).
Los ejemplos del oligómero del compuesto representado por la fórmula (6) incluyen Antigen FR (fabricado por Sumitomo Chemical Co., Ltd.) y Antigen RD (fabricado por Sumitomo Chemical Co., Ltd.).
La despolimerización se lleva a cabo haciendo reaccionar el compuesto representado por la fórmula (6) con un catalizador ácido.
Los ejemplos del catalizador ácido incluyen ácido clorhídrico, ácido sulfúrico, ácido nítrico, ácido tetrafluorobórico, ácido p-toluenosulfónico, monohidrato del ácido p-toluenosulfónico y el catalizador ácido es preferiblemente monohidrato de ácido p-toluenosulfónico.
La cantidad de catalizador ácido a usar es generalmente de 0,1 partes en peso a 30 partes en peso, preferiblemente de 0,1 partes en peso a 20 partes en peso, y más preferiblemente de 1 parte en peso a 10 partes en peso, con respecto a 100 partes en peso del oligómero del compuesto representado por la fórmula (6).
La temperatura de reacción es generalmente de 100 °C a 250 °C, preferiblemente de 120 °C a 230 °C, y más preferiblemente de 140 °C a 200 °C.
La reacción se puede llevar a cabo a una presión normal o a presión reducida, y la reacción se lleva a cabo preferiblemente a presión reducida. En el caso de que la reacción se lleve a cabo a presión reducida, la presión es habitualmente de 0,1 kPa a 10 kPa, preferiblemente de 0,3 kPa a 7 kPa, y más preferiblemente de 0,5 kPa a 5 kPa. La despolimerización se lleva a cabo preferiblemente evaporando el compuesto obtenido representado por la fórmula (6) del sistema de reacción. El compuesto así obtenido tiene una pureza relativamente alta.
La etapa (A) es preferiblemente una etapa que incluye las etapas (A1), (A2), (A3) y (A4). Más preferiblemente, el agua se mezcla en el sistema de reacción antes de la etapa (A2).
<Etapa (A1)>
Los ejemplos de ácido D-tartárico generalmente incluyen productos comercializados.
La cantidad de ácido D-tartárico a usar es generalmente de 0,3 moles a 0,7 moles, preferiblemente de 0,4 moles a 0,6 moles, y más preferiblemente de 0,45 moles a 0,55 moles, con respecto a 1 mol de 1,1,3-trimetil-4-aminoindano. La cantidad de metanol a usar es generalmente de 0,5 partes en peso a 3 partes en peso, preferiblemente de 0,6 partes en peso a 2 partes en peso, y más preferiblemente de 0,8 partes en peso a 2 partes en peso, con respecto a 1 parte en peso de 1,1,3-trimetil-4-aminoindano.
La etapa (A1) se puede llevar a cabo en presencia de agua y un disolvente distinto de metanol y agua, según sea necesario, además del metanol. Los ejemplos del disolvente distinto del metanol y el agua incluyen un disolvente alcohólico distinto del metanol, tal como etanol y 2-propanol; un disolvente éter tal como tetrahidrofurano; un disolvente nitrilo tal como acetonitrilo; un disolvente éster tal como acetato de etilo; un disolvente hidrocarburo aromático tal como tolueno, xileno y etilbenceno; un disolvente hidrocarbonado aromático halogenado tal como monoclorobenceno; un disolvente hidrocarburo alifático tal como heptano y hexano; y un disolvente hidrocarburo alicíclico tal como ciclopentano y ciclohexano, y el disolvente es preferiblemente el disolvente hidrocarburo aromático.
Estos disolventes pueden combinarse y la cantidad total de los disolventes distintos del metanol y el agua que se va a usar es generalmente de 10 partes en peso o menos con respecto a 1 parte en peso de 1,1,3-trimetil-4-aminoindano.
La cantidad de agua a usar es generalmente de 0,01 partes en peso a 0,15 partes en peso, y preferiblemente de 0,01 partes en peso a 0,1 partes en peso, con respecto a 1 parte en peso de 1,1,3-trimetil-4-aminoindano.
La etapa (A1) se lleva a cabo preferiblemente mezclando 1,1,3-trimetil-4-aminoindano, ácido D-tartárico, metanol y agua.
La temperatura de mezclado es habitualmente de 20 °C a 70 °C, y preferiblemente de 30 °C a 50 °C.
Para el orden de mezcla, 1,1,3-trimetil-4-aminoindano, ácido D-tartárico y metanol se pueden mezclar a la vez; o el ácido D-tartárico y el metanol se pueden mezclar y luego se puede agregar el 1,1,3-trimetil-4-aminoindano a la mezcla así obtenida. Se puede añadir una mezcla de ácido D-tartárico y metanol al 1,1,3-trimetil-4-aminoindano. Alternativamente, se puede mezclar 1,1,3-trimetil-4-aminoindano y metanol, y se puede agregar ácido D-tartárico a una mezcla así obtenida. Sobre todo, el ácido D-tartárico se agrega preferiblemente a una mezcla de 1,1,3-trimetil-4-aminoindano y metanol.
Para la etapa (A1), en el caso de mezclar agua, el orden de mezcla es el siguiente: 1,1,3-trimetil-4-aminoindano, ácido D-tartárico, metanol y agua pueden mezclarse de una vez; o se pueden mezclar ácido D-tartárico, metanol y agua, y luego se puede agregar 1,1,3-trimetil-4-aminoindano a una mezcla así obtenida. Se puede agregar una mezcla de ácido D-tartárico, metanol y agua al 1,1,3-trimetil-4-aminoindano; o se pueden mezclar ácido D-tartárico, metanol y 1.1.3- trimetil-4-aminoindano y luego se puede agregar agua a la mezcla así obtenida. Además, se puede mezclar 1.1.3- trimetil-4-aminoindano, metanol y agua y se puede agregar ácido D-tartárico a la mezcla así obtenida. Sobre todo, el ácido D-tartárico se agrega preferiblemente a una mezcla de 1,1,3-trimetil-4-aminoindano, metanol y agua.
La adición se puede llevar a cabo en una porción o en porciones divididas. En el caso de agregar ácido D-tartárico a una mezcla de 1,1,3-trimetil-4-aminoindano y metanol, el ácido D-tartárico se puede agregar en una porción, pero preferiblemente se agregan en porciones divididas.
Mezclando 1,1,3-trimetil-4-aminoindano, ácido D-tartárico y metanol, se obtiene una mezcla que incluye un solvato de metanol de D-tartrato de (R)-1,1,3-trimetil-4-aminoindano, y (S)-1,1,3-trimetil-4-aminoindano y D-tartrato del mismo. Dependiendo de la cantidad de metanol que se use y de la temperatura de mezcla, en algunos casos, una parte del solvato de metanol del D-tartrato de (R)-1,1,3-trimetil-4-aminoindano así producido puede precipitarse en la mezcla.
El (R)-1,1,3-trimetil-4-aminoindano se representa mediante la siguiente fórmula:

Después de la etapa (A1), se puede incluir una etapa en la que se elimine el metanol y, según sea necesario, una parte del disolvente que no sea metanol de la mezcla obtenida en la etapa (A1). La eliminación se realiza habitualmente concentrando la mezcla obtenida a presión reducida. Después de eliminar el metanol y, según sea necesario, se puede agregar una parte del disolvente que no sea metanol, metanol, agua y un disolvente que no sea metanol y agua a un residuo de la mezcla obtenida en la etapa (A1).
<Etapa (A2)>
Al enfriar la mezcla obtenida en la etapa (A1), el solvato de metanol de D-tartrato de (R)-1,1,3-trimetil-4-aminoindano puede precipitar y el solvato de metanol precipitado se puede filtrar para separar el solvato de metanol de D-tartrato de (R)-1,1,3-trimetil-4-aminoindano y la solución que incluye (S)-1, 1,3-trimetil-4-aminoindano y D-tartrato del mismo.
La temperatura después del enfriamiento es una temperatura más baja que la temperatura de mezcla en la etapa (A1), y es preferiblemente de -20 °C a 30 °C, y más preferiblemente de -10 °C a 20 °C.
La velocidad de enfriamiento es habitualmente de 1 °C/hora a 10 °C/hora, y enfriando la mezcla obtenida en la etapa (A1) con la velocidad de enfriamiento, los cristales del solvato de metanol de D-tartrato del (R)-1,1,3-trimetil-4-aminoindano se precipita con alta pureza óptica. La velocidad de enfriamiento es preferiblemente de 1 °C/hora a 8 °C/hora, y más preferiblemente de 3 °C/hora a 6 °C/hora.
La mezcla obtenida en la etapa (A1) se puede mezclar con agua mientras se enfría, y la refrigeración se puede detener una vez y se puede agregar agua, y luego se vuelve a enfriar.
El solvato de metanol extraído del D-tartrato de (R)-1,1,3-trimetil-4-aminoindano se lava generalmente con al menos uno seleccionado del grupo que consiste en metanol, agua y el disolvente que no sea metanol y agua y cuando sea necesario, se puede secar.
Al mezclar con agua, se mejora la propiedad de filtrado durante la extracción de los cristales del solvato de metanol de D-tartrato de (R)-1,1,3-trimetil-4-aminoindano.
<Etapa (A3)>
Los ejemplos del hidróxido de metal alcalino incluyen hidróxido de sodio e hidróxido de potasio, y los ejemplos del carbonato de metal alcalino incluyen carbonato de sodio.
La cantidad de la solución acuosa de hidróxido de metal alcalino o solución acuosa de carbonato de metal alcalino que se va a utilizar, en términos de metales alcalinos, es generalmente de 0,5 a 3 moles, con respecto a 1 mol de ácido D-tartárico utilizado en la etapa (A1). La temperatura de mezcla es habitualmente de 10 °C a 80 °C.
La concentración del hidróxido de metal alcalino o del carbonato de metal alcalino en la solución acuosa de hidróxido de metal alcalino o la solución acuosa de carbonato de metal alcalino es preferiblemente tal que el pH de la capa acuosa después de la mezcla es 9 o más, y más preferiblemente tal que el pH es de 10 a 14.
La mezcla de la solución acuosa de hidróxido de metal alcalino o solución acuosa de carbonato de metal alcalino se puede llevar a cabo en presencia de un disolvente orgánico. Los ejemplos del disolvente orgánico incluyen un disolvente hidrocarbonado aromático tal como tolueno, xileno y etilbenceno; un disolvente hidrocarbonado aromático halogenado tal como monoclorobenceno; un disolvente hidrocarburo alifático tal como heptano y hexano; un disolvente hidrógenocarbonato alicíclico tal como ciclopentano y ciclohexano; un disolvente éter tal como éter dietílico y terc-butil metil éter; y un disolvente éster tal como acetato de etilo.
La cantidad de disolvente orgánico a usar es generalmente de 10 partes en peso o menos con respecto a 1 parte en peso del solvato de metanol.
Para el orden de mezcla, el solvato de metanol del D-tartrato de (R)-1,1,3-trimetil-4-aminoindano, el hidróxido de metal alcalino o la solución acuosa de carbonato de metal alcalino, y si es necesario, el disolvente orgánico se puede mezclar de una vez; o una mezcla del solvato de metanol y, si es necesario, se puede mezclar el disolvente orgánico, el hidróxido de metal alcalino o la solución acuosa de carbonato de metal alcalino. Alternativamente, el solvato de metanol se puede agregar a una mezcla de la solución acuosa de hidróxido de metal alcalino o solución acuosa de carbonato de metal alcalino y, según sea necesario, el disolvente orgánico. Sobre todo, el solvato de metanol se agrega preferiblemente a una mezcla del disolvente orgánico y el hidróxido de metal alcalino o la solución acuosa de carbonato de metal alcalino.
Después de completar la mezcla, la mezcla obtenida puede someterse a separación líquida para obtener el (R)-1,1,3-trimetil-4-aminoindano.
<Etapa (A4)>
La solución que incluye (S)-1,1,3-trimetil-4-aminoindano obtenida en la etapa (A2) y D-tartrato del mismo se concentra, según sea necesario, y luego se mezcla con una solución acuosa de hidróxido de metal alcalino o solución acuosa de carbonato metal alcalino.
La etapa (A4) es igual que la etapa (A3), excepto que se usa la solución que incluye (S)-1,1,3-trimetil-4-aminoindano obtenido en la etapa (A2) y D-tartrato del mismo en lugar del solvato de metanol del D-tartrato de (R)-1,1,3-trimetil-4-aminoindano obtenido en la etapa (A2).
La pureza óptica del (S)-1,1,3-trimetil-4-aminoindano obtenido en la etapa (A4) es generalmente de 20 % ee a 100 % ee, y preferiblemente de 50 % ee a 100 % ee.
<Etapa (B)>
La etapa (B) es preferiblemente una etapa en la que se pone en contacto el (S)-1,1,3-trimetil-4-aminoindano obtenido en la etapa (A) o (C) con un catalizador de metal de transición para realizar la racemización.
La pureza óptica del 1,1,3-trimetil-4-aminoindano obtenido en la etapa (B) es habitualmente de 0 % ee a 25 % ee, preferiblemente de 0 % ee a 10 % ee, y más preferiblemente de 0 % ee al 5 % ee.
Los ejemplos del catalizador de metal de transición incluyen un catalizador de platino tal como negro de platino, platino coloidal, óxido de platino y sulfato de platino-bario; un catalizador de níquel tal como níquel reducido, níquel
Urushihara, formiato de níquel, níquel Raney y tierra de diatomeas de níquel; un catalizador de paladio tal como paladio-carbono, paladio- carbonato de calcio, paladio-alúmina y paladio-platino-carbono; un catalizador de cobalto tal como cobalto Raney; un catalizador de hierro tal como hierro Raney; y un catalizador de cobre tal como cromita de cobre, y pueden utilizarse al menos dos tipos de catalizadores de metales de transición. El catalizador de metal de transición es preferiblemente un catalizador de paladio, y más preferiblemente paladio-carbono, paladio-alúmina, o paladio-platino-carbono.
El catalizador de metal de transición puede estar soportado sobre un soporte. Los ejemplos de soporte incluyen carbón activado, sílice, zeolita y Celite (marca registrada).
El catalizador de metal de transición se puede usar después de hacer que el catalizador de metal de transición y el hidrógeno coexistan para permitir que el catalizador de metal de transición absorba hidrógeno, y se prefiere el catalizador de metal de transición que tiene hidrógeno absorbido.
La cantidad del catalizador de metal de transición que se va a usar es generalmente de 0,0001 partes en peso a 1 parte en peso, y preferiblemente de 0,0005 partes en peso a 0,5 partes en peso, con respecto a 1 parte en peso de (S)-1,1,3-trimetil-4-aminoindano.
El contacto entre el catalizador de metal de transición y el (S)-1,1,3-trimetil-4-aminoindano se puede llevar a cabo en presencia de un disolvente o sin un disolvente.
Los ejemplos del disolvente incluyen un disolvente aromático tal como benceno, clorobenceno, tolueno, xileno, etilbenceno y piridina; un disolvente hidrocarburo que contiene halógeno, tal como cloroformo y diclorometano; un disolvente éster tal como acetato de etilo; un disolvente cetona tal como acetona, metil etil cetona y metil isobutil cetona; un disolvente éter tal como 1,2-dimetoxietano, dietilenglicol dimetil éter, polietilenglicol, tetrahidrofurano y dioxano; un disolvente nitrilo tal como acetonitrilo y propilnitrilo; un disolvente sulfóxido tal como dimetilsulfóxido; un disolvente amida tal como dimetilacetamida y N-metilpirrolidona; un disolvente alcohólico tal como metanol, etanol y 2-propanol; un disolvente acuoso tal como agua, una solución acuosa de hidróxido de sodio y amoníaco acuoso; y una mezcla de disolventes de los mismos, y el disolvente es preferiblemente un disolvente alcohólico, y más preferiblemente 2-propanol.
La cantidad de disolvente a usar es habitualmente 100 partes en peso o menos, y preferiblemente 5 partes en peso o menos, con respecto a 1 parte en peso de (S)-1,1,3-trimetil-4-aminoindano.
El contacto entre el catalizador de metal de transición y el (S)-1,1,3-trimetil-4-aminoindano también se puede llevar a cabo en presencia de un aditivo que será una fuente de hidrógeno.
Los ejemplos del aditivo incluyen ácido fórmico; formiatos tales como formiato de amonio y formiato de sodio; ciclohexeno; un compuesto de ciclohexeno tal como 3-metil-1-ciclohexeno y 4-metil-1-ciclohexeno; 1,3-ciclohexadieno; 1,4-ciclohexadieno; un compuesto octalino tal como 1,2,3,4,4aa, 5,8,8ap-octahidronaftaleno, 1,2,3,4,5,6,7,8-octahidronaftaleno, 1-metiloctalina y trans-2-metiloctalina; tetralina; 1,6-dimetiltetralina; 6-metiltetralina; limoneno; pineno; 3-careno; felandreno; terpinoleno; 1-p-menteno; cadaleno; pulegona; selineno; un compuesto alcohólico tal como metanol, etanol, 2-propanol y ciclohexanol; o una mezcla de los mismos, preferiblemente un compuesto de ciclohexeno, y más preferiblemente ciclohexeno.
La cantidad de aditivo a usar es generalmente de 10 partes en peso o menos, preferiblemente de 5 partes en peso o menos, y más preferiblemente de 2 partes en peso o menos, con respecto a 1 parte en peso de (S)-1,1,3-trimetil-4-aminoindano.
Se puede usar un compuesto en combinación con un aditivo que será una fuente de hidrógeno y un disolvente. El compuesto en combinación con un aditivo que será una fuente de hidrógeno y un disolvente es preferiblemente un compuesto alcohólico, y más preferiblemente 2-propanol.
La etapa (B) es más preferiblemente una etapa en la que un catalizador de metal de transición y (S)-1,1,3-trimetil-4-aminoindano se mezclan en presencia de hidrógeno, y una mezcla así obtenida se calienta para la racemización.
El contacto entre el catalizador de metal de transición y el (S)-1,1,3-trimetil-4-aminoindano se puede llevar a cabo en un recipiente sellado, como un autoclave, o en un recipiente abierto, como un matraz. El contacto entre el catalizador de metal de transición y el (S)-1,1,3-trimetil-4-aminoindano se puede llevar a cabo al aire, en una atmósfera de nitrógeno o en una atmósfera de hidrógeno, y preferiblemente en una atmósfera de nitrógeno o en una atmósfera de hidrógeno.
La temperatura de contacto entre el catalizador de metal de transición y el (S)-1,1,3-trimetil-4-aminoindano es generalmente de 20 °C a 250 °C, preferiblemente de 80 °C a 200 °C, y más preferiblemente de 100 °C a 190 °C.
El contacto entre el catalizador de metal de transición y el (S)-1,1,3-trimetil-4-aminoindano se realiza preferiblemente mezclándolos de 50 °C a 80 °C y calentando a 100 °C a 200 °C, y preferiblemente de 150 °C a 200 °C en una atmósfera
de hidrógeno, y más preferiblemente se lleva a cabo mezclándolos de 50 °C a 80 °C en una atmósfera de hidrógeno, reemplazando el hidrógeno con nitrógeno y luego calentando a 100 °C a 200 °C, y preferiblemente de 150 °C a 200 °C.
El tiempo de contacto entre el catalizador de metal de transición y el (S)-1,1,3-trimetil-4-aminoindano es generalmente de 0,1 horas a 100 horas, y preferiblemente de 0,1 horas a 24 horas.
Al eliminar el catalizador de la mezcla obtenida después del contacto por filtración o similar, se puede extraer el 1,1,3-trimetil-4-aminoindano. El 1,1,3-trimetil-4-aminoindano obtenido también se puede purificar por métodos conocidos tales como concentración, extracción, disolución de transferencia, recristalización y cromatografía.
El catalizador eliminado por filtración o similar se puede recuperar y usar nuevamente para la producción de 1,1,3-trimetil-4-aminoindano. Los ejemplos del método de recuperación incluyen un método de soporte de catalizador sobre un soporte.
El catalizador recuperado se lava preferiblemente con un disolvente. Los ejemplos del disolvente incluyen un disolvente alcohólico tal como metanol, etanol, 2-propanol y butanol; una solución acuosa alcalina tal como hidróxido de sodio, hidróxido de potasio, hidróxido de litio y carbonato de sodio; agua; o una mezcla de disolvente de los mismos.
<Etapa (C)>
La etapa (C) es igual que la etapa de obtener (R)-1,1,3-trimetil-4-aminoindano y (S)-1,1,3-trimetil-4-aminoindano en la etapa (A) excepto que se usa el 1,1,3-trimetil-4-aminoindano obtenido en la etapa (B) en lugar del 1,1,3-trimetil-4-aminoindano.
La etapa (C) es preferiblemente una etapa en la que el 1,1,3-trimetil-4-aminoindano obtenido en la etapa (B) y el 1,1,3-trimetil-4-aminoindano obtenido en una etapa diferente a la etapa (B) se resuelven ópticamente para obtener (R)-1,1,3-trimetil-4-aminoindano.
Repitiendo la etapa (B) y la etapa (C), se puede producir (R)-1,1,3-trimetil-4-aminoindano con un mayor rendimiento.
El método de producción de la presente invención incluye preferiblemente las etapas (A), (B'), (D) y (E). La etapa (A) es como se describe anteriormente.
<Etapa (B')>
La etapa (B') es igual que la etapa de obtener 1,1,3-trimetil-4-aminoindano en la etapa (B) excepto que se usa el (S)-1.1.3- trimetil-4-aminoindano obtenido en la etapa (A) o etapa (E) en lugar del (S)-1,1,3-trimetil-4-aminoindano obtenido en la etapa (A) o etapa (C).
<Etapa (D)>
La etapa (D) es preferiblemente una etapa que incluye las etapas (D1), (D2), (D3) y (D4).
La etapa (D) puede ser una etapa de purificación del 1,1,3-trimetil-4-aminoindano obtenido en la etapa (B') y el 1,1,3-trimetil-4-aminoindano obtenido en la etapa distinta de la etapa (B').
<Etapa (D1)>
Los ejemplos del disolvente orgánico insoluble en agua incluyen un disolvente hidrocarburo alifático tal como hexano y heptano; un disolvente hidrocarburo aromático tal como tolueno, xileno y etilbenceno; un disolvente éster hidrófobo tal como acetato de etilo; un disolvente éter hidrófobo tal como éter dietílico, éter terc-butilmetílico y metilciclopentil éter; y un disolvente hidrófobo de cetona tal como metil isobutil cetona; y se prefieren el disolvente hidrocarburo alifático y el disolvente hidrocarburo aromático, y se prefiere el disolvente hidrocarburo aromático.
La relación entre la cantidad de agua y la cantidad del disolvente orgánico insoluble en agua que se va a utilizar (relación en peso; agua/disolvente orgánico insoluble en agua) es generalmente de 1/99 a 99/1, preferiblemente de 5/95 a 95/5, y más preferiblemente de 10/90 a 90/10.
Los ejemplos del haluro de hidrógeno incluyen cloruro de hidrógeno, bromuro de hidrógeno y yoduro de hidrógeno, y el haluro de hidrógeno es preferiblemente cloruro de hidrógeno o bromuro de hidrógeno, y más preferiblemente cloruro de hidrógeno. El haluro de hidrógeno se puede usar tal cual y puede estar en forma de una solución acuosa.
La cantidad de haluro de hidrógeno que se va a utilizar es generalmente de 1 mol a 2 moles con respecto a 1 mol de 1.1.3- trimetil-4-aminoindano.
La reacción del 1,1,3-trimetil-4-aminoindano con haluro de hidrógeno se lleva a cabo generalmente mezclando 1,1,3-trimetil-4-aminoindano con haluro de hidrógeno.
La temperatura de reacción es generalmente de 0 °C a 100 °C, preferiblemente de 5 °C a 90 °C, y más preferiblemente de 10 °C a 80 °C.
El tiempo de reacción es generalmente de 0,1 horas a 24 horas, preferiblemente de 0,1 horas a 12 horas, y más preferiblemente de 0,1 horas a 6 horas.
<Etapa (D2)>
La separación se lleva a cabo preferiblemente dejando reposar la mezcla obtenida en la etapa (D1) y sometiendo la mezcla a un tratamiento de separación de líquidos.
En el caso de que una capa que tiene una sal de haluro de hidrógeno de 1,1,3-trimetil-4-aminoindano separado sea una capa orgánica, la capa orgánica se lava con agua, según sea necesario. En el caso de que una capa en la que se disuelva la sal de haluro de hidrógeno de 1,1,3-trimetil-4-aminoindano separado sea una capa acuosa, la capa acuosa se lava con el disolvente orgánico insoluble en agua, según sea necesario.
La cantidad de haluro de hidrógeno en la etapa (D1) es 1,15 moles o más, y preferiblemente 1,2 moles o más, con respecto a 1 mol de 1,1,3-trimetil-4-aminoindano, y la sal de haluro de hidrógeno de 1,1,3-trimetil-4-aminoindano generalmente se disuelve en la capa orgánica. Si la cantidad de haluro de hidrógeno que se va a usar es inferior a 1,15 moles con respecto a 1 mol de 1,1,3-trimetil-4-aminoindano, la sal de haluro de hidrógeno de 1,1,3-trimetil-4-aminoindano se disuelve habitualmente en la capa acuosa. De este modo, controlando la cantidad de haluro de hidrógeno en la etapa (D1), se puede controlar la capa en la que se disuelve la sal de haluro de hidrógeno de 1,1,3-trimetil-4-aminoindano.
En el caso de que la concentración de ion haluro de la mezcla obtenida en la etapa (D1) en la capa acuosa sea de 0,8 moles/l o más, la sal de haluro de hidrógeno de 1,1,3-trimetil-4-aminoindano generalmente se disuelve en una capa orgánica. En el caso de que la concentración de ion haluro de la mezcla obtenida en la etapa (D1) en la capa acuosa sea inferior a 0,8 moles/l, la sal de haluro de hidrógeno de 1,1,3-trimetil-4-aminoindano se disuelve generalmente en una capa acuosa. Por lo tanto, controlando la concentración de ion haluro en la capa acuosa de la mezcla, se puede controlar una capa en la que se disuelve la sal de haluro de hidrógeno de 1,1,3-trimetil-4-aminoindano. Los ejemplos del método para controlar la concentración de ion haluro en la capa acuosa incluyen un método en el que se mezclan la mezcla obtenida en la etapa (D1) y un haluro inorgánico soluble en agua tal como cloruro de sodio.
La temperatura para la separación de la capa en la que se disuelve la sal de haluro de hidrógeno de 1,1,3-trimetil-4-aminoindano y la otra capa es generalmente de 0 °C a 100 °C, preferiblemente de 5 °C a 90 °C, y más preferiblemente de 10 °C a 80 °C.
<Etapa (D3)>
La capa en la que se disuelve la sal de haluro de hidrógeno de 1,1,3-trimetil-4-aminoindano se puede usar tal cual o puede concentrarse y luego enfriarse para extraer la sal de haluro de hidrógeno de 1,1,3-trimetil- 4-aminoindano.
La temperatura de enfriamiento es preferiblemente una temperatura que es más baja que la temperatura de separación de la etapa (D2) en 5 °C o más, más preferiblemente de -15 °C a 50 °C, aún más preferiblemente de -5 °C a 40 °C, y de manera particularmente preferible de 0 °C a 30 °C. El tiempo de enfriamiento es habitualmente de 1 minuto a 24 horas.
<Etapa (D4)>
La sal de haluro de hidrógeno precipitada de 1,1,3-trimetil-4-aminoindano puede extraerse filtrando una mezcla en la que precipita la sal. La sal de haluro de hidrógeno de 1,1,3-trimetil-4-aminoindano extraída se lava con un disolvente, según sea necesario.
Ejemplos de la base incluyen amoníaco; un hidróxido de metal alcalino tal como hidróxido de litio, hidróxido de sodio e hidróxido de potasio; un hidróxido de metal alcalinotérreo tal como hidróxido de magnesio, hidróxido de calcio e hidróxido de bario; una sal de hidrógenocarbonato de metal alcalino tal como hidrógenocarbonato de sodio e hidrógenocarbonato de potasio; un carbonato de metal alcalino tal como carbonato de sodio y carbonato de potasio; y una base orgánica tal como trimetilamina, trietilamina, etildiisopropilamina, piridina y quinolina. Entre estos, se prefieren amoníaco, hidróxido de sodio, hidróxido de potasio, hidróxido de calcio, hidróxido de bario, hidrógenocarbonato de sodio, trimetilamina, trietilamina y piridina, son más preferidos amoníaco, hidróxido de sodio, hidróxido de potasio e hidrógenocarbonato de sodio y son aún más preferidos hidróxido de sodio e hidróxido de potasio. Estas bases pueden usarse tal cual o pueden usarse en forma de una solución tal como una solución acuosa.
La reacción de la sal de haluro de hidrógeno de 1,1,3-trimetil-4-aminoindano con una base generalmente se lleva a cabo mezclando ambos. La reacción se lleva a cabo preferiblemente en agua.
La cantidad de la base a usar es generalmente de 1 mol a 2 moles con respecto a 1 mol de la sal de haluro de hidrógeno de 1,1,3-trimetil-4-aminoindano.
La temperatura de reacción es habitualmente de 0 °C a 100 °C. El tiempo de reacción es habitualmente de 0,1 horas a 5 horas.
Una vez completada la reacción, preferiblemente, la mezcla de reacción y el disolvente orgánico insoluble en agua se mezclan para obtener una capa orgánica y la capa orgánica se concentra. La pureza del 1,1,3-trimetil-4-aminoindano obtenido es generalmente del 97,5 % o más.
<Etapa (E)>
La etapa (E) es igual que la etapa (C), excepto que se usa el 1,1,3-trimetil-4-aminoindano obtenido en la etapa (D) en lugar del 1,1,3-trimetil-4-aminoindano obtenido en la etapa (B).
Al repetir las etapas (B1), (D) y (E), se puede producir (R)-1,1,3-trimetil-4-aminoindano con un mayor rendimiento. La presente invención se refiere a un método para producir un compuesto representado por la fórmula (1) (en lo sucesivo, a veces denominado compuesto (1)), que incluye las etapas (A), (B), (C) y (F) y es preferiblemente un método para producir el compuesto (1), que incluye las etapas (A), (B1), (D), (E) y (F). Las etapas (A), (B), (C), (B1), (D) y (E) se describen como anteriormente.
<Etapa (F)>
Ejemplos del átomo de halógeno en R1, R2 y R3 incluye un átomo de flúor, un átomo de cloro, un átomo de bromo y un átomo de yodo.
Ejemplos del grupo alquilo que puede estar sustituido con un átomo de halógeno en R1 y R2 incluye un grupo alquilo que tiene de 1 a 6 átomos de carbono, que puede estar sustituido con un átomo de halógeno, tal como un grupo metilo, un grupo etilo, un grupo n-propilo, un grupo isopropilo, un grupo n-butilo, un grupo sec-butilo, un grupo terc-butilo, un grupo n-pentilo, un grupo n-hexilo, un grupo trifluorometilo, un grupo difluorometilo, un grupo monofluorometilo, un grupo perfluoroetilo, un grupo perfluoro-n-propilo, un grupo perfluoroisopropilo, un perfluoro-n-butilo, un grupo perfluorosec-butilo, un grupo perfluoro-terc-butilo, un grupo perfluoro-n-pentilo, un grupo perfluoro-n-hexilo, un grupo triclorometilo, un grupo tribromometilo y un grupo triyodometilo.
R1 es preferiblemente un átomo de hidrógeno o un grupo metilo, y más preferiblemente un átomo de hidrógeno. R2 es preferiblemente un grupo metilo, un grupo monofluorometilo, un grupo difluorometilo o un grupo trifluorometilo, y más preferiblemente un grupo difluorometilo.
Ejemplos del grupo alcoxi que puede estar sustituido con un átomo de halógeno en R3 incluyen un grupo alcoxi que tiene de 1 a 6 átomos de carbono, que puede estar sustituido con un átomo de halógeno, tal como un grupo metoxi, un grupo etoxi, un grupo n-propoxi, un grupo isopropoxi, un grupo n-butoxi, un grupo sec-butoxi, un grupo terc-butoxi, un grupo n-pentiloxi, un grupo n-hexiloxi, un grupo trifluorometoxi, un grupo difluorometoxi, un grupo perfluoroetoxi, un grupo perfluoro-n-propoxi, un grupo perfluoroisopropoxi, un grupo perfluoro-n-butoxi, un grupo perfluorosec-butoxi, un grupo perfluoro-terc-butoxi, un grupo perfluoro-n-pentiloxi, un grupo perfluoro-n-hexiloxi, un grupo triclorometoxi, un grupo tribromometoxi y un grupo triyodometoxi.
R3 es preferiblemente un átomo de cloro, un grupo etoxi o un grupo hidroxi, y más preferiblemente un átomo de cloro. Los ejemplos del compuesto representado por la fórmula (2) (en lo sucesivo, a veces referido como un compuesto (2)) incluyen 1-metil-3-difluorometilpirazol-4-carboxilato de etilo, ácido 1-metil-3-difluorometilpirazol-4-carboxílico, y cloruro de ácido 1-metil-3-difluorometilpirazol-4-carboxílico.
Los ejemplos del compuesto representado por la fórmula (1) (en lo sucesivo, a veces referido como un compuesto (1)) incluyen amida del ácido (R)-(-)-N-(1,1,3-trimetilindan-4-il)-1-metil-3-difluorometilpirazol-4-carboxílico.
La etapa (F) es preferiblemente una etapa (F-1), (F-2), (F-3) o (F-4).
<Etapa (F-1)>
La etapa (F-1) es una etapa en la que (R)-1,1,3-trimetil-4-aminoindano obtenido en la etapa (C) o la etapa (E) se hace reaccionar con el compuesto (2) en el que R3 es un grupo hidroxi (en lo sucesivo denominado a veces compuesto (21)) en presencia de un agente de deshidratación-condensación para obtener el compuesto (1).
en el que R1 y
Los ejemplos del agente de deshidratación-condensación incluyen un compuesto de carbodiimida tal como hidrocloruro de 1-etil-3-(3-dimetilaminopropil)carbodiimida,1,3-diciclohexilcarbodiimida, y hexafluorato de (benzotriazol-1-iloxi)tris(dimetilamino)fosfonio.
La cantidad del agente de deshidratación-condensación a utilizar es generalmente de 1 mol a 5 moles con respecto a 1 mol del compuesto (2-1).
La cantidad de (R)-1,1,3-trimetil-4-aminoindano a usar es generalmente de 0,5 moles a 3 moles con respecto a 1 mol del compuesto (2-1).
La reacción del compuesto (2-1) con (R)-1,1,3-trimetil-4-aminoindano generalmente se lleva a cabo en presencia de un disolvente inerte para la reacción. Los ejemplos del disolvente incluyen un disolvente éter tal como tetrahidrofurano, dioxano, etilenglicol dimetil éter y éter terc-butilmetílico; un disolvente hidrocarburo alifático tal como hexano, heptano y octano; un disolvente hidrocarburo aromático tal como tolueno, xileno y etilbenceno; un disolvente hidrógenocarbonato halogenado tal como clorobenceno; un disolvente éster tal como acetato de butilo y acetato de etilo; un disolvente nitrilo tal como acetonitrilo; un disolvente amida ácida tal como N, N-dimetilformamida; un disolvente sulfóxido tal como dimetilsulfóxido; un disolvente aromático que contiene nitrógeno, tal como piridina; y una solución mixta de los mismos. La cantidad de disolvente a usar es generalmente de 1 parte en peso a 20 partes en peso con 1 parte en peso del compuesto (2-1). La temperatura de reacción es habitualmente de -20 °C a 150 °C y el tiempo de reacción es habitualmente de 1 hora a 24 horas.
Una vez completada la reacción, se mezcla a mezcla de reacción obtenida, agua, una solución acuosa de hidrógenocarbonato de sodio, una solución acuosa de carbonato de sodio, una solución acuosa de cloruro de amonio, una solución acuosa de hidróxido de sodio, una solución acuosa de hidróxido de potasio o una solución acuosa de un ácido como el ácido clorhídrico, ácido sulfúrico, ácido fosfórico y ácido acético, precipita el sólido y se filtra la mezcla así obtenida para obtener el compuesto (1). En el caso de que el sólido no precipite, la mezcla obtenida se extrae con un disolvente orgánico y la capa orgánica se somete a un tratamiento posterior, como la separación, el secado y la concentración, para extraer el compuesto (1). La capa orgánica se puede lavar con agua; una solución acuosa de sal de hidrógenocarbonato de metal alcalino tal como una solución acuosa de hidrógenocarbonato de sodio; una solución acuosa de carbonato de metal alcalino tal como una solución acuosa de carbonato de sodio; una solución acuosa de cloruro de amonio; una solución acuosa de hidróxido de metal alcalino tal como una solución acuosa de hidróxido de sodio y una solución acuosa de hidróxido de potasio; o una solución acuosa de un ácido tal como ácido clorhídrico, ácido sulfúrico, ácido fosfórico y ácido acético. El lavado de la capa orgánica se lleva a cabo habitualmente entre 0 °C y 70 °C, y preferiblemente entre 20 °C y 60 °C. El compuesto extraído (1) puede purificarse adicionalmente mediante cromatografía en columna, recristalización o similares.
<Etapa (F-2)>
La etapa (F-2) es una etapa en la que se hace reaccionar el (R)-1,1,3-trimetil-4-aminoindano obtenido en la etapa (C) o la etapa (E) con el compuesto (2-1) en la presencia de un ácido de Lewis para obtener el compuesto (1).
Los ejemplos del ácido de Lewis incluyen un cloruro de metal tal como tetracloruro de titanio, tetracloruro de zirconio y cloruro de aluminio; un compuesto de alcóxido metálico tal como etóxido de titanio, propóxido de titanio, etóxido de zirconio, propóxido de zirconio, etóxido de aluminio, propóxido de aluminio, etóxido de antimonio y propóxido de antimonio; un compuesto de la amida metálica tal como tetrakis(dimetilamino)titanio, diclorobis(dimetilamino)titanio y tetrakis(dietilamino)titanio; un compuesto de borano tal como borano, ácido 3,5-bis(trifluorometil)fenil-bórico, ácido 2,4-bis(trifluorometil)fenil-bórico y ácido pentafluorofenil-bórico; y un compuesto de borato tal como trifenilmetil tetrakis(pentafluorofenil)borato, trifenilmetil tetrakis(3,5-bistrifluorometilfenil)borato y N,N-dimetilanilinio tetrakis(pentafluorofenil)borato.
La cantidad de ácido de Lewis a usar es generalmente de 0,001 moles a 3 moles con respecto a 1 mol del compuesto (2-1).
La cantidad de (R)-1,1,3-trimetil-4-aminoindano a usar es generalmente de 0,5 moles a 3 moles con respecto a 1 mol del compuesto (2-1).
La reacción del compuesto (2-1) con (R)-1,1,3-trimetil-4-aminoindano generalmente se lleva a cabo en presencia de un disolvente inerte para la reacción. Los ejemplos del disolvente incluyen los disolventes mencionados en la etapa (F-1). La cantidad de disolvente a usar es generalmente de 1 parte en peso a 20 partes en peso con respecto a 1 parte en peso del compuesto (2-1). La temperatura de reacción es habitualmente de -20 °C a 150 °C y el tiempo de reacción es habitualmente de 1 hora a 120 horas. La reacción se lleva a cabo preferiblemente eliminando el agua producida de este modo.
Una vez completada la reacción, el tratamiento como en la etapa (F-1) se puede llevar a cabo para extraer el compuesto (1).
<Etapa (F-3)>
La etapa (F-3) es una etapa en la que se hace reaccionar el (R)-1,1,3-trimetil-4-aminoindano obtenido en la etapa (C) o la etapa (E) con el compuesto (2) en el que R3 es un grupo alcoxi que tiene de 1 a 6 átomos de carbono, que puede estar sustituido con un átomo de halógeno (en lo sucesivo, a veces denominado compuesto (2-2)) en presencia de un ácido de Lewis o una base de Lewis para obtener el compuesto (1 ).

en el que R1 y R2 tienen los mismos significados que antes y R3' representa un grupo alcoxi que tiene de 1 a 6 átomos de carbono, que puede estar sustituido con un átomo de halógeno.
Los ejemplos del ácido de Lewis incluyen un cloruro de metal tal como tetracloruro de titanio, tetracloruro de zirconio y cloruro de aluminio; y un compuesto alcóxido metálico tal como etóxido de titanio, propóxido de titanio, etóxido de circonio, propóxido de circonio, etóxido de aluminio, propóxido de aluminio, etóxido de antimonio y propóxido de antimonio.
La cantidad de ácido de Lewis a usar es generalmente de 0,01 moles a 3 moles con respecto a 1 mol del compuesto (2-2).
La cantidad de (R)-1,1,3-trimetil-4-aminoindano a usar es generalmente de 0,5 moles a 3 moles con respecto a 1 mol del compuesto (2-2).
Los ejemplos de la base de Lewis incluyen un compuesto de alcóxido metálico tal como metóxido de sodio, etóxido de sodio, terc-butóxido de sodio, metóxido de potasio, etóxido de potasio y terc-butóxido de potasio; un hidruro metálico tal como hidruro de sodio; un compuesto de litio tal como diisopropil amida de litio y terc-butil litio; un compuesto de silicio tal como hexametil disilazano de sodio y hexametil disilazano de potasio; y un compuesto de aluminio tal como trimetil aluminio, trietil aluminio y triisobutil aluminio.
La cantidad de la base de Lewis a usar es generalmente de 0,01 moles a 3 moles con respecto a 1 mol del compuesto (2-2).
La cantidad de (R)-1,1,3-trimetil-4-aminoindano a usar es generalmente de 0,5 moles a 3 moles con respecto a 1 mol del compuesto (2-2).
La reacción del compuesto (2-2) con (R)-1,1,3-trimetil-4-aminoindano se lleva a cabo habitualmente en presencia de un disolvente inerte para la reacción. Los ejemplos del disolvente incluyen los disolventes mencionados en la etapa (F-1). La cantidad de disolvente a usar es generalmente de 1 parte en peso a 20 partes en peso con respecto a 1 parte en peso del compuesto (2-2). La temperatura de reacción es habitualmente de -20 °C a 150 °C y el tiempo de reacción es habitualmente de 1 hora a 110 horas. La reacción se lleva a cabo preferiblemente eliminando los alcoholes producidos de este modo.
Una vez completada la reacción, el tratamiento como en la etapa (F-1) se puede llevar a cabo para extraer el compuesto (1).
<Etapa (F-4)>
La etapa (F-4) es una etapa en la que se hace reaccionar el (R)-1,1,3-trimetil-4-aminoindano obtenido en la etapa (C) o la etapa (E) con el compuesto (2) en el que R3 es un átomo de halógeno (en lo sucesivo denominado a veces compuesto (2-3)) en la presencia de una base para obtener el compuesto (1).

en el que R1 y R2 tienen los mismos significados que antes y R3" representa un átomo de halógeno.
Los ejemplos de la base incluyen un carbonato de metal alcalino tal como carbonato de sodio y carbonato de potasio; y un compuesto aromático que contiene nitrógeno, tal como aminas terciarias, tales como trietilamina y diisopropiletilamina, piridina y 4-dimetilaminopiridina.
La cantidad de la base a usar es generalmente de una cantidad catalítica a 5 moles, y preferiblemente de 1 mol a 3 moles con respecto a 1 mol de (R)-1,1,3-trimetil-4-aminoindano.
La cantidad del compuesto (2-3) a usar es generalmente de 0,5 moles a 1,5 moles, preferiblemente de 0,8 moles a 1,3 moles, y más preferiblemente de 1,0 mol a 1,2 moles, con respecto a 1 mol de (R) 1,1,3-trimetil-4-aminoindano. La reacción del compuesto (2-3) con (R)-1,1,3-trimetil-4-aminoindano generalmente se lleva a cabo en presencia de un disolvente. El disolvente puede ser cualquiera que sea inerte a la reacción, y los ejemplos del mismo incluyen un disolvente hidrocarburo alifático tal como pentano, hexano, heptano, octano y ciclohexano; un disolvente hidrocarburo aromático tal como tolueno, xileno y etilbenceno; un disolvente hidrocarbonado alifático halogenado tal como diclorometano, cloroformo, 1,2-dicloroetano y tetracloruro de carbono; un disolvente hidrocarbonado aromático halogenado tal como clorobenceno, diclorobenceno y triclorobenceno; un disolvente éter tal como éter dietílico, éter diisopropílico, éter terc-butilmetílico, ciclohexilmetil éter, etilenglicol dimetil éter, tetrahidrofurano y dioxano; un disolvente éster tal como acetato de etilo y acetato de butilo; un disolvente nitrilo tal como acetonitrilo; y una solución mixta de los mismos, preferiblemente un disolvente hidrocarburo aromático, un disolvente hidrocarburo aromático halogenado y un disolvente éter, y más preferiblemente tolueno, xileno, etilbenceno, clorobenceno y tetrahidrofurano. La cantidad de disolvente a usar es preferiblemente de 1 parte en peso a 20 partes en peso, y más preferiblemente de 2 partes en peso a 10 partes en peso, con respecto a 1 parte en peso de (R)-1,1,3-trimetil-4-aminoindano.
La temperatura para la reacción del compuesto (2-3) con (R)-1,1,3-trimetil-4-aminoindano es generalmente de -20 °C a 80 °C, preferiblemente de 0 °C a 70 °C , y más preferiblemente de 20 °C a 60 °C y el tiempo de reacción es generalmente de 0,1 horas a 24 horas.
Una vez completada la reacción, el tratamiento como en la etapa (F-1) se puede llevar a cabo para extraer el compuesto (1).
La presente invención se refiere a un método para producir el compuesto (1), que incluye las etapas (A), (B), (C), (G) y (H), y preferiblemente un método para producir el compuesto (1), que incluye las etapas (A), (B1), (D), (E), (G) y (H). Las etapas (A), (B), (C), (B1), (D) y (E) son las mismas que se describieron anteriormente.
<Etapa (G)>
La etapa (G) es preferiblemente una etapa de reacción de un compuesto representado por la fórmula (3) (en lo sucesivo, a veces denominado como un compuesto (3)) con un agente de cloración para obtener un compuesto representado por la fórmula (4) (en lo sucesivo, a veces denominado como un compuesto (4)).
Los ejemplos del agente de cloración incluyen cloruro de tionilo, cloruro de oxalilo y fosgeno. La cantidad de agente de cloración a usar es generalmente de 1 mol a 2 moles, y preferiblemente de 1 mol a 1,5 moles, con respecto a 1 mol del compuesto (3).
La reacción del compuesto (3) con el agente de cloración también se puede llevar a cabo en presencia de amina terciaria o amida. Los ejemplos de la amina terciaria o amida incluyen piridina, picolina, N,N-dimetilformamida y N-metil-N-fenilformamida. La cantidad de amina terciaria o amida a usar es generalmente de 0,001 moles a 0,05 moles, y preferiblemente de 0,003 moles a 0,03 moles, con respecto a 1 mol del compuesto (3).
La reacción del compuesto (3) con el agente de cloración se lleva a cabo generalmente en presencia de un disolvente.
El disolvente puede ser cualquiera que sea un inserto de la reacción, y los ejemplos del mismo incluyen un disolvente hidrocarburo alifático tal como pentano, hexano, heptano y ciclohexano; un disolvente hidrocarburo aromático tal como tolueno, xileno y etilbenceno; un disolvente hidrocarbonado alifático halogenado tal como diclorometano, cloroformo, 1,2-dicloroetano y tetracloruro de carbono; un disolvente hidrocarbonado aromático halogenado tal como clorobenceno, diclorobenceno y triclorobenceno; un disolvente éter tal como éter dietílico, éter diisopropílico, éter tercbutil metílico, éter ciclohexilmetílico, etilenglicol dimetil éter y dioxano; y una solución mixta de los mismos. El disolvente es preferiblemente el disolvente hidrocarburo aromático o el disolvente hidrocarburo aromático halogenado, y más preferiblemente tolueno, xileno, etilbenceno o clorobenceno.
La cantidad de disolvente a usar es preferiblemente de 0,5 partes en peso a 20 partes en peso, y más preferiblemente de 1 parte en peso a 10 partes en peso, con respecto a 1 parte en peso del compuesto (3).
La temperatura de reacción entre el compuesto (3) y el agente de cloración es generalmente de 10 °C a 120 °C, y preferiblemente de 40 °C a 110 °C, y el tiempo de reacción es generalmente de 0,1 horas a 24 horas.
Una vez completada la reacción, la mezcla de reacción obtenida se puede concentrar para obtener un compuesto (4). El compuesto obtenido (4) se puede purificar por destilación o similar.
<Etapa (H)>
La etapa (H) es igual que la etapa (F-4), excepto que se usa el compuesto (4) en lugar del compuesto (2-3).
<Etapa de purificación>
El compuesto obtenido (1) se puede purificar adicionalmente mediante cromatografía en columna, recristalización o similar, y se prefiere la purificación.
Como método de purificación, se prefiere un método en el que un compuesto (1) se disuelve en un disolvente para preparar una solución y se recristaliza utilizando la solución. Se pueden usar cristales de siembra en la recristalización. Los ejemplos del disolvente incluyen un disolvente hidrógenocarbonato alifático tal como pentano, hexano, heptano, octano y ciclohexano; un disolvente hidrocarburo aromático tal como tolueno, xileno y etilbenceno; un disolvente hidrógenocarbonato alifático halogenado, tal como diclorometano, cloroformo, 1,2-dicloroetano y tetracloruro de carbono; un disolvente hidrocarbonado aromático halogenado tal como clorobenceno, diclorobenceno y triclorobenceno; un disolvente éter tal como éter dietílico, éter diisopropílico, éter terc-butilmetílico, ciclohexilmetil éter, etilenglicol dimetil éter, tetrahidrofurano y dioxano; un disolvente éster tal como acetato de etilo y acetato de butilo; un disolvente nitrilo tal como acetonitrilo; un disolvente alcohólico tal como metanol, etanol y 2-propanol; y una solución mixta de los mismos. El disolvente es preferiblemente el disolvente hidrocarburo alifático, el disolvente hidrocarburo aromático, el disolvente hidrocarburo aromático halogenado o el disolvente éster, y más preferiblemente tolueno, xileno, etilbenceno, hexano, heptano o acetato de etilo.
Ejemplos
“%” y “parte(s)” en los ejemplos son “% en peso" y “parte(s) en peso" a menos que se especifique lo contrario.
En los ejemplos, la proporción de isómeros R/isómeros S se analizó mediante cromatografía líquida de alto rendimiento (método de porcentaje de área) utilizando columnas quirales. El contenido de cada uno de 1,1,3-trimetil-4-aminoindano y amida del ácido (R)-(-)-N-(1,1,3-trimetilindan-4-il)-1-metil-3-difluorometilpirazol-4-carboxílico se analizó mediante cromatografía líquida (método del patrón interno).
En los ejemplos, la XRD se midió en las siguientes condiciones.
<Ejemplo 3 y Ejemplo de referencia 14>
Dispositivo: SmartLab (Rigaku Corporation))
Rendimiento del tubo de rayos X: CuKa, 45 kV, 200 mA
Ancho de muestreo: 0,02 °
Rango de escaneo: 5 ° a 50 °
<Ejemplos de referencia 1 a 13>
Dispositivo: Difractómetro X' Pert Pro (PANalytical)
Rendimiento del tubo de rayos X: CuKa, 45 kV, 40 mA
Ancho de muestreo: 0,02 °
Rango de escaneo: 2 ° a 40 °
En los ejemplos, el espectro FT-Raman se midió en las siguientes condiciones.
Dispositivo: espectrómetro Nicolet NXR9650 y NXR960 (Thermo Electron)
Láser de excitación: 1064 nm
Resolución: 4 cm-1
Número de escaneo: 128
Función de apodización: Happ-Genzel.
Relleno cero: 2 niveles.
En los ejemplos, el análisis térmico (DSC) se midió en las siguientes condiciones.
Dispositivo: calorímetro diferencial de barrido Q100
(TA instruments)
Atmósfera: Nitrógeno.
Caudal de gas: 40 ml/min
Velocidad de calentamiento: 15 °C/min.
En los ejemplos, el análisis térmico (TGA) se midió en las siguientes condiciones.
Dispositivo: analizador termogravimétrico Q500 (TA instruments)
Atmósfera: Nitrógeno.
Caudal de gas: 40 ml/min
Velocidad de calentamiento: 15 °C/min.
En los ejemplos, el análisis térmico (TG-DTA) se midió en las siguientes condiciones.
Dispositivo: TG-DTA2000SR (BRUKER)
Atmósfera: Nitrógeno.
Caudal de gas: 150 ml/min
Velocidad de calentamiento: 5 °C/min.
Ejemplo 1
<Etapa (A)>
[Etapa (A1)]
En una atmósfera de nitrógeno, 80,5 partes (pureza: 62,1 %) de 1,1,3-trimetil-4-aminoindano, 31,5 partes (0,63 partes con respecto a 1 parte de 1,1,3-trimetil-4-aminoindano) de metanol, 2,0 partes (0,04 partes con respecto a 1 parte de 1,1,3-trimetil-4-aminoindano) de agua y 9,5 partes de tolueno se mezclaron a temperatura ambiente. La mezcla obtenida se calentó a 40 °C y luego se añadieron 6,5 partes (0,15 moles con respecto a 1 mol de 1,1,3-trimetil-4-aminoindano) de ácido D-tartárico. A la mezcla obtenida se le añadió una pequeña cantidad de cristales de siembra, seguido de agitación durante 1 hora, y luego a esta se añadió 15,1 partes (0,35 moles con respecto a 1 mol de 1,1,3-trimetil-4-aminoindano) de ácido D-tartárico en 7 porciones divididas en un intervalo de 10 minutos.
La mezcla obtenida se agitó a 40 °C durante 3 horas, luego se enfrió a 0 °C a una velocidad de enfriamiento de 5 °C/hora, y se agitó a 0 °C durante 10 horas.
[Etapa (A2)]
La mezcla obtenida se filtró para obtener cada uno de los cristales y un filtrado.
Los cristales obtenidos se lavaron secuencialmente una vez con 35,0 partes de un disolvente mixto de metanol y tolueno enfriados con hielo a 1:9 (relación en peso) y una vez con 50,0 partes de tolueno enfriado con hielo para obtener cada uno de un líquido de lavado y cristales.
Los cristales obtenidos se secaron a presión reducida para obtener 39,9 partes de un solvato de metanol de D-tartrato de (R)-1,1,3-trimetil-4-aminoindano. La relación de isómeros R/isómeros S del solvato de metanol de 1,1,3-trimetil-4-aminoindano fue de 98,1/1,9.
El filtrado y el líquido de lavado se recuperaron todos y se mezclaron para obtener una solución que incluía D-tartrato de (S)-1,1,3-trimetil-4-aminoindano.
[Etapa (A3)]
A una solución formada mezclando 39,5 partes de xileno y 78,9 partes de una solución acuosa de hidróxido de sodio al 14 %, se agregaron 39,5 partes del solvato de metanol obtenido de D-tartrato de (R)-1,1,3-trimetil-4-aminoindano.
La mezcla obtenida se agitó y se sometió a separación líquida. La capa orgánica obtenida se lavó con agua y luego se concentró a presión reducida para obtener 19,9 partes (contenido: 93,9 %) de (R)-1,1,3-trimetil-4-aminoindano. El rendimiento de la etapa (A1) fue del 37,3 %. La relación de isómeros R/isómeros S de 1,1,3-trimetil-4-aminoindano fue de 98,1/1,9.
[Etapa (A4)]
Una solución formada mezclando 10,8 partes de una solución acuosa de hidróxido de sodio al 24 % y 41,7 partes de agua se calentó a 30 °C. A la mezcla obtenida se agregaron gota a gota 160,8 partes de una solución que incluía el D-tartrato de (S)-1,1,3-trimetil-4-aminoindano (contenido de (S)-1,1,3-trimetil-4-aminoindano: 17,6 %) durante 2 horas. La mezcla obtenida se agitó a 30 °C durante 1 hora y se sometió a separación líquida. La capa orgánica obtenida se lavó con agua y luego se concentró a presión reducida para obtener 35,4 partes de (S)-1,1,3-trimetil-4-aminoindano. El contenido de 1,1,3-trimetil-4-aminoindano fue del 76,7 %. Además, la proporción de isómeros R/isómeros S de 1,1,3-trimetil-4-aminoindano fue de 17,7/82,3 (pureza óptica de 64,6 % ee (S)).
<Etapa (B)>
En un recipiente de reacción de autoclave se colocaron 31,0 partes del (S)-1,1,3-trimetil-4-aminoindano obtenido en la etapa (A), 3,25 partes de un tipo E de paladio sobre carbono al 5 % (fabricado por NE CHEMCAT) Corporation, 50 % húmedo) y 1,1 partes de agua para obtener una mezcla. El recipiente de reacción se cerró herméticamente y el gas en el recipiente de reacción se reemplazó con nitrógeno. Mientras se agitaba la mezcla, se incluyó hidrógeno en el recipiente de reacción hasta que la presión interna de hidrógeno en el recipiente de reacción se convirtió en 0,8 MPa, seguido de agitación a una temperatura interna de 80 °C durante 3 horas. El gas en el recipiente de reacción se reemplazó con nitrógeno y la mezcla se agitó a una temperatura interna de 180 °C y una presión interna de 0,80 MPa durante 24 horas. La mezcla de reacción obtenida se enfrió y se filtró usando Celite para obtener cada uno de un sólido y un filtrado. El sólido obtenido se lavó con 10 partes de tolueno para obtener un líquido de lavado. El líquido de lavado obtenido y el filtrado obtenido se mezclaron para obtener 37,9 partes de una solución de 1,1,3-trimetil-4-aminoindano en tolueno (contenido de 1,1,3-trimetil-4-aminoindano: 54,6 %). El 1,1,3-trimetil-4-aminoindano tenía una pureza óptica de 0,90 % ee y una tasa de recuperación de 87,0 %.
<Etapa (C)>
A temperatura ambiente en una atmósfera de nitrógeno se mezclaron 33,0 partes de la solución de 1,1,3-trimetil-4-aminoindano en la etapa (B) en tolueno, 27,9 partes (pureza: 62,1 %) de 1,1,3- trimetil-4-aminoindano, 22,2 partes (0,63 partes con respecto a 1 parte de 1,1,3-trimetil-4-aminoindano) de metanol, 1,4 partes (0,04 partes con respecto a 1 parte de 1,1,3-trimetil-4-aminoindano) de agua y 9,5 partes de tolueno. La mezcla obtenida se calentó a 40 °C y luego se agregaron 4,6 partes (0,15 moles con respecto a 1 mol de 1,1,3-trimetil-4-aminoindano) de ácido D-tartárico. A la solución obtenida se le añadió una pequeña cantidad de cristales de siembre seguido de agitación durante 1 hora y luego se añadió a la misma 10,7 partes (0,35 moles con respecto a 1 mol de 1,1,3-trimetil-4-aminoindano) de ácido D-tartárico en 7 porciones divididas en un intervalo de 10 minutos.
La mezcla obtenida se agitó a 40 °C durante 3 horas, luego se enfrió a 0 °C a una velocidad de enfriamiento de 5 °C/hora, y se agitó adicionalmente a 0 °C durante 8 horas. La mezcla obtenida se filtró para obtener cada uno de los cristales y un filtrado.
Los cristales obtenidos se lavaron secuencialmente una vez con 24,7 partes de un disolvente mixto de metanol y tolueno enfriados con hielo a 1:9 (relación en peso) y una vez con 35,3 partes de tolueno enfriado con hielo para obtener cada uno de un líquido de lavado y cristales.
Los cristales obtenidos se secaron a presión reducida para obtener 28,7 partes de un solvato de metanol de D-tartrato de (R)-1,1,3-trimetil-4-aminoindano. La relación de isómeros R/isómeros S de 1,1,3-trimetil-4-aminoindano del solvato de metanol fue de 97,8/2,2.
El filtrado y el líquido de lavado se recuperaron todos y se mezclaron para obtener una solución que incluye D-tartrato de (S)-1,1,3-trimetil-4-aminoindano y (S)-1,1,3-trimetil-4-aminoindano.
A una solución formada mezclando 28,0 partes de xileno y 56,0 partes de una solución acuosa de hidróxido de sodio al 14 %, se agregaron 28,0 partes del solvato de metanol obtenido de D-tartrato de (R)-1,1,3-trimetil-4- aminoindano. La mezcla obtenida se agitó y se sometió a separación líquida. La capa orgánica obtenida se lavó con 28,0 partes de agua y luego se concentró a presión reducida para obtener 14,9 partes de (R)-1,1,3-trimetil-4-aminoindano. El contenido de 1,1,3-trimetil-4-aminoindano fue del 90,0 %. Además, la relación de isómeros R/isómeros S de 1,1,3-trimetil-4-aminoindano fue de 97,7/2,3.
La cantidad total de (R)-1,1,3-trimetil-4-aminoindano obtenido en la etapa (A) y el (R)-1,1,3-trimetil-4-aminoindano obtenido en la etapa (C) fue de 32,1 partes como una fracción pura, y el rendimiento total de las etapas (A), (B) y (C) fue de 47,7 %.
Ejemplo 2
<Etapa (A)>
[Etapa (A1)]
A temperatura ambiente en una atmósfera de nitrógeno se mezclaron 112,7 partes (pureza: 62,1 %) de 1,1,3-trimetil-4-aminoindano, 44,1 partes (0,63 partes con respecto a 1 parte de 1,1,3-trimetil-4-aminoindano) de metanol, 2,8 partes (0,04 partes con respecto a 1 parte de 1,1,3-trimetil-4-aminoindano) de agua y 13,3 partes de tolueno. La mezcla obtenida se calentó a 40 °C y luego se añadieron 9,1 partes (0,15 moles con respecto a 1 mol de 1,1,3-trimetil-4-aminoindano) de ácido D-tartárico para obtener una solución. A la solución obtenida se le añadió una pequeña cantidad de cristales de siembra, seguido de agitación durante 1 hora, y a la misma se agregaron 21,1 partes (0,35 moles con respecto a 1 mol de 1,1,3-trimetil-4-aminoindano) de ácido D-tartárico en 7 porciones divididas en un intervalo de 10 minutos.
La mezcla obtenida se agitó a 40 °C durante 3 horas, luego se enfrió a 0 °C a una velocidad de enfriamiento de 5 °C/hora, y se agitó adicionalmente a 0 °C durante 2 horas.
[Etapa (A2)]
La mezcla obtenida se filtró para obtener cada uno de los cristales y un filtrado.
Los cristales obtenidos se lavaron secuencialmente una vez con 49,0 partes de un disolvente mixto de metanol y tolueno enfriados con hielo a 1:9 (relación en peso) y una vez con 70,0 partes de tolueno enfriado con hielo para obtener cada uno de un líquido de lavado y cristales.
Los cristales después del lavado se secaron a presión reducida para obtener 56,7 partes de un solvato de metanol de D-tartrato de (R)-1,1,3-trimetil-4-aminoindano. La relación de isómeros R/isómeros S de 1,1,3-trimetil-4-aminoindano del solvato de metanol fue de 98,0/2,0.
El filtrado y el líquido de lavado se recuperaron todos y se mezclaron para obtener una solución que incluye D-tartrato de (S)-1,1,3-trimetil-4-aminoindano y (S)-1,1,3-trimetil-4- aminoindano.
[Etapa (A3)]
A una solución formada mezclando 56,0 partes de xileno y 111,9 partes de una solución acuosa de hidróxido de sodio al 14 %, se añadieron 56,0 partes del solvato de metanol obtenido de D-tartrato de (R)-1,1,3-trimetil-4-aminoindano. La mezcla obtenida se agitó y se sometió a separación líquida. La capa orgánica obtenida se lavó con agua y luego se concentró a presión reducida para obtener 27,5 partes (contenido: 97,3 %) de (R)-1,1,3-trimetil-4-aminoindano. El rendimiento de la etapa (A1) fue del 38,2 %. La relación de isómeros R/isómeros S de 1,1,3-trimetil-4-aminoindano fue de 98,0/2,0.
[Etapa (A4)]
Una solución formada mezclando 14,4 partes de una solución acuosa de hidróxido de sodio al 24 % y 55,5 partes de agua se calentó a 30 °C. A la mezcla obtenida se agregaron gota a gota 188,9 partes (contenido de (S)-1,1,3-trimetil-4-aminoindano: 21,2 %) de una solución que incluye el D-tartrato de (S)-1,1,3, -trimetil-4-aminoindano obtenido durante 2 horas y media. La mezcla obtenida se agitó a 30 °C durante 1 hora y se sometió a separación líquida. La capa orgánica obtenida se lavó con 55,4 partes de agua y luego se concentró a presión reducida para obtener 51,8 partes de (S)-1,1,3-trimetil-4-aminoindano. El contenido de 1,1,3-trimetil-4-aminoindano fue del 76,5 %. Además, la relación de isómeros R/isómeros S de 1,1,3-trimetil-4-aminoindano fue de 17,4/82,6 (pureza óptica de 65,2 % ee (S)).
<Etapa (B')>
En un recipiente de reacción de autoclave se colocaron 47,0 partes del (S)-1,1,3-trimetil-4-aminoindano obtenido en la etapa (A), 4,87 partes de un tipo E de paladio sobre carbono al 5 % (fabricado por NE CHEMCAT). Corporation, 50 % húmedo) y 1,5 partes de agua para obtener una mezcla. El recipiente de reacción se cerró herméticamente y el gas en el recipiente de reacción se reemplazó con nitrógeno. Mientras se agitaba la mezcla, se incluyó hidrógeno en el recipiente de reacción hasta que la presión interna de hidrógeno en el recipiente de reacción se convirtió en 0,8 MPa, seguido de agitación a una temperatura interna de 80 °C durante 3 horas. El gas en el recipiente de reacción se reemplazó con nitrógeno y la mezcla se agitó a una temperatura interna de 180 °C y una presión interna de 0,85 MPa durante 24 horas. La mezcla de reacción obtenida se enfrió y se filtró usando Celite para obtener cada uno de un sólido y un filtrado. El sólido obtenido se lavó con 8 partes de tolueno para obtener un líquido de lavado. El líquido de lavado obtenido y el filtrado obtenido se mezclaron para obtener 50,8 partes de una solución de 1,1,3-trimetil-4-aminoindano en tolueno (contenido de 1,1,3-trimetil-4-aminoindano: 64,9 %). El 1,1,3-trimetil-4-aminoindano tenía una pureza óptica de 0,35 % ee y una tasa de recuperación de 91,7 %.
<Etapa (D)>
En una atmósfera de nitrógeno se mezclaron 41,87 partes de la solución de 1,1,3-trimetil-4-aminoindano obtenido en la etapa (B1) en tolueno, 43,77 partes de 1,1,3-trimetil-4-aminoindano (pureza: 62,1 %), 95,52 partes de tolueno y 24,46 partes de agua. La mezcla obtenida se calentó a 65 °C y luego se añadieron 41,98 partes de ácido clorhídrico concentrado. La mezcla obtenida se agitó a 65 °C durante 1 hora y luego se separó en una capa acuosa y una capa orgánica en la que se había disuelto el hidrocloruro de 1,1,3-trimetil-4-aminoindano. La capa orgánica obtenida se enfrió a 10 °C mientras se agitaba, y precipitaron los cristales de hidrocloruro de 1,1,3-trimetil-4-aminoindano. Los cristales precipitados se extrajeron por filtración para obtener hidrocloruro de 1,1,3-trimetil-4-aminoindano. El hidrocloruro de 1,1,3-trimetil-4-aminoindano obtenido se había disuelto en agua caliente. A la solución obtenida se le añadió una solución acuosa de hidróxido de sodio. A la mezcla obtenida se le añadió tolueno, y luego se separó la capa orgánica. La capa orgánica se lavó con agua y luego se concentró a presión reducida para obtener 73,74 partes de una solución de 1,1,3-trimetil-4-aminoindano en tolueno en forma de un líquido marrón pálido. La tasa de recuperación fue del 93,1 % y el contenido de 1,1,3-trimetil-4-aminoindano fue del 99,4 %.
<Etapa (E)>
A temperatura ambiente en una atmósfera de nitrógeno se mezclaron 72,4 partes (contenido de 1,1,3-trimetil-4-aminoindano: 99,4 %) de la solución de 1,1,3-trimetil-4-aminoindano obtenida en la etapa (D) en tolueno, 45,4 partes (0,63 partes con respecto a 1 parte de 1,1,3-trimetil-4-aminoindano) de metanol, 2,9 partes (0,04 partes con respecto a 1 parte de 1,1,3-trimetil-4-aminoindano) de agua y 57,2 partes de tolueno. La mezcla obtenida se calentó a 40 °C y luego se añadieron 9,3 partes de ácido D-tartárico (0,15 moles con respecto a 1 mol de 1,1,3-trimetil-4-aminoindano) para obtener una solución. A la solución obtenida se le añadió una pequeña cantidad de cristales de siembra, seguido de agitación durante 1 hora. Luego, se agregaron 21,8 partes (0,35 moles con respecto a 1 mol de 1,1,3-trimetil-4-aminoindano) de ácido D-tartárico en 7 porciones divididas en un intervalo de 10 minutos.
La mezcla obtenida se agitó a 40 °C durante 3 horas, luego se enfrió a 0 °C a una velocidad de enfriamiento de 5 °C/hora, y se agitó adicionalmente a 0 °C durante 24 horas. La mezcla obtenida se filtró para obtener cada uno de los cristales y un filtrado. Los cristales obtenidos se lavaron secuencialmente una vez con 50,4 partes de un disolvente mixto de metanol y tolueno enfriados con hielo a 1:9 (relación en peso) y luego se lavaron una vez con 72,0 partes de tolueno enfriado con hielo para obtener cada uno de un líquido de lavado y cristales. Los cristales después del lavado se secaron a presión reducida para obtener 60,8 partes de un solvato de metanol de D-tartrato de (R)-1,1,3-trimetil-4-aminoindano.
A una solución formada mezclando 121,6 partes de xileno y 121,5 partes de una solución acuosa de hidróxido de sodio al 14 %, se agregaron 60,8 partes del solvato de metanol obtenido de D-tartrato de (R)-1,1,3-trimetil-4-aminoindano. La mezcla obtenida se agitó y se sometió a separación líquida. La capa orgánica obtenida se lavó con 91,2 partes de agua y luego se concentró a presión reducida para obtener 32,5 partes de (R)-1,1,3-trimetil-4-aminoindano. El contenido de 1,1,3-trimetil-4-aminoindano fue del 91,3 %. Además, la relación de isómeros R/isómeros S de 1,1,3-trimetil-4-aminoindano fue de 97,3/2,7 y el rendimiento fue de 41,2 %.
La cantidad total de (R)-1,1,3-trimetil-4-aminoindano obtenida en la etapa (A) y el (R)-1,1,3-trimetil-4-aminoindano obtenido en la etapa (E) como fracción pura fue de 56,4 partes, y el rendimiento total de las etapas (A), (B1), (D) y (E) fue de 58,0 %.
Ejemplo 3
<Etapa (G)>
A temperatura ambiente en una atmósfera de nitrógeno, se mezclaron 14,0 partes de ácido 1-metil-3-difluorometilpirazol-4-carboxílico y 35,1 partes de xileno. La mezcla obtenida se calentó a 100 °C. A la mezcla obtenida se añadieron gota a gota 11,2 partes de cloruro de tionilo a lo largo de 5 horas. La mezcla obtenida se agitó a 100 °C durante 15 horas y luego se enfrió a 40 °C. El cloruro de tionilo y el xileno se evaporaron de la mezcla de reacción obtenida a presión reducida para obtener cloruro del ácido 1-metil-3-difluorometilpirazol-4-carboxílico marrón.
<Etapa (H)>
Se mezclaron 14,6 partes del (R)-1,1,3-trimetil-4-aminoindano obtenido en la etapa (C), 9,2 partes de trietilamina y 38,1 partes de xileno para preparar una solución. A la solución obtenida se le añadió gota a gota una solución en la que el cloruro del ácido 1-metil-3-difluorometilpirazol-4-carboxílico obtenido en la etapa (G) se había disuelto en 13,2 partes de xileno a una temperatura de 45 °C a 50 °C durante 2 horas. La mezcla obtenida se agitó a una temperatura de 45 °C a 50 °C durante 15 horas. Se mezcló la mezcla de reacción obtenida y una solución acuosa de hidróxido de sodio al 20 % y luego se separó la capa orgánica. La capa orgánica obtenida se lavó secuencialmente con agua, ácido clorhídrico al 18 %, agua, una solución acuosa de hidróxido de sodio al 1 % y agua, y luego se concentró en condiciones de presión reducida para obtener 27,5 partes de la amida del ácido (R)-(-)-N-(1,1,3-trimetilindan-4-il)-1
metil-3-difluorometilpirazol-4-carboxílico. El contenido de la amida del ácido (RM-)-N-(1,1,3-trimetilindan-4-il)-1-metil-3-difluorometilpirazol-4-carboxílico fue de 89,7 % (un rendimiento con respecto a (R)-1,1,3-trimetil-4-aminoindano de 98,5 %). Además, la proporción de isómeros R/isómeros S de la amida del ácido (RM-)-N-(1,1,3-trimetilindan-4-il)-1-metil-3-difluorometilpirazol-4-carboxílico fue de 97,7 /2,3.
<Etapa de purificación>
A temperatura ambiente en una atmósfera de nitrógeno se mezclaron 27,5 partes de la amida del ácido (R)-(-)-N-(1,1,3-trimetilindan-4-il)-1-metil-3-difluorometilpirazol-4-carboxílico obtenida, 22,0 partes de xileno y 37,1 partes de heptano. La mezcla obtenida se calentó a 75 °C para obtener una solución homogénea. La solución homogénea obtenida se enfrió a 63 °C y luego se agregaron 0,02 partes de cristales de siembra, seguido de agitación a 63 °C durante 1 hora. La mezcla obtenida se enfrió a -5 °C a una velocidad de enfriamiento de 10 °C/hora y se agitó a -5 °C durante 12 horas. La mezcla obtenida se filtró y el sólido obtenido se lavó con 37,3 partes de heptano enfriado con hielo y luego se secó a presión reducida para obtener 24,1 partes de cristales blancos de la amida del ácido (R)-(-)-N-(1,1,3-trimetilindan-4-il)-1-metil-3-difluorometilpirazol-4-carboxílico. La tasa de recuperación fue del 97,6 % y la proporción de isómeros R/isómeros S fue de 98,2/1,8.
La gráfica XRD de los cristales blancos de la amida del ácido
(RM-)-N-(1,1,3-trimetilindan-4-il)-1-metil-3-difluorometilpirazol-4-carboxílico obtenidos se muestra en la Fig. 1,
Ejemplo 4
<Etapa (F-1)>
A temperatura ambiente en una atmósfera de nitrógeno se mezclaron 79,4 partes de ácido 1-metil-3-difluorometilpirazol-4-carboxílico, 79,6 partes (isómeros R/100 isómeros S = 100,0/0,0) de (R)-1,1,3-trimetil-4-aminoindano, 113,0 partes de hidrocloruro de 1-etil-3-(3-dimetilaminopropil)carbodiimida, 11,0 partes de dimetilaminopiridina, 203,7 partes de piridina y 1059,3 partes de dimetilformamida. La mezcla obtenida se agitó a 125 °C durante 5 horas. La mezcla de reacción obtenida se enfrió a temperatura ambiente. La mezcla obtenida se añadió gota a gota a una mezcla de 2.500 partes de agua con hielo y 170 partes de ácido clorhídrico al 36 %, seguido de extracción con acetato de etilo tres veces. La capa orgánica obtenida se lavó secuencialmente con ácido clorhídrico al 5 %, agua, una solución acuosa de hidróxido de sodio al 5 %, agua, solución salina fisiológica saturada y agua, se secó sobre sulfato de magnesio y luego se concentró a presión reducida para obtener 109,0 partes de la amida del ácido (R)-(-)-N-(1,1,3-trimetilindan-4-il)-1-metil-3-difluorometilpirazol-4-carboxílico. El producto obtenido se purificó por cromatografía en gel de sílice, se recristalizó con acetato de etilo/hexano, y luego se secó a presión reducida para obtener 78,3 partes de cristales blancos de la amida del ácido (R)-(-)-N-(1,1,3-trimetilindan-4-il)-1 -metil-3-difluorometilpirazol-4-carboxílico. La relación de isómeros R/isómeros S fue 100.0/0,0 y la pureza fue 99,9 %.
Ejemplo 5
<Etapa (F-2)>
A temperatura ambiente en una atmósfera de nitrógeno se mezclaron (R)-1,1,3-trimetil-4-aminoindano 4,39 partes (pureza: 90,7 %, isómeros R/isómeros S = 96,0/4,0), 2,02 partes (pureza: 99,0 %) de ácido 1-metil-3-difluorometilpirazol-4-carboxílico, 6,07 partes de xileno y 0,29 partes (pureza: 99 %) de etóxido de antimonio (III). Se eliminó el agua de la mezcla obtenida usando un aparato Dean-Stark y el residuo se calentó y se calentó a reflujo durante 60 horas. El líquido de reacción obtenido se enfrió a temperatura ambiente. El rendimiento de la amida del ácido (R)-(-)-N-(1,1,3-trimetilindan-4-il)-1-metil-3-difluorometilpirazol-4-carboxílico con respecto al ácido 1-metil-3-difluorometilpirazol-4-carboxílico fue de 75,9 %. Además, la relación de isómeros R/isómeros S de la amida del ácido (R)-(-)-N-(1,1,3-trimetilindan-4-il)-1-metil-3-difluorometilpirazol-4-carboxílico fue de 96,0 /4,0.
Ejemplo 6
<Etapa (F-2)>
A temperatura ambiente en una atmósfera de nitrógeno se mezclaron 6,58 partes (pureza: 90,7 %, isómeros R/isómeros S = 96,0/4,0) de (R)-1,1,3-trimetil-4-aminoindano, 3,03 partes (pureza: 99,0 % ) de ácido 1-metil-3-difluorometilpirazol-4-carboxílico, 9,10 partes de tolueno y 0,44 partes de ácido 3,5-bis(trifluorometil)fenil bórico.
Se eliminó el agua de la mezcla obtenida usando un aparato Dean-Stark y el residuo se calentó y se llevó a reflujo durante 120 horas. El líquido de reacción obtenido se enfrió a temperatura ambiente. El rendimiento de la amida del ácido (R)-(-)-N-(1,1,3-trimetilindan-4-il)-1-metil-3-difluorometilpirazol-4-carboxílico con respecto al ácido 1-metil-3-difluorometilpirazol-4-carboxílico fue del 78,4 %. Además, la relación de isómeros R/isómeros S de la amida del ácido (R)-(-)-N-(1,1,3-trimetilindan-4-il)-1-metil-3-difluorometilpirazol-4-carboxílico fue de 96,0/4,0.
Ejemplo 7
<Etapa (F-2)>
A temperatura ambiente en una atmósfera de nitrógeno se mezclaron 6,59 partes (pureza: 90,7 %, isómeros R/isómeros S = 96,0/4,0) de (R)-1,1,3-trimetil-4-aminoindano, 3,03 partes (pureza: 99,0 % ) de ácido 1-metil-3-difluorometilpirazol-4-carboxílico, 9,11 partes de tolueno y 0,11 partes (pureza: 99,5 %) de ácido bórico. Se eliminó el agua de la mezcla obtenida usando un aparato Dean-Stark y el residuo se calentó y se llevó a reflujo durante 96 horas. El líquido de reacción obtenido se enfrió a temperatura ambiente. El rendimiento de la amida del ácido (R)-(-)-N-(1,1,3-trimetilindan-4-il)-1-metil-3-difluorometilpirazol-4-carboxílico con respecto al ácido 1-metil-3-difluorometilpirazol-4-carboxílico fue del 71,3 %. Además, la relación de isómeros R/isómeros S de la amida del ácido (R)-(-)-N-(1,1,3-trimetilindan-4-il)-1-metil-3-difluorometilpirazol-4-carboxílico fue de 96,0 /4,0.
Ejemplo 8
<Etapa (F-3)>
A temperatura ambiente en una atmósfera de nitrógeno se mezclaron y agitaron 5,91 partes (pureza: 95,9 %, isómeros R/isómeros S = 95,4/4,6) de (R)-1,1,3-trimetil-4-aminoindano, 6,13 partes (pureza: 97,8 %) de 1-metil-3-difluorometilpirazol-4-carboxilato de etilo, y 30,7 partes de tetrahidrofurano. A la mezcla obtenida se añadieron 1,75 partes de metóxido de sodio. La mezcla obtenida se calentó en un baño de aceite a 90 °C y se agitó durante 10 horas mientras se evaporaba el tetrahidrofurano. En este momento, se añadió a la misma en varias ocasiones tetrahidrofurano fresco en la misma cantidad que el tetrahidrofurano evaporado, de modo que la concentración del líquido de reacción se mantuvo constante. La mezcla obtenida se enfrió a temperatura ambiente y luego se añadieron 92,0 partes de tolueno. La mezcla obtenida se lavó secuencialmente con ácido clorhídrico al 5 %, una solución saturada de bicarbonato de sodio y solución salina fisiológica saturada. La capa orgánica obtenida se concentró a presión reducida para obtener 12,7 partes de la amida del ácido (R)-(-)-N-(1,1,3-trimetilindan-4-il)-1-metil-3-difluorometilpirazol-4-carboxílico. El contenido de la amida del ácido (R)-(-)-N-(1,1,3-trimetilindan-4-il)-1-metil-3-difluorometilpirazol-4-carboxílico fue del 65,7 % (el rendimiento con respecto al 1-metil-3-difluorometilpirazol-4-carboxilato de etilo de 85,0 %). Además, la relación de los isómeros R/isómeros S de la amida del ácido (R)-(-)-N-(1,1,3-trimetilindan-4-il)-1-metil-3-difluorometilpirazol-4-carboxílico fue de 95,7/4,3.
Ejemplo 9
<Etapa (F-3)>
A temperatura ambiente en una atmósfera de nitrógeno se mezclaron 5,91 partes (pureza: 95,9 %, isómeros R/isómeros S = 95,4/4,6) de (R)-1,1,3-trimetil-4-aminoindano, 21,5 partes de tetrahidrofurano y 1,29 partes (pureza: 60 %) de hidruro de sodio. La mezcla obtenida se calentó y se llevó a reflujo durante 1 hora. El líquido de reacción se enfrió a temperatura ambiente. A la solución obtenida se le añadió gota a gota una solución en la que se habían disuelto 6,13 partes (pureza: 97,8 %) de 1-metil-3-difluorometilpirazol-4-carboxilato de etilo en 9,2 partes de tetrahidrofurano. La mezcla obtenida se calentó en un baño de aceite a 90 °C y se agitó durante 8 horas mientras se evaporaba el tetrahidrofurano. En este momento, se añadió a la misma en varias ocasiones tetrahidrofurano fresco en la misma cantidad que el tetrahidrofurano evaporado, de modo que la concentración del líquido de reacción se mantuvo constante. La mezcla obtenida se enfrió a temperatura ambiente y luego se añadieron 92,0 partes de tolueno. La mezcla obtenida se lavó secuencialmente con ácido clorhídrico al 5 %, una solución saturada de bicarbonato de sodio y solución salina fisiológica saturada. La capa orgánica obtenida se concentró a presión reducida para obtener 13,1 partes de la amida del ácido (R)-(-)-N-(1,1,3-trimetilindan-4-il)-1-metil-3-difluorometilpirazol-4-carboxílico. El contenido de la amida del ácido (R)-(-)-N-(1,1,3-trimetilindan-4-il)-1-metil-3-difluorometilpirazol-4-carboxílico fue de 64,8 % (el rendimiento con respecto al 1-metil-3-difluorometilpirazol-4-carboxilato de etilo de 86,4 %). Además, la relación de los isómeros R/isómeros S de la amida del ácido (R)-(-)-N-(1,1,3-trimetilindan-4-il)-1-metil-3-difluorometilpirazol-4-carboxílico fue de 95,4 /4,6.
Ejemplo 10
<Etapa F-3>
A temperatura ambiente en una atmósfera de nitrógeno se mezclaron 5,92 partes (pureza: 95,9 %, isómeros R/isómeros S = 95,4/4,6) de (R)-1,1,3-trimetil-4-aminoindano, 42,9 partes de tolueno y 2,59 partes (pureza: 60 %) de hidruro de sodio. La mezcla obtenida se calentó y se llevó a reflujo durante 1 hora. El líquido de reacción obtenido se enfrió a temperatura ambiente. A la solución obtenida se le añadió gota a gota una solución en la cual se habían disuelto 6,13 partes (pureza: 97,8 %) de 1-metil-3-difluorometilpirazol-4-carboxilato de etilo en 18,4 partes de tolueno. La mezcla obtenida se calentó y se llevó a reflujo durante 2 horas. El líquido de reacción obtenido se enfrió a temperatura ambiente, y luego se le añadieron 30,7 partes de tolueno. La capa orgánica se lavó secuencialmente con ácido clorhídrico al 5 %, una solución saturada de bicarbonato de sodio y solución salina fisiológica saturada. La capa orgánica obtenida se concentró a presión reducida para obtener 14,7 partes de la amida del ácido (R)-(-)-N-(1,1,3-trimetilindan-4-il)-1-metil-3-difluorometilpirazol-4-carboxílico. El contenido de la amida del ácido (R)-(-)-N-(1,1,3-trimetilindan-4-il)-1-metil-3-difluorometilpirazol-4-carboxílico fue del 54,5 % (el rendimiento con respecto al 1-metil-3-difluorometilpirazol-4-carboxilato de metilo del 81,6 %). Además, la relación de isómeros R/isómeros S de la amida del ácido (R)-(-)-N-(1,1,3-trimetilindan-4-il)-1-metil-3-difluorometilpirazol-4-carboxílico fue de 95,6 /4,4.
Ejemplo 11
<Etapa F-3>
A temperatura ambiente en una atmósfera de nitrógeno se mezclaron 5,02 partes (pureza: 98,5 %) de 1-metil-3-difluorometilpirazol-4-carboxilato de etilo, 7,00 partes (pureza: 90,7 %, isómeros R/isómeros S = 97,6/2,4) de (R)-1,1,3-trimetil-4-aminoindano y 20,3 partes de xileno, seguido de agitación. A la mezcla obtenida se le agregaron 1,12 partes de etóxido de titanio (IV). La mezcla obtenida se calentó en un baño de aceite a 150 °C y se agitó durante 23 horas mientras se evaporaba el xileno. En este momento, se añadió a la misma en varias ocasiones xileno fresco en la misma cantidad que el xileno evaporado, de modo que la concentración del líquido de reacción se mantuvo constante. La mezcla obtenida se enfrió a temperatura ambiente y se lavó secuencialmente con agua, ácido clorhídrico al 10 %, una solución acuosa de hidróxido de sodio al 10 % y agua. La capa orgánica obtenida se concentró a presión reducida para obtener 10,98 partes de la amida del ácido (R)-(-)-N-(1,1,3-trimetilindan-4-il)-1-metil-3-difluorometilpirazol-4-carboxílico. El contenido de la amida del ácido (R)-(-)-N-(1,1,3-trimetilindan-4-il)-1-metil-3-difluorometilpirazol-4-carboxílico fue del 58,6 % (el rendimiento con respecto al 1-metil-3-difluorometilpirazol-4-carboxilato de etilo de 80,0 %). Además, la relación de isómeros R/isómeros S de la amida del ácido (R)-(-)-N-(1,1,3-trimetilindan-4-il)-1-metil-3-difluorometilpirazol-4-carboxílico fue de 97,8/2,2.
Ejemplo 12
<Etapa (F-3)>
A temperatura ambiente en una atmósfera de nitrógeno se mezclaron y se agitaron 5,01 partes (pureza: 98,5 %) de 1-metil-3-difluorometilpirazol-4-carboxilato de etilo, 7,01 partes (pureza: 90,7 %, isómeros R/isómeros S = 97,6/2,4) de (R)-1,1,3-trimetil-4-aminoindano y 20,2 partes de xileno. A la mezcla obtenida se le añadieron 1,04 partes de propóxido de titanio (IV). La mezcla obtenida se calentó en un baño de aceite a 150 °C y se agitó durante 24 horas mientras se evaporaba el xileno. En este momento, se añadió a la misma en varias ocasiones xileno fresco en la misma cantidad que el xileno evaporado, de modo que la concentración del líquido de reacción se mantuvo constante. La mezcla obtenida se enfrió a temperatura ambiente y se lavó secuencialmente con agua, ácido clorhídrico al 10 %, una solución acuosa de hidróxido de sodio al 10 % y agua. La capa orgánica obtenida se concentró a presión reducida para obtener 11,37 partes de la amida del ácido (R)-(-)-N-(1,1,3-trimetilindan-4-il)-1-metil-3-difluorometilpirazol-4-carboxílico. El contenido de la amida del ácido (R)-(-)-N-(1,1,3-trimetilindan-4-il)-1-metil-3-difluorometilpirazol-4-carboxílico fue del 55,9 % (el rendimiento con respecto al 1-metil-3-difluorometilpirazol-4-carboxilato de etilo del 79,1 %). Además, la relación de isómeros R/isómeros S de la amida del ácido (R)-(-)-N-(1,1,3-trimetilindan-4-il)-1 -metil-3-difluorometilpirazol-4-carboxílico fue de 97,8/2,2.
Ejemplo 13
<Etapa (F-3)>
A temperatura ambiente en una atmósfera de nitrógeno se mezclaron y se agitaron 3,02 partes (pureza: 98,5 %) de 1-metil-3-difluorometilpirazol-4-carboxilato de etilo, 4,22 partes (pureza: 90,7 %, isómeros R/isómeros S = 97,6/2,4) de (R)-1,1,3-trimetil-4-aminoindano, y 12,2 partes de xileno. A la mezcla obtenida se añadieron 0,75 partes de etóxido de antimonio (III). La mezcla obtenida se calentó en un baño de aceite a 150 °C y se agitó durante 110 horas mientras se evaporaba el xileno. En este momento, se añadió a la misma en varias ocasiones xileno fresco en la misma cantidad que el xileno evaporado, de modo que la concentración del líquido de reacción se mantuvo constante. La mezcla obtenida se enfrió a temperatura ambiente y luego se filtró usando Celite. La capa orgánica obtenida se lavó secuencialmente con agua, ácido clorhídrico al 10 %, una solución acuosa de hidróxido de sodio al 10 % y agua. La
capa orgánica obtenida se concentró a presión reducida para obtener 6,04 partes de la amida del ácido (R)-(-)-N-(1,1,3-trimetilindan-4-il)-1-metil-3-difluorometilpirazol-4-carboxílico. El contenido de la amida del ácido (R)-(-)-N-(1,1,3-trimetilindan-4-il)-1-metil-3-difluorometilpirazol-4-carboxílico fue del 69,1 % (el rendimiento con respecto al 1-metil-3-difluorometilpirazol-4-carboxilato de etilo de 86,3 %). Además, la relación de isómeros R/isómeros S de la amida del ácido (R)-(-)-N-(1,1,3-trimetilindan-4-il)-1-metil-3-difluorometilpirazol-4-carboxílico fue de 97,8 /2,2.
Ejemplo 14
<Etapa (F-3)>
A temperatura ambiente en una atmósfera de nitrógeno se mezclaron y agitaron 3,01 partes (pureza: 98,5 %) de 1-metil-3-difluorometilpirazol-4-carboxilato de etilo, 4,21 partes (pureza: 90,7 %, isómeros R/isómeros S = 97,6/2,4) de (R)-1,1,3-trimetil-4-aminoindano y 21,3 partes de clorobenceno. A la mezcla obtenida se añadieron 2,36 partes de etóxido de aluminio (III). La mezcla obtenida se calentó en un baño de aceite a 150 °C y se agitó durante 82 horas mientras se evaporaba el clorobenceno. En este momento, se añadió a la misma en varias ocasiones clorobenceno fresco en la misma cantidad que el clorobenceno evaporado, de modo que la concentración del líquido de reacción se mantuvo constante. La mezcla obtenida se enfrió a temperatura ambiente y luego se filtró usando Celite. La capa orgánica obtenida se lavó secuencialmente con agua, ácido clorhídrico al 10 %, una solución acuosa de hidróxido de sodio al 10 % y agua. La capa orgánica obtenida se concentró a presión reducida para obtener 6,50 partes de la amida del ácido (R)-(-)-N-(1,1,3-trimetilindan-4-il)-1-metil-3-difluorometilpirazol-4-carboxílico. El contenido de la amida del ácido (R)-(-)-N-(1,1,3-trimetilindan-4-il)-1-metil-3-difluorometilpirazol-4-carboxílico fue del 35,0 % (el rendimiento con respecto al 1-metil-3-difluorometilpirazol-4-carboxilato de etilo de 47,0 %). Además, la proporción de isómeros R/isómeros S de la amida del ácido (R)-(-)-N-(1,1,3-trimetilindan-4-il)-1-metil-3-difluorometilpirazol-4-carboxílico fue de 97,7 /2,3.
<Ejemplo de referencia 1>
El gráfico XRD, el espectro de FT-Raman y el gráfico de DSC/TGA de los cristales blancos de la amida del ácido (R)-(-)-N-(1,1,3-trimetilindan-4-il)-1-metil-3-difluorometilpirazol-4-carboxílico (una relación de isómeros R/isómeros S de 100,0/0,0) sintetizada realizando la misma operación que en el Ejemplo 3, se muestran en la Fig. 2, Fig. 3 y Fig. 4, respectivamente.
<Ejemplo de referencia 2>
A 180 mg de la amida del ácido (R)-(-)-N-(1,1,3-trimetilindan-4-il)-1-metil-3-difluorometilpirazol-4-carboxílico sintetizada en el Ejemplo de referencia 1 se agregaron 250 |jl de etanol/agua (relación de volumen: 95/5). La suspensión obtenida se agitó durante 40 horas mientras se repetía el calentamiento y enfriamiento en un rango de 30 °C a 5 °C. La suspensión obtenida se filtró y el filtrado obtenido se enfrió a 4 °C durante 5 días y luego se enfrió a -20 °C durante 24 horas. La solución obtenida se calentó a temperatura ambiente y luego el disolvente se concentró lentamente durante 6 días. La mezcla obtenida se filtró y el sólido obtenido se secó al aire durante 1 hora para obtener un sólido del etanol/hidrato de la amida del ácido (R)-(-)-N-(1,1,3-trimetilindan-4-il)-1-metil-3-difluorometilpirazol-4-carboxílico. La gráfica XRD, el espectro FT-Raman y la gráfica DSC/TGA del sólido obtenido se muestran en la Fig. 5, Fig. 6 y Fig. 7, respectivamente.
<Ejemplo de referencia 3>
A 150 mg de la amida del ácido (R)-(-)-N-(1,1,3-trimetilindan-4-il)-1-metil-3-difluorometilpirazol-4-carboxílico sintetizada en el Ejemplo de referencia 1 se agregaron 200 j l de etanol/agua (relación de volumen: 95/5). La suspensión obtenida se agitó a 50°C durante 30 minutos para obtener una solución homogénea. La solución obtenida se filtró en un recipiente con 1 mg de etanol/hidrato de la amida del ácido (R)-(-)-N-(1,1,3-trimetilindan-4-il)-1-metil-3-difluorometilpirazol-4-carboxílico obtenida en el Ejemplo de referencia 2, y la suspensión obtenida se agitó a temperatura ambiente durante 1 hora. La mezcla obtenida se filtró y luego el sólido obtenido se secó al aire durante 1 hora para obtener un sólido del etanol/hidrato de la amida del ácido (R)-(-)-N-(1,1,3-trimetilindan-4-il)-1-metil-3-difluorometilpirazol-4-carboxílico. El gráfico de XRD del sólido obtenido fue el mismo que en la Fig. 5.
<Ejemplo de referencia 4>
Veinte mg del sólido del etanol/hidrato de la amida del ácido (R)-(-)-N-(1,1,3-trimetilindan-4-il)-1-metil-3-difluorometilpirazol-4-carboxílico obtenida en el Ejemplo de referencia 2 se secaron a 50 °C durante 24 horas mientras se burbujeaba con nitrógeno a presión reducida. El gráfico XRD, el espectro FT-Raman y el gráfico DSC/TGA del sólido obtenido de la amida del ácido (R)-(-)-N-(1,1,3-trimetilindan-4-il)-1-metil-3-difluorometilpirazol-4-carboxílico se muestran en la Fig. 8, Fig. 9 y Fig. 10, respectivamente.
<Ejemplo de referencia 5>
Veinte mg del sólido del etanol/hidrato de la amida del ácido (R)-(-)-N-(1,1,3-trimetilindan-4-il)-1-metil-3difluorometilpirazol-4-carboxílico obtenida en el Ejemplo de referencia 2 se dejó reposar durante 48 horas. El gráfico XRD, el espectro FT-Raman y el gráfico DSC/TGA del hidrato de la amida del ácido (RM-)-N-(1,1,3-trimetilindan-4-il)-1-metil-3-difluorometilpirazol-4-carboxílico obtenido se muestran en la Fig. 11, la Fig. 12 y la Fig. 13 respectivamente.
<Ejemplo de referencia 6>
A 60 mg de la amida del ácido (R)-(-)-N-(1,1,3-trimetilindan-4-il)-1-metil-3-difluorometilpirazol-4-carboxílico sintetizada en el Ejemplo de referencia 1 se agregaron 750 |jl de hexano y 30 |jl de 2-metoxietanol. La suspensión obtenida se agitó durante 40 horas mientras se repetía el calentamiento y enfriamiento en un rango de 30 °C a 5 °C. La mezcla obtenida se filtró y el sólido obtenido se secó al aire durante 3 horas para obtener un sólido del 2-metoxietanol/hidrato de la amida del ácido (R)-(-)-N-(1,1,3-trimetilindan-4-il)-1-metil-3-difluorometilpirazol-4-carboxílico. El gráfico de XRD del sólido obtenido fue el mismo que en la Fig. 14.
<Ejemplo de referencia 7>
A 60 mg de la amida del ácido (R)-(-)-N-(1,1,3-trimetilindan-4-il)-1-metil-3-difluorometilpirazol-4-carboxílico sintetizada en el Ejemplo de referencia 1 se agregaron 800 j l de hexano y 30 j l de 2-metoxietanol. La suspensión obtenida se agitó a 30 °C durante 1 hora. La suspensión obtenida se enfrió a 5 °C y se añadió a la misma 2 mg del 2-metoxietanol/hidrato de la amida del ácido (R)-(-)-N-(1,1,3-trimetilindan-4-il)-1-metil-3-difluorometilpirazol-4-carboxílico obtenida en el Ejemplo de referencia 6, seguido de agitación durante 20 horas. La mezcla obtenida se filtró y el sólido obtenido se secó al aire durante 3 horas para obtener un sólido del 2-metoxietanol/hidrato de la amida del ácido (R)-(-)-N-(1,1,3-trimetilindan-4-il)-1-metil-3-difluorometilpirazol-4-carboxílico. La gráfica XRD, el espectro FT-Raman y la gráfica DSC/TGA del sólido obtenido se muestran en la Fig. 14, Fig. 15 y Fig. 16, respectivamente.
<Ejemplo de referencia 8>
A 120 mg de la amida del ácido (R)-(-)-N-(1,1,3-trimetilindan-4-il)-1-metil-3-difluorometilpirazol-4-carboxílico sintetizada en el Ejemplo de referencia 1 se agregaron 750 j l de ciclohexano y 60 j l de 1-propanol. La suspensión obtenida se agitó durante 40 horas mientras se repetía el calentamiento y enfriamiento en un rango de 30 °C a 5 °C. La suspensión obtenida se filtró y el filtrado obtenido se enfrió a 4 °C durante 5 días y se enfrió a -20 °C durante 24 horas. La mezcla obtenida se filtró y el sólido obtenido se secó al aire durante 1 hora para obtener un sólido del 1-propanol/ciclohexano/hidrato de la amida del ácido (R)-(-)-N-(1,1,3-trimetilindan-4-il)-1-metil-3-difluorometilpirazol-4-carboxílico. El gráfico XRD del sólido obtenido es el mismo que en la Fig. 17.
<Ejemplo de referencia 9>
A 151 mg de la amida del ácido (R)-(-)-N-(1,1,3-trimetilindan-4-il)-1-metil-3-difluorometilpirazol-4-carboxílico sintetizada en el Ejemplo de referencia 1 se agregaron 750 j l de ciclohexano y 60 j l de 1-propanol. La suspensión obtenida se agitó a 50 °C durante 70 minutos para obtener una solución homogénea. La solución obtenida se filtró en un recipiente enfriado a 5 °C que contenía 1 mg del 1-propanol/ciclohexano/hidrato de la amida del ácido (R)-(-)-N-(1,1,3-trimetilindan-4-il)-1-metil-3-difluorometilpirazol-4-carboxílico obtenida en el Ejemplo de referencia 8, y la suspensión obtenida se agitó a 5 °C durante 2 horas. La mezcla obtenida se filtró y el sólido obtenido se secó al aire durante 1 hora para obtener un sólido del 1-propanol/ciclohexano/hidrato de la amida del ácido (R)-(-)-N-(1,1,3-trimetilindan-4-il)-1-metil-3-difluorometilpirazol-4-carboxílico. La gráfica XRD, el espectro FT-Raman y la gráfica DSC/TGA del sólido obtenido se muestran en la Fig. 17, Fig. 18 y Fig. 19, respectivamente.
<Ejemplo de referencia 10>
A 100 mg de la amida del ácido (R)-(-)-N-(1,1,3-trimetilindan-4-il)-1-metil-3-difluorometilpirazol-4-carboxílico sintetizada en el Ejemplo de referencia 1 se agregaron 750 j l de tetrahidrofurano/agua (relación de volumen: 20/80). La suspensión obtenida se agitó durante 40 horas mientras se repetía el calentamiento y enfriamiento en un rango de 30 °C a 5 °C. La suspensión obtenida se filtró y el filtrado obtenido se enfrió a 4 °C durante 5 días y se enfrió a -20 °C durante 24 horas. La mezcla obtenida se filtró y el sólido obtenido se secó al aire durante 1 hora para obtener un sólido del tetrahidrofurano/hidrato de la amida del ácido (R)-(-)-N-(1,1,3-trimetilindan-4-il)-1-metil-3-difluorometilpirazol-4-carboxílico. El gráfico XRD del sólido obtenido es el mismo que en la Fig. 20.
<Ejemplo de referencia 11>
A 301 mg de la amida del ácido (R)-(-)-N-(1,1,3-trimetilindan-4-il)-1-metil-3-difluorometilpirazol-4-carboxílico sintetizada en el Ejemplo de referencia 1, se agregó 1,6 ml de tetrahidrofurano/agua (relación de volumen: 20/80). La suspensión obtenida se agitó a 30 °C durante 1 hora. La suspensión obtenida se enfrió a 5 °C. A la mezcla obtenida se le agregaron 2 mg del tetrahidrofurano/hidrato de la amida del ácido (R)-(-)-N-(1,1,3-trimetilindan-4-il)-1-metil-3-difluorometilpirazol-4-carboxílico obtenida en el Ejemplo de referencia 10, seguido de agitación durante 20 horas. La mezcla obtenida se filtró y el sólido obtenido se secó al aire durante 1 hora para obtener un sólido del tetrahidrofurano/hidrato de la amida del ácido (R)-(-)-N-(1,1,3-trimetilindan-4-il)-1-metil-3-difluorometilpirazol-4-carboxílico. La gráfica XRD, el espectro FT-Raman y la gráfica DSC/TGA del sólido obtenido se muestran en la Fig.
20, Fig. 21 y Fig. 22, respectivamente.
<Ejemplo de referencia 12>
A 100 mg de la amida del ácido (RM-)-N-(1,1,3-trimetilindan-4-il)-1-metil-3-difluorometilpirazol-4-carboxNico sintetizada en el Ejemplo de referencia 1 se agregaron 750 pl de heptano y 30 pl de dimetilsulfóxido. La suspensión obtenida se agitó durante 40 horas mientras se repetía el calentamiento y enfriamiento en un rango de 30 °C a 5 °C. La mezcla obtenida se filtró y el sólido obtenido se secó al aire durante 1 hora para obtener un sólido del solvato de dimetilsulfóxido de la amida del ácido (R)-(-)-N-(1,1,3-trimetilindan-4-il)-1-metil-3-difluorometilpirazol-4-carboxílico. El gráfico XRD del sólido obtenido es el mismo que en la Fig. 23.
<Ejemplo de referencia 13>
A 301 mg de la amida del ácido (R)-(-)-N-(1,1,3-trimetilindan-4-il)-1-metil-3-difluorometilpirazol-4-carboxílico sintetizada en el Ejemplo de referencia 1 se agregaron 2,2 ml de heptano y 90 pl de dimetilsulfóxido. La suspensión obtenida se agitó a 30 °C durante 1 hora. La suspensión obtenida se enfrió a 5 °C. A la mezcla obtenida se le agregaron 2 mg de solvato de dimetilsulfóxido de la amida del ácido (R)-(-)-N-(1,1,3-trimetilindan-4-il)-1-metil-3-difluorometilpirazol-4-carboxílico obtenida en el Ejemplo de referencia 12, seguido de agitación durante 20 horas. La mezcla obtenida se filtró y el sólido obtenido se secó al aire durante 15 minutos. Al sólido obtenido se añadieron 2,0 ml de heptano, seguido de agitación a temperatura ambiente durante 4 horas. La mezcla obtenida se filtró y el sólido obtenido se lavó con 3,0 ml de heptano. El sólido obtenido se secó a 50 °C durante 20 horas mientras se burbujeaba con nitrógeno a presión reducida para obtener un sólido del solvato de dimetilsulfóxido de la amida del ácido (R)-(-)-N-(1,1,3-trimetilindan-4-il)-1-metil-3-difluorometilpirazol-4-carboxílico. La gráfica XRD, el espectro FT-Raman y la gráfica DSC/TGA del sólido obtenido se muestran en la Fig. 23, Fig. 24 y Fig. 25, respectivamente.
<Ejemplo de referencia 14>
A 5,1 g de la amida del ácido (R)-(-)-N-(1,1,3-trimetilindan-4-il)-1-metil-3-difluorometilpirazol-4-carboxílico sintetizada en el Ejemplo 3 se agregaron 6,5 g de xileno La suspensión obtenida se agitó a 75 °C durante 1 hora para obtener una solución homogénea. La solución obtenida se enfrió a 40 °C y se agitó durante 22 horas. La mezcla obtenida se filtró y el sólido obtenido se lavó con 4,7 g de xileno. El sólido obtenido se secó a 50 °C durante 3 horas a presión reducida para obtener un sólido del solvato de xileno de la amida del ácido (R)-(-)-N-(1,1,3-trimetilindan-4-il)-1 -metil-3-difluorometilpirazol-4-carboxílico. El gráfico XRD y el gráfico TG/DTA del sólido obtenido se muestran en la Fig. 26 y la Fig. 27, respectivamente.
Aplicabilidad industrial
De acuerdo con la presente invención, se puede obtener (R)-1,1,3-trimetil-4-aminoindano con un alto rendimiento.
Claims (13)
1. Un método para producir (R)-1,1,3-trimetil-4-aminoindano, que comprende las siguientes etapas (A), (B) y (C):
(A) : una etapa de resolución óptica del 1,1,3-trimetil-4-aminoindano con ácido D-tartárico para obtener (R)-1,1,3-trimetil-4-aminoindano y (S)-1,1,3-trimetil-4-aminoindano;
(B) : una etapa de racemización del (S)-1,1,3-trimetil-4-aminoindano obtenido en las etapas (A) o (C) para obtener 1.1.3- trimetil-4-aminoindano; y
(C) : una etapa de resolución óptica del 1,1,3-trimetil-4-aminoindano obtenido en la etapa (B) para obtener (R)-1.1.3- trimetil-4-aminoindano y (S)-1,1,3-trimetil-4-aminoindano,
en el que la etapa (B) es una etapa en la que se pone en contacto el (S)-1,1,3-trimetil-4-aminoindano obtenido en las etapas (A) o (C) con un catalizador de metal de transición para realizar la racemización.
2. El método de producción de acuerdo con la reivindicación 1, en el que se repiten las etapas (B) y (C).
3. El método de producción de acuerdo con las reivindicaciones 1 o 2, en el que la etapa (C) es una etapa de resolución óptica del 1,1,3-trimetil-4-aminoindano obtenido en la etapa (B) y el 1,1,3-trimetil-4-aminoindano obtenido en una etapa diferente a la etapa (B) con ácido D-tartárico para obtener (R)-1,1,3-trimetil-4-aminoindano.
4. El método de producción de acuerdo con la reivindicación 1, que comprende las siguientes etapas (A), (B1), (D) y (E):
(A): una etapa de resolución óptica del 1,1,3-trimetil-4-aminoindano con ácido D-tartárico para obtener (R)-1,1,3-trimetil-4-aminoindano y (S)-1,1,3-trimetil-4-aminoindano;
(B'): una etapa de racemización del (S)-1,1,3-trimetil-4-aminoindano obtenido en las etapas (A) o (E) para obtener 1.1.3- trimetil-4-aminoindano;
(D) : una etapa de purificación del 1,1,3-trimetil-4-aminoindano obtenido en la etapa (B1); y
(E) : una etapa de resolución óptica del 1,1,3-trimetil-4-aminoindano obtenido en la etapa (D) para obtener (R)-1.1.3- trimetil-4-aminoindano y (S)-1,1,3-trimetil-4-aminoindano.
5. El método de producción de acuerdo con la reivindicación 4, en el que se repiten las etapas (B'), (D) y (E).
6. El método de producción de acuerdo con una cualquiera de las reivindicaciones 1 a 5, en el que las etapas (B) o (B1) son una etapa en la que se pone en contacto el (S)-1,1,3-trimetil-4-aminoindano obtenido en la etapa (A) con un catalizador de metal de transición para realizar la racemización.
7. El método de producción de acuerdo con una cualquiera de las reivindicaciones 1 a 6, en el que la etapa (A) es una etapa que comprende las siguientes etapas (A1), (A2), (A3) y (A4):
(A1): una etapa de mezclar 1,1,3-trimetil-4-aminoindano con ácido D-tartárico y metanol para obtener una mezcla que incluye un solvato de metanol de D-tartrato de (R)-1,1,3-trimetil-4-aminoindano y D-tartrato de (S)-1,1,3-trimetil-4-aminoindano;
(A2): una etapa de separación de una solución que contiene D-tartrato de (S)-1,1,3-trimetil-4-aminoindano y el solvato de metanol de D-tartrato de (R)-1,1,3-trimetil-4-aminoindano de la mezcla obtenida en la etapa (A1);
(A3): una etapa de mezclar el solvato de metanol de D-tartrato de (R)-1,1,3-trimetil-4-aminoindano obtenido en la etapa (A2) y una solución acuosa de hidróxido de metal alcalino o una solución acuosa de carbonato de metal alcalino para obtener (R)-1,1,3-trimetil-4-aminoindano; y
(A4): una etapa de mezclar la solución que incluye el D-tartrato de (S)-1,1,3-trimetil-4-aminoindano obtenido en la etapa (A2) y una solución acuosa de hidróxido de metal alcalino o una solución acuosa de carbonato de metal alcalino para obtener (S)-1,1,3-trimetil-4-aminoindano.
8. El método de producción de acuerdo con la reivindicación 7, en el que el agua se mezcla con el sistema de reacción antes de la etapa (A2).
9. El método de producción de acuerdo con una cualquiera de las reivindicaciones 4 a 8, en el que la etapa (D) es una etapa que comprende las siguientes etapas (D1), (D2), (D3) y (D4):
(D1): una etapa en la que se hace reaccionar el 1,1,3-trimetil-4-aminoindano obtenido en la etapa (B') con hidruro de hidrógeno en presencia de agua y un disolvente orgánico insoluble en agua;
(D2): una etapa de separar una capa disolviendo en la misma una sal de haluro de hidrógeno de 1,1,3-trimetil-4-aminoindano incluida en la mezcla obtenida en la etapa (D1) de otra capa;
(D3): una etapa de precipitación de la sal de haluro de hidrógeno de 1,1,3-trimetil-4-aminoindano de la capa disolviendo en la misma la sal de haluro de hidrógeno de 1,1,3-trimetil-4-aminoindano obtenida en la etapa (D2); y (D4): una etapa de extracción de la sal de haluro de hidrógeno de 1,1,3-trimetil-4-aminoindano obtenida en la etapa (D3) y hacer reaccionar la sal de haluro de hidrógeno obtenida de esta manera con una base.
10. El método de producción de acuerdo con la reivindicación 9, en el que el haluro de hidrógeno es cloruro de hidrógeno.
11. Un método de producción de acuerdo con la reivindicación 1, que comprende además la etapa (F):
(F): una etapa en la que se hace reaccionar el (R)-1,1,3-trimetil-4-aminoindano obtenido en la etapa (C) con un compuesto representado por la fórmula (2):

en la que R1 y R2 tienen los mismos significados que se definen abajo, y R3 representa un átomo de halógeno, un grupo hidroxilo o un grupo alcoxi que puede estar sustituido con un átomo de halógeno
para obtener el compuesto representado por la fórmula (1):

en la que cada uno de R1 y R2 representa independientemente un grupo alquilo que puede estar sustituido con un átomo de halógeno o un átomo de hidrógeno.
12. Un método de acuerdo con la reivindicación 1, que comprende además las siguientes etapas (G) y (H):
(G): una etapa para obtener un compuesto representado por la fórmula (4) a partir de un compuesto representado por la fórmula (3):

en la que R1 y R2 tienen los mismos significados que abajo

en la que R1 y R2 tienen los mismos significados que abajo; y
(H): una etapa en la que se hace reaccionar el compuesto representado por la fórmula (4) obtenido en la etapa (G) con el (R)-1,1,3-trimetil-4-aminoindano obtenido en la etapa (C) en presencia de una base para obtener el compuesto representado por la fórmula (1):
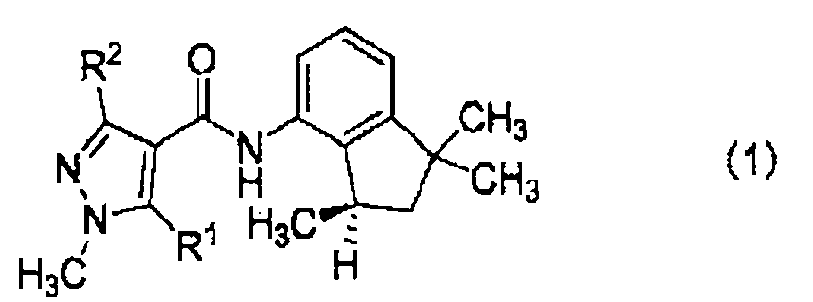
átomo de halógeno o un átomo de hidrógeno.
13. El método de producción de acuerdo con las reivindicaciones 11 o 12, en el que R1 es un átomo de hidrógeno o un grupo metilo, y R2 es un grupo metilo, un grupo monofluorometilo, un grupo difluorometilo o un grupo trifluorometilo.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2014021976 | 2014-02-07 | ||
| PCT/JP2014/084766 WO2015118793A1 (ja) | 2014-02-07 | 2014-12-26 | (r)-1,1,3-トリメチル-4-アミノインダンの製造方法 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2702891T3 true ES2702891T3 (es) | 2019-03-06 |
Family
ID=53777608
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES14882034T Active ES2702891T3 (es) | 2014-02-07 | 2014-12-26 | Método para producir (R)-1,1,3-Trimetil-4-aminoindano |
Country Status (14)
| Country | Link |
|---|---|
| US (1) | US9765032B2 (es) |
| EP (1) | EP3103789B1 (es) |
| JP (1) | JP6451652B2 (es) |
| KR (1) | KR102264639B1 (es) |
| CN (1) | CN105992755B (es) |
| AR (1) | AR101434A1 (es) |
| AU (1) | AU2014381760B2 (es) |
| BR (1) | BR112016017648B1 (es) |
| DK (1) | DK3103789T3 (es) |
| ES (1) | ES2702891T3 (es) |
| IL (1) | IL246923B (es) |
| RU (1) | RU2016135762A (es) |
| TW (1) | TWI646071B (es) |
| WO (1) | WO2015118793A1 (es) |
Families Citing this family (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR102204520B1 (ko) | 2012-10-12 | 2021-01-20 | 엑셀리시스, 인코포레이티드 | 암의 치료에 사용하기 위한 화합물의 신규 제조 방법 |
| EP3677572B1 (en) * | 2017-08-28 | 2023-06-07 | Japan Finechem Company, Inc. | Production method for pyrazole-4-carboxamide derivative |
| DK3774735T3 (da) | 2018-03-26 | 2023-12-11 | Bayer Ag | Enantioselektiv hydrering af 4-substitueret 1,2-dihydroquinoliner i nærvær af en chiral iridiumkatalysator |
| KR102886811B1 (ko) | 2019-09-25 | 2025-11-14 | 바이엘 악티엔게젤샤프트 | 4-치환된 1,2-디히드로퀴놀린의 거울상이성질체선택적 수소화를 위한 신규한 이리듐 촉매의 사용을 포함하는 방법 |
| MX2022003448A (es) | 2019-09-25 | 2022-04-19 | Bayer Ag | Mejora de la hidrogenacion enantioselectiva de 1,2-dihidroquinolinas 4-sustituidas en presencia de un catalizador quiral de iridio y un aditivo. |
| IT201900017330A1 (it) * | 2019-09-26 | 2021-03-26 | Isagro Spa | Processo per la preparazione di (r)-4-amminoindani e corrispondenti ammidi. |
| IL292513B2 (en) * | 2019-11-01 | 2025-11-01 | Sumitomo Chemical Co | Crystal of 3-(difluoromethyl)-1-methyl-n-(1,1,3-trimethyl-2,3-dihydro-1h-inden-4-yl)-1h-pyrazole-4-carboxamide |
| JP7662640B2 (ja) | 2020-07-17 | 2025-04-15 | 住友化学株式会社 | 光学活性な化合物の製造方法 |
| JP2023120459A (ja) * | 2020-07-17 | 2023-08-30 | 住友化学株式会社 | 光学活性な1,1,3-トリメチル-4-アミノインダンの製造方法 |
| CN114773263B (zh) * | 2022-05-16 | 2023-09-05 | 江苏百康德医药科技有限公司 | (r)-2,2,4-三甲基-1,2,3,4-四氢喹啉的制备方法 |
| CN116396189B (zh) * | 2023-04-06 | 2024-11-05 | 无锡科华生物科技有限公司 | 一种(s)-(+)-1-茚满基异氰酸酯的制备方法 |
Family Cites Families (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5093347A (en) | 1991-01-28 | 1992-03-03 | Monsanto Company | 3-difluoromethylpyrazolecarboxamide fungicides, compositions and use |
| US5521317A (en) | 1993-10-22 | 1996-05-28 | American Cyanamid Co. | Processes for the preparation of pesticides and intermediates |
| JP3892503B2 (ja) * | 1996-09-02 | 2007-03-14 | 長瀬産業株式会社 | 接触還元触媒を用いる光学活性アミンのラセミ化法 |
| JP4834333B2 (ja) * | 2005-06-27 | 2011-12-14 | アルプス薬品工業株式会社 | 光学活性ベンジルアミン誘導体の製造方法 |
| JP2011012032A (ja) * | 2009-07-03 | 2011-01-20 | Yamakawa Yakuhin Kogyo Kk | 光学活性な3−アミノピペリジンの製造方法および製造の中間体 |
| JP2012025735A (ja) | 2010-06-24 | 2012-02-09 | Sumitomo Chemical Co Ltd | 植物病害防除組成物及び植物病害防除方法 |
| JP6106976B2 (ja) * | 2012-07-20 | 2017-04-05 | 住友化学株式会社 | 植物病害防除組成物およびその用途 |
| BR112015003520B1 (pt) | 2012-08-31 | 2020-10-13 | Sumitomo Chemical Company, Limited | método para a produção de (r)-1,1,3-trimetil-4-aminoindano |
| BR112015009202B1 (pt) | 2012-11-02 | 2021-01-19 | Sumitomo Chemical Company, Limited | método para produzir racemato de composto |
| WO2014103812A1 (ja) | 2012-12-25 | 2014-07-03 | 住友化学株式会社 | ピラゾール化合物の結晶の製造方法 |
| WO2014103811A1 (ja) | 2012-12-27 | 2014-07-03 | 住友化学株式会社 | 精製されたアミン化合物の製造方法 |
-
2014
- 2014-12-26 US US15/117,058 patent/US9765032B2/en active Active
- 2014-12-26 KR KR1020167021175A patent/KR102264639B1/ko active Active
- 2014-12-26 JP JP2015561187A patent/JP6451652B2/ja active Active
- 2014-12-26 WO PCT/JP2014/084766 patent/WO2015118793A1/ja not_active Ceased
- 2014-12-26 ES ES14882034T patent/ES2702891T3/es active Active
- 2014-12-26 CN CN201480075004.4A patent/CN105992755B/zh active Active
- 2014-12-26 BR BR112016017648-0A patent/BR112016017648B1/pt active IP Right Grant
- 2014-12-26 AU AU2014381760A patent/AU2014381760B2/en active Active
- 2014-12-26 EP EP14882034.3A patent/EP3103789B1/en active Active
- 2014-12-26 RU RU2016135762A patent/RU2016135762A/ru not_active Application Discontinuation
- 2014-12-26 DK DK14882034.3T patent/DK3103789T3/en active
-
2015
- 2015-01-09 TW TW104100693A patent/TWI646071B/zh active
- 2015-01-30 AR ARP150100281A patent/AR101434A1/es active IP Right Grant
-
2016
- 2016-07-25 IL IL246923A patent/IL246923B/en active IP Right Grant
Also Published As
| Publication number | Publication date |
|---|---|
| AR101434A1 (es) | 2016-12-21 |
| US20170166532A1 (en) | 2017-06-15 |
| IL246923A0 (en) | 2016-09-29 |
| CN105992755A (zh) | 2016-10-05 |
| EP3103789B1 (en) | 2018-10-31 |
| DK3103789T3 (en) | 2019-01-28 |
| EP3103789A1 (en) | 2016-12-14 |
| AU2014381760A1 (en) | 2016-08-18 |
| EP3103789A4 (en) | 2017-07-05 |
| RU2016135762A3 (es) | 2018-08-01 |
| JPWO2015118793A1 (ja) | 2017-03-23 |
| AU2014381760B2 (en) | 2018-08-02 |
| RU2016135762A (ru) | 2018-03-14 |
| IL246923B (en) | 2020-08-31 |
| CN105992755B (zh) | 2018-07-13 |
| BR112016017648B1 (pt) | 2021-01-19 |
| US9765032B2 (en) | 2017-09-19 |
| TW201612153A (en) | 2016-04-01 |
| KR20160118256A (ko) | 2016-10-11 |
| KR102264639B1 (ko) | 2021-06-11 |
| JP6451652B2 (ja) | 2019-01-16 |
| TWI646071B (zh) | 2019-01-01 |
| WO2015118793A1 (ja) | 2015-08-13 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2702891T3 (es) | Método para producir (R)-1,1,3-Trimetil-4-aminoindano | |
| KR102593431B1 (ko) | C5aR 길항제의 제조 방법 및 이러한 제조에서의 중간체 | |
| BRPI0407904B1 (pt) | processo para preparação de sal de 5-[(r)-2-(5,6-dietil-indan-2-ilamino) -1-hidróxi-etil]-8-hidróxi-(1h)-quinolin-2-ona, intermediários, bem como sal assim obtido | |
| JP6269508B2 (ja) | 精製されたアミン化合物の製造方法 | |
| ES2637744T3 (es) | Proceso para la síntesis industrial de lurasidona | |
| US20080058523A1 (en) | Processes for synthesizing piperazine-piperidine compounds | |
| US8013158B2 (en) | Crystalline forms of topotecan hydrochloride and processes for making the same | |
| JP7662640B2 (ja) | 光学活性な化合物の製造方法 | |
| TWI628181B (zh) | 一種dpp-iv抑制劑的中間體、其製備方法和藉由其製備dpp-iv抑制劑的方法 | |
| CN101638385A (zh) | 一种新型喹诺酮类衍生物及其制备和应用 | |
| CN106458965A (zh) | 杂芳基羧酸酯衍生物的制造方法及其制造中间体 | |
| CN118005619A (zh) | 用于生产杂环化合物的方法 | |
| CN103974929B (zh) | 卤化烷基胺的氢卤酸盐的制造方法 | |
| CN120549923A (zh) | 一种喹啉酮类双位配体衍生物作为单胺类神经递质转运体抑制剂的应用 | |
| WO2022014413A1 (ja) | 光学活性な1,1,3-トリメチル-4-アミノインダンの製造方法 | |
| AU2004319901A1 (en) | Stereoselective method for the production of clopidogrel | |
| JPH0474353B2 (es) | ||
| CN101638384A (zh) | 一种新型喹诺酮类化合物及其制备和应用 |