ES2703253T3 - Nueva combinación y uso - Google Patents
Nueva combinación y uso Download PDFInfo
- Publication number
- ES2703253T3 ES2703253T3 ES11714379T ES11714379T ES2703253T3 ES 2703253 T3 ES2703253 T3 ES 2703253T3 ES 11714379 T ES11714379 T ES 11714379T ES 11714379 T ES11714379 T ES 11714379T ES 2703253 T3 ES2703253 T3 ES 2703253T3
- Authority
- ES
- Spain
- Prior art keywords
- infections
- combination
- pharmaceutically acceptable
- methyl
- benzylpyrrolidin
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
- A61K31/4453—Non condensed piperidines, e.g. piperocaine only substituted in position 1, e.g. propipocaine, diperodon
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/335—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin
- A61K31/35—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin having six-membered rings with one oxygen as the only ring hetero atom
- A61K31/351—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin having six-membered rings with one oxygen as the only ring hetero atom not condensed with another ring
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4427—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems
- A61K31/4439—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems containing a five-membered ring with nitrogen as a ring hetero atom, e.g. omeprazole
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/47—Quinolines; Isoquinolines
- A61K31/4709—Non-condensed quinolines and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/47—Quinolines; Isoquinolines
- A61K31/4738—Quinolines; Isoquinolines ortho- or peri-condensed with heterocyclic ring systems
- A61K31/4745—Quinolines; Isoquinolines ortho- or peri-condensed with heterocyclic ring systems condensed with ring systems having nitrogen as a ring hetero atom, e.g. phenantrolines
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P23/00—Anaesthetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P23/00—Anaesthetics
- A61P23/02—Local anaesthetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
- A61P31/06—Antibacterial agents for tuberculosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/10—Antimycotics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02A—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE
- Y02A50/00—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE in human health protection, e.g. against extreme weather
- Y02A50/30—Against vector-borne diseases, e.g. mosquito-borne, fly-borne, tick-borne or waterborne diseases whose impact is exacerbated by climate change
Landscapes
- Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Chemical & Material Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Medicinal Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Epidemiology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Organic Chemistry (AREA)
- Oncology (AREA)
- Communicable Diseases (AREA)
- Anesthesiology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Pulmonology (AREA)
- Engineering & Computer Science (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
Abstract
Diclonina o una sal farmacéuticamente acceptable de la misma en combinación con un agente antimicrobiano seleccionado de mupirocina, 4-metil-1-(2-feniletil)-8-fenoxi-2,3-dihidro-1H-pirrolo[3,2-c]-quinolina, 4-(3- bencilpirrolidin-1-il)-2-metil-6-fenoxiquinolina, y N-[4-(3-bencilpirrolidin-1-il)-2-metilquinolin-6-il]benzamida o sales faracéuticamente aceptables de los mismos para su uso en la eliminación de microorganismos clínicamente latentes asociados con una infección microbiana.
Description
DESCRIPCIÓN
Nueva combinación y uso
La presente invención se refiere al uso de un agente anestésico para eliminar microorganismos clínicamente latentes asociados con infecciones microbianas y a nuevas combinaciones que comprenden un agente anestésico y un agente antimicrobiano para el tratamiento de infecciones microbianas.
Antes de la introducción de los antibióticos, los pacientes que padecían infecciones microbianas agudas (por ejemplo, tuberculosis o neumonía) tenían pocas posibilidades de sobrevivir. Por ejemplo, la mortalidad por tuberculosis era de aproximadamente 50 %. Aunque la introducción de agentes antimicrobianos en la década de 1940 y 1950 cambió rápidamente este escenario, las bacterias han respondido ganando progresivamente resistencia a los antibióticos de uso habitual. Actualmente, todos los países del mundo tienen bacterias resistentes a los antibióticos. De hecho, en los Estados Unidos más del 70 % de las bacterias que causan infecciones nosocomiales son resistentes a al menos uno de los principales agentes antimicrobianos que normalmente se utilizan para combatir infecciones (Nature Reviews, Drug Discovery 1; 895-910 (2002)).
Una manera de abordar el creciente problema de las bacterias resistentes, es el desarrollo de nuevas clases de agentes antimicrobianos. Sin embargo, hasta la introducción de linezolid en 2000, no había habido ninguna nueva clase de antibióticos comercializada durante más de 37 años. Además, incluso el desarrollo de nuevas clases de antibióticos proporciona solo una solución temporal, y de hecho ya existen informes de resistencia de determinadas bacterias a linezolid (Lancet 357, 1179 (2001) y Lancet 358, 207-208 (2001)).
Para desarrollar soluciones a más largo plazo al problema de la resistencia bacteriana, es evidente que se requieren enfoques alternativos. Un enfoque alternativo de este tipo es minimizar, en la medida de lo posible, las oportunidades que brindan las bacterias para desarrollar resistencia a antibióticos importantes. Por lo tanto, las estrategias que pueden adoptarse incluyen limitar el uso de antibióticos para el tratamiento de infecciones no agudas, así como controlar qué antibióticos se suministran a los animales para promover el crecimiento.
Sin embargo, para abordar el problema de una manera más eficaz, es necesario obtener una comprensión de los mecanismos reales por los cuales las bacterias generan resistencia a los agentes antibióticos. Para ello primero se debe considerar como actúan los agentes antibióticos actuales para eliminar las bacterias.
Los agentes antimicrobianos se dirigen a los componentes esenciales del metabolismo bacteriano. Por ejemplo, las p-lactamas (por ejemplo, penicilinas y cefalosporinas) inhiben la síntesis de la pared celular, mientras que otros agentes inhiben una serie diversa de dianas, tales como la ADN girasa (quinolonas) y síntesis de proteínas (por ejemplo, macrólidos, aminoglucósidos, tetraciclinas y oxazolidinonas). El intervalo de organismos contra los cuales los agentes antimicrobianos son eficaces varía dependiendo de qué organismos se basan en gran medida de la etapa o etapas metabólicas que están inhibidas. Además, el efecto sobre las bacterias puede variar desde una mera inhibición del crecimiento (es decir, un efecto bacteriostático, como se observa con agentes tales como las tetraciclinas) hasta la eliminación total (es decir, un efecto bactericida, como se observa, por ejemplo, con la penicilina).
Las bacterias han estado desarrollándose en la tierra durante más de 3 mil millones de años y, en ese momento, han tenido que responder a un gran número de tensiones ambientales. Por lo tanto, tal vez no sea sorprendente que las bacterias hayan desarrollado una variedad aparentemente inagotable de mecanismos mediante los cuales pueden responder al estrés metabólico que les imponen los agentes antibióticos. De hecho, los mecanismos mediante los cuales las bacterias pueden generar resistencia incluyen estrategias tan diversas como la inactivación del fármaco, la modificación del sitio de acción, la modificación de la permeabilidad de la pared celular, la sobreproducción de la enzima diana y la evitación de las etapas inhibidas. Sin embargo, se ha observado que la tasa de resistencia que surge para un agente en particular varía en gran medida, dependiendo de factores tales como el mecanismo de acción del agente, de si el modo de eliminación del agente depende del tiempo y de la concentración, de la fuerza contra la población de bacterias y la magnitud y duración de la concentración en suero disponible. Se ha propuesto (Science 264, 388-393 (1994)) que los agentes que se dirigen a enzimas individuales (por ejemplo, rifampicina) son los más propensos a desarrollar resistencia. Además, cuanto más elevados sean los niveles subóptimos del agente antimicrobiano en contacto con la bacteria, más probable será que aparezca la resistencia. Además, ahora se sabe que muchas infecciones microbianas incluyen subpoblaciones de bacterias que son fenotípicamente resistentes a los agentes antimicrobianos (J. Antimicrob. Chemother. 4, 395-404 (1988); J. Med. Microbiol. 38, 197-202 (1993); J. Bacteriol. 182, 1794-1801 (2000); ibid. 182, 6358-6365 (2000); ibid. 183, 6746-6751 (2001); FEMS Microbiol. Lett 202, 59-65 (2001); y Trends in Microbiology 13, 34-40 (2005)). Parece haber varios tipos de bacterias de este tipo fenotípicamente resistentes, incluyendo bacterias persistentes en fase estacionaria, así como aquellas en las profundidades de biopelículas. Sin embargo, cada uno de estos tipos se caracteriza por su tasa baja de crecimiento en comparación con las bacterias en fase logarítmica en las mismas condiciones. La inanición nutricional y las altas densidades celulares también son características comunes de dichas bacterias.
Aunque resistentes a los agentes antimicrobianos en su estado de crecimiento lento, las bacterias fenotípicamente resistentes difieren de las genotípicamente resistentes en que recuperan su susceptibilidad a los antibióticos cuando vuelven a un estado de rápido crecimiento (por ejemplo, cuando los nutrientes se vuelven más accesibles).
La presencia de bacterias fenotípicamente resistentes en una infección conduce a la necesidad de ciclos prolongados de agentes antimicrobianos que comprenden dosis múltiples. Esto se debe a que las bacterias resistentes, que se multiplican lentamente, proporcionan un conjunto de organismos “latentes” que puede convertirse a un estado de crecimiento rápido cuando las condiciones lo permiten (reiniciando así la infección de manera eficaz). Múltiples dosis a lo largo del tiempo afrontan este problema al eliminar gradualmente las bacterias “latentes” que se convierten en formas “activas”.
Sin embargo, el afrontar bacterias “latentes” a través de la administración de ciclos prolongados de agentes antimicrobianos plantea sus propios problemas. Es decir, la exposición prolongada de bacterias a concentraciones subóptimas de agente antimicrobiano puede conducir a la aparición de bacterias genotípicamente resistentes que después pueden multiplicarse rápidamente en presencia de concentraciones incluso altas del agente antimicrobiano. Los ciclos prolongados de agentes antimicrobianos tienen más probabilidades de favorecer la aparición de resistencia genotípica que los ciclos más cortos, ya que las bacterias que no se multiplican tenderán a sobrevivir y, curiosamente, es probable que tengan una mayor capacidad de mutación a la resistencia (Proc. Natl. Acad. Sci. USA 92, 11736-11740 (1995); J. Bacteriol. 179, 6688-6691 (1997); y Antimicrob. Agents Chemother. 44, 1771-1777 (2000)).
A la luz de lo anterior, podría seleccionarse un nuevo enfoque para combatir el problema de la resistencia bacteriana y desarrollar agentes antimicrobianos en función de su capacidad para eliminar microorganismos “latentes”. La producción de dichos agentes permitiría, entre otras cosas, acortar los regímenes de quimioterapia en el tratamiento de infecciones microbianas, reduciendo así la frecuencia con la que surge la resistencia genotípica en los microorganismos.
La solicitud de patente internacional, número de publicación WO2000028074, describe un método de exploración de compuestos para determinar su capacidad de eliminar microorganismos clínicamente latentes. Utilizando este método, el solicitante ha observado que muchos agentes antimicrobianos convencionales, tales como augmentina, azitromicina, levofloxacina, linezolid y mupirocina, que por otro lado presentan una excelente actividad biológica contra bacterias en fase logarítmica (es decir, que se multiplican), muestran poca o ninguna actividad contra microorganismos clínicamente latentes. Esta observación ha requerido el desarrollo de nuevos agentes antimicrobianos que pueden utilizarse para eliminar microorganismos clínicamente latentes.
Las solicitudes de patente internacional, números de publicación WO2007054693, WO2008117079 y WO2008142384, describen compuestos que presentan actividad biológica contra microorganismos clínicamente latentes. Los ejemplos de dichos compuestos incluyen 4-metil-1-(2-feniletil)-8-fenoxi-2,3-dihidro-1H-pirrolo[3,2-c]-quinolina, 4-(3-bencilipirroliIdin-1-il)-2-metil-6-fenoxiquinolina; N-[4-(3-bencilpirrolidin-1-il)-2-metilquinolin-6-il]benzamida y sus derivados farmacéuticamente aceptables.
Se conoce una amplia variedad de agentes anestésicos tanto generales como locales y están disponibles en el comercio. Los anestésicos locales inician sus actividades analgésicas bloqueando de manera reversible los canales de Na+ dependientes del voltaje neural en la membrana celular y, por lo tanto, impidiendo la entrada de Na+. Esto impide posteriormente la producción de potenciales de acción y conducción nerviosa.
Son estos mecanismos de acción los que pueden permitir que los anestésicos locales influyan en una amplia gama de tejidos, lo que reduce la inflamación.
Además de la actividad analgésica, se ha demostrado que ciertos anestésicos locales poseen actividad antimicrobiana. Estudios limitados atribuyen el mecanismo de acción de la actividad antimicrobiana de los anestésicos locales a una alteración de la permeabilidad de la membrana de las células microbianas, lo que conduce a la filtración de componentes celulares y a la subsiguiente lisis celular (Surg. Infect. (Larchmt)., 9(2), 205-213, (2008)).
Un ejemplo de un anestésico local que se ha demostrado que posee actividad antimicrobiana es el clorhidrato de diclonina. (Florestano H J et al., Journal of the American Pharmaceutical Association, Vol. XLV, No.5, 320 - 324 (1956)).Se demostró que el clorhidrato de diclonina mostraba actividad in vitro contra una variedad de bacterias en fase logarítmica (es decir, que se multiplican). Adicionalmente, las preparaciones que contenían una combinación de clorhidrato de diclonina con clorobutanol mostraron un efecto antimicrobiano sinérgico in vitro contra Staphylococcus aureus y Escherichia coli (ibid.), en fase logarítmica.
Sin embargo, por lo que se sabe, el efecto antimicrobiano de los agentes anestésicos como el clorhidrato de diclonina contra microorganismos clínicamente latentes no se ha informado hasta la fecha. Por consiguiente, en una realización de la presente invención, se proporciona diclonina o una de sus sales farmacéuticamente aceptables en combinación con un agente antimicrobiano seleccionado de mupirocina, 4-metil-1-(2-feniletil)-8-fenoxi-2,3-dihidro1H-pirrolo[3,2-c]-quinolina, 4-(3-bencilipirroliIdin-1-il)-2-metil-6-fenoxiquinolina; N-[4-(3-bencilpirrolidin-1-il)-2-metilquinolin-6-il]benzamida o una de sus sales farmacéuticamente aceptables, para uso en el tratamiento de una infección microbiana.
En una realización adicional de la invención, se proporciona una composición farmacéutica que comprende diclonina o una sal farmacéuticamente aceptable de la misma, un agente antimicrobiano seleccionado de mupirocina, 4-metil-1-(2-feniletil)-8-fenoxi-2,3-dihidro-1H-pirrolo [3,2-c] -quinolina, 4-(3-bencilpirrolidin-1-il)-2-metil-6-fenoxiquinolina y N-[4-(3-bencilpirrolidin-1-il)-2-metilquinolin-6-il]benzamida o una de sus sales farmacéuticamente aceptables y un vehículo farmacéuticamente aceptable para su uso en la eliminación de microorganismos clínicamente latentes asociados con una infección microbiana.
La presente invención también se basa en el hallazgo inesperado de que la actividad de ciertos agentes antimicrobianos mejora sustancialmente si dichos agentes se administran en combinación con diclonina o una sal de la misma farmacéuticamente aceptable. Además, se ha demostrado sorprendentemente que las combinaciones de ciertos agentes muestran actividad antimicrobiana sinérgica contra microorganismos en fase logarítmica (es decir, que se multiplican) y/o clínicamente latentes. La sorprendente actividad biológica de las combinaciones de la presente invención, ofrecen la oportunidad acortar los regímenes de quimioterapia y pueden dar como resultado una reducción en la aparición de resistencia microbiana asociada con el uso de dichas combinaciones.
Como se usa en este documento, la expresión "en combinación con" cubre la administración individual y secuencial de un agente antimicrobiano y el agente anestésico. Cuando los agentes se administran secuencialmente, ya sea el agente antimicrobiano o el agente anestésico se puede administrar primero. Cuando la administración es simultánea, los agentes pueden administrarse ya sea en la misma composición farmacéutica o en una diferente. La terapia complementaria, es decir, cuando un agente se usa como tratamiento primario y el otro agente para ayudar a ese tratamiento primario, es también una realización de la presente invención.
Según una realización adicional de la invención, se proporciona un producto que comprende diclonina o una de sus sales farmacéuticamente aceptables y un agente antimicrobiano seleccionado de mupirocina, 4-metil-1-(2-feniletil)-8-fenoxi-2,3-dihidro-1H-pirrolo[3,2-c]-quinolina, 4-(3-bencilpirrolidin-1-il)-2-metil-6-fenoxiquinolina y N-[4- (3-bencilpirrolidin-1-il)-2-metilquinolin-6-il]benzamida o una de sus sales farmacéuticamente aceptables como una preparación combinada para uso simultáneo, individual o secuencial en la eliminación de microorganismos clínicamente latentes asociados con infección microbiana.
Las combinaciones de la presente invención pueden utilizarse para tratar infecciones microbianas. En particular pueden utilizarse para eliminar microorganismos que se multiplican y/o clínicamente latentes asociados con infecciones microbianas. Por lo tanto, las referencias de este documento al tratamiento de una infección microbiana incluyen eliminar microorganismos que se multiplican y/o clínicamente latentes asociados con dichas infecciones. Preferentemente, las combinaciones de la presente invención se utilizan para eliminar microorganismos clínicamente latentes asociados con infecciones microbianas.
Como se usa en el presente documento, “eliminar” significa una pérdida de viabilidad evaluada por la ausencia de actividad metabólica.
Como se usa en el presente documento, “microorganismo clínicamente latente” significa un microorganismo que es metabólicamente activo pero que tiene una tasa de crecimiento que está por debajo del umbral de expresión de enfermedad infecciosa. El umbral de expresión de enfermedad infecciosa se refiere al umbral de la tasa de crecimiento por debajo del cual no aparecen los síntomas de la enfermedad infecciosa en un hospedador.
La actividad metabólica de los microorganismos clínicamente latentes se puede determinar mediante diversos métodos conocidos por los expertos en la técnica; por ejemplo, midiendo los niveles de ARNm en los microorganismos o determinando su tasa de captación de uridina. A este respecto, los microorganismos clínicamente latentes, cuando se les compara con microorganismos en condiciones de crecimiento logarítmico (in vitro o in vivo), poseen niveles reducidos, pero aún significativos de:
(i) ARNm (por ejemplo, de 0,0001 a 50 %, tal como de 1 a 30, de 5 a 25 o de 10 a 20 % del nivel de ARNm); y/o (ii) captación de uridina (por ejemplo, [3H]uridina) (por ejemplo, de 0,0005 a 50 %, tal como de 1 a 40, de 15 a 35 o de 20 a 30 % del nivel de captación de [3H]uridina).
Los microorganismos clínicamente latentes poseen generalmente diversas características identificables. Por ejemplo, pueden ser viables, pero no cultivables; es decir, generalmente no pueden detectarse mediante técnicas de cultivo convencionales, pero son detectables y cuantificables mediante técnicas tales como recuento en caldo de dilución, microscopía o técnicas moleculares tales como reacción en cadena de la polimerasa. Además, los microorganismos clínicamente latentes son fenotípicamente tolerantes, y como tales son sensibles (en fase logarítmica) a los efectos bioestáticos de los agentes antimicrobianos convencionales (es decir, microorganismos para los cuales la concentración inhibidora mínima (CIM) de un agente antimicrobiano convencional está
sustancialmente inalterada); pero poseen una susceptibilidad drásticamente disminuida a la eliminación inducida por fármacos (por ejemplo, microorganismos para los cuales, con cualquier agente antimicrobiano convencional determinado, la proporción de concentración microbicida mínima (por ejemplo concentración bactericida mínima, CBM) con respecto a la CIM es de 10 o mayor).
Como se usa en este documento, el término “microorganismo" significa hongos y bacterias. Las referencias en este documento a “microbiano(a)” y “antimicrobiano(a)" se interpretarán en consecuencia. Por ejemplo, el término “microbiano" significa hongos o bacterias e “infección microbiana" significa cualquier infección fúngica o bacteriana.
Como se usa en este documento, el término “bacteria" (y sus derivados, tales como “infección microbiana"), incluye, pero sin limitación, referencias a organismos (o infecciones debidas a organismos) de las siguientes clases y tipos específicos:
Cocos gram positivos, tales como Estafilococos (por ejemplo, Staphylococcus, aureus, Staph. epidermidis, Staph. saprophyticus, Staph, auricularis, Staph, capitis capitis, Staph, c. ureolyticus, Staph, caprae, Staph, cohnii cohnii, Staph, c. urealyticus, Staph, equorum, Staph, gallinarum, Staph. haemolyticus, Staph, hominis hominis, Staph, h. novobiosepticius, Staph, hyicus, Staph. intermedius, Staph, lugdunensis, Staph, pasteuri, Staph, saccharolyticus, Staph, schleiferi schleiferi, Staph, s. coagulans, Staph, sciuri, Staph, simulans, Staph, warneri y Staph. xylosus);
Estreptococos (por ejemplo, estreptococo piógeno beta hemolítico (tal como Streptococcus agalactiae, Strept. canis, Strept. dysgalactiae dysgalactiae, Strept dysgalactiae equisimilis, Strept. equi equi, Strept equi zooepidemicus, Strept. iniae, Strept. porcinus y Strept. pyogenes), estreptococos piógenos, microaerófilos (Streptococcus “milleri”, tal como Strept. anginosus, Strept. constellatus, Strept. constellatus pharyngidis y Strept. intermedius), estreptococos bucales de los grupos “mitis” (alfa hemolíticos - Streptococcus “viridans”, tal como Strept. mitis, Strept. oralis, Strept sanguinis, Strept cristatus, Strept. gordonii y Strept. parasanguinis), “salivarius” (no hemolíticos, tal como Strept. salivarius y Strept. vestibularis) y “mutans” (estreptococos de la superficie dental, tales como Strept. criceti, Strept mutans, Strept. ratti y Strept. sobrinus), Strept. acidominimus, Strept bovis, Strept. faecalis, Strept equinus, Strept pneumoniae y Strept suis, o estreptococos clasificados de manera alternativa como Estreptococos del Grupo A, B, C, D, E, G, L, P, U o V);
Cocos gram negativos, tales como Neisseria gonorrhoeae, Neisseria meningitidis, Neisseria cinerea, Neisseria elongata, Neisseria flavescens, Neisseria lactamica, Neisseria mucosa, Neisseria sicca, Neisseria subflava and Neisseria weaveri;
Bacilos, tales como Bacillus anthracis, Bacillus subtilis, Bacillus thuringiensis, Bacillus stearothermophilus y Bacillus cereus;
Enterobacterias, tales como Escherichia coli, Enterobacter (por ejemplo, Enterobacter aerogenes, Enterobacter agglomerans y Enterobacter cloacae), Citrobacter (tal como Citrob. Freundii y Citrob. divernis), Hafnia (por ejemplo, Hafnia alvei), Erwinia (por ejemplo, Erwinia persicinus), Morganella morganii, Salmonella (Salmonella enterica y Salmonella typhi), Shigella (por ejemplo, Shigella dysenteriae, Shigella flexneri, Shigella boydii y Shigella sonnei), Klebsiella (por ejemplo, Klebs. pneumoniae, Klebs. oxytoca, Klebs. ornitholytica, Klebs. planticola, Klebs. ozaenae, Klebs. terrigena, Klebs. granulomatis (Calymmatobacterium granulomatis) y Klebs. rhinoscleromatis), Proteus (por ejemplo, Pr. mirabilis, Pr. rettgeri y Pr. vulgaris), Providencia (por ejemplo, Providencia alcalifaciens, Providencia rettgeri y Providencia stuartii), Serratia (por ejemplo, Serratia marcescens y Serratia liquifaciens) y Yersinia (por ejemplo, Yersinia enterocolitica, Yersinia pestis y Yersinia pseudotuberculosis);
Enterococos (por ejemplo, Enterococcus avium, Enterococcus casseliflavus, Enterococcus cecorum, Enterococcus dispar, Enterococcus durans, Enterococcus faecalis, Enterococcus faecium, Enterococcus flavescens, Enterococcus gallinarum, Enterococcus hirae, Enterococcus malodoratus, Enterococcus mundtii, Enterococcus pseudoavium, Enterococcus raffinosus y Enterococcus solitarius);
Helicobacter (por ejemplo, Helicobacterpylori, Helicobacter cinaedi y Helicobacter fennelliae);
Acinetobacter (por ejemplo, A. baumanii, A. calcoaceticus, A. haemolyticus, A. johnsonii, A. junii, A. Iwoffi y A. radioresistens);
Pseudomonas (por ejemplo, Ps. aeruginosa, Ps. maltophilia {Stenotrophomonas maltophilia), Ps. alcaligenes, Ps. chlororaphis, Ps. fluorescens, Ps. luteola. Ps. mendocina, Ps. monteilii, Ps. oryzihabitans, Ps. pertocinogena, Ps. pseudalcaligenes, Ps. putida y Ps. stutzeri); Bacteriodes fragilis;
Peptococcus (por ejemplo, Peptococcus niger);
Peptoestreptococos;
Clostridium (por ejemplo, C. perfringens, C. difficile, C. botulinum, C. tetani, C. absonum, C. argentinense, C. baratii, C. bifermentans, C. beijerinckii, C. butyricum, C. cadaveris, C. carnis, C. celatum, C. clostridioforme, C. cochlearium, C. cocleatum, C. fallax, C. ghonii, C. glycolicum, C. haemolyticum, C. hastiforme, C. histolyticum, C. indolis, C. innocuum, C. irregulare, C. leptum, C. limosum, C. malenominatum, C. novyi, C. oroticum, C. paraputrificum, C. piliforme, C. putrefasciens, C. ramosum, C. septicum, C. sordelii, C. sphenoides, C. sporogenes, C. subterminale, C. symbiosum y C. tedium); Mycoplasma (por ejemplo, M. pneumoniae, M. hominis, M. genitalium y M. urealyticum); Mycobacteria (por ejemplo, Mycobacterium tuberculosis, Mycobacterium avium, Mycobacterium fortuitum, Mycobacterium marinum, Mycobacterium kansasii, Mycobacterium chelonae, Mycobacterium abscessus, Mycobacterium leprae, Mycobacterium smegmitis, Mycobacterium africanum, Mycobacterium alvei, Mycobacterium asiaticum, Mycobacterium aurum, Mycobacterium bohemicum, Mycobacterium bovis, Mycobacterium branderi, Mycobacterium brumae, Mycobacterium celatum, Mycobacterium chubense, Mycobacterium confluentis, Mycobacterium conspicuum, Mycobacterium cookii, Mycobacterium flavescens, Mycobacterium gadium, Mycobacterium gastri, Mycobacterium genavense, Mycobacterium gordonae, Mycobacterium goodii, Mycobacterium haemophilum, Mycobacterium hassicum, Mycobacterium intracellulare, Mycobacterium interjectum, Mycobacterium heidelberense, Mycobacterium lentiflavum, Mycobacterium malmoense, Mycobacterium microgenicum, Mycobacterium microti, Mycobacterium mucogenicum, Mycobacterium neoaurum, Mycobacterium nonchromogenicum, Mycobacterium peregrinum, Mycobacterium phlei, Mycobacterium scrofulaceum, Mycobacterium shimoidei, Mycobacterium simiae, Mycobacterium szulgai, Mycobacterium terrae, Mycobacterium thermoresistabile, Mycobacterium triplex, Mycobacterium triviale, Mycobacterium tusciae, Mycobacterium ulcerans, Mycobacterium vaccae, Mycobacterium wolinskyi y Mycobacterium xenopi); Haemofilus (por ejemplo, Haemophilus influenzae, Haemophilus ducreyi, Haemophilus aegyptius, Haemophilus parainfluenzae, Haemophilus haemolyticus y Haemophilus parahaemolyticus);
Actinobacillus (por ejemplo, Actinobacillus actinomycetemcomitans, Actinobacillus equuli, Actinobacillus hominis, Actinobacillus Hgnieresii, Actinobacillus suis y Actinobacillus ureae);
Actinomyces (por ejemplo, Actinomyces israelii);
Brucella (por ejemplo, Brucella abortus, Brucella canis, Brucella melintensis y Brucella suis);
Campylobacter (por ejemplo, Campylobacter jejuni, Campylobacter coli, Campylobacter lari y Campylobacter fetus);
Listeria monocytogenes;
Vibrio (por ejemplo, Vibrio cholerae y Vibrio parahaemolyticus, Vibrio alginolyticus, Vibrio carchariae, Vibrio fluvialis, Vibrio furnissii, Vibrio hollisae, Vibrio metschnikovii, Vibrio mimicus y Vibrio vulnificus);
Erysipelothrix rhusopathiae;
Corinobacterias (por ejemplo, Corynebacterium diphtheriae, Corynebacterium jeikeum y Corynebacterium urealyticum);
Espiroquetas, tales como Borrelia (por ejemplo, Borrelia recurrentis, Borrelia burgdorferi, Borrelia afzelii, Borrelia andersonii, Borrelia bissettii, Borrelia garinii, Borrelia japonica, Borrelia lusitaniae, Borrelia tanukii, Borrelia turdi, Borrelia valaisiana, Borrelia caucasica, Borrelia crocidurae, Borrelia duttoni, Borrelia graingeri, Borrelia hermsii, Borrelia hispanica, Borrelia latyschewii, Borrelia mazzottii, Borrelia parkeri, Borrelia persica, Borrelia turicatae y Borrelia venezuelensis) y Treponema (Treponema pallidum ssp. pallidum, Treponema pallidum ssp. endemicum, Treponema pallidum ssp. pertenue y Treponema carateum);
Pasteurella (por ejemplo, Pasteurella aerogenes, Pasteurella bettyae, Pasteurella canis, Pasteurella dagmatis, Pasteurella gallinarum, Pasteurella haemolytica, Pasteurella multocida multocida, Pasteurella multocida gallicida, Pasteurella multocida septica, Pasteurella pneumotropica y Pasteurella stomatis);
Bordetella (por ejemplo, Bordetella bronchiseptica, Bordetella hinzii, Bordetella holmseii, Bordetella parapertussis, Bordetella pertussis y Bordetella trematum);
Nocardiáceas, tal como Nocardia (por ejemplo, Nocardia asteroides y Nocardia brasiliensis);
Rickettsias (por ejemplo, Rickettsia rickettsii o Coxiella burnetii);
Legionela (por ejemplo, Legionella anisa, Legionella birminghamensis, Legionella bozemanii, Legionella cincinnatiensis, Legionella dumoffii, Legionella feeleii, Legionella gormanii, Legionella hackeliae, Legionella israelensis, Legionella jordanis, Legionella lansingensis, Legionella longbeachae, Legionella maceachernii, Legionella micdadei, Legionella oakridgensis, Legionella pneumophila, Legionella sainthelensi, Legionella tucsonensis y Legionella wadsworthii);
Moraxella catarrhalis;
Cyclospora cayetanensis;
Entamoeba histolytica;
Giardia lamblia;
Trichomonas vaginalis;
Toxoplasma gondii;
Stenotrophomonas maltophilia;
Burkholderia cepacia; Burkholderia mallei y Burkholderia pseudomallei;
Francisella tularensis;
Gardnerela (por ejemplo, Gardneralla vaginalis y Gardneralla mobiluncus);
Streptobacillus moniliformis;
Flavobacterias tales como Capnocytophaga (por ejemplo, Capnocytophaga canimorsus, Capnocytophaga cynodegmi, Capnocytophaga gingivalis, Capnocytophaga granulosa, Capnocytophaga haemolytica, Capnocytophaga ochracea y Capnocytophaga sputigena);
Bartonella (Bartonella bacilliformis, Bartonella clarridgeiae, Bartonella elizabethae, Bartonella henselae, Bartonella quintana y Bartonella vinsonii arupensis);
Leptospira (por ejemplo, Leptospira biflexa, Leptospira borgpetersenii, Leptospira inadai, Leptospira interrogans, Leptospira kirschneri, Leptospira noguchii, Leptospira santarosai y Leptospira weilii);
Spirilium (por ejemplo, Spirillum minus);
Bacteroides (por ejemplo, Bacteroides caccae, Bacteroides capillosus, Bacteroides coagulans, Bacteroides distasonis, Bacteroides eggerthii, Bacteroides forsythus, Bacteroides fragilis, Bacteroides merdae, Bacteroides ovatus, Bacteroides putredinis, Bacteroides pyogenes, Bacteroides splanchinicus, Bacteroides stercoris, Bacteroides tectus, Bacteroides thetaiotaomicron, Bacteroides uniformis, Bacteroides ureolyticus y Bacteroides vulgatus);
Prevotela (por ejemplo, Prevotella bivia, Prevotella buccae, Prevotella corporis, Prevotella dentalis (Mitsuokella dentalis), Prevotella denticola, Prevotella disiens, Prevotella enoeca, Prevotella heparinolytica, Prevotella intermedia, Prevotella loeschii, Prevotella melaninogenica, Prevotella nigrescens, Prevotella oralis, Prevotella oris, Prevotella oulora, Prevotella tannerae, Prevotella venoralis y Prevotella zoogleoformans);
Porfiromonas (por ejemplo, Porphyromonas asaccharolytica, Porphyromonas cangingivalis, Porphyromonas canoris, Porphyromonas cansulci, Porphyromonas catoniae, Porphyromonas circumdentaria, Porphyromonas crevioricanis, Porphyromonas endodontalis, Porphyromonas gingivalis, Porphyromonas gingivicanis, Porphyromonas levii y Porphyromonas macacae);
Fusobacterium (por ejemplo, F. gonadiaformans, F. mortiferum, F. naviforme, F. necrogenes, F. necrophorum necrophorum, F. necrophorum fundiliforme, F. nucleatum nucleatum, F. nucleatum fusiforme, F. nucleatum polymorphum, F. nucleatum vincentii, F. periodonticum, F. russii, F. ulcerans y F. varium);
Chlamydia (por ejemplo, Chlamydia trachomatis);
Cryptosporidium (por ejemplo, C. parvum, C. hominis, C. canis, C. felis, C. meleagridis y C. muris);
Chlamydofila (por ejemplo, Chlamydophila abortus (Chlamydia psittaci), Chlamydophila pneumoniae (Chlamydia pneumoniae) y Chlamydophila psittaci (Chlamydia psittaci));
Leuconostoc (por ejemplo, Leuconostoc citreum, Leuconostoc cremoris, Leuconostoc dextranicum, Leuconostoc lactis, Leuconostoc mesenteroides y Leuconostoc pseudomesenteroides);
Gemella (por ejemplo, Gemella bergeri, Gemella haemolysans, Gemella morbillorum y Gemella sanguinis); y Ureaplasma (por ejemplo, Ureaplasma parvum y Ureaplasma urealyticum).
Como se usa en el presente documento, el término “fúngico” (y sus derivados, tal como “infección fúngica”) incluye, pero sin limitación, referencias a organismos (o infecciones debidas a organismos) de las siguientes clases y tipos específicos:
Absidia (por ejemplo, Absidia corymbifera);
Ajellomyces (por ejemplo, Ajellomyces capsulatus y Ajellomyces dermatitidis);
Arthroderma (por ejemplo, Arthroderma benhamiae, Arthroderma fulvum, Arthroderma gypseum,
Arthroderma incurvatum, Arthroderma otae y Arthroderma vanbreuseghemii);
Aspergillus (por ejemplo, Aspergillus flavus, Aspergillus fumigatus y Aspergillus niger);
Blastomyces (por ejemplo, Blastomyces dermatitidis);
Candida (por ejemplo, Candida albicans, Candida glabrata, Candida guilliermondii, Candida krusei,
Candida parapsilosis, Candida tropicalis y Candida pelliculosa);
Cladpphialophora (por ejemplo, Cladophialophora carrionii);
Coccidioides (por ejemplo, Coccidioides immitis y Coccidioides posadasii);
Cryptococcus (por ejemplo, Cryptococcus neoformans);
Cunninghamella (por ejemplo, Cunninghamella sp.)
Epidermophyton (por ejemplo, Epidermophyton floccosum);
Exophiala (por ejemplo, Exophiala dermatitidis);
Filobasidiella (por ejemplo, Filobasidiella neoformans);
Fonsecaea (por ejemplo, Fonsecaea pedrosoi);
Fusarium (por ejemplo, Fusarium solani);
Geotrichum (por ejemplo, Geotrichum candidum);
Histoplasma (por ejemplo, Histoplasma capsulatum);
Hortaea (por ejemplo, Hortaea werneckii);
Issatschenkia (por ejemplo, Issatschenkia orientalis);
Madurella (por ejemplo, Madurella grisae);
Malassezia (por ejemplo, Malassezia furfur, Malassezia globosa, Malassezia obtusa, Malassezia
pachydermatis, Malassezia restricta, Malassezia slooffiae y Malassezia sympodialis);
Microsporum (por ejemplo, Microsporum canis, Microsporum fulvum y Microsporum gypseum);
Microsporidia;
Mucor (por ejemplo, Mucor circinelloides);
Nectria (por ejemplo, Nectria haematococca);
Paecilomyces (por ejemplo, Paecilomyces variotii);
Paracoccidioides (por ejemplo, Paracoccidioides brasiliensis);
Penicillium (por ejemplo, Penicillium marneffei);
Pichia (por ejemplo, Pichia anomala y Pichia guilliermondii);
Pneumocystis (por ejemplo, Pneumocystis jiroveci (Pneumocystis carinii));
Pseudallescheria (por ejemplo, Pseudallescheria boydii);
Rhizopus (por ejemplo, Rhizopus oryzae);
Rhodotorula (por ejemplo, Rhodotorula rubra);
Scedosporium (por ejemplo, Scedosporium apiospermum);
Schizophyllum (por ejemplo, Schizophyllum commune);
Sporothrix (por ejemplo, Sporothrix schenckii);
Trichophyton (por ejemplo, Trichophyton mentagrophytes, Trichophyton rubrum, Trichophyton verrucosum y Trichophyton violaceum); y
Trichosporon (por ejemplo, Trichosporon asahii, Trichosporon cutaneum, Trichosporon inkin y Trichosporon mucoides).
Las bacterias particulares que pueden tratarse utilizando una combinación de la invención incluyen:
Estafilococos, tal como Staph. aureus (ya sea sensible a la meticilina (es decir, MSSA) o resistente a la meticilina (es decir, MRSA)) y Staph. epidermidis;
Estreptococos, tal como Strept. agalactiae y Strept. pyogenes;
Bacilos, tal como Bacillus anthracis;
Enterobacterias, tal como Escherichia coli, Klebsiella (por ejemplo, Klebs. pneumoniae y Klebs. oxytoca) y Proteus (por ejemplo, Pr. mirabilis, Pr. rettgeri y Pr. vulgaris);
Haemophilus influenzae;
Enterococos, tal como Enterococcus faecalis y Enterococcus faecium; y
Micobacterias, tal como Mycobacterium tuberculosis.
Preferentemente, la bacteria es Staph. aureus; ya sea MSSA o MRSA.
Los hongos particulares que pueden tratarse con una combinación de la invención incluyen Aspergillus fumigatus, Candida albicans, Cryptococcus neoformans, Histoplasma capsulatum y Pneumocystis jiroveci.
Las combinaciones de la presente invención pueden utilizarse para tratar infecciones asociadas con cualquiera de los organismos bacterianos o fúngicos mencionados anteriormente, y en particular pueden utilizarse para eliminar microorganismos que se multiplican y / o clínicamente latentes asociados con dicha infección. En un aspecto, la invención proporciona diclonina o una de sus sales farmacéuticamente aceptable en combinación con un agente antimicrobiano seleccionado de mupirocina, 4-metil-1-(2-feniletil)-8-fenoxi-2,3-dihidro-1H-pirrolo[3,2-c]-quinolina, 4-3-bencilpirrolidin-1-il)-2-metil-6-fenoxiquinolina y N-[4-(3-bencilpirrolidin-1-il)-2-metilquinolin-6-il]benzamida o una sal farmacéuticamente aceptable para su uso en la eliminación de microorganismos clínicamente latentes asociados con una infección microbiana.
Las afecciones particulares que pueden tratarse utilizando la combinación de la presente invención incluyen tuberculosis (por ejemplo, tuberculosis pulmonar, tuberculosis no pulmonar (tal como tuberculosis de ganglios linfáticos, tuberculosis genitourinaria, tuberculosis de huesos y articulaciones, meningitis tuberculosa) y tuberculosis miliar), carbunco, abscesos, acné vulgar, actinomicosis, asma, disentería bacilar, conjuntivitis bacteriana, queratitis bacteriana, vaginosis bacteriana, botulismo, úlcera de Buruli, infecciones de huesos y articulaciones, bronquitis (aguda o crónica), brucelosis, heridas por quemadura, fiebre por arañazo de gato, celulitis, chancro, colangitis, colecistitis, difteria cutánea, fibrosis quística, cistitis, panbronquiolitis difusa, difteria, caries dental, enfermedades del tracto respiratorio superior, eccema, empiema, endocarditis, endometritis, fiebre entérica, enteritis, epididimitis, epiglotitis, erisipela, erisipclas, erisipeloide, eritrasma, infecciones oculares, furúnculos, vaginosis Gardnerella, infecciones gastrointestinales (gastroenteritis), infecciones genitales, gingivitis, gonorrea, granuloma inguinal, fiebre de Haverhill, quemaduras infectadas, infecciones después de intervenciones dentales, infecciones en la región bucal, infecciones relacionadas con prótesis, abscesos intraabdominales, enfermedad del legionario, lepra, leptospirosis, listeriosis, abscesos hepáticos, enfermedad de Lyme, linfogranuloma venéreo, mastitis, mastoiditis, meningitis e infecciones del sistema nervioso, micetoma, nocardiosis (por ejemplo pie de Madura), uretritis no específica, oftalmía (por ejemplo oftalmía del neonato), osteomielitis, otitis (por ejemplo, otitis externa y otitis media), orquitis, pancreatitis, paroniquia, pelviperitonitis, peritonitis, peritonitis con apendicitis, faringitis, flemones, pinta, peste, derrame pleural, neumonía, infecciones por heridas postoperatorias, gangrena gaseosa postoperatoria, prostatitis, colitis pseudomembranosa, pitacosis, enfisema pulmonar, pielonefritis, pioderma (por ejemplo, impétigo), fiebre Q, fiebre por mordedura de rata, reticulosis, envenenamiento con ricina, enfermedad de Ritter, salmonelosis, salpingitis, artritis septicémica, infecciones septicémicas, septicemia, sinusitis, infecciones cutáneas (por ejemplo, granulomas cutáneos, impétigo, foliculitis y forunculosis), sífilis, infecciones sistémicas, amigdalitis, síndrome de choque tóxico, tracoma, tularemia, fiebre tifoidea, tifus (por ejemplo, tifus epidémico, tifus murino, tifus de los matorrales y fiebre maculosa), uretritis, infecciones por heridas, pian, aspergilosis, candidiasis (por ejemplo, candidiasis orofaríngea, candidiasis vaginal o balanitis), criptococosis, favo, histoplasmosis, intértrigo, mucomicosis, tiña (por ejemplo, tiña corporal, tiña del cuero cabelludo, tiña crural, tiña del pie y tiña ungueal), onicomicosis, pitiriasis versicolor, dermatofitosis y esporotricosis; o infecciones causadas por SASM, SARM, Staph. epidermidis, Strept. agalactiae, Strept. pyogenes, Escherichia coli, Klebs. pneumoniae, Klebs. oxytoca, Pr. mirabilis, Pr. rettgeri, Pr. vulgaris, Haemophilis influenzae, Enterococcus faecalis y Enterococcus faecium.
En una realización preferida de la invención, se proporciona diclonina o una de sus sales farmacéuticamente aceptables en combinación con un agente antimicrobiano seleccionado de mupirocina, 4-metil-1-(2-feniletil)-8-fenoxi-2,3-dihidro-1H-pirrolo [3,2-c]-quinolina, 4-(3-bencilpirrolidin-1-il)-2-metil-6-fenoxiquinolina y N-[4-(3-bencilpirrolidin-1-il)-2-metilquinolin-6-il]benzamida o una de sus sales farmacéuticamente aceptables, preferentemente 4-metil-1-(2-feniletil)-8-fenoxi-2,3-dihidro-1H-pirrolo[3,2-c]-quinolina o una de sus sales farmacéuticamente aceptables, para uso en la descolonización nasal de MSSA o MRSA, preferentemente MRSA, en particular para uso en la eliminación de microorganismos clínicamente latentes asociados con dicha infección.
Se apreciará que las referencias del presente documento a “tratamiento” se extienden a profilaxis así como al tratamiento de enfermedades o síntomas establecidos.
Un compuesto antimicrobiano particularmente preferido es la mupirocina o una de sus sales farmacéuticamente aceptables.
Otros compuestos antimicrobianos preferidos para uso en la presente invención son aquellos capaces de eliminar microorganismos clínicamente latentes. Los métodos para determinar la actividad contra bacterias clínicamente latentes incluyen una determinación, en condiciones conocidas por los expertos en la técnica (tales como las descritas en Nature Reviews, Drug Discovery 1, 895-910 (2002), de la Concentración Bactericida Mínima (“CBM”) estacionaria o Concentración Dormicida Mínima (“CDM”) de un compuesto de ensayo. En el documento WO2000028074 se describe un método de exploración de compuestos adecuado contra microorganismos clínicamente latentes.
Los ejemplos de compuestos capaces de eliminar microorganismos clínicamente latentes incluyen aquellos compuestos desvelados en la solicitud de patente internacional, números de publicación WO2007054693, WO2008117079 y WO2008142384. Estas solicitudes describen métodos adecuados para la preparación de dichos compuestos y dosis para su administración.
Los agentes antimicrobianos particularmente preferidos para su uso en la presente invención son 4-metil-1-(2-feniletil)-8-fenoxi-2,3-dihidro-1H-pirrolo[3,2-c]-quinolina (Ejemplo 9, WO2007054693), 4-(3-bencilpirrolidin-1-il)-2-metil-6-fenoxiquinolina (Ejemplo 8, WO2008142384) y N-[4-(3-bencilpirrolidin-1-il)-2-metilquinolin-6-il]benzamida (Ejemplo 38, WO2008142384), y sus sales farmacéuticamente aceptables. En una realización de la invención, el agente antimicrobiano es 4-(3-bencilpirrolidin-1-il)-2-metil-6-fenoxiquinolina, N-[4-(3-bencilpirrolidin-1-il)-2-metilquinolin-6-il]benzamida o una de sus sales farmacéuticamente aceptables. Un agente antimicrobiano más preferido es 4-metil-1-(2-feniletil)-8-fenoxi-2,3-dihidro-1H-pirrolo[3,2-c]-quinolina o una de sus sales farmacéuticamente aceptables.
Las referencias en este documento a 4-metil-1-(2-feniletil)-8-fenoxi-2,3-dihidro-1H-pirrolo[3,2-c] -quinolina significan un compuesto que tiene la siguiente estructura química:

Las referencias en este documento a 4-(3-bencilpirrolidin-1-il)-2-metil-6-fenoxiquinolina significan un compuesto que tiene la siguiente estructura química:

Las referencias en este documento a N-[4-(3-bencilpirrolidin-1-il)-2-metilquinolin-6-il]benzamida significan un compuesto que tiene la siguiente estructura química:

Los agentes antimicrobianos preferidos de la presente invención pueden prepararse según los métodos desvelados en la solicitud de patente internacional, números de publicación WO2007054693, WO2008117079 y WO2008142384.
Como se utiliza en el presente documento, la expresión “derivado farmacéuticamente aceptable" significa:
(a) sales farmacéuticamente aceptables con ácidos o bases (por ejemplo, sales de adición de ácido); y/o (b) solvatos (incluyendo hidratos).
Las sales de adición de ácido que pueden mencionarse incluyen sales de carboxilato (por ejemplo, sales de formato, acetato, trifluoroacetato, propionato, isobutirato, heptanoato, decanoato, caprato, caprilato, estearato, acrilato, caproato, propiolato, ascorbato, citrato, glucuronato, glutamato, glicolato, a-hidroxibutirato, lactato, tartrato, fenilacetato, mandelato, fenilpropionato, fenilbutirato, benzoato, clorobenzoato, metilbenzoato, hidroxibenzoato, metoxibenzoato, dinitrobenzoato, o-acetoxibenzoato, salicilato, nicotinato, isonicotinato, cinamato, oxalato, malonato, succinato, suberato, sebacato, fumarato, malato, maleato, hidroximaleato, hipurato, ftalato o tereftalato), sales de haluro (por ejemplo sales de cloruro, bromuro o yoduro), sales de sulfonato (por ejemplo, sales de bencensulfonato, metil-, bromo- o cloro-bencensulfonato, xilensulfonato, metansulfonato, etansulfonato, propansulfonato, hidroxietansulfonato, 1- o 2-naftalensulfonato o 1,5-naftalendisulfonato) o sales de sulfato, pirosulfato, bisulfato, sulfito, bisulfito, fosfato, monohidrogenofosfato, dihidrogenofosfato, metafosfato, pirofosfato o nitrato, y similares. Una sal de adición de ácido preferida de la 4-metil-1-(2-feniletil)-8-fenoxi-2,3-dihidro-1H-pirrolo[3,2-c]-quinolina es la sal de clorhidrato de la misma.
Los agentes anestésicos adecuados para su uso en la presente invención incluyen aquellos capaces de eliminar microorganismos que se multiplican y/o clínicamente latentes. El agente anestésico es la diclonina o una sal farmacéuticamente aceptable de la misma.
Un anestésico local particularmente preferido es el clorhidrato de (1-(4-butoxifenil)-3-(1-piperidinil)-1-propanona). El clorhidrato de diclonina está disponible en el comercio de Sigma-Aldrich Co. (Fluka). Por lo tanto, la presente invención también proporciona la diclonina o una sal farmacéuticamente aceptable de la misma, particularmente clorhidrato de diclonina, para su uso en la eliminación de microorganismos clínicamente latentes asociados con una infección microbiana.
Según una realización preferida de la invención, se proporciona una combinación que comprende un agente antimicrobiano seleccionado del grupo que consiste en 4-metil-1-(2-feniletil)-8-fenoxi-2,3-dihidro-1H-pirrolo [3,2-c]-quinolina, 4-(3-bencilpirrolidin-1-il)-2-metil-6-fenoxiquinolina y N-[4-(3-bencilpirrolidin-1-il)-2-metilquinolin-6-il] benzamida o una sal farmacéuticamente aceptable de la misma, y diclonina o una sal farmacéuticamente aceptable de la misma.
De acuerdo con una realización adicional de la invención, se proporciona un agente antimicrobiano seleccionado del grupo que consiste en 4-metil-1-(2-feniletil)-8-fenoxi-2,3-dihidro-1H-pirrolo[3,2-c]-quinolina, 4-(3-bencilpirrolidin-1-il)-2-metil-6-fenoxiquinolina, y N-[4-(3-bencilpirrolidin-1-il)-2-metilquinolin-6-il]benzamida o una sal farmacéuticamente aceptable de los mismos, en combinación con diclonina o una sal farmacéuticamente aceptable de la misma, para su uso en el tratamiento de una infección microbiana, en particular, para la eliminación de microorganismos que se multiplican y/o clínicamente latentes asociados con tal infección.
Adicionalmente se proporciona una composición farmacéutica que comprende un agente antimicrobiano seleccionado del grupo que consiste en 4-metil-1-(2-feniletil)-8-fenoxi-2,3-dihidro-1H-pirrolo[3,2-c]-quinolina, 4-(3-bencilpirrolidin-1-il)-2-metil-6-fenoxiquinolina, y N-[4-(3-bencilpirrolidin-1-il)-2-metilquinolin-6-il]benzamida o una sal farmacéuticamente aceptable de los mismos, diclonina o una sal farmacéuticamente de la misma, y un adyuvante,
diluyente o vehículo farmacéuticamente aceptable; en particular, para su uso en la eliminación de microorganismos que se multiplican y/o clínicamente latentes asociados con una infección microbiana.
Los compuestos para su uso de acuerdo con la invención se pueden administrar de manera simultánea o secuencial y, cuando se administran de manera secuencial, se puede administrar primero o bien el agente antimicrobiano que tiene actividad biológica frente a los microorganismos clínicamente latentes o bien el agente anestésico. Cuando la administración es simultánea, la combinación se puede administrar bien en la misma composición farmacéutica o en una diferente.
De acuerdo con una realización adicional de la invención, se proporciona un producto que comprende un agente antimicrobiano seleccionado del grupo que consiste en 4-metiM-(2-feniletil)-8-fenoxi-2,3-dihidro-1H-pirrolo[3,2-c]-quinolina, 4-(3-bencilpirrolidin-1-il)-2-metil-6-fenoxiquinolina, y N-[4-(3-bencilpirrolidin-1-il)-2-metilquinolin-6-il] benzamida o una sal farmacéuticamente aceptable de los mismos, y diclonina o una sal farmacéuticamente aceptable de los mismos como una preparación combinada para su uso simultáneo, separado o secuencial para su uso en el tratamiento de una infección microbiana, en particular, para eliminar microorganismos de fase log (que se multiplican) y/o clínicamente latentes asociados con tal infección.
En una realización preferida de la invención, el agente antimicrobiano es 4-metil-1-(2-feniletil)-8-fenoxi2,3-dihidro-1H-pirrolo[3,2-c]-quinolina o una sal farmacéuticamente aceptable del mismo, preferentemente la sal de clorhidrato del mismo, que se usa con diclonina o una sal farmacéuticamente aceptable de la misma, , tal como clorhidrato de diclonina.
En una realizaciónn adicional preferida de la invención, el agente antimicrobiano es 4-(3-bencilpirrolidin-1-il)-2- metil-6-fenoxiquinolina o N-[4-(3-bencilpirrolidin-1-il)-2-metilquinolin-6-il]benzamida o una sal farmacéuticamente aceptable de la misma, que se usa con diclonina o una sal farmacéuticamente aceptable de la misma, tal como clorhidrato de diclonina.
En una realización adicional preferida más de la invención, el agente antimicrobiano es mupirocina o una sal farmacéuticamente aceptable de la misma, que se usa con diclonina o una sal farmacéuticamente aceptable de la misma, tal como clorhidrato de diclonina.
Los compuestos para su uso según la invención pueden administrarse como sustancia de partida aunque, preferentemente, los principios activos se proporcionan en forma de composiciones farmacéuticas.
Los principios activos pueden utilizarse como formulaciones individuales o como una sola formulación combinada. Cuando se combinan en la misma formulación, se apreciará que los dos compuestos deben ser estables y compatibles entre sí y con los otros componentes de la formulación.
Las formulaciones de la invención incluyen las que son adecuadas para la administración oral, parenteral (incluyendo administración subcutánea, por ejemplo, por inyección o por comprimido en depósito (depot), intradérmica, intratecal, intramuscular, por ejemplo, por depósito e intravenosa), rectal y tópica (incluyendo dérmica, bucal y sublingual) o en una forma adecuada para la administración por inhalación o administración por insuflación. La vía de administración más adecuada puede depender de la afección y del trastorno del paciente.
Preferentemente, las composiciones de la invención se formulan para administración oral o tópica. En una realización preferida, la composición es una crema o una pomada adaptada para la administración nasal, en particular para el suministro a las fosas nasales anteriores.
Las formulaciones se pueden presentar convenientemente en forma de dosificación unitaria y se pueden preparar mediante cualquiera de los métodos bien conocidos en la técnica de la farmacia, por ejemplo, como se describe en “Remington: The Science and Practice of Pharmacy", Lippincott Williams and Wilkins, 21a Edición, (2005). Los métodos adecuados incluyen la etapa de asociar los principios activos con un vehículo que constituye uno o más excipientes. En general, las formulaciones se preparan asociando de manera uniforme e íntima los principios activos con vehículos líquidos o vehículos sólidos finamente divididos, o ambos, y después, si es necesario, dando forma al producto en la formulación deseada. Se apreciará que cuando los dos principios activos se administran independientemente, cada uno de ellos puede administrarse por un medio diferente.
Cuando se formulan con excipientes, los principios activos pueden estar presentes en una concentración de 0,1 a 99,5 % (tal como de 0,5 a 95 %) en peso de la mezcla total; convenientemente de 30 a 95 % para comprimidos y cápsulas y de 0,01 a 50 % (tal como de 3 a 50 %) para preparaciones líquidas. Una concentración adecuada para la 4-metil-1-(2-feniletil)-8-fenoxi-2,3-dihidro-1H-pirrolo[3,2-c]-quinolina o una sal de la misma farmacéuticamente aceptable es de 0,1 a 5 % (p/v) de la mezcla total.
Las formulaciones adecuadas para administración oral se pueden presentar como unidades distintas, tales como cápsulas, sellos o comprimidos (por ejemplo, comprimidos masticables, en particular, para administración infantil), conteniendo cada una de ellas una unidad predeterminada de principio activo; como polvo o gránulos; como una
solución o suspensión en un líquido acuoso o no acuoso; o como una emulsión líquida de aceite en agua o de agua en aceite. Los principios activos también se pueden presentar en bolo, electuario o pasta.
Un comprimido puede prepararse mediante compresión o moldeo, opcionalmente con uno o más excipientes. Los comprimidos que se preparan con compresión pueden prepararse comprimiendo en una compresora adecuada el principio activo, tal como polvos o gránulos, en forma fluida, opcionalmente mezclado con otros excipientes convencionales tales como agentes aglutinantes (por ejemplo, jarabe, goma arábiga, gelatina, sorbitol, tragacanto, mucílago de almidón, polivinilpirrolidona y/o hidroximetilcelulosa), cargas (por ejemplo, lactosa, azúcar, celulosa microcristalina, almidón de maíz, fosfato de calcio y/o sorbitol), lubricantes (por ejemplo, estearato de magnesio, ácido esteárico, talco, polietilenglicol y/o sílice ), disgregantes (por ejemplo, almidón de patata, croscarmelosa sódica y/o glicolato sódico de almidón) y agentes humectantes (por ejemplo, lauril sulfato sódico). Los comprimidos moldeados pueden prepararse moldeando en una moldeadora adecuada una mezcla del principio activo en polvo con un diluyente líquido inerte. Los comprimidos pueden estar recubiertos o ranurados opcionalmente y pueden formularse para proporcionar una liberación controlada (por ejemplo, liberación retardada, sostenida o pulsada, o una combinación de liberación inmediata y liberación controlada) de los principios activos.
Como alternativa, los principios activos pueden incorporarse en preparaciones líquidas orales, tales como suspensiones, soluciones, emulsiones, jarabes o elixires acuosos u oleaginosos. Las formulaciones que contienen los principios activos también pueden presentarse como un producto seco para su constitución con agua u otro vehículo adecuado antes de su uso. Dichas preparaciones líquidas pueden contener aditivos convencionales, tales como agentes de suspensión (por ejemplo, jarabe de sorbitol, metilcelulosa, jarabe de glucosa/azúcar, gelatina, hidroximetilcelulosa, carboximetilcelulosa, gel de estearato de aluminio y/o grasas comestibles hidrogenadas), agentes emulsionantes (por ejemplo, lecitina, monooleato de sorbitán y/o goma arábiga), vehículos no acuosos (por ejemplo, aceites comestibles, tales como aceite de almendra, aceite de coco fraccionado, ésteres oleaginosos, propilenglicol y/o alcohol etílico) y conservantes (por ejemplo, metil o propil p-hidroxibenzoatos y/o ácido sórbico).
Las composiciones tópicas, que son útiles para el tratamiento de trastornos de la piel o de membranas accesibles por digitación (tales como la membrana de la boca, vagina, cuello uterino, ano y recto), incluyen cremas, pomadas, lociones, aerosoles, geles y soluciones o suspensiones acuosas estériles. Como tales, las composiciones tópicas incluyen aquellas en las que los principios activos se disuelven o dispersan en un vehículo dermatológico conocido en la técnica (por ejemplo, geles acuosos o no acuosos, pomadas, emulsiones de agua en aceite o de aceite en agua). Los constituyentes de dichos vehículos pueden comprender agua, soluciones de tampón acuosas, disolventes no acuosos (tales como etanol, isopropanol, alcohol bencílico, 2-(2-etoxietoxi)etanol, propilenglicol, monolaurato de propilenglicol, glicofurol o glicerol), aceites (por ejemplo, un aceite mineral como una parafina líquida, triglicéridos naturales o sintéticos, tal como Miglyol™, o aceites de silicona, tal como dimeticona). Dependiendo, entre otras cosas, de la naturaleza de la formulación, así como su uso y sitio de aplicación previstos, el vehículo dermatológico empleado puede contener uno o más componentes seleccionados de la siguiente lista: un agente o disolvente solubilizante (por ejemplo, una p-ciclodextrina, tal como hidroxipropil p-ciclodextrina, o un alcohol o poliol, tal como etanol, propilenglicol o glicerol); un agente espesante (por ejemplo, hidroximetilcelulosa, hidroxipropilcelulosa, carboximetilcelulosa o carbómero); un agente gelificante (por ejemplo, un copolímero de polioxietileno-polioxipropileno); un conservante (por ejemplo, alcohol bencílico, cloruro de benzalconio, clorhexidina, clorbutol, un benzoato, sorbato de potasio o EDTA o sal del mismo); y uno o más agentes tamponadores de pH (por ejemplo, una mezcla de dihidrogenofosfato y sales de hidrogenofosfato, o una mezcla de ácido cítrico y una sal de hidrogenofosfato). Las formulaciones tópicas también pueden formularse como un parche transdérmico.
En la técnica se conocen bien métodos de producción de composiciones farmacéuticas tópicas, tales como cremas, pomadas, lociones, aerosoles y soluciones o suspensiones acuosas estériles. Por ejemplo, en los documentos WO9510999, US 6974585, WO2006048747, así como en documentos citados en cualquiera de estas referencias, se describen métodos adecuados para la preparación de composiciones farmacéuticas tópicas.
Las composiciones farmacéuticas tópicas según la presente invención pueden utilizarse para tratar diversos trastornos de la piel o membranas, tales como infecciones de la piel o membranas (por ejemplo, infecciones de las membranas nasales, axila, ingle, perineo, recto, piel dermatítica, úlceras cutáneas y zonas de inserción de instrumental médico, tal como agujas i.v., catéteres y sondas de traqueotomía o nasogástricas) con cualquiera de las bacterias y hongos descritos anteriormente (por ejemplo, cualquiera de los organismos de Staphylococci, Streptococci, Mycobacteria o Pseudomonas mencionados anteriormente en el presente documento, tal como S. aureus (por ejemplo S. aureus resistente a meticilina (SARM))).
Las afecciones bacterianas particulares que pueden tratarse con las composiciones farmacéuticas tópicas de la presente invención también incluyen las afecciones relacionadas con la piel y membranas desveladas anteriormente en el presente documento, así como: acné vulgar; rosácea (incluyendo rosácea eritematotelangiectásica, rosácea papulopustular, rosácea fimatosa y rosácea ocular); erisipela; eritrasma; ectima; ectima gangrenoso; impétigo; paroniquia; celulitis; foliculitis (incluida la foliculitis del jacuzzi); forunculosis; carbunculosis; síndrome de la piel escaldada estafilocócica; escarlatina quirúrgica; enfermedad estreptocócica perianal; síndrome de choque tóxico estreptocócico; queratolisis punctata; tricomicosis axilar; pioderma; otitis del canal externo; síndrome de las uñas verdes; espiroquetosis; fascitis necrotizante; infecciones cutáneas micobacterianas (tales como lupus vulgar,
escrofulodermia, tuberculosis verrugosa, tubercúlide, eritema nudoso, eritema indurado, manifestaciones cutáneas de lepra tuberculoide o lepromatosa, eritema nudoso leproso, infecciones cutáneas causadas por M. kansasii, M. malmoense, M. szulgai, M. simiae, M. gordonae, M. haemophilum, M. avium, M. intracellulare, M. chelonae (incluyendo M. abscessus) o M. fortuitum, granuloma de las piscinas (o de los acuarios), linfadenitis y úlcera de Buruli (úlcera de Bairnsdale, úlcera de Searles, úlcera de Kakerifu o úlcera de Toro)); así como eczema infectado, quemaduras, abrasiones y heridas cutáneas.
Las afecciones fúngicas particulares que pueden tratarse con las composiciones farmacéuticas tópicas de la presente invención también incluyen las afecciones relacionadas con la piel y membranas desveladas anteriormente en el presente documento, así como: candidiasis; esporotricosis; dermatofitosis (por ejemplo, tiña del pie, tiña crural, tiña del cuero cabelludo, tiña ungueal o tiña corporal); tiña versicolor; e infecciones causadas por hongos Trichophyton, Microsporum, Epidermophyton o Pityrosporum ovale. Las composiciones para su uso según la invención, pueden presentarse en un envase o dispositivo dosificador que puede contener una o más formas de dosificación unitaria que contienen los principios activos. El envase puede comprender, por ejemplo, una lámina de metal o plástico, tal como un envase blíster. Cuando las composiciones son para su administración como dos composiciones distintas, estas pueden presentarse en forma de un envase doble.
Las composiciones farmacéuticas también se pueden prescribir al paciente en “envases para pacientes” que contienen el ciclo de tratamiento completo en un solo envase, normalmente un envase blíster. Los envases para pacientes tienen una ventaja sobre las recetas tradicionales ya que el farmacéutico divide un suministro de un producto farmacéutico para el paciente de un suministro a granel, con lo que el paciente siempre puede disponer del prospecto incluido en envase del paciente, que normalmente no está en las recetas tradicionales. Se ha demostrado que la inclusión del prospecto favorece el cumplimiento por parte del paciente al disponer de las instrucciones dadas por el médico tratante.
La administración de la combinación de la invención mediante un solo envase para pacientes, o envases para pacientes de cada composición, que incluye un prospecto que indica al paciente el uso correcto de la invención, es una característica deseable de la presente invención.
En la presente invención también se describe un envase para pacientes que comprende al menos un principio activo de la combinación según la invención y un prospecto con información que contiene instrucciones sobre el uso de la combinación de la invención.
En el presente documento también se describe un envase doble que comprende en asociación para administración individual, el agente antimicrobiano que tiene preferentemente actividad biológica contra microorganismos clínicamente latentes y diclonina o una de sus sales farmacéuticamente aceptables.
La cantidad de principio activo requerida para su uso en el tratamiento variará según la naturaleza de la afección que se vaya a tratar, la edad, y el estado del paciente y, en última instancia, quedará a criterio del médico o veterinario tratante. Sin embargo, en general, la dosis empleada para el tratamiento en un ser humano adulto generalmente estará en el intervalo de 0,02 a 5000 mg al día, preferentemente de 1 a 1500 mg al día. La dosis deseada se puede presentar convenientemente en una sola dosis o en dosis divididas, administradas a intervalos apropiados, por ejemplo, como dos, tres, cuatro o más subdosis al día.
Ensayos biológicos
Los procedimientos de ensayo que pueden emplearse para determinar la actividad biológica (por ejemplo, bactericida o antimicrobiana) de los principios activos, incluyen los procedimientos conocidos por los expertos en la técnica para determinar:
(a) la actividad bactericida contra bacterias clínicamente latentes; y
(b) la actividad antimicrobiana contra bacterias en fase logarítmica.
En relación al apartado (a) anterior, los métodos para determinar la actividad contra bacterias clínicamente latentes incluyen una determinación, en condiciones conocidas por los expertos en la técnica (tales como las descritas en Nature Reviews, Drug Discovery 1, 895-910 (2002), cuyas divulgaciones se incorporan en el presente documento a modo de referencia), de la Concentración Bactericida Mínima (“CBM”) estacionaria o Concentración Dormicida Mínima (“CDM”) de un compuesto de ensayo.
Como ejemplo, el documento WO2000028074 describe un método de exploración de compuestos adecuado para determinar su capacidad para eliminar microorganismos clínicamente latentes. Un método típico puede incluir las siguientes etapas:
(1) cultivar un cultivo bacteriano hasta la fase estacionaria;
(2) tratar el cultivo en fase estacionaria con uno o más agentes antimicrobianos a una concentración y/o durante un tiempo suficiente para eliminar las bacterias en cultivo, seleccionando de este modo una subpoblación fenotípicamente resistente;
(3) incubar una muestra de la subpoblación fenotípicamente resistente con uno o más compuestos o agentes de ensayo; y
(4) evaluar cualquier efecto antimicrobiano contra la subpoblación fenotípicamente resistente.
Según este método, la subpoblación fenotípicamente resistente puede verse como representativa de bacterias clínicamente latentes, que permanecen metabólicamente activas in vivo y que pueden dar como resultado una recidiva o aparición de la enfermedad.
En relación con apartado (b) anterior, los métodos para determinar la actividad contra bacterias en fase logarítmica incluyen una determinación, en condiciones estándar (es decir, condiciones conocidas por los expertos en la técnica, tales como las descritas en el documento WO 2005014585, cuyas divulgaciones del documento se incorporan en la presente memoria por referencia), de la Concentración Inhibidora Mínima (“CIM”) o Concentración Bactericida Mínima (“CBM”) de un compuesto de ensayo. A continuación, se describen ejemplos específicos de dichos métodos.
Ejemplos
Ejemplo 1: ensayo de eficacia in vitro de clorhidrato de diclonina (HT00800059), 4-metil-1-(2-feniletil)-8-fenoxi-2,3-dihidro-1H-pirrolo[3,2-c]-quinolina (HT61) y ambos fármacos en combinación frente a Staphylococcus aureus en fase estacionaria.
Cepa bacteriana
Staphylococcus aureus (Oxford); bacteria Gram positiva; cepa de referencia.
Cultivo de las bacterias
El cultivo en fase estacionaria de Staphylococcus aureus sensible a la meticilina (SASM) se llevó a cabo tal como sigue:
Subcultivo de las cepas bacterianas sobre placas de agar: Durante toda la noche, los cultivos bacterianos de S. aureus se almacenaron a -70 °C con glicerol al 20 % (crioprotector). Se hicieron estrías con el asa de Henle con las células congeladas sobre una placa de agar sanfre que se incubó a 37 °C toda la noche (o más) como placa de cultivo A. Se sembró en estrías una única colonia de cada cepa de a placa de cultivo A sobre la placa de agar sangre que se había incubado a 37 °C toda la noche (o más) como placa de cultivo B. La placa de cultivo B s almacenó a 4 °C durante una semana y sirvió como la placa de inoculación inicial.
Cultivo bacteriano en caldo de cultivo: Se inoculó una única coloni a de la placa de cultivo B en 10 ml de caldo nutritivo (N.° 2, (Oxoid)), que se incubó toda la noche a 37 °C con agitación continua a 120 rpm. Se añadieron 200 pl del cultivo de toda la noche a un frasco de 500 ml que contiene 100 ml de caldo nutritivo. El cultivo de 100 ml se incubó a 37 °C con agitación continua durante 6 a 7 días. La viabilidad de los cultivos se estima mediante recuentos de unidades formadoras de colonias (UFC) en intervalos de 2 horas durante las primeras 24 horas y de 12 a 24 horas después.
Los recuentos de unidades formadoras de colonias (UFC) se realizaron tal y como sigue: A partir de diluciones seriadas 10 veces de los cultivos, se añadieron muestras de 100 pl a un tercio de las placas de agar sangre (Oxoid) por triplicado. Se hizo un recuento de las UFC usando un contador de colonias aCoLyte (Synbiosis) tras la incubación de las placas a 37 °C durante 24 horas o más.
Cultivos bacterianos: se añadieron 10 pl del cultivo de toda la noche a 10 ml de caldo iso-sensitest reciente para hacer la inoculación a 106 UFC/ml. Se añadieron 290 pl de la suspensión celular a cada pocillo de la placa de 96 pocillos, que se incubó a 37 °C durante 24 horas. Se añadieron 300 pl de caldo iso-sensitest sin células bacterianas a los pocillos de la placa como un control no bacteriano.
Determinación de la CIM: La densidad óptica de las células bacterianas se leyó a 405 nm usando un lector de placa (Bio TEK). Se determinó la concentración CIM como la concentración más baja de fármaco que inhibe el crecimiento bacteriano.
Ensayo de fármaco de fase estacionaria
Se usaron cultivos de fase estacionaria de 7 días. Los cultivos de fase estacionaria se diluyeron para obtener recuentos de UFC en la suspensión celular de 106 a 107 UFC/ml. Se usó la suspensión celular para probar la sensibilidad al fármaco.
Compuestos y preparación
Se disolvió HT61 en DMSO a la concentración madre de 10 mg/ml.
Se disolvió HT00800059 en H2O a la solución madre de 10 mg/ml.
Los fármacos se diluyeron en primer lugar tal como sigue:
Se añadieron 7,2 j l de HT61 a 10 mg/ml en 292,8 j l de H2O seguido por una dilución de 2 veces (2x), 3 veces. Se añadieron 57,6 j l de HT00800059 a 10 mg/ml en 242,4 j l de H2O seguido por una dilución de 2 veces (2x), 2 veces.
Se añadieron 10 j l de HT00800059 de cada dilución en los pocillos de una placa de 96 pocillos seguido por la adición de 290 j l de suspensión de células bacterianas para preparar las concentraciones finales de HT00800059 a 64, 32 y 0 jg/ml.
Se añadieron 10 j l de HT00800059 de cada dilución a los pocillos de una placa de 96 pocillos seguida por la adición de 290 j l de suspensión de células bacterianas para preparar las concentraciones finales de HT00800059 a 64, 32 y 0 jg/ml.
En combinación de dos fármacos: se añadieron 10 j l de HT61 de cada dilución a los pocillos de una placa de 96 pocillos seguido por la adición de 10 j l de HT00800059 de cada dilución. Tras la adición de 280 j l de suspensión de células bacterianas para preparar las concentraciones finales de estos dos fármacos tal como se muestra a continuación en la tabla.

La incubación de los compuestos con la suspensión bacteriana se llevó a cabo durante 8 horas. Los recuentos de UFC se llevaron a cabo en intervalos de 1 hora.
Resultados
Los resultados de estos experimentos se resumen en las Figuras 1 a 6.
Conclusiones
1. HT61 a una concentración de 8 jg/ml presentó una baja actividad frente a S. aureus de fase estacionaria.
2. HT61 a una concentración de 4 y 2 jg/ml no presentó actividad observada frente a S. aureus de fase estacionaria.
3. HT00800059 a una concentración de 64 jg/ml eliminó aproximadamente 107 UFC durante 6 horas.
4. HT00800059 a una concentración de 32 jg/ml eliminó aproximadamente 104 UFC durante 8 horas.
5. HT61 en combinación con HT00800059 presentó actividad sinérgica frente a S. aureus de fase estacionaria.
Ejemplo 2: Actividad in vitro de HT00800059, HT61 y ambos fármacos en combinación frente a Staphylococcus aureus en fase log mediante análisis de dameros
Cepa bacteriana
Staphylococcus aureus (Oxford); Gram positiva; Cepa de referencia.
Cultivo de las bacterias
El cultivo en fase log de Staphylococcus aureus sensible a la meticilina (SASM) se llevó a cabo tal como sigue: Las bacterias se cultivaron en 10 ml de caldo nutritivo (N.° 2 (Oxoid)) durante toda la noche a 37 °C con agitación continua a 120 rpm. Los cultivos de toda la noche se diluyeron a 1000 X con caldo iso-sensitest. Los cultivos se incubaron a 37 °C con agitación durante 1-2 horas hasta alcanzar la log UFC 6. La viabilidad de las bacterias se estimó mediante recuentos de unidades formadoras de colonias (UFC). A partir de diluciones seriadas 10 veces de los cultivos experimentales, se añadieron muestras de 100 j l a las placas por triplicado de placas de agar nutritivo (Oxoide) o placas de agar sangre (Oxoide). Se hicieron los recuentos de UFC tras la incubación de las placas a 37 °C durante 24 horas.
Cultivos bacterianos: se añadiéronlo |jl de cultivo de toda la noche a 10 ml de caldo iso-sensitest reciente para hacer la inoculación hasta 106 UFC/ml. Se añadieron 290 j l de la suspensión celular a cada pocillo de la placa de 96 pocillos, que se incubó a 37 °C durante 24 horas. Se añadieron 300 j l de caldo iso-sensitest sin células bacterianas a los pocillos de la placa como un control no bacteriano.
Determinación de la CIM: Se leyó la densidad óptica de las células bacterianas a 405 nm usando un lector de placa (Bio TEK). La concentración CIM se determinó como la concentración más baja de fármaco que inhibe el crecimiento bacteriano.
Compuestos y preparación
El HT61 se disolvió en DMSO hasta la concentración madre de 10 mg/ml.
El HT00800059 se disolvió en H2O hasta la solución madre de 30,07 mg/ml.
Los fármacos se diluyeron en primer lugar tal como sigue:
Se añadieron 28,8 j l de HT61 a 10 mg/ml en 271,2 j l de H2O seguido por la dilución de 2 veces (2x) durante 6 veces.
El HT00800059 a 30,07 mg/ml se diluyó 2 veces (2x) durante 11 veces.
Ambos fármacos se mezclaron como el siguiente patrón: se añadieron 10 j l de HT00800059 de cada dilución a los pocillos de una placa de 96 pocillos desde la parte superior hasta la parte inferior y se añadieron 10 j l de HT61 de cada dilución a la misma placa de 96 pocillos de izquierda a derecha. Tras la adición de 280 j l de suspensión celular en fase log, las concentraciones finales de los fármacos para cada pocillo en la placa de 96 pocillos se muestran en la tabla a continuación.

La incubación de los compuestos con la suspensión bacteriana se llevó a cabo durante 24 horas. El efecto de eliminación de la combinación de fármacos y la CIM del fármaco señal se midió mediante lectura de la densidad óptica a 405 nm con un lector de placa (BioTEK). La concentración CIM se determinó como la concentración más baja de fármaco que inhibe el crecimiento bacteriano.
El índice de concentración inhibitoria fraccionada (CIF) se llevó a cabo tal como sigue: (CIM de fármaco A, probado en combinación) /(CIM de fármaco A, probado solo) (CIM de fármaco B, probado en combinación)/(CIM de fármaco B, probado solo). La interacción se definió como sinérgica si el índice CIF era <1, aditiva si el índice CIF era = 1 y antagonista si el índice CIF era >1.
Resultados

Placa 2
HTOOSQ0059

CIF = 0,75
CIM HTÜ03QQ059 512
HTMS0D059+HT614ug.'ml 12S
HT61 8
HT61+HTOOSO 0059126 Uff'ml 4
1. El CIM de HT61 solo es de 16 pg/ml.
2. El CIM de HT00800059 solo es de 512 pg/ml.
3. Con 4 pg/ml de HT61, la CIM de HT00800059 se redujo a 128 pg/ml.
4. Con HT00800059 a 128 pg/ml, la CIM de HT61 se redujo a 8 pg/ml.
Conclusión
El CIF para HT61 en combinación con HT00800059 era 0,75, lo que es indicativo de un efecto sinérgico frente a Staphylococcus aureus de fase log.
Ejemplo 3: actividad in vitro de clorhidrato de diclonina (HT00800059), 4-(3-bencillpyrrolidin-1-il)-2-metil-6-fenoxiquinolina (HT230) y N-[4-(3-bencillpirrolidin-1-il)-2-metilquinolin-6-il]benzamida (HT281), y combinaciones de los mismos, frente a Staphylococcus aureus de fase log mediante análisis de dameros
Cepa bacteriana
Staphylococcus aureus (Oxford); Gram positiva; Cepa de referencia.
Cultivo de bacterias
El cultivo en fase log de Staphylococcus aureus sensible a la meticilina (SASM) se llevó a cabo como sigue:
Las bacterias se cultivaron en 10 ml de caldo nutritivo (N.° 2 (Oxoid)) durante toda la noche a 37 °C con agitación continua a 120 rpm. Los cultivos de toda la noche se diluyeron a 1000 X con caldo iso-sensitest. Los cultivos se incubaron a 37 °C con agitación durante 1-2 horas hasta alcanzar la log UFC 6. La viabilidad de las bacterias se estimó mediante recuentos de unidades formadoras de colonias (UFC). A partir de diluciones seriadas 10 veces de los cultivos experimentales, se añadieron muestras de 100 pl a las placas por triplicado de placas de agar nutritivo (Oxoide) o placas de agar sangre (Oxoide). Se hicieron los recuentos de UFC tras la incubación de las placas a 37 °C durante 24 horas.
Cultivos bacterianos: se añadiéronlo |jl de cultivo de toda la noche a 10 ml de caldo iso-sensitest reciente para hacer la inoculación hasta 106 UFC/ml. Se añadieron 290 j l de la suspensión celular a cada pocillo de la placa de 96 pocillos, que se incubó a 37 °C durante 24 horas. Se añadieron 300 j l de caldo iso-sensitest sin células bacterianas a los pocillos de la placa como un control no bacteriano.
Determinación de la CIM: Se leyó la densidad óptica de las células bacterianas a 405 nm usando un lector de placa (Bio TEK). La concentración CIM se determinó como la concentración más baja de fármaco que inhibe el crecimiento bacteriano.
Compuestos y preparación
El HT230 se disolvió en DMSO hasta la concentración madre de 10 mg/ml.
El HT281 se disolvió en DMSO hasta la concentración madre de 10 mg/ml.
El HT00800059 se disolvió en H2O hasta la solución madre de 30,07 mg/ml.
El HT230 o el HT281 (10 mg/ml) se diluyó en primer lugar hasta 0,96 mg/ml, despés se llevó a cabo una dilución seriada de 2 veces.
El HT00800059 (30,7 mg/ml) se diluyó en una dilución seriada de 2 veces
Se añadieron 10 j l de cada dilución de cada fármaco a las placas de 96 pocillos en una matriz de dos dimensiones mostrada en la tabla a continuación. Esto fue seguido por la adición de 280 j l de cultivo en fase log a 106 UFC/ml para preparar las concentraciones finales de cada fármaco tal como se muestra.
Combinación de HT230 o HT281 con HT00800059:

En la tabla anterior, los valores en negrita son para el HT00800059. Los valores sin negrita son para HT230 o HT281. Los números mostrados en cada pocillo son jg/ml.
La incubación de los compuestos con la suspensión bacteriana se llevó a cabo durante 24 horas. Los efectos de eliminación de la combinación de fármacos se midieron mediante lectura de la densidad óptica a 405 nm con un lector de placa (Bio TEK).
Análisis de dameros
El índice de concentración inhibitoria fraccionada (CIF) se llevó a cabo tal como sigue: (CIM de fármaco A, probado en combinación) /(CIM de fármaco A, probado solo) (CIM de fármaco B, probado en combinación)/(CIM de fármaco B, probado solo). La interacción se definió como sinérgica si el índice CIF era <1, aditiva si el índice CIF era = 1 y antagonista si el índice CIF era >1.
Resultados
(1) HT230
Análisis de dameros del efecto sinérgico entre HT320 y HT00800059:

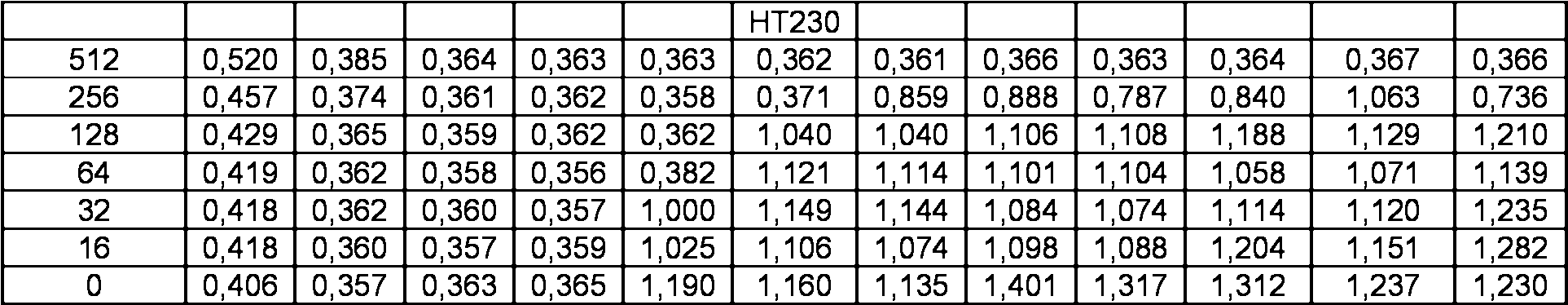
Los valores en negrita representan el crecimiento. CIM: el de HT00800059 antes de la combinación fue 512 y después de la combinación fue de 64; en HT230 antes de la combinación fue 4 y después de la combinación fue de 1. CIF: 64/512 1/4 = 0,375.
(2) HT281
Análisis de dameros del efecto sinérgico entre HT281 y HT00800059:

Los valores en negrita representan el crecimiento. CIM: el de HT00800059 antes de la combinación fue 512 y después de la combinación fue de 128; en HT281 antes de la combinación fue 8 y después de la combinación fue de 4. CIF: 128/512 4/8 = 0,75.
1. El CIM de HT230 solo es de 4 pg/ml.
2. El CIM de HT00800059 solo es de 512 pg/ml.
3. Con HT230 a 2 pg/ml, la CIM de HT00800059 se redujo a 64 pg/ml.
4. Con HT00800059 a 256 pg/ml, la CIM de HT230 se redujo a 1 pg/ml.
5. El CIM de HT281 solo es de 8 pg/ml.
6. Con HT281 a 4 pg/ml, la CIM de HT00800059 se redujo a 128 pg/ml.
7. Con HT00800059 a 128 pg/ml, la CIM de HT230 se redujo a 4 pg/ml.
Conclusiones
El CIF para HT230 en combinación con HT00800059 fue de 0,375, lo que es indicativo de un efecto sinérgico frente a Staphylococcus aureus en fase log.
El CIF para HT281 en combinación con HT00800059 fue de 0,75, lo que es indicativo de un efecto sinérgico frente a Staphylococcus aureus en fase log.
Ejemplo 4: actividad in vitro de clorhidrato de diclonina (HT00800059), muropicina y ambos fármacos en combinación frente a Staphylococcus aureus de fase log mediante análisis de dameros
Cepa bacteriana
Staphylococcus aureus (Oxford); Gram positiva; Cepa de referencia.
Cultivo de bacterias
El cultivo en fase log de Staphylococcus aureus sensible a la meticilina (SASM) se llevó a cabo como sigue:
Las bacterias se cultivaron en 10 ml de caldo nutritivo (N.° 2 (Oxoid)) durante toda la noche a 37 °C con agitación continua a 120 rpm. Los cultivos de toda la noche se diluyeron a 1000 X con caldo iso-sensitest. Los cultivos se incubaron a 37 °C con agitación durante 1-2 horas hasta alcanzar la log UFC 6. La viabilidad de las bacterias se estimó mediante recuentos de unidades formadoras de colonias (UFC). A partir de diluciones seriadas 10 veces de los cultivos experimentales, se añadieron muestras de 100 pl a las placas por triplicado de placas de agar nutritivo
(Oxoide) o placas de agar sangre (Oxoide). Se hicieron los recuentos de UFC tras la incubación de las placas a 37 °C durante 24 horas.
Cultivos bacterianos: se añadieron10 j l de cultivo de toda la noche a 10 ml de caldo iso-sensitest reciente para hacer la inoculación hasta 106 UFC/ml. Se añadieron 290 j l de la suspensión celular a cada pocillo de la placa de 96 pocillos, que se incubó a 37 °C durante 24 horas. Se añadieron 300 j l de caldo iso-sensitest sin células bacterianas a los pocillos de la placa como un control no bacteriano.
Determinación de la CIM: Se leyó la densidad óptica de las células bacterianas a 405 nm usando un lector de placa (Bio TEK). La concentración CIM se determinó como la concentración más baja de fármaco que inhibe el crecimiento bacteriano.
Compuestos y preparación
La mupirocina se disolvió en DMSO a la concentración madre de 10 mg/ml, después se diluyó 10 veces con DMSO hasta 1 mg/ml.
El HT00800059 se disolvió en H2O a la solución madre de 30,07 mg/ml.
La mupirocina (1 mg/ml) se preparó en una serie de diluciones de 2 veces.
El HT00800059 (30,7 mg/ml) se preparó en una serie de diluciones de 2 veces.
Se añadieron 10 j l de cada dilución de cada fármaco a las placas de 96 pocillos en una matriz de dos dimensiones tal como se muestra en la tabla a continuación. Esto fue seguido por la adición de 280 j l de cultivo en fase log a 106 UFC/ml para preparar las concentraciones finales de cada fármaco tal como se muestra.
Combinación de mupirocina con HT00800059:

En la tabla anterior, los valores en negrita son para HT00800059. Los valores sin negrita son para la mupirocina. Los números mostrados en cada pocillo son jg/ml.
La incubación de los compuestos con la suspensión bacteriana se llevó a cabo durante 24 horas. Los efectos de eliminación de la combinación de los fármacos se midieron mediante lectura de densidad óptica a 405 nm con un lector de placa (Bio TEK).
Análisis de dameros
El índice de concentración inhibitoria fraccionada (CIF) se llevó a cabo tal como sigue: (CIM de fármaco A, probado en combinación) /(CIM de fármaco A, probado solo) (CIM de fármaco B, probado en combinación)/(CIM de fármaco B, probado solo). La interacción se definió como sinérgica si el índice CIF era <1, aditiva si el índice CIF era = 1 y antagonista si el índice CIF era >1.
Resultados
(1) HT00800059
Análisis de dameros del efecto sinérgico entre mupirocina y HT00800059 usando caldo nutritivo

Los valores en negrita representan el crecimiento. CIM: para el HT00800059 antes de la combinación fue de 512 y después de la combinación fue de 64. En la mupirocina antes de la combinación era de 0,25 y después de la combinación era de 0,0625. CIF: 64/512 0,0625/0,25 = 0,375.
Análisis de dameros del efecto sinérgico entre la mupirocina y HT00800059 usando caldo iso-sesitest

Los valores en negrita representan el crecimiento. CIM: para el HT00800059 antes de la combinación fue de 512 y después de la combinación fue de 64. En la mupirocina antes de la combinación era de 0,25 y después de la combinación era de 0,0625. CIF: 64/512 0,0625/0,25 = 0,375.
1. El CIM de la mupirocina sola es de 0,25 pg/ml.
2. El CIM de HT00800059 solo es de 512 pg/ml.
3. Con mupirocina a 0,125 pg/ml, la CIM de HT00800059 se redujo a 64 pg/ml.
4. Con HT00800059 a 256 pg/ml, la CIM de mupirocina se redujo a 0,0625 pg/ml.
Conclusión
El CIF para la mupirocina en combinación con HT00800059 fue de 0,375, lo que es indicativo de un efecto sinérgico frente al Staphylococcus aureus de fase log. Los resultados fueron reproducibles usando dos caldos diferentes. Ejemplo 5: actividad in vitro de clorhidrato de diclonina (HT00800059), mupirocina y de ambos fármacos en combinación frente a Staphylococcus aureus resistente a la mupirocina en fase log mediante análisis de dameros Cepa bacteriana
Staphylococcus aureus (Oxford); Gram positiva; Cepa de referencia.
Cultivo de bacterias
El cultivo en fase log de Staphylococcus aureus sensible a la mupirocina (SASM) se llevó a cabo como sigue:
Las bacterias se cultivaron en 10 ml de caldo nutritivo (N.° 2 (Oxoid)) durante toda la noche a 37 °C con agitación continua a 120 rpm. Los cultivos de toda la noche se diluyeron a 1000 X con caldo iso-sensitest. Los cultivos se incubaron a 37 °C con agitación durante 1-2 horas hasta alcanzar la log UFC 6. La viabilidad de las bacterias se estimó mediante recuentos de unidades formadoras de colonias (UFC). A partir de diluciones seriadas 10 veces de los cultivos experimentales, se añadieron muestras de 100 pl a las placas por triplicado de placas de agar nutritivo (Oxoide) o placas de agar sangre (Oxoide). Se hicieron los recuentos de UFC tras la incubación de las placas a 37 °C durante 24 horas.
Cultivos bacterianos: se añadieron10 pl de cultivo de toda la noche a 10 ml de caldo iso-sensitest reciente para hacer la inoculación hasta 106 UFC/ml. Se añadieron 290 pl de la suspensión celular a cada pocillo de la placa de 96 pocillos, que se incubó a 37 °C durante 24 horas. Se añadieron 300 pl de caldo iso-sensitest sin células bacterianas a los pocillos de la placa como un control no bacteriano.
Determinación de la CIM: Se leyó la densidad óptica de las células bacterianas a 405 nm usando un lector de placa (Bio TEK). La concentración CIM se determinó como la concentración más baja de fármaco que inhibe el crecimiento bacteriano.
Compuestos y preparación
La mupirocina se disolvió en DMSO a la concentración madre de 10 mg/ml. El HT00800059 se disolvió en H2O a la solución madre de 30,07 mg/ml.
La mupirocina (10 mg/ml) se diluyó inicialmente a 3,84 mg/ml, después se diluyó adicionalmente en una dilución seriada de diluciones de 2 veces. El HT00800059 (30,7 mg/ml) se realizó en una solución seriada de diluciones de 2 veces.
Se añadieron 10 pl de cada dilución de cada fármaco a las placas de 96 pocillos en una matriz de dos dimensiones tal como se muestra a continuación en la tabla. Esto fue seguido de la adición de 280 pl de cultivo en fase log a 106 UFC/ml para preparar las concentraciones finales de cada fármaco tal como se muestra en la Tabla 1 y la Tabla 2. Combinación de mupirocina con HT00800059:

En la tabla anterior, los valores en negrita son para HT00800059. Los valores sin negrita son para la mupirocina. Los números mostrados en cada pocillo son pg/ml.
La incubación de los compuestos con la suspensión bacteriana se llevó a cabo durante 24 horas. Los efectos de eliminación de la conjugación del fármaco se midieron mediante lectura de densidad óptica a 405 nm con un lector de placa (Bio TEK).
El índice de concentración inhibitoria fraccionada (CIF) se llevó a cabo tal como sigue: (CIM de fármaco A, probado en combinación) /(CIM de fármaco A, probado solo) (CIM de fármaco B, probado en combinación)/(CIM de fármaco B, probado solo). La interacción se definió como sinérgica si el índice CIF era <1, aditiva si el índice CIF era = 1 y antagonista si el índice CIF era >1.
Resultados
Cepa resistente a mupirocina (28 traspasos)
Análisis de dameros del efecto sinérgico entre la mupirocina y el HT00800059 frente a S. aureus resistente a la mupirocina (28 traspasos)

Los valores en negrita representan el crecimiento. CIM: para el HT00800059 antes de la combinación era de 512 y para después de la combinación era de 64. Para la mupirocina antes de la combinación era de 64 y después de la combinación era de 4. CIF: 64/512 4/64 = 0,375.
Cepa resistente a la mupirocina (24 traspasos)
Análisis de dameros del efecto sinérgico entre la mupirocina y el HT00800059 frente a S. aureus resistente a la mupirocina (24 traspasos)

Los valores en negrita representan el crecimiento. CIM: para el HT00800059 antes de la combinación era de 512 y después de la combinación era de 64. Para la mupirocina antes de la combinación era de 16 y después de la combinación era de 1. CIF: 64/512 1/16 = 0,1875.
Conclusiones
Cepa resistente de 28 traspasos
1. El CIM de la mupirocina sola es de 64 pg/ml.
2. El CIM de HT00800059 solo es de 512 pg/ml.
3. Con mupirocina a 32 pg/ml, la CIM de HT00800059 se redujo a 64 pg/ml.
4. Con HT00800059 a 256 pg/ml, la CIM de mupirocina se redujo a 4 pg/ml.
5. Hubo un efecto sinérgico frente a la cepa resistente a la mupirocina con estos dos fármacos, ya que el CIF fue de 0,375.
Cepa resistente de 24 traspasos
6. El CIM de la mupirocina sola es de 16 pg/ml.
7. El CIM de HT00800059 solo es de 512 pg/ml.
8. Con mupirocina a 8 pg/ml, la CIM de hT00800059 se redujo a 64 pg/ml.
9. Con HT00800059 a 256 pg/ml, la CIM de mupirocina se redujo a 1 pg/ml.
10. Hubo un efecto sinérgico frente a la cepa resistente a la mupirocina con estos dos fármacos dado que el CIF fue de 0,1875
Claims (10)
1. Diclonina o una sal farmacéuticamente acceptable de la misma en combinación con un agente antimicrobiano seleccionado de mupirocina, 4-metil-1-(2-feniletil)-8-fenoxi-2,3-dihidro-1H-pirrolo[3,2-c]-quinolina, 4-(3-bencilpirrolidin-1-il)-2-metil-6-fenoxiquinolina, y N-[4-(3-bencilpirrolidin-1-il)-2-metilquinolin-6-il]benzamida o sales faracéuticamente aceptables de los mismos para su uso en la eliminación de microorganismos clínicamente latentes asociados con una infección microbiana.
2. La combinación para el uso de acuerdo con la reivindicación 1 en donde el agente antimicrobiano es 4-metil-1-(2-feniletil)-8-fenoxi-2,3-dihidro-1H-pirrolo[3,2-c]-quinolina o una sal farmacéuticamente aceptable del mismo.
3. La combinación para su uso de acuerdo con cualquiera de las reivindicaciones anteriores en donde la infección microbiana está causada por Staphylococci, Streptococci, Bacillaceae, Enterobacteriaceae, Haemophilis influenzae, Enterococci, o Mycobacteria.
4. La combinación para el uso de acuerdo con la reivindicación 3, en donde la infección está causada por Staphylococcus aureus.
5. La combinación para el uso de acuerdo con una cualquiera de las reivindicaciones 1 a 3 en donde la infección microbiana es una infección fúngica.
6. La combinación para el uso de acuerdo con la reivindicación 5 en donde la infección está causada por Aspergillus fumigatus, Candida albicans, Cryptococcus neoformans, Histoplasma capsulatum o Pneumocystis jiroveci.
7. La combinación para el uso según una cualquiera de las reivindicaciones 1 a 3, en la que el uso es para el tratamiento de tuberculosis, carbunco, abscesos, acné vulgar, actinomicosis, asma, disentería bacilar, conjuntivitis bacteriana, queratitis bacteriana, vaginosis bacteriana, botulismo, úlcera de Buruli, infecciones de huesos y articulaciones, bronquitis (aguda o crónica), brucelosis, heridas por quemadura, fiebre por arañazo de gato, celulitis, chancro, colangitis, colecistitis, difteria cutánea, fibrosis quística, cistitis, panbronquiolitis difusa, difteria, caries dental, enfermedades del tracto respiratorio superior, eccema, empiema, endocarditis, endometritis, fiebre entérica, enteritis, epididimitis, epiglotitis, erisipela, erisipclas, erisipeloide, eritrasma, infecciones oculares, furúnculos, vaginosis Gardnerella, infecciones gastrointestinales (gastroenteritis), infecciones genitales, gingivitis, gonorrea, granuloma inguinal, fiebre de Haverhill, quemaduras infectadas, infecciones después de intervenciones dentales, infecciones en la región bucal, infecciones relacionadas con prótesis, abscesos intraabdominales, enfermedad del legionario, lepra, leptospirosis, listeriosis, abscesos hepáticos, enfermedad de Lyme, linfogranuloma venéreo, mastitis, mastoiditis, meningitis e infecciones del sistema nervioso, micetoma, nocardiosis, uretritis no específica, oftalmía, osteomielitis, otitis, orquitis, pancreatitis, paroniquia, pelviperitonitis, peritonitis, peritonitis con apendicitis, faringitis, flemones, pinta, peste, derrame pleural, neumonía, infecciones por heridas postoperatorias, gangrena gaseosa postoperatoria, prostatitis, colitis pseudomembranosa, pitacosis, enfisema pulmonar, pielonefritis, pioderma, fiebre Q, fiebre por mordedura de rata, reticulosis, envenenamiento con ricina, enfermedad de Ritter, salmonelosis, salpingitis, artritis septicémica, infecciones septicémicas, septicemia, sinusitis, infecciones cutáneas, sífilis, infecciones sistémicas, amigdalitis, síndrome de choque tóxico, tracoma, tularemia, fiebre tifoidea, tifus, uretritis, infecciones por heridas, pian, aspergilosis, candidiasis, criptococosis, favo, histoplasmosis, intértrigo, mucomicosis, tiña, onicomicosis, pitiriasis versicolor, dermatofitosis y esporotricosis; o infecciones causadas por SASM, SARM, Staph. epidermidis, Strept. agalactiae, Strept. pyogenes, Escherichia coli, Klebs. pneumoniae, Klebs. oxytoca, Pr. mirabilis, Pr. rettgeri, Pr. vulgaris, Haemophilis influenzae, Enterococcus faecalis y Enterococcus faecium.
8. Una composición farmacéutica que comprende diclonina o una sal farmacéuticamente aceptable de la misma, un agente antimicrobiano seleccionado de mupirocina, 4-metil-1-(2-feniletil)-8-fenoxi-2,3-dihidro-1H-pirrolo[3,2-c]-quinolina, 4-(3-bencilpirrolidin-1-il)-2-metil-6-fenoxiquinolina, y N-[4-(3-bencilpirrolidin-1-il)-2-metilquinolin-6-il]benzamida o una sal farmacéuticamente aceptable de los mismos, y un vehículo farmacéuticamente aceptable para su uso en la eliminación de microorganismos clínicamente latentes asociados con una infección microbiana.
9. La composición farmacéutica para su uso de acuerdo con la reivindicación 8, en donde la composición se formula para administración oral o tópica.
10. Un producto que comprende diclonina o una sal farmacéuticamente aceptable de la misma y un agente antimicrobiano seleccionado de mupirocina, 4-metil-1-(2-feniletil)-8-fenoxi-2,3-dihidro-1H-pirrolo[3,2-c]-quinolina, 4-(3-bencilpirrolidin-1-il)-2-metil-6-fenoxiquinolina, y N-[4-(3-bencilpirrolidin-1-il)-2-metilquinolin-6-il]benzamida o una sal farmacéuticamente aceptable de los mismos como una preparación combinada para el uso simultáneo, separado o secuencial en la eliminación de microorganismos asociados con una infección microbiana.
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GBGB1005318.9A GB201005318D0 (en) | 2010-03-30 | 2010-03-30 | Novel combination |
| GBGB1013211.6A GB201013211D0 (en) | 2010-08-05 | 2010-08-05 | Novel combination |
| PCT/GB2011/050641 WO2011121345A1 (en) | 2010-03-30 | 2011-03-29 | Novel combination and use |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2703253T3 true ES2703253T3 (es) | 2019-03-07 |
Family
ID=43982400
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES11714379T Active ES2703253T3 (es) | 2010-03-30 | 2011-03-29 | Nueva combinación y uso |
Country Status (18)
| Country | Link |
|---|---|
| US (1) | US9789101B2 (es) |
| EP (1) | EP2552440B1 (es) |
| JP (1) | JP5932762B2 (es) |
| KR (1) | KR20130065649A (es) |
| CN (1) | CN102933212B (es) |
| AU (1) | AU2011234219A1 (es) |
| BR (1) | BR112012024999A2 (es) |
| CA (1) | CA2794570C (es) |
| DK (1) | DK2552440T3 (es) |
| ES (1) | ES2703253T3 (es) |
| HR (1) | HRP20182083T1 (es) |
| HU (1) | HUE041512T2 (es) |
| MX (1) | MX2012011236A (es) |
| PL (1) | PL2552440T3 (es) |
| PT (1) | PT2552440T (es) |
| RU (1) | RU2012146093A (es) |
| SI (1) | SI2552440T1 (es) |
| WO (1) | WO2011121345A1 (es) |
Families Citing this family (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20180028494A1 (en) * | 2015-02-26 | 2018-02-01 | Limited Liability Company "Konsortsium-Pik" | Pharmaceutical composition for treatment of inflammatory ear diseases, method for producing same and method for treatment using said composition |
| GB201518969D0 (en) * | 2015-10-27 | 2015-12-09 | Helperby Therapeutics Ltd | Triple combination |
| EP3703686A4 (en) * | 2017-10-30 | 2021-11-17 | Theracaine LLC | HYDROPHOBIC ARENE SULFONATE SALTS |
| KR102461715B1 (ko) * | 2020-10-12 | 2022-11-02 | 대한민국 | 항균 및 항진균 활성을 나타내는 신균주 페니실리움 비세티 및 이의 용도 |
| CN115643955B (zh) * | 2022-11-06 | 2024-04-26 | 华中农业大学 | 阿霉素在抑制植物病原菌中的应用 |
| CN116083290B (zh) * | 2022-11-21 | 2024-06-14 | 陕西省微生物研究所 | 一种铅锌和/或抗生素和/或抗性基因污染土壤修复的复合微生物菌剂及其应用 |
| US11926627B1 (en) | 2023-08-30 | 2024-03-12 | King Faisal University | Substituted 7-methyl quinoline derivatives as antitubercular agents |
| CN119656167B (zh) * | 2024-09-30 | 2025-09-05 | 天津大学 | 2-氨基喹啉在制备抑制鼠伤寒沙门氏菌药物的应用 |
Family Cites Families (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB843133A (en) * | 1957-07-16 | 1960-08-04 | Biorex Laboratories Ltd | Glycyrrhetinic acid derivatives |
| US4808410A (en) * | 1987-12-16 | 1989-02-28 | Richardson-Vicks Inc. | Pharmaceutical compositions containing dyclonine HC1 and phenol |
| IL95952A0 (en) * | 1989-10-19 | 1991-07-18 | Sterling Drug Inc | Aerosol composition for topical medicament |
| CA2024220A1 (en) * | 1990-08-29 | 1992-03-01 | Don Simmons | Composition containing silver sulfadiazine and benzocaine for use in the treatment of wounds following laser surgery |
| GB9026873D0 (en) * | 1990-12-11 | 1991-01-30 | Beecham Group Plc | Novel compounds |
| DK0727979T3 (da) | 1993-10-22 | 2001-10-01 | Smithkline Beecham Corp | Nyt præparat |
| AU1061100A (en) | 1998-11-09 | 2000-05-29 | St. George's Enterprises Limited | Screening process for antibacterial agents |
| PT1395289E (pt) * | 2000-06-08 | 2011-03-16 | Sang Dr Christine | Tratamento da dor neuropática com antagonistas do receptor de n-metil-d-aspartato (nmda) |
| US6974585B2 (en) | 2001-08-01 | 2005-12-13 | Medlogic Global Limited | Durable multi-component antibiotic formulation for topical use |
| JP3876232B2 (ja) * | 2003-04-18 | 2007-01-31 | 有限会社ユーワ商事 | 眼科用製剤、点眼液、人工涙液、コンタクトレンズケアー用品、洗眼液、および眼軟膏 |
| EP1660486A4 (en) | 2003-08-08 | 2006-12-13 | Ulysses Pharmaceutical Product | HALOGENATED CHINAZOLINYL NITROFURANES AS ANTIBACTERIAL AGENTS |
| JP5122278B2 (ja) * | 2004-05-13 | 2013-01-16 | ユニバーシティ オブ バージニア パテント ファウンデーション | 眼細胞生存の促進におけるラクリチンの使用 |
| KR20070091613A (ko) | 2004-11-08 | 2007-09-11 | 그렌마크 파머수티칼스 엘티디. | 항여드름 화합물 및 항생제 화합물을 함유하는 국소적 제약조성물 |
| GB0522715D0 (en) * | 2005-11-08 | 2005-12-14 | Helperby Therapeutics Ltd | New use |
| US7767217B2 (en) * | 2006-03-14 | 2010-08-03 | Foresight Biotherapeutics | Ophthalmic compositions comprising povidone-iodine |
| GB0705915D0 (en) | 2007-03-28 | 2007-05-09 | Helperby Therapeutics Ltd | New use |
| ES2637999T3 (es) * | 2007-05-17 | 2017-10-18 | Helperby Therapeutics Limited | Uso de compuestos de 4-(pirrolidin-1-il)quinolina para eliminar microorganismos clínicamente latentes |
| US9248093B2 (en) * | 2009-06-11 | 2016-02-02 | Becton, Dickinson And Company | Catheter locking solution having antimicrobial and anticoagulation properties |
-
2011
- 2011-03-29 ES ES11714379T patent/ES2703253T3/es active Active
- 2011-03-29 CN CN201180026672.4A patent/CN102933212B/zh not_active Expired - Fee Related
- 2011-03-29 BR BR112012024999A patent/BR112012024999A2/pt not_active IP Right Cessation
- 2011-03-29 US US13/624,488 patent/US9789101B2/en not_active Expired - Fee Related
- 2011-03-29 CA CA2794570A patent/CA2794570C/en not_active Expired - Fee Related
- 2011-03-29 HU HUE11714379A patent/HUE041512T2/hu unknown
- 2011-03-29 RU RU2012146093/15A patent/RU2012146093A/ru not_active Application Discontinuation
- 2011-03-29 SI SI201131637T patent/SI2552440T1/sl unknown
- 2011-03-29 KR KR1020127028381A patent/KR20130065649A/ko not_active Withdrawn
- 2011-03-29 PL PL11714379T patent/PL2552440T3/pl unknown
- 2011-03-29 HR HRP20182083TT patent/HRP20182083T1/hr unknown
- 2011-03-29 JP JP2013501944A patent/JP5932762B2/ja not_active Expired - Fee Related
- 2011-03-29 WO PCT/GB2011/050641 patent/WO2011121345A1/en not_active Ceased
- 2011-03-29 EP EP11714379.2A patent/EP2552440B1/en not_active Not-in-force
- 2011-03-29 DK DK11714379.2T patent/DK2552440T3/en active
- 2011-03-29 PT PT11714379T patent/PT2552440T/pt unknown
- 2011-03-29 AU AU2011234219A patent/AU2011234219A1/en not_active Abandoned
- 2011-03-29 MX MX2012011236A patent/MX2012011236A/es not_active Application Discontinuation
Also Published As
| Publication number | Publication date |
|---|---|
| HRP20182083T1 (hr) | 2019-03-08 |
| US20130096151A1 (en) | 2013-04-18 |
| JP5932762B2 (ja) | 2016-06-15 |
| EP2552440A1 (en) | 2013-02-06 |
| US9789101B2 (en) | 2017-10-17 |
| CA2794570A1 (en) | 2011-10-06 |
| WO2011121345A1 (en) | 2011-10-06 |
| RU2012146093A (ru) | 2014-05-10 |
| SI2552440T1 (sl) | 2019-03-29 |
| PT2552440T (pt) | 2019-01-10 |
| PL2552440T3 (pl) | 2019-04-30 |
| CN102933212A (zh) | 2013-02-13 |
| BR112012024999A2 (pt) | 2016-07-12 |
| CN102933212B (zh) | 2015-07-22 |
| AU2011234219A1 (en) | 2012-10-18 |
| KR20130065649A (ko) | 2013-06-19 |
| MX2012011236A (es) | 2013-03-05 |
| EP2552440B1 (en) | 2018-10-10 |
| DK2552440T3 (en) | 2019-01-21 |
| HUE041512T2 (hu) | 2019-05-28 |
| CA2794570C (en) | 2018-03-06 |
| JP2013523707A (ja) | 2013-06-17 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2668168T3 (es) | Combinación de fenoxibenzamina y poliximina E para el tratamiento de infecciones microbianas | |
| ES2719557T3 (es) | Tratamientos combinados de zidovudina para tratar infecciones microbianas | |
| ES2703253T3 (es) | Nueva combinación y uso | |
| CA2906263C (en) | Combination comprising zidovudine and polymyxin | |
| US10335454B2 (en) | Combination and use | |
| US20160038438A1 (en) | Combination of nordihydroguaiaretic acid and an aminoglycoside | |
| ES2907506T3 (es) | Combinación triple que comprende 4-metil-8-fenoxi-1-(2-feniletil)-2,3-dihidro-1H-pirrolo[3,2-c]quinolina, mupirocina y neomicina | |
| CA3066043A1 (en) | Combination comprising a particular polymyxin | |
| HK1180210B (en) | Novel combination and use | |
| HK1180210A (en) | Novel combination and use | |
| HK1260003B (en) | Triple combination comprising 4-methyl-8-phenoxy-1-(2-phenylethyl)-2,3-dihydro-1h-pyrrolo[3,2-c]quinoline, mupirocin and neomycin | |
| HK1260003A1 (en) | Triple combination comprising 4-methyl-8-phenoxy-1-(2-phenylethyl)-2,3-dihydro-1h-pyrrolo[3,2-c]quinoline, mupirocin and neomycin | |
| HK1243950B (en) | Novel combination and use | |
| HK1216716B (zh) | 包含齐多夫定和多粘菌素的组合 |