ES2712303T3 - Dominio de unión a ADN del sistema CRISPR para la producción de proteínas no fucosiladas y parcialmente fucosiladas - Google Patents
Dominio de unión a ADN del sistema CRISPR para la producción de proteínas no fucosiladas y parcialmente fucosiladas Download PDFInfo
- Publication number
- ES2712303T3 ES2712303T3 ES15816521T ES15816521T ES2712303T3 ES 2712303 T3 ES2712303 T3 ES 2712303T3 ES 15816521 T ES15816521 T ES 15816521T ES 15816521 T ES15816521 T ES 15816521T ES 2712303 T3 ES2712303 T3 ES 2712303T3
- Authority
- ES
- Spain
- Prior art keywords
- cell
- crispr
- gmd
- dna
- sequence
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies
- C07K16/06—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies from serum
- C07K16/065—Purification, fragmentation
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Y—ENZYMES
- C12Y402/00—Carbon-oxygen lyases (4.2)
- C12Y402/01—Hydro-lyases (4.2.1)
- C12Y402/01047—GDP-mannose 4,6-dehydratase (4.2.1.47), i.e. GMD
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K48/00—Medicinal preparations containing genetic material which is inserted into cells of the living body to treat genetic diseases; Gene therapy
- A61K48/0008—Medicinal preparations containing genetic material which is inserted into cells of the living body to treat genetic diseases; Gene therapy characterised by an aspect of the 'non-active' part of the composition delivered, e.g. wherein such 'non-active' part is not delivered simultaneously with the 'active' part of the composition
- A61K48/0016—Medicinal preparations containing genetic material which is inserted into cells of the living body to treat genetic diseases; Gene therapy characterised by an aspect of the 'non-active' part of the composition delivered, e.g. wherein such 'non-active' part is not delivered simultaneously with the 'active' part of the composition wherein the nucleic acid is delivered as a 'naked' nucleic acid, i.e. not combined with an entity such as a cationic lipid
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/10—Processes for the isolation, preparation or purification of DNA or RNA
- C12N15/102—Mutagenizing nucleic acids
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/11—DNA or RNA fragments; Modified forms thereof; Non-coding nucleic acids having a biological activity
- C12N15/113—Non-coding nucleic acids modulating the expression of genes, e.g. antisense oligonucleotides; Antisense DNA or RNA; Triplex- forming oligonucleotides; Catalytic nucleic acids, e.g. ribozymes; Nucleic acids used in co-suppression or gene silencing
- C12N15/1137—Non-coding nucleic acids modulating the expression of genes, e.g. antisense oligonucleotides; Antisense DNA or RNA; Triplex- forming oligonucleotides; Catalytic nucleic acids, e.g. ribozymes; Nucleic acids used in co-suppression or gene silencing against enzymes
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/06—Animal cells or tissues; Human cells or tissues
- C12N5/0601—Invertebrate cells or tissues, e.g. insect cells; Culture media therefor
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/10—Cells modified by introduction of foreign genetic material
- C12N5/12—Fused cells, e.g. hybridomas
- C12N5/16—Animal cells
- C12N5/163—Animal cells one of the fusion partners being a B or a T lymphocyte
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Y—ENZYMES
- C12Y204/00—Glycosyltransferases (2.4)
- C12Y204/01—Hexosyltransferases (2.4.1)
- C12Y204/01068—Glycoprotein 6-alpha-L-fucosyltransferase (2.4.1.68), i.e. FUT8
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/40—Immunoglobulins specific features characterized by post-translational modification
- C07K2317/41—Glycosylation, sialylation, or fucosylation
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2310/00—Structure or type of the nucleic acid
- C12N2310/10—Type of nucleic acid
- C12N2310/20—Type of nucleic acid involving clustered regularly interspaced short palindromic repeats [CRISPR]
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Engineering & Computer Science (AREA)
- Genetics & Genomics (AREA)
- Organic Chemistry (AREA)
- Biomedical Technology (AREA)
- Wood Science & Technology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Zoology (AREA)
- Biochemistry (AREA)
- General Health & Medical Sciences (AREA)
- General Engineering & Computer Science (AREA)
- Biotechnology (AREA)
- Molecular Biology (AREA)
- Biophysics (AREA)
- Microbiology (AREA)
- Immunology (AREA)
- Medicinal Chemistry (AREA)
- Plant Pathology (AREA)
- Physics & Mathematics (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Cell Biology (AREA)
- Virology (AREA)
- Crystallography & Structural Chemistry (AREA)
- Epidemiology (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Micro-Organisms Or Cultivation Processes Thereof (AREA)
- Enzymes And Modification Thereof (AREA)
- Preparation Of Compounds By Using Micro-Organisms (AREA)
- Peptides Or Proteins (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Measuring Or Testing Involving Enzymes Or Micro-Organisms (AREA)
Abstract
Un dominio de unión a ADN del sistema CRISPR, en donde el dominio de unión a ADN comprende una secuencia de ARN transcrita a partir de la secuencia de nucleótidos seleccionada del grupo que consiste en la SEQ ID NO: 41, la SEQ ID NO: 43 y la SEQ ID NO: 45.
Description
DESCRIPCION
Dominio de union a ADN del sistema CRISPR para la produccion de protemas no fucosiladas y parcialmente fucosiladas
Campo tecnico
La presente divulgacion pertenece al campo de la biotecnolog^a, la ingenieria genetica y la inmunologia. Particularmente, la presente divulgacion se refiere al desarrollo de lineas celulares en las que se modifican las v^as biologicas especificas. Tales modificaciones estan en las enzimas de la celula, en particular, las enzimas implicadas en la glucosilacion de proteinas. La presente divulgacion desarrolla sistemas de expresion de proteinas en los que se logra la modification de la cadena de glicano de la proteina. La modification especifica de la cadena de glicano produce proteinas parcialmente fucosiladas y no fucosiladas, que incluyen anticuerpos. Tales productos se usan en el desarrollo de farmacos y biomarcadores, y en diagnostico y pronostico de enfermedades. La presente divulgacion emplea la tecnologia de las repeticiones palindromicas cortas agrupadas y espaciadas de forma regular (CRISPR, del ingles Clustered Regularly Interspaced Short Palindromic Repeats).
Antecedentes y tecnica anterior de la divulgacion
La glucosilacion en eucariotas se ha estudiado a fondo durante decadas como el mecanismo de modificacion covalente postraduccional de proteinas. Se predice que aproximadamente el 1-2 % del transcriptoma humano (aproximadamente 250-500 glucogenos) traduce las proteinas que son responsables de la glucosilacion (Campbell y Yarema 2005). La glucosilacion de las proteinas celulares desempena muchas funciones importantes tales como el plegamiento y la estabilidad de la proteina, el trafico intracelular e intercelular, la interaction celula-celula y celulamatriz.
Hay cuatro grupos diferentes de glucoproteinas: N-ligadas, O-ligadas, glucosaminoglicanos y proteinas ancladas a glucosilfosfatidilinositol. La glucosilacion N-ligada tiene lugar a traves del nitrogeno de la amida de la cadena lateral de los restos de asparagina, mientras que la glucosilacion O-ligada usa el atomo de oxigeno en la cadena lateral de los restos de serina o treonina. La glucosilacion N-ligada tiene lugar en la secuencia de aminoacidos de Asn-X-Ser/Thr, en donde X puede ser cualquier aminoacido salvo prolina y acido aspartico (Helenius y Aebi 2004).
La fucosa (6-desoxi-L-galactosa) es un monosacarido que esta presente en muchas glucoproteinas y glucolipidos presentes en vertebrados, invertebrados, plantas y bacterias. La fucosilacion es el proceso de transferencia de un resto de fucosa a diversas proteinas y oligosacaridos. La fucosilacion esta regulada por varias moleculas, incluyendo fucosiltransferasas, enzimas sinteticas de guanosina difosfato (GDP)-fucosa, y transportador(es) de GDP-fucosa. Un gran numero de glucoproteinas fucosiladas son proteinas secretoras o proteinas de membrana sobre la superficie celular.
Existen 14,1 millones de casos nuevos de cancer, 8,2 millones de muertes por cancer y 32,6 millones de personas que viven con cancer (dentro de los 5 anos de diagnostico) en 2014 en todo el mundo. La elevada tasa de mortalidad del cancer sirve como un recordatorio de la necesidad de terapias mas eficaces. El cambio mas destacable en el desarrollo de farmacos de oncologia en los ultimos 20 anos ha sido el desplazamiento de los citotoxicos clasicos a farmacos que afectan a las vias de senalizacion implicadas en el cancer, conocidos como "anticuerpos monoclonales" o mAb. Hace una decada, solo habia dos mAb en el mercado y actualmente hay aproximadamente 30 mAb aprobados por la FDA de diversas modalidades terapeuticas, como Adalimumab, Infliximab, Rituximab etc. Los mAbs son el segmento de crecimiento mas rapido en la industria farmaceutica y se espera que esta rapida expansion continue. Hay mas de 100 farmacos biologicos basados en anticuerpos monoclonales en ensayos clinicos. Muchos de estos estan en ensayos de fase II y de fase III y se presentaran ante las agencias reguladoras para su aprobacion. La mejora de medicamentos de anticuerpos monoclonales a traves de las tecnologias descritas en el presente documento allanara el camino para un mejor resultado clinico para los pacientes.
El anticuerpo IgG1 humano es una glucoproteina altamente fucosilada. Dos oligosacaridos biantenarios N-ligados que consisten en un nucleo de heptasacarido con adicion variable de fucosa, galactosa, N-acetilglucosamina de bisection y acido sialico estan presentes en el Asn-297 del IgG1. La glucosilacion del anticuerpo lleva a funciones biologicas unicas conocidas como "funciones efectoras" - citotoxicidad celular dependiente de anticuerpo (ADCC, del ingles Antibody Dependent Cellular Cytotoxicity) y citotoxicidad dependiente del complemento (CDC, del ingles Complement Dependent Cytotoxicity). La ADCC es un sistema inmunitario mediado por celulas en donde las celulas inmunitarias (como celulas citoliticas naturales) lisan las celulas diana identificadas a traves de anticuerpos frente a antigenos de superficie celular.
La funcion efectora de la molecula de IgG se define mediante la interaccion de la region la region Fc del anticuerpo con receptores del leucocito, conocido como FcyRs, o interacciones con componentes del complemento. La composition de la estructura del oligosacarido es, de manera critica, importante para la funcion efectora a traves de la union de FcyR (Shields et al. 2002; Shinkawa et al. 2003; Niwa et al. 2004; Niwa, Shoji-Hosaka, et al. 2004;
Yamane-Ohnuki et al. 2004;). El analisis de la estructura cristalina de la IgG 1 humana ha revelado la interaccion intrincada de las cadenas de oligosacaridos con el dominio CH2 (Harris et al. 1998; Radaev et al. 2001).
La eficacia del mecanismo de ADCC es considerablemente dependiente del nivel de fucosilacion del anticuerpo. Cuanto menor es la fucosilacion, mayor es la tasa de ADCC. Por lo tanto, la perdida de fucosilacion tiene consecuencias biologicas significativas. La perdida podria deberse a las enzimas no funcionales de fucosiltransferasa, dando como resultado la no fucosilacion de proteinas celulares. La ausencia de fucosa de la N-acetilgucosamina principal da como resultado el anticuerpo IgG1 que tiene una elevada afinidad de union por el receptor FcYRIIIa, con el consecuente aumento de 50 - 100 veces mayor eficacia de ADCC. La mejora de ADCC con IgG no fucosilada es directamente proporcional a la elevada afinidad por FcYRIIIa, lo que permite que la Fc de la IgG no fucosilada supere la competicion de las altas concentraciones de IgG fucosilado en suero normal. La razon plausible para la elevada afinidad de la Fc de la IgG no fucosilada por FcYRIIIa puede ser la reduccion o la ausencia de inhibicion esterica en la zona de contacto receptor-ligando (Harris, 1998; Radaev, 2001).
En el sistema de expresion de mamiferos, la enzima a1,6-fucosiltransferasa codificada por el gen Fut8 es responsable de la transferencia del resto de fucosa desde la GDP-fucosa a la N-acetilglucosamina de la cadena de N-glicano en proteinas (Miyoshi, 1999). La alteracion de esta funcion genica a traves de diversos medios lleva a la produccion de proteinas no fucosiladas, que incluyen anticuerpos (Naoko Yamane-Ohnuki, 2004).
La GDP-D-manosa 4,6-deshidratasa (GMD) es un miembro de la subfamilia modificadora de azucar de los nucleotidos de la familia de deshidrogenasa/reductasa de cadena corta (SDR) (Webb, Mulichak et al. 2004).
En sistemas de expresion en mamiferos, la GDP-fucosa, un sustrato esencial de la fucosilacion, se sintetiza en el citoplasma a traves de las vias de novo y de rescate. En la via de fucosilacion de novo, se sintetiza GDP-fucosa a traves de la conversion de GDP-manosa a GDP-4-ceto-6-desoxi-manosa, catalizado por la enzima GDP-manosa 4,6-deshidratasa (GMD). Esta GDP-fucosa se transporta despues a dentro del golgi y se usa como un sustrato para la fucosilacion de proteina por la enzima a1-6 fucosiltransferasa (FUT8). La enzima transfiere el resto de fucosa desde la GDP-fucosa a la N-acetilglucosamina de la cadena de N-glicano (Miyoshi, 1999). Estas enzimas criticas, GDP-manosa 4,6-deshidratasa y a,1-6 fucosiltransferasa estan codificadas por los genes GMD y FUT8, respectivamente.
Las formas no fucosiladas de los anticuerpos terapeuticos desarrollados en soportes de mamiferos, en los que la biosintesis de fucosa esta alterada, pueden tener una ventaja clinica sobre las formas fucosiladas debido a una eficacia mejorada de ADCC frente a las celulas tumorales diana.
Historicamente, los sistemas de inactivacion genica dependian por completo de la mutacion, delecion y/o insertion mediadas por recombination homologa (HR, del ingles homologous recombination). El sistema de HR, aunque es muy especifico, es altamente ineficaz, ya que se necesitan cribar miles de clones para encontrar un clon mutado. Por otra parte, la delecion de variaciones alelicas podria llevar mas tiempo e incluso un cribado mucho mayor. En la ultima decada han evolucionado multiples tecnologias para lograr una modification genica dirigida usando una combination de un dominio de reconocimiento de secuencia de ADN y un dominio nucleasa. Estos sistemas son altamente eficientes en la identification de sitios especificos de interes y la introduction posterior de roturas en la cadena de ADN. La rotura de ADN de cadena doble (DBS, del ingles DNA double-strand break) en el locus genomico dirigido activa la reparation del ADN, que se utiliza para la modificacion de genes. La respuesta al dano del ADN esta altamente conservada en celulas eucariotas. El concepto de diseno del genoma basado en DSB es altamente transferible entre organismos altamente diversos. La creation de una rotura de cadena doble aumenta la frecuencia de inactivacion genica en loci dirigidos en miles de veces a traves de mecanismos de recombinacion homologa y de union de extremos no homologos.
La nucleasa con dedos de zinc (ZFN, del ingles Zinc Finger Nuclease) es una de las tecnicas mas frecuentemente usadas para la alteracion genica. Requiere tres bases a nivel de ADN para cada matriz en tandem de dedos de zinc. Por otra parte, el solapamiento del sitio diana y la interferencia entre los dedos individuales en una matriz de dedos de zinc complica considerablemente la produccion de ZFN especificos de secuencia. Adicionalmente, el principal inconveniente de los ZFN incluye un proceso de selection experimental elaborado y lento para identificar los motivos de ZFN para el reconocimiento de secuencias de ADN especificas.
Existen metodos en la tecnica anterior para la alteracion de los loci genomicos Fut8 y GMD. Sin embargo, ninguno de los metodos dirige la localization especifica en los loci genomicos de FUT8 y GMD mediante la tecnologia CRISPR.
Ronda et al 2014 Biotechnology and Bioengineering 111: 1604-1616 trata una herramienta basada en la red para su uso en la modificacion del genoma en celulas CHO usando CRISPR Cas9 y CRISPy.
Malphettes et al 2010 Biotechnology and Bioengineering 106: 774-783 trata la delecion de FUt8 en lineas celulares CHO usando nucleasas de dedos de zinc y la produccion de anticuerpos no fucosilados.
El documento WO 2013/013013 se refiere a composiciones y metodos para producir glucoproteinas que tienen una estructura de glucano alterada.
El documento WO 03/035835 describe una variante de lmea celular de CHO, las celulas Lecl3, con capacidad reducida para unir fucosa a los hidratos de carbono enlazados a Asn(297).
Kanda et al 2007 J Biotech 130: 300-310 trata el establecimiento de una linea celular de inactivacion de GMD.
El documento WO 2015/010114 trata un metodo y composiciones que utilizan el sistema CRISPR para alterar un gen diana en celulas eucarioticas para producir inactivaciones dobles de alelos.
El documento WO 2015/052231 describe un sistema que permite la modificacion multiple de secuencias de acido nucleico tales como secuencias genomicas.
La presente divulgacion supera las desventajas o limitaciones asociadas con los metodos de la tecnica anterior usando la tecnologia CRISpR para dirigir una localizacion especifica en el loci genomico FUT8 o el loci genomico GMD, que da como resultado una alteration completa del gen y la funcion relacionada, proporcionando una celula que produce proteinas no fucosiladas.
EXPOSICION DE LA DIVULGACION
La invencion es tal como se define en las reivindicaciones y proporciona:
En un primer aspecto: Un dominio de union a ADN del sistema CRISPR, en el que el dominio de union a ADN comprende una secuencia de ARN transcrita a partir de la secuencia de nucleotidos seleccionada del grupo que consiste en la SEQ ID NO: 41, SEQ ID NO: 43, s Eq ID NO: 45 y combinaciones de las mismas;
En un segundo aspecto: Un complejo CRISPR-nucleasa que comprende el dominio de union a ADN de acuerdo con el primer aspecto y nucleasa;
En un tercer aspecto: Un vector que comprende la secuencia de nucleotidos que codifica el dominio de union a ADN de acuerdo con el primer aspecto;
En un cuarto aspecto: Una celula que comprende un vector de acuerdo con el tercer aspecto;
En un quinto aspecto: Un procedimiento in vitro de obtencion de una celula de inactivacion de fucosa, comprendiendo dicho metodo las etapas de:
Obtener una construction CRISPR-nucleasa que comprende la secuencia de nucleotidos que codifica el dominio de union a ADN de acuerdo con el primer aspecto; y
Transfectar una celula con la construccion de la etapa (a) para obtener una celula de inactivacion de fucosa, en la que la construccion CRISPR-nucleasa proporciona el complejo CRISPR-nucleasa que comprende el dominio de union a ADN y nucleasa; y en donde el complejo escinde la secuencia del gen GMD en la celula;
En un sexto aspecto: Un metodo in vitro de obtencion de proteina con fucosilacion que varia del 0 % al 100 %, comprendiendo dicho metodo las etapas de:
a) Cultivar la celula obtenida mediante el metodo de acuerdo con el quinto aspecto, en donde la celula tiene actividad de fucosilacion que varia del 0 % al 100 %; y
b) Obtener la proteina expresada por la celula de la etapa (a).
La presente divulgacion se refiere a un dominio de union a ADN del sistema CRISPR, en donde el dominio de union a ADN comprende la secuencia seleccionada del grupo que consiste en la SEQ ID NO: 13, SEQ ID NO: 15, SEQ ID NO: 17 a la SEQ ID NO: 37, SEQ ID NO: 39, SEQ ID NO: 41, SEQ ID NO: 43, SEQ ID NO: 45, SEQ ID NO: 47 a la SEQ ID NO: 93 y combinaciones de los mismos; un complejo CRISPR-nucleasa que comprende el dominio de union a ADN tal y como se menciona anteriormente y nucleasa; un vector que comprende un dominio de union a ADN tal como se menciona anteriormente; una celula que comprende un vector tal como se menciono anteriormente; un metodo de obtencion de una celula de inactivacion de fucosa, comprendiendo dicho metodo las etapas de a) Obtener una construccion CRISPR-nucleasa, y b) Transfectar una celula con la construccion de la etapa (a) para obtener una celula de inactivacion de fucosa; un metodo de obtencion de proteina con fucosilacion que varia del 0 % al 100 %, comprendiendo dicho metodo las etapas de - a) Obtener una construccion CRISPR-nucleasa, b) Transfectar una celula con la construccion de la etapa (a) para obtener una celula con actividad de fucosilacion que varia del 0% al 100 % y c) Obtener la proteina expresada por la celula de la etapa (b); una proteina con fucosilacion del 0 % al 100 %, obtenido por el metodo tal como se menciona anteriormente; y una composition que comprende la proteina tal como se menciona anteriormente, opcionalmente junto con excipiente farmaceuticamente aceptable. Breve descripcion de las figuras adjuntas
La Figura 1A representa la secuencia codificante del gen Fut8 y la secuencia de la proteina.
La Figura 1B representa la organizacion del gen GMD.
La Figura 2A representa la secuencia de aminoacidos de Fut8 en CHOK1.
La Figura 2B representa la secuencia de aminoacidos completa del gen GMD.
La Figura 3A representa el mapa de construccion para el ARNg de la construction del vector de CRISPR/Cas pD1401.
La Figura 3B representa la secuencia diana del exon 7 de Fut8.
La Figura 4A representa la construccion CRISPR/Cas de GMD pD1401 (ARNg 167-207) que se dirige al Exon 3 del gen GMD.
La Figura 4B representa la construccion CRISPR/Cas de GMD pD1301 (ARNg 404) que se dirige al Exon 4 del gen GMD.
La Figura 4C representa la secuencia diana del exon 3 de GMD.
La Figura 4D representa la secuencia diana del exon 4 de GMD.
La Figura 5 representa las celulas CHOK1 de control y las lineas celulares clonales de CHOK1 transfectadas con la construccion CRISPR/Cas pD1401 (ARNg 514-553) que se dirige al gen Fut8, observadas en el dia 1. La Figura 6A representa las celulas CHOK1 de control y las lineas celulares de CHOK1 transfectadas con la construccion CRISPR/Cas pD1401 (ARNg 514-553) que se dirige al gen Fut8, observadas en el dia 4.
La Figura 6B representa las lineas celulares de CHOK1 transfectadas con la construccion CRISPR/Cas pD1401 (ARNg 167-207) que se dirige al exon 3 del gen GMD.
La Figura 7A representa el ensayo de citometria de flujo con LCA-FITC de las celulas CHOK1 clonales transfectadas con la construccion de CRISPR/Cas pD1401 (ARNg 514-553) que se dirige al exon 7 de FUT8. La Figura 7B representa el ensayo de citometria de flujo de las celulas CHOK1 clonales transfectadas con las construcciones de CRISPR/Cas pD1401 (ARNg 167-207) o pD1301 (ARNg 404) o pD1401 (ARNg 167-207) pD1301 (ARNg 404) que se dirigen al exon 3 y/o al exon 4 de GMD.
La Figura 7C representa el ensayo de citometria de flujo con LCA-FITC de las celulas CHOK1 clonales transfectadas con la construccion de CRISPR/Cas pD1401 (ARNg 167-207) que se dirige al exon 3 de GMD. La Figura 8A representa el perfil de fluorescencia en el ensayo de citometria de flujo con LCA-FITC de las celulas CHOK1 clonales transfectadas con la construccion de CRISPR/Cas pD1401 (ARNg 514-553) que se dirige al exon 7 de FUT8.
La Figura 8B representa el perfil de fluorescencia en el ensayo de citometria de flujo de las celulas CHOK1 clonales transfectadas con las construcciones de CRISPR/Cas pD1401 (ARNg 167-207) o pD1301 (ARNg 404) o pD1401 (ARNg 167-207) pD1301 (ARNg 404) que se dirigen al exon 3 y/o al exon 4 de GMD.
La Figura 8C representa el perfil de fluorescencia en el ensayo de citometria de flujo con LCA-FITC de las celulas CHOK1 clonales transfectadas con la construccion de CRISPR/Cas pD1401 (ARNg 167-207) que se dirige al exon 3 de GMD.
La Figura 9A representa el locus genomico del exon 7 de Fut 8, la respectiva secuencia de aminoacidos, motivos importantes de la enzima como la cadena beta 2 y la helice 3H2 y la secuencia de reconocimiento CRISPR.
La Figura 9B representa el locus genomico del exon 3 y el exon 4 de GMD, la correspondiente secuencia de aminoacidos y las secuencias de reconocimiento de CRISPR.
La Figura 10 representa el ensayo de citometria de flujo con LCA-FITC de las celulas CHOK1 clonales transfectadas con la construccion de CRISPR/Cas pD1401 (ARNg 514-553) que se dirige al exon 7 de FUT8. La Figura 11 representa el perfil de fluorescencia en el ensayo de citometria de flujo con LCA-FITC de las celulas CHOK1 clonales transfectadas con la construccion de CRISPR/Cas pD1401 (ARNg 514-553) que se dirige al exon 7 de FUT8.
La Figura 12 representa el ensayo de citometria de flujo con LCA-FITC de las celulas CHOK1 clonales transfectadas con la construccion de CRISPR/Cas pD1401 (ARNg 514-553) que se dirige al exon 7 de FUT8. La Figura 13A a 13C representan la curva de crecimiento de las celulas CHOK1 clonales transfectadas con la construccion de CRISPR/Cas pD1401 (ARNg 514-553) que se dirige al exon 7 de FUT8.
La Figura 14 representa la comparacion de las celulas CHOK1 clonales transfectadas con la construccion de CRISPR/Cas pD1401 (ARNg 514-553) que se dirige al exon 7 de FUT8 del ensayo de citometria de flujo con LCA-FITC y Strep-FITC.
La Figura 15A representa la figura representativa del producto amplificado por PCR del clon de Fut8 en CRISPR/Cas (CR1-KI-T1 n.° 022) cuando corre en gel de agarosa al 1 %.
La Figura 15B representa la figura representativa del producto amplificado por PCR del clon de GMD en CRISPR/Cas (GMD_1.12 y GMD_1.27) cuando corre en gel de agarosa al 1 %.
La Figura 15C representa la ejecucion representativa en gel de agarosa al 1 % con amplification por PCR de ADN genomico de la linea celular clonal GMD 2.30 con cebadores especificos para el locus del exon 4 de GMD. Las Figuras 16A a 16C representan la digestion por enzimas de restriction representativas del producto amplificado por PCR en el vector pTZ57R/T para confirmar la presencia de inserciones de diferentes lineas celulares de inactivacion.
Las Figuras 17A a 17G representan la alineacion representativa de la secuencia de ADN genomico en los clones de lineas celulares de inactivation de FUT8 que presentan deletion en la secuencia genica de FUT8 en el exon 7.
Las Figuras 17H a 17L representan las alineaciones de la secuencia de nucleotidos con lineas celulares clonales de inactivacion de GMD.
Las Figuras 18A y 18B representan la construccion CRISPR/Cas de FUT8, pD1401 (ARNg 514-553), que da
como resultado la delecion, codones de parada prematura en el locus diana del exon 7 de FUT8.
La Figura 18C representa la construction CRISPR/Cas de GMD, pD1401 (ARNg 167-207), que da como resultado la delecion, codones de parada prematura y mutaciones de desplazamiento del marco en el locus diana del exon 3 de GMD.
La Figura 18D representa la construccion CRISPR/Cas de GMD, pD1401 (ARNg 167-207), que da como resultado la insertion, codones de parada prematura y mutaciones de desplazamiento del marco en el locus diana del exon 3 de GMD.
La Figura 18E representa la construccion CRISPR/Cas de GMD, pD1401 (ARNg 167-207), que da como resultado la insercion, codones de parada prematura en el locus diana del exon 3 de GMD.
La Figura 18F representa la construccion CRISPR/Cas de GMD, pD1301 (ARNg 404), que da como resultado la insercion, codones de parada prematura y mutaciones de desplazamiento del marco en el locus diana del exon 4 de GMD.
La Figura 18G representa que la linea celular transfectada con ambas construcciones CRISPR/Cas de GMD, pD1301 (ARNg 404) y pD1401 (ARNg 167-207) revelan la delecion de los aminoacidos en el locus del exon 4 y el locus del exon 3 permanecio sin cambios.
La Figura 19 representa la comparacion de la secuencia de aminoacidos de FUT8 en varios eucariotas.
La Figura 20 representa la eficacia de la transfection de la linea celular CHOK1 usando diferentes protocolos.
Descripcion detallada de la divulgacion
La presente divulgacion se refiere a un dominio de union a ADN del sistema CRISPR, en donde el dominio de union a ADN comprende la secuencia seleccionada del grupo que consiste en la SEQ ID NO: 13, SEQ ID NO: 15, SEQ ID NO: 17 a la SEQ ID NO: 37, SEQ ID NO: 39, SEQ ID NO: 41, SEQ ID NO: 43, SEQ ID NO: 45, SEQ ID NO: 47 a la SEQ ID NO: 93 y combinaciones de las mismas.
En una divulgacion, la SEQ ID NO: 13, SEQ ID NO: 15, SEQ ID NO: 39 y SEQ ID NO: 17 a la SEQ ID NO: 37 se unen a la secuencia del gen Fut8; y la SEQ ID NO: 41, SEQ ID NO: 43, SEQ ID NO: 45 y SEQ ID NO: 47 a la SEQ ID NO: 93 se unen a la secuencia del gen GMD.
En otra divulgacion, la SEQ ID NO: 13 transcribe a la SEQ ID NO: 14; la SEQ ID NO: 15 transcribe a la SEQ ID NO: 16; la SEQ ID NO: 37 transcribe a la SEQ ID NO: 38; la SEQ ID NO: 39 transcribe a la SEQ ID NO: 40; la SEQ ID NO: 41 transcribe a la SEQ ID NO: 42; la SEQ ID NO: 43 transcribe a la SEQ ID NO: 44 y la SEQ ID NO: 45 transcribe a la SEQ ID NO: 46.
La presente divulgacion tambien se refiere a un complejo CRISPR-nucleasa que comprende el dominio de union a ADN tal y como se menciona anteriormente y nucleasa.
En una divulgacion, la nucleasa es endonucleasa Cas9.
En una divulgacion, la nucleasa es endonucleasa Cas9n.
La presente divulgacion tambien se refiere a un vector que comprende el dominio de union a ADN tal y como se menciona anteriormente.
En una divulgacion, el vector comprende adicionalmente una secuencia que codifica la nucleasa.
La presente divulgacion tambien se refiere a una celula que comprende un vector tal como se menciona anteriormente.
En una divulgacion, la celula se selecciona del grupo que consiste en COS, CHO-S, CHO-K1, CHO-K1 GS (-/-), CHO-DG44, CHO-DUXB11, CHO-DUKX, CHOK1SV, VERO, MDCK, W138, V79, B14AF28-G3, BHK, HaK, NSO, SP2/0-Ag14, HeLa, HEK293-F, HEK293-H, HEK293 -T, YB23HL.P2.G11.16Ag.20, perC6, celula de hibridoma productora de anticuerpos, celula madre embrionaria, celula Namalwa, linea celular de insecto de Spodoptera fugiperda (Sf), Pichia, Saccharomyces y Schizosaccharomyces.
La presente divulgacion tambien se refiere a un metodo de obtencion de una celula de inactivation de fucosa, comprendiendo dicho metodo las etapas de:
a) Obtener una construccion de CRISPR-nucleasa; y
b) Transfectar una celula con la construccion de la etapa (a) para obtener una celula de inactivacion de fucosa. La presente divulgacion tambien se refiere a un metodo de obtencion de proteina con fucosilacion que varia del 0 % al 100 %, comprendiendo dicho metodo las etapas de:
a) Obtener una construccion de CRISPR-nucleasa;
b) Transfectar una celula con la construccion de la etapa (a) para obtener una celula con actividad de
fucosilacion que varia del 0% al 100 %; y
c) Obtener la proteina expresada por la celula de la etapa (b).
En una divulgacion, la construccion CRISPR-nucleasa proporciona el complejo tal como se menciona anteriormente; y el complejo escinde la secuencia genica en la celula, dicho gen seleccionado del grupo que consiste en Fut8, GMD y combinaciones de los mismos.
En otra divulgacion, la secuencia del gen Fut8 que codifica la enzima a-1,6 Fucosiltransferasa se escinde en el Exon 7.
En otra divulgacion mas, la secuencia del gen GMD que codifica la enzima a GDP-D-manosa 4,6-deshidratasa se escinde en el exon seleccionado del grupo que consiste en el Exon 3, el Exon 4 y una combinacion de los mismos. En otra divulgacion mas, la celula se selecciona del grupo que consiste en COS, CHO-S, CHO-K1, CHO-K1 GS (-/-), CHO-DG44, CHO -DUXB11, CHO-DUKX, CHOK1SV, VERO, MDCK, W138, V79, B14AF28-G3, BHK, HaK, NS0, SP2/0-Ag14, HeLa, HEK293-F, HEK293-H, HEK293-T, YB23HL.P2.G11.16Ag.20, perC6, celula de hibridoma productora de anticuerpos, celula madre embrionaria, celula Namalwa, linea celular de insecto de Spodoptera fugiperda (Sf), Pichia, Saccharomyces y Schizosaccharomyces.
En otra divulgacion mas, la proteina esta fucosilada al 0 %, y la proteina se obtiene por alteracion del gen Fut8 en la celula.
En otra divulgacion mas, la proteina tiene del 0 % al 100 % de fucosilacion, y la proteina se obtiene por alteracion del gen GMD en la celula; y el metodo comprende ademas la adicion de L-Fucosa en medio de cultivo.
En otra divulgacion mas, la proteina es un anticuerpo.
En otra divulgacion mas, el anticuerpo es un anticuerpo monoclonal.
En otra divulgacion mas, la celula produce una proteina endogena.
En otra divulgacion mas, el procedimiento comprende adicionalmente una etapa de introduccion de un gen que codifica una proteina en la celula y la obtencion de la proteina.
La presente divulgacion tambien se refiere a una proteina con fucosilacion del 0 % al 100 %, obtenido por el metodo tal como se menciona anteriormente.
En una divulgacion, la proteina es un anticuerpo.
La presente divulgacion tambien se refiere a una composition que comprende la proteina tal y como se menciona anteriormente, opcionalmente junto con excipiente farmaceuticamente aceptable.
En una divulgacion, la proteina es un anticuerpo.
La presente divulgacion se refiere a la production de proteinas no fucosiladas, incluyendo anticuerpos no fucosilados, a partir de la celula.
La presente divulgacion se refiere a la produccion de proteinas parcialmente fucosiladas, que incluye anticuerpos parcialmente fucosilados, a partir de la celula.
La presente divulgacion tambien se refiere al direccionamiento y a la alteracion de genes aguas arriba y aguas abajo de las etapas bioquimicas clave que implican GDP-Fucosa.
La presente divulgacion emplea la tecnologia CRISPR para producir proteinas no fucosiladas.
En la presente divulgacion, una celula sin actividad de fucosilacion tambien se denomina celula de "inactivation de fucosa" o celula "FKO".
El sistema CRISPR (repeticiones palindromicas cortas agrupadas y espaciadas de forma regular) es un mecanismo inmunitario de origen natural, adaptable, usado por muchas bacterias para protegerse a ellas mismas de acidos nucleicos extranos, tales como virus o plasmidos. Las CRISPR son segmentos de ADN procariota que contienen repeticiones cortas de secuencias de bases, seguido de segmentos cortos de "ADN espaciador". Este ADN espaciador es ADN extrano obtenido de exposiciones previas a virus o plasmido bacteriano. Un conjunto de enzimas denominadas enzimas Cas (del ingles CRISPR-associated proteins, proteinas asociadas a CRlSPR) estan en asociacion con estas secuencias CRISPR, y las Cas son nucleasas que pueden cortar ADN.
La bacteria copia el material genetico en cada ADN espaciador en una molecula de ARN. Las enzimas Cas luego toman una de las moleculas de ARN, que se denominan ARN gma (ARNg). En conjunto forman el sistema CRISPR-Cas. Cuando el sistema encuentra ADN de un virus que se empareja con el ARN de CRISPR, el ARN hibrida con la secuencia de ADN y la enzima Cas entonces escinde el ADN en dos, evitando que el virus se replique.
Hay diversas enzimas Cas que funcionan en conjunto con CRISPR, pero lo mejor conocido y empleado con frecuencia en ingenieria genetica es la nucleasa Cas9, que deriva de Streptococcus pyogenes. Conjuntamente, forman el sistema CRISPR/Cas9, llamado el sistema CRISPR de tipo II.
Se ha demostrado que Cas9 es un elemento clave en determinados mecanismos de CRISPR, especificamente, sistemas CRISPR de tipo II en donde solo se requiere una proteina Cas. En este sistema, la endonucleasa Cas9 participa en el procesamiento de los ARNcr que da como resultado la destruccion del ADN diana. La funcion Cas9 es dependiente de la presencia de dos dominios de nucleasa, un dominio de nucleasa de tipo RuvC localizado en el extremo amino terminal y un dominio de nucleasa de tipo HNH que reside en la region media de la proteina.
Para el reconocimiento y escision de ADN especifico de sitio, la nucleasa Cas9 debe complejarse con dos secuencias de ARN, un ARNcr (ARN de CRISPR) y un ARNcr de transactivacion separado (ARNtracr o ARNtr), que es parcialmente complementario al ARNcr. El ARNtracr se requiere para la maduracion de ARNcr a partir de un transcrito primario que codifica multiples pre-ARNcr. Esto tiene lugar en presencia de RNasa III y Cas9. Durante la escision de ADN diana, los dominios de nucleasa de tipo HNH y RuvC de la nucleasa Cas9 cortan ambas cadenas de ADN, generando roturas de cadena doble (DSB). Los sitios de reconocimiento se definen por secuencia diana de 20 nucleotidos dentro de un transcrito de ARNcr asociado. El dominio HNH escinde la cadena complementaria, mientras que el dominio RuvC escinde la cadena no complementaria. La actividad de endonucleasa de cadena doble de Cas9 tambien requiere que una secuencia corta conservada, (2-5) conocida como motivo asociado al protoespaciador (PAM, del ingles Protospacer-Associated Motif), sigue inmediatamente en 3'- de la secuencia complementaria de ARNcr en el ADN diana. El requisito de la secuencia PAM es obligatorio para la funcion de CRISPR/Cas.
En general, se usa un sistema de dos vectores para la modification de genes mediada por CRISPR, 1) una endonucleasa Cas9 y 2) un complejo de ARNcr (ARN de CRISPR) y ARNtracr (ARNcr de transactivacion). Cuando estas dos construcciones se coexpresan en celulas de mamifero, forman un complejo y se reclutan hacia la secuencia de ADN diana. El ARNcr y el ARNtracr se combinan para formar un ARN guia quimerico (ARNg) con la misma funcion - para guiar Cas9 hacia secuencias genicas diana.
Las tecnologias de modificacion genica mediada por recombination son las primeras de su tipo que se usan para la modificacion de genes. Sin embargo, es muy rara la frecuencia de eventos exitosos usando HR, 1 de cada 3x104 celulas.
En la actualidad, la nucleasa con dedos de zinc se esta volviendo popular, ya que permiten una alta especificidad de direccionamiento con mayor frecuencia de eventos mutantes con exito. Usa proteinas de union a ADN con actividad nucleasa que se unen al ADN y crean DSB especificos de sitio. Aunque son eficaces, estos metodos requieren potentes herramientas de ingenieria de proteinas para ser exitosas y por lo tanto, limitan la flexibilidad en el direccionamiento de secuencias del genoma del complejo. La adaptation de CRISPR para las celulas de mamifero ha revolucionado la modificacion genomica con una mayor precision y facilidad del diseno. A diferencia de la ZFN, la CRISPR/Cas no requiere el diseno genetico de proteinas para todos los genes que se direccionan.
El sistema CRISPR solo requiere unas pocas construcciones de ADN simple para codificar el ARNg y Cas9. Ademas, se direccionan multiples genes de manera simultanea. En la divulgation, el sistema CRISPR/Cas se aplica para direccionar dos genes separados, FUT8 y GMD, en la via de la biosintesis de la fucosa. Aunque se produce information para el desarrollo de la linea celular CHOK de inactivation con el complejo individual CRISPR/Cas para los genes FUT8 y GMD, esta claro que el complejo se podria usar junto con la inactivacion de manera simultanea de ambos genes en lineas celulares CHOK y otras lineas celulares relevantes.
Aunque es raro para una secuencia de ARNg de 20 pb tener el 100 % de homologia en multiples sitios a lo largo del genoma, los complejos ARNsg-Cas9 son tolerantes a varios desajustes en sus dianas. Se ha documentado que Cas9 se une a multiples localizaciones en el genoma de forma no especifica, sin embargo solo crea rotura de cadena doble de ADN en un punado de esos sitios. Los datos experimentales tambien sugieren que determinados niveles de desajustes en el sitio diana de ADN permite la rotura de ADN de cadena doble. Por lo tanto, se siguen estrategias para aumentar la especificidad de CRISPR/Cas.
Una de estas observaciones es que una mutation puntual de Aspartato a Alanina (D10A) en el dominio catalitico RuvC dio como resultado roturas de cadena simple (incisiones) en lugar de roturas de cadena doble. La Cas9 mutante se conoce como Cas9n. El uso de Cas9n en dos sitios adyacente de diana de ADN permite la formation de incisiones de ADN en estrecha proximidad, y si los sitios diana estan separados de manera apropiada, genera una rotura de cadena doble.
Por lo tanto, la especificidad de la creacion de DSB es mayor, lo que eventualmente se repara mediante el mecanismo de NHEJ. La union inespedfica de Cas9n solo genera incisiones que generalmente se reparan a traves de la reparacion mediada por HR y raramente genera mutacion o efectos fuera de la diana. En la presente divulgacion, se usan Cas9n y CRISPR para inactivar los genes Fut8 y GMD. En uno de los locus diana de GMD, tambien se usa la endonucleasa Cas9 de tipo silvestre.
En la presente divulgacion, la construccion CRISPR-Cas tras la expresion en una celula proporciona el complejo CRISPR-Cas.
En la presente divulgacion, las expresiones complejo CRISPR-Cas y sistema CRISPR-Cas se usan de manera intercambiable.
La presente divulgacion se refiere a un metodo para la obtencion de proteinas no fucosiladas, mediante la alteracion o la inactivacion de la maquinaria fucosilante en una celula.
La presente divulgacion se refiere a un metodo para la obtencion de proteinas parcialmente fucosiladas, mediante la alteracion o la inactivacion de la maquinaria fucosilante en una celula.
En una divulgacion, la proteina es un anticuerpo.
En una divulgacion preferida pero no limitante, el anticuerpo es un anticuerpo monoclonal.
En la presente divulgacion, las expresiones "anticuerpo no fucosilado", "anticuerpo afucosilado", "anticuerpo fucosilado al 0 %" y "anticuerpo no fucosilado al 100 %" se usan de manera intercambiable y tienen el mismo significado y alcance.
La presente divulgacion se refiere en particular a la alteracion o a la inactivacion del gen FUT8 o del gen GMD en una celula. Cualquier experto en la materia entiende que ambos genes FUT8 y GMD se podrian alterar juntos en la misma linea celular para lograr una linea celular de inactivacion de fucosa que usa las construcciones CRISPR/Cas descritas en la presente divulgacion.
El gen FUT8 codifica la enzima a-1,6 fucosiltransferasa. El gen GMD codifica la GDP-D-manosa 4,6-deshidratasa. En una divulgacion, la celula es una celula que de manera natural produce una proteina.
En una divulgacion, la celula es una celula que de manera natural produce un anticuerpo.
En una divulgacion, la celula es una celula que no produce de forma natura una proteina dada, y se introduce en la celula un gen que codifica la proteina.
En una divulgacion, la celula es una celula que no produce de forma natural un anticuerpo, y se introduce en la celula un gen que codifica un anticuerpo.
En una divulgacion, la celula es una celula que produce de forma natural un anticuerpo, y se introduce en la celula un gen que codifica un anticuerpo.
En una divulgacion, la celula es una celula eucariota.
En una divulgacion, la celula es una celula de mamifero.
En una divulgacion, la celula es una celula de ovario de hamster chino.
En una divulgacion, la celula es una celula de ovario de hamster chino K1 (CHOK1).
En una divulgacion, la celula CHOK1 es una celula productora de anticuerpos.
En una divulgacion, el anticuerpo producido por el metodo de la presente divulgacion es un anticuerpo terapeutico. En una divulgacion, la celula CHOK1 no es una celula productora de anticuerpo, y en la celula se introduce un gen que codifica un anticuerpo.
En algunas divulgaciones, la linea celular se selecciona del grupo que consiste en COS, CHO-S, CHO-K1, CHO-DG44, CHO -DUXB11, CHO-DUKX, CHOK1SV, VERO, MDCK, W138, V79, B14AF28-G3, BHK, HaK, NSO, SP2/0-Ag14, HeLa, HEK293-F, HEK293-H, HEK293-T, YB23HL.P2.G11.16Ag.20, perC6, celula de hibridoma que produce anticuerpos, celula madre embrionaria, celula Namalwa, linea celular de insecto de Spodoptera fugiperda (Sf), Pichia, Saccharomyces y Schizosaccharomyces.
En una divulgacion no limitante, la celula es una celula con inactivacion de glutamina sintetasa (GS-/-), preferentemente una celula CHOK1 con inactivacion de glutamina sintetasa (GS-/-).
En una divulgacion, la celula se cita como una celula de "inactivacion de fucosa" o una celula "FKO" o un soporte de "inactivacion de fucosa" o un soporte "FKO".
En una divulgacion, la celula se cita como una celula recombinante.
En una divulgacion, el complejo CRISPR (repeticiones palindromicas cortas agrupadas y espaciadas de forma regular)-Cas se usa para alterar o inactivar la via de fucosilacion de una celula.
En una divulgacion, el complejo CRISPR (repeticiones palindromicas cortas agrupadas y espaciadas de forma regular)-Cas se usa para alterar o inactivar uno o mas genes de la via de fucosilacion de una celula.
En una divulgacion, el complejo CRISPR (repeticiones palindromicas cortas agrupadas y espaciadas de forma regular)-Cas se usa para alterar o inactivar o mutar el gen seleccionado del grupo que comprende a 1,6 Fucosil transferasa (gen Fut8), GDP manosa 4, 6 deshidratasa (gen GMD), GDP-ceto-6 desoximanosa 3,5 epimerasa 4-reductasa (gen FX), GDP-beta-L-fucosa pirofosforilasa (gen GEPP), y gen de Fucosa cinasa.
En una divulgacion, la presente divulgacion se refiere a la alteracion de una combinacion de gen Fut8 y gen GMD en una celula mediante el complejo CRISPR/Cas de la presente divulgacion.
En la via de fucosilacion de novo, se sintetiza GDP-fucosa a traves de la conversion de GDP-manosa a GDP-4-ceto-6-desoxi-manosa, catalizado por la enzima GDP-manosa 4,6-deshidratasa (GMD). Esta GDP-fucosa se transporta despues a dentro del golgi y se usa como un sustrato para la fucosilacion de proteina por la enzima a-(1-6) fucosiltransferasa. La enzima transfiere el resto de fucosa de la GDP-fucosa a la N-acetil glucosamina de la cadena de N-glicano.
En una divulgacion, el complejo CRISPR (repeticiones palindromicas cortas agrupadas y espaciadas de forma regular)-Cas se usa para alterar el gen Fut8 que codifica la enzima a-1,6 fucosiltransferasa.
En una divulgacion, el complejo CRISPR (repeticiones palindromicas cortas agrupadas y espaciadas de forma regular)-Cas se usa para alterar el gen GMD que codifica la enzima GDP-manosa 4, 6-deshidratasa. En una divulgacion, la region catalitica N-terminal de la enzima fucosil transferasa se direcciona mediante el complejo CRISPR/Cas.
En una divulgacion, el sitio activo de la enzima GDP-manosa 4, 6-deshidratasa se direcciona mediante el complejo CRISPR/Cas.
En una divulgacion particular, el exon 7 de la secuencia genica de Fut8 se direcciona mediante el complejo el complejo CRISPR (repeticiones palindromicas cortas agrupadas y espaciadas de forma regular)-Cas.
En una divulgacion, la enzima fucosiltransferasa se muta a una posicion de aminoacido seleccionada de las secuencias de aminoacidos en la hebra beta 2 y la region de la helice 3H2 codificada por la secuencia codificante del exon 7. Los clones resultantes pueden dar como resultado la parada prematura de la traduccion, y por lo tanto, la ausencia de secuencias aguas abajo tales como Arg-365, Arg-366, Asp-368, Lys-369, Glu-373, Tyr-382, Asp-409, Asp-410, Asp-453, Ser-469 y combinaciones de los mismos.
En una divulgacion particular, el exon 3 o el exon 4 de la secuencia genica de GMD se direcciona mediante construcciones CRISPR /Cas (repeticiones palindromicas cortas agrupadas y espaciadas de forma regular).
Las construcciones CRISPR/Cas estan disenadas como un sistema de dos vectores, en general. Una construccion codifica la expresion de endonucleasa Cas9 y el segundo vector expresa el ARNg - que esta hecho de ARNcr y ARNtracr. El ARNcr normalmente esta disenado como un fragmento de 20 nucleotidos de longitud que reconoce la secuencia diana en funcion del posicionamiento correcto del ARNtracr, la secuencia PAM y el complejo funcional de ARNcr-Cas9-ARNtracr. En determinados casos, un unico vector expresa tanto ARNg y la proteina Cas9 para una mayor actividad y facilidad de uso. La especificidad del reconocimiento de la diana proviene del diseno de ARNcr. En algunas divulgaciones, el dominio de union a ADN tambien se cita como el dominio de reconocimiento de ADN. En una divulgacion, tambien se proporcionan los polinucleotidos que codifican dicho complejo CRISPR/Cas, ya que son celulas que comprenden dichos polinucleotidos.
En una divulgacion, se proporcionan los nucleotidos que codifican el dominio de union a ADN del complejo CRISPR/Cas9. En otra divulgacion, se proporcionan los nucleotidos que codifican el dominio de nucleasa del complejo CRISPR/Cas9.
En una divulgacion, la nucleasa es Cas9.
En otra divulgacion, la nucleasa es el mutante D10A de Cas9n (nickasa, de nick, incision).
En una divulgacion, el complejo CRISPR/Cas reconoce el sitio diana en el gen FUT8 o el gen GMD. En una divulgacion, la nucleasa es una endonucleasa de asentamiento. En otra divulgacion, la nucleasa es una meganucleasa. Tambien se sabe que la especificidad de las endonucleasas de asentamiento y de las meganucleasas se pueden disenar para la union a sitios diana no naturales. Ademas, en divulgaciones a modo de ejemplo, las endonucleasas de asentamiento incluyen I-Scel, I-CeuI, Pl-PspI, Pl-Sce, I-ScelY, I-CsmI, I-PanI, I-SceII, I-Ppol, I-SceIII, I-CreI, I-TevI, I-TevII y I-TevIII. Se conocen sus secuencias de reconocimiento.
En una divulgacion, se usa una combinacion de una o mas de las nucleasas mencionadas anteriormente con el dominio de union a ADN del complejo de proteina CRISPR-Cas.
En una divulgacion, se usa la transfeccion para introducir un complejo CRISPR/Cas en una celula. Aunque se proporciona un protocolo de lipofeccion como una divulgacion a modo de ejemplo, cualquier metodo de transfeccion conocido para un experto en la materia es igualmente aplicable a los metodos de la presente divulgacion.
En otra divulgacion, la presente divulgacion proporciona metodologias para producir proteinas recombinantes en cualquier celula hospedadora en donde la celula hospedadora tiene expresion de gen FUT8 o GMD endogeno que se direcciona a traves de la tecnologia de CRISPR/Cas para alterar el gen FUT8 o GMD endogeno tal como se describe en el presente documento. La linea celular resultante es nula para la expresion del gen FUT8 o del gen GMD y se usa adicionalmente para la expresion del gen de interes.
En la presente divulgacion, se crean diecisiete lineas celulares clonales de inactivacion de FUT8 a partir de un cribado de menos de 60 lineas celulares clonales generadas tras la transfeccion con el complejo CRISPR/Cas pD1401 (ARNg 514-553). En comparacion, solo tres lineas celulares FUT8-/- se podrian seleccionar a partir de aproximadamente 120.000 lineas celulares clonales tal como se documenta en la tecnica anterior.
En la presente divulgacion, se crean treinta lineas celulares clonales de inactivacion de GMD a partir de un cribado de menos de 200 lineas celulares clonales generadas tras la transfeccion con el complejo CRISPR/Cas pD1401 (ARNg 167-207) y pD1301 (ARNg 404).
La especificidad, seguridad y simplicidad del protocolo son algunas de las ventajas ofrecidas por el complejo CRISPR/Cas y el metodo de la presente divulgacion sobre los metodos de la tecnica anterior. La alteration genica mediada por CRISPR proporciona una ventaja unica de especificidad del locus diana que permite que el complejo CRISPR/Cas hecho a medida reconozca la secuencia diana de cualquier complejidad definida por el usuario. El complejo CRISPR/Cas es mas eficaz que la ZFB en terminos de eficacia de modification del genoma y es significativamente menos toxico, lo que permite por lo tanto una mayor eficacia en la generation de clones mutantes frente a un locus particular. En la presente divulgacion, los loci genomicos de FUT8 y los loci genomicos de GMD se direccionan para la modificacion especifica de la secuencia a traves de los ARNg de CRISPR.
La metodologia descrita en el presente documento ha logrado una tasa de eficacia de mas del 28 % de exito de generacion de lineas celulares de inactivacion de FUT8 en CHOK1 (17 lineas celulares de inactivacion en CHOK1 de un cribado de menos de 60 poblaciones celulares clonales) y una tasa de exito del 15 % de generacion de lineas celulares de inactivacion de GMD en CHOK1 (30 lineas celulares de inactivacion de GMD en CHOK1 de 200 poblaciones de celulas clonales). Este logro imprevisto siguiendo la metodologia y las construcciones especificas de CRISPR de la presente divulgacion ha mejorado enormemente el desarrollo de lineas celulares de inactivacion de FUT8 y GMD.
Ademas, la presente divulgacion ha usado solamente un conjunto de construcciones de CRISPR que se direccionan a una localization genomica muy especifica en la secuencia de ADN de FUT8 en CHOK1 y dos sitios separados en los loci genomicos de GMD en CHOK1. Sorprendentemente, el complejo CRISPR/Cas da como resultado no solo la alteracion de los aminoacidos direccionados sino que tambien produce deleciones largas que introducen mutaciones de desplazamiento del marco y codon de parada prematura. De este modo, la presente divulgacion ha logrado muchas lineas celulares de inactivacion de FUT8 en CHOK1 y multiples lineas celulares de inactivacion GMD con modificaciones muy pequenas del ADN en el locus diana asi como grandes modificaciones a nivel genomico en los loci de FUT8 y GMD direccionados. La generacion de tal numero grande de lineas celulares de inactivacion de FUT8 y GMD en CHOK1 es inesperada, considerando el pequeno numero de poblaciones clonales cribadas para el fenotipo de inactivacion de fucosa. Este logro sorprendente proporciona la selection de multiples lineas celulares de inactivacion de FUT8 en CHOK1 y lineas celulares de inactivacion de GMD para establecer las lineas celulares de mejor rendimiento para la sobreexpresion de anticuerpo monoclonal.
En una divulgacion, el gen de interes se introduce en la linea celular resultante a traves de un vector de expresion que comprende secuencias de ADN que codifican la proteina de interes, produciendo de este modo la proteina
recombinante.
En otra divulgacion, la protema expresada de interes incluye anticuerpos, incluyendo anticuerpos monoclonales. En algunas divulgaciones, la inactivacion de un gen FUT8 da como resultado una lmea celular que produce proteinas recombinantes a mayores niveles.
En algunas divulgaciones, la inactivacion de un gen GMD da como resultado una linea celular que produce proteinas recombinantes a mayores niveles.
En determinadas divulgaciones, la inactivacion de un gen FUT8 proporciona una linea celular en la que aumenta una o mas actividades (funciones) de una proteina, en comparacion con proteinas producidas en celulas en donde el gen FUT8 no esta inactivado.
En determinadas divulgaciones, la inactivacion de un gen GMD proporciona una linea celular en la que aumenta una o mas actividades (funciones) de una proteina, en comparacion con proteinas producidas en celulas en donde el gen GMD no esta inactivado.
En una divulgacion, la proteina no fucosilada producida por la celula es un anticuerpo no fucosilado.
En una divulgacion no limitante, la proteina no fucosilada es un anticuerpo IgG1 no fucosilado, y preferentemente un anticuerpo IgG1 monoclonal no fucosilado.
En una divulgacion, el anticuerpo no fucosilado presenta una mayor funcion efectora que un anticuerpo fucosilado correspondiente.
En una divulgacion, el anticuerpo no fucosilado presenta propiedades terapeuticas mas eficaces que un anticuerpo fucosilado correspondiente.
En una divulgacion, el anticuerpo no fucosilado presenta una mejor toxicidad celular dependiente de anticuerpos (ADCC) que un anticuerpo fucosilado correspondiente.
En la presente divulgacion, los metodos, la preparacion y el uso de las proteinas desveladas emplean, a menos que se indique otra cosa, tecnicas convencionales en biologia molecular, bioquimica, quimica computacional, cultivo celular, tecnologia del ADN recombinante, reaccion en cadena de la polimerasa (PCR) y tecnicas relacionadas. Estas tecnicas, sus principios y sus requisitos se explican en la bibliografia y son conocidas para un experto en la materia. Las tecnicas para determinar la identidad de secuencia del acido nucleico y de los aminoacidos son conocidas para un experto en la materia.
La celula con la maquinaria de fucosilacion alterada es una celula que produce anticuerpos de forma natural, o una celula en la que se introduce un gen que codifica un anticuerpo antes o despues de la alteracion de la fucosilacion. Un "fragmento funcional" de una proteina, polipeptido o acido nucleico es una proteina, polipeptido o acido nucleico cuya secuencia no es identica a la proteina, polipeptido o acido nucleico de longitud completa, todavia mantiene la misma funcion que la proteina, polipeptido o acido nucleico de longitud completa.
El termino "anticuerpo" usado en el presente documento incluye tanto preparaciones de anticuerpo policlonal como monoclonal y tambien incluye lo siguiente: moleculas de anticuerpos quimericos, fragmentos F(ab')2 y F(ab), moleculas de Fv, moleculas Fv de cadena simple (scFv), fragmentos de anticuerpos dimericos y trimericos, minicuerpos, moleculas de anticuerpos monoclonales humanizados, anticuerpos humanos, proteinas de fusion que comprenden la region Fc del anticuerpo y cualquiera de los fragmentos funcionales que surgen de estas moleculas, en donde las moleculas derivadas mantienen la funcionalidad inmunologica de la molecula de anticuerpo parental. La expresion "anticuerpo monoclonal" en la presente divulgacion, se refiere a una composicion de anticuerpo que tiene una poblacion de anticuerpo homogenea. El anticuerpo no se limita a la especie o fuente del anticuerpo o por la manera en que se produce. El termino abarca inmunoglobulinas completas, asi como fragmentos tales como Fab, F(ab')2, Fv y otros fragmentos, asi como poblaciones homogeneas de anticuerpos quimericos y humanizados que presentan propiedades de union inmunologica de la molecula de anticuerpo monoclonal parental.
Cabe destacar que los clones/celulas de la presente divulgacion se citan con terminos tales como CR1KOT1#06, CR1KOT1#23 etc., que son denominaciones internas y no representan ninguna caracteristica particular de la celula. Estas lineas celulares se desarrollan usando el complejo CRlSPR/Cas pD1401 (ARNg 514-553).
Cabe destacar que los clones/celulas de la presente divulgacion se citan con terminos tales como C1GMD1.12, C1GMD1.27, etc., que son denominaciones internas y no representan ninguna caracteristica particular de la celula. Estas lineas celulares se desarrollan usando el complejo CRISPR/Cas pD1401 (ARNg 167-207).
Cabe destacar que los clones/celulas de la presente divulgacion se citan con terminos tales como CIGMD2.30, CIGMD2.34, etc., que son denominaciones internas y no representan ninguna caracteristica particular de la celula. Estas lineas celulares se desarrollan usando el complejo CRISPR/Cas pD1301 (ARNg 404).
Cabe destacar que los clones/celulas de la presente divulgacion se citan con terminos tales como CIGMD3.36, CIGMD3.43, etc., que son denominaciones internas y no representan ninguna caracteristica particular de la celula. Estas lineas celulares se desarrollan usando una combinacion del complejo CRISPR/Cas pD1401 (ARNg 167-207) y pD1301 (ARNg 404).
En una divulgacion, se proporciona una composicion que comprende el anticuerpo no fucosilado, opcionalmente junto con un vehiculo o aditivo o excipiente farmaceuticamente aceptable. El vehiculo o aditivo o excipiente farmaceuticamente aceptable se determina mediante la composicion que se esta administrando, asi como mediante el metodo particular usado para administrar la composicion y es conocido por un experto en la materia.
Todas las secuencias proporcionadas por la presente divulgacion se leen en la direccion 5' a 3', a menos que se indique lo contrario.
Los excipientes son importantes para lograr la estabilizacion de la proteina y mejorar otras cualidades de las biologias. Se anade varios excipientes a composiciones para estabilizar proteinas, actuar como agentes antimicrobianos, ayudar en la fabricacion de la forma de dosificacion, controlar o dirigir la administracion del farmaco, y minimizar el dolor tras la inyeccion.
Los excipientes se pueden dividir ampliamente en cinco categorias basandose en sus modos de accion:
1. Estabilizantes de proteinas: Estos excipientes estabilizan la conformation natural de la proteina. Los ejemplos incluyen polioles, azucares, aminoacidos, aminas y sales de precipitation. La sacarosa y trehalosa son los azucares usados con mas frecuencia y los polioles largos son mejores estabilizantes que los polioles mas pequenos.
2. Polimeros y proteinas: Los polimeros hidrofilos, tales como polietilenglicoles (PEG), polisacaridos y proteinas inertes, se usan de manera inespecifica para estabilizar proteinas y mejorar el ensamblaje de las proteinas. Los ejemplos incluyen dextrano, almidon hidroxietilico (HETA), PEG-4000 y gelatina.
3. Tensioactivos: Los tensioactivos no ionicos se usan ampliamente para estabilizar proteinas, suprimir la agregacion y ayudar en el replegamiento proteico. Polisorbato 80 y polisorbato 20, tambien conocidos como Tween 80 y Tween 20, respectivamente, se usan generalmente en agentes terapeuticos de mAb. otros ejemplos incluyen Brij 35, Triton X-l0, Pluronic F127, dodecilsulfato de sodio (SDS).
4. Aminoacidos: Estos excipientes estabilizan las proteinas mediante varios mecanismos. Los ejemplos incluyen histidina, arginina y glicina. Otros aminoacidos usados como excipientes de formulation incluyen mezclas de metionina, prolina, lisina, acido glutamico y arginina.
5. Conservantes: Estos compuestos se incluyen en formulaciones para evitar el crecimiento microbiano. Los ejemplos incluyen alcohol bencilico, m-Cresol y fenol.
El material biologico usado en la presente divulgacion se obtiene de fuera de la India.
Fundamentos para el direccionamiento de secuencia genomica especifica en el locus de FUT 8
FUT8 esta comprendido de tres dominios, un dominio de superhelice en N-terminal, un dominio catalitico y un dominio SH3 en C-terminal.
La estructura de la proteina Fut8 se estudia a fondo para entender el dominio funcional de la secuencia de aminoacidos de la enzima. La estructura cristalina tridimensional de la enzima FUT8 revelo 15 hebras y 16 helices. Hay al menos tres regiones, el extremo N-terminal (restos 68-107), el extremo C-terminal (573-575) y los restos 368 372 que estan desordenados.
El presunto dominio catalitico de la enzima FUT8 consiste en dos dominios, un dominio alfa/beta de lamina abierta y el pliegue de Rossmann ampliamente conocido por la region de union a nucleotidos. El dominio alfa/beta consistio en cinco helices y tres cadenas beta, que son alfa 4, 3H1, 3H2, 3H3, y las cadenas beta 1, beta 2 y beta 3. El dominio se localiza en el extremo N-terminal de la secuencia de la proteina. No hay evidencias claras de como el dominio catalitico del extremo N-terminal es responsable de la funcionalidad de la enzima.
El pliegue de Rossmann se localiza aguas abajo, en los restos 359-492 y contiene varias helices alfa y cadenas beta. Una serie de restos, Arg 365, Arg366, Asp-368, Lys-369, Glu-373, Tyr-382, Asp-409, Asp-410, Asp-453 y Ser-469 desempena un papel importante en el dominio catalitico de la enzima FUT8.
Diez restos de aminoacidos, Arg365, Arg366, Asp-368, Lys-369, Glu-373, Tyr-382, Asp-409, Asp-410, Asp-453 y Ser-469 de la proteina enzimatica FUT8 humana se conservan entre diversas especies, que incluyen los
vertebrados, insectos, nematodos y ascidias tal como se observa en la figura 19 de la presente divulgacion.
Para entender la contribucion de la secuencia de aminoacidos espedfica en el gen FUT8 en la actividad a 1,6 fucosiltransferasa, las regiones de la secuencia de aminoacidos de FUT8 se comparan entre multiples especies. El alineamiento muestra que las secuencias de la enzima constituyen restos de aminoacidos altamente conservados en la cadena beta 2 y en la region de helice 3H2. Por tanto, estas posiciones de aminoacidos son la diana del complejo CRISPR/Cas en el metodo de la presente divulgacion.
Fundamentos para el direccionamiento de los genes GMD y FUT8 en la linea celular CHOK1
El soporte de inactivacion de fucosa es util para lograr el desarrollo de la molecula de anticuerpo monoclonal no fucosilado. En muchos casos, el desarrollo de un anticuerpo completamente no fucosilado es un resultado preferido y, por lo tanto, se preparan estrategias en la presente divulgacion para crear inactivaciones completas de los genes de la via de la biosintesis de la fucosa. En determinados casos, el producto de farmaco terapeutico de anticuerpo monoclonal puede requerir la fucosilacion parcial, que no esta disponible de forma natural. Para crear versiones disenadas de anticuerpos monoclonales fucosilados para fines terapeuticos, la linea celular CHOK1 de inactivacion de GMD es muy util.
El gen GMD esta implicado en la via de fucosilacion, aguas arriba del gen FUT8 y es responsable de la sintesis de GDP-fucosa a traves de la conversion de GDP-manosa a GDP-4-ceto-6-desoxi-manosa. Esta etapa es una de las etapas criticas de la via de biosintesis de fucosa de novo. La GDP fucosa tambien se produce en celulas a traves de la via de rescate y se usa para la fucosilacion de proteinas celulares. En la via de rescate, las celulas captan fucosa del medio de crecimiento. La via de novo para la biosintesis de fucosa se detiene por completo si el gen GMD esta inactivo y completamente no funcional. La biosintesis de GDP-fucosa aun permanece activa a traves de la via de rescate si el medio de crecimiento se suplementa con fucosa. Por lo tanto, la via de biosintesis de fucosa y la fucosilacion de proteina celular aun permanece activa.
Las lineas celulares CHOK1 de inactivacion de GMD proporcionan una ventaja unica si el anticuerpo monoclonal necesita estar desfucosilado al 100 %, se utiliza el soporte celular de inactivacion doble de GMD. En casos, en donde los anticuerpos monoclonales requieren un nivel especifico de fucosilacion, se utiliza la via de rescate para generar GDP-Fucosa a traves de la suplementacion del medio de crecimiento con L-fucosa. Esencialmente, el nivel de fucosilacion de anticuerpo monoclonal se logra a traves de niveles de titulacion de L-fucosa en medio de crecimiento. Por lo tanto, la estrategia de inactivacion de GMD proporciona un producto no fucosilado al 100 % a niveles variables de fucosilacion mediante titulacion simple de L-fucosa en medio de cultivo de CHOK1. Esta es una estrategia unica para controlar la fucosilacion de la produccion de anticuerpo monoclonal en celulas CHOK1.
Por otro lado, la enzima Fu8 funciona aguas abajo de la etapa de biosintesis de GDP-fucosa y es la ultima etapa enzimatica para la fucosilacion de proteinas celulares en el golgi. Los precursores de fucosilacion de la via de novo y de rescate usan la enzima FUT8 para la transferencia del resto a la fucosa final. Por lo tanto, la inactivacion del gen Fut8 esencialmente detiene tanto la via de novo como la de rescate de la fucosilacion de proteina celular. Esta estrategia da como resultado una desfucosilacion del 100 % de anticuerpos monoclonales producidos en la linea celular CHOK1 de inactivacion de Fut8.
Direccionamiento del sitio activo de GMD:
La enzima GDP-D-manosa 4,6-deshidratasa (GMD) cataliza la conversion de GDP-D-manosa al intermediario GDP-4-ceto-6-desoxi-D-manosa. Esto funciona como un punto de ramificacion para diferentes desoxihexosas, incluyendo GDP-D-ramnosa, GDP-L-fucosa, GDP-6-desoxi-D-talosa,y GDPdidesoxi aminoazucar GDP-D-perosamina. Entre estos, la GDP-L-fucosa es un intermediario importante en la via de la biosintesis de la fucosa. GMD es un miembro de la subfamilia modificadora de NDP-azucar de las deshidrogenasas/reductasas de cadena corta (SDR).
Como miembro de esta subfamilia, GMD se une a su cofactor NADP(H) en la porcion de N-terminal de la molecula en la que esta presente una region comun rica en glicina. La triada catalitica se ha identificado como Tyr-XXX-Lys y Ser/Thr, que son todas importantes para la catalisis. Aunque hay una variabilidad significativa en la secuencia de aminoacidos en miembros de este grupo de enzimas, existen similitudes en la estructura tridimensional.
El analisis estructural de GMD de E. coli sugiere que la molecula activa esta en configuracion dimerica. Mientras que el homologo de Arabidopsis thaliana es tetramerico, y el sitio de union de NADP(H) esta intimamente implicado en la creacion de la superficie de contacto del tetramero. Lo mas probable es que la forma funcional de la enzima GMD en eucariotas consista en la configuracion tetramerica. La GMD cristaliza con cuatro unidades monomericas y los monomeros interaccionan entre si para formar el dominio catalitico. Los monomeros opuestos interaccionan a traves de enlaces de hidrogeno entre Asn 163, Arg147, Glu166, Tyr145 y Arg47. La tetramerizacion de GMD da como resultado sitios de union de cofactor adecuados (NADPH) en la zona de contacto. Ser85 desempena un papel crucial en los enlaces de hidrogeno con pirofosfato en el sitio activo. Ademas, los hidroxilos de nicotinamida ribosa estan dentro de la distancia del enlace de hidrogeno con los restos cataliticos Tyr150 y Lys154, interacciones que estan altamente conservadas en enzimas SDR.
El bucle RR, un segmento de nueve restos (Ag35-Arg43), se estira en el monomero adyacente haciendo interacciones protema-protema y contactos con el cofactor adyacente. Las interacciones protema-protema incluyen enlaces de hidrogeno de Arg35 con Ser85 y Glu188. Para la union al sustrato, se ha documentado que la interaction de GDP-D-manosa podria depender de la capacidad para hacer posibles enlaces de hidrogeno con Thr126, Ser127 y Glu128. Ademas, los restos cataltticos Thr126 y Tyr150 as^ como Ser85 podrian hacer enlaces de hidrogeno con el hidroxilo 04 de la hexosa. El mecanismo catalitico propuesto para GMD implica unos pocos restos clave como Thr126, Ser127, Glu128, Tyr150 entre otros.
Considerando la importancia de estos restos, se han direccionado multiples complejos CRISPR/Cas que potencialmente alteran la configuration tetramerica de la enzima activa asi como afectan a la region de union al cofactor y a los motivos de interaccion con el sustrato. Se disena una construction de CRISPR en la zona de contacto dimerica propuesta en la secuencia de aminoacidos ADVDGVGTLRLL. Esta region es parte del Exon 4 del gen GMD. La construccion CRISPR se direcciona a la endonucleasa Cas9 para crear una rotura de ADN de cadena doble en el exon 4. El sitio de la rotura se localiza antes de los restos clave de aminoacidos en el motivo ADVDGVGTLRLL con el supuesto de que cualquier modification en estos aminoacido afecta directamente al mecanismo catalitico de la enzima GMD.
Un segundo conjunto de complejos CRISPR/Cas se disena en el exon 3 del gen GMD. Este diseno de CRISPR es unico para alta especificidad, cuando un mutante de Cas9, conocido como mutante de nickasa D10A Cas9 (Cas9n) se elige, provocando una rotura de ADN de cadena simple. Los dos complejos CRISPR/Cas disenados para dos roturas de ADN de cadena simple permiten un alto nivel de especificidad. Las construcciones se disenan en un motivo de secuencia de aminoacidos de zona de contacto tetramerica propuesto YGDLTDSTCLVK. Las dos roturas de cadena simple permiten la reparation del ADN por el mecanismo NHEJ e introducen mutaciones en esta region. Estas mutaciones afectan al resto importante Ser85 implicado en el mantenimiento de interacciones de los monomeros en la configuracion tetramerica.
La posicion de motivos estructurales importantes en el exon 3 y en el exon 4 del gen MD y en posiciones diana de CRISPR se ilustran en la figura 9B.
Ambos disenos de CRISPR/Cas son unicos y logran un alto potencial en la generation de lineas celulares CHOK1 de inactivation de fucosa.
Direccionamiento del sitio activo de Fut8:
Uno de los aspectos mas importantes de la presente divulgation es el direccionamiento del sitio catalitico de la enzima a 1,6-fucosiltransferasa, codificado por el gen Fut8. La estructura de la proteina Fut8 se estudia a fondo para entender el dominio funcional de la secuencia de aminoacidos de la enzima. La estructura cristalina tridimensional de la enzima FUT8 revelo 15 hebras y 16 helices. Hay al menos tres regiones, el extremo N-terminal (restos 68-107), el extremo C-terminal (573-575) y los restos 368-372 que estan desordenados.
El presunto dominio catalitico de la enzima FUT8 consiste en dos dominios, un dominio alfa/beta de lamina abierta y el pliegue de Rossmann ampliamente conocido por la region de union a nucleotidos. El dominio alfa/beta consiste en cinco helices y tres cadenas beta, que son alfa 4, 3H1, 3H2, 3H3, y las cadenas beta 1, beta 2 y beta 3. El dominio se localiza en el extremo N-terminal de la secuencia de la proteina. No hay evidencias claras de como el dominio catalitico del extremo N-terminal es responsable de la funcionalidad de la enzima. Las secuencias diana CRISPR/Cas se direccionan en esta region. El locus genomico del exon 7 de Fut 8, la respectiva secuencia de aminoacidos y la position de los motivos estructurales importantes y las posiciones diana de CRISPR se ilustran en la figura 9A.
Este direccionamiento no es una selection aleatoria, pero se ha llegado a ella, en la presente divulgacion, mediante experimentation para determinar la posicion altamente especifica en el gen o enzima, cuya alteration asegura que la fucosilacion parcial que esta causada por la enzima truncada o parcialmente funcional se evita.
El plegamiento Rossmann, por otro lado, se localiza aguas abajo en el resto 359-492 y contiene varias helices alfa y cadenas beta. Una serie de restos, Arg 365, Arg366, Asp-368, Lys-369, Glu-373, Tyr-382, Asp-409, Asp-410, Asp-453 y Ser-469 desempena un papel importante en el dominio catalitico de la enzima FUT8.
Por tanto, el direccionamiento de la region equivalente al sitio activo de la enzima asegura la alteracion completa del gen Fut8 y proporciona resultados eficaces en comparacion cualquier tecnica que es incapaz de direccionar una posicion concreta en el gen Fut8 o una tecnica que direcciona otra localization en el gen Fut8, que podria dar como resultado una alteracion parcial del gen Fut8 y de la actividad de la enzima. Una celula con una maquinaria fucosilada parcialmente funcional produce proteinas parcialmente fucosiladas, que muestran menores funciones terapeuticas en comparacion con las proteinas no fucosiladas. Las celulas producidas por el metodo de la presente divulgacion produce proteinas completamente o al 100 % no fucosiladas, incluyendo anticuerpos no fucosilados al 100 %.
La presente divulgacion introduce mutaciones en posiciones criticas de aminoacidos en el sitio catalitico de la secuencia de codon de FUT8 a traves del complejo CRISPR/Cas. El diseno de CRISPR se centra principalmente en el direccionamiento del dominio catalttico de N-terminal, espedficamente la cadena beta 2 y la region de la helice 3H2 mediante la incorporacion de roturas de cadena simple. El sistema de reparacion de ADN celular introduce cambios de nucleotidos mientras lleva a cabo la reparacion de la cadena simple y crea una enzima FUT8 no funcional.
El sistema CRISPR es bien conocido por la delecion e insercion de manera localizada y por lo tanto crea una mutacion de desplazamiento del marco en el exon 7 direccionado e inserta codones de parada. La introduction de codones de parada asegura la finalization prematura de la traduction y el plegamiento Rossmann de aguas abajo se excluye de la estructura de la enzima, dando como resultado una enzima FUT8 no funcional.
En una divulgacion, el posterior analisis de ADN genomico de las lineas celulares de CHOK1 modificadas revela la delecion, insercion, el codon de parada asi como mutaciones de desplazamiento del marco. Por tanto, la presente divulgacion contempla la alteration del gen Fut8 y la enzima fucosiltransferasa mediante el direccionamiento de posiciones de aminoacidos en la cadena beta 2 y en la helice 3H2 a traves de deleciones, inserciones y/o mutaciones de desplazamiento del marco.
Los clones resultantes pueden dar como resultado la parada prematura de la traduccion, provocando de este modo grandes cambios en secuencias criticas aguas abajo, tales como Arg-365, Arg-366, Asp-368, Lys-369, Glu-373, Tyr-382, Asp-409, Asp-410, Asp-453, Ser-469 y combinaciones de los mismos.
La Figura 19 de la presente divulgacion ilustra la alineacion de la secuencia de aminoacidos de FUT8 de rata, ser humano, raton, vaca y hamster chino. Los aminoacidos en el plegamiento de Rossmann 365, 366, 368, 369 y 373 estan marcados con asteriscos. Los aminoacidos en el recuadro sombreado indican restos no alineados con la secuencia consenso. Los aminoacidos en la region del exon 7 tambien estan marcados. Las secuencias de reconocimiento de CRISPR estan marcadas por lineas gruesas.
En la presente divulgacion, se analiza la secuencia de aminoacidos de FUT8 de la base de datos genomica de CHOK1 y se confirma que estos aminoacidos criticos se conservan en el gen FUT8 derivado de la linea celular de CHOK1. Se diseno el complejo CRISPR/Cas especifico de secuencia, se direccionando las secuencias genicas aguas arriba de estos motivos de aminoacidos para introducir modificaciones genomicas. Se analizo como la alteracion de las secuencias de aminoacidos aguas arriba del dominio catalitico critico de la enzima FUT8 altera la funcion enzimatica.
Se indica que la mutacion de estos aminoacidos criticos proporciona la alteracion completa de la funcionalidad del gen FUT8. El direccionamiento genico usando la tecnologia CRISPR/Cas es una nueva estrategia para crear un soporte de linea celular de inactivation de fucosa. Las celulas transfectadas por CRISPR/Cas se criban a traves de ensayos de funcionalidad del gen FUT8. Los clones seleccionados se confirman a traves de la secuenciacion de los loci de FUT8 genomicos para mutaciones. La linea celular mutante CHOK1 de inactivacion de fucosa se usa entonces para la expresion de proteinas terapeuticas no fucosiladas, incluyendo anticuerpos monoclonales terapeuticos no fucosilados o parte de los anticuerpos.
Se disenan construcciones CRISPR/Cas que se dirigen especificamente a las secuencias de codones de aminoacidos en localizaciones genomicas y se clonan en vectores de expresion, por ejemplo, pD1401 o pD1301, en funcion del tipo de gen Cas9. El complejo CRISPR/Cas se transfecta de manera transitoria en celulas CHOK1; las celulas se colocan en placas de 96 pocillos para la generation de colonias individuales. Cada clon se criba despues para la fucosilacion de proteinas celulares usando el ensayo de aglutinina de Lens culinaris (LCA, del ingles Lens culinaris agglutinin). Los clones positivos para la alteracion del gen FUT8 o GMD se ensayan adicionalmente a traves de ensayos enzimaticos y el analisis cinetico de los alelos mutantes del gen FUT8 o del gen GMD. Por ultimo, se analiza cualquier mutacion llevada a cabo a traves de CRISPR/Cas en la secuencia genomica en los loci de FUT8 y GMD. Estas mutaciones implican deleciones o inserciones, que introducen de este modo mutaciones de desplazamiento de marco de la secuencia de codon de FUT8 y GMD, y produciendo la secuencia alterada y las enzimas no funcionales.
La linea celular CHOK1 de inactivacion de fucosa derivada del proceso mencionado anteriormente se usa como un soporte de linea celular para la expresion de proteinas, anticuerpos monoclonales, peptidos, proteinas de fusion de fines terapeuticos, desarrollo de biomarcadores, usos de diagnostico y de pronostico.
La presente divulgacion se describe adicionalmente con referencia a los siguiente ejemplos, que son solo de naturaleza ilustrativa y de no deberian interpretarse en ningun caso como limitantes del alcance de la presente divulgacion.
Preparation del reactivo
Medio de crecimiento completo DMEM avanzado - 500 ml
1. Se anaden 50 ml de FBS (10 % de concentracion final) a la camara superior de la unidad de filtro de 500 ml.
2. Se anaden 10 ml de glutamina 200 mM (4 mM de concentracion final).
3. Se anaden 5 ml de solucion Pen-strep a 100X (1X de concentracion final).
4. Se ajusta el volumen a 500 ml con medio DMEM avanzado.
5. El medio completo se filtra a traves de un filtro de 0,22 |jm.
6. La camara superior se desmonta y se cierra el reservorio o recipiente del medio.
7. El medio se puede usar en 30 dias de preparacion.
8. El medio se almacena a 2 °C a 8 °C y lejos de la exposicion continua a la luz.
9. En los casos en los que se prepara el medio de seleccion de LCA, se mezclan 10 ml de 10 mg/ml de reactivo LCA del reservorio con 500 ml de medio DMEM preparado para lograr una concentracion final de LCA de 200 jg/ml en medio DMEM.
Materiales y equipo
1. Cabina de bioseguridad
2. Centrifuga Sorvall ST 16R
3. Sistema de bano de agua
4. Microscopio de contraste de fase inversa
5. Incubador de CO2
6. Citometro de flujo de mesa de trabajo Millipore GUAVA 8HT easyCyte
7. Analizador de viabilidad celular Vi-cell XR
8. Hemocitometro
9. Refrigerador
10. Centrifugadora de Eppendorf Minispin
11. Micropipetas
12. Micropuntas
13. Placas de cultivo tisular de 96 pocillos
14. placas de cultivo tisular de 12 pocillos 15. Placas de cultivo tisular de 6 pocillos
16. Pipetas serologicas (de 10 ml, 25 ml y 50 ml)
17. Unidad de filtracion de 1000 ml (tamano de poro de 0,22 jm)
18. Etanol al 70 %
19. DMEM avanzado
20. Solucion salina tamponada con fosfato de Dulbecco (DMEM)
21. Suero fetal bovino (FBS)
22. Penicilina-Estreptomicina (Pen-strep)
23. Glutamina
24. Tripsina-EDTA al 0,05 %
25. Azul de tripano al 0,4 %
26. Tubos de microfuga (de 1,5 ml y 2 ml)
27. Tubos Falcon (de 15 ml y 50 ml)
28. Fraccion V de albumina de suero bovino
29. Aglutinina de Lens culinaris-Fluorescema (LCA-FITC)
30. Estreptavidina-Fluoresceina (Strep-FITC)
T l 1: R iv n l r n iv l i n


T l 2: M i m n n l r n iv l i n

Ejemplo 1: DISENO DE CONSTRUCCIONES CRISPR/CAS
El objetivo del presente ejemplo es disenar un complejo CRISPR/Cas para la inactivacion espedfica de los alelos de FUT8 y GMD.
1.1 - Construcciones de CRISPR
La CRISPR se basa en una clase de endonucleasas guiadas por ARN conocidas como Cas9 del sistema inmunitario adaptativo microbiano hallado en Streptococcus pyogenes. La nucleasa Cas9 se dirige a sitios espedficos en el genoma por ARN gma (ARNg). El complejo Cas9/ARNg se une a una secuencia diana de 20 pb que esta seguida por un motivo de activacion de protoespaciador (PAM) de 3 pb NGG o NAG en el gen especifico que se necesita modificar (Jinek, 2012; Mali, 2013). Por tanto, la union de este complejo completo genera roturas de cadena doble (DSB).
Una etapa crucial en la modification dirigida del genoma en los loci genomicos que necesitan ser modificados, es la introduction de estas DSB. Una vez que se han introducido las DSB, se reparan bien mediante la union de extremos no homologos (NHEJ) o la reparation dirigida por homologia (HDR).
Se sabe que la NHEJ introduce de forma eficaz mutaciones de insercion/delecion (indels) que, a su vez, provocan la alteration de un marco de lectura traduccional de la secuencia codificante diana o en sitios de union de factores de action en trans en promotores o potenciadores. Por otro lado, la reparacion mediada por HDR puede insertar mutaciones puntuales especificas o secuencias en el locus diana. Por tanto, la cotransfeccion de tipos celulares con vectores que expresan la nucleasa Cas9 y los ARNg dirigidos a un locus especifico del gen pueden reducir la expresion de genes diana. La frecuencia esperada de mutaciones en estos sitios especificos varia de >1 % al 50 % (Sander 2014).
La selection de mutantes se realiza mediante cribado simple, usando secuenciacion, sin el uso de la selection de marcadores de resistencia a farmacos. Con el fin de aumentar la especificidad de la alteracion genica, la presente divulgacion usa el mutante de Cas9 (D10A) que esta guiado por dos ARN guia para un solo locus del gen y que introduce dos roturas de cadena simple o incisiones. Esto tambien reduce las oportunidades de uniones no especificas en otros sitios aleatorios. Se usa un vector que codifica la Cas9 D10A y los 2 ARNg para provocar la inactivacion eficaz del gen.
Los loci genomicos de GMD y Fut8 se direccionan para deleciones especificas de secuencia a traves de la tecnologia CRISPR/CAS9 y generan sistemas de expresion de mamiferos defucosilados.
1.2 - El proceso completo de obtencion de la construction CRISPR se compone de las siguientes etapas: 1. Diseno de CRISPR.
2. Diseno del cebador.
3. Sintesis de oligonucleotidos.
4. Transformation de construcciones de CRISPR pD1401 (ARNg 514-553), pD1401 (ARNg 167-207) y pD1301 (ARNg 404) en placa de LB (caldo Luria) ampicilina.
5. Inoculation de celulas transformadas (construcciones de CRISPR) en caldo LB con ampicilina.
6. Aislamiento del plasmido pD1401 (ARNg 514-553), pD1401 (ARNg 167-207) y pD1301 (ARNg 404) de celulas DH10B o DH5alpha.
7. Transfection en celulas CHOK1; cribado y seleccion por ensayo de LCA.
8. Aislamiento de ADN genomico de clones seleccionados usando el kit QIAGEN DNeasy Blood & Tissue.
9. Cuantificacion mediante espectrofotometria.
10. Optimization de la condition de la PCR.
11. Comprobacion cruzada de la muestra de ADN genomico mediante PCR.
12. Electroforesis en gel de agarosa.
13. Amplification por PCR usando polimerasa Phusion y adicion de ADN por cola homopolimerica usando Taq polimerasa.
14. Elucion de gel de producto de PCR usando el kit QIAGEN.
15. Clonacion de TA usando el vector pTZ57R/T.
16. Transformacion de muestra ligada pTZ57R/T+CRISPR(PCR) en celulas DH10B o DH5alpha.
17. Inoculacion de celulas transformadas (pTZ57R/T+CRISPR(PCR)) en caldo LB con ampicilina.
18. Aislamiento de ADN plasm^dico (pTZ57R/T+CRISPR(PCR)) de celulas DH5alpha y Dh 10B usando el kit de aislamiento de ADN plasm^dico de QIAGEN.
19. Comprobacion cruzada de la presencia de inserciones por digestion de restriction (sitios).
20. Secuenciacion de cebadores; y
21. Confirmation de las INDEL mediante secuenciacion.
La Figura 1A de la presente divulgation representa la secuencia codificante de Fut8 y la secuencia de la proteina. La secuencia genomica de FUT8 se analiza a partir de la secuencia de la secuencia de la base de datos, ID de secuencia NW 003613860. La secuencia genomica de FUT8 abarca 570171 - 731804 bases y contiene once exones representados como E1 a E11 en la figura. Tambien se indican las posiciones de los pares de bases para cada exon. E1, E2 y parte de E3 constituyen la region no traducida en la secuencia de aguas arriba, y parte de E11 es tambien parte de la region no traducida. Las regiones traducidas se describen como CDS 1 a CDS 9. La longitud de cada CDS se indica debajo del numero de la CDS. Las CD1 a CDS9 codifican las secuencias de aminoacidos que varian de 38 aminoacidos a 105 aminoacidos.
El ARNm de la fucosiltransferasa 8 (Fut8) (3126 pb) de Cricetulus griseus o de hamster chino deriva de la secuencia de referencia del NCBI:
XM_003501735.1, tambien representada por la SEQ ID NO: 1 de la presente divulgacion.
Los exones alternativos se representan en mayusculas y en minusculas.
La estructura de la proteina Fut8 se estudia a fondo para entender el dominio funcional de la enzima. La estructura cristalina tridimensional de la enzima FUT8 revelo 15 hebras y 16 helices.
La secuencia de aminoacidos del gen FUT8 se proporciona en la Figura 2A.
Las regiones de union de CRISPR/Cas se disenan de tal forma que la especificidad del sitio de reconocimiento es alta y, al mismo tiempo, el complejo CRISPR/Cas lleva a cabo la rotura de cadena simple de ADN pretendida.
En una divulgacion, se usa Cas9n (mutante D10A de la endonucleasa Cas9) para el complejo CRISPR/Cas. La endonucleasa Cas9n provoca la rotura de cadena simple de ADN. Los dos sitios de reconocimiento de CRISPR (sitio de reconocimiento 5' y sitio de reconocimiento 3' estan espaciados por una distancia de 5 pares de bases, lo que permite dos roturas de cadena simple en estrecha proximidad. Las roturas resultantes permiten el proceso de NHEJ de reparation de rotura de ADN que introduce mutaciones en esta region.
La construction CRISPR tiene dos unicas secuencias de reconocimiento de CRISPR de 20 pares de bases flanqueadas por andamiajes de ARNg en tandem con elementos promotores de U6 para la expresion eficaz de las secuencias de ARNg. El diseno unico permite que un solo vector exprese dos andamiajes de ARNg separados y dos secuencias unicas de reconocimiento de CRISPR en el ADN genomico.
La secuencia de nucleotidos y de aminoacidos del gen Cas9 de tipo silvestre se proporciona en las SEQ ID NO: 3 y 4 respectivamente.
La secuencia de nucleotidos y de aminoacidos de la endonucleasa Cas9n de se proporciona en las SEQ ID NO: 5 y 6 respectivamente.
El diseno de CRISPR/Cas se coloca unicamente en la cadena beta 2 diana y en la region de la helice 3H2 incorporando roturas de cadena simple. El diseno es compatible con dos roturas de cadena simple en estrecha proximidad, proporcionando de ese modo una mayor especificidad de reconocimiento de la diana ta que el mecanismo de reparacion de NHEJ solo tiene lugar en estas localizaciones genomicas. Las roturas de cadena simple no especificas, si se crean, normalmente se reparan mediante recombination homologa, que es precisa y raramente crea ninguna mutacion.
El principal objetivo de la presente divulgacion es crear mutaciones en el dominio catalitico de N-terminal, la cadena beta 2 y la helice 3H2. La insertion y las deleciones a traves de CRISPR/Cas en esta position hace que la enzima FUT8 sea no funcional. Ademas, las mutaciones de desplazamiento del marco tambien provocan codones de parada prematura de la traduction, el plegamiento de Rossmann que esta aguas abajo de esta region no se expresa entonces. Los restos de aminoacidos en el plegamiento de Rossmann tales como Arg 365, Arg366, Asp-368, Lys-369, Glu-373, Tyr-382, Asp-409, Asp-410, Asp-453 y Ser-469 son muy importantes para la funcionalidad de FuT8. La enzima truncada sera no funcional y lleva a una linea celular de inactivation de fucosa.
La Figura 2A de la presente divulgacion representa la secuencia de aminoacidos de Fut8 en CHOK1. Se proporciona la secuencia de aminoacidos completa del gen FUT8. La secuencia de aminoacidos de cada CDS se indica con puntas de flecha grandes. Las flechas pequenas indican los aminoacidos criticos presentes en el Exon 7 (CDS5) que
se direccionan en el gen Fut8 por construcciones de CRISPR.
Los datos de secuenciacion aleatoria del genoma celular completo de CHO con numero de registro NW 003613860 para el gen Fut8 se corresponde con un total de 161634 pb. El numero de registro de Pubmed para la region codificante de ARNm del gen Fut8 es XM_003501735.1. La secuencia de ARNm, tal como se muestra en la figura 1A, abarca la secuencia codificante completa para la expresion del producto del gen FUT8, que es a-1,6 fucosiltransferasa.
La herramienta de alineacion Spidey (http://www.ncbi.nlm.nih.gov/spidey/spideyweb.cgi) se usa para identificar los exones en el ADN mediante alineacion de la secuencia de ARNm con la secuencia de ADN genomico. Se identificaron un total de 11 exones con los limites que se muestran en la Tabla 3.
T l : r riz i n ARNm F

Se han confirmado las funcionalidades de la enzima FUT8 a traves de estudios de mutagenesis de sitio dirigido de restos de aminoacidos criticamente importantes en el dominio catalitico.
Se observa una identidad del 100 % entre el ADN genomico y la secuencia de ARNm. La organizacion del gen Fut8 que muestra los 11 exones y la posicion de los ARNg que direccionan el exon 7 se muestra en la figura 1A de la presente divulgacion. Se disena la construccion con la nucleasa Cas9 mutante (Cas9n), que crea roturas de cadena simple (incision) en el sitio diana. Se disenan dos ARNg en estrecha proximidad en el exon 7 para crear dos incisiones para la reparacion eventual de ADN.
En la presente divulgacion, los sitios diana de la tecnologia de CRISPR/Cas9 se localizan en los primeros pocos exones del gen Fut8. Esto se hace para evitar la fucosilacion parcial que puede estar causada por una enzima truncada o parcialmente funcional.
La secuencia de nucleotidos del exon 7 (CDS 5) de Fut8 esta representada por la SEQ ID NO: 7 de la presente divulgacion.

La secuencia de aminoacidos del exon 7 (CDS 5) de Fut8 de la celula CHO esta representada por la SEQ ID NO: 8 de la presente divulgacion. Las posiciones de aminoacidos direccionadas en la protema/secuencia del peptido estan subrayadas.

De forma similar a la estrategia descrita anteriormente para el gen Fut8, la herramienta de alineacion Spidey
(http://www.ncbi.nlm.nih.gov/spidey/spideyweb.cgi) se usa para identificar los exones de GMD en el ADN genomico mediante alineacion de la secuencia de ARNm de GMD con la secuencia de ADN genomico. Se han identificado un total de 10 exones con una region no traducida en 5’ y una cola de poli A y se tabulan en la Tabla 4.
La organizacion del gen GMD que muestra los 10 exones se proporciona en la Tabla 4 de la presente divulgacion. Las otras dianas de CRISPR/Cas en el gen GMD, que se consideran para el direccionamiento tambien se proporcionan en la Tabla 6.
T l 4: r riz i n ARNm MD

La secuencia de nucleotidos del exon 3 de GMD esta representada por la SEQ ID NO: 9 de la presente divulgacion.

La secuencia de nucleotidos del exon 4 de GMD esta representada por la SEQ ID NO: 10 de la presente divulgacion.

Las posiciones de aminoacidos direccionadas en la proteina/secuencia del peptido estan en negrita (figura 2B). La secuencia de aminoacidos del exon 3 de GMD esta representada por la SEQ ID NO: 11 de la presente divulgacion.
M K LH Y GDLTD STCL V K IIN E V K PTEIY N LG A Q SH V K
La secuencia de aminoacidos del exon 4 de GMD esta representada por la SEQ ID NO: 12 de la presente divulgacion.

1.3 - Secuencia de interes en el gen Fut8 para el direccionamiento usando CRISPR
En el exon 7 del gen Fut8, las secuencias proporcionadas a continuacion se usan para la union a ADN diana.
La secuencia de reconocimiento 1 de CRISPR se representa por la SEQ ID NO 13.

El ARNg1 -esta representado por la SEQ ID NO: 14,

La secuencia de reconocimiento 2 de CRISPR se representa por la SEQ ID NO 15.

El ARNg2 -esta representado por la SEQ ID NO: 16.

Se han disenado multiples sitios posibles para CRISPR/Cas, todos a lo largo de las secuencias genomicas de FUT8 y GMD. La siguiente tabla indica sitios importantes en las secuencias genomicas de FUT8.


Todos estos sitios son unicos y se usan para crear una posible estrategia de inactivacion del gen en CHO y otras lineas celulares. Todas las secuencias proporcionadas en la tabla anterior se representan en la direction 5’ a 3’. La correspondiente secuencia diana espedfica de ARNcr de 20 pares de bases provendra de la secuencia de reconocimiento de CRISPR proporcionada en cada diseno mencionado en la tabla 5 anterior.
La Tabla 5 de la presente divulgacion enumera diferentes secuencias diana de Fut8 que se consideran para el direccionamiento de inactivacion por CRISPR. Inicialmente se consideraron un total de veinte secuencias diferentes. Se asegura que ninguno de los ARNg se extienda a un limite exon-intron ya que esto puede producir la inactivacion de los ARNg. Basandose en esta tecnica, se elige un tramo de 57 pb en el exon 7 como la diana para el objetivo de inactivacion mediada por CRISPR/Cas. Este incluye dos ARNg, uno en cada cadena que provoca dos roturas de cadena simple.
La secuencia diana en el gen Fut8 que se usa en una divulgacion del presente metodo se muestra a continuacion en la figura 3B de la presente divulgacion como el ARNg 1120-1176. Los sitios de escision se indican con una flecha. La distancia entre los dos ARNg es de 5 bases. La secuencia de reconocimiento de ARNg 1120-1176 esta subrayada en la Figura 3B. El correspondiente fragmento sintetizado se incorpora en el vector pD1401 y se nombra como ARNg 514-553 de pD1401, cuyas caracteristicas se describen posteriormente en la divulgacion.
Este metodo de la presente divulgacion usa Cas9n (mutante de nickasa) en el direccionamiento de la secuencia genomica de Fut8, exon 7 con el sistema CRISPR/Cas. La endonucleasa Cas9n genera roturas de cadena simple (SSB, del ingles single strand break) en la cadena opuesta de ADN. Las secuencias de reconocimiento de CRISPR/Cas en las cadenas superior e inferior estan subrayadas. Los correspondientes sitios de rotura de cadena simple se indican con las puntas de las flechas negras. Las tres secuencias de nucleotidos PAM se indican en negrita.
En la presente divulgacion, se usa uno de los disenos para el direccionamiento en el ARNg 1120-1176 del exon 7. La construccion del vector CRISPR/Cas para este diseno se nombra como ARNg (514-553) de pD1401.
La secuencia de reconocimiento de CRISPR de 5’ y 3’ se indica en pequeno y en cursiva, se reconocen dos sitios separados complementarios a esta secuencia de reconocimiento en la secuencia genomica de FUT8. La secuencia representada en negrita indica la secuencia de andamiaje de ARNg para la union del complejo CRISPR/Cas.
SEQ ID NO: 100 - ARNg andamiaje para el exon 7 de Fut8

Secuencia de reconocimiento de CRISPR en 5’ en el ADN sintetizado a partir de ADN2.0 SEQ ID NO: 37

Secuencia de ARNcr especifica de diana (direccion 5’ a 3’): SEQ ID NO: 38

Secuencia de reconocimiento de CRISPR en 3’ en el ADN sintetizado a partir de ADN2.0- SEQ ID NO: 39

Secuencia de ARNcr especifica de diana (direccion 5’ a 3’): SEQ ID NO: 40

ARNg de andamiaje de ADN2.0-

El mapa de construccion se proporciona en la figura 3A y se marcan las regiones importantes de la secuencia.
1.4 - DISENO DE LA CONSTRUCCION DE CRISPR DE GMD
La Figura 9B de la presente divulgacion proporciona el locus genomico de GMD y las secuencias de reconocimiento de CRISPR.
Se obtuvieron de Pubmed los datos de secuenciacion aleatoria del genoma celular completo de CHO con numero de registro NW 003613635.1 para el locus del gen GMD que consiste en 442215 pb. El numero de registro de Pubmed para la region codificante de ARNm del gen GMD es NM 001246696.1.
GMD es un miembro de la subfamilia modificadora de NDP-azucar de las deshidrogenasas/reductasas de cadena corta (SDR). Como miembro de esta subfamilia, GMD se une a su cofactor NADP (H) en la parte de N-terminal de la molecula en la que esta presente una region comun rica en glicina. La triada catalitica se ha identificado como Tyr-XXX-Lys y Ser/Thr, que son todas importantes para la catalisis. El analisis estructural de GMD de E. coli sugiere que la molecula activa esta en configuracion dimerica. Mientras que el homologo de Arabidopsis thaliana es tetramerico, y el sitio de union de NADP(H) esta intimamente implicado en la creacion de la superficie de contacto del tetramero. Lo mas probable es que la forma funcional de la enzima GMD en eucariotas consista en la configuracion tetramerica. Las regiones de union de CRISPR/Cas se disenan de tal forma que la especificidad del sitio de reconocimiento es alta y, al mismo tiempo, el complejo CRISPR/Cas lleva a cabo la rotura de cadena simple de ADN pretendida.
En una divulgacion, se usa Cas9n (mutante D10A de la endonucleasa Cas9) para el complejo CRISPR/Cas. La endonucleasa Cas9n provoca la rotura de cadena simple de ADN. Los dos sitios de reconocimiento de CRISPR (sitio de reconocimiento 5' y sitio de reconocimiento 3' estan espaciados por una distancia de 5 pares de bases, lo que permite dos roturas de cadena simple en estrecha proximidad. Las roturas resultantes permiten el proceso de NHEJ de reparacion de rotura de ADN que introduce mutaciones en esta region.
La construccion CRISPR tiene dos unicas secuencias de reconocimiento de CRISPR de 20 pares de bases flanqueadas por andamiajes de ARNg en tandem con elementos promotores de U6 para la expresion eficaz de las secuencias de ARNg. El diseno unico permite que un solo vector exprese dos andamiajes de ARNg separados y dos secuencias unicas de reconocimiento de CRISPR en el ADN genomico.
El diseno de CRISPR/Cas se coloca unicamente para dirigir el motivo YGDLTDSTCLVK y el motivo DLAEYT responsable de la zona de contacto del tetramero de la estructura proteica multimerica funcional de GMD. Las dos roturas de cadena simple inducidas por la endonucleasa Cas9n en esta region permite la reparacion de ADN mediada por NHEJ. Las mutaciones incorporadas durante la reparacion de ADN dan como resultado una mutacion de desplazamiento del marco, delecion, insertion asi como codones de parada prematura. Tal mutacion no solo altera el motivo critico para la tetramerizacion, sino que tambien crea mutaciones aguas abajo del resto Ser85, que esta implicado en el mantenimiento de interacciones de los monomeros en la configuracion tetramerica. El diseno es compatible con dos roturas de cadena simple en estrecha proximidad, proporcionando de ese modo una mayor especificidad de reconocimiento de la diana ta que el mecanismo de reparacion de NHEJ solo tiene lugar en estas localizaciones genomicas. Las roturas de cadena simple no especificas, si se crean, normalmente se reparan mediante recombination homologa, que es precisa y raramente crea ninguna mutacion.
La Figura 1B de la presente divulgacion representa la organization genomica de GMD en CHOK1.
Se proporciona la secuencia de aminoacidos completa del gen GMD en la figura 2B. La secuencia de aminoacidos de cada CDS se indica con puntas de flecha grandes. Las dos regiones diana de CRISPR estan subrayadas en el exon 3 y en el exon 4, que se direccionan en el gen GMD. Cada exon esta representado con marcas de flechas. Las lineas finas indican los sitios de reconocimiento de CRISPR para la localization de roturas de cadena simple y la linea gruesa indica el sitio de reconocimiento de CRISPR para la localizacion de la rotura de cadena doble.
Se usan os estrategias diferentes para la inactivation genica, una es el uso del mutante Cas9 (Cas9n) que genera roturas de cadena simple (SSB) y la segunda es el uso de Cas9 de tipo silvestre que genera roturas de cadena
doble (DSB).
En el locus del exon 3 de GMD,
El metodo de la presente divulgacion usa Cas9n (mutante de nickasa) en el direccionamiento de la secuencia genomica de GMD, exon 3 con el sistema CRISPR/Cas. La endonucleasa Cas9n genera roturas de cadena simple (SSB, del ingles single strand break) en la cadena opuesta de ADN. La construccion se nombra como pD1401 (ARNg 167-207) y se representa por la Figura 4A de la presente divulgacion.
La secuencia de reconocimiento de CRISPR de 5’ y 3’ se indica en pequeno y en cursiva, se reconocen dos sitios separados complementarios a esta secuencia en la secuencia genomica de GMD. La secuencia representada en negrita indica la secuencia de andamiaje de ARNg para la union del complejo CRISPR/Cas.
SEQ ID NO: 101 -

Secuencia de reconocimiento de CRISPR en 5’ en el ADN sintetizado a partir de ADN2.0 = SEQ ID NO: 41

Secuencia de ARNcr especifica de diana (direccion 5’ a 3’): SEQ ID NO: 42

Secuencia de reconocimiento de CRISPR en 3’ en el ADN sintetizado a partir de ADN2.0 - SEQ ID NO: 43-
Secuencia de ARNcr especifica de diana (direccion 5’ a 3’): SEQ ID NO: 44

En el locus del exon 4 de GMD,
el exon 4 del gen GMD se direcciona con la endonucleasa Cas9 de tipo silvestre. Esta Cas9 de tipo silvestre genera roturas de cadena doble (DSB) en el sitio diana. La construccion se nombra como pD1301 (ARNg 404) y se representa por la Figura 4B de la presente divulgacion.
La secuencia de reconocimiento de CRISPR se indica en pequeno y en cursiva. La secuencia de ADN genomico de cadena doble se reconoce basandose en esta secuencia por el sistema CRISPR/Cas. La secuencia representada en negrita indica la secuencia de andamiaje de ARNg para la union del complejo CRISPR/Cas.
SEQ ID NO: 102-

Secuencia de reconocimiento de CRISPR en el ADN sintetizado a partir de ADN2.0 - SEQ ID NO: 45-
Secuencia de ARNcr especifica de diana (direccion 5’ a 3’): SEQ ID NO: 46

Secuencias de reconocimiento de CRISPR para el gen GMD
La siguiente tabla representa secuencias de reconocimiento de CRISPR/Cas a lo largo de secuencias codificantes de GMD para posibles sitios de rotura de cadena simple. Cualquiera de estas secuencias de reconocimiento se usa para la rotura de cadena simple mediada por la endonucleasa Cas9n y la estrategia de reparacion del sistema CRISPR/Cas para inactivar el gen GMD.
L ^


En este caso, se disenan un total de diecinueve secuencias diana en la secuencia del gen GMD para los sitios de reconocimiento de CRISPR. Todas las secuencias se representan en la direccion 5’ a 3’; la correspondiente secuencia diana especifica de ARNcr de 20 pares de bases provendra de la secuencia de reconocimiento de CRISPR proporcionada en cada diseno mencionado en la tabla 6 anterior.
Uno de los disenos mencionados anteriormente, El ARNg 394-434, se uso para crear el complejo CRISPR/Cas pD1401 (ARNg 167-207) para la transfeccion de celulas CHOK1. El complejo CRISPR/Cas crea dos roturas de cadena simple en el ADN en la cadena complementaria de la secuencia codificante reconocida del gen GMD. La reparacion exitosa de ADN en el sitio diana crea un gen GMD no funcional y, por lo tanto, se desarrollan lineas celulares CHOK1 con inactivacion de fucosa. Las caracteristicas de pD1401 (ARNg 167-207) se describen posteriormente en la divulgacion.
Aunque la presente divulgacion usa uno de los disenos, uno cualquiera de los sitios de reconocimiento de CRISPR mencionados anteriormente crea un gen GMD no funcional. Por lo tanto, cualquiera de estos posibles sitios solo o en combinacion se usa para el desarrollo de la linea celular CHOK1 con inactivacion de fucosa.
La secuencia diana en el exon 3 de GMD que se usa en una divulgacion del presente metodo se menciona en la tabla 6 como ARNg 394-434. La Figura 4C describe la secuencia de reconocimiento de CRISPR. Los sitios de escision se indican con una flecha. La distancia entre los dos ARNg es de 6 bases. La secuencia de ARNg 394-434 esta subrayada en la Figura 4C. El correspondiente fragmento sintetizado se incorpora en el vector pD1401 y se nombra como ARNg 167-207 de pD1401, cuyas caracteristicas se describen posteriormente en la divulgacion. Este metodo de la presente divulgacion usa Cas9n (mutante de nickasa) en el direccionamiento de la secuencia genomica de GMD, exon 3 con el sistema CRISPR/Cas. La endonucleasa Cas9n genera roturas de cadena simple (SSB, del ingles single strand break) en la cadena opuesta de ADN.
La siguiente tabla representa secuencias de reconocimiento de CRISPR/Cas a lo largo de secuencias codificantes de GMD para posibles sitios de rotura de cadena doble. Cualquiera de estas secuencias de reconocimiento se usa para la rotura de cadena simple mediada por la endonucleasa Cas9 de tipo silvestre y la estrategia de reparacion del sistema CRISPR/Cas para inactivar el gen GMD.
Tabla 7 - secuencias de reconocimiento de CRISPR/Cas a lo largo de secuencias codificantes de GMD para osibles sitios de rotura de cadena doble


Se disenaron veintitres secuencias unicas (ARNg) de reconocimiento de CRISPR a lo largo de la secuencia del gen GMD. Todas las secuencias se representan en la direccion 5’ a 3’; la correspondiente secuencia diana espedfica de ARNcr de 20 pares de bases provendra de la secuencia de reconocimiento de CRISPR proporcionada en cada diseno mencionado en la tabla 7 anterior.
Uno de los disenos mencionados anteriormente, El ARNg 306, se uso para crear el complejo CRISPR/Cas pD1301 (ARNg 404) para la transfeccion de celulas CHOK1. El complejo CRISPR/Cas crea una rotura de cadena doble en el ADN en la secuencia codificante reconocida del gen GMD. La reparacion exitosa de ADN en el sitio diana crea un gen GMD no funcional y, por lo tanto, se desarrollan lineas celulares CHOK1 con inactivacion de fucosa.
Aunque uno de los disenos se usa en la presente divulgacion, uno cualquiera de los sitios de reconocimiento de CRISPR mencionados anteriormente crea un gen GMD no funcional. Por lo tanto, cualquiera de estos posibles sitios solo o en combinacion se usan para el desarrollo de la linea celular CHOK1 con inactivacion de fucosa.
La secuencia diana en el exon 4 de GMD que se usa en una divulgacion del presente metodo se muestra en la tabla 7 anterior como ARNg 306. La Figura 4D describe la secuencia de reconocimiento de CRISPR. Los sitios de escision se indican con una flecha. El correspondiente fragmento sintetizado se incorpora en el vector pD1301 y se nombra como ARNg 404 de pD1301, cuyas caracteristicas se describen posteriormente en la divulgacion. Este metodo de la presente divulgacion usa endonucleasa Cas9 de tipo silvestre en el direccionamiento de la secuencia genomica de GMD, exon 4 con el sistema CRISPR/Cas. La endonucleasa Cas9 de tipo silvestre genera roturas de cadena doble (DSB) en ambas cadenas de ADN.
1.5- Sintesis del complejo CRISPR/Cas
La tecnologia CRISPR se basa en una clase de endonucleasas guiadas por ARN conocidas como Cas9 del sistema inmunitario adaptativo microbiano hallado en Streptococcus pyogenes. La nucleasa Cas9 se dirige a sitios especificos en el genoma por ARN guia (ARNg). Se deben introducir y/o expresar dos componentes en celulas o en un organismo para realizar la modificacion del genoma basado en CRISPR: la nucleasa Cas9; y un "ARN guia" (ARNg).
Veinte secuencias de reconocimiento de nucleotidos en el extremo 5’ del ARNg dirigen la Cas9 a un sitio de diana de ADN especifico usando las reglas de alineacion de bases complementarias de ARN-ADN. Estos sitios diana deben caer inmediatamente en 5’ de una secuencia PAM que coincide con la forma convencional 5-NGG.
La presente divulgacion usa dos tipos diferentes de endonucleasa Cas9 en la presente divulgacion tal como se describe a continuation. En ambos casos se usa un solo vector de transfection que codifica el ARNg y la nucleasa, aumentando de este modo la eficacia de la transfeccion de las celulas CHOK1.
a) La nucleasa Cas9 de tipo silvestre se usa para el direccionamiento del gen GMD en el exon 4. La construction permite la rotura de cadena doble (DSB) en el sitio dirigido.
b) Una nucleasa Cas9 mutante (D10A), conocida como Cas9n se usa para dirigir el locus del exon 3 de GMD y el locus del exon 7 de Fut8, las construcciones crean roturas de cadena simple en lugar de una rotura de cadena doble en el ADN. Este diseno se centra en mejorar la especificidad de construcciones CRISPR/Cas.
En el caso de roturas de cadena simple, se direccionan dos sitios diana en el ADN en estrecha proximidad en donde tiene lugar la rotura de cadena simple o incision en cadenas opuestas de ADN. De este modo, se recluta la maquinaria de reparation del ADN (NHEJ) para reparar el dano del ADN. El reclutamiento de dos complejos ARNg/Cas9n en un intervalo especifico para iniciar la reparacion de ADN mejora la especificidad hacia el sitio dirigido. La union no especifica de solo uno de los complejos ARNg/Cas9n a sitios no relacionados provoca incisiones que normalmente se reparan a traves de reparacion basada en la recombination homologa con una tasa de mutation muy baja. Por lo tanto, esta estrategia aumenta la especificidad del direccionamiento del gen Fut8 y GMD.
Las regiones unicas de ambos genes se direccionan basandose en la information estructural de la enzima de tal forma que anulan la funcion catalitica de la enzima o mediante la alteration de una estructura de mayor orden.
Las caracteristicas importantes de los vectores son,
a) Cas9 - una nucleasa que primero se descubre como un componente del sistema CRISPR en Streptococcus pyogenes y se ha adaptado para su utilidad en celulas de mamifero. La Cas9 guiada por ARN es capaz de introducir de manera eficaz roturas precisas de cadena doble en loci genomicos endogenos en celulas de mamifero con alta eficacia.
Cas9-D10A - Un mutante D10A de nucleasa Cas9 (Cas9n) hace incisiones en cadena simple y se combina con un par de ARN guia de compensation complementarios a las cadenas opuestas de los loci genomicos diana. Esto ayuda a reducir la actividad fuera de diana observada con la Cas9 de tipo silvestre.
b) Andamiaje de ARNg quimerico - El andamiaje de ARN guia quimerico (ARNg) consiste en una region complementaria especifica de diana de 20 nucleotidos, una estructura de ARN de union a Cas9 de 42 nucleotidos, y un finalizador de la transcription de 40 nucleotidos que proviene de S. pyogenes que dirige la nucleasa Cas9 al sitio diana para la modification del genoma. En este caso hay dos andamiajes de ARNg, uno para cada ARNg.
c) Kanamicina-r - Un agente bactericida eficaz que inhibe la translocation ribosomica provocando de este modo errores de codification. El gen que codifica para la resistencia a la Kanamicina es Neomicina fosfotransferasa II (NPT II/Neo). La E. coli transformada con el plasmido que contiene el gen de resistencia a la kanamicina se puede cultivar en medios que contienen 25 |jg/ml de kanamicina.
d) P CMV - El promotor de CMV es un promotor constitutivo de mamiferos y media una fuerte expresion en diversos sistemas celulares.
e) P_hU6.1 - Promotor central humano A de tipo 3 para la expresion de ARN.
1.6 - El proceso completo de obtencion de la construccion CRISPR se compone de las siguientes etapas:
22. Diseno de la diana de CRISPR.
23. Construcciones de vector con dos estructuras principales del vector separadas, a saber, los vectores pD1401 y pD1301 con insertion de ARNg.
24. Transformation de las construcciones de CRISPR pD1401 (ARNg 514-553), pD1401 (ARNg 167-207) y pD1301 (ARNg 404) en celulas competentes de E. coli (DH10B o DH5alpha) y colocacion en LB (Luria Bertani)-Agar suplementado con kanamicina.
25. Inoculation de celulas transformadas (construcciones de CRISPR) en caldo LB con kanamicina.
26. Aislamiento del plasmido de ADN pD1401 (ARNg 514-553), pD1401 (ARNg 167-207) y pD1301 (ARNg 404) de celulas DH10B o DH5alpha.
27. Transfeccion en celulas CHOK1; cribado y selection por ensayo de LCA.
28. Aislamiento de ADN genomico de clones seleccionados usando el kit QIAGEN DNeasy Blood & Tissue. 29. Cuantificacion mediante espectrofotometria.
30. Optimizacion de la condicion de la PCR.
31. Comprobacion cruzada de la muestra de ADN genomico mediante PCR.
32. Electroforesis en gel de agarosa.
33. Amplification por PCR usando polimerasa Phusion y adicion de ADN por cola homopolimerica usando Taq polimerasa.
34. Elucion de gel de producto de PCR usando el kit QIAGEN.
35. donation de TA usando el vector pTZ57R/T.
36. Transformacion de muestra ligada pTZ57R/T+CRISPR(PCR) en celulas DH10B o DH5alpha.
37. Inoculacion de celulas transformadas (pTZ57R/T+CRISPR(PCR)) en caldo LB con ampicilina.
38. Aislamiento de ADN plasmidico (pTZ57R/T+CRISPR(PCR)) de celulas DH5alpha y Dh 10B usando el kit de aislamiento de ADN plasmidico de QIAGEN.
39. Comprobacion cruzada de la presencia de inserciones por digestion de restriction.
40. Secuenciacion de cebadores; y
41. Confirmation de las INDEL mediante secuenciacion.
Ejemplo 2: TRANSFECCION DE CELULAS CON CONSTRUCCIONES TALEN
El presente ejemplo contiene un procedimiento para la transfection de celulas CHOK1 con construcciones CRISPR. Tambien se proporciona la selection y confirmacion de lineas celulares estables unicas para el desarrollo de lineas celulares CHOK1 de inactivation de FUT8 usando la tecnologia CRISPR y la seleccion de clones positivo por ensayo funcional basado en la citometria de flujo.
Protocolo de transfeccion
La transfeccion se isoptimizo usando celulas CHOK1 de tipo adherente y de suspension. Se usaron reactivos de transfeccion mediada por liposoma y liposoma modificado, por ejemplo, Lipofectamina 2000, Lipofectamina 3000, Lipofectamina LTX con reactivo PlusTM, MIRUS TransIT X2, MIRUS TransIT 2020, MIRUS TransIT 293, kit de transfeccion en CHO MIRUS TransIT. Se ensayo una concentration de ADN que varia de 0,5 |jg a 5 |jg para diversos periodos de incubation, por ejemplo, 4 horas, 24 horas y 48 horas. Tambien se ensayaron multiples proporciones de ADN frente a reactivo de transfeccion (jg : jl). Se logra una eficacia de transfeccion optima usando una proportion de ADN frente a reactivo de transfeccion de 1:3, incubacion de 24 horas y Lipofectamina LTX con reactivo PlusTM. Se realizaron experimentos de optimization realizados con ADN plasmidico que expresa GFP. La Figura 20 representa la eficacia de transfeccion de la linea celular CHOK1 usando el protocolo descrito en la divulgation. La eficacia de transfeccion se determino usando una construction de plasmido que expresa proteina verde fluorescente. El numero de celulas verdes observadas tras la transfeccion en comparacion con el numero total de celulas viables determina la eficacia de transfeccion del protocolo establecido. El panel A representa la imagen en campo claro y el panel B representa el mismo campo del microscopio para el canal de fluorescencia rojo.
La eficacia de la transfeccion se calcula mediante la siguiente formula:
Eficacia de la transfeccion = (Numero de celulas que expresan GFP / Numero total de celulas)*100 La eficacia de transfeccion transitoria optimizada es del 40-50 % en celulas CHOK1.
Transfeccion:
Las celulas CHOK1 se inocularon a mas del 90 % de viabilidad y a una densidad de 0,25 x 106 celulas/pocillo en una placa de cultivo tisular de 6 pocillos y se permitio que se adheriesen durante 24 horas. Se usaron las construcciones de CRISPR pD1401 (ARNg 514-553), pD1401 (ARNg 167-207), pD1301 (ARNg 404), la combination de pD1401 (ARNg 167-207) pD1301 (ARNg 404) para la transfeccion usando Lipofectamina Lt X con reactivo PlusTM. Se usaron 2,5 jg de la construccion con una proporcion de ADN frente a reactivo de transfeccion de 1:3. Se incubaron las celulas durante 20-24 horas tras la transfeccion. Antes de la transfeccion, se estimo la cantidad y la calidad de ADN por espectrofotometria de UV. Un ADN de valor A260/280 representa calidad y contamination de proteina. La proporcion de absorbancia a 260nm y 280 nm se usa para evaluar la pureza del ADN. Generalmente, se acepta un A260/280 >1,8 como ADN "puro" o de buena calidad. Se colocaron 3-4 j l de muestra de ADN en la microcubeta y se estimo la concentracion de ADN usando el Biofotometro de Eppendorf D30 frente a un blanco adecuado.
Tabla 8 - Dilucion de ADN de CRISPR:

Dilution de Lipofectamina LTX:

A las celulas se les proporciono un cambio del medio con medio sin suero, 1 hora antes de la transfeccion. Las
construcciones de CRISPR y la solucion de Lipofectamina LTX se diluyeron, se mezclaron suavemente y se incubaron durante 5-10 minutos a 20-25 °C. Las diluciones de ADN y de reactivo de transfeccion (3 ml) se mezclan y se incuban durante 20-30 minutos a temperatura ambiente durante la formation del complejo. Se aspiro el medio de los pocillos. Se anaden gota a gota 1,5 ml de complejo de ADN y reactivo de transfeccion a las celulas colocadas en placa.
Las celulas se incubaron durante 4 horas a 37 °C en una incubadora con CO2 al 5 %. El medio completo se anadio a 1,5 ml/pocillo y se incubo durante 20-24 horas a 37 °C en una incubadora de CO2 al 5 %. Despues de 20-24 horas de transfeccion, se tripsinizan las celulas y se prepara una dilution unica de celulas.
La dilucion unica de celulas se obtiene mediante dilucion de las celulas a una concentration de 0,5 celulas/100 |jl. Se hizo un recuento de celulas usando el hemocitometro. Se permitio que las celulas creciesen durante unos pocos dias a 37 °C en una incubadora con CO2 al 5 %. Se hizo el barrido de la placa para identificar colonias de celulas unicas en microscopio de contraste de fase inversa. Las celulas que crecieron en colonias individuales distintivamente pequenas se marcaron para una amplification posterior. Despues de 2-3 semanas, los clones de celulas individuales se amplificaron de un pocillo de la placa de 96 pocillos a un pocillo de la placa de 6 pocillos por tripsinizacion. Se permitio que crecieran las celulas durante 2-3 dias a 37 °C al 5 % de CO2 en un incubador de CO2. Las celulas se amplifican posteriormente de un pocillo a dos pocillos en una placa de 24 pocillos (colocation de la replica) para el cribado adicional.
Ensayo de union a LCA-FITC (Aglutinina de Lens culinaris - isotiocianato de fluorescema)
El isotiocianato de fluoresceina (FITC) es un fluorocromo conjugado con LCA. Por lo tanto, la presencia de proteinas fucosiladas sobre la membrana celular de las celulas CHOK1 de control se reconoce mediante LCA conjugada con fluoresceina. Estas celulas fluorescen con mas brillo en el canal del citometro de flujo especifico. La fluorescencia observada se representa como unidad de fluorescencia. En las celulas en las que la via de la fucosa esta alterada, las lineas de inactivation no son capaces de producir proteinas celulares fucosiladas y, por lo tanto, las proteinas de la membrana celular no estan fucosiladas. El ensayo de estas celulas con el conjugado fluoresceina-LCA da como resultado una fluorescencia comparable con el fondo. Por lo tanto, las celulas de inactivacion de fucosa fluorescen a un nivel mucho mas bajo (menos de 100 UFR) en comparacion con la linea celular CHOK1 de control.
Las celulas se tripsinizaron, se transfirieron a un tubo de microfuga y se agitaron vorticialmente a 1500 rpm (revoluciones por minuto) durante 5 minutos usando la centrifugadora de Eppendorf Minispin. Se retiro el medio y se anadio medio nuevo a los tubos. Tanto las celulas CHOK1 transfectadas como las no transfectadas se procesaron de manera simultanea. Se ensayaron las celulas en analisis basado en citometria de flujo de LCA-FITC usando el citometro de flujo de mesa de trabajo "Millipore GUAVA 8HT easyCyte".
Se seleccionaron 54 clones de la transfeccion con pD1401 (ARNg 514-553) con perfil de inactivacion de fucosa. De manera similar, se seleccionaron 200 clones de la transfeccion con pD1401 (ARNg 167-207) o pD1301 (ARNg 404) o una combination de pD1401 (ARNg 167-207) pD1301 (ARNg 404) con perfil de inactivacion de fucosa.
Se diluyeron 5 mg/ml de la solucion madre de aglutinina de Lens culinaris-Fluorescema (LCA-FITC) para obtener una concentracion final de 2 jg/ml en tampon de ensayo (DPBS que contiene BSA al 2 %). Se agitaron vorticialmente las celulas a 1500 rpm durante 5 minutos usando la centrifuga minispin de Eppendorf. Se aspiro el medio y se resuspendio el sedimento en 0,25-1 ml de tampon de ensayo que contiene 2 jg/ml de LCA-FITC. Se resuspendieron las celulas CHOK1 de control en 0,25-1 ml de tampon de ensayo solo (control sin tincion) y 0,25-1 ml de tampon de ensayo que contiene 2 jg/ml de LCA-FITC (control con tincion). Todas las muestras se diluyeron para obtener 0,1-0,2 x106 celulas/ml en tampon de ensayo final. Despues se incubaron las muestras en la oscuridad y sobre hielo durante 30 minutos. Despues, 200 j l de cada muestra se dividieron en alicuotas en una placa de 96 pocillos. La placa se cargo despues en el citometro de flujo de mesa de trabajo Millipore GUAVA easyCyte 8HT para la adquisicion y el analisis de datos. El analisis de datos se hizo usando el programa informatico Incyte. En algunos experimentos se uso el citometro de flujo Accuri C6 para la adquisicion y el analisis de datos.
Tambien se realizo una tincion negativa de estreptavidina-fluoresceina (Strep-FITC). Se diluyeron 5 mg/ml de la solucion madre de aglutinina de Lens culinaris-Fluorescema (LCA-FITC) para obtener una concentracion final de 2 jg/ml en tampon de ensayo (DPBS que contiene BSA al 2 %). Se diluyeron 1 mg/ml de la solucion madre de estreptavidina-fluoresceina (Strep-FITC) para obtener una concentracion final de 2 jg/ml en tampon de ensayo (DPBS que contiene BSA al 2 %). Se tomaron suspensiones celulares por duplicado y se agitaron vorticialmente las celulas a 1500 rpm durante 5 minutos usando la centrifuga minispin de Eppendorf. Se aspiro el medio y, en un tubo, se resuspendio el sedimento en tampon de ensayo que contiene 2 jg/ml de LCA-FITC y la muestra duplicada en tampon de ensayo que contiene 2 jg/ml de Strep-FITC. Las celulas CHOK1 de control se resuspendieron en tampon de ensayo solo (control sin tincion), en tampon de ensayo que contiene 2 jg/ml de LCA-FITC (control con tincion) y tampon de ensayo que contiene 2 jg/ml de Strep-FITC.
Todas las muestras se diluyeron para obtener 0,1-0,2 x 106 celulas/ml en 0,25-1 ml de tampon de ensayo. Despues se incubaron las muestras en la oscuridad y sobre hielo durante 30 minutos. Despues, 200 j l de cada muestra se
dividieron en alicuotas en una placa de 96 pocillos. La placa se cargo en el citometro de flujo de mesa de trabajo Millipore GUAVA easyCyte 8HT para la adquisicion y el analisis de datos. El analisis de datos se hizo usando el programa informatico Incyte. En algunos experimentos se uso el citometro de flujo Accuri C6 para la adquisicion y el analisis de datos.
Las celulas CHOK1 se transfectaron con la construccion pD1401 (ARNg 514-553) que direcciona al locus Fut8 y los resultados se proporcionan a continuacion.
Tabla 9

La UFR se refiere al valor medio de unidades de florescencia relativa.
Resultados -Los resultados graficos y el perfil de fluorescencia se proporcionan en la tabla anterior y tambien se representa en la figura 7A y en la figura 8A de la presente divulgacion. Las figuras representan el resultado grafico y el perfil de fluorescencia observado para las lineas celulares de CHOK1 CR1KOT1 n.° 6, CR1KOT1 n.° 18, CRi Ko t I n.° 22, CR1KOT1 n.° 23, CR1KOT1 n.° 26, CR1KOT1 n.° 31, CR1KOT1 n.° 34, CR1KOT1 n.° 37, en el ensayo de union a LCA-FITC basado en citometria de flujo. Este ensayo de citometria de flujo detecta las proteinas fucosiladas sobre la superficie celular. Por lo tanto, las celulas CHOK1 de control fluorescen altamente, ya que muchas proteinas fucosiladas estan presentes en la linea celular CHOK1 de control. En casos en los que el complejo CRISPR/Cas es capaz de alterar el gen FUT8 en lineas celulares transfectadas, la fluorescencia de LCA-FITC se minimiza, ya que no hay proteina fucosilada en la superficie de estas lineas celulares. La figura revela la perdida significativa de fluorescencia cuando las CR1KOT1 n.° 6, CR1KOT1 n.° 18, CR1KOT1 n.° 22, CR1KOT1 n.° 23, CR1KOT1 n.° 26, CR1KOT1 n.° 31, CR1KOT1 n.° 34 y CR1KOT1 n.° 37, se prueban en este ensayo, lo que indica que estas lineas celulares son lineas de CHOK1 con inactivacion de FUT8.
Las celulas CHOK1 se transfectaron con las construcciones pD1401 (ARNg 167-207), pD1301 (ARNg 404), pD1401
(ARNg 167-207)+ pD1301 (ARNg 404) que direccionan al locus GMD y los resultados se proporcionan en la tabla a continuacion.
Tabla 10

Los resultados graficos y el perfil de fluorescencia se proporcionan en la tabla anterior y tambien se representa en las figuras 7B y 8B de la presente divulgacion. Las figuras representan el resultado grafico y el perfil de fluorescencia observado para las CHOK1, C1GMD1.12, C1GMD1.27, C1GMD2.30, C1GMD2.34, C1GMD3.4, C1GMD3.36, C1GMD3.43, C1GMD3.49, C1GMD3.51 en el ensayo de union a LCA-FITC basado en citometria de flujo. Este ensayo de citometria de flujo detecta las proteinas fucosiladas sobre la superficie celular. Por lo tanto, las celulas CHOK1 de control fluorescen altamente, ya que muchas proteinas fucosiladas estan presentes en la linea celular CHOK1 de control. En casos en los que el complejo CRISPR/Cas es capaz de alterar el gen GMD en lineas celulares transfectadas, la fluorescencia de LCA-FITC se minimiza, ya que no hay proteina fucosilada en la superficie de estas lineas celulares. La figura revela la perdida significativa de fluorescencia cuando las C1GMD1.12, C1GMD1.27, C1GMD2.30, C1GMD3.4, C1GMD3.36, C1GMD3.43, C1GMD3.49, C1GMD3.51 se probaron en este ensayo, lo que indica que estas lineas celulares son posibles lineas celulares CHOK1 de inactivacion de GMD. Tambien se probo otro conjunto de transfeccion con el ensayo de fluorescencia de LCA-FITC. Los datos de la citometria de flujo se proporcionan en la tabla a continuacion.
Tabla 11

Los resultados graficos y el perfil de fluorescencia se proporcionan en la tabla anterior y tambien se representa en las figuras 7C y 8C de la presente divulgacion. Las figuras representan el resultado grafico y el perfil de fluorescencia observado para las CHOK1, C1GMD1.37, C1GMD1.4, C1GMD1.41, C1GMD1.43, y C1GMD1.44, en el ensayo de union a LCA-FITC basado en citometria de flujo. Este ensayo de citometria de flujo detecta las proteinas fucosiladas sobre la superficie celular. Por lo tanto, las celulas CHOK1 de control fluorescen altamente, ya que muchas proteinas fucosiladas estan presentes en la linea celular CHOK1 de control. En casos en los que el complejo CRISPR/Cas es capaz de alterar el gen GMD en lineas celulares transfectadas, la fluorescencia de LCA-FITC se minimiza, ya que no hay proteina fucosilada en la superficie de estas lineas celulares. La figura revela la perdida significativa de fluorescencia cuando las C1GMD1.37, C1GMD1.4, C1GMD1.41, C1GMD1.43, C1GMD1.44 se probaron en este ensayo, lo que indica que estas lineas celulares son posibles lineas celulares CHOK1 de inactivacion de GMD. Ensayo de seleccion de LCA (aglutinina de Lens culinaris)
Se separaron multiples poblaciones de lineas celulares clonales unicelulares en placas de replica tras la transfeccion con el complejo CRISPR/Cas. Estas lineas celulares se probaron despues con 200 |jg de reactivo LCA en el medio
de cultivo. Se observaron las celulas cada d^ a para conformar la salud y la morfolog^a de las celulas y se tomaron fotografias en los puntos temporales apropiados. La Figura 5 indica las fotografias tomadas despues de un dia de cultivo tras el punto de partida de seleccion por LCA y la figura 6 indica las fotografias tomadas despues de 4 dias de cultivo. Las lmeas celulares indicadas en este caso muestran resistencia frente a LCA representadas por celulas resistentes en el dia uno que se han multiplicado y cultivado en grandes colonias de celulas tras el dia 4 de cultivo en presencia de reactivo LCA. Se observo la morfologia celular con microscopio y las observaciones se registraron en los dias 1 y 4.
Las celulas se observaron regularmente con el microscopio de contraste de fase inversa y se controlo la morfologia de la colonia. Las fotografias tomadas en diferentes puntos temporales del ensayo de seleccion por LCA con 200 |jg/ml de LCA claramente muestran que las celulas CHOK1 de control estan completamente muertas en el dia 4 de cultivo, mientras que los clones seleccionados muestran un crecimiento celular continuo y morfologia celular sana incluso despues de 4 dias de cultivo.
De estas figuras se observa que los siguientes clones - CR1KOT1 n.° 44, CR1KOT1 n.° 52, CR1KOT1 n.° 55, CR1KOT1 n.° 61 y CR1KOT1 n.° 67, conservan la morfologia de la colonia incluso despues del tratamiento con 200 jg/ml de LCA. Por lo tanto, se considera que estos clones son posibles fenotipos de desactivacion de FUT 8.
Durante la seleccion por LCA de clones transfectados con pD1401 (ARNg 167-207), se comparo la viabilidad de los clones en el dia 1, el dia 4 y el dia 6, en la figura 6B de la presente divulgacion. De estas figuras se observa que los siguientes clones C1GMD1.12, C1GMD1.27, C1GMD1.40, C1GMD1.41, C1GMD1.43 y C1GMD1.44 conservan la morfologia de la colonia incluso despues del tratamiento con 200 jg/ml de LCA. Por lo tanto, se considera que estos clones son posibles fenotipos de desactivacion de FUT 8. Las lineas celulares indicadas en este caso muestran resistencia frente a LCA en el dia uno que se han multiplicado y cultivado en grandes colonias de celulas tras el dia 4 y el dia 6 de cultivo en presencia de reactivo LCA. Se observo la morfologia celular con microscopio y las observaciones se registraron en los dias 1, 4 y 6. Las celulas se observaron regularmente con el microscopio de contraste de fase inversa y se controlo la morfologia de la colonia. Las fotografias tomadas en diferentes puntos temporales del ensayo de seleccion por LCA con 200 jg/ml de LCA claramente muestran que las celulas CHOK1 de control estan completamente muertas en el dia 4 de cultivo, mientras que los clones seleccionados muestran un crecimiento celular continuo y morfologia celular sana incluso despues de 4 dias de cultivo.
Ensayo de union de LCA-FITC:
Para el segundo conjunto de ensayo de union de LCA-FITC, los siguientes clones han demostrado un perfil de citometria de flujo de inactivacion de fucosa: CR1KOT1 n.° 44, CR1KOT1 n.° 46, CR1KOT1 n.° 48, CR1KOT1 n.° 49, CR1KOT1 n.° 51, CR1KOT1 n.° 52, CR1KOT1 n.° 55, CR1KOT1 n.° 59, CR1KOT1 n.° 61, CR1KOT1 n.° 66, CR1KOT1 n.° 67 (Figura 10). Se diluyeron 5 mg/ml de la solucion madre de aglutinina de Lens culinaris-Fluorescema (LCA-FITC) para obtener una concentracion final de 2 jg/ml en tampon de ensayo (DPBS que contiene BSA al 2 %). Se agitaron vorticialmente las celulas a 1500 rpm durante 5 minutos usando la centrifuga minispin de Eppendorf. Se aspiro el medio y se resuspendio el sedimento en 0,25-1 ml de tampon de ensayo que contiene 2 jg/ml de LCA-FITC.
Se resuspendieron las celulas CHOK1 de control en 0,25-1 ml de tampon de ensayo solo (control sin tincion) y 0,25 1 ml de tampon de ensayo que contiene 2 jg/ml de LCA-FITC (control con tincion). Todas las muestras se diluyeron para obtener 0,1-0,2 x 106 celulas/ml en tampon de ensayo final. Despues se incubaron las muestras en la oscuridad y sobre hielo durante 30 minutos. Despues, 200 j l de cada muestra se dividieron en alicuotas en una placa de 96 pocillos. La placa se cargo despues en el citometro de flujo de mesa de trabajo Millipore GUAVA easyCyte 8HT para la adquisicion y el analisis de datos. El analisis de datos se hizo usando el programa informatico Incyte.
Tabla 12


Los resultados proporcionados en la tabla anterior tambien se representan en la representacion grafica en la figura 10 y el perfil de fluorescencia en la figura 11 de la presente divulgacion.
La Figura 10 representa el resultado grafico observado para las lineas celulares de CHOK1 CR1KOT1 n.° 44, CR1KOT1 n.° 46, CR1KOT1 n.° 48, CR1KOT1 n.° 49, CR1KOT1 n.° 51, CR1KOT1 n.° 52, CR1KOT1 n.° 55, CR1KOT1 n.° 59, CR1KOT1 n.° 61, CR1KOT1 n.° 66, CR1KOT1 n.° 67 en el ensayo de union a LCA-FITC basado en citometria de flujo. La Figura 11 representa los perfiles de fluorescencia representativos observados en las lineas celulares CR1KOT1 n.° 44, CR1KOT1 n.° 49, CR1KOT1 n.° 51, CR1KOT1 n.° 52, CR1KOT1 n.° 55, CR1KOT1 n.° 59, CR1KOT1 n.° 61, CR1KOT1 n.° 67. Todos los clones se traspasaron durante los dias adicionales antes de ser analizados en el presente experimento. Este ensayo de citometria de flujo detecta las proteinas fucosiladas sobre la superficie celular. Por lo tanto, las celulas CHOK1 de control fluorescen altamente, ya que muchas proteinas fucosiladas estan presentes en la linea celular CHOK1 de control. En casos en los que el complejo CRISPR/Cas es capaz de alterar el gen FUT8 en lineas celulares transfectadas, la fluorescencia de LCA-FITC se minimiza, ya que no hay proteina fucosilada en la superficie de estas lineas celulares. La figura revela la perdida significativa de fluorescencia cuando las CR1KOT1 n.° 44, CR1KOT1 n.° 46, CR1KOT1 n.° 48, CR1KOT1 n.° 49, CR1KOT1 n.° 51, CR1KOT1 n.° 52, CR1KOT1 n.° 55, CR1KOT1 n.° 59, CR1KOT1 n.° 61, CR1KOT1 n.° 66, CR1KOT1 n.° 67 se probaron en este ensayo, lo que indica que estas lineas celulares son lineas celulares CHOK1 de inactivacion de FUT8.
Conclusion - se han identificado 17 posibles candidatos para la linea celular CHOK1 de inactivacion de fut8. Estos 17 clones son como sigue:
CR1KOT1 n.° 006, CR1KOT1 n.° 018, CR1KOT1 n.° 022, CR1KOT1 n.° 023, CR1KOT1 n.° 026, CR1KOT1 n.° 031, CR1KOT1 n.° 034, CR1KOT1 n.° 036, CR1KOT1 n.° 037, CR1KOT1 n.° 044, CR1KOT1 n.° 049, CR1KOT1 n.° 051, CR1KOT1 n.° 052, CR1KOT1 n.° 055, CR1KOT1 n.° 059, CR1KOT1 n.° 061 y CR1KOT1 n.° 067.
Se han identificado 13 posibles candidatos para la linea celular CHOK1 de inactivacion GMD. Los clones son los siguientes:
C1GMD1.12, C1GMD1.27, C1GMD1.37, C1GMD1.4, C1GMD1.41, C1GMD1.43, C1GMD1.44, C1GMD2.30, C1GMD3.4, C1GMD3.36, C1GMD3.43, C1GMD3.49, C1GMD3.51
Ejemplo 3: ENSAYO DE UNION A LCA-FITC
Las lineas celulares clonales CHOK1 de inactivacion de fucosa se probaron en experimentos de repeticion independientes usando el ensayo de union LCA-FITC:
Se probaron las siguientes lineas celulares clonales para la repetibilidad del ensayo de union a LCA-FITC.
CR1KOT1 n.° 006, CR1KOT1 n.° 018, CR1KOT1 n.° 022, CR1KOT1 n.° 023, CR1KOT1 n.° 026, CR1KOT1 n.° 031, CR1KOT1 n.° 034, CR1KOT1 n.° 036, CR1KOT1 n.° 037, CR1KOT1 n.° 044, CR1KOT1 n.° 049, CR1KOT1 n.° 051, CR1KOT1 n.° 052, CR1KOT1 n.° 055, CR1KOT1 n.° 059, CR1KOT1 n.° 061 y CR1KOT1 n.° 067.
Se diluyeron 5 mg/ml de la solucion madre de aglutinina de Lens culinaris-Fluorescema (LCA-FITC) para obtener una concentracion final de 2 |jg/ml en tampon de ensayo (DPBS que contiene BSA al 2 %). Se agitaron vorticialmente las celulas a 1500 rpm durante 5 minutos usando la centrifuga minispin de Eppendorf. Se aspiro el
medio y se resuspendio el sedimento en 0,25-1 ml de tampon de ensayo que contiene 2 |jg/ml de LCA-FITC. Se resuspendieron las celulas CHOK1 de control en 0,25-1 ml de tampon de ensayo solo (control sin tincion) y 0,25-1 ml de tampon de ensayo que contiene 2 jg/ml de LCA-FITC (control con tincion). Todas las muestras se diluyeron para obtener 0,1-0,2 x 106 celulas/ml en tampon de ensayo final. Despues se incubaron las muestras en la oscuridad y sobre hielo durante 30 minutos. Despues, 200 j l de cada muestra se dividieron en alicuotas en una placa de 96 pocillos. La placa se cargo despues en el citometro de flujo de mesa de trabajo Millipore GUAVA easyCyte 8HT para la adquisicion y el analisis de datos. El analisis de datos se hizo usando el programa informatico Incyte.
Resultados:
La siguiente tabla describe los datos de repetibilidad del ensayo de union de LCA-FITC para todos los clones seleccionados.
Tabla 13

La representacion grafica del ensayo de union a LCA-FITC se presenta en la figura 12. El analisis de los datos sugiere una alta reproducibilidad del patron de union a LCA-FITC para las lineas celulares clonales CHOK1 de inactivacion de fucosa. Estas lineas celulares CHOK1 de inactivacion de fucosa se prueban adicionalmente para las caracteristicas del cultivo y se compararon con la linea celular CHOK1 parental no transfectada.
Ejemplo 4: ENSAYO DE ESTREPTAVIDINA-FITC
Las lineas celulares clonales CHOK1 de inactivacion de fucosa se probaron con el conjugado de estreptavidina-FITC para asegurar la interaction especifica de la union a LCA-FITC:
La tincion de estreptavidina conjugada con FITC (Strep-FITC) de los clones se llevo a cabo para asegurar que no hay union no especifica del colorante FITC. Las proteinas de la membrana celular no se unen al conjugado de estreptavidina-FITC, mientras que las proteinas de membrana fucosilada se unen de forma especifica al conjugado LCA-FITC. Las celulas CHOK1 de control se tineron tanto con LCA-FITC como con Strep-FITC en reacciones separadas para confirmar esta especificidad. Todos los clones se tineron de forma similar con conjugados de estreptavidina-FITC y LCA-FITC para determinar la union no especifica.
La Figura 14 de la presente divulgation representa el ensayo de citometria de flujo de LCA, y la comparacion de los clones a traves del ensayo de LCA-FITC y Strep-FITC, respectivamente. Para los siguientes clones se observo el fenotipo de inactivacion de fucosa: CR1KOT1 n.° 006, CR1KOT1 n.° 018, CR1KOT1 n.° 022, CR1KOT1 n.° 023, CR1KOT1 n.° 026, CR1KOT1 n.° 031, CR1KOT1 n.° 034, CR1KOT1 n.° 036, CR1KOT1 n.° 037, CR1KOT1 n.° 044, CR1KOT1 n.° 049, CR1KOT1 n.° 051, CR1KOT1 n.° 052, CR1KOT1 n.° 055, CR1KOT1 n.° 059, CR1KOT1 n.° 061 y CR1KOT1 n.° 067. En todos los clones, se observo una reduction significativa de la fluorescencia en comparacion con el control y, por lo tanto, esto demuestra la ausencia de proteina fucosilada en la superficie celular para los clones. Estos datos proporcionan la evidencia funcional de que estas lineas funcionales carecen de la proteina fucosilada debido a la alteration del gen FUT8 llevada a cabo usando el complejo en el metodo de la presente
divulgacion.
Se diluyeron 1 mg/ml de la solucion madre de estreptavidina-fluorescema (Strep-FITC) para obtener una concentracion final de 2 jg/ml en tampon de ensayo (DPBS que contiene BSA al 2 %). Se agitaron vorticialmente las celulas a 1500 rpm durante 5 minutos usando la centrifuga minispin de Eppendorf. Se aspiro el medio y se resuspendio el sedimento en 0,25-1 ml de tampon de ensayo que contiene 2 |jg/ml de Estreptavidina-FITC. Se resuspendieron las celulas CHOK1 de control en 0,25-1 ml de tampon de ensayo solo (control sin tincion) y 0,25-1 ml de tampon de ensayo que contiene 2 jg/ml de Estreptavidina-FITC (control con tincion). Todas las muestras se diluyeron para obtener 0,1-0,2 x 106 celulas/ml en tampon de ensayo final. Despues se incubaron las muestras en la oscuridad y sobre hielo durante 30 minutos.
Despues, 200 j l de cada muestra se dividieron en alicuotas en una placa de 96 pocillos. La placa se cargo despues en el citometro de flujo de mesa de trabajo Millipore GUAVA easyCyte 8HT para la adquisicion y el analisis de datos. El analisis de datos se hizo usando el programa informatico Incyte.
Se llevo a cabo a comparacion con Estreptavidina-FITC para asegurar la interaccion especifica del conjugado LCA-FITC. Los datos sugieren solo fluorescencia de fondo observada con el conjugado Estreptavidina-FITC cuando se probo con la linea celular de control CHOK1 y cualquiera de las lineas celulares transfectadas con CRISPR/Cas.
La Figura 14 representa la especificidad del ensayo de citometria de flujo de LCA-FITC, desarrollado para cribar las lineas celulares CHOK1 de inactivacion de fucosa. Los experimentos de citometria de flujo se llevaron a cabo con el conjugado de Estreptavidina-FITC. La Estreptavidina-FITC no reconoce proteinas de superficie celular en la linea celular de CHOK1 de control, lo que indica la interaccion especifica del conjugado LCA-FITC. Adicionalmente, el conjugado de estreptavidina-FITC revela niveles similares de fluorescencia de fondo entre lineas celulares positivas y negativas identificadas por el conjugado LCA-FITC. La Figura 14 no muestra interaccion no especifica del conjugado de LCA-FITC usado en este estudio cuando se comparo con el conjugado de Estreptavidina-FITC.
Determinacion de la curva de crecimiento para las lineas celulares CHOK1 de inactivacion de fucosa:
La determinacion de la curva de crecimiento de los clones seleccionados se realizo para asegurar que el perfil de crecimiento no se altera significativamente en comparacion con las celulas CHOK1 de tipo silvestre durante el proceso de desarrollo de la linea celular de inactivacion. Se inocularon 0,1 X 106 celulas CHOK1 en placas de cultivo tisular de 6 pocillos. La inoculacion se hizo para 5 puntos temporales para cada clon. Para cada punto temporal, se hizo una inoculacion por triplicado (por ejemplo, 15 pocillo para 5 puntos temporales). En cada punto temporal, se hicieron recuentos celulares por triplicado. El recuento de celulas viables se realizo usando un hemocitometro o un analizador de viabilidad celular Vi-cell XR. Se generaron las respectivas curvas de crecimiento con EEM como barra de error. La Tabla 14 describe los datos de crecimiento representativos de una de las lineas celulares de inactivacion de FUT8.
Tabla 14




Resultados: El recuento de celulas viables para cada una de las lmeas celulares se probo y se uso para la determinacion de la curva de crecimiento. Las siguientes lmeas celulares CHOK1 de inactivacion de fucosa se usan para el desarrollo de la curva de crecimiento, CR1KOT1 n.° 006, CR1KOT1 n.° 018, CR1KOT1 n.° 022, CR1KOT1 n.° 023, CR1KOT1 n.° 026, CR1KOT1 n.° 031, CR1KOT1 n.° 034, CR1KOT1 n.° 036, CR1KOT1 n.° 037, CR1KOT1 n.° 044, CR1KOT1 n.° 049, CR1KOT1 n.° 051, CR1KOT1 n.° 052, CR1KOT1 n.° 055, CR1KOT1 n.° 061 y CR1KOT1 n.° 067.
Los datos se analizaron y se representaron graficamente en la curva de crecimiento. Las curvas de crecimiento de las respectivas lineas celulares se proporcionan en las figuras 13A, 13B y 13C de la presente divulgacion.
De las figuras se observa que la mayoria de las lineas celulares clonales tienen un potencial de crecimiento comparable con respecto a la linea celular parental de CHOK1. Estas lineas celulares clonales se san para la sobreexpresion de las proteinas y/o anticuerpos monoclonales terapeuticos. Se hizo un recuento de celulas viables todos los dias durante 5 dias en condiciones de cultivo optimas, usando el contador Vi-Cell. Unas pocas lineas celulares CHOK1 de inactivacion de fucosa, CR1KOT1 n.° 018, CR1KOT1 n.° 026, CR1KOT1 n.° 034, CR1KOT1 n.° 052 y CR1KOT1 n.° 055 han demostrado tener un potencial de crecimiento ligeramente mas lento en comparacion con otras lineas celulares.
Ejemplo 5: ENSAYOS DE SECUENCIACION GENOMICA
Los clones transfectados con CRISPR seleccionados a traves del ensayo funcional, es decir, se usaron ensayos de citometria de flujo con LCA-FITC para el analisis de secuencia genomica. El locus genomico de FUT8 en hamster chino esta bien documentado en la bibliografia (NW 003613860) y se usa como una secuencia de tipo silvestre para comprender el tipo de modificacion genica en cada clon de linea celular. De manera similar, el locus genomico de Gm D de hamster chino se obtuvo de la base de datos de la secuencia, NW_003613635.1, NP_001233625.1, NM_001246696.1 y se uso como secuencia de tipo silvestre para entender el tipo de modificacion genica en cada linea celular. El objetivo de este ejemplo es analizar los resultados de secuenciacion de ADN genomico obtenidos de las lineas celulares CHOK1 de inactivacion de FUT8 transfectadas con CRISPR/Cas y las lineas celulares CHOK1 de inactivacion de GMD. Todas las lineas celulares documentadas en este caso son lineas celulares clonales y se seleccionan del ensayo de selection del medio LCA y del ensayo de citometria de flujo con LCA-FITC.
En resumen, las lineas celulares clonales seleccionadas se cultivan en condiciones de cultivo apropiadas para el aislamiento de ADN genomico, el ADN genomico purificado se uso para la amplification por PCR usando cebadores que flanquean los loci diana de FUT8 y GMD, el producto amplificado por PCR se purifica despues y se clona en un vector adecuado usando celulas E. coli competentes, las colonias de E. coli resistentes a ampicilina resultantes se seleccionan y se cultivan, los ADN plasmidicos se aislan de cada clon bacteriano, aproximadamente se prueban 5 10 colonias bacterianas individuales por lineas celulares clonales a traves de la secuenciacion automatica para comprender el tipo de modificacion en el locus genomico diana de FUT8.
Los siguientes reactivos y soluciones se usaron para llevar a cabo la secuenciacion genomica de los clones seleccionados
Todo el protocolo de secuenciacion genomica se divide en los siguientes cuatro procesos
A. Aislamiento de ADN genomico de los clones seleccionados
B. Estrategia de PCR para amplificar locus genomico especifico para cada linea celular.
C. donation de los productos de PCR en vectores de secuenciacion
D. Analisis de datos de secuencia e identification de INDEL
Aislamiento de ADN genomico de los clones seleccionados
Las lineas celulares CHOK1 clonales se cultivaron en medio DMEM avanzado con suero bovino fetal al 10 %, glutamina 4 mM, 100 unidades/ml de penicilina y 100 |jg/ml de estreptomicina en matraces T175 a 37 °C en presencia de CO2 al 5 % y humedad relativa del 75 % en incubadoras de condiciones controladas. El cultivo celular se observo cada dia y se controlo la viabilidad. Se recolectaron celulas con una confluencia del 80 % y mas del 95 % de viabilidad con tripsinizacion. El dia del aislamiento, se retiro el medio de cultivo y las celulas adherentes se lavaron primero con 10 ml de DPBS seguido por la adicion de 4 ml de solution de tripsina-EDTA al 0,05 % para la tripsinizacion. Se incubaron las celulas a 37 °C durante 2-3 minutos y se recolectaron. Despues se mezclaron las celulas con 10 ml de DPBS y se centrifugaron a 1500 rpm durante 5 minutos. El medio gastado se retiro y el sedimento celular se resuspendio en 10 ml de DPBS. Las celulas se lavaron de nuevo usando la centrifugation a 1500 rpm durante 5 minutos. El DPBS se retiro por completo mediante aspiration. El sedimento celular final se uso
para el aislamiento del ADN genomico.
El ADN genomico se aislo de las celulas CHOK1 de control, y las lmeas celulares clonales CHOK1 transfectadas con CRISPR/Cas que presentan resistencia a LCA y se selecciono a traves del ensayo de citometria de flujo con LCA. El kit de extraccion de ADNg de QIAGEN comercialmente disponible se uso para el aislamiento del ADN genomico siguiendo el protocolo del fabricante.
Diseno de estrategia de PCR
La secuencia de ADN genomico de hamster chino se analizo a partir de la secuencia de la base de datos publicamente disponible NW_003613860. Las secuencias de ADN del exon 7 de FUT8 y la secuencia del intron parcial se uso para disenar la estrategia de PCR para amplificar el locus diana FUT8.
Las secuencias de intron y de exon de GMD se obtuvieron de NW 003613635.1 y NM_001246696.1 y los analisis de secuencia se llevaron a cabo para el exon 3 y el exon 4.
Los cebadores se disenaron basandose en la longitud del cebador, en la longitud del producto de PCR, en el contenido de GC, el la temperatura de fusion y en el potencial de formacion de homoduplex y de heteroduplex. Los cebadores se disenaron flanqueando el locus diana de FUT8 tal como se proporciona a continuation. El producto de PCR amplificado esta pensado para el analisis de mutation debido a la s Sb mediada por CRISPR y la posterior reparation de ADN. La siguiente secuencia de nucleotidos representa la region de interes con secuencias del cebador en negrita.
El exon 7 de Fut8 y las secuencias de intron asociadas se usaron para el diseno del cebador de PCR:

Los intrones estan representados desde la base 21 hasta la base 106 y desde la base 345 hasta la base 371 en minusculas. El exon 7 se representa desde la base 107 hasta la base 344 en mayusculas. Los sitios de union del cebador de izquierda y derecha estan subrayados.
El exon 3 y el exon 4 de GMD y las secuencias de intron asociadas se usaron para el diseno del cebador de PCR: Se usaron las siguientes secuencias.
Exon 3 de GMD e intrones circundantes

Los intrones estan representados desde la base 23 hasta la base 166 y desde la base 277 hasta la base 447 en minusculas. El exon 3 se representa desde la base 167 hasta la base 276 en mayusculas. Los sitios de union del cebador de izquierda y derecha estan subrayados.
Exon 4 de GMD e intrones circundantes

Los intrones estan representados desde la base 21 hasta la base 187 y desde la base 381 hasta la base 432 en minusculas. El exon 4 se representa desde la base 188 hasta la base 380 en mayusculas. Los sitios de union del cebador de izquierda y derecha estan subrayados.
Diseno del cebador para la identificacion de INDEL por PCR
La PCR genomica se realizo usando el kit de extraccion de ADNg de QIAGEN usando los siguientes cebadores mencionados en la tabla 15.
T l 1

La siguiente seccion proporciona detalles experimentales para la generacion de producto de PCR a partir de ADN genomico de CHOK1 de lineas celulares de control y lineas celulares clonales seleccionadas de LCA, la clonacion de productos de PCR en celulas competentes de E. coli y secuenciacion de productos de PCR clonados.
Optimizacion de las condiciones de PCR -Se disenaron los experimentos para estandarizar las condiciones de PCR. Los parametros probados incluyeron, la concentracion de ADN genomico (de 100 ng a 1000 ng), las concentraciones del cebador (2 nmol a 20 nmol), la temperatura de hibridacion de PCR (de 55,8 °C a 62,9 °C) y el tiempo (20 segundos a 50 segundos), el tiempo de extension del producto de PCR (de 30 seg a 60 seg) y el numero de ciclos de PCR se establece a 30 ciclos. La condicion optimizada de llegada se describe en la siguiente seccion.
Las reacciones de PCR se llevaron a cabo usando la prueba de lectura de la polimerasa Phusion para asegurar que las mutaciones mediadas por PCR estan limitadas. Tras los ciclos de amplificacion por PCR, se anade la enzima Taq polimerasa en la mezcla para la adicion de ADN por cola homopolimerica. La etapa de adicion de ADN por cola
homopolimerica es importante, ya que las bases extra anadidas a los productos de PCR permiten la clonacion directa en el vector de secuenciacion descrito en la siguiente seccion. Con el fin de anadir proyecciones de dATP al producto de PCR para el vector de clonacion en TA, el producto amplificado de la polimerasa Phusion se incubo con la ADN polimerasa Taq durante 20 minutos a 72 °C.
Comprobacion cruzada de la muestra de ADN genomico mediante PCR-Los productos de PCR de ADN genomico se analizaron en electroforesis en gel de agarosa y la longitud del producto se confirmo usando un patron de peso molecular. Las muestras de PCR con un perfil de amplificacion claro se usaron en la proxima etapa de procesamiento.
Elucion de gel de producto de PCR usando el kit QIAGEN-Los productos de PCR amplificados se cargaron en gel de agarosa al 1 % de preparacion reciente y se sometieron a electroforesis a 100 V durante una hora para separar los productos de pCr amplificados de los cebadores no usados y cualquier otro dimero producido durante el proceso de amplificacion. Los productos amplificados se escindieron del gel y se eluyeron usando el kit de elucion en gel de Qiagen comercialmente disponible. El ADN se eluyo con agua de grado de biologia molecular altamente pura.
Clonacion de los productos de PCR en vectores de secuenciacion-Los productos amplificados por PCR y purificados en gel de agarosa se usaron despues para la clonacion en vector pTZ57R/T comercialmente disponible a traves de un proceso de ligamiento de ADN. Las condiciones para el ligamiento de ADN se estandarizaron previamente.
Transformacion de muestra ligada pTZ57R/T+CRISPR(PCR) en celulas competentes de E.coli DH5alpha - El ADN ligado se transformo en celulas competentes de E. coli DH5alpha, comercialmente disponibles. Se siguio el protocolo de transformacion tal como se describe por el fabricante para lograr una eficacia de transformacion de alto nivel. Tras la transformacion, las celulas de E. coli se cultivaron en presencia de antibiotico ampicilina para el crecimiento de colonias transformadas.
Inoculacion de celulas transformadas (pTZ57R/T+CRISPR(PCR)) en medio LB con ampicilina - Cada colonia separada se inoculo en caldo LB Ampicilina en un volumen de cultivo de 5 ml y se cultivo toda la noche para el aislamiento de ADN plasmidico.
Aislamiento de ADN plasmidico (pTZ57R/T+CRISPR(PCR) de celulas transformadas DH5alpha - se usaron 4,5 ml del cultivo de toda la noche para el aislamiento de ADN plasmidico usando el kit de aislamiento de ADN plasmidico de QIAGEN comercialmente disponible siguiendo el protocolo del fabricante. El ADN plasmidico se eluyo con agua de grado de biologia molecular altamente pura.
4.3.5 Comprobacion cruzada de los plasmidos para la presencia de inserciones - Se probo la presencia de inserciones en la preparacion de cada plasmido usando enzimas de restriction adecuadas seguido por la electroforesis en gel de agarosa. El tamano de la insertion se comparo con los patrones de peso molecular adecuados.
Analisis de datos de secuencia e identification de INDEL
Secuenciacion - Los plasmidos confirmados se secuenciaron despues con cebadores de secuenciacion especificos presentes en la estructura principal del vector pTZ57R/T. Los datos de la secuencia se generaron en instrumentos de secuenciacion automatica de ADN siguiendo los protocolos apropiados. La secuenciacion se llevo a cabo con cebadores de secuenciacion directos e inversos para asegurar la information de secuencia apropiada.
Analisis de secuencia de ADN - Se analizaron los datos de secuenciacion de ADN de todos los plasmidos. Se comparo la secuencia de ADN de todos los ADN plasmidicos derivados de la linea celular CHOK1 de control y diversas lineas celulares clonales CHOK1 de inactivation de FUT8 mediada por CRISPR y lineas celulares clonales CHOK1 de inactivacion de GMD mediada por CRISPR y se identificaron las diferencias en las secuencias de ADN. De cada clon de linea celular CHOK1, se generaron productos de PCR y se clonaron en E. coli. Se secuenciaron multiples clones de E. coli para confirmar la modification de la secuencia de nucleotidos en el locus genomico diana. Se uso el analisis combinado de los datos de secuencia para identificar posibles lineas celulares CHOK1 de inactivacion de FUT8 y GMD en donde los loci genomicos de FUT8 y GMD se modifican a traves de deletion y/o inserciones (INDEL). Las secuencias de ADN se alinearon despues para mostrar diferencias distintivas. Las Figuras 17A a 17G proporcionan la alineacion de las secuencias de nucleotidos de la linea celular CHOK1 de control y FUT8 y las Figuras (17H a 17L) proporcionan las alineaciones de la secuencia de nucleotidos con las lineas celulares clonales de inactivacion de GMD u la linea celular CHOK1 de control. Se uso la informacion de la secuencia de ADN para atribuir la secuencia de aminoacidos del gen FUT8 (exon 7). Usando el patron de codones para el gen GMD, se analizaron las regiones de exon 3 y exon 4. Despues se analizaron las secuencias de aminoacidos para identificar
deleciones, mutaciones de desplazamiento del marco, inserciones de codones de parada asi como sustituciones de aminoacidos en posiciones espedficas. Las figuras 17A a 17G representan el grado de modificacion de nucleotidos observado en las lmeas celulares CHOK1 de inactivacion de FUT8 y las figuras 17H a 17L en las lineas celulares CHOK1 de inactivacion de GMD cuando se compara con la linea celular CHOK1 de control. Los datos proporcionan una representacion de la organizacion del ADN genomico de FUT8 y GMD entre multiples lineas celulares CHOK1 de inactivacion de FUT8 y GMD.
Reaccion de PCR
En primer lugar, se desnaturalizo el molde de ADN de cadena doble a una temperatura elevada de 94 °C. Los cebadores especificos de secuencia mencionados en la Tabla 15 se hibridaron posteriormente (a 60,4 °C) con sitios que flanquean la secuencia diana. Una ADN polimerasa termoestable (polimerasa Phusion) prolongo (a 72 °C) los cebadores hibridados, duplicando de este modo la cantidad de secuencia de ADN original. Este producto recien sintetizado llego entonces a ser un molde adicional para posteriores ciclos de amplificacion. Estas tres etapas se repitieron durante 30 ciclos, dando como resultado un aumento de 109 veces en la concentracion de ADN diana. Con el fin de anadir proyecciones de dATP al producto de PCR para el vector de clonacion en TA, el producto amplificado por PCR de la polimerasa Phusion se incubo con la polimerasa Taq durante 20 minutos a 72 °C.
Tabla 16

T l 17 - n i i n P R

La Figura 15A de la presente divulgacion representa la figura representativa del producto amplificado por PCR del clon de Fut8 en CRISPR/Cas (CR1-KI-T1 n.° 022) cuando corre en gel de agarosa al 1 %. El a Dn genomico se aisla y se amplifica con los cebadores CRP_P1_Directo y CRP_P01_Inverso en condiciones de PCR estandarizadas. El producto amplificado se somete a electroforesis en gel de agarosa al 1 %. El carril 1 proporciona ADN genomico del clon CR1-KO-T1 n.° 022 amplificado con cebadores especificos y el carril 2 proporciona un patron de peso molecular de ADN.
El resultado revela el tamano del producto esperado del producto amplificado. El producto amplificado por PCR se purifica en gel y se clona en clones bacterianos y se secuencia para confirmar el estado del FUT8 genomico. La Figura 15B de la presente divulgacion representa la figura representativa del producto amplificado por PCR de los clones de GMD con CRISPR/Cas representativos (GMD_1.12 y GMD_1.27) cuando corren en gel de agarosa al 1 %. El ADN genomico se aislo y se amplifico con el conjunto de cebadores GMD_P01_Directo y GMD_P01_Inverso y el conjunto de cebadores GMD_P03_Directo e GMD_P03_Inverso, en condiciones de PCR estandarizadas. El producto amplificado se somete a electroforesis en gel de agarosa al 1 %. El resultado revela el tamano del producto esperado del producto amplificado. El producto amplificado por PCR se purifica en gel y se clona en clones bacterianos y se secuencia para confirmar el estado de las secuencias del exon 3 y el exon 4 de GMD.
En la Figura 15B, el carril 1 representa el ADN genomico del clon GMD_1.12 amplificado con cebadores especificos, el carril 2 representa el ADN genomico del clon GMD_1.27 amplificado con cebadores especificos, el carril 3 representa el patron de peso molecular de ADN del marcador de ADN de 1 kb y el carril 4 representa el patron de peso molecular de ADN del marcador de ADN de 100 pb.
Esta figura representativa describe la amplificacion por PCR de los loci genomicos FUT8 y GMD usando las secuencias de cebador en la Tabla 15 y la polimerasa Phusion. El producto de PCR se modifica adicionalmente con
la ADN polimerasa Taq para la adicion de ADN por cola homopolimerica. El producto final de la PCR se somete despues a electroforesis en gel de agarosa para la elucion del fragmento amplificado.
La figura 15C representa la ejecucion representativa en gel de agarosa al 1 % con amplificacion por PCR de ADN genomico de la linea celular clonal GMD 2.30 con cebadores especificos para el locus del exon 4 de GMD. el carril 1 representa el patron de peso molecular de ADN con el marcador de ADN de 1 kb, el carril 2 representa el patron de peso molecular de ADN del marcador de ADN de 100 pb y el carril 3 representa el ADN genomico del clon GMD2.30 amplificado con cebadores especificos.
Las figuras 15A, 15B y 15C revelan productos amplificados por PCR de tamano apropiado y sometidos a electroforesis para elucion en gel. Se aplico el mismo proceso para amplificar el producto amplificado por PCR de las lineas celulares CHOK1 de control y de los clones CHOK1 de inactivacion de Fut8 y de inactivacion de GMD, que se extrajeron en gel usando el kit de extraccion en gel QIAEX II.
Ligamiento
Los productos amplificados por PCR y eluidos en gel se unen en el vector comercialmente disponible pTZ57R/T. El protocolo de ligamiento se describe tal como sigue
T l 1 - M z l li mi n

La mezcla de ligamiento anterior se incubo a 4 °C toda la noche y el 50 % de la mezcla de ligamiento se transformo en celulas competentes de E. coli DH5alpha mediante el metodo del choque termico.
Transformacion de la muestra ligada en celulas bacterianas mediante el metodo del choque termico
El fin de transformar celulas bacterianas es clonar y propagar el ADN plasmidico. Se tomaron alicuotas de 20 |jl de E. coli competentes (DH5alpha) del congelador a -80 °C y se descongelaron en hielo durante 5 minutos. El 50 % de la muestra ligada (pTZ57R/T+CRISPR(PCR) se anadio a las celulas competentes y se mezclo suavemente y se incubo en hielo durante 20 minutos. El tubo que contiene la mezcla se coloco en bano de agua/ bano seo a 42 °C durante 50 segundos. El tubo se volvio a colocar en hielo durante 2 minutos. Se incubaron 0,950 ml de caldo LB calentado a 37 °C (sin antibiotico ampicilina), se incubo a 37 °C, a 220 rpm durante 1 hora, en el agitador. Se diseminaron 100 j l del cultivo resultante en placas de cultivo de LB caliente ampicilina. Las placas se incubaron durante la noche a 37 °C.
Aislamiento de ADN plasmidico de celulas bacterianas usando QIAPrep spin miniprep
El fin de este procedimiento es crecer/cultivar bacterias que contienen un ADN plasmidico especifico, que se usa en los siguientes experimentos. Se anadieron 5 ml de caldo LB ampicilina en tubos esterilizados por autoclave, las colonias bacterianas aisladas se inocularon de las placas de cultivo a tubos de cultivo con caldo LB ampicilina. Los tubos se incubaron a 220 rpm, a 37 °C toda la noche (aproximadamente 16-18 horas, en funcion del crecimiento de la bacteria). El cultivo de toda la noche de 4,5 ml se centrifugo a 13 rpm durante 1 minuto. El ADN plasmidico se aislo usando el kit de aislamiento de plasmido de QIAGEN. El ADN plasmidico se eluyo con agua de grado de biologia molecular altamente pura y se almaceno en un congelador a -20 °C hasta su uso posterior.
Clones positivos seleccionados usando la digestion por restriccion con enzimas EcoR I-HFy Hind III-HF
El ADN plasmidico aislado de este modo se probo para la presencia de inserciones, en este caso, el fragmento amplificado por PCR. El vector pTZ57R/T contiene multiples sitios de enzimas de restriccion que flanquean el producto de PCR clonado. Los sitios de restriccion EcoRI y H in d i se seleccionan para la digestion por restriccion tal como se describe en la tabla a continuacion. La reaccion se llevo a cabo a 37 °C durante 2 horas para la digestion completa de ADN plasmidico. Tras la digestion por restriccion, la mezcla se sometio a electroforesis en gel de agarosa al 1 % durante 1 hora. La insercion del producto de PCR, si esta presente, esta separada de la estructura principal del vector pTZ57R/T y los clones bacterianos confirmados se usaron para la secuenciacion de ADN.
Tab - - ion

Las figuras 16 A, 16B y 16C de la presente divulgacion representan la digestion por enzimas de restriccion representativas del producto amplificado por PCR en el vector pTZ57R/T para confirmar la presencia de inserciones de diferentes lineas celulares de inactivacion. Las preparaciones de ADN plasmidico de clones bacterianos independientes se digirieron con las enzimas de restriccion EcoRI y HindIII que flanquean el fragmento de PCR clonado en el vector y la mezcla se sometio a electroforesis en gel de agarosa al 1 %. El tamano de los fragmentos de ADN resultantes se estimo a partir de los patrones de peso molecular de ADN.
La figura 16A representa la ejecucion representativa en gel de agarosa al 1 % de digestion por restriccion del plasmido pTZ57R/T con inserciones del producto de PCR. La digestion por restriccion con EcoRI y HindIII da como resultado una insercion de 500 pb. La insercion representa el producto de PCR obtenido de la amplificacion del ADN genomico de la linea celular CR1-KO-T1 n.° 022 con cebadores especificos para el locus del exon 7 de FUT8.
En la figura,
Carril 1 - Marcador de ADN de 100 pb
Carril 2- pTZ57R/T+ CR1-KO-T1 n.°022 a [EcoRI-HF y HindIII-HF]
Carril 3- pTZ57R/T+ CR1-KO-T1 n.° 022 b [EcoRI-HF y HindIII-HF]
Carril 4- pTZ57R/T+ CR1-KO-T1 n.° 022 c [EcoRI-HF y HindIII-HF]
Carril 5- pTZ57R/T+ CR1-KO-T1 n.° 022 d [EcoRI-HF y HindIII-HF]
Carril 6- pTZ57R/T+ CR1-KO-T1 n.° 022 d [Sin corte]
La figura 16B representa la ejecucion representativa en gel de agarosa al 1 % de digestion por restriccion del plasmido pTZ57R/T con inserciones del producto de PCR. La digestion por restriccion con EcoRI y HindIII da como resultado una insercion de 500 pb. La insercion representa el producto de PCR obtenido de la amplificacion del ADN genomico de la linea celular GMD1.27 con cebadores especificos para el locus del exon 3 de Gm D.
En la figura,
Carril 1- Marcador de ADN de 1 kb GeneRuler (Thermoscientific)
Carril 2- pTZ57R/T+(CHO_GMD_1.27) a [BamHI-HF y XbaI]
Carril 3- pTZ57R/T+(CHO_GMD_1.27) b [BamHI-HF y XbaI]
Carril 4- pTZ57R/T+(CHO_GMD_1.27) c [BamHI-HF y XbaI]
Carril 5- pTZ57R/T+(CHO_GMD_1.27) d [BamHI-HF y XbaI]
Carril 6- pTZ57R/T+(CHO_GMD_1.27) d [Sin corte]
La figura 16C representa la ejecucion representativa en gel de agarosa al 1 % de digestion por restriccion del plasmido pTZ57R/T con inserciones del producto de PCR. La digestion por restriccion con EcoRI y HindIII da como resultado una insercion de 500 pb. La insercion representa el producto de PCR obtenido de la amplificacion del ADN genomico de la linea celular GMD 2.30 con cebadores especificos para el locus del exon 4 de GMD.
En la figura,
Carril 1- pTZ57R/T+( GMD_2.30) a [Sin corte]
Carril 2- pTZ57R/T+(GMD_2.30) a [BamHI-HF y XbaI]
Carril 3- pTZ57R/T+(GMD_2.30) b [BamHI-HF y XbaI]
Carril 4- pTZ57R/T+(GMD_2.30) c [BamHI-HF y XbaI]
Carril 5- pTZ57R/T+(GMD_2.30) d [BamHI-HF y XbaI]
Carril 6- Marcador de ADN de 1 kb
Los resultados revelan que todos los clones que portaban inserciones de producto de PCR de longitud prolongada. La estructura principal del vector pTZ57R/T se representa mediante el fragmento observado a una posicion de banda de aproximadamente 5,4 Kb. Basandose en estos datos, se seleccionaron muestras individuales de ADN plasmidico y se usaron para la secuenciacion de ADN. Se aplico el mismo proceso a todos los productos de PCR clonados en el vector pTZ57R/T y los clones confirmados se seleccionaron para la secuenciacion de ADN. El resultado indica la presencia de inserciones que se secuenciaron con los cebadores de secuenciacion presentes en la estructura principal del vector.
Ejemplo 6: CONFIRMACION DE LAS INDEL POR SECUENCIACION
La secuenciacion del ADN plasmidico bacteriano seleccionado se realizo con cebadores de secuenciacion aguas arriba y aguas abajo localizados en la estructura principal del vector pTZ57R/T. Los datos de secuenciacion se recopilaron usando ambos cebadores y se analizaron para determinar la informacion de secuencia de ADN adecuada. Se secuenciaron multiples plasmidos bacterianos y se secuenciaron para generar informacion combinada de secuencias de ADN en los loci genomicos diana de FUT8 y GMD para la linea celular CHOK1 de control y las lineas celulares clonales CHOK1 de inactivacion de FUT8 y las lineas celulares clonales de inactivacion de GMD logradas a traves del complejo CRISPR/Cas.
A continuacion se proporcionan secuencias de ADN genomico de la linea celular CHOK1 de control y las lineas celulares clonales ChOk i de inactivacion de FUT8 y GMD, lo que confirma la presencia de mutaciones de insercion y/o delecion en el gen Fut8 y en el gen GMD, respectivamente, mediante construcciones de CRISPR, de acuerdo con el metodo de la presente divulgacion. El locus genomico dirigido amplificado de cada linea celular que incluye la linea celular CHOK1 de control se clono como productos de PCR en multiples clone bacterianos independientes. La verificacion de la secuencia se llevo a cabo tanto con cebadores de secuenciacion directos e inversos de multiples clones bacterianos independientes (que varian de 5-15) para entender la variabilidad alelica de los loci diana FUT8 y GMD. Los datos de secuencia de ADN a continuacion son representativos de secuencias genomicas en el locus dirigido FUT8 de diversas lineas celulares de inactivacion de FUT8.
Analisis de secuencia de ADN
Linea celular CHOK1 de control (tipo silvestre) - la secuencia del Exon 7 del gen FUT8 esta en mayusculas. La secuencia del intron esta en minusculas y subrayado.

Las secuencias de la linea celular clonal CHOK1 de inactivacion de FUT8 se proporcionan a continuacion. Se observa que la secuencia del exon 7 esta mutada en las lineas celulares.
CR1KOT1 n.° 023

CR1KOT1 n.° 018

CR1KOT1 n.° 055

CR1KOT1 n.° 044

CR1KOT1 n.° 022

CR1KOT1 n.° 036

CR1KOT1 n.° 037

CR1KOT1 n.° 051

CR1KOT1 n.° 052

CR1KOT1 n.° 059
C
 R1KOT1 n.° 061
R1KOT1 n.° 061

CR1KOT1 n.° 067

En la figura 17 de la presente divulgacion se proporciona la alineacion representativa de secuencia de ADN genomico en clones de la linea celular de inactivacion de FUT8 que muestra delecion en la secuencia del gen FUT8. Las figuras 17A a 17G representan el analisis de secuencia de nucleotidos en el locus diana del exon 7 deFut8. El ADN genomico de los clones CHOK1 de inactivacion de FUT8 transfectados con CRISPR/Cas y las lineas celulares CHOK1 de control se usaron para amplificar por PCR el locus FUT8 genomico direccionado. Los datos de la secuencia se recolectaron del analisis de 5-15 clones bacterianos independientes secuenciados con cebadores de secuenciacion directos e inversos. Los datos de la secuenciacion sugieren deleciones de longitudes variables en multiples clones en comparacion con la linea celular CHOK1 de control. La delecion de bases mas grande se observo en el clon CR1KOT1 n.° 061 y la delecion mas pequena es de solo 2 bases en el clon CR1KOT1 n.° 055. Todas las deleciones se localizaron en el sitio diana de CRISPR/Cas.
En la figura 17A, los sitios de union a ADN de CRISPR/Cas a la izquierda y a la derecha se indican mediante recuadros abiertos. Las figuras 17B a 17G indican multiples clones en donde los datos de secuencia revelaron insercion de nueva secuencia de ADN en comparacion con la linea celular CHOK1 de control. La Figura 17B y la Figura 17F que representan los numeros de clones CR1KOT1 n.° 023 y CR1KOT1 n.° 052 relevaron tanto insercion como deletion de secuencias en comparacion con la secuencia genomica de CHOK1. El clon numero CR1KOT1 n.° 037 (figura 17E) revelo una insercion unica y un extenso error de emparejamiento de bases en comparacion con las secuencia genomica de CHOK1. La insercion de bases tambien vario en longitud tal como se muestra en las figuras 17B a 17G.
Los datos sugieren diversos INDEL presentes en el locus genomico FUT8 en las lineas celulares CHOK1 de inactivation de FUT8. En muchos casos, se observo que hay modificaciones muy especificas en las bases direccionadas, y en otros casos, los cambios son amplios e implican tramos mas largos de ADN. Tal diversidad de modification genomica a lo largo del complejo CRISPR/Cas es posible debido a las roturas de cadena simple de ADN endogeno en estrecha cercania y a la reparation a traves de la union de extremos no homologos. Todas estas lineas celulares se seleccionaron a traves de un ensayo de cribado funcional, a saber, un ensayo de citometria de flujo con LCA-FITC. Los resultados tambien implican una alta eficacia de los ensayos funcionales para aislar e identificar la linea celular CHOK1 de inactivacion de FUT8.
Tambien se revela que el diseno del complejo CRISPR/Cas representado en la presente divulgation es unico, ya que este par de complejos CRISPR/Cas con la endonucleasa Cas9n proporciona una alteration de secuencia genica altamente especifica en el locus direccionado FUT8 de las lineas celulares CHOK1.
Analisis de secuencia de ADN de celulas CHOK1 transfectadas con el complejo CRISPR/Cas pD1401 (ARNg 167-207) para el direccionamiento del exon 3 del gen GMD
Linea celular CHOK1 de control (tipo silvestre) - la secuencia del Exon 3 del gen GMD esta en mayusculas. La secuencia del intron esta en minusculas y subrayado.

Las secuencias de la linea celular clonal CHOK1 de inactivacion de GMD se proporcionan a continuation. Se observa que la secuencia del exon 3 esta mutada en las lineas celulares.
GMD_1.12

GMD_1.27

GMD_1.37

GMD_1.41

GMD_1.43

GMD 1.44

Analisis de secuencia de ADN de celulas CHOK1 transfectadas con el complejo CRISPR/Cas pD1301 (ARNg 404) para el direccionamiento del exon 4 del gen GMD
Lmea celular CHOK1 de control (tipo silvestre) - la secuencia del Exon 4 del gen GMD esta en mayusculas. La secuencia del intron esta en minusculas y las localizaciones del cebador estan subrayadas.

La secuencia de la linea celular clonal CHOK1 de inactivacion de GMD se proporciona a continuacion. Se observa que la secuencia del exon 4 esta mutada en la linea celular clonal.
GMD 2.30

Analisis de secuencia de ADN de celulas CHOK1 transfectadas con el complejo CRISPR/Cas pD1401 (ARNg 167-207) y pD1301 (ARNg 404) para el direccionamiento del Exon 3 y el Exon 4 del gen GMD
La secuencia de la lmea celular clonal CHOK1 de inactivacion de GMD se proporciona a continuacion. Se observa que aunque se transfectaron ambos complejos CRISPR/Cas, solo se observaron mutaciones de nucleotidos en el exon 4. El analisis de la secuencia del Exon 3 revelo el Exon 3 de tipo silvestre en la linea celular clonal de inactivacion.
GMD_3.51
Secuencia del Exon 3 -
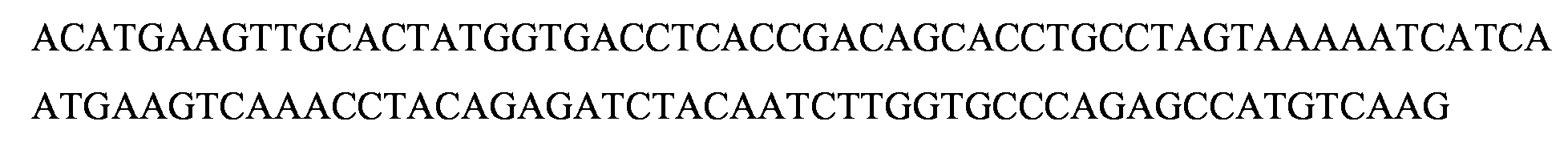
Secuencia del Exon 4 -
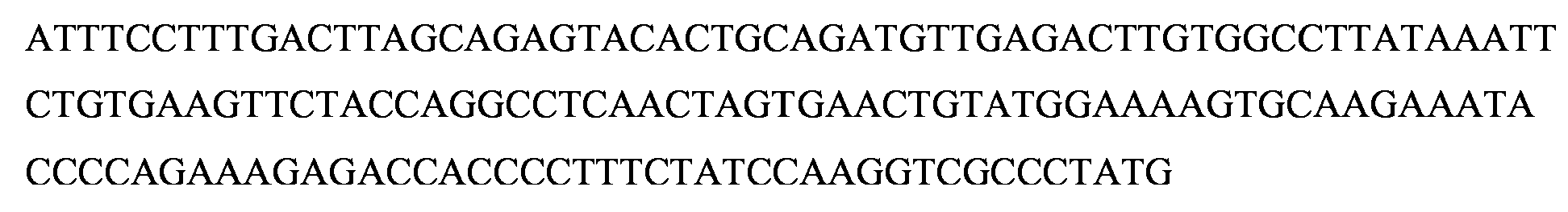
Tabla que muestra e listado de secuencias de las lineas celulares de inactivacion de GMD desarrolladas mediante transfeccion con el complejo CRISPR/Cas pD1401 (ARNg 167-207).
Tabla 20


Tabla que muestra e listado de secuencias de las lineas celulares de inactivacion de GMD desarrolladas mediante transfeccion con el complejo CRISPR/Cas pD1301 (ARNg 404).
Tabla 21
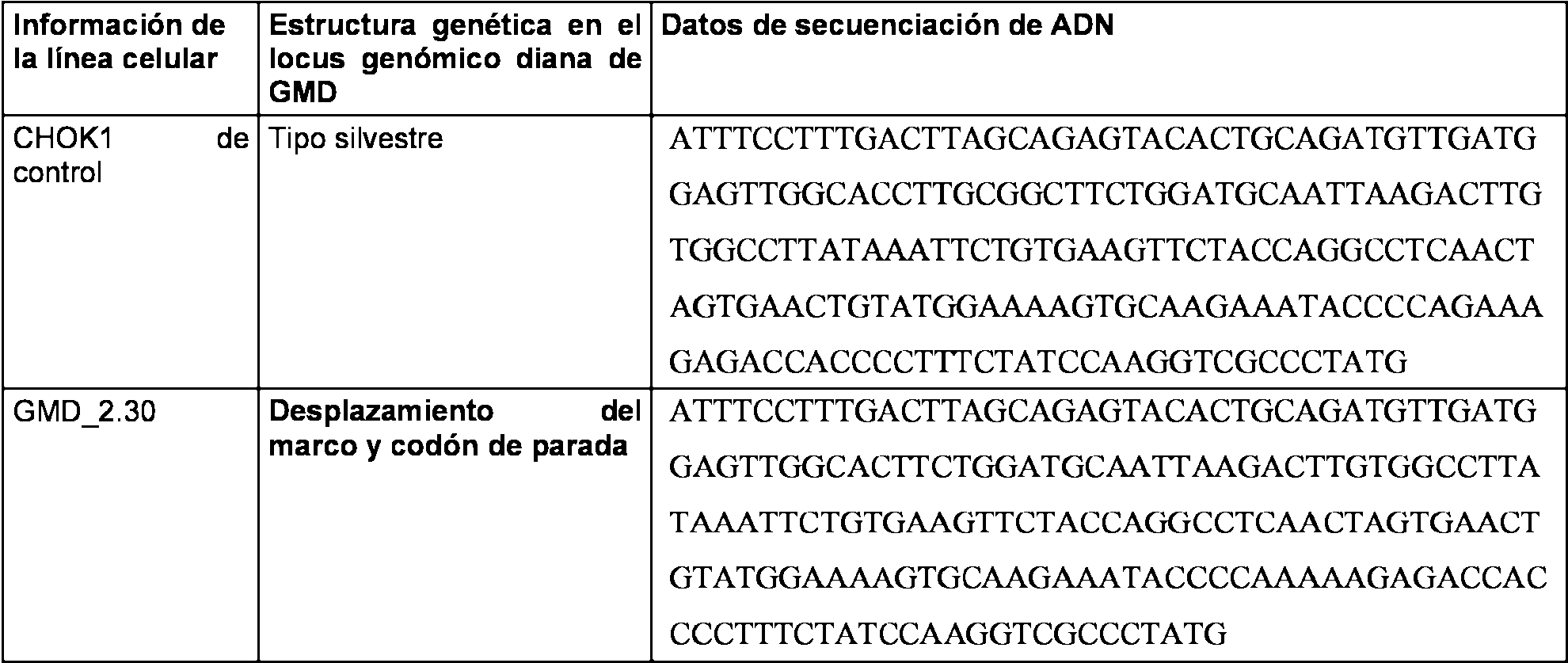
Tabla que muestra e listado de secuencias de las lineas celulares de inactivacion de GMD desarrolladas mediante transfeccion con el complejo CRISPR/Cas pD1401 (ARNg 167-207) y pD1301 (ARNg 404).
Tabla 22


En la figura 17H a17L de la presente divulgacion se proporciona la alineacion representativa de secuencia de ADN genomico en clones de la linea celular CHOK1 de inactivacion de GMD que muestran delecion en la secuencia del gen GMD. Las figuras 17H a 17L representan el analisis de secuencia de nucleotidos en el locus diana de GMD. El ADN genomico de los clones CHOK1 de inactivacion de GMD transfectados con CRISPR/Cas y las lineas celulares CHOK1 de control se usaron para amplificar por PCR los loci de GMD genomicos direccionados, tanto las secuencias del exon 3 como del exon 4. Los datos de la secuencia se recolectaron del analisis de 5-15 clones bacterianos independientes secuenciados con cebadores de secuenciacion directos e inversos. Los datos de la secuenciacion sugieren deleciones de longitudes variables en multiples clones en comparacion con la linea celular CHOK1 de control. La delecion de bases mas grande se observo en el clon GMD1.41 y la delecion mas pequena es de solo 8 bases en el clon GMD 2.30. Todas las deleciones se localizaron en el sitio diana de CRISPR/Cas.
En la figura 17H, los sitios de union a ADN de CRISPR/Cas a la izquierda y a la derecha se indican mediante recuadros abiertos. En la figura 17K, el sitio de union a CRISPR/Cas se indica mediante un recuadro abierto. Las figuras 17I a 17J indican clones en donde los datos de secuencia revelaron insercion de nueva secuencia de ADN en comparacion con la linea celular CHOK1 de control. La figura 17K releva una pequena delecion en el sitio diana del exon 4 de GMD con insercion de CRISPR/Cas pD1301 (ARNg 404) que es el resultado de una rotura de ADN de cadena doble. La figura 17L representa los datos de secuencia de la linea celular CHOK1 de inactivacion de GMD transfectada con dos construcciones de CRISPR/Cas, pD1401 (ARNg 167-207) y pD1301 (ARNg 404) en donde solo la secuencia del exon 4 de GMD se modifica con una delecion de 36 pares de bases.
Los datos sugieren diversos INDEL presentes en el locus genomico GMD en las lineas celulares CHOK1 de inactivacion de GMD. En muchos casos, se observo que hay modificaciones muy especificas en las bases direccionadas, y en otros casos, los cambios son amplios e implican tramos mas largos de ADN. Tal diversidad de modificacion genomica a lo largo del complejo CRISPR/Cas es posible debido a las roturas de cadena simple de ADN y roturas de cadena doble de ADN y la posterior reparacion de ADN. Todas estas lineas celulares se seleccionaron a traves de un ensayo de cribado funcional, a saber, un ensayo de citometria de flujo con LCA-FITC. Los resultados tambien implican una alta eficacia de los ensayos funcionales para aislar e identificar la linea celular CHOK1 de inactivacion de GMD.
Tambien se revela que el diseno del complejo CRISPR/Cas representado en la presente divulgacion es unico, ya que este par de complejos CRISPR/Cas con la endonucleasa Cas9n proporciona una alteracion de secuencia genica altamente especifica en el locus direccionado de GMD de las lineas celulares CHOK1.
Analisis de secuencia de aminoacidos de las lineas celulares CHOK1 de inactivacion de FUT8
Las secuencias de ADN genomico de FUT8 de las lineas celulares CHOK1 de control y CJOK1 de inactivacion de FUT8 se analizaron posteriormente para comprender el impacto de la INDEL de la secuencia de ADN sobre el estado de la proteina FUT8. Las secuencias de ADN en el locus de FUT8 se traducen a secuencias de aminoacidos que usan el sesgo de codon de vertebrados. Se estudio de cerca la secuencia de aminoacidos de la region del exon 7 y los resultados se resumen en la tabla 23. Cuando se comparo con la linea celular CHOK1 de control, las lineas celulares de inactivacion de FUT8 revelaron modificaciones que implican deleciones e insercion de aminoacidos asi como introduction de codones de parada y mutaciones de desplazamiento del marco. Se observo que se obtenian deleciones de 10 aminoacidos o de tramos mas largos de secuencias de aminoacidos en comparacion con la secuencia de la proteina FUT8 de CHOK1.
En muchos casos, se observaron mutaciones de desplazamiento del marco, que alteran la region de C-terminal de la proteina FUT8 para hacer no funcional a la enzima. Ademas, en varios casos, se introdujo un codon de parada como un efecto de la mutation de desplazamiento del marco y, por lo tanto, la proteina FUT8 era truncada y no funcional en estos clones.
T l 2

Asimismo, se observo que la seleccion de los aminoacidos diana en la secuencia de la protema FUT8 es altamente eficaz. El direccionamiento de los aminoacidos conservados en las posiciones de la proteina FUT8 de tipo silvestre con solo un par de complejos CRISPR/Cas ha creado mutaciones en el locus dirigido en multiples lineas celulares de inactivacion.
La alineacion de secuencias de aminoacidos representativas en las lineas celulares CHOK1 de control y CHOK1 transfectada con CRISPR/Cas que presentan la delecion en la secuencia genica de FUT8 se proporcionan en la Figura 18A y 18B de la presente divulgacion. La secuencia de aminoacidos traducida se predijo usando el patron de codones. Los datos indicaron diversos efectos sobre la secuencia de aminoacidos de FUT8 debido a la delecion y/o insercion de nucleotidos observada. Los clones CR1KOT1 n.° 018, CR1KOT1 n.° 044, CR1KOT1 n.° 061, CR1KOT1 n.° 055, CR1KOT1 n.° 067 y CR1KOT1 n.° 051 revelaron la delecion dirigida de posiciones especificas de aminoacidos. En los numeros de clones CR1KOT1 n.° 052, CR1KOT1 n.° 022, CR1KOT1 n.° 036, CR1KOT1 n.° 059, CR1KOT1 n.° 023 y CR1KOT1 n.° 037, se observa la adicion de aminoacidos en la region dirigida (figura 18B). Algunos de los clones revelaron delecion seguida por mutaciones de desplazamiento de marco en codones de parada temprana. Todas estas modificaciones indicaron proteina FUT8 no funcional en las lineas celulares CHOK1 de inactivacion de FUT8 transfectadas pro CRISPR/Cas.
Ademas, el complejo CRISPR/Cas crea mutaciones de desplazamiento del marco seguidas por codones de parada que alteran la region de C-terminal de la enzima FUT8 que contiene un motivo importante II y un motivo III en el plegamiento de Rossmann. Las posiciones de los aminoacidos especificos Tyr-382, Asp-409, Asp-410, Asp-453 y Ser-469 que estan implicados en el dominio catalitico de la enzima FUT8, por lo tanto, no se expresan en estas versiones truncadas del gen FUT8. El resultado final de estas mutaciones criticas es una enzima a-1,6 fucosiltransferasa no funcional, el producto de proteina del gen FUT8 en las lineas celulares CHOK1 de inactivacion de FUT8.
Analisis del locus del exon 3 del gen GMD en lineas celulares clonales transfectadas con el complejo CRISPR/Cas pD1401 (ARNg 167-207).
Las lineas clonales de inactivacion revelaron diferentes tipos de mutaciones en la secuencia de proteina del gen GMD en la region diana. La tabla de a continuacion enumera todas las mutaciones observadas en las lineas celulares clonales de inactivacion de GMD.
Tabla 24

Analisis del locus del exon 4 del gen GMD en lineas celulares clonales transfectadas con el complejo CRISPR/Cas pD1301 (ARNg 404):
Las lineas celulares clonales de inactivacion revelaron mutaciones en la secuencia de proteina del gen GMD en la region diana. La tabla de a continuacion enumera todas las mutaciones observadas en la linea celular clonal de
inactivacion de GMD.
Tabla 25

Analisis de los loci del exon 3 y el exon 4 del gen GMD en la linea celular clonal transfectada con el complejo CRISPR/Cas pD1401 (ARNg 167-207) y pD1301 (ARNg 404):
La lmea celular clonal de inactivacion revelo mutaciones solo en la secuencia de proteina del exon 4 del gen GMD entre las regiones diana. La tabla de a continuacion enumera todas las mutaciones observadas en la linea celular clonal de inactivacion de GMD.
Tabla 26

La Figura 18C representa el analisis de aminoacidos de los clones CHOK1 de inactivacion de GMD generados mediante el uso de pD1401 (ARNg 167-207). Se observan multiples tipos de mutaciones en el locus diana del exon 3 de GMD que incluyen la delecion de restos de aminoacidos, la sustitucion asi como codones de parada prematura. Tales modificaciones producen el gen GMD no funcional y, por lo tanto, dan como resultado lineas celulares de inactivacion de fucosa. Los clones numero GMD 1.12 y GMD 1.37 revelaron la insercion de restos de aminoacidos y mutaciones de desplazamiento del marco que introdujeron codones de parada prematura (figura 18D y 18E). En el caso del clon GMD 2.30, cuando se uso pD1301 (ARNg 404) para la introduccion de rotura de cadena doble de ADN se revelo la insercion y mutaciones de desplazamiento del marco en el locus diana del exon 4 de GMD (figura 18 F). De manera similar, en el clon GMD 3.51 (figura 18G) los datos revelaron delecion de aminoacidos solo en el exon 4 de GMD aunque la linea celular se genero mediante transfeccion con las construcciones CRISPR/Cas pD1401 (ARNg 167-207) y pD1301 (ARNg 404).
Estos datos revelan que el diseno de CRISPR/Cas hecho para direccionar dos sitios diana del exon especificos del gen GMD es muy especifico y ambas construcciones son eficaces en el direccionamiento especifico. La linea celular CHOK1 de inactivacion de GMD desarrollada de este modo se uso para el desarrollo de anticuerpos monoclonales no fucosilados.
Ejemplo 7 - USO DE LiNEA CELULAR DE INACTIVACION DE FUCOSA PARA PRODUCIR ANTICUERPOS PARCIALMENTE FUCOSILADOS Y NO FUCOSILADOS
El soporte de expresion de celulas CHOK1 de inactivacion de fucosa se uso para la expresion de anticuerpos no fucosilados, en particular, anticuerpos monoclonales parcialmente no fucosilados. Los genes de anticuerpos que codifican la cadena pesada y la cadena ligera de los anticuerpos monoclonales se clonaron en los plasmidos adecuados de expresion de genes y se transfectaron en el soporte celular CHOK1 de inactivacion de fucosa descrito en los ejemplos anteriores. El anticuerpo monoclonal producido usando este soporte/metodo se expreso como un anticuerpo no fucosilado. El producto se purifico siguiendo los protocolos y las directrices establecidas para desarrollar un producto de anticuerpo monoclonal biomejorado para uso terapeutico. El anticuerpo no fucosilado biomejorado producido usando este soporte da como resultado un nivel mayor de ADCC y, por lo tanto, mejores resultados terapeuticos.
Los datos de citometria de flujo con LCA-FITC y los posteriores experimentos de secuenciacion de la presente divulgacion confirman que las lineas FKO son incapaces de fucosilar proteinas de membrana. Por tanto, la celula obtenida en la presente divulgacion produce proteinas no fucosiladas, especificamente anticuerpos o fucosilados. Los aspectos caracteristicos y las ventajas terapeuticas de los anticuerpos no fucosilados, tales como mayor ADCC, con conocidas para un experto en la materia.
Las lineas celulares CHOK1 de inactivacion de GMD son utiles en aplicaciones unicas en programas de desarrollo de anticuerpos monoclonales no fucosilados. El gen GMD esta aguas arriba e la etapa critica de GDP-Fucosa en la via de la biosintesis de fucosa. Se puede producir GDP-fucosa en celulas CHOK1 bien por la via de novo que depende por completo de la funcion del gen GMD o a traves de la via de rescate que es independiente de la funcion del gen GMD pero requiere la presencia de L-Fucosa en medio de cultivo. Por lo tanto, es posible lograr una regulacion condicional de la fucosilacion de anticuerpos monoclonales producidos en las lineas celulares de inactivacion de GMD.
Supuesto 1: Expresion genica de anticuerpos monoclonales en celulas CHOK1 de inactivacion de GMD sin L-Fucosa en el medio de cultivo. El anticuerpo monoclonal producido esta afucosilado al 100 %. En este caso, tanto la via de la biosintesis de fucosa de novo como la de rescate son no funcionales.
Supuesto 2: Expresion genica de anticuerpos monoclonales en celulas CHOK1 de inactivacion de GMD con L-Fucosa optima en el medio de cultivo. El anticuerpo monoclonal producido esta fucosilado al 100 %. En este caso, la via de novo esta completamente bloqueada pero la via de rescate es funcional. Esto permite la fucosilacion completa del gen del anticuerpo monoclonal producido en la linea celular CHOK1 de inactivacion de GMD
Supuesto 3: Expresion genica de anticuerpos monoclonales en celulas CHOK1 de inactivacion de GMD con diversos niveles de L-Fucosa en el medio de cultivo. El anticuerpo monoclonal producido en esta condicion esta parcialmente fucosilado. La dosificacion de L-Fucosa en el medio de crecimiento determina el nivel de fucosilacion del anticuerpo monoclonal. Esta dosificacion se titula durante la condicion del cultivo para asegurar el nivel de fucosilacion de anticuerpo monoclonal y, a continuacion, se titula de nuevo para afinar para lograr niveles criticos de fucosilacion de anticuerpos monoclonales diana.
Esto es una ventaja unica con la linea celular CHOK1 de inactivacion del gen GMD y esta caracteristica se describe unicamente en la presente divulgacion.
Aunque la divulgacion y la ejemplificacion se han proporcionado a modo de ilustraciones y ejemplos con el proposito de claridad y comprension, es evidente para un experto en la materia que pueden practicarse diversos cambios y modificaciones sin apartarse del alcance de la divulgacion. Por consiguiente, las descripciones y ejemplos anteriores no deben interpretarse como limitantes del alcance de la presente divulgacion.
Se pretende que el alcance de la divulgacion no este limitado por la presente descripcion detallada, sino mas bien por las reivindicaciones adjuntas. Tambien debe entenderse que las siguientes reivindicaciones pretenden cubrir todas las caracteristicas genericas y especificas de la divulgacion descrita en este documento.
Son posibles muchas modificaciones y variaciones a la luz de las ensenanzas anteriores. Por lo tanto, debe entenderse que la materia reivindicada puede practicarse de otra manera que la descrita especificamente. Cualquier referencia a elementos reivindicados en singular, por ejemplo, usando los articulos "uno", "una", "el", "la", o "dicho" no debe interpretarse como limitante de la divulgacion.
A lo largo de la presente divulgacion, la palabra "comprender" o variaciones tales como "comprende" o "que comprende", donde se use, debe entenderse que implican la inclusion de un elemento indicado, un numero entero o etapa, o grupo de elementos, numeros enteros o etapas, pero no la exclusion de cualquier otro elemento, un numero entero o etapa, o grupo de elementos, numeros enteros o etapas.
Cuando se indique un limite numerico o un intervalo en el presente documento, los puntos extremos se incluyen. Ademas, los valores y subintervalos dentro de un limite o intervalo numerico se incluyen especificamente como si se escribieran explicitamente.
Claims (13)
1. Un dominio de union a ADN del sistema CRISPR, en donde el dominio de union a ADN comprende una secuencia de ARN transcrita a partir de la secuencia de nucleotidos seleccionada del grupo que consiste en la SEQ ID NO: 41, la SEQ ID NO: 43 y la SEQ ID NO: 45.
2. El dominio de union a ADN tal y como se reivindica en la reivindicacion 1, en donde la secuencia de ARN transcrita de las SEQ ID NO: 41, la SEQ ID NO: 43 y la SEQ ID NO: 45 se une a la secuencia del gen GMD; y en donde la SEQ ID NO: 41 transcribe a la SEQ ID NO: 42; la SEQ ID NO: 43 transcribe a la SEQ ID NO: 44 y la SeQ ID NO: 45 transcribe a la SEQ ID NO: 46.
3. Un complejo CRISPR-nucleasa que comprende el dominio de union a ADN tal y como se reivindica en la reivindicacion 1, y nucleasa.
4. El complejo tal como se reivindica en la reivindicacion 3, en donde la nucleasa es endonucleasa Cas9 o endonucleasa Cas9n.
5. Un vector que comprende la secuencia de nucleotidos que codifica el dominio de union a ADN tal como se reivindica en la reivindicacion 1.
6. El vector tal como se reivindica en la reivindicacion 5, que comprende ademas una secuencia de nucleotidos que codifica la nucleasa.
7. Una celula que comprende un vector tal y como se reivindica en la reivindicacion 5.
8. La celula tal como se reivindica en la reivindicacion 7, en donde la celula se selecciona del grupo que consiste en COS, CHO-S, CHO-K1, CHO-K1 GS (-/-), CHO-DG44, CHO -DUXB11, CHO-DUKX, CHOK1SV, VERO, MDCK, W138, V79, B14AF28-G3, BHK, HaK, NS0, SP2/0-Ag14, HeLa, HEK293-F, HEK293-H, HEK293 -T, YB23HL.P2.G11.16Ag.20, perC6, celula de hibridoma productora de anticuerpos, celula madre embrionaria, celula Namalwa, linea celular de insecto de Spodoptera fugiperda (Sf), Pichia, Saccharomyces y Schizosaccharomyces.
9. Un procedimiento in vitro de obtencion de una celula de inactivacion de fucosa, comprendiendo dicho metodo las etapas de:
a) Obtener una construccion CRISPR-nucleasa que comprende la secuencia de nucleotidos que codifica el dominio de union a ADN tal y como se reivindica en la reivindicacion 1; y
b) Transfectar una celula con la construccion de la etapa (a) para obtener una celula de inactivacion de fucosa, en la que la construccion CRISPR-nucleasa proporciona el complejo CRISPR-nucleasa que comprende el dominio de union a ADN y nucleasa; y en donde el complejo escinde la secuencia del gen GMD en la celula.
10. Un metodo in vitro de obtencion de proteina con fucosilacion que varia del 0 % al 100 %, comprendiendo dicho metodo las etapas de:
a) Cultivar la celula obtenida mediante el metodo de la reivindicacion 9, en donde la celula tiene actividad de fucosilacion que varia del 0 % al 100 %; y
b) Obtener la proteina expresada por la celula de la etapa (a).
11. El metodo de acuerdo con la reivindicacion 9, en donde la secuencia del gen GMD que codifica la enzima a GDP-D-manosa 4,6-deshidratasa se escinde en el exon seleccionado del grupo que consiste en el Exon 3, el Exon 4 y una combinacion de los mismos: y en donde la celula se selecciona del grupo que consiste en COS, CHO-S, CHO-K1, CHO-K1 GS (-/-), CHO-DG44, CHO -DUXB11, CHO-DUKX, CHOK1SV, VERO, MDCK, W138, V79, B14AF28-G3, BHK, HaK, NS0, SP2/0-Ag14, HeLa, HEK293-F, HEK293-H, HEK293 -T, YB23HL.P2.G11.16Ag.20, perC6, celula de hibridoma productora de anticuerpos, celula madre embrionaria, celula Namalwa, linea celular de insecto de Spodoptera fugiperda (Sf), Pichia, Saccharomyces y Schizosaccharomyces.
12. El metodo de acuerdo con la reivindicacion 10, en donde el metodo comprende ademas la adicion de L-Fucosa al medio de cultivo.
13. El metodo de acuerdo con la reivindicacion 10, en donde la proteina es un anticuerpo; en donde el anticuerpo es un anticuerpo monoclonal; o en donde la celula produce una proteina endogena o una proteina codificada por un gen introducido en la celula.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| IN5767CH2014 | 2014-11-15 | ||
| PCT/IB2015/058777 WO2016075662A2 (en) | 2014-11-15 | 2015-11-13 | Dna-binding domain, non-fucosylated and partially fucosylated proteins, and methods thereof |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2712303T3 true ES2712303T3 (es) | 2019-05-10 |
Family
ID=55024178
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES15816521T Active ES2712303T3 (es) | 2014-11-15 | 2015-11-13 | Dominio de unión a ADN del sistema CRISPR para la producción de proteínas no fucosiladas y parcialmente fucosiladas |
Country Status (6)
| Country | Link |
|---|---|
| US (2) | US10752674B2 (es) |
| EP (2) | EP3467110A1 (es) |
| JP (2) | JP7090421B2 (es) |
| DK (1) | DK3218490T3 (es) |
| ES (1) | ES2712303T3 (es) |
| WO (1) | WO2016075662A2 (es) |
Families Citing this family (38)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP6261500B2 (ja) | 2011-07-22 | 2018-01-17 | プレジデント アンド フェローズ オブ ハーバード カレッジ | ヌクレアーゼ切断特異性の評価および改善 |
| US20150044192A1 (en) | 2013-08-09 | 2015-02-12 | President And Fellows Of Harvard College | Methods for identifying a target site of a cas9 nuclease |
| US9359599B2 (en) | 2013-08-22 | 2016-06-07 | President And Fellows Of Harvard College | Engineered transcription activator-like effector (TALE) domains and uses thereof |
| US9340799B2 (en) | 2013-09-06 | 2016-05-17 | President And Fellows Of Harvard College | MRNA-sensing switchable gRNAs |
| US9388430B2 (en) | 2013-09-06 | 2016-07-12 | President And Fellows Of Harvard College | Cas9-recombinase fusion proteins and uses thereof |
| US9526784B2 (en) | 2013-09-06 | 2016-12-27 | President And Fellows Of Harvard College | Delivery system for functional nucleases |
| US20150165054A1 (en) | 2013-12-12 | 2015-06-18 | President And Fellows Of Harvard College | Methods for correcting caspase-9 point mutations |
| WO2016022363A2 (en) | 2014-07-30 | 2016-02-11 | President And Fellows Of Harvard College | Cas9 proteins including ligand-dependent inteins |
| EP3467110A1 (en) | 2014-11-15 | 2019-04-10 | Zumutor Biologics Inc. | Dna-binding domain, non-fucosylated and partially fucosylated proteins, and methods thereof |
| EP3294774B1 (en) * | 2015-05-13 | 2024-08-28 | Zumutor Biologics, Inc. | Afucosylated protein, cell expressing said protein and associated methods |
| CA2993474A1 (en) * | 2015-07-25 | 2017-02-02 | Habib FROST | A system, device and a method for providing a therapy or a cure for cancer and other pathological states |
| JP7109784B2 (ja) | 2015-10-23 | 2022-08-01 | プレジデント アンド フェローズ オブ ハーバード カレッジ | 遺伝子編集のための進化したCas9蛋白質 |
| WO2017079165A1 (en) * | 2015-11-02 | 2017-05-11 | Genentech, Inc. | Methods of making fucosylated and afucosylated forms of a protein |
| KR102547316B1 (ko) | 2016-08-03 | 2023-06-23 | 프레지던트 앤드 펠로우즈 오브 하바드 칼리지 | 아데노신 핵염기 편집제 및 그의 용도 |
| US11661590B2 (en) | 2016-08-09 | 2023-05-30 | President And Fellows Of Harvard College | Programmable CAS9-recombinase fusion proteins and uses thereof |
| US11542509B2 (en) | 2016-08-24 | 2023-01-03 | President And Fellows Of Harvard College | Incorporation of unnatural amino acids into proteins using base editing |
| EP3526320A1 (en) | 2016-10-14 | 2019-08-21 | President and Fellows of Harvard College | Aav delivery of nucleobase editors |
| WO2018119359A1 (en) | 2016-12-23 | 2018-06-28 | President And Fellows Of Harvard College | Editing of ccr5 receptor gene to protect against hiv infection |
| JP2020510038A (ja) | 2017-03-09 | 2020-04-02 | プレジデント アンド フェローズ オブ ハーバード カレッジ | がんワクチン |
| EP3592853A1 (en) | 2017-03-09 | 2020-01-15 | President and Fellows of Harvard College | Suppression of pain by gene editing |
| JP2020510439A (ja) | 2017-03-10 | 2020-04-09 | プレジデント アンド フェローズ オブ ハーバード カレッジ | シトシンからグアニンへの塩基編集因子 |
| CA3057192A1 (en) | 2017-03-23 | 2018-09-27 | President And Fellows Of Harvard College | Nucleobase editors comprising nucleic acid programmable dna binding proteins |
| WO2018209320A1 (en) | 2017-05-12 | 2018-11-15 | President And Fellows Of Harvard College | Aptazyme-embedded guide rnas for use with crispr-cas9 in genome editing and transcriptional activation |
| WO2019023680A1 (en) | 2017-07-28 | 2019-01-31 | President And Fellows Of Harvard College | METHODS AND COMPOSITIONS FOR EVOLUTION OF BASIC EDITORS USING PHAGE-ASSISTED CONTINUOUS EVOLUTION (PACE) |
| EP3676376B1 (en) | 2017-08-30 | 2025-01-15 | President and Fellows of Harvard College | High efficiency base editors comprising gam |
| KR20250107288A (ko) | 2017-10-16 | 2025-07-11 | 더 브로드 인스티튜트, 인코퍼레이티드 | 아데노신 염기 편집제의 용도 |
| KR102061251B1 (ko) | 2017-10-31 | 2019-12-31 | 주식회사 에이치유비바이오텍 | 내인성 폴리펩타이드 생산을 위한 재조합 세포 및 방법 |
| WO2019118949A1 (en) | 2017-12-15 | 2019-06-20 | The Broad Institute, Inc. | Systems and methods for predicting repair outcomes in genetic engineering |
| EP3797160A1 (en) * | 2018-05-23 | 2021-03-31 | The Broad Institute Inc. | Base editors and uses thereof |
| US12522807B2 (en) | 2018-07-09 | 2026-01-13 | The Broad Institute, Inc. | RNA programmable epigenetic RNA modifiers and uses thereof |
| WO2020092453A1 (en) | 2018-10-29 | 2020-05-07 | The Broad Institute, Inc. | Nucleobase editors comprising geocas9 and uses thereof |
| US12351837B2 (en) | 2019-01-23 | 2025-07-08 | The Broad Institute, Inc. | Supernegatively charged proteins and uses thereof |
| JP7657726B2 (ja) | 2019-03-19 | 2025-04-07 | ザ ブロード インスティテュート,インコーポレーテッド | 編集ヌクレオチド配列を編集するための方法および組成物 |
| US12473543B2 (en) | 2019-04-17 | 2025-11-18 | The Broad Institute, Inc. | Adenine base editors with reduced off-target effects |
| US12435330B2 (en) | 2019-10-10 | 2025-10-07 | The Broad Institute, Inc. | Methods and compositions for prime editing RNA |
| DE112021002672T5 (de) | 2020-05-08 | 2023-04-13 | President And Fellows Of Harvard College | Vefahren und zusammensetzungen zum gleichzeitigen editieren beider stränge einer doppelsträngigen nukleotid-zielsequenz |
| CN113061605B (zh) * | 2020-07-13 | 2022-11-29 | 中山大学 | 一种岩藻糖转移酶8(fut8)功能缺失细胞株的构建方法及其应用 |
| EP4352094A1 (en) | 2021-06-07 | 2024-04-17 | Amgen Inc. | Using fucosidase to control afucosylation level of glycosylated proteins |
Family Cites Families (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| HUP0600342A3 (en) | 2001-10-25 | 2011-03-28 | Genentech Inc | Glycoprotein compositions |
| JPWO2005035778A1 (ja) | 2003-10-09 | 2006-12-21 | 協和醗酵工業株式会社 | α1,6−フコシルトランスフェラーゼの機能を抑制するRNAを用いた抗体組成物の製造法 |
| CN1890262B (zh) * | 2003-10-09 | 2015-01-28 | 协和发酵麒麟株式会社 | 抗凝血酶ⅲ组合物的制造方法 |
| US20060223147A1 (en) | 2004-08-05 | 2006-10-05 | Kyowa Hakko Kogyo Co., Ltd., | Process for producing glycoprotein composition |
| WO2012120500A2 (en) * | 2011-03-06 | 2012-09-13 | Merck Serono S.A. | Low fucose cell lines and uses thereof |
| WO2013013013A2 (en) | 2011-07-21 | 2013-01-24 | Alnylam Pharmaceuticals, Inc. | Compositions and methods for producing modified glycoproteins |
| US9663782B2 (en) * | 2013-07-19 | 2017-05-30 | Larix Bioscience Llc | Methods and compositions for producing double allele knock outs |
| WO2015052231A2 (en) * | 2013-10-08 | 2015-04-16 | Technical University Of Denmark | Multiplex editing system |
| EP3467110A1 (en) | 2014-11-15 | 2019-04-10 | Zumutor Biologics Inc. | Dna-binding domain, non-fucosylated and partially fucosylated proteins, and methods thereof |
-
2015
- 2015-11-13 EP EP18192111.5A patent/EP3467110A1/en active Pending
- 2015-11-13 DK DK15816521.7T patent/DK3218490T3/en active
- 2015-11-13 WO PCT/IB2015/058777 patent/WO2016075662A2/en not_active Ceased
- 2015-11-13 ES ES15816521T patent/ES2712303T3/es active Active
- 2015-11-13 US US15/526,971 patent/US10752674B2/en active Active
- 2015-11-13 JP JP2017545006A patent/JP7090421B2/ja active Active
- 2015-11-13 EP EP15816521.7A patent/EP3218490B1/en active Active
-
2020
- 2020-07-06 US US16/921,491 patent/US11548937B2/en active Active
- 2020-09-28 JP JP2020162750A patent/JP7134207B2/ja active Active
Also Published As
| Publication number | Publication date |
|---|---|
| JP7090421B2 (ja) | 2022-06-24 |
| WO2016075662A3 (en) | 2016-07-07 |
| US20200339666A1 (en) | 2020-10-29 |
| JP7134207B2 (ja) | 2022-09-09 |
| US10752674B2 (en) | 2020-08-25 |
| WO2016075662A2 (en) | 2016-05-19 |
| EP3218490B1 (en) | 2018-10-31 |
| JP2018502595A (ja) | 2018-02-01 |
| DK3218490T3 (en) | 2019-02-18 |
| EP3218490A2 (en) | 2017-09-20 |
| US20190112358A1 (en) | 2019-04-18 |
| EP3467110A1 (en) | 2019-04-10 |
| JP2021007397A (ja) | 2021-01-28 |
| US11548937B2 (en) | 2023-01-10 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2712303T3 (es) | Dominio de unión a ADN del sistema CRISPR para la producción de proteínas no fucosiladas y parcialmente fucosiladas | |
| US11267899B2 (en) | Afucosylated protein, cell expressing said protein and associated methods | |
| EP3022304B1 (en) | Methods and compositions for producing double allele knock outs | |
| ES2901128T3 (es) | Proteína no fucosilada y métodos de la misma | |
| ES2738641T3 (es) | Producción de proteínas terapéuticas en células de mamífero modificadas genéticamente | |
| US9376484B2 (en) | Production of recombinant proteins with simple glycoforms | |
| US11060083B2 (en) | Methods and compositions for producing double allele knock outs | |
| ES2659118T3 (es) | N-glicosilación en Phaeodactylum tricornutum transformada | |
| JP2020202832A (ja) | 非フコシル化タンパク質および方法 | |
| US20250333690A1 (en) | Cells for glycoengineering and methods of use | |
| NAVARRE | Engineering N-glycosylation profile of pharmaceutical glycoproteins produced in Nicotiana tabacum BY-2 cells |