ES2726664T3 - Compuestos de quinina e isómeros ópticos, procedimiento de preparación y uso médico de los mismos - Google Patents
Compuestos de quinina e isómeros ópticos, procedimiento de preparación y uso médico de los mismos Download PDFInfo
- Publication number
- ES2726664T3 ES2726664T3 ES14825941T ES14825941T ES2726664T3 ES 2726664 T3 ES2726664 T3 ES 2726664T3 ES 14825941 T ES14825941 T ES 14825941T ES 14825941 T ES14825941 T ES 14825941T ES 2726664 T3 ES2726664 T3 ES 2726664T3
- Authority
- ES
- Spain
- Prior art keywords
- acid
- hydroxyl
- azabicyclo
- ethoxy
- octane
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 238000000034 method Methods 0.000 title claims description 19
- 230000003287 optical effect Effects 0.000 title abstract description 22
- 238000002360 preparation method Methods 0.000 title description 40
- LOUPRKONTZGTKE-WZBLMQSHSA-N Quinine Chemical class C([C@H]([C@H](C1)C=C)C2)C[N@@]1[C@@H]2[C@H](O)C1=CC=NC2=CC=C(OC)C=C21 LOUPRKONTZGTKE-WZBLMQSHSA-N 0.000 title description 3
- 150000003839 salts Chemical class 0.000 claims abstract description 175
- 150000001875 compounds Chemical class 0.000 claims abstract description 142
- -1 alkoxyhydrocarbyl Chemical group 0.000 claims abstract description 58
- 229910052736 halogen Inorganic materials 0.000 claims abstract description 17
- 150000002367 halogens Chemical group 0.000 claims abstract description 17
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims abstract description 16
- 125000003545 alkoxy group Chemical group 0.000 claims abstract description 15
- 125000003118 aryl group Chemical group 0.000 claims abstract description 13
- 125000001183 hydrocarbyl group Chemical group 0.000 claims abstract description 13
- 125000000623 heterocyclic group Chemical group 0.000 claims abstract description 10
- 125000004423 acyloxy group Chemical group 0.000 claims abstract description 6
- 125000005842 heteroatom Chemical group 0.000 claims abstract description 4
- 125000002947 alkylene group Chemical group 0.000 claims abstract description 3
- 125000001072 heteroaryl group Chemical group 0.000 claims abstract description 3
- TUJKJAMUKRIRHC-UHFFFAOYSA-N hydroxyl Chemical compound [OH] TUJKJAMUKRIRHC-UHFFFAOYSA-N 0.000 claims abstract 2
- 125000001301 ethoxy group Chemical group [H]C([H])([H])C([H])([H])O* 0.000 claims description 285
- 239000000203 mixture Substances 0.000 claims description 145
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 claims description 144
- 238000006243 chemical reaction Methods 0.000 claims description 143
- 239000002253 acid Substances 0.000 claims description 112
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 claims description 108
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 claims description 108
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 claims description 74
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 claims description 72
- OFOBLEOULBTSOW-UHFFFAOYSA-N Malonic acid Chemical compound OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 claims description 72
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 claims description 72
- XBDQKXXYIPTUBI-UHFFFAOYSA-N dimethylselenoniopropionate Natural products CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 claims description 72
- KQNPFQTWMSNSAP-UHFFFAOYSA-N isobutyric acid Chemical compound CC(C)C(O)=O KQNPFQTWMSNSAP-UHFFFAOYSA-N 0.000 claims description 72
- XNGIFLGASWRNHJ-UHFFFAOYSA-N phthalic acid Chemical compound OC(=O)C1=CC=CC=C1C(O)=O XNGIFLGASWRNHJ-UHFFFAOYSA-N 0.000 claims description 72
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 claims description 39
- 150000001413 amino acids Chemical class 0.000 claims description 38
- 150000007524 organic acids Chemical class 0.000 claims description 38
- 239000005711 Benzoic acid Substances 0.000 claims description 37
- 235000010233 benzoic acid Nutrition 0.000 claims description 37
- XLYOFNOQVPJJNP-UHFFFAOYSA-M hydroxide Chemical compound [OH-] XLYOFNOQVPJJNP-UHFFFAOYSA-M 0.000 claims description 37
- 150000007522 mineralic acids Chemical class 0.000 claims description 37
- QBYIENPQHBMVBV-HFEGYEGKSA-N (2R)-2-hydroxy-2-phenylacetic acid Chemical compound O[C@@H](C(O)=O)c1ccccc1.O[C@@H](C(O)=O)c1ccccc1 QBYIENPQHBMVBV-HFEGYEGKSA-N 0.000 claims description 36
- BVKZGUZCCUSVTD-UHFFFAOYSA-M Bicarbonate Chemical compound OC([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-M 0.000 claims description 36
- BVKZGUZCCUSVTD-UHFFFAOYSA-L Carbonate Chemical compound [O-]C([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-L 0.000 claims description 36
- RGHNJXZEOKUKBD-MGCNEYSASA-N D-galactonic acid Chemical compound OC[C@@H](O)[C@H](O)[C@H](O)[C@@H](O)C(O)=O RGHNJXZEOKUKBD-MGCNEYSASA-N 0.000 claims description 36
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 claims description 36
- IAJILQKETJEXLJ-UHFFFAOYSA-N Galacturonsaeure Natural products O=CC(O)C(O)C(O)C(O)C(O)=O IAJILQKETJEXLJ-UHFFFAOYSA-N 0.000 claims description 36
- 229910002651 NO3 Inorganic materials 0.000 claims description 36
- NHNBFGGVMKEFGY-UHFFFAOYSA-N Nitrate Chemical compound [O-][N+]([O-])=O NHNBFGGVMKEFGY-UHFFFAOYSA-N 0.000 claims description 36
- 229910019142 PO4 Inorganic materials 0.000 claims description 36
- IWYDHOAUDWTVEP-UHFFFAOYSA-N R-2-phenyl-2-hydroxyacetic acid Natural products OC(=O)C(O)C1=CC=CC=C1 IWYDHOAUDWTVEP-UHFFFAOYSA-N 0.000 claims description 36
- KDYFGRWQOYBRFD-UHFFFAOYSA-N Succinic acid Natural products OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 claims description 36
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 claims description 36
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 claims description 36
- IAJILQKETJEXLJ-QTBDOELSSA-N aldehydo-D-glucuronic acid Chemical compound O=C[C@H](O)[C@@H](O)[C@H](O)[C@H](O)C(O)=O IAJILQKETJEXLJ-QTBDOELSSA-N 0.000 claims description 36
- SRSXLGNVWSONIS-UHFFFAOYSA-N benzenesulfonic acid Chemical compound OS(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-N 0.000 claims description 36
- 229940092714 benzenesulfonic acid Drugs 0.000 claims description 36
- KDYFGRWQOYBRFD-NUQCWPJISA-N butanedioic acid Chemical compound O[14C](=O)CC[14C](O)=O KDYFGRWQOYBRFD-NUQCWPJISA-N 0.000 claims description 36
- 229960004106 citric acid Drugs 0.000 claims description 36
- VFNGKCDDZUSWLR-UHFFFAOYSA-L disulfate(2-) Chemical compound [O-]S(=O)(=O)OS([O-])(=O)=O VFNGKCDDZUSWLR-UHFFFAOYSA-L 0.000 claims description 36
- 239000001530 fumaric acid Substances 0.000 claims description 36
- 229940097043 glucuronic acid Drugs 0.000 claims description 36
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 claims description 36
- 229960002510 mandelic acid Drugs 0.000 claims description 36
- 229940098779 methanesulfonic acid Drugs 0.000 claims description 36
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 claims description 36
- 239000010452 phosphate Substances 0.000 claims description 36
- 150000003013 phosphoric acid derivatives Chemical class 0.000 claims description 36
- OJMIONKXNSYLSR-UHFFFAOYSA-N phosphorous acid Chemical compound OP(O)O OJMIONKXNSYLSR-UHFFFAOYSA-N 0.000 claims description 36
- 235000019260 propionic acid Nutrition 0.000 claims description 36
- IUVKMZGDUIUOCP-BTNSXGMBSA-N quinbolone Chemical compound O([C@H]1CC[C@H]2[C@H]3[C@@H]([C@]4(C=CC(=O)C=C4CC3)C)CC[C@@]21C)C1=CCCC1 IUVKMZGDUIUOCP-BTNSXGMBSA-N 0.000 claims description 36
- 239000011975 tartaric acid Substances 0.000 claims description 36
- 235000002906 tartaric acid Nutrition 0.000 claims description 36
- 229960001367 tartaric acid Drugs 0.000 claims description 36
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 claims description 36
- 238000000746 purification Methods 0.000 claims description 35
- 125000003349 3-pyridyl group Chemical group N1=C([H])C([*])=C([H])C([H])=C1[H] 0.000 claims description 20
- 239000003814 drug Substances 0.000 claims description 16
- 125000002941 2-furyl group Chemical group O1C([*])=C([H])C([H])=C1[H] 0.000 claims description 15
- 229940079593 drug Drugs 0.000 claims description 13
- 238000004519 manufacturing process Methods 0.000 claims description 12
- 239000008194 pharmaceutical composition Substances 0.000 claims description 11
- 206010039083 rhinitis Diseases 0.000 claims description 11
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 9
- 208000006545 Chronic Obstructive Pulmonary Disease Diseases 0.000 claims description 8
- 210000002345 respiratory system Anatomy 0.000 claims description 7
- 208000006673 asthma Diseases 0.000 claims description 6
- 230000001684 chronic effect Effects 0.000 claims description 6
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims description 6
- 206010020751 Hypersensitivity Diseases 0.000 claims description 5
- 201000010099 disease Diseases 0.000 claims description 5
- 230000009610 hypersensitivity Effects 0.000 claims description 5
- 230000008569 process Effects 0.000 claims description 5
- 208000026935 allergic disease Diseases 0.000 claims description 4
- ZUOUZKKEUPVFJK-UHFFFAOYSA-N diphenyl Chemical group C1=CC=CC=C1C1=CC=CC=C1 ZUOUZKKEUPVFJK-UHFFFAOYSA-N 0.000 claims description 4
- 238000000926 separation method Methods 0.000 claims description 4
- 125000001544 thienyl group Chemical group 0.000 claims description 4
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 claims description 3
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 claims description 3
- 239000002552 dosage form Substances 0.000 claims description 3
- 239000003937 drug carrier Substances 0.000 claims description 3
- 125000005843 halogen group Chemical group 0.000 claims description 3
- 229910000108 silver(I,III) oxide Inorganic materials 0.000 claims description 3
- 125000004182 2-chlorophenyl group Chemical group [H]C1=C([H])C(Cl)=C(*)C([H])=C1[H] 0.000 claims description 2
- 206010048994 Bladder spasm Diseases 0.000 claims description 2
- 206010011224 Cough Diseases 0.000 claims description 2
- 206010063057 Cystitis noninfective Diseases 0.000 claims description 2
- VGGSQFUCUMXWEO-UHFFFAOYSA-N Ethene Chemical compound C=C VGGSQFUCUMXWEO-UHFFFAOYSA-N 0.000 claims description 2
- 239000005977 Ethylene Substances 0.000 claims description 2
- 208000018522 Gastrointestinal disease Diseases 0.000 claims description 2
- 206010020853 Hypertonic bladder Diseases 0.000 claims description 2
- 208000007107 Stomach Ulcer Diseases 0.000 claims description 2
- 206010046543 Urinary incontinence Diseases 0.000 claims description 2
- 239000004305 biphenyl Chemical group 0.000 claims description 2
- 235000010290 biphenyl Nutrition 0.000 claims description 2
- 206010009887 colitis Diseases 0.000 claims description 2
- 125000000753 cycloalkyl group Chemical group 0.000 claims description 2
- 230000002183 duodenal effect Effects 0.000 claims description 2
- 208000000718 duodenal ulcer Diseases 0.000 claims description 2
- 125000002541 furyl group Chemical group 0.000 claims description 2
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims description 2
- 208000002551 irritable bowel syndrome Diseases 0.000 claims description 2
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 claims description 2
- 230000027939 micturition Effects 0.000 claims description 2
- 125000001624 naphthyl group Chemical group 0.000 claims description 2
- QQONPFPTGQHPMA-UHFFFAOYSA-N propylene Natural products CC=C QQONPFPTGQHPMA-UHFFFAOYSA-N 0.000 claims description 2
- 125000004805 propylene group Chemical group [H]C([H])([H])C([H])([*:1])C([H])([H])[*:2] 0.000 claims description 2
- 125000004076 pyridyl group Chemical group 0.000 claims description 2
- 230000001148 spastic effect Effects 0.000 claims description 2
- 208000011580 syndromic disease Diseases 0.000 claims description 2
- KJIFKLIQANRMOU-UHFFFAOYSA-N oxidanium;4-methylbenzenesulfonate Chemical compound O.CC1=CC=C(S(O)(=O)=O)C=C1 KJIFKLIQANRMOU-UHFFFAOYSA-N 0.000 claims 1
- 150000003242 quaternary ammonium salts Chemical class 0.000 claims 1
- 239000000126 substance Substances 0.000 claims 1
- 230000002378 acidificating effect Effects 0.000 abstract 1
- 239000012453 solvate Substances 0.000 abstract 1
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 156
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 151
- 239000000243 solution Substances 0.000 description 151
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 148
- 239000007787 solid Substances 0.000 description 131
- 239000003513 alkali Substances 0.000 description 128
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 123
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Chemical compound O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 108
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 90
- 229960001701 chloroform Drugs 0.000 description 84
- 239000002904 solvent Substances 0.000 description 82
- 230000002829 reductive effect Effects 0.000 description 78
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 70
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 68
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 51
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 43
- 239000003921 oil Substances 0.000 description 42
- 235000019198 oils Nutrition 0.000 description 42
- 239000002585 base Substances 0.000 description 41
- 238000002390 rotary evaporation Methods 0.000 description 38
- 238000010828 elution Methods 0.000 description 36
- 231100000252 nontoxic Toxicity 0.000 description 36
- 230000003000 nontoxic effect Effects 0.000 description 36
- 239000004475 Arginine Substances 0.000 description 35
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 35
- 229910052757 nitrogen Inorganic materials 0.000 description 34
- 238000000967 suction filtration Methods 0.000 description 34
- NIDWUZTTXGJFNN-UHFFFAOYSA-N 3-bromopropoxybenzene Chemical compound BrCCCOC1=CC=CC=C1 NIDWUZTTXGJFNN-UHFFFAOYSA-N 0.000 description 33
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 33
- 150000007513 acids Chemical class 0.000 description 33
- 235000011114 ammonium hydroxide Nutrition 0.000 description 33
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical group [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 32
- 239000010410 layer Substances 0.000 description 29
- 238000005160 1H NMR spectroscopy Methods 0.000 description 26
- ZAFNJMIOTHYJRJ-UHFFFAOYSA-N Diisopropyl ether Chemical compound CC(C)OC(C)C ZAFNJMIOTHYJRJ-UHFFFAOYSA-N 0.000 description 25
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 25
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 25
- 239000002274 desiccant Substances 0.000 description 25
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 25
- 239000005457 ice water Substances 0.000 description 25
- 229910000104 sodium hydride Inorganic materials 0.000 description 25
- TVMXDCGIABBOFY-UHFFFAOYSA-N octane Chemical compound CCCCCCCC TVMXDCGIABBOFY-UHFFFAOYSA-N 0.000 description 24
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 23
- 239000012044 organic layer Substances 0.000 description 23
- 239000011780 sodium chloride Substances 0.000 description 20
- 238000005481 NMR spectroscopy Methods 0.000 description 19
- 238000004440 column chromatography Methods 0.000 description 19
- 239000000047 product Substances 0.000 description 19
- 239000011541 reaction mixture Substances 0.000 description 19
- IVLICPVPXWEGCA-UHFFFAOYSA-N 3-quinuclidinol Chemical compound C1C[C@@H]2C(O)C[N@]1CC2 IVLICPVPXWEGCA-UHFFFAOYSA-N 0.000 description 18
- IVLICPVPXWEGCA-ZETCQYMHSA-N (3r)-1-azabicyclo[2.2.2]octan-3-ol Chemical compound C1CC2[C@@H](O)CN1CC2 IVLICPVPXWEGCA-ZETCQYMHSA-N 0.000 description 17
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 17
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 17
- 239000012071 phase Substances 0.000 description 17
- 229920006395 saturated elastomer Polymers 0.000 description 17
- 239000000741 silica gel Substances 0.000 description 17
- 229910002027 silica gel Inorganic materials 0.000 description 17
- 239000012312 sodium hydride Substances 0.000 description 17
- 238000001914 filtration Methods 0.000 description 16
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 16
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 15
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 15
- 230000005764 inhibitory process Effects 0.000 description 15
- 102000005962 receptors Human genes 0.000 description 15
- 108020003175 receptors Proteins 0.000 description 15
- 238000010992 reflux Methods 0.000 description 14
- 241000700198 Cavia Species 0.000 description 11
- 241000700199 Cavia porcellus Species 0.000 description 11
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 11
- 239000008101 lactose Substances 0.000 description 11
- 239000007788 liquid Substances 0.000 description 11
- 230000002685 pulmonary effect Effects 0.000 description 11
- 239000000443 aerosol Substances 0.000 description 10
- 229940008126 aerosol Drugs 0.000 description 10
- 239000000706 filtrate Substances 0.000 description 10
- 239000003208 petroleum Substances 0.000 description 10
- 239000000725 suspension Substances 0.000 description 10
- UXVMQQNJUSDDNG-UHFFFAOYSA-L Calcium chloride Chemical compound [Cl-].[Cl-].[Ca+2] UXVMQQNJUSDDNG-UHFFFAOYSA-L 0.000 description 9
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 9
- 210000004027 cell Anatomy 0.000 description 9
- 230000000694 effects Effects 0.000 description 9
- 210000003437 trachea Anatomy 0.000 description 9
- IVLICPVPXWEGCA-SSDOTTSWSA-N (3s)-1-azabicyclo[2.2.2]octan-3-ol Chemical compound C1CC2[C@H](O)CN1CC2 IVLICPVPXWEGCA-SSDOTTSWSA-N 0.000 description 8
- 0 C*C(COC1C(CC2)CCN2C1)(O)I Chemical compound C*C(COC1C(CC2)CCN2C1)(O)I 0.000 description 8
- 239000002775 capsule Substances 0.000 description 8
- 238000001035 drying Methods 0.000 description 8
- 239000007789 gas Substances 0.000 description 8
- 239000000314 lubricant Substances 0.000 description 8
- 235000019359 magnesium stearate Nutrition 0.000 description 8
- 239000007858 starting material Substances 0.000 description 8
- 239000003826 tablet Substances 0.000 description 8
- 210000005090 tracheal smooth muscle Anatomy 0.000 description 7
- LERNTVKEWCAPOY-VOGVJGKGSA-N C[N+]1(C)[C@H]2C[C@H](C[C@@H]1[C@H]1O[C@@H]21)OC(=O)C(O)(c1cccs1)c1cccs1 Chemical compound C[N+]1(C)[C@H]2C[C@H](C[C@@H]1[C@H]1O[C@@H]21)OC(=O)C(O)(c1cccs1)c1cccs1 LERNTVKEWCAPOY-VOGVJGKGSA-N 0.000 description 6
- QMMFVYPAHWMCMS-UHFFFAOYSA-N Dimethyl sulfide Chemical compound CSC QMMFVYPAHWMCMS-UHFFFAOYSA-N 0.000 description 6
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 6
- 229960000686 benzalkonium chloride Drugs 0.000 description 6
- CADWTSSKOVRVJC-UHFFFAOYSA-N benzyl(dimethyl)azanium;chloride Chemical compound [Cl-].C[NH+](C)CC1=CC=CC=C1 CADWTSSKOVRVJC-UHFFFAOYSA-N 0.000 description 6
- 230000015572 biosynthetic process Effects 0.000 description 6
- VAYGXNSJCAHWJZ-UHFFFAOYSA-N dimethyl sulfate Chemical compound COS(=O)(=O)OC VAYGXNSJCAHWJZ-UHFFFAOYSA-N 0.000 description 6
- 239000008103 glucose Substances 0.000 description 6
- 239000000463 material Substances 0.000 description 6
- 230000007935 neutral effect Effects 0.000 description 6
- 238000003756 stirring Methods 0.000 description 6
- 238000003786 synthesis reaction Methods 0.000 description 6
- 229960000257 tiotropium bromide Drugs 0.000 description 6
- 238000005406 washing Methods 0.000 description 6
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 5
- 229930006000 Sucrose Natural products 0.000 description 5
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 5
- 239000004480 active ingredient Substances 0.000 description 5
- 230000037396 body weight Effects 0.000 description 5
- 239000003085 diluting agent Substances 0.000 description 5
- 238000009472 formulation Methods 0.000 description 5
- 238000010438 heat treatment Methods 0.000 description 5
- 238000002156 mixing Methods 0.000 description 5
- 229940097496 nasal spray Drugs 0.000 description 5
- 239000007922 nasal spray Substances 0.000 description 5
- 239000000546 pharmaceutical excipient Substances 0.000 description 5
- 239000006187 pill Substances 0.000 description 5
- 239000000843 powder Substances 0.000 description 5
- 230000001376 precipitating effect Effects 0.000 description 5
- 239000005720 sucrose Substances 0.000 description 5
- WRIDQFICGBMAFQ-UHFFFAOYSA-N (E)-8-Octadecenoic acid Natural products CCCCCCCCCC=CCCCCCCC(O)=O WRIDQFICGBMAFQ-UHFFFAOYSA-N 0.000 description 4
- JJFOBACUIRKUPN-UHFFFAOYSA-N 2-bromoethoxybenzene Chemical compound BrCCOC1=CC=CC=C1 JJFOBACUIRKUPN-UHFFFAOYSA-N 0.000 description 4
- LQJBNNIYVWPHFW-UHFFFAOYSA-N 20:1omega9c fatty acid Natural products CCCCCCCCCCC=CCCCCCCCC(O)=O LQJBNNIYVWPHFW-UHFFFAOYSA-N 0.000 description 4
- QSBYPNXLFMSGKH-UHFFFAOYSA-N 9-Heptadecensaeure Natural products CCCCCCCC=CCCCCCCCC(O)=O QSBYPNXLFMSGKH-UHFFFAOYSA-N 0.000 description 4
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 4
- 241000792859 Enema Species 0.000 description 4
- 229930195725 Mannitol Natural products 0.000 description 4
- 229920000168 Microcrystalline cellulose Polymers 0.000 description 4
- 239000005642 Oleic acid Substances 0.000 description 4
- ZQPPMHVWECSIRJ-UHFFFAOYSA-N Oleic acid Natural products CCCCCCCCC=CCCCCCCCC(O)=O ZQPPMHVWECSIRJ-UHFFFAOYSA-N 0.000 description 4
- 229920002472 Starch Polymers 0.000 description 4
- 230000008485 antagonism Effects 0.000 description 4
- 230000000844 anti-bacterial effect Effects 0.000 description 4
- 239000003899 bactericide agent Substances 0.000 description 4
- BRTFVKHPEHKBQF-UHFFFAOYSA-N bromocyclopentane Chemical compound BrC1CCCC1 BRTFVKHPEHKBQF-UHFFFAOYSA-N 0.000 description 4
- 230000009989 contractile response Effects 0.000 description 4
- 239000007920 enema Substances 0.000 description 4
- 238000001990 intravenous administration Methods 0.000 description 4
- 229960001361 ipratropium bromide Drugs 0.000 description 4
- KEWHKYJURDBRMN-ZEODDXGYSA-M ipratropium bromide hydrate Chemical compound O.[Br-].O([C@H]1C[C@H]2CC[C@@H](C1)[N@@+]2(C)C(C)C)C(=O)C(CO)C1=CC=CC=C1 KEWHKYJURDBRMN-ZEODDXGYSA-M 0.000 description 4
- QXJSBBXBKPUZAA-UHFFFAOYSA-N isooleic acid Natural products CCCCCCCC=CCCCCCCCCC(O)=O QXJSBBXBKPUZAA-UHFFFAOYSA-N 0.000 description 4
- 239000000594 mannitol Substances 0.000 description 4
- 235000010355 mannitol Nutrition 0.000 description 4
- 239000012528 membrane Substances 0.000 description 4
- 235000019813 microcrystalline cellulose Nutrition 0.000 description 4
- 239000008108 microcrystalline cellulose Substances 0.000 description 4
- 229940016286 microcrystalline cellulose Drugs 0.000 description 4
- ZQPPMHVWECSIRJ-KTKRTIGZSA-N oleic acid Chemical compound CCCCCCCC\C=C/CCCCCCCC(O)=O ZQPPMHVWECSIRJ-KTKRTIGZSA-N 0.000 description 4
- 229940098458 powder spray Drugs 0.000 description 4
- 239000003380 propellant Substances 0.000 description 4
- 238000005956 quaternization reaction Methods 0.000 description 4
- WXMKPNITSTVMEF-UHFFFAOYSA-M sodium benzoate Chemical compound [Na+].[O-]C(=O)C1=CC=CC=C1 WXMKPNITSTVMEF-UHFFFAOYSA-M 0.000 description 4
- 239000004299 sodium benzoate Substances 0.000 description 4
- 235000010234 sodium benzoate Nutrition 0.000 description 4
- 239000007921 spray Substances 0.000 description 4
- 239000008107 starch Substances 0.000 description 4
- 235000019698 starch Nutrition 0.000 description 4
- 239000004094 surface-active agent Substances 0.000 description 4
- 238000012360 testing method Methods 0.000 description 4
- 238000011200 topical administration Methods 0.000 description 4
- LVGUZGTVOIAKKC-UHFFFAOYSA-N 1,1,1,2-tetrafluoroethane Chemical compound FCC(F)(F)F LVGUZGTVOIAKKC-UHFFFAOYSA-N 0.000 description 3
- WWJHRSCUAQPFQO-UHFFFAOYSA-M 4-DAMP methiodide Chemical compound [I-].C1C[N+](C)(C)CCC1OC(=O)C(C=1C=CC=CC=1)C1=CC=CC=C1 WWJHRSCUAQPFQO-UHFFFAOYSA-M 0.000 description 3
- LSLYOANBFKQKPT-DIFFPNOSSA-N 5-[(1r)-1-hydroxy-2-[[(2r)-1-(4-hydroxyphenyl)propan-2-yl]amino]ethyl]benzene-1,3-diol Chemical compound C([C@@H](C)NC[C@H](O)C=1C=C(O)C=C(O)C=1)C1=CC=C(O)C=C1 LSLYOANBFKQKPT-DIFFPNOSSA-N 0.000 description 3
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 3
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 3
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 3
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical compound OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 3
- 108010010803 Gelatin Proteins 0.000 description 3
- 239000002202 Polyethylene glycol Substances 0.000 description 3
- 230000009471 action Effects 0.000 description 3
- 230000001154 acute effect Effects 0.000 description 3
- 239000005557 antagonist Substances 0.000 description 3
- PYMYPHUHKUWMLA-UHFFFAOYSA-N arabinose Natural products OCC(O)C(O)C(O)C=O PYMYPHUHKUWMLA-UHFFFAOYSA-N 0.000 description 3
- SRBFZHDQGSBBOR-UHFFFAOYSA-N beta-D-Pyranose-Lyxose Natural products OC1COC(O)C(O)C1O SRBFZHDQGSBBOR-UHFFFAOYSA-N 0.000 description 3
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 3
- 235000010980 cellulose Nutrition 0.000 description 3
- 239000001913 cellulose Substances 0.000 description 3
- 229920002678 cellulose Polymers 0.000 description 3
- 239000003795 chemical substances by application Substances 0.000 description 3
- HRVCQBYLXUYLBL-UHFFFAOYSA-N cyclopentyl(pyridin-3-yl)methanone Chemical compound C=1C=CN=CC=1C(=O)C1CCCC1 HRVCQBYLXUYLBL-UHFFFAOYSA-N 0.000 description 3
- 239000003995 emulsifying agent Substances 0.000 description 3
- 229940095399 enema Drugs 0.000 description 3
- 230000005284 excitation Effects 0.000 description 3
- 229960001022 fenoterol Drugs 0.000 description 3
- 239000008273 gelatin Substances 0.000 description 3
- 229920000159 gelatin Polymers 0.000 description 3
- 229940014259 gelatin Drugs 0.000 description 3
- 235000019322 gelatine Nutrition 0.000 description 3
- 235000011852 gelatine desserts Nutrition 0.000 description 3
- HEBKCHPVOIAQTA-UHFFFAOYSA-N meso ribitol Natural products OCC(O)C(O)C(O)CO HEBKCHPVOIAQTA-UHFFFAOYSA-N 0.000 description 3
- 230000009871 nonspecific binding Effects 0.000 description 3
- 210000000056 organ Anatomy 0.000 description 3
- 229920001223 polyethylene glycol Polymers 0.000 description 3
- 150000003254 radicals Chemical class 0.000 description 3
- 239000011535 reaction buffer Substances 0.000 description 3
- 230000009291 secondary effect Effects 0.000 description 3
- 230000016160 smooth muscle contraction Effects 0.000 description 3
- 239000000600 sorbitol Substances 0.000 description 3
- 235000010356 sorbitol Nutrition 0.000 description 3
- 239000000829 suppository Substances 0.000 description 3
- 239000000375 suspending agent Substances 0.000 description 3
- 230000000699 topical effect Effects 0.000 description 3
- QIJMMRNZBJHXRI-UHFFFAOYSA-N (2-chlorophenyl)-cyclopentylmethanone Chemical compound ClC1=CC=CC=C1C(=O)C1CCCC1 QIJMMRNZBJHXRI-UHFFFAOYSA-N 0.000 description 2
- PAAZPARNPHGIKF-UHFFFAOYSA-N 1,2-dibromoethane Chemical compound BrCCBr PAAZPARNPHGIKF-UHFFFAOYSA-N 0.000 description 2
- IIZPXYDJLKNOIY-JXPKJXOSSA-N 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCC\C=C/C\C=C/C\C=C/C\C=C/CCCCC IIZPXYDJLKNOIY-JXPKJXOSSA-N 0.000 description 2
- OWEGMIWEEQEYGQ-UHFFFAOYSA-N 100676-05-9 Natural products OC1C(O)C(O)C(CO)OC1OCC1C(O)C(O)C(O)C(OC2C(OC(O)C(O)C2O)CO)O1 OWEGMIWEEQEYGQ-UHFFFAOYSA-N 0.000 description 2
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 2
- 244000215068 Acacia senegal Species 0.000 description 2
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 2
- 229930003347 Atropine Natural products 0.000 description 2
- WVDDGKGOMKODPV-UHFFFAOYSA-N Benzyl alcohol Chemical compound OCC1=CC=CC=C1 WVDDGKGOMKODPV-UHFFFAOYSA-N 0.000 description 2
- JOYRKODLDBILNP-UHFFFAOYSA-N Ethyl urethane Chemical compound CCOC(N)=O JOYRKODLDBILNP-UHFFFAOYSA-N 0.000 description 2
- 229930091371 Fructose Natural products 0.000 description 2
- 239000005715 Fructose Substances 0.000 description 2
- RFSUNEUAIZKAJO-ARQDHWQXSA-N Fructose Chemical compound OC[C@H]1O[C@](O)(CO)[C@@H](O)[C@@H]1O RFSUNEUAIZKAJO-ARQDHWQXSA-N 0.000 description 2
- 229920001503 Glucan Polymers 0.000 description 2
- 229920000084 Gum arabic Polymers 0.000 description 2
- RKUNBYITZUJHSG-UHFFFAOYSA-N Hyosciamin-hydrochlorid Natural products CN1C(C2)CCC1CC2OC(=O)C(CO)C1=CC=CC=C1 RKUNBYITZUJHSG-UHFFFAOYSA-N 0.000 description 2
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 2
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 2
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 2
- GUBGYTABKSRVRQ-PICCSMPSSA-N Maltose Natural products O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@@H]1O[C@@H]1[C@@H](CO)OC(O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-PICCSMPSSA-N 0.000 description 2
- 241000124008 Mammalia Species 0.000 description 2
- 102000018697 Membrane Proteins Human genes 0.000 description 2
- 108010052285 Membrane Proteins Proteins 0.000 description 2
- 229920003171 Poly (ethylene oxide) Polymers 0.000 description 2
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 2
- 238000010161 Student-Newman-Keuls test Methods 0.000 description 2
- TVXBFESIOXBWNM-UHFFFAOYSA-N Xylitol Natural products OCCC(O)C(O)C(O)CCO TVXBFESIOXBWNM-UHFFFAOYSA-N 0.000 description 2
- 235000010489 acacia gum Nutrition 0.000 description 2
- 239000000205 acacia gum Substances 0.000 description 2
- 238000000540 analysis of variance Methods 0.000 description 2
- 238000013459 approach Methods 0.000 description 2
- 239000008346 aqueous phase Substances 0.000 description 2
- PYMYPHUHKUWMLA-WDCZJNDASA-N arabinose Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)C=O PYMYPHUHKUWMLA-WDCZJNDASA-N 0.000 description 2
- RKUNBYITZUJHSG-SPUOUPEWSA-N atropine Chemical compound O([C@H]1C[C@H]2CC[C@@H](C1)N2C)C(=O)C(CO)C1=CC=CC=C1 RKUNBYITZUJHSG-SPUOUPEWSA-N 0.000 description 2
- 229960000396 atropine Drugs 0.000 description 2
- 235000019445 benzyl alcohol Nutrition 0.000 description 2
- 229960004217 benzyl alcohol Drugs 0.000 description 2
- GUBGYTABKSRVRQ-QUYVBRFLSA-N beta-maltose Chemical compound OC[C@H]1O[C@H](O[C@H]2[C@H](O)[C@@H](O)[C@H](O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@@H]1O GUBGYTABKSRVRQ-QUYVBRFLSA-N 0.000 description 2
- 238000009739 binding Methods 0.000 description 2
- CHIWXYAWZYPFJS-UHFFFAOYSA-N bromomethoxybenzene Chemical compound BrCOC1=CC=CC=C1 CHIWXYAWZYPFJS-UHFFFAOYSA-N 0.000 description 2
- 239000001506 calcium phosphate Substances 0.000 description 2
- 238000004364 calculation method Methods 0.000 description 2
- AIXAANGOTKPUOY-UHFFFAOYSA-N carbachol Chemical compound [Cl-].C[N+](C)(C)CCOC(N)=O AIXAANGOTKPUOY-UHFFFAOYSA-N 0.000 description 2
- 229910052799 carbon Inorganic materials 0.000 description 2
- 125000004432 carbon atom Chemical group C* 0.000 description 2
- RZTJGUNEEZTTCJ-UHFFFAOYSA-N chloromethoxy(chloromethoxymethoxy)methane Chemical compound ClCOCOCOCCl RZTJGUNEEZTTCJ-UHFFFAOYSA-N 0.000 description 2
- 210000001072 colon Anatomy 0.000 description 2
- 230000001186 cumulative effect Effects 0.000 description 2
- MVEBDOSCXOQNAR-UHFFFAOYSA-N cyclobutyl(phenyl)methanone Chemical compound C=1C=CC=CC=1C(=O)C1CCC1 MVEBDOSCXOQNAR-UHFFFAOYSA-N 0.000 description 2
- BMFYCFSWWDXEPB-UHFFFAOYSA-N cyclohexyl(phenyl)methanone Chemical compound C=1C=CC=CC=1C(=O)C1CCCCC1 BMFYCFSWWDXEPB-UHFFFAOYSA-N 0.000 description 2
- RZTNVJSRFRHASB-UHFFFAOYSA-N cyclopentyl(furan-2-yl)methanone Chemical compound C=1C=COC=1C(=O)C1CCCC1 RZTNVJSRFRHASB-UHFFFAOYSA-N 0.000 description 2
- PJRHFTYXYCVOSJ-UHFFFAOYSA-N cyclopropyl(phenyl)methanone Chemical compound C=1C=CC=CC=1C(=O)C1CC1 PJRHFTYXYCVOSJ-UHFFFAOYSA-N 0.000 description 2
- 230000003205 diastolic effect Effects 0.000 description 2
- 150000002009 diols Chemical class 0.000 description 2
- 238000004821 distillation Methods 0.000 description 2
- 239000000839 emulsion Substances 0.000 description 2
- 238000001704 evaporation Methods 0.000 description 2
- 230000008020 evaporation Effects 0.000 description 2
- 239000011152 fibreglass Substances 0.000 description 2
- 125000005456 glyceride group Chemical group 0.000 description 2
- 239000001963 growth medium Substances 0.000 description 2
- 238000000338 in vitro Methods 0.000 description 2
- 208000030603 inherited susceptibility to asthma Diseases 0.000 description 2
- 238000002347 injection Methods 0.000 description 2
- 239000007924 injection Substances 0.000 description 2
- PNDPGZBMCMUPRI-UHFFFAOYSA-N iodine Chemical compound II PNDPGZBMCMUPRI-UHFFFAOYSA-N 0.000 description 2
- JVTAAEKCZFNVCJ-UHFFFAOYSA-N lactic acid Chemical compound CC(O)C(O)=O JVTAAEKCZFNVCJ-UHFFFAOYSA-N 0.000 description 2
- 235000010445 lecithin Nutrition 0.000 description 2
- 239000000787 lecithin Substances 0.000 description 2
- 229940067606 lecithin Drugs 0.000 description 2
- 239000011777 magnesium Substances 0.000 description 2
- 229910052749 magnesium Inorganic materials 0.000 description 2
- 238000005259 measurement Methods 0.000 description 2
- NZWOPGCLSHLLPA-UHFFFAOYSA-N methacholine Chemical compound C[N+](C)(C)CC(C)OC(C)=O NZWOPGCLSHLLPA-UHFFFAOYSA-N 0.000 description 2
- 239000002480 mineral oil Substances 0.000 description 2
- 235000010446 mineral oil Nutrition 0.000 description 2
- 230000000414 obstructive effect Effects 0.000 description 2
- 235000019271 petrolatum Nutrition 0.000 description 2
- RMHMFHUVIITRHF-UHFFFAOYSA-N pirenzepine Chemical compound C1CN(C)CCN1CC(=O)N1C2=NC=CC=C2NC(=O)C2=CC=CC=C21 RMHMFHUVIITRHF-UHFFFAOYSA-N 0.000 description 2
- 229960004633 pirenzepine Drugs 0.000 description 2
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 2
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 2
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 2
- 239000003755 preservative agent Substances 0.000 description 2
- ULWHHBHJGPPBCO-UHFFFAOYSA-N propane-1,1-diol Chemical compound CCC(O)O ULWHHBHJGPPBCO-UHFFFAOYSA-N 0.000 description 2
- SBYHFKPVCBCYGV-UHFFFAOYSA-N quinuclidine Chemical compound C1CC2CCN1CC2 SBYHFKPVCBCYGV-UHFFFAOYSA-N 0.000 description 2
- 239000002464 receptor antagonist Substances 0.000 description 2
- 229940044551 receptor antagonist Drugs 0.000 description 2
- 210000000664 rectum Anatomy 0.000 description 2
- 230000000241 respiratory effect Effects 0.000 description 2
- 208000023504 respiratory system disease Diseases 0.000 description 2
- 230000004044 response Effects 0.000 description 2
- 238000005507 spraying Methods 0.000 description 2
- TYFQFVWCELRYAO-UHFFFAOYSA-N suberic acid Chemical compound OC(=O)CCCCCCC(O)=O TYFQFVWCELRYAO-UHFFFAOYSA-N 0.000 description 2
- 239000006188 syrup Substances 0.000 description 2
- 235000020357 syrup Nutrition 0.000 description 2
- 239000000454 talc Substances 0.000 description 2
- 229910052623 talc Inorganic materials 0.000 description 2
- 235000012222 talc Nutrition 0.000 description 2
- 230000001225 therapeutic effect Effects 0.000 description 2
- MGSRCZKZVOBKFT-UHFFFAOYSA-N thymol Chemical compound CC(C)C1=CC=C(C)C=C1O MGSRCZKZVOBKFT-UHFFFAOYSA-N 0.000 description 2
- 231100000331 toxic Toxicity 0.000 description 2
- 230000002588 toxic effect Effects 0.000 description 2
- 238000002627 tracheal intubation Methods 0.000 description 2
- 239000003981 vehicle Substances 0.000 description 2
- 239000000811 xylitol Substances 0.000 description 2
- 235000010447 xylitol Nutrition 0.000 description 2
- HEBKCHPVOIAQTA-SCDXWVJYSA-N xylitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)CO HEBKCHPVOIAQTA-SCDXWVJYSA-N 0.000 description 2
- 229960002675 xylitol Drugs 0.000 description 2
- JWZZKOKVBUJMES-UHFFFAOYSA-N (+-)-Isoprenaline Chemical compound CC(C)NCC(O)C1=CC=C(O)C(O)=C1 JWZZKOKVBUJMES-UHFFFAOYSA-N 0.000 description 1
- LNAZSHAWQACDHT-XIYTZBAFSA-N (2r,3r,4s,5r,6s)-4,5-dimethoxy-2-(methoxymethyl)-3-[(2s,3r,4s,5r,6r)-3,4,5-trimethoxy-6-(methoxymethyl)oxan-2-yl]oxy-6-[(2r,3r,4s,5r,6r)-4,5,6-trimethoxy-2-(methoxymethyl)oxan-3-yl]oxyoxane Chemical compound CO[C@@H]1[C@@H](OC)[C@H](OC)[C@@H](COC)O[C@H]1O[C@H]1[C@H](OC)[C@@H](OC)[C@H](O[C@H]2[C@@H]([C@@H](OC)[C@H](OC)O[C@@H]2COC)OC)O[C@@H]1COC LNAZSHAWQACDHT-XIYTZBAFSA-N 0.000 description 1
- VEFLKXRACNJHOV-UHFFFAOYSA-N 1,3-dibromopropane Chemical compound BrCCCBr VEFLKXRACNJHOV-UHFFFAOYSA-N 0.000 description 1
- VBICKXHEKHSIBG-UHFFFAOYSA-N 1-monostearoylglycerol Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCC(O)CO VBICKXHEKHSIBG-UHFFFAOYSA-N 0.000 description 1
- ZIIUUSVHCHPIQD-UHFFFAOYSA-N 2,4,6-trimethyl-N-[3-(trifluoromethyl)phenyl]benzenesulfonamide Chemical compound CC1=CC(C)=CC(C)=C1S(=O)(=O)NC1=CC=CC(C(F)(F)F)=C1 ZIIUUSVHCHPIQD-UHFFFAOYSA-N 0.000 description 1
- CNIIGCLFLJGOGP-UHFFFAOYSA-N 2-(1-naphthalenylmethyl)-4,5-dihydro-1H-imidazole Chemical compound C=1C=CC2=CC=CC=C2C=1CC1=NCCN1 CNIIGCLFLJGOGP-UHFFFAOYSA-N 0.000 description 1
- JVKUCNQGESRUCL-UHFFFAOYSA-N 2-Hydroxyethyl 12-hydroxyoctadecanoate Chemical compound CCCCCCC(O)CCCCCCCCCCC(=O)OCCO JVKUCNQGESRUCL-UHFFFAOYSA-N 0.000 description 1
- IEQAICDLOKRSRL-UHFFFAOYSA-N 2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-(2-dodecoxyethoxy)ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethanol Chemical compound CCCCCCCCCCCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCO IEQAICDLOKRSRL-UHFFFAOYSA-N 0.000 description 1
- AUVALWUPUHHNQV-UHFFFAOYSA-N 2-hydroxy-3-propylbenzoic acid Chemical compound CCCC1=CC=CC(C(O)=O)=C1O AUVALWUPUHHNQV-UHFFFAOYSA-N 0.000 description 1
- RFVNOJDQRGSOEL-UHFFFAOYSA-N 2-hydroxyethyl octadecanoate Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCCO RFVNOJDQRGSOEL-UHFFFAOYSA-N 0.000 description 1
- GGWDOKCSOSEGQB-UHFFFAOYSA-N 3-(2-cyclopentyl-2-phenylethoxy)-1-azabicyclo[2.2.2]octane hydrochloride Chemical compound Cl.C(OC1CN2CCC1CC2)C(C1CCCC1)c1ccccc1 GGWDOKCSOSEGQB-UHFFFAOYSA-N 0.000 description 1
- GZPHSAQLYPIAIN-UHFFFAOYSA-N 3-pyridinecarbonitrile Chemical compound N#CC1=CC=CN=C1 GZPHSAQLYPIAIN-UHFFFAOYSA-N 0.000 description 1
- HIQIXEFWDLTDED-UHFFFAOYSA-N 4-hydroxy-1-piperidin-4-ylpyrrolidin-2-one Chemical compound O=C1CC(O)CN1C1CCNCC1 HIQIXEFWDLTDED-UHFFFAOYSA-N 0.000 description 1
- FHVDTGUDJYJELY-UHFFFAOYSA-N 6-{[2-carboxy-4,5-dihydroxy-6-(phosphanyloxy)oxan-3-yl]oxy}-4,5-dihydroxy-3-phosphanyloxane-2-carboxylic acid Chemical compound O1C(C(O)=O)C(P)C(O)C(O)C1OC1C(C(O)=O)OC(OP)C(O)C1O FHVDTGUDJYJELY-UHFFFAOYSA-N 0.000 description 1
- RZVAJINKPMORJF-UHFFFAOYSA-N Acetaminophen Chemical compound CC(=O)NC1=CC=C(O)C=C1 RZVAJINKPMORJF-UHFFFAOYSA-N 0.000 description 1
- 208000030090 Acute Disease Diseases 0.000 description 1
- 241000416162 Astragalus gummifer Species 0.000 description 1
- KUVIULQEHSCUHY-XYWKZLDCSA-N Beclometasone Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(Cl)[C@@H]1[C@@H]1C[C@H](C)[C@@](C(=O)COC(=O)CC)(OC(=O)CC)[C@@]1(C)C[C@@H]2O KUVIULQEHSCUHY-XYWKZLDCSA-N 0.000 description 1
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 1
- GIWBRBVGLOZHKF-UHFFFAOYSA-N C1OC1(C1CCCC1)C1=CCCO1 Chemical compound C1OC1(C1CCCC1)C1=CCCO1 GIWBRBVGLOZHKF-UHFFFAOYSA-N 0.000 description 1
- HTYBMJGZGGMGOF-UHFFFAOYSA-N CCN(CCC1C)CC1O Chemical compound CCN(CCC1C)CC1O HTYBMJGZGGMGOF-UHFFFAOYSA-N 0.000 description 1
- 102000017927 CHRM1 Human genes 0.000 description 1
- 102000017926 CHRM2 Human genes 0.000 description 1
- 102000017925 CHRM3 Human genes 0.000 description 1
- 101150060249 CHRM3 gene Proteins 0.000 description 1
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 1
- 101150073075 Chrm1 gene Proteins 0.000 description 1
- 101150012960 Chrm2 gene Proteins 0.000 description 1
- 208000017667 Chronic Disease Diseases 0.000 description 1
- 229920002261 Corn starch Polymers 0.000 description 1
- 241000699802 Cricetulus griseus Species 0.000 description 1
- 235000019739 Dicalciumphosphate Nutrition 0.000 description 1
- 239000006144 Dulbecco’s modified Eagle's medium Substances 0.000 description 1
- LVGKNOAMLMIIKO-UHFFFAOYSA-N Elaidinsaeure-aethylester Natural products CCCCCCCCC=CCCCCCCCC(=O)OCC LVGKNOAMLMIIKO-UHFFFAOYSA-N 0.000 description 1
- 239000001856 Ethyl cellulose Substances 0.000 description 1
- ZZSNKZQZMQGXPY-UHFFFAOYSA-N Ethyl cellulose Chemical compound CCOCC1OC(OC)C(OCC)C(OCC)C1OC1C(O)C(O)C(OC)C(CO)O1 ZZSNKZQZMQGXPY-UHFFFAOYSA-N 0.000 description 1
- LHDMBYWDIBGTAR-UHFFFAOYSA-N IOc1ccccc1 Chemical compound IOc1ccccc1 LHDMBYWDIBGTAR-UHFFFAOYSA-N 0.000 description 1
- 239000007836 KH2PO4 Substances 0.000 description 1
- ZDXPYRJPNDTMRX-VKHMYHEASA-N L-glutamine Chemical compound OC(=O)[C@@H](N)CCC(N)=O ZDXPYRJPNDTMRX-VKHMYHEASA-N 0.000 description 1
- 229930182816 L-glutamine Natural products 0.000 description 1
- 208000019693 Lung disease Diseases 0.000 description 1
- 206010049816 Muscle tightness Diseases 0.000 description 1
- HOZDQHPELMDYEM-RGBJRUIASA-N N[C@@](CO[C@@H]1C(CC2)CCN2C1)(C1CCCC1)C1OCCC1 Chemical compound N[C@@](CO[C@@H]1C(CC2)CCN2C1)(C1CCCC1)C1OCCC1 HOZDQHPELMDYEM-RGBJRUIASA-N 0.000 description 1
- 241000475481 Nebula Species 0.000 description 1
- 235000019483 Peanut oil Nutrition 0.000 description 1
- 102000015439 Phospholipases Human genes 0.000 description 1
- 108010064785 Phospholipases Proteins 0.000 description 1
- RVGRUAULSDPKGF-UHFFFAOYSA-N Poloxamer Chemical compound C1CO1.CC1CO1 RVGRUAULSDPKGF-UHFFFAOYSA-N 0.000 description 1
- 239000004698 Polyethylene Substances 0.000 description 1
- 229920001213 Polysorbate 20 Polymers 0.000 description 1
- 229920001214 Polysorbate 60 Polymers 0.000 description 1
- 208000036071 Rhinorrhea Diseases 0.000 description 1
- 206010039101 Rhinorrhoea Diseases 0.000 description 1
- 229920002125 Sokalan® Polymers 0.000 description 1
- 229920001304 Solutol HS 15 Polymers 0.000 description 1
- NWGKJDSIEKMTRX-AAZCQSIUSA-N Sorbitan monooleate Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OC[C@@H](O)[C@H]1OC[C@H](O)[C@H]1O NWGKJDSIEKMTRX-AAZCQSIUSA-N 0.000 description 1
- PRXRUNOAOLTIEF-ADSICKODSA-N Sorbitan trioleate Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OC[C@@H](OC(=O)CCCCCCC\C=C/CCCCCCCC)[C@H]1OC[C@H](O)[C@H]1OC(=O)CCCCCCC\C=C/CCCCCCCC PRXRUNOAOLTIEF-ADSICKODSA-N 0.000 description 1
- 239000004809 Teflon Substances 0.000 description 1
- 229920006362 Teflon® Polymers 0.000 description 1
- WPMWEFXCIYCJSA-UHFFFAOYSA-N Tetraethylene glycol monododecyl ether Chemical compound CCCCCCCCCCCCOCCOCCOCCOCCO WPMWEFXCIYCJSA-UHFFFAOYSA-N 0.000 description 1
- 239000005844 Thymol Substances 0.000 description 1
- 229920001615 Tragacanth Polymers 0.000 description 1
- 208000025865 Ulcer Diseases 0.000 description 1
- IJCWFDPJFXGQBN-RYNSOKOISA-N [(2R)-2-[(2R,3R,4S)-4-hydroxy-3-octadecanoyloxyoxolan-2-yl]-2-octadecanoyloxyethyl] octadecanoate Chemical compound CCCCCCCCCCCCCCCCCC(=O)OC[C@@H](OC(=O)CCCCCCCCCCCCCCCCC)[C@H]1OC[C@H](O)[C@H]1OC(=O)CCCCCCCCCCCCCCCCC IJCWFDPJFXGQBN-RYNSOKOISA-N 0.000 description 1
- LWZFANDGMFTDAV-BURFUSLBSA-N [(2r)-2-[(2r,3r,4s)-3,4-dihydroxyoxolan-2-yl]-2-hydroxyethyl] dodecanoate Chemical compound CCCCCCCCCCCC(=O)OC[C@@H](O)[C@H]1OC[C@H](O)[C@H]1O LWZFANDGMFTDAV-BURFUSLBSA-N 0.000 description 1
- 239000002250 absorbent Substances 0.000 description 1
- 230000002745 absorbent Effects 0.000 description 1
- WRJPSSPFHGNBMG-UHFFFAOYSA-N acetic acid 1-azabicyclo[2.2.2]octan-3-yl ester Chemical compound C1CC2C(OC(=O)C)CN1CC2 WRJPSSPFHGNBMG-UHFFFAOYSA-N 0.000 description 1
- 229940072056 alginate Drugs 0.000 description 1
- 235000010443 alginic acid Nutrition 0.000 description 1
- 229920000615 alginic acid Polymers 0.000 description 1
- 125000004183 alkoxy alkyl group Chemical group 0.000 description 1
- 229910021529 ammonia Inorganic materials 0.000 description 1
- 239000003708 ampul Substances 0.000 description 1
- 230000003042 antagnostic effect Effects 0.000 description 1
- 239000003242 anti bacterial agent Substances 0.000 description 1
- 230000001078 anti-cholinergic effect Effects 0.000 description 1
- 230000002924 anti-infective effect Effects 0.000 description 1
- 229940124599 anti-inflammatory drug Drugs 0.000 description 1
- 239000000043 antiallergic agent Substances 0.000 description 1
- 229940088710 antibiotic agent Drugs 0.000 description 1
- 239000003429 antifungal agent Substances 0.000 description 1
- 229940121375 antifungal agent Drugs 0.000 description 1
- 210000000436 anus Anatomy 0.000 description 1
- 238000003556 assay Methods 0.000 description 1
- 229950000210 beclometasone dipropionate Drugs 0.000 description 1
- 235000013871 bee wax Nutrition 0.000 description 1
- 239000012166 beeswax Substances 0.000 description 1
- 229960001716 benzalkonium Drugs 0.000 description 1
- 229960002233 benzalkonium bromide Drugs 0.000 description 1
- 229940082436 benzalkonium chloride 3 mg Drugs 0.000 description 1
- CYDRXTMLKJDRQH-UHFFFAOYSA-N benzododecinium Chemical compound CCCCCCCCCCCC[N+](C)(C)CC1=CC=CC=C1 CYDRXTMLKJDRQH-UHFFFAOYSA-N 0.000 description 1
- KHSLHYAUZSPBIU-UHFFFAOYSA-M benzododecinium bromide Chemical compound [Br-].CCCCCCCCCCCC[N+](C)(C)CC1=CC=CC=C1 KHSLHYAUZSPBIU-UHFFFAOYSA-M 0.000 description 1
- 229960004365 benzoic acid Drugs 0.000 description 1
- 150000001558 benzoic acid derivatives Chemical class 0.000 description 1
- 230000002146 bilateral effect Effects 0.000 description 1
- 230000027455 binding Effects 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 230000003115 biocidal effect Effects 0.000 description 1
- 210000004204 blood vessel Anatomy 0.000 description 1
- GZUXJHMPEANEGY-UHFFFAOYSA-N bromomethane Chemical compound BrC GZUXJHMPEANEGY-UHFFFAOYSA-N 0.000 description 1
- 210000000621 bronchi Anatomy 0.000 description 1
- 239000007853 buffer solution Substances 0.000 description 1
- 239000001110 calcium chloride Substances 0.000 description 1
- 229910001628 calcium chloride Inorganic materials 0.000 description 1
- 235000011148 calcium chloride Nutrition 0.000 description 1
- 229910000389 calcium phosphate Inorganic materials 0.000 description 1
- 229960001714 calcium phosphate Drugs 0.000 description 1
- 235000011010 calcium phosphates Nutrition 0.000 description 1
- 239000000378 calcium silicate Substances 0.000 description 1
- 229910052918 calcium silicate Inorganic materials 0.000 description 1
- 229960003340 calcium silicate Drugs 0.000 description 1
- 235000012241 calcium silicate Nutrition 0.000 description 1
- OYACROKNLOSFPA-UHFFFAOYSA-N calcium;dioxido(oxo)silane Chemical compound [Ca+2].[O-][Si]([O-])=O OYACROKNLOSFPA-UHFFFAOYSA-N 0.000 description 1
- 239000000969 carrier Substances 0.000 description 1
- 230000008859 change Effects 0.000 description 1
- 210000004978 chinese hamster ovary cell Anatomy 0.000 description 1
- 229910052801 chlorine Inorganic materials 0.000 description 1
- 125000001309 chloro group Chemical group Cl* 0.000 description 1
- OSASVXMJTNOKOY-UHFFFAOYSA-N chlorobutanol Chemical compound CC(C)(O)C(Cl)(Cl)Cl OSASVXMJTNOKOY-UHFFFAOYSA-N 0.000 description 1
- 201000009151 chronic rhinitis Diseases 0.000 description 1
- 229940110456 cocoa butter Drugs 0.000 description 1
- 235000019868 cocoa butter Nutrition 0.000 description 1
- 239000003086 colorant Substances 0.000 description 1
- 229940000425 combination drug Drugs 0.000 description 1
- 230000009137 competitive binding Effects 0.000 description 1
- 230000002860 competitive effect Effects 0.000 description 1
- 230000006957 competitive inhibition Effects 0.000 description 1
- 239000002299 complementary DNA Substances 0.000 description 1
- 229940125773 compound 10 Drugs 0.000 description 1
- 210000002808 connective tissue Anatomy 0.000 description 1
- 230000008602 contraction Effects 0.000 description 1
- 235000005687 corn oil Nutrition 0.000 description 1
- 239000002285 corn oil Substances 0.000 description 1
- 239000008120 corn starch Substances 0.000 description 1
- 235000012343 cottonseed oil Nutrition 0.000 description 1
- 239000002385 cottonseed oil Substances 0.000 description 1
- VYDIMQRLNMMJBW-UHFFFAOYSA-N cyclopentyl(phenyl)methanone Chemical compound C=1C=CC=CC=1C(=O)C1CCCC1 VYDIMQRLNMMJBW-UHFFFAOYSA-N 0.000 description 1
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 1
- 230000003247 decreasing effect Effects 0.000 description 1
- 238000003745 diagnosis Methods 0.000 description 1
- NEFBYIFKOOEVPA-UHFFFAOYSA-K dicalcium phosphate Chemical compound [Ca+2].[Ca+2].[O-]P([O-])([O-])=O NEFBYIFKOOEVPA-UHFFFAOYSA-K 0.000 description 1
- 229910000390 dicalcium phosphate Inorganic materials 0.000 description 1
- 229940038472 dicalcium phosphate Drugs 0.000 description 1
- 235000005911 diet Nutrition 0.000 description 1
- 230000000378 dietary effect Effects 0.000 description 1
- 235000014113 dietary fatty acids Nutrition 0.000 description 1
- 239000007884 disintegrant Substances 0.000 description 1
- 239000002270 dispersing agent Substances 0.000 description 1
- GRWZHXKQBITJKP-UHFFFAOYSA-L dithionite(2-) Chemical compound [O-]S(=O)S([O-])=O GRWZHXKQBITJKP-UHFFFAOYSA-L 0.000 description 1
- 231100000673 dose–response relationship Toxicity 0.000 description 1
- 229940079360 enema for constipation Drugs 0.000 description 1
- 238000005516 engineering process Methods 0.000 description 1
- 239000003797 essential amino acid Substances 0.000 description 1
- 235000020776 essential amino acid Nutrition 0.000 description 1
- 150000002148 esters Chemical class 0.000 description 1
- BEFDCLMNVWHSGT-UHFFFAOYSA-N ethenylcyclopentane Chemical compound C=CC1CCCC1 BEFDCLMNVWHSGT-UHFFFAOYSA-N 0.000 description 1
- 235000019325 ethyl cellulose Nutrition 0.000 description 1
- 229920001249 ethyl cellulose Polymers 0.000 description 1
- 229960003750 ethyl chloride Drugs 0.000 description 1
- LVGKNOAMLMIIKO-QXMHVHEDSA-N ethyl oleate Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OCC LVGKNOAMLMIIKO-QXMHVHEDSA-N 0.000 description 1
- 229940093471 ethyl oleate Drugs 0.000 description 1
- 230000029142 excretion Effects 0.000 description 1
- 239000003925 fat Substances 0.000 description 1
- 235000019197 fats Nutrition 0.000 description 1
- 239000000194 fatty acid Substances 0.000 description 1
- 229930195729 fatty acid Natural products 0.000 description 1
- 150000004665 fatty acids Chemical class 0.000 description 1
- 150000002191 fatty alcohols Chemical class 0.000 description 1
- 239000012091 fetal bovine serum Substances 0.000 description 1
- 238000011049 filling Methods 0.000 description 1
- NBVXSUQYWXRMNV-UHFFFAOYSA-N fluoromethane Chemical compound FC NBVXSUQYWXRMNV-UHFFFAOYSA-N 0.000 description 1
- 235000003599 food sweetener Nutrition 0.000 description 1
- 210000001035 gastrointestinal tract Anatomy 0.000 description 1
- 239000011521 glass Substances 0.000 description 1
- 235000001727 glucose Nutrition 0.000 description 1
- 229940075529 glyceryl stearate Drugs 0.000 description 1
- 229940100242 glycol stearate Drugs 0.000 description 1
- 239000008187 granular material Substances 0.000 description 1
- 230000003862 health status Effects 0.000 description 1
- UKACHOXRXFQJFN-UHFFFAOYSA-N heptafluoropropane Chemical compound FC(F)C(F)(F)C(F)(F)F UKACHOXRXFQJFN-UHFFFAOYSA-N 0.000 description 1
- BXWNKGSJHAJOGX-UHFFFAOYSA-N hexadecan-1-ol Chemical compound CCCCCCCCCCCCCCCCO BXWNKGSJHAJOGX-UHFFFAOYSA-N 0.000 description 1
- 229940049290 hydrogenated coco-glycerides Drugs 0.000 description 1
- 235000010979 hydroxypropyl methyl cellulose Nutrition 0.000 description 1
- 239000001866 hydroxypropyl methyl cellulose Substances 0.000 description 1
- 229920003088 hydroxypropyl methyl cellulose Polymers 0.000 description 1
- UFVKGYZPFZQRLF-UHFFFAOYSA-N hydroxypropyl methyl cellulose Chemical compound OC1C(O)C(OC)OC(CO)C1OC1C(O)C(O)C(OC2C(C(O)C(OC3C(C(O)C(O)C(CO)O3)O)C(CO)O2)O)C(CO)O1 UFVKGYZPFZQRLF-UHFFFAOYSA-N 0.000 description 1
- 230000006872 improvement Effects 0.000 description 1
- 239000004615 ingredient Substances 0.000 description 1
- 229940041682 inhalant solution Drugs 0.000 description 1
- 239000003112 inhibitor Substances 0.000 description 1
- 210000000936 intestine Anatomy 0.000 description 1
- 238000007918 intramuscular administration Methods 0.000 description 1
- 238000007912 intraperitoneal administration Methods 0.000 description 1
- 229960001317 isoprenaline Drugs 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- ZLVXBBHTMQJRSX-VMGNSXQWSA-N jdtic Chemical compound C1([C@]2(C)CCN(C[C@@H]2C)C[C@H](C(C)C)NC(=O)[C@@H]2NCC3=CC(O)=CC=C3C2)=CC=CC(O)=C1 ZLVXBBHTMQJRSX-VMGNSXQWSA-N 0.000 description 1
- 210000004731 jugular vein Anatomy 0.000 description 1
- 239000004310 lactic acid Substances 0.000 description 1
- 235000014655 lactic acid Nutrition 0.000 description 1
- 239000003446 ligand Substances 0.000 description 1
- 239000006193 liquid solution Substances 0.000 description 1
- 239000006194 liquid suspension Substances 0.000 description 1
- 230000005923 long-lasting effect Effects 0.000 description 1
- 210000004072 lung Anatomy 0.000 description 1
- ZLNQQNXFFQJAID-UHFFFAOYSA-L magnesium carbonate Chemical compound [Mg+2].[O-]C([O-])=O ZLNQQNXFFQJAID-UHFFFAOYSA-L 0.000 description 1
- 239000001095 magnesium carbonate Substances 0.000 description 1
- 229910000021 magnesium carbonate Inorganic materials 0.000 description 1
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 1
- 235000019341 magnesium sulphate Nutrition 0.000 description 1
- 229960002329 methacholine Drugs 0.000 description 1
- 229920000609 methyl cellulose Polymers 0.000 description 1
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 1
- 239000001923 methylcellulose Substances 0.000 description 1
- 235000010981 methylcellulose Nutrition 0.000 description 1
- 229910000402 monopotassium phosphate Inorganic materials 0.000 description 1
- 235000019796 monopotassium phosphate Nutrition 0.000 description 1
- 210000003205 muscle Anatomy 0.000 description 1
- 210000003928 nasal cavity Anatomy 0.000 description 1
- 210000002850 nasal mucosa Anatomy 0.000 description 1
- 201000009240 nasopharyngitis Diseases 0.000 description 1
- 231100000344 non-irritating Toxicity 0.000 description 1
- 239000004006 olive oil Substances 0.000 description 1
- 235000008390 olive oil Nutrition 0.000 description 1
- 210000000287 oocyte Anatomy 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 229910052760 oxygen Inorganic materials 0.000 description 1
- 239000012057 packaged powder Substances 0.000 description 1
- 238000007911 parenteral administration Methods 0.000 description 1
- 239000002245 particle Substances 0.000 description 1
- 239000000312 peanut oil Substances 0.000 description 1
- 239000003209 petroleum derivative Substances 0.000 description 1
- 230000000144 pharmacologic effect Effects 0.000 description 1
- 229960003742 phenol Drugs 0.000 description 1
- 239000008055 phosphate buffer solution Substances 0.000 description 1
- 150000003904 phospholipids Chemical class 0.000 description 1
- 229920001993 poloxamer 188 Polymers 0.000 description 1
- 239000008389 polyethoxylated castor oil Substances 0.000 description 1
- 229920000573 polyethylene Polymers 0.000 description 1
- 229940113115 polyethylene glycol 200 Drugs 0.000 description 1
- 229940068886 polyethylene glycol 300 Drugs 0.000 description 1
- 229940068918 polyethylene glycol 400 Drugs 0.000 description 1
- 229940057847 polyethylene glycol 600 Drugs 0.000 description 1
- 229940085675 polyethylene glycol 800 Drugs 0.000 description 1
- 235000010486 polyoxyethylene sorbitan monolaurate Nutrition 0.000 description 1
- 239000000256 polyoxyethylene sorbitan monolaurate Substances 0.000 description 1
- 235000010482 polyoxyethylene sorbitan monooleate Nutrition 0.000 description 1
- 229920001451 polypropylene glycol Polymers 0.000 description 1
- 229920000053 polysorbate 80 Polymers 0.000 description 1
- 239000013641 positive control Substances 0.000 description 1
- GNSKLFRGEWLPPA-UHFFFAOYSA-M potassium dihydrogen phosphate Chemical compound [K+].OP(O)([O-])=O GNSKLFRGEWLPPA-UHFFFAOYSA-M 0.000 description 1
- 239000002244 precipitate Substances 0.000 description 1
- 230000002335 preservative effect Effects 0.000 description 1
- 238000012545 processing Methods 0.000 description 1
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 102000004169 proteins and genes Human genes 0.000 description 1
- 108090000623 proteins and genes Proteins 0.000 description 1
- 125000003410 quininyl group Chemical group 0.000 description 1
- 239000002994 raw material Substances 0.000 description 1
- 229940044601 receptor agonist Drugs 0.000 description 1
- 239000000018 receptor agonist Substances 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 230000036387 respiratory rate Effects 0.000 description 1
- 230000000284 resting effect Effects 0.000 description 1
- CVHZOJJKTDOEJC-UHFFFAOYSA-N saccharin Chemical compound C1=CC=C2C(=O)NS(=O)(=O)C2=C1 CVHZOJJKTDOEJC-UHFFFAOYSA-N 0.000 description 1
- 239000008159 sesame oil Substances 0.000 description 1
- 235000011803 sesame oil Nutrition 0.000 description 1
- 239000002356 single layer Substances 0.000 description 1
- 235000017557 sodium bicarbonate Nutrition 0.000 description 1
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 1
- 239000004334 sorbic acid Substances 0.000 description 1
- 235000010199 sorbic acid Nutrition 0.000 description 1
- 229940075582 sorbic acid Drugs 0.000 description 1
- 235000011067 sorbitan monolaureate Nutrition 0.000 description 1
- 235000011078 sorbitan tristearate Nutrition 0.000 description 1
- 230000006641 stabilisation Effects 0.000 description 1
- 238000011105 stabilization Methods 0.000 description 1
- 230000000087 stabilizing effect Effects 0.000 description 1
- 230000000707 stereoselective effect Effects 0.000 description 1
- 239000003270 steroid hormone Substances 0.000 description 1
- 210000002784 stomach Anatomy 0.000 description 1
- 125000001424 substituent group Chemical group 0.000 description 1
- LSNNMFCWUKXFEE-UHFFFAOYSA-L sulfite Chemical compound [O-]S([O-])=O LSNNMFCWUKXFEE-UHFFFAOYSA-L 0.000 description 1
- 229910052717 sulfur Inorganic materials 0.000 description 1
- 238000013268 sustained release Methods 0.000 description 1
- 239000012730 sustained-release form Substances 0.000 description 1
- 239000003765 sweetening agent Substances 0.000 description 1
- 238000001308 synthesis method Methods 0.000 description 1
- 230000002194 synthesizing effect Effects 0.000 description 1
- 230000009885 systemic effect Effects 0.000 description 1
- 229940126585 therapeutic drug Drugs 0.000 description 1
- 238000011285 therapeutic regimen Methods 0.000 description 1
- 210000000115 thoracic cavity Anatomy 0.000 description 1
- 229960000790 thymol Drugs 0.000 description 1
- 230000001550 time effect Effects 0.000 description 1
- 239000012049 topical pharmaceutical composition Substances 0.000 description 1
- 235000010487 tragacanth Nutrition 0.000 description 1
- 239000000196 tragacanth Substances 0.000 description 1
- 229940116362 tragacanth Drugs 0.000 description 1
- QORWJWZARLRLPR-UHFFFAOYSA-H tricalcium bis(phosphate) Chemical compound [Ca+2].[Ca+2].[Ca+2].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O QORWJWZARLRLPR-UHFFFAOYSA-H 0.000 description 1
- JSPLKZUTYZBBKA-UHFFFAOYSA-N trioxidane Chemical class OOO JSPLKZUTYZBBKA-UHFFFAOYSA-N 0.000 description 1
- 239000000341 volatile oil Substances 0.000 description 1
- 239000000080 wetting agent Substances 0.000 description 1
- 239000003871 white petrolatum Substances 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D453/00—Heterocyclic compounds containing quinuclidine or iso-quinuclidine ring systems, e.g. quinine alkaloids
- C07D453/02—Heterocyclic compounds containing quinuclidine or iso-quinuclidine ring systems, e.g. quinine alkaloids containing not further condensed quinuclidine ring systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/13—Amines
- A61K31/135—Amines having aromatic rings, e.g. ketamine, nortriptyline
- A61K31/137—Arylalkylamines, e.g. amphetamine, epinephrine, salbutamol, ephedrine or methadone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/439—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom the ring forming part of a bridged ring system, e.g. quinuclidine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0043—Nose
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/007—Pulmonary tract; Aromatherapy
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/007—Pulmonary tract; Aromatherapy
- A61K9/0073—Sprays or powders for inhalation; Aerolised or nebulised preparations generated by other means than thermal energy
- A61K9/008—Sprays or powders for inhalation; Aerolised or nebulised preparations generated by other means than thermal energy comprising drug dissolved or suspended in liquid propellant for inhalation via a pressurized metered dose inhaler [MDI]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
- A61K9/4808—Preparations in capsules, e.g. of gelatin, of chocolate characterised by the form of the capsule or the structure of the filling; Capsules containing small tablets; Capsules with outer layer for immediate drug release
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/02—Nasal agents, e.g. decongestants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/06—Antiasthmatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/14—Antitussive agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/02—Drugs for disorders of the urinary system of urine or of the urinary tract, e.g. urine acidifiers
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/10—Drugs for disorders of the urinary system of the bladder
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- Public Health (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Organic Chemistry (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Epidemiology (AREA)
- Pulmonology (AREA)
- Otolaryngology (AREA)
- Urology & Nephrology (AREA)
- Emergency Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
Compuesto de fórmula I:**Fórmula** o una sal, solvato o isómero óptico de la misma farmacéuticamente aceptables, en donde en la fórmula I: n se selecciona entre 1-7, R1 es un hidrocarbilo C3-C7, que puede estar no sustituido u opcionalmente sustituido por halógeno, alcoxi, alcoxihidrocarbilo, heterociclilo o arilo, R2 es un arilo o un heteroarilo que contiene uno o más heteroátomos, que puede estar no sustituido u opcionalmente sustituido, R3 es un hidroxilo, halógeno, alcoxi o aciloxi, en donde el alcoxi o aciloxi puede estar no sustituido u opcionalmente sustituido por halógeno, hidroxilo, alcoxi, hidrocarbilo, alcoxihidrocarbilo, heterociclilo o arilo; R4 y R5 pueden estar presentes o ausentes, y se seleccionan de forma independiente de un grupo que consiste en halógeno, hidroxilo, alcoxi, hidrocarbilo, alcoxihidrocarbilo, heterociclilo o arilo, cuando están presentes; Y es un alquileno C1-C7, lineal o ramificado, o -(CH2-O-CH2)m-, que puede estar opcionalmente sustituido, en donde m es igual a 1-3, X- es un radical ácido o hidroxilo.
Description
DESCRIPCIÓN
Compuestos de quinina e isómeros ópticos, procedimiento de preparación y uso médico de los mismos Campo de la técnica
La presente invención se refiere a compuestos de quinina, isómeros ópticos y procedimientos de fabricación de los mismos y composiciones que comprenden estos compuestos con fines medicinales y, en particular, a nuevos antagonistas del receptor M que tienen un efecto selectivo sobre subtipos del receptor M, que tienen un fuerte efecto sobre los subtipos receptores M3 y M1, pero no tienen ningún efecto significativo sobre el subtipo receptor M2.
Antecedentes de la técnica relacionada
En algunos documentos de patente se encuentra que un compuesto que comprende una estructura de quinina se utiliza para el efecto anti-colinérgico. Por ejemplo, un compuesto divulgado en la patente de invención china CN200810112248.1 y en la patente de invención china CN200910223255.3 (número de publicación CN102070631 A) tiene una estructura como la siguiente,
en donde: R es metilo, etilo, propilo, isopropilo, o ciclopropilo; y X representa un átomo de halógeno.

La patente francesa FR2012964 divulgó una estructura como la siguiente, en donde: R es un átomo de H, hidroxilo o alquilo con 1-4 átomos de carbono; R1 es fenilo o tienilo; y R2 es ciclohexilo, ciclopentilo o tienilo.

La patente de EE.UU. documento US5654314 divulgó una estructura como la siguiente:

La patente WO con documento WO01/04118 divulgó una estructura como la siguiente:

Los compuestos anteriores tienen desventajas significativas, tales como eficacia de corta duración, acción lenta, o efectos tóxicos y secundarios significativos, o similares, en el tratamiento de rinitis, rinitis después de un resfriado, traquitis crónica, hipersensibilidad de las vías respiratorias, asma, enfermedades pulmonares obstructivas crónicas, tos, incontinencia urinaria, micción frecuente, síndrome de vejiga inestable, espasmos vesicales, inflamación de la vejiga y enfermedades gastrointestinales como el síndrome del intestino irritable, colitis espástica, así como úlceras duodenales y gástricas.
Los compuestos de la presente invención superan las desventajas de los compuestos anteriores, y en particular, se caracterizan por una mayor eficacia, acción rápida y menores efectos tóxicos y secundarios en el tratamiento de la traquitis crónica, las hipersensibilidades de las vías respiratorias, asma y enfermedades pulmonares obstructivas crónicas en comparación con los compuestos de la técnica anterior. Debido a su buena estabilidad, los compuestos de la presente invención son adecuados para la fabricación de un inhalante que es administrado una vez al día para tratar enfermedades pulmonares obstructivas crónicas y, en particular, adecuado para fabricar un aerosol de
inhalación de dosis medida de tipo solución que es administrado una vez al día. La presente invención se refiere a la síntesis de dicho compuesto, comprendiendo la fabricación de una composición farmacéutica que comprende dicho compuesto y los usos farmacéuticos del mismo.
El compuesto de la presente invención puede utilizarse también para tratar las enfermedades respiratorias anteriores como rinitis, rinitis después de un resfriado, traquitis crónica, hipersensibilidad de las vías respiratorias, asma, enfermedades pulmonares obstructivas crónicas, y similares, en combinación con agonistas del receptor p2, hormona esteroidea, fármacos antialérgicos, fármacos antiinflamatorios, fármacos antiinfecciosos, antagonistas de la fosfolipasa i.v. y similares.
Contenido de la invención
Un nuevo compuesto antagonista selectivo de subtipos del receptor M de la presente invención puede representarse por la estructura de la fórmula (I):

en donde en la fórmula (I):
n se selecciona entre 1-7, preferiblemente 1-3, lo más preferiblemente 1.
R1 es un hidrocarbilo C3-C7 , que puede estar no sustituido u opcionalmente sustituido por halógenos, alcoxi, alcoxihidrocarbilo, heterociclilo o arilo; preferiblemente un cicloalquilo no sustituido, y lo más preferiblemente un ciclopentilo o ciclohexilo.
R2 es un arilo o un heteroarilo que contiene uno o más heteroátomos (el heteroátomo puede ser N, O o S), por ejemplo, R2 es fenilo, naftilo o bifenilo, que puede estar no sustituido u opcionalmente sustituido por uno o más de entre halógeno, hidroxilo, fenilo, -OR6 , -SR6 , -NR6R7 , -NHCOR6 , -CONR6R7 , -CN, -NO2 , -COOR6 , -CF3 o hidrocarbilo C1-C4 , lineal o ramificado; se prefieren fenilo, piridilo, furilo y tienilo no sustituidos; R6 y R7 pueden ser un átomo de hidrógeno, un hidrocarbilo C1-C4 lineal o ramificado, o pueden formar un ciclohidrocarbilo entre sí.
R3 es un hidroxilo, halógeno, alcoxi o aciloxi, en donde el alcoxi o aciloxi puede estar no sustituido u opcionalmente sustituido por halógeno, hidroxilo, alcoxi, hidrocarbilo, alcoxihidrocarbilo, ciclohidrocarbilo, heterociclilo o arilo; preferiblemente un hidroxilo o metoxilo y lo más preferiblemente un hidroxilo.
R4 y el R5 pueden estar presentes o ausentes, y respectivamente, cuando están presentes, pueden ser sustituyentes como halógenos, hidroxilo, hidrocarbiloxi, hidrocarbilo, hidrocarbiloxihidrocarbilo, heterociclilo y arilo.
Y es un alquileno C1-C7 , lineal o ramificado, o -(CH2-O-CH2V (donde m es igual a 1-3), que puede sustituirse opcionalmente, preferiblemente, sustituirse por halógeno, hidroxilo, alcoxi, alcoxialquilo, hidrocarbilo insaturado, ciclohidrocarbilo, o heterociclilo; preferiblemente un metileno, etileno, propileno o -(CH2-O-CH2)-; y, lo más preferiblemente, un etileno o propileno.
X- es un radical ácido o un hidróxido, preferiblemente un radical ácido farmacéuticamente aceptable, cuyos ejemplos incluyen una sal derivada de un ácido inorgánico, como hidrocloruro, bromuro, yoduro, nitrato, carbonato, bicarbonato, fosfato, hidrofosfato, fosfato dihidroxilado, sulfato, disulfato, sulfito, hidrosulfito o fosfito; y una sal derivada de un ácido orgánico relativamente no tóxico como, pero no limitado a, ácido acético, ácido propiónico, ácido isobutírico, ácido malónico, ácido benzoico, ácido succínico, ácido subérico, ácido fumárico, ácido mandélico, ácido ftálico, ácido bencensulfónico, ácido p-toluensulfónico, ácido cítrico, ácido tartárico, ácido metansulfónico, ácido glucurónico o ácido galactónico o similar. Los ejemplos incluyen además una sal de un aminoácido como arginina o similar.
El compuesto representado por la fórmula (I) puede comprender uno o más centros quirales, en los que un único isómero óptico o una mezcla de varios isómeros ópticos entran dentro del alcance reivindicado por la presente invención.
Los siguientes compuestos pueden ilustrar específicamente el contenido de la presente invención pero no limitan el alcance de la presente invención.
1. Bromuro de (2S,3R)-3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano
2. Bromuro de (2S,3S)-3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano
3. Bromuro de (2R,3R)-3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano
4. Bromuro de (2R,3S)-3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano
5. Bromuro de (2S,3R),(2R,3S)-3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano 6. Bromuro de (2R,3R),(2S,3S)-3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano 7. Bromuro de (2S,3R)-3-[(2-ciclohexil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1 -azabiciclo[2,2,2]octano
8. Bromuro de (2S,3S)-3-[(2-ciclohexil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1 -azabiciclo[2,2,2]octano
9. Bromuro de (2R,3R)-3-[(2-ciclohexil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1 -azabiciclo[2,2,2]octano
10. Bromuro de (2R,3S)-3-[(2-ciclohexil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1 -azabiciclo[2,2,2]octano
11. Bromuro de (2R,3S),(2S,3R)-3-[(2-ciclohexil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano 12. Bromuro de (2R,3R),(2S,3S)-3-[(2-ciclohexil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano 13. Bromuro de (2S,3R)-3-[(2-ciclobutil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano
14. Bromuro de (2S,3S)-3-[(2-ciclobutil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano
15. Bromuro de (2R,3S)-3-[(2-ciclobutil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano
16. Bromuro de (2R,3R)-3-[(2-ciclobutil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano
17. Bromuro de (2R,3S),(2S,3R)-3-[(2-ciclobutil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano 18. Bromuro de (2R,3R),(2S,3S)-3-[(2-ciclobutil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano 19. Bromuro de (2S,3R)-3-[(2-ciclopropil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1 -azabiciclo[2,2,2]octano 20. Bromuro de (2R,3S)-3-[(2-ciclopropil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1 -azabiciclo[2,2,2]octano 21. Bromuro de (2S,3S)-3-[(2-ciclopropil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano 22. Bromuro de (2R,3R)-3-[(2-ciclopropil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1 -azabiciclo[2,2,2]octano 23. Bromuro de (2R,3S),(2S,3R)-3-[(2-ciclopropil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano 24. Bromuro de (2R,3R),(2S,3S)-3-[(2-ciclopropil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano 25. Bromuro de (2S,3R)-3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-(2-fenoxietil)-1-azabiciclo[2,2,2]octano
26. Bromuro de (2R,3R)-3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-(2-fenoxietil)-1-azabiciclo[2,2,2]octano
27. Bromuro de (2R,3R)-3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-fenoximetil-1-azabiciclo[2,2,2]octano
28. Cloruro de (2R,3R)-3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-fenoximetoximetil-1 -azabiciclo[2,2,2]octano 29. Bromuro de (2R,3R)-3-[(2-ciclopentil-2-hidroxil-2-naftil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano 30. Bromuro de (2R,3R)-3-[(2-ciclopentil-2-hidroxil-2-(o-clorofenil))etoxil]-1-(3-fenoxipropil)-1 -azabiciclo[2,2,2]octano 31. Bromuro de (2R,3R)-3-[(2-ciclopentil-2-hidroxil-2-(3-piridil))etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano 32. Bromuro de (2R,3R)-3-[(2-ciclopentil-2-hidroxil-2-(2-furil))etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano 33. Bromuro de (2R,3R)-3-[(2-ciclopentil-2-metoxil-2-(3-piridil))etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano
La presente invención proporciona un esquema de proceso para sintetizar el compuesto de fórmula estructural (I) de la siguiente manera:
Etapa 1: (1-R1-1-R2)-oxirano se preparó según los procedimientos descritos en la bibliografía (1-2) (1. Guangling Wen, Peijin Wu, Improvement on the synthesis method of 3-(2-phenyl-2-cyclopentylethoxyl) quinuclidine hydrochloride, Bullet of the Academy of Millitary Medical Sciences, 1988: 470 de vuelta a 402; 2. Peijin Wu, Liuhong Yun, Synthesis of anticholinergic drug of 2-(1-naftil)-2-cyclopentyl-2-hydroxylethoxyl cyclohydrocarbyl amine compounds, Chinese Journal of Medicinal Chemistry 1999.6, 9(2): pág. 102-105), y arilhidrocarbilcetona (algunos tipos de arilhidrocarbilcetona se generaron por reacción de cianuro de arilo con un reactivo de Grignard preparado por reacción de bromuro de hidrocarbilo con magnesio en THF, véase la fórmula (1) a continuación) se hizo reaccionar con presencia de sulfato de dimetilo, sulfuro de dimetilo e hidruro de sodio para generar 1-aril-1-hidrocarbiloxirano, es decir, el intermedio 1 (véase la fórmula (2) a continuación).
(1)
R?^CN Ri— Br

(2)

Etapa 2: Preparación de 3-[(2-R1-2-R2-2-hidroxil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali
El intermedio 2 pudo obtenerse por reacción del intermedio 1 con quinuclidinol (o quinuclidinol sustituido con R4) bajo la condición de NaH.

A los derivados de 3-quinuclidinol comercialmente disponibles se añadió DMSO, seguido por la adición de hidruro de sodio, y la mezcla se hizo reaccionar a 20-60 °C, y después se enfrió a temperatura ambiente, y a ello se añadió una solución de (1-R1-1-R2)-oxirano (auto-preparado) en DMSO, y una vez finalizada la adición, la mezcla resultante se calentó a 20-70 °C en baño de aceite para reaccionar durante 0,5-12 horas. Bajo la condición de baño de hielo, se añadió agua helada a una temperatura interna de 30 °C o menor. La mezcla de reacción se extrajo con éter isopropílico, y la capa de éter se combinó y se lavó con una solución acuosa saturada de NaCl. La capa orgánica se secó sobre sulfato de sodio anhidro durante la noche, el agente desecante se retiró por filtración, y el disolvente se retiró por evaporación rotativa, para obtener el intermedio 2 como una materia aceitosa de color rojo.
Etapa 3: Purificación de 3-[(2-R1-2-R2-2-hidroxil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali por cromatografía de columna y el tratamiento de purificación relacionado
Una muestra del intermedio 2 anterior se separó en una columna de gel de sílice, utilizando diclorometano o triclorometano y metanol, amoniacados, como fase móvil y una placa de TLC para controlar la pureza de la muestra. El intermedio 2 sería una mezcla que contiene diferentes estructuras ópticas dependiendo de la estructura de isómeros ópticos de quinuclidinol, y bajo el sistema de elución anterior, si el quinuclidinol estaba en configuración S, el intermedio 2 comprendería dos tipos de configuraciones (2R,3S) y (2S,3S), y podía ser purificado en dos tipos libre de álcali, (2R,3S) y (2S,3S), dependiendo de la secuencia de elución; si quinuclidinol estaba en configuración R, el intermedio 2 comprendería dos tipos de configuraciones (2R,3R) y (2S,3R), y podía ser purificado en dos tipos libre de álcali, (2S,3r ) y (2R,3R), dependiendo de la secuencia de elución; si quinuclidinol era un racemato, el intermedio 2 comprendería cuatro tipos de configuraciones (2R,3S), (2S,3S), (2R,3R) y (2S,3R), y podía ser purificada en dos tipos libre de álcali, (2R,3S),(2S,3R) y (2S,3S),(2R,3R), dependiendo de la secuencia de elución. El producto separado libre de álcali se refirió como intermedio 3.
Etapa 4: Preparación de 3-Z-Y-oxilbenceno (intermedio 4)

En un matraz de tres bocas se añadió fenol, seguido de la adición de hidróxido de sodio y una solución de Z-Y-Z (donde Z era un átomo de halógeno) en etanol absoluto, y después la mezcla resultante se hizo reaccionar bajo calentamiento y reflujo en baño de aceite, precipitándose un sólido blanco, hasta reacción esencialmente completa de fenol controlada por TLC (condiciones de la TLC: éter de petróleo/acetato de etilo = 5,0 ml/1,0 ml). Una vez finalizada la reacción, el sólido se retiró por filtración, y el disolvente se retiró del filtrado a 50 °C o menor bajo presión reducida mediante una bomba de agua, obteniéndose de ese modo una materia aceitosa que contenía un sólido blanco, al que se añadió éter de petróleo y después se dejó reposar durante la noche. El sólido precipitado se retiró por filtración, y el disolvente se retiró del filtrado a 25-40 °C bajo presión reducida mediante una bomba de agua, obteniéndose de ese modo una materia aceitosa de color amarillo. La materia aceitosa se destiló bajo presión reducida, para recoger una materia aceitosa incolora, transparente, es decir, el intermedio 4.
Etapa 5: Preparación del compuesto de la fórmula (I)
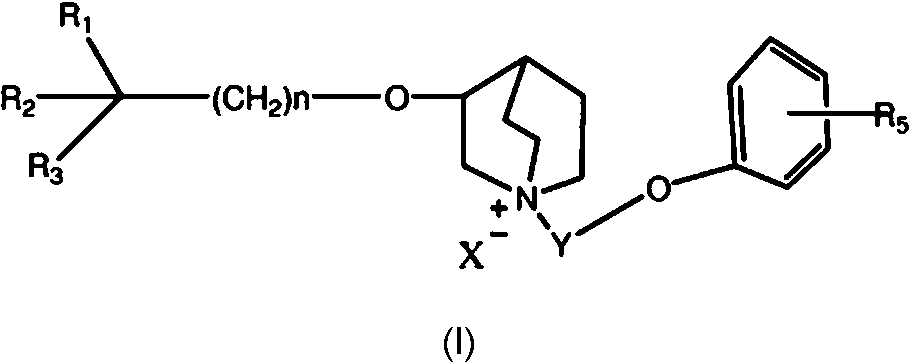
El intermedio 2 o el intermedio 3 se añadieron en un matraz, con forma de berenjena, seguido de la adición de cloroformo, para obtener una solución transparente de color amarillo, a la que se añadieron intermedio 4 y acetonitrilo, y la mezcla resultante se agitó a continuación a temperatura ambiente para reaccionar durante 10-90 h
bajo la protección de nitrógeno, siendo controlado el final de la reacción mediante TLC (condiciones de TLC: cloroformo/metanol/amoníaco en agua = 5,0 ml/1,5 ml/2 d). Una vez finalizada la reacción, el disolvente se retiró de la solución de reacción por evaporación rotativa a 25-40 °C bajo presión reducida mediante una bomba de agua, obteniéndose de ese modo una materia aceitosa de color amarillo, a la que se añadió éter etílico para precipitar una gran cantidad de sólido, y el sólido se recogió mediante filtración por succión para obtener un sólido de color blancuzco, es decir, el compuesto del título de fórmula (I).
El compuesto anterior de fórmula (I) se hizo reaccionar con Ag2Ü para reemplazar el halógeno con hidróxido, que podría después transformarse en otro radical ácido al reaccionar con otro ácido. Ejemplos de una sal de un ácido farmacéuticamente aceptable incluyen una sal derivada de un ácido inorgánico, como hidrocloruro, bromuro, yoduro, nitrato, carbonato, bicarbonato, fosfato, hidrofosfato, fosfato dihidroxilado, sulfato, disulfato o fosfito o similar; y una sal derivada de un ácido orgánico relativamente no tóxico como ácido acético, ácido propiónico, ácido isobutírico, ácido malónico, ácido benzoico, ácido succínico, ácido subérico, ácido fumárico, ácido mandélico, ácido ftálico, ácido bencensulfónico, ácido p-toluensulfónico, ácido cítrico, ácido tartárico, ácido metansulfónico, ácido glucurónico o ácido galactónico o similar. Los ejemplos incluyen además una sal de un aminoácido como arginina o similar.
Cualquier composición farmacéutica que comprenda uno o más compuestos de la fórmula (I) descritos anteriormente está dentro del alcance reivindicado por la presente invención, y la ruta de administración puede ser, por ejemplo, administración oral, tópica, intravenosa, intramuscular, endoarterial, intraperitoneal, rectal, vaginal, endonasal o por inhalación. La formulación de la presente invención puede diseñarse para que sea de acción rápida, liberación rápida o de acción prolongada. Además, el compuesto puede ser administrado por una ruta tópica en lugar de una ruta sistémica y según formas de realización representativas, la composición de la presente invención puede formularse para fármacos administrados para mamíferos, preferentemente humanos.
La composición que comprende uno o más compuestos de la presente invención y excipientes apropiados puede administrarse repetidamente, o la composición se puede administrar de forma continua. Los sitios adecuados de administración incluyen, pero no se limitan a, cavidad nasal, pulmón, vasos sanguíneos, músculos, bronquios, e intestinos y estómago. La formulación puede estar en la forma de una forma de administración líquida, polvo liofilizado, sólido o semisólido, tal como solución, suspensión, emulsión, comprimido, píldora, cápsula, productos pulverizados, supositorio, enemas retentivos, aerosol, aerosol en polvo, o similar, preferiblemente una forma de administración unitaria adecuada para simplemente administrar una dosis exacta. Los ejemplos de excipientes apropiados incluyen, pero no se limitan a, agua, solución salina, lactosa, glucosa, sacarosa, sorbitol, manitol, almidón, goma arábiga, fosfato de calcio, alginato, tragacanto, gelatina, silicato de calcio, celulosa microcristalina, polivinilpirrolidona, celulosa, jarabe, metilcelulosa, etilcelulosa, hidroxipropilmetilcelulosa y poli(ácido acrílico). La composición puede comprender además lubricante como talco, estearato de magnesio y aceite mineral; agente humectante; emulgente; agente de suspensión; conservante tal como metil-, etil-y propil-hidroxilbenzoato; regulador del pH como ácidos y bases inorgánicos y orgánicos; agente edulcorante; y corrector.
Para la administración parenteral, la composición puede estar en la forma de una inyección estéril y polvo envasado asépticamente. Preferiblemente, la inyección se formula a pH 4,5-7,5.
La composición farmacéutica de la presente invención puede ser cualquiera de las formas de administración aceptables por vía oral, incluyendo comprimidos, cápsulas, pastillas, emulsión, suspensión, solución, jarabe, elixir, nebulosa, píldora, troche, productos pulverizados, gránulo y preparado de liberación sostenida. Excipientes adecuados para la administración oral incluyen manitol de calidad farmacéutica, lactosa, almidón, estearato de magnesio, sacarina sódica, talco, celulosa, glucosa, gelatina, sacarosa, carbonato de magnesio y similares. En el caso de comprimidos para administración oral, los portadores normalmente usados incluyen lactosa y celulosa microcristalina, y generalmente se añade un lubricante como estearato de magnesio; para las cápsulas, los diluyentes útiles incluyen lactosa y almidón de maíz seco; cuando se requiere suspensión para administración oral, los ingredientes activos se mezclan con emulgente y agente de suspensión, y también se pueden añadir algunos agentes edulcorantes, correctores o colorantes según corresponda.
Por ejemplo, polvo, solución o suspensión para inhalación pulmonar, pulverizador nasal, administración oral, tópica o intravenosa pueden formarse disolviendo o dispersando uno o más compuestos de la presente invención y, opcionalmente, uno o más materiales auxiliares farmacéuticamente aceptables en un vehículo como una solución salina, solución acuosa de glucosa, glicerol, etanol o similar. Se prepara la composición líquida, y la formulación farmacéutica en forma de suspensión líquida o solución puede prepararse utilizando líquido estéril como aceite, agua, etanol y una combinación de los mismos; para inhalación pulmonar, pulverizador nasal, administración oral o intravenosa, se puede añadir un tensioactivo, agente de suspensión o emulgente farmacéuticamente adecuados; una suspensión puede comprender aceite, como aceite de cacahuete, aceite de sésamo, aceite de semilla de algodón, aceite de maíz y aceite de oliva; una formulación de suspensión también puede comprender ésteres de ácidos grasos, como el oleato de etilo, miristato de isopropilo, glicérido graso y glicérido graso acetilado. Una formulación de suspensión puede comprender alcohol, como etanol, isopropanol, hexadecanol, glicerol y propanodiol; éter, como el polietilenglicol; hidrocarburo del petróleo, como aceite mineral y vaselina. También se puede utilizar agua en una formulación de suspensión.
La composición puede estar en forma de píldora, comprimido o cápsula y, por lo tanto, la composición puede comprender uno o más diluyentes, tales como lactosa, sacarosa, fosfato dicálcico y similares; un disgregante, como
almidón o derivados del mismo; un lubricante, como estearato de magnesio y similares; y/o un aglutinante, como almidón, goma arábiga, polivinilpirrolidona, gelatina, celulosa y derivados de los mismos. La píldora, comprimido o cápsula puede ser preparada por cualquier procedimiento conocido por los expertos en la técnica.
Alternativamente, la composición farmacéutica de la presente invención puede estar en forma de supositorio para la administración rectal. Estos supositorios pueden prepararse mezclando un medicamento con un excipiente no irritante adecuado que sea sólido a temperatura ambiente pero líquido a la temperatura rectal y, así, liberar el medicamento en el recto. Estos materiales incluyen manteca de cacao, cera de abejas, polietilenglicol, estearato de glicerilo y/o cocoglicéridos hidrogenados. La composición adecuada para administración rectal puede comprender también una unidad de enema rectal, que comprende uno o más compuestos de la presente invención y un vehículo farmacéuticamente aceptable (p. ej., solución acuosa de etanol al 50 % o solución salina), y tal vehículo es fisiológicamente compatible con el recto y/o el colon. Una unidad de enema rectal comprende una punta de aplicador protegida por una cubierta inerte, que está preferiblemente compuesta de polietileno, lubricada con un lubricante como vaselina blanca y, preferiblemente, protegida por una válvula antirretorno para evitar el reflujo del medicamento distribuido. La unidad de enema rectal tiene además una longitud suficiente, preferiblemente de 2 pulgadas (5,08 cm), para que se inserte en el colon a través del ano.
La composición farmacéutica de la presente invención también puede estar en forma de administración tópica, especialmente cuando los objetivos terapéuticos incluyen regiones u órganos en los que la composición farmacéutica puede administrarse fácilmente mediante la administración tópica, y las enfermedades de estos órganos incluyen enfermedades de pulmón, de la mucosa nasal y de la tráquea. Las formulaciones tópicas adecuadas para estas regiones u órganos se preparan fácilmente. Para la administración tópica, la composición que comprende uno o más compuestos de la presente invención puede estar en forma de pulverizador nasal, inhalación de solución, inhalación de polvo con dosis medida, inhalación de solución con dosis medida, inhalación de suspensión con dosis medidas y similares.
Para la administración por inhalación, la composición en forma de polvo seco o líquido puede ser entregada mediante un pulverizador. Tales composiciones se preparan según las tecnologías conocidas en el campo de la formulación farmacéutica, y la composición en forma de líquido se puede preparar en solución salina con fenilcarbinol u otros conservantes adecuados, absorbentes que realzan la biodisponibilidad, fluorocarbono y/u otro solubilizante o dispersante convencional.
En el aerosol para inhalación de dosis medida de tipo solución que comprende uno o más compuestos de fórmula (I), el disolvente latente incluye uno o más de entre etanol absoluto, glicerol y dioles, o una mezcla de los mismos, en donde los dioles incluyen, pero no se limitan a, etilenglicol, propanodiol, polietilenglicol 200, polietilenglicol 300, polietilenglicol 400, polietilenglicol 600, polietilenglicol 800 y similares. El propelente en el aerosol anterior incluye uno de entre tetrafluoroetano (HFA-134a) y heptafluoropropano (HFA-227ea), o una mezcla de los mismos. El tensioactivo en el aerosol anterior incluye uno o más de entre ácido oleico; oligómero de ácido láctico (OLA); sorbitanos, tales como Span 20, Span 65, Span 80, Span 85; polioxietilensorbitanos, como Tween 20, Tween 80; alcoholes grasos de polioxietileno, como Brij 30, Brij 35, Cremophor; copolímero de polioxietileno y polioxipropileno, como Pluronic F-68; estearato de polietilenglicol, como Solutol HS15; fosfolípidos, como granulestén y lecitina. Se prefieren ácido oleico, lecitina o una mezcla de los mismos. En el aerosol, el contenido de dicho compuesto de fórmula I es 0,005~1 % en peso, preferiblemente 0,02~0,5 %. El contenido del disolvente latente en el aerosol para inhalación es de 5~40 % en peso, preferiblemente 17,5-29,975 %. El contenido del tensioactivo en el aerosol para inhalación es de 0~5 % en peso, preferiblemente 0,005~2 %. El contenido del propelente en el aerosol para inhalación es de 54~90 % en peso, preferiblemente 70~80 %.
En la inhalación en polvo de dosis medida que comprende uno o más compuestos de la fórmula (I), dicho portador inerte comprende un diluyente y un lubricante, en donde dicho diluyente es uno o más de entre glucano, arabinosa, lactosa, manitol, xilitol, sacarosa, fructosa, sorbitol, maltosa, aminoácido y glucosa, o una mezcla de los mismos, y dicho lubricante es estearato de magnesio o benzoato de sodio.
En naristillae o aerosol nasal de dosis medida que comprende uno o más compuestos de fórmula (I), dicho portador inerte es uno o más seleccionados de cloruro de benzalconio, bromuro de benzalconio, fenilcarbinol, ácido benzoico, tricloro-t-butanol, benzoatos de p-hidroxilo, ácido sórbico, fenol, timol y aceite volátil, o una mezcla de los mismos.
Además, la presente invención proporciona usos de dicha composición farmacéutica que pueden utilizarse para la fabricación de medicamentos para prevenir y tratar diversas enfermedades obstructivas agudas o crónicas de las vías respiratorias, como las enfermedades pulmonares obstructivas crónicas, asma bronquial; así como rinitis aguda o crónica y rinitis después de un resfriado en mamífero y humano.
El compuesto de fórmula (I) y otros ingredientes activos (p. ej., dipropionato de beclometasona, clortrimetón, nafazolina o fenoterol) se formulan en una combinación, que se puede utilizar para el tratamiento de varias enfermedades obstructivas, agudas o crónicas, de las vías respiratorias, como las enfermedades pulmonares obstructivas crónicas, el asma bronquial y varias rinitis.
Para obtener efectos terapéuticos de forma rápida y eficaz, y garantizar los efectos no tóxicos y secundarios en uso, se sugiere que la dosificación diaria del compuesto de la presente invención utilizada debería ser 10-1.000 pg y, de forma óptima, 40-500 pg.
Además de las formas de dosificación representativas, los expertos en la técnica saben también que, en la presente invención, se incluyen, generalmente, otros excipientes, portadores y formas de administración farmacéuticamente aceptables . Se debe entender que la administración específica y el régimen terapéutico para cualquier paciente concreto dependen de diversos factores que incluyen la actividad del compuesto concreto utilizado, la edad, peso corporal, estado de salud general, género, estado dietético del paciente, tiempo de administración, y la tasa de excreción, fármacos de combinación, diagnóstico de terapeutas y la gravedad de la enfermedad concreta a tratar. La cantidad de ingredientes activos puede depender además del compuesto específico y otros fármacos terapéuticos (si están presentes) en la composición.
Descripción detallada de las formas de realización
El contenido de la invención se describe, a continuación, en detalle, mediante los ejemplos de los compuestos e isómeros ópticos puros de los mismos. Debe aclararse que la presente invención no se limita a los ejemplos que se indican a continuación. La proporción de componentes en los ejemplos se expresa en peso, a menos que se indique específicamente lo contrario.
Preparación de compuestos de Ejemplo:
Ejemplo 1. Bromuro de (2S,3R)-3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxi propil)-1-azabiciclo[2,2,2]octano
Etapa 1: Preparación de 1-fenil-1-ciclopentiloxirano
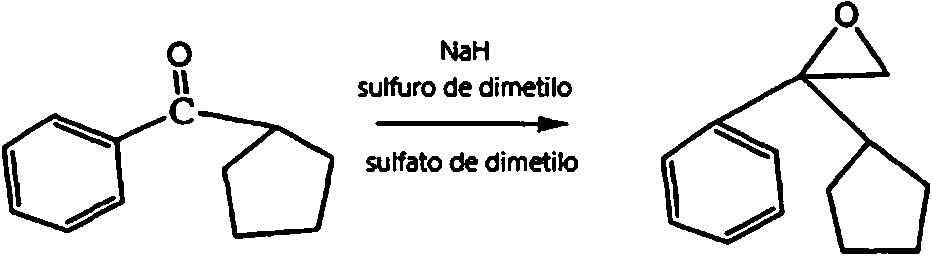
1-fenilo-1-ciclopentiloxirano se obtuvo por reacción de ciclopentilfenilcetona comercialmente disponible como material de partida según la bibliografía(1).
Etapa 2: Preparación de 3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali a partir de S-3-quinuclidinol

A 18,72 g (147 mmol) de S-3-quinuclidinol disponible comercialmente se añadieron 190 ml de DMSO, seguido de la adición de 7,56 g (190 mmol) de hidruro de sodio, y la mezcla se hizo reaccionar a 20-60 °C durante 0,5-12 h, y después se enfrió a temperatura ambiente, y a ello se añadió una solución de 35,72 g (190 mmol) de 1 -fenil-1 -ciclopentiloxirano (autopreparado) en 45 ml de DMSO, y una vez finalizada la adición, la mezcla resultante se calentó a 20-70 °C en baño de aceite para reaccionar durante 0,5-12 horas. Bajo la condición de baño de hielo, se añadieron 120 ml de agua helada a una temperatura interna de 30 °C o menor. La mezcla de reacción se extrajo con éter isopropílico, 100 ml x 3; y la capa de éter se combinó y se lavó con una solución acuosa saturada de NaCl, 100 ml x 3. La capa orgánica se secó sobre sulfato de sodio anhidro durante la noche, el agente desecante se retiró por filtración, y el disolvente se retiró a presión reducida, para obtener 55,7 g de 3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali como materia aceitosa de color rojo, con un rendimiento de 97,39 %. El producto obtenido fue el de las configuraciones (2R,3S),(2S,3S) de 3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali.
Etapa 3: Purificación de 3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali por cromatografía de columna y el tratamiento de purificación relacionado
La bibliografía(3) (3. Bingdahl B, Resul B y Dahlbom R. Facile preparation of the enantiomers of 3-acetoxiquinuclidine and 3-quinuclidinol. Acta Pharm Suec, 1979; 16: 281-283), y otra bibliografía(4-5) (4. Jianhua Gao, Guangling Wen, Qikai Zhang, Synthesis and separation of optically pure hydroxyl ether compounds. Acta Pharmaceutica Sinica, 1987; 22(9): 708-710; 5. Xiangyu Han. Study on the stereoselective synthesis of chiral M receptor antagonists. Informes de Postdoctoral Research Station en la Academy of Military Medical Sciences, pág. 39-40, 2005, Pekín (Medical Library of Chinese PLA: R914, 20050537)) informaron que la configuración quiral R o S de 3-quinuclidinol como materia prima se hizo reaccionar con racemato de 2-fenil-2-ciclopentiloxirano o enantiómeros R o S del mismo, y se podían obtener cuatro isómeros ópticos por cromatografía de columna. La bibliografía anterior ha determinado sus configuraciones absolutas, y según la nomenclatura, el átomo de carbono quiral en la quinuclidina de la molécula se designa como "3", y el carbono quiral ligado a arilo se designa como "2". La Tabla 1 son datos de rotación específica de cuatro tipos de isómeros ópticos quirales puros de 3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali.
Tabla 1. Datos de configuración absoluta y rotación específica del compuesto

Condiciones de medición de rotación específica: la temperatura es de 26 °C, el disolvente es metanol, y la concentración de medición es 0,4 %-1 %.
La muestra de la etapa 2 anterior se separó en columna de gel de sílice, utilizando diclorometano o triclorometano y metanol, amoniacados, como fase móvil y una placa de TLC para controlar la pureza de la muestra. Bajo el sistema de elución anterior, una muestra de una mezcla de ambas configuraciones (2R,3S) y (2S,3S) de 3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali podía ser purificada en dos tipos, (2R,3S) y (2S,3S), libre de álcali dependiendo de la secuencia de elución, obteniéndose de ese modo 23,5 g de (2R,3S)-3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali con un rendimiento de 86,46 %, y 21,1 g de (2S,3S)-3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali con un rendimiento de 77,63 %. El valor medido [a]D26 de la rotación específica de (2R,3S) fue de 43,95, y el valor medido de la rotación específica [a]D26 de (2S,3S) fue de -9,33.
La síntesis e identificación de otras bases de isómeros ópticos puros fueron conforme a los procedimientos de síntesis y separación de los anteriores isómeros ópticos puros de 3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali, y una vez que la base fue cuaternizada, se obtendría el compuesto de título, y el procedimiento de preparación de varios compuestos del título se aclararía, a continuación, en detalle mediante los ejemplos específicos de la síntesis de los compuestos específicos a continuación. Además, cuando se utilizó quinuclidinol racémico como material de partida, la base obtenida fue una mezcla de cuatro tipos de isómeros ópticos antes de la separación por cromatografía de columna, y el compuesto final del título fue también una mezcla de cuatro tipos de isómeros ópticos. Todos estos isómeros ópticos puros y mezclas de isómeros ópticos en diferentes proporciones están dentro del alcance de la presente invención.
Etapa 4: Preparación de 3-bromopropoxibenceno

A un matraz de 150 ml, con tres bocas, se añadieron 9,507 g (101 mmol) de fenol, seguido de la adición de 4,253 g (106 mmol) de hidróxido de sodio y una solución de 52,17 g (258 mmol) de 1,3-dibromopropano en 30 ml de etanol absoluto, y la mezcla resultante se hizo reaccionar después bajo calentamiento y reflujo en baño de aceite, precipitándose un sólido blanco, hasta reacción esencialmente completa de fenol controlada por TLC (condiciones de TLC: éter de petróleo/acetato de etilo = 5,0 ml/1,0 ml). Una vez finalizada la reacción, el sólido se retiró por filtración, y el disolvente se retiró del filtrado a 50 °C o menor bajo presión reducida mediante una bomba de agua, obteniéndose de ese modo una materia aceitosa que contenía un sólido blanco, al que se añadió éter de petróleo y después se dejó reposar durante la noche. El sólido precipitado se retiró por filtración, y el disolvente se retiró del filtrado a 25-40 °C bajo presión reducida mediante una bomba de agua, obteniéndose de ese modo una materia
aceitosa de color amarillo. La materia aceitosa fue destilada bajo presión reducida, para recoger una fracción a 121 123 °C/8 mmHg, obteniéndose de ese modo 12,786 g de una materia aceitosa incolora, transparente, con un rendimiento de 58,9 % y pureza de 95,60 % detectada por CG.
RMN de 1H (CDCls) (ppm): 57,17-6,77 (m, 5H), 63,96 (t, 2H), 63,32 (t, 2H), 62,21 (m, 2H).
Etapa 5: Bromuro de (2R,3S)-3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano

A un matraz de 100 ml, con forma de berenjena, se añadieron 2,871 g (9,1 mmol) de la configuración (2R,3S) de la base preparada en la etapa 3 y se disolvieron añadiendo 18 ml de cloroformo para obtener una solución transparente de color amarillo, a la que se añadieron 11,034 g (51,3 mmol) de 3-bromopropoxibenceno y 50 ml de acetonitrilo, y la mezcla resultante se agitó entonces a temperatura ambiente para reaccionar durante 20-90 h bajo la protección de nitrógeno, siendo el final de la reacción controlada por TLC (condiciones de la TLC: cloroformo/metanol/agua amoniacal = 5,0 ml/1,5 ml/2 d). Una vez finalizada la reacción, el disolvente se retiró de la solución de reacción por evaporación rotativa a 25-40 °C bajo presión reducida mediante una bomba de agua, obteniéndose de ese modo una materia aceitosa de color amarillo, a la que se añadió éter etílico para precipitar una gran cantidad de sólido, y el sólido se recogió mediante filtración por succión para obtener 3,886 g de (2R,3S)-3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano como un sólido de color blancuzco, con un rendimiento de 80,5 %. El valor medido de la rotación específica [a]ü 26 de (2R,3S) fue de 53,56.
El compuesto preparado en el Ejemplo 1 se hizo reaccionar con Ag2Ü para retirar el átomo de bromo para obtener hidróxido, que podía hacerse reaccionar con otros ácidos para transformarlos en las sales correspondientes.
Ejemplos de una sal de un ácido farmacéuticamente aceptable incluyen una sal derivada de un ácido inorgánico, como hidrocloruro, bromuro, yoduro, nitrato, carbonato, bicarbonato, fosfato, hidrofosfato, fosfato dihidroxilado, sulfato, disulfato o fosfito o similar; y una sal derivada de un ácido orgánico relativamente no tóxico como ácido acético, ácido propiónico, ácido isobutírico, ácido malónico, ácido benzoico, ácido succínico, ácido subérico, ácido fumárico, ácido mandélico, ácido ftálico, ácido bencensulfónico, ácido p-toluensulfónico, ácido cítrico, ácido tartárico, ácido metansulfónico, ácido glucurónico o ácido galactónico o similar. Los ejemplos incluyen, además, una sal de un aminoácido como arginina o similar.
RMN de 1H (D2O) (ppm): 57,20-6,65 (m, 10H), 64,15-3,66 (m, 5H), 53,43-3,12 (m, 8H), 52,13 (m, 2H), 2,01 (m, 1H), 51,81-1,40 (m, 13H).
Ejemplo 2. Bromuro de (2S,3S)-3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1 -(3-fenoxipropil)-1 -azabiciclo[2,2,2]octano
Las etapas 1, 2, 3 y 4 eran las mismas que las etapas 1, 2, 3 y 4 en el Ejemplo 1.
Etapa 5: Bromuro de (2S,3S)-3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano

A un matraz de 100 ml, con forma de berenjena, se añadieron 2,872 g (9,1 mmol) de la configuración (2R,3S) de la base y se disolvieron añadiendo 18 ml de cloroformo para obtener una solución transparente de color amarillo, a la que se añadieron 11,032 g (51,3 mmol) de 3-bromopropoxibenceno y 50 ml de acetonitrilo, y la mezcla resultante se agitó entonces a temperatura ambiente para reaccionar durante 20-90 h bajo la protección de nitrógeno, siendo el final de la reacción controlada por TLC (condiciones de la TLC: cloroformo/metanol/agua amoniacal = 5,0 ml/1,5 ml/2 d). Una vez finalizada la reacción, el disolvente se retiró de la solución de reacción por evaporación rotativa a 25-40 °C bajo presión reducida mediante una bomba de agua, obteniéndose de ese modo una materia aceitosa de color amarillo, a la que se añadió éter etílico para precipitar una gran cantidad de sólido, y el sólido se recogió mediante filtración por succión para obtener 3,685 g de bromuro de (2S,3S)-3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano como un sólido de color blancuzco, con un rendimiento de 75,96 %. El valor medido de la rotación específica [a]ü26 de (2S,3S) fue 31,71.
El compuesto preparado en el Ejemplo 2 se hizo reaccionar con Ag2Ü para retirar el átomo de bromo para obtener hidróxido, que podía hacerse reaccionar con otros ácidos para transformarlos en las sales correspondientes. Ejemplos de una sal de un ácido farmacéuticamente aceptable incluyen una sal derivada de un ácido inorgánico, como hidrocloruro, bromuro, yoduro, nitrato, carbonato, bicarbonato, fosfato, hidrofosfato, fosfato dihidroxilado, sulfato, disulfato o fosfito o similar; y una sal derivada de un ácido orgánico relativamente no tóxico como ácido acético, ácido propiónico, ácido isobutírico, ácido malónico, ácido benzoico, ácido succínico, ácido subérico, ácido fumárico, ácido mandélico, ácido ftálico, ácido bencensulfónico, ácido p-toluensulfónico, ácido cítrico, ácido tartárico, ácido metansulfónico, ácido glucurónico o ácido galactónico o similar. Los ejemplos incluyen además una sal de un aminoácido como arginina o similar.
RMN de 1H (D2O) (ppm): 57,22-6,66 (m, 10H), 64,17-3,67 (m, 5H), 53,45-3,14 (m, 8H), 52,15 (m, 2H), 2,03 (m, 1H), 51,83-1,41 (m, 13H).
Ejemplo 3. Bromuro de (2S,3R)-3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano
Etapa 1: igual que la etapa 1 del Ejemplo 1
Etapa 2: Preparación de 3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali a partir de R-3-quinuclidinol

A 18,721 g (147 mmol) de R-3-quinuclidinol disponible comercialmente se añadieron 190 ml de DMSO, seguido de la adición de 7,558 g (190 mmol) de hidruro de sodio, y la mezcla se hizo reaccionar a 20-60 °C durante 0,5-12 h, y después se enfrió a temperatura ambiente, y a ello se añadió una solución de 35,75 g (190 mmol) de 1 -fenil-1-ciclopentiloxirano (autopreparado) en 45 ml de DMSO, y una vez finalizada la adición, la mezcla resultante se calentó a 20-70 °C en baño de aceite para reaccionar durante 0,5-12 horas. Bajo la condición de baño de hielo, se añadieron 120 ml de agua helada a una temperatura interna de 30 °C o menor. La mezcla de reacción se extrajo con éter isopropílico, 100 ml x 3; y la capa de éter se combinó y se lavó con una solución acuosa saturada de NaCl, 100 ml x 3. La capa orgánica se secó sobre sulfato de sodio anhidro durante la noche, el agente desecante se retiró por filtración, y el disolvente se retiró bajo presión reducida, para obtener 54,67 g de 3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali como materia aceitosa de color rojo, con un rendimiento de 95,6 %. El producto obtenido fue (2R,3R),(2S,3R)-3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali.
Etapa 3: Purificación de 3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali por cromatografía de columna y el tratamiento de purificación relacionado
La muestra de la etapa 2 anterior se separó en columna de gel de sílice, utilizando diclorometano o triclorometano y metanol, amoniacados, como fase móvil y una placa de TLC para controlar la pureza de la muestra. Bajo el sistema de elución anterior, una muestra de una mezcla de ambas configuraciones (2R,3R) y (2S,3R) de 3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali podía ser purificada en dos tipos de (2R,3R) y (2S,3R), libre de álcali dependiendo de la secuencia de elución, obteniéndose de ese modo 21,5 g de la configuración (2R,3R) de 3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali con un rendimiento de 79,1 % y el valor medido de la rotación específica [a]ü26 de 9,01, y 21,3 g de la configuración (2S,3R) de 3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali con un rendimiento de 78,37 % y el valor medido de la rotación específica [a]ü26 de -44,20.
Etapa 4: igual que la etapa 4 en el Ejemplo 1
Etapa 5: Bromuro de (2S,3R)-3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano

A un matraz de 100 ml, con forma de berenjena, se añadieron 2,871 g (9,1 mmol) de la configuración (2S,3R) de la base y se disolvieron añadiendo 18 ml de cloroformo para obtener una solución transparente de color amarillo, a la que se añadieron 11,0340 g (51,3 mmol) de 3-bromopropoxibenceno y 50 ml de acetonitrilo, y la mezcla resultante se agitó entonces a temperatura ambiente para reaccionar durante 20-90 h bajo la protección de nitrógeno, siendo el final de la reacción controlado mediante TLC (condiciones de la TLC: cloroformo/metanol/agua amoniacal = 5,0 ml/1,5 ml/2 d). Una vez finalizada la reacción, el disolvente se retiró de la solución de reacción por evaporación rotativa a 25-40 °C bajo presión reducida mediante una bomba de agua, obteniéndose de ese modo una materia aceitosa de color amarillo, a la que se añadió éter etílico para precipitar una gran cantidad de sólido, y el sólido se recogió mediante filtración por succión para obtener 3,86 g de la configuración (2S,3R) de bromuro de 3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano como un sólido de color blancuzco, con un rendimiento de 79,95 %. El valor medido de la rotación específica [a]ü 26 fue de -58,16.
El compuesto preparado en el Ejemplo 3 se hizo reaccionar con Ag2Ü para retirar el átomo de bromo para obtener hidróxido, que podía hacerse reaccionar con otros ácidos para transformarlos en las sales correspondientes. Ejemplos de una sal de un ácido farmacéuticamente aceptable incluyen una sal derivada de un ácido inorgánico, como hidrocloruro, bromuro, yoduro, nitrato, carbonato, bicarbonato, fosfato, hidrofosfato, fosfato dihidroxilado, sulfato, disulfato o fosfito o similar; y una sal derivada de un ácido orgánico relativamente no tóxico como ácido acético, ácido propiónico, ácido isobutírico, ácido malónico, ácido benzoico, ácido succínico, ácido subérico, ácido fumárico, ácido mandélico, ácido ftálico, ácido bencensulfónico, ácido p-toluensulfónico, ácido cítrico, ácido tartárico, ácido metansulfónico, ácido glucurónico o ácido galactónico o similar. Los ejemplos incluyen además una sal de un aminoácido como arginina o similar.
RMN 1H (D2O) (ppm): 57,21-6,86 (m, 10H), 54,15-3,66 (m, 5H), 63,43-3,13 (m, 8H), 52,16 (m, 2H), 2,04 (m, 1H), 6 1,79-1,23 (m, 13H).
Ejemplo 4. Bromuro de (2R,3R)-3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1 -(3-fenoxipropil)-1 -azabiciclo[2,2,2]octano
Etapa 1: igual que la etapa 1 en el Ejemplo 1
Etapas 2 y 3: igual que las etapas 2 y 3 en el Ejemplo 3
Etapa 4: igual que la etapa 4 en el Ejemplo 1
Etapa 5: Bromuro de (2R,3R)-3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano

A un matraz de 100 ml, con forma de berenjena, se añadieron 2,87 g (9,1 mmol) de la configuración (2R,3R) de la base y se disolvieron añadiendo 18 ml de cloroformo para obtener una solución transparente de color amarillo, a la que se añadieron 11,034 g (51,3 mmol) de 3-bromopropoxibenceno y 50 ml de acetonitrilo, y la mezcla resultante se agitó entonces a temperatura ambiente para reaccionar durante 20-90 h bajo la protección de nitrógeno, siendo el final de la reacción controlado mediante TLC (condiciones de la TLC: cloroformo/metanol/agua amoniacal = 5,0 ml/1,5 ml/2 d). Una vez finalizada la reacción, el disolvente se retiró de la solución de reacción por evaporación rotativa a 25-40 °C bajo presión reducida mediante una bomba de agua, obteniéndose de ese modo una materia aceitosa de color amarillo, a la que se añadió éter etílico para precipitar una gran cantidad de sólido, y el sólido se recogió mediante filtración por succión para obtener 3,685 g de la configuración (2R,3R) de bromuro de 3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano como un sólido de color blancuzco, con un rendimiento de 76,34 %. El valor medido de la rotación específica [a]D26 fue de -31,18.
El compuesto preparado en el Ejemplo 4 se hizo reaccionar con Ag2O para retirar el átomo de bromo para obtener hidróxido, que podía hacerse reaccionar con otros ácidos para transformarlos en las sales correspondientes.
Ejemplos de una sal de un ácido farmacéuticamente aceptable incluyen una sal derivada de un ácido inorgánico, como hidrocloruro, bromuro, yoduro, nitrato, carbonato, bicarbonato, fosfato, hidrofosfato, fosfato dihidroxilado, sulfato, disulfato o fosfito o similar; y una sal derivada de un ácido orgánico relativamente no tóxico como ácido acético, ácido propiónico, ácido isobutírico, ácido malónico, ácido benzoico, ácido succínico, ácido subérico, ácido fumárico, ácido mandélico, ácido ftálico, ácido bencensulfónico, ácido p-toluensulfónico, ácido cítrico, ácido tartárico, ácido metansulfónico, ácido glucurónico o ácido galactónico o similar. Los ejemplos incluyen además una sal de un aminoácido como arginina o similar.
RMN de 1H (D2O) (ppm): 57,22-6,87 (m, 10H), 54,17-3,65 (m, 5H), 53,45-3,15 (m, 8H), 52,17 (m, 2H), 2,05 (m, 1H), 51,80-1,26 (m, 13H).
Ejemplo 5. Bromuro de (2R,3S),(2S,3R)-3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1 -(3-fenoxipropil)-1 -azabiciclo[2,2,2]octano
Etapa 1: igual que la etapa 1 en el Ejemplo 1
Etapa 2: Preparación de 3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali a partir de quinuclidinol racémico

A 18,72 g (147 mmol) de quinuclidinol racémico disponible comercialmente se añadieron 190 ml de DMSO, seguido de la adición de 7,59 g (190 mmol) de hidruro de sodio, y la mezcla se hizo reaccionar a 20-60 °C durante 0,5-12 h, y después se enfrió a temperatura ambiente, y a ello se añadió una solución de 35,68 g (190 mmol) de 1 -fenil-1-ciclopentiloxirano (autopreparado) en 45 ml de DMSO, y una vez finalizada la adición, la mezcla resultante se calentó a 20-70 °C en baño de aceite para reaccionar durante 0,5-12 horas. Bajo la condición de baño de hielo, 120 ml de agua helada se añadieron a una temperatura interna de 30 °C o menor. La mezcla de reacción se extrajo con éter isopropílico, 100 ml x 3; y la capa de éter se combinó y se lavó con una solución acuosa saturada de NaCl, 100 ml x 3. La capa orgánica se secó sobre sulfato de sodio anhidro durante la noche, el agente desecante se retiró por filtración, y el disolvente se retiró bajo presión reducida, para obtener 51,99 g de 3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali como materia aceitosa de color rojo, con un rendimiento de 90,92 %. El producto obtenido fue (2R,2S),(2R,3R),(2S,3R),(2S,3S)-3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali.
Etapa 3: Purificación de 3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali por cromatografía de columna y el tratamiento de purificación relacionado
La muestra de la etapa 2 anterior se separó en columna de gel de sílice, utilizando diclorometano o triclorometano y metanol, amoniacados, como fase móvil y una placa de TLC para controlar la pureza de la muestra. Bajo el sistema de elución anterior, una muestra de una mezcla de cuatro configuraciones de (2R,3S),(2R,3R),(2S,3R),(2S,3S) de 3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali podía ser purificada en dos tipos
racémicos, (2R,3S),(2S,3R) y (2R,3R),(2S,3S), libres de álcali, dependiendo de la secuencia de elución, en donde la fracción eluida en primer lugar de la columna eran 22,05 g de configuraciones racémicas (2R,3S),(2S,3R) de 3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali, con un rendimiento de 81,12 %; y la fracción eluida en segundo lugar de la columna eran 21,29 g de configuraciones (2R,3R),(2S,3S) de 3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali, con un rendimiento de 78,33 %.
Etapa 4: igual que la etapa 4 en el Ejemplo 1
Etapa 5: Configuración (2R,3S),(2S,3R) de bromuro de 3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano

A un matraz de 100 ml, con forma de berenjena, se añadieron 2,871 g (9,1 mmol) de la configuración (2R,3S),(2S,3R) de la base y se disolvieron añadiendo 18 ml de cloroformo para obtener una solución transparente de color amarillo, a la que se añadieron 11,035 g (51,3 mmol) de 3-bromopropoxibenceno y 50 ml de acetonitrilo, y la mezcla resultante se agitó entonces a temperatura ambiente para reaccionar durante 20-90 h bajo la protección de nitrógeno, siendo el final de la reacción controlado mediante TLC (condiciones de la TLC: cloroformo/metanol/agua amoniacal = 5,0 ml/1,5 ml/2 d). Una vez finalizada la reacción, el disolvente se retiró de la solución de reacción por evaporación rotativa a 25-40 °C bajo presión reducida mediante una bomba de agua, obteniéndose de ese modo una materia aceitosa de color amarillo, a la que se añadió éter etílico para precipitar una gran cantidad de sólido, y el sólido se recogió mediante filtración por succión para obtener 3,46 g de bromuro de (2R,3S),(2S,3R)-3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano como un sólido de color blancuzco, con un rendimiento de 71,66 %.
El compuesto preparado en el Ejemplo 5 se hizo reaccionar con Ag2Ü para retirar el átomo de bromo para obtener hidróxido, que podía hacerse reaccionar con otros ácidos para transformarlos en las sales correspondientes. Ejemplos de una sal de un ácido farmacéuticamente aceptable incluyen una sal derivada de un ácido inorgánico, como hidrocloruro, bromuro, yoduro, nitrato, carbonato, bicarbonato, fosfato, hidrofosfato, fosfato dihidroxilado, sulfato, disulfato o fosfito o similar; y una sal derivada de un ácido orgánico relativamente no tóxico como ácido acético, ácido propiónico, ácido isobutírico, ácido malónico, ácido benzoico, ácido succínico, ácido subérico, ácido fumárico, ácido mandélico, ácido ftálico, ácido bencensulfónico, ácido p-toluensulfónico, ácido cítrico, ácido tartárico, ácido metansulfónico, ácido glucurónico o ácido galactónico o similar. Los ejemplos incluyen además una sal de un aminoácido como arginina o similar.
RMN de 1H (D2O) 5 (ppm): 7,19-7,15 (m, 7H), 6,82-6,77 (m, 3H), 3,94-3,45 (m, 5H), 3,24-3,12 (m, 8H), 2,15 (m, 2H), 1,96-1,79 (m, 2H), 1,57-1,25 (m, 12H).
Ejemplo 6. Configuración (2R,3R),(2S,3S) de bromuro de 3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano
Etapa 1: igual que la etapa 1 en el Ejemplo 1
Etapas 2 y 3: igual que las etapas 2 y 3 en el Ejemplo 5
Etapa 4: igual que la etapa 4 en el Ejemplo 1
Etapa 5: Configuración (2R,3R),(2S,3S) de bromuro de 3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano

A un matraz de 100 ml, con forma de berenjena, se añadieron 2,87 g (9,1 mmol) de la configuración (2R,3R),(2S,3S) de la base y se disolvieron añadiendo 18 ml de cloroformo para obtener una solución transparente de color amarillo, a la que se añadieron 11,034 g (51,3 mmol) de 3-bromopropoxibenceno y 50 ml de acetonitrilo, y la mezcla resultante se agitó entonces a temperatura ambiente para reaccionar durante 20-90 h bajo la protección de nitrógeno, siendo el final de la reacción controlado mediante TLC (condiciones de la TLC: cloroformo/metanol/agua amoniacal = 5,0 ml/1,5 ml/2 d). Una vez finalizada la reacción, el disolvente se retiró de la solución de reacción por evaporación rotativa a 25-40 °C bajo presión reducida mediante una bomba de agua, obteniéndose de ese modo una materia aceitosa de color amarillo, a la que se añadió éter etílico para precipitar una gran cantidad de sólido, y el sólido se recogió mediante filtración por succión para obtener 3,58 g de la configuración (2R,3R),(2S,3S) de bromuro de 3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano como un sólido de color blancuzco, con un rendimiento de 74,17 %.
Los isómeros ópticos puros de cuatro configuraciones de (2R,3S),(2R,3R),(2S,3R),(2S,3S) de 3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali podían obtenerse de los Ejemplos 1 y 3, y las mezclas de las configuraciones (2R,3S),(2R,3R),(2S,3R),(2S,3S) de bromuro de 3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano en diferentes proporciones se obtuvieron mezclando estos isómeros en cualquier cantidad y cualquier proporción seguida de cuaternización añadiendo 3-bromopropoxibenceno.
El compuesto preparado en el Ejemplo 6 se hizo reaccionar con Ag2Ü para retirar el átomo de bromo para obtener hidróxido, que podría reaccionar con otros ácidos para transformarlos en las sales correspondientes. Ejemplos de una sal de un ácido farmacéuticamente aceptable incluyen una sal derivada de un ácido inorgánico, como hidrocloruro, bromuro, yoduro, nitrato, carbonato, bicarbonato, fosfato, hidrofosfato, fosfato dihidroxilado, sulfato, disulfato o fosfito o similar; y una sal derivada de un ácido orgánico relativamente no tóxico como ácido acético, ácido propiónico, ácido isobutírico, ácido malónico, ácido benzoico, ácido succínico, ácido subérico, ácido fumárico, ácido mandélico, ácido ftálico, ácido bencensulfónico, ácido p-toluensulfónico, ácido cítrico, ácido tartárico, ácido metansulfónico, ácido glucurónico o ácido galactónico o similar. Los ejemplos incluyen además una sal de un aminoácido como arginina o similar.
RMN de 1H (D2O) 5 (ppm): 7,18-7,16 (m, 7H), 6,81-6,75 (m, 3H), 3,94-3,45 (m, 5H), 3,25-3,11 (m, 8H), 2,13 (m, 2H), 1,97-1,78 (m, 2H), 1,56-1,24 (m, 12H).
Ejemplo 7. Bromuro de (2S,3R)-3-[(2-ciclohexil-2-hidroxil-2-fenil)etoxil]-1 -(3-fenoxi propil)-1 -azabiciclo[2,2,2]octano
Etapa 1: Preparación de 1-fenil-1-ciclohexiloxirano
Ciclohexilfenilcetona comercialmente disponible como material de partida se hizo reaccionar para obtener 1 -fenil-1 -ciclohexiloxirano según la bibliografía(1).
A un matraz de 3 litros, con tres bocas, se añadieron 1.880 ml de acetonitrilo, seguido de la adición de sulfuro de dimetilo y sulfato de dimetilo, con un leve desprendimiento de calor, y la mezcla resultante se agitó entonces a temperatura ambiente durante 1,5 h y después se dejó reposar durante la noche. A la solución de reacción se añadió NaH (60 %) en porciones adecuadas agitando durante 30-40 min, generándose un gas (préstese atención a tener el matraz externamente conectado a un condensador Allihn y un tubo de secado con cloruro de calcio anhidro), y después se agitó a temperatura ambiente durante 1 hora, seguido por la adición gota a gota de 332,26 g de ciclohexilfenilcetona durante aproximadamente 10 min, y la mezcla resultante se hizo reaccionar posteriormente en baño de aceite a 40-45 °C durante 90 min después de ser agitada a temperatura ambiente durante 20 min, y después se enfrió a temperatura ambiente y se dejó reposar durante la noche. La solución de reacción se transfirió a un matraz con forma de berenjena, el disolvente se retiró por succión usando una bomba de agua (40-42 °C, -0,095 MPa), y el sólido restante se enfrió entonces a 0-5 °C en baño de hielo, seguido de la adición gota a gota de 2.630 ml de agua helada (siendo la temperatura interior controlada a 0-5 °C) durante aproximadamente 1 hora, y la solución de reacción se extrajo con éter isopropílico (650 ml x 3), la capa de éter se combinó y se lavó con agua hasta que es neutra (siendo el pH de los lavados con agua de 7,0), y la capa de éter se secó sobre sulfato de sodio anhidro durante la noche. El agente desecante se retiró por filtración, el disolvente se retiró bajo presión reducida por succión usando una bomba de agua (40-42 °C, -0,095 MPa), y posteriormente se recogieron 291,13 g de fracción a 117-127 °C/3 mmHg bajo presión reducida usando una bomba de aceite.
RMN de 1H (CDCls) 5 (ppm): 7,18-7,10 (m, 5H), 2,91-2,66 (m, 2H), 2,13-1,27 (m, 11H).
Etapa 2: Preparación de 3-[(2-ciclohexil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali a partir de S-3-quinuclidinol

A 18,72 g (147 mmol) de S-3-quinuclidinol disponible comercialmente se añadieron 190 ml de DMSO, seguido de la adición de 7,59 g (190 mmol) de hidruro de sodio, y la mezcla se hizo reaccionar a 20-60 °C durante 0,5-12 h, y después se enfrió a temperatura ambiente, y a ello se añadió una solución de 38,38 g (190 mmol) de 1 -fenil-1 -ciclohexiloxirano (autopreparado) en 45 ml de DMSO, y una vez finalizada la adición, la mezcla resultante se calentó a 20-70 °C en baño de aceite para reaccionar durante 0,5-12 horas. Bajo la condición de baño de hielo, se añadieron 120 ml de agua helada a una temperatura interna de 30 °C o menor. La mezcla de reacción se extrajo con éter isopropílico, 100 ml x 3; y la capa de éter se combinó y se lavó con una solución acuosa saturada de NaCl, 100 ml x 3. La capa orgánica se secó sobre sulfato de sodio anhidro durante la noche, el agente desecante se retiró por filtración, y el disolvente se retiró para obtener 47,102 g de 3-[(2-ciclohexil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali como una materia aceitosa de color rojo, con un rendimiento de 97,39 %. El producto obtenido fue (2R,3S),(2S,3S)-3-[(2-ciclohexil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali.
Etapa 3: Purificación de 3-[(2-ciclohexil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali por cromatografía de columna y el tratamiento de purificación relacionado
La muestra de la etapa 2 anterior se separó en columna de gel de sílice, utilizando diclorometano o triclorometano y metanol, amoniacados, como fase móvil y una placa de TLC para controlar la pureza de la muestra. Bajo el sistema de elución anterior, una muestra de una mezcla de ambas configuraciones (2R,3S) y (2S,3S) de 3-[(2-ciclohexil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali podía ser purificado en dos tipos (2R,3S) y (2S,3S) libres de álcali, dependiendo de la secuencia de elución, obteniéndose de ese modo 20,282 g de la configuración (2R,3S) de 3-[(2-ciclohexil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali con un rendimiento de 86,12 %, y 20,63 g de la configuración (2S,3S) de 3-[(2-ciclohexil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali con un rendimiento de 87,6 %.
Etapa 4: igual que la etapa 4 en el Ejemplo 1
Etapa 5: Bromuro de (2R,3S)-3-[(2-ciclohexil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano

A un matraz de 100 ml, con forma de berenjena, se añadieron 2,994 g (9,1 mmol) de la configuración (2R,3S) de la base y se disolvieron añadiendo 18 ml de cloroformo para obtener una solución transparente de color amarillo, a la que se añadieron 11,034 g (51,3 mmol) de 3-bromopropoxibenceno y 50 ml de acetonitrilo, y la mezcla resultante se agitó entonces a temperatura ambiente para reaccionar durante 20-90 h bajo la protección de nitrógeno, siendo el final de la reacción controlado mediante TLC (condiciones de la TLC: cloroformo/metanol/agua amoniacal = 5,0 ml/1,5 ml/2 d). Una vez finalizada la reacción, el disolvente se retiró de la solución de reacción por evaporación rotativa a 25-40 °C bajo presión reducida mediante una bomba de agua, obteniéndose de ese modo una materia aceitosa de color amarillo, a la que se añadió éter etílico para precipitar una gran cantidad de sólido, y el sólido se recogió mediante filtración por succión para obtener 4,050 g de bromuro de (2R,3S)-3-[(2-ciclohexil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano como un sólido de color blancuzco, con un rendimiento de 81,82 %.
El compuesto preparado en el Ejemplo 7 se hizo reaccionar con Ag2Ü para retirar el átomo de bromo para obtener hidróxido, que podía hacerse reaccionar con otros ácidos para transformarlos en las sales correspondientes. Ejemplos de una sal de un ácido farmacéuticamente aceptable incluyen una sal derivada de un ácido inorgánico, como hidrocloruro, bromuro, yoduro, nitrato, carbonato, bicarbonato, fosfato, hidrofosfato, fosfato dihidroxilado, sulfato, disulfato o fosfito o similar; y una sal derivada de un ácido orgánico relativamente no tóxico como ácido acético, ácido propiónico, ácido isobutírico, ácido malónico, ácido benzoico, ácido succínico, ácido subérico, ácido fumárico, ácido mandélico, ácido ftálico, ácido bencensulfónico, ácido p-toluensulfónico, ácido cítrico, ácido tartárico, ácido metansulfónico, ácido glucurónico o ácido galactónico o similar. Los ejemplos incluyen además una sal de un aminoácido como arginina o similar.
RMN de 1H (D2O) 5 (ppm): 7,16-7,14 (m, 7H), 6,80-6,77 (m, 3H), 3,93-3,42 (m, 5H), 3,24-3,1 9 (m, 8H), 2,14 (m, 2H), 1,89-1,79 (m, 2H), 1,57-1,27 (m, 14H).
Ejemplo 8. Bromuro de (2S,3S)-3-[(2-ciclohexil-2-hidroxil-2-fenil)etoxil]-1 -(3-fenoxipropil)-1 -azabiciclo[2,2,2]octano
Las etapas 1, 2, 3 y 4 eran iguales que las etapas 1, 2, 3 y 4 en el Ejemplo 7
La etapa 4 era igual que la etapa 4 en el Ejemplo 1
Etapa 5: Bromuro de (2S,3S)-3-[(2-ciclohexil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano

A un matraz de 100 ml, con forma de berenjena, se añadieron 2,993 g (9,1 mmol) de la configuración (2S,3S) de la base y se disolvieron añadiendo 18 ml de cloroformo para obtener una solución transparente de color amarillo, a la que se añadieron 11,035 g (51,3 mmol) de 3-bromopropoxibenceno y 50 ml de acetonitrilo, y la mezcla resultante se agitó entonces a temperatura ambiente para reaccionar durante 20-90 h bajo la protección de nitrógeno, siendo el final de la reacción controlado mediante TLC (condiciones de la TLC: cloroformo/metanol/agua amoniacal = 5,0 ml/1,5 ml/2 d). Una vez finalizada la reacción, el disolvente se retiró de la solución de reacción por evaporación rotativa a 25-40 °C bajo presión reducida mediante una bomba de agua, obteniéndose de ese modo una materia aceitosa de color amarillo, a la que se añadió éter etílico para precipitar una gran cantidad de sólido, y el sólido se recogió mediante filtración por succión para obtener 4,115 g de bromuro de (2S,3S)-3-[(2-ciclohexil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano como un sólido de color blancuzco, con un rendimiento de 83,13 %.
El compuesto preparado en el Ejemplo 8 se hizo reaccionar con Ag2O para retirar el átomo de bromo para obtener hidróxido, que podía hacerse reaccionar con otros ácidos para transformarlos en las sales correspondientes.
Ejemplos de una sal de un ácido farmacéuticamente aceptable incluyen una sal derivada de un ácido inorgánico, como hidrocloruro, bromuro, yoduro, nitrato, carbonato, bicarbonato, fosfato, hidrofosfato, fosfato dihidroxilado, sulfato, disulfato o fosfito o similar; y una sal derivada de un ácido orgánico relativamente no tóxico como ácido acético, ácido propiónico, ácido isobutírico, ácido malónico, ácido benzoico, ácido succínico, ácido subérico, ácido fumárico, ácido mandélico, ácido ftálico, ácido bencensulfónico, ácido p-toluensulfónico, ácido cítrico, ácido tartárico, ácido metansulfónico, ácido glucurónico o ácido galactónico o similar. Los ejemplos incluyen además una sal de un aminoácido como arginina o similar.
RMN de 1H (D2O) 5 (ppm): 7,15-7,14 (m, 7H), 6,82-6,76 (m, 3H), 3,94-3,41 (m, 5H), 3,23-3,18 (m, 8H), 2,14 (m, 2H), 1,89-1,79 (m, 2H), 1,58-1,27 (m, 14H).
Ejemplo 9. Bromuro de (2R,3R)-3-[(2-ciclohexil-2-hidroxil-2-fenil)etoxil]-1 -(3-fenoxipropil)-1 -azabiciclo[2,2,2]octano
Etapa 1: igual que la etapa 1 en el Ejemplo 7
Etapa 2: Preparación de 3-[(2-ciclohexil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali a partir de R-3-quinuclidinol

A 18,72 g (147 mmol) de R-3-quinuclidinol disponible comercialmente se añadieron 190 ml de DMSO, seguido de la adición de 7,591 g (190 mmol) de hidruro de sodio, y la mezcla se hizo reaccionar a 20-60 °C durante 0,5-12 h, y después se enfrió a temperatura ambiente, y a ello se añadió una solución de 38,38 g (190 mmol) de 1 -fenil-1 -ciclohexiloxirano (autopreparado) en 45 ml de DMSO, y una vez finalizada la adición, la mezcla resultante se calentó a 20-70 °C en baño de aceite para reaccionar durante 0,5-12 horas. Bajo la condición de baño de hielo, se añadieron 120 ml de agua helada a una temperatura interna de 30 °C o menor. La mezcla de reacción se extrajo con éter isopropílico, 100 ml x 3; y la capa de éter se combinó y se lavó con una solución acuosa saturada de NaCl, 100 ml x 3. La capa orgánica se secó sobre sulfato de sodio anhidro durante la noche, el agente desecante se retiró por filtración, y el disolvente se retiró por evaporación rotativa, para obtener 45,89 g de 3-[(2-ciclohexil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo [2,2,2]octano libre de álcali como materia aceitosa de color rojo, con un rendimiento de 96,19 %. El producto obtenido fue la configuración (2R,3R),(2S,3R) de 3-[(2-ciclohexil-2-hidroxil-2-fenil)etoxil]-1 -azabiciclo[2,2,2]octano libre de álcali.
Etapa 3: Purificación de 3-[(2-ciclohexil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali por cromatografía de columna y el tratamiento de purificación relacionado
La muestra de la etapa 2 anterior se separó en columna de gel de sílice, utilizando diclorometano o triclorometano y metanol, amoniacados, como fase móvil y una placa de TLC para controlar la pureza de la muestra. Bajo el sistema de elución anterior, una muestra de una mezcla de ambas configuraciones (2R,3R) y (2S,3R) de 3-[(2-ciclohexil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali podía ser purificada en dos tipos de (2R,3R) y (2S,3R) libre de álcali, dependiendo de la secuencia de elución, obteniéndose de ese modo 19,393 g de (2R,3R)-3-[(2-ciclohexil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali con un rendimiento de 84,52 %, y 18,186 g de (2S,3R)-3-[(2-ciclohexil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali con un rendimiento de 79,26 %.
Etapa 4: igual que la etapa 4 en el Ejemplo 1
Etapa 5: Configuración (2S,3R) de bromuro de 3-[(2-ciclohexil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano

A un matraz de 100 ml, con forma de berenjena, se añadieron 2,993 g (9,1 mmol) de la configuración (2S,3R) de la base y se disolvieron añadiendo 18 ml de cloroformo para obtener una solución transparente de color amarillo, a la que se añadieron 11,0340 g (51,3 mmol) de 3-bromopropoxibenceno y 50 ml de acetonitrilo, y la mezcla resultante se agitó entonces a temperatura ambiente para reaccionar durante 20-90 h bajo la protección de nitrógeno, siendo el final de la reacción controlado mediante TLC (condiciones de la TLC: cloroformo/metanol/agua amoniacal = 5,0 ml/1,5 ml/2 d). Una vez finalizada la reacción, el disolvente se retiró de la solución de reacción por evaporación rotativa a 25-40 °C bajo presión reducida mediante una bomba de agua, obteniéndose de ese modo una materia aceitosa de color amarillo, a la que se añadió éter etílico para precipitar una gran cantidad de sólido, y el sólido se recogió mediante filtración por succión para obtener 4,11 g de bromuro de (2S,3R)-3-[(2-ciclohexil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano como un sólido de color blancuzco, con un rendimiento de 83,03 %.
El compuesto preparado en el Ejemplo 9 se hizo reaccionar con Ag2Ü para retirar el átomo de bromo para obtener hidróxido, que podía hacerse reaccionar con otros ácidos para transformarlos en las sales correspondientes. Ejemplos de una sal de un ácido farmacéuticamente aceptable incluyen una sal derivada de un ácido inorgánico, como hidrocloruro, bromuro, yoduro, nitrato, carbonato, bicarbonato, fosfato, hidrofosfato, fosfato dihidroxilado, sulfato, disulfato o fosfito o similar; y una sal derivada de un ácido orgánico relativamente no tóxico como ácido acético, ácido propiónico, ácido isobutírico, ácido malónico, ácido benzoico, ácido succínico, ácido subérico, ácido fumárico, ácido mandélico, ácido ftálico, ácido bencensulfónico, ácido p-toluensulfónico, ácido cítrico, ácido tartárico, ácido metansulfónico, ácido glucurónico o ácido galactónico o similar. Los ejemplos incluyen además una sal de un aminoácido como arginina o similar.
RMN de 1H (D2O) 5 (ppm): 7,16-7,14 (m, 7H), 6,80-6,77 (m, 3H), 3,93-3,42 (m, 5H), 3,24-3,19 (m, 8H), 2,14 (m, 2H), 1,89-1,79 (m, 2H), 1,57-1,27 (m, 14H).
Ejemplo 10. Bromuro de (2R,3R)-3-[(2-ciclohexil-2-hidroxil-2-fenil)etoxil]-1 -(3-fenoxi propil)-1 -azabiciclo[2,2,2]octano
Etapa 1: igual que la etapa 1 en el Ejemplo 7
Las etapas 2 y 3 eran iguales a las etapas 2 y 3 en el Ejemplo 9
Etapa 4: igual que la etapa 4 en el Ejemplo 1
Etapa 5: Bromuro de (2R,3R)-3-[(2-ciclohexil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano

A un matraz de 100 ml, con forma de berenjena, se añadieron 2,995 g (9,1 mmol) de la configuración (2R,3R) de la base y se disolvieron añadiendo 18 ml de cloroformo para obtener una solución transparente de color amarillo, a la que se añadieron 11,034 g (51,3 mmol) de 3-bromopropoxibenceno y 50 ml de acetonitrilo, y la mezcla resultante se agitó entonces a temperatura ambiente para reaccionar durante 20-90 h bajo la protección de nitrógeno, siendo el final de la reacción controlado mediante TLC (condiciones de la TLC: cloroformo/metanol/agua amoniacal = 5,0 ml/1,5 ml/2 d). Una vez finalizada la reacción, el disolvente se retiró de la solución de reacción a 25-40 °C bajo presión reducida mediante una bomba de agua, obteniéndose de ese modo una materia aceitosa de color amarillo, a la que se añadió éter etílico para precipitar una gran cantidad de sólido, y el sólido se recogió mediante filtración por succión para obtener 4,205 g de bromuro de (2R,3R)-3-[(2-ciclohexil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano como un sólido de color blancuzco, con un rendimiento de 84,95 %.
El compuesto preparado en el Ejemplo 10 se hizo reaccionar con Ag2O para retirar el átomo de bromo para obtener hidróxido, que podía hacerse reaccionar con otros ácidos para transformarlos en las sales correspondientes. Ejemplos de una sal de un ácido farmacéuticamente aceptable incluyen una sal derivada de un ácido inorgánico,
como hidrocloruro, bromuro, yoduro, nitrato, carbonato, bicarbonato, fosfato, hidrofosfato, fosfato dihidroxilado, sulfato, disulfato o fosfito o similar; y una sal derivada de un ácido orgánico relativamente no tóxico como ácido acético, ácido propiónico, ácido isobutírico, ácido malónico, ácido benzoico, ácido succínico, ácido subérico, ácido fumárico, ácido mandélico, ácido ftálico, ácido bencensulfónico, ácido p-toluensulfónico, ácido cítrico, ácido tartárico, ácido metansulfónico, ácido glucurónico o ácido galactónico o similar. Los ejemplos incluyen además una sal de un aminoácido como arginina o similar.
RMN de 1H (D2O) 5 (ppm): 7,16-7,14 (m, 7H), 6,81-6,77 (m, 3H), 3,93-3,42 (m, 5H), 3,24-3,19 (m, 8H), 2,14 (m, 2H), 1,89-1,78 (m, 2H), 1,57-1,27 (m, 14H).
Ejemplo 11. Configuración (2R,3S),(2S,3R) de bromuro de 3-[(2-ciclohexil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1 -azabiciclo[2,2,2]octano
Etapa 1: igual que la etapa 1 en el Ejemplo 7
Etapa 2: Preparación de 3-[(2-ciclohexil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali a partir de quinuclidinol racémico

A 18,72 g (147 mmol) de quinuclidinol racémico disponible comercialmente se añadieron 190 ml de DMSO, seguido de la adición de 7,59 g (190 mmol) de hidruro de sodio, y la mezcla se hizo reaccionar a 20-60 °C durante 0,5-12 h, y después se enfrió a temperatura ambiente, y a ello se añadió una solución de 38,42 g (190 mmol) de 1 -fenil-1 -ciclohexiloxirano (autopreparado) en 45 ml de DMSO, y una vez finalizada la adición, la mezcla resultante se calentó a 20-70 °C en baño de aceite para reaccionar durante 0,5-12 horas. Bajo la condición de baño de hielo, se añadieron 120 ml de agua helada a una temperatura interna de 30 °C o menor. La mezcla de reacción se extrajo con éter isopropílico, 100 ml x 3; y la capa de éter se combinó y se lavó con una solución acuosa saturada de NaCl, 100 ml x 3. La capa orgánica se secó sobre sulfato de sodio anhidro durante la noche, el agente desecante se retiró por filtración, y el disolvente se retiró bajo presión reducida, para obtener 43,58 g de 3-[(2-ciclohexil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo [2,2,2]octano libre de álcali como materia aceitosa de color rojo, con un rendimiento de 90,12 %. El producto obtenido fue la configuración (2R,3S),(2R,3R),(2S,3R),(2S,3S) de 3-[(2-ciclohexil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali.
Etapa 3: Purificación de 3-[(2-ciclohexil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali por cromatografía de columna y el tratamiento de purificación relacionado
La muestra de la etapa 2 anterior se separó en columna de gel de sílice, utilizando diclorometano o triclorometano y metanol, amoniacados, como fase móvil y una placa de TLC para controlar la pureza de la muestra. Bajo el sistema de elución anterior, una muestra de una mezcla de cuatro configuraciones de (2R,3S),(2R,3R),(2S,3R),(2S,3S) de 3-[(2-ciclohexil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali podía ser purificada en dos tipos (2R,3S),(2S,3R) y (2R,3R),(2S,3S) racémicos, libres de álcali, dependiendo de la secuencia de elución, obteniéndose de ese modo la fracción eluida en primer lugar de 18,06 g de la configuración (2R,3S),(2S,3R) racémica de 3-[(2-ciclohexil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali con un rendimiento de 82,88 %, y la fracción eluida en segundo lugar de 17,29 g de la configuración (2R,3R),(2S,3S) de 3-[(2-ciclohexil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali con un rendimiento de 79,35 %.
Etapa 4: igual que la etapa 4 en el Ejemplo 1
Etapa 5: Configuración (2R,3S),(2S,3R) de bromuro de 3-[(2-ciclohexil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano

A un matraz de 100 ml, con forma de berenjena, se añadieron 2,993 g (9,1 mmol) de la configuración (2R,3S),(2S,3R) de la base y se disolvieron añadiendo 18 ml de cloroformo para obtener una solución transparente de color amarillo, a la que se añadieron 11,034 g (51,3 mmol) de 3-bromopropoxibenceno y 50 ml de acetonitrilo, y la mezcla resultante se agitó entonces a temperatura ambiente para reaccionar durante 20-90 h bajo la protección de nitrógeno, siendo el final de la reacción controlado mediante TLC (condiciones de la TLC: cloroformo/metanol/agua amoniacal = 5,0 ml/1,5 ml/2 d). Una vez finalizada la reacción, el disolvente se retiró de la solución de reacción a 25 40 °C por evaporación rotativa bajo presión reducida mediante una bomba de agua, obteniéndose de ese modo una materia aceitosa de color amarillo, a la que se añadió éter etílico para precipitar una gran cantidad de sólido, y el sólido se recogió mediante filtración por succión para obtener 4,026 g de la configuración (2R,3S),(2S,3R) de bromuro de 3-[(2-ciclohexil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano como un sólido de color blancuzco, con un rendimiento de 81,36 %.
El compuesto preparado en el Ejemplo 11 se hizo reaccionar con Ag2Ü para retirar el átomo de bromo para obtener hidróxido, que podía hacerse reaccionar con otros ácidos para transformarlos en las sales correspondientes. Ejemplos de una sal de un ácido farmacéuticamente aceptable incluyen una sal derivada de un ácido inorgánico, como hidrocloruro, bromuro, yoduro, nitrato, carbonato, bicarbonato, fosfato, hidrofosfato, fosfato dihidroxilado, sulfato, disulfato o fosfito o similar; y una sal derivada de un ácido orgánico relativamente no tóxico como ácido acético, ácido propiónico, ácido isobutírico, ácido malónico, ácido benzoico, ácido succínico, ácido subérico, ácido fumárico, ácido mandélico, ácido ftálico, ácido bencensulfónico, ácido p-toluensulfónico, ácido cítrico, ácido tartárico, ácido metansulfónico, ácido glucurónico o ácido galactónico o similar. Los ejemplos incluyen además una sal de un aminoácido como arginina o similar.
RMN de 1H (D2O) 5 (ppm): 7,19-7,15 (m, 7H), 6,82-6,78 (m, 3H), 3,93-3,42 (m, 5H), 3,24-3,19 (m, 8H), 2,14 (m, 2H), 1,88-1,79 (m, 2H), 1,58-1,26 (m, 14H).
Ejemplo 12. Bromuro de (2R,3R),(2S,3S)-3-[(2-ciclohexil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano
Etapa 1: igual que la etapa 1 en el Ejemplo 7
Etapas 2 y 3: igual que las etapas 2 y 3 en el Ejemplo 11
Etapa 4: igual que la etapa 4 en el Ejemplo 1
Etapa 5: Configuración (2R,3R),(2S,3S) de bromuro de 3-[(2-ciclohexil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano

A un matraz de 100 ml, con forma de berenjena, se añadieron 2,994 g (9,1 mmol) de la configuración (2R,3R),(2S,3S) de la base y se disolvieron añadiendo 18 ml de cloroformo para obtener una solución transparente de color amarillo, a la que se añadieron 11,034 g (51,3 mmol) de 3-bromopropoxibenceno y 50 ml de acetonitrilo, y la mezcla resultante se agitó entonces a temperatura ambiente para reaccionar durante 20-90 h bajo la protección de nitrógeno, siendo el final de la reacción controlado mediante TLC (condiciones de la TLC: cloroformo/metanol/agua amoniacal = 5,0 ml/1,5 ml/2 d). Una vez finalizada la reacción, el disolvente se retiró de la solución de reacción a 25 40 °C bajo presión reducida mediante una bomba de agua, obteniéndose de ese modo una materia aceitosa de color amarillo, a la que se añadió éter etílico para precipitar una gran cantidad de sólido, y el sólido se recogió mediante filtración por succión para obtener 4,125 g de bromuro de (2R,3R),(2S,3S)-3-[(2-ciclohexil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano como un sólido de color blancuzco, con un rendimiento de 83,36 %.
Los isómeros ópticos puros de cuatro configuraciones de (2R,3S),(2R,3R),(2S,3R),(2S,3S) de 3-[(2-ciclohexil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali podían obtenerse a partir de los Ejemplos 7 y 9, y las mezclas de las configuraciones (2R,3S),(2R,3R),(2S,3R),(2S,3S) de bromuro de 3-[(2-ciclohexil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano en diferentes proporciones se obtuvieron mezclando estos isómeros en cualquier cantidad y cualquier proporción seguido por cuaternización añadiendo 3-bromopropoxibenceno.
El compuesto preparado en el Ejemplo 12 se hizo reaccionar con Ag2Ü para retirar el átomo de bromo para obtener hidróxido, que podía hacerse reaccionar con otros ácidos para transformarlos en las sales correspondientes. Ejemplos de una sal de un ácido farmacéuticamente aceptable incluyen una sal derivada de un ácido inorgánico, como hidrocloruro, bromuro, yoduro, nitrato, carbonato, bicarbonato, fosfato, hidrofosfato, fosfato dihidroxilado, sulfato, disulfato o fosfito o similar; y una sal derivada de un ácido orgánico relativamente no tóxico como ácido acético, ácido propiónico, ácido isobutírico, ácido malónico, ácido benzoico, ácido succínico, ácido subérico, ácido fumárico, ácido mandélico, ácido ftálico, ácido bencensulfónico, ácido p-toluensulfónico, ácido cítrico, ácido tartárico, ácido metansulfónico, ácido glucurónico o ácido galactónico o similar. Los ejemplos incluyen además una sal de un aminoácido como arginina o similar.
RMN de 1H (D2O) 5 (ppm): 7,16-7,14 (m, 7H), 6,80-6,77 (m, 3H), 3,93-3,42 (m, 5H), 3,24-3,19 (m, 8H), 2,14 (m, 2H), 1,89-1,79 (m, 2H), 1,57-1,27 (m, 14H).
Ejemplo 13. Bromuro de (2R,3S)-3-[(2-ciclobutil-2-hidroxil-2-fenil)etoxil]-1 -(3-fenoxi propil)-1 -azabiciclo[2,2,2]octano
Etapa 1: Preparación de 1-fenil-1-ciclobutiloxirano

Se hizo reaccionar ciclobutilfenilcetona comercialmente disponible como material de partida para obtener 1 -fenil-1 -ciclobutiloxirano según la bibliografía(1).
A un matraz de 3 litros, con tres bocas, se añadieron 1.880 ml de acetonitrilo, seguido de la adición de sulfuro de dimetilo y sulfato de dimetilo, con un leve desprendimiento de calor, y la mezcla resultante se agitó entonces a
temperatura ambiente durante 1,5 h y después se dejó reposar durante la noche. Se añadió NaH (60 %) en porciones adecuadas en la solución de reacción con agitación durante 30-40 min, generándose un gas (préstese atención a tener el matraz externamente conectado a un condensador Allihn y un tubo de secado de cloruro de calcio anhidro), y después se agitó a temperatura ambiente durante 1 hora, seguido de la adición gota a gota de 315,6 g de ciclobutilfenilcetona durante aproximadamente 10 min, y la mezcla resultante se hizo reaccionar posteriormente en baño de aceite a 40-45 °C durante 90 min, siendo después agitada a temperatura ambiente durante 20 min, y después se enfrió a temperatura ambiente y se dejó reposar durante la noche. La solución de reacción se transfirió a un matraz con forma de berenjena, el disolvente se retiró por succión usando una bomba de agua (40-42 °C, -0,095 MPa), y el sólido restante se enfrió a 0-5 °C en baño de hielo, seguido de la adición gota a gota de 2.630 ml de agua helada (siendo la temperatura interior controlada a 0-5 °C) durante aproximadamente 1 hora, y la solución de reacción se extrajo con éter isopropílico (650 ml x 3), la capa de éter se combinó y se lavó con agua hasta que es neutra (siendo el pH de los lavados con agua de 7,0), y la capa de éter se secó sobre sulfato de sodio anhidro durante la noche. El agente desecante se retiró por filtración, el disolvente se retiró bajo presión reducida por succión usando una bomba de agua (40-42 °C, -0,095 MPa), y posteriormente se recogieron 282,7 g de fracción a 113-126 °C/3 mmHg bajo presión reducida usando una bomba de aceite, con un rendimiento de 82,37 %.
RMN de 1H (CDCls) 5 (ppm): 7,19 (m, 5H), 2,92-2,65 (m, 2H), 2,61 (m, 1H), 2,03-1,77 (m, 6H).
Etapa 2: Preparación de 3-[(2-ciclobutil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali a partir de S-3-quinuclidinol

A 18,72 g (147 mmol) de S-3-quinuclidinol disponible comercialmente se añadieron 190 ml de DMSO, seguido de la adición de 7,59 g (190 mmol) de hidruro de sodio, y la mezcla se hizo reaccionar a 20-60 °C durante 0,5-12 h, y después se enfrió a temperatura ambiente, y a ello se añadió una solución de 33,054 g (190 mmol) de 1 -fenil-1-ciclobutiloxirano (autopreparado) en 45 ml de DMSO, y una vez finalizada la adición, la mezcla resultante se calentó a 20-70 °C en baño de aceite para reaccionar durante 0,5-12 horas. Bajo la condición de baño de hielo, se añadieron 120 ml de agua helada a una temperatura interna de 30 °C o menor. La mezcla de reacción se extrajo con éter isopropílico, 100 ml x 3; y la capa de éter se combinó y se lavó con una solución acuosa saturada de NaCl, 100 ml x 3. La capa orgánica se secó sobre sulfato de sodio anhidro durante la noche, el agente desecante se retiró por filtración, y el disolvente se retiró bajo presión reducida para obtener 43,473 g de 3-[(2-ciclobutil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali como materia aceitosa de color rojo, con un rendimiento de 98,25 %. El producto obtenido fue bromuro de (2R,3S),(2S,3S)-3-[(2-ciclobutil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali.
Etapa 3: Purificación de 3-[(2-ciclobutil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali por cromatografía de columna y el tratamiento de purificación relacionado
La muestra de la etapa 2 anterior se separó en columna de gel de sílice, utilizando diclorometano o triclorometano y metanol, amoniacados, como fase móvil y una placa de TLC para controlar la pureza de la muestra. Bajo el sistema de elución anterior, una muestra de una mezcla de ambas configuraciones (2r ,3S) y (2S,3S) de 3-[(2-ciclobutil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali podía ser purificada en dos tipos de (2R,3S) y (2S,3S) libre de álcali, dependiendo de la secuencia de elución, obteniéndose de ese modo 17,5548 g de (2R,3S)-3-[(2-ciclobutil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali con un rendimiento de 80,42 %, y 17,31 g de bromuro de (2S,3S)-3-[(2-ciclobutil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali con un rendimiento de 79,62 %.
Etapa 4: igual que la etapa 4 en el Ejemplo 1
Etapa 5: Bromuro de (2R,3S)-3-[(2-ciclobutil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano

A un matraz de 100 ml, con forma de berenjena, se añadieron 2,739 g (9,1 mmol) de la configuración (2R,3S) de la base y se disolvieron añadiendo 18 ml de cloroformo para obtener una solución transparente de color amarillo, a la que se añadieron 11,034 g (51,3 mmol) de 3-bromopropoxibenceno y 50 ml de acetonitrilo, y la mezcla resultante se agitó entonces a temperatura ambiente para reaccionar durante 20-90 h bajo la protección de nitrógeno, siendo el final de la reacción controlado mediante TLC (condiciones de la TLC: cloroformo/metanol/agua amoniacal = 5,0 ml/1,5 ml/2 d). Una vez finalizada la reacción, el disolvente se retiró de la solución de reacción a 25-40 °C bajo presión reducida mediante una bomba de agua, obteniéndose de ese modo una materia aceitosa de color amarillo, a la que se añadió éter etílico para precipitar una gran cantidad de sólido, y el sólido se recogió mediante filtración por succión para obtener 3,828 g de la configuración (2R,3S) de 3-[(2-ciclobutil-2-hidroxil-2-fenil)etoxil]-1 -(3-fenoxipropil)-1-azabiciclo[2,2,2]octano como un sólido de color blancuzco, con un rendimiento de 81,52 %.
El compuesto preparado en el Ejemplo 13 se hizo reaccionar con Ag2Ü para retirar el átomo de bromo para obtener hidróxido, que podía hacerse reaccionar con otros ácidos para transformarlos en las sales correspondientes.
Ejemplos de una sal de un ácido farmacéuticamente aceptable incluyen una sal derivada de un ácido inorgánico, como hidrocloruro, bromuro, yoduro, nitrato, carbonato, bicarbonato, fosfato, hidrofosfato, fosfato dihidroxilado, sulfato, disulfato o fosfito o similar; y una sal derivada de un ácido orgánico relativamente no tóxico como ácido acético, ácido propiónico, ácido isobutírico, ácido malónico, ácido benzoico, ácido succínico, ácido subérico, ácido fumárico, ácido mandélico, ácido ftálico, ácido bencensulfónico, ácido p-toluensulfónico, ácido cítrico, ácido tartárico, ácido metansulfónico, ácido glucurónico o ácido galactónico o similar. Los ejemplos incluyen además una sal de un aminoácido como arginina o similar.
RMN de 1H (D2O) 5 (ppm): 7,19-7,15 (m, 7H), 6,82-6,77 (m, 3H), 3,94-3,68 (m, 5H), 3,49-3,19 (m, 9H), 2,15 (m, 2H), 2,0-1,79 (m, 11H).
Ejemplo 14. Bromuro de (2S,3S)-3-[(2-ciclobutil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano
Las etapas 1, 2 y 3 eran iguales que las etapas 1, 2 y 3 en el Ejemplo 13
La etapa 4 era igual que la etapa 4 en el Ejemplo 1
Etapa 5: Bromuro de (2S,3S)-3-[(2-ciclobutil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano

A un matraz de 100 ml, con forma de berenjena, se añadieron 2,739 g (9,1 mmol) de la configuración (2S,3S) de la base y se disolvieron añadiendo 18 ml de cloroformo para obtener una solución transparente de color amarillo, a la que se añadieron 11,034 g (51,3 mmol) de 3-bromopropoxibenceno y 50 ml de acetonitrilo, y la mezcla resultante se agitó entonces a temperatura ambiente para reaccionar durante 20-90 h bajo la protección de nitrógeno, siendo el final de la reacción controlado mediante TLC (condiciones de la TLC: cloroformo/metanol/agua amoniacal = 5,0 ml/1,5 ml/2 d). Una vez finalizada la reacción, el disolvente se retiró de la solución de reacción a 25-40 °C bajo presión reducida mediante una bomba de agua, obteniéndose de ese modo una materia aceitosa de color amarillo, a la que se añadió éter etílico para precipitar una gran cantidad de sólido, y el sólido se recogió mediante filtración por succión para obtener 3,726 g de bromuro de (2S,3S)-3-[(2-ciclobutil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano como un sólido de color blancuzco, con un rendimiento de 79,34 %.
El compuesto preparado en el Ejemplo 14 se hizo reaccionar con Ag2Ü para retirar el átomo de bromo para obtener hidróxido, que podía hacerse reaccionar con otros ácidos para transformarlos en las sales correspondientes.
Ejemplos de una sal de un ácido farmacéuticamente aceptable incluyen una sal derivada de un ácido inorgánico, como hidrocloruro, bromuro, yoduro, nitrato, carbonato, bicarbonato, fosfato, hidrofosfato, fosfato dihidroxilado,
sulfato, disulfato o fosfito o similar; y una sal derivada de un ácido orgánico relativamente no tóxico como ácido acético, ácido propiónico, ácido isobutírico, ácido malónico, ácido benzoico, ácido succínico, ácido subérico, ácido fumárico, ácido mandélico, ácido ftálico, ácido bencensulfónico, ácido p-toluensulfónico, ácido cítrico, ácido tartárico, ácido metansulfónico, ácido glucurónico o ácido galactónico o similar. Los ejemplos incluyen además una sal de un aminoácido como arginina o similar.
RMN de 1H (D2O) 5 (ppm): 7,19-7,15 (m, 7H), 6,82-6,77 (m, 3H), 3,94-3,68 (m, 5H), 3,49-3,19 (m, 9H), 2,14 (m, 2H), 2,0-1,79 (m, 11H).
Ejemplo 15. Bromuro de (2S,3R)-3-[(2-ciclobutil-2-hidroxil-2-fenil)etoxil]-1 -(3-fenoxipropil)-1 -azabiciclo[2,2,2]octano
La etapa 1 era igual que la etapa 1 en el Ejemplo 13
Etapa 2: Preparación de 3-[(2-ciclobutil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali a partir de R-3-quinuclidinol

A 18,721 g (147 mmol) de R-3-quinuclidinol disponible comercialmente se añadieron 190 ml de DMSO, seguido de la adición de 7,591 g (190 mmol) de hidruro de sodio, y la mezcla se hizo reaccionar a 20-60 °C durante 0,5-12 h, y después se enfrió a temperatura ambiente, y a ello se añadió una solución de 33,054 g (190 mmol) de 1 -fenil-1-ciclobutiloxirano (autopreparado) en 45 ml de DMSO, y una vez finalizada la adición, la mezcla resultante se calentó a 20-70 °C en baño de aceite para reaccionar durante 0,5-12 horas. Bajo la condición de baño de hielo, se añadieron 120 ml de agua helada a una temperatura interna de 30 °C o menor. La mezcla de reacción se extrajo con éter isopropílico, 100 ml x 3; y la capa de éter se combinó y se lavó con una solución acuosa saturada de NaCl, 100 ml x 3. La capa orgánica se secó sobre sulfato de sodio anhidro durante la noche, el agente desecante se retiró por filtración, y el disolvente se retiró bajo presión reducida para obtener 45,91 g de 3-[(2-ciclobutil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali como materia aceitosa de color rojo, con un rendimiento de 97,77 %. El producto obtenido fue la configuración (2R,3R),(2S,3R) de 3-[(2-ciclobutil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali.
Etapa 3: Purificación de 3-[(2-ciclobutil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali por cromatografía de columna y el tratamiento de purificación relacionado
La muestra de la etapa 2 anterior se separó en columna de gel de sílice, utilizando diclorometano o triclorometano y metanol, amoniacados, como fase móvil y una placa de TLC para controlar la pureza de la muestra. Bajo el sistema de elución anterior, una muestra de una mezcla de ambas configuraciones (2r ,3R) y (2S,3R) de 3-[(2-ciclobutil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali podía ser purificada en dos tipos de (2R,3R) y (2S,3R) libres de álcali, dependiendo de la secuencia de elución, obteniéndose de ese modo 18,41 g de (2R,3R)-3-[(2-ciclobutil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali con un rendimiento de 80,22 %, y 18,27 g de (2S,3R)-3-[(2-ciclobutil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali con un rendimiento de 79,59 %.
Etapa 4: igual que la etapa 4 en el Ejemplo 1
Etapa 5: Bromuro de (2S,3R)-3-[(2-ciclobutil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano

A un matraz de 100 ml, con forma de berenjena, se añadieron 2,739 g (9,1 mmol) de la configuración (2S,3R) de la base y se disolvieron añadiendo 18 ml de cloroformo para obtener una solución transparente de color amarillo, a la que se añadieron 11,0340 g (51,3 mmol) de 3-bromopropoxibenceno y 50 ml de acetonitrilo, y la mezcla resultante se agitó entonces a temperatura ambiente para reaccionar durante 20-90 h bajo la protección de nitrógeno, siendo el final de la reacción controlado mediante TLC (condiciones de la TLC: cloroformo/metanol/agua amoniacal = 5,0 ml/1,5 ml/2 d). Una vez finalizada la reacción, el disolvente se retiró de la solución de reacción a 25-40 °C bajo presión reducida mediante una bomba de agua, obteniéndose de ese modo una materia aceitosa de color amarillo, a la que se añadió éter etílico para precipitar una gran cantidad de sólido, y el sólido se recogió mediante filtración por succión para obtener 3,790 g de (2S,3R)-3-[(2-ciclobutil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano como un sólido de color blancuzco, con un rendimiento de 80,72 %.
El compuesto preparado en el Ejemplo 15 se hizo reaccionar con Ag2Ü para retirar el átomo de bromo para obtener hidróxido, que podía hacerse reaccionar con otros ácidos para transformarlos en las sales correspondientes.
Ejemplos de una sal de un ácido farmacéuticamente aceptable incluyen una sal derivada de un ácido inorgánico, como hidrocloruro, bromuro, yoduro, nitrato, carbonato, bicarbonato, fosfato, hidrofosfato, fosfato dihidroxilado, sulfato, disulfato o fosfito o similar; y una sal derivada de un ácido orgánico relativamente no tóxico como ácido acético, ácido propiónico, ácido isobutírico, ácido malónico, ácido benzoico, ácido succínico, ácido subérico, ácido fumárico, ácido mandélico, ácido ftálico, ácido bencensulfónico, ácido p-toluensulfónico, ácido cítrico, ácido tartárico, ácido metansulfónico, ácido glucurónico o ácido galactónico o similar. Los ejemplos incluyen además una sal de un aminoácido como arginina o similar.
RMN de 1H (D2O) 5 (ppm): 7,19-7,15 (m, 7H), 6,82-6,77 (m, 3H), 3,94-3,68 (m, 5H), 3,49-3,19 (m, 9H), 2,15 (m, 2H), 2,0-1,79 (m, 11H).
Ejemplo 16. Bromuro de (2R,3R)-3-[(2-ciclobutil-2-hidroxil-2-fenil)etoxil]-1 -(3-fenoxipropil)-1 -azabiciclo[2,2,2]octano
Etapa 1: igual que la etapa 1 en el Ejemplo 13
Las etapas 2 y 3 eran iguales que las etapas 2 y 3 en el Ejemplo 15
Etapa 4: igual que la etapa 4 en el Ejemplo 1
Etapa 5: Bromuro de (2R,3R)-3-[(2-ciclobutil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano

A un matraz de 100 ml, con forma de berenjena, se añadieron 2,739 g (9,1 mmol) de la configuración (2R,3R) de la base y se disolvieron añadiendo 18 ml de cloroformo para obtener una solución transparente de color amarillo, a la que se añadieron 11,034 g (51,3 mmol) de 3-bromopropoxibenceno y 50 ml de acetonitrilo, y la mezcla resultante se agitó entonces a temperatura ambiente para reaccionar durante 20-90 h bajo la protección de nitrógeno, siendo el final de la reacción controlado mediante TLC (condiciones de la TLC: cloroformo/metanol/agua amoniacal = 5,0 ml/1,5 ml/2 d). Una vez finalizada la reacción, el disolvente se retiró de la solución de reacción por evaporación rotativa a 25-40 °C bajo presión reducida mediante una bomba de agua, obteniéndose de ese modo una materia aceitosa de color amarillo, a la que se añadió éter etílico para precipitar una gran cantidad de sólido, y el sólido se recogió mediante filtración por succión para obtener 3,805 g de la configuración (2R,3R) de 3-[(2-ciclobutil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano como un sólido de color blancuzco, con un rendimiento de 81,02 %.
El compuesto preparado en el Ejemplo 16 se hizo reaccionar con Ag2Ü para retirar el átomo de bromo para obtener hidróxido, que podía hacerse reaccionar con otros ácidos para transformarlos en las sales correspondientes. Ejemplos de una sal de un ácido farmacéuticamente aceptable incluyen una sal derivada de un ácido inorgánico, como hidrocloruro, bromuro, yoduro, nitrato, carbonato, bicarbonato, fosfato, hidrofosfato, fosfato dihidroxilado, sulfato, disulfato o fosfito o similar; y una sal derivada de un ácido orgánico relativamente no tóxico como ácido acético, ácido propiónico, ácido isobutírico, ácido malónico, ácido benzoico, ácido succínico, ácido subérico, ácido fumárico, ácido mandélico, ácido ftálico, ácido bencensulfónico, ácido p-toluensulfónico, ácido cítrico, ácido tartárico, ácido metansulfónico, ácido glucurónico o ácido galactónico o similar. Los ejemplos incluyen además una sal de un aminoácido como arginina o similar.
RMN de 1H (D2O) 5 (ppm): 7,19-7,14 (m, 7H), 6,81-6,77 (m, 3H), 3,94-3,68 (m, 5H), 3,49-3,19 (m, 9H), 2,15 (m, 2H), 2,0-1,77 (m, 11H).
Ejemplo 17. Bromuro de (2R,3S),(2S,3R)-3-[(2-ciclobutil-2-hidroxil-2-fenil)etoxil]-1 -(3-fenoxipropil)-1 -azabiciclo[2,2,2]octano
Etapa 1: igual que la etapa 1 en el Ejemplo 13
Etapa 2: Preparación de 3-[(2-ciclobutil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali a partir de quinuclidinol racémico

A 18,72 g (147 mmol) de quinuclidinol racémico disponible comercialmente se añadieron 190 ml de DMSÜ, seguido de la adición de 7,59 g (190 mmol) de hidruro de sodio, y la mezcla se hizo reaccionar a 20-60 °C durante 0,5-12 h, y después se enfrió a temperatura ambiente, y a ello se añadió una solución de 33,06 g (190 mmol) de 1 -fenil-1-ciclobutiloxirano (autopreparado) en 45 ml de DMSÜ, y una vez finalizada la adición, la mezcla resultante se calentó a 20-70 °C en baño de aceite para reaccionar durante 0,5-12 horas. Bajo la condición de baño de hielo, se añadieron 120 ml de agua helada a una temperatura interna de 30 °C o menor. La mezcla de reacción se extrajo con éter isopropílico, 100 ml x 3; y la capa de éter se combinó y se lavó con una solución acuosa saturada de NaCl, 100 ml x 3. La capa orgánica se secó sobre sulfato de sodio anhidro durante la noche, el agente desecante se retiró por filtración, y el disolvente se retiró bajo presión reducida, para obtener 42,16 g de 3-[(2-ciclobutil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo [2,2,2]octano libre de álcali como materia aceitosa de color rojo, con un rendimiento de 95,28 %. El producto obtenido fue (2R,3S),(2R,3R),(2S,3R),(2S,3S)-3-[(2-ciclobutil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]-octano libre de álcali.
Etapa 3: Purificación de 3-[(2-ciclobutil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali por cromatografía de columna y el tratamiento de purificación relacionado
La muestra de la etapa 2 anterior se separó en columna de gel de sílice, utilizando diclorometano o triclorometano y metanol, amoniacados, como fase móvil y una placa de TLC para controlar la pureza de la muestra. Bajo el sistema de elución anterior, una muestra de una mezcla de cuatro configuraciones de (2R,3S),(2R,3R),(2S,3R),(2S,3S) de 3 [(2-ciclobutil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali podía ser purificada en dos tipos
(2R,3S),(2S,3R) y (2R,3R),(2S,3S) racémicos, libres de álcali, dependiendo de la secuencia de elución, obteniéndose de ese modo la fracción eluida en primer lugar de 17,44 g de configuración racémica (2R,3S),(2S,3R) de 3-[(2-ciclobutil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali con un rendimiento de 82,72 %, y la fracción eluida en segundo lugar de 16,73 g de la configuración (2R,3R),(2S,3S) de 3-[(2-ciclobutil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali con un rendimiento de 79,36 %.
Etapa 4: igual que la etapa 4 en el Ejemplo 1
Etapa 5: Bromuro de (2R,3S),(2S,3R)-3-[(2-ciclobutil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano

A un matraz de 100 ml, con forma de berenjena, se añadieron 2,739 g (9,1 mmol) de la configuración (2R,3S),(2S,3R) de la base y se disolvieron añadiendo 18 ml de cloroformo para obtener una solución transparente de color amarillo, a la que se añadieron 11,0342 g (51,3 mmol) de 3-bromopropoxibenceno y 50 ml de acetonitrilo, y la mezcla resultante se agitó entonces a temperatura ambiente para reaccionar durante 20-90 h bajo la protección de nitrógeno, siendo el final de la reacción controlado mediante TLC (condiciones de la TLC: cloroformo/metanol/agua amoniacal = 5,0 ml/1,5 ml/2 d). Una vez finalizada la reacción, el disolvente se retiró de la solución de reacción a 25-40 °C bajo presión reducida mediante una bomba de agua, obteniéndose de ese modo una materia aceitosa de color amarillo, a la que se añadió éter etílico para precipitar una gran cantidad de sólido, y el sólido se recogió mediante filtración por succión para obtener 3,916 g de la configuración (2R,3S),(2S,3R) de 3-[(2-ciclobutil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]-octano como un sólido de color blancuzco, con un rendimiento de 83,38 %.
El compuesto preparado en el Ejemplo 17 se hizo reaccionar con Ag2Ü para retirar el átomo de bromo para obtener hidróxido, que podía hacerse reaccionar con otros ácidos para transformarlos en las sales correspondientes. Ejemplos de una sal de un ácido farmacéuticamente aceptable incluyen una sal derivada de un ácido inorgánico, como hidrocloruro, bromuro, yoduro, nitrato, carbonato, bicarbonato, fosfato, hidrofosfato, fosfato dihidroxilado, sulfato, disulfato o fosfito o similar; y una sal derivada de un ácido orgánico relativamente no tóxico como ácido acético, ácido propiónico, ácido isobutírico, ácido malónico, ácido benzoico, ácido succínico, ácido subérico, ácido fumárico, ácido mandélico, ácido ftálico, ácido bencensulfónico, ácido p-toluensulfónico, ácido cítrico, ácido tartárico, ácido metansulfónico, ácido glucurónico o ácido galactónico o similar. Los ejemplos incluyen además una sal de un aminoácido como arginina o similar.
RMN de 1H (D2O) 5 (ppm): 7,19-7,15 (m, 7H), 6,82-6,77 (m, 3H), 3,94-3,68 (m, 5H), 3,49-3,19 (m, 9H), 2,15 (m, 2H), 2,0-1,79 (m, 11H).
Ejemplo 18. Bromuro de (2R,3R),(2S,3S)-3-[(2-ciclobutil-2-hidroxil-2-fenil)etoxil]-1 -(3-fenoxipropil)-1 -azabiciclo[2,2,2]-octano
Las etapas 1, 2 y 3 eran iguales que las etapas 1, 2 y 3 en el Ejemplo 17
Etapa 4: igual que la etapa 4 en el Ejemplo 1
Etapa 5: Bromuro de (2R,3R),(2S,3S)-3-[(2-ciclobutil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]-octano
A un matraz de 100 ml, con forma de berenjena, se añadieron 2,739 g (9,1 mmol) de la configuración (2R,3R),(2S,3S) de la base y se disolvieron añadiendo 18 ml de cloroformo para obtener una solución transparente de color amarillo, a la que se añadieron 11,034 g (51,3 mmol) de 3-bromopropoxibenceno y 50 ml de acetonitrilo, y la mezcla resultante se agitó entonces a temperatura ambiente para reaccionar durante 20-90 h bajo la protección de
nitrógeno, siendo el final de la reacción controlado mediante TLC (condiciones de la TLC: cloroformo/metanol/agua amoniacal = 5,0 ml/1,5 ml/2 d). Una vez finalizada la reacción, el disolvente se retiró de la solución de reacción a 25 40 °C por evaporación rotativa bajo presión reducida mediante una bomba de agua, obteniéndose de ese modo una materia aceitosa de color amarillo, a la que se añadió éter etílico para precipitar una gran cantidad de sólido, y el sólido se recogió mediante filtración por succión para obtener 3,918 g de la configuración (2R,3R),(2S,3S) de bromuro de 3-[(2-ciclobutil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano como un sólido de color blancuzco, con un rendimiento de 83,42 %.
Los isómeros ópticos puros de cuatro configuraciones de (2R,3S),(2R,3R),(2S,3R),(2S,3S) de 3-[(2-ciclobutil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali podían obtenerse a partir de los Ejemplos 13 y 15, y mezclas de configuraciones (2R,3S),(2R,3R),(2S,3R),(2S,3S) de bromuro de 3-[(2-ciclobutil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano en diferentes proporciones se obtuvieron mezclando estos isómeros en cualquier cantidad y cualquier proporción seguido por cuaternización añadiendo 3-bromopropoxibenceno.
El compuesto preparado en el Ejemplo 18 se hizo reaccionar con Ag2Ü para retirar el átomo de bromo para obtener hidróxido, que podía hacerse reaccionar con otros ácidos para transformarlos en las sales correspondientes. Ejemplos de una sal de un ácido farmacéuticamente aceptable incluyen una sal derivada de un ácido inorgánico, como hidrocloruro, bromuro, yoduro, nitrato, carbonato, bicarbonato, fosfato, hidrofosfato, fosfato dihidroxilado, sulfato, disulfato o fosfito o similar; y una sal derivada de un ácido orgánico relativamente no tóxico como ácido acético, ácido propiónico, ácido isobutírico, ácido malónico, ácido benzoico, ácido succínico, ácido subérico, ácido fumárico, ácido mandélico, ácido ftálico, ácido bencensulfónico, ácido p-toluensulfónico, ácido cítrico, ácido tartárico, ácido metansulfónico, ácido glucurónico o ácido galactónico o similar. Los ejemplos incluyen además una sal de un aminoácido como arginina o similar.
RMN de 1H (D2O) 5 (ppm): 7,19-7,15 (m, 7H), 6,82-6,77 (m, 3H), 3,94-3,68 (m, 5H), 3,49-3,19 (m, 9H), 2,15 (m, 2H), 2,0-1,79 (m, 11H).
Ejemplo 19. Bromuro de (2R,3S)-3-[(2-ciclopropil-2-hidroxil-2-fenil)etoxil]-1 -(3-fenoxi propil)-1 -azabiciclo[2,2,2]octano
Etapa 1: Preparación de 1-fenil-1-ciclopropiloxirano

Se hizo reaccionar ciclopropilfenilcetona comercialmente disponible como material de partida para obtener 1 -fenil-1-ciclopropiloxirano según la bibliografía(1).
A un matraz de 3 litros, con tres bocas, se añadieron 1.880 ml de acetonitrilo, seguido de la adición de sulfuro de dimetilo y sulfato de dimetilo, con un leve desprendimiento de calor, y la mezcla resultante se agitó entonces a temperatura ambiente durante 1,5 h y después se dejó reposar durante la noche. Se añadió NaH (60 %) en porciones adecuadas en la solución de reacción con agitación durante 30-40 min, generándose un gas (préstese atención a tener el matraz externamente conectado a un condensador Allihn y un tubo de secado de cloruro de calcio anhidro), y después se agitó a temperatura ambiente durante 1 hora, seguido de la adición gota a gota de 300,5 g de ciclpropilfenilcetona durante aproximadamente 10 min, y la mezcla resultante se hizo reaccionar posteriormente en baño de aceite a 40-45 °C durante 90 min siendo después agitada a temperatura ambiente durante 20 min, y después se enfrió a temperatura ambiente y se dejó reposar durante la noche. La solución de reacción se transfirió a un matraz con forma de berenjena, el disolvente se retiró por succión usando una bomba de agua (40-42 °C, -0,095 MPa), y el sólido restante se enfrió a 0-5 °C en baño de hielo, seguido de la adición gota a gota de 2.630 ml de agua helada (siendo la temperatura interior controlada a 0-5 °C) durante aproximadamente 1 hora, y la solución de reacción se extrajo con éter isopropílico (650 ml * 3), la capa de éter se combinó y se lavó con agua hasta que es neutra (siendo el pH de los lavados con agua de 7,0), y la capa de éter se secó sobre sulfato de sodio anhidro durante la noche. El agente desecante se retiró por filtración, el disolvente se retiró bajo presión reducida por succión usando una bomba de agua (40-42 °C, -0,095 MPa), y posteriormente se recogieron 275,8 g de fracción a 111-124 °C/3 mmHg bajo presión reducida usando una bomba de aceite, con un rendimiento de 83,75 %.
RMN de 1H (CDCls) 5 (ppm): 7,19 (m, 5H), 2,91-2,66 (m, 2H), 0,91 (m, 1H), 0,31-0,07 (m, 4H).
Etapa 2: Preparación de 3-[(2-ciclopropil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali a partir de S-3-quinuclidinol

A 18,72 g (147 mmol) de S-3-quinuclidinol disponible comercialmente se añadieron 190 ml de DMSO, seguido de la adición de 7,59 g (190 mmol) de hidruro de sodio, y la mezcla se hizo reaccionar a 20-60 °C durante 0,5-12 h, y después se enfrió a temperatura ambiente, y a ello se añadió una solución de 30,35 g (190 mmol) de 1 -fenil-1-ciclopropiloxirano (autopreparado) en 45 ml de DMSO, y una vez finalizada la adición, la mezcla resultante se calentó a 20-70 °C en baño de aceite para reaccionar durante 0,5-12 horas. Bajo la condición de baño de hielo, se añadieron 120 ml de agua helada a una temperatura interna de 30 °C o menor. La mezcla de reacción se extrajo con éter isopropílico, 100 ml x 3; y la capa de éter se combinó y se lavó con una solución acuosa saturada de NaCl, 100 ml x 3. La capa orgánica se secó sobre sulfato de sodio anhidro durante la noche, el agente desecante se retiró por filtración, y el disolvente se retiró bajo presión reducida para obtener 41,40 g de 3-[(2-ciclopropil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali como materia aceitosa de color rojo, con un rendimiento de 98,13 %. El producto obtenido fue configuración (2R,3S),(2S,3S) de 3-[(2-ciclopropil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali.
Etapa 3: Purificación de 3-[(2-ciclopropil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali por cromatografía de columna y el tratamiento de purificación relacionado
La muestra de la etapa 2 anterior se separó en columna de gel de sílice, utilizando diclorometano o triclorometano y metanol, amoniacados, como fase móvil y una placa de TLC para controlar la pureza de la muestra. Bajo el sistema de elución anterior, una muestra de una mezcla de ambas configuraciones (2R,3S) y (2S,3S) de 3-[(2-ciclopropil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali podía ser purificada en dos tipos de (2R,3S) y (2S,3S) libre de álcali, dependiendo de la secuencia de elución, obteniéndose de ese modo 16,44 g de (2R,3S)-3-[(2-ciclopropil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali con un rendimiento de 79,43 %, y 16,49 g de bromuro de (2S,3S)-3-[(2-ciclopropil-2-hidroxilo-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali con un rendimiento de 79,68 %.
Etapa 4: igual que la etapa 4 en el Ejemplo 1
Etapa 5: Bromuro de (2R,3S)-3-[(2-ciclopropil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano

A un matraz de 100 ml, con forma de berenjena, se añadieron 2,612 g (9,1 mmol) de la configuración (2R,3S) de la base y se disolvieron añadiendo 18 ml de cloroformo para obtener una solución transparente de color amarillo, a la que se añadieron 11,034 g (51,3 mmol) de 3-bromopropoxibenceno y 50 ml de acetonitrilo, y la mezcla resultante se agitó entonces a temperatura ambiente para reaccionar durante 20-90 h bajo la protección de nitrógeno, siendo el final de la reacción controlado mediante TLC (condiciones de la TLC: cloroformo/metanol/agua amoniacal = 5,0 ml/1,5 ml/2 d). Una vez finalizada la reacción, el disolvente se retiró de la solución de reacción a 25-40 °C por evaporación rotativa bajo presión reducida mediante una bomba de agua, obteniéndose de ese modo una materia aceitosa de color amarillo, a la que se añadió éter etílico para precipitar una gran cantidad de sólido, y el sólido se recogió mediante filtración por succión para obtener 3,725 g de bromuro de (2R,3S)-3-[(2-ciclopropil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano como un sólido de color blancuzco, con un rendimiento de 81,55 %.
El compuesto preparado en el Ejemplo 19 se hizo reaccionar con Ag2Ü para retirar el átomo de bromo para obtener hidróxido, que podía hacerse reaccionar con otros ácidos para transformarlos en las sales correspondientes.
Ejemplos de una sal de un ácido farmacéuticamente aceptable incluyen una sal derivada de un ácido inorgánico, como hidrocloruro, bromuro, yoduro, nitrato, carbonato, bicarbonato, fosfato, hidrofosfato, fosfato dihidroxilado, sulfato, disulfato o fosfito o similar; y una sal derivada de un ácido orgánico relativamente no tóxico como ácido acético, ácido propiónico, ácido isobutírico, ácido malónico, ácido benzoico, ácido succínico, ácido subérico, ácido fumárico, ácido mandélico, ácido ftálico, ácido bencensulfónico, ácido p-toluensulfónico, ácido cítrico, ácido tartárico, ácido metansulfónico, ácido glucurónico o ácido galactónico o similar. Los ejemplos incluyen además una sal de un aminoácido como arginina o similar.
RMN de 1H (D2O) 5 (ppm): 7,19-7,15 (m, 7H), 6,82-6,77 (m, 3H), 3,94-3,68 (m, 4H), 3,49-3,19 (m, 9H), 2,15 (m, 2H), 1,82-1,57 (m, 5H), 0,67-0,06 (m, 5H).
Ejemplo 20. Bromuro de (2S,3S)-3-[(2-ciclopropil-2-hidroxil-2-fenil)etoxil]-1 -(3-fenoxi propil)-1 -azabiciclo[2,2,2]octano
Las etapas 1, 2 y 3 eran iguales que las etapas 1, 2 y 3 en el Ejemplo 19
La etapa 4 era igual que la etapa 4 en el Ejemplo 1
Etapa 5: Bromuro de (2S,3S)-3-[(2-ciclopropil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano
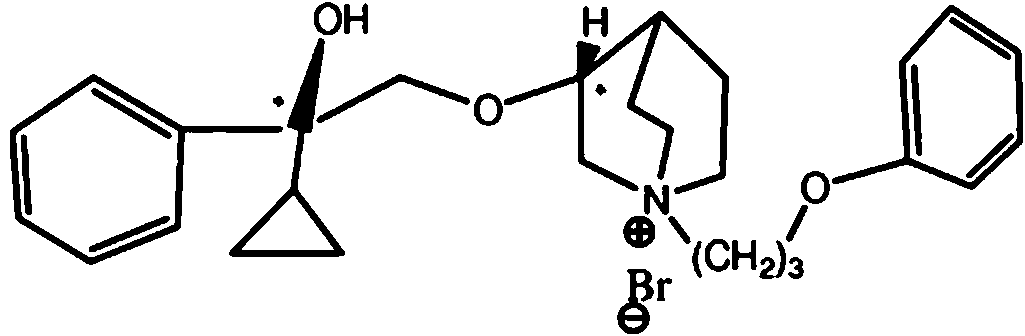
A un matraz de 100 ml, con forma de berenjena, se añadieron 2,612 g (9,1 mmol) de la configuración (2S,3S) de la base y se disolvieron añadiendo 18 ml de cloroformo para obtener una solución transparente de color amarillo, a la que se añadieron 11,034 g (51,3 mmol) de 3-bromopropoxibenceno y 50 ml de acetonitrilo, y la mezcla resultante se agitó entonces a temperatura ambiente para reaccionar durante 20-90 h bajo la protección de nitrógeno, siendo el final de la reacción controlado mediante TLC (condiciones de la TLC: cloroformo/metanol/agua amoniacal = 5,0 ml/1,5 ml/2 d). Una vez finalizada la reacción, el disolvente se retiró de la solución de reacción a 25-40 °C por evaporación rotativa bajo presión reducida mediante una bomba de agua, obteniéndose de ese modo una materia aceitosa de color amarillo, a la que se añadió éter etílico para precipitar una gran cantidad de sólido, y el sólido se recogió mediante filtración por succión para obtener 3,776 g de la configuración (2S,3S) de bromuro de 3-[(2-ciclopropil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano como un sólido de color blancuzco, con un rendimiento de 82,66 %.
El compuesto preparado en el Ejemplo 20 se hizo reaccionar con Ag2Ü para retirar el átomo de bromo para obtener hidróxido, que podía hacerse reaccionar con otros ácidos para transformarlos en las sales correspondientes.
Ejemplos de una sal de un ácido farmacéuticamente aceptable incluyen una sal derivada de un ácido inorgánico, como hidrocloruro, bromuro, yoduro, nitrato, carbonato, bicarbonato, fosfato, hidrofosfato, fosfato dihidroxilado, sulfato, disulfato o fosfito o similar; y una sal derivada de un ácido orgánico relativamente no tóxico como ácido acético, ácido propiónico, ácido isobutírico, ácido malónico, ácido benzoico, ácido succínico, ácido subérico, ácido fumárico, ácido mandélico, ácido ftálico, ácido bencensulfónico, ácido p-toluensulfónico, ácido cítrico, ácido tartárico, ácido metansulfónico, ácido glucurónico o ácido galactónico o similar. Los ejemplos incluyen además una sal de un aminoácido como arginina o similar.
RMN de 1H (D2O) 5 (ppm): 7,19-7,15 (m, 7H), 6,81-6,77 (m, 3H), 3,94-3,68 (m, 4H), 3,49-3,19 (m, 9H), 2,15 (m, 2H), 1,82-1,57 (m, 5H), 0,66-0,06 (m, 5H).
Ejemplo 21. Bromuro de (2S,3R)-3-[(2-ciclopropil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxi propil)-1-azabiciclo[2,2,2]octano
La etapa 1 era igual que la etapa 1 en el Ejemplo 19
Etapa 2: Preparación de 3-[(2-ciclopropil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali a partir de R-3-quinuclidinol

A 18,721 g (147 mmol) de R-3-quinuclidinol disponible comercialmente se añadieron 190 ml de DMSO, seguido de la adición de 7,591 g (190 mmol) de hidruro de sodio, y la mezcla se hizo reaccionar a 20-60 °C durante 0,5-12 h, y después se enfrió a temperatura ambiente, y a ello se añadió una solución de 30,36 g (190 mmol) de 1 -fenil-1 -ciclopropiloxirano (autopreparado) en 45 ml de DMSO, y una vez finalizada la adición, la mezcla resultante se calentó a 20-70 °C en baño de aceite para reaccionar durante 0,5-12 horas. Bajo la condición de baño de hielo, se añadieron 120 ml de agua helada a una temperatura interna de 30 °C o menor. La mezcla de reacción se extrajo con éter isopropílico, 100 ml x 3; y la capa de éter se combinó y se lavó con una solución acuosa saturada de NaCl, 100 ml x 3. La capa orgánica se secó sobre sulfato de sodio anhidro durante la noche, el agente desecante se retiró por filtración, y el disolvente se retiró por evaporación rotativa para obtener 40,99 g de 3-[(2-ciclopropil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo [2,2,2]octano libre de álcali como materia aceitosa de color rojo, con un rendimiento de 97,15 %. El producto obtenido fue la configuración (2R,3R),(2S,3R) de 3-[(2-ciclopropil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali.
Etapa 3: Purificación de 3-[(2-ciclopropil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali por cromatografía de columna y el tratamiento de purificación relacionado
La muestra de la etapa 2 anterior se separó en columna de gel de sílice, utilizando diclorometano o triclorometano y metanol, amoniacados, como fase móvil y una placa de TLC para controlar la pureza de la muestra. Bajo el sistema de elución anterior, una muestra de una mezcla de ambas configuraciones (2R,3R) y (2S,3R) de 3-[(2-ciclopropil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali podía ser purificada en dos tipos de (2R,3R) y (2S,3R) libres de álcali, dependiendo de la secuencia de elución, obteniéndose de ese modo 16,45 g de (2R,3R)-3-[(2-ciclopropil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali con un rendimiento de 80,25 %, y 16,26 g de la configuración (2S,3R) de 3-[(2-ciclopropil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali con un rendimiento de 79,34 %.
Etapa 4: igual que la etapa 4 en el Ejemplo 1
Etapa 5: Bromuro de (2S,3R)-3-[(2-ciclopropil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano

A un matraz de 100 ml, con forma de berenjena, se añadieron 2,612 g (9,1 mmol) de la configuración (2S,3R) de la base y se disolvieron añadiendo 18 ml de cloroformo para obtener una solución transparente de color amarillo, a la que se añadieron 11,0340 g (51,3 mmol) de 3-bromopropoxibenceno y 50 ml de acetonitrilo, y la mezcla resultante se agitó entonces a temperatura ambiente para reaccionar durante 20-90 h bajo la protección de nitrógeno, siendo el final de la reacción controlado mediante TLC (condiciones de la TLC: cloroformo/metanol/agua amoniacal = 5,0 ml/1,5 ml/2 d). Una vez finalizada la reacción, el disolvente se retiró de la solución de reacción a 25-40 °C por evaporación rotativa bajo presión reducida mediante una bomba de agua, obteniéndose de ese modo una materia aceitosa de color amarillo, a la que se añadió éter etílico para precipitar una gran cantidad de sólido, y el sólido se recogió mediante filtración por succión para obtener 3,599 g de (2S,3R)-3-[(2-ciclopropil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano como un sólido de color blancuzco, con un rendimiento de 78,78 %.
El compuesto preparado en el Ejemplo 21 se hizo reaccionar con Ag2Ü para retirar el átomo de bromo para obtener hidróxido, que podía hacerse reaccionar con otros ácidos para transformarlos en las sales correspondientes.
Ejemplos de una sal de un ácido farmacéuticamente aceptable incluyen una sal derivada de un ácido inorgánico, como hidrocloruro, bromuro, yoduro, nitrato, carbonato, bicarbonato, fosfato, hidrofosfato, fosfato dihidroxilado, sulfato, disulfato o fosfito o similar; y una sal derivada de un ácido orgánico relativamente no tóxico como ácido acético, ácido propiónico, ácido isobutírico, ácido malónico, ácido benzoico, ácido succínico, ácido subérico, ácido fumárico, ácido mandélico, ácido ftálico, ácido bencensulfónico, ácido p-toluensulfónico, ácido cítrico, ácido tartárico, ácido metansulfónico, ácido glucurónico o ácido galactónico o similar. Los ejemplos incluyen además una sal de un aminoácido como arginina o similar.
RMN de 1H (D2O) 5 (ppm): 7,19-7,15 (m, 7H), 6,82-6,77 (m, 3H), 3,94-3,68 (m, 4H), 3,49-3,19 (m, 9H), 2,15 (m, 2H), 1,82-1,57 (m, 5H), 0,67-0,06 (m, 5H).
Ejemplo 22. Bromuro de (2R,3R)-3-[(2-ciclopropil-2-hidroxil-2-fenil)etoxil]-1 -(3-fenoxipropil)-1 -azabiciclo[2,2,2]octano
La etapa 1 era igual que la etapa 1 en el Ejemplo 19
Las etapas 2 y 3 eran iguales que las etapas 2 y 3 en el Ejemplo 21
Etapa 4: igual que la etapa 4 en el Ejemplo 1
Etapa 5: Bromuro de (2R,3R)-3-[(2-ciclopropil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano

A un matraz de 100 ml, con forma de berenjena, se añadieron 2,612 g (9,1 mmol) de la configuración (2R,3R) de la base y se disolvieron añadiendo 18 ml de cloroformo para obtener una solución transparente de color amarillo, a la que se añadieron 11,034 g (51,3 mmol) de 3-bromopropoxibenceno y 50 ml de acetonitrilo, y la mezcla resultante se agitó entonces a temperatura ambiente para reaccionar durante 20-90 h bajo la protección de nitrógeno, siendo el final de la reacción controlado mediante TLC (condiciones de la TLC: cloroformo/metanol/agua amoniacal = 5,0 ml/1,5 ml/2 d). Una vez finalizada la reacción, el disolvente se retiró de la solución de reacción a 25-40 °C por evaporación rotativa bajo presión reducida mediante una bomba de agua, obteniéndose de ese modo una materia aceitosa de color amarillo, a la que se añadió éter etílico para precipitar una gran cantidad de sólido, y el sólido se recogió mediante filtración por succión para obtener 3,795 g de bromuro de (2R,3R)-3-[(2-ciclopropil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano como un sólido de color blancuzco, con un rendimiento de 83,07 %.
El compuesto preparado en el Ejemplo 22 se hizo reaccionar con Ag2Ü para retirar el átomo de bromo para obtener hidróxido, que podía hacerse reaccionar con otros ácidos para transformarlos en las sales correspondientes.
Ejemplos de una sal de un ácido farmacéuticamente aceptable incluyen una sal derivada de un ácido inorgánico, como hidrocloruro, bromuro, yoduro, nitrato, carbonato, bicarbonato, fosfato, hidrofosfato, fosfato dihidroxilado, sulfato, disulfato o fosfito o similar; y una sal derivada de un ácido orgánico relativamente no tóxico como ácido acético, ácido propiónico, ácido isobutírico, ácido malónico, ácido benzoico, ácido succínico, ácido subérico, ácido fumárico, ácido mandélico, ácido ftálico, ácido bencensulfónico, ácido p-toluensulfónico, ácido cítrico, ácido tartárico, ácido metansulfónico, ácido glucurónico o ácido galactónico o similar. Los ejemplos incluyen además una sal de un aminoácido como arginina o similar.
RMN de 1H (D2O) 5 (ppm): 7,19-7,15 (m, 7H), 6,82-6,77 (m, 3H), 3,94-3,68 (m, 4H), 3,49-3,19 (m, 9H), 2,15 (m, 2H), 1,82-1,57 (m, 5H), 0,67-0,06 (m, 5H).
Ejemplo 23. Bromuro de (2R,3S),(2S,3R)-3-[(2-ciclopropil-2-hidroxil-2-fenil)etoxil]-1 -(3-fenoxipropil)-1 -azabiciclo[2,2,2]octano
La etapa 1 era igual que la etapa 1 en el Ejemplo 19
Etapa 2: Preparación de 3-[(2-ciclopropil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali a partir de quinuclidinol racémico

A 18,72 g (147 mmol) de quinuclidinol racémico disponible comercialmente se añadieron 190 ml de DMSO, seguido de la adición de 7,59 g (190 mmol) de hidruro de sodio, y la mezcla se hizo reaccionar a 20-60 °C durante 0,5-12 h, y después se enfrió a temperatura ambiente, y a ello se añadió una solución de 30,4 g (190 mmol) de 1 -fenil-1 -ciclopropiloxirano (autopreparado) en 45 ml de DMSO, y una vez finalizada la adición, la mezcla resultante se calentó a 20-70 °C en baño de aceite para reaccionar durante 0,5-12 horas. Bajo la condición de baño de hielo, se añadieron 120 ml de agua helada a una temperatura interna de 30 °C o menor. La mezcla de reacción se extrajo con éter isopropílico, 100 ml x 3; y la capa de éter se combinó y se lavó con una solución acuosa saturada de NaCl, 100 ml x 3. La capa orgánica se secó sobre sulfato de sodio anhidro durante la noche, el agente desecante se retiró por filtración, y el disolvente se retiró por evaporación rotativa, para obtener 42,02 g de 3-[(2-ciclopropil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali como materia aceitosa de color rojo, con un rendimiento de 99,6 %. El producto obtenido fue la configuración (2R,3S),(2R,3R),(2S,3R),(2S,3S) de 3-[(2-ciclopropil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali.
Etapa 3: Purificación de 3-[(2-ciclopropil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali por cromatografía de columna y el tratamiento de purificación relacionado
La muestra de la etapa 2 anterior se separó en columna de gel de sílice, utilizando diclorometano o triclorometano y metanol, amoniacados, como fase móvil y una placa de TLC para controlar la pureza de la muestra. Bajo el sistema de elución anterior, una muestra de una mezcla de cuatro configuraciones de (2R,3S),(2R,3R),(2S,3R),(2S,3S) de 3-[(2-ciclopropil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali podía ser purificada en dos tipos racémicos, (2R,3S),(2S,3R) y (2R,3R),(2S,3S), libres de álcali, dependiendo de la secuencia de elución, obteniéndose de ese modo la fracción eluida en primer lugar de 17,06 g de la configuración (2R,3S),(2S,3R) racémica de 3-[(2-ciclopropil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali con un rendimiento de 81,22 %, y la fracción eluida en segundo lugar de 16,63 g de la configuración (2R,3R),(2S,3S) de 3-[(2-ciclopropil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali con un rendimiento de 79,13 %.
Etapa 4: igual que la etapa 4 en el Ejemplo 1
Etapa 5: Configuración (2R,3S),(2S,3R) de bromuro de 3-[(2-ciclopropil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano

A un matraz de 100 ml, con forma de berenjena, se añadieron 2,612 g (9,1 mmol) de la configuración (2R,3S),(2S,3R) de la base y se disolvieron añadiendo 18 ml de cloroformo para obtener una solución transparente de color amarillo, a la que se añadieron 11,0342 g (51,3 mmol) de 3-bromopropoxibenceno y 50 ml de acetonitrilo, y la mezcla resultante se agitó entonces a temperatura ambiente para reaccionar durante 20-90 h bajo la protección de nitrógeno, siendo el final de la reacción controlado mediante TLC (condiciones de la TLC: cloroformo/metanol/agua amoniacal = 5,0 ml/1,5 ml/2 d). Una vez finalizada la reacción, el disolvente se retiró de la solución de reacción a 25-40 °C por evaporación rotativa bajo presión reducida mediante una bomba de agua, obteniéndose de ese modo una materia aceitosa de color amarillo, a la que se añadió éter etílico para precipitar una gran cantidad de sólido, y el sólido se recogió mediante filtración por succión para obtener 3,76 g de la configuración (2R,3S),(2S,3R) de bromuro de 3-[(2-ciclopropil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano como un sólido de color blancuzco, con un rendimiento de 82,31 %.
El compuesto preparado en el Ejemplo 23 se hizo reaccionar con Ag2Ü para retirar el átomo de bromo para obtener hidróxido, que podía hacerse reaccionar con otros ácidos para transformarlos en las sales correspondientes. Ejemplos de una sal de un ácido farmacéuticamente aceptable incluyen una sal derivada de un ácido inorgánico, como hidrocloruro, bromuro, yoduro, nitrato, carbonato, bicarbonato, fosfato, hidrofosfato, fosfato dihidroxilado, sulfato, disulfato o fosfito o similar; y una sal derivada de un ácido orgánico relativamente no tóxico como ácido acético, ácido propiónico, ácido isobutírico, ácido malónico, ácido benzoico, ácido succínico, ácido subérico, ácido fumárico, ácido mandélico, ácido ftálico, ácido bencensulfónico, ácido p-toluensulfónico, ácido cítrico, ácido tartárico, ácido metansulfónico, ácido glucurónico o ácido galactónico o similar. Los ejemplos incluyen además una sal de un aminoácido como arginina o similar.
RMN de 1H (D2O) 5 (ppm): 7,19-7,15 (m, 7H), 6,82-6,77 (m, 3H), 3,94-3,68 (m, 4H), 3,49-3,19 (m, 9H), 2,15 (m, 2H), 1,82-1,57 (m, 5H), 0,67-0,06 (m, 5H).
Ejemplo 24. Configuración (2R,3R),(2S,3S) de bromuro de 3-[(2-ciclopropil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano
La etapa 1 era igual que la etapa 1 en el Ejemplo 19
Las etapas 2 y 3 eran iguales que las etapas 2 y 3 en el Ejemplo 23
Etapa 4: igual que la etapa 4 en el Ejemplo 1
Etapa 5: Configuración (2R,3R),(2S,3S) de bromuro de 3-[(2-ciclopropil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano

A un matraz de 100 ml, con forma de berenjena, se añadieron 2,994 g (9,1 mmol) de la configuración (2R,3R),(2S,3S) de la base y se disolvieron añadiendo 18 ml de cloroformo para obtener una solución transparente de color amarillo, a la que se añadieron 11,034 g (51,3 mmol) de 3-bromopropoxibenceno y 50 ml de acetonitrilo, y la mezcla resultante se agitó entonces a temperatura ambiente para reaccionar durante 20-90 h bajo la protección de nitrógeno, siendo el final de la reacción controlado mediante TLC (condiciones de la TLC: cloroformo/metanol/agua amoniacal = 5,0 ml/1,5 ml/2 d). Una vez finalizada la reacción, el disolvente se retiró de la solución de reacción a 25 40 °C por evaporación rotativa bajo presión reducida mediante una bomba de agua, obteniéndose de ese modo una materia aceitosa de color amarillo, a la que se añadió éter etílico para precipitar una gran cantidad de sólido, y el sólido se recogió mediante filtración por succión para obtener 3,54 g de la configuración (2R,3R),(2S,3S) de bromuro de 3-[(2-ciclopropil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano como un sólido de color blancuzco, con un rendimiento de 77,49 %.
Los isómeros ópticos puros de cuatro configuraciones de (2R,3S),(2R,3R),(2S,3R),(2S,3S) de 3-[(2-ciclopropil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali podían obtenerse a partir de los Ejemplos 19 y 21, y las mezclas de las configuraciones (2R,3S),(2R,3R),(2S,3R),(2S,3S) de bromuro de 3-[(2-ciclopropil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano en diferentes proporciones se obtuvieron mezclando estos isómeros en cualquier cantidad y cualquier proporción seguida por cuaternización añadiendo 3-bromopropoxibenceno.
El compuesto preparado en el Ejemplo 24 se hizo reaccionar con Ag2Ü para retirar el átomo de bromo para obtener hidróxido, que podía hacerse reaccionar con otros ácidos para transformarlos en las sales correspondientes. Ejemplos de una sal de un ácido farmacéuticamente aceptable incluyen una sal derivada de un ácido inorgánico, como hidrocloruro, bromuro, yoduro, nitrato, carbonato, bicarbonato, fosfato, hidrofosfato, fosfato dihidroxilado, sulfato, disulfato o fosfito o similar; y una sal derivada de un ácido orgánico relativamente no tóxico como ácido acético, ácido propiónico, ácido isobutírico, ácido malónico, ácido benzoico, ácido succínico, ácido subérico, ácido fumárico, ácido mandélico, ácido ftálico, ácido bencensulfónico, ácido p-toluensulfónico, ácido cítrico, ácido tartárico, ácido metansulfónico, ácido glucurónico o ácido galactónico o similar. Los ejemplos incluyen además una sal de un aminoácido como arginina o similar.
RMN de 1H (D2O) 5 (ppm): 7,19-7,15 (m, 7H), 6,82-6,77 (m, 3H), 3,94-3,68 (m, 4H), 3,49-3,19 (m, 9H), 2,15 (m, 2H), 1,82-1,57 (m, 5H), 0,67-0,06 (m, 5H).
Ejemplo 25. Bromuro de (2S,3R)-3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-(2-fenoxi Etil)-1-azabiciclo[2,2,2]octano
Etapa 1: igual que la etapa 1 en el Ejemplo 1
Etapas 2 y 3: igual que las etapas 2 y 3 en el Ejemplo 3
Etapa 4: Preparación de 2-bromoetoxilbenceno

A un matraz de 150 ml, con tres bocas, se añadieron 9,5 g (101 mmol) de fenol, seguido de la adición de 4,25 g (106 mmol) de hidróxido de sodio y una solución de 47,11 g (250 mmol) de 1,2-dibromoetano en 30 ml de etanol absoluto, y la mezcla resultante se hizo reaccionar entonces bajo calentamiento y reflujo en baño de aceite, precipitándose un sólido blanco, hasta reacción esencialmente completa de fenol controlada por TLC (condiciones de la TLC: éter de petróleo/acetato de etilo = 5,0 ml/1,0 ml). Una vez finalizada la reacción, el sólido se retiró por filtración, y el disolvente se retiró del filtrado a 50 °C o menor por evaporación rotativa bajo presión reducida mediante una bomba de agua, obteniéndose de ese modo una materia aceitosa que contenía un sólido blanco, al que se le añadió éter de petróleo y después se dejó reposar durante la noche. El sólido precipitado se retiró por filtración, y el disolvente se retiró del filtrado a 25-40 °C por evaporación rotativa bajo presión reducida mediante una bomba de agua, obteniéndose de ese modo una materia aceitosa de color amarillo. La materia aceitosa fue destilada bajo presión reducida, para recoger una fracción a 8 mmHg, 118-122 °C, obteniéndose de ese modo 11,31 g de una materia aceitosa incolora, transparente, con un rendimiento de 55,7 % y pureza de 95,6065 % detectada por CG.
RMN de 1H (CDCls) 5 (ppm): 7,15-6,77 (m, 5H), 4,45 (t, 2H), 3,79 (t, 2H).
Etapa 5: Bromuro de (2S,3R)-3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-(2-fenoxietil)-1-azabiciclo[2,2,2]octano

A un matraz de 50 ml, con forma de berenjena, se añadieron 1,436 g (4,55 mmol) de la configuración (2S,3R) de la base y se disolvieron añadiendo 9 ml de cloroformo para obtener una solución transparente de color amarillo, a la que se añadieron 5,65 g (28,1 mmol) de 2-bromoetoxibenceno y 25 ml de acetonitrilo, y la mezcla resultante se agitó entonces a temperatura ambiente para reaccionar durante 20-90 h bajo la protección de nitrógeno, siendo el final de la reacción controlado mediante TLC (condiciones de la TLC: cloroformo/metanol/agua amoniacal = 5,0 ml/1,5 ml/2 d). Una vez finalizada la reacción, el disolvente se retiró de la solución de reacción a 25-40 °C por evaporación rotativa bajo presión reducida mediante una bomba de agua, obteniéndose de ese modo una materia aceitosa de color amarillo, a la que se añadió éter etílico para precipitar una gran cantidad de sólido, y el sólido se recogió mediante filtración por succión para obtener 1,857 g de (2S,3R)-3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxietil)-1-azabiciclo[2,2,2]octano como un sólido de color blancuzco, con un rendimiento de 79,12 %.
El compuesto preparado en el Ejemplo 25 se hizo reaccionar con Ag2Ü para retirar el átomo de bromo para obtener hidróxido, que podía reaccionar con otros ácidos para transformarlos en las sales correspondientes. Ejemplos de una sal de un ácido farmacéuticamente aceptable incluyen una sal derivada de un ácido inorgánico, como hidrocloruro, bromuro, yoduro, nitrato, carbonato, bicarbonato, fosfato, hidrofosfato, fosfato dihidroxilado, sulfato, disulfato o fosfito o similar; y una sal derivada de un ácido orgánico relativamente no tóxico como ácido acético, ácido propiónico, ácido isobutírico, ácido malónico, ácido benzoico, ácido succínico, ácido subérico, ácido fumárico, ácido mandélico, ácido ftálico, ácido bencensulfónico, ácido p-toluensulfónico, ácido cítrico, ácido tartárico, ácido metansulfónico, ácido glucurónico o ácido galactónico o similar. Los ejemplos incluyen además una sal de un aminoácido como arginina o similar.
RMN de 1H (D2O) 5 (ppm): 7,18-6,76 (m, 10H), 5,86 (t, 2H), 4,39 (t, 2H), 3,98-3,71 (s, 2H), 3,47-3,18 (m, 7H), 1,92 (m, 1H), 1,81-1,66 (m, 5H), 1,59-1,36 (m, 8H).
Ejemplo 26. Bromuro de (2R,3R)-3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1 -(2-fenoxi Etil)-1 -azabiciclo[2,2,2]octano
Etapa 1: igual que la etapa 1 en el Ejemplo 1
Las etapas 2 y 3 eran iguales que las etapas 2 y 3 en el Ejemplo 3
La etapa 4 era igual que la etapa 4 en el Ejemplo 25
Etapa 5: Bromuro de (2R,3R)-3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-(2-fenoxietil)-1-azabiciclo[2,2,2]octano

A un matraz de 50 ml, con forma de berenjena, se añadieron 1,435 g (4,55 mmol) de la configuración (2R,3R) de la base y se disolvieron añadiendo 9 ml de cloroformo para obtener una solución de color amarillo transparente, a la que se añadieron 5,65 g (28,1 mmol) de 2-bromoetoxibenceno y 25 ml de acetonitrilo, y la mezcla resultante se agitó entonces a temperatura ambiente para reaccionar durante 20-90 h bajo la protección de nitrógeno, siendo el final de la reacción controlado mediante TLC (condiciones de TLC: cloroformo/metanol/agua amoniacal = 5,0 ml/1,5 ml/2 d). Una vez finalizada la reacción, el disolvente se retiró de la solución de reacción a 25-40 °C por evaporación rotativa bajo presión reducida mediante una bomba de agua, obteniéndose de ese modo una materia aceitosa de color amarillo, a la que se añadió éter etílico para precipitar una gran cantidad de sólido, y el sólido se recogió mediante filtración por succión para obtener 1,797 g de (2R,3R)-3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxietil)-1-azabiciclo[2,2,2]octano como un sólido de color blancuzco, con un rendimiento de 76,56 %.
El compuesto preparado en el Ejemplo 26 se hizo reaccionar con Ag2Ü para retirar el átomo de bromo para obtener hidróxido, que podía hacerse reaccionar con otros ácidos para transformarlos en las sales correspondientes. Ejemplos de una sal de un ácido farmacéuticamente aceptable incluyen una sal derivada de un ácido inorgánico, como hidrocloruro, bromuro, yoduro, nitrato, carbonato, bicarbonato, fosfato, hidrofosfato, fosfato dihidroxilado, sulfato, disulfato o fosfito o similar; y una sal derivada de un ácido orgánico relativamente no tóxico como ácido acético, ácido propiónico, ácido isobutírico, ácido malónico, ácido benzoico, ácido succínico, ácido subérico, ácido fumárico, ácido mandélico, ácido ftálico, ácido bencensulfónico, ácido p-toluensulfónico, ácido cítrico, ácido tartárico, ácido metansulfónico, ácido glucurónico o ácido galactónico o similar. Los ejemplos incluyen además una sal de un aminoácido como arginina o similar.
RMN de 1H (D2O) 5 (ppm): 7,19-6,78 (m, 10H), 5,88 (t, 2H), 4,38 (t, 2H), 3,99-3,70 (s, 2H), 3,47-3,17 (m, 7H), 1,93 (m, 1H), 1,81-1,67 (m, 5H), 1,60-1,37 (m, 8H).
Ejemplo 27. Bromuro de (2R,3R)-3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1 -fenoxime thyl-1 -azabiciclo[2,2,2]octano
Etapa 1: igual que la etapa 1 en el Ejemplo 1
Las etapas 2 y 3 eran iguales que las etapas 2 y 3 en el Ejemplo 3
Etapa 4: Preparación del bromometoxilbenceno

A un matraz de 150 ml, con tres bocas, se añadieron 9,5 g (101 mmol) de fenol, seguido de la adición de 4,25 g (106 mmol) de hidróxido de sodio y una solución de 43,51 g (250 mmol) de 1,2-dibromometano en 30 ml de etanol absoluto, y la mezcla resultante se hizo reaccionar después bajo calentamiento y reflujo en baño de aceite, precipitándose un sólido blanco, hasta reacción esencialmente completa de fenol controlada por TLC (condiciones de la TLC: éter de petróleo/acetato de etilo = 5,0 ml/1,0 ml). Una vez finalizada la reacción, el sólido se retiró por filtración, y el disolvente se retiró del filtrado a 50 °C o menor por evaporación rotativa bajo presión reducida mediante una bomba de agua, obteniéndose de ese modo una materia aceitosa que contenía un sólido blanco, al que se le añadió éter de petróleo y después se dejó reposar durante la noche. El sólido precipitado se retiró por filtración, y el disolvente se retiró del filtrado a 25-40 °C por evaporación rotativa bajo presión reducida mediante una bomba de agua, obteniéndose de ese modo una materia aceitosa de color amarillo. La materia aceitosa fue destilada bajo presión reducida, para recoger una fracción a 8 mmHg, 116-120 °C, obteniéndose de ese modo 10,75 g de una materia aceitosa incolora, transparente, con un rendimiento de 56,9 % y pureza de 95,6065 % detectada por CG.
RMN de 1H (CDCls) 5 (ppm): 7,16-6,76 (m, 5H), 5,95 (s, 2H)
Etapa 5: Bromuro de (2R,3R)-3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-fenoximetil-1-azabiciclo[2,2,2]octano

A un matraz de 100 ml, con forma de berenjena, se añadieron 2,871 g (9,1 mmol) de la configuración (2R,3R) de la base y se disolvieron añadiendo 18 ml de cloroformo para obtener una solución de color amarillo transparente, a la que se añadieron 10,29 g (55 mmol) de bromometoxilbenceno y 25 ml de acetonitrilo, y la mezcla resultante se agitó entonces a temperatura ambiente para reaccionar durante 20-90 h bajo la protección de nitrógeno, siendo el final de la reacción controlado mediante TLC (condiciones de TLC: cloroformo/metanol/agua amoniacal = 5,0 ml/1,5 ml/2 d). Una vez finalizada la reacción, el disolvente se retiró de la solución de reacción a 25-40 °C por evaporación rotativa bajo presión reducida mediante una bomba de agua, obteniéndose de ese modo una materia aceitosa de color amarillo, a la que se añadió éter etílico para precipitar una gran cantidad de sólido, y el sólido se recogió mediante filtración por succión para obtener 3,751 g de (2R,3R)-3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-(fenoximetil)-1-azabiciclo[2,2,2]octano como un sólido de color blancuzco, con un rendimiento de 80,2 %.
El compuesto preparado en el Ejemplo 27 se hizo reaccionar con Ag2Ü para retirar el átomo de bromo para obtener hidróxido, que podía hacerse reaccionar con otros ácidos para transformarlos en las sales correspondientes. Ejemplos de una sal de un ácido farmacéuticamente aceptable incluyen una sal derivada de un ácido inorgánico, como hidrocloruro, bromuro, yoduro, nitrato, carbonato, bicarbonato, fosfato, hidrofosfato, fosfato dihidroxilado, sulfato, disulfato o fosfito o similar; y una sal derivada de un ácido orgánico relativamente no tóxico como ácido acético, ácido propiónico, ácido isobutírico, ácido malónico, ácido benzoico, ácido succínico, ácido subérico, ácido fumárico, ácido mandélico, ácido ftálico, ácido bencensulfónico, ácido p-toluensulfónico, ácido cítrico, ácido tartárico, ácido metansulfónico, ácido glucurónico o ácido galactónico o similar. Los ejemplos incluyen además una sal de un aminoácido como arginina o similar.
RMN de 1H (D2O) 5 (ppm): 7,21-6,76 (m, 10H), 5,89 (s, 2H), 3,97-3,73 (s, 2H), 3,48-3,18 (m, 7H), 1,95 (m, 1H), 1,80 1,58 (m, 5H), 1,61-1,38 (m, 8H).
Ejemplo 28. Cloruro de (2R,3R)-3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1 -fenoximetoximetil-1 -azabiciclo[2,2,2]octano
La etapa 1 era igual que la etapa 1 en el Ejemplo 1
Las etapas 2 y 3 eran iguales que las etapas 2 y 3 en el Ejemplo 3
Etapa 4: Preparación del éter de clorometoximetilo y fenilo

A un matraz de 150 ml, con tres bocas, se añadieron 9,5 g (101 mmol) de fenol, seguido de la adición de 4,25 g (106 mmol) de hidróxido de sodio y una solución de 28,75 g (250 mmol) de 1,3-diclorometiléter en 30 ml de etanol absoluto, y la mezcla resultante se hizo reaccionar entonces bajo calentamiento y reflujo en baño de aceite, precipitándose un sólido blanco, hasta reacción esencialmente completa de fenol controlada por TLC (condiciones de la TLC: éter de petróleo/acetato de etilo = 5,0 ml/1,0 ml). Una vez finalizada la reacción, el sólido se retiró por filtración, y el disolvente se retiró del filtrado a 50 °C o menor por evaporación rotativa bajo presión reducida mediante una bomba de agua, obteniéndose de ese modo una materia aceitosa que contenía un sólido blanco, al cual se le añadió éter de petróleo y después se dejó reposar durante la noche. El sólido precipitado se retiró por filtración, y el disolvente se retiró del filtrado a 25-40 °C por evaporación rotativa bajo presión reducida mediante una bomba de agua, obteniéndose de ese modo una materia aceitosa de color amarillo. La materia aceitosa fue destilada bajo presión reducida, para recoger una fracción a 8 mmHg, 106-119 °C, obteniéndose de ese modo 8,38 g de una materia aceitosa incolora, transparente, con un rendimiento de 48,1 % y pureza de 97,62 % detectada por CG.
RMN de 1H (CDCls) 5 (ppm): 7,19-6,79 (m, 5H), 6,01 (s, 2H), 5,47 (s, 2H).
Etapa 5: Cloruro de (2R,3R)-3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-fenoximetoximetil-1-azabiciclo[2,2,2]octano

A un matraz de 100 ml, con forma de berenjena, se añadieron 2,871 g (9,1 mmol) de la configuración (2R,3R) de la base y se disolvieron añadiendo 18 ml de cloroformo para obtener una solución transparente de color amarillo, a la que se añadieron 9,49 g (55 mmol) de éter de clorometoximetilo y fenilo y 25 ml de acetonitrilo, y la mezcla resultante se agitó entonces a temperatura ambiente para reaccionar durante 20-90 h bajo la protección de nitrógeno, siendo el final de la reacción controlado mediante TLC (condiciones de la TLC: cloroformo/metanol/agua amoniacal = 5,0 ml/1,5 ml/2 d). Una vez finalizada la reacción, el disolvente se retiró de la solución de reacción a 25 40 °C por evaporación rotativa bajo presión reducida mediante una bomba de agua, obteniéndose de ese modo una materia aceitosa de color amarillo, a la que se añadió éter etílico para precipitar una gran cantidad de sólido, y el sólido se recogió mediante filtración por succión para obtener 3,13 g de cloruro de (2R,3R)-3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-fenoximetoximetil)-1-azabiciclo[2,2,2]octano como un sólido de color blancuzco, con un rendimiento de 69,2 %.
El compuesto preparado en el Ejemplo 28 se hizo reaccionar con Ag2Ü para retirar el átomo de cloro para obtener hidróxido, que podía hacerse reaccionar con otros ácidos para transformarlos en las sales correspondientes. Ejemplos de una sal de un ácido farmacéuticamente aceptable incluyen una sal derivada de un ácido inorgánico, como hidrocloruro, bromuro, yoduro, nitrato, carbonato, bicarbonato, fosfato, hidrofosfato, fosfato dihidroxilado, sulfato, disulfato o fosfito o similar; y una sal derivada de un ácido orgánico relativamente no tóxico como ácido acético, ácido propiónico, ácido isobutírico, ácido malónico, ácido benzoico, ácido succínico, ácido subérico, ácido fumárico, ácido mandélico, ácido ftálico, ácido bencensulfónico, ácido p-toluensulfónico, ácido cítrico, ácido tartárico, ácido metansulfónico, ácido glucurónico o ácido galactónico o similar. Los ejemplos incluyen además una sal de un aminoácido como arginina o similar.
RMN de 1H (D2O) 6 (ppm): 7,19-6,77 (m, 10H), 6,02 (s, 2H), 5,32 (s, 2H), 3,98-3,68 (s, 2H), 3,49-3,19 (m, 7H), 1,96 (m, 1H), 1,82-1,57 (m, 5H), 1,60-1,36 (m, 8H).
Ejemplo 29. Bromuro de (2R,3R)-3-[(2-ciclopentil-2-hidroxil-2-naftil)etoxil]-1 -(3-fenoxipropil)-1 -azabiciclo[2,2,2]octano
Etapa 1: Preparación de 230 g de 1-naftil-1-ciclopentiloxirano según el procedimiento en la bibliografía(2)
Etapa 2: Preparación de 3-[(2-ciclopentil-2-hidroxil-2-naftil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali a partir de R-3-quinuclidinol

A 18,72 g (147 mmol) de R-3-quinuclidinol disponible comercialmente se añadieron 190 ml de DMSÜ, seguido de la adición de 7,59 g (190 mmol) de hidruro de sodio, y la mezcla se hizo reaccionar a 20-60 °C durante 0,5-12 h, y después se enfrió a temperatura ambiente, y a ello se añadió una solución de 45,15 g (190 mmol) de 1 -naftil-1 -ciclopentiloxirano (autopreparado) en 45 ml de DMSÜ, y una vez finalizada la adición, la mezcla resultante se calentó a 20-70 °C en baño de aceite para reaccionar durante 0,5-12 horas. Bajo la condición de baño de hielo, se añadieron 120 ml de agua helada a una temperatura interna de 30 °C o menor. La mezcla de reacción se extrajo con éter isopropílico, 100 ml x 3; y la capa de éter se combinó y se lavó con una solución acuosa saturada de NaCl, 100 ml x 3. La capa orgánica se secó sobre sulfato de sodio anhidro durante la noche, el agente desecante se retiró por filtración, y el disolvente se retiró bajo presión reducida para obtener 51,71 g de 3-[(2-ciclopentil-2-hidroxil-2naftil)etoxil]-1-azabiciclo [2,2,2]octano libre de álcali como materia aceitosa de color rojo, con un rendimiento de 96,38 %. El producto obtenido fue la configuración (2R,3R),(2S,3R) de 3-[(2-ciclopentil-2-hidroxil-2-naftil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali.
Etapa 3: Purificación de 3-[(2-ciclopentil-2-hidroxil-2-naftil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali por cromatografía de columna y el tratamiento de purificación relacionado
La muestra de la etapa 2 anterior se separó en columna de gel de sílice, utilizando diclorometano o triclorometano y metanol, amoniacados, como fase móvil y una placa de TLC para controlar la pureza de la muestra. Bajo el sistema de elución anterior, una muestra de una mezcla de ambas configuraciones (2R,3R) y (2S,3R) de 3-[(2-ciclopentil-2-hidroxil-2-naftil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali podía ser purificada en dos tipos de (2R,3R) y (2S,3R) libres de álcali, dependiendo de la secuencia de elución, obteniéndose de ese modo 22,36 g de la configuración (2R,3R) de 3-[(2-ciclopentil-2-hidroxil-2-naftil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali con un rendimiento de 86,5 %, y 20,05 g de la configuración (2S,3R) de 3-[(2-ciclopentil-2-hidroxil-2-naftil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali con un rendimiento de 77,55 %.
Etapa 4: igual que la etapa 4 en el Ejemplo 1
Etapa 5: Bromuro de (2R,3R)-3-[(2-ciclopentil-2-hidroxil-2-naftil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano

A un matraz de 100 ml, con forma de berenjena, se añadieron 3,322 g (9,1 mmol) de la configuración (2R,3R) de la base y se disolvieron añadiendo 18 ml de cloroformo para obtener una solución transparente de color amarillo, a la que se añadieron 11,033 g (51,3 mmol) de 3-bromopropoxibenceno y 50 ml de acetonitrilo, y la mezcla resultante se agitó entonces a temperatura ambiente para reaccionar durante 20-90 h bajo la protección de nitrógeno, siendo el final de la reacción controlado mediante TLC (condiciones de la TLC: cloroformo/metanol/agua amoniacal = 5,0 ml/1,5 ml/2 d). Una vez finalizada la reacción, el disolvente se retiró de la solución de reacción a 25-40 °C por evaporación rotativa bajo presión reducida mediante una bomba de agua, obteniéndose de ese modo una materia aceitosa de color amarillo, a la que se añadió éter etílico para precipitar una gran cantidad de sólido, y el sólido se recogió mediante filtración por succión para obtener 4,238 g de (2R,3R)-3-[(2-ciclopentil-2-hidroxil-2-naftil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano como un sólido de color blancuzco, con un rendimiento de 80,3 %.
El compuesto preparado en el Ejemplo 29 se hizo reaccionar con Ag2Ü para retirar el átomo de bromo para obtener hidróxido, que podía hacerse reaccionar con otros ácidos para transformarlos en las sales correspondientes.
Ejemplos de una sal de un ácido farmacéuticamente aceptable incluyen una sal derivada de un ácido inorgánico, como hidrocloruro, bromuro, yoduro, nitrato, carbonato, bicarbonato, fosfato, hidrofosfato, fosfato dihidroxilado, sulfato, disulfato o fosfito o similar; y una sal derivada de un ácido orgánico relativamente no tóxico como ácido acético, ácido propiónico, ácido isobutírico, ácido malónico, ácido benzoico, ácido succínico, ácido subérico, ácido fumárico, ácido mandélico, ácido ftálico, ácido bencensulfónico, ácido p-toluensulfónico, ácido cítrico, ácido tartárico, ácido metansulfónico, ácido glucurónico o ácido galactónico o similar. Los ejemplos incluyen además una sal de un aminoácido como arginina o similar.
RMN de 1H (D2O) (ppm): 57,21-6,66 (m, 12H), 54,17-3,65 (m, 5H), 53,44-3,13 (m, 8H), 52,15 (m, 2H), 2,00 (m, 1H), 51,84-1,42 (m, 13H).
Ejemplo 30. Bromuro de (2R,3R)-3-[(2-ciclopentil-2-hidroxil-2-o-clorofenil)etoxil]-1 -(3-fenoxipropil)-1 -azabiciclo[2,2,2]octano
Etapa 1: 1-o-clorofenil-1-ciclopentiloxirano

Se hizo reaccionar ciclopentil-o-clorofenilcetona comercialmente disponible como material de partida para obtener 1-o-clorofenil-1-ciclopentiloxirano según la bibliografía(1).
A un matraz de 3 litros, con tres bocas, se añadieron 1.880 ml de acetonitrilo, seguido de la adición de sulfuro de dimetilo y sulfato de dimetilo, con un leve desprendimiento de calor, y la mezcla resultante se agitó entonces a temperatura ambiente durante 1,5 h y después se dejó reposar durante la noche. Se añadió NaH (60 %) en porciones adecuadas a la solución de reacción con agitación durante 30-40 min, generándose un gas (préstese atención a tener el matraz externamente conectado a un condensador Allihn y un tubo de secado de cloruro de calcio anhidro), y se agitó entonces a temperatura ambiente durante 1 hora, seguido de la adición gota a gota de 355 g de ciclopentil-o-clorofenil-cetona durante aproximadamente 10 min, y la mezcla resultante se hizo reaccionar posteriormente en baño de aceite a 40-45 °C durante 90 min siendo después agitada a temperatura ambiente durante 20 min, y después se enfrió a temperatura ambiente y se dejó durante la noche. La solución de reacción se transfirió a un matraz con forma de berenjena, el disolvente se retiró por succión usando una bomba de agua (40 42 °C, -0,095 MPa), y el sólido restante se enfrió a 0-5 °C en baño de hielo, seguido de la adición gota a gota de 2.630 ml de agua helada (siendo la temperatura interior controlada a 0-5 °C) durante aproximadamente 1 hora, y la solución de reacción se extrajo con éter isopropílico (650 ml x 3), la capa de éter se combinó y se lavó con agua hasta que es neutra (siendo el pH de los lavados con agua de 7,0), y la capa de éter se secó sobre sulfato de sodio anhidro durante la noche. El agente desecante se retiró por filtración, el disolvente se retiró bajo presión reducida por succión usando una bomba de agua (40-42 °C, -0,095 MPa) y, posteriormente, se recogieron 315,8 g de fracción a 119-132 °C/3 mmHg bajo presión reducida usando una bomba de aceite, con un rendimiento de 83,36 %.
RMN de 1H (CDCls) 5 (ppm): 7,19-7,07 (m, 4H), 2,92-2,64 (m, 1H), 2,21 (s, 2H), 1,61-1,37 (m, 8H).
Etapa 2: Preparación de 3-[(2-ciclopentil-2-hidroxil-2-o-clorofenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali a partir de R-3-quinuclidinol

A 18,72 g (147 mmol) de R-3-quinuclidinol disponible comercialmente se añadieron 190 ml de DMSO, seguido de la adición de 7,59 g (190 mmol) de hidruro de sodio, y la mezcla se hizo reaccionar a 20-60 °C durante 0,5-12 h, y después se enfrió a temperatura ambiente, y a ello se añadió una solución de 42,317 g (190 mmol) de 1-o-clorofenil-1-ciclopentiloxirano (autopreparado) en 45 ml de DMSO, y una vez finalizada la adición, la mezcla resultante se calentó a 20-70 °C en baño de aceite para reaccionar durante 0,5-12 horas. Bajo la condición de baño de hielo, se añadieron 120 ml de agua helada a una temperatura interna de 30 °C o menor. La mezcla de reacción se extrajo con éter isopropílico, 100 ml x 3; y la capa de éter se combinó y se lavó con una solución acuosa saturada de NaCl, 100 ml x 3. La capa orgánica se secó sobre sulfato de sodio anhidro durante la noche, el agente desecante se retiró por filtración, y el disolvente se retiró bajo presión reducida para obtener 49,48 g de 3-[(2-ciclopentil-2-hidroxil-2-oclorofenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali como materia aceitosa de color rojo, con un rendimiento de 96,31 %. El producto obtenido fue (2R,3R),(2S,3R)-3-[(2-ciclopentil-2-hidroxil-2-o-clorofenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali.
Etapa 3: Purificación de 3-[(2-ciclopentil-2-hidroxil-2-o-clorofenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali por cromatografía de columna y el tratamiento de purificación relacionado
La muestra de la etapa 2 anterior se separó en columna de gel de sílice, utilizando diclorometano o triclorometano y metanol, amoniacados, como fase móvil y una placa de TLC para controlar la pureza de la muestra. Bajo el sistema de elución anterior, una muestra de una mezcla de ambas configuraciones (2R,3R) y (2S,3R) de 3-[(2-ciclopentil-2-hidroxil-2-o-clorofenil)etoxil]-1-azabiciclo[2,2,2]octano libres de álcali podía ser purificada en dos tipos (2R,3R) y (2S, 3R) libres de álcali, dependiendo de la secuencia de elución, obteniendo de este modo 21,03 g de (2R,3R)-3-[(2-ciclopentil-2-hidroxil-2-o-clorofenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali con un rendimiento de 85,0 %, y 21,15 g de (2S,3R)-3-[(2-ciclopentil-2-hidroxil-2-o-clorofenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali con un rendimiento de 85,485 %.
Etapa 4: igual que la etapa 4 en el Ejemplo 1
Etapa 5: Bromuro de (2R,3R)-3-[(2-ciclopentil-2-hidroxil-2-o-clorofenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano

A un matraz de 100 ml, con forma de berenjena, se añadieron 3,18 g (9,1 mmol) de la configuración (2R,3R) de la base y se disolvieron añadiendo 18 ml de cloroformo para obtener una solución transparente de color amarillo, a la que se añadieron 11,033 g (51,3 mmol) de 3-bromopropoxibenceno y 50 ml de acetonitrilo, y la mezcla resultante se agitó entonces a temperatura ambiente para reaccionar durante 20-90 h bajo la protección de nitrógeno, siendo el final de la reacción controlado mediante TLC (condiciones de la TLC: cloroformo/metanol/agua amoniacal = 5,0 ml/1,5 ml/2 d). Una vez finalizada la reacción, el disolvente se retiró de la solución de reacción a 25-40 °C por evaporación rotativa bajo presión reducida mediante una bomba de agua, obteniéndose de ese modo una materia aceitosa de color amarillo, a la que se añadió éter etílico para precipitar una gran cantidad de sólido, y el sólido se recogió mediante filtración por succión para obtener 4,25 g de bromuro de (2R,3R)-3-[(2-ciclopentil-2-hidroxil-2-oclorofenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano como un sólido de color blancuzco, con un rendimiento de 82,73 %.
El compuesto preparado en el Ejemplo 30 se hizo reaccionar con Ag2Ü para retirar el átomo de bromo para obtener hidróxido, que podía hacerse reaccionar con otros ácidos para transformarlos en las sales correspondientes.
Ejemplos de una sal de un ácido farmacéuticamente aceptable incluyen una sal derivada de un ácido inorgánico, como hidrocloruro, bromuro, yoduro, nitrato, carbonato, bicarbonato, fosfato, hidrofosfato, fosfato dihidroxilado, sulfato, disulfato o fosfito o similar; y una sal derivada de un ácido orgánico relativamente no tóxico como ácido acético, ácido propiónico, ácido isobutírico, ácido malónico, ácido benzoico, ácido succínico, ácido subérico, ácido fumárico, ácido mandélico, ácido ftálico, ácido bencensulfónico, ácido p-toluensulfónico, ácido cítrico, ácido tartárico, ácido metansulfónico, ácido glucurónico o ácido galactónico o similar. Los ejemplos incluyen además una sal de un aminoácido como arginina o similar.
RMN de 1H (D2O) (ppm): 57,33-6,65 (m, 9H), 54,16-3,63 (m, 5H), 53,45-3,12 (m, 8H), 52,13 (m, 2H), 2,02 (m, 1H), 5 1,83-1,43 (m, 13H).
Ejemplo 31. Bromuro de (2R,3R)-3-[(2-ciclopentil-2-hidroxil-2-(3-piridil))etoxil]-1 -(3-fenoxipropil)-1 -azabiciclo[2,2,2]octano
Etapa 1: Preparación de ciclopentil-3-piridil-cetona

En un matraz de 5 litros, con tres bocas, conectado externamente a un condensador Allihn y un tubo de secado de cloruro de calcio anhidro y un termómetro se colocaron 102,06 g de virutas de magnesio, a ello se añadieron y agitaron 1.600 ml de THF y 1,06 g de yodo, y después, a la solución de reacción, se añadieron gota a gota 78,28 g
de bromociclopentano. A aproximadamente 5 minutos después de que la reacción se iniciara por completo, la solución de reacción pasó gradualmente de tener un color marrón rojizo claro a ser incolora, aumentando la temperatura interna automáticamente a 63-65 °C. Después, se añadieron gota a gota 547,93 g de bromociclopentano adicional durante aproximadamente 35 min, aumentando su temperatura de reflujo gradualmente a 75-77 °C. La mezcla de reacción se hizo reaccionar bajo reflujo en baño de aceite durante 2 horas.
A la solución de reacción anterior se añadieron gota a gota 412,74 g de 3-piridilcarbonitrilo diluido en 1.600 ml de THF, y la mezcla resultante se hizo entonces reaccionar bajo reflujo en baño de aceite durante 4 horas. La temperatura interior se redujo a 5-10 °C, se añadieron, gota a gota, 96 ml de agua helada, y después se agitó durante 20 minutos, seguido de la adición de HCl para ajustar el pH = 2,0, y la mezcla resultante se calentó a reflujo durante 3 horas, la capa orgánica fue entonces separada, y una vez enfriada, la fase acuosa se extrajo con éter isopropílico (200 ml x 3). La capa orgánica se combinó y se lavó con Na2CÜ3 1 % (500 x 3) y agua (500 x 3) de forma consecutiva, y la capa orgánica se secó sobre sulfato de sodio anhidro durante la noche. El agente desecante se retiró por filtración, el disolvente se retiró por evaporación (42 °C, -0,095 MPa) y posteriormente se recogieron 332,26 g de materia aceitosa a 122-130 °C/6-7 mmHg por destilación bajo presión reducida usando una bomba de aceite.
RMN de 1H (CDCls) 5 (ppm): 9,31-7,70 (m, 4H), 2,36 (m, 1H), 1,63-1,47 (m, 8H).
Etapa 2: preparación de 1-(3-piridil)-1-ciclopentiloxirano
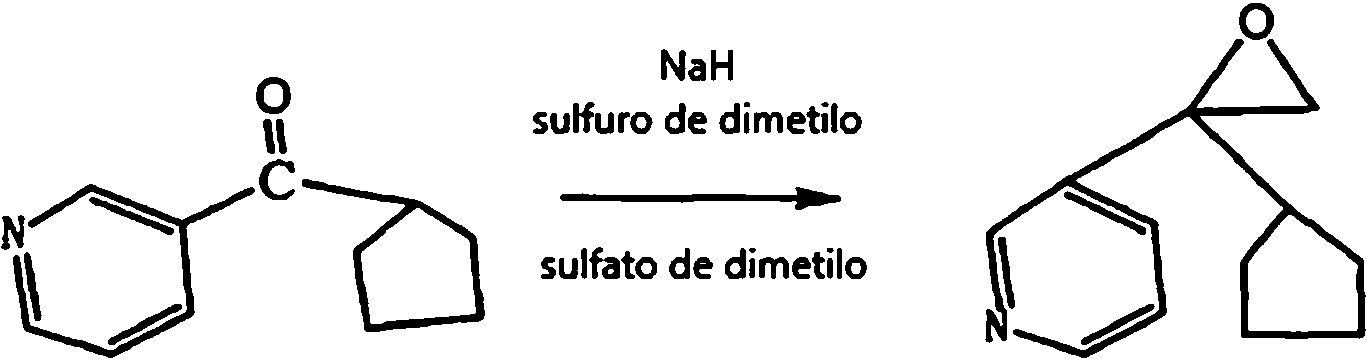
Se hizo reaccionar ciclopentil-3-piridil-cetona como material de partida para obtener 1 -(3-piridil)-1 -ciclopentiloxirano según la bibliografía(1).
A un matraz de 3 litros, con tres bocas, se añadieron 1.880 ml de acetonitrilo, seguido de la adición de sulfuro de dimetilo y sulfato de dimetilo, con un leve desprendimiento de calor, y la mezcla resultante se agitó entonces a temperatura ambiente durante 1,5 h y después se dejó reposar durante la noche. A la solución de reacción se añadió NaH (60 %) en porciones adecuadas con agitación durante 30-40 min, generándose un gas (préstese atención a tener el matraz externamente conectado a un condensador Allihn y un tubo de secado de cloruro de calcio anhidro), y después se agitó a temperatura ambiente durante 1 hora, seguido de la adición gota a gota de 300 g de ciclopentil-3-piridil-cetona durante aproximadamente 10 min, y la mezcla resultante se hizo reaccionar en baño de aceite a 40-45 °C durante 90 min siendo después agitada a temperatura ambiente durante 20 min, y después se enfrió a temperatura ambiente y se dejó reposar durante la noche. La solución de reacción se transfirió a un matraz con forma de berenjena, el disolvente se retiró por succión usando una bomba de agua (40-42 °C, -0,095 MPa), y el sólido restante se enfrió a 0-5 °C en baño de hielo, seguido de la adición gota a gota de 2.630 ml de agua helada (siendo la temperatura interior controlada a 0-5 °C) durante aproximadamente 1 hora, y la solución de reacción se extrajo con éter isopropílico (650 ml x 3), la capa de éter se combinó y se lavó con agua hasta que es neutra (siendo el pH de los lavados con agua de 7,0), y la capa de éter se secó sobre sulfato de sodio anhidro durante la noche. El agente desecante se retiró por filtración, el disolvente se retiró bajo presión reducida por succión usando una bomba de agua (40-42 °C, -0,095 MPa), y posteriormente se recogieron 285,5 g de fracción a 121-131 °C/3 mmHg bajo presión reducida usando una bomba de aceite.
RMN de 1H (CDCls) 5 (ppm): 8,68-7,50 (m, 4H), 2,91-2,66 (m, 2H), 2,23-1,29 (m, 9H).
Etapa 3: Preparación de 3-[(2-ciclopentil-2-hidroxil-2-(3-piridil))etoxil]-1-azabiciclo[2,2,2]octano libre de álcali a partir de R-3-quinuclidinol

A 18,72 g (147 mmol) de R-3-quinuclidinol disponible comercialmente se añadieron 190 ml de DMSO, seguido de la adición de 7,59 g (190 mmol) de hidruro de sodio, y la mezcla se hizo reaccionar a 20-60 °C durante 0,5-12 h, y después se enfrió a temperatura ambiente, y a ello se añadió una solución de 35,91 g (190 mmol) de 1-(3-piridil)-1-ciclopentiloxirano (autopreparado) en 45 ml de DMSO, y una vez finalizada la adición, la mezcla resultante se calentó a 20-70 °C en baño de aceite para reaccionar durante 0,5-12 horas. Bajo la condición de baño de hielo, se añadieron 120 ml de agua helada a una temperatura interna de 30 °C o menor. La mezcla de reacción se extrajo con éter isopropílico, 100 ml x 3; y la capa de éter se combinó y se lavó con una solución acuosa saturada de NaCl, 100 ml x 3. La capa orgánica se secó sobre sulfato de sodio anhidro durante la noche, el agente desecante se retiró por filtración, y el disolvente se retiró bajo presión reducida para obtener 45,66 g de 3-[(2-ciclopentil-2-hidroxil-2-(3-piridil))etoxil]-1-azabiciclo [2,2,2] octano libre de álcali como materia aceitosa de color rojo, con un rendimiento de 98,3 %. El producto obtenido fue (2R,3R),(2S,3R)-3-[(2-ciclopentil-2-hidroxil-2-(3-piridil))etoxil]-1-azabiciclo[2,2,2]octano libre de álcali.
Etapa 4: Purificación de 3-[(2-ciclopentil-2-hidroxil-2-(3-piridil))etoxil]-1-azabiciclo[2,2,2]octano libre de álcali por cromatografía de columna y el tratamiento de purificación relacionado
La muestra de la etapa 3 anterior se separó en columna de gel de sílice, utilizando diclorometano o triclorometano y metanol, amoniacados, como fase móvil y una placa de TLC para controlar la pureza de la muestra. Bajo el sistema de elución anterior, una muestra de una mezcla de ambas configuraciones (2R,3R) y (2S,3R) de 3-[(2-ciclopentil-2-hidroxil-2-(3-piridil))etoxil]-1-azabiciclo[2,2,2]octano álcali libre podía ser purificada en dos tipos de (2R,3R) y (2S,3R) libre de álcali, dependiendo de la secuencia de elución, obteniéndose de ese modo 19,52 g de (2R,3R)-3-[(2-ciclopentil-2-hidroxil-2-(3-piridil))etoxil]-1-azabiciclo[2,2,2]octano libre de álcali con un rendimiento de 85,5 %, y 19,06 g de la configuración (2S,3R) de 3-[(2-ciclopentil-2-hidroxil-2-(3-piridil))etoxil]-1-azabiciclo[2,2,2]octano libre de álcali con un rendimiento de 83,47 %.
Etapa 5: igual que la etapa 4 en el Ejemplo 1
Etapa 6: Bromuro de (2R,3R)-3-[(2-ciclopentil-2-hidroxil-2-(3-piridil))etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano

A un matraz de 100 ml, con forma de berenjena, se añadieron 3,18 g (9,1 mmol) de la configuración (2R,3R) de la base y se disolvieron añadiendo 18 ml de cloroformo para obtener una solución transparente de color amarillo, a la que se añadieron 11,033 g (51,3 mmol) de 3-bromopropoxibenceno y 50 ml de acetonitrilo, y la mezcla resultante se agitó entonces a temperatura ambiente para reaccionar durante 20-90 h bajo la protección de nitrógeno, siendo el final de la reacción controlado mediante TLC (condiciones de la TLC: cloroformo/metanol/agua amoniacal = 5,0 ml/1,5 ml/2 d). Una vez finalizada la reacción, el disolvente se retiró de la solución de reacción a 25-40 °C por evaporación rotativa bajo presión reducida mediante una bomba de agua, obteniéndose de ese modo una materia aceitosa de color amarillo, a la que se añadió éter etílico para precipitar una gran cantidad de sólido, y el sólido se recogió mediante filtración por succión para obtener 3,95 g de (2R,3R)-3-[(2-ciclopentil-2-hidroxil-2-(3-piridil))etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]-octano como un sólido de color blancuzco, con un rendimiento de 81,7 %.
El compuesto preparado en el Ejemplo 31 se hizo reaccionar con Ag2Ü para retirar el átomo de bromo para obtener hidróxido, que podía hacerse reaccionar con otros ácidos para transformarlos en las sales correspondientes. Ejemplos de una sal de un ácido farmacéuticamente aceptable incluyen una sal derivada de un ácido inorgánico, como hidrocloruro, bromuro, yoduro, nitrato, carbonato, bicarbonato, fosfato, hidrofosfato, fosfato dihidroxilado, sulfato, disulfato o fosfito o similar; y una sal derivada de un ácido orgánico relativamente no tóxico como ácido acético, ácido propiónico, ácido isobutírico, ácido malónico, ácido benzoico, ácido succínico, ácido subérico, ácido fumárico, ácido mandélico, ácido ftálico, ácido bencensulfónico, ácido p-toluensulfónico, ácido cítrico, ácido tartárico, ácido metansulfónico, ácido glucurónico o ácido galactónico o similar. Los ejemplos incluyen además una sal de un aminoácido como arginina o similar.
RMN de 1H (D2O) (ppm): 68,72-6,63 (m, 9H), 64,13-3,61 (m, 5H), 63,44-3,13 (m, 8H), 52,14 (m, 2H), 2,01 (m, 1H), 6 1,82-1,45 (m, 13H).
Ejemplo 32. Bromuro de (2R,3R)-3-[(2-ciclopentil-2-hidroxil-2-(2-furil))etoxil]-1 -(3-fenoxipropil)-1 -azabiciclo[2,2,2]octano
Etapa 1: Preparación de ciclopentil-2-furil-cetona

En un matraz de 5 litros, con tres bocas, conectado externamente a un condensador Allihn y a un tubo de secado de cloruro de calcio anhidro y un termómetro, se colocaron 101,5 g de virutas de magnesio, a ello se añadieron y agitaron 1.600 ml de THF y 1,05 g de yodo, y a la solución de reacción se añadieron gota a gota 78,28 g de bromociclopentano. Aproximadamente 5 minutos después de que la reacción se iniciara por completo, la solución de reacción pasó gradualmente de tener un color marrón rojizo claro a ser incolora, aumentando la temperatura interna automáticamente a 62-65 °C. Entonces se añadieron, gota a gota, 548 g de bromociclopentano adicional durante 35 min, aumentando su temperatura de reflujo gradualmente a 75-77 °C. La mezcla de reacción se hizo reaccionar bajo reflujo en baño de aceite durante 2 horas.
A la solución de reacción anterior se añadieron, gota a gota, 404 g de 2-furilcarbonitrilo diluido en 1.600 ml de THF y la mezcla resultante se hizo reaccionar entonces bajo reflujo en baño de aceite durante 4 horas. La temperatura interior se redujo a 5-10 °C, se añadieron, gota a gota, 96 ml de agua helada y después se agitó durante 20 minutos, seguido de la adición de HCl para ajustar el pH = 2,0, y la mezcla resultante se calentó a reflujo durante 3 horas, después se separó la capa orgánica, y una vez enfriada, la fase acuosa se extrajo con éter isopropílico (200 ml x 3). La capa orgánica se combinó y se lavó con Na2CÜ31 % (500 x 3) y agua (500 x 3) de forma consecutiva, y la capa orgánica se secó sobre sulfato de sodio anhidro durante la noche. El agente desecante se retiró por filtración, el disolvente se retiró por evaporación (42 °C, -0,095 MPa), y posteriormente se recogieron 272,6 g de materia aceitosa a 108-119 °C/6-7 mmHg por destilación bajo presión reducida usando una bomba de aceite.
RMN de 1H (CDCls) 5 (ppm): 7,39-6,70 (m, 3H), 2,36 (m, 1H), 1,65-1,45 (m, 8H).
Etapa 2: preparación de 1-(2-furil)-1-ciclopentiloxirano

Se hizo reaccionar ciclopentil-2-furil-cetona como material de partida para obtener 1-(2-furil)-1-ciclopentiloxirano según la bibliografía(1).
A un matraz de 3 litros, con tres bocas, se añadieron 1.780 ml de acetonitrilo seguido de la adición de sulfuro de dimetilo y sulfato de dimetilo, con un leve desprendimiento de calor, y la mezcla resultante se agitó entonces a temperatura ambiente durante 1,5 h y después se dejó reposar durante la noche. A la solución de reacción se añadió NaH (60 %) en porciones adecuadas con agitación durante 30-40 min, generándose un gas (préstese
atención a tener el matraz externamente conectado a un condensador Allihn y un tubo de secado de cloruro de calcio anhidro), y después se agitó a temperatura ambiente durante 1 hora, seguido de la adición gota a gota de 250 g de ciclopentil-2-furil-cetona durante aproximadamente 10 minutos, y la mezcla resultante se hizo reaccionar en baño de aceite a 40-45 °C durante 90 min siendo después agitada a temperatura ambiente durante 20 min, y después se enfrió a temperatura ambiente y se dejó reposar durante la noche. La solución de reacción se transfirió a un matraz con forma de berenjena, el disolvente se retiró por succión usando una bomba de agua (40-42 °C, -0,095 MPa), y el sólido restante se enfrió a 0-5 °C en baño de hielo, seguido de la adición gota a gota de 2.630 ml de agua helada (siendo la temperatura interior controlada a 0-5 °C) durante aproximadamente 1 hora, y la solución de reacción se extrajo con éter isopropílico (650 ml x 3), la capa de éter se combinó y se lavó con agua hasta que es neutra (siendo el pH de los lavados con agua de 7,0), y la capa de éter se secó sobre sulfato de sodio anhidro durante la noche. El agente desecante se retiró por filtración, el disolvente se retiró bajo presión reducida por succión usando una bomba de agua (40-42 °C, -0,095 MPa), y posteriormente se recogieron 231,6 g de fracción a 111-121 °C/3 mmHg bajo presión reducida usando una bomba de aceite.
RMN de 1H (CDCls) 5 (ppm): 7,35-6,50 (m, 3H), 2,93-2,65 (m, 2H), 2,26-1,27 (m, 9H).
Etapa 3: Preparación de 3-[(2-ciclopentil-2-hidroxil-2-(2-furil))etoxil]-1-azabiciclo[2,2,2]octano libre de álcali a partir de R-3-quinuclidinol

A 18,72 g (147 mmol) de R-3-quinuclidinol disponible comercialmente se añadieron 190 ml de DMSO, seguido de la adición de 7,59 g (190 mmol) de hidruro de sodio, y la mezcla se hizo reaccionar a 20-60 °C durante 0,5-12 h, y después se enfrió a temperatura ambiente, y a ello se añadió una solución de 33,88 g (190 mmol) de 1-(2-furil)-1-ciclopentiloxirano (autopreparado) en 45 ml de DMSO, y una vez finalizada la adición, la mezcla resultante se calentó a 20-70 °C en baño de aceite para reaccionar durante 0,5-12 horas. Bajo la condición de baño de hielo, se añadieron 120 ml de agua helada a una temperatura interna de 30 °C o menor. La mezcla de reacción se extrajo con éter isopropílico, 100 ml x 3; y la capa de éter se combinó y se lavó con una solución acuosa saturada de NaCl, 100 ml x 3. La capa orgánica se secó sobre sulfato de sodio anhidro durante la noche, el agente desecante se retiró por filtración, y el disolvente se retiró bajo presión reducida para obtener 42,74 g de 3-[(2-ciclopentil-2-hidroxil-2-(2-furil))etoxil]-1-azabiciclo [2,2,2]octano libre de álcali como materia aceitosa de color rojo, con un rendimiento de 95,32 %. El producto obtenido fue (2R,3R),(2S,3R)-3-[(2-ciclopentil-2-hidroxil-2-(2-furil))etoxil]-1-azabiciclo[2,2,2]octano libre de álcali.
Etapa 4: Purificación de 3-[(2-ciclopentil-2-hidroxil-2-(2-furil))etoxil]-1-azabiciclo[2,2,2]octano libre de álcali por cromatografía de columna y el tratamiento de purificación relacionado
La muestra de la etapa 3 anterior se separó en columna de gel de sílice, utilizando diclorometano o triclorometano y metanol, amoniacados, como fase móvil y una placa de TLC para controlar la pureza de la muestra. Bajo el sistema de elución anterior, una muestra de una mezcla de ambas configuraciones (2R,3R) y (2S,3R) de 3-[(2-ciclopentil-2-hidroxil-2-(2-furil))etoxil]-1-azabiciclo[2,2,2]octano libre de álcali podía ser purificada en dos tipos de (2R,3R) y (2S,3R) libre de álcali, dependiendo de la secuencia de elución, obteniéndose de ese modo 17,84 g de (2R,3R)-3-[(2-ciclopentil-2-hidroxil-2-(2-furil))etoxil]-1-azabiciclo[2,2,2]octano libre de álcali con un rendimiento de 83,5 %, y 18,03 g de la configuración (2S,3R) de 3-[(2-ciclopentil-2-hidroxil-2-(2-furil))etoxil]-1-azabiciclo[2,2,2]octano libre de álcali con un rendimiento de 84,37 %.
Etapa 5: igual que la etapa 3 en el Ejemplo 1
Etapa 6: Bromuro de (2R,3R)-3-[(2-ciclopentil-2-hidroxil-2-(2-furil))etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano

A un matraz de 100 ml, con forma de berenjena, se añadieron 2,78 g (9,1 mmol) de la configuración (2R,3R) de la base y se disolvieron añadiendo 18 ml de cloroformo para obtener una solución transparente de color amarillo, a la que se añadieron 11,033 g (51,3 mmol) de 3-bromopropoxibenceno y 50 ml de acetonitrilo, y la mezcla resultante se agitó entonces a temperatura ambiente para reaccionar durante 20-90 h bajo la protección de nitrógeno, siendo el final de la reacción controlado mediante TLC (condiciones de la TLC: cloroformo/metanol/agua amoniacal = 5,0 ml/1,5 ml/2 d). Una vez finalizada la reacción, el disolvente se retiró de la solución de reacción a 25-40 °C por evaporación rotativa bajo presión reducida mediante una bomba de agua, obteniéndose de ese modo una materia aceitosa de color amarillo, a la que se añadió éter etílico para precipitar una gran cantidad de sólido, y el sólido se recogió mediante filtración por succión para obtener 3,97 g de (2R,3R)-3-[(2-ciclopentil-2-hidroxil-2-(2-furil))etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano como un sólido de color blancuzco, con un rendimiento de 83,9 %.
El compuesto preparado en el Ejemplo 32 se hizo reaccionar con Ag2Ü para retirar el átomo de bromo para obtener hidróxido, que podía hacerse reaccionar con otros ácidos para transformarlos en las sales correspondientes.
Ejemplos de una sal de un ácido farmacéuticamente aceptable incluyen una sal derivada de un ácido inorgánico, como hidrocloruro, bromuro, yoduro, nitrato, carbonato, bicarbonato, fosfato, hidrofosfato, fosfato dihidroxilado, sulfato, disulfato o fosfito o similar; y una sal derivada de un ácido orgánico relativamente no tóxico como ácido acético, ácido propiónico, ácido isobutírico, ácido malónico, ácido benzoico, ácido succínico, ácido subérico, ácido fumárico, ácido mandélico, ácido ftálico, ácido bencensulfónico, ácido p-toluensulfónico, ácido cítrico, ácido tartárico, ácido metansulfónico, ácido glucurónico o ácido galactónico o similar. Los ejemplos incluyen además una sal de un aminoácido como arginina o similar.
RMN de 1H (D2O) (ppm): 57,32-6,19 (m, 8H), 54,14-3,60 (m, 5H), 53,42-3,11 (m, 8H), 52,11 (m, 2H), 1,97 (m, 1H), 5 1,83-1,46 (m, 13H).
Ejemplo 33. Bromuro de (2R,3R)-3-[(2-ciclopentil-2-metoxil-2-(3-piridil))etoxil]-1 -(3-fenoxipropil)-1 -azabiciclo[2,2,2]octano
Etapas 1,2, 3 y 4: igual que las etapas 1,2, 3 y 4 en el Ejemplo 31
Etapa 5: igual que la etapa 4 en el Ejemplo 1
Etapa 6: igual que la etapa 6 en Ejemplo 31
Etapa 7: Bromuro de (2R,3R)-3-[(2-ciclopentil-2-metoxil-2-(3-piridil))etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano

A un matraz de 100 ml, con forma de berenjena, se añadieron 2,655 g (4,87 mmol) de bromuro de (2R,3R)-3-[(2-ciclopentil-2-hidroxil-2-(3-piridil))etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano y se disolvieron añadiendo 30 ml de acetonitrilo, y a ello se añadieron 0,5 g de NaH, seguido de la adición gota a gota de una solución de bromometano (0,5 g) en 10 ml de acetonitrilo, y la mezcla resultante se agitó entonces a temperatura ambiente para reaccionar durante 20-90 horas bajo la protección de nitrógeno, siendo el final de la reacción controlada por TLC (condiciones de la TLC: cloroformo/metanol/agua amoniacal = 5,0 ml/1,5 ml/2 d). Una vez finalizada la reacción, el disolvente se retiró de la solución de reacción a 25-40 °C por evaporación rotativa bajo presión reducida mediante una bomba de agua, obteniéndose de ese modo una materia aceitosa de color amarillo, a la que se añadió éter etílico para precipitar una gran cantidad de sólido, y el sólido se recogió mediante filtración por succión para obtener 2,19 g de bromuro de (2R,3R)-3-[(2-ciclopentil-2-metoxil-2-(3-piridil))etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano como un sólido de color blancuzco, con un rendimiento de 80,37 %.
El compuesto preparado en el Ejemplo 33 se hizo reaccionar con Ag2O para retirar el átomo de bromo para obtener hidróxido, que podía hacerse reaccionar con otros ácidos para transformarlos en las sales correspondientes. Ejemplos de una sal de un ácido farmacéuticamente aceptable incluyen una sal derivada de un ácido inorgánico, como hidrocloruro, bromuro, yoduro, nitrato, carbonato, bicarbonato, fosfato, hidrofosfato, fosfato dihidroxilado, sulfato, disulfato o fosfito o similar; y una sal derivada de un ácido orgánico relativamente no tóxico como ácido acético, ácido propiónico, ácido isobutírico, ácido malónico, ácido benzoico, ácido succínico, ácido subérico, ácido fumárico, ácido mandélico, ácido ftálico, ácido bencensulfónico, ácido p-toluensulfónico, ácido cítrico, ácido tartárico, ácido metansulfónico, ácido glucurónico o ácido galactónico o similar. Los ejemplos incluyen además una sal de un aminoácido como arginina o similar.
RMN de 1H (D2O) (ppm): 58,77-6,66 (m, 9H), 54,15-3,65 (m, 5H), 53,44-3,13 (m, 11H), 52,16 (m, 2H), 2,03 (m, 1H), 51,85-1,43 (m, 13H).
Ejemplo 1: Intensidad del antagonismo de los compuestos de ejemplo en la respuesta contráctil del músculo liso traqueal de conejillos de Indias inducidos por el carbacol (CCh)
1. Procedimiento:
Preparación del espécimen aislado de músculo liso traqueal de un conejillo de Indias:
Después de que un conejillo de Indias fuera narcotizado con uretano, la tráquea entre la garganta y la carina del conejillo de Indias fue extirpada rápidamente, y colocada en solución Krebs-Henseleit (K-H) (composición (g/l): NaCl 6,92, KCl 0,35, CaCl2 0,28, KH2PO4 0,16, MgSO4'7H2O 0,4568, NaHCO3 2,1, glucosa 2,0) purgada con un gas mezcla de 5 % CO2 y 95 % O2. Después de separar los tejidos conectivos y las grasas alrededor de la tráquea, la tráquea se cortó en trozos de tráquea con una anchura de aproximadamente 3 mm y una longitud de 20 mm con unas tijeras quirúrgicas, con ambos extremos de la pieza de la tráquea ligados con sutura de seda de 4~0, y después se colocaron en un baño termostático conteniendo 5 ml de solución K-H (pH 7,4) a 37 °C, purgada continuamente con el gas mezcla de 5 % CO2 y 95 % de O2. Un transductor de la tensión muscular se conectó al extremo superior para aplicar 1,0 g de tensión de reposo al espécimen, el cambio de tensión se registró, la solución de cultivo se reemplazó cada 20 min, y el ensayo se inició después de estar equilibrado durante 60 min.
Procedimiento de administración: Después de estabilizar la pieza de la tráquea, se añadieron 3 * 10-6 mol/l de CCh en un baño, y después de que la tensión contráctil de la pieza de la tráquea alcanzó un nivel máximo, los compuestos de ejemplo, bromuro de ipratropio y bromuro de tiotropio se añadieron a un baño Magnus mediante un régimen de dosificación acumulativa con una dosificación de 10-9 ~ 10-5 mol, respectivamente, y se observó el estado diastólico de la pieza de la tráquea: Si no se producía respuesta (inferior a la concentración de umbral), continuó añadiéndose una siguiente dosis en secuencia; si se producía respuesta, después de que se alcanzara una meseta diastólica, se añadió posteriormente una siguiente dosis. La operación anterior se repitió hasta que la curva contráctil alcanzaba el valor mínimo. Finalmente, se añadieron 10-6 mol/l de isoprenalina para que se alcanzara la máxima relajación y se registró la curva.
Enfoque estadístico: El resultado se expresó como media ± desviación estándar, y el recuento se hizo mediante el paquete estadístico Sigma stat. Los datos se analizaron mediante análisis de varianza, la comparación entre las medias de una pluralidad de especímenes se realizó mediante la prueba Student-Newman-Keuls (SNK); nivel estadístico significativo a = 0,05 (bilateral). EC50 (95 % de límite de confianza) se calculó mediante el software POMS versión 2.0 de Shanghai Scientific and Technical Publishers.
2. Conclusión:
3 * 10-6 mol/l de CCh podría causar que el músculo liso traqueal del conejillo de Indias generara una respuesta contráctil duradera y estable. En el régimen de dosificación acumulativa, los compuestos de ejemplo y los fármacos de control de bromuro de ipratropio y de bromuro de tiotropio se añadieron en el baño Magnus, y cada uno de los compuestos de ejemplo y cada uno de los fármacos de control podían relajar la contracción del músculo liso traqueal inducido por CCh. Los resultados se mostraron en la tabla 2.
3. Resultados
Tabla 2: Antagonismo in vitro de los compuestos de la presente invención contra la contracción del músculo liso traqueal inducido por una concentración de 3 * 10-6 mol/l de CCh, IC50 (pM)



Los resultados de la Tabla 2 mostraron que cada uno de los compuestos de ejemplo tenía un efecto antagonista significativo en el receptor M, en donde la configuración (2R,3R) del compuesto tenía el mayor efecto, y la intensidad del efecto de una pluralidad de compuestos era comparable a la de los agentes de control positivo de bromuro de ipratropio y bromuro de tiotropio. Los compuestos de la presente invención y bromuro de ipratropio tenían un tiempo de comienzo más corto que el bromuro de tiotropio, y por el contrario, el bromuro de tiotropio actuó más lentamente. Los compuestos de la presente invención y el bromuro de tiotropio tenían una mayor duración de la acción.
Ejemplo 2: Efecto ligante y selectivo de los compuestos de la presente invención en tres subtipos de receptor M
1. Procedimiento: las células mn, m2 y m3 del ovocito de hámster chino (CHO) transfectado se colocaron en un medio de cultivo DMEM (que contenía un 15 % de suero fetal bovino, L-glutamina, 1 % de aminoácido no esencial, 1 % de antibiótico/agente antifúngico), respectivamente, y después se incubaron en una incubadora con 5 % de CO2 a 37 °C. Cuando las células de un matraz de cultivo se cultivaron y proliferaron para formar células en monocapa que se difundieron sobre aproximadamente 90 % del fondo del matraz, se desechó el medio de cultivo, y el matraz de cultivo se lavó dos veces con una solución tampón de PBS (pH 7,4), y después las células fueron raspadas con una solución helada de tampón fosfato (pH 7,7, que contenía 5 mmol/l de MgCh). Las células recogidas fueron homogeneizadas con un homogeneizador de vidrio con Teflón, la solución homogeneizada se centrifugó a baja temperatura, 20.000 r x 20 min, y el precipitado se homogeneizó con una solución tampón de reacción (pH 7,7, que contenía 5 mmol/l de MgCh) para formar una suspensión proteica de membrana. La cantidad de proteína añadida en cada uno de los tubos de reacción era: mu (aproximadamente 0,05 mg), m2 (0,05 mg), m3 (0,1 mg), respectivamente; la concentración de [3H]-QNB era 0,1-2,16 nmol/l; se añadió 1 pmol/l de atropina al tubo de unión no específico; y el volumen total de reacción fue de 300 pl. La solución resultante se hizo reaccionar entonces a 25 °C durante 30 minutos y se enfrió con una solución tampón de reacción helada y, posteriormente, se recogió en una membrana de filtro de fibra de vidrio mediante un colector de células multicabezal. Después de ser secada a 80 °C, la lámina de membrana filtrante se colocó en un vial de centelleo de líquidos, seguido de la adición de 5 ml de agente de centelleo de líquidos, y después se mantuvo en lugar oscuro durante la noche, con cpm medido por un espectrómetro de centelleo de líquidos. Los valores Bmax y Kd se calcularon mediante el software GraphPad Prism. En cada uno de los tubos de ensayo se añadieron ligandos marcados con 3H-QNB que no tiene selectividad sobre el receptor M, en los que las concentraciones eran: mu y m3 (1,042 nmol/l), m2 (1,81 nmol/l), repectivamente; diferentes concentraciones de competidor no marcado, Pirenzepina (PZ, competidor selectivo de mn) o Galamina (GI, competidor selectivo de m2) o 4-DAMP (competidor selectivo de m3) o los compuestos de la presente invención, se añadieron simultáneamente, la concentración final fue de 10"10~10"4 mol/l, con 11 dosis en total; posteriormente se añadió la misma cantidad de espécimen de proteína de membrana; el volumen total de reacción fue de 300 pl, y después se realizó una reacción de unión competitiva. Se proporcionó además un tubo de unión no específico, y la unión no específica se midió por una gran cantidad de atropina (la concentración final fue de 1 pmol/l). La solución resultante se hizo reaccionar a 25 °C durante 30 minutos y se enfrió con una solución tampón de reacción, y después se recogió en una membrana de filtro de fibra de vidrio mediante un colector de células multicabezal. Después de ser secada a 80 °C, la lámina de membrana filtrante se colocó en un vial de centelleo de líquidos, seguido de la adición de 5 ml de agente de centelleo de líquidos, y después se mantuvo en lugar oscuro durante la noche, con cpm medido por un espectrómetro de centelleo de líquidos. Los respectivos valores de Ki de los competidores en tres subtipos de receptor M se calcularon mediante el software GraphPad Prism, y se transformaron en valores pKi, para comparar la selectividad de PZ, GI, 4-DAMP y los compuestos a ensayar en los subtipos de receptor M.
2. Resultados
Tabla 3: Comparación de la selectividad del receptor de tres inhibidores competitivos conocidos y los compuestos de la presente invención en tres subtipos de receptor M (x ± s, n = 6)

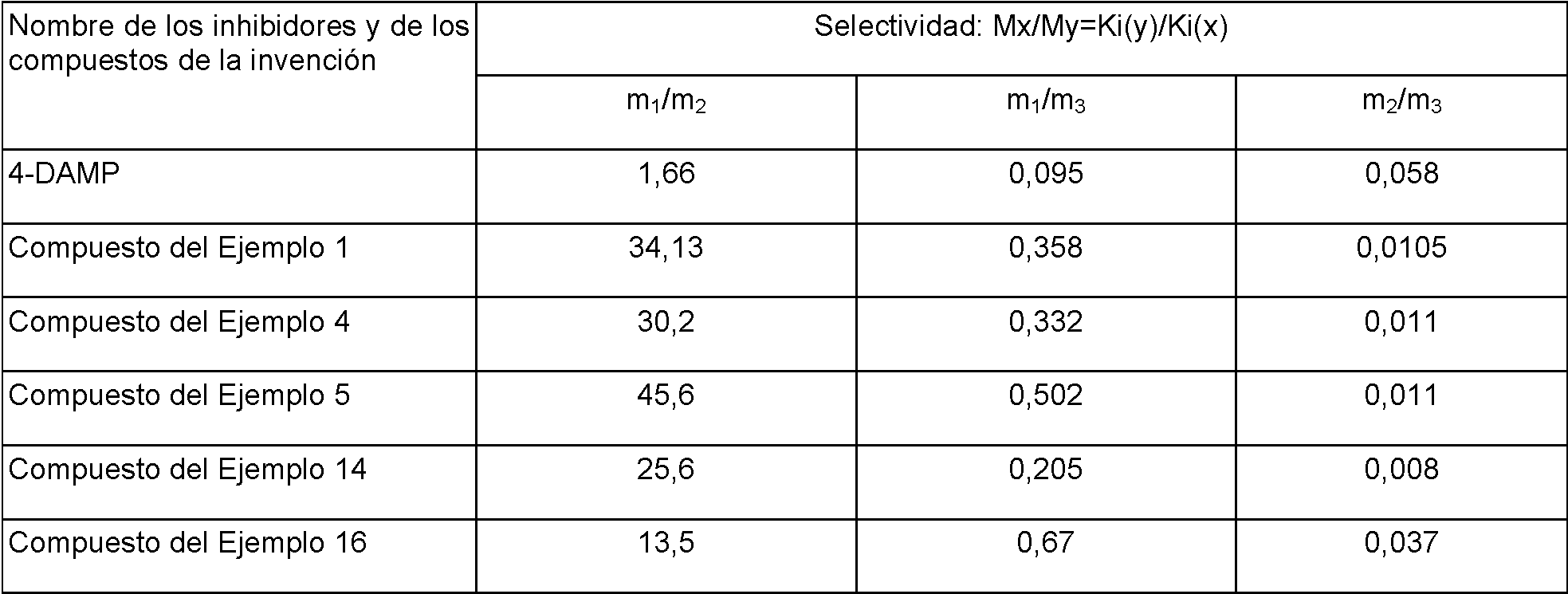
3. Conclusión
La prueba anterior con células de CHO transfectadas mediante ADNc de tres subtipos m1, m2 y m3 de receptor M se utilizó para identificar la selectividad de los compuestos de la presente invención en los subtipos de receptor M. Teniendo en cuenta las características farmacológicas de Pirenzepina, Galamina, y 4-DAMP, los resultados de los ensayos de inhibición competitiva de los tres compuestos con tres tipos de células demostraron que los tres tipos de células podían utilizarse para identificar una selectividad de receptor de un fármaco. Los resultados del ensayo anterior mostraron que los compuestos probados de la presente invención tenían la selectividad más alta en el receptor m3, la segunda selectividad más elevada en el receptor mp, y la selectividad más baja en el receptor m2; en donde tenían los mayores efectos en los receptores m3 y m1. Tenían ventajas significativas en el tratamiento de la rinitis, hipersensibilidad de las vías respiratorias, traquitis crónica senil, EPOC y enfermedades ulcerativas del tubo digestivo, en comparación con la técnica anterior.
Ejemplo 3: Determinación de la intensidad y duración del antagonismo de los compuestos de ejemplo en la respuesta contráctil de los bronquios en conejillos de Indias inducidos por metacolina (Mch)
1. Procedimiento:
Determinación del volumen tidal, caudal de las vías respiratorias y presión transpulmonar:
Se inyectaron intraperitonealmente 1,5 g/kg de uretano para narcotizar al conejillo de Indias. El conejillo de Indias se fijó en posición supina y se trató con intubación traqueal, y la vena yugular externa se separó y se insertó una aguja residente; el conejillo de Indias estaba encerrado en un pletismógrafo corporal, se insertó una aguja sin punta para intubación en la cavidad torácica entre las costillas 4~5 del protórax del conejillo de Indias, y se podía medir la presión intratorácica (con un valor negativo de una columna de agua de un manómetro de agua y una fluctuación con la respiración del conejillo de Indias como marcas). Después de la estabilización, los valores del volumen tidal, el caudal de las vías respiratorias y la presión transpulmonar del conejillo de Indias antes de la administración de Mch se registraron como valores base mediante un sistema MedLab de recogida y procesamiento de señales biológicas. Se inyectaron de forma intravenosa 10 pg de Mch/kg de peso corporal. Los cambios en caudal de las vías respiratorias, volumen tidal y presión transpulmonar del conejillo de Indias se observaron a los 5 minutos. Cálculo de Raw y Cdin: los cambios del aumento en porcentaje del valor Raw y de la disminución en porcentaje de Cdin se calcularon después de la inhalación de Mch.
Las fórmulas de cálculo de Raw y Cdin fueron respectivamente:
Raw después de la inhalación de Mch - Raw antes de la inhalación (valor base)
% de aumento de Raw = ---------------------------------------------------------------------------------------------------------- x 100 % Raw antes de la inhalación (valor base)
Raw antes de la inhalación de Mch (valor base) - Raw después de la inhalación
% de disminución de Cdin = ---------------------------------------------------------------------------------------------------------- x 100 %
Raw antes de la inhalación de Mch (valor base)
Relación dosis-efecto: Para cada uno de los compuestos de ejemplo, 27 conejillos de Indias se dividieron aleatoriamente en 3 grupos: un grupo de control de disolventes con 15 conejillos de Indias, un grupo con 1 pg/kg de los compuestos de ejemplo 4, 10, 14 y 26, un grupo con 3 pg/kg de los compuestos de ejemplo 4, 10, 14 y 26 y un grupo con 10 pg/kg de los compuestos de Ejemplo 4, 10, 14 y 26; después de 30 minutos del goteo en las vías respiratorias de la concentración anterior de fármacos, 10 pg de Mch/kg de peso corporal se inyectaron por vía
intravenosa para la excitación, y se determinó la resistencia de las vías respiratorias (Raw) y la compliancia dinámica pulmonar (Cdin) dentro de 5 min.
Relación tiempo-efecto: Después de que los conejillos de Indias fueron narcotizados, se introdujeron por goteo 10 pg/kg y 5 pg/kg de los compuestos de ejemplo 4, 10, 14, y 26 en las vías respiratorias. Se inyectaron por vía intravenosa 10 pg de Mch/kg de peso corporal para la excitación después de 0,25 h, 0,5 h, 1 h, 1,5 h, 2 h, 4 h, 6 h, 12 h y 24 h después de la dosificación, respectivamente, y se determinaron la resistencia de las vías respiratorias (Raw) y la compliancia dinámica pulmonar (Cdin) a los 5 min.
2. Resultados:
Relación dosis-efecto: las dosis de los compuestos de ejemplo 4, 10, 14, y 26 fueron 1 pg/kg, 3 pg/kg, y 10 pg/kg, respectivamente, y después de 30 min del goteo en las vías respiratorias, se inyectaron 10 pg de Mch/kg de peso corporal por vía intravenosa para la excitación, y se determinó la resistencia de las vías respiratorias (Rw) y la compliancia dinámica pulmonar (Cdin) a los 5 minutos, y los resultados se mostraron en la Tabla siguiente:
Tabla 4. Antagonismo de los compuestos de ejemplo 4, 10, 14 y 26 en la respuesta contráctil de los bronquios en conejillos de Indias inducidos por la relación (Media ± S.E.M) efecto-dosis con Mch

Enfoque estadístico: se usó ANOVA unidireccional, y la comparación entre varios grupos se probó mediante el procedimiento Bonferroni; se realizó la comparación con el grupo de control de disolventes, * P < 0,05, * * P < 0,01, * * * P < 0,001.
Cuando se inyectaron 10 pg de Mch/kg por vía intravenosa, la resistencia de las vías respiratorias de los conejillos de Indias aumentó en un 328 %, la compliancia dinámica pulmonar disminuyó en un 73 %. Una vez 1, 3, y 10 pg/kg de los compuestos en el Ejemplo 4, 10, 14, y 26 fueron sometidos a goteo en las vías respiratorias de los conejillos de Indias, el aumento de la resistencia y la disminución de la complianza dinámica pulmonar fueron inhibidas de forma dependiente de la dosis. Las relaciones de inhibición de los tres grupos de dosificación del compuesto del ejemplo 4 contra el aumento de la resistencia en las vías respiratorias fueron 64,2 % (p<0,01), 86,1 % (p<0,001) y 90,8 % (p<0,001), respectivamente; las relaciones de la inhibición contra la disminución de la complianza dinámica pulmonar fueron 11,2 % (p>0,05), 46,6 % (p<0,001) y 50,0 % (p<0,001), respectivamente. Las relaciones de inhibición de los tres grupos de dosificación del compuesto de ejemplo 10 contra el aumento de la resistencia en las vías respiratorias fueron 63,7 % (p<0,01), 84,5 % (p<0,001) y 91,3 % (p<0,001), respectivamente; las relaciones de
la inhibición contra la disminución de la complianza dinámica pulmonar fueron 10,9 % (p>0,05), 45,9 % (p<0,001) and 49,8 % (p<0,001), respectivamente. Las relaciones de inhibición de los tres grupos de dosificación del compuesto del ejemplo 14 frente al aumento de la resistencia en las vías respiratorias fueron 63,5 % (p<0,01), 85,4 % (p<0,001) y 90,5 % (p<0,001), respectivamente; las relaciones de la inhibición contra la disminución de la complianza dinámica pulmonar fueron 11,0 % (p>0,05), 46,1 % (p<0,001) y 49,5 % (p<0,001), respectivamente. Las relaciones de inhibición de los tres grupos de dosificación del compuesto del ejemplo 26 frente al aumento de la resistencia en las vías respiratorias fueron 64,4 % (p<0,01), 87,2 % (p<0,001) y 92,1 % (p<0,001), respectivamente; las relaciones de la inhibición contra la disminución de la complianza dinámica pulmonar fueron 11,0 % (p>0,05), 47,1 % (p<0,001) y 49,9 % (p<0,001), respectivamente.
Relación tiempo-efecto: Cuando se inyectaron por vía intravenosa 10 pg de Mch/kg, la resistencia de las vías respiratorias de los conejillos de Indias aumentó en un 328 %. Después de 0,25 h del goteo de 10 pg/kg de los compuestos de los ejemplos 4, 10, 14, y 26 en las vías respiratorias, la relación de inhibición contra el aumento de la resistencia de las vías respiratorias fue de 80 % o más; y podía alcanzar el 90 % o más de la relación de inhibición máxima inmediatamente después de 1 h. Con el tiempo, la relación de inhibición contra el aumento de la resistencia de las vías respiratorias fue todavía 85 % o más después de 24 h, y hubo diferencias estadísticas en comparación con el grupo de control de disolventes (p < 0,01-0,001). Para confirmar la exactitud de los resultados, la dosificación de los compuestos de ejemplo 4, 10, 14, y 26 se redujo a 5 pg/kg, cuyos resultados mostraron que cuando 5 pg/kg de los compuestos de ejemplo 4, 10, 14, y 26 se sometieron a goteo en las vías respiratorias, la relación de inhibición contra la resistencia de las vías respiratorias fue de 85 % o más (p < 0,001) después de 12 h, y la relación de inhibición contra la resistencia de las vías respiratorias fue de 65 % o más (p < 0,01) después de 24 h. Después de 12 h del goteo de 10 pg/kg y 5 pg/kg de los compuestos de ejemplo 4, 10, 14, y 26 en las vías respiratorias, no hubo diferencia significativa en la relación de inhibición contra la resistencia de las vías respiratorias entre las dosis de 10 pg/kg y 5 pg/kg. Sin embargo, después de 24 h, hubo diferencias significativas en la relación de inhibición contra la resistencia de las vías respiratorias (p < 0,05) entre 10 pg/kg y 5 pg/kg de los compuestos de ejemplo 4, 10, 14 y 26. Los resultados anteriores indicaron que: los tiempos de acción de 10 pg/kg y 5 pg/kg de los compuestos de ejemplo 4, 10, 14, y 26 administrados por goteo en las vías respiratorias eran más de 24 h, y estos compuestos eran antagonistas de los receptores M superduraderos.
Los compuestos que se muestran en la Tabla 5 se prepararon además según la presente invención, y se determinó su IC50 (mM) de resistencia a la contracción del músculo liso traqueal en conejillos de indias inducidos por una concentración de 3 * 10-6 mol/l de CCh.
Tabla 5. Rotación óptica y la IC50 (mM) de resistencia in vitro para la contracción del músculo liso traqueal de conejillos de indias inducidos por una concentración de 3 * 10-6 mol/l de CCh, de otros compuestos sintetizados según la presente invención




Ejemplo 5: Composiciones y preparación de las mismas
Preparación y uso de naristillae, pulverizador nasal, aerosol, aerosol en polvo, inhalación con atomización y comprimidos
1. Naristillae de prescripción de un solo fármaco
Compuesto del Ejemplo 16 3 mg
Cloruro de benzalconio 3 mg
H2O 10 ml
En condiciones de producción de clase 100.000, el ingrediente activo del compuesto del ejemplo 10 y un bactericida (cloruro de benzalconio) se disolvieron en H2O, y después se rellenaron en un vial de color marrón equipado con un tapón fusiforme que podía ser utilizado para el goteo nasal.
Para este naristillae de prescripción de un solo fármaco, 2,0-20 mg podría disolverse en cada 10 ml de agua, para ser formulado en diferentes concentraciones de naristillae; y la cantidad de bactericida de cloruro de benzalconio podía variar de 1-5 mg.
La dosis de naristillae de la prescripción de un solo fármaco fue de 2-3 gotitas (0,1 ml-0,15 ml)/fosa nasal, aproximadamente 20-300 pg/fosa nasal cada vez.
2. Compuesto naristillae
Compuesto del Ejemplo 10 2,5 mg
Fenoterol 10 mg
Cloruro de benzalconio 5 mg
H2O 10 ml
En condiciones de producción de clase 100.000, los ingredientes activos (compuesto del ejemplo 10 y fenoterol) y un bactericida (cloruro de benzalconio) se disolvieron en H2O, y después se llenaron en un vial de color marrón equipado con un tapón fusiforme que podía ser utilizado para goteo nasal. La cantidad de bactericida de cloruro de
benzalconio podría oscilar entre 1-5 mg. La dosis del compuesto naristillae fue de 2-3 gotitas (0,1 ml-0,15 ml)/fosa nasal, aproximadamente 25 pg/fosa nasal cada vez.
3. Pulverización nasal de la bomba dosificadora
Compuesto del Ejemplo 4 3,33 mg
Cloruro de benzalconio 5.00 mg
HCl 1N añadido hasta pH 4-6
H2O pura 2.0 ml
En condiciones de producción de clase 100.000, los componentes anteriores se disolvieron en H2O pura, y después se llenaron en un frasco equipado con una bomba dosificadora adecuada para pulverización nasal, y el volumen pulverizado de esta bomba cada vez fue de 70-90 pl. La dosis fue 1-2 pulverizaciones/fosa nasal, aproximadamente 21-60 pg/fosa nasal cada vez.
Para obtener una dosis más grande en el tratamiento de pacientes graves, la cantidad de compuesto 4 podría aumentarse.
4. Aerosol de dosis medida que contiene un propulsor
Componente Peso
Compuesto del Ejemplo 4 10 mg
Etanol 2,4 g
Ácido oleico 7,0 mg
HFA-134a 11,75 g
Después de que el compuesto del Ejemplo 4 se disolviera con etanol y ácido oleico, se llenó e1HFA-134a mediante un procedimiento de una sola etapa. Luego, en condiciones de producción de clase 100.000, la mezcla resultante se mezcló con un tensioactivo y propelentes de 1-fluro-3-clorometano, 2-fluro-2-clorometano y 4-fluro-2-cloroetano en proporción, y después se encapsularon en un recipiente a presión cuantitativa mediante una máquina de llenado a presión. Cada frasco contenía 10 g, cada aerosol con 100 mg, y un contenido de 20 pg del compuesto del Ejemplo 4.
5. Aerosol de polvo de tipo cápsula
Un aerosol de polvo de tipo cápsula se componía de los siguientes ingredientes:
Compuesto del Ejemplo 4 0,05 g
Lactosa 98,95 g
Benzoato de sodio 1,0 g
En condiciones de producción de clase 100.000, el compuesto del Ejemplo 4 junto con lactosa y benzoato de sodio se sometieron a micronización, con un diámetro de partícula de 5 pm o menor, y después se mezclaron a fondo y homogéneamente, y finalmente se encapsularon en una cápsula. Cada cápsula contenía 40 mg, y cada inhalación contenía 20 pg del compuesto de la presente invención. En esta composición farmacéutica para el tratamiento de una enfermedad del sistema respiratorio, la lactosa era un diluyente que podría seleccionarse adicionalmente de arabinosa, glucano, manitol, xilitol, sacarosa, fructosa, sorbitol, maltosa, aminoácido o glucosa o similar; el benzoato de sodio era un lubricante, y el estearato de magnesio también podía utilizarse como lubricante.
6. Comprimido
Compuesto del Ejemplo 6 5 g
Lactosa 71,5 g
Celulosa microcristalina 22 g
Estearato de magnesio 1,5 g
El compuesto del Ejemplo 6 se mezcló homogéneamente con lactosa y celulosa microcristalina, y además se mezcló homogéneamente después de añadir estearato de magnesio, y la mezcla resultante se prensó en seco en comprimidos, con un peso por comprimido de 100 mg.
7. Solución de inhalación
Compuesto del Ejemplo 10 100 mg
Solución salina Normal 1.000 ml
El compuesto del Ejemplo 10 se disolvió en 800 ml de solución salina normal, y después se transfirió a un matraz aforado de 1.000 ml, y se complementó con solución salina normal hasta la marca de escala, y después la solución resultante se dividió y se cargó en ampollas, 1 ml/ampolla, y se calentó a 115 °C durante 30 min.
Claims (12)
1. Compuesto de fórmula I:

en donde en la fórmula I:
n se selecciona entre 1-7,
R1 es un hidrocarbilo C3-C7, que puede estar no sustituido u opcionalmente sustituido por halógeno, alcoxi, alcoxihidrocarbilo, heterociclilo o arilo,
R2 es un arilo o un heteroarilo que contiene uno o más heteroátomos, que puede estar no sustituido u opcionalmente sustituido,
R3 es un hidroxilo, halógeno, alcoxi o aciloxi, en donde el alcoxi o aciloxi puede estar no sustituido u opcionalmente sustituido por halógeno, hidroxilo, alcoxi, hidrocarbilo, alcoxihidrocarbilo, heterociclilo o arilo;
R4 y R5 pueden estar presentes o ausentes, y se seleccionan de forma independiente de un grupo que consiste en halógeno, hidroxilo, alcoxi, hidrocarbilo, alcoxihidrocarbilo, heterociclilo o arilo, cuando están presentes;
Y es un alquileno C1-C7, lineal o ramificado, o -(CH2-O-CH2V, que puede estar opcionalmente sustituido, en donde m es igual a 1-3,
X- es un radical ácido o hidroxilo.
2. Compuesto según la reivindicación 1, en donde:
n es 1-3,
R1 es un cicloalquilo no sustituido,
R2 es un fenilo, naftilo o bifenilo, que puede estar no sustituido u opcionalmente sustituido por uno o más halógenos, hidroxilo, fenilo, -OR6, -SR6, -NR6R7, -NHCOR6, -CONR6R7, -CN, -NO2, -COOR6, -CF3 o hidrocarbilo C1-C4, lineal o ramificado, R6 y R7 puede ser un hidrógeno átomo, un hidrocarbilo C1-C4, lineal o ramificado, o puede formar un ciclohidrocarbilo entre sí,
R3 es un hidroxilo o metoxilo
Y es un metileno, etileno, propileno o -(CH2-O-CH2)-.
3. Compuesto según la reivindicación 1 o 2, en donde
n es 1,
R1 es un ciclopentilo o ciclohexilo,
R2 es un fenilo, piridilo, furilo o tienilo, no sustituidos,
Y es un etileno o propileno.
4. Compuesto según una cualquiera de las reivindicaciones anteriores, en donde X-es un radical ácido de un ácido inorgánico farmacéuticamente aceptable o hidróxido, de manera que el compuesto de la fórmula I está en forma de una sal o base de amonio cuaternario farmacéuticamente aceptable, la sal farmacéuticamente aceptable resultante se selecciona de un grupo que consiste en hidrocloruro, bromuro, yoduro, nitrato, carbonato, bicarbonato, fosfato, hidrofosfato, fosfato dihidroxilado, sulfato, disulfato o fosfito.
5. Compuesto según una cualquiera de las reivindicaciones 1-3, en donde X-es un radical ácido de un ácido orgánico farmacéuticamente aceptable, de manera que el compuesto de la fórmula I está en forma de una sal de un ácido orgánico, dicho ácido orgánico es ácido acético, ácido propiónico, ácido isobutírico, ácido malónico, ácido benzoico, ácido succínico, ácido subérico, ácido fumárico, ácido mandélico, ácido ftálico, ácido bencensulfónico, ácido ptoluensulfónico, ácido cítrico, ácido tartárico, ácido metansulfónico, ácido glucurónico, ácido galactónico o aminoácido.
6. Compuesto según la reivindicación 1, que es:
Bromuro de (2S,3R)-3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano;
Bromuro de (2S,3S)-3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano;
Bromuro de (2R,3R)-3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano;
Bromuro de (2R,3S)-3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano;
Bromuro de (2S,3R),(2R,3S)-3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano; Bromuro de (2R,3R),(2S,3S)-3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano; Bromuro de (2S,3R)-3-[(2-ciclohexil-2-hidroxil-2-fenil)etoxil]-1 -(3-fenoxipropil)-1-azabiciclo[2,2,2]octano;
Bromuro de (2S,3S)-3-[(2-ciclohexil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano;
Bromuro de (2R,3R)-3-[(2-ciclohexil-2-hidroxil-2-fenil)etoxil]-1 -(3-fenoxipropil)-1-azabiciclo[2,2,2]octano;
Bromuro de (2R,3S)-3-[(2-ciclohexil-2-hidroxil-2-fenil)etoxil]-1 -(3-fenoxipropil)-1-azabiciclo[2,2,2]octano;
Bromuro de (2R,3S),(2S,3R)-3-[(2-ciclohexil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano; Bromuro de (2R,3R),(2S,3S)-3-[(2-ciclohexil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano; Bromuro de (2S,3R)-3-[(2-ciclobutil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano;
Bromuro de (2S,3S)-3-[(2-ciclobutil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano;
Bromuro de (2R,3S)-3-[(2-ciclobutil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano;
Bromuro de (2R,3R)-3-[(2-ciclobutil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano;
Bromuro de (2R,3S),(2S,3R)-3-[(2-ciclobutil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano; Bromuro de (2R,3R),(2S,3S)-3-[(2-ciclobutil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano; Bromuro de (2S,3R)-3-[(2-ciclopropil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano;
Bromuro de (2R,3S)-3-[(2-ciclopropil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano;
Bromuro de (2S,3S)-3-[(2-ciclopropil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano;
Bromuro de (2R,3R)-3-[(2-ciclopropil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano;
Bromuro de (2R,3S),(2S,3R)-3-[(2-ciclopropil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano; Bromuro de (2R,3R),(2S,3S)-3-[(2-ciclopropil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano; Bromuro de (2S,3R)-3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-(2-fenoxietil)-1-azabiciclo[2,2,2]octano;
Bromuro de (2R,3R)-3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-(2-fenoxietil)-1-azabiciclo[2,2,2]octano;
Bromuro de (2R,3R)-3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-fenoximetil-1-azabiciclo[2,2,2]octano;
Cloruro de (2R,3R)-3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-fenoximetoximetil-1-azabiciclo[2,2,2] cloruro de octano; Bromuro de (2R,3R)-3-[(2-ciclopentil-2-hidroxil-2-naftil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano;
Bromuro de (2R,3R)-3-[(2-ciclopentil-2-hidroxil-2-(o-clorofenil))etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano; Bromuro de (2R,3R)-3-[(2-ciclopentil-2-hidroxil-2-(3-piridil))etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano;
Bromuro de (2R,3R)-3-[(2-ciclopentil-2-hidroxil-2-(2-furil))etoxil]-1-(3-fenoxipropil)-1 -azabiciclo[2,2,2]octano; o Bromuro de (2R,3R)-3-[(2-ciclopentil-2-metoxil-2-(3-piridil))etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano.
7. Una composición farmacéutica que comprende el compuesto según una cualquiera de las reivindicaciones 1-5 y un portador farmacéuticamente aceptable, en donde la composición puede ser cualquier forma de dosificación adecuada, preferiblemente la forma de dosificación para la administración de inhalación o administración nasal; y la composición puede, además, comprender o no comprender, uno o más de los otros fármacos disponibles para la administración combinada.
8. Un procedimiento para la fabricación del compuesto según la reivindicación 1 que comprende las etapas de:
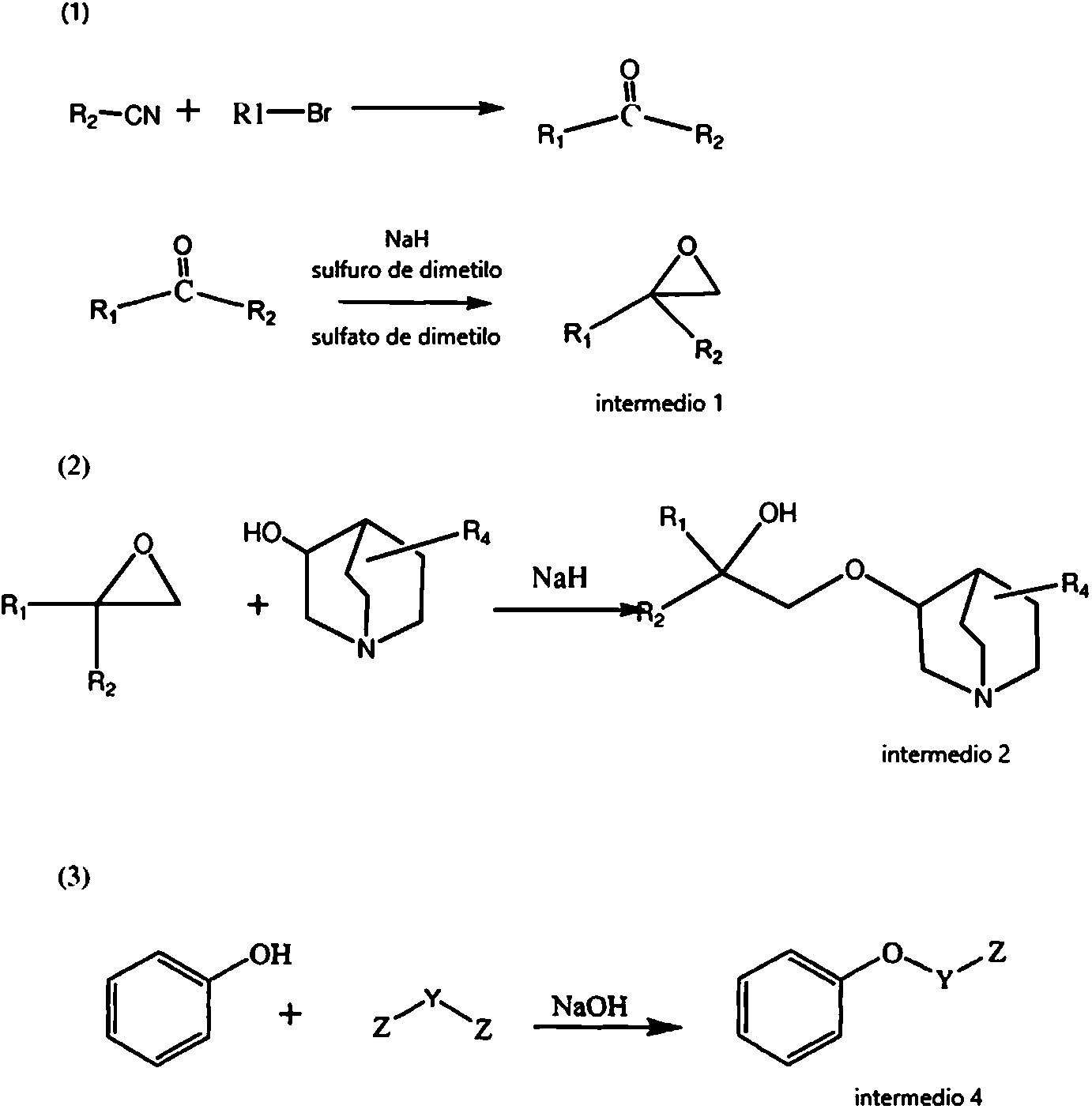
en donde Z es un átomo de halógeno,
(4) hacer reaccionar el intermedio 2 con el intermedio 4, para obtener el compuesto de fórmula (I).
9. El procedimiento según la reivindicación 8, que comprende además la etapa de purificación y separación de los isómeros químicos del intermedio 2 después de terminar la etapa (2).
10. El procedimiento según la reivindicación 8 o 9, en donde después de terminar la etapa (4), el compuesto se hace reaccionar con Ag2O para reemplazar el halógeno con hidróxido, que se puede transformar después en otro radical ácido mediante la reacción con otro ácido.
11. Uso del compuesto según una cualquiera de las reivindicaciones 1-5 para la fabricación de un medicamento capaz de tratar enfermedades asociadas con receptores M.
12. Uso según la reivindicación 11, en donde dichas enfermedades se seleccionan de un grupo que consiste en rinitis, rinitis después de un resfriado, traquitis crónica, hipersensibilidad de las vías respiratorias, asma, enfermedades pulmonares obstructivas crónicas, tos, incontinencia urinaria, micción frecuente, síndrome de vejiga inestable, espasmos vesicales, inflamación de la vejiga y enfermedades gastrointestinales como el síndrome de intestino irritable, colitis espástica, así como úlceras duodenales y gástricas.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201310297901 | 2013-07-13 | ||
| PCT/CN2014/000669 WO2015007073A1 (zh) | 2013-07-13 | 2014-07-11 | 奎宁类化合物、其光学异构体及其制备方法和医药用途 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2726664T3 true ES2726664T3 (es) | 2019-10-08 |
Family
ID=52345746
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES14825941T Active ES2726664T3 (es) | 2013-07-13 | 2014-07-11 | Compuestos de quinina e isómeros ópticos, procedimiento de preparación y uso médico de los mismos |
Country Status (13)
| Country | Link |
|---|---|
| US (1) | US9751875B2 (es) |
| EP (1) | EP3023424B1 (es) |
| JP (1) | JP6218940B2 (es) |
| KR (1) | KR101823451B1 (es) |
| CN (1) | CN105431432B (es) |
| AU (1) | AU2014292722B2 (es) |
| BR (1) | BR112016000629B1 (es) |
| CA (1) | CA2921621C (es) |
| ES (1) | ES2726664T3 (es) |
| RU (1) | RU2641285C2 (es) |
| TR (1) | TR201906532T4 (es) |
| WO (1) | WO2015007073A1 (es) |
| ZA (1) | ZA201600997B (es) |
Families Citing this family (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP6693943B2 (ja) * | 2015-03-02 | 2020-05-13 | 株式会社Lttバイオファーマ | キヌクリジン誘導体 |
| CA3047023C (en) | 2016-12-14 | 2022-05-31 | Beijing Showby Pharmaceutical Co., Ltd. | Class of bifunctional compounds with quaternary ammonium salt structure |
| CN109824661B (zh) * | 2019-03-30 | 2020-06-05 | 山东博洛德生物科技有限公司 | 一种盐酸戊乙奎醚中杂质的制备方法 |
| CN109824662A (zh) * | 2019-03-30 | 2019-05-31 | 山东博洛德生物科技有限公司 | 一种奎宁类化合物及其制备方法 |
| CN109776521A (zh) * | 2019-03-30 | 2019-05-21 | 山东博洛德生物科技有限公司 | 一种含有季铵基团的奎宁类化合物及其制备方法 |
| CA3181333A1 (en) * | 2020-04-26 | 2021-11-04 | Beijing Showby Pharmaceutical Co., Ltd. | Crystals, preparation method and application of a muscarinic receptor antagonist |
| CN114306331B (zh) * | 2020-10-10 | 2023-07-18 | 远大生命科学(武汉)有限公司 | 戊乙奎醚在治疗或预防视力损伤性眼部疾病中的用途 |
| CN118555954B (zh) * | 2022-01-19 | 2025-11-04 | 北京硕佰医药科技有限责任公司 | 一种治疗鼻炎的溶液型鼻喷雾剂及制备方法 |
| CN116514800A (zh) * | 2023-05-06 | 2023-08-01 | 湖南一格制药有限公司 | 盐酸戊乙奎醚的制备方法 |
Family Cites Families (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB1219606A (en) | 1968-07-15 | 1971-01-20 | Rech S Et D Applic Scient Soge | Quinuclidinol derivatives and preparation thereof |
| DE4108393A1 (de) | 1991-03-15 | 1992-09-17 | Boehringer Ingelheim Kg | Neue ester bi- und tricyclischer aminoalkohole, ihre herstellung und ihre verwendung in arzneimitteln |
| NO2005012I1 (no) * | 1994-12-28 | 2005-06-06 | Debio Rech Pharma Sa | Triptorelin og farmasoytisk akseptable salter derav |
| ES2165768B1 (es) * | 1999-07-14 | 2003-04-01 | Almirall Prodesfarma Sa | Nuevos derivados de quinuclidina y composiciones farmaceuticas que los contienen. |
| CZ294251B6 (cs) * | 2000-06-27 | 2004-11-10 | Laboratorios S. A. L. V. A. T., S. A. | Karbamáty a jejich použití pro výrobu farmaceutického prostředku |
| IL156558A0 (en) * | 2000-12-28 | 2004-01-04 | Almirall Prodesfarma Ag | Novel quinuclidine derivatives and medicinal compositions containing the same |
| ES2203327B1 (es) * | 2002-06-21 | 2005-06-16 | Almirall Prodesfarma, S.A. | Nuevos carbamatos de quinuclidina y composiciones farmaceuticas que los contienen. |
| ES2204295B1 (es) * | 2002-07-02 | 2005-08-01 | Almirall Prodesfarma, S.A. | Nuevos derivados de quinuclidina-amida. |
| PE20060259A1 (es) | 2004-04-27 | 2006-03-25 | Glaxo Group Ltd | Compuestos de quinuclidina como antagonistas del receptor de acetilcolina muscarinico |
| JP4637715B2 (ja) * | 2005-10-17 | 2011-02-23 | 日信工業株式会社 | 車両用支持構造体の製造方法 |
| CN1951938A (zh) * | 2005-10-21 | 2007-04-25 | 刘丽娅 | 戊乙奎醚季铵盐及其衍生物 |
| AU2007336054A1 (en) * | 2006-12-19 | 2008-06-26 | Astrazeneca Ab | Quinuclidinol derivatives as muscarinic receptor antagonists |
| KR20090107567A (ko) * | 2007-02-09 | 2009-10-13 | 아스텔라스세이야쿠 가부시키가이샤 | 아자 가교환 화합물 |
| CN102070631B (zh) * | 2009-11-20 | 2016-09-07 | 中国人民解放军军事医学科学院毒物药物研究所 | 戊乙奎醚光学异构体的季铵盐、药物组合物及其医药用途 |
| CN102850344A (zh) * | 2011-06-28 | 2013-01-02 | 中国人民解放军军事医学科学院毒物药物研究所 | 戊乙奎醚光学异构体衍生物用于抗肿瘤的医药用途 |
-
2014
- 2014-07-11 EP EP14825941.9A patent/EP3023424B1/en active Active
- 2014-07-11 RU RU2016104437A patent/RU2641285C2/ru active
- 2014-07-11 US US14/904,766 patent/US9751875B2/en active Active
- 2014-07-11 CN CN201480020342.8A patent/CN105431432B/zh active Active
- 2014-07-11 AU AU2014292722A patent/AU2014292722B2/en active Active
- 2014-07-11 TR TR2019/06532T patent/TR201906532T4/tr unknown
- 2014-07-11 ES ES14825941T patent/ES2726664T3/es active Active
- 2014-07-11 CA CA2921621A patent/CA2921621C/en active Active
- 2014-07-11 JP JP2016524657A patent/JP6218940B2/ja active Active
- 2014-07-11 KR KR1020167003638A patent/KR101823451B1/ko active Active
- 2014-07-11 BR BR112016000629-1A patent/BR112016000629B1/pt active IP Right Grant
- 2014-07-11 WO PCT/CN2014/000669 patent/WO2015007073A1/zh not_active Ceased
-
2016
- 2016-02-12 ZA ZA2016/00997A patent/ZA201600997B/en unknown
Also Published As
| Publication number | Publication date |
|---|---|
| BR112016000629B1 (pt) | 2023-10-31 |
| CA2921621A1 (en) | 2015-01-22 |
| CA2921621C (en) | 2018-08-28 |
| EP3023424A4 (en) | 2017-01-18 |
| CN105431432B (zh) | 2018-01-09 |
| EP3023424A1 (en) | 2016-05-25 |
| US9751875B2 (en) | 2017-09-05 |
| KR20160033146A (ko) | 2016-03-25 |
| CN105431432A (zh) | 2016-03-23 |
| WO2015007073A1 (zh) | 2015-01-22 |
| KR101823451B1 (ko) | 2018-01-30 |
| EP3023424B1 (en) | 2019-02-27 |
| BR112016000629A2 (es) | 2017-07-25 |
| RU2641285C2 (ru) | 2018-01-17 |
| TR201906532T4 (tr) | 2019-05-21 |
| ZA201600997B (en) | 2018-05-30 |
| RU2016104437A (ru) | 2017-08-16 |
| JP6218940B2 (ja) | 2017-10-25 |
| US20160244439A1 (en) | 2016-08-25 |
| AU2014292722A1 (en) | 2016-03-03 |
| AU2014292722B2 (en) | 2017-03-09 |
| JP2016528199A (ja) | 2016-09-15 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2726664T3 (es) | Compuestos de quinina e isómeros ópticos, procedimiento de preparación y uso médico de los mismos | |
| ES2324746T3 (es) | Compuestos de ciclopentano y ciclopenteno sustituidos utiles como inhibidores de neuraminidasa. | |
| CN103249697B (zh) | 适用作多巴胺能稳定剂的普利多匹定(pridopidine)氘化类似物 | |
| JP5596680B2 (ja) | 経口抗がん製剤 | |
| RU2624446C2 (ru) | Трициклические соединения, композиции, содержащие указанные соединения, и их применения | |
| CN113272298B (zh) | 用于治疗呼吸系统疾病的化合物和组合物 | |
| ES2899367T3 (es) | Forma amorfa de trifenatato de vilanterol y procedimientos para su preparación | |
| US9822117B2 (en) | Pyridoindolobenz[b,d]azepine derivatives and uses thereof | |
| WO2020183227A1 (en) | Treating influenza using substituted polycyclic pyridone derivatives and prodrugs thereof in a subject having influenza and a complication risk factor | |
| JP7498504B2 (ja) | 脳神経または心臓保護剤としてのアミノチオール系化合物の使用 | |
| JPH0269417A (ja) | 精神分裂病治療用組成物 | |
| US20150141519A1 (en) | Agomelatine sulfuric acid complex, and preparation method and application thereof | |
| US20240261283A1 (en) | Pharmaceutical preparation for preventing or treating pulmonary fibrosis | |
| US20230100890A1 (en) | Use of combined inhalant cannabinoid therapy in the treatment of migraine | |
| CN115025080B (zh) | 一种抗流感病毒的药物组合物及其用途 | |
| CN104262292A (zh) | 苯乙酸酯化合物及其应用 | |
| WO2025259966A1 (en) | Pharmaceutical formulations comprising a salt of a docosahexaenoic acid (dha) analog | |
| WO2024078387A1 (zh) | 取代3-氟苯丙酸酯类化合物及其制备方法、药物组合及用途 | |
| JP2005060311A (ja) | N−(ベンゾイル)アミノ酸誘導体を有効成分とするニューロパシー性疼痛治療剤 | |
| HK40114273A (zh) | 一种曲前列环素衍生物、其制备方法及应用 |