ES2728201T3 - Inhibidores de Mnk y métodos relacionados con los mismos - Google Patents
Inhibidores de Mnk y métodos relacionados con los mismos Download PDFInfo
- Publication number
- ES2728201T3 ES2728201T3 ES15742147T ES15742147T ES2728201T3 ES 2728201 T3 ES2728201 T3 ES 2728201T3 ES 15742147 T ES15742147 T ES 15742147T ES 15742147 T ES15742147 T ES 15742147T ES 2728201 T3 ES2728201 T3 ES 2728201T3
- Authority
- ES
- Spain
- Prior art keywords
- synthesis
- cycloalkyl
- compound
- alkyl
- pyridin
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 0 CCC*(N(C(C(CC)N(C1=C(*=C)C=C2N(*)c3c(*)c(*)nc(*)n3)C2=O)=*)C1=O)=C Chemical compound CCC*(N(C(C(CC)N(C1=C(*=C)C=C2N(*)c3c(*)c(*)nc(*)n3)C2=O)=*)C1=O)=C 0.000 description 47
- OYRRZWATULMEPF-UHFFFAOYSA-N Nc1ccncn1 Chemical compound Nc1ccncn1 OYRRZWATULMEPF-UHFFFAOYSA-N 0.000 description 4
- DDNRPKSPICFQDZ-UHFFFAOYSA-N O=C(C1=C(C=C2Br)Cl)NC3(CCCCC3)N1C2=O Chemical compound O=C(C1=C(C=C2Br)Cl)NC3(CCCCC3)N1C2=O DDNRPKSPICFQDZ-UHFFFAOYSA-N 0.000 description 2
- OEVBYHNIUPOXDX-UHFFFAOYSA-N O=C(C1=C(C=C2Nc3ccncn3)Cl)NC3(CCCCC3)N1C2=O Chemical compound O=C(C1=C(C=C2Nc3ccncn3)Cl)NC3(CCCCC3)N1C2=O OEVBYHNIUPOXDX-UHFFFAOYSA-N 0.000 description 2
- WUTUVDYXAUYFFK-ARJAWSKDSA-N C=C/N=C(\C=C/N)/N Chemical compound C=C/N=C(\C=C/N)/N WUTUVDYXAUYFFK-ARJAWSKDSA-N 0.000 description 1
- GLRJSUCRQIZARD-UHFFFAOYSA-N CC(C#CC)(NC(C1=C(C)C=C2Nc3ncncc3)=O)N1C2=O Chemical compound CC(C#CC)(NC(C1=C(C)C=C2Nc3ncncc3)=O)N1C2=O GLRJSUCRQIZARD-UHFFFAOYSA-N 0.000 description 1
- ZMSSRIVKYWEXKL-UHFFFAOYSA-N CC(C)(C)OC(NC1=CC(N)NC=N1)=O Chemical compound CC(C)(C)OC(NC1=CC(N)NC=N1)=O ZMSSRIVKYWEXKL-UHFFFAOYSA-N 0.000 description 1
- NMHRIPOIIGVQLO-UHFFFAOYSA-N CC(C)(CCC1)C1(NC(C1=C(C=C2Br)Cl)=O)N1C2=O Chemical compound CC(C)(CCC1)C1(NC(C1=C(C=C2Br)Cl)=O)N1C2=O NMHRIPOIIGVQLO-UHFFFAOYSA-N 0.000 description 1
- YRWGYLSIDZELMS-UHFFFAOYSA-N CC(C)(CCC1)C1(NC(C1=C(C=C2Nc3ncncc3)Cl)=O)N1C2=O Chemical compound CC(C)(CCC1)C1(NC(C1=C(C=C2Nc3ncncc3)Cl)=O)N1C2=O YRWGYLSIDZELMS-UHFFFAOYSA-N 0.000 description 1
- FTGZMZBYOHMEPS-UHFFFAOYSA-N CC(C)(CCC1)C1=O Chemical compound CC(C)(CCC1)C1=O FTGZMZBYOHMEPS-UHFFFAOYSA-N 0.000 description 1
- CIEXJDJHKQURRH-UHFFFAOYSA-N CC(C)(NC(C1=CC=C2Cl)=O)N1C2=O Chemical compound CC(C)(NC(C1=CC=C2Cl)=O)N1C2=O CIEXJDJHKQURRH-UHFFFAOYSA-N 0.000 description 1
- IEZSRTFZJXWJSC-UHFFFAOYSA-N CC(C)(NC(C1=CC=C2[n]3c4ncncc4cc3)=O)N1C2=O Chemical compound CC(C)(NC(C1=CC=C2[n]3c4ncncc4cc3)=O)N1C2=O IEZSRTFZJXWJSC-UHFFFAOYSA-N 0.000 description 1
- FGXZMJWSYKFHRO-UHFFFAOYSA-N CC(C)C(C)(NC(C1=CC=C2Cl)=O)N1C2=O Chemical compound CC(C)C(C)(NC(C1=CC=C2Cl)=O)N1C2=O FGXZMJWSYKFHRO-UHFFFAOYSA-N 0.000 description 1
- MTMFRAIOFPWKIH-UHFFFAOYSA-N CC(C)C(C)(NC(C1=CC=C2Nc3ccncn3)=O)N1C2=O Chemical compound CC(C)C(C)(NC(C1=CC=C2Nc3ccncn3)=O)N1C2=O MTMFRAIOFPWKIH-UHFFFAOYSA-N 0.000 description 1
- UJBNTDMFKAHGKW-UHFFFAOYSA-N CC(C)c1c(N)ncnc1NC1=CC(Cl)=C(C(NC23CCCCC2)=O)N3C1=O Chemical compound CC(C)c1c(N)ncnc1NC1=CC(Cl)=C(C(NC23CCCCC2)=O)N3C1=O UJBNTDMFKAHGKW-UHFFFAOYSA-N 0.000 description 1
- UWDVYDSWRKPMFA-UHFFFAOYSA-N CC(C1C=CCC1)=O Chemical compound CC(C1C=CCC1)=O UWDVYDSWRKPMFA-UHFFFAOYSA-N 0.000 description 1
- SQCWJQLQJURJCB-UHFFFAOYSA-N CC(C1C=NC(NC(C2C=C2)=O)=C2OC)C12N Chemical compound CC(C1C=NC(NC(C2C=C2)=O)=C2OC)C12N SQCWJQLQJURJCB-UHFFFAOYSA-N 0.000 description 1
- CIKAKOKWQCNYGZ-UHFFFAOYSA-N CC(C=C1NC2=NCN(C)C(N)=C2OC)=C(C(NC23CCN(CC#N)CC2)=O)N3C1=O Chemical compound CC(C=C1NC2=NCN(C)C(N)=C2OC)=C(C(NC23CCN(CC#N)CC2)=O)N3C1=O CIKAKOKWQCNYGZ-UHFFFAOYSA-N 0.000 description 1
- XIBOMGGMZFMRQO-UHFFFAOYSA-N CC(C=C1NC2=NCNC(NC)=C2OC)=C(C[NH+](C23CCNCC2)[O-])N3C1=O Chemical compound CC(C=C1NC2=NCNC(NC)=C2OC)=C(C[NH+](C23CCNCC2)[O-])N3C1=O XIBOMGGMZFMRQO-UHFFFAOYSA-N 0.000 description 1
- GMDJOHXJFKCMCN-UHFFFAOYSA-N CC(C=C1Nc(ncnc2N)c2OC)=C(C(NC23CCN(CC#N)CC2)=O)N3C1=O Chemical compound CC(C=C1Nc(ncnc2N)c2OC)=C(C(NC23CCN(CC#N)CC2)=O)N3C1=O GMDJOHXJFKCMCN-UHFFFAOYSA-N 0.000 description 1
- YQSSEHCGGFFNFD-UHFFFAOYSA-N CC(C=C1Nc(ncnc2NC)c2OC)=C(C(NC23CCNCC2)=O)N3C1=O Chemical compound CC(C=C1Nc(ncnc2NC)c2OC)=C(C(NC23CCNCC2)=O)N3C1=O YQSSEHCGGFFNFD-UHFFFAOYSA-N 0.000 description 1
- LTUHLWFACNSIFM-UHFFFAOYSA-N CC(c1cccc(F)c1)(NC(C1=C(C=C2Br)Cl)=O)N1C2=O Chemical compound CC(c1cccc(F)c1)(NC(C1=C(C=C2Br)Cl)=O)N1C2=O LTUHLWFACNSIFM-UHFFFAOYSA-N 0.000 description 1
- BGYBGAPNGLNRFU-UHFFFAOYSA-N CC(c1ccncc1)(NC(C1=C(C=C2Nc3cc(N)ncn3)Cl)=O)N1C2=O Chemical compound CC(c1ccncc1)(NC(C1=C(C=C2Nc3cc(N)ncn3)Cl)=O)N1C2=O BGYBGAPNGLNRFU-UHFFFAOYSA-N 0.000 description 1
- JPAINVMTDOLEKC-UHFFFAOYSA-N CCNC(C(NC(C(Br)=C1)=O)=C1Cl)=O Chemical compound CCNC(C(NC(C(Br)=C1)=O)=C1Cl)=O JPAINVMTDOLEKC-UHFFFAOYSA-N 0.000 description 1
- KTTAPNZPPVHZSE-UHFFFAOYSA-N C[Si+](C)(C)C#Cc1c(N)ncnc1 Chemical compound C[Si+](C)(C)C#Cc1c(N)ncnc1 KTTAPNZPPVHZSE-UHFFFAOYSA-N 0.000 description 1
- WIEGBQIAYAGEEN-UHFFFAOYSA-N Cc1cc(NC2=CC(Cl)=C(C(N)=O)NC2=O)ncn1 Chemical compound Cc1cc(NC2=CC(Cl)=C(C(N)=O)NC2=O)ncn1 WIEGBQIAYAGEEN-UHFFFAOYSA-N 0.000 description 1
- VEQDVIFAAZPSDS-UHFFFAOYSA-N NC(C(NC(C(Br)=C1)=O)=C1Cl)=O Chemical compound NC(C(NC(C(Br)=C1)=O)=C1Cl)=O VEQDVIFAAZPSDS-UHFFFAOYSA-N 0.000 description 1
- NJKPNICNQITSRE-UHFFFAOYSA-N NC(C(NC(C(Nc(ncnc1)c1C#N)=C1)=O)=C1Cl)=O Chemical compound NC(C(NC(C(Nc(ncnc1)c1C#N)=C1)=O)=C1Cl)=O NJKPNICNQITSRE-UHFFFAOYSA-N 0.000 description 1
- GPZKOMXEYWKSBE-UHFFFAOYSA-N NC(C(NC(C(Nc1cc(N)ncn1)=C1)=O)=C1Cl)=O Chemical compound NC(C(NC(C(Nc1cc(N)ncn1)=C1)=O)=C1Cl)=O GPZKOMXEYWKSBE-UHFFFAOYSA-N 0.000 description 1
- ZXUYFPVDZZZEIW-UHFFFAOYSA-N NC(C(NC1=O)=CC=C1Nc1ccncn1)O Chemical compound NC(C(NC1=O)=CC=C1Nc1ccncn1)O ZXUYFPVDZZZEIW-UHFFFAOYSA-N 0.000 description 1
- LHCPRYRLDOSKHK-UHFFFAOYSA-N Nc1c(cn[nH]2)c2ncn1 Chemical compound Nc1c(cn[nH]2)c2ncn1 LHCPRYRLDOSKHK-UHFFFAOYSA-N 0.000 description 1
- OUSCNSVPOGOLLX-BAQGIRSFSA-N Nc1cc(/C=C(/C2CC2)\C(F)(F)F)ncn1 Chemical compound Nc1cc(/C=C(/C2CC2)\C(F)(F)F)ncn1 OUSCNSVPOGOLLX-BAQGIRSFSA-N 0.000 description 1
- JAIYUIOGVNRXEW-UHFFFAOYSA-N Nc1ncncc1C#N Chemical compound Nc1ncncc1C#N JAIYUIOGVNRXEW-UHFFFAOYSA-N 0.000 description 1
- VIDIHVFXUZJRBN-UHFFFAOYSA-N O=C(C1=C(C2OCC2)C=C2Nc3ccncn3)NC3(CCCCC3)N1C2=O Chemical compound O=C(C1=C(C2OCC2)C=C2Nc3ccncn3)NC3(CCCCC3)N1C2=O VIDIHVFXUZJRBN-UHFFFAOYSA-N 0.000 description 1
- RWALTXNCWCAKGV-UHFFFAOYSA-N O=C(C1=C(C=C2Nc3ncnc4c3cn[nH]4)Cl)NC3(CCCCC3)N1C2=O Chemical compound O=C(C1=C(C=C2Nc3ncnc4c3cn[nH]4)Cl)NC3(CCCCC3)N1C2=O RWALTXNCWCAKGV-UHFFFAOYSA-N 0.000 description 1
- AVWGOFIGHOXGPM-UHFFFAOYSA-N O=C(C1=C(C=C2Nc3ncncc3)Cl)NC(CCCC3)(C3F)N1C2=O Chemical compound O=C(C1=C(C=C2Nc3ncncc3)Cl)NC(CCCC3)(C3F)N1C2=O AVWGOFIGHOXGPM-UHFFFAOYSA-N 0.000 description 1
- VQYOFTVCYSPHPG-UHFFFAOYSA-N O=C(CCCC1)C1F Chemical compound O=C(CCCC1)C1F VQYOFTVCYSPHPG-UHFFFAOYSA-N 0.000 description 1
- RMMZGPSYPUIIRO-UHFFFAOYSA-N O=C(c1c[s]cc1)c1c[s]cc1 Chemical compound O=C(c1c[s]cc1)c1c[s]cc1 RMMZGPSYPUIIRO-UHFFFAOYSA-N 0.000 description 1
- HMAHOYGQHDTHHM-UHFFFAOYSA-N OC1N(C2(CCCCC2)NC2=O)C2=C(C2OCC2)C=C1Nc1ccncn1 Chemical compound OC1N(C2(CCCCC2)NC2=O)C2=C(C2OCC2)C=C1Nc1ccncn1 HMAHOYGQHDTHHM-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/4353—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems
- A61K31/437—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems the heterocyclic ring system containing a five-membered ring having nitrogen as a ring hetero atom, e.g. indolizine, beta-carboline
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/506—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/519—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/519—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with heterocyclic rings
- A61K31/52—Purines, e.g. adenine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/55—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole
- A61K31/551—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole having two nitrogen atoms, e.g. dilazep
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/10—Spiro-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/12—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains three hetero rings
- C07D471/20—Spiro-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D473/00—Heterocyclic compounds containing purine ring systems
- C07D473/26—Heterocyclic compounds containing purine ring systems with an oxygen, sulphur, or nitrogen atom directly attached in position 2 or 6, but not in both
- C07D473/32—Nitrogen atom
- C07D473/34—Nitrogen atom attached in position 6, e.g. adenine
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/12—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains three hetero rings
- C07D491/20—Spiro-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D495/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having sulfur atoms as the only ring hetero atoms
- C07D495/12—Heterocyclic compounds containing in the condensed system at least one hetero ring having sulfur atoms as the only ring hetero atoms in which the condensed system contains three hetero rings
- C07D495/20—Spiro-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D519/00—Heterocyclic compounds containing more than one system of two or more relevant hetero rings condensed among themselves or condensed with a common carbocyclic ring system not provided for in groups C07D453/00 or C07D455/00
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- Epidemiology (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Hematology (AREA)
- Oncology (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Oxygen Or Sulfur (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Abstract
Un compuesto de acuerdo con la Fórmula (I): **Fórmula** o un estereoisómero, tautómero o una sal farmacéuticamente aceptable del mismo, en la que: W1 y W2 son independientemente O, S o N-OR', donde R' es alquilo C1-C4; Y es -N(R5)-, -O-, -S-, -C(O)-, -S=O, -S(O)2- o -CHR9-; R1 es hidrógeno, alquilo inferior, cicloalquilo o heterociclilo, donde cualquier alquilo C1-C4, cicloalquilo o heterociclilo está opcionalmente sustituido con 1, 2 o 3 grupos J; n es 1, 2 o 3; R2 y R3 son cada uno independientemente hidrógeno, alquilo, alquenilo, alquinilo, arilo, aralquileno, heteroarilo, heteroarilalquileno, cicloalquilo, cicloalquilalquileno, heterociclilo o heterociclilalquileno, donde cualquier alquilo, arilo, aralquileno, heteroarilo, heteroarilalquileno, cicloalquilo, cicloalquilalquileno, heterociclilo, o heterociclilalquileno, está opcionalmente sustituido con 1, 2 o 3 grupos J; o R2 y R3 tomados junto con el átomo de carbono al que están unidos forman un cicloalquilo o heterociclilo, donde cualquier cicloalquilo o heterociclilo está opcionalmente sustituido con 1, 2 o 3 grupos J; R4a y R4b son cada uno independientemente hidrógeno, halógeno, hidroxilo, tiol, hidroxialquileno, ciano, alquilo, alcoxi, acilo, tioalquilo, alquenilo, alquinilo, cicloalquilo, arilo o heterociclilo; R5 es hidrógeno, ciano o alquilo C1-C4; o R5 y R8 tomados junto con los átomos a los que están unidos forman un heterociclilo condensado opcionalmente sustituido con 1, 2 o 3 grupos J; R6, R7 y R8 son cada uno independientemente hidrógeno, hidroxi, halógeno, ciano, amino, alquilo, alquenilo, alquinilo, alcoxi, cicloalquilo, cicloalquilalquileno, cicloalquilalquenileno, alquilaminilo, alquilcarbonilaminilo, cicloalquilcarbonilaminilo, cicloalquilaminilo, heterociclilaminilo, heteroarilo o heterociclilo, y donde cualquier amino, alquilo, alquenilo, alquinilo, alcoxi, cicloalquilo, cicloalquilalquileno, cicloalquilalquenileno, amino, alquilaminilo, alquilcarbonilaminilo, cicloalquilcarbonilaminilo, cicloalquilaminilo, heterociclilaminilo, heteroarilo, o heterociclilo está opcionalmente sustituido con 1, 2 o 3 grupos J; o R7 y R8 tomados junto con los átomos a los que están unidos forman un heterociclilo condensado o heteroarilo opcionalmente sustituido con 1, 2 o 3 grupos J; J es -SH, -SR9, -S(O)R9, -S(O)2R9, -S(O)NH2, -S(O)NR9R9, -NH2, -NR9R9, -COOH, -C(O)OR9, -C(O)R9, -C(O)-NH2, -C(O)-NR9R9, hidroxi, ciano, halógeno, acetilo, alquilo, alquilo inferior, alquenilo, alquinilo, alcoxi, haloalquilo, tioalquilo, cianoalquileno, alquilaminilo, NH2-C(O)-alquileno, NR9R9-C(O)-alquileno, -CHR9-C(O)-alquilo inferior, -C(O)-alquilo inferior, alquilcarbonilaminilo, cicloalquilo, cicloalquilalquileno, cicloalquilalquenileno, cicloalquilcarbonilaminilo, cicloalquilaminilo, -CHR9-C(O)-cicloalquilo, -C(O)-cicloalquilo, -CHR9-C(O)-arilo, -CHR9-arilo, -C(O)-arilo, -CHR9-C(O)-heterocicloalquilo, -C(O)-heterocicloalquilo, heterociclilaminilo o heterociclilo; o cualesquiera dos grupos J unidos al mismo carbono o heteroátomo pueden tomarse juntos para formar oxo; y R9 es hidrógeno, alquilo C1-C4 o -OH.
Description
DESCRIPCIÓN
Inhibidores de Mnk y métodos relacionados con los mismos
Campo
La presente invención se refiere en general a compuestos que tienen actividad como inhibidores de cinasa de interacción con MAP cinasa (Mnk), así como a composiciones relacionadas y métodos que contienen o usan los mismos. Dichos compuestos encuentran utilidad en cualquier número de aplicaciones terapéuticas, incluyendo el tratamiento del cáncer.
Antecedentes
El factor de iniciación eucariota 4E (eIF4E) es un factor de traducción general, pero tiene el potencial de potenciar preferentemente la traducción de moléculas de ARN mensajero (ARNm) que conducen a la producción de proteínas asociadas a malignidad. Esta selectividad puede estar relacionada con una mayor necesidad de eIF4E y sus socios de unión para la traducción de moléculas de ARNm que contienen estructura secundaria extensa en sus regiones 5' no traducidas (5'-UTR). Estas moléculas de ARNm incluyen las que codifican para determinadas proteínas que controlan la progresión del ciclo celular y la carcinogénesis. En condiciones celulares normales, la traducción de estas moléculas de ARNm asociadas a malignidad se suprime a medida que la disponibilidad de eIF4E activo se ve limitada; sin embargo, sus niveles pueden aumentar cuando eIF4E se sobreexpresa o se hiperactiva. Se han encontrado niveles elevados de eIF4E en muchos tipos de tumores y estirpes celulares de cáncer incluyendo cánceres de colon, mama, vejiga, pulmón, próstata, tracto gastrointestinal, cabeza y cuello, linfomas de Hodgkin y neuroblastomas.
Se cree que la iniciación de la traducción dependiente de la caperuza depende del montaje de eIF4F, un complejo de factor de iniciación que incluye eIF4E, la proteína de andamio eIF4G y la ARN helicasa eIF4A. Debido a que eIF4E es la única de estas proteínas que se une directamente a la estructura de la caperuza de ARNm, es el factor clave para el montaje de eIF4F en la caperuza 5'. La proteína de andamio, eIF4G, también recluta la subunidad ribosómica 40S al ARNm a través de su interacción con eIF3 y se une a eIF4B, una proteína que ayuda a la función ARN-helicasa de eIF4A, facilitando de este modo la traducción de moléculas de ARNm que contienen 5'-UTR estructuradas. La disponibilidad de eIF4E como parte del complejo eIF4F es un factor limitante en el control de la velocidad de traducción, por lo que eIF4E es un regulador importante de la traducción de ARNm.
La regulación de la actividad de eIF4E forma un nodo de convergencia de las vías de señalización PI3K/Akt/mTOR y Ras/Raf/MAPK. La vía de PI3K (fosfoinosítido 3-cinasa)/PTEN (homólogo de fosfatasa y tensina suprimido en el cromosoma diez)/Akt/mTOR (diana de mamífero de la rapamicina) suele estar implicada en la carcinogénesis y en la sensibilidad y resistencia al tratamiento del cáncer. La señalización desregulada a través de la vía PI3K/PTEN/Akt/mTOR es con frecuencia el resultado de alteraciones genéticas en componentes críticos de esta vía y/o mutaciones en receptores de factor de crecimiento corriente arriba o componentes de señalización. PI3K inicia una cascada de eventos cuando se activa, por ejemplo, factores de crecimiento extracelular, mitógenos, citocinas y/o receptores, PDK1 activa Akt, que a su vez fosforila e inactiva el complejo supresor tumoral que comprende TSC1 y 2 (complejo de esclerosis tuberosa 1/2), dando como resultado la activación de mTORC1 (diana del complejo de rapamicina 1) por Rheb-GTP. La activación de PDK1 y Akt por PI3Ks está regulada negativamente por PTEN.
PTEN es un gen supresor tumoral crítico y con frecuencia está mutado o silenciado en los cánceres humanos. Su pérdida da como resultado la activación de Akt y aumenta la señalización de mTORC1 corriente abajo. La implicación del complejo mTOR 1 (mTORC1) en la transformación neoplásica parece depender de su papel regulador hacia el complejo eIF4F; la sobreexpresión de eIF4E puede conferir resistencia a la rapamicina. mTORC 1 regula el ensamble del complejo eIF4F que es crítico para la traducción de moléculas de ARNm asociadas al crecimiento celular, la prevención de la apoptosis y la transformación. mTORC1 consigue esto mediante la fosforilación y la inactivación de 4E-BP y la posterior disociación de 4E-BP de eIF4E. Esto permite entonces que eIF4E interactúe con la proteína eIF4G de andamio, permitiendo el ensamblaje del complejo eIF4F para la traducción de moléculas de ARNm estructuradas. mTORC1 también promueve la activación del activador de la traducción, S6K, que fosforila la proteína ribosómica S6 y otros sustratos, incluyendo eIF4B. La señalización de mTORC1 es inhibida por la rapamicina y sus análogos (rapálogos), aunque estos compuestos actúan alostéricamente, en lugar de inhibir directamente la actividad de mTOR cinasa.
Dada la importancia de la vía PI3K/Akt/mTOR en la regulación de la traducción de ARNm de genes que codifican para proteínas pro-oncogénicas y señalización de mTORC1 activada en una alta proporción de cánceres, estas cinasas se han perseguido activamente como dianas de fármacos oncológicos. Se han identificado varios inhibidores farmacológicos, algunos de los cuales han alcanzado etapas clínicas avanzadas. Sin embargo, recientemente se ha puesto de manifiesto que la vía mTOR participa en un circuito de retroalimentación complicado que puede perjudicar la activación de Akt. Se ha demostrado que el tratamiento prolongado de células cancerosas o pacientes con inhibidores de mTOR provoca una actividad elevada de PI3K que conduce a la fosforilación de Akt y eIF4E y promueve la supervivencia de células cancerígenas. EIF4E, actuando corriente abajo de Akt y mTOR,
recapitula la acción de Akt en la carcinogénesis y resistencia a fármacos, y la señalización de Akt a través de eIF4E es un importante mecanismo de oncogénesis y resistencia a fármacos in vivo.
Además de la vía PDK/Akt/mTOR, eIF4E también es la diana de la cascada de señalización Ras/Raf/MAP que es activada por factores de crecimiento y para la vía de MAP cinasa p38 activada por estrés. Erkl/2 y p38 fosforilan entonces la cinasa 1 que interactúa con la MAP cinasa (Mnk1) y la cinasa 2 que interactúa con la MAP cinasa (Mnk2). La vía de Erk también se activa en muchos cánceres, lo que refleja, por ejemplo, la activación de mutaciones en Ras (que se encuentra en alrededor del 20 % de los tumores) o la pérdida de la función de la proteína activadora Ras GTPasa NF1. Mnk1 y Mnk2 son proteínas cinasa treonina/serina y específicamente fosforilan la serina 209 (Ser209) de eIF4E dentro del complejo eIF4F, en virtud de la interacción entre eIF4E y Mnk, que sirve para reclutar Mnk para actuar sobre eIF4E. Los ratones con eIF4E mutado, en los que la Ser209 se reemplaza por alanina, no muestran fosforilación de eIF4E y atenúan significativamente el crecimiento tumoral. Significativamente, mientras que la actividad de las Mnk es necesaria para la transformación oncogénica mediada por eIF4E, es dispensable para el desarrollo normal. Las Mnk farmacológicamente inhibidoras presentan de este modo una atractiva estrategia terapéutica para el cáncer.
A pesar del aumento de la comprensión de la estructura y función de Mnk, se han hecho pocos progresos con respecto al descubrimiento de inhibidores farmacológicos de Mnk y se han notificado relativamente pocos inhibidores de Mnk: CGP052088 (Tschopp et al., Mol Cell Biol Res Commun. 3(4): 205-211, 2000); CGP57380 (Rowlett et al., Am J Physiol Gastrointest Liver Physiol. 294 (2): G452-459, 2008); y Cercosporamida (Konicek et al., Cáncer Res. 71(5): 1849-1857, 2011). Estos compuestos, sin embargo, se han usado principalmente con el fin de validación de la diana de Mnk. Más recientemente, los investigadores han propuesto compuestos adicionales para tratar enfermedades influenciadas por la inhibición de la actividad cinasa Mnk1 y/o Mnk2, incluyendo, por ejemplo, los compuestos desvelados en el documento WO 2014/044691 y los diversos documentos de patente citados en el mismo, los compuestos desvelados por Oyarzabal et al., Journal of Med. Chem., 53: 6618-6628, 2010 y las 4-dihidropiridinon-3-il)amino-5-metiltien[2,3-d]pirimidinas desveladas por Yu et al., European Journal of Med. Chem., 95: 116-126, 2015.
En consecuencia, aunque se han hecho avances en este campo, sigue existiendo una necesidad significativa en la técnica de compuestos que inhiben específicamente la actividad de Mnk cinasa, en particular con respecto al papel de Mnk en la regulación de vías de cáncer, así como para la composición y métodos asociados. La presente invención satisface esta necesidad y proporciona otras ventajas relacionadas.
Sumario
La presente invención se refiere a compuestos que inhiben o modulan la actividad de Mnk, así como a estereoisómeros, tautómeros y sales farmacéuticamente aceptables de dichos compuestos como agentes terapéuticos candidatos. La presente invención también se refiere a composiciones que contienen dichos compuestos y métodos asociados para tratar afecciones que se beneficiarían de la inhibición de Mnk, tal como el cáncer.
En una realización, la invención se refiere a compuestos que se ajustan a la Fórmula I así como a un estereoisómero, tautómero o sal farmacéuticamente aceptable de dichos compuestos.

Para los compuestos de Fórmula I, R1, R2, R3, R4a, R4b, R6, R7, R8, W1, W2, Y y "n" son como se definen después.
En otra realización, se desvelan composiciones que comprenden un compuesto de estructura (I) en combinación con un vehículo, diluyente o excipiente farmacéuticamente aceptable.
En una realización adicional, se proporcionan métodos para tratar una afección dependiente de Mnk en un mamífero que lo necesite. Dichos métodos comprenden administrar una cantidad eficaz de un compuesto de estructura (I), o composiciones que lo comprenden, al mamífero. Las afecciones incluyen, pero sin limitación, diversas formas de
cáncer como se describe con más detalle después.
Estos y otros aspectos de la invención serán evidentes haciendo referencia a la siguiente descripción detallada. Con este fin, se describen en el presente documento diversas referencias que describen con más detalle determinada información de antecedentes, procedimientos, compuestos y/o composiciones, y cada una de ellas se incorpora en el presente documento por referencia en su totalidad.
Figuras
La Figura 1 ilustra datos de DRXP para la forma de sal de cloruro de hidrógeno de un compuesto de Fórmula I de ejemplo.
Descripción detallada
En la siguiente descripción, se exponen determinados detalles específicos con el fin de proporcionar una comprensión completa de diversas realizaciones de la invención. Sin embargo, un experto en la materia entenderá que la invención se puede poner en práctica sin estos detalles. A menos que el contexto lo exija de otro modo, a lo largo de la presente memoria descriptiva y reivindicaciones, la palabra "comprender" y las variaciones de la misma, tales como "comprende" y "comprendiendo" deben interpretarse en un sentido abierto e inclusivo (es decir, “incluyendo, pero no limitado a").
La referencia en toda la presente memoria descriptiva a "una sola realización" o "una realización" significa que se incluye una característica, estructura o característica particular descrita en relación con la realización en al menos una realización de la presente invención. Por tanto, las apariciones de las frases "en una sola realización" o "en una realización" en diversos lugares a lo largo de la presente memoria descriptiva no se refieren necesariamente a la misma realización. Además, las características, estructuras o características particulares pueden combinarse de cualquier manera adecuada en una o más realizaciones.
Definiciones
Como se usan en el presente documento, y a menos que se indique lo contrario, los siguientes términos y frases tienen el significado que se indica después.
"Amino" se refiere al sustituyente -NH2.
"Aminocarbonilo" se refiere al sustituyente -C(O)NH2.
Carboxilo se refiere al sustituyente -CO2H.
"Carbonilo" se refiere a un grupo -C(O)- o -C(=O)-. Ambas anotaciones se usan indistintamente dentro de la memoria descriptiva.
"Ciano" se refiere al sustituyente -CeN.
"Cianoalquileno" se refiere al sustituyente -(alquilen)CEN.
"Acetilo" se refiere al sustituyente -C(O)CH3.
"Hidroxi" o "hidroxilo" se refiere al sustituyente -OH.
"Hidroxialquileno" se refiere al sustituyente -(alquilen)OH.
"Oxo" se refiere a un oxígeno de sustituyente -O-.
"Tio" o "tiol" se refieren a un sustituyente -SH.
La frase “cinasa que interactúa con MAP cinasa” o el término “Mnk” se refieren a todas las isoformas de la proteína cinasa que interactúa con MAP cinasa incluyendo Mnk-1 y Mnk-2.
"Alquilo" se refiere a un radical de cadena hidrocarbonada saturado, , lineal o ramificado que consiste únicamente en átomos de carbono e hidrógeno, que tiene de uno a doce átomos de carbono (alquilo C1-C12), de uno a ocho átomos de carbono (alquilo C1-C8) o de uno a seis átomos de carbono (alquilo C1-C6), y que está unido al resto de la molécula por un enlace sencillo. Los ejemplos de grupos alquilo incluyen metilo, etilo, n-propilo, 1-metiletil(isopropilo), n-butilo, n-pentilo, 1,1 -dimetiletil(t-butilo), 3-metilhexilo, 2-metilhexilo, y similares.
"Alquilo inferior" tiene el mismo significado que el alquilo definido anteriormente pero que tiene de uno a cuatro
átomos de carbono (alquilo C1-C4).
Alquenilo se refiere a un grupo alquilo insaturado que tiene al menos un doble enlace y de dos a doce átomos de carbono (alquenilo C2-C12), de dos a ocho átomos de carbono (alquenilo C2-C8) o de dos a seis átomos de carbono (alquenilo C2-C6), y que está unido al resto de la molécula por un enlace sencillo, por ejemplo, etenilo, propenilo, butenilo, pentenilo, hexenilo y similares.
"Alquinilo" se refiere a un grupo alquilo insaturado que tiene al menos un enlace triple y de dos a doce átomos de carbono (alquinilo C2-C12), de dos a diez átomos de carbono (alquinilo C2-C10), de dos a ocho átomos de carbono (alquinilo C2-C8) o de dos a seis átomos de carbono (alquinilo C2-C6), y que está unido al resto de la molécula por un enlace sencillo, por ejemplo, etinilo, propinilo, butinilo, pentinilo, hexinilo y similares.
"Alquileno" o "cadena alquileno" se refiere a una cadena hidrocarbonada divalente (alquilo) lineal o ramificada que une el resto de la molécula a un grupo radical, que consiste únicamente en carbono e hidrógeno, respectivamente. Los alquilenos pueden tener de uno a doce átomos de carbono, por ejemplo, metileno, etileno, propileno, n-butileno y similares. La cadena de alquileno se une al resto de la molécula a través de un enlace sencillo o doble. Los puntos de unión de la cadena de alquileno al resto de la molécula pueden ser a través de un carbono o dos carbonos dentro de la cadena. "Alquileno opcionalmente sustituido" se refiere a alquileno o alquileno sustituido.
"Alquenileno" se refiere a alqueno divalente. Los ejemplos de alquenileno incluyen, sin limitación, etenileno (-CH=CH-) y todos los estereoisómeros y formas isoméricas conformacionales de los mismos. "Alquenileno sustituido" se refiere a alqueno sustituido divalente. "Alquenileno opcionalmente sustituido" se refiere a alquenileno o alquenileno sustituido.
“Alquinileno” se refiere al alquino divalente. Los ejemplos de alquinileno incluyen, sin limitación, etinileno, propinileno. “Alquinileno sustituido” se refiere a alquino sustituido divalente.
“Alcoxi” se refiere a un radical de fórmula -ORa en la que Ra es un alquilo que tiene el número indicado de átomos de carbono como se ha definido anteriormente. Los ejemplos de grupos alcoxi incluyen sin limitación, -O-metil(metoxi), -O-etil(etoxi), -O-propil(propoxi), -O-isopropil(isopropoxi) y similares.
"Acilo" se refiere a un radical de fórmula -C(O)Ra en la que Ra es un alquilo que tiene el número indicado de átomos de carbono.
"Alquilaminilo" se refiere a un radical de fórmula -NHRa o -NRaRa en la que cada Ra es, independientemente, un radical alquilo que tiene el número indicado de átomos de carbono como se ha definido anteriormente.
"Cicloalquilaminilo" se refiere a un radical de fórmula -NHRa en la que Ra es un radical cicloalquilo como se define en el presente documento.
"Alquilcarbonilaminilo" se refiere a un radical de fórmula -NHC(O)Ra, en la que Ra es un radical alquilo que tiene el número indicado de átomos de carbono como se define en el presente documento.
"Cicloalquilcarbonilaminilo" se refiere a un radical de fórmula -NHC(O)Ra, en el que Ra es un radical cicloalquilo como se define en el presente documento.
"Alquilaminocarbonilo" se refiere a un radical de fórmula -C(O)NHRa o -C(O)NRaRa, donde cada Ra es independientemente un radical alquilo que tiene el número indicado de átomos de carbono como se define en el presente documento.
"Cicloalquilaminocarbonilo" se refiere a un radical de fórmula -C(O)NHRa, en el que Ra es un radical cicloalquilo como se define en el presente documento.
“Arilo” se refiere a un radical de sistema de anillo hidrocarbonado que comprende hidrógeno, 6 a 18 átomos de carbono y al menos un anillo aromático. Son arilos de ejemplo un radical de sistema de anillo hidrocarbonado que comprende hidrógeno y de 6 a 9 átomos de carbono y al menos un anillo aromático; un radical de sistema de anillo hidrocarbonado que comprende hidrógeno y de 9 a 12 átomos de carbono y al menos un anillo aromático; un radical de sistema de anillo hidrocarbonado que comprende hidrógeno y de 12 a 15 átomos de carbono y al menos un anillo aromático; o un radical de sistema de anillo hidrocarbonado que comprende hidrógeno y de 15 a 18 átomos de carbono y al menos un anillo aromático. Para los fines de la presente invención, el radical arilo puede ser un sistema de anillos monocíclico, bicíclico, tricíclico o tetracíclico, que puede incluir sistemas de anillo condensados o unidos. Los radicales arilo incluyen, pero sin limitación, radicales arilo derivados de aceantrileno, acenaftileno, acefenantrileno, antraceno, azuleno, benceno, criseno, fluoranteno, fluoreno, as-indaceno, s-indaceno, indano, indeno, naftaleno, fenaleno, fenantreno, pleyadeno, pireno y trifenileno. “Arilo opcionalmente sustituido” se refiere a un grupo arilo o un grupo arilo sustituido.
“Arileno” significa arilo divalente y “arileno sustituido” se refiere a arilo sustituido divalente.
"Aralquilo" o "aralquileno" se pueden usar indistintamente y se refieren a un radical de fórmula -Rb-Rc en la que Rb es una cadena alquileno como se define en el presente documento y Rc es uno o más radicales arilo como se definen en el presente documento, por ejemplo, bencilo, difenilmetilo y similares.
"Cicloalquilo" se refiere a un radical hidrocarbonado monocíclico o policíclico no aromático estable que consiste únicamente en átomos de carbono e hidrógeno, que pueden incluir sistemas de anillos condensados o unidos, que tienen de tres a quince átomos de carbono, preferentemente que tiene de tres a diez átomos de carbono, tres a nueve átomos de carbono, tres a ocho átomos de carbono, tres a siete átomos de carbono, tres a seis átomos de carbono, tres a cinco átomos de carbono, un anillo con cuatro átomos de carbono o un anillo con tres átomos de carbono. El anillo cicloalquilo puede estar saturado o insaturado y unido al resto de la molécula por un enlace sencillo. Los radicales monocíclicos incluyen, por ejemplo, ciclopropilo, ciclobutilo, ciclopentilo, ciclohexilo, cicloheptilo y ciclooctilo. Los radicales policíclicos incluyen, por ejemplo, adamantilo, norbornilo, decalinilo, 7,7-dimetil-biciclo[2.2.1]heptanilo y similares.
"Cicloalquilalquileno" o "cicloalquilalquilo" se pueden usar indistintamente y se refieren a un radical de fórmula -RbRe donde Rb es una cadena alquileno como se define en el presente documento y Re es un radical cicloalquilo como se define en el presente documento. En determinadas realizaciones, Rb está además sustituido con un grupo cicloalquilo, de manera que el cicloalquilalquileno comprende dos restos cicloalquilo. Ciclopropilalquileno y ciclobutilalquileno son ejemplos de grupos cicloalquilalquileno, que comprenden al menos un grupo ciclopropilo o al menos un grupo ciclobutilo, respectivamente.
"Condensado" se refiere a cualquier estructura de anillo descrita en el presente documento que se condensa con una estructura de anillo existente en los compuestos de la invención. Cuando el anillo condensado es un anillo heterociclilo o un anillo heteroarilo, cualquier átomo de carbono en la estructura de anillo existente que se convierte en parte del anillo heterociclilo condensado o el anillo heteroarilo condensado puede reemplazarse con un átomo de nitrógeno.
"Halo" o "halógeno" se refiere a bromo (bromo), cloro (cloro), fluoro (flúor), o yodo (yodo).
"Haloalquilo" se refiere a un radical alquilo que tiene el número indicado de átomos de carbono, como se define en el presente documento, en el que uno o más átomos de hidrógeno del grupo alquilo están sustituidos con un halógeno (radicales halo), como se ha definido anteriormente. Los átomos de halógeno pueden ser iguales o diferentes. Son ejemplos de haloalquilos trifluorometilo, difluorometilo, triclorometilo, 2,2,2-trifluoroetilo, 1,2-difluoroetilo, 3-bromo-2-fluoropropilo, 1,2-dibromoetilo, y similares.
“Heterociclilo”, “heterociclo” o “anillo heterocíclico” se refieren a un radical saturado o insaturado estable de 3 a 18 miembros que consiste en dos a doce átomos de carbono y de uno a seis heteroátomos, por ejemplo, uno a cinco heteroátomos, uno a cuatro heteroátomos, uno a tres heteroátomos o uno a dos heteroátomos seleccionados entre el grupo que consiste en nitrógeno, oxígeno y azufre. Los ejemplos de heterociclos incluyen, sin limitación, radicales saturados o insaturados estables de 3-15 miembros, radicales saturados o insaturados estables de 3-12 miembros, radicales saturados o insaturados estables de 3 a 9 miembros, radicales saturados o insaturados estables de 8 miembros, radicales saturados o insaturados estables de 7 miembros, radicales saturados o insaturados estables de 6 miembros o radicales saturados o insaturados estables de 5 miembros.
A menos que se indique lo contrario específicamente en la memoria descriptiva, el radical heterociclilo puede ser un sistema de anillos monocíclico, bicíclico, tricíclico o tetracíclico, que puede incluir sistemas de anillo condensados o unidos; y los átomos de nitrógeno, carbono o azufre en el radical heterociclilo pueden estar opcionalmente oxidados; el átomo de nitrógeno puede estar opcionalmente cuaternizado; y el radical heterociclilo puede estar parcial o totalmente saturado. Los ejemplos de radicales heterociclilo no aromáticos incluyen, pero sin limitación, azetidinilo, dioxolanilo, tienil[1,3]ditianilo, decahidroisoquinolilo, imidazolinilo, imidazolidinilo, isotiazolidinilo, isoxazolidinilo, morfolinilo, octahidroindolilo, octahidroisoindolilo, 2-oxopiperazinilo, 2-oxopiperidinilo, 2-oxopirrolidinilo, oxazolidinilo, piperidinilo, piperazinilo, 4-piperidonilo, pirrolidinilo, pirazolidinilo, quinuclidinilo, tiazolidinilo, tetrahidrofurilo, tietanilo, tritianilo, tetrahidropiranilo, tiomorfolinilo, tiamorfolinilo, 1-oxo-tiomorfolinilo y 1,1-dioxo-tiomorfolinilo. Los heterocíclicos incluyen heteroarilos como se definen en el presente documento y se enumeran ejemplos de heterociclilos aromáticos en la definición de heteroarilos que se indican después.
"Heterociclilalquilo" o "heterociclilalquileno" se refiere a un radical de fórmula -RbRf en la que Rb es una cadena alquileno como se define en el presente documento y Rf es un radical heterociclilo como se ha definido anteriormente y si el heterociclilo es un heterociclilo que contiene nitrógeno, el heterociclilo puede unirse al radical alquilo en el átomo de nitrógeno.
"Heteroarilo" o "heteroarileno" se refiere a un radical de sistema de anillo de 5 a 14 miembros que comprende átomos de hidrógeno, de uno a trece átomos de carbono, de uno a seis heteroátomos seleccionados entre el grupo que consiste en nitrógeno, oxígeno y azufre y al menos un anillo aromático. Para los fines de la presente invención,
el radical heteroarilo puede ser un anillo estable de 5-12 miembros, un anillo estable de 5-10 miembros, un anillo estable de 5-9 miembros, un anillo estable de 5-8 miembros, un anillo estable de 5-7 miembros, o un anillo estable de 6 miembros que comprende al menos 1 heteroátomo, al menos 2 heteroátomos, al menos 3 heteroátomos, al menos 4 heteroátomos, al menos 5 heteroátomos o al menos 6 heteroátomos. Los heteroarilos pueden ser un sistema de anillos monocíclico, bicíclico, tricíclico o tetracíclico, que puede incluir sistemas de anillos condensados o unidos; y los átomos de nitrógeno, carbono o azufre en el radical heteroarilo pueden estar opcionalmente oxidados; el átomo de nitrógeno puede estar opcionalmente cuaternizado. El heteroátomo puede ser un miembro de un anillo aromático o no aromático, a condición de que al menos un anillo en el heteroarilo sea aromático. Los ejemplos incluyen, pero sin limitación, azepinilo, acridinilo, bencimidazolilo, benzotiazolilo, benzindolilo, benzodioxolilo, benzofuranilo, benzooxazolilo, benzotiazolilo, benzotiadiazolilo, benzo[b][1,4]dioxepinilo, 1,4-benzodioxanilo, benzonaftauranilo, benzoxazolilo, Benzodioxolilo, benzodioxinilo, benzopiranilo, benzopiranonilo, benzofuranilo, benzofuranonilo, benzotienilo (benzotiofenilo), benzotriazolilo, benzo[4,6]imidazo[1,2-a]piridinilo, carbazolilo, cinolinilo, dibenzofuranilo, dibenzotiofenilo, furanilo, furanonilo, isotiazolilo, imidazolilo, Indazolilo, indolilo, indazolilo, isoindolilo, indolinilo, isoindolinilo, isoquinolilo, indolizinilo, isoxazolilo, naftiridinilo, oxadiazolilo, 2-oxoazepinilo, oxazolilo, oxiranilo, 1-oxopiridinilo, 1 -oxidopirimidinilo, 1 -oxidopirazinilo, 1 -oxidopiridazinilo, 1 -fenil-1 H-pirrolilo, fenazinilo, fenotiazinilo, fenoxazinilo, ftalazinilo, pteridinilo, purinilo, pirrolilo, pirazolilo, piridinilo, pirazinilo, pirimidinilo, piridazinilo, quinazolinilo, quinoxalinilo, quinolinilo, quinuclidinilo, isoquinolinilo, tetrahidroquinolinilo, tiazolilo, tiadiazolilo, triazolilo, tetrazolilo, triazinilo y tiofenilo (es decir, tienilo).
“Heteroarilalquilo” o “heteroarilalquileno” se refiere a un radical de fórmula -RbRg en la que Rb es una cadena alquileno como se ha definido anteriormente y Rg es un radical heteroarilo como se ha definido anteriormente.
"Tioalquilo" se refiere a un radical de fórmula -SRa en la que Ra es un radical alquilo como se ha definido anteriormente que contiene de uno a doce átomos de carbono, al menos 1-10 átomos de carbono, al menos 1-8 átomos de carbono, al menos 1-6 átomos de carbono, o al menos 1-4 átomos de carbono.
"Heterociclilaminilo" se refiere a un radical de fórmula -NHRf en la que Rf es un radical heterociclilo como se ha definido anteriormente.
"Tiona" se refiere a un grupo =S unido a un átomo de carbono de un grupo cíclico saturado o insaturado (C3-C8) o un resto acíclico (C1-C8).
"Sulfóxido" se refiere a un grupo -S(O)- en el que el átomo de azufre está unido covalentemente a dos átomos de carbono.
"Sulfona" se refiere a un grupo -S(O)2- en el que un azufre hexavalente está unido a cada uno de los dos átomos de oxígeno a través de enlaces dobles y está además unido a dos átomos de carbono a través de enlaces covalentes sencillos.
El término "oxima" se refiere a un radical -C(Ra)=N-ORa en el que Ra es hidrógeno, alquilo inferior, un grupo alquileno o arileno como se ha definido anteriormente.
El compuesto de la invención puede existir en diversas formas isoméricas, así como en una o más formas tautoméricas, incluyendo tanto tautómeros individuales como mezclas de tautómeros. El término "isómero" pretende abarcar todas las formas isoméricas de un compuesto de la presente invención, incluyendo formas tautoméricas del compuesto.
Algunos compuestos que se describen en el presente documento pueden tener centros asimétricos y por tanto existen en diferentes formas enantioméricas y diastereoméricas. Un compuesto de la invención puede estar en forma de un isómero óptico o un diastereómero. En consecuencia, la invención abarca compuestos de la invención y sus usos como se describen en el presente documento en forma de sus isómeros ópticos, diastereoisómeros y mezclas de los mismos, incluyendo una mezcla racémica. Los isómeros ópticos de los compuestos de la invención pueden obtenerse mediante técnicas conocidas tales como síntesis asimétrica, cromatografía quiral o a través de separación química de estereoisómeros mediante el empleo de agentes de resolución ópticamente activos.
A menos que se indique lo contrario, “estereoisómero” significa un estereoisómero de un compuesto que está sustancialmente libre de otros estereoisómeros de ese compuesto. Por tanto, un compuesto estereoméricamente puro que tiene un centro quiral estará sustancialmente libre del enantiómero opuesto del compuesto. Un compuesto estereoméricamente puro que tiene dos centros quirales estará sustancialmente libre de otros diastereómeros del compuesto. Un compuesto normalmente estereoméricamente puro comprende más de aproximadamente el 80 % en peso de un estereoisómero del compuesto y menos de aproximadamente el 20 % en peso de otros estereoisómeros del compuesto, por ejemplo, más de aproximadamente el 90 % en peso de un estereoisómero del compuesto y menos de aproximadamente el 10 % en peso de los otros estereoisómeros del compuesto, o más de aproximadamente el 95 % en peso de un estereoisómero del compuesto y menos de aproximadamente el 5 % en peso de los otros estereoisómeros del compuesto, o más de aproximadamente el 97 % en peso de un estereoisómero del compuesto y menos de aproximadamente el 3 % en peso de los otros estereoisómeros del
compuesto.
Si hay una discrepancia entre una estructura representada y un nombre dado a esa estructura, entonces la estructura representada prevalece. Adicionalmente, si la estereoquímica de una estructura o una porción de una estructura no está indicada, por ejemplo, con líneas en negrita o discontinuas, la estructura o porción de la estructura debe interpretarse como que abarca todos los estereoisómeros de la misma. En algunos casos, sin embargo, donde existe más de un centro quiral, las estructuras y nombres pueden representarse como enantiómeros individuales para ayudar a describir la estereoquímica relativa. Los expertos en la técnica de la síntesis orgánica sabrán si los compuestos se preparan como enantiómeros individuales a partir de los métodos usados para prepararlos.
En la presente descripción, una "sal farmacéuticamente aceptable" es una sal de ácido o base orgánica o inorgánica farmacéuticamente aceptable de un compuesto de la invención. Las sales farmacéuticamente aceptables representativas incluyen, por ejemplo, sales de metales alcalinos, sales alcalinotérreas, sales de amonio, sales hidrosolubles e insolubles en agua, tales como las sales de acetato, amsonato (4,4-diaminostilbeno-2,2-disulfonato), bencenosulfonato, benzoato, bicarbonato, bisulfato, bitartrato, borato, bromuro, butirato, calcio, edetato de calcio, camsilato, carbonato, cloruro, citrato, clavulanato, diclorhidrato, edetato, edisilato, estolato, esilato, fumarato, gluceptato, gluconato, glutamato, glicolilarsanilato, hexafluorofosfato, hexilresorcinato, hidrabamina, bromhidrato, clorhidrato, hidroxinaftoato, yoduro, isotionato, lactato, lactobionato, laurato, malato, maleato, mandelato, mesilato, metilbromuro, metilnitrato, metilsulfato, mucato, napsilato, nitrato, sal de N-metilglucamina amonio, 3-hidroxi-2-naftoato, oleato, oxalato, palmitato, pamoato (1,1-metilen-bis-2-hidroxi-3-naftoato, einbonato), pantotenato, fosfato/difosfato, picrato, poligalacturonato, propionato, p-toluenosulfonato, salicilato, estearato, subacetato, succinato, sulfato, sulfosalicilato, suramato, tannato, tartrato, teoclato, tosilato, trietioduro y valerato. Una sal farmacéuticamente aceptable puede tener más de un átomo cargado en su estructura. En este caso, la sal farmacéuticamente aceptable puede tener múltiples contraiones. Por tanto, una sal farmacéuticamente aceptable puede tener uno o más átomos cargados y/o uno o más contraiones.
Los términos "tratar", "tratando" y "tratamiento" se refieren a la mejora o la erradicación de una enfermedad o síntomas asociados a una enfermedad. En determinadas realizaciones, dichos términos se refieren a minimizar la propagación o el empeoramiento de la enfermedad resultantes de la administración de uno o más agentes profilácticos o terapéuticos a un paciente con una enfermedad de este tipo.
La expresión “cantidad eficaz” se refiere a una cantidad de un compuesto de la invención u otro ingrediente activo suficiente para proporcionar un beneficio terapéutico o profiláctico en el tratamiento o la prevención de una enfermedad o para retardar o minimizar los síntomas asociados a una enfermedad. Adicionalmente, una cantidad terapéuticamente eficaz con respecto a un compuesto de la invención significa aquella cantidad de agente terapéutico solo, o en combinación con otras terapias, que proporciona un beneficio terapéutico en el tratamiento o la prevención de una enfermedad. Usado en conexión con un compuesto de la invención, la expresión puede abarcar una cantidad que mejora la terapia global, reduce o evita los síntomas o causas de la enfermedad, o potencia la eficacia terapéutica o sinergias con otro agente terapéutico.
Los términos "modular', "modulación" y similares se refieren a la capacidad de un compuesto para aumentar o disminuir la función, o actividad de, por ejemplo, la cinasa que interactúa con MAP cinasa (Mnk). La "modulación", en sus diversas formas, pretende abarcar la inhibición, el antagonismo, el antagonismo parcial, la activación, el agonismo y/o el agonismo parcial de la actividad asociada a Mnk. Los inhibidores de Mnk son compuestos que se unen para bloquear parcial o totalmente la estimulación, disminuir, prevenir, retrasar la activación, inactivar, desensibilizar o regular negativamente la transducción de señales. La capacidad de un compuesto para modular la actividad de Mnk se puede demostrar en un ensayo enzimático o en un ensayo a base de células.
Un "paciente" o “sujeto” incluye un animal, tal como un ser humano, vaca, caballo, oveja, cordero, cerdo, pollo, pavo, codorniz, gato, perro, ratón, rata, conejo o cobaya. El animal puede ser un mamífero tal como un no primate y un primate (por ejemplo, mono y ser humano). En una realización, un paciente es un ser humano, tal como un bebé, un niño, un adolescente o un adulto humanos.
El término “profármaco” se refiere a un precursor de un fármaco que es un compuesto que tras la administración a un paciente debe experimentar una conversión química por procesos metabólicos antes de convertirse en un agente farmacológico activo. Son ejemplos de profármacos de compuestos de acuerdo con la Fórmula I ésteres, acetamidas y amidas.
Compuestos de la invención
La presente invención se refiere generalmente a compuestos incluidos en el género de Fórmula I

o un estereoisómero, tautómero o una sal farmacéuticamente aceptable de los mismos, en la que:
W1 y W2 son independientemente O, S o N-OR', donde R' es alquilo C1-C4 ;
Y es -N(R5)-, -O-, -S-, -C(O)-, -S=O, -S(O)2- o -CHR9-;
R1 es hidrógeno, alquilo C1-C4 , cicloalquilo o heterociclilo, en el que cualquier alquilo C1-C4 , cicloalquilo o heterociclilo está opcionalmente sustituido con 1, 2 o 3 grupos J;
n es 1, 2 o 3;
R2 y R3 son cada uno independientemente hidrógeno, alquilo, alquenilo, alquinilo, arilo, aralquileno, heteroarilo, heteroarilalquileno, cicloalquilo, cicloalquilalquileno, heterociclilo o heterociclilalquileno, en los que cualquier alquilo, arilo, aralquileno, heteroarilo, heteroarilalquileno, cicloalquilo, cicloalquilalquileno, heterociclilo o heterociclilalquileno, está opcionalmente sustituido con 1, 2 o 3 grupos J;
o R2 y R3 tomados junto con el átomo de carbono al que están unidos forman un cicloalquilo o heterociclilo, en los que cualquier cicloalquilo o heterociclilo está opcionalmente sustituido con 1, 2 o 3 grupos J;
R4a y R4b son cada uno independientemente hidrógeno, halógeno, hidroxilo, tiol, hidroxialquileno, ciano, alquilo, alcoxi, acilo, tioalquilo, alquenilo, alquinilo, cicloalquilo, arilo o heterociclilo;
R5 es hidrógeno, ciano o alquilo inferior;
o R5 y R8 tomados junto con los átomos a los que están unidos forman un heterociclilo condensado opcionalmente sustituido con 1, 2 o 3 grupos J;
R6, R7 y R8 son cada uno independientemente hidrógeno, hidroxi, halógeno, ciano, amino, alquilo, alquenilo, alquinilo, alcoxi, cicloalquilo, cicloalquilalquileno, cicloalquilalquenileno, alquilaminilo, alquilcarbonilaminilo, cicloalquilcarbonilaminilo, cicloalquilaminilo, heterociclilaminilo, heteroarilo o heterociclilo, y en el que cualquier amino, alquilo, alquenilo, alquinilo, alcoxi, cicloalquilo, cicloalquilalquileno, cicloalquilalquenileno, amino, alquilaminilo, alquilcarbonilaminilo, cicloalquilcarbonilaminilo, cicloalquilaminilo, heterociclilaminilo, heteroarilo, o heterociclilo está opcionalmente sustituido con 1, 2 o 3 grupos J;
o R7 y R8 tomados junto con los átomos a los que están unidos forman un heterociclilo condensado o heteroarilo opcionalmente sustituido con 1, 2 o 3 grupos J;
J es -SH, -SR9, -S(O)R9, -S(O)2R9, -S(O)NH2 , -S(O)NR9R9, -NH2 , -NR9R9, -COOH, -C(O)OR9, -C(O)R9, -C(O)-NH2 , -C(O)-NR9R9, hidroxi, ciano, halógeno, acetilo, alquilo, alquilo C1-C4 , alquenilo, alquinilo, alcoxi, haloalquilo, tioalquilo, cianoalquileno, alquilaminilo, NH2-C(O)-alquileno, NR9R9-C(O)-alquileno, -CHR9-C(O)-alquilo C1-C4 , -C(O)-alquilo C1-C4 , alquilcarbonilaminilo, cicloalquilo, cicloalquilalquileno, cicloalquilalquenileno, cicloalquilcarbonilaminilo, cicloalquilaminilo, -CHR9-C(O)-cicloalquilo, -C(O)-cicloalquilo, -CHR9-C(O)-arilo, -CHR9-arilo, -C(O)-arilo, -CHR9-C(O)-heterocicloalquilo, -C(O)-heterocicloalquilo, heterociclilaminilo o heterociclilo; o cualesquiera dos grupos J unidos al mismo carbono o heteroátomo pueden tomarse juntos para formar oxo; y R9 es hidrógeno, alquilo C1-C4 o -OH.
En una realización de la estructura (I), la presente divulgación proporciona un compuesto que tiene la siguiente estructura (la), así como estereoisómeros, tautómeros o sales farmacéuticamente aceptables de los mismos.

Para los compuestos de Fórmula la, el sustituyente R1 es hidrógeno o alquilo C1-C4 y el índice n es 1, 2 o 3. Los
sustituyentes R2 y R3 en la Fórmula la son cada uno independientemente hidrógeno, alquilo, cicloalquilo, cicloalquilalquileno, heterociclilo o heterociclilalquilo, y cualquiera de dichos alquilo, cicloalquilo, cicloalquilalquileno, heterociclilo o heterociclilalquilo opcionalmente puede estar sustituido con 1, 2 o 3 grupos J.
Los sustituyentes R2 y R3 en la Fórmula Ia cuando se toman junto con el átomo de carbono al que están unidos pueden formar un cicloalquilo o heterociclilo, en el que cualquiera de dichos cicloalquilo o heterociclilo está opcionalmente sustituido con 1, 2 o 3 grupos J. En la Fórmula Ia, R4a es hidrógeno, halógeno, hidroxi, alquilo, alcoxi, tioalquilo, alquenilo o cicloalquilo y el sustituyente R5 es hidrógeno o alquilo C1-C4.
Como alternativa, los grupos sustituyentes R5 y R8 tomados junto con los átomos a los que están unidos forman un heterociclilo condensado que está opcionalmente sustituido con 1, 2 o 3 grupos J.
En una realización, los sustituyentes R6, R7 y R8 son independientemente y en cada aparición hidrógeno, halógeno, alquilo, alquenilo, cicloalquilo, cicloalquilalquilo, cicloalquilalquenilo, amino, alquilaminilo, alquilcarbonilaminilo, cicloalquilcarbonilaminilo, alquilaminilo o cicloalquilaminilo y cualquiera de dichos alquilo, alquenilo, cicloalquilo, cicloalquilalquilo, cicloalquilalquenilo, amino, alquilaminilo, alquilcarbonilaminilo, cicloalquilcarbonilaminilo, alquilaminilo o cicloalquilaminilo está opcionalmente sustituido con 1, 2 o 3 grupos J. Para algunos compuestos de acuerdo con la Fórmula Ia, R7 y R8 tomados junto con los átomos a los que están unidos forman un heterociclilo condensado no sustituido o sustituido con 1, 2 o 3 grupos J.
La variable J en la Fórmula Ia es -SH, -SR9, -S(O)R9, -S(O)2 R9, -S(O)NH2, -S(O)NR9R9, -NH2 , -NR9R9, -COOH, -C(O)OR9, -C(O)R9, -C(O)-NH2 , -C(O)-NR9R9, hidroxi, ciano, halógeno, acetilo, alquilo, alquilo C1-C4 , alquenilo, alquinilo, alcoxi, haloalquilo, tioalquilo, cianoalquileno, alquilaminilo, NH2-C(O)-alquileno, NR9R9-C(O)-alquileno, -CHR9-C(O)-alquilo C1-C4 , -C(O)-alquilo C1-C4 , alquilcarbonilaminilo, cicloalquilo, cicloalquilalquileno, cicloalquilalquenileno, cicloalquilcarbonilaminilo, cicloalquilaminilo, -CHR9-C(O)-cicloalquilo, -C(O)-cicloalquilo, -CHR9-C(O)-arilo, -CHR9-arilo, -C(O)-arilo, -CHR9-C(O)-heterocicloalquilo, -C(O)-heterocicloalquilo, heterociclilaminilo o heterociclilo. Para algunos de los compuestos de la invención de acuerdo con la Fórmula Ia, cualesquiera dos grupos J unidos al mismo carbono o heteroátomo pueden tomarse juntos para formar un grupo oxo.
En algunas realizaciones, la variable J en la fórmula Ia es halógeno, amino, alquilo, haloalquilo, alquilaminilo, cicloalquilo o heterociclilo. Como alternativa, para determinados compuestos de Fórmula Ia, cualesquiera dos grupos J cuando están unidos al mismo carbono o heteroátomo se pueden tomar juntos para formar el grupo oxo.
La presente invención se refiere adicionalmente a compuestos de acuerdo con la Fórmula IIa, ilustrada después, donde la variable Y es -N(R5)- y el subíndice "n" es 1.

De acuerdo con una realización, la variable Y en la fórmula I es -O-, -S-, -C(O)-, sulfóxido, sulfona, -CHR9- o -CH2-, el subíndice "n" es 1 y los compuestos de la invención se ajustan a Formula IIb. Cuando "Y" es -CHR9- en la Fórmula I Ib, el sustituyente R9 es hidrógeno, alquilo C1-C4 o hidroxi.
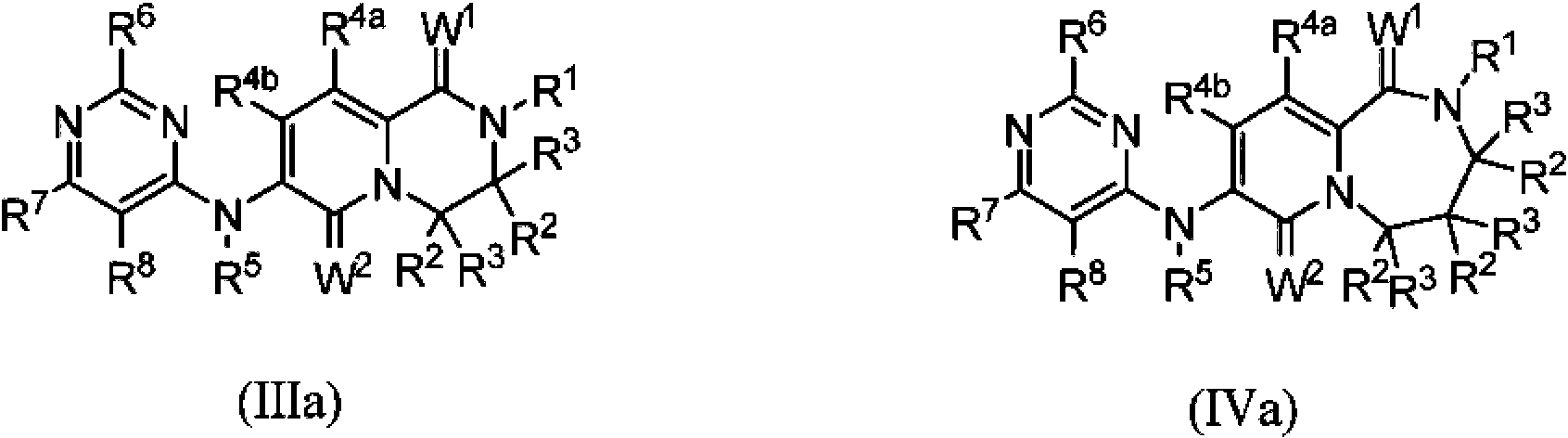
En otra realización de la invención, la variable "Y" en la Fórmula I es -N(R5)-, el subíndice "n" es 2 o 3 y los compuestos de la invención se ajustan a la Fórmula IIIa o la Fórmula IVa, respectivamente:
Como alternativa, en determinadas realizaciones la variable “Y” en la Fórmula I es -O-, -S-, -C(O)-, sulfóxido, sulfona, -CHR9- o -CH2-, "n" es 2 o 3 y los compuestos de la invención se ajustan a la Fórmula IIIb y la Fórmula IVb, respectivamente: Cuando "Y" es -CHR9- en la Fórmula IIIb o Fórmula IVb, el sustituyente R9 es hidrógeno, alquilo inferior o hidroxi.

Para los compuestos de acuerdo con las Fórmulas Ila, I Ib, IlIa, IIIb, IVa y IVb, las variables W1 y W2 son ambas oxo.
En determinadas realizaciones para compuestos de acuerdo con las Fórmulas Ila, IIb, IIIa, IIIb, IVa y IVb, W1 es oxo y W2 es grupo tiona. De acuerdo con una realización, los compuestos de las Fórmulas Ila, IIb, IlIa, IIIb, IVa y IVb comprenden un oxo en W1 y un grupo =N-OR' en W2. También se incluyen dentro del alcance de la presente invención los compuestos de las Fórmulas Ila, IIb, IlIa, IIIb, IVa y IVb que tienen un grupo tiona en W1 y un grupo oxo en W2.
Para los compuestos de las Fórmulas Ila, IIb, IIIa, IIIb, IVa y IVb, cada uno de los sustituyentes R2 y R3 puede ser el mismo, en cuyo caso el átomo de carbono que R2 y R3 están unidos no es un carbono quiral. En determinadas realizaciones, sin embargo, los sustituyentes R2 y R3 son diferentes. Por tanto, el átomo de carbono en el que R2 y R3 están unidos es quiral y el compuesto resultante tendrá estereoisómeros.
En una realización de la invención, cada R2 y R3 en las Fórmulas Ila, IIb, IIIa, IIIb, IVa y IVb es hidrógeno. Como alternativa, uno de los grupos R2 y R3 en las Fórmulas Ila, IIb, IIIa, IIIb, IVa y IVb es hidrógeno y el otro grupo es alquilo opcionalmente sustituido con 1, 2 o 3 grupos J. Para determinados compuestos de acuerdo con las Fórmulas Ila, lIb, IIIa, IIIb, IVa y IVb, R2 y R3 son ambos grupos alquilo que están opcionalmente sustituidos con 1, 2 o 3 grupos J.
Para algunos compuestos de acuerdo con la Fórmula Ila o Fórmula IIb, R2 es alquilo y R3 es alquilo sustituido con 1, 2 o 3 grupos J. Los ejemplos de esta categoría de compuestos de Fórmula Ila y Fórmula IIb son los siguientes compuestos con el sustituyente R2 como alquilo y R3 es haloalquilo; compuestos con compuestos sustituyentes con el sustituyente R2 como alquilo y R3 es cicloalquilo opcionalmente sustituido con 1, 2 o 3 grupos J; compuestos con el sustituyente R2 como alquilo y R3 es ciclopentilo opcionalmente sustituido con 1, 2 o 3 grupos J; compuestos con el sustituyente R2 como alquilo y R3 es arilo opcionalmente sustituido con 1, 2 o 3 grupos J; compuestos con el sustituyente R2 como alquilo y R3 es fenilo opcionalmente sustituido con grupos 1, 2 o 3; compuestos con el sustituyente R2 como alquilo y R3 es cicloalquilalquileno opcionalmente sustituido con 1, 2 o 3 grupos J; compuestos con el sustituyente R2 como alquilo y R3 es aralquileno opcionalmente sustituido con 1, 2 o 3 grupos J; compuestos con el sustituyente R2 como alquilo y R3 es bencilo opcionalmente sustituido con 1, 2 o 3 grupos J; compuestos con el sustituyente R2 como alquilo y R3 es heterociclilo opcionalmente sustituido con 1, 2 o 3 grupos J; compuestos con el sustituyente R2 como alquilo y R3 es heteroarilo opcionalmente sustituido con 1,2 o 3 grupos J; compuestos con el sustituyente R2 como alquilo y R3 es tiofenilo, tiazolilo o piridinilo; compuestos con el sustituyente R2 como alquilo y
R3 es heterociclilalquileno sustituido con 1, 2 o 3 grupos J; o compuestos con el sustituyente R2 como alquilo y R3 es heteroarilalquileno opcionalmente sustituido con 1, 2 o 3 grupos J.
En una realización, para compuestos de acuerdo con las Fórmulas Ila, I Ib, IIIa, IIIb, IVa y IVb cada R2 y R3 son independientemente hidrógeno, alquilo, cicloalquilo, cicloalquilalquileno, heterociclilo o heterociclilalquileno, y cualquiera de dichos alquilo, cicloalquilo, cicloalquilalquileno, heterociclilo o heterociclilalquileno opcionalmente pueden estar sustituidos con 1, 2 o 3 grupos J, elegidos independientemente del grupo que consiste en halógeno, amino, alquilaminilo y alquilo.
Para determinadas Fórmulas IIIa, IIIb, IVa y IVb, R2 y R3 junto con el átomo de carbono al que están unidos forman un anillo cicloalquilo o heterociclilo.
También se contemplan compuestos de Fórmula I en los que Y es -N(R5)-, el subíndice "n" es 1 y R2 y R3 junto con el átomo de carbono al que están unidos forman un anillo cicloalquilo o heterociclilo "A". Dichos compuestos se ajustan a la Fórmula Va y el anillo cicloalquilo o heterociclilo "A" puede estar opcionalmente sustituido con 1, 2 o 3 grupos J.

Como alternativa, en algunas realizaciones Y en la Fórmula I es -O-, -S-, -C(O)-, sulfóxido, sulfona, -CHR- o -CH2-, “n” es 1 y R2 y R3 junto con el resto átomo de carbono al que están unidos forman un anillo cicloalquilo o heterociclilo A. Dichos compuestos se ajustan a la Fórmula Vb y el anillo cicloalquilo o heterociclilo "A" puede estar opcionalmente sustituido con 1, 2 o 3 grupos J. Cuando "Y" es -CHR9- en la Fórmula Vb, el sustituyente R9 es hidrógeno, alquilo inferior o hidroxi.

Para los compuestos de Fórmula Va y Fórmula Vb W1 y W2 son ambos oxo y el anillo A es un cicloalquilo opcionalmente sustituido con 1, 2 o 3 grupos J. También se contemplan compuestos de Fórmula Va y Fórmula Vb para los que el anillo A es un cicloalquilo condensado opcionalmente sustituido con 1, 2 o 3 grupos J; el anillo A es un cicloalquilo opcionalmente sustituido con 1, 2 o 3 grupos J; el anillo A es un ciclobutilo, ciclopentilo o ciclohexilo opcionalmente sustituido con 1, 2 o 3 grupos J, por ejemplo, grupos J seleccionados entre el grupo que consiste en halógeno, amino, alquilaminilo y alquilo.
Para algunas realizaciones, el anillo A de una Fórmula Va o una Fórmula Vb es un heterociclilo opcionalmente sustituido con 1, 2 o 3 grupos J. Son ejemplos de dichos grupos heterociclilo pirrolidinilo, piperidinilo, tetrahidropiranilo, tietanilo o azetidinilo. En una realización, cada uno de los heterociclilos ejemplificados anteriormente puede estar opcionalmente sustituido con 1, 2 o 3 grupos J. Para determinados compuestos de Fórmula Va o Fórmula Vb, el anillo A es un cicloalquilo sustituido con al menos 2 grupos J unidos al mismo átomo de carbono del cicloalquilo, y los dos grupos J unidos al mismo carbono juntos forman un grupo oxo. En otra realización, el anillo A de una Fórmula Va o una Fórmula Vb es un heterociclilo sustituido con al menos 2 grupos J que están unidos al mismo heteroátomo y en el que dichos 2 grupos J tomados juntos para formar oxo. Para algunos compuestos de Fórmula Va o de Fórmula Vb, el anillo A de cicloalquilo o heterociclilo está sustituido con grupos J seleccionados entre el grupo que consiste en halógeno, ciano, hidroxi, trifluorometilo, N-metilamino, metilo,
difluoroetileno y metilen-nitrilo.
La presente invención también proporciona compuestos de acuerdo con la Fórmula VI o sus estereoisómeros, tautómeros o sales farmacéuticamente aceptables. La Fórmula VI es un subgénero de Fórmula I en la que Y es -N(R5)- y los grupos sustituyentes R5 y R8 junto con los átomos a los que están unidos forman un anillo heterocíclico B que puede estar opcionalmente sustituido con 1,2 o 3 grupos J.

También se incluyen dentro del alcance de la presente invención los compuestos de Fórmula I en los que la variable "Y" es -N(R5)- y los grupos sustituyentes R7 y R8 junto con los átomos a los que están unidos forman un anillo condensado C. Dichos compuestos o el estereoisómero, tautómero o sal farmacéuticamente aceptable se ajustan a la Fórmula VIIa. Para los compuestos de Fórmula VIIa, el anillo C puede estar opcionalmente sustituido con 1, 2 o 3 grupos J.

De acuerdo con una realización, la variable "Y" en la Fórmula I es -O-, -S-, -C(O)-, sulfóxido, sulfona, -CHR- o -CH2-, y los grupos sustituyentes R7 y R8 junto con los átomos a los que están unidos forman un anillo condensado C. Dichos compuestos y sus estereoisómeros, tautómeros o sales farmacéuticamente aceptables se ajustan a la Fórmula VI Ib. Para los compuestos de Fórmula VIIb en los que "Y" es -CHR9-, el sustituyente R9 puede ser hidrógeno, alquilo inferior o hidroxi.

Para los compuestos de Fórmula VIIb, el anillo C condensado puede estar opcionalmente sustituido con 1, 2 o 3 grupos J. En una realización de la invención, W1 y W2 son ambos oxo para los compuestos de Fórmula VI, Fórmula VIIa y Fórmula VIIb.
La presente invención se refiere adicionalmente a compuestos de las Fórmulas I, Ia, Ila, I Ib, 1 Ia, IIIb, IVa, IVb, Va, Vb, VI, VIIa y VIIb donde R1 es hidrógeno o un grupo alquilo C1-C4 seleccionado entre metilo, etilo, propilo, butilo,
isopropilo, sec-butilo o terc-butilo, por ejemplo, compuestos con R1 como metilo.
Para determinados compuestos de las Fórmulas I, la, Ila, IIb, IIIa, IIIb, IVa, IVb, Va, Vb, VI, VIIa y VIIb, R4a se selecciona entre el grupo que consiste en hidrógeno, halógeno, alquilo, alcoxi, tioalquilo, alquenilo, y cicloalquilo mientras que el sustituyente R4b es hidrógeno o halógeno. R5 en las Fórmulas I, la, Ila, IIb, IIIa, IIIb, IVa, IVb, Va, Vb,
VI, VIIa y VIIb es hidrógeno o alquilo C1-C4, mientras que los sustituyentes R6, R7 y R8 son hidrógeno.
En una realización de la invención, R6 y R7 en la Fórmula VI son ambos hidrógeno, mientras que para determinados compuestos de Fórmula VIIa y Fórmula VIIb R6 es hidrógeno.
La presente invención se refiere adicionalmente a compuestos de las Fórmulas I, Ia, Ila, IIb, IIIa, IIIb, IVa, IVb, Va y
Vb en los que los grupos sustituyentes R6 y R8 son ambos hidrógeno y R7 se selecciona entre el grupo que consiste en hidroxi, halógeno, ciano, alquilo, alquenilo, alquinilo, alcoxi, cicloalquilo, cicloalquilalquileno, cicloalquilalquenileno, amino, alquilaminilo, alquilcarbonilaminilo, cicloalquilcarbonilaminilo, cicloalquilaminilo, heterociclilaminilo, heteroarilo y heterociclilo. Para estos compuestos de la invención, cualquier alquilo, alquenilo, alquinilo, alcoxi, cicloalquilo, cicloalquilalquileno, cicloalquilalquenileno, amino, alquilaminilo, alquilcarbonilaminilo, cicloalquilcarbonilaminilo, cicloalquilaminilo, heterociclilaminilo, heteroarilo o heterociclilo está opcionalmente sustituido con 1, 2 o 3 grupos J.
En una realización R7 se selecciona entre el grupo que consiste en alquilo, cicloalquilo, cicloalquilalquileno, cicloalquilalquenileno, amino, alquilaminilo, alquilcarbonilaminilo, cicloalquilcarbonilaminilo, heterociclilaminilo, heteroarilo, heterociclilo y cicloalquilaminilo. Para dichos compuestos, cualquier alquilo, alquenilo, cicloalquilo, cicloalquilalquileno, cicloalquilalquenileno, amino, alquilaminilo, alquilcarbonilaminilo, cicloalquilcarbonilaminilo, heterociclilaminilo, heteroarilo, heterociclilo o cicloalquilaminilo puede estar opcionalmente sustituido con 1, 2 o 3 grupos J. Por tanto, la invención proporciona compuestos de las Fórmulas I, la, Ila, IIb, IIIa, IIIb, IVa, IVb, Va y Vb en las que los grupos sustituyentes R6 y R8 son ambos hidrógeno y R7 es amino; los grupos sustituyentes R6 y R8 son ambos hidrógeno, y R7 es alquilaminilo; los grupos sustituyentes R6 y R8 son ambos hidrógeno y R7 es -NHCH3 ; los grupos sustituyentes R6 y R8 son ambos hidrógeno, y R7 es cicloalquilo, por ejemplo, ciclopropilo; los grupos sustituyentes R6 y R8 son ambos hidrógeno y R7 es cicloalquilaminilo sustituido con 1 a 3 grupos J, por ejemplo, halógenos.
En una realización, para los compuestos de acuerdo con las Fórmulas I, la, Ila, IIb, IIIa, IIIb, IVa, IVb, Va y Vb, los grupos sustituyentes R6 y R8 son ambos hidrógeno y R7 se selecciona entre el grupo que consiste en -NHCH(CF3)ciclopropilo, cicloalquilcarbonilaminilo, -NHC(O)ciclopropilo, cicloalquilalquenileno, y -CH=CHciclopropilo.
Para cualquier compuesto de acuerdo con las Fórmulas I, la, Ila, IIb, IIIa, IIIb, IVa, IVb, Va, Vb, VI, VIIa y VIIb, J es -SH, -SR9, -S(O)R9, -S(O)2R9, -S(O)NH2, -S(O)NR9R9, -NH2 , -NR9R9, -COOH, -C(O)OR9, -C(O NR9R9, hidroxi, ciano, halógeno, acetilo, alquilo, alquilo C1-C4, alquenilo, alquinilo, alcoxi, haloalquilo, tioalquilo, cianoalquileno, alquilaminilo, NH2-C(O)-alquileno, NR9R9-C(O)-alquileno, -CHR9-C(O)-alquilo C1-C4, -C(O)-alquilo C1-C4 , alquilcarbonilaminilo, cicloalquilo, cicloalquilalquileno, cicloalquilalquenileno, cicloalquilcarbonilaminilo, cicloalquilaminilo, -CHR9-C(O)-cicloalquilo, -C(O)-cicloalquilo, -CHR9-C(O)-arilo, -CHR9-arilo, -C(O)-arilo, -CHR9-C(O)-heterocicloalquilo, -C(O)-heterocicloalquilo, heterociclilaminilo o heterociclilo y R9 es hidrógeno, alquilo C1-C4 o
-OH. Adicionalmente, cuando dos grupos J unidos al mismo carbono o heteroátomo pueden tomarse juntos para formar oxo.
Para determinados compuestos de acuerdo con las Fórmulas I, la, Ila, IIb, IIIa, IIIb, IVa, IVb, Va, Vb, VI, VIIa y VIIb, J es halógeno, hidroxi, alquilo, alquenilo, alquinilo o cianoalquileno. Son cadenas de alquilo o alquileno ilustrativas aquellas que tienen C1-C10 átomos de carbono, C1-C8 átomos de carbono, C1-C6 átomos de carbono, C1-C4 átomos de carbono, C1-C3 átomos de carbono, así como grupos etilo y metilo. Como alternativa, cuando J es alquenilo o alquinilo, la cadena de carbono tiene al menos un enlace doble o triple respectivamente y C2-C10 átomos de carbono,
C2-C8 átomos de carbono, C2-C6 átomos de carbono, C2-C4 átomos de carbono o C2-C3 átomos de carbono.
Los compuestos de la invención de acuerdo con la Fórmula I, así como las Fórmulas Ia, Ila, IIb, IIIa, IIIb, IVa, IVb,
Va, Vb VI, VIIa y VIIb pueden marcarse isotópicamente al tener uno o más átomos reemplazados por un átomo que tiene una masa atómica o un número de masa diferente. Los ejemplos de isótopos que se pueden incorporar en compuestos de acuerdo con la Fórmula I incluyen isótopos de hidrógeno, carbono, nitrógeno, oxígeno, fósforo, flúor, cloro o yodo. Son ilustrativos de dichos isótopos 2H, 3H, 11C, 13C, 14C, 13N, 15N, 15O, 17O, 18O, 31P, 32P, 35S, 18F, 36Cl,
123I y 125I, respectivamente. Estos compuestos radiomarcados pueden usarse para medir la biodistribución, la concentración tisular y la cinética de transporte y excreción de tejidos biológicos incluyendo un sujeto al que se le administra dicho compuesto marcado. Los compuestos marcados también se usan para determinar la efectividad terapéutica, el sitio o modo de acción, y la afinidad de unión de un candidato terapéutico a una diana farmacológicamente importante. Determinados compuestos marcados radioactivos de acuerdo con la Fórmula I, por tanto, son útiles en estudios de distribución tisular y/o de fármacos. Los isótopos radioactivos tritio, es decir 3H, y carbono 14, es decir, 14C, son particularmente útiles para este fin en vista de su facilidad de incorporación y medios listos de detección.
La sustitución con isótopos más pesados tales como deuterio, es decir, 2H, proporciona determinadas ventajas terapéuticas resultantes de la mayor estabilidad metabólica, por ejemplo, aumento de la semivida in vivo de
compuestos que contienen deuterio. La sustitución de hidrógeno con deuterio puede reducir la dosis requerida para el efecto terapéutico y, por tanto, puede preferirse en un descubrimiento o en un escenario clínico.
La sustitución con isótopos emisores de positrones, tales como 11C, 18F, 15O y 13N, proporciona análogos marcados de los compuestos de la invención que son útiles en estudios de tomografía de emisión de positrones (PET), por ejemplo, para examinar la ocupación del receptor de sustrato. Los compuestos marcados isotópicamente de acuerdo con la Fórmula I, así como las Fórmulas Ia, Ila, IIb, IIIa, IIIb, IVa, IVb, Va, Vb VI, VIIa y VIIb se pueden preparar generalmente por técnicas convencionales conocidas por los expertos en la técnica o por procesos análogos a los descritos en la sección de Preparaciones y Ejemplos como se describe después usando un reactivo de marcado isotópico apropiado.
También se pretende que las realizaciones de la invención que se desvelan en el presente documento abarquen los productos metabólicos in vivo de compuestos de acuerdo con las Fórmulas I, la, Ila, IIb, IIIa, IIIb, IVa, IVb, Va, Vb VI, VIIa y VIIb. Dichos productos pueden ser resultado, por ejemplo, de los procesos de oxidación, reducción, hidrólisis, amidación, esterificación y similares debido principalmente a la actividad enzimática tras la administración de un compuesto de la invención. En consecuencia, la invención incluye compuestos que se producen como subproductos de actividad enzimática o no enzimática sobre un compuesto de la invención después de la administración de un compuesto de este tipo a un mamífero durante un periodo de tiempo suficiente para producir un producto metabólico. Los productos metabólicos, en particular los metabolitos farmacéuticamente activos, se identifican normalmente mediante la administración de un compuesto radiomarcado de la invención en una dosis detectable a un sujeto, tal como rata, ratón, conejillo de indias, mono o humano, durante un período de tiempo suficiente durante el cual se produce el metabolismo, y aislando los productos metabólicos de orina, sangre u otras muestras biológicas que se obtienen del sujeto que recibe el compuesto radiomarcado.
La invención también proporciona formas de sal farmacéuticamente aceptables de los compuestos de las Fórmulas I, Ia, Ila, IIb, IIIa, IIIb, IVa, IVb, Va, Vb VI, VIIa y VIIb. En el ámbito de la invención se incluyen tanto sales de adición de ácido como de base que se forman poniendo en contacto un ácido farmacéuticamente adecuado o una base farmacéuticamente adecuada con un compuesto de la invención.
Con este fin, una "sal de adición de ácido farmacéuticamente aceptable" se refiere a aquellas sales que conservan la eficacia biológica y las propiedades de las bases libres, que no son biológicamente o de otro modo indeseables, y que están formadas con ácidos inorgánicos tales como, pero sin limitación, ácido clorhídrico, ácido bromhídrico, ácido sulfúrico, ácido nítrico, ácido fosfórico y similares, y ácidos orgánicos tales como, pero sin limitación, ácido acético, ácido 2,2-dicloroacético, ácido adípico, ácido algínico, ácido ascórbico, ácido aspártico, ácido bencenosulfónico, ácido benzoico, ácido 4-acetamidobenzoico, ácido alcanfórico, ácido alcanfor-10-sulfónico, ácido cáprico, ácido caproico, ácido caprílico, ácido carbónico, ácido cinámico, ácido cítrico, ácido ciclámico, ácido dodecilsulfúrico, ácido 2-disulfónico, ácido etanosulfónico, ácido 2-hidroxietanosulfónico, ácido fórmico, ácido fumárico, ácido galactárico, ácido gentísico, ácido glucoheptónico, ácido glucónico, ácido glucurónico, ácido glutámico, ácido glutárico, ácido 2-oxo-glutárico, ácido glicerofosfórico, ácido acético, ácido glicólico, ácido hipúrico, ácido isobutírico, ácido láctico, ácido lactobiónico, ácido láurico, ácido maleico, ácido málico, ácido malónico, ácido mandélico, ácido metanosulfónico, ácido múcico, ácido naftaleno-1,5-disulfónico, ácido naftaleno-2-sulfónico, ácido 1-hidroxi-2-naftoico, ácido nicotínico, ácido oleico, ácido orótico, ácido oxálico, ácido palmítico, ácido pamoico, ácido propiónico, ácido piroglutámico, ácido pirúvico, ácido salicílico, ácido 4-aminosalicílico, ácido sebácico, ácido esteárico, ácido succínico, ácido tartárico, ácido tiociánico, ácido p-toluenosulfónico, ácido trifluoroacético, ácido undecilénico y similares.
De manera similar, una "sal de adición de base farmacéuticamente aceptable" se refiere a aquellas sales que conservan la eficacia biológica y las propiedades de los ácidos libres, que no son biológicamente o de otro modo indeseables. Estas sales se preparan por adición de una base inorgánica o una base orgánica al ácido libre. Las sales derivadas de bases inorgánicas incluyen, pero sin limitación, sales de sodio, potasio, litio, amonio, calcio, magnesio, hierro, cinc, cobre, manganeso, aluminio y similares. Son sales inorgánicas preferidas las sales de amonio, sodio, potasio, calcio y magnesio. Las sales derivadas de bases orgánicas incluyen, pero sin limitación, sales de aminas primarias, secundarias y terciarias, aminas sustituidas que incluyen aminas sustituidas naturales, aminas cíclicas y resinas básicas de intercambio iónico, tales como amoníaco, isopropilamina, trimetilamina, dietilamina, trietilamina, tripropilamina, dietanolamina, etanolamina, etanol, 2-dimetilaminoetanol, 2-dietilaminoetanol, diciclohexilamina, lisina, arginina, histidina, cafeína, procaína, hidrabamina, colina, betaína, benetamina, benzatina, etilendiamina, glucosamina, metilglucamina, teobromina, trietanolamina, trometamina, purinas, piperazina, piperidina, etilpiperidina, resinas de poliamina y similares. Son bases orgánicas particularmente preferidas isopropilamina, dietilamina, etanolamina, trimetilamina, diciclohexilamina, colina y cafeína.
Con frecuencia, las cristalizaciones producen un solvato del compuesto de la invención. Como se usa en el presente documento, el término "solvato" se refiere a un agregado que comprende una o más moléculas de un compuesto de la invención con una o más moléculas de disolvente. El disolvente puede ser agua, en cuyo caso el solvato puede ser un hidrato. Como alternativa, el disolvente puede ser un disolvente orgánico. Por tanto, los compuestos de la presente invención pueden existir como un hidrato, incluyendo un monohidrato, dihidrato, hemihidrato, sesquihidrato, trihidrato, tetrahidrato y similares, así como las formas solvatadas correspondientes. Los compuestos de la invención
pueden ser solvatos verdaderos, mientras que, en otros casos, los compuestos de la invención pueden simplemente retener agua adventicia o ser una mezcla de agua más algún disolvente adventicio.
Un "estereoisómero" se refiere a un compuesto formado por los mismos átomos unidos por los mismos enlaces pero que tienen estructuras tridimensionales diferentes, que no son intercambiables. La presente invención contempla diversos estereoisómeros y mezclas de los mismos e incluye "enantiómeros", que se refiere a dos estereoisómeros cuyas moléculas son imágenes especulares no superponibles entre sí.
Los compuestos de la invención o sus sales farmacéuticamente aceptables pueden contener uno o más centros asimétricos y pueden dar lugar, de este modo, a enantiómeros, diastereoisómeros y otras formas estereoisoméricas que pueden definirse, en términos de estereoquímica absoluta, como (R)- o (S)-o, como (D)- o (L)- para los aminoácidos. La presente invención pretende incluir todos estos posibles isómeros, así como sus formas racémicas y ópticamente puras. Los isómeros ópticamente activos (+) y (-), (R)- y (S)- o (D)- y (L) pueden prepararse usando sintones quirales o reactivos quirales, o se resuelven usando técnicas convencionales, por ejemplo, cromatografía y cristalización fraccionada. Las técnicas convencionales para la preparación/aislamiento de enantiómeros individuales incluyen la síntesis quiral a partir de un precursor ópticamente puro adecuado o la resolución del racemato (o el racemato de una sal o derivado) usando, por ejemplo, cromatografía líquida de alta presión quiral (HPLC). Cuando los compuestos que se describen en el presente documento contienen dobles enlaces olefínicos u otros centros de asimetría geométrica y, a menos que se especifique lo contrario, se pretende que los compuestos incluyan ambos isómeros geométricos E y Z. Análogamente, también se pretende incluir todas las formas tautoméricas.
El término “tautómero” se refiere a un desplazamiento de protón desde un átomo de una molécula a otro átomo de la misma molécula. Por ejemplo, cuando W1 es oxo y R1 es H, la presente invención proporciona tautómeros de un compuesto de Fórmula I como se ilustra después:

Existen tautómeros similares para compuestos de las Fórmulas I, la, Ila, 1 b, 1 Ia, IIIb, IVa, IVb, Va, Vb VI, VIIa y VIIb. Los compuestos de la invención se sintetizan usando métodos de síntesis convencionales y, más específicamente, usando los métodos generales que se indican después. Los protocolos sintéticos específicos para varios compuestos de acuerdo con la presente invención se describen en los Ejemplos.
Métodos de síntesis generales
Método 1:

La formación de IXa, cuando n = 1 y X = halógeno u otro grupo saliente, tal como -OTf, -OTs o -OMs, se consiguió exponiendo el intermedio Villa a un aldehído o cetona Xa, o un equivalente de aldehído o cetona Xb-d en condiciones ácidas en las que R2-R3 son como se ha definido anteriormente y Rm = H, CH3 , CH2CH3 o alquilo. Más específicamente, exponer VIIIa donde X es CI o Br a un aldehído o cetona Xa en 1,4-dioxano y ácido sulfúrico concentrado con calentamiento produce el intermedio IXa (n = 1).

Los compuestos de tipo pirimidina de acuerdo con la Fórmula XlIa o Xllb donde Y es N(R5), O o S y P es un grupo protector se pueden adquirir o preparar a partir de Xla mediante diversos métodos, por ejemplo, desplazando el grupo saliente X del compuesto Xla con un nucleófilo N, O o S apropiado. El compuesto Xllb resultante puede desprotegerse para proporcionar Xlla.

Los compuestos de la invención de acuerdo con la Fórmula I cuando Y es N(R5), O o S se sintetizaron poniendo en contacto el intermedio IXa con un compuesto de pirimidina Xlla donde R6-R8 son como se ha definido anteriormente, en condiciones de reacción apropiadas como se describe adicionalmente más adelante.

Más específicamente, los compuestos de Fórmula I cuando Y es N(R5), O o S se sintetizaron usando acoplamiento de Buchwald-Hartwig, acoplamiento de tipo Ullmann o sustitución aromática nucleófila. Por tanto, poner en contacto el intermedio lXa donde X = Cl o Br y n = 1 con un compuesto de Fórmula Xlla donde Y es N(R5), O o S en condiciones adecuadas para el acoplamiento o la sustitución aromática nucleófila proporcionó compuestos de Fórmula I.
Como alternativa, el grupo saliente X del intermedio lXa se puede desplazar con un nucleófilo N, O o S apropiado en condiciones similares a las descritas anteriormente para la síntesis de Xlla de manera que proporcione el intermedio Xllla o el intermedio Xlllb protegido donde Y es N(R5), O, o S. Xlllb pueden desprotegerse para producir Xllla.

Los compuestos de Fórmula I cuando Y es N(R5), O o S se sintetizan fácilmente poniendo en contacto el intermedio Xllla donde Y es N(R5), O o S con un compuesto de pirimidina Xla donde X = halógeno u otro grupo saliente tal como -OTf, -OTs o -OMs en condiciones de acoplamiento de Buchwald-Hartwig, acoplamiento de tipo Ullmann o sustitución aromática nucleófila.

A continuación, se exponen métodos de síntesis más específicos para varios compuestos de Fórmula I. Se entiende que, si se usan grupos protectores durante la síntesis de intermedios o si un compuesto de Fórmula I contiene uno o más grupos protectores, entonces dichos grupos protectores se retiran por métodos conocidos en la técnica química. Otras transformaciones, tales como el desplazamiento de un halógeno, por ejemplo, la conversión de R4a, o R4b = CI en R4a, o R4b = OMe, SMe, CH=CH2 o Me, la conversión de W1, W2 o ambos grupos de O a S; la formación de una oxima convirtiendo W1, W2 o ambos grupos de un grupo oxo (=O) en un grupo =NHOR', la conversión de Y a partir de S en S=O o S(=O)2 , y la conversión de un compuesto intermedio o de Fórmula I en una sal farmacéuticamente aceptable se realiza usando métodos convencionales conocidos en la técnica química.
Método 2:

VIIIb IXa IXb
La formación de IXa o IXb, cuando n = 2 o 3, la variable "X" es un halógeno u otro grupo saliente tal como -OTf, -OTs o -OMs, y la variable "V" es O o n puede conseguirse poniendo en contacto el intermedio VlIIb con un intermedio de etilo 1,2-difuncionalizado XlVa, o un intermedio de propilo 1,3-difuncionalizado XlVb en condiciones adecuadas para la síntesis de los compuestos IXa y IXb, respectivamente. Las variables Z1 y Z2 en XlVa y XlVb pueden ser un halógeno u otros grupos salientes tales como -OTf, -OTs o -OMs, o OH, NHR1 o NHP, donde P es un grupo protector apropiado.

De acuerdo con esta estrategia sintética, el intermedio VIIIb donde la variable "X" es CI o Br y la variable "V" es O se pone en contacto con XlVa donde R2 y R3 son H, Z1 es OH y Z2 es NH-(4-metoxibencilo) en acetonitrilo en presencia de un reactivo formador de enlace amida tal como HATU. La mezcla de reacción se calienta para promover el acoplamiento y produce el intermediado IXb donde n = 2 y P es 4-metoxibencilo.
Los compuestos de Fórmula I de la invención cuando Y es N(R5), O o S se sintetizan poniendo en contacto los intermedios IXa o IXb (n = 2 o 3 y X = halógeno u otro grupo saliente tal como -OTf, -OTs o -OM), con pirimidina XIIa donde Y es N(R5), O, o S y R6-R8 son como se ha definido anteriormente, en condiciones apropiadas seguido de desprotección del compuesto de Fórmula I resultante si se requiere.

Por tanto, poner en contacto el intermedio IXb donde n = 2, X es CI o Br y P es 4-metoxibencilo con pirimidina XlIa, donde Y es N(R5), O o S, en las condiciones adecuadas para el acoplamiento de Buchwald-Hartwig, acoplamiento de tipo Ullmann, o la sustitución aromática nucleófila da como resultado un compuesto de Fórmula I protegido con 4-metoxibencilo. El compuesto de Fórmula I terapéuticamente activo se puede obtener fácilmente por desprotección del grupo 4-metoxibencilo con ácido trifluoroacético.
Como alternativa, se sintetizaron determinados compuestos de Fórmula I desplazando el grupo saliente "X" del intermedio IXa con nucleófilos N, O o S adecuados en condiciones similares a las que se describen en el Método 1 para proporcionar Xllla o el intermedio protegido Xlllb que puede desprotegerse para producir Xllla. El grupo saliente "X" del intermedio lXa puede ser halógeno, -OTf, -OTs o -OMs y el subíndice n es ya sea 2 o 3.

Como se ha descrito anteriormente, los compuestos de Fórmula I de la invención se sintetizan fácilmente poniendo en contacto el intermedio Xllla cuando Y = N(R5), O, S y n = 2 o 3 con pirimidina Xla donde X = halógeno, -OTf, -OTs o -OMs en condiciones adecuadas para el acoplamiento de Buchwald-Hartwig, el acoplamiento de tipo Ullmann o la sustitución aromática nucleófila.

La síntesis de algunos compuestos de Fórmula I se realizó poniendo en contacto el intermedio protegido lXb (n = 2 o 3), con un nucleófilo N, O o S apropiado para proporcionar Xlllc en condiciones similares descritas anteriormente con lXa.

El intermedio Xlllc, obtenido de este modo, se puso entonces en contacto con pirimidina Xla donde X es halógeno o un grupo saliente seleccionado entre el grupo que consiste en -OTf, -OTs y -OMs en condiciones adecuadas para el acoplamiento de Buchwald-Hartwig, el acoplamiento de tipo Ullmann o la sustitución aromática nucleófila. La desprotección por métodos conocidos en la técnica química proporcionó los compuestos de Fórmula I candidatos a inhibidores de MnK.

La síntesis de los compuestos de Fórmula VI, donde n = 1, 2 o 3 se consiguió poniendo en contacto los intermedios IXa o IXb donde la variable "X" es un grupo saliente seleccionado entre el grupo que consiste en halógeno, -OTf, -OTs y -OMs y P es un grupo protector con un intermedio bicíclico XV en presencia de un catalizador de paladiofosfina homogéneo y una base tal como carbonato de cesio o t-butóxido de sodio usando 1,4-dioxano como disolvente. La mezcla de reacción se calentó para promover el acoplamiento, seguido de desprotección si era necesario. Los ejemplos de compuestos intermedios XV sin limitación son 6,7-dihidro-5H-pirrolo [2,3-d]pirimidina, 7H-pirrolo[2,3-d]pirimidina, 9H-purina, 1H-pirazolo[3,4-d]pirimidina, y 5,6,7,8-tetrahidropirido[2,3-d]pirimidina sustituida o no sustituida).

Como alternativa, los compuestos de Fórmula VI donde B es un anillo insaturado de 5 miembros y n = 1, 2 o 3 pueden sintetizarse poniendo en contacto los intermedios IXa o IXb con un intermedio de 5-etinil-4-amino-pirimidina XIIc (donde Ra es un J como se define en el presente documento) en presencia de un catalizador de paladio-fosfina homogéneo y una base tal como carbonato de cesio o t-butóxido de sodio usando 1,4-dioxano como disolvente para la reacción de acoplamiento. La mezcla de reacción se puede calentar para promover el acoplamiento, seguido de desprotección si se requiere. Los intermedios de 5-etinil-4-amino-pirimidina XIIc sintetizados mediante una reacción de acoplamiento cruzado entre XlIa en la que Y = N(R5) y R8 es un grupo saliente tal como halógeno o -OTf y un alquino adecuado que usa catalizadores homogéneos de cobre y/o paladio.
Método 4:

Los compuestos de acuerdo con la Fórmula VlIa o la Fórmula VlIb, donde Y = N(R5), O o S y n es 1, 2 o 3 pueden sintetizarse poniendo en contacto los intermedios IXa o IXb donde X es un grupo saliente tal como halógeno, -OTf, -OTs o -OMs y P es un grupo protector con el intermedio bicíclico XVIa en condiciones de acoplamiento de Buchwald-Hartwig, acoplamiento de tipo Ullmann o sustitución aromática nucleófila. El anillo C del intermedio bicíclico es como se ha definido anteriormente y la variable Y puede ser -N(R5), O o S. Los ejemplos representativos de los intermedios XVIa incluyen sin limitación 6-amino-purina, 4-amino-1H-pirazolo[3,4-d]pirimidina, 4-amino-7H-pirrolo[2,3-d]pirimidina, 4-amino-6,7-dihidro-5H-pirrolo[2,3-d]pirimidina, 7-amino-3H-[1,2,3]triazolo[4,5-d]pirimidina, 4-aminoquinazolina, 4-amino-5,6,7,8-tetrahidropirido[2,3-d]pirimidina, diversas 4-amino-pirido[d]pirimidinas, pirimido[5,4-d]pirimidin-4-amina y 7,8-dihidro-6H-pirimido[5,4-d][1,4]oxazin-4-amina sustituida o no sustituida).
Los compuestos intermedios de tipo pirimidina bicíclicos de acuerdo con la Fórmula XVIa donde Y = N(R5), O o S pueden adquirirse o prepararse a partir de XVI desplazando el grupo saliente X con un nucleófilo N, O o S apropiado usando métodos conocidos en la técnica química. El grupo protector "P" en un intermedio de acuerdo con XVIb puede retirarse para proporcionar el intermedio XVIa.

Como alternativa, los intermedios XHIa o XHIb pueden ponerse en contacto con la pirimidina XVI condensada donde X = halógeno u otro grupo saliente tal como -OTf, -OTs o -OM en condiciones adecuadas para acoplamientos de tipo Buchwald-Hartwig o Ullmann, o condiciones adecuadas para sustitución aromática nucleófila seguida de desprotección si es necesario para proporcionar compuestos de Fórmula VIIa o Fórmula VIIb.
Los métodos para sintetizar una Fórmula I, cuando Y es -C(O) y n = 1, 2 o 3, comprenden poner en contacto el intermedio IXa o intermedio IXb (donde X es un grupo saliente, tal como halógeno, -OTf, -OTs o -OMs, y P es un grupo protector) con el intermedio XlId en presencia de una base tal como n-butil-litio. El producto resultante puede desprotegerse si se requiere para proporcionar compuestos de Fórmula I.
La formación de Fórmula I, donde Y = CH y n = 1, 2 o 3 puede realizarse reduciendo el carbonilo (-C(O)) en "Y" en condiciones de reducción de Wolff-Kishner.
Método 6:

La síntesis de compuestos de Fórmula VIIb, cuando Y = C(O), n = 1, 2 o 3 y C es como se ha definido anteriormente, se realiza poniendo en contacto el intermedio IXa o el intermedio IXb (donde X es un grupo saliente, tal como halógeno, OTf, -OTs o -OMs, y P es un grupo protector) con el intermedio XVIc en presencia de una base tal como n-butil-litio. El producto resultante puede desprotegerse si se requiere para proporcionar compuestos de Fórmula VIIb.
La formación de VIIb, cuando Y = CH y n = 1, 2 o 3 puede conseguirse reduciendo el carbonilo (-C(O)) en "Y" en condiciones de reducción de Wolff-Kishner.
Formulaciones farmacéuticas
En una realización, se formulan compuestos de las Fórmulas I a VII como composiciones farmacéuticamente aceptables que contienen un compuesto de las Fórmulas I-VII en una cantidad eficaz para tratar una enfermedad o afección particular de interés tras la administración de la composición farmacéutica a un mamífero. Las composiciones farmacéuticas de acuerdo con la presente invención pueden comprender un compuesto de las Fórmulas I-VII en combinación con un vehículo, diluyente o excipiente farmacéuticamente aceptable.
A este respecto, un "vehículo, diluyente o excipiente farmacéuticamente aceptable" incluye sin limitación cualquier adyuvante, vehículo, excipiente, sustancia de deslizamiento, agente edulcorante, diluyente, conservante, colorante/colorante, potenciador del sabor, tensioactivo, agente humectante, agente dispersante, agente de suspensión, estabilizador, agente isotónico, disolvente o emulsionante que ha sido aprobado por la Administración de Alimentos y Fármacos de los Estados Unidos como aceptable para uso en seres humanos o animales domésticos.
Además, un "mamífero" incluye seres humanos y animales domésticos tales como animales de laboratorio y animales domésticos (por ejemplo, gatos, perros, cerdos, ganado, ovejas, cabras, caballos, conejos) y animales no domésticos tales como vida silvestre y similares.
Las composiciones farmacéuticas de la invención pueden prepararse combinando un compuesto de la invención con un vehículo, diluyente o excipiente farmacéuticamente aceptable apropiado, y pueden formularse en preparaciones en forma sólida, semisólida, líquida o gaseosa, tales como tabletas, cápsulas, polvos, gránulos, pomadas, soluciones, supositorios, inyecciones, inhalantes, geles, microesferas y aerosoles. Las vías típicas de administración de dichas composiciones farmacéuticas incluyen, sin limitación, oral, tópica, transdérmica, inhalatoria, parenteral, sublingual, bucal, rectal, vaginal e intranasal. El término parenteral como se usa en el presente documento incluye inyecciones subcutáneas, inyección intravenosa, intramuscular, intraesternal o técnicas de infusión. Las composiciones farmacéuticas de la invención se formulan de manera que permitan que los ingredientes activos contenidos en los mismos sean biodisponibles tras la administración de la composición a un paciente. Las composiciones que se administrarán a un sujeto o paciente toman la forma de una o más unidades de dosificación, donde, por ejemplo, un comprimido puede ser una unidad de dosificación única, y un recipiente de un compuesto de la invención en forma de aerosol puede contener una pluralidad de unidades de dosificación. Los métodos reales de preparación de dichas formas de dosificación son conocidos o serán evidentes para los expertos en esta técnica; por ejemplo, véase Remington: The Science and Practice of Pharmacy, 20a edición (Philadelphia College of Pharmacy and Science, 2000). La composición que se ha de administrar contendrá, en cualquier caso, una cantidad terapéuticamente eficaz de un compuesto de la invención, o una sal farmacéuticamente aceptable del mismo, para el tratamiento de una enfermedad o afección de interés de acuerdo con el contenido de la presente invención.
Una composición farmacéutica de la invención puede estar en forma de un sólido de color o líquido. En un aspecto, el o los vehículos son particulados, de manera que las composiciones son, por ejemplo, en forma de comprimido o polvo. El o los vehículos pueden ser líquidos, siendo las composiciones, por ejemplo, un jarabe oral, un líquido inyectable o un aerosol, que es útil en, por ejemplo, la administración por inhalación. Cuando está destinada a la administración oral, la composición farmacéutica está preferentemente en forma sólida o líquida, en la que se incluyen formas semisólidas, semilíquidas, de suspensión y gel dentro de las formas consideradas en el presente documento como sólidas o líquidas.
Como composición sólida para la administración oral, la composición farmacéutica puede formularse en forma de polvo, gránulo, comprimido, píldora, cápsula, chicle, oblea o forma similar. Dicha composición sólida contendrá normalmente uno o más diluyentes inertes o vehículos comestibles. Además, pueden estar presentes uno o más de los siguientes: aglutinantes tales como carboximetilcelulosa, etilcelulosa, celulosa microcristalina, goma de tragacanto o gelatina; excipientes tales como almidón, lactosa o dextrinas, agentes disgregantes tales como ácido algínico, alginato de sodio, Primogel, almidón de maíz y similares; lubricantes tales como estearato de magnesio o Sterotex; sustancias de deslizamiento tales como dióxido de silicio coloidal; agentes edulcorantes tales como sacarosa o sacarina; un agente aromatizante tal como menta, salicilato de metilo o aroma de naranja; y un agente colorante.
Cuando la composición farmacéutica está en forma de una cápsula, por ejemplo, una cápsula de gelatina puede contener, además de materiales del tipo anterior, un vehículo líquido tal como polietilenglicol o aceite.
La composición farmacéutica puede estar en forma de un líquido, por ejemplo, un elixir, un jarabe, una solución, una emulsión o una suspensión. El líquido puede ser para la administración oral o para la administración por inyección, como dos ejemplos. Cuando se destinan a la administración oral, la composición preferida contiene, además de los presentes compuestos, uno o más de entre un agente edulcorante, conservantes, tinte/colorante y potenciador del sabor. En una composición destinada a ser administrada por inyección, pueden incluirse uno o más de entre un tensioactivo, conservante, agente humectante, agente dispersante, agente de suspensión, tampón, estabilizador e agente isotónico.
Las composiciones farmacéuticas líquidas de la invención, ya sean soluciones, suspensiones u otras formas similares, pueden incluir uno o más de los siguientes adyuvantes: diluyentes estériles tales como agua para inyección, solución salina, preferentemente solución salina fisiológica, solución de Ringer, cloruro de sodio isotónico, aceites no volátiles tales como mono o diglicéridos sintéticos que pueden servir como disolvente o medio de suspensión, polietilenglicoles, glicerina, propilenglicol u otros disolventes; agentes antibacterianos tales como alcohol bencílico o metilparabeno; antioxidantes tales como ácido ascórbico o bisulfito de sodio; agentes quelantes tales como ácido etilendiaminotetraacético; tampones tales como acetatos, citratos o fosfatos y agentes para el ajuste de la tonicidad tales como cloruro de sodio o dextrosa. La preparación parenteral puede encerrarse en ampollas, jeringuillas desechables o viales de dosis múltiples hechos de vidrio o plástico. La solución salina fisiológica es un adyuvante preferido. Una composición farmacéutica inyectable es preferentemente estéril.
Una composición farmacéutica líquida de la invención destinada a la administración parenteral u oral debe contener una cantidad de un compuesto de la invención de manera que se obtenga una dosificación adecuada.
La composición farmacéutica de la invención puede estar destinada a la administración tópica, en cuyo caso el vehículo puede comprender adecuadamente una base de solución, emulsión, pomada o gel. La base, por ejemplo, puede comprender uno o más de los siguientes: vaselina, lanolina, polietilenglicoles, cera de abeja, aceite mineral, diluyentes tales como agua y alcohol, y emulsionantes y estabilizadores. Puede haber presentes agentes espesantes en una composición farmacéutica para la administración tópica. Si se destina para la administración transdérmica, la composición puede incluir un parche transdérmico o un dispositivo de iontoforesis.
La composición farmacéutica de la invención puede estar destinada a la administración rectal, en la forma, por ejemplo, de un supositorio, que se fundirá en el recto y liberará el fármaco. La composición para la administración rectal puede contener una base oleaginosa como un excipiente no irritante adecuado. Dichas bases incluyen, sin limitación, lanolina, manteca de cacao y polietilenglicol.
La composición farmacéutica de la invención puede incluir diversos materiales, que modifican la forma física de una unidad de dosificación sólida o líquida. Por ejemplo, la composición puede incluir materiales que formen una cubierta de recubrimiento alrededor de los ingredientes activos. Los materiales que forman la cubierta de recubrimiento son normalmente inertes, y pueden seleccionarse entre, por ejemplo, azúcar, goma laca, y otros agentes de recubrimiento entérico. Como alternativa, los ingredientes activos pueden encerrarse en una cápsula de gelatina. La composición farmacéutica de la invención en forma sólida o líquida puede incluir un agente que se une al compuesto de la invención y, de este modo, ayuda a la entrega del compuesto. Los agentes adecuados que pueden actuar en esta capacidad incluyen un anticuerpo monoclonal o policlonal, una proteína o un liposoma.
La composición farmacéutica de la invención puede consistir en unidades de dosificación que se pueden administrar
como un aerosol. El término aerosol se usa para denotar una diversidad de sistemas que varían de los de naturaleza coloidal a sistemas que consisten en envases presurizados. La entrega puede ser mediante un gas licuado o comprimido o mediante un sistema de bombeo adecuado que dispensa los ingredientes activos. Los aerosoles de compuestos de la invención pueden entregarse en sistemas monofásicos, bifásicos o trifásicos con el fin de liberar el o los ingredientes activos. La entrega del aerosol incluye el recipiente necesario, activadores, válvulas, subrecipientes y similares, que juntos pueden formar un kit. Un experto en la materia, sin experimentación indebida, puede determinar aerosoles preferidos.
Las composiciones farmacéuticas de la invención pueden prepararse mediante cualquier metodología bien conocida en la técnica farmacéutica. Por ejemplo, una composición farmacéutica destinada a ser administrada por inyección puede prepararse combinando un compuesto de la invención con agua destilada estéril para formar una solución. Se puede añadir un tensioactivo para facilitar la formación de una solución o suspensión homogénea. Los tensioactivos son compuestos que interactúan de forma no covalente con el compuesto de la invención con el fin de facilitar la disolución o suspensión homogénea del compuesto en el sistema de entrega acuoso.
En determinadas realizaciones, se administra una composición farmacéutica que comprende un compuesto de Fórmula I a un mamífero en una cantidad suficiente para inhibir la actividad de Mnk tras la administración, y preferentemente con toxicidad aceptable para el mismo. La actividad Mnk de los compuestos de Fórmula I puede ser determinada por un experto en la materia, por ejemplo, como se describe en los Ejemplos después. Las concentraciones y dosificaciones apropiadas pueden ser determinadas fácilmente por un experto en la materia.
Uso terapéutico
Los compuestos de la invención, o sus sales farmacéuticamente aceptables, se administran en una cantidad terapéuticamente eficaz, que variará dependiendo de una diversidad de factores incluyendo la actividad del compuesto específico empleado; la estabilidad metabólica y la longitud de acción del compuesto; la edad, el peso corporal, la salud general, el sexo y la dieta del paciente; el modo y el tiempo de administración; la tasa de excreción; la combinación de fármacos; la gravedad del trastorno o afección particular; y el sujeto sometido a terapia.
"Cantidad eficaz" o "cantidad terapéuticamente eficaz" se refiere a la cantidad de un compuesto de la invención que, cuando se administra a un mamífero, preferentemente un ser humano, es suficiente para efectuar el tratamiento, como se define después, de una afección o enfermedad relacionada con Mnk en el mamífero, preferentemente un ser humano. La cantidad de un compuesto de la invención que constituye una "cantidad terapéuticamente eficaz" variará dependiendo del compuesto, la afección y su gravedad, la forma de administración y la edad del mamífero que se ha de tratar, pero puede determinarse habitualmente por un experto en la materia teniendo en cuenta su propio conocimiento y la presente invención.
Los compuestos de la invención, o sales farmacéuticamente aceptables de los mismos, también se pueden administrar simultáneamente con, antes o después de la administración de uno o más agentes terapéuticos. Dicha terapia de combinación incluye la administración de una única formulación de dosificación farmacéutica que contiene un compuesto de la invención y uno o más agentes activos adicionales, así como la administración del compuesto de la invención y cada agente activo en su propia formulación de dosificación farmacéutica separada. Por ejemplo, un compuesto de la invención y el otro agente activo pueden administrarse al paciente en una sola composición de dosificación oral tal como un comprimido o cápsula o cada agente administrado en formulaciones de dosificación oral separadas. Cuando se usan formulaciones de dosificación separadas, los compuestos de la invención y uno o más agentes activos adicionales pueden administrarse esencialmente al mismo tiempo, es decir, simultáneamente o en tiempos escalonados separadamente, es decir, secuencialmente; se entiende que la terapia de combinación incluye todas estas posologías.
En determinadas realizaciones, los compuestos desvelados son útiles para inhibir la actividad de Mnk y/o pueden ser útiles en el análisis de la actividad de señalización de Mnk en sistemas modelo y/o para prevenir, tratar o mejorar un síntoma asociado a una enfermedad, trastorno o afección patológica que implica Mnk, preferentemente uno que afecta a seres humanos. Un compuesto que inhibe la actividad de Mnk será útil para prevenir, tratar, mejorar o reducir los síntomas o la progresión de enfermedades de crecimiento, proliferación y/o supervivencia de células descontroladas, respuestas inmunitarias celulares inadecuadas o respuestas o enfermedades inflamatorias celulares inadecuadas que se acompañan de crecimiento celular no controlado, proliferación y/o supervivencia, respuestas inmunitarias celulares inapropiadas, o respuestas inflamatorias celulares inapropiadas, en particular en las que el crecimiento, proliferación y/o supervivencia celular incontrolada, las respuestas inmunitarias celulares inapropiadas o las respuestas inflamatorias celulares inapropiadas están mediadas por ejemplo, por tumores hemáticos, tumores sólidos y/o metástasis de los mismos, incluyendo leucemias y síndrome de mielodisplasia, linfomas malignos, por ejemplo, linfoma de células B, linfoma de linfocitos T, linfoma de células pilosas, linfoma de Hodgkins, linfoma no Hodgkins y linfoma de Burkitt, tumores de cabeza y cuello incluyendo tumores cerebrales y metástasis cerebrales, tumores del tórax incluyendo tumores de células no microcíticas y de células microcíticas, tumores gastrointestinales, tumores endocrinos, tumores mamarios y otros tumores ginecológicos, tumores urológicos, tumores de vejiga y próstata, tumores de piel y sarcomas, y/o metástasis de los mismos.
Además, los compuestos de la invención y sus composiciones farmacéuticas son terapias candidatas para la profilaxis y/o terapia de enfermedades relacionadas con citocinas, tales como enfermedades inflamatorias, alergias u otras afecciones asociadas a citocinas proinflamatorias. Las enfermedades inflamatorias de ejemplo incluyen sin limitación, inflamación crónica o aguda, inflamación de las articulaciones tal como la artritis inflamatoria crónica, artritis reumatoide, artritis psoriásica, osteoartritis, artritis reumatoide juvenil, síndrome de Reiter, artritis traumática reumatoide, artritis por rubéola, sinovitis aguda y artritis gotosa; enfermedades inflamatorias de la piel tales como quemaduras solares, psoriasis, psoriasis eritrodérmica, psoriasis pustular, eccema, dermatitis, formación de injerto aguda o crónica, dermatitis atópica, dermatitis de contacto, urticaria y esclerodermia; inflamación del tracto gastrointestinal tal como enfermedad intestinal inflamatoria, enfermedad de Crohn y afecciones relacionadas, colitis ulcerosa, colitis y diverticulitis; nefritis, uretritis, salpingitis, ovaritis, endomiometritis, espondilitis, lupus eritematoso sistémico y trastornos relacionados, esclerosis múltiple, asma, meningitis, mielitis, encefalomielitis, encefalitis, flebitis, tromboflebitis, enfermedades respiratorias tales como asma, bronquitis, enfermedad pulmonar obstructiva crónica (EPOC), enfermedad pulmonar inflamatoria y síndrome de dificultad respiratoria del adulto, y rinitis alérgica; endocarditis, osteomielitis, fiebre reumática, pericarditis reumática, endocarditis reumática, miocarditis reumática, enfermedad de la válvula mitral reumática, enfermedad de la válvula aórtica reumática, prostatitis, prostatocistitis, espondoartropatías, espondilitis anquilosante, sinovitis, tenosinovitis, miositis, faringitis, polimialgia reumática, tendinitis o bursitis, gota, pseudogota, vasculitis, enfermedades inflamatorias de la tiroides seleccionadas entre el grupo que consiste en tiroiditis granulomatosa, tiroiditis linfocítica, tiroiditis fibrosa invasiva, tiroiditis aguda; tiroiditis de Hashimoto, enfermedad de Kawasaki, fenómeno de Raynaud, síndrome de Sjogren, enfermedad neuroinflamatoria, sepsis, conjuntivitis, queratitis, iridociclitis, neuritis óptica, otitis, linfoadenitis, nasofaringitis, sinusitis, faringitis, amigdalitis, laringitis, epiglotitis, bronquitis, neumonitis, estomatitis, gingivitis, esofagitis, gastritis, peritonitis, hepatitis, colelitiasis, colecistitis, glomerulonefritis, enfermedad de Goodpasture, glomerulonefritis, pancreatitis, endomiometritis, miometritis, metritis, cervicitis, endocervicitis, exocervicitis, parametritis, tuberculosis, vaginitis, vulvitis, silicosis, sarcoidosis, neumoconiosis, pirexia, poliartropatías inflamatorias, artropatías psoriásicas, fibrosis intestinal, bronquiectasias y artropatías enteropáticas.
Aunque la inflamación es el proceso patogénico unificador de estas enfermedades, las terapias actuales solo tratan los síntomas de la enfermedad y no la causa subyacente de la inflamación. Las composiciones de la presente invención son útiles para el tratamiento y/o la profilaxis de enfermedades inflamatorias y complicaciones y trastornos relacionados.
En consecuencia, determinadas realizaciones se refieren a un método para tratar una afección dependiente de Mnk en un mamífero que lo necesite, comprendiendo el método administrar una cantidad eficaz de una composición farmacéutica como se ha descrito anteriormente (es decir, una composición farmacéutica que comprende uno o más compuestos de Fórmula I) a un mamífero.
"Tratar" o "tratamiento" como se usa en la presente invención cubre el tratamiento de la enfermedad o afección de interés en un mamífero, preferentemente un ser humano, que tiene la enfermedad o afección de interés e incluye:
(i) prevenir que la enfermedad o afección se produzca en un mamífero, en particular, cuando dicho mamífero está predispuesto a la afección, pero todavía no se ha diagnosticado que la tiene;
(ii) inhibir la enfermedad o afección, es decir, detener su desarrollo;
(iii) aliviar la enfermedad o afección, es decir, provocar la regresión de la enfermedad o afección; o
(iv) aliviar los síntomas resultado de la enfermedad o afección, es decir, aliviar el dolor sin abordar la enfermedad o afección subyacente. Como se usa en el presente documento, los términos “enfermedad” y “afección” pueden usarse indistintamente o pueden ser diferentes en que la enfermedad o afección particular puede no tener un agente causal conocido (de modo que la etiología aún no se ha elaborado) y por tanto aún no se reconoce como una enfermedad, sino solo como una afección o un síndrome indeseable, en los que se ha identificado un conjunto más o menos específico de síntomas por los médicos.
Como se ha descrito anteriormente, la desregulación de la síntesis de proteínas es un evento común en los cánceres humanos. Un regulador clave del control traslacional es eIF4E cuya actividad es un determinante clave de la carcinogenicidad. Debido a que la activación de eIF4E implica fosforilación de una serina clave (Ser209) específicamente por cinasas que interactúan con MAP cinasa (Mnk), los inhibidores de Mnk son fármacos candidatos adecuados para tratar trastornos proliferativos celulares tales como cáncer. Una amplia diversidad de cánceres, incluyendo tumores sólidos, linfomas y leucemias, son susceptibles a las composiciones y métodos que se describen en el presente documento. Los tipos de cáncer que se pueden tratar incluyen, pero sin limitación, adenocarcinoma de mama, próstata y colon; todas las formas de carcinoma broncogénico del pulmón; mieloide; melanoma; hepatoma; neuroblastoma; papiloma; apudoma; coristoma; branquioma; síndrome carcinoide maligno; enfermedad cardiaca carcinoide; y carcinoma (por ejemplo, Walker, células basales, basoescamoso, Brown-Pearce, ductal, tumor de Ehrlich, Krebs 2, célula de merkel, mucinosa, pulmón de células no microcíticas, célula de avena, papilar, scirrosa, bronquiolar, broncogénica, célula de transición). Otros tipos de cánceres que pueden tratarse incluyen trastornos histiocíticos; leucemia; histiocitosis maligna; enfermedad de Hodgkin; pequeño inmunoproliferativo; linfoma no Hodgkin; linfoma difuso de células B grandes, linfoma de linfocitos T, linfoma de células B, linfoma de células pilosas, linfoma de Burkitt, plasmacitoma; reticuloendoteliosis; melanoma; condroblastoma; condroma; condrosarcoma; fibroma; fibrosarcoma; tumores de células gigantes; histiocitoma;
lipoma; liposarcoma; mesotelioma; mixoma; mirosarcoma; osteoma; osteosarcoma; cordoma; craneofaringioma; disgerminoma; hamartoma; mesenquimoma; mesonefroma; miosarcoma; ameloblastoma; cementoma; odontoma; teratoma; timoma; tumor trofoblástico.
Otros cánceres que pueden tratarse usando los compuestos de la invención incluyen, sin limitación, adenoma; colangioma; colesteatoma; ciclindroma; cistadenocarcinoma; cistoadenoma; tumor de células granulosas; ginandroblastoma; hepatoma; hidradenoma; tumor de células de islote; tumor de células de Leydig; papiloma; tumor de células de Sertoli; tumor de células de la teca; leimioma; leiomiosarcoma; mioblastoma; mioma miosarcoma; rabdomioma; rabdomiosarcoma; ependimoma; ganglioneuroma; glioma; meduloblastoma; meningioma; neurilemoma; neuroblastoma; neuroepitelioma; neurofibroma; neuroma; paraganglioma; paraganglioma noncromafina.
En una realización, los compuestos de la invención son agentes terapéuticos candidatos para el tratamiento de cánceres tales como angioqueratoma; hiperplasia angiolinfoidal con eosinofilia; esclerosante angioma; angiomatosis; glomangioma; hemangioendotelioma; hemangioma; hemangiopericitoma; hemangiosarcoma; linfangioma; linfangiomioma; linfangiosarcoma; pinealoma; carcinosarcoma; condrosarcoma; cistosarcoma filodes; fibrosarcoma; hemangiosarcoma; leiomiosarcoma; leucosarcoma; liposarcoma; linfangiosarcoma; miosarcoma; mirosarcoma; carcinoma ovárico; rabdomiosarcoma; sarcoma; neoplasias; nerofibromatosis; y displasia cervical.
En una realización particular, la presente divulgación proporciona métodos para tratar tumores sólidos, cáncer de colon, cáncer de recto, cáncer colorrectal, cáncer de vejiga, cáncer gástrico, cáncer de esófago, cáncer de cabeza y cuello, síndrome de mielodisplasia, cáncer de cerebro, cáncer de SNC, glioma maligno, glioblastoma, cáncer hepatocelular, carcinoma hepatocelular, cáncer de tiroides, cáncer de pulmón, cáncer de pulmón no microcítico (CPNM), cáncer hemático, leucemia, linfoma de células B, linfoma de linfocitos T, linfoma de células pilosas, linfoma de células B difuso de grandes dimensiones, linfoma de Hodgkin, linfoma no Hodgkin, linfoma de Burkitt, cáncer de páncreas, melanoma, mieloma, mieloma múltiple, carcinoma pancreático, carcinoma de células renales, cáncer renal, cáncer cervical, cáncer urotelial, cáncer de próstata, cáncer de próstata resistente a la castración, cáncer de ovario, cáncer de mama o cáncer de mama triple negativo. De acuerdo con un método de este tipo, se puede administrar una cantidad terapéuticamente eficaz de al menos un compuesto de acuerdo con la Fórmula I o un estereoisómero, tautómero o una sal farmacéuticamente aceptable del mismo a un sujeto que ha sido diagnosticado con una enfermedad proliferativa celular, tal como un cáncer. Como alternativa, se puede administrar una composición farmacéutica que comprenda al menos un compuesto de acuerdo con la Fórmula I o un estereoisómero, tautómero o una sal farmacéuticamente aceptable del mismo a un sujeto que ha sido diagnosticado con cáncer.
En determinadas realizaciones, los compuestos de acuerdo con la invención se administran a un sujeto con cáncer junto con otras terapias de cáncer convencionales tales como radioterapia o cirugía. La radioterapia es bien conocida en la técnica e incluye terapias de rayos X, tales como radiación gamma, y terapias radiofarmacéuticas.
En determinadas realizaciones, los compuestos inhibidores de Mnk de la invención se usan con al menos un agente antineoplásico. Los agentes antineoplásicos incluyen fármacos quimioterapéuticos. Un agente quimioterapéutico incluye, pero no se limita a, un inhibidor de la función de la cromatina, un inhibidor de la topoisomerasa, un fármaco inhibidor de los microtúbulos, un agente que daña el ADN, un antimetabolito (por ejemplo, antagonistas de folato, análogos de pirimidina, análogos de purina, y análogos de azúcares modificados), un inhibidor de la síntesis de ADN, un agente interactivo con el ADN (tal como un agente intercalante) y un inhibidor de la reparación del ADN.
Los agentes quimioterapéuticos ilustrativos incluyen, sin limitación, los siguientes grupos: antimetabolitos/agentes contra el cáncer, tales como análogos de pirimidina (5-fluorouracilo, floxuridina, capecitabina, gemcitabina y citarabina) y análogos de la purina, antagonistas de folato e inhibidores relacionados (mercaptopurina, tioguanina, pentostatina y 2-clorodesoxiadenosina (cladribina)); agentes antimitóticos antiproliferativos que incluyen productos naturales tales como alcaloides de la vinca (vinblastina, vincristina y vinorelbina), disrruptores de microtúbulos tales como taxanos (paclitaxel, docetaxel), vincristina, vinblastina, nocodazol, epotilonas y navelbina, epidipodofilotoxinas (etopósido, tenipósido), agentes que dañan el ADN (actinomicina, amsacrina, antraciclinas, bleomicina, busulfán, camptotecina, carboplatino, clorambucilo, cisplatino, ciclofosfamida, Cytoxan, dactinomicina, daunorrubicina, doxorrubicina, epirrubicina, hexametilmelaminaoxaliplatino, ifosfamida, melfalán, mecloretamina, mitomicina, mitoxantrona, nitrosourea, plicamicina, procarbazina, Taxol, taxotere, temozolamida, tenipósido, trietilenotiofosforamida y etopósido (VP16)); antibióticos tales como dactinomicina (actinomicina D), daunorrubicina, doxorrubicina (adriamicina), idarrubicina, antraciclinas, mitoxantrona, bleomicinas, plicamicina (mitramicina) y mitomicina; enzimas (L-asparaginasa que metaboliza sistemáticamente la L-asparagina y priva a las células que no tienen la capacidad de sintetizar su propia asparagina); agentes antiplaquetarios; agentes alquilantes antiproliferativos/antimitóticos tales como mostazas nitrogenadas (mecloretamina, ciclofosfamida y análogos, melfalán, clorambucilo), etileniminas y metilmelaminas (hexametilmelamina y tiotepa), alquil sulfonatos-busulfán, nitrosoureas (carmustina (BCNU) y análogos, estreptozocina), trazenos-dacarbazinina (DTIC); antimetabolitos antiproliferativos/antimitóticos tales como análogos de ácido fólico (metotrexato); complejos de coordinación de platino (cisplatino, carboplatino), procarbazina, hidroxiurea, mitotano, aminoglutetimida; hormonas, análogos hormonales (estrógeno, tamoxifeno, goserelina, bicalutamida, nilutamida) e inhibidores de la aromatasa (letrozol,
anastrozol); anticoagulantes (heparina, sales de heparina sintéticas y otros inhibidores de la trombina); agentes fibrinolíticos (tales como activador del plasminógeno tisular, estreptocinasa y urocinasa), aspirina, dipiridamol, ticlopidina, clopidogrel, abciximab; agentes antimigratorios; agentes antisecretorios (breveldin); inmunosupresores (ciclosporina, tacrolimus (FK-506), sirolimus (rapamicina), azatioprina, micofenolato de mofetilo); compuestos antiangiogénicos (TNP470, genisteína) e inhibidores del factor de crecimiento (inhibidores del factor de crecimiento endotelial vascular (VEGF), inhibidores del factor de crecimiento de fibroblastos (FGF)); bloqueador de los receptores de la angiotensina; donantes de óxido nítrico; oligonucleótidos antisentido; anticuerpos (trastuzumab, rituximab); receptores de antígeno quimérico; inhibidores del ciclo celular e inductores de diferenciación (tretinoína); inhibidores de mTOR, inhibidores de topoisomerasa (doxorrubicina (adriamicina), amsacrina, camptotecina, daunorrubicina, dactinomicina, tenipósido, epirrubicina, etopósido, idarrubicina, irinotecán (CPT-11) y mitoxantrona, topotecán, irinotecán), corticosteroides (cortisona, dexametasona, hidrocortisona, metilprednisolona, prednisona y prednisolona); inhibidores de la cinasa de la transducción de señal del factor de crecimiento; inductores de disfunción mitocondrial, toxinas tales como toxina de cólera, ricina, exotoxina de Pseudomonas, toxina de adenilato ciclasa de Bordetella pertussis o toxina diftérica y activadores de caspasas; y disruptores de cromatina.
En determinadas realizaciones, un inhibidor de Mnk de acuerdo con la presente invención se usa simultáneamente, en la misma formulación o en formulaciones separadas, o secuencialmente con un agente o agentes adicionales como parte de una posología de terapia de combinación.
También son eficaces inhibidores de Mnk de acuerdo con la Fórmula I, la, Ila, IIb, IIIa, IIIb, IVa, IVb, Va, Vb, VI, VIIa y VIIb, incluyendo sus correspondientes sales y composiciones farmacéuticas de compuestos de Fórmula I, Ia, Ila, IIIb, IVa, IVb, Va, Vb, VI, VIIa y VIIb como agentes terapéuticos para tratar o prevenir trastornos mediados por citocinas, tales como inflamación en un paciente, preferentemente en seres humanos. En una realización, un compuesto o composición de acuerdo con la invención es particularmente útil para tratar o prevenir una enfermedad seleccionada entre el grupo que consiste en inflamación crónica o aguda, artritis inflamatoria crónica, artritis reumatoide, psoriasis, EPOC, enfermedad inflamatoria intestinal, choque séptico, enfermedad de Crohn, colitis ulcerosa, esclerosis múltiple y asma.
En un aspecto adicional de la invención, los compuestos de la invención o las formulaciones farmacéuticamente aceptables de los compuestos de la invención se proporcionan como inhibidores de la actividad de Mnk. Dicha inhibición se consigue poniendo en contacto una célula que expresa Mnk con un compuesto o una formulación farmacéuticamente aceptable, para disminuir o inhibir la actividad de Mnk, para proporcionar una eficacia terapéutica para una afección dependiente de Mnk en un mamífero que lo necesite.
Las dosis terapéuticamente eficaces de un compuesto de acuerdo con la Fórmula I o una composición de un compuesto de Fórmula I variarán generalmente de aproximadamente 1 a 2000 mg/día, de aproximadamente 10 a aproximadamente 1000 mg/día, de aproximadamente 10 a aproximadamente 500 mg/día, de aproximadamente 10 a aproximadamente 250 mg/día, de aproximadamente 10 a aproximadamente 100 mg/día, o de aproximadamente 10 a aproximadamente 50 mg/día. Las dosificaciones terapéuticamente eficaces se pueden administrar en una o múltiples dosis. Sin embargo, se apreciará que las dosis específicas de los compuestos de la invención para cualquier paciente particular dependerán de una diversidad de factores tales como edad, sexo, peso corporal, estado general de salud, dieta, respuesta individual del paciente a tratar, tiempo de administración, gravedad de la enfermedad a tratar, actividad del compuesto particular aplicado, forma de dosificación, modo de aplicación y medicación concomitante. La cantidad terapéuticamente eficaz para una situación dada se determinará fácilmente por experimentación de rutina y está dentro de las habilidades y criterio del médico o médico ordinario. En cualquier caso, el compuesto o composición se administrará a dosis y de una manera que permita administrar una cantidad terapéuticamente eficaz basándose en la afección única del paciente.
Síntesis
Los siguientes ejemplos se proporcionan con fines ilustrativos y no limitante.
Ejemplo 1
Síntesis de 7-(pirimidin-4-ilamino)-3,4-dihidro-1H-pirido[1,2-a]pirazin-1,6(2H)-diona (Comp. N.° 1)

Síntesis de 7-cloro-2-(4-metoxibencil)-3,4-dihidro-1H-pirido[1,2-a]pirazin-1,6-(2H)-diona (3)
A una solución de ácido 5-cloro-6-oxo-1,6-dihidropiridin-2-carboxílico (1, 0,25 g, 1,44 mmol) en acetonitrilo (10 ml), se le añadieron 2-((4-metoxibencil)amino)etanol (2, 0,31 g, 1,73 mmol) seguido de carbonato de cesio (1,18 g, 3,61 mmol) y HATU (1,26 g, 3,32 mmol) y la mezcla de reacción se agitó a 50 °C durante 16 h. La mezcla de reacción se diluyó con agua y el compuesto se extrajo en acetato de etilo. La capa orgánica se lavó con salmuera, se separó, se secó sobre sulfato de sodio y se concentró a presión reducida. El residuo obtenido se purificó por cromatografía en columna (sílice, acetato de etilo/hexanos = 60 %) para proporcionar 7-cloro-2-(4-metoxibencil)-3,4-dihidro-1 H-pirido[1,2-a]pirazin-1,6(2H)-diona (3). Rendimiento: 0,24 g, 52 %; EM (IEN) m/z 319 [M 1]+.
Síntesis de (4-metoxibencil)-7-(pirimidin-4-ilamino)-3,4-dihidro-1H-pirido[1,2-a]pirazin-1,6(2H)-diona (5)
A una solución de 7-cloro-2-(4-metoxibencil)-3,4-dihidro-1H-pirido[1,2-a]pirazin-1,6-(2H)-diona (3, 0,45 g, 1,41 mmol) en dioxano (10 ml), pirimidin-4-amina (4, 0,13 g, 1,41 mmol), se le añadieron terc-butóxido de sodio (0,41 g, 4,2 mmol) seguido de X-Phos (0,14 g, 0,28 mmol) y la mezcla de reacción se desgasificó con argón durante 5 min. Se añadió tris(dibencilidenacetona)dipaladio (0) (0,13 g, 0,14 mmol) y la mezcla de reacción se desgasificó con argón durante otros 5 minutos y se agitó a 110 °C durante 16 h. La mezcla de reacción se diluyó con agua y el compuesto se extrajo en acetato de etilo. La capa orgánica se lavó con salmuera, se separó, se secó sobre sulfato de sodio y se concentró a presión reducida. El residuo obtenido se purificó por cromatografía en columna (sílice, metanol/diclorometano = 5 %) para proporcionar 2-(4-metoxibencil)-7-(pirimidin-4-ilamino)-3,4-dihidro-1H-pirido[1,2-a]pirazin-1,6(2H)-diona (5) en forma de un sólido de color amarillo. Rendimiento: 0,42 g, 79 %; EM (IEN) m/z 378 [M 1]+.
Síntesis de 7-(pirimidin-4-ilamino)-3,4-dihidro-1H-pirido[1,2-a]pirazin-1,6(2H)-diona (Comp. N.° 1)
Una solución de 2-(4-metoxibencil)-7-(pirimidin-4-ilamino)-3,4-dihidro-1H-pirido[1,2-a]pirazin-1,6(2H)-diona (5, 0,3 g, 0,79 mmol) en ácido trifluoroacético (5 ml) se calentó a 90 °C durante 48 h. La mezcla de reacción se enfrió a temperatura ambiente y se concentró a presión reducida. El residuo se neutralizó con solución saturada de bicarbonato de sodio y el compuesto se extrajo en metanol al 10 % en acetato de etilo. La capa orgánica se separó, se secó sobre sulfato de sodio, se concentró a presión reducida y el residuo obtenido se purificó por cromatografía en columna (sílice, metanol/diclorometano = 10 %) para proporcionar 7-(pirimidin-4-ilamino)-3,4-dihidro-1H-pirido[1,2-a]pirazin-1,6(2H)-diona (Comp. N.° 1) en forma de un sólido de color beige. Rendimiento: 0,12 g, 59 %; EM (IEN) m/z 258 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 9,45 (s, 1H), 8,79-8,64 (m, 2H), 8,48 (s, 1H), 8,38 (d, J = 5,9 Hz, 1H), 7,41 (dd, J = 6,0, 1,3 Hz, 1H), 7,11 (d, J = 7,9 Hz, 1H), 4,23-4,16 (m, 2H), 3,55-3,48 (m, 2H).
Ejemplo 2
Síntesis de 3,3-dimetil-6-(pirimidin-4-ilamino)-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona (Comp. N.° 2)

Síntesis de 5-cloro-6-oxo-1,6-dihidropiridin-2-carboxamida (2)
Se añadió amoníaco acuoso (15 ml, solución al 30 %) a 5-cloro-6-oxo-1,6-dihidropiridin-2-carboxilato de etilo (1, 0,65 g, 3,2 mmol) a 0 °C y se permitió que la mezcla de reacción se agitara a temperatura ambiente durante 16 h. La mezcla de reacción se concentró a presión reducida y el residuo se trituró con éter dietílico, se filtró y se secó para proporcionar 5-cloro-6-oxo-1,6-dihidropiridin-2-carboxamida (2). Rendimiento: 0,43 g, 75 %; EM (IEN) m/z 173 [M 1]+.
Síntesis de 6-cloro-3,3-dimetil-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona (4)
Procedimiento A: A una solución de 5-cloro-6-oxo-1,6-dihidropiridin-2-carboxamida (2, 1,4 g, 7,9 mmol) en 1,4-dioxano (20 ml), se le añadieron acetona (3,6 g, 79 mmol) y ácido sulfúrico concentrado (0,038 g, 0,39 mmol) a temperatura ambiente y la mezcla de reacción se dejó calentar a 100 °C durante 8 h. La mezcla de reacción se concentró a presión reducida y el residuo se trituró con éter dietílico y hexano, se filtró y se secó para proporcionar 6-cloro-3,3-dimetil-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona (4). Rendimiento: 1,4 g, 83 %; EM (IEN) m/z 213 [M 1]+.
Síntesis de 3,3-dimetil-6-(pirimidin-4-ilamino)-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona (Comp. N.° 2)
Procedimiento B: A una solución de 6-cloro-3,3-dimetil-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona (4, 0,25 g, 1,18 mmol) en 1,4-dioxano (8 ml), se le añadieron pirimidin-4-amina (5, 0,14 g, 1,41 mmol), Brettphos (0,19 g, 0,23 mmol) y carbonato de cesio (0,76 g, 2,36 mmol) y la mezcla de reacción se desgasificó con argón durante 5 minutos. Se añadió tris-dibencilidenacetona dipaladio (0) (0,11 g, 0,12 mmol). La reacción se desgasificó con argón durante otros 5 minutos y después se agitó a 100 °C durante 10 h. La mezcla de reacción se enfrió a temperatura ambiente, se filtró a través de celite y el filtrado se concentró a presión reducida. El residuo se purificó por cromatografía en columna de gel de sílice usando metanol al 5 % en diclorometano para proporcionar 3,3-dimetil-6-(pirimidin-4-ilamino)-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona (Comp. N.° 2) en forma de un sólido de color amarillo claro. Rendimiento: 0,036 g, 11 %; EM (IEN) m/z 272 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 9,70 (s, 1H), 9,42 (s, 1H), 8,81-8,73 (m, 2H), 8,37 (d, J = 5,9 Hz, 1H), 7,40-7,34 M, 1H), 6,87 (d, J = 7,7 Hz, 1H), 1,82 (s, 6H).
Ejemplo 3
Síntesis de 3-(4-fluorobencil)-3-metil-6-(pirimidin-4-ilamino)-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona (Comp. N.° 3)

Síntesis de 6-cloro-3-(4-fluorobencil)-3-metil-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona (3)
La síntesis del intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento A. Rendimiento: 0,17 g, 49 %; EM (IEN) m/z 307 [M 1]+.
Síntesis de 3-(4-fluorobencil)-3-metil-6-(pirimidin-4-ilamino)-2,3-dihidroimidazol-1,5-a]piridin-1,5-diona (Comp. N ° 3) La síntesis del compuesto 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento B. Sólido de color blanquecino; Rendimiento: 0,012 g, 40 %; EM (IEN) m/z 366 [M 1]+; RMN 1H (400 MHz, DMSO-d6) 59,66 (s, 1H), 9,50 (s, 1H), 8,75 (s, 1H), 8,68 (d, J = 7,6 Hz, 1H), 8,40 (d, J = 5,9 Hz, 1H), 7,04-6,92 (m, 4H), 6,57 (d, J = 7,7 Hz, 1H), 3,99 (d, J = 13,9 Hz, 1H), 3,07 (d, J = 13,9 Hz, 1H), 1,97 (s, 3H).
Ejemplo 4
Síntesis de 3-(4-clorobencil)-3-metil-6-(pirimidin-4-ilamino)-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona (Comp. N.° 4)
Síntesis de 6-cloro-3-(4-clorobencil)-3-metil-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona (3)
La síntesis del intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento A. Rendimiento: 0,278 g, 59 %; EM (IEN) m/z 323 [M 1]+.
Síntesis de 3-(4-clorobencil)-3-metil-6-(pirimidin-4-ilamino)-2,3-dihidroimidazo-[1,5-a]piridin-1,5-diona (Comp. N ° 4) La síntesis del compuesto 4 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento B. Sólido de color castaño; Rendimiento: 0,18 g, 59 %; EM (IEN) m/z 382 [M 1]+; RMN 1H (400 MHz, DMSO-d6) 59,68 (s, 1H), 9,51 (s, 1H), 8,75 (s, 1H), 8,68 (d, J = 7,6 Hz, 1H), 8,40 (d, J= 5,9 Hz, 1H), 7,41 (d, J= 6,1 Hz, 1H), 7,20-7,01 (m, 1H), 6,99-6,92 (m, 2H), 6,77 (dd, J= 8,6, 5,0 Hz, 1H), 6,58 (d, J= 7,6 Hz, 1H), 4,02 (d, J = 13,8 Hz, 1H), 3,12 (d, J = 13,8 Hz, 1H), 1,98 (s, 3H).
Ejemplo 5
Síntesis de 3-(3-fluorobencil)-3-metil-6-(pirimidin-4-ilamino)-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona (Comp. N.° 5)
Síntesis de 6-cloro-3-(3-fluorobencil)-3-metil-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona (3)
La síntesis del intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento A. Rendimiento: 0,265 g, 59 %; EM (IEN) m/z 307 [M 1]+.
Síntesis de 3-(3-fluorobencil)-3-metil-6-(pirimidin-4-ilamino)-2,3-dihidroimidazo-[1,5-a]piridin-1,5-diona (Comp. N ° 5) La síntesis del compuesto 5 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento B. Sólido de color beige; Rendimiento: 0,17 g, 56 %; EM (IEN) m/z 366 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 9,68 (s, 1H), 9,51 (s, 1H), 8,75 (s, 1H), 8,68 (d, J = 7,6 Hz, 1H), 8,40 (d, J= 5,9 Hz, 1H), 7,41 (d,/ = 6,1 Hz, 1H), 7,2-7,08 (m, 1H), 6,99-6,91 (m, 1H), 6,77 (dd, J= 8,6, 5,0 Hz, 2H), 6,58 (d, J= 7,6 Hz, 1H), 4,02 (d, J= 13,8 Hz, 1H), 3,12 (d, J = 13,8 Hz, 1H), 1,98 (s, 3H).
Ejemplo 6
Síntesis de 6'-(pirimidin-4-ilamino)-1'H-espiro[ciclopentan-1,3'-imidazo[1,5-a]piridin]-1',5'(2'H)-diona (Comp. N.° 6)
Síntesis de 6'-cloro-1 ’H-espiro[ciclopentan-1,3'-imidazo[1,5-a]piridin]-15'(2'H)-diona (3)
La síntesis del producto intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento A. Rendimiento: 0,18 g, 42 %; EM (IEN) m/z 239 [M 1]+.
Síntesis de 6'-(pirimidin-4-ilamino)-1-espiro[ciclopentano-1,3'-imidazo[1,5-a]-piridina]-1',5'(2'H)-diona (Comp. N ° 6) La síntesis del compuesto 6 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento B. Sólido de color marrón claro; Rendimiento: 0,1 g, 45 %; EM (IEN) m/z 298,10 [M 1]+; RMN 1H
(400 MHz, DMSO-cfe) 810,01 (s, 1H), 9,43 (s, 1H), 8,82-8,73 (m, 2H), 8,37 (d, J = 5,9 Hz, 1H), 7,37 (d, J = 5,9 Hz, 1H), 6,88 (d, J = 7,6 Hz, 1H), 2,89-2,81 (m, 2H), 2,08-1,92 (m, 2H), 1,96-1,70 (m, 4H).
Ejemplo 7
Síntesis de 3-metil-6-(pirimidin-4-ilamino)-3-(2,2,2-trifluoroetil)-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona (Comp. N.° 7)


Síntesis de 6-cloro-3-metil-3-(2',2,2-trifluoroetil)-2,3-dihidroimidazo[1,5-a]piridina-1,5-diona (3)
La síntesis del intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento A. Rendimiento: 0,12 g, 22 %.
Síntesis de 3-metil-6-(pirimidin-4-ilamino)-3-(2,2,2-trifluoroetil)-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona (Comp. N.° 7)
La síntesis del compuesto 7 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento B. Sólido de color verde-amarillo; Rendimiento: 0,020 g, 15 %; EM (IEN) m/z 340 [M 1]+; RMN 1H (400 MHz, DMSO-d6) 8 10,69 (s, 1H), 9,42 (s, 1H), 8,70 (dd, J = 4,5, 1,7 Hz, 1H), 8,79 (s, 1H), 8,30-8,20 (m, 1H), 7,73 (d, J = 1,1 Hz, 1H), 7,32 (dd, J = 9,2, 4,4 Hz, 1H), 3,38-3,75 (m, 1H), 3,19-3,08 (m, 1H), 1,81 (s, 3H).
Ejemplo 8
Síntesis de 3-isopropil-3-metil-6-(pirimidin-4-ilamino)-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona (Comp. N.° 8)

Síntesis de 6-cloro-3-isopropil-3-metil-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona (3)
La síntesis del intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento A. Rendimiento: 0,32 g, 77 %.
Síntesis de 3-isopropil-3-metil-6-(pirimidin-4-ilamino)-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona (Comp. N.° 8) La síntesis del compuesto 8 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento B. Sólido de color blanquecino; Rendimiento: 0,036 g, 19 %; EM (IEN) m/z 300 [M 1]+; RMN 1H (400 MHz, DMSO-cfe) ó 9,67 (s, 1H), 9,41 (s, 1H), 8,83-8,73 (m, 2H), 8,37 (d, J = 5,9 Hz, 1H), 7,37 (d, J = 5,9 Hz, 1H), 6,87 (d, J = 7,6 Hz, 1H), 3,11-2,90 (m, 1H), 1,83 (s, 3H), 1,05 (d, J = 7,0 Hz, 3H), 0,46 (d, J = 6,6 Hz, 3H).
Ejemplo 9
Síntesis de 3-ciclopentil-3-metil-6-(pirimidin-4-ilamino)-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona (Comp. N.° 9)


Síntesis de 6-cloro-3-ciclopentil-3-metil-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona (3)
La síntesis del intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento A. Rendimiento: 0,2 g, 47 %; EM (IEN) m/z 267 [M 1]+.
Síntesis de 3-ciclopentil-3-metil-6-(pirimidin-4-ilamino)-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona (Comp. N.° 9) La síntesis del compuesto 9 se realizó como se ha descrito anteriormente usando el protocolo general del procedimiento B. Sólido de color beige; Rendimiento: 0,12 g, 47 %; EM (IEN) m/z 326 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 9,72 (s, 1H), 9,39 (s, 1H), 8,81-8,73 (m, 2H), 8,37 (d, J = 5,9 Hz, 1H), 7,39-7,32 (m, 1H), 6,86 (d, J = 7,7 Hz, 1H), 3,45-3,40 (m, 1H), 1,84 (s, 3H), 1,68- 1,35 (m, 4H), 1,18-1,1 (m, 1H), 0,85-0,80 (m, 1H).
Ejemplo 10
Síntesis de N-(6,3-dimetil-1,5-dioxo-1,2,5,5-tetrahidroimidazo[1,5-a]piridin-6-il)amino)pirimidin-4-il)ciclopropancarboxamida (Comp. N.° 10)



Síntesis de N-(6-(3,3-dimetil-1,5-dioxo-1,2,3,5-tetrahidroimidazo[1,5-a]piridin-6-il)-amino)pirimidin-4-il)ciclopropancarboxamida (Comp. N.° 10)
La síntesis del compuesto 10 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento B. Sólido de color beige; Rendimiento: 0,075 g, 15 %; EM (IEN) m/z 355 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,87 (s, 1H), 9,68 (s, 1H), 9,20 (s, 1H), 8,64 (d, J = 7,7 Hz, 1H), 8,51 (s, 1H), 7,88 (d, J = 1,0 Hz, 1H), 6,85 (d, J = 7,6 Hz, 1H), 2,02 (m, J = 1H), 1,80 (s, 6H), 0,84 (d, J = 6,1 Hz, 4H).
Ejemplo 11
Síntesis de 3-(4-fluorofenil)-3-metil-6-(pirimidin-4-ilamino)-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona (Comp. N.° 11)

Síntesis de 6-cloro-3-(4-fluorofenil)-3-metil-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona (3)
La síntesis del intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento A. Rendimiento: 0,216 g, 52 %; EM (IEN) m/z 293 [M 1]+.
Síntesis de 3-(4-fluorofenil)-3-metil-6-(pirimidin-4-ilamino)-2,3-dihidroimidazo-[1,5-a]piridin-1,5-diona (Comp. N ° 11) La síntesis del compuesto 11 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento B. Sólido de color marrón; Rendimiento: 0,14 g, 41 %; EM (IEN) m/z 252,15 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,05 (s, 1H), 9,35 (s, 1H), 8,83-8,73 (m, 2H), 8,35 (d, J = 5,9 Hz, 1H), 7,44 (d, J = 8,5, 5,2 Hz, 2H), 7,31-7,16 (m, 3H), 6,99 (d, J = 7,6 Hz, 1H), 2,26 (s, 3H).
Ejemplo 12
Síntesis de 3-(3-fluorofenil)-3-metil-6-(pirimidin-4-ilamino)-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona (Comp. N.° 12)

Síntesis de 6-cloro-3-(3-fluorofenil)-3-metil-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona (3)
La síntesis del intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento A. Rendimiento: 0,208 g, 50 %; EM (IEN) m/z 293 [M 1]+.
Síntesis de 3-(3-fluorofenil)-3-metil-6-(pirimidin-4-ilamino)-2,3-dihidroimidazo-[1,5-a]piridin-1,5-diona (Comp. N ° 12) La síntesis del compuesto 12 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento B. Sólido de color amarillo; Rendimiento: 0,08 g, 34 %; EM (IEN) m/z 252,25 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,07 (s, 1H), 9,34 (s, 1H), 8,84-8,73 (m, 2H), 8,35 (d, J = 5,9 Hz, 1H), 7,43 (m, 1H), 7,32 7,12 (m, 4H), 7,00 (d, J = 7,6 Hz, 1H), 2,26 (s, 3H).
Ejemplo 13
Síntesis de 3-(4-clorofenil)-3-metil-6-(pirimidin-4-ilamino)-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona (Comp. N.° 13)

Síntesis de 6-cloro-3-(4-clorofenil)-3-metil-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona (3)
La síntesis del intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento A. Rendimiento: 0,17 g, 31 %; EM (IEN) m/z 309 [M 1]+.
Síntesis de 3-(4-clorofenil)-3-metil-6-(pirimidin-4-ilamino)-2,3-dihidroimidazol-[1,5-a]piridin-1,5-diona (Comp. N ° 13) La síntesis del compuesto 13 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento B. Sólido de color amarillo; Rendimiento: 0,11 g, 55 %; EM (IEN) m/z 368,15 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,05 (s, 1H), 9,32 (s, 1H), 8,84-8,73 (m, 2H), 8,35 (d, J = 5,9 Hz, 4H), 7,42 (m, 4H), 7,27 (dd, J = 5,9, 1,3 Hz, 1H), 7,00 (d, J = 7,7 Hz, 1H), 2,25 (s, 3H).
Ejemplo 14
Síntesis de 3-metil-6-(pirimidin-4-ilamino)-3-(trifluorometil)-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona (Comp. N.° 14)


Síntesis de 6-cloro-3-metil-3-(trifluorometil)-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona (3)
La síntesis del intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento A. Rendimiento: 0,255 g, 66 %; EM (IEN) m/z 267 [M 1]+
Síntesis de 3-metil-6-(pirimidin-4-ilamino)-3-(trifluorometil)-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona (Comp. N ° 14) La síntesis del compuesto 14 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento B. Sólido de color blanquecino; Rendimiento: 0,06 g, 19 %; EM (IEN) m/z 326 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,50 (s, 1H), 9,58 (s, 1H), 8,84-8,76 (m, 2H), 8,41 (d, J = 5,9 Hz, 1H), 7,40 (dd, J = 5,9, 1,4 Hz, 1H), 7,02 (d, J = 7,7 Hz, 1H), 2,14 (s, 3H).
Ejemplo 15
Síntesis de 3-(aminometil)-3-metil-6-(pirimidin-4-ilamino)-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona (Comp. N.° 15)

Síntesis de 6-cloro-3-((1,3-dioxoisoindolin-2-il)metil)-3-metil-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona (3)
La síntesis del intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento A. Rendimiento: 0,5 g, 50 %; EM (IEN) m/z 358 [M 1]+.
Síntesis de 3-((1,3-dioxoisoindolin-2-il)metil)-3-metil-6-(pirimidin-4-ilamino)-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona (5)
La síntesis del intermedio 5 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento B. Rendimiento: 0,09 g, 26 %; EM (IEN) m/z 417 [M 1]+.
Síntesis de 3-(aminometil)-3-metil-6-(pirimidin-4-ilamino)-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona (Comp. N.° 15) Procedimiento C: A una solución de 3-((1,3-dioxoisoindolin-2-il)metil)-3-metil-6-(pirimidin-4-ilamino)-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona (5, 0,085 g, 0,29 mmol) en metanol (20 ml), se le añadió hidrato de hidrazina (2 ml) y la mezcla de reacción se agitó a temperatura ambiente durante 16 h. La mezcla de reacción se filtró y el filtrado se concentró a presión reducida. El residuo se disolvió en diclorometano y se lavó con agua, se separó, se secó sobre sulfato de sodio y se concentró a presión reducida, que se purificó mediante lavado repetido con éter dietílico y hexano para obtener 3-(aminometil)-3-metil-6-(pirimidin-4-ilamino)-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona (Comp. N.° 15) en forma de un sólido de color blanquecino. Rendimiento: 0,008 g, 9 %; EM (IEN) m/z 287,05 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 9,35 (s, 1H), 8,76 (d, J = 7,8 Hz, 2H), 8,36 (d, J = 5,9 Hz, 1H), 7,36 (d, J= 6,0 Hz, 1H), 6,83 (d, J = 7,6 Hz, 1H), 3,55 (d, J = 13,6 Hz, 1H), 2,92 (d, J = 13,7 Hz, 1H), 1,75 (s, 3H), 1,44 (s, 1H), 1,41-1,34 (m, 2H).
Ejemplo 16
Síntesis de 3-metil-6-(pirimidin-4-ilamino)-3-(tiofen-3-il)-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona (Comp. N.° 16)

A una solución de 5-cloro-6-oxo-1,6-dihidropiridin-2-carboxamida (1, 0,1 g, 0,58 mmol) en acetonitrilo (4 ml), se le añadieron 1-(tiofen-3-il)etanona (2, 0,37 g, 2,9 mmol) y cloruro férrico (0,094 g, 0,58 mmol) y la mezcla de reacción se dejó calentar a 90 °C durante 18 h. La mezcla de reacción se filtró y el filtrado se concentró a presión reducida. El residuo se purificó por cromatografía en columna (sílice, acetato de etilo/hexanos = 50 %) para proporcionar 6-cloro-3-metil-3-(tiofen-3-il)-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona (3). Rendimiento: 0,021 g, 13 %; EM (IEN) m/z 281 [M 1]+
Síntesis de 3-metil-6-(pirimidin-4-ilamino)-3-(tiofen-3-il)-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona (Comp. N.° 16) La síntesis del compuesto 16 se realizó como se ha descrito anteriormente usando el protocolo general del procedimiento B. Sólido de color beige; Rendimiento: 0,008 g, 12 %; EM (IEN) m/z 340 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,05 (s, 1H), 9,35 (s, 1H), 8,81-8,73 (m, 2H), 8,35 (d, J = 5,9 Hz, 1H), 7,74-7,68 M, 1H), 7,49 (dd, J = 5,1, 2,9 Hz, 1H), 7,29 (d, J = 5,9 Hz, 1H), 7,00-6,92 (m, 2H), 2,25 (s, 3H).
Ejemplo 17
Síntesis de 3-metil-6-(pirimidin-4-ilamino)-3-(tiazol-4-il)-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona (Comp. N.° 17)

Síntesis de 6-cloro-3-metil-3-(tiazol-4-il)-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona (3)
La síntesis del intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento A. Rendimiento: 0,2 g, 49 %; EM (IEN) m/z 282 [M 1]+.
Síntesis de 3-metil-6-(pirimidin-4-ilamino)-3-(tiazol-4-il)-2,3-dihidroimidazo[1,5-a]-piridin-1,5-diona (Comp. N.° 17) La síntesis del compuesto 17 se realizó como se ha descrito anteriormente usando el protocolo general del procedimiento B. Sólido de color amarillo. Rendimiento: 0,013 g, 21 %; EM (IEN) m/z 341 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 9,97 (s, 1H), 9,43 (s, 1H), 8,99 (d, J = 1,8 Hz, 1H), 8,83-8,71 (m, 2H), 8,35 (d, J = 6,0 Hz, 1H), 8,07 (d, J = 1,9 Hz, 1H), 7,27 (d, J = 6,0 Hz, 1H), 6,94 (d, J = 7,6 Hz, 1H), 2,30 (s, 3H).
Ejemplo 18
Síntesis de 3-(3-clorofenil)-3-metil-6-(pirimidin-4-ilamino)-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona (Comp. N.° 18)
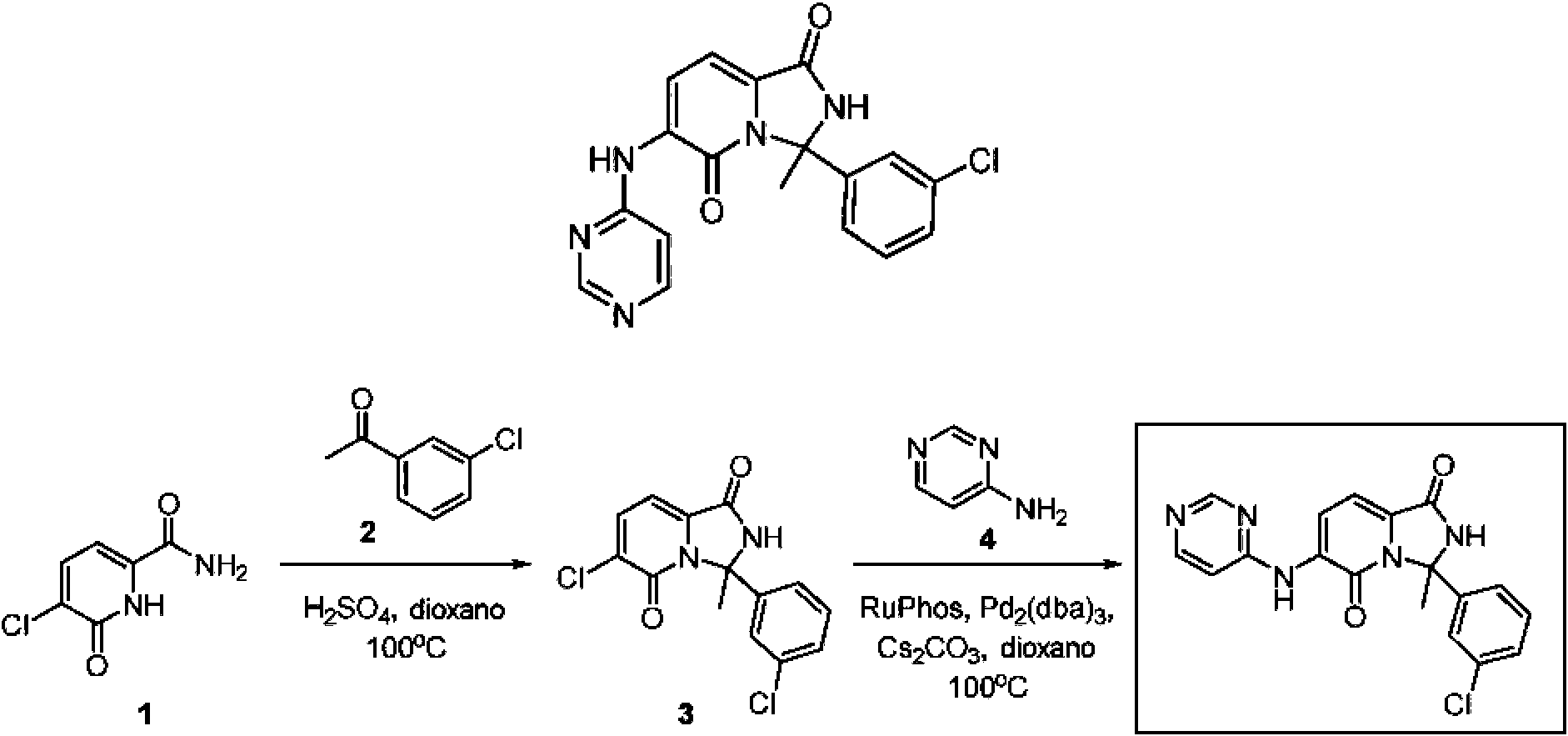
Síntesis de 6-cloro-3-(3-clorofenil)-3-metil-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona (3)
La síntesis del intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento A. Rendimiento: 0,210 g, 39 %; EM (IEN) m/z 309 [M 1]+.
Síntesis de 3-(3-clorofenil)-3-metil-6-(pirimidin-4-ilamino)-2,3-dihidroimidazo-[1,5-a]piridin-1,5-diona (Comp. N ° 18) La síntesis del compuesto 18 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento B. Sólido de color amarillo claro; Rendimiento: 0,053 g, 22 %; EM (IEN) m/z 368 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,07 (s, 1H), 9,35 (s, 1H), 8,86-8,73 (m, 2H), 8,35 (d, J = 5,9 Hz, 1H), 7,51-7,34 M, 3H), 7,34-7,23 (m, 2H), 7,09 (s, 1H), 7,01 (d, J = 7,6 Hz, 1H), 2,25 (s, 3H).
Ejemplo 19
Síntesis de 3-(3-clorobencil)-3-metil-6-(pirimidin-4-ilamino)-2,3-dihidroimidazo[1,5-a]piridin-1, 5-diona (Comp. N.° 19)

S ín te s is de 6 -c lo ro -3 -(3 -c lo ro b e n c il)-3 -m e til-2 ,3 -d ih id ro im id a z o [1 ,5 -a ]p ir id in -1 ,5 -d io n a ( 3 )
La síntesis del intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento A. Rendimiento: 0,4 g, 70 %; EM (IEN) m/z 324 [M 1]+
Síntesis de 3-(3-clorobencil)-3-metil-6-(pirimidin-4-ilamino)-2,3-dihidroimidazo-[1,5-a]piridin-1,5-diona (Comp. N ° 19) La síntesis del compuesto 19 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento B. Sólido de color blanquecino; Rendimiento: 0,2 g, 56 %; EM (IEN) m/z 3 8 2 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 9,67 (s, 1H), 9,49 (s, 1H), 8,75 (s, 1H), 8,68 (d, J = 7,7 Hz, 1H), 8,39 (d, J = 5,9 Hz, 1H), 7,41 (d, J = 5,9 Hz, 1H), 7,24-7,12 (m, 2H), 7,04 (d, J = 2,3 Hz, 1H), 6,87 (dd, J = 6,8, 2,1 Hz, 1H), 6,58 (d, J = 7,7 Hz, 1H), 4,00 (d, J = 13,8 Hz, 1H), 3,11 (d, J = 13,8 Hz, 1H), 1,97 (s, 3H).
Ejemplo 20
Síntesis de 6-(pirimidin-4-ilamino)-1H-espiro[imidazo[1,5-a]piridin-3,3'-piperidin]-1,5(2H)-diona (Comp. N.° 20)

Síntesis de 6-cloro-2H-espiro[imidazo[1,5-a]piridina-3,3'-piperidin]-1, 5-diona (3)
La síntesis del intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento A. Rendimiento: 0,504 g, producto en bruto; EM (IEN) m/z 254 [M 1]+.
Síntesis de 6-cloro-1,5-dioxo-2,5-dihidro-1H-espiro[imidazo[1,5-a]piridin-3,3’-piperidin-1’-carboxilato de terc-butilo (4) A una solución de 1'-bencil-6-cloro-1H-espiro[imidazo[1,5-a]piridin-3,3'-piperidin]-1,5(2H)-diona (3, 0,5 g, 1,9 mmol) en tetrahidrofurano (8 ml) y agua (4 ml), se le añadieron bicarbonato de sodio (0,66 g, 7,8 mmol) y dicarbonato de diterciariobutilo (1,31 ml, 5,9 mmol) a 0 °C y la mezcla de reacción se agitó a temperatura ambiente a 2 h. La mezcla de reacción se diluyó con agua y el compuesto se extrajo en acetato de etilo. La capa orgánica se separó, se secó sobre sulfato de sodio y se concentró a presión reducida. El residuo se purificó por cromatografía en columna (sílice, metanol/diclorometano = 2 %) para proporcionar 6-cloro-1,5-dioxo-2,5-dihidro-1H-espiro[imidazo[1,5-a]piridin-3,3'-piperidin]-carboxilato de terc-butilo 4. Rendimiento: 0,33 g, 47 %; EM (IEN) m/z 354 [M 1]+.
Síntesis de 1,5-dioxo-6-(pirimidin-4-ilamino)-1,5-dihidro-2H-espiro[imidazo[1,5-a]piridin-3,3'-piperidin]-1 ’-carboxilato de terc-butilo (6)
La síntesis del intermedio 6 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento B. Rendimiento: 0,126 g, 31 %; EM (IEN) m/z 413 [M 1]+.
Síntesis de 6-(pirimidin-4-ilamino)-1H-espiro[imidazo[1,5-a]piridin-3,3'-piperidin-1,5(2H)-diona (Comp. N ° 20)
Procedimiento D: A una solución agitada de 6-(pirimidin-4-ilamino)-1H-espiro[imidazo[1,5-a]piridin-3,3'-piperidin]-1,5(2H)-diona (5, 0,12 g, 0,29 mmol) en dioxano (1 ml), se le añadió cloruro de hidrogeno 4 M en dioxano (2 ml) a 0 °C y la mezcla de reacción se agitó a temperatura ambiente durante 3 h. El disolvente se retiró a presión reducida y el residuo se purificó por lavado repetido con pentano. Se disolvió el compuesto en agua y se hizo pasar a través de una columna de estratos para obtener 6-(pirimidin-4-ilamino)-1H-espiro[imidazo[1,5-a]piridin-3,3'-piperidin]-1,5(2H)-diona (Comp. N.° 20) en forma de un sólido de color marrón claro. Rendimiento: 0 , 0 6 2 g, 67 %; EM (IEN) m/z 313,15 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,13 (s, 1H), 9,40 (s, 1H), 8,80-8,73 (m, 2H), 8,38 (d, J = 5,9 Hz, 1H), 7,37 (dd, J = 5,9, 1,3 Hz, 1H), 6,89 (d, J = 7,7 Hz, 1H), 3,94 (d, J = 12,5 Hz, 1H), 3,16-3,12 (m, 1H), 2,95 (d, J = 13,1 Hz, 1H), 2,74-2,65 (m, 1H), 2,60 (s, 1H), 2,46 (d, J = 11,7 Hz, 1H), 1,72 (d, J = 8,2 Hz, 2H), 1,61 (d, J = 13,0 Hz, 1H).
Ejemplo 21
Síntesis de W-[6-[(1,5-dioxospiro[2H-imidazo[1,5-a]piridin-3,1'-ciclopentan]-6-il)amino]pirimidin-4-il]ciclopropancarboxamida (Comp. N.° 21)


Síntesis de 6-doroespiro[2H-imidazo[1,5-a]piridin-3,1'-ciclopentano]-1,5-diona (3)
La síntesis del intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento A. Sólido de color blanquecino; Rendimiento: 0,8 g, 58 %; RMN 1H (400 MHz, DMSO-d6) ó 10,33 (s, 1H), 7,94 (m, 1H), 6,72 (m, 1H), 2,76 (m, 2H), 1,95 (m, 2H), 1,82 (m, 2H), 1,70 (m, 2H).
Síntesis de N-[6-[(1,5-dioxospiro[2H-imidazo[1,5-a]piridin-3,1 '-ciclopentano-6-il)amino]pirimidin-4-il]ciclopropancarboxamida (Comp. N ° 21)
La síntesis del compuesto 21 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento B. Sólido de color blanquecino; Rendimiento: 0,12 g, 15 %; EM (IEN) m/z 381,13 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,87 (s, 1H), 10,01 (s, 1H), 9,24 (s, 1H), 8,65 (d, J = 7,6 Hz, 1H), 8,50 (s, 1H), 7,87 (s, 1H), 6,85 (d, J = 7,6 Hz, 1H), 2,80 (m, 2H), 2,01 (m, 3H), 1,86 (s, 2H) M, 4H).
Ejemplo 22
Síntesis de W-(6-((3-metil-1,5-dioxo-3-(2,2,2-trifluoroetil)-2,3,5-tetrahidroimidazo[1,5-a]piridin-6-il)amino)pirimidin-4-il)ciclopropancarboxamida (Comp. N.° 22)


Síntesis de 6-cloro-3-metil-3-(2,2,2-trifluoroetil)-2H-imidazo[1,5-a]piridin-1,5-diona (3)
La síntesis del intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento A. Sólido de color marrón; Rendimiento: 0,35 g, 43 %; RMN 1H (400 MHz, DMSO-d6) 5 10,15 (s ancho, 1H), 7,99 (d, J = 4 Hz, 1H), 6,79 (d, J = 4 Hz, 1H), 3,74-3,65 (m, 1H), 3,17-3,01 (m, 1H), 1,84 (s, 3H).
Síntesis de N-[6-[[3-metil-1,5-dioxo-3-(2,2,2-trifluoroetil)-2H-imidazo[1,5-a]piridin-6-il]amino]pirimidin-4-il]ciclopropancarboxamida (Comp. N.° 22)
La síntesis del compuesto 22 se realizó como se ha descrito anteriormente usando el protocolo general del procedimiento B. Sólido de color marrón; Rendimiento: 0,2 g, 28 %; EM (IEN) m/z 423,1 [M 1]+; RMN 1H (400 MHz, DMSO-d6) 510,88 (s, 1H), 9,80 (s, 1H), 9,29 (s, 1H), 8,69 (d, J = 7,6 Hz, 1H), 8,51 (s, 1H), 7,90 (s, 1H), 6,93 (d, J = 7,6 Hz, 1H), 3,82-3,70 (m, 1H), 3,16-3,04 (m, 1H), 2,07-1,99 (m, 1H), 1,80 (s, 3H), 0,84 (d, J = 6,0 Hz, 4H).
Ejemplo 23
Síntesis de N-[6-[(3-ciclopentil-3-metil-1,5-dioxo-2H-imidazo[1,5-a]piridin-6-il]amino]pirimidin-4-il]ciclopropancarboxamida (Comp. N.° 23)


Síntesis de 6-cloro-3-ciclopentil-3-metil-2H-imidazo[1,5-a]-piridin-1,5-diona (3)
La síntesis del producto intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento A. Sólido de color blanquecino; Rendimiento: 1,61 g, 52 %; EM (IEN) m/z 267,0 [M 1]+; RMN 1H (400 MHz, CDCl3) ó 8,61 (s, 1H), 7,71 (d, J = 7,2 Hz, 1H), 6,77 (d, J = 7,2 Hz, 1H), 3,54 (m, 1H), 1,98 (s, 3H), 1,68 (m, 2H), 1,55 (m, 2H), 1,34 (m, 2H), 0,91 (m, 1H).
Síntesis de N-[6-[(3-ciclopentil-3-metil-1,5-dioxo-2H-imidazo[1,5-a]piridin-6-il]-amino]pirimidin-4-il]ciclopropancarboxamida (Comp. N.° 23)
La síntesis del compuesto 23 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento B. Sólido de color blanquecino; Rendimiento: 1,4 g, 57 %; EM (IEN) m/z 409,36 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,88 (s, 1H), 9,69 (s, 1H), 9,17 (s, 1H), 8,63 (d, J = 7,6 Hz, 1H), 7,85 (s, 1H), 6,83 (d, J = 7,6 Hz, 1H), 3,39 (m, 1H), 2,02 (m, 1H), 1,83 (s, 3H), 1,82 (m, 1H), 1,51 (m, 5H), 1,10 (m, 1H), 0,83 (d, J = 6,0 Hz, 4H), 0,78 (m, 1H).
Ejemplo 24
Síntesis de W-[6-[[3-metil-1,5-dioxo-3-(trifluorometil)-2H-imidazo[1,5-a]piridin-6-il]amino]pirimidin-4-il]ciclopropancarboxamida (Comp. N.° 24)

Síntesis de 6-cloro-3-metil-3-(trifluorometil)-2H-imidazo[1,5-a]piridin-1,5-diona (3)
La síntesis del producto intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento A. Sólido de color blanquecino; Rendimiento: 6,50 g, 84 %; EM (IEN) m/z 267,11 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,84 (s, 1H), 8,10 (d, J = 7,2 Hz, 1H), 6,85 (d, J = 7,2 Hz, 1H), 2,09 (s, 3H).
Síntesis de N[6-[[3-metil-1,5-dioxo-3-(trifluorometil)-2H-imidazo[1,5-a]piridin-6-il]amino]pirimidin-4-il]ciclopropancarboxamida (Comp. N ° 24)
La síntesis del compuesto 24 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento B. Sólido de color amarillo claro; Rendimiento: 1,75 g, 64 %; EM (IEN) m/z 409,29 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,90 (s, 1H), 10,48 (s, 1H), 9,43 (s, 1H), 8 , 6 8 (d, J = 7,6 Hz, 1H), 8,53 (s, 1H), 7,94 (s, 1H), 6,97 (d, J = 8,0 Hz, 1H), 2,17 (s, 3H), 2,02 (m, 1H), 0,84 (d, J = 6,0 Hz, 4H).
Ejemplo 25
Síntesis de 3-metil-3-((metilamino)metil)-6-(pirimidin-4-ilamino)-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona (Comp. N.° 25)

Síntesis de 6-cloro-3-metil-3-((metilamino)metil)-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona (3)
La síntesis del intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento A. Rendimiento: 0,56 g, producto en bruto; EM (IEN) m/z 242 [M 1]+.
Síntesis de ((6-cloro-3-metil-1,5-dioxo-1,2,3,5-tetrahidroimidazo[1,5-a]piridin-3-il)metil)(metil)carbamato de terc-butilo (4)
A una solución agitada de 6-cloro-3-metil-3-((metilamino)metil)-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona (3, 0,56 g, 2,3 mmol) en tetrahidrofurano (2 ml), se le añadieron bicarbonato de sodio (2,91 g, 34,0 mmol) seguido de dicarbonato de di-terciario butilo (2,58 ml, 11,0 mmol) y la mezcla de reacción se calentó a 100 °C durante 16 h. La mezcla de reacción se inactivó con agua y el compuesto se extrajo en acetato de etilo. La capa orgánica se lavó con ácido clorhídrico 0,5 M y salmuera, se separó, se secó sobre sulfato de sodio y se concentró a presión reducida, y el residuo se purificó lavando repetidamente con pentano para obtener ((6-cloro-3-metil-1,5-dioxo-1,2,3,5-tetrahidroimidazo[1,5-a]piridin-3-il)metil)(metil)carbamato de terc-butilo (4). Rendimiento: 0,67 g, producto en bruto; EM (IEN) m/z 343 [M 1]+.
Síntesis de ((3-metil-1,5-dioxo-6-(pirimidin-4-ilamino)-1,2,3,5-tetrahidroimidazo[1,5-a]piridin-3-il)metil)carbamato de terc-butil metilo (6)
La síntesis del intermedio 6 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento B. Rendimiento: 0,340 g, 72 %; EM (IEN) m/z 401 [M 1]+.
Síntesis de 3-metil-3-((metilamino) metil)-6-(pirimidin-4-ilamino)-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona (Comp. N.° 25)
La síntesis del compuesto 25 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento D. Sólido de color amarillo claro; Rendimiento: 0,11 g, 44 %; EM (IEN) m/z 301,20 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 9,37 (s, 2H), 8,75 (d, J = 6,8 Hz, 2H), 8,36 (d, J = 5,8 Hz, 1H), 7,36 (d, J = 5,9 Hz 1H), 6,82 (d, J = 7,1 Hz, 1H), 3,58 (d, J = 13,1 Hz, 1H), 2,82 (d, J = 13,1 Hz, 1H),2,16(s, 3H), 1,76 (s, 3H).
Ejemplo 26
Síntesis de 6-(pirimidin-4-ilamino)-1H-espiro[imidazo[1,5-a]piridin-3,3'-pirrolidin]-1,5(2H)-diona (Comp. N.° 26)

La síntesis del intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento A. Rendimiento: 0,4 g, 37 %; EM (IEN) m/z 374 [M 1]+.
Síntesis de 1,5-dioxo-6-(pirimidin-4-ilamino)-2,5-dihidro-1H-espiro[imidazo[1,5-a]piridin-3,3’-pirrolidin]-1 '-carboxilato de bencilo (5)
La síntesis del intermedio 5 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento B. Rendimiento: 0,1 g, 34 %; EM (IEN) m/z 432 [M 1]+.
Síntesis de 6-(pirimidin-4-ilamino)-1H-espiro[imidazo[1,5-a]piridin-3,3'-pirrolidin]-1,5(2H)-diona (Comp. N ° 26)
A una solución agitada de 1,5-dioxo-6-(pirimidin-4-ilamino)-2,5-dihidro-1H-espiro[imidazo[1,5-a]piridin-3,3'-pirrolidin]carboxilato de bencilo (5, 0,06 g, 0,138 mmol) en acetato de etilo:metanol (10:1, 33 ml), se le añadió hidróxido de paladio al 20 % (0,03 g). La mezcla de reacción se hidrogenó a presión de balón durante 4 h. El progreso de la reacción se controló por CCF. Después del consumo completo de material de partida, la mezcla de reacción se filtró a través de un lecho de celite y el filtrado se concentró a presión reducida. El residuo se purificó mediante CCF preparativa y se lavó repetidamente con éter para proporcionar 6-(pirimidin-4-ilamino)-1H-espiro[imidazo[1,5-a]piridin-3,3'-pirrolidin]-1,5(2H)-diona (Comp. N.° 26) en forma de un sólido de color blanquecino. Rendimiento: 0,025 g, 60 %; EM (IEN) m/z 299,20 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 9,47 (s, 1H), 8,83-8,74 (m, 2H), 8,38 (d, J = 5,9 Hz, 1H), 7,37 (d, J = 5,9 Hz, 1H), 6,92 (d, J = 7,7 Hz, 1H), 3,67 (d, J = 12,6 Hz, 1H), 3,19 3,03 (m, 2H), 2,95-2,75 (m, 2H), 2,05-1,88 (m, 1H).
Ejemplo 27
Síntesis de 3-(3,5-difluorofenil)-3-metil-6-(pirimidin-4-ilamino)-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona (Comp. N.° 27)

Síntesis de 6-cloro-3-(3,5-dtfluorofenil)-3-metil-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona (3)
La síntesis del intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento A. Sólido de color blanquecino; Rendimiento: 0,21 g, 23 %; EM (IEN) m/z 311 [M 1 ]+.
Síntesis de 3-(3,5-difluorofenil)-3-metil-6-(pirimidin-4-ilamino)-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona (Comp. N° 27)
La síntesis del compuesto 27 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento B. Sólido de color amarillo; Rendimiento: 0,12 g, 50 %; EM (IEN) m/z 370 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,09 (s, 1H), 9,37 (s, 1H), 8,85-8,74 (m, 2H), 8,36 (d, J = 5,9 Hz, 1H), 7,33-7,23 (m, 2H), 7,13 (d, J = 8,0 Hz, 2H), 7,01 (d, J = 7,7 Hz, 1H), 2,23 (s, 3H).
Ejemplo 28
Síntesis de 3-(3-cloro-5-fluorofenil)-3-metil-6-(pirimidin-4-ilamino)-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona (Comp. N.° 28)
S ín te s is de 6 -c lo ro -3 -(3 -c lo ro -5 -flu o ro fe n il)-3 -m e til-2 ,3 -d ih id ro im id a z o [1 ,5 -a ]p ir id in -1 ,5 -d io n a ( 3 )
La síntesis del producto intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento A. Rendimiento: 0,2 g, 21 %; EM (IEN) m/z 326 [M 1]+.
Síntesis de 3-(3-doro-5-fluorofenil)-3-metil-6-(pirimidin-4-ilamino)-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona (Comp. N ° 28)
La síntesis del compuesto 28 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento B. Sólido de color amarillo; Rendimiento: 0,065 g, 27 %; EM (IEN) m/z 286,25 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,10 (s, 1H), 9,39 (s, 1H), 8,86-8,74 (m, 2H), 8,36 (d, J = 5,8 Hz, 1H), 7,53-7,45 (m, 1H), 7,33-7,24 (m, 3H), 7,22 (s, 1H), 2,23 (s, 3H).
Ejemplo 29
Síntesis de N-[6-[(3-isopropil-3-metil-1,5-dioxo-2H-imidazo[1,5-a]piridin-6-il)amino]pirimidin-4-il]ciclopropancarboxamida (Comp. N.° 29)


Síntesis de 6-cloro-3-isopropil-3-metil-2H-imidazo[1,5-a]piridin-1,5-diona (3)
La síntesis del producto intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento A. Sólido de color blanquecino. Rendimiento: 0,80 g, 82 %; EM (IEN) m/z 241,08 [M 1]+; RMN 1H (400 MHz, DMSO-d6): ó 9,94 (s, 1H), 7,95 (d, J = 8,0 Hz, 1H), 6,71 (m, 1H), 3,01 (m, 1H), 1,79 (s, 3H ), 1,03 (d, J = 6,8 Hz, 3H), 0,42 (d, J = 6,4 Hz, 3H).
Síntesis de N-[6-[(3-isopropil-3-metil-1,5-dioxo-2H-imidazo[1,5-a]piridin-6-il]-amino]pirimidin-4-il]ciclopropancarboxamida (Comp. N ° 29)
La síntesis del compuesto 29 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento B. Sólido de color blanquecino; Rendimiento: 0,060 g, 13 %; EM (IEN) m/z 383,30 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,88 (s, 1H), 9,65 (s, 1H), 9,20 (s, 1H), 8,66 (d, J = 7,6 Hz, 1H), 8,51 (s, 1H), 7,87 (s, 1H), 6,84 (d, J =7,6Hz, 1H), 3,08 (m, 1H), 2,02 (m, 1H), 1,82 (s, 3H), 1,04 (d, J = 6,8 Hz, 3H), 0,83 (d, J = 6,0 Hz, 4H), 0,46 (d, J = 6,4 Hz, 3H).
Ejemplo 30
Síntesis de W-[6-[[3-(3-fluorofenil)-3-metil-1,5-dioxo-2H-imidazo[1,5-a]piridin-6-il]amino]pirimidin-4-il]ciclopropancarboxamida (Comp. N.° 30)



Síntesis de 6-cloro-3-metil-3-(trifluorometil)-2H-imidazo[1,5-a]piridin-1,5-diona (3)
La síntesis del intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento A. Sólido de color blanquecino; Rendimiento: 300 mg, 59 %; EM (IEN) m/z 293,13 [M 1]+.
Síntesis de N-[6-[3-(3-fluorofenil)-3-metil-1,5-dioxo-2H-imidazo[1,5-a]piridin-6-il]amino]pirimidin-4-il]ciclopropancarboxamida (Comp. N.° 30)
La síntesis del compuesto 30 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento B. Sólido de color blanquecino; Rendimiento: 100 mg, 22 %; EM (IEN) m/z 435,28 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,86 (s, 1H), 10,03 (s, 1H), 9,17 (s, 1H), 8,69 (d, J = 7,6 Hz, 1H), 8,50 (s, 1H), 7,81 (s, 1H), 7,43 (m, 1H), 7,23 (m, 2H), 7,14 (m, 1H), 6,85 (d, J = 7,6 Hz, 1H), 2,24 (s, 3H), 2,01 (m, 1H) M, 1H), 0,82 (d, J = 6,0 Hz, 4H).
Ejemplo 31
Síntesis de 6-[[6-[(£)-2-ciclopropilvinil]pirimidin-4-il]amino]-3-metil-3-(trifluorometil)-2H-imidazo[1,5-a]-diona (Comp. N.° 31)

Una mezcla de ácido [(E)-2-ciclopropilvinil]borónico (1, 1,0 g, 8,93 mmol), N-terc-butoxicarbonil-N-(6-cloropirimidin-4-il)carbamato de terc-butilo (2, 3,24 g, 9,83 mmol), carbonato de sodio (2,84 g, 26,8 mmol), 1,4-dioxano (10 ml) y agua (3 ml) en un vial de microondas se desgasificó con argón durante 10 minutos. A esta mezcla se le añadió complejo de dicloruro de 1,1-bis(difenilfosfino)ferroceno-paladio (II) diclorometano (0,73 g, 0,89 mmol) y se purgó con argón durante 5 minutos. Después de sellar, el vial se irradió en un reactor de microondas a 130 °C durante 45 minutos. La mezcla de reacción se diluyó con agua (20 ml) y se extrajo con acetato de etilo (30 ml, 3 veces). Los extractos orgánicos se lavaron con agua (10 ml, 2 veces) y solución saturada de salmuera (10 ml, 1 veces). Los compuestos orgánicos se separaron después, se secaron con sulfato de sodio y se filtraron antes de la concentración a sequedad. El residuo en bruto se purificó por cromatografía en columna (sílice, acetato de etilo/hexanos = 25 %) para obtener N-[6-[(E)-2-ciclopropilvinil]pirimidin-4-il]carbamato de ferc-butilo (3) en forma de un sólido de color marrón claro. Rendimiento: 370 mg, 16 %; Em (IEN) m/z 262,22 [M 1-Boc]+; RMN 1H (400 MHz, DMSO-cfe) 810,23 (s, 1H), 8,60 (s, 1H), 7,63 (s, 1H), 6,50 (m, 2H), 1,48 (s, 9H), 0,91 (m, 2H), 0,64 (s, 1H) (m, 2H)
Síntesis de 6-[(E)-2-ciclopropilvinil]pirimidin-4-amina (4)
Una solución de N-[6-[(E)-2-ciclopropilvinil]pirimidin-4-il]carbamato de terc-butilo (3,350 mg, 1,34 mmol) en diclorometano (10 ml) (1,5 ml, 1,34 mmol) se añadió y se agitó a 25 °C durante 16 h. El disolvente se retiró a presión reducida y la mezcla se hizo básica con el residuo con amoníaco acuoso. La filtración y el lavado con éter dietílico proporcionaron (6-[(E)-2-ciclopropilvinil]pirimidin-4-amina (4) en forma de un sólido de color marrón. Rendimiento: 170 mg, 79 % EM (IEN) m/z 162,10 [M+1]+; RMN 1H (400 MHz, DMSO-d6) 68,19 (s, 1H), 6,70 (s ancho, 2H), 6,32 6,27 (m, 2H), 6,17 (s, 1H), 1,60 (m, 1H), 0,84 (m, 2H), 0,55 (m, 2H).
Síntesis de 6-[[6-[(E)-2-ciclopropilvinil]pirimidin-4-il]amino]-3-metil-3-(trifluorometil)-2H-imidazo[1,5-a]piridin-5-diona (Comp. N.° 31)
La síntesis del compuesto 31 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento B. Sólido de color marrón claro; Rendimiento: 70 mg, 19 %; EM (IEN) m/z 392,26 [M 1]+; RMN 1H (400 MHz, DMSO-d6) 610,50 (s ancho, 1H), 9,42 (s, 1H), 8,78 (d, J = 7,6 Hz, 1H), 8,63 (s, 1H), 7,00 (d, J = 8,0 Hz, 1H), 6,46 (m, 2H), 2,13 (s, 3H), 1,69 (m, 1H), 0,90 (m, 2H), 0,62 (m, 2H).
Ejemplo 32
Síntesis de 3,3-dimetil-6-(7H-pirrolo[2,3-d]pirimidin-7-il)-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona (Comp. N.° 32)



Síntesis de 3,3-dimetil-6-(7H-pirrolo[2,3-d]pirimidin-7-il)-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona (Comp. N ° 32) La síntesis del compuesto 32 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento B. Sólido de color amarillo; Rendimiento: 0,13 g, 42 %; EM (IEN) m/z 296 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,1 (s, 1H), 9,15 (s, 1H), 8,83 (s, 1H), 8,18-8,27 (m, 1H), 7,98-7,87 (m, 1H) 6,99 (d, J = 5,9 Hz, 1H), 6,87 (d, J = 7,7 Hz, 1H), 1,88 (s, 6H).
Ejemplo 33
Síntesis de 3-(3,5-diclorofenil)-3-metil-6-(pirimidin-4-ilamino)-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona (Comp. N.° 33)


Síntesis de 6-cloro-3-(3,5-diclorofenil)-3-metil-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona (3)
La síntesis del intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento A. Rendimiento: 0,21 g, 21 %.
Síntesis de 3-(3,5-diclorofenil)-3-metil-6-(pirimidin-4-ilamino)-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona (Comp. N° 33)
La síntesis del compuesto 33 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento B. Sólido de color amarillo; Rendimiento: 0,042 g, 17 %; EM (IEN) m/z 402 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,09 (s, 1H), 9,38 (s, 1H), 8,85-8,74 (m, 2H), 8,36 (d, J = 5,9 Hz, 1H), 7,66 (t, J = 1,9 Hz, 1H), 7,44 (d, J = 1,9 Hz, 2H), 7,29 (d, J = 6,0 Hz, 1H), 7,02 (d, J = 7,7 Hz, 1H), 2,23 (s, 3H).
Ejemplo 34
Síntesis de 3-metil-3-(piridin-2-il)-6-(pirimidin-4-ilamino)-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona (Comp. N.° 34)


Síntesis de 6-cloro-3-metil-3-(piridin-2-il)-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona (3)
La síntesis del intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento A. Rendimiento: 0,26 g, 33 %; EM (IEN) m/z 276 [M 1]+.
Síntesis de 3-metil-3-(piridin-2-il)-6-(pirimidin-4-ilamino)-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona (Comp. N ° 34) La síntesis del compuesto 34 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento B. Sólido de color blanquecino; Rendimiento: 0,012 g, 5 %; EM (IEN) m/z 335,10 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 9,96 (s, 1H), 9,28 (s, 1H), 8,83-8,73 (m, 2H), 8,55-8,48 (m, 1H), 8,34 (d, J = 5,9 1H), 7,88 7,84 (m, 1H), 7,57 (d, J = 8,0 Hz, 1H), 7,37 (dd, J = 7,6, 4,7 Hz, 1H), 7,25 (d, J = 5,9 Hz, 1H), 6,97 (d, J = 7,6 Hz, 1H), 2,29 (s, 3H).
Ejemplo 35
Síntesis de 8-cloro-3-metil-6-(pirimidin-4-ilamino)-3-(trifluorometil)-2H-imidazo[1,5-]piridin-1,5-diona (Comp. N.° 35)
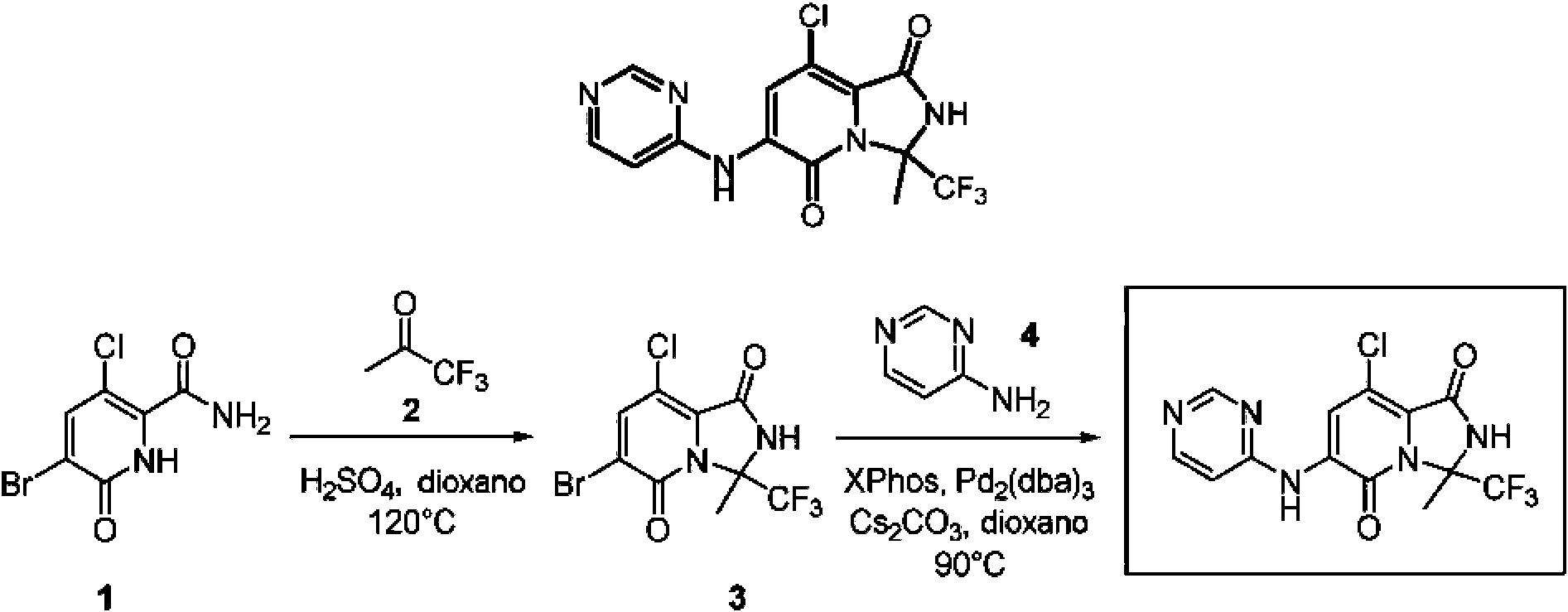
Síntesis de 6-bromo-8-cloro-3-metil-3-(trifluorometil)-2H-imidazo[1,5-a]piridin-1,5-diona (3)
La síntesis del intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del
Procedimiento A. Sólido de color blanquecino; Rendimiento: 1,8 g, 66 %; EM (IEN) m/z 346,6 [M 1]+; RMN 1H (400 MHz, DMSO-cfe) ó 10,90 (s, 1H), 8,36 (s, 1H), 2,06 (s, 3H).
Síntesis de 8-cloro-3-metil-6-(pirimidin-4-ilamino)-3-(trifluorometil)-2H-imidazo-[1,5-a]piridin-1,5-diona (Comp. N.° 35) La síntesis del compuesto 35 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento B. Sólido de color blanquecino; Rendimiento: 0,52 g, 35 %; EM (IEN) m/z 360,22 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,60 (s, 1H), 9,82 (s, 1H), 8,87 (s, 1H), 8,81 (m, 1H), 8,47 (d, J = 6,0 Hz, 1H), 7,46 (d, J = 6,0 Hz, 1H), 2,12 (s, 3H).
Ejemplo 36
Síntesis de 2',2'-dimetil-6-(pirimidin-4-ilamino)espiro[2H-imidazo[1,5-a]piridin-3,1 '-ciclopentano]-1,5-diona (Comp. N.° 36)

La síntesis del intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento A. Sólido de color crema; Rendimiento: 0,12 g, 16 %; EM (IEN) m/z 267,15 [M 1]+.
Síntesis de 2',2'-dimetil-6-(pirimidin-4-ilamino)espiro[2H-imidazo[1,5-a]piridin-3,1 ’-ciclopentano]-1,5-diona (Comp. N.° 36)
La síntesis del compuesto 36 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento B. Sólido de color blanquecino; Rendimiento: 17 mg, 9 %; EM (IEN) m/z 326,30 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 9,66 (s, 1H), 9,48 (s, 1H), 8,78 (d, J = 7,6 Hz, 1H) 6,0 Hz, 1H), 2,94 (m, 1H), 2,50-1,66 (m, 6H), 1,03 (s, 1H) 3H), 0,72 (s, 3H).
Ejemplo 37
Síntesis de 6-[(8-ciclopropil-9H-purin-6-il)amino]-3-metil-3-(trifluorometil)-2H-imidazo[1,5-a]piridin-1,5-diona (Comp. N.° 37)

A una solución de 6-cloropirimidin-4,5-diamina (1, 2,0 g, 13,84 mmol) y ciclopropanocarbaldehído (2, 1,16 g, 16,6 mmol) en metanol (100 ml), se le añadió ácido acético (1,0 ml) y se agitó a temperatura ambiente durante 2 h. A esta mezcla se le añadió cloruro de hierro (III) (11,22 g, 69,18 mmol) y se continuó la agitación durante 24 h. Los disolventes se retiraron a presión reducida y el residuo en bruto se purificó mediante cromatografía en columna (sílice, acetato de etilo/hexanos = 70 %) para proporcionar 6-cloro-8-ciclopropil-9H-purina (3) en forma de un sólido de color blanquecino. Rendimiento: 1,2 g, 45 %; EM (IEN) m/z 195,14 [M 1]+; RMN 1H (400 MHz, DMSO-cfe) ó 13,65 (s, 1H), 8,60 (s, 1H), 2,19 (m, 1H), 1,18 (m, 4H).
Síntesis de (5)-[(6-cloro-8-ciclopropil-purin-9-il)metoxi]etil-trimetil-silano (5)
A una suspensión agitada de hidruro de sodio (0,18 g, 7,71 mmol) en dimetilformamida (10 ml) a 0 °C, se le añadió 6-cloro-8-ciclopropil-9H-purina (3, 1,0 g, 5,14 mmol) y se agitó durante 30 m. A esta mezcla, se le añadió gota a gota 2-(clorometoxi)etil-trimetil-silano (4, 2,0 ml, 6,17 mmol) y se dejó agitar a temperatura ambiente durante 16 h. La mezcla de reacción se inactivó con agua enfriada con hielo (20 ml) y se extrajo con acetato de etilo (50 ml, 3 veces). Las capas orgánicas combinadas se separaron y se secaron con sulfato de magnesio antes de la concentración a sequedad. El residuo en bruto se purificó mediante cromatografía en columna de acetato de etilo/hexanos = 70 %) para proporcionar 2-(6-cloro-8-ciclopropil-purin-9-il) metoxi] etil-trimetil-silano (5) en forma de un sólido de color blanquecino. Rendimiento: 0,84 g, 50 %; EM (IEN) m/z 325,30 [M 1]+; RMN 1H (400 MHz, CDCl3) ó 8,65 (s, 1H), 5,73 (s, 2H), 3,61 (m, 2H), 2,44 (m, 1H), 1,43 (m, 2H), 1,26 (m, 2H), 0,88 (m, 2H), 0,05 (s, 9H).
Síntesis de 8-ciclopropil-9-(2-trimetilsililetoximetil)purin-6-amina (6)
En un tubo sellado una suspensión agitada de 2-[(6-cloro-8-ciclopropil-purin-9-il)metoxi]etil-trimetil-silano (5, 0,8 g, 2,46 mmol) y amoníaco acuoso al 30 % (10 ml) en etanol (10 ml) se calentó a 120 °C durante 14 h. Después de que la CCF mostrara el consumo de 5, se retiró el etanol a presión reducida y el residuo se lavó con agua (10 ml) y éter dietílico (10 ml) para obtener 8-ciclopropil-9-(2-trimetilsililetoximetil)purin-6-amina (6) en forma de un sólido de color blanquecino. Rendimiento: 703 mg, 93 %; EM (IEN) m/z 306,30 [M 1]+; RMN 1H (400 MHz, CDCla) ó 8,00 (s, 1H), 7,10 (s, 2H), 5,64 (s, 2H), 3,57 (m, 2H), 2,24 (m, 1H), 1,08 (m, 4H), 0,83 (m, 2H), 0,09 (s, 9H).
Síntesis de 6-[[8-ciclopropil-9-(2-trimetilsililetoximetil)purin-6-il]amino]-3-metil-3-(trifluorometil)-2H-imidazo[1,5-a]piridin-5-diona (8)
La síntesis del intermedio 8 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento B. Sólido de color amarillo; Rendimiento: 300 mg, 50 %; EM (IEN) m/z 536,26 [M 1]+.
Síntesis de 6-[(8-ciclopropil-9H-purin-6-il)amino]-3-metil-3-(trifluorometil)-2H-imidazo[1,5-a]piridin-1,5-diona (Comp. N.° 37)
A una solución de 6-[[8-ciclopropil-9-(2-trimetilsililetoximetil)purin-6-il]amino]-3-metil-3-(trifluorometil)-2H-imidazo[1,5-a]piridin-1,5-diona (8, 0,25 g, 0,47 mmol) en tetrahidrofurano (50 ml) se le añadió fluoruro de tetrabutilamonio hidratado (0,61 g, 2,33 mmol). Después de que la CCF mostrara el consumo de 8, la mezcla de reacción se diluyó
con agua (50 ml) y se extrajo con acetato de etilo (50 ml, 2 veces). Las capas orgánicas combinadas se secaron sobre sulfato de sodio y se concentraron a presión reducida. Se purificó la mezcla mediante cromatografía en columna (sílice, metanol/diclorometano = 2 %) para proporcionar 6-[(8-ciclopropil-9H-purin-6-il)amino]-3-metil-3-(trifluorometil)-2H-imidazo[1,5-a]piridin-1,5-diona (Comp. N.° 37) en forma de un sólido de color amarillo. Rendimiento: 135 mg, 71 % EM (IEN) m/z 406,29 [M 1]+ (400 MHz, DMSO efe) 813,24 (s, 1H), 10,55 (s, 1H), 8,80 (d, J = 7,6 Hz, 1H), 8,64 (s, 1H), 8,49 (s, 1H), 7,10 (D, J = 7,6 Hz, 1H), 2,16 (m, 1H), 2,15 (s, 3H), 1,12 (m, 4H). Ejemplo 38
Síntesis de W-[6-[(8-cloro-1,5-dioxo-espiro[2H-imidazo[1,5-a]piridin-3,1,-ciclopentan]-6-il)amino]pirimidin-4-il]ciclopropancarboxamida (Comp. N.° 38)

Síntesis de 5-bromo-3-cloro-piridin-2-carboxilato de etilo (2)
A una solución agitada de ácido 5-bromo-3-cloro-piridin-2-carboxílico (1, 150,0 g, 634,38 mmol) en etanol (1,5 l) se le añadió ácido sulfúrico (93,26 g, 951,58 mmol) a temperatura ambiente. La masa de reacción se agitó a 80 °C durante una noche. Después del consumo del material de partida tal como se indica por CCF, la mezcla de reacción se enfrió a temperatura ambiente y el disolvente se retiró a presión reducida. El residuo resultante se neutralizó con solución acuosa saturada de bicarbonato de sodio y se extrajo con acetato de etilo (1 l, 2 veces). Las capas orgánicas se separaron después, se combinaron, se secaron con sulfato de magnesio y se concentró a sequedad en vacío para proporcionar 5-bromo-3-cloro-piridina-2-carboxilato de etilo (2) en forma de un sólido de color blanquecino. Rendimiento: 163 g, 97 %; RMN 1H (400 MHz, DMSO-d6) 88,69 (m, 1H), 8,08 (m, 1H), 7,81 (m, 1H), 4.47 (m, 2H), 1,43 (t, J = 7,2 Hz, 3H).
Síntesis de 5-bromo-3-cloro-1-oxido-piridin-1-io-2-carboxilato de etilo (3)
A una solución agitada de 5-bromo-3-cloro-piridina-2-carboxilato de etilo (2, 151,0 g, 570,89 mmol) en diclorometano (1,73 l) se le añadió anhídrido trifluoroacético (30,0 ml, 1,14 mol) y peróxido de hidrógeno de urea (112,69 g, 1,20 mol) a 0 °C. La reacción se agitó durante una noche a temperatura ambiente. Una vez completada la reacción, la mezcla de reacción se neutralizó con una solución dibásica de fosfato de potasio. Se añadió una disolución de bisulfito de sodio seguido de extracción con diclorometano (100 ml, 2 veces). Las capas orgánicas se separaron, se combinaron, se secaron con sulfato de magnesio, se filtraron y se concentraron a sequedad al vacío para proporcionar 5-bromo-3-cloro-1-oxido-piridin-1-io-2-carboxilato de etilo 3 en forma de un sólido de color blanquecino. Rendimiento: 150,5 g, 94 %; EM (IEN) m/z 281,8 [M 1]+; RMN 1H (400 MHz, DMSO-d6) 88,26 (d, J = 1,2 Hz, 1H), 7.48 (d, J = 1,2 Hz, 1H), 4,50 (c, J = 7,12 Hz, 2H), 1,42 (t, J = 12,12 Hz, 3H).
S ín te s is de 5 -b ro m o -3 -c lo ro -6 -o x o -1 H -p ir id in -2 -c a rb o x ila to de e tilo ( 4 )
A una solución agitada de 3-cloro-1-oxo-piridin-1-io-2-carboxilato de etilo (3,150 g, 534,8 mmol) en dimetilformamida (900 ml) a 0 °C se le añadió anhídrido trifluoroacético (224,63 g, 1,07 mmol). La temperatura de la mezcla de reacción se elevó a 50 °C y se continuó la agitación durante 1 h. Una vez completada la oxidación, la masa de reacción se inactivó con una solución acuosa saturada de bicarbonato de sodio y el producto se extrajo con diclorometano (100 ml, 2 veces). Las capas orgánicas se separaron, combinaron, se secaron con sulfato de magnesio y se concentraron a sequedad al vacío para proporcionar 5-bromo-3-cloro-6-oxo-1H-piridin-2-carboxilato de etilo (4) en forma de un sólido de color amarillo. Rendimiento: 75 g, 50 %; EM (IEN) m/z 281,8 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,44-10,02 (m, 1H), 7,86 (s, 1H), 4,47 (c, J = 7,2 Hz, 2H), 1,43 (t, J = 5,6 Hz, 3H).
Síntesis de 5-bromo-3-cloro-6-oxo-1H-piridina-2-carboxamida (5)
A un matraz de fondo redondo que contenía 5-bromo-3-cloro-6-oxo-1H-piridin-2-carboxilato de etilo (4, 75,0 g, 267,38 mmol) se le añadió amoníaco líquido (150,0 ml, 267,38 mmol) en etanol (100 ml) a 0 °C. La mezcla de reacción se agitó a 45 °C durante 2 h. En este momento, la mezcla se concentró para retirar el amoníaco etanólico. Los sólidos en bruto se lavaron con éter dietílico (500 ml) y se disolvieron en metanol a reflujo (1 l) y se filtraron en caliente. El filtrado se concentró a presión reducida hasta que quedó 1/3 de volumen de disolvente. Se añadió éter dietílico hasta que todos los sólidos precipitaron. El sólido se filtró y se secó al vacío para proporcionar 5-bromo-3-cloro-6-oxo-1H-piridin-2-carboxamida (5) en forma de un sólido de color marrón claro. Rendimiento: 45 g, 69 %; EM (IEN) m/z 248,9 [M-1]-; RMN 1H (400 MHz, DMSO-d6) ó 7,92-7,82 (m, 1H), 7,61-7,59 (m, 1H), 7,36 (s, 1H).
Síntesis 6-bromo-8-cloro-espiro[2H-imidazo[1,5-a]piridin-3,1 '-ciclopentano]-1,5-diona (7)
La síntesis del intermedio 7 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento A. Sólido de color blanquecino; Rendimiento: 450 mg, 87 %; EM (IEN) m/z 317,03 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,39 (s, 1H), 8,25 (s, 1H), 2,73 (m, 4H), 2,21 (m, 2H), 1,93 (m, 2H).
Síntesis de N-[6-[(8-cloro-1,5-dioxo-espiro[2H-imidazo[1,5-a]piridin-3,1 '-ciclopentan]-6-il)amino]pirimidin-4-il]ciclopropancarboxamida (Comp. N.° 38)
La síntesis del compuesto 38 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento B. Sólido de color blanquecino; Rendimiento: 50 mg, 17 %; EM (IEN) m/z 415,32 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,92 (s, 1H), 10,08 (s, 1H), 9,52 (s, 1H), 8,72 (s, 1H), 8,59 (s, 1H), 7,98 (s, 1H), 2,78 (m, 2H), 2,02 (m, 3H), 1,83 (m, 2H), 1,72 (m, 2H), 0,84 (d, J = 6,0 Hz, 4H).
Ejemplo 39
Síntesis de 8,-cloro-6,-(pirimidin-4-ilamino)-2,H-espiro[ciclopentano-1,3,-imidazo[1,5-a]piridin]-1,,5,-diona (Comp. N.° 39)

Síntesis de 8'-cloro-6'-(pirimidin-4-ilamino)-2'H-espiro[ciclopentan-1,3’-imidazo[1,5-a]piridin]-1',5'-diona (Comp. N.° 39)
La síntesis del compuesto 39 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento B. Sólido de color blanquecino; Rendimiento: 0,07 g, 30 %; EM (IEN) m/z 332,28 [M 1]; RMN 1H (400 MHz, DMSO-d6) ó 10,11 (s, 1H), 9,67 (s, 1H), 8,84 (s, 1H), 8,79 (s, 1H), 8,43 (d, J = 5,6 Hz, 1H), 7,43 (d, J =
5,6, 1H), 2,77 (m, 2H), 1,98 (m, 2H), 1,82 (m, 2H), 1,74 (m, 2H).
Ejemplo 40
Síntesis de 3,3-dimetil-6-(5H-pirrolo[2,3-d]pirimidin-7(6H)-il)-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona (Comp. N.° 40)

Síntesis de 3,3-dimetil-6-(5H-pirrolo[2,3-d]pirimidin-7'(6H)-il)-2,3-dihidroimidazo-[1,5-a]piridin-1,5-diona (Comp. N° 40)
A una solución agitada de 3,3-dimetil-6-(7H-pirrolo[2,3-d]pirimidin-7-il)-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona (1, 0,095 g, 0,32 mmol) en metanol (25 ml), se le añadió paladio al 10 % sobre carbono (25 mg). La mezcla de reacción se hidrogenó a presión de balón de gas hidrógeno durante 24 h. El progreso de la reacción se controló por CCF. Después del consumo completo del material de partida, la mezcla de reacción se filtró a través de un lecho de celite y el filtrado se concentró a presión reducida para obtener un residuo en bruto. El residuo se purificó por HPLC preparativa para proporcionar el producto en forma de un sólido de color amarillo claro. Rendimiento: 8,0 mg, 8 %; EM (IEN) m/z 298,20 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 8,35 (s, 1H), 8,15 (s, 1H), 7,96 (d, J = 7,4 Hz, 1H), 6,81 (d, J = 7,5 Hz, 4,17 (t, J = 8,6 Hz, 2H), 3,13 (t, J = 8,6 Hz, 2H), 2,59 (s, 2H), 1,79 (s, 6H).
Ejemplo 41
Síntesis de 3-(2-aminoetil)-3-metil-6-(pirimidin-4-ilamino)-2H-imidazo[1,5-a]piridin-1,5-diona (Comp. N.° 41)

S ín te s is de 3 -c lo ro -3 -[2 -(1 ,3 -d io x o is o in d o lin -2 - il)e til]-3 -m e til-2 H -im id a z o [1 ,5 -a ]p ir id in -1 ,5 -d io n a ( 3 ) La síntesis del intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento A. Sólido de color marrón; Rendimiento: 3,01 g, 59 %; EM (IEN) m/z 372,0 [M 1]+.
Síntesis de 3-[2-(1,3-dioxoisoindolin-2-il)etil]-3-metil-6-(pirimidin-4-ilamino)-2H-imidazo[1,5-a]piridin-1,5-diona (5) La síntesis del intermedio 5 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento B. Sólido de color amarillo; Rendimiento: 1,03 g, 36 %; EM (IEN) m/z 431,31 [M 1]+.
Síntesis de 3-(2-aminoetil)-3-metil-6-(pirimidin-4-ilamino)-2H-imidazo[1,5-a]piridin-1,5-diona (Comp. N.° 41) La síntesis del compuesto 41 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento C. Sólido de color crema; Rendimiento: 300 mg, 54 %; EM (IEN) m/z 301,32 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 9,41 (s ancho, 1H), 8,77 (d, J = 7,6 Hz, 1H), 8,76 (s, 1H), 8,37 (d, J = 6,0 Hz, 1H), 7,37 (d, J = 5,6 Hz, 1H), 6,86 (d, J = 7,6 Hz, 1H), 4,39 (s ancho, 2H), 2,59 (m, 1H), 2,41 (m, 1H), 2,22 (m, 1H), 2,15 (m, 1H), 1,80 (s, 3H).
Ejemplo 42
Síntesis de W-[6-[(8-cloro-3-ciclopentil-3-metil-1,5-dioxo-2H-imidazo[1,5-a]piridin-6-il)amino]pirimidin-4-il]ciclopropancarboxamida (Comp. N.° 42)

Síntesis de 6-bromo-8-cloro-3-ciclopentil-3-metil-2H-imidazo[1,5-a]piridin-1,5-diona (3)
La síntesis del intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento A. Sólido de color marrón claro; Rendimiento: 605 mg, 54 %; EM (IEN) m/z 346,98 [M 1]+.
Síntesis de N-[6-[(8-cloro-3-ciclopentil-3-metil-1,5-dioxo-2H-imidazo[1,5-a]piridin-6-il)amino]pirimidin-4-il]ciclopropancarboxamida (Comp. N.° 42)
La síntesis del compuesto 42 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento B. Sólido de color blanquecino; Rendimiento: 16 mg, 5 %; EM (IEN) m/z 443,35 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,93 (s, 1H), 9,78 (s, 1H), 9,46 (s, 1H), 8,70 (s, 1H), 8,59 (s, 1H), 7,97 (s, 1H), 3,39 (m, 1H), 2,01 (m, 1H), 1,82 (s, 3H), 1,77 (m, 1H), 1,57-1,37 (m, 5H), 1,14 (m, 1H), 0,83 (m, 5H).
Ejemplo 43
Síntesis de W-[6-[(8-cloro-3,3-dimetil-1,5-dioxo-2H-imidazo[1,5-a]piridin-6-il)amino]pirimidin-4-il]ciclopropancarboxamida (Comp. N.° 43)

Síntesis de 6-bromo-8-cloro-3,3-dimetil-2H-imidazo[1,5-a]piridin-1,5-diona (3)
La síntesis del intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento A. Sólido de color blanquecino; Rendimiento: 390 mg, 48 %; EM (IEN) m/z 288,93 [M-1]-; RMN 1H (400 MHz, DMSO-d6) ó 10,03 (s, 1H), 8,24 (s, 1H), 1,75 (s, 6H).
Síntesis de N-[6-[(8-cloro-3,3-dimetil-1,5-dioxo-2H-imidazo[1,5-a]piridin-6-il]-amino]pirimidin-4-il]ciclopropancarboxamida (Comp. N.° 43)
La síntesis del compuesto 43 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento B. Sólido de color amarillo claro; Rendimiento: 42 mg, 17 %; EM (IEN) m/z 389,28 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,92 (s, 1H), 9,74 (s, 1H), 9,49 (s, 1H), 8,70 (s, 1H), 8,59 (s, 1H), 7,98 (s, 1H), 2,02 (m, 1H), 1,79 (s, 6H), 0,84 (d, J = 6,0 Hz, 4H).
Ejemplo 44
Síntesis de N-(6-((8'-cloro-1',5'-dioxo-1',5'-dihidro-2'H-espiro[ciclobutano-1,3'-imidazo[1,5-a]piridin]-6'-il)amino)pirimidin-4-il)ciclopropancarboxamida (Comp. N.° 44)
S ín te s is de 6 '-b ro m o -8 '-c lo ro -2 'H -e s p iro [c ic lo b u ta n -1 ,3 '- im id a z o [1 ,5 -a ]p ir id in ]-1 ',5 -d iona ( 3 ) '
La síntesis del intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento A. Sólido de color marrón; Rendimiento: 0,260 g, 72 %; RMN 1H (400 MHz, DMSO-d6) ó 10,5 (s, 1H), 8,24 (s, 1H), 3,45 (m, 2H), 2,28, 2H), 2,10 (m, 1H), 1,89 (m, 1H).
Síntesis de N-(6-((8 '-cloro-1 ',5 '-dioxo-15 '-dihidro-2 H-espiro[ciclobutan-1,3 '-imidazo-[1,5-a]piridin]-6 '-il)amino)pirimidin-4-il)ciclopropancarboxamida (Comp. N.° 44)
La síntesis del compuesto 44 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento B. Sólido de color amarillo claro; Rendimiento: 0,110 g, 33 %; EM (IEN) m/z 401,27 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,94 (s, 1H), 10,24 (s, 1H), 9,59 (s, 1H), 8,71 (s, 1H), 8,59 (s, 1H), 8,00 (s, 1H), 3,26 (m, 2H), 2,34 (m, 2H), 2,15 (m, 1H), 2,04 (m, 1H), 1,89 (m, 1H), 0,82 (m, 4H).
Ejemplo 45
Síntesis de W-(6'-((8'-cloro-1',5'-dioxo-1',5'-dihidro-2'H-espiro[ciclohexano-1,3'-imidazo[1,5-a]piridin]-6'-il)amino)pirimidin-4-il)ciclopropancarboxamida (Comp. N.° 45)

La síntesis del intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento A. Sólido de color blanquecino; Rendimiento: 1,93 g, 64 %; EM (IEN) m/z 330,99 [M 1]; RMN 1H (400 MHz, DMSO-d6) ó 10,59 (s, 1H), 8,24 (s, 1H), 2,84 (t, J = 10,74, 2H), 1,74 (m, 2H), 1,63 (m, 3H) 1,53 (m, 2H), I , 20 (m, 1H).
Síntesis de N-(6-((8'-cloro-1',5'-dioxo-1',5'-dihidro-2'H-espiro[ciclohexano-1,3,3-imidazo[1,5-a]piridin]-6'-il)amino)pirimidin-4-il)ciclopropancarboxamida (Comp. N.° 45)
La síntesis del compuesto 45 se realizó como se ha descrito anteriormente usando el protocolo general del procedimiento B. Sólido de color amarillo; Rendimiento: 0,051 g, 2,07 %; EM (IEN) m/z 429,34 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,93 (s, 1H), 10,29 (s, 1H), 9,43 (s, 1H), 8,70 (s, 1H), 8,58 (s, 1H), 7,97 (s, 1H), 2,93 (t, J = I I , 16 Hz, 2H), 2,02 (m, 1H), 1,75 (m, 2H), 1,64 (m, 3H), 1,54 (m, 2H), 1,21 (m, 1H), 0,84 (m, 4H).
Ejemplo 46
Síntesis de W-[6-[[8-cloro-3-metil-1,5-dioxo-3-(2,2,2-trifluoroetil)-2H-imidazo[1,5-a]piridin-6-il]amino]pirimidin-4-il]ciclopropancarboxamida (Comp. N.° 46)

Síntesis de 6-bromo-8-cloro-3-metil-3-(2,2,2-trifluoroetil)-2H-imidazo[1,5-a]piridin-1,5-diona (3)
La síntesis del intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento A. Sólido de color marrón; Rendimiento: 325 mg, 57 %; EM (IEN) m/z 358,95 [M 1]+.
Síntesis de N-[6-[[8-cloro-3-metil-1,5-dioxo-3-(2,2,2-trifluoroetil)-2H-imidazo[1,5-a]piridin-6-il]amino]pirimidin-4-il]ciclopropancarboxamida (Comp. N.° 46)
La síntesis del compuesto 46 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento B. Sólido de color marrón claro; Rendimiento: 120 mg, 32 %; EM (IEN) m/z 457,31 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,95 (s, 1H), 9,92 (s, 1H), 9,59 (s, 1H), 8,75 (s, 1H), 8,60 (s, 1H), 8,01 (s, 1H), 3,69 (m, 1H), 3,12 (m, 1H), 2,02 (m, 1H), 1,87 (s, 3H), 0,84 (d, J = 6,0 Hz, 4H).
Ejemplo 47
Síntesis de 8-cloro-3,3-dimetil-6-(pirimidin-4-ilamino)-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona (Comp. N.° 47)

Síntesis de 8-cloro-3,3-dimetil-6-(pirimidin-4-ilamino)-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona (Comp. N ° 47) La síntesis del compuesto 47 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento B. Sólido de color blanquecino; Rendimiento: 0,020 g, 10 %; EM (IEN) m/z 306,25 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 9,78 (s, 1H), 9,64 (s, 1H), 8,84 (s, 1H), 8,79 (s, 1H), 8,43 (d, J = 5,6 Hz, 1H), 7,44 (d, J = 5,2 Hz, 1H), 1,81 (s, 6H).
Ejemplo 48
Síntesis de 8-cloro-3-ciclopentil-3-metil-6-(pirimidin-4-ilamino)-2H-imidazo[1,5-a]piridin-1,5-diona (Comp. N.° 48)

Síntesis de 8-cloro-3-ciclopentil-3-metil-6-(pirimidin-4-ilamino)-2H-imidazo[1,5-a]piridin-1,5-diona (Comp. N ° 48) La síntesis del compuesto 48 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento B. Sólido de color amarillo claro; Rendimiento: 0,019 g, 6 %; EM (IEN) m/z 360,23 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 9,83 (s, 1H), 9,63 (s, 1H), 8,84 (s, 1H), 8,79 (s, 1H), 8,44 (d, J = 5,7 Hz, 1H), 7,43 (d, J = 5,6 Hz, 1H), 3,38 (m, 1H), 1,84 (s, 3H), 1,79 (m, 1H), 1,51 (m, 5H), 1,13 (m, 1H), 0,83 (m, 1H).
Ejemplo 49
Síntesis de W-(6-((8-cloro-3-metil-1,5-dioxo-3-(trifluorometil)-1,2,3,5-tetrahidroimidazo[1,5-a]piridin-6-il)amino)pirimidin-4-il)ciclopropancarboxamida (Comp. N.° 49)

Síntesis de 3-bromo-8-cloro-3-metil-3-(trifluorometil)-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona (3)
La síntesis del intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento A. Sólido de color marrón claro; Rendimiento: 0,7 g, producto en bruto; EM (IEN) m/z 342,97 [M-1]-Síntesis de N-(6-((8-cloro-3-metil-1,5-dioxo-3-(trifluorometil)-1,2,3,5-tetrahidroimidazo[1,5-a]piridin-6-il)amino)pirimidin-4-il)ciclopropancarboxamida (Comp. N ° 49)
La síntesis del compuesto 49 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento B. Sólido de color amarillo claro. Rendimiento: 0,16 g, 42 %; EM (IEN) m/z 443,27 [M 1]+; RMN 1H
(400 MHz, DMSO-cfe) 810,95 (s, 1H), 10,55 (s ancho, 1H), 9,72 (s, 1H), 8,74 (s, 1H), 8,61 (s, 1H), 8,04 (s, 1H), 2,11 (s, 3H), 2,03 (m, 1H), 0,84 (d, J = 6,0 Hz, 4H).
Ejemplo 50
Síntesis de W-[6-[(8-cloro-4,,4,-difluoro-1,5-dioxoespiro[2H-imidazo[1,5-a]piridin-3,1,-ciclohexan]-il)amino]pirimidin-4-il]ciclopropancarboxamida (Comp. N.° 50)

Síntesis de 6-bromo-8-cloro-4',4'-difluoroespiro[2H-imidazo[1,5-a]piperidin-3,1 '-ciclohexano]-1,5-diona (3)
La síntesis del producto intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento A. Sólido de color blanquecino; Rendimiento: 0,41 g; 93 %; EM (IEN) m/z 366,94 [M 1]+; RMN 1H (400 MHz, DMSO-d6) 89,79 (s, 1H), 7,89 (s, 1H), 2,12 (m, 6H), 1,66 (m, 2H).
Síntesis de N-[6-[(8-cloro-4',4'-difluoro-1,5-dioxoespiro[2H-imidazo[1,5-a]piridin-3,1'-ciclohexan]-6-il)amino]pirimidin-4-il]ciclopropancarboxamida (Comp. N.° 50)
La síntesis del compuesto 50 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento B. Sólido de color amarillo claro; Rendimiento: 0,098 g, 39 %; EM (IEN) m/z 465,16 [M 1]+; RMN 1H (400 MHz, DMSO-d6) 810,92 (s, 1H), 10,47 (s, 1H), 9,52 (s, 1H), 8,72 (s, 1H), 8,59 (s, 1H), 7,98 (s, 1H), 3,30-3,20 (m, 2H), 2,35-2,15 (m, 4H), 2,05-1,98 (m, 1H), 1,75-1,67 (m, 2H), 0,97-0,80 (m, 4H).
Ejemplo 51
Síntesis de W-[6-[[8-cloro-3-(3-clorofenil)-3-metil-1,5-dioxo-2H-imidazo[1,5-a]piridin-6-il]amino]pirimidin-4-il]ciclopropancarboxamida (Comp. N.° 51)

Síntesis de 6-bromo-8-cloro-3-(3-clorofenil)-3-metil-2H-imidazo[1,5-a]piridin-1,5-diona (3)
La síntesis del intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento A. Sólido de color blanquecino; Rendimiento: 0,13 g, 17 %; EM (IEN) m/z 386,83 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,38 (s, 1H), 8,32 (s, 1H), 7,52 (s, 1H), 7,43 (m, 2H), 7,34 (d, J = 7,6 Hz, 1H), 2,18 (s, 3H). Síntesis de N-[6-[[8-doro-3-(3-dorofenil)-3-metil-1,5-dioxo-2H-imidazo[1,5-a]piridin-6-il]amino]pirimidin-4-il]ciclopropancarboxamida (Comp. N.° 51)
La síntesis del compuesto 51 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento B. Sólido de color amarillo claro; Rendimiento: 0,04 g, 25 %; EM (IEN) m/z 485,30 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,90 (s, 1H), 10,09 (s, 1H), 9,44 (s, 1H), 8,75 (s, 1H), 8,59 (s, 1H), 7,90 (s, 1H), 7,52 (s, 1H), 7,43 (m, 2H), 7,34 (d, J = 7,2 Hz, 1H), 2,23 (s, 3H), 2,00 (m, 1H), 0,82 (m, 4H).
Ejemplo 52
Síntesis de W-[6-[[8-cloro-3-(3-fluorofenil)-3-metil-1,5-dioxo-2H-imidazo[1,5-a]piridin-6-il]amino]pirimidin-4-il]ciclopropancarboxamida (Comp. N.° 52)

Síntesis de 6-bromo-8-cloro-3-(3-fluorofenil)-3-metil-2H-imidazo[1,5-a]piridin-1,5-diona (3)
La síntesis del intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento A. Sólido de color blanquecino; Rendimiento: 0,18 g, 20 %; EM (IEN) m/z 373,01 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,38 (s, 1H), 8,31 (s, 1H), 7,43 (m, 1H), 7,31 (d, J = 10,3 Hz, 1H), 7,22 (m, 2H), 2,19 (s, 3H). Síntesis de N-[6-[[8-cloro-3-(3-fluorofenil)-3-metil-1,5-dioxo-2H-imidazo[1,5-a]piridin-6-il]amino]pirimidin-4-il]ciclopropancarboxamida (Comp. N.° 52)
La síntesis del compuesto 52 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento B. Sólido de color blanquecino; Rendimiento: 0,035 g, 15 %; EM (IEN) m/z 469,34 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,91 (s, 1H), 10,10 (s, 1H), 9,44 (s, 1H), 8,75 (s, 1H), 8,59 (s, 1H), 7,90 (s, 1H), 7,42 (m, 1H), 7,32 (m, 1H), 7,23 (m, 2H), 2,23 (s, 3H), 2,00 (m, 1H), 0,823 (m, 4H).
Ejemplo 53
Síntesis de (3,S)-3,-amino-6-(pirimidin-4-ilamino)espiro[2H-imidazo[1,5-a]piridin-3,1,-ciclohexan]-1,5-diona (Comp. N.° 53)

Síntesis de 2-[(1S,3R)-3-hidroxiciclohexil]isoindolin-1,3-diona (3)
A una solución de clorhidrato de (1R,3S)-3-aminociclohexanol (2, 1,0 g, 6,57 mmol) en agua (50 ml) a 0 °C, se le añadió 1,3-dioxoisoindolin-2-carboxilato de etilo (1, 1,6 g, 7,3 mmol). La mezcla se dejó agitar a 25 °C durante 3 horas mientras se controlaba por CCF. Después de la terminación, la suspensión se filtró y el residuo se lavó con agua (50 ml) y se secó para obtener 2-[(3R)-3-hidroxiciclohexil]isoindolin-1,3-diona (3) en forma de un sólido de color blanquecino. Rendimiento: 1,45 g, 81 %; RMN 1H (400 MHz, DMSO-d6) ó 7,83 (m, 4H), 4,77 (m, 1H), 3,99 (m, 1H), 2.04- 1,29 (m, 9H).
Síntesis de (S)-2-(3-oxociclohexil) isoindolino-1,3-diona (4)
A una solución agitada de 2-[(3R)-3-hidroxiciclohexil]isoindolin-1,3-diona (3, 1,4 g, 5,71 mmol) en diclorometano (50 ml) a 0 °C, se le añadió clorocromato de piridinio (3,69 g, 17,12 mmol). La mezcla se dejó agitar a 25 °C durante 5 horas mientras se controlaba por CCF. La mezcla de reacción se hizo básica (pH 8) con solución acuosa saturada de bicarbonato de sodio y se extrajo con diclorometano (150 ml, 2 veces). Las capas orgánicas combinadas se secaron sobre sulfato de sodio y se concentraron a presión reducida para obtener 2-(3-oxociclohexil)isoindolin-1,3-diona (4) en forma de un sólido de color marrón. Rendimiento: 1,31 g, 94 %; EM (IEN) m/z 244,15 [M 1]+.
Síntesis de (3'S)-6-cloro-3'-(1,3-dioxoisoindolin-2-il)espiro[2H-imidazo[1,5-a]piridin-3,1 ’-ciclohexan]-1,5-diona (6)
La síntesis del intermedio 6 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento A. Sólido de color crema; Rendimiento: 1,51 g, 71 %; EM (IEN) m/z 398,19 [M 1]+.
Síntesis de (3'S)-3'-(1,3-dioxoisoindolin-2-il)-6-(pirimidin-4-ilamino)espiro[2H-imidazo[1,5-a]piridin-3,1 '-ciclohexano-1.5- diona (8)
La síntesis del intermedio 8 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento B. Sólido de color amarillo; Rendimiento: 104 mg, 45 %; EM (IEN) m/z 457,0 [M 1]+; RMN 1H (400 MHz, DMSO-cfe) ó 10,54 (s, 1H), 9,47 (s, 1H), 8,76 (s, 2H), 8,37 (s, 1H), 7,84 (m, 4H) 7,38 (s, 1H), 6,91 (m, 1H), 4,60 (m, 1H), 3,96 (m, 1H), 5H), 3,32 (m, 1H), 3,06 (m, 1H), 2,32 (m, 1H), 1,99-1,56 (m, 5H,).
Síntesis de (3'S)-3'-amino-6-(pirimidin-4-ilamino)espiro[2H-imidazo[1,5-a]piridin-3,1'-ciclohexan]-1,5-diona (Comp. N.° 53)
La síntesis del compuesto 53 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento C. Sólido de color verdoso pálido; Rendimiento: 60,0 mg, 21 %; EM (IEN) m/z 327,35 [M 1]+; RMN 1H (400 MHz, DMSO-d6): ó 9,37 (s, 1H), 8,78 (d, J = 7,6 Hz, 1H), 8,76 (s, 1H), 8,37 (d, J = 6,0 Hz, 1H), 7,37 (d, J = 5,6 Hz, 1H), 6,89 (d, J = 7,6 Hz, 1H), 2,89 (m, 2H), 2,72 (m, 1H), 1,82 (m, 2H), 1,65 (m, 2H), 1,46 (m, 1H), 1,05 (m, 1H).
Ejemplo 54
Síntesis de clorhidrato de 8-cloro-6-(pirimidin-4-ilamino)espiro[2H-imidazo[1,5-a]piridin-3,3'-piperidin]-1,5-diona (Comp. N.° 54)

Síntesis de 8-cloro-1,5-dioxo-6-(pirimidin-4-ilamino)espiro[2H-imidazo[1,5-a]piridin-3,3'-piperidin-1'-carboxilato de terc-butilo (3)
La síntesis del intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento B. Sólido de color amarillo claro; Rendimiento: 0,08 g, 50 %; EM (IEN) m/z 447,17 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,51 (s, 1H), 9,63 (s, 1H), 8,85 (s, 1H), 8,79 (s, 1H), 8,44 (d, J = 5,8 Hz, 1H), 7,45 (d, J = 5,6 Hz, 1H), 4,14 (m, 2H), 3,89 (m, 1H), 3,05 (m, 1H), 2,90 (m, 1H), 1,93 (m, 3H), 1,41 (s, 9H).
Síntesis de clorhidrato de 8-cloro-6-(pirimidin-4-ilamino)espiro[2H-imidazo[1,5-a]piridin-3,3’-piperidin]-1,5-diona (Comp. N.° 54)
La síntesis del compuesto 54 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento D. Sólido de color marrón claro; Rendimiento: 0,024 g, 35 %; EM (IEN) m/z 347,31 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,23 (s, 1H), 9,99 (s, 1H), 9,72 (s ancho, 1H), 9,48 (s ancho, 1H), 8,99 (s ancho 1H), 8,78 (s, 1H), 8,53 (s ancho, 1H), 7,54 (s ancho, 1H), 4,15 (m, 1H), 3,52 (m, 1H), 3,38 (m, 1H), 3,03 (m, 1H), 2,92 (m, 1H), 2,17 (m, 1H), 2,04 (m, 1H), 1,84 (m, 1H).
Ejemplo 55
Síntesis de 8-cloro-3-metil-6-(pirimidin-4-ilamino)-3-(2,2,2-trifluoroetil)-2H-imidazo[1,5-]piridin-1,5-diona (Comp. N.° 55)

Síntesis de 8-cloro-3-metil-6-(pirimidin-4-ilamino)-3-(2,2,2-trifluoroetil)-2H-imidazo[1,5-a]piridin-1,5-diona (Comp. N° 55)
La síntesis del compuesto 55 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento B. Sólido de color amarillo claro; Rendimiento: 0,1 g, 30 %; EM (IEN) m/z 374,25 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 9,95 (s, 1H), 9,70 (s, 1H), 8,86 (s, 1H), 8,83 (s, 1H), 8,45 (d, J = 5,6 Hz, 1H), 7,45 (d, J = 5,6 Hz, 1H), 3,73 (m, 1H), 3,14 (m, 1H), 1,89 (s, 3H).
Ejemplo 56
Síntesis de W-(6-((8-cloro-1,5-dioxo-1,5-dihidro-2H-espiro[imidazo[1,5-a]piridin-3,4'-piperidin]-6-il)amino)pirimidin-4-il)ciclopropancarboxamida (Comp. N.° 56)
S ín te s is de 6 -b ro m o -8 -c lo ro -1 ,5 -d io x o -1 ,5 -d ih id ro -2 H -e s p iro [im id a z o [1 ,5 -a ]p ir id in -3 ,4 '-p ip e r id in -1 ’-c a rb o x ila to de te rc -b u tilo ( 3 )
Procedimiento E: A una solución agitada de 5-bromo-3-cloro-6-oxo-1H-piridin-2-carboxamida (1, 0,4 g, 1,59 mmol) en 1,4-dioxano (12 ml) se le añadió 4-oxopiperidin-1-carboxilato de terc-butilo (2, 1,58 g, 7,95 mmol) en un vial a temperatura ambiente. Después, se añadió gota a gota ácido sulfúrico concentrado (0,16 g, 1,59 mmol). El vial se selló y se calentó a 120 °C durante 3 h. La mezcla de reacción se inactivó con agua (20 ml) y se extrajo con acetato de etilo (15 ml, 2 veces). Las capas orgánicas se separaron y la capa acuosa se diluyó con 1,4-dioxano (30 ml), agua (25 ml) y se trató con dicarbonato de di-terc-butilo (0,44 g, 2,03 mmol) gota a gota a 0 °C. La mezcla de reacción se agitó a temperatura ambiente durante una noche. Después del consumo del material de partida, la reacción se extrajo con metanol al 10 % en diclorometano (25 ml, 2 veces). Las capas orgánicas se secaron con sulfato de magnesio, se filtraron y se concentraron a sequedad. El residuo en bruto se purificó después por cromatografía en columna ultrarrápida usando alúmina neutra eluyendo con metanol al 2 % en diclorometano. Las fracciones deseadas se concentraron a sequedad al vacío para proporcionar 6-bromo-8-cloro-1,5-dioxo-1,5-dihidro-2H-espiro[imidazo[1,5-a]piridin-3,4'-piperidin]-1'-carboxilato de terc-butilo (3) en forma de un sólido de color amarillo. Rendimiento: 0,26 g, 44 %; EM (IEN) m/z 430,11 [M-1]-; RMN 1H (400 MHz, DMSO-d6) ó 10,77 (s, 1H), 8,27 (m, 1H), 4.04 (s, 2H), 2,92 (m, 4H), 1,57 (m, 2H), 1,42 (s, 9H).
Síntesis de 8-cloro-6-((6-(ciclopropancarboxamido)pirimidin-4-il)amino)-1,5-dioxo-1,5-dihidro-2H-espiro[imidazo[1,5-a]piridin-3,4'-piperidin-1'-carboxilato de terc-butilo (5)
La síntesis del intermedio 5 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento B. Sólido de color amarillo; Rendimiento: 0,14 g, 44 %; EM (IEN) m/z 530,36 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,93 (s, 1H), 10,49 (s, 1H), 9,51 (s, 1H), 8,72 (s, 1H), 8,59 (s, 1H), 8,00 (s, 1H), 4,05 (m, 2H), 3,20 (m, 1H), 3,09 (m, 3H), 2,02 (m, 1H), 1,58 (m, 2H), 1,43 (s, 9H), 0,84 (m, 4H).
Síntesis de N-(6-(8-cloro-1,5-dioxo-1,5-dihidro-2H-espiro[imidazo[1,5-a]piridin-3,4'-piperidin]-6-il)amino)pirimidin-4-il)ciclopropancarboxamida (Comp. N.° 56)
La síntesis del compuesto 56 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento D. Sólido de color amarillo; Rendimiento: 0,10 g, 87 %; EM (IEN) m/z 430,38 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,98 (s, 1H), 10,54 (s, 1H), 9,43 (s, 1H), 9,33 (d, J = 9,6 Hz, (s, 1H), 8,77 (d, J = 11,6 Hz, 1H), 8,73 (s, 1H), 8,60 (s, 1H), 8,00 (s, 1H), 3,48 (s, 2H), 3,32 (m, 2H), 3,18 (m, 2H), 2,02 (m, M, 1H), 1,86 (d, J = 12.4 Hz, 2H), 0,84 (m, 4H).
Ejemplo 57
Síntesis de 8-cloro-3-(3-fluorofenil)-3-metil-6-(pirimidin-4-ilamino)-2H-imidazo[1,5-]piridin-1,5-diona (Comp. N.° 57)

Síntesis de 8-cloro-3-(3-fluorofenil)-3-metil-6-(pirimidin-4-ilamino)-2H-imidazo]1,5-a]piridin-1,5-diona (Comp. N.° 57)
La síntesis del compuesto 57 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento B. Sólido de color blanquecino; Rendimiento: 0,025 g, 10 %; EM (IEN) m/z 386,31 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,14 (s, 1H), 9,56 (s, 1H), 8,84 (s, 1H), 8,82 (s, 1H), 8,41 (d, J = 5,9 Hz, 1H), 7,42 (m, 1H), 7,33 (m, 2H), 7,22 (m, 2H), 2,24 (s, 3H).
Ejemplo 58
Síntesis de 8-cloro-3-(3-clorofenil)-3-metil-6-(pirimidin-4-ilamino)-2H-imidazo[1,5-]piridin-1,5-diona (Comp. 58)

Síntesis de 8-cloro-3-(3-clorofenil)-3-metil-6-(pirimidin-4-ilamino)-2H-imidazo[1,5-a]piridin-1,5-diona (Comp. N ° 58) La síntesis del compuesto 58 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento B. Sólido de color amarillo claro; Rendimiento: 0,07 g, 32 %; EM (IEN) m/z 402,25 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,14 (s, 1H), 9,57 (s, 1H), 8,84 (s, 1H), 8,82 (s, 1H), 8,41 (d, J = 5,9 Hz, 1H), 7,54 (s, 1H), 7,43 (m, 2H), 7,35 (m, 2H), 2,24 (s, 3H).
Ejemplo 59
Síntesis de 6-((6-((1-ciclopropil-2,2,2-trifluoroetil)amino)pirimidin-4-il)amino)-3-metil-3-(trifluorometil)-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona (Comp. N.° 59)

S ín te s is de N -te rc -b u to x ic a rb o n il-N -[6 -[(1 -c ic lo p ro p il-2 ,2 ,2 -tr iflu o ro e til)a m in o ]p ir im id in -4 -il]c a rb a m a to de te rc -b u tilo ( 3 )
A una solución de N-terc-butoxicarbonil-N-(6-cloropirimidin-4-il)carbamato de terc-butilo (0,50 g, 1,52 mmol) y clorhidrato de 1-ciclopropil-2,2,2-trifluoro-etanamina (2, 0,40 g, 2,27 mmol) en monoglima (15 ml) en un vial, se le añadió terc-butóxido de sodio (0,729 g, 7,58 mmol). Después de purgar con gas argón durante 30 minutos, se añadieron BINAP (0,14 g, 0,23 mmol) y tris(dibencilidenacetona)dipaladio (0) (69 mg, 0,080 mmol). El vial se selló y el contenido se calentó a 90 °C durante 24 h. Se retiró el disolvente y la mezcla en bruto se sometió a cromatografía ultrarrápida eluyendo con un gradiente de 10-12 % de acetato de etilo en hexano. Las fracciones deseadas se concentraron a presión para obtener N-terc-butoxicarbonil-N-[6-[(1-ciclopropil-2,2,2-trifluoroetil)amino]pirimidin-4-il]-carbamato de terc-butilo (3) en forma de un sólido de color blanquecino. Rendimiento: 110 mg, 17 %; RMN 1H (400 MHz, DMSO-d6) ó 9,78 (s, 1H), 8,15 (s, 1H), 7,79 (d, J = 9,2 Hz, 1H), 7,10 (s, 1H), 4,47 (m, 1H), 1,46 (s, 1H), 0,61 (m, 1H), 0,49 (m, 2H), 0,39 (m, 1H).
Síntesis de clorhidrato de N4-(1-ciclopropil-2',2,2-trifluoroetil)pirimidin-4,6-diamina (4)
Procedimiento F: A una solución de N-terc-butoxicarbonil-N-[6-[(1-ciclopropil-2,2,2-trifluoroetil)amino]pirimidin-4-il]-carbamato de terc-butilo (3, 250 mg, 0,58 mmol) en diclorometano (2 ml) y metanol (2 ml) se le añadió 4 M de cloruro de hidrogeno en 1,4-dioxano (5 ml). La reacción se agitó a temperatura ambiente durante una noche. Los disolventes se evaporaron a presión reducida, el residuo resultante se trituró con diclorometano. Los sólidos se colocaron en un matraz de fondo redondo y se añadió amoníaco acuoso ajustando el pH a 8-9. Se redujo el contenido de disolvente y se añadió agua. Los sólidos se filtraron y se secaron para proporcionar clorhidrato de N4-(1-ciclopropil-2,2,2-trifluoroetil)pirimidin-4,6-diamina (4) en forma de un sólido de color marrón claro. Rendimiento: 130 mg, producto en bruto; EM (IEN) m/z 233 [M 1-HCl]+.
Síntesis de 6-[[6-[(1-ciclopropil-2,2,2-trifluoroetil)amino]pirimidin-4-il]amino]-3-metil-3-(trifluorometil)-2H-imidazo[1,5-a]piridin-1,5-diona (Comp. N.° 59)
La síntesis del compuesto 59 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento B. Sólido de color blanquecino; Rendimiento: 33 mg, 19 %; EM (IEN) m/z 463,36 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,44 (s ancho, 1H), 9,05 (s, 1H), 8,62 (d, J = 7,6 Hz, 1H), 6,36 (s, 1H), 7,26 (s, 1H), 8,26 (s, 1H), 7,80 (d, J = 8,8 Hz, 1H), 6,96 (d, J = 8,0 Hz, 1H), 6,43 (s, 1H), 4,36 (s ancho, 1H), 2,12 (s, 3H), 1,12 (m, 1H), 0,67 (m, 1H), 0,51 (m, 2H), 0,35 (m, 1H).
Ejemplo 60
Síntesis de N-(6-((8-cloro-1,5-dioxo-1,5-dihidro-2H-espiro[imidazo[1,5-a]piridin-3,3'-piperidin]-6-il)amino)pirimidin-4-il)ciclopropancarboxamida (Comp. N.° 60)

S ín te s is de 6 -b ro m o -8 -c lo ro -1 ,5 -d io x o e s p iro [2 H -im id a z o [1 ,5 -a ]p ir id in -3 ,3 '-p ip e r id in ]-1 ’-c a rb o x ila to de te rc -b u tilo ( 3 )
A una solución agitada de 5-bromo-3-cloro-6-oxo-1H-piridin-2-carboxamida (1, 0,7 g, 2,78 mmol) en 1,4-dioxano (10 ml) en un vial, se le añadió clorhidrato de piperidin-3-ona (2, 1,13 g, 8,35 mmol) a temperatura ambiente. A esta solución se le añadió por goteo ácido sulfúrico concentrado (0,27 g, 2,78 mmol). El vial de reacción se selló y se calentó a 100 °C durante 48 h. Después de la terminación, el disolvente se retiró a presión reducida y la masa de reacción resultante se diluyó con agua (20 ml) y la capa acuosa se lavó con acetato de etilo (20 ml, 3 veces). La solución acuosa (20,0 ml) de 6-bromo-8-cloroespiro[2H-imidazo[1,5-a]piridin-3,3'-piperidin]-1,5-diona (~0,5 g) se trató con hidróxido de sodio (90 mg, 2,26 mmol). Se añadió dicarbonato de di-terc-butilo (0,39 g, 1,80 mmol) en 1,4-dioxano (20 ml) y la mezcla de reacción se agitó a 25 °C durante 6 h. Una vez completado el disolvente se retiró a presión reducida. La capa acuosa se extrajo con acetato de etilo (25 ml, 2 veces) y las capas orgánicas se lavaron con agua (10 ml, 2 veces). Las capas orgánicas se separaron y se secaron con sulfato de sodio, se filtraron y se concentraron a sequedad. El residuo en bruto se purificó por cromatografía en columna ultrarrápida eluyendo con metanol al 2 % en diclorometano. Las fracciones deseadas se concentraron a sequedad para proporcionar 6-bromo-8-cloro-1,5-dioxo-espiro[2H-imidazo[1,5-a]piridin-3,3'-piperidin]-carboxilato de terc-butilo (3) en forma de un sólido de color blanquecino. Rendimiento: 0,4 g; EM (IEN) m/z 432 [M 1]+.
Síntesis de 8-cloro-6-((6-(ciclopropancarboxamido)pirimidin-4-il)amino)-1,5-dioxo-1,5-dihidro-2H-espiro[imidazo[1,5-a]piridin-3,3’-piperidin-1’-carboxilato de etilo (5)
La síntesis del intermedio 5 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento B. Sólido de color amarillo claro; Rendimiento: 0,07 g, 19 %; EM (IEN) m/z 530,0 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,94 (s, 1H), 9,48 (s, 1H), 8,72 (s, 1H), 8,60 (s, 1H), 7,99 (s, 1H), 4,05 (m, 2H), 3,9 (m, 1H), 3,32 (m, 2H), 2,95 (m, 1H), 2,01 (m, 3H), 1,35 (s, 9H), 0,84 (m, 4H).
Síntesis de N-(6-(8-cloro-1,5-dioxo-1,5-dihidro-2H-espiro[imidazo[1,5-a]piridin-3,3’-piperidin]-6-il)amino)pirimidin-4-il)ciclopropancarboxamida (Comp. N.° 60)
La síntesis del compuesto 60 se realizó como se ha descrito anteriormente usando el protocolo general del procedimiento F. Sólido de color marrón claro; Rendimiento: 22 mg, 35 %; EM (IEN) m/z 430,39 [M 1]+; RMN 1H (400 MHz, DMSO-d6): ó 10,98 (s, 1H), 10,04 (s, 1H), 9,55 (s, 1H), 9,19 (s ancho, 1H), 8,61 (s, 1H), 8,02 (s, 1H), 4,17 (m,1H), 3,35 (m, 2H), 3,02 (m, 2H), 2,08 (m, 3H), 1,81 (m, 1H), 0,84 (s, 4H).
Ejemplo 61
Síntesis de W-(6-((8-cloro-1,5-dioxo-1,2',3',5,5',6'-hexahidro-2H-espiro[imidazo[1,5-a]piridin-3,4'-piran]-6-il)amino)pirimidin-4-il)ciclopropancarboxamida (Comp. N.° 61)

Síntesis de 6-bromo-8-cloro-2',3',5',6'-tetrahidro-2H-espiro[imidazo[1,5-a]piridin-3,4'-piranil-1,5-diona (3)
La síntesis del intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento A. Sólido de color marrón claro; Rendimiento: 1,0 g, producto en bruto; RMN 1H (400 MHz, DMSO-d6) ó 10,84 (s, 1H), 8,28 (s, 1H), 3,92 (m, 2H), 3,64 (m, 2H), 3,09 (m, 2H), 1,52 (m, 2H).
S ín te s is de N -(6 -((8 -c lo ro -1 ,5 -d io x o -1 ,2 ,3 ,3 ,5 ,5 ',6 '-h e x a h id ro -2 H -e s p iro [im id a z o [1 ,5 -a ]p ir id in -3 ,4 '-p ira n ]-6 -il)a m in o )p ir im id in -4 - il)c ic lo p ro p a n c a rb o x a m id a (C om p. N ° 61)
La síntesis del compuesto 61 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento B. Sólido de color amarillo claro; Rendimiento: 0,11 g, 21 %; EM (IEN) m/z 431,36 [M 1 ]+; RMN 1H (400 MHz, DMSO-d6) ó 10,94 (s, 1H), 10,54 (s, 1H), 9,51 (s, 1H), 8,72 (s, 1H), 8,59 (s, 1H), 8,0 (s, 1H), 3,94 (m, 2H), 3,68 (t, J = 12,4 Hz, 2H) D, J = 6,0 Hz, 4H).
Ejemplo 62
Síntesis de W-[6-[(8-cloro-1',1'-difluoro-1,5-dioxoespiro[2H-imidazo[1,5-a]piridin-3,3'-ciclobutan]-6-il)amino]pirimidin-4-il]ciclopropancarboxamida (Comp. N.° 62)

Síntesis de 6-bromo-8-cloro-11 ’-difluoro-espiro[2H-imidazo[1,5-a]piridin-3',3'-ciclobutan]-1,5-diona (3)
La síntesis del intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento A. Sólido de color marrón; Rendimiento: 0,4 g, 59 %; EM (IEN) m/z 337 [M 1]+.
Síntesis de N-[6-[(8-cloro-11 '-difluoro-1,5-dioxoespiro[2H-imidazo[1,5-a]piridin-3,3’-ciclobutan]-6-il)amino]pirimidin-4-il]ciclopropancarboxamida (Comp. N.° 62)
La síntesis del compuesto 62 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento B. Sólido de color blanquecino; Rendimiento: 0,025 g, 11 %; EM (IEN) m/z 437,4 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,92 (s, 1H), 10,39 (s, 1H), 9,74 (s, 1H), 8,74 (s, 1H), 8,59 (s, 1H), 8,03 (s, 1H), 4,22-4,15 (m, 1H), 3,13-2,90 (m, 1H), 2,07-1,96 (m, 1H), 0,85-0,77 (s, 4H).
Ejemplo 63
Síntesis de 8-cloro-2',2'-dimetil-6-(pirimidin-4-ilamino)espiro[2H-imidazo[1,5-a]piridin-3,1 '-ciclopentan]-1,5-diona (Comp. N.° 63)

La síntesis del intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del procedimiento A. Sólido de color crema; Rendimiento: 0,2 g, 14 %; RMN 1H (400 MHz, DMSO-cfe) ó 10,03 (s, 1H), 8,29 (s, 1H), 2,96-2,91 (m, 1H), 2,67 (m, 1H), 2,05-1,92 (m, 4H) 1,63-1,58 (m, 1H), 0,99 (s, 3H), 0,65 (s, 3H).
Síntesis de 8-cloro-2’,2'-dimetil-6-(pirimidin-4-ilamino)espiro[2H-imidazo[1,5-a]piridin-3,1'-ciclopentano-1,2-diona (Comp. N.° 63)
La síntesis del compuesto 63 se realizó como se ha descrito anteriormente usando el protocolo general del procedimiento B. Se usó un reactor de microondas para calentar en lugar del calentamiento con baño de aceite convencional. Sólido de color blanquecino; Rendimiento: 0,03 g, 29 %; EM (IEN) m/z 360,29 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 9,76 (s, 1H), 9,66 (s, 1H), 8,84 (s, 1H), 8,81 (s, 1H), 8,44 (d, 1H), 7,47 (d, 1H), 3,00-2,92 (m, 1H), 2,44-2,42 (m, 1H), 2,14-2,10 (m, 1H), 1,99-1,90 (m,lH), 1,65-1,62 (m, 1H), 1,02 (s, 3H), 0,67, (s, 3H).
Ejemplo 64
Síntesis de 8-cloro-6-(pirimidin-4-ilamino)-2,3',5,6,6-tetrahidro-2H-espiro[imidazo[1,5-a]piridin-3,4'-piran]-1,5-diona (Comp. N.° 64)
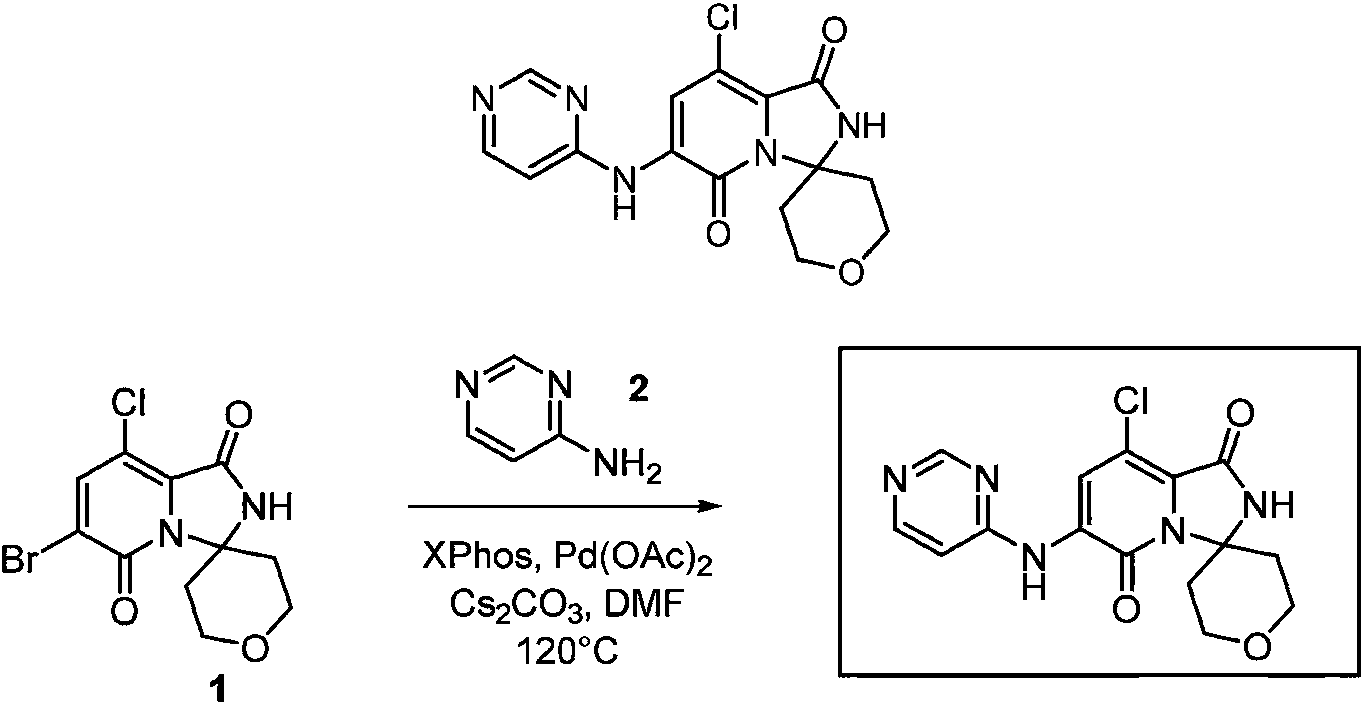
Síntesis de 8-cloro-6-(pirimidin-4-ilamino)-2',3',5',6'-tetrahidro-2H-espiro[imidazo[1,5-a]piridin-3,4'-piran]-1,5-diona (Comp. N ° 64)
La síntesis del compuesto 64 se realizó como se ha descrito anteriormente usando el protocolo general del procedimiento B. Sólido de color amarillo claro; Rendimiento: 0,10 g, 32 %; EM (IEN) m/z 348,27 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,56 (s, 1H), 9,65 (s, 1H), 8,84 (d, J = 12,0 Hz, 2H), 8,44 (s, 1H), 7,47 (s, 1H), 3,94 (m, 2H), 3,70 (m, 2H), 3,19 (m, 2H), 1,54 (d, J = 11,4 Hz, 2H).
Ejemplo 65
Síntesis de W-(6-((8-cloro-1,5-dioxo-1,5-dihidro-2H-espiro[imidazo[1,5-a]piridin-3,3'-tietan]-6-il)amino)pirimidin-4-il)ciclopropancarboxamida (Comp. N.° 65)

Síntesis de 6-bromo-8-cloro-2H-espiro[imidazo[1,5-a]piridin-3,3'-tietan]-1,5-diona (3)
La síntesis del intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento A. Sólido de color marrón oscuro; Rendimiento: 0,25 g, 20 %; RMN 1H (400 m Hz , DMSO-d6) 511,10 (s ancho, 1H), 8,27 (s, 1H), 4,65 (s, 2H), 3,32 (s), 2H).
Síntesis de N-(6-(8-cloro-1,5-dioxo-1,5-dihidro-2H-espiro[imidazo[1,5-a]piridin-3,3’-tietan]-6-il)amino)pirimidin-4-il)ciclopropancarboxamida (Comp. N.° 65)
La síntesis del compuesto 65 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento B. Sólido de color blanquecino; Rendimiento: 0,05 g, 30 %; EM (IEN) m/z 419,26 [M 1]+; RMN 1H (400 MHz, DMSO-d6) 510,95 (s, 1H), 10,80 (s, 1H), 9,65 (s, 1H), 8,69 (s, 1H), 8,59 (s, 1H), 8,02 (s, 1H), 4,66 (s, 2H), 3,32 (s, 2H), 2,03 (m, 1H), 0,85 (s, 4H).
Ejemplo 66
Síntesis de 8-cloro-2'-fluoro-6-(pirimidin-4-ilamino)espiro[2H-imidazo[1,5-a]piridin-3,1'-ciclohexano]-1,5-diona (Comp. N.° 66)

S ín te s is de 6 -b ro m o -8 -c lo ro -2 '- flu o ro -e s p iro [2 H -im id a z o [1 ,5 -a ]p ir id in -3 ,1 '-c ic lo h e xa n o ]-1 ,5 -d io n a ( 3 )
La síntesis del intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento A. Sólido de color marrón claro; Rendimiento: 0,40 g, 57 %; EM (IEN) m/z 349 [M-1].
Síntesis de 8-cloro-2'-fluoro-6-(pirimidin-4-ilamino)espiro[2H-imidazo[1,5-a]piridin-3,1 ’-ciclohexano-1,5-diona (Comp. N ° 66)
La síntesis del compuesto 66 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento B. Sólido de color blanquecino; Rendimiento: 0,1 g, 38 %; EM (IEN) m/z 364,29 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,31 (s, 1H), 9,65 (s, 1H), 8,85 (d, J = 12 Hz, 1H) 8,45 (d, J = 4 Hz, 1H), 7,46 (d, J = 8 Hz, 1H), 5,74-5,58 (m, 1H), 2,98-2,92 (m, 1H), 2,14-2,12 (m, 1H), 1,80-1,23 (m, 1H).
Ejemplo 67
Síntesis de 3,3,8-trimetil-6-(pirimidin-4-ilamino)-2H-imidazo[1,5-a]piridin-1,5-diona (Comp. N.° 67)

Síntesis de 3,3,8-trimetil-6-(pirimidin-4-ilamino)-2H-imidazo[1,5-a]piridin-1,5-diona (Comp. N ° 67)
Procedimiento G: Un vial se cargó con 8-cloro-3,3-dimetil-6-(pirimidin-4-ilamino)-2H-imidazo[1,5-a]piridin-1,5-diona (1,20 g, 0,65 mmol), trimetilboroxina (2,16 g, 1,31 mmol) y fosfato de potasio (0,28 g, 1,31 mmol) en 1,4-dioxano (10 ml) a temperatura ambiente en argón. Después, la mezcla de reacción se purgó con argón durante 10 min, seguida de la adición de tris(dibencilidenacetona)dipaladio (0) (60 mg, 0,07 mmol) y triciclohexilfosfina (18 mg, 0,07 mmol). El vial se selló y se calentó a 140 °C en un reactor de microondas durante 1 h. La mezcla de reacción se concentró a sequedad y el residuo en bruto se sometió a cromatografía en columna ultrarrápida usando gel de sílice de malla 100-200 con un gradiente de disolvente de 0,2-0,5 % de metanol en diclorometano. Las fracciones deseadas se recogieron y se concentraron a sequedad al vacío. El sólido obtenido se agitó en n-pentano y se filtró. Se obtuvo el producto resultante 3,3,8-trimetil-6-(pirimidin-4-ilamino)-2H-imidazo[1,5-a]piridin-1,5-diona (Comp. N.° 67) en forma de un sólido de color blanquecino. Rendimiento: 0,035 g, 18 %; EM (IEN) m/z 286,3 [M 1]+; r Mn 1H (400 MHz, DMSO-d6) ó 9,57 (s, 1H), 9,39 (s, 1H), 8,77 (s, 1H), 8,60 (s, 1H), 8,37 (d, J = 5,76 Hz, 1H), 7,77 (d, J = 5,64 Hz, 1H), 2,44 (s, 3H), 1,79 (s, 6H).
Ejemplo 68
Síntesis 8-ciclopropil-3,3-dimetil-6-(pirimidin-4-ilamino)-2H-imidazo[1,5-a]piridin-1,5-diona (Comp. N.° 68)

Síntesis 8-ciclopropil-3,3-dimetil-6-(pirimidin-4-ilamino)-2H-midazo[1,5-a]piridin-1,5-diona (Comp. N ° 68)
La síntesis del compuesto 68 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento G. Sólido de color blanquecino; Rendimiento: 0,032 g, 16 %; EM (IEN) m/z 312,3 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 9,57 (s, 1H), 9,40 (s, 1H), 8,75 (s, 1H), 8,37 (d, J = 5,6 Hz, 1H), 8,26 (s, 1H), 7,35 (d, J = 5,8 Hz, 1H), 3,17-3,15 (m, 1H), 1,80 (s, 6H), 1,10-0,98 (m, 2H), 0,69-0,68 (m, 2H).
Ejemplo 69
Síntesis de 8-fluoro-3,3-dimetil-6-(pirimidin-4-ilamino)-2H-imidazo[1,5-a]pi ridin-1,5-diona (Comp. N.° 69)

Síntesis de 5-bromo-3-fluoro-piridin-2-carboxilato de etilo (2)
A una solución agitada de ácido 5-bromo-3-fluoro-piridin-2-carboxílico (1,0 g, 4,55 mmol) en etanol (20 ml) se le añadió ácido sulfúrico (0,67 g, 6,82 mmol) a temperatura ambiente. La mezcla de reacción se agitó a reflujo durante la noche. Después del consumo de los materiales de partida como se indicó por CCF, la mezcla de reacción se enfrió a temperatura ambiente y el disolvente se retiró al vacío. El residuo se neutralizó con una solución acuosa saturada de bicarbonato de sodio y se extrajo con acetato de etilo (100 ml, 2 veces). Las capas orgánicas se separaron y se secaron con sulfato de magnesio, se filtraron y se concentraron a sequedad al vacío para proporcionar 5-bromo-3-fluoro-piridin-2-carboxilato de etilo (2) en forma de un sólido de color blanquecino. Rendimiento: 1,0 g, 89 %; EM (IEN) m/z 249,9 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 8,61 (m, 1H), 7,77-7,75 (m, 1H), 4,51 (c, J = 7,16 Hz, 2H), 1,43 (t, J = 7,12 Hz, 3H).
Síntesis de 5-bromo-3-fluoro-1-oxido-piridin-1-oxi-2-carboxilato de etilo (3)
A una solución agitada de 5-bromo-3-fiuoro-piridin-2-carboxilato de etilo (2, 0,9 g, 3,63 mmol) en diclorometano (50 ml) a 0 °C se le añadió anhídrido trifluoroacético (1,52 g, 7,26 mmol), peróxido de hidrógeno de urea (0,72 g, 7,62 mmol). La mezcla de reacción se agitó a temperatura ambiente durante una noche. Una vez completada la oxidación, la mezcla de reacción se neutralizó con una solución de dihidrogenofosfato de potasio y se inactivó con una solución de bisulfito de sodio. El producto se extrajo con diclorometano (100 ml, 2 veces). Las capas orgánicas se separaron, se secaron con sulfato de magnesio, se filtraron y se concentraron a sequedad al vacío para
proporcionar 5-bromo-3-fiuoro-1-oxido-piridin-1-io-2-carboxilato de etilo (3) en forma de un sólido de color blanquecino. Rendimiento: 0,9 g, 89 %; EM (IEN) m/z 265,95 [M 1]+; RMN 1H (400 MHz, DMSO-cfe) ó: 8,22 (s, 1H), 7,30-7,26 (m, 1H), 4,50 (c, J = 7,2 Hz, 2H), 1,42 (t, J = 7,2 Hz, 3H).
Síntesis de 5-bromo-3-fluoro-6-oxo-1H-piridin-2-carboxilato de etilo (4)
A una solución agitada de 5-bromo-3-fluoro-1-oxido-piridin-1-io-2-carboxilato de etilo (3, 0,85 g, 3,21 mmol) en dimetilformamida (15 ml) se le añadió anhídrido trifluoroacético 1,35 g, 6,42 mmol) a 0 °C. La mezcla de reacción se calentó a 50 °C y se agitó durante 1 h, se inactivó con solución acuosa saturada de bicarbonato de sodio y se extrajo con diclorometano (100 ml, 2 veces). Las capas orgánicas se separaron, se secaron con sulfato de magnesio, se filtraron y se concentraron a sequedad al vacío para proporcionar 5-bromo-3-fluoro-6-oxo-1H-piridin-2-carboxilato de etilo (4) en forma de un sólido de color amarillo. Rendimiento: 0,8 g, 94 %; EM (IEN) m/z 264 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 7,86 (m, 1H), 4,47 (c, J = 7,2 Hz, 2H), 1,43 (t, J = 7,2 Hz, 3H).
Síntesis de 5-bromo-3-fluoro-6-oxo-1H-piridin-2-carboxamida (5)
A un matraz de fondo redondo cargado con 5-bromo-3-fluoro-6-oxo-1H-piridin-2-carboxilato de etilo (4, 0,8 g, 3,03 mmol) a 0 °C se le añadió amoníaco líquido (15 ml, 3,03 mmol) en etanol (5 ml). La mezcla de reacción agitada se calentó a 45 °C durante 2 h. Después de consumir completamente el éster se evaporó el amoníaco líquido y el etanol a presión reducida. Se añadió metanol y la mezcla se sometió a reflujo durante 2 h y se filtró mientras estaba caliente. El volumen del filtrado se redujo en 2/3 y al metanol restante se le añadió éter dietílico hasta que precipitó un sólido. El sólido se filtró y se secó al vacío para proporcionar 5-bromo-3-fluoro-6-oxo-1H-piridin-2-carboxamida (5) en forma de un sólido de color marrón claro. Rendimiento: 0,6 g, 85 %; RMN 1H (400 MHz, DMSO-d6) ó 7,88-7,86 (m, 1H), 7,67 (s, 1H), 7,50 (s, 1H).
Síntesis de 6-bromo-8-fluoro-3,3-dimetil-2H-imidazo[1,5-a]piridin-1,5-diona (7)
La síntesis del compuesto intermedio 7 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento A. Sólido de color blanquecino; Rendimiento: 0,24 g, 34 %; EM (IEN) m/z 275,07 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 7,87 (d, J = 7,44 Hz, 1H), 7,06 (s, 1H), 1,96 (s, 6H).
Síntesis de 8-fluoro-3,3-dimetil-6-(pirimidin-4-ilamino)-2H-imidazo[1,5-a]piridin-1,5-diona (Comp. N ° 69)
La síntesis del compuesto 69 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento B. Sólido de color blanquecino; Rendimiento: 0,032 g, 13 %; EM (IEN) m/z 290,32 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 9,72 (s, 1H), 9,61 (s, 1H), 8,83 (d, J = 5,1 Hz, 1H), 8,79 (s, 1H), 8,45 (d, J = 5,7 Hz, 1H), 7,46 (d, J = 5,6 Hz, 1H), 1,82 (s, 6H).
Ejemplo 70
Síntesis de 6'-((6-aminopirimidin-4-il)amino)-8,-cloro-2'H-espiro[ciclohexano-1,3,3-imidazo[1,5-a]piridin]-1'5'-diona (Comp. N.° 70)

Síntesis de 6’-((6-aminopirimidin-4-il)amino)-8'-cloro-2'H-espiro[ciclohexano-1,3,3-imidazo[1,5-a]piridin]-1’,5’-diona (Comp. N ° 70)
La síntesis del compuesto 70 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento B. Rendimiento: 22 mg; EM (IEN) m /z 361,33 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,22 (s, 1H),
8,90 (s, 1H), 8,63 (s, 1H), 8,20 (s, 1H), 6,61 (s, 2H), 6,24 (s, 1H), 2,94 (t, J = 11,36 Hz, 2H), 1,65 (m, 5H), 1,51 (d, J = 12,1 Hz, 2H), 1,21 (m, 1H).
Ejemplo 71
Síntesis de 8-etil-3,3-dimetil-6-(pirimidin-4-ilamino)-2H-imidazo[1,5-a]piridin-1,5-diona (Comp. N.° 71)

Síntesis de 8-etil-3,3-dimetil-6-(pirimidin-4-ilamino)-2H-imidazo[1,5-a]-piridin-1,5-diona (Comp. N ° 71)
La síntesis del compuesto 71 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento G. Sólido de color blanquecino; Rendimiento: 0,04 g, 12 %; EM (IEN) m/z 300,3 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 9,58 (s, 1H), 9,40 (s, 1H), 8,78 (s, 1H), 8,67 (s, 1H), 8,38 (d, J = 5,84 Hz, 1H), 7,38 (d, J = 5,36 Hz, 1H), 2,89 (c, J = 7,76 Hz, 2H), 1,79 (s, 6H), 1,11 (t, J = 7,36 Hz, 3H).
Ejemplo 72
Síntesis de 8-cloro-3-metil-3-(3-piridil)-6-(pirimidin-4-ilamino)-2H-imidazo[1,5-a]piridin-1,5-diona (Comp. N.° 72)


Síntesis de 6-bromo-8-cloro-3-metil-3-(3-piridil)-2H-imidazo[1,5-a]piridin-1,5-diona (3)
La síntesis del intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento A. Sólido de color amarillo; Rendimiento: 0,4 g, 57 %; EM (IEN) m/z 354,02 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,4 (s ancho, 1H), 8,70 (s, 1H), 8,57 (d, J = 4 Hz, 1H), 8,31 (s, 1H) 8,0 Hz, 1H), 7,42-7,39 (m, 1H), 2,22 (s, 3H).
Síntesis de 8-cloro-3-metil-3-(3-piridil)-6-(pirimidin-4-ilamino)-2H-imidazo[1,5-a]piridin-1,5-diona (Comp. N ° 72) Procedimiento H: A una solución de 6-bromo-8-cloro-3-metil-3-(3-piridil)-2H-imidazo[1,5-a]piridin-1,5-diona (100 mg, 0,28 mmol) y pirimidin-4-amina (30 mg, 0,31 mmol) en 1,4-dioxano (12 ml) se le añadió carbonato de cesio (276 mg, 0,85 mmol). La reacción se purgó con argón durante 15 min. Se añadieron XPhos (13 mg, 0,03 mmol), XantPhos (16 mg, 0,030 mmol), acetato de paladio (6 mg, 0,030 mmol) y tris(dibencilidenacetona)dipaladio (0) (26 mg, 0,030 mmol) y se continuó la purga durante otros 10 min. La reacción se agitó a 100 °C durante 16 h. Después de la terminación, la masa de reacción se diluyó con metanol al 10 % en diclorometano y se hizo pasar a través de un lecho de celite. El filtrado en bruto se purificó después por cromatografía en columna ultrarrápida usando metanol al 10 % en diclorometano. Las fracciones deseadas se concentraron a sequedad al vacío para obtener 8-cloro-3-metil-3-(3-piridil)-6-(pirimidin-4-ilamino)-2H-imidazo[1,5-a]piridin-1,5-diona en forma de un sólido de color amarillo pálido. Rendimiento: 0,059 g, 57 %; EM (IEN) m/z 369,29 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,16 (s, 1H), 9,57 (s, 1H), 8,84 (d, J = 6,2 Hz, 1H), 8,75 (d, J = 2,24 Hz, 1H), 8,56-8,55 (m, 1H), 7,82 (d, J = 8,2 Hz, 1H), 7,42-7,38 (m, 1H), 2,27 (s, 3H).
Ejemplo 73
Síntesis de W-(6-((8-cloro-3-metil-1,5-dioxo-3-(piridin-4-il)-1,2,3,5-tetrahidroimidazo[1,5-a]piridin-6-il)amino)pirimidin-4-il)ciclopropanocarboxamida (Comp. N.° 73)

Síntesis de N-(6-((8-cloro-3-metil-1,5-dioxo-3-(piridin-4-il)-1,2,3,5-tetrahidroimidazo[1,5-a]piridin-6-il)amino)pirimidin-4-il)ciclopropanocarboxamida (Comp. N ° 73)
La síntesis del compuesto 73 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento B. Sólido de color blanquecino; Rendimiento: 0,030 g, 16 %; EM (IEN) m/z 352,34 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,91 (s ancho, 1H), 10,15 (s, 1H), 9,45 (s ancho, 1H), 8,76 (s, 1H), 8,58 (m, 3H), 7,90 (s, 1H), 7,40 (s ancho, 2H), 2,21 (s, 3H), 2,05-1,95 (m, 1H), 0,90-0,75 (m, 4H).
Ejemplo 74
Síntesis de 8-cloro-3-metil-3-(2-piridil)-6-(pirimidin-4-ilamino)-2H-imidazo[1,5-a]piridin-1,5-diona (Comp. N.° 74)


Síntesis de 6-bromo-8-cloro-3-metil-3-(piridin-2-il)-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona (3)
La síntesis del intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento A. Sólido de color blanquecino; Rendimiento: 0,30 g, 42 %; EM (IEN) m/z 354 [M 1 ]+.
Síntesis de 8-cloro-3-metil-3-(2-piridil)-6-(pirimidin-4-ilamino)-2H-imidazo[1,5-a]piridin-1,5-diona (Comp. N ° 74) La síntesis del compuesto 74 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento H. Sólido de color blanquecino; Rendimiento: 55 mg, 35 %; RMN 1H (400 MHz, DMSO-d6) ó 10,05 (s ancho, 1H), 9,51 (s ancho, 1H), 8,84 (d, J = 11,1 Hz, 2H), 8,52 (d, J = 3,28 Hz, 1H), 8,40 (d, J = 5,72 Hz, 1H), 7,88 7,84 (m, 1H), 7,66 (d, J = 8,12 Hz, 1H), 7,38-7,35 (m, 1H), 7,24 (d, J = 5,84 Hz, 1H), 2,26 (s, 3H).
Ejemplo 75
Síntesis de 8-cloro-3-metil-3-(4-piridil)-6-(pirimidin-4-ilamino)-2H-imidazo[1,5-a]piridin-1,5-diona (Comp. N.° 75)

La síntesis del producto intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento A. Sólido de color blanquecino; Rendimiento: 0,36 g, 51 %; EM (IEN) m/z 354,02 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,4 (s ancho, 1H), 8,57 (d, J = 5,2 Hz, 2H), 8,33 (s, 1H), 7,40 (d, J = 4,5 Hz, 2H) (s, 3H). Síntesis de 8-cloro-3-metil-3-(4-piridil)-6-(pirimidin-4-ilamino)-2H-imidazo[1,5-a]piridin-1,5-diona (Comp. N ° 75) La síntesis del compuesto 75 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento B. Sólido de color blanquecino; Rendimiento: 59 mg, 57 %; EM (IEN) m/z 369,29 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,18 (s ancho, 1H), 9,58 (s, 1H), 8,84 (d, J = 2,88 Hz, 2H), 8,59 (d, J = 5,64 Hz, 2H), 8,41 (d, J = 5,88 Hz, 1H), 7,43 (d, J = 5,76 Hz, 2H), 7,34 (d, J = 5,76 Hz, 1H), 2,23 (s, 3H).
Ejemplo 76
Síntesis de W-(6-((8-cloro-3-metil-1,5-dioxo-3-(piridin-2-il)-1,2,3,5-tetrahidroimidazo[1,5-a]piridin-6-il)amino)pirimidin-4-il)ciclopropanocarboxamida (Comp. N.° 76)

Síntesis de N-(6-((8-cloro-3-metil-1,5-dioxo-3-(piridin-2-il)-1,2,3,5-tetrahidroimidazo[1,5-ajpiridin-6-il)amino)pirimidin-4-il)ciclopropancarboxamida (Comp. N ° 76)
La síntesis del compuesto 76 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento H. Sólido de color blanquecino; Rendimiento: 15 mg, 8 %; EM (IEN) m/z 452,35 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,90 (s, 1H), 10,01 (s, 1H), 9,36 (s, 1H), 8,73 (s, 1H), 8,58 (s, 1H), 8,51 (d, J = 4,32 Hz, 1H), 7,85 (m, 2H), 7,63 (d, J = 7,96, 1H), 7,37 (m, 1H), 2,27 (s, 3H), 1,99 (m, 1H), 0,81 (m, 4H).
Ejemplo 77
Síntesis de W-[6-[[8-cloro-3-metil-1,5-dioxo-3-(3-piridil)-2H-imidazo[1,5-a]piridin-6-il]amino]pirimidin-4-il]ciclopropanocarboxamida (Comp. N.° 77)

Síntesis de N-[6-[[8-cloro-3-metil-1,5-dioxo-3-(3-piridil)-2H-imidazo[1,5-a]piridin-6-il]amino]pirimidin-4-il]ciclopropanocarboxamida (Comp. N.° 77)
La síntesis del compuesto 77 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento H. Sólido de color amarillo pálido; Rendimiento: 58 mg, 15 %; EM (IEN) m/z 452,36 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,99 (s ancho, 1H), 10,12 (s ancho, 1H), 9,44 (s, 1H), 8,75 (s, 1H), 8,73 (d, J = 2,0 Hz, 1H), 8,58 (s, 1H), 8,56 (d, J = 3,7 Hz, 1H), 7,90 (s, 1H), 7,42-7,38 (m, 1H), 2,26 (s, 3H), 2,02-1,96 (m, 1H), 0,82-0,81 (m, 4H).
Ejemplo 78
Síntesis de W-(6-((8'-cloro-2-fluoro-1 ',5'-dioxo-1 ',5'-dihidro-2'H-espiro[ciclohexano-1,3,3-imidazo[1,5-a]piridin]-6'-il)amino)pirimidin-4-il)ciclopropanocarboxamida (Comp. N.° 78)

Síntesis de 6’-bromo-8'-cloro-2-fluoro-2’H-espiro[ciclohexano-1,3-imidazo[1,5-a]piridin]-1’,5'-diona (3)
La síntesis del producto intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento A. Sólido de color blanquecino; Rendimiento: 0,31 g; EM (IEN) m/z 348,8 [M 1]+.
Síntesis de N-(6-((8’- cloro-2-fluoro-15’- dioxo-1', 5’- dihidro-2' H-espiro[ciclohexano-1,3’-imidazo[1,5-a]piridin]-6’-il)amino)pirimidin-4-il)ciclopropanocarboxamida (Comp. N.° 78)
La síntesis del compuesto 78 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento B. Sólido de color amarillo claro; Rendimiento: 50 mg, 16 %; EM (IEN) m/z 447,34 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,95 (s, 1H), 10,27 (s, 1H), 9,51 (s, 1H), 8,74 (s, 1H), 8,60 (s, 1H), 7,99 (s, 1H), 5,64 (m, 1H), 2,95 (m, 1H), 2,1 (m, 1H), 2,02 (m, 1H), 1,72 (m, 4H), 1,57 (m, 1H), 1,39 (m, 1H), 0,84 (d, J = 5,12 Hz, 4H). Ejemplo 79
Síntesis de 3,3-dimetil-6-(pirimidin-4-ilamino)-8-vinil-2H-imidazo[1,5-a]piridin-1,5-diona (Comp. N.° 79)

Síntesis de 3,3-dimetil-6-(pirimidin-4-ilamino)-8-vinil-2H-imidazo[1,5-a]piridin-1,5-diona (Comp. N ° 79)
La síntesis del compuesto 79 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento G. Sólido de color amarillo pálido; Rendimiento: 50 mg, 17 %; EM (IEN) m /z 298,3 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 9,74 (s, 1H), 9,47 (s, 1H), 9,14 (s, 1H), 8,81 (s, 1H), 8,40 (d, J = 5,88 Hz, 1H), 7,85 (dd, J =
11,04 Hz, 1H), 7,40 (d, J = 5,88 Hz, 1H), 5,75 (d, J = 6,12 Hz, 1H), 5,37 (d, .7=11,24 Hz, 1H), 1,81 (s, 6H).
Ejemplo 80
Síntesis de W-(6-((8-cloro-3-metil-1,5-dioxo-3-(3-(trifluorometil)fenil)-1,2,5,5-tetrahidroimidazo[1,5-a]piridin-6-il)amino)pirimidin-4-il)ciclopropanocarboxamida (Comp. N.° 80)

Síntesis de 6-bromo-8-cloro-3-metil-3-(3-(trifluorometil)fenil)-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona (3)
La síntesis del intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento A. Sólido de color blanquecino; Rendimiento: 0,12 g, 14 %; EM (IEN) m/z 419 [M-1]-.
Síntesis de N-(6-((8-cloro-3-metil-1,5-dioxo-3-(3-(rifluorometil) fenil)-1,2,3,5-tetrahidroimidazo[1,5-a]piridin-6-il)amino)pirimidin-4-il)ciclopropanocarboxamida (Comp. N.° 80)
La síntesis del compuesto 80 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento B. Sólido de color blanquecino; Rendimiento: 16 mg, 11 %; EM (IEN) m/z 519 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 8,75 (s, 1H), 8,58 (s, 1H), 7,90 (s, 1H), 7,82 (s, 1H), 7,75 (d, J = 7,4 Hz, 1H), 7,68 (d, J = 7,92 Hz, 1H), 7,62 (d, J = 7,7 Hz, 1H), 2,27 (s, 3H), 1,98 (m, 1H), 0,81 (m, 4H).
Ejemplo 81
Síntesis de 8-metoxi-3,3-dimetil-6-(pirimidin-4-ilamino)-2H-imidazo[1,5-a]piridin-1,5-diona (Comp. N.° 81)

La síntesis del intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento A. Sólido de color marrón claro; Rendimiento: 0,5 g, 41 %; RMN 1H (400 MHz, DMSO-d6) ó 7,87 (s,
1H), 6,75 (s ancho, 1H), 3,94 (s, 3H), 1,93 (s, 6H).
Síntesis de 8-metoxi-3,3-dimetil-6-(pirimidin-4-ilamino)-2H-imidazo[1,5-a]piridin-1,5-diona (Comp. N ° 81)
La síntesis del compuesto 81 se realizó como se ha descrito anteriormente usando el protocolo general del procedimiento B. Sólido de color amarillo; Rendimiento: 0,060 g, 15 %; RMN 1H (400 MHz, DMSO-d6) ó 9,50-9,41 (s ancho, 1H), 8,79 (s, 1H), 8,39 (d, J = 4,0 Hz, 1H), 7,40 (d, J = 8,0Hz, 1H), 7,13 (s ancho, 1H), 3,85 (s, 3H), 1,76 (s, 6H).
Ejemplo 82
Síntesis de 3,3-dimetil-8-metilsulfanil-6-(pirimidin-4-ilamino)-2H-imidazo[1,5-a]piridin-1,5-diona (Comp. N.° 82)

Síntesis de 3,3-dimetil-8-metilsulfanil-6-(pirimidin-4-ilamino)-2H-imidazo[1,5-a]piridin-1,5-diona (Comp. N ° 82) A un vial se le añadió 8-cloro-3,3-dimetil-6-(pirimidin-4-ilamino)-2H-imidazo[1,5-a]piridin-1,5-diona (0,30 g, 0,98 mmol) en dimetilformamida (5 ml). Se añadió tiometóxido de sodio (0,14 g, 1,96 mmol) a temperatura ambiente, se selló el vial y se calentó a 110 °C durante 36 h. La mezcla de reacción se inactivó con una solución acuosa de cloruro de amonio y se extrajo con metanol al 10 % en diclorometano. Las capas orgánicas se separaron, combinaron, se secaron sobre sulfato de sodio, se filtraron y se concentraron a sequedad. Los sólidos se precipitaron mediante la adición de hielo, se filtraron y se secaron. El material sólido se sometió a HPLC preparativa para proporcionar 3,3-dimetil-8-metilsulfanil-6-(pirimidin-4-ilamino)-2H-imidazo[1,5-a]piridin-1,5-diona (Comp. N.° 82) en forma de un sólido de color amarillo. Rendimiento: 0,060 g, 19 %; EM (IEN) m/z 318,42 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,02 (s, 1H), 9,66 (s, 1H), 8,92 (s, 1H), 8,84 (s, 1H), 8,45 (d, J = 6,0 Hz, 1H), 7,47 (d, J = 6,4 Hz, 1H), 2,47 (s, 3H), 1,80 (s, 6H).
Ejemplo 83
Síntesis de W-(6-((8-cloro-3-metil-1,5-dioxo-3-(m-tolil)-1,2,3,5-tetrahidroimidazo[1,5-a]piridin-6-il)amino)pirimidin-4-il)ciclopropanocarboxamida (Comp. N.° 83)



Síntesis de 6-bromo-8-cloro-3-metil-3-(m-tolil)-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona (3)
La síntesis del producto intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento A. Sólido de color blanquecino; Rendimiento: 0,30 g, 41 %; EM (IEN) m/z 367 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,32 (s, 1H), 8,29 (s, 1H), 7,26 (m, 1H), 7,16 (m, 3H), 2,30 (s, 3H), 2,18 (s, 3H).
Síntesis de N-(6-((8-cloro-3-metil-1,5-dioxo-3-(m-tolil)-1,2,3,5-tetrahidroimidazo-[1,5-a]piridin-6-il)amino)pirimidin-4-il)ciclopropanocarboxamida (Comp. N.° 83)
La síntesis del compuesto 83 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento H. Sólido de color amarillo pálido; Rendimiento: 60 mg, 16 %; EM (IEN) m/z 465,49 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,90 (s, 1H), 10,04 (s, 1H), 9,40 (s, 1H), 8,73 (s, 1H), 7,88 (s, 1H), 7,26 (m, 1H), 7,21 (m, 1H), 7,12 (m, 2H), 2,29 (s, 3H), 2,28 (s, 3H) 1,99 (m, 1H), 0,82 (m, 4H).
Ejemplo 84
Síntesis de W-[6-[(3-íerc-butil-8-cloro-3-metil-1,5-dioxo-2H-imidazo[1,5-a]piridin-6-il)amino]pirimidin-4-il]ciclopropanocarboxamida (Comp. N.° 84)

Síntesis de (6-bromo-3-terc-butil-8-cloro-3-metil-2H-imidazo[1,5-a]piridin-1,5-diona (3)
La síntesis del intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento A. Sólido de color amarillo; Rendimiento: 150 mg, 28 %; EM (IEN) m/z 332,8 [M 1]+.
Síntesis de N-[6-[(3-terc-butil-8-cloro-3-metil-1,5-dioxo-2H-imidazo[1,5-a]piridin-6-il)amino]pirimidin-Il]ciclopropanocarboxamida (Comp. N.° 84)
La síntesis del compuesto 84 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento H. Sólido de color marrón; Rendimiento: 0,060 g, 31 %; EM (IEN) m/z 431,42 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,91 (s, 1H), 9,73 (s, 1H), 9,49 (s, 1H), 8,67 (s, 1H), 8,58 (s, 1H), 7,98 (s, 1H), 2,02 (m, 1H), 1,92 (s, 1H), 0,98 (s, 9H), 0,83 (m, 4H).
Ejemplo 85
Síntesis de N-[6-[[8-cloro-1,5-dioxo-3-(trifluorometil)-2,3-dihidroimidazo[1,5-a]piridin-6-il]amino]pirimidin-4-il]ciclopropanocarboxamida (Comp. N.° 85)


Síntesis de 6-bromo-8-cloro-3-(trifluorometil)-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona (3)
La síntesis del producto intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento A. Sólido de color marrón; Rendimiento: 200 mg, 61 %; RMN 1H (400 MHz, DMSO-d6) 5 10,93 (s, 1H), 8,34 (m, 1H), 6,57 (m, 1H).
Síntesis de N-[6-[[8-doro-1,5-dioxo-3-(trifluorometil)-2,3-dihidroimidazo[1,5-a]piridin-6-il]amino]pirimidin-4-il]ciclopropanocarboxamida (Comp. N.° 85)
La síntesis del compuesto 85 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento B. Sólido de color amarillo pálido; Rendimiento: 0,035 g, 14 %; EM (IEN) m/z 429,33 [M 1]+; RMN 1H (400 MHz, DMSO-d6) 510,95 (s, 1H), 10,58 (s, 1H), 9,80 (s, 1H), 8,76 (s, 1H), 8,61 (s, 1H), 8,06 (s, 1H), 6,61 (d, J = 3,0 Hz, 1H), 2,02 (m, 1H), 0,84 (m, 4H).
Ejemplo 86
Síntesis de N-[6-[(8-cloro-1,5-dioxo-espiro[2H-imidazo[1,5-a]piridin-3,3'-azetidin]-6-il)amino]pirimidin-4-il]ciclopropanocarboxamida (Comp. N.° 86)


Síntesis de 6-bromo-8-cloro-1,5-dioxoespiro[2H-imidazo[1,5-a]piridin-3,3’-azetidin]-1 ’-carboxilato de terc-butilo (3)
A una disolución agitada de 5-bromo-3-cloro-6-oxo-1H-piridin-2-carboxamida (1,0 g, 3,98 mmol) en 1,4-dioxano (12 ml) en un vial se le añadió 3-oxoazetidin-1-carboxilato de terc-butilo (2, 2,72 g, 15,91 mmol) a temperatura ambiente. Después, se añadió gota a gota ácido sulfúrico concentrado (0,39 g, 3,98 mmol). El vial se selló y se calentó a 95 °C durante 16 h. La mezcla de reacción se concentró, se añadió agua (30 ml) y se extrajo con acetato de etilo (30 ml, 2 veces). Las capas orgánicas se separaron y se desecharon.
A una solución agitada de 6-bromo-8-cloroespiro[2H-imidazo[1,5-a]piridin-3,3'-azetidin]-1,5-diona (0,41 g, 1,35 mmol) en 1,4-dioxano (30 ml) y agua (25 ml, 1,35 mmol) se le añadió dicarbonato de di-terc-butilo (0,59 g, 2,71 mmol). La solución se enfrió a 0 °C y se añadió gota a gota una solución acuosa 2 M de hidróxido de sodio con agitación. La mezcla de reacción se agitó a temperatura ambiente durante una noche. Después del consumo de material de partida como se indicó por CCF, la mezcla se extrajo con metanol al 10 % en diclorometano (25 ml, 2 veces). Las capas orgánicas se separaron y se secaron sobre sulfato de magnesio, se filtraron y se concentraron a sequedad. El residuo en bruto se purificó después por cromatografía en columna ultrarrápida usando alúmina neutra con metanol al 2 % en diclorometano. Las fracciones deseadas se concentraron a sequedad al vacío para proporcionar 6 -bromo-8-cloro-1,5-dioxo-espiro[2H-imidazo[1,5-a]piridin-3,3'-azetidin]carboxilato de etilo (3) en forma de un sólido de color amarillo. Rendimiento: 180 mg, 33 %; EM (IEN) m/z 404,03 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,53 (s, 1H), 8,28 (s, 1H), 4,88-4,68 (m, 2H), 3,97-3,89 (m, 2H), 1,42 (s, 9H).
Síntesis de 8-cloro-6-[[6-(ciclopropanocarbonilamino)pirimidin-4-il]amino]-1,5-dioxoespiro[2H-imidazo[1,5-a]piridin-3’-azetidin]-1’-carboxilato de etilo (5)
La síntesis del intermedio 5 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento B. Sólido de color amarillo; Rendimiento: 0,065 g; 29 %; RMN 1H (400 MHz, DMSO-d6) ó 10,93 (s, 1H), 10,46 (s, 1H), 9,63 (s, 1H), 8,71 (s, 1H), 8,59 (s, 1H), 8,10 (s, 1H), 4,89-4,84 (m, 2H), 3,97-3,95 (m, 2H), 2,02 2,0 (m, 1H), 1,42 (s, 9H), 0,79-0,75 (m, 4H).
Síntesis de N-[6-[(8-cloro-1,5-dioxo-espiro[2H-imidazo[1,5-a]piridin-3,3’-azetidin]-6-il)amino]pirimidin-4-il]ciclopropanocarboxamida (Comp. N.° 86)
La síntesis del compuesto 86 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento F. Sólido de color amarillo claro; Rendimiento: 0,03 g; 61 %; EM (IEN) m/z 402,34 [M 1]+; RMN 1H (400 MHz, DMSO-de) ó 11,04 (s, 1H), 10,41 (s, 1H), 9,67 (s, 1H), 9,5 (s ancho, 1H), 8,79 (s, 1H), 8,74 (s, 1H), 8,06 (s, 1 H).
Ejemplo 87
Síntesis de 6-[(6-aminopirimidin-4-il)amino]-8-cloro-3-(3-fluorofenil)-3-metil-2H-imidazo[1,5-a]pi ridin-1,5-diona (Comp. N.° 87)

Síntesis de 6-[(6-aminopirimidin-4-il)amino]-8-cloro-3-(3-fluorofenil)-3-metil-2H-imidazo[1,5-a]piridin-1,5-diona (Comp. N ° 88)
Procedimiento I: En un vial que contenía N-[6-[[8-cloro-3-(3-fluorofenil)-3-metil-1,5-dioxo-2H-imidazo[1,5-a]piridin-6-il]amino]pirimidin-4-il]ciclopropanocarboxamida (1,23 g, 0,49 mmol) en tetrahidrofurano (5 ml), agua (5 ml) y etanol (10 ml) a temperatura ambiente se añadió una solución acuosa concentrada de hidróxido de potasio (0,14 g, 2,45 mmol). Después, la mezcla de reacción se agitó durante 16 h y se extrajo con acetato de etilo (50 ml, 3 veces). Las capas orgánicas se separaron después y se secaron sobre sulfato de sodio, se filtraron y se concentraron a sequedad. El residuo en bruto se sometió a cromatografía en columna ultrarrápida con metanol al 2 % en diclorometano. Las fracciones deseadas se concentraron a sequedad. El sólido resultante se lavó con pentano y éter dietílico para proporcionar el producto en forma de un sólido de color amarillo. Rendimiento: 0,095 g, 48 %; EM (IEN) m/z 401,40 [M 1]+; RMN 1H (400 MHz, DMSO-d6) 610,02 (s ancho, 1H), 8,86 (s, 1H), 8,68 (s, 1H), 8,19 (s, 1H), 7,45-7,38 (m, 1H), 7,30 (d, J = 8,8 Hz, 1H), 7,23-7,15 (m, 2H), 6,58 (s ancho, 2H), 6,17 (s, 1H), 2,21 (s, 1H).
Ejemplo 88
Síntesis de 6-[(6-aminopirimidin-4-il)amino]-8-cloro-3,3-dimetil-2H-imidazo[1,5-a]piridin-1,5-diona (Comp. N.° 88)

Síntesis de 6-[(6-aminopirimidin-4-il)amino]-8-cloro-3,3-dimetil-2H-imidazo[1,5-a]piridin-1,5-diona (Comp. N ° 88) La síntesis del compuesto 88 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento I. Sólido de color amarillo claro; Rendimiento: 0,14 g, 68 %; EM (IEN) m/z 321,34 [M 1]+; RMN 1H (400 MHz, DMSO-d6) 69,64 (s, 1H), 8,92 (s, 1H), 8,64 (s, 1H), 8,20 (s, 1H), 6,60 (s, 2H), 6,25 (s, 1H), 1,79 (s, 6H). Síntesis de clorhidrato de 6-[(6-aminopirimidin-4-il)amino]-8-cloro-3,3-dimetil-2H-imidazo[1,5-a]piridin-1,5-diona (sal cloruro de hidrogeno de Comp. N ° 88)
A 6-[(6-aminopirimidin-4-il)amino]-8-cloro-3,3-dimetil-2H-imidazo[1,5-a]piridin-1,5-diona (Comp. N.° 88, 0,55 g, 1,71 mmol), en diclorometano (10 ml), se le añadió metanol (10 ml) en un vial cloruro de hidrogeno 4 M en 1,4
dioxano (10 ml). Después de agitar durante la noche a temperatura ambiente, la mezcla de reacción se concentró al vacío, se filtró, se lavó con metanol después con éter dietílico y se secó al vacío para proporcionar el producto en forma de un sólido de color amarillo claro. Rendimiento: 0,61 g, 100 %; EM (IEN) m/z 319,43 [M 1]+; RMN 1H (400 MHz, DMSO-cfe) ó 9,83 (s, 1H), 9,77 (s, 1H), 8,48 (s, 1H), 8,42 (s, 1H), 7,75 (s, 2H), 6,49 (s, 1H), 1,79 (s, 6H). Ejemplo 89
Síntesis de 8-cloro-3-(3-clorofenil)-3-metil-6-[[6-(metilamino)pirimidin-4-il]amino]-2H-imidazo[1,5-a]pi ridin-1,5-diona (Comp. N.° 89)

Síntesis de 8-cloro-3-(3-clorofenil)-3-metil-6-[[6-(metilamino)pirimidin-4-il]amino]-2H-imidazo[1,5-a]piridin-5-diona (Comp. N.° 89)
La síntesis del compuesto 89 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento H. Sólido de color amarillo; Rendimiento: 50 mg, 16 %; EM (IEN) m/z 431,32 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,03 (s, 1H), 8,89 (s, 1H), 8,70 (s, 1H), 8,25 (s, 1H), 7,51 (s, 1H), 7,41 (m, 2H), 7,37 (m, 1H), 7,05 (s, 1H), 6,24 (s, 1H), 2,68 (s, 3H), 2,19 (s, 3H).
Ejemplo 90
Síntesis de 8-cloro-3-metil-3-(6-metil-2-piridil)-6-(pirimidin-4-ilamino)-2H-imidazo[1,5-a]pi ridin-1,5-diona (Comp. N.° 90)


Síntesis de 6-bromo-8-cloro-3-metil-3-(6-metil-2-piridil)-2H-imidazo[1,5-a]piridin-1,5-diona (3)
La síntesis del intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento A. Sólido de color marrón; Rendimiento: 0,41 g, 56 %; EM (IEN) m/z 370 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,28 (m, 1H), 8,30 (s, 1H), 7,73 (m, 1H), 7,39 (m, 1H), 7,24 (m, 1H), 2,29 (s, 3H), 2,20 (s, 3H).
Síntesis de 8-cloro-3-metil-3-(6-metil-2-piridil)-6-(pirimidin-4-ilamino)-2H-imidazo[1,5-a]piridin-1,5-diona (Comp. N° 90)
La síntesis del compuesto 90 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento B. Sólido de color amarillo claro; Rendimiento: 0,15 g, 48 %; EM (IEN) m/z 383,37. [M 1]+; RMN 1H (400 MHz, DMSO-cfe) ó 10,00 (s, 1H), 9,48 (s, 1H), 8,83 (s, 1H), 8,81 (s, 1H), 8,39 (d, J = 11,92 Hz, 7,72 (t, J = 7,78 Hz, 1H), 7,37 (m, 1H), 7,31 (m, 1H), 7,22 (m, 1H), 2,38 (s, 3H), 2,25 (s, 3H).
Ejemplo 91
Síntesis de W-[6-[[8-cloro-3-(3-fluorofenil)-1,5-dioxo-2,3-dihidroimidazo[1,5-a]piridin-6-il]amino]pirimidin-4-il]ciclopropanocarboxamida (Comp. N.° 91)

Una mezcla de 5-bromo-3-cloro-6-oxo-1H-piridin-2-carboxamida (1, 0,4 g, 1,59 mmol), 3-fluorobenzaldehído (2, 0,69 g, 5,57 mmol), acetonitrilo 15 ml) y cloruro de hierro (III) (1,81 g, 11,13 mmol) en un tubo sellado se calentó a 90 °C durante 16 h. Una vez que el análisis por CCF mostrara el consumo del material de partida, la mezcla se enfrió y se filtró a través de un lecho de celite. El celite se lavó con acetonitrilo y el filtrado se concentró al vacío. El residuo en bruto se sometió a cromatografía en columna ultrarrápida eluyendo con acetato de etilo al 35 % en hexano. Se recogieron las fracciones deseadas, se concentraron y se secaron a alto vacío para proporcionar 6-bromo-8-cloro-3-(3-fluorofenil)-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona (3) en forma de un sólido de color amarillo. Rendimiento: 0,35 g, 61 %; EM (IEN) m/z 357,20 [M 1]+.
Síntesis de N-[6-[[8-cloro-3-(3-fluorofenil)-1,5-dioxo-2,3-dihidroimidazo[1,5-a]-piridin-6-il]amino]pirimidin-4-il]ciclopropanocarboxamida (Comp. N.° 91)
La síntesis del compuesto 91 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento H. Sólido de color amarillo; Rendimiento: 46 mg, 18 %. EM (IEN) m/z 455,33 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,89 (s, 1H), 10,06 (s, 1H), 9,47 (s, 1H), 8,76 (s, 1H), 8,58 (s, 1H), 7,90 (s, 1H), 7,44 (m, 1H), 7,41 (m, 1H), 7,24 (m, 2H), 6,62 (s, 1H), 2,00 (t, J = 5,68 Hz, 1H), 0,81 (m, 4H).
Ejemplo 92
Síntesis de 6-[(6-aminopirimidin-4-il)amino]-3-(3-clorofenil)-3,8-dimetil-2H-imidazo[1,5-a]pi ridin-1,5-diona (Comp. N.° 92)

Síntesis de 6-bromo-3-(3-clorofenil)-3,8-dimetil-2H-imidazo[1,5-a]piridin-1,5-diona (3)
La síntesis del producto intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del procedimiento A. Sólido de color crema; Rendimiento: 0,25 g, 39 %; EM (IEN) m/z 397,23 [M 1]+.
Síntesis de N-[6-[[3-(3-clorofenil)-3,8-dimetil-1,5-dioxo-2H-imidazo[1,5-a]piridin-6-il]amino]pirimidin-4-il] carbamato de terc-butilo (5)
La síntesis del producto intermedio 5 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento H. Semisólido blanquecino; Rendimiento: 0,26 g, 31 %; EM (IEN) m/z 497,06 [M 1]+.
Síntesis de 6-[(6-aminopirimidin-4-il)amino]-3-(3-clorofenil)-3,8-dimetil-2H-imidazo[1,5-a]piridin-1,5-diona (Comp. N.° 92)
La síntesis del compuesto 92 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento F. Sólido de color amarillo; Rendimiento: 0,077 g, 37 %; EM (IEN) m/z 397,13 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 9,80 (s, 1H), 8,57 (s, 1H), 8,46 (s, 1H), 8,15 (s, 1H) J = 6,3 Hz, 1H), 7,41 (m, 3H), 7,27 (d, J = 6,3 Hz, 1H), 6,50 (s, 2H), 6,10 (s, 1H), 2,47 (s, 3H), 2,20 (s, 3H).
Ejemplo 93
Síntesis de 6-[(6-aminopirimidin-4-il)amino]-8-cloro-3-(3-clorofenil)-3-metil-2H-imidazo[1,5-a]pi ridin-1,5-diona (Comp. N.° 93)

Síntesis de 6-[(6-aminopirimidin-4-il)amino]-8-cloro-3-(3-clorofenil)-3-metil-2H-imidazo[1,5-a]piridin-1,5-diona (Comp. N.° 93)
La síntesis del compuesto 93 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento I. Sólido de color amarillo; Rendimiento: 0,17 g, 57 %; EM (IEN) m/z 417,08 [M 1]+; RMN 1H
(400 MHz, DMSO-cfe) 88,55 (s, 1H), 8,15 (s, 1H), 7,39-7,35 (m, 3H), 6,08 (s, 1H), 2,17 (s, 3H).
Ejemplo 94
Síntesis de 6-[(6-aminopirimidin-4-il)amino]-8-cloro-espiro[2H-imidazo[1,5-a]piridin-3,1'-ciclopentano]-1,5-diona (Comp. N.° 94)

4 M H C I/d io x a n o
DCM/MeOH
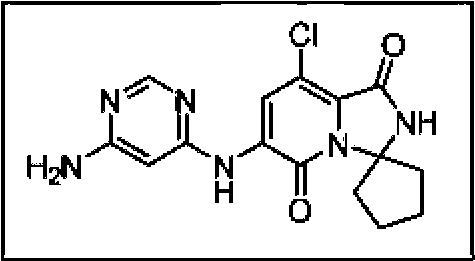
Síntesis de 6-bromo-8-cloro-espiro[2H-imidazo[1,5-a]piridin-3,1 ’-ciclopentano]-1,5-diona (3)
La síntesis del producto intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento A. Sólido de color blanquecino; Rendimiento: 380 mg; 60 %; EM (IEN) m/z 315,06 [M-1]-.
Síntesis de N-[6-[(8-cloro-1,5-dioxoespiro[2H-imidazo[1,5-a]piridin-3,1 ’-ciclopentan]-6-il)amino]pirimidin-4-il]carbamato de terc-butilo (5)
La síntesis del intermedio 5 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento H. Sólido de color amarillo claro; Rendimiento: 0,30 g, 71 %. EM (IEN) m/z 447,03 [M 1]+.
Síntesis de 6-[(6-aminopirimidin-4-il)amino]-8-cloro-espiro[2H-imidazo[1,5-a]ppiridin-3,1 ’-ciclopentano]-1,5-diona (Comp. N.° 94)
La síntesis del compuesto 94 se realizó como se ha descrito anteriormente usando el protocolo general del procedimiento F. Sólido de color amarillo claro; Rendimiento: 0,07 g, 45 %. EM (IEN) m/z 347,14 [M 1]+; RMN 1H (400 MHz, DMSO-d6) 810,04 (s, 1H), 9,15 (s, 1H), 8,60 (s, 1H), 8,26 (s, 1H), 6,85 (s, 2H), 6,30 (s, 1H), 2,77 (s, 2H), 1,97 (s, 2H), 1,77 (m, 4H).
Ejemplo 95
Síntesis de W-[6-[(8-cloro-1,1',5-trioxo-espiro[2H-imidazo[1,5-a]piridin-3,3'-tietan]-6-il)amino]pirimidin-4-il]ciclopropanocarboxamida (Comp. N.° 95)

Una solución al 30 % de peróxido de hidrógeno (0,53 g, 15,55 mmol) se añadió por goteo a 6-bromo-8-cloroespiro[2H-imidazo[1,5-a]piridin-3,3'-tietano]-1,5-diona (1, 1,0 g, 3,11 mmol) disuelto en acetona (15 ml). Se añadió ácido acético (18,66 mg, 0,31 mmol) y la mezcla se agitó a temperatura ambiente durante 16 h. Después de que el análisis por CCF mostrara el consumo de materiales de partida, la mezcla de reacción se concentró a presión reducida. Los sólidos se lavaron con éter dietílico y diclorometano. El sólido se filtró y se secó al vacío para proporcionar 6-bromo-8-cloro-1-oxo-espiro[2H-imidazo[1,5-a]piridin-3,3'-tietano]-1,5-diona (2) en forma de un sólido de color blanquecino. Rendimiento: 0,4 g, 38 %; EM (IEN) m/z 335 [M 1]+; RMN 1H (400 MHz, DMSO-cfe) ó 8,32 (s, 1H), 4,62 (d, J = 12 Hz, 2H), 3,54 (d, J = 16 Hz, 2H).
Síntesis de N-[6-[(8-cloro-1,1 5-trioxo-espiro[2H-imidazo[1,5-a]piridin-3,3’-tietan-6-il)amino]pirimidinil]ciclopropanocarboxamida (Comp. N.° 95)
La síntesis del compuesto 95 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento H. Sólido de color amarillo claro; Rendimiento: 0,08 g, 24 %; EM (IEN) m/z 435,4 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,91 (s ancho, 1H), 10,45-10,32 (m, 1H), 8,76 (s ancho, 1H), 8,59 (s ancho, 1H), 8,04 (s ancho, 1H), 4,66-4,63 (m, 2H), 3,59-3,56 (m, 2H), 2,01 (s ancho, 1H), 0,84 (s, 4H).
Ejemplo 96
Síntesis de 8-cloro-1',1'-dimetil-6-(pirimidin-4-ilamino)espiro[2H-imidazo[1,5-a]piridin-3,2,-ciclohexano]-1,5-diona (Comp. N.° 96)

Síntesis de 6-bromo-8-cloro-1’,1 ’-dimetil-espiro[2H-imidazo[1,5-a]piridin-3,2’-ciclohexano]-1,5-diona (3)
La síntesis del intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento A. Sólido de color marrón claro; Rendimiento: 0,41 g, 57 %; EM (IEN) m/z 360,8 [M 1]+.
Síntesis de 8-cloro-1 1 '-dimetil-6-(pirimidin-4-ilamino)espiro[2H-imidazo[1,5-a]piridin-3,2'-ciclohexano]-1,5-diona (Comp. N ° 96)
La síntesis del compuesto 96 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento B. Sólidos de color blanquecino; Rendimiento: 25 mg, 10 %; EM (IEN) m/z 374,19 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 9,98 (s ancho, 1H), 9,60 (s, 1H), 8,84 (s, 1H) 8,76 (s, 1H) 8,43 (d, J = 4 Hz, 1H), 7,47 (d, J =
4 Hz, 1H), 3,61-3,50 (m, 1H), 1,75-1,36 (m, 7H), 1,25 (s, 3H), 0,64 (s, 3H).
Ejemplo 97
Síntesis de W-(6-((8-cloro-3-metil-1,5-dioxo-3-fenil-1,2,5,5-tetrahidroimidazo[1,5-a]piridin-6-il)amino)pirimidin-4-il)ciclopropanocarboxamida (Comp. N.° 97)

Síntesis de 3-bromo-8-cloro-3-metil-3-fenil-2H-imidazo[1,5-a]piridin-1,5-diona (3)
La síntesis del intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento A. Sólido de color verde; Rendimiento: 525 mg, 75 %; EM (IEN) m/z 353,21 [M 1]+.
Síntesis de N-[6-[(8-cloro-3-metil-1,5-dioxo-3-fenil-2H-imidazo[1,5-a]piridin-6-il]amino]pirimidin-4-il]ciclopropanocarboxamida (Comp. N.° 97)
La síntesis del compuesto 97 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento H. Sólido de color amarillo claro; Rendimiento: 97 mg, 15 %. EM (IEN) m/z 451,13 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,89 (s, 1H), 10,06 (s, 1H), 9,39 (s, 1H), 8,73 (s, 1H), 8,58 (s, 1H), 7,88 (s, 1H), 7,39 (s, 5H), 2,24 (s, 3H), 1,99 (s, 1H), 0,81 (s, 4H).
Ejemplo 98
Síntesis de clorhidrato de 6-[(6-aminopirimidin-4-il)amino]-8-metil-espiro[2H-imidazo[1,5-a]piridin-3,1'-ciclopentano]-1,5-diona (Comp. N.° 98)


KOH
EtOH THF H ,0

Síntesis de 6-bromo-8-metil-espiro[2H-imidazo[1,5-a]piridin-3,1 '-ciclopentano]-1,5-diona (3)
La síntesis del producto intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento A. Sólido de color blanquecino; Rendimiento: 1,8 g, 70 %; EM (IEN) m/z 297,15 [M 1]+.
Síntesis de N-[6-[(8-metil-1,5-dioxoespiro[2H-imidazo[1,5-a]piridin-3,1 ’-ciclopentano]-6-il)amino]pirimidin-4-il]-ciclopropanocarboxamida (5)
La síntesis del intermedio 5 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento H. Sólido de color blanquecino; Rendimiento: 1,30 g, 58 %; EM (IEN) m/z 395,37 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,86 (s, 1H), 9,88 (s, 1H), 9,20 (s, 1H), 8,51 (d, J = 15,96 Hz, 2H), 7,86 (s, 1H), 2,81 (m, 2H), 2,45 (m, 3H), 2,09 (m, 3H), 1,82 (m, 2H), 1,67 (m, 2H), 0,72 (m, 4H).
Síntesis de clorhidrato de 6-[(6-aminopirimidin-4-il)amino]-8-metil-espiro[2H-imidazo[1,5-a]-piridin-3,1 ’-ciclopentano]-1,5-diona (Comp. N.° 98)
La síntesis del compuesto 98 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento I. Sólido de color blanquecino; Rendimiento: 0,76 g, 87 %; EM (IEN) m/z 327,49 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,02 (s, 1H), 9,75 (s, 1H), 8,46 (s, 1H), 8,12 (s, 1H), 7,88 (s, 2H), 6,39 (s, 1H), 2,77 (m, 2H), 2,41 (s, 3H), 1,96 (m, 2H), 1,83 (m, 2H), 1,70 (m, 2H).
Ejemplo 99
Síntesis de 6-[(6-aminopirimidin-4-il)amino]-8-cloro-4',4'-difluoro-espiro[2H-imidazo[1,5-a]piridin-3,1 '-ciclohexano]-1,5-diona (Comp. N.° 99)

Síntesis de 6-bromo-8-cloro-4’,4'-difluoroespiro[2H-imidazo[1,5-a]piridin-3,1’-ciclohexano]-1,5-diona (3)
La síntesis del producto intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento A. Sólido de color blanquecino; Rendimiento: 5,9 g. 80 %; EM (IEN) m/z 364,92 [M-1 ]-.
Síntesis de N-[6-[(8-cloro-4’,4'-difluoro-1,5-dioxoespiro[2H-imidazo[1,5-a]piridin-3,1’-ciclohexan]-6-i)amino]pirimidin-4-il]ciclopropanocarboxamida (5)
La síntesis del intermedio 5 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento B. Sólido de color blanquecino; Rendimiento: 4,71 g, 63 %; EM (IEN) m/z 465,38 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,92 (s, 1H), 10,47 (s, 1H), 9,51 (s, 1H), 8,71 (s, 1H) 8,59 (s, 1H), 7,98 (s, 1H), 3,32 (m, 2H), 2,24 (m, 4H), 2,02 (m, 1H), 1,71 (m, 2H), 0,64 (m, 4H).
Síntesis de 6-[(6-aminopirimidin-4-il)amino]-8-cloro-4’,4'-difluoro-espiro[2H-imidazo[1,5-a]piridin-3,1’-ciclohexano-5-diona (6)
La síntesis del intermedio 6 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento I. Sólido de color amarillo claro; Rendimiento: 0,35 g, 41 %; EM (IEN) m/z 397,33 [M 1]+.
Síntesis de 6-[(6-aminopirimidin-4-il)amino]-8-cloro-4’,4’-difluoro-espiro[2H-imidazo-[1,5-a]piridin-3,1’-ciclohexano-1,5-diona (Comp. N.° 99)
La síntesis del compuesto 99 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento F. Sólido de color amarillo; Rendimiento: 0,66 g, 92 %; EM (IEN) m/z 397,17 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,56 (s, 1H), 9,87 (s, 1H), 8,51 (s, 1H), 8,43 (s, 1H), 7,89 (s, 2H), 6,52 (s, 1H), 3,22 (m, 2H), 2,21 (m, 4H), 1,74 (d, J = 12,12 Hz, 2H).
Ejemplo 100
Síntesis de 8-cloro-6-((7-ciclopropilpirido[4,3-d]pirimidin-4-il)amino)-3,3-dimetil-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona (Comp. N.° 100)


A una solución bien agitada de 4,6-dicloropiridin-3-carboxamida (11,0 g, 57,59 mmol) en 1,4-dioxano (20 ml) en una bomba de acero se le añadió amoníaco líquido (50 ml, 57,59 mmol). La bomba de acero se cerró y la reacción se calentó a 100 °C durante 9 h. El progreso de la reacción de desplazamiento se controló por CCF y CLEM. Después de la terminación, el sólido formado se filtró. El filtrado también se concentró ya que también contenía un 50 % de producto. Los sólidos combinados se secaron al vacío para proporcionar 4-amino-6-cloro-piridin-3-carboxamida (2) en forma de un sólido de color marrón claro. Rendimiento: 10,6 g, producto en bruto, 47 %; RMN 1H (400 MHz, DMSO-cfe) ó 8,36 (s, 1H), 7,97 (s, 1H), 7,50 (s, 1H), 7,37 (s, 1H), 6,65 (s, 1H).
Síntesis de 7-cloro-3H-pirido[4,3-d]pirimidin-4-ona (3)
Se añadió 4-amino-6-cloro-piridin-3-carboxamida (2, 4,5 g, 26,23 mmol) a un tubo de presión y se añadió ortoformiato de trietilo (30 ml). El recipiente de reacción se selló y se calentó a 140 °C durante 11 h. Una vez completada la ciclación, la mezcla de reacción se concentró a presión reducida y al sólido obtenido se le añadió éter dietílico. El sólido se filtró, se lavó con éter dietílico y se secó al vacío para proporcionar 7-cloro-3H-pirido [4,3-d]pirimidin-4-ona (3) en forma de un sólido de color marrón. EM (IEN) m/z 181,90 [M 1]+.
Síntesis de 7-cloro-3-(2-trimetilsililetoximetil)pirido[4,3-d]pirimidin-4-ona (4)
En un matraz de fondo redondo seco en atmósfera de N2 se añadió 7-cloro-3H-pirido[4,3-d]pirimidin-4-ona (3,9.0 g, 49,56 mmol) a dimetilformamida (50 ml). La solución se agitó a 0 °C y se añadió hidruro de sodio (1,78 g, 74,35 mmol) en porciones durante 10 min. La suspensión aniónica se agitó durante 1 h a 0 °C seguido de la adición de cloruro de 2-(trimetilsilil) etoximetilo (12,4 g, 74,35 mmol) a 0 °C. La mezcla de reacción se agitó a temperatura ambiente durante 5 h. Después del consumo de los materiales de partida como se indicó por CCF, la mezcla de reacción se inactivó con hielo. Se precipita un sólido. Se filtró y se lavó con exceso de n-pentano. El sólido se secó al vacío para proporcionar 7-cloro-3-(2-trimetilsililetoximetil)pirido[4,3-d]pirimidin-4-ona (4) en forma de un sólido de color marrón. Rendimiento: 9,1 g, 59 % RMN 1H (400 MHz, DMSO-d6) ó 9,17 (s, 1H), 8,72 (s, 1H), 7,79 (s, 1H), 5,49 (s, 2H), 3,65 (t, J = 16 Hz, 2H) 0,90 (t, J = 16 Hz, 2H), -0,03 (s, 9H).
Síntesis de 7-ciclopropil-3-(2-trimetilsihietoximetil)pirido[4,3-d]pirimidin-4-ona (6)
La síntesis del intermedio 6 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento G. Líquido amarillo; Rendimiento: 2,1 g, 52 %; EM (IEN) m/z 319 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 9,16 (s, 1H), 8,57 (s, 1H), 7,52 (s, 1H), 7,27 (s, 1H), 5,33 (s, 2H), 3,63-3,59 M, 2H), 2,32-2,26 (m, 1H), 1,07-1,05 (m, 4H), 0,89 (t, J = 8,0 Hz, 1H), 0,04 (s, 9H).
Síntesis de 7-ciclopropil-3H-pirido[4,3-d]pirimidin-4-ona (7)
A 7-ciclopropil-3-(2-trimetilsililetoximetil)pirido[4,3-d]pirimidin-4-ona (6, 2,0 g, 6,3 mmol) en diclorometano (10 ml) a 0 °C se le añadió una solución de ácido trifluoroacético al 20 % en diclorometano (10 ml, 6,3 mmol). La mezcla de reacción se calentó a temperatura ambiente y se agitó durante 3 h. Después de la desprotección completa como se indicó por CCF, la mezcla de reacción se concentró, se recogió en diclorometano y se concentró. Este proceso se repitió 2-3 veces. Se añadió tetrahidrofurano (20 ml) al residuo en bruto y se enfrió a 0 °C. Se añadió solución de hidróxido de potasio 3 M (10 ml, 6,3 mmol) a la solución, llevando el pH a 9-10. La suspensión se agitó durante 6
7 h. La reacción se concentró, el residuo se diluyó con metanol al 10 % en diclorometano y las capas orgánicas se lavaron con 10 ml de agua y 10 ml de una solución saturada de salmuera. Las capas orgánicas se separaron, se secaron sobre sulfato de magnesio, se filtraron y se concentraron a sequedad para proporcionar 7-ciclopropil-3H-pirido[4,3-d]pirimidin-4-ona (7) en forma de un sólido de color marrón. Rendimiento: 1,00 g, 85 %; EM (IEN) m/z 188,18 [M 1]+; RMN 1H (400 MHz, DMSO-da) ó 12,57 (s ancho, 1H), 9,15 (s, 1H), 8,32 (s, 1H), 7,38 (s, 1H), 2,27 (s ancho, 1h), 1,03 (s ancho, 4H).
Síntesis de 7-ciclopropilpirido [4,3-d]pirimidin-4-amina (8)
A 7-ciclopropil-3H-pirido[4,3-d]pirimidin-4-ona (7,0 g, 5,34 mmol) en un matraz de fondo redondo en atmósfera de nitrógeno se le añadió N,N-diisopropiletilamina (9,45 ml, 53,42 mmol) a 0 °C. A esta mezcla agitada se añadió lentamente oxicloruro de fósforo (V) (7,48 ml, 80,13 mmol). La mezcla de reacción se agitó a temperatura ambiente durante 2-3 horas hasta que se obtuvo una solución transparente. La solución se concentró a presión reducida en condiciones inertes, se recogió en exceso de tolueno y se volvió a concentrar. Este procedimiento se repitió varias veces con N,N-diisopropiletilamina. El residuo se disolvió en acetonitrilo a lo que se añadió amoníaco acuoso al 30 %. La mezcla de reacción se diluyó con metanol al 10 % en diclorometano y se extrajo del amoníaco acuoso. Las capas orgánicas se separaron, se secaron sobre sulfato de sodio, se filtraron y se secaron sobre sulfato de sodio. Se secó el sólido obtenido para proporcionar 7-ciclopropilpirido [4,3-d]pirimidin-4-amina (8) en forma de un sólido de color marrón. Rendimiento: 0,60 g, 60 %; EM (IEN) m/z 375,8 [M 1]+; RMN 1H (400 MHz, DMSO-da) ó 9,34 (s, 1H), 8,43 (s, 1H), 8,26-7,99 (m, 2H), 7,39 (s, 1H), 1,75 (s, 1H), 1,01-0,84 (s, 4H).
Síntesis de 8-doro-6-[(7-dclopropilpirido[4,3-d]pirimidin-4-il)amino]-3,3-dimetil-2H-imidazo[1,5-a]piridin-1,5-diona (Comp. N ° 100)
La síntesis del compuesto 100 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento H. Sólido de color amarillo pálido; Rendimiento: 0,37 g, 58 %; EM (IEN) m/z 397,48 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 9,46 (s, 1H), 8,85 (s, 1H), 8,75 (s, 1H), 7,58 (s, 1H), 2,32-2,29 (m, 6H), 1,82 (s, 6H), 1,08 1,02 (m, 4H).
Ejemplo 101
Síntesis de 6-((6-aminopirimidin-4-il)amino)-3-(3-fluorofenil)-3,8-dimetil-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona (Comp. N.° 101)

Síntesis de 6-bromo-3-(3-fluorofenil)-3’, 8-dimetil-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona (3)
La síntesis del intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del
Procedimiento A. Sólido de color amarillo; Rendimiento: 0,35 g, 46 %; EM (IEN) m/z: 351,17 [M 1]+; RMN 1H (400 MHz, DMSO-cfe) ó 10,17 (s, 1H), 8,08 (s, 1H), 7,42 (m, 1H), 7,21 (m, 2H), 7,13 (d, J = 8,0 Hz, 1H), 2,43 (s, 3H), 2,1 (s, 3H).
Síntesis de (6-((3-(3-fluorofenil)-3,8-dimetil-1,5-dioxo-1,2,3,5-tetrahidroimidazo[1,5-a]piridin-6-Il)amino)pirimidin-4-il) carbamato de terc-butilo (5)
La síntesis del intermedio 5 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento H. Sólido de color amarillo; Rendimiento: 0,15 g, 34 %; EM (IEN) m/z 481,47 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,0-9,80 (s ancho, 2H), 9,01 (s ancho, 1H), 8,47 (s, 1H), 8,43 (s, 1H), 7,52 (s, 1H), 7,38 (m, 1H), 7,10-7,22 (m, 3H), 2,43 (s, 3H), 2,16 (s, 3H), 1,46 (s, 9H).
Síntesis de 6-((6-aminopirimidin-4-il)amino)-3-(fluorfenil)-3,8-dimetil-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona (Comp. N.° 101)
La síntesis del compuesto 101 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento F. Sólido de color blanquecino; Rendimiento: 0,09 g, 76 %; EM (IEN) m/z 381,50 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 9,82 (s, 1H), 8,60 (s, 1H), 8,45 (s, 1H), 8,17 (s, 1H), 7,42 (m, 1H), 7,25-7,10 (m, 3H), 6,54 (s ancho, 2H), 6,10 (s, 1H), 2,47 (s, 3H), 2,20 (s, 3H).
Ejemplo 102
Síntesis de clorhidrato de 6-((6-aminopirimidin-4-il)amino)-3,3,8-trimetil-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona (Comp. N.° 102)

Síntesis de N-(6-((3,3,8-trimetil-1,5-dioxo-1,2,3,5-tetrahidroimidazo[1,5-a]piridin-6-il)amino)pirimidin-4-il)ciclopropanocarboxamida (3)
La síntesis del producto intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento H. Sólido de color amarillo; Rendimiento: 0,85 g, 50 %; EM (IEN) m/z 369,0 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,86 (s, 1H), 9,52 (s, 1H), 9,16 (s, 1H), 8,53 (s, 1H), 8,47 (s, 1H), 7,86 (s, 1H), 5,75 (s, 1H), 2,42 (s, 3H), 2,02 (m, 1H), 1,77 (s, 6H), 0,88-0,80 (m, 4H).
Síntesis de 6-((6-aminopirimidin-4-il)amino)-3,3,8-trimetil-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona (4)
La síntesis del intermedio 4 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento I. Sólido de color amarillo; Rendimiento: 0,55 g, 80 %; EM (IEN) m/z 301,29 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 9,46 (s, 1H), 8,61 (s, 1H), 8,38 (s, 1H), 8,16 (s, 1H), 6,53 (s ancho, 2H), 6,16 (s, 1H), 2,40 (s, 3H), 1,77 (s, 6H).
S ín te s is de c lo rh id ra to de 6 -((6 -a m in o p ir im id in -4 - il)a m in o )-3 ,3 ,8 -tr im e til-2 ,3 -d ih id ro im id a z o [1 ,5 -a ]p ir id in -1 ,5 -d io n a (C om p. N .° 102)
La síntesis del compuesto 102 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento F. Sólido de color amarillo; Rendimiento: 0,60 g, 97 %; EM (IEN) m/z 301,25 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 9,71 (s, 1H), 9,67 (s, 1H), 8,45 (s, 1H), 8,12 (s, 1H), 7,95-7,70 (s ancho, 2H), 6,39 (s, 1H), 2,41 (s, 3H), 1,77 (s, 6H).
Ejemplo 103
Síntesis de 6-[(6-aminopirimidin-4-il)amino]-4',4'-difluoro-8-metil-espiro[2H-imidazo[1,5-a]piridin-3,1'-ciclohexano]-1,5-diona (Comp. N.° 103)

Síntesis de 6-bromo-4’,4’-difluoro-8-metil-espiro[2H-imidazo[1,5-a]piridin-3,1 ’-ciclohexano]-1,5-diona (3)
La síntesis del producto intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento A. Sólido de color blanquecino; Rendimiento: 1,2 g, 53 %; EM (IEN) m/z 346,99 [M 1]+.
Síntesis de N-[6-[(4’,4’-difluoro-8-metil-1,5-dioxo-espiro[2H-imidazo[1,5-a]piridin-3,1’-ciclohexano]-6-il)amino]pirimidin-4-il]carbamato de terc-butilo (5)
La síntesis del intermedio 5 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento H. Sólido de color amarillo; Rendimiento: 0,8 g, 58 %; EM (IEN) m/z 477,48 [M 1]+.
Síntesis de 6-[(6-aminopirimidin-4-il)amino]-4’,4’-difluoro-8-metil-espiro[2H-imidazo[1,5-a]piridin-3,1 ’-ciclohexano]-1,5-diona (Comp. N ° 103)
La síntesis del compuesto 103 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento D. Sólido de color amarillo; Rendimiento: 0,75 g, 97 %; EM (IEN) m/z 377,40 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,40 (s, 1H), 9,73 (s, 1H), 8,46 (s, 1H), 8,15 (s, 1H), 7,87 (s ancho, 2H), 6,40 (s, 1H), 3,30 (m, 2H), 2,43 (s, 3H), 2,32-2,29 (m, 1H), 2,20-2,10 (m, 3H), 1,65 (m, 2H).
Ejemplo 104
Síntesis de clorhidrato de 8-cloro-6-(pirimidin-4-ilamino)espiro[2H-imidazo[1,5-a]piridin-3,4'-piperidin]-1,5-diona (Comp. N.° 104)

Síntesis de 8-cloro-1,5-dioxo-6-(pirimidin-4-ilamino)espiro[2H-imidazo[1,5-a]piridin-3,4’-piperidin-1 ’-carboxilato de terc-butilo (3)
La síntesis del intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento H. Sólido de color amarillo; Rendimiento: 0,8 g, 52 %; EM (IEN) m/z 447 [M 1]+.
Síntesis de clorhidrato de 8-cloro-6-(pirimidin-4-ilamino)espiro[2H-imidazo[1,5-a]piridin-3,4'-piperidin]-1,5-diona (Comp. N ° 104)
La síntesis del compuesto 104 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento F. Sólido de color amarillo pálido; Rendimiento: 0,6 g, 93 %; EM (IEN) m/z 347,37 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,69 (s, 1H), 10,52 (s ancho, 1H), 9,46 (m, 1H), 8,72 (s, 1H), 8,52 (d, J = 6,0 Hz, 1H), 7,57 (d, J = 5,6 Hz, 1H), 3,50-3,45 (m, 2H), 3,36-3,12 (m, 4H), 1,91-1,86 (m, 2H).
Ejemplo 105
Síntesis de clorhidrato de 6-[(6-aminopirimidin-4-il)amino]-3-íerc-butil-8-cloro-3-metil-2H-imidazo[1,5-a]piridin-1,5-diona (Comp. N.° 105)

Síntesis de N-[6-[(3-terc-butil-8-cloro-3-metil-1,5-dioxo-2H-imidazo[1,5-a]piridin-6-il)amino]pirimidin-4-il]-carbamato de terc-butilo (3)
La síntesis del intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento H. Sólido de color amarillo; Rendimiento: 0,21 g, 50 %; EM (IEN) m/z 431,39 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,04 (s, 1H), 9,73 (s, 1H), 9,50 (s, 1H), 8,57 (s, 1H), 8,51 (s, 1H), 7,77 (s, 1H), 1,92 (s, 3H), 1,48 (s, 9H), 0,99 (s, 9H).
S ín te s is de c lo rh id ra to de 6 -[(6 -a m in o p ir im id in -4 - il)a m in o ]-3 -te rc -b u til-8 -c lo ro -3 -m e til-2 H -im id a z o [1 ,5 -a ]p ir id in -1 ,5-d io n a (C om p. N ° 105)
La síntesis del compuesto 105 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento F. Sólido de color amarillo; Rendimiento: 0,08 g, 54 %; EM (IEN) m/z 363,46 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 9,82 (s, 2H), 8,51 (s, 1H), 8,40 (s, 1H), 7,89 (s ancho, 2H), 6,54 (s, 1H), 1,92 (s, 3H), 0,99 (s, 9H).
Ejemplo 106
Síntesis de diclorhidrato de 6-((6-aminopirimidin-4-il)amino)-8-metil-2H-espiro[imidazo[1,5-a]piridin-3,4'-piperidin]-1,5-diona (Comp. N.° 106)

Síntesis de 6-bromo-8-metil-1,5-dioxo-1,5-dihidro-2H-espiro[imidazo[1,5-a]piridin-3,4’-piperidin-1 ’-carboxilato de tercbutilo (3)
La síntesis del intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento E. Sólido de color blanquecino; Rendimiento: 1,7 g, 43 %; RMN 1H (400 MHz, DMSO-d6) ó 10,57 (s, 1H), 8,04 (s, 1H), 4,03 (s ancho, 2H), 3,56 (s ancho), 1H), 3,11 (s ancho, 4H), 2,23 (s, 3H), 1,42 (s, 9H).
Síntesis de 6-((6-((terc-butoxicarbonil)amino)pirimidin-4-il)amino)-8-metil-1,5-dioxo-1,5-dihidro-2H-espiro[imidazo[1,5-a]piridin-3,4’-piperidin-1'-carboxilato de terc-butilo (5)
La síntesis del intermedio 5 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento H. Sólido de color amarillo; Rendimiento: 1,6 g, 60 %; EM (IEN) m/z 542,2 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,27 (s ancho, 1H), 9,98 (s, 1H), 9,19 (s, 1H), 8,50-8,45 (m, 2H), 7,66 (s, 1H), 4,05 (s ancho, 1H), 3,32 (m, 2H), 2,24 (s, 3H), 1,48 (s, 9H), 1,43 (s, 9H).
Síntesis de diclorhidrato de 6-((6-aminopirimidin-4-il)amino)-8-metil-2H-espiro[imidazo[1,5-a]-piridin-3,4’-piperidin]-1,5-diona (Comp. N ° 106)
La síntesis del compuesto 106 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento F. Sólido de color amarillo pálido; Rendimiento: 1,02 g, 83 %; EM (IEN) m/z 342,2 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,42 (s ancho, 1H), 9,55 (s ancho, 1H), 9,24 (s ancho, 1H), 8,78 (s ancho, 1H), 8,41 (s, 1H), 8,16 (s, 1H), 7,58 (s ancho, 2H), 6,37 (s, 1H), 3,48-3,40 (m, 2H), 3,40-3,28 (m, 2H), 3,20-3,10 (m, 1H), 2,38 (s, 3H), 1,73 (m, 2H).
Ejemplo 107
Síntesis de 6'-((6-aminopirimidin-4-il)amino)-8,-metil-2'H-espiro[ciclohexano-1,3-imidazo[1,5-a]piridin]-1'5'-diona (Comp. N.° 107)

100"C

Síntesis de 6’-bromo-8'-metil-2’H-espiro[ciclohexan-1,3’-imidazo[1,5-a]piridin]-1 ',5’-diona (3)
La síntesis del producto intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento A. Sólido de color blanquecino; Rendimiento: 2,4 g, 71 %; EM (IEN) m/z 311,18 [M 1]+.
Síntesis de (6-((8’-metil-1',5’-dioxo-1',5’-dihidro-2'H-espiro[ciclohexano-1,3-imidazo[1,5-a]piridin]-6’-il)amino)pirimidin-4-il)carbamato de terc-butilo (5)
La síntesis del intermedio 5 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento H. Sólido de color blanquecino; Rendimiento: 2,10 g, 30 %.
Síntesis de clorhidrato de 6’-((6-aminopirimidin-4-il)amino)-8'-metil-2’H-espiro[ciclohexano-1,3,3-imidazo[1,5-a] (Comp. N ° 107)
La síntesis del compuesto 107 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento F. Sólido de color blanquecino; Rendimiento: 0,55 g, 64 %; EM (IEN) m/z 341,50; [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,20 (s, 1H), 9,68 (s, 1H), 8,45 (s, 1H), 8,10 (s, 1H), 7,88 (s ancho, 2H), 6,39 (s, 1H), 3,00 2,90 (m, 2H), 2,42 (s, 3H), 1,80-1,60 (m, 5H), 1,42 (d, J = 12 Hz, 2H), 1,25-1,12 (m, 1H).
Ejemplo 108
Síntesis de diclorhidrato de 6-((6-aminopirimidin-4-il)amino)-8-cloro-2H-espiro[imidazo[1,5-a]piridin-3,4'-piperidin]-1,5-diona (Comp. N.° 108)


Síntesis de 6-[[6-(terc-butoxicarbonilamino)pirimidin-4-il]amino]-8-cloro-1,5-dioxo-espiro[2H-imidazo[1,5-a]piridin-3,4’-piperidin-1-carboxilato de terc-butilo (3)
La síntesis del producto intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento H. Sólido de color blanquecino; Rendimiento: 1,6 g, 54 %; EM (IEN) m/z 562,2 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,47 (s, 1H), 10,05 (s, 1H), 9,49 (s, 1H), 8,72 (s, 1H), 8,51 (s, 1H), 7,78 (s, 1H), 4,09 (s ancho, 1H), 3,33 (m, 4H), 1,74 (s ancho, 1H), 1,48 (s, 9H), 1,43 (s, 9H).
Síntesis de diclorhidrato de 6-((6-aminopirimidin-4-il)amino)-8-cloro-2H-espiro[imidazo[1,5-a]piridin-3,4'-piperidin-1,5-diona (Comp. N.° 108)
La síntesis del compuesto 108 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento F. Sólido de color amarillo pálido; Rendimiento: 0,8 g, 96 %; EM (IEN) m/z 362,16 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,64 (s, 1H), 9,82 (s, 1H), 9,49-9,46 (m, 1H), 9,01-8,98 (m, 1H), 8,51 (s, 1H), 8,40 (s, 1H), 7,91 (s ancho, 2H), 6,54 (s, 1H), 3,54-3,47 (m, 2H), 3,38-3,18 (m, 4H), 1,88-1,85 (m, 2H).
Ejemplo 109
Síntesis de clorhidrato de 8-cloro-6-(pirimidin-4-ilamino)espiro[2H-imidazo[1,5-a]piridin-3,1'-ciclohexano]-1,5-diona (Comp. N.° 109)

Síntesis de 8-cloro-6-(pirimidin-4-ilamino)espiro[2H-imidazo[1,5-a]piridin-3,1 ’-ciclohexano]-1,5-diona (3)
La síntesis del producto intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento H. Sólido de color blanquecino; Rendimiento: 0,8 g, 64 %; EM (IEN) m/z 346,04 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,32 (s, 1H), 9,61 (s, 1H), 8,84 (s, 1H), 8,43 (d, J = 6,4 Hz, 1H), 7,45 (d, J = 6,4 Hz, 1H), 2,9 (t, J = 9,2 Hz, 2H), 1,65-1,54 (m, 6H), 1,25-1,23 (m, 2H).
S ín te s is de c lo rh id ra to de 8 -c lo ro -6 -(p ir im id in -4 - ila m in o )e s p iro [2 H -im id a z o [1 ,5 -a ]p ir id in -3 ,1 ’-c ic lo h e x a n o -1 ,5 -d io n a (C om p. N .° 109)
La síntesis del compuesto 109 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento F. Sólido de color amarillo claro; Rendimiento: 0,6 g, 77 %; EM (IEN) m/z 346,04 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,41 (s, 1H), 10,23 (s, 1H), 9,0 (s, 1H), 8,71 (s, 1H), 8,48 (d, J = 6,4 Hz, 1H), 7,52 (d, J = 6,4 Hz, 1H), 2,9 (t, J = 9,2 Hz, 2H), 1,78-1,54 (m, 6H), 1,23-1,27 (m, 2H).
Ejemplo 110
Síntesis de clorhidrato de 8-cloro-4',4'-difluoro-6-(pirimidin-4-ilamino)espiro[2H-imidazo[1,5-a]piridin-3,1'-ciclohexano]-1,5-diona (Comp. N.° 110)

Síntesis de 8-cloro-4’,4’-difluoro-6-(pirimidin-4-ilamino)espiro[2H-imidazo[1,5-a]piridin-3,1 ’-ciclohexano]-1,5-diona (3) La síntesis del intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento H. Sólido de color amarillo; Rendimiento: 0,85 g, 35 %; EM (IEN) m/z 382,36 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,51 (s, 1H), 9,68 (s, 1H), 8,80-8,84 (m, 2H), 8,44 (d, J = 6,8 Hz, 1H), 7,44 (d, J = 6,4 Hz, 1H), 3,34-3,17 (m, 2H), 2,32-2,17 (m, 4H), 1,76-1,72 (m 2H).
Síntesis de clorhidrato de 8-cloro-4’,4’-dtfluoro-6-(pirimidin-4-ilamino)espiro[2H-imidazo[1,5-a]piridin-3,1’-ciclohexano]-1,5-diona (Comp. N ° 110)
La síntesis del compuesto 110 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento F. Sólido de color amarillo; Rendimiento: 0,81 g, 87 %; EM (IEN) m/z 382,4 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,64 (s, 1H), 10,45 (s, 1H), 9,05 (s, 1H), 8,71 (s, 1H), 8,52 (d, J = 6,8 Hz, 1H), 7,56 (d, J = 6,4 Hz, 1H), 3,30-3,20 (m, 2H), 2,35-2,15 (m, 4H), 1,80-1,72 (m, 2H).
Ejemplo 111
Síntesis de clorhidrato de 6-[(6-aminopirimidin-4-il)amino]-6',8-dicloro-espiro[2H-imidazo[1,5-a]piridin-3,1'-indan-]-1,5-diona (Comp. N.° 111)


La síntesis del producto intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento A. Sólido de color blanquecino; Rendimiento: 0,52 g, 35 %; EM (IEN) m/z: 399,23 [M 1]+; RMN 1H (400 MHz, DMSO-cfe) ó 10,35 (s, 1H), 8,28 (s, 1H), 7,39 (m, 3H), 3,23 (m, 1H), 3,08 (m, 2H), 2,43 (m, 1H).
Síntesis de N-[6-[(6-8-dicloro-1,5-dioxo-espiro[2H-imidazo[1,5-a]pirídin-3,1 ’-indan]-6-il)amino]pirimidin-4-il]carbamato de terc-butilo (5)
La síntesis del intermedio 5 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento H. Sólido de color blanquecino; Rendimiento: 0,38 g, 56 %; EM (IEN) m/z 529,2 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,07 (s, 1H), 9,99 (s, 1H), 9,42 (s, 1H), 8,72 (s, 1H), 8,50 (s, 1H), 7,65 (s, 1H), 7,37 (m, 3H), 3,10 (m, 2H), 3,01 (m, 2H), 1,45 (s, 9H).
Síntesis de clorhidrato de 6-[(6-aminopiriniidin-4-il)amino]-6,8-dicloro-espiro[2H-imidazo[1,5-indol]-1,5-diona (Comp. N.° 111)
La síntesis del compuesto 111 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento F. Sólido de color amarillo pálido; Rendimiento: 0,17 g, 55 %; EM (IEN) m/z 429,3 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,18 (s, 1H), 9,78 (s, 1H), 8,52 (s, 1H), 8,44 (s, 1H), 7,91 (s ancho, 2H), 7,38 (m, 3H), 6,42 (s, 1H), 3,30 (m, 1H), 3,21 (m, 1H), 2,96 (m, 1H), 2,47 (m, 1H).
Ejemplo 112
Síntesis de 8-cloro-6-[(5-fluoropirimidin-4-il)amino]espiro[2H-imidazo[1,5-a]piridin-3,1'-ciclohexano]-1,5-diona (Comp. N.° 112)

Síntesis de 8-cloro-6-[(5-fluoropirimidin-4-il)amino]espiro[2H-imidazo[1,5-a]piridin-3,1'-ciclohexano]-1,5-diona (Comp. N ° 112)
La síntesis del compuesto 112 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento H. Sólido de color amarillo; Rendimiento: 0,14 g, 43 %; EM (IEN) m/z 364,3 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,41 (s, 1H), 8,73 (s, 1H), 8,62 (s, 2H), 8,54 (s, 1H), 2,95-2,87 (m, 2H), 1,80-1,54 (m, 7H), 1,30-1,18 (m, 1H).
Ejemplo 113
Síntesis de 8-cloro-6-(pirimidin-4-ilamino)espiro[2H-imidazo[1,5-a]piridin-3,2'-indan]-1,5-triona (Comp. N.° 113)

La síntesis del intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento A. Sólido de color marrón; Rendimiento: 0,12 g, 28 %; EM (IEN) m/z 378,94 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,40 (s, 1H), 8,40 (s, 1H), 7,88-7,81 (m, 2H), 7,70 (d, J = 7,32, 1H), 7,57 (t, J = 7,36, 1H), 4,12 (d, J = 18,32, 1H), 3,43 (d, J = 18,08, 1H).
Síntesis de 8-cloro-6-(pirimidin-4-ilamino)espiro[2H-imidazo[1,5-a]piridin-3,2'-indano]-1,5-triona (Comp. N ° 113)
La síntesis del compuesto 113 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento H. Sólido de color amarillo; Rendimiento: 0,065 g, 26 %; EM (IEN) m/z 394,06 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,05 (s ancho, 1H), 9,64 (s, 1H), 8,85 (s, 2H), 8,41 (d, J = 5,68 Hz; 1H), 7,86-7,80 (m, 2H), 7,69 (d, J = 7,44 Hz, 1H), 7,55 (t, J = 7,44, 1H), 7,30 (d, J = 5,92 Hz, 1H), 4,15 (d,/= 18,2 Hz, 1H), 3,50 (d. 7 =18,44 Hz, 1H).
Ejemplo 114
Síntesis de 6-((6-aminopirimidin-4-il)amino)-8-cloro-6'-fluoro-2',3'-dihidro-2H-espiro[imidazo[1,5-a]piridin-3,1 '-indeno]-1,5-diona (Comp. N.° 114)


Síntesis de 6-bromo-8-cloro-6’-fluoro-2',3'-dihidro-2H-espiro[imidazo[1,5-a]piridin-3,1’-inden]-1,5-diona (3)
La síntesis del intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento A. Sólido de color blanquecino; Rendimiento: 0,46 g, 32 %; EM (IEN) m/z 381,19 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,37 (s, 1H), 8,31-8,28 (s, 1H), 7,37-7,34 (m, 1H), 7,20-7,16 (m, 2H), 3,22 (m, 1H), 3,06 2,97 (m, 2H), 2,42-2,35 (m, 1H).
Síntesis de (6-((8-cloro-6’-fluoro-1,5-dioxo-1,2’,3',5-tetrahidro-2H-espiro[imidazo[1,5-a]piridin-3,1’-inden]-6-il)amino)pirimidin-4-il)carbamato de terc-butilo (5)
La síntesis del intermedio 5 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento H. Sólido de color blanquecino; Rendimiento: 320 mg, 53 %; EM (IEN) m/z 513,35 [M 1]+.
Síntesis de 6-((6-aminopirimidin-4-il)amino)-8-cloro-6’-fluoro-2',3’-dihidro-2H-espiro[imidazo[1,5-a]piridin-3,1’-indeno]-1,5-diona (Comp. N.° 114)
La síntesis del compuesto 114 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento F. Sólido de color amarillo; Rendimiento: 0,089 g, 37 %; EM (IEN) m/z 413,32 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,16 (s, 1H), 9,72 (s, 1H), 8,49-8,46 (d, 2H), 7,86-7,69 (s ancho, 2H), 7,36 (m, 1H), 7,19 (m, 1H), 7,11 (m, 1H), 6,41 (s, 1H), 3,31-3,35 (m, 2H), 3,17-2,96 (m, 2H).
Ejemplo 115
Síntesis de 8-cloro-6-[(5-cloropirimidin-4-il)amino]espiro[2H-imidazo[1,5-a]piridin-3,1'-ciclohexano]-1,5-diona (Comp. N.° 115)

Síntesis de 8-cloro-6-[(5-cloropirimidin-4-il)amino]espiro[2H-imidazo[1,5-a]piridin-3,1’-ciclohexano-1,5-diona (Comp. N ° 115)
La síntesis del compuesto 115 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento H. Sólido de color amarillo; Rendimiento: 75 mg, 13 %; EM (IEN) m/z 380,23 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,44 (s, 1H), 8,85 (s, 1H), 8,80 (s, 1H), 8,72 (s, 1H), 8,66 (s, 1H), 2,93-2,88 M, 2H), 1,77 1,74 (m, 2H), 1,65-1,61 (m, 3H), 1,57-1,57 (m, 2H), 1,04-1,02 (m, 1H).
Ejemplo 116
Síntesis de 8-cloro-6-[(6-metilpirimidin-4-il)amino]espiro[2H-imidazo[1,5-a]piridin-3,1,-ciclohexano]-1,5-diona (Comp. N.° 116)

Síntesis de 8-cloro-6-[(6-metilpirimidin-4-il)amino]espiro[2H-imidazo[1,5-a]piridin-3,1 ’-ciclohexano-1,5-diona (Comp. N ° 116)
La síntesis del compuesto 116 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento H. Sólido de color amarillo; Rendimiento: 190 mg, 58 %; EM (IEN) m/z 359,81 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,29 (s, 1H), 10,08 (s, 1H), 9,41 (s, 1H), 8,76 (s, 1H), 8,71 (s, 1H), 7,27 (s, 1H), 2,98-2,91 (m, 2H), 2,33 (s, 3H), 1,77-1,74 (m, 2H), 1,67-1,58 (m, 3H), 1,55-1,52 (m, 2H), 1,26-1,19 (m, 1H).
Ejemplo 117
Síntesis de 6'-((7H-purin-6-il)amino)-8,-cloro-2'H-espiro[ciclohexano-1,3,3-imidazo[1,5-a]piridin]-1',5'-diona (Comp. N.° 117)

Síntesis de N-(7-((2-(trimetilsilil)etoxi)metil)-7H-purin-6-il)isobutiramida (2)
A una solución agitada de 2-metil-N-(7H-purin-6-il)propanamida (1, 5 g, 24,36 mmol) en dimetilformamida (20 ml) se le añadió hidruro de sodio (0,88 g, 36,55 mmol) en porciones adicionales en 10 minutos a 0 °C. La suspensión anterior se agitó durante 10 minutos a 0 °C seguida de la adición de cloruro de 2-(trimetilsilil)etoximetilo (4,87 g, 29,24 mmol) lentamente a 0 °C en atmósfera de nitrógeno. La reacción se agitó a temperatura ambiente durante 16 h. Una vez completada la reacción, la mezcla se inactivó con solución acuosa saturada de cloruro de amonio y el producto se extrajo con diclorometano (50 ml, 2 veces). Los compuestos orgánicos se separaron después, se secaron (sulfato de magnesio) y se concentraron a sequedad al vacío y el producto en bruto se purificó por cromatografía ultrarrápida eluyendo con metanol al 2 % en diclorometano. La concentración de las fracciones deseadas proporcionó 2-metil-N-[7-(2-trimetilsililetoximetil)purin-6-il]propanamida (2) en forma de un sólido de color blanquecino. Rendimiento: 2 g, 24 %.
Síntesis de clorhidrato de 7-((2-(trimetilsilil)etoxi)metil)-7H-purin-6-amina (3)
La síntesis del intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento I. Sólido de color blanquecino; Rendimiento: 3,0 g, 73 %; EM (IEN) m/z 266,31 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 8,26 (s, 1H), 8,16 (s, 1H), 7,26 (s ancho, 2H), 5,50 (s, 2H), 3,52 (t, J = 1,56 Hz, 2H), 0,835 (t, J = 1,56 Hz, 2H), 0,088 (s, 9H).
Síntesis de 8’-cloro-6'-((7-((2-(trimetilsilil)etoxi)metil)-7H-purin-6-il)amino)-2-espiro[ddohexan-1,3'-imidazo[1,5-a]piridin]-1’,5'-diona (5)
La síntesis del intermedio 5 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento B. Sólido de color amarillo; Rendimiento: 0,10 g, 26 %; EM (IEN) m/z 516,44 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,42 (s, 1H), 9,07 (s, 1H), 9,03 (s, 1H), 8,71 (s, 1H), 8,58 (s, 1H), 5,74 (s, 2H), 3,61 (t, J = 1,6 Hz, 2H), 0,852 (t, J = 1,64 Hz, 2H), 0,081 (s, 9H).
Síntesis de 6’-((7H-purin-6-il)amino)-8'-doro-2'-espiro[ddohexano-1,3,3-imidazo[1,5-a]piridin]-1’,5'-diona (Comp. N.° 117)
Se disolvió 8-cloro-6-[[7-(2-trimetilsililetoxi)purin-6-il]amino]espiro[2H-imidazo[1,5-a]piridin-3,1 '-ciclohexano]-1,5-diona (5, 0,6 g, 1,2 mmol) en diclorometano (10 ml) en un matraz y se añadió ácido trifluoroacético (1,36 g, 11,95 mmol) gota a gota y se agitó la mezcla a temperatura ambiente durante una noche. Después de la terminación, se evaporó el disolvente a presión reducida y el producto en bruto se basificó mediante solución acuosa saturada de bicarbonato de sodio hasta pH 8 y se extrajo con diclorometano (50 ml, 2 veces). La capa orgánica se secó sobre una capa anhidra. Sulfato de sodio y se concentró para proporcionar producto en bruto. El producto en bruto se purificó por cromatografía en columna ultrarrápida eluyendo con metanol al 2,5 % en diclorometano. Las fracciones deseadas se concentraron a sequedad al vacío para proporcionar 8-cloro-6-(7H-purin-6-ilamino)espiro[2H-imidazo[1,5-a]piridin-3-ciclohexano]-1,5-diona (Comp. N.° 117) en forma de un sólido de color amarillo. Rendimiento: 0,06 g, 13 %; EM (IEN) m/z 386,39 [M 1]+; RMN 1H: 400 MHz, DMSO d6) ó 10,43 (m, 1H), 10,37 (s, 1H), 8,88 (s, 1H), 8,64 (s, 1H), 8,43 (s, 1H), 2,94 (t, J = 2,32 Hz, 2H), 1,77 (m, 2H), 1,63 (m, 3H), 1,57 (d, J = 12,8 Hz, 2H), 1,23 (m, 1H).
Ejemplo 118
Síntesis de 6-[(6-aminopirimidin-4-il)amino]-3-ciclopentil-3-metil-1,5-dioxo-imidazo[1,5-a]piridin-2-carbonitrilo (Comp. N.° 118)

Síntesis de 6-[(6-aminopirimidin-4-il)amino]-3-ciclopentil-3-metil-2H-imidazo[1,5-s]piridin-1,5-diona (2)
La síntesis del intermedio 2 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento I. Sólido de color amarillo; Rendimiento: 0,36 g, 71 %; EM (IEN) m/z 341,16 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 9,61 (s, 1H), 8,62 (s, 1H), 8,57 (d, J = 7,64, 1H), 8,15 (s, 1H), 6,80 (d, J = 7,68 1H), 6,51 (s, 2H), 6,16 (s, 1H), 3,4 (m, 1H), 1,82 (s, 3H), 1,63-1,41 (m, 5H), 1,1 (m, 1H), 0,87-0,75 (m, 2H).
Síntesis de 6-[(6-aminopirimidin-4-il)amino]-3-ciclopentil-3-metil-1,5-dioxo-imidazo[1,5-a]piridin-2-carbonitrilo (Comp. N.° 118)
En un matraz de fondo redondo de dos cuellos se preparó 6-[(6-aminopirimidin-4-il)amino]-3-ciclopentil-3-metil-2H-imidazo[1,5-a]piridin-1,5-diona (2, 0,28 g, 0,82 mmol) en tetrahidrofurano seco (15 ml). La mezcla de reacción se enfrió a 0 °C y se añadió en porciones hidruro de sodio (164 mg, 4,11 mmol). Después de agitar durante 10 min a temperatura ambiente, se añadió bromuro de cianógeno (436 mg, 4,11 mmol) y la mezcla de reacción resultante se agitó a temperatura ambiente durante 20 h. La mezcla de reacción se inactivó con una solución acuosa de cloruro de amonio y después se concentró para obtener la masa bruta. Este producto en bruto se purificó mediante cromatografía ultrarrápida eluyendo con metanol al 2 % en diclorometano. El compuesto se lavó con pentano y se secó al vacío para proporcionar 6-[(6-aminopirimidin-4-il)amino]-3-ciclopentil-3-metil-1,5-dioxo-imidazo[1,5-a]piridin-2-carbonitrilo (Comp. N.° 118) en forma de un sólido de color amarillo. Rendimiento: 0,018 g, 6 %; EM (IEN) m/z 366,16 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 9,08 (s, 1H), 8,71 (d, J = 7,84, 1H), 8,19 (s, 1H), 7,28 (d, J = 7,96, 1H), 6,65 (s, 2H), 6,29 (s, 1H), 3,49-3,43 (m, 1H), 2,21 (s, 3H), 1,94 (m, 1H), 1,7-1,58 (m, 4H), 1,48 (m, 1H), 1,23 (m, 1H), 1,06-1,02 (m, 1H).
Ejemplo 119
Síntesis de 6-[(6-aminopirimidin-4-il)amino]-8-cloro-3-(3-fluorofenil)-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona (Comp. N.° 119)

Síntesis de 6-bromo-8-cloro-3-(3-fluorofenil)-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona (3)
A una suspensión de 5-bromo-3-cloro-6-oxo-1H-piridin-2-carboxamida (1,5 g, 1,99 mmol) en acetonitrilo (15 ml), se le añadieron 3-fluorobenzaldehído (0,86 g, 6,96 mmol) y cloruro férrico (2,25 g, 13,92 mmol) en un vial. Se selló el vial y se calentó la masa de reacción a 85 °C durante 16 h. Después de la terminación, la masa de reacción se enfrió a temperatura ambiente, se filtró a través de lecho de celite, se lavó con metanol al 5 % en diclorometano seguido de concentración para obtener producto en bruto. El producto en bruto se purificó después por cromatografía en columna eluyendo con metanol al 5 % en diclorometano para proporcionar 8-cloro-3-(3-fluorofenil)-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona en forma de un sólido de color blanquecino. Rendimiento: 0,45 g, 63 %; EM (IEN) m/z 256,17 [M 1]+.
Síntesis de (6-((8-cloro-3-(3-fluorofenil)-1,5-dioxo-1,2,3,5-tetrahidroimidazo[1,5-a]piridin-6-il)amino)pirimidin-4-il)carbamato de terc-butilo (5)
La síntesis del intermedio 5 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento H. Sólido de color amarillo; Rendimiento: 0,15 g, 33 %; EM (IEN) m/z 487,43 [M 1]+.
Síntesis de 6-[(6-aminopirimidin-4-il)amino]-8-cloro-3-(3-fluorofenil)-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona (Comp. N.° 119)
La síntesis del compuesto 119 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento F. Sólido de color blanquecino; Rendimiento: 0,012 g, 10 %; EM (IEN) m/z 387,29 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 9,98 (s, 1H), 8,90 (s, 1H), 8,70 (s, 1H), 8,20 (s, 1H), 7,43 (m, 1H), 7,33 (m, 1H), 7,19 (s, 2H), 6,61 (s, 1H), 6,58 (s, 2H), 6,17 (s, 1H).
Ejemplo 120
Síntesis de 8'-cloro-2,2-dimetil-6,-(pirimidin-4-ilamino)-2'H-espiro[ciclobutan-1,3-imidazo[1,5-a]piridin]-1',5'-diona (Comp. N.° 120)

La síntesis del intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento A. Sólido de color blanquecino; Rendimiento: 0,51 g; 64 %; EM (IEN) m/z 331,59 [M 1]+.
Síntesis de 8’-cloro-2,2-dimetil-6'-(pirimidin-4-ilamino)-2’H-espiro[ciclobutano-1,3,3-imidazo[1,5-a]piridin]-1,5’-diona (Comp. N.° 120)
La síntesis del compuesto 120 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento H. Sólido de color amarillo claro; Rendimiento: 0,12 g, 39 %; EM (IEN) m/z 346,80. RMN 1H (400 MHz, DMSO-d6) ó 10,03 (s, 1H), 9,75 (s, 1H), 8,85 (s, 1H), 8,80 (s, 1H), 8,84 (d, J = 16,88 Hz, 1H), 7,48 (d, J = 5,72 Hz, 2H), 3,13-3,08 (m, J = 9,44 Hz, 1H), 2,66-2,59 (m, J = 9,64 Hz, 1H), 1,20 (s, 3H), 0,98 (s, 3H).
Ejemplo 121
Síntesis de 6'-((6-aminopirimidin-4-il)amino)-8,-cloro-2,2-dimetil-2,H-espirociclobutan-1,3'-imidazoI[1,5-a]piridinil-1',5'-diona (Comp. N.° 121)

La síntesis del intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento H. Sólido de color blanquecino; Rendimiento: 0,31 g, producto en bruto; EM (IEN) m/z 461,90 [M 1]+.
Síntesis de 6’-((6-aminopirimidin-4-il)amino)-8'-cloro-2,2-dimetil-2'H-espiro[ciclobutan-1,3'-imidazo[1,5-a]piridin]-1’,5’-diona (Comp. N ° 121)
La síntesis del compuesto 121 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento F. Sólido de color amarillo; Rendimiento: 0,070 g, 30 %; EM (IEN) m/z 361,92 [M 1]+; RMN 1H: (400 MHz, DMSO-d6) ó 9,94 (s, 1H), 9,07 (s, 1H), 8,67 (s, 1H), 8,21 (s, 1H), 6,61 (s, 2H), 3,12-3,05 (m, J = 2,48 Hz, 1H), 2,66-2,58 (m, J = 5,4 Hz, 1H), 1,55-1,50 (m, J = 3,0 Hz, 1H), 1,19 (s, 3H), 0,96 (s, 3H).
Ejemplo 122
Síntesis de 6-[(6-amino-5-cloro-pirimidin-4-il)amino]-8-cloro-espiro[2H-imidazo[1,5-a]piridin-3,1,5-diona (Comp. N.° 122)

Síntesis N-terc-butoxicarbonil-N-(5,6-dicloropirimidin-4-il)carbamato de terc-butilo (2)
Procedimiento J: A una solución agitada de 5,6-dicloropirimidin-4-amina (1,0 g, 18,29 mmol) en tetrahidrofurano (30 ml), se le añadió 4-dimetilaminopiridina (0,16 g, 1,31 mmol) y dicarbonato de di-terc-butilo (8,77 g, 40,2 mmol) a temperatura ambiente. La masa de reacción se agitó a temperatura ambiente durante una noche. Después de la terminación, se destiló el disolvente. El residuo anterior se diluyó con agua y se extrajo con acetato de etilo (50 ml, 2 veces). Los compuestos orgánicos se separaron y secaron (sulfato de magnesio) y se concentraron a sequedad al vacío para proporcionar N-terc-butoxicarbonil-N-(5,6-dicloropirimidin-4-il)carbamato de terc-butilo (2) en forma de un sólido de color blanquecino. Rendimiento: 3,1 g, 47 %; EM (IEN) m/z 364,3 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 9,04 (s, 1H), 1,40 (s, 18H).
Síntesis de N-terc-butoxicarbonil-N-[5-cloro-6-[(8-cloro-1,5-dioxoespiro[2H-imidazo[1,5-a]piridin-3,1 ’-ciclohexano]-6-il)amino)-pirimidin-4-i]carbamato de terc-butilo (4)
La síntesis del intermedio 4 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento B. Sólido de color amarillo; Rendimiento: 0,10 g, 26 %; EM (IEN) m/z 595,45 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,46 (s, 1H), 8,99 (s, 1H), 8,95 (s, 1H), 8,66 (s, 1H), 2,90 (m, 2H), 1,65 (m, 7H), 1,46 (s, 18H), 1,20 (m, 1H).
Síntesis de 6-[(6-amino-5-cloro-pirimidin-4-il)amino]-8-cloro-espiro[2H-imidazo[1,5-a]piridin-3,1 ’-ciclohexano-5-diona (Comp. N.° 122)
La síntesis del compuesto 122 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento F. Sólido de color amarillo; Rendimiento: 0,059 g, 81 %; EM (IEN) m/z 395,35 [M 1]+; RMN 1H: (400 MHz, DMSO-d6) ó 10,35 (s, 1H), 8,62 (s, 1H), 8,55 (s, 1H), 8,23 (s, 2H), 7,27 (s, 1H), 2,91 (t, J = 2,28, 2H), 1,75 (m, 2H), 1,63 (m, 3H), 1,53 (d, J = 12,8 Hz, 2H), 1,23 (m, 1H).
Ejemplo 123
Síntesis de clorhidrato de 6'-((7H-pirrolo[2,3-c/]pirimidin-4-il)amino)-8'-cloro-2'H-espiro[ciclohexano-1,3'-imidazo[1,5-a]piridinil-1',5'-diona (Comp. N.° 123)

Síntesis de 6’-((7H-pirrolo[2,3-d]pirimidin-4-il)amino)-8'-cloro-2’H-espiro[ciclohexano-1,3,3-imidazo[1,5-a]piridin]-1’,5'-diona (3)
La síntesis del intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento H. Sólido de color amarillo; Rendimiento: 0,60 g, producto en bruto; EM (IEN) m/z 385,19 [M 1]+ Síntesis de 6’-((7H-pirrolo[2,3-d]pirimidin-4-il)amino)-8'-cloro-2’H-espiro[ciclohexano-1,3-imidazo[1,5-a]piridin]-1’,5'-diona (Comp. N ° 123)
A una solución agitada de 8-cloro-6-(7H-pirrolo [2,3-d]pirimidin-4-ilamino)espiro[2H-imidazo[1,5-a]piridin-3,1'-ciclohexano]-1,5-diona (3, 0,6 g, 1,56 mmol) en metanol (6 ml) se le añadió cloruro de hidrogeno 4 M en dioxano (4 ml) con enfriamiento y se agitó la mezcla de reacción a temperatura ambiente durante una noche. Después de la terminación, se filtró la masa de reacción usando un embudo sinterizado y se lavó con etanol (10 ml). El sólido obtenido se secó a alto vacío para proporcionar 8-cloro-6-(7H-pirrolo[2,3-d]pirimidin-4-ilamino)espiro[2H-imidazo[1,5-a]piridin-3,1'-ciclohexano]-1,5-diona (Comp. N.° 123) en forma de un sólido de color amarillo claro; Rendimiento: 0,21 g, 32 %; EM (IEN) m/z 385,31 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 12,24 (s, 1H), 10,39 (s, 1H), 9,12 (s, 1H), 8,75 (s, 1H), 8,50 (s, 1H), 7,43 (s, 1H), 6,88 (s, 1H), 2,95 (t, J = 11,14 Hz, 2H), 1,76 (m, 2H), 1,62 (m, 5H), 1,22 (m, 1H).
Ejemplo 124
Síntesis de 8-cloro-3,3-dimetil-6-(pirido[4,3-d]pirimidin-4-ilamino)-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona (Comp. N.° 124)


Síntesis de 8-cloro-3,3-dimetil-6-(pirido[4,3-d]pirimidin-4-ilamino)-2,3-dihidroimidazo[1,5-d]piridin-1,5-diona (Comp. N ° 124)
La síntesis del compuesto 124 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento H. Sólido de color amarillo; Rendimiento: 0,060 g, 16 %; EM (IEN) m/z 357,77 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 9,91 (s, 2H), 9,69 (s, 1H), 8,96 (s, 1H), 8,90-8,86 (d, 1H), 8,87 (s, 1H), 7,73 (d, 1H), 1,84 (s, 6H).
Ejemplo 125
Síntesis de 8'-cloro-6'-((2-metilpirimidin-4-il)amino)-2'H-espiro[ciclohexano-1,3-imidazo[1,5-a]piridin]-1'5'-diona (Comp. N.° 125)

Síntesis de 8’-cloro-6'-((2-metilpirimidin-4-il)amino)-2'H-espiro[ciclohexano-1,3,3-imidazo[1,5-a]piridin]-1’5’-diona (Comp. N ° 125)
La síntesis del compuesto 125 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento H. Sólido de color blanquecino; Rendimiento: 0,16 g, 45 %; EM (IEN) m/z 360,38 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,55 (s, 1H), 10,47 (s, 1H), 8,67 (s, 1H), 8,45 (m, 1H), 7,44 (d, J = 4,8 Hz, 1H), 2,91 (t, J = 11,74 Hz, 2H), 2,65 (s, 3H), 1,76 (m, 2H), 1,65 (m, 3H), 1,56 (m, 2H), 1,19 (m, 1H).
Ejemplo 126
Síntesis de 6-((6-aminopirimidin-4-il)amino)-8-cloro-2H-espiro[imidazo[1,5-a]piridin-3,6'-[1,4]diazepan]-1,5-diona (Comp. N.° 126)

Síntesis de 6-bromo-8-cloro-1’,4'-ditosil-2H-espiro[imidazo[1,5-a]piridin-3,6’-[1,4]diazepan]-1,5-diona (3)
La síntesis del intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento A. Sólido de color blanquecino; Rendimiento: 1,7 g, 43 %; EM (IEN) m/z 442,31 [M 1]+.
Síntesis de 6-bromo-8-cloro-2H-espiro[imidazo[1,5-a]piridin-3,6'-[1,4]diazepan]-1,5-diona (4)
A una solución agitada de bromuro de hidrógeno (1,43 g, 15,24 mmol) en ácido acético en 1,4-dioxano (30 ml), se le añadió 6-bromo-8-cloro-1,4-ditosil-2H-espiro[imidazo[1,5-a]piridin-3,6'-[1,4]diazepan]-1,5-diona (1 g, 1,52 mmol) a temperatura ambiente. Se selló el vial y se calentó la masa de reacción a 100 °C durante 16 h. Después de la terminación, el disolvente se retiró a presión reducida y el producto en bruto se trituró con diclorometano para proporcionar 6-bromo-8-cloro-2H-espiro[imidazo[1,5-a]piridin-3,6'-[1,4]diazepan]-1,5-diona (4) en forma de un sólido de color marrón. Rendimiento: 0,5 g, 94 %; EM (IEN) m/z 362,28 [M 1]+.
Síntesis de 6-bromo-8-cloro-1,5-dioxo-1,5-dihidro-2H-espiro[imidazo[1,5-a]piridin-3,6'-[1,4]diazepan]-1’,4'-dicarboxilato de di-terc-butilo (5)
A una solución agitada de dicarbonato de di-terc-butilo (0,94 g, 4,32 mmol) en diclorometano (30 ml) en un vial a temperatura ambiente, se le añadió 6-bromo-8-cloro-2H-espiro[imidazo[1,5-a]piridin-3,6'-[1,4]diazepan]-diona (4, 0,5 g, 1,44 mmol) y trietilamina (0,73 g, 7,19 mmol). Se selló el vial y se agitó la masa de reacción a temperatura ambiente durante 16 h. Después de terminado, el disolvente se retiró y el producto en bruto se trituró con hexano para proporcionar 6-bromo-8-cloro-1,5-dioxo-1,5-dihidro-2H-espiro[imidazo[1,5-a]piridin-3,6'-[1,4]diazepan]-1',4'-dicarboxilato de di-terc-butilo (5) en forma de un sólido de color amarillo. Rendimiento: 0,45 g, 57 %; EM (IEN) m/z 547,13 [M 1]+.
Síntesis de 6-((6-((terc-butoxicarbonil)amino)pirimidin-4-il)amino)-8-cloro-1,5-dioxo-1,5-dihidro-2H-espiro[imidaz[]1,5-a]piridin-3,6’-[1,4]diazepan]-1',4’-dicarboxilato de di-terc-butilo (7)
La síntesis del intermedio 7 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento H. Sólido de color amarillo; Rendimiento: 0,41 g, 74 %; EM (IEN) m/z 677,32 [M 1]+.
Síntesis de 6-((6-aminopirimidin-4-il)amino)-8-cloro-2H-espiro[imidazo[1,5-a]piridin-3,6’-[1,4]diazepan]-1,5-diona (Comp. N.° 126)
La síntesis del compuesto 126 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento F. Sólido de color blanquecino; Rendimiento: 0,04 g, 18 %; EM (IEN) m/z 377,32 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 9,87 (s, 1H), 8,98 (s, 1H), 8,66 (s, 1H), 8,20 (s, 1H), 6,61 (s, 2H), 3,61-3,58 (m, 2H), 2,95 2,91 (m, 2H), 2,82 (s, 4H), 2,66-2,61 (m, 2H).
Ejemplo 127
Síntesis de 6'-((1H-pirazolo[3,4-c/]pirimidin-4-il)amino)-8'-cloro-2'H-espiro[ciclohexano-1,3-imidazo[1,5-a]piridin]-1 ',5'-diona (Comp. N.° 127)


Síntesis de 6’-((1H-pirazolo[3,4-d]pirimidin-4-il)amino)-8'-cloro-2-espiro[ciclohexan-1,3-imidazo[1,5-a]piridin]-1’,5’-diona (Comp. N.° 127)
La síntesis del compuesto 127 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento B. Rendimiento: 0,037 g, 6 %; EM (IEN) m/z 386,36 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,39 (s, 1H), 9,55 (s, 1H), 8,91 (s, 1H), 8,61 (s, 2H), 2,97 (m, 2H), 1,76-1,56 M, 7H), 1,23 (m, 1H).
Ejemplo 128
Síntesis de clorhidrato de 6-[(6-aminopirimidin-4-il)amino]-8-cloro-1'-metil-espiro[2H-imidazo[1,5-a]piridin-3,4'-piperidin]-1,5-diona (Comp. N.° 128)

La síntesis del intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento A. Sólido de color marrón claro; Rendimiento: 1,0 g, 36 %; EM (IEN) m/z 346,13 [M 1]+
Síntesis de N-[6-[(8-cloro-1’-metil-1,5-dioxoespiro[2H-imidazo[1,5-a]piridin-3,4'-piperidin]-6-il)amino]pirimidin-4-il]carbamato de terc-butilo (5)
La síntesis del intermedio 5 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento H. Sólido de color blanquecino; Rendimiento: 60 mg, 14 %; RMN 1H (400 MHz, DMSO-d6) ó 10,33 (s, 1H), 9,63 (s, 1H), 8,84 (s, 1H), 8,80 (s, 1H), 8,44-8,42 (d, J = 8,0 Hz, 1H), 7,47-7,45 (d, J = 8,0 Hz, 1H), 7,39-7,38 (m, 1H), 5,75 (s, 2H), 3,24-3,17 (m, 2H), 2,80-2,66 (m, 2H), 7,47-7,45 (d, J = 8,0 Hz, 2,39-2,35 (m, 2H), 2,32 (s, 3H), 1,50-1,47 (m, 2H), 1,23 (s, 9H).
Síntesis de 6-[(6-aminopirimidin-4-il)amino]-8-cloro-1’-metil-espiro[2H-imidazo[1,5-a]piridin-3,4'-piperidin]-1,5-diona (Comp. N.° 128)
La síntesis del compuesto 128 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento F. Sólido de color blanquecino; Rendimiento: 0,032 g, 62 %; EM (IEN) m/z 375,82 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,72 (s ancho, 1H), 10,60 (s, 1H), 9,80 (s, 1H), 8,49 (s, 1H), 8,44 (s, 1H), 7,81 (s ancho, 2H), 6,53 (s, 1H), 3,66-3,33 (m, 6H), 2,78 (s, 3H), 1,91-1,88 (m, 2H).
Ejemplo 129
Síntesis de 8-cloro-1 '-metil-6-(pirimidin-4-ilamino)espiro[2H-imidazo[1,5-a]piridin-3,4'-piperidin]-1,5-diona (Comp. N.° 129)


Síntesis de 8-cloro-1’-metil-6-(pirimidin-4-ilamino)espiro[2H-imidazo[1,5-a]piridin-3,4'-piperidin]-1,5-diona (Comp. N° 129)
La síntesis del compuesto 129 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento H. Sólido de color blanquecino; Rendimiento: 65 mg, 31 %; EM (IEN) m/z 360,80 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,32 (s, 1H), 9,62 (s, 1H), 8,43-8,42 (d, J = 5,6 Hz, 1H), 7,47-7,45 (d, J = 4,8 Hz, 1H), 3,25 3,17 (m, 2H), 2,80-2,78 (m, 2H), 2,39-2,33 (m, 2H), 2,24 (s, 3H), 1,50-1,47 (m, 2H).
Ejemplo 130
Síntesis de 1',8-dimetil-6-(pirimidin-4-ilamino)-2H-espiro[imidazo[1,5-a]piridin-3,3'-piperidin]-1,5-diona (Comp. N.° 130)

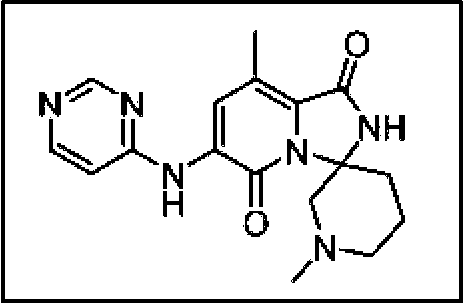
Síntesis de 6-bromo-1' 8-dimetil-espiro[2H-imidazo[1,5-a]piridin-3,3’-piperidin-1,5-diona (3)
La síntesis del intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento A. Sólido de color blanquecino; Rendimiento: 0,7 g, 25 %; EM (IEN) m/z 326,19 [M 1]+.
Síntesis de 1' 8-dimetil-6-(pirimidin-4-ilamino)-2H-espiro[imidazo[1,5-a]piridin-3,3’-piperidin]-1,5-diona (Comp. N. ° 130)
La síntesis del compuesto 130 se realizó como se ha descrito anteriormente usando el protocolo general del
Procedimiento H. Sólido de color amarillo; Rendimiento: 190 mg, 58 %; EM (IEN) m/z 359,81 [M 1]+; RMN 1H (400 MHz, DMSO-cfe) ó 9,93 (s, 1H), 9,35 (s, 1H), 8,77 (s, 1H), 8,59 (s, 1H), 8,37-8,38 (d, J = 5,88 Hz, 1H), 7,35-7,36 (d, J = 5,88 Hz, 1H), 3,01 (m, 2H), 2,95 (m, 1H), 2,50 (s, 2H), 2,45 (s, 3H), 2,25 (s, 3H), 1,91-1,97 (m, 2H), 1,71-1,73 (m, 1H), 1,48-1,51 (m, 1H).
Ejemplo 131
Síntesis de 6'-(pirimidin-4-ilamino)-1'-tioxol-1',2'-dihidro-5'H-espiro[ciclohexano-1,3-imidazo[1,5-a]piridin]-5'-ona (Comp. N.° 131)

Síntesis de 6’-(pirimidin-4-ilamino)-1 '-tioxo-1’,2'-dihidro-5'H-espiro[ciclohexano-1,3,3-imidazo[1,5-a]piridin]-5'-ona (Comp. N.° 131)
Un vial que contenía tetrahidrofurano (10 ml) se cargó con 6-(pirimidin-4-ilamino)espiro[2H-imidazo[1,5-a]piridin-3,1'-ciclohexano]-1,5-diona (1, 0,6 g, 1,93 mmol) y pentasulfuro de fósforo (857 mg, 3,85 mmol). Sellar el vial y calentar la mezcla de reacción a 50 °C durante 16 h. Después de terminado, la masa de reacción se diluyó con metanol al 5 % en diclorometano (100 ml), se lavó con agua (100 ml, 2 veces) y solución salina (100 ml), se secó sobre sulfato de sodio anhidro y se concentró al vacío. El material en bruto se purificó por cromatografía ultrarrápida en gel de sílice en metanol al 3-4 % en diclorometano, se combinaron las fracciones concentradas a presión reducida. El producto se trituró con metanol, se filtró se lavó con metanol (3 ml), éter (5 ml), se secó a alto vacío para obtener el producto deseado 6'-(pirimidin-4-ilamino)-1 '-tioxo-, 1',2'-dihidro-5'H-espiro[ciclohexano-1,3'-imidazo[1,5-a]piridin]-5'-ona (Comp. N.° 131) en forma de un sólido de color amarillo claro. Rendimiento: 0,050 g, 8 %; EM (IEN) m/z 328,41 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 12,26 (s, 1H), 9,46 (s, 1H), 8,80-8,77 (d, 2H), 8,39-8,38 (d, 1H), 7,39-7,38 (d, 1H), 7,07-7,06 (d, 1H), 2,97 (t, 2H), 1,78-1,72 (m, 5H), 1,59-1,56 (d, 1H), 1,26 (m, 1H).
Ejemplo 132
Síntesis de 8'-cloro-1'-(metoxiimino)-6'-(pirimidin-4-ilamino)-1',2'- dihidro-5'H-espiro[ciclohexano-1,3-imidazo[1,5-a]piridin]-5'-ona (Comp. N.° 132)



Síntesis de 8'-cloro-1’-(metoxiimino)-6'-(pirimidin-4-ilamino)-1’,2'-dihidro-5'H-espiro[ciclohexano-1,3'-imidazo[1,5-a]piridin]-5'-ona (Comp. N.° 132)
A una solución de 8'-cloro-6'-(pirimidin-4-ilamino)-2'H-espiro[ciclohexano-1,3-imidazo[1,5-a]piridin]-diona (1,1 g, 2,89 mmol) en cloroformo (30 ml) se le añadió trietilamina (1,21 ml, 8,67 mmol) y clorhidrato de O-metilhidroxilamina (241 mg, 2,89 mmol). La reacción se agitó a reflujo durante la noche. La mezcla resultante se enfrió a temperatura ambiente y se lavó con agua. La capa orgánica se secó sobre sulfato de magnesio, se filtró y se concentró. El producto en bruto se purificó por cromatografía en columna ultrarrápida para proporcionar 8'-cloro-1'-(metoxiimino)-6'-(pirimidin-4-ilamino)-1',2'-dihidro-5'H-espiro[ciclohexano-1,3'-imidazo[1,5-a]piridin]-5'-ona (Comp. N.° 132).
Ejemplo 133
Síntesis de 8'-cloro-6,-(pirimidin-4-ilamino)-5'-tioxo-2,H-espiro[ciclohexano-1,3'-imidazo[1,5-a]piridin]-1'(5'H)-ona (Comp. N.° 133)

Síntesis de 3-cloro-6-oxo-5-(pirimidin-4-ilamino)-1,6-dihidropiridin-2-carboxilato de etilo (3)
La síntesis del intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento H. Sólido de color amarillo; Rendimiento: 0,42 g, 20 %; EM (IEN) m/z 295 [M 1]+
Síntesis de 3-cloro-5-(pirimidin-4-ilamino)-6-tioxo-1,6-dihidropiridin-2-carboxilato de etilo (4)
Un vial se cargó con 3-cloro-6-oxo-5-(pirimidin-4-ilamino)-1,6-dihidropiridin-2-carboxilato (3, 0,35, 1,2 mmol) y piridina (10 ml) fue añadida. A la mezcla anterior se le añadió pentasulfuro de fósforo (0,53 g, 2,4 mmol) y la reacción se calentó a 110 °C durante 16 h. El análisis por CCF mostró presencia de material de partida y se añadió pentasulfuro de fósforo (0,265 g, 1,2 mmol) y se calentó la reacción a 115 °C durante 24 h. La piridina se retiró a presión reducida y se añadió agua (20 ml) y se extrajo con metanol al 5 % en diclorometano (100 ml). La capa orgánica se lavó con bicarbonato de sodio y salmuera y la capa se concentró a sequedad para proporcionar 3-cloro-5-(pirimidin-4-ilamino)-6-tioxo-1,6-dihidropiridin-2-carboxilato de etilo (4) en forma de un sólido de color amarillo marrón y se usó
directamente sin purificación adicional. Rendimiento: 350 mg, producto en bruto; EM (IEN) m/z 310,94 [M 1]+.
Síntesis de 3-cloro-5-(pirimidin-4-ilamino)-6-tioxo-1,6-dihidropiridin-2-carboxamida (5)
Procedimiento K: Un vial se cargó un 3-cloro-5-(pirimidin-4-ilamino)-6-tioxo-1,6-dihidropiridin-2-carboxilato de etilo (4, 0,35 g, 1,2 mmol) y amoníaco metanólico (12 ml). La mezcla se calentó lentamente a 60-65 °C durante 40 h cuando la CCF mostró una conversión completa del material de partida. La mezcla se enfrió y se concentró a presión reducida y se trituró con éter dietílico (10 ml) para proporcionar 3-cloro-5-(pirimidin-4-ilamino)-6-tioxo-1,6-dihidropiridin-2-carboxamida (5) en forma de un sólido de color marrón rojizo. Rendimiento: 350 mg, producto en bruto; EM (IEN) m/z 282,04 [M 1]+.
Síntesis de 8’-cloro-6'-(pirimidin-4-ilamino)-5’-tioxo-2'H-espiro[ciclohexano-1,3’-imidazo[1,5-a]piridin]-1’(5'H)-ona (Comp. N ° 133)
La síntesis del compuesto 133 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento A. Sólido de color amarillo; Rendimiento: 17 mg, 4 %; EM (IEN) m/z 362,06 [M 1]+. RMN 1H (400 MHz, DMSO-d6) ó 10,64 (s ancho, 1H), 9,93 (s, 1H), 9,00 (s, 1H), 8,90 (s, 1H), 8,51 (d, J = 5,6 Hz, 1H), 7,43 (d, J = 6,0 Hz, 1H), 4,22-3,12 (m, 2H), 1,84-1,78 (m, 2H), 1,75-1,60 (m, 3H), 1,54-1,47 (m, 2H), 1,30-1,22 (m, 1H).
Ejemplo 134
Síntesis de 8'-cloro-5,-(metoxiimino)-6'-(pirimidin-4-ilamino)-2'H-espiro[clohexano-1,3'-imidazo[1,5-a]piridin]-1'(5'H)-ona (Comp. N.° 134)

Síntesis de 8’-cloro-5'-(metoxiimino)-6’-(pirimidin-4-ilamino)-2’H-espiro[ciclohexano-1,3’-imidazo[1,5-a]piridin]-1’(5'H)-ona (Comp. N ° 134)
A una solución de 8'-cloro-6'-(pirimidin-4-ilamino)-2'H-espiro[ciclohexano-1,3-imidazo[1,5-a]piridin]-diona (1, 1 g, 2,89 mmol) en cloroformo (30 ml) se le añaden trietilamina (1,21 ml, 8,67 mmol) y clorhidrato de O-metilhidroxilamina (241 mg, 2,89 mmol). La reacción se agita a reflujo durante la noche. La mezcla resultante se enfría a temperatura ambiente y se lava con agua. La capa orgánica se seca sobre sulfato de magnesio, se filtra y se concentra. El producto en bruto se purifica por cromatografía en columna ultrarrápida para proporcionar 8'-cloro-5'-(metoxiimino)-6'-(pirimidin-4-ilamino)-2'H-espiro[ciclohexano-1,3,3-imidazo[1,5-a]piridin]-1'(5'H)-ona (Comp. N.° 134)
Ejemplo 135
Síntesis de 8'-cloro-2,-ciclopropil-6'-(pirimidin-4-ilamino)-2,H-espiro[ciclohexano-1,3-imidazo[1,5-a]piridin]-1',5'-diona (Comp. N.° 135)

A una solución de 6'-bromo-8'-cloro-2'H-espiro[ciclohexano-1,3-imidazo[1,5-a]piridin]1’,5'-diona (1, 1 g, 3,02 mmol) en 1,2-dicloroetano (15 ml) se le añade ácido ciclopropilborónico (2, 0,58 g, 6,80 mmol), acetato de cobre (II) (0,59 g, 3,23 mmol), 2,2'-bi-piridilo (0,50 g, 3,23 mmol) y carbonato de sodio (0,73 g, 6,86 mmol). La reacción se agita a 70 °C durante una noche. La reacción se enfría a temperatura ambiente. La mezcla resultante se enfría con solución acuosa saturada de cloruro de amonio y se extrae con diclorometano. El orgánico se seca sobre sulfato de magnesio, se filtra y se concentra. El producto en bruto se purifica por cromatografía en columna para proporcionar 6’-bromo-8'-cloro-2’-ciclopropil-2'H-espiro[ciclohexano-1,3'-imidazo[1,5-a]piridin]-1'5'-diona (3).
Síntesis de 8’-cloro-2'-ciclopropil-6’-(pirimidin-4-ilamino)-2'H-espiro[ciclohexano-1,3-imidazo[1,5-a]piridin]-1’,5’-diona (Comp. N.° 135)
La síntesis del compuesto 135 se realiza como se ha descrito anteriormente usando el protocolo general del Procedimiento H.
Ejemplo 136
Síntesis de 8'-cloro-2,-(piridin-3-il)-6'-(pirimidin-4-ilamino)-2'H-espiro[ciclohexano-1,3,-imidazoi[1,5-a]piridin]-1',5'-diona (Comp. N.° 136)

Síntesis de 8’-cloro-2'-(piridin-3-il)-6’-(pirimidin-4-ilamino)-2'H-espiro[ciclohexano-1,3-imidazo[1,5-a]piridin]-1 5'-diona (Comp. N ° 136)
A una solución de 8’-cloro-6'-(pirimidin-4-ilamino)-2’H-espiro[ciclohexano-1,3-imidazo[1,5-a]piridin]-1’,5’-diona (1,
450 mg, 1,3 mmol) y 3-yodopiridina (800 mg, 3,9 mmol) en dimetilformamida (10 ml) en un vial, se le añadió carbonato de cesio (550 mg, 1,6 mmol) y la mezcla se desgasificó con argón durante 15 min. A esta mezcla se le añadieron 1,10,-fenantrolina (37 mg, 0,2 mmol), yoduro de cobre (I) (12 mg, 0,05 mmol), XantPhos (26 mg, 0,065 mmol) y la mezcla de reacción se calentó a 130 °C durante 26 h. El análisis por CCF mostró el consumo de material de partida, la mezcla de reacción se filtró sobre lecho de celite y se lavó con metanol al 5 % en diclorometano seguido de la concentración del filtrado. El sólido obtenido se purificó por cromatografía Combi-Flash sobre alúmina neutra usando metanol al 3 %/diclorometano como eluyente, las fracciones apropiadas se concentraron a presión reducida para proporcionar 8'-cloro-2'-(piridin-3-il)-6'-(pirimidin-4-ilamino)-2'H-espiro[ciclohexano-1,3-imidazo[1,5-a]piridin]-1',5'-diona (Comp. N.° 136) en forma de un sólido de color amarillo. Rendimiento: 0,025 g, 4,6 %; EM (IEN) m/z 423,12 [M 1]+; RMN 1H (400 MHz, DIVISOR) ó 10,59 (s, 1H), 8,61 (d, J = 2,0 Hz, 1H), 8,58 (s, 1H), 8,49 (d, J = 4,8 Hz, 1H), 8,33 (d, J =6,0 Hz, 1H), 8,03 (s, 1H), 7,82 (d, J = 8,8 Hz, 1H), 7,47 (dd, J = 4,4, 8,8 Hz, 1H), 6,65 (d, J = 6,0 Hz, 1H), 2,85-2,79 (m, 2H), 1,74-1,53 (m, 6H), 1,17-1,14 (m, 1H).
Ejemplo 137
Síntesis de 7,-fluoro-6'-(pirimidin-4-ilamino)-2,H-espiro[ciclohexano-1,3,3-imidazo[1,5-a]piridin]-1',5,-diona (Comp. N.° 137)

Síntesis de 4-fluoropicolinato de metilo (2)
A una solución de ácido 4-fluoropicolínico (1, 1 g, 7,09 mmol) en tolueno (20 ml) y metanol (5 ml) a temperatura ambiente se le añade (trimetilsilil)diazometano (2 M en hexanos, 5,32 ml, 10,64 mmol) gota a gota. La reacción se agita a 80 °C durante 2 h. La mezcla resultante se concentra y purifica por cromatografía ultrarrápida para proporcionar 4-fluoropicolinato de metilo (2).
Síntesis de 1-óxido de 4-fluoro-2-(metoxicarbonil)piridina (3)
A una solución de 4-fluoropicolinato de metilo (2, 1 g, 6,45 mmol) en diclorometano (30 ml) se le añade peróxido de hidrógeno urea (1,27 g, 13,54 mmol) y anhídrido trifluoroacético (1,79 ml, 12,9 mmol). La reacción se agita a temperatura ambiente durante 2 h. La mezcla resultante se vierte en ácido clorhídrico 0,5 M y se extrae con
diclorometano. La capa orgánica se lava con solución acuosa saturada de bicarbonato de sodio, se seca sobre sulfato de magnesio, se filtra y se concentra. El producto en bruto se purifica por cromatografía ultrarrápida para proporcionar el 1-óxido de 4-fluoro-2-(metoxicarbonil)piridina (3).
Síntesis de 4-fluoro-6-oxo-1,6-dihidropiridin-2-carboxilato de metilo (4)
A una solución de 1-óxido de 4-fluoro-2-(metoxicarbonil)piridina (3, 1 g, 5,84 mmol) en dimetilformamida (25 ml) se le añade anhídrido trifluoroacético (1,62 ml, 11,68 mmol). La reacción se agita a 50 °C durante 3 h. La mezcla resultante se concentra y purifica por cromatografía ultrarrápida para proporcionar 4-fluoro-6-oxo-1,6-dihidropiridin-2-carboxilato de metilo (4).
Síntesis de 5-bromo-4-fluoro-6-oxo-1,6-dihidropiridin-2-carboxilato de metilo (5)
A una solución de 4-fluoro-6-oxo-1,6-dihidropiridin-2-carboxilato de metilo (4,1 g, 5,84 mmol) en acetonitrilo (25 ml) se le añadió N-bromosuccinimida (1,56 g, 8,76 mmol). La reacción se agita a reflujo durante 2 h. La mezcla resultante se vierte en bisulfito de sodio acuoso medio saturado y se extrae con acetato de etilo. Las capas orgánicas se combinan, se secan sobre sulfato de magnesio, se filtran y se concentran. El producto en bruto se purifica vía cromatografía ultrarrápida para proporcionar 5-bromo-4-fluoro-6-oxo-1,6-dihidropiridin-2-carboxilato de metilo (5).
Síntesis de 5-bromo-4-fluoro-6-oxo-1,6-dihidropiridin-2-carboxamida (6)
La síntesis del intermedio 6 se realiza como se ha descrito anteriormente usando el protocolo general del Procedimiento K.
Síntesis de 6'-bromo-7'-fluoro-2'H-espiro[cidohexano-1,3’-imidazo[1,5-a]piridin]-V,5’-diona (8)
La síntesis del intermedio 8 se realiza como se ha descrito anteriormente usando el protocolo general del procedimiento A.
Síntesis de 7’-fluoro-6'-(pirimidin-4-ilamino)-2’H-espiro[ciclohexano-1,3-imidazo[1,5-a]piridin]-1 5'-diona (Comp. N° 137)
La síntesis del compuesto 137 se realiza como se ha descrito anteriormente usando el protocolo general del Procedimiento H.
Ejemplo 138
Síntesis de 8-fenil-6-(pirimidin-4-ilamino)espiro[2H-imidazo[1,5-a]piridin-3,1'-ciclohexano]-1,5-diona (Comp. N.° 138)

Síntesis de 8-fenil-6-(pirimidin-4-ilamino)espiro[2H-imidazo[1,5-a]piridin-3,1’-ciclohexano-1,5-diona (Comp. N ° 138) La síntesis del compuesto 138 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento G. Sólido de color blanquecino; Rendimiento: 0,15 g, 67 %; EM (IEN) m/z 388,48 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,16 (s, 1H), 9,49 (s, 1H), 8,74 (s, 1H), 8,71 (s, 1H), 8,38 (d, J = 5,84 Hz, 1H), 7,47-7,35 (m,
6H), 3,10-3,04 (m, 2H), 1,76-1,49 (m, 7H), 1,04-1,02 (m, 1H).
Ejemplo 139
Síntesis de 8'-(oxetan-2-il)-6,-(pirimidin-4-ilamino)-2'H-espiro[ciclohexano-1,3-imidazo[1,5-a]piridin]-1 ',5'-diona (Comp. N.° 139)

Síntesis de 8’-cloro-2'-(4-metoxibencil)-6’-((4-metoxibencil)(pirimidin-4-il)amino)-2'H-espiro[ciclohexano-1,3’-imidazo[1,5-a]piridin]-1' 5'-diona (2)
A una solución de 8'-cloro-6l-(pirimidin-4-ilamino)-2'H-espiro[ciclohexano-1,3-imidazo[1,5-a]piridin]-1',5'-diona (1, 1 g, 2,89 mmol) en dimetilformamida (15 ml) se le añadió hidruro de sodio (0,21 g, 8,67 mmol) y cloruro de 4-metoxibencilo (1,57 ml, 11,56 mmol). La reacción se agita a temperatura ambiente durante la noche. La mezcla resultante se vierte en agua helada y se extrae con diclorometano. La capa orgánica se seca sobre sulfato de magnesio, se filtra y se concentra. El producto en bruto se purifica por cromatografía en columna para proporcionar 8'-cloro-2’-(4-metox¡benc¡l)-6'-((4-metox¡benc¡l)(p¡r¡mid¡n-4-¡l)am¡no)-2’H-esp¡ro[c¡clohexano-1,3’-imidazo[1,5-a]pir¡d¡n]-1',5’-diona (2).
Síntesis de 2’-(4-metoxibencil)-6'-((4-metoxibencil)(pirimidin-4-il)amino)-1’,5'-dioxo-1’,5'-dihidro-2-[ciclohexano-1,3’-imidazo[1,5-a]piridin]-8'-carbonitrilo (3)
A una solución de 8'-cloro-2’-(4-metox¡benc¡l)-6'-((4-metox¡benc¡l)(p¡r¡mid¡n-4-¡l)am¡no)-2'H-esp¡ro[c¡clohexano-1,3'-imidazo[1,5-a]piridin]-1’,5’-diona (2, 1 g, 1,71 mmol) en acetonitrilo (20 ml) se le añade cianuro de potasio (0,17 g, 2,56 mmol), cloruro de tributilestaño 0,038 ml, 0,14 mmol). La mezcla se desgasifica y se sigue la adición de tris (dibencilidenacetona) dipaladio (0) (64 mg, 0,07 mmol) y tri-terc-butilfosfina (63 mg, 0,31 mmol). La mezcla se desgasifica dos veces más. La reacción se agita a 80 °C durante una noche. La mezcla resultante se enfría a
temperatura ambiente y se filtra a través de un lecho de celite. El filtrado se concentra y purifica por cromatografía en columna para proporcionar 2'-(4-metoxibencil)-6'-((4-metoxibencil) (pirimidin-4-il)amino)-1’,5'-dioxo-1’5’-dihidro-2'H-espiro[ciclohexano-1,3-imidazo[1,5-a]piridin]-8’-carbonitrilo (3).
Síntesis de 2’-(4-metoxibencil)-6'-((4-metoxibencil)(pirimidin-4-il)amino)-1’,5'-dioxo-1’,5’-dihidro-ciclohexano-1,3’-imidazo[1,5-a]piridin]-8'-carbaldehído (4)
A una solución de 2’-(4-metoxibencil)-6'-((4-metoxibencil) (pirimidin-4-il)amino)-1’,5’-dioxo-1’,5’-dihidro-2’H-espiro[ciclohexano-1,3'-imidazo[1,5-a]piridin]-8’-carbonitrilo (3,1 g, 1,73 mmol) en diclorometano (20 ml) a 0 °C se le añade hidruro de diisobutilaluminio (1 M en diclorometano, 3,46 ml, 3,46 mmol) gota a gota. El reactor se agita a 0 °C durante 2 h. La mezcla resultante se vierte en solución salina acuosa saturada de Rochelle. La mezcla bifásica se agita durante la noche y se filtra. El filtrado se extrae con diclorometano. El orgánico se seca sobre sulfato de magnesio, se filtra y se concentra. El producto en bruto se purifica por cromatografía en columna para proporcionar 2’-(4-metoxibencil)-6'-((4-metoxibencil)(pirimidin-4-il)amino)-1’,5’-dioxo-1’,5'-dihidro-2’H-espiro[ciclohexano-1,3-imidazo[1,5-a]piridin]-8'-carbaldehído (4).
Síntesis de 2’-(4-metoxibencil)-6'-((4-metoxibencil)(pirimidin-4-il)amino)-8’-(oxiran-2-il)-2'H-espiro[ciclohexano-3’-imidazo[1,5-a]piridin]-1 ’,5’-diona (5)
Se calienta una suspensión de hidruro de sodio (dispersión al 50 %, lavada sin aceite mineral, 90 mg, 1,87 mmol) en dimetilsulfóxido (2 ml) a 65 °C en argón durante 1 h. El baño de aceite se retira y a la solución transparente se le añade tetrahidrofurano (2 ml). La solución se enfría a -15 °C y se trata con una solución de yoduro de trimetilsulfonio (0,35 g, 1,72 mmol) en dimetilsulfóxido (2 ml). Después de 3 min, se añadió gota a gota una solución de 2’-(4-metoxibencil)-6'-((4-metoxibencil)(pirimidin-4-il)amino)-1’,5'-dioxo-1’,5'-dihidro-espiro[ciclohexano-1,3-imidazo[1,5-a]piridin]-8’-carbaldehído (4, 1 g, 1,72 mmol) en 3 ml de tetrahidrofurano. La reacción se agita durante una noche. La mezcla resultante se vierte en agua y se extrae con acetato de etilo. Los extractos se lavan con agua, se secan y se evaporan para proporcionar 2’-(4-metoxibencil)-6'-((4-metoxibencil)(pirimidin-4-il)amino)-8’-(oxiran-2-il)-2’H-espiro[ciclohexano-1,3-imidazo[1,5-a]piridin]-1’,5'-diona (5).
Síntesis de 2’-(4-metoxibencil)-6'-((4-metoxibencil)(pirimidin-4-il)amino)-8’-(oxetan-2-il)-2'H-espiro[ciclohexano-1,3’-imidazo[1,5-a]piridin]-1 ’,5’-diona (6)
Se añade terc-butóxido de potasio (0,23 g, 2,02 mmol) a una solución de yoduro de trimetilsulfoxonio (0,44 g, 2,02 mmol) en terc-butanol (10 ml) a temperatura ambiente. Después de 15 minutos, se añade una solución de 2’-(4-metoxibencil)-6'-((4-metoxibencil)(pirimidin-4-il)amino)-8’-(oxiran-2-il)espiro[ciclohexano-1,3-imidazo[1,5-a]piridin]-1’,5’-diona (5, 1 g, 1,68 mmol) en dimetilsulfóxido (5 ml). La reacción se agita a 50 °C durante una noche. La mezcla resultante se inactiva con salmuera y se extrae con acetato de etilo. Los extractos combinados se secan sobre sulfato de magnesio. Después de la filtración y la concentración, el residuo se purifica por columna ultrarrápida para proporcionar 2’-(4-metoxibencil)-6'-((4-metoxibencil)(pirimidin-4-il)amino)-8’-(oxetan-il)-2’H-espiro[ciclohexano-1,3'-imidazo[1,5-a]piridin]-1’,5’-diona (6).
Síntesis de 8’-(oxetan-2-il)-6'-(pirimidin-4-ilamino)-2’H-espiro[ciclohexano-1,3'-imidazo[1,5-a]piridin]1’,5'-diona (Comp. N.° 139)
Se disuelve 2’-(4-metoxibencil)-6'-((4-metoxibencil)(pirimidin-4-il)amino)-8’-(oxetan-2-il)-2-espiro[ciclohexano-1,3’-imidazo[1,5-a]piridin]-1’,5'-diona (6, 100 mg, 0,16 mmol) en ácido trifluoroacético (5 ml). La reacción se agita a temperatura ambiente durante la noche. La mezcla resultante se concentra y purifica por cromatografía en columna para proporcionar 8’-(oxetan-2-il)-6'-(pirimidin-4-ilamino)-2’H-espiro[ciclohexano-1,3-imidazo[1,5-a]piridin]-1’,5'-diona (Comp. N.° 139).
Ejemplo 140
Síntesis de 8-cloro-3-metil-6-(pirimidin-4-ilamino)-3-vinil-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona (Comp. N.° 140)


La síntesis del producto intermedio 3 se realiza como se ha descrito anteriormente usando el protocolo general del procedimiento A.
Síntesis de 8-cloro-3-metil-6-(pirimidin-4-ilamino)-3-vinil-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona (Comp. N ° 140) La síntesis del compuesto 140 se realiza como se ha descrito anteriormente usando el protocolo general del Procedimiento H.
Ejemplo 141
Síntesis de 8-cloro-3-metil-3-(prop-1-in-1-il)-6-(pirimidin-4-ilamino)-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona (Comp. N.° 141)

Síntesis de 6-bromo-8-cloro-3-metil-3-(prop-1-in-1-il)-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona (3)
La síntesis del producto intermedio 3 se realiza como se ha descrito anteriormente usando el protocolo general del procedimiento A.
Síntesis de 8-cloro-3-metil-3-(prop-1-in-1-il)-6-(pirimidin-4-ilamino)-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona (Comp. N ° 141)
La síntesis del compuesto 141 se realiza como se ha descrito anteriormente usando el protocolo general del Procedimiento H.
Ejemplo 142
Síntesis de 4-((8'-cloro-1',5'-dioxo-1',5'-dihidro-2'H-espiro[ciclohexano-1,3,3-imidazo[1,5-a]piridin]-6'-il)amino)pirimidin-5-carbonitrilo (Comp. N.° 142)

Síntesis de 4-((8'-cloro-1 ’,5 ’-dioxo-15'-dihidro-2'H-espiro[ciclohexano-1,3,3-imidazo[1,5-a]piridin]-6’-il)amino)pirimidin-5-carbonitrilo (Comp. N ° 142)
La síntesis del compuesto 142 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento H. Sólido de color amarillo; Rendimiento: 0,25 g, 76 %; EM (IEN) m/z 371,38 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,48 (s, 1H), 9,06 (s, 1H), 9,03 (s, 1H), 8,98 (s, 1H), 8,59 (s, 1H), 2,93-2,87 M, 2H), 1,93 1,88 (m, 2H), 1,75-1,54 (m, 5H), 1,27-1,23 (m, 1H).
Ejemplo 143
Síntesis de 8'-cloro-6'-((5-(1-metil-1H-pirazol-4-il)pirimidin-4-il)amino)-2'H-espiroiciclohexano-1,3'-imidazo[1,5-a]piridin]-1',5'-diona (Comp. N.° 143)
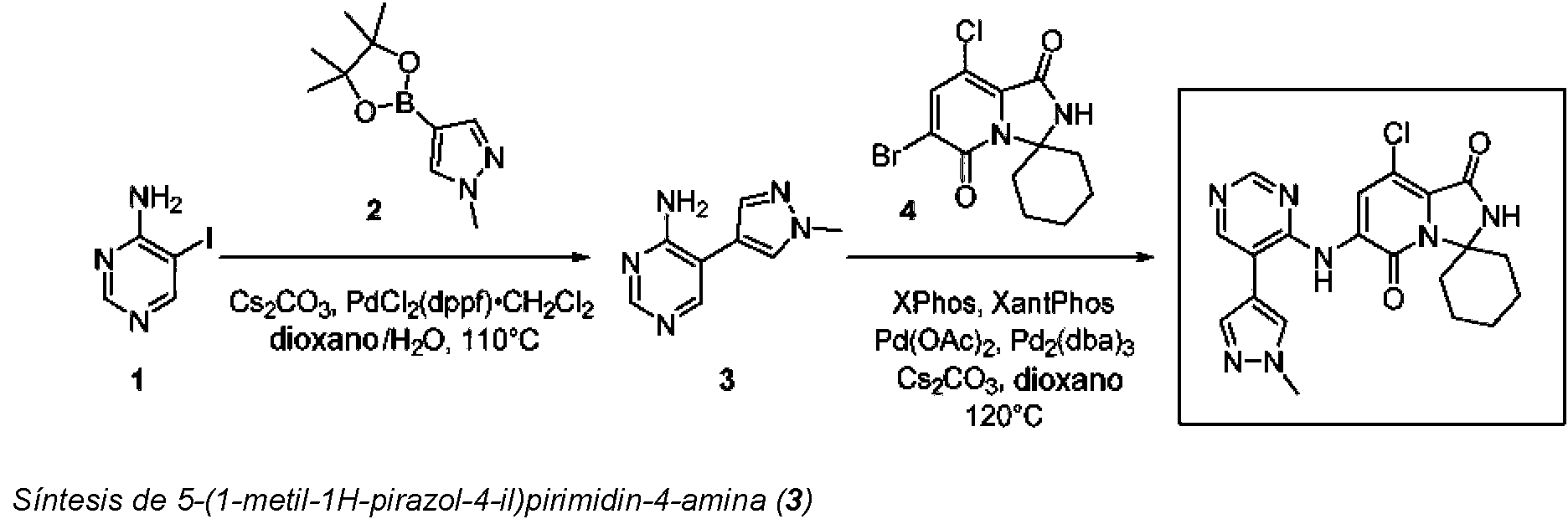
A una solución de 5-yodopirimidin-4-amina (1, 0,5 g, 2,26 mmol) y 1-metil-4-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)pirazol (2, 0,56 g, 2,71 mmol) en dioxano/agua (15 ml, 9: 1,5) en un vial, se le añadió carbonato de cesio (1,84 g, 5,66 mmol) y la mezcla se desgasificó con argón durante 15 min. A esta mezcla se le añadió [1,1'-bis(difenilfosfino)ferroceno]dicloropaladio (II), complejo con diclorometano (0,09 g, 0,11 mmol) y después se calentó a 110 °C durante 16 h. El análisis por CCF mostró el consumo de material de partida, se enfrió la mezcla de reacción a temperatura ambiente, se filtró la masa sobre lecho de celite, se lavó con diclorometano (30 ml) seguido de concentración de filtrado. El compuesto en bruto se purificó por cromatografía de columna ultrarrápida eluyendo con metanol al 3 % en diclorometano. Las fracciones deseadas se concentraron a sequedad para proporcionar como 5-(1-metilpirazol-4-il)pirimidin-4-amina (3) en forma de un sólido de color blanquecino; Rendimiento: 0,2 g, 50 %; EM (IEN) m/z 176,08 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 8,28 (s, 1H), 8,13 (s, 1H), 8,01 (s, 1H), 7,70 (s, 1H), 6,61 (s, 2H), 3,87 (s, 3H).
Síntesis de 8'-cloro-6’-((5-(1-metil-1H-pirazol-4-il)pirimidin-4-il)amino)-2'H-espiro[ciclohexano-1,3'-imidazo[1,5-a]piridin]-1’,5’-diona (Comp. N ° 143).
La síntesis del compuesto 143 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento H. Sólido de color amarillo pálido; Rendimiento: 0,14 g, 36 %; EM (IEN) m/z 426,44 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,35 (s, 1H), 8,83 (s, 1H), 8,76-8,73 (s, 2H), 8,46 (s, 1H), 8,17 (s, 1H), 7,85 (s, 1H), 3,96 (s, 3H), 2,86 (m, 2H), 1,74-1,49 (m, 7H), 1,25 (m, 1H).
Ejemplo 144
Síntesis de 8'-cloro-6,-((5-etinilpirimidin-4-il)amino)-2'H-espiro[ciclohexano-1,3-imidazo[1,5-a]piridin]-1'5'-diona (Comp. N.° 144)
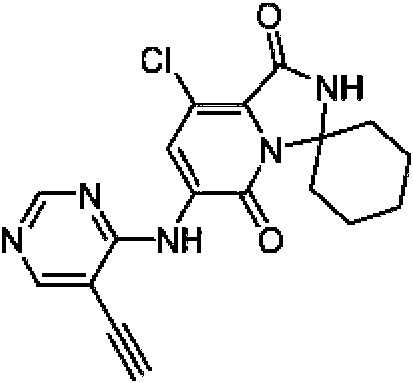

Síntesis de 5-((trimetilsilil)etinil)pirimidin-4-amina (3)
Se tomaron 5-yodopirimidin-l-amina (1, 1 g, 4,52 mmol), yoduro de cobre (I) (172 mg, 0,90 mmol), etinil(trimetil)silano (2, 0,67 g, 6,79 mmol), trifenilfosfina (119 mg, 0,45 mmol), trietilamina (0,914 mg, 9,04 mmol) y cloruro de paladio (II) (80 mg, 0,45 mmol) en un matraz y se añadió tetrahidrofurano seguido de desgasificación con argón durante 5 minutos. La mezcla de reacción se agitó a 40 °C durante 16 h. Después de la terminación, la mezcla de reacción se filtró sobre lecho de celite y el filtrado resultante se concentró para proporcionar 5-(2-trimetilsililetinil)pirimidin-4-amina (3) en forma de un sólido de color marrón. Rendimiento: 0,76 g, 8 8 %; EM (IEN) m/z 192,1 [M 1]+.
Síntesis de 8'-cloro-6’-((5-((trimetilsilil)etinil)pirimidin-4-il)amino)-2'H-espiro[ciclohexano-1,3'-imidazo[1,5-a]piridin]-1’,5’-diona (5)
La síntesis del intermedio 5 se realizó como se ha descrito anteriormente usando el protocolo general del procedimiento B. Sólido de color amarillo. Rendimiento: 0,30 g, 45 %; EM (IEN) m/z 441,99 [M 1]+; RMN 1H (400 MHz, DMSO-d6) 6 10,40 (s, 1H), 9,06 (s, 1H), 8 , 88 (s, 1H), 8,67 (m, 2H), 5,80 (m, 1H), 2,94-2,89 M, 2H), 1,74 1,57 (m, 7H), 1,23 (m, 1H), 0,34 (s, 9H).
Síntesis de 8’-cloro-6'-((5-etinilpirimidin-4-il)amino)-2’H-espiro[ciclohexano-1,3'-imidazo[1,5-a]piridin]-1’,5’-diona (Comp. N.° 144)
Se cargó un matraz con 8'-cloro-6'-((5-((trimetilsilil)etinil)pirimidin-4-il)amino)-2'H-espiro[ciclohexano-1,3'-imidazo[1,5-a]piridin-1',5'-diona (5, 300 mg, 0,67 mmol) y metanol (20 ml) seguido de adición de carbonato de potasio (469 mg, 3,39 mmol) a temperatura ambiente y se agitó la mezcla de reacción Durante 16 h. Después de la terminación, el disolvente se concentró a presión reducida y el residuo resultante se lavó adicionalmente con agua seguido de éter dietílico y pentano para proporcionar 8'-cloro-6'-((5-etinilpirimidin-4-il)amino)-2'H-espiro[ciclohexano-1,3'-imidazo[1,5-a]piridin]-1',5'-diona (Comp. N.° 144) en forma de un sólido de color amarillo. Rendimiento: 100 mg, 40 %; EM (IEN) m/z 370,09 [M 1]+; RMN 1H (400 MHz, DMSO-d6 ) 6 10,42 (s, 1H), 8,99 (s, 1H), 8,90 (s, 1H), 8,70 (s, 2H), 5,22 (s, 2H), 2,90 (s ancho, 2H), 1,73-1,64 (m, 2H), 1,61-1,56 (m, 3H), 1,56-1,53 (m, 2H), 1,27 (m, 1H).
Ejemplo 145
Síntesis de 6'-((1H-pirazolo[3,4-rf]pirimidin-4-il)amino)-8,-cloro-2'H-espiro[ciclohexano-1,3'-imidazo[1,5-a]piridin]-1',5'-diona (Comp. N.° 145)

Síntesis de 6’-((1H-pirazolo[3,4-d]pirimidin-4-il)amino)-8'-cloro-2’H-espiro[ciclohexano-1,3’-imidazo[1,5-a]piridin]-1 ',5'-diona (Comp. N ° 145)
A una suspensión de 6-bromo-8-cloroespiro[2H-imidazo[1,5-a]piridin-3,1'-ciclohexano]-1,5-diona (1, 0,5 g, 1,51 mmol), 1H-pirazolo[3,4-d]pirimidin-4-amina (2, 0,2 g, 1,51 mmol) en terc-butanol (20 ml) en un vial, se le añadió fosfato de potasio (0,96 g, 4,54 mmol), y la mezcla reacción se desgasificó con argón durante 15 min. A esta mezcla se le añadió XantPhos (4 mg, 0,08 mmol) y Pd2(dba)3 (7 mg, 0,08 mmol) y la mezcla de reacción se desgasificó adicionalmente con argón durante 5 min. La mezcla de reacción se calentó a 90 °C durante 18 h. El análisis por CCF mostrara el consumo de material de partida, la mezcla de reacción se filtró sobre lecho de celite y se lavó con diclorometano seguido de concentración del filtrado. El producto en bruto se agitó con metanol (10 ml) y se filtró. El sólido resultante se lavó adicionalmente con éter dietílico (20 ml) y se secó al vacío para proporcionar 8-cloro-6-(1H-pirazolo[3,4-d]pirimidin-4-ilamino)espiro[2H-imidazo[1,5-a]piridin-3,1'-ciclohexano]-1,5-diona (Comp. N.° 145). Rendimiento: 0,037 g, 6 %; EM (IEN) m/z 386,36 [M 1]+; RMN 1H (400 MHz, DMSO-de) ó 10,39 (s, 1H), 9,55 (s, 1H), 8,91 (s, 1H), 8,61 (s, 2H), 2,97 (m, 2H), 1,76-1,56 (m, 7H), 1,23 (m, 1H).
Ejemplo 146
Síntesis de 8-cloro-6-(3H-triazolo[4,5-d]pirimidin-7-ilamino)espiro[2H-imidazo[1,5-a]piridin-3,1'-ciclohexano]-1,5-diona (Comp. N.° 146)


Síntesis de 2-metil-N-(3H-triazolo[4,5-d]pirimidin-7-il)propanamida (3)
A un vial se le añadió 3H-triazolo[4,5-d]pirimidin-7-amina (1, 2,0 g, 14,69 mmol) en dimetilformamida (20 ml) seguido de la adición de 2-metilpropanoato de 2-metilpropanoílo (2, 6,97 g, 44,08 mmol). La mezcla de reacción se agitó a 160 °C durante 1 h. Después de la terminación, la reacción se enfrió a temperatura ambiente y se diluyó con agua (100 ml). El sólido blanquecino precipitado se filtró y se secó al vacío para proporcionar 2-metil-N-(3H-triazolo[4,5-d]pirimidin-7-il)propanamida (3) en forma de un sólido de color blanquecino. Rendimiento: 2,5 g, 82 %; EM (IEN) m/z 205,2 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 15,75 (m, 2H), 11,57 (s, 1H), 8,80 (s, 1H), 2,94-2,97 (m, 1H), 1,20 (d, J = 6 , 8 Hz, 6 H).
Síntesis de N-((2-(trimetilsilil)etoxi)metil)-N-(3-((2-(trimetilsilil)etoxi)metil)-3H-[1,2,3]triazolo[4,5-d]pirimidin-7-il)isobutiramida (4)
A una solución agitada de 2-metil-N-(3H-triazolo[4,5-d]pirimidin-7-il)propanamida (3, 1,5 g, 7,27 mmol) en dimetilformamida (20 ml), hidruro de sodio (0,26 g, 10,91 mmol) se le añadió en porciones en 10 minutos a 0 °C. La suspensión anterior se agitó durante 10 min a 0 °C y se añadió cloruro de 2-(trimetilsilil)etoximetilo (1,82 g, 10,91 mmol) a la misma temperatura en nitrógeno. La reacción se agitó a temperatura ambiente durante 6 h. Después de la terminación, la masa de reacción se inactivó con solución acuosa saturada de cloruro amónico y el extracto en bruto se extrajo con diclorometano (50 ml, 2 veces). Los compuestos orgánicos se separaron después, se secaron (sulfato de magnesio) y se concentraron a sequedad al vacío y el producto en bruto se purificó por cromatografía ultrarrápida eluyendo con acetato de etilo al 5 % en hexano. La concentración de las fracciones deseadas proporciona 2-metil-N-(2-trimetilsililetoxi)-N-[3-(2-trimetilsililetoxi)triazolo[4,5-d]pirimidin-7-il]propanamida (4) como aceite viscoso transparente. Rendimiento: 1,1 g, 44 %, EM (IEN) m/z 467,42 [M 1]+.
Síntesis de N,3-bis((2-(trimetilsihi)etoxi)metil)-3H-[1,2,3]triazolo[4,5-d]pirimidin-7-amina (5)
La síntesis del intermedio 5 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento I. Sólido de color amarillo; Rendimiento: 0,8 g, 94 %; EM (IEN) m/z 397 [M 1]+.
Síntesis de 8’-cloro-6'-((2-(trimetilsihi)etoxi)metil)(3-((2-(trimetilsihi)etoxi)metil)-3H-[1,2,3]triazolo[4,5-d]pirimidin-7-il)amino)-2’H-espiro[ciclohexano-1,3'-imidazo[1,5-a]piridin]-1’, 5'-diona (7)
La síntesis del intermedio 7 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento H. Sólido de color marrón; Rendimiento: 0,3 g, en bruto.
Síntesis de 8-cloro-6-(3H-triazolo[4,5-d]pirimidin-7-ilamino)espiro[2H-imidazo[1,5-a]piridin-3,1 ’-ciclohexano]-1,5-diona (Comp. N.° 146)
A una solución agitada de 8-cloro-6-[2-trimetilsililetoximetil-[3-(2-trimetilsililetoximetil)triazolo[4,5-d]pirimidin-7-il]amino]espiro[2H-imidazo[1,5-a]piridin-3,1'-ciclohexano]-1,5-diona (7, 0,3 g, 0,46 mmol) en diclorometano (15 ml), se añadió gota a gota ácido trifluoroacético (5 ml, 4,63 mmol) a 0 °C. La masa de reacción se agitó durante una noche a temperatura ambiente. Después de la terminación, la masa de reacción se concentró y coevaporó con éter dietílico. El producto en bruto se disolvió después en tetrahidrofurano/etanol y solución de hidróxido de potasio (5 ml, 0,46 mmol) (3 M en agua) y se agitó la mezcla durante 16 h. Una vez completada la reacción, la capa acuosa se separó y la capa orgánica se secó sobre sulfato de sodio, se filtró y se concentró para obtener producto en bruto. El producto en bruto se lavó con metanol y n-pentano y se secó para proporcionar 8-cloro-6-(3H-triazolo [4,5-d]pirimidin-7-ilamino)espiro[2H-imidazo[1,5-a]piridin-3,1'-ciclohexano]-1,5-diona (Comp. N.° 146) en forma de un
sólido de color marrón. Rendimiento: 70 mg, 39 %; EM (IEN) m/z 387,39 [M 1]+; RMN 1H (400 MHz, DMSO-da) ó 10,45 (s ancho, 1H), 9,29 (s, 1H), 8 , 8 8 (s, 1H), 7,77 (s, 1H), 2,94 (t, J = 2,32, 2H), 1,79-1,76 (m, 2H), 1,67-1,51 (m, 5H), 1,27-1,23 (m, 1H).
Ejemplo 147
Síntesis de 8-((6-aminopirimidin-4-il)amino)-10-metil-2,3,4,5-tetrahidropirido[1,2-a][1,4]diazepin-1,7-diona (Comp. N.° 147)

TFA
150 C, m o

Síntesis de N-bencil-5-bromo-N-3-(3-((terc-butildimetilsilil)oxi)propil)-3-metil-6-oxo-1,6-dihidropiridin-2-carboxamida (3)
A una solución agitada de N-bencil-3-(terc-butildimetilsililoxi)propan-1-amina (2, 3,0 g, 12,9 mmol) en dimetilformamida (50 ml), se le añadieron ácido 5-bromo-3-metil-6-oxo-1,6-dihidropiridin-2-carboxílico (1, 3,6 g, 12,9 mmol), HBTU (6,4 g, 16,9 mmol) y N,N-diisopropiletilamina (2,2 g, 16,9 mmol) en un vial a temperatura ambiente y se agitó la mezcla durante 16 h. El análisis por CCF mostró la terminación de la reacción, la mezcla de reacción se inactivó con solución acuosa de bicarbonato de sodio y se extrajo con acetato de etilo (250 ml). La capa orgánica se secó sobre sulfato de sodio y el disolvente se retiró a presión reducida para proporcionar N-bencil-5-bromo-N-(3-((terc-butildimetilsilil)oxi)propil)-3-metil-6-oxo-1,6-dihidropiridin-2-carboxamida (3) en forma de un líquido de color amarillo. Rendimiento: 3,0 g, 47 %; EM (IEN) m/z 495,24 [M-1]\
Síntesis de N-bencil-5-bromo-N-(3-hidroxipropil)-3-metil-6-oxo-1,6-dihidropiridin-2-carboxamida (4)
A una solución agitada de N-bencil-5-bromo-N-(3-((terc-butildimetilsilil)oxi)propil)-3-metil-6-oxo-1,6-dihidropiridin-2-carboxamida (3, 3,0 g, 6,0 mmol) en dioxano (20 ml), se le añadió cloruro de hidrogeno en dioxano (20 ml) a temperatura ambiente y la mezcla se agitó durante 16 h. Después de la terminación, el disolvente se retiró y la
reacción se basificó con solución acuosa de bicarbonato de sodio y se extrajo con 5 % de metanol/diclorometano (200 ml, 3 veces). La capa orgánica se secó sobre sulfato de sodio y el disolvente se retiró a presión reducida para obtener N-bencil-5-bromo-N-(3-hidroxipropil)-3-metil-6-oxo-1,6-dihidropiridin-2-carboxamida (4) en forma de un líquido de color marrón. Rendimiento: 2,2 g, 95 %; EM (IEN) m/z 381,22 [M-1 ]-.
Síntesis de 2-bencil-8-bromo-10-metil-2,3,4,5-tetrahidropirido[1,2-a][1,4]diazepin-1,7-diona (5)
A una solución agitada de N-bencil-5-bromo-N-(3-hidroxipropil)-3-metil-6-oxo-1,6-dihidropiridin-2-carboxamida (4, 1,5 g, 3,9 mmol) en tetrahidrofurano (30 ml), se le añadieron trifenilfosfina (1,5 g, 5,9 mmol) y azodicarboxilato de diisopropilo (1,2 g, 5,9 mmol) a 0 °C. La reacción se agitó a temperatura ambiente durante 16 h. Después de la terminación, el disolvente se retiró a presión reducida y el producto en bruto se purificó por cromatografía ultrarrápida eluyendo con acetato de etilo al 40 % en hexano. Las fracciones apropiadas se concentraron a presión reducida para proporcionar 2-bencil-8-bromo-10-metil-2,3,4,5-tetrahidropirido[1,2-a][1,4]diazepin-1,7-diona (5) en forma de un sólido de color amarillo. Rendimiento: 0,9 g, 64 %; EM (IEN) m/z 361,18 [M 1]+.
Síntesis de 8-((6-(di-(terc-butoxicarbonil)-amino)pirimidin-4-il)amino)-2-bencil-10-metil-2,3,4,5-tetrahidropirido[1,2-a][1,4]diazepin-1,7-diona (7)
La síntesis del intermedio 7 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento H. Sólido de color amarillo; Rendimiento: 0,9 g, 69 %; EM (IEN) m/z 591,66 [M 1 ]+.
Síntesis de 8-((6-aminopirimidin-4-il)amino)-2-bencil-10-metil-2,3,4,5-tetrahidropirido[1,2-a][1,4]diazepin-1,7-diona (8) La síntesis del intermedio 8 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento F. Sólido de color amarillo; Rendimiento: 0,59 g, 99 %; EM (IEN) m/z 391,32 [M 1]+.
Síntesis de 8-((6-aminopirimidin-4-il)amino)-10-metil-2,3,4,5-tetrahidropirido[1,2-a][1,4]diazepin-1,7-diona (Comp. N.° 147)
Se cargó un vial con 8-((6-aminopirimidin-4-il)amino)-2-bencil-10-metil-2,3,4,5-tetrahidropirido[1,2-a][1,4]diazepin-1,7-diona (8 , 0,3 g, 76,9 mmol) y ácido trifluoroacético (7,0 ml) y la mezcla de reacción se calentó en microondas a 150 °C durante 20 min. El análisis por CCF mostró que se completó la reacción y la mezcla se enfrió a temperatura ambiente y después se basificó con solución acuosa de bicarbonato de sodio y se extrajo con metanol al 5 %/diclorometano (200 ml, 3 veces). La capa orgánica se secó sobre sulfato de sodio y el disolvente se retiró a presión reducida para proporcionar 8-((6-aminopirimidin-4-il)amino)-10-metil-2,3,4,5-tetrahidropirido[1,2-a][1,4]diazepin-1,7-diona (Comp. N.° 147) en forma de un sólido de color marrón. Rendimiento: 0,06 g, 26 %; EM (IEN) m/z 301,15 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 8,51 (s, 1H), 8,44 (m, 2H), 8,15 (s, 1H), 6,52 (m, 2H), 6,13 (s, 1H), 5,05 (m, 1H), 3,17 (m, 2H), 2,95 (m, 1H), 2,13 (s, 3H), 1,87 (m, 2H).
Ejemplo 148
Síntesis de 3,3-di-ferc-butil-8-cloro-6-(pirimidin-4-ilamino)-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona (Comp. N.° 148)

Síntesis de 6-bromo-3,3-di-terc-butil-8-cloro-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona (3)
La síntesis del intermedio 3 se realiza como se ha descrito anteriormente usando el protocolo general del
procedimiento A.
Síntesis de 3,3-di-terc-butil-8-cloro-6-(pirimidin-4-ilamino)-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona (Comp. N ° 148) La síntesis del compuesto 148 se realiza como se ha descrito anteriormente usando el protocolo general del Procedimiento H.
Ejemplo 149
Síntesis de 8-cloro-6-(pirimidin-4-ilamino)-3,3-di(tiofen-2-il)-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona (Comp. N.° 149)

Síntesis de 6-bromo-8-cloro-3,3-di(tiofen-2-il)-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona (3)
La síntesis del producto intermedio 3 se realiza como se ha descrito anteriormente usando el protocolo general del procedimiento A.
Síntesis de 8-cloro-6-(pirimidin-4-ilamino)-3,3-di(tiofen-2-il)-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona (Comp. N ° 149) La síntesis del compuesto 149 se realiza como se ha descrito anteriormente usando el protocolo general del Procedimiento H.
Ejemplo 150
Síntesis de 8-cloro-6-(pirimidin-4-ilamino)-3,3-di(tiofen-3-il)-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona (Comp. N.° 150)

Síntesis de 3-bromo-8-cloro-3,3-di(tiofen-3-il)-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona (3)
La síntesis del intermedio 3 se realiza como se ha descrito anteriormente usando el protocolo general del procedimiento A.
Síntesis de 8-cloro-6-(pirimidin-4-ilamino)-3,3-di(tiofen-3-il)-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona (Comp. N ° 150) La síntesis del compuesto 150 se realiza como se ha descrito anteriormente usando el protocolo general del Procedimiento H.
Ejemplo 151
Síntesis de 8-cloro-3,3-dipropil-6-(pirimidin-4-ilamino)-2H-imidazo[1,5-a]piridin-1,5-diona (Comp. N.° 151)

Síntesis de 6-bromo-8-cloro-3,3-dipropil-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona (3)
La síntesis del intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento A. Sólido de color blanquecino; Rendimiento: 0,23 g, 6 6 %; EM (IEN) m/z 347,01 [M 1]+.
Síntesis de 8-cloro-3,3-dipropil-6-(pirimidin-4-ilamino)-2H-imidazo[1,5-aj’piridin-1,5-diona (Comp. N ° 151)
La síntesis del compuesto 151 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento H. Sólido de color blanquecino; Rendimiento: 0,14 g, 57 %; EM (IEN) m/z 362,13 [M 1]+; RMN 1H (400 MHz, DMSO-d6) 6 9,64 (s, 1H), 9,59 (s, 1H), 8,85 (s, 1H), 8,81 (s, 1H), 8,44 (d, J = 5,88 Hz, 1H), 7,42 (d, J = 5,88, 1H), 2,54 (m, 2H), 1,86 (t, J = 10,9 Hz, 2H), 1,15 (m, 2H), 0,82 (m, 8 H).
Ejemplo 152
Síntesis de 3,3-bis(2-aminoetil)-8-cloro-6-(pirimidin-4-ilamino)-2,3-dihidroimidazo[1,5-a]pi ridin-1,5-diona (Comp. N.° 152)

Síntesis de 2,2’-((6-bromo-8-cloro-1,5-dioxo-1,2,3,5-tetrahidroimidazo[1,5-a]piridin-3,3-diil)bis(etano-2,1-diil))bis(isoindolino-1,3-diona) (3)
La síntesis del intermedio 3 se realiza como se ha descrito anteriormente usando el protocolo general del procedimiento A.
Síntesis de 2,2'-((8-cloro-1,5-dioxo-6-(pirimidin-4-ilamino)-1,2,3,5-tetrahidroimidazo[1,5-a]piridin-3,3-diil)bis(etano-2,1-diil))bis(isoindolin-1,3-diona) (5)
La síntesis del intermedio 5 se realiza como se ha descrito anteriormente usando el protocolo general del Procedimiento H.
Síntesis de 3,3-bis(2-aminoetil)-8-cloro-6-(pirimidin-4-ilamino)-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona (Comp. N. ° 152)
La síntesis del compuesto 152 se realiza como se ha descrito anteriormente usando el protocolo del procedimiento C.
Ejemplo 153
Síntesis de 8-cloro-3,3-bis(2-hidroxietil)-6-(pirimidin-4-ilamino)-2,3-dihidroimidazo[1,5-a]pi ridin-1,5-diona (Comp. N.° 153)

Síntesis de 2,2’-(6-bromo-8-cloro-1,5-dioxo-1,2,3,5-tetrahidroimidazo[1,5-a]piridin-3,3-diil)diacetato de dimetilo (3) La síntesis del intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento A. Sólido de color blanquecino; Rendimiento: 0,65 g, 27 %; EM (IEN) m/z 407,18 [M-1]-.
Síntesis de 2,2’-(8-cloro-1,5-dioxo-6-(pirimidin-4-ilamino)-1,2,3,5-tetrahidroimidazol[1,5-a]piridin-3,3-diil)diacetato de dimetilo (5)
La síntesis del intermedio 5 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento H. Sólido de color blanquecino; Rendimiento: 0,34 g, 51 %; EM (IEN) m/z 422,49 [M 1]+.
Síntesis de 8-cloro-3,3-bis(2-hidroxietil)-6-(pirimidin-4-ilamino)-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona (Comp. N° 153)
A una suspensión de hidruro de litio y aluminio (71 mg, 1,9 mmol) en tetrahidrofurano (3 ml) a 0 °C se le añadió lentamente una solución de 2,2'-(8-cloro-1,5-dioxo-6-(pirimidin-4-ilamino)-1,2,3,5-tetrahidroimidazo[1,5-a]piridin-3,3-diil)diacetato de dimetilo (5, 280 mg, 0,66 mmol) en tetrahidrofurano (2 ml). Después del consumo del material de partida, la mezcla de reacción se inactivó con solución de hidróxido de sodio al 10 % (1 ml), se diluyó con 10 ml de acetato de etilo. La capa orgánica se separó y se secó sobre sulfato de sodio anhidro y se concentró a presión reducida para proporcionar el residuo. El residuo se purificó mediante HPLC preparativa para proporcionar 8 -cloro-3,3-bis(2-hidroxietil)-6-(pirimidin-4-ilamino)-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona (Comp. N.° 153) en forma de un sólido de color amarillo. Rendimiento: 0,047 g, 19 %; EM (IEN) m/z 366,09 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 9,58-9,57 (s ancho, 1H), 8,83 (s, 1H), 8,75 (s, 1H), 8,42 (d, J = 5,6 Hz, 1H), 7,44-7,42 D, J = 5,6 Hz, 1H), 4,44 (s ancho, 2H), 3,34-3,26 (m, 4H), 2,79-3,73 (m, 2H), 2,11-2,08 (m, 2H).
Ejemplo 154
Síntesis de 3,3-bis(aminometil)-8-cloro-6-(pirimidin-4-ilamino)-2,3-dihidroimidazo[1,5-a]pi ridin-1,5-diona (Comp. N.° 154)

Síntesis de 2,2’-((6-bromo-8-cloro-1,5-dioxo-1,2,3,3-tetrahidroimidazo[1,5-a]piridin-3,3-diil)bis(metilen)bis(isoindolino-1,3-diona) (3)
La síntesis del intermedio 3 se realiza como se ha descrito anteriormente usando el protocolo general del procedimiento A.
Síntesis de 2,2’-((8-cloro-1,5-dioxo-6-(pirimidin-4-ilamino)-1,2,3,5-tetrahidroimidazo[1,5-a]piridin-3,3-diil)bis(metilen))bis(isoindolino-1,3-diona) (5)
La síntesis del intermedio 5 se realiza como se ha descrito anteriormente usando el protocolo general del Procedimiento H.
Síntesis de 3,3-bis(aminometil)-8-cloro-6-(pirimidin-4-ilamino)-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona (Comp. N° 154)
La síntesis del compuesto 154 se realiza como se ha descrito anteriormente usando el protocolo general del procedimiento C.
Ejemplo 155
Síntesis de 8-cloro-1'-(2-hidroxietil)-6-(pirimidin-4-ilamino)-2H-espiro[imidazo[1,5-a]piridin-3,4,-piperidin]-1,5-diona (Comp. N.° 155)

La síntesis del intermedio 2 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento F. Sólido de color blanquecino; Rendimiento: 3,5 g, 95 %; EM (IEN) m/z 332,1 [M 1]+; RMN 1H (400 MHz, DMSO-cfe) ó 10,89 (s, 1H), 9,45 (s ancho, 1H), 8,67 (s ancho, 1H), 8,30 (s, 1H), 3,70-3,36 (m, 4H), 2,59 2,57 (m, 2H), 1,89-1,85 (m, 2H).
Síntesis de 6-bromo-1’-[2-[terc-butil(dimetil)silil]oxietil]-8-cloroespiro[2H-imidazo[1,5-a]pirídin-3,4’-piperídin]-1,5-diona (4)
Se cargó un matraz con clorhidrato de 6-bromo-8-cloro-espiro[2H-imidazo[1,5-a]piridin-3,4'-piperidin]-1,5-diona (2, 0,5 g, 1,35 mmol) y acetonitrilo (15 ml). La masa de reacción se enfrió a 0 °C y se añadieron carbonato de potasio (281 mg, 2,03 mmol) y (2-bromoetoxi)(terc-butil)dimetilsilano (3, 388 mg, 1,62 mmol) y la masa de reacción se calentó a 80 °C C durante 2 días. Después de la terminación, el disolvente se retiró para obtener el compuesto en bruto. El compuesto en bruto se purificó por columna ultrarrápida con 0,2 % de metanol en diclorometano. Las fracciones deseadas se concentraron para proporcionar 6-bromo-1'-[2-[terc-butil(dimetil)silil]oxietil]-8-cloro-espiro[2H-imidazo[1,5-a]piridin-3,4'-piperidin]-1,5-diona (4) en forma de un sólido de color marrón. Rendimiento: 0,3 g, 45 %; EM (IEN) m/z 373,01 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,62 (s, 1H), 8,25 (s, 1H), 3,69-3,63 (m, 4H), 3,10 2,83 (m, 4H), 1,52 (m, 2H) 1,04 (s, 9H), 0,058 (s, 6 H).
Síntesis de 1’-[2-[terc-butil(dimetil)sihi]oxietil]-8-cloro-6-(pirimidin-4-ilamino)espiro[2H-imidazo[1,5-a]piridin-3,4’-piperidin-1,5-diona (6)
La síntesis del intermedio 6 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento H. Sólido de color amarillo; Rendimiento: 0,15 g, 49 %; EM (IEN) m/z 505 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,37 (s, 1H), 9,63- 9,60 (m, 1H), 8,84- 8,75 (m, 2H), 8,43-8,36 (d, J = 5,8 Hz, 7,47 (d, J = 5,8 Hz, 1H), 3,72-3,70 (m, 2H), 3,26-3,22 (m, 4H), 2,98-2,90 (m, 4H), 1,53-1,46 (m, 2H), 1,33-1,29 (m, 2H), 0,95 (s, 9H), 0,058 (s, 6 H).
Síntesis de 8-cloro-1’-(2-hidroxietil)-6-(pirimidin-4-ilamino)espiro[2H-imidazo[1,5-a]piridin-3,4'-piperidin-1,5-diona (Comp. N.° 155)
La síntesis del compuesto 155 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento F. Sólido de color amarillo; Rendimiento: 0,035 g, 49 %; EM (IEN) m/z 391,34 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,50 (s, 1H), 9,59 (m, 1H), 8,84 (d, J = 16,48 Hz, 2H), 8,43-8,36 (d, J = 5,8Hz, 1H), 7,47 (d, J = 5,8 Hz, 1H), 4,43 (s ancho, 2H), 3,53 (s ancho, 2H), 3,21-3,16 (m, 2H), 2,98-2,90 (m, 2H), 2H), 1,48-1,46 (m, 2H).
Ejemplo 156
Síntesis de 8-cloro-1'-(2,2-difluoroetil)-6-(pirimidin-4-ilamino)espiro[2H-imidazo[1,5-a]piridin-3,4'-piperidin]-1,5-diona (Comp. N.° 156)


Síntesis de 6-bromo-8-cloro-1’-(2,2-difluoroetil)espiro[2H-imidazo[1,5-a]piridin-3,4'-piperidin]-1,5-diona (3)
Se cargó un matraz con clorhidrato de 6-bromo-8-cloro-espiro[2H-imidazo[1,5-a]piridin-3,4'-piperidin]-1,5-diona (1, 0,5 g, 1,35 mmol) y acetonitrilo (15 ml). La reacción se enfrió a 0 °C y se añadió gota a gota N,N-diisopropiletilamina (1,17 ml, 6,77 mmol) seguido de adición de 2-bromo-1,1-difluoroetano (2, 589 mg, 4,06 mmol). La reacción se agitó a 60-110 °C durante 48 h. Después de la terminación, el disolvente se retiró a presión reducida para obtener el producto en bruto. El producto en bruto se purificó mediante columna ultrarrápida usando metanol al 1-3 % en diclorometano. Las fracciones deseadas se concentraron para obtener 6-bromo-8-cloro-1'-(2,2-difluoroetil)espiro[2H-imidazo[1,5-a]piridin-3,4'-piperidin]-diona (3) en forma de un sólido de color marrón. Rendimiento: 0,27 g; 50 %; EM (IEN) m/z 396,11 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,66 (s, 1H), 8,25 (s, 1H), 6,29 (t, J = 55,68 Hz, 1H), 3,12-3,06 (m, 2H), 2,84-2,79 (m, 2H), 2,66-2,60 (m, 2H), 1,49-1,46 (m, 2H).
Síntesis de 8-cloro-1’-(2,2-difluoroetil)-6-(pirimidin-4-ilamino)espiro[2H-imidazo[1,5-a]piridin-3,4'-piperidin]-1,5-diona (Comp. N.° 156)
La síntesis del compuesto 156 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento H. Sólido de color amarillo; Rendimiento: 0,13 g, 50 %; EM (IEN) m/z 411,38 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,39 (s, 1H), 9,60 (s, 1H), 8,84 (s, 1H), 8,80 (s, 1H), 8,43-8,36 (d, J = 6,28 Hz, 1H), 7,47 (d, J = 5,8 Hz, 1H), 6,32 (t, J = 56,0 Hz, 1H), 3,26-2,22 (m, 2H), 2,98-2,90 (m, 2H), 2,88-2,79 (m, 2H), 2,70-2,64 (m, 2H), 1,51-1,48 (m, 2H).
Ejemplo 157
Síntesis de 8-cloro-6-(pirimidin-4-ilamino)-1'-(2,2,2-trifluoroetil)-2H-espiro[imidazo[1,5-a]piridin-3,4'-piperidin]-1,5-diona (Comp. N.° 157)

Síntesis de 6-bromo-8-cloro-1’-(2,2,2-trifluoroetil)-2H-espiro[imidazo[1,5-a]piridin-3,4'-piperidin-1,5-diona (3) La síntesis del producto intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento A. Sólido de color blanquecino; Rendimiento: 1,1 g, 50 %; EM (IEN) m/z 414,28 [M-1]-.
Síntesis de 8-cloro-6-(pirimidin-4-ilamino)-1’-(2,2,2-trifluoroetil)-2H-espiro[imidazo[1,5-a]piridin-3,4'-piperidin]-1,5-diona (Comp. N ° 157)
La síntesis del compuesto 157 se realizó como se ha descrito anteriormente usando el protocolo general del procedimiento B. Sólido de color amarillo; Rendimiento: 0,21 g, 41 %; EM (IEN) m/z 429,24 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,44 (s, 1H), 9,61 (s, 1H), 8,84-8,80 (m, 2H), 8,44-8,43 (m, 1H), 7,46-7,44 (m, 1H), 3,28 3,17 (m, 4H), 2,97-2,95 (m, 2H), 2,86-2,79 (m, 2H), 1,51-1,48 (d, 2H).
Ejemplo 158
Síntesis de 8,-cloro-8-metil-6'-(pirimidin-4-ilamino)-2'H-8-azaspiro[biciclo[3.2.1]ctan-3,3'-imidazoI[1,5-a]piridin]-1',5'-diona (Comp. N.° 158)

La síntesis del intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento A. Sólido de color marrón; Rendimiento: 0,15 g, 10 %; EM (IEN) m/z 372,21 [M 1]+.
Síntesis de 8’-cloro-8-metil-6'-(pirimidin-4-ilamino)-2’H-8-azaspiro[biciclo[3.2.1]octano-3,3’-imidazo[1,5-a]piridin]-1’,5'-diona (Comp. N ° 158)
La síntesis del compuesto 158 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento B. Sólido de color amarillo; Rendimiento: 0,029 g, 20 %; EM (IEN) m/z 387,35 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 9,64 (s, 1H), 9,53 (s, 1H), 8,84 (s, 1H), 8,80 (s, 1H), 8,43 (d, J = 5,88 Hz, 7,46 (d, J = 5,84 Hz, 1H), 3,23 (m, 4H), 2,59 (s, 3H), 1,94 (s, 4H), 1,40 (d, J = 12,8 Hz, 2H).
Ejemplo 159
Síntesis de 8'-cloro-6'-(pirimidin-4-ilamino)-2'H-8-azaspiro[biciclo [3,2.1]octano-3,3'-imidazo[1,5-a]piridin]-1',5'-diona (Comp. N.° 159)

Síntesis de 6'-bromo-8'-cloro-1’,5’-dioxo-1',5'-dihidro-2'H-8-azaspiro[biciclo[3.2.1]octano-3,3’-imidazo[1,5-a]piridin]-8-carboxilato de terc-butilo (3)
La síntesis del intermedio 3 se realiza como se ha descrito anteriormente usando el protocolo general del Procedimiento E.
Síntesis de 8'-cloro-1’,5’-dioxo-6'-(pirimidin-4-ilamino)-1’,5'-dihidro-2'H-8-azaspiro[biciclo[3.2.1]coctano-3,3’-imidazo[1,5-a]piridin]-8-carboxilato de terc-butilo (5)
La síntesis del intermedio 5 se realiza como se ha descrito anteriormente usando el protocolo general del Procedimiento H.
Síntesis de 8'-cloro-6’-(pirimidin-4-ilamino)-2'H-8-azaspiro[biciclo[3.2.1]octano-3,3’-imidazo[1,5-a]piridin]-1’,5'-diona (Comp. N ° 159)
La síntesis del compuesto 159 se realiza como se ha descrito anteriormente usando el protocolo general del Procedimiento F.
Ejemplo 160
Síntesis de 2-(8-cloro-1,5-dioxo-6-(pirimidin-4-ilamino)-1,5-dihidro-2H-espiro[imidazo[1,5-a]piridin-3,4'-piperidin]-1-il)acetonitrilo (Comp. N.° 160)

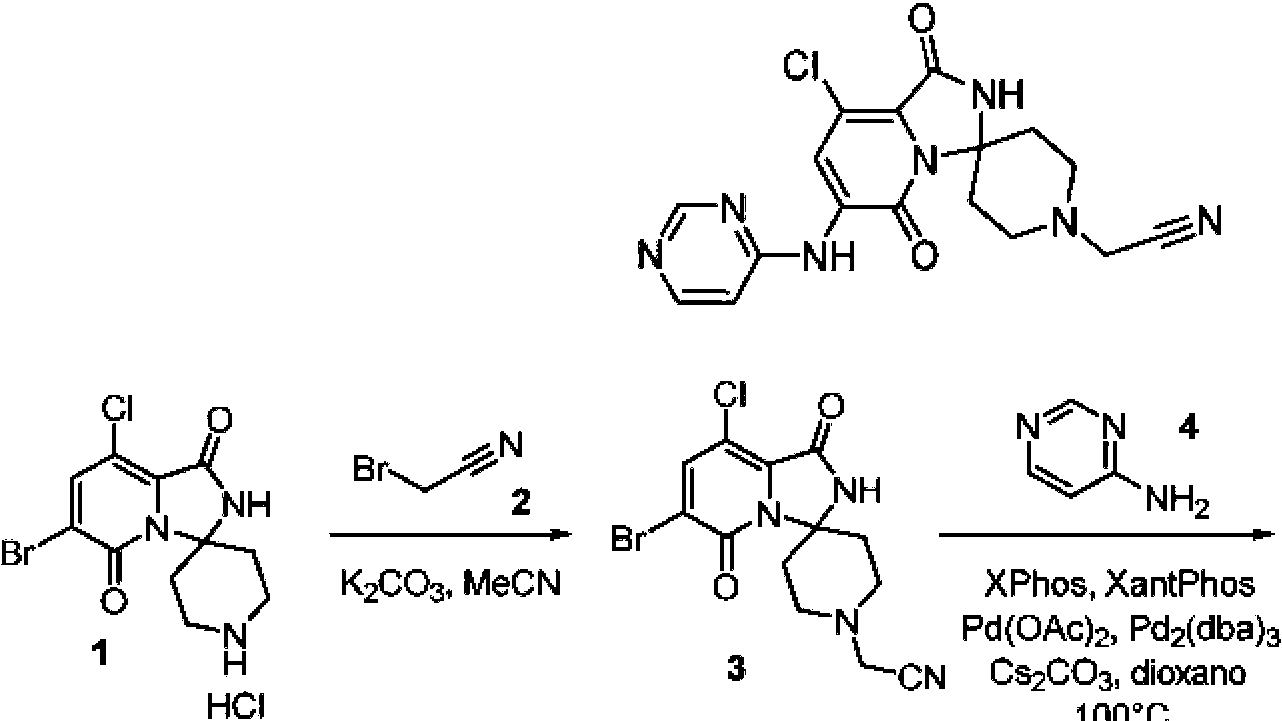
Síntesis de 2-(6-bromo-8-cloro-1,5-dioxo-1,5-dihidro-2H-espiro[imidazo[1,5-a]piridin-3,4’-piperidin]-1 '-il)acetonitrilo (3) Se cargó un matraz con clorhidrato de 6-bromo-8-cloro-espiro[2H-imidazo[1,5-a]piridin-3,4'-piperidin]-1,5-diona (1, 0,5 g, 1,35 mmol) y acetonitrilo (15 ml). La masa de reacción se enfrió a 0 °C y se añadió carbonato de potasio (281 mg, 2,03 mmol) seguido de la adición de 2-bromoacetonitrilo (2, 218 mg, 1,63 mmol). La masa de reacción se agitó a temperatura ambiente durante 10 h. Después de la terminación, el disolvente se retiró a presión reducida para obtener el producto en bruto. El producto en bruto se purificó mediante presión rápida Biotage usando metanol al 1-3 % en diclorometano. Las fracciones deseadas se concentraron para obtener 3-(6-bromo-8-cloro-1,5-dioxoespiro[2H-imidazo[1,5-a]piridin-3,4'-piperidin]-il)propanonitrilo (3) en forma de un sólido de color marrón. Rendimiento: 0,42 g, 83 %; EM (IEN) m/z 373,18 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,83 (s, 1H), 8,25 (s, 1H), 3,75 (s, 2H), 3,15-3,10 (m, 2H), 2,87-2,72 (m, 2H) 2,66-2,60 (m, 2H), 1,57-1,54 (m, 2H).
Síntesis de 2-[8-cloro-1,5-dioxo-6-(pirimidin-4-ilamino)espiro[2H-imidazo[1,5-a]piridin-3,4’-piperidin]-1 ’-il]acetonitrilo (Comp. N ° 160)
La síntesis del compuesto 160 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento H. Sólido de color amarillo; Rendimiento: 0,2 g, 49 %; EM (IEN) m/z 385,97 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,50 (s, 1H), 9,62 (s, 1H), 8,84 (s, 1H), 8,80 (s, 1H), 8,43-8,36 (d, J = 6,28 Hz, 1H), 7,47 (d, J = 5,8 Hz, 1H), 3,75 (s, 2H), 3,26-3,22 (m, 2H), 2,98-2,90 (m, 2H), 2,67-2,61 (m, 2H), 1,60-1,57 (m, 2H).
Ejemplo 161
Síntesis de 8-cloro-1'-(pirimidin-4-il)-6-(pirimidin-4-ilamino)-2H-espiro[imidazo[1,5-a]piridin-3,4'-piperidin]-1,5-diona (Comp. N.° 161)

Síntesis de 6-bromo-8-cloro-1’-(pirimidin-4-il)-2H-espiro[imidazo[1,5-a]piridin-3,4'-piperidin]-1,5-diona (3)
La síntesis del intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del procedimiento A. Sólido de color amarillo. Rendimiento: 0,1 g, 61 %; EM (IEN) m/z 410,02 [M 1]+.
Síntesis de 8-cloro-1 ’-(pirimidin-4-il)-6-(pirimidin-4-ilamino)-2H-espiro[imidazo[1,5-a]piridin-3',4’-piperidin]-1,5-diona (Comp. N ° 161)
La síntesis del compuesto 161 se realizó como se ha descrito anteriormente usando el protocolo general del procedimiento B. Sólido de color amarillo. Rendimiento: 0,06 g, 23 %; EM (IEN) m/z 425,34 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,65 (s, 1H), 9,59 (s, 1H), 8,83 (s, 1H), 8,79 (s, 1H), 8,56 (s, 1H), 8,41 (d, J = 5,84 Hz, 1H), 8,25 (d, J = 6,08 Hz, 1H), 7,39 (d, J =, 3,28 (m, 2H), 3,05 (m, 2H), 1,68 (d, J = 12,6 Hz, 1H).
Ejemplo 162
Síntesis de clorhidrato de 8-cloro-4'-(metilamino)-6-(pirimidin-4-ilamino)espiro[2H-imidazo[1,5-a]piridin-3,1'-ciclohexano]-1,5-diona (Comp. N.° 162)

Síntesis de 6'-bromo-4'-cloro-4-(metilamino)espiro[ciclohexano-1,1 ’-isoindol]-3', 7’(2'H,7a'H)-diona (3)
La síntesis del intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento A. Rendimiento: 0,72 g, producto en bruto; EM (IEN) m/z 359,65 [M-1]-.
Síntesis de N-(6-bromo-8-cloro-1,5-dioxo-espiro[2H-imidazo[1,5-a]piridin-3,4’-ciclohexan]-1'-il)-N-metil-carbamato de terc-butilo (4)
A una mezcla de 6-bromo-8-cloro-4'-(metilamino)espiro[2H-imidazo[1,5-a]piridin-3,1'-ciclohexano]-1,5-diona, (3, 0,72 g, 1,99 mmol) en 1,4-dioxano (10 ml) y agua (10 ml), se le añadió hidróxido de potasio (0,56 g, 9,94 mmol) seguido de la adición de carbonato de terc-butilo de terc-butoxicarbonilo (651 mg, 2,98 mmol) y se agitó la mezcla a temperatura ambiente durante 24 h. Una vez completada la reacción, la mezcla resultante se filtró. El precipitado se disolvió en metanol al 10 % en diclorometano. La capa orgánica se lavó con agua y salmuera, se secó sobre sulfato de sodio anhidro, se filtró y se concentró a presión reducida para obtener N-(6-bromo-8-cloro-1,5-dioxo-espiro[2H-imidazo[1,5-a]piridin-3,4'-ciclohexan]-il)-N-metil-carbamato de terc-butilo (4) en forma de un sólido de color blanquecino. Rendimiento: 0,9 g, 98 %; RMN 1H (400 MHz, DMSO-cfe) 6 10,76 (s, 1H), 8,26 (s, 1H), 4,00-3,97 (m, 1H), 2,98 (s ancho, 2H), 2,73 (s, 3H), 1,98-1,81 (m, 2H), 1,73-1,60 (m, 4H), 1,41 (s, 9H).
Síntesis de N-[8-cloro-1,5-dioxo-6-(pirimidin-4-ilamino)espiro[2H-imidazo[1,5-a]piridin-3,4’-ciclohexan-ill]-N-metilcarbamato de terc-butilo (6)
La síntesis del intermedio 6 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento H. Sólido de color blanquecino; Rendimiento: 0,11 g, 21 %; EM (IEN) m/z 475,31 [M 1]+; RMN 1H (400 MHz, DMSO-d6) 6 10,51 (s, 1H), 9,67 (s, 1H), 8,84 (s, 1H), 8,80 (s, 1H), 8,44 (d, J = 5,6, 1H), 7,46 (s, 1H), 3,94 3,87 (m, 1H), 3,12-3,06 (m, 2H) 2,75 (s, 3H), 1,89-1,85 (m, 3H), 1,66-1,64 (m, 4H), 1,41 (m, 1H) (s, 3H).
Síntesis de clorhidrato de 8-cloro-4’-(metilamino)-6-(pirimidin-4-ilamino)espiro[2H-imidazo[1,5-a]piridin-3,1 '-ciclohexano-1,5-diona (Comp. N.° 162)
La síntesis del compuesto 162 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento F. Sólido de color amarillo; Rendimiento: 0,060 g, 69 %; EM (IEN) m/z 375,26 [M 1]+; RMN 1H (400 MHz, DMSO-d6) 6 9,07 (s, 1H), 8,60 (s, 1H), 8,46 (d, J = 6 , 8 Hz, 1H), 7,58 (d, J = 6,0 Hz, 1H), 3,16-3,13 (m, 1H), 3,04-2,98 (m, 2H), 2,57 (s, 3H), 2,13-2,10 (m, 2H), 1,72-1,66 (m, 4H).
Ejemplo 163
Síntesis de 1 '-acetil-8-cloro-6-(pirimidin-4-ilamino)-2H-espiro[imidazo[1,5-a]piridin-3,4'-piperidin]-1,5-diona (Comp. N.° 163)

Síntesis de 1’-acetil-6-bromo-8-cloro-2H-espiro[imidazo[1,5-a]piridin-3,4'-piperidin]-1,5-diona (3)
La síntesis del intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento A. Sólido de color blanquecino; Rendimiento: 0,60 g, 81 %; EM (IEN) m/z 375,41 [M 1]+.
Síntesis de 1’-acetil-8-cloro-6-(pirimidin-4-ilamino)-2H-espiro[imidazo[1,5-a]piridin-3,4'-piperidin]-1,5-diona (Comp. N.° 163)
La síntesis del compuesto 163 se realizó como se ha descrito anteriormente usando el protocolo general del
Procedimiento H. Sólido de color amarillo; Rendimiento: 0,35 g, 43 %; EM (IEN) m/z 389 [M 1]+; RMN 1H (400 MHz, DMSO-cfe) ó 10,55 (s, 1H), 9,66 (s, 1H), 8,84 (s, 1H), 8,79 (s, 1H), 8,43 (d, J = 5,84 Hz, 1H), 7,43 (d, J = 5,92 Hz, 1H), 4,50 (d, J = 12,64 Hz, 1H), 3,96 (d, J = 2,95 Hz, 1H), 3,10-3,07 (m, 1H), 3,06-3,03 M, 1H), 2,06 (s, 3H), 1,68 1,59 (m, 2H).
Ejemplo 164
Síntesis de 8'-cloro-1,,5'-dioxo-6,-(pirimidin-4-ilamino)-1',5'-dihidro-2'H-espiro[ciclohexano-1,3-imidazo[1,5-a]piridin]-4-carbonitrilo (Comp. N.° 1 6 4 )

Síntesis de 6'-bromo-8'-cloro-1' 5'-dioxo-15'-dihidro-2'H-espiro[ciclohexano-1,3'-imidazo[1,5-a]piridin]-4-carbonitrilo (3)
La síntesis del intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento A. Sólido de color blanquecino; Rendimiento: 0,75 g, 53 %; EM (IEN) m/z 354,08 [M-1]-.
Síntesis de 8'-cloro-1’,5’-dioxo-6'-(pirimidin-4-ilamino)-1’,5'-dihidro-2'H-espiro[ciclohexano-1,3'-imidazo[1,5-a]piridin]-4-carbonitrilo (Comp. N ° 164)
La síntesis del compuesto 164 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento H. Sólido de color amarillo; Rendimiento: 0,21 g, 29 %; EM (IEN) m/z 371,18 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,36 (s, 1H), 9,58 (s, 1H), 8,84-8,78 (m, 2H), 8,44-8,39 (m, 1H), 7,44-7,43 (m, 1H), 3,00 2,94 (m, 2H), 2,82-2,76 (m, 1H), 2,15-2,12 (m, 2H), 1,99-1,90 (m, 3H), 1,67-1,64 (m, 2H).
Ejemplo 165
Síntesis de 8-cloro-3,3-dimetil-6-(pirido[3,4-d]pirimidin-4-ilamino)-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona (Comp. N.° 165)



Síntesis de 8-cloro-3,3-dimetil-6-(pirido[3,4-d]pirimidin-4-ilamino)-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona (Comp. N ° 165)
La síntesis del compuesto 165 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento B. Sólido de color amarillo; Rendimiento: 25 mg, 8 %; EM (IEN) m/z 357,34 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,05 (s ancho, 1H), 10,37 (s, 1H), 9,17 (s, 1H), 8,61 (m, 2H), 8,18 (d, J = 5,24 Hz, 1H), 1,82 (s, 6 H).
Ejemplo 166
Síntesis de 8-cloro-3,3-dimetil-6-(pirimido[5,4-c]piridazin-8-ilamino)-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona (Comp. N.° 166)


Síntesis de 8-cloro-3,3-dimetil-6-(pirimido[5,4-c]piridazin-8-ilamino)-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona (Comp. N ° 166)
La síntesis del compuesto 166 se realiza como se ha descrito anteriormente usando el protocolo general del Procedimiento H.
Ejemplo 167
Síntesis de 8-cloro-3,3-dimetil-6-(pirimido[5,4-d]pirimidin-4-ilamino)-2,3-dihidroimidazo[1,5-a]pi ridin-1,5-diona (Comp. N.° 167)


Síntesis de 8-cloro-3,3-dimetil-6-(pirimido[5,4-d]pirimidin-4-ilamino)-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona (Comp. N ° 167)
La síntesis del compuesto 167 se realiza como se ha descrito anteriormente usando el protocolo general del Procedimiento H.
Ejemplo 168
Síntesis de (Z)-8-cloro-6-((6-(2-ciclopropil-3,3,3-trifluoroprop-1-en-1-il)pirimidin-4-il)amino)-3-(3-fluorofenil)-3-metil-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona (Comp. N.° 168)



Síntesis de (Z)-8-cloro-6-((6-(2-ciclopropil-3,3,3-trifluoroprop-1-en-1-il)pirimidin-4-il)amino)-3-fluorofenil)-3-metil-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona (Comp. N ° 168)
La síntesis del compuesto 168 se realiza como se ha descrito anteriormente usando el protocolo general del Procedimiento H.
Ejemplo 169
Síntesis de clorhidrato de 6'-((6-amino-5-fluoropirimidin-4-il)amino)-8'-cloro-2'H-espirociclohexano-1,3'-imidazoil[1,5-a]piridin]-1',5'-diona (Comp. N.° 169)

La síntesis del intermedio 2 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento J. Sólido de color blanquecino; Rendimiento: 2,3 g, 97 %; EM (IEN) m/z 348 [M 1]+.
Síntesis de N-terc-butox¡carbon¡l-N-[6-(c¡clopropanocarbon¡lam¡no)-5-fluoro-p¡rim¡d¡n-4-¡l]carbamato de terc-butilo (4) La síntesis del intermedio 4 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento H. Sólido de color blanquecino; Rendimiento: 0,9 g, 38 %; EM (IEN) m/z 397,29 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 8 , 6 (s, 1H), 7,83 (s, 1H), 2,41 (s ancho, 1H), 1,46 (s, 18H), 1,30 (m, 4H).
Síntesis de N-(6-amino-5-fluoro-pirimidin-4-il)ciclopropanocarboxamida (5)
A una solución agitada de N-terc-butoxicarbonil-N-[6-(ciclopropanocarbonilamino)-5-fluoro-pirimidin-4-il]carbamato de terc-butilo (4, 0,89 g, 2,25 mmol) en diclorometano (20 ml), se le añadió ácido trifluoroacético (20 ml, 2,25 mmol) a 0 °C y se agitó la masa de reacción a temperatura ambiente durante 16 h. Después de la terminación la reacción, el ácido trifluoroacético se destiló y el compuesto en bruto se basificó con amoníaco líquido. El sólido precipitado se filtró y se secó para proporcionar N-(6-amino-5-fluoro-pirimidin-4-il)ciclopropanocarboxamida (5) en forma de un sólido de color blanquecino. Rendimiento: 0,4 g, 90 %; EM (IEN) m/z 197,06 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,37 (s, 1H), 7,99 (s, 1H), 7,16 (s, 2H), 7,04 (s, 1H), 2,06 (m, 1H), 0,82-0,78 m, 4H).
Síntes¡s de N-[6-[(8-cloro-1,5-dioxo-espiro[2H-imidazo[1,5-a]p¡r¡d¡n-3,1 ’-ciclohexan-6-il)amino]-5-fluoro-pirimidin-4-¡l]c¡clopropanocarboxam¡da (7)
La síntesis del intermedio 7 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento H. Sólido de color amarillo; Rendimiento: 0,22 g, 40 %; EM (IEN) m/z 447 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,87 (s, 1H), 10,45 (s, 1H), 8,59-8,53 (m, 1H), 8,50-8,42 (m, 2H), 2,90 (m, 2H), 1,98-1,98 (m, 1H), 1,77-1,53 (m, 8 H), 0,82-0,84 (m, 4H).
Síntes¡s de 6’-((6-amino-5-fluoropirimidin-4-il)amino)-8'-cloro-2’H-espiro[ciclohexano-1,3-imidazo[1,5-a]piridin]-1’,5’- diona (Comp. N ° 169)
La síntesis del compuesto 169 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento I. Sólido de color blanquecino; Rendimiento: 110 mg, 65 %; EM (IEN) m/z 378,9 [M 1]+; RMN 1H: (400 MHz, DMSO-d6) ó 10,34 (s ancho, 1H), 8,51 (s, 1H), 8,16 (s, 1H), 8,08 (s, 1H), 7,10 (s, 1H), 2,94 (t, J = 2,32, 2H), 1,79-1,76 (m, 2H), 1,67-1,51 (m, 5H), 1,27-1,23 (m, 1H).
Ejemplo 170
Síntesis de 6-[(6-aminopirimidin-4-il)amino]-1 ',8-dimetil-espiro[2H-imidazo[1,5-a]piridin-3,3'-pi peridin]-1,5-diona (Comp. N.° 170)

La síntesis del intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento A. Sólido de color blanquecino; Rendimiento: 0,7 g, 25 %; EM (IEN) m/z 326,19 [M 1]+.
Síntesis de 6-((1’,8-dimetil-1,5-dioxo-1,5-dihidro-2H-espiro[imidazo[1,5-a]piridin-3,3’-piperidin]-6-il)amino)pirimidin-4-il)carbamato de terc-butilo (5)
La síntesis del intermedio 5 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento H. Sólido de color amarillo; Rendimiento: 0,25 g, 64 %; RMN 1H (400 MHz, DMSO-d6) ó 10,92 (s, 1H), 9,80 (s, 1H), 9,10 (s, 1H), 8,52-8,46 (m, 2H), 7,84 (m, 1H), 3,00-2,98 (m, 2H), 2,81-2,79 (m, 1H), 2,43 (s, 3H), 2,21 (s, 3H), 2,03-1,90 (m, 3H), 1,70 (m, 1H), 1,48-1,4 ( s, 9H).
Síntesis de 6-[(6-aminopirimidin-4-il)amino]-1 8-dimetil-espiro[2H-imidazo[1,5-a]piridin-3,3’-piperidin]-1,5-diona (Comp. N ° 170)
La síntesis del compuesto 170 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento F. Sólido de color amarillo; Rendimiento: 0,1 g, 34 %; EM (IEN) m/z 356,47 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 9,82 (s, 1H), 8,59 (s, 1H), 8,38 (s, 1H), 8,16 (s, 1H), 6,52 (s, 2H), 6,14 (s, 1H), 3,32-3,30 (m, 1H), 2,98 (s, 1H), 2,80 (s, 1H), 2,50 (s, 1H), 2,41 (s, 3H), 2,20 (s, 3H), 1,91 (s, 2H), 1,76-1,69 (m, 1H), 1,48-1,45 (m, 1 H).
Ejemplo 171
Síntesis de 8-cloro-6-(pirimidin-4-ilamino)-2',3'-dihidro-1 'H,2H-espiro[imidazo[1,5-a]piridin-3,4'-isoquinolin]-1,5-diona (Comp. N.° 171)
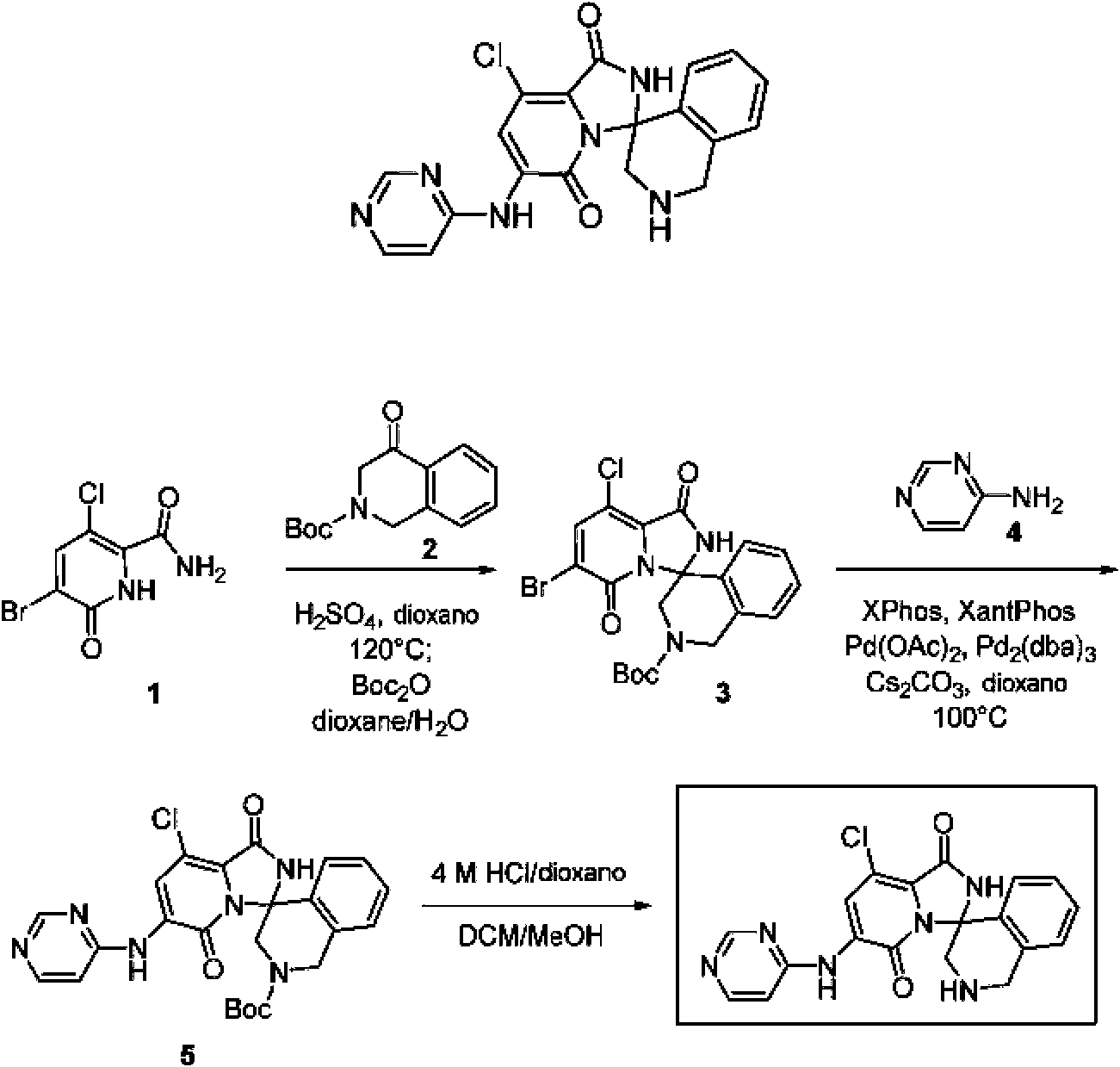
Síntesis de 6-bromo-8-cloro-1,5-dioxo-1,5-dihidro-1’H,2H-espiro[imidazo[1,5-a]piridin-3,4'-isoquinolino]-2’(3'H)-carboxilato de terc-butilo (3)
La síntesis del intermedio 3 se realiza como se ha descrito anteriormente usando el protocolo general del Procedimiento E.
Síntesis de 8-cloro-1,5-dioxo-6-(pirimidin-4-ilamino)-1,5-dihidro-1’H,2H-espiro[imidazo[1,5-a]piridin-3,4’-isoquinolin]-2'(3’H)-carboxilato de terc-butilo (5)
La síntesis del intermedio 5 se realiza como se ha descrito anteriormente usando el protocolo general del Procedimiento H.
Síntesis de 8-cloro-6-(pirimidin-4-ilamino)-2’,3'-dihidro-1’H,2H-espiro[imidazo[1,5-a]piridin-3,4'-isoquinolin]-1,5-diona (Comp. N ° 171)
La síntesis del compuesto 171 se realiza como se ha descrito anteriormente usando el protocolo general del Procedimiento F.
Ejemplo 172
Síntesis de 6-((6-aminopirimidin-4-il)amino)-8-cloro-2',3'-dihidro-1 'H,2H-espiro[imidazo[1,5-a]piridin-3,4'-isoquinolino]-1,5-diona (Comp. N.° 172)

Síntesis de 6-((6-((terc-butoxicarbonil)amino)pirimidin-4-il)amino)-8-cloro-1,5-dioxo-1,5-dihidro-1’H,2H-espiro[imidazo[1,5-a]piridin-3,4’-isoquinolin]-2'(3’H)-carboxilato de terc-butilo (3)
La síntesis del intermedio 3 se realiza como se ha descrito anteriormente usando el protocolo general del Procedimiento H.
Síntesis de 6-((6-aminopirimidin-4-il)amino)-8-cloro-2’,3'-dihidro-1’H,2H-espiro[imidazo[1,5-a]piridin-3,4’-isoquinolino]-1,5-diona (Comp. N.° 172)
La síntesis del compuesto 172 se realiza como se ha descrito anteriormente usando el protocolo g del Procedimiento F.
Ejemplo 173
Síntesis de 8-cloro-6'-fluoro-6-(pirimidin-4-ilamino)-2',3'-dihidro-1 'H,2H-espiro[imidazo[1,5-a]piridin-3,4'-isoquinolino]-1,5-diona (Comp. N.° 173)


Síntesis de 6-bromo-8-cloro-6’-fluoro-1,5-dioxo-1,5-dihidro-1 'H,2H-espiro[imidazo[1,5-a]piridin-3,4’-isoquinolino]-2’(3'H)-carboxilato de terc-butilo (3)
La síntesis del intermedio 3 se realiza como se ha descrito anteriormente usando el protocolo general del Procedimiento E.
Síntesis de 8-cloro-6'-fluoro-1,5-dioxo-6-(pirimidin-4-ilamino)-1,5-dihidro-1 ’H, 2H-espiro[imidazo[1,5-a]piridin-3,4’-isoquinolino]-2'(3’H)-carboxilato de terc-butilo (5)
La síntesis del intermedio 5 se realiza como se ha descrito anteriormente usando el protocolo general del Procedimiento H.
Síntesis de 8-cloro-6'-fluoro-6-(pirimidin-4-ilamino)-2’,3’-dihidro-1’H,2H-espiro[imidazo[1,5-a]piridin-3,4’-isoquinolino]-1,5-diona (Comp. N ° 173)
La síntesis del compuesto 173 se realiza como se ha descrito anteriormente usando el protocolo general del Procedimiento F.
Ejemplo 174
Síntesis de 6-((6-aminopirimidin-4-il)amino)-8-cloro-6'-fluoro-2',3'-dihidro-1 'H,2H-espiro[imidazo[1,5-a]piridin-3,4'-isoquinolino]-1,5-diona (Comp. N.° 174)

Síntesis de 6-((6-((terc-butoxicarbonil)amino)pirimidin-4-il)amino)-8-cloro-6’-fluoro-1,5-dioxo-1,5-dihidro-1’H,2H-espiro[imidazo[1,5-a]piridin-3,4'-isoquinolino]-2’(3'H)-carboxilato de terc-butilo (3)
La síntesis del intermedio 3 se realiza como se ha descrito anteriormente usando el protocolo general del Procedimiento H.
Síntesis de 6-((6-aminopirimidin-4-il)amino)-8-cloro-6’-fluoro-2',3’-dihidro-1,2H-espiro[imidazo[1,5-a]piridin-3,4’-isoquinolino]-1,5-diona (Comp. N ° 174):
La síntesis del compuesto 174 se realiza como se ha descrito anteriormente usando el protocolo general del Procedimiento F.
Ejemplo 175
Síntesis de 8-cloro-6-(pirimidin-4-ilamino)-2H-espiro[imidazo[1,5-a]piridin-3,3'-indolino]-1,5-diona (Comp. N.° 175)

Síntesis de 6-bromo-8-cloro-1,5-dioxo-1,5-dihidro-2H-espiro[imidazo[1,5-a]piridin-3,3’-indolino-1 ’-carboxilato de tercbutilo (3)
La síntesis del intermedio 3 se realiza como se ha descrito anteriormente usando el protocolo general del Procedimiento E.
Síntesis de 8-cloro-1,5-dioxo-6-(pirimidin-4-ilamino)-1,5-dihidro-2H-espiro[imidazo[1,5-a]piridin-3,3’-indolino]-1 ’-carboxilato de terc-butilo (5)
La síntesis del intermedio 5 se realiza como se ha descrito anteriormente usando el protocolo general del Procedimiento H.
Síntesis de 8-cloro-6-(pirimidin-4-ilamino)-2H-espiro[imidazo[1,5-a]piridin-3,3’-indolino]-1,5-diona (Comp. N ° 175) La síntesis del compuesto 175 se realiza como se ha descrito anteriormente usando el protocolo g del Procedimiento F.
Ejemplo 176
Síntesis de 8-cloro-5'-fluoro-6-(pirimidin-4-ilamino)-2H-espiro[imidazo[1,5-a]piridin-3,3'-indolino]-1,5-diona (Comp. N.° 176)
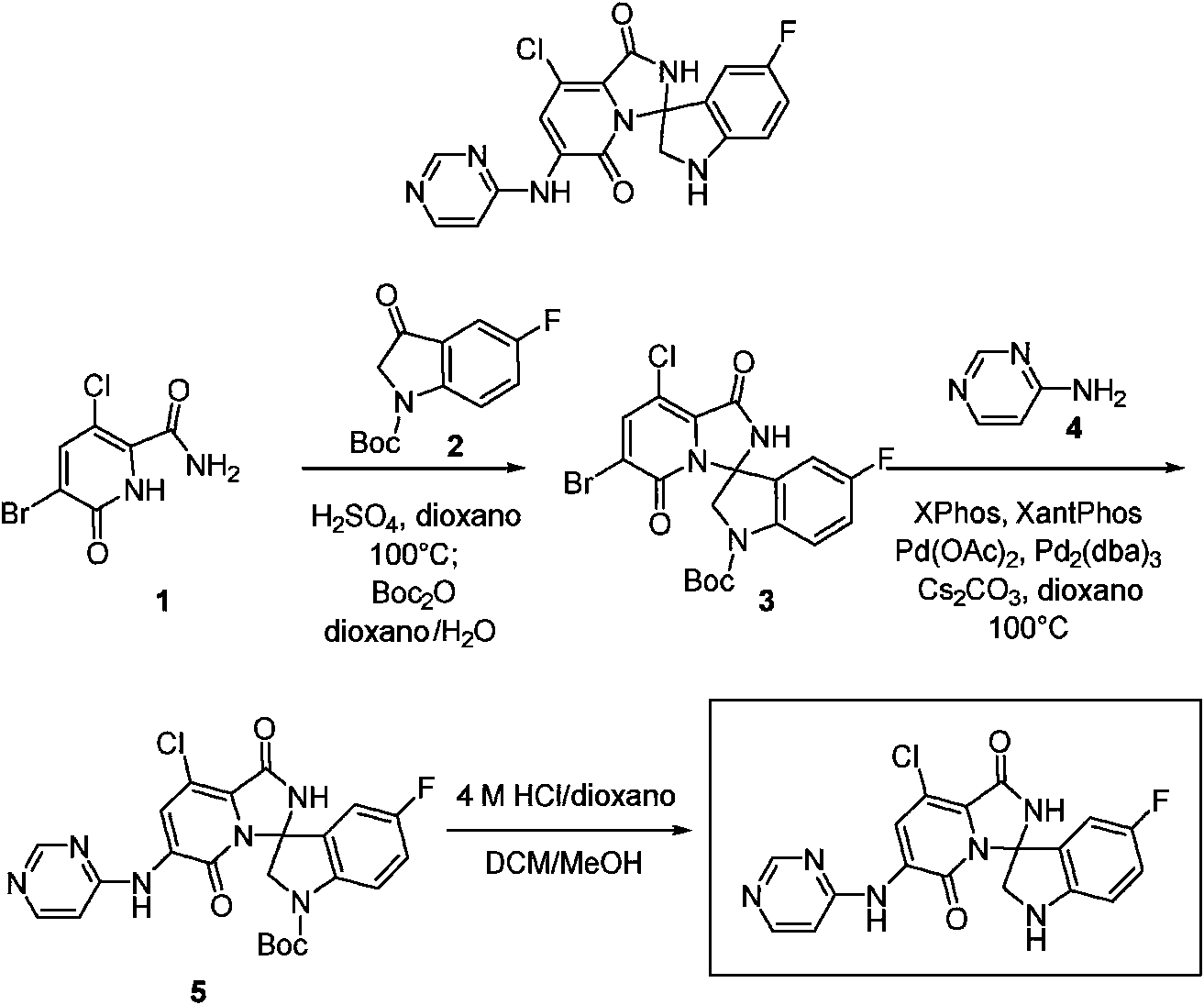
Síntesis de 6-bromo-8-cloro-5’-fluoro-1,5-dioxo-1,5-dihidro-2H-espiro[imidazo[1,5-a]piridin-3,3’-indolino]-1 ’-carboxilato de terc-butilo (3)
La síntesis del producto intermedio 3 se realiza como se ha descrito anteriormente usando el protocolo general del Procedimiento E.
Síntesis de 8-cloro-5'-fluoro-1,5-dioxo-6-(pirimidin-4-ilamino)-1,5-dihidro-2H-espiro[imidazo[1,5-a]piridin-3,3’-indolino]-1'-carboxilato de terc-butilo (5)
La síntesis del intermedio 5 se realiza como se ha descrito anteriormente usando el protocolo general del Procedimiento H.
Síntesis de 8-cloro-5'-fluoro-6-(pirimidin-4-ilamino)-2H-espiro[imidazo[1,5-a]piridin-3,3’-indolino]-1,5-diona (Comp. N° 176)
La síntesis del compuesto 176 se realiza como se ha descrito anteriormente usando el protocolo general del Procedimiento F.
Ejemplo 177
Síntesis de 8-cloro-4',4,-difluoro-6-(pirimidin-4-ilamino)-2H-espiro[imidazo[1,5-a]piridin-3,3'-piperidin]-1,5-diona (Comp. N.° 177)


Síntesis de 6-bromo-8-cloro-4’,4'-difluoro-1,5-dioxo-1,5-dihidro-2H-espiro[imidazo[1,5-a]piridin-3,3’-piperidin]-1 carboxilato de terc-butilo (3)
La síntesis del intermedio 3 se realiza como se ha descrito anteriormente usando el protocolo general del Procedimiento E.
Síntesis de 8-cloro-4’,4’-difluoro-1,5-dioxo-6-(pirimidin-4-ilamino)-1,5-dihidro-2H-espiro[imidazo[1,5-a]piridin-3,3’-piperidin-1 '-carboxilato de terc-butilo(5)
La síntesis del intermedio 5 se realiza como se ha descrito anteriormente usando el protocolo general del Procedimiento H.
Síntesis de 8-cloro-4’,4'-difluoro-6-(pirimidin-4-ilamino)-2H-espiro[imidazo[1,5-a]piridin-3,3’-piperidin]-1,5-diona (Comp. N ° 177)
La síntesis del compuesto 177 se realiza como se ha descrito anteriormente usando el protocolo general del Procedimiento F.
Ejemplo 178
Síntesis de 6-((6-aminopirimidin-4-il)amino)-8-cloro-4,4-difluoro-2H-espiro[imidazo[1,5-a]piridin-3,3'-piperidin]-1,5-diona (Comp. N.° 178)

Síntesis de 6-((6-((terc-butoxicarbonil)amino)pirimidin-4-il)amino)-8-cloro-4’,4’-difluoro-1,5-dioxo-1,5-dihidro-2H-espiro[imidazo[1,5-a]piridin-3,3’-piperidin-1'-carboxilato de terc-butilo (3)
La síntesis del intermedio 3 se realiza como se ha descrito anteriormente usando el protocolo general del Procedimiento H.
Síntesis de 6-((6-aminopirimidin-4-il)amino)-8-cloro-4’,4’-dtfluoro-2H-espiro[imidazo[1,5-a]piridin-3,3’-piperidino]-1,5-diona (Comp. N ° 178)
La síntesis del compuesto 178 se realiza como se ha descrito anteriormente usando el protocolo general del Procedimiento F.
Ejemplo 179
Síntesis de 8'-cloro-2,2-dimetil-6'-(pirimidin-4-ilamino)-4,5-dihidro-2H,2'H-espiro[furan-3,3-midazo[1,5-a]piridin]-1,5'-diona (Comp. N.° 179)

La síntesis del intermedio 3 se realiza como se ha descrito anteriormente usando el protocolo general del Procedimiento A.
S ín te s is de 8 '-c lo ro -2 ,2 -d im e til-6 ’- (p ir im id in -4 - ila m in o )-4 ,5 -d ih id ro -2 H ,2 ’H -e s p iro [fu ra n -3 ,3 ’- im id a z o [1 ,5 -a ]p ir id in ]-1 ,5 ’-d io n a (C om p. N ° 179)
La síntesis del compuesto 179 se realiza como se ha descrito anteriormente usando el protocolo general del Procedimiento H.
Ejemplo 180
Síntesis de 6'-((6-aminopirimidin-4-il)amino)-8'-cloro-2,2-dimetil-4,5-dihidro-2H,2'H-espiro[furan-3,3'-imidazo[1,5-a]piridin]-1',5'-diona (Comp. N.° 1 8 0 )

Síntesis de (6-((8'-cloro-2,2-dimetil-1’,5'-dioxo-1’,4,5,5'-tetrahidro-2H,2’H-espiro[furan-3,3’-imidazo[1,5-a]piridin]-6'-il)amino)pirimidin-4-il)carbamato de terc-butilo (3)
La síntesis del producto intermedio 3 se realiza como se ha descrito anteriormente usando el protocolo general del Procedimiento H.
Síntesis de 6’-((6-aminopirimidin-4-il)amino)-8'-cloro-2,2-dimetil-4,5-dihidro-2H,2’H-espiro[furan-3,3’-imidazo[1,5-a]piridin]-1’,5'-diona (Comp. N ° 180)
La síntesis del compuesto 180 se realiza como se ha descrito anteriormente usando el protocolo general del Procedimiento F.
Ejemplo 181
Síntesis de 3,3-dimetil-1,5-dioxo-6-(pirimidin-4-ilamino)-2H-imidazo[1,5-a]piridin-8-carbonitrilo (Comp. N.° 181)



Síntesis de 3,3-dimetil-1,5-dioxo-6-(pirimidin-4-ilamino)-2H-imidazo[1,5-a]piridin-8-carbonitrilo (Comp. N ° 181)
A un vial se le añadió 8-cloro-3,3-dimetil-6-(pirimidin-4-ilamino)-2H-imidazo[1,5-a]piridin-1,5-diona (1, 1000 mg, 3,27 mmol), cianuro de cobre (I) (293 mg, 3,27 mmol) y cianuro de sodio (160 mg, 3,27 mmol) en 1,4-dioxano (10 ml) a temperatura ambiente en argón. La reacción se purgó con argón durante 5-10 min, seguido de la adición de triciclohexilfosfina (92 mg, 0,33 mmol) y tris(dibencilidenacetona)dipaladio (0) (299 mg, 0,33 mmol) en argón. El vial se selló entonces y se calentó a 150 °C durante 48 h. Después de la terminación, la reacción se inactivó con solución saturada de permanganato de potasio y se extrajo el compuesto en bruto con metanol al 10 % en diclorometano. La capa orgánica se concentró a sequedad y el producto en bruto se purificó por cromatografía en columna ultrarrápida (gel de sílice de malla 100-200) usando metanol al 2 % en diclorometano. Las fracciones deseadas se concentraron a sequedad al vacío para obtener 3,3-dimetil-1,5-dioxo-6-(pirimidin-4-ilamino)-2H-imidazo[1,5-a]piridin-8-carbonitrilo (Comp. N.° 181) en forma de un sólido de color blanquecino. Rendimiento: 0,2 g, 20 %; EM (IEN) m/z 297,36 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,20 (s, 1H), 9,69 (s, 1H), 8,90 (s, 1H), 8,85 (s, 1H), 8,44 (d, J = 5,84 Hz, 1H), 7,44 (d, J = 5,36 Hz, 1H), 1,82 (s, 6H).
Ejemplo 182
Síntesis de 6’-((2-aminopiridin-4-il)amino)-8'-metil-2’H-espiro[ciclohexano-1,3,3-imidazo[1,5-a]pirazin]-1 ’,5’-diona (Comp. N.° 182)

La síntesis del intermedio 2 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento J. Sólido de color marrón claro; Rendimiento: 5,3 g, 70 %; EM (IEN) m/z 329,21 [M 1]+.
Síntesis de N,N-d¡-terc-butox¡carbon¡l(2-(c¡clopropanocarboxam¡do))p¡ríd¡n-4-am¡na (4)
La síntesis del intermedio 4 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento B. Sólido de color marrón claro; Rendimiento: 2,5 g, 51 %; EM (IEN) m/z 378,61 [M 1]+.
Síntesis de clorh¡drato de N-(4-aminopi^idin-2-il)ciclopropanocarboxamida (5)
La síntesis del intermedio 5 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento F. Sólido de color blanquecino; Rendimiento: 1,6 g, producto en bruto; EM (IEN) m/z 178,45 [M 1]+. Síntes¡s de 5-((2-(ciclopropanocarboxamido)piridin-4-il)amino)-3-metil-6-oxo-1,6-dihidropirazin-2-carboxilato de met¡lo (7)
La síntesis del intermedio 7 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento H. Sólido de color amarillo; Rendimiento: 1,5 g, 48 %; EM (IEN) m/z 344,05 [M 1 ]+.
Síntes¡s de 5-((2-(ciclopropanocarboxamido)piridin-4-il)amino)-3-metil-6-oxo-1,6-dihidropirazin-2-carboxamida (8) La síntesis del intermedio 8 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento K. Rendimiento: 600 mg, producto en bruto; EM (IEN) m/z 329,06 [M 1]+.
Síntesis de N-(4-((8’-metil-1',5’-dioxo-1',5’-dihidro-2'H-espiro[ciclohexano-1,3-imidazo[1,5-a]pirazin]-6’-il)amino)piridin-2-il)ciclopropanocarboxamida (10)
5-((2-(Ciclopropanocarboxamido)piridin-4-il)amino)-3-metil-6-oxo-1,6-dihidropirazina-2-carboxamida (8,600 mg, 1,83 mmol) y ciclohexanona 538 mg, 5,48 mmol) se cargaron en acetonitrilo en un vial de microondas de 20 ml. Se añadió cloruro de hierro (III) (889 mg, 1,83 mmol) y se calentó la mezcla de reacción a 80 °C durante 16 h. Al final de la reacción, el disolvente se retiró al vacío y se purificó el compuesto mediante cromatografía en columna de gel de sílice (malla 200-400) eluyendo con metanol al 5 % en diclorometano. Las fracciones apropiadas de las columnas se concentraron a presión reducida para proporcionar N-(4-((8'-metil-1',5'-dioxo-1',5'-dihidro-2'H-espiro[ciclohexanoimidazo[1,5-a]pirazin]-6'-il)amino)piridin-2-il)ciclopropanocarboxamida (10, 100 mg, producto en bruto) en forma de un sólido de color en bruto de color marrón claro que se reenvió directamente a la siguiente etapa. EM (IEN) m/z 409,43 [M 1]+.
Síntesis de 6 ’-((2-aminopiridin-4-il)amino)-8 '-metil-2 'H-espiro[ciclohexano-1, 3 '-imidazo[1,5-a]pirazin]-1'5 ’-diona (Comp. N.° 182)
La síntesis del compuesto 182 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento I. Sólido de color blanquecino; Rendimiento: 2 mg; EM (IEN) m/z 341,21 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 12,59 (s, 1H), 10,36 (s, 1H), 10,18 (s, 1H), 8,09 (s, 1H), 7,79 (s, 1H), 7,77(s, 2H), 7,27 (s, 1H), 2,82-2,76 (m, 2H), 2,50 (s, 3H), 1,77-1,74 (m, 2H), 1,70-1,60 (m, 3H), 1,55-1,52 (m, 2H), 1,23-1,19 (m, 1H).
Ejemplo 183
Síntesis de 3'-amino-6-((6-aminopirimidin-4-il)amino)-8-cloro-6'-fluoro-2,3'-dihidro-2H-espiro[imidazo[1,5-a]-piridin-3,1'inden]-1,5-diona (Comp. N.° 183)

La síntesis del producto intermedio 3 se realiza como se ha descrito anteriormente usando el protocolo general del procedimiento A.
Síntesis de N-(6-(8-cloro-6’-fluoro-1,3’,5-trioxo-1,2,3,5-tetrahidro-2H-espiro[imidazo[1,5-a]-piridin-3,1’-inden]-6-il)amino)pirimidin-4-il)ciclopropanocarboxamida (5)
La síntesis del intermedio 5 se realiza como se ha descrito anteriormente usando el protocolo general del Procedimiento H.
Síntesis de N-(6-((3’-amino-8-cloro-6'-fluoro-1,5-dioxo-1,2,3,3,5-tetrahidro-2H-espiro[imidazo[1,5-aJpirídin-3,1’-indenJ-6-il)amino)pirimidin-4-il)ciclopropanocarboxamida (6)
A una solución de N-(6-((3'-amino-8-cloro-6'-fluoro-1,5-dioxo-1,2,3,4,5-tetrahidro-2H-espiro[imidazo[1,5-a]piridin-3,1'-inden]-6-il)amino)pirimidin-4-il)ciclopropanocarboxamida (6,1 g, 2,02 mmol) en 2-propanol (20 ml) se le añadieron acetato de amonio (0,47 g, 6,06 mmol) y cianoborohidruro de sodio (1,02 g, 16,16 mmol). La reacción se agitó a 110 °C durante una noche. La mezcla resultante se enfrió a temperatura ambiente y se vertió en solución acuosa saturada de bicarbonato de sodio. La mezcla se extrajo con diclorometano. La capa orgánica se secó sobre sulfato de magnesio, se filtró y se concentró. El producto en bruto se purificó por cromatografía en columna para proporcionar N-(6-((3'-amino-8-cloro-6'-fluoro-1,5-dioxo-1,2,3,3,5-tetrahidro-2H-espiro[imidazo[1,5-a]piridin-3,1'-inden]-6-il)amino)pirimidin-4-il)ciclopropanocarboxamida (6).
Síntesis de 3’-amino-6-((6-aminopirimidin-4-il)amino)-8-cloro-6'-fluoro-2’,3’-dihidro-2H-espiro[imidazo[1,5-]piridin-3’,1’-inden]-1,5-diona (Comp. N.° 183)
La síntesis del compuesto 183 se realiza como se ha descrito anteriormente usando el protocolo general del Procedimiento I.
Ejemplo 184
Síntesis de W-(6-((8-cloro-3-(3-clorofenil)-3-metil-1,5-dioxo-1,2,5,5-tetrahidroimidazo[1,5-a]piridin-6-il)amino)pirimidin-4-il)cianamida (Comp. N.° 184)

Síntesis de 8-cloro-3-(3-clorofenil)-6-((6-fluoropirimidin-4-il)amino)-3-metil-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona (3)
La síntesis del intermedio 3 se realiza como se ha descrito anteriormente usando el protocolo general del Procedimiento H.
Síntesis de N-(6-((8-cloro-3-(3-clorofenil)-3-metil-1,5-dioxo-1,2,5,5-tetrahidroimidazo[1,5-a]piridin-6-il)amino)pirimidin-4-il)cianamida (Comp. N.° 184)
A una solución de 8-cloro-3-(3-clorofenil)-6-((6-fluoropirimidin-4-il)amino)-3-metil-2,3-dihidroimidazo[1,5-a]piridin-1,5-diona (3, 100 mg, 0,24 mmol) en N-metil-2-pirrolidinona (4 ml) se le añadió hidrogencianamida de sodio (46 mg, 0,72 mmol). La reacción se agitó a 50 °C durante 2 h. La mezcla resultante se enfrió a temperatura ambiente, se vertió en agua y se acidificó a pH = 5,5 con ácido clorhídrico concentrado. La mezcla se filtró. El producto en bruto sólido se purificó por HPLC para proporcionar N-(6-((8-cloro-3-(3-clorofenil)-3-metil-1,5-dioxol-2,3,5-tetrahidroimidazo[1,5-a]piridin-6-il)amino)pirimidin-4-il)cianamida (Comp. N.° 184)
Ejemplo 185
Síntesis de 8'-cloro-6,-(pirimidin-4-ilamino)-2'H-espiro[biciclo[2,2.1]heptano-7,3,-imidazo[1,5-a]piridin]-1',5'-diona (Comp. N.° 185)

La síntesis del intermedio 3 se realiza como se ha descrito anteriormente usando el protocolo general del procedimiento A.
Síntesis de 8’-cloro-6'-(pirimidin-4-ilamino)-2'H-espiro[biciclo[2,2.1]heptano-7,3’-imidazo[1,5-a]piridin]-1',5'-diona (Comp. N.° 185)
La síntesis del compuesto 185 se realiza como se ha descrito anteriormente usando el protocolo general del Procedimiento H.
Ejemplo 186
Síntesis de 6'-((6-aminopirimidin-4-il)amino)-8'-cloro-2'H-espiro[biciclo[2,2.1]heptano-7,3'-imidazo[1,5-a]piridin]-1',5'-diona (Comp. N.° 186)

Síntesis de N-(6-((8’-cloro-1',5’-dioxo-1',5’-dihidro-2'H-espiro[biciclo[2,2.1]heptano-7,3'imidazo[1,5-a]piridin]-6’-il)amino)pirimidin-4-il)ciclopropanocarboxamida (3)
La síntesis del intermedio 3 se realiza como se ha descrito anteriormente usando el protocolo general del Procedimiento H.
S ín te s is de 6 ’-((6 -a m in o p ir im id in -4 - il)a m in o )-8 '-c lo ro -2 -e s p iro [b ic ic lo [2 ,2.1 ]h e p ta n o -7 ,3 '- im id a z o [1 ,5 -a ]p ir id in ]-1 ’,5 '-d io n a (C om p. N ° 186)
La síntesis del compuesto 186 se realiza como se ha descrito anteriormente usando el protocolo general del Procedimiento I.
Ejemplo 187
Síntesis de 8-cloro-6-[(5-metoxipirimidin-4-il)amino]espiro[2H-imidazo[1,5-a]piridin-3,1,-ciclohexano]-1,5-diona (Comp. N.° 187)

Síntesis de 8-cloro-6-[(5-metoxipirimidin-4-il)amino]espiro[2H-imidazo[1,5-a]piridin-3,1 ’-ciclohexano]-1,5-diona (Comp. N ° 187)
La síntesis del compuesto 187 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento H. Sólido de color gris; Rendimiento: 0,052 g, 17 %; EM (IEN) m/z 376,31 [M 1]+; RMN 1H (400 MHz, DMSO-d6) 610,36 (s, 1H), 8,69 (s, 1H), 8,64 (s, 1H), 8,53 (s, 1H), 8,29 (s, 1H), 4,01 (s, 3H), 3,46-3,40 (m, 1H), 2,93-2,87 (m, 2H) 1,77-1,74 (m, 2H), 1,64-1,61 (m, 3H), 1,55-1,52 (m, 2H), 1,23 (m, 1H).
Ejemplo 188
Síntesis de clorhidrato de 6'-((6-amino-5-etilpirimidin-4-il)amino)-8'-cloro-2'-espiro[ciclohexano-1'-imidazo[1,5-a]piridin]-1',5'-diona (Comp. N.° 188)


4 M HCI/dioxano
DCM/MeOH

Síntesis N-terc-butoxicarbonil-N-(6-cloro-5-etil-pirimidin-4-il)carbamato de terc-butilo (2)
La síntesis del intermedio 2 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento J. Sólido de color marrón claro. Rendimiento: 3,5 g, 96 %; EM (IEN) m/z 358,5 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 8,71 (s, 1H), 2,70 (m, 3H), 1,4 (m, 18H), 1,20 (m, 3H).
Síntesis de N-terc-butoxicarbonil-N-[6-[(8-cloro-1,5-dioxo-espiro[2H-imidazo[1,5-a]piridin-3,1 ’-ciclohexan-6-il)amino]-5-etil-pirimidin-4-il]carbamato de terc-butilo (4)
La síntesis del intermedio 4 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento B. Sólido de color amarillo. Rendimiento: 0,48 g, 42 %; EM (IEN) m/z 589,45 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,40 (s, 1H), 8,75 (s, 1H), 8,71 (s, 1H), 8,54 (s, 1H), 2,94 (m, 2H), 2,59 (m, 2H), 1,74 (m, 2H), 1,65 (m, 2H), 1,57 (m, 2H), 1,14 (m, 18H), 1,25 (m, 1H), 1,22 (m, 4H).
Síntesis de 6’-((6-amino-5-etilpirimidin-4-il)amino)-8'-cloro-2’H-espiro[ciclohexano-1,3-imidazo[1,5-a]piridin]-1 ’,5'-diona (Comp. N.° 188)
La síntesis del compuesto 188 se realizó como se ha descrito anteriormente usando el protocolo general del procedimiento F. Sólido de color amarillo. Rendimiento: 0,32 g, 94 %; EM (IEN) m/z 389,06 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,49 (s, 1H), 8,59 (s, 1H), 8,44-8,33 (m, 2H), 7,74 (s ancho, 2H), 2,93-2,90 (m, 2H), 2,66 (m, 2H), 1,77-1,74 (m, 3H), 1,55-1,52 (m, 2H), 1,25-1,22 (m, 1H), 1,10 (t, J = 14,8 Hz, 3H).
Ejemplo 189
Síntesis de 6'-((6-amino-5-isopropilpirimidin-4-il)amino)-8,-cloro-2'-espirociclohexano-1,-imidazo[1,5-a]piridinil-1',5'-diona (Comp. N.° 189)


4 M HCI/ dioxano
DCIWMeOH

Síntesis de N-terc-butoxicarbonil-N-(6-cloro-5-isopropil-pirimidin-4-il)carbamato de terc-butilo (2)
La síntesis del intermedio 2 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento J. Sólido de color blanquecino; Rendimiento: 4,5 g, 90 %; EM (IEN) m/z 372,3 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 8,92 (s, 1H), 3,32-3,20 (m, 1H), 1,52-1,20 (m, 24H).
Síntesis de N-terc-butoxicarbonil-N-[6-[(8-cloro-1,5-dioxo-espiro[2H-imidazo[1,5-a]piridin-3,1 ’-ciclohexan-6-il)amino]-5-isopropil-pirimidin-4-il]carbamato de terc-butilo (4)
La síntesis del intermedio 4 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento B. Sólido de color amarillo; Rendimiento: 0,41 g, 30 %; EM (IEN) m/z 603,55 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,40 (s, 1H), 8,75-8,72 (m, 2H), 8,67 (s, 1H), 3,32-3,19 (m, 1H), 2,94 (m, 2H), 1,74 (m, 2H), 1,65 (m, 3H), 1,57 (m, 2H), 1,14 (m, 24H), 1,20-1,00 (m, 1H).
Síntesis de 6’-((6-amino-5-isopropilpirimidin-4-il)amino)-8'-cloro-2'H-espiro[ciclohexan-1,3’-imidazo[1,5-a]piridin]-1' 5’-diona (Comp. N.° 189)
La síntesis del compuesto 189 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento F. Sólido de color amarillo; Rendimiento: 0,30 g, 93 %; EM (IEN) m/z 403,17 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,38 (s, 1H), 9,51 (s, 1H), 8,45 (s, 2H), 7,69-7,68 (s ancho, 2H), 3,59-3,56 (m, 2H), 2,93 2,87 (m, 2H), 1,77-1,74 (m, 2H), 1,68-1,65 (m, 3H), 1,56 (m, 2H), 1,36 (m, 2H), 1,77-1,74 (m, 2H) 6H), 1,26 (m, 1H). Ejemplo 190
Síntesis de 8'-cloro-6'-(7H-pirrolo[2,3-c/]pirimidin-7-il)-2'H-espiro[ciclohexano-1,3-imidazo[1,5-3]piridin]-1',5'-diona (Comp. N.° 190)


Síntesis de 8’-cloro-6'-(7H-pirrolo[2’,3-d]pirimidin-7-il)-2-espiro[ciclohexano-1,3,3-imidazo[1,5-a]piridin]-1’,5’-diona (Comp. N.° 190)
La síntesis del compuesto 190 se realizó como se ha descrito anteriormente usando el protocolo general del procedimiento B. Sólido de color amarillo. Rendimiento: 0,070 g, 7 %; EM (IEN) m/z 370,09 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,66 (s, 1H), 9,11 (s, 1H), 8,87 (s, 1H), 8,26 (s, 1H), 7,93 (d, J = 3,56 Hz, 2H), 6,82 (d, J = 3,56 Hz, 2H), 2,94-2,89 (m, 2H), 1,74-1,57 (m, 7H), 1,23 (m, 1H).
Ejemplo 191
Síntesis de 8'-cloro-6'-(pirimidin-4-ilamino)-2-(trifluorometil)-2'H-espiro[ciclohexano-1,3-imidazo[1,5-a]piridin]-1 ',5'-diona (Comp. N.° 191)

Síntesis de 6’-bromo-8'-cloro-2-(trifluorometil)-2’H-espiro[ciclohexano-1,3-imidazo[1,5-a]piridin]-1',5’-diona (3) La síntesis del intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento A. Sólido de color blanquecino; Rendimiento: 0,40 g, 43 %; EM (IEN) m/z 400,59 [M 1]+.
Síntesis de 8’-cloro-6'-(pirimidin-4-ilamino)-2-(trifluorometil)-2’H-espiro[ciclohexano-1,3,3-imidazo]-1,5-a]piridin]-diona (Comp. N ° 191)
La síntesis del compuesto 191 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento B. Sólido de color blanquecino; Rendimiento: 0,030 g, 8 %; EM (IEN) m/z 346,80 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,53 (s, 1H), 9,66 (s, 1H), 8,84 (d, J = 9,6 Hz, 2H), 8,45 (d, J = 5,92 Hz, 1H), 7,44 (d, J = 4,8 Hz, 1H), 4,03 (m, 1H), 2,88 (m, 1H), 2,04 (m, 1H), 1,80 (m, 3H), 1,67 (m, 1H), 1,41 (s, 1H).
Ejemplo 192
Síntesis de 8-cloro-3-metil-3-(2-metilprop-1-enil)-6-(pirimidin-4-ilamino)-2H-imidazo[1,5-a]piridin-1,5-diona (Comp. N.° 192)
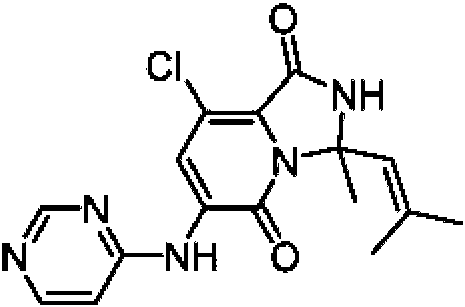

La síntesis del producto intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento A. Líquido espeso marrón claro. Rendimiento: 1,5 g, 28 %; EM (IEN) m/z 329 [M-1]-.
Síntesis de 8-cloro-3-metil-3-(2-metilprop-1-enil)-6-(pirimidin-4-ilamino)-2H-imidazo[1,5-a]piridin-1,5-diona (Comp. N ° 192)
La síntesis del compuesto 192 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento H. Sólido de color blanquecino; Rendimiento: 65 mg, 31 %; EM (IEN) m/z 360,80 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,32 (s, 1H), 9,62 (s, 1H), 8,43 (d, J = 5,6 Hz, 1H), 7,47 (d, J = 4,8 Hz, 1H), 3,25-3,17 (m, 2H), 280-2,78 (m, 2H), 2,39-2,33 (m, 2H), 2,24 (s, 3H), 1,50-1,47 (m, 2H).
Ejemplo 193
Síntesis de clorhidrato de 6'-((6-amino-5-metilpirimidin-4-il)amino)-8'-cloro-2-espiroIciclohexano-1,3'-imidazo[1,5-a]piridini]-1',5'-diona (Comp. N.° 193)

Síntesis de N-terc-butoxicarbonil-N-(6-cloro-5-metil-pirimidin-4-il)carbamato de terc-butilo (2)
La síntesis del intermedio 2 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento J. Sólido de color blanquecino; Rendimiento: 1,1 g, 94 %; EM (IEN) m/z 344,27 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 8,90 (s, 1H), 2,22 (s, 1H), 1,38 (s, 18H).
Síntesis de N-terc-butoxicarbonil-N-[6-[(8-cloro-1,5-dioxo-espiro[2H-imidazo[1,5-a]piridin-3,1 ’-ciclohexan-6-il)amino]-5-metil-pirimidin-4-il]carbamato de terc-butilo (4)
La síntesis del intermedio 4 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento B. Sólido de color amarillo; Rendimiento: 0,20 g, 35 %; EM (IEN) m/z 575,32 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,41 (s, 1H), 8,73 (s, 2H), 8,46 (s, 1H), 2,14 (s, 3H), 1,77-1,66 (m, 7H), 1,46 (m,18H), 1,20 (m, 1H).
Síntesis de 6’-((6-amino-5-metilpirimidin-4-il)amino)-8'-cloro-2’-espirof ciclohexano-1,3-imidazo[1,5-a]piridin]-1’,5'-diona (Comp. N.° 193)
La síntesis del compuesto 193 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento F. Sólido de color amarillo; Rendimiento: 0,11 g, 80 %; EM (IEN) m/z 375,26 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,41 (s, 1H), 8,47 (s, 2H), 8,40 (s, 1H), 7,79 (s ancho, 2H), 2,93-2,87 (m, S, 1H), 1,77-1,74 (m, 2H), 1,65-1,56 (m, 3H), 1,56-1,53 (m, 2H), 1,09 (m, 1H).
Ejemplo 194
Síntesis de 8'-cloro-6'-((5-etilpirimidin-4-il)amino)-2'H-espiro[ciclohexano-1,3,3-imidazo[1,5-a]piridin]-1',5'-diona (Comp. N.° 194)


Síntesis de 8’-cloro-6’-((5-etilpirimidin-4-il)amino)-2'H-espiro[ciclohexano-1,3-imidazo[1,5-a]piridin]-1' 5’-diona (Comp. N.° 194)
La síntesis del compuesto 194 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento B. Sólido de color amarillo; Rendimiento: 0,026 g, 22 %, EM (IEN) m/z 374,21 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,37 (s, 1H), 8,78 (s, 1H), 8,72 (s, 1H), 8,39 (s, 1H), 8,34 (s, 1H), 2,95-2,89 (m, 2H), 2,70 2,64 (m, 2H), 1,77-1,74 (m, 2H), 1,69-1,65 (m, 3H), 1,57-1,54 (m, 2H), 1,26 (m, 4H).
Ejemplo 195
Síntesis de clorhidrato de 6'-((6-amino-5-metoxipirimidin-4-il)amino)-8'-cloro-2-espiro[ciclohexano-1,3-imidazo[1,5-a]piridin]-1',5'-diona (Comp. N.° 195)


Síntesis de N-terc-butoxicarbonil-N-(6-cloro-5-metoxi-pirimidin-4-il)carbamato de terc-butilo (2)
La síntesis del intermedio 2 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento J. Sólido de color blanquecino; Rendimiento: 092 g, 59 %; EM (IEN) m/z 360,12 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 1,44 (s, 18H), 3,92 (s, 3H), 8,64 (s, 1H).
Síntesis de N-terc-butoxicarbonil-N-[6-[(8-cloro-1,5-dioxo-espiro[2H-imidazo[1,5-a]piridin-3,1 ’-ciclohexan]-6-il)amino]-5-metoxi-pirimidin-4-il]carbamato de terc-butilo (4)
La síntesis del intermedio 4 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento B. Sólido de color amarillo; Rendimiento: 0,14 g; 23 %; EM (IEN) m/z 591,23 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,40 (s, 1H), 8,78 (s, 1H), 8,67 (s, 1H), 8,6 (s, 1H), 3,87 (s, 3H), 2,92 (m, 2H), 1,81-1,54 (m, 7H), 1,40 (s, 18H), 1,23 (m, 1H).
Síntesis de 6’-((6-amino-5-metoxipirimidin-4-il)amino)-8'-cloro-2-espiro[ciclohexano-1,3,3-imidazo[1,5-a]piridin]-1',5'-diona (Comp. N.° 195)
La síntesis del compuesto 195 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento F. Sólido de color blanquecino; Rendimiento: 0,075 g, 80 %; EM (IEN) m/z 391,12 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,35 (s, 1H), 8,53 (s, 1H), 8,48 (s, 1H), 8,19 (s, 1H), 7,33 (s ancho, 3H), 2,91 (m, 2H), 1,76 1,52 (m, 7H), 1,26 (m, 1H).
Ejemplo 196
Síntesis de 8’-cloro-6'-((5-etoxipirimidin-4-il)amino)-2’H-espiro[ciclohexano-1,3,3-imidazo[1,5-a]piridin]-1 ',5'-diona (Comp. N.° 196)


Síntesis de 8'-cloro-6’-((5-etoxipirimidin-4-il)amino)-2'H-espiro[ciclohexano-1,3,3-imidazo[1,5-a]piridin]-1’,5’-diona (Comp. N.° 196)
La síntesis del compuesto 196 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento B. Sólido de color amarillo; Rendimiento: 0,055 g, 20 %; EM (IEN) m/z 390,13 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,42 (s, 1H), 8,82 (s, 1H), 8,75 (s, 1H), 8,62 (s, 1H), 8,33 (s, 1H), 4,30 (c, J = 6,96 Hz, 2H), 2,90 (m, 2H), 1,74-1,53 (m, 7H), 1,43 (t, J = 6,96 Hz, 3H), 1,25 (m, 1H).
Ejemplo 197
Síntesis de 8'-cloro-6'-((5-isopropoxipirimidin-4-il)amino)-2'H-espiro[ciclohexano-1,3-imidazo[1,5-a]piridin]-1',5'-diona (Comp. N.° 197)

Síntesis de N,N-di-boc-4-aminopirimidin-5-ol (2)
La síntesis del intermedio 2 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento J. Sólido de color marrón; Rendimiento: 1,2 g, 86 %; EM (IEN) m/z 312,15 [M 1]+; RMN 1H (400 MHz, CDCl3) ó 8,96 (s, 1H), 8,76 (s, 1H), 1,42 (s, 18H).
Síntesis de N,N-di-boc-5-isopropoxipirimidin-4-amina (3)
A una solución de N,N-di-boc-4-aminopirimidin-5-ol (2,1 g, 3,21 mmol) en dimetilformamida (15 ml), se le añadió carbonato de potasio (1,11 g, 8,03 mmol) seguido de la adición de 2-yodo propano (1,64 g, 9,64 mmol). La mezcla de reacción se agitó a 60 °C durante 3 h. La mezcla de reacción se enfrió, se diluyó con acetato de etilo (50 ml) y se lavó con agua fría (20 ml, 3 veces) y salmuera, se secó sobre sulfato de sodio y se concentró a presión reducida para proporcionar N,N-diboc-5-isopropoxipirimidin-4-amina (3) en forma de un sólido de color blanquecino. Rendimiento: 1,1 g, 97 %; EM (IEN) m/z 354,20 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 8,75 (s, 1H), 8,70 (s, 1H), 4,89 (m, 1H), 1,37 (s, 18H), 1,27 (d, J = 6,0 Hz, 6H).
Síntesis de clorhidrato de 5-isopropoxipirimidin-4-amina (4)
La síntesis del intermedio 4 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento F. Sólido de color blanquecino; Rendimiento: 0,55 g, 93 %; EM (IEN) m/z 154,09 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 14,32 (s ancho, 1H), 9,10-8,60 (m, 2H), 8,48 (s, 1H), 8,31 (s ancho, 1H), 8,06 (s, 1H), 4,72 (m, 1H), 1,32 (d, J = 6,0 Hz, 6H)
Síntesis de 8’-cloro-6'-((5-isopropoxipirimidin-4-il)amino)-2’H-espiro[ciclohexano-1,3,3-imidazo[1,5-a]piridin]-1’,5’-diona (Comp. N.° 197)
La síntesis del compuesto 197 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento H. Sólido de color amarillo; Rendimiento: 0,22 g, 61 %; EM (IEN) m/z 404,14 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,4 (s, 1H), 8,71 (s, 2H), 8,56 (s, 1H), 8,34 (s, 1H), 4,87 (m, 1H), 2,92 (m, 2H), 1,77-1,53 (m, 7H), 1,37 (d, J = 6,0 Hz, 6H), 1,23 (m, 1H).
Ejemplo 198
Síntesis de 8'-cloro-2,-ciclopentil-6'-(pirimidin-4-ilamino)-2,H-espiro[ciclohexano-1,3-imidazo[1,5-a]piridin]-1',5'-diona (Comp. N.° 198)

Síntesis de 8’-cloro-2'-ciclopentil-6’-(pirimidin-4-ilamino)-2'H-espiro[ciclohexano-1,3’-imidazo[1,5-a]piridin]-1’,5’-diona (Comp. N.° 198)
En un vial 8'-cloro-6'-(pirimidin-4-ilamino)-2-espiro[ciclohexano-1,3-imidazo[1,5-a]piridin]-1',5'-diona (0,5 g, 1,44 mmol), bromociclopentano (0,26 g, 1,73 mmol) y fosfato de potasio (0,49 g, 3,62 mmol) se tomaron en 1,4-dioxano (10 ml). La mezcla de reacción se purgó con argón durante 10 min y se añadió yoduro de cobre (I) (0,027 g, 0,14 mmol), trans-N,N'-dimetilciclohexano-1,2-diamina (0,041 g, 0,14 mmol) y se continuó purgando durante otros 10 min. La reacción se selló y se calentó a 110 °C durante 24 h. Después de la terminación, la reacción se diluyó con metanol al 5 % en diclorometano (300 ml) y se filtró a través de un lecho de celite. El filtrado se concentró. El compuesto en bruto se purificó por cromatografía en columna usando alúmina neutra y el compuesto se eluyó con diclorometano. El disolvente se retiró a presión reducida para obtener un sólido que se secó a alto vacío para proporcionar 8-cloro-2-ciclopentil-6'-(pirimidin-4-ilamino)-2'H-espiro[ciclohexano-1,3'-imidazo[1,5-a]piridin]-1',5-diona (Comp. N.° 198) en forma de un sólido de color blanquecino. Rendimiento: 0,035 g, 6 %; EM (IEN) m/z 414,14 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 9,50 (s, 1H), 8,81 (s, 1H), 8,77 (s, 1H), 8,40 (d, J = 5,84 Hz, 5,32 Hz, 1H), 7,41 (d, J = 5,32 Hz, 1H), 5,33 (s, 1H), 2,93 (m, 2H), 1,92 (m, 12H), 1,62 (m, 2H), 1,37 (m, 2H).
Ejemplo 199
Síntesis de 6'-((6-aminopirimidin-4-il)amino)-8'-metil-5'-tioxo-2-espiro[ciclopentano-1,3,-imidazo[1,5-a]piridin]-1'(5'H)-ona (Comp. N.° 199)
H2S04, dioxano
90 C

Síntesis de 5-((6-(ciclopropanocarboxamido)pirimidin-4-il)amino)-3-metil-6-oxo-1,6-dihidropiridin-2-carboxilato de nbutilo (3)
La síntesis del intermedio 3 se realiza como se ha descrito anteriormente usando el protocolo general del Procedimiento H.
Síntesis de ácido 4-(6-aminopirimidin-4-il)amino)-3-metil-6-oxo-1,6-dihidropiridin-2-carboxílico (4)
La síntesis del intermedio 4 se realiza como se ha descrito anteriormente usando el protocolo general del Procedimiento I.
Síntesis de ácido 5-((6-aminopirimidin-4-il)amino)-3-metil-6-tioxo-1,6-dihidropiridin-2-carboxílico (5)
A una solución de ácido 5-((6-aminopirimidin-4-il)amino)-3-metil-6-oxo-1,6-dihidropiridin-2-carboxílico (4, 0,5 g, 1,91 mmol) en piridina (10 ml) se le añadió pentasulfuro de fósforo (1,27 g, 5,73 mmol). La reacción se refluye toda la noche. La mezcla resultante se enfría a temperatura ambiente y se concentra. El producto en bruto se purifica mediante cromatografía en columna para proporcionar ácido 5-((6-aminopirimidin-4-il)amino)-3-metil-6-tioxo-1,6-dihidropiridin-2-carboxílico (5).
Síntesis de 5-((6-aminopirimidin-4-il)amino)-3-metil-6-tioxo-1,6-dihidropiridin-2-carboxilato de metilo (6)
A una solución de ácido 5-((6-aminopirimidin-4-il)amino)-3-metil-6-tioxo-1,6-dihidropiridin-2-carboxílico (5, 0,5 g, 1,72 mmol) en tetrahidrofurano (10 ml) y metanol, se le añade (trimetilsilil)diazometano (2 M en hexanos, 1,29 ml, 2,58 mmol). La reacción se agita a temperatura ambiente durante 1 h. La mezcla resultante se cocentró y purificó por cromatografía en columna para proporcionar 5-((6-aminopirimidin-4-il)amino)-3-metil-6-tioxo-1,6-dihidropiridin-2-carboxilato de metilo (6 ).
Síntesis de 5-((6-aminopirimidin-4-il)amino)-3-metil-6-tioxo-1,6-dihidropiridin-2-carboxamida (7)
La síntesis del intermedio 7 se realiza como se ha descrito anteriormente usando el protocolo general del Procedimiento K.
S ín te s is de 6 ’- ( (6 -a m in o p ir im id in -4 - il)a m in o )-8 '-m e til-5 ’-tio xo -2 'H -e s p iro [c ic lo p e n ta n o -1 ,3 - im id a z o [1 ,5 -a ]p ir id in ]-1 ’(5 'H )-o n a (C om p. N .° 199)
La síntesis del compuesto 199 se realiza como se ha descrito anteriormente usando el protocolo general del Procedimiento A.
Ejemplo 200
Síntesis de 6'-((6-aminopirimidin-4-il)(metil)amino)-8'-cloro-2'H-espiro[ciclopentano-1,3'-imidazo[1,5-a]piridin]-1',5'-diona (Comp. N.° 200)

Síntesis de (6-(ciclopropanocarboxamido)pirimidin-4-il)(metil)carbamato de terc-butilo (3)
La síntesis del intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento B. Sólido de color amarillo; Rendimiento: 2,5 g, 80 %; EM (IEN) m/z 293,51 [M 1]+; RMN 1H (400 MHz, DMSO-d6) 6 11,06 (s, 1H), 8,63 (s, 1H), 8,59 (s, 1H), 2,05-1,99 (m, 1H), 1,49 (s, 9H), 0,85-0,83 (m, 4H). Síntesis de clorhidrato de N-(6-(metilamino)pirimidin-4-il)ciclopropanocarboxamida (4)
La síntesis del intermedio 4 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento F. Sólido de color blanquecino; Rendimiento: 1,4 g, 90 %; EM (IEN) m/z 193,30 [M 1]+; RMN 1H (400 MHz, DMSO-d6) 6 11,87 (s ancho, 1H), 9,10 (s ancho, 1H), 8,45 (s, 1H), 7,05 (s ancho, 1H), 2,91 (s, 3H), 2,01 (m, 1H), 0,93-0,89 (m, 4H).
Síntesis de N-(6-((8'-cloro-1' 5'-dioxo-15'-dihidro-2'H-espiro[ciclopentano-1,3,3-imidazo[1,5-a]piridin-6’-il)(metil)amino)pirimidin-4-il)ciclopropanocarboxamida (6)
La síntesis del intermedio 6 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento H. Sólido de color amarillo; Rendimiento: 120 mg, 18 %; EM (IEN) m/z 427,01 [M-1]-.

La síntesis del compuesto 200 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento I. Un sólido marrón claro; Rendimiento: 27 mg, 32 %; EM (IEN) m/z 361,12 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,46 (s, 1H), 8,29 (s, 1H), 8,04 (s, 1H), 7,59 (s ancho, 2H), 5,72 (s, 3H), 2,76-2,69 (m, 2H), 1,91-1,84 (m, 2H), 1,82-1,79 (m, 2H), 1,74-1,69 (m, 2H).
Ejemplo 201
Síntesis de 6'-(pirimidin-4-ilamino)-8'-(tetrahidro-2H-piran-4-il)-2-espirociclohexano-1 '-imidazo[1,5-a]piridinil-1',5'-diona (Comp. N.° 201)

Síntesis de 8’-(3,6-dihidro-2H-piran-4-il)-6'-(pirimidin-4-ilamino)-2-espiro[ciclohexano-1,3'-imidazo[1,5-a ]piridin]-1’,5'-diona (3)
Se cargó un vial con 8'-cloro-6'-(pirimidin-4-ilamino)-2'H-espiro[ciclohexano-1,3-imidazo[1,5-a]piridin]-1',5'-diona (1, 0,50 g, 1,44 mmol) y 2-(3,6-dihidro-2H-piran-4-il)-4,4,5,5-tetrametil-1,3,2-dioxaborolano (2, 0,36 g, 1,73 mmol) en 1,4-dioxano (10 ml). Se añadió carbonato de sodio (0,46 g, 4,33 mmol) seguido de agua (1,44 ml) y se purgó la mezcla con argón durante 10 min. Se añadió después [1,1'-bis(difenilfosfino)ferroceno] dicloropaladio (II), complejo con diclorometano (0,1 lg, 0,144 mmol) y se prosiguió la purga durante 5 min más. La reacción se selló y se calentó a 110 °C durante 16 h. Después de completar la reacción por CCF y CLEM. Se añadió agua (100 ml) y se extrajo con metanol al 10 % en diclorometano (150 ml, 3 veces). Los compuestos orgánicos se lavaron con salmuera (100 ml, 1 veces). Los compuestos orgánicos se separaron y secaron (sulfato de sodio) antes de la concentración a sequedad. El producto en bruto se purificó después por cromatografía en columna ultrarrápida eluyendo con metanol al 2-3 % en diclorometano. Las fracciones deseadas se concentraron a sequedad al vacío para proporcionar 8'-(3,6-dihidro-2H-piran-4-il)-6'-(pirimidin-4-ilamino)-2'H-espiro[ciclohexano-1,3'-imidazo[1,5-a]piridin]-1',5'-diona (3) en forma de un sólido de color blanquecino. Rendimiento: 0,35 g, 61 %; EM (IEN) m/z 394 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,24 (s, 1H), 9,41 (s, 1H), 8,78 (s, 1H), 8,61 (s, 1H), 8,43 (d, J = 6,08 Hz, 1H), 7,44 (d, J = 5,72 Hz, 1H), 5,75 (s, 1H), 4,18 (s, 2H), 3,79 (s, 2H), 3,05 (m, 2H), 2,45 (m, 2H), 1,84 (m, 6H), 1,56 (s, 2H).
Síntesis de 6’-(pirimidin-4-ilamino)-8'-(tetrahidro-2H-piran-4-il)-2-espiro[ciclohexano-1,3’-imidazo[1,5-a]piridin]-1 5'-diona (Comp. N ° 201)
Se cargó un matraz de fondo redondo de doble cuello con 8'-(3,6-dihidro-2H-piran-4-il)-6'-(pirimidin-4-ilamino)-2-espiro[ciclohexano-1,3'-imidazo[1,5-a]piridin]-1',5'-diona (3, 0,22 g, 0,57 mmol) en metanol (10 ml) y tetrahidrofurano (10 ml). Se añadió paladio sobre carbono (0,10 g) seguido de hidróxido amónico (1,0 ml) en atmósfera de nitrógeno. La reacción se llenó con hidrógeno y se agitó a temperatura ambiente durante 3 d. Después de la terminación la reacción controlada por CCF y CLEM, la masa de reacción se diluyó con metanol al 5 % en diclorometano (100 ml) y se hizo pasar a través de lecho de celite y se lavó con un 10 % de metanol/diclorometano (50 ml, 3 veces). El disolvente se retiró al vacío y el material en bruto se purificó por HPLC preparativa para proporcionar 6'-(pirimidin-4-ilamino)-8'-(tetrahidro-2H-piran-4-il)-2'H-espiro[ciclohexano-1,3'-imidazo[1,5-a]piridin]-1',5'-diona (Comp. N.° 201) en forma de un sólido de color blanquecino. Rendimiento: 0,11 g, 49 %; EM (IEN) m/z 396,4 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,16 (6, 1H), 9,40 (s, 1H), 8,85 (s, 1H), 8,81 (s, 1H), 8,38 (d, J = 5,88 Hz, 1H), 7,37 (d, J = 5,88 Hz, 1H), 4,20 (m, 1H), 3,96 (m, 2H), 3,42 (t, J = 11,3 Hz, 2H), 3,01 (m, 2H), 1,70 (m, 9H), 1,58 (m, 2H), 1,22 (m,
1H).
Ejemplo 202
Síntesis de 6'-((6-amino-5-hidroxipirimidin-4-il)amino)-8'-metil-2'H-espiro[ciclohexano-1,3'-imidazo[1,5-a]piridin]-1',5'-diona (Comp. N.° 202)
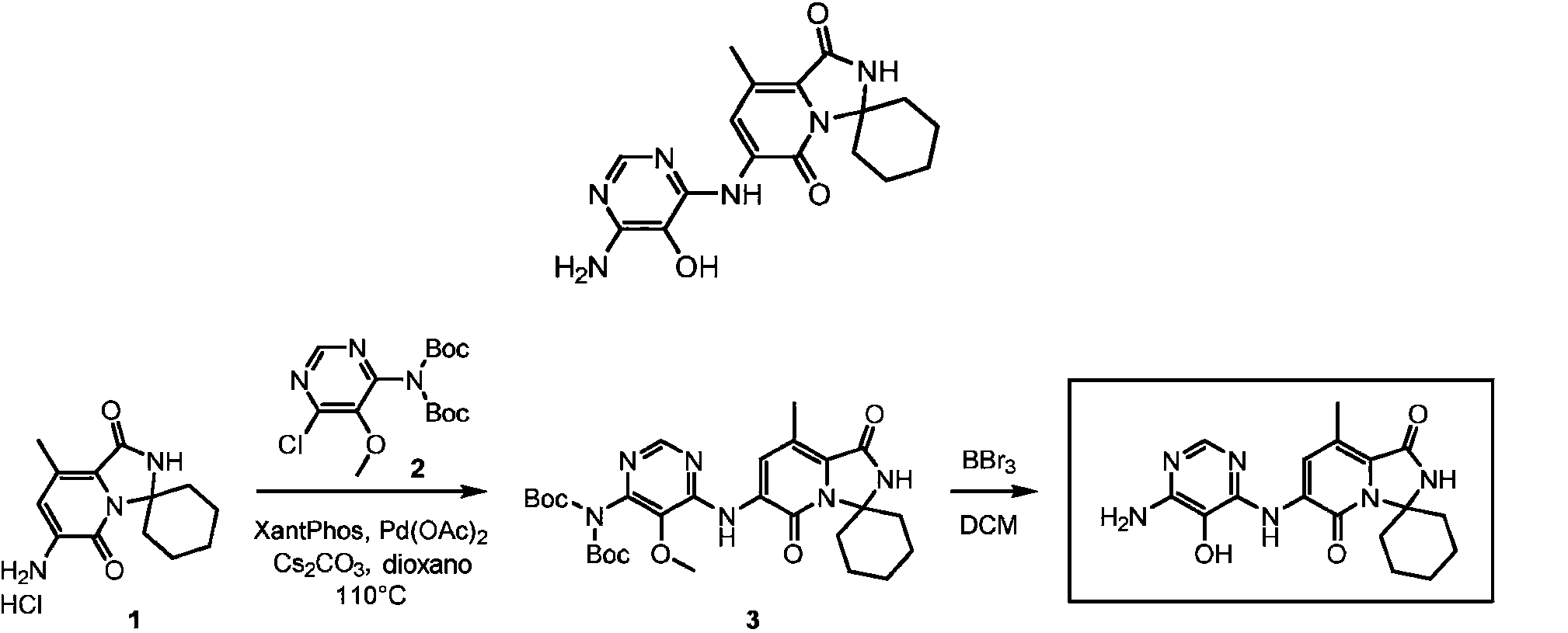
Síntesis de N-terc-butoxicarbonil-N-(5-metoxi-6-((8’-metil-15’-dioxo-15’-dihidro-2'H-espiro[ciclohexano-1 ',3'-imidazo[1,5-a]piridin]-6’-il)amino)pirimidin-4-il)carbamato de terc-butilo (3)
La síntesis del intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del
Procedimiento B. Sólido de color amarillo; Rendimiento: 0,45 g, 64 %; EM (IEN) m/z 571,15 [M 1]+.
Síntesis de 6’-((6-amino-5-hidroxipirimidin-4-il)amino)-8'-metil-2’H-espiro[ciclohexano-1,3-imidazo[1,5-a]piridin]-1’,5'-diona (Comp. N.° 202)
Se cargó un matraz con N-terc-butoxicarbonil-N-(5-metoxi-6-((8'-metil-1',5'-dioxo-1',5'-dihidro-2'H-espiro[ciclohexano-1,3'[1,5-a]piridin]-6'-il)amino)pirimidin-4-il)carbamato de terc-butilo (3, 0,40 g, 0,70 mmol) y se añadió diclorometano
(20 ml), y la mezcla se enfrió a -20 °C. Se añadió gota a gota tribromuro de boro (0,87 g, 3,50 mmol). La masa de
reacción se agitó a temperatura ambiente durante una noche. Después de la terminación, se añadió agua a la
mezcla de reacción y se inactivó con solución acuosa saturada de bicarbonato de sodio hasta pH 8. El sólido
amarillo se precipitó, se filtró y se lavó con agua (20 ml) seguido de éter dietílico y finalmente se secó a alto vacío
para proporcionar 6'-((6-amino-5-hidroxipirimidin-4-il)amino)-8'-metil-2'H-espiro[ciclohexano-1,3-imidazo[1,5-a]piridin]-1',5'-diona (Comp. N.° 202) en forma de un sólido de color amarillo. Rendimiento: 0,085 g, 34 %; EM (IEN)
m/z 357,16 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,06 (s, 1H), 9,15 (s ancho, 1H), 8,50 (s, 1H), 8,40 (m, 1H),
8,08 (s, 1H), 6,65 (s ancho, 2H), 3,00-2,94 (m, 2H), 2,44 (s, 3H), 1,73-1,62 (m, 5H), 1,46-1,43 (m, 2H), 1,24-1,21 (m,
1H).
Ejemplo 203
Síntesis de 6'-((6-amino-2-hidroxipirimidin-4-il)amino)-8'-metil-2'H-espirociclohexano-1,3'-imidazo[1,5-a]piridin]-1',5'-diona (Comp. N.° 203)



Síntesis de 6’-((6-(di-(terc-butoxicarbonil)-amino)-2-metoxipirimidin-4-il)amino)-8’-metil-2’H-espiro[ciclohexano-1,3'-imidazo[1,5-a]piridin]-1' 5'-diona (3)
La síntesis del intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento B. Sólido de color amarillo; Rendimiento: 0,77 g, 51 %; EM (IEN) m/z 571,21 [M 1]+.
Síntesis de 6’-((6-amino-2-hidroxipirimidin-4-il)amino)-8'-metil-2'H-espiro[ciclohexano-1,3'-imidazo[1,5-a]piridin]-1’,5'-diona (Comp. N ° 203)
A una solución agitada de 6'-((6-(di-(terc-butoxicarbonil)-amino)-2-metoxipirimidin-4-il)amino)-8'-metil-2'H-espiro[ciclohexano-1,3'-imidazo[1,5-a]piridin]-1',5'-diona (3, 0,77 g, 1,35 mmol) en diclorometano (15 ml) a -20 °C, se le añadió tribromuro de boro (1 ml) adicional. La mezcla se agitó durante otros 20 minutos a la misma temperatura y después se agitó durante 48 h a temperatura ambiente cuando la CCF mostró una conversión completa del material de partida. La mezcla se inactivó por adición de metanol (2 ml) y el disolvente se retiró a presión reducida para proporcionar el producto en bruto. El producto en bruto se purificó mediante lavado con metanol (5 ml), diclorometano (5 ml) y pentano (25 ml) para proporcionar 6'-((6-amino-2-hidroxipirimidin-4-il)amino)-8'-metil-2'H-espiro[ciclohexano-1,3'-imidazo[1,5-a]piridin]-1',5'-diona (Comp. N.° 203) en forma de un sólido de color amarillo pálido. Rendimiento: 0,28 g, 58 %; EM (IEN) m/z 357,16 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,03 (s ancho, 2H), 8,58 (s ancho, 1H), 8,47 (s ancho, 1H), 6,46 (s ancho, 2H), 5,39 (s ancho, 1H), 3,05-2,92 (m, 2H), 2,40 (s, 3H), 1,80-1,54 (m, 6H), 1,48-1,50 (m, 2H), 1,28-1,16 (m, 1H).
Ejemplo 204
Síntesis de 6'-((6-aminopirimidin-4-il)amino)-2-hidroxi-8'-metil-2'H-espirociclohexano-1,3'-imidazo[1,5-a]piridin]-1',5'-diona (Comp. N.° 204)
S ín te s is de 6 ’-b ro m o -2 -h id ro x i-8 '-m e til-2 ’H -e s p iro [c ic lo h e x a n o -1 ,3 - im id a z o [1 ,5 -a ]p ir id in ]-1 ’,5 '-d io n a ( 3 )
La síntesis del intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento A. Sólido de color amarillo pálido; Rendimiento: 1,3 g, 62 %; EM (IEN) m/z 326,97 [M 1]+.
Síntesis de (6-((2-hidroxi-8'-metil-1’,5'-dioxo-1 ',5'-dihidro-2'H-espiro[ciclohexano-1,3’-imidazo[1,5-a]piridin]-6'-il)amino)pirimidin-4-il)carbamato de terc-butilo (5)
La síntesis del intermedio 5 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento H. Sólido de color amarillo pálido; Rendimiento: 1,1 g, 79 %; EM (IEN) m/z 457,34 [M 1]+.
Síntesis de 6’-((6-aminopirimidin-4-il)amino)-2-hidroxi-8'-metil-2’H-espiro[ciclohexano-1,3’-imidazo[1,5-a]piridin]-1’,5'-diona (Comp. N.° 204)
La síntesis del compuesto 204 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento F. Sólido de color amarillo; Rendimiento: 0,21 g, 25 %; EM (IEN) m/z 357,16 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 9,55 (s, 1H), 8,60 (s ancho, 1H), 8,30 (s, 1H), 8,19 (s ancho, 1H), 6,62 (s ancho, 2H), 6,17 (s, 1H), 5,00-4,95 (m, 1H), 4,70-4,62 (m, 1H), 3,10-3,00 (m, 1H), 1,82-1,48 (m, 6H), 1,38-1,28 (m, 1H).
Ejemplo 205
Síntesis de 6'-((6-aminopirimidin-4-il)amino)-3-hidroxi-8'-metil-2'H-espirociclohexano-1,3'-imidazo[1,5-a]piridin]-1',5'-diona (Comp. N.° 205)

La síntesis del intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento A. Sólido de color blanquecino; Rendimiento: 1,6 g, 22 %; EM (IEN) m/z 329,9 [M 1]+.
Síntesis de N-(6-((8'-metil-1’,3,5'-trioxo-1 ’,5'-dihidro-2'H-espiro[ciclohexano-1,3'-imidazo[1,5-a]piridin]-6’-il)amino)pirimidin-4-il)ciclopropanocarboxamida (5)
La síntesis del intermedio 5 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento H. Sólido de color blanquecino; Rendimiento: 0,40 g, 51 %; EM (IEN) m/z 457,31 [M 1]+.
S ín te s is de 6 ’-((6 -a m in o p irim id in -4 -il)a m in o )-3 -h id ro x i-8 '-m e til-2 -e s p iro [c ic lo h e x a n o -1 ,3 ,3 - im id a z o [1 ,5 -a ]-1 ',5 '-d io n a (C om p. N ° 205)
Se cargó un matraz que contenía tetrahidrofurano y etanol (1:1, 20 ml) con N-(6-((8'-metil-1,3'-trioxo-1',3,5'-trioxo-1',5'-dihidro-2'H-espiro[ciclohexano-1,3-imidazo[1,5-a]piridin]-6'-il)amino)pirimidin-4-il)ciclopropanocarboxamida (5, 0,4 g, 0,9 mmol) y solución 3 M de hidróxido de potasio 8,0 ml) a la reacción anterior. La reacción se agitó a temperatura ambiente durante 18 h. Después de la hidrólisis completa, se añadió borohidruro de sodio (0,18 g, 0,4 mmol) a la reacción anterior a temperatura ambiente. La masa de reacción se agitó durante 2 h cuando la CCF mostró la terminación del material de partida. Los disolventes se retiraron a presión reducida y el en bruto se disolvió en metanol al 10 % en diclorometano y se neutralizó con ácido cítrico al 10 %. La capa orgánica se separó y se secó sobre sulfato de sodio, se filtró y se concentró para obtener sólido. El sólido se filtró y se lavó con metanol (5 ml) y pentano (20 ml) y se secó a alto vacío para proporcionar 6'-((6-aminopirimidin-4-il)amino)-4-hidroxi-8'-metil-2'H-espiro[ciclohexano-1,3'-imidazo[1,5-a]piridin]-1',5'-diona (Comp. N.° 205) en forma de un sólido de color amarillo como una mezcla de diastereómeros. Rendimiento: 0,045 g, 13 %; EM ( iEn ) m/z 357,19 [M 1]+; RMN 1H: 400 MHz, DMSO-cfe) 59,89 y 8,48 (2 s, 1H cada uno, isómero A y B), 8,59 y 8,57 (2s, 1H cada uno, isómero A y B), 8,42 (s ancho, 1H), 8,16 (s ancho, 1H), 6,16 (s ancho, 1H), 5,16 y 4,85 (2 s, 1H cada uno, isómero A y B), 4,22 y 3,73 (2 m, 1H cada uno, isómero A & B), 2,99 y 2,88 (2 m, 2H cada uno, isómero A y B), 2,42 (s, 3H), 1,98-1,11 (m, 6H).
Ejemplo 206
Síntesis de clorhidrato de 6'-((6-aminopirimidin-4-il)amino)-4-hidroxi-8'-metil-2'H-espiro[ciclohexano-1,3'-imidazo[1,5-a]piridin]-1',5'-diona (Comp. N.° 206)

La síntesis del intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento A. Sólido de color blanquecino; Rendimiento: 1,0 g, 35 %; EM (IEN) m/z 326,91 [M 1]+.
Síntesis de (6-((4-hidroxi-8’-metil-1 ',5’-dioxo-1 ',5’-dihidro-2'H-espiro[ciclohexano-1,3’-imidazo[1,5-a]piridin]-6’-il)aminno)pirimidin-4-il)carbamato de terc-butilo (5)
La síntesis del intermedio 5 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento H. Sólido de color blanquecino; Rendimiento: 0,45 g, 81 %; EM (IEN) m/z 457,31 [M 1]+.
S ín te s is de 6 ’- ( (6 -a m in o p ir im id in -4 - il)a m in o )-4 -h id ro x i-8 '-m e til-2 ’H -e s p iro [c ic lo h e x a n o -1 ,3 - im id a z o [1 ,5 -a ]p ir id in ]-1 ’,5 '- diona (Comp. N ° 206)
La síntesis del compuesto 206 se realizó como se ha descrito anteriormente usando el protocolo general del procedimiento F. Sólido de color amarillo como una mezcla de diastereómeros; Rendimiento: 0,049 g, 12 %; EM (IEN) m/z 357,09 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,04 y 9,97 (2 s, 1H cada uno, isómero A y B), 8,58 y 8,56 (2s, 1H cada uno, isómero A y B), 8,38 y 8,35 (2s, 1H cada uno, isómero A y B), 8,16 (s, 1H), 6,51 (s ancho, 2H), 6,15 y 6,14 (2 s, 1H cada uno, isómero A y B), 4,78 y 4,46 (2 brs, 1H cada uno, isómero A & B), 3,86 y 3,53 (2 m, 1H cada uno, isómero A y B), 3,41 y 3,09 (2 m, 2H cada uno, isómero A y B), 2,42 (s, 3H), 1,85-1,56 (m, 4H), 143 y 1,17 (2 m, 2H cada uno, isómero A y B).
Ejemplo 207
Síntesis de 6'-((6-aminopirimidin-4-il)amino)-8'-(hidroximetil)-2-espirociclohexano-1,3'-imidazo[1,5-a]piridin]-1',5'-diona (Comp. N.° 207)

Síntesis de N-(6-(( 1 5’-dioxo-8’-vinil-1' 5’-dihidro-2'H-espiro[ciclohexano-1,3’-imidazo[1,5-a]piridin]-6’-il)amino)pirimidin-4-il)ciclopropanocarboxamida (3)
La síntesis del intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del procedimiento G. sólido de roedor; Rendimiento: 1,5 g, 51 %; EM (IEN) m/z 421,22 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,88 (s, 1H), 10,25 (s, 1H), 9,20 (s, 1H), 8,99 (s, 1H), 8,56 (s, 1H), 7,92-7,84 M), 5,70 (d, J = 17,6 Hz, 1H), 5,34 (d, J = 11,6 Hz, 1H), 3,32 (m, 1H), 2,99-2,93 (m, 2H), 2,05-1,98, 2H), 1,72-1,66 (m, 5H), 1,43-1,40 (m, 2H), 1,34-1,16 (m, 1H), 0,85 (m, 4H).
Síntesis de N-(6-((8'-formil-1 ’,5’-dioxo-1 5’-dihidro-2'H-espiro[ciclohexano-1,3'-imidazo[1,5-a]piridin]-il)amino)pirimidin-4-il)ciclopropanocarboxamida (4)
A una solución agitada de N-(6-((1',5'-dioxo-8'-vinil-1',5'-dihidro-2'H-espiro[ciclohexano-1,3'-imidazo[1,5-a]piridin]-6'-il)amino)pirimidin-4-il)ciclopropanocarboxamida (3, 2,0 g, 14,26 mmol) en dioxano y agua (2:1, 30 ml), se añadió gota a gota una solución de tetraóxido de osmio en butanol (0,60 g, 2,38 mmol) a 0 °C. La masa de reacción se agitó a temperatura ambiente durante una noche. Después de que se terminó la CCF, el disolvente se evaporó a presión reducida y se añadió agua (100 ml). La mezcla se extrajo con metanol al 10 % en diclorometano (50 ml, 2 veces). Los compuestos orgánicos se separaron después y se secaron (sulfato de magnesio) y se concentraron a sequedad al vacío para proporcionar N-(6-((8'-formil-1',5'-dioxo-1',5'-dihidro-2'H-espiro[ciclohexano-1,3'-imidazo[1,5-a]piridin]-il)amino)pirimidin-4-il)ciclopropanocarboxamida (4) en forma de un sólido de color marrón. Rendimiento: 1,2 g, 60 %; EM (IEN) m/z 11,26 [M 1]+.
Síntesis de N-(6-((8’-(hidroximetil)-1' 5’-dioxo-1' 5’-dihidro-2'H-espiro[ciclohexano-1,3’-imidazo[1,5-a]piridin]-6’-il)amino)pirimidin-4-il)ciclopropanocarboxamida (5)
A una solución agitada de N-(6-((8'-formil-1',5'-dioxo-1',5'-dihidro-2'H-espiro[ciclohexano-1,3'-imidazo[1,5-a]piridin]-il)amino)pirimidin-4-il)ciclopropanocarboxamida (1,2 g, 2,85 mmol) en metanol/tetrahidrofurano (1:2, 30 ml), se añadió borohidruro de sodio en porciones a 0 °C. La masa de reacción se agitó a 0 °C durante 2 h. Después de la terminación, la mezcla de reacción se diluyó con agua (100 ml) y la mezcla se extrajo con metanol al 10 % en diclorometano (50 ml, 2 veces). Los compuestos orgánicos se separaron después y se secaron (sulfato de magnesio) y se concentraron a sequedad al vacío para proporcionar N-(6-((8'-(hidroximetil)-1',5'-dioxo-1',5'-dihidro-2'H-espiro[ciclohexano-1,3'-imidazo[1,5-a]piridin]-6'-il)amino)pirimidin-4-il)ciclopropanocarboxamida (5) en forma de un sólido de color marrón. Rendimiento: 0,7 g, 62 %; EM (IEN) m/z 423,43 [M-1 ]-; RMN 1H (400 MHz, DMSO-d6) ó 10,86 (s, 1H), 10,24-10,18 (m, 1H), 8,71-8,62 (m, 1H), 8,56-8,52 (m, 1H), 7,84 (s, 1H), 6,13-5,33 M, 1H), 4,77-4,33 (m, 3H), 3,16-2,84 (m, 4H), 2,01-1,86 (m, 2H), 1,72-1,66 (m, 5H), 1,43-1,40 (m, 2H), 1,34-1,16 (m, 1H).
Síntesis de 6’-((6-aminopirimidin-4-il)amino)-8'-(hidroximetil)-2’H-espiro[ciclohexano-1,3’-imidazo[1,5-a]piridin]-1’,5’-diona (Comp. N.° 207)
La síntesis del compuesto 207 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento I. Sólido de color marrón; Rendimiento: 0,18 g, 29 %; EM (IEN) m/z 357,35 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,23 (s, 1H), 9,23 (m, 1H), 8,47 (m, 1H), 8,33 (s, 1H), 7,25 (s ancho, 2H), 6,27 (s, 1H), 5,18 (s ancho, 1H), 4,81 (s, 2H), 3,01 (m, 2H), 1,73-1,62 (m, 5H), 1,46-1,44 (m, 2H), 1,23 (m, 1H).
Ejemplo 208
Síntesis de 6'-((5-ciclopropilpirimidin-4-il)amino)-8,-metil-2,H-espiroIciclohexano-1,3'-imidazo[1,5-a]piridin]-1',5'-diona (Comp. N.° 208)

Síntesis de 5-ciclopropilpirimidin-4-amina (3)
La síntesis del producto intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento G. Sólido de color marrón; Rendimiento: 0,41 g, 78 %; EM (IEN) m/z 136,08 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 8,02 (s, 1H), 7,81 (s, 1H), 6,75 (s ancho, 2H), 1,57-1,53 (m, 1H) 0,56-0,53 (m, 2H).
Síntesis de 6’-((5-ciclopropilpirimidin-4-il)amino)-8'-metil-2’H-espiro[ciclohexano-1,3’-imidazo[1,5-a]piridin]-1’,5’-diona (Comp. N.° 208)
La síntesis del compuesto 208 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento H. Sólido de color blanquecino; Rendimiento: 0,14 g, 24 %; EM (IEN) m/z 366,20 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,16 (s, 1H), 8,88 (s, 1H), 8,72 (s, 1H), 8,62 (s, 1H), 8,29 (s, 1H), 3,01-2,96 (m, 2H), 2,5 (s, 3H), 1,84-1,59 (m, 6H), 1,48-1,45 (m, 2H), 1,30-1,27 (m, 1H), 1,05-1,01 (m, 2H), 0,73-0,69 (m, 2H).
Ejemplo 209
Síntesis de 8'-metil-6,-(pirimidin-4-iloxi)-2'H-espiro[ciclohexano-1,3'-imidazo[1,5-a]piridin]-1,,5'-diona (Comp. N.° 209)

Síntesis de 6’-((4-metoxibencil) oxi)-8'-metil-2’H-espiro[ciclohexano-1,3-imidazo[1,5-a]piridin]-15’-diona (2)
A una solución de 6'-bromo-8'-metil-2'H-espiro[ciclohexano-1,3-imidazo[1,5-a]piridin]-1',5'-diona (2 g, 6,43 mmol) en dimetilformamida (30 ml) se le añadió hidruro de sodio (0,46 g, 19,29 mmol) y alcohol 4-metoxibencílico (3,19 ml, 25,72 mmol). La reacción se agita a temperatura ambiente durante la noche. La mezcla resultante se vierte en agua helada y se extrae con diclorometano. La capa orgánica se seca sobre sulfato de magnesio, se filtró y se concentró. El producto en bruto se purifica por cromatografía en columna para proporcionar 6'-((4-metoxibencil)oxi)-8'-metil-2'H-espiro[ciclohexano-1,3-imidazo[1,5-a]piridin]-1',5'-diona (2).
Síntesis de 6’-hidroxi-8'-metil-2’H-espiro[ciclohexano-1,3’-imidazol[1,5-a]piridin]-1 ',5’-diona (3)
A una solución de 6'-((4-metoxibencil)oxi)-8'-metil-2'H-espiro[ciclohexano-1,3'-imidazo[1,5-a]piridin]-1',5'-diona (2, 1,5 g, 4,07 mmol) en diclorometano (20 ml) se añade 2,3-dicloro-5,6-diciano-p-benzoquinona (1,38 g, 6,10 mmol). La reacción se agita a temperatura ambiente durante 2 h. La mezcla resultante se concentra y purifica por cromatografía en columna para proporcionar 6'-hidroxi-8'-metil-2'H-espiro[ciclohexano-1,3'-imidazo[1,5-a]piridin]-1',5'-diona (3).
Síntesis de 8’-metil-6'-(pirimidin-4-iloxi)-2’H-espiro[ciclohexano-1,3'-imidazo[1,5-a]piridin]-1’,5'-diona (Comp. N.° 209)
A una solución de 6'-hidroxi-8'-metil-2'H-espiro[ciclohexano-1,3-imidazo[1,5-a]piridin]-1',5'-diona (3, 1 g, 4,03 mmol) en dimetilacetamida (20 ml) se le añadió carbonato de potasio (1,67 g, 12,09 mmol) y 4-bromopirimidina (4, 0,77 g, 4,84 mmol). La reacción se agita a 130 °C durante una noche. La mezcla resultante se enfría a temperatura ambiente, se vierte en agua y se extrae con diclorometano. La capa orgánica se seca sobre sulfato de magnesio, se filtra y se concentra. El producto en bruto se purifica por cromatografía en columna para proporcionar 8'-metil-6'-(pirimidin-4-iloxi)-2'H-espiro[ciclohexano-1,3'-imidazo[1,5-a]piridin]-1',5'-diona (Comp. N.° 209).
Ejemplo 210
Síntesis de 8'-metil-6'-(pirimidin-4-iltio)-2'H-espiro[ciclohexano-1,3'-imidazo[1,5-ajpiridin-1 ',5'-diona (Comp. N.° 210)

Síntesis de 6’-((4-metoxibencil)tio)-8'-metil-2’H-espiro[ciclohexano-1,3-imidazo[1,5-a]piridin]-1',5’-diona (3)
A una solución de 6'-bromo-8'-metil-2'H-espiro[ciclohexano-1,3-imidazo[1,5-a]piridin]-1',5'-diona (1, 2 g, 6,43 mmol) en dimetilformamida (30 ml) se le añaden carbonato de cesio (6,28 g, 19,29 mmol) y (4-metoxifenil)metanotiol (2,19 g, 7,72 mmol). La reacción se agita a 100 °C durante una noche. La mezcla resultante se enfría a temperatura ambiente, se vierte en agua y se extrae con diclorometano. La capa orgánica se seca sobre sulfato de magnesio, se filtra y se concentra. El producto en bruto se purifica por cromatografía en columna para proporcionar 6'-((4-metoxibencil) tio)-8'-metil-2'H-espiro[ciclohexano-1,3-imidazo[1,5-a]piridin]-1',5'-diona (3).
Síntesis de 6'-mercapto-8’-metil-2'H-espiro[ciclohexano-1,3’-imidazo[1,5-a]piridin]-1’,5’-diona (4)
A una solución de 6'-((4-metoxibencil)tio)-8'-metil-2'H-espiro[ciclohexano-1,3-imidazo[1,5-a]piridin]-1',5'-diona (3, 3,3 g, 8,58 mmol) en cloroformo (40 ml) se le añade ácido metanosulfónico (3 ml, 46,23 mmol). La reacción se agita a 50 °C durante una noche. La mezcla resultante se enfría a temperatura ambiente, se vierte en agua y se extrae con diclorometano. La capa orgánica se seca sobre sulfato de magnesio, se filtra y se concentra. El producto en bruto se purifica por cromatografía en columna para proporcionar 6'-mercapto-8'-metil-2'H-espiro[ciclohexano-1,3-imidazo[1,5-a]piridin]-1',5'-diona (4).
Síntesis de 6’-((2-cloropirimidin-4-il)tio)-8'-metil-2’H-espiro[ciclohexano-1,3'-imidazo[1,5-a]piridin]-1' 5’-diona (6) A una solución de 6'-mercapto-8'-metil-2'H-espiro[ciclohexano-1,3-imidazo[1,5-a]piridin]-1',5'-diona (4, 0,50 g, 1,89 mmol) en 2-propanol (10 ml) se le añadió N,N-diisopropiletilamina (0,99 ml, 5,67 mmol) y 2,4-dicloropirimidina (5, 0,34 g, 2,27 mmol). La reacción se agita a 70 °C durante una noche. La mezcla resultante se enfría a temperatura ambiente, se vierte en agua y se extrae con diclorometano. La capa orgánica se seca sobre sulfato de magnesio, se filtra y se concentra. El producto en bruto se purifica por cromatografía en columna para proporcionar 6'-((2-cloropirimidin-4-il)tio)-8'-metil-2'H-espiro[ciclohexano-1,3'-imidazo[1,5-a]piridin]-1',5'-diona (6).
S ín te s is de 8 ’-m e til-6 '-(p ir im id in -4 - ilt io )-2 -e s p iro [c ic lo h e x a n o -1 ,3 ’- im id a z o [1 ,5 -a ]p ir id in ]-1 ’, 5 ’-d io n a (C om p. N .° 210) A una solución de 6’-((2-cloropirimidin-4-il)tio)-8'-metil-2'H-espiro[ciclohexano-1,3-imidazo[1,5-a]piridin]-1’,5'-diona (6, 0,3 g, 0,80 mmol) en ácido acético (4 ml) se le añade pareja de cobre de cinc (0,5 g). La reacción se agita a reflujo durante 4 h. La mezcla resultante se enfría a temperatura ambiente, se filtra y se concentra. El producto en bruto se purifica por cromatografía en columna para proporcionar 8’-metil-6'-(pirimidin-4-iltio)-2’H-espiro[ciclohexano-1,3’-imidazo[1,5-a]piridin]-1',5'-diona (Comp. N.° 210).
Ejemplo 211
Síntesis de 8'-metil-6,-(pirimidin-4-carbonil)-2'H-espiro[ciclohexano-1,3'-imidazo[1,5-a]piridin]-1',5,-diona (Comp. N.° 211)

Síntesis de 8’-metil-6'-(pirimidin-4-carbonil)-2’H-espiro[ciclohexano-1,3-imidazo[1,5-a]piridin]-1',5’-diona (Comp. N° 211)
A una solución de 6'-bromo-8'-metil-2'H-espiro[ciclohexano-1,3-imidazo[1,5-a]piridin]-1’,5'-diona (2, 0,3 g, 0,96 mmol) en tetrahidrofurano (25 ml), se añadió n-butil-litio (0,58 g, 2,89 mmol) a -78 °C. La mezcla de reacción se agitó durante 30 min. A la mezcla se le añadió N-metoxi-N-metilpirimidin-4-carboxamida (1,25 g, 1,44 mmol) a -78 °C y después la mezcla se agitó a temperatura ambiente durante 16 h. Después de la terminación, la reacción se inactivó con solución acuosa de cloruro de amonio (50 ml) y se extrajo con diclorometano (50 ml, 2 veces). La capa orgánica se separó y se secó con salmuera (25 ml) y el disolvente se evaporó a presión reducida. El producto en bruto se purificó por cromatografía en columna de gel de sílice (malla 220-400) usando metanol al 0,5 % en diclorometano como eluyente. Las fracciones se concentraron para proporcionar 8’-metil-6'-(pirimidin-4-carbonil)-2’H-espiro[ciclohexano-1,3-imidazo[1,5-a]piridin]-1’,5’-diona en forma de un sólido de color amarillo. Rendimiento: 0,001 g, 3,0 %; EM (IEN) m/z 339,13 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,58 (s, 1H), 9,26 (s, 1H), 9,07 (d, J = 5,08 Hz, 1H), 9,97 (s, 1H), 7,83 (d, J = 5,2 Hz, 1H), 2,85-2,75 (m, 2H), 2,45 (s, 3H), 1,68-162 (m, 2H), 1,60-1,53 (m, 3H), 1,46-1,43 (m, 2H), 1,10- 1,09 (m, 1H).
Ejemplo 212
Síntesis de 8'-metil-6'-(pirimidin-4-ilmetil)-2'H-espiro[ciclohexano-1,3'-imidazo[1,5-a]piridin]-1',5'-diona (Comp. N.° 212)


Síntesis de 8’-metil-6'-(pirimidin-4-carbonil)-2’H-espiro[ciclohexano-1,3’-imidazo[1,5-a]piridin]-1’,5’-diona (3)
A una solución de 6'-bromo-8'-metil-2'H-espiro[ciclohexano-1,3-imidazo[1,5-a]piridin]-1',5'-diona (2, 2 g, 6,43 mmol) en tetrahidrofurano (30 ml) a -78 °C se añade gota a gota n-butil-litio (1,6 M en hexanos, 12,06 ml, 19,29 mmol), seguido de N-metoxi-N-metilpirimidin-4-carboxamida (1, 1,29 g, 7,72 mmol). La reacción se calienta lentamente a temperatura ambiente y se agita durante 4 h. La reacción se inactiva mediante la adición lenta de agua. La mezcla se calienta a temperatura ambiente y se extrae con acetato de etilo. La capa orgánica se seca sobre sulfato de magnesio, se filtra y se concentra. El producto en bruto se purifica por cromatografía en columna para proporcionar 8'-metil-6'-(pirimidin-4-carbonil)-2'H-espiro[ciclohexano-1,3'-imidazo[1,5-a]piridin]-1',5'-diona (3).
Síntesis de 6’-(hidrazono(pirimidin-4-il)metil)-8'-metil-2’H-espiro[ciclohexano-1,3’-imidazo[1,5-a]piridin]-1' 5’-diona (4)
A una solución de 8'-metil-6'-(pirimidin-4-carbonil)-2'H-espiro[ciclohexano-1,3'-imidazo[1,5-a]piridin]-1',5'-diona (3, 0,7 g, 2,07 mmol) en etanol (4 ml), ácido acético (4 ml) y agua (4 ml) se le añadió hidrato de hidrazina (0,13 g, 4,14 mmol). La reacción se agita a 80 °C durante una noche. La reacción se enfría a temperatura ambiente y se concentra. El material en bruto se resuspende en diclorometano y se lava con solución acuosa saturada de bicarbonato de sodio. La capa orgánica se seca sobre sulfato de magnesio, se filtra y se concentra. El producto en bruto se purifica por cromatografía en columna para proporcionar 6'-(hidrazono(pirimidin-4-il)metil)-8'-metil-2'H-espiro[ciclohexano-1,3'-imidazo[1,5-a]piridin]-1 ',5'-diona (4).
Síntesis de 8’-metil-6'-(pirimidin-4-ilmetil)-2-espiro[ciclohexano-1,3’-imidazo[1,5-a]piridin]-1’,5’-diona (Comp. N ° 212)
A una solución de 6'-(hidrazono(pirimidin-4-il)metil)-8'-metil-2'H-espiro[ciclohexano-1,3'-imidazo[1,5-a]piridin]-1',5'-diona (4, 100 mg, 0,28 mmol) en tolueno (4 ml) se le añadió terc-butóxido de potasio (94 mg, 0,84 mmol). La reacción se refluye toda la noche. La mezcla resultante se enfría a temperatura ambiente, se diluye con diclorometano y se trata con solución de cloruro de amonio 1 M. La capa orgánica se seca sobre sulfato de magnesio, se filtra y se concentra. El producto en bruto se purifica por HPLC para proporcionar 8'-metil-6'-(pirimidin-4-ilmetil)-2'H-espiro[ciclohexano-1,3'-imidazo[1,5-a]piridin]-1',5'-diona (Comp. N.° 212).
Ejemplo 213
Síntesis de 5-((6-aminopirimidin-4-il)amino)-3-cloro-1-isobutil-6-oxo-1,6-dihidropiridin-2-carboxamida (Comp. N.° 213)


Síntesis de 5-bromo-3-cloro-1-isobutil-6-oxo-1,6-dihidropiridin-2-carboxilato de etilo (3)
A una solución de 5-bromo-3-cloro-6-oxo-1,6-dihidropiridin-2-carboxilato de etilo (1, 1,0 g, 3,56 mmol) y 1-yodo-2-metilpropano (2, 1,31 g, 7,13 mmol) en dimetilformamida (12 ml) en un vial, se le añadió carbonato de potasio (261 mg, 1,89 mmol) y la mezcla se agitó a temperatura ambiente durante 16 h. Después de la terminación, la masa de reacción se diluyó con agua (50 ml) y se extrajo con acetato de etilo (50 ml, 2 veces). La capa orgánica combinada se lavó con agua, salmuera, se secó sobre sulfato de sodio anhidro y se concentró al vacío. El producto en bruto se purificó mediante cromatografía en columna de gel de sílice (malla 220-400) usando acetato de etilo al 50 % en hexano como eluyente para producir 5-bromo-3-cloro-1-isobutil-6-oxo-1,6-dihidropiridin- 2-carboxilato de etilo (3) en forma de un sólido de color amarillo claro. Rendimiento: 0,70 g, 58 %; e M (IEN) m/z 336,3 [M 1]+. Síntesis de 5-((6-((terc-butoxicarbonil)amino)pirimidin-4-il)amino)-3-cloro-1-isobutil-6-oxo-1,6-dihidropiridin-2-carboxilato de etilo (5)
La síntesis del intermedio 5 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento H. Sólido de color blanquecino; Rendimiento: 0,36 g, 40 %; EM (IEN) m/z 466,2 [M 1]+.
Síntesis de 5-((6-aminopirimidin-4-il)amino)-3-cloro-1-isobutil-6-oxo-1,6-dihidropiridin-2-carboxamida (Comp. N.° 213) A una solución de 5-((6-((terc-butoxicarbonil)amino)pirimidin-4-il)amino)-3-cloro-1-isobutil-6-oxo-1,6-dihidropiridin-2-carboxilato de etilo (5, 300 mg, 0,64 mmol) en metanol (20 ml) se le añadió nitruro de magnesio (3,25 g, 3,21 mmol) y la reacción se calentó a reflujo durante 16 h. Después de la terminación, el disolvente se retiró a presión reducida y el residuo resultante se agitó en ácido clorhídrico 2 N durante 10 m. La mezcla de reacción se filtró y el sólido obtenido se secó a presión reducida. El producto en bruto se purificó mediante purificación previa, para proporcionar 5-((6-aminopirimidin-4-il)amino)-3-cloro-1-isobutil-6-oxo-1,6-dihidropiridin-2-carboxamida (Comp. N.° 213) en forma de un sólido de color blanquecino. Rendimiento: 70 mg, 32 %; EM (IEN) m/z 336,99 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 9,46 (s, 1H), 8,33 (s, 1H), 8,27 (s, 1H), 7,87 (s, 1H), 7,59-7,54 (m, 3H), 6,05 (s, 1H), 4,17-4,16 (d, J = 6,8 Hz, 2H), 2,08-2,01 (m, 1H), 0,95-0,93 (d, J = 6,8 Hz, 6H).
Ejemplo 214
Síntesis de 5-((6-aminopirimidin-4-il)amino)-3-cloro-6-oxo-1,6-dihidropiridin-2-carboxamida (Comp. N.° 214)

Síntesis de 5-bromo-3-cloro-1-(4-metoxibencil)-6-oxo-1,6-dihidropiridin-2-carboxilato de etilo (2)
Una solución de 5-bromo-3-cloro-6-oxo-1,6-dihidropiridin-2-carboxilato de etilo (3,0 g, 10,7 mmol), cloruro de 4-metoxibutilo (4,19, 26,74 mmol) y carbonato de potasio 4,43 g, 32,09 mmol) en dimetilformamida (40 ml) se agitó a temperatura ambiente durante 16 h. Después de la terminación, la mezcla de reacción se diluyó con agua fría (20 ml) y se extrajo con acetato de etilo (30 ml, 3 veces). La capa orgánica se lavó de nuevo con salmuera, se separó, se secó sobre sulfato de sodio, se filtró y se concentró a presión reducida. El residuo se purificó finalmente por cromatografía en columna ultrarrápida para proporcionar 5-bromo-3-cloro-1-(4-metoxibencil)-6-oxo-1,6-dihidropiridin-2-carboxilato de etilo (2) en forma de un sólido de color blanquecino. Rendimiento: 1,2 g, 28 %; EM (IEN) m/z 399,99 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 8,27 (s, 1H), 7,11 (d, J = 8,64 Hz, 2H), 6,89 (d, J = 8,6 Hz, 2H), 5,11 (s, 2H), 4,27 (c, J = 7,12 Hz, 2H), 3,72 (s, 3H), 1,13 (t, J = 7,12 Hz, 3H).
Síntesis de 5-((6-((terc-butoxicarbonil)amino)pirimidin-4-il)amino)-3-cloro-1-(4-metoxibencil)-6-oxo-1,6-dihidropiridin-2-carboxilato de etilo (4)
La síntesis del intermedio 4 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento H. Sólido de color marrón; Rendimiento: 0,70 g, 44 %; EM (IEN) m/z 530,17 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,04 (s, 1H), 9,49 (s, 1H), 8,64 (s, 1H), 8,50 (s, 1H), 7,76 (s, 1H), 7,11 (d, J = 8,2 Hz, 2H), 6,89 (d, J = 8,44 Hz, 2H), 5,21 (s, 2H), 4,23 (c, J = 6,92 Hz, 2H), 3,72 (s, 3H), 1,48 (s, 9H), 1,13 (t, J = 7,04 Hz, 3H). Síntesis de ácido (5)-5-((6-aminopirimidin-4-il)amino)-3-cloro-1-(4-metoxibencil)-6-oxo-1,6-dihidropiridin-2-carboxílico (5)
Se añadió hidróxido de sodio (0,26 g, 6,6 mmol) a una suspensión de 5-((6-((terc-butoxicarbonil)amino)pirimidin-4-il)amino)-3-cloro-1-(4-metoxibencil)-6-oxo-1,6-dihidropiridin-2-carboxilato de etilo (0,70 g, 1,32 mmol) en metanol/tetrahidrofurano/agua (2:1:1, 30 ml). La mezcla se agitó a 80 °C durante 16 h. Después de la terminación, el disolvente se evaporó a sequedad a presión reducida. El producto en bruto se diluyó con ácido clorhídrico 1 N. El precipitado obtenido se recogió por filtración, se secó, se lavó con pentano y se secó adicionalmente para proporcionar ácido 5-((6-aminopirimidin-4-il)amino)-3-cloro-1-(4-metoxibencil)-6-oxo-1,6-dihidropiridin-2-carboxílico (5) en forma de un sólido de color marrón. Rendimiento: 0,45 g, 85 %; EM (IEN) m/z 402,09 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 9,62 (s, 1H), 8,43 (s, 1H), 8,33 (s, 1H), 7,67 (s ancho, 2H), 7,19 (d, J = 8,16 Hz, 2H), 6,87 (d, J = 8,12 Hz, 2H), 6,39 (s, 1H), 5,18 (s, 2H), 3,71 (s, 3H).
Síntesis de 6-aminopirimidin-4-il)amino)-3-cloro-N, 1-bis(4-metoxibencil)-6-oxo-1,6-dihidropiridin-2-carboxamida (6)
A una solución de ácido 5-((6-aminopirimidin-4-il)amino)-3-cloro-1-(4-metoxibencil)-6-oxo-1,6-dihidropiridin-2-carboxílico (5, 0,40 g, 0,99 mmol) y 4-metoxibencilamina (0,16 g, 1,19 mmol) en dimetilformamida (20 ml) se le añadió N,N-diisopropiletilamina (0,39 g, 2,99 mmol) y HATU (0,57 g, 1,99 mmol) a temperatura ambiente. La mezcla de reacción se agitó durante 40 h. El progreso de la reacción se controló mediante c Le M. A la mezcla de reacción se le añadió tetrahidrofurano (20 ml) y la reacción se calentó a reflujo durante 7 h. Después de la terminación, la reacción se diluyó con agua (20 ml) y se extrajo con metanol al 10 % en diclorometano (30 ml, 2 veces). La capa orgánica combinada se secó sobre sulfato de sodio, se filtró y se evaporó a presión reducida para obtener el producto en bruto. El producto en bruto se purificó por cromatografía en columna ultrarrápida eluyendo con metanol al 1 % en diclorometano. Las mejores fracciones se concentraron para proporcionar 5-((6-aminopirimidin-4-il)amino)-3-cloro-N,1-bis(4-metoxibencil)-6-oxo-1,6-dihidropiridin-2-carboxamida (6) en forma de un sólido de color amarillo. Rendimiento: 0,44 g, 84 %; EM (IEN) m/z 521,16 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 9,35 (m, 1H), 8,74 (s, 1H), 8,53 (s, 1H), 8,17 (s, 1H), 7,19-7,16 (m, 4H), 6,87-6,80 (m, 4H), 6,53 (s, 2H), 6,17 (s, 1H), 5,07 (s, 2H), 4,34 (s, 2H), 3,73 (s, 6H).
Síntesis de 5-((6-aminopirimidin-4-il)amino)-3-cloro-6-oxo-1,6-dihidropiridin-2-carboxamida (Comp. N.° 214)
A una solución de 5-((6-aminopirimidin-4-il)amino)-3-cloro-N,1-bis(4-metoxibencil)-6-oxo-1,6-dihidropiridin-2-carboxamida (6, 0,40 g, 0,77 mmol) en diclorometano (10 ml) se le añadió ácido trifluoroacético (20 ml) y ácido trifluorometanosulfónico (1 ml) a 0 °C. La mezcla de reacción se agitó a 70 °C durante 2 h. Después de la terminación, la mezcla de reacción se concentró y basificó mediante acetato de etilo acuoso. Amoníaco a 0 °C. El precipitado obtenido se recogió por filtración, se lavó con agua y se secó para obtener el producto en bruto. El producto en bruto se agitó con metanol, se filtró, se lavó con pentano y se secó para proporcionar 5-((6-aminopirimidin-4-il)amino)-3-cloro-6-oxo-1,6-dihidropiridin-2-carboxamida (Comp. N.° 214) en forma de un sólido de color amarillo. Rendimiento: 0,035 g, 16 %; EM (IEN) m/z 281,05 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 12,02 (s, 1H), 8,66 (s, 1H), 8,50 (s, 1H), 8,17 (s, 1H), 7,92 (s, 1H), 7,86 (s, 1H), 6,53 (s, 2H), 6,18 (s, 1H).
Ejemplo 215
Síntesis de 3'-(pirimidin-4-ilamino)-4,H-espiro[ciclohexano-1,6,-imidazo[1,5-a]pirimidin]-4',8'(7'H)-diona (Comp. N.° 215)


XPhos. XantPhos
Pd(OAc)2, Pd2(dba)3


95 C
Síntesis de 5-bromopirimidin-2-carboxilato de etilo (2)
A una solución de ácido 5-bromopirimidin-2-carboxílico (1,96 g, 19,8 mmol) en etanol (70 ml) a temperatura ambiente se le añadió ácido sulfúrico (0,5 ml). La mezcla de reacción se calentó a 80 °C durante 16 h. El análisis por CCF mostró el consumo de material de partida. Se concentró la mezcla de reacción a presión reducida para proporcionar un residuo que se diluyó con agua (30 ml) y se extrajo con acetato de etilo (100 ml, 2 veces). La capa orgánica combinada se lavó con solución de bicarbonato de sodio (50 ml) y después se lavó con salmuera (50 ml), se secó sobre sulfato de sodio anhidro, se filtró y se concentró a presión reducida para proporcionar 5-bromopirimidin-2-carboxilato de etilo. Sólido de color blanquecino. Rendimiento: 3,5 g, 77 %; EM (IEN) m/z 230,91 [M 1]+.
Síntesis de 1-óxido de 5-bromo-2-(etoxicarbonil)pirimidina (3)
A una solución enfriada a 0 °C de 1-etil-5-bromopirimidin-2-carboxilato de 1 -etilo (2, 1,5 g, 6,5 mmol) en diclorometano (30 ml), se le añadieron anhídrido trifluoroacético (13,69 g, 65 mmol) y peróxido de hidrógeno de urea (6,1 g, 65 mmol). La mezcla de reacción se agitó a temperatura ambiente durante 16 h. El análisis por CCF mostrara el consumo de material de partida, la mezcla de reacción se diluyó con agua (10 ml) y se neutralizó con bicarbonato de sodio sólido. La solución se extrajo con diclorometano (40 ml, 2 veces). La capa orgánica se secó sobre sulfato de sodio anhidro, se filtró y se concentró a presión reducida para proporcionar 1-óxido de 5-bromo-2-(etoxicarbonil)pirimidina (3) como líquido amarillo que se usó sin purificación adicional. Rendimiento: 0,64 g, producto en bruto; EM (IEN) m/z 247,13 [M 1]+.
Síntesis de 5-bromo-6-oxo-1,6-dihidropirimidin-2-carboxilato de etilo (4)
A una solución enfriada a 0 °C de 1-óxido de 5-bromo-2-(etoxicarbonil)pirimidina (3, 0,62 g, 2,5 mmol) en dimetilformamida (6 ml), se le añadió gota a gota anhídrido trifluoroacético (3,1 g, 15 mmol). La mezcla de reacción se calentó a 50 °C durante 18 h. El análisis por CCF mostró el consumo de material de partida y el disolvente se retiró a presión reducida. El residuo se trituró con metanol (2 ml) y se filtró. El sólido se lavó con éter dietílico y se secó a presión reducida para proporcionar 5-bromo-6-oxo-1,6-dihidropirimidin-2-carboxilato de etilo (4) en forma de un sólido de color blanquecino. Rendimiento: 0,21 g, 34 %; EM (IEN) m/z 245,09 [M- 1]'.
Síntesis de 5-bromo-6-oxo-1,6-dihidropirimidin-2-carboxamida (5)
A una solución de 5-bromo-6-oxo-1,6-dihidropirimidin-2-carboxilato de etilo (4, 0,2 g, 0,81 mmol) en etanol (4 ml), se le añadió amoníaco líquido (4 ml) gota a gota a temperatura ambiente. La mezcla de reacción se calentó a 50 °C durante 16 h. El análisis por CCF mostró el consumo de material de partida. El disolvente se retiró a presión reducida y el residuo se trató con metanol (1 ml) y se filtró. El sólido se lavó con éter dietílico y se secó a presión reducida para proporcionar 5-bromo-6-oxo-1,6-dihidropirimidin-2-carboxamida (5) en forma de un sólido de color blanquecino. Rendimiento: 0,14 g, 76 %; EM (IEN) m/z 218,87 [M 1]+.
Síntesis de 3’-bromo-4-espiro[ciclohexano-1,6'-imidazo[1,5-a]pirimidin]-4’,8'(7’H)-diona (7)
La síntesis del intermedio 7 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento A. Sólido de color blanquecino; Rendimiento: 0,085 g, 50 %; RMN 1H (400 MHz, DMSO-d6) ó 10,96 (s, 1H), 8,55 (s, 1H), 6,94-7,20 (m, 2H), 3,36 (s, 1H), 2,65-2,71 (m, 2H) 1,61-1,73 (m, 2H), 1,19-1,22 (m, 1H).
Síntesis de 3’-(pirimidin-4-ilamino)-4'H-espiro[ciclohexano-1,6'-imidazo[1,5-a]pirimidin]-4',8’(7'H)-diona (Comp. N.° 215)
La síntesis del compuesto 215 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento H. Sólido de color amarillo; Rendimiento: 0,014 g, 17 %; EM (IEN) m/z 313,14 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,77 (s, 1H), 9,42 (s, 1H), 9,34 (s, 1H), 8,77 (s, 1H), 8,38 (s, 1H), 7,35 (s, 1H), 2,85-2,72 (m, 2H), 1,85-1,54 (m, 7H), 1,30-1,20 (m, 1H).
Ejemplo 216
Síntesis de 8'-metil-6'-(pirimidin-4-ilamino)-2'H-espiro[ciclohexano-1,3'-imidazo[1,5-a]piridin]-1 ',5'-diona (Comp. N.° 216)


Síntesis de 5-bromo-3-metil-6-oxo-1,6-dihidropirazin-2-carboxilato de metilo (2)
Se disolvió 3-metil-6-oxo-1,6-dihidropirazin-2-carboxilato de metilo (2,0 g, 11,89 mmol) en diclorometano (30 ml) y se añadió N-bromosuccinimida (2,12 g, 1,89 mmol). La reacción se dejó agitar a temperatura ambiente durante 16 h. Al finalizar, la mezcla de reacción se lavó con agua y salmuera, se secó sobre sulfato de sodio anhidro, se filtró y se concentró. El sólido se lavó con éter para proporcionar 5-bromo-3-metil-6-oxo-1,6-dihidropirazin-2-carboxilato de metilo (2) en forma de un sólido de color marrón claro. Rendimiento: 1,25 g, 42 %; EM (IEN) m/z 247,04 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 12,92 (s ancho, 1H), 3,87 (s, 4H), 2,50 (s, 3H).
Síntesis de 3-metil-6-oxo-5-(pirimidin-4-ilamino)-1,6-dihidropirazin-2-carboxilato de metilo (4)
La síntesis del intermedio 4 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento H. Sólido de color amarillo; Rendimiento: 500 mg, 48 %; EM (IEN) m/z 262,22 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 12,16 (s ancho, 1H), 9,14 (s, 1H), 8,69-8,68 (d, J = 5,6 Hz, 1H), 8,49-8,47 (d, J = 5,6 Hz, 1H), 3,80 (s, 3H), 2,50 (s, 3 H).
Síntesis de 3-metil-6-oxo-5-(pirimidin-4-ilamino)-1,6-dihidropirazin-2-carboxamida (5)
La síntesis del intermedio 5 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento K. Sólido de color marrón. Rendimiento: 350 mg, 74 %; EM (IEN) m/z 247,01 [M 1]+.
Síntesis de 8’-metil-6'-(pirimidin-4-ilamino)-2’H-espiro[ciclohexano-1,3’-imidazo[1,5-a]piridin]-1’,5’-diona (Comp. N.° 216)
A una solución de 3-metil-6-oxo-5-(pirimidin-4-ilamino)-1,6-dihidropirazin-2-carboxamida (5, 100 mg, 0,41 mmol) y ciclohexanona (199 mg, 2,03 mmol) en acetonitrilo en un vial de microondas de 20 ml se le añadió cloruro de hierro (III) (197 mg, 1,21 mmol). La reacción se calentó a 80 °C durante 16 h. Una vez completada la reacción, el disolvente se retiró al vacío y el producto en bruto se purificó por cromatografía en columna de gel de sílice (200-400 malla) eluyendo con metanol al 5 % en diclorometano. Las fracciones apropiadas de la columna se concentraron a presión reducida para proporcionar 8'-metil-6'-(pirimidin-4-ilamino)-2'H-espiro[ciclohexano-1,3'-imidazo[1,5-a]pirazin]-1',5'-diona (Comp. N.° 216) en forma de un sólido de color blanquecino. Rendimiento: 8,5 mg, 6 %; e M (IEN) m/z 327,13 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,17 (s, 1H), 9,11 (s, 1H), 8,88 (s, 1H), 8,72 (d, J = 4,0 Hz 1H), 8,47 (d, J = Hz, 1H), 2,85-2,74 (m, 2H), 2,54 (s, 3H), 1,80-1,50 (m, 7H), 1,30-120 (m, 1H).
Ejemplo 217
Síntesis de 8-((6-aminopirimidin-4-il)amino)-10-metil-2,3,4,5-tetrahidropirido[1,2-a][1,4]diazepin-1,7-diona (Comp. N.° 217)

Síntesis de N-bencil-3-((terc-butildimetilsilil)oxi)propan-1-amina (2)
A una solución agitada de 3-(bencilamino)propanol (1, 2,0 g, 12,1 mmol) en diclorometano (20 ml), se le añadieron imidazol (2,47 g, 36,0 mmol) y cloruro de terc-butildimetilsililo (1,1 g, 3,9 mmol) a temperatura ambiente. La masa de reacción se agitó a temperatura ambiente durante 16 h. Se añadió agua a la mezcla de reacción y se separaron las capas. La capa orgánica se secó sobre sulfato de sodio, se filtró y se concentró para proporcionar N-bencil-3-((tercbutildimetilsilil)oxi)propan-1-amina (2) en forma de un sólido de color marrón. Rendimiento: 3,2 g, 94 %; EM (IEN) m/z 280,29 [M-1]-.
Síntesis de N-bencil-5-bromo-N-(3-((terc-butildimetilsilil)oxi)propil)-3-metil-6-oxo-1,6-dihidropiridin-2-carboxamida (4) A una solución agitada de N-bencil-3-(terc-butildimetilsililoxi)propan-1-amina (2, 3,0 g, 12,9 mmol) en dimetilformamida (50 ml), se le añadieron a temperatura ambiente 5-bromo-3-metil (2, 3,0 g, 12,9 mmol) en dimetilformamida (50 ml), ácido 5-bromo-3-metil-6-oxo-1,6-dihidropiridin-2-carboxílico (3, 3,6 g, 12,9 mmol), HBTU (6,4 g, 16,9 mmol) y N,N-diisopropiletilamina (2,2 g, 16,9 mmol). La mezcla se agitó durante 16 h. El análisis por CCF mostró la finalización de la reacción. La mezcla de reacción se inactivó con una solución acuosa de bicarbonato de sodio y se extrajo con acetato de etilo (250 ml). La capa orgánica se secó sobre sulfato de sodio, se filtró y se concentró para proporcionar N-bencil-5-bromo-N-(3-((terc-butildimetilsilil)oxi)propil)-3-metil-6-oxo-1,6-dihidropiridin-2-carboxamida (4) en forma de un líquido de color amarillo. Rendimiento: 3,0 g, 47 %; EM (IEN) m/z 495,24 [M-1]'. Síntesis de N-bencil-5-bromo-N-(3-hidroxipropil)-3-metil-6-oxo-1,6-dihidropiridin-2-carboxamida (5)
A una solución agitada de N-bencil-5-bromo-N-(3-(terc-butildimetilsilil)oxi)propil)-3-metil-6-oxo-1,6-dihidropiridin-2-carboxamida (4, 3,0 g, 6,0 mmol) en dioxano (20 ml), se le añadió ácido clorhídrico en dioxano (20 ml) a temperatura ambiente. La mezcla se agitó durante 16 h. Después de la terminación, se retiró el disolvente. El residuo se diluyó con bicarbonato de sodio acuoso y se extrajo con metanol al 5 %/diclorometano (200 ml, 3 veces). La capa orgánica se secó sobre sulfato de sodio, se filtró y se concentró para obtener N-bencil-5-bromo-N-(3-hidroxipropil)-3-metil-6-oxo-1,6-dihidropiridin-2-carboxamida (5) Como líquido marrón. Rendimiento: 2,2 g, 95 %; EM (IEN) m/z 381,22 [M-1 ]-.
Síntesis de 2-bencil-8-bromo-10-metil-2,3,4,5-tetrahidropirido[1,2-a][1,4]diazepin-1,7-diona (6)
A una solución agitada de N-bencil-5-bromo-N-(3-hidroxipropil)-3-metil-6-oxo-1,6-dihidropiridin-2-carboxamida (5, 1,5 g, 3,9 mmol) en tetrahidrofurano (30 ml), se le añadieron trifenilfosfina (1,5 g, 5,9 mmol) y azodicarboxilato de diisopropilo (1,2 g, 5,9 mmol) a 0 °C. La mezcla se agitó a temperatura ambiente durante 16 h. Después de la terminación, el disolvente se retiró a presión reducida y el producto en bruto se purificó por medio de cromatografía eluyendo con acetato de etilo al 40 % en hexano. Las fracciones apropiadas se concentraron a presión reducida para proporcionar 2-bencil-8-bromo-10-metil-2,3,4,5-tetrahidropirido[1,2-a][1,4]diazepin-1,7-diona (6) en forma de un sólido de color amarillo. Rendimiento: 0,9 g, 64 %; EM (IEN) m/z 361,18 [M 1]+.
Síntesis de 8-((6-(di-(terc-butoxicarbonil)-amino)pirimidin-4-il)amino)-2-bencil-1-metil-2,3,4,5-tetrahidropirido[1,2-a][1,4]diazepin-1,7-diona (8)
La síntesis del intermedio 8 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento H. Sólido de color amarillo; Rendimiento: 0,9 g, 69 %; EM (IEN) m/z 591,66 [M 1 ]+.
Síntesis de 8-((6-aminopirimidin-4-il)amino)-2-bencil-10-metil-2,3,4,5-tetrahidropirido[1,2-a][1,4]diazepin-1,7-diona (9)
La síntesis del intermedio 9 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento D. Sólido de color amarillo. Rendimiento: 0,59 g, 99 %; EM (IEN) m/z 391,32 [M 1]+.
Síntesis de 8-((6-aminopirimidin-4-il)amino)-10-metil-2,3,4,5-tetrahidropirido[1,2-a][1,4]diazepin-1,7-diona (Comp. N.° 217)
Se cargó un vial con 8-((6-aminopirimidin-4-il)amino)-2-bencil-10-metil-2,3,4,5-tetrahidropirido[1,2-a][1,4]diazepin-1,7-diona (9, 0,3 g, 76,9 mmol) y ácido tríflico (7,0 ml). La mezcla de reacción se calentó en microondas a 150 °C durante 20 min. El análisis por CCF mostró la finalización de la reacción y la mezcla se enfrió a temperatura ambiente. Después se basificó con una solución acuosa de bicarbonato de sodio y se extrajo con metanol al 5 %/diclorometano (200 ml, 3 veces). La capa orgánica se secó sobre sulfato de sodio y el disolvente se retiró a presión reducida para proporcionar 8-((6-aminopirimidin-4-il)amino)-10-metil-2,3,4,5-tetrahidropirido[1,2-a][1,4]diazepin-1,7-diona (Comp. N.° 217) en forma de un sólido de color marrón. Rendimiento: 0,06 g, 26 %; EM (IEN) m/z 301,15 [M 1]+; RMN 1H (400 MHz, DMSO-de) ó 8,51 (s, 1H), 8,36-8,20 (m, 2H), 8,15 (s, 1H), 6,52 (s ancho, 2H), 6,13 Sa, 1H), 5,05 (s ancho, 1H), 3,26-3,04 (m, 2H), 2,95-2,80 (m, 1H), 2,13 (s, 3H), 1,87 (s ancho, 2H).
Ejemplo 218
Síntesis 1-óxido de 6-amino-4-((8'-metil-1',5'-dioxo-1',5'-dihidro-2'H-espiro[ciclohexano-1,3'-imidazo[1,5-a]piridin]-6'-il)amino)pirimidina (Comp. N.° 218)

Síntesis de 1-oxido de 6-amino-4-((8’-metil-1’,5’-dioxo-1’,5’-dihidro-2-espiro[ciclohexano-1,3'-imidazo[1,5-a]piridin]-6’-il)amino)pirimidina (Comp. N.° 218)
A una solución de 6’-((6-aminopirimidin-4-il)amino)-8'-metil-2'H-espiro[ciclohexano-1,3’-imidazo[1,5-a]piridin]-1’,5'-diona (1, 0,1 g, 0,29 mmol) en diclorometano (25 ml), se le añadió ácido 3-cloroperbenzoico (0,10 g, 0,59 mmol). La mezcla de reacción se agitó a temperatura ambiente durante 16 h. Después de la terminación, la mezcla de reacción se diluyó con solución saturada de bicarbonato de sodio (50 ml) y se agitó durante 30 M a temperatura ambiente. Se precipitó sólido amarillo y se filtró y se secó para obtener el producto en bruto. El producto en bruto se purificó por purificación previa para proporcionar 6-amino-4-((8'-metil-1’,5'-dioxo-1’,5'-dihidro-2'H-espiro[ciclohexan-1,3’-imidazo[1,5-a]piridin]-6'-il)amino)pirimidina (Comp. N.° 218) en forma de un sólido de color amarillo. Rendimiento: 50 mg, 50 %; EM (IEN) m/z 357,18 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,02 (s, 1H), 9,01 (s, 1H), 8,53 (s, 1H), 8,23 (s, 1H), 7,48 (s ancho, 1H), 6,65 (s, 1H), 3,02-2,96 (m, 2H), 2,42 (s, 3H), 1,75-1,58 (m, 5H), 1,45-1,42 (m, 2H), 1,22-1,19 (m, 1H).
Ejemplo 219
Síntesis de 6-((6-aminopirimidin-4-il)amino)-1 ',8-dimetil-2H-espiro[imidazo[1,5-a]piridin-3,4'-piperidin]-1,5-diona (Comp. N.° 219)

Síntesis de N-terc-butoxicarbonil-N-(6-((1' 8-dimetil-1,5-dioxo-1,5-dihidro-2H-espiro[imidazo[1,5-a]piridin-3,4’-piperidin]-6-il)amino)pirimidin-4-il)carbamato de terc-butilo (3)
La síntesis del intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento H. Sólido de color blanquecino; Rendimiento: 320 mg, 38 %; EM (IEN) m/z 556,67 [M 1]+.
Síntesis de clorhidrato de 6-((6-aminopirimidin-4-il)amino)-1' 8-dimetil-2H-espiro[imidazo[1,5-a]piridin-3,4'-piperidin-1,5-diona (Comp. N.° 219)
La síntesis del compuesto 219 se realizó como se ha descrito anteriormente usando el protocolo general del procedimiento F. Sólido de color amarillo claro. Rendimiento: 274 mg, 97 %; EM (IEN) m/z 356,18 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,64 (s ancho, 1H), 10,45 (s, 1H), 9,73 (s, 1H), 8,46 (s, 1H), 8,15 (s, 1H), 7,91 (s ancho, 2H), 6,45 (s, 1H), 3,62-3,42 (m, 6H), 2,82 (s, 3H), 2,46 (s, 3H), 1,81-1,78 (m, 2H).
Ejemplo 220
Síntesis de 6-((6-amino-5-metilpirimidin-4-il)amino)-1',8-dimetil-2H-espiro[imidazo[1,5-a]piridin-3,4,-piperidin]-1,5-diona (Comp. N.° 220)
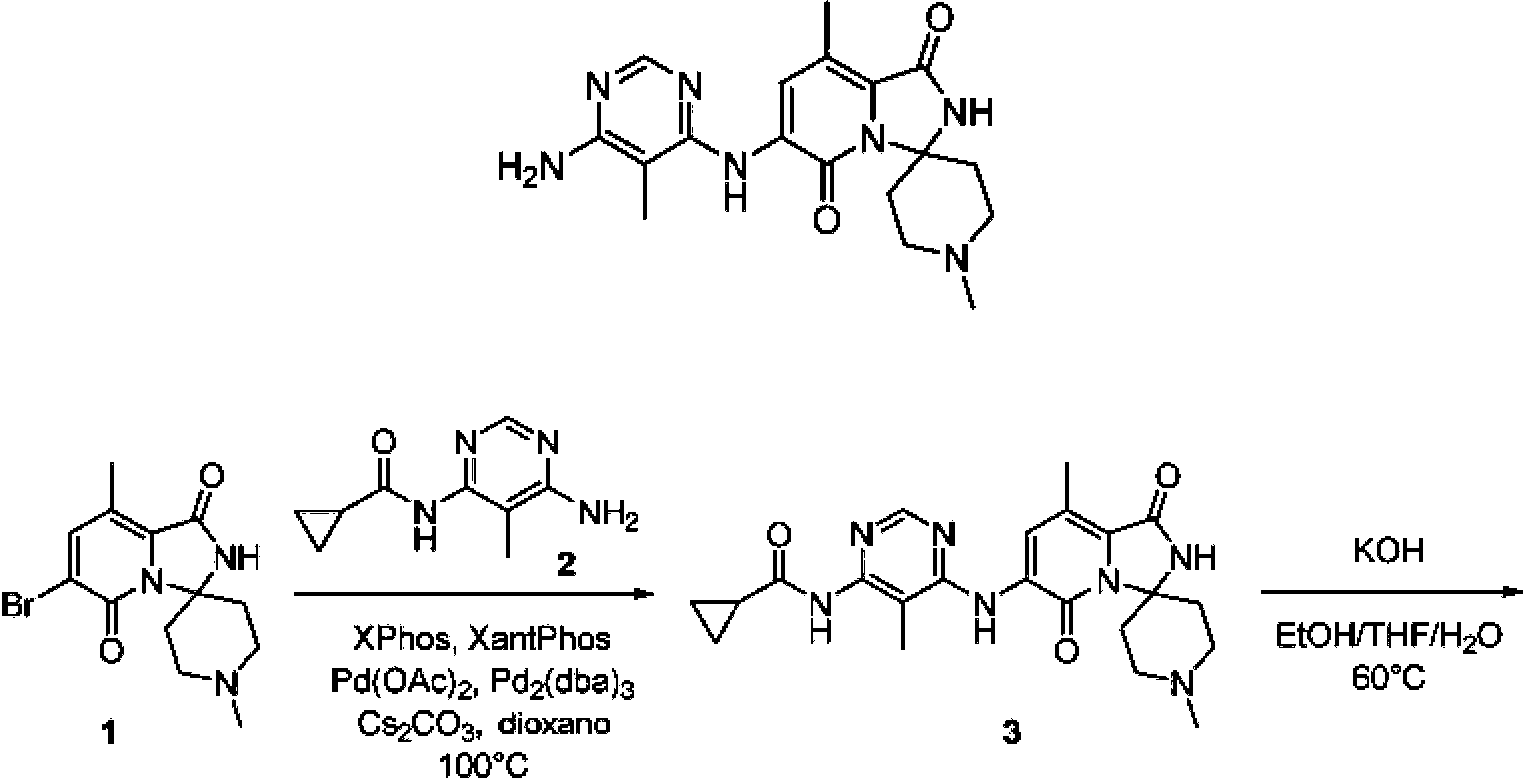

Síntesis de N-(6-((1,8-dimetil-1,5-dioxo-1,5-dihidro-2H-espiro[imidazo[1,5-a]piridin-3,4'-piperidin]-6-il)amino)-5-metilpirimidin-4-il)ciclopropanocarboxamida (3)
La síntesis del intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento H. Sólido de color amarillo. Rendimiento: 450 mg, 67 %; EM (IEN) m/z 438,36 [M 1]+.
Síntesis de 6-((6-amino-5-metilpirimidin-4-il)amino)-1’,8-dimetil-2H-espiro[imidazo[1,5-a]piridin-3,4'-piperidin]-1,5-diona (Comp. N.° 220)
La síntesis del compuesto 220 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento I. Sólido de color blanquecino. Rendimiento: 150 mg, 39 %; EM (IEN) m/z 370,20 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,07 (s, 1H), 8,47 (s, 1H), 8,11 (s, 1H), 8,00 (s, 1H), 6,48 (s, 2H), 3,30-3,20 (m, 2H), 2,78 2,75 (m, 2H), 2,44 (s, 3H), 2,40-2,32 (m, 2H), 2,24 (s, 3H), 1,98 (s, 3H), 1,40-1,36 (m, 2H).
Ejemplo 221
Síntesis de 6-((2-cloropirimidin-4-il)tio)-8'-metil-2'H-espiro[ciclohexano-1,3'-imidazo[1,5-a]piridin]-1',5,-diona (Comp. N.° 221)

Síntesis de 6’-((4-metoxibencil)tio)-8'-metil-2’H-espiro[ciclohexano-1,3’-imidazo[1,5-a]piridin]-1’,5'-diona (3)
6'-Bromo-8'-metil-2'H-espiro[ciclohexano-1,3'-imidazo[1,5-a]piridin]-1',5'-diona (1, 2,0 g, 6,42 mmol ) se disolvió en 2-propanol (20 ml). A esta mezcla se le añadió carbonato de cesio (6,28 g, 19,28 mmol), seguido de (4-metoxifenil)metanotiol (1,18 g, 7,71 mmol). La mezcla de reacción se agitó a 70 °C durante 16 h. Después de la terminación, el disolvente se evaporó a presión reducida y el producto en bruto se lavó con agua (50 ml) seguido de éter dietílico y después se secó a presión reducida para proporcionar 6'-((4-metoxibencil) tio)-8'-metil-2'H-espiro[ciclohexano-1,3'-imidazo[1,5-a]piridin]-1',5'-diona (3) en forma de un sólido de color gris. Rendimiento: 2,15 g, 87 %; EM (IEN) m/z 385,5 [M 1]+.
Síntesis de 6'-mercapto-8’-metil-2'H-espiro[ciclohexano-1,3’-imidazo[1,5-a]piridin]-1’,5’-diona (4)
6'-((4-Metoxibencil)tio)-8'-metil-2'H-espiro[ciclohexano-1,3'-imidazo[1,5-a]piridin]-1',5'-diona (3, 3,3 g, 8,59 mmol) en cloroformo (20 ml) y se añadió ácido metanosulfónico (10 ml). La mezcla de reacción se agitó a 50 °C durante 16 h. Después de la terminación, el disolvente se evaporó a presión reducida. El producto en bruto obtenido se lavó con agua (50 ml) seguido de acetato de etilo y después se secó a presión reducida para proporcionar 6'-mercapto-8'-metil-2'H-espiro[ciclohexano-1,3'-imidazo[1,5-a]piridin]-1',5'-diona en forma de un sólido de color gris. Rendimiento: 1,3 g, 59 %; EM (IEN) m/z 265,6 [M 1]+.
Síntesis de 6’-((2-doropirímidin-4-il)tio)-8'-metil-2'H-espiro[cidohexano-1,3’-imidazo[1,5-a]piridin]-1’,5’-diona (Comp. N.° 221)
La síntesis del compuesto 221 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento B. Sólido de color blanquecino; Rendimiento: 0,4 g, 56 %; EM (IEN) m/z 2,1121 [M 1]+; RMN 1H (400 MHz, DMSO-6) ó 10,52 (s, 1H), 8,45 (d, J = 5,2 Hz, 1H), 8,08 (s, 1H), 7,32 (d, J = 5,6 Hz, 1H), 2,92-2,84 (m, 2H), 2,42 (s, 3H), 1,74-1,71 (m, 2H), 1,65-1,52 (m, 3H), 1,47-1,44 (m, 2H), 1,18-1,15 (m, 1H).
Ejemplo 222
Síntesis de 6'-((6-amino-5-metilpirimidin-4-il)amino)-8'-metil-2'H-espiro[ciclohexano-1,3'-imidazo[1,5-a]piridin]-1',5'-diona (Comp. N.° 222)


100°C

Síntesis de N-(5-metil-6-((8'-metil-1’,5’-dioxo-1’,5’-dihidro-2'H-espiro[ciclohexano-1,3’-imidazo[1,5-aj’piridin]-6’-il)amino)pirimidin-4-il)ciclopropanocarboxamida (3)
La síntesis del intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento H. Sólido de color amarillo; Rendimiento: 0,17 g, 31 %; EM (IEN) m/z 423,21 [M 1]+.
Síntesis de 6’-((6-amino-5-metilpirimidin-4-il)amino)-8'-metil-2’H-espiro[ciclohexano-1,3’-imidazo[1,5-a]piridin]-1’,5’-diona (Comp. N.° 222)
La síntesis del compuesto 222 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento I. Sólido de color blanquecino; Rendimiento: 0,10 g, 70 %; EM (IEN) m/z 355,18 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,05 (s, 1H), 8,48 (s, 1H), 8,12 (s, 1H), 8,01 (s, 1H), 6,48 (s, 2H), 2,98 (t, J = 9,2 Hz, 2H), 2,45 (s, 3H), 1,98 (s, 3H), 1,73-1,58 (m, 5H), 1,48-1,42 (m, 2H), 1,30-1,23 (m, 1H).
Ejemplo 223
Síntesis de clorhidrato de 6'-((6-amino-5-cloropirimidin-4-il)amino)-8'-metil-2'-espirociclohexano-1,3'-imidazo[1,5-a]piridin]-1',5'-diona (Comp. N.°223)


Síntesis de N-terc-butoxicarbonil-N-(5-cloro-6-((8'-metil-1 5’-dioxo-1' 5’-dihidro-2’H-espiro[ciclohexano-1 ’,3'-imidazo[1,5-a]piridin]-6’-il)amino)pirimidin-4-il)carbamato de terc-butilo (3)
La síntesis del intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento B. Sólido de color blanquecino; Rendimiento: 0,32 g, 39 %; EM (IEN) m/z 575,23 [M 1]+; RMN 1H (400 MHz, DMSO-6) ó 10,26 (s, 1H), 9,03 (s, 1H), 8,79 (s, 1H), 8,55 (s, 1H), 2,96 (m, 2H), 2,5 (s, 3H), 1,76-1,58 (m, 5H), 1,49 (m, 2H), 1,39 (s, 18H), 1,25 (m, 1H).
Síntesis de clorhidrato de 6’-((6-amino-5-cloropirimidin-4-il)amino)-8'-metil-2'H-espiro[ciclohexano-1,3’-imidazo[1,5-a]piridin]-1',5’-diona (Comp. N.° 223)
La síntesis del compuesto 223 se realizó como se ha descrito anteriormente usando el protocolo general del procedimiento F. Sólido de color amarillo. Rendimiento: 0,20 g, 88 %; EM (IEN) m/z 375,13 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,15 (s, 1H), 8,60 (s, 1H), 8,47 (s, 1H), 8,21 (s, 1H), 7,23 (s ancho, 2H), 2,97 (t, J = 9,6 Hz, 2H), 2,46 (s, 3H), 1,76-1,58 (m, 5H), 1,48-1,42 (m, 2H), 1,30-1,21 (m, 1H).
Ejemplo 224
Síntesis de 6'-((2-cloropirimidin-4-il)oxi)-8'-metil-2'H-espiro[ciclohexano-1,3'-imidazo[1,5-a]piridin]-1 ',5'-diona (Comp. N.° 224)

Síntesis de 6’-((4-metoxibencil)oxi)-8'-metil-2’H-espiro[ciclohexano-1,3’-imidazo[1,5-a]piridin]-15’-diona (3)
A una solución de 6'-bromo-8'-metil-2'H-espiro[ciclohexano-1,3'-imidazo[1,5-a]piridin]-1',5'-diona (1, 2,5 g, 8,03 mmol) en dimetilformamida (20 ml), se añadió hidruro de sodio (1,15 g, 48,23 mmol) en porciones. La mezcla de reacción se agitó a 0 °C durante 30 m. A la mezcla de reacción se le añadió (4-metoxifenil)metanol (3,34 g, 24,11 mmol) y la reacción se agitó a 80 °C durante 16 h. Después de la terminación, el disolvente se evaporó a presión reducida. El producto en bruto obtenido se lavó con agua (50 ml) seguido de éter dietílico y se secó a presión reducida para proporcionar 6'-((4-metoxibencil)oxi)-8'-metil-2'H-espiro[ciclohexano-1,3'-imidazo[1,5-a]piridin]-1',5'-diona (3) en forma de un sólido de color amarillo. Rendimiento: 1,4 g, 48 %; EM (IEN) m/z 369,15 [M 1 ]+.
Síntesis de 6’-hidroxi-8'-metil-2’H-espiro[ciclohexano-1,3'-imidazo[1,5-a]piridin]-1’,5'-diona (4)
6'-((4-Metoxibencil)oxi)-8'-metil-2'H-espiro[ciclohexano-1,3'-imidazo[1,5-a]piridin]-1',5'-diona (3, 3,3 g, 8,59 mmol) se disolvió en 1,2-dicloroetano (25 ml) y se añadió ácido trifluoroacético (10 ml). La mezcla de reacción se agitó a 50 °C durante 2 h. Después de la terminación, el disolvente se evaporó a presión reducida. El producto en bruto obtenido se lavó con agua (50 ml) seguido de n-pentano y se secó a presión reducida para proporcionar 6'-hidroxi-8'-metil-2'H-espiro[ciclohexano-1,3'-imidazo[1,5-a]piridin]-1',5'-diona (4) en forma de un sólido de color amarillo. Rendimiento: 0,85 g, 90 %; EM (IEN) m/z 249,07 [M 1]+.
Síntesis de 6’-((2-cloropirimidin-4-il)oxi)-8'-metil-2’H-espiro[ciclohexano-1,3’-imidazo[1,5-a]piridin]-1’,5’-diona (Comp. N.° 224)
6'-Hidroxi-8'-metil-2'H-espiro[ciclohexano-1,3'-imidazo[1,5-a]piridin]-1',5'-diona (4, 0,5 g, 2,01 mmol) se disolvió en 2-propanol (20 ml) y se añadieron N,N-diisopropiletilamina (780 mg, 6,05 mmol) y 2,4-dicloropirimidina (0,30 g, 2,83 mmol). La mezcla de reacción se agitó a 120 °C durante 36 h. Después de la terminación, el disolvente se evaporó a presión reducida y se añadió agua (100 ml). El sólido amarillo precipitado se filtró y se secó a presión reducida. Este producto en bruto se purificó mediante cromatografía en columna de gel de sílice (malla 220-400) usando metanol al 2-5 % en diclorometano como eluyente para proporcionar 6'-((2-cloropirimidin-4-il)oxi)-8'-metil-2'H-espiro[ciclohexano-1,3'-imidazo[1,5-a]piridin]-1',5'-diona (Comp. N.° 224). Rendimiento: 0,33 g, 46 %; EM (IEN) m/z 361,12 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,37 (s, 1H), 8,65 (d, J = 5,6 Hz, 1H), 7,61 (s, 1H), 7,28 (d, J = 5,6 Hz, 1H), 6,55 (s, 2H), 2,88-2,72 (m, 2H), 2,44 (m, 2H), 1,72-1,70 (m, 2H), 1,64-1,58 (m, 3H), 1,45-1,42 (m, 2H), 1,17 -1,13 (m, 1H).
Ejemplo 225
Síntesis de 2-(6-aminopirimidin-4-il)amino)-8-metil-1,5-dioxo-1,5-dihidro-2H-espiro[imidazo[1,5-a]piridin-3,4'-piperidin]-1'-il)acetonitrilo (Comp. N.° 225)

Síntesis de (6-((1’-(cianometil)-8-metil-1,5-dioxo-1,5-dihidro-2H-espiro[imidazo[1,5-a]piridin-3,4’-piperidin]-6-il)amino)pirimidin-4-il)-2-azanocarboxilato de etilo (3)
La síntesis del intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento B. Sólido de color amarillo; Rendimiento: 0,25 g, 60 %; EM (IEN) m/z 581,49 [M 1]+.
S ín te s is de 2 -(6 -((6 -a m in o p ir im id in -4 - il)a m in o )-8 -m e til-1 ,5 -d io x o -1 ,5 -d ih id ro -2 H -e s p iro [im id a z o [1 ,5 -a ]p ir id in -3 ,4 ’- piperídin]-1’-il)acetonitrílo (Comp. N ° 225)
La síntesis del compuesto 225 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento F. Sólido de color blanquecino; Rendimiento: 37 mg, 38 %; EM (IEN) m/z 381,19 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,40 (s, 1H), 9,47 (s, 1H), 8,38 (s, 1H), 8,18 (s, 1H), 7,54 (s ancho, 2H), 6,34 (s, 1H), 3,74 (s, 2H), 3,30-3,23 (m, 2H), 2,89-2,87 (m, 2H), (2,66-2,64 (s, 2H), 2,42 (s, 3H), 1,50-1,47 (m, 2H).
Ejemplo 226
Síntesis de 6-((6-aminopirimidin-4-il)amino)-1 '-(2,2-difluoroetil)-8-metil-2H-espiro[imidazo[1,5-a]piridin-3,4'-piperidin]-1,5-diona (Comp. N.° 226)

Síntesis de 6-bromo-1’-(2,2-difluoroetil)-8-metil-2H-espiro[imidazo[1,5-a]piridin-3,4'-piperidin]-1,5-diona (3)
A una solución de clorhidrato de 6-bromo-8-metil-2H-espiro[imidazo[1,5-a]piridin-3,4'-piperidin]-1,5-diona (1, 300 mg, 0,96 mmol) en acetonitrilo se le añadieron 2-bromo-1,1-difluoroetano (348 mg, 2,40 mmol) y carbonato de potasio (397 mg, 2,88 mmol). La reacción se calentó a 90 °C durante 18 h. Una vez completada la reacción, se retiró el disolvente y el producto en bruto se purificó por cromatografía en columna de gel de sílice (malla 200-400) eluyendo con metanol al 5-7 % en diclorometano. Las fracciones apropiadas de la columna se concentraron a presión reducida para proporcionar 6-bromo-(2,2-difluoroetil)-8-metil-2H-espiro[imidazo[1,5-a]piridin-3,4'-piperidin]-1,5-diona (3) en forma de un sólido de color amarillo claro. Rendimiento: 180 mg, 37 %; e M (IEn ) m/z 378,22 [M 1]+.
Síntesis de N-terc-butoxicarbonil-N-(6-(( 1 ’-(2,2-difluoroetil)-8-metil-1,5-dioxo-1,5-dihidro-2H-espiro[imidazo[5-a]piridin-3,4’-piperidin]-6-il)amino)pirimidin-4-il)carbamato de terc-butilo (5)
La síntesis del intermedio 5 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento H. Sólido de color blanquecino; Rendimiento: 200 mg, 69 %; EM (IEN) m/z 606,18.
Síntesis de 6-((6-aminopirimidin-4-il)amino)-1’-(2,2-difluoroetil)-8-metil-2H-espiro[imidazo[1,5-a]piridin-3,4'-piperidina]-1,5-diona (Comp. N ° 226)
La síntesis del compuesto 226 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento F. Sólido de color blanquecino; Rendimiento: 14 mg, 8 %; EM (IEN) m/z 406,20 [M 1]+; RMN 1H (400 MHz, DMSO-cfe) ó 10,08 (s, 1H), 8,57 (s, 1H), 8,38 (s, 1H), 8,16 (s, 1H), 6,51 (s, 2H), 6,30-6,00 (m, 2H), 3,25 3,22 (m, 2H), 2,94-2,91 (m, 2H), 2,87-2,79 (m, 2H), 2,69-2,63 (m, 2H), 2,42 (s, 3H), 1,39-1,36 (m, 2H).
Ejemplo 227
Síntesis de 6-((5-metoxipirimidin-4-il)amino)-1 ',8-dimetil-2H-espiro[imidazo[1,5-a]piridin-3,4'-piperidin]-1,5-diona (Comp. N.° 227)

Síntesis de 6-bromo-1’,8-dimetil-2H-espiro[imidazo[1,5-a]piridin-3,4'-piperidin-1,5-diona (3)
A una solución agitada de 5-bromo-3-metil-6-oxo-1,6-dihidropiridin-2-carboxamida (1, 3 g, 12,98 mmol) en 1,4-dioxano (25 ml), se le añadió 1-metilpiperidin-4-ona (2,2 g, 19,47 mmol) a temperatura ambiente. A la mezcla, se añadió gota a gota cloruro de hidrógeno 4 M en dioxano (6,5 ml, 2,59 mmol). La reacción se calentó hasta 110 °C durante 16 h. Después de la terminación, el disolvente se retiró a presión reducida y el sólido obtenido se lavó con agua caliente seguido de pentano y éter para proporcionar 6-bromo-1,8-dimetil-2H-espiro[imidazo[1,5-a]piridin-3,4'-piperidin]-1,5-diona (3) en forma de un sólido de color blanquecino. Rendimiento: 2,5 g, 59 %; EM (IEN) m/z 326 [M 1]+.
Síntesis de 6-((5-metoxipirimidin-4-il)amino)-1’,8-dimetil-2H-espiro[imidazo[1,5-a]piridin-3,4'-piperidin-1,5-diona (Comp. N.° 227)
La síntesis del compuesto 227 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento B. Sólido de color blanquecino; Rendimiento: 103 mg, 18 %; EM (IEN) m/z 371,16 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,16 (s ancho, 1H), 8,62 (s, 1H), 8,58 (s, 1H), 8,48 (s, 1H), 8,24 (s, 1H), 4,00 (s, 3H), 3,25 3,21 (m, 2H), 2,84-2,81 (m, 2H), 2,45 (s, 3H), 2,48-2,42 (m, 2H), 2,28 (s, 3H), 1,44-1,41 (m, 2H).
Ejemplo 228
Síntesis de 6-((6-amino-5-metoxipirimidin-4-il)amino)-1 ',8-dimetil-2H-espiro[imidazo[1,5-a]piridin-3,4'-piperidin]-1,5-diona (Comp. N.° 228)

Síntesis de N-(6-(( 1 8-dimetil-1,5-dioxo-1,5-dihidro-2H-espiro[imidazo[1,5-a]piridin-3,4’-piperidin]-6-il)amino)-5-metoxipirimidin-4-il)ciclopropanocarboxamida (3)
La síntesis del intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del procedimiento B. Sólido de color amarillo claro; Rendimiento: 103 mg, 31 %; EM (IEN) m/z 454,15 [M 1]+.
Síntesis de 6-((6-amino-5-metoxipirimidin-4-il)amino)-1’,8-dimetil-2H-espiro[imidazo[1,5-a]piridin-3,4'-piperidin]-1,5-diona (Comp. N.° 228)
Un matraz que contenía tetrahidrofurano, etanol y agua (1:1:1, 5 ml cada uno) se cargó con 6-((6-amino-5-metoxipirimidin-4-il)amino)-1',8-dimetil-2H-espiro[imidazo[1,5-a]piridin-3,4'-piperidin]-1,5-diona (3, 0,2 g, 0,441 mmol) e hidróxido de sodio (88 mg, 2,2 mmol). La reacción se agitó a 60 °C durante 16 h. Después de la terminación, la masa de reacción se extrajo con 2-propanol al 10 % en cloroformo (50 ml, 5 veces). La capa orgánica combinada se concentró y el sólido obtenido se lavó con metanol y se secó al vacío para proporcionar 6-((6-amino-5-metoxipirimidin-4-il)amino)-1',8-dimetil-2H-espiro[imidazo[1,5-a]piridin-3,4'-piperidin]-1,5-diona (Comp. N.° 228) en forma de un sólido de color amarillo claro. Rendimiento: 88 mg, 52 %; EM (IEN) m/z 386,19 [M 1]+; Rm N 1H (400 MHz, DMSO-d6) ó 10,11 (s, 1H), 8,45 (s, 1H), 8,32 (s, 1H), 8,00 (s, 2H), 6,67 (s, 2H), 3,69 (s, 3H), 3,30-3,20 (m, 2H), 2,90 (s ancho, 2H), 2,50-2,46 (m, 2H), 2,45 (s, 3H), 2,33 (s, 3H), 1,47-1,45 (m, 2H), 2,30 (m, 2H), 2,90 (s ancho, 2H), 2,50-2,46 (m, 2H).
Ejemplo 229
Síntesis de 2-(6-(5-metoxipirimidin-4-il)amino)-8-metil-1,5-dioxo-1,5-dihidro-2H-espiro[imidazo[1,5-a]piridin-3,4-piperidin]-1'-il)acetonitrilo (Comp. N.° 229)

Síntesis de 2-(6-bromo-8-metil-1,5-dioxo-1,5-dihidro-2H-espiro[imidazo[1,5-aj’piridin-3,4'-piperidin-il)acetonitrilo (3)
Se cargó un matraz con 6-bromo-8-metil-2H-espiro[imidazo[1,5-a]piridin-3,4'-piperidin]-1,5-diona (1, 0,50 g, 1,6 mmol) y acetonitrilo (15 ml). La masa de reacción se enfrió a 0 °C y se añadió carbonato de potasio (664 mg, 4,8 mmol) seguido de la adición de 2-bromoacetonitrilo (288,18 mg, 2,40 mmol). La masa de reacción se agitó a 70 °C durante 16 h. Después de la terminación, la mezcla de reacción se diluyó con solución saturada de cloruro de amonio (50 ml). El precipitado se filtró y se secó a presión reducida para proporcionar 2-(6-bromo-8-metil-1,5-dioxol-1,5-dihidro-2H-espiro[imidazo[1,5-a]piridin-3,4'-piperidin]-1'-il)acetonitrilo (3) en forma de un sólido de color blanquecino. Rendimiento: 0,32 g, 58 %; EM (lEN) m/z 351,26 [M 1]+.
Síntesis de 2-(6-((5-metoxipirimidin-4-il)amino)-8-metil-1,5-dioxo-1,5-dihidro-2H-espiro[imidazo[1,5-a]piridin-3,4’-piperidin]-1'-il)acetonitrilo (Comp. N ° 229)
La síntesis del compuesto 229 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento B. Sólido de color amarillo. Rendimiento: 0,068 g, 20 %; EM (IEN) m/z 396,5 [M 1]+; RMN 1H (400 MHz, DMSO-d6 con-TFA) ó 8,94 (s, 1H), 8,53 (s, 1H), 8,44 (s, 1H), 4,47 (s, 2H), 4,07 (s, 3H), 3,71-3,68 (m, 2H), 3,47-3,39 (m, 4H), 2,47 (s, 3H), 1,92-1,89 (m, 2H).
Ejemplo 230
Síntesis de 1 '-(2,2-difluoroetil)-6-((5-metoxipirimidin-4-il)amino)-8-metil-2H-espiro[imidazo[1,5-a]piridin-3,4'-piperidin]-1,5-diona (Comp. N.° 230)

Síntesis de 1’-(2,2-difluoroetil)-6-((5-metoxipirimidin-4-il)amino)-8-metil-2H-espiro[imidazo[1,5-a]piridin-3,4'-piperidin]-1,5-diona (Comp. N ° 230)
La síntesis del compuesto 230 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento H. Sólido de color blanquecino; Rendimiento: 65 mg, 45 %; EM (IEN) m/z 421,13 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,22 (s, 1H), 8,64 (s, 1H), 8,58 (s, 1H), 8,48 (s, 1H), 8,24 (s, 2H), 6,30-6,00 (m, 1H), 4,00 (s, 1H), 3,31-3,17 (m, 2H), 2,95-2,92 (m, 2H), 2,87-2,78 (m, 2H), 2,68-2,62 (m, 2H), 2,49 (s, 3H), 1,43-1,40 (m, 2H).
Ejemplo 231
Síntesis de 6'-((6-amino-5-metoxipirimidin-4-il)amino)-8'-metil-2'H-espiro[ciclohexano-1,3 midazo[1,5-a]piridin]-1',5'-diona (Comp. N.° 231)

Síntesis de N-(5-metoxi-6-((8'-metil-1' 5’-dioxo-1 ’,5’-dihidro-2'H-espiro[ciclohexano-1,3’-imidazo[1,5-a]piridin]-6’-il)amino)pirimidin-4-il)ciclopropanocarboxamida (3)
La síntesis del intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento H. Sólido de color amarillo claro; Rendimiento: 0,3 g, 71 %; EM (IEN) m/z 439,14 [M 1]+.
Síntesis de 6’-((6-amino-5-metoxipirimidin-4-il)amino)-8'-metil-2’H-espiro[ciclohexano-1,3’-imidazo[1,5-a]piridin]-1’,5'-diona (Comp. N.° 231)
La síntesis del compuesto 231 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento I. Sólido de color amarillo; Rendimiento: 0,085 g, 27 %; EM (IEN) m/z 371,16 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,06 (s, 1H), 8,47 (s, 1H), 8,33 (s, 1H), 8,00 (s, 1H), 6,67 (s, 2H), 3,69 (s, 3H), 3,02-2,95 (m, 2H), 2,45 (s, 3H), 1,78-1,58 (m, 5H), 1,47-1,43 (m, 2H), 1,29-1,20 (m, 1H).
Ejemplo 232
Síntesis de 6-((6-aminopirimidin-4-il)amino)-8-metil-1'-(2,2,2-trifluoroetil)-2H-espiro[imidazo[1,5-a]piridin-3,4'-piperidin]-1,5-diona (Comp. N.° 232)

S ín te s is de N -(6 -((8 -m e til-1 ,5 -d io x o -1 ’-(2 ,2 ,2 -tr if lu o ro e til) -1 ,5 -d ih id ro -2 H -e s p iro [im id a z o [1 ,5 -a ]p ir id in -3 ,4 '-p ip e r id in ]-6 -il)a m in o )p ir im id in -4 - il)c ic lo p ro p a n o c a rb o x a m id a ( 3 )
La síntesis del intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento H. Sólido de color amarillo. Rendimiento: 300 mg, 60 %; EM (IEN) m/z 492,10 [M 1]+.
Síntesis de 6-((6-aminopirimidin-4-il)amino)-8-metil-1’-(2,2,2-trifluoroetil)-2H-espiro[imidazo[1,5-a]piridin-3,4'-piperidin]-1,5-diona (Comp. N.° 232)
La síntesis del compuesto 232 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento I. Sólido de color blanquecino; Rendimiento: 60 mg, 23 %; EM (IEN) m/z 424,19 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,14 (s, 1H), 8,65 (s, 1H), 8,37 (s, 1H), 8,18 (s, 1H), 6,60 (s, 2H), 6,18 (s, 1H), 3,27-3,22 (m, 4H), 2,95-2,90 (m, 2H), 2,85-2,76 (m, 2H), 2,42 (s, 3H), 1,41-1,36 (m, 2H).
Ejemplo 233
Síntesis de 6-((6-amino-5-metilpirimidin-4-il)amino)-1 '-(2,2-difluoroetil)-8-metil-2H-espiro[imidazo[1,5-a]piridin-3,4'-piperidin]-1,5-diona (Comp. N.° 233)

Síntesis de N-(6-((1’-(2,2-dtfluoroetil)-8-metil-1,5-dioxo-1,5-dihidro-2H-espiro[imidazo[1,5-a]piridin-3,4’-piperidin]-6-il)amino)-5-metilpirimidin-4-il)ciclopropanocarboxamida (3)
La síntesis del intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento H. Sólido de color amarillo claro; Rendimiento: 0,35 g, 65 %; EM (IEN) m/z 488,27 [M 1]+.
Síntesis de 6-((6-amino-5-metilpirimidin-4-il)amino)-1’-(2,2-difluoroetil)-8-metil-2H-espiro[imidazo[1,5-a]piridin-3,4'-piperidin]-1,5-diona (Comp. N.° 233)
La síntesis del compuesto 233 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento I. Sólido de color blanquecino; Rendimiento: 80 mg, 26 %; EM (IEN) m/z 420,20 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,13 (s, 1H), 8,48 (s, 1H), 8,12 (s, 1H), 8,00 (s, 1H), 6,48 (s, 2H), 6,30-6,00 (m, 1H), 3,27 3,21 (m, 2H), 2,94-2,91 (m, 2H), 2,86-2,77 (m, 2H), 2,67- 2,61 (m, 2H), 2,44 (s, 3H), 1,98 (s, 3H), 1,41-1,38 (m, 2H).
Ejemplo 234
Síntesis de 6-((6-amino-5-metoxipirimidin-4-il)amino)-8-metil-1 '-(2,2,2-trifluoroetil)-2H-espiro[imidazo[1,5-a]piridin-3,4'-piperidin]-1,5-diona (Comp. N.° 234)

Síntesis N-(5-metoxi-6-((8-metil-1,5-dioxo-1’-(2,2,2-trifluoroetil)-1,5-dihidro-2H-espiro[imidazo[1,5-a]piridin-3,4’-piperidin]-6-il)amino)pirimidin-4-il)ciclopropanocarboxamida (3)
La síntesis del intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del procedimiento B. Sólido de color amarillo claro; Rendimiento: 400 mg, 76 %; EM (IEN) m/z 522,19 [M 1]+.
Síntesis de 6-((6-amino-5-metoxipirimidin-4-il)amino)-8-metil-1’-(2,2,2-trifluoroetil)-2H-espiro[imidazo[1,5-a]piridin-3,4’-piperidin-1,5-diona (Comp. N.° 234)
La síntesis del compuesto 234 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento I. Sólido de color amarillo claro; Rendimiento: 100 mg, 29 %; EM (IEN) m/z 354,12 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,19 (s, 1H), 8,45 (s, 1H), 8,32 (s, 1H), 8,00 (s, 1H), 6,67 (s, 2H), 3,69 (s, 3H), 3,27-2,21 (m, 4H), 2,97-2,90 (m, 2H), 2,83-2,77 (m, 2H), 2,45 (s, 3H), 1,42-1,38 (m, 2H).
Ejemplo 235
Síntesis de 6-((5-metoxipirimidin-4-il)amino)-8-metil-1'-(2,2,2-trifluoroetil)-2H-espiro[imidazo[1,5-a]piridin-3,4'-piperidin]-1,5-diona (Comp. N.° 235)
S ín te s is de 6 -((5 -m e to x ip ir im id in -4 - il)a m in o )-8 -m e til-1 ’-(2 ,2 ,2 -tr if lu o ro e til) -2 H -e s p iro [im id a z o [1 ,5 -a ]p ir id in -3 ,4 '-p ip e r id in ]-1 ,5 -d io n a (C om p. N ° 235 )
La síntesis del compuesto 235 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento B. Sólido de color blanquecino; Rendimiento: 0,13 g, 39 %; EM (IEN) m/z 439,21 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,29 (s, 1H), 8,65 (s, 1H), 8,58 (s, 1H), 8,48 (s, 1H), 8,24 (s, 1H), 4,00 (s, 3H), 3,32-3,22 (m, 4H), 2,96-2,90 (m, 2H), 2,85-2,79 (m, 2H), 2,45 (s, 3H), 1,42-1,39 (m, 2H).
Ejemplo 236
Síntesis de 6-((6-amino-5-metilpirimidin-4-il)amino)-8-metil-1 '-(2,2,2-trifluoroetil)-2H-espiro[imidazo[1,5-a]piridin-3,4'-piperidin]-1,5-diona (Comp. N.° 236)

Síntesis de 6-bromo-8-metil-1’-(2,2,2-trifluoroetil)-2H-espiro[imidazo[1,5-a]piridin-3,4'-piperidin-1,5-diona (3) La síntesis del producto intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento A. Rendimiento: 5,0 g, 58 %; EM (IEN) m/z 394,08 [M 1]+.
Síntesis de N-(5-metil-6-((8-metil-1,5-dioxo-1’-(2,2,2-trifluoroetil)-1,5-dihidro-2H-espiro[imidazo[1,5-a]piridin-3,4’-piperidin]-6-il)amino)pirimidin-4-il)ciclopropanocarboxamida (5)
La síntesis del intermedio 5 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento H. Sólido de color marrón; Rendimiento: 0,32 g, 62 %; EM (IEN) m/z 506,23 [M 1]+.
Síntesis de 6-((6-amino-5-metilpirimidin-4-il)amino)-8-metil-1’-(2,2,2-trifluoroetil)-2H-espiro[imidazo[1,5-a]piridin-3,4’-piperidin]-1,5-diona (Comp. N ° 236)
La síntesis del compuesto 236 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento I. Rendimiento: 70 mg, 27 %; EM (IEN) m/z 438,16 [M 1]+; RMN 1H (400 MHz, DMSO-d6 con d1-TFA) ó 10,17 (s, 1H), 8,48 (s, 1H), 8,12 (s, 1H), 8,00 (s, 1H), 6,48 (s, 2H), 3,32-3,20 (m, 4H), 2,95-2,93 (m, 2H), 2,85 2,75 (m, 2H), 2,45 (s, 3H), 1,98 (s, 3H), 1,41-1,38 (m, 2H).
Ejemplo 237
Síntesis de 6-((6-amino-5-cloropirimidin-4-il)amino)-1',8-dimetil-2H-espiro[imidazo[1,5-a]piridin-3,4'-piperidin]-1,5-diona (Comp. N.° 237)

Síntesis de N-(5-cloro-6-((1’,8-dimetil-1,5-dioxo-1,5-dihidro-2H-espiro[imidazo[1,5-a]piridin-3,4'-piperidin]-6-il)amino)pirimidin-4-il)ciclopropanocarboxamida (3)
La síntesis del producto intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento H. Sólido de color marrón claro; Rendimiento: 210 mg, 50 %; EM (IEN) m/z 458,12 [M 1]+.
Síntesis de 6-((6-amino-5-cloropirimidin-4-il)amino)-1’,8-dimetil-2H-espiro[imidazo[1,5-a]piridin-3,4'-piperidin]-1,5-diona (Comp. N.° 237)
Un matraz que contenía tetrahidrofurano, etanol y agua (1:1:1, 5 ml cada uno) se cargó con 6-((6-amino-5-metoxipirimidin-4-il)amino)-1',8-dimetil-2H-espiro[imidazo[1,5-a]piridin-3,4'-piperidin]-1,5-diona (3, 0,17 g, 0,371 mmol) e hidróxido de sodio (75 mg, 1,85 mmol). La reacción se agitó a 50 °C durante 16 h. Después de la terminación, la masa de reacción se extrajo con metanol al 10 % en diclorometano. La capa orgánica combinada se lavó con agua y salmuera, se secó sobre sulfato de sodio anhidro, se concentró en vacío, se lavó el sólido obtenido con éter y se secó al vacío para proporcionar 6-((6-amino-5-cloropirimidin-4-il)amino)-1',8-dimetil-2H-espiro[imidazo[1,5-a]piridin-3,4'-piperidin]-1,5-diona en forma de un sólido de color amarillo claro. Rendimiento: 88 mg, 31 %; EM (IEN) m/z 390,14 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,16 (s, 1H), 8,52 (s, 1H), 8,50 (s, 1H), 8,17 (s, 1H), 7,10 (s ancho, 2H), 3,30-3,20 (m, 2H), 2,90-2,82 (s ancho, 2H), 2,49-2,40 (m, 2H), 2,46 (s, 3H), 2,30 (s, 3H), 1,46-1,40 (m, 2H).
Ejemplo 238
Síntesis de 2-(6-((6-amino-5-cloropirimidin-4-il)amino)-8-metil-1,5-dioxo-1,5-dihidro-2H-espiro[imidazo[1,5-a]piridin-3,4'-piperidin]-1'-il)acetonitrilo (Comp. N.° 238)

Síntesis de 6-((5-cloro-6-(ciclopropanocarboxamido)pirimidin-4-il)amino)-8-metil-1,5-dioxo-1,5-dihidro-2H-espiro[imidazo[1,5-a]pirídin-3,4’-piperidin-1'-carboxilato de terc-butilo (3)
La síntesis del intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento B. Sólido de color amarillo claro; Rendimiento: 0,35 g, producto en bruto; EM (IEN) m/z 544,27 [M 1]+.
Síntesis de 6-((6-amino-5-cloropirimidin-4-il)amino)-8-metil-1,5-dioxo-1,5-dihidro-2H-espiro[imidazo[1,5-a)piridin-3,4’-piperidin-1 '-carboxilato de terc-butilo (4)
La síntesis del intermedio 4 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento I. Sólido de color amarillo; Rendimiento: 300 mg, producto en bruto; EM (IEN) m/z 476,23 [M 1]+.
Síntesis de clorhidrato de 6-((6-amino-5-cloropirimidin-4-il)amino)-8-metil-2H-espiro[imidazo[1,5-a]piridin-3,4’-piperidin]-1,5-diona (5)
La síntesis del intermedio 5 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento F. Sólido de color amarillo claro; Rendimiento: 0,075 g, producto en bruto; EM (IEN) m/z 116,21 [M 1]+.
Síntesis de 2-(6-(6-amino-5-cloropirimidin-4-il)amino)-8-metil-1,5-dioxo-1,5-dihidro-2H-espiro[imidazo[1,5-a]piridin-3,4'-piperidin-1’-ilacetonitrilo (Comp. N.° 238)
6-((6-Amino-5-cloropirimidin-4-il)amino)-8-metil-2H-espiro[imidazo[1,5-a]piridin-3,4'-piperidin]-diona (5, 75 mg, 0,20 mmol) se disolvió en dimetilformamida (5 ml). A esta mezcla se añadieron N,N-diisopropiletilamina (77 mg, 0,60 mmol) y bromoacetonitrilo (36 mg, 0,30 mmol). La mezcla de reacción se agitó a temperatura ambiente durante 2 h. Después de la terminación, la mezcla de reacción se diluyó con solución saturada de cloruro de amonio (50 ml). El precipitado amarillo se filtró y se secó a presión reducida. El producto en bruto se purificó mediante purificación previa para proporcionar 2-(6-(6-amino-5-cloropirimidin-4-il)amino)-8-metil-1,5-dioxo-1,5-dihidro-2H-espiro[imidazo[1,5-a]piridin-3,4'-piperidin]-il)acetonitrilo (Comp. N.° 238) en forma de un sólido de color blanquecino. Rendimiento: 14 mg, 17 %; EM (IEN) m/z 414,85 [M 1]+; RMN 1H (400 MHz, DMSO-d6 con d1-TFA) ó 8,49 (s, 1H), 8,33 (s, 1H), 4,49 (s, 2H), 3,76-3,71 (m, 2H), 3,58-3,41 (m, 4H), 2,42 (s, 3H), 1,90-1,87 (m, 2H).
Ejemplo 239
Síntesis de 6-((6-amino-5-metilpirimidin-4-il)amino)-8-metil-2',3',5',6'-tetrahidro-2H-espiro[imidazo[1,5-]piridin-3,4'-piran]-1,5-diona (Comp. N.° 239)

La síntesis del intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento A. Sólido de color blanquecino; Rendimiento: 1,4 g, 69 %; EM (IEN) m/z 313 [M 1]+.
Síntesis de (8-metil-1,5-dioxo-1’,2’,3',5’,6'-tetrahidro-2H-espiro[imidazo[1,5-a]piridin-3,4]piran]-6-il)carbamato de tercbutilo (5)
La síntesis del intermedio 5 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento H. Sólido de color blanquecino; Rendimiento: 1,2 g, producto en bruto; EM (IEN) m/z 350 [M 1]+. Síntesis de clorhidrato de 6-amino-8-metil-2’,3',5’,6'-tetrahidro-2H-espiro[imidazo[1,5-a]piridin-3,4'-piran]-1,5-diona (6) La síntesis del intermedio 6 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento F. Sólido de color blanquecino; Rendimiento: 0,45 g, 42 %; EM (IEN) m/z 285 [M 1]+.
Síntesis de 6-((6-(di-(terc-butoxicarbonil)-amino)-5-metilpirimidin-4-il)amino)-8-metil-2’,3',5’,6'-tetrahidro-2H-espiro[imidazo[1,5-a]piridin-3,4'-piran]-1,5-diona (8)
La síntesis del intermedio 8 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento H. Sólido de color blanquecino; Rendimiento: 0,26 g, 44 %; EM (IEN) m/z 557 [M 1]+.
Síntesis de 6-((6-amino-5-metilpirimidin-4-il)amino)-8-metil-2’,3',5’,6'-tetrahidro-2H-espiro[imidazo[1,5-]piridin-3,4’-piran]-1,5-diona (Comp. N.° 239)
La síntesis del compuesto 239 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento F. Sólido de color blanquecino; Rendimiento: 0,12 g, 72 %; EM (IEN) m/z 357 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,47 (s, 1H), 8,54 (s, 1H), 8,41 (s, 1H), 8,21 (s, 1H), 7,73 (s ancho, 2H), 3,96-3,90 (m, 2H), 3,69 (t, J = 12,44 Hz, 2H), 3,25-3,15 (m, 2H), 2,46 (m, 3H), 2,07 (s, 3H), 1,46-1,41 (m, 2H).
Ejemplo 240
Síntesis de 2-(6-((6-amino-5-metilpirimidin-4-il)amino)-8-metil-1,5-dioxo-1,5-dihidro-2H-espiro[imidazo[1,5-a]piridin-3,4'-piperidin]-1'-il)acetonitrilo (Comp. N.° 240)


,

90 C


Síntesis de 6-((6-(ciclopropanocarboxamido)-5-metilpirimidin-4-il)amino)-8-metil-1,5-dioxo-1,5-dihidro-2H-espiro[imidazo[1,5-a]pirídin-3,4’-piperidin-1'-carboxilato de terc-butilo (3)
La síntesis del intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento H. Sólido de color amarillo claro; Rendimiento: 0,75 g, 83 %; EM (IEN) m/z 524,41 [M 1]+.
Síntesis de 6-((6-amino-5-metilpirimidin-4-il)amino)-8-metil-1,5-dioxo-1,5-dihidro-2H-espiro[imidazo[1,5-a]piridin-3,4'-piperidin-1-carboxilato de terc-butilo (4)
La síntesis del intermedio 4 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento I. Sólido de color amarillo. Rendimiento: 650 mg, 33 %; EM (IEN) m/z 456,33 [M 1]+.
Síntesis de 6-((6-amino-5-metilpirimidin-4-il)amino)-8-metil-2H-espiro[imidazo[1,5-a]piridin-3,4'-piperidin]-1,5-diona (5)
La síntesis del intermedio 5 se realizó como se ha descrito anteriormente usando el protocolo general del procedimiento F. Sólido de color marrón claro; Rendimiento: 0,41 g, 80 %; EM (IEN) m/z 356,12 [M 1]+.
Síntesis de 2-(6-(6-amino-5-metilpirimidin-4-il)amino)-8-metil-1,5-dioxo-1,5-dihidro-2H-espiro[imidazo[1,5-a]piridin-3,4’-piperidin]-1’-ilacetonitrilo (Comp. N ° 240)
Clorhidrato de 6-((6-amino-5-metilpyrimidin-4-il)amino)-8-metil-2H-espiro[imidazo[1,5-a]piridin-3,4'-piperidin]-1,5-diona (5, 0,2 g, 0,56 mmol) se disolvió en dimetilformamida (10 ml). Se añadió N,N-diisopropiletilamina (0,22 g, 1,68 mmol), seguida de bromoacetonitrilo (0,10 g, 0,84 mmol). La mezcla de reacción se agitó a temperatura ambiente durante 30 min. Después de la terminación, la mezcla de reacción se diluyó con 50 ml de acetato de etilo, se lavó con solución saturada de cloruro de amonio y solución salina, se retiró el disolvente a presión reducida y se lavó el residuo resultante con metanol éter y pentano, se secó al vacío para proporcionar (6-((6-amino-5-metilpirimidin-4-il)amino)-8-metil-1,5-dioxol-1,5-dihidro-2H-espiro[imidazo[1,5-a]-3,4'-piperidin]-il)acetonitrilo (Comp. N.° 240) en forma de un sólido de color amarillo claro. Rendimiento: 95 mg, 43 %; Em (Ie N) m/z 395,18 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,26 (s, 1H), 8,48 (s, 1H), 8,12 (s, 1H), 7,99 (s, 1H), 6,47 (s, 2H), 3,74 (s, 2H), 3,33 3,30 (m, 2H), 2,92-285 (m, 2H), 2,70-2,58 (m, 2H), 2,44 (s, 3H), 1,98 (s, 3H), 1,53-1,46 (m, 2H).
Ejemplo 241
Síntesis de 6-((6-amino-5-cloropirimidin-4-il)amino)-1 '-(2,2-difluoroetil)-8-metil-2H-espiro[imidazo[1,5-a]piridin-3,4'-piperidin]-1,5-diona (Comp. N.° 241)


Síntesis de N-(5-cloro-6-(1-(2,2-difluoroetil)-8-metil-1,5-dioxo-1,5-dihidro-2H-espiro[imidazo[1,5-a]piridin-3,4’-piperidin]-6-il)amino)pirimidin-4-il)ciclopropanocarboxamida (3)
La síntesis del intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento H. Sólido de color amarillo claro; Rendimiento: 0,38 g, 70 %; EM (IEN) m/z 508,18 [M 1]+.
Síntesis de 6-((6-amino-5-cloropirimidin-4-il)amino)-1’-(2,2-difluoroetil)-8-metil-2H-espiro[imidazo[1,5-a]piridin-3,4'-piperidin]-1,5-diona (Comp. N.° 241)
La síntesis del compuesto 241 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento I. Sólido de color blanquecino; Rendimiento: 25 mg, 8 %; EM (IEN) m/z 410,14 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,26 (s, 1H), 8,53 (s, 1H), 8,51 (s, 1H), 8,18 (s, 1H), 7,21 (s ancho, 2H), 6,46 (t, J = 56,0 Hz, 1H), 3,35-2,98 (m, 8H), 2,46 (s, 3H), 1,71-1,52 (m, 2H).
Ejemplo 242
Síntesis de 6-((6-amino-5-cloropirimidin-4-il)amino)-8-metil-1 '-(2,2,2-trifluoroetil)-2H-espiro[imidazo[1,5-a]piridin-3,4'-piperidin]-1,5-diona (Comp. N.° 242)

Síntesis de N-(5-cloro-6-((8-metil-1,5-dioxo-1’-(2,2,2-trifluoroetil)-5-dihidro-2H-espiro[imidazo[1,5-a]piridin-3,4’-piperidin-1-il-aminopirimidin-3-ilciclopropanocarboxamida (3)
La síntesis del intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento H. Sólido de color blanquecino; Rendimiento: 0,40 g; EM (IEN) m/z 524,1 [M-1]-.
Síntesis de 6-((6-amino-5-cloropirimidin-4-il)amino)-8-metil-1’-(2,2,2-trifluoroetil)-2H-espiro[imidazo[1,5-a]ppiridin-3,4'-piperidin-1,5-diona (Comp. N.° 242)
La síntesis del compuesto 242 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento I. Rendimiento: 60 mg, 17 %; EM (IEN) m/z 458,11 [M 1]+; RMN 1H (400 MHz, DMSO-d6) ó 10,26 (s ancho, 1H), 8,53 (s, 1H), 8,50 (s, 1H), 8,17 (s, 1H), 7,10 (s ancho, 2H), 3,40-3,22 (m, 4H), 2,96-2,93 (m, 2H), 2,84 2,78 (m, 2H), 2,46 (s, 3H), 1,42-1,39 (m, 2H).
Ejemplo 243
Síntesis de 2-(6-(6-amino-5-metoxipirimidin-4-il)amino)-8-metil-1,5-dioxo-1,5-dihidro-2H-espiro[imidazo[1,5-a]piridin-3,4'-piperidin]-1'-il)acetonitrilo (Comp. N.° 243)

Síntesis de 6-((6-(ciclopropanocarboxamido)-5-metoxipirimidin-4-il)amino)-8-metil-1,5-dioxo-1,5-dihidro-2H-espiro[imidazo[1,5-a]pirídin-3,4’-piperidin-1'-carboxilato de terc-butilo (3)
La síntesis del intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento B. Sólido de color amarillo claro; Rendimiento: 0,55 g; EM (IEN) m/z 540,31 [M 1]+.
Síntesis de 6-((6-amino-5-metoxipirimidin-4-il)amino)-8-metil-1,5-dioxo-1,5-dihidro-2H-espiro[imidazo[1,5-a]piridin-3,4’-piperidin-1'-carboxilato de terc-butilo (4)
La síntesis del intermedio 4 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento I. Sólido de color amarillo; Rendimiento: 250 mg; EM (IEN) m/z 472,28 [M 1]+.
Síntesis de clorhidrato de 6-((6-amino-5-metoxipirimidin-4-il)amino)-8-metil-2H-espiro[imidazo[1,5-a]piridin-3,4'-piperidin-1,5-diona (5)
La síntesis del intermedio 5 se realizó como se ha descrito anteriormente usando el protocolo general del procedimiento F. Sólido de color amarillo; Rendimiento: 0,18 g; EM (IEN) m/z 372,22 [M 1]+.
Síntesis de 2-(6-(6-amino-5-metoxipirimidin-4-il)amino)-8-metil-1,5-dioxo-1,5-dihidro-2H-espiro[imidazo[1,5-a]piridin-3,4’-piperidin]-1'-ilacetonitrilo (Comp. N.° 243)
6-((6-Amino-5-metoxipirimidin-4-il)amino)-8-metil-2H-espiro[imidazo[1,5-a]piridin-3,4’-piperidin]-1,5-diona (5, 0,18 g, 0,44 mmol) se disolvió en dimetilformamida (5 ml). A esta mezcla se le añadió N,N-diisopropiletilamina (0,23 g, 1,76 mmol) seguida de bromoacetonitrilo (79 mg, 0,66 mmol). La mezcla de reacción se agitó a temperatura ambiente durante 2 horas. Después de la terminación, la mezcla de reacción se diluyó con solución saturada de cloruro de amonio (50 ml). El precipitado amarillo se filtró y se secó a presión reducida y después se purificó por purificación previa para proporcionar 2-(6-((6-amino-5-metoxipirimidin-4-il)amino)-8-metil-dioxo-1,5-dihidro-2H-espiro[imidazo[1,5-a]piridin-3,4'-piperidin]-1'-il)acetonitrilo (Comp. N.° 243) en forma de un sólido de color blanquecino. Rendimiento 40 mg, 22 %, EM (IEN) m/z 411,20 [M 1]+; RMN 1H (400 MHz, DMSO-d6 con d1-TFA) ó 8,37 (s, 1H), 8,27 (s, 1H), 4,49 (s, 2H), 3,76 (s, 3H), 3,74-3,71 (m, 2H ), 3,56-3,40 (m, 4H), 2,43 (s, 3H), 1,90-1,87 (m, 2H).
Ejemplo 244
Síntesis de 1'-(2,2-difluoroetil)-6-((5-metoxipirimidin-4-il)amino)-8-metil-2H-espiro[imidazo[1,5-a]piridin-3,4'-piperidin]-1,5-diona (Comp. N.° 244)

Síntesis de N-(6-((1’-(2,2-dtfluoroetil)-8-metil-1,5-dioxo-1,5-dihidro-2H-espiro[imidazo[1,5-a]piridin-3,4’-piperidin]-6-il)amino)-5-metoxipirimidin-4-il)ciclopropanocarboxamida (3)
La síntesis del intermedio 3 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento H. Sólido de color amarillo claro; Rendimiento: 0,30 g, 26 %; EM (IEN) m/z 504,14 [M 1]+.
Síntesis de 1’-(2,2-difluoroetil)-6-((5-metoxipirimidin-4-il)amino)-8-metil-2H-espiro[imidazo[1,5-a]piridin-3,4'-piperidin]-1,5-diona (Comp. N.° 244)
La síntesis del compuesto 244 se realizó como se ha descrito anteriormente usando el protocolo general del Procedimiento I. Sólido de color marrón claro; Rendimiento: 27 mg, 10 %; EM (IEN) m/z [M 1]+; RMN 1H (400 MHz, DMSO-d6 con d1-TFA) ó 8,42 (s, 1H), 8,26 (s, 1H), 6,56 (t, J = 56 Hz, 1H), 384-3,64 (m, 7H) 3,40 (m, 4H), 2,46 (s, 3H), 1,92-1,86 (m, 2H).
Ejemplo 245
Síntesis de 3'-((6-aminopirimidin-4-il)amino)-1'-metilespiro[ciclohexano-1,5'-pirrolo[3,4-¿]piridin]-4',7'(1'H,6'H)-diona (Comp. N.° 245)

Síntesis de 4-metoxi-6-(4-metoxibencil)-5,6-dihidro-7H-pirrolo[3,4-b]piridin-7-ona (2)
Tratar una solución de 3-(bromometil)-4-metoxipicolinato de metilo (1,0 mmol, 1 eq) en tetrahidrofurano con 4-metoxibencilamina (2,0 mmol, 2 eq) y se agita la reacción durante 16 h. Después de la terminación, diluir la mezcla con acetato de etilo y agua y separar las capas. Lavar la capa orgánica con ácido clorhídrico 1 M y agua. Concentrar la capa orgánica para proporcionar producto en bruto. Purificar el producto en bruto por cromatografía en columna de gel de sílice para proporcionar 4-metoxi-6-(4-metoxibencil)-5,6-dihidro-7H-pirrolo[3,4-b]piridin-7-ona (2).
Síntesis de 4’-metoxi-6'-(4-metoxibencil)espiro[ciclohexano-1,5'-pirrolo[3,4-b]piridin]-7’(6'H)-ona (4)
A una solución de 4-metoxi-6-(4-metoxibencil)-5,6-dihidro-7H-pirrolo[3,4-b]piridin-7-ona (2, 1,0 mmol, 1 eq) en tetrahidrofurano (25 ml) se le añade hidruro de sodio (2,5 mmol, 2,5 eq) a 0 °C. Se agita la mezcla durante 20 min y después se añade 1,5-dibromo-pentano (3, 1,5 mmol, 1,5 eq) y se agita durante 8 h. Una vez completado, se enfría la mezcla con agua a 0 °C y se añade acetato de etilo. Separar la capa y eliminar el disolvente para obtener producto en bruto. Se purifica el producto en bruto por cromatografía en columna de gel de sílice para proporcionar 4'-metoxi-6'-(4-metoxibencil)espiro[ciclohexano-1, 5’-pirrolo[3,4-b]piridin]-7’(6’H)-ona (4).
Síntesis de 4’-hidroxi-6'-(4-metoxibencil)espiro[ciclohexano-1,5’-pirrolo[3,4-b]piridin]-7'(6'H)-ona (5)
Se trata una solución de 4’-metoxi-6'-(4-metoxibencil)espiro[ciclohexano-1,5'-pirrolo[3,4-b]piridin]-7'(6’H)-ona (4, 1,0 mmol, 1 eq) en diclorometano (25 ml) con tribromuro de boro (2,0 mmol, 2 eq) a 0 °C. Se agita la mezcla durante 2 h a temperatura ambiente y se enfría con agua a 0 °C. Se extrae la mezcla con acetato de etilo y se elimina el disolvente a presión reducida para obtener el producto en bruto. Pureza del producto en bruto por cromatografía en columna de gel de sílice para obtener 4'-hidroxi-6’-(4-metoxibencil)espiro[ciclohexano-1,5'-pirrolo[3,4-b]piridin-7'(6'H)-ona (5).
Síntesis de 6’-(4-metoxibencil)-1 '-metilspiro[ciclohexano-1,5'-pirrolo[3,4-b]piridin]-4’, 7(1 ’H,6'H)-diona (6)
A una solución de 4'-hidroxi-6’-(4-metoxibencil)espiro[ciclohexano-1,5'-pirrolo[3,4-b]piridin]-7’(6’H)-ona (1,0 mmol, 1 eq) en tetrahidrofurano (25 ml), se le añadió hidruro de sodio (2,5 mmol, 2,5 equivalentes) a 0 °C y se agitó durante 20 min. Añadir yodometano (2,5 mmol, 2,5 eq) a la mezcla anterior y agitar durante 16 h. Una vez terminado, añadir agua a la reacción y extraer con acetato de etilo. Eliminar el disolvente a presión reducida para obtener el producto en bruto que se purifica por cromatografía en columna para proporcionar 6'-(4-metoxibencil)-metilspiro[ciclohexano-1,5’-pirrolo[3,4-b]piridin]-4',7'(1'H,6'H)-diona (6).
Síntesis de 3'-bromo-6’-(4-metoxibencil)-1 ’-metilspiro[ciclohexano-1,3,5-pirrolo[3,4-b]piridin] -4’, 7’(1'H,6’H)-diona (7) A una solución de 6'-(4-metoxibencil)-1'-metilespiro[ciclohexano-1,5'-pirrolo[3,4-b]piridin]-4',7'(1'H,6'H)-diona
(1,0 mmol, 1 eq.) en tetracloruro de carbono (25 ml), añadir N-bromosuccinimida y calentar la mezcla a 90 °C durante 16 h. Después de la terminación, la mezcla se diluyó con agua y se extrajo con acetato de etilo. Eliminar el disolvente a presión para obtener el producto en bruto. Se purifica el producto en bruto por cromatografía en columna para proporcionar 3'-bromo-6’-(4-metoxibencil)-metilespiro[ciclohexano-1,5'-pirrolo[3,4-b]piridin]-4',7’(1’H,6’H)-diona (7).
Síntesis de N-(6-((1 ’-metil-4', 7’-dioxo-1 ',4’,6', 7’-tetrahidrospiro[ciclohexano-1,5'-pirrolo[3,4-b]piridin-3’-il)amino)pirimidin-4-il)ciclopropanocarboxamida (9)
La síntesis del intermedio 9 se realiza como se ha descrito anteriormente usando el protocolo general del procedimiento B.
Síntesis de 3’-((6-aminopirimidin-4-il)amino)-6'-(4-metoxibencil)-1’-metilspiro[ciclohexano-1,5'-pirrolo[3,4-b]piridin]-4',7'(1'H,6'H)-diona (10)
La síntesis del intermedio 10 se realiza como se ha descrito anteriormente usando el protocolo general del Procedimiento I.
Síntesis de 3’-((6-aminopirimidin-4-il)amino)-1'-metilespiro[ciclohexano-1,5'-pirrolo[3,4-b]piridin]-4’,7',6'H)-diona (Comp. N ° 245)
Se trata una solución de 3’-((6-aminopirimidin-4-il)amino)-6'-(4-metoxibencil)-metilspiro[ciclohexano-1,5'-pirrolo[3,4-b]piridin]-4’,7’(1’H,6'H)-diona (1,0 mmol, 1 eq) en 1,2-dicloroetano (15 ml) con ácido trifluoroacético (20 mmol, 20 eq) C durante 5 h. Una vez terminado, enfriar la mezcla y eliminar el disolvente. Triturar la mezcla con éter para obtener sólido. El sólido se vuelve a triturar con metanol para obtener 3’-((6-aminopirimidin-4-il)amino)-1’-metilrspiro[ciclohexano-1,5’-pirrolo[3,4-b]piridin]-4’,7’(1’H,6’H)-diona (Comp. N.° 245).
Ejemplo 246
Síntesis de 6'-((6-aminopirimidin-4-il)amino)-8'-metil-2'H-espiro[ciclohexano-1,3'-indolizin]-1 ',5'-diona (Comp. N.° 246)

Síntesis de 5-bromo-N-metoxi-N,3-dimetil-6-oxo-1,6-dihidropiridin-2-carboxamida (2)
A una solución de ácido 5-bromo-3-metil-6-oxo-1,6-dihidropiridin-2-carboxílico (1, 1,0 mmol, 1 eq) en dimetilformamida (25 ml), añadir N-(3-dimetilaminopropil)-N’-etilcarbodiimida (2 mmol, 2 eq), trietilamina (3,0 mmol, 3 eq) y clorhidrato de N,0-dimetilhidroxilamina (1,5 mmol, 1,5 eq) y se agita la reacción durante 6 h. Después de la terminación agregar agua a la mezcla y extraer con acetato de etilo. Lavar la capa de acetato de etilo con agua y salmuera, eliminar el disolvente a presión reducida para obtener 5-bromo-N-metoxi-N,3-dimetil-6-oxo-1,6-dihidropiridin-2-carboxamida (2).
Síntesis de 3-bromo-N-metoxi-1-(4-metoxibencil)-N, 3-dimetil-6-oxo-1,6-dihidropiridin-2-carboxamida (3)
Se añaden hidruro de sodio (2,5 mmol, 2,5 equivalentes) a una solución enfriada de clorhidrato de 5-bromo-N-metoxi-N,3-dimetil-6-oxo-1,6-dihidropiridin-2-carboxamida (2,1 mmol, 1 eq) en dimetilformamida (25 ml) y se agita la reacción durante 20 min. Se añade cloruro de 4-metoxibencilo (1,2 mmol, 1,2 eq) y se agita la reacción durante 16 h. Una vez completada la reacción, se enfría con agua y se extrae con acetato de etilo. Eliminar el disolvente y purificar el producto en bruto por cromatografía en columna para proporcionar 5-bromo-N-metoxi-1-(4-metoxibencil)-N,3-dimetil-6-oxo-1,6-dihidropiridin-2-carboxamida (3).
Síntesis de 3-(5-bromo-1-(4-metoxibencil)-3-metil-6-oxo-1,6-dihidropiridin-2-il)-2-(1-hidroxiciclohexil)-3-oxopropanoato de etilo (5)
A una solución de 2-(1-hidroxiciclohexil)acetato de etilo (4, 1,2 mmol, 1,2 eq.) en tetrahidrofurano (15 ml), se le añade diisopropilamida de litio (2,5 mmol, 2,5 eq) a -78 °C y se agita la reacción durante 20 min. Se añade una solución en tetrahidrofurano (3,1 mmol, 1,0 equiv.) de 5-bromo-N-metoxi-1-(4-metoxibencil)-N,3-dimetil-6-oxo-1,6-dihidropiridin-2-carboxamida (10 ml) a -78 °C en 10 min y continuar agitando durante otras 3 h. Una vez finalizado, añadir una solución acuosa saturada de cloruro de amonio y extraer la masa de reacción con acetato de etilo. Evaporar el disolvente a presión reducida y obtener el producto en bruto que se hace pasar a través de un lecho de gel de sílice para obtener 3-(5-bromo-1-(4-metoxibencil)-3-metil-6-oxo-1,6-dihidropiridin-2-il)-2-(1-hidroxiciclohexil)-3-oxopropanoato de metilo (5).
Síntesis de 6'-bromo-8'-metil-1’,5’-dioxo-1’,5’-dihidro-2’H-espiro[ciclohexano-1,3'-indolizin]-2'-carboxilato de etilo (6)
A una solución de 3-(5-bromo-1-(4-metoxibencil)-3-metil-6-oxo-1,6-dihidropiridin-2-il)-2-(1-hidroxiciclohexil)-3-oxopropanoato de metilo (5, 1,0 mmol, 1,0 eq) en 1,2-dicloroetano (15 ml), añadir ácido trifluoroacético (10 mmol, 10 eq) y calentar la reacción a 60 °C durante 16 h. Después de la terminación, se elimina el disolvente y se calma la reacción con amoníaco y se extrae con diclorometano. Se elimina el disolvente a presión reducida y se purifica el producto en bruto usando cromatografía en columna de gel de sílice para proporcionar 6'-bromo-8'-metil-1',5'-dioxo-1',5'-dihidro-2'H-espiro[ciclohexano-1,3'-indolizin]-2'-carboxilato de etilo (6).
Síntesis de 6’-bromo-8'-metil-2’H-espiro[ciclohexano-1,3'-indolizn]-1,’5’-diona (7)
A una solución de etil 6'-bromo-8'-metil-1',5'-dioxo-1',5'-dihidro-2'H-espiro[ciclohexano-1,3-indolizina]-2'-carboxilato de etilo (6, 1,0 mmol, 1,0 eq) en dimetilsulfóxido (15 ml), se añade cloruro de litio (5,0 mmol, 5 eq) y se calienta la reacción a 140 °C durante 16 h. Después de la terminación, enfriar la reacción, añadir agua y extraer con diclorometano. Eliminar el disolvente a presión reducida y purificar el producto en bruto por cromatografía en columna de gel de sílice para obtener 6'-bromo-8'-metil-2'H-espiro[ciclohexano-1,3-indolizin]-1',5'-diona (7).
Síntesis de N-(6-((8’-metil-1' 5’-dioxo-1 5’-dihidro-2'H-espiro[ciclohexano-1,3-indoliz'n]-6’-il)amino)pirimidin-4-il)ciclopropanocarboxamida (9)
La síntesis del intermedio 9 se realiza como se ha descrito anteriormente usando el protocolo general del Procedimiento H.
Síntesis de 6’-((6-aminopirimidin-4-il)amino)-8'-metil-2’H-espiro[ciclohexano-1,3'-indolizn]-1’,5’-diona (Comp. N.° 246)
La síntesis del compuesto 246 se realiza como se ha descrito anteriormente usando el protocolo general del Procedimiento I.
Ejemplo 247
Síntesis de 6'-((6-amino-5-metilpirimidin-4-il)amino)-8'-metil-2'H-espiro[ciclohexano-1,3-indolizin]-1',5'-diona (Comp. N.° 247)

Síntesis de N-(5-metil-6-((8’-metil-1’,5’-dioxo-1’,5’-dihidro-2'H-espiro[ciclohexano-1,3-indolizin]6’-il)amino)pirimidin-4-il)ciclopropanocarboxamida (3)
La síntesis del intermedio 3 se realiza como se ha descrito anteriormente usando el protocolo general del Procedimiento B.
Síntesis de 6’-((6-amino-5-metilpirimidin-4-il)amino)-8'-metil-2’H-espiro[ciclohexano-1,3-indolizin]-1’,5’-diona (Comp. N.° 247)
La síntesis del compuesto 247 se realiza como se ha descrito anteriormente usando el protocolo general del Procedimiento I.
Ejemplo 248
Síntesis a gran escala de compuestos intermedios y compuestos de Fórmula I
Los compuestos de acuerdo con la presente invención son agentes terapéuticos candidatos para tratar trastornos relacionados con Mnk, tales como trastornos inflamatorios y cáncer. Para proporcionar cantidades comerciales de los compuestos de la invención, la presente invención ilustra un protocolo de síntesis a gran escala para un compuesto de Fórmula I de ejemplo, así como métodos para la fabricación y caracterización de la forma de sal de clorhidrato (forma de sal HCl) de un compuesto de este tipo.
Preparación de N-(6-aminopirimidin-4-il)ciclopropanocarboxamida:

A una solución agitada de 6-cloropirimidin-4-amina (4900 g, 1 equiv., 37,08 mol) en tetrahidrofurano (10 V, 50 l), a 0 °C se le añadió N,N-dimetilaminopiridina (463 g, 0,1 equiv, 3,70 mol). Después se añadió dicarbonato de di-tercbutilo (24,8 l, 3 equiv, 113,9 mol) lentamente durante 1 h (se observó desprendimiento de gas) a la reacción resultante. La mezcla de reacción se volvió de color marrón oscuro con agitación a temperatura ambiente durante un periodo de 16 h. El progreso de la reacción se controló mediante CCF y CLEM. El análisis por CLEM mostró la desaparición completa del MP, así como los picos correspondientes al producto, 73,89 % a TA-2,55 ((M 1) -330,3); (6-cloropirimidin-4-il)carbamato de terc-butilo, 4,09 % a TA-1,98 ((M 1) - 330,3).
Después de la terminación la reacción, la mezcla de reacción se vertió en una mezcla de hielo/agua (30 l) y se agitó adicionalmente durante 30 minutos antes de la extirpación del disolvente de la fase acuosa con acetato de etilo (10 l). Se separaron las fases orgánica y acuosa y la capa acuosa resultante se extrajo dos veces con acetato de etilo (10 l, 2 veces). La capa orgánica combinada se lavó dos veces con agua (10 l, 2 veces), después con salmuera (10 l, 1 veces), y se secó sobre sulfato de sodio anhidro. La capa orgánica seca se concentró a presión reducida a 50 °C para obtener el producto en bruto que se suspendió con hexano (10 l) durante 1 h, se filtró y se secó a presión reducida a 50 °C para obtener un sólido de color rojo ladrillo. Rendimiento: 10,3 kg, (82,6 %). e M (IEN) m/z 329,78 [M 1]+; Pureza CLEM: 99,37 %; RMN 1H (400 MHz, DMSO-d6) 8: 8,86 (s, 1H), 7,85 (s, 1H), 1,48 (s, 18H).
B. Preparación de (6-(ciclopropanocarboxamido)pirimidin-4-il)carbamato de di-terc-butilo (3):
A una solución en agitación de (6-cloropirimidin-4-il)carbamato de di-terc-butilo (5000 g, 1 equiv., 15,20 mol) en dioxano (5 V, 25 l), a temperatura ambiente se le añadió ciclopropanocarboxamida (1291 g, 1 equiv, 15,20 mol) seguida de la adición de carbonato de cesio (3950 g, 0,8 equiv, 12,15 mol). Después de purgar la mezcla de reacción (solución marrón oscuro) con argón durante 30 minutos, se añadieron Xantphos (120 g, 0,015 equiv., 0,23 mol) y acetato de paladio (II) (51 g, 0,015 equiv., 0,23 mol). La purga de la masa de reacción con argón se continuó durante otros 15 min y la mezcla de reacción se calentó después hasta 90 °C y se mantuvo a esa temperatura durante 4 h, tiempo durante el cual el color de la masa de reacción cambió a naranja. El progreso de la reacción se controló mediante CCF y CLEM. El análisis por CLEM mostró la desaparición completa del MP y un pico correspondiente al producto a TA-2,32 min, 86,92 %, ((M 1 )-379,15). Además, el análisis por CLEM mostró picos correspondientes a (6-(ciclopropanocarboxamido)pirimidin-4-il)carbamato de terc-butilo 3,83 %; a TA-1,94 ((M 1) -279,08) y algunos subproductos desconocidos (5,18 % a TA-2,43 (M 1)-605,2, 1,48 % a TA-2,83 (M 1)-589,2).
Después de completar la reacción según se juzgó por CCF y CLEM, la mezcla de reacción se enfrió a 50 °C y se filtró a través de lecho de celite. El lecho de celite se lavó con EtOAc (10 l, 3 veces) para asegurar la extracción completa del producto. Las capas orgánicas combinadas se lavaron con agua (10 l, 2 veces), se secaron sobre sulfato de sodio anhidro y se concentraron a presión reducida para obtener 6,2 kg. Se añadió éter dietílico (6,0 l) al material en bruto y la mezcla se agitó durante 30 minutos para obtener un sólido que fluye libremente. El sólido se filtró, se lavó con éter (1 l, 2 veces) y después se secó para proporcionar (6-(ciclopropanocarboxamido)pirimidin-4-il)carbamato de di-terc-butilo en forma de un sólido de color naranja. Este compuesto se usó en la siguiente etapa sin purificación adicional. Rendimiento: 4,5 kg, (78,2 %); EM (IEN) m/z 378,43 [M 1]+; Pureza CLEM: 95,10 %; RMN 1H (400 MHz, DMSO-d6) 8: 11,30 (s, 1H), 8,66 (s, 1H), 8,25 (s, 1H), 2,02 (m, 1H), 1,46 (s, 18H), 0,85 (m, 4H).
C. Preparación de N-(6-aminopirimidin-4-il)ciclopropanocarboxamida (4):
Se añadió lentamente ácido trifluoroacético (16 l, 10 equiv, 212 mol) durante 1 hora a una solución en agitación de (6-(ciclopropanocarboxamido)pirimidin-4-il)carbamato de di-terc-butilo ((3), 8050 g, 1 equiv, 21,20 mol) en diclorometano (5 V, 40 l). Se observó el desprendimiento del gas durante la adición de ácido trifluoroacético y la
reacción se hizo de color marrón oscuro cuando se agitó continuamente durante 4 h a temperatura ambiente. El progreso de la reacción se controló por CCF.
Después de la terminación la reacción, la masa de reacción se concentró a sequedad a presión reducida (el TFA se debe separar por destilación tanto como sea posible antes de la adición de amoníaco) y se añadió diclorometano (25 l) al residuo. La mezcla se enfrió a 0 °C y se añadió lentamente (pH-10) NH4OH (solución acuosa al 25 %, 6 l) durante 30 min mientras se agitaba la mezcla de reacción continuamente. La mezcla resultante se agitó a 0 °C durante 30 min adicionales y el sólido formado se filtró y se lavó con agua (10 l, 2 veces) seguido de lavado con metanol (2 l, 2 veces) y diclorometano (15 l). El sólido lavado se secó a alto vacío durante la noche para proporcionar N-(6-aminopirimidin-4-il)ciclopropanocarboxamida en forma de un sólido de color amarillo cremoso. Rendimiento: 2,32 Kg (61,02 %); EM (IEN) m/z 178,19 [M 1]+; UPLC: 99,80 %; RMN 1H (400 MHz, DMSO-d6) 8: 10,54 (s, 1H), 8,10 (s, 1H), 7,10 (s, 1H), 6,72 (s ancho, 2H), 1,79 (m, 1H), 0,79 (m, 4H).
El material final de todos los lotes se combinó, se suspendió con dicrometano (10 l) durante 20 min, se filtró y se secó al vacío durante 6 h para obtener una sola carga de N-(6-aminopirimidin-4-il)ciclopropanocarboxamida 3665,3). Preparación de 5-bromo-3-metil-6-oxo-1,6-dihidropiridin-2-carboxamida de etilo:


en mento prome o- , Rendimiento promedio- 37 ,5 % 
A. Preparación de 5-bromo-3-metilpicolinato de etilo (2):
A una solución en agitación de ácido 5-bromo-3-metilpicolínico (10000 g, 1 equiv., 46,29 mol) en etanol (50 l) a 0 °C se le añadió ácido sulfúrico (2,52 l, 1 equiv., 46,29 mol) durante 1 h. Después de la adición de ácido sulfúrico, la mezcla se calentó a 95 °C durante 20 h. El progreso de la reacción se controló usando CCF y CLEM. Después de 20 horas, el análisis por CLEM mostró 12,61 % del MP sin reaccionar, a TA-0,67 ((M-1)-215,92) y 86,95 % de producto a TA-2,04 ((M l)-244,2). La masa de reacción calentada durante 6 h adicionales y el análisis de CLEM después del calentamiento adicional mostró 10,07 % del MP sin reaccionar, a TA-0,7 y 89,93 % de producto a TA-2,08 (M 1)-244,2. La reacción se detuvo concentrando la mezcla de reacción a presión reducida para separar el máximo de disolvente posible. Al residuo se añadió diclorometano (DCM) (25 l) y la mezcla de reacción se vertió en Agua helada (20 l). La capa orgánica (DCM) se separó de la capa acuosa y esta última se extrajo adicionalmente dos veces con DCM (10 l). Las capas orgánicas combinadas se lavaron con solución saturada de bicarbonato de sodio (30 l), se secaron sobre sulfato de sodio y se concentraron a presión reducida para obtener un aceite marrón. Este aceite marrón se usó en la siguiente etapa sin purificación adicional. Rendimiento: 10,1 kg, (89,4 %); EM (IEN) m/z 244,2 [M 1]+; Pureza CLEM: 99,11 %; RMN 1H (400 MHz, DMSO-d6) 8: 8,61 (s, 1H), 8,13 (s, 1H), 4,29 (c, 2H), 2,44 (s, 1H), 1,28 (t, 3H).
B. Preparación de 1-óxido de 5-bromo-2-(etoxicarbonil)-3-metilpiridina (3):
A una solución agitada de 5-bromo-3-metilpicolinato de etilo (9,49 kg, 1 equiv., 38,88 mol) en diclorometano (47,5 l) a 0 °C se le añadió peróxido de hidrógeno de urea (6,3 kg, 1,75 equiv., 68,04 mol), seguido de la adición de anhídrido trifluoroacético (9,7 l, 1,75 equiv, 68,04 mol) a 0 °C durante un período de 3 h. Esta reacción es fuertemente exotérmica (0-30 °C). Después de la adición, la mezcla de reacción se calentó suavemente a temperatura ambiente (TA) y se dejó agitar a TA durante 16 h. El progreso de la reacción se controló usando análisis de CCF y CLEM. Al final de las 16 horas, el análisis por CLEM mostró 0,74 % del MP sin reaccionar a TA-2,04 ((M 1)-244,02); 91,61 % de producto a TA-1,38 ((M 1)-260,01) y algunos subproductos desconocidos (4,67 % a TA-1,28 (M 1 )-188,1, 2,98 % a TA-1,90 (M 1)-217,11).
La mezcla de reacción en bruto se vertió en una mezcla de hielo/agua (30 l) y se permitió que las fases orgánica y acuosa se separaran. La capa acuosa se extrajo con diclorometano (10 l), se separó de la capa acuosa y se
combinó con la primera capa orgánica. Las capas orgánicas combinadas se lavaron después con una solución acuosa de bicarbonato de sodio (50 l), seguido de agua (20 l), se secaron sobre sulfato de sodio y se concentraron para proporcionar el 1-óxido de 5-bromo-2-(etoxicarbonil)-3-metilpiridina como un aceite amarillo rojizo. Este compuesto se usó para la siguiente etapa sin purificación adicional. Rendimiento: 9 kg, (89 %); EM (IEN) m/z 260,09 [M 1]+; Pureza CLEM: 99,34 %; RMN 1H (400 MHz, DMSO-d6) 8: 8,56 (s, 1H), 7,67 (s, 1H), 4,32 (c, 2H), 2,21 (s, 3H), 1,27 (t, 3H).
C. Preparación de 5-bromo-3-metil-6-oxo-1,6-dihidropiridin-2-carboxilato de etilo (4):
A una solución en agitación de 1-óxido de 5-bromo-2-(etoxicarbonil)-3-metilpiridina (9 kg, 1 equiv., 34,62 mol) en N,N-dimetilformamida (35 l) a TA se le añadió anhídrido trifluoroacético (8,65 l, 1,75 equiv, 60,58 mol), durante un período de 3 h. La temperatura de la reacción se mantuvo a 35-40 °C durante la adición de anhídrido trifluoroacético. La mezcla de reacción se agitó a 40 °C (interno) durante 2 horas y el progreso de la reacción se controló usando análisis de CCF y CLEM. El análisis de CLEM mostró 6,26 % del MP sin reaccionar a TA-1,37 ((M 1)-260,01), 31,33 % de producto a TA-1,58 ((M 1)-259,97), 25,88 % de DMF a TA-0,31 y algunos subproductos desconocidos 8,06 % a TA-1,06 (M 1)-259,97, 22,42 % a TA-2,29 (M 1)-323,86).
Después del análisis de CLEM, la masa de reacción se agitó durante 1 h adicional, después se vertió en agua enfriada con hielo (100 l), para proporcionar un sólido blanquecino que se filtró y se lavó con agua (20 l). El compuesto sólido en bruto se lavó adicionalmente con n-hexano (20 l) y se secó a alto vacío para proporcionar 5-bromo-3-metil-6-oxo-1,6-dihidropiridin-2-carboxilato de etilo en forma de un sólido de color blanquecino. Rendimiento: 6 Kg (66,6 %); EM (IEN) m/z 260,09 [M 1]+; UPLC: 66,49 % (96,85 % por HPLC); RMN 1H (400 MHz, DMSO-d6) 8: 11,53 (s ancho, 1H), 7,99 (s, 1H), 4,23 (c, 2H), 2,27 (s ancho, 3H), 1,28 (t, 3H).
D. Preparación de 5-bromo-3-metil-6-oxo-1,6-dihidropiridin-2-carboxamida de etilo (5):
A una solución en agitación de 5-bromo-3-metil-6-oxo-1,6-dihidropiridin-2-carboxilato de etilo (1 kg, 1 equiv., 3,84 mol) en etanol (5 l) se añadió 30 % de amoníaco ac. (7 l). El matraz de fondo redondo se cerró después y se selló para evitar que el amoníaco saliera de la masa de reacción. La mezcla de reacción se agitó después a 40 °C durante 16 h y se controló el progreso de la reacción usando análisis de CCF y CLEM. El análisis de CLEM de la mezcla de reacción mostró la desaparición del MP, 64,82 % de producto a TA-1,09 (M 1 )-231,17), y algunos subproductos desconocidos 18,2 % a TA-0,83 (M 1 )-232,11), 4,74 % A TA-1,76 ((M 1)-262,19).
Después de CLEM, la masa de reacción se concentró a presión reducida para separar etanol y agua para proporcionar un material sólido en bruto. El material en bruto (húmedo) obtenido a partir de varios lotes sintéticos se combinó y se suspendió con solución acuosa saturada de bicarbonato de sodio (50 l) durante 2 h, se filtró y después se lavó con agua (10 l, 2 veces). El sólido blanquecino obtenido de este modo se lavó con diclorometano (20 l) y después se suspendió con metanol al 20 % en diclorometano (30 l) durante 1 hora. La suspensión se filtró, se lavó con diclorometano (15 l) y se secó al vacío durante 6 horas a 60 °C para proporcionar 2,69 kg de 5-bromo-3-metil-6-oxo-1,6-dihidropiridin-2-carboxamida (5) en forma de un sólido de color blanquecino. Rendimiento: 2,69 Kg (50,94 %); EM (IEN) m/z 231 [M 1]+; UPLC: 99,80 %; RMN 1H (400 MHz, DMSO-d6) 8: 11,85 (s ancho, 1H), 7,88 (s, 2H), 7,75 (s, 1H), 2,14 (s, 1H).
En un experimento separado, se suspendió 5-bromo-3-metil-6-oxo-1,6-dihidropiridin-2-carboxamida, 2,2 kg, obtenida combinando el sólido de varios lotes sintéticos en diclorometano (5,0 l) durante 30 minutos, se filtró y se secó al vacío para proporcionar 4554,1 g de 5-bromo-3-metil-6-oxo-1,6-dihidropiridin-2-carboxamida.
Síntesis de 6'-((6-aminopirimidin-4-il)amino)-8,-metil-2,H-espiro[ciclohexano-1,3'-imidazo[1,5-a]piridin]-1 ',5'-diona (Comp. 107):

Se cargaron bicarbonato de sodio sólido (3,3 kg) y agua (40,0 l, DI) en un cartucho de 45 l y se agitó hasta que se disolvió el sólido. Se colocó 5-bromo-3-metil-6-oxo-1,6-dihidropiridin-2-carboxamida (1,5 kg) en un reactor de 50 l y se añadió la solución de bicarbonato de sodio saturado mientras se mantenía la temperatura de esta mezcla de reacción a 18 °C. El análisis por HPLC de la mezcla de reacción después de 16 h mostró que la 5-bromo-3-metil-6-oxo-1,6-dihidropiridin-2-carboxamida (1) tenía una pureza del 92,8 %. La 5-bromo-3-metil-6-oxo-1,6-dihidropiridin-2-carboxamida sólida se filtró a través de un filtro de Nutsche (tejido de tejido apretado de polipropileno de 18” (45,72 cm)). El reactor y torta de filtrado se aclararon con agua (3,0 l) y el sólido se acondicionó a temperatura ambiente hasta que el líquido no fluyó más. El material sólido se transfirió a bandejas de secado y se secó al vacío a 45-50 °C. El material seco pesó 1,16 kg (rendimiento -77 %) y tenía una pureza del 92,8 %. El análisis KF mostró 2,6 %) de agua residual.
B. Síntesis de 6'-bromo-8'-metil-2-espiro[ciclohexano-1,3’-imidazo[1,5-a]piridin]-1’,5'-diona (2):
Se añadieron 5-bromo-3-metil-6-oxo-1,6-dihidropiridin-2-carboxamida (1), (1,16 kg, 1,0 equiv.); 1,4-dioxano (13,8 l); y ciclohexanona (1,96 kg) a un reactor de 50 l y se agitaron a 75-125 rpm. Se añadió ácido sulfúrico (0,13 l) al reactor usando una bomba dosificadora. La temperatura de la imagen de reacción fue de 19,5 °C al comienzo de la adición de ácido sulfúrico y aumentó a 21,8 °C tras la adición completa de ácido sulfúrico. La temperatura de la mezcla de reacción (temperatura del lote) se elevó a 95 °C. Después de agitar durante 3 h, el análisis por HPLC de la mezcla de reacción indicó que se había completado la reacción. La temperatura del lote se ajustó después a 20-30 °C y el disolvente se destiló al vacío (28,5"/Hg (965,12 kPa), temperatura de la camisa de 80 °C) hasta no menos del 75 % del volumen de reacción inicial.
Después de la destilación, la temperatura del lote se ajustó a 25 °C y se mantuvo a esa temperatura durante 13 h. La mezcla de reacción (lote) se filtró a través de un filtro de Nutsche (18” (45,72 cm)), y el licor madre se añadió de nuevo al reactor como un aclarado y después se añadió a la torta de filtro. Se añadió agua (12 l, DI) al reactor como un aclarado y después se transfirió a la torta de filtro. La torta de filtro se acondicionó hasta que el líquido ya no fluyó. La materia sólida obtenida de este modo se transfirió a bandejas de secado y se secó al vacío a 45-50 °C. El material seco pesó 1,28 kg (82 % de rendimiento) y tuvo una pureza >99 %. El análisis por KF mostró 1,4 % de agua. RMN 1H (500 MHz, DMSO-d6) ó 10,37 (s, 1H), 8,01 (s, 1H), 2,82-2,92 (m, 2H), 2,38 (s, 3H), 1,75-1,65 (m, 5H), 1,43 (d, J = 24 Hz, 2H), 1,25-1,15 (m, 1H).
C. Síntesis de N-(6-((8’-metil-1 5’-dioxo-1 5’-dihidro-2'H-espiro[ciclohexano-1,3’-imidazo[1,5]piridin-4’-il)amino)pirimidin-4-il)ciclopropanocarboxamida (4):
A un reactor de 50 l se le añadió 6'-bromo-8'-metil-2'H-espiro[ciclohexano-1,3'-imidazo[1,5-a]piridin]-1',5'-diona (2) (1,23 kg); 1,4-dioxano (15,4 l), N-(6-aminopirimidin-4-il)ciclopropanocarboxamida (3), (0,65 kg); y carbonato de cesio (1,03 kg) y la mezcla de reacción se agitó mientras se rociaba con argón durante 20 minutos. A temperatura
ambiente. Al reactor se le añadió acetato de paladio (II) (18,0 g) y Xantphos (46,0 g) y se continuó con burbujeo con argón durante 20 minutos adicionales. El tubo de burbujeo se retiró después y la temperatura del lote se ajustó a 95 °C. La mezcla de reacción (carga) se agitó durante 18 h, momento en el que el análisis por HPLC indicó que la reacción estaba completa. Una vez completada la reacción, la temperatura del lote se ajustó a 20 °C. Se añadió agua (24,6 l, DI) al reactor y el lote se agitó durante 1 h, seguido de filtración a través de un filtro Nutsche (18” (45,72 cm)). Se usó el licor madre para aclarar el reactor y después se añadió a la torta de filtración. Después, se añadió acetona (6,15 l) en forma de aclarado al reactor y después se transfirió a la torta de filtración. La torta de filtración se acondicionó hasta que no fluyó líquido del filtro. Después de esto, la torta se añadió de nuevo al reactor, se suspendió usando metanol (12,0 l) y se agitó a 125 rpm. La temperatura del lote se ajustó a 20 °C y se continuó la agitación durante 10 minutos. El lote se filtró de nuevo a través de un filtro Nutsche (18” (45,72 cm)) y se acondicionó hasta que el líquido ya no fluía del filtro. El material sólido obtenido de este modo se transfirió a bandejas de secado y se secó al vacío a 45 °C. El material seco pesó 1,42 kg, (98 % de rendimiento) y tenía una pureza de 97,5 %. RMN 1H (500 MHz, DIVISOR) ó 10,85 (s ancho, 1H), 10,07 (s ancho, 1H), 9,09 (s, 1H), 8,53 (s, 1H), 8,46 (s, 1H), 7,85 (s, 1H), 3,95-3,05 (m, 2H), 2,45 (s, 3H), 1,95-2,05 (m, 1H), 1,80-1,60 (m, 5H), 1,44 (d, J = 24 Hz, 2H), 1,25-1,15 (m, 1H), 0,89-0,80 (m, 4H).
D. Síntesis de 6’-((6-aminopirimidin-4-il)amino)-8'-metil-2'H-espiro[ciclohexano-1,3,3-imidazo[1,5-a]piridin]-1 '5'-diona (107):
Se añadieron W-(6-((8’-metil-T,5’-dioxo-T,5’-dihidro-2,H-espiro[ciclohexano-1,3-imidazo[1,5-a]piridin]-6’-il)amino)pirimidin-4-il)ciclopropanocarboxamida (comp. 4), (1,42 kg); tetrahidrofurano (5,7 l); y EtOH (5,7 l) a un reactor de 50 l y se agitaron a 100 rpm. La temperatura del lote se ajustó a 20 °C. A una garrafa de 45 l se le añadió agua (5,7 l, DI) y KOH (1,17 kg) y el contenido de la garrafa se agitó hasta que se formó una solución. Después, se añadió la solución de KOH al reactor de 50 l seguido de la adición de etilendiamina (2,83 l). La temperatura del lote aumentó a 33 °C tras la adición de etilendiamina y se reajustó a 20 °C. Después de agitar durante 16 h, el análisis de HPLC indicó que quedaba 18,6 % del compuesto (4) sin reaccionar. Por tanto, se añadió una solución de KOH (1,17 kg) y agua (5,7 l) al reactor y se continuó la agitación a 20 °C durante 16 h adicionales. Tras la agitación, el análisis por HPLC indicó un 1,3 % de compuesto no reaccionado (4). El pH del lote se ajustó a 2 mediante la adición de HCl concentrado (11,8 kg), durante un periodo de tiempo de 2,5 h, y un sólido comienza a formarse cuando el lote (mezcla de reacción) está a un pH de 12,7. La temperatura del lote se ajustó a 20 °C y la mezcla se agitó durante 10 minutos, después de lo cual el lote que contenía material sólido se filtró a través de un filtro Nutsche (18” (45,72 cm)).
Después, el reactor se aclaró con agua (14,15 l, DI) y el aclarado acuoso se transfirió al filtro mientras se suspendía manualmente el sólido en el lavado. Se realizó un segundo aclarado con agua (14,15 l, DI) y el aclarado se transfirió de nuevo al filtro mientras se suspendía manualmente el sólido en el lavado. Después se añadieron bicarbonato de sodio (1,3 kg) y agua (26,0 l, DI) al reactor de 50 l lavado y la torta de filtración se introdujo lentamente en el reactor durante un periodo de tiempo de aproximadamente 30 minutos para evitar la liberación de gas en exceso. La suspensión resultante se agitó durante 2 h, seguido de filtración a través de un filtro de Nutsche (18” (45,72 cm)), La torta de filtración se volvió a suspender en una solución acuosa de bicarbonato de sodio, Se agitó durante 2 h y se filtró a través de un filtro Nutsche (18” (45,72 cm)). Después del lavado con agua, la torta de filtración se dejó acondicionar durante la noche y después se transfirió a bandejas de secado y se secó al vacío a 45 °C. El lote seco pesaba 1,05 kg, (80 % de rendimiento) y tenía una pureza del 98,5 %. El análisis de IC mostró 0,7 % de cloruro. RMN 1H (500 MHz, DMSO-d6) ó 10,20 (s, 1H), 9,68 (s, 1H), 8,47 (s, 1H), 8,09 (s, 1H), 7,97 (s ancho, 2H) 1H), 3,00 2,90 (m, 2H), 2,43 (s, 3H), 1,80-1,60 (m, 5H), 1,5 (d, J = 24 Hz, 2H), 1,25-1,12 (m, 1H).
E. Síntesis de 6’-((6-aminopirimidin-4-il)amino)-8'-metil-2'H-espiro[ciclohexano-1,3’-imidazo[1,5-a]piridin]-1',5'-diona (107 HCl):
Se añadió 6’-((6-Aminopirimidin-4-il)amino)-8'-metil-2'H-espiro[ciclohexano-1,3’-imidazo[1,5-a]piridin]-1’,5’-diona ((107), (0,99 kg)) a un recipiente de 50 l. Se añadieron tetrahidrofurano (8,22 l), etanol (8,22 l) y agua (8,22 l, DI) a un cartucho de 45 l separado. Esta solución se transfirió después al recipiente de 50 l que contenía el compuesto 9107) y la temperatura de la mezcla de reacción se ajustó a 5 °C. A la mezcla de reacción fría se le añadió KOH (0,45 kg) y toda la mezcla se agitó hasta que se formó una solución. La solución se transfirió después a la garrafa de 45 l y después se pasó, al vacío, a través de un filtro de pulido (filtro de abrillantador Hepa Cap de 0,3 pm) de vuelta al recipiente de 50 l.
La temperatura de la solución (lote) se ajustó a 5 °C y el pH se ajustó a pH 1 mediante la adición de HCl concentrado (37 %, 1,17 l). La temperatura del lote ácido se ajustó a 20 °C, después de lo cual el lote se agitó durante 16 h. Se añadieron cristales de semillas (9,2 g) al lote y se continuó la agitación durante 16 h. Después de la agitación, el lote se filtró a través de un filtro Nutsche (18” (45,72 cm)) y el reactor se aclaró una vez con el licor madre. El aclarado se añadió a la torta de filtración, seguido de aclarado de la torta de filtro con una solución de THF, etanol y agua (1:1:1). Después de acondicionar la torta de filtración durante la noche, se transfirió a bandejas de secado y se secó al vacío a 45 °C. El lote seco pesó 1,12 kg, rendimiento del 102 %, con 4,0 % de agua (horno KF) RMN 1H (500 MHz, DMSO-d6) ó 10,20 (s, 1H), 9,68 (s, 1H), 8,47 (s, 1H), 8,09 (s, 1H), 7,97 (s ancho, 2H), 6,42 ), 1,00-1,90 (m, 2H), 2,43 (s, 3H), 1,80-1,60 (m, 5H), 1,5 (d, J = 24 Hz, 2H) Cu, valores de °20 (Theta)): 3,5 (s), 8,5 (1), 10,5 (m), 14 (s), 17 (s), 19,5 (s),
27 (m).
Estudios biológicos
Ejemplo 249: Ensayo enzimático bioquímico de Mnk
Los compuestos se seleccionan para detectar la inhibición de Mnk usando el kit de ensayo de cinasa ADP-Glo (Promega, catálogo N.° V9101). Todas las reacciones de cinasa se realizan en el tampón de reacción E (HEPES 15 mM pH 7,4, NaCl 20 mM, EGTA 1 mM, MgCh 10 mM, BGG 0,1 mg/ml y Tween-20 al 0,02 %). Las reacciones finales de Mnk1 contenían Mnk1 10 pM recombinante (Life Technologies, PR9138A), péptido de sustrato Mnk Ac-TATKSGSTTKNR-NH2 100 pM (American Peptide Company), ATP 300 pM y concentraciones variables del compuesto inhibidor de interés. Las reacciones Mnk2 finales contenían Mnk2 recombinante 3 nM (Life Technologies, PV5607), péptido de sustrato Mnk Ac-TATKSGSTTKNR-NH2 50 pM (American Peptide Company), ATP 10 pM y concentraciones variables del compuesto inhibidor de interés. La concentración final de DMSO en cada reacción es del 1 %.
Las reacciones de cinasa se realizan en placas de poliestireno de fondo plano de color blanco de 96 pocillos de superficie media en un volumen final de 25 pl. Las enzimas Mnk1/2 se preincubaron con el compuesto y el sustrato peptídico durante 5 minutos antes de la adición de ATP. Después de la adición de ATP, las reacciones de cinasa se incuban a temperatura ambiente durante 40 minutos. Posteriormente las reacciones se detienen mediante la adición de 25 pl de Reactivo ADP-Glo y se incuban durante 40 minutos adicionales. La señal luminiscente final usada para la lectura de la actividad de cinasa se produce por la adición de 45 pl de reactivo de detección de cinasa (kit ADP-Glo, Promega) e incubación durante 40 minutos. La señal luminiscente se detecta usando un contador de marcado múltiple Victor 2 (Perkin Elmer) y se calcula la concentración del compuesto necesario para conseguir la inhibición de la actividad de la enzima en un 50 % (CI50) usando señales de una serie de dilución de 8 puntos.
Los resultados de estos ensayos se exponen en la Tabla 1 a continuación. Con este fin, los valores de CI50 inferiores a 0,01 pM se etiquetan como "+++", de 0,01 a 0,1 pM se etiquetan como "++", y más de 0,1 a 10,0 pM se etiquetan como “+” (ND significa “no disponible”).
Tabla 1
Ensa o Enzimático Bio uímico de Mnk CI





Ejemplo 250: Ensayo celular de señalización peIF4E
El eIF4E fosforilado se somete a ensayo usando el kit de ensayo CISBio peIF4E HTRF® (CisBio, número de catálogo 64EF4PEG). Las células se sembraron en una placa tratada con cultivo de tejidos de 96 pocillos en medio de crecimiento apropiado (90 pl). Los compuestos (10X) se diluyen usando diluciones en serie con factor de dilución 3 en medio de cultivo celular y se añaden a las células. Las placas se incuban durante 2 horas a 37 °C. El sobrenadante de células se retira cuidadosamente mediante la aspiración del sobrenadante o por el chasquido de la placa. Inmediatamente se añaden 50 pl de regulador de pH de lisis complementado (IX) y se incuba durante al menos 30 minutos a temperatura ambiente con agitación. Después de la homogeneización por pipeteo arriba y abajo, se transfieren 16 pl de lisado celular desde la placa de cultivo celular de 96 pocillos a una placa de color blanco de pequeño volumen de 384 pocillos. Se preparan 4 pl de soluciones de anticuerpos premezcladas (vol/vol) en el regulador de pH de detección y se añaden. La placa se cubre con un sellador de placas y se incuba durante la noche a temperatura ambiente. Las emisiones de fluorescencia a dos longitudes de onda diferentes se leen (665 nm y 620 nm) en un Wallac Victor2. Las relaciones de emisión se convierten en porcentajes de inhibición e importan en el software GraphPad Prism. La concentración de compuesto necesaria para conseguir la inhibición de la actividad enzimática en un 50 % (CI50) se calcula usando concentraciones que varían de 20 pM a 0,1 nM (curva de 12 puntos). Los valores de CI50 se determinan usando un modelo de regresión no lineal disponible en GraphPad Prism 5.
Los resultados de estos ensayos se exponen en la Tabla 2 a continuación. Con este fin, los valores de CI50 inferiores a 0,05 pM se etiquetan como "+++", de 0,05 a 1,0 pM se etiquetan como "++", y más de 1,0 a 100 pM se etiquetan como “+” y ND significa “no disponible”.
Tabla 2
En l lr ñliz in IF4E I



Ejemplo 251: Estudios farmacocinéticos
A grupos de ratones Balb/c o ratas Sprague-Dawley (n > 3 por grupo de dosis) se les administran a dosis únicas del compuesto de ensayo. Los compuestos se formulan como soluciones en N-metilpirrolidona al 10 %, 90 % polietilenglicol 400 o como suspensiones en metilcelulosa al 0,5 % en agua para la administración oral por sonda a un nivel de dosis nominal de 10 mg/kg. Los compuestos se formulan en dimetilisosorbida al 10 %, etanol al 15 %, propilenglicol al 35 % y solución salina al 40 % (o D5W al 40 %) para la administración intravenosa a un nivel de dosis nominal de 1 mg/kg. Para animales tratados por vía intravenosa, las muestras de sangre se recogen a las 0,083, 0,25, 0,5, 1, 2, 4, 8 y 24 h después de la dosis. Para animales tratados por vía oral, se recogen muestras de sangre a las 0,25, 0,5, 1, 2, 4, 8 y 24 h después de la dosis. Se recogen muestras de sangre de ratones ya sea en serie a través de vena submandibular (aproximadamente 0,1 ml cada una) o terminalmente a través de punción cardiaca (aproximadamente 0,5 ml cada una). Las muestras de sangre se recogen en serie de ratas a través de un catéter de vena yugular (aproximadamente 0,2 ml cada uno). Cada muestra de sangre se recoge en un tubo que se enfría y contiene EDTA de potasio como anticoagulante. El plasma se separa y se almacena a aproximadamente -80 °C hasta su análisis. Después de la precipitación de la proteína con acetonitrilo que contiene un patrón interno, las muestras de plasma se analizan usando una cromatografía líquida/espectrometría de masas de alta resolución (CL-EMAR) para determinar las concentraciones plasmáticas. Los datos de concentración plasmática frente al
tiempo se someten a análisis de farmacocinética no-compartimental usando Phoenix™ Winnonlin® (Certara LP) para determinar los parámetros farmacocinéticos, incluyendo Área bajo la curva (AUC), Aclaramiento (CI), volumen de distribución en estado estacionario (Vss) y semivida terminal (T1/2). Los datos de dosis oral e intravenosa se destacan en las Tablas 3 y 4, respectivamente.
Tabla 3
Parámetros farmacocinéticos en ratones Balb/c después de una administración única de sonda oral a 10 mg/kg


Abreviaturas: NMP - N-metilpirrolidona; PEG400 - polietilenglicol 400; MC - metilcelulosa
Tabla 4
Parámetros farmacocinéticos en ratones Balb/c después de una sola administración en bolo intravenosa a 1 mg/kg


Abreviaturas: DMI-dimetilisosorbida; EtOH-etanol; PG-propilenglicol; D5W-dextrosa al 5 % en agua
Claims (20)
1. Un compuesto de acuerdo con la Fórmula (I):

o un estereoisómero, tautómero o una sal farmacéuticamente aceptable del mismo, en la que:
W1 y W2 son independientemente O, S o N-OR', donde R' es alquilo C1-C4 ;
Y es -N(R5)-, -O-, -S-, -C(O)-, -S=O, -S(O)2- o -CHR9-;
R1 es hidrógeno, alquilo inferior, cicloalquilo o heterociclilo, donde cualquier alquilo C1-C4 , cicloalquilo o heterociclilo está opcionalmente sustituido con 1, 2 o 3 grupos J;
n es 1, 2 o 3;
R2 y R3 son cada uno independientemente hidrógeno, alquilo, alquenilo, alquinilo, arilo, aralquileno, heteroarilo, heteroarilalquileno, cicloalquilo, cicloalquilalquileno, heterociclilo o heterociclilalquileno, donde cualquier alquilo, arilo, aralquileno, heteroarilo, heteroarilalquileno, cicloalquilo, cicloalquilalquileno, heterociclilo, o heterociclilalquileno, está opcionalmente sustituido con 1, 2 o 3 grupos J;
o R2 y R3 tomados junto con el átomo de carbono al que están unidos forman un cicloalquilo o heterociclilo, donde cualquier cicloalquilo o heterociclilo está opcionalmente sustituido con 1, 2 o 3 grupos J;
R4a y R4b son cada uno independientemente hidrógeno, halógeno, hidroxilo, tiol, hidroxialquileno, ciano, alquilo, alcoxi, acilo, tioalquilo, alquenilo, alquinilo, cicloalquilo, arilo o heterociclilo;
R5 es hidrógeno, ciano o alquilo C1-C4 ;
o R5 y R8 tomados junto con los átomos a los que están unidos forman un heterociclilo condensado opcionalmente sustituido con 1, 2 o 3 grupos J;
R6, R7 y R8 son cada uno independientemente hidrógeno, hidroxi, halógeno, ciano, amino, alquilo, alquenilo, alquinilo, alcoxi, cicloalquilo, cicloalquilalquileno, cicloalquilalquenileno, alquilaminilo, alquilcarbonilaminilo, cicloalquilcarbonilaminilo, cicloalquilaminilo, heterociclilaminilo, heteroarilo o heterociclilo, y donde cualquier amino, alquilo, alquenilo, alquinilo, alcoxi, cicloalquilo, cicloalquilalquileno, cicloalquilalquenileno, amino, alquilaminilo, alquilcarbonilaminilo, cicloalquilcarbonilaminilo, cicloalquilaminilo, heterociclilaminilo, heteroarilo, o heterociclilo está opcionalmente sustituido con 1, 2 o 3 grupos J;
o R7 y R8 tomados junto con los átomos a los que están unidos forman un heterociclilo condensado o heteroarilo opcionalmente sustituido con 1, 2 o 3 grupos J;
J es -SH, -SR9, -S(O)R9, -S(O)2R9, -S(O)NH2 , -S(O)NR9R9, -NH2 , -NR9R9, -COOH, -C(O)OR9, -C(O)R9, -C(O)-NH2 , -C(O)-NR9R9, hidroxi, ciano, halógeno, acetilo, alquilo, alquilo inferior, alquenilo, alquinilo, alcoxi, haloalquilo, tioalquilo, cianoalquileno, alquilaminilo, NH2-C(O)-alquileno, NR9R9-C(O)-alquileno, -CHR9-C(O)-alquilo inferior, -C(O)-alquilo inferior, alquilcarbonilaminilo, cicloalquilo, cicloalquilalquileno, cicloalquilalquenileno, cicloalquilcarbonilaminilo, cicloalquilaminilo, -CHR9-C(O)-cicloalquilo, -C(O)-cicloalquilo, -CHR9-C(O)-arilo, -CHR9-arilo, -C(O)-arilo, -CHR9-C(O)-heterocicloalquilo, -C(O)-heterocicloalquilo, heterociclilaminilo o heterociclilo; o cualesquiera dos grupos J unidos al mismo carbono o heteroátomo pueden tomarse juntos para formar oxo; y R9 es hidrógeno, alquilo C1-C4 o -OH.
2. El compuesto o un estereoisómero, tautómero o sal farmacéuticamente aceptable según la reivindicación 1, en los que n es 1 e Y es -N(R5)-.
3. El compuesto o un estereoisómero, tautómero o sal farmacéuticamente aceptable según la reivindicación 1, donde W1 y W2 son O.
4. El compuesto o un estereoisómero, tautómero o sal farmacéuticamente aceptable según la reivindicación 1, donde R2 y R3 son cada uno independientemente metilo, trifluorometilo, 1,1,1 -trifluoroetileno, ciclopentilo, ciclohexilo, difluorociclohexilo, clorofenilo o fluorofenilo.
5. El compuesto o un estereoisómero, tautómero o sal farmacéuticamente aceptable según la reivindicación 1, donde n es 1 y R2 y R3 junto con el átomo de carbono al que están unidos forman un cicloalquilo o un anillo heterociclilo que está opcionalmente sustituido con 1, 2 o 3 grupos J.
6. El compuesto o un estereoisómero, tautómero o sal farmacéuticamente aceptable según la reivindicación 5, donde R2 y R3 junto con el átomo de carbono al que están unidos forman un anillo heterociclilo que está opcionalmente sustituido con 1, 2 o 3 grupos J seleccionados entre el grupo que consiste en halógeno, -CN, N-metil amino, metilo, difluoroetileno y metilen-nitrilo.
7. El compuesto o un estereoisómero, tautómero o sal farmacéuticamente aceptable según la reivindicación 6, donde el anillo heterociclilo es piperidina.
8. El compuesto o un estereoisómero, tautómero o sal farmacéuticamente aceptable según la reivindicación 1, donde R7 y R8 junto con los átomos a los que están unidos forman un anillo heteroarilo condensado que está opcionalmente sustituido con 1, 2 o 3 grupos J.
9. El compuesto o un estereoisómero, tautómero o sal farmacéuticamente aceptable según la reivindicación 1, donde R4a y R4b son cada uno independientemente hidrógeno, halógeno o alquilo.
10. El compuesto o un estereoisómero, tautómero o sal farmacéuticamente aceptable según la reivindicación 1, donde R5 es hidrógeno.
11. El compuesto o un estereoisómero, tautómero o sal farmacéuticamente aceptable según la reivindicación 1, donde R6 y R8 son hidrógeno y R7 es hidroxi, halógeno, ciano, alquilo, alquenilo, alquinilo, alcoxi, cicloalquil cicloalquileno, cicloalquilalquenileno, amino, alquilaminilo, alquilcarbonilaminilo, cicloalquilcarbonilaminilo, cicloalquilaminilo, heterociclilaminilo, heteroarilo o heterociclilo, y donde cualquier alquilo, alquenilo, alquinilo, alcoxi, cicloalquilo, cicloalquilalquileno, cicloalquilalquenileno, amino, alquilaminilo, alquilcarbonilaminilo, cicloalquilcarbonilaminilo, cicloalquilaminilo, heterociclilaminilo, heteroarilo o heterociclilo está opcionalmente sustituido con 1, 2 o 3 grupos J.
12. El compuesto o un estereoisómero, tautómero o sal farmacéuticamente aceptable según la reivindicación 11, donde R6 y R8 son hidrógeno y R7 es amino, cicloalquilcarbonilaminilo, heterociclilaminilo o cicloalquilalquileno.
13. El compuesto o un estereoisómero, tautómero o sal farmacéuticamente aceptable según la reivindicación 1 seleccionados entre el grupo que consiste en:




14. El compuesto según la reivindicación 1, donde la sal farmacéuticamente aceptable del compuesto de Fórmula I es una sal de ácido orgánico o inorgánico seleccionada entre el grupo que consiste en acetato, mesilato, sulfato, citrato, oxalato, clorhidrato, diclorhidrato, isotionato, lactato y laurato.
15. Una composición farmacéutica que comprende (i) una cantidad terapéuticamente eficaz de al menos un compuesto o un estereoisómero, tautómero o sal farmacéuticamente aceptable del mismo según la reivindicación 1; (ii) en combinación con un vehículo, diluyente o excipiente farmacéuticamente aceptable.
16. El compuesto o un estereoisómero, tautómero o sal farmacéuticamente aceptable del mismo según la reivindicación 13 o 15, seleccionados entre

17. Un compuesto según la reivindicación 16

o un estereoisómero, tautómero o sal farmacéuticamente aceptable del mismo.
18. El compuesto o un estereoisómero, tautómero o sal farmacéuticamente aceptable del mismo según la reivindicación 14 o 17, donde la sal farmacéuticamente aceptable se selecciona entre el grupo que consiste en mesilato, sulfato y clorhidrato.
19. El compuesto o un estereoisómero, tautómero o sal farmacéuticamente aceptable del mismo según la reivindicación 18, donde la sal farmacéuticamente aceptable es clorhidrato.
20. Un compuesto
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201462017112P | 2014-06-25 | 2014-06-25 | |
| PCT/US2015/037416 WO2015200481A1 (en) | 2014-06-25 | 2015-06-24 | Mnk inhibitors and methods related thereto |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2728201T3 true ES2728201T3 (es) | 2019-10-22 |
Family
ID=53724447
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES15742147T Active ES2728201T3 (es) | 2014-06-25 | 2015-06-24 | Inhibidores de Mnk y métodos relacionados con los mismos |
| ES19158973T Active ES2967536T3 (es) | 2014-06-25 | 2015-06-24 | Inhibidores de Mnk y métodos relacionados con los mismos |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES19158973T Active ES2967536T3 (es) | 2014-06-25 | 2015-06-24 | Inhibidores de Mnk y métodos relacionados con los mismos |
Country Status (24)
| Country | Link |
|---|---|
| US (6) | US9382248B2 (es) |
| EP (2) | EP3521289B1 (es) |
| JP (2) | JP6615797B2 (es) |
| KR (1) | KR102460389B1 (es) |
| CN (2) | CN110183449B (es) |
| AU (1) | AU2015279984B2 (es) |
| BR (1) | BR112016029916B1 (es) |
| CA (1) | CA2953365C (es) |
| CL (1) | CL2016003293A1 (es) |
| CO (1) | CO2017000399A2 (es) |
| DK (2) | DK3521289T3 (es) |
| EA (1) | EA033920B1 (es) |
| ES (2) | ES2728201T3 (es) |
| FI (1) | FI3521289T3 (es) |
| IL (1) | IL249557B (es) |
| MX (1) | MX375323B (es) |
| MY (1) | MY189363A (es) |
| NZ (1) | NZ728007A (es) |
| PE (1) | PE20170321A1 (es) |
| PH (1) | PH12016502504B1 (es) |
| SG (2) | SG11201610511YA (es) |
| TW (1) | TWI713455B (es) |
| WO (1) | WO2015200481A1 (es) |
| ZA (1) | ZA201700241B (es) |
Families Citing this family (59)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| TWI713455B (zh) * | 2014-06-25 | 2020-12-21 | 美商伊凡克特治療公司 | MnK抑制劑及其相關方法 |
| EP3285809B1 (en) * | 2015-04-20 | 2019-09-11 | eFFECTOR Therapeutics, Inc. | Inhibitors of immune checkpoint modulators for use in treating cancer and infections |
| US10112955B2 (en) | 2015-10-29 | 2018-10-30 | Effector Therapeutics, Inc. | Isoindoline, azaisoindoline, dihydroindenone and dihydroazaindenone inhibitors of Mnk1 and Mnk2 |
| BR112018008711A2 (pt) | 2015-10-29 | 2018-10-30 | Effector Therapeutics Inc | compostos de pirrolo-, pirazolo-, imidazo-pirimidina e piridina que inibem mnk1 e mnk2 |
| WO2017087808A1 (en) | 2015-11-20 | 2017-05-26 | Effector Therapeutics, Inc. | Heterocyclic compounds that inhibit the kinase activity of mnk useful for treating various cancers |
| WO2017117052A1 (en) * | 2015-12-31 | 2017-07-06 | Effector Therapeutics, Inc. | Mnk biomarkers and uses thereof |
| WO2018098491A1 (en) | 2016-11-28 | 2018-05-31 | Praxis Precision Medicines, Inc. | Compounds and their methods of use |
| DK3548033T3 (da) | 2016-11-28 | 2024-07-15 | Praxis Prec Medicines Inc | Forbindelser og deres fremgangsmåde til anvendelse |
| US20190388425A1 (en) * | 2017-01-20 | 2019-12-26 | Bayer Pharma Aktiengesellschaft | Substituted imidazopyridinpyrimidines |
| TW201831479A (zh) * | 2017-01-20 | 2018-09-01 | 德商拜耳製藥公司 | 經取代二氫咪唑并吡啶二酮 |
| US11492345B2 (en) | 2017-02-13 | 2022-11-08 | Praxis Precision Medicines, Inc. | Compounds and their methods of use |
| CA3053493A1 (en) | 2017-02-14 | 2018-08-23 | Effector Therapeutics, Inc. | Piperidine-substituted mnk inhibitors and methods related thereto |
| WO2018187480A1 (en) | 2017-04-04 | 2018-10-11 | Praxis Precision Medicines, Inc. | Compounds and their methods of use |
| CA3064000A1 (en) | 2017-05-24 | 2018-11-29 | Effector Therapeutics, Inc. | Methods and compositions for cellular immunotherapy |
| CN109020957B (zh) * | 2017-06-12 | 2023-01-13 | 南京天印健华医药科技有限公司 | 作为mnk抑制剂的杂环化合物 |
| US11278535B2 (en) | 2017-08-15 | 2022-03-22 | Praxis Precision Medicines, Inc. | Compounds and their methods of use |
| WO2019079369A1 (en) | 2017-10-19 | 2019-04-25 | Effector Therapeutics, Inc. | BENZIMIDAZOLE INDOLE INHIBITORS OF MNK1 AND MNK2 |
| MX2020012838A (es) | 2018-05-30 | 2021-07-16 | Praxis Prec Medicines Inc | Moduladores de los canales ionicos. |
| EA202092908A1 (ru) | 2018-09-28 | 2021-05-14 | Праксис Пресижн Медсинз, Инк. | Модуляторы ионных каналов |
| CN113286592A (zh) | 2018-10-24 | 2021-08-20 | 效应治疗股份有限公司 | Mnk抑制剂的结晶形式 |
| WO2020108619A1 (zh) * | 2018-11-30 | 2020-06-04 | 上海迪诺医药科技有限公司 | Mnk抑制剂 |
| CN111484494B (zh) * | 2019-01-29 | 2022-09-13 | 诺沃斯达药业(上海)有限公司 | 抑制mnk1和mnk2的多环化合物 |
| EP3972593B1 (en) * | 2019-05-23 | 2025-07-02 | Board of Regents, The University of Texas System | Inhibitor of mnk for the treatment of neuropathic pain |
| US11773099B2 (en) | 2019-05-28 | 2023-10-03 | Praxis Precision Medicines, Inc. | Compounds and their methods of use |
| US11505554B2 (en) | 2019-05-31 | 2022-11-22 | Praxis Precision Medicines, Inc. | Substituted pyridines as ion channel modulators |
| US11279700B2 (en) | 2019-05-31 | 2022-03-22 | Praxis Precision Medicines, Inc. | Ion channel modulators |
| TW202114681A (zh) | 2019-07-02 | 2021-04-16 | 美商eFFECTOR醫療公司 | 轉譯抑制劑及其用途 |
| AU2020299362B2 (en) | 2019-07-02 | 2026-02-26 | Effector Therapeutics, Inc. | EiF4e-inhibiting 4-OXO-3,4-dihydropyrido(3,4-d)pyrimidine compounds |
| US20220251085A1 (en) * | 2019-07-21 | 2022-08-11 | University Of Virginia Patent Foundation | Cysteine binding compositions and methods of use thereof |
| FI4063364T3 (fi) | 2019-11-18 | 2024-11-19 | Jumbo Drug Bank Co Ltd | Mnk-inhibiittoreina toimivat pyrrolotriatsiiniyhdisteet |
| US11767325B2 (en) | 2019-11-26 | 2023-09-26 | Praxis Precision Medicines, Inc. | Substituted [1,2,4]triazolo[4,3-a]pyrazines as ion channel modulators |
| PE20221280A1 (es) | 2019-11-27 | 2022-09-05 | Praxis Prec Medicines Inc | Formulaciones de moduladores de canales ionicos y metodos de preparacion y uso de moduladores de canales ionicos |
| JP2023533616A (ja) * | 2020-06-30 | 2023-08-03 | 4イー セラピューティクス, インコーポレイテッド | Mnk阻害を示すピリジン-1,5-ジオン及びそれらの使用方法 |
| EP4200296A1 (en) * | 2020-08-20 | 2023-06-28 | Hepagene Therapeutics (HK) Limited | Mnk inhibitors |
| US11925642B2 (en) | 2020-10-08 | 2024-03-12 | Board Of Regents, The University Of Texas System | Regulation of eIF4E activity for migraine therapy |
| BR112023023189A2 (pt) * | 2021-05-08 | 2024-01-30 | Jumbo Drug Bank Co Ltd | Composto, formas cristalinas a e b de um composto, método de preparação para a forma cristalina a de um composto, e, uso do composto, das formas cristalinas a e b e de formas cristalinas preparadas pelo método |
| CA3225747A1 (en) * | 2021-06-30 | 2023-01-05 | 4E Therapeutics, Inc. | Spirocyclic pyridine-1,5-diones exhibiting mnk inhibition and their method of use |
| EP4380566A4 (en) * | 2021-08-05 | 2025-08-27 | 4E Therapeutics Inc | METHODS OF TREATING MIGRAINE WITH MNK INHIBITORS |
| CN114671869B (zh) * | 2022-03-31 | 2023-06-30 | 武汉九州钰民医药科技有限公司 | 化合物Tomivosertib的合成方法 |
| CN114853756B (zh) * | 2022-03-31 | 2023-03-28 | 武汉九州钰民医药科技有限公司 | 化合物Tomivosertib的制备工艺 |
| CN114736205B (zh) * | 2022-03-31 | 2023-03-03 | 武汉九州钰民医药科技有限公司 | 化合物Tomivosertib的制备方法 |
| KR20250005348A (ko) | 2022-04-26 | 2025-01-09 | 프락시스 프리시젼 메디신즈, 인크. | 신경 장애의 치료 |
| WO2024017229A1 (zh) * | 2022-07-19 | 2024-01-25 | 成都嘉葆药银医药科技有限公司 | 一种吡咯并三嗪类化合物在制备抗肿瘤药物中的应用 |
| CN116425750B (zh) * | 2023-02-10 | 2026-02-24 | 诺沃斯达药业(上海)有限公司 | 一种mnk抑制剂化合物的盐、晶体及其制备方法 |
| TW202508595A (zh) | 2023-05-04 | 2025-03-01 | 美商銳新醫藥公司 | 用於ras相關疾病或病症之組合療法 |
| US20250049810A1 (en) | 2023-08-07 | 2025-02-13 | Revolution Medicines, Inc. | Methods of treating a ras protein-related disease or disorder |
| AU2024360465A1 (en) | 2023-10-12 | 2026-04-09 | Revolution Medicines, Inc. | Macrocyclic ras inhibitors |
| WO2025171296A1 (en) | 2024-02-09 | 2025-08-14 | Revolution Medicines, Inc. | Ras inhibitors |
| WO2025190364A1 (zh) * | 2024-03-14 | 2025-09-18 | 山东轩竹医药科技有限公司 | Kif18a抑制剂及其用途 |
| TW202547461A (zh) | 2024-05-17 | 2025-12-16 | 美商銳新醫藥公司 | Ras抑制劑 |
| WO2025255438A1 (en) | 2024-06-07 | 2025-12-11 | Revolution Medicines, Inc. | Methods of treating a ras protein-related disease or disorder |
| WO2025265060A1 (en) | 2024-06-21 | 2025-12-26 | Revolution Medicines, Inc. | Therapeutic compositions and methods for managing treatment-related effects |
| WO2026006747A1 (en) | 2024-06-28 | 2026-01-02 | Revolution Medicines, Inc. | Ras inhibitors |
| WO2026015801A1 (en) | 2024-07-12 | 2026-01-15 | Revolution Medicines, Inc. | Methods of treating a ras related disease or disorder |
| WO2026015790A1 (en) | 2024-07-12 | 2026-01-15 | Revolution Medicines, Inc. | Methods of treating a ras related disease or disorder |
| WO2026015825A1 (en) | 2024-07-12 | 2026-01-15 | Revolution Medicines, Inc. | Use of ras inhibitor for treating pancreatic cancer |
| WO2026015796A1 (en) | 2024-07-12 | 2026-01-15 | Revolution Medicines, Inc. | Methods of treating a ras related disease or disorder |
| WO2026050446A1 (en) | 2024-08-29 | 2026-03-05 | Revolution Medicines, Inc. | Ras inhibitors |
| WO2026072904A2 (en) | 2024-09-26 | 2026-04-02 | Revolution Medicines, Inc. | Compositions and methods for treating lung cancer |
Family Cites Families (41)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5491144A (en) | 1991-05-30 | 1996-02-13 | Ciba-Geigy Corporation | Substituted diaminophthalimides and analogues |
| US9357797B2 (en) * | 1999-11-06 | 2016-06-07 | Janmarie Hornack | Dietary supplement containing alkaline electrolyte buffers |
| ATE360422T1 (de) | 2001-06-05 | 2007-05-15 | Lilly Icos Llc | Tetrazyklische verbindungen als pde5-inhibitoren |
| TWI301760B (en) | 2004-02-27 | 2008-10-11 | Merz Pharma Gmbh & Co Kgaa | Tetrahydroquinolinones and their use as antagonists of metabotropic glutamate receptors |
| US7488745B2 (en) | 2004-07-16 | 2009-02-10 | Schering Corporation | Compounds for the treatment of inflammatory disorders |
| TW200613272A (en) | 2004-08-13 | 2006-05-01 | Astrazeneca Ab | Isoindolone compounds and their use as metabotropic glutamate receptor potentiators |
| JP5031745B2 (ja) | 2005-08-12 | 2012-09-26 | アストラゼネカ アクチボラグ | 代謝型グルタミン酸受容体増強性イソインドロン |
| US20080039450A1 (en) * | 2006-06-22 | 2008-02-14 | Jensen Annika J | Compounds |
| EP1889847A1 (en) | 2006-07-10 | 2008-02-20 | DeveloGen Aktiengesellschaft | Pyrrolopyrimidines for pharmaceutical compositions |
| EP2727909A1 (en) | 2007-03-16 | 2014-05-07 | The Scripps Research Institute | Inhibitors of focal adhesion kinase |
| GB0706072D0 (en) | 2007-03-28 | 2007-05-09 | Sterix Ltd | Compound |
| JP2009173629A (ja) | 2007-12-21 | 2009-08-06 | Banyu Pharmaceut Co Ltd | Rsk1阻害作用を有する新規スピロインダン誘導体 |
| WO2009112445A1 (en) | 2008-03-10 | 2009-09-17 | Novartis Ag | Method of increasing cellular phosphatidyl choline by dgat1 inhibition |
| UY32072A (es) * | 2008-08-26 | 2010-03-26 | Boehringer Ingelheim Int | Tienopirimidinas para composiciones farmacéuticas |
| TWI468402B (zh) | 2009-07-31 | 2015-01-11 | 必治妥美雅史谷比公司 | 降低β-類澱粉生成之化合物 |
| US8637525B2 (en) | 2009-07-31 | 2014-01-28 | Bristol-Myers Squibb Company | Compounds for the reduction of beta-amyloid production |
| WO2011017296A1 (en) | 2009-08-04 | 2011-02-10 | Schering Corporation | 4, 5, 6-trisubstituted pyrimidine derivatives as factor ixa inhibitors |
| WO2011106168A1 (en) | 2010-02-24 | 2011-09-01 | Dcam Pharma Inc | Purine compounds for treating autoimmune and demyelinating diseases |
| US9381177B2 (en) | 2010-10-01 | 2016-07-05 | Bayer Intellectual Property Gmbh | Substituted N-(2-arylamino)aryl sulfonamide-containing combinations |
| CA2818853A1 (en) | 2010-11-30 | 2012-06-07 | Gilead Pharmasset Llc | 2'-spirocyclo-nucleosides for use in therapy of hcv or dengue virus |
| US9938269B2 (en) | 2011-06-30 | 2018-04-10 | Abbvie Inc. | Inhibitor compounds of phosphodiesterase type 10A |
| WO2013043192A1 (en) | 2011-09-23 | 2013-03-28 | The United States Of America, As Represented By The Secretary, Department Of Health And Human Services | Galactokinase inhibitors for the treatment and prevention of associated diseases and disorders |
| BR112014015720B1 (pt) | 2011-12-30 | 2020-03-17 | Hanmi Pharm. Co., Ltd. | Derivados de tieno[3,2-d]pirimidina, composição farmacêutica e uso dos mesmos para a prevenção ou tratamento de uma doença causada por ativação anormal de uma proteína quinase |
| CA2868966C (en) | 2012-03-29 | 2021-01-26 | Francis Xavier Tavares | Lactam kinase inhibitors |
| GB201205669D0 (en) | 2012-03-30 | 2012-05-16 | Agency Science Tech & Res | Bicyclic heterocyclic derivatives as mnk2 and mnk2 modulators and uses thereof |
| US10280168B2 (en) | 2012-03-30 | 2019-05-07 | Agency For Science, Technology And Research | Bicyclic heteroaryl derivatives as MNK1 and MNK2 modulators and uses thereof |
| WO2013151975A1 (en) | 2012-04-02 | 2013-10-10 | Northeastern University | Compositions and methods for the inhibition of methyltransferases |
| EP2852593A1 (en) | 2012-05-21 | 2015-04-01 | Bayer Pharma Aktiengesellschaft | Substituted pyrrolopyrimidines |
| TW201412740A (zh) * | 2012-09-20 | 2014-04-01 | Bayer Pharma AG | 經取代之吡咯并嘧啶胺基苯并噻唑酮 |
| EP2917185B1 (en) * | 2012-11-09 | 2017-05-10 | Evotec International GmbH | Sulfoximine substituted quinazolines for pharmaceutical compositions |
| GB2508652A (en) | 2012-12-07 | 2014-06-11 | Agency Science Tech & Res | Heterocyclic piperazine derivatives |
| EP2935304A1 (en) | 2012-12-19 | 2015-10-28 | IDENIX Pharmaceuticals, Inc. | 4'-fluoro nucleosides for the treatment of hcv |
| HK1212699A1 (zh) | 2013-02-20 | 2016-06-17 | 拜耳医药股份公司 | 作为mknk1抑制剂的取代的咪唑并[1,2-b]哒嗪类化合物 |
| TW201605867A (zh) | 2013-11-20 | 2016-02-16 | 拜耳製藥公司 | 噻吩并嘧啶 |
| TWI713455B (zh) | 2014-06-25 | 2020-12-21 | 美商伊凡克特治療公司 | MnK抑制劑及其相關方法 |
| KR20170095985A (ko) | 2014-12-19 | 2017-08-23 | 바이엘 파마 악티엔게젤샤프트 | Mknk1 및 mknk2 억제제로서의 피라졸로피리딘아민 |
| BR112018008711A2 (pt) | 2015-10-29 | 2018-10-30 | Effector Therapeutics Inc | compostos de pirrolo-, pirazolo-, imidazo-pirimidina e piridina que inibem mnk1 e mnk2 |
| US10112955B2 (en) | 2015-10-29 | 2018-10-30 | Effector Therapeutics, Inc. | Isoindoline, azaisoindoline, dihydroindenone and dihydroazaindenone inhibitors of Mnk1 and Mnk2 |
| GB201520499D0 (en) | 2015-11-20 | 2016-01-06 | Medical Res Council Technology | Compounds |
| GB201520500D0 (en) | 2015-11-20 | 2016-01-06 | Medical Res Council Technology | Compounds |
| WO2017087808A1 (en) | 2015-11-20 | 2017-05-26 | Effector Therapeutics, Inc. | Heterocyclic compounds that inhibit the kinase activity of mnk useful for treating various cancers |
-
2015
- 2015-06-23 TW TW104120090A patent/TWI713455B/zh active
- 2015-06-24 MX MX2016017030A patent/MX375323B/es active IP Right Grant
- 2015-06-24 EA EA201790078A patent/EA033920B1/ru not_active IP Right Cessation
- 2015-06-24 DK DK19158973.8T patent/DK3521289T3/da active
- 2015-06-24 JP JP2016575379A patent/JP6615797B2/ja active Active
- 2015-06-24 ES ES15742147T patent/ES2728201T3/es active Active
- 2015-06-24 NZ NZ728007A patent/NZ728007A/en unknown
- 2015-06-24 KR KR1020177002139A patent/KR102460389B1/ko active Active
- 2015-06-24 EP EP19158973.8A patent/EP3521289B1/en active Active
- 2015-06-24 AU AU2015279984A patent/AU2015279984B2/en active Active
- 2015-06-24 WO PCT/US2015/037416 patent/WO2015200481A1/en not_active Ceased
- 2015-06-24 US US14/748,990 patent/US9382248B2/en active Active
- 2015-06-24 SG SG11201610511YA patent/SG11201610511YA/en unknown
- 2015-06-24 ES ES19158973T patent/ES2967536T3/es active Active
- 2015-06-24 DK DK15742147.0T patent/DK3160966T3/da active
- 2015-06-24 MY MYPI2016704761A patent/MY189363A/en unknown
- 2015-06-24 EP EP15742147.0A patent/EP3160966B1/en active Active
- 2015-06-24 CA CA2953365A patent/CA2953365C/en active Active
- 2015-06-24 PH PH1/2016/502504A patent/PH12016502504B1/en unknown
- 2015-06-24 FI FIEP19158973.8T patent/FI3521289T3/fi active
- 2015-06-24 SG SG10201811384TA patent/SG10201811384TA/en unknown
- 2015-06-24 CN CN201910556784.9A patent/CN110183449B/zh active Active
- 2015-06-24 PE PE2016002761A patent/PE20170321A1/es unknown
- 2015-06-24 BR BR112016029916-7A patent/BR112016029916B1/pt active IP Right Grant
- 2015-06-24 CN CN201580046203.7A patent/CN106795162B/zh active Active
-
2016
- 2016-06-21 US US15/187,854 patent/US9669031B2/en active Active
- 2016-12-14 IL IL249557A patent/IL249557B/en unknown
- 2016-12-22 CL CL2016003293A patent/CL2016003293A1/es unknown
-
2017
- 2017-01-11 ZA ZA2017/00241A patent/ZA201700241B/en unknown
- 2017-01-18 CO CONC2017/0000399A patent/CO2017000399A2/es unknown
- 2017-06-02 US US15/611,966 patent/US9814718B2/en active Active
- 2017-11-10 US US15/809,011 patent/US20180085368A1/en not_active Abandoned
-
2018
- 2018-11-02 US US16/178,678 patent/US20190209560A1/en not_active Abandoned
-
2019
- 2019-11-06 JP JP2019201631A patent/JP6979439B2/ja active Active
-
2021
- 2021-05-05 US US17/308,486 patent/US20210338673A1/en not_active Abandoned
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2728201T3 (es) | Inhibidores de Mnk y métodos relacionados con los mismos | |
| CN116003441B (zh) | 杂芳类衍生物调节剂、其制备方法和应用 | |
| US11014926B2 (en) | Pyrrolo-, pyrazolo-, imidazo-pyrimidine and pyridine compounds that inhibit MNK1 and MNK2 | |
| CA3002877A1 (en) | Isoindoline, azaisoindoline, dihydroindenone and dihydroazaindenone inhibitors of mnk1 and mnk2 | |
| HK40012635B (en) | Mnk inhibitors and methods related thereto | |
| HK40012635A (en) | Mnk inhibitors and methods related thereto | |
| HK1236951B (en) | Mnk inhibitors and methods related thereto | |
| HK1236951A1 (en) | Mnk inhibitors and methods related thereto |