ES2733344T3 - Profármacos antagonistas de NMDA - Google Patents
Profármacos antagonistas de NMDA Download PDFInfo
- Publication number
- ES2733344T3 ES2733344T3 ES14802476T ES14802476T ES2733344T3 ES 2733344 T3 ES2733344 T3 ES 2733344T3 ES 14802476 T ES14802476 T ES 14802476T ES 14802476 T ES14802476 T ES 14802476T ES 2733344 T3 ES2733344 T3 ES 2733344T3
- Authority
- ES
- Spain
- Prior art keywords
- phenyl
- pyridin
- ethanamine
- mmol
- pyrrolidin
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 239000000651 prodrug Substances 0.000 title description 69
- 229940002612 prodrug Drugs 0.000 title description 69
- 239000003703 n methyl dextro aspartic acid receptor blocking agent Substances 0.000 title description 3
- 150000001875 compounds Chemical class 0.000 claims abstract description 88
- 150000003839 salts Chemical class 0.000 claims abstract description 36
- 150000001413 amino acids Chemical class 0.000 claims abstract description 4
- FWUQWDCOOWEXRY-ZDUSSCGKSA-N lanicemine Chemical compound C([C@H](N)C=1C=CC=CC=1)C1=CC=CC=N1 FWUQWDCOOWEXRY-ZDUSSCGKSA-N 0.000 claims description 105
- -1 (S) -2-Amino-3-methylbutanoyl Chemical group 0.000 claims description 101
- VLJNHYLEOZPXFW-UHFFFAOYSA-N pyrrolidine-2-carboxamide Chemical compound NC(=O)C1CCCN1 VLJNHYLEOZPXFW-UHFFFAOYSA-N 0.000 claims description 21
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical class OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 claims description 9
- 101000684208 Homo sapiens Prolyl endopeptidase FAP Proteins 0.000 claims description 9
- IGDLGBXVQVNZDV-URORMMCBSA-N (2S)-1-[(2S)-2-amino-3-(1H-indol-3-yl)propanoyl]-N-[(1S)-1-phenyl-2-pyridin-2-ylethyl]pyrrolidine-2-carboxamide Chemical compound N[C@@H](Cc1c[nH]c2ccccc12)C(=O)N1CCC[C@H]1C(=O)N[C@@H](Cc1ccccn1)c1ccccc1 IGDLGBXVQVNZDV-URORMMCBSA-N 0.000 claims description 2
- YXNDGQHSSKBCFK-SDHOMARFSA-N (2S)-1-[(2S)-2-amino-3-(4-hydroxyphenyl)propanoyl]-N-[(1S)-1-phenyl-2-pyridin-2-ylethyl]pyrrolidine-2-carboxamide Chemical compound N[C@@H](Cc1ccc(O)cc1)C(=O)N1CCC[C@H]1C(=O)N[C@@H](Cc1ccccn1)c1ccccc1 YXNDGQHSSKBCFK-SDHOMARFSA-N 0.000 claims description 2
- 239000003112 inhibitor Substances 0.000 claims description 2
- 102100023832 Prolyl endopeptidase FAP Human genes 0.000 claims 1
- 238000003776 cleavage reaction Methods 0.000 claims 1
- 230000007017 scission Effects 0.000 claims 1
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 149
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Natural products CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 92
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 52
- 239000000203 mixture Substances 0.000 description 48
- 235000019439 ethyl acetate Nutrition 0.000 description 46
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 38
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 32
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 30
- BZLVMXJERCGZMT-UHFFFAOYSA-N Methyl tert-butyl ether Chemical compound COC(C)(C)C BZLVMXJERCGZMT-UHFFFAOYSA-N 0.000 description 28
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 26
- 238000006243 chemical reaction Methods 0.000 description 25
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 24
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 23
- 239000007832 Na2SO4 Substances 0.000 description 23
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 23
- 229910052938 sodium sulfate Inorganic materials 0.000 description 23
- 235000011152 sodium sulphate Nutrition 0.000 description 23
- 238000005160 1H NMR spectroscopy Methods 0.000 description 22
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 22
- 239000000047 product Substances 0.000 description 22
- 239000000243 solution Substances 0.000 description 22
- 238000000034 method Methods 0.000 description 21
- 239000011541 reaction mixture Substances 0.000 description 20
- 239000007787 solid Substances 0.000 description 19
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 18
- ZHNUHDYFZUAESO-UHFFFAOYSA-N Formamide Chemical compound NC=O ZHNUHDYFZUAESO-UHFFFAOYSA-N 0.000 description 18
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 17
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 17
- 238000011282 treatment Methods 0.000 description 17
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 15
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 15
- 235000017557 sodium bicarbonate Nutrition 0.000 description 15
- 208000002193 Pain Diseases 0.000 description 14
- 239000012267 brine Substances 0.000 description 13
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 13
- NXFFJDQHYLNEJK-UHFFFAOYSA-N 2-[4-[(4-chlorophenyl)methyl]-7-fluoro-5-methylsulfonyl-2,3-dihydro-1h-cyclopenta[b]indol-3-yl]acetic acid Chemical compound C1=2C(S(=O)(=O)C)=CC(F)=CC=2C=2CCC(CC(O)=O)C=2N1CC1=CC=C(Cl)C=C1 NXFFJDQHYLNEJK-UHFFFAOYSA-N 0.000 description 12
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 12
- 235000011089 carbon dioxide Nutrition 0.000 description 11
- 238000001914 filtration Methods 0.000 description 11
- 229910002092 carbon dioxide Inorganic materials 0.000 description 10
- 239000012458 free base Substances 0.000 description 10
- 238000000746 purification Methods 0.000 description 10
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical group CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 9
- 102100025012 Dipeptidyl peptidase 4 Human genes 0.000 description 9
- 239000007821 HATU Substances 0.000 description 9
- QTBSBXVTEAMEQO-UHFFFAOYSA-N acetic acid Substances CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 9
- 239000008280 blood Substances 0.000 description 9
- 210000004369 blood Anatomy 0.000 description 9
- 230000015572 biosynthetic process Effects 0.000 description 8
- 125000003917 carbamoyl group Chemical group [H]N([H])C(*)=O 0.000 description 8
- 229940125904 compound 1 Drugs 0.000 description 8
- 239000012530 fluid Substances 0.000 description 8
- 239000012074 organic phase Substances 0.000 description 8
- 239000008194 pharmaceutical composition Substances 0.000 description 8
- 239000012071 phase Substances 0.000 description 8
- 238000005481 NMR spectroscopy Methods 0.000 description 7
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 7
- 230000000968 intestinal effect Effects 0.000 description 7
- WDAXFOBOLVPGLV-UHFFFAOYSA-N isobutyric acid ethyl ester Natural products CCOC(=O)C(C)C WDAXFOBOLVPGLV-UHFFFAOYSA-N 0.000 description 7
- 238000000926 separation method Methods 0.000 description 7
- 238000001228 spectrum Methods 0.000 description 7
- KHJHFYAGQZYCLC-GXKRWWSZSA-N (1s)-1-phenyl-2-pyridin-2-ylethanamine;dihydrochloride Chemical compound Cl.Cl.C([C@H](N)C=1C=CC=CC=1)C1=CC=CC=N1 KHJHFYAGQZYCLC-GXKRWWSZSA-N 0.000 description 6
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 6
- 125000000217 alkyl group Chemical group 0.000 description 6
- 238000004458 analytical method Methods 0.000 description 6
- 230000037396 body weight Effects 0.000 description 6
- 229910052681 coesite Inorganic materials 0.000 description 6
- 229910052906 cristobalite Inorganic materials 0.000 description 6
- HPNMFZURTQLUMO-UHFFFAOYSA-N diethylamine Chemical compound CCNCC HPNMFZURTQLUMO-UHFFFAOYSA-N 0.000 description 6
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 6
- 239000000706 filtrate Substances 0.000 description 6
- 208000024714 major depressive disease Diseases 0.000 description 6
- 230000003287 optical effect Effects 0.000 description 6
- 239000012044 organic layer Substances 0.000 description 6
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 6
- 239000000377 silicon dioxide Substances 0.000 description 6
- 235000012239 silicon dioxide Nutrition 0.000 description 6
- NDVLTYZPCACLMA-UHFFFAOYSA-N silver oxide Chemical compound [O-2].[Ag+].[Ag+] NDVLTYZPCACLMA-UHFFFAOYSA-N 0.000 description 6
- 238000003756 stirring Methods 0.000 description 6
- 229910052682 stishovite Inorganic materials 0.000 description 6
- 229910052905 tridymite Inorganic materials 0.000 description 6
- 239000003039 volatile agent Substances 0.000 description 6
- 125000004485 2-pyrrolidinyl group Chemical group [H]N1C([H])([H])C([H])([H])C([H])([H])C1([H])* 0.000 description 5
- 241000700159 Rattus Species 0.000 description 5
- 125000004432 carbon atom Chemical group C* 0.000 description 5
- 230000002354 daily effect Effects 0.000 description 5
- 201000010099 disease Diseases 0.000 description 5
- 238000009472 formulation Methods 0.000 description 5
- 238000011534 incubation Methods 0.000 description 5
- 238000001802 infusion Methods 0.000 description 5
- KQNPFQTWMSNSAP-UHFFFAOYSA-N isobutyric acid Chemical compound CC(C)C(O)=O KQNPFQTWMSNSAP-UHFFFAOYSA-N 0.000 description 5
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 5
- 238000002360 preparation method Methods 0.000 description 5
- 206010010904 Convulsion Diseases 0.000 description 4
- 208000020401 Depressive disease Diseases 0.000 description 4
- OKKJLVBELUTLKV-MZCSYVLQSA-N Deuterated methanol Chemical compound [2H]OC([2H])([2H])[2H] OKKJLVBELUTLKV-MZCSYVLQSA-N 0.000 description 4
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 4
- QUSNBJAOOMFDIB-UHFFFAOYSA-N Ethylamine Chemical compound CCN QUSNBJAOOMFDIB-UHFFFAOYSA-N 0.000 description 4
- KZSNJWFQEVHDMF-BYPYZUCNSA-N L-valine Chemical compound CC(C)[C@H](N)C(O)=O KZSNJWFQEVHDMF-BYPYZUCNSA-N 0.000 description 4
- 239000004472 Lysine Substances 0.000 description 4
- SJRJJKPEHAURKC-UHFFFAOYSA-N N-Methylmorpholine Chemical compound CN1CCOCC1 SJRJJKPEHAURKC-UHFFFAOYSA-N 0.000 description 4
- KZSNJWFQEVHDMF-UHFFFAOYSA-N Valine Natural products CC(C)C(N)C(O)=O KZSNJWFQEVHDMF-UHFFFAOYSA-N 0.000 description 4
- 239000004480 active ingredient Substances 0.000 description 4
- 239000002671 adjuvant Substances 0.000 description 4
- 125000003545 alkoxy group Chemical group 0.000 description 4
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 4
- 238000004440 column chromatography Methods 0.000 description 4
- 239000003814 drug Substances 0.000 description 4
- 230000008014 freezing Effects 0.000 description 4
- 238000007710 freezing Methods 0.000 description 4
- 239000007789 gas Substances 0.000 description 4
- 238000004128 high performance liquid chromatography Methods 0.000 description 4
- 238000000338 in vitro Methods 0.000 description 4
- 238000001727 in vivo Methods 0.000 description 4
- 208000004296 neuralgia Diseases 0.000 description 4
- 208000021722 neuropathic pain Diseases 0.000 description 4
- 239000006186 oral dosage form Substances 0.000 description 4
- 125000001325 propanoyl group Chemical group O=C([*])C([H])([H])C([H])([H])[H] 0.000 description 4
- FBNCDBZHLBYPGY-UHFFFAOYSA-N pyridine;dihydrochloride Chemical compound [Cl-].[Cl-].C1=CC=[NH+]C=C1.C1=CC=[NH+]C=C1 FBNCDBZHLBYPGY-UHFFFAOYSA-N 0.000 description 4
- 239000000523 sample Substances 0.000 description 4
- 239000011734 sodium Substances 0.000 description 4
- 230000001225 therapeutic effect Effects 0.000 description 4
- 229960004295 valine Drugs 0.000 description 4
- 239000004474 valine Substances 0.000 description 4
- 125000004105 2-pyridyl group Chemical group N1=C([*])C([H])=C([H])C([H])=C1[H] 0.000 description 3
- 125000004203 4-hydroxyphenyl group Chemical group [H]OC1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 3
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 3
- 239000004475 Arginine Substances 0.000 description 3
- 208000020925 Bipolar disease Diseases 0.000 description 3
- SJXONSOJBHDFCJ-KRWDZBQOSA-N C(C)C(C(=O)OOC(N[C@@H](CC1=NC=CC=C1)C1=CC=CC=C1)=O)(C)C Chemical compound C(C)C(C(=O)OOC(N[C@@H](CC1=NC=CC=C1)C1=CC=CC=C1)=O)(C)C SJXONSOJBHDFCJ-KRWDZBQOSA-N 0.000 description 3
- 241000282472 Canis lupus familiaris Species 0.000 description 3
- RGHNJXZEOKUKBD-UHFFFAOYSA-N D-gluconic acid Natural products OCC(O)C(O)C(O)C(O)C(O)=O RGHNJXZEOKUKBD-UHFFFAOYSA-N 0.000 description 3
- 108090000790 Enzymes Proteins 0.000 description 3
- 102000004190 Enzymes Human genes 0.000 description 3
- RGHNJXZEOKUKBD-SQOUGZDYSA-N Gluconic acid Natural products OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C(O)=O RGHNJXZEOKUKBD-SQOUGZDYSA-N 0.000 description 3
- 239000005909 Kieselgur Substances 0.000 description 3
- ODKSFYDXXFIFQN-BYPYZUCNSA-P L-argininium(2+) Chemical compound NC(=[NH2+])NCCC[C@H]([NH3+])C(O)=O ODKSFYDXXFIFQN-BYPYZUCNSA-P 0.000 description 3
- HNDVDQJCIGZPNO-YFKPBYRVSA-N L-histidine Chemical compound OC(=O)[C@@H](N)CC1=CN=CN1 HNDVDQJCIGZPNO-YFKPBYRVSA-N 0.000 description 3
- KDXKERNSBIXSRK-YFKPBYRVSA-N L-lysine Chemical compound NCCCC[C@H](N)C(O)=O KDXKERNSBIXSRK-YFKPBYRVSA-N 0.000 description 3
- OUYCCCASQSFEME-QMMMGPOBSA-N L-tyrosine Chemical compound OC(=O)[C@@H](N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-QMMMGPOBSA-N 0.000 description 3
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 3
- RWRDLPDLKQPQOW-UHFFFAOYSA-N Pyrrolidine Chemical compound C1CCNC1 RWRDLPDLKQPQOW-UHFFFAOYSA-N 0.000 description 3
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 3
- 239000012300 argon atmosphere Substances 0.000 description 3
- 229910052799 carbon Inorganic materials 0.000 description 3
- ILUWVORABZTBIU-UHFFFAOYSA-N chloromethyl 2-methylpropanoate Chemical compound CC(C)C(=O)OCCl ILUWVORABZTBIU-UHFFFAOYSA-N 0.000 description 3
- 239000003085 diluting agent Substances 0.000 description 3
- 239000000284 extract Substances 0.000 description 3
- 238000004817 gas chromatography Methods 0.000 description 3
- 239000007903 gelatin capsule Substances 0.000 description 3
- 239000000174 gluconic acid Substances 0.000 description 3
- 235000012208 gluconic acid Nutrition 0.000 description 3
- HNDVDQJCIGZPNO-UHFFFAOYSA-N histidine Natural products OC(=O)C(N)CC1=CN=CN1 HNDVDQJCIGZPNO-UHFFFAOYSA-N 0.000 description 3
- 230000002401 inhibitory effect Effects 0.000 description 3
- 238000001990 intravenous administration Methods 0.000 description 3
- BHIWKHZACMWKOJ-UHFFFAOYSA-N isobutyric acid methyl ester Natural products COC(=O)C(C)C BHIWKHZACMWKOJ-UHFFFAOYSA-N 0.000 description 3
- 239000010410 layer Substances 0.000 description 3
- 229960003136 leucine Drugs 0.000 description 3
- 210000004185 liver Anatomy 0.000 description 3
- 235000018977 lysine Nutrition 0.000 description 3
- VNWKTOKETHGBQD-UHFFFAOYSA-N methane Natural products C VNWKTOKETHGBQD-UHFFFAOYSA-N 0.000 description 3
- 231100000706 no observed effect level Toxicity 0.000 description 3
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 3
- JUJWROOIHBZHMG-UHFFFAOYSA-O pyridinium Chemical compound C1=CC=[NH+]C=C1 JUJWROOIHBZHMG-UHFFFAOYSA-O 0.000 description 3
- 229910001923 silver oxide Inorganic materials 0.000 description 3
- 239000012258 stirred mixture Substances 0.000 description 3
- 238000002560 therapeutic procedure Methods 0.000 description 3
- OUYCCCASQSFEME-UHFFFAOYSA-N tyrosine Natural products OC(=O)C(N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-UHFFFAOYSA-N 0.000 description 3
- QGGHOUZHWORCTD-IRXDYDNUSA-N (2S)-N-[(1S)-1-phenyl-2-pyridin-2-ylethyl]pyrrolidine-2-carboxamide Chemical compound O=C(N[C@@H](Cc1ccccn1)c1ccccc1)[C@@H]1CCCN1 QGGHOUZHWORCTD-IRXDYDNUSA-N 0.000 description 2
- ZQEBQGAAWMOMAI-ZETCQYMHSA-N (2s)-1-[(2-methylpropan-2-yl)oxycarbonyl]pyrrolidine-2-carboxylic acid Chemical compound CC(C)(C)OC(=O)N1CCC[C@H]1C(O)=O ZQEBQGAAWMOMAI-ZETCQYMHSA-N 0.000 description 2
- KPZGRMZPZLOPBS-UHFFFAOYSA-N 1,3-dichloro-2,2-bis(chloromethyl)propane Chemical compound ClCC(CCl)(CCl)CCl KPZGRMZPZLOPBS-UHFFFAOYSA-N 0.000 description 2
- 238000001644 13C nuclear magnetic resonance spectroscopy Methods 0.000 description 2
- XPQIPUZPSLAZDV-UHFFFAOYSA-N 2-pyridylethylamine Chemical compound NCCC1=CC=CC=N1 XPQIPUZPSLAZDV-UHFFFAOYSA-N 0.000 description 2
- QASBCTGZKABPKX-UHFFFAOYSA-N 4-(methylsulfanyl)phenol Chemical compound CSC1=CC=C(O)C=C1 QASBCTGZKABPKX-UHFFFAOYSA-N 0.000 description 2
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 2
- LSNNMFCWUKXFEE-UHFFFAOYSA-M Bisulfite Chemical compound OS([O-])=O LSNNMFCWUKXFEE-UHFFFAOYSA-M 0.000 description 2
- 208000000003 Breakthrough pain Diseases 0.000 description 2
- 208000000094 Chronic Pain Diseases 0.000 description 2
- 206010013654 Drug abuse Diseases 0.000 description 2
- BDAGIHXWWSANSR-UHFFFAOYSA-M Formate Chemical compound [O-]C=O BDAGIHXWWSANSR-UHFFFAOYSA-M 0.000 description 2
- 206010017740 Gas poisoning Diseases 0.000 description 2
- 108010010803 Gelatin Proteins 0.000 description 2
- 101000930822 Giardia intestinalis Dipeptidyl-peptidase 4 Proteins 0.000 description 2
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 2
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 2
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 2
- ROHFNLRQFUQHCH-YFKPBYRVSA-N L-leucine Chemical compound CC(C)C[C@H](N)C(O)=O ROHFNLRQFUQHCH-YFKPBYRVSA-N 0.000 description 2
- COLNVLDHVKWLRT-QMMMGPOBSA-N L-phenylalanine Chemical compound OC(=O)[C@@H](N)CC1=CC=CC=C1 COLNVLDHVKWLRT-QMMMGPOBSA-N 0.000 description 2
- QIVBCDIJIAJPQS-VIFPVBQESA-N L-tryptophane Chemical compound C1=CC=C2C(C[C@H](N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-VIFPVBQESA-N 0.000 description 2
- ROHFNLRQFUQHCH-UHFFFAOYSA-N Leucine Natural products CC(C)CC(N)C(O)=O ROHFNLRQFUQHCH-UHFFFAOYSA-N 0.000 description 2
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 2
- 208000001294 Nociceptive Pain Diseases 0.000 description 2
- 208000021384 Obsessive-Compulsive disease Diseases 0.000 description 2
- 229910019142 PO4 Inorganic materials 0.000 description 2
- 208000004983 Phantom Limb Diseases 0.000 description 2
- 206010056238 Phantom pain Diseases 0.000 description 2
- 239000002202 Polyethylene glycol Substances 0.000 description 2
- ONIBWKKTOPOVIA-UHFFFAOYSA-N Proline Natural products OC(=O)C1CCCN1 ONIBWKKTOPOVIA-UHFFFAOYSA-N 0.000 description 2
- ATUOYWHBWRKTHZ-UHFFFAOYSA-N Propane Chemical compound CCC ATUOYWHBWRKTHZ-UHFFFAOYSA-N 0.000 description 2
- 208000006289 Rett Syndrome Diseases 0.000 description 2
- DYAHQFWOVKZOOW-UHFFFAOYSA-N Sarin Chemical compound CC(C)OP(C)(F)=O DYAHQFWOVKZOOW-UHFFFAOYSA-N 0.000 description 2
- 229940124639 Selective inhibitor Drugs 0.000 description 2
- 206010042458 Suicidal ideation Diseases 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 2
- GWEVSGVZZGPLCZ-UHFFFAOYSA-N Titan oxide Chemical compound O=[Ti]=O GWEVSGVZZGPLCZ-UHFFFAOYSA-N 0.000 description 2
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 2
- QIVBCDIJIAJPQS-UHFFFAOYSA-N Tryptophan Natural products C1=CC=C2C(CC(N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-UHFFFAOYSA-N 0.000 description 2
- YRKCREAYFQTBPV-UHFFFAOYSA-N acetylacetone Chemical compound CC(=O)CC(C)=O YRKCREAYFQTBPV-UHFFFAOYSA-N 0.000 description 2
- 239000002253 acid Substances 0.000 description 2
- 229940024606 amino acid Drugs 0.000 description 2
- 235000001014 amino acid Nutrition 0.000 description 2
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 2
- 230000036765 blood level Effects 0.000 description 2
- 238000013461 design Methods 0.000 description 2
- 229940079593 drug Drugs 0.000 description 2
- 230000000694 effects Effects 0.000 description 2
- 230000007071 enzymatic hydrolysis Effects 0.000 description 2
- 238000006047 enzymatic hydrolysis reaction Methods 0.000 description 2
- 230000001037 epileptic effect Effects 0.000 description 2
- 150000002148 esters Chemical class 0.000 description 2
- 229920000159 gelatin Polymers 0.000 description 2
- 239000008273 gelatin Substances 0.000 description 2
- 235000019322 gelatine Nutrition 0.000 description 2
- 235000011852 gelatine desserts Nutrition 0.000 description 2
- 239000011521 glass Substances 0.000 description 2
- SHFJWMWCIHQNCP-UHFFFAOYSA-M hydron;tetrabutylazanium;sulfate Chemical compound OS([O-])(=O)=O.CCCC[N+](CCCC)(CCCC)CCCC SHFJWMWCIHQNCP-UHFFFAOYSA-M 0.000 description 2
- 125000002140 imidazol-4-yl group Chemical group [H]N1C([H])=NC([*])=C1[H] 0.000 description 2
- 125000001841 imino group Chemical group [H]N=* 0.000 description 2
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 2
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 2
- 231100000822 oral exposure Toxicity 0.000 description 2
- 125000001147 pentyl group Chemical group C(CCCC)* 0.000 description 2
- 230000035699 permeability Effects 0.000 description 2
- COLNVLDHVKWLRT-UHFFFAOYSA-N phenylalanine Natural products OC(=O)C(N)CC1=CC=CC=C1 COLNVLDHVKWLRT-UHFFFAOYSA-N 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 2
- 239000010452 phosphate Substances 0.000 description 2
- 229920001223 polyethylene glycol Polymers 0.000 description 2
- 229960002429 proline Drugs 0.000 description 2
- 238000011321 prophylaxis Methods 0.000 description 2
- 230000001107 psychogenic effect Effects 0.000 description 2
- AOJFQRQNPXYVLM-UHFFFAOYSA-N pyridin-1-ium;chloride Chemical compound [Cl-].C1=CC=[NH+]C=C1 AOJFQRQNPXYVLM-UHFFFAOYSA-N 0.000 description 2
- NYCVCXMSZNOGDH-UHFFFAOYSA-N pyrrolidine-1-carboxylic acid Chemical compound OC(=O)N1CCCC1 NYCVCXMSZNOGDH-UHFFFAOYSA-N 0.000 description 2
- 239000011550 stock solution Substances 0.000 description 2
- 208000011117 substance-related disease Diseases 0.000 description 2
- JRFZNGUHKPPRHG-UHFFFAOYSA-N tert-butyl n-propan-2-ylcarbamate Chemical compound CC(C)NC(=O)OC(C)(C)C JRFZNGUHKPPRHG-UHFFFAOYSA-N 0.000 description 2
- 238000012360 testing method Methods 0.000 description 2
- 238000011287 therapeutic dose Methods 0.000 description 2
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 2
- 231100000607 toxicokinetics Toxicity 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- NPVKVVBMSCQGAS-ZDUSSCGKSA-N (1s)-1-phenyl-2-pyridin-2-ylethanol Chemical compound C([C@H](O)C=1C=CC=CC=1)C1=CC=CC=N1 NPVKVVBMSCQGAS-ZDUSSCGKSA-N 0.000 description 1
- CTPUSMONJOUDPK-KUVIDSNGSA-N (2S)-1-[(2S)-2-amino-3-(4-hydroxyphenyl)propanoyl]-N-[(1S)-1-phenyl-2-pyridin-2-ylethyl]pyrrolidine-2-carboxamide dihydrochloride Chemical compound Cl.Cl.N[C@@H](Cc1ccc(O)cc1)C(=O)N1CCC[C@H]1C(=O)N[C@@H](Cc1ccccn1)c1ccccc1 CTPUSMONJOUDPK-KUVIDSNGSA-N 0.000 description 1
- MTCFGRXMJLQNBG-REOHCLBHSA-N (2S)-2-Amino-3-hydroxypropansäure Chemical compound OC[C@H](N)C(O)=O MTCFGRXMJLQNBG-REOHCLBHSA-N 0.000 description 1
- PJCLWHJUWFZUQV-SZKOKKDDSA-N (2S)-N-[(1S)-1-phenyl-2-pyridin-1-ium-2-ylethyl]pyrrolidin-1-ium-2-carboxamide dichloride Chemical compound [Cl-].[Cl-].O=C(N[C@@H](Cc1cccc[nH+]1)c1ccccc1)[C@@H]1CCC[NH2+]1 PJCLWHJUWFZUQV-SZKOKKDDSA-N 0.000 description 1
- ZYJPUMXJBDHSIF-NSHDSACASA-N (2s)-2-[(2-methylpropan-2-yl)oxycarbonylamino]-3-phenylpropanoic acid Chemical compound CC(C)(C)OC(=O)N[C@H](C(O)=O)CC1=CC=CC=C1 ZYJPUMXJBDHSIF-NSHDSACASA-N 0.000 description 1
- AYMLQYFMYHISQO-QMMMGPOBSA-N (2s)-3-(1h-imidazol-3-ium-5-yl)-2-[(2-methylpropan-2-yl)oxycarbonylamino]propanoate Chemical compound CC(C)(C)OC(=O)N[C@H](C(O)=O)CC1=CN=CN1 AYMLQYFMYHISQO-QMMMGPOBSA-N 0.000 description 1
- CNBUSIJNWNXLQQ-NSHDSACASA-N (2s)-3-(4-hydroxyphenyl)-2-[(2-methylpropan-2-yl)oxycarbonylamino]propanoic acid Chemical compound CC(C)(C)OC(=O)N[C@H](C(O)=O)CC1=CC=C(O)C=C1 CNBUSIJNWNXLQQ-NSHDSACASA-N 0.000 description 1
- SZXBQTSZISFIAO-ZETCQYMHSA-N (2s)-3-methyl-2-[(2-methylpropan-2-yl)oxycarbonylamino]butanoic acid Chemical compound CC(C)[C@@H](C(O)=O)NC(=O)OC(C)(C)C SZXBQTSZISFIAO-ZETCQYMHSA-N 0.000 description 1
- BGOFOVHACSQSIE-ZDUSSCGKSA-N (2s)-5-[bis[(2-methylpropan-2-yl)oxycarbonylamino]methylideneamino]-2-[(2-methylpropan-2-yl)oxycarbonylamino]pentanoic acid Chemical compound CC(C)(C)OC(=O)N[C@H](C(O)=O)CCCNC(NC(=O)OC(C)(C)C)=NC(=O)OC(C)(C)C BGOFOVHACSQSIE-ZDUSSCGKSA-N 0.000 description 1
- 125000004191 (C1-C6) alkoxy group Chemical group 0.000 description 1
- JVTAAEKCZFNVCJ-UWTATZPHSA-M (R)-lactate Chemical compound C[C@@H](O)C([O-])=O JVTAAEKCZFNVCJ-UWTATZPHSA-M 0.000 description 1
- IWYDHOAUDWTVEP-SSDOTTSWSA-M (R)-mandelate Chemical compound [O-]C(=O)[C@H](O)C1=CC=CC=C1 IWYDHOAUDWTVEP-SSDOTTSWSA-M 0.000 description 1
- IWYDHOAUDWTVEP-ZETCQYMHSA-N (S)-mandelic acid Chemical compound OC(=O)[C@@H](O)C1=CC=CC=C1 IWYDHOAUDWTVEP-ZETCQYMHSA-N 0.000 description 1
- KNTZCGBYFGEMFR-UHFFFAOYSA-N (propan-2-ylazaniumyl)formate Chemical compound CC(C)NC(O)=O KNTZCGBYFGEMFR-UHFFFAOYSA-N 0.000 description 1
- CVHZOJJKTDOEJC-UHFFFAOYSA-M 1,1-dioxo-1,2-benzothiazol-3-olate Chemical compound C1=CC=C2C([O-])=NS(=O)(=O)C2=C1 CVHZOJJKTDOEJC-UHFFFAOYSA-M 0.000 description 1
- FWUQWDCOOWEXRY-UHFFFAOYSA-N 1-phenyl-2-pyridin-2-ylethanamine Chemical compound C=1C=CC=CC=1C(N)CC1=CC=CC=N1 FWUQWDCOOWEXRY-UHFFFAOYSA-N 0.000 description 1
- KHJHFYAGQZYCLC-UHFFFAOYSA-N 1-phenyl-2-pyridin-2-ylethanamine;dihydrochloride Chemical compound Cl.Cl.C=1C=CC=CC=1C(N)CC1=CC=CC=N1 KHJHFYAGQZYCLC-UHFFFAOYSA-N 0.000 description 1
- LXFQSRIDYRFTJW-UHFFFAOYSA-N 2,4,6-trimethylbenzenesulfonic acid Chemical compound CC1=CC(C)=C(S(O)(=O)=O)C(C)=C1 LXFQSRIDYRFTJW-UHFFFAOYSA-N 0.000 description 1
- LFXZSGVZSSMCMB-UHFFFAOYSA-M 2,5-dichlorobenzenesulfonate Chemical compound [O-]S(=O)(=O)C1=CC(Cl)=CC=C1Cl LFXZSGVZSSMCMB-UHFFFAOYSA-M 0.000 description 1
- SMNDYUVBFMFKNZ-UHFFFAOYSA-M 2-furoate Chemical compound [O-]C(=O)C1=CC=CO1 SMNDYUVBFMFKNZ-UHFFFAOYSA-M 0.000 description 1
- QKRMFCXDTFLKKT-UHFFFAOYSA-N 2-hydroxyethanesulfonic acid Chemical compound OCCS(O)(=O)=O.OCCS(O)(=O)=O QKRMFCXDTFLKKT-UHFFFAOYSA-N 0.000 description 1
- CVBUNHURBRMLHC-UHFFFAOYSA-N 2-methylpropanoic acid;sodium Chemical compound [Na].CC(C)C(O)=O CVBUNHURBRMLHC-UHFFFAOYSA-N 0.000 description 1
- 229940080296 2-naphthalenesulfonate Drugs 0.000 description 1
- IHCCAYCGZOLTEU-UHFFFAOYSA-M 3-furoate Chemical compound [O-]C(=O)C=1C=COC=1 IHCCAYCGZOLTEU-UHFFFAOYSA-M 0.000 description 1
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 1
- 244000215068 Acacia senegal Species 0.000 description 1
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 1
- USFZMSVCRYTOJT-UHFFFAOYSA-N Ammonium acetate Chemical compound N.CC(O)=O USFZMSVCRYTOJT-UHFFFAOYSA-N 0.000 description 1
- 102100022704 Amyloid-beta precursor protein Human genes 0.000 description 1
- 229920000945 Amylopectin Polymers 0.000 description 1
- DCXYFEDJOCDNAF-UHFFFAOYSA-N Asparagine Natural products OC(=O)C(N)CC(N)=O DCXYFEDJOCDNAF-UHFFFAOYSA-N 0.000 description 1
- KQQGZRYBXLFQEP-ZLBDCPSSSA-N CC(C)(C)OC(NCCCC[C@@H](C(N(CCC1)C1C(N[C@@H](Cc1ncccc1)c1ccccc1)=O)=O)NC(OC(C)(C)C)=O)=O Chemical compound CC(C)(C)OC(NCCCC[C@@H](C(N(CCC1)C1C(N[C@@H](Cc1ncccc1)c1ccccc1)=O)=O)NC(OC(C)(C)C)=O)=O KQQGZRYBXLFQEP-ZLBDCPSSSA-N 0.000 description 1
- LDGAUNPHGCXAKV-ZLBDCPSSSA-N CC(C)(C)OC(N[C@@H](Cc(cc1)ccc1O)C(N(CCC1)C1C(N[C@@H](Cc1ncccc1)c1ccccc1)=O)=O)=O Chemical compound CC(C)(C)OC(N[C@@H](Cc(cc1)ccc1O)C(N(CCC1)C1C(N[C@@H](Cc1ncccc1)c1ccccc1)=O)=O)=O LDGAUNPHGCXAKV-ZLBDCPSSSA-N 0.000 description 1
- PAIOLVDSVXYHAB-SDHOMARFSA-N CC(C)(C)OC(N[C@@H](Cc1c[nH]cn1)C(N(CCC1)[C@@H]1C(N[C@@H](Cc1ncccc1)c1ccccc1)=O)=O)=O Chemical compound CC(C)(C)OC(N[C@@H](Cc1c[nH]cn1)C(N(CCC1)[C@@H]1C(N[C@@H](Cc1ncccc1)c1ccccc1)=O)=O)=O PAIOLVDSVXYHAB-SDHOMARFSA-N 0.000 description 1
- LDKBRLUCVYIOJE-UHFFFAOYSA-N CC(C)C(OC(C)OC(NC(Cc1ncccc1)C1C=CC=CC1)=O)=O Chemical compound CC(C)C(OC(C)OC(NC(Cc1ncccc1)C1C=CC=CC1)=O)=O LDKBRLUCVYIOJE-UHFFFAOYSA-N 0.000 description 1
- CBUYISPEFZMHBR-KRWDZBQOSA-N CC(C)C(OCOC(N[C@@H](Cc1ccccn1)c1ccccc1)=O)=O Chemical compound CC(C)C(OCOC(N[C@@H](Cc1ccccn1)c1ccccc1)=O)=O CBUYISPEFZMHBR-KRWDZBQOSA-N 0.000 description 1
- HDEPIBOBWYNTAW-ACRUOGEOSA-N CC(C)[C@@H](C(N(CCC1)[C@@H]1C(N[C@@H](Cc1ncccc1)c1ccccc1)=O)=O)N Chemical compound CC(C)[C@@H](C(N(CCC1)[C@@H]1C(N[C@@H](Cc1ncccc1)c1ccccc1)=O)=O)N HDEPIBOBWYNTAW-ACRUOGEOSA-N 0.000 description 1
- DTIBPQWTEQBVHL-HJOGWXRNSA-N CC(C)[C@@H](C(N(CCC1)[C@@H]1C(N[C@@H](Cc1ncccc1)c1ccccc1)=O)=O)NC(OC(C)(C)C)=O Chemical compound CC(C)[C@@H](C(N(CCC1)[C@@H]1C(N[C@@H](Cc1ncccc1)c1ccccc1)=O)=O)NC(OC(C)(C)C)=O DTIBPQWTEQBVHL-HJOGWXRNSA-N 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 1
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 1
- 229920002261 Corn starch Polymers 0.000 description 1
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 1
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 1
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 1
- FEWJPZIEWOKRBE-LWMBPPNESA-L D-tartrate(2-) Chemical compound [O-]C(=O)[C@@H](O)[C@H](O)C([O-])=O FEWJPZIEWOKRBE-LWMBPPNESA-L 0.000 description 1
- 206010011971 Decreased interest Diseases 0.000 description 1
- 206010012374 Depressed mood Diseases 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- 208000032131 Diabetic Neuropathies Diseases 0.000 description 1
- 108010016626 Dipeptides Proteins 0.000 description 1
- 206010013496 Disturbance in attention Diseases 0.000 description 1
- 229940122601 Esterase inhibitor Drugs 0.000 description 1
- 108090000371 Esterases Proteins 0.000 description 1
- WHUUTDBJXJRKMK-UHFFFAOYSA-N Glutamic acid Natural products OC(=O)C(N)CCC(O)=O WHUUTDBJXJRKMK-UHFFFAOYSA-N 0.000 description 1
- 239000004471 Glycine Substances 0.000 description 1
- AEMRFAOFKBGASW-UHFFFAOYSA-M Glycolate Chemical compound OCC([O-])=O AEMRFAOFKBGASW-UHFFFAOYSA-M 0.000 description 1
- 229920000084 Gum arabic Polymers 0.000 description 1
- 101000823051 Homo sapiens Amyloid-beta precursor protein Proteins 0.000 description 1
- LCWXJXMHJVIJFK-UHFFFAOYSA-N Hydroxylysine Natural products NCC(O)CC(N)CC(O)=O LCWXJXMHJVIJFK-UHFFFAOYSA-N 0.000 description 1
- PMMYEEVYMWASQN-DMTCNVIQSA-N Hydroxyproline Chemical compound O[C@H]1CN[C@H](C(O)=O)C1 PMMYEEVYMWASQN-DMTCNVIQSA-N 0.000 description 1
- XUJNEKJLAYXESH-REOHCLBHSA-N L-Cysteine Chemical compound SC[C@H](N)C(O)=O XUJNEKJLAYXESH-REOHCLBHSA-N 0.000 description 1
- ONIBWKKTOPOVIA-BYPYZUCNSA-N L-Proline Chemical compound OC(=O)[C@@H]1CCCN1 ONIBWKKTOPOVIA-BYPYZUCNSA-N 0.000 description 1
- QNAYBMKLOCPYGJ-REOHCLBHSA-N L-alanine Chemical compound C[C@H](N)C(O)=O QNAYBMKLOCPYGJ-REOHCLBHSA-N 0.000 description 1
- DCXYFEDJOCDNAF-REOHCLBHSA-N L-asparagine Chemical compound OC(=O)[C@@H](N)CC(N)=O DCXYFEDJOCDNAF-REOHCLBHSA-N 0.000 description 1
- LEVWYRKDKASIDU-IMJSIDKUSA-N L-cystine Chemical compound [O-]C(=O)[C@@H]([NH3+])CSSC[C@H]([NH3+])C([O-])=O LEVWYRKDKASIDU-IMJSIDKUSA-N 0.000 description 1
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 1
- ZDXPYRJPNDTMRX-VKHMYHEASA-N L-glutamine Chemical compound OC(=O)[C@@H](N)CCC(N)=O ZDXPYRJPNDTMRX-VKHMYHEASA-N 0.000 description 1
- AGPKZVBTJJNPAG-WHFBIAKZSA-N L-isoleucine Chemical compound CC[C@H](C)[C@H](N)C(O)=O AGPKZVBTJJNPAG-WHFBIAKZSA-N 0.000 description 1
- JVTAAEKCZFNVCJ-REOHCLBHSA-N L-lactic acid Chemical compound C[C@H](O)C(O)=O JVTAAEKCZFNVCJ-REOHCLBHSA-N 0.000 description 1
- 239000004395 L-leucine Substances 0.000 description 1
- FFEARJCKVFRZRR-BYPYZUCNSA-N L-methionine Chemical compound CSCC[C@H](N)C(O)=O FFEARJCKVFRZRR-BYPYZUCNSA-N 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-L L-tartrate(2-) Chemical compound [O-]C(=O)[C@H](O)[C@@H](O)C([O-])=O FEWJPZIEWOKRBE-JCYAYHJZSA-L 0.000 description 1
- AYFVYJQAPQTCCC-GBXIJSLDSA-N L-threonine Chemical compound C[C@@H](O)[C@H](N)C(O)=O AYFVYJQAPQTCCC-GBXIJSLDSA-N 0.000 description 1
- JVTAAEKCZFNVCJ-UHFFFAOYSA-M Lactate Chemical compound CC(O)C([O-])=O JVTAAEKCZFNVCJ-UHFFFAOYSA-M 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- OFOBLEOULBTSOW-UHFFFAOYSA-L Malonate Chemical compound [O-]C(=O)CC([O-])=O OFOBLEOULBTSOW-UHFFFAOYSA-L 0.000 description 1
- 229930195725 Mannitol Natural products 0.000 description 1
- 241001465754 Metazoa Species 0.000 description 1
- NFVNYBJCJGKVQK-ZDUSSCGKSA-N N-[(Tert-butoxy)carbonyl]-L-tryptophan Chemical compound C1=CC=C2C(C[C@H](NC(=O)OC(C)(C)C)C(O)=O)=CNC2=C1 NFVNYBJCJGKVQK-ZDUSSCGKSA-N 0.000 description 1
- BEEHQODUONYQIO-FKBYEOEOSA-N NCCCC[C@@H](C(N(CCC1)[C@@H]1C(N[C@@H](Cc1ncccc1)c1ccccc1)=O)=O)N Chemical compound NCCCC[C@@H](C(N(CCC1)[C@@H]1C(N[C@@H](Cc1ncccc1)c1ccccc1)=O)=O)N BEEHQODUONYQIO-FKBYEOEOSA-N 0.000 description 1
- PVNIIMVLHYAWGP-UHFFFAOYSA-N Niacin Chemical compound OC(=O)C1=CC=CN=C1 PVNIIMVLHYAWGP-UHFFFAOYSA-N 0.000 description 1
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 1
- 206010036376 Postherpetic Neuralgia Diseases 0.000 description 1
- 208000004550 Postoperative Pain Diseases 0.000 description 1
- AZFUASHXSOTBNU-UHFFFAOYSA-N Propyl 2-methylpropanoate Chemical compound CCCOC(=O)C(C)C AZFUASHXSOTBNU-UHFFFAOYSA-N 0.000 description 1
- LCTONWCANYUPML-UHFFFAOYSA-M Pyruvate Chemical compound CC(=O)C([O-])=O LCTONWCANYUPML-UHFFFAOYSA-M 0.000 description 1
- MTCFGRXMJLQNBG-UHFFFAOYSA-N Serine Natural products OCC(N)C(O)=O MTCFGRXMJLQNBG-UHFFFAOYSA-N 0.000 description 1
- 229920002472 Starch Polymers 0.000 description 1
- 229930006000 Sucrose Natural products 0.000 description 1
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 1
- AYFVYJQAPQTCCC-UHFFFAOYSA-N Threonine Natural products CC(O)C(N)C(O)=O AYFVYJQAPQTCCC-UHFFFAOYSA-N 0.000 description 1
- 239000004473 Threonine Substances 0.000 description 1
- 208000027418 Wounds and injury Diseases 0.000 description 1
- WXIONIWNXBAHRU-UHFFFAOYSA-N [dimethylamino(triazolo[4,5-b]pyridin-3-yloxy)methylidene]-dimethylazanium Chemical compound C1=CN=C2N(OC(N(C)C)=[N+](C)C)N=NC2=C1 WXIONIWNXBAHRU-UHFFFAOYSA-N 0.000 description 1
- 238000010521 absorption reaction Methods 0.000 description 1
- 239000000205 acacia gum Substances 0.000 description 1
- 235000010489 acacia gum Nutrition 0.000 description 1
- CSCPPACGZOOCGX-WFGJKAKNSA-N acetone d6 Chemical compound [2H]C([2H])([2H])C(=O)C([2H])([2H])[2H] CSCPPACGZOOCGX-WFGJKAKNSA-N 0.000 description 1
- WNLRTRBMVRJNCN-UHFFFAOYSA-L adipate(2-) Chemical compound [O-]C(=O)CCCCC([O-])=O WNLRTRBMVRJNCN-UHFFFAOYSA-L 0.000 description 1
- 235000004279 alanine Nutrition 0.000 description 1
- 235000012538 ammonium bicarbonate Nutrition 0.000 description 1
- DZHSAHHDTRWUTF-SIQRNXPUSA-N amyloid-beta polypeptide 42 Chemical compound C([C@@H](C(=O)N[C@@H](C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@H](C(=O)NCC(=O)N[C@@H](CO)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CCCCN)C(=O)NCC(=O)N[C@@H](C)C(=O)N[C@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)NCC(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](C(C)C)C(=O)NCC(=O)NCC(=O)N[C@@H](C(C)C)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](C)C(O)=O)[C@@H](C)CC)C(C)C)NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@@H](NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CC=1N=CNC=1)NC(=O)[C@H](CC=1N=CNC=1)NC(=O)[C@@H](NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)CNC(=O)[C@H](CO)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC=1N=CNC=1)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](C)NC(=O)[C@@H](N)CC(O)=O)C(C)C)C(C)C)C1=CC=CC=C1 DZHSAHHDTRWUTF-SIQRNXPUSA-N 0.000 description 1
- 230000036528 appetite Effects 0.000 description 1
- 235000019789 appetite Nutrition 0.000 description 1
- 229940072107 ascorbate Drugs 0.000 description 1
- 235000010323 ascorbic acid Nutrition 0.000 description 1
- 239000011668 ascorbic acid Substances 0.000 description 1
- 229960001230 asparagine Drugs 0.000 description 1
- 235000009582 asparagine Nutrition 0.000 description 1
- 229940009098 aspartate Drugs 0.000 description 1
- 235000003704 aspartic acid Nutrition 0.000 description 1
- 238000000065 atmospheric pressure chemical ionisation Methods 0.000 description 1
- 229940077388 benzenesulfonate Drugs 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-M benzenesulfonate Chemical compound [O-]S(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-M 0.000 description 1
- OQFSQFPPLPISGP-UHFFFAOYSA-N beta-carboxyaspartic acid Natural products OC(=O)C(N)C(C(O)=O)C(O)=O OQFSQFPPLPISGP-UHFFFAOYSA-N 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 230000004071 biological effect Effects 0.000 description 1
- 208000028683 bipolar I disease Diseases 0.000 description 1
- 208000025307 bipolar depression Diseases 0.000 description 1
- 238000009534 blood test Methods 0.000 description 1
- 230000005587 bubbling Effects 0.000 description 1
- QCIGLPLDNRDLQQ-UHFFFAOYSA-N butan-2-ylcarbamic acid Chemical compound CCC(C)NC(O)=O QCIGLPLDNRDLQQ-UHFFFAOYSA-N 0.000 description 1
- 210000004899 c-terminal region Anatomy 0.000 description 1
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 description 1
- 229910000024 caesium carbonate Inorganic materials 0.000 description 1
- CJZGTCYPCWQAJB-UHFFFAOYSA-L calcium stearate Chemical compound [Ca+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O CJZGTCYPCWQAJB-UHFFFAOYSA-L 0.000 description 1
- 235000013539 calcium stearate Nutrition 0.000 description 1
- 239000008116 calcium stearate Substances 0.000 description 1
- 239000001569 carbon dioxide Substances 0.000 description 1
- 210000004027 cell Anatomy 0.000 description 1
- 239000001913 cellulose Substances 0.000 description 1
- 229920002678 cellulose Polymers 0.000 description 1
- 238000000451 chemical ionisation Methods 0.000 description 1
- 238000001311 chemical methods and process Methods 0.000 description 1
- QOPVNWQGBQYBBP-UHFFFAOYSA-N chloroethyl chloroformate Chemical compound CC(Cl)OC(Cl)=O QOPVNWQGBQYBBP-UHFFFAOYSA-N 0.000 description 1
- 125000004218 chloromethyl group Chemical group [H]C([H])(Cl)* 0.000 description 1
- 230000001684 chronic effect Effects 0.000 description 1
- 229940114081 cinnamate Drugs 0.000 description 1
- 229940125782 compound 2 Drugs 0.000 description 1
- 239000008120 corn starch Substances 0.000 description 1
- 235000018417 cysteine Nutrition 0.000 description 1
- XUJNEKJLAYXESH-UHFFFAOYSA-N cysteine Natural products SCC(N)C(O)=O XUJNEKJLAYXESH-UHFFFAOYSA-N 0.000 description 1
- 229960003067 cystine Drugs 0.000 description 1
- 230000006378 damage Effects 0.000 description 1
- DEZRYPDIMOWBDS-UHFFFAOYSA-N dcm dichloromethane Chemical compound ClCCl.ClCCl DEZRYPDIMOWBDS-UHFFFAOYSA-N 0.000 description 1
- YSMODUONRAFBET-UHFFFAOYSA-N delta-DL-hydroxylysine Natural products NCC(O)CCC(N)C(O)=O YSMODUONRAFBET-UHFFFAOYSA-N 0.000 description 1
- 230000006866 deterioration Effects 0.000 description 1
- UXGNZZKBCMGWAZ-UHFFFAOYSA-N dimethylformamide dmf Chemical compound CN(C)C=O.CN(C)C=O UXGNZZKBCMGWAZ-UHFFFAOYSA-N 0.000 description 1
- 208000035475 disorder Diseases 0.000 description 1
- PMMYEEVYMWASQN-UHFFFAOYSA-N dl-hydroxyproline Natural products OC1C[NH2+]C(C([O-])=O)C1 PMMYEEVYMWASQN-UHFFFAOYSA-N 0.000 description 1
- CETRZFQIITUQQL-UHFFFAOYSA-N dmso dimethylsulfoxide Chemical compound CS(C)=O.CS(C)=O CETRZFQIITUQQL-UHFFFAOYSA-N 0.000 description 1
- 239000002552 dosage form Substances 0.000 description 1
- 229950008913 edisilate Drugs 0.000 description 1
- 238000000132 electrospray ionisation Methods 0.000 description 1
- YSMODUONRAFBET-UHNVWZDZSA-N erythro-5-hydroxy-L-lysine Chemical compound NC[C@H](O)CC[C@H](N)C(O)=O YSMODUONRAFBET-UHNVWZDZSA-N 0.000 description 1
- 239000002329 esterase inhibitor Substances 0.000 description 1
- CCIVGXIOQKPBKL-UHFFFAOYSA-M ethanesulfonate Chemical compound CCS([O-])(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-M 0.000 description 1
- 229940031098 ethanolamine Drugs 0.000 description 1
- OJCSPXHYDFONPU-UHFFFAOYSA-N etoac etoac Chemical compound CCOC(C)=O.CCOC(C)=O OJCSPXHYDFONPU-UHFFFAOYSA-N 0.000 description 1
- 230000003203 everyday effect Effects 0.000 description 1
- 238000002474 experimental method Methods 0.000 description 1
- 235000019253 formic acid Nutrition 0.000 description 1
- 125000000524 functional group Chemical group 0.000 description 1
- 230000002068 genetic effect Effects 0.000 description 1
- 235000013922 glutamic acid Nutrition 0.000 description 1
- 239000004220 glutamic acid Substances 0.000 description 1
- ZDXPYRJPNDTMRX-UHFFFAOYSA-N glutamine Natural products OC(=O)C(N)CCC(N)=O ZDXPYRJPNDTMRX-UHFFFAOYSA-N 0.000 description 1
- 235000004554 glutamine Nutrition 0.000 description 1
- 239000008187 granular material Substances 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-M hydrogensulfate Chemical compound OS([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-M 0.000 description 1
- QJHBJHUKURJDLG-UHFFFAOYSA-N hydroxy-L-lysine Natural products NCCCCC(NO)C(O)=O QJHBJHUKURJDLG-UHFFFAOYSA-N 0.000 description 1
- 229960002591 hydroxyproline Drugs 0.000 description 1
- 238000000099 in vitro assay Methods 0.000 description 1
- 238000002347 injection Methods 0.000 description 1
- 239000007924 injection Substances 0.000 description 1
- 208000014674 injury Diseases 0.000 description 1
- SYJRVVFAAIUVDH-UHFFFAOYSA-N ipa isopropanol Chemical compound CC(C)O.CC(C)O SYJRVVFAAIUVDH-UHFFFAOYSA-N 0.000 description 1
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- 229960000310 isoleucine Drugs 0.000 description 1
- AGPKZVBTJJNPAG-UHFFFAOYSA-N isoleucine Natural products CCC(C)C(N)C(O)=O AGPKZVBTJJNPAG-UHFFFAOYSA-N 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 229940116871 l-lactate Drugs 0.000 description 1
- 229940001447 lactate Drugs 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- 229950003165 lanicemine Drugs 0.000 description 1
- 239000007788 liquid Substances 0.000 description 1
- 239000000314 lubricant Substances 0.000 description 1
- 235000019359 magnesium stearate Nutrition 0.000 description 1
- 229940049920 malate Drugs 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- BJEPYKJPYRNKOW-UHFFFAOYSA-N malic acid Chemical compound OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 description 1
- 239000000594 mannitol Substances 0.000 description 1
- 235000010355 mannitol Nutrition 0.000 description 1
- 238000004519 manufacturing process Methods 0.000 description 1
- 239000000463 material Substances 0.000 description 1
- 239000011159 matrix material Substances 0.000 description 1
- 229930182817 methionine Natural products 0.000 description 1
- 235000006109 methionine Nutrition 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- HZAXFHJVJLSVMW-UHFFFAOYSA-N monoethanolamine hydrochloride Natural products NCCO HZAXFHJVJLSVMW-UHFFFAOYSA-N 0.000 description 1
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- WOOWBQQQJXZGIE-UHFFFAOYSA-N n-ethyl-n-propan-2-ylpropan-2-amine Chemical compound CCN(C(C)C)C(C)C.CCN(C(C)C)C(C)C WOOWBQQQJXZGIE-UHFFFAOYSA-N 0.000 description 1
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 229950005216 napadisilate Drugs 0.000 description 1
- XTEGVFVZDVNBPF-UHFFFAOYSA-N naphthalene-1,5-disulfonic acid Chemical compound C1=CC=C2C(S(=O)(=O)O)=CC=CC2=C1S(O)(=O)=O XTEGVFVZDVNBPF-UHFFFAOYSA-N 0.000 description 1
- KVBGVZZKJNLNJU-UHFFFAOYSA-M naphthalene-2-sulfonate Chemical compound C1=CC=CC2=CC(S(=O)(=O)[O-])=CC=C21 KVBGVZZKJNLNJU-UHFFFAOYSA-M 0.000 description 1
- 230000007935 neutral effect Effects 0.000 description 1
- 235000001968 nicotinic acid Nutrition 0.000 description 1
- 239000011664 nicotinic acid Substances 0.000 description 1
- 238000000655 nuclear magnetic resonance spectrum Methods 0.000 description 1
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 1
- 229940049964 oleate Drugs 0.000 description 1
- ZQPPMHVWECSIRJ-KTKRTIGZSA-N oleic acid Chemical compound CCCCCCCC\C=C/CCCCCCCC(O)=O ZQPPMHVWECSIRJ-KTKRTIGZSA-N 0.000 description 1
- 239000003960 organic solvent Substances 0.000 description 1
- AICOOMRHRUFYCM-ZRRPKQBOSA-N oxazine, 1 Chemical compound C([C@@H]1[C@H](C(C[C@]2(C)[C@@H]([C@H](C)N(C)C)[C@H](O)C[C@]21C)=O)CC1=CC2)C[C@H]1[C@@]1(C)[C@H]2N=C(C(C)C)OC1 AICOOMRHRUFYCM-ZRRPKQBOSA-N 0.000 description 1
- 239000012188 paraffin wax Substances 0.000 description 1
- 239000012466 permeate Substances 0.000 description 1
- 239000000546 pharmaceutical excipient Substances 0.000 description 1
- 125000000286 phenylethyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])C([H])([H])* 0.000 description 1
- XNQULTQRGBXLIA-UHFFFAOYSA-O phosphonic anhydride Chemical compound O[P+](O)=O XNQULTQRGBXLIA-UHFFFAOYSA-O 0.000 description 1
- 239000006187 pill Substances 0.000 description 1
- 238000012123 point-of-care testing Methods 0.000 description 1
- 229920000642 polymer Polymers 0.000 description 1
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 1
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 1
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 1
- 229920001592 potato starch Polymers 0.000 description 1
- 238000011045 prefiltration Methods 0.000 description 1
- 230000002028 premature Effects 0.000 description 1
- 239000001294 propane Substances 0.000 description 1
- MPYTZVJDKGTHDK-UHFFFAOYSA-N pyrrolidine-2-carboxamide;dihydrochloride Chemical compound Cl.Cl.NC(=O)C1CCCN1 MPYTZVJDKGTHDK-UHFFFAOYSA-N 0.000 description 1
- 239000013557 residual solvent Substances 0.000 description 1
- 230000002441 reversible effect Effects 0.000 description 1
- 229920006395 saturated elastomer Polymers 0.000 description 1
- 238000012216 screening Methods 0.000 description 1
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 239000001509 sodium citrate Substances 0.000 description 1
- NLJMYIDDQXHKNR-UHFFFAOYSA-K sodium citrate Chemical compound O.O.[Na+].[Na+].[Na+].[O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O NLJMYIDDQXHKNR-UHFFFAOYSA-K 0.000 description 1
- 239000000600 sorbitol Substances 0.000 description 1
- 230000002269 spontaneous effect Effects 0.000 description 1
- 239000008107 starch Substances 0.000 description 1
- 235000019698 starch Nutrition 0.000 description 1
- 239000000126 substance Substances 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-L succinate(2-) Chemical compound [O-]C(=O)CCC([O-])=O KDYFGRWQOYBRFD-UHFFFAOYSA-L 0.000 description 1
- 239000005720 sucrose Substances 0.000 description 1
- 238000004808 supercritical fluid chromatography Methods 0.000 description 1
- 239000006228 supernatant Substances 0.000 description 1
- 208000024891 symptom Diseases 0.000 description 1
- 238000003786 synthesis reaction Methods 0.000 description 1
- 230000009885 systemic effect Effects 0.000 description 1
- 239000000454 talc Substances 0.000 description 1
- 235000012222 talc Nutrition 0.000 description 1
- 229910052623 talc Inorganic materials 0.000 description 1
- 229940095064 tartrate Drugs 0.000 description 1
- 125000004213 tert-butoxy group Chemical group [H]C([H])([H])C(O*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- DPKBAXPHAYBPRL-UHFFFAOYSA-M tetrabutylazanium;iodide Chemical compound [I-].CCCC[N+](CCCC)(CCCC)CCCC DPKBAXPHAYBPRL-UHFFFAOYSA-M 0.000 description 1
- 239000004408 titanium dioxide Substances 0.000 description 1
- 235000010215 titanium dioxide Nutrition 0.000 description 1
- FGMPLJWBKKVCDB-UHFFFAOYSA-N trans-L-hydroxy-proline Natural products ON1CCCC1C(O)=O FGMPLJWBKKVCDB-UHFFFAOYSA-N 0.000 description 1
- WBYWAXJHAXSJNI-VOTSOKGWSA-M trans-cinnamate Chemical compound [O-]C(=O)\C=C\C1=CC=CC=C1 WBYWAXJHAXSJNI-VOTSOKGWSA-M 0.000 description 1
- 230000007704 transition Effects 0.000 description 1
- 238000004704 ultra performance liquid chromatography Methods 0.000 description 1
- 235000015112 vegetable and seed oil Nutrition 0.000 description 1
- 239000008158 vegetable oil Substances 0.000 description 1
- 210000003462 vein Anatomy 0.000 description 1
- 239000003643 water by type Substances 0.000 description 1
- 239000001993 wax Substances 0.000 description 1
- 229950000339 xinafoate Drugs 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/24—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with substituted hydrocarbon radicals attached to ring carbon atoms
- C07D213/36—Radicals substituted by singly-bound nitrogen atoms
- C07D213/40—Acylated substituent nitrogen atom
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4402—Non condensed pyridines; Hydrogenated derivatives thereof only substituted in position 2, e.g. pheniramine, bisacodyl
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4427—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems
- A61K31/4439—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems containing a five-membered ring with nitrogen as a ring hetero atom, e.g. omeprazole
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/24—Antidepressants
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/06—Dipeptides
- C07K5/06008—Dipeptides with the first amino acid being neutral
- C07K5/06017—Dipeptides with the first amino acid being neutral and aliphatic
- C07K5/06034—Dipeptides with the first amino acid being neutral and aliphatic the side chain containing 2 to 4 carbon atoms
- C07K5/06043—Leu-amino acid
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/06—Dipeptides
- C07K5/06008—Dipeptides with the first amino acid being neutral
- C07K5/06017—Dipeptides with the first amino acid being neutral and aliphatic
- C07K5/06034—Dipeptides with the first amino acid being neutral and aliphatic the side chain containing 2 to 4 carbon atoms
- C07K5/06052—Val-amino acid
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/06—Dipeptides
- C07K5/06008—Dipeptides with the first amino acid being neutral
- C07K5/06078—Dipeptides with the first amino acid being neutral and aromatic or cycloaliphatic
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/06—Dipeptides
- C07K5/06086—Dipeptides with the first amino acid being basic
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/06—Dipeptides
- C07K5/06086—Dipeptides with the first amino acid being basic
- C07K5/06095—Arg-amino acid
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/06—Dipeptides
- C07K5/06104—Dipeptides with the first amino acid being acidic
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/06—Dipeptides
- C07K5/06139—Dipeptides with the first amino acid being heterocyclic
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/06—Dipeptides
- C07K5/06139—Dipeptides with the first amino acid being heterocyclic
- C07K5/06147—Dipeptides with the first amino acid being heterocyclic and His-amino acid; Derivatives thereof
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/06—Dipeptides
- C07K5/06139—Dipeptides with the first amino acid being heterocyclic
- C07K5/06156—Dipeptides with the first amino acid being heterocyclic and Trp-amino acid; Derivatives thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Biochemistry (AREA)
- Biophysics (AREA)
- Genetics & Genomics (AREA)
- Molecular Biology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Veterinary Medicine (AREA)
- Pharmacology & Pharmacy (AREA)
- Public Health (AREA)
- Animal Behavior & Ethology (AREA)
- Epidemiology (AREA)
- Neurosurgery (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Neurology (AREA)
- Biomedical Technology (AREA)
- Psychiatry (AREA)
- Pain & Pain Management (AREA)
- Engineering & Computer Science (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Pyridine Compounds (AREA)
- Medicinal Preparation (AREA)
- Peptides Or Proteins (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Abstract
Un compuesto de fórmula (I):**Fórmula** en donde R1 es**Fórmula** en donde AA es un aminoácido natural enlazado con enlaces peptídicos, o una sal farmacéuticamente aceptable del mismo.
Description
DESCRIPCIÓN
Profármacos antagonistas de NMDA
La presente invención se dirige a profármacos de un antagonista de NMDA, (S)-1-fenil-2-(piridin-2-il)etanamina y se desvela en el presente documento su uso en el tratamiento de depresión y trastornos depresivos, particularmente trastorno depresivo mayor (MDD) y también para el tratamiento de dolor (tales como dolor neuropático). Los profármacos de (S)-1-fenil-2-(piridin-2-il)etanamina también pueden usarse para tratar el Síndrome de Rett, ideación suicida, trastorno bipolar (incluyendo depresión bipolar), trastorno obsesivo compulsivo, envenenamiento por gas sarín y estados epilépticos. Se desvelan además en el presente documento composiciones farmacéuticas que comprenden los profármacos y procesos para prepararlos.
El dolor en una forma u otra es una parte de la vida humana que lo impregna todo. El dolor por lesiones y el dolor post-quirúrgico a menudo son temporales, pero pueden ser graves y pueden persistir. El dolor neuropático tal como la neuropatía diabética y la neuralgia post-herpética impactan gravemente en quienes lo padecen. Cada año, decenas de millones de personas en todo el mundo, incluyendo pacientes al final de sus vidas, padecen dolor con tratamiento inadecuado.
La depresión afecta aproximadamente a 120 millones de personas en todo el mundo. Los síntomas de la depresión incluyen, pero no se limitan a, estado de ánimo deprimido, pérdida de interés o placer, sentimientos de culpa o baja autoestima, sueño o apetito perturbados, baja energía y escasa concentración o cualquier combinación de los mismos. Estos problemas pueden ser crónicos o recurrentes y pueden dar lugar a un deterioro sustancial de una capacidad de un individuo para cuidar de sus responsabilidades del día a día. La depresión es la causa principal de discapacidad según se mide por Años Vividos con una Discapacidad (YLD) y el cuarto contribuyente a la carga global de enfermedad según se mide por Años de Vida Ajustados por Discapacidad (DALY; es decir, la suma de los años de vida potencial perdidos debido a la mortalidad prematura y los años de vida productiva perdidos debido a la discapacidad) en 2000. Por el año 2020, se proyecta que la depresión alcance el segundo lugar en la clasificación de DALY calculada para todas las edades, tanto en hombres como en mujeres. En la actualidad, la depresión ya es la segunda causa de DALY en la categoría de edad de 15-44 años para ambos sexos combinados.
Se ha desvelado el diclorhidrato de (S)-1-fenil-2-(piridin-2-il)etanamina para el tratamiento de MDD a través de tratamiento por infusión intravenosa (Gerard Sanacora et al, póster presentado el 6 de diciembre de 2012 en el 51st Annual Meeting of the American College of Neuropsychopharmacology en Hollywood, Florida, EE.UU. Otras divulgaciones relacionadas incluyen los documentos WO1993/020052, WO2000/056324 y WO2000/63175. Por conveniencia sería útil ser capaz de administrar este fármaco como una forma de dosificación oral. Sin embargo, una preocupación con una forma de dosificación oral tal de diclorhidrato de (S)-1-fenil-2-(piridin-2-il)etanamina sería que podría abrirse para su mal uso intravenoso, por ejemplo, aplastando un comprimido de una forma de dosificación oral de diclorhidrato de (S)-1-fenil-2-(piridin-2-il)etanamina seguido de la inyección inmediata de la forma de dosificación oral aplastada resultante de diclorhidrato de (S)-1-fenil-2-(piridin-2-il)etanamina. Se predice que los profármacos de (S)-1-fenil-2-(piridin-2-il)etanamina se reducen en un cuerpo humano para proporcionar (S)-1-fenil-2-(piridin-2-il)etanamina, de modo que, cuando los profármacos de (S)-1-fenil-2-(piridin-2-il)etanamina se administran por vía oral, se reducirían para liberar una dosis terapéuticamente eficaz de (S)-1-fenil-2-(piridin-2-il)etanamina. Pero si un profármaco de la presente invención se administra por vía intravenosa se predice que el profármaco liberaría (S)-1-fenil-2-(piridin-2-il)etanamina para dar una Cmáx menor, a una velocidad más lenta, que si se hubiera administrado por vía intravenosa la dosis correspondiente de (S)-1-fenil-2-(piridin-2-il)etanamina. El uso de profármacos de (S)-1-fenil-2-(piridin-2-il)etanamina se predeciría que mejoraría el perfil de seguridad clínico de (S)-1-fenil-2-(piridin-2-il)etanamina (por ejemplo en condiciones sobredosis o abuso de drogas (por ejemplo, aplastando píldoras)). Por consiguiente, en resumen, poner (S)-1-fenil-2-(piridin-2-il)etanamina directamente en una formulación oral podría dar lugar a abusos. Los compuestos de la presente invención se metabolizan in vivo para dar (S)-1-fenil-2-(piridin-2-il)etanamina, pero a una velocidad más lenta que cuando se administra (S)-1 -fenil-2-(piridin-2-il)etanamina por vía venosa y por lo tanto no alentaría el abuso potencial de (S)-1-fenil-2-(piridin-2-il)etanamina. Descripción de los dibujos
La Figura 1a y 1b ilustra la conversión de profármaco de (S)-1-fenil-2-(piridin-2-il)etanamina (Ejemplo 5) a (S)-1 -fenil-2-(piridin-2-il)etanamina durante un periodo de incubación en el fluido intestinal humano. En particular, la figura 1a ilustra la concentración del profármaco de (S)-1-fenil-2-(piridin-2-il)etanamina (Ejemplo 5) (concentraciones iniciales de 30, 100, 300 y 600 |j M), conforme se transforma en (S)-1-fenil-2-(piridin-2-il)etanamina, durante un periodo de incubación en el fluido intestinal humano. La Figura 1b ilustra la concentración de (S)-1-fenil-2-(piridin-2-il)etanamina, conforme se convierte desde profármaco de (S)-1-fenil-2-(piridin-2-il)etanamina (Ejemplo 5, concentraciones iniciales de 30, 100, 300 y 600 j M), durante un periodo de incubación en el fluido intestinal humano.
Sumario de la invención
El alcance de la invención se define en las reivindicaciones. Por consiguiente, la invención reivindicada se refiere a un compuesto de fórmula (I):

en donde R1 es:

en donde AA es un aminoácido natural enlazado con enlaces peptídicos,
o una sal farmacéuticamente aceptable del mismo.
Se desvela además en el presente documento un compuesto de fórmula (I):

o una sal farmacéuticamente aceptable del mismo.
Alquilo es una cadena lineal o ramificada y contiene 1-6, por ejemplo, 1-4, átomos de carbono. Alquilo es, por ejemplo, metilo, etilo, n-propilo, iso-propilo, n-butilo, sec-butilo, iso-butilo o /ere-butilo.
Alcoxi es una cadena lineal o ramificada y contiene 1-6, por ejemplo, 1-4, átomos de carbono. Alcoxi es, por ejemplo, metoxi, etoxi, n-propoxi, iso-propoxi, n-butoxi, sec-butoxi, iso-butoxi o /ere-butoxi.
Descripción detallada de la invención
Se desvela en el presente documento un compuesto de fórmula (I) en donde R1 es alquil C1-6C(O)O(alcoxi C1-6), por ejemplo es alquil C1-4C(O)O(alcoxi C1-4). Los ejemplos incluyen: (CH3)2ChC(O)OCH2O, (CHs)2CHC(O)OCH(CH(CHs)2)O, CH3C(O)OCH(CH3)O o (CH3)2CHC(O)OCH(CH3)O.
La presente invención proporciona un compuesto de fórmula (I) en donde R1 es

y AA es un aminoácido natural enlazado con enlaces peptídicos.
En otro aspecto la presente invención proporciona un compuesto de fórmula (I) en donde R1 es
(por lo que el centro quiral * tiene configuración absoluta S).
En un aspecto adicional la presente invención proporciona un compuesto de fórmula (I) en donde AA es, por ejemplo, glicina, alanina, valina, leucina, isoleucina, serina, treonina, cisteína, cistina, metionina, ácido aspártico, ácido glutámico, asparagina, glutamina, lisina, hidroxilisina, arginina, histidina, fenilalanina, tirosina, triptófano, prolina o hidroxiprolina. En un aspecto adicional más AA se selecciona del grupo que consiste en tirosina, triptófano, fenilalanina, leucina, arginina, histidina, lisina y valina. En otro aspecto AA se selecciona del grupo que consiste en tirosina, arginina, histidina, lisina y valina. En un aspecto más adicional AA es valina.
Una sal farmacéuticamente aceptable es, por ejemplo, una sal de adición de ácidos tales como clorhidrato, bromhidrato, sulfato, fosfato, acetato, fumarato, maleato, tartrato, lactato, citrato, piruvato, succinato, oxalato, metanosulfonato, p-toluenosulfonato, bisulfato, bencenosulfonato, etanosulfonato, malonato, xinafoato, ascorbato, oleato, nicotinato, sacarinato, adipato, formiato, glicolato, L-lactato, D-lactato, aspartato, malato, L-tartrato, D-tartrato, estearato, 2-furoato, 3-furoato, napadisilato (naftalen-1,5-disulfonato o naftalen-1-(ácido sulfónico)-5-sulfonato), edisilato (etan-1,2-disulfonato o etan-1-(ácido sulfónico)-2-sulfonato), isetionato (2-hidroxietilsulfonato), 2-mesitilensulfonato, 2-naftalensulfonato, D-mandelato, L-mandelato, 2,5-diclorobencenosulfonato, cinamato o benzoato.
Se desvela en el presente documento un compuesto de fórmula (I) que es:
Isobutirato de (S)-(1-fenil-2-(piridin-2-il)etilcarbamoiloxi)metilo;
Isobutirato de 2-metil-1-((S)-1-fenil-2-(piridin-2-il)etilcarbamoiloxi)propilo;
Diastereómero 1 de isobutirato de 2-metil-1-((S)-1-fenil-2-(piridin-2-il)etilcarbamoiloxi)propilo;
Diastereómero 2 de isobutirato de 2-metil-1-((S)-1-fenil-2-(piridin-2-il)etilcarbamoiloxi)propilo;
Acetato de 1-((S)-1-fenil-2-(piridin-2-il)etilcarbamoiloxi)etilo;
Diastereómero 1 de acetato de 1-((S)-1-fenil-2-(piridin-2-il)etilcarbamoiloxi)etilo;
Diastereómero 2 de acetato de 1-((S)-1-fenil-2-(piridin-2-il)etilcarbamoiloxi)etilo;
1- ((S)-1-fenil-2-(piridin-2-il)etilcarbamoiloxi)etilisobutirato;
Diastereómero 1 de 1-((S)-1-fenil-2-(piridin-2-il)etilcarbamoiloxi)etilisobutirato; o
Diastereómero 2 de 1-((S)-1-fenil-2-(piridin-2-il)etilcarbamoiloxi)etilisobutirato,
o una sal farmacéuticamente aceptable de uno cualquiera de los anteriores.
En un aspecto, la invención proporciona un compuesto seleccionado del grupo que consiste en:
(S)-1-((S)-2-Amino-3-metilbutanoil)-N-((S)-1-fenil-2-(piridin-2-il)etil)pirrolidin-2-carboxamida;
(s)-1-((s)-2,6-Diaminohexanoil)-N-((S)-1-fenil-2-(piridin-2-il)etil)pirrolidin-2-carboxamida;
(S)-1-((S)-2-Amino-3-(1H-imidazol-4-il)propanoil)-N-((S)-1-fenil-2-(piridin-2-il)etil)pirrolidin-2-carboxamida;
2- ((S)-2-((S)-1-((S)-2-Amino-3-(4-hidroxifenil)propanoil)pirrolidin-2-carboxamido)-2-feniletil)piridina;
2-((S)-2-((S)-1-((S)-2-Amino-3-(1H-indol-3-il)propanoil)pirrolidin-2-carboxamido)-2-feniletil)piridina;
(S)-1-((S)-2-Amino-3-fenilpropanoil)-N-((S)-1-fenil-2-(piridin-2-il)etil)pirrolidin-2-carboxamida; y
(S)-1-((S)-2-Amino-4-metilpentanoil)-N-((S)-1-fenil-2-(piridin-2-il)etil)pirrolidin-2-carboxamida,
o una sal farmacéuticamente aceptable de uno cualquiera de los anteriores.
Los compuestos de la invención y la divulgación adicional pueden prepararse adaptando métodos descritos en la técnica o adaptando métodos descritos en los Ejemplos. El compuesto (S)-1-fenil-2-(piridin-2-il)etanamina puede prepararse, por ejemplo, por la metodología de proceso del documento EP-0633879 y el contenido de ese documento se incorpora por referencia.
Los compuestos de la presente invención en donde R1 es

pueden sintetizarse usando un proceso químico que avanza a través de un intermedio 2-((S)-2-fenil-2-((S)-pirrolid-2-incarboxamido)etil)piridina.
Por consiguiente, se desvela además en el presente documento el compuesto 2-((S)-2-fenil-2-((S)-pirrolid-2-incarboxamido)etil)piridina:

o una sal del mismo, en donde dicha sal es, por ejemplo, una sal de clorhidrato, bromhidrato, sulfato, fosfato, acetato, metanosulfonato, p-toluenosulfonato, formiato o benzoato.
Se desvela además en el presente documento el compuesto intermedio cloruro de 2-((S)-2-fenil-2-((S)-pirrolidin-2-iocarboxamido)etil)piridinio

Un compuesto de fórmula (I), o una sal farmacéuticamente aceptable del mismo, puede usarse en el tratamiento de depresión (tales como trastorno depresivo mayor, por ejemplo, tratamiento de trastorno depresivo mayor resistente).
Un compuesto de fórmula (I), o una sal farmacéuticamente aceptable del mismo, puede usarse en el tratamiento de dolor (tales como dolor neuropático, dolor crónico, dolor del miembro fantasma, dolor nociceptivo, dolor psicogénico, dolor incidental o dolor irruptivo).
Un compuesto de fórmula (I), o una sal farmacéuticamente aceptable del mismo, puede usarse en el tratamiento de Síndrome de Rett, ideación suicida, trastorno bipolar, trastorno obsesivo compulsivo, envenenamiento por gas sarín o estados epilépticos.
Por consiguiente, se desvela en el presente documento un compuesto de fórmula (I) o una sal farmacéuticamente aceptable del mismo como se define en el presente documento para su uso en terapia. Por lo tanto, el término "profármaco" como se usa en el presente documento puede referirse a un compuesto de fórmula (I) en forma de una sal o en forma de una base libre.
Se desvela además en el presente documento el uso de un compuesto de fórmula (I) o una sal farmacéuticamente aceptable del mismo como se define en el presente documento en la fabricación de un medicamento para su uso en terapia.
Se desvela además en el presente documento un método de administración de (S)-1-fenil-2-(piridin-2-il)etanamina a un paciente, comprendiendo dicho método administrar un compuesto de fórmula (I) o una sal farmacéuticamente aceptable del mismo, a un paciente en donde dicho compuesto de fórmula (I) se metaboliza en dicho paciente para producir (S)-1-fenil-2-(piridin-2-il)etanamina.
En el contexto de la presente memoria descriptiva, el término "terapia" también incluye "profilaxis" salvo que haya indicaciones específicas al contrario. Los términos "terapéutico" y "terapéuticamente" deben construirse en consecuencia.
Se espera que la profilaxis sea particularmente relevante al tratamiento de personas que han padecido un episodio previo de, o de otra manera se considera que están en un riesgo aumentado de, la enfermedad o afección en cuestión. Las personas en riesgo de desarrollar una enfermedad o afección particular incluyen generalmente aquellas que tienen un historial familiar de la enfermedad o afección, o a aquellas que se han identificado mediante un ensayo genético o pruebas de detección como que son particularmente susceptibles a desarrollar la enfermedad o afección.
Se desvela además en el presente documento un método de tratamiento de depresión que comprende administrar a un paciente que lo necesite una cantidad terapéuticamente eficaz de un compuesto de fórmula (I) o una sal farmacéuticamente aceptable del mismo como se define en el presente documento.
Se desvela además en el presente documento un método de tratamiento de MDD que comprende administrar a un paciente que lo necesite una cantidad terapéuticamente eficaz de un compuesto de fórmula (I) o una sal farmacéuticamente aceptable del mismo como se define en el presente documento.
Se desvela además en el presente documento un método de tratamiento de dolor que comprende administrar a un paciente que lo necesite una cantidad terapéuticamente eficaz de un compuesto de fórmula (I) o una sal farmacéuticamente aceptable del mismo como se define en el presente documento.
Se desvela además en el presente documento un método de tratamiento de dolor neuropático, dolor crónico, dolor del miembro fantasma, dolor nociceptivo, dolor psicogénico, dolor incidental o dolor irruptivo que comprende administrar a un paciente que lo necesite una cantidad terapéuticamente eficaz de un compuesto de fórmula (I) o una sal farmacéuticamente aceptable del mismo como se define en el presente documento.
Se desvela además en el presente documento el uso de un compuesto de fórmula (I) o una sal farmacéuticamente aceptable del mismo para el tratamiento de depresión.
Se desvela además en el presente documento el uso de un compuesto de fórmula (I) o una sal farmacéuticamente aceptable del mismo para el tratamiento de dolor.
Para los usos terapéuticos anteriormente mencionados la dosificación administrada, por supuesto, variará con el compuesto empleado, el modo de administración, el tratamiento deseado y el trastorno indicado. Por ejemplo, la dosificación diaria del compuesto de la invención, si se inhala, puede estar en el intervalo de 0,05 microgramos por kilogramo de peso corporal (mg/kg) a 100 microgramos por kilogramo de peso corporal (mg/kg). Como alternativa, si el compuesto se administra por vía oral, entonces la dosificación diaria del compuesto de la invención puede estar en el intervalo de 0,01 microgramos por kilogramo de peso corporal (mg/kg) a 100 microgramos por kilogramo de peso corporal (mg/kg).
Los compuestos de fórmula (I) y las sales farmacéuticamente aceptables de los mismos pueden usarse solos pero en general se administrarán en forma de una composición farmacéutica en donde el compuesto/la sal de fórmula (I) (principio activo) está en asociación con un adyuvante, diluyente o vehículo farmacéuticamente aceptables. Se describen procedimientos convencionales para la selección y preparación de formulaciones farmacéuticas adecuadas en, por ejemplo, "Pharmaceuticals - The Science of Dosage Form Designs", M. E. Aulton, Churchill Livingstone, 1988.
Dependiendo del modo de administración, la composición farmacéutica comprenderá preferentemente del 0,05 al 99 % (por ciento en peso), más preferentemente del 0,05 al 80 % en peso, aún más preferentemente del 0,10 al 70 % en peso y aún más preferentemente del 0,10 al 50 % en peso, de principio activo, basándose todos los porcentajes en peso en la composición total. En algunas realizaciones, la composición farmacéutica comprende un 0,5 % en peso de principio activo. En algunas realizaciones, la composición farmacéutica comprende un 20 % en peso de principio activo.
Se desvela además en el presente documento una composición farmacéutica que comprende un compuesto de fórmula (I) o una sal farmacéuticamente aceptable del mismo como se define en el presente documento, en asociación con un adyuvante, diluyente o vehículo farmacéuticamente aceptables.
Se desvela además en el presente documento un proceso para la preparación de una composición farmacéutica de la invención que comprende mezclar un compuesto de fórmula (I) o una sal farmacéuticamente aceptable del mismo como se define en el presente documento con un adyuvante, diluyente o vehículo farmacéuticamente aceptables. Para la administración oral el compuesto de la invención puede mezclarse con un adyuvante o un vehículo, por ejemplo, lactosa, sacarosa, sorbitol, manitol; un almidón, por ejemplo, almidón de patata, almidón de maíz o amilopectina; un derivado de celulosa; un aglutinante, por ejemplo, gelatina o polivinilpirrolidona; y/o un lubricante, por ejemplo, estearato magnésico, estearato de calcio, polietilenglicol, una cera, parafina y similares y después se comprime en comprimidos. Si se requieren comprimidos recubiertos, los núcleos, preparados como se ha descrito anteriormente, pueden recubrirse con una solución de azúcar concentrada que puede contener, por ejemplo, goma arábiga, gelatina, talco y dióxido de titanio. Como alternativa, el comprimido puede recubrirse con un polímero adecuado disuelto en un disolvente orgánico fácilmente volátil.
Para la preparación de cápsulas de gelatina blanda, el compuesto de la invención puede mezclarse con, por ejemplo, un aceite vegetal o un polietilenglicol. Las cápsulas de gelatina dura pueden contener gránulos del compuesto usando cualquiera de los excipientes anteriormente mencionados para comprimidos. También pueden cargarse formulaciones líquidas o semisólidas del compuesto de la invención en las cápsulas de gelatina dura. Los compuestos de la invención también pueden administrarse junto con otros compuestos usados para el tratamiento de las afecciones anteriores.
Los siguientes Ejemplos ilustran adicionalmente la presente invención. En los Ejemplos se usaron ciertas técnicas y estas se describen a continuación.
La cromatografía líquida de alta presión (HPLC) se realizó en una columna de fase inversa (RP). Se aplicó un gradiente lineal usando por ejemplo fase móvil A (ácido fórmico al 0,1 % en H2O MiliQ o NH3 al 0,1 % en H2O MiliQ o NH4OAc 10 mM y CH3CN al 5 % en H2O MiliQ, o ácido trifluórico al 0,05 % en H2O MiliQ o NH4HCO3 en H2O MiliQ (10 mM)) y B (CH3OH o CH3CN). Los análisis de espectrómetro de masas (EM) se realizaron en modo iónico positivo y/o negativo usando ionización por electropulverizado (ESI+/-), foto ionización a presión atmosférica (APPI+/-) y/o ionización química a presión atmosférica (APCI+/-).
La cromatografía de gases (GC) se realizó en un GC equipado con un espectrómetro de masas (EM) o un detector de ionización a la llama (FID). La fuente iónica de EM fue impacto de electrones (EI) o bien una ionización química (CI, gas reactivo: metano). Para la separación se usó una columna capilar, por ejemplo DB-5MS (J&W Scientific). Se aplicó un gradiente de temperatura lineal.
Se realizó una cromatografía de fluidos supercríticos (SFC) en una columna de fase recta. Se aplicó un flujo isocrático usando la fase móvil A (CO2) y por ejemplo la fase móvil B (MeOH, EtOH o IPA).
Como alternativa, la cromatografía líquida de alta presión (HPLC) se realizó en una columna de fase recta. Se aplicó un gradiente lineal o un flujo isocrático usando por ejemplo la fase móvil A (heptano) y B (EtOH o IPA).
Los espectros de RMN se registraron en un espectrómetro de RMN de 300 MHz (o campo más alto) equipado con una sonda de configuración adecuada. Los espectros se registraron a temperatura ambiente salvo que se indique de otra manera. Los desplazamientos químicos se dan en ppm campo abajo y arriba a partir de TMS (0,00 ppm). Se usaron las siguientes señales de referencia: TMS 5 0,00, o la señal de disolvente residual de DMSO-d6 5 2,49, CD3OD 5 3,30, acetona-d6 2,04, CDCl3 5 7,25 o D2O 5 4,79 (salvo que se indique de otra manera). Las multiplicidades de resonancia se denotan s, d, t, q, m, a y ap para singlete, doblete, triplete, cuadruplete, multiplete, amplio y aparente, respectivamente.
Lista de abreviaturas
DBU 1,8-Diazabiciclo[5,4,0]undec-7-eno
DCM Diclorometano
DEA Dietilamina
DIPEA Diisopropil etil amina
DMF Dimetilformamida
DMSO Dimetilsulfóxido
EtOAc Acetato de etilo
HATU Hexafluorofosfato de 2-(1H-7-azabenzotriazol-1-il)-1,1,3,3-tetrametiluronio metanaminio
IPA iso-propanol
MTBE Metil terc-butil éter
ta a temperatura ambiente o temperatura ambiental, aproximadamente 20-25 °C
sat saturado
T3P Anhídrido fosfónico de propano
Ejemplo de referencia 1
Isobutirato de (S)-(1-fenil-2-(piridin-2-il)etilcarbamoiloxi)metilo
Etapa A
Isobutirato de clorometilo

A ácido isobutírico (0,600 ml, 6,47 mmol) en DCM (6 ml) se añadió bicarbonato sódico (2092 mg, 24,91 mmol), hidrógeno sulfato de tetrabutilamonio (220 mg, 0,65 mmol) y agua (6 ml). Con agitación rápida, se añadió sulfoclorhidrato de clorometilo (0,767 ml, 7,44 mmol) a ta y la mezcla de reacción se agitó después a ta durante toda la noche. La mezcla de reacción se diluyó después con DCM (10 ml), se lavó con agua (2 x 10 ml), se secó sobre Na2SO4, se filtró y se concentró para dar isobutirato de clorometilo (746 mg, 84 %), que se usó en la siguiente etapa sin purificación adicional. RMN 1H (500 MHz, CDCh) S ppm 1,17 (m, 6 H), 2,62 (m, 1 H), 5,72 (s, 2 H).
Etapa B
Isobutirato de (S)-(1 -fenil-2-(piridin-2-il)etilcarbamoiloxi)metilo

Se añadieron carbonato de cesio (1543 mg, 4,74 mmol) y yoduro de tetrabutilamonio (1749 mg, 4,74 mmol) a (S)-(1-fenil-2-(piridin-2-il)etanamina (313 mg, 1,58 mmol) en DMF anhidro (8 ml) a ta. Se burbujeó dióxido de carbono gaseoso a la mezcla de reacción durante 30 min, seguido de la adición de isobutirato de clorometilo (647 mg, 4,74 mmol) en DMF (2 ml). La mezcla de reacción se agitó a ta con burbujeo continuado de CO2 gaseoso durante toda la noche y se continuó agitando durante el fin de semana sin adición de CO2 gaseoso adicional. La mezcla de reacción se diluyó con agua y se extrajo con EtOAc (3x), las capas orgánicas combinadas se lavaron con agua (2x), salmuera, se secaron sobre Na2SO4 y se concentraron. La purificación se realizó por cromatografía en columna usando un gradiente de EtOAc en heptano (0-60 %) para dar el compuesto del título (239 mg, 44,2 %).
RMN 1H (500 MHz, DMSO-d6) S ppm 1,00 (m, 6 H), 2,46 (m, 1 H, parcialmente oculto en DMSO-d6), 3,09 (m, 2 H), 5,04 (m, 1 H), 5,52 (m, 2 H), 7,17 - 7,24 (m, 3 H), 7,27 - 7,34 (m, 4 H), 7,65 (td, 1 H), 8,24 (d, 1 H), 8,49 (m, 1 H).
Ejemplo de referencia 2
Isobutirato de 2-metil-1-((S)-1-fenil-2-(piridin-2-il)etilcarbamoiloxi)propilo
Hay dos diastereómeros diferentes del Ejemplo de referencia 2 debido a las dos posibles configuraciones en el átomo de carbono identificado con *. Estos se denominan diastereómero 1 del Ejemplo de referencia 2 y diastereómero 2 del Ejemplo de referencia 2. Sus configuraciones absolutas no se han determinado.
Etapa A
Isobutirato de 2-metil-1-((4-(metilsulfonil)fenoxi)carboniloxi)propilo.

(i) Se tomó 4-(metilmercapto)fenol (8,46 g, 57,30 mmol) en DCM (60 ml) y después el matraz de reacción se enfrió a 0 °C, seguido de la adición de carbonoclorhidrato de 1-cloro-2-metilpropilo (4,27 mg, 28,65 mmol). Se añadió una solución de 4-metilmorfolina (7,87 g, 71,63 mmol) en DCM (40 ml) gota a gota, durante 50 min a 0 °C y la mezcla resultante se agitó a esta temperatura durante 5 minutos y se agitó finalmente a ta durante 150 minutos. La mezcla de reacción se lavó con agua (2x), se secó sobre Na2SO4, se filtró y se evaporó para dar carbonato de 1-cloro-2-metilpropil 4-(metiltio)fenilo (13,84 g), que se usó en la siguiente etapa sin purificación adicional.
(ii) Una mezcla de carbonato de 1-cloro-2-metilpropil 4-(metiltio)fenilo (3,50 g, 12,74 mmol), óxido de plata(I) (2,95 g, 12,74 mmol) y ácido isobutírico (13,00 ml, 140,12 mmol) en una atmósfera de argón se calentó a 95 °C durante 2 horas. La mezcla de reacción se enfrió a ta y se agitó a ta durante toda la noche, después se diluyó con MTBE, se filtró a través de tierra de diatomeas y se lavó con más MTBE. Los filtrados combinados se lavaron con agua (4x25 ml), bicarbonato sódico sat. ac. (2x25 ml), se secaron (Na2SO4) y se evaporaron para dar 3,56 g de isobutirato de 2-metil-1-((4-(metiltio)fenoxi)carboniloxi)propilo, que se usó en la siguiente etapa sin purificación adicional.
(iii) Isobutirato de 2-metil-1-((4-(metiltio)fenoxi)carboniloxi)propilo (3,56 g, 10,91 mmol) se tomó en una mezcla de acetona (30 ml) y agua (7,50 ml), seguido de la adición de oxona (13,41 g, 21,81 mmol) en porciones durante 5 min, después se agitó a ta durante 2 horas. La mezcla de reacción se filtró y el filtrado se lavó con MTBE (2x50 ml), el volumen se redujo a 50 ml (destilando la acetona) y después la mezcla resultante se separó entre MTBE y agua. La capa acuosa se extrajo con MTBE y los extractos orgánicos combinados se secaron sobre Na2SO4, se filtraron y se evaporaron para dar 1,89 g del compuesto del título, que se usó en la siguiente etapa sin purificación adicional.
Etapa B:
Isobutirato de 2-metil-1-((S)-1-fenil-2-(piridin-2-il)etilcarbamoiloxi)propilo
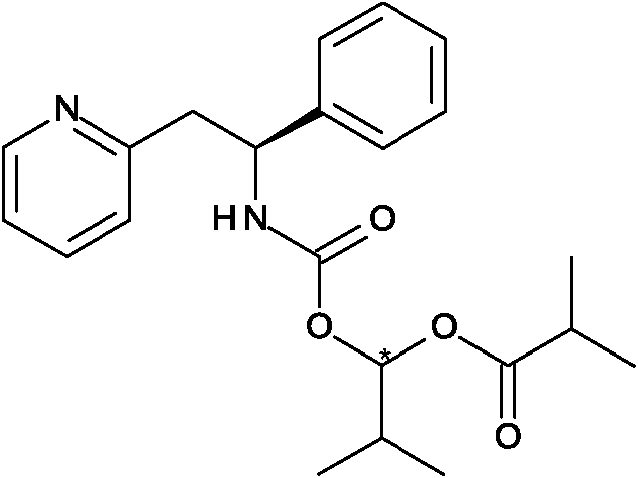
A una mezcla agitada de (S)-1-fenil-2-(piridin-2-il)etanamina (0,215 g, 1,08 mmol) y bicarbonato sódico (0,182 g, 2,17 mmol) en acetonitrilo (3 ml) se añadió isobutirato de 2-metil-1-((4-(metilsulfonil)fenoxi)carboniloxi)propilo (0,389 g, l , 08 mmol) en acetonitrilo (2 ml) y la reacción se agitó a ta durante 2 horas. Separada entre EtOAc y NaHCO3 sat. ac. , la capa orgánica se lavó con NaHCO3 sat. ac., se secó (Na2SO4) y se evaporó para dar 413 mg de material, que se purificó por cromatografía en columna usando un gradiente de EtOAc en heptano (0-50 %) para dar 235 mg como una mezcla de los dos diastereómeros de 2-metil-1-(S)-1-fenil-2-(piridin-2-il)etilcarbamoiloxi)propilisobutirato.
El análisis y la separación de los diastereómeros se realizó en Chiralpak AD-H, 4,6*250 mm; 5 pm, usando MeOH al 10 %/CO2 al 90 % a un flujo de 3 ml/min y Chiralpak AD-H, 20*250 mm; 5 pm, usando MeOH al 10 %/CO2 al 90 % a un flujo de 50 ml/min, respectivamente.
Ejemplo de referencia 2, diastereómero 1
Se obtuvieron 105 mg de diastereómero 1 por separación quiral como el primer diastereómero que eluye con una pureza óptica del 99 %. Mezcla de rotámeros:
RMN 1H (500 MHz, DMSO-d6) 8 ppm 0,66 - 1,07 (m), 1,86 (m, 2,39 (m, 2,98 - 3,21 (m), 5,01 (m, 6,31 (m, 7,10 - 7,26 (m), 7,26 - 7,37 (m), 7,56 - 7,73 (m), 8,10 (d), 8,43 - 8,55 (m). N.° total de protones en el espectro: 28. Relación de rotámeros mayor/menor: 1/0,15.
EM (ES+APCI+) m/z = 385 (M+H)+
Ejemplo de referencia 2, diastereómero 2
Se obtuvieron 104 mg de diastereómero 2 como el segundo diastereómero que eluye. Mezcla de rotámeros:
RMN 1H (500 MHz, DMSO-d6) 8 ppm 0,48 - 0,69 (m), 0,75 - 0,87 (m), 0,87 - 1,07 (m), 1,73 (m, 1,85 (m, 2,40 (m, 2,98 (m, 3,03 - 3,18 (m), 4,97 (m, 6,22 - 6,38 (m), 7,13 - 7,26 (m), 7,26 - 7,37 (m), 7,65 (m, 7,78 (d), 8,06 (d), 8,48 (m). N.° total de protones en el espectro: 28. Relación de rotámeros mayor/menor: 1/0,17.
EM (ES+APCI+) m/z = 385 (M+H)+.
Pureza óptica = 99 %.
Ejemplo de referencia 3
Acetato de 1-((S)-1-fenil-2-(piridin-2-il)etilcarbamoiloxi)etilo
Hay dos diastereómeros diferentes del Ejemplo de referencia 3 debido a las dos posibles configuraciones en el átomo de carbono identificado con *. Estos se denominan diastereómero 1 del Ejemplo de referencia 3 y diastereómero 2 del Ejemplo de referencia 3. Sus configuraciones absolutas no se han determinado.
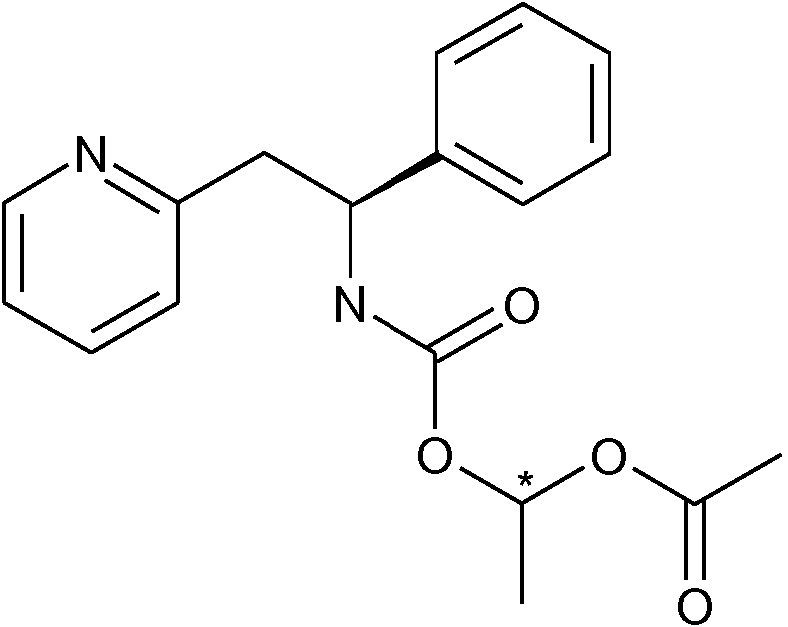
Etapa A:
Acetato de 1-((4-(metilsulfonil)fenoxi)carboniloxi)etilo

(i) Una mezcla de carbonato de 1 -cloroetil 4-(metiltio)fenilo (4,6 g, 18,65 mmol), óxido de plata (I) (4,32 g, 18,65 mmol) y ácido acético (11,75 ml, 205,1 mmol) en una atmósfera de argón se calentó a 95 °C durante 2 h. La mezcla de reacción se enfrió a ta y se diluyó con MTBE, se filtró a través de tierra de diatomeas y se lavó con más MTBE. Los filtrados combinados se lavaron con agua (4 x 25 ml), bicarbonato sódico sat. ac. (2 x 25 ml), se secaron (Na2SO4) y se evaporaron para dar 1,79 g de acetato de 1-((4-(metiltio)fenoxi)carboniloxi)etilo que se usó en la siguiente etapa sin purificación adicional.
(ii) Acetato de 1-((4-(metiltio)fenoxi)carboniloxi)etilo (1,79 g, 6,62 mmol) se tomó en una mezcla de acetona (16 ml) y agua (4,00 ml). Se añadió oxona (8,14 g, 13,24 mmol) en porciones durante 5 min, después se agitó a ta durante toda la noche. La mezcla de reacción se filtró y el filtrado se lavó con MTBE (2x50 ml), el volumen se redujo a aproximadamente 50 ml (destilando la acetona). El producto se separó entre MTBE y agua. La capa acuosa se extrajo con MTBE y los extractos orgánicos combinados se secaron (Na2SO4), se filtraron y se evaporaron para dar 832 mg del compuesto del título y se usaron en la siguiente etapa sin purificación adicional.
Etapa B:
Acetato de 1-((S)-1-fenil-2-(piridin-2-il)etilcarbamoiloxi)etilo

A una mezcla agitada de (S)-1-fenil-2-(piridin-2-il)etanamina (198 mg, 1 mmol) y bicarbonato sódico (168 mg, 2,00 mmol) en acetonitrilo (4 ml) se añadió acetato de 1-((4-(metilsulfonil)fenoxi)carboniloxi)etilo (302 mg, 1,00 mmol) en acetonitrilo (1 ml) y la reacción se agitó a ta durante 4 horas. La mezcla de reacción se separó después entre EtOAc y NaHCO3 sat. ac., la capa orgánica se lavó con NaHCO3 sat. ac., salmuera, se secó sobre Na2SO4 y se evaporó. esta mezcla se pre-purificó por cromatografía en columna usando un gradiente de EtOAc en heptano (0 50 %) para dar 160 mg del compuesto del título como una mezcla de sus dos diastereómeros.
El análisis y la separación de los diastereómeros se realizó en Phenomenex LuxC4, 4,6*250 mm; 5 ^m, usando MeOH+DEA al 30 %/CO2 al 70 % a un flujo de 3 ml/min y Phenomenex LuxC4, 20*250 mm; 5 ^m, usando MeOH+DEA al 25 %/CO2 al 75 % a un flujo de 50 ml/min, respectivamente. Ejemplo de referencia 3, diastereómero 1
Se obtuvieron 29 mg de diastereómero 1 por separación quiral como el primer diastereómero que eluye con una pureza óptica del 99 %.
RMN 1H (500 MHz, DMSO-d6) 5 ppm 1,31 (d, 3H), 1,92 (s, 3 H), 3,00 - 3,16 (m, 2 H), 5,00 (td, 1 H), 6,52 (q, 1 H), 7,14 - 7,25 (m, 3 H), 7,30 (d, 4 H), 7,65 (t, 1 H), 8,13 (d, 1 H), 8,50 (d, 1 H). Las señales en el espectro se ampliaron y no pudo verse la división de la señal de DMSO.
RMN 13C (126 MHz, DMSO-d6) 519,6, 20,7, 44,6, 54,9, 88,7, 121,6, 123,8, 126,4, 126,9, 128,3, 136,2, 143,0, 149,0, 153,2, 158,0, 168,6 ppm. EM (Es +) m/z 328 (M+H)+. Ejemplo de referencia 3, diastereómero 2
Se obtuvieron 39 mg de diastereómero 2 como el segundo diastereómero que eluye.
RMN 1H (500 MHz, DMSO-d6) 5 ppm 1,32 (d, 3 H), 1,95 (s, 3 H), 3,01 - 3,18 (m, 2 H), 5,00 (td, 1 H), 6,53 (q, 1 H), 7,13 - 7,24 (m, 3 H), 7,29 (d, 4 H), 7,65 (t, 1 H), 8,17 (d, 1 H), 8,49 (d, 1 H). Las señales en el espectro se ampliaron y no pudo verse la división de la señal de DMSO.
RMN 13C (126 MHz, DMSO-d6) 519,6, 20,7, 44,4, 54,9, 88,6, 121,6, 123,7, 126,4, 126,9, 128,3, 136,2, 143,0, 149,0, 153,1, 158,0, 168,5 ppm. EM (ES+) m/z 329 (M+H)+.
Pureza óptica 98 %.
Ejemplo de referencia 4
Isobutirato de 1-((S)-1-fenil-2-(piridin-2-il)etilcarbamoiloxi)etilo
Hay dos diastereómeros diferentes del Ejemplo de referencia 4 debido a las dos posibles configuraciones en el átomo de carbono identificado con *. Estos se denominan diastereómero 1 del Ejemplo de referencia 4 y diastereómero 2 del Ejemplo de referencia 4. Sus configuraciones absolutas no se han determinado.

Etapa A:
Isobutirato de 1-((4-(metilsulfonil)fenoxi)carboniloxi)etilo

(i) Se tomó 4-(metilmercapto)fenol (4,91 g, 35,00 mmol) en DCM (100 ml) y se enfrió a 0 °C. Se añadió cloroformiato de 1-cloroetilo (1,888 ml, 17,50 mmol). Una solución de 4-metilmorfolina (4,81 ml, 43,74 mmol) en DCM (20 ml) se añadió gota a gota durante 10 min a 0 °C y la mezcla resultante se agitó a esta temperatura durante 5 minutos. La mezcla de reacción se agitó a ta durante 180 min, después se diluyó con DCM, se lavó con agua (2x), se secó sobre Na2SO4, se filtró y se evaporó para dar carbonato de 1 -cloroetil 4-(metiltio)fenilo (6,94 g), que se usó en la siguiente etapa sin purificación adicional.
(ii) Una mezcla de carbonato de 1 -cloroetil 4-(metiltio)fenilo (2,3 g, 9,32 mmol), óxido de plata (I) (2,160 g, 9,32 mmol) y ácido isobutírico (9,51 ml, 102,55 mmol) en una atmósfera de argón se calentó a 95 °C durante 2 h. La mezcla de reacción se enfrió a ta y se diluyó con MTBE, se filtró a través de tierra de diatomeas y se lavó con más MTBE. Los filtrados combinados se lavaron con agua (4x25 ml), bicarbonato sódico sat. ac. (2x25 ml), se secaron (Na2SO4) y se evaporaron para dar 1,16 g de producto, que se usó en la siguiente etapa sin purificación adicional.
(ii) Isobutirato de 1-((4-(metiltio)fenoxi)carboniloxi)etilo (1,16 g, 3,89 mmol) se tomó en una mezcla de acetona (12 ml) y agua (3,00 ml). Se añadió oxona (4,78 g, 7,78 mmol) en porciones durante 5 min, después se agitó a ta durante toda la noche. La mezcla de reacción se filtró y el filtrado se lavó con MTBE (2x50 ml), el volumen se redujo a aproximadamente 50 ml (destilando la acetona), después se repartió entre MTBE y agua. La capa acuosa se extrajo con MTBE y los extractos orgánicos combinados se secaron sobre Na2SO4, se filtraron y se evaporaron para dar 531 mg del compuesto del título, que se usó en la siguiente etapa sin purificación adicional.
Etapa B
Isobutirato de 1-((S)-1-fenil-2-(piridin-2-il)etilcarbamoiloxi)etilo

A una mezcla agitada de (S)-1-fenil-2-(piridin-2-il)etanamina (0,300 g, 1,51 mmol) y bicarbonato sódico (0,254 g, 3,03 mmol) en acetonitrilo (6 ml) se añadió isobutirato de 1-((4-(metilsulfonil)fenoxi)carboniloxi)etilo (0,500 g, 1,51 mmol) en acetonitrilo (2 ml) y la reacción se agitó durante toda la noche. La mezcla se separó entre EtOAc y NaHCO3 sat. ac., la capa orgánica se lavó con NaHCO3 sat. ac., salmuera, se secó sobre Na2SO4 y se evaporó para dar 0,70 g, que se purificó por cromatografía en columna usando un gradiente de EtOAc en heptano (0-50 %) para dar el compuesto del título (0,309 g, 57,2 %) como mezcla de los dos diastereómeros.
El análisis y la separación de los diastereómeros se realizó en Phenomenex LuxC4, 4,6*250 mm; 5 pm, usando MeOH+DEA al 15%/CO2 al 85% a un flujo de 3 ml/min y Phenomenex LuxC4, 20*250 mm; 5 pm, usando MeOH+DEA al 15 %/CO2 al 85 % a un flujo de 50 ml/min, respectivamente. Ejemplo de referencia 4, diastereómero 1 Se obtuvieron 25 mg de diastereómero 1 por separación quiral como el primer diastereómero que eluye con una pureza óptica del 99 %. Mezcla de rotámeros:
RMN 1H (500 MHz, DMSO-d6) 8 ppm 0,86 - 1,05 (m), 1,19 (d), 1,32 (d), 2,31 - 2,44 (m), 2,98 - 3,20 (m), 4,94 - 5,07 (m), 6,48 (d), 6,52 (q), 7,15 - 7,25 (m), 7,25 - 7,34 (m), 7,60 - 7,70 (m), 7,77 (d), 8,06 - 8,18 (m), 8,44 - 8,55 (m). N.° total de protones en el espectro: 24. Relación mayor/menor: 1/0,07.
EM (ES+APCI+) m/z = 357 (M+H)+.
Pureza de UV = 100%.
Ejemplo de referencia 4, diastereómero 2
Se obtuvieron 25 mg de diastereómero 2 como el segundo isómero que eluye. Mezcla de rotámeros:
RMN 1H (500 MHz, DMSO-d6) 8 ppm 0,82 - 0,94 (m), 0,94 - 1,04 (m), 1,25 (d), 1,32 (d), 2,24 - 2,32 (m), 2,34 - 2,44 (m), 3,00 - 3,17 (m), 5,02 (td), 6,48 (d), 6,52 (q), 7,15 - 7,25 (m), 7,25 - 7,38 (m), 7,55 - 7,77 (m), 8,16 (d), 8,44 - 8,57 (m). N.° total de protones en el espectro: 24. Relación mayor/menor: 1/0,08.
EM (ES+APCI+) m/z = 357 (M+H)+.
Pureza de UV = 100%.
Pureza óptica = 99 %.
Los ejemplos 5 a 12 usan un intermedio común, la preparación del cual se describe ahora.
Preparación del intermedio común para los Ejemplos 5 a 12
Dicloruro de 2-((S)-2-fenil-2-((S)-pirrolidin-2-iocarboxamido)etil)piridinio (que también puede nombrarse como: diclorhidrato de (S)-N-((S)-1-fenil-2-(piridin-2-il)etil)pirrolidin-2-carboxamida)

ETAPA A
2-((S)-1-Fenil-2-(piridin-2-il)etilcarbamoil)pirrolidin-1-carboxilato de (S)-terc-butilo

T3P (50 % en peso en DMF, 0,86 ml, 1,47 mmol) se añadió gota a gota a una solución de (1S)-1-fenil-2-(piridin-2-il)etan-1-amina (200 mg, 0,737 mmol), Boc-L-prolina (159 mg, 0,737 mmol) y DIPEA (0,64 ml, 3,69 mmol) en CH2Cl2 (7,4 ml) a 0 °C. La reacción se calentó gradualmente a ta mientras se agitaba durante toda la noche. la capa orgánica se lavó dos veces con NaHCO3 ac. al 5 %, una vez con salmuera, se secó sobre Na2SO4, se filtró y se concentró. El producto se purificó usando una columna de ajuste de 50 g, eluyendo con un gradiente de MeOH y CH2Cl2 (MeOH al 0 % / CH2Cl2 al 100 % ^ MeOH al 10 % / CH2Cl2 al 90 %), produciendo el compuesto del título 305 mg (>100 %).
RMN 1H (300 MHz, CDCla): ppm 1,35 (s, 9 H), 1,70 - 1,90 (m, 2 H), 2,05 - 2,19 (m, 2 H), 3,13 (dd, 1 H), 3,29 - 3,39 (m, 1 H), 3,46 - 3,62 (m, 2 H), 4,20 - 4,29 (m, 2 H), 5,39 (q, 1 H), 6,91 (d, 1 H), 7,09 - 7,14 (m, 1 H), 7,16 - 7,23 (m, 5 H), 7,49 (td, 1 H), 8,45 - 8,59 (m, 1 H).
Método alternativo:
Se disolvió Boc-L-Pro-OH (2,0 g, 9,29 mmol) en DMF (15 ml). Se añadieron HATU (3,7 g, 9,76 mmol) y base de Hunig (5,3 ml, 30,66 mmol) y la mezcla se agitó a ta durante 30 minutos. Después, se añadió dicloruro de 2-[(2S)-2-azaniomil-2-feniletil]piridin-1-io (2,5 g, 9,29 mmol) a la solución y la mezcla se agitó durante 3 horas 30 minutos a ta. Se añadió agua y la mezcla se extrajo 3 x con EtOAc. La fase orgánica se lavó con salmuera, se secó sobre Na2SO4, se filtró y se concentró a presión reducida. El producto se purificó en una columna de SiO2, eluyendo con un gradiente (MeOH al 0 % / EtOAc al 50 % / heptano al 50 % ^ MeOH al 0 % / EtOAc al 100 % / heptano al 0 % ^ MeOH al 10 % / EtOAc al 90 % / heptano al 0 %) y después en una columna C-18, eluyendo con un gradiente de MeOH y agua (0 al 100 % de MeOH), produciendo 2,9 g (78 %) del compuesto del título.
RMN 1H (300 MHz, CDCI3): ppm 1,35 (s, 9 H), 1,70 - 1,90 (m, 2 H), 2,05 - 2,19 (m, 2 H), 3,13 (dd, J = 13,8, 7,1 Hz, 1 H), 3,29 - 3,39 (m, 1 H), 3,46 - 3,62 (m, 2 H), 4,20 - 4,29 (m, 1 H), 5,39 (c, J = 6,8 Hz, 1 H), 6,91 (d, J = 7,3 Hz, 1 H), 7,09 - 7,14 (m, 1 H), 7,16 - 7,23 (m, 5 H), 7,49 (td, J = 7,63, 1,8 Hz, 1 H), 8,45 - 8,59 (m, 1 H).
Etapa B
Diclorhidrato de (S)-N-((S)-1-fenil-2-(piridin-2-il)etil)pirrolidin-2-carboxamida

A 0 °C, 2-((S)-1-Fenil-2-(piridin-2-il)etilcarbamoil)pirrolidin-1-carboxilato de (S)-ferc-butilo (305 mg, 0,771 mmol) se disolvió en una solución de HCl 4 M en 1,4-dioxano (7,7 ml, 30,8 mmol). Se calentó la reacción gradualmente a ta mientras se agitaba durante toda la noche. Los volátiles se retiraron al vacío y el producto se trituró en MTBE. El sólido se recuperó por filtración en un embudo Büchner, se lavó con MTBE y se secó al vacío proporcionando 263 mg (93 %) del compuesto del título como sal de diclorhidrato.
RMN 1H (300 MHz, DMSO-d6): ppm 1,61 - 1,84 (m, 3 H), 2,30 - 2,36 (m, 1 H), 3,05 - 3,11 (m, 2 H), 3,39 - 3,47 (m, 2 H), 4,05 - 4,20 (m, 2 H), 5,34 (q, 1 H), 7,17 - 7,41 (m, 5 H), 8,20 - 8,42 (m, 1 H), 8,67 - 8,75 (m, 1 H), 9,45 - 9,62 (m, 2 H).
Método alternativo:
(2S)-2-{[(1S)-1-Fenil-2-(piridin-2-il)etil]carbamoil}pirrolidin-1 -carboxilato de ferc-butilo (2,9 g, 7,33 mmol) se disolvió en una solución de HCl 4 M en 1,4-dioxano (73 ml, 293,31 mmol). La mezcla de reacción se agitó a ta durante 3 h. Los volátiles se retiraron al vacío y el producto se trituró en MTBE. El sólido se recuperó por filtración en un embudo Büchner, se lavó con MTBE y se secó al vacío proporcionando 2,7 g (100 %) de (2S)-2-{[(1S)-1-fenil- 2-(piridin-1-io-2-il)etil]carbamoil}pirrolidin-1-io.
RMN 1H (300 MHz, D2O): ppm 1,66 - 1,83 (m, 3 H), 2,20 - 2,26 (m, 1 H), 3,14 (t, J = 6,9 Hz, 2 H), 3,34 (dd, J = 14,1, 8,2 Hz, 1 H), 3,51 (dd, J = 14,4, 7,3 Hz, 1 H), 4,18 (dd, J = 8,5, 6,2 Hz, 1 H), 5,11 (t, J = 7,9 Hz, 1 H), 7,06 - 7,10 (m, 2 H), 7,15 - 7,21 (m, 3 H), 7,61 - 7,70 (m, 2 H), 8,23 (dt, J = 7,9, 1,5 Hz, 1 H), 8,37 (d, J = 5,8 Hz, 1 H).
Ejemplo 5
(S)-1-((S)-2-Amino-3-metilbutanoil)-N-((S)-1-fenil-2-(piridin-2-il)etil)pirrolidin-2-carboxamida

El compuesto del título se preparó como el diclorhidrato de (S)-1-((S)-2-amino-3-metilbutanoil)-N-((S)-1-fenil-2-(piridin-2-il)etil)pirrolidin-2-carboxamida
ETAPA A
(S)-3-Metil-1-oxo-1-((S)-2-((S)-1-fenil-2-(piridin-2-il)etilcarbamoil)pirrolidin-1-il)butan-2-ilcarbamato de ferc-butilo

T3P (50 % en peso en DMF, 0,83 ml, 1,42 mmol) se añadió gota a gota a una solución de sal de diclorhidrato de (S)-N-((S)-1-fenil-2-(piridin-2-il)etil)pirrolidin-2-carboxamida (263 mg, 0,714 mmol), Boc-L-Valina (155 mg, 0,714 mmol) y DIPEA (0,62 ml, 3,57 mmol) en DCM (7 ml) a 0 °C. La reacción se calentó gradualmente a ta mientras se agitaba durante toda la noche. La capa orgánica se lavó dos veces con NaHCO3 ac. al 5 %, una vez con salmuera, se secó sobre Na2SO4, se filtró y se concentró. El producto se purificó usando una columna de ajuste de 50 g, eluyendo con un gradiente de MeOH y CH2Cl2 (MeOH al 5 % / CH2Cl2 al 95 % ^ MeOH al 10 % / CH2Cl2 al 90 %), produciendo el compuesto del título 258 mg (73 %).
RMN 1H (300 MHz, CDCh): ppm 0,91 (d, 3 H), 1,00 (d, 3 H), 1,43 (s, 9 H), 1,85 - 1,99 (m, 2 H), 2,00 -2,16 (m, 1 H), 2,17 - 2,21 (m, 1 H), 3,12 - 3,27 (m, 2 H), 3,47 - 3,62 (m, 1 H), 3,63 - 3,78 (m, 1 H), 4,32 (dd, 1 H), 4,58 (d, 1 H), 5,23 - 5,35 (m, 2 H), 6,97 (d, 1 H), 7,10 (dd, 1 H), 7,14 - 7,30 (m, 5 H), 7,50 (td, 1 H), 7,84 (d, 1 H), 8,48 (dd, 1 H).
ETAPA B
Diclorhidrato de (S)-1-((S)-2-Amino-3-metilbutanoil)-N-((S)-1-fenil-2-(piridin-2-il)etil)pirrolidin-2-carboxamida

(S)-3-metil-1-oxo-1-((S)-2-((S)-1-fenil-2-(piridin-2-il)etilcarbamoil)pirrolidin-1-il)butan-2-ilcarbamato de ferc-butilo (258 mg, 0,522 mmol) se disolvió en una solución de HCl 4 M en 1,4-dioxano (5,2 ml, 30,8 mmol). La mezcla de reacción se agitó durante una noche. Los volátiles se retiraron al vacío y el producto se trituró en EtOAc. El sólido se recuperó por filtración en un embudo Büchner, después se trituró en Et2O. Después de la filtración en un embudo Büchner, el sólido se secó al vacío proporcionando 200 mg (82 %) del compuesto del título como sal de diclorhidrato. El compuesto del título como la sal de diclorhidrato se convirtió después en la base libre por técnicas conocidas por un experto ordinario en la materia. Como alternativa, el compuesto del título puede prepararse como una sal de fumarato por métodos conocidos por un experto ordinario en la materia.
RMN 1H (300 MHz, CD3OD): ppm 0,97 (d, 3 H), 1,04 (d, 3 H), 1,68 - 1,79 (m, 1 H), 1,86 - 2,05 (m, 2 H), 2,13 - 2,27 (m, 2 H), 3,50 - 3,61 (m, 3 H), 3,68 - 3,75 (m, 1 H), 4,02 (d, 1 H), 4,45 (dd, 1 H), 5,40 (dd, 1 H), 7,31 - 7,43 (m, 5 H), 7,90 (t, 1 H), 8,02 (d, 1 H), 8,51 (td, 1 H), 8,75 (d, 1 H).
[M+H]+ = 395,27
Ejemplo 6
(S)-1-((S)-2,6-Diaminohexanoil)-N-((S)-1-fenil-2-(piridin-2-il)etil)pirrolidin-2-carboxamida

El compuesto del título se preparó como el tricloruro, tricloruro de 2-[(2S)-2-{[(2S)-1-[(2S)-2,5-diazinioilhexanoil]pirrolidin-2-il]formamido}-2-feniletiN]piridin-1-io
ETAPA A
(S)-6-oxo-6-((S)-2-((S)-1-fenil-2-(piridin-2-il)etilcarbamoil)pirrolidin-1-il)hexan-1,5-diilcarbamato de ferc-butilo

Sal de diciclohexilamonio de N-alfa,N-épsilon-bis(ferc-butoxicarbonil)-L-lisina (287 mg, 0,54 mmol) se disolvió en DMF anhidro (4 ml). Se añadieron HATU (217 mg, 0,57 mmol) y DIp Ea (0,21 ml, 1,19 mmol) y la mezcla se agitó a ta durante 30 minutos. Después, se añadió dicloruro de (2S)-2-{[(1S)-1-fenil-2-(piridin-1-io-2-il)etil]carbamoil}pirrolidin-1-io (200 mg, 0,54 mmol) a la solución y la mezcla se agitó durante 18 h a ta. Se añadió agua y la mezcla se extrajo 3 x con EtOAc. La fase orgánica se lavó con salmuera, se secó sobre Na2SO4, se filtró y se concentró a presión reducida. El producto se purificó en una columna de SiO2, eluyendo con un gradiente (MeOH al 0 % / EtOAc al 50 % / heptano al 50 % ^ MeOH al 0 % / EtOAc al 100 % / heptano al 0 % ^ MeOH al 10 % / EtOAc al 90 % / heptano al 0 %) después en una columna C-18, eluyendo con un gradiente de MeOH y agua (0 al 100 % de MeOH), produciendo 80 mg (24 %) del compuesto del título.
RMN 1H (300 MHz, CDCla): ppm 1,34 - 1,39 (m, 2 H), 1,41 (s, 9 H), 1,43 (s, 9 H), 1,52 - 1,64 (m, 3 H), 1,68 -2,02 (m, 4 H), 2,08 - 2,22 (m, 2 H), 3,02 - 3,16 (m, 3 H), 3,20 - 3,28 (m, 1 H), 3,48 - 3,57 (m, 1 H), 3,60 - 3,70 (m, 1 H), 4,40 -4,51 (m, 1 H), 4,54 - 4,57 (m, 1 H), 4,97 - 5,08 (m, 1 H), 5,29 - 5,39 (m, 2 H), 7,00 (d, 1 H), 7,10 - 7,32 (m, 6 H), 7,52 (td, 1 H), 7,86 (d, 1 H), 8,49 (d, 1 H).
ETAPA B
Síntesis de tricloruro de 2-[(2S)-2-{[(2S)-1-[(2S)-2,5-diazinioilhexanoil]pirrolidin-2-il]formamido}-2-feniletill]piridin-1-io

(S)-6-oxo-6-((S)-2-((S)-1-fenil-2-(piridin-2-il)etilcarbamoil)pirrolidin-1-il)hexan-1,5-diilcarbamato de ferc-butilo (80 mg, 0,13 mmol) se disolvió en una solución de HCl 4 M en 1,4-dioxano (1,6 ml, 6,4 mmol). La reacción se agitó a ta
durante 18 horas. Los volátiles se retiraron al vacío y el producto se trituró en MTBE. El sólido se recuperó por filtración en un embudo Büchner, se lavó con MTBE y se secó al vacío. El sólido se disolvió en agua y se secó por congelación para proporcionar 60 mg (88 %) del compuesto del título.
RMN 1H (300 MHz, D2O): ppm 1,23 - 1,35 (m, 2 H), 1,46 - 1,59 (m, 3 H), 1,65 - 1,80 (m, 4 H), 2,04 - 2,13 (m, 1 H), 2,80 (t, 2 H), 3,31 - 3,43 (m, 2 H), 3,47 - 3,57 (m, 2 H), 4,16 (t, 1 H), 4,28 (t, 1 H), 5,11 (t, 1 H), 7,10 - 7,13 (m, 2 H), 7,16 - 7,26 (m, 3 H), 7,68 - 7,73 (m, 2 H), 8,31 (td, 1 H), 8,42 (dd, 1 H);
[M+H]+ = 424,2.
Ejemplo 7
(S)-1-((S)-2-Amino-3-(1H-imidazol-4-il)propanoil)-N-((S)-1-fenil-2-(piridin-2-il)etil)pirrolidin-2-carboxamida

El compuesto del título se preparó como el tricloruro, tricloruro de 2-[(2S)-2-{[(2S)-1-[(2S)-2-azanioil-3-(1H-imidazol-1-io-4-il)propanoil]pirrolidin-2-il]formamido}-2-fenil etil]piridin-1-io
ETAPA A
(S)-3-(1H-imidazol-4-il)-1-oxo-1-((S)-2-((S)-1-fenil-2-(piridin-2-il)etilcarbamoil)pirrolidin-1-il)propan-2-ilcarbamato de ferc-butilo

Se disolvió Boc-His-OH (166 mg, 0,65 mmol) en DMF anhidro (4 ml). Se añadieron HATU (260 mg, 0,68 mmol) y DIPEA (0,38 ml, 2,85 mmol) y la mezcla se agitó a ta durante 30 minutos. Después, se añadió cloruro de 2-((S)-2-fenil-2-((S)-pirrolidin-2-iocarboxamido)etil)piridinio (240 mg, 0,65 mmol) y la mezcla se agitó durante 66 horas a ta. Se añadió agua y la mezcla se extrajo 3x con EtOAc. La fase orgánica se lavó con salmuera, se secó sobre Na2SO4, se filtró y se concentró a presión reducida. El producto se purificó en una columna de SiO2, eluyendo con un gradiente de MeOH y CH2Cl2 (0 % a 13 % de MeOH) después en una columna C-18, eluyendo con un gradiente de MeOH y agua (0 al 100 % de MeOH), produciendo 100 mg (29 %) del compuesto del título.
RMN 1H (300 MHz, CDCla): ppm 1,44 (s, 9 H), 1,77 - 2,00 (m, 3 H), 2,05 - 2,19 (m, 1 H), 3,03 - 3,14 (m, 2 H), 3,23 (dd, 1 H), 3,40 (dd, 1 H), 3,52 - 3,58 (m, 1 H), 4,50 - 4,62 (m, 2 H), 5,43 - 5,50 (m, 2 H), 6,91 - 6,97 (m, 2 H), 7,13 -7,27 (m, 5 H), 7,53 (t, 1 H), 7,65 (s, 1 H), 8,53 (d, 1 H), 8,67 (d, 1 H).
ETAPA B
Tricloruro de 2-((S)-2-((S)-1-((S)-2-amonio-3-(1H-imidazol-1-io-4-il)propanoil)pirrolidin-2-carboxamido)-2-feniletil)piridinio

(S)-3-(1H-imidazol-4-il)-1-oxo-1-((S)-2-((S)-1-fenil-2-(piridin-2-il)etilcarbamoil)pirrolidin-1-il)butan-2-ilcarbamato de tere-butilo (100 mg, 0,19 mmol) se disolvió en una solución de HCl 4 M en 1,4-dioxano (2,3 ml, 9,39 mmol). La mezcla de reacción se agitó a ta durante 18 h. Los volátiles se retiraron al vacío y el producto se trituró en MTBE. El sólido se recuperó por filtración en un embudo Büchner, se lavó con MTBE y se secó al vacío. El sólido se disolvió en agua y se secó por congelación para proporcionar 85 mg (69 %) del compuesto del título.
RMN 1H (300 MHz, D2O): ppm 1,50 - 1,58 (m, 1 H), 1,69 - 1,76 (m, 2 H), 2,07 - 2,14 (m, 1 H), 3,12 - 3,21 (m, 3 H), 3,38 (dd, 7,3 Hz, 1 H), 3,48 - 3,56 (m, 2 H), 4,32 (t, 1 H), 4,43 (t, 1 H), 5,14 (t, 1 H), 7,08 - 7,12 (m, 2 H), 7,15 - 7,23 (m, 3 H), 7,26 (s, 1 H), 7,65 - 7,70 (m, 2 H), 8,26 (td, 1 H), 8,39 (d, 1 H), 8,49 (d, 1 H);
[M+H]+ = 433,2;
[M+Na]+ = 455,1.
Ejemplo 8
Diclorhidrato de 2-((S)-2-((S)-1-((S)-2-amino-3-(4-hidroxifenil)propanoil)pirrolidin-2-carboxamido)-2-feniletil)piridina

El compuesto del título también puede nombrarse como: dicloruro de 2-[(2S)-2-{[(2S)-1-[(2S)-2-azanioil-3-(4-hidroxifenil)propanoil]pirrolidin-2-il]formamido}-2-feniletil]piridin-1-io
ETAPA A
(S)-3-(4-hidroxifenil)-1-oxo-1-((S)-2-((S)-1-fenil-2-(piridin-2-il)etilcarbamoil)pirrolidin-1-il)propan-2-ilcarbamato de tere-butilo

Se disolvió Boc-Tyr-OH (229 mg, 0,81 mmol) en DMF anhidro (5 ml). Se añadieron HATU (325 mg, 0,86 mmol) y DIPEA (0,47 ml, 2,69 mmol) y la mezcla se agitó a ta durante 30 minutos. Después, se añadió cloruro de 2-((S)-2
fenil-2-((S)-pirrolidin-2-iocarboxamido)etil)piridinio (300 mg, 0,81 mmol) a la solución y la mezcla se agitó durante 66 h a ta. Se añadió agua y la mezcla se extrajo 3x con EtOAc. La fase orgánica se lavó con salmuera, se secó sobre Na2SO4, se filtró y se concentró a presión reducida. El producto se purificó en una columna de SO2, eluyendo con un (MeOH al 0 % / EtOAc al 50 % / heptano al 50 % ^ MeOH al 0 % / EtOAc al 100 % / heptano al 0 % ^ MeOH al 15 % / EtOAc al 85 % / heptano al 0 %) después en una columna C-18, eluyendo con un gradiente de MeOH y agua (0 al 100 % de MeOH), produciendo 180 mg (40 %) del compuesto del título.
RMN 1H (300 MHz, CDCla): ppm 1,44 (s, 9 H), 1,73 - 2,02 (m, 3 H), 2,16 - 2,22 (m, 1 H), 2,92 - 3,09 (m, 4 H), 3,35 -3,42 (m, 1 H), 3,48 - 3,51 (m, 1 H), 3,53 - 3,68 (m, 1 H), 4,48 - 4,51 (m, 1 H), 4,65 - 4,73 (m, 1 H), 5,06 - 5,14 (m, 1 H), 5,43 (d, 1 H), 6,61 (d, 1 H), 6,86 (d, 2 H), 6,94 - 6,99 (m, 2 H), 7,08 - 7,26 (m, 6 H), 7,42 (dd, 1 H), 8,49 (d, 1 H), 8,63 (s, 1 H).
ETAPA B:
Dicloruro de 2-((S)-2-((S)-1-((S)-2-amonio-3-(4-hidroxifenil)propanoil)pirrolidin-2-carboxamido)-2-feniletil)piridinio

(S)-3-(4-hidroxifenil)-1-oxo-1-((S)-2-((S)-1-fenil-2-(piridin-2-il)etilcarbamoil)pirrolidin-1-il)propan-2-ilcarbamato de terc-butilo (180 mg, 0,32 mmol) se disolvió en una solución de HCl 4 M en 1,4-dioxano (3,2 ml, 12,89 mmol). La reacción se agitó a ta durante 18 horas. Los volátiles se retiraron al vacío y el producto se trituró en MTBE. El sólido se recuperó por filtración en un embudo Büchner, se lavó con MTBE y se secó al vacío. El sólido se disolvió en agua y se secó por congelación para proporcionar 150 mg (88 %) del compuesto del título.
RMN 1H (300 MHz, D2O): ppm 1,48 - 1,57 (m, 1 H), 1,68 - 1,73 (m, 2 H), 1,98 - 2,05 (m, 1 H), 2,68 - 2,76 (m, 1 H), 2,93 - 3,00 (m, 1 H), 3,10 - 3,19 (m, 1 H), 3,40 - 3,63 (m, 3 H), 4,20 - 4,28 (m, 2 H), 5,14 (t, 1 H), 6,65 (d, 2 H), 6,96 (d, 2 H), 7,13 - 7,22 (m, 5 H), 7,63 - 7,66 (m, 2 H), 8,19 - 8,22 (m, 1 H), 8,40 (d, 1 H);
[M+H]+ = 459,2.
Ejemplo 9
Diclorhidrato de 2-((S)-2-((S)-1-((S)-2-amino-3-(1H-indol-3-il)propanoil)pirrolidin-2-carboxamido)-2-feniletil)piridina

El compuesto del título también puede nombrarse: dicloruro de 2-[(2S)-2-{[(2S)-1-[(2S)-2-azanioil-3-(1H-indol-3-il)propanoil]pirrolidin-2-il]formamido}-2-feniletil]piridin-1-io
ETAPA A
(S)-3-(1H-indol-3-il)-1-oxo-1-((S)-2-((S)-1-fenil-2-(piridin-2-il)etilcarbamoil)pirrolidin-1-il)propan-2-ilcarbamato de terc-butilo

Se disolvió Boc-Trp-OH (207 mg, 0,68 mmol) en DMF anhidro (4 ml). Se añadieron HATU (271 mg, 0,71 mmol) y DIPEA (0,39 ml, 2,24 mmol) y la mezcla se agitó a ta durante 30 minutos. Después, se añadió dicloruro de (2S)-2-{[(1S)-1-fenil-2-(piridin-1-io-2-il)etil]carbamoil}pirrolidin-1-io (250 mg, 0,68 mmol) y la mezcla se agitó durante 18 horas a ta. Se añadió agua y la mezcla se extrajo 3x con EtOAc. La fase orgánica se lavó con salmuera, se secó sobre Na2SO4, se filtró y se concentró a presión reducida. El producto se purificó en una columna de SiO2, eluyendo con un gradiente (MeOH al 0 % / EtOAc al 50 % / heptano al 50 % ^ MeOH al 0 % / EtOAc al 100 % / heptano al 0 % ^ MeOH al 15 % / EtOAc al 85 % / heptano al 0 %), produciendo 410 mg (Cuantitativo) del compuesto del título. RMN 1H (300 MHz, CDCla): ppm 1,44 (s, 9 H), 1,77 - 1,89 (m, 3 H), 2,14 - 2,24 (m, 1 H), 3,03 - 3,24 (m, 4 H), 3,28 -3,36 (m, 2 H), 3,54 - 3,65 (m, 1 H), 4,52 - 4,58 (m, 1 H), 4,79 - 4,88 (m, 1 H), 5,12 - 5,27 (m, 1 H), 5,34 - 5,41 (m, 1 H), 6,63 (d, 1 H), 6,94 - 7,31 (m, 8 H), 7,38 - 7,53 (m, 3 H), 7,62 - 7,67 (m, 1 H), 8,59 (d, 1 H), 9,87 (s, 1 H).
ETAPA B
Dicloruro de 2-((S)-2-((S)-1-((S)-2-amonio-3-(1H-indol-3-il)propanoil)pirrolidin-2-carboxamido)-2-feniletil)piridinio

(S)-3-(1H-indol-3-il)-1-oxo-1-((S)-2-((S)-1-fenil-2-(piridin-2-il)etilcarbamoil)pirrolidin-1-il)propan-2-ilcarbamato de terc-butilo (395 mg, 0,68 mmol) se disolvió en una solución de HCl 1 M en Et2O (27,0 ml, 27,20 mmol). La mezcla de reacción se agitó a ta durante 4 h. El producto se trituró en Et2O y el sólido se recuperó por filtración en un embudo Büchner, se lavó con Et2O y se secó al vacío. El sólido se disolvió en agua y se secó por congelación para proporcionar 175 mg (44 %) del compuesto del título.
RMN 1H (300 MHz, D2O): ppm 1,41 - 1,53 (m, 1 H), 1,63 - 1,72 (m, 2 H), 1,93 - 2,04 (m, 1 H), 2,85 (dd, 1 H), 2,94 -3,55 (m, 5 H), 4,19 - 4,31 (m, 2 H), 5,08 (t, 1 H), 6,90 - 7,31 (m, 9 H), 7,38 (d, 1 H), 7,55 - 7,58 (m, 2 H), 8,15 (t, 1 H), 8,31 (d, 1 H);
[M+H]+ = 482,2.
[M+Na]+ = 504,1.
Ejemplo 10
(S)-1-((S)-2-Amino-3-fenilpropanoil)-N-((S)-1-fenil-2-(piridin-2-il)etil)pirrolidin-2-carboxamida
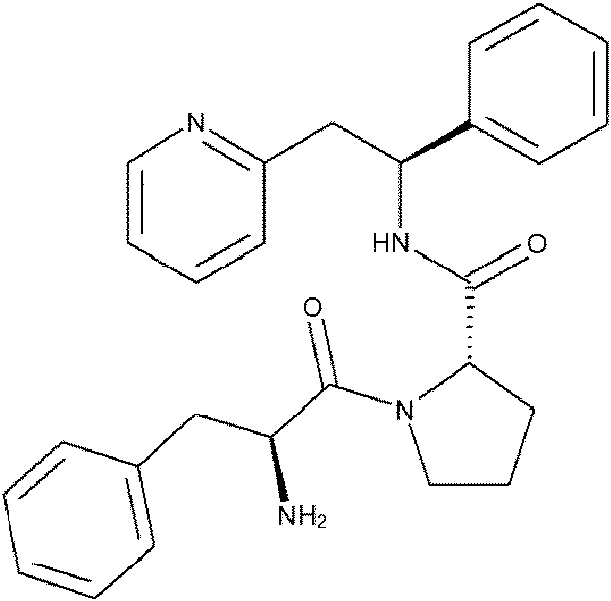
El compuesto del título se preparó como dicloruro de 2-[(2S)-2-{[(2S)-1-[(2S)-2-azanioil-3-fenilpropanoil]pirrolidin-2-il]formamido}-2-feniletil]piridin-1-io
ETAPA A
(S)-1-Oxo-3-fenil-1-((S)-2-((S)-1-fenil-2-(piridin-2-il)etilcarbamoil)pirrolidin-1-il)propan-2-ilcarbamato de ferc-butilo
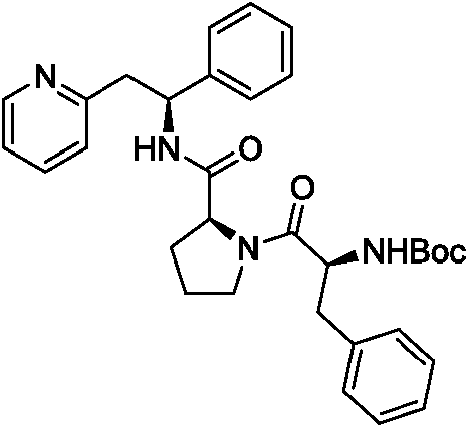
Se disolvió Boc-Phe-OH (180 mg, 0,68 mmol) en DMF anhidro (4 ml). Se añadieron HATU (271 mg, 0,71 mmol) y DIPEA (0,39 ml, 2,24 mmol) y la mezcla se agitó a ta durante 30 minutos. Después, se añadió dicloruro de (2S)-2-{[(1S)-1-fenil-2-(piridin-1-io-2-il)etil]carbamoil}pirrolidin-1-io (250 mg, 0,68 mmol) a la solución y la mezcla se agitó durante 18 h a ta. Se añadió agua y la mezcla se extrajo 3 x con EtOAc. La fase orgánica se lavó con salmuera, se secó sobre Na2SO4, se filtró y se concentró a presión reducida. El producto se purificó en una columna de SiO2, eluyendo con un gradiente (MeOH al 0 % / EtOAc al 50 % / heptano al 50 % ^ MeOH al 0 % / EtOAc al 100 % / heptano al 0 % ^ MeOH al 15 % / EtOAc al 85 % / heptano al 0 %), después en una columna C-18, eluyendo con un gradiente de MeOH y agua (0 al 100 % de MeOH), produciendo 170 mg (46 %) del compuesto del título.
RMN 1H (300 MHz, CDCla): ppm 1,44 (s, 9 H), 1,73 - 1,91 (m, 3 H), 2,10 - 2,19 (m, 1 H), 3,10 - 3,40 (m, 3 H), 3,45 -3,61 (m, 2 H), 4,27 - 4,35 (m, 1 H), 4,50 - 4,56 (m, 1 H), 4,60 - 4,69 (m, 1 H), 5,17 - 5,41 (m, 3 H), 6,93 - 7,03 (m, 2 H), 7,06 - 7,32 (m, 9 H), 7,47 - 7,56 (m, 1 H), 7,82 (d, 1 H), 8,48 (d, 1 H).
ETAPA B
Dicloruro de 2-((S)-2-((S)-1-((S)-2-amonio-3-fenilpropanoil)pirrolidin-2-carboxamido)-2-feniletil)piridinio
(S)-1-Oxo-3-fenil-1-((S)-2-((S)-1-fenil-2-(piridin-2-il)etilcarbamoil)pirrolidin-1-il)propan-2-ilcarbamato de ferc-butilo (170 mg, 0,31 mmol) se disolvió en una solución de HCl 1 M en Et2O (12,5 ml, 12,53 mmol). La mezcla de reacción se agitó a ta durante 18 horas. El producto se trituró en Et2O. El sólido se recuperó por filtración en un embudo Büchner, se lavó con Et2O y se secó al vacío. El sólido se disolvió en agua y se secó por congelación para proporcionar 150 mg (93 %) del compuesto del título. RMN 1H (300 MHz, D2O): ppm 1,39 - 1,48 (m, 1 H), 1,58 -1,65 (m, 2 H), 1,92 - 1,99 (m, 1 H), 2,66 - 2,74 (m, 1 H), 2,95 - 3,10 (m, 2 H), 3,29 - 3,45 (m, 3 H), 4,15 - 4,27 (m, 2 H), 5,09 (t, 1 H), 6,97 - 7,17 (m, 10 H), 7,59 - 7,64 (m, 2 H), 8,18 (t, 1 H), 8,35 (d, 1 H);
[M+H]+ = 443,3,
[M+Na]+ = 465,2.
Ejemplo 11
(S)-1-((S)-2-Amino-4-metilbutanoil)-N-((S)-1-fenil-2-(piridin-2-il)etil)pirrolidin-2-carboxamida

El compuesto del título se preparó como el dicloruro dicloruro de 2-[(2S)-2-{[(2S)-1-[(2S)-2-azanioil-4-metilpentanoil]pirrolidin-2-il]formamido}-2-feniletil]piridin-1-io
ETAPA A
(S)-4-Metil-1-oxo-1-((S)-2-((S)-1-fenil-2-(piridin-2-il)etilcarbamoil)pirrolidin-1-il)pentan-2-ilcarbamato de ferc-butilo

Se disolvió N-(ferc-butoxicarbonil)-L-leucina (157 mg, 0,68 mmol) en DMF anhidro (4 ml). Se añadieron HATU (271 mg, 0,71 mmol) y DIPEA (0,39 ml, 2,24 mmol) y la mezcla se agitó a ta durante 30 minutos. Después, se añadió dicloruro de (2S)-2-{[(1S)-1-fenil-2-(piridin-1-io-2-il)etil]carbamoil}pirrolidin-1-io (250 mg, 0,68 mmol) a la solución y la mezcla se agitó durante 18 horas a ta. Se añadió agua y la mezcla se extrajo 3x con EtOAc. La fase orgánica se lavó con salmuera, se secó sobre Na2SO4, se filtró y se concentró a presión reducida. El producto se purificó en una columna de SiO2, eluyendo con un gradiente (MeOH al 0 % / EtOAc al 50 % / heptano al 50 % ^ MeOH al 0 % / EtOAc al 100 % / heptano al 0 % ^ MeOH al 15 % / EtOAc al 85 % / heptano al 0 %) después en una columna C-18, eluyendo con un gradiente de MeOH y agua (0 al 100 % de MeOH), produciendo 200 mg (58 %) del compuesto del título.
RMN 1H (300 MHz, CDCla): ppm 0,91 (d, 3 H), 0,98 (d, 3 H), 1,34 - 1,52 (m, 1 H), 1,41 (s, 9 H), 1,70 - 1,84 (m, 2 H), 1,85 - 1,96 (m, 3 H), 2,11 -2,18 (m, 1 H), 3,07 - 3,27 (m, 2 H), 3,47 - 3,56 (m, 1 H), 3,61 - 3,71 (m, 1 H), 4,43 -4,56 (m, 2 H), 5,14 - 5,18 (m, 1 H), 5,26 - 5,34 (m, 1 H), 6,98 (d, J = 7,7 Hz, 1 H), 7,08 - 7,25 (m, 5 H), 7,52 (t, 1 H), 7,83 (d, 1 H), 8,48 (d, 1 H).
ETAPA B
Dicloruro de 2-((S)-2-((S)-1-((S)-2-amonio-4-metilpentanoil)pirrolidin-2-carboxamido)-2-feniletil)piridinio

(S)-4-metil-1-oxo-1-((S)-2-((S)-1-fenil-2-(piridin-2-il)etilcarbamoil)pirrolidin-1-il)pentan-2-ilcarbamato de ferc-butilo (200 mg, 0,39 mmol) se disolvió en una solución de HCl 1 M en Et2O (15,7 ml, 15,7 mmol). La mezcla de reacción se agitó a ta durante 18 h. El producto se trituró en Et2O. El sólido se recuperó por filtración en un embudo Büchner, se lavó con Et2O y se secó al vacío. El sólido se disolvió en agua y se secó por congelación para proporcionar 130 mg (69 %) del compuesto del título.
RMN 1H (300 MHz, D2O): ppm 0,72 (d, 6 H), 1,31 - 1,49 (m, 4 H), 1,65 - 1,74 (m, 2 H), 1,93 - 2,02 (m, 1 H), 3,23 -3,49 (m, 4 H), 3,99 - 4,04 (m, 1 H), 4,16 - 4,21 (m, 1 H), 5,07 (t, 1 H), 7,08 - 7,20 (m, 5 H), 7,63 - 7,67 (m, 2 H), 8,22 (td, 1 H), 8,37 (dd, 1 H);
[M+H]+ = 409,2,
[M+Na]+ = 431,2.
Ejemplo 12
Tetracloruro de 2-[(2S)-2-{[(2S)-1-[(2S)-2-Azanioil-5-{[azanioil(iminioil)metil]amino}pentanoil]-pirrolidin-2-il]formamido}-2-feniletil]piridin-1-io

ETAPA A
N-[(1Z)-{[(4S)-4-{[(ferc-butoxi)carbonil]amino}-5-oxo-5-[(2S)-2-{[(1S)-1-fenil-2-(piridin-2-il)etil]carbamoil}pirrolidin-1-il]pentil]amino}({[(ferc-butoxi)carbonil]imino})metil]carbamato de ferc-butilo

Se disolvió Boc-Arg(Boc)2-OH (644 mg, 1,36 mmol) en DMF anhidro (7 ml). Se añadieron HATU (542 mg, 1,43 mmol) y DIPEA (0,78 ml, 4,48 mmol) y la mezcla se agitó a ta durante 30 minutos. Después, se añadió dicloruro de (2S)-2-{[(1S)-1-fenil-2-(piridin-1-io-2-il)etil]carbamoil}pirrolidin-1-io (500 mg, 1,36 mmol) a la solución y la mezcla se agitó durante 18 h a ta. Se añadió agua y la mezcla se extrajo 3x con EtOAc. La fase orgánica se lavó con salmuera, se secó sobre Na2SO4, se filtró y se concentró a presión reducida. El producto se purificó en una columna de SO2, eluyendo con un gradiente (MeoH al 0 % / EtOAc al 50 % / heptano al 50 % ^ MeOH al 0 % / EtOAc al 100 % / heptano al 0 % ^ MeOH al 10 % / EtOAc al 90 % / heptano al 0 %) después en una columna C-18, eluyendo con un gradiente de MeOH y agua (0 al 100 % de MeOH), produciendo 200 mg (20 %) del compuesto del título. RMN 1H (300 MHz, CDCla): ppm 1,43 (s, 9 H), 1,46 (s, 9 H), 1,51 (s, 9 H), 1,57 - 1,76 (m, 4 H), 1,86 - 1,99 (m, 3 H), 2,12 -2,25 (m, 1 H), 3,11 (dd, 1 H), 3,23 - 3,30 (m, 1 H), 3,58 - 3,95 (m, 4 H), 4,42 (t, 1 H), 4,56 (d, 1 H), 5,31 - 5,38
(m, 2 H), 6,98 (d, 1 H), 7,17 - 7,33 (m, 5 H), 7,54 (t, 1 H), 7,93 (d, 1 H), 8,56 (d, 1 H).
ETAPA B
Tetracloruro de 2-[(2S)-2-{[(2S)-1-[(2S)-2-azamumil-5-{[azanioil(iminioil)metil]amino}pentanoil]-pirrolidin-2-il]formamido}-2-feniletil]piridin-1-io

N-[(1Z)-{[(46)-4-{[(ferc-butoxi)carbonil]amino}-5-oxo-5-[(2S)-2-{[(1S)-1-fenil-2-(pindin-2-il)etil]carbamoil}pirrolidin-1-il]pentil]amino}({[(terc-butoxi)carbonil]imino})metil]carbamato de ferc-butilo (200 mg, 0,27 mmol) se disolvió en una solución de HCl 1 M en Et2O (5,3 ml, 10,64 mmol). La reacción se agitó a ta durante 18 horas. El producto se trituró en Et2O. El sólido se recuperó por filtración en un embudo Büchner, se lavó con Et2O y se secó al vacío. El sólido se disolvió en agua y se secó por congelación para proporcionar 140 mg (88 %) del compuesto del título.
RMN 1H (300 MHz, D2O): ppm 1,31 - 1,57 (m, 3 H), 1,66 - 1,78 (m, 4 H), 2,00 - 2,10 (m, 1 H), 2,99 (t, 2 H), 3,28 -3,40 (m, 4 H), 4,16 (t, 1 H), 4,26 (t, 1 H), 5,09 (t, 1 H), 7,10 - 7,13 (m, 2 H), 7,16 - 7,21 (m, 3 H), 7,67 - 7,71 (m, 2 H), 8,27 (t, 1 H), 8,40 (d, 1 H);
[M+H]+ = 452,2.
ACTIVIDAD BIOLÓGICA
Los profármacos descritos en el presente documento se contemplan para administrarse por vía oral por aquellos que padecen depresión o dolor.
Ejemplo 13
Este ejemplo ilustra que diferentes profármacos de (S)-1-fenil-2-(piridin-2-il)etanamina, en donde R1 es alquil C1-6 C(O)O(alcoxi C1-6 ), pueden usarse para lograr diferentes velocidades de conversión (es decir, lenta o rápida) en (S)-1-fenil-2-(piridin-2-il)etanamina para asegurar un perfil de PK adecuado. Por lo tanto, variar R1 para producir diferentes profármacos da como resultado diferentes perfiles farmacocinéticos de (S)-1-fenil-2-(piridin-2-il)etanamina. Además, este Ejemplo ilustra que no se espera pérdida en la exposición oral cuando un profármaco se convierte en una base libre. La conversión de un profármaco en donde R1 es alquil C-i_6C(O)O(alcoxi C1-6), en (S)-1 -fenil-2-(piridin-2-il)etanamina se espera que se produzca a través de una hidrólisis enzimática inicial de un grupo funcional, tales como un éster, seguido de una conversión espontánea en (S)-1-fenil-2-(piridin-2-il)etanamina. Las enzimas implicadas se espera que sean varias esterasas no específicas de alta capacidad, que puedan mostrarse inhibiendo la conversión con un inhibidor de esterasa no selectivo pero no inhibiendo la conversión usando inhibidores selectivos. Se espera que tales esterasas se distribuyan a lo largo de todo el cuerpo humano. El fluido intestinal humano (HIF), la fracción S9 de hígado humano y la sangre entera humana se han usado para examinar las propiedades farmacocinéticas de los profármacos de (S)-1-fenil-2-(piridin-2-il)etanamina así como la velocidad de formación y el grado de formación de la (S)-1-fenil-2-(piridin-2-il)etanamina en diferentes compartimentos corporales. La velocidad de formación de (S)-1-fenil-2-(piridin-2-il)etanamina a partir de diferentes profármacos de (S)-1-fenil-2-(piridin-2-il)etanamina difiere marcadamente en todos los ensayos probados, como se muestra en la Tabla 1.
Tabla 1

continuación

Los resultados in vitro mostrados en la Tabla 1 muestran que pueden lograrse diferentes perfiles farmacocinéticos en plasma de (S)-1-fenil-2-(piridin-2-il)etanamina usando diferentes profármacos de (S)-1-fenil-2-(piridin-2-il)etanamina. Por lo tanto, la selección de un profármaco particular permitirá que se logren diferentes perfiles farmacocinéticos de (S)-1-fenil-2-(piridin-2-il)etanamina, esto es, pueden usarse diferentes profármacos para lograr una conversión lenta o rápida en (S)-1-fenil-2-(piridin-2-il)etanamina. Los ensayos de fluido intestinal humano (HIF), hígado humano y sangre humana usados para generar los resultados mostrados en la Tabla 1 se han descrito en Malmborg J & Ploeger BA, J Pharmacol Toxicol Methods (2013) May-Jun 67(3) 203-13, que se incorpora por referencia en el presente documento.
Ejemplo 14
Este ejemplo ilustra que diferentes profármacos de (S)-1-fenil-2-(piridin-2-il)etanamina, en donde R1 es

pueden usarse para lograr diferentes velocidades de conversión (es decir, lenta o rápida) en (S)-1-fenil-2-(piridin-2-il)etanamina para asegurar un perfil de PK adecuado. Por lo tanto, variar R1 para producir diferentes profármacos da como resultado diferentes perfiles farmacocinéticos de (S)-1-fenil-2-(piridin-2-il)etanamina. Además, este Ejemplo ilustra que no se espera pérdida en la exposición oral cuando un profármaco se convierte en una base libre.
La conversión de un profármaco en (S)-1-fenil-2-(piridin-2-il)etanamina de un compuesto de fórmula (I) en donde R1 es:

se espera que se produzca a través de hidrólisis enzimática en el C-terminal de la prolina, liberando un dipéptido y (S)-1-fenil-2-(piridin-2-il)etanamina. La enzima implicada se espera que sea dipeptidil peptidasa IV (DPpIV), que puede mostrarse inhibiendo la conversión con un inhibidor selectivo de esta enzima. Se espera que DPPIV se distribuya a lo largo de todo el cuerpo humano. El fluido intestinal humano (HIF), la fracción S9 de hígado humano y la sangre entera humana se han usado para examinar las propiedades farmacocinéticas de los profármacos de (S)-1- fenil-2-(piridin-2-il)etanamina así como la velocidad de formación y el grado de formación de la (S)-1-fenil-2-(piridin-2- il)etanamina en diferentes compartimentos corporales. Las células Caco-2 se han usado para evaluar la permeabilidad y la absorción potenciales de los profármacos de (S)-1-fenil-2-(piridin-2-il)etanamina. La velocidad de formación de (S)-1-fenil-2-(piridin-2-il)etanamina a partir de diferentes profármacos de (S)-1-fenil-2-(piridin-2-il)etanamina difirió en los ensayos probados, especialmente en sangre humana, como se muestra en la Tabla 2. La permeabilidad diferente mostrada por diferentes profármacos de (S)-1-fenil-2-(piridin-2-il)etanamina sugiere que la exposición sistémica de profármaco de (S)-1-fenil-2-(piridin-2-il)etanamina variará dependiendo del profármaco elegido.
Tabla 2

Los ensayos in vitro muestran que pueden lograrse diferentes perfiles farmacocinéticos en plasma de (S)-1 -fenil-2-(piridin-2-il)etanamina usando diferentes profármacos. Por lo tanto, pueden lograrse diferentes perfiles farmacocinéticos de (S)-1-fenil-2-(piridin-2-il)etanamina mediante el uso de diferentes profármacos.
Ejemplo 15
Este ejemplo ilustra que las dosis orales de profármaco de (S)-1-fenil-2-(piridin-2-il)etanamina tienen un perfil farmacocinético similar a una infusión IV de 1 hora de (S)-1-fenil-2-(piridin-2-il)etanamina.
Basándose en datos in vitro, se proyecta que (S)-1-fenil-2-(piridin-2-il)etanamina (lanicemina) tenga buena biodisponibilidad oral (> 75 %) en el hombre, pero una Cmáx más alta que aceptable cuando se da como un bolo intravenoso (con respecto a su perfil de seguridad como una infusión de 1 h). Un criterio de selección usado para evaluar la utilidad y la adecuabilidad de un profármaco de (S)-1-fenil-2-(piridin-2-il)etanamina fue el potencial del profármaco de preservar las características de la infusión oral / 1 h iv de la (S)-1-fenil-2-(piridin-2-il)etanamina (por ejemplo, Tmáx, Cmáx, AUC) mientras se embota la Cmáx del resto activo cuando el profármaco se administra directamente en el torrente sanguíneo atenuando de esta manera el mal uso potencial (por ejemplo, responsabilidad de abuso) y / o preocupaciones de seguridad dirigidas por Cmáx. Como un medio de identificación de profármacos con perfiles farmacocinéticos apropiados en el hombre, se construyeron modelos farmacocinéticos multicompartimentales con base fisiológica (PBPK) tanto para (S)-1-fenil-2-(piridin-2-il)etanamina como para cada profármaco en consideración. Los modelos de PBPK se validaron usando un conjunto de profármacos para los que había datos de PK humano disponibles (véase Malmborg J & Ploeger BA, J Pharmacol Toxicol Methods (2013) May-Jun 67(3) 203-13, que se incorpora en el presente documento por referencia). Las constantes de velocidad usadas en el modelo se basaron en estudios de estabilidad in vitro (es decir, fluido intestinal, sangre humana, etc.) llevados a cabo tanto con fármacos como (S)-1-fenil-2-(piridin-2-il)etanamina (valores de ejemplo mostrados en las Tablas 1 y 2).
La Tabla 3 muestra predicciones farmacocinéticas clave para el Ejemplo 5 - uno de los profármacos escindió DPPIV. Como se ejemplifica en la Tabla 3 la exposición Cmáx de (S)-1-fenil-2-(piridin-2-il)etanamina se predice que aumente en >2x cuando una forma de dosis oral dada se aplaste y se administre ilícitamente como una administración en bolo i.v. Por el contrario, la (S)-1-fenil-2-(piridin-2-il)etanamina liberada de un profámaco administrado oral tiene un perfil de PJ similar a una infusión de 1 h de (S)-1-fenil-2-(piridin-2-il)etanamina sin que doble la Cmáx cuando se da como un bolo i.v. Además, dado que la conversión a (S)-1-fenil-2-(piridin-2-il)etanamina se dirige selectivamente por DPPIV, puede usarse un inhibidor de DPPIV para detener la conversión en el caso de mal uso a propósito o inadvertido - un rasgo de seguridad añadido dentro del diseño de profármaco de DPPIV.
En la Tabla 3 el Compuesto 1 es (S)-1-fenil-2-(piridin-2-il)etanamina. En la Tabla 3 todas las dosis se seleccionan para contener cantidades equivalentes de Compuesto 1 [base libre de (S)-1-fenil-2-(piridin-2-il)etanamina,] es decir, cada dosis contiene la misma cantidad de Compuesto 1 dentro y está potencialmente disponible en forma de la base libre (S)-1-fenil-2-(piridin-2-il)etanamina. Por consiguiente, las concentraciones Cmáx modeladas de Compuesto 1 de base libre pueden compararse directamente a través de diferentes administraciones de (S)-1-fenil-2-(piridin-2-il)etanamina y profármaco de (S)-1-fenil-2-(piridin-2-il)etanamina, como se muestra en la Tabla 3.
Tabla 3

Ejemplo 16
Este ejemplo ilustra que cuando se administraron dosis de profármaco de diclorhidrato de (S)-1-fenil-2-(piridin-2-il)etanamina (Ejemplo 5) y diclorhidrato de (S)-1-fenil-2-(piridin-2-il)etanamina a perros, el riesgo de convulsiones, según se representa por NOEL y LOEL, fue menor con el profármaco.
El diclorhidrato de (S)-1-fenil-2-(piridin-2-il)etanamina y el vehículo se administraron una vez al día por sonda oral (agua). Las formulaciones se prepararon dos veces, una vez a la semana. Las formulaciones de dosificación se almacenaron y se refrigeraron (2-8 °C) en recipientes de vidrio ámbar hasta que se requiriera para su dosificación. Se preparó un intervalo de concentraciones de diclorhidrato de (S)-1-fenil-2-(piridin-2-il)etanamina en vehiculo para mantener el volumen de dosis para todas las dosis (1 ml/kg). Las dosis individuales se basaron en los pesos corporales más recientes de los perros usados en el estudio.
Un profármaco de (S)-1-fenil-2-(piridin-2-il)etanamina como sal de fumarato (ejemplo 5) y vehículo se administraron una vez al día durante 14 días a través de sonda oral en aproximadamente el mismo momento cada día (± 1 hora), (el vehículo es ácido glucónico 0,3 M pH 3,0). Se preparó un intervalo de concentraciones de profármaco de (S)-1-fenil-2-(piridin-2-il)etanamina como sal de fumarato en vehículo (por ejemplo 3, 10 y 30 mg/ml de profármaco de (S)-1-fenil-2-(piridin-2-il)etanamina) para mantener el volumen de dosis para todas las dosis (2 ml/kg). Las formulaciones de dosificación se calentaron a temperatura ambiente agitando durante al menos 30 minutos antes de la dosificación y continuamente a lo largo de todo el procedimiento de administración. Las dosis individuales se basaron en los pesos corporales más recientes de los perros usados en el estudio.
En la Tabla 4 el Compuesto 1 es (S)-1-fenil-2-(piridin-2-il)etanamina. La Tabla 4 ilustra los niveles en sangre más altos en los que no se producen convulsiones (Límite de Efecto No Observado, NOEL) y los niveles en sangre más bajos en los que se producen convulsiones (Límite de Efecto Observado Más bajo, LOEL), después de la administración de diclorhidrato de Compuesto 1 y después de la administración de un profármaco de sal de fumarato de Compuesto 1. En cada caso se mide la concentración en sangre más alta (Cmáx) de base libre (S)-1-fenil-2-(piridin-2-il)etanamina. En el primer caso, la base libre (S)-1-fenil-2-(piridin-2-il)etanamina se genera directamente del Compuesto 1, administrado como la sal de diclorhidrato. En el segundo caso, la base libre (S)-1-fenil-2-(piridin-2-il)etanamina se genera a través de la conversión in vivo a partir de un profármaco de Compuesto 1, administrado como la sal de fumarato. El NOEL para las convulsiones fue más alto cuando se administró el profármaco de (S)-1-fenil-2-(piridin-2-il)etanamina como sal de fumarato en comparación con cuando se administró (S)-1-fenil-2-(piridin-2-il)etanamina como sal de diclorhidrato.
Tabla 4

Ejemplo 17
Este ejemplo ilustra que las dosis en aumento de profármaco de (S)-1-fenil-2-(piridin-2-il)etanamina, Ejemplo 5, produce aumentos menos que proporcionales en la concentración de (S)-1-fenil-2-(piridin-2-il)etanamina en fluido
intestinal humano, como se muestra en la Figura 1a y la Figura 1b. Como consecuencia, hace más difícil obtener concentraciones altas de (S)-1-fenil-2-(piridin-2-il)etanamina a partir de la administración de un profármaco de (S)-1-fenil-2-(piridin-2-il)etanamina con respecto a la administracion de (S)-1-fenil-2-(piridin-2-il)etanamina directamente, que se busca para el abuso de drogas. Sin pretender quedar ligados a teoría alguna, los profármacos de (S)-1-fenil-2-(piridin-2-il)etanamina, en donde R1 es, se escinden principalmente por DPPIV. Hay una posibilidad de modulación endógena y / o exógena de DPPIV que impacte en la conversión del profármaco en (S)-1-fenil-2-(piridin-2-il)etanamina.
Las soluciones madre de profármaco de (S)-1 -fenil-2-(piridin-2-il)etanamina (0,33, 1, 3 y 6 mmol/l) se prepararon añadiendo un volumen (8,25, 25, 75 y 150 pl) de solución 20 mmol/l de profármaco de (S)-1-fenil-2-(piridin-2-il)etanamina disuelto en DMSO, a FaSSIF-v2 (491,75, 475, 425 y 350 pl). Posteriormente, se añadieron 10 pl de solución madre de profármaco de (S)-1-fenil-2-(piridin-2-il)etanamina al sobrenadante (90 p1HIF) en un vial de vidrio, se mezcló durante 1 min y se inyectó en el sistema LC-UV. La concentración inicial en las incubadoras fue 33, 100, 300 y 600 pmol/l y las muestras se tomaron a 5, 20, 35, 65 y 125 min desde el inicio de la incubación. Se usó un experimento de incubación con (S)-1-fenil-2-(piridin-2-il)etanamina pmol/l como la muestra patrón (calibración de un punto) para determinar la velocidad de formación y la concentración de (S)-1-fenil-2-(piridin-2-il)etanamina. El análisis se realizó en un detector de matriz de fotodiodo (la longitud de onda analítica usada fue 261 nm) acoplado a UPLC Waters Acquity. La columna usada fue una BEH C-18, 1,7 pm, 2,1 X50 mm ID mantenida a 40 °C y usando un pre-filtro en línea de 0,2 pm. Las fases móviles usadas consistieron en A: TFA al 0,03 % en H2O (v/v) y B: TFA al 0,03 % en acetonitrilo (v/v). Gradiente de ejemplo: El 1 % de B inicial se subió en rampa al 95 % de B durante 7 min y después se mantuvo al 95 % de B durante 0,4 min a un caudal de 0,6 ml/min. La velocidad de conversión de profármaco a (S)-1-fenil-2-(piridin-2-il)etanamina se determinó ajustando las concentraciones medidas de profármaco y (S)-1-fenil-2-(piridin-2-il)etanamina frente al tiempo.
Ejemplo 18
Consistente con las observaciones in vitro a partir del Ejemplo 17, los estudios in vivo (Ejemplo 18 a continuación) en ratas ilustran que las exposiciones Cmáx de (S)-1-fenil-2-(piridin-2-il)etanamina, como se ha descrito anteriormente, aumentan menos que proporcionalmente tras la transición entre las dosis terapéuticas y supra-terapéuticas de un profármaco de (S)-1-fenil-2-(piridin-2-il)etanamina. Los resultados de estos estudios in vivo se tabulan en la Tabla 5. A 52 ratas macho y 52 ratas hembra se administró bien (a) un profármaco de (S)-1-fenil-2-(piridin-2-il)etanamina como una sal de fumarato en vehículo (Ácido Glucónico 0,3 M, pH 3,0) o (b) vehículo (Ácido Glucónico 0,3 M, pH 3,0) una vez al día durante 14 días en aproximadamente el mismo momento cada día (09:26 /-124 minutos). Las dosis (30, 100 y 300 mg/kg de profármaco de (S)-1-fenil-2-(piridin-2-il)etanamina) se administraron en un volumen de dosis de 10 ml/kg. Después de 14 días, las muestras de sangre se recogieron 24 horas post-dosis a partir de la vena lateral de la cola en animales restringidos. Las muestras de sangre se recogieron usando tubos capilares POCT Sarstedt Minivette neutros de 50 ul. La sangre se transfirió después a tubos pre-enfriados que contienen citrato sódico enfriado en hielo, se agitaron 5-10 veces a mano y se congelaron en hielo seco tras 10 segundos de recogida. Las muestras de sangre completa se enviaron congeladas en hielo seco a Covance Laboratories Inc. para su análisis. Las muestras se analizaron para las concentraciones de profármaco de (S)-1-fenil-2-(piridin-2-il)etanamina y (S)-1-fenil-2-(piridin-2-il)etanamina. Todo el trabajo analítico se llevó a cabo por Covance Laboratories, Inc., Madison, Wisconsin usando un método analítico CL/EM/EM desarrollado y validado por ese laboratorio.
La Tabla 5 ilustra parámetros toxicocinéticos siguiendo la administración de dosis terapéuticas y supra-terapéuticas de profármaco de (S)-1-fenil-2-(piridin-2-il)etanamina a ratas.
Tabla 5
Parámetros toxicocinéticos medios para (S)-1-fenil-2-(piridin-2-il)etanamina en ratas después de la administración oral de fumarato de (S)-1-((S)-2-Amino-3-metilbutanoil)-N-((S)-1-fenil-2-(piridin-2-_____________________ il)etil)pirrolidin-2-carboxamida (Ejemplo 5) durante 14 días______________________
(S)-1-fenil-2-(piridin-2-il)etanamina - Macho (Día 14)
Dosis 30 mg/kg/día 100 mg/kg/día (250 pmol/kg/día) 300 mg/kg/día
(75 pmol/kg/día) (750 pmol/kg/día) Cmáx (nmol/l) 9980 ± 890 20700 ± 7660 50700± 18700
AUCü-t (nmolh/l) 133000± 11100 305000 ± 96600 784000± 180000 Tmáx (horas) 3,33 ± 2,52 3,00 ± 0 1,54 ± 1,27
(continuación)
(S)-1-fenil-2-(piridin-2-il)etanamina - Hembra (Día 14)
Dosis 30 mg/kg/día 100 mg/kg/día (250 pmol/kg/día) 300 mg/kg/día (75 pmol/kg/día) (750 pmol/kg/día) Cmáx (nmo|/|) 11900± 1990 32400 ± 2750 83800± 8810 AUCo-t (nmol h/l) 14600± 10000 402000± 14000 1150000 ± 319000 Tmáx (horas) 3,00 ± 0 1,67 ± 1,15 2,67 ± 2,89
Claims (7)
1. Un compuesto de fórmula (I):

en donde R1 es

en donde AA es un aminoácido natural enlazado con enlaces peptídicos,
o una sal farmacéuticamente aceptable del mismo.
2. El compuesto de acuerdo con la reivindicación 1, en donde el compuesto se selecciona del grupo que consiste en: (S)-1-((S)-2-Amino-3-metilbutanoil)-N-((S)-1-fenil-2-(piridin-2-il)etil)pirrolidin-2-carboxamida;
(s)-1-((s)-2,6-Diaminohexanoil)-N-((S)-1-fenil-2-(piridin-2-il)etil)pirrolidin-2-carboxamida;
(S)-1-((S)-2-Amino-3-(1H-imidazol-4-il)propanoil)-N-((S)-1-fenil-2-(piridin-2-il)etil)pirrolidin-2-carboxamida;
2-((S)-2-((S)-1-((S)-2-Amino-3-(4-hidroxifenil)propanoil)pirrolidin-2-carboxamido)-2-feniletil)piridina;
2-((S)-2-((S)-1-((S)-2-Amino-3-(1H-indol-3-il)propanoil)pirrolidin-2-carboxamido)-2-feniletil)piridina;
(S)-1-((S)-2-Amino-3-fenilpropanoil)-N-((S)-1-fenil-2-(piridin-2-il)etil)pirrolidin-2-carboxamida; y
(S)-1-((S)-2-Amino-4-metilpentanoil)-N-((S)-1-fenil-2-(piridin-2-il)etil)pirrolidin-2-carboxamida,
o una sal farmacéuticamente aceptable de uno cualquiera de los anteriores.
3. El compuesto de acuerdo con la reivindicación 1, en donde el compuesto es (S)-1-((S)-2-amino-3-metilbutanoil)-N-((S)-1-fenil-2-(piridin-2-il)etil)pirrolidin-2-carboxamida:

o una sal farmacéuticamente aceptable del mismo.
4. El compuesto de acuerdo con la reivindicación 2, en donde el compuesto es la sal de diclorhidrato de (S)-1-((S)-2-amino-3-metilbutanoil)-N-((S)-1-fenil-2-(piridin-2-il)etil)pirrolidin-2-carboxamida:

5. El compuesto de acuerdo con la reivindicación 2, en donde el compuesto es la sal de fumarato de (S)-1-((S)-2-amino-3-metilbutanoil)-N-((S)-1-fenil-2-(piridin-2-il)etil)pirrolidin-2-carboxamida:
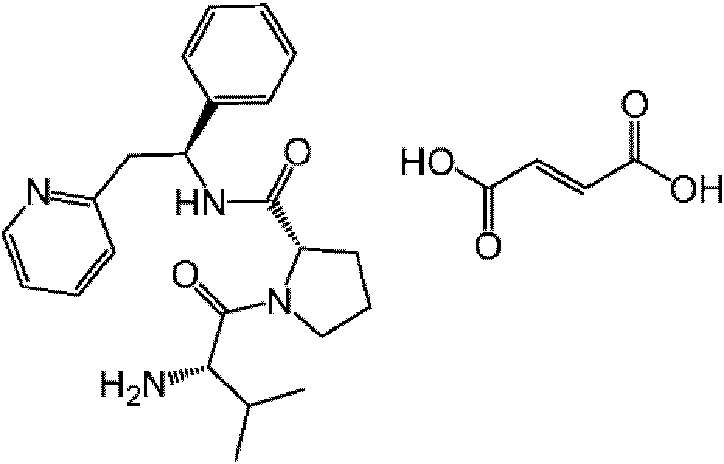
6. El compuesto de acuerdo con una cualquiera de las reivindicaciones 1 a 5, en donde una enzima DPPIV puede escindir R1 para formar (S)-1-fenil-2-(piridin-2-il)etanamina.
7. El compuesto de acuerdo con la reivindicación 6 en donde la escisión de R1 para formar (S)-1-fenil-2-(piridin-2-il)etanamina puede modularse mediante el uso de un inhibidor de DPPIV.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201361899903P | 2013-11-05 | 2013-11-05 | |
| PCT/GB2014/053236 WO2015067923A1 (en) | 2013-11-05 | 2014-10-30 | Nmda antagonist prodrugs |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2733344T3 true ES2733344T3 (es) | 2019-11-28 |
Family
ID=51947383
Family Applications (3)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES14802476T Active ES2733344T3 (es) | 2013-11-05 | 2014-10-30 | Profármacos antagonistas de NMDA |
| ES21196371T Active ES2999064T3 (en) | 2013-11-05 | 2014-10-30 | Nmda antagonist prodrugs |
| ES19166031T Active ES2894903T3 (es) | 2013-11-05 | 2014-10-30 | Profármacos antagonistas de NMDA |
Family Applications After (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES21196371T Active ES2999064T3 (en) | 2013-11-05 | 2014-10-30 | Nmda antagonist prodrugs |
| ES19166031T Active ES2894903T3 (es) | 2013-11-05 | 2014-10-30 | Profármacos antagonistas de NMDA |
Country Status (25)
| Country | Link |
|---|---|
| US (4) | US9822075B2 (es) |
| EP (3) | EP3066091B1 (es) |
| JP (4) | JP6479834B2 (es) |
| KR (2) | KR102336426B1 (es) |
| CN (2) | CN111187202B (es) |
| AR (1) | AR098319A1 (es) |
| AU (1) | AU2014345407B2 (es) |
| CA (1) | CA2928004C (es) |
| CY (1) | CY1121749T1 (es) |
| DK (1) | DK3066091T3 (es) |
| ES (3) | ES2733344T3 (es) |
| HR (2) | HRP20191159T1 (es) |
| HU (2) | HUE070532T2 (es) |
| LT (1) | LT3066091T (es) |
| MX (2) | MX377276B (es) |
| PL (2) | PL4001272T3 (es) |
| PT (1) | PT3066091T (es) |
| RS (2) | RS66369B1 (es) |
| RU (1) | RU2695372C2 (es) |
| SI (1) | SI3066091T1 (es) |
| SM (1) | SMT202500023T1 (es) |
| TR (1) | TR201909632T4 (es) |
| TW (1) | TW201609653A (es) |
| UY (1) | UY35823A (es) |
| WO (1) | WO2015067923A1 (es) |
Families Citing this family (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| PL4001272T3 (pl) | 2013-11-05 | 2025-02-10 | Astrazeneca Ab | Proleki antagonisty nmda |
| US11358935B2 (en) | 2016-11-28 | 2022-06-14 | Biohaven Pharmaceutical Holding Company Ltd. | Prodrugs of lanicemine and their method of use |
| CN107578281B (zh) * | 2017-08-31 | 2021-03-30 | 湖南大学 | 电子商务环境下用户优惠券行为预测方法及模型构建方法 |
| AU2021297823A1 (en) * | 2020-06-23 | 2023-02-16 | Biohaven Therapeutics Ltd. | Topical formulations of (1S)-1-phenyl-2-pyridin-2-ylethanamine |
| WO2025137661A1 (en) * | 2023-12-22 | 2025-06-26 | Biohaven Therapeutics Ltd. | Combination treatment of neurological disorders |
Family Cites Families (19)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| SE7909514L (sv) | 1979-11-16 | 1981-05-17 | Astra Laekemedel Ab | Nya halofenyl-pyridyl-allylaminderivat |
| US4769466A (en) * | 1987-02-06 | 1988-09-06 | Pennwalt Corporation | 2-aminoacetamide pyridinyl derivatives |
| ES2083972T3 (es) * | 1988-08-12 | 1996-05-01 | Astra Ab | Arilalquil-aminas y -amidas con propiedades anticonvulsivas y neuroprotectoras. |
| SG47949A1 (en) | 1991-10-30 | 1998-04-17 | Astra Ab | 2-Heterocyclicethylamine derivatives and their use as pharmaceuticals |
| EP0633879B1 (en) * | 1992-04-03 | 1998-03-11 | Astra Aktiebolag | Enantiomeric 1-phenyl-2-(2-pyridinyl)ethylamine for the treatment of neurodegenerative disorders |
| SE9901077D0 (sv) | 1999-03-23 | 1999-03-23 | Astra Ab | Novel use |
| SE9901340D0 (sv) | 1999-04-15 | 1999-04-15 | Astra Pharma Prod | Novel process |
| US6613879B1 (en) * | 1999-05-14 | 2003-09-02 | Boehringer Ingelheim Pharma Kg | FAP-activated anti-tumour compounds |
| PE20020291A1 (es) | 2000-08-21 | 2002-04-17 | Hoffmann La Roche | Profarmacos para ligandos del receptor nmda |
| ATE540678T1 (de) | 2001-06-11 | 2012-01-15 | Xenoport Inc | Prodrugs aus gaba-analoga, zusammensetzungen und ihre verwendungen |
| GB0310593D0 (en) * | 2003-05-08 | 2003-06-11 | Leuven K U Res & Dev | Peptidic prodrugs |
| EP2371815A1 (en) | 2003-12-30 | 2011-10-05 | XenoPort, Inc. | Sythesis of acyloxyalkyl carbamate prodrugs and intermediates thereof |
| EP1929030A2 (en) * | 2005-09-08 | 2008-06-11 | Shire LLC | Prodrugs of t3 and t4 with enhanced bioavailability |
| CN101246417B (zh) | 2007-02-13 | 2010-09-29 | 艾威梯科技(北京)有限公司 | 音频数据流输入/输出无间断软件切换的方法和系统 |
| CA2706575C (en) | 2008-01-25 | 2015-07-14 | Xenoport, Inc. | Enantiomerically resolving acyloxyalkyl thiocarbonates used in synthesizing acyloxyalkyl carbamate prodrugs |
| WO2010017504A1 (en) * | 2008-08-07 | 2010-02-11 | Xenoport, Inc. | Methods of synthesizing 1-(acyloxy)-alkyl carbamate prodrugs |
| EP2525830B1 (en) * | 2010-01-22 | 2016-05-11 | Ascendis Pharma A/S | Dipeptide-based prodrug linkers for aliphatic amine-containing drugs |
| US8580850B2 (en) * | 2011-08-11 | 2013-11-12 | Xenoport, Inc. | Anhydrous and hemihydrate crystalline forms of an (R)-baclofen prodrug, methods of synthesis and methods of use |
| PL4001272T3 (pl) | 2013-11-05 | 2025-02-10 | Astrazeneca Ab | Proleki antagonisty nmda |
-
2014
- 2014-10-30 PL PL21196371.5T patent/PL4001272T3/pl unknown
- 2014-10-30 SM SM20250023T patent/SMT202500023T1/it unknown
- 2014-10-30 CN CN202010014023.3A patent/CN111187202B/zh active Active
- 2014-10-30 EP EP14802476.3A patent/EP3066091B1/en active Active
- 2014-10-30 MX MX2016005567A patent/MX377276B/es active IP Right Grant
- 2014-10-30 RS RS20241459A patent/RS66369B1/sr unknown
- 2014-10-30 ES ES14802476T patent/ES2733344T3/es active Active
- 2014-10-30 JP JP2016551069A patent/JP6479834B2/ja active Active
- 2014-10-30 KR KR1020167014625A patent/KR102336426B1/ko active Active
- 2014-10-30 DK DK14802476.3T patent/DK3066091T3/da active
- 2014-10-30 LT LTEP14802476.3T patent/LT3066091T/lt unknown
- 2014-10-30 ES ES21196371T patent/ES2999064T3/es active Active
- 2014-10-30 PL PL14802476T patent/PL3066091T3/pl unknown
- 2014-10-30 US US15/034,727 patent/US9822075B2/en active Active
- 2014-10-30 CN CN201480060416.0A patent/CN105683180B/zh active Active
- 2014-10-30 RS RS20190821A patent/RS58991B1/sr unknown
- 2014-10-30 EP EP21196371.5A patent/EP4001272B1/en active Active
- 2014-10-30 RU RU2016119493A patent/RU2695372C2/ru active
- 2014-10-30 HR HRP20191159TT patent/HRP20191159T1/hr unknown
- 2014-10-30 WO PCT/GB2014/053236 patent/WO2015067923A1/en not_active Ceased
- 2014-10-30 ES ES19166031T patent/ES2894903T3/es active Active
- 2014-10-30 AU AU2014345407A patent/AU2014345407B2/en active Active
- 2014-10-30 EP EP19166031.5A patent/EP3564234B1/en active Active
- 2014-10-30 SI SI201431242T patent/SI3066091T1/sl unknown
- 2014-10-30 HU HUE21196371A patent/HUE070532T2/hu unknown
- 2014-10-30 TR TR2019/09632T patent/TR201909632T4/tr unknown
- 2014-10-30 CA CA2928004A patent/CA2928004C/en active Active
- 2014-10-30 HR HRP20241784TT patent/HRP20241784T1/hr unknown
- 2014-10-30 KR KR1020217039470A patent/KR102469167B1/ko active Active
- 2014-10-30 MX MX2020011946A patent/MX394633B/es unknown
- 2014-10-30 PT PT14802476T patent/PT3066091T/pt unknown
- 2014-10-30 HU HUE14802476A patent/HUE045001T2/hu unknown
- 2014-11-05 TW TW103138423A patent/TW201609653A/zh unknown
- 2014-11-05 AR ARP140104166A patent/AR098319A1/es unknown
- 2014-11-05 UY UY0001035823A patent/UY35823A/es not_active Application Discontinuation
-
2017
- 2017-10-17 US US15/785,654 patent/US10207994B2/en active Active
-
2019
- 2019-01-20 US US16/252,648 patent/US10815199B2/en active Active
- 2019-02-05 JP JP2019018440A patent/JP6633233B2/ja active Active
- 2019-07-02 CY CY20191100691T patent/CY1121749T1/el unknown
- 2019-12-10 JP JP2019222567A patent/JP6856737B2/ja active Active
-
2020
- 2020-10-26 US US17/079,553 patent/US12516023B2/en active Active
-
2021
- 2021-03-18 JP JP2021044193A patent/JP7234278B2/ja active Active
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US12516023B2 (en) | NMDA antagonist prodrugs | |
| HK40076024A (en) | Nmda antagonist prodrugs | |
| HK40076024B (en) | Nmda antagonist prodrugs | |
| HK40014361B (en) | Nmda antagonist prodrugs | |
| HK40014361A (en) | Nmda antagonist prodrugs | |
| WO2019217465A1 (en) | Calpain modulators and therapeutic uses thereof background | |
| HK1226721B (en) | Nmda antagonist prodrugs | |
| HK1226721A1 (en) | Nmda antagonist prodrugs | |
| BR112016009949B1 (pt) | Composto pró-fármaco antagonista de nmda, composição farmacêutica, e, uso de um composto |