ES2746081T3 - Procedimiento para la preparación de (1S,2R)-milnaciprán - Google Patents
Procedimiento para la preparación de (1S,2R)-milnaciprán Download PDFInfo
- Publication number
- ES2746081T3 ES2746081T3 ES15804325T ES15804325T ES2746081T3 ES 2746081 T3 ES2746081 T3 ES 2746081T3 ES 15804325 T ES15804325 T ES 15804325T ES 15804325 T ES15804325 T ES 15804325T ES 2746081 T3 ES2746081 T3 ES 2746081T3
- Authority
- ES
- Spain
- Prior art keywords
- alcohol
- enantiomerically enriched
- process according
- enriched form
- levomilnacipran
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C231/00—Preparation of carboxylic acid amides
- C07C231/12—Preparation of carboxylic acid amides by reactions not involving the formation of carboxamide groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C231/00—Preparation of carboxylic acid amides
- C07C231/02—Preparation of carboxylic acid amides from carboxylic acids or from esters, anhydrides, or halides thereof by reaction with ammonia or amines
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C231/00—Preparation of carboxylic acid amides
- C07C231/16—Preparation of optical isomers
- C07C231/18—Preparation of optical isomers by stereospecific synthesis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/07—Optical isomers
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2601/00—Systems containing only non-condensed rings
- C07C2601/02—Systems containing only non-condensed rings with a three-membered ring
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
Un procedimiento para la preparación de levomilnaciprán ((1S,2R)-2-(aminometil)-N,N-dietil-1- fenilciclopropanocarboxamida) que comprende las siguientes etapas: a) convertir directamente la forma enantioméricamente enriquecida del alcohol (D) en la forma enantioméricamente enriquecida del derivado ftalimido (C) mediante tratamiento con ftalimida en presencia de una trialquil- o triarilfosfina y de un azodicarboxilato de dialquilo: **Fórmula** en el que la cantidad de ftalimida está comprendida entre 1 y 1,3 equivalentes con respecto a la cantidad molar de alcohol (D) usada, y las cantidades de fosfina y de azodicarboxilato están comprendidas, independientemente entre sí, entre 1 y 1,5 equivalentes con respecto a la cantidad molar de alcohol (D) usada; b) desbloquear la forma enantioméricamente enriquecida del derivado ftalimido (C) para obtener levomilnaciprán:**Fórmula**
Description
DESCRIPCIÓN
Procedimiento para la preparación de (1S,2R)-milnaciprán
Campo de la invención
[0001] La presente invención se refiere a un procedimiento industrialmente aplicable y ventajoso para la preparación de (1 S,2fí)-milnaciprán (conocido generalmente como levomilnaciprán) o una sal del mismo.
Estado de la técnica
[0002] La (1S,2fí)-2-(aminometil)-W,W-dietil-1-fenilciclopropanocarboxamida, conocida también como levomilnaciprán, es un principio activo útil en el tratamiento de la depresión por su capacidad para actuar como inhibidor de la recaptación de la serotonina-norepinefrina. Dicho compuesto se caracteriza por la siguiente fórmula estructural:

[0003] La preparación del milnaciprán (es decir, la forma racémica del levomilnaciprán) o sales del mismo se ha descrito extensamente en la bibliografía. Por ejemplo, la patente europea EP 0377381 B1 describe un procedimiento para la preparación de un intermedio útil en la síntesis del milnaciprán, que proporciona la transformación de la lactona racémica (±-A) en el alcohol correspondiente (±-D) el cual, tras su activación como cloruro (±-F), se convierte en la amina correspondiente (±-C) mediante tratamiento con ftalimida de potasio:
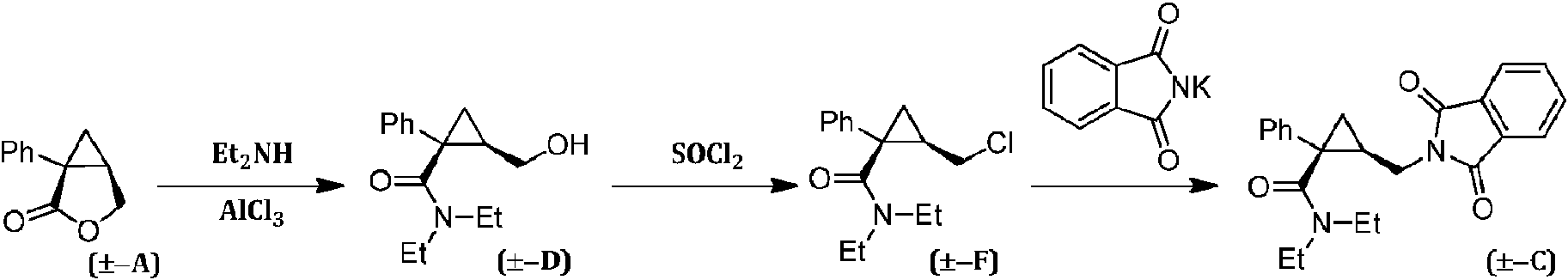
[0004] Como alternativa, tal como se describe en la patente europea EP 200638 B1, la lactona racémica (±-A) se puede tratar con ftalimida de potasio para obtener el ácido (±-B) que se convierte después en la amida (±-C) mediante tratamiento con cloruro de tionilo seguido de dietilamina.

[0005] El alcohol (±-D) se puede convertir también en la amida (±-C) usando el procedimiento descrito en la patente europea EP 1845084 B1, es decir, mediante tratamiento con ácido metanosulfónico en presencia de ortoformiato de trietilo y conversión posterior de la sal (±-E) en el producto deseado mediante tratamiento con ftalimida de potasio.
[0006] Un primer método para obtener levomilnaciprán enantioméricamente enriquecido, mediante separación de la mezcla racémica por cromatografía en una columna quiral, ha sido descrito en J. Chromatogr. 1985, 318, 398-403.
[0007] La patente europea EP 2123628 B1 y la solicitud internacional WO 2012/059933 A1 describen un procedimiento para la preparación del levomilnaciprán por resolución óptica de milnaciprán mediante tratamiento con un derivado de ácido tartárico y con ácido mandélico, respectivamente.
[0008] Una desventaja asociada a estos métodos procede del uso de la cristalización fraccionada o una cromatografía en una columna quiral para la separación de los dos enantiómeros de milnaciprán. De hecho, el primer procedimiento conduce a una disminución drástica del rendimiento total, ya que se debe descartar al menos un 50 % del producto, mientras que el segundo no es industrialmente aplicable, o apenas lo es.
[0009] Se han desarrollado posteriormente algunos planteamientos de síntesis para preparar la forma enantioméricamente enriquecida de milnaciprán, tal como se describe, por ejemplo, en Tetrahedron Letters 1996, 37, 641-644.

[0010] Sin embargo, estas rutas sintéticas proporcionan el empleo de azida de sodio, un reactivo cuyo uso a escala industrial debe limitarse debido a su toxicidad y al hecho de que puede dar lugar a productos y subproductos que se descomponen de forma explosiva.
[0011] Una ruta de síntesis adicional para la preparación del levomilnaciprán se ha descrito en la patente EP 2391599 B1. Esta ruta proporciona el uso del mismo esquema de síntesis descrito en la patente EP 200638 B1 previamente indicada, empleando una forma enantioméricamente enriquecida de la lactona (A) como compuesto de partida, a fin de obtener levomilnaciprán en lugar de milnaciprán.

[0012] Un último planteamiento de síntesis, descrito en la solicitud internacional WO 2014/009767 A1, proporciona la preparación del levomilnaciprán mediante reacción de un alcohol enantioméricamente enriquecido (D) con azida de sodio en presencia de azodicarboxilato de dietilo seguido de la reducción de la azida así obtenida mediante tratamiento con trifenilfosfina. Esta reacción implica el uso de ácido hidrazoico (y no de azida de sodio tal como se indica erróneamente en la solicitud) y, por tanto, se ha de evitar a escala industrial debido a la elevada toxicidad y explosividad de este producto tal como se describe en el Handbook of reactive Chemical hazards de Breterick, Elsevier 7a edición, páginas 1669-1670.
[0013] El objeto de la presente invención es proporcionar un método para la síntesis del levomilnaciprán que se efectúa con elevados rendimientos y proporciona los productos deseados con un grado de pureza adecuado para su uso farmacéutico.
Sumario de la invención
[0014] Estos objetos se consiguen con la presente invención que, en un primer aspecto de la misma, se refiere a un procedimiento para la preparación de (1 S,2R)-milnaciprán que comprende las etapas siguientes:
a) convertir directamente la forma enantioméricamente enriquecida del alcohol (D) en la forma enantioméricamente enriquecida del derivado ftalimido (C) mediante tratamiento con una ftalimida en presencia de una trialquil- o triarilfosfina y de un azodicarboxilato de dialquilo:

en el que la cantidad de ftalimida está comprendida entre 1 y 1,3 equivalentes con respecto a la cantidad molar de alcohol (D) usada, y las cantidades de la fosfina y del azodicarboxilato están comprendidas, independientemente entre sí, entre 1 y 1,5 equivalentes con respecto a la cantidad molar de alcohol (D) usada; b) desbloquear la forma enantioméricamente enriquecida del derivado ftalimido (C) para obtener el levomllnaclprán:

[0015] En este primer aspecto, la invención puede comprender una etapa adicional b') opcional que consiste en el tratamiento con una base inorgánica a fin de aumentar el rendimiento del aislamiento de levomilnaciprán.
[0016] En un segundo aspecto de la misma, la invención consiste en la transformación del levomilnaciprán así obtenido en una sal farmacéuticamente aceptable del mismo, con una etapa de procedimiento c) que consiste en transformar el levomilnaciprán, obtenido de acuerdo con las secuencias de etapas a)-b) o a)-b’) descritas anteriormente, en una sal mediante tratamiento con un ácido farmacéuticamente aceptable (HY):

en la que Ye es la base conjugada de dicho ácido.
Descripción detallada de la invención
[0017] Todos los términos y expresiones usados en esta solicitud, a menos que se indique lo contrario, se han de entender según su significado habitual conocido en el campo. A continuación se enumeran especificaciones más detalladas para determinados términos y expresiones usados en esta solicitud y se deben aplicar uniformemente en toda la memoria descriptiva y reivindicaciones, a menos que se indique lo contrario.
[0018] La pureza enantiomérica se expresa normalmente como el "exceso enantiomérico" o ee que se define, por ejemplo, para el enantiómero (S) como:
ee = [(S-R)/(R+S)] x 100
en la que S y R son las cantidades respectivas de los dos enantiómeros (S) y (R) (tal como se determinan, por ejemplo, mediante HPLC o GC en fase estacionaria quiral).
[0019] La expresión "mezcla racémica" y términos y expresiones derivados se refiere a una muestra de un compuesto quiral que contiene cantidades iguales de los dos isómeros ópticos (+) y (-).
[0020] Las expresiones "forma enantioméricamente enriquecida" o "mezcla enantioméricamente enriquecida" tal como se usan en la presente solicitud indican mezclas en las que uno de los dos enantiómeros está presente en exceso con respecto al otro.
[0021] La expresión "enantiómero (S) o (R) en forma enantioméricamente pura" se refiere a una muestra de un compuesto quiral cuya pureza enantiomérica es de al menos un 99 %, incluso más preferentemente de un 99,5 %.
[0022] El símbolo ••■III (enlace de rayas) presente en algunas de las fórmulas de la memoria descriptiva y
reivindicaciones indica que el sustituyente se encuentra por debajo del plano de la página. El símbolo (enlace de cuña) presente en algunas de las fórmulas de la memoria descriptiva y reivindicaciones indica que el sustituyente se encuentra por encima del plano de la página. En general, la nomenclatura usada en la presente solicitud se basa en el sistema computarizado AUTONOM® v.4.0 del Instituto Beilstein para asignar la nomenclatura sistemática de la IUPAC. En caso de discrepancia entre una estructura dibujada y el nombre asignado a esta estructura, la fórmula especificada debe considerarse correcta. Además, si la estereoquímica de una estructura o una porción de la estructura no está indicada, por ejemplo con un enlace de cuña o de rayas, tal estructura o porción de la misma debe entenderse que engloba todos los estereoisómeros.
[0023] De acuerdo con su aspecto más general, la presente invención se refiere a la preparación del levomilnaciprán o sales del mismo.
[0024] La primera etapa del procedimiento de la invención, a), proporciona la conversión directa de la forma enantioméricamente enriquecida del alcohol (D) en la forma enantioméricamente enriquecida del derivado ftalimido (C) mediante tratamiento con ftalimida en una cantidad de entre 1 y 1,3 equivalentes, preferentemente de entre 1 y 1.05 equivalentes, con respecto a la cantidad molar de alcohol (D) usada, en presencia de una trialquil- o triarilfosfina y un azodicarboxilato de dialquilo. Esta reacción, conocida como reacción de Mitsunobu, normalmente se lleva a cabo mediante tratamiento de un alcohol con un azodicarboxilato de dialquilo (por ejemplo azodicarboxilato de dietilo o, preferentemente, azodicarboxilato de diisopropilo) y un nucleófilo (la ftalimida) en presencia de una trialquil- o triarilfosfina (por ejemplo, tri-n-butilfosfina o, preferentemente, trifenilfosfina) en un disolvente aprótico polar tal como un disolvente clorado (por ejemplo diclorometano) o un éter (por ejemplo tetrahidrofurano, 1,4-dioxano o, preferentemente, 2-metiltetrahidrofurano), a una temperatura de entre -30 y 30 °C, preferentemente de entre 0 y 5 °C.
[0025] Las cantidades de fosfina y de azodicarboxilato están comprendidas, independientemente entre sí, entre 1 y 1.5 equivalentes con respecto a la cantidad molar de alcohol (D) usada; preferentemente estas cantidades son de 1,2 y 1,1 equivalentes, respectivamente.
[0026] La forma enantioméricamente enriquecida del alcohol (D) opcionalmente purificada y aislada se puede preparar en una etapa a') de forma preparatoria y previa al procedimiento de la invención de acuerdo con técnicas conocidas en el campo, por ejemplo, tal como se describe en la patente europea EP 2391599 B1, esto es, mediante tratamiento de una forma enantioméricamente enriquecida de la lactona (A) con dietilamina en presencia de un ácido de Lewis, preferentemente tricloruro de aluminio.
[0027] La etapa siguiente b) consiste en desbloquear la forma enantioméricamente enriquecida del derivado ftalimido (C) para obtener el levomilnaciprán de acuerdo con uno de los procedimientos descritos en Theodora W. Green, Protective Groups in Organic Synthesis, John Wiley & Sons (1999), página 565, que se incorpora por referencia a la presente solicitud. Esta etapa se puede llevar a cabo preferentemente mediante tratamiento con hidrazina, una alquilamina (preferentemente una solución acuosa de metilamina al 40 % p/p) o una hidroxialquilamina, por ejemplo etanolamina, en un disolvente aprótico polar, tal como un éter (por ejemplo tetrahidrofurano, 1,4-dioxano o, preferentemente, 2-metiltetrahidrofurano), a una temperatura de entre 0 y 50 0C, preferentemente de entre 20 y 30 °C.
[0028] La cantidad de hidrazina, alquilamina o hidroxialquilamina puede variar en un intervalo muy amplio; preferentemente, dicha cantidad puede variar entre 2 y 25 equivalentes con respecto a la cantidad molar del derivado ftalimido (C) usada; incluso más preferentemente, dicha cantidad puede variar entre 5 y 15 equivalentes con respecto a la cantidad molar del derivado ftalimido (C) usada, por ejemplo 11 equivalentes.
[0029] En una posible variación de este primer aspecto de la invención, posteriormente a la etapa b) se efectúa una etapa b') adicional que incluye un tratamiento con una base inorgánica, por ejemplo un carbonato (preferentemente una solución acuosa de Na2CO3 , Li2CO3 o K2CO3) o un hidróxido (preferentemente una solución acuosa de LiOH, KOH o NaOH). Esta etapa tiene como fin aumentar el rendimiento de la reacción de desbloqueamiento de la ftalimida y evitar la formación del derivado ftalimido (C) de nuevo durante las etapas de aislamiento del levomilnaciprán.
[0030] En su segundo aspecto, la invención incluye la conversión del levomilnaciprán obtenido tal como se ha descrito anteriormente, con o sin la implementación de la etapa b') opcional, en una sal farmacéuticamente aceptable del mismo, preferentemente su clorhidrato. Esta etapa c) se consigue mediante tratamiento con un ácido prótico farmacéuticamente aceptable HY, en el caso preferente mediante el uso de una solución acuosa de ácido clorhídrico o una solución de cloruro de hidrógeno en un disolvente orgánico (por ejemplo un alcohol, preferentemente metanol).
[0031] De acuerdo con una realización preferente, todas las etapas anteriores del procedimiento de la invención se llevan a cabo sin aislamiento de los compuestos intermedios. Cuando el levomilnaciprán, o cualquier otro compuesto descrito en la presente solicitud, se obtiene con un grado de pureza química no adecuado para su inclusión en un
medicamento, el procedimiento incluye una etapa de purificación adicional, por ejemplo mediante cromatografía o cristalización, opcionalmente tras la formación de un compuesto de adición, tal como una sal o un co-cristal, o mediante lavado con un disolvente orgánico o una solución acuosa, opcionalmente tras modificación del pH.
[0032] La invención se ilustrará adicionalmente mediante los siguientes ejemplos.
[0033] En los ejemplos siguientes, el control del progreso y la finalización de las reacciones descritas se efectuó mediante HPLC.
Método HPLC
[0034] Columna: Symmetry C18250 x 4,6 mm 5 pm o equivalente
Caudal: 1,0 ml/min
Volumen de inyección: 10 pl
Longitud de onda: 210 nm
T 8 Columna: 35 °C
Fase móvil:
Fase A: solución acuosa de H3 PO4 al 0,1 %
Fase B: acetonitrilo
[0035] Gradiente:

EJEMPLO 1
Síntesis de (1 S,2fí)-W,W-dietil-2-(hidroximetil)-1-fenilciclopropano-carboxamida (D).
[0036]

[0037] Se añade dietilamina (1,56 g, 21,3 mmol) gota a gota a una suspensión de tricloruro de aluminio (1,38 g, 10,4 mmol) en tolueno (9,6 ml) y se enfría hasta 0 °C con agitación magnética y en atmósfera de nitrógeno, comprobando que la temperatura interna del sistema no supere los 15 °C. La solución obtenida se mantiene a 25 °C durante aproximadamente 30 minutos y después se añade lentamente una solución de lactona A (1,50 g, 8,6 mmol) en tolueno (2 ml). La mezcla se mantiene con agitación magnética a 25 °C hasta la conversión completa (aproximadamente 2 horas) y se vierte después en agua (7,2 ml) previamente enfriada a 5 °C, comprobando que la temperatura interna no supere los 25 °C. Las fases obtenidas se separan y la fase acuosa se extrae con tolueno. La fase orgánica se filtra mediante un filtro de panel de carbón activo y celita, y se concentra a presión reducida hasta obtener un residuo de 2,50 g (que contiene aproximadamente 2,10 g del producto deseado), que se usa en la etapa siguiente sin ninguna purificación adicional.
EJEMPLO 2
Síntesis de (1 S,2R)-2-((1,3-dioxoisoindolin-2-il)metil)-W,W-dietil-1 -fenilciclopropanocarboxamida (C).
[0038]

[0039] Se añade 2-metiltetrahidrofurano (9 ml) al residuo preparado tal como se describe en el Ejemplo 1 (2,50 g, 8,6 mmol) con agitación magnética y en atmósfera inerte de nitrógeno hasta que se obtiene una solución, y se añaden después polvo de ftalimida (1,30 g, 8,8 mmol) y trifenilfosfina (2,60 g, 9,9 mmol). A la suspensión resultante, enfriada a -15 °C, se le añade lentamente una solución de azodicarboxilato de diisopropilo (DIAD) (1,80 g, 8,9 mmol) en 2-metiltetrahidrofurano (4,5 ml). Al final de la adición, la temperatura de la mezcla se eleva hasta 5 °C y se mantiene en las mismas condiciones hasta la conversión completa (aproximadamente 2 horas). La temperatura de la mezcla se lleva hasta 25 °C, y después se añade agua (4,5 ml) y las fases se separan. La fase orgánica se usa en la etapa siguiente sin ninguna purificación adicional.
EJEMPLO 3
Síntesis de clorhidrato de (1 S,2fí)-2-(aminometil)-N,N-dietil-1-fenilciclopropanocarboxamida (clorhidrato de levomilnaciprán).
[0040]

[0041] Se añade una solución acuosa de metilamina al 40 % p/p (7,50 g, 96,6 mmol) a la solución preparada tal como se describe en el Ejemplo 2 (18,00 g de solución que contiene 8,6 mmol de C) con agitación magnética y en atmósfera de nitrógeno. La mezcla resultante se mantiene con agitación magnética a 25 °C hasta la conversión completa (aproximadamente 3 horas), y después se añade una solución acuosa de hidróxido de sodio al 30 % p/p (4,50 g, 33,7 mmol) y la mezcla se deja en agitación durante 30 minutos más. Las fases se separan y la fase acuosa se extrae con 2-metiltetrahidrofurano (4,5 ml). Las fases orgánicas combinadas se concentran para reducir el volumen a aproximadamente 10 ml, después se añaden agua (6 ml), 2-metiltetrahidrofurano (20 ml) y una solución acuosa de ácido clorhídrico al 37 % p/p (1,30 g, 12,8 mmol) hasta un pH de aproximadamente 1, y comprobando que la temperatura interna del sistema no supere los 30 °C. Al final de la adición, las fases se separan y la fase orgánica se extrae dos veces con agua (4,5 ml) y una solución acuosa de ácido clorhídrico al 37 % p/p (0,60 g, 5,9 mmol). A las fases acuosas combinadas se les añaden 2-metiltetrahidrofurano (9 ml) y una solución acuosa de hidróxido de sodio al 30 % p/p (3,50 g, 26,0 mmol), y comprobando que la temperatura del sistema no supere los 30 °C. Las fases se separan y la fase acuosa se extrae tres veces con 2-metiltetrahidrofurano (4,5 ml). Las fases orgánicas combinadas se concentran a presión reducida hasta que se obtiene un residuo. El aceite obtenido se solubiliza en acetato de isopropilo (4,5 ml) y se mantiene con agitación magnética hasta que se obtiene una solución que se filtra mediante un filtro de panel de carbón activo y celita. Se añade una solución de ácido clorhídrico al 15 % p/p (2,10 g, 8,6 mmol) en metanol al filtrado, y se mantiene a 30 °C, hasta que se obtiene un pH entre 1 y 2. La solución se calienta a reflujo a presión atmosférica para eliminar el metanol hasta que la temperatura interna está en el intervalo de 84 a 85 °C, añadiendo a la vez acetato de isopropilo a fin de mantener constante el volumen de la mezcla. La solución se deja enfriar hasta una temperatura de entre 15 y 20 °C, y después se filtra la suspensión y el sólido se lava con acetato de isopropilo. El producto se seca a 40 °C a presión reducida hasta que se obtiene un peso constante. Se obtienen 2,30 g de clorhidrato de levomilnaciprán con un rendimiento del 94 % con respecto a la cantidad de compuesto (A) usada.
EJEMPLO 4
Síntesis de (1 S,5fí)-1-fenil-3-oxabiciclo[3.1.0]hexan-2-ona (A).
[0042]

[0043] Se añade fenilacetonitrilo (7,00 g, 59,6 mmol) a una solución de (R)-epiclorohidrina (2,80 g, 30,3 mmol) en tolueno (28 ml). Se añade amida de sodio (2,80 g, 71,7 mmol) en porciones a la solución obtenida, se enfría hasta -40 °C comprobando la evolución de gas y que la temperatura interna del sistema no supere los -30 °C. Cuando la reacción ha finalizado (aproximadamente 30 minutos después de la última adición) la mezcla se vierte lentamente en agua, comprobando que la temperatura del sistema no supere los 20 °C. Las fases obtenidas se separan y la fase orgánica se concentra a presión reducida hasta que se obtiene un aceite. Se añade una solución acuosa de hidróxido de sodio al 30 % p/p (12,60 g, 94,4 mmol) al residuo con agitación magnética, después la mezcla se calienta a reflujo hasta la conversión completa (aproximadamente 3 horas). La mezcla se enfría hasta 25 °C y se vierte en una mezcla de agua (14 ml) y tolueno (20 ml). Las fases se separan, lavando la fase acuosa con tolueno. Se añaden tolueno (22 ml) y una solución acuosa de ácido clorhídrico al 36 % p/p (14,00 g, 138,3 mmol) a la mezcla obtenida hasta que se obtiene un pH por debajo de 1. La mezcla se calienta hasta 65 °C con agitación magnética y se mantiene en las mismas condiciones hasta la conversión completa (aproximadamente 2 horas), y después las fases se separan. La fase orgánica se lava con agua y después, tras enfriar hasta 30 °C, se lava adicionalmente con una solución acuosa de bicarbonato de sodio al 5 % p/p y con agua. La fase orgánica se concentra a presión reducida hasta que se obtiene un residuo que se recoge en isopropanol calentando a reflujo hasta que se obtiene una solución. La mezcla se enfría hasta 5 °C hasta la precipitación completa, después el sólido se filtra y se lava con isopropanol. Se obtienen 2,70 g de (A) con un rendimiento del 51 % con respecto a la cantidad de (R)-epiclorohidrina usada.
Comentarios a los Ejemplos 1 a 3
[0044] El procedimiento descrito en los ejemplos 1 a 3, materia de las etapas a')-c) descritas anteriormente, permite obtener el clorhidrato de levomilnaciprán con un rendimiento de aislamiento del 94 % con respecto a la cantidad de compuesto (A) usada. Este rendimiento y, en consecuencia, el método que permite obtenerlo debe ser considerado particularmente significativo cuando se compara con los procedimientos descritos en los documentos de la técnica anterior, que permiten una recuperación del clorhidrato de levomilnaciprán con un rendimiento del 66 % (EP 2391599 B1) o del milnaciprán con un rendimiento del 81 % (EP 1845084 B1).
Claims (10)
1. Un procedimiento para la preparación de levomilnaciprán ((1S,2fí)-2-(aminometil)-N,N-dietil-1-fenilciclopropanocarboxamida) que comprende las siguientes etapas:
a) convertir directamente la forma enantioméricamente enriquecida del alcohol (D) en la forma enantioméricamente enriquecida del derivado ftalimido (C) mediante tratamiento con ftalimida en presencia de una trialquil- o triarilfosfina y de un azodicarboxilato de dialquilo:

en el que la cantidad de ftalimida está comprendida entre 1 y 1,3 equivalentes con respecto a la cantidad molar de alcohol (D) usada, y las cantidades de fosfina y de azodicarboxilato están comprendidas, independientemente entre sí, entre 1 y 1,5 equivalentes con respecto a la cantidad molar de alcohol (D) usada;
b) desbloquear la forma enantioméricamente enriquecida del derivado ftalimido (C) para obtener levomilnaciprán:

2. Un procedimiento de acuerdo con la reivindicación 1, que comprende una etapa b') adicional efectuada tras la etapa b), que consiste en el tratamiento del levomilnaciprán con una base inorgánica.
3. Un procedimiento de acuerdo con una cualquiera de las reivindicaciones 1 o 2, en el que en la etapa a) la cantidad de ftalimida está comprendida entre 1 y 1,05 equivalentes con respecto a la cantidad molar de alcohol (D) usada.
4. Un procedimiento de acuerdo con una cualquiera de las reivindicaciones anteriores, en el que la etapa a) se lleva a cabo en un disolvente aprótico polar seleccionado entre disolvente clorados y éteres.
5. Un procedimiento de acuerdo con una cualquiera de las reivindicaciones anteriores, en el que la etapa b) se lleva a cabo mediante tratamiento con un compuesto seleccionado entre hidrazina, una alquilamina y una hidroxialquilamina.
6. Un procedimiento de acuerdo con la reivindicación 5, en el que la cantidad de hidrazina, alquilamina e hidroxialquilamina está comprendida entre 2 y 25 equivalentes con respecto a la cantidad molar del derivado ftalimido (C).
7. Un procedimiento de acuerdo con una cualquiera de las reivindicaciones 2 a 6, en el que la base inorgánica usada en la etapa b') se selecciona entre carbonatos e hidróxidos.
8. Un procedimiento de acuerdo con una cualquiera de las reivindicaciones anteriores, que incluye adicionalmente la conversión del levomilnaciprán en una sal farmacéuticamente aceptable del mismo mediante tratamiento con un ácido prótico HY, de acuerdo con la etapa c):

en la que Ye es la base conjugada de dicho ácido.
9. Un procedimiento de acuerdo con una cualquiera de las reivindicaciones anteriores, en el que la forma enantioméricamente enriquecida del alcohol (D) se prepara en una etapa a') previa a la etapa a), mediante tratamiento de una forma enantioméricamente enriquecida de lactona (A) con dietilamina en presencia de un ácido
de Lewis:

10. Un procedimiento de acuerdo con una cualquiera de las reivindicaciones anteriores, en el que las etapas a')-c) del procedimiento se llevan a cabo sin aislamiento de los compuestos intermedios.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| ITMI20141883 | 2014-11-04 | ||
| PCT/EP2015/075513 WO2016071303A1 (en) | 2014-11-04 | 2015-11-03 | Process for the preparation of (1s,2r)-milnacipran |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2746081T3 true ES2746081T3 (es) | 2020-03-04 |
Family
ID=52014246
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES15804325T Active ES2746081T3 (es) | 2014-11-04 | 2015-11-03 | Procedimiento para la preparación de (1S,2R)-milnaciprán |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US10131624B2 (es) |
| EP (1) | EP3230258B1 (es) |
| CA (1) | CA2966437A1 (es) |
| ES (1) | ES2746081T3 (es) |
| WO (1) | WO2016071303A1 (es) |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN111333566A (zh) * | 2018-12-19 | 2020-06-26 | 北京万全德众医药生物技术有限公司 | 左旋米那普仑关键中间体的制备方法 |
| CN111620807A (zh) * | 2019-02-27 | 2020-09-04 | 北京万全德众医药生物技术有限公司 | 一种盐酸左旋米那普仑杂质的制备方法 |
Family Cites Families (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| FR2581059B1 (fr) | 1985-04-25 | 1988-04-22 | Pf Medicament | Procede de preparation du chlorhydrate de phenyl-1 diethyl amino carbonyl-1 aminomethyl-2 cyclopropane (z) |
| FR2640972B1 (es) | 1988-12-28 | 1991-04-19 | Pf Medicament | |
| JP4828863B2 (ja) * | 2005-01-28 | 2011-11-30 | 住友化学株式会社 | (z)−1−フェニル−1−(n,n−ジエチルアミノカルボニル)−2−フタルイミドメチルシクロプロパンの製造方法 |
| CN101195583B (zh) | 2006-12-04 | 2012-07-04 | 四川抗菌素工业研究所有限公司 | 一种光学纯米那普仑及其盐的制备方法 |
| FR2941454B1 (fr) | 2009-01-29 | 2011-04-01 | Pf Medicament | Proced de synthese du (1s,2r)-milnacipran |
| US20120289744A1 (en) | 2010-11-03 | 2012-11-15 | Arch Pharmalabs Limited | Process for preparing optically pure milnacipran and its pharmaceutically acceptable salts |
| WO2014009767A1 (en) | 2012-07-07 | 2014-01-16 | Micro Labs Limited | An improved process for the preparation of 1-aryl 2-aminomethyl cyclopropane carboxyamide (z) derivatives, their isomers and salts |
-
2015
- 2015-11-03 CA CA2966437A patent/CA2966437A1/en not_active Abandoned
- 2015-11-03 WO PCT/EP2015/075513 patent/WO2016071303A1/en not_active Ceased
- 2015-11-03 ES ES15804325T patent/ES2746081T3/es active Active
- 2015-11-03 EP EP15804325.7A patent/EP3230258B1/en active Active
- 2015-11-03 US US15/523,560 patent/US10131624B2/en active Active
Also Published As
| Publication number | Publication date |
|---|---|
| CA2966437A1 (en) | 2016-05-12 |
| EP3230258A1 (en) | 2017-10-18 |
| US20180273468A1 (en) | 2018-09-27 |
| WO2016071303A1 (en) | 2016-05-12 |
| EP3230258B1 (en) | 2019-06-26 |
| US10131624B2 (en) | 2018-11-20 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP3077404B1 (en) | Purification of tenofovir alafenamide and its intermediates | |
| US9676803B2 (en) | Efficient process for separation of diastereomers of 9-[(R)-2-[[(R,S)-[[(S)-1-(isopropoxycarbonyl)ethyl]amino]-phenoxyphosphinyl]methoxy]propyl]adenine | |
| WO2009064476A1 (en) | Preparation of sitagliptin intermediate | |
| ES2706314T3 (es) | Método para la producción de praziquantel y precursores del mismo | |
| WO2014157612A1 (ja) | (1s,4s,5s)-4-ブロモ-6-オキサビシクロ[3.2.1]オクタン-7-オンの製造方法 | |
| JP2017523168A (ja) | 3−(3−クロロ−1h−ピラゾール−1−イル)ピリジンの製造方法 | |
| JP7161466B2 (ja) | グリコピロニウム化合物を作製するための方法及び使用する方法 | |
| WO2022232948A1 (en) | Processes for the preparation of the enantiomers of 3,4-methylenedioxymethamphetamine (mdma) and n-methyl-1,3-benzodioxolylbutanamine (mbdb) | |
| WO2015007897A1 (en) | Method of racemisation of undesired enantiomers | |
| CN105777544A (zh) | 一种高光学纯度s-(+)-氟比洛芬酯的制备方法 | |
| ES2746081T3 (es) | Procedimiento para la preparación de (1S,2R)-milnaciprán | |
| US20140296569A1 (en) | Optical Resolution Method for Bicyclic Compound Using Asymmetric Catalyst | |
| CN107108607B (zh) | 制备吡喹酮的方法 | |
| EA010912B1 (ru) | Способ получения производных 2-оксо-1-пирролидина внутримолекулярным аллилированием | |
| ES2811271T3 (es) | Método para la producción de praziquantel y sus precursores | |
| TW202229229A (zh) | 4-硼-l-苯基丙胺酸及其中間體之製造方法 | |
| ES2891549T3 (es) | Proceso para la síntesis de intermedios de nebivolol | |
| CN107646037A (zh) | 选择性合成核苷氨基磷酸酯类化合物的方法 | |
| JP2012522804A (ja) | パラジウム触媒によるオルトフッ素化 | |
| US9056817B2 (en) | Arylated β-dicarbonyl compounds and process for the preparation thereof | |
| RU2326885C1 (ru) | Способ получения диизопропил ((1-(гидроксиметил)-циклопропил)окси) метилфосфоната | |
| JPWO2009142194A1 (ja) | 光学活性アミノアルコール誘導体の製造方法 | |
| US11091436B2 (en) | Process for the separation of optical isomers of racemic 3-alkylpiperidine-carboxylic acid ethyl esters | |
| US20070129443A1 (en) | Production method of aminochlorohydrin sulfate | |
| US9828334B2 (en) | Process for preparing levomilnacipran |