ES2749800T3 - Formulación de liberación sostenida de naltrexona - Google Patents
Formulación de liberación sostenida de naltrexona Download PDFInfo
- Publication number
- ES2749800T3 ES2749800T3 ES07795652T ES07795652T ES2749800T3 ES 2749800 T3 ES2749800 T3 ES 2749800T3 ES 07795652 T ES07795652 T ES 07795652T ES 07795652 T ES07795652 T ES 07795652T ES 2749800 T3 ES2749800 T3 ES 2749800T3
- Authority
- ES
- Spain
- Prior art keywords
- naltrexone
- dosage form
- bupropion
- release
- sustained release
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- DQCKKXVULJGBQN-XFWGSAIBSA-N naltrexone Chemical compound N1([C@@H]2CC3=CC=C(C=4O[C@@H]5[C@](C3=4)([C@]2(CCC5=O)O)CC1)O)CC1CC1 DQCKKXVULJGBQN-XFWGSAIBSA-N 0.000 title claims abstract description 342
- 229960003086 naltrexone Drugs 0.000 title claims abstract description 305
- 238000013268 sustained release Methods 0.000 title claims abstract description 197
- 239000012730 sustained-release form Substances 0.000 title claims abstract description 197
- 239000000203 mixture Substances 0.000 title claims abstract description 106
- 238000009472 formulation Methods 0.000 title claims abstract description 83
- SNPPWIUOZRMYNY-UHFFFAOYSA-N bupropion Chemical compound CC(C)(C)NC(C)C(=O)C1=CC=CC(Cl)=C1 SNPPWIUOZRMYNY-UHFFFAOYSA-N 0.000 claims abstract description 88
- 239000002552 dosage form Substances 0.000 claims abstract description 75
- 229960001058 bupropion Drugs 0.000 claims abstract description 40
- 239000006186 oral dosage form Substances 0.000 claims abstract description 33
- 150000003839 salts Chemical class 0.000 claims abstract description 33
- 238000000338 in vitro Methods 0.000 claims abstract description 12
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 claims abstract description 10
- 239000012738 dissolution medium Substances 0.000 claims abstract description 6
- 238000007922 dissolution test Methods 0.000 claims abstract description 6
- 238000011282 treatment Methods 0.000 claims description 41
- 239000003814 drug Substances 0.000 claims description 24
- 230000004580 weight loss Effects 0.000 claims description 15
- 239000001866 hydroxypropyl methyl cellulose Substances 0.000 claims description 12
- 235000010979 hydroxypropyl methyl cellulose Nutrition 0.000 claims description 12
- 229920003088 hydroxypropyl methyl cellulose Polymers 0.000 claims description 12
- UFVKGYZPFZQRLF-UHFFFAOYSA-N hydroxypropyl methyl cellulose Chemical compound OC1C(O)C(OC)OC(CO)C1OC1C(O)C(O)C(OC2C(C(O)C(OC3C(C(O)C(O)C(CO)O3)O)C(CO)O2)O)C(CO)O1 UFVKGYZPFZQRLF-UHFFFAOYSA-N 0.000 claims description 12
- 208000008589 Obesity Diseases 0.000 claims description 11
- 235000020824 obesity Nutrition 0.000 claims description 11
- 229920001577 copolymer Polymers 0.000 claims description 8
- 229920003171 Poly (ethylene oxide) Polymers 0.000 claims description 7
- -1 polyoxyethylene Polymers 0.000 claims description 7
- NIXOWILDQLNWCW-UHFFFAOYSA-M Acrylate Chemical compound [O-]C(=O)C=C NIXOWILDQLNWCW-UHFFFAOYSA-M 0.000 claims description 5
- 230000004584 weight gain Effects 0.000 claims description 5
- 235000019786 weight gain Nutrition 0.000 claims description 5
- 229920002153 Hydroxypropyl cellulose Polymers 0.000 claims description 4
- CERQOIWHTDAKMF-UHFFFAOYSA-M Methacrylate Chemical compound CC(=C)C([O-])=O CERQOIWHTDAKMF-UHFFFAOYSA-M 0.000 claims description 4
- 235000010977 hydroxypropyl cellulose Nutrition 0.000 claims description 4
- 229920000642 polymer Polymers 0.000 claims description 4
- 239000004354 Hydroxyethyl cellulose Substances 0.000 claims description 3
- 229920000663 Hydroxyethyl cellulose Polymers 0.000 claims description 3
- 229940071826 hydroxyethyl cellulose Drugs 0.000 claims description 3
- 235000019447 hydroxyethyl cellulose Nutrition 0.000 claims description 3
- 239000001863 hydroxypropyl cellulose Substances 0.000 claims description 3
- 229940071676 hydroxypropylcellulose Drugs 0.000 claims description 3
- 230000005764 inhibitory process Effects 0.000 claims description 3
- 238000002360 preparation method Methods 0.000 claims description 3
- 229920002774 Maltodextrin Polymers 0.000 claims description 2
- 239000005913 Maltodextrin Substances 0.000 claims description 2
- QGZKDVFQNNGYKY-UHFFFAOYSA-O ammonium group Chemical group [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 claims description 2
- 238000009506 drug dissolution testing Methods 0.000 claims description 2
- UACSZOWTRIJIFU-UHFFFAOYSA-N hydroxymethyl 2-methylprop-2-enoate Chemical compound CC(=C)C(=O)OCO UACSZOWTRIJIFU-UHFFFAOYSA-N 0.000 claims description 2
- 229940035034 maltodextrin Drugs 0.000 claims description 2
- 229920000609 methyl cellulose Polymers 0.000 claims description 2
- 239000001923 methylcellulose Substances 0.000 claims description 2
- 235000010981 methylcellulose Nutrition 0.000 claims description 2
- 229920000058 polyacrylate Polymers 0.000 claims description 2
- 239000000230 xanthan gum Substances 0.000 claims description 2
- 235000010493 xanthan gum Nutrition 0.000 claims description 2
- 229920001285 xanthan gum Polymers 0.000 claims description 2
- 229940082509 xanthan gum Drugs 0.000 claims description 2
- 229920001971 elastomer Polymers 0.000 claims 1
- FPYJFEHAWHCUMM-UHFFFAOYSA-N maleic anhydride Chemical compound O=C1OC(=O)C=C1 FPYJFEHAWHCUMM-UHFFFAOYSA-N 0.000 claims 1
- XJRBAMWJDBPFIM-UHFFFAOYSA-N methyl vinyl ether Chemical compound COC=C XJRBAMWJDBPFIM-UHFFFAOYSA-N 0.000 claims 1
- 229960002900 methylcellulose Drugs 0.000 claims 1
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 claims 1
- 239000005060 rubber Substances 0.000 claims 1
- 239000012729 immediate-release (IR) formulation Substances 0.000 description 86
- 230000002411 adverse Effects 0.000 description 55
- 230000000694 effects Effects 0.000 description 50
- 150000001875 compounds Chemical class 0.000 description 46
- JLVNEHKORQFVQJ-PYIJOLGTSA-N 6alpha-Naltrexol Chemical compound C([C@]12[C@H]3OC=4C(O)=CC=C(C2=4)C[C@@H]2[C@]1(O)CC[C@H]3O)CN2CC1CC1 JLVNEHKORQFVQJ-PYIJOLGTSA-N 0.000 description 42
- 230000036470 plasma concentration Effects 0.000 description 38
- ZFSXKSSWYSZPGQ-UHFFFAOYSA-N (2-hydroxycyclopentyl)azanium;chloride Chemical compound Cl.NC1CCCC1O ZFSXKSSWYSZPGQ-UHFFFAOYSA-N 0.000 description 35
- 229960000858 naltrexone hydrochloride Drugs 0.000 description 35
- 239000003826 tablet Substances 0.000 description 31
- 229940110294 revia Drugs 0.000 description 27
- 210000002381 plasma Anatomy 0.000 description 21
- 238000000034 method Methods 0.000 description 20
- 239000003981 vehicle Substances 0.000 description 20
- 229940079593 drug Drugs 0.000 description 19
- 239000000546 pharmaceutical excipient Substances 0.000 description 17
- 238000011260 co-administration Methods 0.000 description 16
- 206010028813 Nausea Diseases 0.000 description 14
- 230000008693 nausea Effects 0.000 description 14
- 208000016261 weight loss Diseases 0.000 description 14
- 238000004090 dissolution Methods 0.000 description 13
- RHBRMCOKKKZVRY-ITLPAZOVSA-N naltrexone hydrochloride Chemical compound Cl.N1([C@@H]2CC3=CC=C(C=4O[C@@H]5[C@](C3=4)([C@]2(CCC5=O)O)CC1)O)CC1CC1 RHBRMCOKKKZVRY-ITLPAZOVSA-N 0.000 description 12
- 230000009467 reduction Effects 0.000 description 12
- 239000002775 capsule Substances 0.000 description 9
- 206010047700 Vomiting Diseases 0.000 description 8
- 210000004369 blood Anatomy 0.000 description 8
- 239000008280 blood Substances 0.000 description 8
- VYFYYTLLBUKUHU-UHFFFAOYSA-N dopamine Chemical compound NCCC1=CC=C(O)C(O)=C1 VYFYYTLLBUKUHU-UHFFFAOYSA-N 0.000 description 8
- 239000008194 pharmaceutical composition Substances 0.000 description 8
- 230000000670 limiting effect Effects 0.000 description 7
- 206010019233 Headaches Diseases 0.000 description 6
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 6
- 239000004615 ingredient Substances 0.000 description 6
- QZAYGJVTTNCVMB-UHFFFAOYSA-N serotonin Chemical compound C1=C(O)C=C2C(CCN)=CNC2=C1 QZAYGJVTTNCVMB-UHFFFAOYSA-N 0.000 description 6
- 239000000243 solution Substances 0.000 description 6
- 239000000126 substance Substances 0.000 description 6
- 230000008673 vomiting Effects 0.000 description 6
- 238000004458 analytical method Methods 0.000 description 5
- 239000003085 diluting agent Substances 0.000 description 5
- 231100000869 headache Toxicity 0.000 description 5
- 238000001727 in vivo Methods 0.000 description 5
- 230000001965 increasing effect Effects 0.000 description 5
- AKOAEVOSDHIVFX-UHFFFAOYSA-N Hydroxybupropion Chemical class OCC(C)(C)NC(C)C(=O)C1=CC=CC(Cl)=C1 AKOAEVOSDHIVFX-UHFFFAOYSA-N 0.000 description 4
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 4
- 239000004480 active ingredient Substances 0.000 description 4
- 239000008186 active pharmaceutical agent Substances 0.000 description 4
- 108091008690 chemoreceptors Proteins 0.000 description 4
- 230000002354 daily effect Effects 0.000 description 4
- 229960003638 dopamine Drugs 0.000 description 4
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 4
- 238000005070 sampling Methods 0.000 description 4
- 239000000725 suspension Substances 0.000 description 4
- 208000007848 Alcoholism Diseases 0.000 description 3
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 3
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 3
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 3
- 206010033307 Overweight Diseases 0.000 description 3
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 3
- 229930006000 Sucrose Natural products 0.000 description 3
- 238000010521 absorption reaction Methods 0.000 description 3
- 201000007930 alcohol dependence Diseases 0.000 description 3
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 3
- 239000011230 binding agent Substances 0.000 description 3
- 230000037396 body weight Effects 0.000 description 3
- 238000004364 calculation method Methods 0.000 description 3
- 239000013066 combination product Substances 0.000 description 3
- 229940127555 combination product Drugs 0.000 description 3
- 206010012601 diabetes mellitus Diseases 0.000 description 3
- 239000012458 free base Substances 0.000 description 3
- 238000002347 injection Methods 0.000 description 3
- 239000007924 injection Substances 0.000 description 3
- 239000008101 lactose Substances 0.000 description 3
- 238000004519 manufacturing process Methods 0.000 description 3
- 238000005259 measurement Methods 0.000 description 3
- 239000002207 metabolite Substances 0.000 description 3
- 229940126403 monoamine reuptake inhibitor Drugs 0.000 description 3
- 239000003401 opiate antagonist Substances 0.000 description 3
- 239000000843 powder Substances 0.000 description 3
- 230000002829 reductive effect Effects 0.000 description 3
- 230000000979 retarding effect Effects 0.000 description 3
- 229940076279 serotonin Drugs 0.000 description 3
- 239000005720 sucrose Substances 0.000 description 3
- 230000001225 therapeutic effect Effects 0.000 description 3
- 229940057739 vivitrol Drugs 0.000 description 3
- RTHCYVBBDHJXIQ-MRXNPFEDSA-N (R)-fluoxetine Chemical compound O([C@H](CCNC)C=1C=CC=CC=1)C1=CC=C(C(F)(F)F)C=C1 RTHCYVBBDHJXIQ-MRXNPFEDSA-N 0.000 description 2
- QCQCHGYLTSGIGX-GHXANHINSA-N 4-[[(3ar,5ar,5br,7ar,9s,11ar,11br,13as)-5a,5b,8,8,11a-pentamethyl-3a-[(5-methylpyridine-3-carbonyl)amino]-2-oxo-1-propan-2-yl-4,5,6,7,7a,9,10,11,11b,12,13,13a-dodecahydro-3h-cyclopenta[a]chrysen-9-yl]oxy]-2,2-dimethyl-4-oxobutanoic acid Chemical compound N([C@@]12CC[C@@]3(C)[C@]4(C)CC[C@H]5C(C)(C)[C@@H](OC(=O)CC(C)(C)C(O)=O)CC[C@]5(C)[C@H]4CC[C@@H]3C1=C(C(C2)=O)C(C)C)C(=O)C1=CN=CC(C)=C1 QCQCHGYLTSGIGX-GHXANHINSA-N 0.000 description 2
- 206010000087 Abdominal pain upper Diseases 0.000 description 2
- 206010067484 Adverse reaction Diseases 0.000 description 2
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 2
- 208000019901 Anxiety disease Diseases 0.000 description 2
- 238000012935 Averaging Methods 0.000 description 2
- HEYVINCGKDONRU-UHFFFAOYSA-N Bupropion hydrochloride Chemical compound Cl.CC(C)(C)NC(C)C(=O)C1=CC=CC(Cl)=C1 HEYVINCGKDONRU-UHFFFAOYSA-N 0.000 description 2
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 2
- VTYYLEPIZMXCLO-UHFFFAOYSA-L Calcium carbonate Chemical compound [Ca+2].[O-]C([O-])=O VTYYLEPIZMXCLO-UHFFFAOYSA-L 0.000 description 2
- 229920002261 Corn starch Polymers 0.000 description 2
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 2
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 2
- 206010020772 Hypertension Diseases 0.000 description 2
- ROHFNLRQFUQHCH-UHFFFAOYSA-N Leucine Chemical compound CC(C)CC(N)C(O)=O ROHFNLRQFUQHCH-UHFFFAOYSA-N 0.000 description 2
- 229930195725 Mannitol Natural products 0.000 description 2
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 2
- 206010029216 Nervousness Diseases 0.000 description 2
- 229940123257 Opioid receptor antagonist Drugs 0.000 description 2
- 208000002193 Pain Diseases 0.000 description 2
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 2
- 229920002472 Starch Polymers 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 2
- 239000002253 acid Substances 0.000 description 2
- 150000007513 acids Chemical class 0.000 description 2
- 239000000853 adhesive Substances 0.000 description 2
- 230000001070 adhesive effect Effects 0.000 description 2
- 230000006838 adverse reaction Effects 0.000 description 2
- 230000001430 anti-depressive effect Effects 0.000 description 2
- 239000000935 antidepressant agent Substances 0.000 description 2
- 229940005513 antidepressants Drugs 0.000 description 2
- 239000003963 antioxidant agent Substances 0.000 description 2
- 230000036506 anxiety Effects 0.000 description 2
- 239000002585 base Substances 0.000 description 2
- 230000004071 biological effect Effects 0.000 description 2
- 230000000903 blocking effect Effects 0.000 description 2
- 239000000872 buffer Substances 0.000 description 2
- 239000011575 calcium Substances 0.000 description 2
- 229910052791 calcium Inorganic materials 0.000 description 2
- 239000011111 cardboard Substances 0.000 description 2
- 229920002678 cellulose Polymers 0.000 description 2
- 210000003169 central nervous system Anatomy 0.000 description 2
- 230000008859 change Effects 0.000 description 2
- 201000001883 cholelithiasis Diseases 0.000 description 2
- 229940075614 colloidal silicon dioxide Drugs 0.000 description 2
- 239000003086 colorant Substances 0.000 description 2
- 239000008120 corn starch Substances 0.000 description 2
- 230000007423 decrease Effects 0.000 description 2
- 230000003247 decreasing effect Effects 0.000 description 2
- 239000008121 dextrose Substances 0.000 description 2
- 238000010586 diagram Methods 0.000 description 2
- 235000005911 diet Nutrition 0.000 description 2
- 230000037213 diet Effects 0.000 description 2
- 239000007884 disintegrant Substances 0.000 description 2
- 208000002173 dizziness Diseases 0.000 description 2
- 238000001647 drug administration Methods 0.000 description 2
- 239000003937 drug carrier Substances 0.000 description 2
- 206010016256 fatigue Diseases 0.000 description 2
- 229960002464 fluoxetine Drugs 0.000 description 2
- 230000037406 food intake Effects 0.000 description 2
- 230000002496 gastric effect Effects 0.000 description 2
- 239000000499 gel Substances 0.000 description 2
- 230000005802 health problem Effects 0.000 description 2
- NOESYZHRGYRDHS-UHFFFAOYSA-N insulin Chemical compound N1C(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(NC(=O)CN)C(C)CC)CSSCC(C(NC(CO)C(=O)NC(CC(C)C)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CCC(N)=O)C(=O)NC(CC(C)C)C(=O)NC(CCC(O)=O)C(=O)NC(CC(N)=O)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CSSCC(NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2C=CC(O)=CC=2)NC(=O)C(CC(C)C)NC(=O)C(C)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2NC=NC=2)NC(=O)C(CO)NC(=O)CNC2=O)C(=O)NCC(=O)NC(CCC(O)=O)C(=O)NC(CCCNC(N)=N)C(=O)NCC(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC(O)=CC=3)C(=O)NC(C(C)O)C(=O)N3C(CCC3)C(=O)NC(CCCCN)C(=O)NC(C)C(O)=O)C(=O)NC(CC(N)=O)C(O)=O)=O)NC(=O)C(C(C)CC)NC(=O)C(CO)NC(=O)C(C(C)O)NC(=O)C1CSSCC2NC(=O)C(CC(C)C)NC(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CC(N)=O)NC(=O)C(NC(=O)C(N)CC=1C=CC=CC=1)C(C)C)CC1=CN=CN1 NOESYZHRGYRDHS-UHFFFAOYSA-N 0.000 description 2
- 239000007788 liquid Substances 0.000 description 2
- 239000000314 lubricant Substances 0.000 description 2
- 235000019359 magnesium stearate Nutrition 0.000 description 2
- 239000000594 mannitol Substances 0.000 description 2
- 235000010355 mannitol Nutrition 0.000 description 2
- 239000000463 material Substances 0.000 description 2
- 239000002609 medium Substances 0.000 description 2
- 229940005483 opioid analgesics Drugs 0.000 description 2
- 201000008482 osteoarthritis Diseases 0.000 description 2
- 230000000144 pharmacologic effect Effects 0.000 description 2
- 239000006187 pill Substances 0.000 description 2
- 229920005862 polyol Polymers 0.000 description 2
- 150000003077 polyols Chemical class 0.000 description 2
- 239000000955 prescription drug Substances 0.000 description 2
- 239000003755 preservative agent Substances 0.000 description 2
- 230000002035 prolonged effect Effects 0.000 description 2
- 230000002441 reversible effect Effects 0.000 description 2
- YGSDEFSMJLZEOE-UHFFFAOYSA-N salicylic acid Chemical compound OC(=O)C1=CC=CC=C1O YGSDEFSMJLZEOE-UHFFFAOYSA-N 0.000 description 2
- 210000002966 serum Anatomy 0.000 description 2
- 159000000000 sodium salts Chemical class 0.000 description 2
- 239000007787 solid Substances 0.000 description 2
- 239000008107 starch Substances 0.000 description 2
- 235000019698 starch Nutrition 0.000 description 2
- 239000007939 sustained release tablet Substances 0.000 description 2
- 239000006188 syrup Substances 0.000 description 2
- 235000020357 syrup Nutrition 0.000 description 2
- 239000000454 talc Substances 0.000 description 2
- 229910052623 talc Inorganic materials 0.000 description 2
- 235000012222 talc Nutrition 0.000 description 2
- 229940124597 therapeutic agent Drugs 0.000 description 2
- 238000004448 titration Methods 0.000 description 2
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 2
- 238000010200 validation analysis Methods 0.000 description 2
- 235000015112 vegetable and seed oil Nutrition 0.000 description 2
- 239000008158 vegetable oil Substances 0.000 description 2
- 239000000080 wetting agent Substances 0.000 description 2
- SFLSHLFXELFNJZ-QMMMGPOBSA-N (-)-norepinephrine Chemical compound NC[C@H](O)C1=CC=C(O)C(O)=C1 SFLSHLFXELFNJZ-QMMMGPOBSA-N 0.000 description 1
- QIJRTFXNRTXDIP-UHFFFAOYSA-N (1-carboxy-2-sulfanylethyl)azanium;chloride;hydrate Chemical compound O.Cl.SCC(N)C(O)=O QIJRTFXNRTXDIP-UHFFFAOYSA-N 0.000 description 1
- IXPNQXFRVYWDDI-UHFFFAOYSA-N 1-methyl-2,4-dioxo-1,3-diazinane-5-carboximidamide Chemical compound CN1CC(C(N)=N)C(=O)NC1=O IXPNQXFRVYWDDI-UHFFFAOYSA-N 0.000 description 1
- IIZPXYDJLKNOIY-JXPKJXOSSA-N 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCC\C=C/C\C=C/C\C=C/C\C=C/CCCCC IIZPXYDJLKNOIY-JXPKJXOSSA-N 0.000 description 1
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 1
- IKBZAUYPBWFMDI-UHFFFAOYSA-N 5-bromo-4-methoxy-7-methyl-2,3-dihydro-1h-indene Chemical compound C1=C(Br)C(OC)=C2CCCC2=C1C IKBZAUYPBWFMDI-UHFFFAOYSA-N 0.000 description 1
- 244000215068 Acacia senegal Species 0.000 description 1
- 235000006491 Acacia senegal Nutrition 0.000 description 1
- 102000009027 Albumins Human genes 0.000 description 1
- 108010088751 Albumins Proteins 0.000 description 1
- 229920000856 Amylose Polymers 0.000 description 1
- 244000105624 Arachis hypogaea Species 0.000 description 1
- 239000004475 Arginine Substances 0.000 description 1
- 206010006187 Breast cancer Diseases 0.000 description 1
- 208000026310 Breast neoplasm Diseases 0.000 description 1
- 206010007559 Cardiac failure congestive Diseases 0.000 description 1
- 206010009944 Colon cancer Diseases 0.000 description 1
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 1
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 1
- XBPCUCUWBYBCDP-UHFFFAOYSA-N Dicyclohexylamine Chemical compound C1CCCCC1NC1CCCCC1 XBPCUCUWBYBCDP-UHFFFAOYSA-N 0.000 description 1
- 229940094659 Dopamine reuptake inhibitor Drugs 0.000 description 1
- 208000032928 Dyslipidaemia Diseases 0.000 description 1
- 206010013975 Dyspnoeas Diseases 0.000 description 1
- LVGKNOAMLMIIKO-UHFFFAOYSA-N Elaidinsaeure-aethylester Natural products CCCCCCCCC=CCCCCCCCC(=O)OCC LVGKNOAMLMIIKO-UHFFFAOYSA-N 0.000 description 1
- 206010014733 Endometrial cancer Diseases 0.000 description 1
- 206010014759 Endometrial neoplasm Diseases 0.000 description 1
- 239000001856 Ethyl cellulose Substances 0.000 description 1
- ZZSNKZQZMQGXPY-UHFFFAOYSA-N Ethyl cellulose Chemical compound CCOCC1OC(OC)C(OCC)C(OCC)C1OC1C(O)C(O)C(OC)C(CO)O1 ZZSNKZQZMQGXPY-UHFFFAOYSA-N 0.000 description 1
- 241001539473 Euphoria Species 0.000 description 1
- 206010015535 Euphoric mood Diseases 0.000 description 1
- 208000010201 Exanthema Diseases 0.000 description 1
- 206010016059 Facial pain Diseases 0.000 description 1
- 108010010803 Gelatin Proteins 0.000 description 1
- 239000004471 Glycine Substances 0.000 description 1
- 229920000084 Gum arabic Polymers 0.000 description 1
- 208000004547 Hallucinations Diseases 0.000 description 1
- 206010019280 Heart failures Diseases 0.000 description 1
- GVGLGOZIDCSQPN-PVHGPHFFSA-N Heroin Chemical compound O([C@H]1[C@H](C=C[C@H]23)OC(C)=O)C4=C5[C@@]12CCN(C)[C@@H]3CC5=CC=C4OC(C)=O GVGLGOZIDCSQPN-PVHGPHFFSA-N 0.000 description 1
- 244000043261 Hevea brasiliensis Species 0.000 description 1
- SQUHHTBVTRBESD-UHFFFAOYSA-N Hexa-Ac-myo-Inositol Natural products CC(=O)OC1C(OC(C)=O)C(OC(C)=O)C(OC(C)=O)C(OC(C)=O)C1OC(C)=O SQUHHTBVTRBESD-UHFFFAOYSA-N 0.000 description 1
- 241000282412 Homo Species 0.000 description 1
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 1
- 208000031226 Hyperlipidaemia Diseases 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- 102000004877 Insulin Human genes 0.000 description 1
- 108090001061 Insulin Proteins 0.000 description 1
- 206010022489 Insulin Resistance Diseases 0.000 description 1
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 1
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 1
- 239000004472 Lysine Substances 0.000 description 1
- 241000124008 Mammalia Species 0.000 description 1
- 229920000168 Microcrystalline cellulose Polymers 0.000 description 1
- 206010027951 Mood swings Diseases 0.000 description 1
- 208000000112 Myalgia Diseases 0.000 description 1
- MBBZMMPHUWSWHV-BDVNFPICSA-N N-methylglucamine Chemical compound CNC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO MBBZMMPHUWSWHV-BDVNFPICSA-N 0.000 description 1
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 1
- 102000003840 Opioid Receptors Human genes 0.000 description 1
- 108090000137 Opioid Receptors Proteins 0.000 description 1
- 208000026251 Opioid-Related disease Diseases 0.000 description 1
- 235000019483 Peanut oil Nutrition 0.000 description 1
- 239000004372 Polyvinyl alcohol Substances 0.000 description 1
- 206010060862 Prostate cancer Diseases 0.000 description 1
- 208000000236 Prostatic Neoplasms Diseases 0.000 description 1
- 206010038776 Retching Diseases 0.000 description 1
- 244000000231 Sesamum indicum Species 0.000 description 1
- 208000013738 Sleep Initiation and Maintenance disease Diseases 0.000 description 1
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 1
- 206010041349 Somnolence Diseases 0.000 description 1
- 235000021355 Stearic acid Nutrition 0.000 description 1
- 208000006011 Stroke Diseases 0.000 description 1
- 206010042434 Sudden death Diseases 0.000 description 1
- 208000012886 Vertigo Diseases 0.000 description 1
- 206010047513 Vision blurred Diseases 0.000 description 1
- 206010000059 abdominal discomfort Diseases 0.000 description 1
- 230000002159 abnormal effect Effects 0.000 description 1
- 235000010489 acacia gum Nutrition 0.000 description 1
- 230000004913 activation Effects 0.000 description 1
- 239000002671 adjuvant Substances 0.000 description 1
- 229940050528 albumin Drugs 0.000 description 1
- 229910052783 alkali metal Inorganic materials 0.000 description 1
- 125000005907 alkyl ester group Chemical group 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- 229940024606 amino acid Drugs 0.000 description 1
- 235000001014 amino acid Nutrition 0.000 description 1
- 150000001413 amino acids Chemical class 0.000 description 1
- 150000003863 ammonium salts Chemical class 0.000 description 1
- 238000000540 analysis of variance Methods 0.000 description 1
- 230000008485 antagonism Effects 0.000 description 1
- 239000005557 antagonist Substances 0.000 description 1
- 238000013459 approach Methods 0.000 description 1
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 1
- 206010003119 arrhythmia Diseases 0.000 description 1
- 239000003693 atypical antipsychotic agent Substances 0.000 description 1
- 229940127236 atypical antipsychotics Drugs 0.000 description 1
- 150000007514 bases Chemical class 0.000 description 1
- 230000008901 benefit Effects 0.000 description 1
- 239000000440 bentonite Substances 0.000 description 1
- 229910000278 bentonite Inorganic materials 0.000 description 1
- SVPXDRXYRYOSEX-UHFFFAOYSA-N bentoquatam Chemical compound O.O=[Si]=O.O=[Al]O[Al]=O SVPXDRXYRYOSEX-UHFFFAOYSA-N 0.000 description 1
- 230000015572 biosynthetic process Effects 0.000 description 1
- 230000036765 blood level Effects 0.000 description 1
- 210000004556 brain Anatomy 0.000 description 1
- 210000000481 breast Anatomy 0.000 description 1
- 229960004367 bupropion hydrochloride Drugs 0.000 description 1
- 229910000019 calcium carbonate Inorganic materials 0.000 description 1
- XAAHAAMILDNBPS-UHFFFAOYSA-L calcium hydrogenphosphate dihydrate Chemical compound O.O.[Ca+2].OP([O-])([O-])=O XAAHAAMILDNBPS-UHFFFAOYSA-L 0.000 description 1
- PASHVRUKOFIRIK-UHFFFAOYSA-L calcium sulfate dihydrate Chemical compound O.O.[Ca+2].[O-]S([O-])(=O)=O PASHVRUKOFIRIK-UHFFFAOYSA-L 0.000 description 1
- UBWYRXFZPXBISJ-UHFFFAOYSA-L calcium;2-hydroxypropanoate;trihydrate Chemical compound O.O.O.[Ca+2].CC(O)C([O-])=O.CC(O)C([O-])=O UBWYRXFZPXBISJ-UHFFFAOYSA-L 0.000 description 1
- ZHZFKLKREFECML-UHFFFAOYSA-L calcium;sulfate;hydrate Chemical compound O.[Ca+2].[O-]S([O-])(=O)=O ZHZFKLKREFECML-UHFFFAOYSA-L 0.000 description 1
- GJYLKIZKRHDRER-UHFFFAOYSA-N calcium;sulfuric acid Chemical compound [Ca].OS(O)(=O)=O GJYLKIZKRHDRER-UHFFFAOYSA-N 0.000 description 1
- 230000000747 cardiac effect Effects 0.000 description 1
- 239000012876 carrier material Substances 0.000 description 1
- 235000010980 cellulose Nutrition 0.000 description 1
- 239000001913 cellulose Substances 0.000 description 1
- 235000013339 cereals Nutrition 0.000 description 1
- 239000011248 coating agent Substances 0.000 description 1
- 238000000576 coating method Methods 0.000 description 1
- 208000029742 colonic neoplasm Diseases 0.000 description 1
- 235000009508 confectionery Nutrition 0.000 description 1
- 238000013270 controlled release Methods 0.000 description 1
- 239000011162 core material Substances 0.000 description 1
- 208000029078 coronary artery disease Diseases 0.000 description 1
- 235000012343 cottonseed oil Nutrition 0.000 description 1
- 239000002385 cottonseed oil Substances 0.000 description 1
- 235000019788 craving Nutrition 0.000 description 1
- 125000004186 cyclopropylmethyl group Chemical group [H]C([H])(*)C1([H])C([H])([H])C1([H])[H] 0.000 description 1
- 229960001305 cysteine hydrochloride Drugs 0.000 description 1
- 238000002716 delivery method Methods 0.000 description 1
- 230000001419 dependent effect Effects 0.000 description 1
- 229940096516 dextrates Drugs 0.000 description 1
- 229960002069 diamorphine Drugs 0.000 description 1
- 235000019700 dicalcium phosphate Nutrition 0.000 description 1
- 229940095079 dicalcium phosphate anhydrous Drugs 0.000 description 1
- XYYVYLMBEZUESM-UHFFFAOYSA-N dihydrocodeine Natural products C1C(N(CCC234)C)C2C=CC(=O)C3OC2=C4C1=CC=C2OC XYYVYLMBEZUESM-UHFFFAOYSA-N 0.000 description 1
- 201000010099 disease Diseases 0.000 description 1
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 1
- PXEDJBXQKAGXNJ-QTNFYWBSSA-L disodium L-glutamate Chemical compound [Na+].[Na+].[O-]C(=O)[C@@H](N)CCC([O-])=O PXEDJBXQKAGXNJ-QTNFYWBSSA-L 0.000 description 1
- 239000000221 dopamine uptake inhibitor Substances 0.000 description 1
- 239000008298 dragée Substances 0.000 description 1
- 230000004064 dysfunction Effects 0.000 description 1
- 210000005069 ears Anatomy 0.000 description 1
- 238000003379 elimination reaction Methods 0.000 description 1
- 239000003995 emulsifying agent Substances 0.000 description 1
- 210000004696 endometrium Anatomy 0.000 description 1
- 230000002708 enhancing effect Effects 0.000 description 1
- 210000003743 erythrocyte Anatomy 0.000 description 1
- CCIVGXIOQKPBKL-UHFFFAOYSA-M ethanesulfonate Chemical compound CCS([O-])(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-M 0.000 description 1
- 235000019325 ethyl cellulose Nutrition 0.000 description 1
- 229920001249 ethyl cellulose Polymers 0.000 description 1
- LVGKNOAMLMIIKO-QXMHVHEDSA-N ethyl oleate Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OCC LVGKNOAMLMIIKO-QXMHVHEDSA-N 0.000 description 1
- 229940093471 ethyl oleate Drugs 0.000 description 1
- 230000003203 everyday effect Effects 0.000 description 1
- 201000005884 exanthem Diseases 0.000 description 1
- 230000005284 excitation Effects 0.000 description 1
- 239000003925 fat Substances 0.000 description 1
- 239000013020 final formulation Substances 0.000 description 1
- 239000000796 flavoring agent Substances 0.000 description 1
- 235000019634 flavors Nutrition 0.000 description 1
- 235000013305 food Nutrition 0.000 description 1
- 235000012631 food intake Nutrition 0.000 description 1
- 235000003599 food sweetener Nutrition 0.000 description 1
- 239000003205 fragrance Substances 0.000 description 1
- 208000001130 gallstones Diseases 0.000 description 1
- 239000008273 gelatin Substances 0.000 description 1
- 229920000159 gelatin Polymers 0.000 description 1
- 239000007903 gelatin capsule Substances 0.000 description 1
- 235000019322 gelatine Nutrition 0.000 description 1
- 235000011852 gelatine desserts Nutrition 0.000 description 1
- 238000007429 general method Methods 0.000 description 1
- 230000002068 genetic effect Effects 0.000 description 1
- 239000011521 glass Substances 0.000 description 1
- 239000008103 glucose Substances 0.000 description 1
- 229960001031 glucose Drugs 0.000 description 1
- 239000008187 granular material Substances 0.000 description 1
- 238000005469 granulation Methods 0.000 description 1
- 230000003179 granulation Effects 0.000 description 1
- 239000008202 granule composition Substances 0.000 description 1
- 238000000227 grinding Methods 0.000 description 1
- 230000036541 health Effects 0.000 description 1
- 208000019622 heart disease Diseases 0.000 description 1
- 150000004677 hydrates Chemical class 0.000 description 1
- LLPOLZWFYMWNKH-CMKMFDCUSA-N hydrocodone Chemical compound C([C@H]1[C@H](N(CC[C@@]112)C)C3)CC(=O)[C@@H]1OC1=C2C3=CC=C1OC LLPOLZWFYMWNKH-CMKMFDCUSA-N 0.000 description 1
- 229960000240 hydrocodone Drugs 0.000 description 1
- OROGSEYTTFOCAN-UHFFFAOYSA-N hydrocodone Natural products C1C(N(CCC234)C)C2C=CC(O)C3OC2=C4C1=CC=C2OC OROGSEYTTFOCAN-UHFFFAOYSA-N 0.000 description 1
- 230000006872 improvement Effects 0.000 description 1
- 238000000099 in vitro assay Methods 0.000 description 1
- 238000005462 in vivo assay Methods 0.000 description 1
- 230000002401 inhibitory effect Effects 0.000 description 1
- CDAISMWEOUEBRE-GPIVLXJGSA-N inositol Chemical compound O[C@H]1[C@H](O)[C@@H](O)[C@H](O)[C@H](O)[C@@H]1O CDAISMWEOUEBRE-GPIVLXJGSA-N 0.000 description 1
- 229960000367 inositol Drugs 0.000 description 1
- 206010022437 insomnia Diseases 0.000 description 1
- 229940125396 insulin Drugs 0.000 description 1
- 229960004903 invert sugar Drugs 0.000 description 1
- 230000007794 irritation Effects 0.000 description 1
- 238000002372 labelling Methods 0.000 description 1
- 229960001375 lactose Drugs 0.000 description 1
- 230000002045 lasting effect Effects 0.000 description 1
- 239000007942 layered tablet Substances 0.000 description 1
- 235000010445 lecithin Nutrition 0.000 description 1
- 239000000787 lecithin Substances 0.000 description 1
- 229940067606 lecithin Drugs 0.000 description 1
- 238000012417 linear regression Methods 0.000 description 1
- 239000002502 liposome Substances 0.000 description 1
- 238000004811 liquid chromatography Methods 0.000 description 1
- 238000001294 liquid chromatography-tandem mass spectrometry Methods 0.000 description 1
- 239000006194 liquid suspension Substances 0.000 description 1
- 229910052744 lithium Inorganic materials 0.000 description 1
- 238000011068 loading method Methods 0.000 description 1
- 230000005923 long-lasting effect Effects 0.000 description 1
- 230000007774 longterm Effects 0.000 description 1
- 239000007937 lozenge Substances 0.000 description 1
- 229940037627 magnesium lauryl sulfate Drugs 0.000 description 1
- 239000000395 magnesium oxide Substances 0.000 description 1
- CPLXHLVBOLITMK-UHFFFAOYSA-N magnesium oxide Inorganic materials [Mg]=O CPLXHLVBOLITMK-UHFFFAOYSA-N 0.000 description 1
- 235000012245 magnesium oxide Nutrition 0.000 description 1
- 159000000003 magnesium salts Chemical class 0.000 description 1
- HBNDBUATLJAUQM-UHFFFAOYSA-L magnesium;dodecyl sulfate Chemical compound [Mg+2].CCCCCCCCCCCCOS([O-])(=O)=O.CCCCCCCCCCCCOS([O-])(=O)=O HBNDBUATLJAUQM-UHFFFAOYSA-L 0.000 description 1
- AXZKOIWUVFPNLO-UHFFFAOYSA-N magnesium;oxygen(2-) Chemical compound [O-2].[Mg+2] AXZKOIWUVFPNLO-UHFFFAOYSA-N 0.000 description 1
- 238000007726 management method Methods 0.000 description 1
- 229960001855 mannitol Drugs 0.000 description 1
- 201000000083 maturity-onset diabetes of the young type 1 Diseases 0.000 description 1
- 229910052751 metal Inorganic materials 0.000 description 1
- 239000002184 metal Substances 0.000 description 1
- 229940098779 methanesulfonic acid Drugs 0.000 description 1
- 239000008108 microcrystalline cellulose Substances 0.000 description 1
- 229940016286 microcrystalline cellulose Drugs 0.000 description 1
- 235000019813 microcrystalline cellulose Nutrition 0.000 description 1
- 239000008185 minitablet Substances 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 238000012544 monitoring process Methods 0.000 description 1
- BAVYZALUXZFZLV-UHFFFAOYSA-N mono-methylamine Natural products NC BAVYZALUXZFZLV-UHFFFAOYSA-N 0.000 description 1
- 235000013923 monosodium glutamate Nutrition 0.000 description 1
- 208000013465 muscle pain Diseases 0.000 description 1
- 208000010125 myocardial infarction Diseases 0.000 description 1
- 239000003887 narcotic antagonist Substances 0.000 description 1
- 229920003052 natural elastomer Polymers 0.000 description 1
- 229920001194 natural rubber Polymers 0.000 description 1
- 239000003367 nicotinic antagonist Substances 0.000 description 1
- 229910017604 nitric acid Inorganic materials 0.000 description 1
- 239000002767 noradrenalin uptake inhibitor Substances 0.000 description 1
- SFLSHLFXELFNJZ-UHFFFAOYSA-N norepinephrine Natural products NCC(O)C1=CC=C(O)C(O)=C1 SFLSHLFXELFNJZ-UHFFFAOYSA-N 0.000 description 1
- 229960002748 norepinephrine Drugs 0.000 description 1
- 229940127221 norepinephrine reuptake inhibitor Drugs 0.000 description 1
- 208000001797 obstructive sleep apnea Diseases 0.000 description 1
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 1
- OQCDKBAXFALNLD-UHFFFAOYSA-N octadecanoic acid Natural products CCCCCCCC(C)CCCCCCCCC(O)=O OQCDKBAXFALNLD-UHFFFAOYSA-N 0.000 description 1
- 201000005040 opiate dependence Diseases 0.000 description 1
- 239000000014 opioid analgesic Substances 0.000 description 1
- FJKROLUGYXJWQN-UHFFFAOYSA-N papa-hydroxy-benzoic acid Natural products OC(=O)C1=CC=C(O)C=C1 FJKROLUGYXJWQN-UHFFFAOYSA-N 0.000 description 1
- 239000011087 paperboard Substances 0.000 description 1
- 239000000312 peanut oil Substances 0.000 description 1
- 239000000825 pharmaceutical preparation Substances 0.000 description 1
- 239000000902 placebo Substances 0.000 description 1
- 229940068196 placebo Drugs 0.000 description 1
- 239000004033 plastic Substances 0.000 description 1
- 229920003023 plastic Polymers 0.000 description 1
- 229920002451 polyvinyl alcohol Polymers 0.000 description 1
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 1
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 1
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 1
- XAEFZNCEHLXOMS-UHFFFAOYSA-M potassium benzoate Chemical compound [K+].[O-]C(=O)C1=CC=CC=C1 XAEFZNCEHLXOMS-UHFFFAOYSA-M 0.000 description 1
- 230000003389 potentiating effect Effects 0.000 description 1
- 230000002028 premature Effects 0.000 description 1
- 230000008569 process Effects 0.000 description 1
- 238000012545 processing Methods 0.000 description 1
- 239000000651 prodrug Substances 0.000 description 1
- 229940002612 prodrug Drugs 0.000 description 1
- 239000000047 product Substances 0.000 description 1
- 210000002307 prostate Anatomy 0.000 description 1
- 230000002685 pulmonary effect Effects 0.000 description 1
- 238000011002 quantification Methods 0.000 description 1
- 206010037844 rash Diseases 0.000 description 1
- 230000000241 respiratory effect Effects 0.000 description 1
- 210000004189 reticular formation Anatomy 0.000 description 1
- 230000033764 rhythmic process Effects 0.000 description 1
- 229960004889 salicylic acid Drugs 0.000 description 1
- CDAISMWEOUEBRE-UHFFFAOYSA-N scyllo-inosotol Natural products OC1C(O)C(O)C(O)C(O)C1O CDAISMWEOUEBRE-UHFFFAOYSA-N 0.000 description 1
- 238000000926 separation method Methods 0.000 description 1
- 229940126570 serotonin reuptake inhibitor Drugs 0.000 description 1
- 239000003772 serotonin uptake inhibitor Substances 0.000 description 1
- 239000008159 sesame oil Substances 0.000 description 1
- 235000011803 sesame oil Nutrition 0.000 description 1
- 208000026775 severe diarrhea Diseases 0.000 description 1
- 230000035946 sexual desire Effects 0.000 description 1
- 231100000046 skin rash Toxicity 0.000 description 1
- 201000002859 sleep apnea Diseases 0.000 description 1
- 239000012748 slip agent Substances 0.000 description 1
- 206010041232 sneezing Diseases 0.000 description 1
- 239000011734 sodium Substances 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 235000010413 sodium alginate Nutrition 0.000 description 1
- 239000000661 sodium alginate Substances 0.000 description 1
- 229940005550 sodium alginate Drugs 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- 229940073490 sodium glutamate Drugs 0.000 description 1
- RYYKJJJTJZKILX-UHFFFAOYSA-M sodium octadecanoate Chemical class [Na+].CCCCCCCCCCCCCCCCCC([O-])=O RYYKJJJTJZKILX-UHFFFAOYSA-M 0.000 description 1
- 239000012439 solid excipient Substances 0.000 description 1
- 239000002904 solvent Substances 0.000 description 1
- 239000000600 sorbitol Substances 0.000 description 1
- 235000010356 sorbitol Nutrition 0.000 description 1
- 239000007921 spray Substances 0.000 description 1
- 230000007480 spreading Effects 0.000 description 1
- 238000003892 spreading Methods 0.000 description 1
- 239000003381 stabilizer Substances 0.000 description 1
- 238000007619 statistical method Methods 0.000 description 1
- 239000008117 stearic acid Substances 0.000 description 1
- 210000002784 stomach Anatomy 0.000 description 1
- 238000003860 storage Methods 0.000 description 1
- 230000002459 sustained effect Effects 0.000 description 1
- 239000003765 sweetening agent Substances 0.000 description 1
- 238000004885 tandem mass spectrometry Methods 0.000 description 1
- 238000012360 testing method Methods 0.000 description 1
- 230000004797 therapeutic response Effects 0.000 description 1
- 230000035922 thirst Effects 0.000 description 1
- 230000003867 tiredness Effects 0.000 description 1
- 208000016255 tiredness Diseases 0.000 description 1
- LLPOLZWFYMWNKH-UHFFFAOYSA-N trans-dihydrocodeinone Natural products C1C(N(CCC234)C)C2CCC(=O)C3OC2=C4C1=CC=C2OC LLPOLZWFYMWNKH-UHFFFAOYSA-N 0.000 description 1
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 1
- 208000001072 type 2 diabetes mellitus Diseases 0.000 description 1
- 231100000889 vertigo Toxicity 0.000 description 1
- 239000001993 wax Substances 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2004—Excipients; Inactive ingredients
- A61K9/2022—Organic macromolecular compounds
- A61K9/205—Polysaccharides, e.g. alginate, gums; Cyclodextrin
- A61K9/2054—Cellulose; Cellulose derivatives, e.g. hydroxypropyl methylcellulose
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/13—Amines
- A61K31/135—Amines having aromatic rings, e.g. ketamine, nortriptyline
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/13—Amines
- A61K31/135—Amines having aromatic rings, e.g. ketamine, nortriptyline
- A61K31/137—Arylalkylamines, e.g. amphetamine, epinephrine, salbutamol, ephedrine or methadone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/47—Quinolines; Isoquinolines
- A61K31/485—Morphinan derivatives, e.g. morphine, codeine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2004—Excipients; Inactive ingredients
- A61K9/2022—Organic macromolecular compounds
- A61K9/2031—Organic macromolecular compounds obtained otherwise than by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyethylene glycol, polyethylene oxide, poloxamers
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2072—Pills, tablets, discs, rods characterised by shape, structure or size; Tablets with holes, special break lines or identification marks; Partially coated tablets; Disintegrating flat shaped forms
- A61K9/2086—Layered tablets, e.g. bilayer tablets; Tablets of the type inert core-active coat
- A61K9/209—Layered tablets, e.g. bilayer tablets; Tablets of the type inert core-active coat containing drug in at least two layers or in the core and in at least one outer layer
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/04—Centrally acting analgesics, e.g. opioids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
Landscapes
- Health & Medical Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- Public Health (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Epidemiology (AREA)
- Engineering & Computer Science (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Organic Chemistry (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Emergency Medicine (AREA)
- Child & Adolescent Psychology (AREA)
- Neurosurgery (AREA)
- Neurology (AREA)
- Diabetes (AREA)
- Hematology (AREA)
- Obesity (AREA)
- Biomedical Technology (AREA)
- Pain & Pain Management (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicinal Preparation (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
Abstract
Una forma de dosificación que comprende una formulación de liberación sostenida de la naltrexona, o una sal farmacéuticamente aceptable del mismo y una formulación de liberación sostenida de bupropión o una sal farmacéuticamente aceptable del mismo, en donde la forma de dosificación es una forma de dosificación oral, en la que la forma de dosificación proporciona una velocidad de liberación in vitro de la naltrexona de menos de 80% o menos de 70% en aproximadamente 1 hora, de menos de 90% o menos de 80% en aproximadamente 2 horas, o de menos de 98% en aproximadamente 4 horas, como se determina usando United States Pharmacopeia 24ª edición (USP 24) Aparato 2 de ensayo de disolución que tiene una velocidad de rotación del husillo de 100 rpm y un medio de disolución de agua, a 37°C.
Description
DESCRIPCIÓN
Formulación de liberación sostenida de naltrexona
Campo de la invención
[0001] La presente invención se refiere a composiciones farmacéuticas como se define en las reivindicaciones que comprenden antagonistas del receptor de opioides de liberación sostenida.
Descripción de la técnica relacionada
[0002] WO 2004/071423 se refiere a un método de tratamiento del dolor en un paciente que comprende administrar por vía oral un antagonista opioide.
[0003] WO 83/03197 se refiere a un método para controlar la dismotilidad gastrointestinal en seres humanos mediante la administración de antagonistas de los opioides.
[0004] US 2003/068371 se refiere a una forma de dosificación oral que comprende una cantidad terapéuticamente eficaz de un analgésico opioide, un antagonista opioide y uno o más excipientes farmacéuticamente aceptables.
[0005] US 2002/198227 se refiere a un método para frenar las ansias de la dieta en un paciente, típicamente un paciente diabético o la obesidad, mediante la administración de una dosis baja de naltrexona para el paciente.
[0006] WO2005/032555 se refiere a una composición farmacéutica que comprende de aproximadamente 5 a aproximadamente 20 mg de hidrocodona o una sal farmacéuticamente aceptable del mismo y de 0,055 a alrededor de 0,56 mg de naltrexona o sal farmacéuticamente aceptable del mismo.
[0007] US 6306436 se refiere a composiciones farmacéuticas de hidrocloruro de bupropión estabilizado.
[0008] La obesidad es un importante problema de salud en las sociedades occidentales. Se estima que cerca de 97 millones de adultos en los Estados Unidos tienen sobrepeso o son obesos. Los estudios epidemiológicos han demostrado que el aumento de grados de sobrepeso y la obesidad son importantes predictores de la disminución de la esperanza de vida. La obesidad causa o exacerba muchos problemas de salud, tanto de forma independiente y en asociación con otras enfermedades. Los problemas médicos asociados con la obesidad, que puede ser grave y potencialmente mortal, incluyen la hipertensión; Diabetes mellitus tipo 2; concentraciones elevadas de insulina en plasma; resistencia a la insulina; dislipidemias; hiperlipidemia; endometrio, mama, próstata y cáncer de colon; osteoartritis; complicaciones respiratorias, tales como apnea obstructiva del sueño; colelitiasis; cálculos biliares; arterioscelerosis; enfermedad del corazón; ritmos cardíacos anormales; y arritmias cardiacas (Kopelman, PG, Nature 404, 635-643 (2000)). La obesidad se asocia más con la muerte prematura y con un aumento significativo de la mortalidad y morbilidad por accidente cerebrovascular, infarto de miocardio, insuficiencia cardíaca congestiva, enfermedad coronaria y muerte súbita.
[0009] La obesidad es a menudo tratada por animar a los pacientes a perder peso mediante la reducción de la ingesta de alimentos o mediante el aumento de su nivel de ejercicio y por lo tanto el aumento de su producción de energía. Una pérdida de peso sostenida de 5% a 10% del peso corporal se ha demostrado que mejora las comorbilidades asociadas con la obesidad, como la diabetes y la hipertensión, y puede conducir a la mejora de las condiciones relacionadas con la obesidad tales como la osteoartritis, apnea del sueño y pulmonar y la disfunción cardiaca.
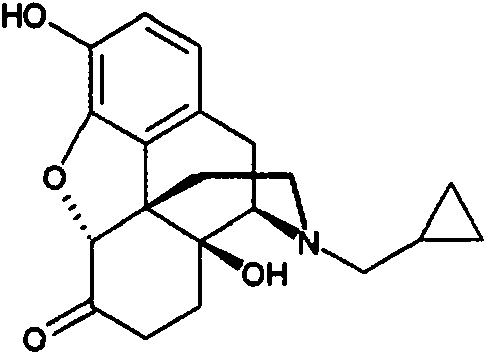
[0010] La naltrexona, tiene el nombre químico (17-(ciclopropilmetil)-4,5a-epoxi-3,14-dihidroximorfinan-6-ona) y la estructura química se muestra a continuación, es un antagonista del receptor opioide utilizado principalmente en la gestión de la dependencia del alcohol y dependencia de opiáceos.
[0011] La naltrexona para la administración oral ha estado disponible comercialmente durante un número de años a partir de diversas fuentes como la sal clorhidrato, clorhidrato de naltrexona, por ejemplo, bajo los nombres comerciales REVIA™ (50 mg) y DEPADE™ (25 mg, 50 mg y 100 mg). Las formas aprobadas de naltrexona oral son formulaciones de liberación inmediata que son eficaces incluso cuando se dosifica tan poca frecuencia como una vez cada 72 horas. Por ejemplo, la etiqueta de la marca DEPADE® de clorhidrato de naltrexona indica que la naltrexona es un antagonista
opioide potente con un efecto farmacológico prolongado (24 a 72 horas) y recomienda una dosis de 50 mg una vez al día. La etiqueta DEPADE® da a conocer que los estudios clínicos indican que 50 mg de clorhidrato de naltrexona bloquearán los efectos farmacológicos de 25 mg de heroína administrada por vía intravenosa para períodos de hasta 24 horas. La etiqueta DEPADE® indica que otros datos sugieren que la duplicación de la dosis de clorhidrato de naltrexona proporciona bloqueo durante aproximadamente 48 horas, y la triplicación de la dosis de clorhidrato de naltrexona proporciona bloqueo durante aproximadamente 72 horas. Por lo tanto, a pesar de alcanzar un pico de concentración sérica rápidamente (Tmax de aproximadamente 1 hora) después de la administración oral, la forma de liberación inmediata de naltrexona tiene efectos relativamente duraderos.
[0012] Los efectos a largo plazo de la forma de liberación inmediata de naltrexona oral se pueden utilizar para fomentar el cumplimiento del paciente mediante la utilización de una frecuencia de dosificación que es menor que una vez por día, por ejemplo, cada dos días o cada tres días. Por ejemplo, la etiqueta DEPADE® indica que un enfoque flexible para un régimen de dosificación se puede emplear para mejorar el cumplimiento. Por lo tanto, la etiqueta DEPADE® da a conocer que los pacientes pueden recibir 50 mg de clorhidrato de naltrexona cada día de la semana con una dosis de 100 mg en sábado o los pacientes pueden recibir 100 mg cada dos días, o 150 mg cada tercer día. La etiqueta DEPADE® se refiere a los estudios clínicos publicados en la literatura que han empleado el siguiente régimen de dosificación: 100 mg el lunes, 100 mg el miércoles, y 150 mg el viernes. Así, el uso de la forma oral de liberación inmediata permite que el paciente tome una dosis relativamente grande de naltrexona en un momento en que la tentación de abusar del alcohol u opioides puede ser menor (por ejemplo, durante la semana), con los efectos duraderos hasta un momento en que la tentación puede ser mayor (por ejemplo, durante el fin de semana)
[0013] La etiqueta DEPADE® indica que la naltrexona no se ha demostrado que causa aumentos significativos de quejas en ensayos controlados con placebo en pacientes que se sabe que están libres de opioides durante más de 7 a 10 días. Aunque un subconjunto de la población de pacientes informa náuseas después de la administración inicial de 25 mg o 50 mg de dosis de la forma oral de liberación inmediata de naltrexona, la náusea a menudo disminuye cuando aquellos pacientes desarrollan una tolerancia. La etiqueta DEPADE® indica que, en un estudio de seguridad de etiqueta abierta con aproximadamente 570 individuos con alcoholismo que reciben naltrexona, las siguientes reacciones adversas de nueva aparición se produjeron en 2% o más de los pacientes: náuseas (10%), dolor de cabeza (7%) , mareo (4%), nerviosismo (4%), fatiga (4%), insomnio (3%), vómitos (3%), ansiedad (2%) y somnolencia (2%). Náusea moderada a severa se informó por 18 pacientes (15%) en un estudio de etiqueta abierta de 10 semanas de la naltrexona entre los participantes dependientes del alcohol, que implica una dosis inicial de 25 mg seguida de una dosis de 50 mg al día durante 10 semanas (O'Malley, S.S. J. Clin. Psychopharmacol. 2000 Feb; 20 (1): 69-76). Ocho de los dieciocho pacientes que experimentaron náuseas moderadas a severas abandonaron el tratamiento, náuseas resueltas dentro de una semana por cinco sujetos y dentro de dos semanas para cuatro, y continuó de vez en cuando a lo largo del período de diez semanas para un sujeto. Los autores relacionaron las náuseas moderadas a severas de pobre cumplimiento de la medicación y mayor consumo de alcohol por los pacientes durante el tratamiento.
[0014] Parece que mientras que algunos pacientes experimentan efectos adversos tras la administración de 25 mg o 50 mg de dosis de naltrexona oral de liberación inmediata, los efectos adversos a menudo desaparecen en un período relativamente corto de tiempo y muchos pacientes son capaces de tolerar las dosis orales significativamente más altos, por ejemplo, 100 mg o 150 mg. Una forma inyectable una vez al mes de naltrexona está disponible comercialmente bajo el nombre comercial Vivitrol® para el tratamiento de alcoholismo. La información de prescripción para Vivitrol® indica que los pacientes deben ser informados de que pueden experimentar náuseas después de la inyección inicial de la naltrexona Vivitrol®; que estos episodios de náuseas tienden a ser leves y desaparecen a los pocos días después de la inyección, y que los pacientes son menos propensos a experimentar náuseas en las inyecciones posteriores.
[0015] Patente de Estados Unidos N° de publicación 2004/0254208 A1 describe el uso de naltrexona en combinación con otros compuestos para afectar la pérdida de peso, pero no da a conocer ningún efecto adverso particulares asociados con las combinaciones.
SUMARIO DE LA INVENCIÓN
[0016] La invención proporciona una forma de dosificación que comprende una formulación de liberación sostenida de la naltrexona, o una sal farmacéuticamente aceptable del mismo y una formulación de liberación sostenida de bupropión o una sal farmacéuticamente aceptable del mismo, en el que la forma de dosificación es una forma de dosificación oral, en la que la forma de dosificación proporciona una velocidad de liberación in vitro de la naltrexona de menos de 80% o menos de 70% en aproximadamente 1 hora, de menos de 90% o menos de 80% en aproximadamente 2 horas, o de menos de 98% en aproximadamente 4 horas, como se determina usando United States Pharmacopeia 24a edición (USP 24) Aparato 2 de ensayo de disolución que tiene una velocidad de rotación del husillo de 100 rpm y un medio de disolución de agua, a 37°C.
[0017] Una incidencia inesperadamente alta de efectos adversos asociados con la co-administración de naltrexona con bupropión y/o la fluoxetina ahora se ha identificado como un problema. Por ejemplo, durante un ensayo clínico descrito en mayor detalle a continuación, la incidencia de eventos adversos de gravedad moderada asociada con la co-administración de naltrexona con liberación inmediata con bupropión fue 30,7%. Esta incidencia de efectos
adversos fue significativamente mayor que la esperada basándose en la incidencia típica de los efectos adversos asociados con la administración de liberación inmediata de naltrexona solo o bupropion solo.
[0018] En una realización, las formulaciones de naltrexona de liberación sostenida tal como se definen en las reivindicaciones ahora han sido desarrollados que proporcionan una solución a este problema.
[0019] Una forma de realización proporciona una forma de dosificación oral tal como se define en las reivindicaciones, que comprende una formulación de naltrexona de liberación sostenida que se formula para proporcionar una reducción de al menos un efecto adverso, en el que el efecto adverso se asocia con co-administración de una formulación de naltrexona de liberación inmediata y un segundo compuesto que es una formulación de liberación sostenida del inhibidor de la recaptación de monoaminas bupropion. También se describe aquí un método de administración de naltrexona, que comprende administrar una formulación de naltrexona de liberación sostenida a un sujeto, por ejemplo, de una manera que sea eficaz para causar pérdida de peso y/o inhibir el aumento de peso.
[0020] Otra realización proporciona una forma de dosificación unitaria oral tal como se define en las reivindicaciones, que comprende una formulación de naltrexona de liberación sostenida que es eficaz para proporcionar, después de la administración, que comprende un perfil de concentración plasmática in vivo en al menos uno seleccionado a partir de:
(a) un naltrexona Cmax que es de aproximadamente 80% o menos de la naltrexona Cmax de REVIA™ clorhidrato de naltrexona de liberación inmediata, y una naltrexona AUCúltimo es decir, en el intervalo de aproximadamente 80% a aproximadamente 125% de la naltrexona AUCúltimo de REVIA™ clorhidrato de naltrexona de liberación inmediata; y
(b) un 6-beta naltrexol Cmax que es de aproximadamente 80% o menos de la 6-beta naltrexol Cmax de clorhidrato de naltrexona de liberación inmediata REVIA™, y un 6-beta naltrexol AUCúltimo es decir, en el intervalo de aproximadamente 80% a aproximadamente 125% de la 6-beta naltrexol AUCúltimo de REVIA™ clorhidrato de naltrexona de liberación inmediata.
[0021] Otra realización proporciona una forma de dosificación oral tal como se define en las reivindicaciones, que comprende naltrexona o una sal farmacéuticamente aceptable del mismo y una composición de vehículo de liberación sostenida, en donde la forma de dosificación oral proporciona una velocidad de liberación in vitro de la naltrexona o sal farmacéuticamente aceptable del mismo de menos de aproximadamente 90% en aproximadamente 4 horas.
[0022] También se describe aquí un método para administrar una formulación de naltrexona de liberación sostenida como se describe en el presente documento, por ejemplo, un método de administración de naltrexona, que comprende administrar una formulación de naltrexona de liberación sostenida a un sujeto en una cantidad que es eficaz para proporcionar un perfil de concentración de plasma in vivo que comprende al menos uno seleccionado de entre:
(a) una naltrexona Cmax que es de aproximadamente 80% o menos de la naltrexona Cmax de REVIA™ clorhidrato de naltrexona de liberación inmediata, y una naltrexona AUCúltimo es decir, en el intervalo de aproximadamente 80% a aproximadamente 125% de la naltrexona AUCúltimo de REVIA™ clorhidrato de naltrexona de liberación inmediata; y
(b) un 6-beta naltrexol Cmax que es de aproximadamente 80% o menos de la 6-beta naltrexol Cmax de clorhidrato de naltrexona de liberación inmediata REVIA™, y un 6-beta naltrexol AUCúltimo es decir, en el intervalo de aproximadamente 80% a aproximadamente 125% de la 6-beta naltrexol AUCúltimo de REVIA™ clorhidrato de naltrexona de liberación inmediata.
[0023] En una realización, una forma de dosificación como se describe en las reivindicaciones es para uso en el tratamiento de una condición relacionada con el peso, por ejemplo, para causar pérdida de peso y/o inhibir el aumento de peso.
[0024] Estas y otras realizaciones se describen en mayor detalle a continuación.
BREVE DESCRIPCIÓN DE LOS DIBUJOS
[0025]
La Figura 1 muestra el perfil de disolución de comprimidos de liberación sostenida de 5 mg de naltrexona que contienen óxido de polietileno.
La Figura 2 muestra el perfil de disolución de comprimidos de liberación sostenida de 5 mg de naltrexona que contienen hidroxipropilmetilcelulosa.
La Figura 3 muestra el perfil de disolución de comprimidos de naltrexona de liberación sostenida y el bupropión contienen hidroxipropilmetilcelulosa.
La Figura 4 muestra las curvas de tiempo de concentración de plasma individuales de naltrexona en sujetos que recibieron naltrexona IR.
Figura 5 muestra las curvas de tiempo de concentración de plasma individuales de naltrexona en sujetos que
recibieron naltrexona SR.
La Figura 6 muestra curvas de tiempo de concentración de plasma individuales de 6-beta naltrexol en sujetos que recibieron naltrexona IR.
La Figura 7 muestra curvas de tiempo de concentración de plasma individuales de 6-beta naltrexol en sujetos que recibieron naltrexona SR.
Las Figuras 8A y 8B muestran las curvas medias de tiempo de concentración en plasma de naltrexona en todos los sujetos que recibieron naltrexona SR (círculos) o naltrexona IR (cuadrados) a través de dos escalas de tiempo.
Figuras 9A y 9B muestran curvas medias de tiempo de concentración plasmática de 6-beta naltrexol en todos los sujetos que recibieron naltrexona SR (círculos) o naltrexona IR (cuadrados) a través de dos escalas de tiempo.
La Figura 10 es un esquema que ilustra la población de sujetos que informaron de náuseas y vómitos usando el Escala de Valoración Eventos Adversos UKU.
La Figura 11 muestra las concentraciones plasmáticas medias de naltrexona en sujetos que recibieron naltrexona IR en combinación con bupropión SR ( • ) o naltrexona SR en combinación con bupropión SR (■)
La Figura 12 muestra las concentraciones plasmáticas medias de 6-beta naltrexol en sujetos que recibieron naltrexona IR en combinación con bupropión SR ( • ) o naltrexona SR en combinación con bupropión SR (■)
La Figura 13 muestra las concentraciones plasmáticas mínimas medias de naltrexona en sujetos que recibieron naltrexona IR en combinación con bupropión SR ( • ) o naltrexona SR en combinación con bupropión SR (■).
La Figura 14 muestra las concentraciones plasmáticas mínimas medias de 6-beta naltrexol en sujetos que recibieron naltrexona IR en combinación con bupropión SR ( • ) o naltrexona SR en combinación con bupropión SR (■).
La Figura 15 muestra las concentraciones plasmáticas medias de bupropión en los sujetos que recibieron bupropión SR en combinación con naltrexona SR ( • ) o en combinación con naltrexona IR (■).
La Figura 16 muestra las concentraciones plasmáticas medias de la hidroxibupropión metabolito de bupropión en los sujetos que recibieron bupropión SR en combinación con naltrexona SR ( • ) o en combinación con naltrexona IR (■).
La Figura 17 muestra las concentraciones plasmáticas medias de la threohydroxybupropion metabolito de bupropión en los sujetos que recibieron bupropión SR en combinación con naltrexona SR ( • ) o en combinación con naltrexona IR (■).
La Figura 18 muestra las concentraciones plasmáticas medias de la eritrohidroxibupropion metabolito de bupropión en los sujetos que recibieron bupropión SR en combinación con naltrexona SR ( • ) o en combinación con naltrexona IR (■).
DESCRIPCION DETALLADA DE LAS REALIZACIONES PREFERIDAS
[0026] Diversas realizaciones proporcionan una forma de dosificación oral tal como se define en las reivindicaciones que comprende una formulación de naltrexona de liberación sostenida. Co-administración de una formulación de naltrexona de liberación sostenida con liberación sostenida de bupropión puede proporcionar una reducción de al menos un efecto adverso en comparación con la co-administración de una formulación de naltrexona de liberación inmediata con liberación sostenida de bupropión. El al menos un efecto adverso puede incluir náusea.
[0027] La forma de dosificación oral puede proporcionar una naltrexona Cmax y/o un 6-beta naltrexol Cmax que es de aproximadamente 80% o menos de la naltrexona Cmax y/o 6-beta naltrexol Cmax de REVIA™ clorhidrato de naltrexona de liberación inmediata. La forma de dosificación oral puede proporcionar unanaltrexona AUCúltimo y/o un 6-beta naltrexol AUCúltimo es decir, en el intervalo de aproximadamente 80% a aproximadamente 125% de la naltrexona AUCúltimo y/o 6-beta naltrexol AUCúltimo de REVIA™ clorhidrato de naltrexona de liberación inmediata.
Definiciones
[0028]
El término "naltrexona" puede ser utilizado de manera general en el presente documento para referirse a una base libre de naltrexona, una sal de naltrexona farmacéuticamente aceptable (incluyendo hidratos y formas anhidras, por ejemplo, dihidrato de clorhidrato de naltrexona y clorhidrato de naltrexona anhidro), un metabolito de la naltrexona, un isómero naltrexona, un profármaco naltrexona o mezclas de los mismos. La referencia aquí a "naltrexona" se entenderá por abarcar todas estas formas, a menos que el contexto indique claramente lo contrario.
El término "sal farmacéuticamente aceptable" se refiere a una formulación de un compuesto que no causa irritación significativa a un organismo al que se administra y no abroga la actividad biológica y propiedades del compuesto. Las sales farmacéuticas de compuestos básicos se pueden obtener haciendo reaccionar el
compuesto con un ácido tal como ácido clorhídrico, ácido bromhídrico, ácido sulfúrico, ácido nítrico, ácido fosfórico, ácido metanosulfónico, ácido etanosulfónico, ácido p-toluenosulfónico, ácido salicílico y similares. Por ejemplo, clorhidrato de naltrexona es una sal farmacéuticamente aceptable de la naltrexona. Las sales farmacéuticas de los compuestos ácidos se pueden obtener haciendo reaccionar el compuesto con una base para formar una sal tal como una sal de amonio, una sal de metal alcalino, tal como una sal de sodio o una sal de potasio, una sal de metal alcalinotérreo, tal como calcio o una sal de magnesio, una sal de bases orgánicas tales como diciclohexilamina, N-metil-D-glucamina, tris(hidroximetil)metilamina, y sales de los mismos con aminoácidos tales como arginina, lisina, y similares.
El término "forma de dosificación oral", como se usa aquí, tiene su significado ordinario como es entendido por los expertos en la técnica y por lo tanto incluye, a modo de ejemplo no limitativo, una formulación de un fármaco o fármacos en una forma administrable a un humano. Las realizaciones ilustrativas de la invención se han descrito principalmente como dirigidas a formas de dosificación oral tales como comprimidos, núcleos, cápsulas, comprimidos oblongos y polvo suelto, pero también se contemplan otras formas de dosificación adecuadas tales como soluciones y suspensiones.
El término "liberación sostenida", como se usa aquí, tiene su significado ordinario tal como se entiende por los expertos en la técnica y por lo tanto incluye, a modo de ejemplo no limitativo, la liberación controlada de un fármaco desde una forma de dosificación durante un período prolongado de tiempo. Por ejemplo, en algunas realizaciones, formas de dosificación de liberación sostenida son aquellas que tienen una velocidad de liberación que es sustancialmente más larga que la de una forma de liberación inmediata comparable, por ejemplo, mayor que 125% de la velocidad de liberación de una forma de dosificación de liberación inmediata. El término "liberación inmediata", como se usa aquí, tiene su significado ordinario tal como se entiende por los expertos en la técnica y por lo tanto incluye, a modo de ejemplo no limitativo, la liberación de un fármaco desde una forma de dosificación en un periodo relativamente breve de tiempo después de la administración. Ejemplos de formas de dosificación de liberación inmediata de clorhidrato de naltrexona incluyen REVIA™ clorhidrato de naltrexona de liberación inmediata y DEPADE™ clorhidrato de naltrexona de liberación inmediata. En algunas realizaciones, formas de dosificación de liberación inmediata son aquellas que tienen una velocidad de liberación que es hasta e incluyendo 125% de la velocidad de liberación para uno o más de clorhidrato de liberación inmediata de naltrexona REVIA™ y DEPADE™ clorhidrato de naltrexona de liberación inmediata. El clorhidrato de naltrexona de liberación inmediata REVIA™ puede ser una forma de dosificación 50 mg. El clorhidrato de naltrexona de liberación inmediata DEPADE™ puede ser una forma de dosificación de 25 mg, 50 mg o 100 mg. Como se usa en el presente documento, una formulación de liberación inmediata puede referirse a REVIA™ clorhidrato de naltrexona de liberación inmediata. En algunas realizaciones, la dosificación de la formulación de liberación inmediata es sustancialmente la misma que una forma de dosificación de liberación sostenida descrita en el presente documento, mientras que en otras realizaciones no lo es.
El término "velocidad de liberación", como se usa aquí, tiene su significado ordinario tal como se entiende por los expertos en la técnica y por lo tanto incluye, a modo de ejemplo no limitativo, una característica relacionada con la cantidad de un ingrediente activo liberado por unidad de tiempo como definido por ensayos in vitro o in vivo. Una velocidad de liberación in vitro se determina por un "ensayo de disolución estándar," realizado de acuerdo con la Farmacopea de Estados Unidos edición 24a (2000) (USP 24), pp. 1941-1943, usando el Aparato 2 descrito en la misma a una velocidad de rotación del husillo de 100 rpm y un medio de disolución de agua, a 37°C, u otras condiciones de ensayo sustancialmente equivalentes a los mismos.
El término "perfil farmacocinético," como se usa aquí, tiene su significado ordinario tal como se entiende por los expertos en la técnica y por lo tanto incluye, a modo de ejemplo no limitativo, una característica de la curva resultante de la gráfica de la concentración de suero sanguíneo de un medicamento con el tiempo, después de la administración del fármaco a un sujeto. Por lo tanto un perfil farmacocinético incluye un parámetro farmacocinético o un conjunto de parámetros que pueden ser utilizados para caracterizar la farmacocinética de una forma de fármaco o de dosificación particular cuando se administra a una población adecuada de pacientes. Varios parámetros farmacocinéticos son conocidos por los expertos en la técnica, incluyendo el área bajo la concentración plasmática en sangre frente a la curva de tiempo (AUC) y la concentración máxima en plasma sanguíneo después de la administración (Cmáx). AUCúltimo indica el área bajo la concentración plasmática en sangre frente a la curva de tiempo desde el momento de la administración hasta el momento de la última concentración mensurable. Los parámetros farmacocinéticos se pueden medir de diversas maneras conocidas por los expertos en la técnica, por ejemplo, dosis única o en estado estacionario. Las diferencias en uno o más de los perfiles farmacocinéticos (por ejemplo, Cmáx) pueden indicar la distinción farmacocinética entre dos formulaciones.
[0029] Los expertos en la técnica entenderán que los parámetros farmacocinéticos pueden ser determinados por comparación con un estándar de referencia utilizando los métodos de ensayos clínicos conocidos y aceptados por los expertos en la técnica, por ejemplo, como se describe en los ejemplos expuestos en este documento. Ya que la farmacocinética de un fármaco puede variar de paciente a paciente, tales ensayos clínicos generalmente implican múltiples pacientes y análisis estadísticos apropiados de los datos resultantes (típicamente ANOVA al 90% de confianza). Las comparaciones de los parámetros farmacocinéticos son sobre una base ajustada de la dosis, como se entiende por los expertos en la técnica.
[0030] En diversas formas de realización relacionadas con las formulaciones de naltrexona de liberación sostenida
descritas en este documento, el patrón de referencia es una formulación de naltrexona de liberación inmediata. Los expertos en la técnica entenderán que una formulación de naltrexona de liberación inmediata apropiada para su uso como patrón de referencia en la determinación de los parámetros farmacocinéticos es la formulación de naltrexona de liberación inmediata de leyenda, ampliamente disponible comercialmente como la marca ReVia® de clorhidrato de naltrexona, o una formulación de liberación inmediata de naltrexona que se formula para que tenga un perfil farmacocinético que es sustancialmente similar a la marca ReVia® de clorhidrato de naltrexona. El gobierno de EE.UU. regula la manera en que los medicamentos de venta con receta pueden ser etiquetados y por lo tanto la referencia a la marca ReVia® de clorhidrato de naltrexona tiene un significado conocido, fijo y definido para los expertos en la técnica.
Co-administración de naltrexona y un segundo compuesto
[0031] En algunas realizaciones, se reconoce que la coadministración de liberación inmediata de naltrexona con un segundo compuesto como se define en las reivindicaciones puede estar asociada con al menos un efecto adverso. El al menos un efecto adverso puede ser de mayor severidad, duración y/o probabilidad de ocurrencia de lo esperado por administración separada de la naltrexona de liberación inmediata y del segundo compuesto como se define en las reivindicaciones. En algunas realizaciones, se cree que el segundo compuesto como se define en las reivindicaciones puede potenciar los efectos de naltrexona inducida en la zona gatillo quimiorreceptora (CTZ) y/o el centro del vómito, potenciando de ese modo un efecto adverso asociado con la administración de naltrexona. Mientras que el efecto adverso en general puede caracterizarse como tolerable cuando la naltrexona de liberación inmediata se administra solo, el efecto adverso potenciado puede caracterizarse como menos tolerable.
[0032] La co-administración de naltrexona de liberación sostenida con el segundo compuesto como se define en las reivindicaciones puede proporcionar una reducción de al menos un efecto adverso en comparación con la co administración de liberación inmediata de naltrexona con el segundo compuesto. La reducción puede incluir una disminución de la gravedad, duración y/o probabilidad de ocurrencia de al menos un efecto adverso. En algunos casos, por ejemplo, la participación de los pacientes individuales, tales reducciones pueden ser en comparación con los efectos adversos que se espera por un experto en la técnica en vista de los efectos secundarios conocidos de una forma de liberación inmediata, y por lo tanto no es necesario que el paciente realmente experimenta efectos secundarios de la forma de liberación inmediata con el fin de beneficiarse de estas reducciones en los efectos adversos. Si bien no se desea estar ligado a ninguna teoría en particular, la reducción en al menos un efecto adverso puede ser el resultado de una concentración en sangre pico más bajo de la naltrexona o un metabolito del mismo asociado con la administración de naltrexona de liberación sostenida en comparación con la que se asocia con la administración de liberación inmediata de naltrexona. Una forma de liberación sostenida de la naltrexona con la disolución durante un período más largo puede proporcionar excitación reducida, antagonismo y/o la inhibición a la zona de disparo de los quimiorreceptores y centro del vómito (VC) en el área postrema del cerebro. Además, formas de dosificación oral de liberación sostenida pueden resultar en una concentración más baja disponible para ser captado por los receptores opioides del estómago.
[0033] En algunas realizaciones, el al menos un efecto adverso se asocia principalmente con la administración del segundo compuesto como se define en las reivindicaciones. Co-administración de naltrexona de liberación sostenida puede reducir el al menos un efecto adverso. En otras realizaciones, el al menos un efecto adverso se asocia principalmente con la administración de la naltrexona de liberación inmediata. naltrexona de liberación sostenida puede proporcionar una reducción en el al menos un efecto adverso en comparación con la formulación de liberación inmediata, y la naltrexona de liberación sostenida puede entonces ser co-administrada con el segundo compuesto. En todavía otras realizaciones, el al menos un efecto adverso se asocia principalmente con la combinación del segundo compuesto y la naltrexona de liberación inmediata. El tipo de efecto adverso asociado con la combinación puede ser uno que también se asocia con la administración de naltrexona sola y/o por la administración del segundo compuesto solo. La gravedad, duración y/o probabilidad de ocurrencia de los efectos adversos pueden ser mayores cuando la naltrexona de liberación inmediata se administra conjuntamente con el segundo compuesto del que se esperaría de las administraciones separadas de liberación inmediata de naltrexona y del segundo compuesto. Co-administración de naltrexona de liberación sostenida y el segundo compuesto puede proporcionar una reducción en el al menos un efecto adverso en comparación con la co-administración de liberación inmediata de naltrexona y el segundo compuesto.
[0034] La naltrexona de liberación inmediata puede comprender REVIA™ clorhidrato de naltrexona de liberación inmediata. La dosis de naltrexona en la naltrexona de liberación inmediata puede ser sustancialmente similar a la de la naltrexona de liberación sostenida. La naltrexona de liberación inmediata es una que está sustancialmente libre de una composición de vehículo de liberación sostenida.
[0035] El segundo compuesto puede incluir un antidepresivo. El segundo compuesto puede incluir un inhibidor de la recaptación de monoaminas y/o un modulador de monoaminas. La monoamina puede incluir la norepinefrina, la dopamina y/o serotonina. El segundo compuesto puede incluir un inhibidor de la recaptación de dopamina y/o un modulador de la dopamina. La modulación de la dopamina puede ser un efecto de la modulación de la serotonina. El segundo compuesto puede incluir un inhibidor de la recaptación de serotonina y/o un modulador de la serotonina. El segundo compuesto puede incluir un compuesto que actúa sobre la zona gatillo quimiorreceptora (CTZ) y/o el centro
del vómito en la formación reticular medular lateral. El segundo es bupropión o una sal farmacéuticamente aceptable del mismo. La sal farmacéuticamente aceptable puede comprender una sal de hidrocloruro. El segundo compuesto es una formulación de liberación sostenida.
[0036] La naltrexona de liberación sostenida se puede proporcionar en una forma de dosificación oral. En algunas realizaciones, la forma de dosificación oral también incluye el segundo compuesto, mientras que en otras realizaciones, no lo hace.
[0037] La co-administración de naltrexona de liberación inmediata o la naltrexona de liberación sostenida con el segundo compuesto, es decir bupropión de liberación sostenida, puede incluir la administración de una forma de dosificación oral que comprende tanto la naltrexona como el segundo compuesto. La co-administración puede incluir la administración de la naltrexona y el segundo compuesto separado, pero sustancialmente al mismo tiempo. La co administración puede incluir la administración de la naltrexona y el segundo compuesto en diferentes momentos (por ejemplo, en diferentes programas de dosificación), pero de tal manera que el plasma sanguíneo incluye simultáneamente las concentraciones de naltrexona o un metabolito de la naltrexona por encima de un primer umbral y concentraciones para el segundo compuesto o para metabolitos del segunda compuesto por encima de un segundo umbral. Estos umbrales pueden ser el nivel detectable mínimo (por ejemplo, cero). Esta última co-administración puede ser adecuada, por ejemplo, si el Tmáx in vivo o la velocidad de disolución de la formulación de naltrexona es sustancialmente diferente de la del segundo compuesto.
[0038] El al menos un efecto adverso puede incluir un efecto asociado con la zona de activación de los quimiorreceptores (CTZ) y/o el centro del vómito. En algunas realizaciones, el al menos un efecto adverso incluye uno o más de náuseas, arcadas, mareos, malestar estomacal, vértigo y vómitos. El al menos un efecto adverso puede incluir uno o más de la ansiedad, nerviosismo, dolor de cabeza, dificultad para dormir, estornudos, congestión nasal, dolor muscular, disminución de la función o deseo sexual, visión borrosa, sed, zumbido en los oídos, debilidad, cansancio, piel sarpullido, confusión, cambios de humor, alucinaciones, diarrea severa y dificultad para respirar.
[0039] El al menos un efecto adverso puede estar asociado con un tratamiento de pérdida de peso. El tratamiento de pérdida de peso puede comprender, por ejemplo, la administración de uno o más de un antipsicótico atípico, un antidepresivo, un inhibidor de la recaptación de monoamina, un modulador de monoamina, un inhibidor de la recaptación de norepinefrina, un inhibidor de la recaptación de dopamina, y/o un antagonista nicotínico. El tratamiento de la pérdida de peso puede comprender la administración de bupropión o una sal farmacéuticamente aceptable del mismo y/o la fluoxetina o una sal farmacéuticamente aceptable del mismo. El tratamiento de pérdida de peso puede comprender la administración de naltrexona. En algunas realizaciones, la forma farmacéutica de naltrexona de liberación sostenida da como resultado la reducción de uno o más efectos adversos atribuidos, parcial o totalmente, a uno o más ingredientes activos no naltrexona de un tratamiento de pérdida de peso. En algunas realizaciones, una forma de dosificación de naltrexona de liberación sostenida da como resultado la reducción de uno o más efectos adversos atribuidos, parcial o totalmente, a principios activos de naltrexona de un tratamiento de pérdida de peso.
Formulaciones de perfil farmacocinético de naltrexona de liberación sostenida
[0040] En algunas realizaciones, una formulación de naltrexona de liberación sostenida tal como se define en las reivindicaciones proporciona un perfil farmacocinético que es farmacocinéticamente distinto de una formulación de naltrexona de liberación inmediata (por ejemplo, REVIA™ clorhidrato de naltrexona de liberación inmediata). La distinción farmacocinética puede ser debido a una diferencia entre los valores Cmax asociados con las formulaciones de liberación inmediata y de liberación sostenida. La formulación de naltrexona de liberación sostenida puede proporcionar una naltrexona Cmax y/o un 6-beta naltrexol Cmax que es de aproximadamente 80% o menos de la naltrexona Cmax y/o 6-beta naltrexol Cmax de una formulación de naltrexona de liberación inmediata (por ejemplo, REVIA™ de clorhidrato de naltrexona de liberación inmediata).
[0041] En algunas realizaciones, la formulación de naltrexona de liberación sostenida tal como se define en las reivindicaciones proporciona una naltrexona AUCúltimo y/o un 6-beta naltrexol AUCúltimo que sustancialmente similar a (por ejemplo, entre aproximadamente 80% a aproximadamente 125% de) una naltrexona AUCúltimo y/o un 6-beta naltrexol AUCúltimo de una formulación de naltrexona de liberación inmediata (por ejemplo, REVIA™ de clorhidrato de naltrexona de liberación inmediata).
[0042] En algunas realizaciones, la dosis de naltrexona de la formulación de naltrexona de liberación inmediata es sustancialmente la misma que la dosis de naltrexona de la formulación de naltrexona de liberación sostenida. En algunas realizaciones en las que un valor de Cmax difiere entre el de liberación inmediata y formulaciones de liberación sostenida, la dosis de naltrexona de la formulación de liberación sostenida naltrexona se identifica como una que proporciona una naltrexona AUCúltimo y/o un 6-beta naltrexol AUCúltimo que es sustancialmente similar a (por ejemplo, entre aproximadamente 80% a aproximadamente 125% de) una naltrexona AUCúltimo y/o un 6-beta naltrexol AUCúltimo de la formulación de naltrexona de liberación sostenida.
[0043] El perfil farmacocinético puede estar relacionado con una eficacia y/o un efecto secundario reducido producido
por la forma de dosificación. Por ejemplo, una formulación de naltrexona de liberación sostenida tal como se define en las reivindicaciones puede producir eficacia sustancialmente similar o mejorada en comparación con una formulación de naltrexona de liberación inmediata debido a una naltrexona sustancialmente similar y/o 6-beta naltrexol AUCúltimo. Como otro ejemplo, una formulación de naltrexona de liberación sostenida puede proporcionar una reducción de un efecto adverso en comparación con la de una formulación de naltrexona de liberación inmediata debido a una naltrexona y/o 6-beta naltrexol inferior Cmáx.
[0044] Los perfiles farmacocinéticos se pueden determinar mediante el análisis del plasma de una población de pacientes que ha recibido formas de dosificación oral de naltrexona de liberación sostenida, y comparándolos con una población de pacientes comparable que haya recibido una formulación de liberación inmediata, utilizando la metodología del ensayo clínico apropiado y análisis estadísticos.
[0045] En algunas realizaciones, las propiedades farmacocinéticas son después de una dosis de fármaco simple, mientras que en otros, están en estado estacionario. Para las mediciones de una sola dosis, los pacientes pueden estar provistos de una única dosis de una composición que comprende la naltrexona de liberación sostenida, y muestras de plasma pueden ser recogidas desde el paciente en diferentes períodos de tiempo relativos a la administración de la composición para determinar los perfiles farmacocinéticos. Para las mediciones de estado estable, los pacientes pueden estar provistos de un régimen de dosificación a través de una pluralidad de días que comprende administrar formas de dosificación orales que comprenden naltrexona de liberación sostenida. Las muestras de plasma pueden entonces ser recogidas del paciente en diferentes períodos de tiempo relativos a una dosificación particular durante el estado estacionario. En algunas realizaciones, el estado estacionario se puede determinar mediante el control de una naltrexona en plasma y/o perfiles de concentración de metabolito naltrexona activo (por ejemplo, 6 betanaltrexol) en momentos específicos del pico esperado y los niveles mínimos en sangre con relación a la administración de una dosificación a través de horas y días y la determinación de cuando el perfil ha alcanzado el estado estacionario. Por ejemplo, la naltrexona in vivo de plasma y/o concentración de metabolito naltrexona activo (por ejemplo, 6-beta naltrexol) se pueden medir una hora después de la dosificación a lo largo de días, hasta que la concentración varía ya no significativamente de día a día. En otras realizaciones, el estado estacionario se puede estimar como un número específico de días después del inicio del régimen de dosificación. Por ejemplo, el estado de equilibrio puede estimarse como seis días después del inicio del régimen de dosificación. En algunas realizaciones, el estado estacionario es estimado después de dosificación alrededor de cinco veces la vida media del fármaco.
[0046] El plasma se puede analizar usando cualquier método apropiado. En algunas realizaciones, se recoge sangre de un paciente. Cualquier cantidad adecuada de sangre se puede recoger. Las muestras de sangre pueden entonces ser centrifugadas hasta que se produce la separación de los glóbulos rojos del plasma. En algunas realizaciones, la naltrexona y/o análisis de metabolito naltrexona se realiza de acuerdo con la validación del método analítico titulado "Validation of a High Performance Liquid Chromatographic Method Using Tandem Mass Spectrometry Lithium Heparinized Plasma".
[0047] Con el fin de medir los parámetros farmacocinéticos antes mencionados, concentraciones de naltrexona in vivo y/o metabolito naltrexona (por ejemplo, 6-beta naltrexol) pueden ser medidas a diversos intervalos de tiempo con respecto a una dosis de naltrexona. En algunas realizaciones, estas concentraciones se miden al menos 10 veces dentro de un período de 24 horas.
[0048] En algunas realizaciones, una población de pacientes experimenta un régimen de dosificación que comprende la administración de una forma de dosificación oral de naltrexona de liberación sostenida como se describe en el presente documento, y los parámetros farmacocinéticos presentados son la media de los parámetros farmacocinéticos a través de los pacientes. El promedio puede ser obtenido mediante el cálculo de los parámetros para cada paciente y luego promediando en todos los pacientes. En algunas realizaciones, el promediado comprende una media aritmética o una media geométrica de mínimos cuadrados. En algunas realizaciones, los parámetros farmacocinéticos se obtuvieron a partir de estudios cruzados.
[0049] se proporciona una formulación de naltrexona de liberación sostenida y/o se administra como una forma de dosificación oral tal como se define en las reivindicaciones. La forma de dosificación oral puede incluir también un segundo compuesto como se define en las reivindicaciones. En una realización, las formas de dosificación oral descritas en este documento son efectivas en el tratamiento de una o más de una condición relacionada con el peso. La condición relacionada con el peso puede comprender una condición caracterizada por un índice de masa corporal mayor de o igual a aproximadamente 25 kg/m2, mayor que o igual a aproximadamente 30 kg/m2 o menos de o igual a aproximadamente 18,5 kg/m2. La condición relacionada con el peso puede comprender la obesidad. La condición relacionada con el peso puede ser causada o se espera que sea causada por la administración de un medicamento. Formas de dosificación oral descritas en esta memoria pueden ser eficaces en que afectan a la pérdida de peso (por ejemplo, causando), la inhibición de la ganancia de peso y/o invertir al menos parcialmente el aumento de peso.
[0050] La forma de dosificación oral puede ser distribuida, proporcionada a una patente para la auto-administración o administrada a un paciente. El paciente puede tener sobrepeso u obesidad. El paciente puede tener un índice de masa corporal (IMC) de aproximadamente 20 kg/m2 o mayor, alrededor de 25 kg/m2 o mayor, o alrededor de 30 kg/m2 o
mayor. El paciente puede ser obeso. El paciente puede estar sufriendo de la diabetes.
[0051] La administración de formas de dosificación orales de liberación sostenida descritas en este documento puede proporcionar sustancialmente la misma o mejor eficacia que las formulaciones de liberación inmediata. Las formas de dosificación oral de liberación sostenida pueden estar asociadas con efectos adversos reducidos en comparación con las formulaciones de liberación inmediata. En algunas realizaciones, la administración de una forma de dosificación oral descrita en el presente documento puede dar como resultado una mejora en el cumplimiento del paciente con un tratamiento (por ejemplo, un tratamiento de pérdida de peso y/o un tratamiento con naltrexona).
[0052] En algunas realizaciones, la presente invención incluye una forma de dosificación tal como se define en las reivindicaciones para su uso en un método de tratamiento, que comprende la identificación de un paciente que sufre de al menos un efecto secundario adverso y/o que es particularmente susceptible a al menos un efecto secundario adverso asociado con un tratamiento de pérdida de peso (por ejemplo, un tratamiento de pérdida de peso que comprende la administración de bupropión) y/o el tratamiento con una formulación de naltrexona de liberación inmediata (por ejemplo, REVIA™ clorhidrato de naltrexona de liberación inmediata), y proporcionar o administrar a la paciente una forma de dosificación de liberación sostenida de naltrexona como se describe en el presente documento.
[0053] Tales realizaciones pueden incluir la etapa de administrar una cantidad terapéuticamente eficaz de la forma de dosificación de liberación sostenida a un mamífero en necesidad del mismo por cualquier vía adecuada o método de entrega, incluyendo los descritos en el presente documento. Niveles de dosificación reales de los compuestos en las formas de dosificación farmacéuticas se pueden variar con el fin de administrar una cantidad de naltrexona que es eficaz para lograr la respuesta terapéutica deseada para un paciente particular.
Dosis
[0054] Se entenderá que el nivel de dosis específica de las formas de dosificación de liberación sostenida descritas en este documento para cualquier paciente particular puede depender de cualquiera de una variedad de factores que incluyen la composición genética, peso corporal, salud general, dieta, tiempo y vía de administración, combinación con otros fármacos y la condición particular a tratar, y su gravedad. En algunas realizaciones, la dosis de naltrexona de las formas de dosificación de liberación sostenida como se describe en las reivindicaciones se determina basándose en la frecuencia de la que se administra la forma de dosificación oral al menos parcialmente. Formas de dosificación oral de liberación sostenida pueden ser administradas con más frecuencia, con menos frecuencia, o sustancialmente a la misma frecuencia que las formulaciones de liberación inmediata (por ejemplo, REVIA™ de clorhidrato de naltrexona de liberación inmediata).
[0055] En algunas realizaciones, una forma de dosificación oral de liberación sostenida tal como se define en las reivindicaciones que comprende naltrexona proporciona dosificaciones en una cantidad de base libre de naltrexona equivalente en el intervalo de aproximadamente 4 mg a aproximadamente 50 mg o en el intervalo de aproximadamente 10 mg a aproximadamente 25 mg. La forma de dosificación oral puede incluir de aproximadamente 4 mg, aproximadamente 8 mg, aproximadamente 12 mg, aproximadamente 16 mg, aproximadamente 32 mg, o aproximadamente 48 mg de naltrexona. La selección de una dosis particular puede estar basada en el peso del paciente. La selección de una dosis particular se puede basar en la identidad, la dosis y/o programa de dosificación de otro compuesto a ser co-administrado. Formas de dosificación unitaria adecuadas para la administración a un ser humano pueden estar configuradas para proporcionar una dosis de base libre de naltrexona equivalente en el intervalo de aproximadamente 0,75 mg/kg a aproximadamente 10 mg/kg, por ejemplo, aproximadamente 2 mg/kg (base es mg de fármaco por kilogramo de peso corporal). Las dosis pueden determinarse al menos parcialmente por un perfil farmacocinético. Por ejemplo, una cantidad de una forma de dosificación oral de naltrexona de liberación sostenida puede ser administrada que es suficiente para proporcionar una AUCúltimo sustancialmente similar a la de una forma de dosificación oral de liberación inmediata de naltrexona.
[0056] Las formas de dosificación de liberación sostenida tal como se definen en las reivindicaciones se pueden administrar una, dos o más veces al día, con o sin una dosis de carga. En algunas realizaciones, el número de administraciones por día es constante (por ejemplo, una vez por día). En otras realizaciones, es variable. El número de administraciones puede cambiar dependiendo de la eficacia de la forma de dosificación, los efectos secundarios observados, los factores externos (por ejemplo, un cambio en otra medicación), o la longitud de tiempo en que se ha administrado la forma de dosificación.
[0057] En una realización, una forma de dosificación de naltrexona de liberación sostenida tal como se define en las reivindicaciones comprende además un segundo compuesto, como se describe en el presente documento. El segundo compuesto puede estar presente en cualquier dosis adecuada. El segundo compuesto comprende el bupropión o una de sus sales farmacéuticamente aceptable en una formulación de liberación sostenida (SR). En una realización, la forma de dosificación comprende tanto la naltrexona como bupropión, por ejemplo, en forma de una tableta de tres capas como se describe en la Solicitud de Patente Provisional de Estados Unidos N° de Serie 60/865.157, presentada el 9 de noviembre, 2006. Los ejemplos de formas de dosificación unitaria adecuadas incluyen aquellos en los que el comprimido de tres capas incluye, por ejemplo, aproximadamente 4 mg de naltrexona SR y 90 mg de bupropión SR; aproximadamente 8 mg de naltrexona SR y 90 mg de bupropión SR; o alrededor de 12 mg de naltrexona SR y 90 mg
de bupropión SR. En una realización, la forma de dosificación se puede administrar como dos tabletas dos veces al día, por ejemplo, para una dosificación diaria de aproximadamente 16 mg, aproximadamente 32 mg, o aproximadamente 48 mg de naltrexona, y alrededor de 360 mg de bupropión o una sal farmacéuticamente aceptable del mismo. En una realización, el segundo compuesto (bupropión de liberación sostenida es decir, o una sal farmacéuticamente aceptable del mismo) y la naltrexona se coadministran por separado, por ejemplo, como formas de dosificación separadas que se coadministran dentro de alrededor de una hora uno del otro. En una realización, las formas de dosificación separadas se administran aproximadamente a aproximadamente el mismo tiempo.
Formulaciones
[0058] Formas de dosificación oral tal como se definen en las reivindicaciones pueden comprender naltrexona y un vehículo de liberación sostenida. Un vehículo de liberación sostenida incluye, a modo de ejemplo no limitativo, un ingrediente o ingredientes que se incluyen en una formulación farmacéutica en cantidades que son eficaces para ampliar la velocidad de liberación de la naltrexona a partir de la formulación en comparación con una formulación de liberación inmediata (por ejemplo, REVIA™ clorhidrato de naltrexona de liberación inmediata). Un vehículo de liberación sostenida puede ser denominado en este documento como un excipiente retardante. Ejemplos de vehículos de liberación sostenida incluyen hidroxipropilmetilcelulosa, óxido de polietileno, poliacrilato, copolímero de acrilato y metacrilato, polímero de metacrilato, copolímero de acrilato y metacrilato, copolímero de acrilato y metacrilato con un grupo amonio, copolímero de anhídrido maleico y metil vinil éter, hidroxi propil celulosa, etil celulosa, hidroxi propil, hidroxi etil celulosa, celulosa de metilo, metacrilato de hidroximetilo, maltodextrina, goma natural y goma de xantano. En algunas realizaciones, la composición de vehículo de liberación sostenida comprende al menos uno de hidroxipropilmetilcelulosa y polioxietileno. Una composición de excipiente de liberación sostenida puede contener uno o más vehículos de liberación sostenida, junto con otros ingredientes adecuados.
[0059] En algunas realizaciones, una forma de dosificación oral tal como se define en las reivindicaciones que comprenden naltrexona comprende una cantidad de una composición de vehículo de liberación sostenida que es eficaz para hacer la forma de dosificación farmacocinéticamente distinta de una formulación de liberación inmediata (por ejemplo, REVIA™ clorhidrato de naltrexona de liberación inmediata). Por ejemplo, en relación con la formulación de liberación inmediata, la cantidad y el tipo de composición de vehículo de liberación sostenida puede ser seleccionada para reducir la naltrexona Cmax y/o 6-beta naltrexol Cmax (Por ejemplo, a aproximadamente 80% o menos de la naltrexona Cmax de o 6-beta naltrexol Cmax de liberación inmediata de naltrexona).
[0060] La cantidad de la composición de vehículo de liberación sostenida puede ser eficaz para proporcionar una velocidad de liberación in vitro de la naltrexona de menos de aproximadamente 90%, o menos de aproximadamente 80%, en aproximadamente 2 horas. La cantidad de la composición de vehículo de liberación sostenida puede ser eficaz para proporcionar una velocidad de liberación in vitro de la naltrexona de menos de aproximadamente 98% en aproximadamente 4 horas. La cantidad de la composición de vehículo de liberación sostenida puede ser eficaz para proporcionar una velocidad de liberación in vitro de la naltrexona de menos de aproximadamente 80% o de aproximadamente el 70% en aproximadamente 1 hora. En la velocidad de liberación in vitro se determina por un ensayo de disolución estándar como se describe anteriormente.
[0061] Una descripción de materiales portadores de liberación sostenida representativos puede encontrarse en Remington: The Science and Practice of Pharmacy (20a ed, Lippincott Williams & Wilkens Publishers (2003)). Los expertos en la técnica pueden formular composiciones de soporte de liberación sostenida utilizando experimentación de rutina informada por la orientación detallada proporcionada en este documento.
[0062] Las formas de dosificación tal como se definen en las reivindicaciones pueden formularse para comprender varios excipientes, aglutinantes, vehículos, disgregantes, recubrimientos, etc. Las preparaciones farmacéuticas se pueden obtener mediante la mezcla de uno o más excipientes sólidos con una composición farmacéutica como se describe en el presente documento, opcionalmente moliendo la mezcla resultante y procesando la mezcla de gránulos, después de añadir auxiliares adecuados, si se desea, para obtener composiciones farmacéuticas adecuadas para uso en diversas formas, por ejemplo, como píldoras, comprimidos, polvos, gránulos, grageas, cápsulas, líquidos, pulverizaciones, geles, jarabes, suspensiones espesas, suspensiones y similares, en formas a granel o de dosificación unitaria, para la ingestión oral por un paciente a tratar. Varios ejemplos de formas de dosificación unitaria se describen en el presente documento; Los ejemplos no limitantes incluyen una píldora, un comprimido, una cápsula, una cápsula de gel, y similares. Ejemplos de excipientes adecuados se enumeran a continuación, algunos de los cuales se mencionan anteriormente como que tiene particulares propiedades de disolución. Vehículos o diluyentes para uso terapéutico farmacéuticamente aceptables son bien conocidos en la técnica farmacéutica, y se describen, por ejemplo, en Remington: The Science and Practice of Pharmacy (2003). El término material de "vehículo" o "excipiente" en la presente memoria puede significar cualquier sustancia, no es en sí un agente terapéutico, usada como un vehículo, diluyente, adyuvante, aglutinante, y/o vehículo para la administración de un agente terapéutico a un sujeto o añadido a una composición farmacéutica para mejorar sus propiedades de manipulación o almacenamiento o para permitir o facilitar la formación de una unidad de dosis de la composición en un artículo discreto tal como una cápsula o comprimido adecuado para la administración oral. Los excipientes pueden incluir, a modo de ilustración y no de limitación, diluyentes, disgregantes, agentes aglutinantes, adhesivos, agentes humectantes, polímeros, lubricantes, deslizantes, sustancias añadidas para enmascarar o contrarrestar un sabor u olor desagradable, aromas, colorantes,
fragancias y sustancias añadidas para mejorar la apariencia de la composición. Los deslizantes pueden ser uno o más de dióxido de silicio coloidal, talco, almidón de maíz, DL-leucina, lauril sulfato de sodio y magnesio, calcio y estearatos de sodio. Los diluyentes pueden ser uno o más de lactosa, almidón, manitol, sorbitol, dextrosa, celulosa microcristalina, fosfato de calcio dibásico, diluyentes basados en sacarosa, azúcar de pastelería, calcio monobásico monohidrato de sulfato, sulfato de calcio dihidratado, calcio trihidrato de lactato, dextratos, inositol, sólidos de cereales hidrolizados, amilosa, celulosa en polvo, carbonato de calcio, glicina o bentonita. Los excipientes aceptables incluyen lactosa, sacarosa, polvo de almidón, almidón de maíz o derivados de los mismos, ésteres de celulosa de ácidos alcanoicos, ésteres alquílicos de celulosa, talco, ácido esteárico, estearato de magnesio, óxido de magnesio, sales de sodio y calcio de ácidos fosfórico y sulfúrico, gelatina, acacia goma, alginato de sodio, polivinil-pirrolidona, y/o alcohol de polivinilo, solución salina, dextrosa, manitol, lactosa, lecitina, albúmina, glutamato de sodio, clorhidrato de cisteína, y similares. Ejemplos de excipientes adecuados para cápsulas de gelatina blanda incluyen aceites vegetales, ceras, grasas, polioles semisólidos y líquidos. Los excipientes adecuados para la preparación de soluciones y jarabes incluyen, sin limitación, agua, polioles, sacarosa, azúcar invertido y glucosa. Las composiciones farmacéuticas pueden incluir adicionalmente conservantes, solubilizantes, estabilizantes, agentes humectantes, emulsionantes, edulcorantes, colorantes, saborizantes, tampones, agentes de revestimiento, o antioxidantes. Disolución o suspensión del ingrediente activo en un vehículo tal como agua o aceite vegetal de origen natural como de sésamo, cacahuete, o aceite de semilla de algodón o un vehículo graso sintético como oleato de etilo o similar puede ser deseado. Tampones, conservantes, antioxidantes y similares pueden incorporarse de acuerdo con la práctica farmacéutica aceptada. El compuesto también se puede hacer en forma microencapsulada. Si se desea, preparaciones de absorción mejorada (por ejemplo, liposomas) pueden ser utilizadas. Los expertos en la técnica pueden formular formas de dosificación de liberación sostenida que contienen uno o más de los ingredientes anteriores por experimentación de rutina informados por las directrices detalladas proporcionadas en este documento.
Kits
[0063] En algunas realizaciones, la presente invención se puede presentar como un kit. El kit puede incluir una o más formas de dosificación unitaria como se define en las reivindicaciones que comprende naltrexona de liberación sostenida. Las formas de dosificación unitaria pueden ser de una formulación oral. Las formas de dosificación unitaria pueden comprender tabletas. El kit puede incluir una pluralidad de formas de dosificación unitaria.
[0064] El kit puede incluir información. La información puede estar en una forma prescrita por una agencia gubernamental que regula la fabricación, uso, o venta de productos farmacéuticos, cuyo aviso refleja la aprobación por la agencia de la forma del fármaco para administración humana o veterinaria. Tal información, por ejemplo, puede ser el etiquetado aprobado por la Administración de Alimentos y Fármacos de Estados Unidos para fármacos con receta, o el inserto de producto aprobado. Las formas de dosificación que comprenden una formulación de naltrexona de liberación sostenida de la invención formulada en un vehículo farmacéutico compatible también pueden prepararse, colocarse en un recipiente apropiado y etiquetarse para el tratamiento de una afección indicada.
[0065] La información puede comprender instrucciones para administrar la forma de dosificación unitaria en una dosis de aproximadamente 4 mg, aproximadamente 8 mg, aproximadamente 12 mg, aproximadamente 16 mg, aproximadamente 32 mg o aproximadamente 48 mg. Estas instrucciones pueden proporcionarse en una variedad de maneras. La información puede comprender instrucciones acerca de cuándo administrar las formas de dosificación de unidad. Por ejemplo, la información puede comprender instrucciones acerca de cuándo administrar las formas de dosificación unitaria en relación con la administración de otro medicamento o comida.
[0066] La información puede ser proporcionada en un medio legible. El medio legible puede comprender una etiqueta. El kit puede comprender un envase terapéutico adecuado para la venta comercial. El kit puede comprender un recipiente. El recipiente puede estar en cualquier conformación o forma convencional como se conoce en la técnica que está hecha de un material farmacéuticamente aceptable, por ejemplo una caja de papel o cartón, una botella de vidrio o de plástico o un tarro, una bolsa resellable (por ejemplo, para sujetar una "recarga" de comprimidos para la colocación en un recipiente diferente), o un envase blíster con dosis individuales para presionar hacia fuera del paquete de acuerdo con un programa terapéutico. El recipiente empleado puede depender de la forma de dosificación exacta implicada, por ejemplo una caja de cartón convencional generalmente no se utiliza para contener una suspensión líquida. Es factible que más de un contenedor se puede utilizar juntos en un solo paquete para comercializar una forma de dosificación única. Por ejemplo, los comprimidos pueden estar contenidos en una botella que a su vez está contenida dentro de una caja.
[0067] La información puede estar asociada con el recipiente, por ejemplo, por estar: escrita en una etiqueta (por ejemplo, la etiqueta del medicamento o una etiqueta separada) fijada de forma adhesiva a una botella que contiene una forma de dosificación descrita en este documento; incluido dentro de un contenedor como un prospecto escrito, como dentro de una caja que contiene los paquetes de dosis unitaria; aplicado directamente al recipiente, tal como está impresa en la pared de una caja; o se adjunta como por estar atado o pegado, por ejemplo como una tarjeta de instrucción fijada al cuello de una botella a través de una cadena, cable u otra línea, cordón o correa de sujeción tipo de dispositivo. La información puede ser impresa directamente sobre un paquete de dosis unitaria o envase blíster o en la tarjeta blister.
EJEMPLOS
[0068] Los siguientes ejemplos no son limitantes y son meramente representativos de varios aspectos de la invención. Ejemplos que no caen dentro del alcance de las reivindicaciones son únicamente para referencia.
EJEMPLO 1
[0069] Se preparó una serie de naltrexona de liberación sostenida (5 mg). Cada una de las formulaciones de liberación sostenida contenía un "excipiente común". La formulación para el excipiente común se muestra en la Tabla 1. La formulación mostrada en la Tabla 2 contenía hidroxipropilmetil celulosa como un vehículo de liberación sostenida o excipiente retardante. La formulación mostrada en la Tabla 3 contenía óxido de polietileno como un excipiente retardante.
[0070] Para hacer el excipiente común, los ingredientes enumerados en la Tabla 1 se colocaron en un granulador hicizallamiento de clave KG-5 (Key International, Englishtown, NJ) y se granula con una solución al 10% de celulosa de hidroxipropilo (HPC). La granulación se secó a temperatura ambiente a una LOD final del 2,71% y luego se tamiza a través de un tamiz de malla 20.
[0071] Para fabricar las formulaciones de naltrexona, el ingrediente farmacéutico activo (API) se mezcló con un dióxido de silicio coloidal como un agente de deslizamiento, el polímero de liberación que se extiende (por ejemplo, ya sea HPMC o Polyox) y el excipiente común usando un mezclador Turbula. El estearato de magnesio se añadió como lubricante a la formulación final.
[0072] Las tabletas con un arnés de 5Kp se comprimieron para determinar las propiedades de desintegración de la tableta. Bajo las condiciones usadas, la tableta 5 kp no se desintegró dentro de 30 minutos en agua USP. Por consiguiente, los comprimidos con una dureza mayor se comprimieron a 9Kp usando estampado cóncavo estándar redondo 1/4" en una prensa Carver.
[0073] Se produjeron las tabletas comprimidas a 9Kp, y las propiedades de las tabletas se enumeran en Tabla 4.
Tabla 1: Formulación para el excipiente Común

Tabla 2: Las formulaciones para comprimidos de 5 mg de fuerza de liberación sostenida que contiene hidroxipropilmetilcelulosa (HPMC)

Tabla 3: Las formulaciones para comprimidos de 5 mg de fuerza de liberación sostenida que contienen óxido de polietileno (Polyox)

Tabla 4: Datos de comprimidos en proceso

[0074] El siguiente ejemplo describe los perfiles de disolución de las formulaciones de naltrexona de liberación sostenida descritos anteriormente.
EJEMPLO 2
[0075] Las mediciones de disolución para los comprimidos se completaron utilizando cestas de malla 10 a 100 rpm. Las muestras se analizaron usando un UV-VIS en Xmáx de 280. Los datos de disolución del ingrediente farmacéutico activo (API) para las formulaciones de HPMC y formulaciones Poliox se presentan en las Tablas 5 y 6, respectivamente. Los datos de disolución para las formulaciones de HPMC y formulaciones Poliox también se representan en las Figuras 1 y 2, respectivamente.


EJEMPLO 3
[0076] Comprimidos de tres capas de naltrexona-bupropión de liberación sostenida se hicieron usando los ingredientes listados en Tabla 7 a Tabla 9, de acuerdo con los métodos generales para la fabricación de comprimidos de tres capas que se describen en la Solicitud de Patente Provisional de Estados Unidos N° de Serie 60/865.157, presentada el 9 de noviembre de 2006. Se fabricó la formulación de naltrexona de liberación sostenida mediante la combinación de los siguientes componentes:
Tabla 7: Formulación de mezcla de naltrexona

[0077] Por lo tanto, la formulación de naltrexona de liberación sostenida incluye 10% de HPMC. Una mezcla de bupropión se hizo combinando los siguientes componentes:
Tabla 8: Formulación de mezcla de bupropión

[0078] Una mezcla de la capa inerte se hizo combinando los siguientes componentes:
Tabla 9: Formulación de mezcla inerte

[0079] La formulación de naltrexona de liberación sostenida, mezcla de bupropión y capa de mezcla inerte se utilizan para formar 150 comprimidos de tres capas con las capas de naltrexona y bupropión en los lados opuestos de la capa inerte, de tal manera que cada tableta era 662,00 mg. Los comprimidos contenían cada una 11,94 mg de naltrexona (13,22 mg de clorhidrato de naltrexona).
[0080] Los datos de disolución de la naltrexona para los comprimidos se presenta en Tabla 10. Los datos de disolución también se representan en la Figura 3.
Tabla 10: Datos de disolución para comprimidos de naltrexona-bupropion

EJEMPLO 4
[0081] Un estudio de solo centro, doble ciego, cruzado, de naltrexona de liberación inmediata y naltrexona de liberación prolongada se llevó a cabo durante 40 voluntarios sanos obesos.
[0082] Los sujetos fueron asignados aleatoriamente para recibir una formulación de naltrexona de liberación sostenida o una formulación de liberación inmediata en una relación 1: 1. A los sujetos se les administró 40 mg de naltrexona SR y 36 mg de naltrexona IR. Los sujetos fueron asignados aleatoriamente para recibir la liberación sostenida y la liberación inmediata en una de dos secuencias: la mitad de los sujetos recibió la formulación de liberación sostenida en el periodo 1, seguido de la formulación de liberación inmediata en el período 2; los sujetos restantes recibieron los tratamientos en el orden inverso.
[0083] La administración del fármaco del estudio en cada período fue separada por un período de lavado de cinco días. Muestras de sangre en serie se recogieron antes de la dosis y a 0,167, 0,33, 0,5, 0,67, 0,83, 1, 1,25, 1,5, 2, 3, 4, 6, 8, 10, 12, 16, 24, 36, 48, 60 y 72 horas después de la dosis. Las muestras se analizaron para las concentraciones de naltrexona y 6-beta naltrexol por métodos LCMS/MS validados. Además de acontecimientos adversos recogidos espontáneamente, información sobre los efectos secundarios se recogió usando la Escala de Evaluación de Efecto Secundario UKU evaluada por el sujeto. La escala se administró antes de la dosis, a las 8, 24 y 72 horas después de la dosis.
[0084] Los datos de concentración-tiempo se importaron directamente en WinNonlin (Versión 5.0, Pharsight Corporation). Los datos se analizaron por métodos no compartimentales utilizando WinNonlin usando el modelo 200 para la entrada extravascular.
[0085] Datos de concentración-tiempo que eran BLLOQ fueron excluidos de los cálculos antes de resumen de datos y el análisis farmacocinético. Los datos fueron resumidos por tiempos de muestreo programados (nominales). Se han usado tiempos reales de muestreo si la desviación de nominal fue considerada significativa.
[0086] Los siguientes parámetros farmacocinéticos se calcularon para concentraciones de naltrexona y 6-beta naltrexol para cada sujeto y cada tratamiento:
Cmax = concentración máxima medida
T max = tiempo para alcanzar la concentración máxima
Ke = constante de velocidad terminal, estimada por regresión log-lineal
AUC (0-t) = área bajo la curva de concentración-tiempo calculada por la regla trapezoidal lineal desde el tiempo 0 al tiempo de la última muestra con una concentración cuantificable (Ct)
AUC (O-oo) = Área bajo la curva de tiempo de concentración desde el tiempo 0 extrapolada hasta el infinito, calculada como AUC (0-t) Ct/Ke
T'/2 = vida media terminal, calculada como Ln(2)/Ke.
[0087] El AUC se utiliza para describir el grado de absorción del fármaco. La velocidad de absorción se caracteriza por Cmax y Tmax. Ke y T1^ describen la cinética en la fase terminal, que, para muchas sustancias, se rige por los procesos de eliminación. Los parámetros farmacocinéticos se calcularon para todos los casos.
[0088] Las Figuras 4 y 5 muestran curvas de tiempo de concentración de plasma individual de naltrexona en sujetos que recibieron naltrexona IR y en los sujetos que recibieron naltrexona SR, respectivamente. Las Figuras 6 y 7 muestran curvas de tiempo de concentración de plasma individual de 6-beta naltrexol en sujetos que recibieron naltrexona IR y en los sujetos que recibieron naltrexona SR, respectivamente.
[0089] La Figura 8A muestra las curvas medias de tiempo de concentración en plasma de naltrexona en sujetos que recibieron naltrexona SR (círculo) o naltrexona IR (cuadrado). La Figura 8B muestra toda la curva de tiempo de concentración de plasma. La naltrexona Cmax es mayor para los pacientes que recibieron naltrexona IR que para los que recibieron naltrexona SR a través del área bajo la curva es comparable entre ambas condiciones. Estos resultados son normalizados a la dosis y cuantificados en la Tabla 11.
Tabla 11. Resumen de los parámetros farmacocinéticos en los sujetos que recibieron naltrexona SR o naltrexona IR

[0090] La Figura 9A muestra las curvas medias de tiempo de concentración de plasma de 6-beta naltrexol en sujetos que recibieron naltrexona SR (círculo) o naltrexona IR (cuadrado). La Figura 9B muestra toda la curva de tiempo de concentración de plasma. El 6-beta naltrexol Cmax es mayor para los pacientes que recibieron naltrexona IR que para los que recibieron naltrexona SR por el área bajo la curva es comparable entre ambas condiciones. Estos resultados son normalizados a la dosis y cuantificados en Tabla 11.
[0091] La Tabla 12 proporciona los eventos adversos informados por los pacientes que recibieron naltrexona SR y por los pacientes que recibieron naltrexona IR. Los eventos adversos se analizan a través de pacientes para cada una de las condiciones de tratamiento. Estos resultados se resumen en la Tabla 13. Los individuos que reciben naltrexona SR eran menos propensos a reportar un evento adverso (45% versus 48%) y también eran menos propensos a reportar más de un evento adverso (13% versus 23%) que los individuos que reciben naltrexona IR.


Tabla 13. Resumen de eventos adversos gastrointestinales, dolor de cabeza, en los sujetos que recibieron naltrexona SR y naltrexona IR.

[0092] La Figura 10 es un esquema que ilustra la población de sujetos que informaron náuseas y vómitos utilizando la Escala de Evaluación de Eventos Adversos UKU. Los individuos que recibieron naltrexona SR eran menos propensos a informar náusea severa (puntuación de 2 o mayor) que los que recibieron naltrexona IR.
[0093] Los expertos en la técnica reconocerán que la incidencia de eventos adversos reportados usando la Escala de Evaluación de Eventos Adversos de UKU es normalmente mayor que los auto-informes. Los pacientes que se han notificado a ser más propensos a reportar eventos adversos cuando se le solicite para informar sobre eventos adversos (Sheftell FD, Feleppa M, Tepper SJ, Rapoport AM, Ciannella L, BigAl ME. La evaluación de los eventos adversos asociados con triptanos-métodos de evaluación de la influencia de resultados de Headache: The Journal of Head and Face Pain 44 (10), 978-982). Por lo tanto, la incidencia de eventos adversos obtenidos por los métodos utilizados en este ejemplo no es directamente comparable a la incidencia de eventos adversos obtenidos por los métodos descritos en el Ejemplo 5.
EJEMPLO 5
[0094] Un estudio doble ciego aleatorizado, estudio de dosis múltiples en paralelo comparó la farmacocinética de un producto de combinación naltrexona SR/bupropion SR con un producto de combinación naltrexona IR/bupropion SR.
[0095] El grupo naltrexona SR/bupropion SR: Cada cápsula contenía dos y una media de 5 mg de mini comprimidos (preparado de acuerdo con el Ejemplo 1) de la naltrexona de liberación sostenida además de un comprimido SR de bupropion HCl 90 mg (dosis total de naltrexona SR 12,5 mg/bupropión SR 90 mg por cápsula) que se valora durante 7 días a. una dosis diaria total de naltrexona SR 37,5 mg/bupropión SR 270 mg.
[0096] Grupo naltrexona IR/bupropión SR: Cada cápsula contenía cápsulas que contienen un comprimido de naltrexona HCl IR de 12 mg más un comprimido de bupropion HCl SR de 90 mg (dosis total de comprimido de naltrexona IR de 12 mg/bupropión SR de 90 mg), que se valora durante 7 días a una dosis diaria final de naltrexona IR de 36 mg/bupropión SR de 270 mg. La formulación naltrexona IR está formulada para tener un perfil farmacocinético que es sustancialmente similar a la marca ReVia® de clorhidrato de naltrexona.
[0097] Voluntarios obesos sanos recibieron al azar una de las dos administraciones de fármacos. Se espera que un total de 60 sujetos se inscriban, 59 sujetos completaron el estudio.
[0098] Se extrajo sangre en el día 1 (0,5, 1,2, 4, 6, 8, 10, y 12 horas) con muestras mínimas extraidas en los días 8 y 15. Las muestras de plasma fueron extraídas de cada sujeto para determinar la naltrexona, 6-beta-naltrexol , las concentraciones de bupropion, hidroxibupropión, treohidroxibupropion, y ertirohidroxibupropion. Las muestras de plasma se analizaron usando los métodos SFBCi/Anapharm validados para la naltrexona y 6-beta naltrexol. Los datos de concentración-tiempo se importaron directamente en WinNonlin (Versión 5.0, Pharsight Corporation). Los datos se analizaron por métodos no compartimentales utilizando WinNonlin utilizando el Modelo 200 para la entrada extravascular.
[0099] Los datos de concentración-tiempo que eran BLLOQ fueron excluidos de los cálculos antes del resumen de datos y el análisis farmacocinético. Los datos fueron resumidos por tiempos de muestreo programados (nominales). Se han usado tiempos reales de muestreo si la desviación de la nominal fue considerado significativa.
[0100] Los parámetros farmacocinéticos medidos en el presente documento son conocidos por los expertos en la técnica. Plasmas de sangre se recogieron a intervalos específicos relativos a la administración de un producto de combinación. Se analizó el plasma para determinar la concentración de un compuesto (por ejemplo, naltrexona). Cmax indica la concentración máxima en plasma sanguíneo del compuesto después de la administración. Tmax indica el tiempo en el que fue alcanzada la concentración plasmática sanguínea máxima del compuesto (Cmáx). AUC indica el área bajo la curva de la concentración como una función del tiempo después de la administración. AUCúltimo indica el área bajo la curva de concentración plasmática desde el momento de la administración hasta el momento de la última concentración mensurable.
[0101] Tabla 14 y Figura 11 comparan las concentraciones plasmáticas promedio de naltrexona en sujetos que recibieron tratamientos de naltrexona IR y bupropión SR (IR/s R; círculos) al promedio de los sujetos que recibieron tratamientos de naltrexona SR y el bupropión SR (SR/SR; cuadrados). La dosis única de la naltrexona en el inicio de la programación de titulación de 12 (IR) o 12,5 (SR) produce concentraciones en plasma durante las primeras 24 horas que son principalmente por debajo del límite inferior de cuantificación (BLLOQ).
Tabla 14. La media de las concentraciones plasmáticas de naltrexona en sujetos que recibieron naltrexona IR en combinación con bupropión SR (IR/s R) o naltrexona SR en combinación con bupropión SR (SR/SR).

[0102] Tabla 15 y Figura 12 comparan las concentraciones plasmáticas promedio 6-beta naltrexol en sujetos que recibieron tratamientos de naltrexona IR y bupropión SR (cuadrados) al promedio de los sujetos que recibieron tratamientos de naltrexona SR y el bupropión SR (círculos). Las concentraciones en plasma asociadas con el tratamiento SR/SR son más bajas durante las primeras horas después de la administración que las asociadas con el tratamiento IR/SR. Como se informó en Tabla 16, el tratamiento SR/SR se asocia con una menor Cmax pero un AUC similar en comparación con el tratamiento IR/SR.
Tabla 15. La media de las concentraciones plasmáticas de 6-beta naltrexol en sujetos que recibieron naltrexona IR en combinación con bupropión SR (IR/SR) o naltrexona SR en combinación con bupropión SR (SR/SR).

Tabla 16. Los parámetros farmacocinéticos medios para 6-beta naltrexol en el Día 1 en sujetos que recibieron naltrexona IR en combinación con bupropión SR (IR/SR) o naltrexona SR en combinación con bupropión SR
(SR/SR).

[0103] Las Figuras 13 y 14 comparan las concentraciones plasmáticas mínimas de naltrexona y 6-beta naltrexol, respectivamente, en sujetos que recibieron tratamientos de naltrexona IR y bupropión SR (círculos) al promedio de los sujetos que recibieron tratamientos de naltrexona SR y el bupropión SR (cuadrados). Para la naltrexona, los niveles valle en el día 8 y el día 15 son principalmente BLLOQ, con la excepción de 1 sujeto en el grupo IR, y 2 sujetos en el grupo SR. Para 6-beta naltrexol, los niveles valle de Día 8 y Día 15 son similares entre los grupos de tratamiento.
[0104] Tabla 17 y Figura 15 comparan las concentraciones plasmáticas promedio de bupropión en los sujetos que recibieron tratamientos de naltrexona IR y bupropión SR (cuadrados) al promedio de los sujetos que recibieron tratamientos de naltrexona SR y bupropión SR (círculos). Los perfiles de concentración de plasma son similares en las condiciones de tratamiento.
Tabla 17. La media de las concentraciones plasmáticas de bupropión en los sujetos que recibieron bupropión SR en combinación con naltrexona IR (IR/SR) o en combinación con naltrexona SR (SR/SR).

[0105] Tablas 18-20 y Figuras 16-18 comparan las concentraciones plasmáticas promedio de los metabolitos de bupropión de hidroxibupropión, treohidroxibupropion y eritrohidroxibupropion en los sujetos que recibieron tratamientos de naltrexona IR y bupropión SR (cuadrados) al promedio de los sujetos que recibieron tratamientos de naltrexona SR y el bupropión SR (círculos). Los perfiles de concentración de plasma son similares en las condiciones de tratamiento.
Tabla 18. Las concentraciones plasmáticas medias del metabolito de bupropión hidroxibupropión en los sujetos que recibieron bupropión SR en combinación con naltrexona IR (IR/SR) o en combinación con naltrexona SR (SR/s R).

Tabla 19. Las concentraciones plasmáticas medias del metabolito de bupropión treohidroxibupropion en los sujetos que recibieron bupropión SR en combinación con naltrexona IR (IR/SR) o en combinación con naltrexona SR
(SR/SR).

Tabla 20. La media de las concentraciones plasmáticas del metabolito de bupropión eritrohidroxibupropion en los sujetos que recibieron bupropión SR en combinación con naltrexona IR (IR/SR) o en combinación con naltrexona SR
(SR/SR).

[0106] Tabla 21 informa el bupropión y los valores de bupropión metabolito AUCúltimo y Cmax para las dos condiciones de tratamiento. Estos valores son similares para ambos tratamientos.
Tabla 21. Los parámetros farmacocinéticos medios para bupropión y sus metabolitos en el Día 1 en sujetos que recibieron bupropión SR en combinación con naltrexona IR (IR/SR) o en combinación con naltrexona SR (SR/SR).

[0107] Se utilizaron listas de eventos adversos, así como registros de dosificación para este análisis. Los datos fueron introducidos en hojas de cálculo, QA'd, y luego se resumió la frecuencia de eventos, % de los sujetos que informaron varios eventos, tiempo hasta la aparición de la primera dosis y días en esquema de titulación. La incidencia de efectos adversos obtenidos utilizando el método no solicitado de este ejemplo no es directamente comparable a la incidencia obtenida usando los métodos del Ejemplo 4.
[0108] Como se muestra en Tabla 22, el porcentaje de sujetos que informaron cualquier evento adverso fue de 26% y 30% para los grupos SR e IR, respectivamente. El porcentaje de sujetos que informaron eventos adversos relacionados con el GI fue del 10% y 16% para los grupos SR e IR; se observaron porcentajes similares para los sujetos que informaron eventos adversos relacionados con el SNC. Más sujetos informaron de más de un evento adverso en el grupo IR (6/30, 20%), que en el grupo SR (2/30, 6,6%). Por último, más sujetos reportaron reacciones adversas que eran moderadas o mayores en la gravedad en el grupo IR (30,7%) en comparación con el grupo SR (16,7%).
Tabla 22: Los eventos adversos asociados con naltrexona IR y SR con el tratamiento con bupropión SR

[0109] Por lo tanto, el grupo que recibió la naltrexona de liberación sostenida con bupropión fue menos propenso a experimentar eventos adversos que el grupo que recibió la naltrexona de liberación inmediata con bupropión. Además, los eventos adversos reportados desde el grupo que recibió la naltrexona de liberación sostenida con bupropión fueron menos graves que los reportados a partir del grupo naltrexona de liberación inmediata.
[0110] 4 sujetos retiraron el consentimiento del estudio; ningunos se enumeran como no haber completado el estudio debido a un evento adverso. Estos sujetos no se eliminan de los cálculos presentados. Tres de los 4 sujetos informaron de un evento adverso, pero ninguno abandonó el día que se reportó el evento. Tres de los sujetos estaban recibiendo IR, el cuarto estaba recibiendo SR.
[0111] Todos los eventos (n = 12) en el grupo SR se informaron como leves, con la excepción de un caso de euforia y una instancia de calambres en el estómago (ambos moderados). La mayoría de los eventos (n = 26) fueron reportados como leves. Había cinco relacionados con GI (emesis, dolor de estómago, náuseas) y 3 eventos relacionados con el SNC que se han descrito como moderados (dolor de cabeza, fatiga).
[0112] Se entenderá por los expertos en la técnica que numerosas y diversas modificaciones pueden hacerse sin apartarse del espíritu de la presente invención. Por lo tanto, debe entenderse claramente que las formas de la presente invención son sólo ilustrativas y no pretenden limitar el alcance de la presente invención.
Claims (13)
1. Una forma de dosificación que comprende una formulación de liberación sostenida de la naltrexona, o una sal farmacéuticamente aceptable del mismo y una formulación de liberación sostenida de bupropión o una sal farmacéuticamente aceptable del mismo, en donde la forma de dosificación es una forma de dosificación oral, en la que la forma de dosificación proporciona una velocidad de liberación in vitro de la naltrexona de menos de 80% o menos de 70% en aproximadamente 1 hora, de menos de 90% o menos de 80% en aproximadamente 2 horas, o de menos de 98% en aproximadamente 4 horas, como se determina usando United States Pharmacopeia 24a edición (USP 24) Aparato 2 de ensayo de disolución que tiene una velocidad de rotación del husillo de 100 rpm y un medio de disolución de agua, a 37°C.
2. La forma de dosificación según la reivindicación 1, en la que la forma de dosificación proporciona una velocidad de liberación in vitro de la naltrexona de menos de 80% o menos de 70% en aproximadamente 1 hora, como se determina usando United States Pharmacopeia 24a edición (USP 24) Aparato 2 de ensayo de disolución que tiene una velocidad de rotación del husillo de 100 rpm y un medio de disolución de agua, a 37°C.
3. La forma de dosificación según la reivindicación 1, en la que la forma de dosificación proporciona una velocidad de liberación in vitro de la naltrexona de menos de 90% o menos de 80% en aproximadamente 2 horas, como se determina usando United States Pharmacopeia 24a edición (USP 24) Aparato 2 de ensayo de disolución que tiene una velocidad de rotación del husillo de 100 rpm y un medio de disolución de agua, a 37°C.
4. La forma de dosificación según la reivindicación 1, en la que la forma de dosificación proporciona una velocidad de liberación in vitro de la naltrexona de menos de 98% en aproximadamente 4 horas, como se determina usando United States Pharmacopeia 24a edición (USP 24) Aparato 2 de ensayo de disolución que tiene una velocidad de rotación de husillo de 100 rpm y un medio de disolución de agua, a 37°C.
5. La forma de dosificación de acuerdo con cualquiera de las reivindicaciones 1-4, que comprende un vehículo de liberación sostenida seleccionado de uno o más de: hidroxipropilmetilcelulosa, polioxietileno, óxido de polietileno, poliacrilato, copolímero de acrilato y metacrilato, polímero de metacrilato, copolímero de acrilato y metacrilato, copolímero de acrilato y metacrilato con un grupo amonio, copolímero de anhídrido maleico y metil vinil éter, hidroxi etil celulosa propilo, hidroxi propil celulosa, hidroxi etil celulosa, metil celulosa, metacrilato de hidroximetilo, maltodextrina, goma natural y goma de xantano.
6. La forma de dosificación de acuerdo con cualquiera de las reivindicaciones 1-5, que comprende aproximadamente 4 mg, aproximadamente 8 mg, aproximadamente 12 mg, aproximadamente 16 mg, aproximadamente 32 mg, o aproximadamente 48 mg de naltrexona.
7. La forma de dosificación de acuerdo con cualquiera de las reivindicaciones 1-6, que comprende aproximadamente 4 mg, aproximadamente 8 mg, o aproximadamente 12 mg de naltrexona y aproximadamente 90 mg de bupropión.
8. La forma de dosificación de acuerdo con cualquiera de las reivindicaciones 1-7, en donde la formulación de liberación sostenida de la naltrexona, o una sal farmacéuticamente aceptable del mismo y la formulación de liberación sostenida de bupropión o una sal farmacéuticamente aceptable del mismo, son adecuadas para la administración por separado.
9. La forma de dosificación de acuerdo con cualquiera de las reivindicaciones 1-7, en donde la forma de dosificación es una forma de dosis unitaria que comprende tanto la formulación de liberación sostenida de la naltrexona, o una sal farmacéuticamente aceptable de la misma como la formulación de liberación sostenida de bupropión o una sal farmacéuticamente aceptable de la misma.
10. Una forma de dosificación según cualquiera de las reivindicaciones 1-9 para uso en el tratamiento de la obesidad.
11. El uso de una forma de dosificación oral según cualquiera de las reivindicaciones 1-9 en la preparación de un medicamento para afectar la pérdida de peso o la inhibición de la ganancia de peso en un paciente.
12. La forma de dosificación según cualquiera de las reivindicaciones 1-8 para uso de acuerdo con la reivindicación 10, o el uso de acuerdo con la reivindicación 11 de una forma de dosificación según cualquiera de las reivindicaciones 1-8, en donde la formulación de liberación sostenida de la naltrexona, o una sal farmacéuticamente aceptable de la misma y la formulación de liberación sostenida de bupropión o una sal farmacéuticamente aceptable de la misma, se administran por separado al paciente.
13. La forma de dosificación para su uso, o el uso de acuerdo con la reivindicación 12, en el que la forma de dosificación se administra como dos tabletas dos veces al día, para una dosis diaria de aproximadamente 16 mg, aproximadamente 32 mg o aproximadamente 48 mg de naltrexona o una sal farmacéuticamente aceptable de las mismas, y alrededor de 360 mg de bupropión o una sal farmacéuticamente aceptable de las mismas.
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US81125106P | 2006-06-05 | 2006-06-05 | |
| US84111406P | 2006-08-29 | 2006-08-29 | |
| PCT/US2007/013030 WO2007145863A2 (en) | 2006-06-05 | 2007-05-31 | Sustained release formulation of naltrexone |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2749800T3 true ES2749800T3 (es) | 2020-03-23 |
Family
ID=38832300
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES07795652T Active ES2749800T3 (es) | 2006-06-05 | 2007-05-31 | Formulación de liberación sostenida de naltrexona |
Country Status (6)
| Country | Link |
|---|---|
| EP (2) | EP2061448B1 (es) |
| JP (3) | JP5478245B2 (es) |
| AR (1) | AR061233A1 (es) |
| ES (1) | ES2749800T3 (es) |
| TW (1) | TW200808319A (es) |
| WO (1) | WO2007145863A2 (es) |
Families Citing this family (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| ES2749800T3 (es) * | 2006-06-05 | 2020-03-23 | Nalpropion Pharmaceuticals Llc | Formulación de liberación sostenida de naltrexona |
| SI2719378T1 (sl) * | 2006-06-19 | 2016-11-30 | Alpharma Pharmaceuticals Llc | Farmacevtski sestavki |
| DK2089005T3 (da) * | 2006-11-09 | 2010-07-19 | Orexigen Therapeutics Inc | Lagdelte farmaceutiske formuleringer omfattende et hurtigt opløsende mellemlag |
| CN110893181A (zh) * | 2012-06-06 | 2020-03-20 | 纳丙药业有限责任公司 | 治疗超重和肥胖症的方法 |
| US8969371B1 (en) | 2013-12-06 | 2015-03-03 | Orexigen Therapeutics, Inc. | Compositions and methods for weight loss in at risk patient populations |
| WO2016125109A1 (en) * | 2015-02-07 | 2016-08-11 | Intas Pharmaceuticals Ltd. | Pharmaceutical composition for the treatment of obesity |
| WO2016125108A1 (en) * | 2015-02-07 | 2016-08-11 | Intas Pharmaceuticals Ltd. | Bilayer pharmaceutical composition for the treatment of obesity |
| CN111836618A (zh) * | 2018-02-08 | 2020-10-27 | 景凱生物科技股份有限公司 | 类阿片受体拮抗剂的固体剂型用的医药剂型 |
Family Cites Families (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0103636B1 (en) * | 1982-03-16 | 1990-09-12 | Rockefeller University | Use of opium antagonists for the manufacture of medicaments for controlling gastrointestinal dysmotility |
| US6306436B1 (en) * | 2000-04-28 | 2001-10-23 | Teva Pharmaceuticals Usa, Inc. | Stabilized, acid-free formulation for sustained release of bupropion hydrochloride |
| US7842307B2 (en) * | 2001-08-06 | 2010-11-30 | Purdue Pharma L.P. | Pharmaceutical formulation containing opioid agonist, opioid antagonist and gelling agent |
| BR0212019A (pt) * | 2001-08-06 | 2005-08-09 | Euro Celtique Sa | Formas de dosagem, métodos para o tratamento da dor, métodos de preparação de uma forma de dosagem e métodos para impedir o abuso de uma forma de dosagem |
| US6972291B2 (en) * | 2002-07-02 | 2005-12-06 | Bernstein Richard K | Method for reducing food intake |
| WO2004071423A2 (en) * | 2003-02-05 | 2004-08-26 | Euro-Celtique S.A. | Methods of administering opioid antagonists and compositions thereof |
| EP1617832B1 (en) | 2003-04-29 | 2008-03-12 | Orexigen Therapeutics, Inc. | Compositions for affecting weight loss |
| DE602004026604D1 (de) * | 2003-09-25 | 2010-05-27 | Euro Celtique Sa | Pharmazeutische kombinationen von hydrocodon und naltrexon |
| CN101370488B (zh) * | 2005-11-22 | 2012-07-18 | 奥雷西根治疗公司 | 增加胰岛素敏感性的组合物 |
| WO2007089318A2 (en) * | 2005-11-23 | 2007-08-09 | Orexigen Therapeutics, Inc. | Compositions and methods for reducing food cravings |
| ES2749800T3 (es) * | 2006-06-05 | 2020-03-23 | Nalpropion Pharmaceuticals Llc | Formulación de liberación sostenida de naltrexona |
-
2007
- 2007-05-31 ES ES07795652T patent/ES2749800T3/es active Active
- 2007-05-31 EP EP07795652.2A patent/EP2061448B1/en active Active
- 2007-05-31 JP JP2009514317A patent/JP5478245B2/ja active Active
- 2007-05-31 WO PCT/US2007/013030 patent/WO2007145863A2/en not_active Ceased
- 2007-05-31 EP EP19186587.2A patent/EP3626236A1/en active Pending
- 2007-06-04 TW TW096119980A patent/TW200808319A/zh unknown
- 2007-06-05 AR ARP070102417A patent/AR061233A1/es unknown
-
2013
- 2013-10-18 JP JP2013217496A patent/JP5921513B2/ja active Active
-
2015
- 2015-11-24 JP JP2015228613A patent/JP6174666B2/ja active Active
Also Published As
| Publication number | Publication date |
|---|---|
| EP2061448A2 (en) | 2009-05-27 |
| JP2014005310A (ja) | 2014-01-16 |
| WO2007145863A2 (en) | 2007-12-21 |
| TW200808319A (en) | 2008-02-16 |
| WO2007145863A3 (en) | 2008-05-29 |
| JP5478245B2 (ja) | 2014-04-23 |
| EP3626236A1 (en) | 2020-03-25 |
| EP2061448B1 (en) | 2019-07-17 |
| JP2009539837A (ja) | 2009-11-19 |
| JP6174666B2 (ja) | 2017-08-02 |
| AR061233A1 (es) | 2008-08-13 |
| JP2016029111A (ja) | 2016-03-03 |
| JP5921513B2 (ja) | 2016-05-24 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US8916195B2 (en) | Sustained release formulation of naltrexone | |
| ES2749800T3 (es) | Formulación de liberación sostenida de naltrexona | |
| DK2182902T3 (en) | Compositions comprising sufentanil and triazolam for procedure sedation and analgesia using oral transmucosal dosage forms | |
| ES2706407T3 (es) | Composiciones farmacéuticas de liberación inmediata que comprenden oxicodona y naloxona | |
| NO338714B1 (no) | Generelt lineær, brusende, oral fentanyl doseringsform som er egnet for administrering av fentanyl over en pasients orale slimhinner ved bukkal, gingival eller sublingval administrering, og fremgangsmåter for fremstilling derav. | |
| JP6511492B2 (ja) | 女性胃不全麻痺に関係する症状の処置 | |
| US20220151960A1 (en) | Treatment of symptoms associated with female gastroparesis | |
| ES2346290T3 (es) | Administracion nasal de metoclopramida para el tratamiento de gastroparesia. | |
| ES2610469T3 (es) | Nuevas formulaciones farmacéuticas útiles en el tratamiento del insomnio | |
| US10548838B1 (en) | Oral liquid compositions including valsartan | |
| Jaiswani et al. | Sublingual tablets: an overview | |
| TW201434464A (zh) | 藥物 | |
| US9364451B1 (en) | Alternating sympathomimetic therapy for the treatment of respiratory ailments | |
| WO2016023822A1 (en) | Afatinib pharmaceutical kit for cancer treatment | |
| Vernet et al. | Gastrointestinal decontamination for acute medication poisoning: implementation of advanced triage for activated charcoal administration | |
| WO2018204040A1 (en) | Oral liquid compositions of valsartan | |
| MXPA06007449A (es) | Forma de dosificacion oral de fentanil efervescente generalmente lineal y metodos para administrarla |