ES2757324T3 - Derivados de (4-hidroxi-2-fenil-1,3-tiazol-5-il)metanona como antagonistas de TRPM8 - Google Patents
Derivados de (4-hidroxi-2-fenil-1,3-tiazol-5-il)metanona como antagonistas de TRPM8 Download PDFInfo
- Publication number
- ES2757324T3 ES2757324T3 ES16823231T ES16823231T ES2757324T3 ES 2757324 T3 ES2757324 T3 ES 2757324T3 ES 16823231 T ES16823231 T ES 16823231T ES 16823231 T ES16823231 T ES 16823231T ES 2757324 T3 ES2757324 T3 ES 2757324T3
- Authority
- ES
- Spain
- Prior art keywords
- thiazol
- methanone
- hydroxy
- fluorophenyl
- oxazolidin
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 102000003610 TRPM8 Human genes 0.000 title description 39
- 101150111302 Trpm8 gene Proteins 0.000 title description 39
- 239000005557 antagonist Substances 0.000 title description 10
- QPPMIEOMODHKRD-UHFFFAOYSA-N OC=1N=C(SC=1C=O)C1=CC=CC=C1 Chemical class OC=1N=C(SC=1C=O)C1=CC=CC=C1 QPPMIEOMODHKRD-UHFFFAOYSA-N 0.000 title description 4
- 150000001875 compounds Chemical class 0.000 claims abstract description 79
- 125000001931 aliphatic group Chemical group 0.000 claims abstract description 7
- 125000004122 cyclic group Chemical group 0.000 claims abstract description 6
- 125000006273 (C1-C3) alkyl group Chemical group 0.000 claims abstract description 5
- 125000000753 cycloalkyl group Chemical group 0.000 claims abstract description 4
- 125000000217 alkyl group Chemical group 0.000 claims abstract description 3
- 125000006615 aromatic heterocyclic group Chemical group 0.000 claims abstract description 3
- 125000005842 heteroatom Chemical group 0.000 claims abstract description 3
- 229910052739 hydrogen Inorganic materials 0.000 claims abstract description 3
- 239000001257 hydrogen Substances 0.000 claims abstract description 3
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims abstract description 3
- 150000003839 salts Chemical class 0.000 claims abstract description 3
- -1 oxazinanyl Chemical group 0.000 claims description 34
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims description 31
- 208000002193 Pain Diseases 0.000 claims description 18
- 201000010099 disease Diseases 0.000 claims description 18
- BAJJMCUHUGXFST-UHFFFAOYSA-N OC=1N=C(SC=1C(=O)N1OCCCC1)C1=C(C=CC=C1)O Chemical compound OC=1N=C(SC=1C(=O)N1OCCCC1)C1=C(C=CC=C1)O BAJJMCUHUGXFST-UHFFFAOYSA-N 0.000 claims description 12
- LVRUKRLHASLAAP-UHFFFAOYSA-N [2-(3-fluorophenyl)-4-hydroxy-1,3-thiazol-5-yl]-(1,2-oxazolidin-2-yl)methanone Chemical compound FC=1C=C(C=CC=1)C=1SC(=C(N=1)O)C(=O)N1OCCC1 LVRUKRLHASLAAP-UHFFFAOYSA-N 0.000 claims description 12
- BKUQCBLRBYEPFS-UHFFFAOYSA-N [4-hydroxy-2-(2-hydroxyphenyl)-1,3-thiazol-5-yl]-(1,2-oxazolidin-2-yl)methanone Chemical compound OC=1N=C(SC=1C(=O)N1OCCC1)C1=C(C=CC=C1)O BKUQCBLRBYEPFS-UHFFFAOYSA-N 0.000 claims description 12
- NBRIJMBZONLPQR-UHFFFAOYSA-N FC=1C=C(C=CC=1)C=1SC(=C(N=1)O)C(=O)N1OCCCC1 Chemical compound FC=1C=C(C=CC=1)C=1SC(=C(N=1)O)C(=O)N1OCCCC1 NBRIJMBZONLPQR-UHFFFAOYSA-N 0.000 claims description 10
- 208000014001 urinary system disease Diseases 0.000 claims description 10
- 206010037211 Psychomotor hyperactivity Diseases 0.000 claims description 9
- KJMHJECRSYTRPH-UHFFFAOYSA-N FC=1C=C(C=CC=1)C=1SC(=C(N=1)O)C(=O)C1=NC=CC=C1 Chemical compound FC=1C=C(C=CC=1)C=1SC(=C(N=1)O)C(=O)C1=NC=CC=C1 KJMHJECRSYTRPH-UHFFFAOYSA-N 0.000 claims description 8
- QVFUCMJRIXYSFE-UHFFFAOYSA-N FC=1C=C(C=CC=1)C=1SC(=C(N=1)O)C(=O)C1NCCC1 Chemical compound FC=1C=C(C=CC=1)C=1SC(=C(N=1)O)C(=O)C1NCCC1 QVFUCMJRIXYSFE-UHFFFAOYSA-N 0.000 claims description 8
- XAUYANGDRWUPFJ-UHFFFAOYSA-N FC=1C=C(C=CC=1)C=1SC(=C(N=1)O)C(=O)C1OCCC1 Chemical compound FC=1C=C(C=CC=1)C=1SC(=C(N=1)O)C(=O)C1OCCC1 XAUYANGDRWUPFJ-UHFFFAOYSA-N 0.000 claims description 8
- NPQFJLHTPOZVJZ-UHFFFAOYSA-N FC=1C=C(C=CC=1)C=1SC(=C(N=1)O)C(=O)N1C=CC=C1 Chemical compound FC=1C=C(C=CC=1)C=1SC(=C(N=1)O)C(=O)N1C=CC=C1 NPQFJLHTPOZVJZ-UHFFFAOYSA-N 0.000 claims description 8
- ORXUXKZHQSHUAG-UHFFFAOYSA-N FC=1C=C(C=CC=1)C=1SC(=C(N=1)O)C(=O)N1CCCC1 Chemical compound FC=1C=C(C=CC=1)C=1SC(=C(N=1)O)C(=O)N1CCCC1 ORXUXKZHQSHUAG-UHFFFAOYSA-N 0.000 claims description 8
- KLDCQEIELFFOIH-UHFFFAOYSA-N FC=1C=C(C=CC=1)C=1SC(=C(N=1)O)C(=O)N1CCCCC1 Chemical compound FC=1C=C(C=CC=1)C=1SC(=C(N=1)O)C(=O)N1CCCCC1 KLDCQEIELFFOIH-UHFFFAOYSA-N 0.000 claims description 8
- NDBZTOVKRDHIGM-UHFFFAOYSA-N FC=1C=C(C=CC=1)C=1SC(=C(N=1)O)C(=O)N1CCOCC1 Chemical compound FC=1C=C(C=CC=1)C=1SC(=C(N=1)O)C(=O)N1CCOCC1 NDBZTOVKRDHIGM-UHFFFAOYSA-N 0.000 claims description 8
- XEVOKCFEKXPSNJ-UHFFFAOYSA-N FC=1C=C(C=CC=1)C=1SC(=C(N=1)O)C(=O)N1COCC1 Chemical compound FC=1C=C(C=CC=1)C=1SC(=C(N=1)O)C(=O)N1COCC1 XEVOKCFEKXPSNJ-UHFFFAOYSA-N 0.000 claims description 8
- OEVVEYKGMZWMKX-UHFFFAOYSA-N FC=1C=C(C=CC=1)C=1SC(=C(N=1)O)C(=O)N1COCCC1 Chemical compound FC=1C=C(C=CC=1)C=1SC(=C(N=1)O)C(=O)N1COCCC1 OEVVEYKGMZWMKX-UHFFFAOYSA-N 0.000 claims description 8
- VEXQZJOQCIAHLQ-UHFFFAOYSA-N OC=1N=C(SC=1C(=O)C1OCCC1)C1=C(C=CC=C1)O Chemical compound OC=1N=C(SC=1C(=O)C1OCCC1)C1=C(C=CC=C1)O VEXQZJOQCIAHLQ-UHFFFAOYSA-N 0.000 claims description 8
- OVYBUTGMADBNMU-UHFFFAOYSA-N OC=1N=C(SC=1C(=O)N1CCOCC1)C1=C(C=CC=C1)O Chemical compound OC=1N=C(SC=1C(=O)N1CCOCC1)C1=C(C=CC=C1)O OVYBUTGMADBNMU-UHFFFAOYSA-N 0.000 claims description 8
- BMMYXUZZAMKIQN-UHFFFAOYSA-N OC=1N=C(SC=1C(=O)N1COCC1)C1=C(C=CC=C1)O Chemical compound OC=1N=C(SC=1C(=O)N1COCC1)C1=C(C=CC=C1)O BMMYXUZZAMKIQN-UHFFFAOYSA-N 0.000 claims description 8
- ZRVSHPMTRPSTNV-UHFFFAOYSA-N OC=1N=C(SC=1C(=O)N1COCCC1)C1=C(C=CC=C1)O Chemical compound OC=1N=C(SC=1C(=O)N1COCCC1)C1=C(C=CC=C1)O ZRVSHPMTRPSTNV-UHFFFAOYSA-N 0.000 claims description 8
- CABSPXRFEQGTLX-UHFFFAOYSA-N [2-(3-fluorophenyl)-4-hydroxy-1,3-thiazol-5-yl]-(1-methylpyrrolidin-2-yl)methanone Chemical compound FC=1C=C(C=CC=1)C=1SC(=C(N=1)O)C(=O)C1N(CCC1)C CABSPXRFEQGTLX-UHFFFAOYSA-N 0.000 claims description 8
- FVTSSUXQMCHMPB-UHFFFAOYSA-N [2-(3-fluorophenyl)-4-hydroxy-1,3-thiazol-5-yl]-(5-methyl-1,2-oxazolidin-2-yl)methanone Chemical compound FC=1C=C(C=CC=1)C=1SC(=C(N=1)O)C(=O)N1OC(CC1)C FVTSSUXQMCHMPB-UHFFFAOYSA-N 0.000 claims description 8
- LGTRCNXYHJUTQS-UHFFFAOYSA-N [2-(3-fluorophenyl)-4-hydroxy-1,3-thiazol-5-yl]-(5-oxa-6-azaspiro[2.4]heptan-6-yl)methanone Chemical compound FC=1C=C(C=CC=1)C=1SC(=C(N=1)O)C(=O)N1OCC2(CC2)C1 LGTRCNXYHJUTQS-UHFFFAOYSA-N 0.000 claims description 8
- OZYWEUPJFWQEEM-UHFFFAOYSA-N [2-(3-fluorophenyl)-4-hydroxy-1,3-thiazol-5-yl]-(oxazepan-2-yl)methanone Chemical compound FC=1C=C(C=CC=1)C=1SC(=C(N=1)O)C(=O)N1OCCCCC1 OZYWEUPJFWQEEM-UHFFFAOYSA-N 0.000 claims description 8
- MCTHFJCJXDGKLU-UHFFFAOYSA-N [2-(3-fluorophenyl)-4-hydroxy-1,3-thiazol-5-yl]-(triazol-1-yl)methanone Chemical compound FC=1C=C(C=CC=1)C=1SC(=C(N=1)O)C(=O)N1N=NC=C1 MCTHFJCJXDGKLU-UHFFFAOYSA-N 0.000 claims description 8
- BOZUYBXFUPNNLM-UHFFFAOYSA-N [2-(3-fluorophenyl)-4-hydroxy-1,3-thiazol-5-yl]-imidazol-1-ylmethanone Chemical compound FC=1C=C(C=CC=1)C=1SC(=C(N=1)O)C(=O)N1C=NC=C1 BOZUYBXFUPNNLM-UHFFFAOYSA-N 0.000 claims description 8
- ACEZHWIESUFSMK-UHFFFAOYSA-N [2-(3-fluorophenyl)-4-hydroxy-1,3-thiazol-5-yl]-pyrazol-1-ylmethanone Chemical compound FC=1C=C(C=CC=1)C=1SC(=C(N=1)O)C(=O)N1N=CC=C1 ACEZHWIESUFSMK-UHFFFAOYSA-N 0.000 claims description 8
- IZAWZPGIRKXFCL-UHFFFAOYSA-N [2-(3-fluorophenyl)-4-hydroxy-1,3-thiazol-5-yl]-pyridin-3-ylmethanone Chemical compound FC=1C=C(C=CC=1)C=1SC(=C(N=1)O)C(=O)C=1C=NC=CC=1 IZAWZPGIRKXFCL-UHFFFAOYSA-N 0.000 claims description 8
- WIJZHWRWLFCSFM-UHFFFAOYSA-N [2-(3-fluorophenyl)-4-hydroxy-1,3-thiazol-5-yl]-pyridin-4-ylmethanone Chemical compound FC=1C=C(C=CC=1)C=1SC(=C(N=1)O)C(=O)C1=CC=NC=C1 WIJZHWRWLFCSFM-UHFFFAOYSA-N 0.000 claims description 8
- 239000008194 pharmaceutical composition Substances 0.000 claims description 8
- MWFIZCYOENJKQD-UHFFFAOYSA-N FC=1C=C(C=CC=1)C=1SC(=C(N=1)O)C(=O)C=1N(C=CC=1)C Chemical compound FC=1C=C(C=CC=1)C=1SC(=C(N=1)O)C(=O)C=1N(C=CC=1)C MWFIZCYOENJKQD-UHFFFAOYSA-N 0.000 claims description 7
- SQVXLBUCEUTBMV-UHFFFAOYSA-N FC=1C=C(C=CC=1)C=1SC(=C(N=1)O)C(=O)C=1NC=CC=1 Chemical compound FC=1C=C(C=CC=1)C=1SC(=C(N=1)O)C(=O)C=1NC=CC=1 SQVXLBUCEUTBMV-UHFFFAOYSA-N 0.000 claims description 7
- XAXNFUCDOJQVKB-UHFFFAOYSA-N FC=1C=C(C=CC=1)C=1SC(=C(N=1)O)C(=O)C=1OC=CC=1 Chemical compound FC=1C=C(C=CC=1)C=1SC(=C(N=1)O)C(=O)C=1OC=CC=1 XAXNFUCDOJQVKB-UHFFFAOYSA-N 0.000 claims description 7
- QCDLGIJVKQNJLZ-UHFFFAOYSA-N FC=1C=C(C=CC=1)C=1SC(=C(N=1)O)C(=O)C=1SC=CC=1 Chemical compound FC=1C=C(C=CC=1)C=1SC(=C(N=1)O)C(=O)C=1SC=CC=1 QCDLGIJVKQNJLZ-UHFFFAOYSA-N 0.000 claims description 7
- 230000002265 prevention Effects 0.000 claims description 7
- XSNIPIPYBWEKRC-UHFFFAOYSA-N 3,3a,4,5,6,6a-hexahydrocyclopenta[d][1,2]oxazol-2-yl-[2-(3-fluorophenyl)-4-hydroxy-1,3-thiazol-5-yl]methanone Chemical compound FC=1C=C(C=CC=1)C=1SC(=C(N=1)O)C(=O)N1OC2C(C1)CCC2 XSNIPIPYBWEKRC-UHFFFAOYSA-N 0.000 claims description 6
- 208000005615 Interstitial Cystitis Diseases 0.000 claims description 6
- 208000003251 Pruritus Diseases 0.000 claims description 6
- OYOXLCIWMWSLLS-UHFFFAOYSA-N [2-(3-fluorophenyl)-4-hydroxy-1,3-thiazol-5-yl]-(1-methyltetrazol-5-yl)methanone Chemical compound FC=1C=C(C=CC=1)C=1SC(=C(N=1)O)C(=O)C1=NN=NN1C OYOXLCIWMWSLLS-UHFFFAOYSA-N 0.000 claims description 6
- 208000002551 irritable bowel syndrome Diseases 0.000 claims description 6
- 208000004296 neuralgia Diseases 0.000 claims description 6
- 208000020016 psychiatric disease Diseases 0.000 claims description 6
- 208000023504 respiratory system disease Diseases 0.000 claims description 6
- 208000003556 Dry Eye Syndromes Diseases 0.000 claims description 5
- 241000909851 Epiphora Species 0.000 claims description 5
- 208000006673 asthma Diseases 0.000 claims description 5
- 230000004397 blinking Effects 0.000 claims description 5
- 208000028867 ischemia Diseases 0.000 claims description 5
- 208000016747 lacrimal apparatus disease Diseases 0.000 claims description 5
- 230000004770 neurodegeneration Effects 0.000 claims description 5
- RGSFGYAAUTVSQA-UHFFFAOYSA-N Cyclopentane Chemical compound C1CCCC1 RGSFGYAAUTVSQA-UHFFFAOYSA-N 0.000 claims description 4
- 208000006011 Stroke Diseases 0.000 claims description 4
- 239000004480 active ingredient Substances 0.000 claims description 4
- 208000021921 corneal disease Diseases 0.000 claims description 4
- 230000004489 tear production Effects 0.000 claims description 4
- 208000019901 Anxiety disease Diseases 0.000 claims description 3
- 206010004446 Benign prostatic hyperplasia Diseases 0.000 claims description 3
- 206010058019 Cancer Pain Diseases 0.000 claims description 3
- 208000000094 Chronic Pain Diseases 0.000 claims description 3
- 208000032131 Diabetic Neuropathies Diseases 0.000 claims description 3
- 208000001640 Fibromyalgia Diseases 0.000 claims description 3
- 206010019233 Headaches Diseases 0.000 claims description 3
- 208000004454 Hyperalgesia Diseases 0.000 claims description 3
- 206010071289 Lower urinary tract symptoms Diseases 0.000 claims description 3
- 208000019695 Migraine disease Diseases 0.000 claims description 3
- 208000028389 Nerve injury Diseases 0.000 claims description 3
- 208000004550 Postoperative Pain Diseases 0.000 claims description 3
- 208000004403 Prostatic Hyperplasia Diseases 0.000 claims description 3
- 206010053552 allodynia Diseases 0.000 claims description 3
- 230000036506 anxiety Effects 0.000 claims description 3
- 125000002541 furyl group Chemical group 0.000 claims description 3
- 231100000869 headache Toxicity 0.000 claims description 3
- 125000002883 imidazolyl group Chemical group 0.000 claims description 3
- 206010027599 migraine Diseases 0.000 claims description 3
- 125000002757 morpholinyl group Chemical group 0.000 claims description 3
- 230000008764 nerve damage Effects 0.000 claims description 3
- 230000001272 neurogenic effect Effects 0.000 claims description 3
- 208000021722 neuropathic pain Diseases 0.000 claims description 3
- 201000001119 neuropathy Diseases 0.000 claims description 3
- 230000007823 neuropathy Effects 0.000 claims description 3
- 125000000160 oxazolidinyl group Chemical group 0.000 claims description 3
- 125000005475 oxolanyl group Chemical group 0.000 claims description 3
- 239000000546 pharmaceutical excipient Substances 0.000 claims description 3
- 125000003386 piperidinyl group Chemical group 0.000 claims description 3
- 208000002815 pulmonary hypertension Diseases 0.000 claims description 3
- 125000003226 pyrazolyl group Chemical group 0.000 claims description 3
- 125000004076 pyridyl group Chemical group 0.000 claims description 3
- 125000000719 pyrrolidinyl group Chemical group 0.000 claims description 3
- 125000000168 pyrrolyl group Chemical group 0.000 claims description 3
- 125000001544 thienyl group Chemical group 0.000 claims description 3
- 125000004214 1-pyrrolidinyl group Chemical group [H]C1([H])N(*)C([H])([H])C([H])([H])C1([H])[H] 0.000 claims description 2
- 125000002941 2-furyl group Chemical group O1C([*])=C([H])C([H])=C1[H] 0.000 claims description 2
- 125000004105 2-pyridyl group Chemical group N1=C([*])C([H])=C([H])C([H])=C1[H] 0.000 claims description 2
- 125000004485 2-pyrrolidinyl group Chemical group [H]N1C([H])([H])C([H])([H])C([H])([H])C1([H])* 0.000 claims description 2
- 125000000339 4-pyridyl group Chemical group N1=C([H])C([H])=C([*])C([H])=C1[H] 0.000 claims description 2
- XDTMQSROBMDMFD-UHFFFAOYSA-N Cyclohexane Chemical compound C1CCCCC1 XDTMQSROBMDMFD-UHFFFAOYSA-N 0.000 claims description 2
- 208000026723 Urinary tract disease Diseases 0.000 claims description 2
- 125000003118 aryl group Chemical group 0.000 claims description 2
- 239000003085 diluting agent Substances 0.000 claims description 2
- DMEGYFMYUHOHGS-UHFFFAOYSA-N heptamethylene Natural products C1CCCCCC1 DMEGYFMYUHOHGS-UHFFFAOYSA-N 0.000 claims description 2
- 125000004573 morpholin-4-yl group Chemical group N1(CCOCC1)* 0.000 claims description 2
- 201000008482 osteoarthritis Diseases 0.000 claims description 2
- 125000000587 piperidin-1-yl group Chemical group [H]C1([H])N(*)C([H])([H])C([H])([H])C([H])([H])C1([H])[H] 0.000 claims description 2
- 206010039073 rheumatoid arthritis Diseases 0.000 claims description 2
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 claims 1
- 125000002962 imidazol-1-yl group Chemical group [*]N1C([H])=NC([H])=C1[H] 0.000 claims 1
- 230000007803 itching Effects 0.000 claims 1
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 115
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 73
- 230000015572 biosynthetic process Effects 0.000 description 44
- 238000003786 synthesis reaction Methods 0.000 description 44
- ILAHWRKJUDSMFH-UHFFFAOYSA-N boron tribromide Chemical compound BrB(Br)Br ILAHWRKJUDSMFH-UHFFFAOYSA-N 0.000 description 40
- 238000005160 1H NMR spectroscopy Methods 0.000 description 37
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 34
- OKKJLVBELUTLKV-MZCSYVLQSA-N Deuterated methanol Chemical compound [2H]OC([2H])([2H])[2H] OKKJLVBELUTLKV-MZCSYVLQSA-N 0.000 description 28
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 27
- 238000006243 chemical reaction Methods 0.000 description 26
- 239000007787 solid Substances 0.000 description 26
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 26
- 235000019439 ethyl acetate Nutrition 0.000 description 25
- 239000012074 organic phase Substances 0.000 description 24
- 239000000243 solution Substances 0.000 description 24
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 23
- 239000000741 silica gel Substances 0.000 description 23
- 229910002027 silica gel Inorganic materials 0.000 description 23
- 239000002904 solvent Substances 0.000 description 23
- PAUVFJUVKIXDJO-UHFFFAOYSA-N 2-(3-fluorophenyl)-4-methoxy-1,3-thiazole-5-carbonyl chloride Chemical compound FC=1C=C(C=CC=1)C=1SC(=C(N=1)OC)C(=O)Cl PAUVFJUVKIXDJO-UHFFFAOYSA-N 0.000 description 22
- 239000012043 crude product Substances 0.000 description 22
- 238000000034 method Methods 0.000 description 21
- 239000000203 mixture Substances 0.000 description 16
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 15
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 15
- 210000004027 cell Anatomy 0.000 description 14
- 230000000694 effects Effects 0.000 description 13
- 239000003921 oil Substances 0.000 description 10
- 235000019198 oils Nutrition 0.000 description 10
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 8
- XRCOFGJOIKSTDP-UHFFFAOYSA-N 4-methoxy-2-(2-methoxyphenyl)-1,3-thiazole-5-carbonyl chloride Chemical compound COC=1N=C(SC=1C(=O)Cl)C1=C(C=CC=C1)OC XRCOFGJOIKSTDP-UHFFFAOYSA-N 0.000 description 7
- 108091006146 Channels Proteins 0.000 description 7
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical class [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 7
- 208000035475 disorder Diseases 0.000 description 7
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 6
- 206010020853 Hypertonic bladder Diseases 0.000 description 6
- 239000003814 drug Substances 0.000 description 6
- 239000003480 eluent Substances 0.000 description 6
- 230000009467 reduction Effects 0.000 description 6
- 238000012360 testing method Methods 0.000 description 6
- 125000004180 3-fluorophenyl group Chemical group [H]C1=C([H])C(*)=C([H])C(F)=C1[H] 0.000 description 5
- 238000005863 Friedel-Crafts acylation reaction Methods 0.000 description 5
- 239000011575 calcium Substances 0.000 description 5
- 229940079593 drug Drugs 0.000 description 5
- 239000000975 dye Substances 0.000 description 5
- 208000013403 hyperactivity Diseases 0.000 description 5
- 238000000338 in vitro Methods 0.000 description 5
- 238000002347 injection Methods 0.000 description 5
- 239000007924 injection Substances 0.000 description 5
- 230000002018 overexpression Effects 0.000 description 5
- AICOOMRHRUFYCM-ZRRPKQBOSA-N oxazine, 1 Chemical compound C([C@@H]1[C@H](C(C[C@]2(C)[C@@H]([C@H](C)N(C)C)[C@H](O)C[C@]21C)=O)CC1=CC2)C[C@H]1[C@@]1(C)[C@H]2N=C(C(C)C)OC1 AICOOMRHRUFYCM-ZRRPKQBOSA-N 0.000 description 5
- JWJGEQSGAYIZRD-UHFFFAOYSA-N 3-fluorobenzenecarbothioamide Chemical compound NC(=S)C1=CC=CC(F)=C1 JWJGEQSGAYIZRD-UHFFFAOYSA-N 0.000 description 4
- NOOLISFMXDJSKH-UHFFFAOYSA-N DL-menthol Natural products CC(C)C1CCC(C)CC1O NOOLISFMXDJSKH-UHFFFAOYSA-N 0.000 description 4
- YNAVUWVOSKDBBP-UHFFFAOYSA-N Morpholine Chemical compound C1COCCN1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 description 4
- 235000019502 Orange oil Nutrition 0.000 description 4
- 208000009722 Overactive Urinary Bladder Diseases 0.000 description 4
- KAESVJOAVNADME-UHFFFAOYSA-N Pyrrole Chemical compound C=1C=CNC=1 KAESVJOAVNADME-UHFFFAOYSA-N 0.000 description 4
- 206010046543 Urinary incontinence Diseases 0.000 description 4
- 239000012267 brine Substances 0.000 description 4
- PFKFTWBEEFSNDU-UHFFFAOYSA-N carbonyldiimidazole Chemical compound C1=CN=CN1C(=O)N1C=CN=C1 PFKFTWBEEFSNDU-UHFFFAOYSA-N 0.000 description 4
- 229940125904 compound 1 Drugs 0.000 description 4
- 238000010511 deprotection reaction Methods 0.000 description 4
- 238000003818 flash chromatography Methods 0.000 description 4
- 230000005764 inhibitory process Effects 0.000 description 4
- 229940041616 menthol Drugs 0.000 description 4
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 4
- 239000010502 orange oil Substances 0.000 description 4
- 208000020629 overactive bladder Diseases 0.000 description 4
- 238000000746 purification Methods 0.000 description 4
- 229910052938 sodium sulfate Inorganic materials 0.000 description 4
- 235000011152 sodium sulphate Nutrition 0.000 description 4
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 4
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical compound ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 description 4
- NOOLISFMXDJSKH-UTLUCORTSA-N (+)-Neomenthol Chemical compound CC(C)[C@@H]1CC[C@@H](C)C[C@@H]1O NOOLISFMXDJSKH-UTLUCORTSA-N 0.000 description 3
- RAXXELZNTBOGNW-UHFFFAOYSA-N 1H-imidazole Chemical compound C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 3
- OISVCGZHLKNMSJ-UHFFFAOYSA-N 2,6-dimethylpyridine Chemical compound CC1=CC=CC(C)=N1 OISVCGZHLKNMSJ-UHFFFAOYSA-N 0.000 description 3
- ZDSVWRMNUIVVDY-UHFFFAOYSA-N 2-(3-fluorophenyl)-4-methoxy-1,3-thiazole-5-carboxylic acid Chemical compound FC=1C=C(C=CC=1)C=1SC(=C(N=1)OC)C(=O)O ZDSVWRMNUIVVDY-UHFFFAOYSA-N 0.000 description 3
- RCEFMOGVOYEGJN-UHFFFAOYSA-N 3-(2-hydroxyphenyl)-6-(3-nitrophenyl)-1,4-dihydropyrimidin-2-one Chemical compound OC1=CC=CC=C1N1C(=O)NC(C=2C=C(C=CC=2)[N+]([O-])=O)=CC1 RCEFMOGVOYEGJN-UHFFFAOYSA-N 0.000 description 3
- MDVYIGJINBYKOM-IBSWDFHHSA-N 3-[(1r,2s,5r)-5-methyl-2-propan-2-ylcyclohexyl]oxypropane-1,2-diol Chemical compound CC(C)[C@@H]1CC[C@@H](C)C[C@H]1OCC(O)CO MDVYIGJINBYKOM-IBSWDFHHSA-N 0.000 description 3
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical class [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 3
- 101100047459 Homo sapiens TRPM8 gene Proteins 0.000 description 3
- RWRDLPDLKQPQOW-UHFFFAOYSA-N Pyrrolidine Chemical compound C1CCNC1 RWRDLPDLKQPQOW-UHFFFAOYSA-N 0.000 description 3
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 3
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 3
- 230000003042 antagnostic effect Effects 0.000 description 3
- 238000003556 assay Methods 0.000 description 3
- 229940125898 compound 5 Drugs 0.000 description 3
- 238000005516 engineering process Methods 0.000 description 3
- NJIJXHPIIHCJKS-UHFFFAOYSA-N ethyl 2-(3-fluorophenyl)-4-hydroxy-1,3-thiazole-5-carboxylate Chemical compound CCOC(=O)c1sc(nc1O)-c1cccc(F)c1 NJIJXHPIIHCJKS-UHFFFAOYSA-N 0.000 description 3
- XGTLHMGXZKTEPO-UHFFFAOYSA-N ethyl 2-(3-fluorophenyl)-4-methoxy-1,3-thiazole-5-carboxylate Chemical compound FC=1C=C(C=CC=1)C=1SC(=C(N=1)OC)C(=O)OCC XGTLHMGXZKTEPO-UHFFFAOYSA-N 0.000 description 3
- 102000045979 human TRPM8 Human genes 0.000 description 3
- 230000006872 improvement Effects 0.000 description 3
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 3
- 239000002609 medium Substances 0.000 description 3
- 239000012071 phase Substances 0.000 description 3
- 102000005962 receptors Human genes 0.000 description 3
- 108020003175 receptors Proteins 0.000 description 3
- 235000017557 sodium bicarbonate Nutrition 0.000 description 3
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 3
- 230000000638 stimulation Effects 0.000 description 3
- 238000002560 therapeutic procedure Methods 0.000 description 3
- 238000004809 thin layer chromatography Methods 0.000 description 3
- 102000042565 transient receptor (TC 1.A.4) family Human genes 0.000 description 3
- 108091053409 transient receptor (TC 1.A.4) family Proteins 0.000 description 3
- WWTBZEKOSBFBEM-SPWPXUSOSA-N (2s)-2-[[2-benzyl-3-[hydroxy-[(1r)-2-phenyl-1-(phenylmethoxycarbonylamino)ethyl]phosphoryl]propanoyl]amino]-3-(1h-indol-3-yl)propanoic acid Chemical compound N([C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)O)C(=O)C(CP(O)(=O)[C@H](CC=1C=CC=CC=1)NC(=O)OCC=1C=CC=CC=1)CC1=CC=CC=C1 WWTBZEKOSBFBEM-SPWPXUSOSA-N 0.000 description 2
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 2
- SGTNSNPWRIOYBX-UHFFFAOYSA-N 2-(3,4-dimethoxyphenyl)-5-{[2-(3,4-dimethoxyphenyl)ethyl](methyl)amino}-2-(propan-2-yl)pentanenitrile Chemical compound C1=C(OC)C(OC)=CC=C1CCN(C)CCCC(C#N)(C(C)C)C1=CC=C(OC)C(OC)=C1 SGTNSNPWRIOYBX-UHFFFAOYSA-N 0.000 description 2
- XMXBHPAUMGOLFT-UHFFFAOYSA-N 2-(3-fluorophenyl)-N,4-dimethoxy-N-methyl-1,3-thiazole-5-carboxamide Chemical compound FC=1C=C(C=CC=1)C=1SC(=C(N=1)OC)C(=O)N(C)OC XMXBHPAUMGOLFT-UHFFFAOYSA-N 0.000 description 2
- CREQWRDCXSWBNK-UHFFFAOYSA-N 2-chloro-1,2-oxazolidine Chemical compound ClN1CCCO1 CREQWRDCXSWBNK-UHFFFAOYSA-N 0.000 description 2
- YNEHBLLUZJTDOL-UHFFFAOYSA-N 2-methoxybenzenecarbothioamide Chemical compound COC1=CC=CC=C1C(N)=S YNEHBLLUZJTDOL-UHFFFAOYSA-N 0.000 description 2
- GSADTPSKXMXLHL-UHFFFAOYSA-N 3-chloro-1,3-oxazinane Chemical compound ClN1CCCOC1 GSADTPSKXMXLHL-UHFFFAOYSA-N 0.000 description 2
- SELJCZDOXAOARL-UHFFFAOYSA-N 3-chloro-1,3-oxazolidine Chemical compound ClN1CCOC1 SELJCZDOXAOARL-UHFFFAOYSA-N 0.000 description 2
- VVEIQXDPBONNOQ-UHFFFAOYSA-N 4-methoxy-2-(2-methoxyphenyl)-1,3-thiazole Chemical compound COC1=CSC(C=2C(=CC=CC=2)OC)=N1 VVEIQXDPBONNOQ-UHFFFAOYSA-N 0.000 description 2
- LIFAQMGORKPVDH-UHFFFAOYSA-N 7-ethoxycoumarin Chemical compound C1=CC(=O)OC2=CC(OCC)=CC=C21 LIFAQMGORKPVDH-UHFFFAOYSA-N 0.000 description 2
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 2
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 2
- 208000006545 Chronic Obstructive Pulmonary Disease Diseases 0.000 description 2
- SGPLLGSDLAWOAU-UHFFFAOYSA-N FC=1C=C(C=CC=1)C=1SC=C(N=1)OC Chemical compound FC=1C=C(C=CC=1)C=1SC=C(N=1)OC SGPLLGSDLAWOAU-UHFFFAOYSA-N 0.000 description 2
- YLQBMQCUIZJEEH-UHFFFAOYSA-N Furan Chemical compound C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 2
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 2
- 229940121948 Muscarinic receptor antagonist Drugs 0.000 description 2
- OPFJDXRVMFKJJO-ZHHKINOHSA-N N-{[3-(2-benzamido-4-methyl-1,3-thiazol-5-yl)-pyrazol-5-yl]carbonyl}-G-dR-G-dD-dD-dD-NH2 Chemical compound S1C(C=2NN=C(C=2)C(=O)NCC(=O)N[C@H](CCCN=C(N)N)C(=O)NCC(=O)N[C@H](CC(O)=O)C(=O)N[C@H](CC(O)=O)C(=O)N[C@H](CC(O)=O)C(N)=O)=C(C)N=C1NC(=O)C1=CC=CC=C1 OPFJDXRVMFKJJO-ZHHKINOHSA-N 0.000 description 2
- 238000005481 NMR spectroscopy Methods 0.000 description 2
- 239000007832 Na2SO4 Substances 0.000 description 2
- PXIPVTKHYLBLMZ-UHFFFAOYSA-N Sodium azide Chemical compound [Na+].[N-]=[N+]=[N-] PXIPVTKHYLBLMZ-UHFFFAOYSA-N 0.000 description 2
- 108091008847 TRPM Proteins 0.000 description 2
- 102000027545 TRPM Human genes 0.000 description 2
- YTPLMLYBLZKORZ-UHFFFAOYSA-N Thiophene Chemical compound C=1C=CSC=1 YTPLMLYBLZKORZ-UHFFFAOYSA-N 0.000 description 2
- HZARZLUEOBIPSR-UHFFFAOYSA-N [2-(3-fluorophenyl)-4-methoxy-1,3-thiazol-5-yl]-(1-methyltetrazol-5-yl)methanone Chemical compound FC=1C=C(C=CC=1)C=1SC(=C(N=1)OC)C(=O)C1=NN=NN1C HZARZLUEOBIPSR-UHFFFAOYSA-N 0.000 description 2
- 230000004075 alteration Effects 0.000 description 2
- 229910052791 calcium Inorganic materials 0.000 description 2
- 238000005119 centrifugation Methods 0.000 description 2
- 239000003153 chemical reaction reagent Substances 0.000 description 2
- 239000000812 cholinergic antagonist Substances 0.000 description 2
- 229940126086 compound 21 Drugs 0.000 description 2
- 229940126208 compound 22 Drugs 0.000 description 2
- 238000000132 electrospray ionisation Methods 0.000 description 2
- 238000002474 experimental method Methods 0.000 description 2
- 230000014509 gene expression Effects 0.000 description 2
- 125000000623 heterocyclic group Chemical group 0.000 description 2
- 230000002757 inflammatory effect Effects 0.000 description 2
- 239000013067 intermediate product Substances 0.000 description 2
- 230000003834 intracellular effect Effects 0.000 description 2
- 210000001853 liver microsome Anatomy 0.000 description 2
- 239000011777 magnesium Substances 0.000 description 2
- 229910052749 magnesium Inorganic materials 0.000 description 2
- 239000000463 material Substances 0.000 description 2
- 239000011159 matrix material Substances 0.000 description 2
- 239000012528 membrane Substances 0.000 description 2
- 230000002503 metabolic effect Effects 0.000 description 2
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 2
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 2
- 210000001589 microsome Anatomy 0.000 description 2
- 239000003068 molecular probe Substances 0.000 description 2
- 210000005036 nerve Anatomy 0.000 description 2
- 238000010899 nucleation Methods 0.000 description 2
- ZRSNZINYAWTAHE-UHFFFAOYSA-N p-methoxybenzaldehyde Chemical compound COC1=CC=C(C=O)C=C1 ZRSNZINYAWTAHE-UHFFFAOYSA-N 0.000 description 2
- 230000007170 pathology Effects 0.000 description 2
- 239000008057 potassium phosphate buffer Substances 0.000 description 2
- 239000000843 powder Substances 0.000 description 2
- 239000000047 product Substances 0.000 description 2
- AQHHHDLHHXJYJD-UHFFFAOYSA-N propranolol Chemical compound C1=CC=C2C(OCC(O)CNC(C)C)=CC=CC2=C1 AQHHHDLHHXJYJD-UHFFFAOYSA-N 0.000 description 2
- 239000011541 reaction mixture Substances 0.000 description 2
- 230000011514 reflex Effects 0.000 description 2
- 229920006395 saturated elastomer Polymers 0.000 description 2
- 230000001953 sensory effect Effects 0.000 description 2
- 238000001228 spectrum Methods 0.000 description 2
- 210000003594 spinal ganglia Anatomy 0.000 description 2
- 239000000126 substance Substances 0.000 description 2
- 239000006228 supernatant Substances 0.000 description 2
- UJJLJRQIPMGXEZ-UHFFFAOYSA-N tetrahydro-2-furoic acid Chemical compound OC(=O)C1CCCO1 UJJLJRQIPMGXEZ-UHFFFAOYSA-N 0.000 description 2
- 230000001225 therapeutic effect Effects 0.000 description 2
- 230000001052 transient effect Effects 0.000 description 2
- 210000001170 unmyelinated nerve fiber Anatomy 0.000 description 2
- 229960001722 verapamil Drugs 0.000 description 2
- AOSZTAHDEDLTLQ-AZKQZHLXSA-N (1S,2S,4R,8S,9S,11S,12R,13S,19S)-6-[(3-chlorophenyl)methyl]-12,19-difluoro-11-hydroxy-8-(2-hydroxyacetyl)-9,13-dimethyl-6-azapentacyclo[10.8.0.02,9.04,8.013,18]icosa-14,17-dien-16-one Chemical compound C([C@@H]1C[C@H]2[C@H]3[C@]([C@]4(C=CC(=O)C=C4[C@@H](F)C3)C)(F)[C@@H](O)C[C@@]2([C@@]1(C1)C(=O)CO)C)N1CC1=CC=CC(Cl)=C1 AOSZTAHDEDLTLQ-AZKQZHLXSA-N 0.000 description 1
- IWZSHWBGHQBIML-ZGGLMWTQSA-N (3S,8S,10R,13S,14S,17S)-17-isoquinolin-7-yl-N,N,10,13-tetramethyl-2,3,4,7,8,9,11,12,14,15,16,17-dodecahydro-1H-cyclopenta[a]phenanthren-3-amine Chemical compound CN(C)[C@H]1CC[C@]2(C)C3CC[C@@]4(C)[C@@H](CC[C@@H]4c4ccc5ccncc5c4)[C@@H]3CC=C2C1 IWZSHWBGHQBIML-ZGGLMWTQSA-N 0.000 description 1
- WEEGYLXZBRQIMU-UHFFFAOYSA-N 1,8-cineole Natural products C1CC2CCC1(C)OC2(C)C WEEGYLXZBRQIMU-UHFFFAOYSA-N 0.000 description 1
- OUKQTRFCDKSEPL-UHFFFAOYSA-N 1-Methyl-2-pyrrolecarboxaldehyde Chemical compound CN1C=CC=C1C=O OUKQTRFCDKSEPL-UHFFFAOYSA-N 0.000 description 1
- OXHNLMTVIGZXSG-UHFFFAOYSA-N 1-Methylpyrrole Chemical compound CN1C=CC=C1 OXHNLMTVIGZXSG-UHFFFAOYSA-N 0.000 description 1
- 125000001462 1-pyrrolyl group Chemical group [*]N1C([H])=C([H])C([H])=C1[H] 0.000 description 1
- QWENRTYMTSOGBR-UHFFFAOYSA-N 1H-1,2,3-Triazole Chemical compound C=1C=NNN=1 QWENRTYMTSOGBR-UHFFFAOYSA-N 0.000 description 1
- 125000000389 2-pyrrolyl group Chemical group [H]N1C([*])=C([H])C([H])=C1[H] 0.000 description 1
- KDNIOKSLVIGAAN-UHFFFAOYSA-N 2-sulfamoylbenzoic acid Chemical class NS(=O)(=O)C1=CC=CC=C1C(O)=O KDNIOKSLVIGAAN-UHFFFAOYSA-N 0.000 description 1
- OZYYDDJMGFFTGA-UHFFFAOYSA-N 3,3a,4,5,6,6a-hexahydro-2h-cyclopenta[d][1,2]oxazole Chemical compound C1NOC2CCCC21 OZYYDDJMGFFTGA-UHFFFAOYSA-N 0.000 description 1
- YPIGHNIIXYSPKF-UHFFFAOYSA-N 3-fluorobenzamide Chemical compound NC(=O)C1=CC=CC(F)=C1 YPIGHNIIXYSPKF-UHFFFAOYSA-N 0.000 description 1
- 125000003349 3-pyridyl group Chemical group N1=C([H])C([*])=C([H])C([H])=C1[H] 0.000 description 1
- ROLMZTIHUMKEAI-UHFFFAOYSA-N 4,5-difluoro-2-hydroxybenzonitrile Chemical compound OC1=CC(F)=C(F)C=C1C#N ROLMZTIHUMKEAI-UHFFFAOYSA-N 0.000 description 1
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 1
- UAVCLEYOCKKASS-UHFFFAOYSA-N 5-methyl-1,2-oxazolidine Chemical compound CC1CCNO1 UAVCLEYOCKKASS-UHFFFAOYSA-N 0.000 description 1
- JGKSDWOZVQWSLG-UHFFFAOYSA-N 5-oxa-6-azaspiro[2.4]heptane Chemical compound C1CC11CONC1 JGKSDWOZVQWSLG-UHFFFAOYSA-N 0.000 description 1
- CJIJXIFQYOPWTF-UHFFFAOYSA-N 7-hydroxycoumarin Natural products O1C(=O)C=CC2=CC(O)=CC=C21 CJIJXIFQYOPWTF-UHFFFAOYSA-N 0.000 description 1
- 108091005462 Cation channels Proteins 0.000 description 1
- 229940126657 Compound 17 Drugs 0.000 description 1
- 206010010774 Constipation Diseases 0.000 description 1
- XFXPMWWXUTWYJX-UHFFFAOYSA-N Cyanide Chemical compound N#[C-] XFXPMWWXUTWYJX-UHFFFAOYSA-N 0.000 description 1
- PMPVIKIVABFJJI-UHFFFAOYSA-N Cyclobutane Chemical compound C1CCC1 PMPVIKIVABFJJI-UHFFFAOYSA-N 0.000 description 1
- NBSCHQHZLSJFNQ-GASJEMHNSA-N D-Glucose 6-phosphate Chemical compound OC1O[C@H](COP(O)(O)=O)[C@@H](O)[C@H](O)[C@H]1O NBSCHQHZLSJFNQ-GASJEMHNSA-N 0.000 description 1
- KDXKERNSBIXSRK-RXMQYKEDSA-N D-lysine Chemical compound NCCCC[C@@H](N)C(O)=O KDXKERNSBIXSRK-RXMQYKEDSA-N 0.000 description 1
- 206010013774 Dry eye Diseases 0.000 description 1
- WEEGYLXZBRQIMU-WAAGHKOSSA-N Eucalyptol Chemical compound C1C[C@H]2CC[C@]1(C)OC2(C)C WEEGYLXZBRQIMU-WAAGHKOSSA-N 0.000 description 1
- OUVXYXNWSVIOSJ-UHFFFAOYSA-N Fluo-4 Chemical compound CC1=CC=C(N(CC(O)=O)CC(O)=O)C(OCCOC=2C(=CC=C(C=2)C2=C3C=C(F)C(=O)C=C3OC3=CC(O)=C(F)C=C32)N(CC(O)=O)CC(O)=O)=C1 OUVXYXNWSVIOSJ-UHFFFAOYSA-N 0.000 description 1
- VFRROHXSMXFLSN-UHFFFAOYSA-N Glc6P Natural products OP(=O)(O)OCC(O)C(O)C(O)C(O)C=O VFRROHXSMXFLSN-UHFFFAOYSA-N 0.000 description 1
- 101001022015 Haloferax volcanii (strain ATCC 29605 / DSM 3757 / JCM 8879 / NBRC 14742 / NCIMB 2012 / VKM B-1768 / DS2) NAD-dependent glucose-6-phosphate dehydrogenase Proteins 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- 102000004310 Ion Channels Human genes 0.000 description 1
- 108090000862 Ion Channels Proteins 0.000 description 1
- ONIBWKKTOPOVIA-BYPYZUCNSA-N L-Proline Chemical compound OC(=O)[C@@H]1CCCN1 ONIBWKKTOPOVIA-BYPYZUCNSA-N 0.000 description 1
- 102100037611 Lysophospholipase Human genes 0.000 description 1
- 150000001200 N-acyl ethanolamides Chemical class 0.000 description 1
- CWLQUGTUXBXTLF-UHFFFAOYSA-N N-methyl-L-proline monohydrate Natural products CN1CCCC1C(O)=O CWLQUGTUXBXTLF-UHFFFAOYSA-N 0.000 description 1
- CWLQUGTUXBXTLF-YFKPBYRVSA-N N-methylproline Chemical compound CN1CCC[C@H]1C(O)=O CWLQUGTUXBXTLF-YFKPBYRVSA-N 0.000 description 1
- 108010058864 Phospholipases A2 Proteins 0.000 description 1
- ONIBWKKTOPOVIA-UHFFFAOYSA-N Proline Natural products OC(=O)C1CCCN1 ONIBWKKTOPOVIA-UHFFFAOYSA-N 0.000 description 1
- WTKZEGDFNFYCGP-UHFFFAOYSA-N Pyrazole Chemical compound C=1C=NNC=1 WTKZEGDFNFYCGP-UHFFFAOYSA-N 0.000 description 1
- 208000027520 Somatoform disease Diseases 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 1
- 108060008646 TRPA Proteins 0.000 description 1
- 108060008648 TRPC Proteins 0.000 description 1
- 102000027549 TRPC Human genes 0.000 description 1
- 229940123537 TRPM8 antagonist Drugs 0.000 description 1
- 108091008846 TRPML Proteins 0.000 description 1
- 102000027544 TRPML Human genes 0.000 description 1
- 108060009332 TRPP Proteins 0.000 description 1
- 102000003563 TRPV Human genes 0.000 description 1
- 108060008564 TRPV Proteins 0.000 description 1
- 102000011753 Transient Receptor Potential Channels Human genes 0.000 description 1
- 108010037150 Transient Receptor Potential Channels Proteins 0.000 description 1
- HDYANYHVCAPMJV-LXQIFKJMSA-N UDP-alpha-D-glucuronic acid Chemical compound C([C@@H]1[C@H]([C@H]([C@@H](O1)N1C(NC(=O)C=C1)=O)O)O)OP(O)(=O)OP(O)(=O)O[C@H]1O[C@H](C(O)=O)[C@@H](O)[C@H](O)[C@H]1O HDYANYHVCAPMJV-LXQIFKJMSA-N 0.000 description 1
- HDYANYHVCAPMJV-UHFFFAOYSA-N Uridine diphospho-D-glucuronic acid Natural products O1C(N2C(NC(=O)C=C2)=O)C(O)C(O)C1COP(O)(=O)OP(O)(=O)OC1OC(C(O)=O)C(O)C(O)C1O HDYANYHVCAPMJV-UHFFFAOYSA-N 0.000 description 1
- 239000009975 Urodyn Substances 0.000 description 1
- 206010047571 Visual impairment Diseases 0.000 description 1
- LNUFLCYMSVYYNW-ZPJMAFJPSA-N [(2r,3r,4s,5r,6r)-2-[(2r,3r,4s,5r,6r)-6-[(2r,3r,4s,5r,6r)-6-[(2r,3r,4s,5r,6r)-6-[[(3s,5s,8r,9s,10s,13r,14s,17r)-10,13-dimethyl-17-[(2r)-6-methylheptan-2-yl]-2,3,4,5,6,7,8,9,11,12,14,15,16,17-tetradecahydro-1h-cyclopenta[a]phenanthren-3-yl]oxy]-4,5-disulfo Chemical compound O([C@@H]1[C@@H](COS(O)(=O)=O)O[C@@H]([C@@H]([C@H]1OS(O)(=O)=O)OS(O)(=O)=O)O[C@@H]1[C@@H](COS(O)(=O)=O)O[C@@H]([C@@H]([C@H]1OS(O)(=O)=O)OS(O)(=O)=O)O[C@@H]1[C@@H](COS(O)(=O)=O)O[C@H]([C@@H]([C@H]1OS(O)(=O)=O)OS(O)(=O)=O)O[C@@H]1C[C@@H]2CC[C@H]3[C@@H]4CC[C@@H]([C@]4(CC[C@@H]3[C@@]2(C)CC1)C)[C@H](C)CCCC(C)C)[C@H]1O[C@H](COS(O)(=O)=O)[C@@H](OS(O)(=O)=O)[C@H](OS(O)(=O)=O)[C@H]1OS(O)(=O)=O LNUFLCYMSVYYNW-ZPJMAFJPSA-N 0.000 description 1
- QWODCTMAIUYXFJ-UHFFFAOYSA-N [2-(3-fluorophenyl)-4-methoxy-1,3-thiazol-5-yl]-(1,2-oxazolidin-2-yl)methanone Chemical compound FC=1C=C(C=CC=1)C=1SC(=C(N=1)OC)C(=O)N1OCCC1 QWODCTMAIUYXFJ-UHFFFAOYSA-N 0.000 description 1
- YXTXYPPADZYEFB-UHFFFAOYSA-N [2-(3-fluorophenyl)-4-methoxy-1,3-thiazol-5-yl]-(1,3-oxazinan-3-yl)methanone Chemical compound FC=1C=C(C=CC=1)C=1SC(=C(N=1)OC)C(=O)N1COCCC1 YXTXYPPADZYEFB-UHFFFAOYSA-N 0.000 description 1
- MSCOLZMXJPERTD-UHFFFAOYSA-N [2-(3-fluorophenyl)-4-methoxy-1,3-thiazol-5-yl]-(1,3-oxazolidin-3-yl)methanone Chemical compound FC=1C=C(C=CC=1)C=1SC(=C(N=1)OC)C(=O)N1COCC1 MSCOLZMXJPERTD-UHFFFAOYSA-N 0.000 description 1
- DJOWCMVJYQTEPN-UHFFFAOYSA-N [2-(3-fluorophenyl)-4-methoxy-1,3-thiazol-5-yl]-(oxazinan-2-yl)methanone Chemical compound FC=1C=C(C=CC=1)C=1SC(=C(N=1)OC)C(=O)N1OCCCC1 DJOWCMVJYQTEPN-UHFFFAOYSA-N 0.000 description 1
- XZKWGGRDFMOZPI-UHFFFAOYSA-N [2-(3-fluorophenyl)-4-methoxy-1,3-thiazol-5-yl]-(oxolan-2-yl)methanone Chemical compound FC=1C=C(C=CC=1)C=1SC(=C(N=1)OC)C(=O)C1OCCC1 XZKWGGRDFMOZPI-UHFFFAOYSA-N 0.000 description 1
- HWXISMWNZLDXOF-UHFFFAOYSA-N [2-(3-fluorophenyl)-4-methoxy-1,3-thiazol-5-yl]-morpholin-4-ylmethanone Chemical compound FC=1C=C(C=CC=1)C=1SC(=C(N=1)OC)C(=O)N1CCOCC1 HWXISMWNZLDXOF-UHFFFAOYSA-N 0.000 description 1
- MNVDYEWPPIPWIA-UHFFFAOYSA-N [2-(3-fluorophenyl)-4-methoxy-1,3-thiazol-5-yl]-piperidin-1-ylmethanone Chemical compound FC=1C=C(C=CC=1)C=1SC(=C(N=1)OC)C(=O)N1CCCCC1 MNVDYEWPPIPWIA-UHFFFAOYSA-N 0.000 description 1
- ZHSODSMJVGIZJX-UHFFFAOYSA-N [2-(3-fluorophenyl)-4-methoxy-1,3-thiazol-5-yl]-pyridin-2-ylmethanone Chemical compound FC=1C=C(C=CC=1)C=1SC(=C(N=1)OC)C(=O)C1=NC=CC=C1 ZHSODSMJVGIZJX-UHFFFAOYSA-N 0.000 description 1
- RFSLDUDLGFOGGV-UHFFFAOYSA-N [2-(3-fluorophenyl)-4-methoxy-1,3-thiazol-5-yl]-pyrrol-1-ylmethanone Chemical compound FC=1C=C(C=CC=1)C=1SC(=C(N=1)OC)C(=O)N1C=CC=C1 RFSLDUDLGFOGGV-UHFFFAOYSA-N 0.000 description 1
- AGBHOBOOEACRRS-UHFFFAOYSA-N [2-(3-fluorophenyl)-4-methoxy-1,3-thiazol-5-yl]-pyrrolidin-1-ylmethanone Chemical compound FC=1C=C(C=CC=1)C=1SC(=C(N=1)OC)C(=O)N1CCCC1 AGBHOBOOEACRRS-UHFFFAOYSA-N 0.000 description 1
- JOSCNYCOYXTLTN-GFCCVEGCSA-N [3-[4-(aminomethyl)-6-(trifluoromethyl)pyridin-2-yl]oxyphenyl]-[(3R)-3-(hydroxymethyl)pyrrolidin-1-yl]methanone Chemical compound NCC1=CC(=NC(=C1)C(F)(F)F)OC=1C=C(C=CC=1)C(=O)N1C[C@@H](CC1)CO JOSCNYCOYXTLTN-GFCCVEGCSA-N 0.000 description 1
- BXVYQYNLSNRGAX-UHFFFAOYSA-N [4-methoxy-2-(2-methoxyphenyl)-1,3-thiazol-5-yl]-(1,2-oxazolidin-2-yl)methanone Chemical compound COC=1N=C(SC=1C(=O)N1OCCC1)C1=C(C=CC=C1)OC BXVYQYNLSNRGAX-UHFFFAOYSA-N 0.000 description 1
- PALCZFJTHZOTMV-UHFFFAOYSA-N [4-methoxy-2-(2-methoxyphenyl)-1,3-thiazol-5-yl]-(1,3-oxazinan-3-yl)methanone Chemical compound COC=1N=C(SC=1C(=O)N1COCCC1)C1=C(C=CC=C1)OC PALCZFJTHZOTMV-UHFFFAOYSA-N 0.000 description 1
- WWSLVUJKAFBJHA-UHFFFAOYSA-N [4-methoxy-2-(2-methoxyphenyl)-1,3-thiazol-5-yl]-(1,3-oxazolidin-3-yl)methanone Chemical compound COC=1N=C(SC=1C(=O)N1COCC1)C1=C(C=CC=C1)OC WWSLVUJKAFBJHA-UHFFFAOYSA-N 0.000 description 1
- LCMHDMVUNXZJGH-UHFFFAOYSA-N [4-methoxy-2-(2-methoxyphenyl)-1,3-thiazol-5-yl]-(oxazinan-2-yl)methanone Chemical compound COC=1N=C(SC=1C(=O)N1OCCCC1)C1=C(C=CC=C1)OC LCMHDMVUNXZJGH-UHFFFAOYSA-N 0.000 description 1
- XMTLXBPXUBXZBT-UHFFFAOYSA-N [4-methoxy-2-(2-methoxyphenyl)-1,3-thiazol-5-yl]-morpholin-4-ylmethanone Chemical compound COC=1N=C(SC=1C(=O)N1CCOCC1)C1=C(C=CC=C1)OC XMTLXBPXUBXZBT-UHFFFAOYSA-N 0.000 description 1
- FPXIHSSYZHFTJX-UHFFFAOYSA-M [Br-].[Mg+]C1=CC=CC=N1 Chemical compound [Br-].[Mg+]C1=CC=CC=N1 FPXIHSSYZHFTJX-UHFFFAOYSA-M 0.000 description 1
- XJLXINKUBYWONI-DQQFMEOOSA-N [[(2r,3r,4r,5r)-5-(6-aminopurin-9-yl)-3-hydroxy-4-phosphonooxyoxolan-2-yl]methoxy-hydroxyphosphoryl] [(2s,3r,4s,5s)-5-(3-carbamoylpyridin-1-ium-1-yl)-3,4-dihydroxyoxolan-2-yl]methyl phosphate Chemical compound NC(=O)C1=CC=C[N+]([C@@H]2[C@H]([C@@H](O)[C@H](COP([O-])(=O)OP(O)(=O)OC[C@@H]3[C@H]([C@@H](OP(O)(O)=O)[C@@H](O3)N3C4=NC=NC(N)=C4N=C3)O)O2)O)=C1 XJLXINKUBYWONI-DQQFMEOOSA-N 0.000 description 1
- 230000002159 abnormal effect Effects 0.000 description 1
- 230000005856 abnormality Effects 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 230000004913 activation Effects 0.000 description 1
- 239000000556 agonist Substances 0.000 description 1
- VREFGVBLTWBCJP-UHFFFAOYSA-N alprazolam Chemical compound C12=CC(Cl)=CC=C2N2C(C)=NN=C2CN=C1C1=CC=CC=C1 VREFGVBLTWBCJP-UHFFFAOYSA-N 0.000 description 1
- 235000019270 ammonium chloride Nutrition 0.000 description 1
- VZTDIZULWFCMLS-UHFFFAOYSA-N ammonium formate Chemical compound [NH4+].[O-]C=O VZTDIZULWFCMLS-UHFFFAOYSA-N 0.000 description 1
- APUPEJJSWDHEBO-UHFFFAOYSA-P ammonium molybdate Chemical compound [NH4+].[NH4+].[O-][Mo]([O-])(=O)=O APUPEJJSWDHEBO-UHFFFAOYSA-P 0.000 description 1
- 235000018660 ammonium molybdate Nutrition 0.000 description 1
- 239000011609 ammonium molybdate Substances 0.000 description 1
- 229940010552 ammonium molybdate Drugs 0.000 description 1
- 238000004458 analytical method Methods 0.000 description 1
- 239000008346 aqueous phase Substances 0.000 description 1
- 150000001491 aromatic compounds Chemical class 0.000 description 1
- 230000002917 arthritic effect Effects 0.000 description 1
- 239000012131 assay buffer Substances 0.000 description 1
- 125000000051 benzyloxy group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])O* 0.000 description 1
- 238000004166 bioassay Methods 0.000 description 1
- 230000033228 biological regulation Effects 0.000 description 1
- 210000004204 blood vessel Anatomy 0.000 description 1
- 210000004556 brain Anatomy 0.000 description 1
- 239000000872 buffer Substances 0.000 description 1
- DRCMAZOSEIMCHM-UHFFFAOYSA-N capsazepine Chemical compound C1C=2C=C(O)C(O)=CC=2CCCN1C(=S)NCCC1=CC=C(Cl)C=C1 DRCMAZOSEIMCHM-UHFFFAOYSA-N 0.000 description 1
- 239000002775 capsule Substances 0.000 description 1
- 230000003197 catalytic effect Effects 0.000 description 1
- 125000002091 cationic group Chemical group 0.000 description 1
- 150000001768 cations Chemical class 0.000 description 1
- 239000006143 cell culture medium Substances 0.000 description 1
- 210000003169 central nervous system Anatomy 0.000 description 1
- 238000011097 chromatography purification Methods 0.000 description 1
- 229960005233 cineole Drugs 0.000 description 1
- 238000004891 communication Methods 0.000 description 1
- 229940125810 compound 20 Drugs 0.000 description 1
- 230000008602 contraction Effects 0.000 description 1
- 239000013068 control sample Substances 0.000 description 1
- 238000013270 controlled release Methods 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- 239000002826 coolant Substances 0.000 description 1
- 238000001816 cooling Methods 0.000 description 1
- 210000004087 cornea Anatomy 0.000 description 1
- 230000008878 coupling Effects 0.000 description 1
- 238000010168 coupling process Methods 0.000 description 1
- 238000005859 coupling reaction Methods 0.000 description 1
- 239000006071 cream Substances 0.000 description 1
- 238000002425 crystallisation Methods 0.000 description 1
- 230000008025 crystallization Effects 0.000 description 1
- WHHGLZMJPXIBIX-UHFFFAOYSA-N decabromodiphenyl ether Chemical compound BrC1=C(Br)C(Br)=C(Br)C(Br)=C1OC1=C(Br)C(Br)=C(Br)C(Br)=C1Br WHHGLZMJPXIBIX-UHFFFAOYSA-N 0.000 description 1
- 230000003247 decreasing effect Effects 0.000 description 1
- 230000001419 dependent effect Effects 0.000 description 1
- 238000001514 detection method Methods 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- FNJVDWXUKLTFFL-UHFFFAOYSA-N diethyl 2-bromopropanedioate Chemical compound CCOC(=O)C(Br)C(=O)OCC FNJVDWXUKLTFFL-UHFFFAOYSA-N 0.000 description 1
- 239000006185 dispersion Substances 0.000 description 1
- 206010013781 dry mouth Diseases 0.000 description 1
- 239000002621 endocannabinoid Substances 0.000 description 1
- 230000005284 excitation Effects 0.000 description 1
- 238000001506 fluorescence spectroscopy Methods 0.000 description 1
- 239000007850 fluorescent dye Substances 0.000 description 1
- 230000004907 flux Effects 0.000 description 1
- 235000019253 formic acid Nutrition 0.000 description 1
- 238000009472 formulation Methods 0.000 description 1
- CNUDBTRUORMMPA-UHFFFAOYSA-N formylthiophene Chemical compound O=CC1=CC=CS1 CNUDBTRUORMMPA-UHFFFAOYSA-N 0.000 description 1
- HYBBIBNJHNGZAN-UHFFFAOYSA-N furfural Chemical compound O=CC1=CC=CO1 HYBBIBNJHNGZAN-UHFFFAOYSA-N 0.000 description 1
- 210000001035 gastrointestinal tract Anatomy 0.000 description 1
- JAXFJECJQZDFJS-XHEPKHHKSA-N gtpl8555 Chemical compound OC(=O)C[C@H](N)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](C(C)C)C(=O)N1CCC[C@@H]1C(=O)N[C@H](B1O[C@@]2(C)[C@H]3C[C@H](C3(C)C)C[C@H]2O1)CCC1=CC=C(F)C=C1 JAXFJECJQZDFJS-XHEPKHHKSA-N 0.000 description 1
- 238000004128 high performance liquid chromatography Methods 0.000 description 1
- 238000000589 high-performance liquid chromatography-mass spectrometry Methods 0.000 description 1
- BHEPBYXIRTUNPN-UHFFFAOYSA-N hydridophosphorus(.) (triplet) Chemical group [PH] BHEPBYXIRTUNPN-UHFFFAOYSA-N 0.000 description 1
- USZLCYNVCCDPLQ-UHFFFAOYSA-N hydron;n-methoxymethanamine;chloride Chemical compound Cl.CNOC USZLCYNVCCDPLQ-UHFFFAOYSA-N 0.000 description 1
- 239000005457 ice water Substances 0.000 description 1
- 238000003384 imaging method Methods 0.000 description 1
- 210000002865 immune cell Anatomy 0.000 description 1
- 230000001771 impaired effect Effects 0.000 description 1
- 208000027866 inflammatory disease Diseases 0.000 description 1
- 238000001990 intravenous administration Methods 0.000 description 1
- 238000010253 intravenous injection Methods 0.000 description 1
- INQOMBQAUSQDDS-UHFFFAOYSA-N iodomethane Chemical compound IC INQOMBQAUSQDDS-UHFFFAOYSA-N 0.000 description 1
- 150000002500 ions Chemical class 0.000 description 1
- CFHGBZLNZZVTAY-UHFFFAOYSA-N lawesson's reagent Chemical compound C1=CC(OC)=CC=C1P1(=S)SP(=S)(C=2C=CC(OC)=CC=2)S1 CFHGBZLNZZVTAY-UHFFFAOYSA-N 0.000 description 1
- 239000006210 lotion Substances 0.000 description 1
- 210000004072 lung Anatomy 0.000 description 1
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 1
- 235000019341 magnesium sulphate Nutrition 0.000 description 1
- 238000004519 manufacturing process Methods 0.000 description 1
- 230000007246 mechanism Effects 0.000 description 1
- 230000001404 mediated effect Effects 0.000 description 1
- 238000002483 medication Methods 0.000 description 1
- VNWKTOKETHGBQD-UHFFFAOYSA-N methane Natural products C VNWKTOKETHGBQD-UHFFFAOYSA-N 0.000 description 1
- HHVOOJDLCVOLKI-VWLOTQADSA-N methyl (2S)-2-(dibenzylamino)-3-(1H-indol-3-yl)propanoate Chemical compound C=1C=CC=CC=1CN([C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)OC)CC1=CC=CC=C1 HHVOOJDLCVOLKI-VWLOTQADSA-N 0.000 description 1
- YDCHPLOFQATIDS-UHFFFAOYSA-N methyl 2-bromoacetate Chemical compound COC(=O)CBr YDCHPLOFQATIDS-UHFFFAOYSA-N 0.000 description 1
- 230000003228 microsomal effect Effects 0.000 description 1
- 230000027939 micturition Effects 0.000 description 1
- 210000003205 muscle Anatomy 0.000 description 1
- 230000003767 neural control Effects 0.000 description 1
- LYGJENNIWJXYER-UHFFFAOYSA-N nitromethane Chemical compound C[N+]([O-])=O LYGJENNIWJXYER-UHFFFAOYSA-N 0.000 description 1
- 210000000929 nociceptor Anatomy 0.000 description 1
- 239000002674 ointment Substances 0.000 description 1
- 230000003287 optical effect Effects 0.000 description 1
- 230000003349 osteoarthritic effect Effects 0.000 description 1
- 125000001715 oxadiazolyl group Chemical group 0.000 description 1
- 208000027753 pain disease Diseases 0.000 description 1
- 210000005037 parasympathetic nerve Anatomy 0.000 description 1
- 230000002093 peripheral effect Effects 0.000 description 1
- 230000000144 pharmacologic effect Effects 0.000 description 1
- 150000003053 piperidines Chemical class 0.000 description 1
- 239000012286 potassium permanganate Substances 0.000 description 1
- 230000003389 potentiating effect Effects 0.000 description 1
- 238000011533 pre-incubation Methods 0.000 description 1
- 238000002360 preparation method Methods 0.000 description 1
- DBABZHXKTCFAPX-UHFFFAOYSA-N probenecid Chemical compound CCCN(CCC)S(=O)(=O)C1=CC=C(C(O)=O)C=C1 DBABZHXKTCFAPX-UHFFFAOYSA-N 0.000 description 1
- 229960003081 probenecid Drugs 0.000 description 1
- 108090000765 processed proteins & peptides Proteins 0.000 description 1
- 229960002429 proline Drugs 0.000 description 1
- 229960003712 propranolol Drugs 0.000 description 1
- 210000005267 prostate cell Anatomy 0.000 description 1
- 102000004169 proteins and genes Human genes 0.000 description 1
- 108090000623 proteins and genes Proteins 0.000 description 1
- ZSKGQVFRTSEPJT-UHFFFAOYSA-N pyrrole-2-carboxaldehyde Chemical compound O=CC1=CC=CN1 ZSKGQVFRTSEPJT-UHFFFAOYSA-N 0.000 description 1
- 230000009158 reflex pathway Effects 0.000 description 1
- 230000002787 reinforcement Effects 0.000 description 1
- 230000004044 response Effects 0.000 description 1
- 230000025488 response to cold Effects 0.000 description 1
- 238000000926 separation method Methods 0.000 description 1
- 239000000377 silicon dioxide Substances 0.000 description 1
- 230000016160 smooth muscle contraction Effects 0.000 description 1
- 239000011734 sodium Substances 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 230000000392 somatic effect Effects 0.000 description 1
- 230000002048 spasmolytic effect Effects 0.000 description 1
- 239000007921 spray Substances 0.000 description 1
- 229940124530 sulfonamide Drugs 0.000 description 1
- 150000003456 sulfonamides Chemical class 0.000 description 1
- 239000000725 suspension Substances 0.000 description 1
- 208000011580 syndromic disease Diseases 0.000 description 1
- 239000006188 syrup Substances 0.000 description 1
- 235000020357 syrup Nutrition 0.000 description 1
- 239000003826 tablet Substances 0.000 description 1
- NQRYJNQNLNOLGT-UHFFFAOYSA-N tetrahydropyridine hydrochloride Natural products C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 1
- CZDYPVPMEAXLPK-UHFFFAOYSA-N tetramethylsilane Chemical compound C[Si](C)(C)C CZDYPVPMEAXLPK-UHFFFAOYSA-N 0.000 description 1
- 150000007979 thiazole derivatives Chemical class 0.000 description 1
- 229930192474 thiophene Natural products 0.000 description 1
- 230000000699 topical effect Effects 0.000 description 1
- 230000009466 transformation Effects 0.000 description 1
- 210000000427 trigeminal ganglion Anatomy 0.000 description 1
- ORHBXUUXSCNDEV-UHFFFAOYSA-N umbelliferone Chemical compound C1=CC(=O)OC2=CC(O)=CC=C21 ORHBXUUXSCNDEV-UHFFFAOYSA-N 0.000 description 1
- 210000003932 urinary bladder Anatomy 0.000 description 1
- 230000002485 urinary effect Effects 0.000 description 1
- 229960004847 urologicals Drugs 0.000 description 1
- 210000003741 urothelium Anatomy 0.000 description 1
- 238000005292 vacuum distillation Methods 0.000 description 1
- 230000004393 visual impairment Effects 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/06—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/425—Thiazoles
- A61K31/427—Thiazoles not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4427—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems
- A61K31/4439—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems containing a five-membered ring with nitrogen as a ring hetero atom, e.g. omeprazole
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/5355—Non-condensed oxazines and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D277/00—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings
- C07D277/02—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings
- C07D277/20—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D277/32—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D277/56—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Medicinal Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Epidemiology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Pain & Pain Management (AREA)
- Pulmonology (AREA)
- Rheumatology (AREA)
- Biomedical Technology (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
Abstract
Compuesto de fórmula (I):**Fórmula** en la que R1 es un grupo heterocíclico alifático o aromático, de 5, 6 o 7 miembros sustituido o no sustituido que contiene 1, 2, 3 o 4 heteroátomos seleccionados de N, O y S, en el que, cuando está sustituido, dicho heterociclo alifático o aromático, de 5, 6 o 7 miembros está sustituido con uno o más grupos independientemente seleccionados de cicloalquilo o alquilo C1-C3, Cl y F o está sustituido con R3 y R4 que tomados juntos forman un resto cíclico saturado; y R2 se selecciona de hidrógeno, alquilo C1-C2, F, Cl y OH; y sales farmacéuticamente aceptables del mismo.
Description
DESCRIPCIÓN
Derivados de (4-hidroxi-2-fenil-1,3-tiazol-5-il)metanona como antagonistas de TRPM8
Breve descripción de la invención
La presente invención se refiere a derivados de 2-fenil-4-hidroxi-1,3-tiazol-5-ilmetanona que son útiles para la prevención, la reducción del riesgo de, la mejora y/o el tratamiento de enfermedades asociadas con la actividad del miembro 8 de la subfamilia M de canal catiónico de potencial de receptor transitorio (a continuación en el presente documento TRPM8) también conocido como receptor de mentol frío (CMR-1), y en particular para la prevención, la reducción del riesgo de, la mejora y/o el tratamiento de prurito, enfermedades del intestino irritable, trastornos respiratorios exacerbados y/o inducidos por frío, isquemia, dolor, neurodegeneración, trastornos psiquiátricos, accidente cerebrovascular, trastornos urológicos, trastornos de la córnea asociados a alteraciones en la producción de las lágrimas y/o parpadeo alterado tal como epífora y enfermedad de ojo seco. La invención se refiere además a composiciones farmacéuticas que contienen los compuestos anteriores.
Antecedentes de la invención
Los canales de potencial de receptor transitorio (TRP) son uno de los grupos más grandes de canales iónicos y, basándose en su homología de secuencia, se clasifican en 6 subfamilias (TRPV, TRPM; TRPA, TRPC, TRPP y TRPML). Los canales de TRP son canales selectivos para cationes activados por varios estímulos físicos (tales como temperatura, osmolaridad y estímulos mecánicos) y químicos. TRPM8, que se clonó en 2002, es un canal catiónico no selectivo de la familia de TRP expresado en una subpoblación de nervios sensoriales somáticos en el ganglio de la raíz dorsal y ganglios trigeminales que provocan excitación de nervios sensoriales. Se activa por temperaturas ligeramente frías y compuestos sintéticos que imitan al frío tales como mentol, eucaliptol e icilina [McKemy D.D. et al., Nature (2002) 416, 52-58; Peier A.M. et al. Cell (2002) 108, 705-715]. Como varios otros canales de TRP, TRPM8 también se activa por voltaje [Nilius B. et al., J. Physiol. (2005) 567, 35-44]. La dependencia del voltaje de TRPM8 se caracteriza por una fuerte rectificación hacia fuera a potencial de transmembrana despolarizado y un cierre rápido y dependiente del potencial a potenciales de membrana negativos. La aplicación de mentol y agentes de enfriamiento desplaza la curva de activación hacia potenciales más negativos, aumentando la posibilidad de abertura del canal y el refuerzo de las corrientes hacia dentro a potenciales de membrana fisiológicos. Otros factores endógenos, tales como productos de fosfolipasa A2 [Vanden Abeele F. et al., J. Biol.Chem. (2006) 281, 40174-40182], endocannabinoides [De Petrocellis L. et al., Exp.Cell. Res. (2007) 313, 1911-1920] y PiP2 [Rohacs T. et al., Nat. Neurosci. (2005) 8, 626-634] también participan en la regulación de los canales.
Hay muchas pruebas directas e indirectas de un papel central de la actividad del canal TRPM8 en enfermedades tales como prurito, enfermedades del intestino irritable, trastornos respiratorios exacerbados y/o inducidos por frío, isquemia, dolor, neurodegeneración, trastornos psiquiátricos, accidente cerebrovascular, trastornos urológicos, síndrome de ojo seco y epífora. Por ejemplo, se ha demostrado que los canales de TRP transducen señales reflejas que están implicadas en la vejiga sobreactiva de pacientes con rutas de reflejos espinales anómalas o dañadas [De Groat W.C. et al., Urology (1997) 50, 36-52]. TRp M8 se activa por temperaturas de entre 8°C y 28°C y se expresa en las neuronas nociceptivas primarias, incluyendo urotelio de la vejiga, ganglios de la raíz dorsal, delta A y fibras C. El agua helada intravesical o el mentol también inducen el reflejo de micción espinal medioado por fibras C en pacientes con urgencia e incontinencia urinaria [Everaerts W. et al., Neurol. Urodyn. (2008) 27, 264-73].
Además, se sabe que TRPM8 regula los flujos de entrada de la concentración de Ca2+ en respuesta a temperatura fría o estímulos farmacológicos. Finalmente, se ha propuesto el posible papel de TRPM8 en asma inducida por frío y en exacerbación del asma, lo que sugiere que t Rp M8 es también una diana relevante para la gestión de estas patologías [Xing H. et al., Molecular Pain (2008), 4, 22-30].
La expresión del canal en cerebro, pulmón, vejiga, tracto gastrointestinal, vasos sanguíneos, próstata y células inmunitarias proporciona además la posibilidad de modulación terapéutica de la actividad de TRPM8 en una amplia gama de patologías. En particular, los trastornos o enfermedades que se ha demostrado que se ven afectados por la modulación de TRPM8 son dolor tal como dolor crónico, dolor neuropático incluyendo alodinia al frío y neuropatía diabética, dolor posoperatorio, dolor osteoartrítico, dolor artrítico reumatoide, dolor por cáncer, neuralgia, neuropatías, algesia, fibromialgia, lesión nerviosa, migraña, cefaleas; isquemia, neurodegeneración, accidente cerebrovascular, trastornos psiquiátricos, incluyendo ansiedad y depresión, y prurito, enfermedades del intestino irritable, trastornos respiratorios exacerbados y/o inducidos por frío tales como EPOC, asma e hipertensión pulmonar exacerbados y/o inducidos por frío; trastornos urológicos tales como síndrome de vejiga dolorosa, cistitis intersticial, sobreactividad del detrusor (vejiga sobreactiva), incontinencia urinaria, sobreactividad del detrusor neurogénica (hiperflexia del detrusor), sobreactividad del detrusor idiopática (inestabilidad del detrusor), hiperplasia prostática benigna, trastornos del tracto urinario inferior y síntomas del tracto urinario inferior [Nilius B. et al. Science STKE (2005), 295, re8; Voets T. et al., Nat. Chem. Biol. (2005), 1, 85-92; Mukerji G. et al., Urology (2006), 6, 31-36; Lazzeri M. et al., Ther. Adv. Urol. (2009), 1, 33-42; Nilius B. et al., Biochim. Biophys. Acta (2007), 1772, 805-12; Wissenbach U. et al., Biol. Cell. (2004), 96, 47-54; Nilius B. et al., Physiol. Rev. (2007), 87, 165-217; Proudfoot C.J. et al., Curr.
Biol. (2006), 16, 1591-1605].
Además, se ha demostrado que la inhibición de TRPM8 es eficaz en el tratamiento de trastornos de la córnea asociados a alteraciones en la producción de las lágrimas y/o parpadeo alterado tal como epífora (documento US20155313854) y enfermedad de ojo seco (Quallo et al, Nature Communications (2015) 6: 7150, DOl:10.1038/ncomms8150, Belmonte et al, Curr Ophtalmol Rep (2015), 3:111-121).
A lo largo de los últimos pocos años, se han dado a conocer varias clases de antagonistas de TRPM8 no peptídicos. Los documentos WO 2006/040136, WO 2007/017092, WO 2007/017093, WO 2007/017094 y WO 2007/080109 describen derivados de benciloxilo como antagonistas de TRPM8 para el tratamiento de trastornos urológicos; el documento WO 2007/134107 describe compuestos que llevan fósforo como antagonistas de TRPM8 para el tratamiento de trastornos relacionados con TRPM8; el documento WO 2009/012430 describe sulfonamidas para el tratamiento de enfermedades asociadas con TRPM8; el documento WO 2010/103381 describe el uso de derivados de piperidina espirocíclicos como moduladores de TRPM8 en la prevención o el tratamiento de enfermedades o trastornos relacionados con TRPM8; el documento WO 2010/125831 describe derivados de ácido sulfamoilbenzoico como moduladores del receptor TRPM8 y su uso en el tratamiento de tratamiento de trastornos inflamatorios, de dolor y urológicos; y el documento WO 2013/092711 describe derivados de 2-ariloxazol y tiazol como moduladores de receptores TRPM8 y su uso en la prevención, la reducción del riesgo de, la mejora y/o el tratamiento de trastornos relacionados con urología.
Un área terapéutica en la que existe todavía una necesidad particularmente alta de desarrollo de antagonistas de TRPM8 es la de trastornos urológicos y dolor asociado. De hecho, los medicamentos y fármacos tradicionales actualmente disponibles para el tratamiento de la incontinencia urinaria y trastornos se caracterizan por varios efectos secundarios. Por ejemplo, por el momento, la terapia del síndrome de vejiga sobreactiva se basa en el uso de fármacos, especialmente agentes anticolinérgicos que afectan a los mecanismos de control neural periférico o a la contracción del músculo liso detrusor de la vejiga. Estos fármacos inhiben nervios parasimpáticos que ejercen un efecto espasmolítico directo sobre el músculo de la vejiga. El resultado de esta acción es la disminución de la presión intravesicular, un aumento en la capacidad y una reducción en la frecuencia de contracción de la vejiga. Sin embargo, el uso de agentes anticolinérgicos está asociado con efectos secundarios graves, tales como boca seca, visiones anómalas, estreñimiento y alteraciones del SNC, que alteran el cumplimiento global del paciente. Las deficiencias de las terapias reales resaltan la necesidad de fármacos novedosos, eficaces y seguros con menos efectos secundarios.
Sumario de la invención
El objetivo de la presente invención es proporcionan antagonistas de TRPM8 novedosos.
Los presentes inventores han encontrado ahora una clase de compuestos de 2-fenil-4-hidroxi-1,3-tiazol-5-ilmetanona compuestos que actúan como antagonistas selectivos del miembro 8 de la subfamilia M de canal catiónico de potencial de receptor transitorio (a continuación en el presente documento denominado TRPM8), adecuados con buena biodisponibilidad oral y que satisfacen deseos anteriores.
Estos compuestos son útiles en el tratamiento de una enfermedad asociada con la actividad de TRPM8, preferiblemente una enfermedad que se deriva de la sobreexpresión y/o hiperactividad del receptor TRPM8.
Descripción detallada de la invención
Un primer objeto de la presente invención es compuestos de fórmula (I):

en la que
R1 es un grupo heterocíclico alifático o aromático, de 5, 6 o 7 miembros sustituido o no sustituido que contiene 1, 2, 3
o 4 heteroátomos seleccionados de N, O y S, en el que, cuando está sustituido, dicho heterociclo alifático o aromático, de 5, 6 o 7 miembros está sustituido con uno o más grupos independientemente seleccionados de cicloalquilo o alquilo C1-C3, Cl y F o está sustituido con R3 y R4 que tomados juntos forman un resto cíclico saturado; R2 se selecciona de hidrógeno, alquilo C1-C2, F, Cl y OH;
y sales farmacéuticamente aceptables de los mismos.
Preferiblemente, dicho heterociclo alifático o aromático, de 5, 6 o 7 miembros no está sustituido o está sustituido con uno o más grupos independientemente seleccionados de cicloalquilo o alquilo C1-C3, Cl y F. Preferiblemente, dicho alquilo C1-C3 es metilo.
Según una realización preferida el heterociclo está sustituido con R3 y R4 que tomados juntos forman un resto cíclico saturado, siendo dicho resto cíclico saturado preferiblemente ciclobutano, ciclopentano o ciclohexano.
Según una realización preferida el heterociclo no está sustituido.
Preferiblemente, R1 se selecciona del grupo que consiste en oxazolidinilo, oxolanilo, pirrolidinilo, oxazinanilo, morfolinilo, piperidinilo, metilpirrolidinilo, pirrolilo, metilpirrolilo, furanilo, tiofenilo, piridinilo, imidazolilo, pirazolilo, oxadiazolilo y oxazolilo.
Más preferiblemente, R1 se selecciona del grupo que consiste en 1,2-oxazolidin-2-ilo, 1,3-oxazolidin-3-ilo, oxolan-2-ilo, pirrolidin-1-ilo, 1,2-oxazinan-2-ilo, 1,3-oxazinan-3-ilo, morfolin-4-ilo, piperidin-1-ilo, pirrolidin-2-ilo, 1-metilpirrolidin-2-ilo, 1H-pirrol-2-ilo, 1-metil-1H-pirrol-2-ilo, furan-2-ilo, tiofen-2-ilo, 1H-pirrol-1-ilo, piridin-2-ilo, piridin-3-ilo, piridin-4-ilo, 1H-imidazol-1-ilo y 1H-pirazol-1-ilo.
Preferiblemente, R2 es F u OH.
Más preferiblemente, R2 es 3-F o 2-OH.
En una realización según la invención, R1
En otra realización según la invención,
En otra realización según la invención,
En otra realización según la invención,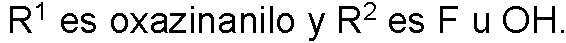
En otra realización según la invención,
En otra realización según la invención,
En otra realización según la invención,
En otra realización según la invención,
En otra realización según la invención,
En otra realización según la invención,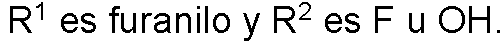
En otra realización según la invención,
En otra realización según la invención,
En otra realización según la invención,
En otra realización según la invención,
Se seleccionan compuestos de fórmula (I) preferidos según la invención de:
[2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-il](1,2-oxazolidin-2-il)metanona (1)
[4-hidroxi-2-(2-hidroxifenil)-1,3-tiazol-5-il](1,2-oxazolidin-2-il)metanona (2)
[2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-il](1,3-oxazolidin-3-il)metanona (3)
[4-hidroxi-2-(2-hidroxifenil)-1,3-tiazol-5-il](1,3-oxazolidin-3-il)metanona (4)
[2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-il](oxolan-2-il)metanona (5)
[4-hidroxi-2-(2-hidroxifenil)-1,3-tiazol-5-il](oxolan-2-il)metanona (6)
[2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-il](pirrolidin-1-il)metanona (7)
[4-hidroxi-2-(2-hidroxifenil)-1,3-tiazol-5-il](1,2-oxazinan-2-il)metanona (8)
[2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-il](1,2-oxazinan-2-il)metanona (9)
[2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-il](1,3-oxazinan-3-il)metanona (10)
[4-hidroxi-2-(2-hidroxifenil)-1,3-tiazol-5-il](1,3-oxazinan-3-il)metanona (11)
[2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-il](morfolin-4-il)metanona (12)
[4-hidroxi-2-(2-hidroxifenil)-1,3-tiazol-5-il](morfolin-4-il)metanona (13)
[2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-il](piperidin-1-il)metanona (14)
[2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-il](pirrolidin-2-il)metanona (15)
[2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-il](1-metilpirrolidin-2-il)metanona (16)
[2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-il](1H-pirrol-2-il)metanona (17)
[2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-il](1-metil-1H-pirrol-2-il)metanona (18)
[2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-il](furan-2-il)metanona (19)
[2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-il](tiofen-2-il)metanona (20)
[2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-il](1H-pirrol-1-il)metanona (21)
[2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-il](piridin-2-il)metanona (22)
[2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-il](piridin-3-il)metanona (23)
[2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-il](piridin-4-il)metanona (24)
[2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-il](1H-imidazol-1-il)metanona (25)
[2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-il](1H-pirazol-1-il)metanona (26)
[2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-il](1,2-oxazepan-2-il)metanona (27)
[2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-il](5-oxa-6-azaspiro[2,4]heptan-6-il)metanona (28)
[2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-il](5-metil-1,2-oxazolidin-2-il)metanona (29)
[2-(3-fluorofenN)-4-hidroxi-1,3-tiazol-5-N](hexahidro-2H-cidopenta[d][1,2]oxazol-2-N)iTietanona (30) [2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-il](1H-1,2,3-triazol-1-il)metanona (31) y
[2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-il](1-metil-1H-tetrazol-5-il)metanona (32).
Los compuestos de fórmula (I) más preferidos según la invención se seleccionan de:
[2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-il](1,2-oxazolidin-2-il)metanona (1)
[4-hidroxi-2-(2-hidroxifenil)-1,3-tiazol-5-il](1,2-oxazolidin-2-il)metanona (2)
[4-hidroxi-2-(2-hidroxifenil)-1,3-tiazol-5-il](1,2-oxazinan-2-il)metanona (8) y
[2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-il](1,2-oxazinan-2-il)metanona (9).
Tal como se mostrará en la sección experimental, los cuatro compuestos anteriores se caracterizan por una potencia muy alta y una semivida de aclaramiento intrínseco alta.
Tal como se describirá en detalles en el ejemplo 33, los presentes inventores han encontrado que los compuestos anteriores 1-32 son antagonistas potentes de TRPM8. La actividad antagonista de TRPM8 de todos los compuestos se determinó in vitro midiendo cambios en los niveles intracelulares de calcio usando un colorante fluorescente sensible a Ca2+ tanto después de la inyección de icilina como después de la inyección de agente de enfriamiento 10; todos los compuestos anteriores han mostrado actividad antagonista con una CI50 por debajo de 2 |iM en ambas pruebas, tal como se muestra en la tabla 1.
Además, tal como se describirá en detalle en el ejemplo 34, todos los compuestos anteriores se sometieron a prueba en el ensayo de estimulación con frío in vitro, mostrando una CI50 por debajo de 2 |iM, tal como se muestra en la tabla 1.
Por tanto, un segundo objeto de la presente invención son los compuestos de fórmula (I) anteriores para su uso como antagonistas de TRPM8, preferiblemente de TRPM8 humano.
Finalmente, tal como se describirá en detalle en el ejemplo 35, todos los compuestos de la invención se sometieron a prueba para determinar la estabilidad metabólica en microsomas de rata, mostrando los compuestos (1 ), (2 ), (8) y (9) la mayor semivida de aclaramiento intrínseco.
Por consiguiente, un tercer objeto de la presente invención es los compuestos anteriores para su uso como medicamentos. Un cuarto objeto de la presente invención es los compuestos anteriores para su uso en la prevención, la reducción del riesgo de, la mejora y/o el tratamiento de una enfermedad asociada con la actividad de TRPM8, preferiblemente una enfermedad que se deriva de la sobreexpresión y/o hiperactividad del receptor TRPM8. Según la presente invención, por “sobreexpresión y/o hiperactividad del receptor TRPM” quiere decirse una expresión y/o actividad del receptor TRPM8 mayor que el nivel fisiológico.
Según la presente invención, por “enfermedad que está asociada con la actividad de TRPM8” quiere decirse preferiblemente una enfermedad seleccionada de dolor, prurito, enfermedades del intestino irritable, trastornos respiratorios exacerbados y/o inducidos por frío, isquemia, neurodegeneración, accidente cerebrovascular, trastornos urológicos, trastornos psiquiátricos y trastornos de la córnea asociados a alteraciones en la producción de las lágrimas y/o parpadeo alterado.
Según una realización preferida, dicho dolor se selecciona de dolor crónico, dolor por cáncer, dolor neuropático, incluyendo alodinia al frío, dolor posoperatorio, neuralgia, neuropatías, incluyendo neuropatía diabética, fibromialgia, algesia, dolor asociado a lesión nerviosa, migraña, cefalea. Según una realización adicional preferida dicho dolor es dolor de origen inflamatorio, preferiblemente asociado a osteoartritis y artritis reumatoide.
Preferiblemente, dicho trastorno respiratorio exacerbado y/o inducido por frío se selecciona de asma, EPOC e hipertensión pulmonar exacerbados y/o inducidos por frío.
Preferiblemente, dichos trastornos urológicos se seleccionan de síndrome de vejiga dolorosa, cistitis intersticial, sobreactividad del detrusor (también conocida como vejiga sobreactiva), incontinencia urinaria, sobreactividad del detrusor neurogénica (también conocida como hiperflexia del detrusor), sobreactividad del detrusor idiopática (también conocida como inestabilidad del detrusor), hiperplasia prostática benigna, trastornos del tracto urinario inferior y síntomas del tracto urinario inferior.
Preferiblemente, dichos trastornos psiquiátricos se seleccionan de ansiedad y depresión.
Preferiblemente, dichos trastornos de la córnea asociados a alteraciones en la producción de las lágrimas y/o parpadeo alterado se seleccionan de epífora y enfermedad de ojo seco.
Un quinto objeto de la presente invención son composiciones farmacéuticas que comprenden el al menos uno de dichos compuestos de fórmula (I) anteriores en combinación con excipientes y/o diluyentes farmacéuticamente aceptables.
Según una realización preferida, dicha composición farmacéutica es para la prevención, la reducción del riesgo de, la mejora y/o el tratamiento de una enfermedad asociada con la actividad de TRPM8, preferiblemente una enfermedad que se deriva de la sobreexpresión y/o hiperactividad del receptor TRPM8.
Según una realización, dicha composición farmacéutica contiene al menos uno de los compuestos de fórmula (I) anteriores como único principio activo. Según una realización alternativa, dicha composición farmacéutica contiene
al menos uno de los compuestos de fórmula (I) anteriores en asociación con al menos otro principio activo.
Según una realización preferida de la invención, también en combinación con las realizaciones anteriores, las composiciones farmacéuticas pueden ser para administración intravesical, intravenosa, tópica u oral.
Los compuestos de la invención de fórmula (I) se formulan convenientemente en composiciones farmacéuticas usando técnicas convencionales y excipientes tales como los descritos en “Remington's Pharmaceutical Sciences Handbook” MACK Publishing, Nueva York, 18a ed., 1990.
Un sexto objeto de la presente invención es un método terapéutico para la prevención, la reducción del riesgo de, la mejora y/o el tratamiento de dicha enfermedad asociada con la actividad de TRPM8, preferiblemente una enfermedad que se deriva de la sobreexpresión y/o hiperactividad del receptor TRPM8, que comprende administrar el compuesto de fórmula (I) anterior en un sujeto que lo necesita.
Los compuestos de la invención pueden administrarse como el único principio activo o en combinación con otros compuestos terapéuticamente activos.
La administración de los compuestos de la invención puede efectuarse mediante instilación intravesical, mediante inyección intravenosa, como un bolo, en preparaciones dermatológicas (cremas, lociones, pulverizaciones y pomadas), mediante inhalación, así como por vía oral en forma de cápsulas, comprimidos, jarabe, formulaciones de liberación controlada y similares.
La dosis diaria promedio depende de varios factores tales como la gravedad de la enfermedad, el estado, la edad, el sexo y el peso del paciente. La dosis variará generalmente desde 1 hasta 1500 mg de compuestos de fórmula (I) al día opcionalmente dividida en múltiples administraciones.
La presente invención se ilustrará por medio de los siguientes ejemplos que no han de considerarse como limitativos del alcance de la invención.
Ejemplos
Síntesis de compuestos preferidos
Los compuestos enumerados en la tabla 1 se han sintetizado siguiendo los procedimientos descritos en los siguientes ejemplos.
Materiales y métodos
Materiales y métodos
Todos los reactivos se adquirieron de Sigma-Aldrich, Fluorochem y Alfa Aesar y se usaron sin purificación adicional. Se registraron espectros de resonancia magnética nuclear (RMN) en el disolvente indicado con tetrametilsilano (TMS) como patrón interno en un instrumento Bruker Avance3 400 MHz. Los desplazamientos químicos se notifian en partes por millón (ppm) en relación con el patrón interno. Se usan abreviaturas de la siguiente manera: s = singlete, d = doblete, t = triplete, q = cuartete, m = multiplete, dd = doblete de dobletes, sa = señal ancha. Las constantes de acoplamiento (valores J) se facilitan en hercios (Hz). Se registraron espectros de HPLC-EM analítica en un instrumento Thermo Finnigan Surveyor acoplado con un aparato Thermo Finnigan LCQ DECA XPPLUS y equipado con una columna de fase inversa C18 (10 |iM, 4,6 mm X 150 mm) Fenomenex Gemini. La mezcla de eluyentes consistía en tampón de ácido fórmico/formiato de amonio 10 mM (pH 4,2) y acetonitrilo usados según el gradiente de desde 90:10 hasta 10:90 a una velocidad de flujo de 0,200 ml/min. Todos los experimentos de EM se realizaron usando ionización por electrospray (ESI) en modo de ión positivo y negativo.
Todas las reacciones se monitorizaron por cromatografía en capa fina (CCF) llevada a cabo en placas de gel de sílice Grace Resolv Davisil de 250 |im de grosor, 60 F254, visualizadas usando UV (254 nm) o manchas tales como KMnO4, p-anisaldehído y molibdato de amonio cérico (CAM). Las purificaciones cromatográficas se llevaron a cabo sobre columnas de gel de sílice con sílice Grace Resolv Davisil 60. Todas las disoluciones orgánicas se secaron sobre Na2SO4 o MgSO4 anhidro y se concentraron en un evaporador rotatorio. Todos los compuestos usados para los ensayos biológicos tienen una pureza de al menos el 98% basándose en los resultados analíticos de HPLC monitorizados con longitudes de onda de 220 y 254 nm, a menos que se indique otra cosa.
Procedimiento general
Síntesis de productos intermedios
Síntesis de 3-fluorobencenocarbotioamida (producto intermedio a)
Un matraz de fondo redondo de 100 ml equipado con un condensador y un agitador magnético se cargó con 3-fluorobenzoamida (2,0 g, 14,4 mmol), que se disolvió en 30 ml de THF, luego se añadió reactivo de Lawesson a la disolución (3,5 g, 8,64 mmol). Se calentó la mezcla hasta 60°C y se agitó durante la noche; se monitorizó la transformación mediante CCF (eluyente: n-hexano/EtOAc 7:3). Se enfrió la disolución a temperatura ambiente y se eliminó el disolvente mediante destilación a vacío.
Se purificó el producto en bruto mediante cromatografía ultrarrápida (eluyente: n-hexano/EtOAc 7:3) a partir de la cual se obtuvo 3-fluorobencenocarbotioamida como un sólido amarillo (2,0 g, 12,9 mmol, R = 89%).
1H-RMN (CDCla): 87,80-7,55 (sa, 1H, NH2), 7,66-7,60 (m, 2H), 7,44-7,37 (m, 1H), 7,27-7,20 (m, 1H), 7,30-7,00 (sa, 1H, NH2).
EM (ES1+) m/z: 156,11 [M+H]+.
Síntesis de 2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-carboxilato de etilo (producto intermedio b)
Un vial de microondas equipado con un agitador magnético se cargó con 3-fluorobencenocarbotioamida (0,5 g, 3,22 mmol) disuelta en etanol seco (8 ml), se añadió bromomalonato de dietilo (0,055 ml, 3,22 mmol) y se tapó el vial herméticamente. Se irradió la disolución en un aparato de microondas a 100°C durante 30 minutos. Se obtuvo 2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-carboxilato de etilo como un sólido amarillo tras la cristalización en etanol (0,439 g, 1,64 mmol, R = 51%).
1H-RMN (CDCla): 89,94 (sa, 1H, OH), 7,80-7,70 (m, 2H), 7,49-7,41 (m, 1H), 7,2-7,17 (m, 1H), 4,43 (q, 2H, J=7,1 Hz), 1,42 (t, 3H, J=7,1 Hz).
EM (ES1+) m/z: 267,81 [M+H]+.
Síntesis de 2-(3-fluorofenil)-4-metoxi-1,3-tiazol-5-carboxilato de etilo (producto intermedio c)
Un matraz de fondo redondo de 25 ml equipado con un agitador magnético se cargó con 2-(3-fluorofenil)-4-hidroxi-1.3- tiazol-5-carboxilato de etilo (0,100 g, 0,374 mmol) que se disolvió en THF seco (3 ml) y DMF (2,5 ml), se trató la disolución con NaH (dispersión en aceite al 60-65%, 0,022 g, 1,5 eq.) y yoduro de metilo (0,140 ml, 7 eq.) y se agitó durante la noche a temperatura ambiente. Se extinguió la reacción en agua y se extrajo en acetato de etilo (20 ml, 3 veces), se recogieron las fases orgánicas y se lavaron con bicarbonato de sodio saturado y salmuera, luego se anhidrificaron sobre sulfato de sodio seco. Se purificó el producto en bruto sobre gel de sílice (eluyente: nhexano/acetato de etilo 9:1). Se obtuvo 2-(3-fluorofenil)-4-metoxi-1,3-tiazol-5-carboxilato de etilo como un sólido amarillo (0,053 g, 0,19 mmol, R = 50%).
1H-RMN (CDCls): 87,69-7,25 (m, 2H), 7,47-7,41 (m, 1H), 7,23-7,16 (m, 1H), 4,36 (q, 2H, J=7,2 Hz), 4,25 (s, 3H), 1,39 (t, 3H, J=7,2 Hz).
EM (ES1+) m/z: 282,08 [M+H]+.
Síntesis de ácido 2-(3-fluorofenil)-4-metoxi-1,3-tiazol-5-carboxílico (producto intermedio d)
Un matraz de fondo redondo de 25 ml equipado con un agitador magnético se cargó con 2-(3-fluorofenil)-4-metoxi-1.3- tiazol-5-carboxilato de etilo (0,097 g, 0,344 mmol) que se disolvió en etanol (3 ml) y agua (0,020 ml). Entonces se añadió KOH (0,193 g, 3,44 mmol) y se agitó la disolución durante la noche a temperatura ambiente. Se diluyó la mezcla en agua (15 ml), se acidificó con HCl 2 N hasta pH 2 y se extrajo en acetato de etilo (20 ml x 2). Se recogieron las fases orgánicas y se lavaron con agua y salmuera, luego se anhidrificaron sobre sulfato de sodio seco. Se obtuvo ácido 2-(3-fluorofenil)-4-metoxi-1,3-tiazol-5-carboxílico como un sólido amarillo (0,077 g, 0,304 mmol, R = 88%).
1H-RMN (CDCls): 87,76-7,71 (m, 2H), 7,50-7,43 (m, 1H), 7,26-7,19 (m, 1H), 4,32 (s, 3H).
EM (ES1') m/z: 252,25 [M-H]'.
Síntesis de cloruro de 2-(3-fluorofenil)-4-metoxi-1,3-tiazol-5-carbonilo (producto intermedio e)
Un matraz de fondo redondo de 25 ml equipado con un agitador magnético y un condensador enfriado con agua se cargó con ácido 2-(3-fluorofenil)-4-metoxi-1,3-tiazol-5-carboxílico (0,049 g, 0,193 mmol) y 5 ml de DCM seco a temperatura ambiente. A la misma temperatura se trató la disolución con un exceso de cloruro de tionilo (0,028 ml, 0,387 mmol) y una cantidad catalítica de DMF (0,002 ml), luego se sometió a reflujo durante 2,5 horas. Se enfrió la disolución, luego se eliminaron los compuestos volátiles a presión reducida. Se separó el residuo oleoso unas cuantas veces con tolueno para eliminar adicionalmente el cloruro de tionilo residual. Se obtuvo cloruro de 2-(3-fluorofenil)-4-metoxi-1,3-tiazol-5-carbonilo como un aceite de color amarillo pálido (0,052 g, 0,0193 mmol, R = 95%) y
se usó sin purificación adicional.
Síntesis de 2-(3-fluorofenil)-N,4-dimetoxi-N-metil-1,3-tiazol-5-carboxamida (producto intermedio f)
En un matraz de fondo redondo de 25 ml equipado con un agitador magnético, se disolvió cloruro de 2-(3-fluorofenil)-4-metoxi-1,3-tiazol-5-carbonilo (0,052 g, 0,193 mmol) en DCM seco (5 ml) y se enfrió hasta 0°C con un baño de hielo. Se trató esta disolución con una mezcla de clorhidrato de N,O-dimetilhidroxilamina (0,038 g, 0,386 mmol), trietilamina (0,1 ml) y DCM (2 ml), y se agitó a la misma temperatura durante 45 minutos. Tal como se comprobó mediante CL-EM, la reacción se completó, por tanto, se extinguió y se sometió a tratamiento final de la siguiente manera: se diluyó la mezcla con DCM (50 ml) y se lavó con agua (10 ml x 2) y salmuera (10 ml), se secó sobre sulfato de sodio anhidro y se destiló el disolvente a vacío. Se obtuvo 2-(3-fluorofenil)-N,4-dimetoxi-N-metil-1,3-tiazol-5-carboxamida (0,060 g, 0,20 mmol, R = 95%) como un sólido oleoso y se usó en la siguiente etapa de síntesis.
1H-RMN (CDCla): 87,79-7,71 (m, 2H), 7,48-7,40 (m, 1H), 7,22-7,14 (m, 1H), 4,25 (s, 3H), 3,77 (s, 3H), 3,35 (s, 3H). EM (ES1+) m/z: 297,32 [M+H]+.
Síntesis de cloruro de 4-metoxi-2-(2-metoxifenil)-1,3-tiazol-5-carbonilo (producto intermedio g)
Partiendo de 2-metoxibenceno-1-carbotioamida (0,152 g, 1,0 mmol), se preparó cloruro de 4-metoxi-2-(2-metoxifenil)-1,3-tiazol-5-carbonilo tal como se describió en la síntesis del producto intermedio e con un rendimiento del 27% a lo largo de 4 etapas.
Síntesis de 2-aril-4-metoxi-1,3-tiazol (producto intermedio h)
Un vial de microondas equipado con un agitador magnético se cargó con 3-fluorobencenocarbotioamida o 2-metoxibenceno-1-carbotioamida (1 mmol) disuelta en etanol seco (3 ml), se añadió bromoacetato de metilo (1 mmol) y se tapó el vial herméticamente. Se irradió la disolución en un aparato de microondas a 100°C durante 15 minutos. Se obtuvo 2-(3-fluorofenil)-4-metoxi-1,3-tiazol (o 4-metoxi-2-(2-metoxifenil)-1,3-tiazol) como un aceite amarillo tras la purificación por cromatografía ultrarrápida (n-hexano:acetato de etilo 9:1).
Procedimiento general para la acilación de Friedel-Crafts de compuestos aromáticos: A una disolución de reactivo aromático (4 mmol) y cloruro de 2-(3-fluorofenil)-4-metoxi-1,3-tiazol-5-carbonilo (6 mmol) en nitrometano (5 ml), se le añadió satisfactoriamente Zn(OTf^6H2O (0,4 mmol). Se agitó la mezcla a temperatura ambiente durante la noche. Entonces se trató la mezcla de reacción con NaHCO3 acuoso saturado (10 ml) y se extrajo con cloroformo (20 ml x 2). Se secó la disolución orgánica combinada sobre Na2SO4 anhidro, se filtró y se concentró a vacío. Se obtuvo el producto correspondiente tras la purificación por cromatografía ultrarrápida.
Ejemplo 1
Síntesis de [2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-il](1,2-oxazolidin-2-il)metanona (1)
Partiendo de cloruro de 2-(3-fluorofenil)-4-metoxi-1,3-tiazol-5-carbonilo (producto intermedio e, 0,110 g, 0,40 mmol) y cloruro de 1,2-oxazolidin-2-io (0,055 g, 0,50 mmol), se preparó [2-(3-fluorofenil)-4-metoxi-1,3-tiazol-5-il](1,2-oxazolidin-2-il)metanona tal como se describió para el producto intermedio f. Se desprotegió el compuesto con 3 equivalentes de tribromuro de boro 1 M en diclorometano, y luego se agitó durante la noche a temperatura ambiente. Se diluyó la reacción con DCM (10 ml) y se agitó con agua (10 ml) durante 10 minutos. Se separó la fase orgánica y se anhidrificó sobre sulfato de sodio anhidro, se destiló el disolvente y se purificó el producto en bruto sobre gel de sílice (n-hexano:acetato de etilo 7:3 como eluyente) para obtener la [2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-il](1,2-oxazolidin-2-il)metanona como un sólido de color marrón pálido (0,055 g, R = 47% a lo largo de dos etapas).
1H-RMN (CDCla): 811,93 (s, 1H), 7,80-7,73 (m, 2H), 7,44-7,39 (m, 1H), 7,19-7,17 (m, 1H), 4,12-4,08 (t, 2H), 3,94 3,89 (t, 2H), 2,47-2,40 (m, 2H).
EM (ES1+) m/z: 294,99 [M+H]+.
Ejemplo 2
Síntesis de [4-hidroxi-2-(2-hidroxifenil)-1,3-tiazol-5-il](1,2-oxazolidin-2-il)metanona (2)
Partiendo de cloruro de 4-metoxi-2-(2-metoxifenil)-1,3-tiazol-5-carbonilo (producto intermedio g, 0,115 g, 0,40 mmol) y cloruro de 1,2-oxazolidin-2-io (0,055 g, 0,50 mmol), se preparó [4-metoxi-2-(2-metoxifenil)-1,3-tiazol-5-il](1,2-oxazolidin-2-il)metanona tal como se describió para el producto intermedio f. Se desprotegió el compuesto con 4 equivalentes de tribromuro de boro 1 M en diclorometano, y luego se agitó durante la noche a temperatura ambiente. Se diluyó la reacción con DCM (10 ml) y se agitó con agua (10 ml) durante 10 minutos. Se separó la fase orgánica y
se anhidrificó sobre sulfato de sodio anhidro, se destiló el disolvente y se purificó el producto en bruto sobre gel de sílice (n-hexano:acetato de etilo 1:1 como eluyente) para obtener la [4-hidroxi-2-(2-hidroxifenil)-1,3-tiazol-5-il](1,2-oxazolidin-2-il)metanona como un sólido de color marrón pálido (0,054 g, R = 46% a lo largo de dos etapas).
1H-RMN (CDCla): 812,14 (a, 1H), 11,65 (s, 1H), 7,66-7,64 (d, 1H), 7,37-7,35 (t, 1H), 7,06-7,04 (d, 1H), 6,91-6,88 (t, 1H), 4,12-4,09 (t, 2H), 3,94-3,92 (t, 2H), 2,48-2,41 (m,2H).
EM (ES1+) m/z: 293,01 [M+H]+.
Ejemplo 3
Síntesis de [2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-il](1,3-oxazolidin-3-il)metanona (3)
Partiendo de cloruro de 2-(3-fluorofenil)-4-metoxi-1,3-tiazol-5-carbonilo (producto intermedio e, 0,110 g, 0,40 mmol) y cloruro de 1,3-oxazolidin-3-io (0,055 g, 0,50 mmol), se preparó [2-(3-fluorofenil)-4-metoxi-1,3-tiazol-5-il](1,3-oxazolidin-3-il)metanona tal como se describió para el producto intermedio f. Se desprotegió el compuesto con 3 equivalentes de tribromuro de boro 1 M en diclorometano, y luego se agitó durante la noche a temperatura ambiente. Se diluyó la reacción con DCM (10 ml) y se agitó con agua (10 ml) durante 10 minutos. Se separó la fase orgánica y se anhidrificó sobre sulfato de sodio anhidro, se destiló el disolvente y se purificó el producto en bruto sobre gel de sílice (n-hexano:acetato de etilo 7:3 como eluyente) para obtener la [2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-il](1,3-oxazolidin-3-il)metanona como un sólido amarillo (0,049 g, R = 42% a lo largo de dos etapas).
1H-RMN (CDCla): 811,91 (s, 1H), 7,81-7,73 (m, 2H), 7,42-7,37 (m, 1H), 7,15-7,10 (m, 1H), 4,36 (s, 2H), 4,10-4,05 (m, 2H), 3,93-3,89 (m, 2H).
EM (ES1+) m/z: 295,09 [M+H]+.
Ejemplo 4
Síntesis de [4-hidroxi-2-(2-hidroxifenil)-1,3-tiazol-5-il](1,3-oxazolidin-3-il)metanona (4)
Partiendo de cloruro de 4-metoxi-2-(2-metoxifenil)-1,3-tiazol-5-carbonilo (producto intermedio g, 0,115 g, 0,40 mmol) y cloruro de 1,3-oxazolidin-3-io (0,055 g, 0,50 mmol), se preparó [4-metoxi-2-(2-metoxifenil)-1,3-tiazol-5-il](1,3-oxazolidin-3-il)metanona tal como se describió para el producto intermedio f. Se desprotegió el compuesto con 4 equivalentes de tribromuro de boro 1 M en diclorometano, y luego se agitó durante la noche a temperatura ambiente. Se diluyó la reacción con DCM (10 ml) y se agitó con agua (10 ml) durante 10 minutos. Se separó la fase orgánica y se anhidrificó sobre sulfato de sodio anhidro, se destiló el disolvente y se purificó el producto en bruto sobre gel de sílice (n-hexano:acetato de etilo 1:1 como eluyente) para obtener la [4-hidroxi-2-(2-hidroxifenil)-1,3-tiazol-5-il](1,3-oxazolidin-3-il)metanona como un sólido de color marrón pálido (0,049 g, R = 42% a lo largo de dos etapas).
1H-RMN (CDCls): 812,11 (a, 1H), 11,62 (s, 1H), 7,61-7,58 (d, 1H), 7,37-7,35 (t, 1H), 7,05-7,04 (d, 1H), 6,93-6,89 (t, 1H), 4,33 (s, 2H), 4,12-4,09 (m, 2H), 3,94-3,92 (m, 2H).
EM (ES1+) m/z: 293,26 [M+H]+.
Ejemplo 5
Síntesis de [2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-il](oxolan-2-il)metanona (5)
A una disolución de ácido tetrahidro-2-furoico (0,135 ml, 1,4 mmol) en dioxano seco (2 ml), se le añadió 1,1'-carbonildiimidazol (CDI, 0,454 g, 2,8 mmol) y se agitó la disolución a temperatura ambiente durante 3 h. Simultáneamente, a una disolución de 0,209 g (1 mmol) de 2-(3-fluorofenil)-4-metoxi-1,3-tiazol (producto intermedio h) en 2 ml de dioxano seco, se le añadieron 0,296 g (4 mmol) de Ca(OH)2 y se agitó la disolución en suspensión resultante a temperatura ambiente durante 30 minutos. A esta disolución, se le añadió la disolución de ácido tetrahidro-2-furoico activado y se calentó la mezcla resultante a 100°C durante 3 h. Se diluyó la reacción con agua (10 ml) y se extrajo con acetato de etilo (10 ml x 2). Se separó la fase orgánica y se anhidrificó sobre sulfato de sodio anhidro, se destiló el disolvente y se purificó el producto en bruto sobre gel de sílice (diclorometano:metanol 98:2 como eluyente) para proporcionar [2-(3-fluorofenil)-4-metoxi-1,3-tiazol-5-il](oxolan-2-il)metanona como un aceite claro (0,117 g, R = 38%). Se desprotegió esta última con 3 equivalentes de tribromuro de boro 1 M en diclorometano, y luego se agitó durante la noche a temperatura ambiente. Se diluyó la reacción con DCM (10 ml) y se agitó con agua (10 ml) durante 10 minutos. Se separó la fase orgánica y se anhidrificó sobre sulfato de sodio anhidro, se destiló el disolvente y se purificó el producto en bruto sobre gel de sílice (diclorometano:metanol 95:5 como eluyente) para obtener la [2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-il](oxolan-2-il)metanona como un aceite claro (0,088 g, R = 79%).
1H-RMN (CDCls): 8 11,90 (s, 1H), 7,81-7,73 (m, 2H), 7,45-7,40 (m, 1H), 7,18-7,16 (m, 1H), 4,56-4,52 (m, 1H), 4,124,08 (m, 2H), 3,91-3,88 (m, 2H), 2,47-2,41 (m, 2H).
EM (ES1+) m/z: 294,33 [M+H]+.
Ejemplo 6
Síntesis de [4-hidroxi-2-(2-hidroxifenil)-1,3-tiazol-5-il](oxolan-2-il)metanona (6)
Se sintetizó [4-hidroxi-2-(2-hidroxifenil)-1,3-tiazol-5-il](oxolan-2-il)metanona siguiendo el procedimiento descrito para el compuesto 5 partiendo de 4-metoxi-2-(2-metoxifenil)-1,3-tiazol (producto intermedio h). Se obtuvo el compuesto como un aceite marrón (0,054 g, R = 33% a lo largo de dos etapas).
1H-RMN (CDCla): 812,10 (a, 1H), 11,66 (s, 1H), 7,65-7,64 (d, 1H), 7,36-7,35 (t, 1H), 7,07-7,04 (d, 1H), 6,91-6,87 (t, 1H), 4,55-4,52 (m, 1H), 4,13-4,10 (m, 2H), 3,92-3,90 (m, 2H), 2,51-2,48 (m, 2H).
EM (ES1+) m/z: 292,29 [M+H]+.
Ejemplo 7
Síntesis de [2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-il](pirrolidin-1-il)metanona (7)
Partiendo de cloruro de 2-(3-fluorofenil)-4-metoxi-1,3-tiazol-5-carbonilo (producto intermedio e, 0,100 g, 0,37 mmol) y pirrolidina (0,046 ml, 0,55 mmol), se preparó [2-(3-fluorofenil)-4-metoxi-1,3-tiazol-5-il](pirrolidin-1-il)metanona tal como se describió para el producto intermedio f. Se desprotegió el compuesto con 3 equivalentes de tribromuro de boro 1 M en diclorometano, luego se agitó durante la noche a temperatura ambiente. Se diluyó la reacción con DCM (10 ml) y se agitó con agua (10 ml) durante 10 minutos. Se separó la fase orgánica y se anhidrificó sobre sulfato de sodio anhidro, se destiló el disolvente y se purificó el producto en bruto sobre gel de sílice (n-hexano:acetato de etilo 9:1 como eluyente) para obtener la [2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-il](pirrolidin-1-il)metanona como un aceite naranja (0,066 g, R = 61% a lo largo de dos etapas).
1H-RMN (CDCla): 811,91 (s, 1H), 7,80-7,71 (m, 2H), 7,44-7,41 (m, 1H), 7,19-7,18 (m, 1H), 3,56-3,52 (m, 2H), 3,51 3,48 (m, 2H), 2,51-2,48 (m, 2H), 2,46-2,41 (m, 2H).
EM (ES1+) m/z: 292,99 [M+H]+.
Ejemplo 8
Síntesis de [4-hidroxi-2-(2-hidroxifenil)-1,3-tiazol-5-il](1,2-oxazinan-2-il)metanona (8)
Partiendo de cloruro de 4-metoxi-2-(2-metoxifenil)-1,3-tiazol-5-carbonilo (producto intermedio g, 0,105 g, 0,37 mmol) y cloruro de 1,2-oxazinan-2-io (0,061 g, 0,50 mmol), se preparó [4-metoxi-2-(2-metoxifenil)-1,3-tiazol-5-il](1,2-oxazinan-2-il)metanona tal como se describió para el producto intermedio f. Se desprotegió el compuesto con 4 equivalentes de tribromuro de boro 1 M en diclorometano, luego se agitó durante la noche a temperatura ambiente. Se diluyó la reacción con DCM (10 ml) y se agitó con agua (10 ml) durante 10 minutos. Se separó la fase orgánica y se anhidrificó sobre sulfato de sodio anhidro, se destiló el disolvente y se purificó el producto en bruto sobre gel de sílice (n-hexano:acetato de etilo 1:1 como eluyente) para obtener la [4-hidroxi-2-(2-hidroxifenil)-1,3-tiazol-5-il](1,2-oxazinan-2-il)metanona como un sólido céreo (0,039 g, R = 34% a lo largo de dos etapas).
1H-RMN (CDCh): 812,33 (a, 1H), 11,62 (s, 1H), 7,65-7,63 (m, 1H), 7,37-7,34 (m, 1H), 7,07-7,05 (m, 1H), 6,91-6,87 (m, 1H), 4,13-4,11 (t, 2H), 3,96-3,93 (t, 2H), 1,97-1,94 (m, 2H), 1,87-1,82 (m, 2H).
EM (ES1+) m/z: 307,04 [M+H]+.
Ejemplo 9
Síntesis de [2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-il](1,2-oxazinan-2-il)metanona (9)
Partiendo de cloruro de 2-(3-fluorofenil)-4-metoxi-1,3-tiazol-5-carbonilo (producto intermedio e, 0,110 g, 0,40 mmol) y cloruro de 1,2-oxazinan-2-io (0,062 g, 0,50 mmol), se preparó [2-(3-fluorofenil)-4-metoxi-1,3-tiazol-5-il](1,2-oxazinan-2-il)metanona tal como se describió para el producto intermedio f. Se desprotegió el compuesto con 3 equivalentes de tribromuro de boro 1 M en diclorometano, luego se agitó durante la noche a temperatura ambiente. Se diluyó la reacción con DCM (10 ml) y se agitó con agua (10 ml) durante 10 minutos. Se separó la fase orgánica y se anhidrificó sobre sulfato de sodio anhidro, se destiló el disolvente y se purificó el producto en bruto sobre gel de sílice (n-hexano:acetato de etilo 7:3 como eluyente) para obtener la [2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-il](1,2-oxazinan-2-il)metanona como un sólido naranja (0,060 g, R = 49% a lo largo de dos etapas).
1H-RMN (CD3OD): 87,81-7,80 (m, 1H), 7,79-7,75 (m, 1H), 7,52-7,50 (m, 1H), 7,28-7,27 (m, 1H), 4,16-4,13 (t, 2H), 3,94-3,91 (t, 2H), 1,96-1,93 (m, 2H), 1,85-1,81 (m, 2H).
EM (ES1+) m/z: 309,26 [M+H]+.
Ejemplo 10
Síntesis de [2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-il](1,3-oxazinan-3-il)metanona (10)
Partiendo de cloruro de 2-(3-fluorofenil)-4-metoxi-1,3-tiazol-5-carbonilo (producto intermedio e, 0,110 g, 0,40 mmol) y cloruro de 1,3-oxazinan-3-io (0,061 g, 0,50 mmol), se preparó [2-(3-fluorofenil)-4-metoxi-1,3-tiazol-5-il](1,3-oxazinan-3-il)metanona tal como se describió para el producto intermedio f. Se desprotegió el compuesto con 3 equivalentes de tribromuro de boro 1 M en diclorometano, luego se agitó durante la noche a temperatura ambiente. Se diluyó la reacción con DCM (10 ml) y se agitó con agua (10 ml) durante 10 minutos. Se separó la fase orgánica y se anhidrificó sobre sulfato de sodio anhidro, se destiló el disolvente y se purificó el producto en bruto sobre gel de sílice (n-hexano:acetato de etilo 8:2 como eluyente) para obtener la [2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-il](1,3-oxazinan-3-il)metanona como un sólido naranja (0,063 g, R = 51% a lo largo de dos etapas).
1H-RMN (CD3OD): 87,80-7,75 (m, 2H), 7,51-7,49 (m, 1H), 7,28-7,26 (m, 1H), 4,34 (s, 2H), 4,15-4,13 (m, 2H), 3,94 3,90 (m, 2H), 1,95-1,91 (m, 2H).
EM (ES1+) m/z: 309,31 [M+H]+.
Ejemplo 11
Síntesis de [4-hidroxi-2-(2-hidroxifenil)-1,3-tiazol-5-il](1,3-oxazinan-3-il)metanona (11)
Partiendo de cloruro de 4-metoxi-2-(2-metoxifenil)-1,3-tiazol-5-carbonilo (producto intermedio g, 0,105 g, 0,37 mmol) y cloruro de 1,3-oxazinan-3-io (0,061 g, 0,50 mmol), se preparó [4-metoxi-2-(2-metoxifenil)-1,3-tiazol-5-il](1,3-oxazinan-3-il)metanona tal como se describió para el producto intermedio f. Se desprotegió el compuesto con 4 equivalentes de tribromuro de boro 1 M en diclorometano, luego se agitó durante la noche a temperatura ambiente. Se diluyó la reacción con DCM (10 ml) y se agitó con agua (10 ml) durante 10 minutos. Se separó la fase orgánica y se anhidrificó sobre sulfato de sodio anhidro, se destiló el disolvente y se purificó el producto en bruto sobre gel de sílice (n-hexano:acetato de etilo 1:1 como eluyente) para obtener la [4-hidroxi-2-(2-hidroxifenil)-1,3-tiazol-5-il](1,3-oxazinan-3-il)metanona como un sólido céreo (0,043 g, R = 38% a lo largo de dos etapas).
1H-RMN (CD3OD): 87,81-7,73 (m, 2H), 7,50-7,48 (m, 1H), 7,29-7,28 (m, 1H), 4,35 (s, 2H), 4,16-4,15 (m, 2H), 3,95 3,93 (m, 2H), 1,92-1,90 (m, 2H).
EM (ES1+) m/z: 307,14 [M+H]+.
Ejemplo 12
Síntesis de [2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-il](morfolin-4-il)metanona (12)
Partiendo de cloruro de 2-(3-fluorofenil)-4-metoxi-1,3-tiazol-5-carbonilo (producto intermedio e, 0,110 g, 0,40 mmol) y morfolina (0,044 ml, 0,50 mmol), se preparó [2-(3-fluorofenil)-4-metoxi-1,3-tiazol-5-il](morfolin-4-il)metanona tal como se describió para el producto intermedio f. Se desprotegió el compuesto con 3 equivalentes de tribromuro de boro 1 M en diclorometano, luego se agitó durante la noche a temperatura ambiente. Se diluyó la reacción con DCM (10 ml) y se agitó con agua (10 ml) durante 10 minutos. Se separó la fase orgánica y se anhidrificó sobre sulfato de sodio anhidro, se destiló el disolvente y se purificó el producto en bruto sobre gel de sílice (n-hexano:acetato de etilo 8:2 como eluyente) para obtener la [2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-il](morfolin-4-il)metanona como un sólido naranja (0,063 g, R = 51% a lo largo de dos etapas).
1H-RMN (CD3OD): 87,81-7,78 (m, 2H), 7,50-7,48 (m, 1H), 7,27-7,25 (m, 1H), 3,80-3,75 (m, 4H), 2,94-2,90 (m, 4H). EM (ES1+) m/z: 309,18 [M+H]+.
Ejemplo 13
Síntesis de [4-hidroxi-2-(2-hidroxifenil)-1,3-tiazol-5-il](morfolin-4-il)metanona (13)
Partiendo de cloruro de 4-metoxi-2-(2-metoxifenil)-1,3-tiazol-5-carbonilo (producto intermedio g, 0,105 g, 0,37 mmol) y morfolina (0,044 ml, 0,50 mmol), se preparó [4-metoxi-2-(2-metoxifenil)-1,3-tiazol-5-il](morfolin-4-il)metanona tal como se describió para el producto intermedio f. Se desprotegió el compuesto con 4 equivalentes de tribromuro de boro 1 M en diclorometano, luego se agitó durante la noche a temperatura ambiente. Se diluyó la reacción con DCM (10 ml) y se agitó con agua (10 ml) durante 10 minutos. Se separó la fase orgánica y se anhidrificó sobre sulfato de
sodio anhidro, se destiló el disolvente y se purificó el producto en bruto sobre gel de sílice (n-hexano:acetato de etilo 1:1 como eluyente) para obtener la [4-hidroxi-2-(2-hidroxifenil)-1,3-tiazol-5-il](morfolin-4-il)metanona como un sólido céreo (0,043 g, R = 38% a lo largo de dos etapas).
1H-RMN (CD3OD): 87,80-7,76 (m, 2H), 7,51-7,49 (m, 1H), 7,28-7,25 (m, 1H), 3,81-3,77 (m, 4H), 2,93-2,91 (m, 4H). EM (ES1+) m/z: 307,14 [M+H]+.
Ejemplo 14
Síntesis de [2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-il](piperidin-1-il)metanona (14)
Partiendo de cloruro de 2-(3-fluorofenil)-4-metoxi-1,3-tiazol-5-carbonilo (producto intermedio e, 0,100 g, 0,37 mmol) y clorhidrato de piperidina (0,067 g, 0,55 mmol), se preparó [2-(3-fluorofenil)-4-metoxi-1,3-tiazol-5-il](piperidin-1-il)metanona tal como se describió para el producto intermedio f. Se desprotegió el compuesto con 3 equivalentes de tribromuro de boro 1 M en diclorometano, luego se agitó durante la noche a temperatura ambiente. Se diluyó la reacción con DCM (10 ml) y se agitó con agua (10 ml) durante 10 minutos. Se separó la fase orgánica y se anhidrificó sobre sulfato de sodio anhidro, se destiló el disolvente y se purificó el producto en bruto sobre gel de sílice (n-hexano:acetato de etilo 9:1 como eluyente) para obtener la [2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-il](piperidin-1-il)metanona como un aceite naranja (0,065 g, R = 58% a lo largo de dos etapas).
1H-RMN (CDCla): 811,91 (s, 1H), 7,80-7,71 (m, 2H), 7,44-7,40 (m, 1H), 7,19-7,17 (m, 1H), 3,55-3,53 (m, 2H), 3,52 3,50 (m, 2H), 2,21-2,18 (m, 2H), 2,15-2,10 (m, 4H).
EM (ES1+) m/z: 307,41 [M+H]+.
Ejemplo 15
Síntesis de [2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-il](pirrolidin-2-il)metanona (15)
Se sintetizó [2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-il](pirrolidin-2-il)metanona siguiendo el procedimiento descrito para el compuesto 5 pero usando la DL-prolina. Se obtuvo el compuesto como un aceite de color rojo claro (0,043 g, R = 31% a lo largo de dos etapas).
1H-RMN (CDCla): 811,91 (s, 1H), 7,81-7,72 (m, 2H), 7,44-7,43 (m, 1H), 7,18-7,15 (m, 1H), 4,14-4,10 (m, 1H), 3,51 3,47 (m, 2H), 2,26-1,80 (m, 4H).
EM (ES1+) m/z: 293,33 [M+H]+.
Ejemplo 16
Síntesis de [2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-il](1-metilpirrolidin-2-il)metanona (16)
Se sintetizó [2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-il](1-metilpirrolidin-2-il)metanona siguiendo el procedimiento descrito para el compuesto 5 pero usando la N-metilprolina. Se obtuvo el compuesto como un aceite naranja (0,047 g, R = 41% a lo largo de dos etapas).
1H-RMN (CDCls): 811,90 (s, 1H), 7,81-7,73 (m, 2H), 7,45-7,43 (m, 1H), 7,18-7,14 (m, 1H), 4,14-4,12 (m, 1H), 3,55 3,43 (m, 5H), 2,29-2,07 (m, 4H).
EM (ES1+) m/z: 307,36 [M+H]+.
Ejemplo 17
Síntesis de [2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-il](1H-pirrol-2-il)metanona (17)
Se obtuvo el compuesto 17 siguiendo el procedimiento general para la acilación de Friedel-Crafts partiendo de cloruro de 2-(3-fluorofenil)-4-metoxi-1,3-tiazol-5-carbonilo (0,145 g, 0,53 mmol) y pirrol (0,055 ml, 0,79 mmol). La desprotección del grupo metoxilo se realizó con 3 equivalentes de tribromuro de boro 1 M en diclorometano tal como se describió para el compuesto 1 y se obtuvo [2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-il](1H-pirrol-2-il)metanona como un sólido marrón (0,088 g, R = 58%).
1H-RMN (CD3OD): 87,85-7,70 (m, 2H), 7,45-7,35 (m, 1H), 7,25-7,19 (m, 1H), 7,05-7,00 (m, 1H), 6,85-6,83 (m, 1H), 6,23-6,22 (m, 1H).
EM (ES1+) m/z: 289,03 [M+H]+.
Ejemplo 18
Síntesis de [2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-il](1-metil-1H-pirrol-2-il)metanona (18)
Se obtuvo el compuesto 18 siguiendo el procedimiento general para la acilación de Friedel-Crafts partiendo de cloruro de 2-(3-fluorofenil)-4-metoxi-1,3-tiazol-5-carbonilo (0,145 g, 0,53 mmol) y N-metilpirrol (0,070 ml, 0,79 mmol). La desprotección del grupo metoxilo se realizó con 3 equivalentes de tribromuro de boro 1 M en diclorometano tal como se describió para el compuesto 1 y se obtuvo [2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-il](1-metil-1H-pirrol-2-il)metanona como un aceite amarillo (0,109 g, R = 68%).
1H-RMN (CD3OD): 87,83-7,74 (m, 2H), 7,45-7,33 (m, 1H), 7,23-7,20 (m, 1H), 7,03-7,01 (m, 1H), 6,82-6,81 (m, 1H), 6,20-6,19 (m, 1H), 3,8 (s, 3H).
EM (ES1+) m/z: 303,43 [M+H]+.
Ejemplo 19
Síntesis de [2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-il](furan-2-il)metanona (19)
Se obtuvo el compuesto 19 siguiendo el procedimiento general para la acilación de Friedel-Crafts partiendo de cloruro de 2-(3-fluorofenil)-4-metoxi-1,3-tiazol-5-carbonilo (0,145 g, 0,53 mmol) y furano (0,058 ml, 0,79 mmol). La desprotección del grupo metoxilo se realizó con 3 equivalentes de tribromuro de boro 1 M en diclorometano tal como se describió para el compuesto 1 y se obtuvo [2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-il](furan-2-il)metanona como un sólido de color verde claro (0,079 g, R = 52%).
1H-RMN (CD3OD): 87,81-7,62 (m, 3H), 7,41-7,39 (m, 1H), 7,25-7,10 (m, 2H), 6,82-6,80 (m, 1H).
EM (ES1+) m/z: 290,31 [M+H]+.
Ejemplo 20
Síntesis de [2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-il](tiofen-2-il)metanona (20)
Se obtuvo el compuesto 20 siguiendo el procedimiento general para la acilación de Friedel-Crafts partiendo de cloruro de 2-(3-fluorofenil)-4-metoxi-1,3-tiazol-5-carbonilo (0,145 g, 0,53 mmol) y tiofeno (0,063 ml, 0,79 mmol). La desprotección del grupo metoxilo se realizó con 3 equivalentes de tribromuro de boro 1 M en diclorometano tal como se describió para el compuesto 1 y se obtuvo [2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-il](tiofen-2-il)metanona como un sólido marrón (0,079 g, R = 49%).
1H-RMN (CD3OD): 87,80-7,79 (m, 1H), 7,77-7,69 (m, 2H), 7,40-7,38 (m, 1H), 7,23-7,20 (m, 2H), 6,83-6,81 (m, 1H). EM (ES1+) m/z: 306,23 [M+H]+.
Ejemplo 21
Síntesis de [2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-il](1H-pirrol-1-il)metanona (21)
A una disolución de pirrol (0,066 ml, 0,95 mmol) y NaH (0,025 g,1,05 mmol) en 4 ml de DMF seco, se le añadió gota a gota cloruro de 2-(3-fluorofenil)-4-metoxi-1,3-tiazol-5-carbonilo (0,171 g, 0,63 mmol) disuelto en 3 ml de DMF seco. Entonces se agitó la mezcla resultante a temperatura ambiente durante la noche. Se extinguió la mezcla con disolución saturada de NH4Cl (10 ml) y se extrajo con acetato de etilo (10 ml x 2). Se separó la fase orgánica y se anhidrificó sobre sulfato de sodio anhidro, se destiló el disolvente y se purificó el producto en bruto sobre gel de sílice (n-hexano:acetato de etilo 9:1 como eluyente) para obtener [2-(3-fluorofenil)-4-metoxi-1,3-tiazol-5-il](1H-pirrol-1-il)metanona (0,157 g, 0,52 mmol, R = 82%). Se desprotegió esta última con 3 equivalentes de tribromuro de boro 1 M en diclorometano, luego se agitó durante la noche a temperatura ambiente. Se diluyó la reacción con DCM (10 ml) y se agitó con agua (10 ml) durante 10 minutos. Se separó la fase orgánica y se anhidrificó sobre sulfato de sodio anhidro, se destiló el disolvente y se purificó el producto en bruto sobre gel de sílice (n-hexano:acetato de etilo 8:2 como eluyente) para obtener la [2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-il](1H-pirrol-1-il)metanona como un aceite claro (0,132 g, R = 88%).
1H-RMN (CD3OD): 87,85-7,70 (m, 2H), 7,45-7,35 (m, 1H), 7,25-7,19 (m, 2H), 7,05-7,00 (m, 1H), 6,85-6,83 (m, 2H). EM (ES1+) m/z: 289,41 [M+H]+.
Ejemplo 22
Síntesis de [2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-il](piridin-2-il)metanona (22)
Un matraz de fondo redondo de 10 ml equipado con un agitador magnético se cargó con cloruro de 2-(3-fluorofenil)-4- metoxi-1,3-tiazol-5-carbonilo (0,060 g, 0,221 mmol) y 5 ml de THF seco y se refrigeró hasta -78°C. Se trató la disolución a la misma temperatura con bromuro de piridin-2-il-magnesio 0,25 M en THF (0,88 ml, 0,221 mmol), luego se agitó a -60°C durante 1 hora. Se retiró el sistema de enfriamiento y se extinguió la reacción a temperatura ambiente con disolución acuosa saturada de cloruro de amonio (10 ml). Se extrajo la mezcla con acetato de etilo (10 ml x 2), se recogieron las fases orgánicas y se lavaron dos veces con agua (10 ml x 2) y una vez con salmuera (10 ml). Entonces se anhidrificó la fase orgánica y se retiraron los disolventes a vacío. Se purificó el producto en bruto sobre gel de sílice mediante cromatografía ultrarrápida para proporcionar [2-(3-fluorofenil)-4-metoxi-1,3-tiazol-5- il](piridin-2-il)metanona con un rendimiento del 54% (0,037 g). Se desprotegió esta última con 3 equivalentes de tribromuro de boro 1 M en diclorometano, luego se agitó durante la noche a temperatura ambiente. Se diluyó la reacción con DCM (10 ml) y se agitó con agua (10 ml) durante 10 minutos. Se separó la fase orgánica y se anhidrificó sobre sulfato de sodio anhidro, se destiló el disolvente y se purificó el producto en bruto sobre gel de sílice (diclorometano: metanol 95:5 como eluyente) para obtener la [2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-il](piridin-2-il)metanona como un aceite de color verde claro (0,024 g, R = 68%).
1H-RMN (CD3OD): 88,80-8,75 (m, 1H), 8,06-8,02 (m, 1H), 7,80-7,71 (m, 3H), 7,54-7,39 (m, 2H), 7,19-7,17 (m, 1H). EM (ES1+) m/z: 301,02 [M+H]+.
Ejemplo 23
Síntesis de [2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-il](piridin-3-il)metanona (23)
Se sintetizó [2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-il](piridin-3-il)metanona según el procedimiento descrito para el compuesto 22 y partiendo de cloruro de piridin-3-il-magnesio. Se obtuvo el compuesto como un aceite naranja (0,055 g, R = 62%).
1H-RMN (CD3OD): 89,1 (s, 1H), 8,81-8,79 (m, 1H), 8,36-8,32 (m, 1H), 7,81-7,73 (m, 2H), 7,64-7,39 (m, 2H), 7,29 7,18 (m, 1H).
EM (ES1+) m/z: 301,03 [M+H]+.
Ejemplo 24
Síntesis de [2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-il](piridin-4-il)metanona (24)
Se sintetizó [2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-il](piridin-4-il)metanona según el procedimiento descrito para el compuesto 22 y partiendo de cloruro de piridin-4-il-magnesio. Se obtuvo el compuesto como un sólido de color marrón claro (0,046 g, R = 68%).
1H-RMN (CD3OD): 89,05-9,03 (m, 2H), 8,71-8,69 (m, 2H), 7,81-7,73 (m, 2H), 7,44-7,39 (m, 1H), 7,19-7,17 (m, 1H). EM (ES1+) m/z: 301,02 [M+H]+.
Ejemplo 25
Síntesis de [2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-il](1H-imidazol-1-il)metanona (25)
Se sintetizó [2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-il](1H-imidazol-1-il)metanona según el procedimiento descrito para el compuesto 21 y partiendo de 1H-imidazol. Se obtuvo el compuesto como un sólido marrón (0,088 g, R = 67%). 1H-RMN (CD3OD): 87,79-7,73 (m, 2H), 7,7 (s, 1H), 7,44-7,39 (m, 1H), 7,19-7,17 (m, 1H), 7,03-7,01 (m, 2H).
EM (ES1+) m/z: 290,31 [M+H]+.
Ejemplo 26
Síntesis de [2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-il](1H-pirazol-1-il)metanona (26)
Se sintetizó [2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-il](1H-pirazol-1-il)metanona según el procedimiento descrito para el compuesto 21 y partiendo de 1H-pirazol. Se obtuvo el compuesto como un sólido marrón (0,095 g, R = 69%).
1H-RMN (CDCla): 811,91 (s, 1H), 7,80-7,73 (m, 2H), 7,72-7,70 (m, 2H), 7,44-7,39 (m, 1H), 7,19-7,17 (m, 1H), 6,34 6,31 (m, 1H).
EM (ES1+) m/z: 290,35 [M+H]+.
Ejemplo 27
Síntesis de [2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-il](1,2-oxazepan-2-il)metanona (27)
Partiendo de cloruro de 2-(3-fluorofenil)-4-metoxi-1,3-tiazol-5-carbonilo (producto intermedio e, 0,110 g, 0,40 mmol) y cloruro de 1,2-oxazepan-2-io (0,069 g, 0,50 mmol), se sintetizó [2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-il](1,2-oxazepan-2-il)metanona según el procedimiento descrito para el compuesto 1. Se obtuvo el compuesto como un sólido amarillo (0,102 g, R = 79%).
1H-RMN (CDCla): 812,22 (s, 1H), 7,79-7,72 (m, 2H), 7,44-7,39 (m, 1H), 7,19-7,17 (m, 1H), 4,18-4,15 (m, 2H), 3,93 3,89 (m, 2H), 1,92-1,85 (m, 6).
EM (ES1+) m/z: 323,37 [M+H]+.
Ejemplo 28
Síntesis de [2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-il](5-oxa-6-azaspiro[2,4]heptan-6-il)metanona (28)
Partiendo de cloruro de 2-(3-fluorofenil)-4-metoxi-1,3-tiazol-5-carbonilo (producto intermedio e, 0,100 g, 0,37 mmol) y 5-oxa-6-azaespiro[2.4]heptano (0,039 g, 0,40 mmol), se sintetizó [2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-il](5-oxa-6-azaespiro[2.4]heptan-6-il)metanona según el procedimiento descrito para el compuesto 1. Se obtuvo el compuesto como un sólido amarillo (0,084 g, R = 71%).
1H-RMN (CDCh): 811,92 (s, 1H), 7,79-7,73 (m, 2H), 7,43-7,40 (m, 1H), 7,17-7,16 (m, 1H), 3,81-3,73 (m, 2H), 3,68 3,59 (m, 2H), 1,92-1,85 (m, 4H).
EM (ES1+) m/z: 321,35 [M+H]+.
Ejemplo 29
Síntesis de [2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-il](5-metil-1,2-oxazolidin-2-il)metanona (29)
Partiendo de cloruro de 2-(3-fluorofenil)-4-metoxi-1,3-tiazol-5-carbonilo (producto intermedio e, 0,100 g, 0,37 mmol) y 5-metil-1,2-oxazolidina (0,035 g, 0,40 mmol), se sintetizó [2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-il](5-metil-1,2-oxazolidin-2-il)metanona según el procedimiento descrito para el compuesto 1. Se obtuvo el compuesto como un sólido amarillo (0,083 g, R = 73%).
1H-RMN (CDCh): 811,90 (s, 1H), 7,79-7,74 (m, 2H), 7,44-7,38 (m, 1H), 7,16-7,11 (m, 1H), 3,68-3,58 (m, 2H), 3,48 3,39 (m, 1H), 2,47-2,23 (m, 2H), 1,87 (d, 3H).
EM (ES1+) m/z: 309,31 [M+H]+.
Ejemplo 30
Síntesis de [2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-il](hexahidro-2H-ciclopenta[d][1,2]oxazol-2-il)metanona (30) Partiendo de cloruro de 2-(3-fluorofenil)-4-metoxi-1,3-tiazol-5-carbonilo (producto intermedio e, 0,100 g, 0,37 mmol) y hexahidro-2H-ciclopenta[d][1,2]oxazol (0,047 g, 0,42 mmol), se sintetizó [2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-il](hexahidro-2H-ciclopenta[d][1,2]oxazol-2-il)metanona según el procedimiento descrito para el compuesto 1. Se obtuvo el compuesto como un sólido amarillo (0,094 g, R = 67%).
1H-RMN (CDCls): 811,97 (s, 1H), 7,80-7,75 (m, 2H), 7,43-7,36 (m, 1H), 7,15-7,12 (m, 1H), 3,90-3,81 (m,1H), 3,71 3,66 (m, 2H), 2,98-2,79 (m, 1H), 1,87-1,81 (m, 2H), 1,67-1,41(m, 4H).
EM (ES1+) m/z: 335,35 [M+H]+.
Ejemplo 31
Síntesis de [2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-il](1H-1,2,3-triazol-1-il)metanona (31)
Partiendo de cloruro de 2-(3-fluorofenil)-4-metoxi-1,3-tiazol-5-carbonilo (producto intermedio e, 0,120 g, 0,44 mmol) y 1H-1,2,3-triazol (0,035 g, 0,50 mmol), se sintetizó [2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-il](1H-1,2,3-triazol-1-il)metanona según el procedimiento descrito para el compuesto 1. Se obtuvo el compuesto como un sólido marrón (0,088 g, R = 69%).
1H-RMN (CDCla): 811,77 (s, 1H), 7,67-7,61 (m, 2H), 7,55 (d, 1H), 7,40-7,32 (m, 1H), 7,17 (d, 1H), 7,07-7,02 (m, 1H). EM (ES1+) m/z: 291,27 [M+H]+.
Ejemplo 32
Síntesis de [2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-il](1-metil-1H-tetrazol-5-il)metanona (32)
A temperatura ambiente, se mezclaron 0,230 g de cloruro de 2-(3-fluorofenil)-4-metoxi-1,3-tiazol-5-carbonilo (0,84 mmol) con 0,035 g (0,84 mmol) de isocianida de metilo. Entonces se calentó la mezcla a 60°C durante 3 horas. Se mezcló la mezcla con 5 ml de acetonitrilo y se enfrió hasta 0°C. Se añadió la mezcla de reacción a 0,055 g de azida de sodio (0,84 mmol) en 0,105 ml de 2,6-dimetilpiridina (0,90 mmol) en 5 ml de acetonitrilo a 0°C. Entonces se calentó la mezcla hasta 60°C y se agitó a esta temperatura durante 1 hora. A temperatura ambiente, se añadieron 10 ml de agua y 10 ml de acetato de etilo a la mezcla. Se eliminó por separación la fase orgánica y se extrajo la fase acuosa con 10 ml de acetato de etilo. Se secaron las fases orgánicas combinadas sobre sulfato de sodio anhidro y se eliminó el disolvente a vacío. Se purificó el producto en bruto sobre gel de sílice (diclorometano:metanol 95:5 como eluyente) para obtener [2-(3-fluorofenil)-4-metoxi-1,3-tiazol-5-il](1-metil-1H-tetrazol-5-il)metanona con un rendimiento del 82% como un polvo marrón. Se desprotegió la [2-(3-fluorofenil)-4-metoxi-1,3-tiazol-5-il](1-metil-1H-tetrazol-5-il)metanona con 3 equivalentes de tribromuro de boro 1 M en diclorometano, y luego se agitó durante la noche a temperatura ambiente. Se diluyó la reacción con DCM (10 ml) y se agitó con agua (10 ml) durante 10 minutos. Se separó la fase orgánica y se anhidrificó sobre sulfato de sodio anhidro, se destiló el disolvente y se purificó el producto en bruto sobre gel de sílice (diclorometano:metanol 90:10 como eluyente) para obtener [2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-il](1-metil-1H-tetrazol-5-il)metanona como un polvo amarillo (0,122 g, rendimiento del 47% a lo largo de dos etapas).
1H-RMN (CDCla): 811,80 (s, 1H), 7,77-7,71 (m, 2H), 7,61-7,52 (m, 1H), 7,47-7,42 (m, 1H), 4,23 (s, 3H).
EM (ES1+) m/z: 306,28 [M+H]+.
Ejemplo 33
Ensayo de TRPM8 in vitro
La actividad antagonista de TRPM8 de los 26 compuestos se determinó midiendo los cambios en los niveles intracelulares de calcio usando un colorante fluorescente sensible a Ca+2. Los experimentos se realizaron usando células HEK-293 que expresaban de manera estable el TRPM8 humano. Se sembraron las células 10000 células/pocillo en placas de 384 recubiertas con poli-D-Lisina MATRIX de fondo negro/transparente (Thermo Scientific, Waltham, MA EE. UU.) en medio completo y se hicieron crecer durante la noche a 37°C, el 5% de CO2.
24h después de la siembra, se retiró el medio de cultivo celular, se lavaron las células con tampón de ensayo de Tyrode y luego se cargaron con el indicador de Ca+2 fluorescente colorante Fluo-4 NW (Molecular Probes, Life Technologies, Paisley, RU) suplementado con probenecid soluble en agua (Molecular Probes). Se incubaron las placas de células cargadas de colorante durante 1 h a temperatura ambiente. Se añadieron los compuestos o vehículo y se monitorizó la respuesta cinética mediante una placa de obtención de imágenes fluorimétricas (FLIPRTETRA; Molecular Devices, Sunnyvale, CA, EE. UU.) a lo largo de un periodo de cinco minutos (300 segundos). Se realizó una segunda inyección 5 minutos después del agonista de referencia agente de enfriamiento 10 o icilina a concentración CEs0. La señal de la fluorescencia emitida se registró durante tres minutos adicionales. Se analizaron los datos mediante Spotfire DecisionSite versión 9.1.1. La bioactividad ejercida por los compuestos o vehículo se expresó como porcentaje de inhibición y entonces se calcularon valores de CI50. La escala en porcentaje se define mediante una inhibición del 100% en la que las unidades de fluorescencia relativa (UFR) de la prueba eran idénticas a los controles MIN en la segunda inyección (capsazepina a CI100, 50 |iM) y el 0% de inhibición en la que las UFR de la prueba eran idénticas a los controles MAX en la segunda inyección (agente de enfriamiento 10 a CEs0, 20-30 |iM). Los resultados obtenidos con cada compuesto sometido a prueba se notifican en la tabla 1.
Ejemplo 34
Ensayo de estimulación con frío in vitro
Se determinó la actividad antagonista de TRPM8 de los 26 compuestos en un ensayo de estimulación con frío in vitro.
En detalle, se sembraron células HEK-293 que expresaban de manera estable el TRPM8 humano (1,5-1,8 x 106) en un frasco T75 en medio completo. 3-4 días después de sembrar (células confluentes a ~ 80%), se eliminó el medio y se cargaron las células mediante una disolución de colorante Screen QuestTM Fluo-8 NW (ABD Bioquest, Sunnyvale, CA, EE. UU.) en la oscuridad. Se incubaron los frascos de células cargadas con colorante durante 45 minutos a temperatura ambiente en la oscuridad y entonces se retiró la disolución de Fluo-8 NW y se sembraron las células en las placas de ensayo 96 (placas de reacción de 96 pocillos MicroAmpTM Optical; Applied Biosystems, Life technologies) a 100.000 células/pocillo (20 |il/pocillo). Se añadió cada compuesto 1-26 y se incubó a temperatura ambiente durante 5 minutos. Se registró la señal durante 2 minutos a 25°C y luego se disminuyó la temperatura hasta niveles subfisiológicos (14°C) y se registró la señal durante 3 minutos mediante el sistema de detección de secuencias ABI Prism® 7900HT (Life Technologies). Se evaluó la diferencia de fluorescencia (AF = fluorescencia a 525 nm a 14°C - fluorescencia a 525 nm a 25°C). Se realizó el análisis calculando AF/F0, donde F0 era la señal fluorescente a la temperatura inicial (25°C). Se generaron curvas de CI50 (mitad de la concentración máxima) ajustando los datos de fluorescencia con una ecuación de curva sigmoidea usando el software GraphPad PRISM® (versión 5, GraphPad Software Inc., La Jolla, CA, EE. UU.). Todas las determinaciones de puntos de datos se realizaron por duplicado antes de que se calculara un valor medio, incluyendo barras de error que representan el error estándar de la media. Los resultados obtenidos con cada compuesto sometido a prueba se notifican en la tabla 1.
Ejemplo 35
Estabilidad metabólica en microsomas de rata
Protocolo del ensayo de fase I
Se disolvieron los compuestos 1, 2, 8 y 9 por duplicado en DMSO y se preincubaron, a la concentración final de 1 |iM, durante 10 min a 37°C en tampón fosfato de potasio 50 mM pH 7,4, MgCl23 mM, con microsomas de hígado de rata (Xenotech) a la concentración final de 0,5 mg/ml.
Tras el periodo de preincubación, se iniciaron las reacciones añadiendo la mezcla de cofactores (NADP, Glc6P, Glc6PDH en bicarbonato de sodio al 2%); se tomaron muestras (50 |il) a los tiempos de 0, 10, 20, 30 y 60 min, se añadieron 150 |il de acetonitrilo con verapamilo 0,02 |iM como patrón interno (ES) para detener la reacción. Tras la centrifugación, se analizaron los sobrenadantes mediante CL-EM/EM.
Se añadió una muestra de control sin cofactores con el fin de comprobar la estabilidad de los compuestos de prueba en la matriz después de 60 min. Se añadieron 7-etoxicumarina y propranolol como patrones de referencia. Los resultados obtenidos con cada compuesto se notifican en la tabla 1.
Protocolo de ensayo de fase II
Se incubaron microsomas de hígado de rata (conc. final de 0,5 mg/ml) en hielo durante 15 min en tampón fosfato de potasio 50 mM pH 7,4, MgCh 3 mM y se activaron con proteína microsomal alameticina 50 |ig/mg. Se transfirieron las muestras a 37°C y se añadieron los compuestos 1, 2, 8 y 9, disueltos en DMSO, por duplicado a la concentración final de 1 |iM. Después de 5 min, se inicia la reacción añadiendo 25 |il de UDPGA (16,15 mg/ml en bicarbonato de sodio al 0,2%). Se tomaron muestras a los tiempos de 0, 10, 20, 30 y 60 min, se añadieron 150 |il de acetonitrilo con verapamilo 0,02 uM como patrón interno (PI) para detener la reacción. Tras la centrifugación, se analizaron los sobrenadantes mediante CL-EM/EM. Se añadió 7-hidroxicumarina como patrón de referencia. El resultado obtenido con cada compuesto se notifica en la tabla 1.
Tabla 1





Claims (8)
1. Compuesto de fórmula (I):

en la que
R1 es un grupo heterocíclico alifático o aromático, de 5, 6 o 7 miembros sustituido o no sustituido que contiene 1, 2, 3 o 4 heteroátomos seleccionados de N, O y S, en el que, cuando está sustituido, dicho heterociclo alifático o aromático, de 5, 6 o 7 miembros está sustituido con uno o más grupos independientemente seleccionados de cicloalquilo o alquilo C1-C3, Cl y F o está sustituido con R3 y R4 que tomados juntos forman un resto cíclico saturado; y
R2 se selecciona de hidrógeno, alquilo C1-C2, F, Cl y OH;
y sales farmacéuticamente aceptables del mismo.
2. Compuesto según la reivindicación 1, en el que R1 se selecciona del grupo que consiste en oxazolidinilo, oxolanilo, pirrolidinilo, oxazinanilo, morfolinilo, piperidinilo, metilpirrolidinilo, pirrolilo, metilpirrolilo, furanilo, tiofenilo, piridinilo, imidazolilo, pirazolilo, oxadiazolilo y oxazolilo.
3. Compuesto según una cualquiera de las reivindicaciones 1 o 2, en el que R1 se selecciona del grupo que consiste en 1,2-oxazolidin-2-ilo, 1,3-oxazolidin-3-ilo, oxolan-2-ilo, pirrolidin-1-ilo, 1,2-oxazinan-2-ilo, 1,3-oxazinan-3-ilo, morfolin-4-ilo, piperidin-1-ilo, pirrolidin-2-ilo, 1 -metilpirrolidin-2-ilo, 1H-pirrol-2-ilo, 1-metil-1H-pirrol-2-ilo, furan-2-ilo, tiofen-2-ilo, 1 H-pirrol-1 -ilo, piridin-2-ilo, piridin-3-ilo, piridin-4-ilo, 1H-imidazol-1-ilo y 1H-pirazol-1-ilo.
4. Compuesto según una cualquiera de las reivindicaciones 1 a 3, en el que dicho heterociclo alifático o aromático, de 5, 6 o 7 miembros no está sustituido.
5. Compuesto según una cualquiera de las reivindicaciones 1 a 3, en el que dicho resto cíclico saturado es ciclobutano, ciclopentano o ciclohexano.
6. Compuesto según una cualquiera de las reivindicaciones 1 a 5, en el que R2 es F u OH.
7. Compuesto según la reivindicación 6, en el que R2 es 3-F o 2-OH.
8. Compuesto según una cualquiera de las reivindicaciones 1 a 7, en el que
R1 es oxazolidinilo y R2 es F u OH,
o
R1 es oxolanilo y R2 es F u OH,
o
R1 es pirrolidinilo y R2 es F u OH,
o
R1 es oxazinanilo y R2 es F u OH,
o
R1 es morfolinilo y R2 es F u OH,
o
R1 es piperidinilo y R2 es F u OH,
o
R1 es metilpirrolidinilo y R2 es F u OH,
o
R1 es pirrolilo y R2 es F u OH,
o
R1 es metilpirrolilo y R2 es F u OH,
o
R1 es furanilo y R2 es F u OH,
o
R1 es tiofenilo y R2 es F u OH,
o
R1 es piridinilo y R2 es F u OH,
o
R1 es imidazolilo y R2 es F u OH,
o
R1 es pirazolilo y R2 es F u OH.
Compuesto según una cualquiera de las reivindicaciones 1 a 8 seleccionado de:
[2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-il](1,2-oxazolidin-2-il)metanona (1)
[4-hidroxi-2-(2-hidroxifenil)-1,3-tiazol-5-il](1,2-oxazolidin-2-il)metanona (2) [2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-il](1,3-oxazolidin-3-il)metanona (3)
[4-hidroxi-2-(2-hidroxifenil)-1,3-tiazol-5-il](1,3-oxazolidin-3-il)metanona (4) [2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-il](oxolan-2-il)metanona (5)
[4-hidroxi-2-(2-hidroxifenil)-1,3-tiazol-5-il](oxolan-2-il)metanona (6)
[2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-il](pirrolidin-1-il)metanona (7)
[4-hidroxi-2-(2-hidroxifenil)-1,3-tiazol-5-il](1,2-oxazinan-2-il)metanona (8)
[2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-il](1,2-oxazinan-2-il)metanona (9)
[2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-il](1,3-oxazinan-3-il)metanona (10)
[4-hidroxi-2-(2-hidroxifenil)-1,3-tiazol-5-il](1,3-oxazinan-3-il)metanona (11) [2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-il](morfolin-4-il)metanona (12)
[4-hidroxi-2-(2-hidroxifenil)-1,3-tiazol-5-il](morfolin-4-il)metanona (13)
[2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-il](piperidin-1-il)metanona (14)
[2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-il](pirrolidin-2-il)metanona (15)
[2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-il](1-metilpirrolidin-2-il)metanona (16)
[2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-il](1H-pirrol-2-il)metanona (17)
[2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-il](1-metil-1H-pirrol-2-il)metanona (18)
[2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-il](furan-2-il)metanona (19)
[2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-il](tiofen-2-il)metanona (20)
[2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-il](1H-pirrol-1-il)metanona (21)
[2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-il](piridin-2-il)metanona (22)
[2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-il](piridin-3-il)metanona (23)
[2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-il](piridin-4-il)metanona (24)
[2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-il](1H-imidazol-1-il)metanona (25)
[2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-il](1H-pirazol-1-il)metanona (26)
[2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-il](1,2-oxazepan-2-il)metanona (27)
[2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-il](5-oxa-6-azaspiro[2,4]heptan-6-il)metanona (28)
[2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-il](5-metil-1,2-oxazolidin-2-il)metanona (29)
[2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-il](hexahidro-2H-ciclopenta[d][1,2]oxazol-2-il)metanona (30)
[2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-il](1H-1,2,3-triazol-1-il)metanona (31) y
[2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-il](1-metil-1H-tetrazol-5-il)metanona (32).
Compuesto según una cualquiera de las reivindicaciones 1 a 9 seleccionado de:
[2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-il](1,2-oxazolidin-2-il)metanona (1)
[4-hidroxi-2-(2-hidroxifenil)-1,3-tiazol-5-il](1,2-oxazolidin-2-il)metanona (2)
[4-hidroxi-2-(2-hidroxifenil)-1,3-tiazol-5-il](1,2-oxazinan-2-il)metanona (8) y
[2-(3-fluorofenil)-4-hidroxi-1,3-tiazol-5-il](1,2-oxazinan-2-il)metanona (9).
Compuesto según una cualquiera de las reivindicaciones 1 a 10, para su uso en la prevención y/o el tratamiento de una enfermedad seleccionada del grupo que consiste en dolor, prurito, enfermedades del intestino irritable, trastornos respiratorios exacerbados y/o inducidos por frío, isquemia, neurodegeneración, accidente cerebrovascular, trastornos urológicos, trastornos psiquiátricos y trastornos de la córnea asociados a alteraciones en la producción de las lágrimas y/o parpadeo alterado.
Compuesto para su uso según la reivindicación 11, en el que dicha enfermedad se selecciona de dolor crónico, dolor por cáncer, dolor neuropático, incluyendo alodinia al frío, dolor posoperatorio, neuralgia, neuropatías, incluyendo neuropatía diabética, fibromialgia, algesia, dolor asociado a lesión nerviosa, migraña, cefalea, dolor asociado a osteoartritis y artritis reumatoide, prurito, enfermedad irritable del intestino, síndrome de vejiga dolorosa, cistitis intersticial, sobreactividad del detrusor, incontinencia urinaria, sobreactividad del detrusor neurogénica, sobreactividad del detrusor idiopática, hiperplasia prostática benigna, trastornos del tracto urinario inferior y síntomas del tracto urinario inferior, ansiedad, depresión y asma, EPOC, hipertensión pulmonar exacerbados y/o inducidos por frío, epífora y enfermedad de ojo seco. Composición farmacéutica que comprende como principio activo al menos un compuesto según una
cualquiera de las reivindicaciones 1 a 10 en combinación con excipientes y/o diluyentes farmacéuticamente aceptables.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP15201788.5A EP3184524A1 (en) | 2015-12-21 | 2015-12-21 | 4-hydroxy-2-phenyl-1,3-thiazol-5-yl methanone derivatives as trpm8 antagonists |
| PCT/EP2016/081567 WO2017108632A1 (en) | 2015-12-21 | 2016-12-16 | 4-hydroxy-2-phenyl-1,3-thiazol-5-yl methanone derivatives as trpm8 antagonists |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2757324T3 true ES2757324T3 (es) | 2020-04-28 |
Family
ID=54883947
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES16823231T Active ES2757324T3 (es) | 2015-12-21 | 2016-12-16 | Derivados de (4-hidroxi-2-fenil-1,3-tiazol-5-il)metanona como antagonistas de TRPM8 |
Country Status (20)
| Country | Link |
|---|---|
| US (1) | US10246448B2 (es) |
| EP (2) | EP3184524A1 (es) |
| JP (1) | JP6908610B2 (es) |
| KR (1) | KR102681427B1 (es) |
| CN (1) | CN108699053B (es) |
| AU (1) | AU2016378108B9 (es) |
| CY (1) | CY1122312T1 (es) |
| DK (1) | DK3394053T3 (es) |
| EA (1) | EA037845B1 (es) |
| ES (1) | ES2757324T3 (es) |
| HK (1) | HK1257896B (es) |
| HR (1) | HRP20192008T1 (es) |
| HU (1) | HUE046284T2 (es) |
| LT (1) | LT3394053T (es) |
| PT (1) | PT3394053T (es) |
| RS (1) | RS59475B1 (es) |
| SI (1) | SI3394053T1 (es) |
| SM (1) | SMT201900644T1 (es) |
| WO (1) | WO2017108632A1 (es) |
| ZA (1) | ZA201803300B (es) |
Family Cites Families (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US2015531A (en) | 1932-07-25 | 1935-09-24 | Champion Paper & Fibre Co | Roll coating machine |
| JP2008515950A (ja) | 2004-10-13 | 2008-05-15 | バイエル・ヘルスケア・アクチェンゲゼルシャフト | 泌尿器系障害の処置用の冷メントール受容体−1(cmr−1)アンタゴニストとしての4−置換ベンジルオキシ−フェニルメチルアミド誘導体 |
| WO2007017093A1 (en) | 2005-08-04 | 2007-02-15 | Bayer Healthcare Ag | Substituted 2-benzyloxy-benzoic acid amide derivatives |
| WO2007017094A1 (en) | 2005-08-04 | 2007-02-15 | Bayer Healthcare Ag | Substituted benzyloxy-phenylmethylcarbamate derivatives |
| WO2007017092A1 (en) | 2005-08-04 | 2007-02-15 | Bayer Healthcare Ag | Substituted 4-benzyloxy-benzoic acid amide derivatives |
| WO2007080109A1 (en) | 2006-01-16 | 2007-07-19 | Bayer Healthcare Ag | Substituded benzyloxy-phenylmethylurea derivatives |
| WO2007134107A2 (en) | 2006-05-10 | 2007-11-22 | Janssen Pharmaceutica N.V. | Cold menthol receptor-1 antagonists |
| BRPI0815096A2 (pt) | 2007-07-18 | 2015-07-14 | Janssen Pharmaceutica Nv | Sulfonamidas como moduladores de trpm8 |
| WO2010103381A1 (en) | 2009-03-13 | 2010-09-16 | Glenmark Pharmaceuticals S.A. | Spirocyclic piperidine derivatives as trpm 8 modulators |
| FR2943672B1 (fr) * | 2009-03-27 | 2011-03-25 | Sanofi Aventis | Derives de 3-alcoxy-4,5-diarylthiophene-2-carboxamide,leur preparation et leur application en therapeutique. |
| RU2011148937A (ru) | 2009-05-01 | 2013-06-10 | Раквалиа Фарма Инк. | Производные сульфамоилбензойной кислоты в качестве антагонистов trpm8 |
| WO2011109551A2 (en) * | 2010-03-03 | 2011-09-09 | Calcimedica Inc. | Compounds that modulate intracellular calcium |
| ES2377785B2 (es) | 2010-09-08 | 2012-09-26 | Universidad Miguel Hernández De Elche | Composición farmacéutica para el tratamiento del ojo seco. |
| EP2481727A1 (en) * | 2011-01-28 | 2012-08-01 | Dompe S.p.A. | TRPM8 receptor antagonists |
| US9585875B2 (en) * | 2011-12-19 | 2017-03-07 | Dompé Farmaceutici S.P.A. | TRPM8 antagonists |
| WO2014181788A1 (ja) * | 2013-05-08 | 2014-11-13 | キッセイ薬品工業株式会社 | α-置換グリシンアミド誘導体 |
| US10196368B2 (en) * | 2014-06-23 | 2019-02-05 | Dompé Farmaceutici S.P.A. | 2-aryl-4-hydroxy-1,3-thiazole derivatives useful as TRPM8-inhibitors in treatment of neuralgia, pain, COPD and asthma |
-
2015
- 2015-12-21 EP EP15201788.5A patent/EP3184524A1/en not_active Withdrawn
-
2016
- 2016-12-16 LT LT16823231T patent/LT3394053T/lt unknown
- 2016-12-16 US US16/061,850 patent/US10246448B2/en active Active
- 2016-12-16 EA EA201891503A patent/EA037845B1/ru unknown
- 2016-12-16 EP EP16823231.2A patent/EP3394053B1/en active Active
- 2016-12-16 AU AU2016378108A patent/AU2016378108B9/en active Active
- 2016-12-16 ES ES16823231T patent/ES2757324T3/es active Active
- 2016-12-16 JP JP2018532605A patent/JP6908610B2/ja active Active
- 2016-12-16 SI SI201630487T patent/SI3394053T1/sl unknown
- 2016-12-16 HU HUE16823231A patent/HUE046284T2/hu unknown
- 2016-12-16 DK DK16823231T patent/DK3394053T3/da active
- 2016-12-16 RS RS20191415A patent/RS59475B1/sr unknown
- 2016-12-16 SM SM20190644T patent/SMT201900644T1/it unknown
- 2016-12-16 HR HRP20192008TT patent/HRP20192008T1/hr unknown
- 2016-12-16 KR KR1020187016746A patent/KR102681427B1/ko active Active
- 2016-12-16 HK HK19100257.7A patent/HK1257896B/en unknown
- 2016-12-16 PT PT168232312T patent/PT3394053T/pt unknown
- 2016-12-16 WO PCT/EP2016/081567 patent/WO2017108632A1/en not_active Ceased
- 2016-12-16 CN CN201680073977.3A patent/CN108699053B/zh active Active
-
2018
- 2018-05-17 ZA ZA2018/03300A patent/ZA201803300B/en unknown
-
2019
- 2019-11-11 CY CY20191101183T patent/CY1122312T1/el unknown
Also Published As
| Publication number | Publication date |
|---|---|
| HK1257896B (en) | 2020-06-19 |
| SI3394053T1 (sl) | 2019-12-31 |
| KR102681427B1 (ko) | 2024-07-05 |
| EP3184524A1 (en) | 2017-06-28 |
| JP6908610B2 (ja) | 2021-07-28 |
| RS59475B1 (sr) | 2019-12-31 |
| CN108699053B (zh) | 2022-01-14 |
| LT3394053T (lt) | 2019-12-10 |
| AU2016378108B2 (en) | 2020-12-17 |
| KR20180095818A (ko) | 2018-08-28 |
| US20180319785A1 (en) | 2018-11-08 |
| WO2017108632A1 (en) | 2017-06-29 |
| AU2016378108A1 (en) | 2018-06-21 |
| AU2016378108B9 (en) | 2021-01-28 |
| EP3394053A1 (en) | 2018-10-31 |
| EP3394053B1 (en) | 2019-08-21 |
| US10246448B2 (en) | 2019-04-02 |
| SMT201900644T1 (it) | 2020-01-14 |
| EA037845B1 (ru) | 2021-05-27 |
| HRP20192008T1 (hr) | 2020-02-07 |
| HUE046284T2 (hu) | 2020-02-28 |
| CN108699053A (zh) | 2018-10-23 |
| PT3394053T (pt) | 2019-10-31 |
| ZA201803300B (en) | 2019-07-31 |
| JP2018538334A (ja) | 2018-12-27 |
| EA201891503A1 (ru) | 2019-01-31 |
| CY1122312T1 (el) | 2021-01-27 |
| DK3394053T3 (da) | 2019-11-18 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US20230303489A1 (en) | Compounds, compositions and methods of use | |
| US10961255B2 (en) | Imidazoles as histone demethylase inhibitors | |
| KR20240159579A (ko) | Parg 억제제 | |
| BRPI0906962B1 (pt) | Composto de aminodiidrotiazina fundido | |
| EP2332943A1 (en) | Novel fused aminodihydrothiazine derivative | |
| US20150011751A1 (en) | Novel triazine derivative | |
| BR112012025496A2 (pt) | composto inibidor quinase, composição farmacêutica que o compreende e seu uso | |
| CA2957048A1 (en) | Optionally fused heterocyclyl-substituted derivatives of pyrimidine useful for the treatment of inflammatory, metabolic, oncologic and autoimmune diseases | |
| US12398097B2 (en) | WDR5-MYC inhibitors | |
| BR112020026653A2 (pt) | Compostos heterocíclicos como inibidores de trk | |
| CA2824460A1 (en) | Azetidine derivatives useful for the treatment of metabolic and inflammatory diseases | |
| WO2023077070A1 (en) | Rxfp1 agonists | |
| CA3194314A1 (en) | 3-[(1h-pyrazol-4-yl)oxy]pyrazin-2-amine compounds as hpk1 inhibitor and use thereof | |
| ES2724111T3 (es) | Derivados de 2-aril-4-hidroxi-1,3-tiazol útiles como inhibidores de TRPM8 en el tratamiento de neuralgia, dolor, EPOC y asma | |
| ES2757324T3 (es) | Derivados de (4-hidroxi-2-fenil-1,3-tiazol-5-il)metanona como antagonistas de TRPM8 | |
| HK1257896A1 (en) | (4-hydroxy-2-phenyl-1,3-thiazol-5-yl) methanone derivatives as trpm8 antagonists | |
| CN119462637A (zh) | Polq抑制剂 | |
| BR112019017514B1 (pt) | Composto sulfoximina, mistura, método para inibir uma glicosidase, e, composição farmacêutica | |
| BR112017008790B1 (pt) | Compostos cromanos substituídos | |
| HK1244786B (zh) | 吲哚和氮杂吲哚衍生物及其用於神经退化性疾病中的用途 |