ES2759060T3 - Administración intraarticular de pepstatina para la artrosis - Google Patents
Administración intraarticular de pepstatina para la artrosis Download PDFInfo
- Publication number
- ES2759060T3 ES2759060T3 ES14741803T ES14741803T ES2759060T3 ES 2759060 T3 ES2759060 T3 ES 2759060T3 ES 14741803 T ES14741803 T ES 14741803T ES 14741803 T ES14741803 T ES 14741803T ES 2759060 T3 ES2759060 T3 ES 2759060T3
- Authority
- ES
- Spain
- Prior art keywords
- pepstatin
- osteoarthritis
- cathepsin
- treatment
- intra
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- FAXGPCHRFPCXOO-LXTPJMTPSA-N pepstatin A Chemical compound OC(=O)C[C@H](O)[C@H](CC(C)C)NC(=O)[C@H](C)NC(=O)C[C@H](O)[C@H](CC(C)C)NC(=O)[C@H](C(C)C)NC(=O)[C@H](C(C)C)NC(=O)CC(C)C FAXGPCHRFPCXOO-LXTPJMTPSA-N 0.000 title claims abstract description 108
- 108010091212 pepstatin Proteins 0.000 title claims abstract description 105
- 229950000964 pepstatin Drugs 0.000 title claims abstract description 100
- 201000008482 osteoarthritis Diseases 0.000 title claims abstract description 60
- 239000003814 drug Substances 0.000 claims abstract description 41
- 150000003839 salts Chemical class 0.000 claims abstract description 32
- 238000011282 treatment Methods 0.000 claims abstract description 32
- 239000000825 pharmaceutical preparation Substances 0.000 claims abstract description 26
- 208000004454 Hyperalgesia Diseases 0.000 claims abstract description 21
- 239000000203 mixture Substances 0.000 claims abstract description 19
- 208000002193 Pain Diseases 0.000 claims abstract description 18
- 230000036407 pain Effects 0.000 claims abstract description 16
- 206010003246 arthritis Diseases 0.000 claims abstract description 15
- 239000000546 pharmaceutical excipient Substances 0.000 claims abstract description 15
- 239000012453 solvate Substances 0.000 claims abstract description 15
- 208000035154 Hyperesthesia Diseases 0.000 claims abstract description 11
- 238000011321 prophylaxis Methods 0.000 claims abstract description 11
- 230000001991 pathophysiological effect Effects 0.000 claims abstract description 8
- 206010053552 allodynia Diseases 0.000 claims abstract description 7
- 230000003902 lesion Effects 0.000 claims abstract description 7
- 230000000472 traumatic effect Effects 0.000 claims abstract description 7
- 230000003442 weekly effect Effects 0.000 claims abstract description 7
- 239000004480 active ingredient Substances 0.000 claims description 25
- 102000003908 Cathepsin D Human genes 0.000 description 44
- 108090000258 Cathepsin D Proteins 0.000 description 44
- 210000001179 synovial fluid Anatomy 0.000 description 34
- 230000000694 effects Effects 0.000 description 30
- 239000000126 substance Substances 0.000 description 29
- 150000001875 compounds Chemical class 0.000 description 26
- 229920002683 Glycosaminoglycan Polymers 0.000 description 22
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 21
- 239000000243 solution Substances 0.000 description 21
- 102000005600 Cathepsins Human genes 0.000 description 20
- 108010084457 Cathepsins Proteins 0.000 description 20
- 201000010099 disease Diseases 0.000 description 20
- 229940079593 drug Drugs 0.000 description 20
- 238000002347 injection Methods 0.000 description 20
- 239000007924 injection Substances 0.000 description 20
- 230000000875 corresponding effect Effects 0.000 description 18
- 108090000765 processed proteins & peptides Proteins 0.000 description 16
- -1 aspartyl Chemical group 0.000 description 14
- 239000003112 inhibitor Substances 0.000 description 14
- 108090000623 proteins and genes Proteins 0.000 description 14
- 241000283690 Bos taurus Species 0.000 description 13
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 13
- 238000000034 method Methods 0.000 description 13
- 238000002360 preparation method Methods 0.000 description 13
- 239000000725 suspension Substances 0.000 description 13
- 238000012360 testing method Methods 0.000 description 13
- 210000001519 tissue Anatomy 0.000 description 13
- 239000003981 vehicle Substances 0.000 description 13
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 12
- 241001465754 Metazoa Species 0.000 description 12
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 12
- 230000015556 catabolic process Effects 0.000 description 12
- 238000002474 experimental method Methods 0.000 description 12
- 239000002609 medium Substances 0.000 description 12
- 102000004169 proteins and genes Human genes 0.000 description 12
- 210000000845 cartilage Anatomy 0.000 description 11
- 210000000281 joint capsule Anatomy 0.000 description 11
- 238000011534 incubation Methods 0.000 description 10
- 102000004580 Aspartic Acid Proteases Human genes 0.000 description 9
- 108010017640 Aspartic Acid Proteases Proteins 0.000 description 9
- 210000001188 articular cartilage Anatomy 0.000 description 9
- 210000000629 knee joint Anatomy 0.000 description 9
- 235000018102 proteins Nutrition 0.000 description 9
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 9
- 206010028980 Neoplasm Diseases 0.000 description 8
- 241000700159 Rattus Species 0.000 description 8
- 239000002585 base Substances 0.000 description 8
- 238000006731 degradation reaction Methods 0.000 description 8
- 238000001035 drying Methods 0.000 description 8
- 230000014509 gene expression Effects 0.000 description 8
- 241000282414 Homo sapiens Species 0.000 description 7
- 239000002253 acid Substances 0.000 description 7
- 210000004369 blood Anatomy 0.000 description 7
- 239000008280 blood Substances 0.000 description 7
- 210000001503 joint Anatomy 0.000 description 7
- 238000001690 micro-dialysis Methods 0.000 description 7
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 6
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 6
- 238000004458 analytical method Methods 0.000 description 6
- 238000003556 assay Methods 0.000 description 6
- 201000011510 cancer Diseases 0.000 description 6
- 230000002797 proteolythic effect Effects 0.000 description 6
- 206010039073 rheumatoid arthritis Diseases 0.000 description 6
- 239000002904 solvent Substances 0.000 description 6
- 238000002560 therapeutic procedure Methods 0.000 description 6
- GFNANZIMVAIWHM-OBYCQNJPSA-N triamcinolone Chemical compound O=C1C=C[C@]2(C)[C@@]3(F)[C@@H](O)C[C@](C)([C@@]([C@H](O)C4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 GFNANZIMVAIWHM-OBYCQNJPSA-N 0.000 description 6
- 229960005294 triamcinolone Drugs 0.000 description 6
- 206010061218 Inflammation Diseases 0.000 description 5
- 150000001412 amines Chemical class 0.000 description 5
- 238000010171 animal model Methods 0.000 description 5
- 230000006378 damage Effects 0.000 description 5
- 238000011161 development Methods 0.000 description 5
- 230000018109 developmental process Effects 0.000 description 5
- 238000009792 diffusion process Methods 0.000 description 5
- 238000009472 formulation Methods 0.000 description 5
- 239000013642 negative control Substances 0.000 description 5
- 239000013641 positive control Substances 0.000 description 5
- 230000009467 reduction Effects 0.000 description 5
- 241000282472 Canis lupus familiaris Species 0.000 description 4
- 102000004190 Enzymes Human genes 0.000 description 4
- 108090000790 Enzymes Proteins 0.000 description 4
- 102000035195 Peptidases Human genes 0.000 description 4
- 108091005804 Peptidases Proteins 0.000 description 4
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 4
- 239000004365 Protease Substances 0.000 description 4
- 102100028255 Renin Human genes 0.000 description 4
- 108090000783 Renin Proteins 0.000 description 4
- 210000000988 bone and bone Anatomy 0.000 description 4
- 210000004027 cell Anatomy 0.000 description 4
- 238000006243 chemical reaction Methods 0.000 description 4
- 239000003795 chemical substances by application Substances 0.000 description 4
- 239000013078 crystal Substances 0.000 description 4
- IPZJQDSFZGZEOY-UHFFFAOYSA-N dimethylmethylene Chemical group C[C]C IPZJQDSFZGZEOY-UHFFFAOYSA-N 0.000 description 4
- 229940088598 enzyme Drugs 0.000 description 4
- 238000011156 evaluation Methods 0.000 description 4
- 230000006870 function Effects 0.000 description 4
- 230000004054 inflammatory process Effects 0.000 description 4
- 210000003141 lower extremity Anatomy 0.000 description 4
- 230000002132 lysosomal effect Effects 0.000 description 4
- 238000005259 measurement Methods 0.000 description 4
- 238000002483 medication Methods 0.000 description 4
- 239000008194 pharmaceutical composition Substances 0.000 description 4
- 229910052700 potassium Inorganic materials 0.000 description 4
- 239000002243 precursor Substances 0.000 description 4
- 230000008569 process Effects 0.000 description 4
- 230000002829 reductive effect Effects 0.000 description 4
- 239000000523 sample Substances 0.000 description 4
- 239000012086 standard solution Substances 0.000 description 4
- 239000006228 supernatant Substances 0.000 description 4
- 230000009885 systemic effect Effects 0.000 description 4
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 3
- 102100021257 Beta-secretase 1 Human genes 0.000 description 3
- 208000026310 Breast neoplasm Diseases 0.000 description 3
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 3
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 3
- 239000006144 Dulbecco’s modified Eagle's medium Substances 0.000 description 3
- 108010037362 Extracellular Matrix Proteins Proteins 0.000 description 3
- 102000010834 Extracellular Matrix Proteins Human genes 0.000 description 3
- 241000282412 Homo Species 0.000 description 3
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 3
- 208000012659 Joint disease Diseases 0.000 description 3
- WMFOQBRAJBCJND-UHFFFAOYSA-M Lithium hydroxide Chemical compound [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 description 3
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 3
- 241000124008 Mammalia Species 0.000 description 3
- MBBZMMPHUWSWHV-BDVNFPICSA-N N-methylglucamine Chemical compound CNC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO MBBZMMPHUWSWHV-BDVNFPICSA-N 0.000 description 3
- 108090000284 Pepsin A Proteins 0.000 description 3
- 102000057297 Pepsin A Human genes 0.000 description 3
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 3
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 3
- 241000219061 Rheum Species 0.000 description 3
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 3
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical group [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 3
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 3
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 3
- LEHOTFFKMJEONL-UHFFFAOYSA-N Uric Acid Chemical compound N1C(=O)NC(=O)C2=C1NC(=O)N2 LEHOTFFKMJEONL-UHFFFAOYSA-N 0.000 description 3
- 238000010521 absorption reaction Methods 0.000 description 3
- 229910052783 alkali metal Inorganic materials 0.000 description 3
- 230000004075 alteration Effects 0.000 description 3
- 230000006907 apoptotic process Effects 0.000 description 3
- 235000010323 ascorbic acid Nutrition 0.000 description 3
- 229960005070 ascorbic acid Drugs 0.000 description 3
- 239000011668 ascorbic acid Substances 0.000 description 3
- JUHORIMYRDESRB-UHFFFAOYSA-N benzathine Chemical compound C=1C=CC=CC=1CNCCNCC1=CC=CC=C1 JUHORIMYRDESRB-UHFFFAOYSA-N 0.000 description 3
- 230000037396 body weight Effects 0.000 description 3
- 229910052791 calcium Inorganic materials 0.000 description 3
- 239000011575 calcium Substances 0.000 description 3
- 238000004113 cell culture Methods 0.000 description 3
- 239000012228 culture supernatant Substances 0.000 description 3
- ZBCBWPMODOFKDW-UHFFFAOYSA-N diethanolamine Chemical compound OCCNCCO ZBCBWPMODOFKDW-UHFFFAOYSA-N 0.000 description 3
- 230000004064 dysfunction Effects 0.000 description 3
- 210000002744 extracellular matrix Anatomy 0.000 description 3
- 239000012530 fluid Substances 0.000 description 3
- 239000011521 glass Substances 0.000 description 3
- 238000001727 in vivo Methods 0.000 description 3
- 230000002757 inflammatory effect Effects 0.000 description 3
- 238000001990 intravenous administration Methods 0.000 description 3
- 210000003127 knee Anatomy 0.000 description 3
- 239000007788 liquid Substances 0.000 description 3
- 229910052749 magnesium Inorganic materials 0.000 description 3
- 239000011777 magnesium Substances 0.000 description 3
- 239000011159 matrix material Substances 0.000 description 3
- 239000012528 membrane Substances 0.000 description 3
- 210000004417 patella Anatomy 0.000 description 3
- 230000036470 plasma concentration Effects 0.000 description 3
- 229920000642 polymer Polymers 0.000 description 3
- 239000011591 potassium Substances 0.000 description 3
- 239000002244 precipitate Substances 0.000 description 3
- 230000002265 prevention Effects 0.000 description 3
- 238000012545 processing Methods 0.000 description 3
- 239000000651 prodrug Substances 0.000 description 3
- 229940002612 prodrug Drugs 0.000 description 3
- 238000011160 research Methods 0.000 description 3
- 229910052708 sodium Inorganic materials 0.000 description 3
- 239000011734 sodium Substances 0.000 description 3
- 238000003860 storage Methods 0.000 description 3
- 239000000758 substrate Substances 0.000 description 3
- 230000008961 swelling Effects 0.000 description 3
- 210000001258 synovial membrane Anatomy 0.000 description 3
- 230000001225 therapeutic effect Effects 0.000 description 3
- HZAXFHJVJLSVMW-UHFFFAOYSA-N 2-Aminoethan-1-ol Chemical compound NCCO HZAXFHJVJLSVMW-UHFFFAOYSA-N 0.000 description 2
- SQDAZGGFXASXDW-UHFFFAOYSA-N 5-bromo-2-(trifluoromethoxy)pyridine Chemical compound FC(F)(F)OC1=CC=C(Br)C=N1 SQDAZGGFXASXDW-UHFFFAOYSA-N 0.000 description 2
- KDCGOANMDULRCW-UHFFFAOYSA-N 7H-purine Chemical compound N1=CNC2=NC=NC2=C1 KDCGOANMDULRCW-UHFFFAOYSA-N 0.000 description 2
- 208000024827 Alzheimer disease Diseases 0.000 description 2
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 2
- 206010006187 Breast cancer Diseases 0.000 description 2
- 229920001287 Chondroitin sulfate Polymers 0.000 description 2
- 102000008186 Collagen Human genes 0.000 description 2
- 108010035532 Collagen Proteins 0.000 description 2
- 206010058314 Dysplasia Diseases 0.000 description 2
- 108010062466 Enzyme Precursors Proteins 0.000 description 2
- 102000010911 Enzyme Precursors Human genes 0.000 description 2
- 241000283086 Equidae Species 0.000 description 2
- PIICEJLVQHRZGT-UHFFFAOYSA-N Ethylenediamine Chemical compound NCCN PIICEJLVQHRZGT-UHFFFAOYSA-N 0.000 description 2
- 241000282326 Felis catus Species 0.000 description 2
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 2
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 2
- 208000031220 Hemophilia Diseases 0.000 description 2
- 208000009292 Hemophilia A Diseases 0.000 description 2
- 101000894895 Homo sapiens Beta-secretase 1 Proteins 0.000 description 2
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 2
- PIWKPBJCKXDKJR-UHFFFAOYSA-N Isoflurane Chemical compound FC(F)OC(Cl)C(F)(F)F PIWKPBJCKXDKJR-UHFFFAOYSA-N 0.000 description 2
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 2
- YNAVUWVOSKDBBP-UHFFFAOYSA-N Morpholine Chemical compound C1COCCN1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 description 2
- 241000699670 Mus sp. Species 0.000 description 2
- SECXISVLQFMRJM-UHFFFAOYSA-N N-Methylpyrrolidone Chemical compound CN1CCCC1=O SECXISVLQFMRJM-UHFFFAOYSA-N 0.000 description 2
- MWUXSHHQAYIFBG-UHFFFAOYSA-N Nitric oxide Chemical compound O=[N] MWUXSHHQAYIFBG-UHFFFAOYSA-N 0.000 description 2
- GLUUGHFHXGJENI-UHFFFAOYSA-N Piperazine Chemical compound C1CNCCN1 GLUUGHFHXGJENI-UHFFFAOYSA-N 0.000 description 2
- NCDNCNXCDXHOMX-UHFFFAOYSA-N Ritonavir Natural products C=1C=CC=CC=1CC(NC(=O)OCC=1SC=NC=1)C(O)CC(CC=1C=CC=CC=1)NC(=O)C(C(C)C)NC(=O)N(C)CC1=CSC(C(C)C)=N1 NCDNCNXCDXHOMX-UHFFFAOYSA-N 0.000 description 2
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 2
- 230000002159 abnormal effect Effects 0.000 description 2
- 150000001298 alcohols Chemical class 0.000 description 2
- 150000001340 alkali metals Chemical class 0.000 description 2
- 229910052784 alkaline earth metal Inorganic materials 0.000 description 2
- 210000001264 anterior cruciate ligament Anatomy 0.000 description 2
- 239000003242 anti bacterial agent Substances 0.000 description 2
- 230000003070 anti-hyperalgesia Effects 0.000 description 2
- 229940088710 antibiotic agent Drugs 0.000 description 2
- 239000003963 antioxidant agent Substances 0.000 description 2
- 235000006708 antioxidants Nutrition 0.000 description 2
- 239000007864 aqueous solution Substances 0.000 description 2
- 239000003696 aspartic proteinase inhibitor Substances 0.000 description 2
- 230000008901 benefit Effects 0.000 description 2
- 230000000903 blocking effect Effects 0.000 description 2
- 210000004556 brain Anatomy 0.000 description 2
- 239000000872 buffer Substances 0.000 description 2
- RYYVLZVUVIJVGH-UHFFFAOYSA-N caffeine Chemical compound CN1C(=O)N(C)C(=O)C2=C1N=CN2C RYYVLZVUVIJVGH-UHFFFAOYSA-N 0.000 description 2
- 238000004364 calculation method Methods 0.000 description 2
- 239000002775 capsule Substances 0.000 description 2
- 229910002091 carbon monoxide Inorganic materials 0.000 description 2
- 239000003153 chemical reaction reagent Substances 0.000 description 2
- VDANGULDQQJODZ-UHFFFAOYSA-N chloroprocaine Chemical compound CCN(CC)CCOC(=O)C1=CC=C(N)C=C1Cl VDANGULDQQJODZ-UHFFFAOYSA-N 0.000 description 2
- 229960002023 chloroprocaine Drugs 0.000 description 2
- HVYWMOMLDIMFJA-DPAQBDIFSA-N cholesterol Chemical compound C1C=C2C[C@@H](O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H]([C@H](C)CCCC(C)C)[C@@]1(C)CC2 HVYWMOMLDIMFJA-DPAQBDIFSA-N 0.000 description 2
- OEYIOHPDSNJKLS-UHFFFAOYSA-N choline Chemical compound C[N+](C)(C)CCO OEYIOHPDSNJKLS-UHFFFAOYSA-N 0.000 description 2
- 229960001231 choline Drugs 0.000 description 2
- 229940059329 chondroitin sulfate Drugs 0.000 description 2
- MYSWGUAQZAJSOK-UHFFFAOYSA-N ciprofloxacin Chemical compound C12=CC(N3CCNCC3)=C(F)C=C2C(=O)C(C(=O)O)=CN1C1CC1 MYSWGUAQZAJSOK-UHFFFAOYSA-N 0.000 description 2
- 229920001436 collagen Polymers 0.000 description 2
- 238000013270 controlled release Methods 0.000 description 2
- 230000003412 degenerative effect Effects 0.000 description 2
- 238000001514 detection method Methods 0.000 description 2
- 238000010790 dilution Methods 0.000 description 2
- 239000012895 dilution Substances 0.000 description 2
- 238000010494 dissociation reaction Methods 0.000 description 2
- 230000005593 dissociations Effects 0.000 description 2
- 238000004090 dissolution Methods 0.000 description 2
- 238000009826 distribution Methods 0.000 description 2
- 230000002526 effect on cardiovascular system Effects 0.000 description 2
- 230000005284 excitation Effects 0.000 description 2
- 239000000284 extract Substances 0.000 description 2
- 238000010265 fast atom bombardment Methods 0.000 description 2
- 230000002068 genetic effect Effects 0.000 description 2
- 239000008103 glucose Substances 0.000 description 2
- KWIUHFFTVRNATP-UHFFFAOYSA-N glycine betaine Chemical compound C[N+](C)(C)CC([O-])=O KWIUHFFTVRNATP-UHFFFAOYSA-N 0.000 description 2
- 201000010536 head and neck cancer Diseases 0.000 description 2
- 208000014829 head and neck neoplasm Diseases 0.000 description 2
- 230000036541 health Effects 0.000 description 2
- 208000002085 hemarthrosis Diseases 0.000 description 2
- IGMNYECMUMZDDF-UHFFFAOYSA-N homogentisic acid Chemical compound OC(=O)CC1=CC(O)=CC=C1O IGMNYECMUMZDDF-UHFFFAOYSA-N 0.000 description 2
- 210000004276 hyalin Anatomy 0.000 description 2
- 150000004677 hydrates Chemical class 0.000 description 2
- 230000006698 induction Effects 0.000 description 2
- 208000014674 injury Diseases 0.000 description 2
- 230000003834 intracellular effect Effects 0.000 description 2
- 229960002725 isoflurane Drugs 0.000 description 2
- 210000003041 ligament Anatomy 0.000 description 2
- 239000002502 liposome Substances 0.000 description 2
- 229910052744 lithium Inorganic materials 0.000 description 2
- 210000004185 liver Anatomy 0.000 description 2
- 238000011068 loading method Methods 0.000 description 2
- 210000004072 lung Anatomy 0.000 description 2
- 210000003712 lysosome Anatomy 0.000 description 2
- 230000001868 lysosomic effect Effects 0.000 description 2
- 238000004949 mass spectrometry Methods 0.000 description 2
- 230000007246 mechanism Effects 0.000 description 2
- 230000005499 meniscus Effects 0.000 description 2
- 229910052751 metal Inorganic materials 0.000 description 2
- 239000002184 metal Substances 0.000 description 2
- 150000002739 metals Chemical class 0.000 description 2
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 2
- 230000004048 modification Effects 0.000 description 2
- 238000012986 modification Methods 0.000 description 2
- 150000007530 organic bases Chemical class 0.000 description 2
- 239000003960 organic solvent Substances 0.000 description 2
- 230000000399 orthopedic effect Effects 0.000 description 2
- 210000000426 patellar ligament Anatomy 0.000 description 2
- 229940111202 pepsin Drugs 0.000 description 2
- 239000012071 phase Substances 0.000 description 2
- 230000035790 physiological processes and functions Effects 0.000 description 2
- 239000004033 plastic Substances 0.000 description 2
- 229920003023 plastic Polymers 0.000 description 2
- 229920000747 poly(lactic acid) Polymers 0.000 description 2
- 229920000768 polyamine Polymers 0.000 description 2
- 239000004626 polylactic acid Substances 0.000 description 2
- 239000011148 porous material Substances 0.000 description 2
- MFDFERRIHVXMIY-UHFFFAOYSA-N procaine Chemical compound CCN(CC)CCOC(=O)C1=CC=C(N)C=C1 MFDFERRIHVXMIY-UHFFFAOYSA-N 0.000 description 2
- 229960004919 procaine Drugs 0.000 description 2
- 239000000047 product Substances 0.000 description 2
- 230000017854 proteolysis Effects 0.000 description 2
- 229960000311 ritonavir Drugs 0.000 description 2
- NCDNCNXCDXHOMX-XGKFQTDJSA-N ritonavir Chemical compound N([C@@H](C(C)C)C(=O)N[C@H](C[C@H](O)[C@H](CC=1C=CC=CC=1)NC(=O)OCC=1SC=NC=1)CC=1C=CC=CC=1)C(=O)N(C)CC1=CSC(C(C)C)=N1 NCDNCNXCDXHOMX-XGKFQTDJSA-N 0.000 description 2
- 238000005070 sampling Methods 0.000 description 2
- 230000035945 sensitivity Effects 0.000 description 2
- 239000000741 silica gel Substances 0.000 description 2
- 229910002027 silica gel Inorganic materials 0.000 description 2
- 239000011780 sodium chloride Substances 0.000 description 2
- 239000007787 solid Substances 0.000 description 2
- 239000003381 stabilizer Substances 0.000 description 2
- 239000011550 stock solution Substances 0.000 description 2
- 235000000346 sugar Nutrition 0.000 description 2
- 150000008163 sugars Chemical class 0.000 description 2
- 230000004083 survival effect Effects 0.000 description 2
- 210000005222 synovial tissue Anatomy 0.000 description 2
- YAPQBXQYLJRXSA-UHFFFAOYSA-N theobromine Chemical compound CN1C(=O)NC(=O)C2=C1N=CN2C YAPQBXQYLJRXSA-UHFFFAOYSA-N 0.000 description 2
- GETQZCLCWQTVFV-UHFFFAOYSA-N trimethylamine Chemical compound CN(C)C GETQZCLCWQTVFV-UHFFFAOYSA-N 0.000 description 2
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 2
- 239000002691 unilamellar liposome Substances 0.000 description 2
- 230000002792 vascular Effects 0.000 description 2
- 210000003462 vein Anatomy 0.000 description 2
- KIUKXJAPPMFGSW-DNGZLQJQSA-N (2S,3S,4S,5R,6R)-6-[(2S,3R,4R,5S,6R)-3-Acetamido-2-[(2S,3S,4R,5R,6R)-6-[(2R,3R,4R,5S,6R)-3-acetamido-2,5-dihydroxy-6-(hydroxymethyl)oxan-4-yl]oxy-2-carboxy-4,5-dihydroxyoxan-3-yl]oxy-5-hydroxy-6-(hydroxymethyl)oxan-4-yl]oxy-3,4,5-trihydroxyoxane-2-carboxylic acid Chemical compound CC(=O)N[C@H]1[C@H](O)O[C@H](CO)[C@@H](O)[C@@H]1O[C@H]1[C@H](O)[C@@H](O)[C@H](O[C@H]2[C@@H]([C@@H](O[C@H]3[C@@H]([C@@H](O)[C@H](O)[C@H](O3)C(O)=O)O)[C@H](O)[C@@H](CO)O2)NC(C)=O)[C@@H](C(O)=O)O1 KIUKXJAPPMFGSW-DNGZLQJQSA-N 0.000 description 1
- 125000001917 2,4-dinitrophenyl group Chemical group [H]C1=C([H])C(=C([H])C(=C1*)[N+]([O-])=O)[N+]([O-])=O 0.000 description 1
- MSWZFWKMSRAUBD-IVMDWMLBSA-N 2-amino-2-deoxy-D-glucopyranose Chemical compound N[C@H]1C(O)O[C@H](CO)[C@@H](O)[C@@H]1O MSWZFWKMSRAUBD-IVMDWMLBSA-N 0.000 description 1
- BFSVOASYOCHEOV-UHFFFAOYSA-N 2-diethylaminoethanol Chemical compound CCN(CC)CCO BFSVOASYOCHEOV-UHFFFAOYSA-N 0.000 description 1
- 229940013085 2-diethylaminoethanol Drugs 0.000 description 1
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 1
- LKDMKWNDBAVNQZ-UHFFFAOYSA-N 4-[[1-[[1-[2-[[1-(4-nitroanilino)-1-oxo-3-phenylpropan-2-yl]carbamoyl]pyrrolidin-1-yl]-1-oxopropan-2-yl]amino]-1-oxopropan-2-yl]amino]-4-oxobutanoic acid Chemical compound OC(=O)CCC(=O)NC(C)C(=O)NC(C)C(=O)N1CCCC1C(=O)NC(C(=O)NC=1C=CC(=CC=1)[N+]([O-])=O)CC1=CC=CC=C1 LKDMKWNDBAVNQZ-UHFFFAOYSA-N 0.000 description 1
- HVCNXQOWACZAFN-UHFFFAOYSA-N 4-ethylmorpholine Chemical compound CCN1CCOCC1 HVCNXQOWACZAFN-UHFFFAOYSA-N 0.000 description 1
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 1
- 108010088751 Albumins Proteins 0.000 description 1
- 102000009027 Albumins Human genes 0.000 description 1
- 102000009091 Amyloidogenic Proteins Human genes 0.000 description 1
- 108010048112 Amyloidogenic Proteins Proteins 0.000 description 1
- 206010002091 Anaesthesia Diseases 0.000 description 1
- 108020000948 Antisense Oligonucleotides Proteins 0.000 description 1
- 239000004475 Arginine Substances 0.000 description 1
- 208000036487 Arthropathies Diseases 0.000 description 1
- 101710110426 Aspartyl protease inhibitor Proteins 0.000 description 1
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 1
- 101710150192 Beta-secretase 1 Proteins 0.000 description 1
- 208000020084 Bone disease Diseases 0.000 description 1
- 208000003174 Brain Neoplasms Diseases 0.000 description 1
- 108090000712 Cathepsin B Proteins 0.000 description 1
- 102000004225 Cathepsin B Human genes 0.000 description 1
- 108090000617 Cathepsin G Proteins 0.000 description 1
- 102000004173 Cathepsin G Human genes 0.000 description 1
- 108090000625 Cathepsin K Proteins 0.000 description 1
- 102000004171 Cathepsin K Human genes 0.000 description 1
- 229940122805 Cathepsin S inhibitor Drugs 0.000 description 1
- 241000700198 Cavia Species 0.000 description 1
- 241000282693 Cercopithecidae Species 0.000 description 1
- 229920001661 Chitosan Polymers 0.000 description 1
- 208000000094 Chronic Pain Diseases 0.000 description 1
- 108090000227 Chymases Proteins 0.000 description 1
- 102000003858 Chymases Human genes 0.000 description 1
- 102000029816 Collagenase Human genes 0.000 description 1
- 108060005980 Collagenase Proteins 0.000 description 1
- 206010010356 Congenital anomaly Diseases 0.000 description 1
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 description 1
- 241000699800 Cricetinae Species 0.000 description 1
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 1
- 206010012289 Dementia Diseases 0.000 description 1
- 229920002307 Dextran Polymers 0.000 description 1
- XBPCUCUWBYBCDP-UHFFFAOYSA-N Dicyclohexylamine Chemical compound C1CCCCC1NC1CCCCC1 XBPCUCUWBYBCDP-UHFFFAOYSA-N 0.000 description 1
- 206010063045 Effusion Diseases 0.000 description 1
- CWYNVVGOOAEACU-UHFFFAOYSA-N Fe2+ Chemical compound [Fe+2] CWYNVVGOOAEACU-UHFFFAOYSA-N 0.000 description 1
- 108010088842 Fibrinolysin Proteins 0.000 description 1
- 102000055441 Gastricsin Human genes 0.000 description 1
- 108090001072 Gastricsin Proteins 0.000 description 1
- 108010010803 Gelatin Proteins 0.000 description 1
- 208000031886 HIV Infections Diseases 0.000 description 1
- 208000037357 HIV infectious disease Diseases 0.000 description 1
- 101710090680 Hemoglobinase Proteins 0.000 description 1
- HTTJABKRGRZYRN-UHFFFAOYSA-N Heparin Chemical compound OC1C(NC(=O)C)C(O)OC(COS(O)(=O)=O)C1OC1C(OS(O)(=O)=O)C(O)C(OC2C(C(OS(O)(=O)=O)C(OC3C(C(O)C(O)C(O3)C(O)=O)OS(O)(=O)=O)C(CO)O2)NS(O)(=O)=O)C(C(O)=O)O1 HTTJABKRGRZYRN-UHFFFAOYSA-N 0.000 description 1
- 241000725303 Human immunodeficiency virus Species 0.000 description 1
- 108010016183 Human immunodeficiency virus 1 p16 protease Proteins 0.000 description 1
- 208000023105 Huntington disease Diseases 0.000 description 1
- LPHGQDQBBGAPDZ-UHFFFAOYSA-N Isocaffeine Natural products CN1C(=O)N(C)C(=O)C2=C1N(C)C=N2 LPHGQDQBBGAPDZ-UHFFFAOYSA-N 0.000 description 1
- 102000001399 Kallikrein Human genes 0.000 description 1
- 108060005987 Kallikrein Proteins 0.000 description 1
- ODKSFYDXXFIFQN-BYPYZUCNSA-P L-argininium(2+) Chemical compound NC(=[NH2+])NCCC[C@H]([NH3+])C(O)=O ODKSFYDXXFIFQN-BYPYZUCNSA-P 0.000 description 1
- HNDVDQJCIGZPNO-YFKPBYRVSA-N L-histidine Chemical compound OC(=O)[C@@H](N)CC1=CN=CN1 HNDVDQJCIGZPNO-YFKPBYRVSA-N 0.000 description 1
- KDXKERNSBIXSRK-YFKPBYRVSA-N L-lysine Chemical compound NCCCC[C@H](N)C(O)=O KDXKERNSBIXSRK-YFKPBYRVSA-N 0.000 description 1
- FBOZXECLQNJBKD-ZDUSSCGKSA-N L-methotrexate Chemical compound C=1N=C2N=C(N)N=C(N)C2=NC=1CN(C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 FBOZXECLQNJBKD-ZDUSSCGKSA-N 0.000 description 1
- 108010028275 Leukocyte Elastase Proteins 0.000 description 1
- GSDSWSVVBLHKDQ-JTQLQIEISA-N Levofloxacin Chemical compound C([C@@H](N1C2=C(C(C(C(O)=O)=C1)=O)C=C1F)C)OC2=C1N1CCN(C)CC1 GSDSWSVVBLHKDQ-JTQLQIEISA-N 0.000 description 1
- NNJVILVZKWQKPM-UHFFFAOYSA-N Lidocaine Chemical compound CCN(CC)CC(=O)NC1=C(C)C=CC=C1C NNJVILVZKWQKPM-UHFFFAOYSA-N 0.000 description 1
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 1
- 239000004472 Lysine Substances 0.000 description 1
- JLVVSXFLKOJNIY-UHFFFAOYSA-N Magnesium ion Chemical compound [Mg+2] JLVVSXFLKOJNIY-UHFFFAOYSA-N 0.000 description 1
- 229930195725 Mannitol Natural products 0.000 description 1
- 102000005741 Metalloproteases Human genes 0.000 description 1
- 108010006035 Metalloproteases Proteins 0.000 description 1
- 206010027476 Metastases Diseases 0.000 description 1
- CERQOIWHTDAKMF-UHFFFAOYSA-M Methacrylate Chemical compound CC(=C)C([O-])=O CERQOIWHTDAKMF-UHFFFAOYSA-M 0.000 description 1
- 229920003094 Methocel™ K4M Polymers 0.000 description 1
- 208000008955 Mucolipidoses Diseases 0.000 description 1
- 241000699666 Mus <mouse, genus> Species 0.000 description 1
- ONXPDKGXOOORHB-BYPYZUCNSA-N N(5)-methyl-L-glutamine Chemical compound CNC(=O)CC[C@H](N)C(O)=O ONXPDKGXOOORHB-BYPYZUCNSA-N 0.000 description 1
- UEEJHVSXFDXPFK-UHFFFAOYSA-N N-dimethylaminoethanol Chemical compound CN(C)CCO UEEJHVSXFDXPFK-UHFFFAOYSA-N 0.000 description 1
- HTLZVHNRZJPSMI-UHFFFAOYSA-N N-ethylpiperidine Chemical compound CCN1CCCCC1 HTLZVHNRZJPSMI-UHFFFAOYSA-N 0.000 description 1
- 102100033174 Neutrophil elastase Human genes 0.000 description 1
- REYJJPSVUYRZGE-UHFFFAOYSA-N Octadecylamine Chemical compound CCCCCCCCCCCCCCCCCCN REYJJPSVUYRZGE-UHFFFAOYSA-N 0.000 description 1
- 108010038807 Oligopeptides Proteins 0.000 description 1
- 102000015636 Oligopeptides Human genes 0.000 description 1
- 241000283973 Oryctolagus cuniculus Species 0.000 description 1
- 208000008558 Osteophyte Diseases 0.000 description 1
- 208000001132 Osteoporosis Diseases 0.000 description 1
- 208000000114 Pain Threshold Diseases 0.000 description 1
- 108090000526 Papain Proteins 0.000 description 1
- 229930040373 Paraformaldehyde Natural products 0.000 description 1
- 102000007079 Peptide Fragments Human genes 0.000 description 1
- 108010033276 Peptide Fragments Proteins 0.000 description 1
- 108010081690 Pertussis Toxin Proteins 0.000 description 1
- 229930182556 Polyacetal Natural products 0.000 description 1
- 229920000604 Polyethylene Glycol 200 Polymers 0.000 description 1
- 229920001710 Polyorthoester Polymers 0.000 description 1
- 229920001213 Polysorbate 20 Polymers 0.000 description 1
- 239000004372 Polyvinyl alcohol Substances 0.000 description 1
- 241000288906 Primates Species 0.000 description 1
- 102000016611 Proteoglycans Human genes 0.000 description 1
- 108010067787 Proteoglycans Proteins 0.000 description 1
- 201000004681 Psoriasis Diseases 0.000 description 1
- 241000700157 Rattus norvegicus Species 0.000 description 1
- 208000025747 Rheumatic disease Diseases 0.000 description 1
- 241000283984 Rodentia Species 0.000 description 1
- MTCFGRXMJLQNBG-UHFFFAOYSA-N Serine Natural products OCC(N)C(O)=O MTCFGRXMJLQNBG-UHFFFAOYSA-N 0.000 description 1
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 1
- 239000004280 Sodium formate Substances 0.000 description 1
- 241000187747 Streptomyces Species 0.000 description 1
- 208000007536 Thrombosis Diseases 0.000 description 1
- GSEJCLTVZPLZKY-UHFFFAOYSA-N Triethanolamine Chemical compound OCCN(CCO)CCO GSEJCLTVZPLZKY-UHFFFAOYSA-N 0.000 description 1
- 102000001400 Tryptase Human genes 0.000 description 1
- 108060005989 Tryptase Proteins 0.000 description 1
- 238000010811 Ultra-Performance Liquid Chromatography-Tandem Mass Spectrometry Methods 0.000 description 1
- TVWHNULVHGKJHS-UHFFFAOYSA-N Uric acid Natural products N1C(=O)NC(=O)C2NC(=O)NC21 TVWHNULVHGKJHS-UHFFFAOYSA-N 0.000 description 1
- 208000027418 Wounds and injury Diseases 0.000 description 1
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 230000004913 activation Effects 0.000 description 1
- 238000001994 activation Methods 0.000 description 1
- 239000013543 active substance Substances 0.000 description 1
- 230000001154 acute effect Effects 0.000 description 1
- 125000002252 acyl group Chemical group 0.000 description 1
- 239000000654 additive Substances 0.000 description 1
- 230000000996 additive effect Effects 0.000 description 1
- 239000002671 adjuvant Substances 0.000 description 1
- 108010003059 aggrecanase Proteins 0.000 description 1
- 230000002776 aggregation Effects 0.000 description 1
- 238000004220 aggregation Methods 0.000 description 1
- 238000007605 air drying Methods 0.000 description 1
- 150000008044 alkali metal hydroxides Chemical class 0.000 description 1
- 229910001860 alkaline earth metal hydroxide Inorganic materials 0.000 description 1
- 150000001342 alkaline earth metals Chemical class 0.000 description 1
- 206010001689 alkaptonuria Diseases 0.000 description 1
- 125000000217 alkyl group Chemical group 0.000 description 1
- VREFGVBLTWBCJP-UHFFFAOYSA-N alprazolam Chemical compound C12=CC(Cl)=CC=C2N2C(C)=NN=C2CN=C1C1=CC=CC=C1 VREFGVBLTWBCJP-UHFFFAOYSA-N 0.000 description 1
- AZDRQVAHHNSJOQ-UHFFFAOYSA-N alumane Chemical class [AlH3] AZDRQVAHHNSJOQ-UHFFFAOYSA-N 0.000 description 1
- 229910052782 aluminium Inorganic materials 0.000 description 1
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 1
- 230000037005 anaesthesia Effects 0.000 description 1
- 230000003698 anagen phase Effects 0.000 description 1
- 239000005557 antagonist Substances 0.000 description 1
- 230000002424 anti-apoptotic effect Effects 0.000 description 1
- 230000030741 antigen processing and presentation Effects 0.000 description 1
- 239000000074 antisense oligonucleotide Substances 0.000 description 1
- 238000012230 antisense oligonucleotides Methods 0.000 description 1
- 238000013459 approach Methods 0.000 description 1
- 239000012736 aqueous medium Substances 0.000 description 1
- 239000003125 aqueous solvent Substances 0.000 description 1
- 239000007900 aqueous suspension Substances 0.000 description 1
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 1
- 229960003121 arginine Drugs 0.000 description 1
- 230000002917 arthritic effect Effects 0.000 description 1
- 230000005784 autoimmunity Effects 0.000 description 1
- 229960002170 azathioprine Drugs 0.000 description 1
- LMEKQMALGUDUQG-UHFFFAOYSA-N azathioprine Chemical compound CN1C=NC([N+]([O-])=O)=C1SC1=NC=NC2=C1NC=N2 LMEKQMALGUDUQG-UHFFFAOYSA-N 0.000 description 1
- 210000003719 b-lymphocyte Anatomy 0.000 description 1
- RQPZNWPYLFFXCP-UHFFFAOYSA-L barium dihydroxide Chemical compound [OH-].[OH-].[Ba+2] RQPZNWPYLFFXCP-UHFFFAOYSA-L 0.000 description 1
- 229910001863 barium hydroxide Inorganic materials 0.000 description 1
- MSWZFWKMSRAUBD-UHFFFAOYSA-N beta-D-galactosamine Natural products NC1C(O)OC(CO)C(O)C1O MSWZFWKMSRAUBD-UHFFFAOYSA-N 0.000 description 1
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 1
- 229960003237 betaine Drugs 0.000 description 1
- 230000004071 biological effect Effects 0.000 description 1
- 238000001574 biopsy Methods 0.000 description 1
- 230000015572 biosynthetic process Effects 0.000 description 1
- 229920001400 block copolymer Polymers 0.000 description 1
- 230000017531 blood circulation Effects 0.000 description 1
- 230000036772 blood pressure Effects 0.000 description 1
- 230000037182 bone density Effects 0.000 description 1
- 210000000481 breast Anatomy 0.000 description 1
- RMRJXGBAOAMLHD-IHFGGWKQSA-N buprenorphine Chemical compound C([C@]12[C@H]3OC=4C(O)=CC=C(C2=4)C[C@@H]2[C@]11CC[C@]3([C@H](C1)[C@](C)(O)C(C)(C)C)OC)CN2CC1CC1 RMRJXGBAOAMLHD-IHFGGWKQSA-N 0.000 description 1
- 229960001736 buprenorphine Drugs 0.000 description 1
- 229960001948 caffeine Drugs 0.000 description 1
- VJEONQKOZGKCAK-UHFFFAOYSA-N caffeine Natural products CN1C(=O)N(C)C(=O)C2=C1C=CN2C VJEONQKOZGKCAK-UHFFFAOYSA-N 0.000 description 1
- AXCZMVOFGPJBDE-UHFFFAOYSA-L calcium dihydroxide Chemical compound [OH-].[OH-].[Ca+2] AXCZMVOFGPJBDE-UHFFFAOYSA-L 0.000 description 1
- 239000000920 calcium hydroxide Substances 0.000 description 1
- 229910001861 calcium hydroxide Inorganic materials 0.000 description 1
- 229910052799 carbon Inorganic materials 0.000 description 1
- 125000002843 carboxylic acid group Chemical group 0.000 description 1
- 239000000679 carrageenan Substances 0.000 description 1
- 235000010418 carrageenan Nutrition 0.000 description 1
- 229920001525 carrageenan Polymers 0.000 description 1
- 229940113118 carrageenan Drugs 0.000 description 1
- 230000010261 cell growth Effects 0.000 description 1
- 230000004709 cell invasion Effects 0.000 description 1
- 210000001175 cerebrospinal fluid Anatomy 0.000 description 1
- 238000012512 characterization method Methods 0.000 description 1
- 235000012000 cholesterol Nutrition 0.000 description 1
- 210000001612 chondrocyte Anatomy 0.000 description 1
- 238000004587 chromatography analysis Methods 0.000 description 1
- 208000022371 chronic pain syndrome Diseases 0.000 description 1
- 229960003405 ciprofloxacin Drugs 0.000 description 1
- 208000010877 cognitive disease Diseases 0.000 description 1
- 229960002424 collagenase Drugs 0.000 description 1
- 238000010668 complexation reaction Methods 0.000 description 1
- 238000003271 compound fluorescence assay Methods 0.000 description 1
- 210000002808 connective tissue Anatomy 0.000 description 1
- 229920001577 copolymer Polymers 0.000 description 1
- 229910052802 copper Inorganic materials 0.000 description 1
- 239000010949 copper Substances 0.000 description 1
- 230000002596 correlated effect Effects 0.000 description 1
- 229940111134 coxibs Drugs 0.000 description 1
- 239000003431 cross linking reagent Substances 0.000 description 1
- 238000002425 crystallisation Methods 0.000 description 1
- 230000008025 crystallization Effects 0.000 description 1
- 239000003255 cyclooxygenase 2 inhibitor Substances 0.000 description 1
- XUJNEKJLAYXESH-UHFFFAOYSA-N cysteine Natural products SCC(N)C(O)=O XUJNEKJLAYXESH-UHFFFAOYSA-N 0.000 description 1
- 235000018417 cysteine Nutrition 0.000 description 1
- 238000000354 decomposition reaction Methods 0.000 description 1
- 230000007850 degeneration Effects 0.000 description 1
- 229920006237 degradable polymer Polymers 0.000 description 1
- 230000003111 delayed effect Effects 0.000 description 1
- 230000001687 destabilization Effects 0.000 description 1
- 238000003745 diagnosis Methods 0.000 description 1
- 239000000032 diagnostic agent Substances 0.000 description 1
- 229940039227 diagnostic agent Drugs 0.000 description 1
- 235000005911 diet Nutrition 0.000 description 1
- 230000037213 diet Effects 0.000 description 1
- 229940043237 diethanolamine Drugs 0.000 description 1
- HPNMFZURTQLUMO-UHFFFAOYSA-N diethylamine Chemical compound CCNCC HPNMFZURTQLUMO-UHFFFAOYSA-N 0.000 description 1
- 150000004683 dihydrates Chemical class 0.000 description 1
- 229910001873 dinitrogen Inorganic materials 0.000 description 1
- 208000035475 disorder Diseases 0.000 description 1
- 239000002270 dispersing agent Substances 0.000 description 1
- 239000002552 dosage form Substances 0.000 description 1
- 238000002036 drum drying Methods 0.000 description 1
- 239000003792 electrolyte Substances 0.000 description 1
- 230000008030 elimination Effects 0.000 description 1
- 238000003379 elimination reaction Methods 0.000 description 1
- 239000003995 emulsifying agent Substances 0.000 description 1
- 230000002255 enzymatic effect Effects 0.000 description 1
- 238000001952 enzyme assay Methods 0.000 description 1
- 229940012017 ethylenediamine Drugs 0.000 description 1
- 238000001704 evaporation Methods 0.000 description 1
- 230000008020 evaporation Effects 0.000 description 1
- 230000029142 excretion Effects 0.000 description 1
- 238000000605 extraction Methods 0.000 description 1
- 102000003977 fibroblast growth factor 18 Human genes 0.000 description 1
- 108090000370 fibroblast growth factor 18 Proteins 0.000 description 1
- 239000012467 final product Substances 0.000 description 1
- 229910052731 fluorine Inorganic materials 0.000 description 1
- 229940124307 fluoroquinolone Drugs 0.000 description 1
- 239000006260 foam Substances 0.000 description 1
- 235000019253 formic acid Nutrition 0.000 description 1
- 238000004108 freeze drying Methods 0.000 description 1
- 230000008014 freezing Effects 0.000 description 1
- 238000007710 freezing Methods 0.000 description 1
- 239000008273 gelatin Substances 0.000 description 1
- 229920000159 gelatin Polymers 0.000 description 1
- 235000019322 gelatine Nutrition 0.000 description 1
- 235000011852 gelatine desserts Nutrition 0.000 description 1
- 231100000025 genetic toxicology Toxicity 0.000 description 1
- 230000001738 genotoxic effect Effects 0.000 description 1
- 208000005017 glioblastoma Diseases 0.000 description 1
- 239000003862 glucocorticoid Substances 0.000 description 1
- 229960002442 glucosamine Drugs 0.000 description 1
- 150000004676 glycans Chemical class 0.000 description 1
- 235000011187 glycerol Nutrition 0.000 description 1
- 239000008187 granular material Substances 0.000 description 1
- 239000002271 gyrase inhibitor Substances 0.000 description 1
- 230000002650 habitual effect Effects 0.000 description 1
- 231100001261 hazardous Toxicity 0.000 description 1
- 230000003862 health status Effects 0.000 description 1
- 229920000669 heparin Polymers 0.000 description 1
- 229960002897 heparin Drugs 0.000 description 1
- HNDVDQJCIGZPNO-UHFFFAOYSA-N histidine Natural products OC(=O)C(N)CC1=CN=CN1 HNDVDQJCIGZPNO-UHFFFAOYSA-N 0.000 description 1
- 210000000003 hoof Anatomy 0.000 description 1
- 230000003054 hormonal effect Effects 0.000 description 1
- 208000033519 human immunodeficiency virus infectious disease Diseases 0.000 description 1
- 229920002674 hyaluronan Polymers 0.000 description 1
- 229960003160 hyaluronic acid Drugs 0.000 description 1
- XGIHQYAWBCFNPY-AZOCGYLKSA-N hydrabamine Chemical compound C([C@@H]12)CC3=CC(C(C)C)=CC=C3[C@@]2(C)CCC[C@@]1(C)CNCCNC[C@@]1(C)[C@@H]2CCC3=CC(C(C)C)=CC=C3[C@@]2(C)CCC1 XGIHQYAWBCFNPY-AZOCGYLKSA-N 0.000 description 1
- 239000000017 hydrogel Substances 0.000 description 1
- 229910052739 hydrogen Inorganic materials 0.000 description 1
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 1
- 238000000338 in vitro Methods 0.000 description 1
- 208000015181 infectious disease Diseases 0.000 description 1
- 208000027866 inflammatory disease Diseases 0.000 description 1
- 239000003978 infusion fluid Substances 0.000 description 1
- 230000002401 inhibitory effect Effects 0.000 description 1
- 230000000977 initiatory effect Effects 0.000 description 1
- 150000007529 inorganic bases Chemical class 0.000 description 1
- 230000003993 interaction Effects 0.000 description 1
- 230000009545 invasion Effects 0.000 description 1
- 239000003456 ion exchange resin Substances 0.000 description 1
- 229920003303 ion-exchange polymer Polymers 0.000 description 1
- 230000002427 irreversible effect Effects 0.000 description 1
- 230000007794 irritation Effects 0.000 description 1
- JJWLVOIRVHMVIS-UHFFFAOYSA-N isopropylamine Chemical compound CC(C)N JJWLVOIRVHMVIS-UHFFFAOYSA-N 0.000 description 1
- 208000018937 joint inflammation Diseases 0.000 description 1
- 210000005067 joint tissue Anatomy 0.000 description 1
- 210000002414 leg Anatomy 0.000 description 1
- 229960003376 levofloxacin Drugs 0.000 description 1
- 229960004194 lidocaine Drugs 0.000 description 1
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 1
- 230000007774 longterm Effects 0.000 description 1
- 231100000053 low toxicity Toxicity 0.000 description 1
- 208000020816 lung neoplasm Diseases 0.000 description 1
- 230000001926 lymphatic effect Effects 0.000 description 1
- 229960003646 lysine Drugs 0.000 description 1
- 229910001425 magnesium ion Inorganic materials 0.000 description 1
- 201000004792 malaria Diseases 0.000 description 1
- 238000013291 male lister hooded rat Methods 0.000 description 1
- 239000000594 mannitol Substances 0.000 description 1
- 235000010355 mannitol Nutrition 0.000 description 1
- 238000004519 manufacturing process Methods 0.000 description 1
- 238000000691 measurement method Methods 0.000 description 1
- 229940126601 medicinal product Drugs 0.000 description 1
- 229960003194 meglumine Drugs 0.000 description 1
- 108020004999 messenger RNA Proteins 0.000 description 1
- 230000002503 metabolic effect Effects 0.000 description 1
- 230000004060 metabolic process Effects 0.000 description 1
- 230000009401 metastasis Effects 0.000 description 1
- 229960000485 methotrexate Drugs 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 230000009456 molecular mechanism Effects 0.000 description 1
- 150000004682 monohydrates Chemical class 0.000 description 1
- 201000007769 mucolipidosis Diseases 0.000 description 1
- 210000003205 muscle Anatomy 0.000 description 1
- 208000004296 neuralgia Diseases 0.000 description 1
- 230000004770 neurodegeneration Effects 0.000 description 1
- 208000015122 neurodegenerative disease Diseases 0.000 description 1
- 230000000926 neurological effect Effects 0.000 description 1
- 208000021722 neuropathic pain Diseases 0.000 description 1
- 239000002547 new drug Substances 0.000 description 1
- 239000000041 non-steroidal anti-inflammatory agent Substances 0.000 description 1
- 229940021182 non-steroidal anti-inflammatory drug Drugs 0.000 description 1
- 231100000252 nontoxic Toxicity 0.000 description 1
- 230000003000 nontoxic effect Effects 0.000 description 1
- 230000003287 optical effect Effects 0.000 description 1
- 239000012074 organic phase Substances 0.000 description 1
- 230000003204 osmotic effect Effects 0.000 description 1
- 230000003349 osteoarthritic effect Effects 0.000 description 1
- 230000002018 overexpression Effects 0.000 description 1
- 229910052760 oxygen Inorganic materials 0.000 description 1
- 230000000242 pagocytic effect Effects 0.000 description 1
- 229940124641 pain reliever Drugs 0.000 description 1
- 230000008058 pain sensation Effects 0.000 description 1
- 230000037040 pain threshold Effects 0.000 description 1
- 125000001312 palmitoyl group Chemical group O=C([*])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 229940055729 papain Drugs 0.000 description 1
- 235000019834 papain Nutrition 0.000 description 1
- 239000012188 paraffin wax Substances 0.000 description 1
- 229920002866 paraformaldehyde Polymers 0.000 description 1
- 238000007911 parenteral administration Methods 0.000 description 1
- 230000001717 pathogenic effect Effects 0.000 description 1
- 230000001575 pathological effect Effects 0.000 description 1
- 230000035515 penetration Effects 0.000 description 1
- 235000019371 penicillin G benzathine Nutrition 0.000 description 1
- 229940066716 pepsin a Drugs 0.000 description 1
- 230000003285 pharmacodynamic effect Effects 0.000 description 1
- 230000000144 pharmacologic effect Effects 0.000 description 1
- 150000008105 phosphatidylcholines Chemical class 0.000 description 1
- 150000003904 phospholipids Chemical class 0.000 description 1
- 230000000704 physical effect Effects 0.000 description 1
- 239000002504 physiological saline solution Substances 0.000 description 1
- 230000007505 plaque formation Effects 0.000 description 1
- 108010020708 plasmepsin Proteins 0.000 description 1
- 229940012957 plasmin Drugs 0.000 description 1
- 239000002798 polar solvent Substances 0.000 description 1
- 239000002745 poly(ortho ester) Substances 0.000 description 1
- 229920001610 polycaprolactone Polymers 0.000 description 1
- 229920002721 polycyanoacrylate Polymers 0.000 description 1
- 229920000656 polylysine Polymers 0.000 description 1
- 239000000256 polyoxyethylene sorbitan monolaurate Substances 0.000 description 1
- 235000010486 polyoxyethylene sorbitan monolaurate Nutrition 0.000 description 1
- 229920006324 polyoxymethylene Polymers 0.000 description 1
- 229920001282 polysaccharide Polymers 0.000 description 1
- 239000005017 polysaccharide Substances 0.000 description 1
- 229920002451 polyvinyl alcohol Polymers 0.000 description 1
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 1
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 1
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 1
- 230000008092 positive effect Effects 0.000 description 1
- 230000002980 postoperative effect Effects 0.000 description 1
- 230000036544 posture Effects 0.000 description 1
- BDAWXSQJJCIFIK-UHFFFAOYSA-N potassium methoxide Chemical compound [K+].[O-]C BDAWXSQJJCIFIK-UHFFFAOYSA-N 0.000 description 1
- 239000000843 powder Substances 0.000 description 1
- 238000001556 precipitation Methods 0.000 description 1
- 230000002028 premature Effects 0.000 description 1
- 239000003755 preservative agent Substances 0.000 description 1
- 238000003825 pressing Methods 0.000 description 1
- 230000003449 preventive effect Effects 0.000 description 1
- 150000003141 primary amines Chemical class 0.000 description 1
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 1
- 239000012602 primary packaging material Substances 0.000 description 1
- 102000004196 processed proteins & peptides Human genes 0.000 description 1
- 230000002035 prolonged effect Effects 0.000 description 1
- BDERNNFJNOPAEC-UHFFFAOYSA-N propan-1-ol Chemical compound CCCO BDERNNFJNOPAEC-UHFFFAOYSA-N 0.000 description 1
- 210000002307 prostate Anatomy 0.000 description 1
- 235000019833 protease Nutrition 0.000 description 1
- 125000006239 protecting group Chemical group 0.000 description 1
- 230000022558 protein metabolic process Effects 0.000 description 1
- 230000006920 protein precipitation Effects 0.000 description 1
- 238000000746 purification Methods 0.000 description 1
- 238000003908 quality control method Methods 0.000 description 1
- 239000010453 quartz Substances 0.000 description 1
- LISFMEBWQUVKPJ-UHFFFAOYSA-N quinolin-2-ol Chemical compound C1=CC=C2NC(=O)C=CC2=C1 LISFMEBWQUVKPJ-UHFFFAOYSA-N 0.000 description 1
- 230000035484 reaction time Effects 0.000 description 1
- 230000001603 reducing effect Effects 0.000 description 1
- 239000013558 reference substance Substances 0.000 description 1
- 230000001105 regulatory effect Effects 0.000 description 1
- 230000036454 renin-angiotensin system Effects 0.000 description 1
- 239000011347 resin Substances 0.000 description 1
- 229920005989 resin Polymers 0.000 description 1
- 230000004044 response Effects 0.000 description 1
- 210000003705 ribosome Anatomy 0.000 description 1
- OARRHUQTFTUEOS-UHFFFAOYSA-N safranin Chemical compound [Cl-].C=12C=C(N)C(C)=CC2=NC2=CC(C)=C(N)C=C2[N+]=1C1=CC=CC=C1 OARRHUQTFTUEOS-UHFFFAOYSA-N 0.000 description 1
- 150000003335 secondary amines Chemical class 0.000 description 1
- 230000009291 secondary effect Effects 0.000 description 1
- 230000028327 secretion Effects 0.000 description 1
- 230000021317 sensory perception Effects 0.000 description 1
- 210000002966 serum Anatomy 0.000 description 1
- 239000007974 sodium acetate buffer Substances 0.000 description 1
- HLBBKKJFGFRGMU-UHFFFAOYSA-M sodium formate Chemical compound [Na+].[O-]C=O HLBBKKJFGFRGMU-UHFFFAOYSA-M 0.000 description 1
- 235000019254 sodium formate Nutrition 0.000 description 1
- 229910052938 sodium sulfate Inorganic materials 0.000 description 1
- 235000011152 sodium sulphate Nutrition 0.000 description 1
- RCOSUMRTSQULBK-UHFFFAOYSA-N sodium;propan-1-olate Chemical compound [Na+].CCC[O-] RCOSUMRTSQULBK-UHFFFAOYSA-N 0.000 description 1
- 230000002269 spontaneous effect Effects 0.000 description 1
- 238000001694 spray drying Methods 0.000 description 1
- 238000010186 staining Methods 0.000 description 1
- 229910001220 stainless steel Inorganic materials 0.000 description 1
- 239000010935 stainless steel Substances 0.000 description 1
- 238000004659 sterilization and disinfection Methods 0.000 description 1
- 230000000638 stimulation Effects 0.000 description 1
- 210000005065 subchondral bone plate Anatomy 0.000 description 1
- 229910052717 sulfur Inorganic materials 0.000 description 1
- 230000001629 suppression Effects 0.000 description 1
- 239000000375 suspending agent Substances 0.000 description 1
- 230000002459 sustained effect Effects 0.000 description 1
- 238000013268 sustained release Methods 0.000 description 1
- 239000012730 sustained-release form Substances 0.000 description 1
- 208000024891 symptom Diseases 0.000 description 1
- 230000002195 synergetic effect Effects 0.000 description 1
- 230000009897 systematic effect Effects 0.000 description 1
- 238000007910 systemic administration Methods 0.000 description 1
- 229940037128 systemic glucocorticoids Drugs 0.000 description 1
- 239000003826 tablet Substances 0.000 description 1
- 210000002435 tendon Anatomy 0.000 description 1
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 1
- 238000010257 thawing Methods 0.000 description 1
- 229960004559 theobromine Drugs 0.000 description 1
- 239000002562 thickening agent Substances 0.000 description 1
- 238000004809 thin layer chromatography Methods 0.000 description 1
- 210000004353 tibial menisci Anatomy 0.000 description 1
- 230000001988 toxicity Effects 0.000 description 1
- 231100000419 toxicity Toxicity 0.000 description 1
- 230000002110 toxicologic effect Effects 0.000 description 1
- 231100000027 toxicology Toxicity 0.000 description 1
- 238000012546 transfer Methods 0.000 description 1
- 230000009466 transformation Effects 0.000 description 1
- 230000032258 transport Effects 0.000 description 1
- 230000008733 trauma Effects 0.000 description 1
- YNDXUCZADRHECN-JNQJZLCISA-N triamcinolone acetonide Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1C[C@H]3OC(C)(C)O[C@@]3(C(=O)CO)[C@@]1(C)C[C@@H]2O YNDXUCZADRHECN-JNQJZLCISA-N 0.000 description 1
- YFTHZRPMJXBUME-UHFFFAOYSA-N tripropylamine Chemical compound CCCN(CCC)CCC YFTHZRPMJXBUME-UHFFFAOYSA-N 0.000 description 1
- 229960000281 trometamol Drugs 0.000 description 1
- 230000001228 trophic effect Effects 0.000 description 1
- 210000004881 tumor cell Anatomy 0.000 description 1
- 229940116269 uric acid Drugs 0.000 description 1
- 229910052720 vanadium Inorganic materials 0.000 description 1
- 238000012795 verification Methods 0.000 description 1
- 239000011782 vitamin Substances 0.000 description 1
- 235000013343 vitamin Nutrition 0.000 description 1
- 229940088594 vitamin Drugs 0.000 description 1
- 229930003231 vitamin Natural products 0.000 description 1
- 238000005406 washing Methods 0.000 description 1
- 239000008215 water for injection Substances 0.000 description 1
- 230000003313 weakening effect Effects 0.000 description 1
- 229910052725 zinc Inorganic materials 0.000 description 1
- 239000011701 zinc Substances 0.000 description 1
- UHVMMEOXYDMDKI-JKYCWFKZSA-L zinc;1-(5-cyanopyridin-2-yl)-3-[(1s,2s)-2-(6-fluoro-2-hydroxy-3-propanoylphenyl)cyclopropyl]urea;diacetate Chemical compound [Zn+2].CC([O-])=O.CC([O-])=O.CCC(=O)C1=CC=C(F)C([C@H]2[C@H](C2)NC(=O)NC=2N=CC(=CC=2)C#N)=C1O UHVMMEOXYDMDKI-JKYCWFKZSA-L 0.000 description 1
- PAPBSGBWRJIAAV-UHFFFAOYSA-N ε-Caprolactone Chemical compound O=C1CCCCCO1 PAPBSGBWRJIAAV-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/04—Peptides having up to 20 amino acids in a fully defined sequence; Derivatives thereof
- A61K38/08—Peptides having 5 to 11 amino acids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0019—Injectable compositions; Intramuscular, intravenous, arterial, subcutaneous administration; Compositions to be administered through the skin in an invasive manner
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/02—Drugs for disorders of the nervous system for peripheral neuropathies
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Veterinary Medicine (AREA)
- Animal Behavior & Ethology (AREA)
- Chemical & Material Sciences (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Epidemiology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Immunology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Gastroenterology & Hepatology (AREA)
- Dermatology (AREA)
- Rheumatology (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Pain & Pain Management (AREA)
- Neurosurgery (AREA)
- Biomedical Technology (AREA)
- Physical Education & Sports Medicine (AREA)
- Neurology (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Abstract
Preparación farmacéutica que contenga pepstatina y/o una de sus sales, solvatos y estereoisómeros fisiológicamente compatibles, incluidas sus mezclas en todas las proporciones, y otros vehículos y/o excipientes, para su uso como medicamento en el tratamiento y/o la profilaxis de estados fisiológicos y/o patofisiológicos seleccionados del grupo compuesto por artrosis, lesiones condrales traumáticas, artritis, dolor, alodinia o hiperalgesia, en los que la preparación farmacéutica se administra por vía intraarticular de la siguiente manera: a) desde semanalmente hasta anualmente, b) desde cada 14 días hasta semestralmente o c) desde mensualmente hasta trimestralmente.
Description
DESCRIPCIÓN
Administración intraarticular de pepstatina para la artrosis
La presente invención se refiere a preparaciones farmacéuticas y medicamentos para la administración intraarticular que contienen pepstatina, así como su preparación y, en particular, su uso en el tratamiento y/o la profilaxis de artrosis, lesiones condrales traumáticas, artritis, dolor, alodinia o hiperalgesia, prefiriéndose en especial para la artrosis.
Antecedentes de la invención
La artrosis es la enfermedad articular más extendida a nivel mundial y se encuentran indicios radiológicos de artrosis en la mayoría de personas de más de 65 años. A pesar de esta importancia fundamental para el sistema sanitario, hasta este momento las causas de la artrosis todavía no se han aclarado y las medidas preventivas eficaces todavía continúan siendo un objetivo lejano. La reducción de la cavidad articular (debido a la destrucción del cartílago articular) junto con alteraciones de los huesos subcondrales y la formación de osteofitos son las características radiológicas de la enfermedad. Para los pacientes, no obstante, la prioridad son los dolores (dolores en reposo nocturnos y relacionados con cargas) con la subsiguiente limitación funcional. Esto es también lo que lleva a los pacientes al aislamiento social con las correspondientes deuteropatías.
El concepto artrosis se refiere, según una definición no oficial en Alemania, a un «desgaste articular» que sobrepasa los niveles habituales de la edad. Se considera que las causas son un exceso de carga (peso corporal algo elevado), causas congénitas o traumáticas, como alineación incorrecta de las articulaciones, o también deformaciones óseas debidas a osteopatías como la osteoporosis. Asimismo, la artrosis puede originarse como consecuencia de otras enfermedades (artrosis secundaria), por ejemplo de una inflamación articular (artritis), o ir acompañada del desarrollo de derrames debido a sobrecargas (reacción inflamatoria secundaria) (artrosis activada). La literatura especializada angloamericana distingue entre la osteoartrosis (osteoarthritis [OA] en inglés), en la que la destrucción de las superficies articulares probablemente se debe fundamentalmente a efectos de carga, y la artritis (arthritis, rheumatoid arthritis [RA] en inglés), en la que tiene prioridad la degeneración articular debido a un componente inflamatorio.
Fundamentalmente, las artrosis se diferencian según su causa. La artrosis alcaptonúrica se basa en un aumento de los depósitos de ácido homogentísico en las articulaciones en caso de la susodicha alcaptonuria. En la artrosis hemofílica se presentan con regularidad hemorragias intraarticulares en caso de hemofilia (hemartrosis). La artrosis úrica se produce por el efecto mecánico de cristales de urato (ácido úrico) en el cartílago sano (W. Pschyrembel y col.: Klinisches Worterbuch mit klinischen Syndromen und einem Anhang Nomina Anatomica. Editorial Walter de Gruyter & Co, 253a edición, 1977).
La displasia de las articulaciones representa la causa clásica de una artrosis. En el ejemplo de la cadera está claro que la zona más cargada mecánicamente en una posición fisiológica de cadera presenta una superficie claramente más grande que en el caso de una cadera displásica. Sin embargo, las cargas debidas a las fuerzas que actúan sobre la articulación son en su mayor parte independientes de la forma de la articulación. Se reparten fundamentalmente sobre la(s) zona(s) de carga principal(es). De esta manera, se produce una mayor carga por presión en una zona pequeña que en una zona más grande. Por lo tanto, en una cadera displásica la carga biomecánica por presión del cartílago articular es mayor que en una posición fisiológica de cadera. En general, se considera que este principio es la causa de la aparición frecuente de alteraciones artrósicas en las articulaciones de soporte que difieren de la forma anatómica ideal.
Si las consecuencias de una lesión son las responsables de un desgaste prematuro, se habla de una artrosis postraumática. Como otras causas de una artrosis secundaria se habla de razones mecánicas, inflamatorias, metabólicas, químicas (quinolonas), tróficas, hormonales, neurológicas y genéticas. En la mayoría de casos, sin embargo, se declara como diagnóstico una artrosis idiopática en la que el médico expresa la aparente falta de una enfermedad causal (H. I. Roach y S. Tilley, Bone and Osteoarthritis F. Bronner y M. C. Farach-Carson (editores), editorial Springer, volumen 4, 2007).
Las causas medicamentosas de una artrosis pueden ser, por ejemplo, antibióticos del tipo inhibidores de girasa (fluoroquinolonas, como ciprofloxacino, levofloxacino). En tejidos con una mala vascularización (cartílago articular hialino, tejidos de tendón), estos medicamentos provocan una complejación de iones magnesio, lo cual tiene como consecuencia que se producen lesiones irreversibles en el tejido conjuntivo. Normalmente estas lesiones son más marcadas en niños y jóvenes en fase de crecimiento. Las tendopatías y artropatías son efectos secundarios conocidos de esta clase de medicamentos. En adultos, según informaciones de farmacólogos y reumatólogos independientes, estos antibióticos provocan una degradación fisiológica acelerada del cartílago articular hialino (M. Menschik y col., Antimicrob. Agents Chemother. 41, 1997, págs. 2562-2565; M. Egerbacher y col., Arch. Toxicol. 73,
2000, págs. 557-563; H. Chang y col., Scand. J. Infect. Dis. 28, 1996, págs. 641-643; A. Chaslerie y col., Therapie 47, 1992, pág. 80). También un tratamiento prolongado con fenprocumón puede favorecer una artrosis a causa de la reducción de la densidad ósea en caso de cargas de la estructura interna de la articulación.
Además de la edad, se conocen como factores de riesgo de la osteoartrosis las sobrecargas mecánicas, (micro)traumatismos, desestabilizaciones de la articulación provocadas por la pérdida de mecanismos de seguridad, así como factores genéticos. Sin embargo, no se han aclarado completamente ni el origen ni las posibilidades de intervención (H. I. Roach y S. Tilley, Bone and Osteoarthritis F. Bronner y M. C. Farach-Carson (editores), editorial Springer, volumen 4, 2007).
En una articulación afectada por artrosis el contenido de monóxido de nitrógeno aumenta temporalmente. De un modo similar, podría observarse a causa de una irritación mecánica elevada del tejido condral (P. Das y col., Journal of Orthopaedic Research 15, 1997, págs. 87-93. A. J. Farrell y col. Annals of the Rheumatic Diseases 51, 1992, págs. 1219-1222; B. Fermor y col., Journal of Orthopaedic Research 19, 2001, págs. 729-737), mientras que una estimulación mecánica moderada repercute de una forma más bien positiva. Con ello, los efectos de fuerza mecánica intervienen como causa en el avance de la osteoartrosis (X. Liu y col., Biorheology 43, 2006, págs. 183 190).
Fundamentalmente, la terapia de la artrosis persigue dos objetivos. Por un lado, la ausencia de dolor bajo una carga habitual y, por otro lado, impedir las limitaciones mecánicas o las alteraciones de una articulación. Estos objetivos no se pueden conseguir a largo plazo mediante un tratamiento del dolor como único método terapéutico sintomático, ya que con este tratamiento no se puede impedir el avance de la enfermedad. Si se quiere conseguir esto último, se debe detener la destrucción del cartílago. Puesto que en pacientes adultos el cartílago articular no se puede regenerar, es de suma importancia, además, la eliminación de factores patogenéticos como displasias articulares o alineaciones incorrectas que provocan un aumento de cargas de presión puntuales del cartílago articular.
Finalmente, con la ayuda de medicamentos se intenta impedir o detener los procesos degenerativos en los tejidos condrales.
La matriz extracelular, que está compuesta en primer lugar de colágeno, proteoglucanos y agua, es fundamental para el estado funcional del cartílago articular y, con ello, para su capacidad de resistencia frente a cargas. Entre las enzimas que intervienen en la degradación de la matriz extracelular se encuentran, en particular, las metaloproteinasas, las agrecanasas y las enzimas catepsinas. Pero también otras enzimas pueden descomponer principalmente la matriz condral, como por ejemplo la plasmina, la calicreína, la elastasa de neutrófilos, la triptasa y la quimasa.
Las catepsinas pertenecen a la superfamilia papaína de las proteasas lisosomales. Las catepsinas participan en la proteólisis normal y en la transformación de proteínas diana y tejidos, así como en la iniciación de cascadas proteolíticas y activaciones de proenzimas. Además, participan en la expresión del CMH clase II (Baldwin (1993) Proc. Natl. Acad. Sci., 90: 6796-6800; Mixuochi (1994) Immunol. Lett., 43: 189-193). En realidad, una expresión anómala de la catepsina puede provocar enfermedades graves. Así, se pudo comprobar una expresión aumentada de catepsina en células cancerosas, por ejemplo en cáncer de mama, de pulmón, de próstata, de glioblastoma y de cabeza y cuello, y se pudo demostrar que la catepsina está asociada con un éxito terapéutico insuficiente en cáncer de mama, de pulmón, de cabeza y cuello, así como en tumores cerebrales (Kos y col. (1998) Oncol. Rep., 5: 1349 1361; Yan y col. (1998) Biol. Chem., 379: 113-123; Mort y col.; (1997) Int. J. Biochem. Cell Biol., 29: 715-720; Friedrick y col. (1999) Eur. J Cancer, 35: 138-144). Además, al parecer una expresión anómala de la catepsina está implicada en el desarrollo de enfermedades inflamatorias y no inflamatorias, como por ejemplo la artritis reumatoide y la osteoartrosis (Keyszer (1995) Arthritis Rheum., 38: 976-984).
El mecanismo molecular de la actividad de la catepsina no se ha esclarecido completamente. Por un lado, se ha descubierto que, por ejemplo, una expresión inducida de catepsina en células B cuyo suero se extrae protege de la apoptosis y que un tratamiento de las células con oligonucleótidos antisentido de la catepsina B induce una apoptosis (Shibata y col. (1998) Biochem. Biophys. Res. Commun., 251: 199-20; Isahara y col. (1999) Neuroscience, 91: 233-249). Estos estudios sugieren un papel antiapoptótico de las catepsinas. Sin embargo, se encuentran en oposición total respecto a estudios previos que describen las catepsinas como mediadoras de la apoptosis (Roberts y col. (1997) Gastroenterology, 113: 1714-1726; Jones y col. (1998) Am. J. Physiol., 275: G723-730).
Las catepsinas se sintetizan como zimógenos inactivos en los ribosomas y se transfieren al sistema lisosomal. Tras la disociación proteolítica del propéptido N terminal aumenta la concentración de catepsina en el medio ácido de los lisosomas hasta 1 mM y los lisosomas liberan las catepsinas hacia el medio extracelular.
Entre las catepsinas se distinguen las cisteína catepsinas B, C, H, F, K, L, O, S, V y W, las aspartil catepsinas D y E y la serina catepsina G.
Ejemplos de inhibidores de catepsina en el desarrollo clínico son inhibidores de catepsina K para el tratamiento de la artrosis e inhibidores de catepsina S para el tratamiento de la artritis, el dolor neuropático y la psoriasis.
Entre las aspartil proteasas también se encuentran, además de la catepsina D, la aspartil proteasa VIH (proteasa VIH-1), la renina, la pepsina A y C, BACE (Asp2, memapsina), la plasmepsina y las aspartil hemoglobinasas (Takahashi, T. y col., Ed. Aspartic Proteinases Structure, Function, Biology and Biomedical Implications (Plenum Press, New York, 1995), Adams, J. y col., Ann. Rep. Med. Chem. 31, 279-288, 1996; Edmunds J. y col., Ann. Rep. Med. Chem. 31,51 -60, 1996; Miller, D. K. y col., Ann. Rep. Med. Chem 31, 249-268, 1996). La catepsina D participa normalmente en la degradación de proteínas intracelulares o fagocitadas y por ello desempeña un papel importante en el metabolismo proteico (Helseth, y col., Proc. Natl. Acad. Sci. USA 81, 3302-3306, 1984), en el catabolismo proteico (Kay, y col., Intracellular Protein Catabolism (eds. Katunuma, y col., 155-162, 1989) y en el procesamiento de antígenos (Guagliardi, y col., Nature, 343, 133-139, 1990; Van Noort, y col., J. Biol. Chem., 264, 14159-14164, 1989).
Los niveles elevados de catepsina D se han relacionado con una serie de enfermedades. Así, los niveles elevados de catepsina D están en correlación con pronósticos malos en caso de cáncer de mama y con invasión celular elevada y mayor riesgo de metástasis, así como con un tiempo de supervivencia menor sin recidivas tras la terapia y una tasa de supervivencia más reducida en conjunto (Westley B. R. y col., Eur. J. Cancer 32, 15-24, 1996; Rochefort, H., Semin. Cancer Biol. 1:153, 1990; Tandon, A. K. y col., N. Engl. J. Med. 322, 297, 1990). La tasa de secreción de catepsina D en caso de cáncer de mama se obtiene a través de una sobreexpresión del gen y a través de un procesamiento modificado de la proteína. Unos niveles elevados de catepsina D y de otras proteasas, como por ejemplo colagenasa, fabricada en las inmediaciones de un tumor en crecimiento, podrían incluso degradar la matriz extracelular alrededor del tumor y así provocar el desprendimiento de células tumorales y la invasión de nuevos tejidos a través del sistema linfático y sanguíneo (Liotta L. A., Scientific American Feb:54, 1992; Liotta L. A. y Stetler-Stevenson W. G., Cancer Biol. 1:99, 1990; Liaudet E., Cell Growth Differ. 6:1045-1052, 1995; Ross J. S., Am. J. Clin. Pathol. 104:36-41, 1995; Dickinson A. J., J. Urol. 154:237-241, 1995).
Además, la catepsina D está relacionada con alteraciones degenerativas del cerebro, como por ejemplo la enfermedad de Alzheimer. Así, la catepsina D está asociada con la descomposición del precursor de la proteína pamiloide o de un precursor mutante que aumenta la expresión de la proteína amiloide en las células transfectadas (Cataldo, A. M. y col., Proc. Natl. Acad. Sci. 87: 3861, 1990; Ladror, U. S. y col., J. Biol. Chem. 269: 18422, 1994, Evin G., Biochemistry 34: 14185-14192, 1995). La proteína p-amiloide, que resulta de la proteólisis del precursor de la proteína p-amiloide, provoca la formación de placas en el cerebro y parece ser la responsable del desarrollo de la enfermedad de Alzheimer. También se han encontrado niveles elevados de catepsina D en el líquido cefalorraquídeo de pacientes de alzhéimer y se pudo demostrar una elevada actividad proteolítica de la catepsina D frente al precursor mutante de la proteína p-amiloide (Schwager, A. L., y col. J. Neurochem. 64:443, 1995). Además, se mide un aumento significativo de la actividad de la catepsina D en biopsias de pacientes con la enfermedad de Huntington (Mantle D., J. Neurol. Sci. 131: 65-70, 1995).
En la manifestación de una artrosis, la catepsina D probablemente desempeña un papel fundamental en varios niveles. Así, se miden niveles más elevados de ARNm de catepsina D en perros con artrosis espontánea en comparación con perros sanos en el cartílago articular del cóndilo de cadera (Clements D. N. y col., Arthritis Res. Ther. 2006; 8(6): R158; Ritchlin C. y col., Scand. J. Immunnol. 40: 292-298, 1994). También Devauchelle V. y col. (Genes Immun. 2004, 5(8): 597-608) muestran tasas de expresión diferentes de catepsina D en pacientes humanos en caso de artrosis en comparación con la artritis reumatoide (véase también Keyszer G. M., Arthritis Rheum. 38: 976-984, 1995). La catepsina D también parece estar implicada en caso de mucolipidosis (Kopitz J., Biochem. J.
295, 2: 577-580, 1993).
La endopeptidasa lisosomal catepsina D es la proteinasa más extendida en los condrocitos (Ruiz-Romero C. y col., Proteomics. 2005, 5(12): 3048-59). La actividad proteolítica de la catepsina D se ha demostrado además en líquidos sinoviales cultivados de pacientes con osteoartrosis (Bo G. P. y col., Clin. Rheumatol. 2009, 28(2): 191-9) y también en tejidos de sinovectomía de pacientes con artritis reumatoide se encuentra una elevada actividad proteolítica (Taubert H. y col., Autoimmunity. 2002, 35(3): 221-4). Lorenz y col. (Proteomics. 2003, 3(6): 991-1002) también escriben que todavía no se ha estudiado con detalle la aspartil proteasa catepsina D lisosomal y secretada en contraste con las catepsinas B y L con respecto a la artritis y artrosis, sin embargo, Lorenz y col. han encontrado niveles proteicos más elevados de catepsina D en tejidos sinoviales de pacientes con artrosis en comparación con pacientes con artritis reumatoide.
Gedikoglu y col. (Ann. Rheum. Dis. 1986, 45(4): 289-92) han podido demostrar igualmente una actividad proteolítica elevada de catepsina D en tejidos sinoviales y Byliss y Ali (Biochem. J. 1978, 171(1): 149-54) en el cartílago de pacientes con artrosis.
En la artrosis se produce una reducción local del valor de pH en las zonas del cartílago. Esta reducción del valor de pH tiene una importancia crucial para entender los procesos catabólicos del cartílago.
En la artrosis también se encuentra una correlación directa del valor de pH reducido en el tejido articular y la gravedad y la evolución de la enfermedad. A un valor de pH de 5,5 se produce una autodigestión del cartílago. Esto se puede inhibir casi completamente en cultivos de explantes (por ejemplo, de ratón, bóvido o humano) mediante pepstatina o ritonavir. Esto sugiere un papel fundamental, si no incluso un papel clave, de la catepsina D en la artrosis, ya que la pepstatina inhibe las aspartil proteasas con una excepción - BACE1 - y hasta ahora solo se han identificado estas dos aspartil proteasas en los tejidos condrales. Así, Bo G. P. y col. (Clin. Rheumatol. 2009, 28(2): 191-9) también describen el importante papel de la catepsina D en las alteraciones patológicas de las articulaciones. El inhibidor de aspartil proteasas más conocido es la pepstatina, un péptido que se aisló originalmente de un cultivo de Streptomyces. La pepstatina es eficaz frente a pepsina, catepsina y renina. Por eso, muchos inhibidores de aspartil proteasas se han basado en el modelo de la estructura de la pepstatina (patente de EE. UU. n.° 4,746,648; Umezawa, H., y col., J. Antibiot (Tokio) 23: 259-62, 1970; Morishima, H., y col., J. Antibiot. (Tokio) 23: 263-5, 1970; Lin, T. y Williams, H. R., J. Biol. Chem. 254: 11875-83, 1979; Jupp, R. A., y col., Biochem. J. 265: 871-8, 1990; Agarwal, N. S. y Rich, D. H., J. Med. Chem. 29: 2519-24, 1986; Baldwin, E. T., y col., Proc. Natl. Acad. Sci., EE. UU.
90: 6796-800, 1993; Francis, S. E. y col., EMBO J 13: 306-17, 1994).
Las aspartil proteasas o la catepsina D se describen con frecuencia como proteínas diana de principios activos para el tratamiento de enfermedades neurodegenerativas, trastornos cognitivos, demencia, alzhéimer, cáncer, malaria, infección por VIH y enfermedades del sistema cardiocirculatorio y vascular y se revelan inhibidores de aspartil proteasas o catepsina D para el tratamiento de estas enfermedades, por ejemplo, en los documentos WO 2009013293, EP 1987834, EP 1872780, EP 1867329, EP 1745778, EP 1745777, EP 1745776, WO 1999002153, WO 1999055687, US 6150416, WO 2003106405, WO 2005087751, WO 2005087215, WO 2005016876, US 2006281729, WO 2008119772, WO 2006074950, WO 2007077004, WO 2005049585, US 6251928 y US 6150416.
El documento WO 2012107153 da a conocer compuestos peptídicos, así como también pepstatina, como antagonistas de la catepsina D que se pueden emplear para el tratamiento de la artrosis y se pueden administrar por vía intraarticular para ello.
Los inhibidores de catepsina D conocidos y los dos compuestos modelo pepstatina y ritonavir inhiben con eficacia la actividad de la catepsina D, sin embargo presentan una selectividad muy reducida frente a otras aspartil proteasas. El papel del sistema renina-angiotensina (RAS, por sus siglas en inglés) en la regulación de la tensión arterial y del contenido de líquido y electrolitos (Oparil, S. y col., N. Engl. J. Med. 1974; 291: 381-401/446-57) y la eficacia de los inhibidores de renina y pepsina en enfermedades del sistema cardiocirculatorio y vascular son suficientemente conocidos y, por tanto, en particular en la administración oral o sistémica de estos inhibidores poco selectivos de catepsina D se debe contar con numerosos efectos secundarios y también en la administración local se debe contar con complicaciones sistémicas debido a la difusión esperada de los compuestos en la sangre.
Además, precisamente los compuestos peptídicos presentan por lo general una estabilidad baja en el plasma, el líquido sinovial y líquidos de otros compartimentos y el metabolismo los degrada muy rápidamente de modo que se espera un tiempo de permanencia reducido en la sangre, en la cápsula articular y en otros compartimentos.
Powell, M. F. y col. (J. Pharm. Sciences, vol. 81, n.° 8, 731-735, 1992) investigaron la estabilidad de compuestos peptídicos en suero humano unificado (mezcla) y en líquido sinovial unificado (mezcla) de pacientes con artritis reumatoide (véase la pág. 731, columna derecha, penúltimo párrafo). En las Tablas 1 y 2, Powell y col., revelaron que la mayoría de los péptidos de ensayo modificados y no modificados con una longitud de 10 a 25 aminoácidos presentan una semivida inferior a una hora en los medios de ensayo, plasma humano (HS, por sus siglas en inglés), líquido sinovial (SF, por sus siglas en inglés), suero de ternera fetal (FCS, por sus siglas en inglés) u homogeneizado de hígado de ratón (MLH, por sus siglas en inglés) (véase la página 735, columna derecha, último párrafo). La estabilidad de los compuestos peptídicos en el suero humano unificado y en el líquido sinovial unificado de pacientes con artritis es básicamente igual de baja (véase la página 733).
En vista de la baja estabilidad y el reducido tiempo de permanencia de los compuestos peptídicos en el plasma y debido a los efectos secundarios esperados descritos anteriormente, una administración sistémica u oral de los inhibidores peptídicos de catepsina D para el tratamiento de la artrosis no entra en consideración.
Por lo general, los especialistas tampoco tienen en cuenta una administración intraarticular de los compuestos peptídicos debido a la reducida semivida esperada en el líquido sinovial, y especialmente debido al reducido tiempo de permanencia esperado en la cápsula articular (difusión mediante la membrana sinovial y degradación) y debido a los efectos secundarios sistémicos que se pueden esperar debido a la difusión al plasma.
Debido sobre todo a la reducida semivida de unas pocas horas de los compuestos peptídicos según Powell y col. (1992), se necesitarían inyecciones intraarticulares frecuentes. No obstante, las inyecciones en la cavidad articular están vinculadas a dolores y a un riesgo de infección considerable para los pacientes y, por consiguiente, dichas inyecciones no se deben realizar con una frecuencia superior a un intervalo de entre dos y cuatro semanas.
Por tanto, el objetivo de la presente invención fue encontrar nuevos medicamentos y preparaciones farmacéuticas que se puedan emplear para la prevención y el tratamiento de la artrosis y que sean lo suficientemente estables para la administración local o intraarticular en el líquido sinovial y que solo tengan una difusión reducida al plasma a través de la membrana sinovial y que, por consiguiente, presenten un tiempo de permanencia elevado en la cápsula articular de modo que la concentración del principio activo se mantenga en un intervalo terapéuticamente eficaz durante el mayor tiempo posible tras la inyección.
Resumen de la invención
Sorprendentemente, pese a su elevado aclaramiento en la administración intravenosa u oral, se ha descubierto que la pepstatina permanece en la cápsula articular o el líquido sinovial con una concentración considerablemente superior a CI50 y, por tanto, con una concentración farmacéuticamente eficaz en caso de inyección intraarticular de una suspensión durante un periodo de tiempo prolongado. Además, pese a su estructura peptídica, la pepstatina muestra una estabilidad sorprendentemente elevada en el líquido sinovial (véase el ejemplo) y, con ello, la pepstatina supera considerablemente incluso las máximas estabilidades de compuestos peptídicos medidas por Powell y col. en el líquido sinovial (véase Powell y col., Tabla II en la página 733: n.° 8 toxina pertussis con una semivida de casi dos días en el líquido sinovial, n.° 15 Myobacterium leprae con una semivida de unas siete horas y media, todos los demás compuestos peptídicos con semividas de unos pocos minutos) y precisamente esta estabilidad sorprendentemente elevada y el tiempo de permanencia sorprendentemente alto en la cápsula articular permiten una aplicación médica relevante de la pepstatina en el tratamiento de la artrosis, ya que la pepstatina no permanece en la cápsula articular solamente unas pocas horas con niveles clínicamente relevantes, como cabría esperar según Powell y col., sino que está presente con una concentración considerablemente superior a CI50 durante más de dos semanas.
De esta forma, la pepstatina es sorprendentemente adecuada para medicamentos y preparaciones farmacéuticas que se administran de forma local o intraarticular para la prevención o el tratamiento de la artrosis y que mantienen niveles elevados durante un periodo de tiempo prolongado, de modo que el medicamento y las preparaciones farmacéuticas según la invención se deben administrar intraarticularmente como máximo de forma semanal, preferentemente en intervalos de uno a varios meses.
La pepstatina inhibe la catepsina D con una elevada eficacia y se esperan pocos efectos secundarios en la administración intraarticular para el tratamiento de la artrosis, puesto que la pepstatina en administración intraarticular alcanza solamente un bajo nivel sistémico (nivel plasmático) debido a su elevado tiempo de permanencia en la cápsula articular y a su lenta liberación desde el líquido sinovial.
En particular, el elevado tiempo de permanencia de la pepstatina en la cápsula articular es sorprendente y terapéuticamente valioso, ya que, por ejemplo, a partir de las investigaciones de Powell y col. (1992) cabría esperar una baja estabilidad de los compuestos peptídicos en el líquido sinovial y habría que contar con una elevada liberación de compuestos peptídicos de pequeño tamaño desde el líquido sinovial al plasma. Estos dos procesos causarían un bajo tiempo de permanencia en la cápsula articular. No obstante, sorprendentemente la pepstatina cuenta con un tiempo de permanencia prolongado en la cápsula articular, pese a lo esperado, ya que su estabilidad en el líquido sinovial es elevada y la liberación desde el líquido sinovial aparentemente es muy baja.
La pepstatina contiene varios centros quirales, de modo que también es objeto de la invención el uso de las formas ópticamente activas (estereoisómeros), los enantiómeros, los racematos, los diastereómeros así como los hidratos y los solvatos de pepstatina.
Se entenderá por derivados farmacéutica o fisiológicamente compatibles, por ejemplo, las sales de pepstatina, así como los compuestos llamados profármacos. Se entenderá por profármacos derivados de pepstatina modificados, por ejemplo, con grupos alquilo o acilo (véanse también los grupos protectores de amino e hidroxi a continuación), azúcares u oligopéptidos que en el organismo se disocian o se liberan rápidamente para dar las moléculas de pepstatina eficaces. Entre estos se encuentran también los derivados poliméricos biodegradables de pepstatina, como se describe, por ejemplo, en Int. J. Pharm. 115 (1995), 61-67.
La pepstatina se puede usar en su forma no salina definitiva. Por otro lado, la presente invención abarca también el empleo de pepstatina en forma de sus sales farmacéuticamente aceptables, que pueden derivarse de diversas bases orgánicas e inorgánicas según las formas de proceder conocidas en el sector. Las formas de sales farmacéuticamente aceptables de la pepstatina se elaboran en su mayor parte de forma convencional. Dado que la
pepstatina contiene un grupo de ácido carboxílico, podrá formarse una de sus sales adecuadas mediante la reacción de la pepstatina con una base adecuada para dar la correspondiente sal de adición de base. Tales bases son, por ejemplo, los hidróxidos de los metales alcalinos, entre los cuales se encuentran el hidróxido de potasio, el hidróxido de sodio y el hidróxido de litio; los hidróxidos de los metales alcalinotérreos tales como el hidróxido de bario y el hidróxido de calcio; los alcoholatos de los metales alcalinos, por ejemplo el metanolato de potasio y el propanolato de sodio; así como diversas bases orgánicas tales como la piperidina, la dietanolamina y la N-metilglutamina. Las sales de aluminio de la pepstatina pertenecen igualmente a este grupo.
De igual modo, a las sales con bases de pepstatina pertenecen las sales de aluminio, de amonio, de calcio, de cobre, férricas(IM), ferrosas(II), de litio, de magnesio, mangánicas(III), manganosas(II), de potasio, de sodio y de cinc, lo cual no debe representar ningún tipo de limitación.
Entre las sales anteriormente citadas se prefieren el amonio; las sales de metales alcalinos de sodio y de potasio, así como las sales de metales alcalinotérreos de calcio y de magnesio. Entre las sales de pepstatina que se derivan de las bases no tóxicas orgánicas farmacéuticamente aceptables se encuentran las sales de aminas primarias, secundarias y terciarias, aminas substituidas, entre las cuales se encuentran también las aminas substituidas de origen natural, las aminas cíclicas así como las resinas intercambiadoras de iones básicas, por ejemplo la arginina, la betaína, la cafeína, la cloroprocaína, la colina, la N,N'-dibenciletilendiamina (benzatina), la diciclohexilamina, la dietanolamina, la dietilamina, el 2-dietilaminoetanol, el 2-dimetilaminoetanol, la etanolamina, la etilendiamina, la N-etilmorfolina, la N-etilpiperidina, la glucamina, la glucosamina, la histidina, la hidrabamina, la iso-propilamina, la lidocaína, la lisina, la meglumina, la N-metil-D-glucamina, la morfolina, la piperazina, la piperidina, las resinas poliamínicas, la procaína, la purina, la teobromina, la trietanolamina, la trietilamina, la trimetilamina, la tripropilamina así como la tris-(hidroximetil)-metilamina (trometamina), lo cual no debe representar ningún tipo de limitación.
Tal como se ha mencionado, se forman las sales de adición de base de pepstatina farmacéuticamente aceptables con metales o con aminas tales como los metales alcalinos y los metales alcalinotérreos o con las aminas orgánicas. Los metales preferentes son el sodio, el potasio, el magnesio y el calcio. Las aminas orgánicas preferentes son la N,N'-dibenciletilendiamina, la cloroprocaína, la colina, la dietanolamina, la etilendiamina, la N-metil-D-glucamina y la procaína.
Las sales de adición de base de pepstatina se preparan poniéndose en contacto la forma de ácido libre con una cantidad suficiente de la base deseada, con lo cual se forma la sal de manera usual. Puede regenerarse el ácido libre poniendo en contacto la forma salina con un ácido y aislando el ácido libre de manera usual. Las formas de ácido libre se diferencian en cierto sentido de sus formas salinas correspondientes en lo que se refiere a determinadas propiedades físicas, tal como la solubilidad en disolventes polares; en el ámbito de la invención las sales corresponden, sin embargo, por lo demás a sus correspondientes formas de ácido libre.
En lo que se refiere a lo que se ha dicho anteriormente, se observa que debe entenderse por el concepto de «sal farmacéuticamente aceptable» en el presente contexto un producto activo que contenga pepstatina en forma de una de sus sales, especialmente cuando esta forma salina proporcione propiedades farmacocinéticas mejoradas al producto activo en comparación con la forma libre del producto activo o en comparación con cualquier otra forma salina del producto activo que hubiera sido empleada con anterioridad. La forma salina farmacéuticamente aceptable del principio activo podrá proporcionar a este principio activo por primera vez una propiedad farmacocinética deseada, de la cual no disponía anteriormente, e incluso puede influenciar positivamente en el cuerpo sobre la farmacodinámica de este principio activo en lo que se refiere a su actividad terapéutica.
Se entenderá por solvatos de pepstatina los compuestos de adición de moléculas inertes de disolventes de pepstatina que se formen debido a su fuerza de atracción mutua. Los solvatos son, por ejemplo, hidratos, como monohidratos o dihidratos, o alcoholatos, es decir, compuestos de adición con alcoholes como, por ejemplo, metanol o etanol.
Se ha descubierto que la pepstatina se tolera bien y posee propiedades farmacológicas valiosas ya que inhibe selectivamente las aspartil proteasas y, en particular, la catepsina D.
Como estados fisiológicos y/o patofisiológicos se entiende estados fisiológicos y/o patofisiológicos que son médicamente relevantes como, por ejemplo, enfermedades o afecciones, disfunciones, dolencias, síntomas o complicaciones médicos y similares, en particular enfermedades.
El dolor es una percepción sensorial compleja que como fenómeno agudo muestra el carácter de una señal de alarma y orientación, pero como dolor crónico ha perdido este carácter y en este caso (como Síndrome del dolor crónico) hoy en día debe considerarse y debe tratarse como un cuadro clínico independiente. En medicina, se denomina hiperalgesia a una sensibilidad y reacción excesivas al dolor en relación a un estímulo generalmente doloroso. Los estímulos que pueden provocar los dolores son, por ejemplo, la presión, el calor, el frío o las
inflamaciones. La hiperalgesia es una forma de hiperestesia, término genérico para una sensibilidad excesiva en relación a un estímulo. En medicina, se denomina alodinia a una sensación de dolor producida por un estímulo que generalmente no provoca dolor.
La pepstatina presenta una actividad biológica ventajosa que puede demostrarse fácilmente en ensayos enzimáticos y experimentos con animales, como se describe en los ejemplos. En este tipo de ensayos basados en enzimas, la pepstatina muestra y provoca un efecto inhibidor que se documenta usualmente por medio de los valores CI50 en un intervalo adecuado, preferentemente en el intervalo micromolar y más preferentemente en el intervalo nanomolar. La pepstatina puede administrarse a humanos o animales, en particular mamíferos tales como monos, caballos, perros, gatos, ratas o ratones, y se puede utilizar en el tratamiento terapéutico del cuerpo humano o del cuerpo animal así como en la lucha contra las enfermedades citadas anteriormente. También puede utilizarse como agente de diagnóstico o como reactivo.
La pepstatina se puede emplear para la elaboración de preparaciones farmacéuticas para la administración intraarticular, en particular de un modo no químico. Para ello, se mezcla con al menos un vehículo o excipiente sólido, líquido y/o semilíquido y, dado el caso, se combina con uno o varios principio(s) activo(s) adicional(es) en una forma de dosificación adecuada.
Las preparaciones farmacéuticas según la invención pueden utilizarse en forma de medicamentos en medicina humana o veterinaria. El paciente o receptor puede pertenecer a cualquier especie de mamífero, por ejemplo, una especie de primate, en especial humanos; roedores, incluidos ratones, ratas y hámsteres; conejos, caballos, bóvidos, perros, gatos, etc. Los modelos animales son de interés para estudios experimentales en los que ofrecen un modelo para el tratamiento de una enfermedad humana.
Se tienen en consideración como vehículos sustancias orgánicas o inorgánicas que son apropiadas para la administración intraarticular y que no reaccionan con los compuestos según la invención. Los excipientes adecuados para la formulación farmacéutica deseada son conocidos por el especialista gracias a sus conocimientos especializados. Además de disolventes como agua, solución salina fisiológica o alcoholes como, por ejemplo, etanol, propanol o glicerina, soluciones de azúcares como glucosa o soluciones de manitol o una mezcla de los disolventes mencionados y otros vehículos para el principio activo, pueden utilizarse estabilizadores y/o agentes reticulantes, emulsionantes, sales para modificar la presión osmótica, antioxidantes, dispersantes, antiespumantes, tampones, conservantes o solubilizantes. Si se desea, las preparaciones o medicamentos según la invención pueden contener uno o varios principios activos, por ejemplo una o varias vitaminas o principios activos que sean eficaces para la profilaxis y/o el tratamiento de las indicaciones médicas mencionadas anteriormente.
Si se desea, las preparaciones o medicamentos según la invención pueden contener uno o varios principios activos adicionales y/o uno o varios adyuvantes.
Los términos «formulación farmacéutica» y «preparación farmacéutica» se utilizan como sinónimos en el marco de la presente invención.
En el presente documento «farmacéuticamente compatible» se refiere a medicamentos, reactivos de precipitación, vehículos, excipientes, estabilizantes, disolventes y otros agentes que permiten la administración de las preparaciones farmacéuticas obtenidas de ellos sin efectos secundarios fisiológicos no deseados en mamíferos. En el caso de preparaciones farmacéuticas para la administración parenteral se exige que sean isotónicas y euhídricas, así como que la formulación, los excipientes utilizados y el material de envasado primario sean tolerables y seguros (baja toxicidad). Sorprendentemente, la pepstatina tiene preferentemente la ventaja de que es posible un uso directo y de que antes del uso en formulaciones farmacéuticas no es necesaria ninguna otra etapa de purificación para eliminar agentes peligrosos desde un punto de vista toxicológico como, por ejemplo, elevadas concentraciones de disolvente orgánico u otros excipientes peligrosos desde un punto de vista toxicológico.
Las preparaciones farmacéuticas según la invención para la administración intraarticular pueden contener pepstatina en forma precipitada no cristalina, precipitada cristalina o en disolución o en suspensión, así como, dado el caso, vehículos y/o excipientes y/o principios activos farmacéuticos adicionales.
Preferentemente, la pepstatina permite elaborar formulaciones con concentraciones elevadas sin que se produzcan agregaciones desfavorables no deseadas de pepstatina. Así, se pueden elaborar soluciones listas para su aplicación con una elevada proporción de principio activo con ayuda de pepstatina con disolventes acuosos o en medios acuosos.
La pepstatina y/o sus sales y solvatos fisiológicamente compatibles también pueden liofilizarse y los liofilizados obtenidos pueden utilizarse, por ejemplo, para la elaboración de preparados para inyección para la administración intraarticular.
Se pueden elaborar preparaciones acuosas para la administración intraarticular disolviendo o suspendiendo la pepstatina en una solución acuosa y, dado el caso, añadiendo excipientes. Para ello, resulta apropiado añadir a una solución o suspensión con una concentración definida de pepstatina volúmenes definidos de solución madre que contiene los excipientes adicionales mencionados en una concentración definida y, dado el caso, diluirla con agua hasta la concentración previamente calculada. Como alternativa, se pueden añadir los excipientes en forma sólida. A continuación, a la solución o suspensión acuosa obtenida se pueden añadir las cantidades necesarias correspondientes de solución madre y/o agua. También resulta apropiado poder disolver o suspender la pepstatina directamente en una solución que contiene todos los demás excipientes.
De un modo ventajoso, las soluciones o suspensiones que contienen pepstatina se elaboran con un valor de pH de 4 a 10, preferentemente con un valor de pH de 5 a 9, y una osmolalidad de 250 a 350 mOsmol/kg. Por consiguiente, la preparación farmacéutica puede administrarse mayoritariamente sin dolor directamente por vía intraarticular. Además, a la preparación para la administración intraarticular también pueden añadirse soluciones de infusión como, por ejemplo, solución de glucosa, solución salina isotónica o solución de Ringer, que pueden contener principios activos adicionales de modo que se pueden administrar mayores cantidades de principio activo.
La pepstatina se tolera bien fisiológicamente, es fácil de elaborar, se puede dosificar con exactitud y, preferentemente, es estable en cuanto al contenido, los productos de descomposición y los agregados a lo largo del almacenamiento y del transporte y en caso de múltiples procesos de congelación y descongelación. Se puede conservar de forma estable preferentemente a lo largo de un periodo de al menos tres meses hasta dos años a temperatura de frigorífico (2-8 °C) y a temperatura ambiente (23-27 °C) y con una humedad relativa (H.r.) del 60 %. A modo de ejemplo, la pepstatina puede conservarse de forma estable mediante secado y cuando se necesita, transferirse a una preparación farmacéutica lista para usar mediante disolución o suspensión. Son métodos posibles para el secado, por ejemplo, sin quedar limitados a estos ejemplos, secado con nitrógeno gas, secado en un horno de vacío, liofilización, lavado con disolvente orgánico y posterior secado al aire, secado en lecho fluido, secado en lecho fluidizado, secado por atomización, secado en tambor, secado a capas; secado al aire a temperatura ambiente y otros métodos.
El concepto «cantidad activa» significa la cantidad de un medicamento o de un principio activo farmacéutico que provoca una respuesta biológica o medicinal en un tejido, en un sistema, en un animal o en un ser humano, que sea buscada o pretendida, por ejemplo, por el investigador o por el médico.
Además, el concepto «cantidad terapéuticamente activa» significa una cantidad que, en comparación con un sujeto correspondiente que no ha recibido esta cantidad, tiene como consecuencia lo siguiente: un tratamiento mejorado, la curación, la prevención o supresión de una enfermedad, de un cuadro clínico, de un estado clínico, de un padecimiento, de una disfunción o evitar efectos secundarios o también la disminución del avance de una enfermedad, de un padecimiento o de una disfunción. El concepto de «cantidad terapéuticamente activa» abarca también aquellas cantidades que son eficaces para aumentar la función fisiológica normal.
Al usar preparaciones o medicamentos según la invención para la administración intraarticular, por lo general se usa pepstatina y/o sus sales y solvatos fisiológicamente compatibles de forma análoga a preparaciones o preparados conocidos y comerciales. La dosificación depende de la edad, el peso, el estado de salud y la constitución del paciente, así como de la gravedad de la enfermedad y de otros factores individuales.
Las preparaciones farmacéuticas según la invención para la administración intraarticular se administran de forma intraarticular preferentemente con una frecuencia entre semanal y anual, prefiriéndose en especial entre cada 14 días y semestralmente, muy en especial, desde mensualmente hasta trimestralmente.
Por tanto, es objeto de la presente invención una preparación farmacéutica que contenga pepstatina y/o una de sus sales, solvatos y estereoisómeros fisiológicamente compatibles, incluidas sus mezclas en todas las proporciones, y otros vehículos y/o excipientes, para su uso como medicamento en el tratamiento y/o la profilaxis de estados fisiológicos y/o patofisiológicos seleccionados del grupo compuesto por artrosis, lesiones condrales traumáticas, artritis, dolor, alodinia o hiperalgesia, en los que la preparación farmacéutica se administra por vía intraarticular de la siguiente manera:
a) desde semanalmente hasta anualmente,
b) desde cada 14 días hasta semestralmente o
c) desde mensualmente hasta trimestralmente.
También es un objeto preferido de la presente invención una preparación farmacéutica citada anteriormente, la cual contenga al menos otro principio activo farmacéutico.
Un objeto especialmente preferido de la presente invención es una preparación farmacéutica citada anteriormente para su uso como medicamento en el tratamiento y/o la profilaxis de la artrosis.
Otro objeto de la presente invención es la pepstatina y/o una de sus sales, solvatos y estereoisómeros fisiológicamente compatibles, incluidas sus mezclas en todas las proporciones, para su uso como medicamento en el tratamiento y/o la profilaxis de estados fisiológicos y/o patofisiológicos seleccionados del grupo compuesto por artrosis, lesiones condrales traumáticas, artritis, dolor, alodinia e hiperalgesia, en los que la pepstatina y/o una de sus sales, solvatos y estereoisómeros fisiológicamente compatibles, incluidas sus mezclas en todas las proporciones, se administra intraarticularmente de la siguiente manera:
a) desde semanalmente hasta anualmente,
b) desde cada 14 días hasta semestralmente o
c) desde mensualmente hasta trimestralmente.
Es un objeto preferido de la invención la pepstatina citada anteriormente y/o sus sales, solvatos y estereoisómeros fisiológicamente compatibles, incluidas sus mezclas en todas las proporciones, para su uso como medicamento en el tratamiento y/o la profilaxis de la artrosis.
Sin embargo, la dosificación individual y los intervalos de administración para un paciente también dependen de un gran número de factores individuales como, por ejemplo, la eficacia del correspondiente compuesto utilizado, la edad, el peso corporal, el estado de salud general, el sexo, la alimentación, del momento y vía de administración, de la velocidad de excreción, de la combinación con otros medicamentos y de la gravedad y duración de la correspondiente enfermedad.
Una medida de la absorción de un principio activo farmacéutico en un organismo es su biodisponibilidad. Si el principio activo farmacéutico se suministra por vía intraarticular al organismo en forma de una solución para inyección, su biodisponibilidad absoluta, es decir, la proporción del fármaco que llega inalterado a la cavidad articular es del 100 %. Se pueden obtener datos respecto a la farmacocinética, es decir respecto a la biodisponibilidad, de forma análoga al método de J. Shaffer y col. (J. Pharm. Sciences, 88 (1999), 313-318).
De igual modo, pueden prepararse tales medicamentos con un procedimiento conocido en general en el sector farmacéutico.
Los medicamentos se pueden adaptar para la administración por vía intraarticular. Tales medicamentos pueden prepararse según todos los procedimientos conocidos en el sector farmacéutico, combinándose, por ejemplo, el principio activo con el o con los vehículos o con el o con los excipientes.
La administración intraarticular tiene la ventaja de que el compuesto según la invención se administra directamente cerca del cartílago articular en el líquido sinovial y desde ahí también puede difundirse hacia el tejido condral. Por lo tanto, las preparaciones farmacéuticas según la invención también pueden inyectarse directamente en la cavidad articular y así desarrollan su efecto directamente en el sitio objetivo previsto. El compuesto según la invención también es adecuado para la elaboración de medicamentos para la administración intraarticular con liberación del principio activo controlada, sostenida y/o retardada (slow-release, sustained-release, controlled release). Por lo tanto, la pepstatina también es adecuada para la elaboración de formulaciones depot, que son ventajosas para los pacientes, ya que es necesaria una aplicación solo a grandes intervalos de tiempo.
A los medicamentos adaptados para la administración intraarticular pertenecen las soluciones para inyección estériles acuosas y no acuosas que contengan antioxidantes, tampones, bacteriostáticos y solutos mediante los cuales la formulación se vuelve isotónica con el líquido sinovial del receptor que debe ser tratado; así como las suspensiones estériles acuosas y no acuosas que pueden contener agentes de suspensión y espesantes. Las formulaciones pueden suministrarse en recipientes que contengan una dosis individual o que contengan dosis múltiples, por ejemplo, ampollas y viales sellados, y pueden conservarse en estado secado por congelación (liofilizado) de tal manera que únicamente se requiera la adición de vehículos líquidos estériles, por ejemplo, agua para inyectables, justo antes de su utilización. Pueden prepararse las soluciones para inyección y las suspensiones, preparadas de acuerdo con una receta, a partir de polvos estériles, de granulados y de tabletas.
La pepstatina puede administrarse también en forma de sistemas de aporte en liposomas, tales como, por ejemplo, vesículas unilamelares pequeñas, vesículas unilamelares grandes y vesículas multilamelares. Los liposomas pueden formarse a partir de diversos fosfolípidos, tales como, por ejemplo, la colesterina, la estearilamina o las fosfatidilcolinas.
La pepstatina puede acoplarse también con polímeros solubles a título de excipientes medicinales orientados a su objetivo. Tales polímeros pueden comprender la polivinilpirrolidona, los copolímeros de pirano, el polihidroxipropilmetacrilamidofenol, el polihidroxietilaspartoamidofenol o el óxido de polietileno-polilisina substituido con restos de palmitoilo. Además, la pepstatina puede estar acoplada a una clase de polímeros biológicamente degradables que son adecuados para conseguir una liberación controlada de un medicamento, por ejemplo, ácido poliláctico, poliepsilon-caprolactona, ácido polihidroxibutírico, poliortoéster, poliacetal, polidihidroxipirano, policianoacrilato, ácido polilácticocoglicólico, polímeros como conjugados entre dextrano y metacrilato, polifosfoéster, distintos polisacáridos y poliaminas tales como poli-e-caprolactona, albúmina, quitosano, colágeno o gelatina modificada y copolímeros de bloque reticulados o anfipáticos de hidrogeles.
Se entiende que el medicamento según la invención puede contener, además de los componentes mencionados en especial anteriormente, otros agentes habituales en el campo en relación al correspondiente tipo de la formulación farmacéutica.
Asimismo, los medicamentos según la invención pueden emplearse para preparar efectos aditivos o sinérgicos en el caso de ciertas terapias conocidas, y/o además podrían emplearse para restablecer la eficacia de ciertas terapias existentes.
Las preparaciones farmacéuticas según la invención pueden contener, además de pepstatina, otros principios activos farmacéuticos adicionales, por ejemplo para el uso en el tratamiento de artrosis, otros inhibidores de catepsina D, AINE, inhibidores de Cox-2, glucocorticoides, ácido hialurónico, azatioprina, metotrexato, anticuerpos anti-CAM, como por ejemplo anticuerpos anti-ICAM-1 y/o FGF-18. Para el tratamiento de las otras enfermedades mencionadas, las preparaciones farmacéuticas según la invención pueden contener, además de pepstatina, otros principios activos farmacéuticos conocidos por el especialista para su tratamiento.
Incluso sin otras formas de realización, se asume que un especialista puede utilizar la descripción anterior en el alcance más amplio
Si no se indica lo contrario, los datos de porcentaje son porcentajes en peso. Todas las temperaturas se indican en grados Celsius. «Tratamiento habitual»: en caso necesario se añade agua, en caso necesario se ajusta el valor de pH entre 2 y 10 según la constitución del producto final, se extrae con acetato de etilo o con diclorometano, se separa, se seca la fase orgánica sobre sulfato de sodio, se filtra, se concentra por evaporación y se purifica mediante cromatografía sobre gel de sílice y/o mediante cristalización.
Valores Rf en gel de sílice; espectrometría de masas: EI (ionización por impacto electrónico, por sus siglas en inglés): M+, FAB (Fast Atom Bombardment): (M+H)+, Th F (tetrahidrofurano), NMP (N-metilpirrolidona), DMSO (dimetilsulfóxido), AE (acetato de etilo), MeOH (metanol), CCF (cromatografía en capa fina)
Se ha sintetizado y caracterizado la pepstatina. No obstante, el especialista también puede llevar a cabo la elaboración y caracterización de la pepstatina por otras vías.
Ejemplo 1: Pepstatina: un inhibidor peptídico de catepsina D
Tabla 1

así como sus sales, derivados, solvatos, profármacos y estereoisómeros fisiológicamente compatibles, incluidas sus mezclas en todas las proporciones.
Además, la pepstatina se caracteriza por su elevada selectividad para la catepsina D frente a la renina (CI50 >10000 nM), su buena penetración en el cartílago y por no presentar toxicidad o genotoxicidad medible.
Ejemplo 2: Ensayo de fluorescencia in vitro para la identificación de inhibidores de catepsina D
Para la identificación de moduladores de la actividad de la catepsina D se llevó a cabo una prueba enzimática continua con un péptido sintético que presenta un grupo fluorescente (MCA=(7-metoxicoumarin-4-il)acetilo) que se extingue mediante transferencia de energía de un grupo Dpn (2,4-dinitrofenilo) de la misma molécula en placas de microtitulación nb de 384 pocillos de Greiner. La disociación del sustrato peptídico mediante catepsina D provoca un aumento de la intensidad de fluorescencia. Para determinar la eficacia de las sustancias se comparó el aumento de intensidad de fluorescencia en función del tiempo en presencia de la sustancia con el incremento de fluorescencia en función del tiempo en ausencia de las sustancias. Como sustancia de referencia se empleó pepstatina A (Sigma-Aldrich). Como sustrato se utilizó MCA-GKPILFFRLK(Dnp)d-R-NH2 (Enzo Life Sciences, Lorrach, Alemania). Como enzima se empleó catepsina D aislada de hígado humano (Sigma-Aldrich) en una concentración final de 1,4 nM. La prueba se realizó en tampón sodio-acetato 100 mM, DMSO al 1,25 % (v/v), Chaps al 0,25 % (p/v), pH 5,5. Por cada 4 pl de solución de catepsina D se añadieron 2 pl de solución de la sustancia con concentraciones de sustancia diluidas en serie y se incubó 10 min a temperatura ambiente. La reacción se inició mediante la adición de 2 pl de solución de sustrato (concentración final 5 pM). Tras realizar una medición de fluorescencia inicial (longitud de onda de excitación 340 nm/longitud de onda de emisión 450 nm) con un lector Envision Multilabel (Perkin Elmer) se incubó la reacción 60 min a temperatura ambiente. A continuación se midió la cantidad de fragmento peptídico disociado durante el tiempo de reacción mediante la determinación del aumento de intensidad de fluorescencia a 450 nm (longitud de onda de excitación 340 nm).
Resultado: La pepstatina inhibe la catepsina D en el intervalo nanomolar (véase la Tabla 1).
Ejemplo 3: Prueba con explante de cartílago
Para estudiar el efecto de potenciales inhibidores de catepsina D sobre la degradación condral se utiliza un modelo inducido por pH que se basa en explantes bovinos. Así, el valor de pH del medio en el que se cultivan los explantes se ajusta al valor de pH patofisiológico de una rodilla artrótica. Este valor de pH es de pH 5,5. A continuación, en este modelo ex vivo se estudian potenciales inhibidores de catepsina D respecto a su eficacia en cuanto a un bloqueo del proceso de debilitamiento condral. Si el cartílago se destruye, se liberan glucosaminoglucanos (GAG) en el sobrenadante del cultivo celular. La cantidad de GAG liberados puede determinarse cuantitativamente con ayuda de DMMB (hidrocloruro de azul de dimetilmetileno). En la comprobación de GAG sulfatados con hidrocloruro de azul de dimetilmetileno se aprovecha la reducción de la absorción a 633 nm. Puesto que también se puede trabajar a concentraciones muy bajas de GAG, no precipita ningún complejo colorante/GAG ni tras una larga incubación de DMMB con GAG, como sucede en otros métodos de medición en ocasiones solo al cabo de poco tiempo. Para determinar la concentración se realiza simultáneamente una gráfica de referencia con sulfato de condroitina. Mediante los valores GAG se pueden calcular los valores CI50, es decir una concentración a la que una sustancia muestra un 50 % de su eficacia.
Soluciones:
Medio de incubación, pH 7,4:
DMEM sin FBS, adición de Pen/Strep al 1 % y 30 pg/ml de ácido ascórbico, el medio no se conserva.
Medio de incubación, pH 5,5:
DMEM sin FBS, el valor de pH se ajusta mediante la adición de MES y se controla con un pH-metro, adición de Pen/Strep al 1 % y 30 pg/ml de ácido ascórbico.
Soluciones para la medición de GAG:
Solución colorante con DMMB (V = 500 ml):
Disolver 8 mg de DMMB (azul de dimetilmetileno) en 2,5 ml de etanol 1 g de formiato sódico+ 1 ml de ácido fórmico, enrasar con agua bidest. a 500 ml.
Medio de incubación: FBS (medio sin FBS)
Soluciones de sulfato de condroitina (curva de referencia)
Lote de soluciones estándar con las siguientes concentraciones: 50 gg/ml; 25 gg/ml; 12,5 gg/ml; 6,25 gg/ml; 3,125 gg/ml; 1,56 gg/ml; 0,78 gg/ml así como un blanco del medio. El lote de la solución estándar se realiza en el medio en el que también se ha llevado a cabo el estudio.
1. ) Ejecución: degradación condral inducida por pH de explantes bovinos
Primero se preparan los explantes bovinos. La inducción de la degradación condral se realiza en placas de 96 pocillos. Para ello, se cultiva un explante por pocillo. Se realiza la adición en cada uno de 200 gl de DMEM (medio de incubación pH 5,5) sin FBS 30 gg/ml de ácido ascórbico. Como control negativo se incuban explantes (n= 4) a pH 7,4 (sin FBS). Este control no entra en el cálculo de los datos, sino que asegura que la modificación del valor de pH tiene el efecto deseado en la liberación de GAG. En este punto se realiza la adición de las sustancias de estudio. No se realiza ninguna incubación previa de los explantes. Los explantes se cultivan con las sustancias correspondientes 3 días en una incubadora a 37 °C y 7,5 % de CO2.
2. ) Desarrollo de la incubación
Para estudiar el efecto de los inhibidores de catepsina D en la liberación de GAG (glucosaminoglucano), se utilizan las sustancias en las concentraciones deseadas y se cultivan durante 3 días. Para ello, los compuestos de estudio se ensayan en un primer experimento a una concentración de 1 gM y DMSO al 1 %. Las sustancias que tienen un efecto de >50 % en la liberación de GAG (que corresponde a <50 % del control en el Assay Explorer), se ensayan en un experimento posterior a 100 nM y DMSO al 1 %. Las sustancias que en estas condiciones tienen un efecto de >50 % en la liberación de GAG (que corresponde a <50 % del control en el Assay Explorer), se ensayan en una relación entre concentración y eficacia. Para ello se estudian los compuestos en las siguientes concentraciones: 30 gM, 10 gM, 3 gM, 1 gM, 0,3 gM, 0,1 gM, 0,03 gM, 0,01 gM.
Como control positivo se utiliza pepstatina A con una concentración de 0,01 gM. La ventana de eficacia (assay window) se define mediante el control (pH 5,5), definido como un 0 % de efecto, y el control pH 5,5 0,01 gM de pepstatina A, definido como un 100 % de efecto. Tras 3 días de incubación, se recogen los sobrenadantes del cultivo celular y se conservan a -20 °C o se miden directamente. Para ello se mide fotométricamente la cantidad de GAG liberado.
Se expresan para concentraciones de 1 gM y 100 nM del efecto (valor 1) de la sustancia correspondiente en % referido al control positivo (pH 5,5 0,01 gM de pepstatina A) y al control negativo (pH 5,5). El valor representa el valor medio de 4 replicados. Para determinar una relación entre concentración y eficacia se expresa un valor CI50 en el banco de datos (Assay Explorer).
3. ) Medición
Los sobrenadantes del cultivo celular (200 gl) se miden directamente o bien se conservan a -20 °C. Para garantizar una determinación exacta de la concentración (gg/ml de GAG en el sobrenadante) de GAG, los valores medidos deben encontrarse en la zona lineal de la curva de referencia. Para garantizar esto, se añaden rutinariamente distintas diluciones (1/5, 1/10, 1/20, 1/40). Las diluciones se preparan con medio y se añaden (15 gl) de modo automatizado (Hamilton) en una placa de 384 pocillos. De un modo igualmente automatizado (o con pipetas multicanal) se añaden 60 gl de solución de DMMB. Se produce una reacción de coloración rápida que seguidamente se mide a 633 nm con un lector de placas (p. ej. Envision).
Según la cantidad de muestra disponible, se lleva a cabo al menos una determinación doble.
Los datos se obtienen del lector MTP en forma de archivos csv o xls y en base a este formato (xls) se guardan como datos primarios o se preparan para calcular el efecto porcentual del compuesto correspondiente.
4. ) Controles de calidad
Como control para la inducción de la degradación condral inducida por el pH se incuban 4 explantes a pH 7,4. Este valor corresponde al valor de pH fisiológico del cartílago y, por lo tanto, en este caso no se debe esperar ningún efecto en la liberación de GAG. Estos valores de GAG (gg/ml de sobrenadante) siempre son, por lo tanto, significativamente más bajos que los valores de GAG en una incubación con pH 5,5.
Otro control que sirve para comprobar el experimento y también es importante para la definición de la ventana de eficacia, es el control con pepstatina (pH 5,5 0,01 gM de pepstatina A). Esta sustancia bloquea de forma no específica la actividad de la mayoría de proteasas y, por lo tanto, establece el efecto máximo posible de un compuesto.
5.) Resultados
En el ensayo de GAG, la pepstatina muestra un valor de CI50 en el intervalo nanomolar (véase la Tabla 1).
(1) Klompmakers, A. y Hendriks, T. (1986) Anal. Biochem. 153, 80-84, Spektrophotometrischer Nachweis für sulfatierte Glycosaminoglycane.
(2) Groves, P.J. Y col. (1997) Anal. Biochem. 245, 247-248
Use of Polyvinyl Alcohol to Stabilize Binding of Sulfated Glycosaminoglycans to Dimethylmethylene Blue.
Ejemplo 4: Estudio del efecto antihiperalgésico en animales
Para inducir una reacción inflamatoria se inyectó intraarticularmente por un lado una solución de carragenano (CAR, 1 %, 50 pl) en una articulación de rata. El lado no inyectado se consultó para fines de control. Se utilizaron seis animales por grupo. La hinchazón se determinó mediante un micrómetro (medial-lateral en la articulación de la rodilla) y la hiperalgesia térmica se determinó mediante una fuente de luz de infrarrojos dirigida de acuerdo con el método de Hargreaves (Hargreaves y col. 1988) en la parte inferior del pie. Puesto que el lugar de la inflamación (articulación de la rodilla) difiere del lugar de la medición (parte inferior de la pata), en este caso se habla de hiperalgesia térmica secundaria, cuyos mecanismos son importantes para encontrar analgésicos eficaces.
Descripción del experimento de hiperalgesia térmica (prueba de Hargreaves): El animal de experimentación se coloca en una cámara de plástico sobre un cristal de cuarzo. Antes del experimento, primero se le dan al animal de experimentación aproximadamente entre 5 y 15 minutos de tiempo para que se acostumbre al entorno. En cuanto el animal de experimentación, tras la fase de exploración, ya no se mueve tan a menudo (fin de la fase de exploración), se coloca la fuente de luz de infrarrojos, cuyo foco se encuentra en el nivel del suelo de cristal, directamente debajo de la pata trasera que debe estimularse. En este momento se inicia una ejecución del experimento pulsando un botón: a través de los infrarrojos se produce el aumento de temperatura de la piel de la pata trasera. El experimento se termina o bien porque el animal de experimentación levanta la pata trasera (como expresión de haber alcanzado el umbral de dolor) o bien porque se alcanza una temperatura máxima fijada mediante el apagado automático de la fuente de luz de infrarrojos. Mientras el animal de experimentación está sentado quieto, se registra la luz que se refleja de la pata. Si retira la pata, se interrumpe esta reflexión, con lo cual se apaga la fuente de luz de infrarrojos y se registra el tiempo desde el encendido hasta el apagado. El aparato está calibrado de modo que la fuente de luz de infrarrojos aumenta la temperatura de la piel hasta aproximadamente 45 grados Celsius en 10 s (Hargreaves y col. 1988). Para el experimento se utiliza un aparato de la empresa Ugo Basile fabricado para esta finalidad.
El CAR se compró a Sigma-Aldrich. La administración de los inhibidores específicos de catepsina D según la invención se realizó intraarticularmente 30 minutos antes del CAR. Como control positivo se utilizó 10 pg/articulación de triamcinolona (TAC) y como control negativo se utilizó el disolvente (vehículo). La hiperalgesia se indica como la diferencia de los tiempos de retirada entre la pata inflamada y la no inflamada.
Resultado: El TAC pudo reducir la hinchazón inducida por CAR, pero no la pepstatina. A diferencia de esto, la pepstatina pudo reducir la magnitud de la hiperalgesia térmica según la dosis.
Valoración: Se ha podido demostrar que la pepstatina ejerce un efecto antihiperalgésico. Esto puede postularse porque la pepstatina no ha demostrado ningún efecto en la hinchazón inflamatoria y, por lo tanto, en el desencadenante de la hiperalgesia. Por consiguiente, puede aceptarse que la pepstatina muestra un efecto reductor del dolor en humanos.
Ejemplo 5: Estabilidad de la pepstatina en líquido sinovial bovino
1. ) Obtención de líquido sinovial bovino
Para la preparación de explantes bovinos (para la cámara de difusión u otras pruebas) se utilizan o bien pezuñas de vacuno (articulaciones metacarpianas) o bien rodillas de vacuno. El líquido sinovial se puede obtener de las dos articulaciones. Para ello, en el orificio de la articulación se retira con cuidado el líquido sinovial de la articulación con una jeringa de 10 ml y una cánula y se vierte en recipientes Eppendorf de 2 ml ya listos. Los recipientes Eppendorf se rotulan según el animal (identificación del vacuno disponible). Para ello se debe prestar atención a que en la preparación de la articulación no entre sangre en la cavidad articular. Si es así, el líquido sinovial se tiñe de rojo y, por lo tanto, debe desecharse. El líquido sinovial en principio es muy viscoso y tiene un color de transparente a amarillo. Se documenta la extracción junto con un análisis macroscópico del líquido sinovial.
2. ) Planteamiento del análisis de estabilidad de las sustancias en LS
Para estudiar la estabilidad de los compuestos individuales se mezcla un conjunto de 4 líquidos sinoviales bovinos distintos. Para ello se utiliza aproximadamente 1 ml de cada LS. La mezcla se coloca directamente en un recipiente de cristal de 5 ml para minimizar los posibles efectos de absorción. Los LS se mezclan concienzuda pero cuidadosamente. Con ello, no deben formarse burbujas de aire ni espuma. Para ello se utiliza un aparato tipo vórtex al nivel mínimo. Los compuestos de estudio se analizan en una concentración inicial de 1 pM (si no se requiere de otro modo). Tras la adición de la sustancia, se vuelve a realizar una mezcla concienzuda y cuidadosa del lote. Para el control óptico se fotografían todos los lotes de LS y las fotografías se guardan en la carpeta eLabBio del experimento correspondiente. Los lotes se incuban durante 48 h a 37 °C y CO2 al 7,5 % en una incubadora.
3. ) Toma de muestras
La toma de muestras se realiza de acuerdo con los tiempos previamente consensuados (si no se requiere de otro modo, véase abajo). Para ello, se toman cuatro veces respectivamente 200 pl del LS de la mezcla y se transfieren directamente a recipientes Eppendorf «Low-binding» de 0,5 ml. Se utilizan recipientes Eppendorf «Low-binding» para minimizar una interacción de las sustancias con el plástico de los recipientes. En el recipiente Eppendorf ya se habían añadido respectivamente 200 pl de acetonitrilo, de modo que luego se obtiene una mezcla 1 1 del LS. Esto facilita el posterior análisis, pero justo tras la adición del LS puede producirse la precipitación de la proteína. Esto debe anotarse en el protocolo. Justo tras la adición de la sustancia se toma la muestra 0 h. Esto corresponde al valor 100 % del cálculo de la estabilidad. Lo ideal sería que aquí se volviera a encontrar la concentración utilizada. Las muestras pueden congelarse a -20 °C.
• 0 h
• 6 h
• 24 h
• 48 h
Como control negativo se utiliza LS sin sustancia. Como control positivo se utiliza LS con 1 pM de sustancia. Esto corresponde al valor 0 h y, por lo tanto, a una estabilidad del 100 %.
La conservación de las muestras se lleva a cabo en recipientes Eppendorf «Low-binding» a -20 °C. A continuación las muestras se analizan cuantitativamente. La comprobación de las sustancias correspondientes se realiza por espectrometría de masas.
4. ) Procesamientos de los datos
Las concentraciones medidas (ng/ml) se representan en una gráfica (GraphPad Prism®) en función del tiempo. Con esto se determina la estabilidad porcentual de la sustancia. Como valor 100 % se utiliza el valor de partida en el LS en el momento 0 h. Los datos se guardan bajo el correspondiente número de experimento en eLabBio y se comunican al banco de datos MSR (en forma de estabilidad porcentual de acuerdo con los correspondientes tiempos de incubación).
5. ) Resultados
La pepstatina permanece estable en el líquido sinovial durante un periodo de tiempo de al menos dos semanas (véase la Figura 1).
Ejemplo 6: Datos farmacocinéticos tras la inyección intraarticular
Para este estudio (KK-Rat-12-003) se han usado 14 ratas Lister Hooded macho. A todas las ratas se les administró una vez una inyección intraarticular en ambas articulaciones de la rodilla en el momento «0». Una inyección estaba compuesta por 30 pl, en los cuales se habían suspendido homogéneamente aprox. 700 pg de compuesto (Suspensión = Compuesto en Methocel K4M 0,5 % con Tween20 0,25 % en PBS). La suspensión se aplicó mediante una cánula 25G. No se registraron particularidades respecto a posibles modificaciones de los parámetros in vivo, como p. ej. el peso corporal, hinchazones de rodilla o posturas para evitar dolores. En cada momento indicado en la Tabla 2 se mataron animales, se prepararon las articulaciones de rodilla (eliminación de tejidos cutáneos y musculares) y se congelaron para el tratamiento ulterior.
Las articulaciones congeladas se descongelaron al poco rato, se desmenuzaron lo máximo posible con el cincel para huesos y a continuación se agregó el cuádruple de volumen de etanol al 80 %. A continuación se homogeneizó con el Ultraturrax, se agitó el extracto durante 20 minutos a TA y después se almacenó al menos 30 minutos a -20 °C. Después se centrifugó 5 minutos a 13000 rpm, se diluyó una alícuota de 10 pl del sobrenadante con solución estándar interna en proporción 1:5000, se transfirió a una placa de PCR y se analizó.
Se combinaron 20 pl de plasma con 20 pl de solución estándar interna, se añadieron 100 pl de metanol y se agitó durante 5 minutos. Después se almacenaron los extractos al menos 30 minutos a -20 °C y a continuación se centrifugó 5 minutos a 13000 rpm. Se transfirieron 80 pl del sobrenadante a una placa de PCR y se analizaron. Todas las muestras se analizaron con ayuda de un sistema UPLC-MS/MS. Los límites de detección para pepstatina fueron 0,1 ng/ml en plasma y 8 pg/g en tejido.
Tabla 2: diámetro de la articulación de la rodilla en mm
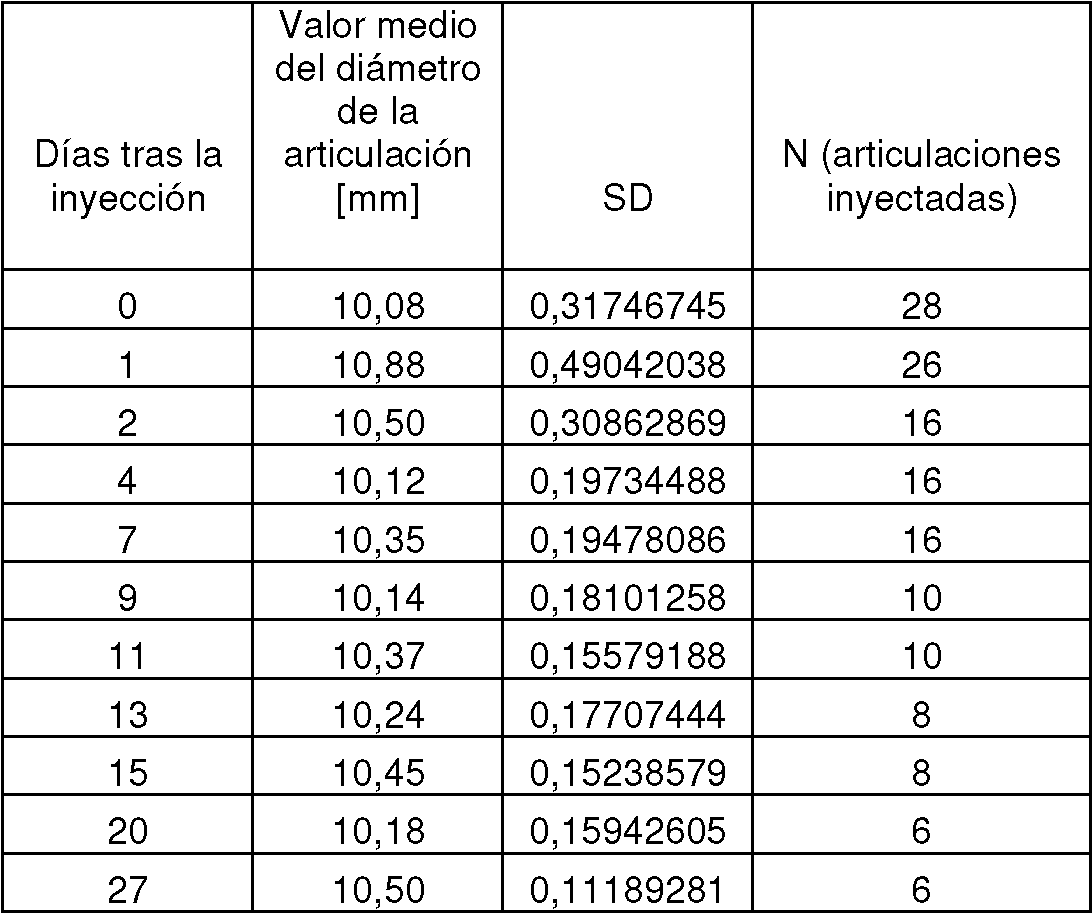
Tras la administración intraarticular de una suspensión, la pepstatina muestra un tiempo de permanencia medio de aprox. 106 horas en la articulación de la rodilla de la rata y, debido a la liberación lenta desde el líquido sinovial y al aclaramiento muy elevado en la sangre, solo una exposición sistemática muy baja. En el plasma, la pepstatina muestra una cinética llamada Flip-Flop, es decir que la semivida del plasma terminal no se determina mediante la eliminación, sino mediante la liberación de pepstatina desde la suspensión y la difusión a través de la membrana sinovial. Se pudo detectar la pepstatina en el plasma, no obstante en muy baja concentración, hasta 28 días tras la administración (véase la Figura 2).
Ejemplo 7: Datos farmacocinéticos tras la administración intravenosa (i.v.) y oral (v.o.)
Los parámetros farmacocinéticos de la pepstatina se determinaron en ratas Wistar (BW de aprox. 250 g), tras la administración de la sustancia de ensayo en un cóctel de hasta 4 sustancias. Se administró pepstatina a las ratas macho (n=3 por tipo de administración) o bien mediante una inyección de bolo i.v. en la vena de la cola o mediante una sonda oral con una cánula de acero inoxidable. Las sustancias de ensayo se disolvieron en DMSO/PEG200/agua (2/60/38 v/v) con una concentración final de 0,8 mg/ml y se administró una dosis de 0,2 mg/kg i.v. y 0,5 mg/kg por vía oral. Las muestras de sangre (200 pl) se tomaron bajo anestesia ligera con isoflurano a través de la vena sublingual en los siguientes momentos tras la administración: iv: 0,1,0,5, 1,2, 4, 6 y 24 h; vo: 0,25, 0,5, 1,2, 4, 6 y 24 h.
Las muestras de sangre se recogieron en tubos de centrifugación con heparina de litio y se centrifugaron 3 minutos a 4 °C con aprox. 10000 g. El plasma resultante se congeló de inmediato a -20 °C y se guardó hasta el análisis. Las concentraciones plasmáticas se determinaron mediante un método LC-MS/MS estándar. Los parámetros farmacocinéticos (Clp, Vee, T1/2, F) se calcularon mediante un análisis NCA.
Tras la administración intravenosa, la pepstatina muestra un aclaramiento muy elevado (> 100 % del flujo sanguíneo del hígado), un volumen de distribución medio y, en consecuencia, una semivida en plasma muy breve (aprox.
0,14 h). Tras la administración oral, todas las concentraciones plasmáticas estaban por debajo del límite de detección (véase la Figura 3).
Ejemplo 8: Eficacia (in vivo ) de la pepstatina en la artrosis: modelo ACLT tMx
Se seleccionaron ratas como animales de ensayo. Tras el afeitado y la desinfección se abre la zona de operación mediante una incisión medial en la piel de aprox. 1 cm de longitud. La cápsula de la articulación de la rodilla se prepara y se presenta el ligamento patelar medial. Tras abrir la cápsula articular, separar los ligamentos parapatelares mediales y desplazar la rótula hacia el lateral, se corta el ligamento cruzado anterior con un bisturí curvo de punta roma (ACLT = Anterior Cruciate Ligament Transection). A continuación, se prepara el ligamento anterior y posterior de fijación del menisco y se corta y se saca el menisco tMx (resectomía del menisco medial). Tras el reposicionamiento de la rótula, finalmente se lava la articulación con solución salina estéril para eliminar los posibles coágulos de sangre formados.
La rótula se reposiciona y el ligamento rotular medial se fija de nuevo mediante sutura continua cerrando la cápsula. La musculatura también se sutura. A continuación se cierra la piel mediante grapas individuales. La duración de la operación es de aprox. 10 minutos. La duración del ensayo postoperatorio fue de 6 semanas.
Para la inyección intraarticular se anestesiaron los animales con isoflurano al 1,5-2 % en volumen. Antes de la inyección, se inyecta buprenorfina por vía subcutánea. La zona de la inyección se afeitó con cuidado y se desinfectó. La articulación de la rodilla se coloca en una posición ligeramente flexionada y se inyecta la sustancia de ensayo o el vehículo en la articulación.
Para el presente ensayo se usó 1 mg de pepstatina en 30 pl. Como control negativo, se inyectó solamente el vehículo en un grupo de ensayo. Como control positivo se usó respectivamente la pata trasera no operada de la rata.
Procesamiento en la histología
Las muestras de tejido tomadas se fijan en paraformaldehído (4 %) durante al menos 72 horas y al cabo de 24 horas se enjuagan con agua corriente. A continuación se descalcifican las muestras mediante Osteosoft durante un periodo de 4 semanas. Después se realiza la infiltración del tejido con parafina y la elaboración de cortes histológicos con 7 pm de grosor. Para la evaluación, se tiñeron los cortes con safranina O Fast Green.
Evaluación
De cada animal se seleccionan dos cortes en la zona de carga y dos personas experimentadas los evaluaron mediante un sistema de evaluación. El sistema de evaluación se basa en el trabajo de V.B. Kraus y col. (Osteoarthritis & Cartilage, 18, pág. 3, 2010).
Resultados
La pepstatina también muestra una eficacia significativa in vivo para la artrosis (véase la Figura 4)
Ejemplo 9: Microdiálisis
Los niveles de medicamento libre en el lugar de actuación son decisivos para el efecto de un medicamento. En las inyecciones realizadas por vía intraarticular, el área de distribución en el líquido sinovial es de gran interés a este respecto.
Se ha usado el procedimiento de microdiálisis, ya que, por una parte, el líquido sinovial representa una matriz compleja para la determinación analítica de péptidos y, por otra parte, solo el medicamento libre, es decir no unido a proteínas, es relevante para el efecto. El eluato obtenido en la microdiálisis permite un análisis de los niveles de medicamento libre de la matriz compleja del líquido sinovial. El motivo es que se usa una membrana de bloqueo (membrana de microdiálisis) que solo pueden atravesar moléculas de hasta un determinado tamaño. Además de la velocidad de flujo del eluato y de la concentración libre disponible del medicamento en el líquido sinovial como fuerza impulsora para el intercambio de sustancias, este «tamaño de poro» determina la concentración del medicamento en el eluato.
Para los presentes experimentos se usaron sondas de microdiálisis de la empresa CMA (número de referencia 000082; CMA 7; muestra de microdiálisis 1 mm 3/pkg) equipadas con una membrana de cuprofano y un tamaño de poro de 6 kDa. El flujo de eluato fue de 0,5 pl/min.
Primero se estudió la cinética de liberación en líquido sinovial de bovino y se comparó con triamcinolona (Triam Injekt® 20 mg), un medicamento aprobado para la inyección intraarticular. Aquí la triamcinolona se usó en suspensión de cristales ya disueltos, mientras que los cristales de pepstatina se añadieron al líquido sinovial directamente y sin disolver. Se emplearon 3 mg de medicamento por ml de líquido sinovial en un recipiente de cristal, lo cual corresponde aproximadamente a las proporciones de concentración tras la inyección intraarticular en
la articulación de la rodilla. Sorprendentemente y a diferencia de la triamcinolona, se midieron niveles muy elevados de pepstatina no unida. Los niveles más o menos constantes también indican una rápida velocidad de disolución con una elevada estabilidad de la pepstatina en el líquido sinovial (véase la Figura 5a).
Debido a la elevada solubilidad de la pepstatina en el líquido sinovial, los resultados de la microdiálisis articular en conejillos de indias (Dunkin Hartley) son sorprendentes: Se pueden encontrar niveles significativos de pepstatina en la articulación hasta 14 días después de la inyección intraarticular de pepstatina [1 mg/articulación administrado en 50 pl como suspensión] y además en casi todos los eluatos examinados (véanse las Figuras 5b y 5c).
Claims (5)
1. Preparación farmacéutica que contenga pepstatina y/o una de sus sales, solvatos y estereoisómeros fisiológicamente compatibles, incluidas sus mezclas en todas las proporciones, y otros vehículos y/o excipientes, para su uso como medicamento en el tratamiento y/o la profilaxis de estados fisiológicos y/o patofisiológicos seleccionados del grupo compuesto por artrosis, lesiones condrales traumáticas, artritis, dolor, alodinia o hiperalgesia, en los que la preparación farmacéutica se administra por vía intraarticular de la siguiente manera: a) desde semanalmente hasta anualmente,
b) desde cada 14 días hasta semestralmente o
c) desde mensualmente hasta trimestralmente.
2. Preparación farmacéutica para el uso de acuerdo con la reivindicación 1 que contiene al menos otro principio activo farmacéutico.
3. Preparación farmacéutica para el uso de acuerdo con las reivindicaciones 1 o 2 para el uso como medicamento en el tratamiento y/o la profilaxis de la artrosis.
4. Pepstatina y/o una de sus sales, solvatos y estereoisómeros fisiológicamente compatibles, incluidas sus mezclas en todas las proporciones, para su uso como medicamento en el tratamiento y/o la profilaxis de estados fisiológicos y/o patofisiológicos seleccionados del grupo compuesto por artrosis, lesiones condrales traumáticas, artritis, dolor, alodinia e hiperalgesia, en los que la pepstatina y/o una de sus sales, solvatos y estereoisómeros fisiológicamente compatibles, incluidas sus mezclas en todas las proporciones, se administra intraarticularmente de la siguiente manera:
a) desde semanalmente hasta anualmente,
b) desde cada 14 días hasta semestralmente o
c) desde mensualmente hasta trimestralmente.
5. La pepstatina y/o una de sus sales, solvatos y estereoisómeros fisiológicamente compatibles, incluidas sus mezclas en todas las proporciones, de acuerdo con la reivindicación 4 para el uso como medicamento en el tratamiento y/o la profilaxis de la artrosis.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP13003923 | 2013-08-06 | ||
| PCT/EP2014/001861 WO2015018472A1 (de) | 2013-08-06 | 2014-07-07 | Intraartikuläre applikation von pepstatin bei arthrose |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2759060T3 true ES2759060T3 (es) | 2020-05-07 |
Family
ID=48948200
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES14741803T Active ES2759060T3 (es) | 2013-08-06 | 2014-07-07 | Administración intraarticular de pepstatina para la artrosis |
Country Status (9)
| Country | Link |
|---|---|
| US (2) | US9750784B2 (es) |
| EP (1) | EP3030250B1 (es) |
| JP (1) | JP6336067B2 (es) |
| CN (1) | CN105451754A (es) |
| AU (1) | AU2014304950B2 (es) |
| CA (1) | CA2920420C (es) |
| ES (1) | ES2759060T3 (es) |
| IL (1) | IL243983B (es) |
| WO (1) | WO2015018472A1 (es) |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP7541746B2 (ja) * | 2019-05-21 | 2024-08-29 | 国立大学法人 東京大学 | 脳腫瘍の検出用蛍光プローブ |
Family Cites Families (26)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3906085A (en) * | 1973-05-21 | 1975-09-16 | Lilly Co Eli | Determination of hypertension associated with elevated renin levels |
| CA1286846C (en) | 1985-02-12 | 1991-07-23 | Catherine Cazaubon | Peptide derivatives which inhibit renin and acid proteases |
| US6251928B1 (en) | 1994-03-16 | 2001-06-26 | Eli Lilly And Company | Treatment of alzheimer's disease employing inhibitors of cathepsin D |
| CA2280096A1 (en) | 1997-02-04 | 1998-08-06 | The Regents Of The University Of California | Nanomolar, non-peptide inhibitors of cathepsin d |
| US5962506A (en) | 1997-07-07 | 1999-10-05 | Pharmacopeia, Inc. | Glycol and hydroxyphosphonate peptidomimetics as inhibitors of aspartyl proteases |
| US5986102A (en) | 1998-04-29 | 1999-11-16 | Pharmacopeia, Inc. | Hydroxypropylamide peptidomimetics as inhibitors of aspartyl proteases |
| JP2000300266A (ja) * | 1999-04-20 | 2000-10-31 | St Marianna Univ School Of Medicine | ポリペプチド及びdna |
| US7132568B2 (en) | 2002-06-17 | 2006-11-07 | Sunesis Pharmaceuticals, Inc. | Aspartyl protease inhibitors |
| ES2323068T3 (es) | 2003-08-08 | 2009-07-06 | Schering Corporation | Inhibidores de bace-1 de aminas ciclicas con un sustituyente de benzamida. |
| EP1654380A4 (en) * | 2003-08-14 | 2009-09-09 | Insight Biopharmaceuticals Ltd | METHODS AND PHARMACEUTICAL COMPOSITIONS FOR MODULATING HEPARANASE ACTIVATION AND USES THEREOF |
| GB0325830D0 (en) | 2003-11-05 | 2003-12-10 | Novartis Ag | Organic compounds |
| JP2007528404A (ja) | 2004-03-09 | 2007-10-11 | エラン ファーマシューティカルズ,インコーポレイテッド | 置換された尿素およびカルバメート、フェナシル−2−ヒドロキシ−3−ジアミノアルカン、ならびにベンズアミド−2−ヒドロキシ−3−ジアミノアルカン系のアスパラギン酸プロテアーゼ阻害薬 |
| US20050239836A1 (en) | 2004-03-09 | 2005-10-27 | Varghese John | Substituted hydroxyethylamine aspartyl protease inhibitors |
| US20080132477A1 (en) | 2005-01-13 | 2008-06-05 | Claudia Betschart | Macrocyclic Compounds Useful as Bace Inhibitors |
| CA2609562A1 (en) | 2005-06-14 | 2006-12-28 | Schering Corporation | Macrocyclic heterocyclic aspartyl protease inhibitors |
| EP1745778A3 (en) | 2005-07-20 | 2007-03-07 | Speedel Experimenta AG | Diaminoalcohols as therapeutic compounds |
| EP1745776A1 (en) | 2005-07-20 | 2007-01-24 | Speedel Experimenta AG | Aminoalcohols as therapeutic compounds |
| EP1745777A1 (en) | 2005-07-20 | 2007-01-24 | Speedel Experimenta AG | Amido-aminoalcohols as therapeutic compounds |
| GB0526614D0 (en) | 2005-12-30 | 2006-02-08 | Novartis Ag | Organic compounds |
| EP1872780A3 (en) | 2006-06-14 | 2008-04-30 | Speedel Experimenta AG | Omega-Phenyloctanamides as therapeutic compounds |
| EP1867329A3 (en) | 2006-06-14 | 2008-05-07 | Speedel Experimenta AG | 5-Amino-4-hydroxy-2-isopropyl-7-[4-methoxy-3-(3-methoxypropoxy)benzyl]-8-methylnonanamides as therapeutic compounds |
| EP1987834A3 (en) | 2007-02-13 | 2008-11-19 | Speedel Experimenta AG | Substituted piperidines as therapeutic compounds |
| WO2008119772A1 (en) | 2007-03-30 | 2008-10-09 | Medivir Ab | Amide derivatives as inhibitors of aspartyl proteases |
| WO2009013293A1 (en) | 2007-07-24 | 2009-01-29 | Novartis Ag | Substituted cyclohexanecarboxamides useful as bace inhibitors |
| MX2013009064A (es) * | 2011-02-08 | 2013-10-01 | Merck Patent Gmbh | Derivados de aminostatina para el tratamiento de artrosis. |
| US20150225369A1 (en) * | 2012-08-29 | 2015-08-13 | Merck Patent Gmbh | Ddr2 inhibitors for the treatment of osteoarthritis |
-
2014
- 2014-07-07 ES ES14741803T patent/ES2759060T3/es active Active
- 2014-07-07 US US14/910,317 patent/US9750784B2/en not_active Expired - Fee Related
- 2014-07-07 AU AU2014304950A patent/AU2014304950B2/en not_active Ceased
- 2014-07-07 WO PCT/EP2014/001861 patent/WO2015018472A1/de not_active Ceased
- 2014-07-07 JP JP2016532244A patent/JP6336067B2/ja not_active Expired - Fee Related
- 2014-07-07 EP EP14741803.2A patent/EP3030250B1/de active Active
- 2014-07-07 CA CA2920420A patent/CA2920420C/en active Active
- 2014-07-07 CN CN201480044142.6A patent/CN105451754A/zh active Pending
-
2016
- 2016-02-04 IL IL243983A patent/IL243983B/en active IP Right Grant
-
2017
- 2017-06-16 US US15/624,777 patent/US20170348380A1/en not_active Abandoned
Also Published As
| Publication number | Publication date |
|---|---|
| EP3030250B1 (de) | 2019-08-28 |
| AU2014304950A1 (en) | 2016-03-17 |
| US20170348380A1 (en) | 2017-12-07 |
| JP6336067B2 (ja) | 2018-06-06 |
| WO2015018472A1 (de) | 2015-02-12 |
| JP2016527284A (ja) | 2016-09-08 |
| AU2014304950B2 (en) | 2019-09-12 |
| EP3030250A1 (de) | 2016-06-15 |
| IL243983B (en) | 2020-04-30 |
| IL243983A0 (en) | 2016-04-21 |
| US20160175381A1 (en) | 2016-06-23 |
| US9750784B2 (en) | 2017-09-05 |
| CA2920420C (en) | 2021-11-23 |
| CN105451754A (zh) | 2016-03-30 |
| CA2920420A1 (en) | 2015-02-12 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US12410170B2 (en) | 2,3,5-trisubstituted pyrazolo[1,5-A]pyrimidine compounds | |
| US7135502B1 (en) | Colchinol derivatives as vascular damaging agents | |
| ES2764452T3 (es) | Inhibidores del proteasoma para tratar un trastorno relacionado con una acumulación de proteína anómala no degradada o un cáncer | |
| ES2711670T3 (es) | Composiciones y métodos para la prevención o tratamiento de la fibrosis pulmonar | |
| ES2509919T3 (es) | Composiciones y métodos para la administración de medicamentos | |
| KR102004559B1 (ko) | 데시타빈 유도체 제제 | |
| ES2847883T3 (es) | Uso de inhibidores de LP-PLA2 en el tratamiento y prevención de enfermedades oculares | |
| US9999655B2 (en) | Use of MKS inhibitor peptide-containing compositions for treating non-small cell lung cancer with same | |
| ES2587863T3 (es) | Uso de un inhibidor de C1 para el tratamiento de un edema secundario del sistema nervioso central | |
| ES2740953T3 (es) | Bloqueo de proteasas inflamatorias con theta-defensinas | |
| US12048750B2 (en) | Tissue-homing peptide conjugates and methods of use thereof | |
| CA2967621A1 (en) | Compositions and methods for preventing or treating diseases, conditions, or processes characterized by aberrant fibroblast proliferation and extracellular matrix deposition | |
| US20120135084A1 (en) | Use of Deuterium Oxide for Treating Viral Diseases of the Eye | |
| WO2021188849A1 (en) | Methods and compositions for treating inflammatory and fibrotic pulmonary disorders | |
| AU2016205125A1 (en) | Formulation of MK2 inhibitor peptides | |
| ES3002189T3 (en) | Para-hydroquinone derivatives as vegf, tnf and/or il inhibitors for the treatment of neuroinflammatory diseases | |
| ES2800162T3 (es) | Uso terapéutico de moduladores funcionales negativos de la eritropoyetina | |
| CN112469411A (zh) | 供在由衰老细胞引起或介导的状况的临床管理中使用和用于治疗癌症的作为Bcl家族拮抗剂的氨基膦酸酯 | |
| ES2759060T3 (es) | Administración intraarticular de pepstatina para la artrosis | |
| ES2627281T3 (es) | Derivados del ácido carboxílico sustituidos como agregados agrecanasa para el tratamiento de la artrosis | |
| CN115120611A (zh) | 一种no供体型胶束组合物及其制备方法与应用 | |
| EP3324950A1 (en) | Treatment for vitiligo | |
| CN104507957B (zh) | 用于治疗关节病的羟基他汀衍生物 | |
| KR20250122469A (ko) | 암 및 대사 질환의 치료를 위한 조성물 및 방법 |