ES2759940T3 - Compuestos de 1,3,4-tiadiazol y su uso para tratar el cáncer - Google Patents
Compuestos de 1,3,4-tiadiazol y su uso para tratar el cáncer Download PDFInfo
- Publication number
- ES2759940T3 ES2759940T3 ES16802102T ES16802102T ES2759940T3 ES 2759940 T3 ES2759940 T3 ES 2759940T3 ES 16802102 T ES16802102 T ES 16802102T ES 16802102 T ES16802102 T ES 16802102T ES 2759940 T3 ES2759940 T3 ES 2759940T3
- Authority
- ES
- Spain
- Prior art keywords
- methoxy
- amino
- pyrrolidin
- thiadiazol
- acetamide
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 206010028980 Neoplasm Diseases 0.000 title claims description 46
- 201000011510 cancer Diseases 0.000 title claims description 40
- 150000004869 1,3,4-thiadiazoles Chemical class 0.000 title description 3
- 150000001875 compounds Chemical class 0.000 claims abstract description 118
- 150000003839 salts Chemical class 0.000 claims abstract description 84
- -1 5-methylpyridazin-3-yl Chemical group 0.000 claims abstract description 63
- 229910052739 hydrogen Inorganic materials 0.000 claims abstract description 15
- 239000001257 hydrogen Substances 0.000 claims abstract description 15
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims abstract description 11
- 125000000876 trifluoromethoxy group Chemical group FC(F)(F)O* 0.000 claims abstract description 9
- 125000001153 fluoro group Chemical group F* 0.000 claims abstract description 7
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 claims abstract description 7
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims abstract description 7
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 claims abstract description 4
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 claims description 26
- 238000011282 treatment Methods 0.000 claims description 25
- DLFVBJFMPXGRIB-UHFFFAOYSA-N Acetamide Chemical compound CC(N)=O DLFVBJFMPXGRIB-UHFFFAOYSA-N 0.000 claims description 16
- 238000002560 therapeutic procedure Methods 0.000 claims description 15
- 125000004207 3-methoxyphenyl group Chemical group [H]C1=C([H])C(*)=C([H])C(OC([H])([H])[H])=C1[H] 0.000 claims description 11
- 125000004575 3-pyrrolidinyl group Chemical group [H]N1C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 claims description 11
- 125000001255 4-fluorophenyl group Chemical group [H]C1=C([H])C(*)=C([H])C([H])=C1F 0.000 claims description 11
- 239000008194 pharmaceutical composition Substances 0.000 claims description 10
- 239000003085 diluting agent Substances 0.000 claims description 8
- 206010006187 Breast cancer Diseases 0.000 claims description 6
- 208000026310 Breast neoplasm Diseases 0.000 claims description 6
- FSLVAZAHHSXHCR-WBMJQRKESA-N (2S)-2-[3-(difluoromethoxy)phenyl]-N-[5-[[(3R)-1-(6-fluoropyridazin-3-yl)pyrrolidin-3-yl]amino]-1,3,4-thiadiazol-2-yl]-2-methoxyacetamide Chemical compound FC(OC=1C=C(C=CC=1)[C@@H](C(=O)NC=1SC(=NN=1)N[C@H]1CN(CC1)C=1N=NC(=CC=1)F)OC)F FSLVAZAHHSXHCR-WBMJQRKESA-N 0.000 claims description 5
- 208000008839 Kidney Neoplasms Diseases 0.000 claims description 5
- 206010058467 Lung neoplasm malignant Diseases 0.000 claims description 5
- 206010061902 Pancreatic neoplasm Diseases 0.000 claims description 5
- 206010038389 Renal cancer Diseases 0.000 claims description 5
- 125000004786 difluoromethoxy group Chemical group [H]C(F)(F)O* 0.000 claims description 5
- 206010073071 hepatocellular carcinoma Diseases 0.000 claims description 5
- 201000010982 kidney cancer Diseases 0.000 claims description 5
- 208000014018 liver neoplasm Diseases 0.000 claims description 5
- 201000005202 lung cancer Diseases 0.000 claims description 5
- 208000020816 lung neoplasm Diseases 0.000 claims description 5
- 208000015486 malignant pancreatic neoplasm Diseases 0.000 claims description 5
- 201000002528 pancreatic cancer Diseases 0.000 claims description 5
- 208000008443 pancreatic carcinoma Diseases 0.000 claims description 5
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 5
- SKUZHMFUXHGQQW-DYVFJYSZSA-N (2S)-N-[5-[[(3R)-1-(6-fluoropyridazin-3-yl)pyrrolidin-3-yl]amino]-1,3,4-thiadiazol-2-yl]-2-methoxy-2-(3-methoxyphenyl)acetamide Chemical compound FC1=CC=C(N=N1)N1C[C@@H](CC1)NC1=NN=C(S1)NC([C@H](C1=CC(=CC=C1)OC)OC)=O SKUZHMFUXHGQQW-DYVFJYSZSA-N 0.000 claims description 3
- BILNGVPSJLOCKW-CJNGLKHVSA-N (2S)-N-[5-[[(3R)-1-(6-fluoropyridazin-3-yl)pyrrolidin-3-yl]amino]-1,3,4-thiadiazol-2-yl]-2-methoxy-2-phenylacetamide Chemical compound FC1=CC=C(N=N1)N1C[C@@H](CC1)NC1=NN=C(S1)NC([C@H](C1=CC=CC=C1)OC)=O BILNGVPSJLOCKW-CJNGLKHVSA-N 0.000 claims description 3
- CQLPOXGXACFBIW-PBHICJAKSA-N (2S)-2-methoxy-N-[5-[[(3R)-1-(6-methylpyridazin-3-yl)pyrrolidin-3-yl]amino]-1,3,4-thiadiazol-2-yl]-2-[3-(trifluoromethoxy)phenyl]acetamide Chemical compound CO[C@H](C(=O)NC=1SC(=NN=1)N[C@H]1CN(CC1)C=1N=NC(=CC=1)C)C1=CC(=CC=C1)OC(F)(F)F CQLPOXGXACFBIW-PBHICJAKSA-N 0.000 claims description 2
- HXFKMUYISOOFNN-WBVHZDCISA-N (2S)-2-(4-fluorophenyl)-2-methoxy-N-[5-[[(3R)-1-(6-methylpyridazin-3-yl)pyrrolidin-3-yl]amino]-1,3,4-thiadiazol-2-yl]acetamide Chemical compound FC1=CC=C(C=C1)[C@@H](C(=O)NC=1SC(=NN=1)N[C@H]1CN(CC1)C=1N=NC(=CC=1)C)OC HXFKMUYISOOFNN-WBVHZDCISA-N 0.000 claims 2
- CJPDRLNNFXCJTE-QAPCUYQASA-N (2S)-2-methoxy-2-(3-methoxyphenyl)-N-[5-[[(3R)-1-(6-methylpyridazin-3-yl)pyrrolidin-3-yl]amino]-1,3,4-thiadiazol-2-yl]acetamide Chemical compound CO[C@H](C(=O)NC=1SC(=NN=1)N[C@H]1CN(CC1)C=1N=NC(=CC=1)C)C1=CC(=CC=C1)OC CJPDRLNNFXCJTE-QAPCUYQASA-N 0.000 claims 2
- QFAHGXFMNKFWKL-KDOFPFPSSA-N (2S)-2-(4-fluoro-3-methoxyphenyl)-2-methoxy-N-[5-[[(3R)-1-(6-methylpyridazin-3-yl)pyrrolidin-3-yl]amino]-1,3,4-thiadiazol-2-yl]acetamide Chemical compound FC1=C(C=C(C=C1)[C@@H](C(=O)NC=1SC(=NN=1)N[C@H]1CN(CC1)C=1N=NC(=CC=1)C)OC)OC QFAHGXFMNKFWKL-KDOFPFPSSA-N 0.000 claims 1
- NKPBNEQDGYAJFW-WBVHZDCISA-N (2S)-2-(4-fluorophenyl)-2-methoxy-N-[5-[[(3R)-1-(5-methylpyridazin-3-yl)pyrrolidin-3-yl]amino]-1,3,4-thiadiazol-2-yl]acetamide Chemical compound FC1=CC=C(C=C1)[C@@H](C(=O)NC=1SC(=NN=1)N[C@H]1CN(CC1)C=1N=NC=C(C=1)C)OC NKPBNEQDGYAJFW-WBVHZDCISA-N 0.000 claims 1
- YNPNZTXNASCQKK-UHFFFAOYSA-N Phenanthrene Natural products C1=CC=C2C3=CC=CC=C3C=CC2=C1 YNPNZTXNASCQKK-UHFFFAOYSA-N 0.000 claims 1
- DGEZNRSVGBDHLK-UHFFFAOYSA-N [1,10]phenanthroline Chemical compound C1=CN=C2C3=NC=CC=C3C=CC2=C1 DGEZNRSVGBDHLK-UHFFFAOYSA-N 0.000 claims 1
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 214
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 71
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 55
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 54
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 49
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 47
- 239000000203 mixture Substances 0.000 description 44
- 239000000543 intermediate Substances 0.000 description 41
- 238000005160 1H NMR spectroscopy Methods 0.000 description 40
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 39
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical group CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 38
- 108010073324 Glutaminase Proteins 0.000 description 35
- 102000009127 Glutaminase Human genes 0.000 description 35
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 35
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 34
- KWYUFKZDYYNOTN-UHFFFAOYSA-M potassium hydroxide Inorganic materials [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 29
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 28
- 239000012043 crude product Substances 0.000 description 27
- 235000019439 ethyl acetate Nutrition 0.000 description 27
- 239000000243 solution Substances 0.000 description 25
- 239000000377 silicon dioxide Substances 0.000 description 23
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 22
- 239000000047 product Substances 0.000 description 22
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 20
- 239000013067 intermediate product Substances 0.000 description 20
- 230000002829 reductive effect Effects 0.000 description 20
- 239000007787 solid Substances 0.000 description 19
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 17
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 17
- 229910052681 coesite Inorganic materials 0.000 description 17
- 229910052906 cristobalite Inorganic materials 0.000 description 17
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 17
- 238000002953 preparative HPLC Methods 0.000 description 17
- 235000012239 silicon dioxide Nutrition 0.000 description 17
- 229910052682 stishovite Inorganic materials 0.000 description 17
- 229910052905 tridymite Inorganic materials 0.000 description 17
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 16
- 201000010099 disease Diseases 0.000 description 16
- 238000012360 testing method Methods 0.000 description 16
- 241001465754 Metazoa Species 0.000 description 15
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 14
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 14
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 14
- 235000019253 formic acid Nutrition 0.000 description 14
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 13
- 239000007821 HATU Substances 0.000 description 13
- 210000004027 cell Anatomy 0.000 description 13
- DIKBFYAXUHHXCS-UHFFFAOYSA-N bromoform Chemical compound BrC(Br)Br DIKBFYAXUHHXCS-UHFFFAOYSA-N 0.000 description 12
- 230000000694 effects Effects 0.000 description 12
- 239000012071 phase Substances 0.000 description 12
- 238000003756 stirring Methods 0.000 description 11
- 239000000126 substance Substances 0.000 description 11
- 230000000259 anti-tumor effect Effects 0.000 description 10
- ZDXPYRJPNDTMRX-UHFFFAOYSA-N glutamine Natural products OC(=O)C(N)CCC(N)=O ZDXPYRJPNDTMRX-UHFFFAOYSA-N 0.000 description 10
- 238000000034 method Methods 0.000 description 10
- 239000011541 reaction mixture Substances 0.000 description 10
- ROSDSFDQCJNGOL-UHFFFAOYSA-N Dimethylamine Chemical compound CNC ROSDSFDQCJNGOL-UHFFFAOYSA-N 0.000 description 9
- WMFOQBRAJBCJND-UHFFFAOYSA-M Lithium hydroxide Chemical compound [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 description 9
- 238000003556 assay Methods 0.000 description 9
- 239000003921 oil Substances 0.000 description 9
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 8
- 235000019341 magnesium sulphate Nutrition 0.000 description 8
- 229910052757 nitrogen Inorganic materials 0.000 description 8
- 229910000069 nitrogen hydride Inorganic materials 0.000 description 8
- HEMHJVSKTPXQMS-UHFFFAOYSA-M sodium hydroxide Inorganic materials [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 8
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 7
- 238000006243 chemical reaction Methods 0.000 description 7
- 238000004255 ion exchange chromatography Methods 0.000 description 7
- 230000001404 mediated effect Effects 0.000 description 7
- 239000002904 solvent Substances 0.000 description 7
- LMDZBCPBFSXMTL-UHFFFAOYSA-N 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide Chemical compound CCN=C=NCCCN(C)C LMDZBCPBFSXMTL-UHFFFAOYSA-N 0.000 description 6
- MJQJGHVSTUVGEA-UHFFFAOYSA-N 2-methoxy-2-(3-methoxyphenyl)acetic acid Chemical compound COC(C(O)=O)C1=CC=CC(OC)=C1 MJQJGHVSTUVGEA-UHFFFAOYSA-N 0.000 description 6
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 6
- BAVYZALUXZFZLV-UHFFFAOYSA-N Methylamine Chemical compound NC BAVYZALUXZFZLV-UHFFFAOYSA-N 0.000 description 6
- YNAVUWVOSKDBBP-UHFFFAOYSA-N Morpholine Chemical compound C1COCCN1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 description 6
- 238000005481 NMR spectroscopy Methods 0.000 description 6
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 6
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 6
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 6
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 6
- 239000002253 acid Substances 0.000 description 6
- 239000008346 aqueous phase Substances 0.000 description 6
- 229950005228 bromoform Drugs 0.000 description 6
- 239000000706 filtrate Substances 0.000 description 6
- 238000004519 manufacturing process Methods 0.000 description 6
- 230000004044 response Effects 0.000 description 6
- 239000012258 stirred mixture Substances 0.000 description 6
- GETQZCLCWQTVFV-UHFFFAOYSA-N trimethylamine Chemical compound CN(C)C GETQZCLCWQTVFV-UHFFFAOYSA-N 0.000 description 6
- ORJYDQMXUNVTGW-ZCFIWIBFSA-N 2-N-[(3R)-1-(6-fluoropyridazin-3-yl)pyrrolidin-3-yl]-1,3,4-thiadiazole-2,5-diamine Chemical compound FC1=CC=C(N=N1)N1C[C@@H](CC1)NC=1SC(=NN=1)N ORJYDQMXUNVTGW-ZCFIWIBFSA-N 0.000 description 5
- XURVCCBHXFSSSU-UHFFFAOYSA-N 2-[3-(difluoromethoxy)phenyl]-2-methoxyacetic acid Chemical compound FC(OC=1C=C(C=CC=1)C(C(=O)O)OC)F XURVCCBHXFSSSU-UHFFFAOYSA-N 0.000 description 5
- WMPDAIZRQDCGFH-UHFFFAOYSA-N 3-methoxybenzaldehyde Chemical compound COC1=CC=CC(C=O)=C1 WMPDAIZRQDCGFH-UHFFFAOYSA-N 0.000 description 5
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 5
- 208000003721 Triple Negative Breast Neoplasms Diseases 0.000 description 5
- 235000011114 ammonium hydroxide Nutrition 0.000 description 5
- 238000004458 analytical method Methods 0.000 description 5
- 239000002585 base Substances 0.000 description 5
- 239000012230 colorless oil Substances 0.000 description 5
- 238000001914 filtration Methods 0.000 description 5
- 229930195712 glutamate Natural products 0.000 description 5
- 230000005764 inhibitory process Effects 0.000 description 5
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 5
- 239000002244 precipitate Substances 0.000 description 5
- DQQJBEAXSOOCPG-SSDOTTSWSA-N tert-butyl n-[(3r)-pyrrolidin-3-yl]carbamate Chemical compound CC(C)(C)OC(=O)N[C@@H]1CCNC1 DQQJBEAXSOOCPG-SSDOTTSWSA-N 0.000 description 5
- 208000022679 triple-negative breast carcinoma Diseases 0.000 description 5
- XSSNXGQLLDQUCF-UHFFFAOYSA-N 2-(4-fluorophenyl)-2-methoxyacetic acid Chemical compound COC(C(O)=O)C1=CC=C(F)C=C1 XSSNXGQLLDQUCF-UHFFFAOYSA-N 0.000 description 4
- PHHFVHIMNXTSKE-MRVPVSSYSA-N 2-N-[(3R)-1-(5-methylpyridazin-3-yl)pyrrolidin-3-yl]-1,3,4-thiadiazole-2,5-diamine Chemical compound CC=1C=C(N=NC=1)N1C[C@@H](CC1)NC=1SC(=NN=1)N PHHFVHIMNXTSKE-MRVPVSSYSA-N 0.000 description 4
- WHGLWFDLHAFGMG-UHFFFAOYSA-N 2-methoxy-2-(4-methoxyphenyl)acetic acid Chemical compound COC(C(O)=O)C1=CC=C(OC)C=C1 WHGLWFDLHAFGMG-UHFFFAOYSA-N 0.000 description 4
- GLYQQFBHCFPEEU-UHFFFAOYSA-N 5-bromo-1,3,4-thiadiazol-2-amine Chemical compound NC1=NN=C(Br)S1 GLYQQFBHCFPEEU-UHFFFAOYSA-N 0.000 description 4
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 4
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 4
- 239000007832 Na2SO4 Substances 0.000 description 4
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 4
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 4
- 239000007983 Tris buffer Substances 0.000 description 4
- 239000000908 ammonium hydroxide Substances 0.000 description 4
- 239000002552 dosage form Substances 0.000 description 4
- 239000003814 drug Substances 0.000 description 4
- 239000003480 eluent Substances 0.000 description 4
- 150000002431 hydrogen Chemical group 0.000 description 4
- 208000037819 metastatic cancer Diseases 0.000 description 4
- 208000011575 metastatic malignant neoplasm Diseases 0.000 description 4
- 208000002154 non-small cell lung carcinoma Diseases 0.000 description 4
- 239000012044 organic layer Substances 0.000 description 4
- BASFCYQUMIYNBI-UHFFFAOYSA-N platinum Chemical group [Pt] BASFCYQUMIYNBI-UHFFFAOYSA-N 0.000 description 4
- 238000011321 prophylaxis Methods 0.000 description 4
- 229910052938 sodium sulfate Inorganic materials 0.000 description 4
- 235000011152 sodium sulphate Nutrition 0.000 description 4
- HWVZIEOTWRUGJU-SECBINFHSA-N tert-butyl N-[(3R)-1-(6-fluoropyridazin-3-yl)pyrrolidin-3-yl]carbamate Chemical compound FC1=CC=C(N=N1)N1C[C@@H](CC1)NC(OC(C)(C)C)=O HWVZIEOTWRUGJU-SECBINFHSA-N 0.000 description 4
- 208000029729 tumor suppressor gene on chromosome 11 Diseases 0.000 description 4
- MJQJGHVSTUVGEA-VIFPVBQESA-N (2S)-2-methoxy-2-(3-methoxyphenyl)acetic acid Chemical compound CO[C@H](C(=O)O)C1=CC(=CC=C1)OC MJQJGHVSTUVGEA-VIFPVBQESA-N 0.000 description 3
- DIWVBIXQCNRCFE-QMMMGPOBSA-N (2s)-2-methoxy-2-phenylacetic acid Chemical compound CO[C@H](C(O)=O)C1=CC=CC=C1 DIWVBIXQCNRCFE-QMMMGPOBSA-N 0.000 description 3
- FVRBISVFTJTIRZ-SSDOTTSWSA-N (3R)-1-(5-chloropyridazin-3-yl)pyrrolidin-3-amine Chemical compound ClC=1C=C(N=NC=1)N1C[C@@H](CC1)N FVRBISVFTJTIRZ-SSDOTTSWSA-N 0.000 description 3
- NKABOLBGITYRPF-ZCFIWIBFSA-N (3R)-1-(6-fluoropyridazin-3-yl)pyrrolidin-3-amine Chemical compound FC1=CC=C(N=N1)N1C[C@@H](CC1)N NKABOLBGITYRPF-ZCFIWIBFSA-N 0.000 description 3
- PKYCWFICOKSIHZ-UHFFFAOYSA-N 1-(3,7-dihydroxyphenoxazin-10-yl)ethanone Chemical compound OC1=CC=C2N(C(=O)C)C3=CC=C(O)C=C3OC2=C1 PKYCWFICOKSIHZ-UHFFFAOYSA-N 0.000 description 3
- QHRHBSMXGXTKKG-UHFFFAOYSA-N 2-(4-fluoro-3-methoxyphenyl)-2-methoxyacetic acid Chemical compound COC(C(O)=O)C1=CC=C(F)C(OC)=C1 QHRHBSMXGXTKKG-UHFFFAOYSA-N 0.000 description 3
- WAHJZZBUOUKKRC-UHFFFAOYSA-N 2-ethoxy-2-(3-methoxyphenyl)acetic acid Chemical compound CCOC(C(O)=O)C1=CC=CC(OC)=C1 WAHJZZBUOUKKRC-UHFFFAOYSA-N 0.000 description 3
- 125000000954 2-hydroxyethyl group Chemical group [H]C([*])([H])C([H])([H])O[H] 0.000 description 3
- XCKBQIZWZWJISI-UHFFFAOYSA-N 2-methoxy-2-[3-(trifluoromethoxy)phenyl]acetic acid Chemical compound COC(C(O)=O)C1=CC=CC(OC(F)(F)F)=C1 XCKBQIZWZWJISI-UHFFFAOYSA-N 0.000 description 3
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 3
- XDTMQSROBMDMFD-UHFFFAOYSA-N Cyclohexane Chemical compound C1CCCCC1 XDTMQSROBMDMFD-UHFFFAOYSA-N 0.000 description 3
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 3
- 102000004316 Oxidoreductases Human genes 0.000 description 3
- 108090000854 Oxidoreductases Proteins 0.000 description 3
- 102000001708 Protein Isoforms Human genes 0.000 description 3
- 108010029485 Protein Isoforms Proteins 0.000 description 3
- RWRDLPDLKQPQOW-UHFFFAOYSA-N Pyrrolidine Chemical compound C1CCNC1 RWRDLPDLKQPQOW-UHFFFAOYSA-N 0.000 description 3
- 229940123237 Taxane Drugs 0.000 description 3
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 3
- 150000001412 amines Chemical class 0.000 description 3
- 150000001413 amino acids Chemical class 0.000 description 3
- 239000012131 assay buffer Substances 0.000 description 3
- SRSXLGNVWSONIS-UHFFFAOYSA-N benzenesulfonic acid Chemical compound OS(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-N 0.000 description 3
- 229940092714 benzenesulfonic acid Drugs 0.000 description 3
- CMRXOLCCAGRRDY-INIZCTEOSA-N benzyl (2S)-2-methoxy-2-(3-methoxyphenyl)acetate Chemical compound CO[C@H](C(=O)OCC1=CC=CC=C1)C1=CC(OC)=CC=C1 CMRXOLCCAGRRDY-INIZCTEOSA-N 0.000 description 3
- AXCZMVOFGPJBDE-UHFFFAOYSA-L calcium dihydroxide Chemical compound [OH-].[OH-].[Ca+2] AXCZMVOFGPJBDE-UHFFFAOYSA-L 0.000 description 3
- 229910052799 carbon Inorganic materials 0.000 description 3
- 230000001413 cellular effect Effects 0.000 description 3
- 239000006071 cream Substances 0.000 description 3
- 238000010790 dilution Methods 0.000 description 3
- 239000012895 dilution Substances 0.000 description 3
- 238000001704 evaporation Methods 0.000 description 3
- 230000008020 evaporation Effects 0.000 description 3
- 239000006260 foam Substances 0.000 description 3
- 239000001963 growth medium Substances 0.000 description 3
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 3
- VTHJTEIRLNZDEV-UHFFFAOYSA-L magnesium dihydroxide Chemical compound [OH-].[OH-].[Mg+2] VTHJTEIRLNZDEV-UHFFFAOYSA-L 0.000 description 3
- 239000000347 magnesium hydroxide Substances 0.000 description 3
- 229910001862 magnesium hydroxide Inorganic materials 0.000 description 3
- 229940098779 methanesulfonic acid Drugs 0.000 description 3
- QYKSWSCZFJBWRQ-UHFFFAOYSA-N methyl 2-(4-fluorophenyl)-2-methoxyacetate Chemical compound COC(=O)C(OC)C1=CC=C(F)C=C1 QYKSWSCZFJBWRQ-UHFFFAOYSA-N 0.000 description 3
- 239000003607 modifier Substances 0.000 description 3
- ZRSNZINYAWTAHE-UHFFFAOYSA-N p-methoxybenzaldehyde Chemical compound COC1=CC=C(C=O)C=C1 ZRSNZINYAWTAHE-UHFFFAOYSA-N 0.000 description 3
- 239000000843 powder Substances 0.000 description 3
- 108090000623 proteins and genes Proteins 0.000 description 3
- 208000024891 symptom Diseases 0.000 description 3
- DKPFODGZWDEEBT-QFIAKTPHSA-N taxane Chemical class C([C@]1(C)CCC[C@@H](C)[C@H]1C1)C[C@H]2[C@H](C)CC[C@@H]1C2(C)C DKPFODGZWDEEBT-QFIAKTPHSA-N 0.000 description 3
- SNGBHKJGYJKESG-SNVBAGLBSA-N tert-butyl N-[(3R)-1-(5-chloropyridazin-3-yl)pyrrolidin-3-yl]carbamate Chemical compound ClC=1C=C(N=NC=1)N1C[C@@H](CC1)NC(OC(C)(C)C)=O SNGBHKJGYJKESG-SNVBAGLBSA-N 0.000 description 3
- JJRZOYMJIJXTMO-LLVKDONJSA-N tert-butyl n-[(3r)-1-(6-methylpyridazin-3-yl)pyrrolidin-3-yl]carbamate Chemical compound N1=NC(C)=CC=C1N1C[C@H](NC(=O)OC(C)(C)C)CC1 JJRZOYMJIJXTMO-LLVKDONJSA-N 0.000 description 3
- 239000003643 water by type Substances 0.000 description 3
- FSLVAZAHHSXHCR-MLGOLLRUSA-N (2R)-2-[3-(difluoromethoxy)phenyl]-N-[5-[[(3R)-1-(6-fluoropyridazin-3-yl)pyrrolidin-3-yl]amino]-1,3,4-thiadiazol-2-yl]-2-methoxyacetamide Chemical compound FC(OC=1C=C(C=CC=1)[C@H](C(=O)NC=1SC(=NN=1)N[C@H]1CN(CC1)C=1N=NC(=CC=1)F)OC)F FSLVAZAHHSXHCR-MLGOLLRUSA-N 0.000 description 2
- FURBCBVZJOABRQ-PBHICJAKSA-N (2S)-N-[5-[[(3R)-1-(5-chloropyridazin-3-yl)pyrrolidin-3-yl]amino]-1,3,4-thiadiazol-2-yl]-2-methoxy-2-(4-methoxyphenyl)acetamide Chemical compound ClC=1C=C(N=NC=1)N1C[C@@H](CC1)NC1=NN=C(S1)NC([C@H](C1=CC=C(C=C1)OC)OC)=O FURBCBVZJOABRQ-PBHICJAKSA-N 0.000 description 2
- QFAHGXFMNKFWKL-IKJXHCRLSA-N 2-(4-fluoro-3-methoxyphenyl)-2-methoxy-N-[5-[[(3R)-1-(6-methylpyridazin-3-yl)pyrrolidin-3-yl]amino]-1,3,4-thiadiazol-2-yl]acetamide Chemical compound FC1=C(C=C(C=C1)C(C(=O)NC=1SC(=NN=1)N[C@H]1CN(CC1)C=1N=NC(=CC=1)C)OC)OC QFAHGXFMNKFWKL-IKJXHCRLSA-N 0.000 description 2
- PWHLVMVNBKXNML-VTBWFHPJSA-N 2-ethoxy-2-(3-methoxyphenyl)-N-[5-[[(3R)-1-(6-methylpyridazin-3-yl)pyrrolidin-3-yl]amino]-1,3,4-thiadiazol-2-yl]acetamide Chemical compound C(C)OC(C(=O)NC=1SC(=NN=1)N[C@H]1CN(CC1)C=1N=NC(=CC=1)C)C1=CC(=CC=C1)OC PWHLVMVNBKXNML-VTBWFHPJSA-N 0.000 description 2
- YEDUAINPPJYDJZ-UHFFFAOYSA-N 2-hydroxybenzothiazole Chemical compound C1=CC=C2SC(O)=NC2=C1 YEDUAINPPJYDJZ-UHFFFAOYSA-N 0.000 description 2
- RVECZHFENFMRHJ-UHFFFAOYSA-N 3,6-difluoropyridazine Chemical compound FC1=CC=C(F)N=N1 RVECZHFENFMRHJ-UHFFFAOYSA-N 0.000 description 2
- HFIUSWPRDIPIPN-UHFFFAOYSA-N 3-(difluoromethoxy)benzaldehyde Chemical compound FC(F)OC1=CC=CC(C=O)=C1 HFIUSWPRDIPIPN-UHFFFAOYSA-N 0.000 description 2
- 125000004172 4-methoxyphenyl group Chemical group [H]C1=C([H])C(OC([H])([H])[H])=C([H])C([H])=C1* 0.000 description 2
- 0 C*c1ccc([C@](C(Nc2nnc(N[C@@](CC3)CN3c3cc(Cl)cnn3)[s]2)=O)OC)cc1 Chemical compound C*c1ccc([C@](C(Nc2nnc(N[C@@](CC3)CN3c3cc(Cl)cnn3)[s]2)=O)OC)cc1 0.000 description 2
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 2
- 102000004190 Enzymes Human genes 0.000 description 2
- 108090000790 Enzymes Proteins 0.000 description 2
- 244000166102 Eucalyptus leucoxylon Species 0.000 description 2
- 235000004694 Eucalyptus leucoxylon Nutrition 0.000 description 2
- 108010001336 Horseradish Peroxidase Proteins 0.000 description 2
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 2
- 206010027476 Metastases Diseases 0.000 description 2
- PCLIMKBDDGJMGD-UHFFFAOYSA-N N-bromosuccinimide Chemical compound BrN1C(=O)CCC1=O PCLIMKBDDGJMGD-UHFFFAOYSA-N 0.000 description 2
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 2
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 2
- DBMJMQXJHONAFJ-UHFFFAOYSA-M Sodium laurylsulphate Chemical compound [Na+].CCCCCCCCCCCCOS([O-])(=O)=O DBMJMQXJHONAFJ-UHFFFAOYSA-M 0.000 description 2
- ZSLZBFCDCINBPY-ZSJPKINUSA-N acetyl-CoA Chemical compound O[C@@H]1[C@H](OP(O)(O)=O)[C@@H](COP(O)(=O)OP(O)(=O)OCC(C)(C)[C@@H](O)C(=O)NCCC(=O)NCCSC(=O)C)O[C@H]1N1C2=NC=NC(N)=C2N=C1 ZSLZBFCDCINBPY-ZSJPKINUSA-N 0.000 description 2
- 229910021529 ammonia Inorganic materials 0.000 description 2
- 230000001093 anti-cancer Effects 0.000 description 2
- 239000012298 atmosphere Substances 0.000 description 2
- AGEZXYOZHKGVCM-UHFFFAOYSA-N benzyl bromide Chemical compound BrCC1=CC=CC=C1 AGEZXYOZHKGVCM-UHFFFAOYSA-N 0.000 description 2
- 239000000872 buffer Substances 0.000 description 2
- 239000000920 calcium hydroxide Substances 0.000 description 2
- 229910001861 calcium hydroxide Inorganic materials 0.000 description 2
- 229910002092 carbon dioxide Inorganic materials 0.000 description 2
- 238000001516 cell proliferation assay Methods 0.000 description 2
- 238000002512 chemotherapy Methods 0.000 description 2
- DEFVIWRASFVYLL-UHFFFAOYSA-N ethylene glycol bis(2-aminoethyl)tetraacetic acid Chemical compound OC(=O)CN(CC(O)=O)CCOCCOCCN(CC(O)=O)CC(O)=O DEFVIWRASFVYLL-UHFFFAOYSA-N 0.000 description 2
- 238000003818 flash chromatography Methods 0.000 description 2
- 230000012010 growth Effects 0.000 description 2
- 238000004128 high performance liquid chromatography Methods 0.000 description 2
- 239000003112 inhibitor Substances 0.000 description 2
- 150000007529 inorganic bases Chemical class 0.000 description 2
- 230000003993 interaction Effects 0.000 description 2
- 230000000155 isotopic effect Effects 0.000 description 2
- JVTAAEKCZFNVCJ-UHFFFAOYSA-N lactic acid Chemical compound CC(O)C(O)=O JVTAAEKCZFNVCJ-UHFFFAOYSA-N 0.000 description 2
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 2
- 238000005259 measurement Methods 0.000 description 2
- 230000004060 metabolic process Effects 0.000 description 2
- 150000007522 mineralic acids Chemical class 0.000 description 2
- 230000000683 nonmetastatic effect Effects 0.000 description 2
- 230000003287 optical effect Effects 0.000 description 2
- 150000007524 organic acids Chemical class 0.000 description 2
- 150000007530 organic bases Chemical class 0.000 description 2
- 239000000546 pharmaceutical excipient Substances 0.000 description 2
- 229910052697 platinum Inorganic materials 0.000 description 2
- 229910000027 potassium carbonate Inorganic materials 0.000 description 2
- 102000004169 proteins and genes Human genes 0.000 description 2
- 238000001959 radiotherapy Methods 0.000 description 2
- 239000011734 sodium Substances 0.000 description 2
- 238000001356 surgical procedure Methods 0.000 description 2
- 239000000725 suspension Substances 0.000 description 2
- 239000003826 tablet Substances 0.000 description 2
- WJKHJLXJJJATHN-UHFFFAOYSA-N triflic anhydride Chemical compound FC(F)(F)S(=O)(=O)OS(=O)(=O)C(F)(F)F WJKHJLXJJJATHN-UHFFFAOYSA-N 0.000 description 2
- 210000004881 tumor cell Anatomy 0.000 description 2
- 230000004614 tumor growth Effects 0.000 description 2
- NKPBNEQDGYAJFW-NVXWUHKLSA-N (2R)-2-(4-fluorophenyl)-2-methoxy-N-[5-[[(3R)-1-(5-methylpyridazin-3-yl)pyrrolidin-3-yl]amino]-1,3,4-thiadiazol-2-yl]acetamide Chemical compound FC1=CC=C(C=C1)[C@H](C(=O)NC=1SC(=NN=1)N[C@H]1CN(CC1)C=1N=NC=C(C=1)C)OC NKPBNEQDGYAJFW-NVXWUHKLSA-N 0.000 description 1
- HXFKMUYISOOFNN-NVXWUHKLSA-N (2R)-2-(4-fluorophenyl)-2-methoxy-N-[5-[[(3R)-1-(6-methylpyridazin-3-yl)pyrrolidin-3-yl]amino]-1,3,4-thiadiazol-2-yl]acetamide Chemical compound FC1=CC=C(C=C1)[C@H](C(=O)NC=1SC(=NN=1)N[C@H]1CN(CC1)C=1N=NC(=CC=1)C)OC HXFKMUYISOOFNN-NVXWUHKLSA-N 0.000 description 1
- PWHLVMVNBKXNML-VQIMIIECSA-N (2R)-2-ethoxy-2-(3-methoxyphenyl)-N-[5-[[(3R)-1-(6-methylpyridazin-3-yl)pyrrolidin-3-yl]amino]-1,3,4-thiadiazol-2-yl]acetamide Chemical compound C(C)O[C@@H](C(=O)NC=1SC(=NN=1)N[C@H]1CN(CC1)C=1N=NC(=CC=1)C)C1=CC(=CC=C1)OC PWHLVMVNBKXNML-VQIMIIECSA-N 0.000 description 1
- IPQZUSSGKYCXIB-PBHICJAKSA-N (2S)-2-[3-(difluoromethoxy)phenyl]-2-methoxy-N-[5-[[(3R)-1-(6-methylpyridazin-3-yl)pyrrolidin-3-yl]amino]-1,3,4-thiadiazol-2-yl]acetamide Chemical compound FC(OC=1C=C(C=CC=1)[C@@H](C(=O)NC=1SC(=NN=1)N[C@H]1CN(CC1)C=1N=NC(=CC=1)C)OC)F IPQZUSSGKYCXIB-PBHICJAKSA-N 0.000 description 1
- YKVDUPZYMXZIGX-WBVHZDCISA-N (2S)-2-methoxy-N-[5-[[(3R)-1-(5-methylpyridazin-3-yl)pyrrolidin-3-yl]amino]-1,3,4-thiadiazol-2-yl]-2-phenylacetamide Chemical compound CO[C@H](C(=O)NC=1SC(=NN=1)N[C@H]1CN(CC1)C=1N=NC=C(C=1)C)C1=CC=CC=C1 YKVDUPZYMXZIGX-WBVHZDCISA-N 0.000 description 1
- MGGFZUWCVPJHDX-MRVPVSSYSA-N (3r)-1-(6-methylpyridazin-3-yl)pyrrolidin-3-amine Chemical compound N1=NC(C)=CC=C1N1C[C@H](N)CC1 MGGFZUWCVPJHDX-MRVPVSSYSA-N 0.000 description 1
- VVZBKOIWKGEJMH-UHFFFAOYSA-N (5-chloropyridazin-3-yl) trifluoromethanesulfonate Chemical compound ClC=1C=C(N=NC=1)OS(=O)(=O)C(F)(F)F VVZBKOIWKGEJMH-UHFFFAOYSA-N 0.000 description 1
- 238000001644 13C nuclear magnetic resonance spectroscopy Methods 0.000 description 1
- 238000004293 19F NMR spectroscopy Methods 0.000 description 1
- YTQQIHUQLOZOJI-UHFFFAOYSA-N 2,3-dihydro-1,2-thiazole Chemical class C1NSC=C1 YTQQIHUQLOZOJI-UHFFFAOYSA-N 0.000 description 1
- OZAIFHULBGXAKX-UHFFFAOYSA-N 2-(2-cyanopropan-2-yldiazenyl)-2-methylpropanenitrile Chemical compound N#CC(C)(C)N=NC(C)(C)C#N OZAIFHULBGXAKX-UHFFFAOYSA-N 0.000 description 1
- RWCMOQXHIDWDDJ-UHFFFAOYSA-N 2-(4-fluorophenyl)-2-hydroxyacetic acid Chemical compound OC(=O)C(O)C1=CC=C(F)C=C1 RWCMOQXHIDWDDJ-UHFFFAOYSA-N 0.000 description 1
- JBCVXARJEBQWRS-SSDOTTSWSA-N 2-N-[(3R)-1-(5-chloropyridazin-3-yl)pyrrolidin-3-yl]-1,3,4-thiadiazole-2,5-diamine Chemical compound ClC=1C=C(N=NC=1)N1C[C@@H](CC1)NC=1SC(=NN=1)N JBCVXARJEBQWRS-SSDOTTSWSA-N 0.000 description 1
- JSMGQCCZONCCBM-MRVPVSSYSA-N 2-N-[(3R)-1-(6-methylpyridazin-3-yl)pyrrolidin-3-yl]-1,3,4-thiadiazole-2,5-diamine Chemical compound CC1=CC=C(N=N1)N1C[C@@H](CC1)NC=1SC(=NN=1)N JSMGQCCZONCCBM-MRVPVSSYSA-N 0.000 description 1
- CQLPOXGXACFBIW-XPCCGILXSA-N 2-methoxy-N-[5-[[(3R)-1-(6-methylpyridazin-3-yl)pyrrolidin-3-yl]amino]-1,3,4-thiadiazol-2-yl]-2-[3-(trifluoromethoxy)phenyl]acetamide Chemical compound COC(C(=O)NC=1SC(=NN=1)N[C@H]1CN(CC1)C=1N=NC(=CC=1)C)C1=CC(=CC=C1)OC(F)(F)F CQLPOXGXACFBIW-XPCCGILXSA-N 0.000 description 1
- JMTMSDXUXJISAY-UHFFFAOYSA-N 2H-benzotriazol-4-ol Chemical group OC1=CC=CC2=C1N=NN2 JMTMSDXUXJISAY-UHFFFAOYSA-N 0.000 description 1
- FQEVHRCPXFKJHF-UHFFFAOYSA-N 3-(trifluoromethoxy)benzaldehyde Chemical compound FC(F)(F)OC1=CC=CC(C=O)=C1 FQEVHRCPXFKJHF-UHFFFAOYSA-N 0.000 description 1
- GXBWRBSCBONTKK-UHFFFAOYSA-N 3-chloro-5-methylpyridazine Chemical compound CC1=CN=NC(Cl)=C1 GXBWRBSCBONTKK-UHFFFAOYSA-N 0.000 description 1
- PRORLQAJNJMGAR-UHFFFAOYSA-N 3-chloro-6-methylpyridazine Chemical compound CC1=CC=C(Cl)N=N1 PRORLQAJNJMGAR-UHFFFAOYSA-N 0.000 description 1
- NHMLZGFOEYLZEN-UHFFFAOYSA-N 4-chloro-1h-pyridazin-6-one Chemical compound ClC=1C=NNC(=O)C=1 NHMLZGFOEYLZEN-UHFFFAOYSA-N 0.000 description 1
- NALVGTOMKSKFFV-UHFFFAOYSA-N 4-fluoro-3-methoxybenzaldehyde Chemical compound COC1=CC(C=O)=CC=C1F NALVGTOMKSKFFV-UHFFFAOYSA-N 0.000 description 1
- SJXIGQMHIZJOGN-UHFFFAOYSA-N 6-(2-bromoethynyl)-2,3-dimethylquinazolin-4-one Chemical compound CN1C(C)=NC2=CC=C(C=C2C1=O)C#CBr SJXIGQMHIZJOGN-UHFFFAOYSA-N 0.000 description 1
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 1
- OZAIFHULBGXAKX-VAWYXSNFSA-N AIBN Substances N#CC(C)(C)\N=N\C(C)(C)C#N OZAIFHULBGXAKX-VAWYXSNFSA-N 0.000 description 1
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 1
- COVZYZSDYWQREU-UHFFFAOYSA-N Busulfan Chemical compound CS(=O)(=O)OCCCCOS(C)(=O)=O COVZYZSDYWQREU-UHFFFAOYSA-N 0.000 description 1
- SGYCIZMJPWFKTJ-UHFFFAOYSA-N COC(C(OC)=O)c(cc1)ccc1OC Chemical compound COC(C(OC)=O)c(cc1)ccc1OC SGYCIZMJPWFKTJ-UHFFFAOYSA-N 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-L Carbonate Chemical compound [O-]C([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-L 0.000 description 1
- YZCKVEUIGOORGS-OUBTZVSYSA-N Deuterium Chemical compound [2H] YZCKVEUIGOORGS-OUBTZVSYSA-N 0.000 description 1
- 206010061818 Disease progression Diseases 0.000 description 1
- 239000006144 Dulbecco’s modified Eagle's medium Substances 0.000 description 1
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 1
- 101150029707 ERBB2 gene Proteins 0.000 description 1
- 241000588724 Escherichia coli Species 0.000 description 1
- 241000400611 Eucalyptus deanei Species 0.000 description 1
- 101710184006 Glutaminase 1 Proteins 0.000 description 1
- 102100034228 Grainyhead-like protein 1 homolog Human genes 0.000 description 1
- 101001069933 Homo sapiens Grainyhead-like protein 1 homolog Proteins 0.000 description 1
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical class Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- ZDZOTLJHXYCWBA-VCVYQWHSSA-N N-debenzoyl-N-(tert-butoxycarbonyl)-10-deacetyltaxol Chemical compound O([C@H]1[C@H]2[C@@](C([C@H](O)C3=C(C)[C@@H](OC(=O)[C@H](O)[C@@H](NC(=O)OC(C)(C)C)C=4C=CC=CC=4)C[C@]1(O)C3(C)C)=O)(C)[C@@H](O)C[C@H]1OC[C@]12OC(=O)C)C(=O)C1=CC=CC=C1 ZDZOTLJHXYCWBA-VCVYQWHSSA-N 0.000 description 1
- 241001460678 Napo <wasp> Species 0.000 description 1
- 108700020796 Oncogene Proteins 0.000 description 1
- 229930012538 Paclitaxel Natural products 0.000 description 1
- BELBBZDIHDAJOR-UHFFFAOYSA-N Phenolsulfonephthalein Chemical compound C1=CC(O)=CC=C1C1(C=2C=CC(O)=CC=2)C2=CC=CC=C2S(=O)(=O)O1 BELBBZDIHDAJOR-UHFFFAOYSA-N 0.000 description 1
- 229920001213 Polysorbate 20 Polymers 0.000 description 1
- LCTONWCANYUPML-UHFFFAOYSA-M Pyruvate Chemical compound CC(=O)C([O-])=O LCTONWCANYUPML-UHFFFAOYSA-M 0.000 description 1
- 239000012980 RPMI-1640 medium Substances 0.000 description 1
- 229910004298 SiO 2 Inorganic materials 0.000 description 1
- OKJPEAGHQZHRQV-UHFFFAOYSA-N Triiodomethane Natural products IC(I)I OKJPEAGHQZHRQV-UHFFFAOYSA-N 0.000 description 1
- YZCKVEUIGOORGS-NJFSPNSNSA-N Tritium Chemical compound [3H] YZCKVEUIGOORGS-NJFSPNSNSA-N 0.000 description 1
- 239000013504 Triton X-100 Substances 0.000 description 1
- 229920004890 Triton X-100 Polymers 0.000 description 1
- 230000006682 Warburg effect Effects 0.000 description 1
- 239000004480 active ingredient Substances 0.000 description 1
- 239000013543 active substance Substances 0.000 description 1
- 238000011374 additional therapy Methods 0.000 description 1
- 150000001299 aldehydes Chemical class 0.000 description 1
- 150000008044 alkali metal hydroxides Chemical class 0.000 description 1
- 229910001860 alkaline earth metal hydroxide Inorganic materials 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- 125000004429 atom Chemical group 0.000 description 1
- CBHOOMGKXCMKIR-UHFFFAOYSA-N azane;methanol Chemical compound N.OC CBHOOMGKXCMKIR-UHFFFAOYSA-N 0.000 description 1
- VSRXQHXAPYXROS-UHFFFAOYSA-N azanide;cyclobutane-1,1-dicarboxylic acid;platinum(2+) Chemical compound [NH2-].[NH2-].[Pt+2].OC(=O)C1(C(O)=O)CCC1 VSRXQHXAPYXROS-UHFFFAOYSA-N 0.000 description 1
- 230000009286 beneficial effect Effects 0.000 description 1
- CMRXOLCCAGRRDY-MRXNPFEDSA-N benzyl (2R)-2-methoxy-2-(3-methoxyphenyl)acetate Chemical compound CO[C@@H](C(=O)OCC1=CC=CC=C1)C1=CC(OC)=CC=C1 CMRXOLCCAGRRDY-MRXNPFEDSA-N 0.000 description 1
- 230000004071 biological effect Effects 0.000 description 1
- 230000015572 biosynthetic process Effects 0.000 description 1
- 210000000988 bone and bone Anatomy 0.000 description 1
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 description 1
- 229910000024 caesium carbonate Inorganic materials 0.000 description 1
- 239000002775 capsule Substances 0.000 description 1
- 239000001569 carbon dioxide Substances 0.000 description 1
- 229910002091 carbon monoxide Inorganic materials 0.000 description 1
- 229960004562 carboplatin Drugs 0.000 description 1
- 239000000969 carrier Substances 0.000 description 1
- 230000015556 catabolic process Effects 0.000 description 1
- 230000010261 cell growth Effects 0.000 description 1
- 239000013592 cell lysate Substances 0.000 description 1
- 230000004663 cell proliferation Effects 0.000 description 1
- 239000003795 chemical substances by application Substances 0.000 description 1
- 238000004296 chiral HPLC Methods 0.000 description 1
- DQLATGHUWYMOKM-UHFFFAOYSA-L cisplatin Chemical compound N[Pt](N)(Cl)Cl DQLATGHUWYMOKM-UHFFFAOYSA-L 0.000 description 1
- 229960004316 cisplatin Drugs 0.000 description 1
- 238000004040 coloring Methods 0.000 description 1
- 238000002648 combination therapy Methods 0.000 description 1
- 239000012141 concentrate Substances 0.000 description 1
- 235000008504 concentrate Nutrition 0.000 description 1
- 238000001816 cooling Methods 0.000 description 1
- 230000003247 decreasing effect Effects 0.000 description 1
- 229940009976 deoxycholate Drugs 0.000 description 1
- KXGVEGMKQFWNSR-LLQZFEROSA-N deoxycholic acid Chemical compound C([C@H]1CC2)[C@H](O)CC[C@]1(C)[C@@H]1[C@@H]2[C@@H]2CC[C@H]([C@@H](CCC(O)=O)C)[C@@]2(C)[C@@H](O)C1 KXGVEGMKQFWNSR-LLQZFEROSA-N 0.000 description 1
- 229910052805 deuterium Inorganic materials 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- 150000004683 dihydrates Chemical class 0.000 description 1
- 125000002147 dimethylamino group Chemical group [H]C([H])([H])N(*)C([H])([H])[H] 0.000 description 1
- 230000005750 disease progression Effects 0.000 description 1
- 208000035475 disorder Diseases 0.000 description 1
- 239000006185 dispersion Substances 0.000 description 1
- 229960003668 docetaxel Drugs 0.000 description 1
- 239000000839 emulsion Substances 0.000 description 1
- 102000015694 estrogen receptors Human genes 0.000 description 1
- 108010038795 estrogen receptors Proteins 0.000 description 1
- 230000005713 exacerbation Effects 0.000 description 1
- 230000005284 excitation Effects 0.000 description 1
- 239000012894 fetal calf serum Substances 0.000 description 1
- 239000012065 filter cake Substances 0.000 description 1
- MHMNJMPURVTYEJ-UHFFFAOYSA-N fluorescein-5-isothiocyanate Chemical compound O1C(=O)C2=CC(N=C=S)=CC=C2C21C1=CC=C(O)C=C1OC1=CC(O)=CC=C21 MHMNJMPURVTYEJ-UHFFFAOYSA-N 0.000 description 1
- 239000012458 free base Substances 0.000 description 1
- 239000000499 gel Substances 0.000 description 1
- 230000002414 glycolytic effect Effects 0.000 description 1
- PCHJSUWPFVWCPO-UHFFFAOYSA-N gold Chemical compound [Au] PCHJSUWPFVWCPO-UHFFFAOYSA-N 0.000 description 1
- 229910052737 gold Inorganic materials 0.000 description 1
- 239000010931 gold Substances 0.000 description 1
- 239000008187 granular material Substances 0.000 description 1
- 239000007902 hard capsule Substances 0.000 description 1
- 238000010438 heat treatment Methods 0.000 description 1
- 230000002440 hepatic effect Effects 0.000 description 1
- 238000000338 in vitro Methods 0.000 description 1
- 238000001727 in vivo Methods 0.000 description 1
- 238000011534 incubation Methods 0.000 description 1
- 239000011261 inert gas Substances 0.000 description 1
- 230000008595 infiltration Effects 0.000 description 1
- 238000001764 infiltration Methods 0.000 description 1
- 238000001802 infusion Methods 0.000 description 1
- 230000002401 inhibitory effect Effects 0.000 description 1
- 238000007918 intramuscular administration Methods 0.000 description 1
- 238000001990 intravenous administration Methods 0.000 description 1
- INQOMBQAUSQDDS-UHFFFAOYSA-N iodomethane Chemical compound IC INQOMBQAUSQDDS-UHFFFAOYSA-N 0.000 description 1
- 238000005342 ion exchange Methods 0.000 description 1
- 150000002500 ions Chemical class 0.000 description 1
- 210000003734 kidney Anatomy 0.000 description 1
- 239000004310 lactic acid Substances 0.000 description 1
- 235000014655 lactic acid Nutrition 0.000 description 1
- 239000010410 layer Substances 0.000 description 1
- 239000008263 liquid aerosol Substances 0.000 description 1
- 238000004811 liquid chromatography Methods 0.000 description 1
- 210000004185 liver Anatomy 0.000 description 1
- 239000007937 lozenge Substances 0.000 description 1
- 238000012423 maintenance Methods 0.000 description 1
- 230000036210 malignancy Effects 0.000 description 1
- 238000004949 mass spectrometry Methods 0.000 description 1
- 230000004066 metabolic change Effects 0.000 description 1
- 230000009401 metastasis Effects 0.000 description 1
- OKKJLVBELUTLKV-VMNATFBRSA-N methanol-d1 Chemical compound [2H]OC OKKJLVBELUTLKV-VMNATFBRSA-N 0.000 description 1
- 125000000325 methylidene group Chemical group [H]C([H])=* 0.000 description 1
- 238000013508 migration Methods 0.000 description 1
- 230000005012 migration Effects 0.000 description 1
- 210000003470 mitochondria Anatomy 0.000 description 1
- 150000004682 monohydrates Chemical class 0.000 description 1
- 239000002773 nucleotide Substances 0.000 description 1
- 125000003729 nucleotide group Chemical group 0.000 description 1
- 239000002674 ointment Substances 0.000 description 1
- 231100000590 oncogenic Toxicity 0.000 description 1
- 230000002246 oncogenic effect Effects 0.000 description 1
- 238000011275 oncology therapy Methods 0.000 description 1
- 210000000056 organ Anatomy 0.000 description 1
- 229940127084 other anti-cancer agent Drugs 0.000 description 1
- 229960001756 oxaliplatin Drugs 0.000 description 1
- DWAFYCQODLXJNR-BNTLRKBRSA-L oxaliplatin Chemical compound O1C(=O)C(=O)O[Pt]11N[C@@H]2CCCC[C@H]2N1 DWAFYCQODLXJNR-BNTLRKBRSA-L 0.000 description 1
- 230000003647 oxidation Effects 0.000 description 1
- 238000007254 oxidation reaction Methods 0.000 description 1
- 229960001592 paclitaxel Drugs 0.000 description 1
- 229910052763 palladium Inorganic materials 0.000 description 1
- 238000007911 parenteral administration Methods 0.000 description 1
- 230000007170 pathology Effects 0.000 description 1
- 230000037361 pathway Effects 0.000 description 1
- 230000002093 peripheral effect Effects 0.000 description 1
- 229940124531 pharmaceutical excipient Drugs 0.000 description 1
- 229960003531 phenolsulfonphthalein Drugs 0.000 description 1
- 239000000256 polyoxyethylene sorbitan monolaurate Substances 0.000 description 1
- 235000010486 polyoxyethylene sorbitan monolaurate Nutrition 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- 235000015320 potassium carbonate Nutrition 0.000 description 1
- 235000011181 potassium carbonates Nutrition 0.000 description 1
- 238000002360 preparation method Methods 0.000 description 1
- 238000004237 preparative chromatography Methods 0.000 description 1
- 239000003755 preservative agent Substances 0.000 description 1
- 238000011324 primary prophylaxis Methods 0.000 description 1
- 102000003998 progesterone receptors Human genes 0.000 description 1
- 108090000468 progesterone receptors Proteins 0.000 description 1
- 238000000746 purification Methods 0.000 description 1
- JUJWROOIHBZHMG-UHFFFAOYSA-O pyridinium Chemical compound C1=CC=[NH+]C=C1 JUJWROOIHBZHMG-UHFFFAOYSA-O 0.000 description 1
- 239000001397 quillaja saponaria molina bark Substances 0.000 description 1
- 230000002441 reversible effect Effects 0.000 description 1
- 238000012552 review Methods 0.000 description 1
- 238000002390 rotary evaporation Methods 0.000 description 1
- 229930182490 saponin Natural products 0.000 description 1
- 150000007949 saponins Chemical class 0.000 description 1
- 229920006395 saturated elastomer Polymers 0.000 description 1
- 238000011125 single therapy Methods 0.000 description 1
- 238000009097 single-agent therapy Methods 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- 239000007901 soft capsule Substances 0.000 description 1
- 210000004872 soft tissue Anatomy 0.000 description 1
- 238000001228 spectrum Methods 0.000 description 1
- 238000010183 spectrum analysis Methods 0.000 description 1
- 238000007920 subcutaneous administration Methods 0.000 description 1
- 239000000829 suppository Substances 0.000 description 1
- 230000004083 survival effect Effects 0.000 description 1
- 238000003786 synthesis reaction Methods 0.000 description 1
- 239000006188 syrup Substances 0.000 description 1
- 235000020357 syrup Nutrition 0.000 description 1
- RCINICONZNJXQF-MZXODVADSA-N taxol Chemical compound O([C@@H]1[C@@]2(C[C@@H](C(C)=C(C2(C)C)[C@H](C([C@]2(C)[C@@H](O)C[C@H]3OC[C@]3([C@H]21)OC(C)=O)=O)OC(=O)C)OC(=O)[C@H](O)[C@@H](NC(=O)C=1C=CC=CC=1)C=1C=CC=CC=1)O)C(=O)C1=CC=CC=C1 RCINICONZNJXQF-MZXODVADSA-N 0.000 description 1
- LJZXNQMAZBFEFK-SECBINFHSA-N tert-butyl (3r)-3-[(6-chloropyridazin-3-yl)amino]pyrrolidine-1-carboxylate Chemical compound C1N(C(=O)OC(C)(C)C)CC[C@H]1NC1=CC=C(Cl)N=N1 LJZXNQMAZBFEFK-SECBINFHSA-N 0.000 description 1
- XVENHGRMEVTBSQ-LLVKDONJSA-N tert-butyl N-[(3R)-1-(5-methylpyridazin-3-yl)pyrrolidin-3-yl]carbamate Chemical compound CC=1C=C(N=NC=1)N1C[C@@H](CC1)NC(OC(C)(C)C)=O XVENHGRMEVTBSQ-LLVKDONJSA-N 0.000 description 1
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 1
- 230000001225 therapeutic effect Effects 0.000 description 1
- 230000000699 topical effect Effects 0.000 description 1
- 230000004102 tricarboxylic acid cycle Effects 0.000 description 1
- 150000004684 trihydrates Chemical class 0.000 description 1
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 1
- 229910052722 tritium Inorganic materials 0.000 description 1
- 229910052721 tungsten Inorganic materials 0.000 description 1
- 238000004704 ultra performance liquid chromatography Methods 0.000 description 1
- 238000010792 warming Methods 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing three or more hetero rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/50—Pyridazines; Hydrogenated pyridazines
- A61K31/501—Pyridazines; Hydrogenated pyridazines not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/53—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with three nitrogens as the only ring hetero atoms, e.g. chlorazanil, melamine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Pharmacology & Pharmacy (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Epidemiology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Peptides Or Proteins (AREA)
- Medicinal Preparation (AREA)
Abstract
Un compuesto de la Fórmula (I):**Fórmula** o una sal farmacéuticamente aceptable de este, en donde: Q es 5-metilpiridazin-3-ilo, 5-cloropiridazin-3-ilo, 6-metilpiridazin-3-ilo, o 6-fluoropiridazin-3-ilo; R es hidrógeno, fluoro, o metoxi; R1 es hidrógeno, metoxi, difluorometoxi, o trifluorometoxi; y R2 es metilo o etilo.
Description
DESCRIPCIÓN
Compuestos de 1,3,4-tiadiazol y su uso para tratar el cáncer.
Campo técnico
La descripción generalmente se refiere a compuestos de 1,3,4-tiadiazol sustituidos y sales farmacéuticamente aceptables de estos. Estos compuestos actúan sobre la enzima glutaminasa 1 ("GLS1") y por lo tanto la descripción se refiere, además, al uso de tales compuestos y sales de estos para tratar o prevenir la enfermedad mediada por GLS1, incluido el cáncer. La descripción se refiere, además, a composiciones farmacéuticas que comprenden tales compuestos y sales; kits que comprenden tales compuestos y sales; métodos de fabricación de tales compuestos y sales; productos intermedios útiles en la fabricación de tales compuestos y sales; y al uso de tales compuestos en métodos para tratar enfermedades mediadas porGLS1, incluido el cáncer.
Antecedentes
La glutamina es el aminoácido plasmático más abundante y participa en muchas rutas que promueven el crecimiento. En particular, la glutamina participa en la oxidación en el ciclo TCA y en el mantenimiento del equilibrio redox celular y, además, proporciona nitrógeno para la síntesis de nucleótidos y aminoácidos (Curi y otros, Front. Biosci.. 2007, 12, 344 57; DeBerardinisy Cheng, Oncogene 2010, 313-324, cada uno de los cuales se incorpora como referencia en su totalidad). Muchas células cancerosas dependen del metabolismo de la glutamina como consecuencia de los cambios metabólicos en la célula, incluido el efecto Warburg donde el piruvato glicolítico se convierte en ácido láctico en lugar de usarse para crear acetil CoA (Koppenol y otros, Nature Reviews 2011, 11,325-337, que se incorpora como referencia en su totalidad). Como consecuencia de esta dependencia del metabolismo de la glutamina, tales células cancerosas son sensibles a los cambios en los niveles de glutamina exógena. Además, la evidencia existente sugiere que la glutaminólisis desempeña una función clave en ciertos tipos de cáncer (Hensley y otros, J. Clin. Invest. 2013, 123, 3678- 3684, que se incorpora como referencia en su totalidad), y se asocia con controladores oncogénicos conocidos tales como Myc (Dang, Cancer Res. 2010, 70, 859- 863, que se incorpora como referencia en su totalidad).
La primera etapa del catabolismo de la glutamina a glutamato es catalizado por la glutaminasa, que existe como dos isoformas, GLS1 y GLS2, originalmente identificadas como expresadas en el riñón y el hígado, respectivamente. Se conoce que la glutaminasa renal (GLS1) se expresa más ubicuamente que la glutaminasa hepática (GLS2), y tiene 2 variantes de corte y empalme, KGA y la isoforma GAC más corta, ambas ubicadas en las mitocondrias. (Elgadi y otros, Physiol. Genomics 1999, 1, 51-62; Cassago y otros, Proc. Natl. Acad. Sci. 2012, 109, 1092-1097, cada uno de los cuales se incorpora como referencia en su totalidad). La expresión de GLS1 se asocia con el crecimiento tumoral y la malignidad en varios tipos de enfermedades (Wang y otros, Cancer Cell 2010, 18, 207-219; van der Heuval y otros, Cancer Bio. Ther.
2012, 13, 1185-1194, cada uno de los cuales se incorpora como referencia en su totalidad). Por lo tanto se espera que los inhibidores de GLS1 sean útiles en el tratamiento del cáncer, como monoterapia o en combinación con otros agentes contra el cáncer. El documento núm. WO 2013/078123 y el documento núm. US 2014/142081 ambos describen derivados de 1,3,4-tiazol como inhibidores de GLS1.
Resumen
En un aspecto, un compuesto de la Fórmula (I):

Q es 5-metilpiridazin-3-ilo, 5-cloropiridazin-3-ilo, 6-metilpiridazin-3-ilo, o 6-fluoropiridazin-3-ilo;
R es hidrógeno, fluoro, o metoxi;
R1 es hidrógeno, metoxi, difluorometoxi, o trifluorometoxi; y
R2 es metilo o etilo.
En otro aspecto, una composición farmacéutica incluye el compuesto de la Fórmula (I), o una sal farmacéuticamente aceptable de este, y al menos un diluyente o portador farmacéuticamente aceptable.
En otro aspecto, el compuesto de la Fórmula (I), o una sal farmacéuticamente aceptable de este, para su uso en terapia. En otro aspecto, el compuesto de la Fórmula (I), o una sal farmacéuticamente aceptable de este, para su uso en el tratamiento del cáncer.
En otro aspecto, el uso del compuesto de la Fórmula (I), o una sal farmacéuticamente aceptable de este, para fabricar un medicamento para el tratamiento del cáncer.
En otro aspecto, un método para tratar el cáncer en un animal de sangre caliente que necesita de tal tratamiento, incluye administrar al animal de sangre caliente una cantidad terapéuticamente efectiva de un compuesto de la Fórmula (I), o una sal farmacéuticamente aceptable de este.
Otros aspectos serán evidentes a partir de la descripción y las reivindicaciones.
Descripción detallada
Muchas modalidades se detallan a lo largo de la descripción y serán evidentes para un lector experto en la técnica. La invención no debe interpretarse como limitada a ninguna(s) modalidad(es) particular(es) de esta.
Se proporciona un compuesto de la Fórmula (I):
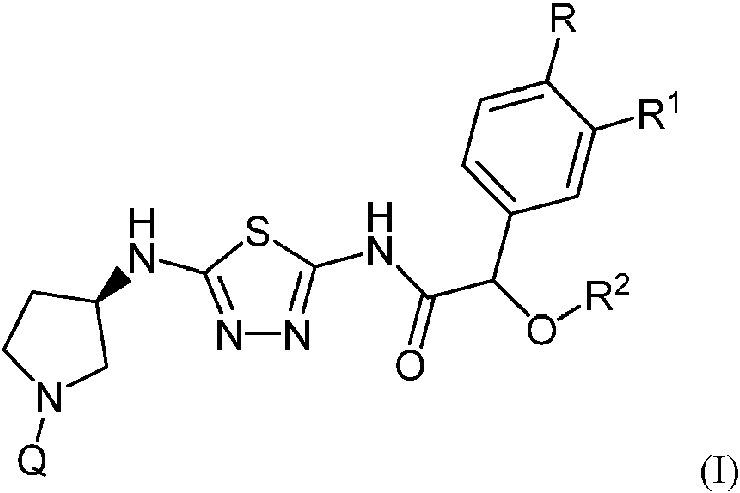
o una sal farmacéuticamente aceptable de este, donde:
Q es 5-metilpiridazin-3-ilo, 5-cloropiridazin-3-ilo, 6-metilpiridazin-3-ilo, o 6-fluoropiridazin-3-ilo;
R es hidrógeno, fluoro, o metoxi;
R1 es hidrógeno, metoxi, difluorometoxi, o trifluorometoxi; y
R2 es metilo o etilo.
Los anillos de 5-metilpiridazin-3-ilo, 5-cloropiridazin-3-ilo, 6-metilpiridazin-3-ilo, o 6-fluoropiridazin-3-ilo tienen las siguientes estructuras:

En algunas modalidades, el compuesto de la Fórmula (I) tiene la siguiente Fórmula

en donde Q, R, R1, y R2 son como se definieron anteriormente.
El término "farmacéuticamente aceptable" se usa para especificar que un objeto (por ejemplo una sal, forma de dosificación, diluyente o portador) es adecuado para su uso en pacientes. Puede encontrarse una lista de ejemplos de sales farmacéuticamente aceptables en el Handbook of Pharmaceutical Salts: Properties, Selection and Use, P. H. Stahl y C. G. Wermuth, editores, Weinheim/Zurich: Wiley-VCH/VHCA, 2002, que se incorporan como referencia en su totalidad. Una sal farmacéuticamente aceptable adecuada de un compuesto de la Fórmula (I) es, por ejemplo, una sal de adición de ácido. Puede formarse una sal de adición de ácido de un compuesto de la Fórmula (I) al poner en contacto el compuesto con un ácido inorgánico u orgánico adecuado bajo condiciones conocidas por el experto. Puede formarse una sal de adición de ácido mediante el uso de, por ejemplo, un ácido inorgánico tal como ácido clorhídrico, ácido bromhídrico, ácido sulfúrico, y ácido fosfórico. Puede formarse una sal de adición de ácido, además, mediante el uso de, por ejemplo, un ácido orgánico tal como ácido trifluoroacético, ácido metanosulfónico, o ácido bencenosulfónico.
Por lo tanto, en una modalidad se proporciona un compuesto de la Fórmula (I) o una sal farmacéuticamente aceptable de este, donde la sal farmacéuticamente aceptable es una sal de ácido clorhídrico, ácido bromhídrico, ácido sulfúrico, ácido fosfórico, ácido trifluoroacético, ácido metanosulfónico, o ácido bencenosulfónico.
En una modalidad se proporciona un compuesto de la Fórmula (I) o una sal farmacéuticamente aceptable de este, donde la sal farmacéuticamente aceptable es una sal de ácido clorhídrico o de ácido bromhídrico.
Una sal farmacéuticamente aceptable adicional adecuada de un compuesto de la Fórmula (I) es una sal de adición de base. Puede formarse una sal de adición de base de un compuesto de la Fórmula (I) al poner en contacto el compuesto con una base inorgánica u orgánica adecuada bajo condiciones conocidas por el experto. Por ejemplo, puede formarse una sal de adición de base mediante el uso de, por ejemplo, una base inorgánica tal como un hidróxido de metal alcalino (tal como hidróxido de sodio, potasio, o litio) o un hidróxido de metal alcalinotérreo (tal como hidróxido de calcio o hidróxido de magnesio). Puede formarse, además, una sal de adición de base mediante el uso de, por ejemplo, una base orgánica tal como metilamina, dimetilamina, trimetilamina, piperidina, morfolina, o tr/s-(2-hidroxietil)amina.
Por lo tanto, en una modalidad se proporciona un compuesto de la Fórmula (I) o una sal farmacéuticamente aceptable de este, donde la sal farmacéuticamente aceptable es una sal de hidróxido de sodio, hidróxido de potasio, hidróxido de litio, hidróxido de calcio, hidróxido de magnesio, metilamina, dimetilamina, trimetilamina, piperidina, morfolina, o tr/s-(2-hidroxietil)amina.
En una modalidad se proporciona un compuesto de la Fórmula (I) o una sal farmacéuticamente aceptable de este, donde la sal farmacéuticamente aceptable es una sal de ácido clorhídrico, ácido bromhídrico, ácido sulfúrico, ácido fosfórico, ácido trifluoroacético, ácido metanosulfónico, ácido bencenosulfónico, hidróxido de sodio, hidróxido de potasio, hidróxido de litio, hidróxido de calcio, hidróxido de magnesio, metilamina, dimetilamina, trimetilamina, piperidina, morfolina, o tr/s-(2-hidroxietil)amina.
Una modalidad adicional proporciona cualquiera de las modalidades definidas en la presente descripción (por ejemplo, la modalidad de la reivindicación 1) con la condición de que uno o más ejemplos específicos (por ejemplo, uno, dos o tres ejemplos específicos, o alternativamente un ejemplo específico) seleccionados del grupo que consiste de los Ejemplos 1(a), 1(b), 2, 3, 4(a), 4(b), 5(a), 5(b), 6(a), 6(b), 7(a), 7(b), 8(a), 8(b), 9(a), 9(b), 10(a), 10(b), 11(a), 11(b), 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33(a) y 33(b) se desreivindique individualmente.
Algunos valores de grupos variables en la Fórmula (I) son los siguientes. Tales valores pueden usarse en combinación con cualquiera de las definiciones, reivindicaciones (por ejemplo, reivindicación 1) o modalidades definidas en la presente descripción para proporcionar modalidades adicionales.
Q puede ser 6-metilpiridazin-3-ilo o 6-fluoropiridazin-3-ilo.
Q puede ser 5-metilpiridazin-3-ilo o 5-cloropiridazin-3-ilo.
Q puede ser 5-metilpiridazin-3-ilo.
Q puede ser 5-cloropiridazin-3-ilo.
Q puede ser 6-metilpiridazin-3-ilo.
Q puede ser o 6-fluoropiridazin-3-ilo.
R puede ser hidrógeno o fluoro.
R puede ser hidrógeno.
R1 puede ser metoxi, difluorometoxi, o trifluorometoxi.
R2 puede ser metilo.
En otra modalidad se proporciona un compuesto de la Fórmula (I), o una sal farmacéuticamente aceptable de este, donde:
Q es 6-metilpiridazin-3-ilo o 6-fluoropiridazin-3-ilo; y
R1 es metoxi o trifluorometoxi.
En otra modalidad se proporciona un compuesto de la Fórmula (I), o una sal farmacéuticamente aceptable de este, donde:
Q es 6-metilpiridazin-3-ilo o 6-fluoropiridazin-3-ilo;
R es hidrógeno; y
R1 es metoxi o trifluorometoxi.
En otra modalidad se proporciona un compuesto de la Fórmula (I), o una sal farmacéuticamente aceptable de este, donde:
Q es 6-metilpiridazin-3-ilo o 6-fluoropiridazin-3-ilo;
R es hidrógeno;
R1 es metoxi o trifluorometoxi; y
R2 es metilo.
En otra modalidad se proporciona un compuesto de la Fórmula (I), o una sal farmacéuticamente aceptable de este, donde:
Q es 6-metilpiridazin-3-ilo o 6-fluoropiridazin-3-ilo; y
R es fluoro.
En una modalidad se proporciona un compuesto de la Fórmula (I), o una sal farmacéuticamente aceptable de este, donde el compuesto se selecciona del grupo que consiste en:
(2s)-2-metoxi-2-(3-metoxifenil)-A/-[5-[[(3R)-1-(6-metilpiridazin-3-il)pirrolidin-3-il]amino]-1,3,4-tiadiazol-2-il]acetamida; (2s)-W-[5-[[(3R)-1-(6-fluoropiridazin-3-il)pirrolidin-3-il]amino]-1,3,4-tiadiazol-2-il]-2-metoxi-2-fenil-acetamida;
(2s)-W-[5-[[(3R)-1-(6-fluoropiridazin-3-il)pirrolidin-3-il]amino]-1,3,4-tiadiazol-2-il]-2-metoxi-2-(3-metoxifenil)acetamida; (2s)-2-etoxi-2-(3-metoxifenil)-A/-[5-[[(3R)-1-(6-metilpiridazin-3-il)pirrolidin-3-il]amino]-1,3,4-tiadiazol-2-il]acetamida;
(2s)-2-(4-fluorofenil)-2-metoxi-W-[5-[[(3R)-1-(6-metilpiridazin-3-il)pirrolidin-3-il]amino]-1,3,4-tiadiazol-2-il]acetamida; (2s)-2-(4-fluoro-3-metoxi-fenil)-2-metoxi-W-[5-[[(3R)-1-(6-metilpiridazin-3-il)pirrolidin-3-il]amino]-1,3,4-tiadiazol-2-il]acetamida;
(2s)-2-metoxi-A/-[5-[[(3R)-1-(6-metilpiridazin-3-il)pirrolidin-3-il]amino]-1,3,4-tiadiazol-2-il]-2-[3-(trifluorometoxi)fenil]acetamida;
(2s)-2-(4-fluorofenil)-2-metoxi-W-[5-[[(3R)-1-(5-metilpiridazin-3-il)pirrolidin-3-il]amino]-1,3,4-tiadiazol-2-il]acetamida; W-[5-[[(3R)-1-(5-cloropiridazin-3-il)pirrolidin-3-il]amino]-1,3,4-tiadiazol-2-il]-2-metoxi-2-(4-metoxifenil)acetamida;
(2s)-[3-(difluorometoxi)fenil]-2-metoxi-W-[5-[[(3R)-1-(6-metilpiridazin-3-il)pirrolidin-3-il]amino]-1,3,4-tiadiazol-2-il]acetamida; y
(2s)-2-[3-(difluorometoxi)fenil]-N-[5-[[(3R)-1-(6-fluoropiridazin-3-il)pirrolidin-3-il]amino]-1,3,4-tiadiazol-2-il]-2-metoxiacetamida.
En una modalidad se proporciona un compuesto de la Fórmula (I), o una sal farmacéuticamente aceptable de este, donde el compuesto se selecciona del grupo que consiste en:
(2s)-2-metoxi-2-(3-metoxifenil)-A/-[5-[[(3R)-1-(6-metilpiridazin-3-il)pirrolidin-3-il]amino]-1,3,4-tiadiazol-2-il]acetamida; (2s)-W-[5-[[(3R)-1-(6-fluoropiridazin-3-il)pirrolidin-3-il]amino]-1,3,4-tiadiazol-2-il]-2-metoxi-2-(3-metoxifenil)acetamida; (2s)-2-etoxi-2-(3-metoxifenil)-A/-[5-[[(3R)-1-(6-metilpiridazin-3-il)pirrolidin-3-il]amino]-1,3,4-tiadiazol-2-il]acetamida;
(2s)-2-(4-fluorofenil)-2-metoxi-W-[5-[[(3R)-1-(6-metilpiridazin-3-il)pirrolidin-3-il]amino]-1,3,4-tiadiazol-2-il]acetamida; (2s)-2-(4-fluoro-3-metoxi-fenil)-2-metoxi-W-[5-[[(3R)-1-(6-metilpiridazin-3-il)pirrolidin-3-il]amino]-1,3,4-tiadiazol-2-il]acetamida;
(2s)-2-metoxi-A/-[5-[[(3R)-1-(6-metilpiridazin-3-il)pirrolidin-3-il]amino]-1,3,4-tiadiazol-2-il]-2-[3-(trifluorometoxi)fenil]acetamida;
(2s)-2-(4-fluorofenil)-2-metoxi-W-[5-[[(3R)-1-(5-metilpiridazin-3-il)pirrolidin-3-il]amino]-1,3,4-tiadiazol-2-il]acetamida; (2s)-[3-(difluorometoxi)fenil]-2-metoxi-W-[5-[[(3R)-1-(6-metilpiridazin-3-il)pirrolidin-3-il]amino]-1,3,4-tiadiazol-2-il]acetamida; y
(2s)-2-[3-(difluorometoxi)fenil]-N-[5-[[(3R)-1-(6-fluoropiridazin-3-il)pirrolidin-3-il]amino]-1,3,4-tiadiazol-2-il]-2-metoxiacetamida.
Los compuestos y sales descritos en esta descripción pueden existir en formas solvatadas y formas no solvatadas. Por ejemplo, una forma solvatada puede ser una forma hidratada, tal como un hemihidrato, un monohidrato, un dihidrato, un trihidrato o una cantidad alternativa de estos. La presente invención abarca todas estas formas solvatadas y no solvatadas de los compuestos de Fórmula (I).
Los átomos de los compuestos y sales descritos en esta descripción pueden existir en diferentes formas isotópicas. La presente invención abarca todas las formas isotópicas de los compuestos de Fórmula (I) que incluyen un carbono 11C o 13C e hidrógeno 1H, 2H (deuterio) o 3H (tritio).
Los compuestos y sales descritos en esta descripción pueden existir como una mezcla de tautómeros. Los "tautómeros" son isómeros estructurales que existen en equilibrio como resultado de la migración de un átomo de hidrógeno. La presente invención incluye todos los tautómeros de los compuestos de Fórmula (I).
Los compuestos de Fórmula (I) pueden prepararse en diferentes formas diastereoméricas. La presente invención incluye todas las formas diastereoméricas de los compuestos de Fórmula (I).
En una modalidad se proporciona un compuesto de la Fórmula (I), o una sal farmacéuticamente aceptable de este, que es un diastereómero único que está en un exceso diastereomérico (% de) de > 95 %, > 98 % o > 99 %. En una modalidad, el diastereómero único está presente en un exceso diastereomérico (% de) de > 99 %.
Los compuestos que se cree que inhiben GLS1, es decir, los compuestos de Fórmula (I), y sales farmacéuticamente aceptables de estos se espera que sean útiles en la terapia, por ejemplo, en el tratamiento de enfermedades o afecciones médicas mediadas al menos en parte porGLSI, incluido el cáncer.
Cuando se menciona "cáncer", esto incluye cáncer no metastásico y, además, cáncer metastásico, tal que el tratamiento del cáncer implica el tratamiento de tanto tumores primarios como también de metástasis tumorales.
En una modalidad el cáncer es cáncer metastásico.
En una modalidad el cáncer es cáncer no metastásico.
"Actividad inhibitoria de GLS1" se refiere a una disminución en la actividad de GLS1 como respuesta directa o indirecta a la presencia de un compuesto de la Fórmula (I), o una sal farmacéuticamente aceptable de este, en relación con la actividad de GLS1 en ausencia del compuesto de la Fórmula (I), o una sal farmacéuticamente aceptable de este. Tal disminución en la actividad puede deberse a la interacción directa del compuesto de la Fórmula (I), o una sal farmacéuticamente aceptable de este con GLS1, o debido a la interacción del compuesto de la Fórmula (I), o una sal farmacéuticamente aceptable de este con uno o más factores que a su vez afectan la actividad de GLS1. Por ejemplo, el compuesto de la Fórmula (I), o una sal farmacéuticamente aceptable de este, puede disminuir GLS1 mediante su unión directa a GLS1; al causar (directa o indirectamente) que otro factor disminuya la actividad de GLS1; o mediante (directa o indirectamente) la disminución de la cantidad de GLS1 presente en la célula u organismo.
El término "terapia" pretende tener el significado normal de tratar una enfermedad o corregir o compensar la patología subyacente. El término "terapia" incluye, además, "profilaxis" a menos que existan indicaciones específicas de lo contrario. Los términos "terapéutico" y "terapéuticamente" deben interpretarse de la manera correspondiente.
El término "cantidad terapéuticamente efectiva" se refiere a una cantidad de un compuesto de la Fórmula (I) como se describe en cualquiera de las modalidades en la presente descripción que es efectiva para proporcionar la terapia en un sujeto. En el caso del cáncer, la cantidad terapéuticamente efectiva puede causar cualquiera de los cambios observables o medibles en un sujeto como se describe en la definición de "terapia", "tratamiento" y "profilaxis" anteriormente. Por ejemplo, la cantidad efectiva puede reducir el número de células cancerosas o tumorales; reducir el tamaño total del tumor; inhibir o detener la infiltración de células tumorales en órganos periféricos que incluyen, por ejemplo, los tejidos blandos y huesos; inhibir y detener la metástasis tumoral; inhibir y detener el crecimiento tumoral; aliviar en cierta medida uno o más de los síntomas asociados con el cáncer; reducir la morbilidad y la mortalidad; mejorar la calidad de vida; o una combinación de tales efectos. Una cantidad efectiva puede ser una cantidad suficiente para disminuir los síntomas de una enfermedad que responde a la inhibición de la actividad de GLS1. Para la terapia del cáncer, la eficacia in vivo puede medirse, por ejemplo, mediante la evaluación de la duración de la supervivencia, el tiempo hasta la progresión de la enfermedad (TTP), las tasas de respuesta (RR), la duración de la respuesta, y/o la calidad de vida. Como reconocen los expertos en la técnica, las cantidades efectivas pueden variar en dependencia de la ruta de administración, uso de excipientes, y el uso conjunto con otros agentes. Por ejemplo, cuando se usa una terapia de combinación, la cantidad del compuesto de la Fórmula (I) o sal farmacéuticamente aceptable descrita en esta descripción y la cantidad del(de los) otro(s) agente(s) farmacéuticamente activo(s) son, cuando se combinan, conjuntamente eficaces para tratar un trastorno diana en el paciente animal. En este contexto, las cantidades combinadas están en una "cantidad terapéuticamente efectiva" si son, cuando se combinan, suficientes para disminuir los síntomas de una enfermedad que responde a la inhibición de la actividad de GLS1 como se describió anteriormente. Típicamente, tales cantidades pueden determinarse por un experto en la técnica, por ejemplo, al iniciar con el intervalo de dosificación descrito en esta descripción para el compuesto de la Fórmula (I) o una sal farmacéuticamente aceptable de este y un(unos) intervalo(s) de dosificación aprobado(s) o publicado(s) de los otros compuestos farmacéuticamente activos.
El término "profilaxis" pretende tener su significado normal e incluye la profilaxis primaria para prevenir el desarrollo de la enfermedad y la profilaxis secundaria en la cual la enfermedad ya se desarrolló y el paciente está protegido temporal o permanentemente contra la exacerbación o empeoramiento de la enfermedad.
El término "tratamiento" se usa como sinónimo de "terapia". Similarmente el término "tratar" puede considerarse como una aplicación de terapia donde "terapia" es como se define en la presente descripción.
En una modalidad se proporciona una composición farmacéutica que incluye el compuesto de la Fórmula (I), o una sal farmacéuticamente aceptable de este, y al menos un diluyente o portador farmacéuticamente aceptable. En una modalidad, la composición farmacéutica incluye un compuesto de la Fórmula (I) como una base libre. En otra modalidad, la composición farmacéutica incluye una sal farmacéuticamente aceptable de un compuesto de la Fórmula (I).
En una modalidad se proporciona un compuesto de Fórmula (I), o una sal farmacéuticamente aceptable de este, para su uso en la terapia.
En una modalidad se proporciona un compuesto de la Fórmula (I), o una sal farmacéuticamente aceptable de este, para su uso en el tratamiento del cáncer.
En una modalidad se proporciona el uso del compuesto de la Fórmula (I), o una sal farmacéuticamente aceptable de este, para la fabricación de un medicamento para el tratamiento del cáncer.
En una modalidad se proporciona un compuesto de la Fórmula (I), o una sal farmacéuticamente aceptable de este, para su uso en el tratamiento de una enfermedad mediada por GLS1. En una modalidad, la enfermedad mediada por GLS1 es cáncer. En algunas modalidades, el cáncer puede ser cáncer de mama (por ejemplo, cáncer de mama triple negativo), cáncer de pulmón (por ejemplo, cáncer de pulmón de células no pequeñas), cáncer pancreático, cáncer renal, o cáncer hepatocelular.
El "cáncer de mama triple negativo" es cualquier cáncer de mama que no expresa, o subexpresa, los genes para el receptor de estrógeno, receptor de progesterona y Her2/neu.
En una modalidad se proporciona el uso del compuesto de la Fórmula (I), o una sal farmacéuticamente aceptable de este, para la fabricación de un medicamento para el tratamiento de una enfermedad mediada porGLS1. En una modalidad, la enfermedad mediada por GLS1 es cáncer. En algunas modalidades, el cáncer puede ser cáncer de mama (por ejemplo, cáncer de mama triple negativo), cáncer de pulmón (por ejemplo, cáncer de pulmón de células no pequeñas), cáncer pancreático, cáncer renal, o cáncer hepatocelular.
En una modalidad se proporciona el uso del compuesto de la Fórmula (I), o una sal farmacéuticamente aceptable de este, para la fabricación de un medicamento para el tratamiento del cáncer.
En una modalidad se proporcionan compuestos para su uso en un método de inhibición de GLS1 que incluye administrar un compuesto de la Fórmula (I).
En una modalidad se proporcionan compuestos para su uso en un método para tratar una enfermedad en la que la inhibición de GLS1 es beneficiosa en un animal de sangre caliente que necesita tal tratamiento, que incluye administrar al animal de sangre caliente una cantidad terapéuticamente efectiva de un compuesto de la Fórmula (I), o una sal farmacéuticamente aceptable de este.
Los "animales de sangre caliente" incluyen, por ejemplo, humanos.
En una modalidad se proporcionan compuestos para su uso en un método para tratar el cáncer en un animal de sangre caliente que necesita tal tratamiento, que incluye administrar al animal de sangre caliente una cantidad terapéuticamente efectiva de un compuesto de la Fórmula (I), o una sal farmacéuticamente aceptable de este. En algunas modalidades, el cáncer puede ser cáncer de mama (por ejemplo, cáncer de mama triple negativo), cáncer de pulmón (por ejemplo, cáncer de pulmón de células no pequeñas), cáncer pancreático, cáncer renal, o cáncer hepatocelular.
El tratamiento para el cáncer descrito en esta descripción puede aplicarse como una terapia única, o puede implicar, en adición a la administración del compuesto de la Fórmula (I), cirugía convencional, radioterapia, o quimioterapia; o una combinación de tales terapias adicionales. Tal cirugía convencional, radioterapia, o quimioterapia pueden administrarse simultáneamente, secuencialmente, o por separado al tratamiento con el compuesto de la Fórmula (I).
Por lo tanto, en una modalidad se proporciona un compuesto de la Fórmula (I), o una sal farmacéuticamente aceptable de este, y al menos una sustancia antitumoral adicional, para su uso en el tratamiento del cáncer.
En una modalidad se proporciona un compuesto de la Fórmula (I), o una sal farmacéuticamente aceptable de este, y al menos una sustancia antitumoral adicional para su uso en el tratamiento simultáneo, separado o secuencial del cáncer.
En una modalidad se proporciona un compuesto de la Fórmula (I), o una sal farmacéuticamente aceptable de este, para su uso en el tratamiento del cáncer, donde el compuesto de la Fórmula (I) se administra simultáneamente, separadamente, o secuencialmente con al menos una sustancia antitumoral adicional.
En una modalidad se proporcionan compuestos para su uso en un método para tratar el cáncer en un animal de sangre caliente que necesita tal tratamiento, que incluye administrar al animal de sangre caliente un compuesto de la Fórmula (I), o una sal farmacéuticamente aceptable de este y al menos una sustancia antitumoral adicional, en donde las cantidades del compuesto de la Fórmula (I), o una sal farmacéuticamente aceptable de este, y la sustancia antitumoral adicional son conjuntamente efectivas para producir un efecto contra el cáncer.
En una modalidad se proporcionan compuestos para su uso en un método para tratar el cáncer en un animal de sangre caliente que necesita tal tratamiento, que incluye administrar al animal de sangre caliente un compuesto de la Fórmula (I), o una sal farmacéuticamente aceptable de este, y simultáneamente, separadamente o secuencialmente administrar al menos una sustancia antitumoral adicional al animal de sangre caliente, en donde las cantidades del compuesto de la Fórmula (I), o una sal farmacéuticamente aceptable de este, y la sustancia antitumoral adicional son conjuntamente efectivas para producir un efecto contra el cáncer.
En cualquier modalidad la sustancia antitumoral adicional es un taxano. En una modalidad el taxano es paclitaxel. En una modalidad el taxano es docetaxel.
En cualquier modalidad la sustancia antitumoral adicional es una terapia con platino. En una modalidad la terapia con platino es cisplatino, oxaliplatino, o carboplatino.
De acuerdo con una modalidad adicional se proporciona un kit que comprende:
a) Un compuesto de la Fórmula (I), o una sal farmacéuticamente aceptable de este, en una primera forma de dosificación unitaria;
b) Una segunda sustancia antitumoral en una segunda forma de dosificación unitaria;
c) Un recipiente para contener las formas de dosificación unitarias primera y segunda; y, opcionalmente,
d) Instrucciones de uso.
Los compuestos de Fórmula (I), y las sales farmacéuticamente aceptables de estos, pueden administrarse como composiciones farmacéuticas, que comprenden uno o más diluyentes o portadores farmacéuticamente aceptables. En consecuencia, en una modalidad se proporciona una composición farmacéutica que comprende un compuesto de la Fórmula (I), o una sal farmacéuticamente aceptable de este, y al menos un diluyente o portador farmacéuticamente aceptable.
Las composiciones pueden estar en una forma adecuada para uso oral (por ejemplo, como tabletas, pastillas, cápsulas duras o blandas, suspensiones acuosas u oleosas, emulsiones, polvos o gránulos dispersables, jarabes o elixires), para uso tópico (por ejemplo, como cremas, ungüentos, geles, o soluciones o suspensiones acuosas u oleosas), para administración por inhalación (por ejemplo, como un polvo finamente dividido o un aerosol líquido), para administración por insuflación (por ejemplo, como un polvo finamente dividido) o para administración parenteral (por ejemplo, como una solución acuosa u oleosa estéril para la dosificación intravenosa, subcutánea, intramuscular), o como un supositorio. Las composiciones pueden obtenerse por procedimientos convencionales mediante el uso de excipientes farmacéuticos convencionales. Por lo tanto, las composiciones que pretenden tener uso oral pueden contener, por ejemplo, uno o más agentes colorantes, edulcorantes, saborizantes, y/o conservantes.
En una modalidad se incluye una composición farmacéutica que comprende un compuesto de la Fórmula (I), o una sal farmacéuticamente aceptable de este, y al menos un diluyente o portador farmacéuticamente aceptable, para su uso en la terapia.
En una modalidad se proporciona una composición farmacéutica que comprende un compuesto de la Fórmula (I), o una sal farmacéuticamente aceptable de este, y al menos un diluyente o portador farmacéuticamente aceptable, para su uso en el tratamiento del cáncer. En algunas modalidades el cáncer puede ser cáncer de mama (por ejemplo, cáncer de mama triple negativo), cáncer de pulmón (por ejemplo, cáncer de pulmón de células no pequeñas), cáncer pancreático, cáncer renal, o cáncer hepatocelular.
El compuesto de la Fórmula (I) normalmente se administrará a un animal de sangre caliente en una dosis unitaria dentro del intervalo de 5-5.000 mg/m2 de área corporal del animal, es decir, aproximadamente 0,1-100 mg/kg, y esto normalmente proporciona una dosis terapéuticamente efectiva. Una forma de dosis unitaria tal como una tableta o cápsula usualmente contendrá, por ejemplo, 1-250 mg de ingrediente activo. La dosis diaria necesariamente variará en dependencia del huésped tratado, la ruta particular de administración, cualquier terapia que se administre conjuntamente, y la gravedad de la enfermedad que se trate. En consecuencia, el profesional que trata a un paciente en particular puede determinar la dosis óptima.
EJEMPLOS
Las diversas modalidades se ilustran mediante los siguientes ejemplos. La invención no debe interpretarse como limitada a los Ejemplos.
Durante la preparación de los ejemplos, generalmente:
a) Las operaciones se llevaron a cabo a temperatura ambiente, es decir en el intervalo de aproximadamente 17 a 30 °C y bajo una atmósfera de un gas inerte tal como nitrógeno a menos que se indique lo contrario;
b) Las evaporaciones se llevaron a cabo mediante evaporación rotativa o mediante el uso de un equipo Genevac al vacío y los procedimientos de tratamiento se llevaron a cabo después de la eliminación de los sólidos residuales por filtración; c) Las purificaciones por cromatografía instantánea se realizaron en un Isco Combiflash Companion automatizado mediante el uso de columnas de sílice preempaquetadas Grace Resolve, e Isco Combiflash Rf (instantánea de fase inversa) mediante el uso de columnas RediSep Gold C18;
d) Los rendimientos, cuando están presentes, no son necesariamente los máximos alcanzables;
e) Las estructuras de los productos finales de Fórmula (I) se confirmaron por espectroscopía de resonancia magnética nuclear (NMR), con valores de desplazamiento químico de NMR medidos en la escala delta. Los espectros de resonancia magnética de protones se determinaron mediante el uso de un instrumento Bruker Avance 700 (700MHz), Bruker Avance 500 (500 MHz), Bruker 400 (400 MHz) o Bruker 300 (300 MHz); 19F NMR se determinaron a 282 MHz o 376 MHz; 13C NMR se determinaron a 75 MHz o 100 MHz; las mediciones se tomaron a aproximadamente 20 - 30 °C a menos que se especifique lo contrario; se usaron las siguientes abreviaturas: s, singulete; d, doblete; t, triplete; q, cuatriplete; m, multiplete; dd, doblete de dobletes; ddd, doblete de doblete de doblete; dt, doblete de tripletes; bs, señal amplia;
f) Los productos finales de Fórmula (I) se caracterizaron, además, por espectroscopía de masas después de cromatografía líquida (LCMS), mediante el uso de un sistema HPLC basado en un sistema de Lc Waters 2790/95 con un 2996 PDA y un espectrómetro de masas de cuadrupolo simple ZQ 2000 amu. Los solventes usados fueron A= agua, B= acetonitrilo, C= 50:50 acetonitrilo:agua 0,1 % de ácido fórmico y D= 50:50 acetonitrilo:agua 0,1 % de hidróxido de amonio. A una tasa de flujo de 1,1 ml/min se inyectaron 5 pL de muestra en una columna Phenomenex Gemini NX de 50 x 2,1 51 pm. El gradiente varió de 95 % de A a 95 % de B durante 4,0 minutos con una infusión de 5 % de C constante (para el análisis de ácido, D se usa para el análisis de base). El flujo se mantuvo al 95 % de B por 0,5 minutos antes de volver a las condiciones de inicio. Los datos se adquirieron de 150 a 850 amu tanto en modo positivo como negativo en el espectrómetro de masas y a 220 -320 nm en el PDA. La LCMS también se realizó en un sistema UPLC mediante el uso de una bomba Waters Acquity Binary con administrador de muestras, Acquity PDA y un espectrómetro de masas SQD. Los solventes usados fueron A1 =0,1 % de ácido fórmico (ac), B1 0,1 % de ácido fórmico en acetonitrilo, A2 = 0,1 % de hidróxido de amonio (ac) y B20,1 % de hidróxido de amonio en acetonitrilo. A una tasa de flujo de 1 ml/min se inyectó 1 pL de muestra en una columna Waters BEH de 50 x 2,1 1,7um (a 40 °C). El gradiente se extendió de 97 % de A1 a 97 % de B1 durante 1,30 minutos antes de mantenerse por 0,2 minutos y volver a las condiciones de inicio (sustituir A1 y B1 por A2 y B2 para el análisis de base). Los datos se obtuvieron de 150 - 1.000 amu en modo de iones positivos y negativos en el espectrómetro de masas y a 245 -320 amu en el PDA;
g) Generalmente los productos intermedios no se caracterizaron por completo y la pureza se evaluó mediante análisis cromatográfico de capa fina, espectral de masas, HPLC y/o NMR;
h) Las siguientes abreviaturas se usaron: h = hora(s); r.t. = temperatura ambiente (-17-30 °C); conc. = concentrado; FCC = cromatografía en columna instantánea mediante el uso de sílice; AIBN = azoóisisobutironitrilo; DCM = diclorometano; DIPEA = di-isopropil etilamina; DMA = W,W-dimetilacetamida; DMF = W,A/-dimetilformamida; DMSO = dimetilsulfóxido; EDC = 1-etil-3-(3-dimetilaminopropil)carbodiimida; Et2O = éter dietílico; EtOAc = acetato de etilo; EtOH = etanol; HATU = 1-[6is(dimetilamino)metilen]-1H-1,2,3-triazolo[4,5-b]piridinio 3-óxido hexafluorofosfato; HOBT = hidroxibenzotriazol; K2CO3 = carbonato de potasio; MeOH = metanol; MeCN = acetonitrilo; MgSO4 = sulfato de magnesio anhidro; Na2SO4 = sulfato de sodio anhidro; NBS = W-bromo succinimida; TFA = ácido trifluoroacético; THF = tetrahidrofurano; sat. = solución acuosa saturada.
En varios de los ejemplos a continuación, se describe un par de compuestos diastereoméricos. Por ejemplo, los compuestos del Ejemplo 1(a) y del Ejemplo 1(b) representan un par de compuestos diastereoméricos, formados como una mezcla en el producto de una reacción única y posteriormente separados. En tales ejemplos, cualquier asignación de estereoquímica no es absoluta. A modo de ilustración, los Ejemplos 1(a) y 1(b) se refieren a los diastereómeros (2S,3R) y 2R,3R) del compuesto nombrado; sin embargo, no se pretende transmitir que el Ejemplo 1(a) se asigne definitivamente como el diastereómero (2S3R) y el Ejemplo 1(b) como el diastereómero (2R,3R).
Ejemplo 1(a) y 1(b)
(2S)-2-metoxi-2-(3-metoxifenil)-W-[5-[[(3R)-1-(6-metilpiridazin-3-il)pirrolidin-3-il]amino]-1,3,4-tiadiazol-2-il]acetamida y (2R)-2-metoxi-2-(3-metoxifenil)-W-[5-[[(3R)-1-(6-metilpiridazin-3-il)pirrolidin-3-il]amino]-1,3,4-tiadiazol-2-il]acetamida

Se adicionó HATU (329 mg, 0,87 mmol) al ácido 2-metoxi-2-(3-metoxifenil)acético (Producto intermedio 14, 141 mg, 0,72 mmol), N2-[(3R)-1-'(6-metilpiridazin-3-il)pirrolidin-3-il]-1,3,4-tiadiazol-2,5-diamina (Producto intermedio 1, 200 mg, 0,72 mmol) y DlpEA (0,25 ml, 1,44 mmol) en DMF (6 ml) a 21 °C bajo nitrógeno. La solución resultante se agitó a 21 °C por 2 horas. La mezcla bruta se purificó mediante cromatografía de intercambio iónico, mediante el uso de una columna SCX. El producto deseado se eluyó de la columna mediante el uso de NH31 M en MeOH y las fracciones puras se evaporaron hasta la sequedad para proporcionar un producto bruto que se purificó adicionalmente por FCC (SiO2, 0 a 12 % de MeOH en DCM). Las fracciones puras se evaporaron hasta la sequedad para proporcionar la mezcla de diastereoisómeros como una goma de color amarillo pálido (95 mg). Los diastereoisómeros se separaron por HPLC preparativa (columna Lux C2, 20 ^m, 50 mm x250 mm, 100 % de MeOH a 200 ml/min) para producir:
Primer isómero eluido Ejemplo 1(a) 2-metoxi-2-(3-metoxifenil)-N-[5-[[(3R)-1-(6-metilpiridazin-3-il)pirrolidin-3-il]amino]-1.3.4- tiadiazol-2-il]acetamida (58 mg, 18 %). 1H NMR (400 MHz, DMSO-d6, 30 °C) 52,05 (1H, td), 2,21 - 2,35 (1H, m), 2.41 (3H, s), 3,40 - 3,47 (1H, m), 3,55 (2H, ddd), 3,69 - 3,75 (4H, m), 4,31 - 4,46 (1H, m), 4,93 (1H, s), 6,82 (1H, d), 6,87 -6.94 (1H, m), 6 ,99-7,07 (2H, m), 7,22 (1H, d), 7,29 (1H, t), 7,61 (1H, d), 12,13 (1H, s). m/z: ES+ [M+H]+ 456.
Segundo isómero eluido Ejemplo 1(b) 2-metoxi-2-(3-metoxifenil)-N-[5-[[(3R)-1-(6-metilpiridazin-3-il)pirrolidin-3-il]amino]-1.3.4- tiadiazol-2-il]acetamida (55 mg, 17 %). 1H NMR (400 MHz, DMSO-d6, 30 °C) 52,05 (1H, td), 2,21 - 2,35 (1H, m), 2.41 (3H, s), 3,4 - 3,47 (1H, m), 3,55 (2H, ddd), 3,69 - 3,75 (4H, m), 4,31 - 4,46 (1H, m), 4,93 (1H, s), 6,82 (1H, d), 6,87 -6.94 (1H, m), 6 ,99-7,07 (2H, m), 7,22 (1H, d), 7,29 (1H, t), 7,61 (1H, d), 12,13 (1H, s). m/z: ES+ [M+H]+456.
Ejemplo 2
(2S)-N-[5-[[(3R)-1-(6-fluoropiridazin-3-il)pirrolidin-3-il]amino]-1,3,4-tiadiazol-2-il]-2-metoxi-2-fenil-acetamida

Se adicionó HOBT (120 mg, 0,78 mmol) a N2-[(3R)-1-(6-fluoropiridazin-3-il)pirrolidin-3-il]-1,3,4-tiadiazol-2,5-diamina (Producto intermedio 4, 220 mg, 0,78 mmol), ácido (2S)-2-metoxi-2-fenil-acético (130 mg, 0,78 mmol) y EDC (300 mg, 1,56 mmol) en DMF (3 ml) a 25 °C. La mezcla resultante se agitó a 25 °C por 3 horas. El producto bruto se purificó por HPLC preparativa (columna XBridge C18 OBD, 5 ^m, 50 mm x 150 mm). Se usaron decrecientemente mezclas polares de agua (que contenían ácido fórmico al 0,05 %) y MeCN como fase móvil. Las fracciones que contenían el compuesto deseado se evaporaron hasta la sequedad para proporcionar el Ejemplo 2 (2S)-N-[5-[[(3R)-1-(6-fluoropiridazin-3-il)pirrolidin-3-il]amino]-1,3,4-tiadiazol-2-il]-2-metoxi-2-fenil-acetamida (95 mg, 27,8 %) como un sólido blanco. 1H NMR (400 MHz, MeOD, 22 °C) 52,16 - 2,23 (m, 1H), 2,36-2,44 (m, 1H), 3,44 (s, 3H), 3,55 - 3,69 (m, 3H), 3,82 - 3,86 (m, 1H), 4,46 -4,51 (m, 1H), 4,93 (s, 1H), 7,15-7,19 (m, 1H), 7,24-7,27 (m, 1H), 7,35-7,43 (m, 3H), 7,47 - 7,49 (m, 2H). m/z: ES+ [M+H]+ 430.
Ejemplo 3
(2S)-N-[5-[[(3R)-1-(6-fluoropiridazin-3-il)pirrolidin-3-il]amino]-1,3,4-tiadiazol-2-il]-2-metoxi-2-(3-metoxifenil)acetamida

Se adicionó HATU (405 mg, 1,07 mmol) al ácido (2S)-2-metoxi-2-(3-metoxifenil)acético (Producto intermedio 12, 174 mg, 0,89 mmol), N2-[(3R)-1-(6-fluoropiridazin-3-il)pirrolidin-3-il]-1,3,4-tiadiazol-2,5-diamina (Producto intermedio 4, 250 mg, 0,89 mmol) y DIPEA (0,155 ml, 0,89 mmol) en DMF (8 ml) a 21 °C bajo N2. La solución resultante se agitó a 0 °C por 45 minutos. El producto bruto se purificó mediante cromatografía de intercambio iónico, mediante el uso de una columna SCX. El producto deseado se eluyó de la columna mediante el uso de NH31 M en MeOH y las fracciones puras se evaporaron hasta la sequedad para proporcionar una goma. El producto bruto se purificó por FCC (SiO2, 0 a 9 % de MeOH en DCM). Las fracciones puras se evaporaron hasta la sequedad, se trituraron con éter/DCM y se filtraron para proporcionar el Ejemplo 3 (2S)-N-[5-[[(3R)-1-(6-fluoropiridazin-3-il)pirrolidin-3-il]amino]-1,3,4-tiadiazol-2-il]-2-metoxi-2-(3-metoxifenil)acetamida (185 mg, 45 %) como un sólido crema. 1H NMR (400 MHz, DMSo-d6, 30 °C) 52,12 (1H, td), 2,3 -2,46 (1H, m), 3,37 (3H, s), 3,52 (1H, dd), 3,59 - 3,67 (2H, m), 3,81 (4H, m), 4,34 - 4,56 (1H, m), 5,00 (1H, s), 6,97 (1H, ddd), 7,03 - 7,14 (2H, m), 7,22 (1H, dd), 7,32 - 7,51 (2H, m), 7,73 (1H, d), 12,20 (1H, s). m/z: ES+ [M+H]+ 486.
Ejemplos 4(a) y 4(b)
(2S)-2-etoxi-2-(3-metoxifenil)-N-[5-[[(3R)-1-(6-metilpiridazin-3-il)pirrolidin-3-il]amino]-1,3,4-tiadiazol-2-il]acetamida y (2R)-2-etoxi-2-(3-metoxifenil)-N-[5-[[(3R)-1-(6-metilpiridazin-3-il)pirrolidin-3-il]amino]-1,3,4-tiadiazol-2-il]acetamida

Se pesaron ácido 2-etoxi-2-(3-metoxifenil)acético (Producto intermedio 15, 0,11 g, 0,54 mmol) y N2-[(3R)-1-(6-metilpiridazin-3-il)pirrolidin-3-il]-1,3,4-tiadiazol-2,5-diamina (Producto intermedio 1, 0,15 g, 0,54 mmol) en un matraz de fondo redondo seguido de DlPEA (0,1 ml, 0,54 mmol) y d Mf (5 ml). La solución resultante se trató después con HATU (0,21 g, 0,54 mmol) y se dejó agitar a r.t. bajo N2 por 24 horas. El solvente se eliminó bajo presión reducida y la goma residual se disolvió en DCM, se adsorbió sobre sílice y se purificó por FCC (SiO2 0-10 % de MeOH en DCM). La evaporación de las fracciones puras bajo presión reducida produjo el compuesto del título como una espuma de color amarillo claro. La espuma se disolvió en metanol y se adicionó a una columna de intercambio iónico SCX que se lavó con DCM, después metanol y después se eluyó con NH32M en MeOH. El solvente se eliminó bajo presión reducida y se purificó adicionalmente mediante HPLC preparativa (columna SunFire C18, 5 ^m, 50 mm x 19 mm, tasa de flujo de 25 ml/min). Se usaron decrecientemente relaciones polares de agua y MeCN que contenían 0,1 % de ácido fórmico como fase móvil. Las fracciones puras se evaporaron hasta la sequedad para proporcionar el producto como una mezcla de diastereoisómeros como un sólido blanco (67 mg). Los diastereoisómeros se separaron por HPLC preparativa (columna Phenomenex Lux C4, 20 ^m, 50 mm x 250 mm, MeOH a 120 ml/min) para producir:
Primer isómero eluido ejemplo 4(a) 2-etoxi-2-(3-metoxifenil)-N-[5-[[(3R)-1-(6-metilpiridazin-3-il)pirrolidin-3-il]amino]-1,3,4-tiadiazol-2-il]acetamida (25 mg, 37 %). 1H NMR (400 MHz, DMSO-d6, 30 °C) 5 1,17 (3H, t), 1,99 - 2,09 (1H, m), 2,22 -2,32 (1H, m), 2,40 (3H, s), 3,40 - 3,56 (5H, m), 3,69 - 3,73 (1H, m), 3,74 (3H, s), 4,32 - 4,40 (1H, m), 5,04 (1H, s), 6,83 (1H, d), 6,89 (1H, dd), 7,00 - 7,05 (2H, m), 7,22 (1H, d), 7,28 (1H, t), 7,65 (1H, d), 12,10 (1H, s). m/z: ES+ [M+H]+ 470. Segundo isómero eluido ejemplo 4(b) 2-etoxi-2-(3-metoxifenil)-N-[5-[[(3R)-1-(6-metilpiridazin-3-il)pirrolidin-3-il]amino]-1,3,4-tiadiazol-2-il]acetamida (25 mg, 37 %). 1H NMR (400 MHz, DMSO-d6, 30 °C) 51,17 (3H, t), 1,97 - 2,09 (1H, m), 2,21 - 2,31 (1H, m), 2,40 (3H, s), 3,39 - 3,56 (5H, m), 3,70 - 3,73 (1H, m), 3,74 (3H, s), 4,33 - 4,40 (1H, m), 5,04 (1H, s), 6,84 (1H, d), 6,87 - 6,92 (1H, m), 7,01 - 7,05 (2H, m), 7,23 (1H, d), 7,28 (1H, t), 7,65 (1H, d), 12,11 (1H, s). m/z: ES+ [M+H]+ 470.
Ejemplos 5(a) y 5(b)
(2S)-2-(4-fluorofenil)-2-metoxi-N-[5-[[(3R)-1-(6-metilpiridazin-3-il)pirrolidin-3-il]amino]-1,3,4-tiadiazol-2-il]acetamiday (2R)-2-(4-fluorofenil)-2-metoxi-N-[5-[[(3R)-1-(6-metilpiridazin-3-il)pirrolidin-3-il]amino]-1,3,4-tiadiazol-2-il]acetamida

N2-[(3R)-1-(6-met¡lp¡r¡daz¡n-3-il)p¡rrol¡d¡n-3-¡l]-1,3,4-t¡ad¡azol-2,5-d¡am¡na (Producto intermedio 1, 0,15 g, 0,541 mmol) y ácido 2-(4-fluorofenil)-2-metoxiacét¡co (Producto intermedio 16, 0,1 g, 0,541 mmol) se disolvieron en DMF (2 ml) a r.t bajo N2. La mezcla se agitó durante 5 minutos antes de la adición de DIPEA (0,34 ml, 1,943 mmol) y HATU (0,21 g, 0,541 mmol), después a r.t. por 2 horas. La mezcla bruta se pasó después a través de una columna SCX de 5 g se lavó con MeoH después se eluyó con NH32N en MeOH. La fracción básica se evaporó bajo presión reducida para producir una goma naranja que se purificó por HPLC preparativa (columna SunFire C18, 5 ^m, 50 mm x 19 mm, tasa de flujo 25 ml/min). Se usaron decrecientemente relaciones polares de agua y MeCN que contenían 0,1 % de ácido fórmico como fase móvil. Las fracciones puras se combinaron, se evaporaron bajo presión reducida y se pasaron a través de una columna SCX de 2 g se lavaron con MeOH y después se eluyeron con NH32N en MeOH. La fracción básica se evaporó hasta la sequedad para proporcionar la mezcla de diastereoisómeros como una espuma blanquecina. Los diastereoisómeros se separaron por HPLC preparativa (columna Amy-C, 5 |jm, 20 mm x250 mm, 2:3 heptano:EtOH que contiene 0,1 % v/vde modificador NH3 a 21 ml/min) para producir:
Primer isómero eluido Ejemplo 5(a) 2-(4-fluorofenil)-2-metoxi-N-[5-[[(3R)-1 -(6-met¡lp¡ridaz¡n-3-¡l)p¡rrol¡d¡n-3-¡l]am¡no]-1,3,4-tiadiazol-2-¡l]acetam¡da (55,3 mg, 23,0 %). 1H NMR (400 MHz, DMSO-d6, 21 °C) 52,04 (1H, dq), 2,26 (1H, dt), 2,41 (3H, s), 3,30 (3H, s), 3,57 - 3,41 (3H, m), 3,72 (1H, dd), 4,36 (1H, q), 4,98 (1H, s), 6,83 (1H, d), 7,22 (3H, ddd), 7,55 - 7,44 (2H, m), 7,66 (1H, d), 12,28 (s, 1H). m/z: ES+ [M+H]+ 444.
Segundo isómero eluido, Ejemplo 5(b) 2-(4-fluorofen¡l)-2-metox¡-N-[5-[[(3R)-1-(6-met¡lp¡ridaz¡n-3-¡l)p¡rrol¡d¡n-3-¡l]am¡no]-1,3,4-tiadiazol-2-¡l]acetam¡da (59,2 mg, 24,7 %). 1H NMR (400 MHz, DMSO-d6, 21 °C) 52,04 (1H, dq), 2,31 - 2,23 (1H, m), 2,41 (3H, s), 3,30 (3H, s), 3,44 (1H, dd), 3,59 - 3,49 (2H, m), 3,71 (1H, dd), 4,36 (1H, q), 4,97 (1H, s), 6,82 (1H, d), 7,22 (3H, ddd), 7,53- 7,46 (2H, m), 7,64 (1H, d), 12,28 (1H, s). m/z: ES+ [M+H]+ 444.
Ejemplos 6(a) y 6(b)
(2S)-2-(4-fluoro-3-metox¡-fen¡l)-2-metox¡-N-[5-[[(3R)-1-(6-met¡lp¡r¡dazin-3-¡l)p¡rrol¡d¡n-3-¡l]am¡no]-1,3,4-t¡ad¡azol-2-il]acetamida y (2R)-2-(4-fluoro-3-metoxi-fenil)-2-metox¡-N-[5-[[(3R)-1 -(6-met¡lp¡ridaz¡n-3-¡l)p¡rrol¡d¡n-3-¡l]am¡no]-1,3,4-tiadiazol-2-¡l]acetam¡da

N2-[(3R)-1-(6-met¡lp¡r¡daz¡n-3-il)p¡rrol¡d¡n-3-¡l]-1,3,4-t¡ad¡azol-2,5-d¡am¡na (Producto intermedio 1, 200 mg, 0,72 mmol) y ácido 2-(4-fluoro-3-metoxi-fenil)-2-metox¡-acét¡co (Producto intermedio 18, 150 mg, 0,72 mmol) se disolvieron en DMF (2 ml) a r.t. bajo nitrógeno. La mezcla se agitó por 5 minutos antes de la adición de DIPEA (0,34 ml, 1,94 mmol) y HATU (0,27 g, 0,72 mmol) y después se agitó a r.t. durante la noche. La mezcla cruda se pasó a través de una columna SCX de 5 g se lavó con MeOH y después se eluyó con NH32M en MeOH. La fracción básica se evaporó para producir el producto bruto como una goma naranja que se purificó por HPLC preparativa (columna SunFire C18, 5 ^m, 50 mm x 19 mm, tasa de flujo 25 ml/min). Se usaron decrecientemente relaciones polares de agua y MeCN que contenían 0,1 % de ácido fórmico como fase móvil. Las fracciones puras se combinaron, se evaporaron y se pasaron a través de una columna SCX de 2 g se lavaron con MeOH y después se eluyeron con NH32M en MeOH. La fracción básica se evaporó para producir la mezcla
de diastereoisómeros como un sólido blanquecino. Los diastereoisómeros se separaron después por SFC (columna Lux C3, 5 ^m, 21,2 mm x 250 mm, MeOH/CO235 % que contiene modificador NH3, 50 ml/min) para producir:
Primer isómero eluido Ejemplo 6(a) 2-(4-fluoro-3-metoxi-fenil)-2-metoxi-N-[5-[[(3R)-1-(6-metilpiridazin-3-il)pirrolidin-3-il]amino]-1,3,4-tiadiazol-2-il]acetamida (34 mg, 10%). 1H NMR (400 MHz, DMSO-d6, 21 °C) 5 1,98-2,12 (1H, m), 2,21 -2,33 (1H, m), 2,41 (3H, s), 3,32 (3H, s), 3,40 - 3,58 (3H, m), 3,69 - 3,79 (1H, m), 3,84 (3H, s), 4,32 - 4,41 (1H, m), 4,95 (s, 1H), 6,83 (1H, d), 6,98 - 7,05 (1H, m), 7,19 - 7,28 (3H, m), 7,68 (1H, d). m/z: ES+[M+H]+ 474.
Segundo isómero eluido Ejemplo 6(b) 2-(4-fluoro-3-metoxi-fenil)-2-metoxi-N-[5-[[(3R)-1-(6-metilpiridazin-3-il)pirrolidin-3-il]amino]-1,3,4-tiadiazol-2-il]acetamida (44 mg, 13%). 1H NMR (400 MHz, DMSO-d6, 21 °C) 5 1,99-2,10 (1H, m), 2 ,23 2,34 (1H, m), 2,41 (3H, s), 3,31 (3H, s), 3,41 - 3,48 (1H, m), 3,50 - 3,56 (2H, m), 3,67 - 3,78 (1H, m), 3,84 (3H, s), 4,32 -4,39 (1H, m), 4,94 (1H, s), 6,82 (1H, d), 6,98 - 7,04 (1H, m), 7,17 - 7,30 (3H, m), 7,65 (1H, d). m/z: ES+[M+H]+ 474.
Ejemplos 7(a) y 7(b)
(2S)-2-metoxi-N-[5-[[(3R)-1-(6-metilpiridazin-3-il)pirrolidin-3-il]amino]-1,3,4-tiadiazol-2-il]-2-[3-(trifluorometoxi)fenil]acetamida y (2R)-2-metoxi-N-[5-[[(3R)-1-(6-metilpiridazin-3-il)pirrolidin-3-il]amino]-1,3,4-tiadiazol-2-il]-2-[3-(trifluorometoxi)fenil]acetamida

DIPEA (0,14 ml, 0,81 mmol), HATU (247 mg, 0,65 mmol) y ácido 2-metoxi-2-[3-(trifluorometoxi)fenil]acético (Producto intermedio 19, 160 mg, 0,65 mmol) se adicionaron a una solución de N2-[(3R)-1-(6-metilpiridazin-3-il)pirrolidin-3-il]-1,3,4-tiadiazol-2,5-diamina (Producto intermedio 1, 150 mg, 0,54 mmol) en d Mf (4 ml). La mezcla se agitó a r.t. por 18 h. Después esta se diluyó con agua (5 ml) y después se extrajo en DCM (10 ml), se evaporó y purificó por HPLC preparativa (columna XBridge o Bd C18, 5 ^m, 50 mm x 19 mm, tasa de flujo de 25 ml/min). Se usaron decrecientemente relaciones polares de agua y MeCN que contienen 0,3 ml/L de NH4OH como una fase móvil. Las fracciones puras se evaporaron después para producir una mezcla de diastereoisómeros. Los diastereoisómeros se separaron por HPLC preparativa (columna Amy-C, 5 ^m, 4,6 mm x 250 mm, heptano/EtOH 7/3 que contiene modificador NH3, 21 ml/min) para producir: Primer isómero eluido Ejemplo 7(a) 2-metoxi-N-[5-[[(3R)-1-(6-metilpiridazin-3-il)pirrolidin-3-il]amino]-1,3,4-tiadiazol-2-il]-2-[3-(trifluorometoxi)fenil]acetamida (50 mg, 18 %). 1H NMR (400 MHz, DMSO-d6, 21 °C) 5 1,96 - 2,11 (1H, m), 2,20 - 2,37 (1H, m), 2,41 (3H, s), 3,34 (3H, s), 3,42 - 3,60 (3H, m), 3,66 - 3,78 (1H, m), 4,37 (1H, q), 5,07 (1H, s), 6,84 (1H, d), 7,23 (1H, d), 7,37 (1H, d), 7,41 - 7,58 (3H, m), 7,72 (1H, d), 12,37 (1H, s). m/z: ES+[M+H]+ 510.
Segundo isómero eluido Ejemplo 7(b) 2-metoxi-N-[5-[[(3R)-1-(6-metilpiridazin-3-il)pirrolidin-3-il]amino]-1,3,4-tiadiazol-2-il]-2-[3-(trifluorometoxi)fenil]acetamida (34 mg, 12 %). 1H NMR (400 MHz, DMSO-d6, 21 °C) 5 1,97 - 2,13 (1H, m), 2,20 -2,34 (1H, m), 2,41 (3H, s), 3,34 (3H, s), 3,39 - 3,60 (3H, m), 3,66-3,77 (1H, m), 4,36 (1H, d), 5,07 (1H, s), 6,83 (1H, d), 7,23 (1H, d), 7,37 (1H, d), 7 ,43-7,60 (3H, m), 7,72 (1H, d), 12,36 (1H, s). m/z: ES+[M+H]+510.
Ejemplos 8(a) y 8(b)
(2S)-[3-(difluorometoxi)fenil]-2-metoxi-N-[5-[[(3R)-1-(6-metilpiridazin-3-il)pirrolidin-3-il]amino]-1,3,4-tiadiazol-2-il]acetamida y (2R)-[3-(difluorometoxi)fenil]-2-metoxi-N-[5-[[(3R)-1-(6-metilpiridazin-3-il)pirrolidin-3-il]amino]-1,3,4-tiadiazol-2-il]acetamida

Se pesaron ácido 2-[3-(difluorometoxi)fenil]-2-metoxi-acético (Producto intermedio 20, 0,11 g, 0,469 mmol) y N2-[(3R)-1-(6-metilpiridazin-3-il)pirrolidin-3-il]-1,3,4-tiadiazol-2,5-diamina (Producto intermedio 1, 0,13 g, 0,469 mmol) en un matraz de fondo redondo. Se adicionaron DMF (3 ml) y DIPEA (0,15 g, 1,172 mmol) seguido de HATU (0,18 g, 0,469 mmol) y la solución resultante se dejó agitar a r.t. bajo nitrógeno por 15 h. El solvente se eliminó bajo presión reducida y la goma residual se disolvió en DCM, se absorbió sobre sílice y se purificó por FCC (SiO2, 1-10 % de MeOH que contiene 0,1 % de NH3 en DCM). La evaporación de las fracciones puras bajo presión reducida dio una goma de color amarillo pálido que se separó porsFC quiral preparativa (columna Lux C3, 5 ^m, 21,2 mmx250 mm, tasa de flujo de 50 ml/min a una longitud de onda de 210 nm con 40:60 MeOH:CO2 0,1 % de NH3 como eluyente) para producir:
Primer isómero eluido Ejemplo 8(a) [3-(difluorometoxi)fenil]-2-metoxi-N-[5-[[(3R)-1-(6-metilpiridazin-3-il)pirrolidin-3-il]amino]-1,3,4-tiadiazol-2-il]acetamida (48 mg, 20 %). 1H NMR (400 MHz, DMSO-d6, 30 °C) 52,07 - 2,00 (1H, m), 2,31 -2,22 (1H, m), 2,40 (3H, s), 3,34 (3H, s), 3,58 - 3,41 (3H, m), 3,71 (1H, dd), 4,38 - 4,34 (1H, m), 5,01 (1H, s), 6,83 (1H, d), 7,49 - 7,04 (6H, m), 7,71 (1H, d), 12,31 (1H, s). m/z: ES+ [M+H]+ 492.
Segundo isómero eluido Ejemplo 8(b) [3-(difluorometoxi)fenil]-2-metoxi-N-[5-[[(3R)-1-(6-metilpiridazin-3-il)pirrolidin-3-il]amino]-1,3,4-tiadiazol-2-il]acetamida (45 mg, 19 %). 1H NMR (400 MHz, DMSO-d6, 30 °C) 52,08 - 2,00 (1H, m), 2,34 -2,21 (1H, m), 2,40 (3H, s), 3,32 (3H, s), 3,57 - 3,42 (3H, m), 3,71 (1H, dd), 4,36 (1H, m), 5,01 (1H, s), 6,82 (1H, d), 7,49 -7,04 (6H, m), 7,69 (1H, d), 12,26 (1H, s). m/z: ES+ [M+H]+ 492.
Ejemplos 9(a) y 9(b)
(2S)-2-[3-(difluorometoxi)fenil]-N-[5-[[(3R)-1-(6-fluoropiridazin-3-il)pirrolidin-3-il]amino]-1,3,4- tiadiazol-2-il]-2-metoxiacetamida y (2R)-2-[3-(difluorometoxi)fenil]-N-[5-[[(3R)-1-(6-fluoropiridazin-3-il)pirrolidin-3-il]amino]-1,3,4-tiadiazol-2-il]-2-metoxi-acetamida

Se adicionó HATU (811 mg, 2,13 mmol) al ácido 2-[3-(difluorometoxi)fenil]-2-metoxi-acético (Producto intermedio 20, 454 mg, 1,96 mmol), N2-[(3R)-1-(6-fluoropiridazin-3-il)pirrolidin-3-il]-1,3,4-tiadiazol-2,5-diamina (Producto intermedio 4, 500 mg, 1,78 mmol) y DIp Ea (0,31 ml, 1,78 mmol) en DMF (12 ml) a 21 °C bajo N2. La solución resultante se agitó a 21 °C por 45 minutos. El producto bruto se purificó mediante cromatografía de intercambio iónico, mediante el uso de una columna SCX. El producto deseado se eluyó de la columna mediante el uso de NH31 M en MeOH y las fracciones se evaporaron hasta una goma. El producto bruto se purificó por FCC (SiO2, 0 - 8 % de MeOH en DCM). Las fracciones se evaporaron hasta la sequedad para producir un sólido gomoso. El producto bruto se purificó adicionalmente por FCC (SiO2, 0 - 9 % de MeOH en EtoAc). Las fracciones puras se evaporaron hasta la sequedad, se trituraron con DCM/étery se filtraron para proporcionar la mezcla de diastereoisómeros como un sólido amarillo (210 mg). Los diastereoisómeros se separaron por HpLC preparativa (columna Phenomenex Lux C2, 20 ^m, 50 mm x 250 mm mediante el uso de EtOH como eluyente a 120 ml/min) para producir:
Primer isómero eluido Ejemplo 9(a) (2S)-2-[3-(difluorometoxi)fenil]-N-[5-[[(3R)-1-(6-fluoropiridazin-3-il)pirrolidin-3-ilo]amino]-1,3,4-tiadiazol-2-il]-2-metoxiacetamida (72 mg, 8 %). 1H NMR (400 MHz, DMSO, 30 °C) 52,02 (1H, dd), 2,25 (1H, dq), 3,29 (3H, s), 3,39 - 3,57 (3H, m), 3,70 (1H, dd), 4,34 (1H, d), 4,98 (1H, s), 6,97 - 7,25 (3H, m), 7,27 - 7,35 (2H, m), 7,38- 7,48 (1H, m), 7,64 (1H, d), 12,19 (1H, s). m/z: ES-[M-H]- 494.
Segundo isómero eluido Ejemplo 9(b) (2R)-2-[3-(difluorometoxi)fenil]-N-[5-[[(3R)-1-(6-fluoropiridazin-3-il)pirrolidin-3-il]amino]-1,3,4-tiadiazol-2-il]-2-metoxiacetamida (95 mg, 11 %). 1H NMR (400 MHz, DMSO, 30 °C) 52,02 (1H, dd), 2,25 (1H, dq), 3,29 (3H, s), 3,39 - 3,57 (3H, m), 3,70 (1H, dd), 4,34 (1H, d), 4,98 (1H, s), 6,97 - 7,25 (3H, m), 7,27 - 7,35 (2H, m), 7,38- 7,48 (1H, m), 7,64 (1H, d), 12,19 (1H, s). m/z: ES-[M-H]- 494.
Ejemplos 10(a) y 10(b)
(2S)-2-(4-fluorofenil)-2-metoxi-N-[5-[[(3R)-1-(5-metilpiridazin-3-il)pirrolidin-3-il]amino]-1,3,4-tiadiazol-2-il]acetamiday (2R)-2-(4-fluorofenil)-2-metoxi-N-[5-[[(3R)-1-(5-metilpiridazin-3-il)pirrolidin-3-il]amino]-1,3,4-tiadiazol-2-il] acetamida

Se disolvieron N2-[(3R)-1-(5-metilpiridazin-3-il)pirrolidin-3-il]-1,3,4-tiadiazol-2,5-diamina (Producto intermedio 7, 100 mg, 0,36 mmol) y ácido 2-(4-fluorofenil)-2-metoxiacético (Producto intermedio 16, 66 mg, 0,36 mmol) en DMF (2,0 ml) a r.t. bajo N2. La mezcla se agitó por 5 minutos antes de la adición de DIPEA (0,09 ml, 0,54 mmol) y HATU (165 mg, 0,43 mmol) y después a r.t. durante la noche. La mezcla bruta se pasó a través de una columna SCX de 5 g, se lavó con MeOH, después se eluyó con NH32M en MeOH. La fracción básica se evaporó para producir una goma naranja. El producto bruto se purificó por HPLC preparativa (columna SunFire C18, 5 ^m, 50 mm x 19 mm a 25 ml/min). Se usaron decrecientemente relaciones polares de agua y MeCN que contenían 0,1 % de ácido fórmico como fase móvil. Las fracciones que contenían la masa deseada se combinaron, se evaporaron y se pasaron a través de una columna SCX de 5 g se lavaron con MeOH y después se eluyeron con NH32M en MeOH. La fracción básica se evaporó para producir la mezcla de diastereoisómeros como un sólido blanquecino (70 mg). Los diastereoisómeros se separaron por HPLC preparativa (columna Lux C4, 20 ^m, 50 mm x250 mm, 100 % de MeOH a 120 ml/min) para producir:
Primer isómero eluido Ejemplo 10(a) 2-(4-fluorofenil)-2-metoxi-N-[5-[[(3R)-1-(5-metilpiridazin-3-il)pirrolidin-3-il]amino]-1.3.4- tiadiazol-2-il]acetamida (25 mg, 16 %). 1H NMR (400 MHz, DMSO, 30 °C) 2,01 - 2,11 (1H, m), 2,20 (3H, s), 2,28 (1H, dt), 3,31 (3H, s), 3,48 (1H, dd), 3,52 - 3,61 (2H, m), 3,74 (1H, dd), 4,33 - 4,41 (1H, m), 4,98 (1H, s), 6,69 (1H, s), 7,21 (2H, t), 7,47 - 7,53 (2H, m), 7,63 (1H, d), 8,36 (1H, s), 12,20 (1H, s). m/z: ES+ [M+H]+ 444.
Segundo isómero eluido, Ejemplo 10(b) 2-(4-fluorofenil)-2-metoxi-N-[5-[[(3R)-1-(5-metilpiridazin-3-il)pirrolidin-3-il]amino]-1.3.4- tiadiazol-2-il]acetamida (26 mg, 17%). 1H NMR (400 MHz, DMSO, 30 °C) 2,00-2,1 (1H, m), 2,21 (3H, s), 2,27 (1H, dt), 3,31 (3H, s), 3,45 - 3,60 (3H, m), 3,74 (1H, dd), 4,32 - 4,40 (1H, m), 4,97 (1H, s), 6,69 (1H, d), 7,17 - 7,25 (2H, m), 7,47 - 7,53 (2H, m), 7,59 (1H, d), 8,36 (1H, d). m/z: ES+ [M+H]+ 444.
Ejemplos 11(a) y 11(b)
(2S)-N-[5-[[(3R)-1-(5-cloropiridazin-3-il)pirrolidin-3-il]amino]-1,3,4-tiadiazol-2-il]-2-metoxi-2-(4-metoxifenil)acetamida y (2R)-N-[5-[[(3R)-1-(5-cloropiridazin-3-il)pirrolidin-3-il]amino]-1,3,4-tiadiazol-2-il]-2-metoxi-2-(4-metoxifenil)acetamida
Se disolvieron N2-[(3R)-1-(5-clorop¡r¡daz¡n-3-il)p¡rrol¡d¡n-3-¡l]-1,3,4-t¡ad¡azol-2,5-d¡am¡na (Producto intermedio 9, 100 mg, 0,34 mmol) y ácido 2-metoxi-2-(4-metoxifen¡l)acét¡co (Producto intermedio 21, 70 mg, 0,34 mmol) en DMF (2,0 ml) y se trataron con DIPEA (0,15 ml, 0,84 mmol) y hAt U (190 mg, 0,50 mmol) después se agitó a r.t. durante la noche. La mezcla bruta se diluyó con agua (10 ml) y se extrajo en DCM (10 ml). Después se evaporó el DCM y el producto bruto se purificó por HPLC preparativa (columna SunFire C18, 5 ^m, 50 m mx 19 mm a 25 ml/min). Se usaron decrecientemente relaciones polares de agua y MeCN que contenían 0,1 % de ácido fórmico como fase móvil. Las fracciones que contenían la masa deseada se combinaron, se evaporaron y se pasaron a través de una columna SCX se lavaron con MeOH y después se eluyeron con NH32M en MeOH. La fracción básica se evaporó para producir la mezcla de diastereoisómeros como un sólido blanquecino (35 mg). Los diastereoisómeros se separaron por HPLC preparativa (columna Lux C4, 20 ^m, 50 mm x 250 mm, 100 % de MeOH a 120 ml/min) para producir:
Primer isómero eluido Ejemplo 11(a) N-[5-[[(3R)-1-(5-clorop¡r¡daz¡n-3-¡l)pirrol¡d¡n-3-¡l]am¡no]-1,3,4-t¡ad¡azol-2-¡l]-2-metox¡-2-(4-metoxifenil)acetamida (12 mg, 7,5 %) 1H NMR (400 MHz, DMSO, 30 °C) 2,03 - 2,13 (1H, m), 2,23 - 2,33 (1H, m), 3,49 - 3,63 (3H, m), 3,75 (3H, s), 3,76 - 3,80 (1H, m), 4,33 - 4,42 (1H, m), 4,89 (1H, s), 6,90 - 6,96 (2H, m), 7,10 (1H, d), 7,34 -7,41 (2H, m), 7,60 (1H, d), 8,55 (1H, d), 12,10 (1H, s); metro/z: ES+ [M H]+ 476. más 3H oscurecido por el pico del agua a 3,3 ppm
Segundo isómero eluido Ejemplo 11(b) N-[5-[[(3R)-1-(5-clorop¡r¡daz¡n-3-il)p¡rrol¡d¡n-3-¡l]am¡no]-1,3,4-t¡ad¡azol-2-¡l]-2-metoxi-2-(4-metoxifen¡l)acetam¡da (15 mg, 9,4%). 1H NMR (400 MHz, DMSO, 30 °C) 2,01 -2,12 (1H, m), 2,22-2,31 (1H, m), 3,27 (3H, s), 3,47 - 3,62 (3H, m), 3,75 (3H, s), 3,76 - 3,79 (1H, m), 4,33 - 4,42 (1H, m), 4,86 (1H, s), 6,89 - 6,96 (2H, m), 7,10 (1H, d), 7,34-7,41 (2H, m), 7,51 (1H, d), 8,55 (1H, d). m/z: ES+ [M+H]+476.
Ejemplo 12
(2S)-N-[5-[[(3R)-1-(5-clorop¡r¡daz¡n-3-¡l)p¡rrol¡din-3-¡l]am¡no]-1,3,4-t¡ad¡azol-2-¡l]-2-metox¡-2-fen¡l-acetam¡da
Se adicionaron DIPEA (0,04 ml, 0,20 mmol), N2-[(3R)-1-(5-clorop¡r¡daz¡n-3-il)p¡rrol¡d¡n-3-¡l]-1,3,4-t¡ad¡azol-2,5-d¡am¡na (Producto intermedio 9, 40 mg, 0,13 mmol) y ácido (2S)-2-metoxi-2-fenilacét¡co (20 mg, 0,13 mmol) a una solución de HATU (61 mg, 0,16 mmol) en DMF (2 ml). La mezcla se agitó a 25 °C por 18 horas. Después esta se diluyó con agua (5 ml) y después se extrajo en DCM (10 ml) y se evaporó bajo presión reducida. El producto bruto se purificó por HPLC preparativa (columna SunFire C18, 5 ^m, 50 mm x 19 mm a 25 ml/min). Se usaron decrecientemente relaciones polares de agua y MeCN que contenían 0,1 % de ácido fórmico como fase móvil. Después las fracciones apropiadas se evaporaron y se purificaron nuevamente por cromatografía preparativa básica. Se usó una columna XBridge (5 micras, C18, 50x19 mm) . Se usaron decrecientemente relaciones polares de agua que contenía 0,1 % de hidróxido de amonio y acetonitrilo como fase móvil. Después las fracciones puras se evaporaron y se secaron en el horno de vacío para proporcionar:
(2S)-N-[5-[[(3R)-1-(5-clorop¡r¡daz¡n-3-¡l)p¡rrol¡din-3-¡l]am¡no]-1,3,4-t¡ad¡azol-2-¡l]-2-metox¡-2-fen¡l-acetam¡da como un sólido blanco (15 mg, 25 %). 1H NMR (400 MHz, DMSO-d6, 25 °C) 51,98 - 2,14 (1H, m), 2,17 - 2,36 (1H, m), 3,30 (3H, s), 3,44 - 3,64 (3H, m), 3,69 - 3,81 (1H, m), 4,28 - 4,42 (1H, m), 4,96 (1H, s), 7,11 (1H, d), 7,30 - 7,47 (5H, m), 7,69 (1H, d), 8,55 (1H, d), 12,24 (1H, s). m/z: ES+ [M+H]+ 446, 448.
Ejemplo 13
(2S)-2-metox¡-N-[5-[[(3R)-1-(5-met¡lp¡r¡daz¡n-3-¡l)p¡rrolid¡n-3-¡l]am¡no]-1,3,4-t¡ad¡azol-2-¡l]-2-fen¡l-acetam¡da

Se adicionaron DIPEA (0,09 ml, 0,54 mmol), HATU (164 mg, 0,43 mmol) y ácido (2S)-2-metoxi-2-fenilacético (60 mg, 0,36 mmol) a una solución de N2-[(3R)-1-(5-metilpiridazin-3-il)pirrolidin-3-il]-1,3,4-tiadiazol-2,5-diamina (Producto intermedio 7, 100 mg, 0,36 mmol) en DMF (2 ml). La mezcla se agitó a temperatura ambiente por 2 horas. Después esta se diluyó con agua (5 ml), se extrajo en DCM (10 ml) y se evaporó bajo presión reducida. El producto bruto se purificó por HPLC preparativa (columna SunFire C18, 5 ^m, 50 mm x 19 mm a 25 ml/min). Se usaron decrecientemente relaciones polares de agua y MeCN que contenían 0,1 % de ácido fórmico como fase móvil. Las fracciones recolectadas se pasaron después por un cartucho SCX que se lavó con metanol antes de eluircon amoniaco 2 M en metanol. El amoníaco en metanol se evaporó y el residuo se secó en un horno de vacío para proporcionar:
(2S)-2-metoxi-N-[5-[[(3R)-1-(5-metilpiridazin-3-il)pirrolidin-3-il]amino]-1,3,4-tiadiazol-2-il]-2-fenil-acetamida como un sólido blanco (28 mg, 18%). 1H NMR(400 MHz, DMSO-d6, 25 °C) 52,01 -2,10 (1H, m), 2,19 (3H, s), 2,22-2,34 (1H, m), 3,31 (3H, s), 3,42 - 3,60 (3H, m), 3,70 - 3,74 (1H, m), 4,30 - 4,42 (1H, m), 4,98 (1H, s), 6,70 (1H, s), 7,30 - 7,42 (3H, m), 7,43 -7,49 (2H, m), 7,70 (1H, d), 8,35 (1H, s), 12,26 (1H, s). m/z: ES+ [M+H]+ 426
Ejemplos adicionales
Los compuestos de los siguientes ejemplos se prepararon de manera similar a los ejemplos anteriores.

Ejemplos 33(a) y 33(b)
(2R)-2-metox¡-2-(4-metox¡feml)-W-[5-[[(3R)-1-(5-met¡lp¡ndaz¡n-3-¡l)p¡n'ol¡d¡n-3-¡l]am¡no]-1,3,4-t¡ad¡azol-2-¡l]acetam¡da y (2S)-2-metox¡-2-(4-metox¡feml)-A/-[5-[[(3R)-1-(5-met¡lp¡ndaz¡n-3-¡l)p¡n'ol¡d¡n-3-¡l]am¡no]-1,3,4-t¡ad¡azol-2-¡l]acetam¡da

Se disolvieron N2-[(3R)-1-(5-met¡lp¡r¡daz¡n-3-il)p¡rrol¡d¡n-3-¡l]-1,3,4-t¡ad¡azol-2,5-d¡am¡na (Producto intermedio 7, 0,05 g, 0,173 mmol) y N-[(d¡met¡lam¡no)(3H-[1,2,3]tr¡azolo[4,5-b]p¡r¡d¡n-3-ilox¡)met¡l¡den]-N-met¡lmet¡lam¡n¡o hexafluorofosfato, HATU (0,08 g, 0,208 mmol) en DMF (2 ml) a r.t. bajo N2. La mezcla se agitó por 5 minutos antes de la adición de ácido 2-metoxi-2-(4-metoxifen¡l)acét¡co (Producto intermedio 21, 0,04 g, 0,173 mmol) y DIPEA (0,05 ml, 0,26 mmol). La reacción se agitó a r.t. durante la noche bajo N2. La mezcla bruta se pasó a través de una columna SCX de 5 g se lavó con MeOH después se eluyó con NH32 M en MeOH. La fracción básica se evaporó bajo presión reducida para producir el producto impuro como una goma naranja. El producto bruto se purificó por HPLC preparativa (columna SunFire C18, 5 ^m, 50 mm x 19 mm a 25 ml/min). Se usaron decrecientemente relaciones polares de agua y MeCN que contenía 0,1 % de ácido fórmico como fase móvil. Las fracciones que contenían la masa deseada se combinaron, se evaporaron bajo presión reducida y se pasaron a través de una columna SCX de 5 g, se lavaron con MeOH, después se eluyeron con NH32 M en MeOH. La fracción básica se evaporó bajo presión reducida para producir la mezcla de diastereoisómeros como un sólido blanquecino. Los diastereoisómeros se separaron mediante SFC quiral preparativa (columna Lux C1, 5 ^m, 21,2 mm x 250 mm, tasa de flujo 50 ml/min a una longitud de onda de 210 nm con 40:60 MeOH:Co2 0,1 % de NH3 como eluyente) para producir:
Primer isómero eluido Ejemplo 33(a) 2-metox¡-2-(4-metox¡fen¡l)-N-[5-[[(3R)-1-(5-met¡lpir¡daz¡n-3-¡l)p¡rrol¡d¡n-3-¡l] amino]-1,3,4-tiadiazol-2-¡l]acetam¡da (6,6 mg, 8 %). 1H NMR (400 MHz, DMSO-d6) 512,18 (1H, s), 8,36 (1H, d), 7,68 (1H, d), 7,41 - 7,34 (2H, m), 6,99 - 6,90 (2H, m), 6,71 (1H, s), 4,90 (1H, s), 4,45 - 4,30 (1H, m), 3,74 (3H, s), 3,64 - 3,37 (4H, m), 3,27 (3H, s), 2,35 - 2,23 (1H, m), 2,20 (3H, s), 2,09 - 1,98 (1H, m). m/z: ES+ [M+H]+ 456.
Segundo eluido Ejemplo 33(b) 2-metox¡-2-(4-metox¡fen¡l)-N-[5-[[(3R)-1-(5-met¡lp¡ridaz¡n-3-¡l)p¡rrol¡d¡n-3-¡l]am¡no]-1,3,4-tiadiazol-2-¡l]acetam¡da (5,7 mg, 6 %). 1H NMR (400 MHz, DMSO-d6) 512,18 (1H, s), 8,35 (1H, d), 7,66 (1H, d), 7,37 (2H, d), 6,94 (2H, d), 6,70 (1H, d), 4,89 (1H, s), 4,43 - 4,29 (1H, m), 3,74 (3H, s), 3,73 - 3,69 (1H, m), 3,60 - 3,45 (3H, m), 3,27 (3H, s), 2,34-2,23 (1H, m), 2,20 (3H, s), 2,10-2,01 (1H, m). m/z: ES+ [M+H]+ 456.
Producto intermedio 1
Se disolvieron 5-bromo-1,3,4-tiadiazol-2-amina (912 mg, 5,07 mmol), (3R)-1-(6-met¡lpir¡daz¡n-3-¡l)p¡rrol¡d¡n-3-am¡na (Producto intermedio 2, 860 mg, 4,83 mmol) y DIp Ea (0,924 ml, 5,31 mmol) en Dm F (10 ml). La reacción se calentó hasta 100 °C por 1 h después se dejó a r.t. durante la noche. El producto bruto se purificó mediante cromatografía de intercambio iónico, mediante el uso de una columna SCX. El producto deseado se eluyó de la columna mediante el uso de NH31 M en MeOH y las fracciones puras se evaporaron hasta la sequedad para proporcionar el producto bruto. Esto se disolvió en DCM/MeOH, se adsorbió sobre sílice y se purificó por FCC (S¡O2, 0 a 20 % de MeOH en DCM). Las fracciones puras se evaporaron hasta la sequedad para proporcionar N2-[(3R)-1-(6-met¡lp¡ridaz¡n-3-¡l)p¡rrol¡d¡n-3-¡l]-1,3,4-t¡ad¡azol-2,5-diamina (350 mg, 26 %) como una goma marrón. m/z: ES+ [M+H]+ 278.
Producto intermedio 2
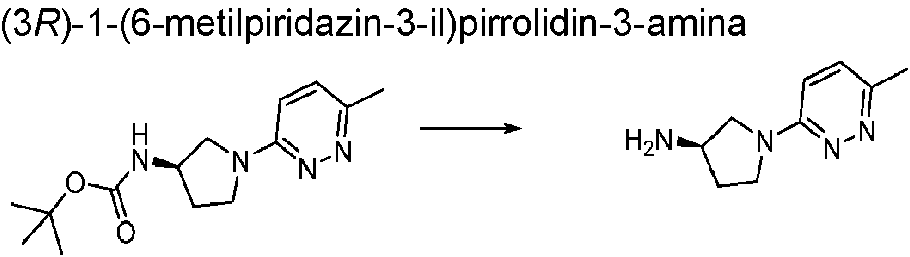
Se adicionó ácido trifluoroacético (12 ml) a terc-butil N-[(3R)-1-(6-metilpiridazin-3-il)pirrolidin-3-il]carbamato (Producto intermedio 3, 2,1 g, 7,54 mmol), en DCM (60 ml) a 21 °C bajo nitrógeno. La solución resultante se agitó a 21 °C por 2 h. El producto bruto se purificó mediante cromatografía de intercambio iónico, mediante el uso de una columna SCX. El producto deseado se eluyó de la columna mediante el uso de NH3 7 M en MeOH y las fracciones puras se evaporaron hasta la sequedad para proporcionar (3R)-1-(6-metilpiridazin-3-il)pirrolidin-3-amina (1,6 g, 119 %) como un aceite amarillo que se solidificó al reposar. 1H NMR (400 MHz, DMSO-d6, 27 °C) 5 1,56 - 1,8 (1H, m), 2,04 (1H, m), 2,39 (3H, s), 3,07 (1H, m), 3,37 - 3,43 (1H, m), 3,47 - 3,66 (3H, m), 4,08 (1H, s), 6,73 (1H, d), 7,19 (1H, d). m/z: ES+ [M+H]+ 179.
Producto intermedio 3
terc-butil N-[(3R)-1-(6-metilpiridazin-3-il)pirrolidin-3-il]carbamato

Una mezcla de DIPEA (8,49 ml, 48,62 mmol), terc-butil N-[(3R)-pirrolidin-3-il]carbamato (3,62 g, 19,45 mmol), 3-cloro-6-metilpiridazina (2,5 g, 19,45 mmol) y n-butanol (30 ml) se agitó a 130 °C por 12 h y después se dejó para refrescarse durante el fin de semana. La mezcla de reacción se evaporó y el producto bruto se purificó por FCC (SiO2, 0 a 10 % de NH31 M en MeOH en EtOAc). Las fracciones puras se evaporaron hasta la sequedad para proporcionar terc-butil N-[(3R)-1-(6-metilpiridazin-3-il)pirrolidin-3-il]carbamato (2,1 g, 38,8 %) como un sólido amarillo. 1H NMR (400 MHz, DMSO-d6, 27 °C) 5 1,39 (9H, s), 1,88 (1H, m), 2,14 (1H, m), 2,40 (3H, s), 3,23 (1H, m), 3,37 - 3,45 (1H, m), 3,47 - 3,58 (1H, m), 3,61 (1H, m), 3,99 - 4,2 (1H, m), 6,77 (1H, d), 7,20 (2H, m); m/z: ES+ [M+H]+ 279.
Producto intermedio 4
N2-[(3R)-1-(6-fluoropiridazin-3-il)pirrolidin-3-il]-1,3,4-tiadiazol-2,5-diamina

Se adicionó DIPEA (3,48 ml, 19,96 mmol) a 5-bromo-1,3,4-tiadiazol-2-amina (1,797 g, 9,98 mmol) y (3R)-1-(6-fluoropiridazin-3-il)pirrolidin-3-amina (Producto intermedio 5, 2 g, 10,98 mmol) en DMF anhidro (40 ml) a r.t. La solución resultante se agitó a 80 °C por 4 h. El producto bruto se purificó mediante cromatografía de intercambio iónico, mediante el uso de una columna SCX. El producto deseado se eluyó de la columna mediante el uso de NH3 1 M en MeOH y las fracciones puras se evaporaron hasta la sequedad para proporcionar N2-[(3R)-1-(6-fluoropiridazin-3-il)pirrolidin-3-il]-1,3,4-tiadiazol-2,5-diamina (2,9 g, 103 %) como un sólido marrón. 1H NMR (400 MHz, DMSO-d6, 30 °C) 5 1,90-2,12 (1H, m), 2,23 (1H, dtd), 3,42 (1H, dd), 3,47 - 3,61 (2H, m), 3,69 (1H, dd), 4,25 (1H, dq), 6,25 (2H, s), 7,04 (1H, d), 7,14 (1H, dd), 7,33 (1H, dd). m/z: ES+ [M+H]+ 282.
Producto intermedio 5
(3R)-1-(6-fluoropiridazin-3-il)pirrolidin-3-amina

Se adicionó terc-butil N-[(3R)-1-(6-fluoropiridazin-3-il)pirrolidin-3-il]carbamato (Producto intermedio 6, 6 g, 21,25 mmol) a DCM (70 ml) y TFA (14,00 ml) a 25 °C. La solución resultante se agitó a 25 °C por 4 h. El producto bruto se purificó mediante cromatografía de intercambio iónico, mediante el uso de una columna SCX. El producto deseado se eluyó de la columna mediante el uso de NH31 M en MeOH y las fracciones puras se evaporaron hasta la sequedad para proporcionar (3R)-1-(6-fluoropiridazin-3-il)pirrolidin-3-amina (2,0 g, 52 %) como un sólido gomoso amarillo pálido. 1H NMR (400 MHz, DMSO-d6, 30 °C) 5 1,55- 1,83 (1H, m), 1,98-2,13 (1H, m), 2 ,89-3,14 (1H, m), 3,29-3,43 (1H, m), 3,54 (3H, ddt), 7,06 (1H, dd), 7,30 (1H, dd). m/z: ES+ [M+H]+ 183.
Producto intermedio 6
terc-butil N-[(3R)-1-(6-fluoropiridazin-3-il)pirrolidin-3-il]carbamato

Una mezcla de 3,6-difluoropiridazina (6,06 g, 52,21 mmol) terc-butil N-[(3R)-pirrolidin-3-il]carbamato (9,72 g, 52,21 mmol), DIPEA (22,80 ml, 130,53 mmol) y n-butanol (140 ml) se agitó a 130 °C por 10 h. La mezcla de reacción se diluyó con EtOAc (750 ml), y se lavó dos veces con agua (150 ml). La capa orgánica se secó sobre Na2SO4, se filtró y se evaporó para proporcionar el producto bruto. Este se disolvió después en DCM y el producto bruto se purificó por FCC (SiO2, 30 -65 % de EtOAc en heptanos). Las fracciones puras se evaporaron hasta la sequedad para proporcionar terc-butil N-[(3R)-1-(6-fluoropiridazin-3-il)pirrolidin-3-il]carbamato (15 g, 102 %) como un sólido crema. 1H n Mr (400 MHz, CDCh, 30 °C) 5 1,46 (9H, s), 1,91 - 2,13 (1H, m), 2,32 (1H, dq), 3,40 (1H, dd), 3,56 - 3,72 (2H, m), 3,78 (1H, dd), 4,37 (1H, s), 4,70 (1H, s), 6,78 (1H, dd), 6,98 (1H, dd). m/z: ES+ [M+H]+ 283.
Producto intermedio 7
N2-[(3R)-1-(5-Metilpiridazin-3-il)pirrolidin-3-il]-1,3,4-tiadiazol-2,5-diamina

Se disolvió terc-6ut//(3R)-3-[(5-metilpiridazin-3-il)amino]pirrolidina-1-carboxilato (Producto intermedio 8, 200 mg, 0,72 mmol) en DCM (5 ml) y se trató con TFA (5 ml). Se dejó agitar a temperatura ambiente por 2 horas, después se evaporó hasta la sequedad para producir un aceite amarillo. Después este se disolvió en acetonitrilo (5 ml) y se trató con DIPEA (0,38 ml, 2,15 mmol) seguido de 5-bromo-1,3,4-tiadiazol-2-ilamina (130 mg, 0,72 mmol) y después se calentó hasta 80 °C por 3 h. La mezcla de reacción se evaporó y se purificó por FCC (SiO2, 0-10 % de Nh32M en MeOH en DCM). Las fracciones que contenían el producto se combinaron y se evaporaron para producir N2-[(3R)-1-(5-metilpiridazin-3-il)pirrolidin-3-il]-1,3,4-tiadiazol-2,5-diamina (100 mg, 50 %). 1H NMR (400 MHz, DMSO-d6, 25 °C) 5 1,99 - 2,05 (1H, m), 2,18-2,31 (4H, m), 3,38- 3,58 (3H, m), 3,67-3,71 (1H, m), 4 ,19-4 ,30 (1H, m), 6,31 (2H, s), 6,70 (1H, s), 7,09 (1H, d), 8,36 (1H, s). m/z: ES+ [M+H]+ 278.
Producto intermedio 8
terc-butil (3R)-3-[(6-cloropiridazin-3-il)amino]pirrolidina-1-carboxilato

Se disolvió terc-butil N-[(3R)-pirrolidin-3-il]carbamato (250 mg, 1,34 mmol) en 1-butanol (2 ml) y se trató con 3-cloro-5-metilpiridazina (0,25 M en DCM, 2,68 ml, 1,34 mmol) seguido de DIPEA (0,48 ml, 2,68 mmol). La mezcla se calentó hasta 140 °C por 2 horas. La mezcla de reacción se enfrió hasta r.t. se diluyó con agua (10 ml) y se extrajo dos veces en DCM (10 ml). El solvente se evaporó y el residuo se purificó por FCC (SiO2, 0 -10 % de metanol en DCM). Las fracciones que contenían el producto se combinaron y se evaporaron para producir terc-butil N-[(3R)-1-(5-metilpiridazin-3-il)pirrolidin-3-il]carbamato (200 mg, 53%). 1H NMR (400 MHz, DMSO-d6, 25 °C) 51,40 (9H, s), 1,85-1,93 (1H, m), 2,07 - 2,18 (1H, m), 2,20 (3H, s), 3,24 - 3,28 (1H, m), 3,38 - 3,48 (1H, m), 3,48 -3,59 (1H, m), 3,60 - 3,65 (1H, m), 4 ,10-4 ,15 (1H, m), 6,67 (1H, s), 7,24 (1H, d), 8,35 (1H, d). m/z: ES+ [M+H]+ 279.
Producto intermedio 9
N2-[(3R)-1-(5-cloropiridazin-3-il)pirrolidin-3-il]-1,3,4-tiadiazol-2,5-diamina

Se disolvió (3R)-1-(5-cloropiridazin-3-il)pirrolidin-3-amina (Producto intermedio 10, 50 mg, 0,25 mmol) en acetonitrilo (5 ml) y se trató con DIPEA (0,07 ml, 0,38 mmol) seguido de 5-bromo-1,3,4-tiadiazol-2-ilamina (50 mg, 0,25 mmol) y se calentó hasta 80 °C por 3 h. Después la mezcla de reacción se evaporó y se purificó por FCC (SiO2, 0-10 % de NH32M
en MeOH en DCM). Las fracciones que contenían el producto se combinaron y se evaporaron para producir N2-[(3R)-1-(5-doropiridazin-3-il)pirroNdin-3-il]-1,3,4-tiadiazol-2,5-diamina que se usó directamente en la siguiente etapa (50 mg, 66 %). m/z: ES+ [M+H]+298, 300.
Producto intermedio 10
(3R)-1-(5-cloropiridazin-3-il)pirrolidin-3-amina

Se disolvió terc-butil N-[(3R)-1-(5-cloropiridazin-3-il)pirrolidin-3-il]carbamato (Producto intermedio 11, 77 mg, 0,25 mmol) en DCM (1 ml), se trató con TFA (1 ml), y se dejó a agitar a r.t. por 2 h. Después se evaporó hasta la sequedad y se pasó por un cartucho SCX se lavó con metanol y se eluyó con amoniaco metanólico 2M. La fracción básica se evaporó para producir (3R)-1-(5-cloropiridazin-3-il)pirrolidin-3-amina, que se usó directamente en la siguiente etapa (50 mg, 99 %). m/z: ES+ [M+H]+ 199, 201.
Producto intermedio 11
terc-butil N-[(3R)-1-(5-cloropiridazin-3-il)pirrolidin-3-il]carbamato

Se disolvió 5-cloropiridazin-3-ol (0,2 g, 1,53 mmol) en DCM (2 ml) y trietilamina (0,47 ml, 3,37 mmol) y se enfrió hasta -20 °C. Después se trató, gota a gota, con anhídrido trifluorometanosulfónico (1 M en DCM, 3,22 ml, 3,22 mmol). Después se permitió que la mezcla de reacción retornara lentamente hasta la temperatura ambiente. Se inactivó mediante la adición de agua (10 ml) y se extrajo en DCM (10 ml). Los orgánicos se lavaron con HCl 1 M (10 ml) se secaron (MgSO4), se filtraron y se evaporaron para producir (5-cloropiridazin-3-il)trifluorometanosulfonato. Después este se disolvió en DMF (2 ml) y se enfrió a 0 °C antes de tratarse con trietilamina (0,21 ml, 1,53 mmol) y terc-butil N-[(3R)-pirrolidin-3-il]carbamato (290 mg, 1,53 mmol). Después se dejó volver a temperatura ambiente, se diluyó con agua (20 ml) y se extrajo en DCM (20 ml). Los orgánicos se evaporaron y purificaron por FCC (SiO2, 0 - 10 % de MeOH en DCM). Las fracciones que contenían el producto se combinaron y se evaporaron para producir terc-butil N-[(3R)-1-(5-cloropiridazin-3-il)pirrolidin-3-il]carbamato, (77 mg, 17%). 1H NMR(400 MHz, DMSO-d6, 25 °C) 51,40 (9H, s), 1,84- 1,99 (1H, m), 2,05 - 2,20 (1H, m), 3,32 - 3,34 (1H, m), 3,42 - 3,60 (2H, m), 3,60 - 3,73 (1H, m), 4,00 - 4,28 (1H, m), 7,08 (1H, d), 7,26 (1H, d), 8,55 (1H, d). m/z: ES+ [M+H]+ 299, 301.
Producto intermedio 12
Ácido (2S)-2-metoxi-2-(3-metoxifenil)acético

Bencil (2S)-2-metoxi-2-(3-metoxifenil)acetato (Producto intermedio 13(a), 7,85 g, 27,42 mmol) y 10 % de Pd en C (1 g, 27,42 mmol) en etanol (250 ml) se agitaron bajo una atmósfera de hidrógeno a temperatura ambiente por 4 horas. La mezcla de reacción se filtró a través de celita que se lavó con MeOH. Los orgánicos se evaporaron para producir ácido (2S)-2-metoxi-2-(3-metoxifenil)acético como un aceite transparente (6,0 g, 112 %, contiene algo de MeOH). 1H NMR (400 MHz, DMSO-d6, 30 °C) 53,29 (3H, s), 3,74 (3H, s), 4,72 (1H, s), 6,77- 7,06 (3H, m), 7,28 (1H, t), 12,77 (1H, s).
Producto intermedio 13(a)
Bencil (2S)-2-metoxi-2-(3-metoxifenil)acetato

Se adicionó bromuro de bencilo (7,81 ml, 65,75 mmol) gota a gota al ácido 2-metoxi-2-(3-metoxifenil)acético (Producto intermedio 14, 10,75 g, 54,79 mmol), carbonato de potasio (11,36 g, 82,19 mmol) en DMF (200 ml) a 21 °C bajo nitrógeno. La mezcla resultante se agitó a 85 °C durante 4 horas y después se dejó en agitación a temperatura ambiente durante el fin de semana. La mezcla de reacción se diluyó con EtOAc (600 ml), y se lavó dos veces con agua (200 ml). La capa orgánica se secó sobre Na2SO4, se filtró y se evaporó para proporcionar un producto bruto que se purificó por FCC (SiO2 1 0 - 25 % de EtOAc en heptanos). Las fracciones puras se evaporaron hasta la sequedad para proporcionar un aceite incoloro. (10,5 g).
Se adicionó bromuro de bencilo (5,59 ml, 47,09 mmol) gota a gota al ácido 2-metoxi-2-(3-metoxifenil)acético (Producto intermedio 14, 7,7 g, 39,25 mmol), carbonato de potasio (8,14 g, 58,87 mmol) en DMF (150 ml) a 21 °C bajo nitrógeno. La mezcla resultante se agitó a 85 °C por 4 horas, después se dejó en agitación a temperatura ambiente durante el fin de semana.
La reacción se calentó después a 85 °C por 4 horas más antes de enfriarse a temperatura ambiente. La mezcla de reacción se diluyó con EtOAc (500 ml), y se lavó dos veces con agua (200 ml). La capa orgánica se secó sobre Na2SO4, se filtró y se evaporó para proporcionar el producto bruto. El producto bruto se purificó por FCC (SiO210 - 25 % de EtOAc en heptanos). Las fracciones puras se evaporaron hasta la sequedad para proporcionar un aceite amarillo pálido (6 g). El producto de las dos reacciones se combinó y los enantiómeros se separaron por HPLC quiral. (Columna Chiralpak OD, 20 ^m, 100 mm x 250 mm, mediante el uso de una mezcla 95/5 de heptano:EtOH como eluyentes a 250 ml/min). Las fracciones que contenían el compuesto deseado se evaporaron hasta la sequedad para proporcionar:
Primer isómero eluido Producto intermedio 13(a) bencil (2S)-2-metoxi-2-(3-metoxifenil)acetato (7,86 g, 39 %). 1H NMR (400 MHz, DMSO-d6, 27 °C) 53,37 (3H, s), 3,78 (3H, s), 5,02 (1H, s), 5,20 (2H, s), 6,75 - 7,10 (3H, m), 7,20 - 7,52 (6H, m).
Segundo isómero eluido Producto intermedio 13(b) bencil (2R)-2-metoxi-2-(3-metoxifenil)acetato (7,9 g, 39 %). 1H NMR (400 MHz, DMSO-d6, 27 °C) 53,37 (3H, s), 3,78 (3H, s), 5,02 (1H, s), 5,20 (2H, s), 6,75 - 7,10 (3H, m), 7,20 - 7,52 (6H, m).
Producto intermedio 14
Ácido 2-metoxi-2-(3-metoxifenil)acético

Se adicionó una solución de hidróxido de potasio (2,267 g, 40,40 mmol) en MeOH (10 ml) durante 2 h en pequeñas porciones a una mezcla agitada de 3-metoxibenzaldehído (1 g, 7,34 mmol) y bromoformo (0,771 ml, 8,81 mmol) en MeOH (5,00 ml) a 0 °C. La mezcla después se dejó calentar hasta r.t. y se dejó agitar durante la noche. Los sólidos se filtraron bajo presión reducida, y se enjuagaron los sólidos con MeOH (15 ml). El filtrado se evaporó hasta una pasta blanca espesa y después se redisolvió en agua (50 ml). Después esta se lavó con Et2O (50 ml) y después la porción acuosa se acidificó hasta pH 2 (~5 ml de solución de HCl 2 M). Después se extrajo la fase acuosa con EtOAc (3 x 50 ml). Los orgánicos combinados se secaron sobre MgSO4 y se filtraron después los solventes se evaporaron bajo presión reducida para producir ácido 2-metoxi-2-(3-metoxifenil)acético como un aceite amarillo (1,4 g, 97 %). 1H NMR (400 MHz, DMSO-d6, 30 °C) 53,18 (3H, s), 3,75 (3H, s), 4,74 (1H, s), 6,82-7,05 (3H, m), 7,29 (1H, m), 12,78 (1H, s).
Producto intermedio 15
Ácido 2-etoxi-2-(3-metoxifenil)acético

A una mezcla agitada de 3-metoxibenzaldehído (5,0 g, 36,72 mmol) y bromoformo (3,85 ml, 44,06 mmol) en etanol (40 ml) a 0 °C se le adicionó gota a gota durante un período de 1 hora una solución de hidróxido de potasio (11,33 g, 201,98 mmol) en etanol (60 ml). Después de completar la adición la mezcla se dejó agitar a r.t. durante la noche. Se formó un precipitado que se eliminó por filtración, y el filtrado se evaporó para producir una pasta que se recolectó en agua (100 ml) y se extrajo con EtOAc (2 x 100 ml). Después se acidificó la fase acuosa hasta pH = 2 con HCl 2 M y se extrajo con EtOAc (2 x 100ml). Los orgánicos combinados se secaron (MgSO4), se filtraron y se evaporaron para producir un aceite marrón pálido. Esto se absorbió sobre sílice y se purificó por FCC (SiO2, 5 % de MeOH en DCM) para producir ácido 2-etoxi-2-(3-metoxifenil)acético (3,1 g, 40 %) como un aceite marrón pálido. 1H NMR (400 MHz, cDCh, 21 °C) 5 1,28 (3H, t), 2,09 (1H, s), 3,64 - 3,52 (2H, m), 3,81 (3H, s) 4,86 (1H, s), 6,89 (1H, ddd), 6,99 (1H, m), 7,03 (1H, m), 7,29 (1H, t). m/z: ES-[M-H]- 209.
Producto intermedio 16
Ácido 2-(4-fluorofenil)-2-metoxi-acético
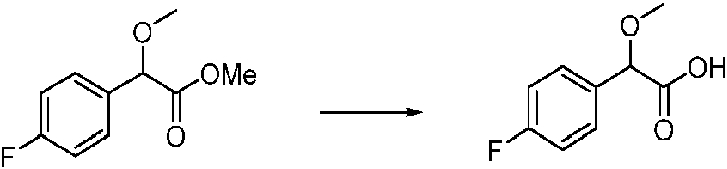
El 2-(4-fluorofenil)-2-metoxi-acetato de metilo (Producto intermedio 17, 1,32 g, 6,66 mmol) se disolvió en metanol (24 ml) y se agitó a r.t. Se adicionó una solución de hidróxido de potasio (0,45 g, 7,992 mmol) en metanol (12 ml), y la mezcla se agitó a r.t. por 5 h.
La mezcla se evaporó hasta la sequedad, se particionó entre agua y EtOAc (70 ml cada uno). El acuoso se lavó con EtOAc (70 ml) después se acidificó con ácido clorhídrico 2 N hasta pH 2. Después se extrajo con EtOAc (2 x 100 ml). Los orgánicos se secaron (MgSO4) y se evaporaron para producir ácido 2-(4-fluorofenil)-2-metoxi-acético como una goma transparente (1,16 g, 94 %). 1H NMR (400 MHz, CDCls) 3,42 (3H, s) 4,77 (1H, s), 7,10-7,04 (2H, m), 7,44-7,40 (2H, m). Producto intermedio 17
2-(4-fluorofenil)-2-metoxiacetato de metilo

Se disolvió carbonato de cesio (7,64 g, 23,45 mmol) en DMF (20 ml) a r.t. y se adicionó yodometano (2,4 ml, 38,55 mmol), seguido de ácido 2-(4-fluorofenil)-2-hidroxiacético (2,0 g, 11,75 mmol), y la mezcla se agitó por 48 h a r.t. La DMF se evaporó bajo presión reducida y el residuo se particionó entre EtOAc y agua (75 ml cada uno). Los orgánicos se lavaron con agua (75 ml), se secaron (MgSO4), se evaporaron bajo presión reducida y se purificaron por FCC (SiO2, 3:1 ciclohexano:EtOAc). Las fracciones puras se evaporaron hasta la sequedad para proporcionar metil 2-(4-fluorofenil)-2-metoxiacetato (1,68 g, 72 %) como un aceite incoloro. 1H NMR (400 m Hz , CDCl3, 20 °C) 53,40 (3H, s), 3,72 (3H, s), 4,76 (1H, s), 7,09 - 7,03 (2H, m), 7,44 - 7,40 (2H, m).
Producto intermedio 18
Ácido 2-(4-fluoro-3-metoxi-fenil)-2-metoxi-acético

A una mezcla agitada de 4-fluoro-3-metoxi-benzaldehído (1,0 g, 6,48 mmol) y bromoformo (0,68 ml, 7,785 mmol) en MeOH (10 ml) a 0 °C se adicionó, gota a gota por 1 h, una solución de hidróxido de potasio (2,0 g, 35,682 mmol) en MeOH (20 ml). Después de la adición la mezcla se agitó y se calentó hasta r.t. durante la noche. El precipitado resultante se eliminó por filtración. El filtrado se evaporó para producir una pasta que se recolectó en agua (100 ml) y se extrajo con EtOAc (2 x 100 ml). La fase acuosa se acidificó después hasta pH=2 con HCl 2N. Se extrajo con EtOAc (2 x 100 ml). Los orgánicos combinados se secaron (MgSO4), se filtraron y se evaporaron bajo presión reducida para proporcionar el producto bruto. El producto bruto se purificó adicionalmente por FCC (SiO2, 0-5 % de MeOH en Dc M) para producir ácido 2-(4-fluoro-3-metoxi-fenil)-2-metoxi-acético (0,66 g, 47 %) como un aceite incoloro. 1H NMR (400 MHz, CDCl3, 30 °C) 53,43 (3H, s), 3,90 (3H, s), 4,75 (1H, s), 6,95- 7,00 (1H, m), 7,10-7,04 (2H, m). m/z: ES-[M-H]-213.
Producto intermedio 19
Ácido 2-metoxi-2-[3-(trifluorometoxi)fenil]acético

Se adicionó una solución de hidróxido de potasio (1,851 g, 33,00 mmol) en MeOH (10 ml) por 2 h en pequeñas porciones a una mezcla agitada de 3-(trifluorometoxi)benzaldehído (1,141 g, 6 mmol) y bromoformo (0,630 ml, 7,20 mmol) en MeOH (5,00 ml) a 0 °C. La mezcla se dejó calentar hasta r.t. y se dejó agitar durante la noche. Se formó un precipitado blanco en la mezcla de reacción. Los sólidos se filtraron bajo presión reducida y se enjuagó la torta del filtro con MeOH (15 ml). El filtrado se evaporó hasta una pasta blanca espesa y después se redisolvió en agua (50 ml). Después esta se lavó con Et2O (50 ml). La fase acuosa se acidificó hasta pH = 2 (~5 ml de solución de HCl 2 M) y después se extrajo en EtOAc (3 x 50 ml). Las capas orgánicas se secaron (MgSO4), se filtraron y se evaporaron bajo presión reducida para producir un aceite transparente. El producto bruto se purificó por FCC (SiO2, 10-50 % de EtOAc en heptanos). Las fracciones puras se evaporaron hasta la sequedad para proporcionar ácido 2-metoxi-2-[3-(trifluorometoxi)fenil]acético (0,832 g, 55 %) como un aceite incoloro. 1H NMR (400 MHz, CDCls, 30 °C) 53,47 (3H, s), 4,81 (1H, s), 7,20 - 7,24 (1H, m), 7,33 (1H, s), 7,37 -7,46 (2H, m). m/z: ES-[M-H]- 249.4.
Producto intermedio 20
Ácido 2-[3-(difluorometoxi)fenil]-2-metoxi-acético

A una mezcla agitada de 3-(difluorometoxi)benzaldehído (2,0 g, 11,61 mmol) y bromoformo (1,22 ml, 13,94 mmol) en MeOH (40 ml) a 0 °C se adicionó gota a gota por un período de 1 hora una solución de hidróxido de potasio (3,59 g, 63,90 mmol) en MeOH (60 ml). Después de la adición la mezcla se dejó agitar mientras se calentaba hasta temperatura ambiente durante la noche. El precipitado se eliminó por filtración y el filtrado se evaporó para producir una pasta que se recolectó en agua (100 ml) y se extrajo con EtOAc (2 x 75 ml). Después la fase acuosa se acidificó hasta pH = 1 con HCl 2M y se extrajo con EtOAc (2 x 75 ml). Los orgánicos combinados se secaron (MgSO4), se filtraron, y se evaporaron para producir un aceite marrón pálido. Este se purificó por FCC (SiO2 , 95:5 ciclohexano:EtOAc 0,1 % de ácido fórmico con aumento a 8:2 EtOAc:ciclohexano 0,1 % de ácido fórmico). Las fracciones apropiadas se evaporaron bajo presión reducida para proporcionar ácido 2-[3-(difluorometoxi)fenil]-2-metoxi-acético como un aceite incoloro (1,3 g 48 %). 1H NMR (400 MHz, CDCls , 21 °C) 53,46 (3H, s), 4,80 (1H, s), 6,53 (1H, t), 7,10-7,15 (1H, m), 7 ,22-7,23 (1H, m), 7,29- 7,32 (1H, m), 7,39 (1H,t). m/z: ES- [M-H]- 231.
Producto intermedio 21
Ácido 2-metoxi-2-(4-metoxifenil)acético

A una mezcla agitada de p-anisaldehído (3,58 g, 26,29 mmol) y bromoformo (2,76 ml, 31,55 mmol) en MeOH (30 ml) a 0 °C se adicionó, gota a gota por 30 minutos, una solución de hidróxido de potasio (8,12 g, 144,65 mmol) en MeOH (60 ml). Después de la adición la mezcla se dejó agitar mientras se calentaba hasta r.t. durante la noche. A la mañana siguiente el precipitado se eliminó por filtración. El filtrado se evaporó para producir una pasta que se recolectó en agua (100 ml) y se extrajo con EtOAc (2 x 100 ml) para eliminar cualquier aldehído de partida sin reaccionar restante. Después la fase acuosa se acidificó hasta pH = 2 con HCl 2N. Se extrajo con EtOAc (2 x 100 ml). Los orgánicos combinados se secaron (MgSO4), se filtraron y se evaporaron para producir ácido 2-metoxi-2-(4-metoxifenil)acético como una goma naranja (3,1 g, 60 %). 1H NMR (400mHz, DMSO, 30 °C) 3,27 (3H, s), 3,75 (3H, s), 4,69 (1H, s), 6,93 (2H, d), 7,30 (2H, d), 12,45 (1H, brs). m/z: ES- [M-H]- 195.
Ensayos biológicos
Los siguientes ensayos se usaron para medir los efectos de los compuestos descritos en la presente descripción: a) ensayo de potencia enzimática de GLS; b) ensayo de potencia celular de GLS; c) Ensayo de proliferación celular con GLS. Durante la descripción de los ensayos, generalmente:
i. Las siguientes abreviaturas se usaron: CO2 = dióxido de carbono; DMEM = medio águila modificada de Dulbecco; DMSO = dimetil sulfóxido; EDTA = ácido etilendiaminotetraacético; EGTA = ácido etilenglicol tetraacético; FCS = suero fetal de ternera; h = hora(s); NBS = superficie no unidora; SDS = dodecil sulfato de sodio; TRIS = Tris(hidroximetil)aminometano. ii. Los valores de IC50 se calcularon mediante el uso de un modelo de ajuste inteligente en Genedata. El valor de IC50 fue la concentración del compuesto de prueba que inhibió el 50 % de la actividad biológica.
Ensayo a): Ensayo de potencia enzimática de GLS
Se usó un ensayo acoplado Glutamato Oxidasa/AmplexRed para medir la capacidad de los compuestos para unirse e inhibir la actividad de GLS1 in vitro. La proteína GLS marcada con 6His (aminoácidos 63-669) expresada en E. Coli se purificó y se almacenó a -80 °C en alícuotas. La GLS1 se diluyó hasta la concentración de trabajo 2 x y se incubó a temperatura ambiente para permitir que las formas tetraméricas/diméricas alcanzaran el estado estacionario. Las mediciones del ensayo se realizaron en un tampón que comprende TRIS 50 mM pH 7,8, NaPO4 100 mM, pH 7,8, 0,001 % v/v de Tween20. La proteína GLS1 recombinante purificada se diluyó en tampón de ensayo hasta 12 nM y se incubó previamente a temperatura ambiente por 30 minutos. Los compuestos de prueba se prepararon por dilución en 100 % de DMSO para producir el intervalo de dosis correcto para una respuesta de concentración de 12 puntos y se dispensó un volumen apropiado (2,5-60 nl) en placas de microensayo de 384 pocillos (código de producto Greiner 784900) mediante el uso de un dispensador acústico Labcyte Echo 555. La concentración de DMSO se mantuvo al 2 % mediante el rellenado de nuevo con solución de DMSO. Después se dispensaron 3 pL de proteína GLS1 diluida (12 nM) en cada pocillo mediante el uso de un dispensador automático BioRaptr (Beckman-Coulter) y se incubaron durante 15 minutos a temperatura ambiente. Después se adicionaron 3 pl de glutamina 100 mM diluida en tampón de ensayo y la reacción se incubó a temperatura ambiente por 60 minutos. Después se detuvo la reacción mediante la adición de 6-(2-bromoetinil)-2,3-dimetilquinazolin-4-ona 45 pM, rojo Amplex 75 pM, 0,375 unidades/ml de peroxidasa de rábano picante, 0,12 unidades/ml de glutamato oxidasa en TRIS pH 7,5 100 mM. Después de 30 minutos a temperatura ambiente en la oscuridad, las placas se leyeron en un Perkin Elmer EnVision mediante el uso de filtros ópticos de 535/590nm y los datos sin procesar se analizaron mediante el uso de Genedata para generar los valores de IC50. Una versión artefacto del ensayo donde la proteína GLS marcada con 6His y la glutamina se reemplazaron con tampón de ensayo se usó, además, para descartar efectos no específicos sobre los componentes del ensayo.
Ensayo b): Ensayo de potencia celular de GLS
Se evaluó el potencial de los compuestos para inhibir la actividad celular de GLS mediante el uso de un ensayo acoplado con PC3 que mide el agotamiento de glutamato celular. Los compuestos de prueba se prepararon por dilución en 100 % de DMSO para producir el intervalo de dosis correcto para una respuesta de concentración de 12 puntos y se dispensó un volumen apropiado (5-120 nl) en placas de microensayo de 384 pocillos (código de producto Corning 3712) mediante el uso de un dispensador acústico Labcyte Echo 555. La concentración de DMSO se mantuvo al 0,3 % mediante el rellenado de nuevo con solución de DMSO. Las células PC3 se cultivaron en DMEM libre de fenol, 10 % de FCS dializado, glutamina 2 mM y después de la dispersión por tripsinización se colocaron en placas a 5,6 x 103 células por pocillo en 40 pl de medio de crecimiento directamente en las placas de ensayo de 384 pocillos que contienen el compuesto dispensado. Después de la incubación por 6 h a 37 °C, 5 % de CO2 se aspiró el medio de crecimiento y las células se lisaron en 15 pl de tampón que contenía TRIS 10 mM, pH 7,4, NaCl 100 mM, EDTA 1 mM, EGTA 1 mM, NaF 1 mM, Na4P2O720 mM, Na3VO42 mM, 1 % de Tritón X-100, 10 % de glicerol, 0,1 % de SDS y 0,5 % de desoxicolato. Después se transfirieron 4 pl de lisado celular a una placa NBS de 384 pocillos (código de producto Corning 3575) y se adicionaron 35 pl de Amplex Red 27,5 pM, 0,1375 U/ml de peroxidasa de rábano picante, 0,044 U/ml de glutamato oxidasa, TRIS pH 7,5 100 mM. Después de 30 minutos a temperatura ambiente en la oscuridad, las placas se leyeron en un Perkin Elmer EnVision mediante el uso de filtros ópticos de 535/590nm y los datos brutos se analizaron mediante el uso de un software patentado para generar los valores de IC50.
Ensayo c): Ensayo de proliferación celular con GLS
La capacidad de los compuestos para inhibir el crecimiento celular se midió mediante el uso de un ensayo de proliferación celular NCI-H1703 en placa de 384 pocillos. Las células NCI-H1703 se cultivaron en RPMI1640 libre de rojo fenol, 10 % de FCS y glutamina 2 mM y se sembraron a una densidad de 750 células por pocillo en 40 pl de medio de crecimiento en placas de ensayo de 384 pocillos de fondo transparente (código de producto Corning 3712) y se incubaron por 24 h a 37 °C, 5 % de CO2. Los compuestos de prueba se prepararon por dilución en 100 % de DMSO para producir el intervalo de dosis correcto para una respuesta de concentración de 12 puntos y se dispensó un volumen apropiado (5-120 nl) directamente en las placas de ensayo que contenían células en placa. La concentración de DMSO se mantuvo al 0,3 % mediante el rellenado de nuevo con solución de DMSO. Las placas se incubaron durante 5 días a 37 °C, 5 % de CO2, se adicionaron Sytox Green y Saponin a una concentración final de 2 pM y 0,25 % respectivamente y se incubaron por 6 h antes del análisis. Las placas se leyeron en un Acumen eX3 (TTP Labtech) mediante el uso de excitación de 488 nm y un conjunto de filtros FITC (500-530 nm) para la emisión. Los valores de IC50 se calcularon mediante el ajuste de la curva a la inhibición máxima del crecimiento del día cero mediante el uso del análisis en el software GeneData.
Los resultados de los ensayos a) - c) se muestran en la Tabla 1.
Tabla 1. Datos del ensa o

(continuación)

Claims (12)
1. Un compuesto de la Fórmula (I):

o una sal farmacéuticamente aceptable de este, en donde:
Q es 5-metilpiridazin-3-ilo, 5-doropiridazin-3-ilo, 6-metilpiridazin-3-ilo, o 6-fluoropiridazin-3-ilo;
R es hidrógeno, fluoro, o metoxi;
R1 es hidrógeno, metoxi, difluorometoxi, o trifluorometoxi; y
R2 es metilo o etilo.
2. El compuesto de la reivindicación 1, o una sal farmacéuticamente aceptable de este, en donde Q es 6-metilpiridazin-3-ilo, o 6-fluoropiridazin-3-ilo.
3. El compuesto de la reivindicación 1, o una sal farmacéuticamente aceptable de este, en donde R es hidrógeno o fluoro.
4. El compuesto de la reivindicación 1, o una sal farmacéuticamente aceptable de este, en donde R es hidrógeno.
5. El compuesto de la reivindicación 1, o una sal farmacéuticamente aceptable de este, en donde R1 es metoxi, difluorometoxi, o trifluorometoxi.
6. El compuesto de la reivindicación 1, o una sal farmacéuticamente aceptable de este, en donde R2 es metilo.
7. El compuesto de la reivindicación 1, o una sal farmacéuticamente aceptable de este, en donde el compuesto se selecciona del grupo que consiste en:
(2S)-2-metoxi-2-(3-metoxifenil)-N-[5-[[(3R)-1-(6-metilpiridazin-3-il)pirrolidin-3-il]amino]-1,3,4-tiadiazol-2-il]acetamida;
(2S)-N-[5-[[(3R)-1-(6-fluoropiridazin-3-il)pirrolidin-3-il]amino]-1,3,4-tiadiazol-2-il]-2-metoxi-2-fenil-acetamida;
(2S)-N-[5-[[(3R)-1-(6-fluoropiridazin-3-il)pirrolidin-3-il]amino]-1,3,4-tiadiazol-2-il]-2-metoxi-2-(3-metoxifenil)acetamida;
(2S)-2-etoxi-2-(3-metoxifenil)-N-[5-[[(3R)-1-(6-metilpiridazin-3-il)pirrolidin-3-il]amino]-1,3,4-tiadiazol-2-il]acetamida; (2s)-2-(4-fluorofenil)-2-metoxi-N-[5-[[(3R)-1-(6-metilpiridazin-3-il)pirrolidin-3-il]amino]-1,3,4-tiadiazol-2-il]acetamida;
(2s)-2-(4-fluoro-3-metoxi-fenil)-2-metoxi-N-[5-[[(3R)-1-(6-metilpiridazin-3-il)pirrolidin-3-il]amino]-1,3,4-tiadiazol-2-il]acetamida;
(2s)-2-metoxi-N-[5-[[(3R)-1-(6-metilpiridazin-3-il)pirrolidin-3-il]amino]-1,3,4-tiadiazol-2-il]-2-[3-(trifluorometoxi)fenil]acetamida;
(2S)-2-(4-fluorofenil)-2-metoxi-N-[5-[[(3R)-1-(5-metilpiridazin-3-il)pirrolidin-3-il]amino]-1,3,4-tiadiazol-2-il]acetamida;
Ñ-[5-[[(3R)-1-(5-cloropiridazin-3-il)pirrolidin-3-il]amino]-1,3,4-tiadiazol-2-il]-2-metoxi-2-(4-metoxifenil)acetamida; (2S)-[3-(difluorometoxi)fenil]-2-metoxi-N-[5-[[(3R)-1-(6-metilpiridazin-3-il)pirrolidin-3-il]amino]-1,3,4-tiadiazol-2-il]acetamida; y
(2S)-2-[3-(difluorometoxi)fenil]-N-[5-[[(3R)-1-(6-fluoropiridazin-3-il)pirrolidin-3-il]amino]-1,3,4-tiadiazol-2-il]-2-metoxi-acetamida.
8. El compuesto de la reivindicación 1, o una sal farmacéuticamente aceptable de este, en donde el compuesto se selecciona del grupo que consiste en:
(2S)-2-metoxi-2-(3-metoxifenil)-N-[5-[[(3R)-1-(6-metilpiridazin-3-il)pirrolidin-3-il]amino]-1,3,4-tiadiazol-2-il]acetamida;
(2S)-N-[5-[[(3R)-1-(6-fluoropiridazin-3-il)pirrolidin-3-il]amino]-1,3,4-tiadiazol-2-il]-2-metoxi-2-(3-metoxifenil)acetamida;
(2S)-2-etoxi-2-(3-metoxifenil)-N-[5-[[(3R)-1-(6-metilpiridazin-3-il)pirrolidin-3-il]amino]-1,3,4-tiadiazol-2-il]acetamida; (2S)-2-(4-fluorofenil)-2-metoxi-N-[5-[[(3R)-1-(6-metilpiridazin-3-il)pirrolidin-3-il]amino]-1,3,4-tiadiazol-2-il]acetamida;
(2S)-2-(4-fluoro-3-metox¡-fen¡l)-2-metox¡-W-[5-[[(3R)-1-(6-met¡lp¡r¡daz¡n-3-¡l)p¡rrol¡d¡n-3-¡l]am¡no]-1,3,4-t¡ad¡azol-2-il]acetamida;
(2s)-2-metox¡-A/-[5-[[(3R)-1-(6-met¡lp¡r¡daz¡n-3-¡l)p¡rrol¡d¡n-3-¡l]am¡no]-1,3,4-t¡ad¡azol-2-¡l]-2-[3-(tr¡fluorometox¡)fen¡l]acetam¡da;
(2S)-2-(4-fluorofen¡l)-2-metox¡-W-[5-[[(3R)-1-(5-met¡lp¡r¡daz¡n-3-¡l)p¡rrol¡d¡n-3-¡l]am¡no]-1,3,4-t¡ad¡azol-2-¡l]acetam¡da;
(2sH3-(d¡fluorometox¡)fen¡l]-2-metox¡-W-[5-[[(3R)-1-(6-met¡lp¡r¡daz¡n-3-¡l)p¡rrol¡d¡n-3-¡l]am¡no]-1,3,4-t¡ad¡azol-2-¡l]acetam¡da; y
(2s)-2-[3-(d¡fluorometox¡)fen¡l]-N-[5-[[(3R)-1-(6-fluorop¡r¡daz¡n-3-¡l)p¡rrol¡d¡n-3-¡l]am¡no]-1,3,4-t¡ad¡azol-2-¡l]-2-metox¡-acetam¡da.
9. Una compos¡c¡ón farmacéut¡ca que comprende un compuesto de acuerdo con cualqu¡er re¡v¡nd¡cac¡ón de la 1 a la 8, o una sal farmacéut¡camente aceptable de este, y al menos un d¡luyente o portador farmacéut¡camente aceptable.
10. El compuesto de acuerdo con cualqu¡er re¡v¡nd¡cac¡ón de la 1 a la 8, o una sal farmacéut¡camente aceptable de este, para su uso en terap¡a.
11. El compuesto para su uso de acuerdo con la re¡v¡nd¡cac¡ón 10 para el tratam¡ento del cáncer.
12. El compuesto para su uso de acuerdo con la re¡v¡nd¡cac¡ón 11, en donde el cánceres cáncer de mama, cáncer de pulmón, cáncer pancreát¡co, cáncer renal, o cáncer hepatocelular.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201562260787P | 2015-11-30 | 2015-11-30 | |
| PCT/EP2016/079251 WO2017093300A1 (en) | 2015-11-30 | 2016-11-30 | 1,3,4-thiadiazole compounds and their use in treating cancer |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| ES2759940T3 true ES2759940T3 (es) | 2020-05-12 |
| ES2759940T8 ES2759940T8 (es) | 2020-05-20 |
Family
ID=57421876
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES16802102T Active ES2759940T3 (es) | 2015-11-30 | 2016-11-30 | Compuestos de 1,3,4-tiadiazol y su uso para tratar el cáncer |
Country Status (25)
| Country | Link |
|---|---|
| US (1) | US9938265B2 (es) |
| EP (1) | EP3383871B1 (es) |
| JP (1) | JP6873132B2 (es) |
| KR (1) | KR20180083412A (es) |
| CN (1) | CN108349967B (es) |
| AR (1) | AR106876A1 (es) |
| AU (1) | AU2016363719B2 (es) |
| BR (1) | BR112018010812A2 (es) |
| CA (1) | CA3005516C (es) |
| CL (1) | CL2018001408A1 (es) |
| CO (1) | CO2018006929A2 (es) |
| DK (1) | DK3383871T3 (es) |
| DO (1) | DOP2018000134A (es) |
| EA (1) | EA201891240A1 (es) |
| ES (1) | ES2759940T3 (es) |
| IL (1) | IL258644A (es) |
| MX (1) | MX2018006528A (es) |
| NI (1) | NI201800065A (es) |
| PE (1) | PE20181450A1 (es) |
| PH (1) | PH12018501132A1 (es) |
| SG (1) | SG11201803813UA (es) |
| SV (1) | SV2018005701A (es) |
| TN (1) | TN2018000126A1 (es) |
| TW (1) | TW201730188A (es) |
| WO (1) | WO2017093300A1 (es) |
Families Citing this family (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB201520959D0 (en) | 2015-11-27 | 2016-01-13 | Astrazeneca Ab And Cancer Res Technology Ltd | Bis-pyridazine compounds and their use in treating cancer |
| TW201731511A (zh) * | 2015-11-30 | 2017-09-16 | 阿斯特捷利康公司 | 1,3,4-噻二唑化合物及其在治療癌症中之用途 |
| US10323028B2 (en) | 2015-11-30 | 2019-06-18 | Astrazeneca Ab | 1,3,4-thiadiazole compounds and their use in treating cancer |
| AU2019363115B2 (en) | 2018-10-16 | 2021-12-09 | Medshine Discovery Inc. | Thiadiazole derivative and uses thereof as a GLS1 inhibitor |
| WO2025196446A1 (en) * | 2024-03-22 | 2025-09-25 | Sitryx Therapeutics Limited | Thiadiazoles as glutaminase 1 inhibitors |
Family Cites Families (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2001038332A1 (en) * | 1999-11-24 | 2001-05-31 | Merck & Co., Inc. | Gamma-hydroxy-2-(fluoroalkylaminocarbonyl)-1-piperazinepentanamides as hiv protease inhibitors |
| US6451828B1 (en) * | 2000-08-10 | 2002-09-17 | Elan Pharmaceuticals, Inc. | Selective inhibition of glutaminase by bis-thiadiazoles |
| MY142655A (en) * | 2003-06-12 | 2010-12-15 | Euro Celtique Sa | Therapeutic agents useful for treating pain |
| WO2006053342A2 (en) * | 2004-11-12 | 2006-05-18 | Osi Pharmaceuticals, Inc. | Integrin antagonists useful as anticancer agents |
| SI2421826T1 (sl) * | 2009-04-20 | 2014-02-28 | F. Hoffmann-La Roche Ag | Derivati prolina kot inhibitorji katepsina |
| ES2761866T3 (es) * | 2011-11-21 | 2020-05-21 | Calithera Biosciences Inc | Inhibidores heterocíclicos de glutaminasa |
| BR112015010955A2 (pt) * | 2012-11-16 | 2017-07-11 | Calithera Biosciences Inc | inibidores de glutaminase heterocíclica |
| US9029531B2 (en) | 2012-11-22 | 2015-05-12 | Agios Pharmaceuticals, Inc. | Compounds and their methods of use |
| CA2934700A1 (en) * | 2014-01-06 | 2015-07-09 | Rhizen Pharmaceuticals Sa | Glutaminase inhibitors |
| EP3116872A4 (en) * | 2014-03-14 | 2017-08-30 | Calithera Biosciences, Inc. | Combination therapy with glutaminase inhibitors |
| GB201409624D0 (en) | 2014-05-30 | 2014-07-16 | Astrazeneca Ab | 1,3,4-thiadiazole compounds and their use in treating cancer |
| US10323028B2 (en) * | 2015-11-30 | 2019-06-18 | Astrazeneca Ab | 1,3,4-thiadiazole compounds and their use in treating cancer |
| TW201731511A (zh) | 2015-11-30 | 2017-09-16 | 阿斯特捷利康公司 | 1,3,4-噻二唑化合物及其在治療癌症中之用途 |
-
2016
- 2016-11-29 US US15/363,018 patent/US9938265B2/en active Active
- 2016-11-29 TW TW105139312A patent/TW201730188A/zh unknown
- 2016-11-30 AR ARP160103665A patent/AR106876A1/es unknown
- 2016-11-30 AU AU2016363719A patent/AU2016363719B2/en active Active
- 2016-11-30 WO PCT/EP2016/079251 patent/WO2017093300A1/en not_active Ceased
- 2016-11-30 TN TNP/2018/000126A patent/TN2018000126A1/en unknown
- 2016-11-30 CA CA3005516A patent/CA3005516C/en active Active
- 2016-11-30 CN CN201680067261.2A patent/CN108349967B/zh active Active
- 2016-11-30 KR KR1020187017164A patent/KR20180083412A/ko not_active Withdrawn
- 2016-11-30 EP EP16802102.0A patent/EP3383871B1/en active Active
- 2016-11-30 BR BR112018010812A patent/BR112018010812A2/pt not_active Application Discontinuation
- 2016-11-30 DK DK16802102.0T patent/DK3383871T3/da active
- 2016-11-30 PE PE2018001021A patent/PE20181450A1/es unknown
- 2016-11-30 ES ES16802102T patent/ES2759940T3/es active Active
- 2016-11-30 JP JP2018526717A patent/JP6873132B2/ja active Active
- 2016-11-30 SG SG11201803813UA patent/SG11201803813UA/en unknown
- 2016-11-30 MX MX2018006528A patent/MX2018006528A/es unknown
- 2016-11-30 EA EA201891240A patent/EA201891240A1/ru unknown
-
2018
- 2018-04-11 IL IL258644A patent/IL258644A/en unknown
- 2018-05-24 CL CL2018001408A patent/CL2018001408A1/es unknown
- 2018-05-29 PH PH12018501132A patent/PH12018501132A1/en unknown
- 2018-05-29 NI NI201800065A patent/NI201800065A/es unknown
- 2018-05-29 SV SV2018005701A patent/SV2018005701A/es unknown
- 2018-05-29 DO DO2018000134A patent/DOP2018000134A/es unknown
- 2018-06-29 CO CONC2018/0006929A patent/CO2018006929A2/es unknown
Also Published As
| Publication number | Publication date |
|---|---|
| CA3005516C (en) | 2024-04-16 |
| TN2018000126A1 (en) | 2019-10-04 |
| US9938265B2 (en) | 2018-04-10 |
| WO2017093300A1 (en) | 2017-06-08 |
| JP2019501134A (ja) | 2019-01-17 |
| US20170152254A1 (en) | 2017-06-01 |
| SG11201803813UA (en) | 2018-06-28 |
| SV2018005701A (es) | 2018-11-27 |
| AU2016363719A1 (en) | 2018-07-05 |
| NI201800065A (es) | 2018-10-18 |
| CN108349967A (zh) | 2018-07-31 |
| IL258644A (en) | 2018-06-28 |
| ES2759940T8 (es) | 2020-05-20 |
| JP6873132B2 (ja) | 2021-05-19 |
| CL2018001408A1 (es) | 2018-10-12 |
| AU2016363719B2 (en) | 2019-11-14 |
| EA201891240A1 (ru) | 2018-11-30 |
| CN108349967B (zh) | 2022-02-15 |
| BR112018010812A2 (pt) | 2018-11-27 |
| EP3383871B1 (en) | 2019-09-11 |
| DOP2018000134A (es) | 2018-06-30 |
| AR106876A1 (es) | 2018-02-28 |
| CO2018006929A2 (es) | 2018-10-10 |
| PE20181450A1 (es) | 2018-09-12 |
| CA3005516A1 (en) | 2017-06-08 |
| AU2016363719A8 (en) | 2018-07-12 |
| KR20180083412A (ko) | 2018-07-20 |
| PH12018501132A1 (en) | 2019-01-21 |
| MX2018006528A (es) | 2019-05-15 |
| DK3383871T3 (da) | 2019-12-16 |
| EP3383871A1 (en) | 2018-10-10 |
| TW201730188A (zh) | 2017-09-01 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2688396T3 (es) | Compuestos de 1,3,4,tiadiazol y su uso en el tratamiento del cáncer | |
| ES2796562T3 (es) | Compuestos de 1,3,4-tiadiazol y su uso en el tratamiento del cáncer | |
| ES2759940T3 (es) | Compuestos de 1,3,4-tiadiazol y su uso para tratar el cáncer | |
| ES2914333T3 (es) | Compuestos de 1,3,4-tiadiazol y su uso para tratar cáncer | |
| US10577354B2 (en) | Bis-pyridazine compounds and their use in treating cancer | |
| HK1260702B (en) | 1,3,4-thiadiazole compounds and their use in treating cancer | |
| HK1260702A1 (en) | 1,3,4-thiadiazole compounds and their use in treating cancer | |
| HK1260707A1 (en) | 1,3,4-thiadiazole compounds and their use in treating cancer |