ES2769876T3 - Derivados de uncialamicina, métodos de síntesis y su uso como agentes antitumorales - Google Patents
Derivados de uncialamicina, métodos de síntesis y su uso como agentes antitumorales Download PDFInfo
- Publication number
- ES2769876T3 ES2769876T3 ES14758223T ES14758223T ES2769876T3 ES 2769876 T3 ES2769876 T3 ES 2769876T3 ES 14758223 T ES14758223 T ES 14758223T ES 14758223 T ES14758223 T ES 14758223T ES 2769876 T3 ES2769876 T3 ES 2769876T3
- Authority
- ES
- Spain
- Prior art keywords
- substituted
- protecting group
- hydrogen
- alkyl
- group
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 0 *OC(c1ccccc1C=O)C#N Chemical compound *OC(c1ccccc1C=O)C#N 0.000 description 14
- QMEOASZKINLLSS-UHFFFAOYSA-N CC(C)(C)OC(Nc1ccc(COC2=O)c2c1)=O Chemical compound CC(C)(C)OC(Nc1ccc(COC2=O)c2c1)=O QMEOASZKINLLSS-UHFFFAOYSA-N 0.000 description 1
- CMVIRMWIDQUAGG-UHFFFAOYSA-N CCN(CC)C(c1cc(cc(C)c(C)c2)c2cc1C=O)=O Chemical compound CCN(CC)C(c1cc(cc(C)c(C)c2)c2cc1C=O)=O CMVIRMWIDQUAGG-UHFFFAOYSA-N 0.000 description 1
- JSOBGEOXZBIWGW-UHFFFAOYSA-N N#CC(c1c2cc(CN(C(c(c3c(c(Cl)c4Cl)Cl)c4Cl)=O)C3=O)cc1)OC2=O Chemical compound N#CC(c1c2cc(CN(C(c(c3c(c(Cl)c4Cl)Cl)c4Cl)=O)C3=O)cc1)OC2=O JSOBGEOXZBIWGW-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/02—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains two hetero rings
- C07D491/08—Bridged systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/4353—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems
- A61K31/4355—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems the heterocyclic ring system containing a five-membered ring having oxygen as a ring hetero atom
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/439—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom the ring forming part of a bridged ring system, e.g. quinuclidine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6801—Drug-antibody or immunoglobulin conjugates defined by the pharmacologically or therapeutically active agent
- A61K47/6803—Drugs conjugated to an antibody or immunoglobulin, e.g. cisplatin-antibody conjugates
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6835—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site
- A61K47/6851—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a determinant of a tumour cell
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0019—Injectable compositions; Intramuscular, intravenous, arterial, subcutaneous administration; Compositions to be administered through the skin in an invasive manner
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0019—Injectable compositions; Intramuscular, intravenous, arterial, subcutaneous administration; Compositions to be administered through the skin in an invasive manner
- A61K9/0021—Intradermal administration, e.g. through microneedle arrays or needleless injectors
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/12—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains three hetero rings
- C07D491/18—Bridged systems
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Epidemiology (AREA)
- Immunology (AREA)
- Dermatology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Cell Biology (AREA)
- Oncology (AREA)
- Communicable Diseases (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Peptides Or Proteins (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Medicinal Preparation (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
- Steroid Compounds (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
Un compuesto de la fórmula: **(Ver fórmula)** en donde: cada uno de R1 y R2 se selecciona independientemente entre hidrógeno, hidroxi, alquilo(C1-12), alquilo sustituido(C1 10 -12), alquenilo(C2-12), alquenilo sustituido(C2-12), alquinilo(C2-12), alquinilo sustituido(C2- 12), arilo(C6-12), arilo sustituido(C6-12), aralquilo(C7-12), aralquilo sustituido(C7-12), heteroarilo(C1-12), heteroarilo sustituido(C1-12), heterocicloalquilo(C2-12), heterocicloalquilo sustituido(C2-12), acilo(C1-12), acilo sustituido(C1-12), aciloxi(C1-12), aciloxi sustituido(C1-12), alquilamino(C1-12), alquilamino sustituido(C1-12); un grupo protector de amina monovalente, -C(O)O(CH2)nS-A1, -C(O)O(CH2)nS(O)-A1 o -C(O)O(CH2)nS(O)2-A1, en donde: A1 es arilo(C6-12) o arilo sustituido(C6-12); y n es 1, 2, 3, 4 o 5; o R1 y R2 se toman juntos y forman un grupo protector de amina divalente, R3 es hidrógeno, hidroxi, halo o alcoxi(C1-12) o alcoxi sustituido(C1-12); o es 1,2 o 3; R4 es hidrógeno, un grupo protector de amina monovalente, alquilo(C1-12) o alquilo sustituido(C1-12); cada uno de R5, R6 y R7 es independientemente hidrógeno, hidroxi, amino, mercapto, -OX1, -NX2X3 o -SX4; o alquilo(C1-12) o alquilo sustituido(C1-12); en donde: X1 es un grupo protector de hidroxi; X2 y X3 se seleccionan independientemente entre hidrógeno, un grupo protector de amina monovalente, o cuando X2 y X3 se toman juntos forman un grupo protector de amina divalente; y X4 es un grupo protector de tiol; y R8 es hidroxi, amino o mercapto; en donde: cada grupo protector de amina se selecciona independientemente entre la lista que consiste en formilo, acetilo, propionilo, pivaloílo, t-butilacetilo, 2-cloroacetilo, 2-bromoacetilo, trifluoroacetilo, tricloroacetilo, o nitrofenoxiacetilo, α-clorobutirilo, benzoílo, 4-clorobenzoílo, 4-bromobenzoílo, 4-nitrobenzoílo, un grupo sulfonilo, un grupo aralquilo y un grupo sililo; el grupo protector de hidroxilo se selecciona entre la lista que consiste en formilo, acetilo, propionilo, pivaloílo, tbutilacetilo, 2-cloroacetilo, 2-bromoacetilo, trifluoroacetilo, tricloroacetilo, o-nitrofenoxiacetilo, α-clorobutirilo, benzoílo, 4-clorobenzoílo, 4-bromobenzoílo, 4-nitrobenzoílo, un grupo sulfonilo, un grupo aciloxi, un grupo aralquilo y un grupo sililo; y el grupo protector de tiol se selecciona entre la lista que consiste en formilo, acetilo, propionilo, pivaloílo, tbutilacetilo, 2-cloroacetilo, 2-bromoacetilo, trifluoroacetilo, tricloroacetilo, o-nitrofenoxiacetilo, α-clorobutirilo, benzoílo, 4-clorobenzoílo, 4-bromobenzoílo, 4-nitrobenzoílo, un grupo sulfonilo, un grupo aciloxi, un grupo aralquilo y un grupo sililo; o una sal farmacéuticamente aceptable del mismo.
Description
DESCRIPCIÓN
Derivados de uncialamicina, métodos de síntesis y su uso como agentes antitumorales
Antecedentes
1. Campo de la invención
La presente divulgación se refiere a los campos de la medicina, la farmacología, la química y la oncología. En particular, se divulgan compuestos, conjugados farmacológicos, métodos de tratamiento y métodos de síntesis relacionados con la uncialamicina y análogos de la misma.
2. Técnica relacionada
La uncialamicina es un antibiótico que ha demostrado ser muy prometedor como un posible reactivo antineoplásico. El uso de la uncialamicina como antineoplásico se ha descrito en Nicolaou, et al., 2007 y Nicolaou, et al., 2008, los cuales se incorporan en el presente documento por referencia. Se ha demostrado que el compuesto es un potente antitumoral para el cáncer con una CI50 en el orden de picomolar. Adicionalmente, el compuesto ha sido concebido como una posible carga útil para anticuerpos. Desafortunadamente, los métodos de síntesis actuales carecen de la flexibilidad para producir numerosos análogos y generar cantidades suficientes de los compuestos deseados. Dado el impacto mundial y generalizado del cáncer, los nuevos agentes terapéuticos son de importancia comercial.
El documento WO 2007/038868 describe un compuesto de enediina, la uncialamicina, incluyendo análogos de la misma. La solicitud también se refiere a métodos de uso del compuesto para tratar infecciones bacterianas, para inhibir el crecimiento bacteriano y para destruir bacterias. El compuesto también puede utilizarse para tratar tumores, para impedir el crecimiento de células tumorales, para destruir células tumorales, para tratar cánceres, para impedir el crecimiento de células cancerosas y para destruir células cancerosas.
El documento WO 2013/122823 describe compuestos de enediina utilizados en fármacos quimioterapéuticos, especialmente en conjugados, para el tratamiento de enfermedades tales como el cáncer.
Sumario
La presente invención proporciona un compuesto de acuerdo con la reivindicación 1.
Como se describe en el presente documento, se proporciona un compuesto de la fórmula:

en donde: Y1 es -O(CH2)mY', -NH(CH2)mY', -S(CH2)mY' o -(CH2)mNR-iR2, o se toma junto con Z1 como se define a continuación; en donde: Y' es hidroxi, halo, mercapto, alquilo(C1-12), alquilo sustituido(C1-12), alquenilo(C2-12), alquenilo sustituido(C2-i2), alquinilo(C2-12), alquinilo sustituido(C2-12), acilo(C<12), acilo sustituido(C1-C2), aciloxi(C1-12), aciloxi sustituido(C1-12), alquilamino(C1-12) o alquilamino sustituido(C1-12); m es 1, 2, 3, 4, 5 o 6 ; y cada uno de R1 y R2 se selecciona independientemente entre hidrógeno, hidroxi, alquilo(01-12), alquilo sustituido(C1-12), alquenilo(C2-12), alquenilo sustituido(C2-12), alquinilo(C2-12), alquinilo sustituido(C2-12), arilo(C6-12), arilo sustituido(C6-12), aralquilo(C7-12), aralquilo sustituido(C7-12), heteroarilo(C1-12), heteroarilo sustituido(C1-12), heterocicloalquilo(C2-12), heterocicloalquilo sustituido(C2-12), acilo(C1-12), acilo sustituido(C1-12), aciloxi(C1-12), aciloxi sustituido(C1-12), alquilamino(C1-12), alquilamino sustituido(C1-12); un grupo protector de amina monovalente, -C(O)O(CH2)nS-A1, -C(O)O(CH2)nS(O)-A1 o -C(O)O(CH2)nS(O)2-A1, en donde: A1 es arilo(C6-12), arilo sustituido(C6-12), o

en donde: A2 es alquenilo(C2-12), alquenilo sustituido(C2-12), alquinilo(C2-12), alquinilo sustituido(C2-12), acilo(C1-12), acilo sustituido(C1-12), aciloxi(C1-12), aciloxi sustituido(C1-12), alcoxi(C1-12), alcoxi sustituido(C1-12), alquilamino(C1-12), alquilamino
sustituidO(ci-i2), dialquilaminO(C2-i2) o dialquilamino sustituido(C2-12); en donde A2 no es -CO2H, -CO2CH3, -OCH3, -OCH2CH3, -C(O)CH3, -NHCH3, -NHCH2CH3, -N(CH3)2, -C(O)NH2 y -OC(O)CH3; n es 1, 2, 3, 4 o 5; o Ri y R2 se toman juntos y son un grupo protector de amina divalente, alcanodiílo(C1-12), alquilaminodiílo(C1-8); alcoxidiílo(C1-8); o una versión sustituida de cualquiera de estos grupos; o Y1 se toma con Z1 y es alquilaminodiílo(C1-8) alquilaminodiílo sustituido(C1-8); -alcanodiil(C1-6)-NZ2-alcanodiílo(C1-6) o -alcanodiil sustituido(C1-6)-NZ2-alcanodiílo sustituido(C1-6), en donde: Z2 es hidrógeno, un grupo protector de amina, acilo(C6-12), acilo sustituido(C6-12), -C(O)O(CH2)nS-A3 o -C(O)O(CH2)nS(O)2-A3, en donde: A3 es arilo(C6-12), arilo sustituido(C6-12), o

en donde: A4 es alquenilo(C2-12), alquenilo sustituido(C2-12), alquinilo(C2-12), alquinilo sustituido(C2-12), acilo(C1-12), acilo sustituido(C1-12), aciloxi(C1-12), aciloxi sustituido(C1-12), alcoxi(C1-12), alcoxi sustituido(C1-12), alquilamino(C1-12), alquilamino sustituido(C1-12), dialquilamino(C2-12), o dialquilamino sustituido(C2-12), en donde A4 no es -CO2H, -CO2CH3, -OCH3, -OCH2CH3, -C(O)CH3, -NHCH3, -NHCH2CH3, -N(CH3)2, -C(O)NH2 y -OC(O)CH3; Z1 está ausente, es hidrógeno o se toma junto con Y1 como se ha definido anteriormente; cada uno de R3 y Z2 se selecciona independientemente entre hidrógeno, hidroxi, halo, amino, ciano, nitro, fosfato o mercapto, o alquilo(C1-12), alquenilo(C2-12), alquinilo(C2-12), arilo(C6-12), aralquilo(C7-12), heteroarilo(C1-12), heterocicloalquilo(C2-12), acilo(C1-12), alcoxi(C1-12), aciloxi(C1-12), alquilamino(C1-12), dialquilamino(C2-12), amido(C1-12) o una versión sustituida de cualquiera de estos grupos; o es 1,2 o 3; R4 es hidrógeno, alquilo(C1-12), un grupo protector de amina monovalente o alquilo sustituido(C1-12); cada uno de R5, R6 y R7 es independientemente hidrógeno, hidroxi, amino, mercapto, -OX1, -NX2X3 o -SX4; o alquilo(C1-12), alcoxi(C1-12), aciloxi(C1-12), alquilamino(C1-12), dialquilamino(C2-12), alquiltio(C1-12), amido(C1-12) o una versión sustituida de cualquiera de estos grupos; en donde: X1 es un grupo protector de hidroxi; X2 y X3 se seleccionan independientemente entre hidrógeno, un grupo protector de amina monovalente, o cuando X2 y X3 se toman juntos forman un grupo protector de amina divalente; X4 es un grupo protector de tiol; R8 es hidroxi, amino o mercapto; o alcoxi(C1-12), aciloxi(C1-12), alquilamino(C1-12), dialquilamino(C2-12), alquiltio(C1-12), amido(C1-12), o una versión sustituida de cualquiera de estos grupos; y cada uno de Rg, R10, R11 y R12 se selecciona independientemente entre hidrógeno, hidroxi, amino, mercapto, -OX1, -NX2X3 o -SX4, o alquilo(C1-12), alquenilo(C2-12), alquinilo(C2-12), arilo(C6-12), aralquilo(C7-12), heteroarilo(C1-12), heterocicloalquilo(C2-12), acilo(C1-12), alcoxi(C1-12), aciloxi(C1-12), alquilamino(C1-12), dialquilamino(C2-12), alquiltio(C1-12), amido(C1-12), o una versión sustituida de cualquiera de estos grupos; o Y2-R13; en donde: X1 es alquilo(C1-12), alquilo sustituido(C1-12), alquenilo(C2-12), alquenilo sustituido(C2-12), alquinilo(C2-12), alquinilo sustituido(C2-12), arilo(C6-12), arilo sustituido(C6-12), aralquilo(C7-12), aralquilo sustituido(C7-12), heteroarilo(C1-12), heteroarilo sustituido(C1-12), heterocicloalquilo(C2-12), heterocicloalquilo sustituido(C2-12), acilo(C1-12), acilo sustituido(C1-12) o un grupo protector de hidroxi; X2 y X3 se seleccionan independientemente entre hidrógeno, alquilo(01-12), alquilo sustituido(C1-12), alquenilo(C2-12), alquenilo sustituido(C2-12), alquinilo(C2-12), alquinilo sustituido(C2-12), arilo(C6-12), arilo sustituido(C6-12), aralquilo(C7-12), aralquilo sustituido(C7-12), heteroarilo(C1-12), heteroarilo sustituido(C1-12), heterocicloalquilo(C2-12), heterocicloalquilo sustituido(C2-12), acilo(C1-12), acilo sustituido(C1-12), un grupo protector de amina monovalente, o cuando X2 y X3 se toman juntos forman un grupo protector de amina divalente; X4 es alquilo(01-12), alquilo sustituido(C1-12), alquenilo(C2-12), alquenilo sustituido(C2-12), alquinilo(C2-12), alquinilo sustituido(C2-12), arilo(C6-12), arilo sustituido(C6-12), aralquilo(C7-12), aralquilo sustituido(C7-12), heteroarilo(C1-12), heteroarilo sustituido(C1-12), heterocicloalquilo(C2-12), heterocicloalquilo sustituido(C2-12), acilo(C1-12), acilo sustituido(C1-12), o un grupo protector de tiol; Y2 es alcanodiílo(C1-12) o alcanodiílo sustituido(C1-12); y R13 es hidroxi, amino, mercapto, -OX1, -NX2X3 o -SX4, o heteroarilo(C1-12), heterocicloalquilo(C2-12), acilo(C1-12), alcoxi(C1-12), aciloxi(C1-12), alquilamino(C1-12), dialquilamino(C2-12), alquiltio(C1-12), amido(C1-12) o una versión sustituida de cualquiera de estos grupos; y cada uno de p y q es independientemente 1 o 2 ; Z3 es hidrógeno, hidroxi, halo, amino, ciano, nitro, fosfato o mercapto, o alquilo(01-12), alquenilo(C2-12), alquinilo(C2-12), arilo(C6-12), aralquilo(C7-12), heteroarilo(C1-12), heterocicloalquilo(C2-12), acilo(C1-12), alcoxi(C1-12), aciloxi(C1-12), alquilamino(C1-12), dialquilamino(C2-12), amido(C1-12) o una versión sustituida de cualquiera de estos grupos; con la condición de que Y1 no sea -NHMe o -NHCH2CH2NH2; o una sal farmacéuticamente aceptable del mismo. Como se describe en el presente documento, el compuesto se define adicionalmente como:

en donde: Y1 es -O(CH2)mY', -NH(CH2)mY', -S(CH2)mY' o -CH2NR1R2, en donde: Y' es alquilo(C1-12), alquilo sustituido(C1-12), alquenilo(C2-12), alquenilo sustituido(C2-12), alquinilo(C2-12), alquinilo sustituido(C2-12), acilo(C<12), acilo sustituido(C1-12), aciloxi(C1-12), aciloxi sustituido(C1-12), alquilamino(C1-12), o alquilamino sustituido(C1-12); m es 1, 2, 3, 4, 5
o 6 ; cada uno de Ri y R2 se selecciona independientemente entre hidrógeno, hidroxi, alquilO(ci-i2), alquenilo(C<12), alquinilO(C<i2), arilo(C<12), aralquilo(C<12), heteroarilo(C<12), heterocicloalquilo(C<12), acilo(C<12), alcoxi(C<12) o una versión sustituida de cualquiera de estos grupos; R1 y R2 se toman juntos y son un grupo protector divalente, alcanodiílo(1-12), alcoxidiílo(C1-8) o una versión sustituida de cualquiera de estos grupos; o -C(O)O(CH2)nS-A1 o -C(O)O(CH2)nS(O)2-A1, en donde: A1 es arilo(C6-12), arilo sustituido(C6-12), o

en donde: A2 es alquenilo(C2-12), alquenilo sustituido(C2-12), alquinilo(C2-12), alquinilo sustituido(C2-12), acilo(C1-12), acilo sustituido(C1-12), aciloxi(C1-12), aciloxi sustituido(C1-12), alcoxi(C1-12), alcoxi sustituido(C1-12), alquilamino(C1-12), alquilamino sustituido(C1-12), dialquilamino(C2-12) o dialquilamino sustituido(C2-12); en donde A2 no es -CO2H, -CO2CH3, -OCH3, -OCH2CH3 , -C(O)CH3, -NHCH3, -NHCH2CH3, -N(CH3)2, -C(O)NH2 y -OC(O)CH3; n es 1, 2, 3, 4 o 5; R3 es hidrógeno, hidroxi, halo, amino, ciano, nitro, fosfato o mercapto, o alquilo(C1-12), alquenilo(C2-12), alquinilo(C2-12), arilo(oa-12), aralquilo(C7-12), heterocicloalquilo(C2-12), acilo(C1-12), alcoxi(C1-12), aciloxi(C1-12), alquilamino(C1-12), dialquilamino(C2-12), amido(C1-12), o una versión sustituida de cualquiera de estos grupos; R4 es hidrógeno, alquilo(C1-12), un grupo protector de amina monovalente o alquilo sustituido(C1-12); cada uno de R5, Ra y R7 es independientemente hidrógeno, hidroxi, amino, mercapto, -OX1, -NX2X3 o -SX4; o alquilo(C1-12), alcoxi(C1-12), aciloxi(C1-12), alquilamino(C1-12), dialquilamino(C2-12), alquiltio(C1-12), amido(C1-12) o una versión sustituida de cualquiera de estos grupos; en donde: X1 es un grupo protector de hidroxi; X2 y X3 se seleccionan independientemente entre hidrógeno, un grupo protector de amina monovalente, o cuando X2 y X3 se toman juntos forman un grupo protector de amina divalente; X4 es un grupo protector de tiol; R8 es hidroxi, amino o mercapto; o alcoxi(C1-12), aciloxi(C1-12), alquilamino(C1-12), dialquilamino(C2-12), alquiltio(C1-12), amido(C1-12), o una versión sustituida de cualquiera de estos grupos; cada uno de Rg, R10 y R11 se selecciona independientemente entre hidrógeno, hidroxi, amino, mercapto, -OX1, -NX2X3 o -SX4 , o alquilo(C1-12), alquenilo(C2-12), alquinilo(C2-12), arilo(oa-12), aralquilo(C7-12), heteroarilo(C1-12), heterocicloalquilo(C2-12), acilo(C1-12), alcoxi(C1-12), aciloxi(C1-12), alquilamino(C1-12), dialquilamino(C2-12), alquiltio(C1-12), amido(C1-12), o una versión sustituida de cualquiera de estos grupos; o Y2-R12; en donde: X 1 es alquilo(C1-12), alquilo sustituido(C1-12), alquenilo(C2-12), alquenilo sustituido(C2-12), alquinilo(C2-12), alquinilo sustituido(C2-12), arilo(Ca-12), arilo sustituido(Ca-12), aralquilo(C7-12), aralquilo sustituido(C7-12), heteroarilo(C1-12), heteroarilo sustituido(C1-12), heterocicloalquilo(C2-12), heterocicloalquilo sustituido(C2-12), acilo(C1-12), acilo sustituido(C1-12) o un grupo protector de hidroxi; X2 y X3 se seleccionan independientemente entre hidrógeno, alquilo(C1-12), alquilo sustituido(C1-12), alquenilo(C2-12), alquenilo sustituido(C2-12), alquinilo(C2-12), alquinilo sustituido(C2-12), arilo(Ca-12), arilo sustituido(Ca-12), aralquilo(C7-12), aralquilo sustituido(C7-12), heteroarilo(C1-12), heteroarilo sustituido(C1-12), heterocicloalquilo(C2-12), heterocicloalquilo sustituido(C2-12), acilo(C1-12), acilo sustituido(C1-12), un grupo protector de amina monovalente, o cuando X2 y X3 se toman juntos forman un grupo protector de amina divalente; X4 es alquilo(C1-12), alquilo sustituido(C1-12), alquenilo(C2-12), alquenilo sustituido(C2-12), alquinilo(C2-12), alquinilo sustituido(C2-12), arilo(Ca-12), arilo sustituido(Ca-12), aralquilo(C7-12), aralquilo sustituido(C7-12), heteroarilo(C1-12), heteroarilo sustituido(C1-12), heterocicloalquilo(C2-12), heterocicloalquilo sustituido(C2-12), acilo(C1-12), acilo sustituido(C1-12), o un grupo protector de tiol; Y2 es alcanodiílo(C1-12) o alcanodiílo sustituido(C1-12); o R12 es hidroxi, amino, mercapto, -OX1, -NX2X3 o -SX4, o heteroarilo(C1-12), heterocicloalquilo(C2-12), acilo(C1-12), alcoxi(C1-12), aciloxi(C1-12), alquilamino(C1-12), dialquilamino(C2-12), alquiltio(C1-12), amido(C1-12) o una versión sustituida de cualquiera de estos grupos; o con la condición de que Y1 no sea NHMe, -NHC(O)CaH4NH2, -NHC(O)CH2NH2, -NHC(O)CH(CH2OH)NH2, -NHC(O)CH(NH2)CH2CH2CH2CH2NH2 o -NHCH2CH2NH2; o una sal farmacéuticamente aceptable del mismo. Como se describe en el presente documento, la fórmula se define adicionalmente como:

en donde: Y1 es -O(CH2)mY', -NH(CH2)mY', -S(CH2)mY' o -(CH2)mNR1R2, o se toma junto con Z1 como se describe a continuación; en donde: Y' es hidroxi, halo, mercapto, alquenilo(C2-12), alquenilo sustituido(C2-12), alquinilo(C2-12), alquinilo sustituido(C2-12), acilo(C<12), acilo sustituido(C1-12), aciloxi(C1-12), aciloxi sustituido(C1-12), alquilamino(C1-12), o alquilamino sustituido(C1-12); m es 1, 2, 3, 4, 5 o a; y cada uno de R1 y R2 se selecciona independientemente entre hidrógeno, hidroxi, alquilo(C1-12), alquilo sustituido(C1-12), alquenilo(C2-12), alquenilo sustituido(C2-12), alquinilo(C2-12), alquinilo sustituido(C2-12), arilo(Ca-12), arilo sustituido(Ca-12), aralquilo(C7-12), aralquilo sustituido(C7-12), heteroarilo(C1-12), heteroarilo sustituido(C1-12), heterocicloalquilo(C2-12), heterocicloalquilo sustituido(C2-12), acilo(C1-12), acilo sustituido(C1-12), aciloxi(C1-12), aciloxi sustituido(C1-12), alquilamino(C1-12), alquilamino sustituido(C1-12); un grupo protector de amina monovalente, -C(O)O(CH2)nS-A1, -C(O)O(CH2)nS(O)-A1 o -C(O)O(CH2)nS(O)2-A1, en donde: A1 es arilo(Ca-12) o arilo sustituido(Ca-12); n es 1, 2, 3, 4 o 5; R1 y R2 se toman juntos y son un grupo protector divalente, alcanodiílo(C<12), alquilaminodiílo(C<8); alcoxidiílo(C<8); o una versión sustituida de cualquiera de estos grupos; o Y1 se toma con Z1 y es alquilaminodiílo(C1-8) alquilaminodiílo sustituido(C1-8); o -alcanodiil(C1-a)-NZ2-alcanodiílo(C1-a), en donde: Z2 es hidrógeno,
un grupo protector de amina, acilO(C6-i2), acilo sustituidO(C6-i2), -C(O)O(CH2)nS-A3 o -C(O)O(CH2)nS(O)2-A3, en donde: A3 es arilo(Ce-12) o arilo sustituido(C6-12); Z1 está ausente, es hidrógeno o se toma junto con Y1 como se ha definido anteriormente; R3 es hidrógeno, hidroxi, halo, amino, ciano, nitro, fosfato o mercapto, o alquilo(C1-12), alquenilo(C2-12), alquinilo(C2-12), arilo(C6-12), aralquilo(C7-12), heteroarilo(C1-12), heterocicloalquilo(C2-12), acilo(C1-12), alcoxi(C1-12), aciloxi(C1-12), alquilamino(C1-12), dialquilamino(C2-12), amido(C1-12) o una versión sustituida de cualquiera de estos grupos; o es 1, 2 o 3; R4 es hidrógeno, alquilo(C1-12), un grupo protector de amina monovalente o alquilo sustituido(C1-12); cada uno de R5, R6 y R7 es independientemente hidrógeno, hidroxi, amino, mercapto, -OX1, -NX2X3 o -SX4; o alquilo(C1-12), alcoxi(C1-12), aciloxi(C1-12), alquilamino(C1-12), dialquilamino(C2-12), alquiltio(C1-12), amido(C1-12) o una versión sustituida de cualquiera de estos grupos; en donde: X 1 es un grupo protector de hidroxi; X2 y X3 se seleccionan independientemente entre hidrógeno, un grupo protector de amina monovalente, o cuando X2 y X3 se toman juntos forman un grupo protector de amina divalente; X4 es un grupo protector de tiol; R8 es hidroxi, amino o mercapto; o alcoxi(C1-12), aciloxi(C1-12), alquilamino(C1-12), dialquilamino(C2-12), alquiltio(C1-12), amido(C1-12), o una versión sustituida de cualquiera de estos grupos; o una sal farmacéuticamente aceptable del mismo. El compuesto de acuerdo con la invención se define como:

en donde: cada uno de R1 y R2 se selecciona independientemente entre hidrógeno, hidroxi, alquilo(C1-12), alquilo sustituido(C1-12), alquenilo(C2-12), alquenilo sustituido(C2-12), alquinilo(C2-12), alquinilo sustituido(C2-12), arilo(C6-12), arilo sustituido(C6-12), aralquilo(C7-12), aralquilo sustituido(C7-12), heteroarilo(C1-12), heteroarilo sustituido(C1-12), heterocicloalquilo(C2-12), heterocicloalquilo sustituido(C2-12), acilo(C1-12), acilo sustituido(C1-12), aciloxi(C1-12), aciloxi sustituido(C1-12), alquilamino(C1-12), alquilamino sustituido(C1-12); un grupo protector de amina monovalente, -C(O)O(CH2)nS-A1, -C(O)O(CH2)nS(O)-A1 o -C(O)O(CH2)nS(O)2-A1, en donde: A1 es arilo(C6-12) o arilo sustituido(C6-12); y n es 1, 2, 3, 4 o 5; R3 es hidrógeno, hidroxi, halo o alcoxi(C1-12) o alcoxi sustituido(C1-12); o es 1,2 o 3; R4 es hidrógeno, un grupo protector de amina monovalente, alquilo(C1 -12) o alquilo sustituido(C1-12); cada uno de R5, R6 y R7 es independientemente hidrógeno, hidroxi, amino, mercapto, -OX1, -NX2X3 o -SX4; o alquilo(C1-12) o alquilo sustituido(C1-12); en donde: X1 es un grupo protector de hidroxi; X2 y X3 se seleccionan independientemente entre hidrógeno, un grupo protector de amina monovalente, o cuando X2 y X3 se toman juntos forman un grupo protector de amina divalente; y X4 es un grupo protector de tiol; y R8 es hidroxi, amino o mercapto; o una sal farmacéuticamente aceptable del mismo. En algunas realizaciones, la fórmula se define adicionalmente como:

en donde: cada uno de R1 y R2 se selecciona independientemente entre hidrógeno, hidroxi, alquilo(C1-12), alquilo sustituido(C1-12), arilo(C6-12), arilo sustituido(C6-12), aralquilo(C7-12), aralquilo sustituido(C7-12), acilo(C1-12), acilo sustituido(C1-12), un grupo protector de amina monovalente, -C(O)O(CH2)nS-C6H5, -C(O)O(CH2)nS(O)-C6H5 o -C(O)O(CH2)nS(O)2-C6H5, un grupo protector de amina monovalente, o R1 y R2 se toman juntos y forman un grupo protector de amina divalente, o alquilo(C1-12), arilo(C6-12), aralquilo(C7-12), acilo(C1-12), o una versión sustituida de cualquiera de estos grupos; R3 es hidrógeno, hidroxi, halo o alcoxi(C1-12) o alcoxi sustituido(C1-12); y o es 1, 2 o 3; o una sal farmacéuticamente aceptable del mismo. Como también se describe en el presente documento, la fórmula se define adicionalmente como:

en donde: R4 es hidrógeno, alquilO(ci-i2), un grupo protector de amina monovalente o alquilo sustituidO(ci-i2)i cada uno de R5, R6 y R7 es independientemente hidrógeno, hidroxi, amino, mercapto, -OX1, -NX2X3 o -SX4; o alquilo(C1-12), alcoxi(C1-12), aciloxi(C1-12), alquilamino(C1-12), dialquilamino(C2-12), alquiltio(C1-12), amido(C1-12) o una versión sustituida de cualquiera de estos grupos; en donde: X1 es un grupo protector de hidroxi; X2 y X3 se seleccionan independientemente entre hidrógeno, un grupo protector de amina monovalente, o cuando X2 y X3 se toman juntos forman un grupo protector de amina divalente; X4 es un grupo protector de tiol; R8 es hidroxi, amino o mercapto; o alcoxi(C1-12), aciloxi(C1-12), alquilamino(C1-12), dialquilamino(C2-12), alquiltio(C1-12), amido(C1-12), o una versión sustituida de cualquiera de estos grupos; cada uno de Rg, R10, R11 y R12 se selecciona independientemente entre hidrógeno, hidroxi, amino, -NX2X3 o heteroarilo(C1-12), heterocicloalquilo(C2-12), acilo(C1-12), alcoxi(C1-12), aciloxi(C1-12), alquilamino(C1-12), dialquilamino(C2-12), amido(C2-12) o una versión sustituida de cualquiera de estos grupos; o Y2-R13; en donde: X2 y X3 se seleccionan independientemente entre hidrógeno, un grupo protector de amina monovalente, o cuando X2 y X3 se toman juntos forman un grupo protector de amina divalente; Y2 es alcanodiílo(C1-12) o alcanodiílo sustituido(C1-12); o R13 es hidroxi, amino o -NX2X3 o alquilamino(C1-12), dialquilamino(C2-12), amido(C1-12), o una versión sustituida de cualquiera de estos grupos; o cada uno de p y q es independientemente 1 o 2 ; una sal farmacéuticamente aceptable del mismo. Como también se describe en el presente documento, la fórmula se define adicionalmente como:

en donde: cada uno de Rg, R10, R11 y R12 se selecciona independientemente entre hidrógeno, hidroxi, amino, -NX2X3 0 alcoxi(C1-12), alquilamino(C1-12), dialquilamino(C2-12), amido(C1-12) o una versión sustituida de cualquiera de estos grupos; o Y2-R13; en donde: X2 y X3 se seleccionan independientemente entre hidrógeno, un grupo protector de amina monovalente, o cuando X2 y X3 se toman juntos forman un grupo protector de amina divalente; Y2 es alcanodiílo(C1-12) o alcanodiílo sustituido(C1-12); o R13 es hidroxi, amino o -NX2X3 o alquilamino(C1-12), dialquilamino(C2-12), amido(C1-12), o una versión sustituida de cualquiera de estos grupos; y cada uno de p y q es independientemente 1 o 2; o una sal farmacéuticamente aceptable del mismo. Como también se describe en el presente documento, el compuesto se define adicionalmente como:

en donde: Z2 es hidrógeno, un grupo protector de amina, acilo(C1-12), acilo sustituido(C1-12), -C(O)O(CH2)nS-A3, -C(O)O(CH2)nS(O)-A3 o -C(O)O(CH2)nS(O)2-A3, en donde: A3 es arilo(C6-12) o arilo sustituido(C6-12) R3 es hidrógeno, hidroxi, halo, amino, ciano, nitro, fosfato o mercapto, o alquilo(C1-12), alquenilo(C2-12), alquinilo(C2-12), arilo(C6-12), aralquilo(C7-12), heteroarilo(C1-12), heterocicloalquilo(C2-12), acilo(C1-12), alcoxi(C1-12), aciloxi(C1-12), alquilamino(C1-12), dialquilamino(C2-12), amido(C1-12) o una versión sustituida de cualquiera de estos grupos; o es 1 o 2; R4 es hidrógeno, un grupo protector de amina monovalente, alquilo(C1-12) o alquilo sustituido(C1-12) cada uno de R5, R6 y R7 es independientemente hidrógeno, hidroxi, amino, mercapto, -OX1, -NX2X3 o -SX4; o alquilo(C1-12) alquilo sustituido(C1-12) en donde: X1 es un grupo protector de hidroxi; X2 y X3 se seleccionan independientemente entre hidrógeno, un grupo protector de amina monovalente, o cuando X2 y X3 se toman juntos forman un grupo protector de amina divalente; X4 es un grupo protector de tiol; R8 es hidroxi, amino o mercapto; o una sal farmacéuticamente aceptable del mismo.
Como también se describe en el presente documento, el compuesto se define adicionalmente como:

en donde: Z2 es hidrógeno, un grupo protector de amina, acilo(C6-i2), acilo sustituido(C6-i2), -C(O)O(CH2)nS-A3, -C(O)O(CH2)nS(O)-A3 o -C(O)O(CH2)nS(O)2-A3, en donde: A3 es arilo(C6-12) o arilo sustituido(C6-12) R3 es hidrógeno, hidroxi, halo, amino, ciano, nitro, fosfato o mercapto, o alquilo(C1-12), alquenilo(C2-12), alquinilo(C2-12), arilo(C6-12), aralquilo(C7-12), heteroarilo(C1-12), heterocicloalquilo(C2-12), acilo(C1-12), alcoxi(C1-12), aciloxi(C1-12), alquilamino(C1-12), dialquilamino(C2-12), amido(C1-12) o una versión sustituida de cualquiera de estos grupos; o es 1 o 2; o una sal farmacéuticamente aceptable del mismo. En algunas realizaciones, la fórmula se define adicionalmente como:

en donde: X1 es un grupo protector de hidroxi; cada uno de R1 y R2 se selecciona independientemente entre hidrógeno, alquilo(C1-12) o alquilo sustituido(C1-12) y R3 es hidrógeno, alcoxi(C1-12) o alcoxi sustituido(C1-12) y o es 2; o una sal farmacéuticamente aceptable del mismo. Como también se describe en el presente documento, la fórmula es la fórmula I. Como se describe en el presente documento, la fórmula es la fórmula II. Como se describe en el presente documento, Y1 se toma junto con Z1 y es -alcanodiil (C<6)-NZ2-alcanodiílo (o<a). Como se describe en el presente documento, Y1 se toma junto con Z1 y es -CH2CH2-NZ2-CH2CH2-. Como se describe en el presente documento, Z2 es hidrógeno. Como se describe en el presente documento, Z2 es un grupo protector de amina. Como se describe en el presente documento, el grupo protector de amina es 2-(trimetilsilil)etoxicarbonilo o t-butoxicarbonilo. Como se describe en el presente documento, Z2 es -C(O)O(CH2)nS-C6H5. Como se describe en el presente documento, Z2 es -C(O)O(CH2)nS(O)2-C6H5. Como se describe en el presente documento, Z1 es hidrógeno. Como se describe en el presente documento, Y1 es -O(CH2)mY', -S(CH2)mY' o -(CH2)mNR1R2. Como se describe en el presente documento, Y1 es -(CH2)mNR1R2. Como se describe en el presente documento, m es 1, 2 o 3. Como se describe en el presente documento, m es 1 o 2. Como se describe en el presente documento, R1 es -C(O)O(CH2)nS-A1 o -C(O)O(CH2)nS(O)2-A1, en donde: A1 es arilo(C<12), arilo sustituido(C<12) o

en donde: A2 es aciloxi(C<12), aciloxi sustituido(C<12), alcoxi(C<12), alcoxi sustituido(C<12), alquilamino(C<12), alquilamino sustituido(C<12), dialquilamino(C<12) o dialquilamino sustituido(C<12), en donde A2 no es -CO2H, -CO2CH3, -OCH3, -OCH2CH3 , -C(O)CH3, -NHCH3, -NHCH2CH3, -N(CH3)2, -C(O)NH2 y -OC(O)CH3; y n es 1, 2, 3, 4 o 5. Como se describe en el presente documento, R1 es hidrógeno. Como se describe en el presente documento, R1 es alquilo(C<12) o alquilo sustituido(C<12). Como se describe en el presente documento, R1 es alquilo(C<12). Como se describe en el presente documento, R1 es metilo. Como se describe en el presente documento, R1 es un grupo protector de amina monovalente. Como se describe en el presente documento, R1 es 2-(trimetilsilil)etoxicarbonilo o t-butoxicarbonilo. Como se describe en el presente documento, R1 es -C(O)O(CH2)2SC6H5. Como se describe en el presente documento, R1 es -C(O)O(CH2)2S(O)2C6H5. Como se describe en el presente documento, R1 se toma junto con R2 y es un grupo protector divalente. Como se describe en el presente documento, R1 y R2 es ftalimida o ftalimida sustituida. Como se describe en el presente documento, R2 es hidrógeno. Como se describe en el presente documento, R2 es alquilo(C<12) o alquilo sustituido(C<12). Como se describe en el presente documento, R2 es alquilo(C<12). Como se describe en el presente documento, R2 es metilo. Como se describe en el presente documento, R2 es un grupo protector de amina monovalente. Como se describe en el presente documento, R2 es 2-(trimetilsilil)etoxicarbonilo o t-butoxicarbonilo. Como se describe en el presente documento, R2 es -C(O)O(CH1)nS-A1
o -C(O)O(CH2)nS(O)2-Ai, en donde: Ai es arilO(c<i2), arilo sustitu idos^) o

en donde: A2 es aciloxi(c<i2), aciloxi sustituidos^), alcoxi(c<i2), alcoxi sustitu idosi2), a lquilam inosi2), alquilamino sustituido(C<12), dialquilamino(C<i2) o dialquilamino sustituido(C<i2), en donde A2 no es -CO2H, -CO2CH3, -OCH3, -OCH2CH3 , -C(O)CH3, -NHCH3, -NHCH2CH3, -N(CH3)2, -C(O)NH2 y -OC(O)CH3; y n es i , 2, 3, 4 o 5. Como se describe en el presente documento, R2 es -C(O)O(CH2)2SC6H5. Como se describe en el presente documento, R2 es -C(O)O(CH2)2S(O)2CaH5. Como se describe en el presente documento, R2 se toma junto con Ri y es un grupo protector divalente. Como se describe en el presente documento, Ri y R2 son ftalimida o ftalimida sustituida. Como se describe en el presente documento, R3 es hidrógeno. Como se describe en el presente documento, R3 es alcoxi(o<i2). Como se describe en el presente documento, R3 es metoxi. Como se describe en el presente documento, R4 es hidrógeno. Como se describe en el presente documento, R4 es un grupo protector de amina monovalente. Como se describe en el presente documento, R4 es -C(O)CH2CH=CH2. Como se describe en el presente documento, R5 es alquilo(C<i2). Como se describe en el presente documento, R5 es metilo. Como se describe en el presente documento, R6 es hidroxi. Como se describe en el presente documento, R7 es hidroxi. Como se describe en el presente documento, R8 es hidroxi. Como se describe en el presente documento, Rg es amino. Como se describe en el presente documento, Rg es a lq u ilo s i2). Como se describe en el presente documento, Rg es metilo. Como se describe en el presente documento, Rg es Y2-R i2. Como se describe en el presente documento, Y2 es alcanodiílo(C<i2). Como se describe en el presente documento, Y2 es -CH2-. Como se describe en el presente documento, R i2 es amino. Como se describe en el presente documento, R i2 es -NX2X3. Como se describe en el presente documento, X2 y X3 se toman juntos y forman una ftalimida. Como se describe en el presente documento, X2 y X3 se toman juntos y forman una ftalimida sustituida. Como se describe en el presente documento, X2 es hidrógeno. Como se describe en el presente documento, X3 es t-butoxicarbonilo. Como se describe en el presente documento, Rio es amino. Como se describe en el presente documento, Rio es alquilo(C<i2). Como se describe en el presente documento, Ri0 es metilo. Como se describe en el presente documento, Ri0 es Y2-Ri2. Como se describe en el presente documento, Y2 es a lcanodiílosi2). Como se describe en el presente documento, Y2 es -CH2-. Como se describe en el presente documento, R i2 es amino. Como se describe en el presente documento, R i2 es -NX2X3. Como se describe en el presente documento, X2 y X3 se toman juntos y forman una ftalimida. Como se describe en el presente documento, X2 y X3 se toman juntos y forman una ftalimida sustituida. Como se describe en el presente documento, X2 es hidrógeno. Como se describe en el presente documento, X3 es t-butoxicarbonilo. Como se describe en el presente documento, R ii es hidrógeno. Como se describe en el presente documento, el átomo de carbono i7 está en la configuración R. Como se describe en el presente documento, el átomo de carbono i7 está en la configuración S. En algunas realizaciones, el compuesto se define adicionalmente como:

o una sal farmacéuticamente aceptable del mismo. En algunas realizaciones, el compuesto se define adicionalmente como:

o una sal farmacéuticamente aceptable del mismo. También se describen en el presente documento:

o una sal farmacéuticamente aceptable de los mismos. En algunas realizaciones, el compuesto se define adicionalmente como:


o una sal farmacéuticamente aceptable del mismo. También se describen en el presente documento:

o una sal farmacéuticamente aceptable del mismo.
En otro aspecto, la presente divulgación proporciona una composición farmacéutica que comprende un compuesto y un excipiente. En algunas realizaciones, la composición se formula para administración oral, intraadiposa, intraarterial, intraarticular, intracraneal, intradérmica, intralesional, intramuscular, intranasal, intraocular,
intrapericárdica, intraperitoneal, intrapleural, intraprostática, intrarrectal, intratecal, intratraqueal, intratumoral, intraumbilical, intravaginal, intravenosa, intravesicular, intravítrea, liposomal, local, mucosal, parenteral, rectal, subconjuntival, subcutánea, sublingual, tópica, transbucal, transdérmica, vaginal, mediante un catéter, mediante un lavado, mediante infusión continua, mediante infusión, mediante inhalación, mediante inyección, mediante liberación local o mediante perfusión localizada.
Se proporciona un método de preparación de un compuesto de la invención de acuerdo con la reivindicación 8 , con realizaciones adicionales descritas en las reivindicaciones dependientes de la reivindicación 8.
La presente divulgación proporciona un método para preparar un compuesto de la fórmula:

en donde: Y1 es -O(CH2)mY', -NH(CH2)mY', -S(CH2)mY' o -(CH2)mNR-iR2, o se toma junto con Z1 como se describe a continuación; en donde: Y' es hidroxi, halo, mercapto, alquilo(C1-12), alquilo sustituido(C1-12), alquenilo(C2-12), alquenilo sustituido(C2-12), alquinilo(C2-12), alquinilo sustituido(C2-12), acilo(C1-12), acilo sustituido(C1-12), aciloxi(C1-12), aciloxi sustituido(C1-12), alquilamino(C1-12), o alquilamino sustituido(C1-12); m es 1, 2, 3, 4, 5 o 6 ; y cada uno de R1 y R2 se selecciona independientemente entre hidrógeno, hidroxi, alquilo(01-12), alquilo sustituido(C1-12), alquenilo(C2-12), alquenilo sustituido(C2-12), alquinilo(C2-12), alquinilo sustituido(C2-12), arilo(C6-12), arilo sustituido(C6-12), aralquilo(C7-12), aralquilo sustituido(C7-12), heteroarilo(C1-12), heteroarilo sustituido(C1-12), heterocicloalquilo(C2-12), heterocicloalquilo sustituido(C2-12), acilo(C1-12), acilo sustituido(C1-12), aciloxi(C1-12), aciloxi sustituido(C1-12), alquilamino(C1-12), alquilamino sustituido(C1-12); un grupo protector de amina monovalente, -C(O)O(CH2)nS-A1, -C(O)O(CH2)nS(O)-A1 o -C(O)O(CH2)nS(O)2-A1, en donde: A1 es arilo(C6-12), arilo sustituido(C6-12), o

en donde: A2 es alquenilo(C2-12), alquenilo sustituido(C2-12), alquinilo(C2-12), alquinilo sustituido(C2-12), acilo(C1-12), acilo sustituido(C1-12), aciloxi(C1-12), aciloxi sustituido(C1-12), alcoxi(C1-12), alcoxi sustituido(C1-12), alquilamino(C1-12), alquilamino sustituido(C1-12), dialquilamino(C2-12) o dialquilamino sustituido(C2-12), en donde A2 no es -CO2H, -CO2CH3, -OCH3, -OCH2CH3, -C(O)CH3, -NHCH3, -NHCH2CH3, -N(CH3)2, -C(O)NH2 y -OC(O)CH3; n es 1, 2, 3, 4 o 5; o R1 y R2 se toman juntos y son un grupo protector de amina divalente, alcanodiílo(C1-12), alquilaminodiílo(C1-8); alcoxidiílo(C1-8); o una versión sustituida de cualquiera de estos grupos; o Y1 se toma con Z1 y es alquilaminodiílo(C1-8) alquilaminodiílo sustituido(C1-8); -alcanodiil(C1-6)-NZ2-alcanodiílo(C1-6) o -alcanodiil sustituido(C1-6)-NZ2-alcanodiílo sustituido(C1-6), en donde: Z2 es hidrógeno, un grupo protector de amina, acilo(C6-12), acilo sustituido(C6-12), -C(O)O(CH2)nS-A3 o -C(O)O(CH2)nS(O)2-A3, en donde: A3 es arilo(C6-12), arilo sustituido(C6-12), o

en donde: A4 es alquenilo(C2-12), alquenilo sustituido(C2-12), alquinilo(C2-12), alquinilo sustituido(C2-12), acilo(C1-12), acilo sustituido(C1-12), aciloxi(C1-12), aciloxi sustituido(C1-12), alcoxi(C1-12), alcoxi sustituido(C1-12), alquilamino(C1-12), alquilamino sustituido(C1-12), dialquilamino(C2-12) o dialquilamino sustituido(C2-12), en donde A4 no es -CO2H, -CO2CH3, -OCH3, -OCH2CH3, -C(O)CH3, -NHCH3, -NHCH2CH3, -N(CH3)2, -C(O)NH2 y -OC(O)CH3; Z1 está ausente, es hidrógeno o se toma junto con Y1 como se ha definido anteriormente; R3 es hidrógeno, hidroxi, halo, amino, ciano, nitro, fosfato o mercapto, o alquilo(C1-12), alquenilo(C2-12), alquinilo(C2-12), arilo(C6-12), aralquilo(C7-12), heteroarilo(C1-12), heterocicloalquilo(C2-12), acilo(C1-12), alcoxi(C1-12), aciloxi(C1-12), alquilamino(C1-12), dialquilamino(C2-12), amido(C1-12) o una versión sustituida de cualquiera de estos grupos; o es 1,2 o 3; R4 es hidrógeno, alquilo(C1-12), un grupo protector de amina monovalente o alquilo sustituido(C1-12); cada uno de R5, R6 y R7 es independientemente hidrógeno, hidroxi, amino, mercapto, -OX1, -NX2X3 o -SX4; o alquilo(C1-12), alcoxi(C1-12), aciloxi(C1-12), alquilamino(C1-12), dialquilamino(C2-12), alquiltio(C1-12), amido(C1-12) o una versión sustituida de cualquiera de estos grupos; en donde: X1 es un grupo protector de hidroxi; X2 y X3 se seleccionan independientemente entre hidrógeno, un grupo protector de amina monovalente, o cuando X2 y X3 se toman juntos forman un grupo protector de amina divalente; X4 es un grupo protector de tiol; R8 es
hidroxi, amino o mercapto; o alcoxi(ci-i2), aciloxi(ci-i2), alquilamino(Ci-i2), dialquilamino(C2-i2), alquiltiO(ci-i2), amidO(ci-i2), o una versión sustituida de cualquiera de estos grupos; y cada uno de R9, Ri0 y Ri i se selecciona independientemente entre hidrógeno, hidroxi, amino, mercapto, -OXi, -NX2X3 o -SX4, o alquilo(Ci-i2), alquenilo(C2-i2), alquinilo(C2-i2), arilo(C6-i2), aralquilo(C7-i2), heteroarilo(Ci-i2), heterocicloalquilo(Ci-i2), acilo(Ci-i2), alcoxi(Ci-i2), aciloxi(Ci-i2), alquilamino(Ci-i2), dialquilamino(C2-i2), alquiltio(Ci-i2), amido(Ci-i2), o una versión sustituida de cualquiera de estos grupos; o Y2-R13; en donde: Xi es alquilo(Ci-i2), alquilo sustituido(Ci-i2), alquenilo(C2-i2), alquenilo sustituido(C2-i2), alquinilo(C2-i2), alquinilo sustituido(C2-i2), arilo(C6-i2), arilo sustituido(C6-i2), aralquilo (C7-12), aralquilo sustituido(C7-i2), heteroarilo(Ci-i2), heteroarilo sustituido(Ci-i2), heterocicloalquilo(Ci-i2), heterocicloalquilo sustituido(Ci-i2), acilo(Ci-i2), acilo sustituido(Ci-i2), o un grupo protector de hidroxi; X2 y X3 se seleccionan independientemente entre hidrógeno, alquilo(Ci-i2), alquilo sustituido(Ci-i2), alquenilo(C2-i2), alquenilo sustituido(C2-i2), alquinilo(C2-i2), alquinilo sustituido(C2-i2), arilo(C6-i2), arilo sustituido(C6-i2), aralquilo(C7-i2), aralquilo sustituido(C7-i2), heteroarilo(Ci-i2), heteroarilo sustituido(Ci-i2), heterocicloalquilo(C2-i2), heterocicloalquilo sustituido(Ci-i2), acilo(Ci-i2), acilo sustituido(Ci-i2), un grupo protector de amina monovalente, o cuando X2 y X3 se toman juntos forman un grupo protector de amina divalente; X4 es alquilo(Ci-i2), alquilo sustituido(Ci-i2), alquenilo(C2-i2), alquenilo sustituido(C2-i2), alquinilo(C2-i2), alquinilo sustituido(C2-i2), arilo(C6-i2), arilo sustituido(C6-i2), aralquilo(C7-i2), aralquilo sustituido(C7-i2), heteroarilo(Ci-i2), heteroarilo sustituido(Ci-i2), heterocicloalquilo(Ci-i2), heterocicloalquilo sustituido(C2-i2), acilo(Ci-i2), acilo sustituido(Ci-i2), o un grupo protector de tiol; Y2 es alcanodiílo(Ci-i2) o alcanodiílo sustituido(Ci-i2) y R13 es hidroxi, amino, mercapto, -OXi, -NX2X3 o -SX4, o heteroarilo(Ci-i2), heterocicloalquilo(C2-i2), acilo(Ci-i2), alcoxi(Ci-i2), aciloxi(Ci-i2), alquilamino(Ci-i2), dialquilamino(C2-i2), alquiltio(Ci-i2), amido(Ci-i2) o una versión sustituida de cualquiera de estos grupos; y cada uno de p y q es independientemente i o 2 ; realizando una reacción que comprende mezclar un compuesto de la fórmula:

en donde: R4 es hidrógeno, alquilo(Ci-i2), un grupo protector de amina monovalente o alquilo sustituido(Ci-i2) cada uno de R5 , R6 y R7 es independientemente hidrógeno, hidroxi, amino, mercapto, -OXi, -NX2X3 o -SX4; o alquilo(Ci-i2), alcoxi(Ci-i2), aciloxi(Ci-i2), alquilamino(Ci-i2), dialquilamino(C2-i2), alquiltio(Ci-i2), amido(Ci-i2) o una versión sustituida de cualquiera de estos grupos; en donde: Xi es un grupo protector de hidroxi; X2 y X3 se seleccionan independientemente entre hidrógeno, un grupo protector de amina monovalente, o cuando X2 y X3 se toman juntos forman un grupo protector de amina divalente; X4 es un grupo protector de tiol; y R i4 es -O-, -S- o -NRi5-; en donde: Ri5 es hidrógeno, alquilo(Ci-6) o alquilo sustituido(Ci-6); Ri5 es ciano, alcoxi(Ci-i2), alcoxi sustituido(Ci-i2), aciloxi(Ci-i2), aciloxi sustituido(Ci-i2), alquiltio(Ci-i2), alquiltio sustituido(Ci-i2), alquilsulfonilo(Ci-i2), alquilsulfonilo sustituido(Ci-i2), arilsulfonilo(Ci-i2), arilsulfonilo sustituido(Ci-i2) alquilsulfoniloxi(Ci-i2), alquilsulfoniloxi sustituido(Ci-i2), arilsulfoniloxi(Ci-i2) o arilsulfoniloxi sustituido(Ci-i2); con un compuesto de la fórmula:

en donde: Yi es --O(CH2)mY', -NH(CH2)mY', -S(CH2)mY' o -(CH2)mNRiR2, o se toma con Zi como se describe a continuación; en donde: Y' es hidroxi, halo, mercapto, alquilo(Ci-i2), alquilo sustituido(Ci-i2), alquenilo(C2-i2), alquenilo sustituido(C2-i2), alquinilo(Ci-i2), alquinilo sustituido(C2-i2), acilo(Ci-i2), acilo sustituido(Ci-i2), aciloxi(Ci-i2), aciloxi sustituido(Ci-i2), alquilamino(Ci-i2) o alquilamino sustituido(Ci-i2); m es i, 2, 3, 4, 5 o 6 ; y cada uno de Ri y R2 se selecciona independientemente entre hidrógeno, hidroxi, alquilo(Ci-i2), alquilo sustituido(Ci-i2), alquenilo(C2-i2), alquenilo sustituido(C2-i2), alquinilo(C2-i2), alquinilo sustituido(C2-i2), arilo(C6-i2), arilo sustituido(C6-i2), aralquilo(C7-i2), aralquilo sustituido(C7-i2), heteroarilo(Ci-i2), heteroarilo sustituido(Ci-i2), heterocicloalquilo(C2-i2), heterocicloalquilo sustituido(Ci-i2), acilo(Ci-i2), acilo sustituido(Ci-i2), aciloxi(Ci-i2), aciloxi sustituido(Ci-i2), alquilamino(Ci-i2), alquilamino sustituido(Ci-i2); un grupo protector de amina monovalente, -C(O)O(CH2)nS-Ai, -C(O)O(CH2)nS(O)-Ai o -CCO)O(CH2)nS(O)2-Ai, en donde: Ai es arilo(C6-i2), arilo sustituido(C6-i2), o

i3
en donde: A2 es alquenilO(C2-i2), alquenilo sustituidO(C2-i2), alquinilO(C2-i2), alquinilo sustituidO(C2-i2), acilO(ci-i2), acilo sustituidO(ci-i2), aciloxi(ci-i2), aciloxi sustituido(ci-i2), alcoxi(ci-i2), alcoxi sustituido(ci-i2), alquilamino(ci-i2), alquilamino sustituido(ci-i2), dialquilamino(C2-i2) o dialquilamino sustituido(C2-i2), en donde A2 no es -CO2H, -CO2CH3, -OCH3, -OCH2CH3, -C(O)CH3, -NHCH3, -NHCH2CH3, -N(CH3)2, -C(O)NH2 y -OC(O)CH3; n es 1, 2, 3, 4 o 5; o Ri y R2 se toman juntos y son un grupo protector de amina divalente, alcanodiílo(Ci-i2), alquilaminodiílo (Ci-8); alcoxidiílo(Ci-8); o una versión sustituida de cualquiera de estos grupos; o Yi se toma con Zi y es alquilaminodiílo(Ci-8) alquilaminodiílo sustituido(Ci-8); -alcanodiil(Ci-6)-NZ2-alcanodiílo(Ci-6) o -alcanodiil sustituido(Ci-6)-NZ2-alcanodiílo sustituido(oi-a), en donde: Z2 es hidrógeno, un grupo protector de amina, acilo(Ci-i2), acilo sustituido(Ci-i2), -C(O)O(CH2)nS-A3 o -C(O)O(CH2)nS(O)2-A3, en donde: A3 es arilo(Ca-i2), arilo sustituido(oa-i2), o

en donde: A4 es alquenilo(C2-i2), alquenilo sustituido(C2-i2), alquinilo(C2-i2), alquinilo sustituido(C2-i2), acilo(Ci-i2), acilo sustituido(Ci-i2), aciloxi(Ci-i2), aciloxi sustituido(Ci-i2), alcoxi(Ci-i2), alcoxi sustituido(Ci-i2), alquilamino(Ci-i2), alquilamino sustituido(Ci-i2), dialquilamino(C2-i2) o dialquilamino sustituido(C2-i2), en donde A4 no es -CO2H, -CO2CH3, -OCH3, -OCH2CH3, -C(O)CH3, -NHCH3, -NHCH2CH3, -N(CH3)2, -C(O)NH2 y -OC(O)CH3; Zi está ausente, es hidrógeno o se toma junto con Yi como se ha definido anteriormente; R3 es hidrógeno, hidroxi, halo, amino, ciano, nitro, fosfato o mercapto, o alquilo(Ci-i2), alquenilo(C2-i2), alquinilo(C2-i2), arilo(Ca-i2), aralquilo(C7-i2), heteroarilo(Ci-i2), heterocicloalquilo(Ci-i2), acilo(Ci-i2), alcoxi(Ci-i2), aciloxi(Ci-i2), alquilamino(Ci-i2), dialquilamino(C2-i2), amido(Ci-i2) o una versión sustituida de cualquiera de estos grupos; y o es i o 2; o cada uno de Rg, R i0, R ii y R i2 se selecciona independientemente entre hidrógeno, hidroxi, amino, mercapto, -OXi, -NX2X3 o -SX4, o alquilo(Ci-i2), alquenilo(C2-i2), alquinilo(C2-i2), arilo(Ca-i2), aralquilo(C7-i2), heteroarilo(Ci-i2), heterocicloalquilo(C2-i2), acilo(Ci-i2), alcoxi(Ci-i2), aciloxi(Ci-i2), alquilamino(Ci-i2), dialquilamino(C2-i2), alquiltio(Ci-i2), amido(Ci-i2), o una versión sustituida de cualquiera de estos grupos; o Y2-R i3; en donde: Xi es un grupo protector de hidroxi; X2 y X3 se seleccionan independientemente entre hidrógeno, un grupo protector de amina monovalente, o cuando X2 y X3 se toman juntos forman un grupo protector de amina divalente; X4 es un grupo protector de tiol; Y2 es alcanodiílo(Ci-i2) o alcanodiílo sustituido(Ci-i2); o Ri3 es hidroxi, amino, mercapto, -OXi, -NX2X3 o -SX4 , o heteroarilo(Ca-i2), heterocicloalquilo(C2-i2), acilo(Ci-i2), alcoxi(Ci-i2), aciloxi(Ci-i2), alquilamino(Ci-i2), dialquilamino(C2-i2), alquiltio(Ci-i2), amido(Ci-i2) o una versión sustituida de cualquiera de estos grupos; y cada uno de p y q es independientemente i o 2 ; en presencia de una base nucleófila fuerte en un disolvente en condiciones suficientes para provocar una reacción. Como se describe en el presente documento, Yi no es -NH2, -NHMe o -NHCH2CH2NH2. En alguna realización, la base es bis(trimetilsilil)amida de litio, bis(trimetilsilil)amida sódica, bis(trimetilsilil)amida potásica, diisopropilamida de litio, diisopropilamida sódica, diisopropilamida potásica, tetrametilpiperiduro de litio, t-butóxido de litio, t-butóxido sódico o t-butóxido potásico, hidruro de litio, hidruro sódico o hidruro potásico. En algunas realizaciones, la base es bis(trimetilsilil)amida de litio. En algunas realizaciones, el disolvente es tetrahidrofurano. En algunas realizaciones, el método comprende adicionalmente mezclar el compuesto de fórmula IV o de fórmula V con la base antes de poner en contacto el compuesto de fórmula III con el compuesto de fórmula IV o de fórmula V. Como se describe en el presente documento, el compuesto de fórmula III se disuelve en THF y se añade mediante una cánula. En algunas realizaciones, el método comprende adicionalmente disolver el compuesto de fórmula I o de fórmula II en un disolvente orgánico y hacer reaccionar el compuesto de la fórmula:

en donde: Yi es -O(CH2)mY', -NH(CH2)mY', -S(CH2)mY' o -(CH2)mNRiR2, o se toma junto con Zi como se describe a continuación; en donde: Y' es hidroxi, halo, mercapto, alquilo(Ci-i2), alquilo sustituido(Ci-i2), alquenilo(C2-i2), alquenilo sustituido(C2-i2), alquinilo(C2-i2), alquinilo sustituido(C2-i2), acilo(Ci-i2), acilo sustituido(Ci-i2), aciloxi(Ci-i2), aciloxi sustituido(Ci-i2), alquilamino(Ci-i2) o alquilamino sustituido(Ci-i2); m es i, 2, 3, 4, 5 o a; y cada uno de Ri y R2 se selecciona independientemente entre hidrógeno, hidroxi, alquilo(Ci-i2), alquilo sustituido(Ci-i2), alquenilo(C2-i2), alquenilo sustituido(C2-i2), alquinilo(C2-i2), alquinilo sustituido(C2-i2), arilo(Ca-i2), arilo sustituido(Ca-i2), aralquilo(C7-i2), aralquilo sustituido(C7-i2), heteroarilo(Ci-i2), heteroarilo sustituido(Ci-i2), heterocicloalquilo(C2-i2), heterocicloalquilo sustituido(C2-i2), acilo(Ci-i2), acilo sustituido(Ci-i2), aciloxi(Ci-i2), aciloxi sustituido(Ci-i2), alquilamino(Ci-i2), alquilamino sustituido(Ci-i2); un grupo protector de amina monovalente, -C(O)O(CH2)nS-Ai, -C(O)O(CH2)nS(O)-Ai o -C(O)O(CH2)nS(O)2-Ai, en donde: Ai es arilo(Ca-i2), arilo sustituido(Ca-i2), o
i4

en donde: A2 es alquenilO(C2-i2), alquenilo sustituidO(C2-i2), alquinilo (C2-12), alquinilo sustituidO(C2-i2), acilO(ci-i2), acilo sustituidO(ci-i2), aciloxi(ci-i2), aciloxi sustituido(ci-i2), alcoxi(ci-i2), alcoxi sustituido(ci-i2), alquilamino(ci-i2), alquilamino sustituido(ci-i2), dialquilamino(C2-i2) o dialquilamino sustituido(C2-i2), en donde A2 no es -CO2H, -CO2CH3, -OCH3, -OCH2CH3, -C(O)CH3, -NHCH3, -NHCH2CH3, -N(CH3)2, -C(O)NH2 y -OC(O)CH3; n es i, 2, 3, 4 o 5; o Ri y R2 se toman juntos y son un grupo protector de amina divalente, alcanodiílo(Ci-i2), alquilaminodiílo(Ci-8) alcoxidiílo(Ci-8) o una versión sustituida de cualquiera de estos grupos; o Yi se toma con Zi y es alquilaminodiílo(Ci-8) alquilaminodiílo sustituido(Ci-8); -alcanodiil(Ci-6)-NZ2-alcanodiílo(Ci-6) o -alcanodiil sustituido(Ci-6)-NZ2-alcanodiílo sustituido(oi-a), en donde: Z2 es hidrógeno, un grupo protector de amina, acilo(Ci-i2), acilo sustituido(Ci-i2), -C(O)O(CH2)nS-A3 o -C(O)O(CH2)nS(O)2-A3, en donde: A3 es arilo(Ca-i2), arilo sustituido(oa-i2), o

en donde: A4 es alquenilo(C2-i2), alquenilo sustituido(C2-i2), alquinilo(C2-i2), alquinilo sustituido(C2-i2), acilo(Ci-i2), acilo sustituido(Ci-i2), aciloxi(Ci-i2), aciloxi sustituido(Ci-i2), alcoxi(Ci-i2), alcoxi sustituido(Ci-i2), alquilamino(Ci-i2), alquilamino sustituido(Ci-i2), dialquilamino(C2-i2) o dialquilamino sustituido(C2-i2), en donde A2 no es -CO2H, -CO2CH3, -OCH3, -OCH2CH3, -C(O)CH3, -NHCH3, -NHCH2CH3, -N(CH3)2, -C(O)NH2 y -OC(O)CH3; Zi está ausente, es hidrógeno o se toma junto con Yi como se ha definido anteriormente; R3 es hidrógeno, hidroxi, halo, amino, ciano, nitro, fosfato o mercapto, o alquilo(Ci-i2), alquenilo(C2-i2), alquinilo(C2-i2), arilo(Ca-i2), aralquilo(C7-i2), heteroarilo(Ci-i2), heterocicloalquilo(Ci-i2), acilo(Ci-i2), alcoxi(Ci-i2), aciloxi(Ci-i2), alquilamino(Ci-i2), dialquilamino(C2-i2), amido(Ci-i2) o una versión sustituida de cualquiera de estos grupos; o es i o 2; R4 es hidrógeno, alquilo(Ci-i2), un grupo protector de amina monovalente o alquilo sustituido(Ci-i2); cada uno de R5, Ra y R7 es independientemente hidrógeno, hidroxi, amino, mercapto, -OXi, -NX2X3 o -SX4; o alquilo(Ci-i2), alcoxi(Ci-i2), aciloxi(Ci-i2), alquilamino(Ci-i2), dialquilamino(C2-i2), alquiltio(Ci-i2) amido(Ci-i2) o una versión sustituida de cualquiera de estos grupos; en donde: Xi es un grupo protector de hidroxi; X2 y X3 se seleccionan independientemente entre hidrógeno, un grupo protector de amina monovalente, o cuando X2 y X3 se toman juntos forman un grupo protector de amina divalente; X4 es un grupo protector de tiol; R8 es hidroxi, amino o mercapto; o alcoxi(Ci-i2), aciloxi(Ci-i2), alquilamino(Ci-i2), dialquilamino(C2-i2), alquiltio(Ci-i2), amido(Ci-i2), o una versión sustituida de cualquiera de estos grupos; y cada uno de Rg, Rio, Rii y R i2 se selecciona independientemente entre hidrógeno, hidroxi, amino, mercapto, -OXi, -NX2X3, o -SX4, o alquilo(Ci-i2), alquenilo(C2-i2), alquinilo(C2-i2), arilo(Ca-i2), aralquilo(C7-i2), heteroarilo(Ci-i2), heterocicloalquilo(C2-i2), acilo(Ci-i2), alcoxi(C2-i2), aciloxi(Ci-i2), alquilamino(Ci-i2), dialquilamino(C2-i2), alquiltio(Ci-i2), amido(Ci-i2), o una versión sustituida de cualquiera de estos grupos; o Y2-Ri3; en donde: Xi es alquilo(Ci-i2), alquilo sustituido(Ci-i2), alquenilo(C2-i2), alquenilo sustituido(C2-i2), alquinilo(C2-i2), alquinilo sustituido(C2-i2), arilo(Ca-i2), arilo sustituido(Ca-i2), aralquilo(C7-i2), aralquilo sustituido(C7-i2), heteroarilo(Ci-i2), heteroarilo sustituido(Ci-i2), heterocicloalquilo(Ci-i2), heterocicloalquilo sustituido(Ci-i2), acilo(Ci-i2), acilo sustituido(Ci-i2), o un grupo protector de hidroxi; X2 y X3 se seleccionan independientemente entre hidrógeno, alquilo(Ci-i2), alquilo sustituido(Ci-i2), alquenilo(C2-i2), alquenilo sustituido(C2-i2), alquinilo(C2-i2), alquinilo sustituido(C2-i2), arilo(Ca-i2), arilo sustituido(Ca-i2), aralquilo(C7-i2), aralquilo sustituido(C7-i2), heteroarilo(Ci-i2), heteroarilo sustituido(Ci-i2), heterocicloalquilo(C2-i2), heterocicloalquilo sustituido(C2-i2), acilo(Ci-i2), acilo sustituido(Ci-i2), un grupo protector de amina monovalente, o cuando X2 y X3 se toman juntos forman un grupo protector de amina divalente; X4 es alquilo(Ci-i2), alquilo sustituido(Ci-i2), alquenilo(C2-i2), alquenilo sustituido(C2-i2), alquinilo(C2-i2), alquinilo sustituido(C2-i2), arilo(Ca-i2), arilo sustituido(Ca-i2), aralquilo(C7-i2), aralquilo sustituido(C7-i2), heteroarilo(Ci-i2), heteroarilo sustituido(Ci-i2), heterocicloalquilo(Ci-i2), heterocicloalquilo sustituido(C2-i2), acilo(Ci-i2), acilo sustituido(Ci-i2), o un grupo protector de tiol; Y2 es alcanodiílo(Ci-i2) o alcanodiílo sustituido(Ci-i2); y Ri3 es hidroxi, amino, mercapto, -OXi, -NX2X3 o -SX4, o heteroarilo(Ci-i2), heterocicloalquilo(C2-i2), acilo(Ci-i2), alcoxi(Ci-i2), aciloxi(Ci-i2), alquilamino(Ci-i2), dialquilamino(C2-i2), alquiltio(Ci-i2), amido(Ci-i2) o una versión sustituida de cualquiera de estos grupos; y cada uno de p y q es independientemente i o 2 ; con una sal de paladio en presencia de una base en condiciones suficientes para provocar una reacción para formar un compuesto de la fórmula:
i5

en donde: Yi es -O(CH2)mY', -NH(CH2)mY', -S(CH2)mY' o -(CH2)mNRiR2, o se toma junto con Zi como se describe a continuación; en donde: Y' es hidroxi, halo, mercapto, alquilo(ci-i2), alquilo sustituido(ci-i2), alquenilo(C2-i2), alquenilo sustituido(C2-i2), alquinilo(C2-12), alquinilo sustituido(C2-12), acilo(C1-12), acilo sustituido(C1-12), aciloxi(C1-12), aciloxi sustituido(C1-12), alquilamino(C1-12) o alquilamino sustituido(C1-12); m es 1, 2, 3, 4, 5 o 6 ; y cada uno de R1 y R2 se selecciona independientemente entre hidrógeno, hidroxi, alquilo(C1-12), alquilo sustituido(C1-12), alquenilo(C2-12), alquenilo sustituido(C2-12), alquinilo(C2-12), alquinilo sustituido(C2-12), arilo(C6-12), arilo sustituido(C6-12), aralquilo(C7-12), aralquilo sustituido(C7-12), heteroarilo(C1-12), heteroarilo sustituido(C1-12), heterocicloalquilo(C1-12), heterocicloalquilo sustituido(C2-12), acilo(C1-12), acilo sustituido(C1-12), aciloxi(C1-12), aciloxi sustituido(C1-12), alquilamino(C1-12), alquilamino sustituido(C1-12); un grupo protector de amina monovalente, -C(O)O(CH2)nS-A1, -C(O)O(CH2)nS(O)-A1 o -C(O)O(CH2)nS(O)2-A1, en donde: A1 es arilo(C6-12), arilo sustituido(C6-12), o

en donde: A2 es alquenilo(C2-12), alquenilo sustituido(C2-12), alquinilo(C2-12), alquinilo sustituido(C2-12), acilo(C1-12), acilo sustituido(C1-12), aciloxi(C1-12), aciloxi sustituido(C1-12), alcoxi(C1-12), alcoxi sustituido(C1-12), alquilamino(C1-12), alquilamino sustituido(C1-12), dialquilamino(C2-12) o dialquilamino sustituido(C2-12), en donde A2 no es -CO2H, -CO2CH3, -OCH3, -OCH2CH3, -C(O)CH3, -NHCH3, -NHCH2CH3, -N(CH3)2, -C(O)NH2 y -OC(O)CH3; n es 1, 2, 3, 4 o 5; o R1 y R2 se toman juntos y son un grupo protector de amina divalente, alcanodiílo(C1-12), alquilaminodiílo (C1-8); alcoxidiílo(C1-8); o una versión sustituida de cualquiera de estos grupos; o Y1 se toma con Z1 y es alquilaminodiílo(C1-8) alquilaminodiílo sustituido(C1-8); -alcanodiil(C1-6)-NZ2-alcanodiílo(C1-6) o -alcanodiil sustituido(C1-6)-NZ2-alcanodiílo sustituido(C1-6), en donde: Z2 es hidrógeno, un grupo protector de amina, acilo(C1-12), acilo sustituido(C1-12), -C(O)O(CH2)nS-A3 o -C(O)O(CH2)nS(O)2-A3, en donde: A3 es arilo(C6-12), arilo sustituido(C6-12), o

en donde: A4 es alquenilo(C2-12), alquenilo sustituido(C2-12), alquinilo(C2-12), alquinilo sustituido(C2-12), acilo(C1-12), acilo sustituido(C1-12), aciloxi(C1-12), aciloxi sustituido(C1-12), alcoxi(C1-12), alcoxi sustituido(C1-12), alquilamino(C1-12), alquilamino sustituido(C1-12), dialquilamino(C2-12) o dialquilamino sustituido(C2-12), en donde A2 no es -CO2H, -CO2CH3, -OCH3, -OCH2CH3, -C(O)CH3, -NHCH3, -NHCH2CH3, -N(CH3)2, -C(O)NH2 y -OC(O)CH3; Z1 está ausente, es hidrógeno o se toma junto con Y1 como se ha definido anteriormente; R3 es hidrógeno, hidroxi, halo, amino, ciano, nitro, fosfato o mercapto, o alquilo(C1-12), alquenilo(C2-12), alquinilo(C2-12), arilo(C6-12), aralquilo(C7-12), heteroarilo(C1-12), heterocicloalquilo(C1-12), acilo(C1-12), alcoxi(C1-12), aciloxi(C1-12), alquilamino(C1-12), dialquilamino(C2-12), amido(C1-12) o una versión sustituida de cualquiera de estos grupos; o es 1 o 2 ; cada uno de R5, R6 y R7 es independientemente hidrógeno, hidroxi, amino, mercapto, -OX1, -NX2X3 o -SX4; o alquilo(C1-12), alcoxi(C1-12), aciloxi(C1-12), alquilamino(C1-12), dialquilamino(C2-12), alquiltio(C1-12), amido(C1-12) o una versión sustituida de cualquiera de estos grupos; en donde: X1 es un grupo protector de hidroxi; X2 y X3 se seleccionan independientemente entre hidrógeno, un grupo protector de amina monovalente, o cuando X2 y X3 se toman juntos forman un grupo protector de amina divalente; X4 es un grupo protector de tiol; R8 es hidroxi, amino o mercapto; o alcoxipd-12), aciloxi(C1-12), alquilamino(C1-12), dialquilamino(C2-12), alquiltio(C1-12), amido(C1-12), o una versión sustituida de cualquiera de estos grupos; y cada uno de Rg, R10, R11 y R12 se selecciona independientemente entre hidrógeno, hidroxi, amino, mercapto, -OX1, -NX2X3 o -SX4, o alquilo(C1-12), alquenilo(C2-12), alquinilo(C2-12), arilo(C6-12), aralquilo(C7-12), heteroarilo(C1-12), heterocicloalquilo(C1-12), acilo(C1-12), alcoxi(C1-12), aciloxi(C1-12), alquilamino(C1-12), dialquilamino(C2-12), alquiltio(C1-12), amido(C1-12), o una versión sustituida de cualquiera de estos grupos; o Y2-R13; en donde: X1 es alquilo(C1-12), alquilo sustituido(C1-12), alquenilo(C2-12), alquenilo sustituido(C2-12), alquinilo(C2-12), alquinilo sustituido(C2-12), arilo(C6-12), arilo sustituido(C6-12), aralquilo(C7-12), aralquilo sustituido(C7-12), heteroarilo(C1-12), heteroarilo sustituido(C1-12), heterocicloalquilo(C2-12), heterocicloalquilo sustituido(C1-12), acilo(C1-12), acilo sustituido(C1-12), o un grupo protector de hidroxi; X2 y X3 se seleccionan independientemente entre hidrógeno, alquilo(C1-12), alquilo sustituido(C1-12), alquenilo(C2-12), alquenilo sustituido(C2-12), alquinilo(C2-12), alquinilo sustituido(C2-12), arilo(C6-12), arilo sustituido(C6-12), aralquilo(C7-12), aralquilo sustituido(C7-12), heteroarilo(C1-12), heteroarilo
sustituidO(ci-i2), heterocicloalquilO(ci-i2), heterocicloalquilo sustituidO(C2-i2), acilO(ci-i2), acilo sustituidO(ci-i2), un grupo protector de amina monovalente, o cuando X2 y X3 se toman juntos forman un grupo protector de amina divalente; X4 es alquilo(Ci-i2), alquilo sustituido(Ci-i2), alquenilo(C2-i2), alquenilo sustituido(C2-i2), alquinilo(C2-i2), alquinilo sustituido(C2-12), arilo(C6-i2), arilo sustituido(C6-i2), aralquilo(C7-i2), aralquilo sustituido(C7-i2), heteroarilo(Ci-i2), heteroarilo sustituido^i-12), heterocicloalquilo(C2-i2), heterocicloalquilo sustituido(Ci-i2), acilo(Ci-i2), acilo sustituido(Ci-i2), o un grupo protector de tiol; Y2 es alcanodiílo(Ci-i2) o alcanodiílo sustituido(Ci-i2); y R13 es hidroxi, amino, mercapto, -OXi, -NX2X3 o -SX4 , o heteroarilo(Ci-i2), heterocicloalquilo(C2-i2), acilo(Ci-i2), alcoxi(Ci-i2), aciloxi(Ci-i2), alquilamino(Ci-i2), dialquilamino(C2-i2), alquiltio(Ci-i2), amido(Ci-i2) o una versión sustituida de cualquiera de estos grupos; y cada uno de p y q es independientemente 1 o 2. En algunas realizaciones, la sal de paladio comprende un complejo de paladio y fosfina o una sal de paladio en presencia de un ligando u otras sales o complejos de metales. En algunas realizaciones, la sal de paladio es Pd(PPh3)4. En algunas realizaciones, la base es trietilamina, diisopropiletilamina, etilendiamina, 1,8-diazabiciclo[5.4.0]undec-7-eno o morfolina. En algunas realizaciones, la base es morfolina. En algunas realizaciones, el disolvente es tetrahidrofurano. En algunas realizaciones, el método comprende adicionalmente retirar uno o más grupos protectores.
En otro aspecto más, la presente divulgación proporciona un conjugado de la fórmula:
(A5-L)r-A6 (XVII)
en donde: A5 es un compuesto de la reivindicación 1; L es un enlazador; r es 1, 2, 3, 4, 5, 6 , 7, 8 , 9, 10, 11 o 12; y A6 es un resto de direccionamiento celular. En algunas realizaciones, L comprende un polipéptido escindible por una enzima intracelular. En algunas realizaciones, la enzima es catepsina B. En algunas realizaciones, L comprende un grupo autoinmolante. En algunas realizaciones, L es:

En algunas realizaciones, A6 es un anticuerpo cuyo antígeno es un antígeno asociado a tumor. En algunas realizaciones, el antígeno es mesotelina, glipicano-3 o CD70. En algunas realizaciones, A5 es un compuesto de acuerdo con la fórmula (I). En algunas realizaciones, el conjugado comprende adicionalmente una estructura de la fórmula:

en donde: A6 es un anticuerpo y r es 1, 2, 3 o 4. En algunas realizaciones, el anticuerpo es un anticuerpo antimesotelina, anti-glipicano-3 o anti-CD70.
En otro aspecto más, la presente divulgación proporciona una composición farmacéutica que comprende un conjugado de la presente divulgación y un excipiente. En algunas realizaciones, el compuesto se formula para administración oral, intraadiposa, intraarterial, intraarticular, intracraneal, intradérmica, intralesional, intramuscular, intranasal, intraocular, intrapericárdica, intraperitoneal, intrapleural, intraprostática, intrarrectal, intratecal, intratraqueal, intratumoral, intraumbilical, intravaginal, intravenosa, intravesicular, intravítrea, liposomal, local, mucosal, parenteral, rectal, subconjuntival, subcutánea, sublingual, tópica, transbucal, transdérmica, vaginal, mediante un catéter, mediante un lavado, mediante infusión continua, mediante infusión, mediante inhalación, mediante inyección, mediante liberación local o mediante perfusión localizada.
En otro aspecto, la presente divulgación proporciona un compuesto o composición para uso en el tratamiento del
cáncer de acuerdo con la reivindicación 16. En algunas realizaciones, el cáncer es de la vejiga, sangre, hueso, cerebro, mama, sistema nervioso central, cuello del útero, colon, endometrio, esófago, vesícula biliar, genitales, tracto genitourinario, cabeza, riñón, laringe, hígado, pulmón, tejido muscular, cuello, mucosa oral o nasal, ovario, páncreas, próstata, piel, bazo, intestino delgado, intestino grueso, estómago, testículo o tiroides. En otras realizaciones, el cáncer es un carcinoma, sarcoma, leucemia, melanoma, mesotelioma, mieloma múltiple o seminoma. En algunas realizaciones, el cáncer es cáncer de pulmón, cáncer gástrico, cáncer de ovario, cáncer de hígado, cáncer renal o cáncer de mama. En algunas realizaciones, el cáncer es cáncer de pulmón H226, cáncer gástrico N87, cáncer de ovario OVCAR3, cáncer de hígado Hep3B, cáncer de hígado HepG2, cáncer renal 786-O o cáncer de mama multirresistente ADR. En algunas realizaciones, el cáncer muestra uno o más marcadores tumorales. En algunas realizaciones, el marcador tumoral es mesotelina, antígeno de membrana específico de la próstata, CD19, CD22, CD30, CD70, B7H4, proteína tirosina cinasa 7, glipicano-3, RG1, CTLA-4 o CD44. En algunas realizaciones, el marcador tumoral es mesotelina, glipicano-3 o CD70. En algunas realizaciones, el método comprende administrar un compuesto o una composición de un compuesto de la presente divulgación. En algunas realizaciones, el método comprende administrar un conjugado o una composición de la presente divulgación. En algunas realizaciones, el compuesto comprende un anticuerpo. En algunas realizaciones, el anticuerpo se une a un antígeno sobre la superficie de la célula cancerosa. En algunas realizaciones, la unión del anticuerpo con el antígeno sobre la superficie de la célula desencadena la internalización del compuesto. En algunas realizaciones, la internalización del compuesto desencadena la escisión de un compuesto terapéutico a partir del anticuerpo. En algunas realizaciones, el método comprende adicionalmente administrar un segundo agente terapéutico. En algunas realizaciones, el segundo agente terapéutico es un segundo agente quimioterapéutico, cirugía, una radioterapia, una terapia genética o una inmunoterapia. En algunas realizaciones, el método comprende administrar el compuesto una vez. En otras realizaciones, el método comprende administrar el compuesto dos o más veces. En algunas realizaciones, la enfermedad o trastorno es una infección bacteriana. En algunas realizaciones, la infección bacteriana es una infección de Staphylococcus aureus, Escherichia coli o Burkholderia cepacia. En algunas realizaciones, la infección bacteriana es una infección de Burkholderia cepacia.
También se describe en el presente documento un método para preparar un compuesto de la fórmula:

en donde: X1, X2, X3 son hidrógeno o un grupo protector de hidroxi; X4 es hidrógeno o un grupo protector de amino; cada uno de R1 y R2 está independientemente ausente, es hidrógeno, o R1 y R2 se toman juntos y son alquilaminodiílo(C1-12), alquilaminodiílo sustituido (C1-12); alquenodiílo(C2-12) o alquenodiílo sustituido(C2-12); R3 es hidrógeno, amino, carboxi, ciano, halo, hidroxi o alquilo(C1-12), acilo(C1-12), amido(C1-12), alcoxi(C1-12), alquilamino(C1-12), dialquilamino(C2-12), o una versión sustituida de cualquiera de estos grupos; o -Y1-A1; en donde: Y1 es alcanodiílo(C1-8) o alcanodiílo sustitu idos^; A1 es un enlazador en donde el enlazador tiene la fórmula:

en donde: R5 es hidrógeno, alquilo(C1-6) o alquilo sustituidos-^; Re es -C(O)-Y3-A3, en donde: Y3 es alcanodiílos-12) o alcanodiílo sustituidos-12) y A3 es un grupo reactivo de tiol; Y2 es -C(O)-, -C(O)-alcanodiílos-12); -C(O)-alcanodiílo sustituido(C1-12) o un grupo autoinmolante; A2 es un enlace covalente, un resto de aminoácido o un polipéptido; y o es 1, 2, 3, 4, 5, 6 , 7 u 8 ; n es 1, 2, 3 o 4; y R4 es a lqu ilos-12) o alquilo sustituidos-12) que comprende a) hacer reaccionar un compuesto de la fórmula:

en donde: Ri, R2, R3, R4, Xi, X2, X3 y X4 son como se han definido anteriormente; n es 0, 1, 2 o 3; R3' es -Y1-A1; en donde: Y1 es alcanodiílo(C1-8) o alcanodiílo sustituido(C1-8); y A1 es -NPhth; con una alquilamina(C1-12), dialquilaminas-18), o una versión sustituida de cualquiera de estos grupos con agua en un disolvente en condiciones suficientes para provocar una reacción para formar un compuesto de la fórmula:

en donde: R1, R2, R3, R4, n, X1, X2, X3 y X4 son como se han definido anteriormente; R3' es -Y1-NH2 ; en donde: Y1 es alcanodiílo(C1-8) o alcanodiílo sustituido(C1-8); b) hacer reaccionar el compuesto de fórmula XX con un enlazador de la fórmula:

en donde: R5 y R6 es hidrógeno, alquilona), alquilo sustituidos-a), o un grupo protector de amina; Y2 es -C(O)OH, HOC(O)-alcanodiílo(C1-12); HOC(O)-alcanodiílo sustituido(C1-12), un grupo autoinmolante o un grupo autoinmolante activado; A2 es un enlace covalente, un resto de aminoácido o un polipéptido; y o es 0, 1,2, 3, 4, 5, 6 , 7 u 8 ; y una base en un disolvente en condiciones suficientes para provocar una reacción para formar un compuesto de la fórmula:

en donde: R1, R2, R3, R4, X1, X2, X3 , X4, n, o, Y1, Y2, A1, A2 y R5 son como se han definido anteriormente; c) desproteger uno o más grupos funcionales en el compuesto de fórmula XXII que comprende hacer reaccionar el compuesto con un reactivo que retira el grupo protector en condiciones suficientes para provocar una reacción para formar un compuesto de la fórmula:

en donde: Ri, R2, R3, R4, X1, X2, X3 , X4, n, o, Y1, Y2, A1 y A2 son como se han definido anteriormente; d) hacer reaccionar el compuesto de fórmula XXIII con un compuesto de la fórmula:
O
X5 ° ^ Y3A3 (XXIV)
en donde: X5 es hidrógeno o un agente de activación; Y3 es alcanodiílo(C1-12) o alcanodiílo sustituido(C1-12) y A3 es un grupo reactivo de tiol; en presencia de una base en un disolvente en condiciones suficientes para provocar una reacción para formar un compuesto de fórmula XVIII. Como se describe en el presente documento, el compuesto de la fórmula se define adicionalmente como:

en donde: X2 y X3 son hidrógeno o un grupo protector de hidroxi; X4 es hidrógeno o un grupo protector de amino; cada uno de R1 y R2 está independientemente ausente, es hidrógeno o R1 y R2 se toman juntos y son alquilaminodiílo(C1-8), alquilaminodiílo sustituidos^), alquenodiílo(C2-8) o alquenodiílo sustituido(C2-8); R3 es hidrógeno, amino, carboxi, ciano, halo, hidroxi, o alcoxi(C1-12) o alcoxi sustituido(C1-12) o Y2 es -C(O)-, -C(O)-alcanodiílo(C1-12), -C(O)-alcanodiílo sustituido(C<12) o un grupo autoinmolante; A2 es un enlace covalente, un resto de aminoácido o un polipéptido; y o es 1, 2, 3, 4, 5, 6, 7 u 8; n es 1, 2 o 3; e Y3 es un enlace covalente, alcanodiílo(C1-12) o alcanodiílo sustituido(C1-12). Como se describe en el presente documento, el compuesto de fórmula XXV se define adicionalmente como:

en donde: X2 y X3 son hidrógeno o un grupo protector de hidroxi; X4 es hidrógeno o un grupo protector de amino;
cada uno de Ri y R2 está independientemente ausente, hidrógeno, o R1 y R2 se toman juntos y son alquilaminodiílO(Ci -8) alquilaminodiílo sustituido(C1-8); alquenodiílo(C2-8) o alquenodiílo sustituido(C2-8); R3 es hidrógeno, amino, carboxi, ciano, halo, hidroxi, o alcoxi(C1-12) o alcoxi sustituido(C1-12) y n es 1, 2 o 3. Como se describe en el presente documento, el compuesto de fórmula XIX se define adicionalmente como:

en donde: R1, R2, R3, n, X2, X3 y X4 son como se han definido anteriormente. Como se describe en el presente documento, el compuesto de fórmula XXI se define adicionalmente como:

en donde: R5 es un grupo protector de amina; A2 es un enlace covalente, un resto de aminoácido o un polipéptido; y o es 1, 2, 3, 4, 5, 6 , 7 u 8. Como se describe en el presente documento, el compuesto de fórmula xXlV se define adicionalmente como:

en donde: X5 es hidrógeno o un agente de activación; e Y3 es alcanodiílo(ci-i2) o alcanodiílo sustituido(ci-i2). Como se describe en el presente documento, el compuesto de fórmula XXIV se define adicionalmente como:

en donde: Y3 es alcanodiílo(Ci-i2) o alcanodiílo sustituido(Ci-i2). Como se describe en el presente documento, Ri es hidrógeno. Como se describe adicionalmente en el presente documento, Ri es alcoxi(Ci-i2). Como también se describe en el presente documento, Ri es metoxi. Como se describe adicionalmente en el presente documento, Ri y R2 se toman juntos y son alquenodiílo(C2-i2) o alquenodiílo sustituido(C2-i2). Como también se describe en el presente documento, la fórmula se define adicionalmente como:
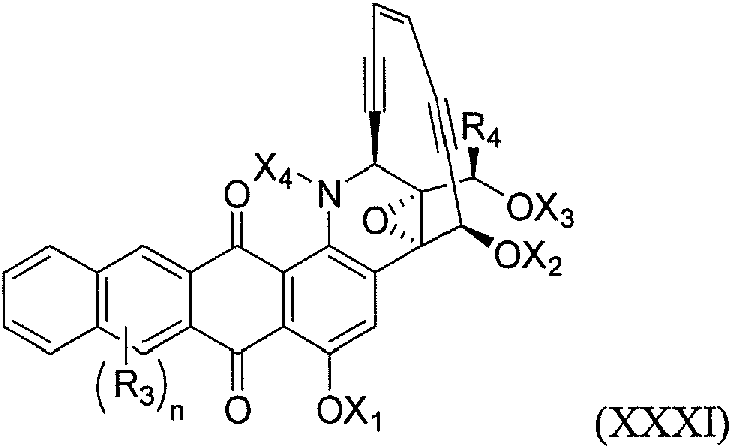
en donde: R3, R4, n, Xi, X2 , X3 y X4 son como se han definido anteriormente. Tal como se analiza en el presente documento, Ri y R2 se toman juntos y son alquilaminodiílo(Ci-i2) o alquilaminodiílo sustituido(Ci-i2). Como se analiza adicionalmente en el presente documento, la fórmula se define adicionalmente como:
2 i

en donde: R3, R4, n, X1, X2, X3 y X4 son como se han definido anteriormente; y R7 es hidrógeno o un grupo protector de amino. Como también se describe en el presente documento, R3 es -Y1-A1 en donde: Y1 es alcanodiílo(C1-8) o alcanodiílo sustituido(C1-8); A 1 es un enlazador en donde el enlazador tiene la fórmula:

en donde: R5 es hidrógeno, alquilona) o alquilo sustituido(C1-6) R6 es -C(O)-Y3-A3, en donde: Y3 es alcanodiílo(C1-12) o alcanodiílo sustituido(C1-12) y A3 es un grupo reactivo de tiol; Y2 es -C(O)-, -C(O)-alcanodiílo(C1-12), -C(O)-alcanodiílo sustituido(C1-12), o un grupo autoinmolante; A2 es un enlace covalente, un resto de aminoácido o un polipéptido; y o es 1, 2, 3, 4, 5, 6 , 7 u 8. Como se describe en el presente documento, R3 es

en donde Y2, A2 e Y3 son como se han definido anteriormente. Como también se describe en el presente documento, A2 es -HN-Val-Cit-C(O)O-. En algunas realizaciones, Y2 es un grupo autoinmolante. Como también se describe en el presente documento, Y2 es

Como también se describe en el presente documento, R4 es metilo. Como también se describe en el presente documento, X1, X2 o X3 son hidrógeno. Como también se describe en el presente documento, X3 es un grupo protector de hidroxi. Como también se describe en el presente documento, X4 es hidrógeno. Como también se describe en el presente documento, X4 es un grupo protector de amina. Como también se describe en el presente documento, la alquilamina(C1-12) o alquilamina sustituida(C1-12) de la etapa a) es metilamina. Como también se describe en el presente documento, la base de la etapa b) es una base nitrogenada y la base es diisopropiletilamina. Como también se describe en el presente documento, el reactivo que retira el grupo protector de la etapa c) es una base y la base es piperidina. Como también se describe en el presente documento, la base de la etapa d) es una base nitrogenada y la base es diisopropiletilamina. Como también se describe en el presente documento, el método comprende adicionalmente una o más etapas de desprotección. Como también se describe en el presente documento, se prevé que estos compuestos de la presente divulgación se unan a un anticuerpo u otro resto de direccionamiento celular como se describe en la patente de Estados Unidos 8.798.431, solicitud de patente de Estados Unidos n.° 2013/0209494 y solicitud de patente PCT WO 2013/122823.
En las realizaciones de esta divulgación, se contempla que las condiciones suficientes para provocar que se produzca una reacción pueden incluir diversos factores que afectan a la eficacia de la reacción. Se prevé su modificación dentro de las realizaciones de cualquiera de los presentes métodos. Dichas condiciones incluyen el número de equivalentes de un compuesto, la elección del disolvente, la elección de la temperatura o el tiempo de reacción. En algunos aspectos de la presente divulgación, las reacciones de la presente divulgación pueden realizarse en cualquier disolvente orgánico. En algunas realizaciones, el disolvente es una amida(C1-12), un éster(C1-12), un alcohol(C1-12), un areno(C1-12), un areno sustituido(C1-12), un alcano(C1-12), un alcano sustituido(C1-12), un heterocicloalcano(C1-12), o un éter(C1-12). En algunas realizaciones, el disolvente de la presente divulgación es éter dietílico, acetonitrilo, dimetilformamida, tetrahidrofurano, dimetilsulfóxido, cloroformo, diclorometano, hexanos,
metanol, etanol, pentanos, benceno, tolueno, acetato de etilo o N-metil-2-pirrolidona. Además, las condiciones pueden afectar a la temperatura de la reacción. En algunas realizaciones, la temperatura es a temperatura ambiente. En algunas realizaciones, a temperatura ambiente o 25 °C, también puede realizarse a una temperatura de aproximadamente 10 °C a aproximadamente 40 °C o más, preferiblemente de aproximadamente 20 °C a aproximadamente 30 °C. En algunas realizaciones, la temperatura puede reducirse para mejorar la eficacia de la reacción. En algunas realizaciones, la temperatura reducida es una temperatura inferior a la temperatura ambiente. En algunas realizaciones, la temperatura es 0 °C o -78 °C. En algunas realizaciones, la temperatura está dentro de un margen de 20 °C de la temperatura establecida y más preferiblemente de 10 °C de la temperatura establecida. En algunas realizaciones, la temperatura puede aumentarse para acelerar la reacción. En algunas realizaciones, la temperatura de reacción es una temperatura de aproximadamente 50 °C a aproximadamente 100 °C. En algunas realizaciones, la temperatura está dentro de un margen de 20 °C de la temperatura establecida y más preferiblemente de 10 °C de la temperatura establecida. Además, el tiempo de reacción puede variarse para aumentar la eficacia de la reacción. En algunas realizaciones, la reacción dura entre aproximadamente la mitad del tiempo ejemplificado y aproximadamente dos veces el tiempo ejemplificado. A modo de ejemplo no limitante, en una reacción que se ha ejemplificado con una duración de 1 hora, el tiempo de reacción es de aproximadamente 30 minutos a 2 horas. Finalmente, la cantidad de equivalentes añadidos también puede alterar la eficacia de la reacción. En algunas realizaciones, se prevé la adición de un reactivo de aproximadamente la mitad de la cantidad ejemplificada a aproximadamente 3 veces la cantidad ejemplificada. A modo de ejemplo no limitante, la adición del reactivo se mide basándose en uno de los materiales de partida que contiene un átomo de carbono que aparece en el producto final. Además, se prevé que los equivalentes de un reactivo que se consume (es decir, no catalítico) no sean inferiores a 1 equivalente.
Se contempla que cualquier método o composición descrita en el presente documento pueda implementarse con respecto a cualquier otro método o composición descrita en el presente documento.
El uso de la palabra "un" o "uno/a", cuando se usa junto con la expresión "que comprende" en las reivindicaciones y/o en la memoria descriptiva puede significar "uno/a", pero también está en consonancia con el significado de "uno/a o más", "al menos uno/a", y "uno/a o más de uno/a". La palabra "aproximadamente" significa más o menos 5 % del número indicado.
Breve descripción de las figuras
Los siguientes dibujos forman parte de la presente memoria descriptiva y se incluyen para demostrar adicionalmente determinados aspectos de la presente divulgación. La divulgación puede entenderse mejor por referencia a uno o más de estos dibujos, en combinación con lo detallado.
FIG. 1 - Síntesis de análogos de uncialamicina
FIG. 2 - Uncialamicina y algunos ejemplos de aminometil uncialamicina, y otros análogos de uncialamicina que contienen amina.
Descripción de realizaciones ilustrativas
La presente divulgación proporciona nuevos análogos de uncialamicina y proporciona métodos mejorados para sintetizar uncialamicina y análogos. Se ha demostrado que estos compuestos tienen una CI50 del orden de picomolar en células cancerosas. Adicionalmente, estos análogos se funcionalizan con un grupo amina, que puede utilizarse para modificar fracciones de direccionamiento celular para generar nuevos tratamientos tales como, pero sin limitación, un conjugado de anticuerpo-fármaco.
I. Uncialamicina y formulaciones de la misma
Los compuestos proporcionados por la presente divulgación se muestran, por ejemplo, anteriormente en la sección Sumario y más adelante en las reivindicaciones. Se pueden fabricar utilizando los métodos descritos en la sección de Ejemplos. La uncialamicina y sus análogos pueden sintetizarse de acuerdo con los métodos descritos, por ejemplo, en la sección de Ejemplos a continuación. Estos métodos pueden modificarse y optimizarse aún más utilizando los principios y técnicas de la química orgánica aplicados por una persona experta en la técnica. Dichos principios y técnicas se muestran, por ejemplo, en March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure (2007).
La uncialamicina y sus análogos de la divulgación pueden contener uno o más átomos de carbono o nitrógeno sustituidos asimétricamente, y pueden aislarse en forma ópticamente activa o racémica. Por lo tanto, se pretende toda forma quiral, diastereomérica, racémica, epimérica y todas las formas isoméricas geométricas de una fórmula química, a menos que se indique específicamente la forma estereoquímica o isomérica específica. Los compuestos pueden aparecer como racematos y mezclas racémicas, enantiómeros individuales, mezclas diastereoméricas y diastereómeros individuales. En algunas realizaciones, se obtiene un único diastereómero. Los centros quirales de la uncialamicina y sus análogos pueden tener configuración S o R.
Las fórmulas químicas utilizadas para representar los compuestos de la divulgación normalmente solo mostrarán uno de posiblemente varios tautómeros distintos. Por ejemplo, se sabe que existen muchos tipos de grupos cetona en equilibrio con los grupos enol correspondientes. De manera similar, existen muchos tipos de grupos imina en equilibrio con los grupos enamina. Independientemente de qué tautómero se represente para un compuesto dado, e independientemente de cuál es el más predominante, se pretenden todos los tautómeros de una fórmula química dada.
Los compuestos de la divulgación también pueden tener la ventaja de que pueden ser más eficaces que, ser menos tóxicos que, actuar durante más tiempo que, ser más potentes que, producir menos efectos secundarios que, ser absorbido más fácilmente que y/o tener un mejor perfil farmacocinético (por ejemplo, una biodisponibilidad oral más elevada y/o una eliminación más baja) que, y/o tener otras propiedades farmacológicas, físicas o químicas útiles con respecto a, los compuestos conocidos en la técnica anterior, ya sea para uso en las indicaciones indicadas en el presente documento o de otra manera.
Además, los átomos componen la uncialamicina y sus análogos están destinados a incluir todas las formas isotópicas de tales átomos. Los isótopos, como se usa en el presente documento, incluyen los átomos que tienen el mismo número atómico pero distintos números másicos. A modo de ejemplo general y sin limitación, los isótopos de hidrógeno incluyen tritio y deuterio, y los isótopos del carbono incluyen 13C y 14C.
Los compuestos de la presente divulgación también pueden existir en forma de profármaco. Dado que se sabe que los profármacos potencian numerosas cualidades convenientes de los fármacos (por ejemplo, solubilidad, biodisponibilidad, fabricación, etc.), los compuestos empleados en algunos métodos de la divulgación pueden, si se desea, suministrarse en forma de profármaco. Por lo tanto, la divulgación contempla profármacos de compuestos de la presente divulgación, así como métodos de suministro profármacos. Los profármacos de los compuestos empleados en la divulgación pueden prepararse modificando grupos funcionales presentes en el compuesto de tal forma que se escindan las modificaciones, ya sea por manipulación rutinaria o in vivo, en el compuesto precursor. Por consiguiente, los profármacos incluyen, por ejemplo, compuestos descritos en el presente documento en los que un grupo hidroxi, amino o carboxilo está unido a cualquier grupo que, cuando el profármaco se administra a un sujeto, se escinde para formar un hidroxi, amino o ácido carboxílico, respectivamente.
Debe reconocerse que el anión o catión particular que forma parte de cualquier sal de la presente divulgación no es crítico, siempre que la sal, como un todo, sea farmacológicamente aceptable. Se presentan ejemplos adicionales de sales farmacéuticamente aceptables y sus métodos de preparación y uso en Handbook of Pharmaceutical Salts: Properties, and Use (2002).
II. Fracciones de direccionamiento celular
En algunos aspectos, la presente divulgación proporciona compuestos conjugados directamente o a través de enlazadores a una fracción de direccionamiento celular. En algunas realizaciones, la conjugación del compuesto con una fracción de direccionamiento celular aumenta la eficacia del compuesto en el tratamiento de una enfermedad o trastorno. Las fracciones de direccionamiento celular de acuerdo con las realizaciones pueden ser, por ejemplo, un anticuerpo, un factor de crecimiento, una hormona, un péptido, un aptámero, una molécula pequeña tal como una hormona, un agente para la formación de imágenes, o un cofactor o una citocina. Por ejemplo, una fracción de direccionamiento celular de acuerdo con las realizaciones puede unirse a una célula de cáncer hepático, tal como una célula Hep3B. Se ha demostrado que el antígeno gp240 se expresa en diversos melanomas, pero no en tejidos normales. Por lo tanto, en algunas realizaciones, los compuestos de la presente divulgación pueden utilizarse en conjugados con un anticuerpo para un antígeno específico que se expresa en una célula cancerosa pero no en tejidos normales.
En determinadas realizaciones adicionales, se prevé que las fracciones de direccionamiento a células cancerosas se unen a múltiples tipos de células cancerosas. Por ejemplo, el anticuerpo monoclonal 8H9 y los anticuerpos monocatenarios derivados del mismo se unen a una glucoproteína que se expresa en los cánceres de mama, sarcomas y neuroblastomas (Onda et al., 2004). Otro ejemplo son los agentes de direccionamiento celular descritos en la Publicación de Patente de Estados Unidos N.° 2004/005647 y en Winthrop et al., 2003, que se unen a MUC-1, un antígeno que se expresa en diversos tipos de cáncer. Por lo tanto, se entenderá que, en determinadas realizaciones, las construcciones de direccionamiento celular de acuerdo con las realizaciones pueden dirigirse contra una pluralidad de tipos de cáncer o tumor.
Adicionalmente, determinadas moléculas de superficie celular se expresan de forma elevada en las células tumorales, incluyendo receptores hormonales tales como el receptor de la gonadotropina coriónica humana y el receptor de la hormona liberadora de gonadotropina (Nechushtan et al., 1997). Por lo tanto, las hormonas correspondientes pueden utilizarse como las fracciones específicas de direccionamiento celular en la terapia contra el cáncer. Adicionalmente, la fracción de direccionamiento celular que puede utilizarse incluye un cofactor, un azúcar, una molécula farmacológica, un agente para la formación de imágenes o un colorante fluorescente. Se sabe que muchas células cancerosas sobreexpresan los receptores de folato y, por lo tanto, el ácido fólico u otros derivados del folato pueden utilizarse como conjugados para desencadenar la interacción específica de célula entre
los conjugados de la presente divulgación y una célula (Campbell, et al. 1991; Weitman, et al., 1992).
Dado que se ha identificado un gran número de receptores de superficie celular en las células hematopoyéticas de diversas estirpes, los ligandos o anticuerpos específicos para estos receptores pueden utilizarse como fracciones de direccionamiento específicos de células. La iL2 también se puede usar como una fracción de direccionamiento específico de células en una proteína quimérica para dirigirse células IL2R+. Como alternativa, otras moléculas tales como B7-1, B7-2 y CD40 pueden utilizarse para dirigirse específicamente linfocitos T activados (The Leucocyte Antigen Facts Book, 1993, Barclay et al. (eds.), Academic Press). Adicionalmente, Los linfocitos B expresan CD19, CD40 y receptor de IL4, y puede ser diana de fracciones que se unen a estos receptores, tales como el ligando de CD40, IL4, IL5, IL6 y CD28. La eliminación de células inmunitarias tales como linfocitos T y linfocitos B es particularmente útil en el tratamiento de tumores linfoides.
Otras citocinas que pueden utilizarse para dirigirse a subconjuntos celulares específicos incluyen las interleucinas (IL1 a IL15), el factor estimulante de colonias de granulocitos, el factor estimulante de colonias de macrófagos, el factor estimulante de colonias de granulocitos-macrófagos, el factor inhibidor de la leucemia, el factor de necrosis tumoral, el factor de crecimiento transformante, el factor de crecimiento epidérmico, los factores de crecimiento insulínicos y/o el factor de crecimiento de fibroblastos (Thompson (ed.), 1994, The Cytokine Handbook, Academic Press, San Diego). En algunos aspectos, el polipéptido de direccionamiento es una citocina que se une al receptor Fnl4, tal como TWEAK (véase, por ejemplo, Winkles, 2008; Zhou et al., 2011 y Burkly et al., 2007).
Un experto en la materia reconoce que hay diversas citocinas conocidas, entre ellas las hematopoyetinas (ovillo de cuatro hélices) (tales como EPO (eritropoyetina), IL-2 (factor de crecimiento de linfocitos T), IL-3 (CSF de multicolonias), IL-4 (BCGF-1, BSF-1), IL-5 (BCGF-2), IL-6 IL-4 (IFN-P2, BSF-2, BCDF), IL-7, IL-8, IL-9, IL-11, IL-13 (P600), G-CSF, IL-15 (factor de crecimiento de linfocitos T), GM-CSF (factor estimulante de colonias de granulocitos y macrófagos), OSM (OM, oncostatina M) y LIF (factor inhibidor de la leucemia)); interferones (tales como IFN-y, IFN-a e IFN-p); superfamilia de las inmunoglobina (como B7.1 (CD80) y B7.2 (B70, CD86)); familia TNF (tal como TNF-a (caquectina), TNF-p (linfotoxina, LT, LT-a), LT-p, ligando de c D40 (Cd 40L), ligando Fas (FasL), ligando de CD27 (CD27L), ligando de CD30 (CD30L) y 41BBL)); y las no asignadas a una familia en particular (tal como TGF-p, IL 1a, IL-1 p, IL-1 RA, IL-10 (inhibidor de la síntesis de citocinas F), IL-12 (factor estimulante de linfocitos NK), MIF, IL-16, IL-17 (mCTLA-8) y/o IL-18 (IGIF, factor inductor de interferón-y)). Adicionalmente, la porción Fc de la cadena pesada de un anticuerpo puede utilizarse para dirigirse a células que expresan el receptor de Fc, tal como el uso de la porción Fc de un anticuerpo IgE para dirigirse a los mastocitos y basófilos.
Adicionalmente, en algunos aspectos, la fracción de direccionamiento celular puede ser una secuencia peptídica o un péptido cíclico. Se proporcionan ejemplos, péptidos de direccionamiento a células y tejidos que pueden utilizarse de acuerdo con las realizaciones, por ejemplo, en las patentes de Estados Unidos n.° 6.232.287; 6.528.481; 7.452.964; 7.671.010; 7.781.565; 8.507.445; y 8.450.278.
Por lo tanto, en algunas realizaciones, las fracciones de direccionamiento celular son anticuerpos o avímeros. Los anticuerpos y avímeros se pueden generar frente a prácticamente cualquier marcador de superficie celular, por lo tanto, lo que proporciona un método para dirigir el suministro de GrB a prácticamente cualquier población celular de interés. A continuación, se detallan los métodos para generar anticuerpos que pueden utilizarse como fracciones de direccionamiento celular. Los métodos para generar avímeros que se unen a un marcador de superficie celular dado se detallan en las publicaciones de Patente de Estados Unidos N.° 2006/0234299 y 2006/0223114.
A. Fracciones de direccionamiento de anticuerpos y de tipo anticuerpos
Como se indicó anteriormente, en algunos aspectos la fracción de direccionamiento celular es un anticuerpo. Como se usa en el presente documento, el término "anticuerpo" pretende incluir inmunoglobulinas y fragmentos de las mismas que son específicamente reactivas a la proteína o péptido designado, o fragmentos de las mismas. Los anticuerpos adecuados incluyen, pero sin limitación, anticuerpos humanos, anticuerpos primatizados, anticuerpos desinmunizados, anticuerpos quiméricos, anticuerpos biespecíficos, anticuerpos humanizados, anticuerpos conjugados (es decir, anticuerpos conjugados o fusionados a otras proteínas, radiomarcadores, citotoxinas), productos inmunofarmacéuticos modulares pequeños (los "SMIP™", forma siglada de Small Modular ImmunoPharmaceuticals), anticuerpos monocatenarios, anticuerpos cameloides, moléculas de tipo anticuerpos (por ejemplo, anticalinas) y fragmentos de anticuerpos. Como se usa en el presente documento, el término "anticuerpos" también incluye anticuerpos monoclonales intactos, anticuerpos policlonales, anticuerpos de dominio único (por ejemplo, anticuerpos de dominio único de tiburón (por ejemplo, IgNAR o fragmentos de los mismos)), anticuerpos multiespecíficos (por ejemplo, anticuerpos biespecíficos) formados a partir de al menos dos anticuerpos intactos, y fragmentos de anticuerpos siempre que presenten la actividad biológica deseada. En algunos aspectos, el anticuerpo puede ser un VHH (es decir, un anticuerpo VHH específico de antígeno) que comprende solo una cadena pesada. Por ejemplo, tales moléculas de anticuerpo pueden obtenerse de una llama u otro anticuerpo de camélido (por ejemplo, una IgG2 o IgG3 de camélido, o un marco que presente CDR de tal Ig de camélido) o de un anticuerpo de tiburón. Los polipéptidos de anticuerpo para su uso en el presente documento pueden ser de cualquier tipo (por ejemplo, IgG, IgM, IgA, IgD e IgE). En general, son preferentes las IgG y/o las IgM debido a que son los anticuerpos más comunes en la situación fisiológica y debido a que son los que se fabrican más fácilmente en un entorno de
laboratorio.
Como se usa en el presente documento, un "fragmento de anticuerpo" incluye una porción de un anticuerpo intacto, tal como, por ejemplo, la región de unión a antígeno o variable de un anticuerpo. Los ejemplos de fragmentos de anticuerpo incluyen fragmentos Fab, Fab', F(ab')2, Fc y Fv; triacuerpos; tetracuerpos; anticuerpos lineales; moléculas de anticuerpo monocatenarias; y anticuerpos multiespecíficos formados a partir de fragmentos de anticuerpo. La expresión "fragmento de anticuerpo" también incluye cualquier proteína sintética o modificada por ingeniería genética, que actúa como un anticuerpo al unirse a un antígeno específico para formar un complejo. Por ejemplo, los fragmentos de anticuerpo incluyen fragmentos aislados, fragmentos "Fv", que consisten en las regiones variables de las cadenas pesada y ligera, moléculas polipeptídicas monocatenarias recombinantes en las cuales las regiones variables de la cadena ligera y la pesada están conectadas por un enlazador peptídico ("proteínas ScFv"), y unidades de reconocimiento mínimas que consisten en los restos de aminoácidos que imitan la región hipervariable.
Además, se contemplan para su uso con las presentes realizaciones los "minianticuerpos" o "minicuerpos". Los minicuerpos son cadenas polipeptídicas sFv que incluyen dominios de oligomerización en sus extremos C, separados del sFv por una región bisagra (Pack et al. 1992). El dominio de oligomerización comprende hélices a autoasociativas, por ejemplo, cremalleras de leucina, que pueden estabilizarse aún más mediante enlaces disulfuro adicionales. El dominio de oligomerización está diseñado para ser compatible con el plegamiento vectorial a través de una membrana, un proceso que se cree que facilita el plegamiento del polipéptido en vivo en una proteína de unión funcional. En general, los minicuerpos se producen utilizando métodos recombinantes bien conocidos en la técnica. Véase, por ejemplo, Pack et al. (1992); Cumber et al. (1992).
En algunos casos, las moléculas de tipo anticuerpo son armazones proteicos que se pueden utilizar para presentar dominios CDR de anticuerpos. El origen de tales armazones proteicos puede ser, pero sin limitación, las estructuras seleccionadas de: fibronectina (ver, por ejemplo, la publicación de Patente de Estados Unidos N.° 2009/0253899, que incluye el dominio 10 de fibronectina tipo III, la proteína Z que se origina en el dominio B de la proteína A de Staphylococcus aureus, la tiorredoxina A o proteínas con un motivo repetido tal como la "repetición de anquirina" (Kohl et al., 2003), la "repetición armadillo", la "repetición rica en leucina" y la "repetición tetratricopeptídica". Las técnicas para preparar y usar diversas construcciones y fragmentos basados en anticuerpos son bien conocidas en la técnica. Se describen en detalle moléculas de tipo anticuerpo adicionales, tales como las anticalinas, en las Publicaciones de patentes de Estados Unidos N.° 2010/0285564, 2006/0058510, 2006/0088908, 2005/0106660, la publicación PCT N.° WO 2006/056464 y (Skerra, 2001).
Los peptidomiméticos de unión de tipo anticuerpo también se contemplan en las presentes realizaciones. Liu et al. (2003) describen "peptidomiméticos de unión de tipo anticuerpos" (ABiP), que son péptidos que actúan como anticuerpos simplificados y tienen determinadas ventajas como una semivida en suero más larga así como métodos de síntesis menos engorrosos. Asimismo, en algunos aspectos, las moléculas de tipo anticuerpo son péptidos cíclicos o bicíclicos. Por ejemplo, se proporcionan métodos para aislar péptidos bicíclicos de unión a antígeno (por ejemplo, por presentación en fagos) y para el uso de tales péptidos, en la publicación de patente de Estados Unidos N.° 2010/0317547.
Se reconoce que los anticuerpos monoclonales (MAb, forma siglada de monoclonal antibodies) tienen determinadas ventajas, por ejemplo, la reproducibilidad y la producción a gran escala. Las realizaciones de la divulgación proporcionan anticuerpos monoclonales de origen humano, murino, de mono, de rata, de hámster, de conejo y de pollo. En algunas realizaciones, se utilizan anticuerpos monoclonales humanos o fragmentos de los mismos.
Los anticuerpos "humanizados" también se contemplan, como son los anticuerpos quiméricos de ratón, rata o de otra especie, que portan dominios humanos de la región constante y/o variable, anticuerpos biespecíficos, anticuerpos recombinantes y modificados por ingeniería genética, y fragmentos de los mismos. Como se usa en el presente documento, el término inmunoglobulina "humanizada" se refiere a una inmunoglobulina que comprende una región marco conservada humana y una o más CDR de una inmunoglobulina no humana (habitualmente una de ratón o rata). La inmunoglobulina no humana que proporciona las CDR se denomina "donante" y la inmunoglobulina humana que proporciona el armazón se denomina "aceptadora". Un "anticuerpo humanizado" es un anticuerpo que comprende una inmunoglobulina de cadena ligera humanizada y cadena pesada humanizada. Los métodos de humanización de anticuerpos son bien conocidos en la técnica, véase, por ejemplo, Harvey et al., 2004.
B. Formulaciones
Los materiales para su uso en la preparación de microesferas y/o microcápsulas son, por ejemplo, los polímeros biodegradables/bioerosionables tales como la poligalactina, poli-(isobutilcianoacrilato), poli(2-hidroxietil-L-glutamina) y poli(ácido láctico). Los transportadores biocompatibles que pueden utilizarse al formular una formulación parenteral de liberación controlada son hidratos de carbono (por ejemplo, dextranos), proteínas (por ejemplo, albúmina), lipoproteínas o anticuerpos. Los materiales para su uso en implantes pueden ser no biodegradables (por ejemplo, polidimetilsiloxano) o biodegradables (por ejemplo, poli(caprolactona), poli(ácido láctico), poli(ácido glicólico) o poli(ortoésteres), o combinaciones de los mismos).
Las formulaciones para el uso oral incluyen comprimidos que contienen el principio (o principios) activo (por ejemplo, uncialamicina y sus análogos) en una mezcla con excipientes no tóxicos farmacéuticamente aceptables. Dichas formulaciones son conocidas por el experto en la materia. Los excipientes pueden ser, por ejemplo, diluyentes o cargas inertes (por ejemplo, sacarosa, sorbitol, azúcar, manitol, celulosa microcristalina, almidones incluyendo almidón de patata, carbonato de calcio, cloruro de sodio, lactosa, fosfato de calcio, sulfato de calcio o fosfato de sodio); agentes de granulación y disgregantes (por ejemplo, derivados de celulosa que incluyen celulosa microcristalina, almidones incluyendo almidón de patata, croscarmelosa de sodio, alginatos o ácido algínico); agentes aglutinantes (por ejemplo, sacarosa, glucosa, sorbitol, goma arábiga, ácido algínico, alginato de sodio, gelatina, almidón, almidón pregelatinizado, celulosa microcristalina, silicato de aluminio y magnesio, carboximetilcelulosa de sodio, metilcelulosa, hidroxipropilmetilcelulosa, etilcelulosa, polivinilpirrolidona o polietilenglicol); y agentes lubricantes, emolientes y antiadherentes (por ejemplo, estearato de magnesio, estearato de zinc, ácido esteárico, sílices, aceites vegetales hidrogenados o talco). Otros excipientes farmacéuticamente aceptables pueden ser colorantes, agentes aromatizantes, plastificantes, humectantes, agentes tamponantes y similares.
Los comprimidos pueden estar sin recubrir o pueden recubrirse mediante técnicas conocidas, opcionalmente para retrasar la disgregación y la absorción en el tubo gastrointestinal y proporcionar de esta forma una acción sostenida durante un periodo más largo. El recubrimiento puede adaptarse para liberar el fármaco activo con un patrón predeterminado (por ejemplo, para lograr una formulación de liberación controlada) o puede adaptarse para no liberar el fármaco activo hasta después del paso por el estómago (recubrimiento entérico). El recubrimiento puede ser un recubrimiento de azúcar, un recubrimiento de película (por ejemplo, a base de hidroxipropilmetilcelulosa, metilcelulosa, metil hidroxietilcelulosa, hidroxipropilcelulosa, carboximetilcelulosa, copolímeros de acrilato, polietilenglicoles y/o polivinilpirrolidona), o un recubrimiento entérico (por ejemplo, a base de copolímero de ácido metacrílico, ftalato acetato de celulosa, ftalato de hidroxipropil metilcelulosa, acetato succinato de hidroxipropil metilcelulosa, ftalato acetato de polivinilo, goma laca y/o etilcelulosa). Adicionalmente, se puede emplear un material con tiempo de retardo, tal como, por ejemplo, monoestearato de glicerilo o diestearato de glicerilo.
III. Terapias
A. Indicaciones
i. Cáncer y otras enfermedades hiperproliferativas
Si bien las enfermedades hiperproliferativas pueden asociarse con cualquier enfermedad que haga que una célula comience a reproducirse sin control, el ejemplo prototípico es el cáncer. Uno de los elementos clave del cáncer es que se interrumpe el ciclo apoptótico normal de la célula y, por lo tanto, los agentes que interrumpen el crecimiento de las células son importantes como agentes terapéuticos para tratar estas enfermedades. En la presente divulgación, se pueden utilizar análogos de uncialamicina para reducir el recuento celular y, como tal, potencialmente se pueden usar para tratar diversos tipos de líneas cancerosas. En diversos aspectos, se anticipa que los análogos de uncialamicina de la presente divulgación pueden utilizarse para tratar prácticamente cualquier neoplasia maligna.
Las células cancerosas que pueden tratarse con los compuestos de la presente divulgación incluyen, pero sin limitación, células de la vejiga, sangre, hueso, médula ósea, cerebro, mama, colon, esófago, origen gastrointestinal, encía, cabeza, riñón, hígado, pulmón, nasofaringe, cuello, ovario, próstata, piel, estómago, páncreas, testículos, lengua, cuello uterino o útero. Además, el cáncer puede ser específicamente del siguiente tipo histológico, aunque no se limita a estos: neoplasia, maligno; carcinoma; carcinoma, indiferenciado; carcinoma de células gigantes y fusiformes; carcinoma microcítico; carcinoma papilar; carcinoma de células escamosas; carcinoma linfoepitelial; carcinoma de células basales; carcinoma pilomatricial; carcinoma de células transicionales; carcinoma papilar de células transicionales; adenocarcinoma; gastrinoma, maligno; colangiocarcinoma; carcinoma hepatocelular; carcinoma hepatocelular y colangiocarcinoma combinados; adenocarcinoma trabecular; carcinoma adenoideo quístico; adenocarcinoma en pólipo adenomatoso; adenocarcinoma, poliposis coli familiar; carcinoma sólido; tumores carcinoides, maligno; adenocarcinoma branquioloalveolar; adenocarcinoma papilar; carcinoma cromófobo; carcinoma acidófilo; adenocarcinoma oxifílico; carcinoma basófilo; adenocarcinoma de células claras; carcinoma de células granulares; adenocarcinoma folicular; adenocarcinoma papilar y folicular; carcinoma esclerosante no encapsulante; carcinoma cortical suprarrenal; carcinoma endometroide; carcinoma de anejos cutáneos; adenocarcinoma apocrino; adenocarcinoma sebáceo; adenocarcinoma ceruminoso; carcinoma mucoepidermoide; cistadenocarcinoma; cistadenocarcinoma papilar; cistadenocarcinoma papilar seroso; cistadenocarcinoma mucinoso; adenocarcinoma mucinoso; carcinoma de células de anillo de sello; carcinoma de conducto infiltrante; carcinoma medular; carcinoma lobular; carcinoma inflamatorio; enfermedad de Paget, mamaria; carcinoma de células acinares; carcinoma adenoescamoso; adenocarcinoma con metaplasia escamosa; timoma, maligno; tumor del estroma ovárico, maligno; tecoma, maligno; tumor de células de la granulosa, maligno; androblastoma, maligno; carcinoma de células de Sertoli; tumor de células de leydig, maligno; tumor de lipocitos, maligno; paraganglioma, maligno; paraganglioma extramamario, maligno; feocromocitoma; glomangiosarcoma; melanoma maligno; melanoma amelánico; melanoma de diseminación superficial; melanoma maligno en nevo gigante pigmentado; melanoma de células epitelioides; nevo azul, maligno; sarcoma; fibrosarcoma; histiocitoma fibroso, maligno; mixosarcoma; liposarcoma; liomiosarcoma; rabdomiosarcoma; rabdomiosarcoma embrionario; rabdomiosarcoma alveolar; sarcoma
estromal; tumor mixto, maligno; tumor mixto mulleriano; nefroblastoma; hepatoblastoma; carcinosarcoma; mesenquimoma, maligno; tumor de Brenner, maligno; tumores filoides, maligno; sarcoma sinovial; mesotelioma, maligno; disgerminoma; carcinoma embrionario; teratoma, maligno; struma ovarii, maligno; coriocarcinoma; mesonefroma, maligno; hemangiosarcoma; hemangioendotelioma, maligno; sarcoma de Kaposi; hemangiopericitoma, maligno; linfangiosarcoma; osteosarcoma; osteosarcoma yuxtacortical; condrosarcoma; condroblastoma, maligno; condrosarcoma mesenquimatoso; tumor óseo de células gigantes; sarcoma de Ewing; tumor odontogénico, maligno; odontosarcoma ameloblástico; ameloblastoma, maligno; fibrosarcoma ameloblástico; pinealoma, maligno; cordoma; glioma, maligno; ependimoma; astrocitoma; astrocitoma protoplásmico; astrocitoma fibrilar; astroblastoma; glioblastoma; oligodendroglioma; oligodendroblastoma; neuroectodérmico primitivo; sarcoma cerebeloso; ganglioneuroblastoma; neuroblastoma; retinoblastoma; tumor neurogénico olfativo; meningioma, maligno; neurofibrosarcoma; neurilemoma, maligno; tumor de células granulares, maligno; linfoma maligno; enfermedad de Hodgkin; de Hodgkin; paragranuloma; linfoma maligno, pequeño linfocítico; linfoma maligno, de células grandes, difuso; linfoma maligno, folicular; micosis fungoide; otros linfomas no hodgkin especificados; histiocitosis maligna; mieloma múltiple; sarcoma de mastocitos; enfermedad inmunoproliferativa del intestino delgado; leucemia; leucemia linfoide; leucemia de células plasmáticas; eritroleucemia; leucemia de células de linfosarcoma; leucemia mieloide; leucemia basófila; leucemia eosinófila; leucemia monocítica; leucemia de mastocitos; leucemia megacarioblástica; sarcoma mieloide; y tricoleucemia.
ii. Infecciones bacterianas
En algunos aspectos de la presente divulgación, los compuestos divulgados en el presente documento pueden utilizarse para tratar una infección bacteriana. Aunque los humanos contienen numerosas bacterias distintas sobre y dentro de su cuerpo, un desequilibrio en los niveles bacterianos o la introducción de bacterias patógenas puede provocar una infección bacteriana sintomática. Las bacterias patógenas provocan diversas enfermedades distintas, entre ellas, pero sin limitación, numerosas enfermedades alimentarias, fiebre tifoidea, tuberculosis, neumonía, sífilis y lepra.
Adicionalmente, las distintas bacterias tienen una amplia gama de interacciones con el cuerpo y esas interacciones pueden modular la capacidad de las bacterias para provocar una infección. Por ejemplo, las bacterias pueden ser patógenas de forma condicional, de modo que solo provocan una infección en condiciones específicas. Por ejemplo, las bacterias Staphylococcus y Streptococcus existen en el bioma bacteriano humano normal, pero cuando a estas bacterias se les permite colonizar otras partes del cuerpo, provocan una infección de la piel, neumonía o sepsis. Otras bacterias se conocen como patógenos oportunistas y solo provocan enfermedades en un paciente con un sistema inmunitario debilitado u otra enfermedad o trastorno.
Las bacterias también pueden ser patógenos intracelulares, los cuales pueden crecer y reproducirse dentro de las células del organismo hospedador. Dichas bacterias se pueden dividir en dos categorías principales como parásitos intracelulares obligados o parásitos intracelulares facultativos. Los parásitos intracelulares obligados precisan a la célula hospedadora para reproducirse, e incluyen a bacterias tales como, pero sin limitación Chlamydophila, Rickettsia y Ehrlichia, que se sabe que provocan neumonía, infecciones de las vías urinarias, tifus y fiebre de las Montañas Rocosas. Los parásitos intracelulares facultativos pueden reproducirse de forma intracelular o extracelular. Algunos ejemplos no limitantes de parásitos intracelulares facultativos incluyen Salmonela, Listeria, Legionella, Mycobacterium y Brucella, que se sabe que provocan intoxicación alimentaria, fiebre tifoidea, sepsis, meningitis, enfermedad del legionario, tuberculosis, lepra y brucelosis.
Para finalizar, las infecciones bacterianas podrían enfocarse en una ubicación específica dentro o sobre el cuerpo. Por ejemplo, las bacterias pueden ser inofensivas si solo se exponen a los órganos específicos, pero cuando entran en contacto con un órgano o tejido específico, las bacterias pueden comenzar a multiplicarse y provocar una infección bacteriana.
En particular, los inventores contemplan el tratamiento de infecciones bacterianas, entre ellas las provocadas por Staphyloccoccus aureus. S. aureus es un importante patógeno humano, que provoca una amplia diversidad de enfermedades que varían de infecciones leves de piel y de partes blandas e intoxicación alimentaria a enfermedades potencialmente mortales, tales como infecciones posquirúrgicas graves, septicemia, endocarditis, neumonía necrotizante y síndrome de choque tóxico. Estos organismos tienen una capacidad notable para acumular determinantes adicionales de resistencia a antibióticos, dando como resultado la formación de cepas resistentes a múltiples fármacos.
Adicionalmente, los compuestos proporcionados por la presente divulgación pueden utilizarse para tratar una infección de Burkholderia cepacia. B. cepacia es un importante patógeno humano, que provoca neumonía, en particular en pacientes inmunodeprimidos. Se sabe que este patógeno en particular es resistente a varios antibióticos y, por lo tanto, los nuevos antibióticos son terapéuticamente importantes.
B. Formulaciones farmacéuticas y vías de administración
Cuando se contemplen aplicaciones clínicas, será necesario preparar composiciones farmacéuticas en una forma
apropiada para la aplicación prevista. En general, esto implicará preparar composiciones que estén esencialmente exentas de pirógenos, así como de otras impurezas que podrían ser dañinas para los seres humanos o los animales.
En general, se deseará emplear sales y tampones apropiados para estabilizar los vectores de suministro y permitir la captación por las células diana. Además, se emplearán tampones cuando se introduzcan células recombinantes en un paciente. Las composiciones acuosas de la presente divulgación comprenden una cantidad eficaz del vector para las células, disuelto o dispersado en un transportador o medio acuoso farmacéuticamente aceptable. Dichas composiciones también se denominan inóculos. La expresión "farmacéutica o farmacológicamente aceptable" se refiere a entidades moleculares y a composiciones que no producen reacciones adversas, alérgicas u otras no deseadas cuando se administran a un animal o un ser humano. Como se usa en el presente documento, "transportador farmacéuticamente aceptable" incluye todos y cada uno de los disolventes, medios de dispersión, recubrimientos, agentes antibacterianos y antifúngicos, agentes isotónicos y retardantes de la absorción, y similares. El uso de tales medios y agentes para sustancias farmacéuticamente activas se conoce en la técnica. Excepto en el caso de que algún agente o medio convencional sea incompatible con los vectores o células de la presente divulgación, se contempla su uso en composiciones terapéuticas. En las composiciones también pueden incorporarse principios activos complementarios.
Las composiciones activas de la presente divulgación pueden incluir preparados farmacéuticos clásicos. La administración de estas composiciones terapéuticas de acuerdo con la presente divulgación se realizará a través de cualquier vía habitual, siempre que el tejido diana esté disponible a través de esa vía. Dichas vías incluyen la vía oral, nasal, bucal, rectal, vaginal o tópica. Como alternativa, la administración puede ser inyección ortotópica, intradérmica, subcutánea, intramuscular, intratumoral, intraperitoneal o intravenosa. Dichas composiciones normalmente se administrarán como composiciones farmacéuticamente aceptables, como se cita anteriormente.
Los compuestos activos también puede administrarse por vía parenteral o intraperitoneal. Las soluciones de los compuestos activos como base libre o sales farmacológicamente aceptables pueden prepararse en agua mezclada adecuadamente con un tensioactivo, tal como hidroxipropilcelulosa. También se pueden preparar dispersiones en glicerol, polietilenglicoles líquidos y mezclas de los mismos, y en aceites. En condiciones normales de almacenamiento y uso, estos preparados contienen un conservante para prevenir el crecimiento de microorganismos.
Las formas farmacéuticas adecuadas para el uso inyectable incluyen soluciones o dispersiones acuosas estériles y polvos estériles para la preparación extemporánea de soluciones o dispersiones inyectables estériles. En todos los casos, la forma debe ser estéril y debe ser fluida en la medida en que exista facilidad de inyección. Debe ser estable en las condiciones de fabricación y almacenamiento, y debe conservarse frente a la acción contaminante de microorganismos, tales como bacterias y hongos. El transportador puede ser un disolvente o medio de dispersión que contenga, por ejemplo, agua, etanol, poliol (por ejemplo, glicerol, propilenglicol y polietilenglicol líquido, y similares), mezclas adecuadas de los mismos, y aceites vegetales. La fluidez apropiada puede mantenerse, por ejemplo, mediante el uso de un recubrimiento, tal como lecitina, mediante el mantenimiento del tamaño de partícula necesario en el caso de una dispersión y mediante el uso de tensioactivos. La prevención de la acción de microorganismos puede conseguirse mediante diversos agentes antibacterianos y antifúngicos, por ejemplo, parabenos, clorobutanol, fenol, ácido sórbico, timerosal y similares. En muchos casos, será preferible incluir agentes isotónicos, por ejemplo, azúcares o cloruro de sodio. La absorción prolongada de las composiciones inyectables puede conseguirse mediante el uso en las composiciones de agentes que retrasan la absorción, por ejemplo, monoestearato de aluminio y gelatina.
Las soluciones inyectables estériles se preparan incorporando los compuestos activos en la cantidad necesaria en el disolvente apropiado con otros diversos ingredientes enumerados anteriormente, según lo necesario, seguido de esterilización por filtración. En general, las dispersiones se preparan incorporando los diversos principios activos esterilizados en un vehículo estéril que contiene el medio de dispersión básico y los otros ingredientes necesarios de los enumerados anteriormente. En el caso de polvos estériles para la preparación de soluciones inyectables estériles, los métodos de preparación preferentes son las técnicas de secado al vacío y liofilización, que producen un polvo del principio activo más cualquier ingrediente adicional deseado, a partir de la solución de los mismos previamente filtrada a esterilidad.
Como se usa en el presente documento, "transportador farmacéuticamente aceptable" incluye todos y cada uno de los disolventes, medios de dispersión, recubrimientos, agentes antibacterianos y antifúngicos, agentes isotónicos y retardantes de la absorción, y similares. En la técnica se conoce bien el uso de tales medios y agentes para sustancias farmacéuticas activas. Excepto en el caso de que algún medio o agente convencional sea incompatible con el principio activo, se contempla su uso en las composiciones terapéuticas. Además, pueden incorporarse en las composiciones principios activos complementarios.
Para la administración oral, los derivados de uncialamicina de la presente divulgación pueden incorporarse con excipientes y utilizarse en forma de enjuagues bucales y dentífricos no ingeribles. Se puede preparar un enjuague bucal incorporando el principio activo en la cantidad necesaria en un disolvente apropiado, tal como una solución de borato de sodio (solución de Dobell). Como alternativa, el principio activo puede incorporarse en un lavado
antiséptico que contenga borato de sodio, glicerina y bicarbonato de potasio. El principio activo también se puede dispersar en dentífricos, incluyendo: geles, pastas, polvos y pastas. El principio activo se puede añadir en una cantidad terapéuticamente eficaz a una pasta dentífrica que puede incluir agua, aglutinantes, abrasivos, agentes aromatizantes, agentes espumantes y humectantes.
Las composiciones de la presente divulgación pueden formularse en forma neutra o de sal. Las sales farmacéuticamente aceptables incluyen sales de adición de ácido (formadas con los grupos amino libres de la proteína) y que se forman con ácidos inorgánicos tales como, por ejemplo, ácidos clorhídrico o fosfórico o ácidos orgánicos tales como acético, oxálico, tartárico, mandélico y similares. Las sales formadas con los grupos carboxilo libres también pueden obtenerse de bases inorgánicas tales como, por ejemplo, sodio, potasio, amonio, calcio o hidróxidos férricos, y bases orgánicas tales como isopropilamina, trimetilamina, histidina, procaína y similares.
Tras la formulación, las soluciones se administrarán de una manera compatible con la formulación de dosificación y en una cantidad tal que sea terapéuticamente eficaz. Las formulaciones se administran fácilmente en diversas formas farmacéuticas, tales como soluciones inyectables, cápsulas de liberación de fármacos y similares. Para la administración parenteral, por ejemplo, en una solución acuosa, la solución debe tamponarse adecuadamente, si fuera necesario, y el diluyente líquido en primer lugar tiene que hacerse isotónico con suficiente solución salina o glucosa. Estas soluciones acuosas particulares son especialmente adecuadas para la administración intravenosa, intramuscular, subcutánea e intraperitoneal. A este respecto, los medios acuosos estériles que pueden emplearse serán conocidos por los expertos en la materia a la luz de la presente divulgación. Por ejemplo, una dosificación podría disolverse en 1 ml de solución de NaCl isotónica y añadirse a 1000 ml de líquido de hipodermoclisis o inyectarse en el sitio propuesto de infusión, (véase, por ejemplo, "Remington's Pharmaceutical Sciences" 15a edición, páginas 1035-1038 y 1570-1580). Se producirá necesariamente alguna variación en la dosificación dependiendo de la afección del sujeto que se está tratando. La persona responsable de la administración, en cualquier caso, determinará la dosis apropiada para el sujeto individual. Además, para la administración a seres humanos, los preparados deben cumplir con los criterios de esterilidad, pirogenicidad, seguridad general y pureza, según lo exigen los criterios de la Oficina de biofármacos de la FDA.
C. Métodos de tratamiento
El cáncer, conocido desde el punto de vista médico como una neoplasia maligna, es un amplio grupo de enfermedades que implican el crecimiento celular no regulado. En el cáncer, las células se dividen y crecen sin control, formando tumores malignos e invadiendo partes cercanas del cuerpo. El cáncer también puede diseminarse a partes más distantes del cuerpo a través del sistema linfático o el torrente sanguíneo. No todos los tumores son cancerosos; los tumores benignos no invaden los tejidos vecinos y no se diseminan por todo el cuerpo. Hay más de 200 cánceres conocidos distintos que afectan a los humanos.
Las causas del cáncer son diversas, complejas y solo parcialmente entendidas. Se sabe que muchas cosas aumentan el riesgo de padecer cáncer, entre ellos el tabaquismo, factores relacionados con la alimentación, determinadas infecciones, la exposición a la radiación, la falta de actividad física, la obesidad y los contaminantes ambientales. Estos factores pueden dañar directamente los genes o combinarse con fallas genéticas existentes dentro de las células para provocar mutaciones que producen cáncer. Se pueden encontrar el origen de aproximadamente el 5-10% de los cánceres directamente en defectos genéticos heredados. Muchos cánceres podrían prevenirse al no fumar, comiendo más vegetales, frutas y cereales integrales, comiendo menos carne e hidratos de carbono refinados, manteniendo un peso saludable, haciendo ejercicio, minimizando la exposición a la luz solar y vacunándose frente a algunas enfermedades infecciosas.
Los métodos terapéuticos de la divulgación (que incluyen el tratamiento profiláctico) en general incluyen la administración a un sujeto que lo necesite de una cantidad terapéuticamente eficaz de las composiciones descritas en el presente documento, incluyendo un mamífero, en particular, un ser humano. Dicho tratamiento se administrará adecuadamente a los sujetos, particularmente seres humanos, que padecen, que tiene, son susceptible o con riesgo de una enfermedad, trastorno o síntoma de la misma. La determinación de los sujetos "en riesgo" puede hacerse mediante cualquier determinación objetiva o subjetiva, por una prueba diagnóstica o la opinión de un sujeto o un médico (por ejemplo, una prueba genética, un marcador enzimático o proteico, un marcador (como se define en el presente documento), la historia familiar y similares).
En algunos aspectos de la presente divulgación, la presente divulgación proporciona compuestos que se administran sin modificación o de administran como profármacos. En algunas realizaciones, los compuestos se administran en combinación con otro agente terapéutico, en donde cada agente se administra de forma independiente o donde los fármacos se combinan mediante modificaciones químicas y un grupo enlazador. En algunas realizaciones, los fármacos se administran como un conjugado con una fracción de direccionamiento celular. En algunas realizaciones, los compuestos de la presente divulgación se administran como un conjugado con un anticuerpo.
i. Conjugados de anticuerpo-fármaco (los ADC, forma siglada de antibody-drug conjúgate)
En algunas realizaciones, los compuestos de la presente divulgación son citotoxinas potentes y se utilizan
terapéuticamente en conjugados de anticuerpo-fármaco (los ADC). Sin quedar ligados a teoría alguna, la conjugación del compuesto de la presente divulgación a un anticuerpo permite el suministro dirigido del compuesto en el sitio de acción previsto, tal como una célula cancerosa, y reduce el riesgo de toxicidad sistémica.
La conjugación de un anticuerpo con el compuesto se puede utilizar con un enlazador de manera que el anticuerpo se escinda enzimáticamente del compuesto de la presente divulgación. En algunas realizaciones, los ADC de la presente divulgación contienen un enlazador escindible por catepsina. Sin quedar ligados a teoría alguna, el enlazador escindible por catepsina tiene el siguiente mecanismo de acción:

Después de la unión del anticuerpo a su antígeno en una célula cancerosa diana, el ADC se internaliza en la célula cancerosa y finalmente llega a un lisosoma, donde la enzima catepsina B está presente. El dipéptido citrulina-valina (Cit-Val) es un motivo sustrato para la catepsina B (se conocen otros motivos sustrato y se pueden usar en su lugar) y la catepsina B lo escinde en la ubicación indicada por la línea de puntos. La molécula resultante de la escisión contiene un grupo p-aminobenciloxicarbonilo (PABC), que es inestable y sufre eliminación en 1,6 (autoinmolación) y descarboxilación para liberar 48aa. Sin quedar ligados a teoría alguna, en el enlazador, el grupo PABC sirve como un espaciador entre la fracción 48aa y el dipéptido Cit-Val, para impedir que 48aa interfiera estéricamente con la acción de la catepsina B. El 48aa daña el a Dn a través de un mecanismo característico de las citotoxinas de enediina y provoca la muerte celular. De esta manera, el 48aa se libera solo dentro de una célula diana pero no mientras el ADC está en la circulación general, evitando la toxicidad sistémica.
Los expertos en la materia entenderán que las condiciones y la metodología descritas anteriormente son a modo de ejemplo y no limitantes, y que en la técnica se conocen otros enfoques para la conjugación y que, usando esos enfoques, pueden prepararse los ADC utilizables de las citotoxinas de la presente divulgación.
Una técnica de conjugación alternativa que se puede utilizar incluye la "química de encaje" sin cobre, en la que un grupo azida se agrega a través del enlace alquino colado de un ciclooctino, para formar un anillo de 1,2,3-triazol. Véase, por ejemplo, Agard et al., J. Amer. Chem. Soc. 2004,126, 15046-15047; Best, Biochemistry 2009, 48, 6571 6584. En algunas realizaciones, la azida se localiza en el anticuerpo y el ciclooctino en la fracción farmacológica, o viceversa. En algunas realizaciones, el grupo ciclooctino es proporcionado por un reactivo DIBO (disponible en Invitrogen/Molecular Probes, Eugene, Oregón).
Otra técnica de conjugación que puede utilizarse implica la introducción de un aminoácido no natural en un anticuerpo, con el aminoácido no natural proporcionando funcionalidad para la conjugación con un grupo funcional reactivo en la fracción farmacológica. Por ejemplo, el aminoácido no natural p-acetilfenilalanina puede incorporarse en un anticuerpo u otro polipéptido, como se muestra en Tian et al., documento WO 2008/030612 A2 (2008). El grupo cetona en p-acetilfenilalanina puede ser un sitio de conjugación mediante la formación de una oxima con un grupo hidroxilamino en fracción farmacológica. Como alternativa, el aminoácido no natural p-azidofenilalanina puede incorporarse en un anticuerpo para proporcionar un grupo funcional azida para la conjugación a través de química de encaje. Los aminoácidos no naturales también pueden incorporarse en un anticuerpo u otro polipéptido utilizando métodos sin de células, como se muestra en Goerke et al., documento US 2010/0093024 A1 (2010) y Chowdari et al., Biotechnol. Bioeng. 2009, 102 (2), 400-416.
Otra técnica de conjugación más que se puede utilizar se basa en un grupo amina (NH2) que puede utilizarse para la conjugación, utilizando la enzima transglutaminasa, como se muestra en Jeger et al., Angew. Chem. Int. Ed. 2010, 49, 9995 -9997.
Adicionalmente, otra técnica de conjugación que se puede utilizar utiliza la enzima Sortasa A, como se muestra en Levary et al., PLoS One 2011,6(4), e18342; Proft, Biotechnol. Lett. 2010, 32, 1-10; Ploegh et al., documento WO 2010/087994 A2 (2010); y Mao et al., documento WO 2005/051976 A2 (2005). El motivo de reconocimiento de la Sortasa A (normalmente, LPXTG, donde X es cualquier aminoácido natural) puede estar unido al anticuerpo y el motivo aceptor nucleófilo (normalmente, GGG) se ubica en la fracción farmacológica, o viceversa.
Se muestran técnicas de conjugación adicionales, por ejemplo, en la Patente US 2013/0209494.
Los expertos en la materia apreciarán que, dado que se pueden introducir múltiples grupos tiol a través de la reacción de tiolación, puede estar unida a un anticuerpo más de una fracción farmacológica (es decir, 116). Esta relación denomina relación de sustitución (RS) o, como alternativa, relación de fármaco-anticuerpo (RFA). En algunas realizaciones, la RFA está entre aproximadamente 1 y aproximadamente 4. En otras realizaciones, la RFA está entre aproximadamente 1,2 y aproximadamente 1,9. Los expertos en la materia también apreciarán que, si bien cada anticuerpo individual se conjuga con un número entero de fracciones farmacológicas, como un todo, una composición particular de un ADC puede tener una RFA no entera, ya que la RFA refleja un promedio estadístico de las moléculas individuales dentro de la composición.
En algunos aspectos, los ADC contienen un anticuerpo desarrollado para unirse específicamente a cualquier antígeno de cáncer. En algunas realizaciones, los anticuerpos que pueden utilizarse en los ADC de la presente divulgación incluyen los que se unen específicamente a los siguientes antígenos: mesotelina, antígeno de membrana específico de la próstata (AMEP), CD19, CD22, CD30, CD70, B7H4 (también conocido como O8 E), proteína tirosina quinasa 7 (PTK7), glipicano-3, RG1, CTLA-4 y CD44. El anticuerpo puede ser animal (por ejemplo, murino), quimérico, humanizado o humano. En algunas realizaciones, el anticuerpo es un anticuerpo humano. En algunos aspectos, el anticuerpo es monoclonal. En algunas realizaciones, el anticuerpo es un anticuerpo monoclonal humano. La preparación de anticuerpos monoclonales humanos contra algunos de los antígenos mencionados anteriormente se divulga en Korman et al., documento US 2009/0074660 A1 (B7H4); Rao-Naik et al., documento 8.097.703 B2 (CD19); King et al., documento US 2010/0143368 A1 (CD22); Keler et al., documento US 7.387.776 B2 (2008) (CD30); Terrett et al., documento US 8.124.738 B2 y Coccia et al., documento US 2010/0150950 (2010) (CD70); Korman et al., documento US 6.984.720 B1 (2006) (CTLA-4); Korman et al., documento US 8.008.449 B2 (2011) (PD-1); Huang et al., documento US 2009/0297438 A1 y Cardarelli et al., documentos US 7.875.278 B2 (PSMA); Terrett et al., documento US 2010/0034826 A1 (PTK7); Terrett et al., documento US 2010/0209432 (A1) y Terrett et al., documento US 8.680.247 (2014) (glipicano-3); Harkins et al., documento US 7.335.748 B2(2008) (RG1); Terrett et al., documento US 8.268.970 B2 (2012) and Terrett et al., documento US 8.268.970 B2 (2012) (mesotelina); y Xu et al., documento US 2010/0092484 A1 (CD44).
D. Terapias de combinación
Es muy común en el campo de la terapia contra el cáncer combinar modalidades terapéuticas. Lo siguiente es una discusión general de terapias que pueden utilizarse junto con las terapias de la presente divulgación.
Para tratar cánceres utilizando los métodos y composiciones de la presente divulgación, generalmente se pondría en contacto una célula tumoral o sujeto con un análogo de uncialamicina o un conjugado de anticuerpo fármaco del análogo de uncialamicina, y al menos otra terapia. Estas terapias se proporcionarían en una cantidad combinada eficaz para lograr una reducción en uno o más parámetros de la enfermedad. Este procedimiento puede implicar poner en contacto las células/sujetos con ambos agentes/terapias al mismo tiempo, por ejemplo, usando una sola composición o formulación farmacológica que incluya ambos agentes, o poner en contacto la célula/sujeto con dos composiciones o formulaciones distintas, al mismo tiempo, en donde una composición incluye el análogo de uncialamicina o el conjugado de anticuerpo fármaco del análogo de uncialamicina y la otra incluye el otro agente.
Como alternativa, el análogo de uncialamicina o el conjugado de anticuerpo fármaco del análogo de uncialamicina pueden preceder o seguir al otro tratamiento en intervalos que varían de minutos a semanas. En general, se
aseguraría de que no pase un período de tiempo significativo entre el momento de cada administración, de tal manera que las terapias aún puedan ejercer un efecto combinado de forma ventajosa sobre la célula/sujeto. En tales casos, se contempla poner en contacto la célula con ambas modalidades dentro de aproximadamente las 12-24 horas, dentro de aproximadamente las 6-12 horas o con un tiempo de retraso de solo aproximadamente 12 horas. En algunas situaciones, sin embargo, puede ser conveniente prolongar significativamente el período de tiempo para el tratamiento, cuando transcurren varios días (2, 3, 4, 5, 6 o 7) a varias semanas (1, 2, 3, 4, 5, 6, 7 u 8) entre las administraciones respectivas.
Además, es concebible que se desee más de una administración del péptido o de la otra terapia. Se pueden emplear diversas combinaciones, donde un compuesto de la presente divulgación es "A" y la otra terapia es "B", como se ejemplifica a continuación:
A/B/A B/A/B B/B/A A/A/B B/A/A A/B/B B/B/B/A B/B/A/B
A/A/B/B A/B/A/B A/B/B/A B/B/A/A B/A/B/A B/A/A/B B/B/B/A
A/A/A/B B/A/A/A A/B/A/A A/A/B/A A/B/B/B B/A/B/B B/B/A/B
Se contemplan otras combinaciones. Lo siguiente es una discusión general de las terapias contra el cáncer que pueden utilizarse en combinación con los péptidos de la presente divulgación.
1. Quimioterapia
El término "quimioterapia" se refiere al uso de fármacos para tratar el cáncer. Se utiliza "agente quimioterapéutico" para denotar un compuesto o composición que se administra en el tratamiento del cáncer. Estos agentes o fármacos se clasifican por su modo de actividad dentro de una célula, por ejemplo, si afectan y en qué fase el ciclo celular. Como alternativa, un agente puede caracterizarse en función de su capacidad de reticular directamente el ADN, de intercalarse en el ADN o de inducir anomalía cromosómica y mitóticas al afectar la síntesis de ácido nucleico. La mayoría de los agentes quimioterapéuticos se dividen en las siguientes categorías: agentes alquilantes, antimetabolitos, antibióticos antitumorales, inhibidores mitóticos y nitrosoureas.
Los ejemplos de agentes quimioterapéuticos incluyen agentes alquilantes tales como tiotepa y ciclofosfamida; alquilsulfonatos tales como busulfán, improsulfán y piposulfán; aziridinas tales como benzodopa, carbocuona, meturedopa y uredopa; etileniminas y metilmelaminas incluyendo altretamina, trietilenmelamina, trietilenfosforamida, trietilentiofosforamida y trimetilolmelamina; acetogeninas (especialmente bullatacina y bullatacinona); una camptotecina (incluyendo el análogo sintético topotecán); briostatina; calistatina; CC-1065 (incluyendo sus análogos sintéticos adozelesina, carzelesina y bizelesina); criptoficinas (particularmente criptoficina 1 y criptoficina 8); dolastatina; duocarmicina (incluyendo los análogos sintéticos, KW-2189 y CB1-TM1); eleuterobina; pancratistatina; una sarcodictiina; espongistatina; mostazas nitrogenadas tales como clorambucilo, clornafazina, colofosfamida, estramustina, ifosfamida, mecloretamina, clorhidrato de óxido de mecloretamina, melfalán, novembiquina, fenesterina, prednimustina, trofosfamida, mostaza de uracilo; nitrosoureas, tales como carmustina, clorozotocina, fotemustina, lomustina, nimustina y ranimnustina; antibióticos tales como los antibióticos de enediina (por ejemplo, caliqueamicina, especialmente calicheamicina gammalI y calicheamicin omegaI1; dinemicina, incluyendo dinemicina A; bisfosfonatos, tal como clodronato; una esperamicina; así como cromóforo de neocarzinostatina y cromóforos de antibióticos de enediina relacionados con cromoproteína), aclacinomisinas, actinomicina, autrarnicina, azaserina, bleomicinas, cactinomicina, carabicina, carminomicina, carzinofilina, cromomicinas, dactinomicina, daunorrubicina, detorrubicina, 6-diazo-5-oxo-L-norleucina, doxorrubicina (incluyendo morfolino-doxorrubicina, cianomorfolinodoxorrubicina, 2-pirrolino-doxorrubicina y desoxidoxorrubicina), epirrubicina, esorrubicina, idarrubicina, marcelomicina, mitomicinas tales como mitomicina C, ácido micofenólico, nogalarnicina, olivomicinas, peplomicina, potfiromicina, puromicina, quelamicina, rodorrubicina, estreptonigrina, estreptozocina, tubercidina, ubenimex, zinostatina, zorrubicina; antimetabolitos tales como metotrexato y 5-fluorouracilo (5-FU); análogos del ácido fólico tales como denopterina, metotrexato, pteropterina, trimetrexato; análogos de purina tales como fludarabina, 6-mercaptopurina, tiamiprina, tioguanina; análogos de pirimidina tales como ancitabina, azacitidina, 6-azauridina, carmofur, citarabina, didesoxiuridina, doxifluridina, enocitabina, floxuridina; andrógenos, tales como calusterona, propionato de dromostanolona, epitiostanol, mepitiostano, testolactona; antisuprarrenales tales como aminoglutetimida, mitotano, trilostano; reabastecedor del ácido fólico tal como ácido frolínico; aceglatona; glucósido de aldofosfamida; ácido aminolevulínico; eniluracilo; amsacrina; bestrabucilo; bisantreno; edatraxato; defofamina; demecolcina; diazicuona; elformitina; acetato de eliptinio; una epotilona; etoglúcido; nitrato de galio; hidroxiurea; lentinano; lonidainina; maitansinoides tales como maitansina y ansamitocinas; mitoguazona; mitoxantrona; mopidanmol; nitraerina; pentostatina; fenamet; pirarrubicina; losoxantrona; ácido podofilínico; 2-etilhidrazida; procarbazina; complejo de polisacárido PSK); razoxana; rizoxina; sizofirano; espirogermanio; ácido tenuazónico; triazicuona; 2,2',2"-triclorotrietilamina; tricotecenos (especialmente toxina T-2, verracurina A, roridina A y anguidina); uretano; vindesina; dacarbazina; manomustina; mitobronitol; mitolactol; pipobromano; gacitosina; arabinósido ("Ara-C"); ciclofosfamida; tiotepa; taxoides, por ejemplo, paclitaxel y doxetaxel; clorambucilo; gemcitabina; 6-tioguanina; mercaptopurina; metotrexato; complejos de coordinación de platino como cisplatino, oxaliplatino y carboplatino; vinblastina; platino; etopósido (VP-16); ifosfamida; mitoxantrona; vincristina; vinorelbina; novantrona; tenipósido; edatrexato; daunomicina; aminopterina; xeloda; ibandronato; irinotecán (por ejemplo, CPT-11); inhibidor de la topoisomerasa RFS 2000; difluorometilomitina (DMFO); retinoides tales como ácido retinoico; capecitabina;
cisplatino (CDDP), carboplatino, procarbazina, mecloretamina, ciclofosfamida, camptotecina, ifosfamida, melfalán, clorambucilo, busulfán, nitrosurea, dactinomicina, daunorrubicina, doxorrubicina, bleomicina, plicomicina, mitomicina, etopósido (VP16), tamoxifeno, raloxifeno, agentes de unión a receptor de estrógeno, taxol, paclitaxel, docetaxel, gemcitabien, navelbina, inhibidores de la farnesil-proteína tansferasa, transplatino, 5-fluorouracilo, vincristina, vinblastina y metotrexato, y sales, ácidos o derivados farmacéuticamente aceptables de cualquiera de los anteriores.
2. Radioterapia
La radioterapia, también llamada terapia de radiación, es el tratamiento del cáncer y otras enfermedades con radiación ionizante. La radiación ionizante deposita energía que daña o destruye las células en el área tratada, al dañar su material genético, haciendo imposible que estas células continúen creciendo. Aunque la radiación daña tanto las células cancerosas como las células normales, estas últimas pueden repararse a sí mismos y funcionar correctamente.
La radioterapia utilizada de acuerdo con la presente divulgación puede incluir, pero sin limitación, el uso de rayos y, rayos X y/o el suministro dirigida de radioisótopos a las células tumorales. Además, se contemplan otras formas de factores que dañan el ADN, tales como las microondas y la radiación UV. Es muy probable que todos estos factores induzcan una amplia gama de daños en el ADN, en los precursores del ADN, en la replicación y reparación de ADN, y en el ensamblaje y mantenimiento de los cromosomas. Los intervalos de dosificación de rayos X varían de dosis diarias de 50 a 200 roentgens durante períodos prolongados de tiempo (3 a 4 semanas), a dosis únicas de 2000 a 6000 roentgens. Los intervalos de dosificación para los radioisótopos varían extensamente y dependen de la semivida del isótopo, de la fuerza y el tipo de radiación emitida, y de la captación por las células neoplásicas.
La radioterapia puede comprender el uso de anticuerpos radiomarcados para el suministro de dosis de radiación directamente al lugar del cáncer (radioinmunoterapia). Los anticuerpos son proteínas altamente específicas que el cuerpo produce en respuesta a la presencia de antígenos (sustancias reconocidas como extrañas por el sistema inmunitario). Algunas células tumorales contienen antígenos específicos que desencadenan la producción de anticuerpos específicos de tumor. Se pueden fabricar grandes cantidades de estos anticuerpos en el laboratorio y unirlos a sustancias radioactivas (un proceso conocido como radiomarcaje). Una vez inyectado en el cuerpo, los anticuerpos buscan activamente las células cancerosas, que son destruidas por la acción de destructora de las células (citotóxica) de la radiación. Este enfoque puede minimizar el riesgo de daño por radiación a las células sanas.
La radioterapia conformada utiliza la misma máquina de radioterapia, un acelerador lineal, que el tratamiento de radioterapia normal, pero se colocan bloques de metal en la trayectoria del haz de rayos X para modificar su forma para que coincida con la del cáncer. Esto garantiza que se está proporcionando al tumor una dosis de radiación más alta. Las células circundantes sanas y las estructuras cercanas reciben una dosis más baja de radiación, por lo que se reduce la posibilidad de efectos secundarios. Se ha desarrollado un dispositivo llamado colimador multiláminas y se puede utilizar como una alternativa a los bloques de metal. El colimador multiláminas consiste en varias láminas de metal que se fijan al acelerador lineal. Cada capa se puede ajustar para que los haces de la radioterapia puedan tener la forma del área de tratamiento, sin necesidad de bloques de metal. El posicionamiento preciso de la máquina de radioterapia es muy importante para el tratamiento de radioterapia conformada y se puede usar una máquina de exploración especial para verificar la posición de los órganos internos al comienzo de cada tratamiento.
La radioterapia de intensidad modulada de alta resolución también utiliza un colimador multiláminas. Durante este tratamiento, las capas del colimador multiláminas se mueven mientras se administra el tratamiento. Es probable que este método logre una conformación aún más precisa de los haces de tratamiento y permita que la dosis de radioterapia sea constante en toda el área de tratamiento.
Aunque los estudios de investigación han demostrado que la radioterapia conformada y la radioterapia de intensidad modulada pueden reducir los efectos secundarios del tratamiento con radioterapia, es posible que conformar el área de tratamiento con tanta precisión pueda mantener las células cancerosas microscópicas justo fuera del área de tratamiento que se está destruyendo. Esto significa que el riesgo de que el cáncer regrese en el futuro puede ser mayor con estas técnicas especializadas de radioterapia.
Los científicos también están buscando formas de aumentar la eficacia de la radioterapia. Se están estudiando dos tipos de fármacos en investigación por su efecto sobre las células que reciben radiación. Los radiosensibilizantes hacen que las células tumorales sean más propensas a sufrir daños, y los radioprotectores protegen a los tejidos normales de los efectos de la radiación. La hipertermia, el uso de calor, también se está estudiando por su eficacia en la sensibilización de los tejidos a la radiación.
3. Inmunoterapia
En el contexto del tratamiento del cáncer, los productos inmunoterápicos, en general, se basan en el uso de células y moléculas efectoras inmunitarias para dirigirse y destruir a las células cancerosas. Trastuzumab (Herceptin™) es un buen ejemplo. El efector inmunitario puede ser, por ejemplo, un anticuerpo específico para algún marcador de
superficie de una célula tumoral. El anticuerpo solo puede servir como un efector de la terapia o puede reunir otras células que efectúan realmente la destrucción celular. El anticuerpo también puede conjugarse con un fármaco o toxina (quimioterápico, radionúclido, la cadena A de ricina, toxina colérica, toxina pertúsica, etc.) y servir simplemente como un agente de direccionamiento. Como alternativa, el efector puede ser un linfocito que lleva una molécula de superficie que interactúa, tanto directa como indirectamente, con una diana de células tumorales. Las diversas células efectoras incluyen a los linfocitos T citotóxicos y los linfocitos NK. La combinación de modalidades terapéuticas, es decir, la actividad citotóxica directa y la inhibición o reducción de ErbB2, proporcionarían un beneficio terapéutico en el tratamiento de los cánceres que sobreexpresan ErbB2.
En un aspecto de la inmunoterapia, la célula tumoral debe portar algún marcador que pueda utilizarse como diana, es decir, no está presente en la mayoría de las otras células. Existen muchos marcadores tumorales y cualquiera de estos puede ser adecuado para el uso como diana en el contexto de la presente divulgación. Los marcadores tumorales comunes incluyen el antígeno carcinoembrionario, el antígeno específico de la próstata, el antígeno asociado a tumor urinario, el antígeno fetal, tirosinasa (p97), gp68, TAG-72, HMFG, el antígeno Sialil Lewis, MucA, MucB, PLAP, receptor de estrógenos, receptor de laminina, erb B y p155. Un aspecto alternativo de la inmunoterapia es combinar los efectos antineoplásicos con los efectos inmunoestimulantes. Además, existen moléculas inmunoestimulantes, entre ellas: citocinas tales como IL-2, IL-4, IL-12, GM-CSF, y-IFN, quimiocinas tales como MIP-1, MCP-1, IL-8 y factores de crecimiento tales como el ligando de FLT3. Se ha demostrado que la combinación de moléculas inmunoestimulantes, ya sea como proteínas o mediante el suministro de genes en combinación con un supresor tumoral, aumenta los efectos antitumorales (Ju et al. 2000). Además, los anticuerpos contra cualquiera de estos compuestos pueden utilizarse para direccionar a los agentes antineoplásicos discutidos en el presente documento.
Los ejemplos de inmunoterapias actualmente bajo investigación o en uso son, los adyuvantes inmunitarios por ejemplo, Mycobacterium bovis, Plasmodium falciparum, dinitroclorobenceno y compuestos aromáticos (Patentes de Estados Unidos 5.801.005 y 5.739.169; Hui y Hashimoto, 1998; Christodoulides et al., 1998), la terapia con citocinas, por ejemplo, interferones a, p y y; IL-1, GM-CSF y TNF (Bukowski et al., 1998; Davidson et al., 1998; Hellstrand et al., 1998), la terapia génica, por ejemplo, TNF, IL-1, lL-2, p53 (Qin et al. 1998; Austin-Ward y Villaseca, 1998; Patentes de Estados Unidos 5.830.880 y 5.846.945) y los anticuerpos monoclonales, por ejemplo, anti-gangliósido GM2, anti-HER-2, anti-p185 (Pietras et al., 1998; Hanibuchi et al., 1998; Patente de Estados Unidos 5.824.311). Se contempla que se puedan emplear una o más terapias contra el cáncer con las terapias de silenciamiento génico descritas en el presente documento.
En la inmunoterapia activa, se administra un péptido, polipéptido o proteína antigénica, o una composición de células tumorales autólogas o alogénicas o "vacuna", generalmente con un adyuvante bacteriano distinto (Ravindranath y Morton, 1991; Morton et al., 1992; Mitchell et al., 1990; Mitchell et al., 1993).
En la inmunoterapia adoptiva, los linfocitos circulantes del paciente, o linfocitos infiltrados en el tumor, se aíslan in vitro, se activan mediante linfocinas tales como IL-2 o se transducen con genes para la necrosis tumoral, y se readministran (Rosenberg et al. 1988; 1989).
4. Cirugía
Aproximadamente el 60 % de las personas con cáncer se someterán a una cirugía de algún tipo, que incluye la cirugía preventiva, diagnóstica o de estadificación, curativa y paliativa. La cirugía curativa es un tratamiento contra el cáncer que se puede usar junto con otras terapias, tales como el tratamiento de la presente divulgación, quimioterapia, radioterapia, terapia hormonal, terapia génica, inmunoterapia y/o terapias alternativas.
La cirugía curativa incluye la resección en la que todo o parte del tejido canceroso se extrae, extirpa y/o destruye físicamente. Resección tumoral se refiere a la extirpación física de al menos parte de un tumor. Además de la resección tumoral, el tratamiento quirúrgico incluye cirugía láser, criocirugía, electrocirugía y cirugía controlada microscópicamente (cirugía de Mohs). Se contempla además que la presente divulgación pueda utilizarse junto con la eliminación de cánceres superficiales, de precánceres o de cantidades menores de tejido normal.
Tras la escisión de parte o de la totalidad de las células, tejidos o tumores cancerosos, se puede formar una cavidad en el cuerpo. El tratamiento puede realizarse por perfusión, inyección directa o aplicación local al área con una terapia antineoplásica adicional. Dicho tratamiento puede repetirse, por ejemplo, cada 1, 2, 3, 4, 5, 6 o 7 días, o cada 1, 2, 3, 4 y 5 semanas o cada 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11 o 12 meses. Estos tratamientos pueden ser, además, de dosificaciones variables.
5. Otros agentes
Se contempla que con la presente divulgación se puedan utilizar otros agentes. Estos agentes adicionales incluyen agentes inmunomoduladores, agentes que afectan la regulación al alza de los receptores de superficie celular y las uniones GAP, agentes citostáticos y de diferenciación, inhibidores de la adhesión celular, agentes que aumentan la sensibilidad de las células hiperproliferativas a inductores apoptóticos u otros agentes biológicos. Los agentes
inmunomoduladores incluyen el factor de necrosis tumoral; interferón alfa, beta y gamma; IL-2 y otras citocinas; F42K y otros análogos de citocina, o MIP-1, MIP-1 p, MCP-1, RANTES y otras quimiocinas. Se contempla además que la regulación al alza de los receptores de superficie celular o sus ligandos, tales como Fas/ligando de Fas, DR4 o DR5/TRAIL (ligando Apo-2) potenciaría las capacidades inductoras apoptóticas de la presente invención mediante el establecimiento de un efecto autocrino o paracrino sobre las células hiperproliferativas. Los aumentos en la señalización intercelular al elevar el número de uniones GAP aumentarían los efectos anti hiperproliferativos en la población de células hiperproliferativas vecinas. En otras realizaciones, los agentes citostáticos o de diferenciación se pueden usar en combinación con la presente divulgación para mejorar la eficacia anti proliferativa de los tratamientos. Se contemplan inhibidores de la adhesión celular para mejorar la eficacia de la presente divulgación. Los ejemplos de inhibidores de la adhesión celular son los inhibidores de la quinasa de adhesión focal (FAK) y la lovastatina. Se contempla además que otros agentes que aumentan la sensibilidad de una célula hiperproliferativa a la apoptosis, tal como el anticuerpo c225, puedan utilizarse en combinación con la presente divulgación para mejorar la eficacia del tratamiento.
Ha habido muchos avances en la terapia del cáncer a continuación de la introducción de los fármacos quimioterapéuticos citotóxicos. Sin embargo, una de las consecuencias de la quimioterapia es el desarrollo/adquisición de fenotipos resistentes a los fármacos y el desarrollo de multirresistencia. El desarrollo de resistencia a los fármacos sigue siendo un obstáculo importante en el tratamiento de tales tumores y, por lo tanto, existe una necesidad obvia de enfoques alternativos, tales como la terapia génica.
Otra forma de terapia para su uso junto con quimioterapia, radioterapia o terapia biológica incluye la hipertermia, que es un procedimiento en el cual el tejido de un paciente está expuesto a altas temperaturas (hasta 41 °C (106 °F)). Pueden estar implicados en la aplicación de hipertermia local, regional o de cuerpo entero los dispositivos de calentamiento externo o interno. La hipertermia local implica la aplicación de calor en un área pequeña, tal como un tumor. El calor puede generarse externamente con ondas de alta frecuencia dirigidas a un tumor desde un dispositivo fuera del cuerpo. El calor interno puede implicar una sonda estéril, que incluye cables delgados con calor, o tubos huecos llenos de agua tibia, antenas de microondas implantadas o electrodos de radiofrecuencia.
El órgano o la extremidad de un paciente se calienta para terapia regional, lo que se logra utilizando dispositivos que producen alta energía, tales como imanes. Como alternativa, parte de la sangre del paciente puede extraerse y calentarse antes de perfundirse en un área, la cual se calentará internamente. También se puede implementar el calentamiento del cuerpo completo en los casos en que el cáncer se haya diseminado por todo el cuerpo. Para este fin, se pueden utilizar mantas de agua tibia, cera caliente, bobinas inductoras y cámaras térmicas.
El experto en la materia consulta la 15a edición de "Remington's Pharmaceutical Sciences", capítulo 33, en particular las páginas 624-652. Se producirá necesariamente alguna variación en la dosificación dependiendo de la afección del sujeto que se está tratando. La persona responsable de la administración, en cualquier caso, determinará la dosis apropiada para el sujeto individual. Además, para la administración a seres humanos, los preparados deben cumplir con los criterios de esterilidad, pirogenicidad, seguridad general y pureza, según lo exigen los criterios de la Oficina de biofármacos de la FDA.
También debe señalarse que cualquiera de las terapias anteriores puede resultar útil por sí misma en el tratamiento del cáncer.
6. Antibióticos
El término "antibióticos" se refiere a fármacos que pueden utilizarse para tratar una infección bacteriana ya sea inhibiendo el crecimiento de las bacterias o destruyendo bacterias. Sin quedar ligados a teoría alguna, se cree que los antibióticos se pueden clasificar en dos clases principales: los agentes bactericidas que destruyen bacterias o los agentes bacteriostáticos que ralentizan o impiden el crecimiento de las bacterias.
En algunas realizaciones, los presentes compuestos se administran en combinación con uno o más antibióticos adicionales. En algunas realizaciones, los antibióticos pueden encontrarse en una amplia gama de clases. En algunas realizaciones, los compuestos de la presente divulgación pueden utilizarse con otro antibiótico. En algunas realizaciones, los compuestos pueden utilizarse junto con un antibiótico de espectro limitado que se dirige a un tipo de bacteria específico. En algunos ejemplos no limitantes de antibióticos bactericidas se incluyen, penicilina, cefalosporina, polimixina, rifamicina, lipiarmicina, quinolonas y sulfonamidas. En algunos ejemplos no limitantes de antibióticos bacteriostáticos se incluyen, macrólidos, lincosamidas o tetraciclinas. En algunas realizaciones, el antibiótico es un aminoglucósido tal como kanamicina y estreptomicina, una ansamicina tal como rifaximina y geldanamicina, un carbacefem tal como loracarbef, un carbapenem tal como ertapenem, imipenem, una cefalosporina tal como cefalexina, cefixima, cefepima y ceftobiprol, un glucopéptido tal como vancomicina o teicoplanina, una lincosamida tal como lincomicina y clindamicina, un lipopéptido tal como la daptomicina, un macrólido tal como la claritromicina, espiramicina, azitromicina y telitromicina, un monobactam tal como aztreonam, un nitrofurano tal como furazolidona y nitrofurantoína, oxazolidononas tales como linezolid, una penicilina tal como la amoxicilina, azlocilina, flucloxacilina y penicilina G, un polipéptido antibiótico tal como la bacitracina, polimixina B y colistina, una quinolona tal como ciprofloxacina, levofloxacina y gatifloxacina, una sulfonamida tal como la
sulfadiazina de plata, mefenida, sulfadimetoxina o sulfasalazina, o una tetraciclina tal como demeclociclina, doxiciclina, minociclina, oxitetraciclina o tetraciclina. En algunas realizaciones, los compuestos podrían combinarse con un fármaco que actúe contra las micobacterias, tal como cicloserina, capreomicina, etionamida, rifampicina, rifabutina, rifapentina y estreptomicina. Otros antibióticos que se contemplan para las terapias combinadas pueden incluir arsfenamina, cloranfenicol, fosfomicina, ácido fusídico, metronidazol, mupirocina, platensimicina, quinupristina, dalfopristina, tianfenicol, tigeciclina, tinidazol o trimetoprima.
IV. Métodos de síntesis
En algunos aspectos, los compuestos de la presente divulgación pueden sintetizarse utilizando los métodos de química orgánica como se describe en la presente solicitud. Estos métodos pueden modificarse y optimizarse aún más utilizando los principios y técnicas de la química orgánica aplicados por una persona experta en la técnica. Dichos principios y técnicas se muestran, por ejemplo, en March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure (2007).
1. Aumento de escala del proceso
Los métodos de síntesis descritos en el presente documento pueden modificarse y optimizarse adicionalmente para la producción preparativa, preliminar o a gran escala, ya sea discontinua o continua, utilizando los principios y técnicas de la química de procesos aplicados por una persona experta en la materia. Dichos principios y técnicas se muestran, por ejemplo, en Practical Process Research & Development (2000). El método de síntesis descrito en el presente documento podría utilizarse para producir cantidades a escala preparativa de uncialamicina y derivados de la misma.
2. Definiciones químicas
Cuando se usa en el contexto de un grupo químico: "hidrógeno" significa -H; "hidroxi" significa -OH; "oxo" significa =O; "carbonilo" significa -C(=O)-; "carboxi" significa -C(=O)OH (también escrito como -COOH o -CO2H); "halo" significa independientemente -F, -Cl, -Br o -I; "amino" significa -NH2; "hidroxiamino" significa -NHOH; "nitro" significa -NO2; imino significa =NH; "ciano" significa -CN; "isocianato" significa -N=C=O; "azido" significa -N3; en un contexto monovalente "fosfato" significa -OP(O)(OH)2 o una forma desprotonada del mismo; en un contexto divalente "fosfato" significa -OP(O)(OH)O- o una forma desprotonada del mismo; "mercapto" significa -SH; y "tio" significa =S; "sulfonilo" significa -S(O)2-; y "sulfinilo" significa -S(O)-.
En el contexto de fórmulas químicas, el símbolo "-" significa un enlace sencillo, "=" significa un doble enlace, y "=" significa un triple enlace. El símbolo "—" representa un enlace opcional, que, si está presente, es sencillo o doble. El símbolo " representa un enlace sencillo o un doble enlace. Por lo tanto, por ejemplo, la fórmula

incluye

Y se entiende que ninguno de dichos átomos de anillo forma parte de más de un doble enlace. Además, se aprecia que el símbolo de enlace covalente "-", cuando conecta uno o dos átomos estereogénicos, no indica ninguna estereoquímica preferida. En su lugar, incluye todos los estereoisómeros así como mezclas de los mismos. El símbolo "'A /v v ', cuando se dibuja perpendicularmente a través de un enlace (por ejemplo,

para metilo) indica un punto de unión del grupo. Se aprecia que el punto de unión solo se identifica normalmente de esta manera para grupos más grandes con el fin de ayudar al lector a identificar de forma inequívoca un punto de unión. El símbolo significa un enlace sencillo donde el grupo unido al extremo más grueso de la cuña está "fuera de la página". El símbolo "’M||II " significa un enlace sencillo donde el grupo unido al extremo más grueso de la cuña está "en la página". El símbolo "JVVV" significa un enlace sencillo donde la geometría alrededor de un doble enlace (por ejemplo, E o Z) está sin definir. Ambas opciones, así como combinaciones de las mismos, están contempladas. Cualquier valencia indefinida en un átomo de una estructura mostrada en esta solicitud representa implícitamente un átomo de hidrógeno unido a ese átomo. Un punto en negrita en un átomo de carbono indica que el
hidrógeno unido a ese carbono está orientado hacia fuera del plano del papel.
Cuando un grupo "R" se representa como un "grupo flotante" en un sistema de anillo, por ejemplo, en la fórmula:

entonces R puede reemplazar a cualquier átomo de hidrógeno unido a cualquiera de los átomos en el anillo, incluyendo un hidrógeno representado, implicado o definido expresamente, siempre que se forme una estructura estable. Cuando un grupo "R" se representa como un "grupo flotante" en un sistema de anillo condensado, como por ejemplo en la fórmula:

entonces R puede reemplazar a cualquier hidrógeno unido a cualquiera de los átomos en el anillo de cualquiera de los anillos condensados, a menos que se especifique lo contrario. Los hidrógenos reemplazables incluyen los hidrógenos representados (por ejemplo, el hidrógeno unido al nitrógeno de la fórmula anterior), hidrógenos implicados (por ejemplo, un hidrógeno de la fórmula anterior que no se ha mostrado pero que se entiende que está presente), hidrógenos definidos expresamente e hidrógenos opcionales cuya presencia depende de la identidad de un átomo del anillo (por ejemplo, un hidrógeno unido al grupo X, cuando X es igual a -CH-), siempre que se forme una estructura estable. En el ejemplo representado, R puede residir en el anillo de 5 miembros o de 6 miembros del sistema de anillo condensado. En la fórmula anterior, el subíndice "y" inmediatamente después del grupo "R" entre paréntesis, representa una variable numérica. A menos que se especifique otra cosa, esta variable puede ser 0, 1, 2 o cualquier número entero mayor de 2, limitado únicamente por el número máximo de átomos de hidrógeno reemplazables del anillo o sistema de anillos.
Para los grupos y clases que se indican a continuación, los siguientes subíndices entre paréntesis definen adicionalmente el grupo/clase como se indica a continuación: "(Cn)" define el número exacto (n) de átomos de carbono en el grupo/clase. "(C<n)" define el número máximo (n) de átomos de carbono que puede haber en el grupo/clase, siendo el número mínimo tan pequeño como sea posible para el grupo en cuestión, por ejemplo, se entiende que el número mínimo de átomos de carbono en el grupo "alquenilo(c<8)" o la clase "alqueno(c<8)" es dos. Por ejemplo, "alcoxi(c<io)" designa aquellos grupos alcoxi que tienen de 1 a 10 átomos de carbono. (Cn-n') define tanto el número mínimo (n) como el máximo (n') de átomos de carbono en el grupo. Análogamente, "alquilo(C2-io)" designa aquellos grupos alquilo que tienen de 2 a 10 átomos de carbono. Además, los límites de carbono también se pueden expresar como intervalos antes de la variable. Por ejemplo, arilo C6-C12 representa un grupo arilo que tiene un mínimo de 6 átomos de carbono y un máximo de doce átomos de carbono. Un experto en la materia apreciará que todas estas expresiones de limitaciones de carbono sin las mismas y pueden usarse indistintamente.
El término "saturado", como se usa en el presente documento, significa que el compuesto o grupo así modificado no tiene dobles enlaces carbono-carbono ni triples enlaces carbono-carbono, excepto como se indica a continuación. En el caso de versiones sustituidas de grupos saturados, pueden estar presentes uno o más dobles enlaces carbono oxígeno o un doble enlace carbono nitrógeno. Y cuando dicho enlace está presente, entonces no se impiden los dobles enlaces carbono-carbono que pueden aparecer como parte de la tautomería ceto-enol o tautomería imina/enamina.
El término "alifático", cuando se usa sin el modificador "sustituido" significa que el compuesto/grupo así modificado es un compuesto o grupo de hidrocarburo, acíclico o cíclico, pero no aromático. En compuestos/grupos alifáticos, los átomos de carbono pueden unirse formando cadenas lineales, cadenas ramificadas o anillos no aromáticos (alicíclicos). Los compuestos/grupos alifáticos pueden estar saturados, es decir, unidos por enlaces simples (alcanos/alquilo), o insaturados, con uno o más dobles enlaces (alquenos/alquenilo) o con uno o más triples enlaces (alquinos/alquinilo).
El término "alquilo", cuando se usa sin el modificador "sustituido", se refiere a un grupo alifático saturado monovalente con un átomo de carbono como punto de unión, una estructura lineal o ramificada, ciclo, cíclica o acíclica, y sin átomos que no sean carbono e hidrógeno. Por lo tanto, como se usa en el presente documento, cicloalquilo es un subconjunto de alquilo, en donde el átomo de carbono que forma el punto de unión también es un miembro de una o más estructuras de anillos no aromáticos en donde el grupo cicloalquilo no consiste en átomos que no sean carbono e hidrógeno. Como se usa en el presente documento, el término no excluye la presencia de uno o más grupos alquilo (lo que permita la limitación del número de carbonos) unidos al anillo o sistema de anillos.
Los grupos -CH3 (Me), -CH2CH3 (Et), -CH2CH2CH3 (n-Pr o propilo), -CH(CH3)2 (i-Pr, 'Pr o isopropilo), -CH(CH2)2 (ciclopropilo), -CH2CH2CH2CH3 (n-Bu), -CH(CH3)CH2CH3 (sec-butilo), -CH2CH(CH3)2 (isobutilo), -C(CH3)3 (ferc-butilo, f-butilo, f-Bu o fBu), -CH2C(CH3)3 (neo-pentilo), ciclobutilo, ciclopentilo, ciclohexilo y ciclohexilmetilo son ejemplos no limitantes de grupos alquilo. El término "alcanodiílo", cuando se usa sin el modificador "sustituido", se refiere a un grupo alifático saturado divalente, con uno o dos átomos de carbono saturados como punto(s) de unión, una estructura lineal o ramificada, ciclo, cíclica o acíclica, sin dobles o triples enlaces carbono-carbono, y sin átomos que no sean carbono e hidrógeno. Los grupos -CH2-(metileno), -CH2CH2-, -CH2C(CH3)2CH2-, -CH2CH2CH2- y

son ejemplos no limitantes de grupos alcanodiílo. El término "alquilideno", cuando se usa sin el modificador "sustituido", se refiere al grupo divalente =CRR' en el que R y R' son independientemente hidrógeno, alquilo, o R y R' se toman juntos para representar un alcanodiílo que tiene al menos dos átomos de carbono. Los ejemplos no limitantes de grupos alquilideno incluyen: =CH2, =c H(CH2CH3) y =C(CH3)2. Un "alcano" se refiere al compuesto H-R, en donde R es alquilo tal como este término se ha definido anteriormente. Cuando cualquiera de estos términos se usa con el modificador "sustituido", uno o más átomos de hidrógeno se han reemplazado independientemente por -OH, -F, -Cl, -Br, -I, -NH2, -NO2, -CO2H, -CO2CH3, -CN, -SH, -OCH3, -OCH2CH3, -C(O)CH3, -NHCH3, -NHCH2CH3, -N(CH3)2, -C(O)NH2, -OC(O)CH3 o -S(O)2NH2. Los siguientes grupos son ejemplos no limitantes de grupos alquilo sustituido: -CH2OH, -CH2Cl, -CF3, -CH2CN, -CH2C(O)OH, -CH2C(O)OCH3, -CH2C(O)NH2, -CH2C(O)CH3, -CH2OCH3, -CH2OC(O)CH3, -CH2NH2, -CH2N(CH3)2 y -CH2CH2Cl. El término "haloalquilo" es un subconjunto de alquilo sustituido, en el que uno o más átomos de hidrógeno se han sustituido por un grupo halo y no está presente ningún otro átomo aparte de carbono, hidrógeno y halógeno. El grupo -CH2Cl es un ejemplo no limitante de un haloalquilo. El término "fluoroalquilo" es un subconjunto de alquilo sustituido, en el que uno o más hidrógenos se han sustituido por un grupo fluoro y no está presente ningún otro átomo aparte de carbono, hidrógeno y flúor. Los grupos -CH2F, -CF3 y -CH2CF3 son ejemplos no limitantes de grupos fluoroalquilo.
El término "alquenilo", cuando se usa sin el modificador "sustituido", se refiere a un grupo alifático insaturado monovalente con un átomo de carbono como punto de unión, una estructura lineal o ramificada, ciclo, cíclica o acíclica, al menos un doble enlace carbono-carbono no aromático, sin triples enlaces carbono-carbono, y sin átomos que no sean carbono e hidrógeno. Los ejemplos no limitantes de grupos alquenilo incluyen: -CH=CH2 (vinilo), -CH=CHCH3, -CH=CHCH2CH3, -CH2CH=CH2 (alilo), -CH2CH=CHCH3 y -CH=CHCH=CH2. El término "alquenodiílo", cuando se usa sin el modificador "sustituido", se refiere a un grupo alifático insaturado divalente, con dos átomos de carbono como puntos de unión, una estructura lineal o ramificada, ciclo, cíclica o acíclica, al menos un doble enlace carbono-carbono no aromático, sin triples enlaces carbono-carbono, y sin átomos que no sean carbono e hidrógeno. Los grupos -CH=CH-, -CH=C(CH3)CH2-, -CH=CHCH2- y

son ejemplos no limitantes de grupos alquenodiílo. Debe apreciarse que, aunque el grupo alquenodiílo es alifático, una vez que se conecta en ambos extremos, este grupo no se excluye de formar parte de una estructura aromática. Los términos "alqueno" u "olefina" son sinónimos y se refieren a un compuesto que tiene la fórmula H-R, en donde R es alquenilo tal como este término se ha definido anteriormente. Un "alqueno terminal" se refiere a un alqueno que tiene solo un doble enlace carbono-carbono, en donde ese enlace forma un grupo vinilo en el extremo de la molécula. Cuando cualquiera de estos términos se usa con el modificador "sustituido", uno o más átomos de hidrógeno se han reemplazado independientemente por -OH, -F, -Cl, -Br, -I, -NH2, -NO2, -CO2H, -CO2CH3, -CN, -SH, -OCH3, -OCH2CH3, -C(O)CH3, -NHCH3, -NHCH2CH3, -N(CH3)2, -C(O)NH2, -OC(O)CH3 o -S(O)2NH2. Los grupos -CH=CHF, -CH=CHCl y -CH=CHBr, son ejemplos no limitantes de grupos alquenilo sustituido.
El término "alquinilo", cuando se usa sin el modificador "sustituido", se refiere a un grupo alifático insaturado monovalente con un átomo de carbono como punto de unión, una estructura lineal o ramificada, ciclo, cíclica o acíclica, al menos un triple enlace carbono-carbono, y sin átomos que no sean carbono e hidrógeno. Como se usa en el presente documento, el término alquinilo no excluye la presencia de uno o más dobles enlaces carbonocarbono no aromáticos. Los grupos -C=CH, -C=CCH3 y -CH2CECCH3, son ejemplos no limitantes de grupos alquinilo. Un "alquino" se refiere al compuesto H-R, en donde R es alquinilo. Cuando cualquiera de estos términos se usa con el modificador "sustituido", uno o más átomos de hidrógeno se han reemplazado independientemente por -OH, -F, -Cl, -Br, -I, -NH2, -NO2, -CO2H, -CO2CH3, -CN, -SH, -OCH3, -OCH2CH3, -C(O)CH3, -NHCH3, -NHCH2CH3, -N(CH3)2, -C(O)NH2, -OC(O)CH3 o -S(O)2NH2.
El término "arilo", cuando se usa sin el modificador "sustituido", se refiere a un grupo aromático insaturado monovalente con un átomo de carbono aromático como punto de unión, formando parte dicho átomo de carbono de una o más estructuras de anillos aromáticos de seis miembros, en donde los átomos en el anillo son todos carbono, y en donde el grupo no consiste en átomos que no sean carbono e hidrógeno. Si está presente más de un anillo, los
anillos pueden estar condensados o no condensados. Como se usa en el presente documento, el término no excluye la presencia de uno o más grupos alquilo o aralquilo (lo que permita la limitación del número de carbonos) unidos al primer anillo aromático o a cualquier anillo aromático adicional presente. Los ejemplos no limitantes de grupos arilo incluyen fenilo (Ph), metilfenilo, (dimetil)fenilo, -C6H4CH2CH3 (etilfenilo), naftilo y un grupo monovalente derivado de bifenilo. El término "arenodiílo", cuando se usa sin el modificador "sustituido", se refiere a un grupo aromático divalente con dos átomos de carbono aromáticos como puntos de unión, formando parte dichos átomos de carbono de una o más estructuras de anillos aromáticos de seis miembros en donde los átomos en el anillo son todos carbono, y en donde el grupo monovalente no consiste en átomos que no sean carbono e hidrógeno. Como se usa en el presente documento, el término no excluye la presencia de uno o más grupos alquilo, arilo o aralquilo (lo que permita la limitación del número de carbonos) unidos al primer anillo aromático o a cualquier anillo aromático adicional presente. Si está presente más de un anillo, los anillos pueden estar condensados o no condensados. Los anillos no condensados pueden conectarse a través de uno o más de los siguientes: un enlace covalente, grupos alcanodiílo o alquenodiílo (lo que permita la limitación del número de carbonos). Los ejemplos no limitantes de grupos arenodiílo grupos incluyen:

Benceno y tolueno son ejemplos no limitantes de arenos. Cuando cualquiera de estos términos se usa con el modificador "sustituido", uno o más átomos de hidrógeno se han reemplazado independientemente por -OH, -F, -Cl, -Br, -I, -NH2, -NO2, -CO2H, -CO2CH3, -CN, -SH, -OCH3, -OCH2CH3, -C(O)CH3, -NHCH3, -NHCH2CH3, -N(CH3)2, -C(O)NH2, -OC(O)CH3 o -S(O)2NH2.
El término "aralquilo", cuando se usa sin el modificador "sustituido", se refiere al grupo monovalente -alcanodiil-arilo, en el que cada uno de los términos alcanodiílo y arilo se usa de manera coherente con las definiciones proporcionadas en el presente documento. Son ejemplos no limitantes de aralquilos: fenilmetilo (bencilo, Bn) y 2-feniletilo. Cuando el término aralquilo se usa con el modificador "sustituido", uno o más átomos de hidrógeno del alcanodiílo y/o del grupo arilo se han reemplazado independientemente por -OH, -F, -Cl, -Br, -I, -NH2, -NO2, -CO2H, -CO2CH3, -CN, -SH, -OCH3, -OCH2CH3, -C(O)CH3, -NHCH3, -NHCH2CH3, -N(CH3)2, -C(O)NH2, -OC(O)CH3 o -S(O)2NH2. Son ejemplos no limitantes de aralquilos sustituidos: (3-clorofenil)-metilo y 2-cloro-2-fenil-et-1-ilo.
El término "heteroarilo", cuando se usa sin el modificador "sustituido", se refiere a un grupo aromático monovalente con un átomo de nitrógeno o un átomo de carbono aromático como punto de unión, formando parte dicho átomo de carbono o átomo de nitrógeno de una o más estructuras de anillos aromáticos en donde al menos uno de los átomos en el anillo es nitrógeno, oxígeno o azufre, y en donde el grupo heteroarilo no consiste en átomos que no sean carbono, hidrógeno, nitrógeno aromático, oxígeno aromático y azufre aromático. Si está presente más de un anillo, los anillos pueden estar condensados o no condensados. Como se usa en el presente documento, el término no excluye la presencia de uno o más grupos alquilo, arilo y/o aralquilo (lo que permita la limitación del número de carbonos) unidos al anillo aromático o al sistema de anillos aromáticos. Los ejemplos no limitantes de grupos heteroarilo incluyen furanilo, imidazolilo, indolilo, indazolilo (Im), isoxazolilo, metilpiridinilo, oxazolilo, fenilpiridinilo, piridinilo, pirrolilo, pirimidinilo, pirazinilo, quinolilo, quinazolilo, quinoxalinilo, triazinilo, tetrazolilo, tiazolilo, tienilo y triazolilo. El término "W-heteroarilo" se refiere a un grupo heteroarilo con un átomo de nitrógeno como punto de unión. El término "heteroarenodiílo", cuando se usa sin el modificador "sustituido", se refiere a un grupo aromático divalente, con dos átomos de carbono aromáticos, dos átomos de nitrógeno aromáticos o un átomo de carbono aromático y un átomo de nitrógeno aromático como los dos puntos de unión, formando parte dichos átomos de una o más estructuras de anillos aromáticos en donde al menos uno de los átomos en el anillo es nitrógeno, oxígeno o azufre, y en donde el grupo divalente no consiste en átomos que no sean carbono, hidrógeno, nitrógeno aromático, oxígeno aromático y azufre aromático. Si está presente más de un anillo, los anillos pueden estar condensados o no condensados. Los anillos no condensados pueden conectarse a través de uno o más de los siguientes: un enlace covalente, grupos alcanodiílo o alquenodiílo (lo que permita la limitación del número de carbonos). Como se usa en el presente documento, el término no excluye la presencia de uno o más grupos alquilo, arilo y/o aralquilo (lo que permita la limitación del número de carbonos) unidos al anillo aromático o al sistema de anillos aromáticos. Los ejemplos no limitantes de grupos heteroarenodiílo incluyen:

Un "heteroareno" se refiere al compuesto H-R, en donde R es heteroarilo. Piridina y quinolina son ejemplos no limitantes de heteroarenos. Cuando estos términos se usan con el modificador "sustituido", uno o más átomos de hidrógeno se han reemplazado independientemente por -OH, -F, -Cl, -Br, -I, -NH2, -NO2, -CO2H, -CO2CH3, -CN, -SH, -OCH3, -OCH2CH3, -C(O)CH3, -NHCH3, -NHCH2CH3, -N(CH3)2, -C(O)NH2, -OC(O)CH3 o -S(O)2NH2. Cuando estos términos se usan con el modificador "sustituido", uno o más átomos de hidrógeno en el heteroátomo se han reemplazado por un grupo protector apropiado, por ejemplo un átomo de hidrógeno unido a una amina puede estar reemplazado por un grupo protector de amina tal como se ha definido ese grupo.
El término "heterocicloalquilo", cuando se usa sin el modificador "sustituido", se refiere a un grupo no aromático monovalente con un átomo de carbono o un átomo de nitrógeno como punto de unión, formando parte dicho átomo de carbono o átomo de nitrógeno de una o más estructuras de anillos no aromáticos en donde al menos uno de los átomos en el anillo es nitrógeno, oxígeno o azufre, y en donde el grupo heterocicloalquilo no consiste en átomos que no sean carbono, hidrógeno, nitrógeno, oxígeno y azufre. Si está presente más de un anillo, los anillos pueden estar condensados o no condensados. Como se usa en el presente documento, el término no excluye la presencia de uno o más grupos alquilo (lo que permita la limitación del número de carbonos) unidos al anillo o sistema de anillos. Asimismo, el término no excluye la presencia de uno o más dobles enlaces en el anillo o sistema de anillos, con la condición de que el grupo resultante siga siendo no aromático. Los ejemplos no limitantes de grupos heterocicloalquilo incluyen aziridinilo, azetidinilo, pirrolidinilo, piperidinilo, piperazinilo, morfolinilo, tiomorfolinilo, tetrahidrofuranilo, tetrahidrotiofuranilo, tetrahidropiranilo, piranilo, oxiranilo y oxetanilo. El término "N-heterocicloalquilo" se refiere a un grupo heterocicloalquilo con un átomo de nitrógeno como punto de unión. El término "heterocicloalcanodiílo", cuando se usa sin el modificador "sustituido", se refiere a un grupo cíclico divalente, con dos átomos de carbono, dos átomos de nitrógeno o un átomo de carbono y un átomo de nitrógeno como los dos puntos de unión, formando parte dichos átomos de una o más estructuras de anillos en donde al menos uno de los átomos en el anillo es nitrógeno, oxígeno o azufre, y en donde el grupo divalente no consiste en átomos que no sean carbono, hidrógeno, nitrógeno, oxígeno y azufre. Si está presente más de un anillo, los anillos pueden estar condensados o no condensados. Los anillos no condensados pueden conectarse a través de uno o más de los siguientes: un enlace covalente, grupos alcanodiílo o alquenodiílo (lo que permita la limitación del número de carbonos). Como se usa en el presente documento, el término no excluye la presencia de uno o más grupos alquilo (lo que permita la limitación del número de carbonos) unidos al anillo o sistema de anillos. Asimismo, el término no excluye la presencia de uno o más dobles enlaces en el anillo o sistema de anillos, con la condición de que el grupo resultante siga siendo no aromático. Los ejemplos no limitantes de grupos heterocicloalcanodiílo incluyen:

Cuando estos términos se usan con el modificador "sustituido", uno o más átomos de hidrógeno se han reemplazado independientemente por -OH, -F, -Cl, -Br, -I, -NH2, -NO2, -CO2H, -CO2CH3, -CN, -SH, -OCH3, -OCH2CH3, -C(O)CH3, -NHCH3, -NHCH2CH3, -N(CH3)2, -C(O)NH2, -OC(O)CH3, -S(O)2NH2 o -C(O)OC(CH3)3 (ferc-butiloxicarbonilo, BOC). Cuando estos términos se usan con el modificador "sustituido", uno o más átomos de hidrógeno en el heteroátomo se han reemplazado por un grupo protector apropiado, por ejemplo un átomo de hidrógeno unido a una amina puede estar reemplazado por un grupo protector de amina tal como se ha definido ese grupo.
El término "acilo", cuando se usa sin el modificador "sustituido", se refiere al grupo -C(O)R, en el que R es un hidrógeno, alquilo, arilo, aralquilo o heteroarilo, tal como estos términos se han definido anteriormente. Los grupos -CHO, -C(O)CH3 (acetilo, Ac), -C(O)CH2CH3, -C(O)CH2CH2CH3, -C(O)CH(CH3)2, -C(O)CH(CH2)2, -C(O)CaH5, -C(O)CaH4CH3, -C(O)CH2CaH5, -C(O)(imidazolilo) son ejemplos no limitantes de grupos acilo. Un "tioacilo" se define de manera análoga, con la excepción de que el átomo de oxígeno del grupo -C(O)R se ha reemplazado por un átomo de azufre, -C(S)R. El término "aldehído" corresponde a un alcano, como se ha definido anteriormente, en donde al menos uno de los átomos de hidrógeno se ha reemplazado por un grupo -CHO. Cuando cualquiera de estos términos se usa con el modificador "sustituido", uno o más átomos de hidrógeno (incluyendo un átomo de hidrógeno directamente unido al grupo carbonilo o tiocarbonilo, si lo hay) se ha reemplazado independientemente por -OH, -F, -Cl, -Br, -I, -NH2, -NO2, -CO2H, -CO2CH3, -CN, -SH, -OCH3, -OCH2CH3, -C(O)CH3, -NHCH3, -NHCH2CH3, -N(CH3)2, -C(O)NH2, -OC(O)CH3 o -S(O)2NH2. Los grupos -C(O)CH2CF3, -CO2H (carboxilo), -CO2CH3 (metilcarboxilo), -CO2CH2CH3, -C(O)NH2 (carbamoílo) y -CON(CH3)2, son ejemplos no limitantes de grupos acilo sustituido.
El término "alcoxi", cuando se usa sin el modificador "sustituido", se refiere al grupo -OR, en el que R es un alquilo, tal como ese término se han definido anteriormente. Los ejemplos no limitantes de grupos alcoxi incluyen: -OCH3
(metoxi), -OCH2CH3 (etoxi), -OCH2CH2CH3, -OCH(CH3)2 (isopropoxi), -OC(CH3)3 (ferc-butoxi), -OCH(CH2)2, -O-ciclopentilo y -O-ciclohexilo. Los términos "alqueniloxi", "alquiniloxi", "ariloxi", "aralcoxi", "heteroariloxi", "heterocicloalcoxi" y "aciloxi", cuando se usan sin el modificador "sustituido", se refieren a grupos, definidos como -OR, en el que R es alquenilo, alquinilo, arilo, aralquilo, heteroarilo, heterocicloalquilo y acilo, respectivamente. El término "alcoxidiílo" se refiere al grupo divalente -O-alcanodiil-, -O-alcanodiil-O- o -alcanodiil-O-alcanodiil-. El término "alquiltio" y "aciltio", cuando se usa sin el modificador "sustituido", se refiere al grupo -SR, en el que R es un alquilo y acilo, respectivamente. El término "alcohol" corresponde a un alcano, como se ha definido anteriormente, en donde al menos uno de los átomos de hidrógeno se ha reemplazado por un grupo hidroxi. El término "éter" corresponde a un alcano, como se ha definido anteriormente, en donde al menos uno de los átomos de hidrógeno se ha reemplazado por un grupo alcoxi. Cuando cualquiera de estos términos se usa con el modificador "sustituido", uno o más átomos de hidrógeno se han reemplazado independientemente por -OH, -F, -Cl, -Br, -I, -NH2, -NO2, -CO2H, -CO2CH3, -CN, -SH, -OCH3, -OCH2CH3, -C(O)CH3, -NHCH3, -NHCH2CH3, -N(CH3)2, -C(O)NH2, -OC(O)CH3 o -S(O)2NH2.
El término "alquilamino", cuando se usa sin el modificador "sustituido", se refiere al grupo -NHR, en el que R es un alquilo, tal como ese término se han definido anteriormente. Los ejemplos no limitantes de grupos alquilamino incluyen: -NHCH3 y -NHCH2CH3. El término "dialquilamino", cuando se usa sin el modificador "sustituido", se refiere al grupo -NRR', en el que R y R' pueden ser el mismo o diferentes grupos alquilo, o R y R' pueden tomarse juntos para representar un alcanodiílo. Los ejemplos no limitantes de grupos dialquilamino incluyen: -N(c H3)2, -N(CH3)(CH2CH3) y W-pirrolidinilo. Los términos "alcoxiamino", "alquenilamino", "alquinilamino", "arilamino", "aralquilamino", "heteroarilamino", "heterocicloalquilamino" y "alquilsulfonilamino", cuando se usan sin el modificador "sustituido", se refieren a grupos, definidos como -NHR, en el que R es alcoxi, alquenilo, alquinilo, arilo, aralquilo, heteroarilo, heterocicloalquilo y alquilsulfonilo, respectivamente. Un ejemplo no limitante de un grupo arilamino es -NHC6H5. El término "amido" (acilamino), cuando se usa sin el modificador "sustituido", se refiere al grupo -NHR, en el que R es acilo, tal como ese término se han definido anteriormente. Un ejemplo no limitante de un grupo amido es -NHC(O)CH3. El término "alquilimino", cuando se usa sin el modificador "sustituido", se refiere al grupo divalente =NR, en el que R es un alquilo, tal como ese término se han definido anteriormente. El término "alquilaminodiílo" se refiere al grupo divalente -NH-alcanodiil-, -NH-alcanodiil-NH- o -alcanodiil-NH-alcanodiil-. Cuando cualquiera de estos términos se usa con el modificador "sustituido", uno o más átomos de hidrógeno se han reemplazado independientemente por -OH, -F, -Cl, -Br, -I, -NH2, -NO2, -CO2H, -CO2CH3, -CN, -SH, -OCH3, -OCH2CH3, -C(O)CH3, -NHCH3, -NHCH2CH3, -N(CH3)2, -C(O)NH2, -OC(O)CH3, -S(O)2NH2 o uno o más de los átomos de hidrógeno directamente unidos al átomo de nitrógeno es un grupo protector de amina. Los grupos -NHC(O)OCH3 y -NHC(O)NHCH3 son ejemplos no limitantes de grupos amido sustituido. El término alquilamina, dialquilamino o trialquilamina representa un compuesto en donde el grupo es NH2R, NHRR' o NRR'R", respectivamente, en donde R, R' y R" son alquilo tal como ese término se ha definido anteriormente.
Los términos "alquilsulfonilo" y "alquilsulfinilo", cuando se usan sin el modificador "sustituido", se refieren a los grupos -S(O)2R y -S(O)R, respectivamente, en el que R es un alquilo, tal como ese término se han definido anteriormente. Los términos "alquenilsulfonilo", "alquinilsulfonilo", "arilsulfonilo", "alquilsulfonilo", "heteroarilsulfonilo" y "heterocicloalquilsulfonilo" se definen de manera análoga a en la que esos términos se han definido anteriormente. Cuando cualquiera de estos términos se usa con el modificador "sustituido", uno o más átomos de hidrógeno se han reemplazado independientemente por -OH, -F, -Cl, -Br, -I, -NH2, -NO2, -CO2H, -CO2CH3, -CN, -SH, -OCH3, -OCH2CH3, -C(O)CH3, -NHCH3, -NHCH2CH3, -N(CH3)2, -C(O)NH2, -OC(O)CH3 o -S(O)2NH2.
El término "alquilsililo", cuando se usa sin el modificador "sustituido", se refiere a un grupo monovalente, definido como -SiH2R, -SiHRR' o -SiRR'R", en el que R, R' y R" pueden ser el mismo o diferentes grupos alquilo, o cualquier combinación de dos de R, R' y R" pueden tomarse juntos para representar un alcanodiílo. Los grupos -SH 2CH3, -SiH(CH3)2, -Si(CH3)3 y -Si(CH3)2C(CH3)3, son ejemplos no limitantes de grupos alquilsililo sin sustituir. El término "alquilsililo sustituido" se refiere a -SH 2R, -S íh Rr ' o -SiRR'R", en el que al menos uno de R, R' y R" es un alquilo sustituido o dos de R, R' y R" pueden tomarse juntos para representar un alcanodiílo sin sustituir. Cuando más de uno de R, R' y R" es un alquilo sustituido, pueden ser iguales o diferentes. Cualquiera de R, R' y R" que no sea alquilo sin sustituir o alcanodiílo sustituido, puede ser alquilo, igual o diferente, o puede tomarse para representar un alcanodiílo junto con dos o más átomos de carbono saturados, estando al menos dos de los cuales unidos al átomo de silicio. El término "arilsililo" o "aralquilsililo" se refiere al grupo como se ha definido anteriormente donde al menos uno de R, R' o R" es un grupo arilo o aralquilo tal como esos grupos se han definido anteriormente.
Un "aminoácido" es un grupo funcional que contiene un grupo -CO2H y un grupo -NH2 en la misma estructura de carbono lineal. En su realización preferida, el término "aminoácido" se refiere a uno de los aminoácidos naturales o disponibles comercialmente así como a sus enantiómeros y diastereómeros. Como se usa en el presente documento, el término "resto de aminoácido" se refiere a un aminoácido divalente que está unido a través del grupo amina y el grupo carboxilato que están conectados por un alcanodiílo^a) que se ha sustituido opcionalmente por -OH, -F, -Cl, -Br, -I, -NH2, -NO2, -CO2H, -CO2CH3, -CN, -SH, -OCH3, -OCH2CH3, -C(O)CH3, -NHCH3, -NHCH2CH3, -N(CH3)2, -C(O)NH2, -OC(O)CH3, -NHC(O)NH2, -NHC(NH)NH2 o -S(O)2NH2 o un alquilo(Ci-i2), alquenilo(C2-i2), alquinilo(C2-i2), arilo(oa-i2), aralquilo(C7-i2), heteroarilo(Ci-i2), heteorcicloalquilo(C2-i2), acilo(Ci-i2), o una versión sustituida de cualquiera de estos grupos en donde uno o más átomos de hidrógeno en el grupo químico se han sustituido con -OH, -F, -Cl, -Br, -I, -NH2, -NO2, -CO2H, -CO2CH3, -CN, -SH, -OCH3, -OCH2CH3, -C(O)CH3, -NHCH3, -NHCH2CH3, N(CHa)2, -C(O)NH2, -OC(O)CHa, -NHC(O)NH2, -NHC(NH)NH2 o -S(O)2NH2, por ejemplo,

En algunas realizaciones, el resto de aminoácido es un a-aminoácido en donde el alcanodiílo es un metileno de tal forma que el carbonilo y la amina se unen mediante un solo carbono. Además, el
Un "enlazador" es un grupo químico bifuncional que permite la reacción de cada extremo de la molécula independientemente del otro ya sea a través de las diferentes relatividades o a través del uso de grupos protectores de forma que dos moléculas puedan unirse entre sí. Algunos ejemplos no limitantes de enlazadores incluyen polipéptidos, polímeros, oligonucleótidos, aminoácidos, polietilenglicol, etilendiamina o etanolamina.
Una "base" en el contexto de esta solicitud es un compuesto que tiene un solo par de electrones. Los ejemplos limitantes de una base pueden incluir trietilamina, un hidróxido de metal, hidruro de metal o un alcano de metal. Un alquil litio es un compuesto de la fórmula alquil(c<i2)-Li. Una base nitrogenada es una alquilamina, dialquilamina, trialquilamina, heterocicloalcano que contiene nitrógeno o heteroareno en donde la base puede aceptar un protón para formar una especie cargada positivamente. Algunos ejemplos no limitantes que incluyen una base nitrogenada podrían ser 4,4-dimetilpiridina, piridina, piperidina, 1,8-diazabiciclo[5.4.0]undec-7-eno, diisopropiletilamina o trietilamina.
Una "fuente de fluoruro" en el contexto de esta solicitud es un reactivo que genera o que contiene un ion fluoruro. Algunos ejemplos no limitantes incluyen ácido fluorhídrico, fluoruro de metal, trihidrofluoruro de trietilamina o fluoruro de tetrabutilamonio.
Un "agente de activación", como se usa en el contexto de esta solicitud, es un compuesto que reacciona con un grupo ácido carboxílico para potenciar su capacidad de reaccionar con un nucleófilo. Dichos reactivos son bien conocidos en la técnica. Los reactivos se emplean habitualmente en la producción de un enlace amida a partir de un grupo amina y un ácido carboxílico. Algunos grupos habituales incluyen N-hidroxisuccinimida, 4-nitrofenol, reactivos de carbodiimida y reactivos de triazol. Dichos reactivos se enseñan, por ejemplo, por Montalbetti and Falque, 2005, que se incorpora en el presente documento por referencia.
Un "metal" en el contexto de esta solicitud es un metal de transición o un metal de los grupos I o II.
Un "grupo autoinmolante" es un grupo que se descompone en condiciones fisiológicas. Dichos grupos son bien conocidos en la técnica. Dichos grupos se enseñan por Kratz et al., 2011 en "Chapter 19: Site-Specific Prodrug Activation and the Concept of Self-Immolation" en Drug Delivery in Oncology: From Basic Research to Cancer Therapy.
Un "grupo reactivo de tiol" es un grupo químico funcional que experimenta una reacción con un grupo -SH tal como en una cadena lateral de un aminoácido para formar un enlace covalente que une el átomo de azufre con el grupo. Dichos grupos son bien conocidos en la técnica e incluyen, pero sin limitación, agentes que pueden experimentar adición de conjugados. Algunos ejemplos no limitantes incluyen una maleimida o una haloacetamida tal como yodoactamida.
Un "grupo protector de amina" es bien conocido en la técnica. Un grupo protector de amina es un grupo que evita la reactividad del grupo amina durante una reacción que modifica alguna otra porción de la molécula y puede eliminarse fácilmente para generar la amina deseada. Pueden encontrarse grupos protectores de amina al menos en Greene and Wuts, 1999. Algunos ejemplos no limitantes de grupos protectores de amino incluyen formilo, acetilo, propionilo, pivaloílo, t-butilacetilo, 2-cloroacetilo, 2-bromoacetilo, trifluoroacetilo, tricloroacetilo, o-nitrofenoxiacetilo, aclorobutirilo, benzoílo, 4-clorobenzoílo, 4-bromobenzoílo, 4-nitrobenzoílo y similares; grupos sulfonilo tales como bencenosulfonilo, p-toluenosulfonilo y similares; grupos alcoxi- o ariloxicarbonilo (que forman uretanos con la amina protegida) tales como benciloxicarbonilo (Cbz), p-clorobenciloxicarbonilo, p-metoxibenciloxicarbonilo, pnitrobenciloxicarbonilo, 2-nitrobenciloxicarbonilo, p-bromobenciloxicarbonilo, 3,4-dimetoxibenciloxicarbonilo, 3,5-dimetoxibenciloxicarbonilo, 2,4-dimetoxibenciloxicarbonilo, 4-metoxibenciloxicarbonilo, 2-nitro-4,5-dimetoxibenciloxicarbonilo, 3,4,5-trimetoxibenciloxicarbonilo, 1 -(p-bifenilil)-1 -metiletoxicarbonilo, a,a-dimetil-3,5-dimetoxibenciloxicarbonilo, benzhidriloxicarbonilo, t-butiloxicarbonilo (Boc), diisopropilmetoxicarbonilo, isopropiloxicarbonilo, etoxicarbonilo, metoxicarbonilo, aliloxicarbonilo (Alloc), 2,2,2-tricloroetoxicarbonilo, 2-trimetilsililetiloxicarbonilo (Teoc), fenoxicarbonilo, 4-nitrofenoxicarbonilo, fluorenil-9-metoxicarbonilo (Fmoc), ciclopentiloxicarbonilo, adamantiloxicarbonilo, ciclohexiloxicarbonilo, feniltiocarbonilo y similares; grupos aralquilo tales como bencilo, trifenilmetilo, benciloximetilo y similares; y grupos sililo tales como trimetilsilil y similares. Además, el "grupo protector de amina" puede ser un grupo protector divalente de tal forma que los dos átomos de hidrógeno en una amina primaria se reemplazan por un solo grupo protector. En esta situación, el grupo protector de amina puede ser ftalimida (phth) o un derivado sustituido de la misma en donde el término "sustituido" es como se ha
definido anteriormente. En algunas realizaciones, el derivado de ftalimida halogenado puede ser be tetracloroftalimida (TCphth).
Un "grupo protector de hidroxilo" es bien conocido en la técnica. Un grupo protector de hidroxilo es un grupo que evita la reactividad del grupo hidroxilo durante una reacción que modifica alguna otra porción de la molécula y puede eliminarse fácilmente para generar el hidroxilo deseado. Pueden encontrarse grupos protectores de hidroxilo al menos en Greene and Wuts, 1999, que se incorpora en el presente documento por referencia. Algunos ejemplos no limitantes de grupos protectores de hidroxilo incluyen grupos acilo tales como formilo, acetilo, propionilo, pivaloílo, tbutilacetilo, 2-cloroacetilo, 2-bromoacetilo, trifluoroacetilo, tricloroacetilo, o-nitrofenoxiacetilo, a-clorobutirilo, benzoílo, 4-clorobenzoílo, 4-bromobenzoílo, 4-nitrobenzoílo y similares; grupos sulfonilo tales como bencenosulfonilo, ptoluenosulfonilo y similares; grupos aciloxi tales como benciloxicarbonilo (Cbz), p-clorobenciloxicarbonilo, pmetoxibenciloxicarbonilo, p-nitrobenciloxicarbonilo, 2-nitrobenciloxicarbonilo, p-bromobenciloxicarbonilo, 3,4-dimetoxibenciloxicarbonilo, 3,5-dimetoxibenciloxicarbonilo, 2,4-dimetoxibenciloxicarbonilo, 4-metoxibenciloxicarbonilo, 2-nitro-4,5-dimetoxibenciloxicarbonilo, 3,4,5-trimetoxibenciloxicarbonilo, 1-(p-bifenilil)-1-metiletoxicarbonilo, a,a--dimetil-3,5-dimetoxibenciloxicarbonilo, benzhidriloxicarbonilo, t-butiloxicarbonilo (Boc), diisopropilmetoxicarbonilo, isopropiloxicarbonilo, etoxicarbonilo, metoxicarbonilo, aliloxicarbonilo (Alloc), 2,2,2-tricloroetoxicarbonilo, 2-trimetilsililetiloxicarbonilo (Teoc), fenoxicarbonilo, 4-nitrofenoxicarbonilo, fluorenil-9-metoxicarbonilo (Fmoc), ciclopentiloxicarbonilo, adamantiloxicarbonilo, ciclohexiloxicarbonilo, feniltiocarbonilo y similares; grupos aralquilo tales como bencilo, trifenilmetilo, benciloximetilo y similares; y grupos sililo tales como trimetilsilil y similares.
Un "grupo protector de tiol" es bien conocido en la técnica. Un grupo protector de tiol es un grupo que evita la reactividad del grupo mercapto durante una reacción que modifica alguna otra porción de la molécula y puede eliminarse fácilmente para generar el grupo mercapto deseado. Pueden encontrarse grupos protectores de tiol al menos en Greene and Wuts, 1999. Algunos ejemplos no limitantes de grupos protectores de tiol incluyen grupos acilo tales como formilo, acetilo, propionilo, pivaloílo, t-butilacetilo, 2-cloroacetilo, 2-bromoacetilo, trifluoroacetilo, tricloroacetilo, o-nitrofenoxiacetilo, a-clorobutirilo, benzoílo, 4-clorobenzoílo, 4-bromobenzoílo, 4-nitrobenzoílo y similares; grupos sulfonilo tales como bencenosulfonilo, p-toluenosulfonilo y similares; grupos aciloxi tales como benciloxicarbonilo (Cbz), p-clorobenciloxicarbonilo, p-metoxibenciloxicarbonilo, p-nitrobenciloxicarbonilo, 2-nitrobenciloxicarbonilo, p-bromobenciloxicarbonilo, 3,4-dimetoxibenciloxicarbonilo, 3,5-dimetoxibenciloxicarbonilo, 2,4-dimetoxibenciloxicarbonilo, 4-metoxibenciloxicarbonilo, 2-nitro-4,5-dimetoxibenciloxicarbonilo, 3,4,5-trimetoxibenciloxicarbonilo, 1-(p-bifenilil)-1-metiletoxicarbonilo, a,a-dimetil-3,5-dimetoxibenciloxicarbonilo, benzhidriloxicarbonilo, t-butiloxicarbonilo (Boc), diisopropilmetoxicarbonilo, isopropiloxicarbonilo, etoxicarbonilo, metoxicarbonilo, aliloxicarbonilo (Alloc), 2,2,2-tricloroetoxicarbonilo, 2-trimetilsililetiloxicarbonilo (Teoc), fenoxicarbonilo, 4-nitrofenoxicarbonilo, fluorenil-9-metoxicarbonilo (Fmoc), ciclopentiloxicarbonilo, adamantiloxicarbonilo, ciclohexiloxicarbonilo, feniltiocarbonilo y similares; grupos aralquilo tales como bencilo, trifenilmetilo, benciloximetilo y similares; y grupos sililo tales como trimetilsilil y similares.
Un "estereoisómero" o "isómero óptico" es un isómero de un compuesto dado en el que los mismos átomos están unidos a los otros mismos átomos, pero donde la configuración de esos átomos en tres dimensiones es diferente. Los "enantiómeros" son estereoisómeros de un compuesto dado que son imágenes especulares entre sí, como las manos izquierda y derecha. Los "diastereómeros" son estereoisómeros de un compuesto dado que no son enantiómeros. Las moléculas quirales contienen un centro quiral, también denominado estereocentro o centro estereogénico, que es cualquier punto, aunque no necesariamente un átomo, en una molécula que tiene grupos de tal forma que un intercambio de dos grupos cualesquiera conduce a un estereoisómero. En compuestos orgánicos, el centro quiral es normalmente un átomo de carbono, fósforo o azufre, aunque también es posible que otros átomos sean estereocentros en compuestos orgánicos e inorgánicos. Una molécula puede tener múltiples estereocentros, dándole muchos estereoisómeros. En compuestos cuya estereoisomería se debe a centros estereogénicos tetraédricos (por ejemplo, carbono tetraédrico), el número total de estereoisómeros hipotéticamente posibles no superará 2n, donde n es el número de estereocentros tetraédricos. Las moléculas con simetría con frecuencia tienen menos que el número máximo posible de estereoisómeros. Una mezcla 50:50 de enantiómeros se denomina mezcla racémica. Como alternativa, una mezcla de enantiómeros puede estar enantioméricamente enriquecida de manera que un enantiómero está presente en una cantidad mayor del 50 %. Normalmente, los enantiómeros y/o diastereómeros pueden resolverse o separarse usando técnicas conocidas en la técnica. Se contempla que para cualquier estereocentro o eje de quiralidad para el cual no se haya definido la estereoquímica, ese estereocentro o eje de quiralidad puede estar presente en su forma R, forma S o como una mezcla de las formas R y S, incluyendo mezclas racémicas y no racémicas. Como se usa en el presente documento, la frase "sustancialmente libre de otros estereoisómeros" significa que la composición contiene < 15%, más preferiblemente < 10%, aún más preferiblemente < 5 %, o lo más preferiblemente < 1 % de otro(s) estereoisómero(s).
V. Ejemplos
Los siguientes ejemplos se incluyen para demostrar realizaciones preferidas de la divulgación. Los expertos en la materia deberían apreciar que las técnicas divulgadas en los siguientes ejemplos representan técnicas descubiertas por el inventor para que funcionen bien en la práctica de la divulgación, y por tanto, se puede considerar que constituyen modos preferidos para su práctica.
Ejemplo 1 - materiales y métodos
Todas las reacciones se realizaron en una atmósfera de argón con disolventes secos en condiciones anhidras, a menos que se indique de otro modo. Tetrahidrofurano seco (THF), tolueno, benceno, metanol (MeOH), éter dietílico (Et2O), N,N-dimetilformamida (DMF) y cloruro de metileno (0H20h) se obtuvieron pasando formulaciones sin oxígeno, secadas previamente, disponibles en el mercado, a través de columnas de alúmina activadas. Los rendimientos se refieren a materiales cromatográfica y espectroscópicamente homogéneos (1H RMN), a menos que se indique otra cosa. Los reactivos se adquirieron con la más alta calidad comercial y se usaron sin purificación adicional, a menos que se indique otra cosa. Las reacciones se controlaron por cromatografía de capa fina (TLC) realizada sobre placas de gel de sílice E. Merck de 0,25 mm (60F-254) usando luz UV como agente de visualización y una solución etanólica de ácido fosfomolíbdico y sulfato de cerio, y calor como agentes de desarrollo. Se usó gel de sílice E. Merck (60, tamaño de partículas 0,040 - 0,063 mm) para la cromatografía en columna ultrarrápida, y se desactivó suspendiéndose en eluyente con H2O añadida al 5 % 12 horas antes de la cromatografía. Los espectros de RMN se registraron en instrumentos Bruker DRX-500 o DRX-600 y se calibraron usando disolvente no deuterado residual (CDCh: Sh = 7,26 ppm, So = 77,0 ppm; o CD3CN: Sh = 1,94 ppm, So = 118,26 ppm) como referencia interna. Se usaron las siguientes abreviaturas para designar las multiplicidades: s = singlete, d = doblete, t = triplete, c = cuadruplete, m = multiplete, a = ancho. Los espectros de infrarrojos (IR) se registraron en un espectrómetro Perkin-Elmer 100 FT-IR. Los espectros de masas de alta resolución (HRMS) se registraron en un espectrómetro de masas Agilent ESI-TOF (tiempo de vuelo) usando MALDI (ionización mediante desorción por láser asistida por matriz) o ESI (ionización por electronebulización). Las rotaciones ópticas se registraron en un polarímetro Perkin-Elmer Model 343 a 589 nm, y se informan en unidades de 10'1 (grados cm2 g_1).
Ejemplo 2 - descripción de métodos de síntesis para la preparación de uncialamicina y sus análogos
La estrategia de síntesis para la síntesis de uncialamicina y sus análogos se destaca en la sección experimental detallada anteriormente y en los esquemas de síntesis (Esquemas 1-20) que se resaltan y analizan a continuación. Se hizo reaccionar dicloroeteno con paladio catalítico y cobre en condiciones de Sonogashira con triisopropilsililacetileno para formar 27 (ejemplo de referencia). El compuesto 27 se hizo reaccionar con trimetilsililacetileno en condiciones de Sonogashira para producir 28 (ejemplo de referencia). El grupo trimetilsililo de 28 se escindió en carbonato potásico para dar 11 (ejemplo de referencia). La preparación de 11 se muestra en el Esquema 1.

isatina desprotegida 2 (ejemplo de referencia). La isatina 2 se hizo reaccionar con trans-4-metoxi-3-buten-2-ona en presencia de una base seguido de acidificación. La mezcla resultante se trató con carbonato potásico, se calentó y después se acidificó para dar 4 (ejemplo de referencia). El cetoácido 4 se protegió con bromuro de dimetoxibencilo para dar el intermedio 6 que se hizo reaccionar con el catalizador de Noyori para dar la lactona 8 (ejemplo de referencia). La lactona 8 se redujo para dar el intermedio de hemiacetal que después se protegió con cloruro de TES para dar 10 (ejemplo de referencia). El TES lactol 10 se hizo reaccionar con 11 para dar el enodiino 12 (ejemplo de referencia). El grupo TES de 12 se escindió usando ácido acético para dar 13 (ejemplo de referencia). El lactol 13 se trató con un agente reductor y después se epoxidó con mCPBA para dar el epóxido 14 (ejemplo de referencia).



ODMB 14 ODMB 13
Esquema 2. Síntesis mejorada de uncialamicina (26a) Construcción del intermedio clave 14
Después de la preparación de 14, el alcohol primario se aciló para dar 15 (ejemplo de referencia). El acetato 15 se oxidó con peryodinano de Dess-Martin para dar la cetona 16 (ejemplo de referencia). Usando TBAF, el enodiino se desprotegió y la cetona se redujo para dar el alcohol secundario 17 (ejemplo de referencia). El alcohol secundario 17 se protegió con cloruro de TES y después el acetato se desprotegió con carbonato potásico para dar el alcohol primario 18 (ejemplo de referencia). El alcohol primario 18 se oxidó para dar el aldehído 19 usando peryodinano de Dess-Martin (ejemplo de referencia). Usando DDQ, el grupo dimetoxibencilo se retiró para dar el fenol libre 20 (ejemplo de referencia). En presencia de la base nucleófila fuerte, KHMDS y CeCl3, 20 se cicla para dar para dar el enodiino cíclico 21a (ejemplo de referencia). El enodiino cíclico 21a se metoxiló con metanol y PhI(OAc)2 para dar la quinona aminal 22a. El material de partida 23 se hizo reaccionar con 22a en condiciones de condensación de Hauser para dar el derivado de uncialamicina protegido 24a (ejemplo de referencia) que se desprotegió con Pd(PPh3)4 y morfolina para dar 25a (ejemplo de referencia). El análogo de uncialamicina protegido con TES 25a se hizo reaccionar con HF para dar la uncialamicina 26a (ejemplo de referencia).

Esquema 3. Síntesis me orada de uncialamicina (26a). Construcción del intermedio clave 22a y terminación de la síntesis
En el Esquema 4, se prepara un análogo de 23 para dar acceso a análogos de 8-aminouncialamicina. Se hizo reaccionar 6-aminoftalida con anhídrido de Boc para dar la ftalida protegida con Boc 29 (ejemplo de referencia). La ftalida protegida 29 se hizo reaccionar con NBS y AIBN para dar el derivado bromado 30 (ejemplo de referencia) que se hidrató para dar el compuesto de hidroxi 31 (ejemplo de referencia) que experimenta sustitución nucleófila con dietilamina después de la activación con SOCh para dar la formilbenzamida 32 (ejemplo de referencia). Se introdujo un grupo ciano en 32 con TMSCN y KCN seguido de ciclación en condiciones ácidas para dar la cianoftalida 33 (ejemplo de referencia). Como se ha descrito previamente en el Esquema 3 para la uncialamicina, la cianoftalida 33 se hizo reaccionar con el enodiino cíclico 22a para dar el análogo de uncialamicina 34a (ejemplo de referencia). Como se muestra en el Esquema 5, el análogo de uncialamicina 34a se desprotegió con morfolina y Pd(PPh3)4 para dar la 8-aminouncialamicina protegida con TES 35a (ejemplo de referencia). Con HF, el grupo TES se retiró para dar la 8-aminouncialamicina 36a (ejemplo de referencia).

70% en 2 etapas
Esquema 4. Síntesis de cianoftalida 33

Esquema 5. S íntesis de 8-am ino-uncia lam ic ina (36a).
De forma similar a las reacciones mostradas en los Esquemas 4 y 5, se prepararon derivados de aminometil de uncialamicina. Se convirtió ácido 2,5-dimetilbenzoico en el éster metílico 37a (ejemplo de referencia) usando MeI seguido de bromación, ciclación y después reacción con ftalimida en condiciones básicas para dar la ftalida 40a (ejemplo de referencia). La ftalida se bromó con NBS y AIBN seguido de hidroxilación, activación y adición nucleófila de dietilamina para dar 43a (ejemplo de referencia). Después, la formilbenzamida 43a se hizo reaccionar con
TMSCN y KCN seguido de ácido acético para dar la cianoftalida 44a (ejemplo de referencia). Análogamente, se prepararon los isómeros 44b, 44c y 44d (ejemplo de referencia). Como se representa en el Esquema 7, la cianoftalida 44a o uno de sus isómeros se hizo reaccionar con 22a o su diastereómero 22b con una base no nucleófila fuerte en condiciones de condensación de Hauser para dar el análogo de aminometiluncialamicina protegido 45aa que se desprotegió con Pd(PPh3)4 y morfolina para dar 46aa y después HF para dar 47aa. El grupo ftalimida se retiró con metilamina para dar la aminometiluncialamicina 48aa que después se protegió con Boc usando anhídrido de Boc para dar 49aa. Usando métodos similares y con rendimientos comparables, se prepararon otros análogos de aminometiluncialamicina incluyendo 48ba, 48ca, 48da y 48ba.

44b, 44c y 44d se preparan de acuerdo con la ruta anterior
con rendimientos comparables
PhthN

Esquema 6. Síntesis de las cianoftalidas 44a, 44b, 44c y 44d.

NPhth O OH NPhth O OH

48aa: 8-aminometM-uncialamicina 
47ba, 47ca, 47da, 47db, 48ba, 48ca y 48da se preparan
de acuerdo con la ruta anterior con rendimientos comparables:

, , ,
48ba: R = H, 17(R) 48ca: R = H, 17(R) 47db: R = Phth, 17(S )
48da: R = H, 17(R)
Esquema 7. Síntesis de aminometil-uncialamicinas (48aa, 48ba, 48ca y 48da).
Como se ha descrito en el Esquema 3 y en el Esquema 5, se empleó un método similar para obtener la cianoftalida 55 (ejemplo de referencia) partiendo de 2,4-dihidroxi-3-metilbenzoato de metilo y se muestra en el Esquema 8. Después de la preparación de la cianoftalida 55, el compuesto se sometió a condensación de Hauser para dar un análogo de la uncialamicina 58b (ejemplo de referencia) que se describe en el Esquema 9.

58a: 17 R , 58b: 17 S 57a: 17{R), 57b: 17($)
Esquema 9. Síntesis de las 8-ftalimidometil-uncialamicinas 58a y 58b
De forma análoga a los otros derivados de uncialamicina descritos anteriormente, se preparó una uncialamicina expandida usando las cianoftalidas con núcleo de naftaleno 63a y 63b (ejemplo de referencia). La preparación se muestra en el Esquema 10. Análogamente, la condensación para formar el análogo de uncialamicina apropiado se describe en el Esquema 11.

n u , ,CH2CI2, 0 ~ 25 °C,
THF, -78 C;
98% después, DMF,

62a: R¡ = PhthN, R2 = H 63a: R< = PhthN, R2 = H 62b: R-i = H, R2= PhthN 63b: Ri = H, R2 = PhthN mezcla 1:1 inseparable Esquema 10. Síntesis de las cianoftalidas 63a y 63b.

66a: R ■, = PhthN, R2 = H a: = , = 66b: R! = H, R2 = PhthN 65b: R! = H, R2 = PhthN
mezcla 1:1 inseparable
Esquema 11. Síntesis de las 8-ftalirmdometil-uncialamicinas 66a/66b.
Además, los esquemas 12-15 también se prevén como rutas de síntesis potenciales que pueden permitir la producción de uncialamicina y sus análogos. Estos esquemas muestran métodos de síntesis adicionales que pueden emplearse para acceder a análogos de uncialamicina descritos en el presente documento.

después HOAc, 
 25 °C, 70%
25 °C, 70%
Esquema 12. Síntesis de la cianoftalida 76


Esquema 15. Síntesis de las 8-aminometil-uncialamicinas 48aa y 49aa.
Se prepararon análogos de uncialamicina con un sistema de anillos de isoindolina partiendo de ácido 2,4,5 trimetilbenzoico que se protegió en forma del éster metílico y después se bromó y se cicló para dar 88 (ejemplo de referencia). Después, se hizo reaccionar 88 con tritilamina en presencia de base de Hunig para dar 89 (ejemplo de referencia). El grupo tritilo se retiró con ácido y la amina se protegió con un grupo Teoc para dar 90 (ejemplo de referencia). La lactona 90 se abrió con hidróxido y después se oxidó con PCC para dar el compuesto hidroxilado 92 (ejemplo de referencia). El compuesto hidroxilado 92 se convierte en la cianoftalida 93 (ejemplo de referencia) como se muestra en el Esquema 16. Como se ha descrito previamente y como se muestra específicamente en los Esquemas 17 y 18, la cianoftalida 93 se hizo reaccionar con 22a para dar el análogo deseado de uncialamicina o una versión derivatizada del mismo (ejemplo de referencia).


: son o n-unca amcna
Esquema 17. Síntesis de las isoindolin-uncialamicinas (96, 97 y 98).

Usando un método similar al empleado en el Esquema 16, se prepara una versión de metilamina de 8-aminometiluncialamicina. Como se muestra en el Esquema 19, la cianoftalida se preparó partiendo con
bromometilftalida que se hizo reaccionar con TeocNHMe en condiciones básicas para dar la ftalida 101 (ejemplo de referencia). Después de la apertura del éster cíclico para dar 102 (ejemplo de referencia), el compuesto se oxidó para dar 103 (ejemplo de referencia) y se introdujo un grupo ciano para dar la cianoftalida 104 (ejemplo de referencia). Después, la cianoftalida se hizo reaccionar en condiciones básicas fuertes con 22a para dar el análogo de metilaminometiluncialamicina y derivados del mismo que se describen en el Esquema 20.

en eapas
Esquema 19. Síntesis de la cianoftalida 104


108
R = SPh; 109 >, DMDO, CH2CI2, R = S02Ph: 110 J -78~25°C , 62%
Esquema 20. Síntesis de las 8-N-metilaminometil-uncialamicinas (107, 108, 109 y 110).
Ejemplo 3 - métodos de síntesis y caracterización

5-Hidroxi isatina (2). A una solución agitada de 5-metoxi isatina (1) (10,0 g, 56,5 mmol, 1,0 equiv.) en CH2CI2 (40 ml) a 0 °C se le añadió gota a gota BBr3 (36,8 g, 147 mmol, 2,6 equiv.) durante 2 h con agitación vigorosa a 0 °C. Después de que se completara la adición, el baño de hielo se retiró y la mezcla de reacción se agitó durante 1 h más a temperatura ambiente, se diluyó con CH2Cl2 (500 ml), se enfrió a 0 °C y se inactivó con la adición secuencial cuidadosa de NaHCO3 sólido (37 g) y H2O (60 ml) fría (0-5 °C) (PRECAUCIÓN, gas HBr). La mezcla resultante se lavó con salmuera (50 ml) y las fases acuosas combinadas se extrajeron con CH2O 2 (50 ml). Las fases orgánicas combinadas se concentraron y el residuo se recristalizó en ácido acético glacial para producir la isatina 2 en forma de un sólido de color rojo oscuro (8 ,8 g, 54 mmol, rendimiento del 95 %). Fr = 0,31 (gel de sílice, CH2Ch:EtOAc 1:1); IR (película) vmáx = 3278, 1720, 1614, 1477, 1291, 1194, 881, 822, 800 cm-1; 1H RMN (500 MHz, DMSO): 8 = 10,73
(s, 1 H), 9,52 (a, 1 H), 7,00 (dd, J = 8,5, 2,6 Hz, 1 H), 6,83 (d, J = 2,6 Hz, 1 H), 6,74 (d, J = 8,3 Hz, 1 H) ppm; 13C RMN (125 MHz, CDCI3): 6 = 184,9, 159,5, 153,2, 143,1, 125,1, 118,2, 113,1, 110,5, 105,1 ppm; HRMS (ESI-TOF): calc. para Ca^NOs* [M+H+]: 164,0342, observado 164,0344.

Cetoácido 4. La isatina 2 (6,7 g, 47 mmol, 1,0 equiv.) se disolvió en NaOH acuoso 2 N (47 ml, 94 mmol, 2,0 equiv.) a temperatura ambiente y se añadió frans-4-metoxi-3-buten-2-ona (pureza del 90 %, 9,1 g, 94 mmol, 2,0 equiv.) en una porción con agitación vigorosa. La mezcla de reacción se agitó durante 15 min, se enfrió a 0 °C y se acidificó con HCl conc. a pH 1. El sólido de color verde resultante se recogió por filtración y se lavó con agua enfriada con hielo (se ensayó el pH del filtrado después de cada lavado, y se mantuvo en lavado hasta que alcanzó pH 5~6). El sólido húmedo se disolvió en NaHCO3 ac. saturado (60 ml) y, después de retirar por filtración una traza de material insoluble oscuro, se añadió K2CO3 ac. saturado (20 ml). La mezcla resultante se agitó a 80 °C durante 30 min y después, a la misma temperatura, se acidificó cuidadosamente con HCl conc. a pH 1 (PRECAUCIÓN, gas CO2). La suspensión resultante se agitó durante 10 min más a 80 °C, después se dejó enfriar a temperatura ambiente y se almacenó a 0 °C durante 12 h. El sólido resultante se recogió por filtración, se lavó con agua enfriada con hielo (se ensayó pH del filtrado después de cada lavado, y se mantuvo en lavado con una pequeña cantidad de hielo-agua hasta que alcanzó pH 5~6) y se secó al vacío para producir el cetoácido 2 en forma de un sólido de color pardo (7,7 g, 33 mmol, rendimiento del 81 %). El material en bruto se usó directamente para la siguiente etapa sin purificación adicional.

Lactona 8. A una solución agitada del cetoácido 4 (7,7 g, 33 mmol, 1,0 equiv.) en DMF (50 ml) a temperatura ambiente se le añadieron secuencialmente Cs2CO3 sólido (10 ,8 g, 167 mmol, 5,0 equiv.) y n-Bu4NI (1 ,8 g, 5,0 mmol, 0,15 equiv.), seguido de una solución de bromuro de 3,4-dimetoxibencilo (5) [DMBBr, 30,7 g, 133 mmol, 4,0 equiv. en DMF (15 ml)]. La mezcla de reacción se agitó a la misma temperatura durante 5 h y después se repartió entre EtOAc (300 ml) y H2O (300 ml). La capa acuosa se extrajo con EtOAc (3 x 150 ml) y las capas orgánicas combinadas se lavaron con H2O (3 x 150 ml) y salmuera (150 ml), se secaron sobre MgSO4 y se concentraron. El residuo se pasó a través de un lecho corto de Celite® eluyendo con hexanos/EtOAc (v/v = 1/2), se concentró y se coevaporó con tolueno (3 x 25) para producir el bis-DMB cetoéster 6 en bruto en forma de un aceite oscuro, que se disolvió en CH2Cl2 (150 ml) y se enfrió a 0 °C. A esta solución se le añadieron secuencialmente catalizador de (S,S)-Noyori 7 (1,0 g, 1,7 mmol, 0,05 equiv.) y una mezcla enfriada previamente (0-5 °C) de Et3N/HCO2H (Et3N: 8,4 g, 83 mmol, 2,5 equiv.; HCO2H: 6 ,6 g, 143 mmol, 4,3 equiv.). La mezcla resultante se agitó a 0 °C durante 24 h, después se inactivó con NaHCO3 ac. saturado (150 ml), la capa acuosa se separó y se extrajo con CH2Cl2 (3 x 150 ml) y las capas orgánicas combinadas se lavaron con NaHCO3 ac. saturado (150 ml) y salmuera (150 ml), se secaron sobre MgSO4 y se concentraron. La cromatografía en columna ultrarrápida (gel de sílice, CH2Ch:EtOAc 15:1 a 10:1) dio un sólido de color pardo oscuro, que se recristalizó en EtOAc para producir la lactona (+)-8 en forma de un sólido de color blanquecino (6,1 g, 17 mmol, rendimiento del 82 %, >99% de ee mediante análisis por HPLC del derivado de bis-TES éter 9 obtenido a partir de reducción con DIBAL-H y protección con TES; columna quiral OD-H 5 |j, 4,6 x 250 mm, hexanos:/-PrOH 98:2, 1 ml min'1; tiempo de retención del enantiómero principal = 14,37 min, tiempo de retención del enantiómero secundario = 16,88 min). 8: [a]o25 = 19 (c = 1,00, CH2O 2), Fr = 0,21 (gel de sílice, hexanos:EtOAc 7:3); IR (película) vmáx = 2935, 2836, 2254, 1756, 1605, 1509, 1460, 1263, 1213 cm-1; RMN 1H (600 MHz, CDCl3): 6 = 8 ,88 (s, 1 H), 8,20 (d, J = 2,8 Hz, 1 H), 8,08 (d, J = 9,2 Hz, 1 H), 7,49 (dd, J = 9,2, 2,8 Hz, 1 H), 7,05 (d, J = 8,2 Hz, 1 H), 7,04 (s, 1 H), 6,87 (d, J = 8,1 Hz, 1 H), 5,70 (c, J = 6 ,8 Hz, 1 H), 5,16 (s, 2 H), 3,91 (s, 3 H), 3,86 (s, 3 H), 1,73 (d, J = 6 ,8 Hz, 3 H) ppm; 13C RMN (150 MHz, CDCh): 6 = 169,5, 159,2, 149,0, 145,0, 144,4, 140,8, 131,1, 128,2, 126,4, 124,2, 123,9, 120,7, 111,3, 111,0, 101,8, 76,4, 70,5, 55,8, 20,0 ppm; HRMS (ESI-TOF): calc. para C21H20NO5+ [M+H+]: 366,1336, observado 366,1338.

TES lactol 10. A una solución agitada de la lactona 8 (3,80 g, 10,4 mmol, 1,0 equiv.) en CH2CI2 (75 ml) a -78 °C se le añadió lentamente DIBAL-H (1,0 M en CH2Cl2, 25 ml, 25 mmol, 2,4 equiv.) durante 2 h. La mezcla de reacción se agitó a -78 °C durante 1 h más, hasta que el análisis por TLC mostró el consumo completo de 8 (hexanos:EtOAc 1:1). La mezcla de reacción se diluyó con CH2O 2 (400 ml) y después se vertió lentamente en tartrato de sodio y potasio ac. saturado (200 ml). La mezcla resultante se agitó vigorosamente a temperatura ambiente durante 6 h hasta que se formaron dos capas, la capa orgánica se separó y la capa acuosa se extrajo con CH2Cl2 (5 x 150 ml) y después con EtOAc (3 x 50 ml). Los extractos orgánicos combinados se secaron sobre MgSO4 y se concentraron. El residuo de color amarillo pálido resultante se recogió en DMF (120 ml). A esta mezcla se le añadió imidazol (1,84 g, 27,0 mmol, 2,6 equiv.) y después TESCl (2,26 ml, 2,04 g, 13,5 mmol, 1,3 equiv.) a 0 °C. La mezcla de reacción se agitó a temperatura ambiente durante 20 min, después se repartió entre Et2O (150 ml) y NaHCO3 ac. saturado (150 ml). La capa acuosa se extrajo con Et2O (2 x 150 ml) y las capas orgánicas combinadas se lavaron con H2O (3 x 150 ml) y salmuera (150 ml), se secaron sobre MgSO4 y se concentraron. La cromatografía en columna ultrarrápida (gel de sílice, hexanos:EtOAc 2:1 a 3:2) proporcionó TES lactol 10 (4,55 g, 9,46 mmol, rendimiento del 91 % en 2 etapas, mezcla aprox. 1:1 de diaestereoisómeros intrascendente) en forma de un aceite de color amarillo pálido. 10 : Fr = 0,31 ("isómero syn) y 0,25 (isómero anti) (gel de sílice, hexanos:EtOAc 3:2); IR (película) vmáx = 2953, 2875, 1625, 1604, 1515, 1461, 1211 cm-1; 1H RMN (600 MHz, CDCla) del isómero syn: 8 = 8,69 (s, 1 H), 8,10 (d, J = 9,2 Hz, 1 H), 7,44 (dd, J = 9,2, 2,7 Hz, 1 H), 7,27 (d, J = 2,8 Hz, 1 H), 7,02 (d, J = 6,9 Hz, 1 H), 7,01 (s, 1 H), 6,90 (d, J = 6,8 Hz, 1 H), 6,90 (s, 1 H), 5,68 (dc, J = 6,5, 2,3 Hz, 1 H), 5,09 (dd, J = 17,1, 10,9 Hz, 1 H), 3,92 (s, 3 H), 3,91 (s, 3 H), 1,59 (d, J = 6,5 Hz, 3 H), 1,02 (t, J = 7,9 Hz, 9 H), 0,74 (c, J = 7,8 Hz, 6 H) ppm; isómero anti: 8 = 8,69 (s, 1 H), 8,11 (d, J = 9,2 Hz, 1 H), 7,43 (dd, J = 9,2, 2,6 Hz, 1 H), 7,32 (d, J = 2,4 Hz, 1 H), 7,01 (d, J = 6,1 Hz, 1 H), 7,00 (s, 1 H), 6,91 (d, J = 8,6 Hz, 1 H), 6,84 (s, 1 H), 5,46 (c, J = 6,5 Hz, 1 H), 5,09 (dd, J = 18,3, 10,8 Hz, 1 H), 3,92 (s, 3 H), 3,91 (s, 3 H), 1,67 (d, J = 6,5 Hz, 3 H), 1,06 (t, J = 8,0 Hz, 9 H), 0,79 (c, J = 7,7 Hz, 6 H) ppm; 13C RMN (150 MHz, CDCla) del isómero syn: 8 = 157,6, 149,2, 149,1, 144,0, 143,6, 141,5, 136,7, 131,1, 128,6, 124,1, 122,5, 120,4, 111,0, 111,0, 102,6, 100,0, 78,5, 70,3, 55,9, 55,9, 21,4, 6,8, 5,1 ppm; isómero anti: 8 = 157,6, 149,2, 149,1, 143,9, 143,1, 141,5, 136,8, 131,0, 128,6, 124,3, 122,6, 120,4, 111,1, 111,0, 102,5, 100,5, 79,5, 70,3, 56,0, 55,9, 23,6, 6,9, 5,1 ppm; HRMS (ESI-TOF): calc. para C27H36NOaSi+ [M+H+]: 482,2357, observado 482,2367.

Enino 27. A una suspensión agitada de Pd(PPh3)4 (380 mg, 0,3 mmol, 3 % en mol) y Cul (60 mg, 0,3 mmol, 3 % en mol) en 50 ml de Et2O a temperatura ambiente se le añadieron secuencialmente n-BuNH2 (1,61 g, 2,20 ml, 22,0 mmol, 2,0 equiv.), (Z)-1,2-dicloroetileno (2,03 g, 1,74 ml, 21,0 mmol, 1,9 equiv.) y triisopropilsililacetileno (2,00 g, 2,55 ml, 11,0 mmol, 1,0 equiv.) y la mezcla de reacción se agitó a temperatura ambiente durante 10 h (materiales volátiles, dejar la menor cantidad de fase de gas que sea posible en un matraz cerrado herméticamente). Después de la evaporación del disolvente, el residuo se recogió con hexanos y se filtró a través de un lecho corto de Celite®, eluyendo con hexanos, y el filtrado se concentró. La cromatografía en columna ultrarrápida (gel de sílice, hexanos) proporcionó un aceite incoloro (2,48 g, 10,2 mmol, rendimiento del 93 %). Fr (hexanos) = 0,77; IR (película) vmáx = 2945, 2154, 1463, 882, 671 cm-1; RMN 1H (500 MHz, CDCh): 8 = 6,39 (d, J = 7,5 Hz, 1H), 5,89 (d, J = 7,5 Hz, 1H), 1,09 (s, 18 H), 1,07 (s, 3 H); 13C RMN (125 MHz, CDCh): 8 = 129,2, 112,4, 100,5, 100,1, 18,6, 11,3 ppm; HRMS (El): calc. para C-i3H23S¡Cl: 242,1257, observado 242,1285.

Enodiino 28. A una suspensión agitada de Pd(PPh3)4 (140 mg, 0,12 mmol, 2,5 % en mol) y Cul (25 mg, 0,13 mmol, 2,5 % en mol) en 25 ml de Et2O a temperatura ambiente se le añadieron secuencialmente n-BuNH2 (731 mg, 1,0 ml, 10.0 mmol, 2,0 equiv.), el enino 27 (1,19 g, 4,9 mmol, 1,0 equiv.) y trimetilsililacetileno (972 mg, 1,40 ml, 9,9 mmol, 2.0 equiv.) y la mezcla de reacción se agitó a temperatura ambiente durante 12 h (materiales volátiles, dejar la menor cantidad de fase de gas que sea posible en un matraz cerrado herméticamente). Después de la evaporación del disolvente, el residuo se recogió con hexanos y se filtró a través de un lecho corto de Celite®, eluyendo con hexanos, y el filtrado se concentró. La cromatografía en columna ultrarrápida (gel de sílice, hexanos) proporcionó un aceite incoloro (1,44 g, 4,7 mmol, rendimiento del 96 %). Fr (hexanos) = 0,48; IR (película) vmáx = 2948, 2153, 2121, 1250, 1069, 848 cm-1; RMN 1H (500 MHz, CDCh): 8 = 5,84 (d, J = 11,3 Hz, 1H), 5,82 (d, J = 11,3 Hz, 1H), 1,09 (s, 18
H), 1,07 (s, 3 H), 0,18 (s, 9 H); 13C RMN (125 MHz, CDCI3): 5 = 120,5, 120,0, 103,7, 103,0, 102,0, 100,0, 18,7, 11,2, -0,3 ppm; HRMS (EI): calc. para C18H32SÍ2 : 304,2042, observado 304,2032.

Enodiino 11. A una solución agitada del enodiino 28 (1,38 g, 4,5 mmol, 1,0 equiv.) en 70 ml de benceno/metanol (1:1) a temperatura ambiente se le añadió K2CO3 (0,69 g, 5,0 mmol, 1,1 equiv.) y la mezcla de reacción se agitó a temperatura ambiente durante 2 h. Se añadió agua (50 ml) y la fase acuosa se extrajo con hexanos (3 x 50 ml). Las fases orgánicas se lavaron con salmuera (50 ml), se secaron sobre MgSO4 anhidro y se evaporaron para dejar un aceite de color amarillento (1,01 g, 4,3 mmol, 96%) (PRECAUCIÓN: 11 se vuelve lentamente de color negro durante el almacenamiento, lo cual conduce a rendimientos reducidos para las siguientes etapas, debe usarse fresco para obtener los mejores rendimientos). Fr (hexanos) = 0,66; IR (película) vmáx = 3303, 2945, 2149, 1463, 1049, 882 cm-1; RMN 1H (500 MHz, CDCla): 5 = 5,91 (dd, J = 11,0, 0,7 Hz, 1H), 5,81 (dd, J = 11,0, 1,2 Hz, 1H), 3,30 (dd, J = 1,2, 0,7 Hz, 1H), 1,08 (s, 18 H), 1,07 (s, 3 H); 13C RMN (125 MHz, CDCla): 5 = 119,3, 103,4, 100,3, 84,8, 80,9, 18,7, 11,2 ppm; HRMS (EI): calc. para C15H24Si: 189,1099, observado 189,1100.

Enodiino 12. A una solución agitada del enodiino recién preparado 11 (11,56 g, 49,76 mmol, 2,0 equiv.) en THF (116 ml) a 0 °C se le añadió cloruro de isopropilmagnesio (2,0 M en THF, 24,9 ml, 49,76 mmol, 2,0 equiv.) gota a gota. La mezcla de reacción se agitó a 25 °C durante 1 h, después se calentó brevemente con una pistola de calor a reflujo. De nuevo, la mezcla de reacción se agitó a 25 °C durante 1 h, después se calentó brevemente con una pistola de calor a reflujo. Finalmente, la mezcla de reacción se agitó a 25 °C durante 45 min y después se enfrió a 0 °C, seguido de la adición de una solución del TES lactol 10 (12,0 g, 24,88 mmol, 1,0 equiv.) en THF (54 ml) mediante una cánula. La mezcla resultante se agitó a 0 °C durante 1 h, seguido de la adición gota a gota de cloroformiato de alilo (5,29 ml, 49,76 mmol, 2,0 equiv.) durante 30 min a 0 °C. La mezcla de reacción se agitó a 25 °C durante 30 min, hasta que el análisis por TLC mostró el consumo completo de 10 (hexanos:EtOAc 3:2). Después, la mezcla de reacción se repartió entre EtOAc (150 ml) y H2O (150 ml). La capa acuosa se extrajo con EtOAc (3 x 150 ml) y las capas orgánicas combinadas se lavaron con NH4Cl ac. saturado (150 ml), NaHCO3 ac. saturado (150 ml), y salmuera (150 ml), después se secaron sobre MgSO4 y se concentraron. La cromatografía en columna ultrarrápida (gel de sílice, hexanos:EtOAc 10:1 a 4:1) proporcionó el enodiino 12 (17,87 g, 22,38 mmol, rendimiento del 90 %) en forma de un sólido de color amarillo pálido. 12: Fr = 0,43 (gel de sílice, hexanos:EtOAc 4:1); IR (película) vmáx = 2957, 2877, 2261, 1705, 1517, 1498, 1381, 1261, 1238 cm-1; 1H RMN (600 MHz, CD3CN): 5 = 7,58 (a, 1 H), 7,1-6,8 (m, 5 H), 6,45-6,40 (m, 1 H), 6,3-6,2 (m, 1 H), 5,99 (m, 1 H), 6,9-6,8 (m, 2 H), 5,35-4,95 (m, 5 H), 4,70 (m, 2 H), 3,80 (s, 3 H), 3,79 (s, 3 H), [1,46 (d, J = 6,5 Hz, 3 H) y 1,37 (d, J = 6,5 Hz, 3 H) par diastereotrópico], 1,11 (m, 21 H), 0,99 (m, 9 H), 0,70 (m, 6 H) ppm; HRMS (Es I-TOF): calc. para C46^ 4NOySi2+ [M+H+]: 798,4216, observado 798,4217.

Lactol 13. A una solución agitada del enodiino 12 (16,68 g, 20,89 mmol) en CH3CN (84 ml) y H2O (21 ml) a temperatura ambiente se le añadió AcOH (42 ml). La mezcla de reacción se agitó a 25 °C durante 2 h, se diluyó con EtOAc (100 ml) y se inactivó con NaHCO3 ac. saturado (100 ml). La capa orgánica se separó y la capa acuosa se extrajo con EtOAc (2 x 150 ml). Las capas orgánicas combinadas se lavaron con NaHCO3 ac. saturado (150 ml) y salmuera (150 ml), se secaron sobre MgSO4 y se concentraron. La cromatografía en columna ultrarrápida (gel de sílice, hexanos:EtOAc 4:1 a 2:1) proporcionó el lactol 13 (12,86 g, 18,79 mmol, rendimiento del 91 %) en forma de un sólido de color amarillo. 13: Fr = 0,28 (gel de sílice, hexanos:EtOAc 3:2); IR (película) vmáx = 3443, 2960, 2261, 2144,
1702, 1516, 1498, 1380, 1260 cm-1; 1H RMN (600 MHz, CD3CN): 5 = 7,56 (a, 1 H), 7,1-6,9 (m, 5 H), 6,3-6,2 (m, 2 H), 6,00 (m, 1 H), 5,9-5,8 (m, 2 H), 5,4-5,3 (m, 1 H), 5,25 (m, 1 H), 5,25-4,9 (m, 3 H), 4,8-4,5 (m, 3 H), 3,80 (s, 3 H), 3,79 (s, 3 H), [1,44 (d, J = 6,5 Hz, 3 H) y 1,35 (d, J = 6,5 Hz, 3 H) par diastereotrópico], 1,11 (m, 21 H), [0,19 (s, 9 H) par diastereotópico] ppm; HRMS (ESI-To F): calc. para C4qH5qN07 SÍ+ [M+H+]: 684,3351, observado 684,3348.

Epóxido 14. A una solución agitada del lactol 13 (12,00 g, 17,5 mmol, 1,0 equiv.) en MeOH (117 ml) a 0 °C se le añadió en porciones NaBH4 (863 mg, 22,8 mmol, 1,3 equiv.) (PRECAUCIÓN: desprendimiento de gas). La mezcla de reacción se agitó a 0 °C durante 20 min, hasta que el análisis por TLC mostró el consumo completo de 13 (hexanos:EtOAc 1:1), después se diluyó con salmuera (200 ml) y se extrajo con CH2O 2 (3 x 200 ml). Los extractos orgánicos combinados se secaron sobre MgSO4, se concentraron y se recogieron en CH2Cl2 (100 ml). A esta solución at 0 °C se le añadió NaHCO3 en polvo (2,94 g, 35,0 mmol, 2,0 equiv.), seguido de la adición gota a gota de una solución fría (0 °C) de m-CPBA (pureza del 90%, 3,47 g, 24,5 mmol, 1,4 equiv.) en CH2Cl2 (17 ml) mediante una cánula. La mezcla de reacción se agitó a 0 °C durante 4 h (PRECAUCIÓN: si la temperatura de la reacción superaba 0 °C, el carbamato de alilo se epoxidaría) y después se inactivó con NaHCO3 ac. saturado (50 ml) y Na2S2O3 ac. saturado (50 ml). La capa orgánica se separó y la capa acuosa se extrajo con CH2O 2 (2 x 200 ml). Las capas orgánicas combinadas se secaron sobre MgSO4 y se concentraron. La cromatografía en columna ultrarrápida (gel de sílice, hexanos:EtOAc 3:2 a 1:1) proporcionó el epóxido 14 (10,01 g, 14,23 mmol, rendimiento del 81 % en dos etapas) en forma de un sólido de color amarillo. 14: Fr = 0,47 (gel de sílice, hexanos:EtOAc 3:7); [a]D25 = 200,0° (c = 1,00, CH2Cl2); IR (película) vmáx = 3475, 2940, 1703, 1505, 1261 cm-1; 1H RMN (600 MHz, CD3CN): 5 = 7,37 (d, J = 2,5 Hz, 1 H), 7,25 (a, 1 H), 7,06 (d, J = 1,9 Hz, 1 H), 7,00 (dd, J = 8,2, 1,9 Hz, 1 H), 6,96 (dd, J = 8,7, 2,6 Hz, 1 H), 6,93 (d, J = 8,2 Hz, 1 H), 6,03 (a, 1 H), 5,89 (a, 1 H), 5,88 (d, J = 11,0 Hz, 1 H), 5,73 (dd, J = 11,0, 1,3 Hz, 1 H), 5,45 5,10 (a, 2 H), 5,02 (sistema AB, 2 H), 4,75-4,45 (a, 2 H), 4,22 (cd, J = 6,8, 4,5 Hz, 1 H), 4,19 (dd, J = 13,0, 4,9 Hz, 1 H), 4,04 (dd, J = 11,9, 5,1 Hz, 1 H), 3,80 (s, 3 H), 3,80 (s, 3 H), 3,64 (d, J = 3,5 Hz, 1 H), 3,35 (t, J = 5,1 Hz, 1 H), 1,43 (d, J = 6,9 Hz, 3 H), 1,10 (s, 21 H) ppm; 13C RMN (150 MHz, CD3CN): 5 = 157,1, 155,4, 149,9, 149,9, 133,4, 130,2, 130,0, 129,6, 129,1, 121,5, 121,4, 120,2, 117,8, 115,7, 114,9, 112,6, 112,2, 104,4, 100,6, 94,1, 82,8, 77,8, 70,8, 68,1, 67,4, 62,2, 60,3, 56,1, 45,0, 21,3, 18,9, 11,8 ppm; HRMS (ESI-TOF): calc. para C40H52NOaSi+ [M+H+]: 702,3457, observado 702,3460.

Acetato 15. A una solución agitada del epóxido 14 (11,0 g, 15,67 mmol, 1,0 equiv.) en CH2Cl2 (261 ml) a -78 °C se le añadieron N,N-diisopropiletilamina (5,45 ml, 31,32 mmol, 2,0 equiv.) y AcCl (1,12 ml, 15,67 mmol, 1,0 equiv.). La mezcla resultante se agitó a -78 °C durante 16 h. La reacción se interrumpió con NaHCO3 ac. saturado (150 ml) y se extrajo con CH2Cl2 (2 x 200 ml). Las capas orgánicas combinadas se secaron sobre MgSO4 y se concentraron. La cromatografía en columna ultrarrápida (gel de sílice, hexanos:EtOAc 3:2 a 1:1) proporcionó el acetato 15 (9,45 g, 13,45 mmol, rendimiento del 81 %) en forma de una espuma de color amarillo. 15: Fr = 0,17 (gel de sílice, hexanos:EtOAc 3:2); [a]D25 = -60,4° (c = 1,00, CH2Ch); IR (película) vmáx = 3485, 2941, 2136, 1708, 1504, 1382, 1236 cm' 1; 1H RMN (600 MHz, CD3CN): 5 = 7,23 (a, 1 H), 7,10 (d, J = 2,8 Hz, 1 H), 7,04 (d, J = 1,9 Hz, 1 H), 7,00 (dd, J = 8,8, 2,7 Hz, 1 H), 6,98 (dd, J = 8,2, 1,9 Hz, 1 H), 6,92 (d, J = 8,2 Hz, 1 H), 6,09 (s, 1 H), 5,90 (a, 1 H), 5,88 (d, J = 11,0 Hz, 1 H), 5,73 (dd, J = 11,3, 1,9 Hz, 1 H), 5,3-5,1 (a, 2 H), 5,02 (s, 2 H), 4,7-4,5 (a, 4 H), 4,18 (cd, J = 6,8, 4,4 Hz, 1 H), 3,80 (s, 3 H), 3,80 (s, 3 H), 3,65 (d, J = 3,5 Hz, 1 H), 2,04 (s, 3 H), 1,43 (d, J = 7,0 Hz, 3 H), 1,11 (s, 21 H) ppm; 13C RMN (150 MHz, CD3CN): 5 = 171,1, 158,2, 157,4, 155,4, 150,2, 150,0, 133,5,130,3, 130,0, 129,6, 128,7, 121.4, 120,2, 115,8, 115,0, 112,6, 112,5, 104,5, 100,8, 93,7, 83,3, 78,4, 71,0, 68,1, 67,6, 62,2, 60,7, 56,3, 56,3, 44,8, 21.4, 20,9, 19,0, 11,9 ppm; HRMS (ESI-TOF): calc. para C42Ha4NOgSi+ [M+H+]: 744,3562, observado 744,3556.

Cetona 16. A una solución agitada del acetato 15 (9,0 g, 12,09 mmol, 1,0 equiv.) en CH2CI2 (128 ml) se le añadieron NaHCO3 en polvo (4,06 g, 48,36 mmol, 4,0 equiv.) y peryodinano de Dess-Martin (10,26 g, 24,18 mmol, 2,0 equiv.) a 0 °C. La mezcla resultante se agitó a 25 °C durante 2 h y después se inactivó con Na2S2O3 ac. saturado (150 ml). La capa orgánica se separó y la capa acuosa se extrajo con CH2Cl2 (2 x 200 ml). Los extractos orgánicos combinados se lavaron con NaHCO3 ac. saturado (150 ml) y salmuera (150 ml), se secaron sobre MgSO4 y se filtraron a través de un lecho corto de Celite® (lavado con EtOAc antes de usarse). Los sólidos se aclararon con EtOAc y los filtrados combinados se concentraron hasta aprox. 5 ml en volumen. La cromatografía en columna ultrarrápida (gel de sílice, hexanos:EtOAc 2:1) proporcionó la cetona 16 (8,35 g, 11,25 mmol, rendimiento del 93 %) en forma de un aceite de color amarillo. 16: Fr = 0,30 (gel de sílice, hexanos:EtOAc 3:2); [a]o25 = 41,8° (c = 1,00, CH2Ch); IR (película) vmáx = 2958, 2142, 1747, 1712, 1505, 1384, 1308, 1222 cm-1; 1H RMN (600 MHz, CD3CN): 8 = 7,33 (d a, J = 7,3 Hz, 1 H), 7,16 (d, J = 2,7 Hz, 1 H), 7,07 (dd, J = 8 ,8 , 2,8 Hz, 1 H), 7,05 (d, J = 1,9 Hz, 1 H), 6,99 (dd, J = 8,1, 1,9 Hz, 1 H), 6,93 (d, J = 8,2 Hz, 1 H), 6,05 (s a, 1 H), 5,89 (a, 1 H), 5,89 (d, J = 11,3 Hz, 1 H), 5,76 (dd, J = 11,2, 1,9 Hz, 1 H), 5,25 5,15 (a, 2 H), 5,04 (s, 2 H), 4,73 (d, J = 12,8 Hz, 1 H), 4,7-4,5 (a, 2 H), 4,55 (d, J = 12,8 Hz, 1 H), 3,80 (s, 3 H), 3,80 (s, 3 H), 2,49 (s, 3 H), 2,01 (s, 3 H), 1,11 (s, 21 H) ppm; 13C RMN (150 MHz, CD3CN): 8 = 200,1, 170,6, 157,6, 155,6, 150,1, 150,0, 133,2, 130,1, 129,9, 129,4, 126,7, 121,9, 121,4, 119,8, 116,6, 114,8, 112,6, 112,4, 104,5, 102,3, 91,5, 83,9, 76,6, 71,0, 67,8, 61,3, 61,1, 56,3, 46,2, 29,9, 20,7, 19,0, 11,9 ppm; HRMS (ESI-TOF): calc. para C42H52NOgSi+ [M+H+]: 742,3406, observado 742,3400.

Alcohol secundario 17. A una solución agitada de la cetona 16 (1,94 g, 2,62 mmol, 1,0 equiv.) en THF (20 ml) se le añadió una solución premezclada de HOAc/TBAF en THF (HOAc: 394 mg, 6,55 mmol, 2,5 equiv.; TBAF: 6,55 ml, 1,0 M en THF, 6,55 mmol, 2,5 equiv.) a 0 °C, la mezcla de reacción se agitó a 15 °C durante 1 h y después se añadió MeOH (25 ml) a 0 °C seguido de la adición en porciones de NaBH4 (200 mg, 5,25 mmol, 2,0 equiv.) (PRECAUCIÓN: desprendimiento de gas, si se añade demasiado rápido, la p.d. disminuirá). La mezcla resultante se agitó a 0 °C durante 30 min y después se inactivó con H2O (10 ml). La mezcla de reacción se evaporó cuidadosamente hasta aprox. 20 ml en volumen, y después se repartió entre EtOAc (100 ml) y H2O (60 ml). La capa acuosa se extrajo con EtOAc (3 x 100 ml) y las capas orgánicas combinadas se secaron sobre MgSO4 y se concentraron para producir el alcohol secundario 17 (1,43 g, 2,44 mmol, rendimiento del 93 %, > 25:1 de p.d.) en forma de un sólido de color amarillo ( PRECAUCIÓN: 17 se vuelve lentamente de color negro durante el almacenamiento, lo cual conduce a rendimientos reducidos para las siguientes etapas, debe usarse fresco para obtener los mejores rendimientos). 17: Fr = 0,19 (gel de sílice, hexanos:EtOAc 3:2); [a]o25 = 158,9° (c = 1,00, CH2O 2); IR (película) vmáx = 3508, 3283, 2931, 1740, 1706, 1507, 1263, 1239, 1027 cm-1; 1H RMN (600 MHz, CD3CN): 8 = 7,27 (d a, J = 8 ,6 Hz, 1 H), 7,14 (d, J = 2,8 Hz, 1 H), 7,05 (d, J = 1,6 Hz, 1 H), 7,01 (dd, J = 8 ,8 , 2,8 Hz, 1 H), 6,99 (dd, J = 8,1, 1,6 Hz, 1 H), 6,93 (d, J = 8,2 Hz, 1 H), 6,05-5,83 (a, 2 H), 5,81 (sistema AB, 2 H), 5,40-5,12 (a, 2 H), 5,03 (sistema AB, 2 H), 4,76 (dd, J = 18,1, 13,0 Hz, 2 H), 4,70-4,50 (a, 2 H), 4,23 (cd, J = 6,2, 4,6 Hz, 1 H), 3,80 (s, 3 H), 3,80 (s, 3 H), 3,47 (s, 1 H), 3,15 (d, J = 4,7 Hz, 1 H), 2,04 (s, 3 H), 1,43 (d, J = 6,4 Hz, 3 H) ppm; 13C RMN (150 MHz, CD3CN): 8 = 171,2, 157,4, 155.7, 150,2, 150,0, 133,4, 130,4, 129,8, 128,8, 121,3, 121,3, 120,7, 118,0, 115,9, 115,1, 112,6, 112,5, 93,3, 86,9, 82.7, 81,0, 77,2, 71,0, 67,6, 67,1, 62,0, 61,2, 60,9, 56,3, 45,8, 21,0, 20,1 ppm; HRMS (ESI-TOF): calc. para C33H34NO9+ [M+H+]: 588,2228, observado 588,2224.

Alcohol primario 18. A una solución agitada del acetato 17 (1,43 g, 2,44 mmol, 1,0 equiv.) en DMF (15 ml) a 0 °C se le añadieron imidazol (332 mg, 4,88 mmol, 2,0 equiv.) y TESCl (551 mg, 3,66 mmol, 1,5 equiv.). La mezcla de reacción se agitó a 0 °C durante 15 min y a 25 °C durante 5 min, después se inactivó con NaHCO 3 ac. saturado (50 ml) y se extrajo con Et 2 O (3 x 50 ml). Los extractos orgánicos combinados se lavaron con H 2 O (3 x 50 ml) y salmuera (50 ml), se secaron sobre MgSO 4 y se concentraron. El residuo resultante se recogió en THF (20 ml) y se enfrió a 0 °C. A esta solución se le añadió una solución saturada fría (0-5 °C) de K 2 CO 3 en MeOH (2,5 ml) y la mezcla de reacción se agitó a -10 °C durante 20 min (PRECAUCIÓN: 18 experimenta lentamente la desprotección de TES a esta temperatura, por lo que la reacción debería controlarse cuidadosamente a intervalos de 2 min). Después, la mezcla resultante se repartió entre EtOAc (50 ml) y tampón pH 6,8 (50 ml). La capa acuosa se extrajo con EtOAc (2 x 50 ml) y después las capas orgánicas combinadas se lavaron con salmuera (50 ml), se secaron sobre MgSO 4 y se concentraron hasta aprox. 5 ml en volumen. La cromatografía en columna ultrarrápida (gel de sílice, hexanos:EtOAc 3:1 a 2:1) proporcionó el alcohol primario 18 (1,36 g, 2,07 mmol, rendimiento del 85 % en dos etapas) en forma de un sólido de color amarillo (PRECAUCIÓN: 18 se vuelve lentamente de color negro durante el almacenamiento, lo cual conduce a rendimientos reducidos para las siguientes etapas, debe usarse fresco para obtener los mejores rendimientos). 18: F r = 0,42 (gel de sílice, hexanos:EtOAc 3:2); [a]o25 = 47,2° (c = 1,00, CH 2 Cl 2 ); IR (película) vmáx = 3493, 3286, 2955, 2876, 1701, 1504, 1387, 1261, 1237 cm -1 ; 1 H RMN (600 MHz, CD 3 CN): 8 = 7,39 (d, J = 2,8 Hz, 1 H), 7,22 (d a, J = 8,8 Hz, 1 H), 7,06 (d, J = 1,6 Hz, 1 H), 7,01 (dd, J = 8,2, 1,9 Hz, 1 H), 6,96 (dd, J = 8,8, 2,8 Hz, 1 H), 6,93 (d, J = 8,2 Hz, 1 H), 6,20-5,80 (a, 2 H), 5,76 (sistema AB, 2 H), 5,47-5,05 (a, 2 H), 5,03 (sistema AB, 2 H), 4,80 4,45 (a, 2 H), 4,38 (c, J = 6,3 Hz, 1 H), 4,24 (dd, J = 12,5, 5,7 Hz, 1 H), 4,17 (dd, J = 12,5, 5,0 Hz, 1 H), 3,81 (s, 3 H), 3,80 (s, 3 H), 3,40 (d, J = 1,3 Hz, 1 H), 3,15 (t, J = 5,5 Hz, 1 H), 1,43 (d, J = 6,3 Hz, 3 H), 0,95 (t, J = 7,9 Hz, 9 H), 0,63 (c, J = 7,9 Hz, 6 H) ppm; 13 C RMN (150 MHz, CD 3 CN): 8 = 157,2, 155,8, 150,1, 150,0, 133,7, 133,5, 130,5, 130,3, 129,8, 129,5, 129,0, 121,6, 121,6, 120,3, 117,9, 116,0, 115,1, 112,8, 112,5, 94,4, 86,6, 81,6, 81,0, 76,8, 70,9, 67,9, 67,5, 63,4, 60,1, 56,3, 45,9, 21,5, 7,2, 5,5 ppm; HRMS (ESI-TOF): calc. para C 37 H 46 NOsSi+ [M+H+]: 660,2987, observado 660,2989.

Aldehido 19. A una solución agitada del alcohol primario 18 (1,25 g, 1,90 mmol, 1,0 equiv.) en CH 2 Cl 2 (20 ml) a 0 °C se le añadieron NaHCO 3 en polvo (639 mg, 7,60 mmol, 4,0 equiv.), y peryodinano de Dess-Martin (3,22 g, 3,80 mmol, 2,0 equiv.). La mezcla de reacción se agitó a 25 °C durante 1,5 h, y después se repartió entre CH 2 Cl 2 (30 ml) y Na 2 S 2 O 3 ac. saturado (30 ml). La capa acuosa se extrajo con CH 2 Cl 2 (2 x 30 ml) y las capas orgánicas combinadas se lavaron con salmuera (30 ml), se secaron sobre MgSO 4 y se filtraron a través de un lecho corto de Celite® (lavado con EtOAc antes de usarse). Los sólidos se aclararon con EtOAc y los filtrados combinados se concentraron hasta aprox. 5 ml en volumen. La cromatografía en columna ultrarrápida (gel de sílice, hexanos:EtOAc 4:1) proporcionó el aldehído 19 (1,12 g, 1,71 mmol, rendimiento del 90 %) en forma de un aceite de color amarillo (PRECAUCIÓN: 19 se vuelve lentamente de color negro durante el almacenamiento, lo cual conduce a rendimientos reducidos para las siguientes etapas, debe usarse fresco para obtener los mejores rendimientos). 19: Fr = 0,47 (gel de sílice, hexanos:EtOAc 3:2); [a]o25 = 86,9° (c = 1,00, CH 2 Ch); IR (película) vmáx = 3282, 2956, 2877, 2261, 1708, 1502, 1385, 1263, 1239 cm ' 1 ; 1 H RMN (600 MHz, CD 3 CN): 8 = 9,83 (s, 1 H), 7,31 (d a, J = 7,4 Hz, 1 H), 7,21 (d, J = 2,8 Hz, 1 H), 7,04 (d, J = 1,9 Hz, 1 H), 7,02 (dd, J = 8,8, 2,8 Hz, 1 H), 6,98 (dd, J = 8,2, 1,9 Hz, 1 H), 6,92 (d, J = 8.2 Hz, 1 H), 6,0-5,8 (a, 2 H), 5,80 (m, 2 H), 5,3-5,1 (a, 2 H), 5,00 (doblete AB, 2 H), 4,7-4,5 (a, 2 H), 4,38 (c, J = 6.3 Hz, 1 H), 3,80 (s, 3 H), 3,80 (s, 3 H), 3,44 (d, J = 1,8 Hz, 1 H), 1,43 (d, J = 6,4 Hz, 3 H), 0,96 (t, J = 8,0 Hz, 9 H), 0,63 (c, J = 8,0 Hz, 6 H) ppm; 13 C RMN (150 MHz, CD 3 CN): 8 = 196,3, 157,0, 155,6, 150,1, 150,0, 133,3, 130,2, 129,9, 125,2, 121,5, 121,1, 121,0, 118,0, 116,2, 115,0, 112,7, 112,4, 92,7, 87,0, 83,1, 80,9, 80,5, 70,9, 67,7, 67,5, 64,6, 56,2, 46,2, 22,2, 7,0, 5,4 ppm; HRMS (ESI-TOF): calc. para C 3yH 44 NOaSi+ [M+H+]: 658,2831, observado 658,2846.

Fenol 20. A una solución agitada del aldehido 19 (1,12 g, 1,71 mmol, 1,0 equiv.) en CH2CI2 (50 ml) a temperatura ambiente se le añadieron tampón pH 6 ,8 (5,0 ml) y DDQ (1,16 g, 5,13 mmol, 3,0 equiv.). El matraz de reacción se envolvió con papel de aluminio y la mezcla resultante se agitó a temperatura ambiente durante 12 h, hasta que el análisis por TLC mostró el consumo completo de 19 (EtOAc al 4% en CH2Ch). Después, la mezcla de reacción se repartió entre CH2Cl2 (100 ml) y tampón pH 6 ,8 (100 ml). La capa acuosa se extrajo con CH2O 2 (2 x 100 ml) y las capas orgánicas combinadas se lavaron con tampón pH 6 ,8 (50 ml), H2O (50 ml) y salmuera (50 ml), se secaron sobre MgSO4 y se filtraron a través de un lecho corto de Celite® (lavado con EtOAc antes de usarse). Los sólidos se aclararon con EtOAc al 5% en CH2Cl2 y los filtrados combinados se concentraron hasta aprox. 5 ml en volumen. La cromatografía en columna ultrarrápida (CH2O 2: EtOAc 1:0 a 30:1, después hexanos: EtOAc 4:1 después de que se eluyera completamente 3,4-dimetoxi-benzaldehído) proporcionó el fenol 20 ( PRECAUCIÓN: 20 era sensible a trazas de HCl en CH 2 CI 2 , se realizó la concentración de las fracciones hasta aprox. 20 ml, después se diluyó con 20 ml de tolueno; se repitieron las mismas operaciones 3 veces y después concentrar a sequedad) (810 mg, 1,59 mmol, rendimiento del 93 %) en forma de un aceite de color amarillo (PRECAUCIÓN: 20 se vuelve lentamente de color negro durante el almacenamiento, lo cual conduce a rendimientos reducidos para las siguientes etapas, debe usarse fresco para obtener los mejores rendimientos). 20: F r = 0,44 (gel de sílice, hexanos:EtOAc 3:2); [a]o25 = 121,2° (c = 1,00, CH2Cl2); IR (película) vmáx = 3296, 2956, 1680, 1505, 1392, 1315 cm-1; 1H RMN (500 MHz, CD3CN): 8 = 9,79 (s, 1 H), 7,22 (d a, J = 8,4 Hz, 1 H), 7,15 (s a, 1 H), 7,06 (d, J = 2,8 Hz, 1 H), 6,84 (dd, J = 8 ,6 , 2,6 Hz, 1 H), 5,90 (s a, 1 H), 5,81 (s, 2 H), 5,3-5,1 (a, 2 H), 4,7-4,5 (a, 2 H), 4,38 (c, J = 6,4 Hz, 1 H), 3,49 (s, 1 H), 1,42 (d, J = 6,4 Hz, 3 H), 0,96 (t, J = 8,0 Hz, 9 H), 0,63 (c, J = 8,0 Hz, 6 H) ppm; 13C RMN (125 MHz, CD3CN): 8 = 196,4, 155,3, 133,4, 130,1, 125,2, 121,1, 121,0, 117,9, 116,7, 115,1, 92,8, 86,9, 83,1, 68,2, 67,6, 67,5, 64,6, 46,3, 22,2, 7,0, 5,4 ppm; HRMS (ESI-TOF): calc. para C2sH34NO6Si+ [M+H+]: 508,2150, observado 508,2140.

Enodiino cíclico 21a. Un matraz de fondo redondo de 250 ml que contenía CeCl3 anhidro disponible comercialmente (perlas, malla -10, 1,57 g, 6,36 mmol, 4,0 equiv.) se calentó a 110 °C (temperatura del baño de aceite) a alto vacío con agitación vigorosa durante 16 h para proporcionar un polvo de color blanco. El matraz se lavó abundantemente con Ar y se añadió THF (50 ml) para formar una suspensión turbia, que se sonicó durante 2 h para proporcionar una suspensión lechosa. Una solución del fenol 20 (810 mg, 1,59 mmol, 1,0 equiv.) en THF (50 ml) se añadió a la suspensión preformada de CeCl3 mediante una cánula con sonicación durante 20 min a temperatura ambiente. La mezcla resultante se agitó a temperatura ambiente durante 30 min y después se enfrió a -78 °C, seguido de la adición gota a gota de KHMDS (1,0 M en THF, 9,60 ml, 9,60 mmol, 6,0 equiv.), tiempo durante el cual el color de la mezcla de reacción se volvió de amarillo claro a pardo, y después pardo oscuro. La mezcla de reacción se agitó a -78 °C durante 1 h, después se calentó lentamente a -40 °C durante 2 h y se inactivó mediante la adición de AcOH (1,0 M en THF, 19,1 ml, 19,1 mmol, 12,0 equiv.) a la misma temperatura. Después de que se completara la adición, la mezcla de reacción se dejó calentar a temperatura ambiente, se concentró cuidadosamente hasta aprox. 40 ml en volumen y después se repartió entre EtOAc (100 ml) y tampón pH 6 ,8 (100 ml). La capa acuosa se extrajo con EtOAc (2 x 100 ml) y las capas orgánicas combinadas se lavaron con H2O (50 ml) y salmuera (50 ml), se secaron sobre MgSO4 y se filtraron a través de un lecho corto de Celite® (lavado con EtOAc antes de usarse). Los sólidos se aclararon con EtOAc y los filtrados combinados se concentraron hasta aprox. 5 ml en volumen. La cromatografía en columna ultrarrápida (gel de sílice, hexanos:EtOAc 4:1 a 3:1) proporcionó el enodiino cíclico 21a (601 mg, 1,18 mmol, rendimiento del 74 %) en forma de un sólido de color blanquecino y una mezcla de 21a y su epímero C 17 21b (154 mg, 21a:21b 1:3, rendimiento del 19 %) en forma de un sólido de color amarillento.
21a: Fr = 0,36 (gel de sílice, hexanos:EtOAc 3:2); [a]o25 = 477,8° (c = 0,50, CH2Ch); IR (película) vmáx = 3395, 2955, 2877, 1682, 1504, 1395, 1281 cm ' 1; 1H RMN (600 MHz, CD3CN): 8 = 7,94 (d, J = 2,8 Hz, 1 H), 7,14 (a, 1 H), 6,95 (s, 1 H), 6,73 (dd, J = 8,7, 2,8 Hz, 1 H), 5,91 (a, 1 H), 5,90 (d, J = 10,0 Hz, 1 H), 5,83 (s, 1 H), 5,75 (d, J = 9,9 Hz, 1 H),
5,35-5,15 (a, 2 H), 5,05 (d, J = 4,7 Hz, 1 H), 4,75-4,55 (a, 2 H), 4,54 (c, J = 6,3 Hz, 1 H), 4,24 (d, J = 4,7 Hz), 1,38 (d, J = 6,2 Hz, 3 H), 0,98 (t, J = 8,0 Hz, 9 H), 0,65 (m, 6 H) ppm; 13C RMN (150 MHz, CD3CN): 8 = 154,3, 133,5, 130,2, 128,3, 124,5, 123,9, 118,3, 117,7, 115,4, 100,3, 97,1, 91,3, 77,2, 67,3, 66,8, 66,0, 64,9, 60,9, 47,4, 22,3, 7,2.
5,6 ppm; HRMS (ESI-TOF): calc. para C2sH34NO6Si+ [M+H+]: 508,2150, observado 508,2140

Quinona aminal 22a. A una solución agitada del enodiino cíclico 21a (601 mg, 1,18 mmol, 1,0 equiv.) en MeOH (40 ml) a 0 °C se le añadió una solución de PhI(OAc)2 (420 mg, 1,30 mmol, 1,1 equiv.) en MeOH (20 ml) mediante una cánula. La mezcla resultante se agitó a 0 °C durante 10 min y a 25 °C durante 5 min, después se repartió entre EtOAc (100 ml) y NaHCO3 ac. semisaturado (100 ml). La capa acuosa se extrajo con EtOAc (2 x 50 ml) y las capas orgánicas combinadas se lavaron con salmuera (50 ml), se secaron sobre MgSO4 y se filtraron a través de un lecho corto de Celite® (lavado con EtOAc antes de usarse). Los sólidos se aclararon con EtOAc y los filtrados combinados se concentraron hasta aprox. 3 ml en volumen. La cromatografía en columna ultrarrápida (gel de sílice, hexanos:EtOAc 4:1 a 3:1) proporcionó la quinona aminal 22a (529 mg, 0,983 mmol, rendimiento del 83 %) en forma de un sólido de color blanco. 22a: Fr = 0,30 (gel de sílice, hexanos:EtOAc 3:2); [a]D25 = 592,2° (c = 0,50, CH2O 2); IR (película) vmáx = 3448, 2954, 2877, 1707, 1666, 1390, 1282 cm-1; 1H RMN (600 MHz, CD3CN): 8 = 7,54 (a, 1 H), 7,04 (d, J = 1,9 Hz, 1 H), 6,27 (dd, J = 10,4, 2,0 Hz, 1 H), 6,05-5,95 (m, 3 H), 5,86 (d, J = 9,9 Hz, 1 H), 5,36 (d, J = 17,0 Hz, 1 H), 5,25 (d, J = 10,5 Hz, 1 H), 4,83 (d, J = 4,8 Hz, 1 H), 4,8-4,6 (a, 2 H), 4,54 (c, J = 6,3 Hz, 1 H), 4,25 (d, J = 5,0 Hz, 1 H), 3,03 (s, 3 H), 1,34 (d, J = 6,3 Hz, 3 H), 0,96 (t, J = 8,0 Hz, 9 H), 0,63 (m, 6 H) ppm; 13C RMN (150 MHz, CD3CN): 8 = 184,8, 138,6, 133,4, 128,7, 124,8, 124,6, 99,7, 98,1, 92,3, 89,0, 70,0, 66,5, 63,8, 51,3, 46,8, 22,4, 7,2, 5,6 ppm; HRMS (ESI-TOF): calc. para C2gH36NOySi+ [M+H+]: 538,2255, observado 538,2247.

Antraquinona 25a. A una solución de 3-ciano-1(3H)-isobenzofuranona (23, 34 mg, 0,22 mmol, 3,0 equiv.) en THF (1,0 ml) a -78 °C se le añadió LiHMDS (1,0 M en Th F, 0,29 ml, 0,29 mmol, 4,0 equiv.). La mezcla resultante se agitó a -78 °C durante 20 min y se añadió una solución enfriada previamente de la quinona aminal 22a (39 mg, 72 pmol, 1,0 equiv.) en THF (1,0 ml) a -78 °C mediante una cánula. Después de agitar durante 5 min a -78 °C, la mezcla de reacción se calentó a temperatura ambiente y se agitó durante 50 min más, tiempo durante el cual la mezcla de reacción se volvió de color rojo oscuro y el análisis por TLC mostró el consumo completo de 22a (EtOAc al 8% en CH2Cl2). Después, la mezcla de reacción se inactivó mediante la adición de tampón pH 6,8 (20 ml) y se extrajo con EtOAc (3 x 20 ml). Los extractos orgánicos combinados se lavaron con salmuera (20 ml), se secaron sobre MgSO4 y se filtraron a través de un lecho corto de Celite® (lavado con EtOAc antes de usarse). Los sólidos se aclararon con EtOAc y los filtrados combinados se concentraron para producir la alloc-antraquinona en bruto 24 en forma de un sólido de color rojo oscuro, que se disolvió en THF desgasificado (1,5 ml) en una atmósfera de Ar y se enfrió a 0 °C. A esta solución se le añadió Pd(PPh3)4 (6 mg, 5,2 pmol, 0,08 equiv.), seguido de la adición gota a gota de morfolina (16 mg, 0,17 mmol, 2,4 equiv.), el matraz de reacción se envolvió con papel de aluminio y la mezcla resultante se dejó en agitación a 0 °C durante 2 h, tiempo durante el cual la mezcla de reacción se volvió de color púrpura oscuro. El baño de refrigeración se retiró y la mezcla de reacción se agitó a temperatura ambiente durante 20 min y después se inactivó mediante la adición de tampón pH 6,8 (20 ml) y se extrajo con EtOAc (3 x 20 ml). Los extractos orgánicos combinados se lavaron con NaHCO3 ac. saturado (20 ml) y salmuera (20 ml), se secaron sobre MgSO4 y se filtraron a través de un lecho corto de Celite® (lavado con EtOAc antes de usarse). Los sólidos se aclararon con EtOAc y los filtrados combinados se concentraron hasta aprox. 1 ml en volumen. La cromatografía en columna ultrarrápida [gel de sílice desactivado (véanse los Métodos Generales), hexanos:EtOAc 2:1 a 1:1] proporcionó la antraquinona 25a (29 mg, 52 pmol, rendimiento del 73 %) en forma de un sólido de color púrpura. 25: Fr = 0,58 (gel de sílice, hexanos:EtOAc 3:2); [a]o25 = 2600° (c = 0,002, EtOAc); IR (película) vmáx = 3429, 2955, 2876, 1788, 1620, 1587, 1487, 1277, 1234 cm ' 1; 1H RMN (600 MHz, CD3CN): 8 = 13,17 (s, 1 H), 9,98 (d, J = 4,0 Hz, 1 H), 8,48 (s, 1 H), 8,29 (d, J = 7,9 Hz, 1 H), 8,27 (d, J = 8,0 Hz, 1 H), 7,86 (t, J = 7,7 Hz, 1 H), 7,81 (t, J = 7,0 Hz, 1 H), 5,95 (d, J = 9,9 Hz, 1 H), 5,88 (d, J = 10,1 Hz, 1 H), 5,12 (d, J = 5,0 Hz, 1 H), 4,99 (d, J = 4,4 Hz, 1 H), 4,56 (c, J = 6,3 Hz, 1 H), 4,43 (d, J =
5,0 Hz, 1 H), 1,39 (d, J = 6,4 Hz, 3 H), 0,99 (t, J = 8,0 Hz, 9 H), 0,67 (c, J = 7,7 Hz, 6 H) ppm; 13C RMN (150 MHz, CD3CN): 8 = 188,5, 184,2, 156,6, 144,7, 136,2, 135,8, 135,5, 134,3, 133,6, 130,9, 127,7, 127,0, 124,8, 123,8, 114,2, 112,3, 100,2, 99,8, 91,3, 88,4, 77,3, 66,8, 64,8, 44,3, 22,6, 7,2, 5,6 ppm; HRMS (ESI-TOF): calc. para C32H32NO6Si+ [M+H+]: 554,1993, observado 554,1992.

Uncialamicina (26a). A una solución de la antraquinona 25a (29 mg, 52 |jmol, 1,0 equiv.) en THF desgasificado (9,0 ml) a temperatura ambiente se le añadió una solución de 1:1 de 3HF^Et3N:THF (3,0 ml). El matraz de reacción cerrado herméticamente se envolvió con papel de aluminio y la mezcla de reacción se agitó a temperatura ambiente durante 1,5 h, y después se repartió entre EtOAc (30 ml) y NaHCO3 ac. saturado (30 ml). La capa orgánica se lavó con salmuera (30 ml), se secó sobre Na2SO4 y se concentró hasta aprox. 1 ml en volumen. La cromatografía en columna ultrarrápida [gel de sílice desactivado (véanse los Métodos Generales), hexanos:EtOAc 1:1 a 1:2] proporcionó la uncialamicina (26a) (23 mg, 52 jmol, rendimiento del 99 %) en forma de un sólido de color púrpura.
26: p.f. = 175 °C (descomp., EtOAc); Fr = 0,14 (gel de sílice, hexanos:EtOAc 3:2); [a]D25 = 23 00° (c = 0,002, EtOAc); IR (película) vmáx = 3429, 2926, 1715, 1620, 1587, 1484, 1355, 1234 cm-1; RMN 1H (600 MHz, DMSO-de): 8 = 13,19 (s, 1 H), 10,01 (d, J = 4,5 Hz, 1 H), 8,53 (s, 1 H), 8,24 (solapamiento de dobletes, 2 H), 7,94 (td, J = 7,4, 1,1 Hz, 1 H), 7,89 (td, J = 7,4, 1,1 Hz, 1 H), 6,68 (d, J = 5,1 Hz, 1 H), 6,06 (d, J = 9,8 Hz, 1 H), 5,98 (d, J = 10,0 Hz, 1 H), 5,39 (d, J = 5,7 Hz, 1 H), 5,16 (d, J = 5,1 Hz, 1 H), 5,07 (d, J = 4,6 Hz, 1 H), 4,33 (quint., J = 6,2 Hz, 1 H), 1,31 (d, J = 6,5 Hz, 3 H) ppm; 13C RMN (150 MHz, DMSO-d6): 8 = 186,8, 182,1, 154,7, 143,5, 135,5, 134,8, 134,3, 133,5, 132,1, 129,8, 126,5, 126,0, 123,9, 123,2, 112,6, 110,3, 100,3, 98,8, 89,6, 87,3, 75,9, 63,5, 62,9, 59,7, 43,1, 21,9 ppm; HRMS (ESI-TOF): calc. para C26H18NO6+ [M+H+]: 440,1129, observado 440,1123.

Ftalida 29. A una solución agitada de 6 -aminoftalida (10,0 g, 67,0 mmol, 1,0 equiv.), DMAP (164 mg, 1,34 mmol, 0,02 equiv.) y Et3N (13,6 g, 18,7 ml, 134 mmol, 2,0 equiv.) en THF (40 ml) a 0 °C, se le añadió en porciones Boc2O (17,6 g, 80,4 mmol, 1,2 equiv.) durante 10 min con agitación vigorosa a 0 °C (PRECAUCIÓN: desprendimiento de gas). Después de que se completara la adición, el baño de hielo se retiró y la mezcla de reacción se agitó durante 3 h a 40 °C, se diluyó con EtOAc (50 ml), se enfrió a 0 °C y se inactivó mediante la adición cuidadosa de NH4Cl ac. saturado (40 ml). La mezcla resultante se extrajo con EtOAc (3 x 50 ml) y las capas orgánicas combinadas se lavaron con agua (50 ml) y salmuera (50 ml), se secaron sobre MgSO4 y se concentraron. La cromatografía en columna ultrarrápida (gel de sílice, hexanos:EtOAc 2:1 a 1:1) dio la ftalida 29 en forma de un sólido de color amarillento (13,4 g, 53,6 mmol, rendimiento del 80 %). 29: Fr = 0,31 (gel de sílice, hexanos:EtOAc 2:1); IR (película) vmáx = 3341, 2979, 2931, 2851, 1741, 1719, 1604, 1538, 1497, 1450, 1423, 1390, 1365, 1316, 1299, 1234, 1126, 1063, 1002 cm-1; RMN 1H (500 MHz, CDCla): 8 = 7,83 (d, J = 2,0, 1 H), 7,76 (d, J = 7,8, 1 H), 7,36 (d, J = 8,3, 1 H), 6,92 (a, 1 H), 5,23 (s, 2 H), 1,47 (s, 9 H) ppm; 13C RMN (125 MHz, CDCla): 8 = 170,9, 152,5, 140,5, 139,6, 126,4, 124,8, 122,5, 114,6, 81,1, 69,5, 28,2 ppm; HRMS (ESI-TOF): calc. para C13H16NO4+ [M+H+]: 250,1074, observado 250,1076.

Formilbenzamida 32. A una suspensión agitada de la ftalida 29 (13,4 g, 53,6 mmol, 1,0 equiv.) en CCU/benceno (200 ml, 1:1) a temperatura ambiente, se le añadió N-bromosuccinimida (11,4 g, 64,3 mmol, 1,2 equiv.) en una porción. La mezcla de reacción se calentó a reflujo y después se añadió azobisisobutironitrilo (1,76 g, 10,7 mmol, 0,2 equiv.) en una porción. La mezcla de reacción se agitó vigorosamente a reflujo durante 2 h más, después se enfrió a temperatura ambiente y se almacenó a 0 °C durante 8 h. La mezcla de reacción se filtró y los precipitados se lavaron con CCl4 (0-5 °C). Los filtrados combinados se concentraron para producir 30 en bruto en forma de una espuma de color amarillo, que se suspendió en H2O/THF (200 ml, 1:1). La suspensión se agitó a 85 °C durante 5 h y se enfrió a
temperatura ambiente. La mezcla de reacción se extrajo con EtOAc (5 x 50 ml) y las capas orgánicas combinadas se secaron sobre MgSO4 y se concentraron para producir 31 en bruto en forma de un sólido higroscópico de color amarillo, que se secó sobre P2O5 durante 8 h más. El ácido 31 en bruto se suspendió en SOCl2 (60 ml) y la mezcla de reacción se calentó a reflujo durante 2 h. Después, la mezcla de reacción se enfrió a temperatura ambiente y se concentró en atmósfera de N2 y el residuo se coevaporó con tolueno (2 x 25 ml) para retirar todas las trazas de SOCl2. El producto en bruto se disolvió en CH2Cl2 (50 ml) y la mezcla de reacción se enfrió a 0 °C. Se añadió gota a gota una solución de dietilamina (7,1 g, 10 ml, 96,7 mmol, 1,8 equiv.) en CH2Cl2 (10 ml). Después de que se completara la adición, el baño de hielo se retiró y la mezcla de reacción se agitó a temperatura ambiente durante 2 h. La reacción se interrumpió con HCl 1 N (100 ml) y la capa acuosa se extrajo con CH2Cl2 (2 x 50 ml). Las capas orgánicas combinadas se lavaron con salmuera (50 ml), se secaron sobre MgSO4 y se concentraron. La cromatografía en columna ultrarrápida (gel de sílice, hexanos:EtOAc 3:1 a 2:1) dio la formilbenzamida 32 en forma de un sólido de color amarillento (10,3 g, 32,2 mmol, rendimiento del 60 %). 32: Fr = 0,31 (gel de sílice, hexanos:EtOAc 2:1); RMN 1H (600 MHz, CDCh): 8 = 9,89 (s, 1 H), 7,82 (d, J = 8,5 Hz, 1 H), 7,52 (dd, J = 8,4, 1,4 Hz, 1 H), 7,33 (d, J = 1,6 Hz, 1 H), 7,33 (a, 1 H), 3,60 (a, 2 H), 3,12 (c, J = 7,1 Hz, 2 H), 1,51 (s, 9 H), 1,30 (t, J = 7,1 Hz, 3 H), 1,04 (t, J = 7,1 Hz, 3 H) ppm; 13C RMN (150 MHz, CDCla): 8 = 189,1, 168,5, 151,9, 144,1, 140,6, 131,6, 126,9, 117,8, 115,7, 81,5, 60,4, 43,0, 39,2, 28,2, 13,8, 12,6 ppm; HRMS (ESI-TOF): calc. para C17H25N2O4+ [M+H+]: 321,1809, observado 321,1810.

Cianoftalida 33. A una solución agitada de la formilbenzamida 32 (1,03 g, 3,22 mmol, 1,0 equiv.) en CH2Cl2 ( 6 ml) a 0 °C se le añadieron TMSCN (640 mg, 0,81 ml, 6,44 mmol, 2,0 equiv.) y una solución de KCN (5,2 mg, 0 ,08 mmol, 0,025 equiv.) y 18-corona-6 (16 mg, 0,06 mmol, 0,02 equiv.) en THF (0,6 ml). La mezcla de reacción se agitó a la misma temperatura durante 1,5 h en un matraz cerrado herméticamente y durante 30 min a temperatura ambiente. Después, la mezcla de reacción se concentró en atmósfera de N2 y el residuo se coevaporó con tolueno (2 x 25 ml) para retirar todas las trazas de TMSCN. El aceite de color pardo resultante se disolvió en AcOH (3 ml) y se agitó durante 36 h a temperatura ambiente. La reacción se interrumpió mediante la adición cuidadosa de NaOH 1 N (10 ml) y la mezcla resultante se repartió entre EtOAc (20 ml) y NaOH 1 N (10 ml). La capa acuosa se extrajo con EtOAc (3 x 20 ml) y las capas orgánicas combinadas se lavaron con H2O (20 ml) y salmuera (20 ml), se secaron sobre MgSO4 y se concentraron para dar el producto en bruto en forma de un sólido cristalino. El producto en bruto se disolvió en C ^C N (20 ml) y se añadió HF ac. (del 48% al 51%, 4 ml) en una porción a temperatura ambiente. Después de agitar durante 16 h a temperatura ambiente, la mezcla de reacción se vertió cuidadosamente en NaHCO3 ac. saturado (150 ml). La capa acuosa se extrajo con EtOAc (3 x 50 ml) y las capas orgánicas combinadas se lavaron con H2O (25 ml) y salmuera (25 ml), se secaron sobre MgSO4 y se concentraron. La cromatografía en columna ultrarrápida (gel de sílice, CH2Ch:EtOAc 4:1) dio la cianoftalida 33 en forma de un sólido de color blanco (393 mg, 2,25 mmol, rendimiento del 70 %). 33: Fr = 0,31 (gel de sílice, CH2Ch:EtOAc 4:1); 1H RMN (400 MHz, CD3CN): 8 = 7,46 (d, J = 8,3, 1 H), 7,09 (dd, J = 8,3, 1,7 Hz, 1 H), 7,02 (d, J = 1,7 Hz, 1 H), 6,15 (s, 1 H), 4,76 (a, 2 H) ppm; 13C RMN (150 MHz, CD3CN): 8 = 169,5, 151,9, 131,2, 126,4, 124,5, 122,9, 116,3, 109,2, 67,1 ppm; HRMS (ESI-TOF): calc. para C9H7N2O2+ [M+H+]: 175,0502, observado 175,0503.

Antraquinona 35a. A una solución de la cianoftalida 33 (38 mg, 0,22 mmol, 3,0 equiv.) en THF (1,0 ml) a -78 °C se le añadió LiHMDS (1,0 M en THF, 0,29 ml, 0,29 mmol, 4,0 equiv.). La mezcla resultante se agitó a -78 °C durante 20 min y se añadió una solución enfriada previamente de la quinona aminal 22a (39 mg, 73 pmol, 1,0 equiv.) en THF (1,0 ml) a -78 °C mediante una cánula. Después de agitar durante 5 min a -78 °C, la mezcla de reacción se calentó a temperatura ambiente y se agitó durante 1 h más, tiempo durante el cual la mezcla de reacción se volvió de color rojo oscuro y el análisis por TLC mostró el consumo completo de 22a (EtOAc al 8 % en CH2O 2). Después, la mezcla de reacción se inactivó mediante la adición de tampón pH 6 ,8 (20 ml) y se extrajo con EtOAc (3 x 20 ml). Los extractos orgánicos combinados se lavaron con salmuera ( 20 ml), se secaron sobre MgSO4 y se filtraron a través de un lecho corto de Celite® (lavado con EtOAc antes de usarse). Los sólidos se aclararon con EtOAc y los filtrados combinados se concentraron para producir la alloc-antraquinona 34a en bruto en forma de un sólido de color rojo
oscuro, que se disolvió en THF desgasificado (1,5 ml) en una atmósfera de Ar y se enfrió a 0 °C. A esta solución se le añadió Pd(PPh3)4 ( 8 mg, 3,5 |jmol, 0,1 equiv.), seguido de la adición gota a gota de morfolina (16 mg, 16 jl, 0,17 mmol, 2,4 equiv.), el matraz de reacción se envolvió con papel de aluminio y la mezcla resultante se dejó en agitación a 0 °C durante 2 h, tiempo durante el cual la mezcla de reacción se volvió de color púrpura oscuro. El baño de refrigeración se retiró y la mezcla de reacción se agitó a temperatura ambiente durante 20 min y después se inactivó mediante la adición de tampón pH 6 ,8 (20 ml) y se extrajo con EtOAc (3 x 20 ml). Los extractos orgánicos combinados se lavaron con NaHCO3 ac. saturado ( 20 ml) y salmuera ( 20 ml), se secaron sobre MgSO4 y se filtraron a través de un lecho corto de Celite® (lavado con EtOAc antes de usarse). Los sólidos se aclararon con EtOAc y los filtrados combinados se concentraron hasta aprox. 1 ml en volumen. La cromatografía en columna ultrarrápida [gel de sílice desactivado (véanse los Métodos Generales), hexanos:EtOAc 2:1 a 1:1] proporcionó la antraquinona 35a (28 mg, 50 jmol, rendimiento del 69 %) en forma de un sólido de color púrpura. 35a: Fr = 0,58 (gel de sílice, hexanos:EtOAc 1:1); [a]D25 = 2600° (c = 0,002, EtOAc); IR (película) vmáx = 3376, 3240, 2955, 2876, 1625, 1580, 1481, 1353, 1322, 1258, 1232 cm-1; 1H RMN (600 MHz, CD3CN): 8 = 13,11 (s, 1 H), 9,94 (d, J = 3,8 Hz, 1 H), 8,39 (s, 1 H), 8,01 (d, J = 8 ,6 Hz, 1 H), 7,35 (d, J = 2,0 Hz, 1 H), 6,99 (dd, J = 8 ,6 , 2,0 Hz, 1 H), 5,93 (d, J = 10,0 Hz, 1 H), 5,86 (d, J = 10,0 Hz, 1 H), 5,16 (a, 2 H), 5,10 (d, J = 4,8 Hz, 1 H), 4,92 (d, J = 4,0 Hz, 1 H), 4,54 (c, J = 6,3 Hz, 1 H), 4,40 (d, J = 4,9 Hz, 1 H), 1,38 (d, J = 6,3 Hz, 3 H), 0,98 (t, J = 8,0 Hz, 9 H), 0,66 (c, J = 7,9 Hz, 6 H) ppm; 13C RMN (150 MHz, CD3CN): 8 = 189,3, 183,8, 156,4, 154,1, 144,0, 136,1, 135,4, 130,2, 129,7, 125,4, 124,9, 123,7, 120,3, 114,8, 112,6, 109,9, 100,3, 100,1, 91,3, 88,3, 77,3, 6 6 ,8 , 65,0, 64,8, 44,3, 22,6, 7,3, 5,6 ppm; HRMS (ESI-TOF): calc. para C32H33^ O 6Si+ [M+H+]: 569,2102, observado 569,2104.

8 -Amino-uncialamicina (36a). A una solución de la antraquinona 35a (28 mg, 50 jmol, 1,0 equiv.) en THF desgasificado (7,5 ml) a temperatura ambiente se le añadió una solución de 1:1 de 3 HF^Et3N:THF (2,5 ml). El matraz de reacción se envolvió con papel de aluminio y la mezcla de reacción se agitó a temperatura ambiente durante 1,5 h, y después se repartió entre EtOAc (25 ml) y NaHCO3 ac. saturado (25 ml). La capa orgánica se lavó con salmuera (25 ml), se secó sobre Na2SO4 y se concentró hasta aprox. 1 ml en volumen. La cromatografía en columna ultrarrápida [gel de sílice desactivado (véanse los Métodos Generales), hexanos:EtOAc 1:1 a 1:3] proporcionó la 8 -amino-uncialamicina (36a) (22 mg, 48 jmol, rendimiento del 98 %) en forma de un sólido de color púrpura. 36a: Fr = 0,24 (gel de sílice, hexanos:EtOAc 1:1); [a]D25 = 870° (c = 0,02, EtOAc); IR (película) vmáx = 3375, 3239, 2934, 1626, 1581, 1480, 1357, 1326, 1258, 1233 cm-1; 1H RMN (400 MHz, CD3CN): 8 = 13,10 (s, 1 H), 9,96 (d, J = 4,0 Hz, 1 H), 8,41 (s, 1 H), 8,00 (d, J = 8 ,6 Hz, 1 H), 7,34 (d, J = 2,4 Hz, 1 H), 6,98 (dd, J = 8 ,6 , 2,4 Hz, 1 H), 5,95 (dd, J = 10,0, 0,6 Hz, 1 H), 5,88 (dt, J = 10,0, 1,2 Hz, 1 H), 5,24 (d, J = 5,0 Hz, 1 H), 5,17 (a, 2 H), 4,86 (dd, J = 4,3, 1,6 Hz, 1 H), 4,50 (d, J = 4,9 Hz, 1 H), 437 (dc, J = 5,0, 6,5 Hz, 1 H), 3,26 (d, J = 4,9 Hz, 1 H), 1,38 (d, J = 6,5 Hz, 3 H) ppm; 13C RMN (150 MHz, CD3CN): 8 = 189,3, 183,9, 156,4, 154,1, 143,8, 136,0, 135,4, 130,2, 129,7, 125,3, 124,5, 123,9, 120,3, 114,8, 112,7, 109,9, 100,4, 99,5, 91,2, 8 8 ,8 , 76,9, 66,0, 65,5, 64,8, 44,2, 21,2 ppm; HRMS (ESI-TOF): calc. para C26H19N2O6+ [M+H+]: 455,1238, observado 455,1239.

Ftalida 40a. A una suspensión de ácido dimetilbenzoico (7,5 g, 49,9 mmol, 1,0 equiv.) y K2CO3 anhidro (10,3 g, 74,8 mmol, 1,5 equiv., secado a alto vacío a 110 °C durante 16 h antes de usarse) en DMF (50 ml) se le añadió gota a gota Mel (7,79 g, 3,42 ml, 54,9 mmol, 1,1 equiv.) durante 10 min con agitación vigorosa a temperatura ambiente. Después de que se completara la adición, la mezcla de reacción se agitó durante 5 h más a temperatura ambiente, después se vertió en H2O (100 ml) y se extrajo con EtOAc (3 x 100 ml). Las capas orgánicas combinadas se lavaron con agua (3 x 50 ml) y salmuera (2 x 50 ml), se secaron sobre MgSO4 y se concentraron para producir el producto en bruto 37a en forma de un aceite incoloro. El éster 37a y W-bromosuccinimida (19,1 g, 100 mmol, 2,1 equiv.) se disolvieron en CCU (50 ml), la mezcla de reacción se calentó a 80 °C y se añadió peróxido de benzoílo (121 mg, 0,5 mmol, 0,01 equiv.) en una porción. El calentamiento se continuó durante 8 h y la mezcla de reacción se enfrió a temperatura ambiente y después se almacenó a 0 °C durante 12 h. Después, la mezcla de reacción se filtró y los precipitados se aclararon con CCU (0-5 °C). Después, los filtrados combinados se lavaron con NaHCO3 ac. saturado (25 ml) y salmuera (25 ml), se secaron sobre MgSO4 y se concentraron para producir el producto en bruto 38a en forma de un sólido de color amarillento. El dibromoéster 38a neto se calentó a 150 °C a ligero vacío durante 10 h. La
mezcla de reacción se enfrió a temperatura ambiente y se obtuvo un sólido de color pardo en forma de producto en bruto 39a. La bromoftalida 39a se disolvió en DMF (50 ml) y se añadieron secuencialmente K2CO3 anhidro (10,3 g, 74,8 mmol, 1,5 equiv., secado a alto vacío a 110 °C durante 16 h antes de usarse) y n-Bu4NI (1,8 g, 5,0 mmol, 0,1 equiv.), seguido de ftalimida (8,0 g, 54,9 mmol, 1,1 equiv.) en una porción. La mezcla de reacción se agitó a 40 °C durante 4 h, después se vertió en H2O (100 ml) y se extrajo con EtOAc (3 x 100 ml). Las capas orgánicas combinadas se lavaron con agua (3 x 50 ml) y salmuera (2 x 50 ml), se secaron sobre MgSO4 y se concentraron. La cromatografía en columna ultrarrápida (gel de sílice, CH2Cf2:EtOAc 10:1 a 8:1) dio la ftalida 40a en forma de un sólido de color amarillento (10,8 g, 36,9 mmol, rendimiento del 74 %). 40a: Fr = 0,31 (gel de sílice, CH2Ch:EtOAc 8:1); 1H RMN (600 MHz, CDCls): 8 = 7,93 (s, 1 H), 7,87 (dd, J = 5,4, 3,1 Hz, 2 H), 7,76 (d, J = 7,9 Hz, 1 H), 7,75 (dd, J = 5,5, 3,0 Hz, 2 H), 7,47 (d, J = 7,8 Hz, 1 H), 5,29 (s, 2 H), 4,96 (s, 2 H) ppm; HRMS (ESI-TOF): calc. para C17H12NO4+ [M+H+]: 294,0761, observado 294,0765.

Ftalida 40b. Se preparó de acuerdo con el procedimiento general descrito anteriormente para la preparación de 40a, a partir de ácido 2,4-dimetilbenzoico (1,52 g, 10,1 mmol, 1,0 equiv.) para producir la ftalida 40b en forma de un sólido de color amarillento (2,08 g, 7,09 mmol, rendimiento del 70 %). 40b: Fr = 0,31 (gel de sílice, CH2Ch:EtOAc 8:1); RMN 1H (400 MHz, CDCla): 8 = 7,86 (dd, J = 5,4, 3,1 Hz, 2 H), 7,85 (d, J = 8,0 Hz, 1 H), 7,74 (dd, J = 5,5, 3,0 Hz, 2 H), 7,57 (d, J = 7,6 Hz, 1 H), 7,51 (s, 1 H), 5,44 (s, 2 H), 4,87 (s, 2 H) ppm; HRMS (ESI-TOF): calc. para C17H12NO4+ [M+H+]: 294,0761, observado 294,0760.

Ftalida 40c. Se preparó de acuerdo con el procedimiento general descrito anteriormente para la preparación de 40a, a partir de ácido 2,3-dimetilbenzoico (2,00 g, 13,3 mmol, 1,0 equiv.) para producir la ftalida 40c en forma de un sólido de color amarillento (2,15 g, 7,33 mmol, rendimiento del 55 %). 40c: Fr = 0,31 (gel de sílice, CH2Ch:EtOAc 8:1); 1H RMN (400 MHz, CDCls): 8 = 7,86 (d, J = 7,6, 1 H), 7,85 (dd, J = 5,4, 3,1 Hz, 2 H), 7,80 (d, J = 7,6 Hz, 1 H), 7,75 (dd, J = 5,5, 3,0 Hz, 2 H), 7,51 (t, J = 7,6 Hz, 1 H), 5,54 (s, 2 H), 4,85 (s, 2 H) ppm; HRMS (ESI-TOF): calc. para C17H12NO4+ [M+H+]: 294,0761, observado 294,0763.

Ftalida 40d. Se preparó de acuerdo con el procedimiento general descrito anteriormente para la preparación de 40a, a partir de ácido 2,6-dimetilbenzoico (2,02 g, 13,5 mmol, 1,0 equiv.) para producir la ftalida 40d en forma de un sólido de color amarillento (2,53 g, 8,63 mmol, rendimiento del 64 %). 40d: Fr = 0,31 (gel de sílice, CH2Ch:EtOAc 8:1); RMN 1H (500 MHz, CDCls): 8 = 7,91 (dd, J = 5,4, 3,1 Hz, 2 H), 7,77 (dd, J = 5,5, 3,0 Hz, 2 H), 7,56 (t, J = 7,7 Hz, 1 H), 7,37 (dd, J = 7,7, 0,8 Hz, 1 H), 7,18 (dd, J = 7,7, 0,8 Hz, 1 H), 5,48 (s, 2 H), 5,33 (s, 2 H) ppm; HRMS (ESI-TOF): calc. para C17H12NO4+ [M+H+]: 294,0761, observado 294,0765.

Formilbenzamida 43a. A una suspensión agitada de la ftalida 40a (10,8 g, 36,9 mmol, 1,0 equiv.) en CCU/benceno (140 ml, 1:1) a temperatura ambiente se le añadió W-bromosuccinimida (7,85 g, 44,3 mmol, 1,2 equiv.) en una porción. La mezcla de reacción se calentó a reflujo y después se añadió azobisisobutironitrilo (1,21 g, 7,37 mmol, 0,2 equiv.) en una porción. La mezcla de reacción se agitó vigorosamente a reflujo durante 2 h más, después se enfrió a
temperatura ambiente y se almacenó a 0 °C durante 8 h. La mezcla de reacción se filtró y los precipitados se aclararon con CCU (0-5 °C). Después, los filtrados combinados se lavaron con NaHCO3 ac. saturado (25 ml) y salmuera (25 ml), se secaron sobre Mg2SO4 y se concentraron para producir 41a en bruto en forma de una espuma de color amarillo, que se suspendió en H2O/THF (140 ml, 1:1). La suspensión se agitó a 85 °C durante 5h y se enfrió a temperatura ambiente. La mezcla de reacción se extrajo con EtOAc (5 x 50 ml) y las capas orgánicas combinadas se secaron sobre MgSO4 y se concentraron para producir 42a en bruto en forma de un sólido higroscópico de color amarillo, que se secó sobre P2O5 durante 8 h más. El ácido en bruto 42a se suspendió en SOCl2 (40 ml) y la mezcla de reacción se calentó a reflujo durante 2 h. La mezcla de reacción se concentró en atmósfera de N2 y el residuo se coevaporó con tolueno (2 x 25 ml) para retirar todas las trazas de SOCl2. El producto en bruto se disolvió en CH2Cl2 (35 ml) y la mezcla de reacción se enfrió a 0 °C. Se añadió gota a gota una solución de dietilamina (5,0 g, 7 ml, 67,7 mmol, 1,8 equiv.) en CH2Ch (7 ml), el baño de hielo se retiró y la mezcla de reacción se agitó a temperatura ambiente durante 2 h. La reacción se interrumpió con HCl 1 N (70 ml), y la mezcla de reacción se extrajo con CH2O 2 (2 x 50 ml). Las capas orgánicas combinadas se lavaron con salmuera (50 ml), se secaron sobre MgSO4 y se concentraron. La cromatografía en columna ultrarrápida (gel de sílice, hexanos:EtOAc 3:1 a 2:1) proporcionó la formilbenzamida 43a en forma de un sólido de color amarillento (6,05 g, 16,6 mmol, rendimiento del 45 %). 43a: Fr = 0,31 (gel de sílice, hexanos:EtOAc 2:1); RMN 1H (400 MHz, CDCls): 8 = 9,99 (s, 1 H), 7,88 (d, J = 8,0 Hz, 1 H), 7,85 (dd, J = 5,4, 3,1 Hz, 2 H), 7,73 (dd, J = 5,5, 3,0 Hz, 2 H), 7,57 (dd, J = 8,0, 1,4 Hz, 1 H), 7,38 (d, J = 1,4 Hz, 1 H), 4,89 (s, 2 H), 3,58 (c, J = 7,1 Hz, 2 H), 3,07 (c, J = 7,1 Hz, 2 H), 1,28 (t, J = 7,1 Hz, 3 H), 1,00 (t, J = 7,1 Hz, 3 H) ppm; 13C RMN (100 MHz, CDCh): 8 = 189,9, 168,2, 167,7, 142,6, 140,0, 134,2, 132,0, 131,8, 130,3, 129,1, 126,7, 123,5, 43,0, 41,0, 39,1, 13,7, 12,6 ppm; HRMS (ESI-TOF): calc. para C21H21N2O4+ [M+H+]: 365,1496, observado 365,1496.

Formilbenzamida 43c. Se preparó de acuerdo con el procedimiento general descrito anteriormente para la preparación de 43a, a partir de la ftalida 40c (2,11 g, 7,21 mmol, 1,0 equiv.) para producir la formilbenzamida 43c en forma de un sólido de color amarillento (1,05 g, 2,88 mmol, rendimiento del 40 %). 43c: Fr = 0,31 (gel de sílice, hexanos:EtOAc 2:1); 1H RMN (400 MHz, CDCla): 8 = 10,36 (s, 1 H), 7,91 (dd, J = 5,4, 3,1 Hz, 2 H), 7,76 (dd, J = 5,5, 3,0 Hz, 2 H), 7,51 (t, J = 7,6 Hz, 1 H), 7,27 (d, J = 7,6 Hz, 1 H), 7,25 (dd, J = 7,6, 1,0 Hz, 1 H), 5,32 (s, 2 H), 3,61 (c, J = 7,2 Hz, 2 H), 3,14 (c, J = 7,2, 2 H), 1,30 (t, J = 7,2 Hz, 3 H), 1,06 (t, J = 7,2 Hz, 3 H) ppm; HRMS (ESI-TOF): calc. para C21H21N2O4+ [M+H+]: 365,1496, observado 365,1496.

Formilbenzamida 43d. Se preparó de acuerdo con el procedimiento general descrito anteriormente para la preparación de 43a, a partir de la ftalida 40d (2,49 g, 8,49 mmol, 1,0 equiv.) para producir la formilbenzamida 43d en forma de un sólido de color amarillento (1,30 g, 3,57 mmol, rendimiento del 42%). 43d: Fr = 0,31 (gel de sílice, hexanos:EtOAc 2:1); 1H RMN (500 MHz, CDCls): 8 = 10,04 (s, 1 H), 7,89 (dd, J = 5,4, 3,1 Hz, 2 H), 7,86 (dd, J = 7,5, 1,4 Hz, 1 H), 7,76 (dd, J = 5,5, 3,0 Hz, 2 H), 7,49 (dt, J = 7,7, 1,4 Hz, 1 H), 7,46 (t, J = 7,6 Hz, 1 H), 5,09 (d, J = 16 Hz, 1 H), 4,68 (d, J = 16 Hz, 1 H), 3,58 (c, J = 7,1 Hz, 2 H), 3,07 (c, J = 7,1 Hz, 2 H), 1,28 (t, J = 7,1 Hz, 3 H), 1,00 (t, J = 7,1 Hz, 3 H) ppm; 13C RMN (100 MHz, CDCls): 8 = 189,9, 168,2, 167,7, 142,6, 140,0, 134,2, 132,0, 131,8, 130,3, 129,1, 126,7, 123,5, 43,0, 41,0, 39,1, 13,7, 12,6 ppm; HRMS (ESI-TOF): calc. para C21H21N2O4+ [M+H+]: 365,1496, observado 365,1496.

Cianoftalida 44a. A una solución agitada de la formil-benzamida 43a (1,17 g, 3,22 mmol, 1,0 equiv.) en CH2Cl2 (6 ml) a 0°C se le añadieron TMSCN (640 mg, 0,81 ml, 6,44 mmol, 2,0 equiv.) y una solución de KCN (5,2 mg, 0,08 mmol, 0,025 equiv.) y 18-corona-6 (16 mg, 0,06 mmol, 0 ,02 equiv.) en THF (0,6 ml). La mezcla de reacción se
agitó a la misma temperatura en un matraz cerrado herméticamente durante 1,5 h y durante 30 min a temperatura ambiente. Después, la mezcla de reacción se concentró en atmósfera de N2 y el residuo se coevaporó con tolueno (2 x 25 ml) para retirar todas las trazas de TMSCN. El aceite de color pardo resultante se disolvió en AcOH (3 ml) y se agitó durante 48 h a temperatura ambiente hasta que el análisis por TLC mostró la conversión completa (hexanos:EtOAc 3:2). La reacción se interrumpió mediante la adición cuidadosa de NaOH 1 N (10 ml) y la mezcla resultante se repartió entre EtOAc (20 ml) y NaOH 1 N (10 ml). La capa acuosa se extrajo con EtOAc (3 x 20 ml) y las capas orgánicas combinadas se lavaron con H2O ( 20 ml) y salmuera ( 20 ml), se secaron sobre MgSO4 y se concentraron. La cromatografía en columna ultrarrápida (gel de sílice, hexanos:EtOAc 2:1 a 3:2) dio un sólido de color amarillento, que se recristalizó en EtOAc para producir la cianoftalida 44a en forma de un sólido de color blanco (902 mg, 2,83 mmol, rendimiento del 88 %). 44a: Fr = 0,31 (gel de sílice, hexanos:EtOAc 3:2); 1H RMN (400 MHz, CDCla): 8 = 7,99 (s, 1 H), 7,91 (d, J = 8,0 Hz, 1 H), 7,86 (dd, J = 5,4, 3,1 Hz, 2 H), 7,74 (dd, J = 5,5, 3,0 Hz, 2 H), 7,67 (d, J = 8,0 Hz, 1 H), 6,06 (s, 1 H), 4,98 (s, 2 H) ppm; 13C RMN (100 MHz, CDCla): 8 = 167,7, 167,0, 141,2, 140,6, 136,0, 134,4, 131,7, 126,2, 125,0, 123,6, 123,1, 113,6, 65,5, 40,7 ppm; HRMS (ESI-TOF): calc. para C18H11N2O4+ [M+H+]: 319,0713, observado 319,0715.

Cianoftalida 44b. Se preparó de acuerdo con el procedimiento general descrito anteriormente para la preparación de 44a, a partir de la formilbenzamida 43b (605 mg, 1,66 mmol, 1,0 equiv.) para producir la cianoftalida 44b en forma de un sólido de color blanco (450 mg, 1,41 mmol, rendimiento del 85 %). 44b: Fr = 0,31 (gel de sílice, hexanos:EtOAc 3:2); 1H RMN (600 MHz, CDCls): 8 = 7,94 (d, J = 7,9 Hz, 1 H), 7,89 (dd, J = 5,4, 3,1 Hz, 2 H), 7,77 (d, J = 7,9 Hz, 1 H), 7,76 (dd, J = 5,5, 3,1 Hz, 2 H), 7,75 (s, 1 H), 6,04 (s, 1 H), 4,99 (dd, J = 15,0, 25,4 Hz, 2 H) ppm; 13C RMN (100 MHz, CDCls): 8 = 167,7, 166,9, 144,8, 142,4, 134,5, 132,0, 131,8, 127,0, 124,0, 123,7, 122,8, 113,6, 65,5, 41,1 ppm; HRMS (ESI-TOF): calc. para C18H11N2O4+ [M+H+]: 319,0713, observado 319,0715.

Cianoftalida 44c. Se preparó de acuerdo con el procedimiento general descrito anteriormente para la preparación de 44a, a partir de la formilbenzamida 43c (992 mg, 2,72 mmol, 1,0 equiv.) para producir la cianoftalida 44c en forma de un sólido de color blanco (650 mg, 2,04 mmol, rendimiento del 75 %). 44a: Fr = 0,30 (gel de sílice, hexanos:EtOAc 3:2); 1H RMN (500 MHz, CD3CN): 8 = 7,88 (d, J = 7,7, 1 H), 7,87 (dd, J = 5,4, 3,1 Hz, 2 H), 7,86 (d, J = 7,7 Hz, 1 H), 7,81 (dd, J = 5,5, 3,0 Hz, 2 H), 7,70 (t, J = 7,7 Hz, 1 H), 6,48 (s, 1 H), 4,98 (dd, J = 15,6, 27,8 Hz, 2 H) ppm; 13C RMN (100 MHz, CDCls): 8 = 169,0, 168,7, 141,4, 137,1, 135,4, 133,1, 132,9, 132,7, 126,5, 126,0, 124,2, 115,1, 66,7, 38,0 ppm; HRMS (ESI-TOF): calc. para C18H11N2O4+ [M+H+]: 319,0713, observado 319,0715.

Cianoftalida 44d. Se preparó de acuerdo con el procedimiento general descrito anteriormente para la preparación de 44a, a partir de la formilbenzamida 43d (1,24 g, 3,41 mmol, 1,0 equiv.) para producir la cianoftalida 44d en forma de un sólido de color blanco (902 mg, 2,83 mmol, rendimiento del 83 %). 44d: Fr = 0,30 (gel de sílice, hexanos:EtOAc 3:2); 1H RMN (600 MHz, CDCls): 8 = 7,91 (dd, J = 5,4, 3,1 Hz, 2 H), 7,78 (dd, J = 5,5, 3,0 Hz, 2 H), 7,72 (t, J = 7,7 Hz, 1 H), 7,60 (d, J = 7,7 Hz, 1 H), 7,39 (d, J = 7,7 Hz, 1 H), 6,10 (s, 1 H), 5,43 (dd, J = 18,1, 25,7 Hz, 2 H) ppm; 13C RMN (100 MHz, CDCls): 8 = 167,8, 167,0, 142,4, 138,4, 135,8, 134,4, 131,9, 129,1, 123,7, 123,6, 121,7, 113,7, 65,5, 36,5 ppm; HRMS (ESI-TOF): calc. para C18H11N2O4+ [M+H+]: 319,0713, observado 319,0715.

Antraquinona 46aa. A una solución de la cianoftalida 44a (91 mg, 0,27 mmol, 2,0 equiv.) en THF (1,2 ml) a -78 °C se le añadió LiHMDS (1,0 M en THF, 0,43 ml, 0,43 mmol, 3,0 equiv.). La mezcla resultante se agitó a -78 °C durante 20 min y se añadió una solución enfriada previamente de la quinona aminal 22a (78 mg, 0,14 mmol, 1,0 equiv.) en THF (1,4 ml) a -78 °C mediante una cánula. Después de agitar durante 5 min a -78 °C, la mezcla de reacción se calentó a temperatura ambiente y se agitó durante 1,5 h más, tiempo durante el cual la mezcla de reacción se volvió de color rojo oscuro y el análisis por TLC mostró el consumo completo de 22a (EtOAc al 8 % en CH2O 2). Después, la mezcla de reacción se inactivó mediante la adición de tampón pH 6 ,8 (30 ml) y se extrajo con EtOAc (3 x 30 ml). Los extractos orgánicos combinados se lavaron con salmuera (30 ml), se secaron sobre MgSO4 y se filtraron a través de un lecho corto de Celite® (lavado con EtOAc antes de usarse). Los sólidos se aclararon con EtOAc y los filtrados combinados se concentraron para producir la alloc-antraquinona 45aa en bruto en forma de un sólido de color rojo oscuro, que se disolvió en THF desgasificado (2,0 ml) en una atmósfera de Ar y se enfrió a 0 °C. A esta solución se le añadió Pd(PPh3)4 (32 mg, 10,4 pmol, 0,16 equiv.), seguido de la adición gota a gota de morfolina (32 mg, 32 pl, 0,34 mmol, 2,4 equiv.). El matraz de reacción se envolvió con papel de aluminio y la mezcla resultante se dejó en agitación a 0 °C durante 2 h, tiempo durante el cual la mezcla de reacción se volvió de color púrpura oscuro. El baño de refrigeración se retiró y la mezcla de reacción se agitó a temperatura ambiente durante 20 min y después se inactivó mediante la adición de tampón pH 6 ,8 (30 ml) y se extrajo con EtOAc (3 x 30 ml). Los extractos orgánicos combinados se lavaron con H2O (30 ml) y salmuera (30 ml), se secaron sobre MgSO4 y se filtraron a través de un lecho corto de Celite® (lavado con EtOAc antes de usarse). Los sólidos se aclararon con EtOAc y los filtrados combinados se concentraron hasta aprox. 1 ml en volumen. La cromatografía en columna ultrarrápida [gel de sílice desactivado (véanse los Métodos Generales), hexanos:EtOAc 2:1 a 1:1] proporcionó la antraquinona 46aa en forma de un sólido de color púrpura (92 mg, 0,13 mmol, rendimiento del 90 %). 46aa: Fr = 0,58 (gel de sílice, hexanos:EtOAc 1:1); [a]o25 = 2600° (c = 0,002, EtOAc); 1H RMN (600 MHz, CD3CN): 8 = 13,11 (s, 1 H), 9,97 (d, J = 4,0 Hz, 1 H), 8,46 (s, 1 H), 8,23 (d, J = 8,0 Hz, 1 H), 8,20 (s, 1 H), 7,87 (d, J = 1,9 Hz, 2 H), 7,81 (d, J = 2,2 Hz, 1 H), 7,80 (d, J = 1,9 Hz, 2 H), 5,94 (d, J = 10,0 Hz, 1 H), 5,87 (d, J = 9,8 Hz, 1 H), 5,10 (s, 1 H), 4,97 (s, 2 H), 4,97 (s, 1H), 4,55 (c, J = 5,8 Hz, 1 H), 4,45 (d, J = 3,7 Hz, 1 H), 1,38 (d, J = 5,8 Hz, 3 H), 0,98 (t, J = 7,6 Hz, 9 H), 0,66 (c, J = 7,6 Hz, 6 H) ppm; 13C RMN (150 MHz, CD3CN): 8 = 188,2, 183,8, 169,0, 156,7, 144,8, 143,5, 136,3, 135,4, 135,1, 134,8, 133,8, 133,0, 130,9, 128,3, 126,1, 124,9, 124,1, 123,8, 114,2, 112,2, 100,2, 99,7, 91,3, 88,4, 77,4, 66,7, 64,9, 64,7, 44,3, 41,8, 22,6, 7,3, 5,6 ppm; HRMS (ESI-TOF): calc. para C4-iH36N2OaSi+ [M+H+]: 712,2241, observado 712,2243.

8-Ftalimidometil-uncialamicina (47aa). A una solución agitada de la antraquinona 46aa (92 mg, 0,13 mmol, 1,0 equiv.) en THF desgasificado (15 ml) a temperatura ambiente se le añadió una solución de 1:1 de 3 HF^Et3N:THF (5,0 ml). El matraz de reacción se envolvió con papel de aluminio y la mezcla de reacción se agitó a temperatura ambiente durante 1,5 h, y después se repartió entre EtOAc (50 ml) y NaHCO3 ac. saturado (50 ml). La capa acuosa se extrajo con EtOAc (2 x 25 ml) y los extractos orgánicos combinados se lavaron con salmuera (25 ml), se secaron sobre Na2SO4 y se concentraron hasta aprox. 1 ml en volumen. La cromatografía en columna ultrarrápida (gel de sílice desactivado, hexanos:EtOAc 1:1 a 1:2) proporcionó la ftalimidometil-uncialamicina (47aa) en forma de un sólido de color púrpura (76 mg, 0,13 mmol, rendimiento del 98 %). 47aa: Fr = 0,24 [gel de sílice desactivado (véanse los Métodos Generales), hexanos:EtOAc 1:1]; [a]o25 = 26000° (c = 0,005, EtOAc) [Este valor (probablemente demasiado elevado), se obtuvo en la dilución a la que el polarímetro mostró una lectura de rotación; a mayor concentración, el instrumento no proporcionaba una lectura debido a la insolubilidad del compuesto]; 1H r Mn (600 MHz, CD3CN): 8 = 13,10 (s, 1 H), 9,99 (d, J = 4,1 Hz, 1 H), 8,48 (s, 1 H), 8,23 (d, J = 8,1 Hz, 1 H), 8,21 (s, 1 H), 7,87 (dd, J = 5,4, 3,1 Hz, 2 H), 7,82 (d, J = 2,2 Hz, 1 H), 7,81 (dd, J = 5,5, 3,0 Hz, 2 H), 5,96 (d, J = 9,9 Hz, 1 H), 5,88 (d, J = 9,9 Hz, 1 H), 5,24 (d, J = 4,9 Hz, 1 H), 5,49 (s, 2 H), 4,91 (dd, J = 4,4, 1,4 Hz, 1 H), 4,44 (d, J = 4,9 Hz, 1 H), 4,38 (dc, J = 5,0, 6,5 Hz, 1 H), 3,27 (d, J = 4,9 Hz, 1 H), 1,38 (d, J = 6,5 Hz, 3 H) ppm; 13C RMN (150 MHz, CD3CN):
5 = 188,2, 183,9, 169,0, 156,7, 144,7, 143,6, 136,2, 135,4, 135,1, 134,8, 133,8, 133,0, 130,9, 128,4, 126,1, 124,5, 124,1, 124,1, 114,3, 112,3, 100,3, 99,1,91,3, 8 8 ,8 , 77,0, 65,8, 65,3, 64,7, 44,2, 41,8, 21,2 ppm; HRMS (ESI-TOF): calc. para C35H23N2O8+ [M+H+]: 599,1449, observado 599,1447.

7-Ftalimidometil-uncialamicina (47ba). Se preparó de acuerdo con el procedimiento general descrito anteriormente para la preparación de 47aa, a partir de la cianoftalida 44b (23 mg, 73 |jmol, 3,0 equiv.) y la quinona aminal 22a (13 mg, 24 jmol, 1,0 equiv.) a través del intermedio 46ba para producir la 7-ftalimido-metil-uncialamicina (47ba) (11 mg, 18 jmol, rendimiento del 76 %) en forma de un sólido de color púrpura. 47ba: Fr = 0,24 (gel de sílice, hexanos:EtOAc 1:1); [a]D25 = 28000° (c = 0,005, EtOAc) [Este valor (probablemente demasiado elevado), se obtuvo en la dilución a la que el polarímetro mostró una lectura de rotación; a mayor concentración, el instrumento no proporcionaba una lectura debido a la insolubilidad del compuesto]; 1H RMN (600 MHz, CD3CN): 5 = 13,13 (s, 1 H), 9,96 (d, J = 4,1 Hz, 1 H), 8,49 (s, 1 H), 8,20 (s, 1 H), 8,19 (d, J = 8,1 Hz, 1 H), 7,87 (dd, J = 5,4, 3,1 Hz, 2 H), 7,81 (dd, J = 5,5, 3,0 Hz, 2 H), 7,77 (d, J = 8,1 Hz, 1 H), 5,96 (d, J = 10,0 Hz, 1 H), 5,87 (d, J = 10,0 Hz, 1 H), 5,24 (d, J = 4,5 Hz, 1 H), 4,98 (s, 2 H), 4,90 (dd, J = 4,4, 1,5 Hz, 1 H), 4,56 (d, J = 4,9 Hz, 1 H), 4,38 (dc, J = 5,0, 6,5 Hz, 1 H), 3,32 (d, J = 5,0 Hz, 1 H), 1,38 (d, J = 6,5 Hz, 3 H) ppm; 13C RMN (150 MHz, CD3CN): 5 = 188,1, 183,8, 169,0, 156,7, 144,8, 144,7, 136,1, 136,0, 135,4, 133,5, 133,0, 132,9, 131,0, 127,7, 126,8, 124,5, 124,1, 124,1, 114,2, 112,4, 100,4, 99,1, 91,3, 88 ,8 , 77,0, 65,9, 65,3, 64,7, 44,3, 41,9, 21,2 ppm; HRMS (ESI-TOF): calc. para C35H23N2O8+ [M+H+]: 599,1449, observado 599,1449.

6-Ftalimidometil-uncialamicina (47ca). Se preparó de acuerdo con el procedimiento general descrito anteriormente para la preparación de 47aa, a partir de la cianoftalida 44c (59 mg, 0,18 mmol, 3,0 equiv.) y la quinona aminal 22a (33 mg, 61 jmol, 1,0 equiv.) a través del intermedio 46ca para producir la 6 -ftalimido-metil-uncialamicina (47ca) (1 mg, 2 jmol, rendimiento del 3 %) en forma de un sólido de color púrpura. 47ca: Fr = 0,23 (gel de sílice, hexanos:EtOAc 1:1); [a]D25 = 28000° (c = 0,005, EtOAc) [Este valor (probablemente demasiado elevado), se obtuvo en la dilución a la que el polarímetro mostró una lectura de rotación; a mayor concentración, el instrumento no proporcionaba una lectura debido a la insolubilidad del compuesto]; 1H RMN (600 MHz, CD3CN): 5 = 13,07 (s, 1 H), 9,92 (d, J = 4,1 Hz, 1 H), 8,52 (s, 1 H), 8,31 (d, J = 7,7 Hz, 1 H), 7,92 (dd, J = 5,4, 3,1 Hz, 2 H), 7,85 (dd, J = 5,5, 3,0 Hz, 2 H), 7,69 (t, J = 7,9 Hz, 1 H), 7,57 (d, J = 7,9 Hz, 1 H), 5,98 (d, J = 9,5 Hz, 1 H), 5,91 (d, J = 9,7 Hz, 1 H), 5,51 (d, J = 2,9 Hz, 2 H), 5,26 (d, J = 4,8 Hz, 1 H), 4,93 (dd, J = 4,2, 1,5 Hz, 1 H), 4,58 (d, J = 5,1 Hz, 1 H), 4,39 (dc, J = 5,0, 6,5 Hz, 1 H), 3,33 (d, J = 4,9 Hz, 1 H), 1,40 (d, J = 6,5 Hz, 3 H) ppm; HRMS (ESI-TOF): calc. para C35H23N2O8+ [M+H+]: 599,1449, observado 599,1444.

9-Ftalimidometil-uncialamicina (47da). Se preparó de acuerdo con el procedimiento general descrito anteriormente para la preparación de 47aa, a partir de la cianoftalida 44d (59 mg, 0,18 mmol, 3,0 equiv.) y la quinona aminal 22a
(33 mg, 61 |jmol, 1,0 equiv.) a través del intermedio 46da para producir la 9-ftalimido-metil-uncialamicina (47da) (26 mg, 43 jmol, rendimiento del 71 %) en forma de un sólido de color púrpura. 47da: Fr = 0,25 (gel de sílice, hexanos:EtOAc 1:1); [a]D25 = 30000° (c = 0,005, EtOAc) [Este valor (probablemente demasiado elevado), se obtuvo en la dilución a la que el polarímetro mostró una lectura de rotación; a mayor concentración, el instrumento no proporcionaba una lectura debido a la insolubilidad del compuesto]; 1H RMN (600 MHz, CD3CN): 5 = 13,19 (s, 1 H), 9,95 (d, J = 4,0 Hz, 1 H), 8,53 (s, 1 H), 8,32 (d, J = 7,9 Hz, 1 H), 7,92 (dd, J = 5,4, 3,1 Hz, 2 H), 7,86 (dd, J = 5,5, 3,0 Hz, 2 H), 7,73 (t, J = 7,8 Hz, 1 H), 7,52 (d, J = 7,8 Hz, 1 H), 5,98 (d, J = 10,0 Hz, 1 H), 5,90 (d, J = 10,0 Hz, 1 H), 5,49 (s, 2 H), 5,24 (d, J = 5,0 Hz, 1 H), 4,93 (dd, J = 4,4, 1,4 Hz, 1 H), 4,58 (d, J = 5,0 Hz, 1 H), 4,39 (dc, J = 5,0, 6,5 Hz, 1 H), 3,33 (d, J = 5,0 Hz, 1 H), 1,39 (d, J = 6,5 Hz, 3 H) ppm; 13C RMN (150 MHz, CD3CN): 5 = 190,9, 184,0, 169,4, 156,6, 144,3, 140,2, 137,7, 135,7, 135,4, 135,1, 133,2, 131,6, 131,2, 130,7, 127,5, 124,5, 124,1, 124,1, 115,1, 112,1, 100,4, 99,1, 91,2, 88 ,8 , 77,1, 65,9, 65,3, 64,7, 44,3, 42,1, 21,2 ppm; HRMS (ESI-TOF): calc. para C35H23N2O8+ [M+H+]: 599,1449, observado 599,1445.

8-Ftalimidometil-1-ep/-uncialamicina (47ab). Se preparó de acuerdo con el procedimiento general descrito anteriormente para la preparación de 47aa, a partir de la cianoftalida 44a (47 mg, 0,15 mmol, 3,0 equiv.) y la quinona aminal 22b (26 mg, 48 jmol, 1,0 equiv.) a través del intermedio 46ab para producir la 8-ftalimidometil-17-ep/-uncialamicina (47ab) (21 mg, 35 jm ol, rendimiento del 73 %) en forma de un sólido de color púrpura. 47ab: Fr = 0,30 (gel de sílice, hexanos:EtoAc 1 :1 ); [a]D25 = 26000° (c = 0,005, EtOAc) [Este valor (probablemente demasiado elevado), se obtuvo en la dilución a la que el polarímetro mostró una lectura de rotación; a mayor concentración, el instrumento no proporcionaba una lectura debido a la insolubilidad del compuesto]; IR (película) vmáx = 3424, 2918, 1770, 1716, 1625, 1597, 1493, 1394, 1329, 1248, 1207 cm-1; 1H RMN (400 MHz, CD3CN): 5 = 13,13 (s, 1 H), 10,04 (d, J = 3,9 Hz, 1 H), 8,25 (d, J = 8,1 Hz, 1 H), 8,21 (d, J = 1,2 Hz, 1 H), 7,88 (dd, J = 5,4, 3,2 Hz, 2 H), 7,83 (d, J = 1,0 Hz, 1 H), 7,81 (dd, J = 5,4, 3,2 Hz, 2 H), 7,62 (s, 1 H), 5,93 (sistema AB, 2 H), 5,86 (d, J = 4,2 Hz, 1 H), 5,11 (dc, J = 5,0, 6,5 Hz, 1 H), 4,98 (s, 2 H), 4,87 (d, J = 4,2 Hz, 1 H), 4,11 (d, J = 4,4 Hz, 1 H), 3,13 (d, J = 4,6 Hz, 1 H), 1,34 (d, J = 6,5 Hz, 3 H) ppm; 13C RMN (150 MHz, CD3CN): 5 = 188,2, 184,0, 169,0, 156,9, 143,8, 143,7, 137,0, 135,4, 135,1, 134,8, 133,8, 133,0, 128,4, 126,6, 126,6, 126,1, 124,1, 123,9, 114,6, 112,2, 100,5, 99,9, 90,4, 88 ,8 , 76,7, 65,6, 65,3, 60,4, 43,8, 41,7, 20,3 ppm; HRMS (ESI-TOF): calc. para C35H23N2O8+ [M+H+]: 599,1449, observado 599,1444.

9-Ftalimidometil-17-ep/'-uncialamicina (47db). Se preparó de acuerdo con el procedimiento general descrito anteriormente para la preparación de 47aa, a partir de la cianoftalida 44d (23 mg, 73 jmol, 3,0 equiv.) y la quinona aminal 22b (13 mg, 24 jmol, 1,0 equiv.) a través de intermedio 46db para producir la 9-ftalimidometil-17-ep/-uncialamicina (47db) (10 mg, 17 jmol, rendimiento del 70 %) en forma de un sólido de color púrpura. 47db: Fr = 0,30 (gel de sílice, hexanos:EtOAc 1:1); 1H RMN (500 MHz, CD3CN): 5 = 13,22 (s, 1 H), 10,00 (d, J= 4,0 Hz, 1 H), 8,32 (dd, J = 7,9, 1,2 Hz, 1 H), 7,92 (dd, J = 5,4, 3,2 Hz, 2 H), 7,85 (dd, J = 5,4, 3,2 Hz, 2 H), 7,72 (t, J = 7,9 Hz, 1 H), 7,65 (s, 1 H), 7,52 (dd, J= 7,9, 1,2 Hz, 1 H), 5,95 (sistema AB, 2 H), 5,88 (d, J= 4,8 Hz, 1 H), 5,48 (s, 2 H), 5,12 (dc, J = 5,0, 6,5 Hz, 1 H), 4,89 (dd, J = 4,5, 1,3 Hz, 1 H), 4,28 (d, J = 4,9 Hz, 1 H), 3,21 (d, J = 4,9 Hz, 1 H), 1,35 (d, J = 6,5 Hz, 3 H) ppm; HRMS (ESI-TOF): calc. para C35H23N2O8+ [M+H+]: 599,1449, observado 599,1444.

8-Aminometil-uncialamicina (48aa). A una solución de 8 -ftalimidometil-uncialamicina (47aa) (3,6 mg, 5 jmol, 1,0 equiv.) en THF desgasificado (1,0 ml) a 0 °C se le añadió gota a gota MeNH2 ac. (al 40%, 1,0 ml). El matraz de reacción se envolvió con papel de aluminio y la mezcla de reacción se agitó a 10 °C durante 18 h, después se enfrió a 0 °C y se diluyó con THF desgasificado (10 ml), se concentró a 5 °C en atmósfera de N2 hasta aprox. 1 ml en volumen y se diluyó con THF desgasificado frío (0 °C, 10 ml). Se repitieron las mismas operaciones 5 veces para producir la 8 -aminometil-uncialamicina en bruto (48aa) en forma de un sólido de color púrpura, que se usó inmediatamente para la siguiente etapa (PRECAUCIÓN: 48aa era extremadamente inestable, sensible tanto a ácidos como a bases, y se descomponía lentamente a -78 °C, debe usarse fresco para obtener los mejores rendimientos). 48aa: Fr = 0,10 (gel de sílice, MeOH:EtOAc 1:1); HRMS (ESI-TOF): calc. para C27H21N2O6+ [M+H+]: 469,1394, observado 469,1397.

7-Aminometil-uncialamicina (48ba). Se preparó de acuerdo con el procedimiento general descrito anteriormente para la preparación de 48aa, a partir de la 7-ftalimidometil-uncialamicina (47ba) (1,5 mg, 2 |jmol, 1 ,0 equiv.) para producir la 7-aminometil-uncialamicina en bruto (48ba) en forma de un sólido de color púrpura, que se usó inmediatamente para la siguiente etapa (PRECAUCIÓN: 48ba era extremadamente inestable, sensible tanto a ácidos como a bases, y se descomponía lentamente a -78 °C, debe usarse fresco para obtener los mejores rendimientos). 48ba: Fr = 0,10 (gel de sílice, MeOH:EtOAc 1:1); HRMS (ESI-TOF): calc. para C27H21N2O6+ [M+H+]: 469,1394, observado 469,1396.

6-Aminometil-uncialamicina (48ca). Se preparó de acuerdo con el procedimiento general descrito anteriormente para la preparación de 48aa, a partir de la 6 -ftalimidometil-uncialamicina (47ca) (1,0 mg, 1 jmol, 1,0 equiv.) para producir la 6 -aminometil-uncialamicina en bruto (48ca) en forma de un sólido de color púrpura, que se usó inmediatamente para la siguiente etapa (PRECAUCIÓN: 48ca era extremadamente inestable, sensible tanto a ácidos como a bases, y se descomponía lentamente a -78 °C, debe usarse fresco para obtener los mejores rendimientos). 48ca: Fr = 0,10 (gel de sílice, MeOH:EtOAc 1:1); HRMS (ESI-TOF): calc. para C27H21N2O6+ [M+H+]: 469,1394, observado 469,1390.

9-Aminometil-uncialamicina (48da). Se preparó de acuerdo con el procedimiento general descrito anteriormente para la preparación de 48aa, a partir de la 9-ftalimidometil-uncialamicina (47da) (3,0 mg, 4 |jmol, 1,0 equiv.) para producir la 9-aminometil-uncialamicina en bruto (48da) en forma de un sólido de color púrpura, que se usó inmediatamente para la siguiente etapa (PRECAUCIÓN: 48da era extremadamente inestable, sensible tanto a ácidos como a bases, y se descomponía lentamente a -78 °C, debe usarse fresco para obtener los mejores rendimientos). 48da: Fr = 0,10 (gel de sílice, MeOH:EtOAc 1:1); HRMS (ESI-TOF): calc. para C 27 H 21 N 2 O 6 + [M+H+]: 469,1394, encontrado 469,1399

8-ferc-ButilcarbamoNmetN-uncialamicma (49aa). La 8-aminometil-uncialamicina en bruto (48aa) [preparada de acuerdo con el procedimiento general descrito anteriormente a partir de la 8-ftalimidometil-uncialamicina (47aa) (3,6 mg, 5 jmol, 1,0 equiv.)] se suspendió en THF desgasificado (1,0 ml) a 0 °C y se añadió NaHCO 3 ac. saturado (1,0 ml) en una porción, seguido de Boc 2 O (1,3 mg, 6 jmol, 1,2 equiv.). El matraz de reacción se envolvió con papel de aluminio y la mezcla de reacción se agitó a 0 °C durante 1,5 h, y después se repartió entre EtOAc (5 ml) y tampón pH 6,8 (5 ml). La capa acuosa se extrajo con EtOAc (2 x 25 ml) y los extractos orgánicos combinados se lavaron con salmuera (5 ml), se secaron sobre Na 2 SO 4 y se concentraron hasta aprox. 1 ml en volumen. La cromatografía en columna ultrarrápida [gel de sílice desactivado (véanse los Métodos Generales), hexanos:EtOAc 1:1 a 1:2] proporcionó la 8-terc-butilcarbamoilmetil-uncialamicina (49aa) (2,7 mg, 4,5 jmol, rendimiento del 95 %) en forma de un sólido de color púrpura. 49aa: Fr = 0,27 (gel de sílice, hexanos:EtOAc 1:1); [a]D25 = 2300° (c = 0,002, EtOAc); 1 H RMN (600 MHz, CD 3 CN): 6 = 13,17 (s, 1 H), 10,00 (d, J = 4,4 Hz, 1 H), 8,50 (s, 1 H), 8,25 (d, J = 8,1 Hz, 1 H), 8,15 (s, 1 H), 7,75 (dd, J = 8,0, 1,8 Hz, 1 H), 6,03 (a, 1 H), 5,97 (d, J = 9,9 Hz, 1 H), 5,89 (dt, J = 9,9, 1,3 Hz, 1 H), 5,24 (d, J = 4,4 Hz, 1 H), 4,92 (dd, J = 4,4, 1,7 Hz, 2 H), 4,65 (d, J = 5,0 Hz, 1 H), 4,39 (d, J = 5,7, 1 H), 4,38 (dc, J = 5,0, 6,5 Hz, 1 H), 3,37 (d, J = 4,8 Hz, 1 H), 1,43 (s, 9 H), 1,38 (d, J = 6,5 Hz, 3 H) ppm; 13 C RMN (150 MHz, CD 3 CN): 6 = 188,5, 184,1, 156,6, 147,3, 144,6, 136,1, 134,6, 134,0, 133,7, 128,1, 125,0, 124,5, 124,1, 114,4, 112,4, 100,4, 99,1, 91,2, 88,8, 79,8, 77,0, 65,8, 65,3, 64,7, 44,3, 44,2, 28,5, 21,2 ppm; HRMS (ESI-TOF): calc. para C 32 H 29 N 2 O 8+ [M+H+]: 569,1918, observado 569,1918.

Sulfuro 49ea. La 8-aminometil-uncialamicina en bruto (48aa) [preparada de acuerdo con el procedimiento general descrito anteriormente a partir de la 8-ftalimidometil-uncialamicina (47aa, 16 mg, 27 jmol, 1,0 equiv.)] se suspendió en THF desgasificado (1,0 ml) a 0 °C y se añadió NaHCO 3 ac. saturado (1,0 ml) en una porción, seguido de la adición gota a gota de una solución de cloroformiato de 2-(feniltio)etilo (14 mg, 66 jmol, 2,5 equiv.) en THF desgasificado (0,5 ml). El matraz de reacción se envolvió con papel de aluminio y la mezcla de reacción se agitó a 0 °C durante 5 h, y después se repartió entre EtOAc (5 ml) y tampón pH 6,8 (5 ml). La capa acuosa se extrajo con EtOAc (2 x 25 ml) y los extractos orgánicos combinados se lavaron con salmuera (5 ml), se secaron sobre Na 2 SO 4 y se concentraron hasta aprox. 1 ml en volumen. La cromatografía en columna ultrarrápida [gel de sílice desactivado (véanse los Métodos Generales), hexanos:EtOAc 1:1 a 1:2] para producir el sulfuro 49ea (9,5 mg, 14 |jmol, rendimiento del 55 %) en forma de un sólido de color púrpura. 49ea: Fr = 0,23 (gel de sílice, hexanos:EtOAc 1:1); 1H RMN (600 MHz, CD3CN): 8 = 13,20 (s, 1 H), 10,01 (d, J = 4,1 Hz, 1 H), 8,50 (s, 1 H), 8,23 (d, J = 8,0 Hz, 1 H), 8,16 (s, 1 H), 7,73 (dd, J = 8,0, 1,8 Hz, 1 H), 6,00 (a, 1 H), 5,98 (d, J = 10,0 Hz, 1 H), 5,89 (dt, J = 10,0, 1,3 Hz, 1 H), 5,24 (d, J = 4,4 Hz, 1 H), 4,94 (dd, J = 4,4, 1,7 Hz, 2 H), 4,65 (d, J = 5,0 Hz, 1 H), 4,4-4,2 (m, 2 H), 4,39 (d, J = 5,0, 1 H), 4,38 (dc, J = 5,0, 6,5 Hz, 1 H), 3,35 (d, J = 4,8 Hz, 1 H), 3,2-3,0 (m, 2 H), 1,38 (d, J = 6,5 Hz, 3 H) ppm; 13C RMN (150 MHz, CD3CN): 8 = 188,6, 184,2, 154,4, 147,3, 144,6, 136,1, 135,1, 134,6, 134,0, 133,8, 129,1, 128,6, 128,1, 125,3 125,1, 124,5, 124,1, 114,3, 112,4, 100,3, 99,1, 91,2, 88 ,8 , 77,1, 65,8, 65,3, 64,7, 64,6, 44,3, 44,2, 32,4, 21,2 ppm; HRMS (ESI-TOF): calc. para C36H29N2O8S+ [M+H+]: 649,1639 encontrado 649,1638.

Sulfona 49fa. A una solución agitada del sulfuro 49ea (6,5 mg, 10 jmol, 1,0 equiv.) en CH2Cl2 (0,5 ml) se le añadió dimetildioxirano recién preparado (2,0 ml, ~0,1 M en acetona, 0,2 mmol, 20 equiv.) a -78 °C, la mezcla de reacción se envolvió en papel de aluminio y se dejó calentar a 0 °C. Después de agitar a 0 °C durante 20 min, se añadió Me2S (84 mg, 0,1 ml, 1,4 mmol, 135 equiv.) en una porción y la agitación se continuó durante 20 min a la misma temperatura. Después, la mezcla de reacción se diluyó con EtOAc (25 ml), se lavó con H2O (5 ml) y salmuera (5 ml), se secó sobre Na2SO4 y se concentró hasta aprox. 1 ml en volumen. La cromatografía en columna ultrarrápida [gel de sílice desactivado (véanse los Métodos Generales), hexanos: EtOAc 1:1 a 1:3] proporcionó la sulfona 49fa (2,7 mg, 4,5 jmol, rendimiento del 55 %) en forma de un sólido de color púrpura. 49fa: Fr = 0,23 [gel de sílice desactivado (véanse los Métodos Generales), hexanos:EtOAc 1:3 y después EtOAc:MeOH 50:1]; 1H RMN (600 MHz, CD3CN): 8 = 13,19 (s, 1 H), 9,99 (d, J = 4,1 Hz, 1 H), 8,51 (s, 1 H), 8,24 (d, J = 8,0 Hz, 1 H), 8,18 (s, 1 H), 7,73 (dd, J = 8,0, 1,8 Hz, 1 H), 6,05 (a, 1 H), 5,97 (d, J = 10,0 Hz, 1 H), 5,88 (dt, J = 10,0, 1,3 Hz, 1 H), 5,26 (d, J = 4,4 Hz, 1 H), 4,97 (dd, J = 4,4, 1,7 Hz, 2 H), 4,66 (d, J = 5,0 Hz, 1 H), 4,6-4,3 (m, 2 H), 4,39 (d, J = 5,1, 1 H), 4,38 (dc, J = 5,0, 6,5 Hz, 1 H), 3,50 (a, 2 H), 3,35 (d, J = 4,9 Hz, 1 H), 1,38 (d, J = 6,5 Hz, 3 H) ppm; 13C RMN (150 MHz, CD3CN): 8 = 188,5, 184,1, 153,7, 147,3, 144,6, 139,1, 136,1, 134,6, 134,1, 134,0, 133,7, 129,5, 128,1, 128,0, 125,0, 124,5, 124,1, 114,4, 112,4, 100,4, 99,1, 91,2, 8 8 ,8 , 77,0, 65,8, 65,3, 64,7, 59,4, 55,2, 44,3, 44,2, 21,2 ppm; HRMS (ESI-TOF): calc. para C36H29N2O10S+ [M+H+]: 681,1537, observado 681,1532.

Formilbenzamida 52. A una solución agitada de 2,4-dihidroxi-3-metilbenzoato de metilo (0,93 g, 5,10 mmol) en DMF (10 ml) se le añadieron K2CO3 anhidro (5,64 g, 40,8 mmol, 8,0 equiv., secado a alto vacío a 110 °C durante 16 h antes de usarse) y yoduro de metilo (925 jl, 2,90 g, 20,4 mmol, 4,0 equiv.) a temperatura ambiente en atmósfera de N2. Después de agitar a 50 °C durante 9 h, la mezcla de reacción se filtró a través de un lecho corto de Celite®. El filtrado se diluyó con EtOAc (25 ml) y se acidificó con HCl 3 N (20 ml). La capa orgánica se separó y la capa acuosa se extrajo con EtOAc (3 x 25 ml). Las capas orgánicas combinadas se lavaron con NaHCO3 ac. saturado (25 ml) y salmuera (3 x 25 ml), se secaron sobre MgSO4 y se concentraron para proporcionar el éter metílico 50 (1,05 g, 5,01 mmol, rendimiento del 98 %) en forma de un aceite incoloro. Se añadió trimetilaluminio (solución 2 ,0 M en hexanos, 5,0 ml, 10,0 mmol, 2,0 equiv.) a una solución enfriada con hielo de dietilamina (2,0 ml, 1,46 g, 20,0 mmol, 4,0 equiv.) en benceno (3,0 ml). Después de 10 min, el baño de refrigeración se retiró y el matraz de reacción se dejó calentar a temperatura ambiente. Una solución del éter metílico 50 (vide supra) en 2,0 ml de benceno se añadió gota a gota a la mezcla de reacción durante 5 min. Después, la mezcla de reacción se calentó a reflujo en un baño de aceite a 120 °C (PRECAUCIÓN: desprendimiento de gas). Después de 7 h, el baño de calentamiento se retiró y el matraz de reacción se dejó enfriar a temperatura ambiente. La mezcla de reacción se vertió cuidadosamente en una mezcla de agua con hielo (25 ml) y HCl conc. (0,5 ml). Las capas se separaron y la capa acuosa se extrajo con EtOAc (3 x 25 l). Las capas orgánicas combinadas se lavaron con H2O (25 ml) y salmuera (25 ml), se secaron sobre MgSO4 y se concentraron. La cromatografía en columna ultrarrápida (gel de sílice, hexanos:EtOAc 3:1 a 1:1) dio la benzamida 51 en forma de un aceite de color amarillento (2,14 g, 8,5 mmol, rendimiento del 85 %). A una solución
agitada de la benzamida 51 en THF (60 ml) a -78 °C se le añadió N.N.N'.N'-tetrametiletilendiamina (2,54 ml, 1,98 g, 17.0 mmol, 2,0 equiv.), seguido de la adición gota a gota de ferc-BuLi (solución 1,7 M en pentano, 10,0 ml, 17.0 mmol, 2,0 equiv.) a -78 °C. Después de 50 min, se añadió DMF (7,85 ml, 102 mmol, 12 equiv.) y después de 50 min más, el baño de refrigeración se retiró y la mezcla se dejó calentar a temperatura ambiente durante 2 h. La mezcla de reacción se diluyó con H2O (25 ml) y, después de 20 min de agitación, la solución diluida se concentró parcialmente para retirar los disolventes orgánicos volátiles. Al residuo acuoso se le añadió EtOAc (50 ml) y las dos capas se separaron. La capa acuosa se extrajo con EtOAc (50 ml). Las capas orgánicas combinadas se lavaron con H2O (25 ml) y salmuera (3 x 25 ml), se secaron sobre MgSO4 y se concentraron. La cromatografía en columna ultrarrápida (gel de sílice, hexanos:EtOAc 2:1 a 1:1) dio la formilbenzamida 52 en forma de un sólido de color amarillo pálido (2,37 g, 8,5 mmol, rendimiento del 99 %). 52: Fr = 0,31 (gel de sílice, hexanos:EtOAc 1:1); RMN 1H (500 MHz, CDCla): 8 = 9,93 (s, 1 H), 7,20 (s, 1 H), 3,90 (s, 3 H), 3,80 (s, 3 H), 3,73 (dc, J = 7,1, 13,7 Hz, 1 H), 3,52 (dc, J = 7,1, 13,7 Hz, 1 H), 3,03 (ddc, J = 7,2, 14,4, 14,4, Hz, 2 H), 2,21 (s, 3 H), 1,29 (t, J = 7,1 Hz, 3 H), 1,00 (t, J = 7,2 Hz, 3 H) ppm; 13C RMN (125 MHz, CDCla): 8 = 189,9, 166,2, 159,0, 155,1, 131,3, 128,1, 120,2, 104,3, 112,3, 62,0, 55,9, 43,2, 39,2, 13,8, 12,7, 9,7 ppm; HRMS (ESI-TOF): calc. para C15H22NO4+ [M+H+]: 280,1543, observado 280,1540.

Cianoftalida 55. A una solución agitada de la formilbenzamida 52 (2,37 g, 8,5 mmol, 1,0 equiv.) en CCU ( 8 ml) se le añadió N-bromosuccinimida (1,95 g, 10,2 mmol, 1,2 equiv.) a temperatura ambiente en atmósfera de N2, la mezcla de reacción se calentó a 80 °C y se añadió peróxido de benzoílo (121 mg, 0,5 mmol, 0,06 equiv.) en una porción. El calentamiento se continuó durante 2 h y la mezcla de reacción se dejó enfriar a temperatura ambiente, se lavó con NaHCO3 ac. saturado (25 ml) y salmuera (25 ml), se secó sobre MgSO4 y se concentró para dar el bromuro de bencilo 53 en bruto en forma de un sólido de color pardo claro. Después, el bromuro de bencilo 53 se disolvió en DMF (7,5 ml), Se añadieron secuencialmente K2CO3 (1,75 g, 12,8 mmol, 1,5 equiv.) y n-Bu4NI (0,31 g, 0,85 mmol, 0,1 equiv.), seguido de ftalimida (1,4 g, 9,4 mmol, 1,1 equiv.) en una porción. La mezcla de reacción se agitó a 40 °C durante 2 h, después se vertió en H2O (25 ml) y se extrajo con EtOAc (3 x 25 ml). Las capas orgánicas combinadas se lavaron con agua (3 x 25 ml) y salmuera (2 x 25 ml), se secaron sobre MgSO4 y se concentraron. La cromatografía en columna ultrarrápida (gel de sílice, hexanos:EtOAc 2:1 a 1:2) dio la ftalimida 54 en forma de un sólido de color amarillento (2,16 g, 5,1 mmol, rendimiento del 60 % en 2 etapas). A una solución agitada de la ftalimida 54 en CH2Cl2 (10 ml) a 0 °C se le añadieron TMSCN (1,01 g, 1,28 ml, 10,2 mmol, 2,0 equiv.) y una solución de KCN (8,2 mg, 0,13 mmol, 0,025 equiv.) y 18-corona-6 (25 mg, 0,09 mmol, 0,02 equiv.) en THF (1,0 ml). La mezcla de reacción se agitó a la misma temperatura en un matraz cerrado herméticamente durante 1,5 h y durante 30 min a temperatura ambiente. Después, la mezcla de reacción se concentró en atmósfera de N2 y el residuo se coevaporó con tolueno (2 x 25 ml) para retirar todas las trazas de TMSCN. El aceite de color pardo resultante se disolvió en AcOH (5 ml) y se agitó durante 80 h a temperatura ambiente hasta que el análisis por TLC mostró la conversión completa (hexanos:EtOAc 1:1). La reacción se interrumpió mediante la adición cuidadosa de NaOH 1 N (15 ml) y la mezcla resultante se repartió entre EtOAc (30 ml) y NaOH 1 N (15 ml). La capa acuosa se extrajo con EtOAc (3 x 30 ml) y las capas orgánicas combinadas se lavaron con H2O (30 ml) y salmuera (30 ml), se secaron sobre MgSO4 y se concentraron. La cromatografía en columna ultrarrápida (gel de sílice, hexanos:EtOAc 3:2 a 1:1) dio un sólido de color amarillento, que se recristalizó en EtOAc para producir la cianoftalida 55 en forma de un sólido de color amarillento. (1,36 g, 3,6 mmol, rendimiento del 70 %). 52: Fr = 0,35 (gel de sílice, hexanos:EtOAc 1:1); RMN 1H (500 MHz, CDCh): 8 = 7,81 (dd, J = 5,5, 3,0 Hz, 2 H), 7,71 (dd, J = 5,5, 3,0 Hz, 2 H), 6,81 (s, 1 H), 5,93 (s, 1 H), 4,97 (s, 2 H), 4,23 (s, 3 H), 3,94 (s, 3 H) ppm; 13C RMN (100 MHz, CDCh): 8 = 167,7, 165,3, 165,0, 159,4, 145,4, 134,0, 131,9, 123,2, 119,7, 114,0, 107,7, 99,4, 64,9, 63,2, 56,8, 31,4 ppm; HRMS (ESI-TOF): calc. para C20H15N2O6+ [M+H+]: 379,0925, observado 379,0926.

8-Ftalimidometil-7,9-dimetoxi-uncialamicina (58a). A una solución agitada de la cianoftalida 55 (72 mg, 0,19 mmol, 3,0 equiv.) en THF (0,8 ml) a -78 °C se le añadió LiHMDS (1,0 M en THF, 0,25 ml, 0,25 mmol, 4,0
equiv.). La mezcla resultante se agitó a -78 °C durante 20 min y se añadió una solución enfriada previamente de la quinona aminal 22a (34 mg, 63 |jmol, 1,0 equiv.) en THF (0,8 ml) a -78 °C mediante una cánula. Después de agitar durante 5 min a -78 °C, la mezcla de reacción se dejó calentar a temperatura ambiente y se agitó durante 90 min más, tiempo durante el cual la mezcla de reacción se volvió de color rojo oscuro. Después, la mezcla de reacción se inactivó con tampón pH 6 ,8 (30 ml) y se extrajo con EtOAc (3 x 30 ml). Los extractos orgánicos combinados se lavaron con salmuera (30 ml), se secaron sobre MgSO4 y se filtraron a través de un lecho corto de Celite® (lavado con EtOAc antes de usarse). Los sólidos se aclararon con EtOAc y los filtrados combinados se concentraron para producir la alloc-antraquinona 56a en bruto en forma de un sólido de color rojo oscuro, que se disolvió en THF desgasificado (1,5 ml) en una atmósfera de Ar y se enfrió a 0 °C. A esta solución se le añadió Pd(PPh3)4 (4 mg, 1,3 jmol, 0,2 equiv.), seguido de la adición gota a gota de morfolina (7 mg, 7 j l , 76 jmol, 1,2 equiv.), el matraz de reacción se envolvió con papel de aluminio y la mezcla resultante se dejó en agitación a 0 °C durante 2 h, tiempo durante el cual se volvió de color púrpura. El baño de refrigeración se retiró y la mezcla de reacción se agitó a temperatura ambiente durante 20 min y después se inactivó mediante la adición de tampón pH 6 ,8 (10 ml) y se extrajo con EtOAc (3 x 10 ml). Los extractos orgánicos combinados se lavaron con H2O (10 ml) y salmuera (10 ml), se secaron sobre MgSO4 y se filtraron a través de un lecho corto de Celite® (lavado con EtOAc antes de usarse). Los sólidos se aclararon con EtOAc y los filtrados combinados se concentraron hasta aprox. 1 ml en volumen. La cromatografía en columna ultrarrápida [gel de sílice desactivado (véanse los Métodos Generales), hexanos:EtOAc 1:1 a 1:2] proporcionó la antraquinona 57a en bruto en forma de un sólido de color púrpura. A una solución de la antraquinona 57a en THF desgasificado (1,5 ml) a temperatura ambiente se le añadió una solución de 1:1 de 3HF^EtaN:THF (0,5 ml). El matraz de reacción se envolvió con papel de aluminio y la mezcla de reacción se agitó a temperatura ambiente durante 1,5 h, y después se repartió entre EtOAc (10 ml) y NaHCO3 ac. saturado (10 ml). La capa orgánica se lavó con salmuera (10 ml), se secó sobre Na2SO4 y se concentró hasta aprox. 1 ml en volumen. La cromatografía en columna ultrarrápida [gel de sílice desactivado (véanse los Métodos Generales), hexanos:EtOAc 1:1 a 1:3] proporcionó la 8-ftalimidometil-7,9-dimetoxi-uncialamicina (58a) (5 mg, 7,6 jmol, rendimiento del 12 %) en forma de un sólido de color púrpura. 58a: Fr = 0,42 (gel de sílice, hexanos:EtOAc 1:2); 1H RMN (600 MHz, CD3CN): 8 = 13,58 (s, 1 H), 9,88 (d, J = 4,2 Hz, 1 H), 8,47 (s, 1 H), 7,80 (dd, J = 5,4, 3,1 Hz, 2 H), 7,77 (dd, J = 5,5, 3,0 Hz, 2 H), 7,67 (s, 1 H), 5,96 (d, J = 10,0 Hz, 1 H), 5,88 (d, J = 9,9 Hz, 1 H), 5,23 (d, J = 4,9 Hz, 1 H), 4,97 (s, 2 H), 4,91 (dd, J = 4.3, 1,4 Hz, 1 H), 4,54 (d, J = 5,0 Hz, 1 H), 4,37 (dc, J = 5,0, 6,5 Hz, 1 H), 3,93 (s, 3 H), 3,92 (s, 3 H), 3,30 (d, J = 4,9 Hz, 1 H), 1,38 (d, J = 6,3 Hz, 3 H) ppm; 13C RMN (150 MHz, CD3CN): 8 = 187,1, 183,4, 168,7, 164,3, 162,2, 156.3, 144,3, 139,0, 135,1, 134,6, 133,0, 131,5, 130,7, 125,2, 124,5, 124,1, 123,8, 119,6, 114,9, 112,3, 100,4, 99,1, 91,2, 88 ,8 , 77,0, 65,9, 65,3, 64,8, 62,7, 57,2, 56,0, 44,3, 21,2 ppm; HRMS (ESI-TOF): calc. para C37H27N2O10+ [M+H+]: 659,1660, observado 659,1664.

8-FtaMmidometN-7,9-dimetoxi-17-ep/-uncialamicma (58b). Se preparó de acuerdo con el procedimiento general descrito anteriormente para la preparación de 58a, a partir de la cianoftalida 55 (30 mg, 78 jmol, 3,0 equiv.) y la quinona aminal 22b (14 mg, 26 jmol, 1 ,0 equiv.) a través del intermedio 57b para producir la 8-ftalimidometil-7,9-dimetoxi-17-ep/-unciatamicina (58b) (2 mg, 44 jmol, rendimiento del 12 %) en forma de un sólido de color púrpura.
58b: Fr = 0,52 (gel de sílice, hexanos:EtOAc 1:2); 1H RMN (500 MHz, CD3CN): 8 = 13,63 (s, 1 H), 9,93 (d, J = 4,0 Hz, 1 H), 7,80 (m, 2 H), 7,77 (m, 2 H), 7,67 (s, 1 H), 7,59 (s, 1 H), 5,93 (sistema AB, 2 H), 5,85 (d, J = 4,9 Hz, 1 H), 5,10 (dc, J = 5,0, 6,5 Hz, 1 H), 4,97 (s, 2 H), 4,87 (dd, J = 4,6, 1,4 Hz, 1 H), 4,27 (d, J = 5,0 Hz, 1 H), 3,19 (d, J = 4,8 Hz, 1 H), 1,34 (d, J = 6,5 Hz, 3 H) ppm; HRMS (ESI-TOF): calc. para C37H27N2O10+ [M+H+]: 659,1660, observado 659,1663.

Formilbenzamida 60. Una solución agitada de ácido 6,7-dimetil-2-naftoico (2,46 g, 12,3 mmol) se suspendió en SOCl2 (15 ml) y la mezcla de reacción se calentó a reflujo durante 2 h. Después, la mezcla de reacción se enfrió a temperatura ambiente y se concentró en atmósfera de N2 y el residuo se coevaporó con tolueno (2 x 25 ml) para retirar todas las trazas de SOCl2. El producto en bruto se disolvió en CH2Cl2 (2 ml) y la mezcla de reacción se enfrió a 0 °C. Se añadió gota a gota una solución de dietilamina (1,62 g, 2,29 ml, 22,1 mmol, 1,8 equiv.) en CH2Cl2 (2 ml).
Después de que se completara la adición, el baño de hielo se retiró y la mezcla de reacción se agitó a temperatura ambiente durante 2 h. La reacción se interrumpió con HCl 1 N (25 ml) y la capa acuosa se extrajo con CH2Cl2 (2 x 20 ml). Las capas orgánicas combinadas se lavaron con salmuera (20 ml), se secaron sobre MgSO4 y se concentraron para proporcionar la benzamida 59 (3,11 g, 12,2 mmol, rendimiento del 99 %) en forma de un sólido de color amarillo. A una solución agitada de la benzamida 59 en THF (75 ml) a -78 °C se le añadió N,N,N',N'-tetrametiletilendiamina (3,64 ml, 2,83 g, 24,4 mmol, 2,0 equiv.), seguido de la adición gota a gota de ferc-BuLi (solución 1,7 M en pentano, 14,3 ml, 24,4 mmol, 2,0 equiv.) a -78 °C. Después de 50 min, se añadió DMF (11,24 ml, 146 mmol, 12 equiv.) y, después de 50 min más de agitación, el baño de refrigeración se retiró y el matraz de reacción se dejó calentar a temperatura ambiente. Después de 2 h, la mezcla de reacción se diluyó con H2O (25 ml). Después de 20 min de agitación adicional, la solución diluida se concentró parcialmente para retirar los disolventes orgánicos volátiles. Al residuo acuoso resultante se le añadió EtOAc (50 ml) y las dos capas se separaron. La capa acuosa se extrajo con EtOAc (50 ml) y las capas orgánicas combinadas se lavaron con H2O (25 ml) y salmuera (3 x 25 ml), se secaron sobre MgSO4 y se concentraron. La cromatografía en columna ultrarrápida (gel de sílice, hexanos:EtOAc 2:1 a 1:1) dio la formilbenzamida 60 en forma de un sólido de color amarillo pálido (1,07 g, 3,78 mmol, rendimiento del 31 %). 52: Fr = 0,31 (gel de sílice, hexanos:EtOAc 1:1); RMN 1H (500 MHz, CDCla): 8 = 10,13 (s, 1 H), 8,65 (s, 1 H), 8,56 (s, 1 H), 7,41 (s, 1 H), 7,20 (s, 1 H), 3,83 (dc, J = 7,1, 13,7 Hz, 1 H), 3,55 (dc, J = 7,1, 13,7 Hz, 1 H), 3,03 (ddc, J = 7,2, 14,4, 14,4, Hz, 2 H), 2,21 (s, 3 H), 2,20 (s, 3 H), 1,25 (t, J = 7,1 Hz, 3 H), 0,98 (t, J = 7,2 Hz, 3 H) ppm; 13C RMN (125 MHz, CDCla): 8 = 190,0, 160,2, 138,8, 138,4, 133,1, 132,9, 132,3, 132,1, 132,0, 128,9, 128,6, 128,5, 120,2, 43,2, 39,2, 13,8, 12,7, 9,7 ppm; HRMS (ESI-TOF): calc. para C1sH22NO2+ [M+H+]: 284,1645, observado 284,1640.

Cianoftalida 63a/63b. A una solución agitada de la formilbenzamida 60 (1,07 g, 3,78 mmol, 1,0 equiv.) en CCU (4 ml) se le añadió N-bromosuccinimida ( 866 mg, 4,53 mmol, 1,2 equiv.) a temperatura ambiente en atmósfera de N2, la mezcla de reacción se calentó a 80 °C y se añadió peróxido de benzoílo (54 mg, 0,222 mmol, 0,06 equiv.) en una porción. El calentamiento se continuó durante 2 h y la mezcla de reacción se enfrió a temperatura ambiente, se lavó con NaHCO3 ac. saturado ( 10 ml) y salmuera ( 10 ml), se secó sobre MgSO4 y se concentró para dar la mezcla de bromuro de bencilo 61a/61b en bruto (aprox. 1:1 por 1H RMN) en forma de un sólido de color pardo claro. Después, el bromuro de bencilo 61a/61b (aprox. 1:1 por 1H RMN) se disolvió en DMF (3,5 ml), y se añadieron secuencialmente K2CO3 (777 mg, 5,69 mmol, 1,5 equiv.) y n-Bu4NI (138 mg, 0,378 mmol, 0,1 equiv.), seguido de ftalimida (622 mg, 4,18 mmol, 1,1 equiv.) en una porción. La mezcla de reacción se agitó a 40 °C durante 2 h, después se vertió en H2O (10 ml) y se extrajo con EtOAc (3 x 10 ml). Las capas orgánicas combinadas se lavaron con agua (3 x 10 ml) y salmuera (2 x 10 ml), se secaron sobre MgSO4 y se concentraron. La cromatografía en columna ultrarrápida (gel de sílice, hexanos:EtOAc 2:1 a 1:2) dio una mezcla de la ftalimida 62a/62b en forma de un sólido de color amarillento (aprox. 1:1 por 1H RMN, 599 mg, 1,40 mmol, rendimiento del 37 % en 2 etapas). A una solución agitada de la ftalimida 62a/62b en CH2Cl2 (3 ml) a 0 °C se le añadieron TMSCN (277 mg, 0,35 ml, 2,80 mmol, 2,0 equiv.) y una solución de KCN (2,2 mg, 35 pmol, 0,025 equiv.) y 18-corona-6 (6 ,8 mg, 25 pmol, 0,02 equiv.) en THF (0,3 ml). La mezcla de reacción se agitó a la misma temperatura en un matraz cerrado herméticamente durante 1,5 h y durante 30 min a temperatura ambiente. Después, la mezcla de reacción se concentró en atmósfera de N2 y el residuo se coevaporó con tolueno (2 x 10 ml) para retirar todas las trazas de TMSCN. El aceite de color pardo resultante se disolvió en AcOH (1,5 ml) y se agitó durante 80 h a temperatura ambiente hasta que el análisis por TLC mostró la conversión completa (hexanos:EtOAc 1:1). La reacción se interrumpió mediante la adición cuidadosa de NaOH 1 N (5 ml) y la mezcla resultante se repartió entre EtOAc (10 ml) y NaOH 1 N (5 ml). La capa acuosa se extrajo con EtOAc (3 x 10 ml) y las capas orgánicas combinadas se lavaron con H2O (10 ml) y salmuera (10 ml), se secaron sobre MgSO4 y se concentraron. La cromatografía en columna ultrarrápida (gel de sílice, CH2Ch:EtOAc 30:1 a 20:1) dio una mezcla de la cianoftalida 63a/63b en forma de un sólido de color amarillento, (aprox. 1:1 por 1H RMN, 375 mg, 0,98 mmol, rendimiento del 70 %). 63a: Fr = 0,35 (gel de sílice, hexanos:EtOAc 1:1); 63b: Fr = 0,33 (gel de sílice, hexanos:EtOAc 1:1); RMN 1H (500 MHz, CDCh): 8 = 8,41 (s, 1H), 8,37 (s, 1H), 7,81 (m, 4 H), 7,71 (m, 4 H), 7,61 (s, 1 H), 7,59 (s, 1 H), 7,56 (s, 1 H), 7,55 (s, 1 H), 7,49 (s, 1 H), 7,47 (s, 1 H), 6,04 (a, 2 H), 5,17 (s, 2 H), 5,15 (s, 2 H), 2,27 (s, 3 H), 2,24 (s, 3 H) ppm; HRMS (ESI-TOF): calc. para C23H-I5N2O4+ [M+H+]: 383,1026, observado 383,1026.

Ftalimidometilbenzo-uncialamicina 66aa/66ba. A una solución de la cianoftalida 63a/63b (aprox. 1:1 por 1H RMN, 73 mg, 0,19 mmol, 3,0 equiv.) en THF (0,8 ml) a -78 °C se le añadió LiHMDS (1,0 M en THF, 0,25 ml, 0,25 mmol, 4,0 equiv.). La mezcla resultante se agitó a -78 °C durante 20 min y se añadió una solución enfriada previamente de la quinona aminal 22a (34 mg, 63 pmol, 1,0 equiv.) en THF (0,8 ml) a -78 °C mediante una cánula. Después de agitar durante 5 min a -78 °C, la mezcla de reacción se calentó a temperatura ambiente y se agitó durante 90 min más, tiempo durante el cual la mezcla de reacción se volvió de color rojo oscuro. Después, la mezcla de reacción se inactivó mediante la adición de tampón pH 6 ,8 (30 ml) y se extrajo con EtOAc (3 x 30 ml). Los extractos orgánicos combinados se lavaron con salmuera (30 ml), se secaron sobre MgSO4 y se filtraron a través de un lecho corto de Celite® (lavado con EtOAc antes de usarse). Los sólidos se aclararon con EtOAc y los filtrados combinados se concentraron para producir la mezcla en bruto de alloc-antraquinona 64aa/64ba (aprox. 1:1 por 1H RMN) en forma de un sólido de color rojo oscuro, que se disolvió en THF desgasificado (1,5 ml) en una atmósfera de Ar y se enfrió a 0 °C. A esta solución se le añadió Pd(PPh3)4 (4 mg, 1,3 pmol, 0,2 equiv.), seguido de la adición gota a gota de morfolina (7 mg, 7 pl, 76 pmol, 1,2 equiv.), el matraz de reacción se envolvió con papel de aluminio y la mezcla resultante se dejó en agitación a 0 °C durante 2 h, tiempo durante el cual se volvió de color púrpura. El baño de refrigeración se retiró y la mezcla de reacción se agitó a temperatura ambiente durante 20 min y después se inactivó mediante la adición de tampón pH 6 ,8 (10 ml) y se extrajo con EtOAc (3 x 10 ml). Los extractos orgánicos combinados se lavaron con H2O ( 10 ml) y salmuera ( 10 ml), se secaron sobre MgSO4 y se filtraron a través de un lecho corto de Celite® (lavado con EtOAc antes de usarse). Los sólidos se aclararon con EtOAc y los filtrados combinados se concentraron hasta aprox. 1 ml en volumen. La cromatografía en columna ultrarrápida [gel de sílice desactivado (véanse los Métodos Generales), hexanos:EtOAc 1:1 a 1:2] proporcionó la mezcla en bruto de antraquinona 65aa/66ba (aprox. 1:1 por 1H RMN) en forma de un sólido de color púrpura. A una solución de la antraquinona 65aa/66ba en THF desgasificado (1,5 ml) a temperatura ambiente se le añadió una solución de 1:1 de 3HF^EtaN:THF (0,5 ml). El matraz de reacción se envolvió con papel de aluminio y la mezcla de reacción se agitó a temperatura ambiente durante 1,5 h, y después se repartió entre EtOAc (10 ml) y NaHCO3 ac. saturado (10 ml). La capa orgánica se lavó con salmuera (10 ml), se secó sobre Na2SO4 y se concentró hasta aprox. 1 ml en volumen. La cromatografía en columna ultrarrápida [gel de sílice desactivado (véanse los Métodos Generales), hexanos:EtOAc 1:1 a 1:3] proporcionó una mezcla 1:1 de la ftalimido-metilbenzo-uncialamicina 66aa/66ba (aprox. 1:1 por 1H RMN, 12 mg, 17,7 pmol, rendimiento del 28 %) en forma de un sólido de color púrpura. 66aa/66ba: Fr = 0,42 (gel de sílice, hexanos:EtOAc 1:2); 1H RMN (600 MHz, CD3CN): 8 = 13,49 (s, 2 H), 9,91 (d, J = 4,1 Hz, 1 H), 9,88 (d, J = 4,2 Hz, 1 H), 8,47 (s, 1 H), 8,31 (s, 1 H), 7,80 (m, 4 H), 7,76 (m, 4 H), 7,67 (s, 1 H), 7,65 (s, 1 H), 7,43 (s, 1 H), 7,41 (s, 1 H), 5,96 (d, J= 10,0 Hz, 2 H), 5,89 (d, J = 9,9 Hz, 2 H), 5,25 (d, J = 5,0 Hz, 1 H), 5,23 (d, J = 4,9 Hz, 1 H), 5,11 (s, 2 H), 5,07 (s, 2 H), 4,91 (dd, J = 4,4, 1,4 Hz, 1 H), 4,89 (dd, J = 4,5, 1,4 Hz, 1 H), 4,53 (d, J = 5,0 Hz, 1 H), 4,51 (d, J = 5,0 Hz, 1 H), 4,37 (dc, J = 5,0, 6,5 Hz, 1 H), 4,35 (dc, J = 5,0, 6,5 Hz, 1 H), 3,30 (d, J = 4,9 Hz, 1 H), 2,11 (s, 3 H), 2,09 (s, 3 H), 1,38 (d, J = 6,5 Hz, 3 H), 1,38 (d, J = 6,5 Hz, 3 H) ppm; HRMS (ESI-TOF): calc. para C40H27N2Ob+ [M+H+]: 663,1762, observado 663,1764.

Ftalida 72. A una solución agitada de ácido 2,7-dihidroxil-2-naftoico (7,50 g, 36,7 mmol, 1,0 equiv.) en MeOH (50 ml) se le añadió H2SO4 conc. (1,0 ml) en una porción a temperatura ambiente y la mezcla de reacción se calentó a reflujo durante 12 h. Después de la refrigeración, el éster 67, que se había separado en forma de agujas, se recogió en un filtro. El filtrado se concentró para obtener un segundo cultivo de cristales. Los dos cultivos se combinaron, se disolvieron en éter y se lavaron con NaHCO3 ac. al 5% y H2O. La capa orgánica se secó sobre MgSO4, se concentró a sequedad y el producto en bruto se recristalizó en metanol para producir el éster 67 en forma de un sólido de color blanco (7,85 g, 36,0 mmol, rendimiento del 98 %). Una solución de N-feniltrifluorometano-sulfonimida (39,8 g, 111 mmol, 3,0 equiv.) en CH2Cl2 (125 ml) se añadió gota a gota a una solución del éster 67 (7,85 g, 36,0 mmol, 1,0 equiv.), N-diisopropiletilamina (25,9 ml, 148 mmol, 4,1 equiv.) y DMAP (452 mg, 3,7 mmol, 0,10 equiv.) en CH2Cl2 (100 ml) a 0 °C. Después de que se completara la adición, el baño de hielo se retiró y la mezcla de reacción se agitó durante 1,5 h más a temperatura ambiente, después se inactivó con NaHCO3 ac. saturado (10 ml) y se diluyó con
H2O (40 ml). La capa acuosa se extrajo con CH2CI2 (3 x 30 ml) y las capas orgánicas combinadas se secaron sobre MgSO4 y se concentraron. La cromatografía en columna ultrarrápida [gel de sílice desactivado (véanse los Métodos Generales), hexanos:EtOAc 10:1] proporcionó el bis-triflato 68 en forma de un sólido de color blanco (14,72 g, 30,5 mmol, rendimiento del 85 %). Se añadió trimetilboroxina (11,26 g, 89,8 mmol, 3,0 equiv.) a una mezcla del triflato 68 (14,72 g, 30,5 mmol, 1,0 equiv.), Pd(PPh3)4 (3,46 g, 3,0 mmol, 0,1 equiv.) y K2CO3 (24,80 g, 179,4 mmol, 6.0 equiv.) en dioxano desgasificado (150 ml) y la mezcla de reacción se agitó a 95 °C durante 1 h. Se añadió H2O (25 ml) y la mezcla se agitó durante 4 h más a 95 °C. La mezcla de reacción enfriada se filtró a través de un lecho corto de Celite® y se concentró. La cromatografía en columna ultrarrápida (gel de sílice, hexanos:EtOAc 10:1 a 5:1) proporcionó el 2-naftoato de metilo 69 en forma de un sólido de color blanco (5,29 g, 24,7 mmol, rendimiento del 81 %). El éster 69 (5,29 g, 24,7 mmol, 1,0 equiv.) y N-bromosuccinimida (19,1 g, 100 mmol, 4,0 equiv.) se disolvieron en CCl4 (50 ml), la mezcla de reacción se calentó a 80 °C y se añadió peróxido de benzoílo (121 mg, 0,5 mmol, 0,02 equiv.) en una porción. El calentamiento se continuó durante 8 h y la mezcla de reacción se enfrió a temperatura ambiente y después se almacenó a 0 °C durante 12 h. Después, la mezcla de reacción se filtró y los precipitados se aclararon con CCU (0-5 °C). Después, los filtrados combinados se lavaron con NaHCO3 ac. saturado (25 ml) y salmuera (25 ml), se secaron sobre MgSO4 y se concentraron para producir el producto en bruto 70 en forma de un sólido de color amarillento. El dibromoéster 7o puro se calentó a 150 °C a ligero vacío durante 10 h. La refrigeración de la mezcla de reacción a temperatura ambiente dio el producto en bruto 71 en forma de un sólido de color pardo claro. La bromoftalida 71 en bruto se disolvió en DMF (50 ml) y se añadieron secuencialmente K2CO3 anhidro (10,3 g, 74,8 mmol, 3,0 equiv., secado a alto vacío a 110 °C durante 16 h antes de usarse) y n-Bu4NI (1,8 g, 5.0 mmol, 0,2 equiv.), seguido de ftalimida (8,0 g, 54,9 mmol, 2,2 equiv.) en una porción. La mezcla de reacción se agitó a 40 °C durante 4 h, después se vertió en H2O (100 ml) y se extrajo con EtOAc (3 x 100 ml). Las capas orgánicas combinadas se lavaron con agua (3 x 50 ml) y salmuera (2 x 50 ml), se secaron sobre MgSO4 y se concentraron. La cromatografía en columna ultrarrápida (gel de sílice, CH2Ch:EtOAc 10:1 a 8:1) dio la ftalida 72 en forma de un sólido de color amarillento (4,94 g, 14,4 mmol, rendimiento del 39 %). 72: Fr = 0,31 (gel de sílice, CH2Cl2:EtOAc 8:1); RMN 1H (600 MHz, CDCh): 8 = 8,48 (s, 1 H), 8,03 (d, J = 8,0 Hz, 1 H), 7,88 (s, 1 H), 7,87 (dd, J = 5,4, 3,1 Hz, 2 H), 7,75 (dd, J = 5,5, 3,0 Hz, 2 H), 7,64 (s, 1 H), 7,47 (d, J = 7,8 Hz, 1 H), 5,45 (s, 2 H), 4,99 (s, 2 H) ppm; HRMS (ESI-TOF): calc. para C21H14NO4+ [M+H+]: 344,0917, observado 344,0915.

Formilbenzamida 75. A una suspensión agitada de la ftalida 72 (4,94 g, 14,4 mmol, 1,0 equiv.) en CCU/benceno (50 ml, 1:1) a temperatura ambiente se le añadió N-bromosuccinimida (3,06 g, 17,3 mmol, 1,2 equiv.) en una porción. La mezcla de reacción se calentó a reflujo y después se añadió azobisisobutironitrilo (0,47 g, 2,87 mmol, 0,2 equiv.) en una porción. La mezcla de reacción se agitó vigorosamente a reflujo durante 2 h más, después se enfrió a temperatura ambiente y se almacenó a 0 °C durante 8 h. La mezcla de reacción se filtró y los precipitados se aclararon con CCU (0-5 °C). Después, los filtrados combinados se lavaron con NaHCO3 ac. saturado (10 ml) y salmuera (10 ml), se secaron sobre Mg2SO4 y se concentraron para producir 73 en bruto en forma de una espuma de color amarillo, que se suspendió en H2O/THF (50 ml, 1:1). La suspensión se agitó a 85 °C durante 5 h y después se dejó enfriar a temperatura ambiente. La mezcla de reacción se extrajo con EtOAc (5 x 20 ml) y las capas orgánicas combinadas se secaron sobre MgSO4 y se concentraron para producir 74 en bruto en forma de un sólido higroscópico de color amarillo, que se secó sobre P2O5 durante 8 h. El ácido en bruto 74 se suspendió en SOCl2 (15 ml) y la mezcla de reacción se calentó a reflujo durante 2 h. La mezcla de reacción se concentró en atmósfera de N2 y las últimas trazas de volátiles (por ejemplo, SOCh) se retiraron azeotrópicamente con tolueno (2 x 10 ml). El producto en bruto se disolvió en CH2Cl2 (15 ml) y la solución se enfrió a 0 °C. Se añadió gota a gota una solución de dietilamina (1,91 g, 2,68 ml, 26,0 mmol, 1,8 equiv.) en CH2Cl2 (3,0 ml), el baño de hielo se retiró y la mezcla de reacción se agitó a temperatura ambiente durante 2 h. La reacción se interrumpió con HCl ac. 1 N (27 ml), y la mezcla de reacción resultante se extrajo con CH2Cl2 (2 x 20 ml). Las capas orgánicas combinadas se lavaron con salmuera (20 ml), se secaron sobre MgSO4 y se concentraron. La cromatografía en columna ultrarrápida (gel de sílice, hexanos:EtOAc 3:1 a 2:1) proporcionó la formilbenzamida 75 en forma de un sólido de color amarillento (2,57 g, 6,19 mmol, rendimiento del 43 %). 75: Fr = 0,31 (gel de sílice, hexanos:EtOAc 2:1); RMN 1H (600 MHz, CDCl3): 8 = 10,13 (s, 1 H), 8,61 (s, 1 H), 8,50 (s, 1 H), 8,00 (d, J = 7,9 Hz, 1 H), 7,81 (dd, J = 5,4, 3,1 Hz, 2 H), 7,71 (dd, J = 5,5, 3,0 Hz, 2 H), 7,64 (s, 1 H), 7,21 (d, J = 7,8 Hz, 1 H), 3,81 (dc, J = 7,1, 13,7 Hz, 1 H), 3,51 (dc, J = 7,1, 13,7 Hz, 1 H), 3,01 (ddc, J = 7,2, 14,4, 14,4, Hz, 2 H), 1,21 (t, J = 7,1 Hz, 3 H), 0,99 (t, J = 7,2 Hz, 3 H) ppm; HRMS (ESI-TOF): calc. para C25H23N2O4+ [M+H+]: 415,1652, observado 415,1658.
Cianoftalida 76. A una solución agitada de la formil-benzamida 75 (1,33 g, 3,20 mmol, 1,0 equiv.) en CH2Cl2 (6 ml) a 0 °C se le añadieron TMSCN (640 mg, 0,81 ml, 6,44 mmol, 2,0 equiv.) y una solución de KCN (5,2 mg, 0,08 mmol, 0,025 equiv.) y 18-corona-6 (16 mg, 0,06 mmol, 0,02 equiv.) en THF (0,6 ml). La mezcla de reacción se agitó a la misma temperatura en un matraz cerrado herméticamente durante 1,5 h, y durante 30 min más a temperatura ambiente. La mezcla de reacción se concentró en atmósfera de N2 y después el residuo se secó azeotrópicamente con tolueno (2 x 25 ml) para retirar todas las trazas de TMSCN. El aceite de color pardo resultante se disolvió en AcOH (3 ml) y se agitó durante 48 h a temperatura ambiente hasta que el análisis por TLC mostró la conversión completa (hexanos:EtOAc 3:2). La reacción se interrumpió mediante la adición cuidadosa de NaOH ac. 1 N (10 ml) y la mezcla resultante se extrajo con EtOAc (20 ml). La capa acuosa se extrajo con EtOAc (3 x 20 ml) y las capas orgánicas combinadas se lavaron con H2O ( 20 ml) y salmuera ( 20 ml), se secaron sobre MgSO4 y se concentraron. La cromatografía en columna ultrarrápida (gel de sílice, hexanos:EtOAc 2:1 a 3:2) dio un sólido de color amarillento, que se recristalizó en EtOAc para producir la cianoftalida 76 (825 mg, 2,24 mmol, rendimiento del 70 %). 76: Fr = 0,33 (gel de sílice, hexanos:EtOAc 3:2); RMN 1H (400 MHz, CDCla): 8 = 8,53 (s, 1 H), 7,99 (d, J = 8,0 Hz, 1 H), 7,86 (dd, J = 5,4, 3,1 Hz, 2 H), 7,74 (dd, J = 5,5, 3,0 Hz, 2 H), 7,64 (s, 1 H), 7,53 (s, 1 H), 7,35 (d, J = 8,0 Hz, 1 H), 6,01 (s, 1 H), 4,99 (s, 2 H) ppm; HRMS (ESI-TOF): calc. para C22H13N2O4IM+H+]: 369,0870, observado 369,0872.

Ftalim idometil-benzo-uncialamicina 79a. A una solución de la cianoftalida 76 (70 mg, 0,19 mmol, 3,0 equiv.) en THF (0,8 ml) a -78 °C se le añadió LiHMDS (1,0 M en THF, 0,25 ml, 0,25 mmol, 4,0 equiv.). La mezcla resultante se agitó a -78 °C durante 20 min y se añadió una solución enfriada previamente de la quinona aminal 22a (véase la última descripción, 34 mg, 63 pmol, 1,0 equiv.) en THF (0,8 ml) a -78 °C mediante una cánula. Después de agitar durante 5 min a -78 °C, la mezcla de reacción se dejó calentar a temperatura ambiente y se agitó durante 110 min más, tiempo durante el cual la mezcla de reacción se volvió de color rojo oscuro. La mezcla resultante se inactivó mediante la adición de tampón fosfato pH 6 ,8 (30 ml) y se extrajo con EtOAc (3 x 30 ml). Los extractos orgánicos combinados se lavaron con salmuera (30 ml), se secaron sobre MgSO4 y se filtraron a través de un lecho corto de Celite® (lavado con EtOAc antes de usarse). Los sólidos se aclararon con EtOAc y los filtrados combinados se concentraron para producir la mezcla en bruto de alloc-naftacenoquinona 77a en forma de un sólido de color rojo oscuro. Sin purificación, este producto (77a) se disolvió en THF desgasificado (1,5 ml) en una atmósfera de Ar y se enfrió a 0 °C. A esta solución en agitación se le añadió Pd(PPh3)4 (4 mg, 1,3 pmol, 0 ,2 equiv.) seguido de la adición gota a gota de morfolina (7 mg, 7 pl, 76 pmol, 1,2 equiv.). El matraz de reacción se envolvió con papel de aluminio y la mezcla resultante se dejó en agitación a 0 °C durante 2 h, tiempo durante el cual se volvió de color azul oscuro. El baño de refrigeración se retiró y la mezcla de reacción se agitó a temperatura ambiente durante 20 min, se inactivó mediante la adición de tampón fosfato pH 6 ,8 (10 ml) y se extrajo con EtOAc (3 x 10 ml). Los extractos orgánicos combinados se lavaron con H2O ( 10 ml) y salmuera ( 10 ml), se secaron sobre MgSO4 y se filtraron a través de un lecho corto de Celite® (lavado con EtOAc antes de usarse). Los sólidos obtenidos se aclararon con EtOAc y los filtrados combinados se concentraron hasta aprox. 1 ml en volumen. La cromatografía en columna ultrarrápida [gel de sílice desactivado (véanse los Métodos Generales), hexanos:EtOAc 1:1 a 1:2] proporcionó la mezcla en bruto de naftacenoquinona 78a en forma de un sólido de color púrpura oscuro. A una solución de la naftacenoquinona 78a en THF desgasificado (1,5 ml) a temperatura ambiente se le añadió una solución de 1:1 de 3HF^EtaN:THF (0,5 ml). El matraz de reacción se envolvió con papel de aluminio y la mezcla de reacción se agitó a temperatura ambiente durante 2 h, y después se repartió entre EtOAc (10 ml) y NaHCO3 ac. saturado (10 ml). La capa orgánica se lavó con salmuera (10 ml), se secó sobre Na2SO4 y se concentró hasta aprox. 1 ml en volumen. La cromatografía en columna ultrarrápida [gel de sílice desactivado (véanse los Métodos Generales), hexanos:EtOAc 1:1 a 1:3] proporcionó una mezcla 1:1 de la ftalimidometil-benzo-uncialamicina 79a (27 mg, 42,2 pmol, rendimiento del 67 %) en forma de un sólido de color púrpura oscuro. 79a: Fr = 0,42 (gel de sílice, hexanos:EtOAc 1:2); 1H RMN (600 MHz, CD3CN): 8 = 13,49 (s, 1 H), 9,89 (d, J = 4,2 Hz, 1 H), 8,47 (s, 1 H), 8,31 (s, 1 H), 7,91 (d, J = 8,0 Hz, 1 H), 7,80 (m, 2 H), 7,76 (m, 2 H), 7,44 (s, 1 H), 7,21 (d, J = 8,0 Hz, 1 H), 5,96 (d, J = 10,0 Hz, 1 H), 5,89 (d, J = 9,9 Hz, 1 H), 5,24 (d, J = 5,0 Hz, 1 H), 5,10 (s, 2 H), 4,90 (dd, J = 4,4, 1,4 Hz, 1 H), 4,52 (d, J = 5,0 Hz, 1 H), 4,37 (dc, J = 5,0, 6,5 Hz, 1 H), 3,30 (d, J= 4,9 Hz, 1 H), 1,38 (d, J = 6,5 Hz, 3 H) ppm; HRMS (ESI-TOF): calc. para C39H25N2O8+ [M+H+]: 649,1605, observado 649,1604.
Formilbenzamida 81. Una solución de ácido 2-bromo-5-metilbenzoico (156 mg, 0,73 mmol, 1,0 equiv.) en SOCI2 (5 ml) se agitó a la temperatura de reflujo durante 2 h. La mezcla de reacción se concentró en atmósfera de N2 y el residuo se coevaporó con tolueno (2x5 ml) para retirar todas las trazas de SOCl2. El producto en bruto se disolvió en CH2Cl2 (5 ml) y la mezcla de reacción se enfrió a 0 °C. Se añadió gota a gota una solución de dietilamina (97 mg, 0,14 ml, 1,31 mmol, 1,8 equiv.) en CH2Cl2 (0,2 ml), el baño de hielo se retiró y la mezcla de reacción se agitó a temperatura ambiente durante 2 h. La reacción se interrumpió con HCl ac. 1 N (1,5 ml), y la mezcla resultante se extrajo con CH2Cl2 (2 x 5 ml). Las capas orgánicas combinadas se lavaron con salmuera (5 ml), se secaron sobre MgSO4 y se concentraron. La cromatografía en columna ultrarrápida (gel de sílice, hexanos:EtOAc 3:1 a 2:1) proporcionó la benzamida 80 en forma de un sólido de color amarillento (194 mg, 0,72 mmol, rendimiento del 98 %). A una solución agitada de /'-PrMgBr (0,85 ml, 1,0 M en THF, 0,85 mmol, 1,2 equiv.) en THF (1,0 ml) a 0 °C se le añadió n-BuLi (0,68 ml, 2,50 M en hexanos, 1,70 mmol, 2,4 equiv.). La solución de color amarillo resultante se enfrió a -78 °C y se añadió gota a gota una solución de bromuro 80 (234 mg, 0,72 mmol, 1,0 equiv.) en THF (1,0 ml). Después de agitar durante 1 h a -78 °C, se añadió DMF (0,23 ml, 2,88 mmol, 4,0 equiv.). Después de agitar durante 3 h a -78 °C, se añadió NH4Cl ac. saturado (5 ml) y se dejó que la mezcla resultante alcanzara la temperatura ambiente. La mezcla se extrajo con EtOAc (10 ml), la capa orgánica se secó sobre MgSO4, se concentró y se purificó por cromatografía en columna (gel de sílice, hexanos:EtOAc = 1:1) para proporcionar el aldehído 81 (156 mg, 0,71 mmol, rendimiento del 99 %) en forma de un polvo de color blanco. 81: Fr = 0,42 (gel de sílice, hexanos:EtOAc 1:2); RMN 1H (500 MHz, CDCla): 8 = 10,01 (s, 1 H), 7,77 (s, 1 H), 7,45 (d, J = 7,7 Hz, 1 H), 7,28 (d, J = 7,7 Hz, 1 H), 3,42 (m, 2 H), 3,13 (m, 2 H), 2,46 (s, 3 H), 1,32 (t, J = 7,1 Hz, 3 H), 1,08 (t, J = 7,2 Hz, 3 H) ppm; HRMS (ESI-TOF): calc. para C15H22NO4+ [M+H+]: 280,1543, observado 280,1540.

Cianoftalida 84. A una solución agitada de la formilbenzamida 81 (156 mg, 0,71 mmol, 1,0 equiv.) en CCU (0,7 ml) se le añadió N-bromosuccinimida (163 mg, 0,85 mmol, 1,2 equiv.) a temperatura ambiente en atmósfera de N2, la mezcla de reacción se calentó a 80 °C y se añadió peróxido de benzoílo (10 mg, 0,04 mmol, 0,06 equiv.) en una porción. El calentamiento se continuó durante 2 h a la misma temperatura y después la mezcla de reacción se dejó enfriar a temperatura ambiente, se lavó con NaHCO3 ac. saturado (2 ml) y salmuera (2 ml), se secó sobre MgSO4 y se concentró para dar el bromuro de bencilo 82 en bruto en forma de un sólido de color pardo claro. El bromuro de bencilo 82 se disolvió en DMF (0,6 ml) y a la solución agitada se le añadieron secuencialmente K2CO3 (146 mg, 1,07 mmol, 1,5 equiv.), n-Bu4NI ( 26 mg, 0,07 mmol, 0,1 equiv.) y 2,3,4,5-tetracloroftalimida (224 mg, 0,79 mmol, 1,1 equiv., en una porción). La mezcla de reacción se calentó a 70 °C y se agitó a esa temperatura durante 2 h, después se vertió en H2O (2 ml) y se extrajo con EtOAc (3x2 ml). Las capas orgánicas combinadas se lavaron con agua (3x2 ml) y salmuera (2x2 ml), se secaron sobre MgSO4 y se concentraron. La cromatografía en columna ultrarrápida (gel de sílice, hexanos:EtOAc 2:1 a 1:2) dio la tetracloroftalimida 83 en forma de un sólido de color amarillento (214 mg, 0,43 mmol, rendimiento del 60 % en 2 etapas). A una solución agitada de la tetracloro-ftalimida 83 en CH2Cl2 (0,8 ml) a 0 °C se le añadieron TMSCN (84 mg, 0,11 ml, 0,85 mmol, 2,0 equiv.) y una solución de KCN (1 mg, 0,02 mmol, 0,025 equiv.) y 18-corona-6 (2 mg, 0,01 mmol, 0,02 equiv.) en THF (0,1 ml). La mezcla de reacción se agitó a la misma temperatura en un matraz cerrado herméticamente durante 1,5 h y durante 30 min a temperatura ambiente. La mezcla resultante se concentró en atmósfera de N2 y el residuo se secó con la retirada azeotrópica de todas las trazas de TMSCN con tolueno (2 x 2 ml). El aceite de color pardo resultante se disolvió en AcOH (0,5 ml) y se agitó durante 48 h a temperatura ambiente hasta que el análisis por TLC mostró la conversión completa (hexanos:EtOAc 1:1). La reacción se interrumpió mediante la adición cuidadosa de NaOH ac. 1 N (1,5 ml), y la mezcla resultante se repartió entre EtOAc (3 ml) y H2O (1,5 ml). La capa acuosa se extrajo con EtOAc (3 x 3 ml) y las capas orgánicas combinadas se lavaron con H2O (3 ml) y salmuera (3 ml), se secaron sobre MgSO4 y se concentraron. La cromatografía en columna ultrarrápida (gel de sílice, hexanos:EtOAc 3:2 a 1:1) dio un sólido de color amarillento, que se recristalizó en EtOAc para producir la cianoftalida 84 en forma de un sólido de color amarillento (136 mg, 0,30 mmol, rendimiento del 70 %). 84: Fr = 0,35 (gel de sílice, hexanos:EtOAc 1:1); RMN 1H (500 MHz, CDCla): 8 = 6,81 (s, 1 H), 5,93 (s, 1 H), 4,97 (s, 2 H), 4,23 (s, 3 H), 3,94 (s, 3 H) ppm; HRMS (ESI-TOF): calc. para C18H7CUN2O4+ [M+H+]: 454,9154, observado 454,9156.

8-ferc-ButNcarbamoNmetM-uncialamicma (49aa). A una solución agitada de la cianoftalida 84 (36 mg, 80 pmol, 3,0 equiv.) en THF (0,35 ml) a -78 °C se le añadió LiHMDS (1,0 M en THF, 0,11 ml, 0,11 mmol, 4,0 equiv.). La mezcla resultante se agitó a -78 °C durante 20 min y se añadió una solución enfriada previamente de la quinona aminal 22a (véase la descripción anterior, 15 mg, 28 pmol, 1,0 equiv.) en THF (0,35 ml) a -78 °C mediante una cánula. Después de agitar durante 5 min a -78 °C, la mezcla de reacción se dejó calentar a temperatura ambiente y se agitó durante 90 min más, tiempo durante el cual la mezcla de reacción se volvió de color rojo oscuro. Después, la mezcla resultante se inactivó con tampón pH 6 ,8 (15 ml) y se extrajo con EtOAc (3 x 15 ml). Los extractos orgánicos combinados se lavaron con salmuera (15 ml), se secaron sobre MgSO4 y se filtraron a través de un lecho corto de Celite® (lavado con EtOAc antes de usarse). Los sólidos se aclararon con EtOAc y los filtrados combinados se concentraron para producir el derivado de alloc-uncialamicina 85a en bruto en forma de un sólido de color rojo oscuro, que se disolvió en THF desgasificado (1,0 ml) en una atmósfera de Ar y se enfrió a 0 °C. A esta solución se le añadió Pd(PPh3)4 (2 mg, 0,7 pmol, 0,2 equiv.), seguido de la adición gota a gota de etilendiamina (5 mg, 6 pl, 84 pmol, 3,0 equiv.), el matraz de reacción se envolvió con papel de aluminio y la mezcla resultante se dejó en agitación a 0 °C durante 2 h, tiempo durante el cual se volvió de color púrpura. Después, se añadió H2O (0,5 ml) y la mezcla resultante se agitó a 50 °C durante 2 h, se diluyó con tampón fosfato pH 7,2 (10 ml) y se extrajo con EtOAc (5 x 10 ml). Los extractos orgánicos combinados se lavaron con H2O (10 ml) y salmuera (10 ml), se secaron sobre MgSO4 y se filtraron. Los filtrados se concentraron hasta aprox. 1 ml en volumen para producir una solución en bruto de la 8 -aminometil-uncialamicina (48aa), a la que con se le añadieron secuencialmente, con agitación a 0 °C, THF desgasificado (1,0 ml), NaHCO3 ac. saturado (1,0 ml, en una porción) y Boc2O (7 mg, 34 pmol, 1,2 equiv.). El matraz de reacción se envolvió con papel de aluminio y la mezcla de reacción se agitó a 0 °C durante 1,5 h y después se repartió entre EtOAc (5 ml) y tampón fosfato pH 6 ,8 (5 ml). La capa acuosa se extrajo con EtOAc (2 x 25 ml) y los extractos orgánicos combinados se lavaron con salmuera (5 ml), se secaron sobre Na2SO4 y se concentraron hasta aprox. 1 ml en volumen. La cromatografía en columna ultrarrápida [gel de sílice desactivado (véanse los Métodos Generales), hexanos:EtOAc 1:1 a 1:2] proporcionó la 8 -ferc-butil-carbamoilmetil-uncialamicina (49aa) (5,0 mg, 8,3 pmol, rendimiento del 30 % en 3 etapas) en forma de un sólido de color púrpura. 49aa: Fr = 0,27 (gel de sílice, hexanos:EtOAc 1:1); [a]o25 = 2300° (c = 0,002, EtOAc); 1H RMN (600 MHz, CD3CN): 8 = 13,17 (s, 1 H), 10,00 (d, J = 4,4 Hz, 1 H), 8,50 (s, 1 H), 8,25 (d, J = 8,1 Hz, 1 H), 8,15 (s, 1 H), 7,75 (dd, J= 8,0, 1,8 Hz, 1 H), 6,03 (a, 1 H), 5,97 (d, J= 9,9 Hz, 1 H), 5,89 (dt, J = 9,9, 1,3 Hz, 1 H), 5,24 (d, J = 4,4 Hz, 1 H), 4,92 (dd, J = 4,4, 1,7 Hz, 2 H), 4,65 (d, J = 5,0 Hz, 1 H), 4,39 (d, J = 5,7, 1 H), 4,38 (dc, J = 5,0, 6,5 Hz, 1 H), 3,37 (d, J = 4,8 Hz, 1 H), 1,43 (s, 9 H), 1,38 (d, J = 6,5 Hz, 3 H) ppm; 13C RMN (150 MHz, CD3CN): 8 = 188,5, 184,1, 156,6, 147,3, 144,6, 136,1, 134,6, 134,0, 133,7, 128,1, 125,0, 124,5, 124,1, 114,4, 112,4, 100,4, 99,1,91,2, 8 8 ,8 , 79,8, 77,0, 65,8, 65,3, 64,7, 44,3, 44,2, 28,5, 21,2 ppm; HRMS (ESI-TOF): calc. para C32H29N2Os+ [M+H+]: 569,1918, observado 569,1918.

Isoindolina 89. A una suspensión de ácido trimetilbenzoico (15,0 g, 91,4 mmol, 1,0 equiv.) y K2CO3 anhidro (18,8 g, 137 mmol, 1,5 equiv.) en Dm F (100 ml) se le añadió gota a gota Mel (14,3 g, 6,27 ml, 100 mmol, 1,1 equiv.) durante 3 min con agitación vigorosa a temperatura ambiente. Después de que se completara la adición, la mezcla de reacción se agitó durante 5 h más a temperatura ambiente, después se vertió en H2O (200 ml) y se extrajo con Et2O (3 x 200 ml). Las capas orgánicas combinadas se lavaron con agua (3 x 100 ml) y salmuera (2 x 100 ml), se secaron sobre MgSO4 y se concentraron para producir el producto en bruto 86 en forma de un aceite incoloro. El éster 86 y W-bromosuccinimida (52,0 g, 292 mmol, 3,2 equiv.) se disolvieron en CCU (200 ml), la mezcla de reacción se calentó a 80 °C y se añadió peróxido de benzoílo (242 mg, 1,0 mmol, 0,02 equiv.) en una porción. El calentamiento se continuó durante 8 h y la mezcla de reacción se enfrió a temperatura ambiente y después se almacenó a 0 °C durante 3 h. Después, la mezcla de reacción se filtró a través de un lecho corto de Celite® y los precipitados se aclararon con CCl4 (0-5 °C). Después, los filtrados combinados se lavaron con NaHCO3 ac. saturado (25 ml) y salmuera (25 ml), se secaron sobre MgSO4 y se concentraron para producir el producto en bruto 87 en forma de un sólido de color blanco. El tribromoéster 87 puro se calentó a 150 °C a ligero vacío durante 10 h. La mezcla de reacción se enfrió a temperatura ambiente y se obtuvo un sólido de color pardo oscuro en forma de producto en bruto 88. La dibromoftalida 88 se disolvió en DMF (100 ml) y se añadió /'-P^NEt (29,5 g, 39,7 ml, 228 mmol, 2,5
equiv.) en una porción, seguido de tritilamina (23,7 g, 91,4 mmol, 1,0 equiv.). La mezcla de reacción se agitó a 60 °C durante 4 h, después se vertió en H2O (300 ml) y se agitó vigorosamente durante 15min. Los precipitados se filtraron, se aclararon con H2O y se secaron al vacío para producir la isoindolina 89 en forma de un sólido de color blanco. Los filtrados combinados se extrajeron con EtOAc (3 x 100 ml) y las capas orgánicas combinadas se lavaron con agua (3 x 50 ml) y salmuera (2 x 50 ml), se secaron sobre MgSO4 y se concentraron. La cromatografía en columna ultrarrápida (gel de sílice, CH2Ch:EtOAc 50:1 a 20:1) dio una segunda porción de la isoindolina 89 en forma de un sólido de color blanco (20,6 g, 49,3 mmol, rendimiento del 54 %). 89: Fr = 0,47 (gel de sílice, CH2Ch:EtOAc 20:1); IR (película) Vmáx = 3708, 3681, 2973, 2939, 2923, 2866, 2844, 2826, 1762, 1709, 1614, 1428, 1394, 1350, 1301, 1150, 1126, 1053, 1032, 1008 cm-1; RMN 1H (600 MHz, CDCls): 8 = 7,59 (m, 6 H), 7,30 (m, 11 H), 5,21 (s, 2 H), 3,99 (d, J = 7,6 Hz, 4 H) ppm; 13C RMN (150 MHz, CDCla): 8 = 171,0, 145,7, 142,1, 130,1, 129,4, 128,5, 128,0, 127,7, 126,5, 126,4, 119,0, 115,6, 75,1, 69,4, 52,2, 51,6 ppm; HRMS (ESI-TOF): calc. para C29H24NO2+ [M+H+]: 418,1802, observado 418,1807.

Isoindolina 90. A una solución agitada de la isoindolina 89 (15,4 g, 36,9 mmol, 1,0 equiv.) en CH2Ch (50 ml) a temperatura ambiente se le añadió gota a gota TFA (50,5 g, 33,9 ml, 443 mmol, 12 equiv.) a 0 °C. La mezcla de reacción se dejó en agitación a temperatura ambiente durante 45 min y después se concentró en atmósfera de N2 y el residuo se coevaporó con tolueno (2 x 25 ml) para retirar todas las trazas de TFA. El aceite de color pardo resultante se disolvió en THF (50 ml) y se añadió NaHCO3 ac. saturado (20 ml) en una porción, seguido de una solución de TeocCl en THF (1,0 M, 50 ml, 50 mmol, 1,4 equiv.) a 0 °C. La mezcla de reacción se agitó vigorosamente a temperatura ambiente durante 12 h más, después se concentró hasta aprox. 25 ml, se diluyó con CH2Cl2 (50 ml) y se inactivó con NaHCO3 ac. saturado (20 ml). La fase acuosa se extrajo con CH2Cl2 (2 x 50 ml) y después las fases orgánicas combinadas se lavaron con H2O (25 ml) y salmuera (25 ml), se secaron sobre Mg2SO4 y se concentraron. La cromatografía en columna ultrarrápida (gel de sílice, hexanos:EtOAc 3:1 a 2:1) proporcionó la isoindolina 90 en forma de un sólido de color amarillento (10,0 g, 31,3 mmol, rendimiento del 85 %). 90: Fr = 0,31 (gel de sílice, hexanos:EtOAc 2:1); RMN 1H (400 MHz, CDCh): 8 = 9,99 (s, 1 H), 7,88 (d, J = 8,0 Hz, 1 H), 7,85 (dd, J = 5,4, 3,1 Hz, 2 H), 7,73 (dd, J = 5,5, 3,0 Hz, 2 H), 7,57 (dd, J = 8,0, 1,4 Hz, 1 H), 7,38 (d, J = 1,4 Hz, 1 H), 4,89 (s, 2 H), 3,58 (c, J = 7,1 Hz, 2 H), 3,07 (c, J = 7,1 Hz, 2 H), 1,28 (t, J = 7,1 Hz, 3 H), 1,00 (t, J = 7,1 Hz, 3 H) ppm; 13C RMN (100 MHz, CDCh): 8 = 189,9, 168,2, 167,7, 142,6, 140,0, 134,2, 132,0, 131,8, 130,3, 129,1, 126,7, 123,5, 43,0, 41,0, 39,1, 13,7, 12,6 ppm; HRMS (ESI-TOF): calc. para C-,6H22NO4Si+ [M+H+]: 320,1313, observado 320,1318.

Cianoftalida 93. A una solución agitada de la isoindolina 90 (1,60 g, 5,0 mmol, 1,0 equiv.) en MeOH ac. (al 85%, 20 ml) se le añadió KOH en polvo (420 mg, 7,5 mmol, 1,5 equiv.) en una porción y la mezcla se agitó a temperatura ambiente durante 12 h. La mezcla de reacción se concentró al vacío para retirar MeOH y el residuo se diluyó con H2O (10 ml). Después, la mezcla se neutralizó (pH = 4) mediante la adición de KHSO4 ac. (1,0 M). Los precipitados así formados se recogieron por filtración y se aclararon con H2O (3 x 5 ml) para dar el hidroxiácido 91 en forma de un sólido de color parduzco. El hidroxiácido 91 se añadió a una suspensión agitada de PCC (1,61 g, 7,5 mmol, 1,5 equiv.) en CH2Cl2 (20 ml) a temperatura ambiente. La mezcla de reacción se agitó durante 3 h a temperatura ambiente, se diluyó con Et2O (50 ml) y se pasó a través de un lecho corto de Celite®. Los sólidos residuales se lavaron con Et2O (3 x 15 ml) y los filtrados combinados se concentraron para dar la hidroxiftalida 92 en bruto en forma de un sólido de color rojo oscuro. La hidroxiftalida 92 se suspendió en acetona cianohidrina (0,7 ml, 7,5 mmol, 1,5 equiv.) y se añadió /-Pr2NEt (13 pl, 75 pmol, 0,015 equiv.) en una porción a 0 °C. La mezcla de reacción se agitó a la misma temperatura en un matraz cerrado herméticamente durante 1,5 h y durante 30 min a temperatura ambiente. Después, la mezcla de reacción se concentró en atmósfera de N2 y el residuo se coevaporó con tolueno (2 x 25 ml) para retirar todas las trazas de acetona cianohidrina. El aceite de color pardo resultante se disolvió en CH2Cl2 (25 ml) y se añadió DCC (1,20 g, 6,0 mmol, 1,2 equiv.) en una porción a 0 °C. La mezcla de reacción se agitó durante 8 h a temperatura ambiente hasta que el análisis por TLC mostró la conversión completa (hexanos:EtOAc 3:2). La reacción se filtró para retirar el subproducto de urea y el filtrado se concentró. La cromatografía en columna ultrarrápida (gel de sílice, hexanos:EtOAc 2:1 a 3:2) dio un sólido de color amarillento, que se recristalizó en EtOAc para producir la cianoftalida 93 en forma de un sólido de color blanco (826 mg, 2,4 mmol, rendimiento del 48 %). 93: Fr = 0,31 (gel de sílice, hexanos:EtOAc 3:2); RMN 1H (400 MHz, CDCls): 8 = 7,99 (s, 1 H), 7,91 (d, J = 8,0 Hz, 1 H), 7,86 (dd, J = 5,4, 3,1 Hz, 2 H), 7,74 (dd, J = 5,5, 3,0 Hz, 2 H), 7,67 (d, J = 8,0 Hz, 1 H), 6,06 (s, 1 H), 4,98 (s, 2 H)
ppm; 13C RMN (100 MHz, CDCI3): 5 = 167,7, 167,0, 141,2, 140,6, 136,0, 134,4, 131,7, 126,2, 125,0, 123,6, 123,1, 113,6, 65,5, 40,7 ppm; HRMS (ESI-TOF): calc. para C-,7H21N2O4Si+ [M+H+]: 345,1265, observado 345,1271.

Antraquinona 94a. A una solución de la cianoftalida 93 (93 mg, 0,27 mmol, 2,0 equiv.) en THF (1,2 ml) a -78 °C se le añadió LiHMDS (1,0 M en THF, 0,43 ml, 0,43 mmol, 3,0 equiv.). La mezcla resultante se agitó a -78 °C durante 20 min y se añadió una solución enfriada previamente de la quinona aminal 22a (78 mg, 0,14 mmol, 1,0 equiv.) en THF (1,4 ml) a -78 °C mediante una cánula. Después de agitar durante 5 min a -78 °C, la mezcla de reacción se calentó a temperatura ambiente y se agitó durante 1,5 h más, tiempo durante el cual la mezcla de reacción se volvió de color rojo oscuro y el análisis por TLC mostró el consumo completo de 22a (EtOAc al 8 % en CH2O 2). Después, la mezcla de reacción se inactivó mediante la adición de tampón pH 6 ,8 (30 ml) y se extrajo con EtOAc (3 x 30 ml). Los extractos orgánicos combinados se lavaron con salmuera (30 ml), se secaron sobre MgSO4 y se filtraron a través de un lecho corto de Celite® (lavado con EtOAc antes de usarse). Los sólidos se aclararon con EtOAc y los filtrados combinados se concentraron para producir la alloc-antraquinona 94a en bruto en forma de un sólido de color rojo oscuro, que se disolvió en THF desgasificado (2,0 ml) en una atmósfera de Ar y se enfrió a 0 °C. A esta solución se le añadió Pd(PPh3)4 (32 mg, 10,4 pmol, 0,16 equiv.), seguido de la adición gota a gota de morfolina (32 mg, 32 pl, 0,34 mmol, 2,4 equiv.). El matraz de reacción se envolvió con papel de aluminio y la mezcla resultante se dejó en agitación a 0 °C durante 2 h, tiempo durante el cual la mezcla de reacción se volvió de color púrpura oscuro. El baño de refrigeración se retiró y la mezcla de reacción se agitó a temperatura ambiente durante 20 min y después se inactivó mediante la adición de tampón pH 6 ,8 (30 ml) y se extrajo con EtOAc (3 x 30 ml). Los extractos orgánicos combinados se lavaron con H2O (30 ml) y salmuera (30 ml), se secaron sobre MgSO4 y se filtraron a través de un lecho corto de Celite® (lavado con EtOAc antes de usarse). Los sólidos se aclararon con EtOAc y los filtrados combinados se concentraron hasta aprox. 1 ml en volumen. La cromatografía en columna ultrarrápida [gel de sílice desactivado (véanse los Métodos Generales), hexanos:EtOAc 2:1 a 1:1] proporcionó la antraquinona 95a en forma de un sólido de color púrpura (93 mg, 0,13 mmol, rendimiento del 90 %). 95a: Fr = 0,58 (gel de sílice, hexanos:EtOAc 1:1); [a]D25 = 2600° (c = 0,002, EtOAc); 1H RMN (600 MHz, CD3CN): 5 = 13,11 (s, 1 H), 9,97 (d, J = 4,0 Hz, 1 H), 8,46 (s, 1 H), 8,23 (d, J = 8,0 Hz, 1 H), 8,20 (s, 1 H), 7,87 (d, J = 1,9 Hz, 2 H), 7,81 (d, J = 2,2 Hz, 1 H), 7,80 (d, J = 1,9 Hz, 2 H), 5,94 (d, J = 10,0 Hz, 1 H), 5,87 (d, J = 9,8 Hz, 1 H), 5,10 (s, 1 H), 4,97 (s, 2 H), 4,97 (s, 1H), 4,55 (c, J = 5,8 Hz, 1 H), 4,45 (d, J = 3,7 Hz, 1 H), 1,38 (d, J = 5,8 Hz, 3 H), 0,98 (t, J = 7,6 Hz, 9 H), 0,66 (c, J = 7,6 Hz, 6 H) ppm; 13C RMN (150 MHz, CD3CN): 5 = 188,2, 183,8, 169,0, 156,7, 144,8, 143,5, 136,3, 135,4, 135,1, 134,8, 133,8, 133,0, 130,9, 128,3, 126,1, 124,9, 124,1, 123,8, 114,2, 112,2, 100,2, 99,7, 91,3, 88,4, 77,4, 66,7, 64,9, 64,7, 44,3, 41,8, 22,6, 7,3, 5,6 ppm; HRMS (ESI-TOF): calc. para C40H47N2ObSÍ2+ [M+H+]: 739,2865, observado 739,2871.

Isoindolin-uncialamicina 96. A una solución agitada de la antraquinona 95a (93 mg, 0,13 mmol, 1,0 equiv.) en THF desgasificado (15 ml) a temperatura ambiente se le añadió una solución de 1:1 de 3 HF^Et3N:THF (5,0 ml). El matraz de reacción se envolvió con papel de aluminio y la mezcla de reacción se agitó a temperatura ambiente durante 1,5 h, y después se repartió entre EtOAc (50 ml) y NaHCO3 ac. saturado (50 ml). La capa acuosa se extrajo con EtOAc (2 x 25 ml) y los extractos orgánicos combinados se lavaron con salmuera (25 ml), se secaron sobre Na2SO4 y se concentraron hasta aprox. 1 ml en volumen. La cromatografía en columna ultrarrápida (gel de sílice desactivado, hexanos:EtOAc 1:1 a 1:2) proporcionó la isoindolin-uncialamicina 96 en forma de un sólido de color púrpura (76 mg, 0,13 mmol, rendimiento del 98 %). 96: Fr = 0,24 [gel de sílice desactivado (véanse los Métodos
Generales), hexanos:EtOAc 1:1]; [a]D25 = 1600° (c = 0,005, EtOAc); 1H RMN (600 MHz, CD3CN): 5 = 13,10 (s, 1 H), 9,99 (d, J = 4,1 Hz, 1 H), 8,48 (s, 1 H), 8,23 (d, J = 8,1 Hz, 1 H), 8,21 (s, 1 H), 7,87 (dd, J = 5,4, 3,1 Hz, 2 H), 7,82 (d, J = 2,2 Hz, 1 H), 7,81 (dd, J = 5,5, 3,0 Hz, 2 H), 5,96 (d, J = 9,9 Hz, 1 H), 5,88 (d, J = 9,9 Hz, 1 H), 5,24 (d, J = 4,9 Hz, 1 H), 5,49 (s, 2 H), 4,91 (dd, J = 4,4, 1,4 Hz, 1 H), 4,44 (d, J = 4,9 Hz, 1 H), 4,38 (dc, J = 5,0, 6,5 Hz, 1 H), 3,27 (d, J = 4,9 Hz, 1 H), 1,38 (d, J = 6,5 Hz, 3 H) ppm; 13C RMN (150 MHz, CD3CN): 5 = 188,2, 183,9, 169,0, 156,7, 144,7, 143,6, 136,2, 135,4, 135,1, 134,8, 133,8, 133,0, 130,9, 128,4, 126,1, 124,5, 124,1, 124,1, 114,3, 112,3, 100,3, 99,1, 91,3, 88,8, 77,0, 65,8, 65,3, 64,7, 44,2, 41,8, 21,2 ppm; HRMS (ESI-TOF): calc. para C34H33N2OsSi+ [M+H+]: 625,2001, observado 625,2006.

Isoindolin-uncialamicina 97. A una solución de la isoindolin-uncialamicina 96 (3,6 mg, 5 |jmol, 1,0 equiv.) en THF desgasificado (1,0 ml) a 0 °C se le añadió gota a gota TBAF/HOAc (1/1, 1,0 M, 0,1 ml). El matraz de reacción se envolvió con papel de aluminio y la mezcla de reacción se agitó a temperatura ambiente durante 6 h, después se inactivó con H2O (1,0 ml), se enfrió a 0 °C y se diluyó con THF desgasificado (10 ml), y la fase orgánica se separó y se concentró a 5 °C en atmósfera de N2 hasta aprox. 1 ml en volumen y se diluyó con THF frío desgasificado (0 °C, 10 ml). Se repitieron las mismas operaciones 5 veces para producir la isoindolin-uncialamicina 97 en bruto en forma de un sólido de color púrpura, que se usó inmediatamente para la siguiente etapa (PRECAUCIÓN: 97 era extremadamente inestable, sensible tanto a ácidos como a bases, y se descomponía lentamente a -78 °C, debe usarse fresco para obtener los mejores rendimientos). 97: Fr = 0,10 (gel de sílice, MeOH: EtOAc 1:1); HRMS (ESI-TOF): calc. para C28H21N2O6+ [M+H+]: 481,1394, observado 481,1397.

ferc-ButilcarbamoN-isoindolm-uncialamicma 98. La isoindolin-uncialamicina 97 en bruto [preparada de acuerdo con el procedimiento descrito anteriormente a partir de la 8-ftalimidometil-uncialamicina 47aa (3,6 mg, 5 jmol, 1,0 equiv.)] se suspendió en THF desgasificado (1,0 ml) a 0 °C y se añadió NaHCO3 ac. saturado (1,0 ml) en una porción, seguido de Boc2O (1,3 mg, 6 jmol, 1,2 equiv.). El matraz de reacción se envolvió con papel de aluminio y la mezcla de reacción se agitó a 0 °C durante 1,5 h, y después se repartió entre EtOAc (5 ml) y tampón pH 6,8 (5 ml). La capa acuosa se extrajo con EtOAc (2 x 25 ml) y los extractos orgánicos combinados se lavaron con salmuera (5 ml), se secaron sobre Na2SO4 y se concentraron hasta aprox. 1 ml en volumen. La cromatografía en columna ultrarrápida [gel de sílice desactivado (véanse los Métodos Generales), hexanos:EtOAc 1:1 a 1:2] proporcionó la 8-terc-butilcarbamoilmetil-isoindolin-uncialamicina 98 (2,7 mg, 4,5 jmol, rendimiento del 95 %) en forma de un sólido de color púrpura. 98: Fr = 0,27 (gel de sílice, hexanos:EtOAc 1:1); [a]D25 = 2300° (c = 0,002, EtOAc); 1H RMN (600 MHz, CD3CN): 5 = 13,17 (s, 1 H), 10,00 (d, J = 4,4 Hz, 1 H), 8,50 (s, 1 H), 8,25 (d, J = 8,1 Hz, 1 H), 8,15 (s, 1 H), 7,75 (dd, J= 8,0, 1,8 Hz, 1 H), 6,03 (a, 1 H), 5,97 (d, J= 9,9 Hz, 1 H), 5,89 (dt, J = 9,9, 1,3 Hz, 1 H), 5,24 (d, J = 4,4 Hz, 1 H), 4,92 (dd, J = 4,4, 1,7 Hz, 2 H), 4,65 (d, J = 5,0 Hz, 1 H), 4,39 (d, J = 5,7, 1 H), 4,38 (dc, J = 5,0, 6,5 Hz, 1 H), 3,37 (d, J = 4,8 Hz, 1 H), 1,43 (s, 9 H), 1,38 (d, J = 6,5 Hz, 3 H) ppm; 13C RMN (150 MHz, CD3CN): 5 = 188,5, 184,1, 156,6, 147,3, 144,6, 136,1, 134,6, 134,0, 133,7, 128,1, 125,0, 124,5, 124,1, 114,4, 112,4, 100,4, 99,1, 91,2, 88,8, 79,8, 77,0, 65,8, 65,3, 64,7, 44,3, 44,2, 28,5, 21,2 ppm; HRMS (ESI-TOF): calc. para C33H29N2O8+ [M+H+]: 581,1918, observado 581,1918.

Sulfuro 99. La 8 -aminometil-uncialamicina 97 en bruto [preparada de acuerdo con el procedimiento general descrito anteriormente a partir de la 8 -ftalimidometil-uncialamicina (96, 16 mg, 27 |jmol, 1,0 equiv.)] se suspendió en THF desgasificado (1,0 ml) a 0 °C y se añadió NaHCO3 ac. saturado (1,0 ml) en una porción, seguido de la adición gota a gota de una solución de cloroformiato de 2-(feniltio)etilo (14 mg, 66 jmol, 2,5 equiv.) en THF desgasificado (0,5 ml). El matraz de reacción se envolvió con papel de aluminio y la mezcla de reacción se agitó a 0 °C durante 5 h, y después se repartió entre EtOAc (5 ml) y tampón pH 6 ,8 (5 ml). La capa acuosa se extrajo con EtOAc (2 x 25 ml) y los extractos orgánicos combinados se lavaron con salmuera (5 ml), se secaron sobre Na2SO4 y se concentraron hasta aprox. 1 ml en volumen. La cromatografía en columna ultrarrápida [gel de sílice desactivado (véanse los Métodos Generales), hexanos:EtOAc 1:1 a 1:2] para producir el sulfuro 99 (9,5 mg, 14 jmol, rendimiento del 55 %) en forma de un sólido de color púrpura. 99: Fr = 0,23 (gel de sílice, hexanos:EtOAc 1:1); 1H RMN (600 MHz, CD3CN): 8 = 13,20 (s, 1 H), 10,01 (d, J = 4,1 Hz, 1 H), 8,50 (s, 1 H), 8,23 (d, J = 8,0 Hz, 1 H), 8,16 (s, 1 H), 7,73 (dd, J = 8,0, 1,8 Hz, 1 H), 6,00 (a, 1 H), 5,98 (d, J = 10,0 Hz, 1 H), 5,89 (dt, J = 10,0, 1,3 Hz, 1 H), 5,24 (d, J = 4,4 Hz, 1 H), 4,94 (dd, J = 4,4, 1,7 Hz, 2 H), 4,65 (d, J = 5,0 Hz, 1 H), 4,4-4,2 (m, 2 H), 4,39 (d, J = 5,0, 1 H), 4,38 (dc, J = 5,0, 6,5 Hz, 1 H), 3,35 (d, J = 4,8 Hz, 1 H), 3,2-3,0 (m, 2 H), 1,38 (d, J = 6,5 Hz, 3 H) ppm; 13C RMN (150 MHz, CD3CN): 8 = 188,6, 184,2, 154,4, 147,3, 144,6, 136,1, 135,1, 134,6, 134,0, 133,8, 129,1, 128,6, 128,1, 125,3 125,1, 124,5, 124,1, 114,3, 112,4, 100,3, 99,1, 91,2, 8 8 ,8 , 77,1,65,8, 65,3, 64,7, 64,6, 44,3, 44,2, 32,4, 21,2 ppm; HRMS (ESI-TOF): calc. para C37H29N2OsS+ [M+H+]: 661,1639 encontrado 661,1638.

Sulfona 100. A una solución agitada del sulfuro 99 (6,5 mg, 10 jmol, 1,0 equiv.) en CH2Cl2 (0,5 ml) se le añadió dimetildioxirano recién preparado (2,0 ml, ~0,1 M en acetona, 0,2 mmol, 20 equiv.) a -78 °C, la mezcla de reacción se envolvió en papel de aluminio y se dejó calentar a 0 °C. Después de agitar a 0 °C durante 20 min, se añadió Me2S (84 mg, 0,1 ml, 1,4 mmol, 135 equiv.) en una porción y la agitación se continuó durante 20 min a la misma temperatura. Después, la mezcla de reacción se diluyó con EtOAc (25 ml), se lavó con H2O (5 ml) y salmuera (5 ml), se secó sobre Na2SO4 y se concentró hasta aprox. 1 ml en volumen. La cromatografía en columna ultrarrápida [gel de sílice desactivado (véanse los Métodos Generales), hexanos:EtOAc 1:1 a 1:3] proporcionó la sulfona 100 (2,7 mg, 4,5 jmol, rendimiento del 55 %) en forma de un sólido de color púrpura. 100: Fr = 0,23 [gel de sílice desactivado (véanse los Métodos Generales), hexanos:EtOAc 1:3 y después EtoAc:MeOH 50:1]; 1H RMN (600 MHz, CD3CN): 8 = 13,19 (s, 1 H), 9,99 (d, J = 4,1 Hz, 1 H), 8,51 (s, 1 H), 8,24 (d, J = 8,0 Hz, 1 H), 8,18 (s, 1 H), 7,73 (dd, J = 8,0, 1,8 Hz, 1 H), 6,05 (a, 1 H), 5,97 (d, J = 10,0 Hz, 1 H), 5,88 (dt, J = 10,0, 1,3 Hz, 1 H), 5,26 (d, J = 4,4 Hz, 1 H), 4,97 (dd, J = 4,4, 1,7 Hz, 2 H), 4,66 (d, J = 5,0 Hz, 1 H), 4,6-4,3 (m, 2 H), 4,39 (d, J = 5,1, 1 H), 4,38 (dc, J = 5,0, 6,5 Hz, 1 H), 3,50 (a, 2 H), 3,35 (d, J = 4,9 Hz, 1 H), 1,38 (d, J = 6,5 Hz, 3 H) ppm; 13C RMN (150 MHz, CD3CN): 8 = 188,5, 184,1, 153,7, 147,3, 144,6, 139,1, 136,1, 134,6, 134,1, 134,0, 133,7, 129,5, 128,1, 128,0, 125,0, 124,5, 124,1, 114,4, 112,4, 100,4, 99,1, 91,2, 8 8 ,8 , 77,0, 65,8, 65,3, 64,7, 59,4, 55,2, 44,3, 44,2, 21,2 ppm; HRMS (ESI-TOF): calc. para C37H29N2O10S+ [M+H+]: 693,1537, observado 693,1532.

Ftalida 101. A una solución de 6 -bromometilftalida (11,3 g, 49,9 mmol, 1,0 equiv.) en DMF (50 ml) se le añadieron
secuencialmente CS2CO3 anhidro (24,3 g, 74,8 mmol, 1,5 equiv.) y n-Bu4NI (1,8 g, 5,0 mmol, 0,1 equiv.), seguido de W-metil-2-(trimetilsilil)etilcarbamato (9,6 g, 54,9 mmol, 1,1 equiv.) en una porción. La mezcla de reacción se agitó a 60 °C durante 2 h, después se vertió en H2O (300 ml) y se agitó vigorosamente durante 15 min. Los precipitados se filtraron y se aclararon con H2O y se secaron al vacío para producir la ftalida 101 en forma de un sólido de color blanco. Los filtrados combinados se extrajeron con EtOAc (3 x 100 ml) y las capas orgánicas combinadas se lavaron con agua (3 x 50 ml) y salmuera (2 x 50 ml), se secaron sobre MgSO4 y se concentraron. La cromatografía en columna ultrarrápida (gel de sílice, CH2Ch:EtOAc 10 :1 a 8 :1 ) dio una segunda porción de la ftalida 101 en forma de un sólido de color blanco (13,0 g, 40,4 mmol, rendimiento del 81 %). 101: Fr = 0,31 (gel de sílice, CH2Ch:EtOAc 8:1); IR (película) vmáx = 3708, 3681, 2973, 2939, 2923, 2866, 2844, 2826, 1762, 1709, 1614, 1428, 1394, 1350, 1301, 1150, 1126, 1053, 1032, 1008 cm-1; RMN 1H (600 MHz, CDCls): 8 = 7,93 (s, 1 H), 7,87 (dd, J= 5,4, 3,1 Hz, 2 H), 7,76 (d, J = 7,9 Hz, 1 H), 7,75 (dd, J = 5,5, 3,0 Hz, 2 H), 7,47 (d, J = 7,8 Hz, 1 H), 5,29 (s, 2 H), 4,96 (s, 2 H) ppm; 13C RMN (150 MHz, CDCla): 8 = 170,6, 167,8, 146,0, 137,9, 134,4, 134,2, 131,9, 126,4, 125,3, 123,6, 122,5, 69,5, 40,9 ppm; HRMS (ESI-TOF): calc. para C ^H 24NO4Si+ [M+H+]:322,1469, observado 322,1465.

Cianoftalida 104. A una solución agitada de la ftalida 101 (1,61 g, 5,0 mmol, 1,0 equiv.) en MeOH ac. (al 85%, 20 ml) se le añadió KOH en polvo (420 mg, 7,5 mmol, 1,5 equiv.) en una porción y la mezcla se agitó a temperatura ambiente durante 12 h. La mezcla de reacción se concentró al vacío para retirar MeOH y el residuo se diluyó con H2O (10 ml). Después, la mezcla se neutralizó (pH = 4) mediante la adición de KHSO4 ac. (1,0 M). Los precipitados así formados se recogieron por filtración y se aclararon con H2O (3 x 5 ml) para dar el hidroxiácido 102 en forma de un sólido de color parduzco. El hidroxiácido 102 se añadió a una suspensión agitada de PCC (1,61 g, 7,5 mmol, 1,5 equiv.) en CH2Cl2 (20 ml) a temperatura ambiente. La mezcla de reacción se agitó durante 3 h a temperatura ambiente, se diluyó con Et2O (50 ml) y se pasó a través de un lecho corto de Celite®. Los sólidos residuales se lavaron con Et2O (3 x 15 ml) y los filtrados combinados se concentraron para dar la hidroxiftalida 103 en bruto en forma de un sólido de color rojo oscuro. La hidroxiftalida 103 se suspendió en acetona cianohidrina (0,7 ml, 7.5 mmol, 1,5 equiv.) y se añadió /'-P^NEt (13 pl, 75 pmol, 0,015 equiv.) en una porción a 0 °C. La mezcla de reacción se agitó a la misma temperatura en un matraz cerrado herméticamente durante 1,5 h y durante 30 min a temperatura ambiente. Después, la mezcla de reacción se concentró en atmósfera de N2 y el residuo se coevaporó con tolueno (2 x 25 ml) para retirar todas las trazas de acetona cianohidrina. El aceite de color pardo resultante se disolvió en CH2Cl2 (25 ml) y se añadió DCC (1,20 g, 6,0 mmol, 1,2 equiv.) en una porción a 0 °C. La mezcla de reacción se agitó durante 8 h a temperatura ambiente hasta que el análisis por TLC mostró la conversión completa (hexanos: EtOAc 3:2). La reacción se filtró para retirar el subproducto de urea y el filtrado se concentró. La cromatografía en columna ultrarrápida (gel de sílice, hexanos:EtoAc 2 :1 a 3:2) dio un sólido de color amarillento, que se recristalizó en EtOAc para producir la cianoftalida 104 en forma de un sólido de color blanco (917 mg, 2,65 mmol, rendimiento del 53 %). 104: Fr = 0,31 (gel de sílice, hexanos:EtOAc 3:2); RMN 1H (400 MHz, CDCls): 8 = 7,99 (s, 1 H), 7,91 (d, J = 8,0 Hz, 1 H), 7,86 (dd, J = 5,4, 3,1 Hz, 2 H), 7,74 (dd, J = 5,5, 3,0 Hz, 2 H), 7,67 (d, J = 8,0 Hz, 1 H), 6.06 (s, 1 H), 4,98 (s, 2 H) ppm; 13C RMN (100 MHz, CDCh): 8 = 167,7, 167,0, 141,2, 140,6, 136,0, 134,4, 131,7, 126,2, 125,0, 123,6, 123,1, 113,6, 65,5, 40,7 ppm; HRMS (ESI-TOF): calc. para C1/H 23N2O4Si+ [M+H+]: 347,1422, observado 347,1425.

Antraquinona 106a. A una solución de la cianoftalida 104 (10 mg, 29 pmol, 2,0 equiv.) en THF (0,12 ml) a -78 °C se le añadió LiHMDS (1,0 M en THF, 43 pl, 43 pmol, 3,0 equiv.). La mezcla resultante se agitó a -78 °C durante 20 min y se añadió una solución enfriada previamente de la quinona aminal 22a (8 mg, 14 pmol, 1,0 equiv.) en THF (0,14 ml) a -78 °C mediante una cánula. Después de agitar durante 5 min a -78 °C, la mezcla de reacción se calentó a temperatura ambiente y se agitó durante 1,5 h más, tiempo durante el cual la mezcla de reacción se volvió de color rojo oscuro y el análisis por TLC mostró el consumo completo de 22a (EtOAc al 8 % en CH2O 2). Después, la mezcla de reacción se inactivó mediante la adición de tampón pH 6 ,8 (3 ml) y se extrajo con EtOAc (3 x 3 ml). Los extractos orgánicos combinados se lavaron con salmuera (3 ml), se secaron sobre MgSO4 y se filtraron a través de un lecho
corto de Celite® (lavado con EtOAc antes de usarse). Los sólidos se aclararon con EtOAc y los filtrados combinados se concentraron para producir la alloc-antraquinona 28a en bruto en forma de un sólido de color rojo oscuro, que se disolvió en THF desgasificado (0,2 ml) en una atmósfera de Ar y se enfrió a 0 °C. A esta solución se le añadió Pd(PPh3)4 (3 mg, 1,0 pmol, 0,16 equiv.), seguido de la adición gota a gota de morfolina (3 mg, 3 pl, 30 pmol, 2,4 equiv.). El matraz de reacción se envolvió con papel de aluminio y la mezcla resultante se dejó en agitación a 0 °C durante 2 h, tiempo durante el cual la mezcla de reacción se volvió de color púrpura oscuro. El baño de refrigeración
se retiró y la mezcla de reacción se agitó a temperatura ambiente durante 20 min y después se inactivó mediante la adición de tampón pH 6 ,8 (3 ml) y se extrajo con EtOAc (3 x 3 ml). Los extractos orgánicos combinados se lavaron con H2O (3 ml) y salmuera (3 ml), se secaron sobre MgSO4 y se filtraron a través de un lecho corto de Celite®
(lavado con EtOAc antes de usarse). Los sólidos se aclararon con EtOAc y los filtrados combinados se concentraron
hasta aprox. 1 ml en volumen. La cromatografía en columna ultrarrápida [gel de sílice desactivado (véanse los Métodos Generales), hexanos:EtOAc 2:1 a 1:1] proporcionó la antraquinona 106a en forma de un sólido de color púrpura (8 mg, 11 pmol, rendimiento del 80 %). l06a: Fr = 0,58 (gel de sílice, hexanos:EtOAc 1:1); [a]D25 = 2600° (c
= 0,002, EtOAc); 1H RMN (600 MHz, CD3CN): 8 = 13,11 (s, 1 H), 9,97 (d, J = 4,0 Hz, 1 H), 8,46 (s, 1 H), 8,23 (d, J =
8.0 Hz, 1 H), 8,20 (s, 1 H), 7,87 (d, J = 1,9 Hz, 2 H), 7,81 (d, J = 2,2 Hz, 1 H), 7,80 (d, J = 1,9 Hz, 2 H), 5,94 (d, J =
10.0 Hz, 1 H), 5,87 (d, J = 9,8 Hz, 1 H), 5,10 (s, 1 H), 4,97 (s, 2 H), 4,97 (s, 1H), 4,55 (c, J = 5,8 Hz, 1 H), 4,45 (d, J = 3,7 Hz, 1 H), 1,38 (d, J = 5,8 Hz, 3 H), 0,98 (t, J = 7,6 Hz, 9 H), 0,66 (c, J = 7,6 Hz, 6 H) ppm; 13C RMN ( CD3CN): 8 = 188,2, 183,8, 169,0, 156,7, 144,8, 143,5, 136,3, 135,4, 135,1, 134,8, 133,8, 133,0, 130,9, 128,3, 126,1, 124,9, 124,1, 123,8, 114,2, 112,2, 100,2, 99,7, 91,3, 88,4, 77,4, 66,7, 64,9, 64,7, 44,3, 41,8, 22,6, 7,3, 5,6 ppm;
HRMS (ESI-TOF): calc. para C40H49N2ObSÍ2+ [M+H+]: 741,3022, observado 741,3023.

N-metN-8-[2-(trímetNsiNl)etM]carbamoMmetM-uncialamicma 107. A una solución agitada de la antraquinona 106a
(7 mg, 10 pmol, 1,0 equiv.) en THF desgasificado (1,5 ml) a temperatura ambiente se le añadió una solución de 1:1
de 3 HF^Et3N:THF (0,5 ml). El matraz de reacción se envolvió con papel de aluminio y la mezcla de reacción se agitó a temperatura ambiente durante 1,5 h, y después se repartió entre EtOAc (5,0 ml) y NaHCO3 ac. saturado (5,0 ml).
La capa acuosa se extrajo con EtOAc (2 x 2,5 ml) y los extractos orgánicos combinados se lavaron con salmuera
(2,5 ml), se secaron sobre Na2SO4 y se concentraron hasta aprox. 1 ml en volumen. La cromatografía en columna ultrarrápida (gel de sílice desactivado, hexanos:EtOAc 1:1 a 1:2) proporcionó el análogo de uncialamicina 107 en forma de un sólido de color púrpura (7 mg, 10 pmol, rendimiento del 99 %). 107: Fr = 0,24 [gel de sílice desactivado (véanse los Métodos Generales), hexanos:EtOAc 1:1]; [a]D25 = 1600° (c = 0,005, EtOAc); 1H RMN (600 MHz, CD3CN): 8 = 13,10 (s, 1 H), 9,99 (d, J= 4,1 Hz, 1 H), 8,48 (s, 1 H), 8,23 (d, J = 8,1 Hz, 1 H), 8,21 (s, 1 H), 7,87 (dd, J
= 5,4, 3,1 Hz, 2 H), 7,82 (d, J = 2,2 Hz, 1 H), 7,81 (dd, J = 5,5, 3,0 Hz, 2 H), 5,96 (d, J = 9,9 Hz, 1 H), 5,88 (d, J=
9,9 Hz, 1 H), 5,24 (d, J= 4,9 Hz, 1 H), 5,49 (s, 2 H), 4,91 (dd, J = 4,4, 1,4 Hz, 1 H), 4,44 (d, J = 4,9 Hz, 1 H), 4,38 (dc,
J = 5,0, 6,5 Hz, 1 H), 3,27 (d, J = 4,9 Hz, 1 H), 1,38 (d, J = 6,5 Hz, 3 H) ppm; 13C RMN (150 MHz, CD3CN): 8 = 188,2,
183,9, 169,0, 156,7, 144,7, 143,6, 136,2, 135,4, 135,1, 134,8, 133,8, 133,0, 130,9, 128,4, 126,1, 124,5, 124,1, 124,1,
114,3, 112,3, 100,3, 99,1, 91,3, 8 8 ,8 , 77,0, 65,8, 65,3, 64,7, 44,2, 41,8, 21,2 ppm; HRMS (ESI-TOF): calc. para C34H35N2OsSi+ [M+H+]: 627,2157, observado 627,2157.

N-metil-8-aminometil-uncialamicina 108. A una solución del análogo de uncialamicina 107 (3,6 mg, 5 pmol, 1,0 equiv.) en THF desgasificado (1,0 ml) a 0 °C se le añadió gota a gota TBAF/HOAc (1/1, 1,0 M, 0,1 ml). El matraz de reacción se envolvió con papel de aluminio y la mezcla de reacción se agitó a temperatura ambiente durante 6 h, después se inactivó con H2O (1,0 ml), se enfrió a 0 °C y se diluyó con THF desgasificado (10 ml), y la fase orgánica
se separó y se concentró a 5 °C en atmósfera de N 2 hasta aprox. 1 ml en volumen y se diluyó con THF frío desgasificado (0 °C, 10 ml). Se repitieron las mismas operaciones 5 veces para producir la N-metil-8-aminometiluncialamicina 108 en bruto en forma de un sólido de color púrpura, que se usó inmediatamente para la siguiente etapa (PRECAUCIÓN: 108 era extremadamente inestable, sensible tanto a ácidos como a bases, y se descomponía lentamente a -78 °C, debe usarse fresco para obtener los mejores rendimientos). 108: Fr = 0,10 (gel de sílice, MeOH:EtOAc 1:1); HRMS (ESI-TOF): calc. para C 2 sH 23 N 2 O 6 + [M+H+]: 483,1551, observado 483,1557.

Sulfuro 109. La N-metil-8-aminometil-uncialamicina 108 en bruto [preparada de acuerdo con el procedimiento general descrito anteriormente a partir del análogo de uncialamicina 107 (6 mg, 9 pmol, 1,0 equiv.)] se suspendió en THF desgasificado (0,3 ml) a 0 °C y se añadió NaHCO 3 ac. saturado (0,3 ml) en una porción, seguido de la adición gota a gota de una solución de cloroformiato de 2-(feniltio)etilo (5 mg, 22 pmol, 2,5 equiv.) en THF desgasificado (0,2 ml). El matraz de reacción se envolvió con papel de aluminio y la mezcla de reacción se agitó a 0 °C durante 5 h, y después se repartió entre EtOAc (2 ml) y tampón pH 6,8 (2 ml). La capa acuosa se extrajo con EtOAc (2 x 8 ml) y los extractos orgánicos combinados se lavaron con salmuera (2 ml), se secaron sobre Na 2 SO 4 y se concentraron hasta aprox. 1 ml en volumen. La cromatografía en columna ultrarrápida [gel de sílice desactivado (véanse los Métodos Generales), hexanos:EtOAc 1:1 a 1:2] para producir el sulfuro 109 (4 mg, 5,5 pmol, rendimiento del 61 %) en forma de un sólido de color púrpura. 109: Fr = 0,23 (gel de sílice, hexanos:EtOAc 1:1); 1 H RMN (600 MHz, CD 3 CN): 8 = 13,20 (s, 1 H), 10,01 (d, J = 4,1 Hz, 1 H), 8,50 (s, 1 H), 8,23 (d, J = 8,0 Hz, 1 H), 8,16 (s, 1 H), 7,73 (dd, J = 8,0, 1,8 Hz, 1 H), 6,00 (a, 1 H), 5,98 (d, J= 10,0 Hz, 1 H), 5,89 (dt, J = 10,0, 1,3 Hz, 1 H), 5,24 (d, J= 4,4 Hz, 1 H), 4,94 (dd, J = 4,4, 1,7 Hz, 2 H), 4,65 (d, J = 5,0 Hz, 1 H), 4,4-4,2 (m, 2 H), 4,39 (d, J = 5,0, 1 H), 4,38 (dc, J = 5,0, 6,5 Hz, 1 H), 3,35 (d, J = 4,8 Hz, 1 H), 3,2-3,0 (m, 2 H), 1,38 (d, J = 6,5 Hz, 3 H) ppm; 13 C RMN (150 MHz, CD 3 CN): 8 = 188,6, 184,2, 154,4, 147,3, 144,6, 136,1, 135,1, 134,6, 134,0, 133,8, 129,1, 128,6, 128,1, 125,3125,1, 124,5, 124,1, 114,3, 112,4, 100,3, 99,1,91,2, 88,8, 77,1, 65,8, 65,3, 64,7, 64,6, 44,3, 44,2, 32,4, 21,2 ppm; HRMS (ESI-TOF): calc. para C 37 H 31 N 2 O 8 S+ [M+H+]: 663,1796 encontrado 663,1798.

Sulfona 110. A una solución agitada del sulfuro 109 (4 mg, 5,5 pmol, 1,0 equiv.) en CH 2 Cl 2 (0,3 ml) se le añadió dimetildioxirano recién preparado (1,0 ml, ~0,1 M en acetona, 0,1 mmol, 18 equiv.) a -78 °C, la mezcla de reacción se envolvió en papel de aluminio y se dejó calentar a 0 °C. Después de agitar a 0 °C durante 20 min, se añadió Me 2 S (42 mg, 50 pl, 0,7 mmol, 135 equiv.) en una porción y la agitación se continuó durante 20 min a la misma temperatura. Después, la mezcla de reacción se diluyó con EtOAc (12 ml), se lavó con H 2 O (3 ml) y salmuera (3 ml), se secó sobre Na 2 SO 4 y se concentró hasta aprox. 1 ml en volumen. La cromatografía en columna ultrarrápida [gel de sílice desactivado (véanse los Métodos Generales), hexanos:EtOAc 1:1 a 1:3] proporcionó la sulfona 110 (20 mg, 3,4 pmol, rendimiento del 62 %) en forma de un sólido de color púrpura. 110: Fr = 0,23 [gel de sílice desactivado (véanse los Métodos Generales), hexanos:EtOAc 1:3 y después EtoAc:MeOH 50:1]; 1 H RMN (600 MHz, CD 3 CN): 8 = 13,19 (s, 1 H), 9,99 (d, J = 4,1 Hz, 1 H), 8,51 (s, 1 H), 8,24 (d, J = 8,0 Hz, 1 H), 8,18 (s, 1 H), 7,73 (dd, J = 8,0, 1,8 Hz, 1 H), 6,05 (a, 1 H), 5,97 (d, J = 10,0 Hz, 1 H), 5,88 (dt, J = 10,0, 1,3 Hz, 1 H), 5,26 (d, J = 4,4 Hz, 1 H), 4,97 (dd, J = 4,4, 1,7 Hz, 2 H), 4,66 (d, J = 5,0 Hz, 1 H), 4,6-4,3 (m, 2 H), 4,39 (d, J = 5,1, 1 H), 4,38 (dc, J = 5,0, 6,5 Hz, 1 H), 3,50 (a, 2 H), 3,35 (d, J = 4,9 Hz, 1 H), 1,38 (d, J = 6,5 Hz, 3 H) ppm; 13 C RMN (150 MHz, CD 3 CN): 8 = 188,5, 184,1, 153,7, 147,3, 144,6, 139,1, 136,1, 134,6, 134,1, 134,0, 133,7, 129,5, 128,1, 128,0, 125,0, 124,5, 124,1, 114,4, 112,4, 100,4, 99,1, 91,2, 88,8, 77,0, 65,8, 65,3, 64,7, 59,4, 55,2, 44,3, 44,2, 21,2 ppm; HRMS (ESI-TOF): calc. para C 37 H 31 N 2 O 10 S+ [M+H+]: 695,1694, observado 695,1692.
Ejemplo 4 - Actividad biológica de derivados de uncialamicina
Se ha demostrado que los compuestos de la presente divulgación son citotóxicos en diversas líneas celulares de cáncer diferentes. Las citotoxicidades son comparables a la citotoxicidad de los compuestos cuando el compuesto se ha conjugado con un anticuerpo (es decir, en forma de un ADC), lo que demuestra que compuestos son citotoxinas independientes del anticuerpo. Esta observación respalda la idea de que la citotoxicidad de los compuestos no puede atribuirse a una función efectora relacionada con anticuerpos, tal como la citotoxicidad mediada por células dependiente de anticuerpos (ADCC).
Se usó un ensayo de luminiscencia ATP, siendo el procedimiento como sigue: Se sembraron células a 1 x 103 células/pocillo en placas de 96 pocillos durante 3 h para los ensayos ATP CellTiterGl°™, respectivamente. Se añadieron diluciones en serie (1:3) de compuestos a los pocillos. Las placas se dejaron incubar durante 72 h. Se usó un kit de viabilidad celular CellTiter^™ de Promega para medir el contenido de ATP de las células tratadas con compuestos siguiendo las instrucciones del fabricante. Una disminución en el contenido de ATP es una medida de disminución en la viabilidad celular. El valor CE50 - la concentración a la cual un agente reduce la viabilidad celular en un 50% del efecto máximo - se determinó usando el software PRISM™, versión 5.0 (GraphPad Software, La Jolla, CA, Estados Unidos).
Los resultados de los ensayos se presentan en la Tabla 1. Los compuestos se ensayaron frente a ADR, una línea celular de cáncer de mama multirresistente, así como a las líneas celulares H226, N87 y OVCAR3.
Tabla 1: Activi i l i l m l r n iv l i n fr n lín l lares de cáncer

(continuación)

En determinadas condiciones, algunos de los compuestos de la presente divulgación son inestables, pero se obtuvieron cantidades suficientes para realizar los ensayos biológicos eliminando el grupo ftalimida del precursor protegido con ftalimida con metanamina (como se describe en el Ejemplo 5) y aislando el compuesto en forma de su sal de formiato.
Ejemplo 5 - Conjugación de uncialamicina con un enlazador para su unión a un anticuerpo
Para preparar un ADC, es necesario proporcionar un enlazador que conecte covalentemente el anticuerpo y el análogo citotóxico de uncialamicina. Una vez que el ADC ha alcanzado el sitio de acción deseada, deseablemente el enlazador se escinde para liberar la citotoxina.
Uno de los compuestos de esta divulgación, 8-aminometil uncialamicina (48aa), es una citotoxina potente pero es inestable y difícil de derivatizar con el enlazador necesario para preparar un conjugado anticuerpo-fármaco.

Por lo tanto, para preparar un derivado de 48aa adecuado para su conjugación, tal como 116 en el Esquema 21 a continuación, se utilizó un enfoque alternativo. En este enfoque, en lugar de derivatizar el propio 48aa, se utilizó un compuesto precursor protegido 77a, que tiene grupos protectores de Alloc y TES en N1 y el grupo hidroxilo de C17. Cuando se retira el grupo protector de ftalimida del compuesto 77a, el compuesto 111 resultante es mucho más estable que 48aa y susceptible a la química para unir un enlazador al mismo. Sin pretender quedar vinculado por teoría alguna, la inestabilidad de 48aa se debe a su grupo metilamino libre pero en 111, los grupos Alloc y TES anulan el efecto desestabilizante del grupo metilamino. Aunque se muestra la conjugación del enlazador con 48aa, se contempla que cualquiera de los compuestos de la presente divulgación puede derivatizarse de forma similar.

El compuesto 77a (48,3 mg, 0,061 mmol) se disolvió en MeOH (5 ml) y se añadió THF (1 ml) para solubilizarlo. La solución se enfrió a 0 °C y se añadió lentamente metanamina ac. al 40% (500 pl, 6,06 mmol) por el lateral del matraz mientras el color se iba oscureciendo. La mezcla se dejó dentro de un congelador a 0 °C durante una noche. El análisis por LCMS mostró una conversión de aproximadamente el 75% en el compuesto 111 ([M+H]: 667,3). La mezcla de reacción se calentó a temperatura ambiente (TA) y se agitó durante 4 h, después de lo cual la conversión fue mayor del 94%. La reacción se diluyó con EtOAc (10 ml) y se lavó con NaHCO
3
acuoso saturado (10 ml) y salmuera (10 ml). La capa orgánica se secó sobre Na
2
SO
4
, se filtró, se concentró, se sometió a alto vacío durante 30 min y se llevó a la siguiente etapa sin purificación adicional.
A una solución del compuesto 111 (0,041 g, 0,061 mmol) en DMF (1 ml) se le añadieron base de Hunig (0,032 ml, 0,183 mmol) y ((s)-3-metil-1-(((S)-1-((4-((((4-nitrofenoxi)carbonil)oxi)metil)fenil)amino)-1-oxo-5-ureido-pentan-2-il)amino)-1-oxobutan-2-il)carbamato de (9H-fluoren-9-il)metilo (0,056 g, 0,073 mmol) a TA. Después de 3 h, El análisis por LCMS mostró una conversión del 85 % en el compuesto 112 ([M+H]: 1295,6). La mezcla de reacción en
bruto se usó en la siguiente etapa sin purificación adicional.
El compuesto 112 se disolvió en THF (1 ml) y la mezcla se enfrió a 0 °C. A esta solución se le añadió Pd(PPh
3 )4
(7,05 mg, 6,10 |jmol) seguido de morfolina (0,013 ml, 0,146 mmol). El color cambio de pardo a púrpura en 5 segundos. La reacción se agitó a 0 °C durante 2 h, después de lo cual el análisis por LCMS mostró la desaparición del material de partida y la aparición del producto deseado 113 ([M+H]: 1210,5). La mezcla de reacción se diluyó con EtOAc (10 ml) y se lavó con tampón fosfato pH 6,8 (10 ml). La capa acuosa se extrajo con EtOAc (3 x 10 ml) y las capas orgánicas combinadas se secaron sobre Na
2
SO
4
, se filtraron y se concentraron. El producto en bruto se purificó usando un sistema de cromatografía líquida COMBIFLASH® (40 g de gel de sílice) con un gradiente de MeOH al 0-20% en CH
2
Cl
2
para dar el compuesto 4 en forma de un sólido de color púrpura. RMN 1H (400 MHz, DMSO-da) 813,17 (s, 1H), 10,07 (s, 1H), 10,00 (d, J = 4,4 Hz, 1H), 8,52 (s, 1H), 8,21 (d, J = 8 Hz, 1H), 8,13-8,10 (m, 2H), 8,01 (t, J = 5,6 Hz, 1H), 7,89 (d, J = 7,2 Hz, 2H), 7,81 (d, J = 7,2 Hz, 1H), 7,74 (t, J = 7,6 Hz, 2H, 7,60 (d, J = 8,4 Hz, 2H), 7,44-7,39 (m, 3H), 7,32 (t, J = 7,2 Hz, 3H), 6,71 (d, J = 5,2 Hz, 1H), 6,10-5,96 (m, 3H), 5,40 (s, 2H), 5,11 (dd, J = 4,8, 1,2 Hz, 1H), 5,05-5,01 (m, 2H), 4,51 (dd, J = 12,8, 6,4 Hz, 1H), 4,41-4,40 (m, 3H), 4,31-4,23 (m, 3H), 3,93 (t, J = 8,8 Hz, 1H), 3,03-2,93 (m, 2H), 2,00-1,99 (m, 1H), 1,69-1,58 (m, 2H), 1,35 (d, J = 6,4 Hz, 6H), 0,97-0,85 (m, 15H), 0,62 (c, J = 8,0 Hz, 6H).
A una solución del compuesto 113 (0,074 g, 0,061 mmol) en DMF (3 ml) se le añadió piperidina (50 jl, 0,506 mmol). La mezcla de reacción se agitó a TA durante 1 h. El análisis por LCMS mostró la formación del compuesto 114 ([M+H]: 988,4). La mezcla de reacción se enfrió a 0 °C y se añadió trietilamina (85 jl, 0,610 mmol), seguido de trihidrofluoruro de trietilamina (99 jl, 0,610 mmol). Después de 5 min, el baño de refrigeración se retiró y la mezcla de reacción se agitó durante 1 h. El análisis por LCMS mostró la formación del compuesto 115 ([M+H]: 874,4). La mezcla de reacción se diluyó con EtOAc (50 ml) y se lavó con salmuera (50 ml). La capa orgánica se secó sobre Na
2
SO
4
, se filtró y se concentró.
El compuesto en bruto 115 se disolvió en NMP (3 ml) y se añadió base de Hunig (0,032 ml, 0,183 mmol), seguido de 6-(2,5-dioxo-2,5-dihidro-1H-pirrol-1-il)hexanoato de 2,5-dioxopirrolidin-1-ilo (22,57 mg, 0,073 mmol). Después de 2 h, el análisis por LCMS mostró la formación del producto 116 ([M+H]: 1067,5). Se añadieron otros 1,2 equiv. de éster de N-hidroxisuccinimida del ácido 6-maleimidocaproico (MC-OSu) y 3 equiv. de base de Hunig (0,032 ml, 0,183 mmol). El matraz de reacción se cubrió con papel de aluminio y se mantuvo a 0 °C durante una noche. El análisis por LCMS mostró que la reacción se había completado. La reacción se diluyó con 2 ml de DMSO y se purificó en una máquina Waters Delta Prep 4000™ con una columna prep C18 X Bridge (30 x 250 mm, 5 mm OBD) usando un gradiente de agua al 5-95%/acetonitrilo (ácido fórmico al 0,05%) durante 40 min. Se descubrió que una fracción recogida a los 20,2 min contenía el producto deseado. Se liofilizó para proporcionar 6,3 mg (rendimiento global del 8,42% a partir de 77a) de 116 en forma de un sólido de color púrpura. 1H RMN (400 MHz, DMSO-D6) 8 13,11 (s, 1H), 9,93 (s, 2H), 8,45 (s, 1H), 8,42 (s, 1H), 8,22-8,13 (m, 2H), 8,08 (d, J = 7,6 Hz, 1H), 8,00 (t, J = 6,0 Hz, 1H), 7,80 (d, J = 8,4 Hz, 2H), 7,60 (d, J = 8,4 Hz, 2H), 7,30 (d, J = 8,4 Hz, 2H), 6,99 (s, 2H), 6,67 (d, J = 4,4 Hz, 2H), 6,07-5,96 (m, 3H), 5,40 (s a, 3H), 5,14 (d, J = 3,6 Hz, 1H), 5,06 (dd, J = 4,8, 1,6 Hz, 1H), 5,00 (s, 2H), 4,40-4,31 (m, 4H), 4,17 (dd, J = 8,4, 6,8 Hz, 1H), 3,07-2,91 (m, 2H), 2,20-2,09 (m, 2H), 1,99-193 (m, 2H), 1,70-1,45 (m, 5H), 1,31 1,14 (m, 7H), 0,85 (d, J = 6,8 Hz, 3H), 0,77 (d, J = 6,8 Hz, 3H).
Ejemplo 6 - Conjugación del compuesto al anticuerpo
Se utilizó el siguiente procedimiento general para preparar los conjugados de anticuerpo-fármaco (los ADC) de 116, que tiene la estructura que se muestra a continuación, donde Ab denota un anticuerpo y la unidad repetitiva m se repite 1, 2, 3, 4, 5, 6, 7, 8, 9 o 10. En algunos ejemplos particulares no limitantes, m es 1, 2, 3 o 4.

Este procedimiento se basa en la introducción de grupos tiol libres en un anticuerpo por reacción de grupos amino en £ de la lisina con 2-iminotiolano, seguido de reacción con una fracción enlazadora de fármaco que contiene maleimida, tal como, pero sin limitación, 116. Inicialmente, se cambia el tampón del anticuerpo a tampón fosfato 0,1 M (pH 8,0) que contiene NaCl 50 mM y ácido dietilentriamino pentaacético 2 mM (DTPA), y se concentra hasta 5 10 mg/ml. La tiolación se logra mediante la adición de 2-iminotiolano al anticuerpo. La cantidad de 2-iminotiolano a añadir se puede determinar mediante un experimento preliminar y varía de un anticuerpo a otro. En el experimento preliminar, se añade una titulación de cantidades crecientes de 2-iminotiolano al anticuerpo, y después de la incubación con el anticuerpo durante 1 h TA (temperatura ambiente, alrededor de 25 °C), el anticuerpo se desala en HEPES 50 mM, glicina 5 mM, DTPA 2 mM, pH 5,5 utilizando una columna SEPHADEX™ G-25 y el número de grupos tiol introducidos se determina rápidamente por reacción con ditiodipiridina (DTDP). La reacción de los grupos tiol con DTDP da como resultado la liberación de tiopiridina, lo puede controlarse espectroscópicamente a 324 nm. Se utilizan normalmente muestras con una concentración de proteína de 0,5-1,0 mg/ml. Se puede utilizar la absorbancia a 280 nm para determinar con precisión la concentración de proteína en las muestras, y luego se incuba una alícuota de cada muestra (0,9 ml) con 0,1 ml de DTDP (solución madre de etanol 5 mM) durante 10 min a TA. También se incuban muestras blanco de tampón solo más DTDP. Después de 10 min, se mide la absorbancia a 324 nm y se cuantifica el número de grupos tiol utilizando un coeficiente de extinción para tiopiridina de 19.800 M-1.
En algunas realizaciones, es conveniente un nivel de tiolación de aproximadamente dos a tres grupos tiol por anticuerpo. Por ejemplo, con algunos anticuerpos esto se puede lograr añadiendo un exceso molar de 15 veces de 2-iminotiolano seguido de incubación a TA durante 1 h. Después, el anticuerpo luego se incuba con 2-iminotiolano a la relación molar deseada y a continuación se desala en tampón de conjugación (HEPES 50 mM, glicina 5 mM, DTPA 2 mM, pH 5,5)). El material tiolado se mantiene en hielo mientras se cuantifica el número de tioles introducidos como se describe anteriormente.
Después de verificar la cantidad de tioles introducidos, la fracción enlazadora de fármaco (citotoxina) se añade en un exceso molar de 2,5 veces por tiol. Se permite proceder la reacción de conjugación en un tampón de conjugación que contiene una concentración final de propilenglicol al 25 % y trehalosa al 5 %. Habitualmente, la solución madre del enlazador de fármaco se disuelve en d Ms O al 100%. La solución madre se añade directamente al anticuerpo tiolado.
La mezcla de reacción de conjugación se incuba a TA durante 2 h con agitación suave. Después se añade a la mezcla de conjugación un exceso molar de 10 veces de N-etil maleimida (solución madre de DMSO 100 mM) y se agita durante una hora adicional para bloquear los tioles sin reaccionar.
Después, la muestra se filtra a través de un filtro de 0,2 p. Se intercambia el tampón del material a través de una membrana PES FFT VivaFlow 50 Sartorius de 30 MWCO a glicina 10 mg/ml, sorbitol 20 mg/ml, acetonitrilo 5 % pH 5,0 (volumen de intercambio de tampón de FFT 5X), para eliminar el fármaco sin reaccionar. La formulación final se lleva a cabo mediante FFT en sorbitol 20 mg/ml, glicina 10 mg/ml, pH 5,0.
Empleando la técnica general anterior, se prepararon tres ADC de 116, como se muestra en la Tabla 2.
Tabl 2: E m l n limi n n n i r -f rm l r n iv l ción

El anticuerpo anti-mesotelina en el ADC I fue 6A4, cuyas regiones determinantes de complementariedad (CDR) y otras características se desvelan en Terrett et al., documento US 8.268.970 B2 (2012), cuya divulgación se incorpora en el presente documento por referencia. El anticuerpo anti-glipicano 3 en el ADC II fue 4A6, cuyas CDR y otras características se desvelan en Terrett et al., documento US 8.680.247 (2014), cuya divulgación se incorpora en el presente documento por referencia. El anticuerpo anti-CD70 en el ADC III fue 2H5, cuyas CDR y otras características se desvelan en Terret et al., documento US 8.124.738 (2012), cuya divulgación se incorpora en el presente documento por referencia.
Ejemplo 7 - Actividad biológica de los conjugados de anticuerpo-fármaco
El siguiente procedimiento se utilizó para ensayos de proliferación de incorporación de 3H-timidina para medir in vitro la actividad de los ADC preparados frente a diversas líneas celulares de cáncer.
Las células se sembraron respectivamente a 1,25 x 104 células/pocillo en placas de 96 pocillos durante 3h para ensayos de 3H timidina. Se añadieron a los pocillos diluciones en serie (1:3) del conjugado. Se incubaron las placas durante 72 h. Las placas se pulsaron con 1,0 pCi de 3H-timidina por pocillo durante las últimas 24 h del período total de incubación, se recogieron y se leyeron en el contador de centelleo Top Count (Packard Instruments, Meriden, CT). Una disminución del grado de incorporación de 3H-timidina es una medida de disminución de la proliferación celular. Se determinó utilizando el software PRISM ™ versión 5.0 (programa informático GraphPad, La Jolla, CA, EE.UU.) el valor de CE50 , la concentración a la cual un agente inhibe o reduce la proliferación celular en un 50 % de la inhibición máxima.
La actividad del ADC I, que es un ADC de un anticuerpo anti-mesotelina, se analizó frente a las células de cáncer H226 (pulmón), N87 (gástrico) y OVCAR3 (ovárico), las cuales expresan mesotelina, sirviendo el ADC II que sirve
como un ADC comparativo. Los resultados se muestran en la Tabla 3. Las actividades del ADC II comparativo contra estas líneas celulares fueron notablemente más bajas.
Tabla 3: Actividad del ADC contra células cancerosas ue ex resan mesotelina

La actividad del ADC II, que es un ADC de un anticuerpo anti-glipicano 3, se analizó frente a las células de cáncer de hígado Hep3B y HepG2, que expresan glipicano 3. Se analizó también el ADC I con fines comparativos. Los resultados se proporcionan en la Tabla 4. La potencia del ADC II fue notablemente mayor que la del ADC I comparativo contra estas líneas celulares.
Tabla 4: Actividad del ADC contra células cancerosas ue ex resan Gli icano 3

La actividad del ADC III, que es un ADC de un anticuerpo anti-CD70, se analizó frente a células 786-O, que son células cancerosas renales que expresan CD70. Se analizó también el ADC I con fines comparativos. Los resultados se proporcionan en la Tabla 5. De nuevo, el ADC comparativo fue mucho menos potente que el ADC diseñado para dirigirse a la línea celular específica.
Tabla 5: Actividad del ADC contra células cancerosas ue ex resan CD70

V. Referencias
Patente de Estados Unidos 5.739.169
Patente de Estados Unidos 5.801.005
Patente de Estados Unidos 5.824.311
Patente de Estados Unidos 5.830.880
Patente de Estados Unidos 5.846.945
Patente de Estados Unidos 6.232.287
Patente de Estados Unidos 6.528.481
Patente de Estados Unidos 6.984.720
Patente de Estados Unidos 7.335.748
Patente de Estados Unidos 7.387.776
Patente de Estados Unidos 7.452.964
Patente de Estados Unidos 7.671.010
Patente de Estados Unidos 7.781.565
Patente de Estados Unidos 7.875.278
Patente de Estados Unidos 8,008449
Patente de Estados Unidos 8.097.703
Patente de Estados Unidos 8.124.738
Patente de Estados Unidos 8.268.970
Patente de Estados Unidos 8.450.278
Patente de Estados Unidos 8.507.445
Patente de Estados Unidos 8.680.247
Patente de Estados Unidos 8.709.431
Publicaciones de patente de Estados Unidos 2004/0005647
Publicaciones de patente de Estados Unidos 2006/0234299
Publicaciones de patente de Estados Unidos 2006/0223114
Publicaciones de patente de Estados Unidos 2006/0058510
Publicaciones de patente de Estados Unidos 2006/0088908
Publicaciones de patente de Estados Unidos 2009/0074660
Publicaciones de patente de Estados Unidos 2009/0253899
Publicaciones de patente de Estados Unidos 2009/0297438
Publicaciones de patente de Estados Unidos 2010/0034826
Publicaciones de patente de Estados Unidos 2010/0143368
Publicaciones de patente de Estados Unidos 2010/0150950
Publicaciones de patente de Estados Unidos 2010/0092484
Publicaciones de patente de Estados Unidos 2010/0209432
Publicaciones de patente de Estados Unidos 2010/0285564
Publicaciones de patente de Estados Unidos 2010/0317547
Publicaciones de patente de Estados Unidos 2010/0093024
Publicaciones de patente de Estados Unidos 2013/0209494
documento WO 2005/051976
documento WO 2006/056464
documento WO 2008/030612
documento WO 2010/087994
documento WO 2013/122823
Agard et al., J. Amer. Chem. Soc. 2004, 126, 15046-15047
Austin-Ward et al., 1998
Barclay et al. (eds.), The Leucocyte Antigen Facts Book, 1993, Academic Press.
Best, Biochemistry 2009, 48, 6571-6584
Bukowski et al., 1998
Burkly et al. Cytokine 40:1-16 (2007).
Campbell, et al., Cancer Research, 51(19):5329-5338, 1991.
Chen, J.S., Tesis doctoral, 2008.
Christodoulides et al., 1998 Cumber et al. (1992)
Davidson et al., 1998
Goerke et al., Biotechnol. Bioeng. 2009, 102 (2), 400-416.
Green et al., 1999
Handbook of Pharmaceutical Salts: Properties, and Use (2002)
Hanibuchi et al., 1998
Harvey et al., 2004
Hellstrand et al., 1998
Hui, et al., 1998
Jeger et al., Angew. Chem. Int. Ed. 2010, 49, 9995 -9997.
Ju et al., 2000 Kohl et al., 2003
Levary et al., PLoS One 2011, 6(4), e18342 Liu et al., Mol. Cancer Ther., 2:1341-1350, 2003. March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure (2007) Mitchell et al., 1990
Mitchell et al., 1993
Montalbetti y Falque, Tetrahedron, 61:10827-10852, 2005.
Morton et al., 1992 Nechushtan et al., 1997
Nicolaou, et al., Angew. Chem. Int. Ed. 2007, 46, 4704.
Nicolaou, et al., Angew. Chem. Int. Ed. 2008, 47, 185.
Onda et al., Cancer Res., 64:1419-1424, 2004.
Pack et al. (1992)
Pietras et al., 1998
Practical Process Research & Development (2000)
Proft, Biotechnol. Lett. 2010, 32, 1-10.
Qin et al., 1998 Ravindranath et al., 1991
Remington's Pharmaceutical Sciences, 15a edición, capítulo 33, en particular las páginas 624-652 Remington's Pharmaceutical Sciences, 15a edición, páginas 1035-1038 y 1570-1580 Rosenberg et al., 1988; 1989 Skerra, 2001.
Thompson (ed.), 1994, The Cytokine Handbook, Academic Press, San Diego
Weitman, et al., Cancer Research, 52(12):3396-3401, 1992.
Winkles, Nat Rev Drug Discov 7:411-425 (2008).
Winthrop et al., Clin. Cancer Res., 9:3845s-3853s, 2003.
Zhou et al., Mol Cancer Ther. 10(7):1276-88, 2011.
Claims (1)
- REIVINDICACIONES1. Un compuesto de la fórmula:
 en donde:cada uno de Ri y R 2 se selecciona independientemente entre hidrógeno, hidroxi,alquilO(ci-i2), alquilo sustituidos- 12 ), alquenilo(C2-12), alquenilo sustituido(C2-12), alquinilo(C2-12), alquinilo sustituido(C2- 12 ), arilo(C6-12), arilo sustituido(C6-12), aralquilo(C7-12), aralquilo sustituido(C7-12), heteroarilo(C1-12), heteroarilo sustituido(C1-12), heterocicloalquilo(C2-12), heterocicloalquilo sustituido(C2-12), acilo(C1-12), acilo sustituidos- 12 ), aciloxi(C1-12), aciloxi sustituido(C1-12), alquilamino(C1-12), alquilamino sustituido(C1-12); un grupo protector de amina monovalente, -C(O)O(CH 2 )nS-A 1 , -C(O)O(CH 2 )nS(O)-A 1 o -C(O)O(CH 2 )nS(O) 2 -A 1 ,en donde:A 1 es arilo(C 6 - 12 ) o arilo sustituido(C 6 - 12 ); yn es 1, 2, 3, 4 o 5; oR 1 y R 2 se toman juntos y forman un grupo protector de amina divalente,R 3 es hidrógeno, hidroxi, halo o alcoxis- 12 ) o alcoxi sustituidos- 12 ); o es 1,2 o 3;R 4 es hidrógeno, un grupo protector de amina monovalente, alquilo(C 1 - 12 ) o alquilo sustituidos- 12 );cada uno de R 5 , R6 y R 7 es independientemente hidrógeno, hidroxi, amino, mercapto, -OX 1 , -NX 2 X 3 o -SX 4 ; o alquilo(C1-12) o alquilo sustituidos- 12 );en donde:X 1 es un grupo protector de hidroxi;X 2 y X 3 se seleccionan independientemente entre hidrógeno, un grupo protector de amina monovalente, o cuando X 2 y X 3 se toman juntos forman un grupo protector de amina divalente; yX 4 es un grupo protector de tiol; yR8 es hidroxi, amino o mercapto;en donde: cada grupo protector de amina se selecciona independientemente entre la lista que consiste en formilo, acetilo, propionilo, pivaloílo, t-butilacetilo, 2-cloroacetilo, 2-bromoacetilo, trifluoroacetilo, tricloroacetilo, onitrofenoxiacetilo, a-clorobutirilo, benzoílo, 4-clorobenzoílo, 4-bromobenzoílo, 4-nitrobenzoílo, un grupo sulfonilo, un grupo aralquilo y un grupo sililo;el grupo protector de hidroxilo se selecciona entre la lista que consiste en formilo, acetilo, propionilo, pivaloílo, tbutilacetilo, 2-cloroacetilo, 2-bromoacetilo, trifluoroacetilo, tricloroacetilo, o-nitrofenoxiacetilo, a-clorobutirilo, benzoílo, 4-clorobenzoílo, 4-bromobenzoílo, 4-nitrobenzoílo, un grupo sulfonilo, un grupo aciloxi, un grupo aralquilo y un grupo sililo; yel grupo protector de tiol se selecciona entre la lista que consiste en formilo, acetilo, propionilo, pivaloílo, tbutilacetilo, 2-cloroacetilo, 2-bromoacetilo, trifluoroacetilo, tricloroacetilo, o-nitrofenoxiacetilo, a-clorobutirilo, benzoílo, 4-clorobenzoílo, 4-bromobenzoílo, 4-nitrobenzoílo, un grupo sulfonilo, un grupo aciloxi, un grupo aralquilo y un grupo sililo; ouna sal farmacéuticamente aceptable del mismo.2. El compuesto de la reivindicación 1, en donde la fórmula se define adicionalmente como:
en donde:cada uno de Ri y R 2 se selecciona independientemente entre hidrógeno, hidroxi,alquilO(ci-i2), alquilo sustituidos- 12 ), alquenilo(C2-12), alquenilo sustituido(C2-12), alquinilo(C2-12), alquinilo sustituido(C2- 12 ), arilo(C6-12), arilo sustituido(C6-12), aralquilo(C7-12), aralquilo sustituido(C7-12), heteroarilo(C1-12), heteroarilo sustituido(C1-12), heterocicloalquilo(C2-12), heterocicloalquilo sustituido(C2-12), acilo(C1-12), acilo sustituidos- 12 ), aciloxi(C1-12), aciloxi sustituido(C1-12), alquilamino(C1-12), alquilamino sustituido(C1-12); un grupo protector de amina monovalente, -C(O)O(CH 2 )nS-A 1 , -C(O)O(CH 2 )nS(O)-A 1 o -C(O)O(CH 2 )nS(O) 2 -A 1 ,en donde:A 1 es arilo(C 6 - 12 ) o arilo sustituido(C 6 - 12 ); yn es 1, 2, 3, 4 o 5; oR 1 y R 2 se toman juntos y forman un grupo protector de amina divalente,R 3 es hidrógeno, hidroxi, halo o alcoxis- 12 ) o alcoxi sustituidos- 12 ); o es 1,2 o 3;R 4 es hidrógeno, un grupo protector de amina monovalente, alquilo(C 1 - 12 ) o alquilo sustituidos- 12 );cada uno de R 5 , R6 y R 7 es independientemente hidrógeno, hidroxi, amino, mercapto, -OX 1 , -NX 2 X 3 o -SX 4 ; o alquilo(C1-12) o alquilo sustituidos- 12 );en donde:X 1 es un grupo protector de hidroxi;X 2 y X 3 se seleccionan independientemente entre hidrógeno, un grupo protector de amina monovalente, o cuando X 2 y X 3 se toman juntos forman un grupo protector de amina divalente; yX 4 es un grupo protector de tiol; yR8 es hidroxi, amino o mercapto;en donde: cada grupo protector de amina se selecciona independientemente entre la lista que consiste en formilo, acetilo, propionilo, pivaloílo, t-butilacetilo, 2-cloroacetilo, 2-bromoacetilo, trifluoroacetilo, tricloroacetilo, onitrofenoxiacetilo, a-clorobutirilo, benzoílo, 4-clorobenzoílo, 4-bromobenzoílo, 4-nitrobenzoílo, un grupo sulfonilo, un grupo aralquilo y un grupo sililo;el grupo protector de hidroxilo se selecciona entre la lista que consiste en formilo, acetilo, propionilo, pivaloílo, tbutilacetilo, 2-cloroacetilo, 2-bromoacetilo, trifluoroacetilo, tricloroacetilo, o-nitrofenoxiacetilo, a-clorobutirilo, benzoílo, 4-clorobenzoílo, 4-bromobenzoílo, 4-nitrobenzoílo, un grupo sulfonilo, un grupo aciloxi, un grupo aralquilo y un grupo sililo; yel grupo protector de tiol se selecciona entre la lista que consiste en formilo, acetilo, propionilo, pivaloílo, tbutilacetilo, 2-cloroacetilo, 2-bromoacetilo, trifluoroacetilo, tricloroacetilo, o-nitrofenoxiacetilo, a-clorobutirilo, benzoílo, 4-clorobenzoílo, 4-bromobenzoílo, 4-nitrobenzoílo, un grupo sulfonilo, un grupo aciloxi, un grupo aralquilo y un grupo sililo; ouna sal farmacéuticamente aceptable del mismo.2. El compuesto de la reivindicación 1, en donde la fórmula se define adicionalmente como: en donde:cada uno de Ri y R 2 se selecciona independientemente entre hidrógeno, hidroxi,alquilO(ci-i2), alquilo sustituidos-^), alquenilo(C2-12), alquenilo sustituido(C2-12), alquinilo(C2-12), alquinilo sustituido(C2- 12 ), arilo(C6-12), arilo sustituido(C6-12), aralquilo(C7-12), aralquilo sustituido(C7-12), heteroarilo(C1-12), heteroarilo sustituido(C1-12), heterocicloalquilo(C2-12), heterocicloalquilo sustituido(C2-12), acilo(C1-12), acilo sustituido(C1-12), aciloxi(C1-12), aciloxi sustituidos- 12 ), alquilamino(C1-12), alquilamino sustituido(C1-12), un grupo protector de amina monovalente, -C(O)O(CH 2 )nS-A 1 , -C(O)O(CH 2 )nS(O)-A 1 o -C(O)O(CH 2 )nS(O) 2 -A 1 ,en donde:A 1 es arilo(C 6 - 12 ) o arilo sustituido(C 6 - 12 ); yn es 1, 2, 3, 4 o 5;R 3 es hidrógeno, hidroxi, halo o alcoxis- 12 ) o alcoxi sustituido(C 1 - 12 )o es 1,2 o 3;R 4 es hidrógeno, un grupo protector de amina monovalente, alquilos- 12 ) o alquilo sustituidos- 12 )cada uno de R 5 , R6 y R 7 es independientemente hidrógeno, hidroxi, amino, mercapto, -OX 1 , -NX 2 X 3 o -SX 4 ; o alquilo(C1-12) o alquilo sustituido(C1-12)en donde:X 1 es un grupo protector de hidroxi;X 2 y X 3 se seleccionan independientemente entre hidrógeno, un grupo protector de amina monovalente, o cuando X 2 y X 3 se toman juntos forman un grupo protector de amina divalente; yX 4 es un grupo protector de tiol; yR8 es hidroxi, amino o mercapto; ouna sal farmacéuticamente aceptable del mismo.3. El compuesto de la reivindicación 1, en donde la fórmula se define adicionalmente como:
en donde:cada uno de Ri y R 2 se selecciona independientemente entre hidrógeno, hidroxi,alquilO(ci-i2), alquilo sustituidos-^), alquenilo(C2-12), alquenilo sustituido(C2-12), alquinilo(C2-12), alquinilo sustituido(C2- 12 ), arilo(C6-12), arilo sustituido(C6-12), aralquilo(C7-12), aralquilo sustituido(C7-12), heteroarilo(C1-12), heteroarilo sustituido(C1-12), heterocicloalquilo(C2-12), heterocicloalquilo sustituido(C2-12), acilo(C1-12), acilo sustituido(C1-12), aciloxi(C1-12), aciloxi sustituidos- 12 ), alquilamino(C1-12), alquilamino sustituido(C1-12), un grupo protector de amina monovalente, -C(O)O(CH 2 )nS-A 1 , -C(O)O(CH 2 )nS(O)-A 1 o -C(O)O(CH 2 )nS(O) 2 -A 1 ,en donde:A 1 es arilo(C 6 - 12 ) o arilo sustituido(C 6 - 12 ); yn es 1, 2, 3, 4 o 5;R 3 es hidrógeno, hidroxi, halo o alcoxis- 12 ) o alcoxi sustituido(C 1 - 12 )o es 1,2 o 3;R 4 es hidrógeno, un grupo protector de amina monovalente, alquilos- 12 ) o alquilo sustituidos- 12 )cada uno de R 5 , R6 y R 7 es independientemente hidrógeno, hidroxi, amino, mercapto, -OX 1 , -NX 2 X 3 o -SX 4 ; o alquilo(C1-12) o alquilo sustituido(C1-12)en donde:X 1 es un grupo protector de hidroxi;X 2 y X 3 se seleccionan independientemente entre hidrógeno, un grupo protector de amina monovalente, o cuando X 2 y X 3 se toman juntos forman un grupo protector de amina divalente; yX 4 es un grupo protector de tiol; yR8 es hidroxi, amino o mercapto; ouna sal farmacéuticamente aceptable del mismo.3. El compuesto de la reivindicación 1, en donde la fórmula se define adicionalmente como: en donde:cada uno de R 1 y R 2 se selecciona independientemente entre hidrógeno, hidroxi,alquilo(C1-12), alquilo sustituidos- 12 ), arilo(C6-12), arilo sustituido(C6-12), aralquilo(C7-12), aralquilo sustituido(C7-12), acilo(C 1 - 12 ), acilo sustituidos- 12 ), un grupo protector de amina monovalente, -C(O)O(CH2)nS-C6H5, -C(O)O(CH2)nS(O)-C6H5 o -C(O)O(CH2)nS(O)2-C6H5,un grupo protector de amina monovalente, o R 1 y R 2 se toman juntos y forman un grupo protector de amina divalente; R 3 es hidrógeno, hidroxi, halo o alcoxi(C1-12) o alcoxi sustituido(C1-12); yo es 1, 2 o 3; ouna sal farmacéuticamente aceptable del mismo.4. El compuesto de la reivindicación 1, en donde la fórmula se define adicionalmente como:
en donde:cada uno de R 1 y R 2 se selecciona independientemente entre hidrógeno, hidroxi,alquilo(C1-12), alquilo sustituidos- 12 ), arilo(C6-12), arilo sustituido(C6-12), aralquilo(C7-12), aralquilo sustituido(C7-12), acilo(C 1 - 12 ), acilo sustituidos- 12 ), un grupo protector de amina monovalente, -C(O)O(CH2)nS-C6H5, -C(O)O(CH2)nS(O)-C6H5 o -C(O)O(CH2)nS(O)2-C6H5,un grupo protector de amina monovalente, o R 1 y R 2 se toman juntos y forman un grupo protector de amina divalente; R 3 es hidrógeno, hidroxi, halo o alcoxi(C1-12) o alcoxi sustituido(C1-12); yo es 1, 2 o 3; ouna sal farmacéuticamente aceptable del mismo.4. El compuesto de la reivindicación 1, en donde la fórmula se define adicionalmente como: en donde:X 1 es un grupo protector de hidroxi;cada uno de R 1 y R 2 se selecciona independientemente entre hidrógeno, alquilo(C 1 - 12 ) o alquilo sustituido(C 1 - 12 ) y R 3 es hidrógeno, alcoxi(C 1 - 12 ) o alcoxi sustituidos-^; yo es 2 ;o una sal farmacéuticamente aceptable del mismo.5. El compuesto de la reivindicación 1, en dondeR 1 o R 2 es hidrógeno, oalquilo(C 1 - 12 ) o alquilo sustituido(C 1 - 12 ), ogrupo protector de amina monovalente, oR 1 se toma junto con R 2 y es un grupo protector de amina divalente, oen donde R 3 es hidrógeno, oalcoxi(C 1 - 12 ), oen donde R 4 es hidrógeno, oun grupo protector de amina monovalente, oen donde R 5 es alquilo(C 1 - 12 ) oen donde R 6 , R 7 o R 8 es hidroxi, oen donde el átomo de carbono 17 está en la configuración R, oen donde el átomo de carbono 17 está en la configuración S.6. El compuesto de la reivindicación 1, en donde el compuesto se define adicionalmente como:
en donde:X 1 es un grupo protector de hidroxi;cada uno de R 1 y R 2 se selecciona independientemente entre hidrógeno, alquilo(C 1 - 12 ) o alquilo sustituido(C 1 - 12 ) y R 3 es hidrógeno, alcoxi(C 1 - 12 ) o alcoxi sustituidos-^; yo es 2 ;o una sal farmacéuticamente aceptable del mismo.5. El compuesto de la reivindicación 1, en dondeR 1 o R 2 es hidrógeno, oalquilo(C 1 - 12 ) o alquilo sustituido(C 1 - 12 ), ogrupo protector de amina monovalente, oR 1 se toma junto con R 2 y es un grupo protector de amina divalente, oen donde R 3 es hidrógeno, oalcoxi(C 1 - 12 ), oen donde R 4 es hidrógeno, oun grupo protector de amina monovalente, oen donde R 5 es alquilo(C 1 - 12 ) oen donde R 6 , R 7 o R 8 es hidroxi, oen donde el átomo de carbono 17 está en la configuración R, oen donde el átomo de carbono 17 está en la configuración S.6. El compuesto de la reivindicación 1, en donde el compuesto se define adicionalmente como:
 o una sal farmacéuticamente aceptable del mismo.7. El compuesto de la reivindicación 1, definido adicionalmente como:
o una sal farmacéuticamente aceptable del mismo.7. El compuesto de la reivindicación 1, definido adicionalmente como:
 o una sal farmacéuticamente aceptable del mismo.8. Un método para preparar un compuesto de acuerdo con cualquier reivindicación anterior: realizando una reacción que comprende mezclar un compuesto de la fórmula:
o una sal farmacéuticamente aceptable del mismo.8. Un método para preparar un compuesto de acuerdo con cualquier reivindicación anterior: realizando una reacción que comprende mezclar un compuesto de la fórmula: en donde:R 4 es hidrógeno, alquilo(C 1 - 12 ), un grupo protector de amina monovalente, o alquilo sustituido(C 1 - 12 )cada uno de R 5 , R 6 y R 7 es independientemente hidrógeno, hidroxi, amino, mercapto, -OX 1 , -NX 2 X 3 o -SX 4 ; o alquilo(C 1 - 12 ) o una versión sustituida de este grupo;en donde:X 1 es un grupo protector de hidroxi;X 2 y X 3 se seleccionan independientemente entre hidrógeno, un grupo protector de amina monovalente, o cuando X 2 y X 3 se toman juntos forman un grupo protector de amina divalente; yX 4 es un grupo protector de tiol; yR 14 es -O-, -S- o -NR 15 -;en donde:R 15 es hidrógeno, alquilona) o alquilo sustituidô -a) yR 15 es ciano, alcoxi(C 1 - 12 ), alcoxi sustituido(C 1 - 12 ), aciloxi(C 1 - 12 ), aciloxi sustituido(C 1 - 12 ), alquiltio(C 1 - 12 ), alquiltio sustituido(C 1 - 12 ), alquilsulfonilo(C 1 - 12 ), alquilsulfonilo sustituido(C 1 - 12 ), arilsulfonilo(C 1 - 12 ), arilsulfonilo sustituido(C 1 - 12 ) alquilsulfoniloxi(ci-i 2 ), alquilsulfoniloxi sustituidO(ci-i 2 ), arilsulfoniloxi(ci-i 2 ) o arilsulfoniloxi sustituidO(ci-i 2 )icon un compuesto de la fórmula:
en donde:R 4 es hidrógeno, alquilo(C 1 - 12 ), un grupo protector de amina monovalente, o alquilo sustituido(C 1 - 12 )cada uno de R 5 , R 6 y R 7 es independientemente hidrógeno, hidroxi, amino, mercapto, -OX 1 , -NX 2 X 3 o -SX 4 ; o alquilo(C 1 - 12 ) o una versión sustituida de este grupo;en donde:X 1 es un grupo protector de hidroxi;X 2 y X 3 se seleccionan independientemente entre hidrógeno, un grupo protector de amina monovalente, o cuando X 2 y X 3 se toman juntos forman un grupo protector de amina divalente; yX 4 es un grupo protector de tiol; yR 14 es -O-, -S- o -NR 15 -;en donde:R 15 es hidrógeno, alquilona) o alquilo sustituidô -a) yR 15 es ciano, alcoxi(C 1 - 12 ), alcoxi sustituido(C 1 - 12 ), aciloxi(C 1 - 12 ), aciloxi sustituido(C 1 - 12 ), alquiltio(C 1 - 12 ), alquiltio sustituido(C 1 - 12 ), alquilsulfonilo(C 1 - 12 ), alquilsulfonilo sustituido(C 1 - 12 ), arilsulfonilo(C 1 - 12 ), arilsulfonilo sustituido(C 1 - 12 ) alquilsulfoniloxi(ci-i 2 ), alquilsulfoniloxi sustituidO(ci-i 2 ), arilsulfoniloxi(ci-i 2 ) o arilsulfoniloxi sustituidO(ci-i 2 )icon un compuesto de la fórmula: en donde:Yi es -(CH 2 )mNRiR 2 ;en donde:m es 1; ycada uno de Ri y R 2 se selecciona independientemente entre hidrógeno, hidroxi, alquilo(ci-i 2 ), alquilo sustituido(ci-i2), alquenilo(C2-i2), alquenilo sustituido(C2-i2), alquinilo(C2-i2), alquinilo sustituido(C2-i2), arilo(C6-i2), arilo sustituido(C6-i2), aralquilo(C7-i2), aralquilo sustituido(C7-i2), heteroarilo(Ci-i2), heteroarilo sustituido(Ci-i2), heterocicloalquilo(C2-i2), heterocicloalquilo sustituido(C2-i2), acilo(Ci-i2), acilo sustituido(Ci-i2), aciloxi(Ci-i2), aciloxi sustituido(Ci-i2), alquilamino(Ci-i2), alquilamino sustituido(Ci-i2); un grupo protector de amina monovalente, -C(O)O(CH 2 )nS-Ai, -C(O)O(CH 2 )nS(O)-Ai o -C(O)O(CH 2 )nS(O) 2 -Ai,en donde:Ai es arilo(C6-i2), arilo sustituido(C6-i2), o
en donde:Yi es -(CH 2 )mNRiR 2 ;en donde:m es 1; ycada uno de Ri y R 2 se selecciona independientemente entre hidrógeno, hidroxi, alquilo(ci-i 2 ), alquilo sustituido(ci-i2), alquenilo(C2-i2), alquenilo sustituido(C2-i2), alquinilo(C2-i2), alquinilo sustituido(C2-i2), arilo(C6-i2), arilo sustituido(C6-i2), aralquilo(C7-i2), aralquilo sustituido(C7-i2), heteroarilo(Ci-i2), heteroarilo sustituido(Ci-i2), heterocicloalquilo(C2-i2), heterocicloalquilo sustituido(C2-i2), acilo(Ci-i2), acilo sustituido(Ci-i2), aciloxi(Ci-i2), aciloxi sustituido(Ci-i2), alquilamino(Ci-i2), alquilamino sustituido(Ci-i2); un grupo protector de amina monovalente, -C(O)O(CH 2 )nS-Ai, -C(O)O(CH 2 )nS(O)-Ai o -C(O)O(CH 2 )nS(O) 2 -Ai,en donde:Ai es arilo(C6-i2), arilo sustituido(C6-i2), o en donde:A 2 es alquenilo(C2-i2), alquenilo sustituido(C2-i2), alquinilo(C2-i2), alquinilo sustituido(C2-i2), acilo(Ci-i2), acilo sustituido(Ci-i2), aciloxi(Ci-i2), aciloxi sustituido(Ci-i2), alcoxi(Ci-i2), alcoxi sustituido(Ci-i2), alquilamino(Ci-i2), alquilamino sustituido(Ci-i2), dialquilamino(C2-i2) o dialquilamino sustituido(C2-i2), en donde A 2 no es -CO 2 H, -CO 2 CH 3 , -OCH 3 , -OCH 2 CH 3 , -C(O)CHa, -NHCH 3 , -NHCH 2 CH 3 , -N(CHa) 2 , -C(O)NH 2 y -OC(O)CH3; y n es i, 2, 3, 4 o 5; oRi y R 2 se toman juntos y son un grupo protector de amina divalente, alcanodiílo(Ci-i2), alquilaminodiílo(Ci-8); alcoxidiílo(Ci-8); o una versión sustituida de cualquiera de estos grupos; oR 3 es hidrógeno, hidroxi, halo o alcoxi(Ci-i2), o una versión sustituida de este grupo; yo es i o 2;en presencia de una base no nucleófila fuerte en un disolvente en condiciones suficientes para provocar una reacción, en donde la base es bis(trimetilsilil)amida de litio, bis(trimetilsilil)amida sódica, bis(trimetilsilil)amida potásica, diisopropilamida de litio, diisopropilamida sódica, diisopropilamida potásica, tetrametilpiperiduro de litio, tbutóxido de litio, t-butóxido sódico o t-butóxido potásico, hidruro de litio, hidruro sódico o hidruro potásico, opcionalmente en donde la base es bis(trimetilsilil)amida de litio, oen donde el disolvente es tetrahidrofurano, oen donde el método comprende adicionalmente mezclar el compuesto de fórmula IV con la base antes de poner en contacto el compuesto de fórmula III con el compuesto de fórmula IV, opcionalmenteen donde el compuesto de fórmula III se disuelve en THF y se añade mediante una cánula.9. El método de la reivindicación 8, que comprende adicionalmente disolver el compuesto de fórmula I o de fórmula II en un disolvente orgánico y hacer reaccionar el compuesto de la fórmula:i i 0
en donde:A 2 es alquenilo(C2-i2), alquenilo sustituido(C2-i2), alquinilo(C2-i2), alquinilo sustituido(C2-i2), acilo(Ci-i2), acilo sustituido(Ci-i2), aciloxi(Ci-i2), aciloxi sustituido(Ci-i2), alcoxi(Ci-i2), alcoxi sustituido(Ci-i2), alquilamino(Ci-i2), alquilamino sustituido(Ci-i2), dialquilamino(C2-i2) o dialquilamino sustituido(C2-i2), en donde A 2 no es -CO 2 H, -CO 2 CH 3 , -OCH 3 , -OCH 2 CH 3 , -C(O)CHa, -NHCH 3 , -NHCH 2 CH 3 , -N(CHa) 2 , -C(O)NH 2 y -OC(O)CH3; y n es i, 2, 3, 4 o 5; oRi y R 2 se toman juntos y son un grupo protector de amina divalente, alcanodiílo(Ci-i2), alquilaminodiílo(Ci-8); alcoxidiílo(Ci-8); o una versión sustituida de cualquiera de estos grupos; oR 3 es hidrógeno, hidroxi, halo o alcoxi(Ci-i2), o una versión sustituida de este grupo; yo es i o 2;en presencia de una base no nucleófila fuerte en un disolvente en condiciones suficientes para provocar una reacción, en donde la base es bis(trimetilsilil)amida de litio, bis(trimetilsilil)amida sódica, bis(trimetilsilil)amida potásica, diisopropilamida de litio, diisopropilamida sódica, diisopropilamida potásica, tetrametilpiperiduro de litio, tbutóxido de litio, t-butóxido sódico o t-butóxido potásico, hidruro de litio, hidruro sódico o hidruro potásico, opcionalmente en donde la base es bis(trimetilsilil)amida de litio, oen donde el disolvente es tetrahidrofurano, oen donde el método comprende adicionalmente mezclar el compuesto de fórmula IV con la base antes de poner en contacto el compuesto de fórmula III con el compuesto de fórmula IV, opcionalmenteen donde el compuesto de fórmula III se disuelve en THF y se añade mediante una cánula.9. El método de la reivindicación 8, que comprende adicionalmente disolver el compuesto de fórmula I o de fórmula II en un disolvente orgánico y hacer reaccionar el compuesto de la fórmula:i i 0 en donde:Yi es -(CH 2 )mNR-,R 2 ;en donde:m es 1; ycada uno de Ri y R 2 se selecciona independientemente entre hidrógeno, hidroxi, alquilO(ci-i 2 ), alquilo sustituidO(ci-i2), alquenilo(C2-12), alquenilo sustituido(C2-12), alquinilo(C2-12), alquinilo sustituido(C2-12), arilo(C6-12), arilo sustituido(C6-12), aralquilo(C7-12), aralquilo sustituido(C7-12), heteroarilo(C1-12), heteroarilo sustituido(C1-12), heterocicloalquilo(C2-12), heterocicloalquilo sustituido(C2-12), acilo(C1-12), acilo sustituido(C1-12), aciloxi(C1-12), aciloxi sustituido(C1-12), alquilamino(C1-12), alquilamino sustituido(C1-12); un grupo protector de amina monovalente, -C(O)O(CH 2 )nS-A 1 , -C(O)O(CH 2 )nS(O)-A 1 o -C(O)O(CH 2 )nS(O) 2 -A 1 ,en donde:A 1 es arilo(C 6 - 12 ) o arilo sustituido(C 6 - 12 ); yn es 1, 2, 3, 4 o 5; oRi y R 2 se toman juntos y son un grupo protector de amina divalente;R 3 es hidrógeno, hidroxi, halo o alcoxi(C 1 - 12 ) o una versión sustituida de este grupo;o es 1 o 2;R 4 es hidrógeno, alquilo(C 1 - 12 ), un grupo protector de amina monovalente o alquilo sustituido(C 1 - 12 );cada uno de R 5 , R6 y R 7 es independientemente hidrógeno, hidroxi, amino, mercapto, -OX 1 , -NX 2 X 3 o -SX 4 ; o alquilo(C 1 - 12 ) o una versión sustituida de cualquiera de estos grupos;en donde:X 1 es un grupo protector de hidroxi;X 2 y X 3 se seleccionan independientemente entre hidrógeno, un grupo protector de amina monovalente, o cuando X 2 y X 3 se toman juntos forman un grupo protector de amina divalente;X 4 es un grupo protector de tiol;R8 es hidroxi, amino o mercapto;con una sal de paladio en presencia de una base en condiciones suficientes para provocar una reacción para formar un compuesto de la fórmula:
en donde:Yi es -(CH 2 )mNR-,R 2 ;en donde:m es 1; ycada uno de Ri y R 2 se selecciona independientemente entre hidrógeno, hidroxi, alquilO(ci-i 2 ), alquilo sustituidO(ci-i2), alquenilo(C2-12), alquenilo sustituido(C2-12), alquinilo(C2-12), alquinilo sustituido(C2-12), arilo(C6-12), arilo sustituido(C6-12), aralquilo(C7-12), aralquilo sustituido(C7-12), heteroarilo(C1-12), heteroarilo sustituido(C1-12), heterocicloalquilo(C2-12), heterocicloalquilo sustituido(C2-12), acilo(C1-12), acilo sustituido(C1-12), aciloxi(C1-12), aciloxi sustituido(C1-12), alquilamino(C1-12), alquilamino sustituido(C1-12); un grupo protector de amina monovalente, -C(O)O(CH 2 )nS-A 1 , -C(O)O(CH 2 )nS(O)-A 1 o -C(O)O(CH 2 )nS(O) 2 -A 1 ,en donde:A 1 es arilo(C 6 - 12 ) o arilo sustituido(C 6 - 12 ); yn es 1, 2, 3, 4 o 5; oRi y R 2 se toman juntos y son un grupo protector de amina divalente;R 3 es hidrógeno, hidroxi, halo o alcoxi(C 1 - 12 ) o una versión sustituida de este grupo;o es 1 o 2;R 4 es hidrógeno, alquilo(C 1 - 12 ), un grupo protector de amina monovalente o alquilo sustituido(C 1 - 12 );cada uno de R 5 , R6 y R 7 es independientemente hidrógeno, hidroxi, amino, mercapto, -OX 1 , -NX 2 X 3 o -SX 4 ; o alquilo(C 1 - 12 ) o una versión sustituida de cualquiera de estos grupos;en donde:X 1 es un grupo protector de hidroxi;X 2 y X 3 se seleccionan independientemente entre hidrógeno, un grupo protector de amina monovalente, o cuando X 2 y X 3 se toman juntos forman un grupo protector de amina divalente;X 4 es un grupo protector de tiol;R8 es hidroxi, amino o mercapto;con una sal de paladio en presencia de una base en condiciones suficientes para provocar una reacción para formar un compuesto de la fórmula: en donde:Y 1 es -(CH 2 )mNR 1 R 2 ;en donde:m es 1; ycada uno de Ri y R 2 se selecciona independientemente entre hidrógeno, hidroxi, alquiÍ 0 (oi-i 2 ), alquilo sustituidO(ci-i 2 ), alquenilo(C 2 -i 2 ), alquenilo sustituido(C 2 -i 2 ), alquinilo(C 2 -i 2 ), alquinilo sustituido(C 2 -i 2 ), arilo(C 6 -i 2 ), arilo sustituido(C 6 -i 2 ), aralquilo(C7-i2), aralquilo sustituido(C7-i2), heteroarilo(ci-i 2 ), heteroarilo sustituido(ci-i 2 ), heterocidoalquilo(C 2 -i 2 ), heterocicloalquilo sustituido(C 2 -i 2 ), acilo(ci-i 2 ), acilo sustituido(ci-i 2 ), aciloxi(ci-i 2 ), aciloxi sustituido(ci-i 2 ), alquilamino(ci-i 2 ), alquilamino sustituido(ci-i 2 ), un grupo protector de amina monovalente, -C(O)O(CH 2 )nS-Ai, -C(O)O(CH 2 )nS(O)-Ai o -C(O)O(CH 2 )nS(O) 2 -Ai,en donde:Ai es arilo(C 6 -i 2 ) o arilo sustituido(C 6 -i 2 ) yn es i, 2, 3, 4 o 5; oRi y R 2 se toman juntos y son un grupo protector de amina divalente;oR 3 es hidrógeno, hidroxi, halo o alcoxi(Ci-i 2 ) o una versión sustituida de este grupo;o es i o 2;cada uno de R 5 , R6 y R 7 es independientemente hidrógeno, hidroxi, amino, mercapto, -OXi, -NX 2 X 3 o -SX 4 ; o alquilo(Ci-i2) o una versión sustituida de cualquiera de este grupo;en donde:Xi es un grupo protector de hidroxi;X 2 y X 3 se seleccionan independientemente entre hidrógeno, un grupo protector de amina monovalente, o cuandoX 2 y X 3 se toman juntos forman un grupo protector de amina divalente; yX 4 es un grupo protector de tiol;R8 es hidroxi, amino o mercapto;opcionalmente en donde la sal de paladio comprende adicionalmente un complejo de paladio y fosfina o una sal de paladio en presencia de un ligando u otras sales o complejos de metales,tales como Pd(PPh 3 ) 4 ,en donde la base es trietilamina, diisopropiletilamina, etilendiamina, i,8-diazabiciclo[5.4.0]undec-7-eno, o morfolina, opcionalmenteen donde la base es morfolina, oen donde el disolvente es tetrahidrofurano, oel método comprende adicionalmente retirar uno o más grupos protectores.i0. Un conjugado de la fórmula:(A5-L)r-A6 (XVII)en donde:A 5 es un compuesto de la reivindicación i;L es un enlazador;r es i, 2, 3, 4, 5, 6, 7, 8, 9, i0, i i o i2; yA6 es un resto de direccionamiento celular.11. El conjugado de la reivindicación i0, en donde L comprende un polipéptido escindible por una enzima intracelular, opcionalmente, en donde la enzima es catepsina B, oen donde L comprende un grupo autoinmolante.12. El conjugado de la reivindicación i i , en donde A6 es un anticuerpo cuyo antígeno es un antígeno asociado a tumor, opcionalmente en donde el antígeno es mesotelina, glipicano-3 o CD70, preferiblemente en donde el anticuerpo es un anticuerpo anti-mesotelina, anti-glipicano-3 o anti-CD70.13. El conjugado de la reivindicación i0, que comprende adicionalmente una estructura de la fórmula:i i 2
en donde:Y 1 es -(CH 2 )mNR 1 R 2 ;en donde:m es 1; ycada uno de Ri y R 2 se selecciona independientemente entre hidrógeno, hidroxi, alquiÍ 0 (oi-i 2 ), alquilo sustituidO(ci-i 2 ), alquenilo(C 2 -i 2 ), alquenilo sustituido(C 2 -i 2 ), alquinilo(C 2 -i 2 ), alquinilo sustituido(C 2 -i 2 ), arilo(C 6 -i 2 ), arilo sustituido(C 6 -i 2 ), aralquilo(C7-i2), aralquilo sustituido(C7-i2), heteroarilo(ci-i 2 ), heteroarilo sustituido(ci-i 2 ), heterocidoalquilo(C 2 -i 2 ), heterocicloalquilo sustituido(C 2 -i 2 ), acilo(ci-i 2 ), acilo sustituido(ci-i 2 ), aciloxi(ci-i 2 ), aciloxi sustituido(ci-i 2 ), alquilamino(ci-i 2 ), alquilamino sustituido(ci-i 2 ), un grupo protector de amina monovalente, -C(O)O(CH 2 )nS-Ai, -C(O)O(CH 2 )nS(O)-Ai o -C(O)O(CH 2 )nS(O) 2 -Ai,en donde:Ai es arilo(C 6 -i 2 ) o arilo sustituido(C 6 -i 2 ) yn es i, 2, 3, 4 o 5; oRi y R 2 se toman juntos y son un grupo protector de amina divalente;oR 3 es hidrógeno, hidroxi, halo o alcoxi(Ci-i 2 ) o una versión sustituida de este grupo;o es i o 2;cada uno de R 5 , R6 y R 7 es independientemente hidrógeno, hidroxi, amino, mercapto, -OXi, -NX 2 X 3 o -SX 4 ; o alquilo(Ci-i2) o una versión sustituida de cualquiera de este grupo;en donde:Xi es un grupo protector de hidroxi;X 2 y X 3 se seleccionan independientemente entre hidrógeno, un grupo protector de amina monovalente, o cuandoX 2 y X 3 se toman juntos forman un grupo protector de amina divalente; yX 4 es un grupo protector de tiol;R8 es hidroxi, amino o mercapto;opcionalmente en donde la sal de paladio comprende adicionalmente un complejo de paladio y fosfina o una sal de paladio en presencia de un ligando u otras sales o complejos de metales,tales como Pd(PPh 3 ) 4 ,en donde la base es trietilamina, diisopropiletilamina, etilendiamina, i,8-diazabiciclo[5.4.0]undec-7-eno, o morfolina, opcionalmenteen donde la base es morfolina, oen donde el disolvente es tetrahidrofurano, oel método comprende adicionalmente retirar uno o más grupos protectores.i0. Un conjugado de la fórmula:(A5-L)r-A6 (XVII)en donde:A 5 es un compuesto de la reivindicación i;L es un enlazador;r es i, 2, 3, 4, 5, 6, 7, 8, 9, i0, i i o i2; yA6 es un resto de direccionamiento celular.11. El conjugado de la reivindicación i0, en donde L comprende un polipéptido escindible por una enzima intracelular, opcionalmente, en donde la enzima es catepsina B, oen donde L comprende un grupo autoinmolante.12. El conjugado de la reivindicación i i , en donde A6 es un anticuerpo cuyo antígeno es un antígeno asociado a tumor, opcionalmente en donde el antígeno es mesotelina, glipicano-3 o CD70, preferiblemente en donde el anticuerpo es un anticuerpo anti-mesotelina, anti-glipicano-3 o anti-CD70.13. El conjugado de la reivindicación i0, que comprende adicionalmente una estructura de la fórmula:i i 2 en donde:A6 es un anticuerpo y r es 1, 2, 3 o 4.14. Una composición farmacéutica que comprende un compuesto de acuerdo con una cualquiera de las reivindicaciones 1 a 6 o un conjugado de acuerdo con una cualquiera de las reivindicaciones 10 a 13 y un excipiente.15. La composición farmacéutica de la reivindicación 14, en donde el compuesto se formula para administración oral, intraadiposa, intraarterial, intraarticular, intracraneal, intradérmica, intralesional, intramuscular, intranasal, intraocular, intrapericárdica, intraperitoneal, intrapleural, intraprostática, intrarrectal, intratecal, intratraqueal, intratumoral, intraumbilical, intravaginal, intravenosa, intravesicular, intravítrea, liposomal, local, mucosal, parenteral, rectal, subconjuntival, subcutánea, sublingual, tópica, transbucal, transdérmica, vaginal, mediante un catéter, mediante un lavado, mediante infusión continua, mediante infusión, mediante inhalación, mediante inyección, mediante liberación local o mediante perfusión localizada.16. El compuesto o la composición de acuerdo con una cualquiera de las reivindicaciones 1 a 7 y 10 a 15 para su uso en el tratamiento del cáncer.17. El compuesto o la composición para el uso de la reivindicación 16 en donde el cáncer es cáncer de pulmón, cáncer gástrico, cáncer de ovario, cáncer de hígado, cáncer renal o cáncer de mama,en donde el cáncer muestra uno o más marcadores tumorales, opcionalmenteen donde el marcador tumoral es mesotelina, glipicano-3 o CD70,en donde el tratamiento comprende adicionalmente administrar un segundo agente terapéutico, opcionalmente en donde el segundo agente terapéutico es un segundo agente quimioterapéutico, cirugía, una radioterapia, una terapia genética o una inmunoterapia.18. El compuesto de acuerdo con la reivindicación 7, definido adicionalmente como:
en donde:A6 es un anticuerpo y r es 1, 2, 3 o 4.14. Una composición farmacéutica que comprende un compuesto de acuerdo con una cualquiera de las reivindicaciones 1 a 6 o un conjugado de acuerdo con una cualquiera de las reivindicaciones 10 a 13 y un excipiente.15. La composición farmacéutica de la reivindicación 14, en donde el compuesto se formula para administración oral, intraadiposa, intraarterial, intraarticular, intracraneal, intradérmica, intralesional, intramuscular, intranasal, intraocular, intrapericárdica, intraperitoneal, intrapleural, intraprostática, intrarrectal, intratecal, intratraqueal, intratumoral, intraumbilical, intravaginal, intravenosa, intravesicular, intravítrea, liposomal, local, mucosal, parenteral, rectal, subconjuntival, subcutánea, sublingual, tópica, transbucal, transdérmica, vaginal, mediante un catéter, mediante un lavado, mediante infusión continua, mediante infusión, mediante inhalación, mediante inyección, mediante liberación local o mediante perfusión localizada.16. El compuesto o la composición de acuerdo con una cualquiera de las reivindicaciones 1 a 7 y 10 a 15 para su uso en el tratamiento del cáncer.17. El compuesto o la composición para el uso de la reivindicación 16 en donde el cáncer es cáncer de pulmón, cáncer gástrico, cáncer de ovario, cáncer de hígado, cáncer renal o cáncer de mama,en donde el cáncer muestra uno o más marcadores tumorales, opcionalmenteen donde el marcador tumoral es mesotelina, glipicano-3 o CD70,en donde el tratamiento comprende adicionalmente administrar un segundo agente terapéutico, opcionalmente en donde el segundo agente terapéutico es un segundo agente quimioterapéutico, cirugía, una radioterapia, una terapia genética o una inmunoterapia.18. El compuesto de acuerdo con la reivindicación 7, definido adicionalmente como: o una sal farmacéuticamente aceptable del mismo.19. El compuesto de acuerdo con la reivindicación 7, definido adicionalmente como:
o una sal farmacéuticamente aceptable del mismo.19. El compuesto de acuerdo con la reivindicación 7, definido adicionalmente como: o una sal farmacéuticamente aceptable del mismo.Ċ
o una sal farmacéuticamente aceptable del mismo.Ċ
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201361865896P | 2013-08-14 | 2013-08-14 | |
| US201361868783P | 2013-08-22 | 2013-08-22 | |
| US201461937003P | 2014-02-07 | 2014-02-07 | |
| PCT/US2014/051127 WO2015023879A1 (en) | 2013-08-14 | 2014-08-14 | Derivatives of uncialamycin, methods of synthesis and their use as antitumor agents |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2769876T3 true ES2769876T3 (es) | 2020-06-29 |
Family
ID=51453866
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES14758223T Active ES2769876T3 (es) | 2013-08-14 | 2014-08-14 | Derivados de uncialamicina, métodos de síntesis y su uso como agentes antitumorales |
Country Status (13)
| Country | Link |
|---|---|
| US (3) | US9777013B2 (es) |
| EP (1) | EP3033346B1 (es) |
| JP (2) | JP6474404B2 (es) |
| KR (1) | KR20160068738A (es) |
| CN (1) | CN105899515B (es) |
| AU (2) | AU2014306592B2 (es) |
| CA (1) | CA2921401A1 (es) |
| EA (1) | EA201690388A1 (es) |
| ES (1) | ES2769876T3 (es) |
| IL (1) | IL244084B (es) |
| MX (2) | MX368016B (es) |
| SG (2) | SG10201810067YA (es) |
| WO (1) | WO2015023879A1 (es) |
Families Citing this family (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| AU2014306592B2 (en) * | 2013-08-14 | 2019-04-04 | Bristol-Myers Squibb Company | Derivatives of uncialamycin, methods of synthesis and their use as antitumor agents |
| EP3079668A1 (en) | 2013-12-09 | 2016-10-19 | Durect Corporation | Pharmaceutically active agent complexes, polymer complexes, and compositions and methods involving the same |
| CN108884147B (zh) | 2015-09-23 | 2024-02-27 | 百时美施贵宝公司 | 结合磷脂酰肌醇蛋白聚糖3的基于纤连蛋白的支架分子 |
| CA3008678A1 (en) | 2015-12-21 | 2017-06-29 | Bristol-Myers Squibb Company | Variant antibodies for site-specific conjugation |
| US20190218294A1 (en) | 2016-09-09 | 2019-07-18 | Bristol-Myers Squibb Company | Use of an anti-pd-1 antibody in combination with an anti-mesothelin antibody in cancer treatment |
| US10738338B2 (en) | 2016-10-18 | 2020-08-11 | The Research Foundation for the State University | Method and composition for biocatalytic protein-oligonucleotide conjugation and protein-oligonucleotide conjugate |
| MX2020012674A (es) | 2018-05-29 | 2021-02-09 | Bristol Myers Squibb Co | Porciones autoinmolantes modificadas para usarse en profarmacos y conjugados y metodos de uso y fabricacion. |
| KR102861586B1 (ko) | 2018-11-30 | 2025-09-17 | 브리스톨-마이어스 스큅 컴퍼니 | 글루타민-함유 경쇄 c-말단 연장부를 포함하는 항체, 그의 접합체, 및 방법 및 용도 |
| US12478686B2 (en) | 2018-12-12 | 2025-11-25 | Bristol-Myers Squibb Company | Antibodies modified for transglutaminase conjugation, conjugates thereof, and methods and uses |
| CN109928981B (zh) * | 2019-04-19 | 2021-06-11 | 长沙天赐生物医药科技有限公司 | 洋浦霉素类似物、制备方法及其应用 |
| WO2021055306A1 (en) | 2019-09-16 | 2021-03-25 | Bristol-Myers Squibb Company | Dual capture method for analysis of antibody-drug conjugates |
| US11027253B2 (en) | 2019-09-26 | 2021-06-08 | Chevron Phillips Chemical Company Lp | Systems and methods for reducing heat exchanger fouling rate |
| CN116813627B (zh) * | 2023-06-15 | 2025-08-12 | 哈药慈航制药股份有限公司 | 一种天赐霉素类似物及其制备方法和应用 |
Family Cites Families (51)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5824311A (en) | 1987-11-30 | 1998-10-20 | Trustees Of The University Of Pennsylvania | Treatment of tumors with monoclonal antibodies against oncogene antigens |
| WO1992000762A1 (en) | 1990-07-05 | 1992-01-23 | Akzo N.V. | Receptor directed-toxin conjugates |
| US5281710A (en) | 1990-08-01 | 1994-01-25 | The Scripps Research Institute | Dynemicin analogs: synthesis, methods of preparation and use |
| CA2054821C (en) | 1990-11-05 | 2001-05-01 | Wilhelm Quittmann | Process for the production of 3-aminocrotononitrile |
| US5162330A (en) | 1990-11-05 | 1992-11-10 | Bristol-Myers Squibb Co. | Dynemicin c antibiotic, its triacetyl derivative and pharmaceutical composition containing same |
| US5801029A (en) | 1993-02-16 | 1998-09-01 | Onyx Pharmaceuticals, Inc. | Cytopathic viruses for therapy and prophylaxis of neoplasia |
| US5801005A (en) | 1993-03-17 | 1998-09-01 | University Of Washington | Immune reactivity to HER-2/neu protein for diagnosis of malignancies in which the HER-2/neu oncogene is associated |
| WO1996003124A1 (en) * | 1994-07-27 | 1996-02-08 | California Institute Of Technology | Dynemicin analogs |
| GB9506466D0 (en) | 1994-08-26 | 1995-05-17 | Prolifix Ltd | Cell cycle regulated repressor and dna element |
| US5739169A (en) | 1996-05-31 | 1998-04-14 | Procept, Incorporated | Aromatic compounds for inhibiting immune response |
| DE19742706B4 (de) | 1997-09-26 | 2013-07-25 | Pieris Proteolab Ag | Lipocalinmuteine |
| US6232287B1 (en) | 1998-03-13 | 2001-05-15 | The Burnham Institute | Molecules that home to various selected organs or tissues |
| US6528481B1 (en) | 1999-02-16 | 2003-03-04 | The Burnam Institute | NG2/HM proteoglycan-binding peptides that home to angiogenic vasculature and related methods |
| EP1212422B1 (en) | 1999-08-24 | 2007-02-21 | Medarex, Inc. | Human ctla-4 antibodies and their uses |
| US7452964B2 (en) | 2001-09-07 | 2008-11-18 | Board Of Regents, The University Of Texas System | Compositions and methods of use of targeting peptides against placenta and adipose tissues |
| US7183388B2 (en) | 2001-03-30 | 2007-02-27 | The Regents Of The University Of California | Anti-MUC-1 single chain antibodies for tumor targeting |
| US20060223114A1 (en) | 2001-04-26 | 2006-10-05 | Avidia Research Institute | Protein scaffolds and uses thereof |
| US7671010B2 (en) | 2002-08-30 | 2010-03-02 | The Board Of Regents Of The University Of Texas System | Compositions and methods of use of targeting peptides for diagnosis and therapy of human cancer |
| US8507445B2 (en) | 2001-09-07 | 2013-08-13 | Board Of Regents, The University Of Texas System | Compositions and methods of use of targeting peptides for diagnosis and therapy of human cancer |
| WO2003029462A1 (en) | 2001-09-27 | 2003-04-10 | Pieris Proteolab Ag | Muteins of human neutrophil gelatinase-associated lipocalin and related proteins |
| WO2003029471A1 (en) | 2001-09-27 | 2003-04-10 | Pieris Proteolab Ag | Muteins of apolipoprotein d |
| IL162835A0 (en) | 2002-01-09 | 2005-11-20 | Medarex Inc | Human monoclonal antibodies against cd30 |
| WO2003104418A2 (en) | 2002-06-06 | 2003-12-18 | Research Corporation Technologies, Inc. | Reconstituted polypeptides |
| MXPA06000798A (es) | 2003-07-22 | 2006-04-07 | Schering Ag | Anticuerpos de rg1 y usos de los mismos. |
| WO2005051976A2 (en) | 2003-11-20 | 2005-06-09 | Ansata Therapeutics, Inc. | Protein and peptide ligation processes and one-step purification processes |
| JP4806680B2 (ja) | 2004-05-19 | 2011-11-02 | メダレックス インコーポレイテッド | 自己犠牲リンカー及び薬剤複合体 |
| US20060234299A1 (en) | 2004-11-16 | 2006-10-19 | Avidia Research Institute | Protein scaffolds and uses thereof |
| US7892827B2 (en) | 2004-11-26 | 2011-02-22 | Pieris Ag | Compound with affinity for the cytotoxic T lymphocyte-associated antigen (CTLA-4) |
| JP2008529556A (ja) | 2005-02-18 | 2008-08-07 | メダレックス, インク. | 前立腺特異的膜抗原(psma)に対するヒトモノクローナル抗体 |
| CN101160324B (zh) | 2005-02-18 | 2013-04-24 | 米德列斯公司 | 缺乏岩藻糖残基的抗前列腺特异性膜抗原(psma)单克隆抗体 |
| DK2439273T3 (da) | 2005-05-09 | 2019-06-03 | Ono Pharmaceutical Co | Humane monoklonale antistoffer til programmeret død-1(pd-1) og fremgangsmåder til behandling af cancer ved anvendelse af anti-pd-1- antistoffer alene eller i kombination med andre immunterapeutika |
| CA2611814A1 (en) | 2005-06-20 | 2007-01-04 | Medarex, Inc. | Cd19 antibodies and their uses |
| NZ566395A (en) | 2005-09-26 | 2012-03-30 | Medarex Inc | Human monoclonal antibodies to CD70 |
| WO2007038868A2 (en) | 2005-10-03 | 2007-04-12 | The University Of British Columbia | Novel enediyne compound and uses thereof |
| WO2007067991A2 (en) | 2005-12-08 | 2007-06-14 | Medarex, Inc. | Human monoclonal antibodies to o8e |
| EA017812B1 (ru) | 2005-12-08 | 2013-03-29 | Медарекс, Инк. | Выделенное моноклональное антитело или его антигенсвязывающий участок, которые связываются с протеинтирозинкиназой 7 ( ptk7) человека, и их применение |
| JP5167155B2 (ja) | 2006-03-09 | 2013-03-21 | ザ ボード オブ リージェンツ オブ ザ ユニバーシティー オブ テキサス システム | ペプチド結合に基づく多数の細胞株のプロファイリングに関連した組成物および方法 |
| EP2032606B1 (en) | 2006-05-30 | 2013-11-27 | Genentech, Inc. | Antibodies and immunoconjugates and uses therefor |
| WO2008066583A2 (en) | 2006-06-29 | 2008-06-05 | The Board Of Trustees Of The Leland Stanford Junior University | Cell-free synthesis of proteins containing unnatural amino acids |
| JP2010502221A (ja) | 2006-09-08 | 2010-01-28 | アンブルックス,インコーポレイテッド | 脊椎動物細胞による非天然アミノ酸の部位特異的組み込み |
| US8481683B2 (en) | 2006-12-01 | 2013-07-09 | Medarex, Inc. | Human antibodies that bind CD22 and uses thereof |
| MX2009006277A (es) | 2006-12-14 | 2009-07-24 | Medarex Inc | Anticuerpos humanos que se enlazan a cd70 y usos de los mismos. |
| UY30776A1 (es) | 2006-12-21 | 2008-07-03 | Medarex Inc | Anticuerpos cd44 |
| ME02345B (me) | 2007-07-17 | 2016-08-31 | Squibb & Sons Llc | MONOKLONSKA ANTlTELA PROTIV GLIPIKANA-3 |
| ES2526355T3 (es) | 2007-10-01 | 2015-01-09 | Bristol-Myers Squibb Company | Anticuerpos humanos que se adhieren a mesotelina, y usos de los mismos |
| EP2653545A1 (en) | 2008-02-05 | 2013-10-23 | Bicycle Therapeutics Limited | Methods and compositions |
| WO2010045469A2 (en) | 2008-10-15 | 2010-04-22 | The Board Of Regents Of The University Of Texas System | Muc18 targeting peptides |
| WO2010087994A2 (en) | 2009-01-30 | 2010-08-05 | Whitehead Institute For Biomedical Research | Methods for ligation and uses thereof |
| HUE033704T2 (en) | 2012-02-13 | 2017-12-28 | Bristol Myers Squibb Co | Enediyne compounds, conjugates thereof, and uses and methods therefor |
| US8798431B2 (en) | 2012-06-01 | 2014-08-05 | Telefonaktiebolaget L M Ericsson (Publ) | Fine-grained optical shuffle interconnect topology migration |
| AU2014306592B2 (en) * | 2013-08-14 | 2019-04-04 | Bristol-Myers Squibb Company | Derivatives of uncialamycin, methods of synthesis and their use as antitumor agents |
-
2014
- 2014-08-14 AU AU2014306592A patent/AU2014306592B2/en not_active Ceased
- 2014-08-14 SG SG10201810067YA patent/SG10201810067YA/en unknown
- 2014-08-14 MX MX2016001975A patent/MX368016B/es active IP Right Grant
- 2014-08-14 JP JP2016534843A patent/JP6474404B2/ja not_active Expired - Fee Related
- 2014-08-14 SG SG11201601047SA patent/SG11201601047SA/en unknown
- 2014-08-14 CN CN201480056718.0A patent/CN105899515B/zh active Active
- 2014-08-14 EP EP14758223.3A patent/EP3033346B1/en active Active
- 2014-08-14 CA CA2921401A patent/CA2921401A1/en not_active Abandoned
- 2014-08-14 EA EA201690388A patent/EA201690388A1/ru unknown
- 2014-08-14 ES ES14758223T patent/ES2769876T3/es active Active
- 2014-08-14 WO PCT/US2014/051127 patent/WO2015023879A1/en not_active Ceased
- 2014-08-14 KR KR1020167006599A patent/KR20160068738A/ko not_active Ceased
- 2014-08-14 US US14/911,881 patent/US9777013B2/en active Active
-
2016
- 2016-02-11 IL IL244084A patent/IL244084B/en not_active IP Right Cessation
- 2016-02-12 MX MX2019010875A patent/MX2019010875A/es unknown
-
2017
- 2017-08-15 US US15/677,959 patent/US10233192B2/en active Active
-
2019
- 2019-01-29 JP JP2019013008A patent/JP2019089804A/ja active Pending
- 2019-03-18 US US16/357,109 patent/US10889590B2/en active Active
- 2019-06-07 AU AU2019204034A patent/AU2019204034A1/en not_active Abandoned
Also Published As
| Publication number | Publication date |
|---|---|
| AU2019204034A1 (en) | 2019-07-04 |
| IL244084B (en) | 2019-08-29 |
| JP2016534105A (ja) | 2016-11-04 |
| AU2014306592B2 (en) | 2019-04-04 |
| MX368016B (es) | 2019-09-13 |
| US10233192B2 (en) | 2019-03-19 |
| US9777013B2 (en) | 2017-10-03 |
| AU2014306592A1 (en) | 2016-03-10 |
| IL244084A0 (en) | 2016-04-21 |
| US20190382412A1 (en) | 2019-12-19 |
| JP2019089804A (ja) | 2019-06-13 |
| CN105899515B (zh) | 2020-01-14 |
| EP3033346B1 (en) | 2020-01-08 |
| US20160185791A1 (en) | 2016-06-30 |
| EP3033346A1 (en) | 2016-06-22 |
| JP6474404B2 (ja) | 2019-02-27 |
| CN105899515A (zh) | 2016-08-24 |
| EA201690388A1 (ru) | 2016-07-29 |
| SG11201601047SA (en) | 2016-03-30 |
| CA2921401A1 (en) | 2015-02-19 |
| US20180065976A1 (en) | 2018-03-08 |
| MX2019010875A (es) | 2019-12-19 |
| WO2015023879A1 (en) | 2015-02-19 |
| US10889590B2 (en) | 2021-01-12 |
| KR20160068738A (ko) | 2016-06-15 |
| MX2016001975A (es) | 2016-11-14 |
| SG10201810067YA (en) | 2018-12-28 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2769876T3 (es) | Derivados de uncialamicina, métodos de síntesis y su uso como agentes antitumorales | |
| JP6385525B2 (ja) | タンパク質−活性剤コンジュゲートおよびこれを調製するための方法 | |
| BR112021001503A2 (pt) | compostos de 1h-pirazolo[4,3-d]pirimidina como agonistas de receptor tipo toll 7 (tlr7) e métodos e usos dos mesmos | |
| JP2018509404A (ja) | デスアセトキシツブリシンhおよびその類似体 | |
| AU2022201000A1 (en) | Chelated PSMA inhibitors | |
| CN119136833A (zh) | 膜易位结构域及其用途 | |
| US11584754B2 (en) | Derivatives of thailanstatin A, methods of treatment and methods of synthesis thereof | |
| US10874646B2 (en) | Epothilone analogs, methods of synthesis, methods of treatment, and drug conjugates thereof | |
| US12077606B2 (en) | Cyclic peptide analogs and conjugates thereof | |
| JP2018525346A (ja) | シシジマイシンa及びその類似体の全合成 | |
| WO2023231988A1 (zh) | 一种药物连接体偶联物的制备方法及其中间体 | |
| RU2801630C2 (ru) | Коньюгаты антитело-лекарственное средство, включающие антитело против человеческого dlk1, и их применение | |
| CA3240164A1 (en) | Tripeptide linkers and methods of use thereof | |
| CN120285217A (zh) | 双特异性抗体-奥瑞他汀类药物偶联物及其用途 | |
| CN107250144A (zh) | 三草癌菌素dc‑45‑a2的全合成及三草癌菌素类似物的制备 | |
| JP2020516626A (ja) | アジリジンを含有するエポチロン類似体、合成方法、治療方法及び薬物コンジュゲート |