ES2770026T3 - Derivados de imidazotiazol e imidazopirazina como inhibidores del receptor activado por proteasa 4 (PAR4) para tratar la agregación plaquetaria - Google Patents
Derivados de imidazotiazol e imidazopirazina como inhibidores del receptor activado por proteasa 4 (PAR4) para tratar la agregación plaquetaria Download PDFInfo
- Publication number
- ES2770026T3 ES2770026T3 ES17172927T ES17172927T ES2770026T3 ES 2770026 T3 ES2770026 T3 ES 2770026T3 ES 17172927 T ES17172927 T ES 17172927T ES 17172927 T ES17172927 T ES 17172927T ES 2770026 T3 ES2770026 T3 ES 2770026T3
- Authority
- ES
- Spain
- Prior art keywords
- alkyl
- group
- halo
- nr11r12
- substituted
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 208000010110 spontaneous platelet aggregation Diseases 0.000 title description 12
- 239000003112 inhibitor Substances 0.000 title description 10
- 102000004885 Protease-activated receptor 4 Human genes 0.000 title description 8
- 108090001010 Protease-activated receptor 4 Proteins 0.000 title description 8
- UMZCLZPXPCNKML-UHFFFAOYSA-N 2h-imidazo[4,5-d][1,3]thiazole Chemical compound C1=NC2=NCSC2=N1 UMZCLZPXPCNKML-UHFFFAOYSA-N 0.000 title description 2
- 150000005235 imidazopyrazines Chemical class 0.000 title description 2
- 150000001875 compounds Chemical class 0.000 claims abstract description 283
- 125000005843 halogen group Chemical group 0.000 claims abstract description 149
- -1 (C = O) NR11R12 Chemical group 0.000 claims abstract description 127
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims abstract description 94
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 claims abstract description 73
- 239000000460 chlorine Substances 0.000 claims abstract description 67
- 150000003839 salts Chemical class 0.000 claims abstract description 63
- 125000002853 C1-C4 hydroxyalkyl group Chemical group 0.000 claims abstract description 59
- 239000012453 solvate Substances 0.000 claims abstract description 58
- 125000006570 (C5-C6) heteroaryl group Chemical group 0.000 claims abstract description 56
- 125000000217 alkyl group Chemical group 0.000 claims abstract description 53
- 229910052757 nitrogen Inorganic materials 0.000 claims abstract description 53
- 229910052739 hydrogen Inorganic materials 0.000 claims abstract description 51
- 125000004093 cyano group Chemical group *C#N 0.000 claims abstract description 44
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 claims abstract description 44
- 125000000623 heterocyclic group Chemical group 0.000 claims abstract description 40
- 229910052731 fluorine Inorganic materials 0.000 claims abstract description 38
- 229910052801 chlorine Inorganic materials 0.000 claims abstract description 37
- 125000004043 oxo group Chemical group O=* 0.000 claims abstract description 37
- 125000003545 alkoxy group Chemical group 0.000 claims abstract description 36
- 125000005913 (C3-C6) cycloalkyl group Chemical group 0.000 claims abstract description 31
- 125000004432 carbon atom Chemical group C* 0.000 claims abstract description 31
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims abstract description 31
- 229910052760 oxygen Inorganic materials 0.000 claims abstract description 29
- 125000004765 (C1-C4) haloalkyl group Chemical group 0.000 claims abstract description 28
- 125000001188 haloalkyl group Chemical group 0.000 claims abstract description 28
- 229910052717 sulfur Inorganic materials 0.000 claims abstract description 28
- 229910052736 halogen Inorganic materials 0.000 claims abstract description 27
- 150000002367 halogens Chemical class 0.000 claims abstract description 27
- 229910052799 carbon Inorganic materials 0.000 claims abstract description 25
- 239000011737 fluorine Substances 0.000 claims abstract description 25
- 125000004453 alkoxycarbonyl group Chemical group 0.000 claims abstract description 24
- 125000005842 heteroatom Chemical group 0.000 claims abstract description 24
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 claims abstract description 24
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 claims abstract description 23
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 claims abstract description 21
- 125000004663 dialkyl amino group Chemical group 0.000 claims abstract description 20
- FZWLAAWBMGSTSO-UHFFFAOYSA-N Thiazole Chemical compound C1=CSC=N1 FZWLAAWBMGSTSO-UHFFFAOYSA-N 0.000 claims abstract description 15
- 125000004414 alkyl thio group Chemical group 0.000 claims abstract description 15
- 125000004767 (C1-C4) haloalkoxy group Chemical group 0.000 claims abstract description 14
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 claims abstract description 9
- 125000006699 (C1-C3) hydroxyalkyl group Chemical group 0.000 claims abstract description 8
- 125000006163 5-membered heteroaryl group Chemical group 0.000 claims abstract description 8
- 101150020251 NR13 gene Proteins 0.000 claims abstract description 7
- JNCMHMUGTWEVOZ-UHFFFAOYSA-N F[CH]F Chemical group F[CH]F JNCMHMUGTWEVOZ-UHFFFAOYSA-N 0.000 claims abstract description 6
- 108010081348 HRT1 protein Hairy Chemical group 0.000 claims abstract description 6
- 102100021881 Hairy/enhancer-of-split related with YRPW motif protein 1 Human genes 0.000 claims abstract description 6
- ZCQWOFVYLHDMMC-UHFFFAOYSA-N Oxazole Chemical compound C1=COC=N1 ZCQWOFVYLHDMMC-UHFFFAOYSA-N 0.000 claims abstract description 6
- 125000005647 linker group Chemical group 0.000 claims abstract description 6
- 125000006656 (C2-C4) alkenyl group Chemical group 0.000 claims abstract description 5
- 125000005330 8 membered heterocyclic group Chemical group 0.000 claims abstract description 5
- 125000003236 benzoyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C(*)=O 0.000 claims abstract description 5
- 150000003852 triazoles Chemical class 0.000 claims abstract description 5
- WCPAKWJPBJAGKN-UHFFFAOYSA-N oxadiazole Chemical compound C1=CON=N1 WCPAKWJPBJAGKN-UHFFFAOYSA-N 0.000 claims abstract description 4
- VLLMWSRANPNYQX-UHFFFAOYSA-N thiadiazole Chemical compound C1=CSN=N1.C1=CSN=N1 VLLMWSRANPNYQX-UHFFFAOYSA-N 0.000 claims abstract description 4
- 125000005129 aryl carbonyl group Chemical group 0.000 claims abstract 2
- 230000009424 thromboembolic effect Effects 0.000 claims description 36
- 125000003118 aryl group Chemical group 0.000 claims description 24
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 claims description 21
- 125000000753 cycloalkyl group Chemical group 0.000 claims description 14
- 238000011282 treatment Methods 0.000 claims description 12
- 125000002768 hydroxyalkyl group Chemical group 0.000 claims description 9
- 125000005196 alkyl carbonyloxy group Chemical group 0.000 claims description 8
- 125000001153 fluoro group Chemical group F* 0.000 claims description 8
- 239000008194 pharmaceutical composition Substances 0.000 claims description 8
- 238000011321 prophylaxis Methods 0.000 claims description 8
- 239000003814 drug Substances 0.000 claims description 7
- 230000002526 effect on cardiovascular system Effects 0.000 claims description 7
- 101100054666 Streptomyces halstedii sch3 gene Proteins 0.000 claims description 5
- 229940124597 therapeutic agent Drugs 0.000 claims description 5
- 238000002560 therapeutic procedure Methods 0.000 claims description 5
- 239000003937 drug carrier Substances 0.000 claims description 4
- 210000005242 cardiac chamber Anatomy 0.000 claims description 3
- 125000001309 chloro group Chemical group Cl* 0.000 claims description 3
- 230000003836 peripheral circulation Effects 0.000 claims description 3
- 125000003341 7 membered heterocyclic group Chemical group 0.000 claims description 2
- 101100243950 Arabidopsis thaliana PIE1 gene Proteins 0.000 claims 12
- PXBRQCKWGAHEHS-UHFFFAOYSA-N dichlorodifluoromethane Chemical compound FC(F)(Cl)Cl PXBRQCKWGAHEHS-UHFFFAOYSA-N 0.000 claims 11
- 125000004356 hydroxy functional group Chemical group O* 0.000 claims 4
- 125000004466 alkoxycarbonylamino group Chemical group 0.000 claims 2
- 125000004178 (C1-C4) alkyl group Chemical group 0.000 abstract description 73
- 125000000876 trifluoromethoxy group Chemical group FC(F)(F)O* 0.000 abstract description 67
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 abstract description 64
- 125000000229 (C1-C4)alkoxy group Chemical group 0.000 abstract description 61
- 125000000041 C6-C10 aryl group Chemical group 0.000 abstract description 25
- 125000006273 (C1-C3) alkyl group Chemical group 0.000 abstract description 18
- 125000004786 difluoromethoxy group Chemical group [H]C(F)(F)O* 0.000 abstract description 17
- 125000001072 heteroaryl group Chemical group 0.000 abstract description 17
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 abstract description 13
- 125000002947 alkylene group Chemical group 0.000 abstract description 12
- 125000004429 atom Chemical group 0.000 abstract description 11
- 125000001313 C5-C10 heteroaryl group Chemical group 0.000 abstract description 7
- 125000004448 alkyl carbonyl group Chemical group 0.000 abstract description 7
- 125000004450 alkenylene group Chemical group 0.000 abstract description 6
- 125000006527 (C1-C5) alkyl group Chemical group 0.000 abstract description 5
- 125000002887 hydroxy group Chemical group [H]O* 0.000 abstract description 5
- 125000006274 (C1-C3)alkoxy group Chemical group 0.000 abstract description 4
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 abstract description 4
- 125000004966 cyanoalkyl group Chemical group 0.000 abstract description 4
- 125000004785 fluoromethoxy group Chemical group [H]C([H])(F)O* 0.000 abstract description 4
- 125000005529 alkyleneoxy group Chemical group 0.000 abstract description 2
- 125000006660 (C3-C4) halocycloalkyl group Chemical group 0.000 abstract 1
- 125000004183 alkoxy alkyl group Chemical group 0.000 abstract 1
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 343
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 284
- 238000000034 method Methods 0.000 description 248
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 244
- 239000000203 mixture Substances 0.000 description 236
- 239000000243 solution Substances 0.000 description 196
- 239000007787 solid Substances 0.000 description 191
- 238000005160 1H NMR spectroscopy Methods 0.000 description 168
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical class OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 161
- 235000019439 ethyl acetate Nutrition 0.000 description 120
- 239000012267 brine Substances 0.000 description 118
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 118
- 239000011541 reaction mixture Substances 0.000 description 102
- 229920006395 saturated elastomer Polymers 0.000 description 99
- 239000000463 material Substances 0.000 description 90
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 85
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 84
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 69
- 239000012074 organic phase Substances 0.000 description 65
- 239000003921 oil Substances 0.000 description 63
- 235000019198 oils Nutrition 0.000 description 63
- 239000000741 silica gel Substances 0.000 description 61
- 229910002027 silica gel Inorganic materials 0.000 description 61
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 60
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 60
- WBKFWQBXFREOFH-UHFFFAOYSA-N dichloromethane;ethyl acetate Chemical compound ClCCl.CCOC(C)=O WBKFWQBXFREOFH-UHFFFAOYSA-N 0.000 description 59
- 239000000047 product Substances 0.000 description 58
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 57
- 238000005481 NMR spectroscopy Methods 0.000 description 55
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical class [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 53
- 238000006243 chemical reaction Methods 0.000 description 53
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 52
- 239000012299 nitrogen atmosphere Substances 0.000 description 50
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 44
- 238000010828 elution Methods 0.000 description 43
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 40
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 39
- 230000002829 reductive effect Effects 0.000 description 39
- 239000012043 crude product Substances 0.000 description 38
- 239000011734 sodium Substances 0.000 description 36
- XUYVSAMSJCSGNW-UHFFFAOYSA-N 6-methoxy-2-(2-methoxyimidazo[2,1-b][1,3,4]thiadiazol-6-yl)-1-benzofuran-4-ol Chemical compound C1=C(OC)C=C2OC(C3=CN4N=C(SC4=N3)OC)=CC2=C1O XUYVSAMSJCSGNW-UHFFFAOYSA-N 0.000 description 35
- 238000004587 chromatography analysis Methods 0.000 description 30
- 238000003818 flash chromatography Methods 0.000 description 30
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 30
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 28
- 239000002904 solvent Substances 0.000 description 28
- 238000001816 cooling Methods 0.000 description 27
- YNAVUWVOSKDBBP-UHFFFAOYSA-N Morpholine Chemical compound C1COCCN1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 description 26
- 239000000725 suspension Substances 0.000 description 26
- 125000000437 thiazol-2-yl group Chemical group [H]C1=C([H])N=C(*)S1 0.000 description 25
- 125000001424 substituent group Chemical group 0.000 description 24
- 239000012230 colorless oil Substances 0.000 description 23
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 23
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 22
- 208000035475 disorder Diseases 0.000 description 22
- 238000003756 stirring Methods 0.000 description 21
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 20
- MZRVEZGGRBJDDB-UHFFFAOYSA-N N-Butyllithium Chemical compound [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 20
- 238000001704 evaporation Methods 0.000 description 19
- TUQOTMZNTHZOKS-UHFFFAOYSA-N tributylphosphine Chemical compound CCCCP(CCCC)CCCC TUQOTMZNTHZOKS-UHFFFAOYSA-N 0.000 description 19
- DTQVDTLACAAQTR-UHFFFAOYSA-N trifluoroacetic acid Substances OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 19
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 18
- 235000017557 sodium bicarbonate Nutrition 0.000 description 18
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 18
- WGLUMOCWFMKWIL-UHFFFAOYSA-N dichloromethane;methanol Chemical compound OC.ClCCl WGLUMOCWFMKWIL-UHFFFAOYSA-N 0.000 description 17
- 230000008020 evaporation Effects 0.000 description 17
- OQJBFFCUFALWQL-UHFFFAOYSA-N n-(piperidine-1-carbonylimino)piperidine-1-carboxamide Chemical compound C1CCCCN1C(=O)N=NC(=O)N1CCCCC1 OQJBFFCUFALWQL-UHFFFAOYSA-N 0.000 description 17
- PCHJSUWPFVWCPO-UHFFFAOYSA-N gold Chemical compound [Au] PCHJSUWPFVWCPO-UHFFFAOYSA-N 0.000 description 16
- 229910052737 gold Inorganic materials 0.000 description 16
- 239000010931 gold Substances 0.000 description 16
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 16
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 15
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid group Chemical group C(C1=CC=CC=C1)(=O)O WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 14
- 125000004433 nitrogen atom Chemical group N* 0.000 description 14
- 238000010992 reflux Methods 0.000 description 14
- 239000008346 aqueous phase Substances 0.000 description 13
- 239000000284 extract Substances 0.000 description 13
- 238000003786 synthesis reaction Methods 0.000 description 13
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 12
- 238000004440 column chromatography Methods 0.000 description 12
- 239000000706 filtrate Substances 0.000 description 12
- IKGLACJFEHSFNN-UHFFFAOYSA-N hydron;triethylazanium;trifluoride Chemical compound F.F.F.CCN(CC)CC IKGLACJFEHSFNN-UHFFFAOYSA-N 0.000 description 12
- HMVYYTRDXNKRBQ-UHFFFAOYSA-N 1,3-thiazole-4-carboxylic acid Chemical compound OC(=O)C1=CSC=N1 HMVYYTRDXNKRBQ-UHFFFAOYSA-N 0.000 description 11
- PBCJIPOGFJYBJE-UHFFFAOYSA-N acetonitrile;hydrate Chemical compound O.CC#N PBCJIPOGFJYBJE-UHFFFAOYSA-N 0.000 description 11
- 230000015572 biosynthetic process Effects 0.000 description 11
- MHYCRLGKOZWVEF-UHFFFAOYSA-N ethyl acetate;hydrate Chemical compound O.CCOC(C)=O MHYCRLGKOZWVEF-UHFFFAOYSA-N 0.000 description 11
- 239000012071 phase Substances 0.000 description 11
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 10
- 229910052796 boron Inorganic materials 0.000 description 10
- 238000010168 coupling process Methods 0.000 description 10
- ICEQLCZWZXUUIJ-UHFFFAOYSA-N decan-3-ol Chemical compound CCCCCCCC(O)CC ICEQLCZWZXUUIJ-UHFFFAOYSA-N 0.000 description 10
- 235000020357 syrup Nutrition 0.000 description 10
- 239000006188 syrup Substances 0.000 description 10
- OAMZXMDZZWGPMH-UHFFFAOYSA-N ethyl acetate;toluene Chemical compound CCOC(C)=O.CC1=CC=CC=C1 OAMZXMDZZWGPMH-UHFFFAOYSA-N 0.000 description 9
- 239000012044 organic layer Substances 0.000 description 9
- 238000002360 preparation method Methods 0.000 description 9
- 238000004809 thin layer chromatography Methods 0.000 description 9
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 8
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 8
- 125000003342 alkenyl group Chemical group 0.000 description 8
- 239000001257 hydrogen Substances 0.000 description 8
- 239000000543 intermediate Substances 0.000 description 8
- 229910000027 potassium carbonate Inorganic materials 0.000 description 8
- 239000007858 starting material Substances 0.000 description 8
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 8
- BOXRDDIXJOUZLV-UHFFFAOYSA-N 2h-imidazo[4,5-d]thiadiazole Chemical compound N1SC2=NC=NC2=N1 BOXRDDIXJOUZLV-UHFFFAOYSA-N 0.000 description 7
- 102100034741 Cyclin-dependent kinase 20 Human genes 0.000 description 7
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 7
- 239000012448 Lithium borohydride Substances 0.000 description 7
- 101500014379 Lymnaea stagnalis Ovulation hormone Proteins 0.000 description 7
- 239000002253 acid Substances 0.000 description 7
- 229910052794 bromium Inorganic materials 0.000 description 7
- 238000001035 drying Methods 0.000 description 7
- 238000004128 high performance liquid chromatography Methods 0.000 description 7
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 6
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 6
- 108090000190 Thrombin Proteins 0.000 description 6
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 6
- 150000001298 alcohols Chemical class 0.000 description 6
- JFDZBHWFFUWGJE-UHFFFAOYSA-N benzonitrile Chemical compound N#CC1=CC=CC=C1 JFDZBHWFFUWGJE-UHFFFAOYSA-N 0.000 description 6
- 238000002425 crystallisation Methods 0.000 description 6
- 230000008025 crystallization Effects 0.000 description 6
- VICYTAYPKBLQFB-UHFFFAOYSA-N ethyl 3-bromo-2-oxopropanoate Chemical compound CCOC(=O)C(=O)CBr VICYTAYPKBLQFB-UHFFFAOYSA-N 0.000 description 6
- 239000006260 foam Substances 0.000 description 6
- 125000004438 haloalkoxy group Chemical group 0.000 description 6
- CFHGBZLNZZVTAY-UHFFFAOYSA-N lawesson's reagent Chemical compound C1=CC(OC)=CC=C1P1(=S)SP(=S)(C=2C=CC(OC)=CC=2)S1 CFHGBZLNZZVTAY-UHFFFAOYSA-N 0.000 description 6
- 230000014759 maintenance of location Effects 0.000 description 6
- VNWKTOKETHGBQD-UHFFFAOYSA-N methane Natural products C VNWKTOKETHGBQD-UHFFFAOYSA-N 0.000 description 6
- YOWKNNKTQWCYNC-UHFFFAOYSA-N methyl 2-bromo-1,3-thiazole-4-carboxylate Chemical compound COC(=O)C1=CSC(Br)=N1 YOWKNNKTQWCYNC-UHFFFAOYSA-N 0.000 description 6
- 125000000896 monocarboxylic acid group Chemical group 0.000 description 6
- 125000002950 monocyclic group Chemical group 0.000 description 6
- CTSLXHKWHWQRSH-UHFFFAOYSA-N oxalyl chloride Chemical compound ClC(=O)C(Cl)=O CTSLXHKWHWQRSH-UHFFFAOYSA-N 0.000 description 6
- LMYJGUNNJIDROI-UHFFFAOYSA-N oxan-4-ol Chemical compound OC1CCOCC1 LMYJGUNNJIDROI-UHFFFAOYSA-N 0.000 description 6
- 238000000746 purification Methods 0.000 description 6
- 239000003039 volatile agent Substances 0.000 description 6
- 208000004476 Acute Coronary Syndrome Diseases 0.000 description 5
- QGJOPFRUJISHPQ-UHFFFAOYSA-N Carbon disulfide Chemical compound S=C=S QGJOPFRUJISHPQ-UHFFFAOYSA-N 0.000 description 5
- UIIMBOGNXHQVGW-DEQYMQKBSA-M Sodium bicarbonate-14C Chemical class [Na+].O[14C]([O-])=O UIIMBOGNXHQVGW-DEQYMQKBSA-M 0.000 description 5
- 208000007536 Thrombosis Diseases 0.000 description 5
- 229960000583 acetic acid Drugs 0.000 description 5
- 125000000304 alkynyl group Chemical group 0.000 description 5
- 150000001408 amides Chemical class 0.000 description 5
- 125000002619 bicyclic group Chemical group 0.000 description 5
- 235000008504 concentrate Nutrition 0.000 description 5
- 239000012141 concentrate Substances 0.000 description 5
- 230000008878 coupling Effects 0.000 description 5
- 238000005859 coupling reaction Methods 0.000 description 5
- OAYLNYINCPYISS-UHFFFAOYSA-N ethyl acetate;hexane Chemical compound CCCCCC.CCOC(C)=O OAYLNYINCPYISS-UHFFFAOYSA-N 0.000 description 5
- 238000001914 filtration Methods 0.000 description 5
- 239000007789 gas Substances 0.000 description 5
- 239000008241 heterogeneous mixture Substances 0.000 description 5
- 229910052740 iodine Inorganic materials 0.000 description 5
- 208000010125 myocardial infarction Diseases 0.000 description 5
- 239000002244 precipitate Substances 0.000 description 5
- 238000002953 preparative HPLC Methods 0.000 description 5
- 125000006239 protecting group Chemical group 0.000 description 5
- BCNZYOJHNLTNEZ-UHFFFAOYSA-N tert-butyldimethylsilyl chloride Chemical compound CC(C)(C)[Si](C)(C)Cl BCNZYOJHNLTNEZ-UHFFFAOYSA-N 0.000 description 5
- 150000003573 thiols Chemical class 0.000 description 5
- WQLGUHSUNQOJSS-UHFFFAOYSA-N (2-bromo-1,3-thiazol-4-yl)methoxy-tert-butyl-dimethylsilane Chemical compound CC(C)(C)[Si](C)(C)OCC1=CSC(Br)=N1 WQLGUHSUNQOJSS-UHFFFAOYSA-N 0.000 description 4
- IVXXMVYYCIITOP-UHFFFAOYSA-N 1-(6-methoxy-4-phenylmethoxy-1-benzofuran-2-yl)ethanone Chemical compound C=12C=C(C(C)=O)OC2=CC(OC)=CC=1OCC1=CC=CC=C1 IVXXMVYYCIITOP-UHFFFAOYSA-N 0.000 description 4
- IANQTJSKSUMEQM-UHFFFAOYSA-N 1-benzofuran Chemical compound C1=CC=C2OC=CC2=C1 IANQTJSKSUMEQM-UHFFFAOYSA-N 0.000 description 4
- GLZIACYLPINALG-UHFFFAOYSA-N 4-[4-[[6-methoxy-2-(2-methoxyimidazo[2,1-b][1,3,4]thiadiazol-6-yl)-1-benzofuran-4-yl]oxymethyl]-1,3-thiazol-2-yl]-2,6-dimethyloxan-4-ol Chemical compound N1=C2SC(OC)=NN2C=C1C(OC1=CC(OC)=C2)=CC1=C2OCC(N=1)=CSC=1C1(O)CC(C)OC(C)C1 GLZIACYLPINALG-UHFFFAOYSA-N 0.000 description 4
- COZNVYCBUNSTGU-UHFFFAOYSA-N 4-[4-[[6-methoxy-2-(2-methoxyimidazo[2,1-b][1,3,4]thiadiazol-6-yl)-1-benzofuran-4-yl]oxymethyl]-5-methyl-1,3-thiazol-2-yl]oxan-4-ol Chemical compound N1=C2SC(OC)=NN2C=C1C(OC1=CC(OC)=C2)=CC1=C2OCC(=C(S1)C)N=C1C1(O)CCOCC1 COZNVYCBUNSTGU-UHFFFAOYSA-N 0.000 description 4
- USFZMSVCRYTOJT-UHFFFAOYSA-N Ammonium acetate Chemical compound N.CC(O)=O USFZMSVCRYTOJT-UHFFFAOYSA-N 0.000 description 4
- 239000005695 Ammonium acetate Substances 0.000 description 4
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 4
- OAKJQQAXSVQMHS-UHFFFAOYSA-N Hydrazine Chemical compound NN OAKJQQAXSVQMHS-UHFFFAOYSA-N 0.000 description 4
- PCLIMKBDDGJMGD-UHFFFAOYSA-N N-bromosuccinimide Chemical compound BrN1C(=O)CCC1=O PCLIMKBDDGJMGD-UHFFFAOYSA-N 0.000 description 4
- JRNVZBWKYDBUCA-UHFFFAOYSA-N N-chlorosuccinimide Chemical compound ClN1C(=O)CCC1=O JRNVZBWKYDBUCA-UHFFFAOYSA-N 0.000 description 4
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 4
- 230000002378 acidificating effect Effects 0.000 description 4
- 150000001299 aldehydes Chemical class 0.000 description 4
- 125000005236 alkanoylamino group Chemical group 0.000 description 4
- 150000001412 amines Chemical class 0.000 description 4
- 235000019257 ammonium acetate Nutrition 0.000 description 4
- 229940043376 ammonium acetate Drugs 0.000 description 4
- 230000002785 anti-thrombosis Effects 0.000 description 4
- 239000003146 anticoagulant agent Substances 0.000 description 4
- 125000003710 aryl alkyl group Chemical group 0.000 description 4
- 125000004658 aryl carbonyl amino group Chemical group 0.000 description 4
- 239000012298 atmosphere Substances 0.000 description 4
- 239000013058 crude material Substances 0.000 description 4
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 4
- CSJLBAMHHLJAAS-UHFFFAOYSA-N diethylaminosulfur trifluoride Substances CCN(CC)S(F)(F)F CSJLBAMHHLJAAS-UHFFFAOYSA-N 0.000 description 4
- 238000007865 diluting Methods 0.000 description 4
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 4
- 125000001624 naphthyl group Chemical group 0.000 description 4
- JMJRYTGVHCAYCT-UHFFFAOYSA-N oxan-4-one Chemical compound O=C1CCOCC1 JMJRYTGVHCAYCT-UHFFFAOYSA-N 0.000 description 4
- BEZDDPMMPIDMGJ-UHFFFAOYSA-N pentamethylbenzene Chemical compound CC1=CC(C)=C(C)C(C)=C1C BEZDDPMMPIDMGJ-UHFFFAOYSA-N 0.000 description 4
- 239000003208 petroleum Substances 0.000 description 4
- CTRLRINCMYICJO-UHFFFAOYSA-N phenyl azide Chemical compound [N-]=[N+]=NC1=CC=CC=C1 CTRLRINCMYICJO-UHFFFAOYSA-N 0.000 description 4
- 239000000843 powder Substances 0.000 description 4
- 230000008569 process Effects 0.000 description 4
- 238000010791 quenching Methods 0.000 description 4
- 230000000171 quenching effect Effects 0.000 description 4
- 102000005962 receptors Human genes 0.000 description 4
- 108020003175 receptors Proteins 0.000 description 4
- 239000011780 sodium chloride Substances 0.000 description 4
- 239000000126 substance Substances 0.000 description 4
- SELIBVBRTKHGSC-UHFFFAOYSA-N (5-methyl-1,3-thiazol-4-yl)methanol Chemical compound CC=1SC=NC=1CO SELIBVBRTKHGSC-UHFFFAOYSA-N 0.000 description 3
- RFFLAFLAYFXFSW-UHFFFAOYSA-N 1,2-dichlorobenzene Chemical compound ClC1=CC=CC=C1Cl RFFLAFLAYFXFSW-UHFFFAOYSA-N 0.000 description 3
- NEEOAQQOLWGHRP-UHFFFAOYSA-N 2-bromo-6-(6-methoxy-4-phenylmethoxy-1-benzofuran-2-yl)imidazo[2,1-b][1,3,4]thiadiazole Chemical compound C=12C=C(C=3N=C4SC(Br)=NN4C=3)OC2=CC(OC)=CC=1OCC1=CC=CC=C1 NEEOAQQOLWGHRP-UHFFFAOYSA-N 0.000 description 3
- OMEHRAPYTUQRCX-UHFFFAOYSA-N 2-methoxy-6-[6-methoxy-4-[(2-piperidin-4-yl-1,3-thiazol-4-yl)methoxy]-1-benzofuran-2-yl]imidazo[2,1-b][1,3,4]thiadiazole Chemical compound N1=C2SC(OC)=NN2C=C1C(OC1=CC(OC)=C2)=CC1=C2OCC(N=1)=CSC=1C1CCNCC1 OMEHRAPYTUQRCX-UHFFFAOYSA-N 0.000 description 3
- DZAPDQOKMQDRNB-UHFFFAOYSA-N 2-methoxy-6-[6-methoxy-4-[[2-(4-methoxyoxan-4-yl)-1,3-thiazol-4-yl]methoxy]-1-benzofuran-2-yl]imidazo[2,1-b][1,3,4]thiadiazole Chemical compound N1=C2SC(OC)=NN2C=C1C(OC1=CC(OC)=C2)=CC1=C2OCC(N=1)=CSC=1C1(OC)CCOCC1 DZAPDQOKMQDRNB-UHFFFAOYSA-N 0.000 description 3
- XYMDLQOGVOFQLS-UHFFFAOYSA-N 3-[4-(hydroxymethyl)-1,3-thiazol-2-yl]oxolan-3-ol Chemical compound OCC1=CSC(C2(O)COCC2)=N1 XYMDLQOGVOFQLS-UHFFFAOYSA-N 0.000 description 3
- HGOGQDNZLKJKFF-UHFFFAOYSA-N 3-[4-[[6-methoxy-2-(2-methoxyimidazo[2,1-b][1,3,4]thiadiazol-6-yl)-1-benzofuran-4-yl]oxymethyl]-1,3-thiazol-2-yl]oxolan-3-ol Chemical compound N1=C2SC(OC)=NN2C=C1C(OC1=CC(OC)=C2)=CC1=C2OCC(N=1)=CSC=1C1(O)CCOC1 HGOGQDNZLKJKFF-UHFFFAOYSA-N 0.000 description 3
- XDPCNPCKDGQBAN-UHFFFAOYSA-N 3-hydroxytetrahydrofuran Chemical compound OC1CCOC1 XDPCNPCKDGQBAN-UHFFFAOYSA-N 0.000 description 3
- OQJNRFJWBROGBB-UHFFFAOYSA-N 4,4-difluorocyclohexane-1-carbothioamide Chemical compound NC(=S)C1CCC(F)(F)CC1 OQJNRFJWBROGBB-UHFFFAOYSA-N 0.000 description 3
- JKPXYSGFTYLPTJ-UHFFFAOYSA-N 4,4-difluorocyclohexane-1-carboxamide Chemical compound NC(=O)C1CCC(F)(F)CC1 JKPXYSGFTYLPTJ-UHFFFAOYSA-N 0.000 description 3
- YZNZCTLWUSVEOJ-UHFFFAOYSA-N 4-(4-methyl-1,3-thiazol-2-yl)oxan-4-ol Chemical compound CC1=CSC(C2(O)CCOCC2)=N1 YZNZCTLWUSVEOJ-UHFFFAOYSA-N 0.000 description 3
- KDCFTJFFLNOYLJ-UHFFFAOYSA-N 4-[4-(hydroxymethyl)-1,3-thiazol-2-yl]-2,6-dimethyloxan-4-ol Chemical compound C1C(C)OC(C)CC1(O)C1=NC(CO)=CS1 KDCFTJFFLNOYLJ-UHFFFAOYSA-N 0.000 description 3
- SNPFOJFOWDTKML-UHFFFAOYSA-N 4-[4-(hydroxymethyl)-1,3-thiazol-2-yl]oxan-4-ol Chemical compound OCC1=CSC(C2(O)CCOCC2)=N1 SNPFOJFOWDTKML-UHFFFAOYSA-N 0.000 description 3
- YMYNOMMNUGLKOT-UHFFFAOYSA-N 4-[4-(hydroxymethyl)-5-methyl-1,3-thiazol-2-yl]oxan-4-ol Chemical compound OCC1=C(C)SC(C2(O)CCOCC2)=N1 YMYNOMMNUGLKOT-UHFFFAOYSA-N 0.000 description 3
- CXAXQTROOVVLBS-UHFFFAOYSA-N 4-[4-[[6-methoxy-2-(2-methoxyimidazo[2,1-b][1,3,4]thiadiazol-6-yl)-1-benzofuran-4-yl]oxymethyl]-1,3-thiazol-2-yl]oxan-4-ol Chemical compound N1=C2SC(OC)=NN2C=C1C(OC1=CC(OC)=C2)=CC1=C2OCC(N=1)=CSC=1C1(O)CCOCC1 CXAXQTROOVVLBS-UHFFFAOYSA-N 0.000 description 3
- PDEMTZGSAVDSEZ-UHFFFAOYSA-N 4-[4-[[tert-butyl(dimethyl)silyl]oxymethyl]-1,3-thiazol-2-yl]-2,6-dimethyloxan-4-ol Chemical compound C1C(C)OC(C)CC1(O)C1=NC(CO[Si](C)(C)C(C)(C)C)=CS1 PDEMTZGSAVDSEZ-UHFFFAOYSA-N 0.000 description 3
- KVCQTKNUUQOELD-UHFFFAOYSA-N 4-amino-n-[1-(3-chloro-2-fluoroanilino)-6-methylisoquinolin-5-yl]thieno[3,2-d]pyrimidine-7-carboxamide Chemical compound N=1C=CC2=C(NC(=O)C=3C4=NC=NC(N)=C4SC=3)C(C)=CC=C2C=1NC1=CC=CC(Cl)=C1F KVCQTKNUUQOELD-UHFFFAOYSA-N 0.000 description 3
- KELDCYKIUJVEPM-UHFFFAOYSA-N 7-methoxy-2,2-dimethyl-5-phenylmethoxy-1,3-benzodioxin-4-one Chemical compound C=12C(=O)OC(C)(C)OC2=CC(OC)=CC=1OCC1=CC=CC=C1 KELDCYKIUJVEPM-UHFFFAOYSA-N 0.000 description 3
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 3
- 206010002388 Angina unstable Diseases 0.000 description 3
- 239000005552 B01AC04 - Clopidogrel Substances 0.000 description 3
- 239000005711 Benzoic acid Substances 0.000 description 3
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 3
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 3
- 101150065749 Churc1 gene Proteins 0.000 description 3
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 3
- 239000007821 HATU Substances 0.000 description 3
- 208000032843 Hemorrhage Diseases 0.000 description 3
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 3
- 229910003827 NRaRb Inorganic materials 0.000 description 3
- 102000002020 Protease-activated receptors Human genes 0.000 description 3
- 108050009310 Protease-activated receptors Proteins 0.000 description 3
- 208000006011 Stroke Diseases 0.000 description 3
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical group [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 3
- 208000007814 Unstable Angina Diseases 0.000 description 3
- DSAOUKUZGDHUAM-UHFFFAOYSA-N [2-(4-methoxyoxan-4-yl)-1,3-thiazol-4-yl]methanol Chemical compound N=1C(CO)=CSC=1C1(OC)CCOCC1 DSAOUKUZGDHUAM-UHFFFAOYSA-N 0.000 description 3
- 125000001931 aliphatic group Chemical group 0.000 description 3
- 229960004676 antithrombotic agent Drugs 0.000 description 3
- 238000003556 assay Methods 0.000 description 3
- 239000002585 base Substances 0.000 description 3
- 125000004244 benzofuran-2-yl group Chemical group [H]C1=C(*)OC2=C([H])C([H])=C([H])C([H])=C12 0.000 description 3
- 235000010233 benzoic acid Nutrition 0.000 description 3
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 3
- 208000034158 bleeding Diseases 0.000 description 3
- 230000000740 bleeding effect Effects 0.000 description 3
- 238000009835 boiling Methods 0.000 description 3
- ZADPBFCGQRWHPN-UHFFFAOYSA-N boronic acid Chemical compound OBO ZADPBFCGQRWHPN-UHFFFAOYSA-N 0.000 description 3
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 3
- 150000001649 bromium compounds Chemical class 0.000 description 3
- 150000001721 carbon Chemical group 0.000 description 3
- 235000011089 carbon dioxide Nutrition 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- GKTWGGQPFAXNFI-HNNXBMFYSA-N clopidogrel Chemical compound C1([C@H](N2CC=3C=CSC=3CC2)C(=O)OC)=CC=CC=C1Cl GKTWGGQPFAXNFI-HNNXBMFYSA-N 0.000 description 3
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 3
- SNRCKKQHDUIRIY-UHFFFAOYSA-L cyclopenta-1,4-dien-1-yl(diphenyl)phosphane;dichloromethane;dichloropalladium;iron(2+) Chemical compound [Fe+2].ClCCl.Cl[Pd]Cl.C1=C[CH-]C(P(C=2C=CC=CC=2)C=2C=CC=CC=2)=C1.C1=C[CH-]C(P(C=2C=CC=CC=2)C=2C=CC=CC=2)=C1 SNRCKKQHDUIRIY-UHFFFAOYSA-L 0.000 description 3
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 3
- 238000010511 deprotection reaction Methods 0.000 description 3
- 238000004821 distillation Methods 0.000 description 3
- 230000000694 effects Effects 0.000 description 3
- 239000003480 eluent Substances 0.000 description 3
- UKKGDCGESAFSJY-UHFFFAOYSA-N ethyl 2-phenyl-1,3-thiazole-4-carboxylate Chemical compound CCOC(=O)C1=CSC(C=2C=CC=CC=2)=N1 UKKGDCGESAFSJY-UHFFFAOYSA-N 0.000 description 3
- XLOSEAOBCVYOAQ-UHFFFAOYSA-N ethyl 5-phenyl-1,3,4-thiadiazole-2-carboxylate Chemical compound S1C(C(=O)OCC)=NN=C1C1=CC=CC=C1 XLOSEAOBCVYOAQ-UHFFFAOYSA-N 0.000 description 3
- 238000010438 heat treatment Methods 0.000 description 3
- KDCIHNCMPUBDKT-UHFFFAOYSA-N hexane;propan-2-one Chemical compound CC(C)=O.CCCCCC KDCIHNCMPUBDKT-UHFFFAOYSA-N 0.000 description 3
- 150000002431 hydrogen Chemical class 0.000 description 3
- UVNXNSUKKOLFBM-UHFFFAOYSA-N imidazo[2,1-b][1,3,4]thiadiazole Chemical compound N1=CSC2=NC=CN21 UVNXNSUKKOLFBM-UHFFFAOYSA-N 0.000 description 3
- 150000005233 imidazopyridazines Chemical class 0.000 description 3
- 201000004332 intermediate coronary syndrome Diseases 0.000 description 3
- INQOMBQAUSQDDS-UHFFFAOYSA-N iodomethane Chemical compound IC INQOMBQAUSQDDS-UHFFFAOYSA-N 0.000 description 3
- 238000004949 mass spectrometry Methods 0.000 description 3
- VGSYPILDFPNKCS-UHFFFAOYSA-N methyl 2-morpholin-4-yl-1,3-thiazole-4-carboxylate Chemical compound COC(=O)C1=CSC(N2CCOCC2)=N1 VGSYPILDFPNKCS-UHFFFAOYSA-N 0.000 description 3
- SWCDUPGTBWYCQM-UHFFFAOYSA-N methyl 5-methyl-2-morpholin-4-yl-1,3-thiazole-4-carboxylate Chemical compound S1C(C)=C(C(=O)OC)N=C1N1CCOCC1 SWCDUPGTBWYCQM-UHFFFAOYSA-N 0.000 description 3
- 150000007522 mineralic acids Chemical class 0.000 description 3
- 125000002757 morpholinyl group Chemical group 0.000 description 3
- 231100000252 nontoxic Toxicity 0.000 description 3
- 230000003000 nontoxic effect Effects 0.000 description 3
- 125000004194 piperazin-1-yl group Chemical group [H]N1C([H])([H])C([H])([H])N(*)C([H])([H])C1([H])[H] 0.000 description 3
- 125000004482 piperidin-4-yl group Chemical group N1CCC(CC1)* 0.000 description 3
- 239000000106 platelet aggregation inhibitor Substances 0.000 description 3
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 3
- VVWRJUBEIPHGQF-UHFFFAOYSA-N propan-2-yl n-propan-2-yloxycarbonyliminocarbamate Chemical compound CC(C)OC(=O)N=NC(=O)OC(C)C VVWRJUBEIPHGQF-UHFFFAOYSA-N 0.000 description 3
- 125000004076 pyridyl group Chemical group 0.000 description 3
- 125000000714 pyrimidinyl group Chemical group 0.000 description 3
- 229910052701 rubidium Inorganic materials 0.000 description 3
- 238000010898 silica gel chromatography Methods 0.000 description 3
- 239000011593 sulfur Substances 0.000 description 3
- 125000004434 sulfur atom Chemical group 0.000 description 3
- MCENSTKCMOSOMH-UHFFFAOYSA-N tert-butyl-[[2-(4-methoxyoxan-4-yl)-1,3-thiazol-4-yl]methoxy]-dimethylsilane Chemical compound N=1C(CO[Si](C)(C)C(C)(C)C)=CSC=1C1(OC)CCOCC1 MCENSTKCMOSOMH-UHFFFAOYSA-N 0.000 description 3
- FPGGTKZVZWFYPV-UHFFFAOYSA-M tetrabutylammonium fluoride Chemical compound [F-].CCCC[N+](CCCC)(CCCC)CCCC FPGGTKZVZWFYPV-UHFFFAOYSA-M 0.000 description 3
- CXWXQJXEFPUFDZ-UHFFFAOYSA-N tetralin Chemical compound C1=CC=C2CCCCC2=C1 CXWXQJXEFPUFDZ-UHFFFAOYSA-N 0.000 description 3
- 125000004495 thiazol-4-yl group Chemical group S1C=NC(=C1)* 0.000 description 3
- 125000004568 thiomorpholinyl group Chemical group 0.000 description 3
- 229960004072 thrombin Drugs 0.000 description 3
- 238000001665 trituration Methods 0.000 description 3
- 238000005406 washing Methods 0.000 description 3
- FRPHIGOLOLSXAI-UHFFFAOYSA-N (2-bromo-1,3-thiazol-4-yl)methanol Chemical compound OCC1=CSC(Br)=N1 FRPHIGOLOLSXAI-UHFFFAOYSA-N 0.000 description 2
- XKOGEZTZTOYLNN-UHFFFAOYSA-N (2-bromo-5-propan-2-yl-1,3-thiazol-4-yl)methoxy-tert-butyl-dimethylsilane Chemical compound CC(C)C=1SC(Br)=NC=1CO[Si](C)(C)C(C)(C)C XKOGEZTZTOYLNN-UHFFFAOYSA-N 0.000 description 2
- KRCIFTBSQKDYMH-UHFFFAOYSA-N (2-methyl-1,3-thiazol-4-yl)methanol Chemical compound CC1=NC(CO)=CS1 KRCIFTBSQKDYMH-UHFFFAOYSA-N 0.000 description 2
- TXJGUOBTJLHIBX-UHFFFAOYSA-N (2-morpholin-4-yl-1,3-oxazol-4-yl)methanol Chemical compound OCC1=COC(N2CCOCC2)=N1 TXJGUOBTJLHIBX-UHFFFAOYSA-N 0.000 description 2
- QJKKFCRFBAXDQE-UHFFFAOYSA-N (2-morpholin-4-yl-1,3-thiazol-4-yl)methanol Chemical compound OCC1=CSC(N2CCOCC2)=N1 QJKKFCRFBAXDQE-UHFFFAOYSA-N 0.000 description 2
- HNFDKPSDOQRBBX-UHFFFAOYSA-N (2-phenyl-1,3-thiazol-4-yl)methanol Chemical compound OCC1=CSC(C=2C=CC=CC=2)=N1 HNFDKPSDOQRBBX-UHFFFAOYSA-N 0.000 description 2
- TYUWZKSJURGPRJ-UHFFFAOYSA-N (4-phenyl-1,3-thiazol-2-yl)methanol Chemical compound S1C(CO)=NC(C=2C=CC=CC=2)=C1 TYUWZKSJURGPRJ-UHFFFAOYSA-N 0.000 description 2
- SORMTKWJNHUEJN-UHFFFAOYSA-N (5-chloro-2-phenyl-1,3-thiazol-4-yl)methanol Chemical compound S1C(Cl)=C(CO)N=C1C1=CC=CC=C1 SORMTKWJNHUEJN-UHFFFAOYSA-N 0.000 description 2
- KDESQLPDNXUCQT-UHFFFAOYSA-N (5-phenyl-1,2,4-thiadiazol-3-yl)methanol Chemical compound OCC1=NSC(C=2C=CC=CC=2)=N1 KDESQLPDNXUCQT-UHFFFAOYSA-N 0.000 description 2
- MBIZXFATKUQOOA-UHFFFAOYSA-N 1,3,4-thiadiazole Chemical compound C1=NN=CS1 MBIZXFATKUQOOA-UHFFFAOYSA-N 0.000 description 2
- BCMCBBGGLRIHSE-UHFFFAOYSA-N 1,3-benzoxazole Chemical class C1=CC=C2OC=NC2=C1 BCMCBBGGLRIHSE-UHFFFAOYSA-N 0.000 description 2
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 2
- PUZXFMNARNVTEK-UHFFFAOYSA-N 1-(2-phenyl-1,3-thiazol-4-yl)ethanol Chemical compound CC(O)C1=CSC(C=2C=CC=CC=2)=N1 PUZXFMNARNVTEK-UHFFFAOYSA-N 0.000 description 2
- ZOOGZFPRAKXWKI-UHFFFAOYSA-N 1-(2-phenyl-1,3-thiazol-4-yl)ethanone Chemical compound CC(=O)C1=CSC(C=2C=CC=CC=2)=N1 ZOOGZFPRAKXWKI-UHFFFAOYSA-N 0.000 description 2
- DNUTZBZXLPWRJG-UHFFFAOYSA-N 1-Piperidine carboxylic acid Chemical compound OC(=O)N1CCCCC1 DNUTZBZXLPWRJG-UHFFFAOYSA-N 0.000 description 2
- ABDDQTDRAHXHOC-QMMMGPOBSA-N 1-[(7s)-5,7-dihydro-4h-thieno[2,3-c]pyran-7-yl]-n-methylmethanamine Chemical compound CNC[C@@H]1OCCC2=C1SC=C2 ABDDQTDRAHXHOC-QMMMGPOBSA-N 0.000 description 2
- DURPTKYDGMDSBL-UHFFFAOYSA-N 1-butoxybutane Chemical compound CCCCOCCCC DURPTKYDGMDSBL-UHFFFAOYSA-N 0.000 description 2
- IGMNPRJAMCOILX-UHFFFAOYSA-N 2-(2-methoxyethoxy)acetamide Chemical compound COCCOCC(N)=O IGMNPRJAMCOILX-UHFFFAOYSA-N 0.000 description 2
- NNVDFLWGCBSTPK-UHFFFAOYSA-N 2-(4-fluorooxan-4-yl)-4-methyl-1,3-thiazole Chemical compound CC1=CSC(C2(F)CCOCC2)=N1 NNVDFLWGCBSTPK-UHFFFAOYSA-N 0.000 description 2
- MYRGZTAFPPWJGI-UHFFFAOYSA-N 2-(bromomethyl)-4-phenyl-1,3-thiazole Chemical compound S1C(CBr)=NC(C=2C=CC=CC=2)=C1 MYRGZTAFPPWJGI-UHFFFAOYSA-N 0.000 description 2
- GVNVAWHJIKLAGL-UHFFFAOYSA-N 2-(cyclohexen-1-yl)cyclohexan-1-one Chemical compound O=C1CCCCC1C1=CCCCC1 GVNVAWHJIKLAGL-UHFFFAOYSA-N 0.000 description 2
- FVNCEASHFRUCEE-UHFFFAOYSA-N 2-[4-(hydroxymethyl)-2-morpholin-4-yl-1,3-thiazol-5-yl]propan-2-ol Chemical compound OCC1=C(C(C)(O)C)SC(N2CCOCC2)=N1 FVNCEASHFRUCEE-UHFFFAOYSA-N 0.000 description 2
- WILKSHHHRGQDLY-UHFFFAOYSA-N 2-[4-[4-[[6-methoxy-2-(2-methoxyimidazo[2,1-b][1,3,4]thiadiazol-6-yl)-1-benzofuran-4-yl]oxymethyl]-1,3-thiazol-2-yl]phenyl]propan-2-ol Chemical compound N1=C2SC(OC)=NN2C=C1C(OC1=CC(OC)=C2)=CC1=C2OCC(N=1)=CSC=1C1=CC=C(C(C)(C)O)C=C1 WILKSHHHRGQDLY-UHFFFAOYSA-N 0.000 description 2
- QGTIVZKGALSYFQ-UHFFFAOYSA-N 2-[4-[[6-methoxy-2-(2-methoxyimidazo[2,1-b][1,3,4]thiadiazol-6-yl)-1-benzofuran-4-yl]oxymethyl]-2-morpholin-4-yl-1,3-thiazol-5-yl]propan-2-ol Chemical compound N1=C2SC(OC)=NN2C=C1C(OC1=CC(OC)=C2)=CC1=C2OCC(=C(S1)C(C)(C)O)N=C1N1CCOCC1 QGTIVZKGALSYFQ-UHFFFAOYSA-N 0.000 description 2
- CZVJQFMVWUCDQY-UHFFFAOYSA-N 2-[4-[[tert-butyl(dimethyl)silyl]oxymethyl]-2-morpholin-4-yl-1,3-thiazol-5-yl]propan-2-ol Chemical compound S1C(C(C)(C)O)=C(CO[Si](C)(C)C(C)(C)C)N=C1N1CCOCC1 CZVJQFMVWUCDQY-UHFFFAOYSA-N 0.000 description 2
- ZMHYNNOYIQZWPD-UHFFFAOYSA-N 2-bromo-1-(6-methoxy-4-phenylmethoxy-1-benzofuran-2-yl)ethanone Chemical compound C=12C=C(C(=O)CBr)OC2=CC(OC)=CC=1OCC1=CC=CC=C1 ZMHYNNOYIQZWPD-UHFFFAOYSA-N 0.000 description 2
- SVVNEEADTVEIMO-UHFFFAOYSA-N 2-hydroxy-4-methoxy-6-phenylmethoxybenzaldehyde Chemical compound COC1=CC(O)=C(C=O)C(OCC=2C=CC=CC=2)=C1 SVVNEEADTVEIMO-UHFFFAOYSA-N 0.000 description 2
- KRDDTIMFUUMXBL-UHFFFAOYSA-N 2-methoxy-6-(6-methoxy-4-phenylmethoxy-1-benzofuran-2-yl)imidazo[2,1-b][1,3,4]thiadiazole Chemical compound N1=C2SC(OC)=NN2C=C1C(OC1=CC(OC)=C2)=CC1=C2OCC1=CC=CC=C1 KRDDTIMFUUMXBL-UHFFFAOYSA-N 0.000 description 2
- UUGPJSSDEZNVQR-UHFFFAOYSA-N 2-methoxy-6-[6-methoxy-4-[(1-phenyltriazol-4-yl)methoxy]-1-benzofuran-2-yl]imidazo[2,1-b][1,3,4]thiadiazole Chemical compound N1=C2SC(OC)=NN2C=C1C(OC1=CC(OC)=C2)=CC1=C2OCC(N=N1)=CN1C1=CC=CC=C1 UUGPJSSDEZNVQR-UHFFFAOYSA-N 0.000 description 2
- MJOYITBVGFBTNC-UHFFFAOYSA-N 2-methoxy-6-[6-methoxy-4-[(2-methyl-1,3-thiazol-4-yl)methoxy]-1-benzofuran-2-yl]imidazo[2,1-b][1,3,4]thiadiazole Chemical compound N1=C2SC(OC)=NN2C=C1C(OC1=CC(OC)=C2)=CC1=C2OCC1=CSC(C)=N1 MJOYITBVGFBTNC-UHFFFAOYSA-N 0.000 description 2
- RROGCGMHOWNSLD-UHFFFAOYSA-N 2-methoxy-6-[6-methoxy-4-[(2-phenyl-1,3-thiazol-4-yl)methoxy]-1-benzofuran-2-yl]imidazo[2,1-b][1,3,4]thiadiazole Chemical compound N1=C2SC(OC)=NN2C=C1C(OC1=CC(OC)=C2)=CC1=C2OCC(N=1)=CSC=1C1=CC=CC=C1 RROGCGMHOWNSLD-UHFFFAOYSA-N 0.000 description 2
- WMFQRGDWGQYTJQ-UHFFFAOYSA-N 2-methoxy-6-[6-methoxy-4-[(4-phenyl-1,3-thiazol-2-yl)methoxy]-1-benzofuran-2-yl]imidazo[2,1-b][1,3,4]thiadiazole Chemical compound N1=C2SC(OC)=NN2C=C1C(OC1=CC(OC)=C2)=CC1=C2OCC(SC=1)=NC=1C1=CC=CC=C1 WMFQRGDWGQYTJQ-UHFFFAOYSA-N 0.000 description 2
- WBIIJNUNSLZONO-UHFFFAOYSA-N 2-methoxy-6-[6-methoxy-4-[(5-phenyl-1,2,4-thiadiazol-3-yl)methoxy]-1-benzofuran-2-yl]imidazo[2,1-b][1,3,4]thiadiazole Chemical compound N1=C2SC(OC)=NN2C=C1C(OC1=CC(OC)=C2)=CC1=C2OCC(N=1)=NSC=1C1=CC=CC=C1 WBIIJNUNSLZONO-UHFFFAOYSA-N 0.000 description 2
- FNGDUOUXBBOESX-UHFFFAOYSA-N 2-methoxy-6-[6-methoxy-4-[(5-phenyl-1,3,4-thiadiazol-2-yl)methoxy]-1-benzofuran-2-yl]imidazo[2,1-b][1,3,4]thiadiazole Chemical compound N1=C2SC(OC)=NN2C=C1C(OC1=CC(OC)=C2)=CC1=C2OCC(S1)=NN=C1C1=CC=CC=C1 FNGDUOUXBBOESX-UHFFFAOYSA-N 0.000 description 2
- HXZKNKZHIQQUGW-UHFFFAOYSA-N 2-methoxy-6-[6-methoxy-4-[1-(2-phenyl-1,3-thiazol-4-yl)ethoxy]-1-benzofuran-2-yl]imidazo[2,1-b][1,3,4]thiadiazole Chemical compound N1=C2SC(OC)=NN2C=C1C(OC1=CC(OC)=C2)=CC1=C2OC(C)C(N=1)=CSC=1C1=CC=CC=C1 HXZKNKZHIQQUGW-UHFFFAOYSA-N 0.000 description 2
- CUQAZVNQNUWZDB-UHFFFAOYSA-N 2-methoxy-6-[6-methoxy-4-[[2-(2-methoxyethoxymethyl)-1,3-thiazol-4-yl]methoxy]-1-benzofuran-2-yl]imidazo[2,1-b][1,3,4]thiadiazole Chemical compound S1C(COCCOC)=NC(COC=2C=3C=C(OC=3C=C(OC)C=2)C=2N=C3SC(OC)=NN3C=2)=C1 CUQAZVNQNUWZDB-UHFFFAOYSA-N 0.000 description 2
- GTGOGXREKPCZFR-UHFFFAOYSA-N 2-methoxy-6-[6-methoxy-4-[[2-(4-methylsulfonylpiperazin-1-yl)-1,3-thiazol-4-yl]methoxy]-1-benzofuran-2-yl]imidazo[2,1-b][1,3,4]thiadiazole Chemical compound N1=C2SC(OC)=NN2C=C1C(OC1=CC(OC)=C2)=CC1=C2OCC(N=1)=CSC=1N1CCN(S(C)(=O)=O)CC1 GTGOGXREKPCZFR-UHFFFAOYSA-N 0.000 description 2
- KCFABGDGMFFCNI-UHFFFAOYSA-N 2-methoxy-6-[6-methoxy-4-[[2-(4-propan-2-ylsulfonylpiperazin-1-yl)-1,3-thiazol-4-yl]methoxy]-1-benzofuran-2-yl]imidazo[2,1-b][1,3,4]thiadiazole Chemical compound N1=C2SC(OC)=NN2C=C1C(OC1=CC(OC)=C2)=CC1=C2OCC(N=1)=CSC=1N1CCN(S(=O)(=O)C(C)C)CC1 KCFABGDGMFFCNI-UHFFFAOYSA-N 0.000 description 2
- NREGDMIGOWLBKQ-UHFFFAOYSA-N 2-methoxy-6-[6-methoxy-4-[[2-(trifluoromethyl)-1,3-thiazol-4-yl]methoxy]-1-benzofuran-2-yl]imidazo[2,1-b][1,3,4]thiadiazole Chemical compound N1=C2SC(OC)=NN2C=C1C(OC1=CC(OC)=C2)=CC1=C2OCC1=CSC(C(F)(F)F)=N1 NREGDMIGOWLBKQ-UHFFFAOYSA-N 0.000 description 2
- MYWNNHYCGXLFNE-HNNXBMFYSA-N 2-methoxy-6-[6-methoxy-4-[[2-[(2s)-2-(methoxymethyl)pyrrolidin-1-yl]-1,3-thiazol-4-yl]methoxy]-1-benzofuran-2-yl]imidazo[2,1-b][1,3,4]thiadiazole Chemical compound COC[C@@H]1CCCN1C1=NC(COC=2C=3C=C(OC=3C=C(OC)C=2)C=2N=C3SC(OC)=NN3C=2)=CS1 MYWNNHYCGXLFNE-HNNXBMFYSA-N 0.000 description 2
- MYJLPJGPNYYZOV-UHFFFAOYSA-N 2-methoxy-6-[6-methoxy-4-[[5-methyl-2-(oxan-4-yl)-1,3-thiazol-4-yl]methoxy]-1-benzofuran-2-yl]imidazo[2,1-b][1,3,4]thiadiazole Chemical compound N1=C2SC(OC)=NN2C=C1C(OC1=CC(OC)=C2)=CC1=C2OCC(=C(S1)C)N=C1C1CCOCC1 MYJLPJGPNYYZOV-UHFFFAOYSA-N 0.000 description 2
- IBUSLNJQKLZPNR-UHFFFAOYSA-N 2-phenyl-1,3-thiazole-4-carboxylic acid Chemical compound OC(=O)C1=CSC(C=2C=CC=CC=2)=N1 IBUSLNJQKLZPNR-UHFFFAOYSA-N 0.000 description 2
- KBDFMVFWPWXXPE-UHFFFAOYSA-N 3-[4-[[6-methoxy-2-(2-methoxyimidazo[2,1-b][1,3,4]thiadiazol-6-yl)-1-benzofuran-4-yl]oxymethyl]-1,3-oxazol-2-yl]-8-oxa-3-azabicyclo[3.2.1]octane Chemical compound C1C(O2)CCC2CN1C1=NC(COC2=C3C=C(OC3=CC(OC)=C2)C2=CN3N=C(SC3=N2)OC)=CO1 KBDFMVFWPWXXPE-UHFFFAOYSA-N 0.000 description 2
- RBNOFBYJQBWEOA-UHFFFAOYSA-N 3-[4-[[6-methoxy-2-(2-methoxyimidazo[2,1-b][1,3,4]thiadiazol-6-yl)-1-benzofuran-4-yl]oxymethyl]-1,3-thiazol-2-yl]-8-oxa-3-azabicyclo[3.2.1]octane Chemical compound C1C(O2)CCC2CN1C1=NC(COC2=C3C=C(OC3=CC(OC)=C2)C2=CN3N=C(SC3=N2)OC)=CS1 RBNOFBYJQBWEOA-UHFFFAOYSA-N 0.000 description 2
- RHAGKGYWNRLLIG-UHFFFAOYSA-N 3-[[4-[[6-methoxy-2-(2-methoxyimidazo[2,1-b][1,3,4]thiadiazol-6-yl)-1-benzofuran-4-yl]oxymethyl]-1,3-thiazol-2-yl]-methylamino]propanenitrile Chemical compound N1=C2SC(OC)=NN2C=C1C(OC1=CC(OC)=C2)=CC1=C2OCC1=CSC(N(C)CCC#N)=N1 RHAGKGYWNRLLIG-UHFFFAOYSA-N 0.000 description 2
- OYQJRVKWZJLOTM-UHFFFAOYSA-N 4-(bromomethyl)-2-(2-methoxyethoxymethyl)-1,3-thiazole Chemical compound COCCOCC1=NC(CBr)=CS1 OYQJRVKWZJLOTM-UHFFFAOYSA-N 0.000 description 2
- HRCRNFICHZTFNU-UHFFFAOYSA-N 4-(bromomethyl)-5-methyl-2-(oxan-4-yl)-1,3-thiazole Chemical compound BrCC1=C(C)SC(C2CCOCC2)=N1 HRCRNFICHZTFNU-UHFFFAOYSA-N 0.000 description 2
- BVKSWTSUANSZLA-UHFFFAOYSA-N 4-(hydroxymethyl)-2-morpholin-4-yl-1,3-thiazole-5-carbaldehyde Chemical compound S1C(C=O)=C(CO)N=C1N1CCOCC1 BVKSWTSUANSZLA-UHFFFAOYSA-N 0.000 description 2
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 2
- PHVWEVDFRGVSTE-UHFFFAOYSA-N 4-[4-(hydroxymethyl)-1,3-thiazol-2-yl]-n,n-dimethylbenzamide Chemical compound C1=CC(C(=O)N(C)C)=CC=C1C1=NC(CO)=CS1 PHVWEVDFRGVSTE-UHFFFAOYSA-N 0.000 description 2
- FOPBIADLNYYSCZ-UHFFFAOYSA-N 4-[4-[[6-methoxy-2-(2-methoxyimidazo[2,1-b][1,3,4]thiadiazol-6-yl)-1-benzofuran-4-yl]oxymethyl]-1,3-oxazol-2-yl]morpholine Chemical compound N1=C2SC(OC)=NN2C=C1C(OC1=CC(OC)=C2)=CC1=C2OCC(N=1)=COC=1N1CCOCC1 FOPBIADLNYYSCZ-UHFFFAOYSA-N 0.000 description 2
- MUGGZJLOVWOLEY-UHFFFAOYSA-N 4-[4-[[6-methoxy-2-(2-methoxyimidazo[2,1-b][1,3,4]thiadiazol-6-yl)-1-benzofuran-4-yl]oxymethyl]-1,3-thiazol-2-yl]morpholine Chemical compound N1=C2SC(OC)=NN2C=C1C(OC1=CC(OC)=C2)=CC1=C2OCC(N=1)=CSC=1N1CCOCC1 MUGGZJLOVWOLEY-UHFFFAOYSA-N 0.000 description 2
- TZDAEAYKWSPUJX-UHFFFAOYSA-N 4-[4-[[6-methoxy-2-(2-methoxyimidazo[2,1-b][1,3,4]thiadiazol-6-yl)-1-benzofuran-4-yl]oxymethyl]-5-(trifluoromethyl)-1,3-thiazol-2-yl]morpholine Chemical compound N1=C2SC(OC)=NN2C=C1C(OC1=CC(OC)=C2)=CC1=C2OCC(=C(S1)C(F)(F)F)N=C1N1CCOCC1 TZDAEAYKWSPUJX-UHFFFAOYSA-N 0.000 description 2
- XIRJAYIXTVGZIP-UHFFFAOYSA-N 4-[4-[[6-methoxy-2-(2-methoxyimidazo[2,1-b][1,3,4]thiadiazol-6-yl)-1-benzofuran-4-yl]oxymethyl]-5-methyl-1,3-oxazol-2-yl]morpholine Chemical compound N1=C2SC(OC)=NN2C=C1C(OC1=CC(OC)=C2)=CC1=C2OCC(=C(O1)C)N=C1N1CCOCC1 XIRJAYIXTVGZIP-UHFFFAOYSA-N 0.000 description 2
- MINMDCMSHDBHKG-UHFFFAOYSA-N 4-[4-[[6-methoxy-2-(2-methoxyimidazo[2,1-b][1,3,4]thiadiazol-6-yl)-1-benzofuran-4-yl]oxymethyl]-5-methyl-1,3-thiazol-2-yl]morpholine Chemical compound N1=C2SC(OC)=NN2C=C1C(OC1=CC(OC)=C2)=CC1=C2OCC(=C(S1)C)N=C1N1CCOCC1 MINMDCMSHDBHKG-UHFFFAOYSA-N 0.000 description 2
- SVVQHWZAWGZLLM-UHFFFAOYSA-N 4-[4-[[6-methoxy-2-(2-methoxyimidazo[2,1-b][1,3,4]thiadiazol-6-yl)-1-benzofuran-4-yl]oxymethyl]-5-propan-2-yl-1,3-thiazol-2-yl]-n,n-dimethylbenzamide Chemical compound N1=C2SC(OC)=NN2C=C1C(OC1=CC(OC)=C2)=CC1=C2OCC(=C(S1)C(C)C)N=C1C1=CC=C(C(=O)N(C)C)C=C1 SVVQHWZAWGZLLM-UHFFFAOYSA-N 0.000 description 2
- ZHKJUSGEGPPZTP-UHFFFAOYSA-N 4-[4-[[tert-butyl(dimethyl)silyl]oxymethyl]-1,3-thiazol-2-yl]oxan-4-ol Chemical compound CC(C)(C)[Si](C)(C)OCC1=CSC(C2(O)CCOCC2)=N1 ZHKJUSGEGPPZTP-UHFFFAOYSA-N 0.000 description 2
- BKKXFHYCELTMFP-UHFFFAOYSA-N 4-[5-ethyl-4-[[6-methoxy-2-(2-methoxyimidazo[2,1-b][1,3,4]thiadiazol-6-yl)-1-benzofuran-4-yl]oxymethyl]-1,3-thiazol-2-yl]-n,n-dimethylbenzamide Chemical compound N=1C(COC=2C=3C=C(OC=3C=C(OC)C=2)C=2N=C3SC(OC)=NN3C=2)=C(CC)SC=1C1=CC=C(C(=O)N(C)C)C=C1 BKKXFHYCELTMFP-UHFFFAOYSA-N 0.000 description 2
- YIKCGURUWNKLFF-UHFFFAOYSA-N 4-[5-ethyl-4-[[6-methoxy-2-(2-methoxyimidazo[2,1-b][1,3,4]thiadiazol-6-yl)-1-benzofuran-4-yl]oxymethyl]-1,3-thiazol-2-yl]morpholine Chemical compound N=1C(COC=2C=3C=C(OC=3C=C(OC)C=2)C=2N=C3SC(OC)=NN3C=2)=C(CC)SC=1N1CCOCC1 YIKCGURUWNKLFF-UHFFFAOYSA-N 0.000 description 2
- NDLGGJGFWMAILW-UHFFFAOYSA-N 4-[[6-methoxy-2-(2-methoxyimidazo[2,1-b][1,3,4]thiadiazol-6-yl)-1-benzofuran-4-yl]oxymethyl]-2-morpholin-4-yl-1,3-thiazole-5-carbaldehyde Chemical compound N1=C2SC(OC)=NN2C=C1C(OC1=CC(OC)=C2)=CC1=C2OCC(=C(S1)C=O)N=C1N1CCOCC1 NDLGGJGFWMAILW-UHFFFAOYSA-N 0.000 description 2
- UUDBZRMHMRVHLK-UHFFFAOYSA-N 4-[[6-methoxy-2-(2-methoxyimidazo[2,1-b][1,3,4]thiadiazol-6-yl)-1-benzofuran-4-yl]oxymethyl]-5-methyl-1,3-thiazol-2-amine Chemical compound N1=C2SC(OC)=NN2C=C1C(OC1=CC(OC)=C2)=CC1=C2OCC=1N=C(N)SC=1C UUDBZRMHMRVHLK-UHFFFAOYSA-N 0.000 description 2
- PHRYRFVFQYBEJG-UHFFFAOYSA-N 4-hydroxy-4-[4-[[6-methoxy-2-(2-methoxyimidazo[2,1-b][1,3,4]thiadiazol-6-yl)-1-benzofuran-4-yl]oxymethyl]-1,3-thiazol-2-yl]cyclohexan-1-one Chemical compound N1=C2SC(OC)=NN2C=C1C(OC1=CC(OC)=C2)=CC1=C2OCC(N=1)=CSC=1C1(O)CCC(=O)CC1 PHRYRFVFQYBEJG-UHFFFAOYSA-N 0.000 description 2
- QMHIMXFNBOYPND-UHFFFAOYSA-N 4-methylthiazole Chemical compound CC1=CSC=N1 QMHIMXFNBOYPND-UHFFFAOYSA-N 0.000 description 2
- HEYYTJUHMHNLMD-UHFFFAOYSA-N 5-[4-[[6-methoxy-2-(2-methoxyimidazo[2,1-b][1,3,4]thiadiazol-6-yl)-1-benzofuran-4-yl]oxymethyl]-1,3-thiazol-2-yl]-1,2-oxazole Chemical compound N1=C2SC(OC)=NN2C=C1C(OC1=CC(OC)=C2)=CC1=C2OCC(N=1)=CSC=1C1=CC=NO1 HEYYTJUHMHNLMD-UHFFFAOYSA-N 0.000 description 2
- FRZGXEFPIGPPJK-UHFFFAOYSA-N 5-[4-[[6-methoxy-2-(2-methoxyimidazo[2,1-b][1,3,4]thiadiazol-6-yl)-1-benzofuran-4-yl]oxymethyl]-1,3-thiazol-2-yl]-1,3-oxazole Chemical compound N1=C2SC(OC)=NN2C=C1C(OC1=CC(OC)=C2)=CC1=C2OCC(N=1)=CSC=1C1=CN=CO1 FRZGXEFPIGPPJK-UHFFFAOYSA-N 0.000 description 2
- GLYQQFBHCFPEEU-UHFFFAOYSA-N 5-bromo-1,3,4-thiadiazol-2-amine Chemical compound NC1=NN=C(Br)S1 GLYQQFBHCFPEEU-UHFFFAOYSA-N 0.000 description 2
- DEABRQRGKCVNEK-UHFFFAOYSA-N 6-[4-[[2-(2-fluoropyridin-4-yl)-5-propan-2-yl-1,3-thiazol-4-yl]methoxy]-6-methoxy-1-benzofuran-2-yl]-2-methoxyimidazo[2,1-b][1,3,4]thiadiazole Chemical compound N1=C2SC(OC)=NN2C=C1C(OC1=CC(OC)=C2)=CC1=C2OCC(=C(S1)C(C)C)N=C1C1=CC=NC(F)=C1 DEABRQRGKCVNEK-UHFFFAOYSA-N 0.000 description 2
- WIJXQXSKKBTTEU-UHFFFAOYSA-N 6-[4-[[2-(4,4-difluorocyclohexyl)-1,3-thiazol-4-yl]methoxy]-6-methoxy-1-benzofuran-2-yl]-2-methoxyimidazo[2,1-b][1,3,4]thiadiazole Chemical compound N1=C2SC(OC)=NN2C=C1C(OC1=CC(OC)=C2)=CC1=C2OCC(N=1)=CSC=1C1CCC(F)(F)CC1 WIJXQXSKKBTTEU-UHFFFAOYSA-N 0.000 description 2
- FQSQEOZFKDUYMF-UHFFFAOYSA-N 6-[4-[[2-(4-fluoro-2,6-dimethyloxan-4-yl)-1,3-thiazol-4-yl]methoxy]-6-methoxy-1-benzofuran-2-yl]-2-methoxyimidazo[2,1-b][1,3,4]thiadiazole Chemical compound N1=C2SC(OC)=NN2C=C1C(OC1=CC(OC)=C2)=CC1=C2OCC(N=1)=CSC=1C1(F)CC(C)OC(C)C1 FQSQEOZFKDUYMF-UHFFFAOYSA-N 0.000 description 2
- XJVQUSDPXYCBGA-UHFFFAOYSA-N 6-[4-[[2-(4-fluorooxan-4-yl)-1,3-thiazol-4-yl]methoxy]-6-methoxy-1-benzofuran-2-yl]-2-methoxyimidazo[2,1-b][1,3,4]thiadiazole Chemical compound N1=C2SC(OC)=NN2C=C1C(OC1=CC(OC)=C2)=CC1=C2OCC(N=1)=CSC=1C1(F)CCOCC1 XJVQUSDPXYCBGA-UHFFFAOYSA-N 0.000 description 2
- ZFYDPZVRBWBDPL-UHFFFAOYSA-N 6-[4-[[2-(4-fluorooxan-4-yl)-5-methyl-1,3-thiazol-4-yl]methoxy]-6-methoxy-1-benzofuran-2-yl]-2-methoxyimidazo[2,1-b][1,3,4]thiadiazole Chemical compound N1=C2SC(OC)=NN2C=C1C(OC1=CC(OC)=C2)=CC1=C2OCC(=C(S1)C)N=C1C1(F)CCOCC1 ZFYDPZVRBWBDPL-UHFFFAOYSA-N 0.000 description 2
- YPODSXMAZNBCFX-UHFFFAOYSA-N 6-[4-[[6-methoxy-2-(2-methoxyimidazo[2,1-b][1,3,4]thiadiazol-6-yl)-1-benzofuran-4-yl]oxymethyl]-1,3-thiazol-2-yl]-2-oxa-6-azaspiro[3.3]heptane Chemical compound N1=C2SC(OC)=NN2C=C1C(OC1=CC(OC)=C2)=CC1=C2OCC(N=1)=CSC=1N(C1)CC21COC2 YPODSXMAZNBCFX-UHFFFAOYSA-N 0.000 description 2
- XJLDYKIEURAVBW-UHFFFAOYSA-N Aethyl-heptyl-keton Natural products CCCCCCCC(=O)CC XJLDYKIEURAVBW-UHFFFAOYSA-N 0.000 description 2
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 2
- PAYRUJLWNCNPSJ-UHFFFAOYSA-N Aniline Chemical compound NC1=CC=CC=C1 PAYRUJLWNCNPSJ-UHFFFAOYSA-N 0.000 description 2
- BSYNRYMUTXBXSQ-UHFFFAOYSA-N Aspirin Chemical compound CC(=O)OC1=CC=CC=C1C(O)=O BSYNRYMUTXBXSQ-UHFFFAOYSA-N 0.000 description 2
- 201000001320 Atherosclerosis Diseases 0.000 description 2
- 206010003658 Atrial Fibrillation Diseases 0.000 description 2
- 125000000882 C2-C6 alkenyl group Chemical group 0.000 description 2
- 206010008088 Cerebral artery embolism Diseases 0.000 description 2
- 206010008092 Cerebral artery thrombosis Diseases 0.000 description 2
- 206010011091 Coronary artery thrombosis Diseases 0.000 description 2
- 206010014513 Embolism arterial Diseases 0.000 description 2
- 240000001414 Eucalyptus viminalis Species 0.000 description 2
- 101001098529 Homo sapiens Proteinase-activated receptor 1 Proteins 0.000 description 2
- 101000713169 Homo sapiens Solute carrier family 52, riboflavin transporter, member 2 Proteins 0.000 description 2
- 208000032382 Ischaemic stroke Diseases 0.000 description 2
- 150000001204 N-oxides Chemical class 0.000 description 2
- 208000005764 Peripheral Arterial Disease Diseases 0.000 description 2
- 208000030831 Peripheral arterial occlusive disease Diseases 0.000 description 2
- 229940123645 Protease-activated receptor-4 antagonist Drugs 0.000 description 2
- 102100038239 Protein Churchill Human genes 0.000 description 2
- 102100037136 Proteinase-activated receptor 1 Human genes 0.000 description 2
- 208000010378 Pulmonary Embolism Diseases 0.000 description 2
- 206010063544 Renal embolism Diseases 0.000 description 2
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 2
- WQDUMFSSJAZKTM-UHFFFAOYSA-N Sodium methoxide Chemical compound [Na+].[O-]C WQDUMFSSJAZKTM-UHFFFAOYSA-N 0.000 description 2
- QIOZLISABUUKJY-UHFFFAOYSA-N Thiobenzamide Chemical compound NC(=S)C1=CC=CC=C1 QIOZLISABUUKJY-UHFFFAOYSA-N 0.000 description 2
- 229940122388 Thrombin inhibitor Drugs 0.000 description 2
- OKJPEAGHQZHRQV-UHFFFAOYSA-N Triiodomethane Natural products IC(I)I OKJPEAGHQZHRQV-UHFFFAOYSA-N 0.000 description 2
- QCIPDKSLARBRND-UHFFFAOYSA-N [2-(1,2-oxazol-5-yl)-1,3-thiazol-4-yl]methanol Chemical compound OCC1=CSC(C=2ON=CC=2)=N1 QCIPDKSLARBRND-UHFFFAOYSA-N 0.000 description 2
- LTBVSFOZNYAENN-UHFFFAOYSA-N [2-(1,3-oxazol-5-yl)-1,3-thiazol-4-yl]methanol Chemical compound OCC1=CSC(C=2OC=NC=2)=N1 LTBVSFOZNYAENN-UHFFFAOYSA-N 0.000 description 2
- JTRKWLWMIHKNMV-UHFFFAOYSA-N [2-(2-methoxyethoxymethyl)-1,3-thiazol-4-yl]methanol Chemical compound COCCOCC1=NC(CO)=CS1 JTRKWLWMIHKNMV-UHFFFAOYSA-N 0.000 description 2
- MRNKITUOBJYNPG-UHFFFAOYSA-N [2-(4,4-difluorocyclohexyl)-1,3-thiazol-4-yl]methanol Chemical compound OCC1=CSC(C2CCC(F)(F)CC2)=N1 MRNKITUOBJYNPG-UHFFFAOYSA-N 0.000 description 2
- BPQZYDHLJFDYQD-UHFFFAOYSA-N [2-(8-oxa-3-azabicyclo[3.2.1]octan-3-yl)-1,3-thiazol-4-yl]methanol Chemical compound OCC1=CSC(N2CC3CCC(O3)C2)=N1 BPQZYDHLJFDYQD-UHFFFAOYSA-N 0.000 description 2
- NGTOSARTLLCFTQ-UHFFFAOYSA-N [2-(trifluoromethyl)-1,3-thiazol-4-yl]methanol Chemical compound OCC1=CSC(C(F)(F)F)=N1 NGTOSARTLLCFTQ-UHFFFAOYSA-N 0.000 description 2
- XANYMJZJBACOHU-UHFFFAOYSA-N [5-(trifluoromethyl)-1,3-thiazol-4-yl]methanol Chemical compound OCC=1N=CSC=1C(F)(F)F XANYMJZJBACOHU-UHFFFAOYSA-N 0.000 description 2
- MCYDCHRKQWQFKG-UHFFFAOYSA-N [5-methyl-2-(8-oxa-3-azabicyclo[3.2.1]octan-3-yl)-1,3-thiazol-4-yl]methanol Chemical compound OCC1=C(C)SC(N2CC3CCC(O3)C2)=N1 MCYDCHRKQWQFKG-UHFFFAOYSA-N 0.000 description 2
- OXWQZJQRWNZFDT-UHFFFAOYSA-N [5-methyl-2-(oxan-4-yl)-1,3-thiazol-4-yl]methanol Chemical compound OCC1=C(C)SC(C2CCOCC2)=N1 OXWQZJQRWNZFDT-UHFFFAOYSA-N 0.000 description 2
- CKUAXEQHGKSLHN-UHFFFAOYSA-N [C].[N] Chemical group [C].[N] CKUAXEQHGKSLHN-UHFFFAOYSA-N 0.000 description 2
- 229960001138 acetylsalicylic acid Drugs 0.000 description 2
- 230000004913 activation Effects 0.000 description 2
- 206010000891 acute myocardial infarction Diseases 0.000 description 2
- 125000002252 acyl group Chemical group 0.000 description 2
- 125000005073 adamantyl group Chemical group C12(CC3CC(CC(C1)C3)C2)* 0.000 description 2
- 230000029936 alkylation Effects 0.000 description 2
- 238000005804 alkylation reaction Methods 0.000 description 2
- 125000004419 alkynylene group Chemical group 0.000 description 2
- 239000005557 antagonist Substances 0.000 description 2
- 229940127219 anticoagulant drug Drugs 0.000 description 2
- 125000004104 aryloxy group Chemical group 0.000 description 2
- 230000008901 benefit Effects 0.000 description 2
- 210000004369 blood Anatomy 0.000 description 2
- 239000008280 blood Substances 0.000 description 2
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 description 2
- 229910000024 caesium carbonate Inorganic materials 0.000 description 2
- 239000001110 calcium chloride Substances 0.000 description 2
- 229910001628 calcium chloride Inorganic materials 0.000 description 2
- 239000011203 carbon fibre reinforced carbon Substances 0.000 description 2
- MOIPGXQKZSZOQX-UHFFFAOYSA-N carbonyl bromide Chemical class BrC(Br)=O MOIPGXQKZSZOQX-UHFFFAOYSA-N 0.000 description 2
- 239000003153 chemical reaction reagent Substances 0.000 description 2
- BULLHNJGPPOUOX-UHFFFAOYSA-N chloroacetone Chemical compound CC(=O)CCl BULLHNJGPPOUOX-UHFFFAOYSA-N 0.000 description 2
- IJOOHPMOJXWVHK-UHFFFAOYSA-N chlorotrimethylsilane Chemical compound C[Si](C)(C)Cl IJOOHPMOJXWVHK-UHFFFAOYSA-N 0.000 description 2
- 229960003009 clopidogrel Drugs 0.000 description 2
- 238000009833 condensation Methods 0.000 description 2
- 230000005494 condensation Effects 0.000 description 2
- QTMDXZNDVAMKGV-UHFFFAOYSA-L copper(ii) bromide Chemical compound [Cu+2].[Br-].[Br-] QTMDXZNDVAMKGV-UHFFFAOYSA-L 0.000 description 2
- 208000002528 coronary thrombosis Diseases 0.000 description 2
- 150000001907 coumarones Chemical class 0.000 description 2
- 239000013078 crystal Substances 0.000 description 2
- 125000001162 cycloheptenyl group Chemical group C1(=CCCCCC1)* 0.000 description 2
- 125000000582 cycloheptyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 2
- 125000000640 cyclooctyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])C1([H])[H] 0.000 description 2
- NLUNLVTVUDIHFE-UHFFFAOYSA-N cyclooctylcyclooctane Chemical compound C1CCCCCCC1C1CCCCCCC1 NLUNLVTVUDIHFE-UHFFFAOYSA-N 0.000 description 2
- NPOMSUOUAZCMBL-UHFFFAOYSA-N dichloromethane;ethoxyethane Chemical compound ClCCl.CCOCC NPOMSUOUAZCMBL-UHFFFAOYSA-N 0.000 description 2
- YDVNLQGCLLPHAH-UHFFFAOYSA-N dichloromethane;hydrate Chemical compound O.ClCCl YDVNLQGCLLPHAH-UHFFFAOYSA-N 0.000 description 2
- 150000002148 esters Chemical class 0.000 description 2
- YMBMCMOZIGSBOA-UHFFFAOYSA-N ethyl 2-amino-2-sulfanylideneacetate Chemical compound CCOC(=O)C(N)=S YMBMCMOZIGSBOA-UHFFFAOYSA-N 0.000 description 2
- KTNLDLYUFMOCFN-UHFFFAOYSA-N ethyl 2-amino-5-iodo-1,3-thiazole-4-carboxylate Chemical compound CCOC(=O)C=1N=C(N)SC=1I KTNLDLYUFMOCFN-UHFFFAOYSA-N 0.000 description 2
- GQKNRLWNPUZFFG-UHFFFAOYSA-N ethyl 2-morpholin-4-yl-1,3-oxazole-4-carboxylate Chemical compound CCOC(=O)C1=COC(N2CCOCC2)=N1 GQKNRLWNPUZFFG-UHFFFAOYSA-N 0.000 description 2
- OINWWRRVBHJAKO-UHFFFAOYSA-N ethyl 2-morpholin-4-yl-1,3-thiazole-4-carboxylate Chemical compound CCOC(=O)C1=CSC(N2CCOCC2)=N1 OINWWRRVBHJAKO-UHFFFAOYSA-N 0.000 description 2
- GWFISTWSEPVSDU-UHFFFAOYSA-N ethyl 4-phenyl-1,3-thiazole-2-carboxylate Chemical compound S1C(C(=O)OCC)=NC(C=2C=CC=CC=2)=C1 GWFISTWSEPVSDU-UHFFFAOYSA-N 0.000 description 2
- MXQXSYXBIGLSQQ-UHFFFAOYSA-N ethyl 5-chloro-2-phenyl-1,3-thiazole-4-carboxylate Chemical compound S1C(Cl)=C(C(=O)OCC)N=C1C1=CC=CC=C1 MXQXSYXBIGLSQQ-UHFFFAOYSA-N 0.000 description 2
- XMDZWPSEQYJMHL-UHFFFAOYSA-N ethyl 5-iodo-1,3-thiazole-4-carboxylate Chemical compound CCOC(=O)C=1N=CSC=1I XMDZWPSEQYJMHL-UHFFFAOYSA-N 0.000 description 2
- SEAHRCSDOPUOKR-UHFFFAOYSA-N ethyl 5-phenyl-1,2,4-thiadiazole-3-carboxylate Chemical compound CCOC(=O)C1=NSC(C=2C=CC=CC=2)=N1 SEAHRCSDOPUOKR-UHFFFAOYSA-N 0.000 description 2
- 239000012065 filter cake Substances 0.000 description 2
- 150000002430 hydrocarbons Chemical group 0.000 description 2
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 2
- 125000001841 imino group Chemical group [H]N=* 0.000 description 2
- 239000007943 implant Substances 0.000 description 2
- 125000003392 indanyl group Chemical group C1(CCC2=CC=CC=C12)* 0.000 description 2
- 230000005764 inhibitory process Effects 0.000 description 2
- 201000010849 intracranial embolism Diseases 0.000 description 2
- 125000000468 ketone group Chemical group 0.000 description 2
- 150000002576 ketones Chemical class 0.000 description 2
- 239000003446 ligand Substances 0.000 description 2
- 239000012280 lithium aluminium hydride Substances 0.000 description 2
- DLEDOFVPSDKWEF-UHFFFAOYSA-N lithium butane Chemical compound [Li+].CCC[CH2-] DLEDOFVPSDKWEF-UHFFFAOYSA-N 0.000 description 2
- 229940127215 low-molecular weight heparin Drugs 0.000 description 2
- VGWOUTBYGICCLO-UHFFFAOYSA-N methyl 2-phenyl-1,3-thiazole-4-carboxylate Chemical compound COC(=O)C1=CSC(C=2C=CC=CC=2)=N1 VGWOUTBYGICCLO-UHFFFAOYSA-N 0.000 description 2
- HYDCQMJFDPQCJH-UHFFFAOYSA-N methyl 3-bromo-2-oxobutanoate Chemical compound COC(=O)C(=O)C(C)Br HYDCQMJFDPQCJH-UHFFFAOYSA-N 0.000 description 2
- KFHFEMYPMZRFDV-UHFFFAOYSA-N methyl 5-ethyl-2-morpholin-4-yl-1,3-thiazole-4-carboxylate Chemical compound COC(=O)C1=C(CC)SC(N2CCOCC2)=N1 KFHFEMYPMZRFDV-UHFFFAOYSA-N 0.000 description 2
- 150000004702 methyl esters Chemical class 0.000 description 2
- 239000004570 mortar (masonry) Substances 0.000 description 2
- IMLVEYAVSNLRRR-UHFFFAOYSA-N n-(2-methoxyethyl)-4-[4-[[6-methoxy-2-(2-methoxyimidazo[2,1-b][1,3,4]thiadiazol-6-yl)-1-benzofuran-4-yl]oxymethyl]-1,3-thiazol-2-yl]-n-methylbenzamide Chemical compound C1=CC(C(=O)N(C)CCOC)=CC=C1C1=NC(COC=2C=3C=C(OC=3C=C(OC)C=2)C=2N=C3SC(OC)=NN3C=2)=CS1 IMLVEYAVSNLRRR-UHFFFAOYSA-N 0.000 description 2
- 150000007524 organic acids Chemical class 0.000 description 2
- 125000004287 oxazol-2-yl group Chemical group [H]C1=C([H])N=C(*)O1 0.000 description 2
- KJIFKLIQANRMOU-UHFFFAOYSA-N oxidanium;4-methylbenzenesulfonate Chemical compound O.CC1=CC=C(S(O)(=O)=O)C=C1 KJIFKLIQANRMOU-UHFFFAOYSA-N 0.000 description 2
- 230000003647 oxidation Effects 0.000 description 2
- 238000007254 oxidation reaction Methods 0.000 description 2
- 239000001301 oxygen Substances 0.000 description 2
- 125000004430 oxygen atom Chemical group O* 0.000 description 2
- LXNAVEXFUKBNMK-UHFFFAOYSA-N palladium(II) acetate Substances [Pd].CC(O)=O.CC(O)=O LXNAVEXFUKBNMK-UHFFFAOYSA-N 0.000 description 2
- YJVFFLUZDVXJQI-UHFFFAOYSA-L palladium(ii) acetate Chemical compound [Pd+2].CC([O-])=O.CC([O-])=O YJVFFLUZDVXJQI-UHFFFAOYSA-L 0.000 description 2
- 230000036961 partial effect Effects 0.000 description 2
- 239000002245 particle Substances 0.000 description 2
- 125000000286 phenylethyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])C([H])([H])* 0.000 description 2
- XHXFXVLFKHQFAL-UHFFFAOYSA-N phosphoryl trichloride Chemical compound ClP(Cl)(Cl)=O XHXFXVLFKHQFAL-UHFFFAOYSA-N 0.000 description 2
- 125000003386 piperidinyl group Chemical group 0.000 description 2
- 230000010118 platelet activation Effects 0.000 description 2
- NROKBHXJSPEDAR-UHFFFAOYSA-M potassium fluoride Chemical compound [F-].[K+] NROKBHXJSPEDAR-UHFFFAOYSA-M 0.000 description 2
- 230000002265 prevention Effects 0.000 description 2
- VVWRJUBEIPHGQF-MDZDMXLPSA-N propan-2-yl (ne)-n-propan-2-yloxycarbonyliminocarbamate Chemical compound CC(C)OC(=O)\N=N\C(=O)OC(C)C VVWRJUBEIPHGQF-MDZDMXLPSA-N 0.000 description 2
- 150000003254 radicals Chemical class 0.000 description 2
- 238000001953 recrystallisation Methods 0.000 description 2
- 238000004007 reversed phase HPLC Methods 0.000 description 2
- 230000002441 reversible effect Effects 0.000 description 2
- 238000007363 ring formation reaction Methods 0.000 description 2
- 230000011664 signaling Effects 0.000 description 2
- XGVXKJKTISMIOW-ZDUSSCGKSA-N simurosertib Chemical compound N1N=CC(C=2SC=3C(=O)NC(=NC=3C=2)[C@H]2N3CCC(CC3)C2)=C1C XGVXKJKTISMIOW-ZDUSSCGKSA-N 0.000 description 2
- 239000012279 sodium borohydride Substances 0.000 description 2
- 229910000033 sodium borohydride Inorganic materials 0.000 description 2
- 229910000029 sodium carbonate Inorganic materials 0.000 description 2
- 229910000104 sodium hydride Inorganic materials 0.000 description 2
- 239000012312 sodium hydride Substances 0.000 description 2
- 238000000638 solvent extraction Methods 0.000 description 2
- 125000000547 substituted alkyl group Chemical group 0.000 description 2
- DYHSDKLCOJIUFX-UHFFFAOYSA-N tert-butoxycarbonyl anhydride Chemical compound CC(C)(C)OC(=O)OC(=O)OC(C)(C)C DYHSDKLCOJIUFX-UHFFFAOYSA-N 0.000 description 2
- TUULDEMXKCQWOR-UHFFFAOYSA-N tert-butyl-dimethyl-[(5-methyl-1,3-thiazol-4-yl)methoxy]silane Chemical compound CC=1SC=NC=1CO[Si](C)(C)C(C)(C)C TUULDEMXKCQWOR-UHFFFAOYSA-N 0.000 description 2
- KCAVYFWOSRVXOY-UHFFFAOYSA-N tert-butyl-dimethyl-[[2-(8-oxa-3-azabicyclo[3.2.1]octan-3-yl)-1,3-thiazol-4-yl]methoxy]silane Chemical compound CC(C)(C)[Si](C)(C)OCC1=CSC(N2CC3CCC(O3)C2)=N1 KCAVYFWOSRVXOY-UHFFFAOYSA-N 0.000 description 2
- IATMYVAHRKLCFX-UHFFFAOYSA-N tert-butyl-dimethyl-[[5-methyl-2-(8-oxa-3-azabicyclo[3.2.1]octan-3-yl)-1,3-thiazol-4-yl]methoxy]silane Chemical compound CC(C)(C)[Si](C)(C)OCC1=C(C)SC(N2CC3CCC(O3)C2)=N1 IATMYVAHRKLCFX-UHFFFAOYSA-N 0.000 description 2
- VZGDMQKNWNREIO-UHFFFAOYSA-N tetrachloromethane Chemical compound ClC(Cl)(Cl)Cl VZGDMQKNWNREIO-UHFFFAOYSA-N 0.000 description 2
- 125000001412 tetrahydropyranyl group Chemical group 0.000 description 2
- UMGDCJDMYOKAJW-UHFFFAOYSA-N thiourea Chemical compound NC(N)=S UMGDCJDMYOKAJW-UHFFFAOYSA-N 0.000 description 2
- 239000003868 thrombin inhibitor Substances 0.000 description 2
- 201000005060 thrombophlebitis Diseases 0.000 description 2
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 2
- 230000009466 transformation Effects 0.000 description 2
- 238000000844 transformation Methods 0.000 description 2
- 230000001052 transient effect Effects 0.000 description 2
- 229960005044 vorapaxar Drugs 0.000 description 2
- ZBGXUVOIWDMMJE-QHNZEKIYSA-N vorapaxar Chemical compound C(/[C@@H]1[C@H]2[C@H](C(O[C@@H]2C)=O)C[C@H]2[C@H]1CC[C@H](C2)NC(=O)OCC)=C\C(N=C1)=CC=C1C1=CC=CC(F)=C1 ZBGXUVOIWDMMJE-QHNZEKIYSA-N 0.000 description 2
- PJVWKTKQMONHTI-UHFFFAOYSA-N warfarin Chemical compound OC=1C2=CC=CC=C2OC(=O)C=1C(CC(=O)C)C1=CC=CC=C1 PJVWKTKQMONHTI-UHFFFAOYSA-N 0.000 description 2
- 239000003643 water by type Substances 0.000 description 2
- CXNIUSPIQKWYAI-UHFFFAOYSA-N xantphos Chemical compound C=12OC3=C(P(C=4C=CC=CC=4)C=4C=CC=CC=4)C=CC=C3C(C)(C)C2=CC=CC=1P(C=1C=CC=CC=1)C1=CC=CC=C1 CXNIUSPIQKWYAI-UHFFFAOYSA-N 0.000 description 2
- GNLJBJNONOOOQC-UHFFFAOYSA-N $l^{3}-carbane;magnesium Chemical compound [Mg]C GNLJBJNONOOOQC-UHFFFAOYSA-N 0.000 description 1
- VCGRFBXVSFAGGA-UHFFFAOYSA-N (1,1-dioxo-1,4-thiazinan-4-yl)-[6-[[3-(4-fluorophenyl)-5-methyl-1,2-oxazol-4-yl]methoxy]pyridin-3-yl]methanone Chemical compound CC=1ON=C(C=2C=CC(F)=CC=2)C=1COC(N=C1)=CC=C1C(=O)N1CCS(=O)(=O)CC1 VCGRFBXVSFAGGA-UHFFFAOYSA-N 0.000 description 1
- RNKOUSCCPHSCFE-UHFFFAOYSA-N (2,4-dimethoxyphenyl)methanol Chemical compound COC1=CC=C(CO)C(OC)=C1 RNKOUSCCPHSCFE-UHFFFAOYSA-N 0.000 description 1
- MQMFHTSXMPGVBI-UHFFFAOYSA-N (2-bromo-5-methyl-1,3-thiazol-4-yl)methanol Chemical compound CC=1SC(Br)=NC=1CO MQMFHTSXMPGVBI-UHFFFAOYSA-N 0.000 description 1
- WKDLXFYTDOLWQW-UHFFFAOYSA-N (2-bromo-5-propan-2-yl-1,3-thiazol-4-yl)methanol Chemical compound CC(C)C=1SC(Br)=NC=1CO WKDLXFYTDOLWQW-UHFFFAOYSA-N 0.000 description 1
- WYYBDMZUGLHCKA-UHFFFAOYSA-N (4-bromo-2-methylphenyl)thiourea Chemical compound CC1=CC(Br)=CC=C1NC(N)=S WYYBDMZUGLHCKA-UHFFFAOYSA-N 0.000 description 1
- MQIVBOZBWLTZHZ-UHFFFAOYSA-N (5-chloro-2-morpholin-4-yl-1,3-thiazol-4-yl)methanol Chemical compound S1C(Cl)=C(CO)N=C1N1CCOCC1 MQIVBOZBWLTZHZ-UHFFFAOYSA-N 0.000 description 1
- QYHQZLTZTNMTAE-UHFFFAOYSA-N (5-ethyl-2-morpholin-4-yl-1,3-thiazol-4-yl)methanol Chemical compound OCC1=C(CC)SC(N2CCOCC2)=N1 QYHQZLTZTNMTAE-UHFFFAOYSA-N 0.000 description 1
- FRYMXKQIKOMSSI-UHFFFAOYSA-N (5-methyl-2-morpholin-4-yl-1,3-oxazol-4-yl)methanol Chemical compound OCC1=C(C)OC(N2CCOCC2)=N1 FRYMXKQIKOMSSI-UHFFFAOYSA-N 0.000 description 1
- UEPBCEHLQBXDNV-UHFFFAOYSA-N (5-methyl-2-morpholin-4-yl-1,3-thiazol-4-yl)methanol Chemical compound OCC1=C(C)SC(N2CCOCC2)=N1 UEPBCEHLQBXDNV-UHFFFAOYSA-N 0.000 description 1
- KDWPCWGJRDPKNX-UHFFFAOYSA-N (5-phenyl-1,3,4-thiadiazol-2-yl)methanol Chemical compound S1C(CO)=NN=C1C1=CC=CC=C1 KDWPCWGJRDPKNX-UHFFFAOYSA-N 0.000 description 1
- 125000000008 (C1-C10) alkyl group Chemical group 0.000 description 1
- 125000004191 (C1-C6) alkoxy group Chemical group 0.000 description 1
- CHPRFKYDQRKRRK-LURJTMIESA-N (S)-2-(methoxymethyl)pyrrolidine Chemical compound COC[C@@H]1CCCN1 CHPRFKYDQRKRRK-LURJTMIESA-N 0.000 description 1
- 125000005869 (methoxyethoxy)methanyl group Chemical group [H]C([H])([H])OC([H])([H])C([H])([H])OC([H])([H])* 0.000 description 1
- KZPYGQFFRCFCPP-UHFFFAOYSA-N 1,1'-bis(diphenylphosphino)ferrocene Chemical compound [Fe+2].C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1 KZPYGQFFRCFCPP-UHFFFAOYSA-N 0.000 description 1
- FIARMZDBEGVMLV-UHFFFAOYSA-N 1,1,2,2,2-pentafluoroethanolate Chemical group [O-]C(F)(F)C(F)(F)F FIARMZDBEGVMLV-UHFFFAOYSA-N 0.000 description 1
- 125000006002 1,1-difluoroethyl group Chemical group 0.000 description 1
- 125000004514 1,2,4-thiadiazolyl group Chemical group 0.000 description 1
- CEDRGDFENMZKCQ-UHFFFAOYSA-N 1,2-oxazole-5-carbothioamide Chemical compound NC(=S)C1=CC=NO1 CEDRGDFENMZKCQ-UHFFFAOYSA-N 0.000 description 1
- LQQKDSXCDXHLLF-UHFFFAOYSA-N 1,3-dibromopropan-2-one Chemical compound BrCC(=O)CBr LQQKDSXCDXHLLF-UHFFFAOYSA-N 0.000 description 1
- WNXJIVFYUVYPPR-UHFFFAOYSA-N 1,3-dioxolane Chemical compound C1COCO1 WNXJIVFYUVYPPR-UHFFFAOYSA-N 0.000 description 1
- TULKTUDUXQCDLC-UHFFFAOYSA-N 1,3-oxazole-5-carbothioamide Chemical compound NC(=S)C1=CN=CO1 TULKTUDUXQCDLC-UHFFFAOYSA-N 0.000 description 1
- RAIPHJJURHTUIC-UHFFFAOYSA-N 1,3-thiazol-2-amine Chemical compound NC1=NC=CS1 RAIPHJJURHTUIC-UHFFFAOYSA-N 0.000 description 1
- 125000004776 1-fluoroethyl group Chemical group [H]C([H])([H])C([H])(F)* 0.000 description 1
- 125000006017 1-propenyl group Chemical group 0.000 description 1
- AAILEWXSEQLMNI-UHFFFAOYSA-N 1h-pyridazin-6-one Chemical class OC1=CC=CN=N1 AAILEWXSEQLMNI-UHFFFAOYSA-N 0.000 description 1
- NRKYWOKHZRQRJR-UHFFFAOYSA-N 2,2,2-trifluoroacetamide Chemical compound NC(=O)C(F)(F)F NRKYWOKHZRQRJR-UHFFFAOYSA-N 0.000 description 1
- 125000004206 2,2,2-trifluoroethyl group Chemical group [H]C([H])(*)C(F)(F)F 0.000 description 1
- CLLLODNOQBVIMS-UHFFFAOYSA-N 2-(2-methoxyethoxy)acetic acid Chemical compound COCCOCC(O)=O CLLLODNOQBVIMS-UHFFFAOYSA-N 0.000 description 1
- HWBFUTJAXKVJNC-UHFFFAOYSA-N 2-[4-[4-(hydroxymethyl)-1,3-thiazol-2-yl]phenyl]propan-2-ol Chemical compound C1=CC(C(C)(O)C)=CC=C1C1=NC(CO)=CS1 HWBFUTJAXKVJNC-UHFFFAOYSA-N 0.000 description 1
- 125000005273 2-acetoxybenzoic acid group Chemical group 0.000 description 1
- CDAWCLOXVUBKRW-UHFFFAOYSA-N 2-aminophenol Chemical class NC1=CC=CC=C1O CDAWCLOXVUBKRW-UHFFFAOYSA-N 0.000 description 1
- 125000004974 2-butenyl group Chemical group C(C=CC)* 0.000 description 1
- PBYFKVQPMPFARB-UHFFFAOYSA-N 2-chloro-1-(6-methoxy-4-phenylmethoxy-1-benzofuran-2-yl)ethanone Chemical compound C=12C=C(C(=O)CCl)OC2=CC(OC)=CC=1OCC1=CC=CC=C1 PBYFKVQPMPFARB-UHFFFAOYSA-N 0.000 description 1
- 125000006040 2-hexenyl group Chemical group 0.000 description 1
- HMDXNKYJONMJPU-UHFFFAOYSA-N 2-methoxy-6-[6-methoxy-4-[(3-phenyltriazol-4-yl)methoxy]-1-benzofuran-2-yl]imidazo[2,1-b][1,3,4]thiadiazole Chemical compound N1=C2SC(OC)=NN2C=C1C(OC1=CC(OC)=C2)=CC1=C2OCC1=CN=NN1C1=CC=CC=C1 HMDXNKYJONMJPU-UHFFFAOYSA-N 0.000 description 1
- YHAFFHWOHFNWTG-UHFFFAOYSA-N 2-methoxy-6-[6-methoxy-4-[[2-(1-methylsulfonylpiperidin-4-yl)-1,3-thiazol-4-yl]methoxy]-1-benzofuran-2-yl]imidazo[2,1-b][1,3,4]thiadiazole Chemical compound N1=C2SC(OC)=NN2C=C1C(OC1=CC(OC)=C2)=CC1=C2OCC(N=1)=CSC=1C1CCN(S(C)(=O)=O)CC1 YHAFFHWOHFNWTG-UHFFFAOYSA-N 0.000 description 1
- QYACVRVQEKUSAK-UHFFFAOYSA-N 2-methoxy-6-[6-methoxy-4-[[2-(1-propan-2-ylsulfonylpiperidin-4-yl)-1,3-thiazol-4-yl]methoxy]-1-benzofuran-2-yl]imidazo[2,1-b][1,3,4]thiadiazole Chemical compound N1=C2SC(OC)=NN2C=C1C(OC1=CC(OC)=C2)=CC1=C2OCC(N=1)=CSC=1C1CCN(S(=O)(=O)C(C)C)CC1 QYACVRVQEKUSAK-UHFFFAOYSA-N 0.000 description 1
- BDZNBNNJQRTJAR-UHFFFAOYSA-N 2-methoxy-6-[6-methoxy-4-[[2-[1-(trifluoromethylsulfonyl)piperidin-4-yl]-1,3-thiazol-4-yl]methoxy]-1-benzofuran-2-yl]imidazo[2,1-b][1,3,4]thiadiazole Chemical compound N1=C2SC(OC)=NN2C=C1C(OC1=CC(OC)=C2)=CC1=C2OCC(N=1)=CSC=1C1CCN(S(=O)(=O)C(F)(F)F)CC1 BDZNBNNJQRTJAR-UHFFFAOYSA-N 0.000 description 1
- KOHBEDRJXKOYHL-UHFFFAOYSA-N 2-methoxy-n-methylethanamine Chemical compound CNCCOC KOHBEDRJXKOYHL-UHFFFAOYSA-N 0.000 description 1
- 125000006022 2-methyl-2-propenyl group Chemical group 0.000 description 1
- JAWKOSRQDZQJMU-UHFFFAOYSA-N 2-methylpropan-2-ol;toluene Chemical compound CC(C)(C)O.CC1=CC=CC=C1 JAWKOSRQDZQJMU-UHFFFAOYSA-N 0.000 description 1
- 125000006088 2-oxoazepinyl group Chemical group 0.000 description 1
- 125000004638 2-oxopiperazinyl group Chemical group O=C1N(CCNC1)* 0.000 description 1
- 125000006087 2-oxopyrrolodinyl group Chemical group 0.000 description 1
- 125000006024 2-pentenyl group Chemical group 0.000 description 1
- 125000003903 2-propenyl group Chemical group [H]C([*])([H])C([H])=C([H])[H] 0.000 description 1
- VFPBFBJZAMZMKJ-UHFFFAOYSA-N 3,5-dimethyloxan-4-one Chemical compound CC1COCC(C)C1=O VFPBFBJZAMZMKJ-UHFFFAOYSA-N 0.000 description 1
- WNEODWDFDXWOLU-QHCPKHFHSA-N 3-[3-(hydroxymethyl)-4-[1-methyl-5-[[5-[(2s)-2-methyl-4-(oxetan-3-yl)piperazin-1-yl]pyridin-2-yl]amino]-6-oxopyridin-3-yl]pyridin-2-yl]-7,7-dimethyl-1,2,6,8-tetrahydrocyclopenta[3,4]pyrrolo[3,5-b]pyrazin-4-one Chemical compound C([C@@H](N(CC1)C=2C=NC(NC=3C(N(C)C=C(C=3)C=3C(=C(N4C(C5=CC=6CC(C)(C)CC=6N5CC4)=O)N=CC=3)CO)=O)=CC=2)C)N1C1COC1 WNEODWDFDXWOLU-QHCPKHFHSA-N 0.000 description 1
- 125000004975 3-butenyl group Chemical group C(CC=C)* 0.000 description 1
- 125000006041 3-hexenyl group Chemical group 0.000 description 1
- HYIUDFLDFSIXTR-UHFFFAOYSA-N 4,4-difluorocyclohexane-1-carboxylic acid Chemical compound OC(=O)C1CCC(F)(F)CC1 HYIUDFLDFSIXTR-UHFFFAOYSA-N 0.000 description 1
- VZLONUKFKRQPKQ-UHFFFAOYSA-N 4-(bromomethyl)-2-(4-fluorooxan-4-yl)-1,3-thiazole Chemical compound N=1C(CBr)=CSC=1C1(F)CCOCC1 VZLONUKFKRQPKQ-UHFFFAOYSA-N 0.000 description 1
- WOLAEYWBLGTSQR-UHFFFAOYSA-N 4-[4-(hydroxymethyl)-5-propan-2-yl-1,3-thiazol-2-yl]-n,n-dimethylbenzamide Chemical compound OCC1=C(C(C)C)SC(C=2C=CC(=CC=2)C(=O)N(C)C)=N1 WOLAEYWBLGTSQR-UHFFFAOYSA-N 0.000 description 1
- RSUYDIKQBCPPFW-UHFFFAOYSA-N 4-[4-[4-(hydroxymethyl)-1,3-thiazol-2-yl]piperazin-1-yl]sulfonylbenzonitrile Chemical compound OCC1=CSC(N2CCN(CC2)S(=O)(=O)C=2C=CC(=CC=2)C#N)=N1 RSUYDIKQBCPPFW-UHFFFAOYSA-N 0.000 description 1
- GWAHHWYTRMGILF-UHFFFAOYSA-N 4-[4-[4-[[6-methoxy-2-(2-methoxyimidazo[2,1-b][1,3,4]thiadiazol-6-yl)-1-benzofuran-4-yl]oxymethyl]-1,3-thiazol-2-yl]piperazin-1-yl]sulfonylbenzonitrile Chemical compound N1=C2SC(OC)=NN2C=C1C(OC1=CC(OC)=C2)=CC1=C2OCC(N=1)=CSC=1N(CC1)CCN1S(=O)(=O)C1=CC=C(C#N)C=C1 GWAHHWYTRMGILF-UHFFFAOYSA-N 0.000 description 1
- KEEBLYWBELVGPQ-UHFFFAOYSA-N 4-[4-[[6-methoxy-2-(2-methoxyimidazo[2,1-b][1,3,4]thiadiazol-6-yl)-1-benzofuran-4-yl]oxymethyl]-1,3-thiazol-2-yl]-n,n-dimethylbenzamide Chemical compound N1=C2SC(OC)=NN2C=C1C(OC1=CC(OC)=C2)=CC1=C2OCC(N=1)=CSC=1C1=CC=C(C(=O)N(C)C)C=C1 KEEBLYWBELVGPQ-UHFFFAOYSA-N 0.000 description 1
- GPISVVPOYSSYDY-UHFFFAOYSA-N 4-[4-[[6-methoxy-2-(2-methoxyimidazo[2,1-b][1,3,4]thiadiazol-6-yl)-1-benzofuran-4-yl]oxymethyl]-1,3-thiazol-2-yl]-n-methylbenzamide Chemical compound C1=CC(C(=O)NC)=CC=C1C1=NC(COC=2C=3C=C(OC=3C=C(OC)C=2)C=2N=C3SC(OC)=NN3C=2)=CS1 GPISVVPOYSSYDY-UHFFFAOYSA-N 0.000 description 1
- AHRKBWBTZMIPIY-UHFFFAOYSA-N 4-[4-[[6-methoxy-2-(2-methoxyimidazo[2,1-b][1,3,4]thiadiazol-6-yl)-1-benzofuran-4-yl]oxymethyl]-1,3-thiazol-2-yl]piperidin-4-ol Chemical compound N1=C2SC(OC)=NN2C=C1C(OC1=CC(OC)=C2)=CC1=C2OCC(N=1)=CSC=1C1(O)CCNCC1 AHRKBWBTZMIPIY-UHFFFAOYSA-N 0.000 description 1
- IZHLHUFOADZHPG-UHFFFAOYSA-N 4-[4-[[6-methoxy-2-(2-methoxyimidazo[2,1-b][1,3,4]thiadiazol-6-yl)-1-benzofuran-4-yl]oxymethyl]-5-(trifluoromethyl)-1,3-thiazol-2-yl]oxan-4-ol Chemical compound N1=C2SC(OC)=NN2C=C1C(OC1=CC(OC)=C2)=CC1=C2OCC(=C(S1)C(F)(F)F)N=C1C1(O)CCOCC1 IZHLHUFOADZHPG-UHFFFAOYSA-N 0.000 description 1
- QSLGJXVTDUNXLH-UHFFFAOYSA-N 4-[4-[[tert-butyl(dimethyl)silyl]oxymethyl]-5-propan-2-yl-1,3-thiazol-2-yl]-n,n-dimethylbenzamide Chemical compound CC(C)(C)[Si](C)(C)OCC1=C(C(C)C)SC(C=2C=CC(=CC=2)C(=O)N(C)C)=N1 QSLGJXVTDUNXLH-UHFFFAOYSA-N 0.000 description 1
- NBNXGLGZVSCCGL-UHFFFAOYSA-N 4-[5-ethyl-4-(hydroxymethyl)-1,3-thiazol-2-yl]-n,n-dimethylbenzamide Chemical compound OCC1=C(CC)SC(C=2C=CC(=CC=2)C(=O)N(C)C)=N1 NBNXGLGZVSCCGL-UHFFFAOYSA-N 0.000 description 1
- 229960000549 4-dimethylaminophenol Drugs 0.000 description 1
- 125000006042 4-hexenyl group Chemical group 0.000 description 1
- 125000003119 4-methyl-3-pentenyl group Chemical group [H]\C(=C(/C([H])([H])[H])C([H])([H])[H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000005986 4-piperidonyl group Chemical group 0.000 description 1
- FLDSMVTWEZKONL-AWEZNQCLSA-N 5,5-dimethyl-N-[(3S)-5-methyl-4-oxo-2,3-dihydro-1,5-benzoxazepin-3-yl]-1,4,7,8-tetrahydrooxepino[4,5-c]pyrazole-3-carboxamide Chemical compound CC1(CC2=C(NN=C2C(=O)N[C@@H]2C(N(C3=C(OC2)C=CC=C3)C)=O)CCO1)C FLDSMVTWEZKONL-AWEZNQCLSA-N 0.000 description 1
- OCKGFTQIICXDQW-ZEQRLZLVSA-N 5-[(1r)-1-hydroxy-2-[4-[(2r)-2-hydroxy-2-(4-methyl-1-oxo-3h-2-benzofuran-5-yl)ethyl]piperazin-1-yl]ethyl]-4-methyl-3h-2-benzofuran-1-one Chemical compound C1=C2C(=O)OCC2=C(C)C([C@@H](O)CN2CCN(CC2)C[C@H](O)C2=CC=C3C(=O)OCC3=C2C)=C1 OCKGFTQIICXDQW-ZEQRLZLVSA-N 0.000 description 1
- XFJBGINZIMNZBW-CRAIPNDOSA-N 5-chloro-2-[4-[(1r,2s)-2-[2-(5-methylsulfonylpyridin-2-yl)oxyethyl]cyclopropyl]piperidin-1-yl]pyrimidine Chemical compound N1=CC(S(=O)(=O)C)=CC=C1OCC[C@H]1[C@@H](C2CCN(CC2)C=2N=CC(Cl)=CN=2)C1 XFJBGINZIMNZBW-CRAIPNDOSA-N 0.000 description 1
- 125000006043 5-hexenyl group Chemical group 0.000 description 1
- WFVAUGFNBVXHIW-UHFFFAOYSA-N 5-hydroxy-7-methoxy-2,2-dimethyl-1,3-benzodioxin-4-one Chemical compound O=C1OC(C)(C)OC2=CC(OC)=CC(O)=C21 WFVAUGFNBVXHIW-UHFFFAOYSA-N 0.000 description 1
- MJQSRSOTRPMVKB-UHFFFAOYSA-N 5h-imidazo[4,5-c]pyridazine Chemical compound C1=NNC2=NC=NC2=C1 MJQSRSOTRPMVKB-UHFFFAOYSA-N 0.000 description 1
- QYCFQNZPVAIRRQ-UHFFFAOYSA-N 6-[4-[(5-chloro-2-phenyl-1,3-thiazol-4-yl)methoxy]-6-methoxy-1-benzofuran-2-yl]-2-methoxyimidazo[2,1-b][1,3,4]thiadiazole Chemical compound N1=C2SC(OC)=NN2C=C1C(OC1=CC(OC)=C2)=CC1=C2OCC(=C(S1)Cl)N=C1C1=CC=CC=C1 QYCFQNZPVAIRRQ-UHFFFAOYSA-N 0.000 description 1
- HZZNYBLWSTVSCP-UHFFFAOYSA-N 6-[4-[[2-[1-(benzenesulfonyl)piperidin-4-yl]-1,3-thiazol-4-yl]methoxy]-6-methoxy-1-benzofuran-2-yl]-2-methoxyimidazo[2,1-b][1,3,4]thiadiazole Chemical compound N1=C2SC(OC)=NN2C=C1C(OC1=CC(OC)=C2)=CC1=C2OCC(N=1)=CSC=1C(CC1)CCN1S(=O)(=O)C1=CC=CC=C1 HZZNYBLWSTVSCP-UHFFFAOYSA-N 0.000 description 1
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 1
- CYJRNFFLTBEQSQ-UHFFFAOYSA-N 8-(3-methyl-1-benzothiophen-5-yl)-N-(4-methylsulfonylpyridin-3-yl)quinoxalin-6-amine Chemical compound CS(=O)(=O)C1=C(C=NC=C1)NC=1C=C2N=CC=NC2=C(C=1)C=1C=CC2=C(C(=CS2)C)C=1 CYJRNFFLTBEQSQ-UHFFFAOYSA-N 0.000 description 1
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 1
- GBTPSLPMUMGGLO-UHFFFAOYSA-N 8-[4-[[6-methoxy-2-(2-methoxyimidazo[2,1-b][1,3,4]thiadiazol-6-yl)-1-benzofuran-4-yl]oxymethyl]-1,3-thiazol-2-yl]-1,4-dioxa-8-azaspiro[4.5]decane Chemical compound N1=C2SC(OC)=NN2C=C1C(OC1=CC(OC)=C2)=CC1=C2OCC(N=1)=CSC=1N(CC1)CCC21OCCO2 GBTPSLPMUMGGLO-UHFFFAOYSA-N 0.000 description 1
- POOPWPIOIMBTOH-UHFFFAOYSA-N 8-oxa-3-azabicyclo[3.2.1]octane Chemical compound C1NCC2CCC1O2 POOPWPIOIMBTOH-UHFFFAOYSA-N 0.000 description 1
- 206010002383 Angina Pectoris Diseases 0.000 description 1
- QNZCBYKSOIHPEH-UHFFFAOYSA-N Apixaban Chemical compound C1=CC(OC)=CC=C1N1C(C(=O)N(CC2)C=3C=CC(=CC=3)N3C(CCCC3)=O)=C2C(C(N)=O)=N1 QNZCBYKSOIHPEH-UHFFFAOYSA-N 0.000 description 1
- 200000000007 Arterial disease Diseases 0.000 description 1
- 206010003178 Arterial thrombosis Diseases 0.000 description 1
- QWKAUGRRIXBIPO-UHFFFAOYSA-N Atopaxar Chemical compound N=C1C=2C(F)=C(OCC)C(OCC)=CC=2CN1CC(=O)C(C=C(C=1OC)C(C)(C)C)=CC=1N1CCOCC1 QWKAUGRRIXBIPO-UHFFFAOYSA-N 0.000 description 1
- 239000005465 B01AC22 - Prasugrel Substances 0.000 description 1
- 239000004342 Benzoyl peroxide Substances 0.000 description 1
- OMPJBNCRMGITSC-UHFFFAOYSA-N Benzoylperoxide Chemical compound C=1C=CC=CC=1C(=O)OOC(=O)C1=CC=CC=C1 OMPJBNCRMGITSC-UHFFFAOYSA-N 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-M Bicarbonate Chemical compound OC([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-M 0.000 description 1
- COVZYZSDYWQREU-UHFFFAOYSA-N Busulfan Chemical compound CS(=O)(=O)OCCCCOS(C)(=O)=O COVZYZSDYWQREU-UHFFFAOYSA-N 0.000 description 1
- 125000003601 C2-C6 alkynyl group Chemical group 0.000 description 1
- UXVMQQNJUSDDNG-UHFFFAOYSA-L Calcium chloride Chemical compound [Cl-].[Cl-].[Ca+2] UXVMQQNJUSDDNG-UHFFFAOYSA-L 0.000 description 1
- 206010007688 Carotid artery thrombosis Diseases 0.000 description 1
- 241000282693 Cercopithecidae Species 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 1
- 229910021589 Copper(I) bromide Inorganic materials 0.000 description 1
- 229910021595 Copper(I) iodide Inorganic materials 0.000 description 1
- 229910021590 Copper(II) bromide Inorganic materials 0.000 description 1
- YZCKVEUIGOORGS-OUBTZVSYSA-N Deuterium Chemical compound [2H] YZCKVEUIGOORGS-OUBTZVSYSA-N 0.000 description 1
- XXRCUYVCPSWGCC-UHFFFAOYSA-N Ethyl pyruvate Chemical compound CCOC(=O)C(C)=O XXRCUYVCPSWGCC-UHFFFAOYSA-N 0.000 description 1
- 244000166102 Eucalyptus leucoxylon Species 0.000 description 1
- 235000004694 Eucalyptus leucoxylon Nutrition 0.000 description 1
- 102000003688 G-Protein-Coupled Receptors Human genes 0.000 description 1
- 108090000045 G-Protein-Coupled Receptors Proteins 0.000 description 1
- HTTJABKRGRZYRN-UHFFFAOYSA-N Heparin Chemical compound OC1C(NC(=O)C)C(O)OC(COS(O)(=O)=O)C1OC1C(OS(O)(=O)=O)C(O)C(OC2C(C(OS(O)(=O)=O)C(OC3C(C(O)C(O)C(O3)C(O)=O)OS(O)(=O)=O)C(CO)O2)NS(O)(=O)=O)C(C(O)=O)O1 HTTJABKRGRZYRN-UHFFFAOYSA-N 0.000 description 1
- 229930194542 Keto Natural products 0.000 description 1
- 229910010082 LiAlH Inorganic materials 0.000 description 1
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 1
- 241000282567 Macaca fascicularis Species 0.000 description 1
- 241001465754 Metazoa Species 0.000 description 1
- 239000012359 Methanesulfonyl chloride Substances 0.000 description 1
- QPJVMBTYPHYUOC-UHFFFAOYSA-N Methyl benzoate Natural products COC(=O)C1=CC=CC=C1 QPJVMBTYPHYUOC-UHFFFAOYSA-N 0.000 description 1
- AYCPARAPKDAOEN-LJQANCHMSA-N N-[(1S)-2-(dimethylamino)-1-phenylethyl]-6,6-dimethyl-3-[(2-methyl-4-thieno[3,2-d]pyrimidinyl)amino]-1,4-dihydropyrrolo[3,4-c]pyrazole-5-carboxamide Chemical compound C1([C@H](NC(=O)N2C(C=3NN=C(NC=4C=5SC=CC=5N=C(C)N=4)C=3C2)(C)C)CN(C)C)=CC=CC=C1 AYCPARAPKDAOEN-LJQANCHMSA-N 0.000 description 1
- WZKRKKDBZCWURF-UHFFFAOYSA-N OC(=O)C(F)(F)F.COc1nn2cc(nc2s1)-c1cc2c(OCc3csc(n3)C3(O)CCNCC3)cc(OC)cc2o1 Chemical compound OC(=O)C(F)(F)F.COc1nn2cc(nc2s1)-c1cc2c(OCc3csc(n3)C3(O)CCNCC3)cc(OC)cc2o1 WZKRKKDBZCWURF-UHFFFAOYSA-N 0.000 description 1
- MEWUOQPYZHTCHE-AKIIRXMFSA-M O[C@@H](CO)[C@@H]1OC(C(=C1CCCCCCCC\C=C/CCCCCCCC(=O)[O-])O)=O.[Na+] Chemical compound O[C@@H](CO)[C@@H]1OC(C(=C1CCCCCCCC\C=C/CCCCCCCC(=O)[O-])O)=O.[Na+] MEWUOQPYZHTCHE-AKIIRXMFSA-M 0.000 description 1
- 235000019502 Orange oil Nutrition 0.000 description 1
- 229940126629 PAR4 antagonist Drugs 0.000 description 1
- 229910019213 POCl3 Inorganic materials 0.000 description 1
- 229910006069 SO3H Inorganic materials 0.000 description 1
- 208000006117 ST-elevation myocardial infarction Diseases 0.000 description 1
- 229940124639 Selective inhibitor Drugs 0.000 description 1
- 229910018557 Si O Inorganic materials 0.000 description 1
- 229910007991 Si-N Inorganic materials 0.000 description 1
- XUIMIQQOPSSXEZ-UHFFFAOYSA-N Silicon Chemical group [Si] XUIMIQQOPSSXEZ-UHFFFAOYSA-N 0.000 description 1
- 229910006294 Si—N Inorganic materials 0.000 description 1
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 1
- 208000007718 Stable Angina Diseases 0.000 description 1
- 206010042434 Sudden death Diseases 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 1
- 101000712605 Theromyzon tessulatum Theromin Proteins 0.000 description 1
- 102000003790 Thrombin receptors Human genes 0.000 description 1
- 108090000166 Thrombin receptors Proteins 0.000 description 1
- 208000001435 Thromboembolism Diseases 0.000 description 1
- 208000032109 Transient ischaemic attack Diseases 0.000 description 1
- YZCKVEUIGOORGS-NJFSPNSNSA-N Tritium Chemical compound [3H] YZCKVEUIGOORGS-NJFSPNSNSA-N 0.000 description 1
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Natural products NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 1
- 206010053648 Vascular occlusion Diseases 0.000 description 1
- 208000027418 Wounds and injury Diseases 0.000 description 1
- REAVSCLQAUEHBS-UHFFFAOYSA-N [2-(1,4-dioxa-8-azaspiro[4.5]decan-8-yl)-1,3-thiazol-4-yl]methanol Chemical compound OCC1=CSC(N2CCC3(CC2)OCCO3)=N1 REAVSCLQAUEHBS-UHFFFAOYSA-N 0.000 description 1
- IECXHAIAUXGZPS-UHFFFAOYSA-N [2-(2-fluoropyridin-4-yl)-5-propan-2-yl-1,3-thiazol-4-yl]methanol Chemical compound OCC1=C(C(C)C)SC(C=2C=C(F)N=CC=2)=N1 IECXHAIAUXGZPS-UHFFFAOYSA-N 0.000 description 1
- FBXUDXUJKBSHSM-UHFFFAOYSA-N [2-(2-oxa-6-azaspiro[3.3]heptan-6-yl)-1,3-thiazol-4-yl]methanol Chemical compound OCC1=CSC(N2CC3(COC3)C2)=N1 FBXUDXUJKBSHSM-UHFFFAOYSA-N 0.000 description 1
- HSMNSHACEUGBAN-UHFFFAOYSA-N [2-(4-methylsulfonylpiperazin-1-yl)-1,3-thiazol-4-yl]methanol Chemical compound C1CN(S(=O)(=O)C)CCN1C1=NC(CO)=CS1 HSMNSHACEUGBAN-UHFFFAOYSA-N 0.000 description 1
- VYZGDLOISUXGMY-UHFFFAOYSA-N [2-(4-propan-2-ylsulfonylpiperazin-1-yl)-1,3-thiazol-4-yl]methanol Chemical compound C1CN(S(=O)(=O)C(C)C)CCN1C1=NC(CO)=CS1 VYZGDLOISUXGMY-UHFFFAOYSA-N 0.000 description 1
- RWHVRVWVWQPASY-UHFFFAOYSA-N [2-(8-oxa-3-azabicyclo[3.2.1]octan-3-yl)-1,3-oxazol-4-yl]methanol Chemical compound OCC1=COC(N2CC3CCC(O3)C2)=N1 RWHVRVWVWQPASY-UHFFFAOYSA-N 0.000 description 1
- KRNAQIPLKRYFIU-UHFFFAOYSA-N [2-(oxan-4-yl)-1,3-thiazol-4-yl]methanol Chemical compound OCC1=CSC(C2CCOCC2)=N1 KRNAQIPLKRYFIU-UHFFFAOYSA-N 0.000 description 1
- TXKFRVMFRUPLSD-VIFPVBQESA-N [2-[(2s)-2-(methoxymethyl)pyrrolidin-1-yl]-1,3-thiazol-4-yl]methanol Chemical compound COC[C@@H]1CCCN1C1=NC(CO)=CS1 TXKFRVMFRUPLSD-VIFPVBQESA-N 0.000 description 1
- YSVZGWAJIHWNQK-UHFFFAOYSA-N [3-(hydroxymethyl)-2-bicyclo[2.2.1]heptanyl]methanol Chemical compound C1CC2C(CO)C(CO)C1C2 YSVZGWAJIHWNQK-UHFFFAOYSA-N 0.000 description 1
- QJYYVSIRDJVQJW-UHFFFAOYSA-N [4-(dimethylcarbamoyl)phenyl]boronic acid Chemical compound CN(C)C(=O)C1=CC=C(B(O)O)C=C1 QJYYVSIRDJVQJW-UHFFFAOYSA-N 0.000 description 1
- UOQXKVSFZJNNRP-UHFFFAOYSA-N [4-[4-[[6-methoxy-2-(2-methoxyimidazo[2,1-b][1,3,4]thiadiazol-6-yl)-1-benzofuran-4-yl]oxymethyl]-1,3-thiazol-2-yl]piperidin-1-yl]-phenylmethanone Chemical compound N1=C2SC(OC)=NN2C=C1C(OC1=CC(OC)=C2)=CC1=C2OCC(N=1)=CSC=1C(CC1)CCN1C(=O)C1=CC=CC=C1 UOQXKVSFZJNNRP-UHFFFAOYSA-N 0.000 description 1
- WZBRVPGYHMADNR-UHFFFAOYSA-N [4-[[6-methoxy-2-(2-methoxyimidazo[2,1-b][1,3,4]thiadiazol-6-yl)-1-benzofuran-4-yl]oxymethyl]-2-morpholin-4-yl-1,3-thiazol-5-yl]methanol Chemical compound N1=C2SC(OC)=NN2C=C1C(OC1=CC(OC)=C2)=CC1=C2OCC(=C(S1)CO)N=C1N1CCOCC1 WZBRVPGYHMADNR-UHFFFAOYSA-N 0.000 description 1
- BACOJTRRFFVSSS-UHFFFAOYSA-N [4-hydroxy-4-[4-[[6-methoxy-2-(2-methoxyimidazo[2,1-b][1,3,4]thiadiazol-6-yl)-1-benzofuran-4-yl]oxymethyl]-1,3-thiazol-2-yl]piperidin-1-yl]-phenylmethanone Chemical compound N1=C2SC(OC)=NN2C=C1C(OC1=CC(OC)=C2)=CC1=C2OCC(N=1)=CSC=1C(CC1)(O)CCN1C(=O)C1=CC=CC=C1 BACOJTRRFFVSSS-UHFFFAOYSA-N 0.000 description 1
- CSCPPACGZOOCGX-WFGJKAKNSA-N acetone d6 Chemical compound [2H]C([2H])([2H])C(=O)C([2H])([2H])[2H] CSCPPACGZOOCGX-WFGJKAKNSA-N 0.000 description 1
- 238000007171 acid catalysis Methods 0.000 description 1
- 150000008043 acidic salts Chemical class 0.000 description 1
- 239000012190 activator Substances 0.000 description 1
- 230000002776 aggregation Effects 0.000 description 1
- 238000004220 aggregation Methods 0.000 description 1
- 150000001447 alkali salts Chemical class 0.000 description 1
- 150000001336 alkenes Chemical class 0.000 description 1
- 125000005081 alkoxyalkoxyalkyl group Chemical group 0.000 description 1
- 208000026935 allergic disease Diseases 0.000 description 1
- HSFWRNGVRCDJHI-UHFFFAOYSA-N alpha-acetylene Natural products C#C HSFWRNGVRCDJHI-UHFFFAOYSA-N 0.000 description 1
- NMPVEAUIHMEAQP-UHFFFAOYSA-N alpha-bromo-acetaldehyde Natural products BrCC=O NMPVEAUIHMEAQP-UHFFFAOYSA-N 0.000 description 1
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 1
- 229910052782 aluminium Inorganic materials 0.000 description 1
- VMWZRHGIAVCFNS-UHFFFAOYSA-J aluminum;lithium;tetrahydroxide Chemical compound [Li+].[OH-].[OH-].[OH-].[OH-].[Al+3] VMWZRHGIAVCFNS-UHFFFAOYSA-J 0.000 description 1
- 125000004103 aminoalkyl group Chemical group 0.000 description 1
- QGZKDVFQNNGYKY-UHFFFAOYSA-N ammonia Natural products N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 1
- 239000000908 ammonium hydroxide Substances 0.000 description 1
- 125000002178 anthracenyl group Chemical group C1(=CC=CC2=CC3=CC=CC=C3C=C12)* 0.000 description 1
- 229940127090 anticoagulant agent Drugs 0.000 description 1
- 229960003886 apixaban Drugs 0.000 description 1
- 125000006615 aromatic heterocyclic group Chemical group 0.000 description 1
- 208000028922 artery disease Diseases 0.000 description 1
- 125000004659 aryl alkyl thio group Chemical group 0.000 description 1
- 125000002102 aryl alkyloxo group Chemical group 0.000 description 1
- 125000005325 aryloxy aryl group Chemical group 0.000 description 1
- 230000007214 atherothrombosis Effects 0.000 description 1
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 1
- 229950007648 atopaxar Drugs 0.000 description 1
- 125000002785 azepinyl group Chemical group 0.000 description 1
- 125000002393 azetidinyl group Chemical group 0.000 description 1
- KXDAEFPNCMNJSK-UHFFFAOYSA-N benzene carboxamide Natural products NC(=O)C1=CC=CC=C1 KXDAEFPNCMNJSK-UHFFFAOYSA-N 0.000 description 1
- 125000003785 benzimidazolyl group Chemical group N1=C(NC2=C1C=CC=C2)* 0.000 description 1
- 125000004618 benzofuryl group Chemical group O1C(=CC2=C1C=CC=C2)* 0.000 description 1
- 125000004196 benzothienyl group Chemical group S1C(=CC2=C1C=CC=C2)* 0.000 description 1
- 235000019400 benzoyl peroxide Nutrition 0.000 description 1
- AGEZXYOZHKGVCM-UHFFFAOYSA-N benzyl bromide Chemical compound BrCC1=CC=CC=C1 AGEZXYOZHKGVCM-UHFFFAOYSA-N 0.000 description 1
- PPDJNZTUDFPAHX-UHFFFAOYSA-N benzyltrimethylammonium dichloroiodate Chemical compound Cl[I-]Cl.C[N+](C)(C)CC1=CC=CC=C1 PPDJNZTUDFPAHX-UHFFFAOYSA-N 0.000 description 1
- 125000002618 bicyclic heterocycle group Chemical group 0.000 description 1
- 150000001602 bicycloalkyls Chemical group 0.000 description 1
- SIPUZPBQZHNSDW-UHFFFAOYSA-N bis(2-methylpropyl)aluminum Chemical compound CC(C)C[Al]CC(C)C SIPUZPBQZHNSDW-UHFFFAOYSA-N 0.000 description 1
- 230000031709 bromination Effects 0.000 description 1
- 238000005893 bromination reaction Methods 0.000 description 1
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000000480 butynyl group Chemical group [*]C#CC([H])([H])C([H])([H])[H] 0.000 description 1
- 239000011575 calcium Substances 0.000 description 1
- 125000000609 carbazolyl group Chemical group C1(=CC=CC=2C3=CC=CC=C3NC12)* 0.000 description 1
- 125000002837 carbocyclic group Chemical group 0.000 description 1
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 1
- 150000001735 carboxylic acids Chemical class 0.000 description 1
- 230000007211 cardiovascular event Effects 0.000 description 1
- 239000003054 catalyst Substances 0.000 description 1
- 125000003636 chemical group Chemical group 0.000 description 1
- 238000005660 chlorination reaction Methods 0.000 description 1
- QSKWJTXWJJOJFP-UHFFFAOYSA-N chloroform;ethoxyethane Chemical compound ClC(Cl)Cl.CCOCC QSKWJTXWJJOJFP-UHFFFAOYSA-N 0.000 description 1
- 238000003776 cleavage reaction Methods 0.000 description 1
- 239000000470 constituent Substances 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- JZCCFEFSEZPSOG-UHFFFAOYSA-L copper(II) sulfate pentahydrate Chemical compound O.O.O.O.O.[Cu+2].[O-]S([O-])(=O)=O JZCCFEFSEZPSOG-UHFFFAOYSA-L 0.000 description 1
- NKNDPYCGAZPOFS-UHFFFAOYSA-M copper(i) bromide Chemical compound Br[Cu] NKNDPYCGAZPOFS-UHFFFAOYSA-M 0.000 description 1
- LSXDOTMGLUJQCM-UHFFFAOYSA-M copper(i) iodide Chemical compound I[Cu] LSXDOTMGLUJQCM-UHFFFAOYSA-M 0.000 description 1
- 230000000875 corresponding effect Effects 0.000 description 1
- 229940072645 coumadin Drugs 0.000 description 1
- 239000010779 crude oil Substances 0.000 description 1
- 125000004122 cyclic group Chemical group 0.000 description 1
- 125000000000 cycloalkoxy group Chemical group 0.000 description 1
- 125000001316 cycloalkyl alkyl group Chemical group 0.000 description 1
- 125000005112 cycloalkylalkoxy group Chemical group 0.000 description 1
- 125000001047 cyclobutenyl group Chemical group C1(=CCC1)* 0.000 description 1
- XSYZCZPCBXYQTE-UHFFFAOYSA-N cyclodecylcyclodecane Chemical compound C1CCCCCCCCC1C1CCCCCCCCC1 XSYZCZPCBXYQTE-UHFFFAOYSA-N 0.000 description 1
- 125000000596 cyclohexenyl group Chemical group C1(=CCCCC1)* 0.000 description 1
- 125000002433 cyclopentenyl group Chemical group C1(=CCCC1)* 0.000 description 1
- 229960003850 dabigatran Drugs 0.000 description 1
- YBSJFWOBGCMAKL-UHFFFAOYSA-N dabigatran Chemical group N=1C2=CC(C(=O)N(CCC(O)=O)C=3N=CC=CC=3)=CC=C2N(C)C=1CNC1=CC=C(C(N)=N)C=C1 YBSJFWOBGCMAKL-UHFFFAOYSA-N 0.000 description 1
- 230000006378 damage Effects 0.000 description 1
- 230000034994 death Effects 0.000 description 1
- 125000002704 decyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 230000001419 dependent effect Effects 0.000 description 1
- 238000001514 detection method Methods 0.000 description 1
- 229910052805 deuterium Inorganic materials 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- 229940111685 dibasic potassium phosphate Drugs 0.000 description 1
- GLUUGHFHXGJENI-UHFFFAOYSA-N diethylenediamine Chemical group C1CNCCN1 GLUUGHFHXGJENI-UHFFFAOYSA-N 0.000 description 1
- 125000001028 difluoromethyl group Chemical group [H]C(F)(F)* 0.000 description 1
- ZPWVASYFFYYZEW-UHFFFAOYSA-L dipotassium hydrogen phosphate Chemical compound [K+].[K+].OP([O-])([O-])=O ZPWVASYFFYYZEW-UHFFFAOYSA-L 0.000 description 1
- 238000006073 displacement reaction Methods 0.000 description 1
- CNXMDTWQWLGCPE-UHFFFAOYSA-N ditert-butyl-(2-phenylphenyl)phosphane Chemical group CC(C)(C)P(C(C)(C)C)C1=CC=CC=C1C1=CC=CC=C1 CNXMDTWQWLGCPE-UHFFFAOYSA-N 0.000 description 1
- 239000002552 dosage form Substances 0.000 description 1
- 238000002330 electrospray ionisation mass spectrometry Methods 0.000 description 1
- NJSUFZNXBBXAAC-UHFFFAOYSA-N ethanol;toluene Chemical compound CCO.CC1=CC=CC=C1 NJSUFZNXBBXAAC-UHFFFAOYSA-N 0.000 description 1
- KZKFLXXDLLPCKS-UHFFFAOYSA-N ethyl 2-(2-benzoylhydrazinyl)-2-oxoacetate Chemical compound CCOC(=O)C(=O)NNC(=O)C1=CC=CC=C1 KZKFLXXDLLPCKS-UHFFFAOYSA-N 0.000 description 1
- YXCVTAILWHWTKY-UHFFFAOYSA-N ethyl 2-[4-bromo-2-methyl-n-[(2-methylpropan-2-yl)oxycarbonyl]anilino]-1,3-thiazole-4-carboxylate Chemical compound CCOC(=O)C1=CSC(N(C(=O)OC(C)(C)C)C=2C(=CC(Br)=CC=2)C)=N1 YXCVTAILWHWTKY-UHFFFAOYSA-N 0.000 description 1
- XHFUVBWCMLLKOZ-UHFFFAOYSA-N ethyl 2-amino-1,3-thiazole-4-carboxylate Chemical compound CCOC(=O)C1=CSC(N)=N1 XHFUVBWCMLLKOZ-UHFFFAOYSA-N 0.000 description 1
- RZMZBHSKPLVQCP-UHFFFAOYSA-N ethyl 2-amino-2-oxoacetate Chemical compound CCOC(=O)C(N)=O RZMZBHSKPLVQCP-UHFFFAOYSA-N 0.000 description 1
- SGIBUKNPAQMWAP-UHFFFAOYSA-N ethyl 2-bromo-1,3-oxazole-4-carboxylate Chemical compound CCOC(=O)C1=COC(Br)=N1 SGIBUKNPAQMWAP-UHFFFAOYSA-N 0.000 description 1
- QWWPUBQHZFHZSF-UHFFFAOYSA-N ethyl 2-methyl-1,3-thiazole-4-carboxylate Chemical compound CCOC(=O)C1=CSC(C)=N1 QWWPUBQHZFHZSF-UHFFFAOYSA-N 0.000 description 1
- DALSSTCLLLLFJN-UHFFFAOYSA-N ethyl 2-oxo-1,3,4-oxathiazole-5-carboxylate Chemical compound CCOC(=O)C1=NSC(=O)O1 DALSSTCLLLLFJN-UHFFFAOYSA-N 0.000 description 1
- LCSWEWCKVNOEBB-UHFFFAOYSA-N ethyl 5-chloro-2-morpholin-4-yl-1,3-thiazole-4-carboxylate Chemical compound S1C(Cl)=C(C(=O)OCC)N=C1N1CCOCC1 LCSWEWCKVNOEBB-UHFFFAOYSA-N 0.000 description 1
- SNOLCJGSEPTCMD-UHFFFAOYSA-N ethyl 5-methyl-1,3-thiazole-4-carboxylate Chemical compound CCOC(=O)C=1N=CSC=1C SNOLCJGSEPTCMD-UHFFFAOYSA-N 0.000 description 1
- 125000004494 ethyl ester group Chemical group 0.000 description 1
- 125000002534 ethynyl group Chemical group [H]C#C* 0.000 description 1
- 125000003983 fluorenyl group Chemical group C1(=CC=CC=2C3=CC=CC=C3CC12)* 0.000 description 1
- 125000003709 fluoroalkyl group Chemical group 0.000 description 1
- 125000004216 fluoromethyl group Chemical group [H]C([H])(F)* 0.000 description 1
- 150000003948 formamides Chemical class 0.000 description 1
- 238000009472 formulation Methods 0.000 description 1
- 238000001640 fractional crystallisation Methods 0.000 description 1
- 239000012458 free base Substances 0.000 description 1
- 238000004108 freeze drying Methods 0.000 description 1
- 125000000524 functional group Chemical group 0.000 description 1
- 125000002541 furyl group Chemical group 0.000 description 1
- 239000011521 glass Substances 0.000 description 1
- 229940093915 gynecological organic acid Drugs 0.000 description 1
- 125000004995 haloalkylthio group Chemical group 0.000 description 1
- 125000001475 halogen functional group Chemical group 0.000 description 1
- 229960002897 heparin Drugs 0.000 description 1
- 229920000669 heparin Polymers 0.000 description 1
- 125000006343 heptafluoro propyl group Chemical group 0.000 description 1
- 125000004446 heteroarylalkyl group Chemical group 0.000 description 1
- 125000005553 heteroaryloxy group Chemical group 0.000 description 1
- 238000004896 high resolution mass spectrometry Methods 0.000 description 1
- 150000004677 hydrates Chemical class 0.000 description 1
- 229930195733 hydrocarbon Natural products 0.000 description 1
- 150000004680 hydrogen peroxides Chemical class 0.000 description 1
- 238000005984 hydrogenation reaction Methods 0.000 description 1
- USZLCYNVCCDPLQ-UHFFFAOYSA-N hydron;n-methoxymethanamine;chloride Chemical compound Cl.CNOC USZLCYNVCCDPLQ-UHFFFAOYSA-N 0.000 description 1
- XLYOFNOQVPJJNP-UHFFFAOYSA-M hydroxide Chemical compound [OH-] XLYOFNOQVPJJNP-UHFFFAOYSA-M 0.000 description 1
- 239000005457 ice water Substances 0.000 description 1
- 125000002636 imidazolinyl group Chemical group 0.000 description 1
- 125000002883 imidazolyl group Chemical group 0.000 description 1
- 238000000338 in vitro Methods 0.000 description 1
- 238000001727 in vivo Methods 0.000 description 1
- 230000002779 inactivation Effects 0.000 description 1
- 125000003387 indolinyl group Chemical group N1(CCC2=CC=CC=C12)* 0.000 description 1
- 125000001041 indolyl group Chemical group 0.000 description 1
- 230000002401 inhibitory effect Effects 0.000 description 1
- 208000014674 injury Diseases 0.000 description 1
- 229910052500 inorganic mineral Inorganic materials 0.000 description 1
- 239000011630 iodine Substances 0.000 description 1
- 230000007794 irritation Effects 0.000 description 1
- 230000000302 ischemic effect Effects 0.000 description 1
- OWFXIOWLTKNBAP-UHFFFAOYSA-N isoamyl nitrite Chemical compound CC(C)CCON=O OWFXIOWLTKNBAP-UHFFFAOYSA-N 0.000 description 1
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- 125000001972 isopentyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000003253 isopropoxy group Chemical group [H]C([H])([H])C([H])(O*)C([H])([H])[H] 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 125000005956 isoquinolyl group Chemical group 0.000 description 1
- 125000004628 isothiazolidinyl group Chemical group S1N(CCC1)* 0.000 description 1
- 125000001786 isothiazolyl group Chemical group 0.000 description 1
- XLTUPERVRFLGLJ-UHFFFAOYSA-N isothiocyanato(trimethyl)silane Chemical compound C[Si](C)(C)N=C=S XLTUPERVRFLGLJ-UHFFFAOYSA-N 0.000 description 1
- 125000003971 isoxazolinyl group Chemical group 0.000 description 1
- 125000000842 isoxazolyl group Chemical group 0.000 description 1
- 239000010410 layer Substances 0.000 description 1
- 238000004811 liquid chromatography Methods 0.000 description 1
- YNESATAKKCNGOF-UHFFFAOYSA-N lithium bis(trimethylsilyl)amide Chemical compound [Li+].C[Si](C)(C)[N-][Si](C)(C)C YNESATAKKCNGOF-UHFFFAOYSA-N 0.000 description 1
- 229910000103 lithium hydride Inorganic materials 0.000 description 1
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 1
- 235000019341 magnesium sulphate Nutrition 0.000 description 1
- NXPHGHWWQRMDIA-UHFFFAOYSA-M magnesium;carbanide;bromide Chemical compound [CH3-].[Mg+2].[Br-] NXPHGHWWQRMDIA-UHFFFAOYSA-M 0.000 description 1
- 238000003760 magnetic stirring Methods 0.000 description 1
- 230000007246 mechanism Effects 0.000 description 1
- 238000002844 melting Methods 0.000 description 1
- 230000008018 melting Effects 0.000 description 1
- IBIKHMZPHNKTHM-RDTXWAMCSA-N merck compound 25 Chemical compound C1C[C@@H](C(O)=O)[C@H](O)CN1C(C1=C(F)C=CC=C11)=NN1C(=O)C1=C(Cl)C=CC=C1C1CC1 IBIKHMZPHNKTHM-RDTXWAMCSA-N 0.000 description 1
- QARBMVPHQWIHKH-UHFFFAOYSA-N methanesulfonyl chloride Chemical compound CS(Cl)(=O)=O QARBMVPHQWIHKH-UHFFFAOYSA-N 0.000 description 1
- GQJCAQADCPTHKN-UHFFFAOYSA-N methyl 2,2-difluoro-2-fluorosulfonylacetate Chemical compound COC(=O)C(F)(F)S(F)(=O)=O GQJCAQADCPTHKN-UHFFFAOYSA-N 0.000 description 1
- ZPFCEYUYBJVUED-UHFFFAOYSA-N methyl 2-amino-5-methyl-1,3-thiazole-4-carboxylate Chemical compound COC(=O)C=1N=C(N)SC=1C ZPFCEYUYBJVUED-UHFFFAOYSA-N 0.000 description 1
- YEJDFNBFDCUMAG-UHFFFAOYSA-N methyl 2-amino-5-propan-2-yl-1,3-thiazole-4-carboxylate Chemical compound COC(=O)C=1N=C(N)SC=1C(C)C YEJDFNBFDCUMAG-UHFFFAOYSA-N 0.000 description 1
- YMKSUNUZQDIVIE-UHFFFAOYSA-N methyl 2-bromo-5-methyl-1,3-thiazole-4-carboxylate Chemical compound COC(=O)C=1N=C(Br)SC=1C YMKSUNUZQDIVIE-UHFFFAOYSA-N 0.000 description 1
- AQOIWGWWWPVWHM-UHFFFAOYSA-N methyl 2-bromo-5-propan-2-yl-1,3-thiazole-4-carboxylate Chemical compound COC(=O)C=1N=C(Br)SC=1C(C)C AQOIWGWWWPVWHM-UHFFFAOYSA-N 0.000 description 1
- XPIWVCAMONZQCP-UHFFFAOYSA-N methyl 2-oxobutanoate Chemical compound CCC(=O)C(=O)OC XPIWVCAMONZQCP-UHFFFAOYSA-N 0.000 description 1
- MQONVZMIFQQQHA-UHFFFAOYSA-N methyl 3-bromo-2-oxopropanoate Chemical compound COC(=O)C(=O)CBr MQONVZMIFQQQHA-UHFFFAOYSA-N 0.000 description 1
- GWCAGRNQSBWSQA-UHFFFAOYSA-N methyl 4-[4-(hydroxymethyl)-1,3-thiazol-2-yl]benzoate Chemical compound C1=CC(C(=O)OC)=CC=C1C1=NC(CO)=CS1 GWCAGRNQSBWSQA-UHFFFAOYSA-N 0.000 description 1
- YNLYLZLGDNLGCF-UHFFFAOYSA-N methyl 4-[4-[[tert-butyl(dimethyl)silyl]oxymethyl]-1,3-thiazol-2-yl]benzoate Chemical compound C1=CC(C(=O)OC)=CC=C1C1=NC(CO[Si](C)(C)C(C)(C)C)=CS1 YNLYLZLGDNLGCF-UHFFFAOYSA-N 0.000 description 1
- JYIXBVQJZAJAAM-UHFFFAOYSA-N methyl 5-chloro-2-morpholin-4-yl-1,3-thiazole-4-carboxylate Chemical compound S1C(Cl)=C(C(=O)OC)N=C1N1CCOCC1 JYIXBVQJZAJAAM-UHFFFAOYSA-N 0.000 description 1
- RTUJXYVGZKFYOX-UHFFFAOYSA-N methyl 5-methyl-2-[(2-methylpropan-2-yl)oxycarbonylamino]-1,3-thiazole-4-carboxylate Chemical compound COC(=O)C=1N=C(NC(=O)OC(C)(C)C)SC=1C RTUJXYVGZKFYOX-UHFFFAOYSA-N 0.000 description 1
- 229940095102 methyl benzoate Drugs 0.000 description 1
- 125000000250 methylamino group Chemical group [H]N(*)C([H])([H])[H] 0.000 description 1
- 230000003278 mimic effect Effects 0.000 description 1
- 235000010755 mineral Nutrition 0.000 description 1
- 239000011707 mineral Substances 0.000 description 1
- 125000002911 monocyclic heterocycle group Chemical group 0.000 description 1
- GSLBUBZXFUYMSW-UHFFFAOYSA-N morpholine-4-carbothioamide Chemical compound NC(=S)N1CCOCC1 GSLBUBZXFUYMSW-UHFFFAOYSA-N 0.000 description 1
- 239000012452 mother liquor Substances 0.000 description 1
- ZUMVAKLNFYFWGK-UHFFFAOYSA-N n-(4-bromo-2-methylphenyl)-4-[[6-methoxy-2-(2-methoxyimidazo[2,1-b][1,3,4]thiadiazol-6-yl)-1-benzofuran-4-yl]oxymethyl]-1,3-thiazol-2-amine Chemical compound N1=C2SC(OC)=NN2C=C1C(OC1=CC(OC)=C2)=CC1=C2OCC(N=1)=CSC=1NC1=CC=C(Br)C=C1C ZUMVAKLNFYFWGK-UHFFFAOYSA-N 0.000 description 1
- RACJERJJTUONSE-UHFFFAOYSA-N n-(cyanomethyl)-4-[4-[[6-methoxy-2-(2-methoxyimidazo[2,1-b][1,3,4]thiadiazol-6-yl)-1-benzofuran-4-yl]oxymethyl]-5-methyl-1,3-thiazol-2-yl]-n-methylbenzamide Chemical compound N1=C2SC(OC)=NN2C=C1C(OC1=CC(OC)=C2)=CC1=C2OCC(=C(S1)C)N=C1C1=CC=C(C(=O)N(C)CC#N)C=C1 RACJERJJTUONSE-UHFFFAOYSA-N 0.000 description 1
- SYSQUGFVNFXIIT-UHFFFAOYSA-N n-[4-(1,3-benzoxazol-2-yl)phenyl]-4-nitrobenzenesulfonamide Chemical class C1=CC([N+](=O)[O-])=CC=C1S(=O)(=O)NC1=CC=C(C=2OC3=CC=CC=C3N=2)C=C1 SYSQUGFVNFXIIT-UHFFFAOYSA-N 0.000 description 1
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- FPCJTUJMONOSAN-UHFFFAOYSA-N n-methoxy-n-methyl-2-phenyl-1,3-thiazole-4-carboxamide Chemical compound CON(C)C(=O)C1=CSC(C=2C=CC=CC=2)=N1 FPCJTUJMONOSAN-UHFFFAOYSA-N 0.000 description 1
- 125000000740 n-pentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000001971 neopentyl group Chemical group [H]C([*])([H])C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- YCWSUKQGVSGXJO-NTUHNPAUSA-N nifuroxazide Chemical group C1=CC(O)=CC=C1C(=O)N\N=C\C1=CC=C([N+]([O-])=O)O1 YCWSUKQGVSGXJO-NTUHNPAUSA-N 0.000 description 1
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 1
- MWUXSHHQAYIFBG-UHFFFAOYSA-N nitrogen oxide Inorganic materials O=[N] MWUXSHHQAYIFBG-UHFFFAOYSA-N 0.000 description 1
- 239000012457 nonaqueous media Substances 0.000 description 1
- 125000002868 norbornyl group Chemical group C12(CCC(CC1)C2)* 0.000 description 1
- 239000012038 nucleophile Substances 0.000 description 1
- 239000010502 orange oil Substances 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 239000003960 organic solvent Substances 0.000 description 1
- 125000000160 oxazolidinyl group Chemical group 0.000 description 1
- 125000002971 oxazolyl group Chemical group 0.000 description 1
- 125000003566 oxetanyl group Chemical group 0.000 description 1
- 239000007800 oxidant agent Substances 0.000 description 1
- JLPJFSCQKHRSQR-UHFFFAOYSA-N oxolan-3-one Chemical compound O=C1CCOC1 JLPJFSCQKHRSQR-UHFFFAOYSA-N 0.000 description 1
- MUJIDPITZJWBSW-UHFFFAOYSA-N palladium(2+) Chemical compound [Pd+2] MUJIDPITZJWBSW-UHFFFAOYSA-N 0.000 description 1
- 230000037361 pathway Effects 0.000 description 1
- 125000006340 pentafluoro ethyl group Chemical group FC(F)(F)C(F)(F)* 0.000 description 1
- 125000006194 pentinyl group Chemical group 0.000 description 1
- 125000001147 pentyl group Chemical group C(CCCC)* 0.000 description 1
- 230000002093 peripheral effect Effects 0.000 description 1
- LIGACIXOYTUXAW-UHFFFAOYSA-N phenacyl bromide Chemical compound BrCC(=O)C1=CC=CC=C1 LIGACIXOYTUXAW-UHFFFAOYSA-N 0.000 description 1
- WLJVXDMOQOGPHL-UHFFFAOYSA-N phenylacetic acid Chemical compound OC(=O)CC1=CC=CC=C1 WLJVXDMOQOGPHL-UHFFFAOYSA-N 0.000 description 1
- 125000002071 phenylalkoxy group Chemical group 0.000 description 1
- 125000003884 phenylalkyl group Chemical group 0.000 description 1
- IPNPIHIZVLFAFP-UHFFFAOYSA-N phosphorus tribromide Chemical compound BrP(Br)Br IPNPIHIZVLFAFP-UHFFFAOYSA-N 0.000 description 1
- 150000004885 piperazines Chemical group 0.000 description 1
- 125000004193 piperazinyl group Chemical group 0.000 description 1
- 125000005936 piperidyl group Chemical group 0.000 description 1
- LYKMMUBOEFYJQG-UHFFFAOYSA-N piperoxan Chemical compound C1OC2=CC=CC=C2OC1CN1CCCCC1 LYKMMUBOEFYJQG-UHFFFAOYSA-N 0.000 description 1
- 229940020573 plavix Drugs 0.000 description 1
- 239000003880 polar aprotic solvent Substances 0.000 description 1
- 125000005575 polycyclic aromatic hydrocarbon group Chemical group 0.000 description 1
- 239000011698 potassium fluoride Substances 0.000 description 1
- 235000003270 potassium fluoride Nutrition 0.000 description 1
- CASUWPDYGGAUQV-UHFFFAOYSA-M potassium;methanol;hydroxide Chemical compound [OH-].[K+].OC CASUWPDYGGAUQV-UHFFFAOYSA-M 0.000 description 1
- 230000003389 potentiating effect Effects 0.000 description 1
- DTGLZDAWLRGWQN-UHFFFAOYSA-N prasugrel Chemical compound C1CC=2SC(OC(=O)C)=CC=2CN1C(C=1C(=CC=CC=1)F)C(=O)C1CC1 DTGLZDAWLRGWQN-UHFFFAOYSA-N 0.000 description 1
- 229960004197 prasugrel Drugs 0.000 description 1
- 108090000765 processed proteins & peptides Proteins 0.000 description 1
- 102000004196 processed proteins & peptides Human genes 0.000 description 1
- TVDSBUOJIPERQY-UHFFFAOYSA-N prop-2-yn-1-ol Chemical compound OCC#C TVDSBUOJIPERQY-UHFFFAOYSA-N 0.000 description 1
- DRINJBFRTLBHNF-UHFFFAOYSA-N propane-2-sulfonyl chloride Chemical compound CC(C)S(Cl)(=O)=O DRINJBFRTLBHNF-UHFFFAOYSA-N 0.000 description 1
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000002568 propynyl group Chemical group [*]C#CC([H])([H])[H] 0.000 description 1
- 230000004224 protection Effects 0.000 description 1
- 102000004169 proteins and genes Human genes 0.000 description 1
- 108090000623 proteins and genes Proteins 0.000 description 1
- 230000017854 proteolysis Effects 0.000 description 1
- 230000002797 proteolythic effect Effects 0.000 description 1
- 125000000561 purinyl group Chemical group N1=C(N=C2N=CNC2=C1)* 0.000 description 1
- 125000003373 pyrazinyl group Chemical group 0.000 description 1
- 150000003217 pyrazoles Chemical class 0.000 description 1
- 125000002098 pyridazinyl group Chemical group 0.000 description 1
- 125000000719 pyrrolidinyl group Chemical group 0.000 description 1
- 150000004728 pyruvic acid derivatives Chemical class 0.000 description 1
- 150000003242 quaternary ammonium salts Chemical class 0.000 description 1
- 125000005493 quinolyl group Chemical group 0.000 description 1
- 125000004621 quinuclidinyl group Chemical group N12C(CC(CC1)CC2)* 0.000 description 1
- 239000000700 radioactive tracer Substances 0.000 description 1
- 238000011084 recovery Methods 0.000 description 1
- 230000000306 recurrent effect Effects 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 238000012552 review Methods 0.000 description 1
- 229910052702 rhenium Inorganic materials 0.000 description 1
- 125000006413 ring segment Chemical group 0.000 description 1
- 229960001148 rivaroxaban Drugs 0.000 description 1
- KGFYHTZWPPHNLQ-AWEZNQCLSA-N rivaroxaban Chemical compound S1C(Cl)=CC=C1C(=O)NC[C@@H]1OC(=O)N(C=2C=CC(=CC=2)N2C(COCC2)=O)C1 KGFYHTZWPPHNLQ-AWEZNQCLSA-N 0.000 description 1
- 238000007127 saponification reaction Methods 0.000 description 1
- 239000012047 saturated solution Substances 0.000 description 1
- 230000007017 scission Effects 0.000 description 1
- 238000000926 separation method Methods 0.000 description 1
- LIVNPJMFVYWSIS-UHFFFAOYSA-N silicon monoxide Inorganic materials [Si-]#[O+] LIVNPJMFVYWSIS-UHFFFAOYSA-N 0.000 description 1
- AKHNMLFCWUSKQB-UHFFFAOYSA-L sodium thiosulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=S AKHNMLFCWUSKQB-UHFFFAOYSA-L 0.000 description 1
- 235000019345 sodium thiosulphate Nutrition 0.000 description 1
- OUGXVMIOSSJAFK-UHFFFAOYSA-M sodium;dichloromethane;hydrogen carbonate Chemical compound [Na+].ClCCl.OC([O-])=O OUGXVMIOSSJAFK-UHFFFAOYSA-M 0.000 description 1
- 239000011343 solid material Substances 0.000 description 1
- 238000007614 solvation Methods 0.000 description 1
- 239000011877 solvent mixture Substances 0.000 description 1
- 238000000527 sonication Methods 0.000 description 1
- 125000003003 spiro group Chemical group 0.000 description 1
- 125000005017 substituted alkenyl group Chemical group 0.000 description 1
- 125000004426 substituted alkynyl group Chemical group 0.000 description 1
- 238000006467 substitution reaction Methods 0.000 description 1
- 125000000020 sulfo group Chemical group O=S(=O)([*])O[H] 0.000 description 1
- 150000003457 sulfones Chemical class 0.000 description 1
- 125000000472 sulfonyl group Chemical group *S(*)(=O)=O 0.000 description 1
- 238000001308 synthesis method Methods 0.000 description 1
- PBOPEUGCAJAJIX-UHFFFAOYSA-N tert-butyl 4-[4-[[tert-butyl(dimethyl)silyl]oxymethyl]-1,3-thiazol-2-yl]benzoate Chemical compound C1=CC(C(=O)OC(C)(C)C)=CC=C1C1=NC(CO[Si](C)(C)C(C)(C)C)=CS1 PBOPEUGCAJAJIX-UHFFFAOYSA-N 0.000 description 1
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- WOXCOXYFCZCSAN-UHFFFAOYSA-N tert-butyl n-(4-bromo-2-methylphenyl)-n-[4-(hydroxymethyl)-1,3-thiazol-2-yl]carbamate Chemical compound CC1=CC(Br)=CC=C1N(C(=O)OC(C)(C)C)C1=NC(CO)=CS1 WOXCOXYFCZCSAN-UHFFFAOYSA-N 0.000 description 1
- KKVALNSGBHZFGP-UHFFFAOYSA-N tert-butyl n-[(2,4-dimethoxyphenyl)methyl]-n-[4-(hydroxymethyl)-5-methyl-1,3-thiazol-2-yl]carbamate Chemical compound COC1=CC(OC)=CC=C1CN(C(=O)OC(C)(C)C)C1=NC(CO)=C(C)S1 KKVALNSGBHZFGP-UHFFFAOYSA-N 0.000 description 1
- 239000012414 tert-butyl nitrite Substances 0.000 description 1
- IOGXOCVLYRDXLW-UHFFFAOYSA-N tert-butyl nitrite Chemical compound CC(C)(C)ON=O IOGXOCVLYRDXLW-UHFFFAOYSA-N 0.000 description 1
- ZQCYJSPBHBUXJF-UHFFFAOYSA-N tert-butyl-[[2-(2-fluoropyridin-4-yl)-5-propan-2-yl-1,3-thiazol-4-yl]methoxy]-dimethylsilane Chemical compound CC(C)(C)[Si](C)(C)OCC1=C(C(C)C)SC(C=2C=C(F)N=CC=2)=N1 ZQCYJSPBHBUXJF-UHFFFAOYSA-N 0.000 description 1
- KSHMSVMVGCQBAV-UHFFFAOYSA-N tert-butyl-dimethyl-[(2-morpholin-4-yl-1,3-thiazol-4-yl)methoxy]silane Chemical compound CC(C)(C)[Si](C)(C)OCC1=CSC(N2CCOCC2)=N1 KSHMSVMVGCQBAV-UHFFFAOYSA-N 0.000 description 1
- KEJSSPMHLXYZLE-UHFFFAOYSA-N tert-butyl-dimethyl-[[5-(trifluoromethyl)-1,3-thiazol-4-yl]methoxy]silane Chemical compound CC(C)(C)[Si](C)(C)OCC=1N=CSC=1C(F)(F)F KEJSSPMHLXYZLE-UHFFFAOYSA-N 0.000 description 1
- VLTWLVXBVKSBTI-UHFFFAOYSA-N tert-butyl-ethyl-methyl-[(5-methyl-2-morpholin-4-yl-1,3-oxazol-4-yl)methoxy]silane Chemical compound O1C(C)=C(CO[Si](C)(CC)C(C)(C)C)N=C1N1CCOCC1 VLTWLVXBVKSBTI-UHFFFAOYSA-N 0.000 description 1
- 125000006092 tetrahydro-1,1-dioxothienyl group Chemical group 0.000 description 1
- 125000003718 tetrahydrofuranyl group Chemical group 0.000 description 1
- 125000001712 tetrahydronaphthyl group Chemical group C1(CCCC2=CC=CC=C12)* 0.000 description 1
- 125000004632 tetrahydrothiopyranyl group Chemical group S1C(CCCC1)* 0.000 description 1
- 125000006090 thiamorpholinyl sulfone group Chemical group 0.000 description 1
- 125000006089 thiamorpholinyl sulfoxide group Chemical group 0.000 description 1
- 125000001984 thiazolidinyl group Chemical group 0.000 description 1
- 125000000335 thiazolyl group Chemical group 0.000 description 1
- 125000001544 thienyl group Chemical group 0.000 description 1
- 125000005309 thioalkoxy group Chemical group 0.000 description 1
- 125000005403 thiohaloalkoxy group Chemical group 0.000 description 1
- 125000004055 thiomethyl group Chemical group [H]SC([H])([H])* 0.000 description 1
- OEKWJQXRCDYSHL-FNOIDJSQSA-N ticagrelor Chemical compound C1([C@@H]2C[C@H]2NC=2N=C(N=C3N([C@H]4[C@@H]([C@H](O)[C@@H](OCCO)C4)O)N=NC3=2)SCCC)=CC=C(F)C(F)=C1 OEKWJQXRCDYSHL-FNOIDJSQSA-N 0.000 description 1
- 229960002528 ticagrelor Drugs 0.000 description 1
- 210000001519 tissue Anatomy 0.000 description 1
- 125000005424 tosyloxy group Chemical group S(=O)(=O)(C1=CC=C(C)C=C1)O* 0.000 description 1
- 231100000419 toxicity Toxicity 0.000 description 1
- 230000001988 toxicity Effects 0.000 description 1
- 201000010875 transient cerebral ischemia Diseases 0.000 description 1
- 125000004306 triazinyl group Chemical group 0.000 description 1
- FAQYAMRNWDIXMY-UHFFFAOYSA-N trichloroborane Chemical compound ClB(Cl)Cl FAQYAMRNWDIXMY-UHFFFAOYSA-N 0.000 description 1
- 125000003866 trichloromethyl group Chemical group ClC(Cl)(Cl)* 0.000 description 1
- GRGCWBWNLSTIEN-UHFFFAOYSA-N trifluoromethanesulfonyl chloride Chemical compound FC(F)(F)S(Cl)(=O)=O GRGCWBWNLSTIEN-UHFFFAOYSA-N 0.000 description 1
- 125000004385 trihaloalkyl group Chemical group 0.000 description 1
- 229910052722 tritium Inorganic materials 0.000 description 1
- 238000001291 vacuum drying Methods 0.000 description 1
- 208000021331 vascular occlusion disease Diseases 0.000 description 1
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 1
- 229960005080 warfarin Drugs 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D513/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for in groups C07D463/00, C07D477/00 or C07D499/00 - C07D507/00
- C07D513/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for in groups C07D463/00, C07D477/00 or C07D499/00 - C07D507/00 in which the condensed system contains two hetero rings
- C07D513/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/433—Thidiazoles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/4353—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems
- A61K31/4355—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems the heterocyclic ring system containing a five-membered ring having oxygen as a ring hetero atom
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4427—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems
- A61K31/4439—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems containing a five-membered ring with nitrogen as a ring hetero atom, e.g. omeprazole
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
- A61K31/4523—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems
- A61K31/454—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems containing a five-membered ring with nitrogen as a ring hetero atom, e.g. pimozide, domperidone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/496—Non-condensed piperazines containing further heterocyclic rings, e.g. rifampin, thiothixene or sparfloxacin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/4965—Non-condensed pyrazines
- A61K31/497—Non-condensed pyrazines containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/50—Pyridazines; Hydrogenated pyridazines
- A61K31/5025—Pyridazines; Hydrogenated pyridazines ortho- or peri-condensed with heterocyclic ring systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/5375—1,4-Oxazines, e.g. morpholine
- A61K31/5377—1,4-Oxazines, e.g. morpholine not condensed and containing further heterocyclic rings, e.g. timolol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/5375—1,4-Oxazines, e.g. morpholine
- A61K31/5386—1,4-Oxazines, e.g. morpholine spiro-condensed or forming part of bridged ring systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/54—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one sulfur as the ring hetero atoms, e.g. sulthiame
- A61K31/541—Non-condensed thiazines containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/55—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/55—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole
- A61K31/553—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole having at least one nitrogen and one oxygen as ring hetero atoms, e.g. loxapine, staurosporine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K51/00—Preparations containing radioactive substances for use in therapy or testing in vivo
- A61K51/02—Preparations containing radioactive substances for use in therapy or testing in vivo characterised by the carrier, i.e. characterised by the agent or material covalently linked or complexing the radioactive nucleus
- A61K51/04—Organic compounds
- A61K51/041—Heterocyclic compounds
- A61K51/044—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine, rifamycins
- A61K51/0453—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine, rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/02—Antithrombotic agents; Anticoagulants; Platelet aggregation inhibitors
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B59/00—Introduction of isotopes of elements into organic compounds ; Labelled organic compounds per se
- C07B59/002—Heterocyclic compounds
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D519/00—Heterocyclic compounds containing more than one system of two or more relevant hetero rings condensed among themselves or condensed with a common carbocyclic ring system not provided for in groups C07D453/00 or C07D455/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/05—Isotopically modified compounds, e.g. labelled
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/55—Design of synthesis routes, e.g. reducing the use of auxiliary or protecting groups
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Epidemiology (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Cardiology (AREA)
- Diabetes (AREA)
- Heart & Thoracic Surgery (AREA)
- Hematology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Physics & Mathematics (AREA)
- Optics & Photonics (AREA)
- Vascular Medicine (AREA)
- Urology & Nephrology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Oxygen Or Sulfur (AREA)
- Plural Heterocyclic Compounds (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
Un compuesto de fórmula I: **(Ver fórmula)** o un estereoisómero, un tautómero, una sal o solvato farmacéuticamente aceptables del mismo, en donde: W es O; R0 es R1; Y es S o -CR8=CR9-; R1 se selecciona independientemente del grupo que consiste en: H, halo alquilo C1-C2, ciclopropilo alcoxi C1-C2, alquiltio C1-C2, halo alquilo C1-C2-, que contiene de 1 a 5 halógenos, en donde halo es F o Cl, y halo cicloalquilo C3-C4 R8 y R9 se seleccionan independientemente del grupo que consiste en: H, flúor, cloro, alquilo C1-C3, alcoxi C1-C2 y haloalquilo C1-C2; siempre que al menos uno de R1, R8 y R9 sea distinto de H; R2 es H; X1 se selecciona del grupo que consiste en CH o N; X2, X3 y X4 se seleccionan independientemente de CR3; R3 se selecciona del grupo que consiste en H, alquilo C1-C3, alcoxi C1-C3, flúor, cloro, OCF3 y haloalquilo C1-C2, que contiene de 1 a 5 halógenos; R4 y R5 se seleccionan independientemente de H y metilo; **(Ver fórmula)** es un anillo heteroarilo de 5 miembros seleccionado entre tiazol, tiadiazol, oxazol, oxadiazol y triazol; R6 se selecciona del grupo que consiste en H, halo, OCF3, OCHF2, OH, CN, NO2, NR11R12, alcoxicarbonilo C1-C4, (C=O)NR11R12 y alquilo C1-C5 sustituido con de 0 a 7 grupos seleccionados independientemente de halo, CF3, OCF3, OH, hidroxialquilo C1-C4, alcoxi C1-C4, alcoxi C1-C4-alcoxi C1-C4, dialquilaminofenil C1-C4-alquilo C1-C4-, (dialcoxi C1-C4-alquil C1-C4)-alquilo C1-C4, dialquilamino C1-C4, cicloalquilo C3-C6 y alquiltio C1-C4 o R6 es B-D-, en donde D es un enlazador que se selecciona entre: un enlace simple, **(Ver fórmula)** alquileno C1-C4 sustituido con de 0 a 4 grupos seleccionados independientemente de halo u OH, alquilenoxi C1-C4, alquenileno C2-C6 y B se selecciona del grupo que consiste en: arilo C6-C10 sustituido con de 0 a 3 grupos seleccionados independientemente del grupo que consiste en halo, alcoxi C1-C4, haloalcoxi -C1-C4, alquilo C1-C4, haloalquilo C1-C4, cicloalquilo C3-C6, ciano, nitro, NR11R12, OH, alquilcarboniloxi C1-C4-alquilo C1-C4, hidroxialquilo C1-C4, COOR14, SO2R14, (C=O)NR11R12, SO2NR11R12, N(R13)(C=O)NR11R12, N(R13)(C=O)OR14, N(R13)(C=O)R14, NR13S(O)R14, NR13SO2R14, O(C=O)NR11R12, O(C=O)OR14, O(C=O)R14, (C=O)OR14 y heteroarilo de 5-6 miembros, heteroarilo de 5 a 10 miembros sustituido con de 0 a 3 grupos seleccionados independientemente del grupo que consiste en halo, alcoxi C1-C4, haloalcoxi C1-C4, alquilo C1-C4, haloalquilo C1-C4, cicloalquilo C3-C6, ciano, nitro, NR11R12, OH, alquilcarboniloxi C1-C4-alquilo C1-C4, hidroxialquilo C1-C4, COOR14, SO2R14, (C=O)NR11R12, SO2NR11R12, N(R13)(C=O)NR11R12, N(R13)(C=O)OR14, N(R13)(C=O)R14, NR13S(O)R14, NR13SO2R14, O(C=O)NR11R12, O(C=O)OR14, O(C=O)R14, (C=O)OR14, heteroarilo de 5-6 miembros y (CH2)fenilo, heterociclilo de 4 a 10 miembros que contiene átomos de carbono y de 1 a 2 heteroátomos adicionales seleccionados entre N, O y S, y sustituido con de 0 a 3 grupos seleccionados independientemente del grupo que consiste en halo, oxo, -(CHR13)n1-heteroarilo de 5 o 6 miembros sustituido con de 0 a 3 grupos seleccionados independientemente del grupo que consiste en halo, alcoxi C1-C4, alquilo C1-C4, ciclopropilo, CF3, OCF3 y CF2CH3; NR13S(O)R14, NR13SO2R14, -(CHR13)n1-heterociclilo de 4 a 10 miembros sustituido con de 0 a 3 grupos seleccionados independientemente del grupo que consiste en halo, alcoxi C1-C4, alquilo C1-C4, ciclopropilo, CF3, OCF3 y CF2CH3; OH, hidroxialquilo C1-C4, alcoxi C1-C4, haloalcoxi C1-C4, dialquilamino C1-C4-alquilo C1-C4, NR11R12, ciano, alquilo C1-C4, haloalquilo C1-C4, cicloalquilo C3-C6, alcoxi C1-C4-alquilo C1-C4, cicloalquil C3-C6-alquilcarbonilo C1-C4, arilcarbonilo C6-C10, alquilcarboniloxi C1-C4-alquilo C1-C4, COOR14, SO2R14, (C=O)NR11R12, SO2NR11R12, N(R13)(C=O)NR11R12, N(R13)(C=O)OR14, N(R13)(C=O)R14, O(C=O)NR11R12, O(C=O)OR14, O(C=O)R14, (C=O)OR14 y arilo C6-C10 sustituido con de 0 a 3 grupos seleccionados independientemente del grupo que consiste en halo, alcoxi C1-C4, alquilo C1-C4, ciclopropilo, alcoxicarbonilo C1-C4, (C=O)NR11R12, CF3, OCF3 y CF2CH3; cicloalquilo C3-C6 que puede contener insaturación, sustituido con de 0 a 3 grupos seleccionados independientemente del grupo que consiste en halo, CF3, OCF3, heteroarilo de 5 o 6 miembros, OH, oxo, hidroxialquilo C1-C4, arilo C6-C10, COOH, oxo, alcoxicarbonilo C1-C4, (C=O)NR11R12 y alquilo C1-C4 y espirocicloalquilo C5-C11 que puede contener insaturación y que opcionalmente contiene de 1 a 3 heteroátomos seleccionados entre O, N o S y que se sustituye con de 0 a 3 grupos seleccionados independientemente del grupo que consiste en halo, CF3, OCF3, heteroarilo de 5 o 6 miembros, OH, hidroxialquilo C1-C4, arilo C6-C10 y alquilo C1-C4; R11 y R12 se seleccionan, en cada caso, independientemente entre el grupo que consiste en: H, alquilo C1-C4, haloalquilo C1-C4, alquenilo C2-C4, -(CR14R14)n1-fenilo sustituido con de 0 a 3 grupos seleccionados independientemente entre el grupo que consiste en halo, alcoxi C1-C4, alquilo C1-C4, ciclopropilo, CF3, OCF3, heteroarilo de 5 o 6 miembros, OH, OCHF2, dialquilamino C1-C4 y ciano, -(CHR13)n1-cicloalquilo C3-C6 sustituido con de 0 a 3 grupos seleccionados independientemente del grupo que consiste en halo, CF3, OCF3, heteroarilo de 5 o 6 miembros, OH, hidroxialquilo C1-C4 y alquilo C1-C4, -(CHR13)n1-heterociclilo de 4 a 10 miembros sustituido con de 0 a 3 grupos seleccionados independientemente del grupo que consiste en halo, CF3, OCF3, heteroarilo de 5 o 6 miembros, OH, oxo, hidroxialquilo C1-C4 y alquilo C1-C4, -(CHR13)n1-heteroarilo de 5 a 10 miembros sustituido con de 0 a 3 grupos seleccionados independientemente del grupo que consiste en halo, CF3, OCF3, heteroarilo de 5 o 6 miembros, OH, hidroxialquilo C1-C4 y alquilo C1-C4, dialquilamino C1-C4-alquilo C1-C4, dialcoxi C1-C4-alquilo C1-C4, hidroxialquilo C1-C4, cianoalquilo C1-C4, alcoxi C1-C4-alquilo C1-C4, alcoxicarbonil C1-C4-alquilo C1-C4, alcoxicarbonilo C1-C4, alquilcarbonilo C1-C4, fenilcarbonilo; alcoxicarbonilamino C1-C4-alquilcarbonilo C1-C4 y dialquilamino C1-C4-alquilcarbonilo C1-C4, de manera alternativa, R11 y R12, cuando se unen al mismo nitrógeno, se combinan para formar un anillo heterocíclico de 4 a 8 miembros que contiene átomos de carbono, sustituido con de 0 a 3 grupos seleccionados independientemente del grupo que consiste en halo, CF3, CHF2, OCF3, OCHF2, OCH2F, heteroarilo de 5 o 6 miembros, OH, oxo, hidroxialquilo C1-C4, alquilo C1-C4 y alcoxi C1-C4, y de 0 a 2 heteroátomos adicionales seleccionados entre N, NR13, O y S(O)p; R13 se selecciona, en cada caso, independientemente del grupo que consiste en H, alquilo C1-C6 y -(CH2)fenilo; R14 se selecciona, en cada caso, independientemente del grupo que consiste en H, alquilo C1-C6, haloalquilo C1-C4-, alcoxicarbonilamino C1-C4- y -(CH2)n1fenilo sustituido con de 0 a 3 grupos seleccionados independientemente del grupo que consiste en halo, alcoxi C1-C4, alquilo C1-C4, ciclopropilo, CF3, OCF3, heteroarilo de 5 o 6 miembros, OH, OCHF2, dialquilamino C1-C4 y ciano; R7 se selecciona entre el grupo que consiste en H, halo, hidroxilo, oxo, alquilo C1-C3, hidroxialquilo C1-C3 y haloalquilo C1-C2, que contiene de 1 a 5 halógenos; n1, en cada caso, se selecciona de 0, 1, 2 o 3 y p, en cada caso, es independientemente 0, 1 y 2.
Description
DESCRIPCIÓN
Derivados de imidazotiazol e imidazopirazina como inhibidores del receptor activado por proteasa 4 (PAR4) para tratar la agregación plaquetaria
Campo de la invención
La presente invención proporciona novedosos inhibidores de imidazotiadiazol y de imidazopiridazina de la agregación plaquetaria, que son útiles para la prevención o el tratamiento de trastornos tromboembólicos. La presente invención también se refiere a composiciones farmacéuticas que contienen estos compuestos y a dichos compuestos para su uso en el tratamiento de trastornos tromboembólicos.
Antecedentes de la invención
Las enfermedades tromboembólicas siguen siendo la principal causa de muerte en los países desarrollados, a pesar de la disponibilidad de anticoagulantes, tales como warfarina (COUMADIN®), heparina, heparinas de bajo peso molecular (LMWH), pentasacáridos sintéticos e inhibidores plaquetarios, tales como aspirina y clopidogrel (PLAVIX®).
Las terapias de inhibición plaquetaria actuales tienen ciertas limitaciones, que incluyen un mayor riesgo de sangrado así como una eficacia parcial (reducción del riesgo cardiovascular relativo en el intervalo del 20 al 30 %). Por ello, sigue siendo un objetivo importante el descubrimiento y el desarrollo de antitrombóticos orales o parenterales seguros y eficaces para la prevención y el tratamiento de un amplio espectro de trastornos tromboembólicos.
La trombina alfa es el activador más potente conocido de agregación plaquetaria y desgranulación. La activación plaquetaria está involucrada causalmente en las oclusiones vasculares aterotrombóticas. La trombina activa las plaquetas mediante la escisión de los receptores acoplados a la proteína G, denominados receptores activados por proteasa (PAR). Los PAR proporcionan sus propios ligandos crípticos presentes en el dominio extracelular del terminal N que son desenmascarados por la proteólisis, con la consecuente fijación intramolecular al receptor para inducir la señalización (mecanismo de ligando unido; Coughlin, S.R., Nature, 407:258-264 (2000)). Los péptidos sintéticos que imitan la secuencia del terminal N recién formado después de la activación proteolítica pueden inducir la señalización independientemente de la escisión del receptor. Las plaquetas cumplen una función clave en los eventos aterotrombóticos. Las plaquetas humanas expresan al menos dos receptores de trombina, generalmente denominados PAR1 y PAR4. Los inhibidores de PAR1 han sido objeto de exhaustivas investigaciones y varios compuestos, que incluyen vorapaxar y atopaxar, han alcanzado etapas más avanzadas de los ensayos clínicos. Recientemente, en el ensayo de fase III TRACER en pacientes con síndrome coronario agudo (ACS), el vorapaxar no redujo de manera considerable los eventos cardiovasculares, sino que aumentó en gran medida el riesgo de sangrado grave (Tricoci, P. et al., N. Eng. J. Med., 366(1):20-33 (2012). Por ello, aun es necesario descubrir nuevos inhibidores plaquetarios que tengan mayor eficacia y menos efectos secundarios de sangrado.
Existen varios informes iniciales de estudios preclínicos de inhibidores de PAR4. Lee, F-Y. et al., "Synthesis of 1-Benzyl-3-(5'-hydroxymethyl-2'-furyl)indazole Analogues as Novel Antiplatelet Agents", J. Med. Chem., 44(22):3746-3749 (2001) divulgan en el resumen, que el compuesto

"se descubrió que es un inhibidor selectivo y potente de la activación plaquetaria dependiente del receptor activado por proteasa tipo 4 (PAR4)".
El compuesto 58 también se denomina YD-3 en Wu, C-C. et al., "Selective Inhibition of Protease-activated Receptor 4-dependent Platelet Activation by YD-3", Thromb. Haemost., 87:1026-1033 (2002). Véase también Chen, H.S. et al., "Synthesis and platelet activity", J. Bioorg. Med. Chem., 16:1262-1278 (2008).
Los documentos EP1166785 A1 y EP0667345 describen varios derivados de pirazol que son útiles como inhibidores de la agregación plaquetaria. El documento EP-A0005783 divulga nuevos compuestos de imidotiadiazol para su uso como antitrombóticos. El documento WO2011/074658 divulga nuevos inhibidores plaquetarios. El documento EP-A0185345 divulga derivados imidazo para su uso como antitrombóticos.
Sumario de la invención
Se ha descubierto que los compuestos de imidazotiadiazol e imidazopiridazina de acuerdo con la presente invención son antagonistas de PAR4 que inhiben la agregación plaquetaria en los ensayos de agregación plaquetaria inducida por la trombina gamma. Además, se ha demostrado que un compuesto o compuestos de la presente invención inhibe la agregación plaquetaria en un ensayo de agregación plaquetaria inducida por la trombina alfa e inhibe la formación de trombos en un modelo de trombosis arterial en monos cinomolgos.
En consecuencia, la presente invención proporciona análogos de imidazotiadiazol y de imidazopiridazina novedosos que son antagonistas de PAR4 y son útiles como inhibidores selectivos de la agregación plaquetaria, incluyendo estereoisómeros, tautómeros, sales o solvatos farmacéuticamente aceptables de los mismos.
La presente invención también proporciona procesos e intermedios para fabricar los compuestos de la presente invención o estereoisómeros, tautómeros, sales o solvatos farmacéuticamente aceptables de los mismos.
La presente invención también proporciona composiciones farmacéuticas que comprenden un vehículo farmacéuticamente aceptable y al menos uno de los compuestos de la presente invención o estereoisómeros, tautómeros, sales o solvatos farmacéuticamente aceptables del mismo.
La presente invención también proporciona compuestos de la presente invención o estereoisómeros, tautómeros, sales o solvatos farmacéuticamente aceptables de los mismos para su uso en el tratamiento o profilaxis de trastornos tromboembólicos.
La presente invención también proporciona los compuestos de la presente invención o estereoisómeros, tautómeros, sales o solvatos farmacéuticamente aceptables de los mismos, para su uso en terapia.
Otras características y ventajas de la invención serán evidentes a partir de la siguiente descripción detallada y las reivindicaciones.
Breve descripción de las figuras
La Figura 1A es un gráfico que muestra la eficacia del ejemplo 3 para inhibir la agregación de plaquetas humanas lavadas estimuladas por trombina alfa 1,5 nM;
La Figura 1B es un gráfico que muestra el CI50 del ejemplo 3 para inhibir la agregación plaquetaria inducida por trombina alfa y
La Figura 1C es un gráfico que muestra la eficacia antitrombótica del ejemplo 3 en el modelo de trombosis de la arteria carótida inducida por lesiones electrolíticas en monos cinomolgos.
Descripción detallada
En una forma de realización, la presente invención proporciona compuestos de imidazotiadiazol o imidazopiridazina, estereoisómeros, tautómeros, sales o solvatos de los mismos, de fórmula I, que tienen la estructura:

I
en donde:
W es O;
R0 es R1;
Y es S o -CR8=CR9-;
R1 se selecciona independientemente del grupo que consiste en:
H,
halo,
alquilo C1-C2,
ciclopropilo,
alcoxi C1-C2,
alquiltio C1-C2,
haloalquilo C1-C2, que contiene de 1 a 5 halógenos, en donde halo es F o Cl,
y
halocicloalquilo-C3-C4;
R8 y R9 se seleccionan independientemente del grupo que consiste en:
H,
flúor,
cloro,
alquilo C1-C3,
alcoxi C1-C2 y
haloalquilo C1-C2,
siempre que al menos uno de R, R8 y R9 sea distinto de H;
R2 es H,
X1 se selecciona del grupo que consiste en CH o N;
X2, X3 y X4 se seleccionan independientemente de CR3;
R3 se selecciona del grupo que consiste en H, alquilo C1-C3, alcoxi C1-C3, flúor, cloro, OCF3 y haloalquilo C1-C2, que contiene de 1 a 5 halógenos;
R4 y R5 se seleccionan independientemente de H y metilo;

es un anillo heteroarilo de 5 miembros seleccionado entre tiazol, tiadiazol, oxazol, oxadiazol y triazol;
R6 se selecciona del grupo que consiste en H, halo, OCF3, OCHF2, OH, CN, NO2, NR11R12, alcoxicarbonilo C1-C4, (C=O)NR11R12 y alquilo C1-C5 sustituido con de 0 a 7 grupos seleccionados independientemente entre halo, CF3, OCF3, OH, hidroxialquilo C1-C4, alcoxi C1-C4, alcoxi C1-C4-alcoxi C1-C4, dialquilaminofenil C1-C4-alquilo C1-C4, (dialcoxi C1-C4-alquil C1-C4)-alquilo C1-C4, dialquilamino C1-C4, cicloalquilo C3-C6 y alquiltio C1-C4 o
R6 es B-D-, en donde D es un enlazador, que se selecciona entre:
un enlace simple,
-O-,
-S-,

alquileno C1-C4 sustituido con de 0 a 4 grupos seleccionados independientemente entre halo u OH, alquilenoxi C1-C4,
alquenileno C2-C6 y
B se selecciona del grupo que consiste en:
arilo C6-C10 sustituido con de 0 a 3 grupos seleccionados independientemente del grupo que consiste en halo, alcoxi C1-C4, haloalcoxi -C1-C4, alquilo C1-C4, haloalquilo C1-C4, cicloalquilo C3-C6, ciano, nitro, NR11R12, OH, alquilcarboniloxi C1-C4-alquilo C1-C4, hidroxialquilo C1-C4, COOR14, SO2R14, (C=O)NR11R12, SO2NR11R12, N(R13)(C=O)NR11R12, N(R13)(C=O)OR14, N(R13)(C=O)R14, NR13S(O)R14, NR13SO2R14, O(C=O)NR11R12, O(C=O)OR14, O(C=O)R14, (C=O)OR14 y heteroarilo de 5-6 miembros,
heteroarilo de 5 a 10 miembros sustituido con de 0 a 3 grupos seleccionados independientemente del grupo que consiste en halo, alcoxi C1-C4, haloalcoxi C1-C4, alquilo C1-C4, haloalquilo C1-C4, cicloalquilo C3-C6, ciano, nitro, NR11R12, OH, alquilcarboniloxi C1-C4-alquilo C1-C4, hidroxialquilo C1-C4, COOR14, SO2R14, (C=O)NR11R12, SO2NR11R12, N(R13)(C=O)NR11R12, N(R13)(C=O)OR14, N(R13)(C=O)R14, NR13S(O)R14, NR13SO2R14, O(C=O)NR11R12, O(C=O)OR14, O(C=O)R14, (C=O)OR14, heteroarilo de 5-6 miembros y (CH2)fenilo,
heterociclilo de 4 a 10 miembros que contiene átomos de carbono y de 1 a 2 heteroátomos adicionales seleccionados entre N, O y S y sustituido con de 0 a 3 grupos seleccionados independientemente del grupo que consiste en halo, oxo, -(CHR13)n1-heteroarilo de 5 o 6 miembros sustituido con de 0 a 3 grupos seleccionados independientemente del grupo que consiste en halo, alcoxi C1-C4, alquilo C1-C4, ciclopropilo, CF3, OCF3 y CF2CH3; NR13S(O)R14, NR13SO2R14, -(CHR13)n1-heterociclilo de 4 a 10 miembros sustituido con de 0 a 3 grupos seleccionados independientemente del grupo que consiste en halo, alcoxi C1-C4, alquilo C1-C4, ciclopropilo, CF3, OCF3 y CF2CH3; OH, hidroxialquilo C1-C4, alcoxi C1-C4, haloalcoxi C1-C4, dialquilamino C1-C4-alquilo C1-C4, NR11R12, ciano, alquilo C1-C4, haloalquilo C1-C4, cicloalquilo C3-C6, alcoxi C1-C4-alquilo C1-C4, cicloalquil C3-C6-alquilcarbonilo C1-C4-, arilcarbonilo C6-C10, alquilcarboniloxi C1-C4-alquilo C1-C4, COOR14, SO2R14, (C=O)NR11R12, SO2NR11R12, N(R13)(C=O)NR11R12, N(R13)(C=O)OR14, N(R13)(C=O)R14, O(C=O)NR11R12, O(C=O)OR14, O(C=O)R14, (C=O)OR14 y arilo C6-C10 sustituido con de 0 a 3 grupos seleccionados independientemente del grupo que consiste en halo, alcoxi C1-C4, alquilo C1-C4, ciclopropilo, alcoxicarbonilo C1-C4, (C=O)NR11R12, CF3, OCF3y CF2CH3;
cicloalquilo C3-C6 que puede contener insaturación, sustituido con de 0 a 3 grupos seleccionados independientemente del grupo que consiste en halo, CF3, OCF3, heteroarilo de 5 o 6 miembros, OH, oxo, hidroxialquilo C1-C4, arilo C6-C10, COOH, oxo, alcoxicarbonilo C1-C4, (C=O)NR11R12 y alquilo C1-C4; y espirocicloalquilo C5-C11 que puede contener insaturación y que opcionalmente contiene de 1 a 3 heteroátomos seleccionados entre O, N o S y que se sustituye con de 0 a 3 grupos seleccionados independientemente del grupo que consiste en halo, CF3, OCF3, heteroarilo de 5 o 6 miembros, OH, hidroxialquilo C1-C4, arilo C6-C10 y alquilo C1-C4;
R11 y R12 se seleccionan, en cada caso, independientemente entre el grupo que consiste en:
H,
alquilo C1-C4,
haloalquilo C1-C4,
alquenilo C2-C4,
-(CR14R14)n1-fenilo sustituido con de 0 a 3 grupos seleccionados independientemente entre el grupo que consiste en halo, alcoxi C1-C4, alquilo C1-C4, ciclopropilo, CF3, OCF3, heteroarilo de 5 o 6 miembros, Oh , OCHF2, dialquilamino C1-C4 y ciano;
-(CHR13)n1-cicloalquilo C3-C6 sustituido con de 0 a 3 grupos seleccionados independientemente del grupo que consiste en halo, CF3, OCF3, heteroarilo de 5 o 6 miembros, OH, hidroxialquilo C1-C4 y alquilo C1-C4;
-(CHR13)n1-heterociclilo de 4 a 10 miembros sustituido con de 0 a 3 grupos seleccionados independientemente del grupo que consiste en halo, CF3, OCF3, heteroarilo de 5 o 6 miembros, OH, oxo, hidroxialquilo C1-C4 y alquilo C1-C4;
-(CHR13)n1-heteroarilo de 5 a 10 miembros sustituido con de 0 a 3 grupos seleccionados independientemente del grupo que consiste en halo, CF3, OCF3, heteroarilo de 5 o 6 miembros, OH, hidroxialquilo C1-C4 y alquilo C1-C4;
dialquilamino C1-C4-alquilo C1-C4,
dialcoxi C1-C4-alquilo C1-C4,
hidroxialquilo C1-C4,
cianoalquilo C1-C4,
alcoxi C1-C4-alquilo C1-C4,
alcoxicarbonil C1-C4-alquilo C1-C4,
alcoxicarbonilo C1-C4,
alquilcarbonilo C1-C4,
fenilcarbonilo;
alcoxicarbonilamino C1-C4-alquilcarbonilo C1-C4- y
dialquilamino C1-C4-alquilcarbonilo C1-C4,
de manera alternativa, R11 y R12, cuando se unen al mismo nitrógeno, se combinan para formar un anillo heterocíclico de 4 a 8 miembros que contiene átomos de carbono, sustituidos con de 0 a 3 grupos seleccionados independientemente del grupo que consiste en halo, CF3, CHF2, OCF3, OCHF2, OCH2F, heteroarilo de 5 o 6 miembros, OH, oxo, hidroxialquilo C1-C4, alquilo C1-C4 y alcoxi C1-C4 y de 0 a 2 heteroátomos adicionales seleccionados entre N, NR13, O y S(O)p;
R13 se selecciona, en cada caso, independientemente del grupo que consiste en H, alquilo C1-C6 y -(CH2)fenilo; R14 se selecciona, en cada caso, independientemente del grupo que consiste en H, alquilo C1-C6, haloalquilo C1-C4-, alcoxicarbonilamino C1-C4- y -(CH2)n1fenilo sustituido con de 0 a 3 grupos seleccionados independientemente del grupo que consiste en halo, alcoxi C1-C4, alquilo C1-C4, ciclopropilo, CF3, OCF3, heteroarilo de 5 o 6 miembros, OH, OCHF2, dialquilamino C1-C4 y ciano;
R7 se selecciona entre el grupo que consiste en H, halo, hidroxilo, oxo, alquilo C1-C3, hidroxialquilo C1-C3 y haloalquilo C1-C2, que contiene de 1 a 5 halógenos;
n1, en cada caso, se selecciona de 0, 1, 2 o 3 y
p, en cada caso, es independientemente 0, 1 y 2.
En otra realización, la presente invención proporciona compuestos, estereoisómeros, tautómeros, sales o solvatos
de los mismos, en donde:
Y es S o -CH=CH-;
R0 es R1;
R1 se selecciona entre el grupo que consiste en:
alquilo C1-C2,
alquiltio C1-C2
alcoxi C1-C2 y
haloalquilo C1-C2, que contiene de 1 a 5 halógenos, en donde halo es F o Cl;
R3 se selecciona entre el grupo que consiste en:
H,
alcoxi C1-C2,
flúor y
cloro y
cada uno de R4 y R5 es H.
Aun en otra realización, la presente invención proporciona compuestos, estereoisómeros, tautómeros, sales o solvatos de los mismos, en donde los compuestos son compuestos de fórmula IA y IB:

En una realización, la presente invención proporciona compuestos, estereoisómeros, tautómeros, sales o solvatos de los mismos, en donde los compuestos son compuestos de fórmula IC, ID, IE y IF:
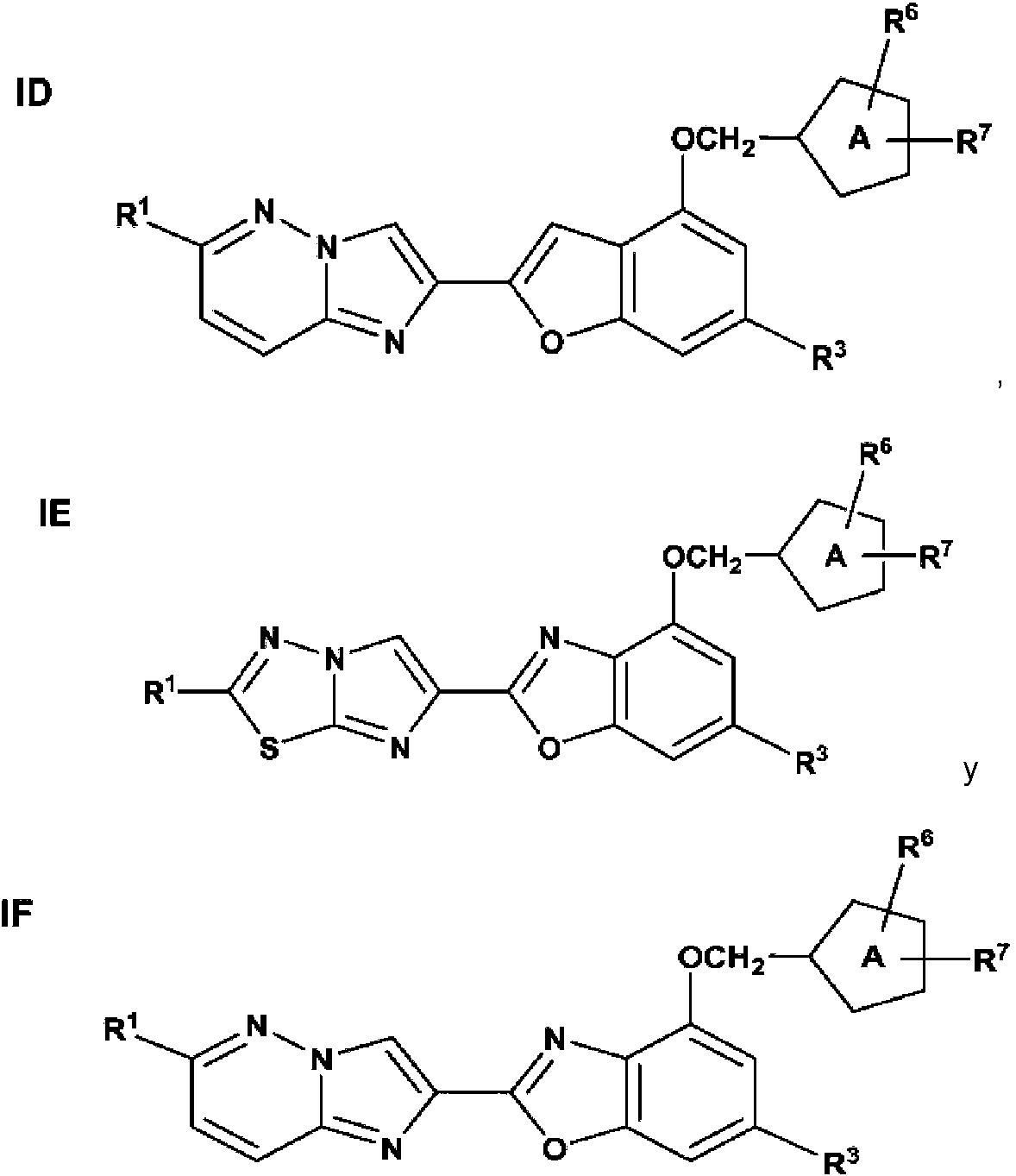
En una realización, la presente invención proporciona compuestos, estereoisómeros, tautómeros, sales o solvatos de los mismos, en donde los compuestos son compuestos de fórmula IA.
En una realización, la presente invención proporciona compuestos, estereoisómeros, tautómeros, sales o solvatos de los mismos, en donde los compuestos son compuestos de fórmula IB.
En una realización, la presente invención proporciona compuestos, estereoisómeros, tautómeros, sales o solvatos de los mismos, en donde los compuestos son compuestos de fórmula IC.
En una realización, la presente invención proporciona compuestos, estereoisómeros, tautómeros, sales o solvatos de los mismos, en donde los compuestos son compuestos de fórmula ID.
En una realización, la presente invención proporciona compuestos, estereoisómeros, tautómeros, sales o solvatos de los mismos, en donde los compuestos son compuestos de fórmula IE.
En una realización, la presente invención proporciona compuestos, estereoisómeros, tautómeros, sales o solvatos de los mismos, en donde los compuestos son compuestos de fórmula IF.
Aun en otra realización, la presente invención proporciona compuestos, estereoisómeros, tautómeros, sales o solvatos de los mismos, en donde R6 es

Y

se selecciona entre el grupo que consiste en:
arilo C6-C10 sustituido con de 0 a 3 grupos seleccionados independientemente del grupo que consiste en halo, alcoxi C1-C4, haloalcoxi C1-C4, alquilo C1-C4, haloalquilo C1-C4, cicloalquilo C3-C6, ciano, nitro, NR11R12, OH, alquilcarboniloxi C1-C4-alquilo C1-C4, hidroxialquilo C1-C4, COOR14, SO2R14, (C=O)NR11R12, SO2NR11R12, N(R13)(C=O)NR11R12, N(R13)(C=O)OR14, N(R13)(C=O)R14, NR13S(O)R14, NR13SO2R14, O(C=O)NR11R12, O(C=O)OR14, O(C=O)R14, (C=O)OR14 y heteroarilo de 5-6 miembros,
heteroarilo de 5 a 10 miembros sustituido con de 0 a 3 grupos seleccionados independientemente del grupo que consiste en halo, alcoxi C1-C4, haloalcoxi C1-C4, alquilo C1-C4, haloalquilo C1-C4, cicloalquilo C3-C6, ciano, nitro, NR11R12, OH, alquilcarboniloxi C1-C4-alquilo C1-C4, hidroxialquilo C1-C4, COOR14, SO2R14, (C=O)NR11R12, SO2NR11R12, N(R13)(C=O)NR11R12, N(R13)(C=O)OR14, N(R13)(C=O)R14, NR13S(O)R14, NR13SO2R14, O(C=O)NR11R12, O(C=O)OR14, O(C=O)R14, (C=O)OR14, heteroarilo de 5-6 miembros y (CH2)fenilo,
heterociclilo de 4 a 10 miembros que contiene átomos de carbono y de 1 a 2 heteroátomos adicionales seleccionados entre N, O y S, y sustituido con de 0 a 3 grupos seleccionados independientemente del grupo que consiste en halo, oxo, -(CHR13)n1-heteroarilo de 5 o 6 miembros sustituido con de 0 a 3 grupos seleccionados independientemente del grupo que consiste en halo, alcoxi C1-C4, alquilo C1-C4, ciclopropilo, CF3, OCF3 y CF2CH3; NR13S(O)R14, NR13SO2R14, -(CHR13)n1-heterociclilo de 4 a 10 miembros sustituido con de 0 a 3 grupos seleccionados independientemente del grupo que consiste en halo, alcoxi C1-C4, alquilo C1-C4, ciclopropilo, CF3, OCF3 y CF2CH3; Oh , hidroxialquilo C1-C4, alcoxi C1-C4, haloalcoxi C1-C4, dialquilamino C1-C4-alq NR11R12, ciano, alquilo C1-C4, haloalquilo C1-C4, cicloalquilo C3-C6, alcoxi C1-C4-alquilo C1-C4, cicloalquil C3-C6-alquilcarbonilo C1-C4, arilcarbonilo C6-C10, alquilcarboniloxi C1-C4-alquilo C1-C4, COOR14, SO2R14, (C=O)NR11R12, SO2NR11R12, N(R13)(C=O)NR11R12, N(R13)(C=O)OR14, N(R13)(C=O)R14, O(C=O)NR11R12, O(C=O)OR14, O(C=O)R14, (C=O)OR14 y arilo C6-C10 sustituido con de 0 a 3 grupos seleccionados independientemente entre el grupo que consiste en halo, alcoxi C1-C4, alquilo C1-C4, ciclopropilo, alcoxicarbonilo C1-C4, (C=O)NR11R12, CF3, OCF3 y CF2CH3 y
cicloalquilo C3-C6 que puede contener insaturación, sustituido con de 0 a 3 grupos seleccionados independientemente del grupo que consiste en halo, CF3, OCF3, heteroarilo de 5 o 6 miembros, OH, oxo, hidroxialquilo C1-C4, arilo C6-C10, COOH, oxo, alcoxicarbonilo C1-C4, (C=O)NR11R12 y alquilo C1-C4.
Aun en otra realización, la presente invención proporciona compuestos, estereoisómeros, tautómeros, sales o solvatos de los mismos, en donde R6 se selecciona entre el grupo que consiste en H, halo, OCF3, OCHF2, NR11R12, alcoxicarbonilo C1-C4, (C=O)NR11R12 y alquilo C1-C5 sustituido con de 0 a 7 grupos seleccionados independientemente entre halo, CF3, OCF3, OH, hidroxi-alquilo C1-C4, alcoxi C1-C4, alcoxi C1-C4-alcoxi C1-C4, dialquilaminofenil C1-C4-alquilo C1-C4, (dialcoxi C1-C4-alquil C1-C4)-alquilo C1-C4, dialquilamino C1-C4 y cicloalquilo
C3-C6.
Aun en otra realización, la presente invención proporciona compuestos, estereoisómeros, tautómeros, sales o solvatos de los mismos, en donde

Aun en otra realización, la presente invención proporciona compuestos, estereoisómeros, tautómeros, sales o solvatos de los mismos, en donde:

se selecciona del grupo que consiste en

se selecciona del grupo que consiste en:
arilo C6-C10 sustituido con de 0 a 3 grupos seleccionados independientemente del grupo que consiste en halo, alcoxi C1-C4, haloalcoxi C1-C4, alquilo C1-C4, haloalquilo C1-C4, cicloalquilo C3-C6, ciano, nitro, NR11R12, OH, alquilcarboniloxi C1-C4-alquilo C1-C4, hidroxialquilo C1-C4, COOR14, SO2R14, (C=O)NR11R12, SO2NR11R12, N(R13)(C=O)NR11R12, N(R13)(C=O)OR14, N(R13)(C=O)R14, NR13S(O)R14, NR13SO2R14, O(C=O)NR11R12, O(C=O)OR14, O(C=O)R14, (C=O)OR14 y heteroarilo de 5-6 miembros,
heteroarilo de 5 a 10 miembros sustituido con de 0 a 3 grupos seleccionados independientemente del grupo que consiste en halo, alcoxi C1-C4, haloalcoxi C1-C4, alquilo C1-C4, haloalquilo C1-C4, cicloalquilo C3-C6, ciano, nitro, NR11R12, OH, alquilcarboniloxi C1-C4-alquilo C1-C4, hidroxialquilo C1-C4, COOR14, SO2R14, (C=O)NR11R12, SO2NR11R12, N(R13)(C=O)NR11R12, N(R13)(C=O)OR14, N(R13)(C=O)R14, NR13S(O)R14, NR13SO2R14, O(C=O)NR11R12, O(C=O)OR14, O(C=O)R14, (C=O)OR14, heteroarilo de 5-6 miembros y (CH2)fenilo,
heterociclilo de 4 a 10 miembros que contiene átomos de carbono y de 1 a 2 heteroátomos adicionales seleccionados de N, O y S, y sustituido con de 0 a 3 grupos seleccionados independientemente del grupo que consiste en halo, oxo, -(CHR13)n1-heteroarilo de 5 o 6 miembros sustituido con de 0 a 3 grupos seleccionados independientemente del grupo que consiste en halo, alcoxi C1-C4, alquilo C1-C4, ciclopropilo, CF3, OCF3 y CF2CH3; NR13S(O)R14, NR13SO2R14, -(CHR13)n1-heterociclilo de 4 a 10 miembros sustituido con de 0 a 3 grupos seleccionados independientemente del grupo que consiste en halo, alcoxi C1-C4, alquilo C1-C4, ciclopropilo, CF3, OCF3 y CF2CH3; OH, hidroxialquilo C1-C4, alcoxi C1-C4, haloalcoxi C1-C4, dialquilamino C1-C4-alquilo C1-C4, NR11R12, ciano, alquilo C1-C4, haloalquilo C1-C4, cicloalquilo C3-C6, alcoxi C1-C4-alquilo C1-C4, cicloalquil C3-C6-alquilcarbonilo C1-C4, arilcarbonilo C6-C10, alquilcarboniloxi C1-C4-alquilo C1-C4, COOR14, SO2R14, (C=O)NR11R12, SO2NR11R12, N(R13)(C=O)NR11R12, N(R13)(C=O)OR14, N(R13)(C=O)R14, O(C=O)NR11R12, O(C=O)OR14, O(C=O)R14, (C=O)OR14 y arilo C6-C10 sustituido con de 0 a 3 grupos seleccionados independientemente del grupo que consiste en halo, alcoxi C1-C4, alquilo C1-C4, ciclopropilo, alcoxicarbonilo C1-C4, (C=O)NR11R12, CF3, OCF3 y CF2CH3 y
cicloalquilo C3-C6 que puede contener insaturación, sustituido con de 0 a 3 grupos seleccionados independientemente del grupo que consiste en halo, CF3, OCF3, heteroarilo de 5 o 6 miembros, OH, oxo, hidroxialquilo C1-C4, arilo C6-C10, COOH, oxo, alcoxicarbonilo C1-C4, (C=O)NR11R12 y alquilo C1-C4.
En una realización, la presente invención proporciona compuestos, estereoisómeros, tautómeros, sales o solvatos de los mismos, en donde:

se selecciona del grupo que consiste en

se selecciona del grupo que consiste en:
arilo C6-C10 sustituido con de 0 a 3 grupos seleccionados independientemente del grupo que consiste en halo, alcoxi C1-C4, haloalcoxi C1-C4, alquilo C1-C4, haloalquilo C1-C4, cicloalquilo C3-C6, ciano, nitro, NR11R12, OH, alquilcarboniloxi C1-C4-alquilo C1-C4, hidroxialquilo C1-C4, COOR14, SO2R14, (C=O)NR11R12, SO2NR11R12, N(R13)(C=O)NR11R12, N(R13)(C=O)OR14, N(R13)(C=O)R14, NR13S(O)R14, NR13SO2R14, O(C=O)NR11R12, O(C=O)OR14, O(C=O)R14, (C=O)OR14 y heteroarilo de 5-6 miembros,
heteroarilo de 5 a 10 miembros sustituido con de 0 a 3 grupos seleccionados independientemente del grupo que consiste en halo, alcoxi C1-C4, haloalcoxi C1-C4, alquilo C1-C4, haloalquilo C1-C4, cicloalquilo C3-C6, ciano, nitro, NR11R12, OH, alquilcarboniloxi C1-C4-alquilo C1-C4, hidroxialquilo C1-C4, COOR14, SO2R14, (C=O)NR11R12, SO2NR11R12, N(R13)(C=O)NR11R12, N(R13)(C=O)OR14, N(R13)(C=O)R14, NR13S(O)R14, NR13SO2R14, O(C=O)NR11R12, O(C=O)OR14, O(C=O)R14, (C=O)OR14, heteroarilo de 5-6 miembros y (CH2)fenilo,
heterociclilo de 4 a 10 miembros que contiene átomos de carbono y de 1 a 2 heteroátomos adicionales seleccionados de N, O y S y sustituido con de 0 a 3 grupos seleccionados independientemente del grupo que consiste en halo, oxo, -(CHR13)n1-heteroarilo de 5 o 6 miembros sustituido con de 0 a 3 grupos seleccionados independientemente del grupo que consiste en halo, alcoxi C1-C4, alquilo C1-C4, ciclopropilo, CF3, OCF3 y CF2CH3; NR13S(O)R14, NR13SO2R14, -(CHR13)n1-heterociclilo de 4 a 10 miembros sustituido con de 0 a 3 grupos seleccionados independientemente del grupo que consiste en halo, alcoxi C1-C4, alquilo C1-C4, ciclopropilo, CF3, OCF3 y CF2CH3; OH, hidroxialquilo C1-C4, alcoxi C1-C4, haloalcoxi C1-C4, dialquilamino C1-C4-alquilo C1-C4, NR11R12, ciano, alquilo C1-C4, haloalquilo C1-C4, cicloalquilo C3-C6, alcoxi C1-C4- alquilo C1-C4, cicloalquil C3-C6-alquilcarbonilo C1-C4, arilcarbonilo C6-C10, alquilcarboniloxi C1-C4-alquilo C1-C4, COOR14, SO2R14, (C=O)NR11R12, SO2NR11R12, N(R13)(C=O)NR11R12, N(R13)(C=O)OR14, N(R13)(C=O)R14, O(C=O)NR11R12, O(C=O)OR14, O(C=O)R14, (C=O)OR14 y arilo C6-C10 sustituido con de 0 a 3 grupos seleccionados independientemente del grupo que consiste en halo, alcoxi C1-C4, alquilo C1-C4, ciclopropilo, alcoxicarbonilo C1-C4-, (C=O)NR11R12, CF3, OCF3 y CF2CH3 y
cicloalquilo C3-C6 que puede contener insaturación, sustituido con de 0 a 3 grupos seleccionados independientemente del grupo que consiste en halo, CF3, OCF3, heteroarilo de 5 o 6 miembros, OH, oxo, hidroxialquilo C1-C4, arilo C6-C10, COOH, oxo, alcoxicarbonilo C1-C4, (C=O)NR11R12 y alquilo C1-C4 y
R7 se selecciona del grupo que consiste en H, halo, alquilo C1-C4 e hidroxialquilo C1-C3.
En aun otra forma de realización, la presente invención proporciona compuestos, estereoisómeros, tautómeros, sales o solvatos de los mismos, en donde:
Y es S o -CH=CH-;
R1 se selecciona del grupo que consiste en:
CH3,
SCH3,
OCH3,
CH(CH)3F,
C(CH3)F2 y
CF3;
cada uno de X2 y X4 es CH y
X3 es CR3 en donde R3 es OCH3, F o Cl;
el anillo heteroarilo de 5 miembros

se selecciona entre el grupo que consiste en

a) fenilo o fenilo sustituido, ue se selecciona entre el ru o ue consiste en

b) heterociclilo, que se selecciona entre el grupo que consiste en

c) heterociclilo sustituido, que se selecciona entre el grupo que consiste en

y v J
d) cicloalquilo, que se selecciona entre el ru o ue consiste en

e) heteroaril-alquilo C1-C3, que es

f) heteroarilo, que se selecciona entre el grupo que consiste en

g) heteroarilo sustituido, que se selecciona entre el grupo que consiste en
h) haloalquilo, que es -CF3,
i) halo, que es Br,
j) alcoxialcoxialquilo, que es -CH2OCH2CH2OCH3,
k) alquilo que se selecciona entre -CH2CH3 o CH3,
l) aril-alquilo C1-C3 que se selecciona entre

m) H,
n ) -NRq11uRe12 s,e selecciona entre el ru o ue consiste en


R7 se selecciona entre el grupo que consiste en H y alquilo C1-C3.
En una realización, la presente invención proporciona compuestos, estereoisómeros, tautómeros, sales o solvatos de los mismos, en donde:
se selecciona entre el grupo que consiste en

fenilo sustituido, que se selecciona entre el grupo que consiste en
heteroarilo, que se selecciona entre el grupo que consiste en

heterociclilo, que se selecciona entre el grupo que consiste en

cicloalquilo, que es

R7 se selecciona entre H o alquilo C1-C3.
En aún otra realización, la presente invención proporciona compuestos, estereoisómeros, tautómeros, sales o solvatos de los mismos, en donde:

se selecciona del ru o ue consiste en

R6 se selecciona del grupo que consiste en H, halo, CF3, OCF3, OCHF2, cicloalquiloxi C3-C6, OH, CN, NO2, NR11R12, alcoxicarbonilo C1-C4, C(=O) NR11R12, feniloxi, feniltio, fenil-alcoxi Ci-C4,heteroaril-alcoxi C1-C4 y fenilalquilo C1-C4 y
R7 es H o CH3.
En aún otra realización, la presente invención proporciona compuestos, estereoisómeros, tautómeros, sales o solvatos de los mismos, en donde:
Y es S;
X1 es CH;
cada uno de R4 y R5 es H;
R1 es OCH3 o CH(CH3) F;
R3 es OCH3 o F;
cada uno de R4 y R5 es H;

R6 es

R7 es H o CH3.
En una realización, la presente invención proporciona compuestos, estereoisómeros, tautómeros, sales o solvatos de los mismos, en donde:
Y es CH=CH;
X1 es CH;
cada uno de R4 y R5 es H;
R1 es CH3;
R3 es OCH3 o F;

R6 se selecciona entre el grupo que consiste en

R7 es H. 
En otra realización, la presente invención proporciona compuestos, estereoisómeros, tautómeros, sales o solvatos de los mismos, en donde:
R0 es alcoxi C1-C4, alquilo Ci-C, haloalquilo C1-C2, que contiene de 1 a 5 halógenos, en donde halo es F o Cl o halo;
R3 es alcoxi C1-C o halo;
R4 es H y
R5 es H.
En aún otra realización, la presente invención proporciona compuestos, estereoisómeros, tautómeros, sales o solvatos del mismo en donde:
X1 es CH;

en donde R1 se selecciona independientemente entre alquilo C1-C4, haloalquilo C1-C2, que contiene de 1 a 5
halógenos, en donde halo es F o Cl, o alcoxi C1-C3 y
cada uno de R8 y R9 es H.
En aún otra realización, la presente invención proporciona compuestos, estereoisómeros, tautómeros, sales o solvatos de los mismos, en donde:
X1 es CH o N;
R3 es OCH3 o flúor y
R6 es
alquilo C1-C3,
haloalquilo C1-C2,
fenilo,
fenilo sustituido con de 0 a 3 sustituyentes seleccionados entre 1 o 2 grupos halo, haloalquilo C1-C2 que contiene de 1 a 5 halógenos, alquilo C 1 - C 3 y alcoxi C 1 - C 3 o

En una realización, la presente invención proporciona compuestos, estereoisómeros, tautómeros, sales o solvatos de los mismos, en donde
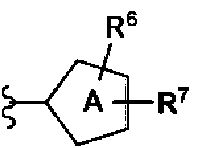
es un anillo heteroarilo de 5 miembros que contiene uno o dos átomos de N y un átomo de S o tres átomos de N. En otra realización, la presente invención proporciona compuestos, estereoisómeros, tautómeros, sales o solvatos de los mismos, en donde:

X1 es CH;
R1 es OCH3, CH3 o C2H5;
R1a es CH3;
R3 es OCH3 o F;

cada uno de R8 y R9 es H.
En aún otra realización, la presente invención proporciona compuestos, estereoisómeros, tautómeros, sales o solvatos de los mismos, en donde:

es
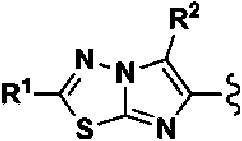
en donde
R1 es OCH3, -CHFCH3 o -CF2CH3;
X1 es CH;
R3 es OCH3 o flúor;

es
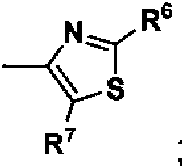
R6 se selecciona del grupo que consiste en:
a) fenilo sustituido con de 0 a 3 grupos seleccionados independientemente entre el grupo que consiste en flúor, cloro, -CH3, - CF3, OH, ciano, -OCH3, -OCF3, -CH2OH, -C(CH3)2OH, -SO2CH3 y (C=O)NR11R12, en donde NR11R12 se selecciona entre:

b) piridinilo o pirimidinilo sustituido con de 0 a 3 grupos seleccionados independientemente del grupo que consiste en flúor, cloro, -CH3 y -OCH3;
c) tetrahidropiranilo, tetrahidrotiopiranilo, piperidinilo, morfolinilo, tiomorfolinilo o piperazina sustituido con de 0 a 3 grupos seleccionados independientemente entre el grupo que consiste en flúor, OH, -CH3 y -NH2 y d) ciclohexilo sustituido con de 0 a 3 grupos seleccionados independientemente entre el grupo que consiste en flúor, OH y -NH2 y
R7 se selecciona entre el grupo que consiste en H y -CH3.
En aún otra realización, la presente invención proporciona compuestos, estereoisómeros, tautómeros, sales o solvatos de los mismos, en donde:

es

en donde
R1 es CH3;
X1 es CH;
R3 es OCH3;

es

R6 se selecciona entre el grupo que consiste en:
a) fenilo sustituido con de 0 a 3 grupos seleccionados independientemente entre el grupo que consiste en cloro, -CF3, ciano, -OCH3, -OCF3, -SO2CH3 y (C=O)N(CH3h;
b) piridinilo o pirimidinilo sustituido con de 0 a 3 grupos seleccionados independientemente del grupo que consiste en flúor, cloro, -CH3 y -OCH3 y
c) piperidinilo, morfolinilo o tiomorfolinilo sustituido con de 0 a 3 grupos seleccionados independientemente entre el grupo que consiste en flúor, OH, -CH3 y -NH2;
R7 se selecciona entre el grupo que consiste en H y -CH3 y
cada uno de R8 y R9 es H.
En una realización, la presente invención proporciona compuestos, estereoisómeros, tautómeros, sales o solvatos de los mismos, en donde:
R1 se selecciona independientemente entre el grupo que consiste en:
H,
flúor,
cloro,
alquilo C1-C2,
alcoxi C1-C2,
alquiltio C1-C2 y
haloalquilo C1-C2, que contiene de 1 a 5 halógenos, en donde halo es F o Cl;
R8 y R9 se seleccionan independientemente del grupo que consiste en:
H,
flúor,
cloro,
CH3,
OCF3,
CF3 y
CHF2;
con la condición de que al menos uno de R1, R8 y R9 sea distinto de H;
X2 y X4 son CH;
X3 es CR3;
R6 se selecciona entre el grupo que consiste en H, halo, OCF3, OCHF2, OH, NR11R12, alcoxicarbonilo C1-C4, (C=O)NR11R12 y alquilo C1-C5 sustituido con de 0 a 7 grupos seleccionados independientemente entre halo, CF3, OCF3, OH, hidroxialquilo C1-C4, alcoxi C1-C4, alcoxi C1-C4-alcoxi C1-C4, dialquilaminofenil C1-C4-alquilo C1-C4, (dialcoxi C1-C4-alquil C1-C4)-alquilo C1-C4, dialquilamino C1-C4, cicloalquilo C3-C6 y alquiltio C1-C4 o
R6 es B-D-, en donde D es un enlazador, que se selecciona entre:
un enlace simple,
-O-,
-S-,

alquileno C1-C4 sustituido con de 0 a 4 grupos seleccionados independientemente de halo u OH y
C2-C6 alquenileno y
B se selecciona del grupo que consiste en:
arilo C6-C10 sustituido con de 0 a 3 grupos seleccionados independientemente del grupo que consiste en halo, alcoxi C1-C4, haloalcoxi C1-C4, alquilo C1-C4, haloalquilo C1-C4, cicloalquilo C3-C6, ciano, nitro, NR11R12, alquilcarboniloxi C1-C4-alquilo C1-C4, hidroxialquilo C1-C4, COOR14, SO2R14, (C=O)NR11R12, SO2NR11R12, N(R13)(C=O)NR11R12, N(R13)(C=O)OR14, N(R13)(C=O)R14, NR13S(O)R14, NR13SO2R14, O(C=O)NR11R12, O(C=O)OR14, O(C=O)R14, (C=O)OR14 y heteroarilo de 5-6 miembros,
heteroarilo de 5 a 10 miembros sustituido con de 0 a 3 grupos seleccionados independientemente del grupo que consiste en halo, alcoxi C1-C4, haloalcoxi C1-C4, alquilo C1-C4, haloalquilo C1-C4, cicloalquilo nitro, NR11R12, OH, alquilcarboniloxi C1-C4-alquilo C1-C4, hidroxialquilo C1-C4, COOR14, SO2R14, (C=O)NR11R12, SO2NR11R12, N(R13)(C=O)NR11R12, N(R13)(C=O)OR14, N(R13)(C=O)R14, NR13S(O)R14, NR13SO2R14, O(C=O)NR11R12, O(C=O)OR14, O(C=O)R14, (C=O)OR14, heteroarilo de 5-6 miembros y (CH2)fenilo,
heterociclilo de 4 a 10 miembros que contiene átomos de carbono y de 1 a 2 heteroátomos adicionales seleccionados de N, O y S y sustituido con de 0 a 3 grupos seleccionados independientemente del grupo que consiste en halo, oxo, -(CHR13)n1-heteroarilo de 5 o 6 miembros sustituido con de 0 a 3 grupos seleccionados independientemente del grupo que consiste en halo, alcoxi C1-C4, alquilo C1-C4, ciclopropilo, CF3, OCF3 y CF2CH3; NR13S(O)R14, NR13SO2R14, -(CHR13)n1-heterociclilo de 4 a 10 miembros sustituido con de 0 a 3 grupos seleccionados independientemente del grupo que consiste en halo, alcoxi C1-C4, alquilo C1-C4, ciclopropilo, CF3, OCF3 y CF2CH3; OH, hidroxialquilo C1-C4, alcoxi C1-C4, haloalcoxi C1-C4, dialquilamino
C1-C4-alquilo C1-C4, NR11R12, ciano, alquilo C1-C4, haloalquilo C1-C4, cicloalquilo C3-C6, alcoxi C1-C4-alquilo
C1-C4, cicloalquil C3-C6-alquilcarbonilo C1-C4, arilcarbonilo C6-C10, alquilcarboniloxi C1-C4-alquilo C1-C4, COOR14, SO2R14, (C=O)NR11R12, SO2NR11R12, N(R13)(C=O)NR11R12, N(R13)(C=O)OR14, N(R13)(C=O)R14, O(C=O)NR11R12, O(C=O)OR14, O(C=O)R14, (C=O)OR14 y arilo C6-C10 sustituido con de 0 a 3 grupos seleccionados independientemente del grupo que consiste en halo, alcoxi C1-C4, alquilo C1-C4, ciclopropilo, alcoxicarbonilo C1-C4-, (C=O)NR11R12, CF3, OCF3 y CF2CH3 y
cicloalquilo C3-C6 que puede contener insaturación, sustituido con de 0 a 3 grupos seleccionados independientemente del grupo que consiste en halo, CF3, OCF3, heteroarilo de 5 o 6 miembros, OH, oxo, hidroxialquilo C1-C4-, arilo C6-C10, COOH, oxo, alcoxicarbonilo C1-C4-, (C=O)NR11R12 y alquilo C1-C4;
R11 y R12 se seleccionan, en cada caso, independientemente del grupo que consiste en:
H,
alquilo C1-C4,
haloalquilo C1-C4,
alquenilo C2-C4,
-(CR14R14)n1-fenilo sustituido con de 0 a 3 grupos seleccionados independientemente del grupo que consiste en halo, alcoxi C1-C4, alquilo C1-C4, ciclopropilo, CF3, OCF3, heteroarilo de 5 o 6 miembros, Oh , OCHF2, dialquilamino C1-C4 y ciano;
-(CHR13)n1-cicloalquilo C3-C6 sustituido con de 0 a 3 grupos seleccionados independientemente del grupo que consiste en halo, CF3, OCF3, heteroarilo de 5 o 6 miembros, OH, hidroxialquilo C1-C4 y alquilo C1-C4;
-(CHR13)n1-heterociclilo de 4 a 10 miembros sustituido con de 0 a 3 grupos seleccionados independientemente del grupo que consiste en halo, CF3, OCF3, heteroarilo de 5 o 6 miembros, OH, oxo, hidroxialquilo C1-C4 y alquilo C1-C4;
-(CHR13)n1-heteroarilo de 5 a 10 miembros sustituido con de 0 a 3 grupos seleccionados independientemente
del grupo que consiste en halo, CF3, OCF3, heteroarilo de 5 o 6 miembros, OH, hidroxialquilo C1-C4 y alquilo C1-C4,
dialquilamino C1-C4-alquilo C1-C4,
dialcoxi C1-C4-alquilo C1-C4,
hidroxialquilo C1-C4,
cianoalquilo C1-C4,
alcoxi C1-C4-alquilo C1-C4,
alcoxicarbonil C1-C4-alquilo C1-C4,
alcoxicarbonilo C1-C4,
alquilcarbonilo C1-C4 y
fenilcarbonilo;
de manera alternativa, R11 y R12, cuando se unen al mismo nitrógeno, se combinan para formar un anillo heterocíclico de 4 a 8 miembros que contiene átomos de carbono, sustituido con de 0 a 3 grupos seleccionados independientemente del grupo que consiste en halo, CF3, CHF2, OCF3, OCHF2, OCH2F, heteroarilo de 5 o 6 miembros, OH, oxo, hidroxialquilo C1-C4, alquilo C1-C4 y alcoxi C1-C4, y de 0 a 2 heteroátomos adicionales seleccionados de N, NR13, O y S(O)p;
R13 se selecciona, en cada caso, independientemente del grupo que consiste en H y alquilo C1-C3;
R14 se selecciona, en cada caso, independientemente del grupo que consiste en H, alquilo C1-C3 y haloalquilo C1-C2;
R7 se selecciona del grupo que consiste en H, flúor, cloro, oxo, alquilo C1-C3, hidroxialquilo C1-C3 y haloalquilo C1-C2, que contiene de 1 a 5 halógenos;
n1, en cada caso, se selecciona entre 0, 1, 2 o 3 y
p, en cada caso, se selecciona entre 0, 1 y 2.
En otra realización, la presente invención proporciona compuestos, estereoisómeros, tautómeros, sales o solvatos de los mismos, en donde:
R1 se selecciona independientemente entre el grupo que consiste en:
cloro,
CH3,
OCH3,
SCH3,
CHFCH3 y
CF2CH3:
X1 es CH;
X2 y X4 son CH;
X3 es CR3;
R3 se selecciona entre el grupo que consiste en OCH3, flúor y cloro;
R4 y R5 se seleccionan independientemente entre H y CH3;

es un anillo heteroarilo de 5 miembros seleccionado entre triazol y oxazol;
R6 se selecciona del grupo que consiste en NR11R12 y alquilo C1-C5 sustituido con de 0 a 3 grupos seleccionados independientemente entre halo, CF3, OCF3, OH, hidroxialquilo C1-C4, alcoxi C1-C4, alcoxi C1-C4-alcoxi C1-C4, dialquilaminofenil C1-C4-alquilo C1-C4, (dialcoxi C1-C4-alquil C1-C4)-alquilo C1-C4, dialquilamino C1-C4, cicloalquilo C3-C6 y alquiltio C1-C4 o
R6 es B-D-, en donde D es un enlace simple;
B se selecciona del grupo que consiste en:
arilo C6-C10 sustituido con de 0 a 3 grupos seleccionados independientemente del grupo que consiste en halo, alcoxi C1-C4, haloalcoxi C1-C4, alquilo C1-C4, haloalquilo C1-C4, cicloalquilo C3-C6, ciano, nitro, NR11R12, OH, alquilcarboniloxi C1-C4-alquilo C1-C4, hidroxialquilo C1-C4, COOR14, SO2R14, (C=O)NR11R12, SO2NR11R12, N(R13)(C=O)NR11R12, N(R13)(C=O)OR14, N(R13)(C=O)R14, NR13S(O)R14, NR13SO2R14, O(C=O)NR11R12, O(C=O)OR14, O(C=O)R14, (C=O)OR14 y heteroarilo de 5-6 miembros;
heteroarilo de 5 a 10 miembros sustituido con de 0 a 3 grupos seleccionados independientemente del grupo que consiste en halo, alcoxi C1-C4, haloalcoxi C1-C4, alquilo C1-C4, haloalquilo C1-C4, cicloalquilo C3-C6, ciano, nitro, NR11R12, OH, alquilcarboniloxi C1-C4-alquilo C1-C4, hidroxialquilo C1-C4, COOR14, SO2R14, (C=O)NR11R12, SO2NR11R12, N(R13)(C=O)NR11R12, N(R13)(C=O)OR14, N(R13)(C=O)R14, NR13S(O)R14, NR13SO2R14, O(C=O)NR11R12, O(C=O)OR14, O(C=O)R14, (C=O)OR14, heteroarilo de 5-6 miembros y
(CH2)fenilo,
heterocicMio de 4 a 10 miembros que contiene átomos de carbono y de 1 a 2 heteroátomos adicionales seleccionados de N, O y S, y sustituido con de 0 a 3 grupos seleccionados independientemente del grupo que consiste en halo, oxo, -(CHR13)n1-heteroarilo de 5 o 6 miembros sustituido con de 0 a 3 grupos seleccionados independientemente del grupo que consiste en halo, alcoxi C1-C4, alquilo C1-C4, ciclopropilo, CF3, OCF3 y CF2CH3; NR13S(O)R14, NR13SO2R14, -(CHR13)n1-heterociclilo de 4 a 10 miembros sustituido con de 0 a 3 grupos seleccionados independientemente del grupo que consiste en halo, alcoxi C1-C4, alquilo C1-C4, ciclopropilo, CF3, OCF3 y CF2CH3; OH, hidroxialquilo C1-C4, alcoxi C1-C4, haloalcoxi C1-C4, dialquilamino C1-C4-alquilo C1-C4, NR11R12, ciano, alquilo C1-C4, haloalquilo C1-C4, cicloalquilo C3-C6, alcoxi C1-C4-alquilo C1-C4, cicloalquil C3-C6-alquilcarbonNo C1-C4, arilcarbonilo C6-C10, alquilcarboniloxi C1-C4-alquilo C1-C4, COOR14, SO2R14, (C=O)NR11R12, SO2NR11R12, N(R13)(C=O)NR11R12, N(R13)(C=O)OR14, N(R13)(C=O)R14, O(C=O)NR11R12, O(C=O)OR14, O(C=O)R14, (C=O)OR14 y arilo C6-C10 sustituido con de 0 a 3 grupos seleccionados independientemente del grupo que consiste en halo, alcoxi C1-C4, alquilo C1-C4, ciclopropilo, alcoxicarbonilo C1-C4, (C=O)NR11R12, CF3, OCF3 y CF2CH3 y
cicloalquilo C3-C6 que puede contener insaturación, sustituido con de 0 a 3 grupos seleccionados independientemente del grupo que consiste en halo, CF3, OCF3, heteroarilo de 5 o 6 miembros, OH, oxo, hidroxialquilo C1-C4, arilo C6-C10, COOH, oxo, alcoxicarbonilo C1-C4, (C=O)NR11R12 y alquilo C1-C4;
R11 y R12 se seleccionan, en cada caso, independientemente del grupo que consiste en:
H,
alquilo C1-C4,
haloalquilo C1-C4,
dialquilamino C1-C4-alquilo C1-C4,
dialcoxi C1-C4-alquilo C1-C4,
hidroxialquilo C1-C4,
cianoalquilo C1-C4,
alcoxi C1-C4-alquilo C1-C4,
alcoxicarbonilo C1-C4-alquilo C1-C4 y
alcoxicarbonilo C1-C4;
de manera alternativa, R11 y R12, cuando se unen al mismo nitrógeno, se combinan para formar un anillo heterocíclico de 4 a 7 miembros que contiene átomos de carbono, sustituidos con de 0 a 2 grupos seleccionados independientemente del grupo que consiste en halo, CF3, CHF2, OCF3, OCHF2, OCH2F, OH, oxo, hidroxialquilo C1-C2, alquilo C1-C3 y alcoxi C1-C3 y de 0 a 2 heteroátomos adicionales seleccionados entre N, NR13, O y S(O)p; R13 se selecciona, en cada caso, independientemente del grupo que consiste en H y alquilo C1-C3;
R14 se selecciona, en cada caso, independientemente del grupo que consiste en H y alquilo C1-C3;
R7 se selecciona del grupo que consiste en H, flúor, cloro, alquilo C1-C3, hidroxialquilo C1-C3 y haloalquilo C1-C2, que contiene de 1 a 5 halógenos;
n1, en cada caso, se selecciona de 0, 1,2 o 3 y
p, en cada caso, se selecciona entre 0, 1 y 2.
Aun en otra realización, la presente invención proporciona compuestos, estereoisómeros, tautómeros, sales o solvatos de los mismos, en donde los compuestos se seleccionan de los ejemplos, preferentemente de los ejemplos 3 a 318.
Aun en otra realización, la presente invención proporciona compuestos, estereoisómeros, tautómeros, sales o solvatos de los mismos, en donde los compuestos se seleccionan de:
Quiral


Ċ



Preferentemente, los compuestos PAR4 de la invención tienen una CI50 en el ensayo FLIPR (descrito más adelante) de alrededor de 10 j M, preferentemente, 5 j M o menos, con mayor preferencia, 500 nM o menos y, aun con mayor preferencia, 10 nM o menos. Los datos de la actividad de los compuestos de la presente invención se indican en las tablas del ejemplo F.
En algunas realizaciones, la presente invención proporciona al menos un compuesto de la presente invención o un estereoisómero, un tautómero, sales o solvatos farmacéuticamente aceptables del mismo.
En algunas realizaciones, la presente invención proporciona una composición farmacéutica, que incluye un portador farmacéuticamente aceptable y una cantidad terapéuticamente eficaz de un compuesto de fórmula I, lA, IB, IC, ID, IE o IF, preferentemente, un compuesto seleccionado de uno de los ejemplos, con mayor preferencia, los ejemplos 3 a 318, o estereoisómeros, tautómeros, sales o solvatos farmacéuticamente aceptables del mismo, solo o en combinación con otro agente terapéutico.
En algunas realizaciones, la presente invención proporciona una composición farmacéutica que también incluye otro u otros agentes terapéuticos. En una realización preferida, la presente invención proporciona una composición farmacéutica, en donde el o los agentes terapéuticos adicionales son un inhibidor plaquetario o una combinación de los mismos. Preferentemente, el o los inhibidores plaquetarios son antagonistas de P2Y12 y/o aspirina. Preferentemente, los antagonistas de P2Y12 son clopidogrel, ticagrelor o prasugrel. En otra realización preferida, la presente invención proporciona una composición farmacéutica, en donde el o los agentes terapéuticos adicionales son un anticoagulante o una combinación de los mismos. Preferentemente, el o los agentes anticoagulantes son
inhibidores de FXa o inhibidores de trombina. Preferentemente, los inhibidores de FXa son apixaban o rivaroxaban. Preferentemente, el inhibidor de trombina es dabigatran.
En algunas realizaciones, la presente invención proporciona compuestos de la presente invención o estereoisómeros, tautómeros, sales o solvatos farmacéuticamente aceptables de los mismos, para su uso en el tratamiento o la profilaxis de un trastorno tromboembólico.
En algunas formas de realización, la presente invención proporciona un compuesto de fórmula I, IA, IB, IC, ID, IE o IF, preferentemente, un compuesto seleccionado de uno de los ejemplos, con mayor preferencia, los ejemplos 3 a 318, o estereoisómeros, tautómeros, sales o solvatos farmacéuticamente aceptables delos mismo, para su uso en el tratamiento de un trastorno tromoboembólico o la profilaxis primaria o secundaria de un trastorno tromboembólico, en donde el trastorno tromboembólico se selecciona del grupo que consiste en trastornos tromboembólicos cardiovasculares arteriales, trastornos tromboembólicos cardiovasculares venosos, trastornos tromboembólicos cerebrovasculares y trastornos tromboembólicos en las cavidades cardíacas o en la circulación periférica.
En algunas realizaciones, la presente invención proporciona un compuesto de fórmula I, IA, IB, IC, ID, IE o IF, preferentemente, un compuesto seleccionado de uno de los ejemplos, con mayor preferencia, los ejemplos 3 a 318, o estereoisómeros, tautómeros, sales o solvatos farmacéuticamente aceptables del mismo, para el tratamiento de un trastorno tromboembólico o la profilaxis primaria o secundaria de un trastorno tromboembólico, en donde el trastorno tromboembólico se selecciona del grupo que consiste en síndrome coronario agudo, angina inestable, angina estable, infarto de miocardio con elevación del segmento ST, infarto de miocardio sin elevación del segmento ST, fibrilación auricular, infarto de miocardio, accidente isquémico transitorio, apoplejía, ateroesclerosis, enfermedad arterial periférica, flebotrombosis, flebotrombosis profunda, tromboflebitis, embolia arterial, trombosis arterial coronaria, trombosis arterial cerebral, embolia cerebral, embolia renal, embolia pulmonar, trombosis cancerosa y trombosis provocada por implantes, dispositivos y procedimientos médicos en los que la sangre está expuesta a una superficie artificial que promueve la trombosis.
En algunas formas de realización, la presente invención proporciona un compuesto de fórmula I, IA, IB, IC, ID, IE o IF, preferentemente, un compuesto seleccionado de uno de los ejemplos, con mayor preferencia, los ejemplos 3 a 318, o estereoisómeros, tautómeros, sales o solvatos farmacéuticamente aceptables del mismo, para el tratamiento de un trastorno tromboembólico o la profilaxis primaria o secundaria de un trastorno tromboembólico, en donde el trastorno tromboembólico se selecciona del grupo que consiste en síndrome coronario agudo, angina inestable, angina estable, infarto de miocardio con elevación del segmento ST e infarto de miocardio sin elevación del segmento ST.
En algunas formas de realización, la presente invención proporciona un compuesto de fórmula I, IA, IB, IC, ID, IE o IF, preferentemente, un compuesto seleccionado de uno de los ejemplos, con mayor preferencia, los ejemplos 3 a 318, o estereoisómeros, tautómeros, sales o solvatos farmacéuticamente aceptables del mismo, en donde el trastorno tromboembólico se selecciona del grupo que consiste en accidente isquémico transitorio y apoplejía.
En algunas formas de realización, la presente invención proporciona un compuesto de fórmula I, IA, IB, IC, ID, IE o IF, preferentemente, un compuesto seleccionado de uno de los ejemplos, con mayor preferencia, los ejemplos 3 a 318, o estereoisómeros, tautómeros, sales o solvatos farmacéuticamente aceptables del mismo, en donde el trastorno tromboembólico es enfermedad arterial periférica.
En algunas formas de realización, la presente invención incluye un compuesto para su uso tal como se describió anteriormente, en donde el trastorno tromboembólico se selecciona de angina inestable, síndrome coronario agudo, fibrilación auricular, primer infarto de miocardio, infarto de miocardio recurrente, muerte súbita isquémica, accidente isquémico transitorio, apoplejía, ateroesclerosis, enfermedad arterial oclusiva periférica, flebotrombosis, flebotrombosis profunda, tromboflebitis, embolia arterial, trombosis arterial coronaria, trombosis arterial cerebral, embolia cerebral, embolia renal, embolia pulmonar y trombosis provocada por implantes, dispositivos o procedimientos médicos en los que la sangre está expuesta a una superficie artificial que promueve la trombosis. En algunas formas de realización, la presente invención incluye un antagonista de PAR4, que es un compuesto de fórmula I, IA, IB, IC, ID, IE o IF, preferentemente, un compuesto seleccionado de uno de los ejemplos, con mayor preferencia, los ejemplos 3 a 318 de la invención, para su uso para inhibir o prevenir la agregación plaquetaria. Otras realizaciones de la invención
En algunas realizaciones, la presente invención proporciona un compuesto de la presente invención o estereoisómeros, tautómeros, sales o solvatos farmacéuticamente aceptables del mismo, para su uso en terapia para el tratamiento o la profilaxis de un trastorno tromboembólico.
Química
Los compuestos de la presente invención pueden tener uno o más centros asimétricos. A menos que se indique lo
contrario, todas las formas quirales (enantioméricas y diastoméricas) y racémicas de los compuestos de la presente invención están incluidas en la presente invención. Muchos isómeros geométricos de olefinas, enlaces dobles C=N y similares también pueden estar presentes en los compuestos y todos esos isómeros estables están contemplados en la presente invención. Los isómeros geométricos cis y trans de los compuestos de la presente invención se describen y se pueden aislar como una mezcla de isómeros o como formas isoméricas separadas. Los presentes compuestos se pueden aislar en formas ópticamente activas o racémicas. Se conoce bien en la técnica cómo preparar formas ópticamente activas, tal como mediante la resolución de formas racémicas o mediante la síntesis de materiales de partida ópticamente activos. Se prevén todas las formas quirales (enantioméricas y diastereoméricas) y racémicas, así como todas las formas isoméricas geométricas de una estructura, a menos que se indique específicamente la forma estereoquímica o isomérica específica. Cuando no se menciona específicamente la configuración (cis, trans, R o S) de un compuesto (o de un carbono asimétrico), entonces se prevé uno cualquiera de los isómeros o una mezcla de más de un isómero. Los procesos de preparación pueden usar racematos, enantiómeros o diastereómeros como materiales de partida. Todos los procesos que se usan para preparar los compuestos de la presente invención y los intermedios allí elaborados se consideran parte de la presente invención. Cuando se preparan productos enantioméricos o diastoméricos, se pueden separar mediante métodos convencionales, por ejemplo, mediante cromatografía o cristalización fraccional. Los compuestos de la presente invención y sus sales pueden existir en múltiples formas tautoméricas, en donde los átomos de hidrógeno se transponen a otras partes de las moléculas y los enlaces químicos entre los átomos de las moléculas se redisponen en consecuencia. Debe entenderse que todas las formas tautoméricas, en caso de que existan, están incluidas en la invención.
El peso molecular de los compuestos de la presente invención es, preferentemente, menos de aproximadamente 800 gramos por mol.
Tal como se usa en el presente documento, el término "alquilo" o "alquileno", solo o como parte de otro grupo, pretende incluir grupos hidrocarburo alifáticos saturados, tanto de cadena lineal como ramificada, que tienen de 1 a 10 carbonos o la cantidad especificada de átomos de carbono. Por ejemplo, " alquilo C1-10" (o alquileno) pretende incluir los grupos alquilo C1, C2, C3, C4, C5, Ce, C7, Ce, Cg y C10. Además, por ejemplo, “alquilo C1-C6” indica que el alquilo tiene de 1 a 6 átomos de carbono. Los grupos alquilo pueden estar sin sustituir o sustituidos con al menos un hidrógeno que es reemplazado por otro grupo químico. Los grupos alquilo de ejemplo incluyen, pero no se limitan a, metilo (Me), etilo (Et), propilo (por ejemplo, n-propilo e isopropilo), butilo (por ejemplo, n-butilo, isobutilo, t-butilo) y pentilo (por ejemplo, n-pentilo, isopentilo, neopentilo), así como cadenas isoméricas de los mismos y similares, así como los grupos que pueden incluir opcionalmente de 1 a 4 sustituyentes, tales como halo, por ejemplo, F, Br, Cl, I o CF3, alquilo, alcoxi, arilo, ariloxi, aril(arilo) o diarilo, arilalquilo, arilalquiloxi, alquenilo, cicloalquilo, cicloalquilalquilo, cicloalquilalquiloxi, amino, hidroxi, hidroxialquilo, acilo, heteroarilo, heteroariloxi, heteroarilalquilo, heteroarilalcoxi, ariloxialquilo, alquiltio, arilalquiltio, ariloxiarilo, alquilamido, alcanoilamino, arilcarbonilamino, nitro, ciano, tiol, haloalquilo, trihaloalquilo y/o alquiltio, así como (=O), ORa, SRa, (=S), -NRaRb, -N(alquilo)3+, -NRaSO2, -NRaSO2Rc, -SO2Rc-SO2NRaRb, -SO2NRaC(=O)Rb, SO3H, -PO(OH)2, -C(=O)Ra, -CO2Ra, -C(=O)NRaRb, -C(=O)(alquilen C1-C4)NRaRb, -C(=O)NRa(SO2)Rb, -CO2(alquilen C1-C4)NRaRb, -NRaC(=O)Rb, -NRaCO2Rb, -NRa(alquilen C1-C4)CO2Rb, =N-OH, =N-O-alquilo, en donde Ra y Rb son iguales o diferentes y se seleccionan independientemente de hidrógeno, alquilo, alquenilo, CO2H, CO2(alquilo), cicloalquilo C3-C7, fenilo, bencilo, feniletilo, naftilo, un heterociclo de 4 a 7 miembros o un heteroarilo de 5 a 6 miembros o, cuando se unen al mismo átomo de nitrógeno se pueden unir para formar un heterociclo o heteroarilo, y Rc se selecciona de los mismos grupos que Ra y Rb, pero no es hidrógeno. Cada grupo Ra y Rb cuando es distinto de hidrógeno, y cada grupo Rc tiene opcionalmente hasta 3 sustituyentes adicionales unidos en cualquier átomo de carbono o nitrógeno disponible de Ra, Rb y/o Rc, dicho sustituyente o sustituyentes son iguales o diferentes y se seleccionan independientemente del grupo que consiste en alquilo (C1-C6), alquenilo (C2-C6), hidroxi, halógeno, ciano, nitro, CF3, O(alquilo C1-C6), OCF3, C(=O)H, C(=O)( alquilo C1-C6), CO2H, CO2(alquilo C1-C6), NHCO2(alquilo C1-C6), -S(alquilo C1-C6), -NH2, NH(alquilo C1-C6), N(alquilo C1-C6)2, N(CH3)3+, SO2(alquilo C1-C6), C(=O)(alquilen C1-C4)NH2, C(=O)(alquilen C1-C4)NH(alquilo), C(=O)(alquilen C1-C4)N(alquilo C1-C4)2, cicloalquilo C3-C7, fenilo, bencilo, feniletilo, feniloxi, benciloxi, naftilo, un heterociclo de 4 a 7 miembros o un heteroarilo de 5 a 6 miembros. Cuando un alquilo sustituido se sustituye con un grupo arilo, heterociclo, cicloalquilo o heteroarilo, dichos sistemas de anillos son como se definen más adelante y por tanto pueden tener cero, uno, dos o tres sustituyentes, tal como se definen más adelante.
"Alquenilo" o "alquenileno", solo o como parte de otro grupo, pretende incluir cadenas de hidrocarburos de configuración tanto lineal como ramificada y que tienen uno o más enlaces dobles de carbono-carbono que pueden ocurrir en cualquier punto estable de la cadena. Por ejemplo, "alquenilo C2-6 " (o alquenileno) pretende incluir grupos alquenilo C2, C3, C4, C5 y C6. Los ejemplos de alquenilo incluyen, pero no se limitan a, etenilo, 1-propenilo, 2-propenilo, 2-butenilo, 3-butenilo, 2-pentenilo, 3-pentenilo, 4-pentenilo, 2-hexenilo, 3-hexenilo, 4-hexenilo, 5-hexenilo, 2-metil-2-propenilo y 4-metil-3-pentenilo y se pueden sustituir opcionalmente con de 1 a 4 sustituyentes, a saber, halógeno, haloalquilo, alquilo, alcoxi, alquenilo, alquinilo, arilo, arilalquilo, cicloalquilo, amino, hidroxi, heteroarilo, cicloheteroalquilo, alcanoilamino, alquilamido, arilcarbonil-amino, nitro, ciano, tiol y/o alquiltio.
"Alquinilo" o "alquinileno", solo o como parte de otro grupo, pretende incluir cadenas de hidrocarburos de configuración tanto lineal como ramificada y que tienen uno o más enlaces triples de carbono-carbono que pueden ocurrir en cualquier punto estable de la cadena. Por ejemplo, "alquinilo C2-6" (o alquinileno) pretende incluir grupos
alquinilo C2, C3, C4, C5 y Ce, tales como etinilo, propinilo, butinilo, pentinilo y hexinilo y que se puede sustituir opcionalmente con de 1 a 4 sustituyentes, a saber, halógeno, haloalquilo, alquilo, alcoxi, alquenilo, alquinilo, arilo, arilalquilo, cicloalquilo, amino, heteroarilo, cicloheteroalquilo, hidroxi, alcanoilamino, alquilamido, arilcarbonilamino, nitro, ciano, tiol y/o alquiltio.
El término "alcoxi" o "alquiloxi", solo o como parte de otro grupo, se refiere a un grupo -O-alquilo, en donde alquilo es como se definió con anterioridad. "Alcoxi C1-6" (o alquiloxi) pretende incluir grupos alcoxi C1, C2, C3, C4, C5 y Ca. Los ejemplos de grupos alcoxi incluyen, pero no se limitan a, metoxi, etoxi, propoxi (por ejemplo, n-propoxi e isopropoxi) y t-butoxi. De manera similar, "alquiltio" o "tioalcoxi", solo o como parte de otro grupo, representa un grupo alquilo o un grupo alcoxi, como se definió anteriormente, con la cantidad indicada de átomos de carbono unidos mediante un puente de azufre; por ejemplo, metil-S- y etil-S-.
"Halo" o "halógeno", solo o como parte de otro grupo, incluye flúor, cloro, bromo y yodo.
"Haloalquilo" pretende incluir grupos hidrocarburo alifáticos saturados, tanto de cadena lineal como ramificada, que tienen la cantidad especificada de átomos de carbono, sustituidos con de 1 a 7 halógenos, preferentemente, de 1 a 4 halógenos, preferentemente, F y/o Cl. Los ejemplos de haloalquilo incluyen, pero no se limitan a, fluorometilo, difluorometilo, trifluorometilo, triclorometilo, pentafluoroetilo, pentacloroetilo, 1,1 -difluoroetilo, 1 -fluoroetilo, 2,2,2-trifluoroetilo, heptafluoropropilo y heptacloropropilo. Los ejemplos de haloalquilo también incluyen "fluoroalquilo", que pretende incluir grupos hidrocarburo alifáticos saturados, tanto de cadena lineal como ramificada, que tienen la cantidad especificada de átomos de carbono, sustituidos con de 1 a 7 átomos de flúor, preferentemente, de 1 a 4 átomos de flúor.
"Haloalcoxi C1-C2" o "haloalquiloxi" representa un grupo haloalquilo, como se definió anteriormente, con la cantidad indicada de átomos de carbono unidos mediante un puente de oxígeno. Por ejemplo, "haloalcoxi C-i-a" pretende incluir grupos haloalcoxi C1, C2, C3, C4, C5 y C6. Los ejemplos de haloalcoxi incluyen, pero no se limitan a, trifluorometoxi, 2,2,2-trifluoroetoxi, pentafluoroetoxi y similares. De manera similar, "haloalquiltio" o "tiohaloalcoxi" representa un grupo haloalquilo, como se definió anteriormente, con la cantidad indicada de átomos de carbono unidos mediante un puente de azufre; por ejemplo, trifluorometil-S- y pentafluoroetil-S-.
A menos que se indique lo contrario, el término "cicloalquilo", como se usa en el presente documento, solo o como parte de otro grupo, incluye grupos hidrocarburo cíclicos, saturados o parcialmente insaturados (que contienen 1 o 2 enlaces dobles), que contienen de 1 a 3 anillos, que incluyen alquilo monocíclico, alquilo bicíclico (o bicicloalquilo) y alquilo tricíclico, que contienen un total de 3 a 10 carbonos que forman el anillo (C3-C10 cicloalquilo) y que se pueden fusionar a 1 o 2 anillos aromáticos, tal como se describe para el arilo, que incluye ciclopropilo, ciclobutilo, ciclopentilo, ciclohexilo, cicloheptilo, ciclooctilo, ciclodecilo, ciclododecilo, ciclohexenilo, norbornilo,

cualquiera de esos grupos se puede sustituir opcionalmente con de 1 a 4 sustituyentes, tales como halógeno, alquilo, alcoxi, hidroxi, arilo, ariloxi, arilalquilo, cicloalquilo, alquilamido, alcanoilamino, oxo, acilo, arilcarbonilamino, amino, nitro, ciano, tiol y/o alquiltio, y/o cualquiera de los sustituyentes de alquilo, así como los grupos que incluyen 2 enlaces libres y, por tanto, son grupos de enlace.
Como se usan en el presente documento, "carbociclo" o "resto carbocíclico" pretende significar cualquier anillo monocíclico o bicíclico estable de 3, 4, 5, 6 o 7 miembros, o cualquier anillo bicíclico o tricíclico de 7, 8, 9, 10, 11, 12 o 13 miembros, cualquiera de los cuales puede estar saturado, parcialmente insaturado, insaturado o ser aromático. Los ejemplos de dichos carbociclos incluyen, pero no se limitan a, ciclopropilo, ciclobutilo, ciclobutenilo, ciclopentilo, ciclopentenilo, ciclohexilo, cicloheptenilo, cicloheptilo, cicloheptenilo, adamantilo, ciclooctilo, ciclooctenilo, ciclooctadienilo, [3.3.0]biciclooctano, [4.3.0]biciclononano, [4.4.0]biciclodecano, [2.2.2]biciclooctano, fluorenilo, fenilo, naftilo, indanilo, adamantilo, antracenilo y tetrahidronaftilo (tetralina). Como se indicó anteriormente, los anillos puenteados también se incluyen en la definición de carbociclo (por ejemplo, [2.2.2]biciclooctano). A menos que se especifique lo contrario, los carbociclos preferidos son ciclopropilo, ciclobutilo, ciclopentilo, ciclohexilo, fenilo e indanilo. Cuando se usa el término "carbociclo", este pretende incluir "arilo". Un anillo puenteado se produce cuando uno o más átomos de carbono se unen a dos átomos de carbono no adyacentes. Los puentes preferidos son uno o dos átomos de carbono. Cabe destacar que un puente siempre convierte un anillo monocíclico en un anillo tricíclico. Cuando un anillo está puenteado, los sustituyentes enumerados para el anillo también pueden estar presentes en el puente.
Los grupos "arilo" se refieren a hidrocarburos monocíclicos o policíclicos aromáticos que incluyen, por ejemplo, fenilo, naftilo y fenantranilo. Los restos arilo se conocen bien y se describen, por ejemplo, en Lewis, R. J., ed., Hawley's Condensed Chemical Dictionary, 13a edición, J. Wiley & Sons, Inc., Nueva York (1997). "Arilo C6-10 " se refiere a fenilo y naftilo. A menos que se especifique lo contrario, "arilo", "arilo C6-10" o "resto aromático" pueden estar sin sustituir o sustituidos con de 1 a 3 grupos seleccionados entre OH, Oalcoxi C1-C3, Cl, F, Br, I, c N, NO2, NH2, N(CHa)H, N(CH3)2, CF3, OCF3, OCHF2, C(=O)CH3, SCH3, S(=O)CH3, S(=O)2CH3, alquilo C1-C3, CO2H y CO2CH3.
Como se usa en el presente documento, el término grupo "heterociclo" o "heterocíclico" pretende significar un anillo heterocíclico monocíclico, bicíclico o tricíclico estable de 4 a 14 miembros, que está saturado o parcialmente insaturado y que consiste en átomos de carbono y 1, 2, 3 o 4 heteroátomos seleccionados independientemente del grupo que consiste en N, NH, O y S y que incluye cualquier grupo bicíclico en el que cualquiera de los anillos heterocíclicos antes definidos está condensado a un anillo de benceno. Los heteroátomos de nitrógeno y de azufre se pueden oxidar opcionalmente (es decir, N ^O y S(O)p, en donde p es 0, 1 o 2). El átomo de nitrógeno puede estar sustituido o no sustituido (es decir, N o NR, en donde R es H u otro sustituyente, si se define). El anillo heterocíclico se puede unir a su grupo colgante en cualquier heteroátomo o átomo de carbono que dé como resultado una estructura estable. Los anillos heterocíclicos descritos en el presente documento se pueden sustituir opcionalmente en un átomo de carbono o de nitrógeno, si el compuesto resultante es estable, con de 1 a 3 grupos seleccionados entre OH, Oalcoxi C1-C3, Cl, F, Br, I, CN, NO2, NH2, N(CH3)H, N(CH3)2, CF3, OCF3, OCHF2, =O, C(=O)CH S(=O)CH3, S(=O)2CH3, alquilo C1-C3, CO2H y CO2CH3. Opcionalmente, se puede cuaternizar un nitrógeno en el heterociclo. Se prefiere que cuando la cantidad total de átomos S y O en el heterociclo excede 1, estos heteroátomos no sean adyacentes entre sí. Se prefiere que la cantidad total de átomos S y O en el heterociclo no sea mayor de 1. Los anillos espiro y puenteados también se incluyen en la definición de heterociclo. Un anillo puenteado se produce cuando uno o más átomos (es decir, C, O, N o S) se unen a dos átomos de carbono o de nitrógeno no adyacentes. Los ejemplos de anillos puenteados incluyen, pero no se limitan a, un átomo de carbono, dos átomos de carbono, un átomo de nitrógeno, dos átomos de nitrógeno y un grupo carbono-nitrógeno. Cabe destacar que un puente siempre convierte un anillo monocíclico en un anillo tricíclico. Cuando un anillo está puenteado, los sustituyentes enumerados para el anillo también pueden estar presentes en el puente. Cuando se usa el término “heterociclo”, no se pretende incluir heteroarilo.
Los grupos heterocíclicos monocíclicos a modo de ejemplo incluyen azetidinilo, pirrolidinilo, oxetanilo, imidazolinilo, oxazolidinilo, isoxazolinilo, tiazolidinilo, isotiazolidinilo, tetrahidrofuranilo, piperidilo, piperazinilo, 2-oxopiperazinilo, 2-oxopiperidilo, 2-oxopirrolodinilo, 2-oxoazepinilo, azepinilo, 4-piperidonilo, tetrahidropiranilo, morfolinilo, tiamorfolinilo, sulfóxido de tiamorfolinilo, tiamorfolinilsulfona, 1,3-dioxolano, tetrahidro-1,1 -dioxotienilo y similares.
Los grupos heterociclo bicíclicos a modo de ejemplo incluyen quinuclidinilo.
Los grupos heterociclo preferidos incluyen:

que se pueden sustituir opcionalmente.
Como se usa en el presente documento, la expresión "grupo heterocíclico aromático" o "heteroarilo" pretende significar hidrocarburos aromáticos, monocíclicos y policíclicos, estables, que incluyen al menos un heteroátomo miembro del anillo, tal como azufre, oxígeno o nitrógeno. Los grupos heteroarilo incluyen, sin limitación, piridilo, pirimidinilo, pirazinilo, piridazinilo, triazinilo, furilo, quinolilo, isoquinolilo, tienilo, imidazolilo, tiazolilo, indolilo, pirroílo, oxazolilo, benzofurilo, benzotienilo, benztiazolilo, isoxazolilo, pirazolilo, triazolilo, tetrazolilo, indazolilo, 1,2,4-tiadiazolilo, isotiazolilo, purinilo, carbazolilo, bencimidazolilo, indolinilo, benzodioxolanilo y benzodioxano. Los grupos heteroarilo están sin sustituir o sustituidos con de 1 a 3 grupos seleccionados de OH, Oalcoxi C1-C3, Cl, F, Br, I, Cn , NO2, NH2, N(CH3)H, N(CH3)2, CF3, OCF3, OCHF2, =O, C(=O)CH3, SCH3, S(=O)CH3, S(=O)2CH3, alquilo C1-C3, CO2H y CO2CH3. El átomo de nitrógeno está sustituido o sin sustituir (es decir, N o NR, en donde R es H u otro sustituyente, si se define). Los heteroátomos de nitrógeno y de azufre se pueden oxidar opcionalmente (es decir,
N ^O y S(O)p, en donde p es 0, 1 o 2). Los anillos puenteados también se incluyen en la definición de heteroarilo. Un anillo puenteado se produce cuando uno o más átomos (es decir, C, O, No S) se unen a dos átomos de carbono o
de nitrógeno no adyacentes. Los ejemplos de anillos puenteados incluyen, pero no se limitan a, un átomo de carbono, dos átomos de carbono, un átomo de nitrógeno, dos átomos de nitrógeno y un grupo carbono-nitrógeno. Cabe destacar que un puente siempre convierte un anillo monocíclico en un anillo tricíclico. Cuando un anillo está puenteado, los sustituyentes indicados para el anillo también pueden estar presentes en el puente.
Los grupos heteroarilo preferidos incluyen:

y similares.
Cuando el término "insaturado" se usa en el presente documento para referirse a un anillo o a un grupo, ese grupo puede estar completa o parcialmente insaturado.
El término "acilo", solo o como parte de otro grupo, se refiere a un grupo carbonilo ligado a un radical orgánico, más en particular, el grupo C(=O)Re, así como los grupos bivalentes -C(=O)- o -C(=O)Re-, que están unidos a radicales orgánicos. El grupo Re se puede seleccionar de alquilo, alquenilo, alquinilo, aminoalquilo, alquilo sustituido, alquenilo sustituido o alquinilo sustituido, tal como se definen en el presente documento, o cuando fuese adecuado, el grupo bivalente correspondiente, por ejemplo, alquileno, alquenileno y similares.
La designación V lp  o " j " unida a un anillo u otro grupo se refiere a un enlace libre o grupo de enlace. A lo largo de la memoria descriptiva, un experto en la técnica puede elegir grupos y sustituyentes de los mismos para proporcionar restos y compuestos estables y compuestos útiles como compuestos farmacéuticamente aceptables y/o compuestos intermedios útiles para fabricar compuestos farmacéuticamente aceptables.
o " j " unida a un anillo u otro grupo se refiere a un enlace libre o grupo de enlace. A lo largo de la memoria descriptiva, un experto en la técnica puede elegir grupos y sustituyentes de los mismos para proporcionar restos y compuestos estables y compuestos útiles como compuestos farmacéuticamente aceptables y/o compuestos intermedios útiles para fabricar compuestos farmacéuticamente aceptables.
El término "contraión" se usa para representar una especie de carga negativa, tal como cloruro, bromuro, hidróxido, acetato y sulfato.
Como se indica en el presente documento, el término "sustituido" significa que al menos un átomo de hidrógeno se reemplaza por un grupo distinto de hidrógeno, siempre que se mantengan las valencias normales y que la sustitución dé como resultado un compuesto estable. Cuando un sustituyente es ceto (es decir, =O), entonces se reemplazan 2 hidrógenos en el átomo. Los sustituyentes ceto no están presentes en los restos aromáticos. Los enlaces dobles del anillo, como se usan en el presente documento, son enlaces dobles que se forman entre dos átomos del anillo adyacentes (por ejemplo, C=C, C=N o N=N).
Cuando existen átomos de nitrógeno (por ejemplo, aminas) en los compuestos de la presente invención, estos se pueden convertir a N-óxidos mediante el tratamiento con un agente oxidante (por ejemplo, mCPBA y/o peróxidos de hidrógeno) para obtener otros compuestos de esta invención. Por ello, se considera que los átomos de nitrógeno indicados y reivindicados incluyen tanto el nitrógeno indicado como su derivado N-óxido (N^O). En los casos en los que hay átomos de carbono cuaternarios en los compuestos de la presente invención, estos se pueden reemplazar por átomos de silicio, siempre que no formen enlaces Si-N o Si-O.
Cuando cualquier variable ocurre más de una vez en cualquier constituyente o fórmula de un compuesto, su definición en cada caso es independiente de su definición en cada uno de los otros casos. Por ello, por ejemplo, si se muestra que un grupo está sustituido con de 0 a 3 R3a, entonces dicho grupo se puede sustituir opcionalmente con hasta 3 grupos R3a, y en cada caso, R3a se selecciona independientemente de la definición de R3a. Además, las combinaciones de los sustituyentes y/o las variables se permiten solo si las combinaciones dan como resultado compuestos estables.
Cuando se muestra que un enlace a un sustituyente cruza un enlace que conecta dos átomos en un anillo, dicho sustituyente puede unirse a cualquier átomo en el anillo. Cuando se enumera un sustituyente sin indicar el átomo en el cual dicho sustituyente está unido al resto del compuesto de una fórmula dada, entonces dicho sustituyente puede unirse mediante cualquier átomo en dicho sustituyente. Las combinaciones de los sustituyentes y/o las variables se permiten solo si las combinaciones dan como resultado en compuestos estables.
La frase "farmacéuticamente aceptable" se usa en el presente documento para referirse a aquellos compuestos, materiales, composiciones y/o formas de dosificación que, dentro del alcance del criterio médico sensato, son adecuados para usar en contacto con los tejidos de seres humanos y animales sin provocar excesiva toxicidad, irritación, respuesta alérgica y/u otros problemas o complicaciones proporcionales con una relación riesgo/beneficio razonable.
Como se usa en el presente documento, “sales farmacéuticamente aceptables” se refiere a derivados de los compuestos divulgados, en donde el compuesto de origen se modifica elaborando sales ácidas o básicas del mismo. Los ejemplos de sales farmacéuticamente aceptables incluyen, pero no se limitan a, sales de ácidos orgánicos o minerales de grupos básicos, tales como aminas; y sales alcalinas u orgánicas de grupos ácidos, tales como ácidos carboxílicos. Las sales farmacéuticamente aceptables incluyen las sales no tóxicas convencionales o las sales de amonio cuaternario del compuesto de origen formadas, por ejemplo, a partir de ácidos orgánicos o inorgánicos no tóxicos. Por ejemplo, dichas sales no tóxicas convencionales incluyen las derivadas de ácidos inorgánicos tales como clorhídrico, bromhídrico, sulfúrico, sulfámico, fosfórico y nítrico; y las sales preparadas a partir de ácidos orgánicos tales como acético, propiónico, succínico, glicólico, esteárico, láctico, málico, tartárico, cítrico, ascórbico, pamoico, maleico, hidroximaleico, fenilacético, glutámico, benzoico, salicílico, sulfanílico, 2-acetoxibenzoico, fumárico, toluensulfónico, metansulfónico, etandisulfónico, oxálico, isetiónico y similares.
Las sales farmacéuticamente aceptables de la presente invención se pueden sintetizar del compuesto de origen que contiene un resto básico o ácido mediante métodos químicos convencionales. En general, dichas sales se pueden preparar haciendo reaccionar las formas básicas o ácidas libres de estos compuestos con una cantidad estequiométrica de la base o del ácido adecuados en agua, en un disolvente orgánico o en una mezcla de los dos; en general, se prefieren medios no acuosos como éter, acetato de etilo, etanol, isopropanol o acetonitrilo. Se encuentran listas de sales adecuadas en Allen, L. V. Jr., ed., Remington: The Science and Practice of Pharmacy, 22.a edición,; Pharmaceutical Press, Londres, Reino Unido (2012).
En el presente documento también se proporcionan compuestos etiquetados isotópicamente de la presente invención, es decir, cuando uno o más de los átomos descritos son reemplazados por un isótopo de ese átomo (por ejemplo, 12C reemplazado por 13C o por 14C e isótopos de hidrógeno que incluyen tritio y deuterio). Dichos compuestos tienen varios usos potenciales, por ejemplo, como estándares y reactivos para determinar la capacidad de un potencial compuesto farmacéutico para unirse a proteínas o receptores diana o para formar imágenes de los compuestos de la presente invención unidos a receptores biológicos in vivo o in vitro.
Preferentemente, los compuestos de la presente invención, después de su preparación, se aíslan y se purifican para obtener una composición que contiene una cantidad en peso igual o superior al 98 %, preferentemente 99 % de un compuesto de la presente invención ("sustancialmente puro"), que después se usa o se formula como se describe en el presente documento. Dichos compuestos "sustancialmente puros" también se contemplan en el presente documento como parte de la presente invención.
Las expresiones "compuesto estable" y "estructura estable" pretenden indicar un compuesto que es lo suficientemente potente para sobrevivir al aislamiento a un grado útil de pureza de una mezcla de reacción, y a la formulación en un agente terapéutico eficaz. Se prefiere que los compuestos de la presente invención no contengan un grupo N-halo, S(O)2H ni S(O)H.
El término "solvato" significa una asociación física de un compuesto de esta invención con una o más moléculas de disolvente, ya sea orgánico o inorgánico. Esta asociación física incluye unión a hidrógeno. En ciertos casos, el solvato será capaz de aislarse, por ejemplo, cuando una o más moléculas de disolvente se incorporan en la red cristalina del sólido cristalino. "Solvato" abarca tanto solvatos en fase de solución como solvatos que se pueden aislar. Los solvatos a modo de ejemplo incluyen, pero no se limitan a, hidratos, etanolatos, metanolatos e isopropanolatos. En la técnica se conocen varios métodos de solvatación.
Las abreviaturas tal como se usan en el presente documento se definen de la siguiente manera: “1 x” para una vez, “2 x” para dos veces, “3 x” para tres veces, "°C" para grados Celsius, “eq” para equivalente o equivalentes, “g” para gramo o gramos, “mg” para miligramo o miligramos, “l” para litro o litros, “ml” para mililitro o mililitros, “|jl” para microlitro o microlitros, “N” para normal, “M” para molar, “mmol” para milimol o milimoles, “min” para minuto o minutos, “h” para hora u horas, “ta” para temperatura ambiente, “TR” para tiempo de retención, “atm” para atmósfera, “psi” para libras por pulgada cuadrada, “conc.” para concentrado, “sat” para saturado, “MW” para peso molecular, “pf” para punto de fusión, “MS” o "espec. de masas" para espectrometría de masas, “ESI” para espectroscopia de masas por ionización por electropulverización, “HR” para alta resolución, “HRMS” para espectroscopia de masas de alta resolución, “LCMS” para cromatografía de líquidos/espectrometría de masas, “HPLC” para cromatografía de líquidos de alta presión, “RP HPLC” para HPLC de fase inversa, “TLC” para cromatografía de capa fina, "SM" para material de partida, “RMN” para espectroscopía de resonancia magnética nuclear, “1H” para protón, “8” para delta, “s” para singlete, “d” para doblete, “t” para triplete, “q” para cuartete, “m” para multiplete, “a” para ancho, “Hz” para hertz, y “tlc” para cromatografía de capa fina. "a", "p", "R", "S", "E" y "Z" son designaciones estereoquímicas conocidas para los expertos en la técnica.

continuación
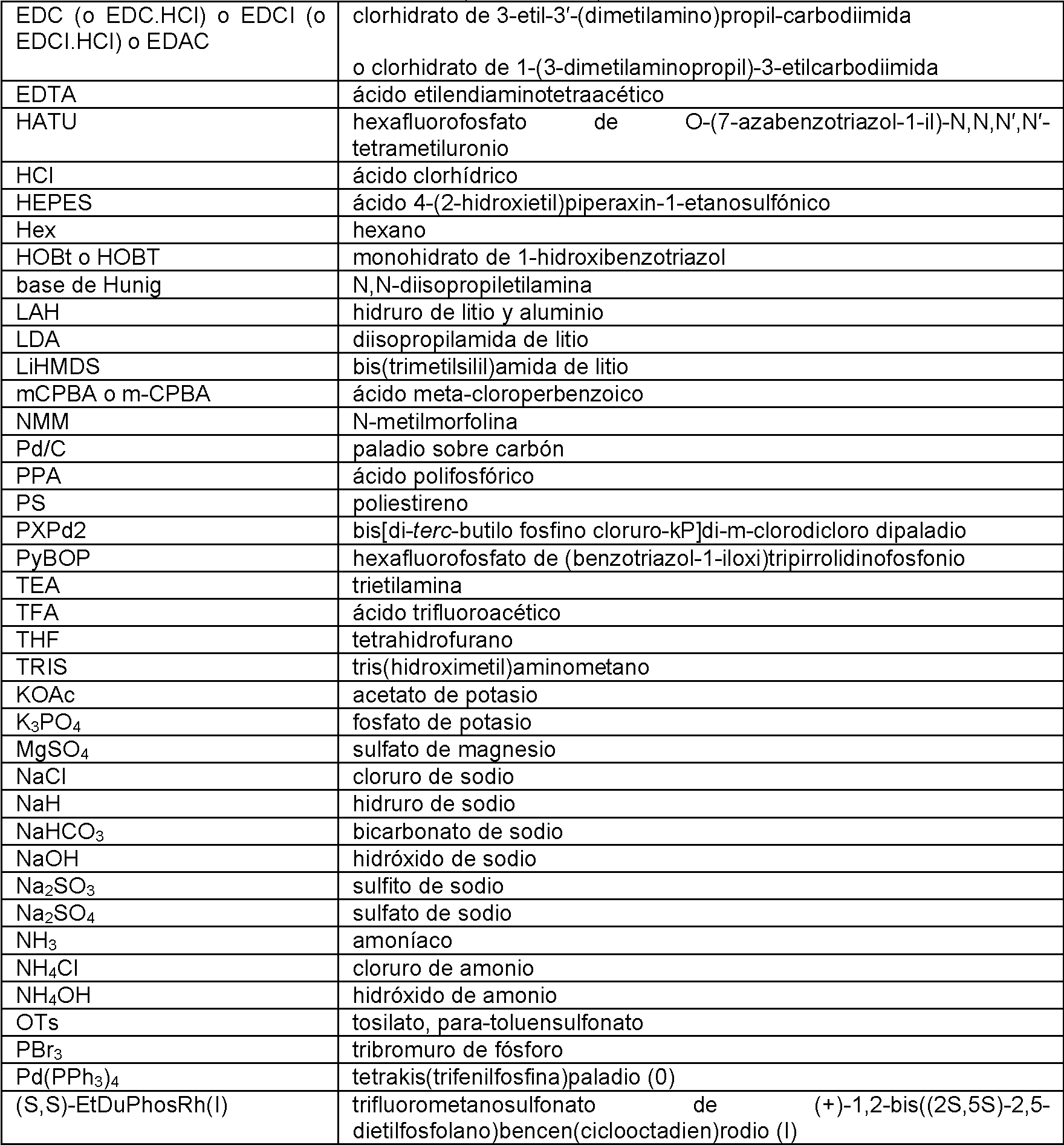
Los compuestos de la presente invención se pueden preparar de varias maneras conocidas por un experto en la técnica de la síntesis orgánica. Los compuestos de la presente invención se pueden sintetizar con los métodos descritos a continuación, junto con los métodos de síntesis conocidos en la técnica de la química orgánica sintética o por variaciones de los mismos, según apreciarán los expertos en la técnica. Los métodos preferidos incluyen, pero no se limitan a, los que se describen a continuación. Las reacciones se realizan en un disolvente o en una mezcla de disolventes adecuados para los reactivos y materiales usados y adecuados para las transformaciones que se llevan a cabo. Los expertos en la técnica de la síntesis orgánica comprenderán que la funcionalidad presente en la molécula debe ser compatible con las transformaciones que se proponen. En ocasiones, esto requerirá cierto criterio para modificar el orden de las etapas de síntesis o para seleccionar un esquema particular de proceso en lugar de otro, a fin de obtener el compuesto de la invención deseado.
También se reconocerá que otra consideración importante en la planificación de cualquier vía de síntesis en este campo es la elección prudente del grupo protector que se usa para la protección de los grupos funcionales reactivos presentes en los compuestos descritos en esta invención. Una reseña confiable que describe las diversas alternativas para el médico capacitado es Wuts e t al. (Greene's Protective Groups In Organic Synthesis, 4.a edición, Wiley-Interscience (2006)).
Los compuestos de fórmula I de la presente invención se pueden obtener mediante condensación de una amina de fórmula III con una cetona de fórmula IV que contiene un grupo saliente Z, tal como bromuro, yoduro o tosilato, y un grupo protector PG, tal como bencilo, como se indica en el esquema 1. Ambos compuestos de fórmula III y IV están
disponibles en el comercio o se pueden preparar mediante medios conocidos por el experto en la técnica. Esta condensación es promovida por calentamiento, ya sea térmico o preferentemente mediante irradiación de microondas. El grupo protector se puede retirar mediante métodos conocidos en la técnica, tales como BCh a -78 °C en presencia de pentametilbenceno. La posterior alquilación usando un alcohol VI en condiciones de Mitsunobu o un bromuro VII en presencia de una base, tal como carbonato de potasio, proporciona los compuestos de fórmula I. Los alcoholes y bromuros VI y VII están disponibles en el comercio o se pueden preparar mediante los métodos conocidos en la técnica.
Esquema 1

De manera alternativa, los compuestos de fórmula I se pueden preparar a partir de compuestos de fórmula IX después de la activación del grupo tiometilo mediante oxidación a una sulfona VII, como se indica en el esquema 2. Esto permite la introducción de varios nucleófilos como grupos R0, tales como alcoholes, tioles y aminas en presencia de una base, tal como carbonato de potasio o hidruro de sodio ya sea puro o en un disolvente aprótico polar, tal como dimetilformamida, para obtener compuestos XI. Los compuestos XI se pueden convertir en compuestos de fórmula I mediante la eliminación del grupo protector (PG) y la alquilación como se analizó en el
esquema 1.
Esquema 2

Los benzofuranos sustituidos que tienen sustituyentes de a-bromocetona en la posición 2 (XV) se pueden preparar como se indica en el esquema 3. Los o-hidroxibenzaldehídos XII se pueden preparar mediante los métodos conocidos por el experto en la técnica de la síntesis orgánica y se pueden condensar con cetonas de fórmula XIII que tienen un grupo saliente Q, tal como cloro, bromo o tosiloxi, para dar benzofuranos XIV. La bromación de los compuestos de fórmula XIV produce bromocetonas XV, que se pueden condensar con un aminoheterociclo III sustituido de acuerdo con el esquema 1 para obtener compuestos de fórmula I. Las bromocetonas XV son un subconjunto específico de los compuestos IV en el esquema 1.
Esquema 3

XIII
Los compuestos de benzoxazol de fórmula I se pueden preparar a partir de aminoheterociclo III sustituido y ésteres de piruvato de fórmula XVI que contienen un grupo saliente Z, tal como bromuro, yoduro o tosilato, como se indica en el esquema 4. Ambos compuestos de fórmula III y XVI están disponibles en el comercio o se pueden obtener mediante medios conocidos por el experto en la técnica. Después de la condensación y saponificación del éster para formar el ácido XVIII, los aminofenoles de fórmula XIX se acoplan para formar amidas de fórmula XX, que se pueden ciclar en condiciones de catálisis ácida para formar compuestos de benzoxazol de fórmula XXI. Estos se pueden desproteger y alquilar como se indica en el esquema 1 para obtener los compuestos de fórmula I.
Esquema 4


Los aminoheterociclos XXIV se pueden preparar a partir de disulfuro de carbono (XXII) mediante el intermedio de tioxantato XXIII. Estos aminoheterociclos son útiles para la preparación de los compuestos de fórmula I.
Esquema 5
1. KOH MeOH
reflujo, 1h CS2
2. Lavado éter
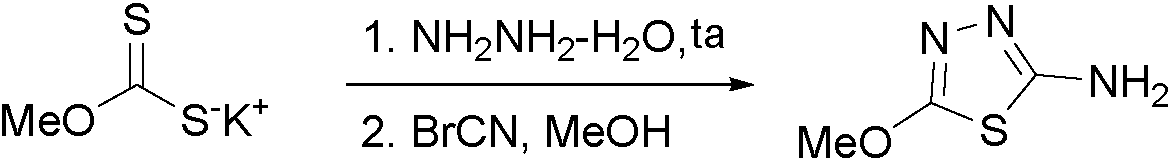
XXII XXIV XXIII
Los aminoheterociclos XXX, que son intermedios útiles para la preparación de los compuestos de fórmula I, en donde Y = -CH2CH2-, se pueden preparar a partir de los cetoésteres XXV. La ciclación con hidrazina seguida de oxidación con bromo da piridazinonas XXVII. La cloración, el desplazamiento con hidrazina y la posterior hidrogenación proporcionan aminoheterociclos XXX, que son un subconjunto específico de compuestos III en el esquema I. Como tales, estos aminoheterociclos son útiles para la preparación de los compuestos de fórmula I.
Esquema 6

Ejemplos
Los siguientes compuestos de la invención se prepararon, se aislaron y se caracterizaron con los métodos divulgados en el presente documento. Muestran un alcance parcial de la invención y no pretenden limitar el alcance de la invención. En los procesos experimentales, las relaciones de la solución expresan una relación en volumen, a menos que se indique lo contrario. Los desplazamientos químicos de RMN (8) se indican en partes por millón (ppm). Los productos se analizaron mediante HPLC analítica de fase inversa usando los siguientes métodos:
Método A: Columna: ZORBAX® XDB-C18 3,5 micrómetros, 4,6 x 30 mm; fase móvil: A=MeOH:H2O:TFA (95:5:05), B=MeOH:H2O:TFA (5:95:05). Grad.: T=0: 100 % de disolvente A; T=2: 100 % de disolvente B; tiempo de detención: 4 min. Caudal = 3,0 ml/min.
Método B: Columna: Agilent POROSHELL® 120; EC-C18, 2,7 um; 2,1 x 30 mm; fase móvil: disolvente A: 5 % de MeOH: 95 % de H2O 0,1 % de AcOH; disolvente B: 95 % de MeOH: 5 % de H2O 0,1 % de AcOH; grad.: T=0: 100 % de disolvente A; T=1: 100 % de disolvente B; tiempo de detención: 4 min. Caudal = 1,0 ml/min.
Método C: columna Sunfire C183,5 micrómetros (4,6 x 30 mm) eluída a 3 ml/min con un gradiente de 2 min de 100 % de A a 100 % de B (A: 5 % de metanol, 94,95 % de agua, 0,05 % de TFA; B: 5 % de agua, 94,95 % de metanol, 0,05 % de TFA, UV 220 nm).
Método D: columna Eclipse XDB-C183,5 micrómetros (4,6 x 30 mm) eluída a 3 ml/min con un gradiente de 2 min de 100 % de A a 100 % de B (A: 5 % de metanol, 94,95 % de agua, 0,05 % de TFA; B: 5 % de agua, 94,95 % de metanol, 0,05 % de TFA, UV 220 nm).
Método E: columna Eclipse XDB-C183,5 micrómetros (4,6 x 30 mm) eluída a 3 ml/min con un gradiente de 2 min de 100 % de A a 100 % de B (A: 5 % de acetonitrilo, 94,95 % de agua, 0,05 % de TFA; B: 5 % de agua, 94,95 % de acetonitrilo, 0,05 % de TFA, UV 220 nm).
Método F: columna ZORBAX® SB-Phenyl 3,5 micrómetros (4,6 x 50 mm) eluída a 3 ml/min con un gradiente de 2 min de 100 % de A a 100 % de B (A: 5 % de metanol, 94,95 % de agua, 0,05 % de TFA; B: 5 % de agua, 94,95 % de metanol, 0,05 % de TFA, UV 220 nm).
Método G: columna Waters BEH C18 (2,0 x 50 mm, partículas de 1,7 pm); fase móvil A: 5:95 metanol:agua con acetato de amonio 10 mM; fase móvil B: 95:5 metanol:agua con acetato de amonio 10 mM ; temperatura: 40 °C; gradiente: 0,5 min de retención a 0 % de B, 0-100 % de B durante 4 min, después, una retención de 0,5 min a 100 % de B; caudal: 0,5 ml/min.
Método H: columna Waters BEH C18 (2,0 x 50 mm, partículas de 1,7 pm); fase móvil A: 5:95 acetonitrilo:agua con acetato de amonio 10 mM ; fase móvil B: 95:5 acetonitrilo:agua con acetato de amonio 10 mM ; temperatura: 40 °C; gradiente: 0,5 min de retención a 0 % de B, 0-100 % de B durante 4 min, después, una retención de 0,5 min a 100 % de B; caudal: 1 ml/min.
La purificación de los productos mediante HPLC preparativa de fase inversa se realizó usando el siguiente método: Método A: Columna: ZORBAX® SB-C18 PrepHT, 5 micrómetros, 21,2 x 100 mm; fase móvil: A=MeOH:H2O:TFA (5:95:0,05), B=MeOH:H2O:TFA (95:5:0,05). Grad.: 0 a 2 min: isocrático 25 % de disolvente B; gradiente de 8 min de 25 a 100 % de disolvente B; tiempo de detención = 15 min. Caudal = 20 ml/min, detección a UV 220 nm.
Ejemplo 1
2-metoxi-6-(6-metoxi-4-((2-metiltiazol-4-il)metoxi)benzofuran-2-il)imidazo[2,1-b][1,3,4]tiadiazol

1A. (2-metiltiazol-4-il)metanol

Una solución de etiléster del ácido 2-metil-tiazol-4-carboxílico (1,26 g, 7,36 mmol) en etiléter (10 ml) se enfrió a -78 °C y se trató con una solución de LAH (0,83 g, 21,9 mmol) en THF seco (30 ml) añadido gota a gota durante 10 min. Después de 3 horas, a -78 °C, la mezcla se inactivó con Na2SO4 saturado (aprox. 20 ml). La mezcla se calentó hasta 22 °C y se extrajo con etiléter (4 x 50 ml). Los extractos combinados se lavaron con salmuera, se secaron en MgSO4 anhidro y se concentraron para obtener un aceite. La filtración en una almohadilla de gel de sílice (3 x 7 cm) y la elución con acetato de etilo produjeron un aceite que se destiló para obtener el material del título (0,664 g, 70 %) en forma de un aceite que se cristalizó. P.e. 60-70 °C / 0,2 torr. HRMS (ESI) calc. para CsHbNOs [M+H]+ m/z 130,0321, encontrado 130,0342. RNM 1H (CDCla, 600 MHz) 8 6,99 (d, J= 0,8 Hz, 1H), 4,70 (s, 1H), 2,98 (a s, 1H), 2,68 (s, 3H).
1B. 5-(benciloxi)-7-metoxi-2,2-dimetil-4H-benzo[d][1,3]dioxin-4-ona

Una solución de 5-hidroxi-7-metoxi-2,2-dimetil-4H-benzo[d][1,3]dioxin-4-ona (30,00 g, 0,134 mol, véase Kamisuki, S. e t al. Tetrahedron, 60, 5695-5700 (2004) para la preparación) en N,N-dimetilformamida (400 ml) se trató con carbonato de potasio anhidro en polvo (19,41 g, 0,14 mol) añadido de una vez. La mezcla resultante se agitó al vacío durante 10 min y después se purgó con nitrógeno. El matraz de reacción se colocó en un baño de agua (22 °C) y se trató con bromuro de bencilo (24,03 g, 0,14 mol), añadido gota a gota durante 15 min. La mezcla resultante se agitó después a 22 °C durante 18 h (la TLC indicó que no hubo material de partida restante). El sólido se filtró y se lavó con N,N-dimetilformamida. El filtrado se evaporó al vacío y el aceite residual se diluyó con acetato de etilo (500 ml) y se lavó con ácido clorhídrico 0,1 N frío, bicarbonato de sodio saturado y salmuera. Después de secar sobre sulfato de magnesio anhidro, la evaporación del disolvente produjo un jarabe espeso. La cristalización a partir de acetato de etilo (50 ml) y hexano (150 ml) produjo 35,17 g de 5-(benciloxi)-7-metoxi-2,2-dimetil-4H-benzo[d][1,3]dioxin-4-ona en forma de prismas incoloros grandes. La cromatografía de los licores madre sobre gel de sílice (4 x 13 cm, elución con tolueno - acetato de etilo 0-5 %) produjo 6,64 g de material adicional para obtener un rendimiento total de 41,81 g (99 %). HRMS (ESI) calc. para C18H19O5 [M+H]+ m/z 315,1227, encontrado 315,1386. RMN 1H (CDCla, 600 MHz) 8 1,68 (s, 6 H), 3,77 (s, 3H), 5,19 (s, 2H), 5,19 (s, 2H), 6,04 (d, J = 2,03 Hz, 1H), 6,15 (d, J = 2,03 Hz, 1H), 7,27 (a t, 1H), 7,36 (at, 2H), 7,52 (a d, 2H).
1C. 2-(benciloxi)-6-hidroxi-4-metoxibenzaldehído

Una solución de 5-(benciloxi)-7-metoxi-2,2-dimetil-4H-benzo[d][1,3]dioxin-4-ona (Ejemplo 1B, 6,76 g, 21,5 mmol) en diclorometano (120 ml) se enfrió a -78 °C y se trató con 43 ml (64,5 mmol) de una solución 1,5 M de hidruro de diisobutilaluminio en tolueno, añadido gota a gota durante 20 min. La mezcla resultante se agitó a -78 °C durante 3 h. La mezcla de reacción se inactivó mediante la adición cuidadosa de metanol (5 ml), añadido gota a gota durante 15 min, seguido de ácido clorhídrico 1 N (50 ml), añadido gota a gota durante 15 min. El baño de enfriamiento se retiró y se agregaron 150 ml adicionales de ácido clorhídrico 1 N durante 20 min. Después, la mezcla se agitó a 22 °C durante 2 h y se diluyó con diclorometano (400 ml). La fase orgánica se recolectó y la fase acuosa (pH ~ 1) se extrajo con diclorometano (3 x 50 ml). Los extractos orgánicos combinados se lavaron con salmuera, se secaron
sobre sulfato de magnesio anhidro y se concentraron al vacío. El aceite residual se diluyó con tetrahidrofurano (70 ml), se trató con 10 ml de ácido clorhídrico 0,1 N y se agitó a 20 °C durante 2 h. La mezcla de reacción se diluyó con acetato de etilo (300 ml), se lavó con salmuera, se secó sobre sulfato de magnesio anhidro y se evaporó al vacío para obtener un aceite transparente. La cromatografía sobre gel de sílice (4 x 13 cm, elución con tolueno) produjo 4,08 g (73 % de rendimiento) del aldehído del título en forma de un aceite transparente que se solidificó en reposo. LC (Método C): 2,237 min. HRMS (ESI) calc. para C15H15O4 [M+H]+ m/z 259,0965, encontrado 259,1153. RMN 1H (CDCla, 600 MHz) 6 3,80 (s, 3H), 5,07 (s, 2H), 5,97 (d, J = 2,1 Hz, 1H), 6,01 (d, J= 2,1 Hz, 1H), 7,3 - 7,4 (m, 5 H), 10,15 (s, 1H), 12,49 (s, 1H).
1D. 1-(4-(benciloxi)-6-metoxibenzofuran-2-il)etanona

Una solución de 2-(benciloxi)-6-hidroxi-4-metoxibenzaldehído (Ejemplo 1C, 3,46 g, 13,4 mmol) en N,N-dimetilformamida (50 ml) se trató con carbonato de cesio anhidro en polvo (4,58 g, 14,05 mmol), añadido de una vez. La mezcla resultante se agitó al vacío durante 10 min y después se purgó con nitrógeno. El matraz de reacción se colocó en un baño de agua (22 °C) y se trató con cloroacetona (1,74 g, 18,7 mmol), añadido gota a gota durante 5 min. La mezcla resultante después se agitó después a 22 °C durante 18 h (la TLC indicó que no hubo aldehído de partida restante y que se formó el intermedio aldehído alquilado). El sólido se filtró y se lavó con N,N-dimetilformamida. El filtrado se evaporó al vacío y el aceite residual se diluyó con acetato de etilo (300 ml) y se lavó con ácido clorhídrico 0,1 N frío, bicarbonato de sodio saturado y salmuera. Después de secar sobre sulfato de magnesio anhidro, la evaporación del disolvente produjo un jarabe espeso. Este jarabe se diluyó con tetrahidrofurano (50 ml) y acetato de etilo (50 ml), se trató con monohidrato de ácido p-toluensulfónico (0,2 g) y se agitó a 20 °C durante 1 h (la TLC indicó la ciclación completa del intermedio aldehído alquilado en benzofurano). La mezcla de reacción se diluyó con acetato de etilo (300 ml) y se lavó con bicarbonato de sodio saturado y salmuera. Después de secar sobre sulfato de magnesio anhidro, la evaporación del disolvente produjo un jarabe espeso. La cromatografía sobre gel de sílice (4 x 12 cm, elución con tolueno - acetato de etilo 2-4 %) produjo 3,51 g ( 88 % de rendimiento) del benzofurano del título en forma de un sólido de color amarillo. La recristalización a partir de acetato de etilo (10 ml) y hexano (20 ml) produjo el material del título en forma de prismas grandes de color amarillo (3,15 g). LC (Método D): 2,148 min. HRMS (ESI) calc. para C18H17O4 [M+H]+ m/z 297,1121, encontrado 297,1092. RMN 1H (CDCla, 600 MHz) 6 2,51 (s, 3H), 3,82 (s, 3H), 5,13 (s, 2H), 6,37 (d, J = 1,77 Hz, 1H), 6,63 (a s, 1H), 7,34 (a t, 1H), 7,39 (a t, 2H), 7,44 (a d, 2H), 7,55 (d, J = 0,7 Hz, 1H).
1E. 1-(4-(benciloxi)-6-metoxibenzofuran-2-il)-2-bromoetanona

Un matraz de tres bocas de 250 ml se equipó con una barra agitadora magnética, se purgó con atmósfera de nitrógeno y se cargó con tetrahidrofurano anhidro (25 ml), seguido de 9,3 ml (9,3 mmol) de una solución 1 M de bis(trimetilsilil)amida de litio en tetrahidrofurano. La mezcla se enfrió a -78 °C y se trató con una solución de 1-(4-(benciloxi)-6-metoxibenzofuran-2-il)etanona (Ejemplo 1D, 2,40 g, 8,1 mmol) en tetrahidrofurano (20 ml), añadida gota a gota durante 10 min. La mezcla resultante se agitó después a -78 °C durante 45 min. Después se agregó gota a gota clorotrimetilsilano (1,18 ml, 9,31 mmol) durante 5 min y la solución resultante se agitó a -78 °C durante otros 20 min. El baño de enfriamiento se retiró después y la mezcla se calentó a temperatura ambiente durante 30 min. La mezcla de reacción se inactivó mediante la adición a una solución fría de acetato de etilo ( 200 ml), bicarbonato de sodio saturado (30 ml) y hielo. La fase orgánica se secó rápidamente sobre sulfato de magnesio anhidro (agitación magnética) y se evaporó al vacío para obtener el sililenoléter en forma de un aceite que se coevaporó con tolueno (20 ml). El sililenoléter se disolvió en tetrahidrofurano seco (40 ml), se enfrió a -20 °C y se trató con bicarbonato de sodio sólido (0,10 g), seguido de N-bromosuccinimida (1,44 g, 8,1 mmol), añadida en pequeñas porciones durante 15 min. La mezcla de reacción se calentó a 0 °C durante 2 h y después se inactivó mediante la adición de acetato de etilo (300 ml) y bicarbonato de sodio saturado. La fase orgánica se lavó con salmuera, se secó sobre sulfato de magnesio anhidro y se evaporó para obtener un aceite naranja. La cromatografía sobre gel de sílice (4 x 12 cm, elución con tolueno - acetato de etilo 0-5 %) produjo 2,62 g ( 86 % de rendimiento) de la bromometilcetona del título en forma de un sólido de color amarillo. La recristalización a partir de acetato de etilo (10 ml) y hexano (20 ml) produjo prismas de color amarillo (2,30 g). LC (Método E): 1,977 min. HRMS (ESI) calc. para C-isH -^ B ^ [M+H]+ m/z 375,0226, encontrado 375,0277. RMN 1H (CDCls, 600 MHz) 6 3,84 (s, 3H), 4,33 (s, 2H), 5,14 (s, 2H), 6,38 (d, J = 1,76 Hz, 1H), 6,64 (a s, 1H), 7,35 (a t, 1H), 7,40 (at, 2H), 7,44 (a d, 2H), 7,70 (s, 1H).
1EE. 1-(4-(bencNoxi)-6-metoxibenzofuran-2-N)-2-doroetanona

Se agregó dicloroyodato de benciltrimetilamonio (117 g, 169mmol) a una solución de 1-(4-(benciloxi)-6-metoxibenzofuran-2-il)etanona (Ejemplo 1D, 50 g, 170 mmol) en THF (500 ml) en un matraz de fondo redondo de 1 l, de múltiples bocas en atmósfera de nitrógeno. La mezcla de reacción se agitó a temperatura ambiente durante 6 h, se enfrió a 0 °C y se inactivó con una solución de NaHCO3 al 10 %. La capa orgánica se lavó con una solución de tiosulfato de sodio 1 M, agua y salmuera, se secó sobre Na2SO4 y se concentró al vacío (temperatura del baño <45 °C). El residuo se trituró con EtOAc al 5 % en éter de petróleo y se secó para obtener la clorometilcetona del título en forma de un sólido de color amarillo claro (48 g, 130 mmol, 78 %). RMN 1H (300 MHz, DMSO-d6) 8 3,84-3,82 (d, J =4,5 Hz, 3H) 4,98 (s, 2H), 5,27(s, 2H), 6,62 -6,61 (d, J =1,8 Hz, 1H), 6,92-6,93 (m, 1H), 7,54-7,36 (m, 5H), 8,10-8,09 (d, J =3 Hz, 1H); MS m/z: [M+H]+ 331,0.
1F. 6-(4-(benciloxi)-6-metoxibenzofuran-2-il)-2-bromoimidazo[2,1-b][1,3,4]tiadiazol

Una mezcla de 1-(4-(benciloxi)-6-metoxibenzofuran-2-il)-2-bromoetanona (Ejemplo 1E, 3,00 g, 8,0 mmol) y 5-bromo-1,3,4-tiadiazol-2-amina (1,65 g, 9,16 mmol) en isopropanol (100 ml) se calentó en un matraz de presión equipado con una barra agitadora magnética a 78-80 °C durante 18 h (homogénea después de 20 min y, después, se formó un precipitado tras 2 h). La mezcla enfriada se transfirió después a cinco viales de 20 ml para microondas y se calentó en un aparato de microondas a 150 °C durante 30 min. Cada vial se diluyó después con diclorometano (250 ml), se lavó con bicarbonato de sodio saturado (25 ml) y salmuera (25 ml) y se secó sobre sulfato de magnesio anhidro . Las fracciones se combinaron y se concentraron al vacío. La cromatografía del sólido residual de color de color pardo anaranjado sobre gel de sílice (4 x 10 cm, elución lenta con diclorometano debido a la escasa solubilidad) produjo 2,96 g del imidazotiadiazol del título contaminado con algo de 1-(4-(benciloxi)-6-metoxibenzofuran-2-il)etanona. El material sólido se trituró con acetato de etilo (20 ml), se filtró, se lavó con acetato de etilo (10 ml) y se secó al vacío para obtener 2,34 g (64 % de rendimiento) del imidazotiadiazol puro del título en forma de un sólido de color blanquecino que se usa tal cual en la siguiente etapa. LC (Método E): 2,188 min. HRMS (ESI) calc. para C20H15BrNaOaS [M+H]+ m/z 456,00175, encontrado 456,00397. RMN 1H (CDCla, 600 MHz) 8 3,82 (s, 3H), 5,16 (s, 2H), 6,38 (d, J = 1,67 Hz, 1H), 6 ,66 (a s, 1H), 7,15 (s, 1H), 7,31 (a t, 1H), 7,38 (a t, 2H), 7,45 (a d, 2H), 8,02 (s, 1H).
De manera alternativa, el ejemplo 1F, 6-(4-(benciloxi)-6-metoxibenzofuran-2-il)-2-bromoimidazo[2,1-b][1,3,4]tiadiazol, se preparó de la siguiente manera:
Un matraz de tres bocas de 1000 ml equipado con una barra agitadora magnética y purgado con atmósfera de nitrógeno, se cargó con NMP seca (200 ml), seguido de 1-(4-(benciloxi)-6-metoxibenzofuran-2-il)-2-cloroetanona (Ejemplo 1EE, 50 g, 150 mmol) y 5-bromo-1,3,4-tiadiazol-2-amina (27,2 g, 151 mmol). La mezcla resultante se agitó a 80 °C durante 8 h. La TLC (8:2 diclorometano/éter de petróleo) y la LC/MS mostraron un material no ciclado intermedio (m/z 476) y la mezcla de reacción se agitó a 120 °C durante 3 h. La mezcla de reacción se enfrió a temperatura ambiente, se inactivó con agua y se extrajo con EtOAc (3x). Las capas orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4 y se concentraron al vacío. El residuo de color pardo espeso se purificó mediante cromatografía sobre gel de sílice (de 0 a 10 0 % de diclorometano en éter de petróleo) para obtener un sólido de color pardo. Este material se trituró con EtOAc y se secó para obtener el imidazotiadiazol del título (24 g, 50 mmol, 33 %) en forma de un sólido de color pardo claro. (Véase el procedimiento descrito anteriormente para los datos analíticos).
1G. 6-(4-(benciloxi)-6-metoxibenzofuran-2-il)-2-metoxiimidazo[2,1-b][1,3,4]tiadiazol

Una solución de 6-(4-(benciloxi)-6-metoxibenzofuran-2-il)-2-bromoimidazo[2,1-b][1,3,4]tiadiazol (Ejemplo 1F, 2,30 g,
5,04 mmol) en una mezcla de diclorometano (180 ml) y metanol (45 ml) se trató a 22 °C con 4,2 ml de una solución de 25 % en peso de metóxido de sodio en metanol (0,2 mmol), añadido en una porción. Se agregó más metanol (45 ml) y la mezcla se agitó durante 1 h. La mezcla de reacción se inactivó mediante la adición de 25 ml de ácido clorhídrico 1 N, seguido de 20 ml de bicarbonato de sodio saturado. El disolvente se evaporó a presión reducida y el residuo se diluyó con diclorometano (400 ml), se lavó con salmuera, se secó sobre sulfato de magnesio anhidro y se evaporó al vacío. La cromatografía del residuo sobre gel de sílice (3 x 10 cm, elución con diclorometano-acetato de etilo 0-4 %) produjo 1,70 g (83 % de rendimiento) del compuesto del título en forma de un sólido de color blanco. Este material se recristalizó de acetato de etilo (30 ml por gramo, 80 % de recuperación) para obtener agujas de color blanco. LC (Método D): 2,293 min. HRMS (ESI) calc. para C21H18N3O4S [M+H]+ m/z 408,1013, encontrado 408,1024. RMN 1H (CDCla, 600 MHz) 8 3,81 (s, 3H), 4,18 (s, 3H), 5,16 (s, 2H), 6,37 (d, J = 1,75 Hz, 1H), 6,67 (a s, 1H), 7,07 (s, 1H), 7,31 (a t, 1H), 7,37 (a t, 2H), 7,45 (a d, 2H), 7,81 (s, 1H).
1H. 6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-ol

Una mezcla de 6-(4-(benciloxi)-6-metoxibenzofuran-2-il)-2-metoxiimidazo[2,1-b][1,3,4]tiadiazol (Ejemplo 1G, 1,250 g, 3,06 mmol) y pentametilbenceno (3,17 g, 21,4 mmol) en diclorometano (200 ml) se enfrió a -78 °C en atmósfera de nitrógeno y después se trató inmediatamente (para evitar la cristalización) con 8 ml (8 mmol) de una solución 1 M de tricloruro de boro en diclorometano, añadida gota a gota durante 3 min. La mezcla resultante se agitó a -78 °C durante 1 h. La mezcla de reacción se inactivó mediante la adición de una solución de bicarbonato de sodio (6 g) en agua (100 ml), añadida en una porción. El baño de enfriamiento se retiró y la mezcla resultante se agitó a temperatura ambiente durante 1 h. El sólido formado se filtró y se lavó sucesivamente con agua (50 ml) y diclorometano (50 ml). La torta de filtro se empapó con etanol anhidro (15 ml) y después se secó por succión. El sólido de color blanco obtenido se secó al vacío durante 24 h para obtener 0,788 g (80 % de rendimiento) del material del título puro (> 95 % mediante HPLC). El filtrado combinado y los lavados se diluyeron con diclorometano (600 ml) y se agitaron en un baño de agua caliente hasta que la fase orgánica se volvió transparente, sin sólidos aparentes en suspensión. La fase orgánica se recolectó, se secó sobre sulfato de magnesio anhidro y se filtró rápidamente mientras aún estaba caliente. El filtrado se evaporó y el residuo (producto y pentametilbenceno) se trituró con tolueno (20 ml), el sólido se recolectó y se lavó con tolueno (20 ml) para obtener 0,186 g (19 % de rendimiento, 99 % de rendimiento combinado) del material del título en forma de un sólido de color tostado (> 95 % mediante HPLC). LC (Método E): 1,444 min. HRMS (ESI) calc. para C14H12 N3O4S [M+H]+ m/z 318,0543, encontrado 318,0578. RMN 1H (DMSO-d6, 600 MHz) 8 3,71 (s, 3H), 4,16 (s, 3H), 6,21 (d, J = 1,87 Hz, 1H), 6,61 (a s, 1H), 6,95 (s, 1H), 8,29 (s, 1H), 9,96 (s, 1H).
Ejemplo 1. 2-metoxi-6-(6-metoxi-4-((2-metiltiazol-4-il)metoxi)benzofuran-2-il)imidazo[2,1-b][1,3,4]tiadiazol

Una mezcla de 6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-ol (Ejemplo 1H, 0,100 g, 0,315 mmol) y trifenilfosfina (0,123 g, 0,47 mmol) se mantuvo al vacío durante 10 minutos. La mezcla se purgó con nitrógeno y después se cargó con THF seco (8 ml) y (2-metiltiazol-4-il)metanol (Ejemplo 1A, 0,049 g, 0,38 mmol). La mezcla se calentó a 50 °C y se sometió a ultrasonidos durante 5 minutos. La mezcla enfriada se trató con una solución de DIAD (0,096 g, 0,47 mmol) en THF seco (2 ml), añadido gota a gota en tres porciones durante 20 minutos. La mezcla se volvió homogénea después de 40 min y se agitó a 22 °C durante 6 h. La mezcla de reacción se diluyó con diclorometano (250 ml), se lavó con bicarbonato de sodio saturado y salmuera y se secó sobre MgSO4 anhidro. La evaporación produjo un residuo semisólido que se purificó mediante cromatografía sobre gel de sílice (2,5 x 10 cm, diclorometano/EtOAc 8 :2 ) para obtener el material del título (0,103 g, 76 %) en forma de cubos de color blanco. LC (Método A): 2,224 min. Hr MS (ESI) calc. para C19H17N4O4S2 [M+H]+ m/z 429,0686, encontrado 429,0605. RMN 1H (CDCla, 600 MHz) 8 7,82 (s, 1H), 7,17 (s, 1H), 7,06 (s, 1H), 6,67 (m, 1H), 6,39 (d, J = 1,89 Hz, 1H), 5,25 (d, J=0,9 Hz, 2H), 4,18 (s, 3H), 3,82 (s, 3H), 2,72 (s, 3H).
Ejemplo 2
2-metoxi-6-(6-metoxi-4-((2-(trifluorometil)tiazol-4-il)metoxi)benzofuran-2-il)imidazo[2,1-b][1,3,4]tiadiazol

2A. 2-(trifluorometil)tiazol-4-carboxilato de etilo

Una mezcla de 2,2,2-trifluoroacetamida (7,12 g, 63 mmol) y reactivo de Lawesson (15,3 g, 37,8 mmol) en THF (60 ml) se calentó a reflujo durante 18 horas. La reacción se enfrió a temperatura ambiente y se trató con bromopiruvato de etilo (8,0 ml, 63 mmol). La reacción se agitó a reflujo durante 18 horas más, después se concentró al vacío y se diluyó con acetato de etilo. Esta mezcla se lavó con agua (1x) y salmuera (1x), se secó sobre sulfato de magnesio anhidro, se filtró y se concentró. El residuo se purificó mediante cromatografía sobre gel de sílice (8 x 1 1 cm, tolueno, la segunda vez con 12 0 g de gel de sílice, hexano/acetato de etilo) para obtener el material del título (4,47 g, 32 %) en forma de un sólido de color amarillo claro. RMN 1H (CDCla, 400 MHz) 8 8,37 (s, 1H), 4,45 (q, J = 7,0 Hz, 2H), 1,41 (t, J = 7,0 Hz, 1H).
2B. (2-(trifluorometil)tiazol-4-il)metanol

Se hizo reaccionar 2-(trifluorometil)tiazol-4-carboxilato de etilo (Ejemplo 2A, 1,50 g, 6 ,66 mmol) como se describe en el ejemplo 1A y se obtuvo el material del título deseado (0,95 g, 78 %) en forma de un aceite transparente después de la destilación (p.e.: 55-65 °C /0,2torr). RMN 1H (CDCla, 400 MHz) 8 7,47 (s, 1H), 4,85 (s, 2H), 2,25 (a s, 1H). Ejemplo 2. 2-metoxi-6-(6-metoxi-4-((2-(trifluorometil)tiazol-4-il)metoxi)benzofuran-2-il)imidazo[2,1-b][1,3,4]tiadiazol

Se hicieron reaccionar 6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-ol (Ejemplo 1H, 0,100 g, 0,315 mmol) y (2-(trifluorometil)tiazol-4-il)metanol (Ejemplo 2B, 0,075 g, 0,409 mmol) como se describe en el ejemplo 1 para obtener el material del título (0,070, 46 %) después de la cristalización en AcOEt. LC (Método B): 2,448 min. HRMS (ESI) calc. para C19H14F3N4O4S2 [M+H]+ m/z 483,0403, encontrado 483,0411. RMN 1H (CDCh, 600 MHz) 8 7.82 (s, 1H), 7,17 (s, 1H), 7,06 (s, 1H), 6,67 (m, 1H), 6,39 (d, J = 1,89 Hz, 1H), 5,25 (d, J=0,9 Hz, 2H), 4,18 (s, 3H), 3.82 (s, 3H), 2,72 (s, 3H).
Ejemplo 3
2-metoxi-6-(6-metoxi-4-((2-feniltiazol-4-il)metoxi)benzofuran-2-il)imidazo[2,1-b][1,3,4]tiadiazol
3A. 2-feniltiazol-4-carboxilato de metilo
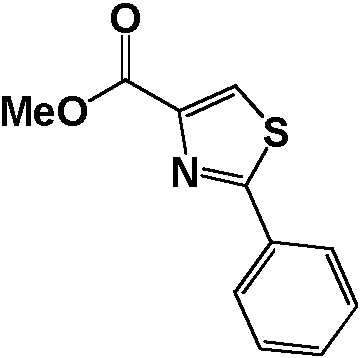
Una solución de benzotioamida (4,0 g, 29,2 mmol) en THF (80 ml) se trató gota a gota con bromopiruvato de metilo (7,6 g, 39 mmol) y se calentó a reflujo durante 18 horas. La reacción se concentró al vacío, se diluyó con acetato de etilo, se lavó con agua (1x) y salmuera (1x) y se secó sobre sulfato de magnesio anhidro. El residuo obtenido después de la concentración se purificó mediante cromatografía sobre gel de sílice (4,5 x 11 cm, AcOEt al 20 %/tolueno), seguido de una segunda purificación con AcOEt al 20 %/hexano. El material del título se obtuvo después de la concentración en forma de un aceite de color amarillo (5,25, 77 %). RMN 1H (CDCh, 400 MHz): 8,14 (s, 1H) 8,00 (m, 2H) 7,46-7,42 (m, 3H) 4,43 (q, J=7,0 Hz, 2H) 1,42 (t, J=7,3 Hz, 3H).
3B. (2-feniltiazol-4-il)metanol

En un matraz de fondo redondo, de 250 ml, se disolvió 2-feniltiazol-4-carboxilato de metilo (Ejemplo 3A, 1,50 g, 6,43 mmol) en etiléter (40 ml). La solución se enfrió a -78 °C y se trató con hidróxido de litio y aluminio (0,75 g, 19,76 mmol) en porciones durante 20 minutos. La reacción se agitó a -78 °C durante 3,5 horas, después se trató con 20 ml de una solución saturada de Na2SO4. Se dejó que la reacción alcanzara la temperatura ambiente y se diluyó con acetato de etilo, se lavó con HCl 1 N (1x) y salmuera (1x), se secó sobre sulfato de magnesio anhidro, se filtró y se concentró. El residuo se purificó mediante cromatografía sobre gel de sílice (3 x 12 cm, acetato de etilo al 30 %/diclorometano) para obtener un aceite de color amarillo claro (1,06 g), que después se destiló (balón a balón, punto de ebullición: 110-120 °C/0,2 torr) y se obtuvo el material del título (0,88 g, 72 %) en forma de un aceite transparente. RMN 1H (CDCla, 400 MHz): 7,95-7,90 (m, 2H) 7,45-7,40 (m, 3H) 7,16 (s, 1H) 4,82 (s, 2H) 2,34 (a s, 1H).
Ejemplo 3. 2-metoxi-6-(6-metoxi-4-((2-feniltiazol-4-il)metoxi)benzofuran-2-il)imidazo[2,1-b][1,3,4]tiadiazol

Una mezcla de 6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-ol (Ejemplo 1H, 0,800 g, 2,52 mmol), trifenilfosfina (0,992 g, 3,78 mmol) y (2-feniltiazol-4-il)metanol (Ejemplo 3B, 0,555 g, 2,90 mmol) en un matraz de 200 ml equipado con un embudo de adición se mantuvo al vacío durante 10 minutos. Después la mezcla se purgó con nitrógeno y se cargó con tetrahidrofurano seco (60 ml, destilado sobre hidruro de litio y aluminio). La solución se calentó a ~ 50 °C y después se sometió a ultrasonidos durante 5 min. La mezcla heterogénea enfriada se trató después a 22 °C con una solución de azodicarboxilato de diisopropilo (0,663 g, 3,28 mmol) en tetrahidrofurano (15 ml), añadido gota a gota durante 2,5 h. La reacción se volvió homogénea (color amarillo claro) al final de la adición. Después, la mezcla se agitó durante 2,5 h más (5 h en total). La mezcla de reacción se diluyó después con diclorometano (400 ml), se lavó con bicarbonato de sodio saturado (20 ml) y salmuera y se secó (sulfato de magnesio anhidro). La evaporación produjo un sólido de color blanco que se sometió a cromatografía sobre gel de sílice (3 x 12 cm, elución con diclorometano - acetato de etilo 98,5:1,5 a 97:3). Las fracciones se recolectaron y se evaporaron para obtener el compuesto deseado (1,40 g) en forma de un sólido de color blanco, contaminado con hidrazida mediante TLC. La cristalización en acetato de etilo (40 ml) produjo el material del título puro (0,838 g, 68 %) en forma de un sólido de color blanco. Los licores madre (0,475 g) se sometieron a cromatografía sobre gel de sílice (3 x 12 cm, elución con diclorometano - acetato de etilo 98,5 : 1,5 a 97 : 3) para obtener, después de la cristalización a partir de acetato de etilo (30 ml), más compuesto deseado (0,160 g, 13 %, 81 % en total) en forma de un sólido de color blanco. LC (Método C): 2,480 min. HRMS (ESI) calc. para C24H19N4O4S2 [M+H]+ m/z 491,0842, encontrado 491,0865. RMN 1H (CDCla, 400 MHz) 3,85 (s, 3H) 4,21 (s, 3H) 5,33
- 5,55 (m, 2H) 6,48 (d, J=1,96 Hz, 1H) 6,72 (dd, J=1,96, 0,78 Hz, 1H) 7,12 (s, 1H) 7,36 - 7,39 (m, 1H) 7,41 - 7,50 (m, 3H) 7,86 (s, 1H) 7,95 - 8,02 (m, 2H).
Ejemplo 4
2-metoxi-6-(6-metoxi-4-((4-feniltiazol-2-il)metoxi)benzofuran-2-il)imidazo[2,1-b][1,3,4]tiadiazol

4A. 2-amino-2-tioxoacetato de etilo

Una solución de 2-amino-2-oxoacetato de etilo (5,00 g, 42,7 mmol) en tetrahidrofurano (150 ml) se trató con reactivo de Lawesson en polvo (mortero y pistilo) (9,50 g, 23,49 mmol), y la solución transparente de color naranja resultante se calentó a reflujo (temperatura del baño 85 °C) durante 4 h (se formó el producto de TLC con un mayor Rf con parte del material de partida restante). La mezcla enfriada se concentró a presión reducida y el residuo se diluyó con acetato de etilo (400 ml), se lavó con bicarbonato de sodio saturado y salmuera y se secó sobre sulfato de magnesio anhidro. La evaporación produjo un sólido de color naranja que se sometió a cromatografía sobre gel de sílice (3 x 10 cm, elución con tolueno - acetato de etilo 9:1) para obtener el material del título (3,189 g, 56 %) en forma de un sólido de color amarillo. LC (Método C): 0,816 min. RMN 1H (CDCla, 400 MHz) 8 ppm: 1,41 (t, J=7,0 Hz, 3H), 4,38 (q, J=7,0 Hz, 2H), 7,69 (a s, 1H) 8,24 (a s, 1H).
4B. 4-feniltiazol-2-carboxilato de etilo

Una mezcla de 2-bromo-1-feniletanona (1,790 g, 8,99 mmol) y 2-amino-2-tioxoacetato de etilo (Ejemplo 4A, 1,20 g, 9,01 mmol) en benceno (80 ml) y etanol (10 ml) se agitó a temperatura ambiente durante 18 h. La mezcla se calentó a 80 °C durante 1 h. El disolvente se evaporó a presión reducida y el residuo se repartió entre acetato de etilo (300 ml) y bicarbonato de sodio acuoso saturado (100 ml). La fase orgánica se lavó con salmuera, se secó sobre sulfato de magnesio anhidro y se concentró al vacío. El aceite transparente residual se sometió a cromatografía sobre gel de sílice (4 x 10 cm, elución con tolueno-acetato de etilo 0-2-4 %) para obtener un aceite de color amarillo (1,588 g). Este se destiló al vacío (punto de ebullición: 105-115 °C/0,1 torr, destilación de balón a balón, temperatura del baño del aire) para obtener el material del título (1,409 g, 67 %) en forma de un jarabe de color amarillo claro que después de reposar se solidificó en un sólido casi incoloro. LC (Método C): 2,009 min. HRMS (ESI) calc. para C12H12NO2S [M+H]+ m/z 234,0583, encontrado 234,0597. RMN 1H (CDCla, 400 MHz) 8 ppm: 1,48 (t, J=7,2 Hz, 3H), 4,52 (q, J=7,2 Hz, 2H), 7,35 - 7,49 (m, 3H), 7,75 (s, 1H), 7,93 - 8,00 (m, 2H).
4C. 2-(hidroximetil)-4-feniltiazol

Una solución de 4-feniltiazol-2-carboxilato de etilo (Ejemplo 4B, 1,300 g, 5,57 mmol) en dietiléter (60 ml) en un matraz de 500 ml en atmósfera de nitrógeno se enfrió a -40 °C (baño de hielo seco - agua - cloruro de calcio) y se trató con LiAlH4 sólido (0,40 g, 10,54 mmol) añadido de una vez. La mezcla se agitó a -40 °C durante 2,5 h. La reacción se inactivó mediante la adición gota a gota de acetato de etilo (1 ml) y agua (0,4 ml), seguido de hidróxido de sodio acuoso al 15 % (0,4 ml) y agua (1,2 ml). Después el baño se retiró y la mezcla se agitó a temperatura ambiente durante 50 min. El sólido formado se filtró y se lavó con éter (50 ml). El filtrado y el lavado combinados se lavaron con salmuera (20 ml) y se secaron sobre sulfato de magnesio anhidro. La evaporación produjo un aceite de
color amarillo que se purificó mediante cromatografía sobre gel de sílice (2,5 x 8 cm, elución con tolueno - acetato de etilo 9 : 1, 8 : 2 a 7 : 3). Después el aceite de color amarillo claro resultante (0,931 g) se destiló al vacío (punto de ebullición: 105-110 °C/0,1 torr, balón a balón, temperatura del baño del aire) para obtener el material del título (0,918 g, 86 %) en forma de un jarabe incoloro. LC (Método C): 1,672 min. HRMS (ESI) calc. para C10H10NOS [M+H]+ m/z 192,0478, encontrado 192,0508. RMN 1H (CDCls, 400 MHz) 8 ppm: 2,90 (a t, 1H), 5,02 (d, J=4,30 Hz, 2H), 7,31 -7,38 (m, 1H), 7,39 - 7,45 (m, 2H), 7,46 (d, J=0,8 Hz, 1H), 7,85 - 7,92 (m, 2H).
4D. 2-(bromometil)-4-feniltiazol

Una solución de (4-feniltiazol-2-il)metanol (Ejemplo 4C, 0,530 g, 2,77 mmol) en diclorometano (10 ml) se enfrió a 0 °C (baño de hielo) y se trató con PBr3 (0,118 ml, 1,247 mmol), añadido gota a gota durante 2 min. Inmediatamente se formó una goma pesada de color blanco. Después de 10 min, el baño se retiró y la solución se agitó a 22 °C durante 4 h. La mezcla de reacción se inactivó con hielo (~ 10 g) y se vertió en una mezcla de acetato de etilo (150 ml) y bicarbonato de sodio saturado (50 ml). La fase orgánica se lavó con salmuera, se secó sobre sulfato de magnesio anhidro y se concentró al vacío. El residuo sólido se sometió a cromatografía sobre gel de sílice (2,5 x 6 cm, elución con tolueno) para obtener el material del título (0,561 g, 80 %) en forma de un aceite de color amarillo claro que se solidificó en el congelador y se convirtió en un sólido de color amarillo claro. LC (Método C): 2,062 min. HRMS (ESI) calc. para C-iüHgBrNS [M+H]+ m/z 253,9634, encontrado 253,9655. RMN 1H (CDCla, 400 MHz) 8 ppm: 4,81 (s, 2H), 7,34 - 7,39 (m, 1H), 7,41 - 7,47 (m, 2H), 7,52 (s, 1H), 7,86 - 7,92 (m, 2H).
Ejemplo 4. 2-metoxi-6-(6-metoxi-4-((4-feniltiazol-2-il)metoxi)benzofuran-2-il)imidazo[2,1-b][1,3,4]tiadiazol

Una suspensión de 6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-ol (Ejemplo 1H, 0,080 g, 0,252 mmol) y 2-(bromometil)-4-feniltiazol (0,128 g, 0,504 mmol) en N,N-dimetilformamida (3 ml) se mantuvo al vacío (1 kPa (10 mbar)) durante 5 minutos. Después el matraz se purgó con nitrógeno y se agregó de una vez carbonato de potasio anhidro recién convertido en polvo (mortero y pistilo) (0,105 g, 0,756 mmol). La mezcla resultante se agitó a temperatura ambiente con unos pocos períodos cortos de sonicación (~ 1 min) durante 1 hora. La mezcla heterogénea se volvió casi homogénea (excepto por el carbonato de potasio) después de 10 min y se comenzó a precipitar de nuevo hasta volverse un sólido de color crema. La mezcla de reacción se inactivó con ácido clorhídrico 1 N (2 ml) y después se repartió entre diclorometano (150 ml) y bicarbonato de sodio saturado (20 ml). La fase orgánica se lavó con salmuera, se secó sobre sulfato de magnesio anhidro y se concentró al vacío. El residuo sólido de color amarillo claro se sometió a cromatografía sobre gel de sílice (2,5 x 6 cm, elución con diclorometano-acetato de etilo 0-2-5 %) para obtener el material del título (0,116 g, 94 %) en forma de un sólido de color amarillo claro. La cristalización en acetato de etilo ( 12 ml) produjo el material del título (0,086 g) en forma de un sólido de color amarillo claro. LC (Método C): 2,474 min. HRMS (ESI) calc. para C24H19N4O4S2 [M+H]+ m/z 491,0842, encontrado 491,0864. RMN 1H (CDCls, 400 MHz) 8 ppm: 3,86 (s, 3H), 4,22 (s, 3H), 5,54 (s, 2H), 6,48 (d, J=1,96 Hz, 1H), 6,75 (a d, 1H), 7,15 (s, 1H), 7,32 - 7,39 (m, 1H), 7,41 - 7,49 (m, 2H), 7,53 (s, 1H), 7,87 (s, 1H), 7,90 - 7,95 (m, 2H).
Ejemplo 5
4-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2-il)morfolina

5A. 2-morfolinotiazol-4-carboxilato de metilo

Una solución de 2-bromotiazol-4-carboxilato de metilo (0,20 g, 0,901 mmol) en THF (10 ml) se trató con morfolina (0,17 ml, 1,94 mmol) y se sometió a reflujo durante 18 h. Después la reacción se diluyó con acetato de etilo y se lavó con NaHCO3 saturado (1x) y salmuera (1x) y se secó sobre sulfato de magnesio anhidro, se filtró y se concentró. El residuo se purificó mediante cromatografía sobre gel de sílice (2,5 x 10 cm, AcOEt al 50 %/CH2Ch) para obtener el material del título (0,192 g, 92 %) en forma de un sólido de color amarillo. RMN 1H (CDCh, 400 MHz) 8 ppm: 7,44 (s, 1H) 3,82 (s, 3H) 3,75 (m, 4H) 3,45 (m, 4H).
5B. (2-morfolinotiazol-4-il)metanol

Una solución de 2-morfolinotiazol-4-carboxilato de metilo (0,76 g, 3,33 mmol) en etiléter (20 ml) se trató con hidruro de litio y aluminio (0,38 g, 10,01 mmol) en porciones durante 10 min. La mezcla se agitó a -78 °C durante 4 horas, después se trató lentamente con acetato de etilo (10 ml) y Na2SO4 saturado (20 ml). Se dejó que la mezcla alcanzara la temperatura ambiente, se diluyó con acetato de etilo, se lavó con NaHCO3 saturado (1 x) y salmuera (1x), se secó sobre sulfato de magnesio anhidro, se filtró y se concentró. El residuo se purificó mediante cromatografía sobre gel de sílice (3 x 10 cm, AcOEt al 25 %/CH2Cl2 a AcOEt al 100 %) para obtener el material del título en forma de un sólido de color beis (0,458 g) que después se destiló (balón a balón, 135-145 °C / 0,2 torr) y produjo el producto deseado (0,455 g, 68 %) en forma de un sólido de color blanco. LC (Método F): 0,873 min. HRMS (ESI) calc. para C8H13N2O2S [M+H]+ m/z 201,07, encontrado 201,2. RMN 1H (CDCh, 400 MHz) 8 ppm: 6,43 (s, 1H) 4,53 (d, J=3,9 Hz, 2H) 3,79 (m, 4H) 3,44 (m, 4H) 2,17 (s, 1H).
Ejemplo 5. 4-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2-il)morfolina
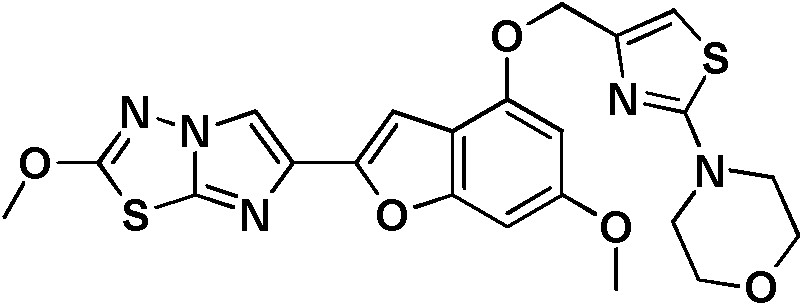
Se agregaron 6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-ol (Ejemplo 1H, 0,10 g, 0,315 mmol), trifenilfosfina (0,124 g, 0,473 mmol) y (2-morfolinotiazol-4-il)metanol (Ejemplo 5B, 0,086 g, 0,429 mmol) a un matraz de fondo redondo de 25 ml y se purgaron al vacío y con nitrógeno. Después se agregó tetrahidrofurano (8 ml) y la mezcla se trató con DIAD (0,083 g, 0,410 mmol) en tetrahidrofurano (10 ml). La mezcla se agitó a 22 °C durante 1 hora y se diluyó con acetato de etilo. Se lavó con NaHCO3 saturado (1x) y salmuera (1x), se secó sobre sulfato de magnesio anhidro, se filtró y se concentró. El residuo se purificó mediante cromatografía en columna sobre gel de sílice (2,5 x 10 cm, 40 % de acetato de etilo en C ^C h) y el residuo obtenido después de la concentración se cristalizó en acetato de etilo para obtener el material del título en forma de cristales (0,083 g, 53 %) y en forma de un sólido impuro amorfo del licor madre (0,169 g). LC (Método F): 2,466 min. HRMS (ESI) calc. para C22H22N5O5S2 [M+H]+ m/z 500,1057, encontrado 500,1075. RMN 1H (CDCh, 400 MHz) 8 ppm: 7,82 (s, 1H) 7,07 (s, 1H) 6,67 (d, J=2 Hz, 1H) 6,62 (s, 1H) 6,40 (d, J=1,5 Hz, 1H) 5,10 (s, 2H) 4,19 (s, 3H) 3,82 (s, 3H) 3,81 (m, 4H) 3,46 (m, 4H).
Ejemplo 6
2-metoxi-6-(6-metoxi-4-((2-((2-metoxietoxi)metil)tiazol-4-il)metoxi)benzofuran-2-il)imidazo[2,1-b][1,3,4]tiadiazol
6A. 2-(2-metoxietoxi)acetamida

Una solución de ácido 2-(2-metoxietoxi)acético (5,0 g, 37,3 mmol) en CH2CI2 (50 ml) se trató con cloruro de oxalilo (9,5 ml, 109 mmol) y DMF (2 gotas) y la reacción se agitó durante 3 horas. Después de la evaporación al vacío, el residuo se coevaporó con CH2O 2 (2x) y después se disolvió en THF (10 ml) y se trató gota a gota con una mezcla de hidróxido de amonio (12 ml), THF (25 ml) y agua (10 ml) durante 5 min. La reacción se agitó a 0-5 °C durante 30 min y después a 22 °C durante 1 h. La reacción se diluyó con CH2Cl2, se lavó con agua (1x), HCl 1 N (1x), NaHCO3 saturado (1x) y salmuera (1x), se secó sobre sulfato de magnesio anhidro, se filtró y se concentró. Como el producto parecía ser soluble en agua, la fase acuosa se evaporó al vacío y se extrajo con CH2Ch (5 x 200 ml), se secó sobre sulfato de magnesio anhidro, se filtró y se concentró para obtener el material del título (3,59 g, 72 %) en forma de un aceite que se solidificó. Este se destiló (balón a balón, 105-115 °C/0,2 torr) para obtener el producto deseado puro (3,39 g) en forma de un aceite transparente que se solidificó en forma de un sólido de color blanco. RMN 1H (cDch, 400 MHz) 8 ppm: 6,93 (muy ancho s, 1H), 5,43 (muy ancho s, 1H), 3,99 (s, 2H), 3,66 - 3,70 (m, 2H), 3,53 - 3,56 (m, 2H), 3,39 (s, 3H).
6B. 2-(2-metoxietoxi)etantioamida

Una solución de 2-(2-metoxietoxi)acetamida (Ejemplo 6A, 3,39 g, 25,5 mmol) en THF (40 ml) se trató con reactivo de Lawesson (6,55 g, 16,19 mmol) y la reacción se sometió a reflujo durante 18 horas. Después la reacción se enfrió a temperatura ambiente, se concentró al vacío, se diluyó con acetato de etilo y se lavó con NaHCO3 saturado (1x) y salmuera (1x). Las fases acuosas se extrajeron con acetato de etilo (2 x 200 ml) y los extractos orgánicos se secaron sobre sulfato de magnesio anhidro, se filtraron y se concentraron. El residuo se purificó mediante cromatografía sobre gel de sílice (3,5 x 11 cm, 30 % de AcOEt/CH2Ch) para obtener el material del título (3,26 g, 86 %) en forma de un aceite de color amarillo. RMN 1H (CDCh, 400 MHz) 8 ppm: 8,58 (muy ancho s, 1H), 7,50 (muy ancho s, 1H), 4,36 (s, 2H), 3,66 - 3,69 (m, 2H), 3,53 - 3,56 (m, 2H), 3,39 (s, 3H).
6C. 2-((2-metoxietoxi)metil)tiazol-4-carboxilato de etilo

A una solución de 2-(2-metoxietoxi)etantioamida (Ejemplo 6B, 3,26 g, 21,85 mmol) en etanol (60 ml), se le agregó gota a gota etilbromopiruvato (3,7 ml, 29,5 mmol) y la mezcla se sometió a reflujo durante 18 horas. La reacción se concentró al vacío, se diluyó con acetato de etilo, se lavó con agua (1x) y salmuera (1x), se secó sobre sulfato de magnesio anhidro, se filtró y se concentró. El residuo se purificó mediante cromatografía de columna sobre gel de sílice (3,5 x 10 cm, 30 % de acetato de etilo/CH2Cl2) para obtener el material del título (4,36 g, 81 %) en forma de un aceite. LC (Método F): 1,791 min. RMN 1H (CDCla, 400 MHz) 8 ppm: 8,16 (s, 1H), 4,87 (s, 2H), 4,41 (q, J = 7,10 Hz, 2H), 3,74 - 3,77 (m, 2H), 3,57 - 3,60 (m, 2H), 3,39 (s, 3H), 1,39 (t, J = 7,10 Hz, 3H).
6D. (2-((2-metoxietoxi)metil)tiazol-4-il)metanol

A una solución de 2-((2-metoxietoxi)metil)tiazol-4-carboxilato de etilo (Ejemplo 6C, 2,27 g, 9,25 mmol) en éter (50 ml), se le agregó en porciones hidruro de litio y aluminio (1,06 g, 27,9 mmol) durante 10 min a -78 °C. Después, la reacción se agitó a 78 °C durante 1 h. Después se agregó acetato de etilo (10 ml) a la reacción seguido de agua (20 ml) y se dejó que la reacción alcanzara la temperatura ambiente. La mezcla se diluyó después con acetato de
etilo y se lavó con HCl 1 N (1x) y salmuera (1x). Las fases acuosas combinadas se extrajeron con acetato de etilo (2 x 300 ml), se secaron sobre sulfato de magnesio anhidro, se filtraron y se concentraron. El residuo se purificó mediante cromatografía sobre gel de sílice (3,5 x 10 cm, acetato de etilo) para obtener el material del título (0,357 g, 19 %) en forma de un aceite de color pardo. LC (Método F): 1,791 min. RMN 1H (CDCh, 400 MHz) 8 ppm: 7,16 (s, 1H), 4,82 (s, 2H), 4,74 (a s, 2H), 3,7-3,75 (m, 2H), 3,54 -3,61 (m, 2H), 3,38 (s, 3H), 2,28 (a s, 1H).
6 E. 4-(bromometil)-2-((2-metoxietoxi)metil)tiazol

Una solución de (2-((2-metoxietoxi)metil)tiazol-4-il)metanol (0,35 g, 1,72 mmol) en éter (15 ml) se trató con PBr3 (0,1 ml, 1,06 mmol) a temperatura ambiente. Se formó un precipitado. La reacción se agitó a temperatura ambiente durante 18 horas, después se diluyó con acetato de etilo, se lavó con NaHCO3 saturado (1 x) y salmuera (1 x), se secó sobre sulfato de magnesio anhidro, se filtró y se concentró. El residuo se purificó mediante cromatografía sobre gel de sílice (3 x 10 cm, 20 % de acetato de etilo/C^Ch) para obtener el material del título (0,233 g, 51 %) en forma de un aceite transparente. RMN 1H (CDCla, 400 MHz) 8 ppm: 7,27 (s, 1H), 4,83 (s, 2H), 4,55 (s, 2H), 3,73 - 3,76 (m, 2H), 3,57 - 3,60 (m, 2H), 3,39 (s, 3H).
Ejemplo 6. 2-metoxi-6-(6-metoxi-4-((2-((2-metoxietoxi)metil)tiazol-4-il)metoxi)benzofuran-2-il)imidazo[2,1-b][1,3,4]tiadiazol

Una suspensión de 6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-ol (Ejemplo 1H, 0,10 g, 0,315 mmol) y 4-(bromometil)-2-((2-metoxietoxi)metil)tiazol (Ejemplo 6 E, 0,10 g, 0,376 mmol) en DMF (5 ml) se purgó al vacío y con nitrógeno durante 10 minutos. Después, la mezcla se trató con carbonato de potasio (0,10 g, 0,724 mmol) y la reacción se agitó a temperatura ambiente durante 2,5 horas. La reacción se diluyó después con diclorometano, se lavó con agua (1 x) y salmuera (1 x), se secó sobre sulfato de magnesio anhidro, se filtró y se concentró. El residuo se purificó mediante cromatografía sobre gel de sílice (2,5 x 10 cm, 50 % de acetato de etilo/CH2Cl2) para obtener el material del título que se cristalizó en acetato de etilo y produjo el material del título deseado (0,055 g, 35 %) junto con material no cristalizado (9 mg, 6 %). LC (Método F): 2,476 min. HRMS (ESI) calc. para C22H23N4O6S2 [M+H]+ m/z 503,1054, encontrado 503,1066. RMN 1H (CDCla, 400 MHz) 8 ppm: 7,82 (s, 1H), 7,35 (s, 1H), 7,05 (s, 1H), 6 ,68 (a s, 1H), 6,39 (d, J = 1,9 Hz, 1H), 5,28 (s, 2H), 4,86 (s, 2H), 4,19 (s, 3H), 3,82 (s, 3H), 3,74 - 3,77 (m, 2H), 3,58 - 3,61 (m, 2H), 3,39 (s, 3H).
Ejemplo 7
2-metoxi-6-(6-metoxi-4-((5-fenil-1,2,4-tiadiazol-3-il)metoxi)benzofuran-2-il)imidazo[2,1-b][1,3,4]tiadiazol

7A. 5-fenil-1,2,4-tiadiazol-3-carboxilato de etilo
Una mezcla de 2-oxo-1,3,4-oxatiazol-5-carboxilato de etilo (publicación estadounidense n.° 2005/0096362) (1,5 g, 8,56 mmol) y benzonitrilo (4,37 ml, 42,8 mmol) en 1,2-diclorobenceno (15,42 ml, 137 mmol) se calentó a 160 °C durante 4 días. Después, la reacción se enfrió a temperatura ambiente y el disolvente se evaporó calentando la reacción a 75 °C en máximo vacío. El residuo se purificó mediante cromatografía sobre gel de sílice (100 % de CH2Cl2 a 3 % de EtOAc en CH2Ch) para obtener el material del título (0,064 g, 3 %). LC (Método B): 2,021 min. HRMS (ESI) calc. para C11 H11 N2O2S [M+H]+ m/z 235,0541, encontrado 235. RMN 1H (CDCla, 400 MHz) 8 ppm: 8,01 - 8,09 (m, 1H), 7,49 -7,59 (m, 2H), 4,56 (q, J=7,17 Hz, 1H), 1,50 (t, J=7,24 Hz, 1H).
7B. (5-fenil-1,2,4-tiadiazol-3-il)metanol

A una solución de 5-fenil-1,2,4-tiadiazol-3-carboxilato de etilo (Ejemplo 7A, 230 mg, 0,982 mmol) en etanol anhidro (3 ml, 51,4 mmol) se le agregó NaBH4 (149 mg, 3,93 mmol) a 0 °C. La mezcla de reacción se calentó a 80 °C durante 30 min, después se agregó HCl 1 N (1 ml) y se evaporó el etanol. Se agregó diclorometano a la reacción seguido de salmuera y esta se extrajo con diclorometano (3x). Las capas orgánicas se secaron sobre sulfato de magnesio anhidro y se concentraron. El residuo se purificó mediante cromatografía en columna sobre gel de sílice (100 % de CH2Cl2 hasta 10 % de EtOAc/CH2Ch) para obtener el material del título (25 mg, 13 %). LC (Método B): 1,858 min. LCMS (APCI) calc. para C9H9N2OS [M+H]+ m/z 193,04, encontrado 193,0. RMN 1H (CDCla, 400 MHz) 8 ppm: 7,89 - 8,03 (m, 2H), 7,46 - 7,62 (m, 3H), 4,99 (d, J=5,87 Hz, 2H), 2,81 (t, J=6,06 Hz, 1H)
Ejemplo 7. 2-metoxi-6-(6-metoxi-4-((5-fenil-1,2,4-tiadiazol-3-il)metoxi)benzofuran-2-il)imidazo[2,1-b][1,3,4]tiadiazol

Se colocaron 6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-ol (Ejemplo 1H, 8,25 mg, 0,026 mmol) y (5-fenil-1,2,4-tiadiazol-3-il)metanol (Ejemplo 7B, 5 mg, 0,025 mmol) en un matraz que se purgó con N2. Se agregó THF seco (4 ml) y a esta suspensión resultante se le agregó tri-n-butilfosfina (0,017 ml, 0,065 mmol) y se le agregó gota a gota una solución de 1,1 '-(azodicarbonil)dipiperidina (16,57 mg, 0,065 mmol) en THF seco (2,5 ml) mediante una bomba de jeringa durante 2 h. La suspensión resultante se agitó durante 2 horas más a temperatura ambiente, en cuyo momento la LC mostró que no había material de partida restante. La mezcla se diluyó con EtOAc, se lavó con HCl 0,2 N, NaHCO3 acuoso saturado y salmuera, se secó sobre sulfato de magnesio anhidro, se filtró y se concentró. El residuo se purificó mediante cromatografía sobre gel de sílice (50 % de diclorometano/hexanos a 100 % de diclorometano a 1 % de EtOAc/CH2Cl2 a 7 % de EtOAc/CH2Ch) y se liofilizó en MeCN/agua para obtener el material del título (6,2 mg, 49 %). LC (Método B): 2,615 min. HRMS (Es I) calc. para C2aH18N5O4S2 [M+H]+ m/z 492,0795, encontrado 492,0828. RMN 1H (CDCh, 400 MHz) 8 ppm: 8,00 (dd, J=8,02, 1,37 Hz, 2H), 7,85 (s, 1H), 7,50 - 7,56 (m, 3H), 7,14 (s, 1H), 6,71 - 6,75 (m, 1H), 6,51 (d, J=1,57 Hz, 1H), 5,51 (s, 2H), 4,21 (s, 3H), 3,84 (s, 3H).
Ejemplo 8
2-metoxi-6-(6-metoxi-4-((5-fenil-1,3,4-tiadiazol-2-il)metoxi)benzofuran-2-il)imidazo[2,1-b][1,3,4]tiadiazol

8A. 5-fenil-1,3,4-tiadiazol-2-carboxilato de etilo

A una solución de 2-(2-benzoilhidrazinil)-2-oxoacetato de etilo (1 g, 4,23 mmol) en THF seco (5 ml, 61,0 mmol) se le agregó el reactivo de Lawesson (1,079 g, 2,67 mmol). La reacción se agitó a temperatura ambiente durante 2 h sin ninguna reacción. La mezcla se calentó después a 50 °C y después se calentó a reflujo. Se agregó más reactivo de Lawesson (1,079 g, 2,67 mmol) y tras 16 h a reflujo, la reacción se había completado a la mitad. La mezcla se evaporó a sequedad y el residuo se purificó mediante cromatografía en columna sobre gel de sílice (50 % de CH2Cl2/hexanos hasta 100 % de CH2Ch) para obtener el material del título (0,35 g, 35 %). LC (Método B): 2,063 min, LCMS (APCI) calc. para C11 H11 N2O2S [M+H]+ m/z 235,05, encontrado 235,0. RMN 1H (CDCla, 400 MHz) 8 ppm: 1,49 (t, J=1,00 Hz, 3H), 4,55 (q, J=1,00 Hz, 2H), 7,45 - 7,65 (m, 3H), 8,02 - 8,07 (m, 2H).
8B. (5-fenil-1,3,4-tiadiazol-2-il)metanol

A una solución de 5-fenil-1,3,4-tiadiazol-2-carboxilato de etilo (350 mg, 1,494 mmol) en metanol anhidro (5 ml, 124 mmol), se le agregó NaBH4 (226 mg, 5,98 mmol) a 0 °C. La mezcla de reacción se agitó a temperatura ambiente durante 16 h. Se agregó AcOH (2 ml) y la reacción se concentró a sequedad. El residuo se disolvió en EtOAc, salmuera y agua y se extrajo con EtOAc (3x). Los extractos orgánicos combinados se lavaron con NaHCO3 acuoso saturado y salmuera y se secaron sobre sulfato de magnesio anhidro . Después de la filtración y evaporación, el residuo se trituró con etiléter para obtener el material del título como un primer cultivo (150 mg, 52 %). LC (Método B): 2,022 min, LCMS (APCI) calc. para C9H9N2OS [M+H]+ m/z 193,04, encontrado 193,2. RMN 1H (CDCla, 400 MHz) 8 ppm: 7,92 -8,03 (m, 2H), 7,44 - 7,59 (m, 3H), 5,14 (a. d, J=3,90 Hz, 2H), 2,63 (a. s., 1H).
Ejemplo 8. 2-metoxi-6-(6-metoxi-4-((5-fenil-1,3,4-tiadiazol-2-il)metoxi)benzofuran-2-il)imidazo[2,1-b][1,3,4]tiadiazol

En un matraz de fondo redondo de 200 ml, se agregó benceno a 5-fenil-1,3,4-tiadiazol-2-carboxilato de etilo (Ejemplo 8B, 80 mg, 0,252 mmol) y 6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-ol (Ejemplo 1H, 58,2 mg, 0,303 mmol) y la mezcla se sometió a ultrasonido durante 30 segundos y se concentró al vacío para retirar los rastros de agua en el material de partida. Se agregó trifenilfosfina (99 mg, 0,378 mmol) y la mezcla se secó a alto vacío durante 10 min. Se agregó THF (40 ml) y la mezcla se sometió a ultrasonido / se calentó durante 5 min. Se agregó gota a gota azodicarboxilato de diisopropilo (68,6 pl, 0,353 mmol) en THF (4 ml) en aprox. 1 h y la LC/MS indicó que la reacción no se había completado. Se agregó de nuevo azodicarboxilato de diisopropilo (2 gotas) y la mezcla se diluyó en CH2Ch, se lavó con NaHCO3 acuoso saturado (1x) y salmuera (1x), se secó sobre MgSO4 anhidro y se concentró. El residuo se purificó mediante cromatografía sobre gel de sílice (100 % de CH2Cl2 hasta 15 % de EtOAc/CH2Cl2) para obtener un residuo que se trituró con MeCN y produjo el material del título (36 mg, 29 %). LC (Método A): 2,901 min. HRMS (ESI) calc. para C23H17N5O4S2 [M+H]+ m/z 492,0722, encontrado 492,0806. RMN 1H (CDCls, 400 MHz) 8 ppm: 7,96 - 8,02 (m, 2H), 7,87 (s, 1H), 7,45 - 7,55 (m, 3H), 7,10 (s, 1H), 6,73 - 6,78 (m, 1H), 6,48 (d, J=1,57 Hz, 1H), 5,63 (s, 2H), 4,22 (s, 3H), 3,86 (s, 3H).
Ejemplo 9
2-metoxi-6-(6-metoxi-4-((1-fenil-1H-1,2,3-triazol-4-il)metoxi)benzofuran-2-il)imidazo[2,1-b][1,3,4]tiadiazol

9A. 2-metoxi-6-(6-metoxi-4-(prop-2-in-1-iloxi)benzofuran-2-il)imidazo[2,1-b][1,3,4]tiadiazol
Una solución de 6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-ol (Ejemplo 1H, 205 mg, 0,646 mmol) en THF (10 ml) se trató, a temperatura ambiente y en atmósfera de nitrógeno, con alcohol de propargilo (0,096 ml, 1,615 mmol), tri-n-butilfosfina (0,398 ml, 1,615 mmol) y gota a gota durante un período de 25 min con una solución de 1,1'-(azodicarbonilo)dipiperidina (408 mg, 1,615 mmol) en THF (10 ml). La mezcla se sometió a ultrasonido en un baño durante 30 min y se agitó a temperatura ambiente durante 30 min más. La mezcla se disolvió después en diclorometano (50 ml) y se lavó con NaHcO3 acuoso saturado y salmuera, y se secó (MgSO4). La evaporación del disolvente produjo un sólido que se purificó mediante cromatografía en columna sobre gel de sílice ISCO para obtener el material del título (180 mg, 0,507 mmol, 78 % de rendimiento). RMN 1H (DMSO-d6, 400 MHz) 8 ppm: 8,38 (s, 1H), 6,92 (s, 1H), 6,86 (dd, J=1,8, 1,0 Hz, 1H), 6,53 (d, J=1,6 Hz, 1H), 4,94 (d, J=2,7 Hz, 2H), 4,21 (s, 3H), 3,77-3,84 (m, 3H), 3,60-3,66 (m, 1H).
9B. Azidobenceno
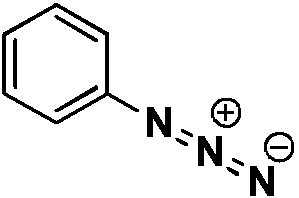
Una solución de anilina (500 mg, 5,37 mmol) en acetonitrilo (10 ml, 191 mmol) se enfrió en un baño de hielo y se trató con nitrito de ferc-butilo (680 mg, 6,59 mmol) y gota a gota con TMS-N3 (0,713 ml, 5,37 mmol). El baño de hielo se retiró y la mezcla se agitó durante una noche a temperatura ambiente en N2. El acetonitrilo se evaporó cuidadosamente (NB: el azidobenceno también es volátil) y el residuo (750 mg) se hizo pasar a través de una almohadilla de gel de sílice (20 g) y se eluyó con éter de petróleo (35-55 °C). La evaporación del disolvente produjo el material del título en forma de un aceite (500 mg, 4,20 mmol, 78 % de rendimiento) que aún contenía rastros del disolvente, como lo indica la RMN 1H. RMN 1H (CDCla, 400 MHz) 8 ppm: 7,32-7,41 (m, 2H), 7,12-7,19 (m, 1H), 7,01 7,09 (m, 2H).
Ejemplo 9. 2-metoxi-6-(6-metoxi-4-((1-fenil-1H-1,2,3-triazol-4-il)metoxi)benzofuran-2-il)imidazo[2,1-b][1,3,4]tiadiazol
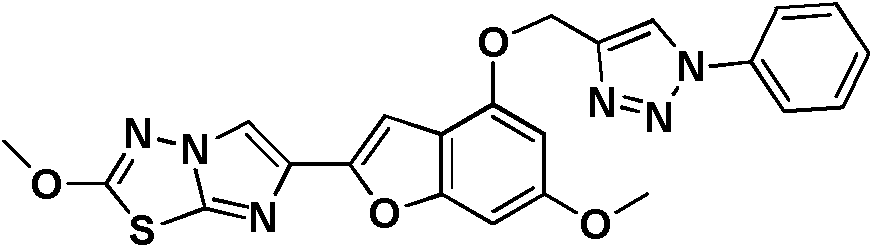
Una solución de 2-metoxi-6-(6-metoxi-4-(prop-2-in-1-iloxi)benzofuran-2-il)imidazo[2,1-b][1,3,4]tiadiazol (Ejemplo 9A, 20 mg, 0,056 mmol) y azidobenceno (Ejemplo 9B, 19 mg, 0,159 mmol) en DMF (4 ml, 51,7 mmol) se trató, a temperatura ambiente y en atmósfera de nitrógeno, con (R)-2-((S)-1,2-dihidroxietil)-4-hidroxi-5-oxo-2,5-dihidrofuran-3-olato de sodio (8 mg, 0,040 mmol) y sulfato de cobre (II) pentahidrato (5 mg, 0,020 mmol). La mezcla se agitó durante 2 horas (reacción seguida de HPLC) y después se diluyó con diclorometano (60 ml) y se lavó con NaHCO3 saturado y salmuera y se secó (MgSO4). El disolvente se evaporó y el residuo sólido se trituró con acetonitrilo (2 x 1 ml) y se liofilizó para obtener el material del título (13 mg, 0,027 mmol, 49 % de rendimiento). LC (Método A): 2,213 min. HRMS (ESI) calc. para C23H19N6O4S [M+H]+ m/z 475,1183, encontrado 475,1204. RMN 1H (DMSO-d6, 400 MHz) 8 ppm: 9,04 (s, 1H), 8,37 (s, 1H), 7,90-7,99 (m, 2H), 7,58-7,67 (m, 2H), 7,51 (tt, J=7,4, 1,2 Hz, 1H), 6,97-7,03 (m, 1H), 6,83-6,88 (m, 1H), 6,71 (d, J=1,6 Hz, 1H), 5,38 (s, 3H), 4,20 (s, 3H), 3,83 (s, 3H).
Ejemplo 10
2-metoxi-6-(6-metoxi-4-((1-fenil-1H-1,2,3-triazol-5-il)metoxi)benzofuran-2-il)imidazo[2,1-b][1,3,4]tiadiazol

En un vial de 5 ml para microondas, se agregó 2-metoxi-6-(6-metoxi-4-(prop-2-in-1-iloxi)benzofuran-2-il)imidazo[2,1-b][1,3,4]tiadiazol (Ejemplo 9A, 27 mg, 0,076 mmol), azidobenceno (Ejemplo 9B, 30 mg, 0,252 mmol), DMF anhidro (2,5 ml, 32,3 mmol) y (Cp*RuCl)4 (12 mg) en atmósfera de nitrógeno. El vial se tapó y se calentó a 110 °C durante 20 min en el aparato de microondas. El disolvente se evaporó y el residuo se purificó mediante cromatografía sobre gel de sílice ISCO, se concentró y se trituró dos veces con metanol (2 x 1 ml). Al sólido se le agregó acetonitrilo (2 ml) y agua (4 ml) y la mezcla se liofilizó durante el fin de semana para obtener el material del título (5 mg, 10,54 |jmol, 14 % de rendimiento). LC (Método F): 2,480 min. HRMS (ESI) calc. para C23H19N6O4S [M+H]+ m/z 475,1183, encontrado 475,1234. RMN 1H (DMSO-da, 400 MHz) 8 ppm: 8,35 (s, 1H), 8,12 (s, 1H), 7,66-7,73 (m, 2H), 7,53-7,63 (m, 3H), 6,84 (dd, J=2,0, 0,8 Hz, 1H), 6,75 (d, J=0,8 Hz, 1H), 5,40 (s, 2H), 4,20 (s, 3H), 3,79 (s, 3H).
Preparación de alcoholes
Los siguientes alcoholes se prepararon de acuerdo con los procedimientos descritos en los ejemplos 3 a 8.





Preparación de bromuros
Los siguientes bromuros se prepararon de acuerdo con el procedimiento descrito en el ejemplo 4.

continuación

continuación

continuación

Ejemplos 11 a 35
Los siguientes ejemplos adicionales se prepararon, aislaron y caracterizaron usando los métodos divulgados anteriormente.

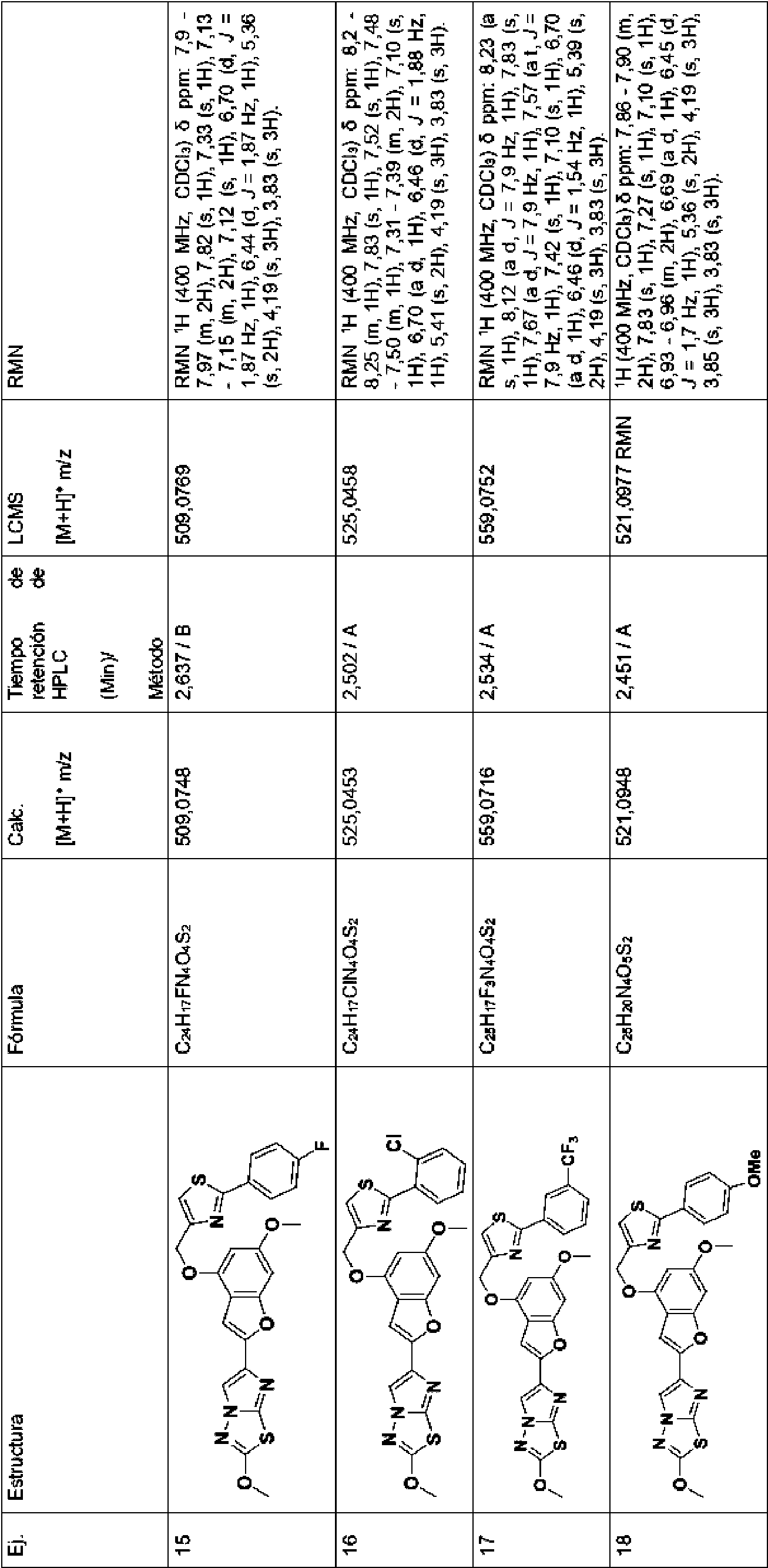

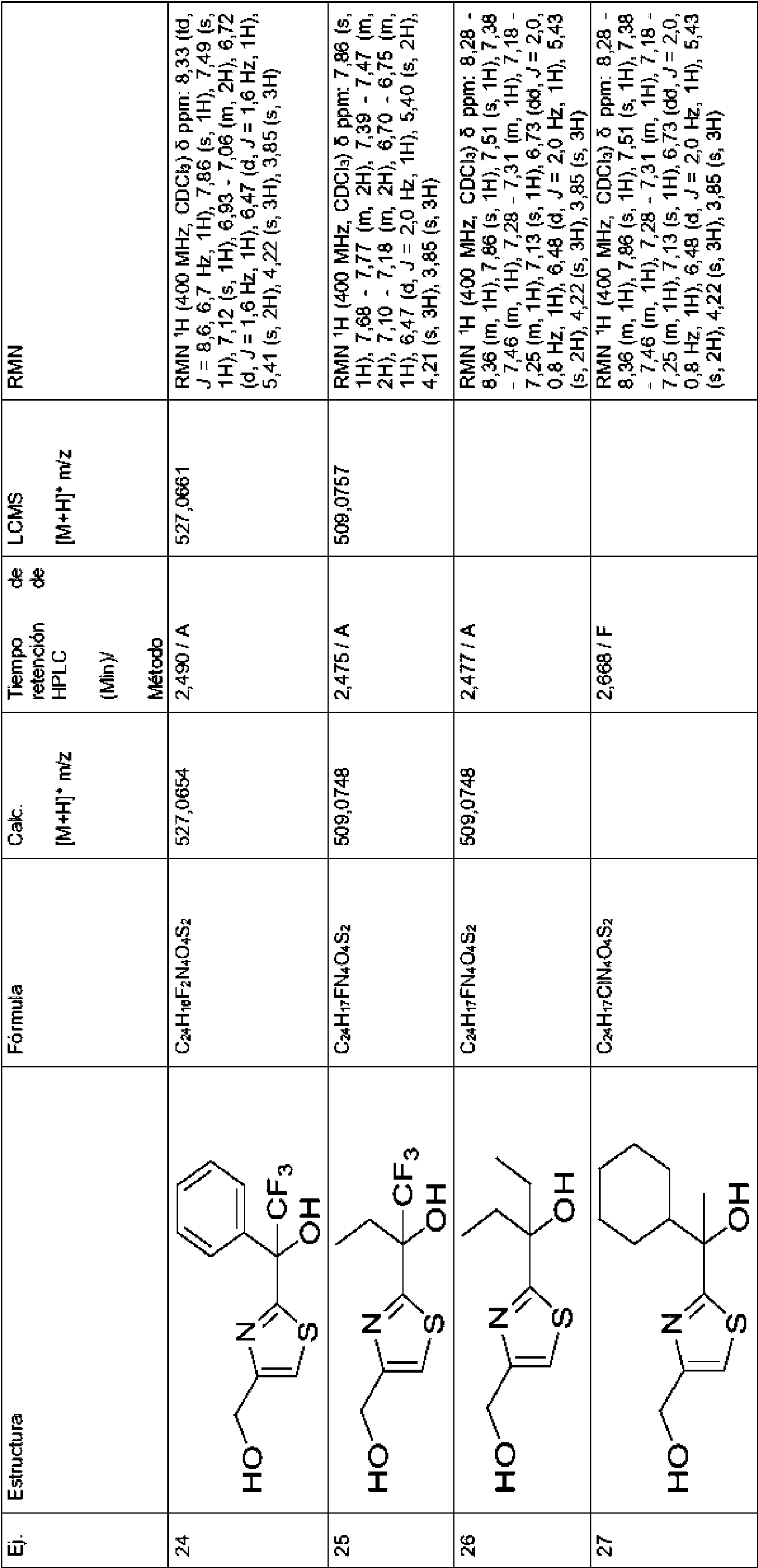


Ejemplo 36
4-(4-(((6-metoxi-2-(2-metoxNmidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2-il)-N,N-dimetilbenzamida

36A. 4-(4-(((ferc-butildimetilsilil)oxi)metil)tiazol-2-il)-N,N-dimetilbenzamida

En un matraz de vidrio a presión de 350 ml, una mezcla de 2-bromo-4-(((ferc-butildimetilsilil)oxi)-metil)tiazol (Ejemplo 37B, 3,00 g, 9,73 mmol), ácido (4-(dimetilcarbamoil)fenil)-borónico (2,82 g, 14,61 mmol) en tolueno (90 ml) y EtOH (30 ml) se trató con Na2CO3 2 M (6,0 ml, 12,0 mmol) y la mezcla heterogénea resultante se purgó con nitrógeno durante 10 min. A esta mezcla se le agregó Pd(dppf)Cl2.DCM (0,50 g, 0,61 mmol) y el vial sellado se calentó a 95 °C durante 2 h. La mezcla de reacción enfriada se repartió después entre acetato de etilo y bicarbonato de sodio saturado. La fase orgánica se separó, se lavó con salmuera, se secó sobre sulfato de magnesio anhidro y se concentró al vacío. El jarabe de color amarillo claro obtenido se sometió a cromatografía sobre gel de sílice (elución con 0-20 % de acetato de etilo-diclorometano) para obtener 3,24 g ( 88 %) del material del título en forma de un jarabe transparente. LC (Método A): 2,401 min. RMN 1 H (DMSO-d6, 400 MHz) 8 ppm: 7,97 (d, J = 8,2 Hz, 2H), 7,52 (d, J = 8,2 Hz, 2H), 7,48 -7,54 (s, 1H), 4,83 (s, 2H), 3,00 (a. s., 3H), 2,93 (a. s., 3H), 0,91 (s, 9H), 0,11 (s, 6 H).
36B. 4-(4-(hidroximetil)tiazol-2-il)-N,N-dimetilbenzamida

Una solución de 4-(4-(((ferc-butildimetilsilil)oxi)metil)tiazol-2-il)-N,N-dimetilbenzamida (3,24 g, 8 ,6 mmol) en tetrahidrofurano (150 ml) se trató con trihidrofluoruro de trietilamina (7,0 ml, 43,0 mmol) y la solución transparente resultante se agitó a 23 °C durante 18 h. La mezcla de reacción se inactivó con bicarbonato de sodio acuoso saturado (100 ml) y se agitó durante 10 min. La mezcla de reacción se extrajo con diclorometano (3 x 250 ml) y la fase orgánica combinada se lavó con bicarbonato de sodio saturado y salmuera, se secó sobre sulfato de magnesio anhidro y se concentró al vacío. El residuo obtenido se sometió a cromatografía sobre gel de sílice (elución con 50 100 % de acetato de etilo-diclorometano) para obtener 1,98 g ( 88 %) del material del título en forma de un sólido de color blanco. LC (Método A): 1,762 min. HRMS (ESI) Anal. calc. para C13H15N2O2S [M+H]+ m/z 263,0849; encontrado 263,0865. RMN 1H (CDCh, 400 MHz) 8 ppm: 7,96 (d, J = 7,8 Hz, 2H), 7,49 (d, J = 7,8 Hz, 2H), 7,22 (s, 1H), 4,82 (s, 2H), 3,13 (a. s., 3H), 3,00 (a. s., 3H), 2,66 (a. s., 1H).
Ejemplo 36. 4-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2-il)-N,N-dimetilbenzamida

Una mezcla de 6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-ol (Ejemplo 1H, 1,00 g, 3,15 mmol) y 4-(4-(hidroximetil)tiazol-2-il)-N,N-dimetilbenzamida (Ejemplo 36B, 0,950 g, 3,62 mmol) en tetrahidrofurano seco (80 ml) se trató a 22 °C y en atmósfera de nitrógeno con tri-n-butilfosfina (2,0 ml, 8,11 mmol), añadida en una porción, seguido de una solución de 1,1'-(azodicarbonil)dipiperidina (2,00 g, 7,93 mmol) en tetrahidrofurano (20 ml), añadida gota a gota durante 40 min. Después de 2 h más a 22 °C, la mezcla de reacción se repartió entre diclorometano y bicarbonato de sodio acuoso saturado. La fase orgánica se lavó con salmuera, se secó sobre sulfato de magnesio anhidro y se concentró al vacío para obtener un residuo vítreo. La cromatografía sobre gel de sílice (gradiente de elución de acetato de etilo en diclorometano) produjo 1,343 g (66 %) del material del título en forma de un sólido de color blanco, después de la trituración en acetonitrilo. LC (Método A): 2,597 min. HRMS (ESI) Anal. calc. para C27H24N5O5S2 [M+H]+ m/z 562,1213; encontrado 562,1216. RMN 1H (CDCla, 400 MHz) 8 ppm: 8,02 (d, J = 8,0 Hz, 2H), 7,86 (s, 1H), 7,52 (d, J = 8,0 Hz, 2H), 7,42 (s, 1H), 7,12 (s, 1H), 6,73 (a s, 1H), 6,48 (a s, 1H), 5,41 (s, 2H), 4,22 (s, 3H), 3,86 (s, 3H), 3,15 (a s, 3H), 3,02 (a s, 3H).
Ejemplo 37
4-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2-il)benzoato de metilo

37A. (2-bromotiazol-4-il)metanol

Una solución de 2-bromotiazol-4-carboxilato de metilo (0,500 g, 2,25 mmol) en EtOH (10 ml) en un matraz de 50 ml en atmósfera de nitrógeno se enfrió a 0 °C y se trató con NaBH4 (170 mg, 4,50 mmol) en porciones durante 5 min. Después de agitar durante 15 min a 0 °C, la mezcla de reacción se calentó a 90 °C durante 1 h. La mezcla enfriada se inactivó con NH4Cl acuoso saturado (15 ml) y la agitación continuó durante 20 min. Después se agregó acetato de etilo (50 ml) y la fase orgánica se separó, se lavó con salmuera, se secó sobre MgSO4 y se concentró para obtener el compuesto del título (0,212 g, 49 %) que se usó tal cual en la siguiente etapa. RMN 1H (CDCl3, 400 MHz) 8 ppm: 7,18 (s, 1H), 4,76 (s, 2H), 2,21 (a s, 1H).
37B. 2-bromo-4-(((terc-butildimetilsilil)oxi)metil)tiazol

A una solución enfriada en hielo de (2-bromotiazol-4-il)metanol (0,212 g, 1,09 mmol) en DMF (10 ml), se le agregó TBDMSCl (0,329 g, 2,19 mmol), seguido de imidazol (0,171 g, 2,51 mmol). La mezcla de reacción se dejó calentar a temperatura ambiente durante 10 min y se agitó durante 18 h en atmósfera de N2. La reacción se inactivó mediante la adición de EtOH a 0 °C y la mezcla se agitó a 20 °C durante 10 min antes de repartirla entre EtOAc y NaHCO3 acuoso saturado. La capa orgánica se separó, se lavó con salmuera, se secó sobre MgSO4 y se filtró. El residuo se purificó en ISCO usando una columna Re DISEP® de 12 g (0 a 5 % de EtOAc-DCM) para obtener el producto deseado en forma de un aceite de color amarillo (0,333 g, 99 %). LCMS (APCI): calc. para C10H-igBrNOSSi [M+H]+ m/z 308,01, encontrado 308,0. RMN 1 H (CDCla, 400 MHz) 8 ppm: 7,14 (t, J = 1,5 Hz, 1H), 4,83 (d, J = 1,6 Hz, 2H),
0,95 (s, 9H), 0,12 (s, 6 H).
37C. 4-(4-(((ferc-butildimetilsilil)oxi)metil)tiazol-2-il)benzoato de metilo

Una mezcla de 2-bromo-4-(((ferc-butildimetilsilil)oxi)metil)tiazol (0,150 g, 0,487 mmol) y ácido (4-(metoxicarbonil)fenil)borónico (0,119 g, 0,662 mmol) en tetrahidrofurano (4 ml) se trató con fluoruro de potasio (0,085 g, 1,460 mmol), 2-(di-t-butilfosfino)bifenilo (0,015 g, 0,049 mmol) y acetato de paladio (II) (5,5 mg, 0,024 mmol). La mezcla de reacción se purgó con nitrógeno durante 5 min y después se calentó a 70 °C durante 16 h. La mezcla de reacción enfriada se concentró y el residuo se sometió a cromatografía sobre gel de sílice (ISCO, gradiente de elución de diclorometano en hexano) para obtener 0,050 g (28 %) del material del título. LC (Método B): 2,793 min. LCMS (APCI): calc. para C-iaH26NO3SSi [M+H]+ m/z 364,14; encontrado 364,2. RMN 1 H (400 MHz, acetona-d6) 8 ppm: 8,11 (s, 4H), 7,51 (s, 1H), 4,92 (d, J= 1,17 Hz, 2H), 3,92 (s, 3H), 0,97 (s, 9H), 0,16 (s, 6 H).
37D. 4-(4-(hidroximetil)tiazol-2-il)benzoato de metilo

Una solución de 4-(4-(((ferc-butildimetilsilil)oxi)metil)tiazol-2-il)benzoato de metilo (0,050 g, 0,138 mmol) en tetrahidrofurano seco ( 10 ml) en atmósfera de nitrógeno se trató gota a gota con trihidrofluoruro de trietilamina (0,112 ml, 0,688 mmol) y la solución se agitó a temperatura ambiente durante 16 h. La mezcla de reacción se repartió después entre diclorometano-bicarbonato de sodio acuoso saturado y la fase orgánica se separó, se lavó con salmuera, se secó sobre sulfato de magnesio anhidro y se concentró al vacío. Esto produjo 0,030 g ( 88 %) del material del título como una espuma de color beis que se usó tal cual en la siguiente etapa. LC (Método B): 2,049 min. LCMS (APCI): calc. para C12 H12 NO3S [M+H]+ m/z 250,05; encontrado 250,2. RMN 1H (400 MHz, acetonad6) 8 ppm: 8,09 (s, 4H), 7,51 (s, 1H), 4,73-4,87 (m, 2H), 3,91 (s, 3H), 2,83 (a s, 1H).
Ejemplo 37. 4-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2-il)benzoato de metilo
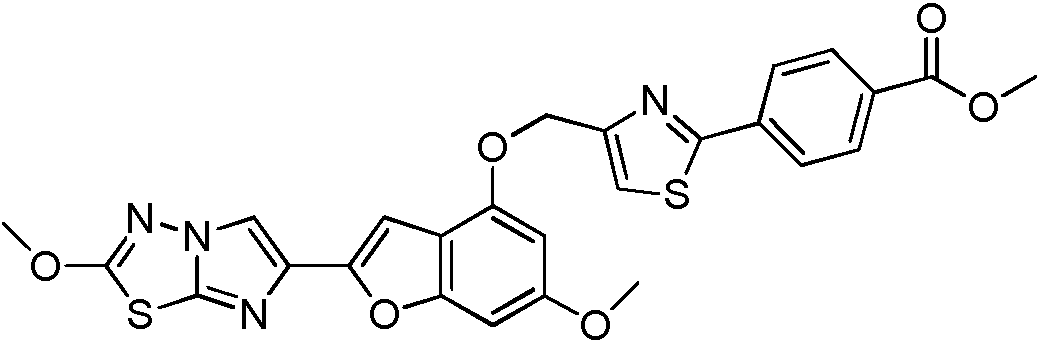
El compuesto del título se preparó de acuerdo con el procedimiento de acoplamiento general descrito en el ejemplo 36 para obtener un sólido. LC (Método A): 2,488 min. HRMS (ESI) calc. para C26H21N4O6S2 [M+H]+ m/z 549,0903; encontrado 549,0913. RMN 1H (400 MHz, DMSO-d6) 8 ppm: 8,37 (s, 1H), 8,05-8,16 (m, 4H), 8,01 (s, 1H), 7,03 (s, 1H), 6,85 ( a s, 1H), 6,65 (d, J= 1,96 Hz, 1H), 5,40 (s, 2H), 4,20 (s, 3H), 3,89 (s, 3H), 3,82 (s, 3H).
Ejemplo 38
2-(4-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2-il)fenil)propan-2-ol

38A. 2-(4-(4-(((ferc-butildimetilsilil)oxi)metil)tiazol-2-il)fenil)propan-2-ol
Una solución helada de 4-(4-(((terc-butildimetilsilil)oxi)metil)tiazol-2-il)benzoato de metilo (Ejemplo 37C, 0,150 g, 0,413 mmol) en tetrahidrofurano (5 ml) se trató con bromuro de metilmagnesio (1 M en butiléter, 1,65 ml, 1,65 mmol). Después el baño de enfriamiento se retiró y la mezcla se agitó a temperatura ambiente durante 30 min antes de repartirla entre acetato de etilo y una solución acuosa de ácido cítrico. La capa orgánica se separó, se lavó con salmuera, se secó sobre sulfato de sodio anhidro y se concentró para obtener 0,100 g (67 %) del material del título, que se usó tal cual en la siguiente etapa. LC (Método B): 2,773 min. LCMS (APCI): calc. para C-igH30NO2SSi [M+H]+ m/z 364,18; encontrado 364,2.
38B. 2-(4-(4-(hidroximetil)tiazol-2-il)fenil)propan-2-ol

Este compuesto se preparó de acuerdo con el procedimiento de desprotección descrito en el ejemplo 37D. LC (Método A): 1,584 min. HRMS (ESI): calc. para C13H16NO2S [M+H]+ m/z 250,0902; encontrado 250,0895. RMN 1H (400 MHz, CDCl3) 8 ppm: 7,92 (m, J= 8,22 Hz, 2H), 7,57 (m, J= 8,22 Hz, 2H), 7,18 (s, 1H), 4,84 (d, J= 5,87 Hz, 2H), 2,20-2,29 (m, 1H), 1,77 (s, 1H), 1,62 (s, 6 H)
Ejemplo 38. 2-(4-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2-il)fenil)propan-2 -ol

El compuesto del título se preparó de acuerdo con el procedimiento de acoplamiento de Mitsunobu descrito en el ejemplo 36. LC (Método A): 2,379 min. HRMS (ESI): calc. para C27H25N4O5S2 [M+H]+ m/z 549,1266; encontrado 549,1221. RMN 1H (400 MHz, CDCla) 8 ppm: 7,95 (m, J=8,4 Hz, 2H), 7,86 (s, 1H), 7,59 (m, J= 8,4 Hz, 2H), 7,36 (s, 1H), 7,12 (s, 1H), 6,72 (s, 1H), 6,45-6,52 (m, 1H), 5,40 (s, 2H), 4,22 (s, 3H), 3,85 (s, 3H), 1,77 (s, 1H), 1,63 (s, 6 H). Ejemplo 39
4-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2-il)-N-(2-metoxietil)-N-metilbenzamida

39A. 4-(4-(((terc-butildimetilsilil)oxi)metil)tiazol-2-il)benzoato de terc-butilo
Una mezcla de 2-bromo-4-(((terc-butildimetilsilil)oxi)metil)tiazol (Ejemplo 37B, 1,542 g, 5,000 mmol) y ácido (4 -( te rc - butoxicarbonil)fenil)borónico (1,388 g, 6,25 mmol) en toluen-terc-butanol (3:1, 60 ml) se purgó con una corriente de burbujas de N2 durante 15 min en un matraz sellable. A esta mezcla se le agregó Pd(dppf)Cl2.DCM (0,204 g, 0,250 mmol) y Na2CO32 M (3,13 ml, 6,25 mmol), el matraz se selló y la mezcla se agitó a 95 °C (temperatura del baño de aceite) durante 4 h. Se agregaron otros 0,25 equiv. de ácido borónico y Na2CO32 M, junto con una pequeña cantidad del catalizador. La mezcla se calentó a 95 °C durante 2 h más antes de dejarla enfriar a temperatura ambiente y después se repartió entre EtOAc-agua. La fase orgánica se separó, se lavó (salmuera), se secó (Na2SO4) y se evaporó para obtener una goma de color de color pardo oscuro. La cromatografía ultrarrápida (Isco/ EtOAc al 0-10 %-hexano) de esta goma produjo el compuesto del título (1,065 g, 52,5 %) en forma de una goma incolora. Este material se usó tal cual en la siguiente etapa. LC (Método B): 3,407 min. LCMS (APCI): calc. para C21H32NO3SSi [M+H]+ m/z 406,19; encontrado 406,2. RMN 1 H (400 MHz, CDCla) 8 ppm: 8,04 (d, J= 8 ,6 Hz, 2H), 7,98 (d, J= 8 ,6 Hz, 2H), 7,26 (s, 1H), 4,79 (s, 2H), 1,47 (s, 9H), 0,82 (s, 9H), 0,10 (s, 6 H).
39B. 4-(4-(hidroximetil)tiazol-2-il)benzoato de ferc-butilo

A una solución de 4-(4-(((ferc-butildimetilsilil)oxi)metil)tiazol-2-il)benzoato de ferc-butilo (1,058 g, 2,61 mmol) en THF seco (25 ml) en atmósfera de N2 se le agregó gota a gota trihidrofluoruro de trietilamina (1,274 ml, 7,82 mmol) y la mezcla se agitó a temperatura ambiente durante 18 h. La mezcla resultante se repartió entre EtOAc-NaHCO3 acuoso saturado y la fase orgánica se separó, se secó (Na2SO4) y se evaporó para obtener el compuesto del título (0,760 g, 100 %) en forma de una goma incolora que se cristalizó en reposo al vacío. Este material era esencialmente puro y se usó tal cual en la siguiente etapa. LC (Método B): 2,239 min. LCMS (APCI): calc. para C15H18NO3S [M+H]+ m/z 292,10; encontrado 292,2. RMN 1 H (400 MHz, CDCla) 8 ppm: 8,18-7,92 (m, 4H), 7,27 (s, 1H), 4,86 (d, J= 5,87 Hz, 2H), 2,41-2,22 (m, 1H), 1,63 (s, 9H).
39C. 4-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2-il)benzoato de ferc-butilo.

El compuesto del título se preparó de acuerdo con el procedimiento de acoplamiento de Mitsunobu general descrito en el ejemplo 36. LC (Método A) 2,599 min. HRMS (ESI): calc. para C29H27N4O6S2 [M+H]+ m/z 591,1372; encontrado 591,1363. RMN 1H (400 MHz, CDCla) 8 ppm: 7,99-8,10 (m, 4H), 7,86 (s, 1H), 7,44 (s, 1H), 7,12 (s, 1H), 6,71-6,75 (m, 1H), 6,46-6,49 (m, 1H), 5,42 (s, 2H), 4,22 (s, 3H), 3,86 (s, 3H), 1,63 (s, 9H).
39D. Ácido 4-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2-il)benzoico

A una solución de 4-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tia-diazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2-il)benzoato de ferc-butilo (0,841 g, 1,424 mmol) en DCM (6 ml), se le agregó TFA (3 ml) y la solución de color amarillo claro resultante se agitó a temperatura ambiente durante 4 h antes de retirar los volátiles al vacío. El residuo se trituró con un volumen mínimo de DCM y el sólido resultante se filtró y después se liofilizó a partir de DMSO. Esto produjo el compuesto del título esencialmente puro (0,701 g, 92 %) en forma de un sólido que se usó tal cual en la siguiente etapa. LC (Método A): 2,347 min. HRMS (ESI): calc. para C25H19N4O6S2 [M+H]+ m/z 535,0746; encontrado 535,0742. RMN 1H (400 MHz, DMSO-d6) 8 ppm: 13,18 (a s, 1H), 8,37 (s, 1H), 8,03-8,13 (m, 4H), 8,00 (s, 1H), 7,01 7,06 (m, 1H), 6,85 (d, J= 0,78 Hz, 1H), 6,65 (d, J= 1,96 Hz, 1H), 5,40 (s, 2H), 4,20 (s, 3H), 3,82 (s, 3H).
Ejemplo 39. 4-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2-il)-N-(2-metoxietil)-N-metilbenzamida

A una solución de ácido 4-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2-il)benzoico (0,050 g, 0,094 mmol) en Dm F (3 ml), se le agregó DIEA (0,082 ml, 0 ,468 mmol), seguido de 2-metoxi-N-metiletanamina (0,0092 g, 0,103 mmol) y, por último, HATU (0,039 g, 0,103 mmol). La mezcla resultante se agitó a temperatura ambiente durante 2 h antes de evaporarla a sequedad. El residuo se repartió entre DCM-agua y la fase orgánica se separó, se lavó (NaHCO3 acuoso saturado), se secó (Na2SO4) y se adsorbió directamente sobre una precolumna de gel de sílice. La cromatografía ultrarrápida (Isco, gradiente de elución 0-80 % de EtOAc-DCM),
seguida de liofilización del material obtenido a partir de MeCN-agua, produjo el compuesto del título (0,044 g, 78 %) en forma de un sólido. LC (Método A): 2,325 min. HRMS (ESI): calc. para C29H28N5O6S2 [M+H]+ m/z 606,1481; encontrado 606,1469. RMN 1H (400 MHz, CDCls) 6 ppm: 8,01 (m, J= 7,83 Hz, 2H), 7,86 (s, 1H), 7,49-7,56 (m, 2H), 7,41 (s, 1H), 7,12 (s, 1H), 6,73 (s, 1H), 6,45 -6,49 (m, 1H), 5,41 (s, 2H), 4,22 (s, 3H), 3,86 (s, 3H), 3,65-3,81 (m, 2H), 3,45-3,53 (m, 2H), 3,37-3,45 (m, 2H), 3,24-3,37 (m, 1H), 3,12-3,21 (m, 2H), 3,06-3,11 (m, 1H).
Ejemplo 40
4-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2-il)-N-metilbenzamida

El compuesto del título se preparó de acuerdo con el método de acoplamiento de amida general descrito en el ejemplo 39. LC (Método A): 2,239 min. HRMS (ESI): calc. para C26H22N5O5S2 [M+H]+ m/z 548,1062; encontrado 548,1058. RMN 1H (400 MHz, CDCla) 6 ppm: 8,04 (d, J= 8,22 Hz, 2H), 7,81-7,89 (m, 3H), 7,44 (s, 1H), 7,12 (s, 1H), 6,66-6,78 (m, 1H), 6,44-6,50 (m, 1H), 6,14-6,24 (m, 1H), 5,41 (s, 2H), 4,22 (s, 3H), 3,85 (s, 3H), 3,06 (d, J= 5,09 Hz, 3H).
Ejemplo 41
N-(ferc-butil)-4-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2-il)benzamida

El compuesto del título se preparó de acuerdo con el método de acoplamiento de amida general descrito en el ejemplo 39. LC (Método A): 2,458 min. HRMS (ESI): calc. para C29H28N5O5S2 [M+H]+ m/z 590,1532; encontrado 590,1536. RMN 1H (400 MHz, CDCh) 6 ppm: 8,03 (m, 2H), 7,86 (s, 1H), 7,81 (m, 2H), 7,43 (s, 1H), 7,12 (s, 1H), 6,73 (s, 1H), 6,48 (a s, 1H), 5,98 (s, 1H), 5,41 (s, 2H), 4,22 (s, 3H), 3,86 (s, 3H), 1,51 (s, 9H).
Ejemplo 42
4-(5-isopropil-4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2-il)-N,N-dimetilbenzamida

42A. 2-bromo-5-isopropiltiazol-4-carboxilato de metilo

A una solución de 2-amino-5-isopropiltiazol-4-carboxilato de metilo (1,000 g, 4,99 mmol) en CH3CN (10 ml), se le agregó nitrito de isopentilo (1,073 ml, 7,99 mmol) y después bromuro de cobre (I) (1,433 g, 9,99 mmol) y la mezcla resultante se calentó a 80 °C durante 3 h. La mezcla de reacción se concentró después a presión reducida y el residuo se repartió entre EtOAc y agua. La fase orgánica se separó, se filtró a través de una almohadilla de CELITE® y se concentró. El residuo se purificó en ISCO usando una columna REDISEP® de 24 g (0 a 40 % de EtOAc-hexanos) para obtener el producto deseado en forma de un aceite de color rojo claro (0,787 g, 60 %). LCMS (APCI): calc. para C8HnBrNO2S [M+H]+ m/z 263,962, encontrado 264,0. RMN 1H (CDCh, 400 MHz) 8 ppm: 4,17 (dt, J= 13,7, 6 ,8 Hz, 1H), 3,94 (s, 3H), 1,35 (s, 3H), 1,33 (s, 3H).
42B. (2-bromo-5-isopropiltiazol-4-il)metanol

El compuesto se preparó de acuerdo con el procedimiento descrito en el ejemplo 37A. La mezcla de reacción se inactivó con MeOH (10 ml) y se agitó a temperatura ambiente durante 10 min antes de concentrarla a presión reducida. El residuo se disolvió en DCM, se lavó con NaHCO3 acuoso saturado, agua y salmuera, se secó sobre MgSO4 y se evaporó. El producto en bruto se purificó en ISCO usando una columna REDlSEP® de 12 g (0 a 15 % de MeOH-DCM) para obtener el producto deseado en forma de un sólido de color blanco (0,509 g, 72 %). LCMS (APCI): calc. para CyH-nBrNOS [M+H]+ m/z 234,97, encontrado 218,0 (M+H-OH). RMN 1H (CDCla, 400 MHz) 8 ppm: 4,64 (d, J = 6,3 Hz, 2H), 3,32 (dt, J = 13,7, 6 ,8 Hz, 1H), 2,26 (t, J = 6,2 Hz, 1H), 1,30 (d, J = 6 ,8 Hz, 6 H).
42C. 2-bromo-4-(((terc-butildimetilsilil)oxi)metil)-5-isopropiltiazol

El compuesto se preparó de acuerdo con el procedimiento descrito en el ejemplo 37B. El producto en bruto se purificó en ISCO usando una columna REDISEP® de 12 g (0 a 50 % de EtOAc-hexanos) para obtener el producto puro en forma de un aceite incoloro (0,744 g, 99 %). LCMS (APCI): calc. para C13H25BrNOSSi [M+H]+ m/z 350,05, encontrado 350,1. RMN 1H (CDCla, 400 MHz) 8 ppm: 4,73 (s, 2H), 3,47 (dt, J= 13,7, 7,0 Hz, 1H), 1,29 (d, J= 7,0 Hz, 6 H), 0,91 (s, 9H), 0,10 (s, 6 H).
42D.4-(4-(((terc-butildimetilsilil)oxi)metil)-5-isopropiltiazol-2-il)-N,N-dimetil benzamida

En un tubo sellable de 75 ml, se agregaron 2-bromo-4-(((terc-butildimetilsilil)oxi)metil)-5-isopropiltiazol (0,200 g, 0,571 mmol), ácido (4-(dimetilcarbamoil)fenil)borónico (0,166 g, 0,859 mmol) y Pd(dppf)Cl2.DCM (0,030 g, 0,037 mmol) en una mezcla de tolueno-etanol (3:1, 6,5 ml). La solución de color naranja resultante se desgasificó con un flujo de burbujas de nitrógeno durante 15 min y después se agregó una solución acuosa 2 M de Na2CO3 (0,342 ml, 0,685 mmol), el recipiente de reacción se selló y la mezcla se calentó a 95 °C (temperatura del baño) durante 4 h. La mezcla de reacción enfriada de color de color pardo oscuro se diluyó con EtOAc, se lavó con NaHCO3 acuoso saturado y salmuera, se secó sobre MgSO4 y se evaporó. El residuo se purificó en ISCO usando una columna REDISEP® de 12 g (0 a 60 % de EtOAc-DCM) para obtener el compuesto del título en forma de un aceite incoloro (0,080 g, 34 %). Lc Ms (APCI): calc. para C22H35N2O2SSi [M+H]+ m/z 419,21, encontrado 419,2. RMN 1 H (CDCl3, 400 MHz) 8 ppm: 7,91 - 7,96 (m, 2H), 7,45 - 7,49 (m, 2H), 4,84 (s, 2H), 3,51 (dt, J = 13,6, 6,7 Hz, 1H), 3,13 (a. s., 3H), 3,01 (a. s., 3H), 1,36 (s, 3H), 1,35 (s, 3H), 0,93 (s, 9H), 0,13 (s, 6 H).
42E. 4-(4-(hidroximetil)-5-isopropiltiazol-2-il)-N,N-dimetilbenzamida

La reacción se realizó de acuerdo con el procedimiento del ejemplo 37D. La mezcla de reacción se diluyó con DCM, se lavó con NaHCÜ3 acuoso saturado, agua y salmuera, se secó sobre MgSÜ4 y se evaporó. El material en bruto se purificó en ISCO usando una columna REDISEP® de 4 g (0 a 15 % de MeOH-DCM) para obtener el producto deseado en forma de un sólido de color amarillo (0,056 g, 96 %). LCMS (APCI): calc. para C16H21N2O2S [M+H]+ m/z 305,125, encontrado 305,1. RMN 1H (CDCla, 400 MHz) 8 ppm: 7,4 (d, J = 8,2 Hz, 2H), 7,49 (d, J = 8,2 Hz, 2H), 4,74 (d, J = 5,9 Hz, 2H), 3,34 (dt, J = 13,6, 6,7 Hz, 1H), 3,14 (a. s., 3H), 3,01 (a. s., 3H), 2,45 (t, J = 5,9 Hz, 1H), 1,36 (d, J = 7,0 Hz, 6H).
Ejemplo 42. 4-(5-isopropil-4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2-il)-N,N-dimetilbenzamida

El compuesto del título se preparó de acuerdo con el procedimiento usado para la síntesis del ejemplo 36. El producto en bruto se suspendió en CH3CN, se sometió a ultrasonido, se filtró y se secó (x2) para obtener el compuesto del título en forma de un sólido de color blanquecino (0,018 g, 16 %). lC (Método C): 2,359 min. HRMS (ESI): calc. para C30H30N5O5S2 [M+H]+ m/z 604,161, encontrado 604,1690. RMN 1 H (CDCh, 400 MHz) 8 ppm: 7,95 8,00 (m, J = 8,2 Hz, 2H), 7,84 (s, 1H), 7,47-7,52 (m, J = 8,2 Hz, 2H), 7,02 (s, 1H), 6,71 (s, 1H), 6,60 (d, J = 1,6 Hz, 1H), 5,32 (s, 2H), 4,21 (s, 3H), 3,86 (s, 3H), 3,50 (dt, J = 13,8, 7,0 Hz, 1H), 3,14 (a s, 3H), 3,02 (a s, 3H), 1,37 (d, J = 7,0 Hz, 6H).
Ejemplo 43
6-(4-((2-(2-fluoropiridin-4-il)-5-isopropiltiazol-4-il)metoxi)-6-metoxibenzofuran-2-il)-2-metoxiimidazo[2,1-b][1,3,4]tiadiazol

43A. 4-(((terc-butildimetilsilil)oxi)metil)-2-(2-fluoropiridin-4-il)-5-isopropiltiazol

El compuesto se preparó de acuerdo con el procedimiento descrito para el ejemplo 42D. El producto en bruto se purificó en ISCO usando una columna REDISEP® de 12 g (0 a 5% de EtOAc-DCM) para obtener el producto en forma de un sólido de color blanco (0,078 g, 38 %). LCMS (APCI): calc. para C1sH28FN2OSSi [M+H]+ m/z 367,16,
encontrado 367,2. RMN 1H (CDCI3, 400 MHz) 5 ppm: 8,26 (d, J = 5 ,3 Hz, 1H), 7,64 (dt, J = 5,3, 1,6 Hz, 1H), 7,42 (s, 1H), 4,85 (s, 2H), 3,54 (dt, J = 13,7, 6 ,8 Hz, 1H), 1,38 (s, 3H), 1,36 (s, 3H), 0,93 (s, 9H), 0,13 (s, 6 H).
43B. (2-(2-fluoropiridin-4-il)-5-isopropiltiazol-4-il)metanol

El compuesto se preparó usando el procedimiento descrito en el ejemplo 37D. La mezcla de reacción se diluyó con DCM, se lavó con NaHCO3 acuoso saturado, agua y salmuera, se secó sobre MgSO4 y se evaporó. El producto en bruto se purificó en ISCO usando una columna REDISEP® de 4 g (0 a 15 % de MeOH-DCM) para obtener el producto deseado en forma de un sólido incoloro (0,048 g, 89 %). LCm S (APCI): calc. para C12H13FN2OS [M+H]+ m/z 253,073, encontrado 253,1. RMN 1H (CDCla, 400 MHz) 5 ppm: 8,28 (d, J = 5,1 Hz, 1H), 7,65 (dt, J = 5,5, 1,6 Hz, 1H), 7,41 - 7,45 (m, 1H), 4,77 (d, J = 5,9 Hz, 2H), 3,38 (dt, J = 13,7, 6 ,8 Hz, 1H), 2,36 (t, J = 5,9 Hz, 1H), 1,38 (d, J = 6,9 Hz, 6 H).
Ejemplo 43. 6-(4-((2-(2-fluoropiridin-4-il)-5-isopropiltiazol-4-il)metoxi)-6-metoxibenzofuran-2-il)-2-metoxiimidazo[2,1-b][1,3,4]tiadiazol

El compuesto del título se preparó de acuerdo con el procedimiento usado para la síntesis del ejemplo 36. El producto en bruto se suspendió en CH3CN, se sometió a ultrasonido y se filtró. El sólido resultante se purificó en ISCO usando una columna REDISEP® de 4 g (0 a 40 % de EtOAc-DCM) para obtener el compuesto del título en forma de un sólido de color amarillo (0,063 g, 60 %). LC (Método C): 2,457 min. HRm S (ESI): calc. para C26H23FN5O4S2 [M+H]+ m/z 552,110, encontrado 552,1181. RMN 1 H (CDCla, 400 MHz) 5 ppm: 8,29 (d, J = 5,5 Hz, 1H), 7,84 (s, 1H), 7,68 (dt, J = 5,5, 1,6 Hz, 1H), 7,46 (s, 1H), 7,02 (s, 1H), 6,72 (d, J = 0,8 Hz, 1H), 6,58 (d, J = 2,0 Hz, 1H), 5,34 (s, 2H), 4,21 (s, 3H), 3,87 (s, 3H), 3,53 (dt, J = 13,6, 7,0 Hz, 1H), 1,39 (d, J = 7,0 Hz, 6 H).
Ejemplo 44
4-(5-etil-4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2-il)-N,N-dimetilbenzamida

El compuesto se preparó usando el procedimiento descrito en el ejemplo 37A. La mezcla de reacción se inactivó con MeOH (10 ml) y se agitó a temperatura ambiente durante 10 min. Después la mezcla se concentró a presión reducida y el residuo se disolvió en DCM, se lavó con NaHCO3 acuoso saturado, agua y salmuera, se secó sobre MgSO4 y se evaporó para obtener el producto deseado en forma de un aceite incoloro (0,156 g, 88 %). LCMS
(APCI): calc. para CaHgBrNOS [M+H]+ m/z 220,95, encontrado 205,9 (M+H-OH). RMN 1H (CDCla, 400 MHz) 5 ppm: 4,63 (a. s., 2H), 2,82 (q, J = 7,4 Hz, 2H), 2,29 (a. s., 1H), 1,28 (t, J = 7,4 Hz, 3H).
44B. 2-bromo-4-(((ferc-butildimetilsilil)oxi)metil)-5-etiltiazol

El compuesto se preparó de acuerdo con el procedimiento descrito en el ejemplo 37B. El producto en bruto se purificó en ISCO usando una columna REDISEP® de 12 g (0 a 50 % de EtOAc-hexanos) para obtener el compuesto del título en forma de un aceite incoloro (0,173 g, 73%). LCMS (APCI): calc. para C12H23BrNOSSi [M+H]+ m/z 336,04, encontrado 337,1. RMN 1H (CDCla, 400 MHz) 5 ppm: 4,73 (s, 2H), 2,88 (q, J = 7,7 Hz, 2H), 1,28 (t, J = 7,6 Hz, 3H), 0,91 (s, 9H), 0,10 (s, 6 H).
44C. 4-(4-(((ferc-butildimetilsilil)oxi)metil)-5-etiltiazol-2-il)-N,N-dimetilbenzamida
El compuesto se preparó de acuerdo con el procedimiento descrito para el ejemplo 36A. El producto en bruto se purificó en ISCO usando una columna REDISEp® de 4 g (0 a 60 % de EtOAc-DCM) para obtener el compuesto del título en forma de un aceite de color parduzco (0,155 g, 75 %). LCMS (APCI): calc. para C2-iH33N2O2SSi [M+H]+ m/z 405,20, encontrado 405,2. RMN 1H (CDCls, 400 MHz) 5 ppm: 7,90-7,96 (m, J = 7,8 Hz, 2H), 7,44-7,50 (m, J = 8,2 Hz, 2H), 4,84 (s, 2H), 3,13 (a s, 3H), 3,01 (a s, 3H), 2,96 (q, J = 7,4 Hz, 2H), 1,34 (t, J = 7,5 Hz, 3H), 0,93 (s, 9H), 0,13 (s, 6 H).
44D. 4-(5-etil-4-(hidroximetil)tiazol-2-il)-N,N-dimetilbenzamida

El compuesto se preparó de acuerdo con el procedimiento descrito en el ejemplo 36B. La mezcla de reacción se diluyó con DCM, se lavó con NaHCO3 acuoso saturado, agua y salmuera, se secó sobre MgSO4 y se evaporó. El producto en bruto se purificó en ISCO usando una columna REDISEP® de 4 g (0 a 15 % de MeOH-DCM) para obtener el producto deseado en forma de un aceite incoloro (0,094 g, 85 %). LCMS (APCI): calc. para C15H19N2O2S [M+H]+ m/z 291,109, encontrado 291,1. RMN 1H (CDCls, 400 MHz) 5 ppm: 7,91 - 7,96 (m, J = 8,2 Hz, 2H), 7,46 -7,51 (m, J = 8,2 Hz, 2H), 4,73 (d, J = 5,9 Hz, 2H), 3,14 (a. s., 3H), 3,01 (a. s., 3H), 2,89 (q, J = 7,7 Hz, 2H), 2,40 (t, J = 5,9 Hz, 1H), 1,34 (t, J = 7,6 Hz, 3H).
Ejemplo 44. 4-(5-etil-4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2-il)-N,N-dimetilbenzamida

El compuesto del título se preparó de acuerdo con el procedimiento descrito para la síntesis del ejemplo 36. El producto en bruto se suspendió en CH3CN, se sometió a ultrasonido y se filtró antes de purificarlo en ISCO usando una columna REDISEP® de 4 g (0 a 70 % de EtOAc-DCM) para obtener el compuesto del título en forma de un sólido de color blanquecino (0,059 g, 49 %). LC (Método C): 2,409 min. HRMS (ESI): calc. para C29H28N5O5S2 [M+H]+ m/z 590,145, encontrado 590,1505. RMN 1 H (CDCls, 400 MHz) 5 ppm: 7,94 - 8,00 (m, J = 8,2 Hz, 2H), 7,84 (s, 1H), 7,46 - 7,52 (m, J = 7,8 Hz, 2H), 7,04 (s, 1H), 6,71 (d, J = 0,8 Hz, 1H), 6,58 (d, J = 2,0 Hz, 1H), 5,32 (s, 2H), 4,21 (s, 3H), 3,86 (s, 3H), 3,14 (a s, 3H), 2,95 - 3,06 (m, 5H), 1,35 (t, J = 7,6 Hz, 3H).
Ejemplo 45
4-(4-(((6-metoxi-2-(2-metoxNmidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)-5-metiltiazol-2-il)benzoato de ferc-butilo

45A. (2-bromo-5-metiltiazol-4-il)metanol
El compuesto se preparó de acuerdo con el procedimiento descrito en el ejemplo 37A. La mezcla de reacción se inactivó con MeOH (10 ml) y se agitó a temperatura ambiente durante 10 min. Después la mezcla se concentró a presión reducida y el residuo se disolvió en DCM, se lavó con NaHCO3 acuoso saturado, agua y salmuera, se secó sobre MgSO4 y se evaporó para obtener el producto deseado en forma de un aceite incoloro (0,843 g, 96 %). LCMS (APCI): calc. para CaHyBrNOS [M+H]+ m/z 207,94, encontrado 208,0.
45B. 2-bromo-4-(((ferc-butildimetilsilil)oxi)metil)-5-metiltiazol

El compuesto se preparó de acuerdo con el procedimiento descrito en el ejemplo 37B. El producto en bruto se purificó en ISCO usando una columna REDISEP® de 40 g (50 a 100 % de DCM-hexanos) para obtener el compuesto del título en forma de un aceite incoloro (0,682 g, 52 %). LCMS (APCI): calc. para C-n^-iBrNOSSi [M+H]+ m/z 322,03, encontrado 322,0. RMN 1H (CDCl3, 400 MHz) 8 ppm: 4,64 (s, 2H), 2,34 (s, 3H), 0,81 (s, 9H), 0,00 (s, 6 H).
45C. 4-(4-(((ferc-butildimetilsilil)oxi)metil)-5-metiltiazol-2-il)benzoato de ferc-butilo

En un vial sellable, una suspensión de ácido (4-(ferc-butoxicarbonil)fenil)borónico (0,611 g, 2,75 mmol) y 2-bromo-4-(((ferc-butildimetilsilil)oxi)metil)-5-metiltiazol (0,682 g, 2,116 mmol) en tolueno (34 ml) y etanol (9,3 ml) se trató con carbonato de sodio acuoso 2 M (1,27 ml, 2,54 mmol) y después se purgó con nitrógeno durante 5 min. A esta mezcla, se le agregó dicloruro de [1,1'-bis(difenilfosfino)ferrocen]paladio (II).DCM (0,091 g, 0,133 mmol), el vial se selló y la mezcla se calentó a 95 °C durante 4 h. La mezcla de reacción enfriada se repartió entre acetato de etilobicarbonato de sodio acuoso saturado y la fase orgánica se separó, se lavó con salmuera, se secó sobre sulfato de magnesio anhidro y se evaporó al vacío. La cromatografía del residuo sobre gel de sílice (ISCO, gradiente de elución de diclorometano en hexano) produjo 0,654 g (74 %) del compuesto del título. LC (Método A): 2,966 min. HRMS (ESI): calc. para C22H34NO3SSi [M+H]+ m/z 420,2029; encontrado 420,2038. RMN 1H (400 MHz, CDCl3) 8 ppm: 7,97 8,10 (m, 2H), 7,85-7,97 (m, 2H), 4,86 (s, 2H), 2,54 (s, 3H), 1,62 (s, 9H), 0,94 (s, 9H), 0,14 (s, 6 H).
45D. 4-(4-(hidroximetil)-5-metiltiazol-2-il)benzoato de ferc-butilo

El compuesto del título se preparó de acuerdo con el método descrito en el ejemplo 37D. LC (Método A): 2,225 min. HRMS (ESI): calc. para C16H20NO3S [M+H]+ m/z 306,1164; encontrado 306,1161. RMN 1H (400 MHz, CDCl3) 8 ppm:
7,99-8,07 (m, 2H), 7,87-7,99 (m, 2H), 4,74 (d, J= 5 ,77 Hz, 2H), 2,50 (s, 3H), 2,35 (t, J= 5 ,77 Hz, 1H), 1,62 (s, 9H). Ejemplo 45.
4-(4-(((6-metoxi-2-(2-metoxNmidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)-5-metiltiazol-2-il)benzoato de ferc-butilo

El compuesto del título se preparó de acuerdo con el procedimiento de acoplamiento general de Mitsunobu descrito en el ejemplo 36. LC (Método A): 2,761 min. HRMS (ESI): calc. para C30H29N4O6S2 [M+H]+ m/z 605,1529; encontrado 605,1518. RMN 1H (400 MHz, CDCh) 8 ppm: 8,00-8,07 (m, 2H), 7,92-7,99 (m, 2H), 7,84 (s, 1H), 7,06 (s, 1H), 6,69 6,73 (m, 1H), 6,55-6,59 (m, 1H), 5,34 (s, 2H), 4,21 (s, 3H), 3,85 (s, 3H), 2,59 (s, 3H), 1,62 (s, 9H).
Ejemplo 46.
Ácido 4-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)-5-metiltiazol-2-il)benzoico

El compuesto del título se preparó a partir del ejemplo 45 anterior de acuerdo con el método general de desprotección descrito en el ejemplo 39D. LC (Método A): 2,436 min. HRMS (ESI): calc. para C26H21N4O6S2 [M+H]+ m/z 549,0903; encontrado 549,0898. RMN 1H (400 MHz, DMSO-d6) 8 ppm: 13,07 (a s, 1H), 8,30 (s, 1H), 7,92-8,01 (m, 4H), 6,87 (s, 1H), 6,76-6,79 (m, 1H), 6,60-6,64 (m, 1H), 5,27 (s, 2H), 4,13 (s, 3H), 3,75 (s, 3H), 2,52 (s, 3H). Ejemplo 47.
4-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)-5-metiltiazol-2-il)-N-(2,2,2-trifluoroetil)benzamida

El compuesto del título se preparó a partir del ejemplo 46 de acuerdo con el método general de acoplamiento de amida descrito en el ejemplo 39. LC (Método a ): 2,412 min. HRMS (ESI): calc. para C28H23F3N5O5S2 [M+H]+ m/z 630,1093; encontrado 630,1092. RMN 1H (400 MHz, CDCh) 8 ppm: 7,99 (d, J= 8,22 Hz, 2H), 7,76-7,88 (m, 3H), 7,03 (s, 1H), 6,69 (s, 1H), 6,54 (s, 1H), 6,32 (t, J= 6,46 Hz, 1H), 5,31 (s, 2H), 4,06-4,22 (m, 5H), 3,83 (s, 3H), 2,57 (s, 3H). Ejemplo 48.
N-(cianometil)-4-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)-5-metiltiazol-2-il)-N-metilbenzamida

El compuesto del título se preparó a partir del ejemplo 46 de acuerdo con el método general de acoplamiento de amida descrito en el ejemplo 39. LC (Método A): 2,327 min. HRMS (ESI): calc. para C29H25N6O5S2 [M+H]+ m/z
601,1328; encontrado 601,1328. RMN 1H (400 MHz, CDCI3) 5 ppm: 8,00 (m, 2H), 7,84 (s, 1H), 7,52-7,59 (m, 2H), 7,05 (s, 1H), 6,69-6,74 (m, 1H), 6,53-6,59 (m, 1H), 5,33 (s, 2H), 4,48 (a s, 2H), 4,21 (s, 3H), 3,86 (s, 3H), 3,19 (s, 3H), 2,59 (s, 3H).
Ejemplo 49.
4-(4-(((6-metoxi-2-(2-metoxNmidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)-5-metiltiazol-2-il)-N-metilbenzamida

El compuesto del título se preparó a partir del ejemplo 46 de acuerdo con el método general de acoplamiento de amida descrito en el ejemplo 39. LC (Método A): 2,364 min. HRMS (ESI): calc. para C27H24N5O5S2 [M+H]+ m/z 562,1219; encontrado 562,1215. RMN 1H (400 MHz, CDCla) 5 ppm: 7,98 (d, J= 8,22 Hz, 2H), 7,78-7,86 (m, 3H), 7,05 (s, 1H), 6,65-6,76 (m, 1H), 6,53-6,59 (m, 1H), 6,12-6,24 (m, 1H), 5,33 (s, 2H), 4,21 (s, 3H), 3,85 (s, 3H), 3,05 (d, J= 5,09 Hz, 3H), 2,58 (s, 3H).
Preparación de alcoholes
Los siguientes alcoholes adicionales se prepararon de acuerdo con los procedimientos descritos en los ejemplos 36 a 49.

continuación

continuación

continuación

continuación

continuación

Ejemplos 50 a 82
Los siguientes ejemplos adicionales se prepararon, se aislaron y se caracterizaron usando los métodos descritos anteriormente.









Ejemplo 83
2-metoxi-6-(6-metoxi-4-(1-(2-feniltiazol-4-il)etoxi)benzofuran-2-il)imidazo[2,1-b][1,3,4]tiadiazol

83A. Ácido 2-feniltiazol-4-carboxílico

Una solución de 2-feniltiazol-4-carboxilato de etilo (Ejemplo 4B, 3,046 g, 13,06 mmol) en metanol (20 ml) se trató con una solución de NaOH (1,044 g, 26,1 mmol) en agua (10 ml), añadida gota a gota durante 2 min y la solución resultante se agitó a temperatura ambiente durante 1 h. Después el metanol se evaporó a presión reducida y la pasta residual se diluyó con una mezcla de agua (30 ml) y acetato de etilo (200 ml). El pH se ajustó a ~ 3 con ácido clorhídrico concentrado, la fase orgánica se separó y la fase acuosa se volvió a extraer con acetato de etilo (2 x 150 ml). El extracto orgánico combinado se lavó con salmuera (3 x 35 ml) y se secó sobre sulfato de magnesio anhidro. Después de la concentración del disolvente a presión reducida, el residuo sólido obtenido se secó al vacío durante 18 h para obtener 2,629 g (98 %) del compuesto del título en forma de un sólido cristalino de color blanco. LC (Método A): 1,842 min. HRMS (ESI) Anal. calc. para C10H8NO2S [M+H]+ m/z 206,027; encontrado 206,0266. RMN 1H (CDCla, 400 MHz) 8 ppm: 8,30 (s, 1H), 7,94 - 8,05 (m, 2H), 7,4 - 7,55 (m, 3H).
83B. A/-metoxi-W-metil-2-feniltiazol-4-carboxamida

Una mezcla de ácido 2-feniltiazol-4-carboxílico (1,00 g, 4,87 mmol) en diclorometano (20 ml) se trató con cloruro de oxalilo (1,237 g, 9,75 mmol) y una gota de N,N-dimetilformamida y la mezcla resultante se agitó a 22 °C durante 4 h. El disolvente se evaporó a presión reducida y el sólido residual se coevaporó con tolueno (10 ml). Este sólido se diluyó con diclorometano ( 10 ml) y se agregó gota a gota durante 2 min a una mezcla enfriada en hielo de clorhidrato de N,O-dimetilhidroxilamina (0,713 g, 7,31 mmol) y trietilamina (2,03 ml, 14,62 mmol) en diclorometano (20 ml). Después el baño de enfriamiento se retiró y la mezcla con un precipitado de color blanco se agitó a temperatura ambiente durante 1 h. La mezcla de reacción se inactivó mediante la adición de bicarbonato de sodio acuoso saturado (30 ml) y diclorometano (200 ml). La fase orgánica se lavó con salmuera, se secó sobre sulfato de magnesio anhidro y se concentró al vacío para obtener un aceite transparente. Este aceite se sometió a cromatografía sobre gel de sílice (elución con tolueno-acetato de etilo; 8:2 a 7:3) para obtener 1,054 g (87 %) del compuesto del título en forma de un aceite transparente. LC (Método A): 2,022 min. HRMS (ESI) Anal. calc. para C12H13N2O2S [M+H]+ m/z 249,0692; encontrado 249,0694. RMN 1H (CDCla, 400 MHz) 8 ppm: 8,03 (s, 1H), 7,91 -8,01 (m, 2H), 7,39 - 7,55 (m, 3H), 3,90 (s, 3H), 3,51 (a s, 3H).
83C. 1-(2-feniltiazol-4-il)etanona

Una solución de N-metoxi-N-metil-2-feniltiazol-4-carboxamida (1,00 g, 4,03 mmol) en tetrahidrofurano seco (20 ml) a 0°C se trató con bromuro de metilmagnesio (1 M en butiléter, 6,0 ml, 6,0 mmol) gota a gota durante 2 min. La
solución de color amarillo claro resultante se agitó a 0 °C durante 30 min y después la mezcla de reacción se inactivó mediante la adición a una mezcla de hielo (ca. 200 g) y ácido clorhídrico concentrado (2 ml). La fase acuosa se extrajo con acetato de etilo y el extracto orgánico se lavó con bicarbonato de sodio acuoso saturado y salmuera. La fase orgánica se secó sobresobre sulfato de magnesio anhidro y se concentró al vacío para obtener un sólido de color blanco. Este sólido se sometió a cromatografía sobre gel de sílice (elución con 0-5% de acetato de etilotolueno) para obtener 0,806 g (98 %) del compuesto del título en forma de prismas incoloros. LC (Método A): 2,017 min. HRMS (ESI) Anal. calc. para C11 H10NOS [M+H]+ m/z 204,0478; encontrado 204,0484. RMN 1H (CDCls, 400 MHz) 8 ppm: 8,14 (d, J = 1,6 Hz, 1H), 7,95 - 8,05 (m, 2H), 7,43 -7,57 (m, 3H), 2,76 (d, J = 1,6 Hz, 3H).
83D. 1-(2-feniltiazol-4-il)etanol

Una solución de 1-(2-feniltiazol-4-il)etanona (0,410 g, 2,017 mmol) en tetrahidrofurano seco (5 ml) se enfrió a 0 °C y se trató con borohidruro de sodio ( 0,114 g, 3,03 mmol) y después con metanol (0,040 ml, 1,0 mmol). La mezcla de color púrpura resultante se agitó a 0 °C durante 15 min y después a 23 °C durante 5 h. La mezcla se volvió a enfriar en hielo y se trató gota a gota con 1 ml de ácido acético acuoso al 50 %. Después la mezcla se repartió entre acetato de etilo (200 ml) y bicarbonato de sodio acuoso saturado. La fase orgánica se separó, se lavó con bicarbonato de sodio saturado y salmuera, se secó sobre sulfato de magnesio anhidro y se evaporó a presión reducida para obtener un jarabe de color amarillo. Este material se sometió a cromatografía sobre gel de sílice (elución con 20-40 % de acetato de etilo-tolueno) para obtener 0,392 g (95 %) del material del título en forma de un jarabe de color amarillo claro. LC (Método A): 1,874 min. HRMS (ESl) Anal. calc. para C11 H12NOS [M+H]+ m/z 206,0634; encontrado 206,0638. RMN 1H (CDCla, 400 MHz) 8 ppm: 7,9 - 8,0 (m, 2H), 7,4 - 7,5 (m, 3H), 7,12 (s, 1H), 5,05 (q, J = 6,3 Hz, 1H), 2,87 (as, 1H), 1,63 (d, J = 6,3 Hz, 3H).
Ejemplo 83. 2-metoxi-6-(6-metoxi-4-(1-(2-feniltiazol-4-il)etoxi)benzofuran-2-il)imidazo[2,1-b][1,3,4]tiadiazol

Una mezcla de 6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-ol (Ejemplo 1H, 0,080 g, 0,252 mmol) y 1-(2-feniltiazol-4-il)etanol (0,062 g, 0,303 mmol) en tetrahidrofurano seco (10 ml) se trató a 22 °C y en nitrógeno con tri-n-butilfosfina (0,157 ml, 0,63 mmol), añadida en una porción, seguido de una solución de 1,1'-(azodicarbonil)dipiperidina (0,083 g, 0,328 mmol) en tetrahidrofurano (4 ml), añadida gota a gota durante 30 min. Después de 2 h más a 22 °C, la mezcla de reacción se repartió entre diclorometano y bicarbonato de sodio saturado. La fase orgánica se lavó con salmuera, se secó sobresobre sulfato de magnesio anhidro y se concentró al vacío para obtener un residuo vítreo. La cromatografía sobre gel de sílice (gradiente de elución de acetato de etilo en diclorometano) produjo 0,078 g (61 %) del compuesto del título en forma de un sólido de color blanco, después de la trituración con acetonitrilo. LC (Método A): 2,596 min. HRMS (ESl) Anal. calc. para C25H20N4O5S2 [M+H]+ m/z 505,0999; encontrado 505,1001. RMN 1H (CDCls, 400 MHz) 8 ppm: 7,95 - 8,0 (m, 2H), 7,86 (s, 1H), 7,40 - 7,50 (m, 3H), 7,23 (s, 1H), 7,14 (s, 1H), 6 ,68 (a d, 1H), 6,39 (d, J = 2,0 Hz, 1H), 5,73 (q, J = 6,5 Hz, 1H), 4,22 (s, 3H), 3,79 (s, 3H), 1,82 (d, J = 6,5 Hz, 3H).
Ejemplo 84
4-(5-cloro-4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b]tiadiazol-6-il)benzofuran-4-il)oxi)tiazol-2-il)morfolina

84A. -2-morfolinotiazol-4-carboxilato de metilo y etilo

Una solución de morfolina (3,0 ml, 34,2 mmol) en EtOH (50 ml) se trató con 2-bromotiazol-4-carboxilato de metilo (1,65 g, 7,43 mmol) y DIEA (6 ,8 ml, 39,4 mmol) y la mezcla resultante se sometió a reflujo durante 18 h en atmósfera de N2. Después la mezcla de reacción se concentró a presión reducida y el residuo se purificó en ISCO usando una columna REDISEP® de 24 g (0 a 30 % de EtOAc-DCM) para obtener el producto (1,22 g, 72 %; mezcla de ésteres de metilo y etilo) en forma de un aceite de color amarillo. Esta mezcla se usó tal cual en la siguiente etapa. LCMS (APCI): calc. para C10H15N2O3S [M+H]+ m/z 277,03, encontrado 277,1; LCMS (APCI): calc. para C9H13N2O3S [M+H]+ m/z 229,06, encontrado 229,1.
84B. Etiléster del ácido 5-cloro-2-morfolinotiazol-4-carboxílico y metiléster del ácido 5-cloro-2-morfolinotiazol-4-carboxílico

Una mezcla de etiléster y metiléster del ácido 2-morfolinotiazol-4-carboxílico (1,22 g, 5,04 mmol) en una mezcla de DCM-CHCl3-ácido acético (1:1:1, 9 ml) se trató a 22 °C con N-clorosuccinimida (0,807 g, 6,04 mmol). La mezcla resultante se agitó a temperatura ambiente durante 2 h, después se agregó más NCS (0,050 g) y la mezcla se agitó a 40 °C durante 18 h. Después se agregó CELITE® y la mezcla se concentró. Se agregó DCM, seguido de NaHCO3 acuoso saturado y la capa orgánica se separó, se lavó con salmuera, se secó sobre MgSO4 y se evaporó a sequedad. El residuo se purificó en ISCO usando una columna REDISEP® de 24 g (0 a 30 % de EtOAc-DCM) para obtener los ésteres del título en forma de un sólido de color blanco (0,746 g, 54 %). Esta mezcla se usó tal cual en la siguiente etapa. LCMS (APCI): calc. para C10H14CIN2O3S [M+H]+ m/z 277,03, encontrado 277,1; LCMS (APCI): calc. para C9H12CIN2O3S [M+H]+ m/z 263,02, encontrado 263,0. RMN 1 H (CDCI3, 400 MHz) 8 ppm: 4,38 (q, J = 7,0 Hz, 1H), 3,91 (s, 1H), 3,76 -3,83 (m, 4H), 3,40 - 3,49 (m, 4H), 1,40 (t, J = 7,0 Hz, 2H).
84C. (5-cloro-2-morfolinotiazol-4-il)metanol

Una mezcla de etiléster y metiléster del ácido 2-morfolinotiazol-4-carboxílico (0,746 g, 2,70 mmol) en Et2O (50 ml) se enfrió a -78 °C y se trató con LAH (0,307 g, 8,09 mmol). Después el baño de enfriamiento se retiró y la mezcla resultante se agitó durante 2,5 h a temperatura ambiente. La mezcla de reacción se volvió a enfriar a -78 °C y se inactivó mediante la adición gota a gota de acetato de etilo (5 ml) durante 5 min. Después de 10 min, se agregó gota a gota agua (8,0 ml) durante 5 min, después una solución acuosa de NaOH 1 N (8,5 ml) y finalmente agua (10 ml). El baño de enfriamiento se retiró y la mezcla heterogénea se agitó a temperatura ambiente hasta que se tornó blanca (ca. 30 min). Después la suspensión se filtró y la torta de filtro se lavó con dietiléter (10 ml). El filtrado combinado se lavó con salmuera y se secó sobre sulfato de magnesio anhidro. La evaporación proporcionó el compuesto deseado en forma de un sólido de color blanquecino (0,249 g, 39 %). Este material se usó tal cual en la siguiente etapa. LCMS (APCI): calc. para C8H12CIN2O2S [M+H]+ m/z 235,02, encontrado 235,1. RMN 1 H (CDCI3, 400 MHz) 8 ppm: 4,52 (d, J = 5,7 Hz, 2H), 3,76 - 3,84 (m, 4H), 3,37 - 3,44 (m, 4H), 2,32 (t, J = 5,7 Hz, 1H)
Ejemplo 84.
4-(5-cloro-4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b]tiadiazol-6-il) benzofuran-4-il)oxi)tiazol-2-il)morfolina

Se agregaron 6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-ol (Ejemplo 1H, 0,070 g, 0,22 mmol) y (5-doro-2-morfolinotiazol-4-il)metanol (0,057 g, 0,24 mmol) a un matraz de fondo redondo de 25 ml que después se purgó con N2. Después se agregaron THF seco (4 ml) y tri-n-butilfosfina (0,14 ml, 0,55 mmol) y la mezcla de reacción se trató gota a gota con una solución de 1,1' (azodicarbonil)dipiperidina (0,139 g, 0,55 mmol) en THF seco (3,5 ml) durante 1 h. La suspensión de color beis resultante se agitó durante 2 h más a temperatura ambiente y después se diluyó con EtOAc, se lavó con NaHCO3 y salmuera, se secó sobre MgSO4 y se evaporó a sequedad. El residuo se purificó en ISCO usando una columna r Ed ISEP® de 4 g (0 a 40 % de EtOAc-DCM). El sólido obtenido se suspendió en MeOH, se sometió a ultrasonido, se filtró y se secó al vacío para obtener el compuesto del título (0,082 g, 70 %) en forma de un sólido de color blanco. LC (Método C): 2,366 min. HRMS (ESI): calc. para C22H21ClN5O5S2 [M+H]+ m/z 534,059, encontrado 534,0719. RMN 1H (CDCla, 400 MHz) 8 ppm: 7,83 (s, 1H), 7,08 (s, 1H), 6,70 (d, J = 1,6 Hz, 1H), 6,51 (d, J = 1,6 Hz, 1H), 5,05 (s, 2H), 4,21 (s, 3H), 3,85 (s, 3H), 3,77 - 3,83 (m, 4H), 3,39 -3 ,46 (m, 4H).
Ejemplo 85
6-(4-((5-cloro-2-feniltiazol-4-il)metoxi)-6-metoxibenzofuran-2-il)-2-metoximidazo[2,1-b][1,3,4]tiadiazol
85A. 2-feniltiazol-4-carboxilato de etilo

Una solución de benzotioamida (3,00 g, 21,87 mmol) en EtOH (70 ml) se trató gota a gota con bromopiruvato de etilo (5,10 g, 26,2 mmol) y se agitó a temperatura ambiente durante 30 min antes de calentarla a reflujo durante 1,5 h. La mezcla enfriada se diluyó con acetato de etilo ( 200 ml), se lavó (NaHCO3 acuoso, salmuera), se secó sobre MgSO4 anhidro y se evaporó. El residuo se purificó en ISCO usando una columna REDISEP® de 80 g (10 a 20 % de EtOAchexano) para obtener el compuesto del título (4,82 g, 94 %) en forma de un aceite de color amarillo. LCMS (APCI): calc. para C12H12NO2S [M+H]+m/z 234,05, encontrado 234,1. RMN 1H (CDCla, 400 MHz) 8 ppm: 8,14 -8,19 (m, 1H), 7,98 - 8,07 (m, 2H), 7,41 - 7,51 (m, 3H), 4,46 (q, J = 7,2 Hz, 2H), 1,44 (t, J = 7,2 Hz, 3H)
85B. Etiléster del ácido 5-cloro-2-feniltiazol-4-carboxílico

Se trató 2-feniltiazol-4-carboxilato de etilo (0,300 g, 1,29 mmol) de acuerdo con el método descrito en el ejemplo 84B anterior. El residuo en bruto se purificó en ISCO usando una columna REDISEP® de 24 g (0 a 30 % de EtOAchexano) para obtener el material del título en forma de un aceite incoloro (0,066 g, 19 %). LCMS (APCI): calc. para C12H-i1 ClNO2S [M+H]+ m/z 268,01, encontrado 268,0. RMN 1H (CDCls, 400 MHz) 8 ppm: 7,87 - 7,94 (m, 2H), 7,42 -7,50 (m, 3H), 4,48 (q, J = 7,0 Hz, 2H), 1,46 (t, J = 7,1 Hz, 3H)
85C. (5-cloro-2-feniltiazol-4-il)metanol

Se redujo etiléster del ácido 5-cloro-2-feniltiazol-4-carboxílico (0,066 g, 0,25 mmol) de acuerdo con el método descrito en el ejemplo 5B para obtener el compuesto del título (0,048 g, 86 %) en forma de un sólido de color amarillo claro. LCMS (APCI): calc. para C-iüHgClNOS [M+H]+ m/z 226,00, encontrado 226,0. RMN 1H (CDCls, 400 MHz) 8 ppm: 7,86 (dd, J = 6,5, 2,2 Hz, 2H), 7,39 - 7,50 (m, 3H), 4,77 (s, 2H), 2,39 (a s, 1H)
Ejemplo 85.
6-(4-((5-cloro-2-feniltiazol-4-il)metoxi)-6-metoxibenzofuran-2-il)-2-metoxiimidazo[2,1-b][1,3,4]tiadiazol

Se hicieron reaccionar 6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-ol (Ejemplo 1H, 0,058 g, 0,18 mmol) y (5-cloro-2-feniltiazol-4-il)metanol (0,041 g, 0,18 mmol), como se describe en el ejemplo 36. El residuo en bruto se purificó en ISCO usando una columna REDISEP® Gold de 12 g (5 a 20 % de EtOAc-DCM) y el producto obtenido se trituró con C^CN-MeOH. El sólido resultante se volvió a purificar en ISCO usando una columna REDISEP® de 4 g (0 a 10 % de EtOAc-DCM) para obtener el compuesto puro del título en forma de un sólido de color amarillo claro (0,048 g, 50 %). LC (Método C): 2,569 min. LCm S (APCI): calc. para C24H1sClN4O4S2 [M+H]+ m/z 525,04, encontrado 525,10. RMN 1H (CDCla, 400 MHz) 8 ppm: 7,86 (dt, J = 3,8, 3,0 Hz, 2H), 7,81 (s, 1H), 7,39 - 7,46 (m, 3H), 7,06 (s, 1H), 6,70 (s, 1H), 6,55 (d, J = 2,0 Hz, 1H), 5,28 (s, 2H), 4,18 (s, 3H), 3,84 (s, 3H).
Ejemplo 86
4-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)-5-(trifluorometil)tiazol-2-il)morfolina

86 A. 2-morfolin-5-(trifluorometil)tiazol-4-carboxilato de metilo

Este producto se preparó adaptando la metodología descrita por Nagib D. A. e t al., (Nature, 480:224 (2011)). Por ello, un matraz de fondo redondo de 50 ml se cargó con 2-morfolinotiazol-4-carboxilato de metilo (0,200 g, 0,876 mmol) [cf. Ejemplo 84A], fosfato de potasio dibásico (1,831 g, 10,51 mmol) [previamente secado a 105 °C al vacío durante la noche] e hidrato de diclorotris(1,10-fenantrolin)-rutenio (II) (0,030 g, 0,04 mmol) y la mezcla se mantuvo al vacío durante 10 min. Después el matraz se purgó con nitrógeno, se cargó con acetonitrilo (10 ml), se desgasificó en vacío leve durante 2 min y después se volvió a purgar con nitrógeno. Después se agregó cloruro de trifluorometansulfonilo (0,742 ml, 7,01 mmol) de una vez y la suspensión de color naranja se agitó y se irradió con una lamparita fluorescente en forma de globo de 13 W durante 24 h. Después la mezcla de reacción se inactivó mediante la adición a una mezcla de acetato de etilo (200 ml) y agua (50 ml). La fase orgánica se separó, se lavó sucesivamente con bicarbonato de sodio acuoso saturado ( 20 ml) y salmuera, se secó sobre sulfato de magnesio anhidro y se evaporó a presión reducida. El residuo oleoso de color amarillo claro obtenido se sometió a cromatografía sobre gel de sílice (elución con diclorometano-acetato de etilo, 90:10) para obtener 0,189 g (72 %) del
material del título en forma de placas largas de color blanco. LC (Método A): 2,047 min. HRMS (ESI): Anal. calc. para C10H12F3N2O3S [M+H]+ m/z 297,0515; encontrado 297,0526. RMN 1 H (CDCh, 400 MHz) 8 ppm: 3,94 (s, 3H), 3,78 -3,87 (m, 4H), 3,50 -3,59 (m, 4H).
8 6 B. (2-morfolin-5-(trifluorometil)tiazol-4-il)metanol

Una solución de 2-morfolin-5-(trifluorometil)tiazol-4-carboxilato de metilo (0,177 g, 0,597 mmol) en tetrahidrofurano (4 ml), en nitrógeno se enfrió a 0 °C y se trató con metanol (0,048 ml, 1,19 mmol) y después con borohidruro de litio (0,026 g, 1,195 mmol), añadido de una vez. Después de 30 min, se retiró el baño de enfriamiento y la solución turbia resultante se agitó a temperatura ambiente durante 2,5 h. La mezcla de reacción se volvió a enfriar en hielo, se inactivó con ácido acético acuoso al 50 % (dos gotas) y se diluyó con acetato de etilo (100 ml). Esta mezcla se lavó con bicarbonato de sodio saturado y salmuera, se secó sobre sulfato de magnesio anhidro y se concentró a presión reducida. El residuo sólido de color blanco obtenido se sometió a cromatografía sobre gel de sílice (elución con acetato de etilo) para obtener 0,144 g (90 %) del material del título en forma de un sólido de color blanco. LC (Método A): 1,833 min. HRMS (ESI): Anal. calc. para C9H12 F3N2O2S [M+H]+ m/z 269,0566; encontrado 269,0573. RMN 1 H (CDCl3, 400 MHz) 8 ppm: 4,62 (d, J = 5,9 Hz, 2H), 3,82 (d, J = 5,0 Hz, 4H), 3,51 (d, J = 5,0 Hz, 4H), 2,57 (t, J = 5,9 Hz, 1H).
Ejemplo 86.
4-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)-5-(trifluorometil)tiazol-2-il)morfolina

Una mezcla de 6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-ol (Ejemplo 1H, 0,080 g, 0,252 mmol) y 2-morfolin-5-(trifluorometil)tiazol-4-il)metanol (0,074 g, 0,277 mmol) en tetrahidrofurano seco (10 ml) se trató a 22 °C y en atmósfera de nitrógeno con tri-n-butilfosfina (0,128 g, 0,63 mmol), añadida en una porción, seguido de una solución de 1,1'-(azodicarbonil)dipiperidina (0,102 g, 0,403 mmol) en tetrahidrofurano (2 ml), añadida gota a gota durante 30 min. Después de 2 h más a 22 °C, la mezcla de reacción se repartió entre diclorometano y bicarbonato de sodio saturado. La fase orgánica se lavó con salmuera, se secó sobre sulfato de magnesio anhidro y se concentró al vacío para obtener un residuo vítreo. La cromatografía sobre gel de sílice (gradiente de elución de acetato de etilo en diclorometano) produjo 0,104 g (73 %) del compuesto del título en forma de un sólido de color blanco, después de la trituración en acetonitrilo. LC (Método A): 2,576 min. HRMS (ESI): Anal. calc. para C23H21F3N5O5S2 [M+H]+ m/z 568,0931; encontrado 568,0978. RMN 1 H (400 MHz, CDCh) 8 ppm: 7,84 (s, 1H), 7,06 (s, 1H), 6,71 (a. s, 1H), 6,47 (d, J = 1,6 Hz, 1H), 5,13 (s, 2H), 4,21 (s, 3H), 3,85 (s, 3H), 3,79 - 3,84 (m, 4H), 3,48 -3,55 (m, 4H).
Ejemplo 87
4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)-5-metiltiazol-2-amina

87A. 2-((terc-butoxicarbonil)amino)-5-metiltiazol-4-carboxilato de metilo
Me C 02Me
sY n
h' Vo Y 1
Una solución de 2-amino-5-metiltiazol-4-carboxilato de metilo (1,00 g, 5,81 mmol) en tetrahidrofurano (20 ml) se trató con dicarbonato de di-ferc-butilo (1,27 g, 5,81 mmol), añadido de una vez, seguido de trietilamina (1,619 ml, 11,61 mmol) y DMAP (0,040 g, 0,327 mmol). La solución resultante se agitó a 22 °C durante 18 h. Después la mezcla de reacción se diluyó con acetato de etilo (200 ml) y agua (50 ml) y la fase orgánica se lavó sucesivamente con agua, ácido clorhídrico 0,1 N frío, bicarbonato de sodio acuoso saturado y salmuera y se secó sobre sulfato de magnesio anhidro. La evaporación del disolvente a presión reducida produjo un aceite transparente que se sometió a cromatografía sobre gel de sílice (elución con tolueno-acetato de etilo, 8:2 a 7: 3) para obtener 1,319 g (83 %) del material del título en forma de un sólido de color amarillo claro. LC (Método A): 2,049 min. LCMS (APCI): Anal. calc. para C11 H15N2O4S [M-H]- m/z 271; encontrado 271. RMN 1H (CDCla, 400 MHz) 8 ppm: 8,24 (a. s., 1H), 3,90 (s, 3H), 2,69 (s, 3H), 1,53 (s, 9H).
87B. 2-((ferc-butoxicarbonil)(2,4-dimetoxibencil)amino)-5-metiltiazol-4-carboxilato de metilo y 2 -((fe rc - butoxicarbonil)imino)-3-(2,4-dimetoxibencil)-5-metil-2,3-dihidrotiazol-4-carboxilato de (Z)-metilo.
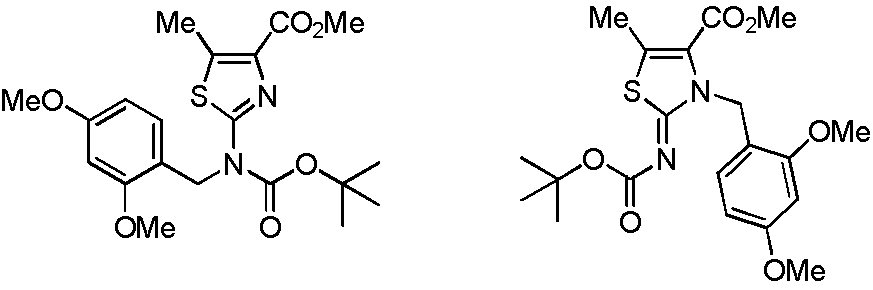
Una mezcla de 2-((ferc-butoxicarbonil)amino)-5-metiltiazol-4-carboxilato de metilo (1,18 g, 4,33 mmol) y (2,4-dimetoxifenil)metanol (0,802 g, 4,77 mmol) en tetrahidrofurano seco (40 ml) se trató a 22 °C con tri-n-butilfosfina (2,67 ml, 10,83 mmol), añadida de una vez, seguido de una solución de 1,1'-(azodicarbonil)dipiperidina (2,187 g, 8,67 mmol) en tetrahidrofurano (25 ml), añadido gota a gota durante 40 min. La mezcla se agitó durante 2 h más y se repartió entre acetato de etilo (250 ml) y bicarbonato de sodio acuoso saturado (20 ml). La fase orgánica se separó, se lavó con salmuera, se secó sobre sulfato de magnesio anhidro y se concentró al vacío para obtener un residuo tipo gel. La cromatografía sobre gel de sílice (elución con 0- 5 % de acetato de etilo-tolueno) produjo 0,993 g (54 %) de 2-((ferc-butoxicarbonil)(2,4-dimetoxibencil)amino)-5-metiltiazol-4-carboxilato de metilo en forma de un sólido de color blanco. LC (Método A): 2,388 min. HRMS (ESI): Anal. calc. para C20H27N2O6S [M+H]+ m/z 423,1584; encontrado 423,1474. RMN 1H (CDCla, 400 MHz) 8 ppm: 6,96 (d, J = 8,2 Hz, 1H), 6,43 (a s, 1H), 6,38 (a d, J = 8,2 Hz, 1H), 5,28 (s, 2H), 3,86 (s, 3H), 3,79 (s, 3H), 3,78 (s, 3H), 2,66 (s, 3H), 1,43 (s, 9H). La elución adicional produjo 0,287 g (16 %) de 2-((ferc-butoxicarbonil)imino)-3-(2,4-dimetoxibencil)-5-metil-2,3-dihidrotiazol-4-carboxilato de (Z)-metilo en forma de un sólido de color blanco. lC (Método A): 2,310 min. HRMS (ESI): Anal. calc. para C20H27N2O6S [M+H]+ m/z 423,1584; encontrado 423,1481. RMN 1H (CDCls, 400 MHz) 8 ppm: 6,84 (d, J = 8,2 Hz, 1H), 6,34 - 6,40 (m, 2H), 5,58 (s, 2H), 3,77 (s, 3H), 3,76 (s, 3H), 3,73 (s, 3H), 2,42 (s, 3H), 1,55 (s, 9H).
87C. 2,4-dimetoxibencil(4-(hidroximetil)-5-metiltiazol-2-il)carbamato de terc-butilo

Una solución de 2-((ferc-butoxicarbonil)(2,4-dimetoxibencil)amino)-5-metiltiazol-4-carboxilato de metilo (0,650 g, 1,538 mmol) en tetrahidrofurano (15 ml) y en nitrógeno se enfrió a 0 °C y se trató con metanol (0,124 ml, 3,08 mmol), seguido de borohidruro de litio (0,134 g, 6,15 mmol), añadido de una vez. Después de 10 min, se retiró el baño de enfriamiento y la solución turbia resultante se agitó a temperatura ambiente durante 6 h. La mezcla de reacción se volvió a enfriar y se inactivó con ácido acético acuoso al 50 % (1 ml). Después de que finalizó la evolución de hidrógeno, la mezcla de reacción se diluyó con diclorometano (250 ml), se lavó con bicarbonato de sodio saturado y salmuera y después se secó sobre sulfato de magnesio anhidro. Después de la concentración a presión reducida, el residuo sólido obtenido se sometió a cromatografía sobre gel de sílice (elución con diclorometano-acetato de etilo 95: 5 a 9: 1) para obtener 0,488 g (80 %) del material del título en forma de un sólido de color blanco. LC (Método A): 2,213 min. HRMS (ESI): Anal. calc. para C19H27N2O5S [M+H]+ m/z 395,1635; encontrado 395,1627. RMN 1H (CDCls, 400 MHz) 8 ppm: 6,91 (d, J = 8,5 Hz, 1H), 6,45 (d, J = 2,1 Hz, 1H), 6,38 (dd, J = 8,5, 2,1 Hz, 1H), 5,23 (s, 2H), 4,49 (d, J = 5,8 Hz, 2H), 3,82 (s, 3H), 3,78 (s, 3H), 2,32 (t, J = 5,8 Hz, 1H), 2,29 (s, 3H), 1,46 (s, 9H).
87D. 2,4-dimetoxibencil(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)-5-metiltiazol-2 -il)carbamato de ferc-butilo

Una mezcla de 6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-ol (Ejemplo 1H, 0,378 g, 1,19 mmol) y 2,4-dimetoxibencil(4-(hidroximetil)-5-metiltiazol-2-il)carbamato de ferc-butilo (0,469 g, 1,19 mmol) en tetrahidrofurano seco (30 ml) se trató a 22 °C con tri-n-butilfosfina (0,88 ml, 3,57 mmol), añadida en una porción, seguido de una solución de 1,1'-(azodicarbonil)dipiperidina (0,450 g, 1,785 mmol) en tetrahidrofurano (10 ml), añadida gota a gota durante 40 min. Después de 2 h más a 22 °C, la mezcla de reacción se repartió entre diclorometano (300 ml) y bicarbonato de sodio acuoso saturado. La fase orgánica se lavó con salmuera, se secó sobre sulfato de magnesio anhidro y se concentró al vacío para obtener un residuo vítreo. La cromatografía sobre gel de sílice (elución con 0-5 % de acetato de etilo-diclorometano) produjo 0,622 g (75 %) del material del título en forma de un sólido de color blanco. LC (Método A): 2,703 min. HRMS (ESI): Anal. calc. para C33H36N5O8S2 [M+H]+ m/z 694,2005; encontrado 694,2006. RMN 1H (CDCla, 400 MHz) 8 ppm: 7,83 (s, 1H), 7,01 (s, 1H), 6,95 (d, J = 8,5 Hz, 1H), 6,67 (s, 1H), 6,47 (a s, 1H), 6,43 (d, J = 2,2 Hz, 1H), 6,37 (dd, J = 8,5, 2,2 Hz, 1H), 5,26 (s, 2H), 5,11 (s, 2H), 4,21 (s, 3H), 3,80 (s, 3H), 3,79 (s, 3H), 3,78 (s, 3H), 2,41 (s, 3H), 1,44 (s, 9H).
Ejemplo 87.
4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)-5-metiltiazol-2-amina

A una mezcla de 2,4-dimetoxibencil(4-(((6-metoxi-2-(2-metoxiimidazo-[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)-5-metiltiazol-2-il)carbamato de ferc-butilo (0,643 g, 0,927 mmol) y 1,2,3,4,5-pentametilbenceno (2,40 g, 16,19 mmol) en diclorometano (10 ml), se le agregó ácido 2,2,2-trifluoroacético (15 ml, 196 mmol) en una porción y la solución transparente resultante se agitó a 23 °C durante 3 h. Después los volátiles se evaporaron a presión reducida y el residuo se repartió entre diclorometano (500 ml) y bicarbonato de sodio acuoso saturado. La fase acuosa se separó y se volvió a extraer con diclorometano ( 2 x 50 ml) y el extracto orgánico combinado se lavó con salmuera, se secó sobre sulfato de magnesio anhidro y se evaporó a presión reducida para obtener un residuo de color blanco. Este residuo sólido se sometió a cromatografía sobre gel de sílice (elución con 1-5 % de MeOH-diclorometano) para obtener 0,351 g (85 %) del compuesto del título en forma de un sólido de color blanco. LC (Método A): 2,189 min. HRMS (ESI): Anal. calc. para C19H18N5O4S2 [M+H]+ m/z 444,0795; encontrado 444,0797. RMN 1 H (DMSO-d6, 400 MHz) 8 ppm: 8,37 (s, 1H), 6,89 (s, 1H), 6,80 (s, 1H), 6,74 (a s, 2H), 6,57 (d, J = 1,6 Hz, 1H), 4,93 (s, 2H), 4,20 (s, 3H), 3,80 (s, 3H), 2,26 (s, 3H).
Ejemplo 88 (Referencia, no reivindicado)
4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)-2-morfolinotiazol-5-carbaldehído

88 A. 4-(4-(((ferc-butildimetilsilil)oxi)metil)tiazol-2-il)morfolina

Una mezcla de 2-bromo-4-(((ferc-butildimetilsilil)oxi)metil)tiazol (Ejemplo 37B, 2,46 g, 7,98 mmol) y morfolina (3,13 ml, 35,9 mmol) en THF (7 ml) en un vial a presión de 50 ml se calentó a 83 °C durante 48 h. Después la mezcla enfriada se evaporó al vacío y el residuo se diluyó con acetato de etilo ( 200 ml) y se lavó sucesivamente con agua, HCl 0,1 N frío, bicarbonato de sodio saturado y salmuera. Después de secarlo sobre sulfato de magnesio anhidro, el disolvente se evaporó y el residuo oleoso de color amarillo claro obtenido se purificó mediante cromatografía ultrarrápida en Isco (cartucho de 40 g, elución con 0-9 % de acetato de etilo-DCM) para obtener un aceite de color amarillo (1,98 g, 79 %). La destilación (Kugelrohr) de este aceite al vacío produjo 1,85 g (74 %) del compuesto del título en forma de un aceite amarillo claro: p.e. 105-115 °C / 0,04 torr. LC (Método F): 2,205 min. HRMS (ESI): calc. para C14H27N2O2SSÍ [M+H]+ m/z 315,156, encontrado 315,158. RMN 1H (CDCla, 400 MHz) 8 ppm: 6,47 (t, J = 1,5 Hz, 1H), 4,67 (d, J = 1,5 Hz, 2H), 3,74-3,90 (m, 4H), 3,39-3,51 (m, 4H), 0,95 (s, 9H), 0,11 (s, 6 H).
8 B. 4-(((ferc-butildimetilsilil)oxi)metil)-2-morfolinotiazol-5-carbaldehído

Una solución de 4-(4-(((ferc-butildimetilsilil)oxi)metil)tiazol-2-il)morfolina (0,400 g, 1,272 mmol) en THF anhidro (10 ml) se enfrió a -78 °C en atmósfera de nitrógeno y después se agregó gota a gota una solución de BuLi (1,6 M en hexanos, 1,11 ml, 1,780 mmol) durante 10 min. La mezcla resultante se agitó a -78 °C durante 30 min y después se agregó gota a gota DMF seco (0,591 ml, 7,63 mmol). La mezcla se agitó durante 2 h a -78 °C y después se calentó a temperatura ambiente. Después de que se completó la reacción (LC), se agregó NH4Cl acuoso saturado (3 ml) y la mezcla resultante se agitó durante 10 min a 20 °C, después se diluyó en DCM (50 ml) y se lavó con salmuera. La fase orgánica se secó (MgSO4) y se evaporó y el residuo se purificó en ISCO usando una columna REDISEP® de 12 g (60 a 100 % de DCM-hexanos) para obtener el producto del título en forma de un sólido de color amarillo brillante (0,20 g, 46 %). LCMS (APCI): calc. para C15H27N2O3SSÍ [M+H]+ m/z 343,14, encontrado 343,1. RMN 1H (CDCla, 400 MHz) 8 ppm: 10,22 (s, 1H), 4,91 (s, 2H), 3,78 - 3,83 (m, 4H), 3,56 - 3,63 (m, 4H), 0,94 (s, 9H), 0,14 (s, 6 H).
88 C. 4-(hidroximetil)-2-morfolinotiazol-5-carbaldehído

A una solución de 4-(((ferc-butildimetilsilil)oxi)metil)-2-morfolinotiazol-5-carbaldehído (0,200 g, 0,584 mmol) en THF seco (5 ml), en atmósfera de N2 , se le agregó gota a gota TBAF (2,7 M en THF, 0,427 ml, 1,168 mmol) y la mezcla se agitó a temperatura ambiente durante 2 h. Después la mezcla de reacción se diluyó con DCM y se lavó con NaHCO3 acuoso y salmuera y después se secó sobre MgSO4 y se evaporó. El producto en bruto se purificó en ISCO usando una columna REDISEP® de 4 g (0 a 15 % de MeOH-DCM) para obtener el producto en forma de un sólido de color amarillo (0,102 g, 77 %). LCMS (APCI): calc. para C9H13N2O3S [M+H]+ m/z 229,06, encontrado 229,1. RMN 1 H (CDCls, 400 MHz) 8 ppm: 9,88 (s, 1H), 4,84 (d, J = 5,9 Hz, 2H), 3,78 - 3,87 (m, 4H), 3,59 - 3,68 (m, 4H), 2,77 (t, J = 5,1 Hz, 1H).
Ejemplo 88.
4-(((6-metoxi-2-(2-metoxNmidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)-2-morfolinotiazol-5-carbaldehído
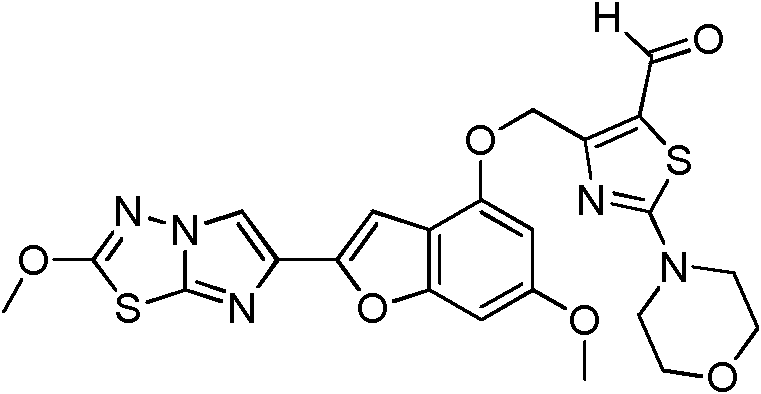
Se hicieron reaccionar 6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-ol (Ejemplo 1H, 0,139 g, 0,438 mmol) y 4-(hidroximetil)-2-morfolinotiazol-5-carbaldehído (0,100 g, 0,438 mmol), como se describe en el ejemplo 86. La mezcla de reacción se concentró al vacío y el residuo en bruto se suspendió en CH3CN, se sometió a ultrasonido y se filtró. El sólido resultante se purificó en ISCO usando una columna REDISEP® Gold de 4 g (0 a 60 % de EtOAc-DCM). El material obtenido se suspendió en CH3CN, se sometió a ultrasonido, se filtró y se secó para obtener el compuesto del título en forma de un sólido de color blanquecino (0,100 g, 43 %). LC (Método C): 2,287 min. HRMS (ESI): calc. para C23H22N5O6S2 [M+H]+ m/z 528,093, encontrado 528,0988. RMN 1 H (CDCla, 400 MHz) 8 ppm: 10,15 (s, 1H), 7,85 (s, 1H), 7,03 (s, 1H), 6,74 (s, 1H), 6,45 (d, J = 1,6 Hz, 1H), 5,36 (s, 2H), 4,22 (s, 3H), 3,85 (s, 3H), 3,81 - 3,85 (m, 4H), 3,65 (t, J = 4,7 Hz, 4H).
Ejemplo 89.
(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)-2-morfolinotiazol-5-il)metanol

Una solución de 4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)-2-morfolinotiazol-5-carbaldehído (0,088 g, 0,167 mmol) en THF seco (10 ml) en un matraz de 50 ml, en atmósfera de nitrógeno se enfrió a 0°C y se trató con MeOH (6,75 pl, 0,167 mmol), seguido de LiBH4 (3,63 mg, 0,167 mmol). La mezcla resultante se agitó a 0 °C durante 1 h antes de inactivarla con MeOH (5 ml) y se agitó a temperatura ambiente durante 10 min. Después la mezcla de reacción se diluyó con DCM, se lavó con NaHCO3 acuoso y salmuera, se secó sobre MgSO4 y se evaporó. El residuo se purificó en ISCO usando una columna REDISEP® de 4 g (0 a 100 % de EtOAc-DCM) para obtener el compuesto del título en forma de un sólido de color blanco (0,084 g, 95 %). LC (Método C): 2,062 min. HRMS (ESI): calc. para C23H24N5O6S2 [M+H]+ m/z 530,109, encontrado 530,114. RMN 1H (CDCla, 400 MHz) 8 ppm: 7,84 (s, 1H), 7,04 (d, J = 0,8 Hz, 1H), 6,72 (d, J = 0,8 Hz, 1H), 6,48 -6,53 (m, 1H), 5,11 - 5,17 (m, 2H), 4,77 -4,83 (m, 2H), 4,26 (s, 1H), 4,21 (s, 2H), 3,79 - 3,87 (m, 7H), 3,45 - 3,52 (m, 4H), 1,91 (t, J = 6,3 Hz, 1H).
Ejemplo 91
2-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)-2-morfolinotiazol-5-il)propan-2 -ol

91A. 2-(4-(((terc-butildimetilsilil)oxi)metil)-2-morfolinotiazol-5-il)propan-2-ol

Una solución de 4-(4-(((terc-butildimetilsilil)oxi)metil)tiazol-2-il)morfolina (Ejemplo 88 A, 0,100 g, 0,318 mmol) en THF seco (5 ml) se enfrió a -78 °C en atmósfera de nitrógeno y después se agregó gota a gota una solución de n-BuLi (1,26 M en hexanos, 0,454 ml, 0,572 mmol) durante 10 min. La mezcla resultante se agitó a -78 °C durante 30 min y después se agregó gota a gota acetona seca (1,0 ml, 13,62 mmol). La mezcla de reacción se calentó a temperatura ambiente y se agitó durante 2 h más antes de inactivarla con NH4Cl acuoso saturado (5 ml). La mezcla se diluyó con DCM (50 ml), se lavó con salmuera y, después de concentrarla a presión reducida, el material en bruto se purificó en ISCO usando una columna REDISEP® de 4 g (0 a 50 % de EtOAc-DCM) para obtener el producto deseado (0,040 g, 25 %) en forma de un aceite incoloro que se usó tal cual en la siguiente etapa. LC (Método A): 2,072 min. LCMS (APCI): calc. para C17H33N2O3SSi [M+H]+ m/z 373,20, encontrado 373,20.
91B. 2-(4-(hidroximetil)-2-morfolinotiazol-5-il)propan-2-ol

A una solución de 2-(4-(((terc-butildimetilsilil)oxi)metil)-2-morfolinotiazol-5-il)propan-2-ol (0,040 g, 0,107 mmol) en THF seco (2 ml), en atmósfera de N2 , se le agregó gota a gota trihidrofluoruro de trietilamina (0,105 ml, 0,644 mmol), y la mezcla se agitó a temperatura ambiente durante una noche. La mezcla de reacción se diluyó después con DCM y la solución se lavó con NaHCO3 acuoso, se secó sobre MgSO4 y se evaporó. El residuo se purificó en ISCO usando una columna REDISEP® de 4 g (0 a 15 % de MeOH-DCM) para obtener el compuesto del título en forma de un sólido de color blanco (0,019 g, 69 %). LCMS (APCI): calc. para C11 H19N2O3S [M+H]+ m/z 259,104, encontrado 259,1. RMN 1H (CDCh, 400 MHz) 8 ppm: 4,70 (s, 2H), 3,76 - 3,83 (m, 4H), 3,36 - 3,43 (m, 4H), 3,07 (a. s., 1H), 2,94 (a. s., 1H), 1,63 (s, 6 H).
Ejemplo 91.
2-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)-2-morfolinotiazol-5-il)propan-2 -ol

Se hicieron reaccionar 6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-ol (Ejemplo 1H, 0,023 g, 0,074 mmol) y 2-(4-(hidroximetil)-2-morfolinotiazol-5-il)propan-2-ol (0,019 g, 0,074 mmol), como se describe en el ejemplo 8 6. La mezcla de reacción se concentró al vacío y el residuo se suspendió en CH3CN, se sometió a ultrasonido y se filtró. El sólido resultante se purificó en ISCO usando una columna REDISEP® Gold de 4 g (0 a 70 % de EtOAc-DCM) y el material obtenido se suspendió en CH3CN, se sometió a ultrasonido, se filtró y se secó para obtener el compuesto del título en forma de un sólido de color blanquecino (0,005 g, 13 %). LC (Método C): 2,047 min. HRMS (ESI): calc. para C25H28N5O6S2 [M+H]+ m/z 558,140, encontrado 558,1482. RMN 1 H (CDCh, 400 MHz) 8 ppm: 7,84 (s, 1H), 7,03 (s, 1H), 6 ,68 - 6,72 (m, 1H), 6,57 (d, J = 2,0 Hz, 1H), 5,24 (s, 2H), 4,21 (s, 3H), 3,85 (s, 3H), 3,78 - 3,84 (m, 4H), 3,41 - 3,47 (m, 4H), 2,89 (s, 1H), 1,64 (s, 6 H).
Ejemplo 92
3-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2-il)-8-oxa-3-azabiciclo[3.2.1]octano

92A. 2-(8-oxa-3-azabiddo[3.2.1]octan-3-il)tiazol-4-carboxNato de metilo

Una solución de biciclomorfolina (0,52 ml, 4,75 mmol) en THF (10 ml) se trató con 2-bromotiazol-4-carboxilato de metilo (1,0 g, 4,24 mmol) y DIEA (1,66 ml, 9,50 mmol) y la mezcla resultante se sometió a reflujo durante 18 h en atmósfera de N2. La mezcla de reacción se concentró después a presión reducida y el residuo se purificó en ISCO usando una columna REDISEP® de 24 g (0 a 40 % de EtOAc-DCM) para obtener el producto deseado como una goma de color amarillo (0,740 g, 62 %). Lc Ms (APCI): calc. para C11 H15N2O3S [M+H]+ m/z 255,07, encontrado 255,1. RMN 1H (CDCfj, 400 MHz) 8 ppm: 7,48 (s, 1H), 4,48 (d, J = 2,7 Hz, 2H), 3,89 (s, 3H), 3,58 (d, J = 12,1 Hz, 2H), 3,37 (dd, J = 11,9, 2,5 Hz, 2H), 1,98 - 2,07 (m, 2H), 1,86 - 1,94 (m, 2H).
92B. (2-(8-oxa-3-azabiciclo[3.2.1]octan-3-il)tiazol-4-il)metanol

Una solución de 2-(8-oxa-3-azabiciclo[3.2.1]octan-3-il)tiazol-4-carboxilato de metilo (0,740 g, 2,91 mmol) en THF seco (15 ml) en un matraz de 50 ml en atmósfera de nitrógeno se enfrió a 0°C y se trató con LiBH4 (0,127 g, 5,82 mmol) y después con MeOH (0,24 ml, 5,82 mmol). Después de 10 min a 0 °C, el baño se retiró y la mezcla de reacción se agitó a temperatura ambiente durante una noche. La mezcla de reacción se inactivó después con MeOH (10 ml), se concentró a presión reducida y el residuo se disolvió en DCM y se secó sobre MgSO4. Los volátiles se retiraron a presión reducida y el residuo se purificó en ISCO usando una columna REDISEP® de 24 g (0 a 15 % de MeOH-DCM) para obtener el alcohol del título (0,60 g, 91 %) en forma de un aceite incoloro que se solidificó en reposo al vacío para obtener un sólido de color blanco. LCm S (APCI): calc. para C10H15N2O2S [M+H]+ m/z 227,08, encontrado 227,1. RMN 1H (CDCla, 400 MHz) 8 ppm: 6,41 (s, 1H), 4,53 (s, 2H), 4,02 (dt, J = 12,2, 6,2 Hz, 2H), 3,49 (d, J = 11,7 Hz, 2H), 3,30 (dd, J = 12,1, 2,0 Hz, 2H), 2,78 (a. s., 1H), 1,96 -2,03 (m, 2H), 1,88 - 1,92 (m, 2H).
Ejemplo 92. 3-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2-il)-8-oxa-3-azabiciclo[3.2.1]octano

Se hicieron reaccionar 6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-ol (Ejemplo 1H, 0,070 g, 0,22 mmol) y (2-(8-oxa-3-azabiciclo[3.2.1]octan-3-il)tiazol-4-il)metanol (0,055 g, 0,24 mmol), como se describe en el ejemplo 86. El material crudo se purificó en ISCO usando una columna REDISEP® Gold de 4 g (0 a 45 % de EtOAc-DCM) y el material obtenido se trituró con CH3CN para obtener el compuesto del título en forma de un sólido color crema (0,032 g, 28 %). LC (Método C): 2,262 min. HRMS (ESI): calc. para C24H24N5O5S2 [M+H]+ m/z 526,114, encontrado 526,124. RMN 1 H (CDCla, 400 MHz) 8 ppm: 8,37 (s, 1H), 6,98 (s, 1H), 6,92 (s, 1H), 6,82 (s, 1H), 6,56 (d, J = 2,0 Hz, 1H), 4,42 (a. s., 2H), 4,20 (s, 3H), 3,80 (s, 3H), 3,47 (d, J = 11,7 Hz, 3H), 3,14 -3,19 (m, 3H), 1,86 (d, J = 8,2 Hz, 2H), 1,76 - 1,80 (m, 2H).
Ejemplo 93
3-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b]tiadiazol-6-il)benzofuran-4-il)oxi)metil)-5-metiltiazol-2-il)-8-oxa-3-azabiciclo
[3.2.1]-octano

93A. 3-(4-(((terc-butildimetilsilil)oxi)metil)tiazol-2-il)-8-oxa-3-azabiciclo[3.2.1]octano

A una solución de (2-(8-oxa-3-azabicido[3.2.1]octan-3-il)tiazol-4-il)metanol (0,490 g, 2,165 mmol) en DMF (10 ml) enfriada a 0 °C en atmósfera de N2 , se le agregó TBDMS-Cl (0,653 g, 4,33 mmol) y después imidazol (0,339 g, 4,98 mmol). La mezcla de reacción se llevó a temperatura ambiente durante 10 min y se agitó a la misma temperatura durante 18 h. Después la mezcla se volvió a enfriar a 0 °C y se agregó EtOH (2 ml). Después de 10 min, la mezcla se calentó a temperatura ambiente y después se repartió entre EtOAc y NaHCO3 acuoso saturado. La capa orgánica se lavó con salmuera, se secó sobre MgSO4, se filtró y se evaporó. El residuo se purificó en ISCO usando una columna REDISEP® de 12 g (0 a 20 % de EtOAc-DCM) para obtener el producto deseado en forma de un aceite incoloro (0,70 g, 95 %). LCMS (APCI): calc. para C-i6H2gN2O2SSi [M+H]+ m/z 341,16, encontrado 341,2. RMN 1H (CDCla, 400 MHz) 8 ppm: 6,30 (s, 1H), 4,55 (s, 2H), 4,32 -4,37 (m, 2H), 3,38 (d, J = 11,7 Hz, 2H), 3,18 (dd, J = 11,7, 2,3 Hz, 2H), 1,84 -1,91 (m, 2H), 1,76 - 1,83 (m, 2H), 0,83 (s, 9H), 0,00 (s, 6H).
93B. 3-(4-(((terc-butildimetilsilil)oxi)metil)-5-metiltiazol-2-il)-8-oxa-3-azabiciclo[3.2.1]octano

Una solución de 3-(4-(((terc-butildimetilsilil)oxi)metil)tiazol-2-il)-8-oxa-3-azabiciclo[3.2.1]octano (0,700 g, 2,055 mmol) en THF anhidro (20 ml) se enfrió a -78 °C en atmósfera de nitrógeno y se agregó gota a gota una solución de BuLi (1,6 M en hexanos, 2,70 ml, 4,32 mmol) durante 10 min. La mezcla resultante se agitó a -78 °C durante 30 min y después se trató gota a gota con yodometano (0,257 ml, 4,11 mmol). El baño de enfriamiento se retiró y la mezcla se agitó durante 2 h a temperatura ambiente antes de inactivarla con NH4Cl acuoso saturado (5 ml). Después de agitarla durante 10 min, la mezcla se diluyó con DCM (100 ml) y se lavó con salmuera. La fase orgánica se secó (MgSO4) y se evaporó, y el residuo se purificó en ISCO usando una columna REDISEP® de 24 g (0 a 40 % de EtOAc-DCM) para obtener el producto deseado en forma de un aceite de color amarillo (0,514 g, 71 %). LCMS (APCI): calc. para C1 /Ha1 N2O2SSi [M+H]+ m/z 355,18, encontrado 355,2. RMN 1H (CDCla, 400 MHz) 8 ppm: 4,55 -4,61 (m, 2H), 4,40 - 4,47 (m, 2H), 3,44 (d, J = 12,1 Hz, 2H), 3,24 (dd, J = 12,1, 2,3 Hz, 2H), 2,31 (s, 3H), 1,93 - 2,01 (m, 2H), 1,85 - 1,93 (m, 2H), 0,92 (s, 9H), 0,10 (s, 6H).
93C. (2-(8-oxa-3-azabiciclo[3.2.1]octan-3-il)-5-metiltiazol-4-il)metanol
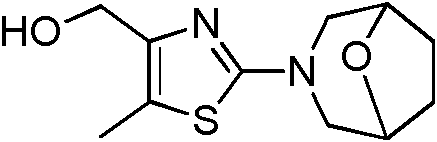
A una solución de 3-(4-(((terc-butildimetilsilil)oxi)metil)-5-metiltiazol-2-il)-8-oxa-3-azabiciclo[3.2.1]octano (0,514 g, 1,450 mmol) en THF seco (10 ml), en atmósfera de N2 , se le agregó gota a gota trihidrofluoruro de trietilamina (1,3 ml, 7,98 mmol) y la mezcla se agitó a temperatura ambiente durante una noche. La mezcla de reacción se inactivó después con MeOH y se concentró a presión reducida. El residuo se purificó en ISCO usando una columna REDISEP® de 12 g (0 a 15 % de MeOH-DCM) para obtener producto del título en forma de un sólido de color blanquecino (0,278 g, 80 %). LCMS (APCI): calc. para C11 H17N2O2S [M+H]+ m/z 241,09, encontrado 241,1. RMN 1H (CDCls, 400 MHz) 8 ppm: 4,35-4,41 (m, 4H), 3,62 (t, J = 5,7 Hz, 1H), 3,45 (d, J = 12,1 Hz, 2H), 3,10 (dd, J = 12,1,
2,3 Hz, 2H), 2,26 (s, 3H), 1,86 - 1,94 (m, 2H), 1,79 - 1,86 (m, 2H).
Ejemplo 93. 3-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b]tiadiazol-6-il) benzofuran-4-il)oxi)metil)-5-metiltiazol-2-il)-8-oxa-3-azabiciclo-[3.2.1 ]-octano

Se hicieron reaccionar 6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-ol (Ejemplo 1H, 0,070 g, 0,22 mmol) y (2-(8-oxa-3-azabicido[3.2.1]octan-3-il)-5-metiltiazol-4-il)metanol (0,058 g, 0,24 mmol), como se describe en el ejemplo 86. La mezcla de reacción se concentró al vacío y el residuo se suspendió en CH3CN, se sometió a ultrasonido y se filtró. El sólido resultante se purificó en ISCO usando una columna REDISEP® Gold de 4 g (0 a 70 % de EtOAc-DCM) para obtener el compuesto del título en forma de un sólido de color blanco (0,056 g, 47%). LC (Método C): 2,182 min. LCMS (APCI): calc. para C25H26N5O5S2 [M+H]+ m/z 540,130, encontrado 540,1. RMN 1 H (CDCl3, 400 MHz) 8 ppm: 8,33 (s, 1H), 6,87 (s, 1H), 6,78 (s, 1H), 6,57 (d, J = 1,6 Hz, 1H), 4,96 (s, 2H), 4,37 (a. s., 2H), 4,17 (s, 3H), 3,77 (s, 3H), 3,37 (d, J = 11,7 Hz, 2H), 3,08 (dd, J = 11,8, 2,2 Hz, 2H), 2,28 (s, 3H), 1,78 -1,87 (m, 2H), 1,68 - 1,78 (m, 2H).
Ejemplo 94
4-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)-5-metiltiazol-2-il)morfolina

94A. 5-metil-2-morfolinotiazol-4-carboxilato de metilo

Una solución de 2-bromo-5-metiltiazol-4-carboxilato de metilo (2,80 g, 11,86 mmol) y morfolina (4,5 ml, 51,7 mmol) en THF (10 ml) se calentó a reflujo en atmósfera de nitrógeno durante 18 h. Los volátiles se retiraron después a presión reducida y el producto en bruto se purificó en ISCO usando una columna REDISEP® de 40 g (0 a 40 % de EtOAc-DCM), para obtener el compuesto del título (2,20 g, 77 %) en forma de un sólido de color amarillo. LCMS (APCI): calc. para C10H15N2O3S [M+H]+ m/z 243,07, encontrado 243,1. RMN 1H (CDCh, 400 MHz) 8 ppm: 3,89 (s, 3H), 3,77-3,83 (m, 4H), 3,41-3,47 (m, 4H), 2,64 (s, 3H).
De manera alternativa, el ejemplo 94A, 5-metil-2-morfolinotiazol-4-carboxilato de metilo, se preparó de la siguiente manera:
94AA. 3-bromo-2-oxobutanoato de metilo

En un matraz de fondo redondo, de 4 bocas, de 5 l equipado con un agitador mecánico, un termopar de temperatura, un condensador y un embudo de adición de 1 l, se cargó bromuro de cobre (II) (962 g, 4310 mmol) y acetato de etilo (2 l). Se agregó gota a gota una solución de 2-cetobutirato de metilo (250 g, 2150 mmol) en CHCH (828 ml). Se conectó un depurador (400 ml de NaOH 1 N) y la mezcla de reacción se calentó a reflujo (75 °C). La reacción comenzó de color verde oscuro y; a medida que se siguió calentándola, se tornó verde claro con formación de un precipitado de color blanco. Después de una hora a reflujo, la RMN indicó que la reacción estaba completa. La reacción se enfrió a temperatura ambiente y se filtró a través de una almohadilla de CELITE®. El filtrado se concentró hasta convertirse en un aceite, se disolvió en cloruro de metileno (500 ml) y se filtró de nuevo a través de CELITE®. El filtrado después se hizo pasar a través de una almohadilla de gel de sílice y se eluyó con acetato de etilo. La concentración del filtrado produjo el bromocetoéster del título (399 g, 2040 mmol, 95 %) en forma de un aceite de color amarillo. RMN 1H (400 MHz, CDCls) 8 5,18 (q, J = 6,7 Hz, 1H), 3,94 (s, 3H), 1,83 (d, J = 6 ,8 Hz, 3H).
94AAA. Morfolin-4-carbotioamida

A una solución de morfolina (199 g, 2280 mmol) en CHCH (1 l), se le agregó gota a gota isotiocianatotrimetilsilano (150 g, 1140 mmol). Casi de inmediato, se formó un precipitado de color blanco y la reacción se agitó durante 1 h a temperatura ambiente. La reacción se filtró después y el sólido resultante se lavó con más CHCh y se secó al vacío para obtener la tiourea del título en forma de un sólido de color blanco. (137 g, 937 mmol, 82 %). RMN 1H (400 MHz, DMSO-da) 8 3,81 - 3,71 (m, 2H), 3,17 - 3,08 (m, 2H).
94A. 5-metil-2-morfolinotiazol-4-carboxilato de metilo

A una solución de morfolin-4-carbotioamida (Ejemplo 94AAA, 175 g, 1200 mmol) en metanol (500 ml), se le cargó 3-bromo-2-oxobutanoato de metilo (Ejemplo 94AA, 233 g, 1200 mmol). La reacción se calentó después a reflujo durante 1 hora, se enfrió a temperatura ambiente y se filtró. El filtrado se concentró y el producto en bruto se purificó mediante cromatografía sobre gel de sílice . El tiazol del título (206 g, 850 mmol, 71 %) se aisló en forma de un aceite de color amarillo. (Véase el procedimiento descrito anteriormente para los datos analíticos).
94B. (5-metil-2-morfolinotiazol-4-il)metanol

El compuesto se preparó de acuerdo con el protocolo descrito para el ejemplo 92B. El producto en bruto se purificó en ISCO usando una columna REDISEP® Gold de 24 g (0 a 50 % de EtoAc-DCM) para obtener el compuesto del título en forma de un sólido de color blanco (0,086 g, 51 %). LCMS (APCI): calc. para C9H15N2O2S [M+H]+ m/z 215,08, encontrado 215,1. RMN 1H (CDCla, 400 MHz) 8 ppm: 4,48 (d, J = 4,7 Hz, 2H), 3,77-3,83 (m, 4H), 3,37-3,43 (m, 4H), 2,30 (t, J = 4,7 Hz, 1H), 2,28 (s, 3H).
Ejemplo 94.
4-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)-5-metiltiazol-2-il)morfolina

El compuesto del título se preparó de acuerdo con el protocolo descrito para el ejemplo 8 6. El producto en bruto se
purificó en ISCO usando una columna REDISEP® de 4 g (0 a 40 % de EtOAc-DCM) y el sólido obtenido se suspendió en MeOH, se sometió a ultrasonido, se filtró y se secó para obtener el compuesto del título en forma de un sólido de color blanquecino (0,094 g, 53 %). LC (Método C): 2,314 min. HRMS (ESI): calc. para C23H24N5O5S2 [M+H]+ m/z 514,122, encontrado 514,126. RMN 1H (CDCla, 400 MHz) 8 ppm: 7,83 (s, 1H), 7,06 (d, J = 0,8 Hz, 1H), 6,69 (d, J = 0,8 Hz, 1H), 6,50 (d, J = 2,0 Hz, 1H), 5,05 (s, 2H), 4,21 (s, 3H), 3,85 (s, 3H), 3,78- 3,84 (m, 4H), 3,39 3,46 (m, 4H), 2,37 (s, 3H).
Ejemplo 95
2-metoxi-6-(6-metoxi-4-((5-metil-2-(tetrahidro-2H-piran-4-il)tiazol-4-il)metoxi)benzofuran-2-il)imidazo[2,1-b][1,3,4]tiadiazol

95A. 4-(((ferc-butildimetilsilil)oxi)metil)-2-(tetrahidro-2H-piran-4-il)tiazol

A una solución de (2-(tetrahidro-2H-piran-4-il)tiazol-4-il)metanol (0,075 g, 0,376 mmol) en diclorometano (5 ml) a temperatura ambiente, se le agregó imidazol (0,0384 g, 0,565 mmol) y después terc-butilclorodimetilsilano (0,071 g, 0,470 mmol). La mezcla de reacción resultante se agitó a temperatura ambiente durante 18 h y después se inactivó con metanol y se concentró a presión reducida. El residuo en bruto se purificó mediante cromatografía en columna (Isco, cartucho de 24 g) eluyendo con un gradiente de acetato de etilo en diclorometano (de 0 a 50 %) para obtener el compuesto puro del título (0,102 g, 86 %). LC (Método A): 2,416 min. LCMS (APCI) calc. para C15H2sNO2SSi [M+H]+ m/z 314,16, encontrado 314,2. RMN 1H (CDCh, 400 MHz) 8 ppm: 0,11 (s, 6 H), 0,94 (s, 9H), 1,82-1,94 (m, 2H), 1,99-2,07 (m, 2H), 3,16-3,26 (m, 1H), 3,53 (td, J= 2,5, 11,7 Hz, 2H), 4,02-4,09 (m, 2H), 4,84 (s, 2H), 7,06 (s, 1H).
95B. (5-metil-2-(tetrahidro-2H-piran-4-il)tiazol-4-il)metanol

A una solución de 4-(((ferc-butildimetilsilil)oxi)metil)-2-(tetrahidro-2H-piran-4-il)tiazol (1,38 g, 4,40 mmol) en THF seco (50 ml), a -78 °C en atmósfera de nitrógeno, se le agregó gota a gota n-BuLi (1,5 M en hexanos, 4,40 ml, 6,60 mmol). La mezcla de reacción se agitó durante 20 min a la misma temperatura antes de que se agregara yoduro de metilo (0,826 ml, 13,20 mmol). La mezcla de reacción resultante se calentó a - 20 °C durante 2 h y después se inactivó con metanol y se concentró a presión reducida. El residuo en bruto obtenido se disolvió en diclorometano, se lavó con agua y salmuera, se secó (MgSO4) y se evaporó. El producto en bruto (1,44 g, 100 %) se usó tal cual en la siguiente etapa. LC (Método A): 2,648 min. LCMS (APCI): calc. para C16H30NO2SSi [M+H]+ m/z 328,18, encontrado 328,2.
A una solución de 4-(((ferc-butildimetilsilil)oxi)metil)-5-metil-2-(tetrahidro-2H-piran-4-il)tiazol (1,44 g, 4,40 mmol) en THF (30 ml) a temperatura ambiente, se le agregó TBAF (75 % de solución en agua, 2,381 ml, 6,60 mmol) y la mezcla resultante se agitó a temperatura ambiente durante 2 h. Después se agregó otro equivalente de TBAF (75 % de solución en agua, 1,587 ml, 4,40 mmol) y después la agitación continuó durante 3 h más a temperatura ambiente. La mezcla de reacción se inactivó con agua y el producto se extrajo con diclorometano (3x). Los extractos orgánicos combinados se lavaron con salmuera, se secaron sobre MgSO4, se filtraron y se concentraron. El residuo en bruto obtenido se purificó mediante cromatografía en columna (Isco, cartucho de 40 g) eluyendo con un gradiente de acetato de etilo en diclorometano (0 a 100 %) para obtener el compuesto del título en forma de un sólido de color blanco (0,782 g, 83%). LC (Método A): 1,255 min. LCMS (APCI): calc. para C10H16NO2S [M+H]+ m/z 214,09, encontrado 214,2. RMN 1 H (DMSO-d6, 400 MHz) 8 ppm: 1,59-1,72 (m, 2H), 1,85-1,94 (m, 2H), 2,38 (s, 3H), 3,08 3,20 (m, 1H), 3,43 (td, J= 1,6, 11,3 Hz, 2H), 3,87-3,93 (m, 2H), 4,42 (s, 2H), 4,98 (a s, 1H).
95C. 4-(bromometil)-5-metil-2-(tetrahidro-2H-piran-4-il)tiazol

A una solución de (5-metil-2-(tetrahidro-2H-piran-4-il)tiazol-4-il)metanol (250 mg, 1,172 mmol) en diclorometano
(25 ml) enfriada a 0°C, se le agregó tribromofosfina (0,055 ml, 0,586 mmol). Después de agitar durante 5 min, se retiró el baño de enfriamiento y la solución se agitó a temperatura ambiente durante 3 h. La mezcla de reacción se vertió en una mezcla de acetato de etilo y bicarbonato de sodio acuoso saturado y la capa orgánica se lavó con salmuera, se secó sobre MgSO4, se filtró y se concentró. El residuo en bruto obtenido se purificó mediante cromatografía en columna (Isco, cartucho de 12 g) eluyendo con una mezcla de acetato de etilo en diclorometano (1:1) para obtener el producto en forma de un sólido de color blanco (0,285 g, 88 %). LC (Método A): 1,828 min. LCMS (APCI): calc. para C ^H ^rN O S [M+H]+ m/z 276,01, encontrado 276,0. RMN 1H (DMSO-da, 400 MHz) 8 ppm: 1,58-1,72 (m, 2H), 1,87-1,96 (m, 2H), 2,39 (s, 3H), 3,10-3,22 (m, 1H), 3,43 (td, J= 2,0, 11,3 Hz, 2H), 3,87-3,94 (m, 2H), 4,70 (s, 2H).
Ejemplo 95.
2-metoxi-6-(6-metoxi-4-((5-metil-2-(tetrahidro-2H-piran-4-il)tiazol-4-il)metoxi)benzofuran-2-il)imidazo[2,1-b][1,3,4]tiadiazol

A una solución de 6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-ol (Ejemplo 1H, 0,060 g, 0,189 mmol) y 4-(bromometil)-5-metil-2-(tetrahidro-2H-piran-4-il)tiazol (0,055 g, 0,199 mmol) en DMF (5 ml), en atmósfera de nitrógeno, se le agregó carbonato de potasio (0,065 g, 0,473 mmol) y la mezcla resultante se agitó a temperatura ambiente durante 1,25 h. La mezcla de reacción en bruto se disolvió con diclorometano, se lavó con agua y salmuera, se secó sobre MgSO4, se filtró y se concentró. El residuo en bruto obtenido se purificó mediante cromatografía en columna (Isco, cartucho de 12 g) eluyendo con un gradiente de acetato de etilo en diclorometano (de 0 a 50 %). El sólido de color blanco obtenido se trituró en acetonitrilo para obtener el compuesto del título en forma de un sólido de color blanco (0,050 g, 52 %). LC (Método A): 2,348 min. LCMS (ESI): calc. para C24H25N4O5S2 [M+H]+ m/z 513,1266, encontrado 513,1299. RMN 1 H (DMSO-d6, 400 MHz) 8 ppm: 1,62-1,75 (m, 2H), 1,91-1,98 (m, 2H), 2,46 (s, 3H), 3,15-3,25 (m, 1H), 3,45 (td, J= 1,6, 11,3 Hz, 2H), 3,81 (s, 3H), 3,87-3,94 (m, 2H), 4,20 (s, 3H), 5,19 (s, 2H), 6,64 (s, 1H), 6,83 (s, 1H), 6,89 (s, 1H), 8,37 (s, 1H).
Ejemplo 96
4-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)oxazol-2-il)morfolina

96A. 2-morfolinooxazol-4-carboxilato de etilo

A una solución de morfolina (1,5 ml, 17,22 mmol) en THF seco (10 ml), se le agregó 2-bromooxazol-4-carboxilato de etilo (1,00 g, 4,55 mmol). Después, la mezcla se calentó a 95 °C durante 18 h en atmósfera de N2. La mezcla de reacción se concentró a presión reducida y el material en bruto se purificó en ISCO usando una columna REDISEP® de 24 g (0 a 60 % de EtOAc-DCM) para obtener el compuesto del título en forma de un aceite de color amarillo (1,02 g, 99 %). LCMS (APCI): calc. para C10H15N2O4 [M+H]+ m/z 227,10, encontrado 227,1. RMN 1H (CDCla, 400 MHz) 8 ppm: 7,80 (s, 1H), 4,36 (q, J = 7,0 Hz, 2H), 3,73- 3,81 (m, 4H), 3,51-3,59 (m, 4H), 1,36 (t, J = 7,1 Hz, 3H).
96B. (2-morfolinooxazol-4-il)metanol
r -
O H
o
X %
El compuesto se preparó a partir de 2-morfolinooxazol-4-carboxilato de etilo (1,029 g, 4,55 mmol) usando el protocolo descrito en el ejemplo 92B. La mezcla del producto en bruto se purificó en ISCO usando una columna REDISEP® Gold de 12 g (0 a 15 % de MeOH-DCM) para obtener el compuesto del título en forma de un sólido de color blanco (0,354 g, 42 %). LCMS (APCI): calc. para C8H13N2O3 [M+H]+ m/z 185,09, encontrado 185,1. RMN 1H (CDCla, 400 MHz) 8 ppm: 7,18 (s, 1H), 4,48 (d, J = 5,7 Hz, 2H), 3,73-3,82 (m, 4H), 3,44-3,54 (m, 4H), 2,65 (t, J = 5,7 Hz, 1H).
Ejemplo 96.
4-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)oxazol-2-il)morfolina

El compuesto del título se preparó de acuerdo con el procedimiento general descrito para el ejemplo 8 6. El producto en bruto se purificó en ISCO usando una columna r Ed ISEP® Gold de 4 g (0 a 80 % de EtOAc-DeM), y el material obtenido se suspendió en CH3CN, se sometió a ultrasonido, se filtró y se secó para obtener el compuesto del título en forma de un sólido de color blanco (0,060 g, 56 %). LC (Método C): 2,173 min. HRMS (ESI): calc. para C22H22N5O6S [M+H]+ m/z 483,129, encontrado 484,132. RMN 1 H (CDCla, 400 MHz) 8 ppm: 7,84 (s, 1H), 7,29 (s, 1H), 7,07 (s, 1H), 6,70 (s, 1H), 6,43 (d, J = 1,6 Hz, 1H), 5,02 (s, 2H), 4,21 (s, 3H), 3,85 (s, 3H), 3,77-3,82 (m, 4H), 3,50 3,55 (m, 4H).
Ejemplo 97
4-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)-5-metiloxazol-2-il)morfolina

7A. 4-(4(((ferc-butildimetilsilil)oxi)metil)oxazol-2-il)morfolina

El compuesto del título se preparó a partir de (2-morfolinooxazol-4-il)metanol (0,250 g, 1,36 mmol) usando el protocolo descrito para el ejemplo 92C. La mezcla del producto en bruto se purificó en ISCO usando una columna REDISEP® Gold de 12 g (0 a 15 % de MeOH-DCM) para obtener el compuesto del título en forma de un sólido de color blanco (0,354 g, 42 %). LCMS (APCI): calc. para C14H27N2O3SÍ [M+H]+ m/z 299,17, encontrado 299,2. RMN 1 H (CDCl3, 400 MHz) 8 ppm: 7,11 (t, J = 1,4 Hz, 1H), 4,57 (d, J = 1,6 Hz, 2H), 3,74-3,81 (m, 4H), 3,44-3,50 (m, 4H), 0,91-0,95 (m, 9H), 0,12 (s, 6 H).
97B. 4-(4-(((ferc-butil(etil)(metil)silil)oxi)metil)-5-metiloxazol-2-il)morfolina

El compuesto del título se preparó a partir de 4-(4(((ferc-butildimetilsilil)oxi)metil)oxazol-2-il)morfolina (0,296 g,
0,99 mmol) de acuerdo con el protocolo descrito en el ejemplo 93B. La mezcla del producto en bruto se purificó en ISCO usando una columna REDISEP® Gold de 12 g (0 a 60 % de EtOAc-DCM) para obtener el compuesto del título en forma de un aceite incoloro (0,127 g, 32 %). LCMS (APCI): calc. para C-i6H3-iN2O3Si [M+H]+ m/z 327,20, encontrado 327,2. RMN 1H (CDCla, 400 MHz) 8 ppm: 4,53 (s, 2H), 3,73-3,80 (m, 4H), 3,39-3,46 (m, 4H), 2,24 (d, J = 0,8 Hz, 3H), 0,97-1,04 (m, 3H), 0,91 -0,96 (m, 9H), 0,73 (s, 1H), 0,60 (s, 1H), 0,11 (s, 3H).
97C. (5-metil-2-morfolinooxazol-4-il)metanol
hcOlVG°
El compuesto del título se preparó a partir de 4-(4(((terc-butil(etil)(metil)silil)oxi)metil)-5-metiloxazol-2-il)morfolina (0,124 g, 0,38 mmol) de acuerdo con el protocolo descrito en el ejemplo 93C. La mezcla del producto en bruto se purificó en ISCO usando una columna REDISEP® Gold de 4 g (0 a 15 % de MeOH-DCM) para obtener un aceite que se disolvió en DCM (100 ml) y se lavó con HCl 1 N (2 x 30 ml). La capa acuosa se basificó con Na2CO3 sólido a pH 8 y después se volvió a extraer con DCM (5 x 20 ml). La fase orgánica combinada se secó sobre MgSO4, se filtró y se concentró para obtener el producto deseado en forma de la base libre (0,035 g, 34 %). LCMS (APCI): calc. para C9H15N2O3 [M+H]+ m/z 199,11, encontrado 199,1. RMN 1H (CDCh, 400 MHz) 8 ppm: 4,42 (a s, 2H), 3,74-3,81 (m, 4H), 3,41-3,48 (m, 4H), 2,18-2,26 (m, 4H).
Ejemplo 97.
4-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4] tiadiazol-6-il)benzofuran-4-il)oxi)metil)-5-metiloxazol-2-il)morfolina

Se hicieron reaccionar 6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-ol (Ejemplo 1H, 0,042 g, 0,13 mmol) y (2-(8-oxa-3-azabiciclo[3,2,1]octan-3-il)-5-metiltiazol-4-il)metanol (0,026 g, 0,13 mmol), como se describe en el ejemplo 86. La mezcla de reacción se concentró al vacío y el residuo se suspendió en CH3CN, se sometió a ultrasonido y se filtró. El sólido resultante se purificó en ISCO usando una columna REDISEP® Gold de 4 g (0 a 70 % de EtOAc-DCM) y el material obtenido se suspendió en CH3CN, se sometió a ultrasonido, se filtró y se secó para obtener el compuesto del título en forma de un sólido de color blanquecino (0,022 g, 34 %). LC (Método C): 2,126 min. LCMS (APCI): calc. para C23H24N5O6S [M+H]+ m/z 498,14, encontrado 498,1. RMN 1 H (CDCh, 400 MHz) 8 ppm: 7,84 (s, 1H), 7,05 (s, 1H), 6,70 (s, 1H), 6,45 (d, J = 2,0 Hz, 1H), 4,95 (s, 2H), 4,21 (s, 3H), 3,85 (s, 3H), 3,77-3,81 (m, 4H), 3,45-3,50 (m, 4H), 2,28 (s, 3H).
Ejemplo 98
3-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)oxazol-2-il)-8-oxa-3-azabiciclo[3.2.1]octano

98A. 2-(8-oxa-3-azabiciclo[3.2.1]octan-3-il)oxazol-4-carboxilato de etilo

A una solución de 8-oxa-3-azabiciclo[3.2.1]octano (0,292 ml, 2,65 mmol) en THF (10 ml), se le agregó 2bromooxazol-4-carboxilato de etilo (0,583 g, 2,65 mmol) seguido de DIEA (0,926 ml, 5,30 mmol). La mezcla se calentó a reflujo durante 18 h en atmósfera de N2. La mezcla de reacción enfriada se concentró después a presión reducida y el residuo en bruto se purificó en ISCO usando una columna REDISEP® de 24 g (0 a 45 % de EtOAc-DCM) para obtener el producto deseado en forma de un sólido de color amarillo claro (0,592 g, 89 %). LCMS (APCI): calc. para C12 H17N2O4 [M+H]+ m/z 253,11, encontrado 253,1. RMN 1H (CDCh, 400 MHz) 8 ppm: 7,78 (s, 1H), 4,43 (d, J = 2,2 Hz, 2H), 4,35 (q, J = 7,2 Hz, 2H), 3,66 (d, J = 12,4 Hz, 2H), 3,34 (dd, J = 12,3, 2,2 Hz, 2H), 1,92-2,04 (m, 2H), 1,84-1,92 (m, 2H), 1,36 (t, J = 7,0 Hz, 3H).
98B. (2-(8-oxa-3-azabiciclo[3.2.1]octan-3-il)oxazol-4-il)metanol

El compuesto del título se preparó a partir de 2-(8-oxa-3-azabiciclo[3.2.1]octan-3-il)oxazol-4-carboxilato de etilo de acuerdo con el método descrito en el ejemplo 92B. La mezcla del producto en bruto se purificó en ISCO usando una columna REDISEP® de 4 g (0 a 15 % de MeOH-DCM) y el material obtenido se suspendió en MeOH, se sometió a ultrasonido, se filtró y se secó para obtener el producto deseado en forma de un sólido de color blanco (0,382 g, 77%). LCMS (APCI): calc. para C10H15N2O3 [M+H]+ m/z 211,11, encontrado 211,1. RMN 1H (CDCla, 400 MHz) 8 ppm: 7,35 (s, 1H), 4,98 (t, J = 5,5 Hz, 1H), 4,34-4,40 (m, 2H), 4,20 (d, J = 5,5 Hz, 2H), 3,45 (d, J = 12,1 Hz, 2H), 3,10 (dd, J =12,1, 2,3 Hz, 2H), 1,79-1,89 (m, 2H), 1,71 - 1,79 (m, 2H).
Ejemplo 98.
3-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)oxazol-2-il)-8-oxa-3-azabiciclo[3.2.1]octano

El compuesto del título se preparó de acuerdo con el procedimiento descrito para la síntesis del ejemplo 92B. La mezcla del producto en bruto se purificó en ISCO usando una columna REDISEP® Gold de 12 g (0 a 80 % de EtOAc-DCM) y el material obtenido se suspendió de nuevo en CH3CN, se sometió a ultrasonido, se filtró y se secó para obtener el compuesto del título en forma de un sólido de color blanco (0,046 g, 41 %). LC (Método C): 2,218 min. LCMS (APCI): calc. para C24H24N5O6S [M+H]+ m/z 510,15, encontrado 510,2. RMN 1 H (DMSO-d6, 400 MHz) 8 ppm: 8,30 (s, 1H), 7,66 (s, 1H), 6,87 (s, 1H), 6,75 (s, 1H), 6,52 (d, J = 1,6 Hz, 1H), 4,89 (s, 2H), 4,32 (a s, 2H), 4,14 (s, 3H), 3,74 (s, 3H), 3,43 (d, J = 12,1 Hz, 2H), 3,09 (dd, J = 12,1, 2,0 Hz, 2H), 1,74-1,84 (m, 2H), 1,66-1,74 (m, 2H). Ejemplo 99
4-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2-il)piperazin-1-carboxilato de tere-butilo

99A. 2-(4-(tere-butoxicarbonil)piperazin-1-il)tiazol-4-carboxilato de metilo

A una solución de piperazin-1-carboxilato de ferc-butilo (3,77 g, 20,26 mmol) en MeOH (40 ml), se le agregó 2-bromotiazol-4-carboxilato de metilo (1,50 g, 6,75 mmol) , seguido de DIEA (6,25 ml, 35,8 mmol). Después la mezcla se calentó a reflujo durante 18 h en atmósfera de N2. La mezcla enfriada se concentró después a presión reducida y el residuo se purificó en ISCO usando una columna REDISEP® de 40 g (0 a 30 % de EtOAc-DCM) para obtener el producto en forma de un sólido color crema (0,579 g, 26 %). LCMS (APCI): calc. para C14H22N3O4S [M+H]+ m/z 328,13, encontrado 328,2. RMN 1H (CDCla, 400 MHz) 8 ppm: 7,50 (s, 1H), 3,90 (s, 3H), 3,55 (a s, 8 H), 1,47-1,51 (m, 9H).
99B. 2-(4-(ferc-butoxicarbonil)piperazin-1-il)tiazol-4-carboxilato de metilo

El compuesto se preparó de acuerdo con el procedimiento descrito en el ejemplo 92. El producto en bruto se purificó en ISCO usando una columna REDISEP® de 24 g (0 a 20 % de MeOH-DcM) para obtener el compuesto del título en forma de una espuma de color blanco (0,494 g, 93 %). LCMS (APCI): calc. para C13H22N3O3S [M+H]+ m/z 300,13, encontrado 300,1. RMN 1H (CDCh, 400 MHz) 8 ppm: 6,45 (s, 1H), 4,56 (a s, 2H), 3,53-3,61 (m, 4H), 3,49 (a s, 4H), 3,35-3,44 (m, 1H), 1,49 (s, 9H).
Ejemplo 99.
4-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2-il)piperazin-1-carboxilato de ferc-butilo

El compuesto del título se preparó de acuerdo con el procedimiento descrito en el ejemplo 86 y se aisló en forma de un sólido de color blanquecino (0,339 g, 38 %). LC (Método C): 1,602 min. HRMS (ESI): calc. para C27H31N6O6S2 [M+H]+ m/z 599,175, encontrado 599,177. RMN 1H (CDCh, 400 MHz) 8 ppm: 7,85 (s, 1H), 7,10 (s, 1H), 6,70 (s, 1H), 6,64 (s, 1H), 6,44 (d, J = 2,0 Hz, 1H), 5,16 (s, 2H), 4,21 (s, 3H), 3,85 (s, 3H), 3,59 (d, J = 5,5 Hz, 4H), 3,55 (a s, 4H), 1,49 (s, 9H).
Ejemplo 100
2-metoxi-6-(6-metoxi-4-((2-(4-(metilsulfonil)piperazin-1-il)tiazol-4-il)metoxi)benzofuran-2-il)imidazo[2,1-b][1,3,4]tiadiazol

100A. 2-(4-(metilsulfonil)piperazin-1-il)tiazol-4-carboxilato de metilo

A una solución de 2-(4-(terc-butoxicarbonil)piperazin-1-il)tiazol-4-carboxilato de metilo (0,20 g, 0,61 mmol) en DCM (5 ml), se le agregó ácido trifluoroacético (2 ml, 26 mmol). La mezcla resultante se agitó a temperatura ambiente durante 3 h y después se concentró a presión reducida y el residuo en bruto se usó tal cual en la siguiente etapa. La sal de TFA de 2-(piperazin-1-il)tiazol-4-carboxilato de metilo resultante (0,32 g, 0,94 mmol) se disolvió en DCM (10 ml), se trató con trietilamina (1,10 ml, 7,92 mmol) y se agitó a 25 °C durante 5 min. Después la mezcla se enfrió a 0 °C, se agregó cloruro de metansulfonilo (0,10 ml, 1,29 mmol) y la mezcla se agitó a 0 °C durante 4 h antes de agitarla durante una noche a 25 °C. La mezcla de reacción se diluyó después con DCM, se lavó con NaHCO3 acuoso saturado y salmuera, se secó sobre MgSO4 y se evaporó. El residuo se purificó en ISCO usando una columna REDISEP® de 24 g (0 a 35 % de EtOAc-DCM) para obtener el producto deseado en forma de un sólido de color blanco (0,281, 98 %). LCMS (APCI): calc. para C10H16N3O4S2 [M+H]+ m/z 306,06, encontrado 306,1. RMN 1 H (CDCl3, 400 MHz) 8 ppm: 7,49-7,53 (m, 1H), 3,85-3,91 (m, 3H), 3,63-3,72 (m, 4H), 3,30-3,39 (m, 4H), 2,80 (s, 3H).
100B. (2-(4-(metilsulfonil)piperazin-1-il)tiazol-4-il)metanol

El compuesto se preparó de acuerdo con el procedimiento descrito en el ejemplo 92B. El producto en bruto se purificó en ISCO usando una columna REDISEP® de 24 g (0 a 20 % de MeOH-DCM) para obtener el compuesto del título en forma de una espuma de color blanco (0,215 g, 84 %). LCMS (APCI): calc. para C9H16N3O3S2 [M+H]+ m/z 278,06, encontrado 278,1. RMN 1H (CDCla, 400 MHz) 8 ppm: 6,50 (s, 1H), 4,59 (s, 2H), 3,80 (a s, 3H), 3,55-3,69 (m, 1H), 3,39-3,48 (m, 4H), 3,33-3,39 (m, 1H), 2,80-2,88 (m, 3H).
Ejemplo 100.
2-metoxi-6-(6-metoxi-4-((2-(4-(metilsulfonil)piperazin-1-il)tiazol-4-il)metoxi)benzofuran-2-il)imidazo[2,1-b][1,3,4]tiadiazol

El compuesto del título se preparó y se purificó de acuerdo con el método descrito en el ejemplo 86 y se aisló en forma de un sólido de color blanquecino (0,038 g, 30 %). LC (Método C): 2,225 min. Hr MS (ESI): calc. para C23H25N6O6S3 [M+H]+ m/z 577,0998, encontrado 577,1017. RMN 1H (CDCla, 400 MHz) 8 ppm: 8,37 (s, 1H), 7,00 (s,
1H), 6,98 (s, 1H), 6,82 (d, J = 0 ,8 Hz, 1H), 6,57 (d, J = 1,6 Hz, 1H), 5,07 (s, 2H), 4,20 (s, 3H), 3,80 (s, 3H), 3,51-3,58 (m, 4H), 3,21-3,28 (m, 4H), 2,92 (s, 3H).
Ejemplo 101
4-((4-(4-(((6-metoxi-2-(2-metoxNmidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2-il)piperazin-1-il)sulfonil)benzonitrilo

101A. 2-(4-((4-cianofenil)sulfonil)piperazin-1-il)tiazol-4-carboxilato de metilo

El compuesto se preparó usando el procedimiento descrito en el ejemplo 100A anterior. El producto en bruto se purificó en ISCO usando una columna REDISEP® de 24 g (0 a 40 % de EtOAc-DCM) para obtener el producto deseado en forma de un sólido de color blanco (0,222 g, 93 %). LCMS (APCI): calc. para C16H17N4O4S2 [M+H]+ m/z 393,07, encontrado 393,1. RMN 1H (CDCla, 400 MHz) 8 ppm: 7,83-7,93 (m, 4H), 7,50 (s, 1H), 3,87-3,91 (m, 3H), 3,66-3,73 (m, 3H), 3,57-3,62 (m, 1H), 3,16-3,25 (m, 4H).
101B. 4-((4-(4-(hidroximetil)tiazol-2-il)piperazin-1-il)sulfonil)benzonitrilo
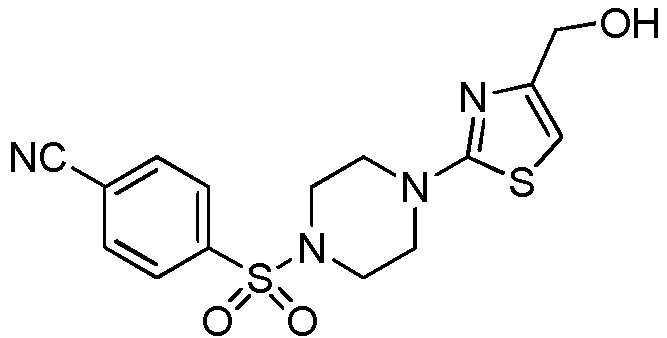
El compuesto se preparó de acuerdo con el procedimiento descrito en el ejemplo 92B. El producto en bruto se purificó en ISCO usando una columna REDISEP® de 24 g (0 a 15 % de MeOH-DCM) para obtener el producto deseado en forma de un sólido de color blanco (0,103 g, 50 %). LCMS (APCI): calc. para C15H17N4O3S2 [M+H]+ m/z 365,06, encontrado 365,1. RMN 1H (CDCla, 400 MHz) 8 ppm: 7,83-7,95 (m, 4H), 6,47 (s, 1H), 4,56 (s, 2H), 3,84 (a s, 3H), 3,54-3,67 (m, 1H), 3,23-3,31 (m, 3H), 3,15-3,22 (m, 1H).
Ejemplo 101. 4-((4-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2-il)piperazin-1 il)sulfonil)benzonitrilo

El compuesto del título se preparó de acuerdo con el procedimiento descrito en el ejemplo 86. El producto en bruto
se purificó en ISCO usando una columna REDISEP® de 4 g (0 a 60 % de EtOAc-DCM) para obtener el compuesto del título en forma de un sólido color crema (0,051 g, 41 %). LC (Método C): 2,339 min. HRMS (ESI): calc. para C29H26N7O6S3 [M+H]+ m/z 664,1107, encontrado 664,1118. RMN 1H (CDCla, 400 MHz) 8 ppm: 7,83-7,94 (m, 5H), 7,20 (s, 1H), 6,69 (s, 2H), 6,42 (d, J = 2,0 Hz, 1H), 5,18 (s, 2H), 4,25 (s, 3H), 3,84 (s, 3H), 3,81 (a s, 4H), 3,23-3,30 (m, 4H).
Ejemplo 102
6-(4-((2-(4-(isopropilsulfonil)piperazin-1-il)tiazol-4-il)metoxi)-6-metoxibenzo-furan-2-il)-2-metoxiimidazo[2,1-b][1,3,4]tiadiazol

102A. 2-(4-(isopropilsulfonil)piperazin-1-il)tiazol-4-carboxilato de metilo

El compuesto se preparó de acuerdo con el procedimiento descrito en el ejemplo 100A. El producto en bruto se purificó en ISCO usando una columna REDlSEP® de 24 g (0 a 55% de EtoAc-DCM) para obtener el producto deseado en forma de un sólido de color blanco (0,080 g, 56 %). LCMS (APCI): calc. para C12H20N3O4S2 [M+H]+ m/z 334,08, encontrado 334,1. RMN 1H (CDCla, 400 MHz) 8 ppm: 7,53 (s, 1H), 3,89-3,94 (m, 3H), 3,64-3,70 (m, 3H), 3,46-3,58 (m, 5H), 3,17-3,27 (m, 1H), 1,37 (d, J = 7,0 Hz, 6 H).
102B. (2-(4-(isopropilsulfonil)piperazin-1-il)tiazol-4-il)metanol

El compuesto se preparó de acuerdo con el procedimiento descrito en el ejemplo 92B. El producto en bruto se purificó en ISCO usando una columna REDISEP® de 4 g (0 a 15 % de MeoH-DCM) para obtener el producto deseado en forma de una espuma de color blanco (0,070 g, 96 %). LCMS (APCI): calc. para C11 H20N3O3S2 [M+H]+ m/z 306,09, encontrado 306,1. RMN 1 H (CDCla, 400 MHz) 8 ppm: 6,49 (s, 1H), 4,61 (s, 1H), 4,53 (s, 1H), 3,81 (a s, 3H), 3,46-3,63 (m, 5H), 3,15-3,29 (m, 1H), 1,61 (a s, 1H), 1,37 (d, J = 6,7 Hz, 6 H).
Ejemplo 102. 6-(4-((2-(4-(isopropilsulfonil)piperazin-1-il)tiazol-4-il)metoxi)-6-metoxibenzofuran-2-il)-2-metoxiimidazo[2,1-b][1,3,4]tiadiazol

El compuesto del título se preparó de acuerdo con el procedimiento descrito en el ejemplo 86. El producto en bruto se purificó en ISCO usando una columna REDISEP® de 4 g (0 a 60 % de EtOAc-DCM) para obtener compuesto del título en forma de un sólido color crema (0,082 g, 71 %). LC (Método C): 2,266 min. HRMS (ESI): calc. para C25H29N6O6S3 [M+H]+ m/z 605,1311, encontrado 605,1327. RMN 1 H (CDCla, 400 MHz) 8 ppm: 8,36 (s, 1H), 6,98 (d, J = 3,5 Hz, 2H), 6,80-6,84 (m, 1H), 6,56 (d, J = 2,0 Hz, 1H), 5,06 (s, 2H), 4,20 (s, 3H), 3,79 (s, 3H), 3,45-3,50 (m, 4H), 3,36-3,39 (m, 4H), 3,29 (s, 1H), 1,23 (d, J = 6,7 Hz, 6 H).
Ejemplo 103
8-(4-(((6-metoxi-2-(2-metoxNmidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2-il)-1,4-dioxa-8-azaespiro[4.5]decano

103A. 2-(1,4-dioxa-8-azaespiro[4.5]decan-8-il)tiazol-4-carboxilato de metilo
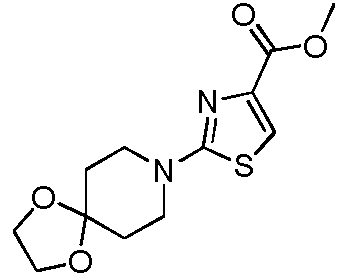
El compuesto se preparó de acuerdo con el procedimiento descrito en el ejemplo 92A. El producto en bruto se purificó en ISCO usando una columna REDISEP® de 24 g (0 a 55 % de EtOAc-DCM) para obtener el producto deseado en forma de un aceite incoloro (0,416 g, 65 %). LCMS (APCI): calc. para C12 H17N2O4S [M+H]+ m/z 285,09, encontrado 285,1. RMN 1H (CDCls, 400 MHz) 8 ppm: 7,46 (s, 1H), 4,00 (s, 4H), 3,89 (s, 3H), 3,64-3,73 (m, 4H), 1,77 1,86 (m, 4H).
103B. (2-(1,4-dioxa-8-azaespiro[4,5]decan-8-il)tiazol-4-il)metanol

El compuesto se preparó de acuerdo con el procedimiento descrito en el ejemplo 92B. El producto en bruto se purificó en ISCO usando una columna REDISEP® de 4 g (0 a 15 % de MeoH-DCM) para obtener el producto deseado en forma de un aceite incoloro (0,124 g, 69 %). LCMS (APCI): calc. para C11 H17N2O3S [M+H]+ m/z 257,10, encontrado 257,1. RMN 1H (CDCls, 400 MHz) 8 ppm: 6,39 (s, 1H), 4,55 (a s, 2H), 3,99-4,02 (m, 5H), 3,65-3,73 (m, 4H), 1,81 -1,88 (m, 4H).
Ejemplo 103. 8-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2-il)-1,4-dioxa-8-azaespiro[4.5]decano

El compuesto del título se preparó de acuerdo con el procedimiento descrito en el ejemplo 86. El producto en bruto se purificó en ISCO usando una REDISEP® de 4 g (0 a 50 % de EtOAc-DCM) para obtener el compuesto del título en forma de un sólido color crema (0,083 g, 38 %). LC (Método C): 2,395 min. Hr MS (ESI): calc. para C25H26N5O6S2 [M+H]+ m/z 556,1325, encontrado 556,1352. RMN 1H (CDCla, 400 MHz) 8 ppm: 7,85 (s, 1H), 7,09 (s, 1H), 6,68-6,73 (m, 1H), 6,60 (s, 1H), 6,46 (d, J = 2,0 Hz, 1H), 5,25 (a s, 2H), 4,21 (s, 3H), 4,01 (s, 4H), 3,83-3,86 (m, 3H), 3,78 (a. s., 3H), 1,84-1,92 (m, 4H).
Ejemplo 104
6-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2-il)-2-oxa-6-azaespiro[3.3]heptano

104A. 2-(2-oxa-6-azaespiro[3.3]heptan-6-il)tiazol-4-carboxilato de metilo
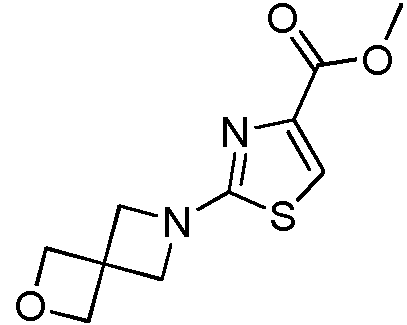
El compuesto se preparó de acuerdo con el procedimiento descrito en el ejemplo 92A. El producto en bruto se purificó en ISCO usando una columna REDISEP® de 24 g (0 a 100% de EtOAc-DCM) para obtener el producto deseado en forma de un sólido de color blanco (0,145 g, 34 %). LCMS (APCI): calc. para C10H13N2O3S [M+H]+ m/z 241,06, encontrado 241,1. RMN 1H (CDCla, 400 MHz) 8 ppm: 7,51 (s, 1H), 4,85 (s, 4H), 4,30 (s, 4H), 3,90 (s, 3H). 104B. (2-(2-oxa-6-azaespiro[3.3]heptan-6-il)tiazol-4-il)metanol

El compuesto se preparó de acuerdo con el procedimiento descrito en el ejemplo 92B. La mezcla de reacción se inactivó con MeOH (10 ml) y se agitó a temperatura ambiente durante 10 min. Después la mezcla se concentró a presión reducida, se disolvió en DCM y se lavó con NaHCO3 acuoso, agua y salmuera, se secó sobre MgSO4 y se evaporó. El producto se aisló en forma de una goma de color blanco (0,053 g, 41 %). RMN 1H (CDCh, 400 MHz) 8 ppm: 6,45-6,47 (m, 1H), 4,85-4,86 (m, 4H), 4,56 (d, J = 6,1 Hz, 2H), 4,24 (s, 4H), 2,11 (t, J = 6,1 Hz, 1H).
Ejemplo 104. 6-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2-il)-2-oxa-6-azaespiro[3.3]heptano
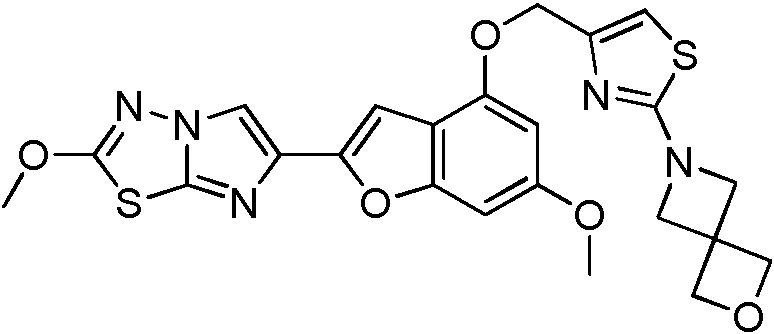
El compuesto del título se preparó de acuerdo con el procedimiento descrito en el ejemplo 86. El producto en bruto se purificó en ISCO usando una columna REDISEP® de 4 g (0 a 100 % de EtOAc-DCM) para obtener el compuesto del título en forma de un sólido de color blanco (0,070 g, 62 %). LC (Método C): 2,081 min. HRMS (ESI): calc. para C23H22N5O5S2 [M+H]+ m/z 512,1062, encontrado 512,1067. RMN 1 H (CDCh, 400 MHz) 8 ppm: 7,85 (s, 1H), 7,09 (s, 1H), 6,70 (s, 1H), 6,63 (s, 1H), 6,41 (d, J = 2,0 Hz, 1H), 5,12 (s, 2H), 4,87 (s, 4H), 4,27 (s, 4H), 4,22 (s, 3H), 3,84 (s, 3H).
Ejemplo 105
4-(5-etil-4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2-il)morfolina

105A. 5-etil-2-morfolinotiazol-4-carboxilato de metilo

El compuesto se preparó de acuerdo con el procedimiento descrito en el ejemplo 92A. El producto en bruto se purificó en ISCO usando una columna REDISEP® de 12 g (0 a 50% de EtOAc-DCM) para obtener el producto deseado en forma de un sólido de color blanco (0,349 g, 68 %). LCMS (APCI): calc. para C11 H17N2O3S [M+H]+ m/z 257,09, encontrado 257,1. RMN 1H (CDCh, 400 MHz) 8 ppm: 3,88 (s, 3H), 3,75 - 3,85 (m, 4H), 3,41 - 3,50 (m, 4H), 3,14 (q, J = 7,6 Hz, 2H), 1,27 (t, J = 7,4 Hz, 3H).
105B. (5-etil-2-morfolinotiazol-4-il)metanol

El compuesto se preparó de acuerdo con el procedimiento descrito en el ejemplo 92B. La mezcla de reacción se inactivó con MeOH (10 ml) y se agitó a temperatura ambiente durante 10 min. Después la mezcla se concentró a presión reducida, se diluyó con DCM, se lavó con NaHCO3, agua y salmuera, se secó sobre MgSO4 y se evaporó. El residuo se purificó en ISCO usando una columna REDISEP® de 12 g (0 a 15 % de MeOH-DCM) para obtener el producto deseado en forma de un sólido de color blanco (0,235 g, 76 %). LCMS (APCI): calc. para C10H17N2O2S [M+H]+ I 229,10, encontrado 229,1. RMN 1H (CDCla, 400 MHz) 8 ppm: 4,48 (d, J = 5,9 Hz, 2H), 3,77-3,85 (m, 4H), 3,37-3,46 (m, 4H), 2,68 (q, J = 7,4 Hz, 2H), 2,25 (t, J = 5,9 Hz, 1H), 1,22 (t, J = 7,4 Hz, 3H).
Ejemplo 105. 4-(5-etil-4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2-il)morfolina

El compuesto del título se preparó de acuerdo con el procedimiento descrito para la síntesis del ejemplo 86. El producto en bruto se purificó en ISCO usando una columna REDISEP® de 4 g (0 a 60 % de EtOAc-DCM), y el sólido obtenido se suspendió en CH3CN, se sometió a ultrasonido, se filtró y se secó para obtener el compuesto del título en forma de un sólido de color blanquecino (0,086 g, 74 %). LC (Método C): 2,263 min. HRMS (ESI): calc. para C24H26N5O5S2 [M+H]+ m/z 528,1375, encontrado 528,1374. RMN 1 H (CDCla, 400 MHz) 8 ppm: 7,83 (s, 1H), 7,04 (s, 1H), 6,69 (dd, J = 2,0, 0,8 Hz, 1H), 6,52 (d, J = 2,0 Hz, 1H), 5,05 (s, 2H), 4,21 (s, 3H), 3,85 (s, 3H), 3,79-3,84 (m, 4H), 3,42-3,47 (m, 4H), 2,78 (q, J = 7,6 Hz, 2H), 1,23 (t, J = 7,6 Hz, 3H).
Ejemplo 106
3-((4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2il)(metil)amino)propanonitrilo

106A. 2-((2-cianoetil)(metil)amino)tiazol-4-carboxilato de metilo

En un tubo sellable, una solución de 2-bromotiazol-4-carboxilato de metilo (0,500 g, 2,252 mmol) y 3-(metilamino)propanonitrilo (0,176 ml, 1,876 mmol) en dioxano (8 ml) se trató con carbonato de cesio (0,611 g, 1,876 mmol), acetato de paladio (II) (0,021 g, 0,094 mmol) y Xantphos (0,065 g, 0,113 mmol). El sistema se purgó con nitrógeno durante 5 min, después el tubo se selló y la mezcla se calentó a 100 °C durante 16 h. La mezcla de reacción enfriada se repartió después entre acetato de etilo y bicarbonato de sodio acuoso saturado. La fase orgánica se lavó con salmuera, se secó sobresobre sulfato de magnesio anhidro y se concentró al vacío. El residuo obtenido se sometió a cromatografía sobre gel de sílice (ISCO, gradiente de elución de metanol en diclorometano) para obtener 0,300 g (59 %) del compuesto del título. LC (Método B): 1,927 min. LCMS (APCI): calc. para C9H12N3O2S [M+H]+ m/z 226,07; encontrado 226,0.
106B. 3-((4-(hidroximetil)tiazol-2-il)(metil)amino)propanonitrilo

Una solución de 2-((2-cianoetil)(metil)amino)tiazol-4-carboxilato de metilo (0,300 g, 1,332 mmol) en etanol (5 ml) a 0 °C se trató con borohidruro de sodio (0,151 g, 4,00 mmol), seguido de cloruro de calcio (0,177 g, 1,598 mmol) y después se agitó a temperatura ambiente durante 4 h. La mezcla de reacción se repartió entre acetato de etilo y bicarbonato de sodio acuoso saturado, la fase orgánica se separó, se lavó con salmuera, se secó sobre sulfato de magnesio anhidro y se concentró al vacío. El residuo obtenido se sometió a cromatografía sobre gel de sílice (ISCO, gradiente de elución de acetato de etilo en diclorometano) para obtener 0,048 g (18 %) del material del título en forma de un aceite que cristalizó en reposo para obtener un sólido de color blanco. LC (Método B): 1,376 min. LCMS (APCI): calc. para C8H12N3OS [M+H]+ m/z 198,07; encontrado 198,0. RMN 1H (400 MHz, CDCla) 8 ppm: 6,43 (s, 1H), 4,52 (s, 2H), 3,80 (t, J= 6,55 Hz, 2H), 3,18 ( as, 1H), 3,15 (s, 3H), 2,73 (t, J= 6,55 Hz, 2H).
Ejemplo 106.
3-((4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2-il)(metil)amino)propanonitrilo

Una mezcla de 6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-ol (Ejemplo 1H, 0,064 g, 0,203 mmol) y 3-((4-(hidroximetil)tiazol-2-il)(metil)-amino)propanonitrilo (0,048 g, 0,243 mmol) en THF seco (3,5 ml) en atmósfera de nitrógeno se trató a 22 °C con tri-n-butilfosfina (0,132 ml, 0,507 mmol), seguido de una solución de 1,1'-(azodicarbonil)dipiperidina (0,129 g, 0,507 mmol) en THF seco (2,5 ml), añadida gota a gota (mediante una bomba de jeringa) durante 1 h. La suspensión de color beis resultante se agitó durante 1 h más y después se repartió entre acetato de etilo y bicarbonato de sodio acuoso saturado. La fase orgánica se separó, se lavó con salmuera, se secó sobre sulfato de magnesio anhidro y se concentró al vacío. El residuo obtenido se sometió a cromatografía sobre gel de sílice (ISCO, gradiente de elución de acetato de etilo en diclorometano) para obtener 0,085 g (85 %) del compuesto del título en forma de un sólido de color blanco. LC (Método A): 2,179 min. HRMS
(ESI): calc. para C22H21N6O4S2 [M+H]+ m/z 497,1066; encontrado: 497,1113. RMN 1 H (400 MHz, CDCI3) 5 ppm: 7,85 (s, 1H), 7,09 (s, 1H), 6,68-6,72 (m, 1H), 6,62 (a s, 1H), 6,46 (d, J= 1,96 Hz, 1H), 5,11 (s, 2H), 4,21 (s, 3H), 3,82-3,89 (m, 5H), 3,20 (s, 3H), 2,80 (t, J= 6,46 Hz, 2H).
Ejemplo 107
(S)-2-metoxi-6-(6-metoxi-4-((2-(2-(metoximetil)pirrolidin-1-il)tiazol-4-il)metoxi)benzofuran-2-il)imidazo[2,1-b][1,3,4]tiadiazol

107A. (S)-4-(((ferc-butildimetilsilil)oxi)metil)-2-(2-(metoximetil)pirrolidin-1-il)tiazol

Una solución de 2-bromo-4-(((ferc-butildimetilsilil)oxi)metil)tiazol (Ejemplo 37B, 0,500 g, 1,622 mmol) y (S)-2-(metoximetil)pirrolidina (0,224 g, 1,946 mmol) en 1,4-dioxano (5 ml) se trató con trietilamina (0,678 ml, 4,87 mmol) y la mezcla resultante se calentó a 100 °C durante 18 h. La mezcla de reacción enfriada se concentró al vacío y el residuo se sometió a cromatografía sobre gel de sílice (ISCO, gradiente de elución de acetato de etilo en diclorometano) para obtener 0,175 g (31 %) del material del título. LC (Método B): 2,685 min. LCMS (APCI): calc. para C-,6H31N2O2SSi [M+H]+ m/z 343,19; encontrado 343,2. RMN 1H (400 MHz, CDCh) 5 ppm: 6,35 (s, 1H), 4,68 (s, 2H), 4,02 (dt, J= 6,65, 3,33 Hz, 1H), 3,61 (dd, J= 9,39, 3,4 Hz, 1H), 3,40-3,56 (m, 2H), 3,28-3,39 (m, 4H), 1,91-2,16 (m, 4H), 0,95 (s, 9H), 0,12 (s, 6H).
107B. (S)-(2-(2-(metoximetil)pirrolidin-1-il)tiazol-4-il)metanol

El compuesto del título se preparó de acuerdo con el procedimiento de desprotección descrito en el ejemplo 93C. LC (Método B): 1,431 min. LCMS (APCI): calc. para C10H17N2O2S [M+H]+ m/z 229,10; encontrado 229,2. RMN 1H (400 MHz, CDCl3) 5 ppm: 6,33 (s, 1H), 4,54 (s, 2H), 3,96-4,08 (m, 1H), 3,38-3,63 (m, 4H), 3,27-3,37 (m, 4H), 1,92-2,19 (m, 4H).
Ejemplo 107. (S)-2-metoxi-6-(6-metoxi-4-((2-(2-(metoximetil)pirrolidin-1-il)tiazol-4-il)metoxi)benzofuran-2-il)imidazo[2,1-b][1,3,4]tiadiazol

El compuesto del título se preparó de acuerdo con el procedimiento general de acoplamiento descrito en el ejemplo 106. LC (Método A): 2,090 min. HRMS (ESI): calc. para C24H26N5O5S2 [M+H]+ m/z 528,1375; encontrado 528,1352. RMN 1H (400 MHz, CDCh) 5 ppm: 7,83 (s, 1H), 7,10 (s, 1H), 6,69 (s, 1H), 6,53 (s, 1H), 6,45 (s, 1H), 5,13 (s, 2H), 4,19 (s, 3H), 4,05 (a s, 1H), 3,84 (s, 3H), 3,58-3,66 (m, 1H), 3,43-3,58 (m, 2H), 3,31-3,43 (m, 4H), 1,94-2,19 (m, 4H). Ejemplo 108
N-(4-bromo-2-metilfenil)-4-(((6-metoxi-2-(2-metoxümidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2-amina

108A. 2-((4-bromo-2-metilfenil)(terc-butoxicarbonil)amino)tiazol-4-carboxilato de etilo

Una suspensión de 2-((4-bromo-2-metilfenil)amino)tiazol-4-carboxilato de etilo (0,083 g, 0,243 mmol; obtenida mediante la condensación de 1-(4-bromo-2-metilfenil)tiourea con bromopiruvato de etilo) en THF (2 ml) se trató, en atmósfera de nitrógeno, con dicarbonato de di-terc-butilo (0,169 ml, 0,730 mmol), DmAp (0,015 g, 0,122 mmol) y trietilamina (0,102 ml, 0,730 mmol). La mezcla se agitó durante 3 h a temperatura ambiente antes de que se concentrara a presión reducida. El residuo sólido obtenido se purificó en ISCO usando una columna REDISEP® Gold de 12 g (elución con hexanos-EtOAc) para obtener el material del título (0,080 g, 74,5 %) en forma de un sólido. LC (Método F): 2,341 min. LCMS (APCI) calc. para C1sH22BrN2O4S [M+H]+ m/z 441,05, encontrado 441,2. RMN 1H (400 MHz, CDCla) 8 ppm: 7,82 (s, 1 H), 7,45 (d, J= 2,0 Hz, 1H), 7,40 (dd, J= 8,2, 2,3 Hz, 1H), 7,06 (d, J= 8,2 Hz, 1H), 4,28 (q, J= 7,2 Hz, 2H), 2,09 (s, 3H), 1,44 (s, 9H), 1,32 (t, J= 7,0 Hz, 3 ).
108B. (4-bromo-2-metilfenil)(4-(hidroximetil)tiazol-2-il)carbamato de terc-butilo

Una solución enfriada en hielo de 2-((4-bromo-2-metilfenil)(terc-butoxicarbonil)amino)-tiazol-4-carboxilato de etilo (0,080 g, 0,181 mmol) en THF (3 ml), en atmósfera de nitrógeno, se trató con NaBH4 (0,0274 g, 0,725 mmol) y metanol (0,147 ml, 3,63 mmol). Después de 30 min, se retiró el baño de hielo y la reacción se agitó a temperatura ambiente durante 5,5 h. En ese momento, se agregó más NaBH4 (0,013 g) y metanol (0,3 ml) y la agitación continuó durante 30 min. La solución turbia resultante se enfrió en un baño de hielo y se inactivó con ácido acético (0,5 ml). La mezcla de reacción se diluyó con acetato de etilo (40 ml), se lavó con bicarbonato de sodio acuoso saturado (2 x 20 ml) y salmuera (20 ml) y después se secó sobre sulfato de magnesio anhidro. La evaporación del disolvente produjo un residuo esponjoso que se purificó en ISCO usando una columna REDISEP® Gold de 4 g (elución con hexanos-EtOAc) para obtener el material del título (0,064 g, 88 %). LC (Método F): 2,216 min. LCMS (APCI): calc. para C^^cjBr^OaS [M+H]+ m/z 399,04, encontrado 399,0. RMN 1H (400 MHz, CDCla) 8 ppm: 7,45 (d, J= 2,3 Hz, 1H), 7,40 (dd, J= 8,4, 2,2 Hz, 1H), 7,02 (d, J= 8 ,6 Hz, 1H), 6,81 (s, 1H), 4,49 (d, J= 6,3 Hz, 2H), 2,09 (s, 3H), 1,96 (t, J= 6,3 Hz, 1H), 1,44 (s, 9H).
108C. (4-bromo-2-metilfenil)(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2 -il)carbamato de terc-butilo

A 6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-ol sólido (Ejemplo 1H, 0,045 g, 0,142 mmol), se le agregó, a temperatura ambiente en atmósfera de nitrógeno, (4-bromo-2-metilfenil)(4-(hidroximetil)tiazol-2-il)carbamato de ferc-butilo (0,062 g, 0,156 mmol) y tri-n-butilfosfina (0,175 ml, 0,709 mmol) y la mezcla se bombeó en alto vacío durante 20 min. Después se agregó THF anhidro (3 ml), seguido de la adición gota a gota una solución de 1,1'-(azodicarbonil)dipiperidina (0,089 g, 0,355 mmol) en THF (3 ml) durante 20 min. La mezcla se agitó a temperatura ambiente durante 3 h más antes de diluirla con diclorometano (75 ml), se lavó con NaHCO3 acuoso saturado (2 x 20 ml), agua (20 ml) y salmuera (20 ml) y finalmente se secó (MgSO4). La evaporación del disolvente produjo un semisólido que se purificó en ISCO usando una columna REDISEP® Gold de 24 g (elución con hexanos-EtOAc) para obtener un sólido que se siguió triturando con acetonitrilo (1 ml) y se liofilizó para obtener el compuesto del título (0,088 g, 89 %) en forma de un sólido. LC (Método F): 2,630 min. HRMS (ESI): calc. para C3üH29BrN5O6S2 [M+H]+ m/z 698,0743, encontrado 698,0753. RMN 1H (400 MHz, DMSO-da) 8 ppm: 8,36 (s, 1H), 7,59 (d, J= 2,0 Hz, 1H), 7,48 (dd, J= 8,4, 2,2 Hz, 1H), 7,40 (s, 1H), 7,25 (d, J= 8,2 Hz, 1H), 6,89- 6,94 (m, 1H), 6,79 (s, 1H), 6,48 (d, J= 1,6 Hz, 1H), 5,02 (s, 2H), 4,21 (s, 3H), 3,77 (s, 3H), 2,00 (s, 3H), 1,37 (s, 9H).
Ejemplo 108. N-(4-bromo-2-metilfenil)-4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2-amina
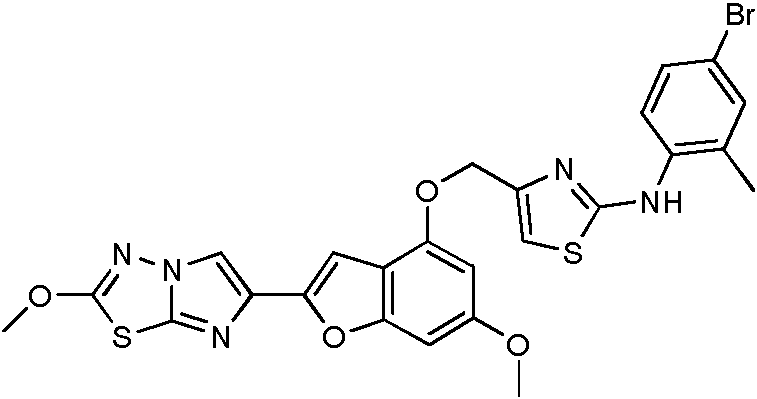
A una solución de (4-bromo-2-metilfenil)(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2-il)carbamato de ferc-butilo (0,030 g, 0,043 mmol) en diclorometano (4 ml), se le agregó una solución de TFA al 95 % (0,5 ml) en agua. La mezcla se agitó a temperatura ambiente durante 3 h, después se agregó tolueno (5 ml) y la mezcla se concentró. Se agregó tolueno (5 ml) al concentrado y los volátiles se evaporaron para obtener un sólido, que posteriormente se trituró con acetonitrilo (1 ml). La mezcla se filtró y el sólido obtenido se liofilizó de MeCN-agua para obtener el compuesto del título (0,025 g, 97 %) en forma de un polvo de color blanco. LC (Método F): 2,535 min. HRMS (ESI): calc. para C25H2-iBrN5O4S2 [M+H]+ m/z 598,0218, encontrado 598,0214. RMN 1H (400 MHz, DMSO-d6) 8 ppm: 9,37 (s, 1H), 8,37 (s, 1H), 7,96 (d, J= 8,6 Hz, 1H), 7,40 (d, J= 2,0 Hz, H), 7,33 (dd, J= 8,6, 2,3 Hz, 1H), 6,98 (d, J= 7,4 Hz, 2H), 6,83 (d, J= 1,2 Hz, 1H), 6,62 (d, J= 2,0 Hz, 1H), 5,11 (s, 2H), 4,20 (s, 3H), 3,80 (s, 3H), 2,26 (s, 3H).
Ejemplo 109
4-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2-il)piperidin-1-carboxilato de ferc-butilo

109A. 2-(1-(ferc-butoxicarbonil)piperidin-4-il)tiazol-4-carboxilato de etilo

A una suspensión de 4-carbamotioilpiperidin-1-carboxilato de ferc-butilo (1,50 g, 6,14 mmol) en etanol (6 ml) a 0 °C, se le agregó gota a gota una solución de 3-bromo-2-oxopropanoato de etilo (0,788 ml, 6,26 mmol) en etanol (6 ml). Después se retiró el baño de hielo y la mezcla de reacción se agitó a temperatura ambiente durante una noche. Después se agregó trietilamina (1,5 ml, 10,76 mmol) y la mezcla se concentró prácticamente a sequedad y el concentrado se diluyó con acetato de etilo, se lavó con salmuera, se secó (MgSO4) y se evaporó a sequedad. El residuo se purificó mediante cromatografía ultrarrápida usando hexanos-acetato de etilo como eluyente para obtener 2-(1-(ferc-butoxicarbonil)piperidin-4-il)tiazol-4-carboxilato de etilo (1,55 g, 74,2 %) en forma de un aceite prácticamente incoloro que cristalizó en reposo para obtener un sólido de color blanco. LC (Método A): 2,115 min. RMN 1 H (DMSO-d6, 400 MHz) 8 ppm: 8,42 (s, 1H), 7,20 (a s, 2H), 4,29 (q, J = 7,0 Hz, 2H), 4,00 (m, 1H), 3,24 (m, 1H), 2,88 (a s, 1H), 2,03 (m, 2H), 1,54 (m, 2H), 1,29 (t, J = 7,2 Hz, 3H).
109B. 4-(4-(hidroximetil)tiazol-2-il)piperidin-1 -carboxilato de ferc-butilo

A una solución en agitación de 2-(1-(ferc-butoxicarbonil)piperidin-4-il)tiazol-4-carboxilato de etilo (1,430 g, 4,20 mmol) en THF (21 ml) a temperatura ambiente, se le agregó borohidruro de litio (0,183 g, 8,40 mmol), seguido de MeOH (0,340 ml, 8,40 mmol). La mezcla resultante se agitó a temperatura ambiente durante 16 h antes de inactivarla con NH4Cl acuoso saturado y se extrajo con EtOAc. La fase orgánica se separó, se secó (MgSO4), se filtró y se concentró a sequedad. El residuo se purificó mediante cromatografía ultrarrápida usando DCM-EtOAc como eluyente para obtener el producto en forma de un aceite transparente e incoloro. Este aceite se absorbió en acetonitrilo-agua y se liofilizó para obtener 4-(4-(hidroximetil)tiazol-2-il)piperidin-1-carboxilato de ferc-butilo (0,996 g, 79 %) en forma de un sólido de color blanco. LC (Método A): 1,875 min. RMN 1 H (DMSO-d6, 400 MHz) 8 ppm: 7,27 (m, 1H), 5,26 (t, J = 5,2 Hz, 1H), 4,52 (d, J = 4,7 Hz, 2H), 3,99 (d, J = 11,3 Hz, 2H), 3,15 (m, 1H), 2,87 (a s, 2H), 2,00 (m, 2H), 1,51 (m, 2H), 1,40 (s, 9H).
Ejemplo 109. 4-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2-il)piperidin-1 -carboxilato de ferc-butilo

A una suspensión de 6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-ol (Ejemplo 1H, 0,339 g, 1,069 mmol) y 4-(4-(hidroximetil)tiazol-2-il)piperidin-1 -carboxilato de ferc-butilo (0,319 g, 1,069 mmol) en THF seco (8 ml), se le agregó tri-n-butilfosfina (0,694 ml, 2,67 mmol), seguido de una solución de ADDP (0,674 g, 2,67 mmol) en THF (2 ml) añadida gota a gota durante 30 min mediante una bomba de jeringa. Después de agitar durante 30 min más, la mezcla de reacción se repartió entre EtOAc y NaHCO3 acuoso saturado. La fase orgánica se separó, se lavó con salmuera, se secó (MgSO4), se filtró y se concentró a sequedad. El residuo se purificó mediante cromatografía ultrarrápida usando DCM-EtOAc como eluyente para obtener el compuesto del título (0,432 g, 67,6 %) en forma de un sólido de color blanco. LC (Método A): 2,479 min. HRMS (ESI): calc. para C28H32N5O6S2 [M+H]+ m/z 598,1794, encontrado 598,1806. RMN 1 H (DMSO-d6, 400 MHz) 8 ppm: 8,37 (s, 1H), 7,70 (s, 1H), 6,98 (d, J = 0,8 Hz, 1H), 6,83 (dd, J = 0,8, 1,6 Hz, 1H), 6,61 (d, J = 2,0 Hz, 1H), 5,26 (s, 2H), 4,20 (s, 3H), 4,00 (m, 2H), 3,80 (s, 3H), 3,22 (m, 1H), 2,90 (a s, 1H), 2,04 (m, 2H), 1,55 (m, 2H), 1,40 (s, 9H).
Ejemplo 110. (4-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2-il)piperidin-1 -il)(fenil)metanona

A una suspensión en agitación de 4-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4
il)oxi)metil)tiazol-2-il)piperidin-1-carboxilato de ferc-butilo (0,406 g, 0,679 mmol) en DCM (8 ml), se le agregó TFA (1 ml) y la mezcla se agitó a temperatura ambiente durante 4 h antes de que se concentrara a sequedad. El residuo se repartió entre EtOAc-NaHCO3 acuoso saturado y la fase orgánica se separó, se secó con MgSO4, se filtró y se concentró a sequedad para obtener 2-metoxi-6-(6-metoxi-4-((2-(piperidin-4-il)tiazol-4-il)metoxi)benzofuran-2-il)imidazo[2,1-b][1,3,4]tiadiazol (sal de TFA, 0,415 g, 100 %), en forma de un sólido de color beis. LC (Método A): 1,990 min. LCMS (APCI): calc. para C23H24N5O4S2 [M+H]+ m/z 498,13, encontrado 498,20.
A una solución en agitación de 2-metoxi-6-(6-metoxi-4-((2-(piperidin-4-il)tiazol-4-il)metoxi)benzofuran-2-il)imidazo[2,1-b][1,3,4]tiadiazol (0,025 g, 0,050 mmol) en DMF (1 ml), se le agregó DIEA (0,044 ml, 0,250 mmol) y ácido benzoico (0,0067 g, 0,055 mmol), seguido de HATU (0,021 g, 0,055 mmol). La mezcla de reacción se agitó durante 1 h antes de diluirla con DMF (1 ml) y someterla directamente a purificación mediante HPLC preparativa (Método A). Las fracciones que contenían el producto se concentraron a sequedad y el residuo se liofilizó a partir de MeCN-agua para obtener el compuesto del título (0,012 g, 39,9 %) en forma de un sólido amorfo de color blanco. LC (Método A): 2,328 min. HRMS (ESI): calc. para C30H28N5O5S2 [M+H]+ m/z 602,1532, encontrado 602,1532. RMN 1 H (DMSO-da, 400 MHz) 8 ppm: 8,37 (s, 1H), 7,72 (s, 1H), 7,46-7,39 (m, 5H), 6,98 (d, J = 0,8 Hz, 1H), 6,83 (dd, J = 0,8, 1,6 Hz, 1H), 6,61 (d, J = 2,0 Hz, 1H), 5,26 (s, 2H), 4,52 (a s, 1H), 4,20 (s, 3H), 3,80 (s, 3H), 3,65 (a s, 1H), 3,08 (m, 3H), 2,10 (m, 2H), 1,69 (m, 2H).
Ejemplo 111
6-(4-((2-(1-(isopropilsulfonil)piperidin-4-il)tiazol-4-il)metoxi)-6-metoxibenzofuran-2-il)-2-metoxiimidazo[2,1-b][1,3,4]tiadiazol

A una solución en agitación de 2-metoxi-6-(6-metoxi-4-((2-(piperidin-4-il)tiazol-4-il)metoxi)benzofuran-2-il)imidazo[2,1-b][1,3,4]tiadiazol (0,025 g, 0,050 mmol) en DMF (1 ml), se le agregó DIEA (0,044 ml, 0,250 mmol) y cloruro de propan-2-sulfonilo (5,61 pl, 0,050 mmol). La mezcla de reacción se agitó a temperatura ambiente durante 1 h antes de diluirla con DMF (1 ml) y purificarla mediante HPLC preparativa (Método A). Las fracciones que contenían el producto se concentraron a sequedad y el residuo se liofilizó a partir de MeCN-agua para obtener el compuesto del título (0,007 g, 0,012 mmol, 23,19 %) en forma de un sólido amorfo de color blanco. LC (Método A): 2,283 min. HRMS (ESI): calc. para C26H30N5O6S3 [M+H]+ m/z 604,1358, encontrado 604,1373. RMN 1 H (DMSO-d6, 400 MHz) 8 ppm: 8,37 (s, 1H), 7,72 (s, 1H), 6,98 (d, J = 0,8 Hz, 1H), 6,83 (dd, J = 0,8, 2,0 Hz, 1H), 6,61 (d, J = 2,0 Hz, 1H), 5,27 (s, 2H), 4,20 (s, 3H), 3,81 (s, 3H), 3,72 (m, 2H), 3,24 (m, 2H), 3,06 (m, 2H), 2,12 (m, 2H), 1,67 (m, 2H), 1,22 (d, J = 7,0 Hz, 6 H).
Ejemplo 112
2-metoxi-6-(6-metoxi-4-((2-(1-(metilsulfonil)piperidin-4-il)tiazol-4-il)metoxi)benzofuran-2-il)imidazo[2,1-b][1,3,4]tiadiazol

El compuesto del título se preparó de acuerdo con el método descrito anteriormente en el ejemplo 111 y se aisló en forma de un sólido. LC (Método A): 2,172 min. HRMS (ESI): calc. para C24H26N5O6S3 [M+H]+ m/z 576,1040, encontrado 576,1041. RMN 1 H (DMSO-d6, 400 MHz) 8 ppm: 8,37 (s, 1H), 7,73 (s, 1H), 6,99 (s, 1H), 6,84 (dd, J = 0,8, 1,6 Hz, 1H), 6,61 (d, J = 2,0 Hz, 1H), 5,27 (s, 2H), 4,20 (s, 3H), 3,81 (s, 3H), 3,64 (m, 2H), 3,18 (m, 1H), 2,90 (m, 2H), 2,89 (s, 3H), 2,18 (m, 2H), 1,75 (m, 2H).
Ejemplo 113
2-metoxi-6-(6-metoxi-4-((2-(1-(fenilsulfonil)piperidin-4-il)tiazol-4-il)metoxi)benzofuran-2-il)imidazo[2,1-b][1,3,4]tiadiazol

El compuesto del título se preparó de acuerdo con el método descrito anteriormente en el ejemplo 111 y se aisló en forma de un sólido. LC (Método A): 2,014 min. HRMS (ESI): calc. para C29H28N5O6S3 [M+H]+ m/z 638,1202, encontrado 638,1422. RMN 1 H (DMSO-d6, 400 MHz) 6 ppm: 8,37 (s, 1H), 7,79 - 7,64 (m, 6 H), 6,97 (d, J = 0,8Hz, 1H), 6,83 (dd, J = 0,8, 1,6 Hz, 1H), 6,59 (d, J = 2,0 Hz, 1H), 5,23 (s, 2H), 4,20 (s, 3H), 3,80 (s, 3H), 3,71 (m, 2H), 3,06 (m, 1H), 2,45 (m, 2H), 2,13 (m, 2H), 1,72 (m, 2H).
Ejemplo 114
2-metoxi-6-(6-metoxi-4-((2-(1-((trifluorometil)sulfonil)piperidin-4-il)tiazol-4-il)metoxi)benzofuran-2-il)imidazo[2,1-b][1,3,4]tiadiazol

El compuesto del título se preparó de acuerdo con el método descrito anteriormente en el ejemplo 111 y se aisló en forma de un sólido. LC (Método A): 2,403 min. HRMS (ESI): calc. para C24H23F3N5O6S3 [M+H]+ m/z 630,0763, encontrado 630,0821. RMN 1 H (DMSO-d6, 400 MHz) 6 ppm: 8,37 (s, 1H), 7,74 (s, 1H), 6,98 (d, J = 0,8 Hz, 1H), 6,84 (dd, J = 0,8, 1,6 Hz, 1H), 6,61 (d, J = 1,6 Hz, 1H), 5,27 (s, 2H), 4,20 (s, 3H), 3,89 (m, 2H), 3,80 (s, 3H), 2,23 (m, 2H), 1,74 (m, 2H).
Ejemplo 115
6-(4-((2-(4,4-difluorociclohexil)tiazol-4-il)metoxi)-6-metoxibenzofuran-2-il)-2-metoxiimidazo[2,1-b][1,3,4]tiadiazol
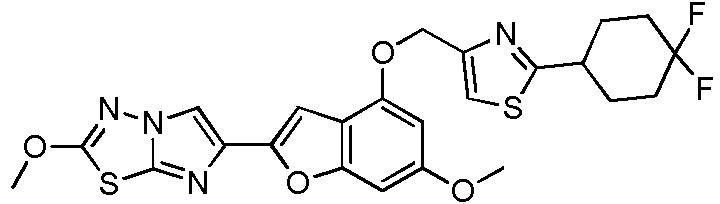
115A. 4,4-difluorociclohexancarboxamida

A una solución en agitación de ácido 4,4-difluorociclohexancarboxílico (1,50 g, 9,14 mmol) en DCM (22 ml), se le agregó cloruro de oxalilo (1,600 ml, 18,28 mmol) y la mezcla de reacción se agitó durante 1 h a temperatura ambiente antes de que se evaporara a sequedad. El residuo se absorbió en THF seco (4,5 ml) y se agregó con agitación a amoníaco acuoso helado concentrado enfriado en hielo (22 ml). La mezcla se agitó a 0 °C durante 2 min y después a temperatura ambiente durante 30 min antes de diluirla con agua y extraerla con EtOAc. La fase orgánica se secó (MgSO4), se filtró y se concentró a sequedad para obtener 4,4-difluorociclohexancarboxamida (1 , 0 2 g, 68,4 %) en forma de un sólido de color blanco. Este material se usó tal cual en la siguiente etapa. LC (Método F): 2,567 min. RMN 1 H (DMSO-d6, 400 MHz) 6 ppm: 7,29 (a s, 1H), 6,79 (a s, 1H), 2,26-2,18 (m, 1H), 2,07 1,97 (m, 2H), 1,86-1,70 (m, 4H), 1,62-1,51 (m, 2H).
115B. 4,4-difluorociclohexancarbotioamida

A una solución de 4,4-difluorociclohexancarboxamida (1,00 g, 6,13 mmol) en THF (10 ml), se le agregó reactivo de Lawesson (1,239 g, 3,06 mmol) y la mezcla se calentó en un recipiente sellado a 65 °C durante 6 h. La mezcla
enfriada se repartió entre EtOAc-NaHCO3 acuoso saturado y la fase orgánica se lavó con salmuera, se secó (MgSO4), se filtró y se concentró a sequedad. El residuo se purificó mediante cromatografía ultrarrápida usando hexanos-EtOAc como eluyente para obtener 4,4-difluorociclohexan-carbotioamida (0,634 g, 3,54 mmol, 57,7 % ) en forma de un sólido de color blanco. LC (Método F): 1,432 min. RMN 1H (DMSO-d6, 400 MHz) 8 ppm: 9,42 (a s, 1h ), 9,15 (a s, 1H), 2,63 (m, 1H), 2,10 -2,01 (m, 2H), 1,90- 1,80 (m, 1H), 1,79 - 1,72 (m, 5H).
115C. 2-(4,4-difluorociclohexil)tiazol-4-carboxilato de etilo

Un recipiente sellable se cargó con 4,4-difluorociclohexancarbotioamida (0,600 g, 3,35 mmol), bromopiruvato de etilo (0,505 ml, 4,02 mmol) e i-PrOH (15 ml) y la mezcla se calentó a 85 °C durante 3 h. La mezcla enfriada se repartió entre EtOAc-NaHCO3 acuoso saturado y la fase orgánica se secó (MgSO4), se filtró y se concentró a sequedad. El residuo se purificó mediante cromatografía en columna usando hexanos-EtOAc como eluyente para obtener 2-(4,4-difluorociclohexil)tiazol-4-carboxilato de etilo (0,600 g, 2,179 mmol, 65,1 %) en forma de un sólido de color blanco. LC (Método F): 2,089 min. RMN 1H (DMSO-d6, 400 MHz) 8 ppm: 8,43 (s, 1H), 4,29 (q, J = 7,0 Hz, 2H), 3,27 (m, 1H), 2,20-1,91 (m, 6 H), 1,75 (m, 2H), 1,30 (t, J = 7,2 Hz, 3H).
115D. (2-(4,4-difluorociclohexil)tiazol-4-il)metanol

A una solución enfriada en hielo de 2-(4,4-difluorociclohexil)tiazol-4-carboxilato de etilo (0,580 g, 2,107 mmol) en THF (11 ml), se le agregó LiBH4 (0,092 g, 4,20 mmol) de una vez, seguido de MeOH (0,170 ml, 4,20 mmol). La mezcla resultante se agitó a 0 °C durante 5 min y después a temperatura ambiente durante 4 h. La mezcla se enfrió en un baño de hielo, se inactivó mediante la adición gota a gota de NH4Cl acuoso saturado y se extrajo con acetato de etilo. La fase orgánica se lavó después con salmuera, se secó (MgSO4), se filtró y se concentró a sequedad. El residuo se purificó mediante cromatografía ultrarrápida usando un gradiente de 0 a 100 % de EtOAc en hexanos para obtener (2-(4,4-difluorociclohexil)tiazol-4-il)metanol (0,398 g, 81 %) en forma de un aceite transparente e incoloro. LC (Método F): 1,776 min. RMN 1H (DMSO-d6, 400 MHz) 8 ppm: 7,28 (m, 1H), 5,26 (a s, 1H), 4,52 (s, 2H), 3,17 (m, 1H), 2,14 -1,90 (m, 6 H), 1,78 - 1,67 (m, 2H).
Ejemplo 115. 6-(4-((2-(4,4-difluorociclohexil)tiazol-4-il)metoxi)-6-metoxibenzofuran-2-il)-2-metoxiimidazo[2,1-b][1,3,4]tiadiazol

El compuesto del título se preparó de acuerdo con el método descrito en el ejemplo 109 y se aisló en forma de un sólido. LC (Método F): 2,556 min. HRMS (ESI): calc. para C24H23FN4O4S2 [M+H]+ m/z 533,1145, encontrado 533,1161. RMN 1H (DMSO-d6, 400 MHz) 8 ppm: 8,37 (s, 1H), 7,71 (s, 1H), 6,98 (m, 1H), 6,61 (m, 1H), 5,28 (m, 2H), 4,20 (s, 3H), 3,80 (s, 3H), 3,24 (m, 1H), 2,18-1,92 (m, 6 H), 1,76 (m, 2H).
Ejemplo 116
5-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2-il)isoxazol

116A. 2-(isoxazol-5-il)tiazol-4-carboxilato de etilo

A una suspensión de isoxazol-5-carbotioamida (0,500 g, 3,90 mmol) en etanol (10 ml), se le agregó 3-bromo-2-oxopropanoato de etilo (0,598 ml, 4,29 mmol) y la mezcla resultante se calentó a 90 °C durante 1,5 h. La mezcla de reacción enfriada se evaporó a sequedad y el residuo se repartió después entre acetato de etilo y bicarbonato de sodio acuoso saturado. La fase orgánica se separó, se lavó con salmuera, se secó sobre sulfato de magnesio anhidro y se concentró al vacío. El residuo obtenido se sometió a cromatografía sobre gel de sílice (ISCO, gradiente de elución de acetato de etilo en diclorometano) para obtener 0,800 g (92 %) del material del título en forma de un sólido de color rojizo. LC (Método B): 2,167 min. Lc MS (ESI): calc. para C9H9N2O3S [M+H]+ m/z 225,03; encontrado: 225,0. RMN 1H (400 MHz, CDCls) 8 ppm: 8,36-8,42 (m, 1H), 8,28-8,34 (m, 1H), 6,94-7,10 (m, 1H), 4,35-4,53 (m, 2H), 1,31-1,48 (m, 3H).
116B. (2-(isoxazol-5-il)tiazol-4-il)metanol

Una solución de 2-(isoxazol-5-il)tiazol-4-carboxilato de etilo (0,087 g, 0,388 mmol) en THF (2 ml) a 0 °C se trató con metanol (0,056 ml, 1,395 mmol) y después con borohidruro de litio (0,030 g, 1,395 mmol). Después de 15 min, se retiró el baño de hielo y la mezcla de reacción se agitó a 22 °C durante 3 h. La mezcla de reacción se inactivó después con NH4Cl acuoso saturado y se diluyó con diclorometano. La fase acuosa se separó y se volvió a extraer (x3) con diclorometano, el extracto orgánico combinado se secó sobre MgSO4 y la evaporación a presión reducida produjo 0,039 g (56 %) del material del título que se usó tal cual en la siguiente etapa. LC (Método B): 1,818 min. LCMS (APCI): calc. para C7H7N2O2S [M+H]+ m/z 183,02; encontrado: 183,0. RMN 1H (400 MHz, CDCls) 8 ppm: 8,36 (d, J= 1,8 Hz, 1H), 7,41 (s, 1H), 6,87 (d, J= 1,8 Hz, 1H), 4,87 (d, J= 5,4 Hz, 2H), 3,00 (t, J= 5,4 Hz, 1H).
Ejemplo 116. 5-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2-il)isoxazol
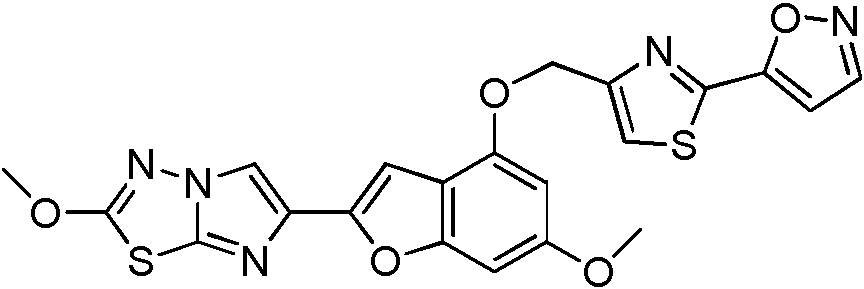
Una mezcla de 6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-ol (Ejemplo 1H, 0,032 g, 0,100 mmol) y (2-(isoxazol-5-il)tiazol-4-il)metanol (0,020 g, 0,110 mmol) en THF seco (2,5 ml), en atmósfera de nitrógeno se trató a 22 °C con tri-n-butilfosfina (0,065 ml, 0,249 mmol), seguido de una solución de 1,1'-(azodicarbonil)dipiperidina (0,064 g, 0,249 mmol) en THF seco (2,5 ml), añadido gota a gota (mediante una bomba de jeringa) durante 1 h. La suspensión de color beis resultante se agitó durante 1 h más a temperatura ambiente y después se repartió entre acetato de etilo y bicarbonato de sodio acuoso saturado. La fase orgánica se separó, se lavó con salmuera, se secó sobre sulfato de magnesio anhidro y se concentró al vacío. El residuo obtenido se sometió a cromatografía sobre gel de sílice (ISCO, gradiente de elución de acetato de etilo en diclorometano) y el material obtenido se trituró con metanol para obtener (después de la filtración y del secado al vacío) 0,013 g (27 %) del compuesto del título. LC (Método A): 2,289 min. Hr Ms (ESI): calc. para C21H16N5O5S2 [M+H]+ m/z 482,0593; encontrado: 482,0602. RMN 1H (400 MHz, CDCla) 8 ppm: 8,38 (d, J= 1,96 Hz, 1H), 7,86 (s, 1H), 7,58 (s, 1H), 7,10 (s, 1H), 6,89 (d, J= 1,96 Hz, 1H), 6,69 - 6,76 (m, 1H), 6,43 (d, J= 1,57 Hz, 1H), 5,37-5,45 (m, 2H), 4,21 (s, 3H), 3,85 (s, 3H).
Ejemplo 117
5-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2-il)oxazol

117A. 2-(oxazol-5-il)tiazol-4-carboxilato de etilo

Se hizo reaccionar oxazol-5-carbotioamida (0,390 g, 3,04 mmol) con 3-bromo-2-oxopropanoato de etilo (0,263 ml, 1,884 mmol), como se describe en el ejemplo 116A anterior, para obtener 0,085 g (22 %) del material del título. LC (Método B): 1,811 min. LCMS (APCI): calc. para C9H9N2O3S [M+H]+ m/z 225,03; encontrado: 225,0. RMN 1 H (400 MHz, CDCl3) 8 ppm: 8,21 (s, 1H), 7,98 (s, 1H), 7,80 (s, 1H), 4,44 (q, J= 7,04 Hz, 2H), 1,41 (t, J= 7,04 Hz, 3H).
117B. (2-(oxazol-5-il)tiazol-4-il)metanol

Se trató 2-(oxazol-5-il)tiazol-4-carboxilato de etilo (0,112 g, 0,499 mmol) con borohidruro de litio (0,022 g, 0,999 mmol), como se describe en el ejemplo 116B anterior, para obtener 0,024 g (26 %) del compuesto del título después de la cromatografía ultrarrápida. lC (Método B): 1,462 min. HRMS (ESI): calc. para C7H7N2O2S [M+H]+ m/z 183,0228; encontrado 183,0222. RMN 1H (400 MHz, CDCls) 8 ppm: 7,99 (s, 1H), 7,80 (s, 1H), 7,32 (s, 1H), 4,86 (s, 2H), 2,90 (as, 1H).
Ejemplo 117. 5-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2-il)oxazol

La reacción de 6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-ol (Ejemplo 1H, 0,079 g, 0,249 mmol) con (2-(oxazol-5-il)tiazol-4-il)metanol (0,050 g, 0,274 mmol), como se describe en el ejemplo 116 anterior, produjo 0,035 g (29 %) del compuesto del título en forma de un sólido de color beis. LC (Método A): 2,278 min. HRMS (ESI): calc. para C21H16N5O5S2 [M+H]+ m/z 482,0593; encontrado 482,0595. RMN 1H (400 MHz, CDCla) 8 ppm: 7,98 (s, 1H), 7,86 (s, 1H), 7,69 (s, 1H), 7,46 (s, 1H), 7,11 (s, 1H), 6,72 (a d, 1H), 6,44 (d, J= 1,96 Hz, 1H), 5,39 (s, 2H), 4,22 (s, 3H), 3,85 (s, 3H).
Ejemplo 118
4-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2-il)tetrahidro-2H-piran-4-ol

118A. 4-(4-(((ferc-butildimetilsilil)oxi)metil)tiazol-2-il)tetrahidro-2H-piran-4-ol

Una solución de 2-bromo-4-(((terc-butildimetilsilil)oxi)metil)tiazol (Ejemplo 37B, 3,39 g, 11,00 mmol) en THF seco (55 ml) se enfrió a -78°C en atmósfera de N2 y después se agregó gota a gota n-butil litio 1,45 M (9,10 ml, 13,20 mmol). La mezcla resultante se agitó durante 30 min para obtener una solución de color de color pardo claro. A esta mezcla se le agregó lentamente una solución de dihidro-2H-piran-4(3H)-ona (1,219 ml, 13,20 mmol) en THF seco (5 ml) y la mezcla se mantuvo a -78 °C durante 1 h para obtener una solución de color de color pardo claro. La reacción se inactivó mediante la adición de NH4Cl acuoso saturado (15 ml) y después se retiró el baño de enfriamiento y la mezcla se repartió entre EtOAc-agua. La fase orgánica se separó, se lavó (salmuera), se secó (Na2SO4) y se evaporó para obtener un aceite de color amarillo claro. La cromatografía ultrarrápida (Isco/ 0-30 % de acetona-hexano) produjo 4-(4-(((terc-butildimetilsilil)oxi)metil)tiazol-2-il)tetrahidro-2H-piran-4-ol (2,97 g, 82 %) en forma de un aceite incoloro que cristalizó en reposo al vacío. LC (Método A): 2,262 min. HRMS (ESI): calc. para C15H28NOaSSi [M+H]+ m/z 330,156; encontrado 330,158. RMN 1H (400 MHz, DMSO-da): 87,23 (s, 1H), 5,99 (s, 1H), 4,64 (s, 2H), 3,64 (m, 4H), 2,00 (ddd, J= 5,48, 11,35, 13,69 Hz, 2H), 1,56 (a d, J= 12,91 Hz, 2H), 0,81 (s, 9H), 0,00 (s, 6H).
118B. 4-(4-(hidroximetil)tiazol-2-il)tetrahidro-2H-piran-4-ol

A una solución de 4-(4-(((terc-butildimetilsilil)oxi)metil)tiazol-2-il)tetrahidro-2H-piran-4-ol (2,96 g, 8,98 mmol) en THF seco (40 ml), en atmósfera de N2 , se le agregó gota a gota trihidrofluoruro de trietilamina (3,66 ml, 22,46 mmol) y la mezcla se agitó a temperatura ambiente durante 16 h. Después la mezcla se concentró a la mitad del volumen, el concentrado se diluyó con DCM y después se agregó NaHcO3 acuoso saturado (¡Precaución: evolución de gas vigorosa!). La fase orgánica se separó, se lavó (NaHCO3 acuoso saturado), se secó (Na2SO4) y se evaporó para obtener solo una pequeña cantidad de un residuo de color amarillo claro. La fase acuosa se saturó posteriormente con NaCl sólido y la mezcla se extrajo con DCM (x6). Los extractos orgánicos combinados se secaron (Na2SO4) y se evaporaron para obtener un sólido de color blanco. Después la fase acuosa se neutralizó con HCl conc. (pH 7) y se volvió a extraer con DCM (x5). El extracto orgánico se volvió a secar (Na2SO4) y se evaporó para obtener material adicional en forma de un sólido de color blanquecino. Estos sólidos se combinaron para obtener 4-(4-(hidroximetil)tiazol-2-il)tetrahidro-2H-piran-4-ol (1,145 g, 59,2 %) en forma de un sólido color crema que se usó tal cual en la siguiente etapa sin más purificación. LC (Método A): 0,861 min. HRMS (ESI): calc. para C9H14NO3S [M+H]+ m/z 216,069; encontrado 216,070. RMN 1 H (400 MHz, DMSO-d6): 87,24 (s, 1H), 6,00 (s, 1H), 5,21 (t, J= 5,87 Hz, 1H),4,48 (d, J= 5,87 Hz, 2H), 3,68 (m, 4H), 2,04 (m, 2H), 1,60 (a d, J= 13,30 Hz, 2H).
Ejemplo 118. 4-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2-i l)tetrahidro-2 H-piran-4-ol

A un matraz secado al fuego se le agregó 6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-ol (Ejemplo 1H, 0,040 g, 0,126 mmol) y 4-(4-(hidroximetil)tiazol-2-il)tetrahidro-2H-piran-4-ol (0,034 g, 0,158 mmol); después el matraz se purgó con N2, y se agregó THF seco (2 ml). A la suspensión resultante se le agregó tri-nbutilfosfina (0,082 ml, 0,315 mmol) y después se agregó gota a gota una solución de 1,1'-(azodicarbonil)dipiperidina (0,080 g, 0,315 mmol) en THF seco (2 ml) (mediante una bomba de jeringa) durante 30 min. La mezcla resultante se agitó a temperatura ambiente durante 1 h más, después se diluyó con EtOAc, se lavó (NaHCO3 acuoso saturado, H2O y salmuera), se secó (Na2SO4) y se evaporó para obtener una goma de color amarillo claro. La cromatografía ultrarrápida (Isco/0-30 % de éter-DCM, después 0-100 % de EtOAc-DCM y finalmente 0-3 % de MeOH-DCM) produjo 4-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2-il)tetrahidro-2H-piran-4-ol (0,049 g, 76 %) en forma de una goma incolora que se liofilizó a partir de MeCN-agua en forma de un sólido de color blanquecino. LC (Método A): 2,106 min. HRMS (ESI): calc. para C23H23N4O6S2 [M+H]+ m/z 515,106; encontrado 515,107. RMN 1 H (400 MHz, DMSO-d6): 88,33 (s, 1H), 7,66 (s, 1H), 6,95 (s, 1H), 6,80 (s, 1H), 6,59 (d, J= 1,57 Hz, 1H), 5,23 (s, 2H), 4,17 (s, 3H), 3,77 (s, 3H), 3,70 (m, 4H), 2,08 (ddd, J= 5,48, 10,96, 13,30 Hz, 1H), 1,64 (a d, J= 12,52 Hz, 2H).
Ejemplo 119
6-(4-((2-(4-fluorotetrahidro-2H-piran-4-il)tiazol-4-il)metoxi)-6-metoxibenzofuran-2-il)-2-metoxiimidazo[2,1-b][1,3,4]tiadiazol
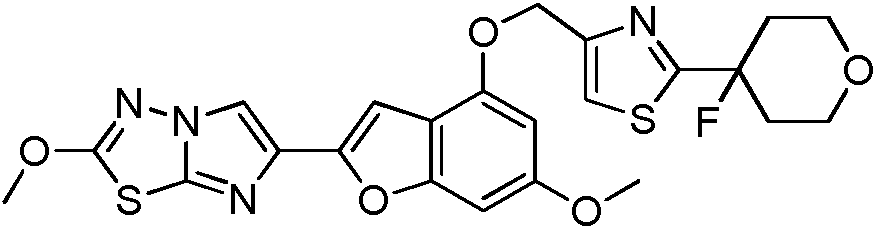
119A. 4-(4-metiltiazol-2-il)tetrahidro-2H-piran-4-ol

Una solución de 4-metiltiazol (0,910 ml, 10,0 mmol) en THF seco (45 ml) se enfrió a -78 °C en atmósfera de N2 y después se agregó gota a gota n-butil litio (1,45 M en hexanos, 7,59 ml, 11,00 mmol). La mezcla resultante se agitó durante 15 min para obtener una solución de color amarillo brillante. A esta mezcla se le agregó lentamente una solución de dihidro-2H-piran-4(3H)-ona (1,108 ml, 12,00 mmol) en THF seco (5 ml) y la mezcla se mantuvo a -78 °C durante 1 h para obtener una solución de color amarillo claro. La reacción se inactivó después mediante la adición de NH4Cl acuoso saturado (5 ml) y la mezcla se repartió entre EtOAc-agua. La fase orgánica se separó, se lavó (salmuera), se secó (Na2SO4) y se evaporó para obtener un aceite de color amarillo claro que solidificó en reposo al vacío. La cromatografía ultrarrápida (Isco/ 0-50 % de acetona-hexano) produjo 4-(4-metiltiazol-2-il)tetrahidro-2H-piran-4-ol (1,535 g, 77 %) en forma de un sólido cristalino de color blanco. Este material se usó tal cual en la siguiente etapa. LC (Método A): 1,091 min. HRMS: calc. para C9H14NO2S [M+1]+ m/z 200,075; encontrado 200,075. RMN 1H (400 MHz, DMSO-da) 8 ppm: 1,59 (a d, J= 12,52 Hz, 2H), 2,04 (ddd, J= 5,87, 10,56, 13,30 Hz, 2H), 2,29 (s, 3H), 3,72-3,62 (m, 4H), 5,95 (s, 1H), 7,08 (s, 1H).
119B. (2-(4-fluorotetrahidro-2H-piran-4-il)-4-metiltiazol

A una solución de 4-(4-metiltiazol-2-il)tetrahidro-2H-piran-4-ol (0,022 g, 0,110 mmol) en diclorometano (1,75 ml), enfriada a 0 °C en nitrógeno, se le agregó gota a gota DAST (0,018 ml, 0,138 mmol). La mezcla de reacción resultante se agitó a 0 °C durante 2 h, después se agregó carbonato de sodio acuoso saturado y la mezcla heterogénea se agitó vigorosamente durante 15 min para garantizar la inactivación completa. La mezcla se repartió después entre diclorometano y solución de bicarbonato acuoso saturado. La capa orgánica se separó, se secó sobre sulfato de magnesio y se concentró a presión reducida. El producto en bruto (0,021 g, 95 %) se usó tal cual en la siguiente etapa. LC (Método A): 1,722 min. LCMS (APCI): calc. para C9H13FN0 S [M+H]+ m/z 202,07, encontrado 202,2. RMN 1H (CDCla, 400 MHz) 8 ppm: 2,00-2,11, (m, 2H), 2,28-2,49 (m, 2H), 2,45 (s, 3H), 3,79-3,98 (m, 4H), 6 ,88 (s, 1H).
119C. 4-(bromometil)-2-(4-fluorotetrahidro-2H-piran-4-il)tiazol

Un tubo sellado se cargó con 2-(4-fluorotetrahidro-2H-piran-4-il)-4-metiltiazol (0,021 g, 0,104 mmol), tetracloruro de carbono (2 ml), NBS (0,0204 g, 0,115 mmol) y peróxido de benzoílo (0,002 g, 8,26 pmol). La mezcla de reacción se agitó después a 85 °C durante 2,5 h. Después de enfriarla, la mezcla de reacción en bruto se absorbió en diclorometano y se retiró el sólido presente mediante filtración. El filtrado se concentró y el residuo en bruto se purificó mediante HPLC preparativa (Método A) para obtener compuesto del título puro (0,010 g, 34 %). LC (Método A): 1,784 min. LCMS (APCI): calc. para CaH-^BrFNOS [M+H]+ m/z 279,98, encontrado 280,0. RMN 1 H (CD3OD, 400 MHz) 8 ppm: 1,96-2,09 (m, 2H), 2,26-2,46 (m, 2H), 3,77-3,96 (m, 4H), 4,62 (s, 2H), 7,60 (s, 1H).
Ejemplo 119. 6-(4-((2-(4-fluorotetrahidro-2H-piran-4-il)tiazol-4-il)metoxi)-6-metoxibenzofuran-2-il)-2-metoxiimidazo[2,1-b][1,3,4]tiadiazol

A una solución de 6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-ol (Ejemplo 1H, 0,009 g, 0,028 mmol) y 4-(bromoirietN)-2-(4-fluorotetrahidro-2H-piran-4-N)tiazol (0,010 g, 0,036 mmol), en agitación en DMF (1 ml) en atmósfera de nitrógeno, se le agregó carbonato de potasio (0,009 g, 0,065 mmol) y la mezcla de reacción resultante se agitó a temperatura ambiente durante 2 h. La mezcla de reacción en bruto se diluyó con diclorometano, se lavó con agua y salmuera, se secó sobre MgSO4, se filtró y se concentró. El residuo en bruto obtenido se purificó mediante HPLC preparativa (Método A) para obtener el compuesto del título puro (0,010 g, 68 %). LC (Método A): 2,376 min. LCMS (ESI): calc. para C23H22FN4O5S2 [M+H]+ m/z 517,1016, encontrado 517,1054. RMN 1H (DMSO-da, 400 MHz) 8 ppm: 2,01-2,15 (m, 2H), 2,17-2,39 (m, 2H), 3,63-3,77 (m, 2H), 3,78-3,90 (m, 2H), 3,81 (s, 3H), 4,20 (s, 3H), 5,32 (s, 2H), 6,62 (d, J= 2,0 Hz, 1H), 6,84 (d, J= 0,8 Hz, 1H), 7,00 (s, 1H), 7,93 (s, 1H), 8,37 (s, 1H).
Ejemplo 120
2-metoxi-6-(6-metoxi-4-((2-(4-metoxitetrahidro-2H-piran-4-il)tiazol-4-il)metoxi)benzofuran-2-il)imidazo[2,1-b][1,3,4]tiadiazol

120A. 4-(((terc-butildimetilsilil)oxi)metil)-2-(4-metoxitetrahidro-2H-piran-4-il)tiazol

A una suspensión de hidruro de sodio (0,097 g, 2,428 mmol) [N o ta : N a H a l 60 % en a ce ite se la vó sin a ce ite con h e x a n o s (x2) a n te s de a g re g a r T H F seco a l m a tra z de re a cc ió n ] en THF seco (5 ml), en atmósfera de N2 , se le agregó una solución de 4-(4-(((terc-butildimetilsilil)oxi)metil)tiazol-2-il)tetrahidro-2H-piran-4-ol (Ejemplo 118A, 0,400 g, 1,214 mmol) en THF seco (3 ml) y la mezcla se agitó a temperatura ambiente hasta que ya no hubo más evolución de gas (ca. 30 min). A la mezcla resultante de color amarillo claro, se le agregó gota a gota yodometano (0,091 ml, 1,457 mmol) y la agitación continuó a temperatura ambiente durante 16 h. La mezcla de reacción después se inactivó mediante la adición cuidadosa de NH4Cl acuoso saturado (5 ml) y posteriormente se repartió entre EtOAc-agua. La fase orgánica se separó, se secó (Na2SO4) y se evaporó para obtener un aceite prácticamente incoloro. La cromatografía ultrarrápida (Isco/ 0-50 % de EtOAc-hexano) produjo 4-(((terc-butildimetilsilil)oxi)metil)-2-(4-metoxitetrahidro-2H-piran-4-il)tiazol (0,362 g, 87 %) en forma de un aceite incoloro que se usó tal cual en la siguiente etapa. LC (Método A): 2,442 min. HRMS (ESI): calc. para C16H3üNO3SSi [M+H]+ m/z 344,171; encontrado 344,173. RMN 1 H (400 MHz, DMSO-d6): 8 7,42 (s, 1H), 4,67 (s, 2H), 3,58 (m, 4H), 2,99 (s, 3H), 2,01 (m, 2H), 1,87 (m, 2H), 0,81 (s, 9H), 0,00 (s, 6 H).
120B. (2-(4-metoxitetrahidro-2H-piran-4-il)tiazol-4-il)metanol

A una solución de 4-(((terc-butildimetilsilil)oxi)metil)-2-(4-metoxitetrahidro-2H-piran-4-il)tiazol (0,358 g, 1,042 mmol) en THF seco (10 ml), en atmósfera de N2 , se le agregó gota a gota trihidrofluoruro de trietilamina (0,848 ml, 5,21 mmol) y la mezcla se agitó a temperatura ambiente durante 5 h. Después la mezcla se diluyó con DCM y la solución se lavó (NaHCO3 acuoso saturado), se secó (Na2SO4) y se evaporó para obtener (2-(4-metoxitetrahidro-2H-piran-4-il)tiazol-4-il)metanol (0,227 g, 95 %) en forma de una goma prácticamente incolora que se usó tal cual en la siguiente etapa. lC (Método A): 1,301 min. HRMS (ESI): calc. para C10H16NO3S [M+H]+ m/z 230,085; encontrado 230,085. RMN 1 H (400 MHz, DMSO-d6): 8 7,38 (t, J= 1,17 Hz, 1H), 5,23 (t, J= 5,48 Hz, 1H), 4,48 (dt, J= 1,17, 5,48 Hz, 2H), 3,58 (m, 4H), 2,99 (s, 3H), 2,01 (m, 2H), 1,87 (m, 2H).
Ejemplo 120. 2-metoxi-6-(6-metoxi-4-((2-(4-metoxitetrahidro-2H-piran-4-il)tiazol-4-il)metoxi)benzofuran-2-il)imidazo[2,1-b][1,3,4]tiadiazol

A un matraz secado al fuego se le agregó 6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-ol (Ejemplo 1H, 0,040 g, 0,126 mmol) y (2-(4-metoxitetrahidro-2H-piran-4-il)tiazol-4-il)metanol (0,036 g, 0,158 mmol); después el matraz se purgó con N2 y se agregó THF seco (2 ml). A la suspensión resultante se le agregó tri-nbutilfosfina (0,082 ml, 0,315 mmol) y después se agregó gota a gota una solución de 1,1'-(azodicarbonil)dipiperidina (0,080 g, 0,315 mmol) en THF seco (2 ml) (mediante una bomba de jeringa) durante 30 min. La mezcla resultante se agitó a temperatura ambiente durante 1 h más, después se diluyó con EtOAc, se lavó (NaHCO3 acuoso saturado, H2O y salmuera), se secó (Na2SO4) y se evaporó para obtener una goma de color amarillo claro. La cromatografía ultrarrápida (Isco/0-50 % de éter-DCM) produjo 2-metoxi-6-(6-metoxi-4-((2-(4-metoxitetrahidro-2H-piran-4-il)tiazol-4-il)metoxi)benzofuran-2-il)imidazo[2,1-b][1,3,4]tiadiazol (0,045 g, 67,5 %) en forma de un sólido de color blanco. LC (Método A): 2,348 min. HRMS (ESI): calc. para C24H25N4O6S2 [M+H]+ m/z 529,122; encontrado 529,124. RMN 1H (400 MHz, DMSO-da): 8 8,33 (s, 1H), 7,84 (s, 1H), 6,95 (s, 1H), 6,80 (s, 1H), 6,59 (d, J= 1,57 Hz, 1H), 5,23 (s, 2H), 4,17 (s, 3H), 3,77 (s, 3H), 3,64 (m, 4H), 3,06 (s, 3H), 2,09 (m, 2H), 1,95 (m, 2H).
Ejemplo 121
4-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2-il)-2,6-dimetiltetrahidro-2H-piran-4-ol

121A. 4-(4-(((terc-butildimetilsilil)oxi)metil)tiazol-2-il)-2,6-dimetiltetrahidro-2H-piran-4-ol

Una solución de 2-bromo-4-(((terc-butildimetilsilil)oxi)metil)tiazol (Ejemplo 37B, 5,00 g, 16,22 mmol) en THF seco (75 ml) se enfrió a -78°C en atmósfera de N2 y después se agregó gota a gota n-butil litio 1,45 M (11,89 ml, 17,84 mmol). La mezcla resultante se agitó durante 15 min para obtener una solución de color de color pardo claro. A esta mezcla se le agregó lentamente una solución de 3,5-dimetildihidro-2H-piran-4(3H)-ona (2,494 g, 19,46 mmol) [Aube, J. e t al. J.Org.Chem., 69: 1716 (2004)] en THF seco (5 ml) y la agitación continuó a -78 °C durante 2 h para obtener una solución de color de color pardo claro. La reacción se inactivó después mediante la adición de NH4Cl acuoso saturado (10 ml), se retiró el baño de enfriamiento y la mezcla se repartió entre EtOAc-agua. La fase orgánica se separó, se lavó (agua y salmuera), se secó (Na2SO4) y se evaporó para obtener un aceite de color amarillo claro. La cromatografía ultrarrápida (Isco/ 0 - 10 0 % de EtOAc-tolueno) permitió obtener el producto impuro en forma de un aceite incoloro (2,11 g). Este material se volvió a someter a cromatografía (Isco/ 0-50 % de étercloroformo) para obtener 4-(4-(((terc-butildimetilsilil)oxi)metil)tiazol-2-il)-2,6-dimetiltetrahidro-2H-piran-4-ol (1,767 g, 30,5 %) en forma de un aceite viscoso que se solidificó en reposo. Este material se usó tal cual en la siguiente etapa. LC (Método A): 2,352 min. HRMS (ESI): calc. para C1yH32NOaSSi [M+H]+ m/z 358,187; encontrado 358,188. RMN 1 H (400 MHz, DMSO-d6): 8 7,30 (s, 1H), 5,94 (s, 1H), 4,64 (s, 2H), 3,78 (ddq, J= 1,57, 6,26, 12,52 Hz, 2H), 1,98 (d, J= 12,52 Hz, 2H), 1,36 (t, J= 12,52 Hz, 2H), 1,00 (d, J= 6,26 Hz, 6 H), 0,80 (s, 9H), 0,00 (s, 6 H).
121B. 4-(4-(hidroximetil)tiazol-2-il)-2,6-dimetiltetrahidro-2H-piran-4-ol

A una solución de 4-(4-(((terc-butildimetilsilil)oxi)metil)tiazol-2-il)-2,6-dimetiltetrahidro-2H-piran-4-ol (1,742 g, 4,87 mmol) en THF seco (30 ml), en atmósfera de N2 , se le agregó gota a gota trihidrofluoruro de trietilamina (2,380 ml, 14,61 mmol) y la mezcla se agitó a temperatura ambiente durante 14 h. Después la mezcla se diluyó con DCM y la solución se lavó (NaHCO3 acuoso saturado), se secó (Na2SO4) y se evaporó para obtener el producto (0,800 g, 68 %) en forma de un sólido de color amarillo claro. La fase acuosa se saturó con NaCl sólido y se extrajo con EtOAc (x2) para obtener (después de la separación, el secado y la evaporación de la fase orgánica combinada) 0,329 g más ( 28 %) del producto en forma de un sólido cristalino de color blanco. Los sólidos se combinaron para obtener 4-(4-(hidroximetil)tiazol-2-il)-2,6-dimetiltetrahidro-2H-piran-4-ol (1,129 g, 95 %) que era esencialmente puro y se usó tal cual en la siguiente etapa. LC (Método A): 1,210 min. Hr Ms (ESI): calc. para C11 H18NO3S [M+H]+ m/z 244,100; encontrado 244,101. RMN 1H (400 MHz, DMSO-da): 8 7,29 (s, 1H), 5,95 (s, 1H), 5,20 (t, J= 5,87 Hz, 1H), 4,48 (d, J= 5,48 Hz, 2H), 3,78 (ddq, J= 1,96, 6,26, 11,35 Hz, 2H), 2,02 (d, J= 12,91 Hz, 2H), 1,39 (t, J= 11,74 Hz, 2H), 1,04 (d, J= 6,26 Hz, 6 H).
Ejemplo 121. 4-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2-il)-2,6-dimetiltetrahidro-2H-piran-4-ol

A un matraz secado al fuego se le agregó 6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-ol (Ejemplo 1H, 0,600 g, 1,891 mmol) y 4-(4-(hidroximetil)tiazol-2-il)-2,6-dimetiltetrahidro-2H-piran-4-ol (0,552 g, 2,269 mmol); después el matraz se purgó con N2 , y se agregó THF seco (20 ml). A la suspensión resultante se le agregó tri-n-butilfosfina (1,228 ml, 4,73 mmol), y después se agregó gota a gota una solución de 1,1'-(azodicarbonil)dipiperidina (1,205 g, 4,73 mmol) en THF seco (8 ml) (mediante una bomba de jeringa) durante 30 min. La mezcla resultante se agitó a temperatura ambiente durante 1 h más, después se inactivó con NaHCO3 acuoso saturado y se repartió entre DCM-agua. El extracto orgánico se separó, se secó (Na2SO4) y se evaporó para obtener un sólido de color amarillo claro. La cromatografía ultrarrápida (Isco/ 0-100 % de EtOAc-DCM) produjo un sólido que se trituró con MeCN para obtener (después de la filtración, el lavado con un volumen mínimo de MeCN y el secado al vacío) 4-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2-il)-2,6-dimetiltetrahidro-2H-piran-4-ol (679 mg, 66 %) en forma de un sólido color crema. LC (Método A): 2,202 min. HRMS (ESI): calc. para C25H27N4O6S2 [M+H]+ m/z 543,137; encontrado 543,140. RMN 1 H (400 MHz, DMSO-d6): 8 8,30 (s, 1H), 7,68 (s, 1H), 6,89 (s, 1H), 6,76 (s, 1H), 6,59 (d, J= 1,96 Hz, 1H), 6,01 (s, 1H), 5,22 (s, 2H), 4,14 (s, 3H), 3,76 (m, 2H), 3,73 (s, 3H), 2,00 (d, J= 11,76 Hz, 2H), 1,37 (t, J= 11,74 Hz, 2H), 0,99 (d, J= 6,26 Hz, 6 H).
Ejemplo 122. 6-(4-((2-(4-fluoro-2,6-dimetiltetrahidro-2H-piran-4-il)tiazol-4-il)metoxi)-6-metoxibenzofuran-2-il)-2-metoxi-imidazo[2,1-b][1,3,4]tiadiazol

A una suspensión enfriada en hielo de 4-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2-il)-2,6-dimetil-tetrahidro-2H-piran-4-ol (0,023 g, 0,042 mmol) en Dc M (3 ml), en atmósfera de N2, se le agregó gota a gota DAST (0,014 ml, 0,106 mmol) y la mezcla resultante se agitó a 0 °C durante 20 min. Se agregó otra alícuota de DAST (0,007 ml, 0,053 mmol), el baño de enfriamiento se retiró y la solución resultante de color amarillo claro se agitó a temperatura ambiente durante 16 h. La mezcla de reacción se volvió a enfriar después a 0 °C y se inactivó mediante la adición gota a gota de NaHCO3 acuoso saturado (3 ml). La mezcla se agitó vigorosamente a 0 °C durante 5 min, después el baño de enfriamiento se retiró y la agitación continuó hasta que no se observó más evolución de gas. Después la fase orgánica se separó y se aplicó directamente a una precolumna
de gel de sílice. La cromatografía ultrarrápida (Isco/ 0-100 % de EtOAc-hexano) produjo 6-(4-((2-(4-fluoro-2,6-dimetiltetrahidro-2H-piran-4-il)tiazol-4-il)metoxi)-6-metoxibenzofuran-2-il)-2-metoxi-imidazo[2,1-b][1,3,4]tiadiazol (0,020 g, 87 % de rendimiento) en forma de una goma incolora que se liofilizó a partir de MeCN-agua para obtener un sólido de color blanco. La RMN indicó que era una mezcla de isómeros 3:2. LC (Método A): 2,470 min. HRMS (ESI): calc. para C25H26FN4O5S2 [M+H]+ m/z 545,133; encontrado 545,135. RMN 1H (400 MHz, DMSO-da): 8 8,31 (s, 0,4H), 8,30 (s, 0,6H), 7,97 (s, 0,6H), 7,84 (s, 0,4H), 6,93 (s, 0,4H), 6,91 (s, 0,6H), 6,77 (m, 1H), 6,58 (d, J= 1,96 Hz, 0,6H), 6,56 (d, J= 1,57 Hz, 0,4H), 5,29 (s, 1,2H), 5,23 (s, 0,8H), 4,14 (s, 3H), 3,76 (m, 0,8H), 3,74 (s, 1,2H), 3,73 (s, 1,8H), 3,53 (m, 1,2H), 2,38 (dd, J= 1,96, 12,52 Hz, 1H), 2,07 (m, 1H), 1,83-1,61 (m, 2H), 1,10 (d, J= 6,26 Hz, 2,4H), 1,08 (d, J= 6,26 Hz, 3,6H).
Ejemplo 123
3-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzo-furan-4-il)oxi)metil)tiazol-2-il)tetrahidrofuran-3-ol
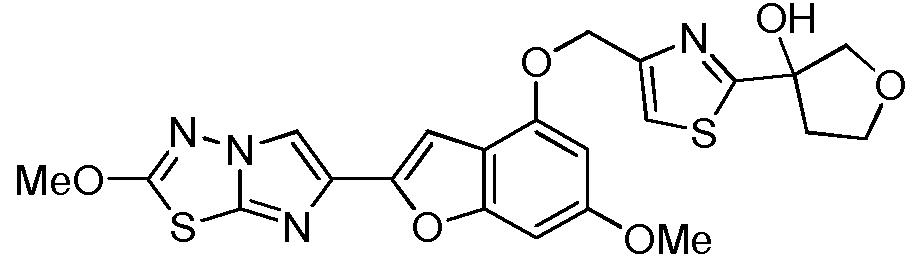
123A. 3-(4-(((ferc-butildimetilsilil)oxi)metil)tiazol-2-il)tetrahidrofuran-3-ol

Una solución de 2-bromo-4-(((ferc-butildimetilsilil)oxi)metil)tiazol (Ejemplo 37B, 1,542 g, 5,000 mmol) en THF seco (20 ml) se enfrió a -78°C en atmósfera de N2 y después se agregó gota a gota n-butil litio 1,45 M (3,79 ml, 5,50 mmol). La mezcla resultante se agitó durante 15 min para obtener una solución de color amarillo parduzco claro. A esta mezcla se le agregó lentamente una solución de dihidrofuran-3(2H)-ona (0,517 g, 6,00 mmol) en THF seco (2,5 ml) y la mezcla se agitó a -78 °C durante 1 h para obtener una solución de color de color pardo claro. La reacción se inactivó después mediante la adición de NH4Cl acuoso saturado (5 ml), se retiró el baño de enfriamiento y la mezcla se repartió entre EtOAc-agua. La fase orgánica se separó, se lavó (salmuera), se secó (Na2SO4) y se evaporó para obtener un aceite de color amarillo claro. La cromatografía ultrarrápida (Isco/ 0-50 % de EtOAchexano) produjo 3-(4-(((ferc-butildimetilsilil)oxi)metil)tiazol-2-il)tetrahidrofuran-3-ol (1,052 g, 66,7 %) en forma de un aceite que cristalizó en reposo al vacío. Este material se usó tal cual en la siguiente etapa. LC (Método A): 2,226 min. HRMS (ESI): calc. para C-mH26N0 3 SSÍ [M+H]+ m/z 316,140; encontrado 316,147. RMN 1H (400 MHz, DMSO-d6): 8 7,28 (s, 1H), 6,32 (s, 1H), 4,65 (s, 2H), 3,92 (m, 2H), 3,78 (q, J= 9,00 Hz, 2H), 2,36 (dt, J= 9,00, 12,52 Hz, 1H), 2,09 (dt, J= 5,09, 12,52 Hz, 1H), 0,82 (s, 9H), 0,00 (s, 6 H).
123B. 3-(4-(hidroximetil)tiazol-2-il)tetrahidrofuran-3-ol

A una solución de 3-(4-(((ferc-butildimetilsilil)oxi)metil)tiazol-2-il)tetrahidrofuran-3-ol (1,022 g, 3,24 mmol) en THF seco (20 ml), en atmósfera de N2 , se le agregó gota a gota trihidrofluoruro de trietilamina (1,319 ml, 8,10 mmol) y la mezcla se agitó a temperatura ambiente durante 16 h. La mezcla se diluyó con DCM y después se agregó NaHCO3 acuoso saturado (Precaución: evolución de gas vigorosa). La fase orgánica se separó, se lavó (NaHCO3 acuoso saturado), se secó (Na2SO4) y se evaporó para obtener una goma incolora que solidificó en reposo al vacío. La fase acuosa se saturó con NaCl sólido y se volvió a extraer con DCM para obtener (después de secarse como se indicó anteriormente) una goma incolora adicional que también solidificó en reposo. Estos sólidos se combinaron para obtener 3-(4-(hidroximetil)tiazol-2-il)tetrahidrofuran-3-ol en forma de un sólido color crema. Este material se usó tal cual en la siguiente etapa sin purificación adicional. LC (Método A): 0,734 min. HRMS (ESI): calc. para C9H12NO3S [M+H]+ m/z 202,054; encontrado 202,055. RMN 1 H (400 MHz, DMSO-d6): 8 7,18 (s, 1H), 6,23 (s, 1H), 5,12 (a s, 1H), 4,37 (s, 2H), 3,85 (dd, J= 1,57, 9,00 Hz, 1H), 3,84 (d, J= 9,00 Hz, 1H), 3,74 (d, J= 9,00 Hz, 1H), 3,68 (d, J= 9,00 Hz, 1H), 2,29 (dt, J= 9,00, 12,91 Hz, 1H), 2,02 (dt, J= 5,09, 12,52 Hz, 1H).
Ejemplo 123. 3-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2-il)tetrahidrofuran-3-ol

A un matraz secado al fuego se le agregó 6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-ol (Ejemplo 1H, 0,063 g, 0,200 mmol) y 3-(4-(hidroximetil)tiazol-2-il)tetrahidrofuran-3-ol (0,040 g, 0,200 mmol); después el matraz se purgó con N2 y se agregó THF seco (5 ml). A la suspensión resultante se le agregó tri-n-butilfosfina (0,123 ml, 0,500 mmol) y después se agregó gota a gota una solución de 1,1'-(azodicarbonil)dipiperidina (0,127 g, 0,500 mmol) en THF seco (2 ml) (mediante una bomba de jeringa) durante 30 min. La mezcla resultante se agitó a temperatura ambiente durante 1 h más, después se diluyó con EtOAc, se lavó (NaHCO3 acuoso saturado, H2O y salmuera), se secó (Na2SO4) y se evaporó para obtener un semisólido de color amarillo claro. La cromatografía ultrarrápida (Isco/ 0 - 10 0 % de EtOAc-DeM) produjo 3-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2-il)tetrahidrofuran-3-ol (0,015 g, 14,98 %) en forma de un sólido. LC (Método A): 2,161 min. HRMS (ESI): calc. para C22H21N4O6S2 [M+H]+ m/z 501,090; encontrado 501,092. RMN 1H (400 MHz, DMSO-da): 8 8,33 (s, 1H), 7,71 (s, 1H), 6,95 (s, 1H), 6,80 (s, 1H), 6,58 (d, J= 1,57 Hz, 1H), 6,44 (s, 1H), 5,23 (s, 2H), 4,17 (s, 3H), 3,98 (d, J= 5,09 Hz, 1H), 3,96 (d, J= 5,48 Hz, 1H), 3,88 (d, J= 9,00 Hz, 1H), 3,83 (d, J= 9,00 Hz, 1H), 3,77 (s, 3H), 2,43 (m, 1H), 2,16 (dt, J= 5,09, 12,52 Hz, 2H).
Ejemplo 124
4-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)-5-metiltiazol-2-il)tetrahidro-2H-piran-4-ol
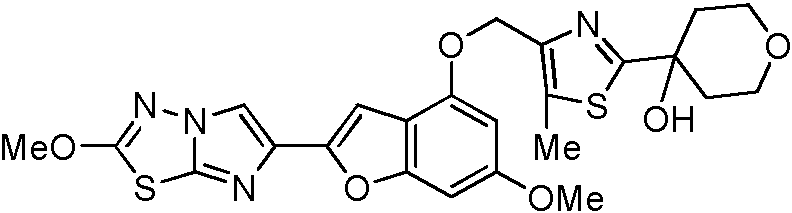
124A. (5-metiltiazol-4-il)metanol

A una solución enfriada en hielo de 5-metiltiazol-4-carboxilato de etilo (2,57 g, 15,00 mmol) en THF seco (50 ml), en atmósfera de N2 , se le agregó borohidruro de litio (0,654 g, 30,0 mmol) de una vez, seguido de MeOH (1,214 ml, 30,0 mmol) gota a gota. Después se retiró el baño de enfriamiento y la mezcla se agitó a temperatura ambiente durante 16 h. La mezcla se volvió a enfriar a 0 °C y después se inactivó cuidadosamente mediante la adición lenta de NH4Cl acuoso saturado (20 ml) con agitación vigorosa. Después se retiró el baño de enfriamiento y la mezcla se repartió entre EtOAc-agua. La fase orgánica se separó y después se lavó con agua y salmuera. La fase acuosa combinada se saturó con NaCl sólido y se volvió a extraer con EtOAc (x4). La fase orgánica combinada se secó (Na2SO4) y se evaporó para obtener un aceite de color amarillo que se purificó mediante cromatografía ultrarrápida (isco/ 0-100 % de EtoAc-DCM) para obtener (5-metiltiazol-4-il)metanol (1,144 g, 59,0 %) en forma de un sólido prácticamente incoloro que cristalizó en reposo al vacío para obtener un sólido. Este material se usó tal cual en la siguiente etapa. LC (Método X): 0,620 min. HRMS (ESI): calc. para CsHaNOS [M+H]+ m/z 130,032; encontrado 130,032. RMN 1 H (400 MHz, DMSO-d6): 8 8,75 (s, 1H), 4,98 (a s, 1H), 4,46 (s, 2H), 2,40 (s, 3H).
124B. 4-(((terc-butildimetilsilil)oxi)metil)-5-metiltiazol

A una solución de (5-metiltiazol-4-il)metanol (1,134 g, 8,78 mmol) e imidazol (1,793 g, 26,3 mmol) en DMF (40 ml), en atmósfera de N2 , se le agregó terc-butildimetilclorosilano (1,455 g, 9,66 mmol) y la mezcla resultante se agitó a temperatura ambiente en N2 durante 16 h. Después la solución se concentró a presión reducida y el aceite residual se repartió entre EtOAc-NH4Cl acuoso saturado. La fase orgánica se separó, se lavó (agua y salmuera), se secó (Na2SO4) y se evaporó para obtener un aceite viscoso de color amarillo claro. La cromatografía ultrarrápida (Isco/ 0 10 % de EtOAc-DCM) produjo 4-(((terc-butildimetilsilil)oxi)metil)-5-metiltiazol (1,484 g, 69,4 %) en forma de un aceite
incoloro que se usó tal cual en la siguiente etapa. LC (Método A ): 2 ,272 min. HRMS (ESI): calc. para C11 H22NOSSÍ [M+H]+ m/z 244,119; encontrado 244,123. RMN 1H (400 MHz, CDCla): 88,46 (s, 1H), 4,74 (s, 2H), 2,43 (s, 3H), 0,82 (s, 9H), 0,00 (s, 6H).
124C. 4-(4-(((ferc-butildimetilsilil)oxi)metil)-5-metiltiazol-2-il)tetrahidro-2H-piran-4-ol

Una solución de 4-(((ferc-butildimetilsilil)oxi)metil)-5-metiltiazol (1,480 g, 6,08 mmol) en THF seco (45 ml) se enfrió a -78 °C en atmósfera de N2 y después se agregó gota a gota n-butil litio (1,45 M en hexanos, 5,03 ml, 7,30 mmol). La solución que al inicio era incolora se tornó púrpura brillante cerca del final de la adición, y la mezcla resultante se agitó durante 15 min a la misma temperatura. A esta solución de color púrpura se le agregó lentamente una solución de dihidro-2H-piran-4(3H)-ona (0,674 ml, 7,30 mmol) en THF seco (3 ml) y la solución se mantuvo a -78 °C durante 1 h para obtener una solución de color naranja claro. La reacción se inactivó después mediante la adición de NH4Cl acuoso saturado (10 ml), después se retiró el baño de enfriamiento y la mezcla se repartió entre EtOAc-agua. La fase orgánica se separó, se lavó (salmuera), se secó (Na2SO4) y se evaporó para obtener un aceite de color amarillo claro. La cromatografía ultrarrápida (Isco/ 0-30 % de acetona-hexano) produjo 4-(4-(((ferc-butildimetilsilil)oxi)metil)-5-metiltiazol-2-il)tetrahidro-2H-piran-4-ol (1,865 g, 89 %) en forma de un aceite incoloro que se usó tal cual en la siguiente etapa. LC (Método A): 2,297 min. HRMS (ESI): calc. para C16H3üNO3SSi [M+H]+ m/z 344,172; encontrado 344,176. RMN 1H (400 MHz, DMSO-d6): 85,92 (s, 1H), 4,59 (s, 2H), 3,66 (m, 4H), 2,34 (s, 3H), 2,00 (m, 2H), 1,57 (d, J= 12,91 Hz, 2H), 0,81 (s, 9H), 0,00 (s, 6H).
124D. 4-(4-(hidroximetil)-5-metiltiazol-2-il)tetrahidro-2H-piran-4-ol

A una solución de 4-(4-(((ferc-butildimetilsilil)oxi)metil)-5-metiltiazol-2-il)tetrahidro-2H-piran-4-ol (1,861 g, 5,42 mmol) en THF seco (15 ml), en atmósfera de N2 , se le agregó gota a gota trihidrofluoruro de trietilamina (2,65 ml, 16,25 mmol y la mezcla se agitó a temperatura ambiente durante 18 h. Después la mezcla se concentró a alrededor de la mitad del volumen a presión reducida, el concentrado se diluyó con DCM y la solución se lavó (NaHCO3 acuoso saturado), se secó (Na2SO4) y se evaporó para obtener 4-(4-(hidroximetil)-5-metiltiazol-2-il)tetrahidro-2H-piran-4-ol (1,114 g, 90 %) en forma de un sólido de color blanco. Este material era esencialmente puro y se usó tal cual en la siguiente etapa. LC (Método A): 1,062 min. HRMS (ESI): calc. para C10H16NO3S [M+H]+ m/z 230,085; encontrado 230,086. RMN 1 H (400 MHz, DMSO-d6): 85,90 (s, 1H), 4,93 (t, J= 5,87 Hz, 1H), 4,38 (d, J= 5,48 Hz, 2H), 3,67 (m, 4H), 2,34 (s, 3H), 2,02 (m, 2H), 1,58 (d, J= 11,74 Hz, 2H).
Ejemplo 124. 4-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)-5-metiltiazol-2-il)tetrahidro-2 H-piran-4-ol

A un matraz secado al fuego se le agregó 6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-ol (Ejemplo 1H, 0,060 g, 0,189 mmol) y 4-(4-(hidroximetil)-5-metiltiazol-2-il)tetrahidro-2H-piran-4-ol (0,054 g, 0,236 mmol); después el matraz se purgó con N2 y se agregó THF seco (3 ml). A la suspensión resultante se le agregó tri-n-butilfosfina (0,123 ml, 0,473 mmol) y después se agregó gota a gota una solución de 1,1'-(azodicarbonil)dipiperidina (0,120 g, 0,473 mmol) en THF seco (2 ml) (mediante una bomba de jeringa) durante 30 min. La mezcla resultante se agitó a temperatura ambiente durante 1 h más, después se diluyó con EtOAc, se lavó (NaHCO3 acuoso saturado, H2O y salmuera), se secó (Na2SO4) y se evaporó para obtener un semisólido de color amarillo claro. La cromatografía ultrarrápida (Isco/0-100 % de EtOAc-DCM) produjo 4-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)-5-metiltiazol-2-il)tetrahidro-2H-piran-4-ol (0,077 g, 77 %) en forma de un sólido de color blanco. LC (Método A): 2,214 min. Hr Ms (ESI): calc. para C24H25N4O6S2 [M+H]+ m/z 529,122; encontrado 529,125. RMN 1 H (400 MHz, DMSO-d6): 88,30 (s, 1H), 6,83 (s, 1H), 6,76 (s, 1H), 6,59 (d, J= 1,57 Hz, 1H), 5,98 (s, 1H), 5,13 (s, 2H), 4,13 (s, 3H), 3,74 (s, 3H), 3,65 (m, 4H), 2,39 (s, 3H), 2,02 (m,
2H), 1,58 (d, J= 12,91 Hz, 2H).
Ejemplo 125
6-(4-((2-(4-fluorotetrahidro-2H-piran-4-il)-5-metiltiazol-4-il)metoxi)-6-metoxibenzofuran-2-il)-2-metoxiimidazo[2,1-b][1,3,4]-tiadiazol

A una suspensión enfriada en hielo de 4-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)-5-metiltiazol-2-il)tetrahidro-2H-piran-4-ol (0,025 g, 0,047 mmol) en DCM (3 ml), en atmósfera de N2 , se le agregó gota a gota DAST (0,016 ml, 0,118 mmol) y la mezcla resultante se agitó a 0°C durante 20 min. Después se retiró el baño de enfriamiento y la solución de color amarillo claro resultante se agitó a temperatura ambiente durante 1 h. Después la mezcla de reacción se volvió a enfriar a 0 °C y se inactivó mediante la adición gota a gota de NaHCO3 acuoso saturado (3 ml). La mezcla se agitó vigorosamente a 0°C durante 5 min y después se retiró el baño de enfriamiento, la mezcla se diluyó con DCM y NaHCO3 acuoso saturado adicional y la agitación continuó hasta que ya no se observó más evolución de gas. La fase orgánica se separó después y se aplicó directamente a una precolumna de gel de sílice. La cromatografía ultrarrápida (Isco/ 0 - 10 0 % de EtOAc-hexano) produjo 6-(4-((2-(4-fluorotetrahidro-2H-piran-4-il)-5-metiltiazol-4-il)metoxi)-6-metoxibenzo-furan-2-il)-2-metoxiimidazo[2,1-b][1,3,4]tiadiazol (0 , 020 g, 80 %) en forma de una goma incolora que se liofilizó a partir de MeCN-agua para obtener un sólido de color blanco. LC (Método A): 2,405 min. HRMS (ESI): calc. para C24H24FN4O5S2 [M+H]+ m/z 531,117; encontrado 531,118. RMN 1H (400 MHz, DMSO-da): 8 8,30 (s, 1H), 6,83 (s, 1H), 6,77 (s, 1H), 6,58 (d, J= 1,96 Hz, 1H), 5,18 (s, 2H), 4,13 (s, 3H), 3,74 (s, 3H), 3,74 (m, 2H), 3,62 (dt, J= 1,96, 10,96 Hz, 2H), 2,45 (s, 3H), 2,23-2,10 (m, 2H), 1,99 (m, 2H).
Ejemplos 126 y 127
8-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)-tiazol-2-il)-1,4-dioxaespiro[4.5]decan-8-ol y 6-(4-((2-(1,4-dioxaespiro-[4.5]dec-7-en-8-il)tiazol-4-il)metoxi)-6-metoxibenzofuran-2-il)-2-metoxiimidazo[2,1-b][1,3,4]tiadiazol, respectivamente.
126A. 8-(4-(((ferc-butildimetilsilil)oxi)metil)tiazol-2-il)-1,4-dioxaespiro-[4.5]decan-8-ol
Una solución de 2-bromo-4-(((ferc-butildimetilsilil)oxi)metil)tiazol (Ejemplo 37B, 1,00 g, 3,24 mmol) en THF seco (15 ml) se enfrió a -78 °C en atmósfera de N2 y después se agregó gota a gota n-butil litio (1,45 M en hexanos, 2,68 ml, 3,89 mmol). La mezcla resultante se agitó durante 30 min para obtener una solución de color amarillo parduzco claro. A esta mezcla se le agregó gota a gota una solución de 1,4-dioxaespiro[4,5]decan-8-ona (0,608 g, 3,89 mmol) en THF seco (4 ml) y la mezcla se mantuvo a -78 °C durante 2 h. La reacción se inactivó mediante la adición de NH4Cl acuoso saturado (5 ml), después se retiró el baño de enfriamiento y la mezcla se repartió entre EtOAc-agua. La fase orgánica se separó, se lavó (salmuera), se secó (Na2SO4) y se evaporó para obtener un aceite de color amarillo claro que se purificó mediante cromatografía ultrarrápida (Isco/0-100 % de EtOAc-hexano) para obtener 8-(4-(((ferc-butildimetilsilil)oxi)-metil)tiazol-2-il)-1,4-dioxaespiro[4,5]decan-8-ol (0,920 g, 73,6 %) en forma de una goma incolora que cristalizó en reposo al vacío. Este material se usó tal cual en la siguiente etapa. LC (Método A): 2,287 min. HRMS (ESI): calc. para C1sH32NO4SS¡ [M+H]+ m/z 386,182; encontrado 386,182. RMN 1 H (400 MHz, DMSO-d6): 8 7,20 (s, 1H), 5,85 (s, 1H), 4,64 (s, 2H), 3,81 (s, 4H), 2,03 (dt, J= 3,91, 12,91 Hz, 2H), 1,78 (dt, J= 4,30, 13,30 Hz, 2H), 1,68 (d, J= 12,91 Hz, 2H), 1,53 (d, J= 12,52 Hz, 2H), 0,82 (s, 9H), 0,00 (s, 6 H).
126B. 8-(4-(hidroximetil)tiazol-2-il)-1,4-dioxaespiro[4.5]decan-8-ol

A una solución de 8-(4-(((terc-butildimetilsilil)oxi)metil)tiazol-2-il)-1,4-dioxaespiro[4.5]decan-8-ol (0,916 g, 2,376 mmol) en THF seco (10 ml), en atmósfera de N2 , se le agregó gota a gota trihidrofluoruro de trietilamina (1,160 ml, 7,13 mmol) y la mezcla se agitó a temperatura ambiente durante 18 h. Después la mezcla se repartió entre EtOAc-NaHCO3 acuoso saturado y la fase orgánica se lavó (salmuera), se secó (Na2SO4) y se evaporó para obtener 8-(4-(hidroximetil)tiazol-2-il)-1,4-dioxaespiro[4.5]decan-8-ol (0,579 g, 90 %) en forma de una goma incolora que cristalizó en reposo al vacío. Este material era esencialmente puro y se usó tal cual en la siguiente etapa. LC (Método A): 1,148 min. HRMS (ESI): calc. para C12 H18NO4S [M+H]+ m/z 272,096; encontrado 272,095. RMN 1 H (400 MHz, DMSO-d6): 8 7,20 (s, 1H), 5,86 (s, 1H), 5,20 (t, J= 5,48 Hz, 1H), 4,46 (d, J= 5,09 Hz, 2H), 3,84 (s, 4H), 2,07 (dt, J= 3,91, 12,91 Hz, 2H), 1,81 (dt, J= 4,30, 13,30 Hz, 2H), 1,70 (d, J= 12,91 Hz, 2H), 1,55 (d, J= 12,52 Hz, 2H).
Ejemplos 126 y 127. 8-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)-tiazol-2-il)-1,4-dioxaespiro[4.5]decan-8-ol y 6-(4-((2-(1,4-dioxaespiro-[4.5]dec-7-en-8-il)tiazol-4-il)metoxi)-6-metoxibenzofuran-2-il)-2-metoxiimidazo[2,1-b][1,3,4]tiadiazoi

A un matraz secado al fuego se le agregó 6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-ol (Ejemplo 1H, 0,600 g, 1,891 mmol) y 8-(4-(hidroximetil)tiazol-2-il)-1,4-dioxaespiro[4.5]decan-8-ol (0,539 g, 1,985 mmol); después el matraz se purgó con N2 y se agregó THF seco (15 ml). A la suspensión resultante se le agregó tri-n-butilfosfina (1,228 ml, 4,73 mmol) y después una solución de 1,1'-(azodicarbonil)dipiperidina (1,205 g, 4,73 mmol) en THF seco (10 ml) durante ca. 30 min (mediante una bomba de jeringa). La mezcla resultante se agitó a temperatura ambiente durante 1 h más y después se diluyó con EtOAc, se lavó (NaHCO3 acuoso saturado, H2O y salmuera), se secó (Na2SO4) y se evaporó para obtener un semisólido de color ámbar claro. La cromatografía ultrarrápida (Isco/ 0-100 % de EtOAc-DCM) produjo 2 productos principales. La fracción 1 se identificó como 6-(4-((2-(1,4-dioxaespiro[4.5]dec-7-en-8-il)tiazol-4-il)metoxi)-6-metoxibenzofuran-2-il)-2-metoxiimidazo[2,1-b][1,3,4]tiadiazoi (0,127 g, 12,15 %) y se aisló en forma de una espuma de color blanco. Este material se liofilizó a partir de MeCN-agua para obtener un sólido color crema. LC (Método A): 2,362 min. HRMS (ESI): calc. para C26H25N4O6S2 [M+H]+ m/z 553,121; encontrado 553,122. RMN 1 H (400 MHz, DMSO-d6): 8 8,34 (s, 1H), 7,67 (s, 1H), 6,97 (s, 1H), 6,80 (s, 1H), 6,57 (s, 1H), 6,50 (a s, 1H), 5,24 (s, 2H), 4,17 (s, 3H), 3,89 (s, 4H), 3,77 (s, 3H), 2,63 (a s, 2H), 2,38 (a s, 2H), 1,79 (t, J= 6,26 Hz, 2H). La fracción 2 se identificó como 8-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2-il)-1,4-dioxaespiro-[4.5]decan-8-ol (0,254 g, 23,54 %) y se aisló en forma de una espuma blanquecina. Este material se liofilizó a partir de MeCN-agua para obtener un sólido color crema. LC (Método A): 2,193 min. HRMS (ESI): calc. para C26H27N4O7S2 [M+H]+ m/z 571,132; encontrado 571,132. RMN 1 H (400 MHz, DMSO-d6): 8 8,29 (s, 1H), 7,58 (s, 1H), 6,89 (s, 1H), 6,75 (s, 1H), 6,53 (s, 1H), 5,92 (s, 1H), 5,16 (s, 2H), 4,12 (s, 3H), 3,80 (s, 4H), 3,73 (s, 3H), 2,06 (dt, J= 3,13, 12,91 Hz, 2H), 1,78 (dt, J= 3,52, 13,30 Hz, 2H), 1,70 (d, J= 13,30 Hz, 2H), 1,53 (d, J= 12,52 Hz, 2H).
Ejemplo 128
4-hidroxi-4-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2-il)ciclohexanona

A una mezcla de 8-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2-il)-1,4-dioxaespiro[4.5]decan-8-ol (0,013 g, 0,023 mmol) en DCM (1 ml), se agregó TfA (0,2 ml), y la solución resultante se agitó a temperatura ambiente en un matraz sellado durante 18 h. Después los volátiles se evaporaron para obtener el producto impuro en forma de un sólido. La cromatografía ultrarrápida (Isco/ 0-100 % de EtOAc-DCM) produjo 4-hidroxi-4-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)-metil)tiazol-2-il)ciclohexanona (0,009 g, 75 %) en forma de un sólido de color blanco. LC (Método A): 2,208 min. HRMS (ESI): calc.
para C24H23N4O6S2 [M+H]+ m/z 527,105; encontrado 527,106. RMN 1H (400 MHz, DMSO-da): 58,30 (s, 1H), 7,66 (s, 1H), 6,91 (s, 1H), 6,76 (s, 1H), 6,54 (d, J= 1,57 Hz, 1H), 6,39 (s, 1H), 5,19 (s, 2H), 4,14 (s, 3H), 3,73 (s, 3H), 2,60 (m, 2H), 2,25 (dt, J= 4,70, 14,09 Hz, 2H), 2,18 (m, 2H), 2,06 (m, 2H).
Ejemplo 129
4-hidroxi-4-(4-(((6-metoxi-2-(2-metoxNmidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2-il)piperidin-1-carboxilato de ferc-butilo

129A. 4-(4-(((ferc-butildimetilsilil)oxi)metil)tiazol-2-il)-4-hidroxipiperidin-1 -carboxilato de ferc-butilo

Una solución de 4-(((ferc-butildimetilsilil)oxi)metil)tiazol (0,738 g, 3,22 mmol) en THF seco (8 ml) se enfrió a -78 °C en atmósfera de N2 y después se agregó gota a gota n-butil litio (1,45 M en hexanos, 2,440 ml, 3,54 mmol). La mezcla resultante se agitó durante 35 min para obtener una solución de color de color pardo claro. A esta mezcla se le agregó lentamente una solución de 4-oxopiperidin-1-carboxilato de ferc-butilo (0,769 g, 3,86 mmol) en THF seco (2 ml) y la mezcla se agitó a -78 °C durante 2 h para obtener una solución de color de color pardo claro. La reacción se inactivó después mediante la adición de NH4Cl acuoso saturado (5 ml), se retiró el baño de enfriamiento y la mezcla se repartió entre EtOAc-agua. La fase orgánica se separó, se lavó con salmuera, se secó (MgSO4) y se evaporó para obtener un aceite de color amarillo. Este aceite se purificó mediante cromatografía ultrarrápida usando hexanos-EtOAc como eluyente para obtener 4-(4-(((ferc-butildimetilsilil)oxi)metil)tiazol-2-il)-4-hidroxipiperidin-1-carboxilato de ferc-butilo (0,850 g, 61,6 %) en forma de una goma transparente e incolora:. LC (Método A): 2,427 min. LCMS (APCI): calc. para C20H37N2O4SSi [M+H]+ m/z 429,22, encontrado 429,20.
129B. 4-hidroxi-4-(4-(hidroximetil)tiazol-2-il)piperidin-1 -carboxilato de ferc-butilo
A una solución en agitación de 4-(4-(((ferc-butildimetilsilil)oxi)metil)tiazol-2-il)-4-hidroxipiperidin-1-carboxilato de fercbutilo (0,850 g, 1,983 mmol) en (11 ml), se le agregó trihidrofluoruro de trietilamina (1,60 ml, 9,83 mmol) y la mezcla de reacción se agitó a temperatura ambiente durante 4 h. Después la mezcla resultante se repartió entre EtOAc-NaHCO3 acuoso saturado y la fase orgánica se separó, se secó (MgSO4), se filtró y se concentró a sequedad. El residuo se purificó mediante cromatografía ultrarrápida usando DCM-EtOAc como eluyente para obtener 4-hidroxi-4-(4-(hidroximetil)tiazol-2-il)piperidin-1 -carboxilato de ferc-butilo (0,476 g, 77 %) en forma de un aceite transparente e incoloro. LC (Método A): 1,670 min. RMN 1 H (DMSO-d6, 400 MHz) 5 ppm: 7,27 (m, 1H), 6,09 (s, 1H), 5,23 (t, J = 5,7 Hz, 1H), 4,50 (d, J = 4,7 Hz, 2H), 6,82 (d, J = 11,3 Hz, 2H), 3,12 (a s, 1H), 1,90 (m, 2H), 1,68 (d, J = 12,9 Hz, 2H), 1,41 (s, 9H).
Ejemplo 129. 4-hidroxi-4-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2-il)piperidin-1 -carboxilato de ferc-butilo
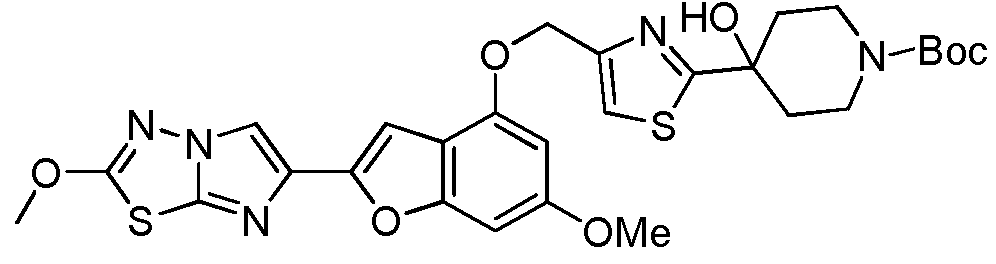
A una suspensión de 6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-ol (Ejemplo 1H, 0,202 g, 0,636 mmol) y 4-hidroxi-4-(4-(hidroximetil)tiazol-2-il)piperidin-1 -carboxilato de ferc-butilo (0,200 g, 0,636 mmol) en THF seco (8 ml), se le agregó tri-n-butilfosfina (0,413 ml, 1,590 mmol), seguido de una solución de ADDP (0,401 g,
1,590 mmol) en THF (2 ml), añadida gota a gota durante 30 min mediante una bomba de jeringa. Después de agitar durante 30 min más, la mezcla de reacción se repartió entre EtOAc y NaHCO3 acuoso saturado. La fase orgánica se separó, se lavó con salmuera, se secó (MgSO4), se filtró y se concentró a sequedad. El residuo se purificó mediante cromatografía ultrarrápida usando DCM-EtOAc como eluyente para obtener el compuesto del título (0,263 g, 0,429 mmol, 67,4 %) en forma de un sólido de color blanco. LC (Método A): 2,387 min. HRMS (ESI): calc. para C28H32N5O7S2 [M+H]+ m/z 614,1743, encontrado 614,1755. RMN 1H (DMSO-da, 400 MHz) 8 ppm: 8,37 (s, 1H), 7,70 (s, 1H), 6,97 (d, J = 0,8 Hz, 1H), 6,83 (dd, J = 0,8, 1,6 Hz, 1H), 6,61 (d, J =1,6 Hz, 1H), 6,19 (a s, 1H), 5,25 (s, 2H), 4,20 (s, 3H), 3,84 (m, 2H), 3,80 (s, 3H), 3,14 (a s, 1H), 1,94 (m, 2H), 1,72 (d, J = 12,9 Hz, 2H), 1,41 (s, 9H).
Ejemplo 130
(4-hidroxi-4-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2-il)piperidin-1-il)(fenil)metanona

A una suspensión en agitación de 4-hidroxi-4-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2-il)piperidin-1-carboxilato de tere-butilo (0,662 g, 1,079 mmol) en Dc M (10 ml), se le agregó TFA (3 ml) y la solución resultante se agitó durante 4 h a temperatura ambiente antes de que se concentrara a sequedad para obtener un aceite de color ámbar. Este aceite en bruto se repartió entre EtOAc-NaHCO3 acuoso saturado y la fase orgánica se separó, se secó (MgSO4), se filtró y se concentró a sequedad para obtener 2 ,2 ,2 -trifluoroacetato de 4-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2-il)piperidin-4-ol (0,554 g, 1,079 mmol, 100 %) en forma de un sólido de color beis. LC (Método A): 1,980 min. h Rm S (ESI): calc. para C23H24N5O5S2 [M+H]+ m/z 514,1219, encontrado 514,1228. A una solución en agitación de 4-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2-il)piperidin-4-ol (0,031 g, 0,050 mmol) en DMF (1 ml), se le agregó DIEA (0,070 ml, 0,400 mmol) y ácido benzoico (0,0073 g, 0,060 mmol), seguido de HATU (0,023 g, 0,060 mmol). La mezcla de reacción se agitó durante 1 h y después se diluyó con DMF (1 ml) y se purificó mediante HPLC preparativa (Método A). Las fracciones que contenían el producto deseado se concentraron a sequedad y el residuo se liofilizó a partir de MeCN-agua para obtener el compuesto del título (0,022 g, 71,2 %) en forma de un sólido amorfo de color blanco. LC (Método A): 2,257 min. HRMS (ESI): calc. para C30H28N5O6S2 [M+H]+ m/z 618,1481, encontrado 618,1484. RMN 1 H (DMSO-d6, 400 MHz) 8 ppm: 8,37 (s, 1H), 7,71 (s, 1H), 7,46 -7 ,40 (m, 5H), 6,98 (d, J = 0,8 Hz, 1H), 6,84 (dd, J = 0,8, 1,6 Hz, 1H), 6,63 (d, J = 1,6 Hz, 1H), 6,30 (a s, 1H), 5,26 (s, 2H), 4,36 (a s, 1H), 4,20 (s, 3H), 3,80 (s, 3H), 3,48 (m, 2H), 3,24 (a s, 1H), 2,03 (a s, 2H), 1,79 (m, 2H).
Ejemplo 131
4-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)-oxi)metil)-5-(trifluorometil)tiazol-2-il)tetrahidro-2H-piran-4-ol

131A. 5-yodotiazol-4-carboxilato de etilo

A una solución de 2-aminotiazol-4-carboxilato de etilo (2,92 g, 16,96 mmol) en diclorometano (100 ml), se le agregó NIS (5,00 g, 22,22 mmol). La mezcla de reacción resultante se agitó a temperatura ambiente durante 24 h y después
se diluyó con acetato de etilo, se lavó con agua y salmuera, se secó sobre MgSO4, se filtró y se concentró para obtener 2-amino-5-yodotiazol-4-carboxilato de etilo (4,75 g, 90 %) que se usó tal cual en la siguiente etapa. LC (Método A): 1,549 min. LCMS (APCI): calc. para C6H8IN2O2S [M+H]+ m/z 298,94, encontrado 299,0.
Una solución de 2-amino-5-yodotiazol-4-carboxilato de etilo (4,75 g, 15,93 mmol) en DMF (70 ml) se enfrió en un baño de hielo en atmósfera de nitrógeno y después se agregó terc-butilnitrito (2,74 ml, 23,04 mmol) y la mezcla se agitó a temperatura ambiente durante 2 h. La mezcla de reacción se vertió en salmuera y la mezcla se extrajo 3 veces con acetato de etilo. Los extractos orgánicos combinados se lavaron con salmuera, se secaron sobre MgSO4, se filtraron y se concentraron. El residuo en bruto obtenido se purificó mediante cromatografía de columna (Isco, cartucho de 8 g) eluyendo con un gradiente de acetato de etilo en hexanos (de 0 a 50 %) para obtener el compuesto puro del título (0,825 g, 18 %). LC (Método A): 1,601 min. LCMS (APCI): calc. para C6H7 INO2S [M+H]+ m/z 283,92, encontrado 283,9. RMN 1H (CDCh, 400 MHz) 6 ppm: 1,45 (t, J= 7,0 Hz, 3H), 4,46 (q, J= 7,0 Hz, 2H), 8,95 (s, 1H). 131B. 5-(trifluorometil)tiazol-4-carboxilato de etilo

A un tubo sellable cargado con 5-yodotiazol-4-carboxilato de etilo (0,825 g, 2,91 mmol) y DMF seco (20 ml), se le agregó 2,2-difluoro-2-(fluorosulfonil)acetato de metilo (0,742 ml, 5,83 mmol) y después yoduro de cobre (I) (1,110 g, 5,83 mmol). El recipiente de reacción se selló y la mezcla de reacción se agitó durante una noche a 85 °C (temperatura del baño). La mezcla de reacción enfriada se absorbió en éter y se filtró a través de una almohadilla CELITE®. Después el filtrado se lavó con agua y salmuera, se secó sobre MgSO4, se filtró y se concentró. El residuo en bruto obtenido se purificó mediante cromatografía en columna (Isco, cartucho de 24 g) eluyendo con un gradiente de EtOAc en hexanos (de 0 a 50 %) para obtener el compuesto deseado en forma de un sólido de color amarillo (0,438 g, 67 %). LC (Método A): 1,750 min. LCMS (APCI): calc. para C7H7F3NO2S [M+H]+ m/z 226,01, encontrado 226,0. RMN 1 H (CDCla, 400 MHz) 6 ppm: 1,43 (t, J= 7,4 Hz, 3H), 4,48 (q, J= 7,4 Hz, 2H), 8,90 (s, 1H).
131C. (5-(trifluorometil)tiazol-4-il)metanol
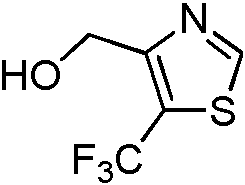
A una solución enfriada en hielo de 5-(trifluorometil)tiazol-4-carboxilato de etilo (0, 425 g, 1,887 mmol) en THF seco (50 ml), en atmósfera de nitrógeno, se le agregó borohidruro de litio (0,082 g, 3,77 mmol) de una vez, seguido de la adición gota a gota de metanol (0,153 ml, 3,77 mmol). Después se retiró el baño de enfriamiento y la mezcla se agitó a temperatura ambiente durante 1 h. La mezcla se volvió a enfriar a 0 °C y después se inactivó cuidadosamente mediante la adición lenta de NH4Cl acuoso saturado (10 ml) con agitación vigorosa. Después se retiró el baño de enfriamiento y la mezcla se repartió entre acetato de etilo-agua. La fase orgánica se separó, se lavó con salmuera, se secó sobre MgSO4, se filtró y se concentró. El residuo en bruto obtenido se purificó mediante cromatografía en columna (Isco, cartucho de 12 g) eluyendo con un gradiente de EtOAc en hexanos (de 0 a 100 %) para obtener el producto deseado en forma de un aceite de color amarillo claro (0,200 g, 58 %). Tiempo de retención de LC (Método A): 1,248 min. LCMS (APCI): calc. para C5H5F3NOS [M+H]+ m/z 184,00, encontrado 184,0. RMN 1 H (CDCh, 400 MHz) 6 ppm: 4,90 (s, 2H), 8,96 (s, 1H).
131D. 4-(((terc-butildimetilsilil)oxi)metil)-5-(trifluorometil)tiazol

A una solución de (5-(trifluorometil)tiazol-4-il)metanol (0,200 g, 1,092 mmol) en diclorometano (10 ml) a temperatura ambiente, se le agregó imidazol (0,112 g, 1,638 mmol) y después terc-butilclorodimetilsilano (0,206 g, 1,365 mmol). La mezcla de reacción resultante se agitó a temperatura ambiente durante 1 h y después la reacción se inactivó con MeOH y se concentró a presión reducida. El residuo en bruto se purificó mediante cromatografía en columna (Isco, cartucho de 12 g), eluyendo con un gradiente de acetato de etilo en hexanos (de 0 a 50 %) para obtener el producto deseado en forma de un aceite incoloro (0,145 g, 45 %). LC (Método A): 2,409 min. LCMS (APCI): calc. para
C11 H19F3NOSSÍ [M+H]+ m/z 298,09, encontrado 298,2. RMN 1 H (CDCI3, 400 MHz) 5 ppm: 0,11 (s, 6H), 0,91 (s, 9H), 4,92 (s, 2H), 8,84 (s, 1H).
131E. 4-(4-(((ferc-butildimetilsilil)oxi)metil)-5-(trifluorometil)tiazol-2-il)tetrahidro-2H-piran-4-ol

Una solución de 4-(((ferc-butildimetilsilil)oxi)metil)-5-(trifluorometil)tiazol (0,145 g, 0,488 mmol) en THF seco (5 ml) se enfrió a -78 °C en atmósfera de nitrógeno antes de agregar gota a gota n-butil litio (1,5 M en hexanos, 0,390 ml, 0,585 mmol). La solución obtenida se agitó durante 15 min a -78 °C antes de que se agregara lentamente una solución de dihidro-2H-piran-4(3H)-ona (0,054 ml, 0,585 mmol) en THF seco (1 ml). La mezcla de reacción resultante se agitó a -78 °C durante 1 h y después se inactivó mediante la adición de NH4Cl acuoso saturado (1,5 ml). Después se retiró el baño de enfriamiento y la mezcla se diluyó con acetato de etilo. La fase orgánica se separó, se lavó (salmuera), se secó (MgSO4), se filtró y se evaporó para obtener un aceite de color amarillo claro. El residuo en bruto obtenido se purificó mediante cromatografía en columna (Isco, cartucho de 12 g) eluyendo con un gradiente de EtOAc en hexanos (de 0 a 50 %) para obtener el compuesto deseado en forma de un aceite de color amarillo claro (0,185 g, 95%). LC (Método A): 2,415 min. LCMS (APCI): calc. para C-,6H27F3NO3SSi [M+H]+ m/z 398,14, encontrado 398,2. RMN 1H (CDCh, 400 MHz) 5 ppm: 0,10 (s, 6H), 0,91 (s, 9H), 1,61 (a s, 1H), 1,75-1,82 (m, 2H), 2,25-2,36 (m, 2H), 3,83-3,97 (m, 4H), 4,84 (d, J= 0,8 Hz, 2H).
131F. 4-(4-(hidroximetil)-5-(trifluorometil)tiazol-2-il)tetrahidro-2H-piran-4-ol

A una solución de 4-(4-(((ferc-butildimetilsilil)oxi)metil)-5-(trifluorometil)tiazol-2-il)tetrahidro-2H-piran-4-ol (0,185 g, 0,465 mmol) en THF (3 ml) a temperatura ambiente, se le agregó TBAF (75 % de solución en agua, 0,252 ml, 0,698 mmol). Después de 30 min de agitación, se agregó otro equivalente de TBAF (75 % de solución en agua, 0,168 ml, 0,465 mmol) y la mezcla se agitó durante 1 h más. La mezcla resultante se inactivó con salmuera y después se agregó diclorometano. La capa orgánica aislada se lavó con salmuera, se secó sobre MgSO4, se filtró y se concentró. El residuo en bruto obtenido se purificó mediante cromatografía en columna (Isco, cartucho de 12 g) eluyendo con un gradiente de acetato de etilo en hexanos (de 0 a 10 0 %) para obtener el compuesto deseado en forma de un aceite incoloro (0,101 g, 77 %). LC (Método A): 1,504 min. LCMS (APCI): calc. para C10H13F3NO3S [M+H]+ m/z 284,06, encontrado 284,0. RMN 1H (CDCh, 400 MHz) 5 ppm: 1,65 (a s, 1H), 1,79 (dd, J= 2,0, 14,0 Hz, 2H), 2,28-2,38 (m, 2H), 2,71 (a s, 1H), 3,82-3,97 (m, 4H), 4,81 (d, J= 1,2 Hz, 2H).
Ejemplo 131. 4-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)-5-(trifluorometil)tiazol-2-il)tetrahidro-2H-piran-4-ol

A una mezcla de 6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-ol (Ejemplo 1H, 0,090 g, 0,284 mmol) y 4-(4-(hidroximetil)-5-(trifluorometil)tiazol-2-il)tetrahidro-2H-piran-4-ol (0,100 g, 0,355 mmol) en atmósfera de nitrógeno, se le agregó THF seco (5 ml). A la suspensión resultante se le agregó tri-n-butilfosfina (0,184 ml, 0,709 mmol) seguido de la adición gota a gota de una solución de 1,1'-(azodicarbonil)dipiperidina (0,181 g, 0,709 mmol) en THF seco (2,5 ml). La mezcla de reacción resultante se agitó a temperatura ambiente durante 45 min y después se diluyó con acetato de etilo, se lavó con NaHCO3 acuoso saturado, se secó sobre MgSO4, se filtró y se concentró. El residuo en bruto obtenido se purificó mediante cromatografía en columna (Isco, cartucho de 24 g) que se eluyó con un gradiente de EtOAc en DCM (de 0 a 50 %) para obtener el compuesto del título en forma de un sólido de color blanco (0,108 g, 65 %). LC (Método A): 2,689 min. HRMS (ESI): calc. para C24H22F3N4O6S2 [M+H]+ m/z 583,0933, encontrado 523,0967. RMN 1 H (DMSO-d6, 400 MHz) 5 ppm: 1,70 (d, J= 13,3 Hz, 2H), 2,06-2,17 (m, 2H), 3,62-3,83 (m, 4H), 3,81 (s, 3H), 4,20 (s, 3H), 5,37 (s, 2H), 6,63 (d, J= 9,0 Hz, 2H), 6 ,86 (s, 2H), 8,38 (s, 1H).
Ejemplo 132
6-(4-((2-(4-fluorotetrahidro-2H-piran-4-il)-5-(trifluorometil)tiazol-4-il)metoxi)-6-metoxibenzofuran-2-il)-2-metoxiimidazo[2,1-b][1,3,4]tiadiazol

A una suspensión enfriada en hielo de 4-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)-5-(trifluorometil)tiazol-2-il)tetrahidro-2H-piran-4-ol (0,052 g, 0,089 mmol) en diclorometano (10 ml), en nitrógeno, se le agregó gota a gota DAST (0,029 ml, 0,223 mmol). La mezcla resultante se agitó a 0 °C durante 20 min y después se retiró el baño de enfriamiento y la solución resultante de color amarillo claro se agitó a temperatura ambiente durante 1 h. La mezcla de reacción se volvió a enfriar a 0 °C, se inactivó mediante la adición gota a gota de NaHCO3 acuoso saturado (5 ml) y se agitó vigorosamente durante 15 min para garantizar la inactivación completa. La mezcla resultante se diluyó con diclorometano y bicarbonato de sodio acuoso saturado después la capa orgánica se separó, se secó sobre sulfato de magnesio y se concentró a presión reducida. El residuo en bruto obtenido se purificó mediante cromatografía en columna (Isco, cartucho de 24 g) eluyendo con un gradiente de EtOAc en DCM (de 0 a 50 %) para obtener el compuesto del título en forma de un sólido de color blanco (0,045 g, 86 %). LC (Método A): 2,492 min. LCMS (ESI): calc. para C24H21F4N4O5S2 [M+H]+ m/z 585,0890, encontrado 585,0904. RMN 1H (DMSO-da, 400 MHz) 8 ppm: 2,03-2,39 (m, 4H), 3,69 (td, J= 2,0, 11,7 Hz, 2H), 3,82 (s, 3H), 3,85-3,92 (m, 2H), 4,20 (s, 3H), 5,43 (s, 2H), 6,65 (d, J= 1,6 Hz, 1H), 6,87 (s, 2H), 8,38 (s, 1H).
Ejemplo 133
1-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2-il)ciclohexanol

133A: 1-(4-(hidroximetil)tiazol-2-il)ciclohexanol

Método general: Una solución de 2-bromo-4-(((terc-butildimetilsilil)oxi)metil)-tiazol (Ejemplo 37B, 0,195 g, 0,632 mmol) en THF seco (5 ml) se enfrió a -78 °C en atmósfera de nitrógeno antes de que se agregara gota a gota n-butil litio (1,5 M en hexanos, 0,506 ml, 0,759 mmol). La mezcla resultante se agitó durante 15 min y después se agregó una solución de ciclohexanona (0,075 mg, 0,759 mmol) en THF seco (1 ml) y la agitación continuó a -78 °C durante 1 h. Después la reacción se inactivó mediante la adición de NH4Cl acuoso saturado (1,5 ml), se retiró el baño de enfriamiento y la mezcla se diluyó con acetato de etilo. La fase orgánica se separó, se lavó (salmuera), se secó (MgSO4), se filtró y se evaporó para obtener un aceite de color amarillo claro. El 1-(4-(((terc-butildimetilsilil)-oxi)metil)tiazol-2-il)ciclohexanol en bruto obtenido (0,185 g, 89 %) se usó tal cual en la siguiente etapa. LC (Método A): 2,391 min. LCMS (APCI): calc. para C16HaüNO2SSi [M+H]+ m/z 328,18, encontrado 328,2.
A una solución de 1-(4-(((terc-butildimetilsilil)oxi)metil)tiazol-2-il)ciclo-hexanol en bruto (0,185 g, 0,480 mmol) en THF (3 m) a temperatura ambiente, se le agregó TBAF (75 % de solución en agua, 0,260 ml, 0,720 mmol). Después de 30 min, se agregó otro equivalente de TBAF (75 % de solución en agua, 0,173 ml, 0,480 mmol) y la mezcla resultante se agitó durante 1 h más. Después la mezcla de reacción se inactivó con salmuera y se agregó DCM. La capa orgánica aislada se lavó con salmuera, se secó sobre MgSO4, se filtró y se concentró. El residuo en bruto obtenido se purificó mediante cromatografía en columna (Isco, cartucho de 12 g) eluyendo con un gradiente de acetato de etilo en hexanos (de 0 a 10 0 %) para obtener el compuesto deseado en forma de un aceite incoloro (0,076 g, 74%). LC (Método A): 1,319 min. LCMS (APCI): calc. para C10H16NO2S [M+H]+ m/z 214,09, encontrado 214,2. RMN 1 H (DMSO-d6, 400 MHz) 8 ppm: 1,18-1,32 (m, 1H), 1,48-1,75 (m, 7H), 1,84 (td, J= 3,9, 12,9 Hz, 2H),
4,50 (dd, J= 0,8, 5,9 Hz, 2H), 5,22 (t, J= 5,9 Hz, 1H), 5,69 (s, 1H), 7,21 (s, 1H).
Ejemplo 133. 1-(4-(((6-metoxi-2-(2-metoxümidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2-il)ciclohexanol

A una mezcla de 6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-ol (Ejemplo 1H, 0,090 g, 0,284 mmol) y 1-(4-(hidroximetN)-tiazol-2-N)cidohexanol (0,076 g, 0,355 mmol) en atmósfera de nitrógeno a temperatura ambiente, se le agregó THF seco (5 ml). A la suspensión resultante, se le agregó tri-n-butilfosfina (0,184 ml, 0,709 mmol) y después se agregó gota a gota una solución de 1,1'-(azodicarbonil)dipiperidina (0,181 g, 0,709 mmol) en THF seco (2,5 ml) y la mezcla resultante se agitó a temperatura ambiente durante 45 min. La mezcla de reacción se diluyó con acetato de etilo y se lavó con NaHCO3 acuoso saturado, se secó sobre MgSO4, se filtró y se concentró. El residuo en bruto obtenido se purificó mediante cromatografía en columna (Isco, cartucho de 24 g) eluyendo con un gradiente de acetato de etilo en diclorometano (de 0 a 50 %) para obtener el compuesto del título en forma de un sólido de color blanco (0,104 g, 72 %). LC (Método A): 2,339 min. LCMS (ESI): calc. para C24H25N4O5S2 [M+H]+ m/z 513,1266, encontrado 513,1256. RMN 1H (DMSO-da, 400 MHz) 8 ppm: 1,20-1,36 (m, 1H), 1,48-1,79 (m, 7H), 1,88 (td, J= 3,5, 12,5 Hz, 2H), 3,81 (s, 3H), 4,20 (s, 3H), 5,24 (s, 2H), 5,80 (s, 1H), 6,61 (d, J= 2,0 Hz, 1H), 6,82-6,84 (m, 1H), 6,98 (s, 1H), 7,64 (s, 1H), 8,37 (s, 1H).
Ejemplo 134
6-(4-((2-(1-fluorociclohexil)tiazol-4-il)metoxi)-6-metoxibenzofuran-2-il)-2-metoxiimidazo[2,1-b][1,3,4]tiadiazol

A una suspensión enfriada en hielo de 1-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2-il)ciclohexanol (0,050 g, 0,098 mmol) en diclorometano (10 ml), en atmósfera de nitrógeno, se le agregó gota a gota DAST (0,032 ml, 0,244 mmol). La mezcla resultante se agitó a 0 °C durante 15 min, después se retiró el baño de enfriamiento y la solución resultante de color amarillo claro se agitó a temperatura ambiente durante 1 h. La mezcla de reacción se volvió a enfriar a 0 °C, se inactivó mediante la adición gota a gota de NaHCO3 acuoso saturado (5 ml) y se agitó vigorosamente durante 15 min para garantizar la inactivación completa. La mezcla resultante diluyéndose volvió a diluir con diclorometano y solución de bicarbonato de sodio acuoso saturado y la capa orgánica se separó, se secó sobre sulfato de magnesio y se concentró a presión reducida. El residuo en bruto obtenido se purificó mediante cromatografía en columna (Isco, cartucho de 24 g) eluyendo con un gradiente de acetato de etilo en diclorometano (de 0 a 50 %) para obtener el compuesto del título en forma de un sólido de color blanco (0,037 g, 74%). LC (Método A): 2,524 min. LCMS (ESI): calc. para C24H24FN4O4S2 [M+H]+ m/z 515,1223, encontrado 515,1203. RMN 1 H (DMSO-d6, 400 MHz) 8 ppm: 1,30-1,46 (m, 1H), 1,56-1,73 (m, 5H), 1,95-2,13 (m, 4H), 3,81 (s, 3H), 4,20 (s, 3H), 5,30 (s, 2H), 6,62 (d, J= 2,0 Hz, 1H), 6,83-6,85 (m, 1H), 6,99 (s, 1H), 7,88 (s, 1H), 8,37 (s, 1H).
Ejemplo 135
4,4-difluoro-1-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2-il)ciclohexanol

135A. 1-(4-(((ferc-butildimetilsilil)oxi)metil)tiazol-2-il)-4,4-difluorociclohexanol

Una solución de 2-bromo-4-(((terc-butildimetilsilil)oxi)metil)tiazol (Ejemplo 37B, 0,500 g, 1,622 mmol) en THF seco (8 ml) se enfrió a -78 °C en atmósfera de N2 y después se agregó gota a gota n-butil litio (1,45 M en hexanos, 0,714 ml, 1,784 mmol). La mezcla resultante se agitó durante 35 min para obtener una solución de color de color pardo claro. A esta mezcla se le agregó lentamente una solución de 4,4-difluorociclohexanona (0,218 g, 1,622 mmol) en THF seco (2 ml) y la mezcla se agitó a -78 °C durante 2 h para obtener una solución de color de color pardo claro. La reacción después se inactivó mediante la adición de NH4Cl acuoso saturado (5 ml), se retiró el baño de enfriamiento y la mezcla se repartió entre EtOAc-agua. La fase orgánica se separó, se lavó con salmuera, se secó (MgSO4) y se evaporó para obtener un aceite de color amarillo claro. Este aceite se purificó mediante cromatografía ultrarrápida usando DCM-EtOAc como eluyente para obtener 1-(4-(((terc-butildimetilsilil)oxi)metil)tiazol-2-il)-4,4-difluorociclohexanol (0,289 g, 49,0 %) en forma de un sólido de color beis. LC (Método A): 2,354 min. RMN 1H (DMSO-da, 400 MHz) 8 ppm: 7,32 (s, 1H), 6,21 (s, 1H), 4,70 (s, 2H), 2,21-1,99 (m, 6 H), 1,86 (m, 2H), 0,88 (s, 9H), 0,06 (s, 6 H).
135B. 4,4-difluoro-1-(4-(hidroximetil)tiazol-2-il)ciclohexanol

A una solución de 1-(4-(((terc-butildimetilsilil)oxi)metil)tiazol-2-il)-4,4-difluorociclohexanol (0,289 g, 0,795 mmol) en THF seco (10 ml), en atmósfera de N2 , se le agregó gota a gota trihidrofluoruro de trietilamina (0,647 ml, 3,97 mmol) y la mezcla se agitó a temperatura ambiente durante 16 h. Después la mezcla se diluyó con EtOAc y la solución se lavó (NaHCO3 acuoso saturado), se secó (MgSO4) y se evaporó para obtener 4,4-difluoro-1-(4-(hidroximetil)tiazol-2-il)ciclohexanol (0,164 g, 83 %) en forma de un sólido de color blanco. Este material se usó tal cual en la siguiente etapa sin purificación adicional. LC (Método A): 1,332 min. RMN 1 H (DMSO-d6, 400 MHz) 8 ppm: 7,28 (s, 1H), 6,18 (s, 1H), 5,25 (t, J = 5,9 Hz, 1H), 4,50 (d, J = 5,5 Hz, 2H), 2,21 - 1,99 (m, 6 H), 1,88 (m, 2H).
Ejemplo 135. 4,4-difluoro-1-(4-(((6-metoxi-2-(2-metoxiimidazo-[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2 -il)ciclohexanol

A una suspensión de 6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-ol (Ejemplo 1H, 0,186 g, 0,586 mmol) y 4,4-difluoro-1-(4-(hidroximetil)tiazol-2-il)ciclohexanol (0,146 g, 0,586 mmol) en t Hf seco (8 ml), se le agregó tri-n-butilfosfina (0,380 ml, 1,464 mmol), seguido de una solución de ADDP (0,369 g, 1,464 mmol) en THF (2 ml) añadida gota a gota durante 30 min mediante una bomba de jeringa. Después de agitar durante 30 min más, la mezcla de reacción se repartió entre EtOAc y NaHCO3 acuoso saturado. La fase orgánica se separó, se lavó con salmuera, se secó (MgSO4), se filtró y se concentró hasta secarse. El residuo se purificó mediante cromatografía ultrarrápida usando DCM-EtOAc como eluyente para obtener un sólido de color beis con un tinte amarillo. Este sólido se volvió a triturar con acetonitrilo y el sólido resultante se filtró, se enjuagó con dietiléter y se secó al vacío para obtener el compuesto del título (0,250 g, 78 %) en forma de un sólido de color beis. LC (Método A): 2,322 min. HRMS (ESI): calc. para C24H23F2N4O5S2 [M+H]+ m/z 549,1078, encontrado 549,1101. RMN 1 H (DMSO-d6, 400 MHz) 8 ppm: 8,37 (s, 1H), 7,71 (s, 1H), 6,98 (s, 1H), 6,83 (s, 1H), 6,62 (d, J = 1,6 Hz, 1H), 6,29 (a s, 1H), 5,26 (s, 2H), 4,20 (s, 3H), 3,80 (s, 3H), 2,20-2,01 (m, 6 H), 1,91 (m, 2H).
Ejemplo 136
2-metoxi-6-(6-metoxi-4-((2-(1,4,4-trifluorociclohexil)tiazol-4-il)metoxi)benzofuran-2-il)imidazo[2,1-b][1,3,4]tiadiazol

A una mezcla enfriada en hielo de 4,4-difluoro-1-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2-il)ciclohexanol (0,050 g, 0,091 mmol) en diclorometano (4 ml), en atmósfera de N2 ,
se le agregó gota a gota DAST (0,036 ml, 0,273 mmol). La mezcla de reacción se agitó a 0 °C durante 1 h antes de inactivarla con NaHCO3 acuoso saturado. La mezcla resultante se extrajo con EtOAc, después de lo cual la fase orgánica se lavó con salmuera, se secó (MgSO4), se filtró y se concentró a sequedad. El residuo se purificó mediante cromatografía ultrarrápida usando un gradiente de 0 a 100 % de EtAOc en DCM para obtener el compuesto del título (0,007 g, 0,013 mmol, 13,95 %) en forma de un sólido de color blanco. LC (Método A): 2,322 min. HRMS (ESI): calc. para C24H22F3N4O4S2 [M+H]+ m/z 551,1035, encontrado 551,1055. RMN 1H (DMSO-da, 400 MHz) 8 ppm: 8,37 (s, 1H), 7,94 (s, 1H), 6,99 (s, 1H), 6,84 (d, J = 0,8 Hz, 1H), 6,62 (d, J = 2,0 Hz, 1H), 5,31 (s, 2H), 4,20 (s, 3H), 3,81 (s, 3H), 2,34-2,07 (m, 8 H).
Preparación de alcoholes
Los siguientes alcoholes adicionales se prepararon de acuerdo con el procedimiento general descrito en el ejemplo 133A;
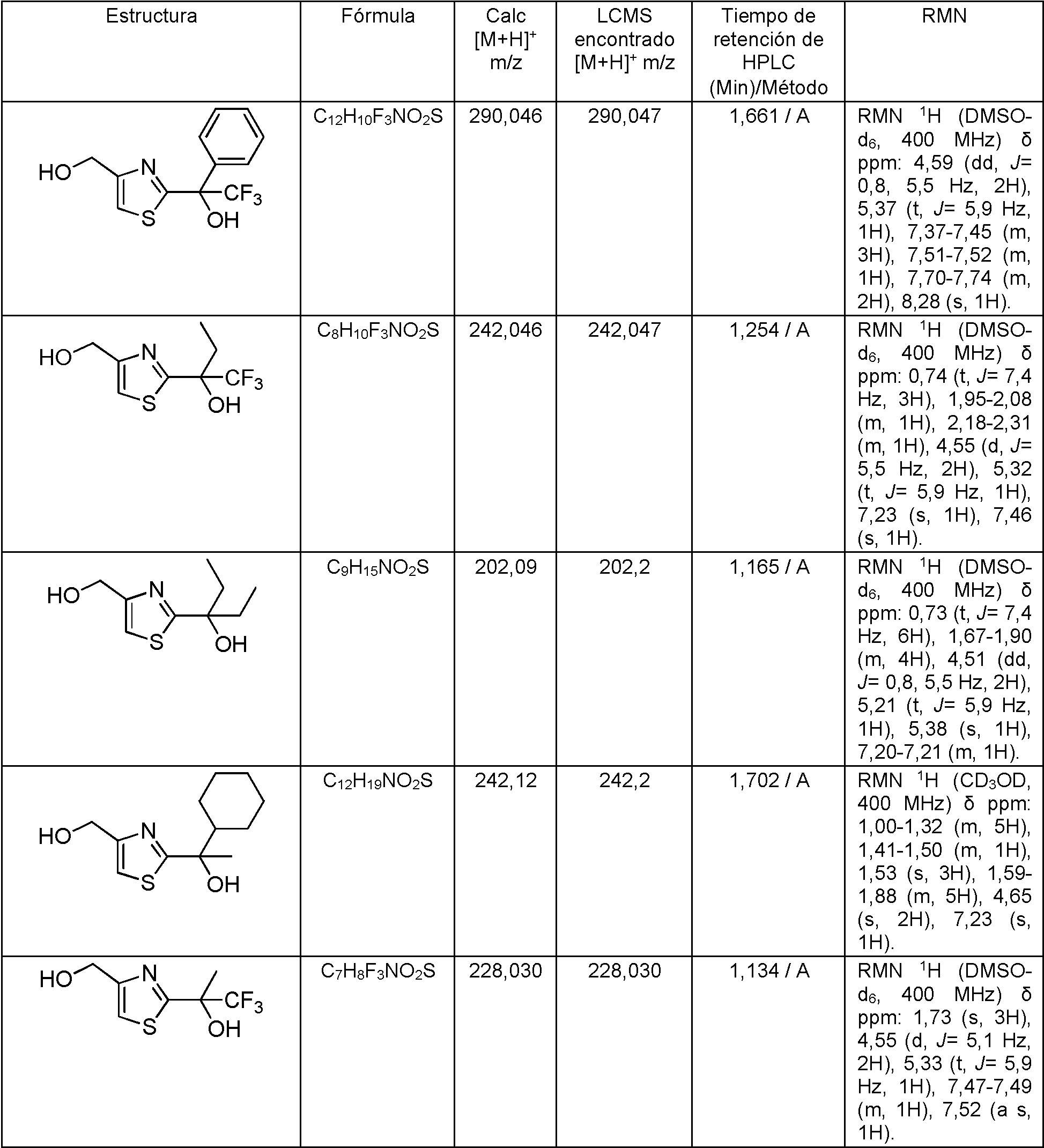
continuación
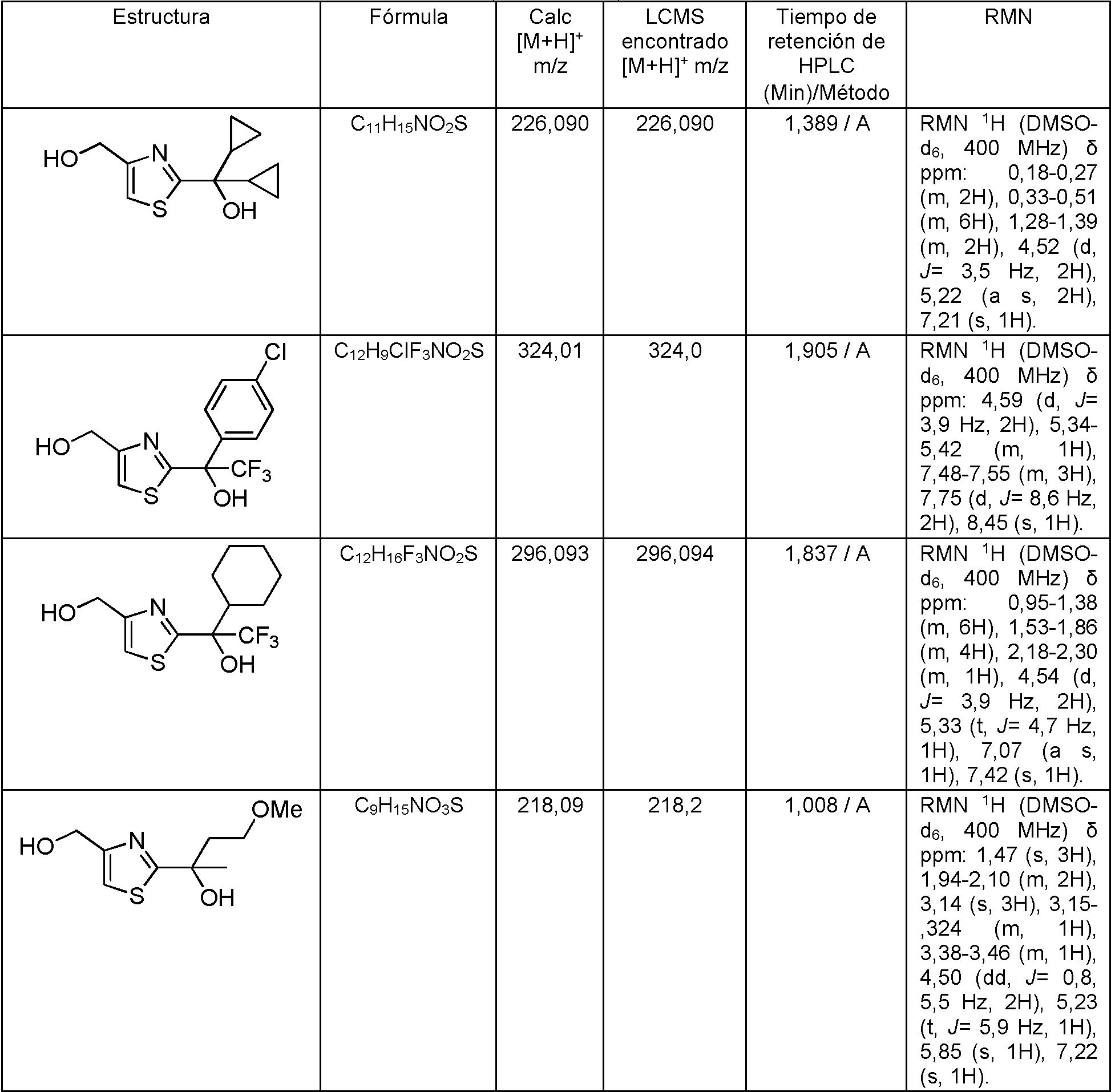
Ejemplos 137 a 147
Los siguientes ejemplos adicionales se prepararon, aislaron y caracterizaron usando los métodos descritos en los Ejemplos 133 y 134 anteriores.
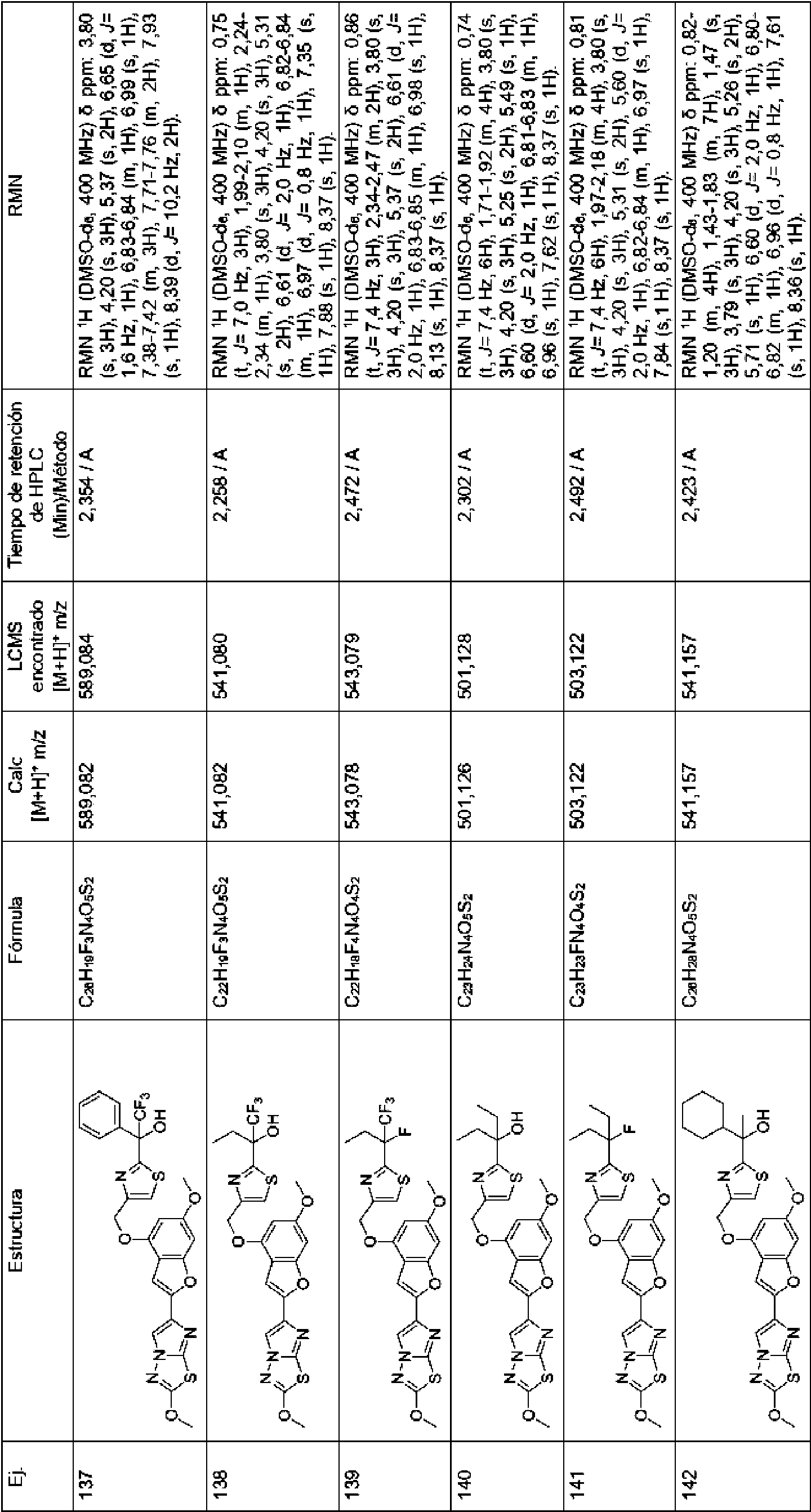

Ejemplos 148 y 149
4-(4-(((6-metoxi-2-(2-metoxNmidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2-il)cidohex-3-encarboxilato de ferc-butilo y 4-hidroxi-4-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-N)oxi)metN)tiazol-2-N)cidohexancarboxNato de ferc-butilo, respectivamente.

148A. Ácido 4-oxociclohexancarboxílico

A una solución de 4-oxociclohexancarboxilato de etilo (5,00 g, 29,4 mmol) en una mezcla de metanol (30 ml) y THF (125 ml), se le agregó una solución acuosa de NaOH (3 N, 29,4 ml, 88 mmol) y la mezcla de reacción resultante se calentó a 60 °C durante 3 h. La mezcla enfriada se concentró a presión reducida, el concentrado acuoso se acidificó (pH 1) con HCl 1 N y la mezcla se extrajo con DCM (x3). El extracto orgánico combinado se lavó con salmuera, se secó sobre MgSO4, se filtró y se concentró para obtener el compuesto del título (3,175 g, 76 %). RMN 1 H (DMSO-d6, 400 MHz) 8 ppm: 1,70-1,86 (m, 2H), 2,03-2,14 (m, 2H), 2,18-2,30 (m, 2H), 2,32-2,46 (m, 2H), 2,66-2,77 (m, 1H), 12,32 (a s, 1H).
148B. 4-oxociclohexancarboxilato de ferc-butilo

A una solución enfriada en hielo de ácido 4-oxociclohexancarboxílico (3,175 g, 22,34 mmol) en piridina (12 ml, 148 mmol) y ferc-butanol (17 ml, 178 mmol), se le agregó POCh puro (3,0 ml, 32,2 mmol). Después se retiró el baño de enfriamiento y la mezcla de reacción se agitó a temperatura ambiente durante 4 h. La mezcla cruda se vertió después en agua y el producto se extrajo con EtOAc (3x). El extracto orgánico combinado se lavó con HCl 2 N (x2) y salmuera, se secó sobre MgSO4, se filtró y se concentró. El residuo en bruto obtenido se usó tal cual sin purificación adicional (3,02 g, 68 %). RMN 1 H (DMSO-d6, 400 MHz) 8 ppm: 1,41 (s, 9H), 1,70-1,85 (m, 2H), 2,01-2,13 (m, 2H), 2,18-2,29 (m, 2H), 2,32-2,45 (m, 2H), 2,63-2,75 (m, 1H).
148C. 4-hidroxi-4-(4-(hidroximetil)tiazol-2-il)ciclohexancarboxilato de ferc-butilo

A una solución de 2-bromo-4-((( fe rc - butildimetilsilil)oxi)metil)tiazol (Ejemplo 37B, 0,200 g, 0,649 mmol) en THF seco (5 ml), enfriada a -78 °C en atmósfera de nitrógeno, se le agregó gota a gota n-butil litio (1,5 M en hexanos, 0,519 ml, 0,778 mmol). La mezcla resultante se agitó durante 5 min antes de que una solución previamente enfriada (-78 °C) de 4-oxociclohexancarboxilato de ferc-butilo (0,129 g, 0,649 mmol) en THF seco (1 ml) se agregara mediante una cánula en la mezcla de reacción. La mezcla resultante se agitó a -78 °C durante 1,5 h, y después la reacción se inactivó mediante la adición de NH4Cl acuoso saturado (3 ml). Después se retiró el baño de enfriamiento y la mezcla se diluyó con acetato de etilo. La fase orgánica se separó, se lavó (salmuera), se secó (MgSO4), se filtró y se evaporó para obtener el producto en bruto, 4-(4-(((ferc-butildimetilsilil)oxi)metil)tiazol-2-il)-4-hidroxiciclohexancarboxilato de ferc-butilo (0,177 g, 64 %), en forma de un aceite de color amarillo claro. Este material se usó tal cual en la siguiente etapa sin purificación adicional. LC (Método A): 2,447 min. LCMS (APCI): calc. para C2-iH3aNO4SSi [M+H]+ m/z 428,23, encontrado 428,2.
A una solución de 4-(4-(((ferc-butildimetilsilil)oxi)metil)tiazol-2-il)-4-hidroxiciclohexancarboxilato de ferc-butilo (0,277 g, 0,415 mmol) en THF (5 ml) a temperatura ambiente, se le agregó TBAF (75 % de solución en agua, 0,299 ml, 0,829 mmol). La mezcla resultante se agitó durante 1 h, después la reacción se inactivó con salmuera y la mezcla resultante se diluyó con diclorometano. La capa orgánica se separó, se lavó con salmuera, se secó sobre MgSO4, se filtró y se concentró. El residuo en bruto obtenido se purificó mediante cromatografía en columna (Isco, cartucho de 1 2 g) eluyendo con un gradiente de acetato de etilo en diclorometano (de 0 a 10 0 %) para obtener el compuesto deseado en forma de un aceite viscoso incoloro que era una mezcla de isómeros cis y trans (0,073 g, 56 %). LC
(Método A ): 1,768, 1,789 min. LCMS (APCI): calc. para C15H24NO4S [M+H]+ m/z 314,14, encontrado 314,2 . RMN 1H (DMSO-da, 400 MHz) 8 ppm: 1,40 (s, 9H), 1,52-1,93 (m, 7H), 2,01-2,12 (m, 1H), 2,17-2,28 (m, 1H), 4,50 (s, 2H), 5,23 (t, J= 5,1 Hz, 1H), 5,78 (d, J= 11,7 Hz, 1H), 7,24 (d, J= 11,3 Hz, 1H).
Ejemplos 148 y 149. 4-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2-N)cidohex-3-encarboxilato de ferc-butilo y 4-hidroxi-4-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2-il)ciclohexancarboxilato de ferc-butilo, respectivamente.

A una mezcla de 6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-ol (Ejemplo 1H, 0,070 g, 0,221 mmol) y 4-hidroxi-4-(4-(hidroximetil)tiazol-2-il)ciclohexancarboxilato de ferc-butilo (0,069 g, 0,221 mmol), se le agregó THF seco (5 ml) y el recipiente se purgó con nitrógeno. A la suspensión resultante se le agregó tri-nbutilfosfina (0,143 ml, 0,551 mmol), seguido de la adición gota a gota de una solución de 1,1'-(azodicarbonil)dipiperidina (0,141 g, 0,551 mmol) en THF seco (2,5 ml). La mezcla de reacción resultante se agitó a temperatura ambiente durante 1,5 h, después se diluyó con acetato de etilo, se lavó con NaHCO3 acuoso saturado, se secó sobre MgSO4, se filtró y se concentró. El residuo en bruto obtenido se purificó mediante cromatografía ultrarrápida (Isco, cartucho de 24 g) usando un gradiente de 0-100 % de EtOAc en DCM para obtener dos fracciones. La fracción 1 se obtuvo en forma de un sólido que se volvió a triturar con MeOH para obtener (después de la filtración, el lavado con MeOH y el secado al vacío) 4-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2-il)ciclohex-3-encarboxilato de ferc-butilo (0,011 g, 8 %) en forma de un sólido de color blanco. LC (Método A): 2,567 min. HRMS (ESI) calc. para C29H31N4O6S2 [M+H]+ m/z 595,168, encontrado 595,168. RMN 1 H (DMSO-d6, 400 MHz) 8 ppm: 1,38 (s, 9H), 1,64 (m, 1H), 2,00 (m, 1H), 2,25-2,65 (m, 5H), 3,77 (s, 3H), 4,17 (s, 3H), 5,23 (s, 2H), 6,57 (d, J= 1,57 Hz, 1H), 6,61 (a s, 1H), 6,80 (s, 1H), 6,97 (s, 1H), 7,66 (s, 1H), 8,33 (s, 1H). La fracción 2 se volvió a purificar mediante HPLC preparativa (Método A) para obtener 4-hidroxi-4-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2-il)ciclohexancarboxilato de fercbutilo en forma de un sólido de color blanco que era una mezcla de isómeros cis y trans (0,010 g, 7 %). LC (Método A): 2,320 min. HRMS (ESI): calc. para C29H33N4O7S2 [M+H]+ m/z 613,1791, encontrado 613,1789. RMN 1 H (DMSO-d6, 400 MHz) 8 ppm: 1,39 (s, 4,5H), 1,41 (s, 4,5H), 1,61-1,98 (m, 7H), 2,04-2,16 (m, 1H), 2,21-2,31 (m, 1H), 3,80 (s, 3H), 4,21 (s, 3H), 5,24 (s, 1H), 5,25 (s, 1H), 5,87 (a s, 1H), 6,60 (d, J= 2,0 Hz, 0,5H), 6,61 (d, J= 2,0 Hz, 0,5H), 6,81 6,84 (m, 1H), 6,97 (d, J= 0,8 Hz, 0,5H), 6,98 (d, J= 0,8 Hz, 0,5H), 7,65 (s, 0,5H), 7,68 (s, 0,5H), 8,36 (s, 0,5H), 8,37 (s, 0,5H).
Ejemplo 150
Ácido 4-hidroxi-4-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2-il)ciclohexancarboxílico

A una solución de 4-hidroxi-4-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2-il)ciclohexancarboxilato de ferc-butilo (0,486 g, 0,587 mmol) en diclorometano (20 ml) a temperatura ambiente, se le agregó TFA (2,261 ml, 29,3 mmol) y la mezcla se agitó durante 1,5 h. La mezcla resultante se diluyó con tolueno y los volátiles se retiraron después a presión reducida. El aceite de color naranja obtenido se coevaporó con metanol (x2) para obtener el compuesto crudo del título (0,297 g, 91 %) en forma de un sólido de color naranja que era una mezcla de isómeros cis y trans. Este material se usó tal cual en la siguiente etapa sin purificación adicional. LC (Método A): 2,110, 2,142 min. HRMS (ESI): calc. para C25H25N4O7S2 [M+H]+ m/z 557,1165, encontrado 557,1169. RMN 1 H (DMSO-d6, 400 MHz) 8 ppm: 1,60-2,15 (m, 8 H), 2,24-2,35 (m, 1H), 2,21 2,31 (m, 1H), 3,81 (s, 3H), 4,21 (s, 3H), 5,24 (d, J= 5,4 Hz, 1H), 5,26 (s, 1H), 5,90 (s, 0,5H), 5,92 (s, 0,5H), 6,61 (d, J= 2,0 Hz, 0,5H), 6,62 (d, J= 2,0 Hz, 0,5H), 6,82-6,85 (m, 1H), 6,99 (d, J= 0,4 Hz, 0,5H), 7,00 (d, J= 0,8 Hz, 0,5H), 7,66 (s, 0,5H), 7,67 (s, 0,5H), 8,38 (s, 1H).
Ejemplo 151
(4-hidroxi-4-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2-il)ciclohexil)(pirrolidin-1 -il)metanona

A una solución de ácido 4-hidroxi-4-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-N)oxi)metN)tiazol-2-N)cidohexancarboxíNco (0,025 g, 0,045 mmol) y pirrolidina (3,71 pl, 0,045 mmol) en DMF (1 ml), se le agregó DIEA (0,039 ml, 0,225 mmol), seguido de HATU (0,0214 g, 0,056 mmol). La mezcla de reacción resultante se agitó a temperatura ambiente durante 3 h, después se diluyó con acetato de etilo y se lavó con NaHCO3 acuoso saturado y salmuera. La capa orgánica se separó, se secó sobre MgSO4, se filtró y se concentró. El residuo en bruto obtenido se purificó mediante HPLC preparativa (Método A) para obtener dos fracciones. La fracción 1 se identificó como el isómero A del compuesto del título (0,008 g, 25 %). LC (Método A): 2,236 min. HRMS (ESI): calc. para C29H32N5O6S2 [M+H]+ m/z 610,1794, encontrado 610,1777. RMN 1 H (DMSO-da, 400 MHz) 8 ppm: 1,60-1,91 (m, 11), 2,28-2,36 (m, 2H), 3,24 (t, J= 7,0 Hz, 2H), 3,45 (t, J= 6,7 Hz, 2H), 3,80 (s, 3H), 4,19 (s, 3H), 5,25 (s, 2H), 5,80 (a s, 1H), 6,61 (d, J= 2,0 Hz, 1H), 6,81-6,84 (m, 1H), 7,00 (s, 1H), 7,72 (s, 1H), 8,38 (s, 1H). La fracción 2 se volvió a purificar mediante HPLC preparativa (Método A) para obtener el isómero B puro del compuesto del título (0,004 g, 14 %). LC (Método A): 2,301 min. HRMS (ESI): calc. para C29H32N5O6S2 [M+H]+ m/z 610,1794, encontrado 610,1791. RMN 1 H (DMSO-d6, 400 MHz) 8 ppm: 1,51-1,60 (m, 2H), 1,70-2,04 (m, 11H), 3,27 (t, J= 6,7 Hz, 2H), 3,48 (t, J= 6,7 Hz, 2H), 3,81 (s, 3H), 4,20 (s, 3H), 5,23 (s, 2H), 5,90 (s, 1H), 6,60 (d, J= 1,6 Hz, 1H), 6,82 6,84 (m, 1H), 6,97 (d, J= 0,8 Hz, 1H), 7,64 (s, 1H), 8,37 (s, 1H).
Ejemplo 152
N-(cianometil)-4-hidroxi-4-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2-il)-N-metilciclohexancarboxamida

El compuesto del título se preparó a partir de ácido 4-hidroxi-4-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2-il)ciclohexancarboxílico y 2-(metilamino)acetonitrilo de acuerdo con el método descrito en el ejemplo 151 anterior. La mezcla del producto en bruto se separó en sus dos isómeros usando HPLC preparativa (Método A). Isómero A. LC (Método A): 2,143 min. HRMS (ESI): calc. para C28H29N6O6S2 [M+H]+ m/z 609,1590, encontrado 609,1581. RMN 1 H (DMSO-d6, 400 MHz) 8 ppm: 1,61-1,88 (m, 7H), 2,27-2,34 (m, 1H), 2,78-2,89 (m, 1H), 3,11 (s, 3H), 3,81 (s, 3H), 4,20 (s, 3H), 4,36 (s, 2H), 5,26 (s, 2H), 5,88 (a s, 1H), 6,62 (d, J= 2,0 Hz, 1H), 6,81-6,85 (m, 1H), 7,00 (d, J= 0,8 Hz, 1H), 7,72 (s, 1H), 8,37 (s, 1H). Isómero B. LC (Método A): 2,177 min. HRMS (ESI): calc. para C28H29N6O6S2 [M+H]+ m/z 609,1590, encontrado 609,1580. RMN 1 H (DMSO-d6, 400 MHz) 8 ppm: 1,49-1,62 (m, 2H), 1,74-1,90 (m, 4H), 1,96-2,10 (m, 2H), 2,72-2,85 (m, 1H), 3,13 (s, 3H), 3,81 (s, 3H), 4,20 (s, 3H), 4,37 (s, 2H), 5,23 (s, 2H), 5,94 (a s, 1H), 6,59 (d, J= 1,6 Hz, 1H), 6,82-6,85 (m, 1H), 6,97 (s, 1H), 7,65 (s, 1H), 8,37 (s, 1H).
Ejemplo 153
6-(4-((2-(4-(4-bromofenil)tetrahidro-2H-piran-4-il)tiazol-4-il)metoxi)-6-metoxibenzofuran-2-il)-2-metoxiimidazo[2,1-b][1,3,4]tiadiazol

153A. 4-(4-bromofenil)tetrahidro-2H-piran-4-carbonitrilo

Una solución de 2-(4-bromofenil)acetonitrilo (1,00 g, 5,10 mmol) en THF seco (5,85 ml) se trató con hidróxido de sodio 17 M (9,00 ml, 153 mmol), sulfato ácido de tetrabutilamonio (0,173 g, 0,510 mmol) y 1-cloro-2-(2-cloroetoxi)etano (0,628 ml, 5,36 mmol). La mezcla de reacción se calentó a reflujo durante 4 h, después la mezcla enfriada se diluyó con EtOAc y se lavó con HCl 1 N, agua y NaHCO3 acuoso saturado. La fase orgánica se secó sobre MgSO4, se filtró y se concentró a sequedad. Después el residuo se purificó mediante cromatografía ultrarrápida usando un gradiente de 0 a 100 % de EtOAc en hexanos para obtener 4-(4-bromofenil)tetrahidro-2H-piran-4-carbonitrilo (1,11 g, 82 %) en forma de un aceite de color amarillo claro. LC (Método F): 2,080 min. RMN 1H (DMSO-da, 400 MHz) 8 ppm: 7,66 (ddd, J = 2,0, 2,7, 8 ,6 Hz, 2H), 7,52 (ddd, J = 2,0, 2,7, 8 ,6 Hz), 4,01 (m, 2H), 3,64 (m, 2H), 2,12 -1,99 (m, 4H).
153B. 4-(4-bromofenil)tetrahidro-2H-piran-4-carboxamida

Se agitó 4-(4-bromofenil)tetrahidro-2H-piran-4-carbonitrilo (1,09 g, 4,10 mmol) en ácido sulfúrico concentrado (3 ml) a temperatura ambiente durante 40 h. Después la mezcla se vertió en hielo, la suspensión resultante se filtró y la torta de filtro se lavó intensamente con agua hasta que el pH del lavado fue neutro. El sólido de color blanco resultante se enjuagó con hexanos y después se secó a presión reducida para obtener 4-(4-bromofenil)tetrahidro-2H-piran-4-carboxamida (1,065 g, 92 %). LC (Método F): 1,835 min. RMN 1 H (DMSO-d6, 400 MHz) 8 ppm: 7,53 (dt, J = 2,5, 9,0 Hz, 2H), 7,32 (dt, J = 2,5, 9,0 Hz, 2H), 7,23 (a s, 1H), 7,06 (a s, 1H), 3,73 (dt, J = 3,7, 11,7 Hz, 2H), 3,46 (dt, J = 2,0, 11,7 Hz, 2H), 2,40 (d, J = 13,3 Hz, 2H), 1,77 (m, 2H).
153C. 4-(4-bromofenil)tetrahidro-2H-piran-4-carbotioamida

A una solución en agitación de 4-(4-bromofenil)tetrahidro-2H-piran-4-carboxamida (1,00 g, 3,52 mmol) en THF (12 ml), se le agregó reactivo de Lawesson (0,712 g, 1,760 mmol) de una vez y la mezcla de reacción se calentó a reflujo durante 6 h. Después la mezcla de reacción enfriada se concentró prácticamente a sequedad y se repartió entre EtOAc-NaHCO3 acuoso saturado. La fase orgánica se secó (MgSO4), se filtró y se concentró a sequedad y el residuo se purificó mediante cromatografía ultrarrápida usando hexanos-EtOAc como eluyente para obtener 4-(4-bromofenil)tetrahidro-2H-piran-4-carbotioamida en forma de un sólido de color blanco (0,771 g, 73,0 %). LC (Método F): 1,993 min. RMN 1 H (DMSO-d6, 400 MHz) 8 ppm: 9,70 (s, 1H), 8,92 (s, 1H), 7,54 (ddd, J = 2,0, 2,7, 8 ,6 Hz, 2H), 7,40 ((ddd, J = 2,0, 2,7, 8 ,6 Hz, 2H), 3,59 (m, 4H), 2,60 (m, 2H), 2,07 (m, 2H).
153D. 2-(4-(4-bromofenil)tetrahidro-2H-piran-4-il)tiazol-4-carboxilato de etilo

A una mezcla de 4-(4-bromofenil)tetrahidro-2H-piran-4-carbotioamida (0,725 g, 2,415 mmol) en isopropanol (10 ml), se le agregó bromopiruvato de etilo (0,365 ml, 2,90 mmol) y la mezcla de reacción se calentó a reflujo durante 2,75 h. La mezcla enfriada se repartió entre EtOAc y NaHCO3 acuoso saturado y la fase orgánica se lavó con salmuera,
se secó sobre (MgSO4), se filtró y se concentró a sequedad. El residuo se purificó mediante cromatografía ultrarrápida usando un gradiente de 0 a 100 % de EtOAc en hexanos para obtener 2-(4-(4-bromofenil)tetrahidro-2H-piran-4-il)tiazol-4-carboxilato de etilo (0,288 g, 30,1 %) en forma de un aceite transparente e incoloro. LC (Método F): 2,320 min. RMN 1H (DMSO-da, 400 MHz) 8 ppm: 8,46 (s, 1H), 7,55 (dt, J = 2,4, 9,0 Hz, 2H), 7,38 (dt, J = 2,4, 9,0 Hz, 2H), 4,29 (q, J = 7,0 Hz, 2H), 3,70 (m, 2H), 3,57 (m, 2H), 2,56 (m, 2H), 2,35 (m, 2H), 1,29 (t, J = 7,0 Hz, 3H).
153E. (2-(4-(4-bromofenil)tetrahidro-2H-piran-4-il)tiazol-4-il)metanol

A una solución enfriada en hielo de 2-(4-(4-bromofenil)tetrahidro-2H-piran-4-il)tiazol-4-carboxilato de etilo (0,288 g, 0,727 mmol) en THF (3,6 ml), se le agregó LiBH4 (0,03l4 g, 1,441 mmol) de una vez y después MeOH (0,058 ml, 1,441 mmol). La mezcla resultante se agitó a 0 °C durante 5 min y después a temperatura ambiente durante 16 h. Después la mezcla se volvió a enfriar a 0 °C y se inactivó cuidadosamente mediante la adición gota a gota de NH4Cl acuoso saturado. La mezcla resultante se extrajo con acetato de etilo, después de lo cual la fase orgánica se lavó con salmuera, se secó (MgSO4), se filtró y se evaporó hasta secarse. El residuo obtenido se purificó mediante cromatografía ultrarrápida usando un gradiente de 0 a 100 % de EtOAc en hexanos para obtener (2-(4-(4-bromofenil)tetrahidro-2H-piran-4-il)tiazol-4-il)metanol (0,226 g, 89 %) en forma de un sólido de color blanco. LC (Método F): 2,111 min. RMN 1 H (DMSO-d6, 400 MHz) 8 ppm: 7,52 (m, 2H), 7,35 (m, 3H), 5,28 (t, J = 5,7 Hz, 1H), 4,53 (dd, J = 1,2, 5,9 Hz, 2H), 3,72 (m, 1H), 3,69 (t, J = 4,3 Hz, 1H), 3,55 (m, 2H), 2,55 (m, 1H), 2,29 (m, 2H).
Ejemplo 153. 6-(4-((2-(4-(4-bromofenil)tetrahidro-2H-piran-4-il)tiazol-4-il)metoxi)-6-metoxibenzofuran-2-il)-2-metoxiimidazo[2 , 1 -b][1,3,4]tiadiazol

A un matraz secado al fuego que contenía 6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-ol (Ejemplo 1 H, 0,035 g, 0 , 1 1 0 mmol) y (2-(4-(4-bromofenil)tetrahidro-2H-piran-4-il)tiazol-4-il)metanol (0,039 g, 0,110 mmol), se le agregó THF seco (4 ml) y después tri-n-butilfosfina (0,072 ml, 0,276 mmol). A la suspensión resultante, se le agregó gota a gota una solución de ADDP (0,070 g, 0,276 mmol) en THF (1 ml) durante 30 min mediante una bomba de jeringa. Después de agitar durante 1,5 h, la mezcla de reacción se diluyó con EtOAc y después se lavó con HCl 1 N, NaHCO3 acuoso saturado, agua y salmuera. La fase orgánica se secó (MgSO4) y se evaporó y el material en bruto se trituró con DMSO, se filtró, se enjuagó con acetonitrilo y se secó al vacío para obtener el compuesto del título (0,035 g, 48,6 %) en forma de un sólido de color beis. LC (Método F): 2,732 min. HRMS (ESI): calc. para C2gH26BrN4OsS2 [M+H]+ m/z 653,0528, encontrado 653,0530. RMN 1 H (DMSO-d6, 400 MHz) 8 ppm: 8,36 (s, 1H), 7,75 (s, 1H), 7,52 (d, J = 8 ,6 Hz, 2H), 7,36 (d, J = 8 ,6 Hz, 2H), 6,98 (s, 1H), 6,83 (s, 1H), 6,62 (d, J = 2,0 Hz, 1H), 5,29 (s, 2H), 4,20 (s, 3H), 3,80 (s, 3H), 3,74 - 3,70 (m, 2H), 3,56 (m, 2H), 2,57 (m, 2H), 2,35 - 2,28 (m, 2H).
Ejemplo 154
6-(4-((2-(4-(4-clorofenil)tetrahidro-2H-piran-4-il)tiazol-4-il)metoxi)-6-metoxibenzofuran-2-il)-2-metoxiimidazo[2,1-b][1,3,4]tiadiazol

154A. (2-(4-(4-dorofenil)tetrahidro-2H-piran-4-il)tiazol-4-il)metanol

El alcohol se preparó de acuerdo con el método descrito anteriormente en el ejemplo 153. LC (Método F): 2,076 min. RMN 1H (DMSO-da, 400 MHz) 8 ppm: 7,40 (m, 4H), 7,34 (m, 1H), 5,28 (t, J = 5,7 Hz, 1H), 4,53 (dd, J = 1,2, 5,9 Hz, 2H), 3,71 (m, 2H), 3,55 (m, 2H), 2,54 (m, 2H), 2,30 (m, 2H).
Ejemplo 154. 6-(4-((2-(4-(4-clorofenil)tetrahidro-2H-piran-4-il)tiazol-4-il)metoxi)-6-metoxibenzofuran-2-il)-2-metoxiimidazo[2,1-b][1,3,4]tiadiazol

El compuesto del título se preparó de acuerdo con el método descrito anteriormente en el ejemplo 153 y se aisló en forma de un sólido. LC (Método F): 2,728 min. HRMS (ESI): calc. para C2gH26ClN4OaS2 [M+H]+ m/z 609,1028, encontrado 609,1077. RMN 1H (DMSO-da, 400 MHz) 8 ppm: 8,37 (s, 1H), 7,75 (s, 1H), 7,41 (m, 4H), 6,98 (d, J = 0,8 Hz, 1H), 6,83 (dd, J = 0,8, 1,6 Hz, 1H), 6,62 (d, J = 1,6 Hz, 1H), 5,29 (s, 2H), 4,20 (s, 3H), 3,80 (s, 3H), 3,75 - 3,70 (m, 2H), 3,56 (m, 2H), 2,58 (m, 2H), 2,32 (m, 2H).
Ejemplo 155
6-(4-((2-(4-(3-clorofenil)tetrahidro-2H-piran-4-il)tiazol-4-il)metoxi)-6-metoxibenzofuran-2-il)-2-metoxiimidazo[2,1-b][1,3,4]tiadiazol

155A. (2-(4-(3-clorofenil)tetrahidro-2H-piran-4-il)tiazol-4-il)metanol

El alcohol se preparó de acuerdo con el método descrito anteriormente en el ejemplo 153. LC (Método F): 2,057 min. RMN 1H (DMSO-da, 400 MHz) 8 ppm: 7,40-7,35 (m, 4H), 7,32-7,29 (m, 1H), 5,29 (m, 1H), 4,53 (s, 1H), 3,74 (m, 1H), 3,71 (t, J = 4,3 Hz, 1H), 3,56 (dd, J = 2,3, 9,4 Hz, 1H), 3,53 (dd, J = 2,3, 9,4 Hz, 1H), 2,56 (dd, J = 2,0, 11,7 Hz, 2H), 2,33 (dd, J= 3,9, 9,4 Hz, 1H), 2,29 (m, 1H).
Ejemplo 155. 6-(4-((2-(4-(3-clorofenil)tetrahidro-2H-piran-4-il)tiazol-4-il)metoxi)-6-metoxibenzofuran-2-il)-2-metoxiimidazo[2,1-b][1,3,4]tiadiazol

El compuesto del título se preparó de acuerdo con el método descrito anteriormente en el ejemplo 153 y se aisló en forma de un sólido. LC (Método F): 2,679 min. HRMS (ESI): calc. para C2gH26ClN4OsS2 [M+H]+ m/z 609,1033, encontrado 609,1042. RMN 1 H (DMSO-d6, 400 MHz) 8 ppm: 8,36 (s, 1H), 7,77 (s, 1H), 7,42 - 7,35 (m, 3H), 7,30 (m, 1H), 6,98 (d, J = 0,8 Hz, 1H), 6,83 (dd, J = 0,8, 1,6 Hz, 1H), 6,62 (d, J = 1,6 Hz, 1H), 5,30 (s, 2H), 4,20 (s, 3H), 3,80 (s, 3H), 3,73 (dt, J = 4,3, 12,1 Hz, 2H), 3,55 (m, 2H), 2,60 (m, 2H), 2,37 -2 ,30 (m, 2H).
Ejemplo 156
6-(4-((2-(4-(2-fluorofenil)tetrahidro-2H-piran-4-il)tiazol-4-il)metoxi)-6-metoxibenzofuran-2-il)-2-metoxiimidazo[2,1-b][1,3,4]tiadiazol

156A. (2-(4-(2-fluorofenil)tetrahidro-2H-piran-4-il)tiazol-4-il)metanol

El alcohol se preparó de acuerdo con el método descrito anteriormente en el ejemplo 153. LC (Método F): 1,948 min. RMN 1 H (DMSO-d6, 400 MHz) 8 ppm: 7,48 (dt, J = 2,0, 8,2 Hz, 1H), 7,38-7,33 (m, 1H), 7,31 (m, 1H), 7,23 (dt, J = 1,6, 7,8 Hz, 1H), 7,13 (ddd, J = 1,2, 8,2, 12,9 Hz, 1H), 5,27 (a s, 1H), 4,52 (s, 2H), 3,73-3,62 (m, 4H), 2,44 (m, 4H). Ejemplo 156. 6-(4-((2-(4-(2-fluorofenil)tetrahidro-2H-piran-4-il)tiazol-4-il)metoxi)-6-metoxibenzofuran-2-il)-2-metoxiimidazo[2,1-b][1,3,4]tiadiazol

El compuesto del título se preparó de acuerdo con el método descrito anteriormente en el ejemplo 153 y se aisló en forma de un sólido. LC (Método F): 2,633 min. HRMS (ESI): calc. para C29H26FN4O5S2 [M+H]+ m/z 593,1329, encontrado 593,1356. RMN 1H (DMSO-da, 400 MHz) 8 ppm: 8,37 (s, 1H), 7,73 (s, 1H), 7,51 (dt, J = 1,6, 8,2 Hz, 1H), 7,37 (m, 1H), 7,25 (dt, J = 1,2, 7,4 Hz, 1H), 7,15 (ddd, J = 1,2, 8,2, 12,9 Hz, 1H), 6,98 (s, 1H), 6,83 (dd, J = 0,8, 2,0 Hz, 1H), 6,61 (d, J = 2,0 Hz, 1H), 5,28 (s, 2H), 4,20 (s, 3H), 3,80 (s, 3H), 3,74 - 3,63 (m, 4H), 2,58 - 2,42 (m, 4H). Ejemplo 157
2-metoxi-6-(6-metoxi-4-((2-(4-(p-tolil)tetrahidro-2H-piran-4-il)tiazol-4-il)metoxi)benzofuran-2-il)imidazo[2,1-b][1,3,4]tiadiazol

157A. (2-(4-(p-tolil)tetrahidro-2H-piran-4-il)tiazol-4-il)metanol

El alcohol se preparó de acuerdo con el método descrito anteriormente en el ejemplo 153. LC (Método F): 2,043 min. RMN 1 H (DMSO-d6, 400 MHz) 8 ppm: 7,29 (t, J = 1,2 Hz, 1H), 7,27 (m, 2H), 7,14 (d, J = 7,8 Hz, 2H), 5,26 (t, J = 5,5 Hz, 1H), 4,53 (d, J = 4,3 Hz, 2H), 3,72 -3 ,66 (m, 2H), 3,57 (m, 1H), 3,55 (dd, J = 2,7, 9,0 Hz, 1H), 2,53 (m, 2H), 2,29 (m, 2H), 2,25 (s, 3H).
Ejemplo 157. 2-metoxi-6-(6-metoxi-4-((2-(4-(p-tolil)tetrahidro-2H-piran-4-il)tiazol-4-il)metoxi)benzofuran-2-il)imidazo[2,1-b][1,3,4]tiadiazol

El compuesto del título se preparó de acuerdo con el método descrito anteriormente en el ejemplo 153 y se aisló en forma de un sólido. LC (Método F): 2,662 min. HRMS (ESI): calc. para C30H29N4O5S2 [M+H]+ m/z 589,1579, encontrado 589,1593. RMN 1 H (DMSO-d6, 400 MHz) 8 ppm: 8,37 (s, 1H), 7,71 (s, 1H), 7,29 (d, J = 8,2 Hz, 2H), 7,14 (d, J = 8,2 Hz, 2H), 6,98 (s, 1H), 6,83 (dd, J = 0,8, 1,6 Hz, 1H), 6,26 (d, J = 2,0 Hz, 1H), 5,28 (s, 2H), 4,20 (s, 3H), 3,80 (s, 3H), 3,71 (m, 2H), 3,57 (m, 2H), 2,56 (m, 2H), 2,32 (m, 2H), 2,25 (s, 3H).
Ejemplo 158
2-metoxi-6-(6-metoxi-4-((2-(4-(3-metoxifenil)tetrahidro-2H-piran-4-il)tiazol-4-il)metoxi)benzofuran-2-il)imidazo[2,1-b][1,3,4]tiadiazol

158A. (2-(4-(3-metoxifenil)tetrahidro-2H-piran-4-il)tiazol-4-il)metanol

El alcohol se preparó de acuerdo con el método descrito anteriormente en el ejemplo 153. LC (Método F): 1,953 min. RMN 1H (DMSO-da, 400 MHz) 8 ppm: 7,32 (s, 1H), 7,26 (t, J = 8,0 Hz, 1H), 6,96 (dd, J = 1,2, 7,8 Hz, 1H), 6 ,88 (m, 1H), 6,80 (dd, J = 2,3, 8,0 Hz, 1H), 5,28 (t, J = 5,7 Hz, 1H), 4,53 (d, J = 5,5 Hz, 2H), 3,72 (m, 2H), 3,72 (s, 3H), 3,54 (m, 2H), 2,53 (m, 2H), 2,29 (ddd, J = 3,5, 9,2, 13,3 Hz, 2H).
Ejemplo 158. 2-metoxi-6-(6-metoxi-4-((2-(4-(3-metoxifenil)tetrahidro-2H-piran-4-il)tiazol-4-il)metoxi)benzofuran-2-il)imidazo[2,1-b][1,3,4]tiadiazol

El compuesto del título se preparó de acuerdo con el método descrito anteriormente en el ejemplo 153 y se aisló en forma de un sólido. LC (Método F): 2,675 min. HRMS (ESI): calc. para C30H29N4O6S2 [M+H]+ m/z 605,1529, encontrado 605,1544. RMN 1 H (DMSO-d6, 400 MHz) 8 ppm: 836 (s, 1H), 7,74 (s, 1H), 7,25 (t, J = 8,0 Hz, 1H), 6,98 (m, 2H), 6,90 (m, 1H), 6,82 (m, 1H), 6,81 (m, 1H), 6,62 (d, J = 1,6 Hz, 1H), 5,29 (s, 2H), 4,20 (s, 3H), 3,80 (s, 3H), 3,73 (m, 2H), 3,71 (s, 3H), 3,56 (m, 2H), 2,57 (m, 2H), 2,32 (m, 2H).
Ejemplo 159
2-metoxi-6-(6-metoxi-4-((2-(4-(4-metoxifenil)tetrahidro-2H-piran-4-il)tiazol-4-il)metoxi)benzofuran-2-il)imidazo[2,1-b][1,3,4]tiadiazol

159A. (2-(4-(4-metoxifenil)tetrahidro-2H-piran-4-il)tiazol-4-il)metanol
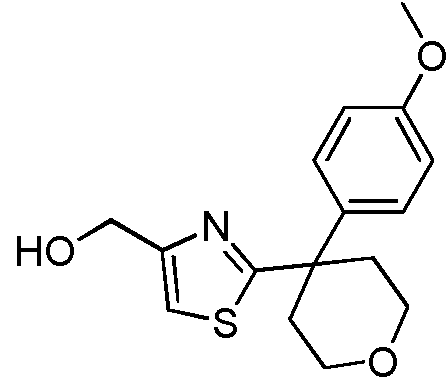
El alcohol se preparó de acuerdo con el método descrito anteriormente en el ejemplo 153. LCMS (APCI): calc. para C16H20NO3S [M+H]+ m/z 306,12, encontrado 306,20.
Ejemplo 159. 2-metoxi-6-(6-metoxi-4-((2-(4-(4-metoxifenil)tetrahidro-2H-piran-4-il)tiazol-4-il)metoxi)benzofuran-2-il)imidazo[2,1-b][1,3,4]tiadiazol

El compuesto del título se preparó de acuerdo con el método descrito anteriormente en el ejemplo 153 y se aisló en forma de un sólido. LC (Método F): 2,440 min. HRMS (ESI): calc. para C30H29N4O6S2 [M+H]+ m/z 605,1529, encontrado 605,1557. RMN 1 H (DMSO-d6, 400 MHz) 6 ppm: 8,36 (s, 1H), 7,70 (s, 1H), 7,32 (d, J = 8 ,6 Hz, 2H), 6,98 (s, 1H), 6 ,88 (d, J = 9,0 Hz, 2H), 6,83 (s, 1H), 6,62 (d, J = 1,6 Hz, 1H), 5,28 (s, 2H), 4,20 (s, 3H), 3,80 (s, 3H), 3,72 (s, 3H), 3,70 (m, 2H), 3,60 (m, 2H), 2,55 (m, 2H), 2,30 (m, 2H).
Ejemplo 160
4-(4-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2-il)tetrahidro-2H-piran-4-il)benzoato de metilo

160A. 2-(4-(4-bromofenil)tetrahidro-2H-piran-4-il)-4-(((terc-butildimetilsilil)-oxi)metil)tiazol

A una solución en agitación de (2-(4-(4-bromofenil)tetrahidro-2H-piran-4-il)tiazol-4-il)metanol (Ejemplo 153E, 0,163 g, 0,460 mmol) e imidazol (0,047 g, 0,690 mmol) en DCM (7,5 ml) a temperatura ambiente, se le agregó te rc - butildorodimetilsilano (0,087 g, 0,575 mmol). La mezcla de reacción se agitó durante una noche, después se inactivó con MeOH y se concentró a sequedad. El residuo se purificó mediante cromatografía de columna usando hexanos-EtOAc como eluyente para obtener 2-(4-(4-bromofenil)tetrahidro-2H-piran-4-il)-4-(((terc-butildimetilsilil)oxi)metil)tiazol (0,214 g, 99 %) en forma de un aceite transparente e incoloro. LC (Método A): 2,683 min. RMN 1 H (DMSO-d6, 400 MHz) 8 ppm: 7,46 (d, J= 8 ,8 Hz, 2H), 7,32 (s, 1H), 7,28 (d, J= 8 ,8 Hz, 2H), 4,66 (s, 2H), 3,65 (m, 2H), 3,49 (m, 2H), 2,48 (m, 2H), 2,23 (m, 2H), 0,81 (s, 9H), 0,00 (s, 6 H).
160B. 4-(4-(4-(((terc-butildimetilsilil)oxi)metil)tiazol-2-il)tetrahidro-2H-piran-4-il)benzoato de metilo y 4-(4-(4-(hidroximetil)tiazol-2-il)tetrahidro-2H-piran-4-il)benzoato de metilo

A una mezcla de Pd(OAc)2 (0,002 g, 9,14 pmol) y Xantphos (0,0106 g, 0,018 mmol), se le agregó una solución de 2-(4-(4-bromofenil)tetrahidro-2H-piran-4-il)-4-(((terc-butildimetilsilil)oxi)metil)tiazol (0,214 g, 0,457 mmol) en Et3N (2 ml, 14,35 mmol), después de lo cual se agregó MeOH (0,185 ml, 4,57 mmol). La mezcla de reacción se colocó en alto vacío y después se volvió a llenar con CO (g) (usando un globo relleno con CO). El recipiente se selló y después se calentó con agitación a 70 °C (temperatura del baño de aceite) durante 24 h. La mezcla enfriada se filtró a través de CELITE® y la torta de filtro se lavó con MeOH adicional. El filtrado se evaporó y el residuo se purificó mediante cromatografía ultrarrápida usando hexanos-EtOAc como eluyente para obtener 4-(4 -(4 -( ( ( te rc - butildimetilsilil)oxi)metil)tiazol-2-il)tetrahidro-2H-piran-4-il)benzoato de metilo (0,052 g, 25,4 %) en forma de un aceite transparente e incoloro. LC (Método A): 2,552 min. lCm S (APCI): calc. para C23H34NO4SSi [M+H]+ m/z 448,20, encontrado 448,20. La elución adicional produjo 4-(4-(4-(hidroximetil)tiazol-2-il)tetrahidro-2H-piran-4-il)benzoato de metilo (0,029 g, 19,04 %) en forma de un sólido de color blanco. LC (Método A): 1,769 min. LCMS (APCl): calc. para C17H21NO4S [M+H]+ m/z 334,11, encontrado 334,20.
Ejemplo 160. 4-(4-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2-il)tetrahidro-2H-piran-4-il)benzoato de metilo

El compuesto del título se preparó de acuerdo con el método descrito anteriormente en el ejemplo 153 y se aisló en forma de un sólido. LC (Método A): 2,588 min. HRMS (ESI): calc. para C31H29N4O7S2 [M+H]+ m/z 633,1478, encontrado 633,1493. RMN 1 H (DMSO-d6, 400 MHz) 8 ppm: 8,36 (s, 1H), 7,91 (d, J = 8 ,6 Hz, 2H), 7,76 (s, 1H), 7,56 (d, J = 8,2 Hz, 2H), 6,97 (s, 1H), 6,82 (m, 1H), 6,61 (d, J = 1,6 Hz, 1H), 5,30 (s, 2H), 4,20 (s, 3H), 3,82 (s, 3H), 3,80 (s, 3H), 3,74 (dt, J = 3,9, 12,1 Hz, 2H), 3,57 (m, 2H), 2,62 (m, 2H), 2,36 (m, 2H).
Ejemplo 161
4-(4-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2-il)tetrahidro-2H-piran-4-il)-N,N-dimetilbenzamida

161A. 4-(4-(4-(((ferc-butildimetilsilil)oxi)metil)tiazol-2-il)tetrahidro-2H-piran-4-il)-N,N-dimetilbenzamida

A una solución en agitación de 4-(4-(4-(((ferc-butildimetilsilil)oxi)metil)tiazol-2-il)tetrahidro-2H-piran-4-il)benzoato de metilo (Ejemplo 160B, 0,863 g, 1,928 mmol) en MeOH (10 ml), se le agregó hidróxido de sodio 1 N (2,121 ml, 2,121 mmol). La mezcla de reacción se agitó a temperatura ambiente durante 2 h y después a 55 °C durante 1 h. La mezcla enfriada se neutralizó (pH 7) con HCl 1 N y la agitación continuó durante 10 min. La suspensión resultante se filtró y el residuo se lavó con agua y se secó al vacío para obtener ácido 4-(4-(4-(((ferc-butildimetilsilil)oxi)metil)tiazol-2-il)tetrahidro-2H-piran-4-il)benzoico (0,305 g, 0,703 mmol, 36,5 %) en forma de un sólido de color blanco. LC (Método A): 2,418 min. LCMS (APCI): calc. para C22Hs2NO4SS¡ [M+H]+ m/z 434,18, encontrado 434,20.
A una solución en agitación de ácido 4-(4-(4-(((ferc-butildimetilsilil)oxi)metil)tiazol-2-il)tetrahidro-2H-piran-4-il)benzoico (0,100 g, 0,231 mmol) en DMF (3 ml), se le agregó dimetilamina (0,115 ml, 0,231 mmol), DIEA (0,201 ml, 1,153 mmol) y HATU (0,088 g, 0,231 mmol). Después de la agitación a temperatura ambiente durante 2 h, la mezcla se repartió entre EtOAc y NaHCO3 acuoso saturado. La fase orgánica se separó, se secó (MgSO4), se filtró y se concentró a sequedad. El residuo se purificó mediante cromatografía ultrarrápida usando DCM-EtOAc como eluyente para obtener 4-(4-(4-(((ferc-butildimetilsilil)oxi)metil)tiazol-2-il)tetrahidro-2H-piran-4-il)-N,N-dimetilbenzamida (0,088 g, 83 %) en forma de un sólido de color blanco. LC (Método A): 2,343 min. RMN 1H (DMSO-d6, 400 MHz) 8 ppm: 7,43 (m, 2H), 7,37 (m, 3H), 4,73 (d, J = 0,8 Hz, 2H), 4,34 (t, J = 5,1 Hz, 1H), 3,74 (m, 2H), 3,56 (m, 2H), 3,44 (m, 1H), 2,95 (a s, 2H), 2,88 (a s, 2H), 2,58 (m, 2H), 2,33 (m, 2H), 0,87 (s, 9H), 0,06 (s, 6 H).
161B. 4-(4-(4-(hidroximetil)tiazol-2-il)tetrahidro-2H-piran-4-il)-N,N-dimetilbenzamida

A una solución en agitación de 4-(4-(4-(((ferc-butildimetilsilil)oxi)metil)tiazol-2-il)tetrahidro-2H-piran-4-il)-N,N-dimetilbenzamida (0,088 g, 0,191 mmol) en THF (1 ml), se le agregó trihidrofluoruro de trietilamina (0,156 ml, 0,955 mmol) y la mezcla se agitó a temperatura ambiente durante 4 h. La mezcla resultante se repartió entre EtOAc y NaHCO3 acuoso saturado y la fase orgánica se separó, se secó (MgSO4), se filtró y se concentró a sequedad. El residuo se purificó mediante cromatografía ultrarrápida usando DCM-EtOAc como eluyente para obtener 4-(4-(4-(hidroximetil)tiazol-2-il)tetrahidro-2H-piran-4-il)-N,N-dimetilbenzamida (0,044 g, 66,5 %) en forma de un aceite transparente e incoloro. LC (Método A): 1,492 min. LCMS (APCI): calc. para C18H23N2O3S [M+H]+ m/z 347,14, encontrado 347,20.
Ejemplo 161. 4-(4-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2-il)tetrahidro-2H-piran-4-il)-N,N-dimetilbenzamida
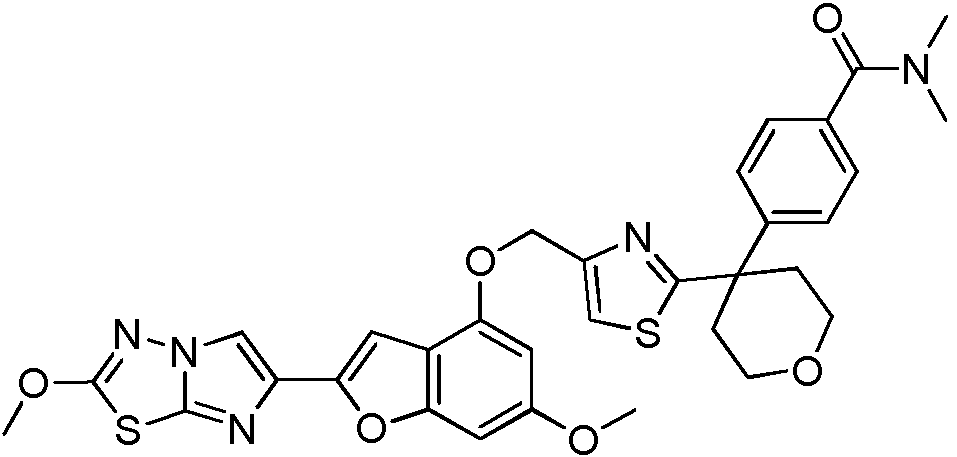
A una suspensión de 6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-ol (Ejemplo 1H, 0,040 g, 0,127 mmol) y 4-(4-(4-(hidroximetil)tiazol-2-il)tetrahidro-2H-piran-4-il)-N,N-dimetilbenzamida (0,044 g, 0,127 mmol) en THF seco (8 ml), se le agregó tri-n-butilfosfina (0,082 ml, 0,318 mmol) y después se agregó gota a gota una solución de ADDP (0,080 g, 0,318 mmol) en THF (2 ml) durante 30 min mediante una bomba de jeringa. Después de agitar durante 30 min más, la mezcla de reacción se repartió entre EtOAc y NaHCO3 acuoso saturado. La fase orgánica se separó, se lavó con salmuera, se secó (MgSO4), se filtró y se concentró hasta secarse. El residuo se purificó mediante cromatografía en columna ultrarrápida usando de 0 a 10 % de MeOH:NH4OH (9:1) en DCM como eluyente para obtener el producto levemente impuro. El material obtenido se volvió a triturar con MeCN y la suspensión resultante se filtró, se lavó con un volumen mínimo de MeCN y se secó a presión reducida para obtener el compuesto del título (0,040 g, 48,8 %) en forma de un sólido de color blanco. LC (Método A): 2,297 min. HRMS (ESI): calc. para C32H32N5O6S2 [M+H]+ m/z 646,1794 encontrado, 646,1870. RMN 1H (DMSO-da, 400 MHz) 8 ppm: 8,36 (s, 1H), 7,76 (s, 1H), 7,40 (dd, J = 8,4, 38,5 Hz, 4H), 6,97 (d, J = 0,8 Hz, 1H), 6,83 (dd, J = 0,8, 1,6 Hz, 1H), 6,62 (d, J = 1,6 Hz, 1H), 5,30 (s, 2h), 4,20 (s, 3H), 3,80 (s, 3H), 3,75 (m, 2H), 3,57 (m, 2H), 2,95 (a s, 3H), 2,87 (a s, 3H), 2,61 (m, 2H), 2,35 (m, 2H).
Ejemplo 162
2-metoxi-6-(6-metoxi-4-((2-(4-feniltetrahidro-2H-piran-4-il)tiazol-4-il)metoxi)benzofuran-2-il)imidazo[2,1-b][1,3,4]tiadiazol

162A. (2-(4-feniltetrahidro-2H-piran-4-il)tiazol-4-il)metanol

El alcohol se preparó de acuerdo con el método descrito anteriormente en el ejemplo 153. LC (Método F): 1,936 min. RMN 1 H (DMSO-d6, 400 MHz) 8 ppm: 7,39 (m, 2H), 7,34 (m, 3H), 7,22 (m, 1H), 5,27 (t, J = 5,9 Hz, 1H), 4,54 (m, 2H), 3,72 (m, 2H), 3,58 (dd, J = 2,3, 9,4 Hz, 1H), 3,55 (dd, J = 2,3, 9,4 Hz, 1H), 2,56 (m, 2H), 2,33 (m, 1H), 2,30 (dd, J = 3,9, 9,4 Hz, 1H).
Ejemplo 162. 2-metoxi-6-(6-metoxi-4-((2-(4-feniltetrahidro-2H-piran-4-il)tiazol-4-il)metoxi)benzofuran-2-il)imidazo[2,1-b][1,3,4]tiadiazol

El compuesto del título se preparó de acuerdo con el método descrito anteriormente en el ejemplo 153 y se aisló en forma de un sólido. LC (Método F): 2,651 min. HRMS (ESI): calc. para C29H27N4O5S2 [M+H]+ m/z 575,1423, encontrado 575,1442. RMN 1H (DMSO-da, 400 MHz) 8 ppm: 8,36 (s, 1H), 7,73 (s, 1H), 7,41 (m, 2H), 7,33 (m, 2H), 7,23 (m, 1H), 6,98 (d, J = 0,8 Hz, 1H), 6,83 (dd, J = 0,8, 1,6 Hz, 1H), 6,63 (d, J = 2,0 Hz, 1H), 5,30 (s, 2H), 4,20 (s, 3H), 3,80 (s, 3H), 3,73 (dt, J = 4,3, 11,3 Hz, 2H), 3,57 (m, 2H), 2,60 (m, 2H), 2,34 (m, 2H).
Ejemplo 163
6-(4-((2-(3-(4-clorofenil)tetrahidrofuran-3-il)tiazol-4-il)metoxi)-6-metoxibenzofuran-2-il)-2-metoxiimidazo[2,1-b][1,3,4]tiadiazol

163A. 3-(4-clorofenil)tetrahidrofuran-3-carbonitrilo

A una suspensión en agitación de NaH (60 % en aceite, 0,792 g, 19,79 mmol) (Nota: previamente se lavó dos veces con hexanos y se secó al vacío) en NMP (17 ml) enfriada a -20 °C, se le agregó gota a gota una mezcla de 2-(4-clorofenil)acetonitrilo (1,00 g, 6,60 mmol) y 1-cloro-2-(clorometoxi)etano (0,851 g, 6,60 mmol) en dietiléter (4 ml). Después la mezcla se calentó a temperatura ambiente durante 24 h. La reacción se inactivó lentamente con agua helada y la mezcla resultante se extrajo con éter (x3). La capa orgánica combinada se lavó con agua y salmuera, se secó (MgSO4), se concentró y se evaporó. El residuo se purificó mediante cromatografía ultrarrápida usando hexanos-EtOAc como eluyente para obtener 3-(4-clorofenil)tetrahidrofuran-3-carbonitrilo (0,621 g, 45,3 %) en forma de un aceite de color naranja. lC (Método F): 1,970 min. r Mn 1 H (DMSO-d6, 400 MHz) 8 ppm: 7,54 (m, 4 H), 4,39 (d, J = 9,0 Hz, 1H), 4,06 (dd, J = 5,7, 8,2 Hz, 2H), 3,84 (d, J = 8,6 Hz, 1H), 2,77 (m, 1H), 2,48 (m, 1H).
163B. 3-(4-clorofenil)tetrahidrofuran-3-carboxamida

Una solución de 3-(4-clorofenil)tetrahidrofuran-3-carbonitrilo (0,615 g, 2,96 mmol) en H2SO4 concentrado (4 ml) se agitó durante la noche a temperatura ambiente. Después la mezcla de reacción se inactivó cuidadosamente con hielo picado y la mezcla se agitó durante 1 h. La suspensión resultante se filtró y la torta de filtro se lavó con agua, después con dietiléter y finalmente se secó a presión reducida para obtener 3-(4-clorofenil)-tetrahidrofuran-3-carboxamida (0,466 g, 69,7 %) en forma de un sólido de color beis. Le (Método F): 1,699 min. RMN 1 H (DMSO-d6,
400 MHz) 8 ppm: 7,40 (m, 2H), 7,32 (m, 2H), 7,30 (a s, 1H), 7,07 (a s, 1H), 4,45 (d, J = 8,2 Hz, 1H), 3,79 (m, 1H), 3,75 (d, J = 7,8 Hz, 1H), 3,72 (d, J = 8 ,6 Hz, 1H), 2,82 (ddd, J = 5,1, 7,4, 12,5 Hz, 1H), 2,10 (m, 1H).
163C. 3-(4-dorofenil)tetrahidrofuran-3-carbotioamida
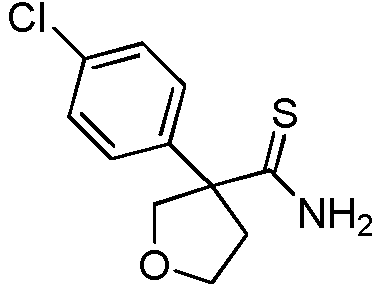
A una solución en agitación de 3-(4-clorofenil)tetrahidrofuran-3-carboxamida (0,450 g, 1,994 mmol) en THF (15 ml), se le agregó 2,4-disulfuro de 2,4-bis(4-metoxifenil)-1,3,2,4-ditiadifosfetano (0,403 g, 0,997 mmol) de una vez y la mezcla se calentó a reflujo durante 2 h. Después la mezcla resultante se concentró prácticamente a sequedad y el concentrado se repartió entre EtOAc y NaHCO3 acuoso saturado. La fase orgánica se separó, se secó (MgSO4), se filtró y se concentró a sequedad. El residuo se purificó mediante cromatografía ultrarrápida usando hexanos-EtOAc como eluyente para obtener 3-(4-clorofenil)-tetrahidrofuran-3-carbotioamida (0,356 g, 73,9 %) en forma de un sólido de color blanco. LC (Método F): 1,864 min. RMN 1H (DMSO-da, 400 MHz) 8 ppm: 9,71 (a s, 1H), 9,00 (a s, 1H), 7,43 (m, 4H), 4,43 (d, J = 9,0 Hz, 1H), 3,99 (d, J = 8 ,6 Hz, 1H), 3,84 -3,71 (m, 2H), 2,91 (m, 1H), 2,33 (m, 1H).
163D. 2-(3-(4-clorofenil)tetrahidrofuran-3-il)tiazol-4-carboxilato de etilo

A una mezcla de 3-(4-clorofenil)tetrahidrofuran-3-carbotioamida (0,345 g, 1,427 mmol) en i-PrOH (10 ml), se le agregó bromopiruvato de etilo (0,215 ml, 1,713 mmol) y la mezcla de reacción se calentó a reflujo durante 3 h. La mezcla enfriada se repartió entre EtOAc y NaHCO3 acuoso saturado y la fase orgánica se lavó con salmuera, se secó (MgSO4), se filtró y se concentró a sequedad. El residuo se purificó mediante cromatografía ultrarrápida usando un gradiente de 0 a 100 % de EtOAc en hexanos para obtener 2-(3-(4-clorofenil)tetrahidrofuran-3-il)tiazol-4-carboxilato de etilo (0,200 g, 41,5 %) en forma de un aceite transparente de color amarillo. LC (Método F): 2,248 min. RMN 1 H (DMSO-d6, 400 MHz) 8 ppm: 8,45 (s, 1H), 7,41 (m, 4H), 4,58 (d, J = 8 ,6 Hz, 1H), 4,29 (q, J = 7,0 Hz, 2H), 4,11 (d, J = 8 ,6 Hz, 1H), 3,97 -3 ,86 (m, 2H), 2,98 (m, 1H), 2,59 (m, 1H), 1,29 (t, J = 7,0 Hz, 3H).
163E. (2-(3-(4-clorofenil)tetrahidrofuran-3-il)tiazol-4-il)metanol

A una solución enfriada en hielo de 2-(3-(4-clorofenil)tetrahidrofuran-3-il)tiazol-4-carboxilato de etilo (0,200 g, 0,592 mmol) en THF (3 ml), se le agregó LiBH4 (0,026 g, 1,184 mmol) de una vez y después MeOH (0,048 ml, 1,184 mmol). La mezcla resultante se agitó a 0 °C durante 5 min y después a temperatura ambiente durante 4 h. Después la mezcla se volvió a enfriar a 0 °C, se inactivó mediante la adición gota a gota de NH4Cl acuoso saturado y después se extrajo con acetato de etilo. La fase orgánica se separó, se lavó con salmuera, se secó (MgSO4), se filtró y se concentró a sequedad. El residuo se purificó mediante cromatografía de columna usando un gradiente de 0 a 100 % de EtOAc en hexanos para obtener (2-(3-(4-clorofenil)tetrahidrofuran-3-il)tiazol-4-il)metanol (0,146 g, 83 %) en forma de un aceite transparente e incoloro. LC (Método F): 1,990 min. RMN 1 H (DMSO-d6, 400 MHz) 8 ppm: 7,39 (m, 4H), 7,32 (s, 1H), 5,28 (a s, 1H), 4,54 (d, J = 8 ,6 Hz, 1H), 4,51 (s, 2H), 4,09 (d, J = 8,2 Hz, 1H), 3,95 - 3,84 (m, 2H), 2,95 (ddd, J = 4,7, 7,0, 12,1 Hz, 1H), 2,55 (m, 1H).
Ejemplo 163. 6-(4-((2-(3-(4-clorofenil)tetrahidrofuran-3-il)tiazol-4-il)metoxi)-6-metoxibenzofuran-2-il)-2-metoxiimidazo[2,1-b][1,3,4]tiadiazol

A una suspensión de 6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-ol (Ejemplo 1H, 0,032 g, 0,101 mmol) y (2-(3-(4-clorofenil)tetrahidrofuran-3-il)tiazol-4-il)metanol (0,030 g, 0,101 mmol) en THF seco (8 ml), se le agregó tri-n-butilfosfina (0,066 ml, 0,254 mmol) y después se agregó gota a gota una solución de ADDP (0,064 g, 0,254 mmol) en THF (2 ml) durante 30 min mediante una bomba de jeringa. Después de agitar durante 30 min más, la mezcla de reacción se repartió entre EtOAc y NaHCO3 acuoso saturado. La fase orgánica se separó, se lavó con salmuera, se secó (MgSO4), se filtró y se concentró a sequedad. El residuo se purificó mediante cromatografía ultrarrápida usando DCM-EtOAc como eluyente para obtener el producto en forma de un sólido de color beis amarillento. Este material se volvió a triturar con acetonitrilo, la mezcla se filtró y la torta de filtro se enjuagó con dietiléter y después se secó al vacío. Esto produjo 6-(4-((2-(3-(4-clorofenil)tetrahidrofuran-3-il)tiazol-4-il)metoxi)-6-metoxibenzofuran-2-il)-2-metoxiimidazo[2,1-b][1,3,4]tiadiazol puro (0,041 g, 67,9 %) en forma de un sólido de color blanco. LC (Método F): 2,619 min. HRMS (ESI): calc. para C2sH24ClN4O5S2 [M+H]+ m/z 595,0877, encontrado 595,0888. RMN 1 H (DMSO-d6, 400 MHz) 8 ppm: 8,37 (s, 1H), 7,74 (s, 1H), 7,40 (s, 4H), 6,98 (s, 1H), 6,83 (d, J = 0,8 Hz, 1H), 6,60 (d, J = 1,6 Hz, 1H), 5,26 (s, 2H), 4,59 (d, J = 8 ,6 Hz, 1H), 4,20 (s, 3H), 4,12 (d, J = 8 ,6 Hz, 1H), 3,98 -3,87 (m, 2H), 3,80 (s, 3H), 2,99 (m, 1H), 2,62 -2 ,55 (m, 1H).
Ejemplo 164
2-metoxi-6-(6-metoxi-4-((2-(3-(p-tolil)tetrahidrofuran-3-il)tiazol-4-il)metoxi)benzofuran-2-il)imidazo[2,1-b][1,3,4]tiadiazol

164A. (2-(3-(p-tolil)tetrahidrofuran-3-il)tiazol-4-il)metanol

El alcohol se preparó de acuerdo con el método descrito anteriormente en el ejemplo 163. LC (Método F): 1,995 min. RMN 1 H (DMSO-d6, 400 MHz) 8 ppm: 7,28 (s, 1H), 7,22 (m, 2H), 7,14 (d, J = 8,2 Hz, 2H), 5,26 (t, J = 5,7 Hz, 1H), 4,55 (d, J = 8,2 Hz, 1H), 4,50 (dd, J = 0,8, 5,9 Hz, 2H), 4,07 (d, J = 8,2 Hz, 1H), 3,93 - 3,83 (m, 2H), 2,94 (ddd, J = 4,7, 7,2, 12,1 Hz, 1H), 2,53 (m, 1H).
Ejemplo 164. 2-metoxi-6-(6-metoxi-4-((2-(3-(p-tolil)tetrahidrofuran-3-il)tiazol-4-il)metoxi)benzofuran-2-il)imidazo[2,1-b][1,3,4]tiadiazol
El compuesto del título se preparó de acuerdo con el método descrito anteriormente en el ejemplo 163 y se aisló en forma de un sólido. LC (Método F): 2,628 min. HRMS (ESI): calc. para C29H27N4O5S2 [M+H]+ m/z 575,1423, encontrado 575,1441. RMN 1H (DMSO-da, 400 MHz) 8 ppm: 8,37 (s, 1H), 7,70 (s, 1H), 7,25 (d, J = 8,2 Hz, 2H), 7,15 (d, J = 7,8 Hz, 2H), 6,98 (s, 1H), 6,83 (s, 1H), 6,61 (d, J = 1,6 Hz, 1H), 5,26 (s, 2H), 4,59 (d, J = 8,2 Hz, 1H), 4,20 (s, 3H), 4,10 (d, J = 8,2 Hz, 1H), 3,96 -3 ,86 (m, 2H), 3,80 (s, 3H), 2,98 (m, 1H), 2,56 (dt, J = 8,2, 12,5 Hz, 1H), 2,26 (s, 3H).
Ejemplo 165
6-(4-((2-(3-(3-clorofenil)tetrahidrofuran-3-il)tiazol-4-il)metoxi)-6-metoxibenzofuran-2-il)-2-metoxiimidazo[2,1-b][1,3,4]tiadiazol

165A. (2-(3-(3-clorofenil)tetrahidrofuran-3-il)tiazol-4-il)metanol

El alcohol se preparó de acuerdo con el método descrito anteriormente en el ejemplo 163. LC (Método F): 2,004 min. RMN 1 H (DMSO-d6, 400 MHz) 8 ppm: 7,37 -7 ,27 (m, 5H), 5,25 (t, J = 5,7 Hz, 1H), 4,53 (d, J = 8 ,6 Hz, 1H), 4,48 (d, J = 5,1 Hz, 2H), 4,07 (d, J = 8 ,6 Hz, 1H), 3,89 (m, 1H), 3,83 (q, J = 7,8 Hz, 1H), 2,93 (ddd, J = 4,7, 7,4, 12,5 Hz, 1H), 2,54 (dt, J = 8,2, 12,5 Hz, 1H).
Ejemplo 165. 6-(4-((2-(3-(3-clorofenil)tetrahidrofuran-3-il)tiazol-4-il)metoxi)-6-metoxibenzofuran-2-il)-2-metoxiimidazo[2,1-b][1,3,4]tiadiazol

El compuesto del título se preparó de acuerdo con el método descrito anteriormente en el ejemplo 163 y se aisló en forma de un sólido. LC (Método F): 2,650 min. HRMS (ESI): calc. para C2sH24ClN4O5S2 [M+H]+ m/z 595,0877, encontrado 595,0896. RMN 1 H (DMSO-d6, 400 MHz) 8 ppm: 8,65 (s, 1H), 8,02 (d, J = 9,4 Hz, 1H), 7,98 (m, 2H), 7,93 (s, 1H), 7,55 - 7,49 (m, 3H), 7,26 (s, 1H), 7,19 (d, J = 9,0 Hz, 1H), 6,78 (dd, J = 2,1, 10,0 Hz, 2H), 5,44 (s, 2H), 3,80 (s, 3H), 2,53 (s, 3H).
Ejemplo 166
6-(4-((2-(3-(2-fluorofenil)tetrahidrofuran-3-il)tiazol-4-il)metoxi)-6-metoxibenzofuran-2-il)-2-metoxiimidazo[2,1-b][1,3,4]tiadiazol

166A. (2-(3-(2-fluorofenil)tetrahidrofuran-3-il)tiazol-4-il)metanol

El alcohol se preparó de acuerdo con el método descrito anteriormente en el ejemplo 163. LC (Método F): 1,900 min. RMN 1H (DMSO-da, 400 MHz) 8 ppm: 7,52 (dt, J = 1,6, 7,8 Hz, 1H), 7,38 (m, 1H), 7,28 (s, 1H), 7,23 (dt, J = 1,2, 7,6 Hz, 1H), 7,18 (ddd, J = 1,2, 8,2, 11,7 Hz, 1H), 5,26 (t, J = 5,9 Hz, 1H), 4,58 (dd, J = 3,1, 8 ,6 Hz, 1H), 4,48 (d, J = 5,5 Hz, 2H), 4,09 (d, J = 8 ,6 Hz, 1H), 3,91 (m, 2H), 2,97 (m, 1H), 2,55 (m, 1H).
Ejemplo 166. 6-(4-((2-(3-(2-fluorofenil)tetrahidrofuran-3-il)tiazol-4-il)metoxi)-6-metoxibenzofuran-2-il)-2-metoxiimidazo[2,1 -b][1,3,4]tiadiazol

El compuesto del título se preparó de acuerdo con el método descrito anteriormente en el ejemplo 163 y se aisló en forma de un sólido. LC (Método F): 2,627 min. HRMS (ESI): calc. para C28H24FN4O5S2 [M+H]+ m/z 579,1172, encontrado 579,1202. RMN 1 H (DMSO-d6, 400 MHz) 8 ppm: 8,36 (s, 1H), 7,70 (s, 1H), 7,55 (dt, J = 1,6, 7,8 Hz, 1H), 7,39 (m, 1H), 7,25 (dt, J = 1,2, 7,4 Hz, 1H), 7,20 (ddd, J = 0,8, 8,2, 11,7 Hz, 1H), 6,97 (s, 1H), 6,82 (d, J = 0,8 Hz, 1H), 6,59 (d, J = 1,6 Hz, 1H), 5,23 (s, 2H), 4,62 (dd, J = 2,7, 8 ,6 Hz, 1H), 4,20 (s, 3H), 4,12 (d, J = 8 ,6 Hz, 1H), 3,94 (m, 1H), 3,80 (s, 3H), 3,01 (m, 1H), 2,58 (dt, J = 8,4, 12,5 Hz, 1H).
Ejemplo 167
2-metoxi-6-(6-metoxi-4-((2-(4-metiltetrahidro-2H-piran-4-il)tiazol-4-il)metoxi)benzofuran-2-il)imidazo[2,1-b][1,3,4]tiadiazol

167A. 4-metiltetrahidro-2H-piran-4-carboxilato de metilo

A una solución en agitación de tetrahidro-2H-piran-4-carboxilato de metilo (0,926 ml, 6,94 mmol) en THF anhidro (20 ml), a -78 °C en nitrógeno, se le agregó gota a gota LDA (1 M en xx, 8,32 ml, 8,32 mmol) durante 30 min. La
mezcla se calentó a 0 °C durante 15 min, después se volvió a enfriar a -78 °C y se agregó gota a gota yodometano (0,867 ml, 13,87 mmol) durante 5 min. La solución se agitó a -78 °C durante 30 min y después a 0 °C durante 3 h antes de inactivarla con NH4Cl acuoso saturado y se extrajo con acetato de etilo. La fase orgánica se lavó con agua y salmuera, se secó (MgSO4), se filtró y se evaporó para obtener un aceite incoloro. Este material se purificó mediante cromatografía ultrarrápida usando un gradiente de 0 a 100 % de EtOAc en hexanos para obtener 4-metiltetrahidro-2H-piran-4-carboxilato de metilo (0,666 g, 60,7%) en forma de un aceite transparente e incoloro. LC (Método F): 1,549 min. RMN 1H (DMSO-d6, 400 MHz) 8 ppm: 3,67 (dt, J = 4,5, 12,2 Hz, 2H), 3,64 (s, 3H), 3,33 (m, 2H), 1,91 (m, 2H), 1,43 (dd, J = 3,9, 9,8 Hz, 1H), 1,40 (dd, J = 3,9, 9,8 Hz, 1H), 1,16 (s, 3H).
167B. Ácido 4-metiltetrahidro-2H-piran-4-carboxílico

A una solución en agitación de 4-metiltetrahidro-2H-piran-4-carboxilato de metilo (0,600 g, 3,79 mmol) en MeOH (15 ml), se le agregó hidróxido de sodio 1 N (7,59 ml, 7,59 mmol) y la mezcla se agitó a temperatura ambiente durante 24 h. Después la mezcla se concentró para retirar el MeOH y el concentrado acuoso se lavó con EtOAc. La fase acuosa se acidificó a pH 1 usando HCl concentrado y después se extrajo con EtOAc. El extracto orgánico se secó (MgSO4), se filtró y se concentró a sequedad para obtener ácido 4-metiltetrahidro-2H-piran-4-carboxílico (0,316 g, 57,8 %) en forma de un aceite transparente e incoloro. RMN 1H (DMSO-d6, 400 MHz) 8 ppm: 12,21 (s, 1H), 3,67 (dt, J = 4,5, 12,1 Hz, 2H), 3,35 (m, 2H), 1,89 (m, 2H), 1,36 (ddd, J = 3,9, 9,8, 13,7 Hz, 2H), 1,15 (s, 3H).
167C. 4-metiltetrahidro-2H-piran-4-carboxamida

A una solución en agitación de ácido 4-metiltetrahidro-2H-piran-4-carboxílico (0,300 g, 2,081 mmol) en DCM (5 ml), se le agregó cloruro de oxalilo (0,364 ml, 4,16 mmol) y la mezcla de reacción se agitó durante 1 h antes de que se concentrara a sequedad. El residuo se absorbió en t Hf (1 ml) y esta solución se agregó con agitación a amoníaco acuoso concentrado (5 ml) a 0 °C. La mezcla se agitó a 0 °C durante 2 min y después a temperatura ambiente durante 30 min antes de diluirla con agua y extraerla con EtOAc. La fase acuosa se concentró a sequedad para obtener un sólido de color blanco que se suspendió en EtOAc (20 ml) y la mezcla se calentó con agitación a 60 °C durante 20 min y después se filtró en caliente. El filtrado se combinó con el extracto orgánico original y se evaporó a sequedad para obtener 4-metiltetrahidro-2H-piran-4-carboxamida (0,255 g, 86 %) en forma de un sólido de color blanco que se usó tal cual en la siguiente etapa. RMN 1H (DMSO-d6, 400 MHz) 8 ppm: 7,18 (a s, 1H), 6 ,86 (a s, 1H), 3,61 (m, 2H), 3,37 (m, 2H), 1,92 (m, 2H), 1,33 (dd, J = 3,9, 9,4 Hz, 1H), 1,30 (m, 1H), 1,09 (s, 3H).
167D. 4-metiltetrahidro-2H-piran-4-carbotioamida

A una solución en agitación de 4-metiltetrahidro-2H-piran-4-carboxamida (0,250 g, 1,746 mmol) en THF (4 ml), se le agregó reactivo de Lawesson (0,353 g, 0,873 mmol) y la mezcla de reacción se calentó a reflujo durante 6 h. La mezcla enfriada se concentró prácticamente a sequedad y después se repartió entre EtOAc-NaHCO3 acuoso saturado. La fase orgánica se separó y la fase acuosa se saturó con NaCl (sólido) y se volvió a extraer con EtOAc. El extracto orgánico combinado se secó (MgSO4), se filtró y se concentró a sequedad. El residuo se purificó mediante columna ultrarrápida usando hexanos-EtOAc como eluyente para obtener 4-metiltetrahidro-2H-piran-4-carbotioamida (0,052 g, 18,7 %) en forma de un sólido de color blanco. r Mn 1H (DMSO-d6, 400 MHz) 8 ppm: 9,61 (a s, 1H), 8,79 (a s, 1H), 3,58 (m, 2H), 3,48 (m, 2H), 2,13 (m, 2H), 1,52 (m, 2H), 1,18 (s, 3H).
167E. 2-(4-metiltetrahidro-2H-piran-4-il)tiazol-4-carboxilato de etilo

A una mezcla de 4-metiltetrahidro-2H-piran-4-carbotioamida (0,052 g, 0,327 mmol) en i-PrOH (5 ml), se le agregó bromopiruvato de etilo (0,049 ml, 0,392 mmol) y la mezcla de reacción se calentó a reflujo durante 3 h. La mezcla enfriada se repartió entre EtOAc-NaHCO3 acuoso saturado y la fase orgánica se lavó con salmuera, se secó (MgSO4), se filtró y se concentró a sequedad. El residuo se purificó mediante cromatografía ultrarrápida usando un gradiente de 0 a 100 % de EtOAc en hexanos para obtener 2-(4-metiltetrahidro-2H-piran-4-il)tiazol-4-carboxilato de etilo (44 mg, 0,172 mmol, 52,8 % de rendimiento) en forma de un aceite transparente e incoloro. El material crudo se usó tal cual sin purificación adicional. LC (Método F): 2,003 min.
167F. (2-(4-metiltetrahidro-2H-piran-4-il)tiazol-4-il)metanol

A una solución enfriada en hielo de 2-(4-metiltetrahidro-2H-piran-4-il)tiazol-4-carboxilato de etilo (0,045 g, 0,176 mmol) en THF (1 ml), se le agregó LiBH4 (0,008 g, 0,352 mmol) de una vez y después MeOH (0,014 ml, 0,352 mmol). La mezcla resultante se agitó a 0 °C durante 5 min y después a temperatura ambiente durante 16 h. Después la mezcla se volvió a enfriar a 0 °C y se inactivó mediante la adición lenta de NH4Cl acuoso saturado. La mezcla resultante se extrajo con acetato de etilo, después de lo cual la fase orgánica se lavó con salmuera, se secó (MgSO4), se filtró y se concentró a sequedad. El residuo se purificó mediante cromatografía ultrarrápida usando un gradiente de 0 a 100 % de EtOAc en hexanos para obtener (2-(4-metiltetrahidro-2H-piran-4-il)tiazol-4-il)metanol (0,028 g, 74,5 % de rendimiento) en forma de un aceite transparente e incoloro. LC (Método F): 1,564 min.
Ejemplo 167. 2-metoxi-6-(6-metoxi-4-((2-(4-metiltetrahidro-2H-piran-4-il)tiazol-4-il)metoxi)benzofuran-2-il)imidazo[2,1-b][1,3,4]tiadiazol

A una mezcla de 6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-ol (Ejemplo 1H, 0,042 g, 0,131 mmol) y (2-(4-metiltetrahidro-2H-piran-4-il)tiazol-4-il)metanol (0,028 g, 0,131 mmol) en Th F seco (4 ml), se le agregó tri-n-butilfosfina (0,085 ml, 0,328 mmol) y la suspensión resultante se cargó con una solución de ADDP (0,083 g, 0,328 mmol) en THF (1 ml), añadido gota a gota durante 30 min mediante una bomba de jeringa. Después de agitar durante 1,5 h, la mezcla de reacción se diluyó con EtOAc y después se lavó con HCl 1 N, NaHCO3 acuoso saturado, agua y salmuera. La solución orgánica se evaporó y el residuo en bruto obtenido se disolvió en DMSO y se purificó mediante HPLC preparativa (columna Zorbax SB-C1821,2 x 100 mm, que se eluyó con CH3CN-agua-0 , 1 % de TFA). Las fracciones que contenían el producto se concentraron a sequedad y el residuo se liofilizó a partir de CH3CN-agua para obtener el compuesto del título (0,038 g, 0,074 mmol, 56,5 %) en forma de un sólido de color blanco amorfo. LC (Método F): 2,590 min. HRMS (ESI): calc. para C24H25N4O5S2 [M+H]+ m/z 513,1266, encontrado 513,1305. RMN 1H (DMSO-d6, 400 MHz) 8 ppm: 8,37 (s, 1H), 7,74 (s, 1H), 6,99 (d, J = 0,8 Hz, 1H), 6,83 (dd, J = 0,8, 1,6 Hz, 1H), 6,62 (d, J = 2,0 Hz, 1H), 5,28 (s, 2H), 4,20 (s, 3H), 3,81 (s, 3H), 3,70 (m, 2H), 3,53 (dd, J = 3,1, 7,8 Hz, 1H), 3,50 (dd, J = 3,1, 7,8 Hz, 1H), 2,12 (m, 2H), 1,74 (m, 2H), 1,38 (s, 3H).
Ejemplo 168
6-(4-((2-(croman-4-il)tiazol-4-il)metoxi)-6-metoxibenzofuran-2-il)-2-metoxiimidazo[2,1-b][1,3,4]tiadiazol

168A. (2-(croman-4-il)tiazol-4-il)metanol

El alcohol se preparó de acuerdo con el método descrito en el ejemplo 167. LC (Método F): 1,894 min. RMN 1H (DMSO-da, 400 MHz) 8 ppm: 7,30 (m, 1H), 7,16 (m, 1H), 7,09 (dd, J = 1,2, 7,4 Hz, 1H), 6 ,86 (dd, J = 1,2, 7,4 Hz, 1H), 6,82 (m, 1H), 5,29 (t, J = 5,7 Hz, 1H), 4,56 (m, 1H), 4,54 (dd, J = 1,2, 5,9 Hz, 2H), 4,24 (m, 1H), 4,13 -4 ,08 (m, 1H), 2,32 -2 ,28 (m, 2H).
Ejemplo 168. 6-(4-((2-(croman-4-il)tiazol-4-il)metoxi)-6-metoxibenzofuran-2-il)-2-metoxiimidazo[2,1-b][1,3,4]tiadiazol

El compuesto del título se preparó de acuerdo con el método descrito anteriormente en el ejemplo 167 y se aisló en forma de un sólido. LC (Método F): 2,638 min. HRMS (ESI): calc. para C27H23N4O5S2 [M+H]+ m/z 547,1110, encontrado 547,1130. RMN 1 H (DMSO-d6, 400 MHz) 8 ppm: 8,37 (s, 1H), 7,72 (s, 1H), 7,18 (m, 1H), 7,13 (m, 1H), 6,98 (d, J = 0,8 Hz, 1H), 6 ,88 -6 ,82 (m, 3H), 6,61 (d, J = 2,0 Hz, 1H), 5,29 (s, 2H), 4,63 (t, J = 5,1 Hz, 1H), 4,25 (m, 1H), 4,20 (s, 3H), 4,13 -4 ,08 (m, 1H), 3,80 (s, 3H), 2,34 (q, J = 5,5 Hz, 2H).
Ejemplo 169
6-(4-((2-(2H-cromen-4-il)tiazol-4-il)metoxi)-6-metoxibenzofuran-2-il)-2-metoxiimidazo[2,1-b][1,3,4]tiadiazol

169A. 4-(4-(hidroximetil)tiazol-2-il)croman-4-ol

El alcohol se preparó de acuerdo con el método descrito en el ejemplo 118. LC (Método F): 1,519 min. RMN 1 H (DMSO-d6, 400 MHz) 8 ppm: 7,33 (d, J = 1,2 Hz, 1H), 7,15 (m, 1H), 7,02 (m, 1H), 6,81 (m, 2H), 6,73 (s, 1H), 5,22 (t, J = 5,9 Hz, 1H), 4,46 -4,41 (m, 3H), 4,30 (m, 1H), 2,50 (m, 1H), 2,17 (ddd, J = 2,7, 5,7, 13,7 Hz, 1H).
169B. 4-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2-il)croman-4-ol

El compuesto del título se preparó de 6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-ol (Ejemplo 1H) y 4-(4-(hidroximetil)tiazol-2-il)croman-4-ol de acuerdo con el método descrito en el ejemplo 118 y se aisló en forma de un sólido. LC (Método A): 2,478 min. HRMS (ESI): calc. para C27H23N4O6S2 [M+H]+ m/z 563,1059, encontrado 563,1059. RMN 1H (DMSO-da, 400 MHz) 8 ppm: 8,36 (s, 1H), 7,75 (s, 1H), 7,17 (m, 1H), 7,05 (m, 1H), 6,95 (s, 1H), 6,83 (m, 4H), 6,55 (m, 1H), 5,20 (s, 2H), 4,45 (m, 1H), 4,31 (m, 1H), 4,20 (s, 3H), 3,77 (s, 3H), 2,54 (m, 1H), 2,21 (m, 1H).
Ejemplo 169. 6-(4-((2-(2H-cromen-4-il)tiazol-4-il)metoxi)-6-metoxibenzofuran-2-il)-2-metoxiimidazo[2,1-b][1,3,4]tiadiazol

A una solución enfriada en hielo de 4-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2-il)croman-4-ol (0,025 g, 0,044 mmol) en DCM (5 ml), se le agregó DAST (0,018 ml, 0,133 mmol) y la mezcla de reacción se agitó a 0 °C durante 5 min y después a temperatura ambiente durante 20 min. La reacción se inactivó a 0 °C con NaHCO3 acuoso saturado y se diluyó con DCM. La fase orgánica se separó, se secó (MgSO4), se filtró y se concentró a sequedad. El residuo se purificó mediante HPLC preparativa (Método A). Las fracciones que contenían el producto se evaporaron y el residuo se liofilizó de MeCN-agua para obtener 6-(4-((2-(2H-cromen-4-il)tiazol-4-il)metoxi)-6-metoxibenzofuran-2-il)-2-metoxiimidazo[2,1-b][1,3,4]tiadiazol (0,007 g, 28,9 %) en forma de un sólido amorfo de color blanco. LC (Método F): 2,206 min. Hr MS (ESI): calc. para C27H21N4O5S2 [M+H]+ m/z 545,0953, encontrado 545,0956. RMN 1 H (DMSO-d6, 400 MHz) 8 ppm: 8,37 (s, 1H), 7,90 (dd, J = 1,6, 7,8 Hz, 1H), 7,89 (s, 1H), 7,23 (m, 1H), 7,02 (d, J = 0,8 Hz, 1H), 6,94 (dt, J - 1,2, 7,4 Hz, 1H), 6,90 (dd, J = 1,2, 7,8 Hz, 1H), 6,85 (d, J = 0,8 Hz, 1H), 6,67 (d, J = 2,0 Hz, 1H), 6,57 (t, J = 4,1 Hz, 1H), 5,39 (s, 2H), 4,85 (d, J = 3,9 Hz, 2H), 4,20 (s, 3H), 3,81 (s, 3H).
Ejemplo 170
(R)-N-(4-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2-il)tetrahidro-2H-piran-4-il)-2-metilpropan-2-sulfinamida

170A. (R)-N-(dihidro-2H-piran-4(3H)-iliden)-2-metilpropan-2-sulfinamida
A una solución en agitación de dihidro-2H-piran-4(3H)-ona (2,40 g, 23,97 mmol) en THF (50 ml) a temperatura ambiente en atmósfera de nitrógeno, se le agregó etóxido de titanio (IV) (8,80 ml, 42,0 mmol) y después (R)-(+)-2 -metilpropan-2-sulfinamida (2,58 g, 21,29 mmol). La reacción se agitó a temperatura ambiente durante 3 h y después
se inactivó vertiéndola en una mezcla de NaHCO3 acuoso saturado frío (50 ml) y EtOAc (50 ml), con agitación rápida. La suspensión resultante se filtró y el residuo se enjuagó con EtOAc. La capa acuosa del filtrado se separó y se volvió a extraer con EtOAc. Las fases orgánicas combinadas se secaron (Na2SO4) y se concentraron para obtener un aceite de color amarillo claro. La cromatografía ultrarrápida (Isco/ 0-100 % de EtOAc-hexano) produjo (R)-N-(dihidro-2H-piran-4(3H)-iliden)-2-metilpropan-2-sulfinamida (2,045 g, 47,2 %) en forma de un aceite transparente e incoloro que cristalizó en reposo al vacío. Este material se usó tal cual en la siguiente etapa. LC (Método A): 1,199 min. HRMS (ESI): calc. para C9H18NO2S [M+H]+ m/z 204,106; encontrado 204,106.
170B. (R)-N-(4-(4-(((terc-butildimetilsilil)oxi)metil)tiazol-2-il)tetrahidro-2H-piran-4-il)-2-metilpropan-2-sulfinamida

Una solución de 2-bromo-4-(((terc-butildimetilsilil)oxi)metil)tiazol (Ejemplo 37B, 2,58 g, 8,37 mmol) en THF seco (40 ml) se enfrió a -78°C en N2 y después se agregó gota a gota n-butil litio 1,45 M (6,93 ml, 10,04 mmol). La mezcla resultante se agitó durante 30 min para obtener una solución de color amarillo parduzco claro. A esta mezcla se le agregó una solución de (R)-N-(dihidro-2H-piran-4(3H)-iliden)-2-metilpropan-2-sulfinamida (2,042 g, 10,04 mmol) en THF seco (10 ml) durante 5 min y la mezcla se mantuvo a -78 °C durante 2 h. La reacción se inactivó mediante la adición de NH4Cl acuoso saturado ( 10 ml), después se retiró el baño de enfriamiento y la mezcla se repartió entre EtOAc-agua. La fase orgánica se separó, la fase acuosa se volvió a extraer con EtOAc y la fase orgánica combinada se lavó (salmuera), se secó (Na2SO4) y se evaporó para obtener un aceite de color amarillo claro. La cromatografía ultrarrápida (Isco/ 0-5 % [10 % de N^OH-MeOH]-DCM) produjo (R)-N-(4-(4-(((terc-butildimetilsilil)oxi)-metil)tiazol-2-il)tetrahidro-2H-piran-4-il)-2-metilpropan-2-sulfinamida (3,05 g, 84 %) en forma de una goma prácticamente incolora. Este material se usó tal cual en la siguiente etapa. LC (Método A): 2,394 min. HRMS (ESI): calc. para C19H37N2OaS2Si [M+H]+ m/z 433,201; encontrado 433,203. RMN 1H (400 MHz, DMSO-da): 8 7,35 (s, 1H), 5,60 (s, 1H), 4,65 (s, 2H), 3,69 (m, 2H), 3,49 (m, 1H), 3,38 (m, 1H), 2,27 (m, 1H), 2,09 (m, 3H), 1,02 (s, 9H), 0,81 (s, 9H), 0,00 (s, 6 H).
170C. (R)-N-(4-(4-(hidroximetil)tiazol-2-il)tetrahidro-2H-piran-4-il)-2-metilpropan-2-sulfinamida
A una solución de (R)-N-(4-(4-(((terc-butildimetilsilil)oxi)metil)tiazol-2-il)tetrahidro-2H-piran-4-il)-2-metilpropan-2-sulfinamida (3,045 g, 7,04 mmol) en THF seco (25 ml), en atmósfera de N2 , se le agregó gota a gota trihidrofluoruro de trietilamina (3,44 ml, 21,11 mmol) y la mezcla se agitó a temperatura ambiente durante 2 días. Después la mezcla se diluyó con DCM y la solución se lavó (NaHCO3 acuoso saturado), se secó (Na2SO4) y se evaporó para obtener (R)-N-(4-(4-(hidroximetil)tiazol-2-il)tetrahidro-2H-piran-4-il)-2-metilpropan-2-sulfinamida (2,074 g, 93 %) en forma de un aceite incoloro que cristalizó en reposo al vacío. Este material era esencialmente puro y se usó tal cual en la siguiente etapa. LC (Método A): 1,426 min. HRMS (ESI): calc. para C13H23N2O3S2 [M+H]+ m/z 319,115; encontrado 319,115. RMN 1H (400 MHz, DMSO-d6): 8 7,31 (s, 1H), 5,58 (s, 1H), 5,22 (t, J= 5,87 Hz, 1H), 4,47 (d, J= 5,87 Hz, 2H), 3,69 (m, 2H), 3,49 (m, 1H), 3,39 (m, 1H), 2,25 (m, 1H), 2,09 (m, 3H), 1,03 (s, 9H).
Ejemplo 170. (R)-N-(4-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2-il)tetrahidro-2H-piran-4-il)-2-metilpropan-2-sulfinamida

A un matraz secado al fuego se le agregó 6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-ol (Ejemplo 1H, 0,200 g, 0,630 mmol) y (R)-N-(4-(4-(hidroximetil)tiazol-2-il)tetrahidro-2H-piran-4-il)-2-metilpropan-2sulfinamida (0,241 g, 0,756 mmol); después el matraz se purgó con N2 y se agregó THF seco (8 ml). A la suspensión resultante se le agregó tri-n-butilfosfina (0,409 ml, 1,576 mmol) y después se agregó gota a gota una solución de 1,1'-(azodicarbonil)dipiperidina (0,402 g, 1,576 mmol) en THF seco (5 ml) (mediante una bomba de jeringa) durante 30 min. La mezcla resultante se agitó a temperatura ambiente durante 1 h más, después se diluyó con EtOAc, se lavó (NaHCO3 acuoso saturado, H2O y salmuera), se secó (Na2SO4) y se evaporó para obtener una goma de color amarillo claro. La cromatografía ultrarrápida (Isco/ 0-5 % [10 % de NH4OH-MeOH]-DCM) produjo un sólido que se trituró con un volumen mínimo de MeCN. La suspensión resultante se filtró y la torta de filtro se lavó con una pequeña cantidad de MeCN y después se secó al vacío para obtener (R)-N-(4-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2-il)tetrahidro-2H-piran-4-il)-2-metilpropan-2-sulfinamida (0,294 g, 76 %) en forma de un sólido de color blanco. LC (Método A): 2,240 min. HRMS (ESI): calc. para C27H32N5O6S3 [M+H]+ m/z 618,151; encontrado 618,150. RMN 1 H (400 MHz, DMSO-d6): 8 8,33 (s, 1H), 7,77 (s, 1H), 6,95 (s, 1H), 6,80 (s, 1H), 6,59 (d, J= 1,57 Hz, 1H), 5,72 (s, 1H), 5,25 (s, 2H), 4,17 (s, 3H), 3,77 (s, 3H), 3,73 (m, 2H), 3,55 (m, 1H), 3,46 (m, 1H), 2,31 (m, 1H), 2,18 (m, 3H), 1,08 (s, 9H).
Ejemplo 171
N-(4-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2-il)tetrahidro-2H-piran-4-il)-2-metilpropan-2-sulfonamida

A una solución de (R)-N-(4-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2-il)tetrahidro-2H-piran-4-il)-2-metilpropan-2-sulfinamida (0,050 g, 0,081 mmol) en DCM (3 ml), en atmósfera de N2 , se le agregó ácido 3-cloroperoxibenzoico al 70 % (0,030 g, 0,121 mmol) de una vez y la mezcla resultante de color amarillo se agitó a temperatura ambiente durante 6 h. Después la mezcla de reacción se diluyó con DCM y esta mezcla se lavó (NaOH 0,1 N, NaHCO3 acuoso saturado y salmuera), se secó (Na2SO4) y se evaporó para obtener un sólido de color verde oscuro. La cromatografía ultrarrápida (Isco/ 0-100 % de acetonahexano) produjo N-(4-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2-il)tetrahidro-2H-piran-4-il)-2-metilpropan-2-sulfonamida (0,004 g, 7,80 %) en forma de un sólido de color blanquecino. LC (Método A): 2,258 min. HRMS (ESI): calc. para C27H32N5O7S3 [M+H]+ m/z 634,146; encontrado 634,147. RMN 1 H (400 MHz, DMSO-d6): 8 8,33 (s, 1H), 7,75 (s, 1H), 7,17 (s, 1H), 6,90 (s, 1H), 6,76 (s, 1H), 6,55 (d, J= 1,96 Hz, 1H), 5,21 (s, 2H), 4,14 (s, 3H), 3,74 (s, 3H), 3,70 (m, 2H), 3,47 (m, 2H), 2,24 (m, 4H), 1,12 (s, 9H).
Ejemplo 172
4-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2-il)tetrahidro-2H-piran-4-amina, HCl

Método A: A una solución de (R)-N-(4-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2-il)tetrahidro-2H-piran-4-il)-2-metilpropan-2-sulfinamida (0,0103 g, 0,017 mmol) en Th F (1 ml), se le agregó HCl acuoso 6 M (0,050 ml) y la mezcla se agitó a temperatura ambiente en un vial sellado durante 30 min. Después la mezcla se diluyó con DCM, se lavó (NaHCO3 acuoso saturado), se secó (Na2SO4) y se evaporó para obtener una goma. Este material se purificó mediante HPLC preparativa (Método A) para obtener un sólido que se liofilizó a partir de MeCN-agua para obtener 4-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2-il)tetrahidro-2H-piran-4-amina, TFA (0,005 g, 48,0 %) en forma de un sólido de color blanco. LC (Método A): 1,911 min. HRMS (ESI): calc. para C23H24N5O5S2 [M+H]+ m/z 514,122; encontrado 514,122. RMN 1 H (400 MHz, DMSO-d6) 8 8,30 (s, 1H), 7,87 (s, 1H), 6,93 (s, 1H), 6,78 (s, 1H), 6,55 (d, J= 1,96 Hz, 1H), 5,25 (s, 2H), 4,14 (s, 3H), 3,74 (s, 3H), 2,26 (m, 4H), 2,20-2,04 (m, 6 H).
Método B: A una suspensión de (R)-N-(4-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4il)oxi)metil)tiazol-2-il)tetrahidro-2H-piran-4-il)-2-metilpropan-2-sulfinamida (0,280 g, 0,453 mmol) en THF (12 ml), se le agregó HCl acuoso 6 M (0,755 ml, 4,53 mmol) y la mezcla se agitó a temperatura ambiente en un vial sellado durante 30 min. La mezcla se filtró posteriormente, la torta de filtro se lavó con una pequeña cantidad de THF y después se secó al vacío para obtener 4-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2-il)tetrahidro-2H-piran-4-amina, HCl (0,249 g, 100 % de rendimiento) en forma de un sólido de color blanco. Una pequeña porción de este sólido se repartió entre EtOAc-NaHCO3 acuoso saturado (se agregó un volumen mínimo de MeOH para ayudar a la solubilización) y la fase orgánica se separó, se secó (Na2SO4) y se evaporó para obtener un sólido de color blanco. La cromatografía ultrarrápida (Isco/ 0-10 % [10 % de NH4OH-MeOH]-DCM) produjo 4-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2-il)tetrahidro-2H-piran-4-amina en forma de un sólido de color blanco. Lc (Método A): 2,038 min. HRMS (ESI): calc. para C23H24N5O5S2 [M+H]+ m/z 514,122; encontrado 514,120. RMN 1 H (400 MHz, DMSO-da): 8 8,30 (s, 1H), 7,57 (s, 1H), 6,91 (s, 1H), 6,76 (m, 1H), 6,56 (d, J= 1,96 Hz, 1H), 5,18 (s, 2H), 4,14 (s, 3H), 3,74 (s, 3H), 3,70 (dt, J= 2,35, 10,96 Hz, 2H), 3,63 (dt, J= 4,30, 11,35 Hz, 2H), 2,05 (m, 2H), 1,50 (d, J= 12,52 Hz, 2H).
Ejemplo 173
(2-((4-(4-(((6-metoxi-2-(2-metoxiimidazo-[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)-tiazol-2-il)tetrahidro-2H-piran-4-il)amino)-2-oxoetil)carbamato de tere-butilo

A una mezcla de 4-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2-il)tetrahidro-2H-piran-4-amina (0,035 g, 0,068 mmol) y ácido 2-((tere-butoxicarbonil)amino)acético (0,013 g, 0,076 mmol) en DMF (2 ml), se le agregó DIEA (0,048 ml, 0,275 mmol) y después HATU (0,029 g, 0,076 mmol). La solución de color amarillo claro resultante se agitó a temperatura ambiente durante 16 h y después se diluyó con agua (6 ml), la suspensión resultante se filtró y el residuo se lavó con agua. El residuo húmedo se repartió entre DCM-NaHCO3 acuoso saturado y la fase orgánica se separó, se secó (Na2SO4) y se evaporó para obtener una goma incolora. La cromatografía ultrarrápida (Isco/ 0-10 % [10 % de N^OH-MeOH]-DCM) produjo (2-((4-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2-il)tetrahidro-2H-piran-4-il)amino)-2-oxoetil)carbamato de tere-butilo (0,029 g, 62,9 %) en forma de una goma incolora que se liofilizó a partir de MeCN-agua para obtener un sólido de color blanco. LC (Método A): 2,292 min. Hr Ms (ESI): calc. para C30H35N6O8S2 [M+H]+ m/z 671,196; encontrado 671,195. RMN 1 H (400 MHz, DMSO-d6): 8 8,38 (s, 1H), 8,34 (s, 1H), 7,69 (s, 1H), 7,01 (s, 1H), 6,93 (t, J= 5,87 Hz, 1H), 6,84 (m, 1H), 6,20 (t, J= 1,96 Hz, 1H), 5,25 (s, 2H), 4,20 (s, 3H), 3,81 (s, 3H), 3,74 (m, 2H), 3,63-3,57 (m, 4H), 2,32 (m, 2H), 2,12 (m, 2H), 1,38 (s, 9H).
Ejemplo 174
2-(dimetilamino)-N-(4-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)-tiazol-2-il)tetrahidro-2H-piran-4-il)acetamida

A una suspensión de 4-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2il)tetrahidro-2H-piran-4-amina, HCl (Ejemplo 172, 0,035 g, 0,064 mmol) y ácido 2-(dimetilamino)acético (7,44 mg, 0,070 mmol) en DMF (1,5 ml), se le agregó DIEA (0,056 ml, 0,318 mmol) y después Ha TU (0,027 g, 0,070 mmol). La solución resultante de color amarillo claro se agitó a temperatura ambiente durante 1 h, después la mezcla se diluyó con agua (3 ml), la suspensión resultante se filtró y el residuo se lavó con agua. El residuo húmedo se repartió entre DCM-NaHCO3 acuoso saturado y la fase orgánica se separó, se secó (Na2SO4) y se evaporó para obtener una goma incolora. La cromatografía ultrarrápida (Isco/ 0-10 % [10 % de NH4OH-MeOH]-DCM) produjo 2-(dimetilamino)-N-(4-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2-il)tetrahidro-2H-piran-4-il)acetamida (0,026 g, 68,2 %) en forma de un sólido. LC (Método A): 2,026 min. HRMS (ESI): calc. para C27H31N6O6S2 [M+H]+ m/z 599,175; encontrado 599,176. RMN 1 H (400 MHz, DMSO-d6): 8 8,31 (s, 1H), 8,13 (s, 1H), 7,62 (s, 1H), 6,93 (s, 1H), 6,77 (s, 1H), 6,55 (d, J= 1,96 Hz, 1H), 5,18 (s, 2H), 4,14 (s, 3H), 3,75 (s, 3H), 3,70 (m, 2H), 3,51 (t, J= 10,96 Hz, 2H), 2,88 (s, 2H), 2,30 (m, 2H), 2,12 (m, 2H), 2,20 (s, 6 H), 2,07 (m, 2H).
Ejemplo 175
2-metoxi-6-(6-metoxi-4-((2-(2-fenilpropan-2-il)tiazol-4-il)metoxi)benzofuran-2-il)imidazo[2,1-b][1,3,4]tiadiazol

175A. 2-metil-2-fenilpropanamida
A una solución en agitación de ácido 2-metil-2-fenilpropanoico (2,00 g, 12,18 mmol) en DCM (29 ml), se le agregó cloruro de oxalilo (2,132 ml, 24,36 mmol) y la mezcla de reacción se agitó durante 1 h a temperatura ambiente antes de que se evaporara a sequedad. El residuo resultante se absorbió en THF (6 ml) y después se agregó con agitación a amoníaco acuoso helado concentrado (29 ml). Después de 2 min, se retiró el baño de enfriamiento y la mezcla se agitó a temperatura ambiente durante 30 min. La mezcla resultante se diluyó con agua y después se extrajo con EtOAc. La fase orgánica se lavó con salmuera, se secó (MgSO4), se filtró y se concentró a sequedad para obtener 2-metil-2-fenilpropanamida (0,763 g, 38,4 %) en forma de un sólido de color blanco. Este material se usó tal cual en la siguiente etapa. LC (Método F): 1,563 min. RMN 1 H (DMSO-d6, 400 MHz) 8 ppm: 7,35-7,29 (m, 4H), 7,20 (m, 1H), 6 ,86 (a s, 2H), 1,42 (s, 6 H).
175B. 2-metil-2-fenilpropantioamida

Una solución de 2-metil-2-fenilpropanamida (0,756 g, 4,63 mmol) y reactivo de Lawesson (0,937 g, 2,316 mmol) en THF (5 ml) se calentó a reflujo durante 2 h. La mezcla de reacción enfriada se repartió entre EtOAc-NaHCO3 acuoso saturado y la fase orgánica se separó, se secó (MgSO4), se filtró y se evaporó. El residuo se purificó mediante cromatografía ultrarrápida usando hexanos-EtOAc como eluyente para obtener 2-metil-2-fenilpropantioamida (0,452 g, 54,4 %) en forma de un sólido de color blanco. LC (Método F): 1,784 min. RMN 1 H (DMSO-d6, 400 MHz) 8 ppm: 9,54 (a s, 1H), 8,50 (a s, 1H), 7,38-7,35 (m, 2H), 7,30 (m, 2H), 7,21 (m, 1H), 1,56 (s, 6 H).
175C. 2-(2-fenilpropan-2-il)tiazol-4-carboxilato de etilo
A una mezcla de 2-metil-2-fenilpropantioamida (0,445 g, 2,482 mmol) en i-PrOH (5 ml), se le agregó bromopiruvato de etilo (0,375 ml, 2,98 mmol) y la mezcla de reacción se calentó a reflujo durante 3 h. La mezcla enfriada se repartió entre EtOAc-NaHCO3 acuoso saturado y la fase orgánica se separó, se lavó con salmuera, se secó (MgSO4), se filtró y se concentró a sequedad. El residuo se purificó mediante cromatografía de columna usando un gradiente de 0 a 100 % de EtOAc en hexanos para obtener 2-(2-fenilpropan-2-il)tiazol-4-carboxilato de etilo (524 mg, 77 % de rendimiento) en forma de un aceite transparente e incoloro. LC (Método F): 2,206 min. RMN 1 H (DMSO-d6, 400 MHz) 8 ppm: 8,41 (s, 1H), 7,36 - 7,31 (m, 4H), 7,28 - 7,23 (m, 1H), 4,29 (q, J = 7,0 Hz, 2H), 1,79 (s, 6 H), 1,30 (t, J = 7,0 Hz, 3H).
175D. (2-(2-fenilpropan-2-il)tiazol-4-il)metanol

A una solución de 2-(2-fenilpropan-2-il)tiazol-4-carboxilato de etilo (0,515 g, 1,870 mmol) en THF (9 ml), se le agregó LiBH4 (0,081 g, 3,74 mmol), y después MeOH (0,151 ml, 3,74 mmol). Posteriormente, la mezcla de reacción se agitó a temperatura ambiente durante 16 h y después se inactivó mediante la adición gota a gota de NH4Cl acuoso saturado. La mezcla resultante se extrajo con acetato de etilo y después la fase orgánica se lavó con salmuera, se secó (MgSO4), se filtró y se concentró a sequedad. El residuo se purificó mediante cromatografía ultrarrápida usando un gradiente de 0 a 100 % de EtOAc en hexanos para obtener (2-(2-fenilpropan-2-il)tiazol-4-il)metanol (0,350 g, 80 %) en forma de un aceite transparente. LC (Método F): 1,528 min. RMN 1 H (DMSO-d6, 400 MHz) 8 ppm: 7,35-7,26 (m, 5H), 7,22 (m, 1H), 5,26 (t, J = 5,7 Hz, 1H), 4,53 (dd, J = 0,8, 5,5 Hz, 2H), 1,76 (s, 6 H).
Ejemplo 175. 2-metoxi-6-(6-metoxi-4-((2-(2-fenilpropan-2-il)tiazol-4-il)metoxi)benzofuran-2-il)imidazo[2,1-b][1,3,4]tiadiazol

A una suspensión de 6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-ol (Ejemplo 1H, 0,105 g, 0,330 mmol) y (2-(2-fenilpropan-2-il)tiazol-4-il)metanol (0,077 g, 0,330 mmol) en THF seco (8 ml), se le agregó tri-nbutilfosfina (0,214 ml, 0,825 mmol) y después una solución de ADDP (0,208 g, 0,825 mmol) en ThF (2 ml), añadido gota a gota durante 30 min a través de una bomba de jeringa. Después de agitar durante 30 min más, la mezcla de reacción se repartió entre EtOAc y NaHCO3 acuoso saturado. La fase orgánica se separó, se lavó con salmuera, se secó (MgSO4), se filtró y se concentró a sequedad. El residuo se purificó mediante cromatografía en columna usando DCM-EtOAc como eluyente para obtener el compuesto del título (0,097 g, 0,182 mmol, 55,2 %) en forma de un sólido de color beis. LC (Método A): 2,536 min. HRMS (ESI): calc. para C27H25N4O4S2 [M+H]+ m/z 533,1317, encontrado 533,1336. RMN 1 H (DMSO-d6, 400 MHz) 8 ppm: 8,37 (s, 1H), 7,69 (s, 1H), 7,37 - 7,29 (m, 4H), 7,23 (m, 1H), 6,99 (s, 1H), 6,83 (s, 1H), 6,62 (d, J = 2,0 Hz, 1H), 5,28 (s, 2H), 4,20 (s, 3H), 3,80 (s, 3H), 1,80 (s, 6 H).
Ejemplo 176
6-(4-((2-(2-(4-clorofenil)propan-2-il)tiazol-4-il)metoxi)-6-metoxibenzofuran-2-il)-2-metoxiimidazo[2,1-b][1,3,4]tiadiazol

176A. (2-(2-(4-clorofenil)propan-2-il)tiazol-4-il)metanol

El alcohol se preparó de acuerdo con el método descrito anteriormente en el ejemplo 175. LC (Método A): 2,032 min. RMN 1H (DMSO-da, 400 MHz) 8 ppm: 7,36 (m, 4H), 7,29 (s, 1H), 5,27 (t, J = 5,5 Hz, 1H), 4,53 (d, J = 4,7 Hz, 2H), 1,75 (s, 6 H).
Ejemplo 176. 6-(4-((2-(2-(4-dorofenil)propan-2-il)tiazol-4-il)metoxi)-6-metoxibenzofuran-2-il)-2-metoxiimidazo[2,1-b][1,3,4]tiadiazol

El compuesto del título se preparó de acuerdo con el método descrito anteriormente en el ejemplo 175 y se aisló en forma de un sólido. LC (Método A): 2,588 min. HRMS (ESI): calc. para C27H24ClN4O4S2 [M+H]+ m/z 567,0928, encontrado 567,0937. RMN 1 H (DMSO-d6, 400 MHz) 8 ppm: 8,37 (s, 1H)7,70 (s, 1H), 7,37 (s, 4H), 6,98 (s, 1H), 6,83 (d, J = 0,8 Hz, 1H), 6,61 (d, J = 1,6 Hz, 1H), 5,28 (s, 2H), 4,20 (s, 3H), 3,80 (s, 3H), 1,78 (s, 6 H).
Ejemplo 177
2-metoxi-6-(6-metoxi-4-((2-(1-(piridin-3-il)ciclobutil)tiazol-4-il)metoxi)benzofuran-2-il)imidazo[2,1-b][1,3,4]tiadiazol

177A. 1-(piridin-3-il)ciclobutancarbonitrilo
A una solución de 2-(piridin-3-il)acetonitrilo (0,903 ml, 8,46 mmol) en THF seco (10 ml), se le agregó hidróxido de sodio 17 M (14,94 ml, 254 mmol) y sulfato ácido de tetrabutilamonio (0,287 g, 0,846 mmol) y después se agregó gota a gota 1,3-dibromopropano (0,902 ml, 8,89 mmol). La mezcla resultante se calentó a reflujo durante 4 h y después se dejó enfriar y la fase acuosa se decantó cuidadosamente. La fase orgánica se diluyó con EtOAc, se lavó con agua y salmuera, se secó sobre MgSO4, se filtró y se concentró a sequedad. El residuo se purificó mediante columna ultrarrápida usando un gradiente de 0 a 10 % de MeOH: NH4OH (9:1) en DCM para obtener 1 -(piridin-3-il)ciclobutancarbonitrilo (0,659 g, 49,2 %) en forma de un aceite de color amarillo. LC (Método A): 0,746 min. RMN 1 H (DMSO-d6, 400 MHz) 8 ppm: 8,71 (dd, J = 0,8, 2,7 Hz, 1H), 8,57 (dd, J = 1,6, 4,7 Hz, 1H), 7,92 (m, 1H), 7,48 (ddd, J = 0,8, 4,7, 7,8 Hz, 1H), 2,81 - 2,74 (m, 2H), 2,72 - 2,64 (m, 2H), 2,30 (m, 1H), 2,04 (m, 1H).
177B. 1-(piridin-3-il)ciclobutancarboxamida

Una mezcla de 1-(piridin-3-il)cidobutancarbonitrilo (0,659 g, 4,17 mmol) y H2SO4 (4 ml) concentrado se agitó a temperatura ambiente durante 16 h. La mezcla de reacción después se vertió cuidadosamente en hielo picado y la mezcla se basificó con NaHCO3 sólido hasta que cesó la evolución de gas. Después, la mezcla se extrajo con EtOAc y la fase orgánica se separó, se secó (MgSO4), se filtró y se concentró a sequedad. El residuo se purificó mediante columna ultrarrápida usando de 0 a 10 % de MeOH:NH4OH (9:1) en DCM como eluyente para obtener 1 -(piridin-3-il)ciclobutancarboxamida (0,283 g, 34,7 % de rendimiento) en forma de un sólido de color beis. LCMS (APCI): calc. para C10H13N2O [M+H]+ m/z 177,10, encontrado 177,20. RMN 1H (DMSO-da, 400 MHz) 8 ppm: 8,55 (dd, J = 0,8, 2,7 Hz, 1H), 8,43 (m, 1H), 7,70 (m, 1H), 7,35 (ddd, J = 0,8, 4,7, 8,2 Hz, 1H), 7,28 (a s, 1H), 6,96 (a s, 1H), 2,75 - 2,66 (m, 2H), 2,40 -2 ,33 (m, 2H), 1,88 - 1,72 (m, 2H).
177C.1-(piridin-3-il)ciclobutancarbotioamida

A una solución en agitación de 1-(piridin-3-il)ciclobutancarboxamida (0,283 g, 1,445 mmol) en THF (5 ml), se le agregó reactivo de Lawesson (0,292 g, 0,723 mmol) y la mezcla se calentó a reflujo durante 6 h. La mezcla enfriada se concentró después prácticamente a sequedad y el concentrado se repartió entre EtOAc-NaHCO3 acuoso saturado. La fase orgánica se separó, se secó (MgSO4), se filtró y se concentró a sequedad. El residuo se purificó mediante cromatografía en columna, usando 0 a 10 % de MeOH:NH4OH (9:1) en DCM como eluyente para obtener 1-(piridin-3-il)ciclobutancarbotioamida (0,208 g, 74,8 %) en forma de un sólido de color blanco. LC (Método A): 0,929 min. RMN 1 H (DMSO-d6, 400 MHz) 8 ppm: 9,54 (a s, 1H), 9,09 (a s, 1H), 8,73 (dd, J = 0,8, 2,3 Hz 1H), 8,44 (dd, J = 1,6, 4,7 Hz, 1H), 7,90 (m, 1H), 7,36 (ddd, J = 0,8, 4,7, 7,8 Hz, 1H), 2,87 - 2,80 (m, 2H), 2,59 - 2,52 (m, 2H), 1,73 -1,60 (m, 2H).
177D.2-(1-(piridin-3-il)ciclobutil)tiazol-4-carboxilato de etilo

A una suspensión de 1-(piridin-3-il)ciclobutancarbotioamida (0,200 g, 1,040 mmol) en etanol (2 ml) a 0 °C, se le agregó gota a gota una solución de 3-bromo-2-oxopropanoato de etilo (0,124 ml, 0,988 mmol) en etanol (2 ml). Después se retiró el baño de hielo y la mezcla de reacción se agitó a temperatura ambiente durante 24 h, antes de repartirla entre EtOAc y NaHCO3 acuoso saturado. La fase orgánica se separó, se secó (MgSO4), se filtró y se concentró a sequedad. El residuo se purificó mediante columna ultrarrápida usando 0-10 % de MeOH:NH4OH (9:1) en DCM para obtener 2-(1-(piridin-3-il)ciclobutil)tiazol-4-carboxilato de etilo (0,169 g, 53,0 % de rendimiento) en forma de un aceite de color amarillo claro. LC (Método A): 1,456 min. LCMS (APCI): calc. para C15H17N2O2S [M+H]+ m/z 289,10, encontrado 289,20.
177E.(2-(1-(piridin-3-il)ciclobutil)tiazol-4-il)metanol
A una solución en agitación de 2-(1-(piridin-3-il)ciclobutil)tiazol-4-carboxilato de etilo (0,169 g, 0,586 mmol) en THF (3 ml) a temperatura ambiente, se le agregó LiBH4 (0,027 g, 1,232 mmol) y después MeOH (0,050 ml, 1,232). La mezcla de reacción se agitó durante 16 h y después se inactivó mediante la adición de NH4Cl acuoso saturado. La mezcla resultante se extrajo con acetato de etilo y la fase orgánica se secó (MgSO4), se filtró y se concentró a
sequedad. El residuo se purificó mediante columna ultrarrápida usando un gradiente de 0-10 % MeOH:NH4OH (9:1) en DCM como eluyente para obtener (2-(1-(piridin-3-il)cidobutil)tiazol-4-il)metanol (0,053 g, 34,9 %) en forma de un aceite transparente incoloro. LC (Método A): 1,607 min. LCMS (APCI): calc. para C13H15N2OS [M+H]+ m/z 247,09, encontrado 247,20.
Ejemplo 177. 2-metoxi-6-(6-metoxi-4-((2-(1-(piridin-3-il)ciclobutil)tiazol-4-il)metoxi)benzofuran-2-il)imidazo[2,1-b][1,3,4]tiadiazol

El compuesto del título se preparó de acuerdo con el método descrito anteriormente en el ejemplo 175 y se aisló en forma de un sólido. LC (Método A): 2,324 min. HRMS (ESI): calc. para C27H24N5O4S2 [M+H]+ m/z 546,1270, encontrado 546,1251. RMN 1H (DMSO-da, 400 MHz) 8 ppm: 8,51 (d, J = 5,5 Hz, 1H), 8,48 (m, 1H), 8,36 (s, 1H), 8,18 (d, J = 8,2 Hz, 1H), 7,78 (s, 1H), 7,72 (dd, J = 5,7, 8,0 Hz, 1H), 7,18 (a s, 1H), 6,99 (d, J = 0,8 Hz, 1H), 6,83 (dd, J = 0,8, 1,6 Hz, 1H), 6,60 (d, J = 2,0 Hz, 1H), 5,27 (s, 2H), 4,36 (t, J = 5,1 Hz, 1H), 4,20 (s, 3H), 3,80 (s, 3H), 3,44 (m, 2H), 2,92 -2,81 (m, 4H), 2,20 -2 ,13 (m, 1H), 2,02 -1,92 (m, 1H).
Ejemplo 178
2-metoxi-6-(6-metoxi-4-((2-(1-(pirimidin-5-il)vinil)tiazol-4-il)metoxi)benzofuran-2-il)imidazo[2,1-b][1,3,4]tiadiazol

178A. 1-(4-(((ferc-butildimetilsilil)oxi)metil)tiazol-2-il)etanona

Una solución de 2-bromo-4-(((ferc-butildimetilsilil)oxi)metil)tiazol (Ejemplo 37B, 2,00 g, 6,49 mmol) en THF seco (32 ml) se enfrió a -78 °C en atmósfera de N2 y después se agregó gota a gota n-butil litio (1,45 M en hexanos, 4,92 ml, 7,14 mmol). La mezcla resultante se agitó durante 35 min para obtener una solución de color de color pardo claro. A esta mezcla se le agregó lentamente una solución de N,N-dimetilacetamida (0,608 ml, 6,49 mmol) en THF seco (8 ml) y la mezcla se agitó a -78 °C durante 2 h para obtener una solución de color de color pardo claro. La reacción después se inactivó mediante la adición de NH4Cl acuoso saturado; el baño de enfriamiento se retiró y la mezcla se repartió con EtOAc-agua. La fase orgánica se separó, se lavó (salmuera), se secó (MgSO4) y se evaporó para obtener un aceite de color amarillo claro. Este aceite se purificó mediante cromatografía en columna usando un gradiente de 0 a 10 % de éter en hexanos para obtener 1-(4-(((ferc-butildimetilsilil)oxi)metil)tiazol-2-il)etanona (1,07 g, 60,8 % de rendimiento) en forma de un aceite de color amarillo. LC (Método A): 2,368 min. LCMS (APCI): calc. para C-i2H22NO2SSi [M+H]+ m/z 272,11, encontrado 272,20.
178B. trifluorometansulfonato de 1-(4-(((ferc-butildimetilsilil)oxi)metil)tiazol-2-il)vinilo
A una solución en agitación de 1-(4-(((ferc-butildimetilsilil)oxi)metil)tiazol-2-il)etanona (1,068 g, 3,93 mmol) y 1,1,1-trifluoro-N-fenil-N-((trifluorometil)sulfonil)-metansulfonamida (1,546 g, 4,33 mmol) en THF (27 ml) a -78 °C, se le agregó KHMDS (1,1 M en THF, 4,32 ml, 3,93 mmol) gota a gota durante 20 min. La mezcla de reacción se calentó a -20 °C y se agitó a la misma temperatura durante 2 h, antes de inactivarla con NH4Cl acuoso saturado y extraerla con EtOAc. La fase orgánica se lavó con salmuera, se secó (MgSO4), se filtró y se concentró a sequedad. El residuo se purificó mediante cromatografía ultrarrápida usando un gradiente de 0 a 10 % de dietiléter en hexanos para obtener trifluorometansulfonato de 1-(4-(((ferc-butildimetilsilil)oxi)metil)tiazol-2-il)vinilo (0,432 g, 27,2 % de rendimiento) en forma de un aceite transparente e incoloro. lC (Método A): 2,484 min. RMN 1H (DMSO-d6, 400 MHz) 8 ppm: 7,69 (s, 1H), 6,31 (d, J = 5,1 Hz, 1H), 5,84 (d, J = 5,1 Hz, 1H), 4,78 (d, J = 0,8 Hz, 2H), 0,89 (s, 9H), 0,09 (s, 6H).
178C.4-(((ferc-butildimetilsilil)oxi)metil)-2-(1-(pirimidin-5-il)vinil)tiazol

A una mezcla de trifluorometansulfonato de 1-(4-(((ferc-butildimetilsilil)oxi)metil)tiazol-2-il)vinilo (0,272 g, 0,506 mmol) y ácido pirimidin-5-ilborónico (0,094 g, 0,758 mmol) en tolueno (8 ml) y etanol (1,5 ml), se le agregó carbonato de sodio 2 M (0,303 ml, 0,607 mmol) y la mezcla se desgasificó con un flujo de burbujas de nitrógeno durante 5 min. A esta mezcla después se le agregó PdCh(dppf).CH2Cl2 (0,025 g, 0,030 mmol), el recipiente se selló y la mezcla se calentó a 90 °C durante 3 h. La mezcla enfriada se repartió con NaHCO3 acuoso saturado-DCM y la fase orgánica se separó, se lavó con salmuera, se secó (MgSO4), se filtró y se concentró. El residuo se purificó mediante columna ultrarrápida usando DCM-EtOAc como eluyente para obtener 4-(((ferc-butildimetilsilil)oxi)metil)-2-(1-(pirimidin-5-il)vinil)tiazol (0,103 g, 0,309 mmol, 61,1 % de rendimiento) en forma de un aceite de color amarillo. LC (Método A): 2,358 min. LCMS (APCI): calc. para C-i6H24NaOSSi [M+H]+ m/z 334,14, encontrado 334,20.
178D.(2-(1-(pirimidin-5-il)vinil)tiazol-4-il)metanol

A una solución en agitación de 4-(((ferc-butildimetilsilil)oxi)metil)-2-(1-(pirimidin-5-il)vinil)tiazol (0,103 g, 0,309 mmol) en THF (2 ml) a temperatura ambiente, se le agregó trihidrofluoruro de trietilamina (0,251 ml, 1,544 mmol) y la mezcla se agitó durante 16 h. La mezcla resultante se repartió con EtOAc-NaHCO3 acuoso saturado y la fase orgánica se separó, se secó (MgSO4), se filtró y se evaporó a sequedad. El residuo se purificó mediante cromatografía en columna ultrarrápida usando hexanos-acetato de etilo como eluyente para obtener (2 -(1 -(pirimidin-5-il)vinil)tiazol-4-il)metanol (0,017 g, 0,078 mmol, 25,1 %) en forma de un aceite transparente e incoloro. LC (Método A): 1,242 min. LCMS (APCI): calc. para C10H10N3OS [M+H]+ m/z 220,05, encontrado 220,20.
Ejemplo 178. 2-metoxi-6-(6-metoxi-4-((2-(1-(pirimidin-5-il)vinil)tiazol-4-il)metoxi)benzofuran-2-il)imidazo[2,1-b][1,3,4]tiadiazol

A una suspensión de 6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-ol (Ejemplo 1H, 0,0246 g, 0,078 mmol) y (2-(1-(pirimidin-5-il)vinil)tiazol-4-il)metanol (0,017 g, 0,078 mmol) en THF seco (8 ml), se le agregó trin-butilfosfina (0,050 ml, 0,194 mmol) y después se agregó gota a gota una solución de ADDP (0,049 g, 0,194 mmol) en THF (2 ml) durante 30 min a través de una bomba de jeringa. Después de agitar durante otros 30 min, la mezcla de reacción se repartió con EtOAc-NaHCO3acuoso saturado y la fase orgánica se separó, se lavó con salmuera, se secó (MgSO4), se filtró y se concentró a sequedad. El residuo se purificó mediante columna ultrarrápida usando
DCM-EtOAc como eluyente para obtener el producto impuro. Este material se volvió a someter a cromatografía usando hexanos-EtOAc como eluyente para obtener el producto puro en forma de un sólido de color blanco que posteriormente se liofilizó a partir de acetonitrilo-agua para obtener el compuesto del título (0,014 g, 0,024 mmol, 30,6 %) en forma de un sólido amorfo de color blanco. LC (Método a ): 2,288 min. HRMS (ESI): calc. para C24H19N6O4S2 [M+H]+ m/z 519,0904, encontrado 519,0902. RMN 1H (DMSO-da, 400 MHz) 8 ppm: 9,21 (s, 1H), 8,99 (s, 2H), 8,36 (s, 1H), 7,92 (s, 1H), 7,00 (d, J = 0,8 Hz, 1H), 6,83 (m, 1H), 6,61 (d, J = 2,0 Hz, 1H), 6,28 (s, 1H), 6,00 (s, 1H), 5,31 (s, 2H), 4,20 (s, 3H), 3,79 (s, 3H).
Ejemplo 179
6-(6-fluoro-4-((2-feniltiazol-4-il)metoxi)benzofuran-2-il)-2-metoxiimidazo[2,1-b][1,3,4]tiadiazol

179A. 4-fluoro-2-hidroxi-6-metoxibenzaldehído y 2-fluoro-6-hidroxi-4-metoxibenzaldehído

Una mezcla 1:5 de 4-fluoro-2,6-dimetoxibenzaldehído y 2-fluoro-4,6-dimetoxibenzaldehído (Helvetica Chim. Acta, 81:1596-1607 (1998), 1 g, 5,43 mmol) en 30 ml de diclorometano se enfrió a 0-5 °C. A esta mezcla se le agregó gota a gota durante 25 minutos tribromoborano (7,33 ml, 7,33 mmol) en 10 ml de diclorometano y la reacción se agitó a 0 5 °C durante aproximadamente 5-10 min. Después, la mezcla se vertió en hielo, se diluyó con diclorometano y se extrajo dos veces con diclorometano. Las capas orgánicas combinadas se secaron en MgSO4, se filtraron y se concentraron. El residuo se purificó mediante cromatografía en columna sobre gel de sílice en ISCO (columna de 40 g de oro con 90 % de hexanos y 10 % de diclorometano hasta 80 % de hexanos con 10 % de diclorometano y 10 % de acetato de etilo). Ambos isómeros se recolectaron al mismo tiempo para obtener los materiales del título (0,720 g, 78 %) en forma de un sólido cristalino blanco. 2-fluoro-6-hidroxi-4-metoxibenzaldehído (isómero principal, no deseado) - RMN 1H (400 MHz, CDCls) 8 ppm: 11,91 (s, 1H), 10,05 (s, 1H), 6,17 - 6,25 (m, 1H), 3,86 (s, 3H). 4-fluoro-2-hidroxi-6-metoxibenzaldehído (isómero secundario, deseado) - RMN 1H (400 MHz, CDCla) 8 ppm: 12,23 12,42 (m, 1H), 10,22 (s, 1H), 6,23 -6,27 (m, 1H), 6,13 (dd, J=10,96, 2,35 Hz, 1H), 3,90 (s, 3H).
179B. 1-(6-fluoro-4-metoxibenzofuran-2-il)etanona

A una solución de una mezcla de 4-fluoro-2-hidroxi-6-metoxibenzaldehído y 2-fluoro-6-hidroxi-4-metoxibenzaldehído (4,63 g, 27,2 mmol) en acetonitrilo (49,7 ml, 952 mmol), se le agregó yoduro de potasio (0,903 g, 5,44 mmol), carbonato de cesio (9,75 g, 29,9 mmol) y 1-cloropropan-2-ona (2,395 ml, 28,6 mmol). La mezcla se agitó a temperatura ambiente durante 2 h, se trató con 0,1 eq de carbonato de cesio y se calentó a 60 °C durante 1 h y a 80 °C durante otra hora. La reacción se dejó durante la noche a temperatura ambiente, después se filtró en una almohadilla pequeña de sílice y se enjuagó con acetato de etilo (aproximadamente 500 ml). El residuo obtenido después de la concentración se purificó mediante cromatografía sobre gel de sílice (ISCO, 120 g de sílice con 100 % de tolueno usando UV a 315 nm, después, con el tiempo, la polaridad aumentó hasta 10 % de acetato de etilo). Las fracciones se evaporaron para obtener una mezcla 7:1 de los isómeros deseados/no deseados que se volvió a cristalizar durante la noche con acetato de etilo. El material del título se obtuvo (0,216 g, 3,8 %) en forma de cristales incoloros. LC (Método B): 1,928 min. LCMS (APCI) calc. para C11 H-I0F0 3 [M+H]+ m/z 209,06, encontrado 209,1. RMN 1H (CDCls, 400 MHz) 8 ppm: 7,55-7,61 (m, 1H), 6,78- 6,99 (m, 1H), 6,46-6,53 (m, 1H), 3,96 (s, 3H), 2,55-2,60 (m,
3H).
179C. 1-(6-fluoro-4-hidroxibenzofuran-2-il)etanona

A una solución en agitación de 1-(4,6-dimetoxibenzofuran-2-il)etanona (0. 216 g, 1,038 mmol) en clorobenceno (3,69 ml, 36,3 mmol), se le agregó tricloruro de aluminio (0,277 g, 2,075 mmol). Después de calentarse durante 3 h a 85 °C, la mezcla se inactivó con hielo y HCl 1,0 N y se extrajo con acetato de etilo (4x). Las capas orgánicas combinadas se secaron sobre sulfato de magnesio anhidro, se filtraron y se concentraron. El residuo se purificó en cromatografía sobre gel de sílice (BIOTAGE® 24 g, que se eluyó con un gradiente de hexanos y acetato de etilo) para obtener el material del título (0,191 g, 95 %) en forma de un sólido de color blanco. LC (Método B): 1,794 min. LCMS (APCI) calc. para C10H8FO3 [M+H]+ m/z 195,05, encontrado 195,9. RMN 1H (CDCla, 400 MHz) 8 ppm: 7,58 (s, 1H), 6,87 - 6,93 (m, 1H), 6,46 - 6,53 (m, 1H), 5,62 (s, 1H), 2,60 (s, 3H).
179D. 1-(6-fluoro-4-((2-feniltiazol-4-il)metoxi)benzofuran-2-il)etanona

Se agregó benceno a 1-(6-fluoro-4-hidroxibenzofuran-2-il)etanona (0. 178 g, 0,917 mmol); la mezcla se sometió a ultrasonido durante 30 segundos y se concentró al vacío para retirar rastros de agua en el material de partida. Se agregó trifenilfosfina (373 mg, 1,421 mmol) y la mezcla se secó sobre alto vacío durante 10 min. Se agregaron (2-feniltiazol-4-il)metanol (Ejemplo 3B, 0,175 g, 0,917 mmol) y THF (15 ml) y la mezcla se sometió a ultrasonido/se calentó durante 5 min. Se agregó gota a gota azodicarboxilato de diisopropilo (275 pl, 1,412 mmol) en THF (2 ml) durante 1 h y la solución amarilla resultante se sometió a ultrasonido durante 15 min y se agitó durante la noche a temperatura ambiente La mezcla se diluyó en diclorometano, se lavó con NaHCO3 saturado y salmuera, se secó sobre MgSO4 y se concentró. El residuo se purificó en cromatografía sobre gel de sílice (columna de 24 g de oro ISCO, usando 5 % de acetato de etilo en hexanos a 40 % (incrementos del 10 %)) para obtener el material del título (0,132 g, 32 %) en forma de un sólido de color blanco. LC (Método B): 2,613 min. LCMS (APCI) calc. para C20H15FNO3S [M+H]+ m/z 368,07, encontrado 368,2. RMN 1 H (CDCla, 400 MHz) 8 ppm: 7,94-8,02 (m, 2H), 7,62-7,67 (m, 1H), 7,44-7,51 (m, 3H), 7,38 (s, 1H), 6,91-6,96 (m, 1H), 6,64-6,72 (m, 1H), 5,39 (d, J=0,78 Hz, 2H), 2,58 (s, 3H).
179E. 2-bromo-1-(6-fluoro-4-((2-feniltiazol-4-il)metoxi)benzofuran-2-il)etanona

A una suspensión de 1-(6-fluoro-4-((2-feniltiazol-4-il)metoxi)benzofuran-2-il)etanona (0,132 g, 0,359 mmol) en acetato de etilo (5 ml), se le agregó bromuro de cobre (II) (160 mg, 0,719 mmol) y la mezcla se calentó a 80 °C durante 48 h. El sólido se filtró y se enjuagó con EtOAc. El sólido se purificó mediante cromatografía sobre gel de sílice (ISCO de 12 g con diclorometano y acetato de etilo (95:5)) y produjo el material del título (0,055 g, 34 %) en forma de un sólido blanquecino. LC (Método B): 2,424 min. lCm S (ApCi) calc. para C20H-i4BrFNO3S [M+H]+ m/z 445,99, encontrado 446,0.
179F. 2-bromo-6-(6-fluoro-4-((2-feniltiazol-4-il)metoxi)benzofuran-2-il)imidazo[2,1-b][1,3,4]tiadiazol

En un vial de presión para microondas de 2-5 ml, se agregó 2-bromo-1-(6-fluoro-4-((2-feniltiazol-4-il)metoxi)benzofuran-2-il)etanona (0,035 g, 0,078 mmol) en propan-2-ol (2 ml) y después 5-bromo-1,3,4-tiadiazol-2-amina (16,2 mg, 0,09 mmol). La reacción se calentó a 80 °C durante la noche ya 150 °C durante 1 h en un horno de microondas. La mezcla de reacción después se vertió en una mezcla de diclorometano (8 ml) y NaHCO3 saturado (2 ml) y esto se extrajo dos veces con diclorometano. Los extractos orgánicos se secaron en MgSO4, se filtraron y se concentraron. El residuo se purificó mediante cromatografía sobre gel de sílice (columna de oro ISCO de 12 g usando 0 a 2 % de acetato de etilo en diclorometano) para obtener el material del título (0,018 g, 43 %) en forma de un sólido amarillento. LC (Método B): 2,754 min. LCMS (APCI) calc. para C22H-i3BrFN4O2S2 [M+H]+ m/z 526,96, encontrado 527,0.
Ejemplo 179. 6-(6-fluoro-4-((2-feniltiazol-4-il)metoxi)benzofuran-2-il)-2-metoxiimidazo[2,1-b][1,3,4]tiadiazol

Se disolvió 2-bromo-6-(6-fluoro-4-((2-feniltiazol-4-il)metoxi)benzofuran-2-il)imidazo[2,1-b][1,3,4]tiadiazol (0,017 g, 0,032 mmol) en diclorometano (1,3 ml) (se requería un poco de calor y ultrasonido). Se agregó metanol (0,3 ml) y después metóxido de sodio (14,74 pl, 0,064 mmol) de una vez. La reacción se agitó a temperatura ambiente durante 25 min, después se inactivó con HCl 1,0 N y se agitó hasta que el color de la reacción cambió a amarillo. Después se agregó NaHCO3 acuoso saturado y este se extrajo con diclorometano (4x). Las capas orgánicas combinadas se secaron en MgSO4, se filtraron y se concentraron. El residuo se purificó mediante cromatografía en columna sobre gel de sílice (columna BIOTAGE® de 12 g usando 0 a 5 % de acetato de etilo en diclorometano) para obtener el material del título (0,010 g, 64 %) en forma de un sólido amarillento. LC (Método A): 2,488 min. HRMS (ESI) calc. para C23H15FN4O3S2 [M+H]+ m/z 479,0570, encontrado 479,0661. RMN 1 H (CDCla, 400 MHz) 8 ppm: 7,94-8,03 (m, 2H), 7,89 (s, 1H), 7,43 - 7,53 (m, 3H), 7,39 (s, 1H), 7,19 (s, 1H), 6,85 -6,94 (m, 1H), 6,59 - 6 ,68 (m, 1H), 5,40 (s, 2H), 4,23 (s, 3H).
Ejemplo 180
N-(2-cianoetil)-N-etil-4-(4-(((6-fluoro-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2-il)benzamida

180A. 1-(4-(benciloxi)-6-fluorobenzofuran-2-il)etanona
Una solución de 1-(6-fluoro-4-hidroxibenzofuran-2-il)etanona (Ejemplo 179C, 1,00 g, 5,15 mmol) en N,N-dimetilformamida (10 ml) se trató con carbonato de potasio anhidro (0,747 g, 5,41 mmol), después se agregó gota a gota bromuro de bencilo (0,735 ml, 6,18 mmol) durante 5 min. La mezcla heterogénea resultante se sometió a ultrasonido durante 1 h, la suspensión resultante se filtró y la torta de filtro se lavó con N,N-dimetilformamida. El filtrado combinado se evaporó a sequedad y el residuo se repartió entre acetato de etilo y bicarbonato de sodio
acuoso saturado. La fase orgánica se separó, se lavó con salmuera, se secó sobre sulfato de magnesio anhidro y se concentró al vacío. El residuo obtenido se sometió a cromatografía sobre gel de sílice (ISCO, gradiente de elución de diclorometano en hexano) para obtener 1,33 g (91 %) del material del título en forma de un sólido de color blanco. LC (Método A): 2,334 min. HRMS (ESI): calc. para C17H14FO3 [M+H]+ m/z 285,0927; encontrado 285,0927. RMN 1H (400 MHz, CDCl3) 8 ppm: 7,59 (s, 1H), 7,33-7,49 (m, 5H), 6,76-6,94 (m, 1H), 6,51-6,58 (m, 1H), 5,16 (s, 2H), 2,54 (s, 3H).
180B. 1-(4-(benciloxi)-6-fluorobenzofuran-2-il)-2-bromoetanona

A un matraz equipado con una barra agitadora magnética y purgado con atmósfera de nitrógeno se le agregó THF seco (35 ml) y después bis(trimetilsilil)amida de litio (1 M en THF, 6,30 ml, 6,30 mmol). La mezcla se enfrió a -78 °C, y se agregó gota a gota clorotrimetilsilano (0,771 ml, 6,04 mmol) durante 2 min. Después de 5 min, se agregó gota a gota una solución de 1-(4-(benciloxi)-6-fluorobenzofuran-2-il)etanona (1,492 g, 5,25 mmol) en THF seco (14 ml) durante 10 min y la mezcla resultante se agitó a -78 °C durante 30 min. Después se retiró el baño de enfriamiento y la mezcla se calentó a aproximadamente 10 °C durante 20 min. La mezcla de reacción se inactivó agregándola a una mezcla fría de acetato de etilo (300 ml), bicarbonato de sodio acuoso saturado (40 ml) y hielo. La fase orgánica se separó rápidamente, se secó sobre sulfato de magnesio anhidro y se evaporó al vacío para obtener el sililenoléter en forma de un aceite transparente. Este aceite se coevaporó con tolueno (10 ml, 0,2 kPa (2 mbar)) antes de que se absorbiera en tetrahidrofurano seco (35 ml). La mezcla se enfrió a -35 °C en nitrógeno, se trató con bicarbonato de sodio sólido (30 mg) y después N-bromosuccinimida (0,981 g, 5,51 mmol), añadido en porciones pequeñas durante 10 min. La mezcla de reacción se calentó a 5 °C durante 3,5 h y después se inactivó mediante la adición de acetato de etilo (400 ml) y bicarbonato de sodio acuoso saturado (40 ml). La fase orgánica se separó, se lavó con salmuera, se secó sobre sulfato de magnesio anhidro y se evaporó para obtener un sólido de color blanco. La cromatografía sobre gel de sílice (ISCO, gradiente de elución de diclorometano en hexano) produjo 1,509 g (79 %) del material del título en forma de un sólido de color amarillo. LC (Método A): 2,282 min. Hr Ms (ESI): calc. para C-i7H-i3BrFO3 [M+H]+ m/z 363,0032; encontrado 363,004. RMN 1H (400 MHz, CDCh) 8 ppm: 7,75 (d, J= 0,78 Hz, 1H), 7,35-7,51 (m, 5H), 6,91 (m, 1H), 6,58 (dd, J= 11,35, 1,96 Hz, 1H), 5,19 (s, 2H), 4,37 (s, 2H).
180C. 6-(4-(benciloxi)-6-fluorobenzofuran-2-il)-2-bromoimidazo[2,1-b][1,3,4]tiadiazol

Una mezcla de 1-(4-(benciloxi)-6-fluorobenzofuran-2-il)-2-bromoetanona (1,07 g, 2,95 mmol) y 5-bromo-1,3,4-tiadiazol-2-amina (0,610 g, 3,39 mmol) en isopropanol (80 ml) se calentó en un recipiente a presión sellado a 80 °C durante 18 h. La mezcla heterogénea resultante se calentó después en un reactor de microondas a 150 °C durante 30 min. La mezcla de reacción enfriada se repartió con diclorometano-bicarbonato de sodio acuoso saturado y la fase orgánica se separó, se lavó con salmuera, se secó sobre sulfato de magnesio anhidro y se concentró al vacío. El residuo obtenido se sometió a cromatografía sobre gel de sílice (ISCO, gradiente de elución de acetato de etilo en diclorometano) para obtener 0,740 g (57 %) del material del título en forma de un sólido. LC (Método A): 2,456 min. HRMS (ESI): calc. para C-igH12 BrFN3O2S [M+H]+ m/z 443,9818; encontrado 443,9834. RMN 1H (400 MHz, CDCh) 8 ppm: 8,09 (s, 1H), 7,32-7,54 (m, 5H), 7,21 (s, 1H), 6,86-6,91 (m, 1H), 6,52-6,59 (m, 1H), 5,21 (s, 2H).
180D.6-(4-(benciloxi)-6-fluorobenzofuran-2-il)-2-metoxiimidazo[2,1-b][1,3,4]tiadiazol

Una suspensión de 6-(4-(benciloxi)-6-fluorobenzofuran-2-il)-2-bromoimidazo[2,1-b][1,3,4]tiadiazol (0,740 g, 1,666 mmol) en una mezcla de diclorometano (30 ml) y metanol (10 ml) se trató a 22 °C con metóxido de sodio (25 % en peso en MeOH, 1,01 ml, 4,45 mmol), agregado en una porción. Después de 30 min, la mezcla de reacción transparente se inactivó mediante la adición de 5 ml de ácido clorhídrico 1 N y el disolvente se evaporó a presión
reducida. El residuo se repartió entre diclorometano y bicarbonato de sodio acuoso saturado y la fase orgánica se separó, se lavó con salmuera, se secó sobre sulfato de magnesio anhidro y se evaporó al vacío. La cromatografía del residuo sobre gel de sílice (ISCO, gradiente de elución de acetato de etilo en diclorometano) produjo 0,480 g (82 %) del compuesto del título en forma de un sólido de color blanco. LC (Método A): 2,477 min. HRMs (ESI): calc. para C20H15FN3O3S [M+H]+ m/z 396,0818; encontrado 396,0862. RMN 1 H (400 MHz, CDCla) 8 ppm: 7,88 (s, 1H), 7,32 7,55 (m, 5H), 7,13 (s, 1H), 6 ,88 (m, 1H), 6,54 (m, 1H), 5,20 (s, 2H), 4,22 (s, 3H).
180E. 6-fluoro-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-ol

Una mezcla de 6-(4-(benciloxi)-6-fluorobenzofuran-2-il)-2-metoxiimidazo[2,1-b][1,3,4]tiadiazol (0,480 g, 1,21 mmol) y pentametilbenceno (1,26 g, 8,50 mmol) en diclorometano (75 ml) se enfrió a -78 °C en nitrógeno y se trató con tricloruro de boro (1 M en DCM, 3,5 ml, 3,5 mmol), añadido gota a gota durante 2 min. La mezcla resultante se agitó a -78 °C durante 40 min y después se inactivó mediante la adición de bicarbonato de sodio acuoso saturado (45 ml). Se retiró baño de enfriamiento y la mezcla resultante se agitó a temperatura ambiente durante 2 h. La suspensión resultante se filtró y la torta de filtro se lavó sucesivamente con agua y diclorometano y después se secó al vacío (en pentóxido de fósforo) para obtener 0,363 g (98 %) del compuesto del título en forma de un sólido de color tostado. LC (Método A): 2,096 min. HRMS (ESI): calc. para C13H9FN3O3S [M+H]+ m/z 306,0349; encontrado 306,0369. RMN 1 H (400 MHz, DMSO-d6) 8 ppm: 10,51 (a s, 1H), 8,41 (s, 1H), 7,07 (s, 1H), 6,90-6,99 (m, 1H), 6,42-6,52 (m, 1H), 4,21 (s, 3H).
180F. 4-(4-(((6-fluoro-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2-il)benzoato de ferc-butilo

El compuesto del título se preparó de acuerdo con el procedimiento general descrito en el ejemplo 36. LC (Método A): 2,689 min. HRMS (ESI): calc. para C28H24FN4O5S2 [M+H]+ m/z 579,1172; encontrado 579,1159. RMN 1H (400 MHz, CDCl3) 8 ppm: 7,99-8,11 (m, 4H), 7,89 (s, 1H), 7,44 (s, 1H), 7,15 (s, 1H), 6,88-6,94 (m, 1H), 6,63 (dd, J= 11,35, 1,96 Hz, 1H), 5,42 (s, 2H), 4,22 (s, 3H), 1,63 (s, 9H).
180G. Ácido 4-(4-(((6-fluoro-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2-il)benzoico

A una solución de 4-(4-(((6-fluoro-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2-il)benzoato de ferc-butilo (0,345 g, 0,596 mmol) en diclorometano (6 ml), se le agregó ácido trifluoroacético (3 ml) y la mezcla homogénea de color amarillento se agitó a temperatura ambiente durante 4 h. Después, los volátiles se evaporaron a presión reducida y el residuo sólido resultante se trituró con diclorometano, se filtró y se secó al vacío para obtener 0,272 g (87 %) del compuesto del título en forma de un sólido de color beis. LC (Método A): 2,428 min. HRMS (ESI): calc. para C24H16FN4O5S2 [M+H]+ m/z 523,0546; encontrado 523,0541. RMN 1H (400 MHz, DMSO-d6) 8 ppm: 8,46 (s, 1H), 8,00-8,13 (m, 5H), 7,18 (d, J= 9,00 Hz, 1H), 7,12 (m, 1H), 6,99-7,06 (m, 1H), 5,44 (s, 2H), 4,20 (s, 3H).
Ejemplo 180.
N-(2-cianoetil)-N-etil-4-(4-(((6-fluoro-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2-il)benzamida

Una solución de ácido 4-(4-(((6-fluoro-2-(2-metoxümidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2-il)benzoico (0,040 g, 0,077 mmol) en N,N-dimetilformamida (2 ml) se trató con diisopropiletilamina (0,067 ml, 0,383 mmol) y después con 3-(etilamino)propanonitrilo (0,0083 g, 0,084 mmol). Después se agregó HATU (0,032 g, 0,084 mmol) y la mezcla de reacción se agitó a temperatura ambiente durante 3 h. Los volátiles después se evaporaron al vacío y el residuo se repartió entre cloroformo y bicarbonato de sodio acuoso saturado. La fase orgánica se separó, se lavó con salmuera, se secó sobre sulfato de sodio anhidro y se evaporó al vacío. La cromatografía del residuo sobre gel de sílice (ISCO, gradiente de elución de acetato de etilo en cloroformo) produjo 0,034 g (74 %) del compuesto del título en forma de un sólido de color blanco. LC (Método A): 2,374 min. HRMS (ESI): calc. para C29H24FN6O4S2 [M+H]+m/z 603,1285; encontrado 603,1286. RMN 1H (400 MHz, CDCls) 8 ppm: 8,05 (m, 2H), 7,89 (s, 1H), 7,52 (m, 2H), 7,43 (d, J= 0,78 Hz, 1H), 7,15 (s, 1H), 6,91 (d, J= 8,61 Hz, 1H), 6,63 (dd, J= 11,54, 1,76 Hz, 1H), 5,41 (s, 2H), 4,22 (s, 3H), 3,74 (a s, 2H), 3,46 (a s, 2H), 2,87 (a s, 2H), 1,21 (a s, 3H).
Ejemplo 181
4-(4-(((6-fluoro-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2-il)-N-(2,2,2-trifluoroetil)benzamida

El compuesto del título se preparó de acuerdo con el método descrito anteriormente en el ejemplo 180. LC (Método A): 2,369 min. HRMS (ESI): calc. para C26H18F4N5O4S2 [M+H]+ m/z 604,0736; encontrado 604,0725. RMN 1 H (400 MHz, DMSO-d6) 8 ppm: 9,24 (t, J= 6,26 Hz, 1H), 8,46 (s, 1H), 8,06-8,14 (m, 2H), 7,98-8,06 (m, 3H), 7,18 (d, J= 7,83 Hz, 1H), 7,12 (s, 1H), 7,03 (dd, J= 12,13, 1,96 Hz, 1H), 5,44 (s, 2H), 4,20 (s, 3H), 4,04-4,17 (m, 2H).
Ejemplo 182
N-(ferc-butil)-4-(4-(((6-fluoro-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2-il)-N-metilbenzamida

El compuesto del título se preparó de acuerdo con el método descrito anteriormente en el ejemplo 180. LC (Método A): 2,391 min. HRMS (ESI): calc. para C29H27FN5O4S2 [M+H]+ m/z 592,1489; encontrado 592,1500. RMN 1H (400 MHz, CDCla) 8 ppm: 7,88-7,94 (m, 2H), 7,81 (s, 1H), 7,41-7,48 (m, 2H), 7,33 (s, 1H), 7,07 (s, 1H), 6,83 (dd, J= 8,61, 0,78 Hz, 1H), 6,55 (dd, J= 11,35, 1,96 Hz, 1H), 5,32 (s, 2H), 4,14 (s, 3H), 2,82 (s, 3H), 1,46 (s, 9H).
Ejemplo 183
(4-(4-(((6-fluoro-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2-il)fenil)(pirrolidin-1-il)metanona

El compuesto del título se preparó de acuerdo con el método descrito anteriormente en el ejemplo 180. LC (Método A): 2,307 min. HRMS (ESI): calc. para C28H28FNaO4S2 [M+H]+ m/z 576,1176; encontrado 576,1159. RMN 1H (400 MHz, CDCls) 8 ppm: 8,02 (m, 2H), 7,89 (s, 1H), 7,63 (m, 2H), 7,42 (s, 1H), 7,16 (s, 1H), 6,91 (d, J= 8,61 Hz, 1H),
6,63 (dd, J= 11,35, 1,96 Hz, 1H), 5,41 (s, 2H), 4,22 (s, 3H), 3,68 (t, J= 6 ,85 Hz, 2H), 3,47 (t, J= 6 ,46 Hz, 2H), 1,84 -2,05 (m, 4H).
Ejemplo 184
N-(cianometil)-4-(4-(((6-fluoro-2-(2-metoxNmidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2-il)-N-metilbenzamida

El compuesto del título se preparó de acuerdo con el método descrito anteriormente en el ejemplo 180. LC (Método A): 2,274 min. HRMS (ESI): calc. para C27H20FN6O4S2 [M+H]+ m/z 575,0972; encontrado 575,0963. RMN 1H (400 MHz, CDCla) 8 ppm: 8,07 (m, 2H), 7,89 (s, 1H), 7,58 (m, 2H), 7,45 (s, 1H), 7,15 (s, 1H), 6,91 (dd, J= 8,61, 0,78 Hz, 1H), 6,62 (dd, J= 11,35, 1,96 Hz, 1H), 5,41 (s, 2H), 4,49 (a s, 2H), 4,22 (s, 3H), 3,20 (s, 3H).
Ejemplo 185
4-(4-(((6-fluoro-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2-il)-N,N-dimetilbenzamida

El compuesto del título se preparó a partir de 6-fluoro-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-ol (Ejemplo 180E) y 4-(4-(hidroximetil)tiazol-2-il)-N,N-dimetilbenzamida (Ejemplo 36B) de acuerdo con el método general descrito en el ejemplo 36. LC (Método A): 2,327 min. HRMS (ESI): calc. para C26H21FN5O4S2 [M+H]+m/z 550,1019; encontrado: 550,0999. RMN 1H (400 MHz, CDCla) 8 ppm: 8,02 (m, 2 H), 7,89 (s, 1H), 7,53 (m, 2H), 7,42 (s, 1H), 7,15 (s, 1H), 6,91 (d, J= 8,61 Hz, 1H), 6,63 (d, J= 11,35 Hz, 1H), 5,41 (s, 2H), 4,22 (s, 3H), 3,15 (a s, 3H), 3,02 (a s, 3H).
Ejemplo 186
4-(4-(((6-fluoro-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)-5-metiltiazol-2-il)-N-metilbenzamida

186A. 4-(4-(((6-fluoro-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)-5-metiltiazol-2-il)benzoato de tere-butilo

El compuesto del título se preparó a partir de 6-fluoro-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-ol (Ejemplo 180E) y tere-4-(4-(hidroximetil)-5-metiltiazol-2-il)benzoato de butilo (Ejemplo 45D) de acuerdo con el método general descrito en el ejemplo 180F. LC (Método A): 2,788 min. HRMS (ESl): calc. para C29H26FN4O5S2 [M+H]+ m/z 593,1329; encontrado 593,1318. RMN 1H (400 MHz, CDCls) 8 ppm: 8,02-8,07 (m, 2H), 7,93-7,99 (m, 2H), 7,87 (s, 1H), 7,08 (s, 1H), 6,89 (dd, J= 8,61, 0,78 Hz, 1H), 6,73 (dd, J= 11,74, 1,96 Hz, 1H), 5,34 (s, 2H), 4,21 (s, 3H), 2,60 (s, 3H), 1,62 (s, 9H).
186B. Ácido 4-(4-(((6-fluoro-2-(2-metoxNmidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)-5-metiltiazol-2-il)benzoico

El compuesto del título se preparó de acuerdo con el método de desprotección general descrito en el ejemplo 180G. LC (Método A): 2,436 min. HRMS (ESI): calc. para C25H18FN4O5S2 [M+H]+ m/z 537,0703; encontrado 537,0696. RMN 1H (400 MHz, DMSO-da) 8 ppm: 13,14 (a s, 1H), 8,46 (s, 1H), 7,99-8,08 (m, 4H), 7,14-7,21 (m, 1H), 6,99-7,09 (m, 2H), 5,37 (s, 2H), 4,20 (s, 3H), 2,60 (s, 3H).
Ejemplo 186. 4-(4-(((6-fluoro-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)-5-metiltiazol-2-il)-N-metilbenzamida

El compuesto del título se preparó de acuerdo con el método general de acoplamiento de amida descrito en el ejemplo 180. LC (Método a ): 2,389 min. HRMS (ESI): calc. para C26H20FN5O4S2 [M+H]+ m/z 549,0941; encontrado 550,1029. RMN 1H (400 MHz, CDCls) 8 ppm: 7,98 (d, J= 8,0 Hz, 2H), 7,87 (s, 1H), 7,83 (d, J= 8,0 Hz, 2H), 7,08 (s, 1H), 6,89 (d, J= 8,61 Hz, 1H), 6,72 (dd, J= 11,54, 1,76 Hz, 1H), 6,15 (a s, 1H), 5,33 (s, 2H), 4,22 (s, 3H), 3,05 (d, J= 5,09 Hz, 3H), 2,59 (s, 3H).
Ejemplo 187
6-(6-cloro-4-((2-feniltiazol-4-il)metoxi)benzofuran-2-il)-2-metoxiimidazo[2,1-b][1,3,4]tiadiazol

187A. 4-cloro-2,6-dimetoxibenzaldehído

Una solución de 1-cloro-3,5-dimetoxibenceno (5 g, 29,0 mmol) y TMEDA (4,37 ml, 29,0 mmol) en dietiléter (100 ml, 962 mmol) a -78 °C en atmósfera de N2 se cargó con BuLi (19,91 ml, 31,9 mmol) gota a gota durante un período de 30 minutos usando una bomba de jeringa. Después de agitar durante 4 horas a -78 °C, se agregó DMF y la mezcla de reacción se continuó agitando durante 1,5 horas, después de lo cual se agregó HCl 1 N (~30 ml) (a -78 °C). La mezcla de reacción se calentó a temperatura ambiente y se extrajo con acetato de etilo. La fase orgánica se secó (MgSO4), se filtró y se concentró a sequedad. El residuo se purificó mediante ISCO usando hexanos / EtOAc como eluyente. Las fracciones que contenían el producto deseado se concentraron a sequedad para obtener el material del título (1,97 g, 9,82 mmol, 33,9 % de rendimiento) en forma de un sólido de color amarillo claro. LC (Método B): 1,924 min. LCMS (APCI) calc. para C9H1üClO3 [M+H]+ m/z 201,03, encontrado 201,0. RMN 1H (CDCla, 400 MHz) 8 ppm: 10,28 (s, 1H), 6,87 (s, 2H), 3,86 (s, 6 H).
187B. 4-cloro-2-hidroxi-6-metoxibenzaldehído

A una solución en agitación de 4-cloro-2,6-dimetoxibenzaldehído (1,95 g, 9,72 mmol) en DCM (20 ml, 311 mmol) a -78 °C, se le agregó lentamente tribromuro de boro (9,72 ml, 9,72 mmol). La mezcla de reacción se agitó a -78 °C durante 10 minutos, después se calentó a temperatura ambiente y se agitó durante 1 hora mientras se monitoreaba el progreso de la reacción mediante LCMS. Una vez que se consumió la totalidad del material de partida, la reacción se inactivó con agua y se extrajo con DCM. La fase orgánica se lavó con salmuera, se secó (MgSO4), se filtró y se concentró a sequedad para obtener el material del título (1,79 g, 9,59 mmol, 99 % de rendimiento) en forma de un sólido de color púrpura. LC (Método B): 2,199 min. Lc MS (APCI) calc. para CaHeCO [M+H]+ m/z 187,02, encontrado 187,0. RMN 1H (CDCh, 400 MHz) 8 ppm: 11,89 (s, 1H), 10,20 (s, 1H), 6,75 (t, J = 2,0 Hz, 1H), 6,66 (m, 1H), 3,91 (s, 1H).
187C. 1-(6-cloro-4-metoxibenzofuran-2-il)etanona

Una solución en agitación de 4-cloro-2-hidroxi-6-metoxibenzaldehído (1,79 g, 9,59 mmol) en N,N-dimetilformamida (15 ml, 9,59 mmol) se cargó con carbonato de cesio (3,75 g, 11,51 mmol) y 1-cloropropan-2-ona (0,975 ml, 11,51 mmol). La mezcla de reacción se calentó en un recipiente sellable a 65 °C durante 7 horas, se filtró en un papel de filtro Whatman para retirar los insolubles enjuagándola con DCM y después se lavó con NaHCO3 saturado. La fase orgánica se secó (MgSO4), se filtró y se concentró a sequedad. El residuo se purificó mediante ISCO usando hexanos / EtOAc como eluyente. Las fracciones que contenían el producto deseado se concentraron para obtener el material del título (1,43 g, 6,37 mmol, 66% de rendimiento) en forma de un sólido de color amarillo claro. LC (Método A): 1,952 min. LCMS (APCI) calc. para C11 H10ClO3 [M+H]+ m/z 225,03, encontrado 225,0. RMN 1H (CDCl3, 400 MHz) 8 ppm: 7,94 (d, J = 0,8 Hz, 1H), 7,49 (dd, J = 0,8, 1,6 Hz, 1H), 6,97 (d, J = 1,6 Hz, 1H), 3,97 (s, 3H).
187D. 1-(6-cloro-4-hidroxibenzofuran-2-il)etanona

A una solución en agitación de 1-(6-cloro-4-metoxibenzofuran-2-il)etanona (1,43 g, 6,37 mmol) en clorobenceno (15 ml, 148 mmol), se le agregó cloruro de aluminio (3,40 g, 25,5 mmol) en porciones durante un período de 10 minutos. El recipiente de reacción se selló y se calentó a 100 °C durante 40 minutos, después se enfrió a temperatura ambiente y se vertió en hielo picado (la barra agitadora se enjuagó con EtOAc). Esto se agitó durante 30 minutos, después se extrajo con acetato de etilo. La fase orgánica se secó (MgSO4), se filtró y se concentró a sequedad. El residuo se purificó mediante ISCO usando hexanos / EtOAc como eluyente. Las fracciones que contenían el producto deseado se concentraron para obtener el material del título (1,18 g, 5,60 mmol, 88 % de rendimiento) en forma de un sólido de color de color pardo claro. LC (Método A): 1,783 min. LCMS (APCI) calc. para C1 üH8CIO3 [M+H]+ m/z 211,02, encontrado 211,0. RMN 1H (CDCla, 400 MHz) 8 ppm: 11,01 (s, 1H), 7,89 (s, 1H), 6,72 (s, 1H), 2,52 (s, 3H).
187E. 1-(4-(benciloxi)-6-clorobenzofuran-2-il)etanona

Una solución en agitación de 1-(6-cloro-4-hidroxibenzofuran-2-il)etanona (1,18 g, 5,60 mmol) en DMF seco (10 ml, 129 mmol) a temperatura ambiente se cargó con K2CO3 (0,774 g, 5,60 mmol) y DMF. La mezcla de reacción se agitó durante 1,5 horas, después se repartió entre acetato de etilo y agua. La fase orgánica se lavó con salmuera, se secó (MgSO4), se filtró y se concentró a sequedad. El residuo se purificó mediante ISCO usando hexanos / EtOAc como
eluyente. Las fracciones que contenían el producto deseado se concentraron para obtener el material del título (1,57 g, 5,22 mmol, 93 % de rendimiento) en forma de un aceite de color ámbar. LC (Método B): 2,420 min. LCMS (APCI) calc. para C17H14CO3 [M+H]+ m/z 301,06, encontrado 301,0. RMN 1H (CDCla, 400 MHz) 8 ppm: 8,00 (d, J = 0,8 Hz, 1H), 7,53 (m, 3H), 7,44 (m, 2H), 7,38 (m, 1H), 7,10 (d, J = 1,6 Hz, 1H), 5,53 (s, 2H), 2,54 (s, 3H).
187F. 1-(4-(benciloxi)-6-clorobenzofuran-2-il)-2-bromoetanona

Un matraz de fondo redondo, de 200 ml secado al fuego, equipado con una barra agitadora y en atmósfera de nitrógeno se cargó con THF anhidro (12 ml) y después con bis(trimetilsilil)amida de litio (6,22 ml, 6,22 mmol). La mezcla se enfrió a -78 °C y se trató con una solución de 1-(4-(benciloxi)-6-clorobenzofuran-2-il)etanona (1,56 g, 5,19 mmol) en THF (lavado de 6 ml 2 ml), añadido gota a gota durante 10 minutos a través de una bomba de jeringa. La mezcla resultante se agitó a -78 °C durante 45 minutos y después se cargó con trimetilclorosilano (0,769 ml, 6,02 mmol), añadido gota a gota durante 5 minutos a través de una bomba de jeringa y después se agitó durante otros 20 minutos. El baño de enfriamiento se retiró y la mezcla se calentó a 10 °C durante 30 minutos. La mezcla de reacción se inactivó con una mezcla de acetato de etilo frío (80 ml), NaHCO3 saturado (12 ml) y hielo. La fase orgánica se secó ((MgSO4), mientras se agitaba durante ~5 minutos para retirar todo rastro de agua), se filtró y se concentró a sequedad para obtener el sililenoléter en forma de un aceite de color amarillo que se coevaporó con tolueno (4 ml). El sililenoléter se disolvió en THF seco (20 ml), se enfrió a -30 °C (usando un baño de enfriamiento hecho de 1:1 CaCh: agua usando hielo seco; el baño se estabiliza a alrededor -de 30 a -45 °C) y se trató con NaHCO3 (~50 mg) y después N-bromosuccinimida (0,923 g, 5,19 mmol), añadida en pequeñas porciones durante 15 minutos. La mezcla de reacción se calentó a 0 °C durante 2 horas (controlado mediante LCMS) y después se inactivó mediante la adición de acetato de etilo (100 ml) y NaHCO3 saturado. La fase orgánica se lavó con salmuera, se secó (MgSO4) y se evaporó para obtener un sólido de color naranja que se purificó mediante ISCO usando hexanos / EtOAc como eluyente. Las fracciones que contenían el producto deseado se concentraron para obtener el material del título 1,48 g, 3,51 mmol, 67,6 % de rendimiento) en forma de un sólido de color amarillo. LC (Método B): 2,528 min. LCMS (APCI) calc. para C17H13BrClO3 [M+H]+ m/z 378,97, encontrado 379,0.
187G. 6-(4-(benciloxi)-6-clorobenzofuran-2-il)-2-bromoimidazo[2,1-b][1,3,4]tiadiazol

Un recipiente sellable se cargó con 1-(4-(benciloxi)-6-clorobenzofuran-2-il)-2-bromoetanona (1,48 g, 3,51 mmol), 5-bromo-1,3,4-tiadiazol-2-amina (0,632 g, 3,51 mmol) e IPA (25 ml, 324 mmol). La mezcla de reacción se calentó en un baño de aceite a 80 °C durante 6 horas, después se calentó en el microondas a 150 °C durante 1 hora. La mezcla de reacción se dejó reposar durante 1 hora y el material insoluble se filtró y se enjuagó con MeOH para obtener el producto deseado en forma de un sólido de color pardo (1,19 g, 2,58 mmol, 73,6 % de rendimiento). LC (Método A): 2,549 min. LCMS (APCI) calc. para C-igH12BrClN3O2S [M+H]+ m/z 459,95, encontrado 460,0. RMN 1 H (CDCl3, 400 MHz) 8 ppm: 8,74 (s, 1H), 7,55 - 7,50 (m, 2H), 7,45 - 7,34 (m, 4H), 7,17 (d, J = 0,8 Hz, 1H), 7,02 (d, J = 1,6 Hz, 1H), 5,32 (s, 2H).
187H. 6-(4-(benciloxi)-6-clorobenzofuran-2-il)-2-metoxiimidazo[2,1-b][1,3,4]tiadiazol

A una solución en agitación de 6-(4-(benciloxi)-6-clorobenzofuran-2-il)-2-bromoimidazo[2,1-b][1,3,4]tiadiazol (1,18 g, 2,56 mmol) en DCM (40 ml, 622 mmol) y metanol (10 ml, 247 mmol), se le agregó metóxido de sodio (1,164 ml, 5,12 mmol). La mezcla de reacción se agitó a temperatura ambiente durante 1 h 15 min mientras se controlaba mediante TLC (7:3 hexanos : EtOAc). La mezcla de reacción se inactivó con HCl 1 N y se extrajo con DCM. La fase orgánica se lavó con salmuera, se secó (MgSO4), se filtró y se concentró a sequedad. El residuo se trituró con
MeOH (ultrasonido) y el material sólido se filtró, se enjuagó con MeOH y se aspiró seco para obtener el compuesto deseado en forma de un sólido de color pardo (859 mg, 2,086 mmol, 81 % de rendimiento). LC (Método A): 2,478 min. LCMS (APCI) calc. para C20H15ClN3O3S [M+H]+ m/z 412,05, encontrado 412,0. RMN 1H (CDCl3, 400 MHz) 8 ppm: 8,50 (s, 1H), 7,52 (m, 2H), 7,43 (m, 2H), 7,36 (m, 2H), 7,09 (d, J = 0,8 Hz, 1H), 7,00 (d, J = 1,6 Hz, 1H), 5,31 (s, 2H), 4,21 (s, 3H).
187I. 6-cloro-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-ol

Una solución en agitación de 6-(4-(benciloxi)-6-clorobenzofuran-2-il)-2-metoxiimidazo[2,1-b][1,3,4]tiadiazol (0,85 g, 2,064 mmol) y pentametilbenceno (2,142 g, 14,45 mmol) en DCM en atmósfera de N2 se enfrió a -78 °C después de lo cual se agregó gota a gota tricloruro de boro (5,16 ml, 5,16 mmol) durante ~ 4 minutos. La reacción se controló mediante TLC usando 1:1 hexanos-EtOAc como eluyente. La mezcla de reacción se agitó a -78 °C durante 30 minutos, después de lo cual se agregó una mezcla de agua (40 ml) y NaHCO3 saturado (5 ml) (a -78 °C) y la mezcla se agitó hasta alcanzar temperatura ambiente (se retiró del baño de enfriamiento). El precipitado sólido se filtró y se enjuagó con dietiléter, después se secó durante la noche para obtener el material del título (441 mg, 1,371 mmol, 66,4 % de rendimiento) en forma de un sólido de color beis. El filtrado se extrajo con DCM. La fase orgánica se lavó con salmuera, se secó (MgSO4) y se concentró a sequedad. El residuo se purificó mediante ISCO usando DCM / EtOAc como eluyente. Las fracciones que contenían el producto deseado se concentraron para obtener el material del título (25 mg, 0,078 mmol, 3,77 % de rendimiento) en forma de un sólido de color beis. LC (Método A): 2,167 min. LCMS (APCI) calc. para C-isHgClNsOsS [M+H]+ m/z 322,00, encontrado 322,0. RMN 1 H (CDCl3, 400 MHz) 8 ppm: 10,50 (a. S, 1H), 8,45 (s, 1H), 7,17 (dd, J = 0,8, 1,6 Hz, 1H), 7,09 (d, J = 0,8 Hz, 1H), 6,67 (d, J = 2,0 Hz, 2H), 4,21 (s, 3H).
Ejemplo 187. 6-(6-cloro-4-((2-feniltiazol-4-il)metoxi)benzofuran-2-il)-2-metoxiimidazo[2,1-b][1,3,4]tiadiazol

A un matraz de fondo redondo, de 100 ml secado al fuego, que contenía 6-cloro-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-ol (0,025 g, 0,078 mmol) y (2-feniltiazol-4-il)metanol (Ejemplo 3B, 37,2 mg, 0,194 mmol) en THF seco (4 ml), se le agregó tributilfosfina (0,050 ml, 0,194 mmol). La solución resultante se cargó con una solución de ADDP (0,049 g, 0,194 mmol) en THF (1 ml) añadido gota a gota durante 30 minutos a través de una bomba de jeringa. Después de agitar durante 1,5 horas, la mezcla de reacción se diluyó con EtOAc y después se lavó con HCl 1 N, NaHCO3 saturado, agua y salmuera. La fase orgánica se secó (MgSO4) y después se concentró a sequedad. El residuo se purificó mediante ISCO usando 0 a 10 % de dietiléter en DCM. Las fracciones que contenían el producto deseado se concentraron para obtener el material del título en forma de un sólido de color beis (0,020 g, 0,040 mmol, 52,0 % de rendimiento). LC (Método A): 2,534 min. LCMS (APCI) calc. para C23H16ClN4O3S2 [M+H]+ m/z 495,03, encontrado 495,0. RMN 1H (CDCl3, 400 MHz) 8 ppm: 8,49 (s, 1H), 7,99 - 7,96 (m, 2H), 7,93 (s, 1H), 7,55 - 7,50 (m, 3H), 7,40 (dd, J = 0,8, 1,6 Hz, 1H), 7,15 (dd, J = 0,4, 1,6 Hz, 1H), 7,14 (d, J = 0,8 Hz, 1H), 5,43 (s, 2H), 4,21 (s, 3H).
Ejemplo 188
6-(6-cloro-4-((2-(4-clorofenil)tiazol-4-il)metoxi)benzofuran-2-il)-2-metoxiimidazo[2,1-b][1,3,4]tiadiazol
Una suspensión de 6-cloro-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-ol (Ejemplo 187I, 0,030 g, 0,093 mmol) y 2-(bromometil)-4-(4-clorofenil)tiazol (0,0404 g, 0,140 mmol) en N,N-dimetilformamida (3 ml) se mantuvo al vacío (1 kPa (10 mbar)) durante 5 min. El matraz después se purgó con nitrógeno y se agregó carbonato
de potasio anhidro en polvo recién obtenido (0,105 g, 0,756 mmol) de una sola vez. La mezcla resultante se agitó a temperatura ambiente durante 16 h. La mezcla heterogénea se inactivó con ácido clorhídrico 1 N (1 ml) después de lo cual se agregaron agua y MeOH. El material sólido se filtró y la torta de filtro se enjuagó con agua, metanol y acetonitrilo para obtener un sólido de color beis. El material sólido se disolvió en DCM con una pequeña cantidad de metanol, después se absorbió en una precolumna sobre gel de sílice y se purificó mediante cromatografía ultrarrápida (0-100 % de EtOAc-diclorometano). El producto obtenido se liofilizó a partir de acetonitrilo-agua para obtener el compuesto del título en forma de un sólido amorfo de color beis (0,031 g, 0,059 mmol, 62,8 %). LC (Método F): 2,714 min. HRMS (ESI): calc. para C23H15O 2N4O3S2 [M+H]+ m/z 528,9963, encontrado 528,9954. RMN 1H (DMSO-d6, 400 MHz) 8 ppm: 8,47 (s, 1H), 7,98 (m, 2H), 7,94 (s, 1H), 7,58 (m, 2H), 7,38 (s, 1H), 7,12 (d, J = 0,8 Hz, 2H), 5,42 (s, 2H), 4,20 (s, 3H).
Ejemplo 189
6-(6-cloro-4-((2-(m-tolil)tiazol-4-il)metoxi)benzofuran-2-il)-2-metoxiimidazo[2,1-b][1,3,4]tiadiazol

El compuesto del título se preparó de acuerdo con el método descrito anteriormente en el ejemplo 188 para obtener un sólido. LC (Método F): 2,687 min. HRMS (ESI): calc. para C24H1sClN4O3S2 [M+H]+ m/z 509,0509, encontrado 509,0512. RMN 1 H (DMSO-d6, 400 MHz) 8 ppm: 8,49 (s, 1H), 7,91 (s, 1H), 7,80 (s, 1H), 7,76 (d, J = 7,8 Hz, 1H), 7,40 (t, J = 7,6 Hz, 1H), 7,32 (d, J = 7,8 Hz, 1H), 7,15 (d, J = 1,6 Hz, 1H), 7,14 (d, J = 0,8 Hz, 1H), 5,42 (s, 2H), 4,20 (s, 3H), 2,39 (s, 3H).
Ejemplo 190
(6-(6-cloro-4-((2-(4-fluorotetrahidro-2H-pyran-4-il)tiazol-4-il)metoxi)benzofuran-2-il)-2-metoxiimidazo[2,1-b][1,3,4]tiadiazol

A una solución de 4-(bromometil)-2-(4-fluorotetrahidro-2H-piran-4-il)tiazol (Ejemplo 119C, 0,016 g, 0,057 mmol) en DMF (1,5 ml), se le agregó 6-cloro-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-ol (Ejemplo 187I, 0,017 g, 0,052 mmol) y después carbonato de potasio en polvo recién obtenido (0 , 0 22 g, 0,156 mmol). La mezcla se agitó en un vial sellado a temperatura ambiente durante 2 h, después se diluyó con agua, la mezcla resultante se filtró, y la torta de filtro se lavó con NH4Cl acuoso saturado y después con agua. El residuo húmedo se absorbió en DCM, y la solución se lavó (NaHCO3 acuoso saturado), se secó (Na2SO4) y se evaporó para obtener una goma. La cromatografía ultrarrápida (Isco/ 0-30 % de EtOAc-DCM) produjo 6-(6-cloro-4-((2-(4-fluorotetrahidro-2H-piran-4-il)tiazol-4-il)metoxi)benzofuran-2-il)-2-metoxiimidazo[2,1-b][1,3,4]tiadiazol (0,012 g, 44,4 %) en forma de un sólido. LC (Método A): 2,483 min. HRMS (ESI): calc. para C22H19CFN4O4S2 [M+H]+ m/z 521,052; encontrado 521,053. RMN 1H (400 MHz, DMSO-d6): 8 8,46 (s, 1H), 7,94 (s, 1H), 7,36 (a s, 1H), 7,08 (d, J= 1,57 Hz, 1H), 7,07 (s, 1H), 5,35 (s, 2H), 4,17 (s, 3H), 3,81 (m, 2H), 3,67 (dt, J= 1,96, 10,96 Hz, 2H), 2,33-2,16 (m, 2H), 2,05 (m, 2H).
Ejemplo 191
2-metoxi-6-(4-((2-feniltiazol-4-il)metoxi)benzofuran-2-il)imidazo[2,1-b][1,3,4]tiadiazol

191A. 5-(benciloxi)-2,2-dimetil-4H-benzo[d][1,3]dioxin-4-ona

Una solución de 5-hidroxi-2,2-dimetil-4H-benzo[d][1,3]dioxin-4-ona (6,00 g, 30,9 mmol) (Hadfield, A. e t al., Synthetic Communications, 24(7):1025-1028 (1994)) en N,N-dimetilformamida (35 ml) se trató con carbonato de potasio anhidro en polvo (5,15 g, 37,26 mmol) añadido de una sola vez. La mezcla resultante se agitó al vacío durante 10 min y después se purgó con nitrógeno. El matraz de reacción se colocó en un baño de agua (22 °C) y se trató con bromuro de bencilo (5,55 g, 32,16 mmol), añadido gota a gota durante 15 min. La mezcla resultante se agitó a 22 °C durante 18 h. El sólido formado se filtró y se lavó con N,N-dimetilformamida. El filtrado se evaporó al vacío y el aceite residual se diluyó con acetato de etilo (300 ml), se lavó con ácido clorhídrico 0,1 N frío, bicarbonato de sodio saturado y salmuera. Después de secar sobre sulfato de magnesio anhidro, la evaporación del disolvente produjo un jarabe espeso. La cromatografía sobre gel de sílice (4 x 13 cm, elución con tolueno - acetato de etilo 0 - 5 %) produjo 8,78 g (100 % de rendimiento) del material del título en forma de un sólido de color blanco. LC (Método A): 1,982 min. RMN 1H (CDCla, 600 MHz) 8 ppm: 1,69 (s, 6 H), 5,23 (s, 2H), 6,53 (d, J = 8,2 Hz, 1H), 6,62 (d, J = 8,4 Hz, 1H), 7,24 - 7,3 (m, 1H), 7,34 - 7,4 (m, 3 H), 7,52 (d amplio, J = 7,4 Hz 2H).
191B. 2-(benciloxi)-6-hidroxibenzaldehído

Una solución de 5-(benciloxi)-2,2-dimetil-4H-benzo[d][1,3]dioxin-4-ona (4,00 g, 14,07 mmol) en diclorometano (80 ml) se enfrió a -78 °C y se trató con una solución de hidruro de diisobutilalumio (6,00 g, 42,2 mmol) en tolueno (40 ml), añadido gota a gota durante 20 min. La mezcla resultante se agitó a -78 °C durante 3 h. La mezcla de reacción se inactivó mediante la adición cuidadosa de metanol (5 ml), añadido gota a gota durante 15 min y después ácido clorhídrico 4 N (20 ml), añadido gota a gota durante 15 min. El baño de enfriamiento se retiró, se agregaron 80 ml adicionales de ácido clorhídrico 4 N durante 10 min y la mezcla se agitó vigorosamente a 22 °C durante 4 h. La mezcla de reacción se diluyó con acetato de etilo ( 200 ml), se lavó con salmuera, se secó sobre sulfato de magnesio anhidro y se evaporó al vacío. El aceite resultante se sometió a cromatografía sobre gel de sílice (4 x 10 cm, tolueno de elución) para obtener 2,25 g (70 % de rendimiento) del material del título en forma de un sólido de color amarillo claro. LC (Método A): 2,219 min. HRMS (ESI) calc. para C14H13O3 [M H]+ m/z 229,0859, encontrado 229,0859. RMN 1H (CDCla, 600 MHz) 8 ppm: 5,12 (s, 2H), 6,43 (d, J = 8,25 Hz, 1H), 6,52 (d, J = 8,46 Hz, 1H), 7,34 - 7,4 (m, 6 H), 10,39 (s, 1H), 11,95 (s, 1H).
191C. 1-(4-(benciloxi)benzofuran-2-il)etanona

Una solución de 2-(benciloxi)-6-hidroxibenzaldehído (11,10 g, 48,63 mmol) en N,N-dimetilformamida (120 ml) se trató con carbonato de cesio anhidro en polvo (15,8 g, 48,63 mmol), añadido de una vez. La mezcla resultante se agitó al vacío durante 10 min y después se purgó con nitrógeno. El matraz de reacción se colocó en un baño de agua (22 °C) y se trató con cloroacetona (4,65 ml, 58,4 mmol), añadida gota a gota durante 10 min. La mezcla resultante se agitó después a 22 °C durante 18 h (la TLC indicó que no hubo aldehído de inicio restante y se formó el intermedio aldehído alquilado). La mezcla de reacción después se mantuvo al vacío (1 kPa (10 mbar)) durante 15 min para retirar cualquier cloroacetona sin reaccionar y después se purgó con nitrógeno. Después se agregó carbonato de cesio anhidro (1,0 g, 3,1 mmol), y la mezcla se calentó a 55 °C y se agitó durante 40 h (se agregó más carbonato de cesio, 1 g, después de 24 h y 32 h) hasta que se completó la conversión del intermedio aldehído alquilado en benzofurano, según se controló mediante TLC. El sólido se filtró y se lavó con N,N-dimetilformamida. El filtrado se evaporó al vacío y el aceite residual se diluyó con acetato de etilo (400 ml), se lavó con ácido clorhídrico frío 0,1 N, bicarbonato de sodio saturado y salmuera. Después de secar sobre sulfato de magnesio anhidro, la evaporación del disolvente produjo un jarabe espeso. La cromatografía sobre gel de sílice (4,5 x 12 cm, elución con tolueno - acetato de etilo 2 - 4 %) produjo 11,67 g (90 % de rendimiento) del benzofurano del título en forma de un sólido de color amarillo claro. La recristalización de una mezcla de acetato de etilo (40 ml) y hexano (40 ml) produjo prismas incoloros (10,50 g). LC (Método A): 2,162 min. HRMS (ESI) calc. para C17H15O3 [M H]+ m/z 267,1016, encontrado 267,1022. RMN 1H (CDCh, 600 MHz) 8 ppm: 2,56 (s, 3H), 5,20 (s, 2H), 6,73 (d, J = 8,0 Hz, 1H), 7,17 (d,
J = 8 ,4 Hz, 1H), 7,3 - 7,5 (m, 6 H), 7,63 (s, 1H).
191D. 1-(4-(benciloxi)benzofuran-2-il)-2-bromoetanona

Un matraz de tres bocas, de 250 ml se equipó con una barra agitadora magnética, se purgó con atmósfera de nitrógeno y se cargó con tetrahidrofurano anhidro (40 ml) y después con 21,6 ml (21,6 mmol) de una solución 1 M de bis(trimetilsilil)amida de litio en tetrahidrofurano. La mezcla se enfrió a -78 °C y se trató con una solución de 1-(4-(benciloxi)benzofuran-2-il)etanona (5,00 g, 18,77 mmol) en tetrahidrofurano (20 ml), añadida gota a gota durante 10 min. La mezcla resultante se agitó a -78 °C durante 45 min. Después se agregó gota a gota clorotrimetilsilano (2,74 ml, 21,6 mmol) durante 5 min y la solución resultante se agitó a -78 °C durante otros 20 min. El baño de enfriamiento se retiró después y la mezcla se calentó a temperatura ambiente durante 30 min. La mezcla de reacción se inactivó mediante la adición a una solución fría de acetato de etilo (300 ml), bicarbonato de sodio saturado (40 ml) y hielo. La fase orgánica se secó rápidamente sobre sulfato de magnesio anhidro (agitación magnética) y se evaporó al vacío para obtener el sililenoléter en forma de un aceite que se coevaporó con tolueno (20 ml). El sililenoléter se disolvió en tetrahidrofurano seco (80 ml), se enfrió a -25 °C y se trató con bicarbonato de sodio sólido (0 , 10 g) y después N-bromosuccinimida (3,34 g, 18,8 mmol), añadida en pequeñas porciones durante 10 min. La mezcla de reacción se calentó a 0 °C durante 2 h y después se inactivó mediante la adición de acetato de etilo (350 ml) y bicarbonato de sodio saturado. La fase orgánica se lavó con salmuera, se secó sobre sulfato de magnesio anhidro y se evaporó para obtener un aceite de color naranja. La cromatografía sobre gel de sílice (4,5 x 12 cm, elución con tolueno - acetato de etilo 0 - 1 %) produjo 6,13 g de la bromometilcetona del título en forma de un sólido de color amarillo. La recristalización de acetato de etilo (20 ml) y hexano (40 ml) produjo prismas de color amarillo claro (4,93 g, 76 % de rendimiento). LC (Método A): 2,215 min. HRMS (ESI) calc. para C17H14B1O [M H]+ m/z 345,0121, encontrado 345,0109. RMN 1H (CDCla, 600 MHz) 8 ppm: 4,39 (s, 2H), 5,20 (s, 2H), 6,75 (d, J = 7,86 Hz, 1H), 7,17 (d, J = 8,25 Hz, 1H), 7,34 - 7,46 (m, 6 H), 7,78 (s, 1H).
191E. 6-(4-(benciloxi)benzofuran-2-il)-2-bromoimidazo[2,1-b][1,3,4]tiadiazol

Una mezcla de 1-(4-(benciloxi)benzofuran-2-il)-2-bromoetanona (3,00 g, 8,69 mmol) y 5-bromo-1,3,4-tiadiazol-2-amina (1,80 g, 10 , 0 mmol) en isopropanol ( 10 0 ml) se calentó en un matraz de presión equipado con una barra agitadora magnética a 80 °C durante 20 h (homogénea después de 20 min y se formó un precipitado después de 2 h). La mezcla enfriada después se transfirió a cinco viales de 20 ml para microondas y se calentó en un aparato de microondas a 150 °C durante 30 min. Cada vial se diluyó con diclorometano (250 ml), se lavó con bicarbonato de sodio saturado (25 ml) y salmuera (25 ml) y se secó sobre sulfato de magnesio anhidro. Las fracciones se combinaron y se concentraron al vacío. La cromatografía del sólido residual de color pardo anaranjado sobre gel de sílice (4 x 10 cm, elución lenta con diclorometano) produjo 2,82 g del imidazotiadiazol del título contaminado con 1-(4-(benciloxi)benzofuran-2-il)etanona. El material sólido se trituró con acetato de etilo (15 ml), se filtró, se lavó con acetato de etilo (10 ml) y se secó al vacío para obtener 2,37 g (64 % de rendimiento) del imidazotiadiazol puro del título en forma de un sólido blanquecino que se usa como tal en la siguiente etapa. LC (Método A): 2,425 min. HRMS (ESI) calc. para C19H13BrNaO2S [M H]+ m/z 425,9906, encontrado 425,9893. RMN 1H (CDCla, 600 MHz) 8 ppm: 5,21 (s, 2H), 6,72 (d, J = 8,07 Hz, 1H), 7,13 (d, J = 8,26 Hz, 1H), 7,18 (a t, 1H), 7,25 (s, 1H), 7,32 (a t, 1H), 7,38 (a t, 2H), 7,47 (a d, 2H), 8,09 (s, 1H).
191F. 6-(4-(benciloxi)benzofuran-2-il)-2-metoxiimidazo[2,1-b][1,3,4]tiadiazol

Una solución de 6-(4-(benciloxi)benzofuran-2-il)-2-bromoimidazo[2,1-b][1,3,4]tiadiazol (3,22 g, 7,55 mmol) en una mezcla de diclorometano (400 ml) y metanol (50 ml) se trató a 22 °C con 6,3 ml de una solución de 25 % en peso de metóxido de sodio en metanol (30,2 mmol), añadido en una porción. Se agregó más metanol (45 ml) y la mezcla se agitó durante 40 min. La mezcla de reacción se inactivó mediante la adición de 40 ml de ácido clorhídrico 1 N y
después 10 ml de bicarbonato de sodio saturado. El disolvente se evaporó a presión reducida y el residuo se diluyó con diclorometano (400 ml), se lavó con salmuera, se secó sobre sulfato de magnesio anhidro y se evaporó al vacío. La cristalización del residuo sólido de color blanco de 1,2-dicloroetano (30 ml) produjo 2,19 g del material del título en forma de un sólido de color blanco. La cromatografía de los licores madre sobre gel de sílice (3 x 10 cm, elución con 0 -1 % de acetato de etilo- diclorometano) produjo otros 0,46 g de producto (rendimiento total, 2,65 g, 93 %). LC (Método A): 2,379 min. HRMS (ESI) calc. para C20H16N3O3S [M H]+ m/z 378,0907, encontrado 378,0911. RMN 1H (CDCla, 600 MHz) 8 ppm: 4,18 (s, 3H), 5,21 (s, 2H), 6,71 (dd, J = 7,4 Hz and J = 0,95 Hz, 1H), 7,12 - 7,17 (m, 3H), 7,32 (a t, 1H), 7,38 (t amplio, 2H), 7,47 (a d, 2H), 7,88 (s, 1H).
191G. 2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-ol

Una mezcla de 6-(4-(benciloxi)benzofuran-2-il)-2-metoxiimidazo[2,1-b][1,3,4]tiadiazol (2,640 g, 6,99 mmol) y pentametilbenceno (7,25 g, 48,9 mmol) en diclorometano (400 ml) se enfrió a -78 °C en atmósfera de nitrógeno y después se trató inmediatamente con 18,2 ml (8,2 mmol) de una solución 1 M de tricloruro de boro en diclorometano, añadida gota a gota durante 3 min. La mezcla resultante se agitó a -78 °C durante 1 h. La mezcla de reacción se inactivó mediante la adición de una solución de bicarbonato de sodio (10,6 g) en agua (50 ml), añadida en una porción. El baño de enfriamiento se retiró y la mezcla resultante se agitó a temperatura ambiente durante 1 h. El sólido formado se filtró y se lavó sucesivamente con agua (50 ml) y diclorometano (25 ml). La torta de filtro se empapó con etanol anhidro (10 ml) y después se succionó seca. El sólido de color blanco obtenido se secó después al vacío durante algunos días en pentóxido de fósforo hasta alcanzar un peso constante para producir 1,459 g (72 % de rendimiento) del material del título. El filtrado combinado y los lavados (fases orgánica y acuosa de la etapa de desprotección) se diluyeron con diclorometano (500 ml) y se agitaron en un baño de agua caliente hasta que la fase orgánica se volvió transparente, sin sólidos aparentes en suspensión. La fase orgánica se recolectó, se secó sobre sulfato de magnesio anhidro y se filtró rápidamente mientras aún estaba caliente. El filtrado se evaporó y el residuo (producto y pentametilbenceno) se trituró con tolueno (20 ml). El sólido se recolectó mediante filtración y se lavó con tolueno (20 ml) para obtener, después de secar al vacío, 0,239 g (12 % de rendimiento; 84 % de rendimiento combinado) del material del título en forma de un sólido de color tostado. LC (Método A): 1,908 min. HRMS (ESI) calc. para C13H10N3O3S [M H]+ m/z 288,0437, encontrado 288,0446. RMN 1 H (DMSO-d6, 600 MHz) 8 ppm: 4,46 (s, 3H), 6,58 (d, J = 7,8 Hz, 1H), 6,97 (d, J = 8,15 Hz, 1H), 7,0-7,07 (m, 3H), 8,40 (s, 1H).
Ejemplo 191. 2-metoxi-6-(4-((2-feniltiazol-4-il)metoxi)benzofuran-2-il)imidazo[2,1-b][1,3,4]tiadiazol

Una mezcla de 2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-ol (0,100 g, 0,349 mmol), trifenilfosfina (0,165 g, 0,627 mmol) y (2-feniltiazol-4-il)metanol (Ejemplo 3B, 0,080 g, 0,418 mmol) en un matraz de 50 ml se mantuvo al vacío durante 10 min y después se purgó con nitrógeno. Se agregó tetrahidrofurano seco (10 ml) y la mezcla resultante se calentó ligeramente y se mantuvo en un baño ultrasónico durante 5 min. La mezcla enfriada (aún heterogénea) se trató a 22 °C con una solución de azodicarboxilato de diisopropilo (0,113 g, 0,558 mmol) en tetrahidrofurano (2 ml), añadida gota a gota durante 1 h. La mezcla se agitó después a 2 °C durante 4 h. La mezcla de reacción transparente se inactivó mediante la adición de diclorometano ( 10 0 ml) y bicarbonato de sodio saturado (10 ml). La fase orgánica se lavó con salmuera, se secó sobre sulfato de magnesio anhidro y se concentró al vacío. La cromatografía del residuo sobre gel de sílice (2,5 x 12 cm, elución con 0 - 3 % de acetato de etilo-diclorometano) y después la cristalización de la fracción deseada de acetato de etilo (8 ml) produjo 0,028 g (24 % de rendimiento) del material del título en forma de un sólido de color blanco. LC (Método A): 2,426 min. HRMS (ESI) calc. para C23H17N4O3S2 [M H]+ m/z 461,0737, encontrado 461,0926. RMN 1 H (CDCla, 400 MHz) 8 ppm: 4,22 (s, 3H), 5,45 (d, J=0,78 Hz, 2H), 6,80 (dd, J=7,04, 1,57 Hz, 1H), 7,15 - 7,21 (m, 2H), 7,22 (s, 1H), 7,38 (s, 1H), 7,42 - 7,51 (m, 3H), 7,92 (s, 1H), 7,95 -8,03 (m, 2H).
Ejemplo 192
(S)-2-(1-fluoroetil)-6-(6-metoxi-4-((2-feniltiazol-4-il)metoxi)benzofuran-2-il)imidazo[2,1-b][1,3,4]tiadiazol

192A. (S)-5-(1-fluoroetil)-1,3,4-tiadiazol-2-amina

Se cargó un recipiente a presión sellable de 350 ml con tiosemicarbazida (11,17 g, 122,5 mmol) y dioxano seco (100 ml) y la mezcla se enfrió a 0 °C en atmósfera de N2. A esta mezcla, que se agitó rápidamente, se le agregó lentamente una solución de ácido (S)-2-fluoropropanoico (9,40 g, 102,1 mmol, de Fritz-Langhals, E., Tetrahedron Asymmetry, 981 (1994)) en dioxano (10 ml). A la mezcla resultante se le agregó gota a gota POCl3 (11,22 ml, 122,5 mmol), después el baño de enfriamiento se retiró y la suspensión espesa de color blanco se agitó a temperatura ambiente durante 1 h. El recipiente se selló y la mezcla se calentó a 90-95°C (temperatura del baño de aceite) durante 5 h. La mezcla enfriada se agitó a temperatura ambiente durante 14 h y después el sobrenadante (mezcla bifásica) se decantó y se concentró a presión reducida. La fase inferior se vertió lentamente en agua helada (250 ml) y después se agregó también el concentrado. Esta mezcla se agitó rápidamente hasta que se convirtió esencialmente en una solución homogénea (turbia) y después se basificó a pH 9-9,5 usando 40 % de NaOH acuoso. La suspensión resultante se filtró y la torta de filtro se lavó con agua (Nota: La LC de este sólido de color beis mostró que contenía solo un rastro del producto deseado, de modo que no se continuó investigando). El filtrado combinado se extrajo después con EtOAc (x3) y la fase orgánica se secó (Na2SO4) y se evaporó para obtener un sólido de color crema (10,58 g, 70 %) que era el producto esencialmente puro de acuerdo con Lc y LCMS. Este material se usó tal cual sin purificación adicional. Una muestra analítica se purificó mediante cromatografía ultrarrápida [Isco/ 0-20 % de (MeOH-NH4OH, 9:1)-DCM] para obtener un sólido de color blanco. LC (Método B): 0,608 min. MS (ESI) calc. para C4H6FN3S m/z: 147,03; encontrado: 148,05 [M+H]+. RMN 1H (600 MHz, DMSO-da) 8 7,38 (s, 2H), 5,82 (dq, J= 6,4, 48,0 Hz, 1H), 1,65 (dd, J= 6,4, 24,0 Hz, 3H). LC quiral: S:R= 95:5.
192B. (S)-6-(4-(benciloxi)-6-metoxibenzofuran-2-il)-2-(1-fluoroetil)imidazo[2,1-b][1,3,4]tiadiazol

En un vial de 20 ml, se suspendieron 1-(4-(benciloxi)-6-metoxibenzofuran-2-il)-2-bromoetanona (Ejemplo 1E, 1,501 g, 4,000 mmol) y (S)-5-(1-fluoroetil)-1,3,4-tiadiazol-2-amina (0,589 g, 4,000 mmol) en 2-propanol (20 ml) y se calentaron a 80 °C durante 16 h. La mezcla después se calentó en un reactor de microondas a 150 °C durante 30 min. La mezcla enfriada se evaporó a sequedad y el residuo obtenido se absorbió en CH2Cl2 (200 ml), se lavó (NaHCO3 acuoso saturado), se secó (Na2SO4) y se concentró. El residuo se purificó en ISCO (0-30 % de acetonahexanos) para obtener el compuesto del título (0,922 g, 54,4 %) en forma de un sólido claro. LC (Método B): 2,403 min. MS (ESI) calc. para C22H19FN3O3S [M+H]+ m/z: 424,1131; encontrado: 424,1146. RMN 1 H (400 MHz, DMSO-d6 ) d ppm 8,61 (s, 1H) 7,51 (d, J=7,4 Hz, 2H) 7,42 (t, J=7,6 Hz, 2H) 7,35 (t, J=7,0 Hz, 1H) 7,08 (s, 1 H) 6,83 -6,85 (m, 1H) 6,54 (d, J=1,2 Hz, 1H) 6,16 (dq, J=47,1, 6,4 Hz, 1H) 5,26 (s, 2H) 3,80 (s, 3H) 1,79 (dd, J=24,5, 6 ,8 Hz, 3H).
192C. (S)-2-(2-(1-fluoroetil)imidazo[2,1-b][1,3,4]tiadiazol-6-il)-6-metoxibenzofuran-4-ol

Una mezcla de (S)-6-(4-(benciloxi)-6-metoxibenzofuran-2-il)-2-(1-fluoroetil)imidazo[2,1-b][1,3,4]tiadiazol (0,152 g, 0,359 mmol) y pentametilbenceno (0,374 g, 2,52 mmol) en diclorometano (24 ml, 373 mmol) se enfrió a -78 °C en atmósfera de nitrógeno y después se trató inmediatamente con tricloruro de boro 1,0 M en diclorometano (1 ml, 1,000 mmol), añadido gota a gota durante 3 min. La mezcla resultante se agitó a -78 °C durante 1 h. La mezcla de
reacción se inactivó mediante la adición de una solución de bicarbonato de sodio (0,71 g) en agua (12 ml), añadida en una porción. El baño de enfriamiento se retiró y la mezcla resultante se agitó a temperatura ambiente durante 1 h. El sólido formado se filtró, se lavó sucesivamente con agua (8 ml) y diclorometano (8 ml). La torta de filtro se empapó con etanol anhidro y se succionó seca. El sólido de color blanco obtenido se secó al vacío en P2O5 durante 36 h. LC (Método B): 2,038 min. MS (ESI) calc. para C15H13FN3O3S [M+H]+ m/z: 334,0656; encontrado: 334,0680. RMN 1H (400 MHz, DMSO-d6) 8 ppm 10,06 (s, 1H) 8,56 (s, 1H) 7,09 (s, 1H)6,67 (s, 1H) 6,26 -6,28 (m, 1H) 6,16 (dq, J=46,9, 6,4 Hz, 1H) 3,76 (s, 3H) 1,80 (dd, J=24,7, 6,3 Hz, 3H).
Ejemplo 192. (S)-2-(1-fluoroetil)-6-(6-metoxi-4-((2-feniltiazol-4-il)metoxi)benzofuran-2-il)imidazo[2,1-b][1,3,4]tiadiazol

A una mezcla de (S)-2-(2-(1-fluoroetil)imidazo[2,1-b][1,3,4]tiadiazol-6-il)-6-metoxibenzofuran-4-ol (0,050 g, 0,150 mmol), (2-feniltiazol-4-il)metanol (Ejemplo 3B, 0,086 g, 0,450 mmol) y trifenilfosfina (0,118 g, 0,450 mmol) en N2 se le agregó THF seco (3 ml). A la solución resultante de color ámbar claro, se le agregó una solución de DIAD (0,087 ml, 0,450 mmol) en THF seco (2 ml) gota a gota durante 2 h para obtener una solución de color amarillo parduzco claro. Después de agitar a temperatura ambiente durante otros 30 min, la LC mostró que no había material de partida restante. Después los volátiles se retiraron a presión reducida para obtener una goma de color ámbar. La cromatografía ultrarrápida (Isco/ 0-20 % de éter-DCM) produjo el producto en forma de una goma casi incolora. Esta goma se trituró con un volumen mínimo de MeOH, la suspensión resultante se filtró y la torta de filtro se lavó con un volumen mínimo de MeOH y después se secó al vacío para obtener el material del título (0,048 g, 63,2 %) en forma de un sólido que también se liofilizó a partir de MeCN-H2O para obtener un sólido de color crema. LC (Método A): 2,453 min. HRMS (ESI) calc. para C25H20FN4O3S2 [M H]+ m/z 507,096, encontrado 506,098. RMN 1 H (DMSO-d6, 400 MHz) 8 ppm: 8,67 (s, 1H), 8,05-8,02 (m, 2H), 7,96 (s, 1H), 7,60-7,56 (m, 3H), 7,20 (d, J= 0,8 Hz, 1H), 6,93 (dd, J= 0,8, 2,0 Hz, 1H), 6,73 (d, J= 1,6 Hz, 1H), 6,23 (dq, J= 6,7, 47,0 Hz, 1H), 5,45 (s, 2H), 3,89 (s, 3H), 1,86 (d, J= 6,7, 24,6 Hz, 3H).
Ejemplo 193
(S)-4-(4-(((2-(2-(1-fluoroetil)imidazo[2,1-b][1,3,4]tiadiazol-6-il)-6-metoxibenzofuran-4-il)oxi)metil)tiazol-2-il)morfolina

Una solución de (S)-2-(2-(1-fluoroetil)imidazo[2,1-b][1,3,4]tiadiazol-6-il)-6-metoxibenzofuran-4-ol (Ejemplo 192C, 0,050 g, 0,150 mmol) en DMF (2 ml) se agitó en N2 y después se agregó 4-(4-(bromometil)tiazol-2-il)morfolina (0,047 g, 0,180 mmol), seguido de carbonato de potasio en polvo (0,062 g, 0,450 mmol) y la mezcla se agitó a temperatura ambiente durante 4 h. La mezcla se diluyó con NH4Cl acuoso saturado y se extrajo con DCM (x2). El extracto orgánico se lavó (salmuera), se secó (Na2SO4) y se evaporó para obtener una goma de color ámbar claro. La cromatografía ultrarrápida (Isco/ 0-100 % de EtOAc-hexano) produjo (S)-4-(4-(((2-(2-(1-fluoroetil)imidazo[2,1-b][1,3,4]tiadiazol-6-il)-6-metoxibenzofuran-4-il)oxi)metil)tiazol-2-il)morfolina (0,055 g, 71,1 %) en forma de una goma incolora, que se liofilizó a partir de MeCN-agua en forma de un sólido de color crema. LC (Método A): 2,271 min. HRMS (ESI): calc. para C23H23FN5O4S2 [M+H]+ m/z 516,118; encontrado 516,121. RMN 1H (400 MHz, DMSO-d6): 8 8,54 (s, 1H), 7,02 (s, 1H), 6,92 (s, 1H), 6,77 (s, 1H), 6,52 (d, J= 1,96 Hz, 1H), 6,10 (dq, J= 6,26, 46,95 Hz, 1H), 5,01 (s, 2H), 3,74 (s, 3H), 3,64 (t, J= 4,89 Hz, 4H), 3,31 (t, J= 4,89 Hz, 4H), 1,73 (dd, J= 6,26, 24,65 Hz, 3H).
Ejemplo 194
(S)-4-(4-(((2-(2-(1-fluoroetil)imidazo[2,1-b][1,3,4]tiadiazol-6-il)-6-metoxibenzofuran-4-il)oxi)metil)tiazol-2-il)-N,N-dimetilbenzamida
A una matraz secado al fuego se le agregó (S)-2-(2-(1-fluoroetil)imidazo[2,1-b][1,3,4]tiadiazol-6-il)-6-metoxibenzofuran-4-ol (Ejemplo 192C, 0,050 g, 0,150 mmol) y 4-(4-(hidroximetil)tiazol-2-il)-N,N-dimetilbenzamida (Ejemplo 36B, 0,047 g, 0,180 mmol); después el matraz se purgó con N2 y se agregó THF seco (3 ml). A la suspensión resultante se le agregó tri-n-butilfosfina (0,097 ml, 0,375 mmol) y después se agregó gota a gota una solución de 1,1'-(azodicarbonil)dipiperidina (0,096 g, 0,375 mmol) en THF seco (2 ml) (a través de una bomba de jeringa) durante 30 min. La mezcla resultante se agitó a temperatura ambiente durante otros 15 min y la mezcla se diluyó con EtOAc, se lavó (NaHCÜ3 acuoso saturado, H2O, salmuera), se secó (Na2SÜ4) y se evaporó para obtener un semisólido de color amarillo. La cromatografía ultrarrápida (Isco/ 0-100 % de EtOAc-DCM) produjo una goma incolora que se trituró con MeCN. La suspensión resultante se filtró y la torta de filtro se lavó con MeCN y después se secó al vacío para obtener un sólido de color amarillo claro que se liofilizó a partir de MeCN-agua para obtener (S)-4-(4-(((2-(2-(1-fluoroetil)imidazo[2,1-b][1,3,4]tiadiazol-6-il)-6-metoxibenzofuran-4-il)oxi)metil)tiazol-2-il)-N,N-dimetilbenzamida (0,066 g, 76 %) en forma de un sólido color amarillo claro. LC (Método A): 2,345 min. HRMS (ESI): calc. para C28H25FN5O4S2 [M+H]+ m/z 578,133; encontrado 578,133. RMN 1 H (400 MHz, DMSO-d6): 8 8,54 (s, 1H), 7,96 (d, J= 8,61 Hz, 2H), 7,89 (s, 1H), 7,48 (d, J= 8,61 Hz, 2H), 7,07 (s, 1H), 6,80 (s, 1H), 6,60 (d, J= 1,96 Hz, 1H), 6,10 (dq, J= 6,65, 46,95 Hz, 1H), 5,33 (s, 2H), 3,76 (s, 3H), 2,94 (a s, 3H), 2,87 (a s, 3H), 1,73 (dd, J= 6,65, 24,65 Hz, 3H).
Ejemplo 195
(S)-6-(6-fluoro-4-((2-feniltiazol-4-il)metoxi)benzofuran-2-il)-2-(1-fluoroetil)imidazo[2,1-b][1,3,4]tiadiazol

Una solución de 2-bromo-1-(6-fluoro-4-((2-feniltiazol-4-il)metoxi)benzofuran-2-il)etanona (Ejemplo 179E, 0,020 g, 0,045 mmol) y (S)-5-(1-fluoroetil)-1,3,4-tiadiazol-2-amina (Ejemplo 192A, 0,008 g, 0,052 mmol) en propan-2-ol (2 ml) se calentó a 70 °C durante 16 h. La mezcla de reacción enfriada se repartió entre diclorometano y bicarbonato de sodio acuoso saturado. La fase orgánica se separó, se lavó con salmuera, se secó sobre sulfato de magnesio anhidro y se concentró al vacío. El residuo obtenido se sometió a cromatografía sobre gel de sílice (ISCO, gradiente de elución de acetato de etilo in diclorometano) para obtener el compuesto del título (0,015 g, 68 %) en forma de un sólido de color blanco. LC (Método A): 2,493 min. HRMS (ESI) calc. para C24H16F2N4O2S2 [M+H]+ m/z: 494,0761; encontrado 495,0776. RMN 1H (CDCla, 400 MHz) 8 ppm: 8,07 (s, 1H), 7,93-8,02 (m, 2H), 7,42-7,51 (m, 3H), 7,38 (s, 1H), 7,25 (s, 1H), 6,87 -6,94 (m, 1H), 6,64 (dd, J= 11,35, 1,96 Hz, 1H), 5,65-6,04 (m, 1H), 5,41 (s, 2H), 1,80-1,95 (m, 3H).
Ejemplo 196
(S)-4-(4-(((2-(2-(1-fluoroetil)imidazo[2,1-b][1,3,4]tiadiazol-6-il)-6-metoxibenzofuran-4-il)oxi)metil)tiazol-2-il)tetrahidro-2H-piran-4-ol

A un matraz secado al fuego se le agregó (S)-2-(2-(1-fluoroetil)imidazo[2,1-b][1,3,4]tiadiazol-6-il)-6-metoxibenzofuran-4-ol (Ejemplo 192C, 0,040 g, 0,120 mmol) y 4-(4-(hidroximetil)tiazol-2-il)tetrahidro-2H-piran-4-ol (Ejemplo 118B, 0,032 g, 0,150 mmol); después el matraz se purgó con N2 y se agregó THF seco (2 ml). A la suspensión resultante se le agregó tri-n-butilfosfina (0,078 ml, 0,300 mmol) y después se agregó gota a gota una solución de 1,1 '-(azodicarbonil)dipiperidina (0,076 g, 0,300 mmol) en THF seco (2 ml) (a través de una bomba de jeringa) durante 30 min. La mezcla resultante se agitó a temperatura ambiente durante 2 h, en cuyo momento se agregó más tri-n-butilfosfina (0,078 ml, 0,300 mmol) y 1,1'-(azodicarbonil)dipiperidina (0,076 g, 0,300 mmol) y la agitación continuó durante 16 h. La mezcla después se diluyó con EtOAc, se lavó (NaHCO3 acuoso saturado, H2O, salmuera), se secó (Na2SO4) y se evaporó para obtener una goma de color amarillo dorado. La cromatografía ultrarrápida (Isco/ 0-100 % de EtOAc-DCM) produjo el compuesto del título (0,047 g, 73,8 %) en forma de una goma incolora que se liofilizó a partir de MeCN-agua en forma de un sólido de color blanquecino. LC (Método A): 2,236 min. HRMS (ESI): calc. para C24H24FN4O5S2 [M+H]+ m/z 531,117; encontrado 531,120. RMN 1 H (400 MHz, DMSO-d6): 8 8,54 (s, 1H), 7,64 (s, 1H), 7,02 (s, 1H), 6,78 (s, 1H), 6,57 (d, J= 1,96 Hz, 1H), 6,11 (dq, J= 6,65, 46,95 Hz, 1H), 6,08 (s, 1H), 5,21 (s, 2H), 3,75 (s, 3H), 3,67 (m, 4H), 2,05 (ddd, J= 5,48, 11,35, 13,69 Hz, 2H), 1,73 (dd, J= 6,65,
25,04 Hz, 3H), 1,61 (a d, J= 12,91 Hz, 3H).
Ejemplo 197
(S)-2-(1-fluoroetil)-6-(4-((2-(4-fluorotetrahidro-2H-piran-4-il)tiazol-4-il)metoxi)-6-metoxibenzofuran-2-il)imidazo[2,1-b][1,3,4]tiadiazol

A una solución de 4-(bromoirietN)-2-(4-fluorotetrahidro-2H-piran-4-N)tiazol (Ejemplo 119C, 0,016 g, 0,057 mmol) en DMF (1,5 ml), se le agregó (S)-2-(2-(1-fluoroetil)imidazo[2,1-b][1,3,4]tiadiazol-6-il)-6-metoxibenzofuran-4-ol (Ejemplo 192C, 0,017 g, 0,052 mmol) y después carbonato de potasio en polvo recién preparado (0,022 g, 0,156 mmol). La mezcla se agitó en un vial sellado a temperatura ambiente durante 2 h y después se diluyó con NH4Cl acuoso saturado, la mezcla resultante se filtró y la torta de filtro se lavó con agua. El residuo húmedo y gomoso se absorbió en DCM y la solución se lavó (NaHCO3 acuoso saturado), se secó (Na2SO4) y se evaporó para obtener una goma de color amarillo. La cromatografía ultrarrápida (Isco/ 0-40 % de EtOAc-DCM) produjo el producto ligeramente impuro en forma de una goma de color amarillo claro. Este material se volvió a purificar mediante HPLC preparativa (Método A) para obtener (S)-2-(1-fluoroetil)-6-(4-((2-(4-fluorotetrahidro-2H-piran-4-il)tiazol-4-il)metoxi)-6-metoxibenzofuran-2-il)imidazo[2,1-b][1,3,4]tiadiazol puro (0,020 g, 72,3 %) en forma de un sólido. LC (Método A): 2,400 min. HRMS (ESI): calc. para C24H23F2N4O4S2 [M+H]+ m/z 533,113; encontrado 533,115. RMN 1H (400 MHz, DMSO-da): 8 8,57 (s, 1H), 7,91 (s, 1H), 7,07 (s, 1H), 6,83 (m, 1H), 6,61 (d, J= 1,57 Hz, 1H), 6,13 (dq, J= 6,65, 46,56 Hz, 1H), 5,30 (s, 2H), 3,81 (m, 2H), 3,79 (s, 3H), 3,67 (dt, J= 1,57, 10,96 Hz, 2H), 2,33-2,16 (m, 2H), 2,05 (m, 2H), 1,73 (dd, J= 6,65, 24,65 Hz, 3H).
Ejemplo 198
4-(4-(((2-(2-((S)-1-fluoroetil)imidazo[2,1-b][1,3,4]tiadiazol-6-il)-6-metoxibenzofuran-4-il)oxi)metil)tiazol-2-il)-2,6-dimetiltetrahidro-2H-piran-4-ol

A un matraz secado al fuego se le agregó (S)-2-(2-(1-fluoroetil)imidazo[2,1-b][1,3,4]tiadiazol-6-il)-6-metoxibenzofuran-4-ol (Ejemplo 192C, 0,630 g, 1,890 mmol) y 4-(4-(hidroximetil)tiazol-2-il)-2,6-dimetiltetrahidro-2H-piran-4-ol (Ejemplo 121B, 0,552 g, 2,268 mmol), después el matraz se purgó con N2 y se agregó THF seco (20 ml). A la suspensión resultante se le agregó tri-n-butilfosfina (1,227 ml, 4,72 mmol) y después se agregó gota a gota una solución de 1,1'-(azodicarbonil)dipiperidina (1,204 g, 4,72 mmol) en THF seco (10 ml) (a través de una bomba de jeringa) durante 30 min. La mezcla resultante se agitó a temperatura ambiente durante 2 h más y después se diluyó con EtOAc, se lavó (NaHCO3 acuoso saturado, H2O, salmuera), se secó (Na2SO4) y se evaporó para obtener un semisólido de color amarillo. La cromatografía ultrarrápida (Isco/ 0-100 % de EtOAc-DCM) produjo el producto en forma de un sólido. Este sólido se trituró con MeCN, la suspensión resultante se filtró y la torta de filtro se lavó con MeCN y después se secó al vacío para obtener 4-(4-(((2-(2-((S)-1-fluoroetil)imidazo[2,1-b][1,3,4]tiadiazol-6-il)-6-metoxibenzofuran-4-il)oxi)-metil)tiazol-2-il)-2,6-dimetiltetrahidro-2H-piran-4-ol (0,823 g, 78 %) en forma de un sólido blanquecino. LC (Método A): 2,324 min. HRMS (ESI): calc. para C26H28FN4O5S2 [M+H]+ m/z 559,149; encontrado 559,151. RMN 1 H (400 MHz, DMSO-d6): 8 8,54 (s, 1H), 7,70 (s, 1H), 7,01 (s, 1H), 6,79 (s, 1H), 6,62 (d, J= 1,57 Hz, 1H), 6,11 (dq, J= 6,65, 46,95 Hz, 1H), 6,03 (s, 1H), 5,25 (s, 2H), 3,77 (m, 2H), 3,76 (s, 3H), 2,01 (d, J= 12,91 Hz, 2H), 1,74 (dd, J= 6,65, 25,04 Hz, 3H), 1,39 (t, J= 12,91 Hz, 2H), 1,01 (d, J= 6,26 Hz, 6 H).
Ejemplo 199
6-(4-((2-(4-fluoro-2,6-dimetiltetrahidro-2H-piran-4-il)tiazol-4-il)metoxi)-6-metoxibenzofuran-2-il)-2-((S)-1-fluoroetil)imidazo[2,1-b][1,3,4]tiadiazol

A una suspensión enfriada en hielo de 4-(4-(((2-(2-((S)-1-fluoroetil)imidazo[2,1-b][1,3,4]tia-diazol-6-il)-6-metoxibenzofuran-4-il)oxi)metil)tiazol-2-il)-2,6-dimetiltetrahidro-2H-piran-4-ol (Ejemplo 198, 0,021 g, 0,038 mmol) en DCM (3 ml), en atmósfera de N2 , se le agregó DAST (0,012 ml, 0,094 mmol) gota a gota y la mezcla resultante se agitó a 0 °C durante 20 min. Se agregó otra alícuota de DAST (0,007 ml, 0,053 mmol), el baño de enfriamiento se retiró y la solución pálida resultante se agitó a temperatura ambiente durante 16 h. La mezcla de reacción después se volvió a enfriar a 0 °C y se inactivó mediante la adición gota a gota de NaHCO3 acuoso saturado (3 ml). La mezcla se agitó vigorosamente a 0 °C durante 5 min, después el baño de enfriamiento se retiró y se continuó agitando hasta que no se observó más evolución de gas. Después la fase orgánica se separó y se aplicó directamente a una precolumna de gel de sílice. La cromatografía ultrarrápida (Isco/ 0-100 % de EtOAc-hexano) produjo 6-(4-((2-(4-fluoro-2,6-dimetiltetrahidro-2H-piran-4-il)tiazol-4-il)metoxi)-6-metoxibenzofuran-2-il)-2-((S)-1-fluoroetil)imidazo-[2,1-b][1,3,4]tiadiazol (0,014 g, 66,4 %) en forma de una goma incolora que se liofilizó de MeCN-agua para obtener un sólido de color blanco. La RMN indicó que era una mezcla de isómeros 3:2. LC (Método A): 2,487 min. HRMS (ESI): calc. para C26H27F2N4O4S2 [M+H]+ m/z 561,144; encontrado 561,146. RMN 1H (400 MHz, DMSO-da): 8 8,54 (s, 0,4H), 8,53 (s, 0,6H), 7,94 (s, 0,6H), 7,85 (s, 0,4H), 7,03 (s, 0,4H), 7,02 (s, 0,6H), 6,79 (m, 1H), 6,59 (d, J= 1,57 Hz, 0,6H), 6,57 (d, J= 1,96 Hz, 0,4H), 6,10 (dq, J= 6,65, 46,56 Hz, 1H), 5,30 (s, 1,2H), 5,25 (s, 0,8H), 3,76 (m, 0,8H), 3,75 (s, 1,2H), 3,74 (s, 1,8H), 3,53 (m, 1,2H), 2,38 (m, 1H), 2,08 (m, 1H), 1,83-1,61 (m, 2H), 1,73 (dd, J= 6,65, 25,04 Hz, 3H), 1,10 (d, J= 6,26 Hz, 2,4H), 1,01 (d, J= 6,26 Hz, 3,6H).
Ejemplo 200
(S)-4-(4-(((2-(2-(1-fluoroetil)imidazo[2,1-b][1,3,4]tiadiazol-6-il)-6-metoxibenzofuran-4-il)oxi)metil)-5-metiltiazol-2-il)tetrahidro-2 H-piran-4-ol
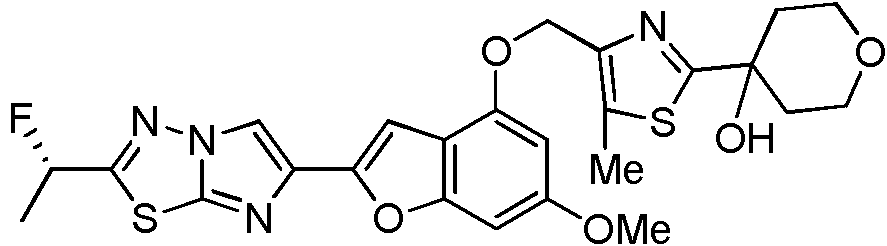
A un matraz secado al fuego se le agregó (S)-2-(2-(1-fluoroetil)imidazo[2,1-b][1,3,4]tiadiazol-6-il)-6-metoxibenzofuran-4-ol (Ejemplo 192C, 0,050 g, 0,150 mmol) y 4-(4-(hidroximetil)-5-metiltiazol-2-il)tetrahidro-2H-piran-4-ol (Ejemplo 124D, 0,038 g, 0,165 mmol), después el matraz se purgó con N2 y se agregó THF seco (3 ml). A la suspensión resultante se le agregó tri-n-butilfosfina (0,097 ml, 0,375 mmol) y después se agregó gota a gota una solución de 1,1'-(azodicarbonil)dipiperidina (0,096 g, 0,375 mmol) en THF seco (2 ml) (a través de una bomba de jeringa) durante 30 min. La mezcla resultante se agitó a temperatura ambiente durante 15 min más y la mezcla se diluyó con EtOAc, se lavó (NaHCO3 acuoso saturado, H2O, salmuera), se secó (Na2SO4) y se evaporó para obtener un sólido ceroso de color amarillo claro. La cromatografía ultrarrápida (Isco/ 0-100 % de EtOAc-DCM) produjo el compuesto del título (0,061 g, 74,7 %) en forma de una goma incolora que se liofilizó a partir de MeCN-agua en forma de un sólido. LC (Método A): 2,256 min. HRMS (ESI): calc. para C25H26FN4O5S2 [M+H]+ m/z 545,133; encontrado 545,132. RMN 1H (400 MHz, DMSO-d6): 8 8,54 (s, 1H), 6,94 (s, 1H), 6,78 (m, 1H), 6,61 (d, J= 1,96 Hz, 1H), 6,09 (dq, J= 6,65, 46,95 Hz, 1H), 5,99 (a s, 1H), 5,14 (s, 2H), 3,75 (s, 3H), 3,65 (m, 4H), 2,40 (s, 3H), 2,02 (m, 2H), 1,72 (dd, J= 6,65, 24,65 Hz, 3H), 1,58 (d, J= 11,74 Hz, 3H).
Ejemplo 201
6-metoxi-2-(2-(metiltio)imidazo[2,1-b][1,3,4]tiadiazol-6-il)-4-((2-feniltiazol-4-il)metoxi)benzo[d]oxazol

201A. 2-(metiltio)imidazo[2,1-b][1,3,4]tiadiazol-6-carboxilato de etilo

Una mezcla de 2-amino-5-metiltio-1,3,4-tiadiazol (25,0 g, 0,17 mol), 3-bromopiruvato de etilo (23,7 ml, 0,189 mol) y etanol (125 ml) en un recipiente sellable de 350 ml se calentó a 150 °C (temperatura del baño de aceite) durante 20 min. La mezcla enfriada se concentró después a sequedad y el residuo se repartió con acetato de etilo-NaHCO3 acuoso saturado. La fase orgánica se separó, se lavó (salmuera), se secó (MgSO4), se filtró y se concentró a sequedad. El residuo obtenido se absorbió en un volumen mínimo de diclorometano, la suspensión resultante se filtró y la torta de filtro se lavó con una pequeña cantidad de diclorometano. El residuo sólido se secó al vacío para obtener amino-5-metiltio-1,3,4-tiadiazol recuperado (3,7 g, 15 %). Posteriormente, el filtrado obtenido se evaporó y el residuo se cristalizó de un volumen mínimo de etanol caliente para obtener el compuesto del título en forma de un sólido cristalino de color beis (10,8 g, 26 %). LC (Método B): 1,267 min. RMN 1 H (600 MHz, DMSO-d6) 8 ppm: 8,76 (s, 1H), 4,27 (q, J= 7,2 Hz, 2H), 2,78 (s, 3H), 1,28 (t, J= 7,2 Hz, 3H).
201B. Ácido 2-(metiltio)imidazo[2,1-b][1,3,4]tiadiazol-6-carboxílico

En un vial sellable para microondas, una solución de 2-(metiltio)imidazo[2,1-b][1,3,4]tiadiazol-6-carboxilato de etilo (1,22 g, 5,0 mmol) en ácido acético glacial (15 ml) se trató con 48 % de HBr (1,42 ml, 12,5 mmol) gota a gota. El vial después se selló y la suspensión resultante se calentó a 150 °C (microondas) durante 1,5 h. La mezcla enfriada se filtró después y la torta de filtro se lavó con un volumen mínimo de ácido acético glacial y después con DCM. Después de secar al vacío, esto produjo la sal de HBr esencialmente pura del compuesto del título (1,37 g, 92 %) en forma de un sólido de color blanco. Este material se usó tal cual en la siguiente etapa. LC (Método A): 1,313 min. LCMS (APCI): calc. para C6H6N3O2S2 [M+H]+ m/z 215,99; encontrado 216,00. RMN 1H (600 MHz, DMSO-d6) 8 ppm: 8,63 (s, 1H), 2,75 (s, 3H).
201C. Cloruro de 2-(metiltio)imidazo[2,1-b][1,3,4]tiadiazol-6-carbonilo

A una suspensión en agitación de ácido 2-(metiltio)imidazo[2,1-b][1,3,4]tiadiazol-6-carboxílico (15,0 g, 0,070 mol) en DCM (350 ml), se le agregó cloruro de oxalilo (29,5 ml, 0,348 mol) y después DMF (1 gota). Se observó la evolución de gas y la mezcla de reacción se agitó a temperatura ambiente durante 3,5 h. La suspensión se concentró después a sequedad para obtener el compuesto del título en forma de un sólido de color amarillo claro (rendimiento cuantitativo asumido) que se usó tal cual en la siguiente etapa. LC (Método A): 1,686 min; RMN 1 H (600 MHz, DMSO-d6) 8 ppm 8 ,68 (s, 1 H) 2,78 (s, 3H).
201D. N-(2,6-dihidroxi-4-metoxifenil)-2-(metiltio)imidazo[2,1-b][1,3,4]tiadiazol-6-carboxamida

A una suspensión enfriada en hielo de cloruro de 2-(metiltio)imidazo[2,1-b][1,3,4]tiadiazol-6-carbonilo (0,378 g, 1,62 mmol) y 2-amino-5-metoxibencen-1,3-diol (0,251 g, 1,62 mmol) en DMF (10 ml), se le agregó trietilamina (1,13 ml, 8,10 mmol). La mezcla de reacción se agitó durante 5 min, después el baño de enfriamiento se retiró y se continuó agitando a temperatura ambiente durante 16 h. La mezcla resultante se evaporó a sequedad y el residuo se repartió con DCM-salmuera-agua (2:1:1). La fase orgánica se separó, se secó (MgSO4) y se evaporó a sequedad. El residuo se trituró con MeOH y el producto se filtró y se secó al vacío para obtener el compuesto del título en forma de un sólido de color beis (0,100 g, 0,284 mmol, 17,5 %). LC (Método E): 1,828 min. HRMS (ESI): calc. para C13H13N4O4S2 [M+H]+ m/z 353,0378, encontrado 353,0603. RMN 1 H (DMSO-d6, 400 MHz) 8 ppm: 10,26 (m, 2H), 9,26 (d, J = 9,4 Hz, 1H), 8,76 (s, 1H), 6,01 (d, J = 2,3 Hz, 2H), 3,66 (s, 3H), 2,81 (s, 3H).
201E. 6-metoxi-2-(2-(metiltio)imidazo[2,1-b][1,3,4]tiadiazol-6-il)benzo[d]oxazol-4-ol

Un recipiente para microondas se cargó con N-(2,6-dihidroxi-4-metoxifenil)-2-(metiltio)imidazo[2,1-b][1,3,4]tiadiazol-6 -carboxamida (0,086 g, 0,244 mmol), TFA (1 ml) y ácido acético (1 ml). Después el recipiente se selló y se calentó en un microondas a 200 °C durante 10 min. La mezcla de reacción se dejó reposar a temperatura ambiente durante 16 h y el sólido cristalino de color pardo resultante se filtró y se secó al vacío para obtener el compuesto del título (0,044 g, 0,098 mmol, 40 %). LC (Método E): 1,961 min. HRMS (ESI): calc. para C13H11 N4O3S2 [M+H]+ m/z 335,0273, encontrado 335,0505. RMN 1H (DMSO-da, 400 MHz) 8 ppm: 8,85 (s, 1H), 6,79 (d, J = 1,8 Hz, 1H), 6,36 (d, J = 1,8 Hz, 1H), 3,77 (s, 3H), 2,81 (s, 3H).
Ejemplo 201. 6-metoxi-2-(2-(metiltio)imidazo[2,1-b][1,3,4]tiadiazol-6-il)-4-((2-feniltiazol-4-il)metoxi)benzo[d]oxazol

Una suspensión de 6-metoxi-2-(2-(metiltio)imidazo[2,1-b][1,3,4]tiadiazol-6-il)benzo[d]oxazol-4-ol (0,080 g, 0,239 mmol) y 2-(bromometil)-4-feniltiazol (0,122 g, 0,479 mmol) en N,N-dimetilformamida (3 ml) se mantuvo al vacío (1 kPa (10 mbar)) durante 5 min, después el matraz se purgó con nitrógeno y se agregó carbonato de potasio anhidro en polvo recién obtenido (0,105 g, 0,756 mmol) de una sola vez. La mezcla resultante se agitó a temperatura ambiente durante 16 h, después la reacción se inactivó con HCl 1 N (2 ml) y finalmente se repartió entre diclorometano (150 ml) y bicarbonato de sodio acuoso saturado (20 ml). La fase orgánica se separó, se lavó con salmuera, se secó sobre sulfato de magnesio anhidro y se concentró al vacío. El residuo se trituró con acetonitrilo y el producto deseado se filtró y se secó al vacío para obtener 6-metoxi-2-(2-(metiltio)imidazo[2,1-b][1,3,4]tiadiazol-6-il)-4-((2-feniltiazol-4-il)metoxi)benzo[d]oxazol (0,068 g, 0,134 mmol, 56,0 %) en forma de un sólido de color blanco. LC (Método F): 2,653 min. HRMS (ESI): calc. para C23H18N5O3S3 [M+H]+ m/z 508,0572, encontrado 508,0607. RMN 1 H (DMSO-d6, 400 MHz) 8 ppm: 8 ,88 (s, 1H), 7,94 (m, 2H), 7,85 (s, 1H), 7,51-7,46 (m, 3H), 6,96 (d, J = 2,0 Hz, 1H), 6,72 (d, J = 2,0 Hz, 1H), 5,44 (s, 2H), 3,80 (s, 3H), 2,77 (s, 3H).
Ejemplo 203
4-(((6-metoxi-2-(6-metilimidazo[1,2-b]piridazin-2-il)benzofuran-4-il)oxi)metil)-2-feniltiazol

203A. 2-(4-(benciloxi)-6-metoxibenzofuran-2-il)-6-metilimidazo[1,2-b]piridazina

Una mezcla de 6-metilpiridazin-3-amina (1,52 g, 13,93 mmol), 1-(4-(benciloxi)-6-metoxibenzofuran-2-il)-2-bromoetanona (Ejemplo 1E, 5,00 g, 13,33 mmol) y 2-propanol (110 ml) en un matraz de presión de 150 ml se calentó a 65 °C. La mezcla era prácticamente homogénea después de calentarse durante 30 min y se volvió a precipitar después de 40 min. La mezcla se calentó durante un total de 48 h. La mezcla de reacción enfriada se diluyó con diclorometano (600 ml), se lavó con bicarbonato de sodio saturado y salmuera y se secó sobre sulfato de magnesio anhidro. La evaporación produjo un sólido de color naranja parduzco que se sometió a cromatografía sobre gel de sílice (4 x 9 cm, elución con diclorometano - acetato de etilo 0 - 5 %) para obtener el material del título (3,64 g) en forma de un sólido de color naranja parduzco. El sólido se hirvió con acetato de etilo (30 ml, parcialmente soluble) y se dejó reposar a temperatura ambiente durante 2 h. Los cristales se recolectaron mediante filtración y se secaron al
vacío durante la noche para obtener el material del título (3,440 g, 67 %) en forma de agujas de color amarillo parduzco claro. LC (Método A): 2,279 min. HRMS (ESI) calc. para C23H20N3O3 [M H]+ m/z 386,1505, encontrado 386,1532. RMN 1H (CDCla, 400 MHz) 8 ppm: 2,59 (s, 3 H), 3,86 (s, 3 H), 5,21 (s, 2 H), 6,43 (d, J=1,96 Hz, 1 H), 6,75 (a d, 1 H), 6,94 (d, J=9,39 Hz, 1 H), 7,31 - 7,38 (m, 2 H), 7,38 - 7,45 (m, 2 H), 7,50 (d amplio, J=7,43 Hz, 2 H), 7,82 (d, J=9,39 Hz, 1 H), 8,19 (s, 1 H).
203B. 6-metoxi-2-(6-metilimidazo[1,2-b]piridazin-2-il)benzofuran-4-ol
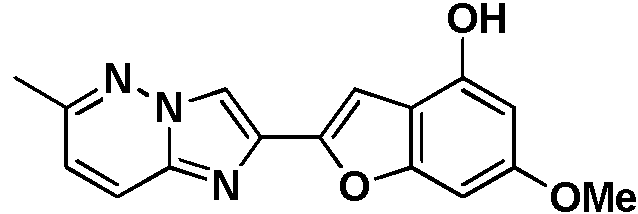
Una solución de 2-(4-(benciloxi)-6-metoxibenzofuran-2-il)-6-metilimidazo[1,2-b]piridazina (Ejemplo 39A, 1,00 g, 2,59 mmol) en una mezcla de diclorometano (420 ml) y metanol (150 ml) en un matraz de 1 l se hidrogenó en 10 % de paladio sobre carbón (0,30 g, es decir, 30 mg de Pd) y en 101,325 kPa (1 atm) de hidrógeno durante 6 h. La mezcla de reacción se mantuvo al vacío durante 2 min y después se purgó con nitrógeno. El catalizador se filtró y se lavó con diclorometano-metanol caliente (8:2, 100 ml) y el filtrado combinado se concentró a presión reducida. El residuo de color amarillo se hirvió con 1,2-dicloroetano (30 ml) y se dejó reposar a temperatura ambiente durante 18 h. El sólido se filtró (contiene metanol según RMN) y se secó al vacío a 120 °C durante 12 h para obtener el material del título (0,760 g, 99 % de rendimiento) de un sólido de color amarillo. LC (Método A): 1,844 min. RMN 1 H (DMSO-d6, 400 MHz) 8 ppm: 2,54 (s, 3H), 3,77 (s, 3H), 6,28 (d, J=1,96 Hz, 1H), 6,70 (dd, J=1,96, 1,17 Hz, 1H), 7,20 (d, J=9,39 Hz, 1H), 7,24 (d, J=0,78 Hz, 1H), 8,03 (d, J=9,78 Hz, 1H), 8,50 (s, 1H), 10,10 (a s, 1H).
Ejemplo 203. 4-(((6-metoxi-2-(6-metilimidazo[1,2-b]piridazin-2-il)benzofuran-4-il)oxi)metil)-2-feniltiazol

En un matraz de fondo redondo, de 100 ml, se purgó una suspensión de 6-metoxi-2-(6-metilimidazo[1,2-b]piridazin-2-il)benzofuran-4-ol (0,190 g, 0,643 mmol) y 4-(bromometil)-2-feniltiazol (0,180 g, 0,708 mmol) en DMF (5 ml) al vacío y en N2 durante 10 min. La reacción se trató con carbonato de potasio (0,24 g, 1,737 mmol) y se agitó a 22 °C durante 18 horas, después se diluyó con DCM y se lavó con agua (1x) y salmuera (1x). Las capas orgánicas se secaron sobre sulfato de magnesio anhidro, se filtraron y se concentraron. El residuo se purificó mediante cromatografía sobre gel de sílice (2,5 x 10 cm, 0 % a 50 % de EtOAc en CH2Ch) para obtener el material del título impuro (0,198 g, 66 %). El sólido se trituró en acetato de etilo caliente para proporcionar el material del título puro (0,176 g). LC (Método A): 2,414 min. HRMS (ESI) calc. para C26H21N4O3S [M H]+ m/z 469,1334, encontrado 469,1379. RMN 1H (CDCh, 400 MHz) 8 ppm: 2,57 (s, 3H), 3,84 (s, 3 H), 5,39 (s, 2 H), 6,46 (d, J=1,96 Hz, 1 H), 6,74 (d a, 1 H), 6,91 (d, J=9,1 Hz, 1 H), 7,34 (s, 1 H), 7,36 (s, 1 H), 7,39 - 7,46 (m, 3 H), 7,80 (d, J=9,1 Hz, 1 H), 7,90 - 8,0 (m, 2 H), 8,19 (s, 1 H).
Ejemplo 204
4-(((6-metoxi-2-(6-metilimidazo[1,2-b]piridazin-2-il)benzofuran-4-il)oxi)metil)-2-(tetrahidro-2H-tiopiran-4-il)tiazol

Una mezcla de 6-metoxi-2-(6-metilimidazo[1,2-b]piridazin-2-il)benzofuran-4-ol (Ejemplo 203B, 0,340 g, 1,15 mmol) y (2-(tetrahidro-2H-tiopiran-4-il)tiazol-4-il)metanol (0,266 g, 1,24 mmol) en un matraz de fondo redondo, de 100 ml equipado con un embudo adicional se mantuvo al vacío durante 5 min. El matraz después se purgó con nitrógeno y se cargó con tetrahidrofurano seco (30 ml), seguido de tributilfosfina (0,74 ml, 3,0 mmol), añadida en una porción. La mezcla heterogénea se trató a 22 °C con una solución de 1,1'-(azodicarbonil)-dipiperidina (0,43 g, 1,70 mmol) en tetrahidrofurano ( 20 ml), añadido gota a gota durante 1 h (durante algunos períodos breves, se sometió a ultrasonido). La mezcla de reacción era homogénea al final de la adición y se agitó durante 2 h más. La mezcla de
reacción se repartió entre diclorometano (250 ml) y bicarbonato de sodio saturado. La fase orgánica se lavó con salmuera, se secó sobre sulfato de magnesio anhidro y se concentró al vacío para obtener un residuo vítreo. El residuo se sometió a cromatografía sobre gel de sílice (elución de 0 - 30 % de acetato de etilo-diclorometano) para obtener 0,420 g ( 74 % de rendimiento ) del material del título en forma de un sólido de color amarillo claro. LC (Método A): 2,561 min. HRMS (ESI): calc. para C25H25N4O3S2 [M H]+ m/z 493,1368, encontrado 493,1388. RMN 1H (400 MHz, DMSO-da) 8 ppm: 8,53 (s, 1H), 8,01 (d, J= 9,4 Hz, 1H), 7,73 (s, 1H), 7,26 (s, 1H), 7,18 (d, J= 9,4 Hz, 1H), 6 ,88 (a d, 1H), 6,63 (d, J= 2,0 Hz, 1H), 5,28 (s, 2H), 3,83 (s, 3H), 3,10-3,18 (m, 1H), 2,75-2,85 (m, 2H), 2,65-2,72 (m, 2H), 2,5 (s, 3H), 2,28-2,43 (m, 2H), 1,70-1,90 (m, 2H).
Ejemplo 205
2-bromo-4-(((6-metoxi-2-(6-metilimidazo[1,2-b]piridazin-2-il)benzofuran-4-il)oxi)metil)tiazol

205A. (2-bromotiazol-4-il)metanol

A una solución de 2-bromotiazol-4-carboxilato de metilo (1,00 g, 4,50 mmol) en etanol (15 ml) a 0 oC, se le agregó NaBH4 (1,022 g, 27,0 mmol). El baño de enfriamiento se retiró y la mezcla de reacción se agitó a temperatura ambiente durante 15 min y después a 90 °C durante 1,5 h. La mezcla de reacción enfriada se trató con ácido acético (5 ml) y después la mezcla se evaporó a sequedad a presión reducida. El residuo obtenido se sometió a cromatografía sobre gel de sílice (ISCO, gradiente de elución de acetato de etilo en diclorometano) para obtener el material del título (0,571 g, 65 %) en forma de un aceite incoloro. LC (Método A): 1,101 min. LRMS (ApCI): calc. para C4HsBrNOS [M+H]+ m/z: 193,93; encontrado 193,90. RMN 1H (400 MHz, CDCla) 8 ppm: 7,18 (d, J= 0,78 Hz, 1H), 4,76 (d, J= 1,00 Hz, 2H), 2,28 - 2,71 (m, 1H).
205B. 2-bromo-4-(bromometil)tiazol

Una solución de (2-bromotiazol-4-il)metanol (0,571 g, 2,94 mmol) en diclorometano (8 ml) se enfrió a 0 °C y se trató con PBr3 (0,128 ml, 1,359 mmol) gota a gota durante 2 min. Después de 5 min, el baño de hielo se retiró y la solución se agitó a 22 °C durante 4 h. La mezcla de reacción se vertió en una mezcla de acetato de etilo y bicarbonato de sodio acuoso saturado, la fase orgánica se separó, se lavó con salmuera, se secó sobre sulfato de magnesio anhidro y se concentró al vacío. El residuo obtenido se sometió a cromatografía sobre gel de sílice (ISCO, gradiente de elución de diclorometano en hexanos) para obtener 0,550 g (72 %) del material del título en forma de un aceite incoloro. LC (Método B): 1,813 min. LRMS (APCI): calc. para C4H4B^Ns [M+H]+ m/z: 255,84; encontrado: 255,80. RMN 1H (400 MHz, CDCla) 8 ppm: 7,27 (s, 1H, debajo de CHCla), 4,54 (s, 2H).
Ejemplo 205. 2-bromo-4-(((6-metoxi-2-(6-metilimidazo[1,2-b]piridazin-2-il)benzofuran-4-il)oxi)metil)tiazol

Una mezcla de 6-metoxi-2-(6-metilimidazo[1,2-b]piridazin-2-il)benzofuran-4-ol (Ejemplo 203B, 0,208 g, 0,704 mmol) y 2-bromo-4-(bromometil)tiazol (0,181 g, 0,704 mmol) en DMF (5 ml) se mantuvo al vacío durante 5 min, después el matraz se purgó con nitrógeno y se agregó, de una sola vez, carbonato de potasio anhidro en polvo (0,291 g, 2,10 mmol). La mezcla resultante se agitó a temperatura ambiente durante 1,5 h, después se inactivó con ácido clorhídrico 1 N (1 ml) y finalmente se repartió entre diclorometano y bicarbonato de sodio acuoso saturado. La fase
orgánica se separó, se lavó con salmuera, se secó sobre sulfato de magnesio anhidro y se concentró al vacío. El residuo obtenido se sometió a cromatografía sobre gel de sílice (ISCO, gradiente de elución de acetato de etilo en diclorometano) para obtener 0,220 g (66 %) del material del título en forma de un sólido. LC (Método A): 2,267 min. HRMS (ESI) calc. para C20Hi6BrN4OaS [M+H]+ m/z: 471,0126; encontrado 471,0121. RMN 1H (400 MHz, CDCla) 8 ppm: 8,18 (s, 1 H), 7,82 (d, J= 9,39 Hz, 1H), 7,31 (s, 1H), 7,34 (s, 1H), 6,93 (d, J= 9,00 Hz, 1H), 6,74-6,79 (m, 1H), 6,40 (d, J= 1,96 Hz, 1H), 5,27-5,34 (m, 2H), 3,86 (s, 3H), 2,59 (s, 3H).
Ejemplo 206. 2-cloro-4-(((6-metoxi-2-(6-metilimidazo[1,2-b]piridazin-2-il)benzofuran-4-il)oxi)metil)tiazol

El compuesto del título se preparó usando 2-cloro-4-(bromometil)tiazol, de acuerdo con el método descrito anteriormente en el ejemplo 205. LC (Método A): 2,314 min. HRMS (ESI) calc. para C20H-i6ClN4OaS [M+H]+ m/z 427,0632; encontrado 427,0611. RMN 1H (400 MHz, CDCla) 8 ppm: 8,19 (s, 1H), 7,82 (d, J= 9,39 Hz, 1H), 7,31 (s, 1H), 7,29 (s, 1H), 6,94 (d, J= 9,39 Hz, 1H), 6,77 (s, 1H), 6,40 (s, 1H), 5,27 (s, 2H), 3,86 (s, 3H), 2,59 (s, 3H).
Ejemplo 207
4-(((6-metoxi-2-(6-metilimidazo[1,2-b]piridazin-2-il)benzofuran-4-il)oxi)metil)-2-(4-metilpiperazin-1-il)tiazol
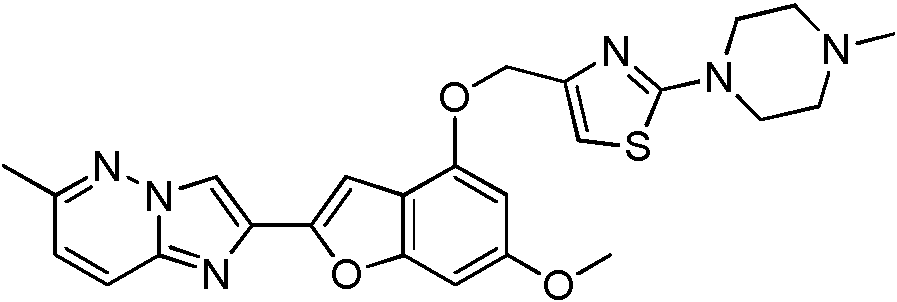
En un tubo sellado, 2-bromo-4-(((6-metoxi-2-(6-metilimidazo[1,2-b]piridazin-2-il)benzofuran-4-il)oxi)metil)tiazol (Ejemplo 205, 0,025 g, 0,053 mmol) en tetrahidrofurano (2 ml) se trató con 1-metilpiperazina (8,85 pl, 0,080 mmol) y la mezcla resultante se calentó a 80 °C durante 16 h. La mezcla de reacción resultante se inactivó con ácido clorhídrico 1 N (0,5 ml) y después se repartió entre diclorometano y bicarbonato de sodio acuoso saturado. La fase orgánica se separó, se lavó con salmuera, se secó sobre sulfato de magnesio anhidro y se concentró al vacío. El residuo obtenido se sometió a cromatografía sobre gel de sílice (ISCO, gradiente de elución de acetato de etilo en diclorometano) y el material obtenido se trituró con acetato de etilo para obtener (después de la filtración y de secar al vacío) 0,016 g (60 %) del compuesto del título en forma de un sólido. LC (Método A): 1,955 min. HRMS (ESI) calc. para C25H27N6O3S [M+H]+m/z: 491,1865; encontrado 491,1876. RMN 1H (400 MHz, CDCh) 8 ppm: 8,19 (s, 1H), 7,82 (d, J= 9,39 Hz, 1H), 7,34 (s, 1H), 6,94 (d, J= 9,39 Hz, 1H), 6,75 (s, 1H), 6,63 (s, 1H), 6,44 (s, 1H), 5,13 (s, 2H), 3,86 (s, 3H), 3,54 (t, J= 1,00 Hz, 4H), 2,59 (s, 3H), 2,54 (t, J= 4,89 Hz, 4H), 2,36 (s, 3H).
Ejemplo 208
4-(((6-metoxi-2-(6-metilimidazo[1,2-b]piridazin-2-il)benzofuran-4-il)oxi) metil)-2-(4-(trifluorometil)fenil)tiazol

208A. 2-(4-(trifluorometil)fenil)tiazol-4-carboxilato de etilo

Una solución de 4-(trifluorometil)benzotioamida (3,00 g, 14,62 mmol) en THF (45 ml) se trató a 22 °C con una solución de 3-bromo-2-oxopropanoato de etilo (2,202 ml, 17,54 mmol) en THF (5 ml), añadido gota a gota durante 5 min. La mezcla resultante se agitó a temperatura ambiente durante 30 min y después se calentó a reflujo durante 18 h. La mezcla enfriada se diluyó con acetato de etilo (200 ml), se lavó con bicarbonato de sodio saturado y salmuera, se secó sobre sulfato de magnesio anhidro y se evaporó. El residuo se purificó en ISCO usando una columna REDISEP® de 80 g (30 a 100 % de DCM-hexanos) para obtener el producto deseado en forma de un sólido de color amarillo (2,52 g, 57 %). LCMS (APCI): calc. para C13H11 F3NO2S [M+H]+ m/z 302,05, encontrado 302,0. RMN 1H (CDCla, 400 MHz) 8 ppm: 8,23 (s, 1H), 8,14 (d, J = 8,4 Hz, 2H), 7,73 (d, J = 8,4 Hz, 2H), 4,47 (q, J = 7,2 Hz, 2H), 1,45 (t, J = 7,2 Hz, 3H).
208B. (2-(4-(trifluorometil)fenil)tiazol-4-il)metanol
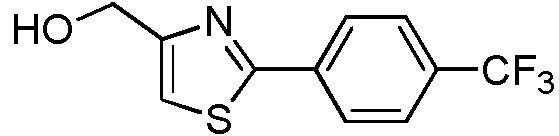
Una solución de 2-(4-(trifluorometil)fenil)tiazol-4-carboxilato de etilo (2,00 g, 6,64 mmol) en dietiléter seco (120 ml) se enfrió a -78 °C en atmósfera de nitrógeno, después se trató con LiAlH4 sólido (0,756 g, 19,9 mmol) por porciones durante 5 min. Después de 3 h a -78 °C, la mezcla de reacción se inactivó mediante la adición gota a gota de acetato de etilo (10 ml) durante 5 min. Después de 10 min, se agregó agua (1 ml) gota a gota durante 10 min, después una solución acuosa al 15 % de NaOH ( 1 ml) y finalmente, se agregó más agua (2 , 1 ml). Después se retiró el baño de enfriamiento y la mezcla heterogénea se agitó a temperatura ambiente durante 30 min para obtener una suspensión de color blanco. La suspensión se filtró y después la torta de filtro se lavó con éter (200 ml). El filtrado combinado se lavó con salmuera, se secó sobre sulfato de magnesio anhidro y se evaporó para obtener el producto deseado en forma de un sólido de color blanco (1,46 g, 85 %). LCMS (APCI): calc. para C11 H9F3NOS [M+H]+ m/z 260,028, encontrado 260,0. RMN 1 H (CDCh, 400 MHz) 8 ppm: 8,07 (d, J = 8,2 Hz, 2H), 7,71 (d, J = 8,2 Hz, 2H), 7,28 (s, 1H), 4,87 (d, J = 5,5 Hz, 2H), 2,31 (t, J = 5,5 Hz, 1H).
208C. 4-(bromometil)-2-(4-(trifluorometil)fenil)tiazol

A una solución en agitación de (2-(4-(trifluorometil)fenil)tiazol-4-il)metanol (0,500 g, 1,93 mmol) en DCM seco (20 ml), en atmósfera de nitrógeno, se le agregó tribromuro de fósforo (0,37 ml, 3,86 mmol) y la mezcla de reacción se agitó a temperatura ambiente durante 16 h. Después de que se completó, la mezcla se concentró y el concentrado se repartió entre bicarbonato de sodio acuoso saturado y DCM. La fase orgánica se separó y la fase acuosa se volvió a extraer dos veces con DCM. Las capas orgánicas combinadas se lavaron con salmuera, se secaron sobre MgSO4 y se concentraron. El residuo se purificó en ISCO usando una columna REDISEP® de 40 g (20 a 100 % de DCM-hexanos) para obtener el producto deseado en forma de un sólido de color blanco (0,196 g, 32 %). LCMS (APCI): calc. para C-nHyB^NS [M+H]+ m/z 321,94, encontrado 321,9. RMN 1H (CDCh, 400 MHz) 8 ppm: 8,05-8,11 (m, 2H), 7,68-7,75 (m, 2H), 7,39 (s, 1H), 4,65 (s, 2H).
Ejemplo 208. 4-(((6-metoxi-2-(6-metilimidazo[1,2-b]piridazin-2-il) benzofuran-4-il)oxi)metil)-2-(4-(trifluorometil)fenil)tiazol

Una mezcla de 4-metoxi-2-(6-metilimidazo[1,2-b]piridazin-2-il)benzofuran-6-ol (Ejemplo 203B, 0,060 g, 0,20 mmol) y 4-(bromometil)-2-(4-(trifluorometil)fenil)-tiazol (0,072 g, 0,22 mmol) en DMF (8 ml) se trató con K2CO3 (0,076 g, 0,55 mmol) y la mezcla homogénea resultante se agitó a 23 °C durante 1 h para obtener una suspensión. Después de 3 h más, la mezcla de reacción se diluyó con diclorometano (100 ml), se lavó con agua y salmuera, se secó sobre sulfato de magnesio anhidro y se evaporó. El polvo blanquecino resultante se suspendió en acetonitrilo, se sometió a ultrasonido, se filtró y se secó para obtener el compuesto del título en forma de un sólido blanquecino (0,090 g, 83%). LC (Método F): 2,453 min. LCMS (APCI): calc. para C27H20F3N4O3S [M+H]+ m/z 537,11, encontrado 537,1.
RMN 1H (CDCla, 400 MHz) 5 ppm: 8,20 (s, 1H), 8,07 - 8,13 (m, J = 8,2 Hz, 2H), 7,83 (d, J = 9,4 Hz, 1H), 7,70 - 7,75 (m, J = 8,2 Hz, 2H), 7,48 (s, 1H), 7,36 (s, 1H), 6,95 (d, J = 9,4 Hz, 1H), 6,78 (dd, J = 2,0, 0,8 Hz, 1H), 6,49 (d, J = 2,0 Hz, 1H), 5,39 - 5,45 (m, 2H), 3,87 (s, 3H), 2,60 (s, 3H).
Ejemplo 209
2-(4-dorofenil)-4-(((6-metoxi-2-(6-metilimidazo[1,2-b]piridazin-2-il)benzofuran-4-il)oxi)metil)tiazol

209A. 2-(4-clorofenil)tiazol-4-carboxilato de etilo

El compuesto se preparó de acuerdo con el método descrito en el ejemplo 208A. El material crudo se purificó en ISCO usando una columna REDISEP® de 120 g (30 a 100 % de DCM-hexanos) para obtener el producto deseado en forma de un sólido de color amarillo (3,18 g, 51 %). LCMS (APCI): calc. para C12H-nClNO2S [M+H]+ m/z 268,01, encontrado 268,0. RMN 1H (CDCla, 400 MHz) 5 ppm: 8,17 (s, 1H), 7,96 (dd, J = 8 ,8 , 4,8 Hz, 2H), 7,44 (dd, J = 9,2, 4,8 Hz, 2H), 4,46 (q, J = 7,0 Hz, 2H), 1,44 (t, J = 7,1 Hz, 3H).
209B. (2-(4-clorofenil)tiazol-4-il)metanol

El compuesto se preparó de acuerdo con el método descrito en el ejemplo 208B. El producto deseado se aisló en forma de un sólido de color amarillo (2,51 g, 93 %). LCMS (APCl): calc. para C10HgClNOS [M+H]+ m/z 226,00, encontrado 226,0. RMN 1H (CDCls, 400 MHz) 5 ppm: 7,87 - 7,91 (m, 2H), 7,39 - 7,45 (m, 2H), 7,21 (t, J = 1,0 Hz, 1H), 4,84 (d, J = 5,7 Hz, 2H), 2,31 (t, J = 5,7 Hz, 1H).
209C. 4-(bromometil)-2-(4-clorofenil)tiazol

El compuesto se preparó de acuerdo con el método descrito en el ejemplo 208C. El producto deseado se aisló en forma de un sólido de color blanco (0,320 g, 83 %). LCMS (APCI): calc. para C10HsBrClNOS [M+H]+ m/z 287,92, encontrado 287,9. RMN 1H (CDCls, 400 MHz) 5 ppm: 7,90 (d, J = 8,0 Hz, 2H), 7,43 (d, J = 8,0 Hz, 2H), 7,32 (s, 1H), 4,63 (s, 2H).
Ejemplo 209. 2-(4-clorofenil)-4-(((6-metoxi-2-(6-metilimidazo[1,2-b]piridazin-2-il)benzofuran-4-il)oxi)metil)tiazol
El compuesto se preparó de acuerdo con el método descrito en el ejemplo 208. El material crudo se purificó en ISCO usando una columna REDISEP® de 12 g (20 a 100 % de EtOAc-DCM) y el polvo amarillo obtenido se suspendió en acetonitrilo, se sometió a ultrasonido, se filtró y se secó para obtener el compuesto del título en forma de un sólido de color amarillo claro (0,087 g, 85 %). LC (Método A): 2,569 min. LCMS (a Pc I): calc. para C26H20ClN4O3S [M+H]+ m/z 503,09, encontrado 503,1. RMN 1H (CDCh, 400 MHz) 8 ppm: 8,20 (s, 1H), 7,92 (d, J = 8,8 Hz, 2H), 7,83 (d, J = 9,4 Hz, 1H), 7,44 (d, J = 8,8 Hz, 2H), 7,40 (s, 1H), 7,36 (s, 1H), 6,95 (d, J = 9,4 Hz, 1H), 6,78 (dd, J = 2,0, 0,8 Hz, 1H), 6,48 (d, J = 2,0 Hz, 1H), 5,40 (s, 2H), 3,87 (s, 3H), 2,60 (s, 3H).
Ejemplo 210
4-(((6-metoxi-2-(6-metilimidazo[1,2-b]piridazin-2-il)benzofuran-4-il)oxi)metil) -2-(4-(trifluorometoxi)fenil)tiazol

210A. 2-(4-(trifluorometoxi)fenil)tiazol-4-carboxilato de etilo

El compuesto se preparó de acuerdo con el método descrito en el ejemplo 208A. El residuo se purificó en ISCO usando una columna REDISEP® de 80 g (30 a 100 % de DCM-hexanos) para obtener el producto deseado en forma de un sólido de color blanco (3,75 g, 87 %). LCMS (APCI): calc. para C13H11 F3NO3S [M+H]+ m/z 318,03, encontrado 318,0. RMN 1H (CDCla, 400 MHz) 8 ppm: 8,18 (s, 1H), 8,06 (d, J = 9,0 Hz, 2H), 7,31 (d, J = 9,0, 2H), 4,46 (q, J = 7,0 Hz, 2H), 1,44 (t, J = 7,0 Hz, 3H).
210B. (2-(4-(trifluorometoxi)fenil)tiazol-4-il)metanol

El compuesto se preparó de acuerdo con el método descrito en el ejemplo 208B. El producto deseado se aisló en forma de un sólido de color blanco (2,63 g, 82 %). LCMS (APCI): calc. para C11 H9F3NO2S [M+H]+ m/z 276,02, encontrado 276,0. RMN 1H (CDCla, 400 MHz) 8 ppm: (d, J = 8,1, 2H), 7,29 (d, J = 8,1 Hz, 2H), 7,22 (s, 1H), 4,84 (d, J = 5,5 Hz, 2H), 2,47 (t, J = 5,5 Hz, 1H).
210C. 4-(bromometil)-2-(4-(trifluorometoxi)fenil)tiazol

El compuesto se preparó de acuerdo con el método descrito en el ejemplo 208C. El residuo se purificó en ISCO usando una columna REDISEP® de 40 g (50 a 100 % de DCM-hexanos) para obtener el compuesto del título en forma de un sólido de color blanco (0,078 g, 22 %). LCMS (APCI): calc. para C^HaB^NOS [M+H]+ m/z 337,94, encontrado 338,0. RMN 1H (CDCh, 400 MHz) 8 ppm: 8,00 (d, J = 9,0, 2H), 7,33 (s, 1H), 7,30 (d, J = 9,0, 2H), 4,64 (s, 2H).
Ejemplo 210. 4-(((6-metoxi-2-(6-metilimidazo[1,2-b]piridazin-2-il)benzofuran-4-il)oxi)metil) -2-(4-(trifluorometoxi)fenil)tiazol

El compuesto se preparó de acuerdo con el método descrito en el ejemplo 208. El residuo se purificó en ISCO usando una columna REDISEP® de 12 g (10 a 80 % de EtOAc-DCM) y el polvo de color amarillo obtenido se suspendió en acetonitrilo, se sometió a ultrasonido y se filtró para obtener el compuesto del título en forma de un sólido de color amarillo (0,079 g, 71 %). LC (Método A): 2,544 min. LCMS (APCI): calc. para C27H20F3N4O4S [M+H]+ m/z 553,11, encontrado 553,1. RMN 1H (CDCla, 400 MHz) 8 ppm: 8,20 (s, 1H), 8,02 (d, J = 9,0 Hz, 2H), 7,83 (d, J = 9,2 Hz, 1H), 7,42 (s, 1H), 7,36 (s, 1H), 7,31 (d, J = 9,0 Hz, 2H), 6,95 (d, J = 9,2 Hz, 1H), 6,78 (s, 1H), 6,49 (d, J = 2,0 Hz, 1H), 5,41 (s, 2H), 3,87 (s, 3H), 2,60 (s, 3H).
Ejemplo 211
4-(((6-metoxi-2-(6-metilimidazo[1,2-b]piridazin-2-il)benzofuran-4-il)oxi)metil)-2-(piridin-4-il)tiazol

211A. 2-(piridin-4-il)tiazol-4-carboxilato de etilo

El compuesto se preparó de acuerdo con el método descrito en el ejemplo 208A. El residuo se purificó en ISCO usando una columna REDISEP® de 120 g (30 a 80 % de EtOAc-hexanos) para obtener el producto deseado en forma de un aceite de color rojo oscuro (2 , 1 8 g, 43 %). LCMS (APCI): calc. para C11 H11 N2O2S [M+H]+ m/z 235,05, encontrado 235,1. RMN 1H (CDCla, 400 MHz) 8 ppm: 8,71 - 8,78 (m, 2H), 8,28 (d, J = 0,8 Hz, 1H), 7,85 - 7,91 (m, 2H), 4,43 - 4,51 (m, 2H), 1,41 -1,48 (m, 3H).
211B. (2-(piridin-4-il)tiazol-4-il)metanol

Una solución de 2-(piridin-4-il)tiazol-4-carboxilato de etilo (2,18 g, 9,31 mmol) en EtOH (70 ml) en un matraz de 250 ml en atmósfera de nitrógeno se enfrió a 0 °C y se trató con NaBH4 (2,11 g, 55,8 mmol), añadido en porciones pequeñas durante 10 min. Después de 10 min a 0 °C, el baño de enfriamiento se retiró y la mezcla de reacción se agitó a 70 °C durante 1 h. La mezcla enfriada se inactivó después con NH4Cl acuoso saturado (15 ml) y se agitó durante 20 min más antes de extraerse con acetato de etilo (100 ml). La fase orgánica se lavó con salmuera, se secó sobre MgSO4 y se concentró para obtener un sólido de color tostado que se trituró en Et2O para obtener el producto en forma de un polvo de color blanco (1,20 g, 67 %). LCMS (APCI): calc. para C9H9N2OS [M+H]+ m/z 193,04, encontrado 193,1. RMN 1 H (DMSO-d6, 400 MHz) 8 ppm: 8,69 (d, J = 4,4, 2H), 7,85 (d, J = 4,4, 2H), 7,66 (d, J = 1,1 Hz, 1H), 5,47 (t, J = 5,7 Hz, 1H), 4,66 (dd, J = 5,7, 1,1 Hz, 2H).
Ejemplo 211. 4-(((6-metoxi-2-(6-metilimidazo[1,2-b]piridazin-2-il)benzofuran-4-il)oxi)metil) -2-(piridin-4-il)tiazol
El compuesto del título se preparó de acuerdo con el método general descrito en el ejemplo 36. El producto en bruto se purificó en ISCO usando una columna REDISEP® de 12 g (0 a 15 % de MeOH-DCM) y el polvo de color amarillo obtenido se suspendió en CH3CN, se sometió a ultrasonido, se filtró y se secó para obtener el compuesto del título en forma de un sólido de color amarillo claro (0,079 g, 71 %). LC (Método A): 2,392 min. LCMS (APCI): calc. para C25H20N5O3S [M+H]+ m/z 470,12, encontrado 470,2. RMN 1 H (CDCla, 400 MHz) 8 ppm: 8,70-8,79 (m, 2H), 8,20 (s, 1H), 7,81-7,87 (m, 3H), 7,54 (s, 1H), 7,36 (s, 1H), 6,95 (d, J = 9,4 Hz, 1H), 6,78 (s, 1H), 6,48 (d, J = 1,6 Hz, 1H), 5,43 (s, 2H), 3,87 (s, 3H), 2,60 (s, 3H).
Ejemplo 212
2-(4-fluorotetrahidro-2H-piran-4-il)-4-(((6-metoxi-2-(6-metilimidazo[1,2-b]piridazin-2-il)benzofuran-4-il)oxi)metil)tiazol

A una solución de 6-metoxi-2-(6-metilimidazo[1,2-b]piridazin-2-il)benzofuran-4-ol (Ejemplo 203B, 0,0285 g, 0,096 mmol) y 4-(bromometil)-2-(4-fluorotetrahidro-2H-piran-4-il)tiazol (Ejemplo 119C, 0,027 g, 0,096 mmol) en DMF (2 ml), en nitrógeno, se le agregó carbonato de potasio (0,0306 g, 0,222 mmol) y la mezcla de reacción resultante se agitó a temperatura ambiente durante 2,5 h. La mezcla de reacción en bruto se diluyó con diclorometano, se lavó con agua y salmuera, se secó sobre MgSO4, se filtró y se concentró. El residuo en bruto obtenido se purificó mediante cromatografía en columna (Isco, cartucho de 12 g) eluyendo con un gradiente de acetato de etilo en diclorometano (de 0 a 100 %). El producto obtenido de la cromatografía en columna se trituró en etanol y el sólido se recolectó mediante filtración y se secó al vacío para obtener el compuesto del título puro (0,034 g, 71 %). LC (Método A): 2,306 min. HRMS (ESI): calc. para C25H24FN4O4S [M+H]+ m/z 495,1502, encontrado 495,1797. RMN 1 H (DMSO-d6, 400 MHz) 8 ppm: 2,03-2,15 (m, 2H), 2,18-2,40 (m, 2H), 2,54 (s, 3H), 3,65-3,76 (m, 2H), 3,81-3,89 (m, 2H), 3,83 (s, 3H), 5,34 (s, 2H), 6,65 (d, J= 2,0 Hz, 1H), 6,89-6,91 (m, 1H), 7,19 (d, J= 9,4 Hz, 1H), 7,28 (s, 1H), 7,97 (s, 1H), 8,01 (d, J= 9,0 Hz, 1H), 8,54 (s, 1H).
Ejemplos 213 a 242
Los siguientes ejemplos se prepararon, se aislaron y se caracterizaron de acuerdo con los métodos descritos anteriormente.









Los Ejemplos 243 a 286 se prepararon de acuerdo con el siguiente procedimiento:

2-bromo-4-(((6-metoxi-2-(6-metilimidazo[1,2-b]piridazin-2-il)benzofuran-4-il)oxi)metil)tiazol (Ejemplo 205, 15 mg, 0,031 mmol) en dioxano (0,6 ml) se agregó en un tubo Wheaton (16 x 100 mm) que contiene una amina (0,108 mmol) y una barra agitadora. Después, a la solución se le agregó butóxido de potasio (8,1 mg, 72 pmol) y aducto de metil-t-butiléter de paladio-cloro(2-diciclohexilfosfino-2',4',6'-tri-i-propil-1,1'-bifenil)[2-(2-aminoetil)fenil] paladio (II) (2,1 mg, 2,9 pmol). Todos los tubos se taparon y se calentaron a 105 °C en un bloque de calentamiento durante la noche. Todas las muestras se secaron mediante un flujo de nitrógeno, se disolvieron en DMF (1,0 ml), se filtraron con un filtro Whatman 0,45 um PVDF y se purificaron mediante HPLC preparativa.
Los Ejemplos 287 a 317 se prepararon de acuerdo con el siguiente procedimiento:

Se agregó HATU (9,71 mg, 26 umol) y DIPEA (14 ul, 79 umol) a una solución de ácido 4-(4-(((6-metoxi-2-(2-metoxiimidazo[2,1-b][1,3,4]tiadiazol-6-il)benzofuran-4-il)oxi)metil)tiazol-2-il)benzoico (Ejemplo 39D, 10,5 mg, 20 umol) en DMF (0,6 ml) en un tubo Wheaton (16 x 100 mm). La mezcla se agitó a temperatura ambiente durante 5 minutos y después se agregó la solución de la amina (200 pmol) en DMF y el tubo se tapó y se agitó durante 2 horas a temperatura ambiente. La mezcla de reacción en bruto se purificó mediante HPLC preparativa.
En las estructuras establecidas a continuación para los Ejemplos 243 a 317, el “-O ” unido a un átomo de carbono se usa para indicar un grupo “-OH”. De manera similar, en las estructuras establecidas a continuación para los Ejemplos 243 a 317, la “N” unida a un átomo de carbono se usa para indicar un resto “NH”.

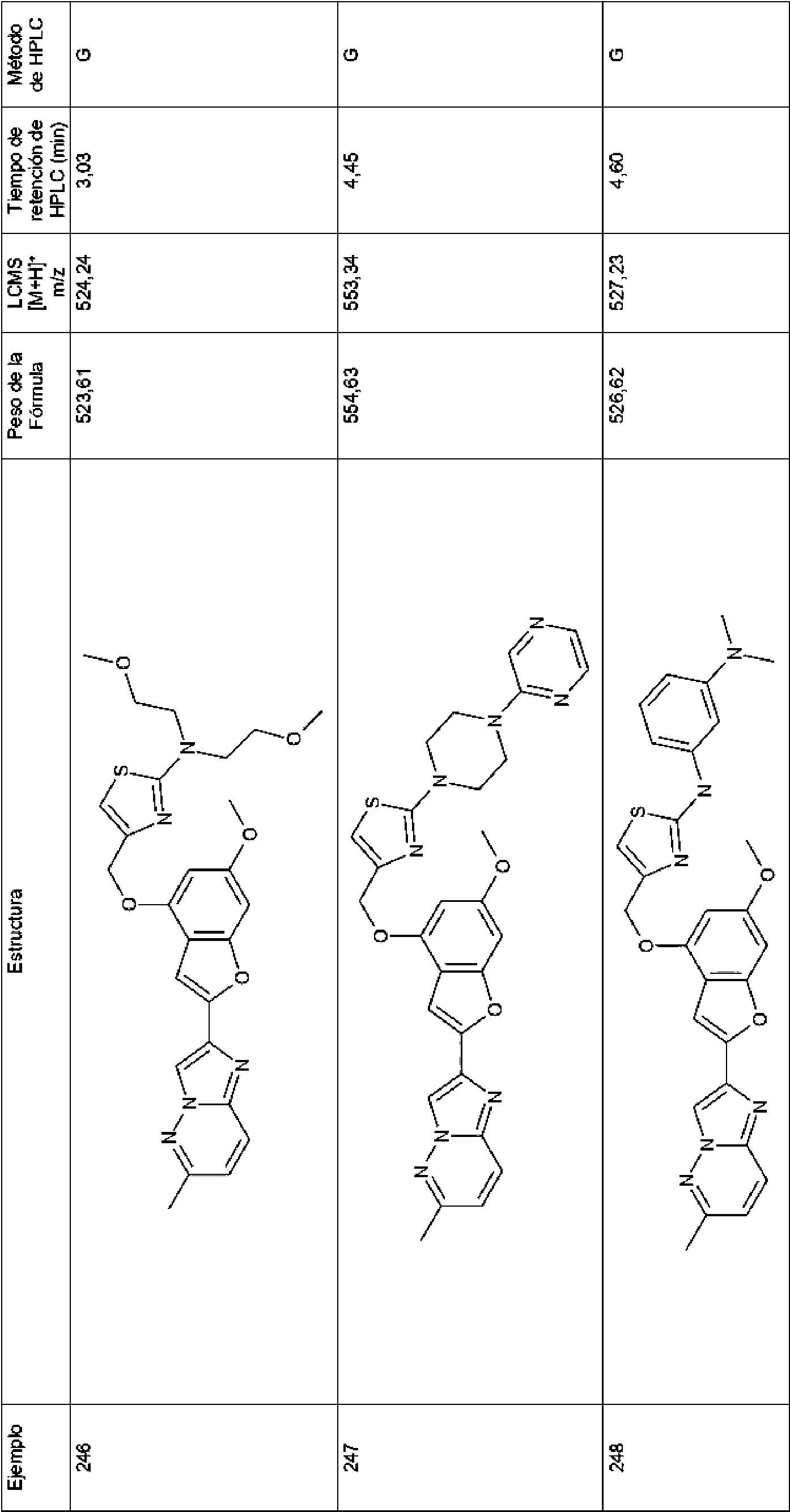





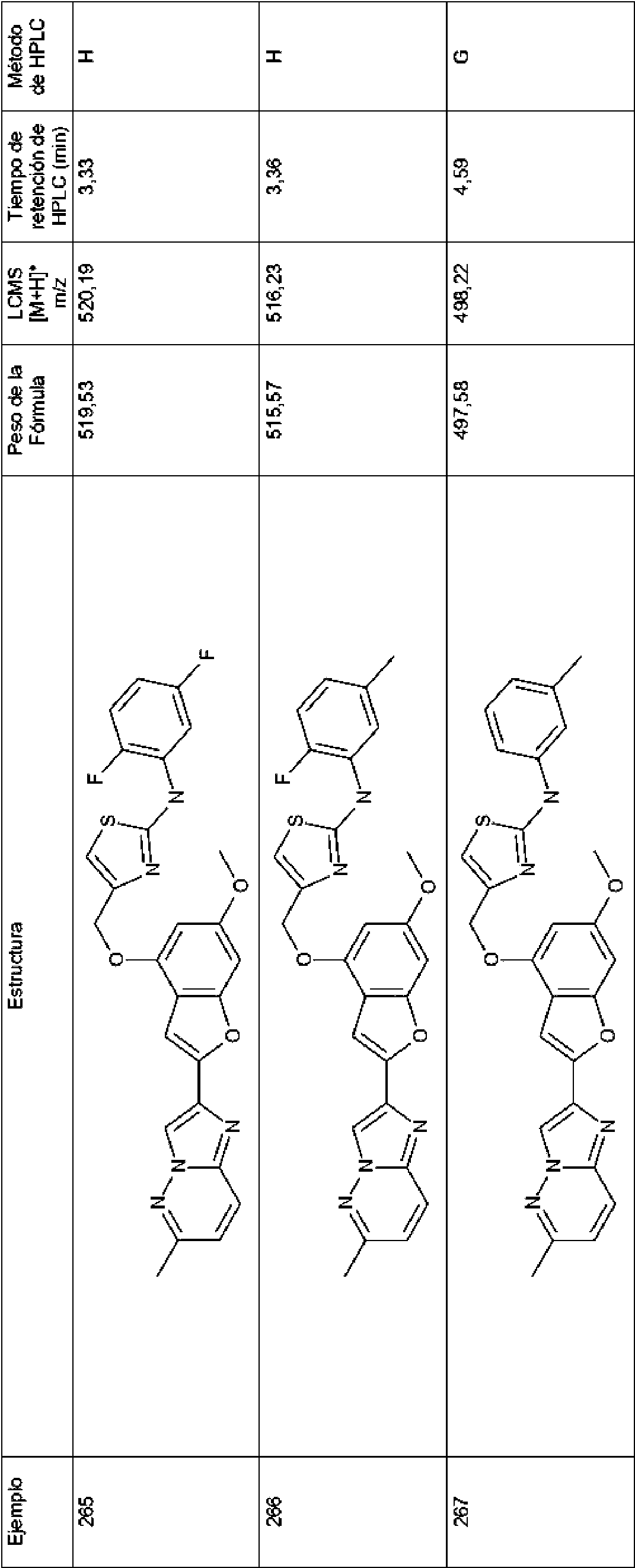












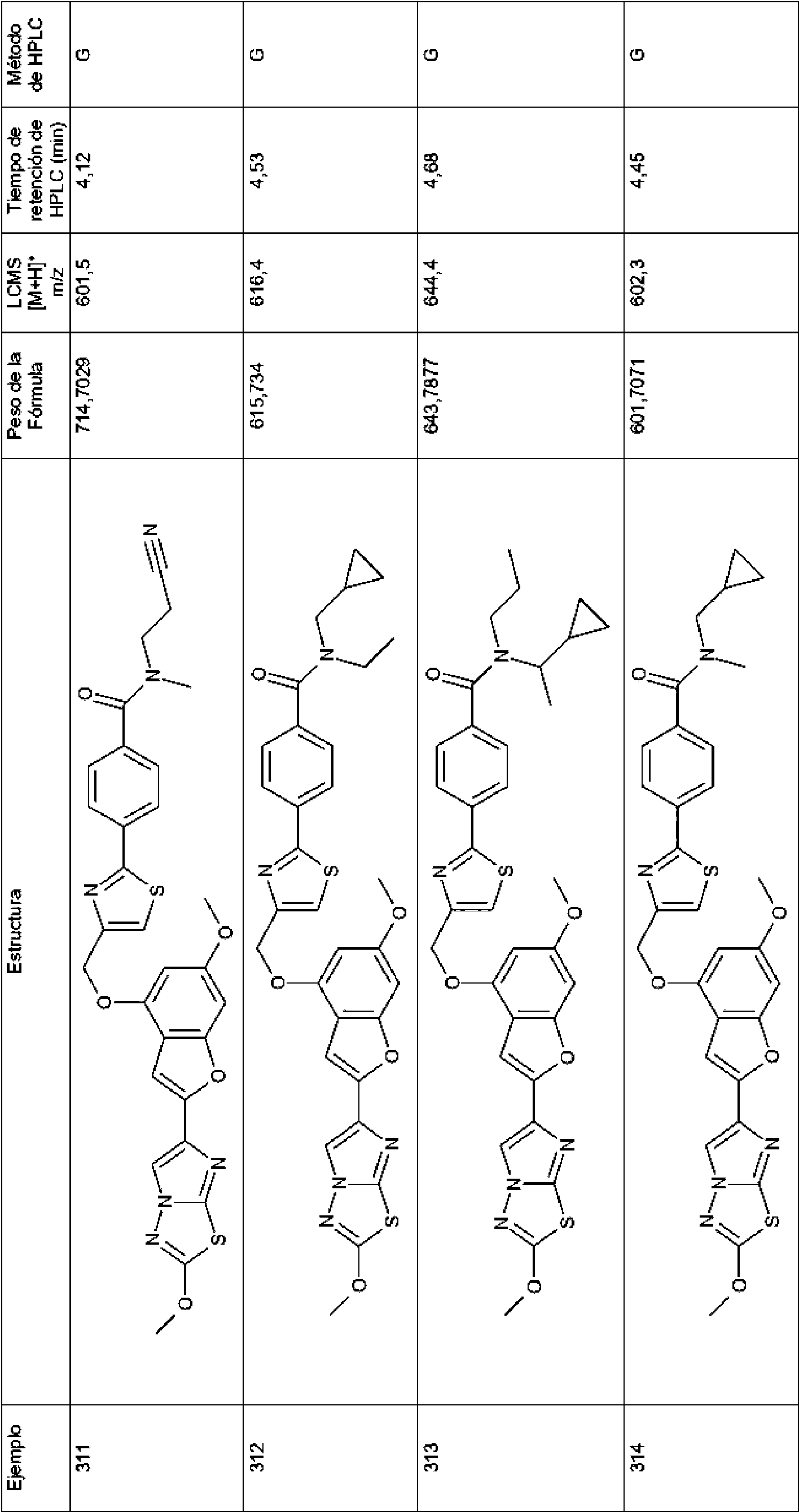

Ejemplo 318
2-metoxi-6-(6-metoxi-4-((2-feniltiazol-4-il)metoxi)benzofuran-2-il)imidazo[2,1-b][1,3,4]tiadiazol etiquetado como tritio

Una matriz Trisorber con una barra agitadora se cargó con 2-metoxi-6-(6-metoxi-4-((2-feniltiazol-4-il)metoxi)benzofuran-2-il)imidazo[2,1-b][1,3,4]tiadiazol (Ejemplo 3, 0,50 mg, 1,02 |jmol) y catalizador de Crabtree (hexafluoridofosfato de ((SP-4)tris(ciclohexil)fosfan[(1-2-r|:5-6-r|)-cicloocta-1,5-dien]piridiniridio) (2,3 mg, 2,86 jmol). A esto se le agregó CH2Cl2 (0,50 ml) mediante jeringa. El matraz se unió al Trisorber y se sometió a ciclos de congelamiento, bombeo y descongelamiento para retirar los gases disueltos. A la mezcla de reacción se le agregó 1,0 Ci de gas tritio y la mezcla se agitó a temperatura ambiente. Después de 18 h, el CH2Cl2 se retiró mediante un evaporador giratorio. El producto en bruto se disolvió en etanol y el tritio lábil se intercambió cuando se retiró el etanol mediante el evaporador giratorio. Esto se repitió dos veces más. El producto en bruto se disolvió en 5,0 ml de etanol. Una alícuota de la mezcla cruda (20 j l) se diluyó en 5,0 ml de etanol. Una alícuota de 20 j l de esta solución diluida se contó mediante conteo de centelleo de líquidos e indicó que había 207 mCi de material sometido a tritio. El análisis mediante HPLC (AGILENT® 1100 HPLC con un detector radioquímico BetaRam, columna = PHENOMENEX® Luna 5 um C18(2), 250 X 4,6 mm, fase móvil A= 100 agua: 1 TFA, fase móvil B = 1000 acetonitrilo: 1 TFA. Gradiente = 0 min 90 % de B, 8 min 90 % de B, 15 min 100 % de B, caudal = 1,0 ml/min) indicó que la mezcla del producto en bruto contenía un 8,5 % del producto deseado (tiempo de retención = 6,4 min). El producto en bruto se purificó mediante HPLC semipreparativa (AGILENT® 1100 HPLC, Columna = PHENOMENEX® Luna, 5 um C18(2), 10 X 250 mm, Fase móvil A = 700 CH3CN : 300 agua con TFA al 0,1 %, fase móvil B = MeOH, Gradiente = 0 min 100 % de A, 25 min 100 % de A, 30 min 100 % de B, caudal = 4,0 ml/min, detección de UV a 305 nm. Las fracciones que contenían producto se recolectaron con tiempos de retención de 20,2 a 23,5 min. Las fracciones recolectadas se agruparon y el disolvente se retiró mediante evaporador giratorio. El producto resultante se disolvió en 8,5 ml de 90:10 etanol : agua para producir una solución 1,0 mCi/ml, 8,5 mCi en total. Análisis mediante HPLC/Rad (sistema AGILENT® 1100 HPLC, columna = PHENOMENEX® Luna 5 um C18(2), 4,6 X 150 mm, fase móvil A = agua 1000 : 1 TFA, fase móvil D = acetonitrilo. Gradiente = 0 min 65 % de D, 20 min 65 % de D, 25 min 100 % de D, 35 min 100 % de D, caudal = 1,2 ml/min; detección de UV a 305 nm, tiempo de retención = 11,95 min, 99,74 % radioquímicamente puro. El análisis mediante LC/MS (+ ion) indicó m/z =491/492/493/494/495/486/497/498/499. La actividad específica se determinó mediante LC/MS a través de la comparación con el análisis mediante LC/MS del ejemplo 3 no etiquetado y fue de 42,5 Ci/mmol. RMN 3H- (320 MHz, DMSO-D6) 8 8,02 (s, T), 7,09 (s, T).
BIOLOGÍA
La expresión "antagonista de PAR4" denota un inhibidor de agregación plaquetaria que fija PAR4 e inhibe la escisión y/o señalización de PAR4. En general, la actividad de PAR4 se reduce de manera dependiente de la dosis en al menos el 10 %, 20 %, 30 %, 40 %, 50 %, 60 %, 70 %, 80 %, 90 % o 100 %, en comparación con dicha actividad en una célula de control. La célula de control es una célula que no se ha tratado con el compuesto. La actividad de PAR4 se determina mediante un método estándar en la técnica, que incluye los métodos descritos en el presente documento (por ejemplo, movilización de calcio en células que expresan pAR4, agregación plaquetaria, ensayos de activación plaquetaria que miden, por ejemplo, la movilización del calcio, la liberación de p-selectina o CD40L, o modelos de trombosis y hemostasia). La expresión "antagonista de PAR4" también incluye un compuesto que inhibe PAR1 y PAR4.
Resulta conveniente encontrar compuestos con características ventajosas y mejoradas en comparación con los inhibidores plaquetarios conocidos, en una o más de las siguientes categorías que se proporcionan como ejemplo y que no pretenden ser limitantes: (a) propiedades farmacocinéticas, que incluyen la biodisponibilidad oral, la semivida y la depuración; (b) propiedades farmacéuticas; (c) requisitos de dosis; (d) factores que disminuyen las características de máximo a mínimo de la concentración en sangre; (e) factores que aumentan la concentración del fármaco activo en el receptor; (f) factores que disminuyen la responsabilidad de interacciones farmacológicas clínicas; (g) factores que disminuyen los posibles efectos secundarios adversos, que incluyen la selectividad frente a otras dianas biológicas; y (h) un índice terapéutico mejorado menos propenso al sangrado; y (h) factores que mejoran los costos de fabricación o la viabilidad.
Como se usa en el presente documento, el término "compuesto" significa una sustancia química natural o derivada artificialmente. Los compuestos pueden incluir, por ejemplo, péptidos, polipéptidos, moléculas orgánicas sintéticas, moléculas orgánicas naturales, moléculas de ácidos nucleicos, moléculas de ácidos nucleicos de péptidos y componentes y derivados de aquellos.
Como se usa en el presente documento, el término "paciente" abarca todas las especies de mamíferos.
Como se usa en el presente documento, el término "sujeto" se refiere a cualquier organismo humano o no humano que podría beneficiarse posiblemente del tratamiento con un antagonista de PAR4. Los sujetos de ejemplo incluyen seres humanos de cualquier edad con factores de riesgo para enfermedades cardiovasculares o pacientes que ya han experimentado un episodio de enfermedad cardiovascular. Los factores de riesgo comunes incluyen, pero no se limitan a, edad, sexo masculino, hipertensión, tabaquismo o antecedentes de tabaquismo, aumento de triglicéridos, aumento del colesterol total o del colesterol LDL.
En algunas formas de realización, el sujeto es una especie que tiene un repertorio de receptores plaquetarios PAR1/PAR4 dual. Como se usa en el presente documento, la expresión "repertorio de receptores plaquetarios PAR1/PAR4 dual" significa que un sujeto expresa PAR1 y PAR4 en plaquetas o sus precursores. Los sujetos de ejemplo que tienen un repertorio de receptores plaquetarios PAR1/PAR4 dual incluyen seres humanos, primates no humanos y conejillos de Indias.
En otras formas de realización, el sujeto es una especie que tiene un repertorio de receptores plaquetarios PAR3/PAR4 dual. Como se usa en el presente documento, la expresión "repertorio de receptores plaquetarios PAR3/PAR4 dual" significa que un sujeto expresa PAR3 y PAR4 en plaquetas o sus precursores. Los sujetos de ejemplo que tienen un repertorio de receptores plaquetarios PAR1/PAR4 dual incluyen roedores y conejos.
Como se usan en el presente documento, "tratar" o "tratamiento" abarcan el tratamiento de un estado de enfermedad en un mamífero, particularmente en un ser humano, e incluyen: (a) inhibir el estado de enfermedad, es decir, detener su desarrollo y/o (b) aliviar el estado de enfermedad, es decir, causar la regresión de un estado de enfermedad.
Como se usan en el presente documento, "profilaxis" o "prevención" abarcan el tratamiento preventivo de un estado de enfermedad subclínico en un mamífero, particularmente en un ser humano, con el fin de reducir la probabilidad de que se produzca un estado de enfermedad clínica. Los pacientes se seleccionan para la terapia preventiva en función de factores que se sabe aumentan el riesgo de sufrir un estado de enfermedad clínica, en comparación con la población general. Las terapias de "profilaxis" se pueden dividir en (a) prevención primaria y (b) prevención secundaria. La prevención primaria se define como el tratamiento en un sujeto que aún no presentó un estado de enfermedad clínica, mientras que la prevención secundaria se define como la prevención de una segunda ocurrencia del mismo estado de enfermedad o de uno similar.
Como se usa en el presente documento, la "reducción del riesgo" abarca terapias que disminuyen la incidencia del desarrollo de un estado de enfermedad clínica. Como tales, las terapias de prevención primaria y secundaria son ejemplos de reducción del riesgo.
"Cantidad terapéuticamente eficaz" pretende incluir una cantidad de un compuesto de la presente invención que es eficaz cuando se administra sola o en combinación para inhibir y/o antagonizar PAR4 y/o para prevenir o tratar los trastornos enumerados en el presente documento. Cuando se aplica a una combinación, la expresión se refiere a cantidades combinadas de ingredientes activos que producen el efecto preventivo o terapéutico, ya sea que se administren de manera combinada, en serie o simultánea.
Como se usa en el presente documento, el término "trombosis" se refiere a la formación o a la presencia de un trombo (pl. trombos) con un vaso sanguíneo que puede causar isquemia o infarto de los tejidos suministrados por el vaso. Como se usa en el presente documento, el término "embolia" se refiere a un bloqueo repentino de una arteria mediante un coágulo o material extraño que fue transportado hasta el sitio de incrustación por el torrente sanguíneo. Como se usa en el presente documento, el término "tromboembolia" se refiere a la obstrucción de un vaso sanguíneo con material trombótico transportado por el flujo sanguíneo desde el sitio de origen para tapar otro vaso. La expresión "trastornos tromboembólicos" implica trastornos tanto "trombóticos" como "embólicos" (definidos anteriormente).
Como se usa en el presente documento, la expresión "trastornos tromboembólicos" incluye trastornos tromboembólicos cardiovasculares arteriales, trastornos tromboembólicos cerebrovasculares o cardiovasculares venosos y trastornos tromboembólicos en las cavidades cardíacas o en la circulación periférica. Como se usa en el presente documento, la expresión "trastornos tromboembólicos" también incluye trastornos específicos seleccionados entre, pero no limitados a, angina inestable u otros síndromes coronarios agudos, fibrilación auricular, primer infarto de miocardio o infarto de miocardio recurrente, muerte súbita isquémica, accidente isquémico transitorio, apoplejía, ateroesclerosis, enfermedad arterial oclusiva periférica, flebotrombosis, flebotrombosis profunda, tromboflebitis, embolia arterial, trombosis arterial coronaria, trombosis arterial cerebral, embolia cerebral, embolia renal, embolia pulmonar y trombosis provocada por implantes, dispositivos o procedimientos médicos en los que la sangre está expuesta a una superficie artificial que promueve la trombosis. Los implantes o dispositivos médicos incluyen, pero no se limitan a: válvulas protésicas, válvulas artificiales, catéteres permanentes, stents, oxigenadores de la sangre, derivaciones, puertos de acceso vasculares, dispositivos de asistencia ventricular y corazones o cavidades cardíacas artificiales e injertos de los vasos. Los procedimientos incluyen, pero no se limitan
a: bypass cardiopulmonar, intervención coronaria percutánea y hemodiálisis. En otra realización, la expresión "trastornos tromboembólicos" incluye síndrome coronario agudo, apoplejía, trombosis venosa profunda y embolia pulmonar.
En otra realización, la presente invención proporciona un compuesto para su uso en el tratamiento de un trastorno tromboembólico, en donde el trastorno tromboembólico se selecciona de angina inestable, un síndrome coronario agudo, fibrilación auricular, infarto de miocardio, accidente isquémico transitorio, apoplejía, ateroesclerosis, enfermedad arterial oclusiva periférica, flebotrombosis, flebotrombosis profunda, tromboflebitis, embolia arterial, trombosis arterial coronaria, trombosis arterial cerebral, embolia cerebral, embolia renal, embolia pulmonar y trombosis provocada por implantes, dispositivos o procedimientos médicos en los que la sangre está expuesta a una superficie artificial que promueve la trombosis. En otra realización, la presente invención proporciona un compuesto para su uso en el tratamiento de un trastorno tromboembólico, en donde el trastorno tromboembólico se selecciona de síndrome coronario agudo, apoplejía, trombosis venosa, fibrilación auricular y trombosis provocada por implantes y procedimientos médicos.
En otra realización, la presente invención proporciona un compuesto para su uso en la profilaxis primaria de un trastorno tromboembólico, en donde el trastorno tromboembólico se selecciona de angina inestable, un síndrome coronario agudo, fibrilación auricular, infarto de miocardio, muerte súbita isquémica, accidente isquémico transitorio, apoplejía, ateroesclerosis, enfermedad arterial oclusiva periférica, flebotrombosis, flebotrombosis profunda, tromboflebitis, embolia arterial, trombosis arterial coronaria, trombosis arterial cerebral, embolia cerebral, embolia renal, embolia pulmonar y trombosis provocada por implantes, dispositivos o procedimientos médicos en los que la sangre está expuesta a una superficie artificial que promueve la trombosis. En otra realización, la presente invención proporciona un compuesto para su uso en la profilaxis primaria de un trastorno tromboembólico, en donde el trastorno tromboembólico se selecciona de un síndrome coronario agudo, apoplejía, trombosis venosa y trombosis provocada por implantes y dispositivos médicos.
En otra realización, la presente invención proporciona un compuesto para su uso en la profilaxis secundaria de un trastorno tromboembólico, en donde el trastorno tromboembólico se selecciona de angina inestable, un síndrome coronario agudo, fibrilación auricular, infarto de miocardio recurrente, accidente isquémico transitorio, apoplejía, ateroesclerosis, enfermedad arterial oclusiva periférica, flebotrombosis, flebotrombosis profunda, tromboflebitis, embolia arterial, trombosis arterial coronaria, trombosis arterial cerebral, embolia cerebral, embolia renal, embolia pulmonar y trombosis provocada por implantes, dispositivos o procedimientos médicos en los que la sangre está expuesta a una superficie artificial que promueve la trombosis. En otra realización, la presente invención proporciona un compuesto para su uso en la profilaxis secundaria de un trastorno tromboembólico, en donde el trastorno tromboembólico se selecciona de síndrome coronario agudo, apoplejía, fibrilación auricular y trombosis venosa. Como se usa en el presente documento, el término "apoplejía" se refiere a apoplejía embólica o apoplejía aterotrombótica que surge de trombosis oclusiva en la arteria carótida primitiva, arteria carótida interna o arterias intracerebrales.
Cabe destacar que la trombosis incluye la oclusión del vaso (por ejemplo, después de un bypass) y la reoclusión (por ejemplo, durante o después de una angioplastía coronaria transluminal percutánea). Los trastornos tromboembólicos pueden ser el resultado de afecciones que incluyen, pero no se limitan a, ateroesclerosis, cirugía o complicaciones quirúrgicas, inmovilización prolongada, fibrilación arterial, trombofilia congénita, cáncer, diabetes, efectos de medicamentos u hormonas y complicaciones del embarazo.
A menudo, los trastornos tromboembólicos se asocian a pacientes con ateroesclerosis. Los factores de riesgo para la ateroesclerosis incluyen, pero no se limitan a, el género masculino, la edad, la hipertensión, los trastornos lipídicos y la diabetes mellitus. Los factores de riesgo para la ateroesclerosis son, al mismo tiempo, factores de riesgo para complicaciones de ateroesclerosis, es decir, trastornos tromboembólicos.
De manera similar, la fibrilación arterial generalmente se asocia a trastornos tromboembólicos. Los factores de riesgo para la fibrilación y los trastornos tromboembólicos posteriores incluyen enfermedad cardiovascular, enfermedad cardíaca reumática, enfermedad no reumática de la válvula mitral, enfermedad cardiovascular hipertensiva, enfermedad pulmonar crónica y varias anomalías cardíacas misceláneas, así como tirotoxicosis.
La diabetes mellitus con frecuencia se asocia a la ateroesclerosis y a trastornos tromboembólicos. Los factores de riesgo para la del tipo 2 más común incluyen, pero no se limitan a, antecedentes familiares, obesidad, inactividad física, raza/etnia, prueba previa de tolerancia a la glucosa o glucosa en ayunas deficiente, antecedentes de diabetes mellitus gestacional o de haber dado a luz a un "bebé grande", hipertensión, colestero1HDL bajo y síndrome del ovario poliquístico.
La trombosis se ha asociado a varios tipos de tumores, por ejemplo, cáncer de páncreas, cáncer de mama, tumores cerebrales, cáncer de pulmón, cáncer de ovarios, cáncer de próstata, cáncer gastrointestinal y linfoma de Hodgkin o de no Hodgkin. Estudios recientes sugieren que la frecuencia del cáncer en pacientes con trombosis reflejan la frecuencia de un tipo particular de cáncer en la población general. (Levitan, N. e t al., Medicine (Baltimore), 78(5):285291 (1999); Levine M. e t al., N. Engl. J. Med., 334(11):677-681 (1996); Blom, J.W. e t al., JAMA, 293(6):715-722 (2005).) Por lo tanto, los tipos de cáncer más comunes asociados a la trombosis en los hombres son cáncer de próstata, cáncer colorrectal, cáncer cerebral y cáncer de pulmón, y en las mujeres, cáncer de mama, cáncer de ovarios y cáncer de pulmón. La tasa tromboembolismo venoso (VTE) observada en pacientes de cáncer es considerable. Las variaciones de las tasas de VTE entre diferentes tipos de tumores se relacionan, más probablemente, con la selección de la población de pacientes. Es posible que los pacientes de cáncer con riesgo de padecer trombosis tengan algunos de los siguientes factores de riesgos o todos ellos: (i) la etapa del cáncer (es decir, presencia de metástasis), (ii) la presencia de catéteres venosos centrales, (iii) cirugía y terapias contra el cáncer, lo que incluye quimioterapia, y (iv) hormonas y fármacos antiangiogénicos. Por ello, la práctica clínica habitual es administrar a los pacientes que tienen tumores avanzados heparina o heparina de bajo peso molecular para prevenir trastornos tromboembólicos. Algunas preparaciones de heparina de bajo peso molecular fueron aprobadas por la FDA para estas indicaciones.
Como se usa en el presente documento, la expresión "composición farmacéutica" significa cualquier composición que contiene al menos un agente terapéutica o biológicamente activo y que es adecuado para la administración al paciente. Cualquiera de estas formulaciones se puede preparar mediante métodos bien conocidos y aceptados en la técnica. Véase, por ejemplo, Gennaro, A.R., ed., Remington: The Science and Practice of Pharmacy, 20.a edición, Mack Publishing Co., Easton, Pa. (2000).
La invención incluye administrar a un sujeto una composición farmacéutica que incluye un compuesto que se fija a PAR4 y que inhibe la escisión y/o la señalización de PAR4 (denominado en el presente documento "antagonista de PAR4" o "compuesto terapéutico").
Los compuestos de esta memoria descriptiva se pueden administrar en forma oral, como comprimidos, cápsulas (cada una de las cuales incluye formulaciones de liberación sostenida o formulaciones de liberación controlada), píldoras, polvos, gránulos, elixires, tinturas, suspensiones, jarabes y emulsiones. También se pueden administrar de manera intravenosa (bolo o infusión), intraperitoneal, subcutánea o intramuscular, en todos estos casos, usando formas de dosificación bien conocidas por los expertos habituales en las técnicas farmacéuticas. Se pueden administrar solos, pero generalmente se administran con un vehículo farmacéutico seleccionado en función de la vía de administración elegida y la práctica farmacéutica estándar.
La dosis preferida del antagonista de PAR4 es una dosis biológicamente activa. Una dosis biológicamente activa es una dosis que inhibe la escisión y/o la señalización de PAR4 y tiene un efecto antitrombótico. Preferentemente, el antagonista de PAR4 puede reducir la actividad de PAR4 en al menos un 5 %, 10 %, 20 %, 30 %, 40 %, 50 %, 60 %, 70 %, 80 %, 90 %, 100 % o más de 100 % por debajo de los niveles de control no tratados. Los niveles de PAR4 en plaquetas se miden mediante cualquier método conocido en la técnica, lo que incluye, por ejemplo, el ensayo de unión al receptor, la agregación plaquetaria, los ensayos de activación plaquetaria (por ejemplo, expresión de pselectina mediante FACS), transferencia Western o análisis ELISA usando los anticuerpos sensibles a la escisión de PAR4. De manera alternativa, la actividad biológica de PAR4 se mide evaluando la señalización celular provocada por PAR4 (por ejemplo, la movilización del calcio u otros ensayos sobre el mensajero secundario).
En algunas formas de realización, una cantidad terapéuticamente eficaz de un compuesto de PAR4 es, preferentemente, de aproximadamente menos de 100 mg/kg, 50 mg/kg, 10 mg/kg, 5 mg/kg, 1 mg/kg o menos de 1 mg/kg. En una forma de realización de mayor preferencia, la cantidad terapéuticamente eficaz del compuesto de PAR4 es de menos de 5 mg/kg. En una forma de realización de máxima preferencia, la cantidad terapéuticamente eficaz del compuesto de PAR4 es de menos de 1 mg/kg. Las dosis eficaces varían, según reconocerán los expertos en la técnica, según la vía de administración y el uso del excipiente.
Los compuestos de esta invención se pueden administrar de manera intranasal mediante el uso tópico de vehículos intranasales adecuados o mediante vías transdérmicas, usando parches transdérmicos para la piel. Cuando se administra en forma de un sistema de administración transdérmico, la administración de las dosis será, por supuesto, continua en lugar de intermitente durante todo el régimen de dosificación.
Generalmente, los compuestos se administran en combinación con diluyentes farmacéuticos adecuados, excipientes o vehículos (que se denominan colectivamente en el presente documento "vehículos farmacéuticos") seleccionados adecuadamente con respecto a la forma de administración pretendida, es decir, comprimidos orales, cápsulas, elixires, jarabes y similares, y de acuerdo con las prácticas farmacéuticas convencionales.
Por ejemplo, para la administración oral en forma de un comprimido o cápsula, el componente del fármaco activo se puede combinar con un vehículo oral, no tóxico, farmacéuticamente aceptable e inerte, tal como lactosa, almidón, sacarosa, glucosa, metilcelulosa, estearato de magnesio, fosfato de dicalcio, sulfato de calcio, manitol, sorbitol y similares; para la administración oral en forma líquida, los componentes del fármaco oral se pueden combinar con cualquier vehículo inerte oral, no tóxico, farmacéuticamente aceptable, tal como etanol, glicerol, agua y similares. Asimismo, cuando sea conveniente o necesario, también se pueden incorporar en la mezcla aglutinantes, lubricantes, agentes desintegrantes y agentes colorantes adecuados. Los aglutinantes adecuados incluyen almidón, gelatina, azúcares naturales, tales como glucosa o beta-lactosa, endulzantes de maíz, gomas naturales y sintéticas,
tales como acacia, tragacanto o alginato de sodio, carboximetilcelulosa, polietilenglicol, ceras y similares. Los lubricantes usados en estas formas de dosificación incluyen oleato de sodio, estearato de sodio, estearato de magnesio, benzoato de sodio, acetato de sodio, cloruro de sodio y similares. Los desintegrantes incluyen, pero no se limitan a, almidón, metilcelulosa, agar, bentonita, goma xantana y similares.
Los compuestos de la presente invención también se pueden administrar en forma de sistemas de administración de liposomas, tales como vesículas unilaminares pequeñas, vesículas unilaminares grandes y vesículas multilaminares. Los liposomas se pueden formar con varios fosfolípidos, tales como colesterol, estearilamina o fosfatidilcolinas. Los compuestos de la presente invención se pueden acoplar a polímeros solubles, como portadores de fármacos dirigibles. Tales polímeros pueden incluir polivinilpirrolidona, copolímero de pirano, polihidroxipropilmetacrilamidafenol, polihidroxietilaspartamidafenol o polietilenóxido-polilisina sustituida con restos de palmitoílo. Asimismo, los compuestos de la presente invención se pueden acoplar a una clase de polímeros biodegradables útiles para lograr la liberación controlada de un fármaco, por ejemplo, ácido poliláctico, ácido poliglicólico, copolímeros de ácido poliláctico y poliglicólico, caprolactona de poliépsilon, ácido polihidroxibutírico, poliortoésteres, poliacetales, polidihidropiranos, policianoacrilatos y copolímeros en bloque reticulados o anfipáticos de hidrogeles.
Las formas de dosificación (composiciones farmacéuticas) adecuadas para la administración pueden contener de aproximadamente 1 miligramo a aproximadamente 10 0 miligramos de ingrediente activo por unidad de dosificación. Generalmente, en estas composiciones farmacéuticas, el ingrediente activo está presente en una cantidad de aproximadamente el 0,5-95 % en peso en función del peso total de la composición.
Las cápsulas de gelatina pueden contener el ingrediente activo y portadores en polvo, tales como lactosa, almidón, derivados de celulosa, estearato de magnesio, ácido esteárico y similares. Se pueden usar diluyentes similares para fabricar comprimidos. Tanto los comprimidos como las cápsulas se pueden fabricar como productos de liberación sostenida para proporcionar una liberación continua del medicamento durante un período de horas. Los comprimidos pueden estar recubiertos con azúcar o con una película para disimular el sabor desagradable y proteger el comprimido de la atmósfera, o pueden estar recubiertos de manera entérica para la desintegración selectiva en el tubo gastrointestinal.
Las formas de dosificación líquidas para la administración oral pueden contener colorantes y saborizantes, a fin de aumentar la aceptación por parte del paciente.
En general, el agua, un aceite adecuado, la solución salina, la dextrosa acuosa (glucosa) y las soluciones de azúcares relacionados, y los glicoles, tales como propilenglicol o polietilenglicoles son portadores adecuados para soluciones parenterales. Las soluciones para la administración parenteral pueden contener una sal hidrosoluble del ingrediente activo, agentes estabilizantes adecuados y, si fuera necesario, sustancias tamponadoras. Los agentes antioxidantes, tales como bisulfito de sodio, sulfito de sodio o ácido ascórbico, ya sean solos o en combinación, son agentes estabilizantes adecuados. También se usan el ácido cítrico y sus sales, y EDTA de sodio. Asimismo, las soluciones parenterales pueden contener conservantes, tales como cloruro de benzalconio, metilparabeno o propilparabeno y clorobutanol.
Los portadores farmacéuticos adecuados se describen en Remington's Pharmaceutical Sciences, Mack Publishing Company, un texto de referencia estándar en este campo.
Las formas de dosificación farmacéuticas útiles y representativas para la administración de los compuestos de esta invención se pueden ilustrar de la siguiente manera:
Cápsulas
Se puede preparar una gran cantidad de unidades de cápsulas llenando cápsulas de gelatina dura de dos piezas estándares, cada una con 100 miligramos de ingrediente activo en polvo, 150 miligramos de lactosa, 50 miligramos de celulosa y 6 miligramos de estearato de magnesio.
Cápsulas de gelatina blandas
Una mezcla de ingrediente activo en un aceite que se puede digerir, tal como aceite de soja, aceite de semillas de algodón o aceite de oliva, se puede preparar e inyectar por medio de una bomba de desplazamiento positivo en gelatina para formar cápsulas de gelatina blandas que contienen 100 miligramos del ingrediente activo. Las cápsulas deben lavarse y secarse.
Comprimidos
Los comprimidos se pueden preparar mediante procedimientos convencionales, de modo que la unidad de dosificación sea de 100 miligramos de ingrediente activo, 0,2 miligramos de dióxido de silicio coloidal, 5 miligramos de estearato de magnesio, 275 miligramos de celulosa microcristalina, 11 miligramos de almidón y 98,8 miligramos
de lactosa. Los recubrimientos adecuados se pueden aplicar para aumentar la palatabilidad o retrasar la absorción. Dispersión
Se puede preparar una dispersión seca en atomizador para la administración oral mediante métodos conocidos por el experto en la técnica.
Inyectable
Una composición parenteral adecuada para la administración mediante inyección se puede preparar agitando un 1,5 % en peso de ingrediente activo en un 10 % en volumen de propilenglicol y agua. La solución debería transformarse en isotónica con cloruro de sodio y debería esterilizarse.
Suspensión
Se puede preparar una suspensión acuosa para administración oral, de modo que cada 5 ml contengan 100 mg de ingrediente activo finamente dividido, 200 mg de carboximetilcelulosa de sodio, 5 mg de benzoato de sodio, 1 , 0 g de solución de sorbitol, U.S.P. y 0,025 ml de vanilina.
Cuando dos o más de los segundos agentes terapéuticos antes mencionados se administran con el compuesto de fórmula I, IA, IB, IC, ID, IE o IF, preferentemente, un compuesto seleccionado de uno de los ejemplos, en general, la cantidad de cada componente en una dosificación diaria típica y una forma de dosificación típica se puede reducir con respecto a la dosificación habitual del agente cuando se administra solo, en vista del efecto aditivo o sinérgico de los agentes terapéuticos cuando se administran en combinación.
En particular, cuando se suministran como una sola unidad de dosis, es posible que se produzca una interacción química entre los ingredientes activos combinados. Por esta razón, cuando el compuesto de los ejemplos y un segundo agente terapéutico se combinan en una sola unidad de dosis, se formulan de manera tal que, si bien los ingredientes activos se combinan en una sola unidad de dosificación, se minimiza (es decir, se reduce) el contacto físico entre los ingredientes activos. Por ejemplo, un ingrediente activo se puede recubrir de manera entérica. Mediante el recubrimiento entérico de uno de los ingredientes activos, es posible no solo minimizar el contacto entre los ingredientes activos combinados, sino que también es posible controlar la liberación de uno de estos componentes en el tracto gastrointestinal, de manera que uno de estos componentes no se libere en el estómago sino en el intestino. Uno de los ingredientes activos también se puede recubrir con un material que realice la liberación sostenida en el tracto gastrointestinal y, además, sirve para minimizar el contacto físico entre los ingredientes activos combinados. Además, el componente de liberación sostenida también se puede recubrir adicionalmente de manera entérica, de manera que la liberación de este componente se produzca solamente en el intestino. Otro enfoque más involucraría la formulación de un producto combinado en el que un componente se recubre con un polímero de liberación sostenida y/o entérica y el otro componente también se recubre con un polímero, tal como hidroxipropilmetilcelulosa (HPMC) de baja viscosidad u otros materiales adecuados conocidos en la técnica, a fin de separar aún más los componentes activos. El recubrimiento polimérico sirve para formar una barrera adicional que evita a interacción con el otro componente.
Estas y otras maneras de minimizar el contacto entre los componentes de los productos combinados de la presente invención, ya sea que se administren en una sola forma de dosificación o en formas de dosificación separadas, pero al mismo tiempo y de la misma manera, serán evidentes para los expertos en la técnica, una vez que lea la presente descripción.
Adicionalmente, ciertos compuestos descritos en el presente documento pueden ser útiles como metabolitos de otros compuestos. Por lo tanto, en una realización, los compuestos pueden ser útiles, ya sea como un compuesto sustancialmente puro, que también se puede incorporar en una composición farmacéutica, o puede ser útil como metabolito que se genera después de la administración del profármaco de ese compuesto. En una realización, un compuesto puede ser útil como un metabolito, al ser útil para tratar trastornos, como se describe en el presente documento.
La actividad de los antagonistas de PAR4 de la presente invención se pude medir en varios ensayos in vitro. Los ensayos de ejemplo se muestran en los ejemplos a continuación.
El ensayo FLIPR es un ensayo in vitro de ejemplo para medir la actividad de los antagonistas de PAR4 de la presente invención. En este ensayo, la movilización del calcio intracelular se induce en las células que expresan PAR4 mediante un agonista de PAR4 y se controla la movilización del calcio. Véase, por ejemplo, el ejemplo A. AYPGKF es un agonista de PAR4 conocido. Un agonista de PAR4 alternativo es H-Ala-Phe(4-F)-Pro-Gly-Trp-Leu-Val-Lys-Asn-Gly-NH2. Como se muestra en el ejemplo B a continuación, H-Ala-Phe(4-F)-Pro-Gly-Trp-Leu-Val-Lys-Asn-Gly-NH2 fue validado como un agonista de PAR4 en el ensayo FLIPR. Se realizó una comparación lado a lado de los valores CI50 de ~180 compuestos usando AYPGKF en comparación con H-Ala-Phe(4-F)-Pro-Gly-Trp-Leu-ValLys-Asn-Gly-NH2. Los resultados demostraron una fuerte correlación entre los dos ensayos. Adicionalmente, H-Ala-Phe(4 -F)-Pro-Gly-Trp-Leu-Val-Lys-Asn-Gly-NH2 ha mejorado la actividad del agonista, en comparación con AYPGKF con un CE50 que es 10 veces más bajo que el CE50 para AYPGKF en el ensayo FLIPR. H-Ala-Phe(4-F)-Pro-Gly-Trp-Leu-Val-Lys-Asn-Gly-NH2 se puede sintetizar usando métodos conocidos por los expertos en la técnica.
El ensayo FLIPR también se puede usar como un sistema de cribado inverso para evaluar la actividad del agonista o la actividad del antagonista de PAR1 en una línea celular que expresa PAR1 y PAR4. La actividad del antagonista de PAR1 se puede evaluar por la capacidad del compuesto para inhibir la movilización del calcio inducida por el péptido agonista de PAR1 SFLLRN u otros péptidos agonistas de PAR1.
Los compuestos de la presente invención se pueden analizar in v itro para determinar su capacidad para inhibir la agregación plaquetaria inducida por la trombina gamma, como se muestra en el ejemplo C. La trombina gamma, un producto proteolítico de la trombina alfa que ya no interactúa con PAR1, escinde y activa PAR4 selectivamente (Soslau, G. e t al., "Unique pathway of thrombin-induced platelet aggregation mediated by glycoprotein Ib", J. Biol. Chem., 276:21173-21183 (2001)). La agregación plaquetaria se puede controlar en un formato de ensayo de agregación de microplacas de 96 pocillos o mediante el uso de un agregómetro de plaquetas estándar. El ensayo de agregación también se puede usar para evaluar la selectividad del compuesto para inhibir la agregación plaquetaria inducida por los péptidos agonistas de PAR4, el péptido agonista de PAR1, ADP o el análogo de tromboxano U46619.
El ejemplo D es un ensayo de agregación plaquetaria inducida por trombina alfa. La trombina alfa activa PAR1 y PAR4. La capacidad de un antagonista de PAR4 selectivo de la presente invención, a saber, el compuesto del ejemplo 3 para inhibir la agregación plaquetaria se midió usando un agregómetro óptico estándar. La inhibición de la agregación plaquetaria inducida por trombina alfa mediante el compuesto del ejemplo 3 se muestra en las Figuras 1A y 1B. Los datos muestran, por primera vez en la técnica, que un antagonista de PAR4 solo puede inhibir eficazmente la agregación plaquetaria. El alcance de la inhibición plaquetaria mediante el antagonista de PAR4 es, al menos, comparable con lo que se describió anteriormente para los antagonistas de PAR1.
El ejemplo E es un ensayo de agregación plaquetaria inducido por el factor tisular. Las condiciones en este ensayo imitan los eventos fisiológicos durante la formación del trombo. En este ensayo, la agregación plaquetaria en PRP humano se inició mediante la adición de factor tisular y CaCl2. El factor tisular, el iniciador de la cascada de coagulación extrínseca, es altamente elevado en la placa ateroesclerótica humana. La exposición de la sangre al factor tisular en el sitio ateroesclerótico desencadena una fuerte generación de trombina e induce la formación de los trombos obstructivos.
La eficacia de los antagonistas de PAR4 de la presente invención para prevenir la trombosis también se puede medir en varios ensayos in vivo. Los mamíferos de ejemplo que pueden proporcionar modelos de trombosis y hemostasis para evaluar la eficacia de los antagonistas de PAR4 de la presente invención como agentes antitrombóticos incluyen, pero no se limitan a, conejillos de Indias y primates. Los modelos de eficacia relevantes incluyen, pero no se limitan a, trombosis de la arteria carótida inducida por lesiones electrolíticas, trombosis de la arteria carótida inducida por FeCh y trombosis de derivación arteriovenosa. Los modelos de tiempo de sangrado del riñón, tiempo de sangrado renal y otras mediciones del tiempo de sangrado se pueden usar para evaluar el riesgo de sangrado de los agentes antitrombóticos descritos en la presente invención. El ejemplo G describe un modelo in v ivo de trombosis arterial en monos cinomolgos. Los compuestos de la presente invención se pueden evaluar en este modelo para determinar su capacidad de inhibir la formación de trombos inducida por la lesión electrolítica de la arteria carótida. La demostración de eficacia en este modelo respalda la utilidad de los antagonistas de PAR4 de la presente invención para el tratamiento de las enfermedades tromboembólicas.
ENSAYOS
Materiales
1) Péptidos agonistas de PAR1 y PAR4
SFFLRR es un conocido péptido agonista selectivo de PAR1 con alta afinidad. (Referencia: Seiler, S.M., "Thrombin receptor antagonists", Seminars in Thrombosis and Hemostasis, 22(3):223-232 (1996).) Se sintetizaron los péptidos agonistas de PAR4 AYPGKF y H-Ala-Phe(4 -F)-Pro-Gly-Trp-Leu-Val-Lys-Asn-Gly-NH2. H-Ala-Phe(4-F)-Pro-Gly-Trp-Leu-Val-Lys-Asn-Gly-NH2 mostró una actividad mejorada del agonista de PAR4 en comparación con AYPGKF en el ensayo fLiPR (CE50 de 8 pM para H-Ala-Phe(4 -F)-Pro-Gly-Trp-Leu-Val-Lys-Asn-Gly-NH2 y 60 pM para AYPGKF) y en el ensayo de agregación de plaquetas lavadas (CE50 de 0,9 pM para H-Ala-Phe(4-F)-Pro-Gly-Trp-Leu-Val-Lys-Asn-Gly-NH2 y 12 pM para AYPGKF).
2) Células que expresan PAR4
Las células HEK293 que expresan de manera estable PAR4 se generaron mediante un método estándar de transfección del vector de expresión de ADNc F2R23 humano o mediante tecnología RAGE de Athersys Inc.
(Cleveland, OH) y se seleccionaron en función de la expresión de la proteína PAR4 de la expresión de ARNm. Aquellas células demostraron respuestas funcionales a la elevación del calcio intracelular inducida por el péptido agonista de PAR4 usando FLIPR (lector de placas de imágenes fluorométricas; Molecular Devices Corp.). Estas células expresan PAR1 endógeno y pueden provocar la señal de calcio ante la estimulación con el péptido agonista de PAR1. Las células se cultivaron en medio de Eagle modificado por Dulbecco (DMEM) (Invitrogen, Carlsbad, CA), 10 % de FBS, 1 % de PSG, 3 pg/ml de puromicina y 25 nM de metotrexato) a 37 °C con 5 % de CO2.
3) Preparación de plasma rico en plaquetas (PRP)
Se recolectó sangre humana en citrato de sodio al 3,8 % en una relación de 1 ml por 9 ml de sangre. El plasma rico en plaquetas se aisló mediante centrifugación a 170 g durante 14 minutos.
4) Preparación de plaquetas lavadas (WP)
Se recolectó sangre humana en ACD (citrato de tri-sodio 85 mM, ácido cítrico 78 mM, D-glucosa 110 mM, pH 4,4) en una relación de 1,4 ml por 10 ml de sangre. La PRP se aisló mediante centrifugación a 170 g durante 14 minutos y las plaquetas se convirtieron en pelets mediante centrifugación a 1300 g durante 6 minutos. Las plaquetas se lavaron una vez con 10 ml de ACD que contenía 1 mg/ml de albúmina de suero bovino. Las plaquetas se volvieron a suspender a ~2,5 X 108/ml en tampón Tyrode (NaCl 137 mM, KCl 2 mM, MgCh 1,0 mM, CaCl21 mM, glucosa 5 mM, HEPES 20 mM pH 7,4).
Ejemplo A
Ensayo FLIPR en células HEK293 que expresan PAR4
Se evaluó la actividad de los antagonistas de PAR4 de la presente invención en células que expresan PAR4 mediante el control de la movilización del calcio intracelular inducida por H-Ala-Phe(4-F)-Pro-Gly-Trp-Leu-Val-Lys-Asn-Gly-NH2 usando FDSS6000 (Hamamatsu Photonics, Japón) mediante fluo-4. También se llevaron a cabo cribados inversos para determinar la actividad del agonista de PAR1. En resumen, las células 20664.1J clones de HEK293 EBNA PAR4 se colocaron en placas durante 24 h antes del experimento en placas negras, recubiertas con poli-D-lisina, de fondo transparente, de 384 pocillos (Greiner Bio-One, Monroe, NC). Las células se colocaron en placas a 20,000 células/pocillo en medio de crecimiento de 20 pl y se incubaron a 37 °C con 5 % de CO2 durante la noche. En el momento del ensayo, el medio se reemplazó por 40 pl 1x de solución salina tamponada de Hank (HBSS) (con HEPES 10 mM) y se agregaron 20 pl del compuesto de prueba a varias concentraciones también diluido en 1X de tampón HBSS, y una concentración final de 0,67 % de DMSO en FDSS para la medición de los agonistas. Después las células se incubaron durante 30 minutos a temperatura ambiente y se agregaron 20 pl del péptido agonista para la medición de antagonistas en FDSS. El péptido agonista H-Ala-Phe(4-F)-Pro-Gly-Trp-Leu-Val-Lys-Asn-Gly-NH2 para el cribado del antagonista de PAR4 o SFFLRR para el cribado inverso de PAR1 se evaluaron en forma rutinaria para asegurar una respuesta a CE50 en el ensayo (~2,5 pM de H-Ala-Phe(4-F)-Pro-Gly-Trp-Leu-Val-Lys-Asn-Gly-NH2 y 600 nM para SFFlRr ).
Ejemplo B
Validación de H-Ala-Phe(4 -F)-Pro-Gly-Trp-Leu-Val-Lys-Asn-Gly-NH2 como agonista de PAR4
Para validar H-Ala-Phe(4 -F)-Pro-Gly-Trp-Leu-Val-Lys-Asn-Gly-NH2 como agonista de PAR4 en el ensayo FLIPR, se realizó una comparación lado a lado de los valores CI50 de ~ 180 compuestos usando AYPGKF frente a H-Ala-Phe(4 -F)-Pro-Gly-Trp-Leu-Val-Lys-Asn-Gly-NH2. Los resultados demostraron una alta correlación entre los dos ensayos (coeficiente de correlación por rangos de Spearman rho= 0,7760, p<0,0001). La relevancia del ensayo FLIPR en las células HEK293 se confirmó mediante la conectividad directa del ensayo con el ensayo de plaquetas lavadas. Los valores CI50 de ~ 200 compuestos del ensayo AYPGKF FLIPR tuvieron una gran correlación con los del ensayo de agregación de plaquetas lavadas AYPGKF (coeficiente de correlación por rangos de Spearman rho= 0,836, p<0,001). Se obtuvieron resultados similares comparando FLIPR y los datos de plaquetas lavadas usando H-Ala-Phe(4 -F)-Pro-Gly-Trp-Leu-Val-Lys-Asn-Gly-NH2.
Ejemplo C
Ensayos de agregación plaquetaria inducida por trombina gamma
La capacidad de los compuestos de la presente invención de inhibir la agregación plaquetaria inducida por trombina gamma se evaluó en un formato de ensayo de agregación de microplacas de 96 pocillos. En resumen, la suspensión de plaquetas lavadas o PRP (100 pl) se preincubó durante 5 minutos a temperatura ambiente con concentraciones variantes de compuestos. La agregación se inició mediante ~ 10-50 nM de trombina gamma (Haematologic Technologies, Essex Junction, VT), que se tituló diariamente para obtener 80 % de agregación plaquetaria. Se agregó 1 U/ml de Refludan (Berlex, Montville, NJ) a la muestra de trombina gamma para evitar la activación de PAR1 inducida por la contaminación de trombina alfa residual. Después la placa se colocó en un lector de placas
SPECTRAMAX® Plus de Molecular Devices (Sunnyvale, CA) a 37 °C. La placa se mezcló durante 10 segundos antes de la primera lectura y 50 segundos entre cada lectura durante hasta 15 minutos a 405 nM. Se recolectaron datos con el software SOFTMAX® 4.71. La placa también incluía una muestra de control no tratado que se utilizó como ODmáx, mientras que el tampón sin contenido de plaquetas era el ODmín. La agregación plaquetaria se determinó sustrayendo el ODmáx del ODmín para obtener el 100 % del valor de agregación. En muestras experimentales, la transmisión observada se sustrajo del valor mínimo y después se comparó con el 100 % del valor de agregación para determinar el porcentaje de agregación. Los valores CI50 se determinan usando el software Excel Fit.
Los ensayos de agregación también se usaron para evaluar la selectividad del compuesto con respecto a otros receptores de plaquetas usando SFFLRR para PAR1, colágeno (Chrono-Log, Havertown, PA) para receptores de colágeno, ADP para P2Y1 y P2Y12 y U46619 (Cayman Chemical, Ann Arbor, MI) para receptores de tromboxano. Ejemplo D
Ensayos de agregación plaquetaria inducida por trombina alfa
La capacidad del antagonista de PAR4 para inhibir la agregación plaquetaria inducida por trombina alfa se evaluó usando plaquetas lavadas humanas. El ejemplo 3 se preincubó con plaquetas lavadas durante 20 min. La agregación se inició mediante la adición de 1,5 nM de trombina alfa (Haematologic Technologies, Essex Junction, VT) a 300 |jl de plaquetas lavadas a una velocidad de agitación de 1000 rpm. La agregación plaquetaria se controló usando un agregómetro óptico (Chrono-Log, Havertown, PA) y se midió el área debajo de la curva (AUC) a los 6 min. Se calculó el CI50 usando el control de vehículo como 0 % de inhibición. Se calculó que el CI50 para la inhibición de la agregación plaquetaria mediante el ejemplo 3 fue de 5 nM (n=3) (Figuras 1A y 1B).
Ejemplo E
Ensayo de agregación plaquetaria inducida por el factor tisular
La capacidad de los antagonistas de PAR1 o PAR4 para inhibir la agregación plaquetaria inducida por trombina endógena se evaluó en un ensayo de agregación dirigido por el factor tisular. La agregación se inicia mediante la adición de CaCl2 y factor tisular humano recombinante, que da como resultado la generación de trombina a través de la activación de la vía de coagulación en el plasma. Los agentes anticoagulantes, tales como el inhibidor de tripsina del maíz (Haematologic Technologies, Essex Junction, VT) a 50 jg/ml y PEFABLOC® FG (Centerchem, Norwalk, CT) también se agregan a la muestra para evitar la formación de coágulos de fibrina durante el tiempo del estudio. La agregación plaquetaria se controla usando instrumentación estándar, que incluye un agregómetro óptico o un agregómetro de impedancia.
Ejemplo F
Las siguientes tablas establecieron los resultados obtenidos usando varios compuestos de la invención evaluados en el ensayo FLIPR y en el ensayo de agregación plaquetaria (ensayo PRP). Como se indicó anteriormente, el ensayo FLIPR, un ensayo in v itro , mide la actividad del antagonista de PAR4 de los compuestos evaluados como se describió en el ejemplo A. El ensayo PRP, un ensayo in vitro, mide la actividad del antagonista de PAR4 de los compuestos evaluados en presencia de proteínas de plasma y del agonista de trombina como se describió en el ejemplo C.
Tabla 1
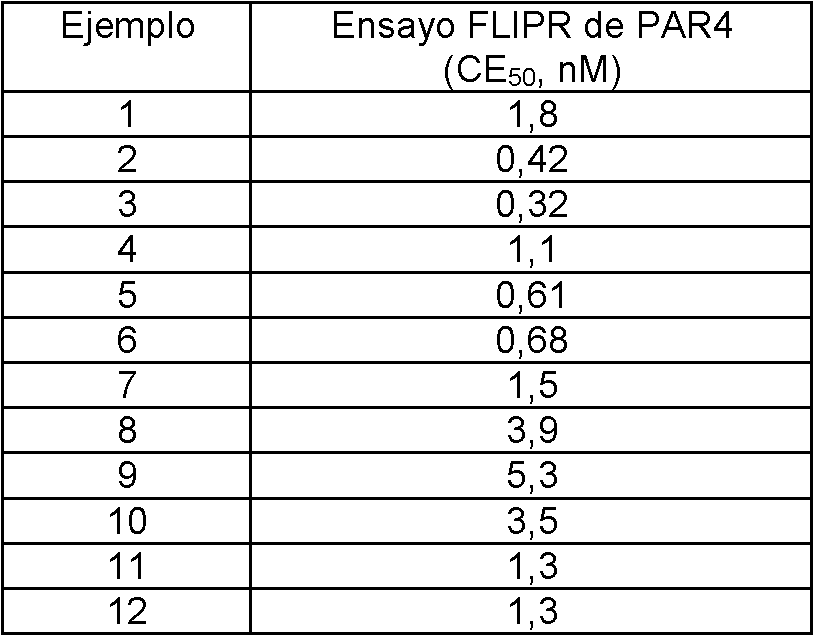
continuación

continuación

continuación

continuación

continuación

continuación

Tabla 2

continuación

Ejemplo G
Modelo de trombosis de la arteria carótida inducida por lesiones electrolíticas en mono cinomolgo
Se usaron monos cinomolgos sanos en el estudio. Estos monos se retiraron de otros estudios farmacocinéticos y farmacodinámicos y tuvieron un período de reposo farmacológico de, al menos, 4 semanas.
El día del estudio, se les administraron compuestos o vehículos de manera oral de 1 a 2 horas antes del experimento. Después, los monos se sedaron mediante la administración intramuscular de 0,2 mg/kg de atropina, 5 mg/kg de Telazol® (tiletamina/zolazepam) y 0,1 mg/kg de hidromorfina para facilitar la colocación de un tubo endotraqueal. Se colocó un catéter intravenoso en la vena cefálica izquierda para la administración de fluidos, a fin de prevenir la deshidratación. Después, los animales recibieron anestesia por inhalación, isoflurano (1-5 % para producir efecto) y oxígeno, se les suministró respiración asistida y se los colocó en una almohadilla de calentamiento controlada por termostato para mantener la temperatura corporal a 37 oC. La anestesia general se mantuvo en un plano quirúrgico usando isoflurano inhalable y oxígeno. Se colocó una cánula en la arterial braquial izquierda para registrar la presión arterial y la frecuencia cardíaca. Se controlaron la presión arterial y la frecuencia cardíaca para mantener los signos vitales normales.
El modelo de trombosis de la arteria carótida en monos se basó en un modelo de trombosis arterial en conejos, como lo describen Wong e t al. (Wong P.C.. et al., Nonpeptide factor Xa inhibitors: II. Antithrombotic evaluation in a rabbit model of electrically induced carotid artery thrombosis. J Pharmacol Exp Ther 295: 212-218, (2002).) La trombosis se indujo mediante estimulación eléctrica de la arteria carótida durante 5 min a 10 mA usando un electrodo bipolar externo de acero inoxidable. El flujo sanguíneo de la carótida se midió con un medidor de flujo transónico de tamaño adecuado y un flujómetro perivascular TRANSONIC® (Modelo TS420, Transonic Systems Inc., Ithaca, NY). Se registró continuamente durante un período de 90 min para controlar la oclusión inducida por trombosis. El flujo sanguíneo integrado de la carótida se midió por el área debajo de la curva de tiempo-flujo. Se expresó como el porcentaje del flujo sanguíneo de la carótida de control total, que daría resultado si el flujo sanguíneo de control se hubiera mantenido continuamente durante 90 min. Asimismo, se retiró el trombo de la arteria lesionada, se coaguló dos veces en un papel de pesaje, a fin de retirar el fluido residual y se pesó. La Figura 1C muestra los resultados de un experimento de respuesta a la dosis con el ejemplo 3 en el modelo de trombo arterial inducido por electricidad en mono cinomolgo, lo que demuestra la eficacia antitrombótica in v ivo de un antagonista de PAR4.
Claims (15)
1. Un compuesto de fórmula I:

I
o un estereoisómero, un tautómero, una sal o solvato farmacéuticamente aceptables del mismo, en donde:
W es O;
R0 es R1 ;
Y es So -CR8=CR9-;
R1 se selecciona independientemente del grupo que consiste en:
H,
halo
alquilo C1-C2 ,
ciclopropilo
alcoxi C1-C2,
alquiltio C1-C2,
halo alquilo C1-C2-, que contiene de 1 a 5 halógenos, en donde halo es F o Cl, y
halo cicloalquilo C3-C4
R8 y R9 se seleccionan independientemente del grupo que consiste en:
H,
flúor,
cloro,
alquilo C1-C3,
alcoxi C1-C2 y
haloalquilo C1-C2 ;
siempre que al menos uno de R1 , R8 y R9 sea distinto de H;
R2 es H;
X1 se selecciona del grupo que consiste en CH o N;
X2 , X3 y X4 se seleccionan independientemente de CR3;
R3 se selecciona del grupo que consiste en H, alquilo C1-C3, alcoxi C1-C3, flúor, cloro, OCF3 y haloalquilo C1-C2, que contiene de 1a 5 halógenos;
R4 y R5 se seleccionan independientemente de H y metilo;

es un anillo heteroarilo de 5 miembros seleccionado entre tiazol, tiadiazol, oxazol, oxadiazol ytriazol;
R6 se selecciona del grupo que consiste en H, halo, OCF3, OCHF2 , OH, CN, NO2 , NR11 R12 , alcoxicarbonilo C1-C4, (C=O)NR11 R12 y alquilo C1-C5 sustituido con de 0 a 7 grupos seleccionados independientemente de halo, CF3, OCF3, OH, hidroxialquilo C1-C4, alcoxi C1-C4, alcoxi C1-C4-alcoxi C1-C4, dialquilaminofenil C1-C4-alquilo C1-C4-, (dialcoxi C1-C4-alquil C1-C4)-alquilo C1-C4, dialquilamino C1-C4, cicloalquilo C3-C6 y alquiltio C1-C4 o
R6 es B-D-, en donde D es un enlazador que se selecciona entre:
un enlace simple,
-O-,
-s-,

alquileno C1-C4 sustituido con de 0 a 4 grupos seleccionados independientemente de halo u OH, alquilenoxi C1-C4,
alquenileno C2-C6 y
B se selecciona del grupo que consiste en:
arilo C6-C10 sustituido con de 0 a 3 grupos seleccionados independientemente del grupo que consiste en halo, alcoxi C1-C4, haloalcoxi -C1-C4, alquilo C1-C4, haloalquilo C1-C4, cicloalquilo C3-C6, ciano, nitro, NR11R12, OH, alquilcarboniloxi C1-C4-alquilo C1-C4, hidroxialquilo C1-C4, COOR14, SO2R14, (C=O)NR11R12, SO2NR11R12, N(R13)(C=O)NR11R12, N(R13)(C=O)OR14, N(R13)(C=O)R14, NR13S(O)R14, NR13SO2R14, O(C=O)NR11R12, O(C=O)OR14, O(C=O)R14, (C=O)OR14 y heteroarilo de 5-6 miembros,
heteroarilo de 5 a 10 miembros sustituido con de 0 a 3 grupos seleccionados independientemente del grupo que consiste en halo, alcoxi C1-C4, haloalcoxi C1-C4, alquilo C1-C4, haloalquilo C1-C4, cicloalquilo C3-C6, ciano, nitro, NR11R12, OH, alquilcarboniloxi C1-C4-alquilo C1-C4, hidroxialquilo C1-C4, COOR14, SO2R14, (C=O)NR11R12, SO2NR11R12, N(R13)(C=O)NR11R12, N(R13)(C=O)OR14, N(R13)(C=O)R14, NR13S(O)R14, NR13SO2R14, O(C=O)NR11R12, O(C=O)OR14, O(C=O)R14, (C=O)OR14, heteroarilo de 5-6 miembros y (CH2)fenilo,
heterociclilo de 4 a 10 miembros que contiene átomos de carbono y de 1 a 2 heteroátomos adicionales seleccionados entre N, O y S, y sustituido con de 0 a 3 grupos seleccionados independientemente del grupo que consiste en halo, oxo, -(CHR13)n1-heteroarilo de 5 o 6 miembros sustituido con de 0 a 3 grupos seleccionados independientemente del grupo que consiste en halo, alcoxi C1-C4, alquilo C1-C4, ciclopropilo, CF3, OCF3 y CF2CH3; NR13S(O)R14, NR13SO2R14, -(CHR13)n1-heterociclilo de 4 a 10 miembros sustituido con de 0 a 3 grupos seleccionados independientemente del grupo que consiste en halo, alcoxi C1-C4, alquilo C1-C4, ciclopropilo, CF3, OCF3 y CF2CH3; OH, hidroxialquilo C1-C4, alcoxi C1-C4, haloalcoxi C1-C4, dialquilamino C1-C4-alquilo C1-C4, NR11 R12 , ciano, alquilo C1-C4, haloalquilo C1-C4, cicloalquilo C3-C6, alcoxi C1-C4-alquilo C1-C4, cicloalquil C3-C6-alquilcarbonilo C1-C4, arilcarbonilo C6-C10, alquilcarboniloxi C1-C4-alquilo C1-C4, COOR14, SO2R14, (C=O)NR11R12, SO2NR11R12, N(R13)(C=O)NR11R12, N(R13)(C=O)OR14, N(R13)(C=O)R14, O(C=O)NR11R12, O(C=O)OR14, O(C=O)R14, (C=O)OR14 y arilo C6-C10 sustituido con de 0 a 3 grupos seleccionados independientemente del grupo que consiste en halo, alcoxi C1-C4, alquilo C1-C4, ciclopropilo, alcoxicarbonilo C1-C4, (C=O)NR11R12, CF3, OCF3 y CF2CH3;
cicloalquilo C3-C6 que puede contener insaturación, sustituido con de 0 a 3 grupos seleccionados independientemente del grupo que consiste en halo, CF3, OCF3, heteroarilo de 5 o 6 miembros, OH, oxo, hidroxialquilo C1-C4, arilo C6-C10, COOH, oxo, alcoxicarbonilo C1-C4, (C=O)NR11R12 y alquilo C1-C4 y espirocicloalquilo C5-C11 que puede contener insaturación y que opcionalmente contiene de 1 a 3 heteroátomos seleccionados entre O, N o S y que se sustituye con de 0 a 3 grupos seleccionados independientemente del grupo que consiste en halo, CF3, OCF3, heteroarilo de 5 o 6 miembros, OH, hidroxialquilo C1-C4, arilo C6-C10 y alquilo C1-C4;
R11 y R12 se seleccionan, en cada caso, independientemente entre el grupo que consiste en:
H,
alquilo C1-C4,
haloalquilo C1-C4,
alquenilo C2-C4,
-(CR14R14)n1-fenilo sustituido con de 0 a 3 grupos seleccionados independientemente entre el grupo que consiste en halo, alcoxi C1-C4, alquilo C1-C4, ciclopropilo, CF3, OCF3, heteroarilo de 5 o 6 miembros, Oh , OCHF2 , dialquilamino C1-C4 y ciano,
-(CHR13)n1-cicloalquilo C3-C6 sustituido con de 0 a 3 grupos seleccionados independientemente del grupo que consiste en halo, CF3, OCF3, heteroarilo de 5 o 6 miembros, OH, hidroxialquilo C1-C4 y alquilo C1-C4, -(CHR13)n1-heterociclilo de 4 a 10 miembros sustituido con de 0 a 3 grupos seleccionados independientemente del grupo que consiste en halo, CF3, OCF3, heteroarilo de 5 o 6 miembros, OH, oxo, hidroxialquilo C1-C4 y alquilo C1-C4,
-(CHR13)n1-heteroarilo de 5 a 10 miembros sustituido con de 0 a 3 grupos seleccionados independientemente del grupo que consiste en halo, CF3, OCF3, heteroarilo de 5 o 6 miembros, OH, hidroxialquilo C1-C4 y alquilo C1-C4,
dialquilamino C1-C4-alquilo C1-C4,
dialcoxi C1-C4-alquilo C1-C4,
hidroxialquilo C1-C4,
cianoalquilo C1-C4,
alcoxi C1-C4-alquilo C1-C4,
alcoxicarbonil C1-C4-alquilo C1-C4,
alcoxicarbonilo C1-C4,
alquilcarbonilo C1-C4,
fenilcarbonilo;
alcoxicarbonilamino C1-C4-alquilcarbonilo C1-C4 y
dialquilamino C1-C4-alquilcarbonilo C1-C4,
de manera alternativa, R11 y R12, cuando se unen al mismo nitrógeno, se combinan para formar un anillo heterocíclico de 4 a 8 miembros que contiene átomos de carbono, sustituido con de 0 a 3 grupos seleccionados independientemente del grupo que consiste en halo, CF3, CHF2 , OCF3, OCHF2 , OCH2F, heteroarilo de 5 o 6 miembros, OH, oxo, hidroxialquilo C1-C4, alquilo C1-C4 y alcoxi C1-C4, y de 0 a 2 heteroátomos adicionales seleccionados entre N, NR13, O y S(O)p;
R13 se selecciona, en cada caso, independientemente del grupo que consiste en H, alquilo C1-C6 y -(CH2)fenilo;
R14 se selecciona, en cada caso, independientemente del grupo que consiste en H, alquilo C1-C6, haloalquilo C1-C4-, alcoxicarbonilamino C1-C4- y -(C ^V fenilo sustituido con de 0 a 3 grupos seleccionados independientemente del grupo que consiste en halo, alcoxi C1-C4, alquilo C1-C4, ciclopropilo, CF3, OCF3, heteroarilo de 5 o 6 miembros, OH, OCHF2 , dialquilamino C1-C4 y ciano;
R7 se selecciona entre el grupo que consiste en H, halo, hidroxilo, oxo, alquilo C1-C3, hidroxialquilo C1-C3 y haloalquilo C1-C2 , que contiene de 1 a 5 halógenos;
n1, en cada caso, se selecciona de 0, 1, 2 o 3 y
p, en cada caso, es independientemente 0 , 1 y 2.
2. El compuesto de la reivindicación 1 o un estereoisómero, un tautómero, una sal o un solvato farmacéuticamente aceptables del mismo, en donde:
W es O;
R0 es R1;
Y es S o -CR8=CR9-;
R1 se selecciona independientemente del grupo que consiste en:
H,
flúor,
cloro,
alquilo C1-C2 ,
alcoxi C1-C2,
alquiltio C1-C2 y
haloalquilo C1-C2 , que contiene de 1 a 5 halógenos, en donde halo es F o Cl;
R8 y R9 se seleccionan independientemente del grupo que consiste en:
H,
flúor,
cloro,
CH3,
COH3,
CF3 y
CHF2
siempre que al menos uno de R1, R8 y R9 sea distinto de H;
R2 es H;
X1 se selecciona del grupo que consiste en CH o N;
X2 y X3 son CH;
X4 es CR3;
R3 se selecciona del grupo que consiste en H, alquilo C1-C3, alcoxi C1-C3, flúor, cloro, OCF3 y haloalquilo C1-C2, que contiene de 1 a 5 halógenos;
R4 y R5 se seleccionan independientemente entre H y metilo;

es un anillo heteroarilo de 5 miembros seleccionado entre tiazol, tiadiazol, oxazol, oxadiazol y triazol;
R6 se selecciona del grupo que consiste en H, halo, OCF3, OCHF2, OH, NR11R12, alcoxicarbonilo C1-C4, (C=O)NR11R12 y alquilo C1-C5 sustituido con de 0 a 7 grupos seleccionados independientemente de halo, CF3, OCF3, OH, hidroxialquilo C1-C4, alcoxi C1-C4, alcoxi C1-C4-alcoxi C1-C4, dialquilaminofenil C1-C4-alquilo C1-C4, (dialcoxi C1-C4-alquil C1-C4)-alquilo C1-C4, dialquilamino C1-C4, cicloalquilo C3-C6 y alquiltio C1-C4 o
R6 es B-D-, en donde D es un enlazador, que se selecciona entre:
un enlace simple,
-O-,
-S-,
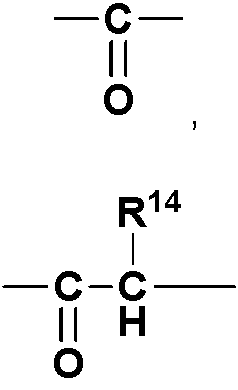
alquileno C1-C4 sustituido con de 0 a 4 grupos seleccionados independientemente de halo u OH y
alquenileno C2-C6 y
B se selecciona del grupo que consiste en:
arilo C6-C10 sustituido con de 0 a 3 grupos seleccionados independientemente del grupo que consiste en halo, alcoxi C1-C4, haloalcoxi C1-C4, alquilo C1-C4, haloalquilo C1-C4, cicloalquilo C3-C6, ciano, nitro, N alquilcarboniloxi C1-C4-alquilo C1-C4, hidroxialquilo C1-C4, COOR14, SO2R14, (C=O)NR11R12, SO2NR11R12, N(R13)(C=O)NR11R12, N(R13)(C=O)OR14, N(R13)(C=O)R14, NR13S(O)R14, NR13SO2R14, O(C=O)NR11R12, O(C=O)OR14, O(C=O)R14, (C=O)OR14 y heteroarilo de 5-6 miembros,
heteroarilo de 5 a 10 miembros sustituido con de 0 a 3 grupos seleccionados independientemente del grupo que consiste en halo, alcoxi C1-C4, haloalcoxi C1-C4, alquilo C1-C4, haloalquilo C1-C4, cicloalquilo C3-C6, ciano, nitro, NR11R12, OH, alquilcarboniloxi C1-C4-alquilo C1-C4, hidroxialquilo C1-C4, COOR14, SO2R14, (C=O)NR11R12, SO2NR11R12, N(R13)(C=O)NR11R12, N(R13)(C=O)OR14, N(R13)(C=O)R14, NR13S(O)R14, NR13SO2R14, O(C=O)NR11R12, O(C=O)OR14, O(C=O)R14, (C=O)OR14, heteroarilo de 5-6 miembros y (CH2)fenilo,
heterociclilo de 4 a 10 miembros que contiene átomos de carbono y de 1 a 2 heteroátomos adicionales seleccionados de N, O y S y sustituido con de 0 a 3 grupos seleccionados independientemente del grupo que consiste en halo, oxo, -(CHR13)n1-heteroarilo de 5 o 6 miembros sustituido con de 0 a 3 grupos seleccionados independientemente del grupo que consiste en halo, alcoxi C1-C4, alquilo C1-C4, ciclopropilo, CF3, OCF3 y CF2CH3; NR13S(O)R14, NR13SO2R14, -(CHR13)n1-heterociclilo de 4 a 10 miembros sustituido con de 0 a 3 grupos seleccionados independientemente del grupo que consiste en halo, alcoxi C1-C4, alquilo C1-C4, ciclopropilo, CF3, OCF3 y CF2CH3; OH, hidroxialquilo C1-C4, alcoxi C1-C4, haloalcoxi C1-C4, dialquilamino
C1-C4-alquilo C1-C4, NR11 R12 , ciano, alquilo C1-C4, haloalquilo C1-C4, cicloalquilo C3-C6, alcoxi C1-C4-alquilo
C1-C4, cicloalquil C3-C6-alquilcarbonilo C1-C4, arilcarbonilo C6-C10, alquilcarboniloxi C1-C4-alquilo C1-C4, COOR14, SO2R14, (C=O)NR11R12, SO2NR11R12, N(R13)(C=O)NR11R12, N(R13)(C=O)OR14, N(R13)(C=O)R14, O(C=O)NR11R12, O(C=O)OR14, O(C=O)R14, (C=O)OR14 y arilo C6-C10 sustituido con de 0 a 3 grupos seleccionados independientemente del grupo que consiste en halo, alcoxi C1-C4, alquilo C1-C4, ciclopropilo, alcoxicarbonilo C1-C4, (C=O)NR11R12, CF3, OCF3 y CF2CH3 y
cicloalquilo C3-C6 que puede contener insaturación, sustituido con de 0 a 3 grupos seleccionados independientemente del grupo que consiste en halo, CF3, OCF3, heteroarilo de 5 o 6 miembros, OH, oxo, hidroxialquilo C1-C4, arilo C6-C10, COOH, oxo, alcoxicarbonilo C1-C4, (C=O)NR11R12 y alquilo C1-C4;
R11 y R12 se seleccionan, en cada caso, independientemente del grupo que consiste en:
H,
alquilo C1-C4,
haloalquilo C1-C4,
alquenilo C2-C4,
-(CR14R14)n1-fenilo sustituido con de 0 a 3 grupos seleccionados independientemente del grupo que consiste en halo, alcoxi C1-C4, alquilo C1-C4, ciclopropilo, CF3, OCF3, heteroarilo de 5 o 6 miembros, Oh , OCHF2, dialquilamino C1-C4 y ciano;
-(CHR13)n1-cicloalquilo C3-C6 sustituido con de 0 a 3 grupos seleccionados independientemente del grupo que consiste en halo, CF3, OCF3, heteroarilo de 5 o 6 miembros, OH, hidroxialquilo C1-C4 y alquilo C1-C4;
-(CHR13)n1-heterociclilo de 4 a 10 miembros sustituido con de 0 a 3 grupos seleccionados independientemente del grupo que consiste en halo, CF3, OCF3, heteroarilo de 5 o 6 miembros, OH, oxo, hidroxialquilo C1-C4 y alquilo C1-C4;
-(CHR13)n1-heteroarilo de 5 a 10 miembros sustituido con de 0 a 3 grupos seleccionados independientemente del grupo que consiste en halo, CF3, OCF3, heteroarilo de 5 o 6 miembros, OH, hidroxialquilo C1-C4 y alquilo C1-C4;
dialquilamino C1-C4-alquilo C1-C4,
dialcoxi C1-C4-alquilo C1-C4,
hidroxialquilo C1-C4,
cianoalquilo C1-C4,
alcoxi C1-C4-alquilo C1-C4,
alcoxicarbonil C1-C4-alquilo C1-C4,
alcoxicarbonilo C1-C4,
alquilcarbonilo C1-C4 y
fenilcarbonilo;
de manera alternativa, R11 y R12, cuando se unen al mismo nitrógeno, se combinan para formar un anillo heterocíclico de 4 a 8 miembros que contiene átomos de carbono, sustituido con de 0 a 3 grupos seleccionados independientemente del grupo que consiste en halo, CF3, CHF2 , OCF3, OCHF2 , OCH2F, heteroarilo de 5 o 6 miembros, OH, oxo, hidroxialquilo C1-C4 alquilo, alquilo C1-C4 y alcoxi C1-C4 y de 0 a 2 heteroátomos adicionales seleccionados de N, NR13, O y S(O)p;
R13 se selecciona, en cada caso, independientemente del grupo que consiste en H y alquilo C1-C3;
R14 se selecciona, en cada caso, independientemente del grupo que consiste en H, alquilo C1-C3 y haloalquilo C1-C2;
R7 se selecciona del grupo que consiste en H, flúor, cloro, oxo, alquilo C1-C3, hidroxialquilo C1-C3 y haloalquilo C1-C2 , que contiene de 1 a 5 halógenos;
n1, en cada caso, se selecciona de 0, 1, 2 o 3 y
p, en cada caso, se selecciona de 0 , 1 y 2.
3. El compuesto de la reivindicación 1, o un estereoisómero, un tautómero, una sal o un solvato farmacéuticamente aceptables del mismo, en donde:
W es O;
R0 es R1;
Y es S o -CH=CH-;
R1 se selecciona independientemente del grupo que consiste en:
cloro
CH3,
OCH3,
SCH3,
CHFCH3 y
CF2CH3,
R2es H;
X1 es CH;
X2 y X4 son CH;
X3 es CR3;
R3 se selecciona del grupo que consiste en OCH3, flúor y cloro;
R4 y R5 se seleccionan independientemente de H y CH3;

es un anillo heteroarilo de 5 miembros seleccionado entre tiazol y oxazol;
R6 se selecciona del grupo que consiste en NR11R12 y alquilo C1-C5 sustituido con de 0 a 3 grupos seleccionados independientemente de halo, CF3, OCF3, OH, hidroxialquilo C1-C4, alcoxi C1-C4, alcoxi C1-C4-alcoxi C1-C4, dialquilaminofenil C1-C4-alquilo -C1-C4, (dialcoxi C1-C4-alquil C1-C4)-alquilo C1-C4, dialquilamino C1-C4, cicloalquilo C3-C6 y alquiltio C1-C4 o
R6 es B-D-, en donde D es un enlace simple,
B se selecciona del grupo que consiste en:
arilo C6-C10 sustituido con de 0 a 3 grupos seleccionados independientemente del grupo que consiste en halo, alcoxi C1-C4, haloalcoxi C1-C4, alquilo C1-C4, haloalquilo C1-C4, cicloalquilo C3-C6, ciano, nitro, NR11R12, OH, alquilcarboniloxi C1-C4-alquilo C1-C4, hidroxialquilo C1-C4, COOR14, SO2R14, (C=O)NR11R12, SO2NR11R12, N(R13)(C=O)NR11R12, N(R13)(C=O)OR14, N(R13)(C=O)R14, NR13S(O)R14, NR13SO2R14, O(C=O)NR11R12, O(C=O)OR14, O(C=O)R14, (C=O)OR14 y heteroarilo de 5-6 miembros,
heteroarilo de 5 a 10 miembros sustituido con de 0 a 3 grupos seleccionados independientemente del grupo que consiste en halo, alcoxi C1-C4, haloalcoxi C1-C4, alquilo C1-C4, haloalquilo C1-C4, cicloalquilo C3-C6, ciano, nitro, NR11R12, OH, alquilcarboniloxi C1-C4-alquilo C1-C4, hidroxialquilo C1-C4, COOR14, SO2R14, (C=O)NR11R12, SO2NR11R12, N(R13)(C=O)NR11R12, N(R13)(C=O)OR14, N(R13)(C=O)R14, NR13S(O)R14, NR13SO2R14, O(C=O)NR11R12, O(C=O)OR14, O(C=O)R14, (C=O)OR14, heteroarilo de 5-6 miembros y (CH2)fenilo,
heterociclilo de 4 a 10 miembros que contiene átomos de carbono y de 1 a 2 heteroátomos adicionales seleccionados de N, O y S y sustituido con de 0 a 3 grupos seleccionados independientemente del grupo que consiste en halo, oxo, -(CHR13)n1-heteroarilo de 5 o 6 miembros sustituido con de 0 a 3 grupos seleccionados independientemente del grupo que consiste en halo, alcoxi C1-C4, alquilo C1-C4, ciclopropilo, CF3, OCF3 y CF2CH3; NR13S(O)R14, NR13SO2R14, -(CHR13)n1-heterociclilo de 4 a 10 miembros sustituido con de 0 a 3 grupos seleccionados independientemente del grupo que consiste en halo, alcoxi C1-C4, alquilo C1-C4, ciclopropilo, CF3, OCF3 y CF2CH3; OH, hidroxialquilo C1-C4, alcoxi C1-C4, haloalcoxi C1-C4, dialquilamino C1-C4-alquilo C1-C4, NR11 R12 , ciano, alquilo C1-C4, haloalquilo C1-C4, cicloalquilo C3-C6, alcoxi C1-C4-alquilo C1-C4, cicloalquil C3-C6-alquilcarbonilo C1-C4, arilcarbonilo C6-C10, alquilcarboniloxi C1-C4-alquilo C1-C4, COOR14, SO2R14, (C=O)NR11R12, SO2NR11R12, N(R13)(C=O)NR11R12, N(R13)(C=O)OR14, N(R13)(C=O)R14, O(C=O)NR11R12, O(C=O)OR14, O(C=O)R14, (C=O)OR14 y arilo C6-C10 sustituido con de 0 a 3 grupos seleccionados independientemente del grupo que consiste en halo, alcoxi C1-C4, alquilo C1-C4, ciclopropilo, alcoxicarbonilo C1-C4, (C=O)NR11R12, CF3, OCF3 y CF2CH3 y
cicloalquilo C3-C6 que puede contener insaturación, sustituido con de 0 a 3 grupos seleccionados independientemente del grupo que consiste en halo, CF3, OCF3, heteroarilo de 5 o 6 miembros, OH, oxo, hidroxialquilo C1-C4, arilo C6-C10, COOH, oxo, alcoxicarbonilo C1-C4, (C=O)NR11R12 y alquilo C1-C4;
R11 y R12 se seleccionan, en cada caso, independientemente del grupo que consiste en:
H,
alquilo C1-C4,
haloalquilo C1-C4,
dialquilamino C1-C4-alquilo C1-C4,
dialcoxi C1-C4-alquilo C1-C4,
hidroxialquilo C1-C4,
cianoalquilo C1-C4,
alcoxi C1-C4-alquilo C1-C4,
alcoxicarbonil C1-C4-alquilo C1-C4 y
alcoxicarbonilo C1-C4;
de manera alternativa, R11 y R12, cuando se unen al mismo nitrógeno, se combinan para formar un anillo heterocíclico de 4 a 7 miembros que contiene átomos de carbono sustituidos con de 0 a 2 grupos seleccionados independientemente del grupo que consiste en halo, CF3, CHF2 , OCF3, OCHF2 , OCH2F, OH, oxo, hidroxialquilo C1-C2 , alquilo C1-C3 y alcoxi C1-C3 y de 0 a 2 heteroátomos adicionales seleccionados de N, NR13, O y S(O)p; R13 se selecciona, en cada caso, independientemente del grupo que consiste en H y alquilo C1-C3;
R14 se selecciona, en cada caso, independientemente del grupo que consiste en H y alquilo C1-C3
R7 se selecciona del grupo que consiste en H, flúor, cloro, alquilo C1-C3, hidroxialquilo C1-C3 y haloalquilo C1-C2 , que contiene de 1 a 5 halógenos;
n1, en cada caso, se selecciona de 0, 1, 2 o 3 y
p, en cada caso, se selecciona entre 0 , 1 y 2.
4. Un compuesto de la reivindicación 1, que tiene la fórmula:

6. Un compuesto de la reivindicación 1 que tiene la fórmula:

7. Un compuesto de la reivindicación 1 que tiene la fórmula:

8. Una composición farmacéutica, que comprende un vehículo farmacéuticamente aceptable y un compuesto tal como se define en cualquiera de las reivindicaciones 1 a 3 o estereoisómeros, tautómeros, sales o solvatos farmacéuticamente aceptables del mismo, solo o en combinación con otro agente terapéutico.
9. Una composición farmacéutica, que comprende un vehículo farmacéuticamente aceptable y un compuesto como se define en cualquiera de las reivindicaciones 4 a 7, solo o en combinación con otro agente terapéutico.
10. Un compuesto como se define en cualquiera de las reivindicaciones 1 a 3 o estereoisómeros, tautómeros, sales o solvatos farmacéuticamente aceptables del mismo, para su uso en terapia.
11. Un compuesto como se define cualquiera de las reivindicaciones 1 a 3, o estereoisómeros, tautómeros, sales o solvatos farmacéuticamente aceptables del mismo, para su uso en el tratamiento de un trastorno tromboembólico o para la profilaxis primaria o secundaria de un trastorno tromboembólico.
12. Un compuesto para su uso como se define en la reivindicación 11, en donde el trastorno tromboembólico se selecciona entre el grupo que consiste en trastornos tromboembólicos cardiovasculares arteriales, trastornos tromboembólicos cardiovasculares venosos, trastornos tromboembólicos cerebrovasculares y trastornos tromboembólicos en las cavidades cardíacas o en la circulación periférica.13
13. Un compuesto como se define en cualquiera de las reivindicaciones 4 a 7 para su uso en terapia.
14. Un compuesto como se define en cualquiera de las reivindicaciones 4 a 7 para su uso en el tratamiento de un trastorno tromboembólico o la profilaxis primaria o secundaria de un trastorno tromboembólico.
15. Un compuesto para su uso como se define en la reivindicación 14, en donde el trastorno tromboembólico se selecciona del grupo que consiste en trastornos tromboembólicos cardiovasculares arteriales, trastornos tromboembólicos cardiovasculares venosos, trastornos tromboembólicos cerebrovasculares y trastornos tromboembólicos en las cavidades cardíacas o en la circulación periférica.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201261638577P | 2012-04-26 | 2012-04-26 | |
| US201361787680P | 2013-03-15 | 2013-03-15 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2770026T3 true ES2770026T3 (es) | 2020-06-30 |
Family
ID=48407806
Family Applications (3)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES19192142T Active ES2935173T3 (es) | 2012-04-26 | 2013-04-24 | Composiciones farmacéuticas que comprenden derivados de imidazotiadiazol e imidazopiridazina como inhibidores del receptor activado por proteasa 4 (PAR4) para tratar la agregación plaquetaria |
| ES17172927T Active ES2770026T3 (es) | 2012-04-26 | 2013-04-24 | Derivados de imidazotiazol e imidazopirazina como inhibidores del receptor activado por proteasa 4 (PAR4) para tratar la agregación plaquetaria |
| ES13721857.4T Active ES2640916T3 (es) | 2012-04-26 | 2013-04-24 | Derivados de imidazotiadiazol e imidazopirazina como inhibidores del receptor 4 activados por proteasa (PAR4) para tratar agregación plaquetaria |
Family Applications Before (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES19192142T Active ES2935173T3 (es) | 2012-04-26 | 2013-04-24 | Composiciones farmacéuticas que comprenden derivados de imidazotiadiazol e imidazopiridazina como inhibidores del receptor activado por proteasa 4 (PAR4) para tratar la agregación plaquetaria |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES13721857.4T Active ES2640916T3 (es) | 2012-04-26 | 2013-04-24 | Derivados de imidazotiadiazol e imidazopirazina como inhibidores del receptor 4 activados por proteasa (PAR4) para tratar agregación plaquetaria |
Country Status (36)
| Country | Link |
|---|---|
| US (6) | US9688695B2 (es) |
| EP (3) | EP3632919B1 (es) |
| JP (1) | JP6073464B2 (es) |
| KR (3) | KR20200127273A (es) |
| CN (2) | CN108383858B (es) |
| AR (1) | AR090834A1 (es) |
| AU (1) | AU2013251632B2 (es) |
| BR (1) | BR112014026643B1 (es) |
| CA (1) | CA2871650C (es) |
| CL (1) | CL2014002915A1 (es) |
| CO (1) | CO7160040A2 (es) |
| CY (2) | CY1119466T1 (es) |
| DK (3) | DK2841437T3 (es) |
| EA (1) | EA026724B1 (es) |
| ES (3) | ES2935173T3 (es) |
| FI (1) | FI3632919T3 (es) |
| HR (3) | HRP20221495T1 (es) |
| HU (3) | HUE034566T2 (es) |
| IL (1) | IL235246B (es) |
| LT (2) | LT3632919T (es) |
| MX (1) | MX354175B (es) |
| MY (1) | MY175061A (es) |
| NZ (1) | NZ631027A (es) |
| PE (1) | PE20142285A1 (es) |
| PH (1) | PH12014502173B1 (es) |
| PL (3) | PL3632919T3 (es) |
| PT (3) | PT3632919T (es) |
| RS (3) | RS59882B1 (es) |
| SG (1) | SG11201406757SA (es) |
| SI (3) | SI2841437T1 (es) |
| SM (3) | SMT202300004T1 (es) |
| TN (1) | TN2014000436A1 (es) |
| TW (1) | TWI572605B (es) |
| UY (1) | UY34763A (es) |
| WO (1) | WO2013163279A1 (es) |
| ZA (1) | ZA201407799B (es) |
Families Citing this family (43)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2855489B1 (en) | 2012-04-26 | 2017-01-04 | Bristol-Myers Squibb Company | Imidazothiadiazole and imidazopyridazine derivatives as protease activated receptor 4 (par4) inhibitors for treating platelet aggregation |
| SG11201406759YA (en) * | 2012-04-26 | 2014-11-27 | Bristol Myers Squibb Co | Imidazothiadiazole derivatives as protease activated receptor 4 (par4) inhibitors for treating platelet aggregation |
| SI2841437T1 (sl) * | 2012-04-26 | 2017-09-29 | Bristol-Myers Squibb Company | Derivati imidazotiadiazola in imidazopirazina kot proteazno aktivirani receptor 4 (PAR4) inhibitorji za zdravljenje agregacije trombocitov |
| TWI568722B (zh) | 2012-06-15 | 2017-02-01 | 葛蘭馬克製藥公司 | 作爲mPGES-1抑制劑之三唑酮化合物 |
| ES2649156T3 (es) | 2013-01-14 | 2018-01-10 | Incyte Holdings Corporation | Compuestos bicíclicos de carboxamida aromática útiles como inhibidores de quinasas Pim |
| PE20191245A1 (es) | 2013-01-15 | 2019-09-18 | Incyte Holdings Corp | Compuestos de tiazolcarboxamidas y piridinacarboxamida utiles como inhibidores de quinasa pim |
| EA201690458A1 (ru) | 2013-08-23 | 2016-07-29 | Инсайт Корпорейшн | Фуро- и тиенопиридинкарбоксамиды, используемые в качестве ингибиторов pim-киназы |
| US9598419B1 (en) | 2014-06-24 | 2017-03-21 | Universite De Montreal | Imidazotriazine and imidazodiazine compounds |
| US9617279B1 (en) | 2014-06-24 | 2017-04-11 | Bristol-Myers Squibb Company | Imidazooxadiazole compounds |
| US9822124B2 (en) | 2014-07-14 | 2017-11-21 | Incyte Corporation | Bicyclic heteroaromatic carboxamide compounds useful as Pim kinase inhibitors |
| US9580418B2 (en) | 2014-07-14 | 2017-02-28 | Incyte Corporation | Bicyclic aromatic carboxamide compounds useful as Pim kinase inhibitors |
| WO2016134450A1 (en) | 2015-02-26 | 2016-09-01 | Université de Montréal | Imidazopyridazine and imidazothiadiazole compounds |
| EP3262052A1 (en) | 2015-02-26 | 2018-01-03 | Bristol-Myers Squibb Company | Benzothiazole and benzothiophne compounds |
| WO2016196244A1 (en) | 2015-05-29 | 2016-12-08 | Incyte Corporation | Pyridineamine compounds useful as pim kinase inhibitors |
| US9789087B2 (en) | 2015-08-03 | 2017-10-17 | Thomas Jefferson University | PAR4 inhibitor therapy for patients with PAR4 polymorphism |
| TWI734699B (zh) | 2015-09-09 | 2021-08-01 | 美商英塞特公司 | Pim激酶抑制劑之鹽 |
| US9920032B2 (en) | 2015-10-02 | 2018-03-20 | Incyte Corporation | Heterocyclic compounds useful as pim kinase inhibitors |
| EP3371196B1 (en) | 2015-10-19 | 2021-10-27 | Université de Montréal | Heterocyclic compounds as inhibitors of platelet aggregation |
| US9963466B2 (en) | 2016-03-07 | 2018-05-08 | Vanderbilt University | Substituted 5-membered heterocyclic analogs as protease activated receptor 4 (PAR-4) antagonists |
| WO2017184520A1 (en) * | 2016-04-18 | 2017-10-26 | Vanderbilt University | Substituted and fused 6-membered protease activated receptor 4 (par-4) antagonists |
| EP3484878B1 (en) | 2016-07-14 | 2020-08-19 | Bristol-Myers Squibb Company | Bicyclic heteroaryl substituted compounds |
| CN109689664B (zh) | 2016-07-14 | 2022-04-15 | 百时美施贵宝公司 | 作为par4抑制剂的三环杂芳基取代的喹啉和氮杂喹啉化合物 |
| CN109689642B (zh) | 2016-07-14 | 2022-04-12 | 百时美施贵宝公司 | 经单环杂芳基取代的化合物 |
| KR102494647B1 (ko) * | 2016-07-14 | 2023-01-31 | 브리스톨-마이어스 스큅 컴퍼니 | 비시클릭 헤테로아릴 치환된 화합물 |
| CN108239083B (zh) | 2016-12-26 | 2021-08-17 | 阿里根公司 | 芳香烃受体调节剂 |
| EP3713937A2 (en) | 2017-11-20 | 2020-09-30 | Ariagen, Inc. | Indole compounds as aryl hydrocarbon receptor (ahr) modulators |
| WO2019113487A1 (en) | 2017-12-08 | 2019-06-13 | Incyte Corporation | Low dose combination therapy for treatment of myeloproliferative neoplasms |
| CN111094300B (zh) * | 2018-01-31 | 2022-09-16 | 江苏恒瑞医药股份有限公司 | 苯并杂芳基类衍生物、其制备方法及其在医药上的应用 |
| CN110218218B (zh) * | 2018-03-01 | 2022-04-08 | 江苏恒瑞医药股份有限公司 | 苯并呋喃类衍生物、其制备方法及其在医药上的应用 |
| CN110272436A (zh) * | 2018-03-14 | 2019-09-24 | 中国药科大学 | 氘代的咪唑并噻二唑衍生物及其医药用途 |
| CN112074522B (zh) * | 2018-05-16 | 2022-08-26 | 深圳信立泰药业股份有限公司 | 作为用于治疗血小板聚集的蛋白酶激活受体4(par4)抑制剂的化合物 |
| WO2019218956A1 (zh) * | 2018-05-16 | 2019-11-21 | 深圳信立泰药业股份有限公司 | 作为用于治疗血小板聚集的蛋白酶激活受体4(par4)抑制剂的化合物 |
| CN110627817B (zh) * | 2018-06-25 | 2022-03-29 | 中国药科大学 | 咪唑并环类par4拮抗剂及其医药用途 |
| JPWO2020045638A1 (ja) * | 2018-08-30 | 2021-08-12 | 日本メジフィジックス株式会社 | 放射性イミダゾチアジアゾール誘導体化合物 |
| US20230054194A1 (en) | 2018-11-19 | 2023-02-23 | Ariagen, Inc. | Methods of Treating Cancer |
| HRP20231527T1 (hr) * | 2018-12-21 | 2024-04-26 | Bristol-Myers Squibb Company | Kristalni oblici par4 inhibitora |
| CN111349105B (zh) * | 2018-12-24 | 2023-04-07 | 江苏恒瑞医药股份有限公司 | 苯并呋喃类衍生物、其制备方法及其在医药上的应用 |
| CN111362970A (zh) * | 2018-12-25 | 2020-07-03 | 天津药物研究院有限公司 | 苯并呋喃类衍生物及其制备方法和用途 |
| IL287177B2 (en) | 2019-04-15 | 2026-04-01 | Ariagen Inc | Indole-based chiral thiazole derivatives and their use |
| EP3782702A1 (en) * | 2019-08-21 | 2021-02-24 | AC BioScience SA | Compounds and use thereof for the treatment of infectious diseases and cancer |
| CN112079791A (zh) * | 2020-08-21 | 2020-12-15 | 宁夏农林科学院农业资源与环境研究所(宁夏土壤与植物营养重点实验室) | 单环β-内酰胺类抗生素侧链酸及其酯、其制备方法和应用 |
| CN113150005B (zh) * | 2021-04-09 | 2022-08-30 | 中国药科大学 | 喹喔啉类化合物、制备方法及其在医药上的应用 |
| US12077607B2 (en) | 2021-09-23 | 2024-09-03 | University Of Utah Research Foundation | Functional and therapeutic effects of PAR4 cleavage by cathepsin G |
Family Cites Families (61)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE2823686A1 (de) * | 1978-05-31 | 1979-12-06 | Bayer Ag | Imidazo eckige klammer auf 2.1-b eckige klammer zu eckige klammer auf 1.3.4 eckige klammer zu -thiadiazole |
| US4444770A (en) | 1980-05-29 | 1984-04-24 | Bayer Aktiengesellschaft | New imidazoazole-alkenoic acid amide compounds, intermediate products for their production, their production, and their medicinal use |
| DE3020421A1 (de) | 1980-05-29 | 1981-12-10 | Bayer Ag, 5090 Leverkusen | Imidazoazolalkensaeureamide, neue zwischenprodukte zu ihrer herstellung, ihre herstellung und ihre verwendung in arzneimitteln |
| DE3446778A1 (de) | 1984-12-21 | 1986-07-03 | Dr. Karl Thomae Gmbh, 7950 Biberach | Neue imidazoderivate, ihre herstellung und diese verbindungen enthaltende arzneimittel |
| US4925849A (en) | 1987-06-15 | 1990-05-15 | Fujisawa Pharmaceutical Company, Ltd. | Pharmaceutically useful pyrazolopyridines |
| GB8901423D0 (en) | 1989-01-23 | 1989-03-15 | Fujisawa Pharmaceutical Co | Pyrazolopyridine compound and processes for preparation thereof |
| DE69232323T2 (de) | 1991-01-29 | 2002-08-08 | Fujisawa Pharmaceutical Co., Ltd. | Verwendung von Adenosinantagonisten zur Vorbeugung und Behandlung von Pankreatitis und Ulcera |
| US5342851A (en) * | 1992-10-07 | 1994-08-30 | Mcneil-Ppc, Inc. | Substituted thiazole derivatives useful as platelet aggregation inhibitors |
| JP2928079B2 (ja) | 1994-02-14 | 1999-07-28 | 永信薬品工業股▲ふん▼有限公司 | 1−(置換ベンジル)−3−(置換アリール)縮合ピラゾール類、その製造法及びその用途 |
| ATE451346T1 (de) | 1998-03-10 | 2009-12-15 | Ono Pharmaceutical Co | Carbonsäurederivate und medikamente die diese als aktiven wirkstoff enthalten |
| PL355019A1 (en) | 1999-10-08 | 2004-03-22 | Grunenthal Gmbh | Bicyclic imidazo-5-yl-amine derivatives |
| DE19948434A1 (de) | 1999-10-08 | 2001-06-07 | Gruenenthal Gmbh | Substanzbibliothek enthaltend bicyclische Imidazo-5-amine und/oder bicyclische Imidazo-3-amine |
| DE10019714A1 (de) | 2000-04-20 | 2002-01-10 | Gruenenthal Gmbh | Salze von bicyclischen, N-acylierten Imidazo-3-aminen und Imidazo-5-aminen |
| US6387942B2 (en) | 2000-06-19 | 2002-05-14 | Yung Shin Pharmaceutical Ind. Co. Ltd | Method of treating disorders related to protease-activated receptors-induced cell activation |
| EP1442028A4 (en) | 2001-11-06 | 2009-11-04 | Bristol Myers Squibb Co | SUBSTITUTED ACID DERIVATIVES, WHICH APPRECIATE AS ANTIDIBILICS AND AGENTS AGAINST OBESITAS, AND METHODS |
| CA2364985A1 (en) | 2001-12-14 | 2003-06-14 | John W. Gillard | Imidazo(2,1-b)thiadiazole sulfonamides |
| EP1582516B1 (en) | 2003-01-10 | 2013-07-17 | Idemitsu Kosan Co., Ltd. | Nitrogenous heterocyclic derivative and organic electroluminescent element employing the same |
| CA2527906A1 (en) | 2003-06-13 | 2004-12-23 | Aegera Therapeutics, Inc. | Acylated imidazo[2,1-b]-1,3,4,-thiadiazole-2-sulfonamides and uses thereof |
| US20060135571A1 (en) | 2003-06-13 | 2006-06-22 | Jaquith James B | Imidazo(2,1-b)-1,3,4-thiadiazole sulfoxides and sulfones |
| JP2007527416A (ja) | 2003-10-31 | 2007-09-27 | ジヤンセン・フアーマシユーチカ・ナームローゼ・フエンノートシヤツプ | ペルオキシソーム増殖因子活性化受容体(ppar)二重作動薬として有用なフェノキシ酢酸誘導体 |
| EP1684762A4 (en) | 2003-11-13 | 2009-06-17 | Ambit Biosciences Corp | UREA DERIVATIVES AS MODULATORS OF KINASE |
| EP1713784A1 (en) | 2004-02-12 | 2006-10-25 | Neurogen Corporation | Imidazo-pyridazines, triazolo-pyridazines and related benzodiazepine receptor ligands |
| EP1867331A4 (en) | 2005-04-06 | 2009-04-08 | Takeda Pharmaceutical | TRIAZOLE DERIVATIVE AND USE THEREOF |
| JP2008546804A (ja) | 2005-06-24 | 2008-12-25 | ザ・トラスティーズ・オブ・ザ・ユニバーシティ・オブ・ペンシルバニア | 造影剤として用いるためのリガンドの放射性標識ペグ化 |
| CA2624102A1 (en) | 2005-09-29 | 2007-04-12 | Sanofi-Aventis | Phenyl- and pyridinyl- 1, 2 , 4 - oxadiazolone derivatives, processes for their preparation and their use as pharmaceuticals |
| NL2000397C2 (nl) | 2006-01-05 | 2007-10-30 | Pfizer Prod Inc | Bicyclische heteroarylverbindingen als PDE10 inhibitoren. |
| MX2008011615A (es) | 2006-03-14 | 2008-09-22 | Amgen Inc | Derivados de acidos carboxilicos biciclicos utiles para tratar trastornos metabolicos. |
| US20090170845A1 (en) | 2006-04-13 | 2009-07-02 | Aegera Therapeutics Inc. | USE OF IMIDAZO[2,1-b]-1,3,4-THIADIAZOLE-2-SULFONAMIDE COMPOUNDS TO TREAT NEUROPATHIC PAIN |
| DE102006054757A1 (de) | 2006-11-21 | 2008-05-29 | Bayer Healthcare Ag | Neue aza-bicyclische Verbindungen und ihre Verwendung |
| US7638541B2 (en) | 2006-12-28 | 2009-12-29 | Metabolex Inc. | 5-ethyl-2-{4-[4-(4-tetrazol-1-yl-phenoxymethyl)-thiazol-2-yl]-piperidin-1-yl}-pyrimidine |
| EP1964841A1 (en) | 2007-02-28 | 2008-09-03 | sanofi-aventis | Imidazo[1,2-a]azine and their use as pharmaceuticals |
| ES2395583T3 (es) | 2007-05-10 | 2013-02-13 | Ge Healthcare Limited | IMIDAZOL (1,2-A)PIRIDINAS y compuestos relacionados con actividad frente a los receptores cannabinoides CB2 |
| WO2009017954A1 (en) | 2007-08-01 | 2009-02-05 | Phenomix Corporation | Inhibitors of jak2 kinase |
| CA2695989A1 (en) | 2007-08-10 | 2009-02-19 | Glaxosmithkline Llc | Certain nitrogen containing bicyclic chemical entities for treating viral infections |
| WO2009027733A1 (en) | 2007-08-24 | 2009-03-05 | Astrazeneca Ab | (2-pyridin-3-ylimidazo[1,2-b]pyridazin-6-yl) urea derivatives as antibacterial agents |
| GB0718735D0 (en) | 2007-09-25 | 2007-11-07 | Prolysis Ltd | Antibacterial agents |
| JP2011506487A (ja) | 2007-12-21 | 2011-03-03 | ザ・ユニバーシティ・オブ・シドニー | トランスロケータータンパク質リガンド |
| AU2009231906A1 (en) | 2008-03-31 | 2009-10-08 | Metabolex, Inc. | Oxymethylene aryl compounds and uses thereof |
| MX2011000211A (es) | 2008-07-15 | 2011-03-03 | Sanofi Aventis | Oxazolopirimidinas como agonistas del receptor edg-1. |
| US9006252B2 (en) | 2008-09-26 | 2015-04-14 | National Health Research Institutes | Fused multicyclic compounds as protein kinase inhibitors |
| SG172352A1 (en) | 2008-12-23 | 2011-07-28 | Abbott Lab | Anti-viral compounds |
| WO2011074658A1 (ja) * | 2009-12-18 | 2011-06-23 | 田辺三菱製薬株式会社 | 新規抗血小板薬 |
| CN102372701A (zh) | 2010-08-10 | 2012-03-14 | 江苏恒瑞医药股份有限公司 | 氮杂双环己烷类衍生物、其制备方法及其在医药上的应用 |
| TW201217365A (en) | 2010-08-11 | 2012-05-01 | Millennium Pharm Inc | Heteroaryls and uses thereof |
| CN103874496A (zh) | 2011-08-22 | 2014-06-18 | 默沙东公司 | 作为bace抑制剂的2-螺-取代的亚氨基噻嗪类及其单和二氧化物、组合物及其用途 |
| SI2841437T1 (sl) * | 2012-04-26 | 2017-09-29 | Bristol-Myers Squibb Company | Derivati imidazotiadiazola in imidazopirazina kot proteazno aktivirani receptor 4 (PAR4) inhibitorji za zdravljenje agregacije trombocitov |
| EP2855489B1 (en) * | 2012-04-26 | 2017-01-04 | Bristol-Myers Squibb Company | Imidazothiadiazole and imidazopyridazine derivatives as protease activated receptor 4 (par4) inhibitors for treating platelet aggregation |
| CA2871640A1 (en) | 2012-04-26 | 2013-10-31 | Bristol-Myers Squibb Company | Par4 agonist peptides |
| SG11201406759YA (en) * | 2012-04-26 | 2014-11-27 | Bristol Myers Squibb Co | Imidazothiadiazole derivatives as protease activated receptor 4 (par4) inhibitors for treating platelet aggregation |
| CA2870336A1 (en) | 2012-04-27 | 2013-10-31 | Novartis Ag | Cyclic bridgehead ether dgat1 inhibitors |
| PE20161416A1 (es) | 2012-05-15 | 2017-01-08 | Novartis Ag | Derivados de benzamida para la inhibicion de la actividad abl1, abl2 y bcr-abl1 |
| WO2013177188A1 (en) | 2012-05-22 | 2013-11-28 | Idenix Pharmaceuticals, Inc. | 3',5'-cyclic phosphoramidate prodrugs for hcv infection |
| TWI568722B (zh) | 2012-06-15 | 2017-02-01 | 葛蘭馬克製藥公司 | 作爲mPGES-1抑制劑之三唑酮化合物 |
| PE20150863A1 (es) | 2012-06-21 | 2015-06-11 | Univ Indiana Res & Tech Corp | Analogos de glucagon que exhiben actividad de receptor de gip |
| TW201408652A (zh) | 2012-07-11 | 2014-03-01 | Hoffmann La Roche | 作爲RORc調節劑之芳基磺內醯胺衍生物 |
| TW201414737A (zh) | 2012-07-13 | 2014-04-16 | 必治妥美雅史谷比公司 | 作爲激酶抑制劑之咪唑并三□甲腈 |
| US9605002B2 (en) | 2012-07-18 | 2017-03-28 | University Of Notre Dame Du Lac | 5,5-heteroaromatic anti-infective compounds |
| TW201422592A (zh) | 2012-08-27 | 2014-06-16 | Boehringer Ingelheim Int | β-分泌酶抑制劑 |
| MY172021A (en) | 2012-10-16 | 2019-11-12 | Janssen Sciences Ireland Uc | Rsv antiviral compounds |
| US9630945B2 (en) | 2012-12-19 | 2017-04-25 | Novartis Ag | Autotaxin inhibitors |
| EA030003B1 (ru) | 2012-12-21 | 2018-06-29 | Джилид Сайэнс, Инк. | Полициклическое карбамоилпиридоновое соединение и его фармацевтическое применение для лечения вич-инфекции |
-
2013
- 2013-04-24 SI SI201330734T patent/SI2841437T1/sl unknown
- 2013-04-24 PE PE2014001782A patent/PE20142285A1/es active IP Right Grant
- 2013-04-24 RS RS20191664A patent/RS59882B1/sr unknown
- 2013-04-24 BR BR112014026643-3A patent/BR112014026643B1/pt active IP Right Grant
- 2013-04-24 RS RS20230041A patent/RS63975B1/sr unknown
- 2013-04-24 KR KR1020207031590A patent/KR20200127273A/ko not_active Ceased
- 2013-04-24 KR KR1020147030174A patent/KR102098804B1/ko active Active
- 2013-04-24 RS RS20170983A patent/RS56395B1/sr unknown
- 2013-04-24 FI FIEP19192142.8T patent/FI3632919T3/fi active
- 2013-04-24 EP EP19192142.8A patent/EP3632919B1/en active Active
- 2013-04-24 KR KR1020207009636A patent/KR102176379B1/ko active Active
- 2013-04-24 ES ES19192142T patent/ES2935173T3/es active Active
- 2013-04-24 NZ NZ631027A patent/NZ631027A/en unknown
- 2013-04-24 SM SM20230004T patent/SMT202300004T1/it unknown
- 2013-04-24 HU HUE13721857A patent/HUE034566T2/hu unknown
- 2013-04-24 WO PCT/US2013/037956 patent/WO2013163279A1/en not_active Ceased
- 2013-04-24 EA EA201491968A patent/EA026724B1/ru not_active IP Right Cessation
- 2013-04-24 TW TW102114695A patent/TWI572605B/zh active
- 2013-04-24 AR ARP130101380A patent/AR090834A1/es active IP Right Grant
- 2013-04-24 CA CA2871650A patent/CA2871650C/en active Active
- 2013-04-24 PL PL19192142.8T patent/PL3632919T3/pl unknown
- 2013-04-24 PL PL17172927T patent/PL3243826T3/pl unknown
- 2013-04-24 DK DK13721857.4T patent/DK2841437T3/en active
- 2013-04-24 UY UY0001034763A patent/UY34763A/es unknown
- 2013-04-24 MY MYPI2014703169A patent/MY175061A/en unknown
- 2013-04-24 AU AU2013251632A patent/AU2013251632B2/en active Active
- 2013-04-24 HU HUE17172927A patent/HUE048654T2/hu unknown
- 2013-04-24 MX MX2014012296A patent/MX354175B/es active IP Right Grant
- 2013-04-24 CN CN201810251394.6A patent/CN108383858B/zh active Active
- 2013-04-24 HU HUE19192142A patent/HUE060743T2/hu unknown
- 2013-04-24 EP EP17172927.0A patent/EP3243826B1/en active Active
- 2013-04-24 SI SI201331625T patent/SI3243826T1/sl unknown
- 2013-04-24 SG SG11201406757SA patent/SG11201406757SA/en unknown
- 2013-04-24 HR HRP20221495TT patent/HRP20221495T1/hr unknown
- 2013-04-24 PT PT191921428T patent/PT3632919T/pt unknown
- 2013-04-24 LT LTEP19192142.8T patent/LT3632919T/lt unknown
- 2013-04-24 EP EP13721857.4A patent/EP2841437B1/en active Active
- 2013-04-24 HR HRP20171453TT patent/HRP20171453T1/hr unknown
- 2013-04-24 DK DK19192142.8T patent/DK3632919T3/da active
- 2013-04-24 LT LTEP13721857.4T patent/LT2841437T/lt unknown
- 2013-04-24 US US14/396,771 patent/US9688695B2/en active Active
- 2013-04-24 SM SM20170457T patent/SMT201700457T1/it unknown
- 2013-04-24 ES ES17172927T patent/ES2770026T3/es active Active
- 2013-04-24 PT PT137218574T patent/PT2841437T/pt unknown
- 2013-04-24 CN CN201380032304.XA patent/CN104583218B/zh active Active
- 2013-04-24 JP JP2015509103A patent/JP6073464B2/ja active Active
- 2013-04-24 PT PT171729270T patent/PT3243826T/pt unknown
- 2013-04-24 PL PL13721857T patent/PL2841437T3/pl unknown
- 2013-04-24 SM SM20200049T patent/SMT202000049T1/it unknown
- 2013-04-24 DK DK17172927.0T patent/DK3243826T3/da active
- 2013-04-24 SI SI201332026T patent/SI3632919T1/sl unknown
- 2013-04-24 ES ES13721857.4T patent/ES2640916T3/es active Active
-
2014
- 2014-09-29 PH PH12014502173A patent/PH12014502173B1/en unknown
- 2014-10-20 TN TN2014000436A patent/TN2014000436A1/fr unknown
- 2014-10-21 CO CO14232423A patent/CO7160040A2/es unknown
- 2014-10-21 IL IL235246A patent/IL235246B/en active IP Right Grant
- 2014-10-24 ZA ZA2014/07799A patent/ZA201407799B/en unknown
- 2014-10-27 CL CL2014002915A patent/CL2014002915A1/es unknown
-
2017
- 2017-05-12 US US15/593,534 patent/US10047103B2/en active Active
- 2017-09-29 CY CY20171101023T patent/CY1119466T1/el unknown
-
2018
- 2018-06-27 US US16/020,043 patent/US10428077B2/en active Active
-
2019
- 2019-08-13 US US16/538,917 patent/US10822343B2/en active Active
- 2019-12-30 HR HRP20192336TT patent/HRP20192336T1/hr unknown
-
2020
- 2020-01-24 CY CY20201100069T patent/CY1122625T1/el unknown
-
2022
- 2022-09-02 US US17/902,618 patent/US12084452B2/en active Active
-
2024
- 2024-09-06 US US18/826,736 patent/US20250002497A1/en active Pending
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2770026T3 (es) | Derivados de imidazotiazol e imidazopirazina como inhibidores del receptor activado por proteasa 4 (PAR4) para tratar la agregación plaquetaria | |
| US9518064B2 (en) | Imidazothiadiazole and imidazopyridazine derivatives as protease activated receptor 4 (PAR4) inhibitors for treating platelet aggregation | |
| HK1203953B (en) | Imidazothiadiazole and imidazopyrazine derivatives as protease activated receptor 4 (par4) inhibitors for treating platelet aggregation |